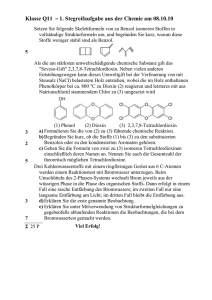
Untersuchungen an Kobalt (II)-und Kobalt (III)
Werbung
-und Kobalt (III)")
Diss. Nr. 4097 Untersuchungen an Kobalt(II)- und Kobalt(III)-Komplexen des Cobyrinsäure-heptamethylesters ABHANDLUNG zur Erlangung der Würde eines Doktors der technischen Wissenschaften der EIDGENÖSSISCHEN TECHNISCHEN HOCHSCHULE ZÜRICH vorgelegt von LUCIUS WERTHEMANN dipl. Ing.-Chem. geboren von am Basel zx. ETH Januar 1940 (Kanton Basel) Angenommen auf Antrag Prof. Dr. A. von Eschenmoser, Referent Prof. Dr. D. Arigoni, Korreferent Juris Druck + Verlag Zürich 1968 Leer - Vide - Empty Meinem sehr verehrten Lehrer, Herrn Prof. unter dessen Leitung ich die Dr. A. Eschenmoser, vorliegende Promotionsarbeit ausführen konnte, danke ich für seine unermüdliche Hilfe und die vielen wertvollen Grossen Dank schulde ich auch Herrn Dr. Arbeit stets tischen entgegengebrachtes, Anregungen. R. Keese Ratschläge. für sein meiner förderndes Interesse und die vielen prak¬ Leer - Vide - Empty - 5 - INHALTSVERZEICHNIS 7 Einleitung Theoretischer Teil 9 A. Dicyano~Co(m)-cobyrinsäure-heptamethylester B. Aquo-cyano-Co(III)-cobyrinsäure-heptamethylester C. Chloro- und D. Reduktion E. Dijodo-Co(III)-cobyrinsäure-heptamethylester F. Kobalt(H)-Komplexe Jodo-cyano-Co(IH)-cobyrinsäure-heptamethylester von Co(m)-cobyrinsäure-heptamethylester-Kbmplexen des 9 18 21 25 28 Cobyrinsäure-heptamethylesters (kristalliner Jodo-Co(II)-cobyrinsäure-heptamethylester) 32 G. Alkyl-Co(m)-cobyrinsäure-heptamethylester-Komplexe 48 H. Versuche zur Methylgruppe Experimenteller Deprotonierung der axialen, kobaltgebundenen in Cobyrinsäure-heptamethylester-Komplexen Teil 60 65 Zusammenfassung 112 Literaturverzeichnis 113 Leer - Vide - Empty 7 - - EINLEITUNG 1948 durch Extraktion eine Röntgenstrukturanalyse aufgeklärt, An diesem genheit der führenden, aus von stellt Vitamin B 12 komplexen neuen Molekül Methode Rindsleber erstmals isoliert D.Hodgkin (2) schon historisch zeigte sich gegenüber zum und 1955 durch in seiner Struktur vollständig gesehen einen Sonderfall dar. ersten Mal die grosse Ueberle- den bei dieser herkömmlichen Verfahren einer (1) Verbindung nicht zum Ziel Strukturaufklärung. ,CONH, 2 ,CONH„ R = CN Vitamin B 12 H2NOC CONH„ Cyanocobalamin HO OH hWh HgNOC R CH,CH, *3 ^g l ) = 'CH. HCH3 N NH„ CONH„ 5 ' -Desoxyadenosin CH„ b^No OH\ OH^ TjT N-^^CH • / Coenzym B 12 HOCH. Figur 1 Chemisch stellt Vitamin B 12 den ersten tallkomplex dral mit Kobalt als koordiniert, isolierten, Zentralatom dar. Das wobei die vier in einer Ebene natürlich vorkommenden Me¬ dreiwertige Metall ist oktahe- liegenden Ligandstellen durch das 8 - Corrinsystem kann, Die Kenntnisse über das Vitamin B 12 derum durch kannte ge B 12 durch Coenzyms des das man Vorliegen 12 diol zu Spekulationen abhangigen Reaktionen, Vitamin B 12 von hgandierte Um die in ist möglichst 12, Synthese wenn in Weg der eine unter Umlagerung in der Folge ungeklärten von 1,2-Propan- einerseits mit der den grossen des Vitamins B 12 An dieser Liganden am Verbindung spielt. Komplexes besseren Einblick nur in zu er¬ polaren Losungsmitteln lösli¬ hiesigen Laboratorium (5 und 6) mit am und heptamethyl-cornn-Komplexe von anderseits aber auch zum zu sollen im re¬ jedoch wurde die andere leicht loslichen Modell besitzt Vorteil, mit von Kobalt-bisdimethylglyoxim-Komple- In dieser Arbeit besten ei¬ synthetisch herzustellen. der Abbau des Vitamins B 12 lekulargewicht physiologische wird dennoch angenom¬ Abspaltung des Nucleotidteils und der Ami¬ Darstellung Dieses so Coenzym B 12 gesteuerten Reaktionen das Co(m)-tetra-, penta- (7) kann, die polare Verbindung umzuwandeln, oder aber weniger wurde heptamethylester gewählt. ler er¬ fur die damali¬ immer noch Aminopropanolgruppe entweder das möglich (sog. Cobaloximen) angestrebt. Möglichkeit, sen der niedrigem Molekulargewicht eingeschlagen, G.Schrauzer xen die beeinflusst werden ahnliche Modellsubstanz Letzterer lativ was System stand über die z.B. chemische Reaktivität dieses nopropanolgruppe, der Hodgkin (4) aufgeklart, Kobalt die entscheidende Rolle wünschenswert, es che Vitamin B ne wie dass bei den durch Vitamin oder Corrin halten, D. bekannte B 12 das an Obschon durch Variation z.B. men, nochmals wesentlich erweitert. Wie¬ von Propionaldehyd. Wirksamkeit vom austau¬ Azamethinkette einer Kobalt-Kohlenstoff-Bindung, einer Mittelpunkt mechanistischer Coenzym-B Ko¬ Neuartiges darstellte. Diese lichtempfindliche Bindung etwas 5'-Desoxyadenosylrestes eines Koordinationspartner -System -n vom wurden 1960 durch die Isolierung (3) H. Barker Rontgenstrukturanalyse eine Komplexchemie im mit dem nicht unzerstort Haupttrager des Elektronenspektrums des Moleküls. dreizehn Zentren ist der von Ligand lassen sich die axialen Cornnligand, Der vierzahmge schen. Wahrend dieser besetzt werden. balt getrennt werden - abgesehen von Cobyrinsauredem hohen Mo¬ den sterischen und elektronischen Verhaltnis¬ entsprechen. Folgenden und das Verhalten reduzierter zweiwertigem Metall untersucht werden. die Reaktivität verschiedener axia¬ Kobalt-corrin-Komplexe mit ein- und - 9 - TEIL THEORETISCHER A. Der Dicyano-Co(III)-cobyrinsaure-heptamethylester partielle Abbau Alkohol wurde schon 1964 in Vitamin B 12 durch starke Saure von K. Bernhauer von Salzsaure und Aethanol konnte pan-2-ol Gruppe abgetrennten optimale Voraussetzungen den sollten, gur 2), 1) wurde es fur er auf welches auch die vom eine an Methylester-Derivat der Harvard Gegenwart 1-AminoproDa isolieren. geschaffen darzustellen University USA und von Durch Erhitzen Nucleotidteil und der Kristallisation des Produkts das in beschrieben. Cobyrinsàure-heptaathylester vorgezogen, stitut gemeinsam bearbeitete den (9) am jedoch wer¬ (vgl. Fi¬ hiesigen In¬ Totalsynthese ausgerichtet ist. ,COR. Rj RjOC = R2 = -OCH3 •Cobyrinsàure-heptamethyl- /^-—CORj ester im Folgenden (Cobester) verwendete Kurz formel: R1OC 2 /Co/ -OH R» -NH, "2 »Cobyrsaure (Faktor -NH2 R2 - = V. ) -NHCH2CH(OH)CH3 (D-l -Aminopropan-2-ol) C obinamid Figur 2 1) Die neueste Zusammenfassung der Literatur über Vitamin B 12 umfasst Veröffentlichungen bis August 1965. F.Wagner (8) von - Erne an diesem nach reduktiver zwei Ringe B und C des (10) R. Keese von molarer, Ruckfluss am Reaktionsansatzes, vermieden werden. gewordene violett thylenchlorid der beiden fraktioniert ausschütteln. circa Salzen nur saurem lichem und letzteres zu von Phase dem Ver¬ ein¬ mit festem Methanols Kaliumcyanid dunkel¬ und Me- Vergleich der NMR-Spektren mit Methylenchlorid waren neben den Es ex¬ anorganischen erwies sich als unvollständig umgesetzten einmal wahrend Dadurch konnten zuganglichen von Cyanid und relativ Reihe, kannt Da die negativ war. Vitamin 1) ein eimgen Fallen in sich mit Tetrachlorkohlenstoff B 12 Derivate. separat noch kochen. tamin B 12 des Gasstrom sich die Farbe der Losung festem ein in unter 90% an Tagen zwei gun¬ Material in schwefel¬ Tetrachlorkohlenstoff-los - Heptamethylester erhalten werden. Die Einfuhrung leicht Tagen vier Ueberhitzung und kann, enthalt das % polarere stig, die 70 % Heptaester Produkt Methanol (60) 15 % weniger Methylestergruppen als das mit Tetrachlor¬ noch ungefähr 2 abzutrennen, von Wie durch werden In der wasserigen kohlenstoff erhaltene. lokale Massnahmen Zugabe Reaktionsgemisch hess Auszuge festgestellt trahierte Produkt eine nach dem Entfernen des Das durch nach carminrot. ziegel- P.Loliger wahrend Neutralisation änderte ausgeführten Natnumbicarbonat von Bei der entsprechenden der von Durch den schwachen gekocht. ohne diese was Ma¬ welche die Imide der entwickelten Methode wird Vitamin B 12 und durch den Sauerstoffausschluss konnten trat, Bogard (39) mit Erfolg durch¬ identifiziert werden. Schwefelsaurelosung methanolischer sauerstoffreiem Stickstoff kohlen des . Imide konnten erhaltene des Vitamin B 12 Abbauprodukten Bei der T Ozonolyse Durch Eduktes darstellten. synthetischen Zwischenprodukte 1 von Aufarbeitung neben viel polarem Chromatographie getrennte Substanzen, durch terial ca. - Cobyrmsaure-heptamethylester geführte Ozonolyse lieferte mit den 10 B 12 und dreiwertigen Fur die von axiale intramolekular Vorteil Analogon in eines der Vi¬ spektroskopische Vergleiche be¬ den Cobalammen kompensiert, im eine positive Ladung Cobyrinsaure-hepta- verschiedenen, sich durch Variation der Seitenket¬ ergebenden B 12 Derivate vgl. den Uebersichtsar(11) und Figur 1 und 2. Nukleohdteils R. Bonnet fur dessen geladene Phosphatgruppe des Nucleotidteils, die beim einigen Derivaten z.B. Kobalts Liganden ergab den Komplexes, Dicyanocobinamid Nomenklatur der ten und des tikel das als stabilen - methylester fehlt, axiale ist der 11 - Komplex bei normaler Oxidationsstufe des Kobalts ohne Liganden zweifach positiv geladen. erhalten, ist zufuhren. Diese notwendig, es zwei Um nun negativ geladene einen ungeladenen Komplex axiale Ionen als Liganden Ladungsdifferenz der Modellsubstanz gegenüber Vitamin B 12 und auch gegenüber den Cobaloximen bildet den wesentlichen Unterschied der sen, sich, zu ein¬ wie im Verlauf der Arbeit Alkylderivaten ungunstig auswirkt. offenkundig wurde, Es zeigte sich Cobyrinsaure-heptamethylester-Komplexe nämlich, dass die¬ zu allem bei den vor nur ungeladene kristallisiert und damit auch fur weitere Untersuchungen zuganglich gemacht werden konnten. Der als Schlusselsubstanz dargestellte Dicyano-Co(HI)-cobyrinsaure-heptamethylester ungeladenen Komplex erfüllt, rung nach einem und ist apolaren Losungsmitteln in wie 1)2) ' der die Forde¬ , kann leicht kristallisiert werden Tetrachlorkohlenstoff oder Benzol gut lös¬ lich. Charakterisierung des Dicyano-Co(HI)-cobesters eingegangen Bevor auf die wird, sollen erst einige bei der Anwendung der Schwierigkeiten spektroskopischen Die Struktur und das hohe ten die aus UV/VIS-, Methoden Molekulargewicht den verschiedenen So hatten die und Besonderheiten von angeführt werden, die dieser Arbeit auftraten. in 1036 des Co(HI)-cobesters schrank¬ Spektren erhältlichen Informationen wesentlich die IR- und die NMR-Spektren und beschreibenden Wert und konnten nicht direkt zur meist nur ein. vergleichenden Identifikation eines Produkts herangezogen werden. Die IR-Spektren aller Derivate zeigten cm" Esterbande bei 1730 Streckschwingungen 1) 2) , 1575, 1100 und 1000 cm" 1150, d.h. bei C-H Schwingungen drei charakteristische Banden . 1490 und 1430 Axiale cm sowie im um 2950 Gebiet der cm , die C=C/C=N drei weitere Banden bei Cyanogruppen erscheinen bei ca. 2125 cm" , bei relativ kleinen Wellenzahlen. Folgenden wird Cobyrinsaure-heptamethylester mit "Cobester" abgekürzt. Nomenklaturvorschlag der IUPAC-Konferrenz 1966 (12), gemäss welchem der Name Cobyrinsaure den mit drei Essigsaure- und vier Propionsauregruppen substituierten Cornnliganden und das dreiwertige Kobalt als Zentral¬ atom einschhesst, wird in der vorliegenden Arbeit nicht angewendet. Es hat sich als gunstig erwiesen, das Metall und dessen Wertigkeit im Namen ge¬ trennt anzuführen. Insbesondere in Anbetracht eines kurzlich am hiesigen La¬ Im Der boratorium von der sich daraus passung A.Fischli (13) synthetisierten, ergebenden Entwicklungen scheint gerechtfertigt. metallfreien Corrins und eine solche Nomenklaturan¬ 12 - - IR-Spektrum des Dicyano-Co(in)-cobesters in Chloroform Figur 3 Die Figur NMR-Spektren aller Co(III)-cobester-Derivate 4 und 5 zeigen das zol resp. sind ähnlich Spektrum des Dicyano-Co(m)-cobesters gegliedert. in Deuteroben¬ Deuterochloroform. NMR-Spektrum von Dicyano-Co(m)-cobester 3 Figur 4 2 in Deuterobenzol 13 NMR-Spektrum von Dicyano-Co(m)-cobester 3 4 2 100 Mc/s Spektrum erlaubt 1 ppm 5 Figur Das in Deuterochloroform folgende Zuordnung der Signale oder Sig¬ nalgruppen: Gruppe a: zwischen 0,9 - Methylgruppen Gruppe b: zwischen 1,6 - 1,6 in ppm fünf scharfe - 3,1 ppm zwei - vinylischen Methylgruppen 22 Signale den sechs tertiären 1, 2, 7, 12, 12, 17-Stellung. Singletts bei 2,15 und 2,40 ppm den Stellung in 5 und 15. Das Multiplett allen der Seitenketten und dem tertiären Proton in Methylenprotonen Stellung 18. Gruppe c: zwischen 3,20 estergruppen in Die scharfen ppm. 8, 13, Signale dieser über den 0,6 4,1 und die zwei Stellung 3, Die - Singletts den sieben Methyl¬ Multipletts den vier Protonen bilden zwei entsprechenden im allylischen Protonen für die Gruppen und sind Pentamethyl-Co(m)-corrin (5) ppm nach tieferem Feld verschoben. terierungsversuche vier 19. um gegen¬ ca. Auf Grund der durch Deu- synthetische Verbindung erhaltenen Infor¬ mationen könnten die beiden Signalgruppen folgendermassen zugeord¬ net werden: Das Multiplett grösserer Das bei 3,60 ppm den Protonen 8 und 13 Elektronendichte im Multiplett bei 3,90 ppm den Protonen 3 und 19 kleinerer Elektronendichte im (auf der Seite (auf der Seite Molekül). Molekül). 14 - Gruppe d: Da zur bei 5,67 Abschätzung Das ppm. So thylesterprotonen entsprechenden durch jene der Die musste ist, erwies und der es vier Auswertung ein sich als Vinylproton das Signal des Vinylprotons auch zu klein anderer Abschnitt als interner Standard die Signalgruppe Bestes, wenn c obigen Spektren 21 der Me¬ Protonen Interpre¬ Signale der allylischen Protonen die Methylestergruppen überdeckt diesem Verfahren gibt Stellung 10. Dieses Verfahren wurde bei der Standard anzunehmen. der beiden in allylischen Protonen als internen, 25 Spektren angewandt, tation aller dem Singlett Gesamtintegration der und deshalb unbrauchbar gewählt werden. - (vgl. Figur 5). waren Dicyano-Co(m)-cobesters des nach folgende Resultate: Spektrum Spektrum in Deuterobenzol Theoretisch Deuterochloroform in Signalgruppe a 20 H 20 H 18 H Signalgruppe b 34 H 34 H 29 Die Stellung des Vinylprotons hangt stark den axialen dieses Proton durch eine der von Liganden und dem Losungsmittel ab. fur die axialen Liganden ihrer Stellung man parallelen Einfluss fest. Diese Beziehung gilt Ladung und wird Tabelle 1 wie positive Ladung erwartungsgemass Serie cher Ladung des Komplexes, Wahrend, stellt hier rein einen nur H in Tabelle 1 entschirmt zeigt, wird, so der spektrochemischen innerhalb Komplexen glei¬ phänomenologisch aufgeführt. Stellung des Vinylprotons in Stellung 10 in S (rel. TMS) (in Deutero¬ chloroform) positiv geladene Cobester-Komplexe ungeladene Cobester-Komplexe Dicyano- 5,6 Aquo-cyano- 6,48 Chloro-cyano- 5,95 Methyl- 6,71 Jodo-cyano- 6,14 Jodmethyl-jodo- 6,30 Spektrochemische Serie: I < Br < Cl < F < OH" < H20 « CN 15 - Das zu Elektronenspektrum MO-Berechnungen, von durch so Uebereinstimmend wurde dabei - Vitamin B 12 (16) G.Schrauzer festgestellt, schon verschiedentlich Anlass gab P.Day (17 und spektrums der Corrinligandchromophor ist, auf den die axialen Zentralatom qualitativ So treten erfassbar einwirken. ester-Verbindungen, die Ausnahme bilden banden auf, die ir die, Die entspricht /3-Bande einer 18). axialen in Liganden Co(IH)-cob- den meisten Alkyldenvate, und das drei Haupt¬ -»w*-Uebergangen zugeordnet werden können. Dabei wird dieser Arbeit ««.-Bande die und der Trager des Elektronen¬ dass auch beim m Vitamin B 12 angewandte Terminologie verwendet: 7tf—»8ir-Uebergang, einem die etwas kurzerwellige Schwingungsfeinstruktur dieses Uebergangs und die y -Bande dem energiereicheren 7-tt-»9tt—Uebergang. wertigen Kobaltion können Die d-d-Uebergange am zentralen, drei¬ nicht beobachtet werden. UV/VIS-Spektrum des Dicyano-Co(III)-cobesters in Benzol ,.104 1- 3Ô0 4u0 esdxT 5u0 Figur 6 Der Einfluss der axialen tals des stems Kobalts. legt, gibt die den. ot - an, und Eine ein Liganden dass " UV/VIS-Spektrum Beanspruchung Day, auf die Ladung der mit zunehmendem Abhängigkeit nach 4 be¬ pz-Orbi- Konjugation des Sy¬ der vier Comn-Stickstoffatome nicht fest¬ nephelauxetischem Effekt Wellenlangen, Verschiebung Stellung der entsprechenden axialen Liganden folgende Aufstellung des der sich über den Mechanismus des Einflusses g- -Banden nach grosseren solche Kobalt-corrin "Durch diese Wechselwirkung werde die herabgemindert. axialen eines Liganden auf auf der unterschiedlichen ruht nach Schrauzer Jorgensen (19)) in der eines Liganden bathochrom verschoben UV/VIS-Spektren von der nephelauxetischen Reihe konnte auch in wer¬ der (vgl. dieser Arbeit bei - F"< H„0 < Gegensatz Im diesen zu Methoden, nichts über die Art der axialen Dies ren, zwischen 165° Es treten dabei Peaks, beim durch gelingt tion. jedoch Da die Intensität der Abspaltung Massenzahl 27 als Kombination eines Bei allen mit Chlor intensivster Gibt entsprechen. man kann keine Deuteriumabstraktion beobachtet der axialen Liganden angenommen. erscheint bei 180 Ionenquellentemperatur die der durch Peak, Dies entspricht Cyanwasserstoff, Cyanidliganden Verbindungen so Liganden auf, sondern Wasserstoff, Methyl oder, mit gebildeten Fragmente temperaturabhängig ist, wird Dicyanoderivats Fall des Im Fällen eine Identifika¬ nicht die Massenzahlen der axialen der Probe schweres Wasser zu, eine thermische in vielen aufgenommenen Spektren derselben Verbindung. Vorliegen eines Perchlorat-Gegenions, werden. i" Cobester-Komplexes ausgesagt eines Vereinigung dieser Fragmente die einer < Vergleich der bei verschiedenen Ionenquellentemperatu- 210°, und Liganden Br Hand deren in den meisten Fällen an Massenspektrum das kann, ermöglicht werden " CN~< -NCS"< Cl"< < NH3 - beobachtet werden. Komplexen den meisten 16 mit Wasserstoff aus dem Restmolekül wurden die Intensitäten der aus den axialen entsteht. Liganden ge¬ bildeten Fragmente mit jener des als internen Standard gewählten Molekülbruch¬ stücks Acetyl (Masse 43) verglichen. Im lich sind nach Arbeiten ist, chemisch beit das strukturell dem Dicyanocobinamid, von von Dicyano-Co(III)-cobester K. Bernhauer unterschiedlicher Reaktivität. Diese, (14) gemachte Beobachtung hat wahrscheinlich folgenden Grund. ist nicht vollständig planar, sondern er hat eine von 161 (11). Während dadurch die Essigsäuregruppen Der Corrinligand verläuft, bei wel¬ mit einem Oeffnungs- grösseren Propionsäuregruppen gegen¬ einander geneigt werden und somit einen axialen den die Cyanidgruppen vorliegenden Ar¬ dachförmige Struktur, cher der First durch das Kobalt und das C-Atom 10 winkel le die beiden auch in der sehr ähn¬ weiter auseinander Liganden stark abschirmen, gerückt, wer¬ wodurch der andere axia¬ Ligand noch mehr exponiert wird. Die Endoseite des Moleküls mit den Pro¬ pionsäuregruppen Vitamin B 12 und der geschützte Ligand Nomenklatur, werden in dieser als untere bezeichnet. Arbeit, wie in der 17 - - Dass die unterschiedliche Reaktivität der beiden Spektrum In diesem gen. Figur 3). chen (vgl. Grund, erkennt man Dicyano-Co(m)-cobester Im wird durch sowohl der Struktur des Cobesters, nommen, dass dadurch die obere (20) E. Winnacker (2), D.Hodgkin (vgl. Versu¬ haben wird ange¬ Arbeiten betroffen wird. Cyanogruppe von dass bei gezeigt, weniger Dicyano-Co(HI)-l,2,2,7,7,12,12-heptamethylcor- wie dem rin die beiden Cyanogruppen gegenüber Labilität einer Cyanogruppe Säure vergleichbar reaktiv sind. Diese dafür, auch der Grund ist wahrscheinlich dass bei Dünnschichtchromatographie analysenreiner violetter Dicyano-Co(HI)-cobester der an späteren in Röntgenstrukturanalyse auch der als durch hiesigen Laboratorium am Corrinen, substituierten Säure, wie nahe zule¬ cm Cyanogruppe abgespalten. Auf eine Chloro-cyano-cobyrsäure (vgl. Figur 2) der bei 2125 Cyanidbande eine nur 18) gezeigt wird, spezifisch S. rein sterisch be¬ IR-Spektrum des Dicyano-Co(in)-cobesters scheint auch das dingt ist, Cyanogruppen Silicagel oder neutralem Alox scheinbar eine rötliche mit wendet Aus dem gleichen Grund wurde dem wurde, jeweils sich die Ausbeute die am Kaliumcyanidlösung mit Tetrachlorkohlenstoff extrahierten graphie des besten um aus als mobile Phase Verunreinigung enthält. Durch Besprühen der Platte Gebrauch mit einer methanolischen niert werden. Benzol-Methylacetat 3 % festes circa 15 % Benzol-Hexan kann dieser Silicagel, das insgesamt 89 %. erfolgt, war zur Chromato¬ Dicyano-Co(HI)-cobesters ver¬ Dadurch erhöhte Kaliumcyanid beigemischt. auf vor Effekt elimi¬ Bei der die Anwesenheit Kristallisation, von Cyanidionen überflüssig. Dem gegenüber erwies sich diese Massnahme bei der Aufnahme des UV/VIS-Spektrums von Säure, wie des sie Chlorid nachweisbar Für die oder in Dicyano-Komplexes absolutem, sind, über in als Feinsprit verringern die Extinktion im Kristallisation der Cobester-Komplexe sind nötig. Schon Spuren destilliertem Phosphorpentoxid Methylen- Elektronenspektrum. die Systeme Benzol-Hexan Methylacetat-Hexan vorzuziehen, da Methylenchlorid-Diisopropyläther oder Methylenchlorid-Hexan zum Teil irreversibel in das Kristallgitter eingebaut wer¬ den. Die in Absorptionsmaxima des UV/VIS-Spektrums Feinsprit (mit 0,1 o/oo Kaliumcyanid) bei 369 nm, Jf-, /3- und «-Bande entsprechend) sind 30'000. Die Extinktion der y- -Bande Dicyano-Co(IH)-cobester 545 gegenüber dem angegebenen Spektrum des Dicyanocobinamids verschoben. von in Wasser und 584 nm von um nm (der K. Bernhauer ca. 3 nm (14) bathochrom beträgt jedoch in beiden Fällen ca. - Im lung NMR-Spektrum £=5,70ppm (vgl. bei grosste Abschirmung, Cyanids B. in der wiederum steht einem 30%-iger Cyanogruppe einer Da der leophilen Liganden der Darstellung Perchlorsaure Ligandaustausch verfolgen. an die Wert, d.h. mit der in Stellung Dicyano-Co(m)-cobesters gegenüber Methylenchlond vibriert, Hand der an des weiteren Cobester Derivate Farbveranderung konnte sehr aktiv ist, mussten bei der Aufarbeitung Produkts erreicht werden. So erhielt man in einem den ra¬ nach orange gut von violett positiv geladene Aquo-cyano-Co(in)-cobester ausgenutzt. man ' gegenüber nuc- Fremdionen ferngehalten werden. Deshalb konnte durch Chromatographie keine des 10-Stel- ungeladenen Cobester Einklang im der Vinylprotons den kleinsten dieser Arbeit bei in Dies des Signal Aquo-cyano-Co(III)-cobyrinsaure-heptamethylester Die Labilität schen das spektrochemischen Serie. Saure wurde bei der Mit liegt - Figur 4). Dies stellt der dar, Komplex beobachtet wurde. des Benzol in 18 Reinigung entsprechenden Versuch Silicagel den Chloro-cyano-Co(m)-cobester, wahrend bei jenem an Alox die Produkte nicht identifiziert wurden. Da das den konnte, rohe wurde Austausch der Spektrum in Aquo-cyano-Co(III)-cobester-perchlorat von einer Aquogruppe Benzol oder nicht kristallisiert mikroanalytischen Bestimmung abgesehen. gegen Aethanol einem 1:1 zu Gemisch verhindern, von wurde das Wasser und Um wer¬ einen UV/VIS- 1,2-Dimethoxyathan (Monoglym) aufgenommen. 1) Hat soll men Cobester Komplex zwei axiale Liganden verschiedener Struktur, so derjenige im Namen an erster Stelle kommen, von welchem angenom¬ wird, dass er die "obere" Koordinationsstelle des Metalls besetzt. ein 19 - UV/VIS-Spektrum - Aquo-cyano-Co(III)-cobester-perchlorats des Benzol in e-io4 /\ 2- i-JA / \ 300 400 500 600 nm Figur 7 Das UV/VIS-Spektrum Aquo-cyano-Co(in)-cobester-perchlorats des zeigt die drei charakteristischen Hauptbanden, und y- bei 356 trum des 498 nm Das nm. P. von Aquo-cyano-cobinamids und 355 (22) George hat die bei et- 521 nm, Wasser in nifikant und wahrscheinlich nm aufgenommene Spek¬ entsprechenden Banden Die Differenz zwischen diesen beiden nm. Benzol in ß- bei 490 531 nm, bei Spektren ist nicht sig¬ Losungsmittel abhangig. vom Gegenüber dem Dicano-Co(m)-cobester Spektrum ist dieses des Saurebe- handlungsprodukts tischen um 15-60 auf die d-Elektronenwolke hypsochrom verschoben. nm Reihe hat Wasser als Ligand des Metalls als Gemäss der nephelauxe- viel kleineren ausdehnenden einen die Einfluss Ob die beobachtete Cyanogruppe. Verschiebung nach kürzeren Wellenlangen allem auf die Stellung des Wassers der nephelauxetischen Reihe (vgl. Komplexes zurückzuführen ist, Im bei 2135 IR-Spektrum cm was , des einer Dicyano-Co(m)-cobesters H.Hill (21), der diesen falls beobachtet <r hat, gibt -Donator Effekt des Cyanid, nimmt der S. muss Aquo-cyano 16) oder auch auf die offen bleiben. 10 cm nach grosseren Wellenzahlen Unterschied bei den dafür Transhganden, in ]ener des entspricht. entsprechenden Cobalaimnen eben¬ folgenden Grund an: "Durch den schwächeren diesem Fall Wasser ionische Charakter der bung der Schwingung liegt die Bande der Cyanogruppe Derivats Verschiebung dieser Schwingung gegenüber von in positive Ladung des Cyanogruppe ab, nach grosseren Wellenzahlen zur verglichen was Folge hat. eine " Es mit Verschie¬ ist jedoch 20 - darauf Verschiebung gegenüber dem ungeladenen Dicyano-Co(m)-cobester der Cyanidschwingung 10-Stellung wegen der positiven Ladung tieferem Feld verschoben. 6,42 Da die ppm auf. Aquo- ausser gegen Wasser Substanz nicht in in der gereinigt ist der durch Säure Ligand vorliegt, ausgetauscht wird. Verunreinigung der deprotonierten Form, d.h. dem die ist in Anbetracht des hohen ergab konjugaten Säure Aquocyano-cobinamid mit einer bathochromen und 360 von pK-Wert einen ist, eine ^11, 11 von konnte der (22) P.George 71). kleinen 5 nm Eine genaue und pK-Wert Bestimmung durch diese Methode Verschiebung der einzig dazu geeigneten, nicht y-Banden nach 547 nm, Aquo-cyano-Co(ni)- des dass die hat, gegenüber /3-Banden Die der wurden dabei weit Es auffiel, P. weniger George (22) folgenden 100%-ige bei den Grund haben. nur nur Reversi¬ zeigte sich dabei auch, von nur «. - 20'500 und beeinflusst. im UV/VIS-Spektrum protonierung des Aquo-cyano-Co(in)-cobester-perchlorats die auch von konjugaten Säure. Die Extinktionen der Verschiebung der Absorptionsmaxima wird y Figur 20, wegen der -Banden deprotonierten Verbindung eine Extinktion 27'000 bei der längen, jedoch aber die Säure-Basengleichgewichts bestätigt werden. y-Bande war scharfen möglich. Durch dieses Verfahren konnte bilität des für angibt. Da die Deprotonierung cobester-perchlorats spektroskopisch angenähert ermittelt werden (vgl. S. pK-Wertes Mikrotitration im Gemisch wobei /3- Verschiebung der ot-, verbunden nm So auszuschliessen. Monoglym-Wasser 1:1 einen pK*-Wert nm 6,35 möglich, es Hydroxo-cyano-Co(III)-cobester entspricht, 524 bei Vinylregion konnte, werden in nach analogen Spek¬ In einem ppm. Signale der das Deutero¬ Protons Kobalt erwartungsgemäss 6,48 noch ein anderer axialer Möglichkeit, dass Die am erscheint bei Es treten zwei schwache Säurezugabe trum ohne dass positive Ladung Signal des ist das Tropfen Trifluoressigsäure chloroform mit einem und auch durch die NMR-Spektrum des Aquo-cyano-Co(m)-cobester-perchlorats Im der die sein kann. bedingt Kobalt am dass hinzuweisen, beobachtete - nach bei der De¬ grösseren Wellen¬ entsprechenden Cobinamid Derivaten Durch den Verlust der positiven Ladung bei der Deprotonierung oder durch den gegenüber der Aquogruppe stärkeren indukti¬ ven Donatoreffekt der stickstoffatome setzten 7 ir Hydroxygruppe verringert, Niveaus des was Corrin wird die Elektronegativität der wiederum eine ir-Systems zur Destabilisierung vier Corrin- des obersten be¬ Folge hat. Die bathochrome Ver- 21 - Schiebung ist 7ir-»9ir und Im Ausdruck der energiearmer ein des die Massenzahl 27 % gebildet jener des als Standard und Wasserstoff Cyanogruppe aus betragt Diese 210 bei 43). auf, deren Intensität mit steigender Temperatur zunimmt. zu Oberhalb 210 43 bei 225 tritt erreicht im sie Ionenquellentempera- Massenspektrum zusatzlich die 34 % wird durch Kombination des Chlors oder mit gewählten Molekulbruchstucks Acetyl (Massen¬ zahl nis 7ir -*8ir Aquo-cyano-Co(in)-cobester-perchlorats tntt,wie temperaturabhangiger Intensität auf. tur 345 gewordenen Uebergange . Massenspektrum erwartet, - 255 bei aus dem . Dieses Massenzahl 9% Von im 50 Verhält¬ Methylchlond, Fragment, mit Gerust- Perchlorat-Gegemon Ester-Methylgruppen gebildet werden. C. Chloro- und Jodo-cyano-Co(III)-cobyrins au re-heptamethy lester Um Komplex aus zu Aquo-cyano-Co(HI)-cobester-perchlorat dem erhalten, genügt es, triumchloridlosung schuttein, zu Um beim anschliessenden wurden dem vorzubeugen, nahmsweise aus Bei deren einem um das Chromatogramm Silicagel 10 % Mikroanalyse analogen Versuch einem festes einem von nicht kristallisiert Gemisch der axial stutzt auf die weiter oben nichtionischen mit wasseriger Na- wobei die Farbe sofort nach weinrot umschlug. isomeren auf der Saule Ligandaustausch Natriumchlorid erhielt beigemischt. Aus¬ man schone rote hexa¬ Chloro-cyano-Co(in)-cobester ent¬ (20), ausgehend von E.Winnacker Dicyano-Co(m)-l,2,2, 7, 7,12,12-heptamethylcornn, pisch ähnliche Produkt einen Methylenchlorid-Losung Methylenchlond-Hexan kristallisiert gonale Plattchen, sprach. die werden, konnte das da es UV/VIS-spektrosko- sich dabei vermutlich Chloro-cyano-Co(HI)-corrine handelte. dargelegten Punkte wird angenommen, dass genden Cobester-Komplex die axialen Liganden gleich angeordnet sind der 1954 durch A. Todd D. Hodgkin (2) in (23) dargestellten der ersten und anschliessend Rontgenstrukturanalyse eines im Ge¬ vorlie¬ wie bei von Vitamin B 12-Kom- plexes untersuchten Chlorocyano-cobyrsaure: Chlorogruppe "oben" und Cyanogrup¬ pe "unten". 22 - UV/VIS-Spektrum Das a—, /3- und y-Banden Chlors in der des Chloro-cyano-Co(m)-cobesters 565 um, bei nephelauxetischen - 531 nm und 370 nm liegt, der Stellung des jenen des Dicyano- zwischen entsprechend, Reihe in Benzol mit Aquo-cyano-Komplexes. und des UV/VTS-Spektrum Chloro-cyano-Co(HI)-cobesters des in Benzol t.104 300 500 400 600 nm Figur 8 Im Ht-Spektrum erscheint die C=N-Streckschwingung auch die kleine Differenz nicht signifikant ist, Effekts eine solche so von 5 ist doch auf Tendenz, eine bei 2120 cm Wenn . Dicyano-Co(HI)-cobester gegenüber dem cm Grund des stärkeren induktiven Donator- Verschiebung nach kleineren Wellenzahlen, zu erwarten. Das Signal des Vinylprotons Deutero-chloroform bei über dem ebenfalls 5,95 der Effekt für diesen Massenspektrum erkennt nach tieferem Feld wonach diese in Verschiebung gegen¬ um 0,35 Verschiebung spektrochemischen Serie abhängt, ppm. von der könnte man die von den beiden axialen Liganden mit gebildeten Fragmente. Während die Cyanogruppe als Cyanwasser¬ Methylgruppe, senzahl NMR-Spektrum Komplex richtig vorausgesagt werden. stoff mit der Masse 27 einer in der tritt im entspricht einer aufgestellten Hypothese, Stellung der axialen Liganden Molekülteilchen 10-Stellung Dies ungeladenen Dicyano-Derivat Gemäss der eingangs Im in der ppm auf. als auftritt, erscheint der Methylchlorid 27 erreicht bei 175 temperatur des Maximum. mit der Chlorligand, Masse 50. jene der Massenzahl 50 Ein nach Kombination mit Die Intensität der Mas¬ erst bei 187 Ionenquellen- gestaffeltes Auftreten der Kombinationsprodukte 23 - - Cobester-Komplexen bei unterschiedlichen axialen Liganden konnte bei allen gestellt werden. Ob dies direkt ein gandbmdungen darstellt, kann mcht Zusammenhang sei darauf fur die thermische Mass mit Sicherheit entschieden werden. dass der hingewiesen, tersuchten Cobester-Denvate unter der im Massenspektrum fur die So wiesen auch die schmolzenen Proben mcht auf Zersetzung hin. Kaliumjodid erhielt Mit um in zu haben. Kaliumjodid beigemischt Jod aus worden war, der ge¬ Aquo-cyano-Co(III)-cobester-perchlorat dem Nach der Dieses Cobester-Derivat wurde dar¬ Chromatographie eine weitere Sihcagel, dem an daraus durch Sauerstoffoxidation um auszuschliessen, kristallisierten un¬ axialen Li¬ UV/VIS-Spektren ungeladenen X-cyano-Co(III)-cobester der Reihe der Vergleichssubstanz detes man Jodo-cyano-Co(III)-cobester. den blauvioletten gestellt, eine In diesem der meisten Schmelzpunkt ganden kritischen Temperatur lag. fest¬ Stabilität der Li- aus kein gebil¬ Benzol-Hexan schwarzviolette Tetraeder. Die spektroskopischen Daten des Jodo-cyano-Co(m)-cobesters wesentlichen mit den erwarteten Werten uberein. Benzol die oc -, chrom nach nen /3 - 596 nm, y-Banden gegenüber und 561 nm und 375 grösseren nephelauxetischen nm So sind jenen des Cyanid, stimmen im UV/VIS-Spektrum in Dicyano-Denvats batho- (vgl. Figur 9). verschoben Effekt hat als im war Da Jodid ei¬ die Tendenz der Ver¬ schiebung vorauszusehen. Wie sich auch bei spateren Versuchen ester ein mit Im gegenüber dem Methanol oder Gegensatz dazu Alkylcobalaminen gehen. zeigte, sehr ist labil. So ein Jodoligand erhalt man am in Cob- Methanol Aquo-cyano-Co(III)-cobester-Komplex entsprechendes UV/VIS-Spektrum Absorptionsmaxima stabil Feinsprit sein bei 352 nm, soll nach erhaltene und erst nach 490 nm und K. Bernhauer 520 (38) Jodo-Co(III)-cobalamin Zugabe von Wasser in nm in das (vgl. Figur 9). das durch Jodspaltung absolutem Methanol von gelost Aquo-Co(III)-cobalamin über¬ 24 - UV/VIS-Spektrum von - Jodo-cyano-Co(III)-cobester i 1 , —i 300 i | 600 500 400 Methanol in Benzol resp. nm Figur 9 Im IR-Spektrum Im NMR-Spektrum erscheint die protons gegenüber jenem über 6,14 Cyanidbande in Deuterochloroform vom der extremen relative Unter schwach bei 2125 Dicyano-Co(IH)-cobester jenem des Chloro-cyano-Derivats ppm verschoben. nur dagegen um 0,25 ist das um 0, cm 5 ppm und gegen¬ ppm nach tieferem Feld, se 127 142) auf, Stellung des Jodids in der spektrochemischen Serie scheint die Lage des Signals verständlich. Liganden entstandenen zu Massenzahl des Gunsten des so den Jodoliganden entsprechende Massenzahl welche höchstens teilweise sekundär Cyanwasserstoffs, aus Cyanwasserstoff (Masse 27) und Methyljodid (Mas¬ auch die einem unkombinierten Ist die Intensität der die des nach Berücksichtigung der aufgestellten Hypothese und Im Massenspektrum tritt neben den erwarteten Massenzahlen der axialen . Signal des Vinyl- aus Methyljodids dem von Methyljodid 185 bis entstanden ist. 205 grösser als ändert sich das Verhältnis bei höherer Temperatur Cyanidfragments. 25 - D. - Reduktion von Co(III)-cobyrinsäure-heptamethylester-Komplexen Kobalt im Cobester erreicht durch Reduktion Das Diese gelingt relativ leicht zur kann Verfügung (vgl. 12-Systems (8)). F.Wagner Natriumborhydrid Die spezifische (24) mit Zinn(II)-Chlorid H.Hill durch katalytische Hydrierung Systems wird mit man 0,1 n Nucleophile Reaktivität (27) A. W. Johnson biologischen Aktivität Bei dem durch die O. Schindler von B 12 nen Derivat, des Kobalts die von von Titration von welches UV/VIS-spektroskopisch NMR-Spektrum, (HI) gelang, hat die Hypothesen aufgestellt, Wasserstoffs betrafen. tat bezeichnet, mit B 12 wie Nachweis (32) für die allem in Anbe¬ vor 1951 erstmals bildete die H. Hill durch bei man ein einem graugrü¬ Wertigkeit (29). Potential In ei¬ von luftempfindliches Produkt, übereinstimmte. am Kobalt im B 12 wurden verschies allem die Existenz eines axial eines G.Wilkinson konnte beim B 12 G. Schrauzer (30 ligandierten derartigen Wasserstoffatoms im und 31) beim Hydridopentacyanokobal bisher nicht erbracht werden. einwertige Stufe einen pK-Wert Am Cobaloxim von 10,54 ge¬ liegt beträchtlich höher als der des Hydridotetracarbonylkobalts, messen. Dieser der W. Hieber von Der er vor einwertige Klarheit brachte erst 1963 Standard, Ueber die Koordinationsverhältnisse dene was erhaltenen, Aquo-cobalamin Kalomelelektrode als mit einer die sauerstoffempfindlichen, sehr Zweielektronen Reduktion bei pH 8 erhielt -1,2 V, wobei Cobalaminen beobachtete an Thiolen, lange Zeit einen umstrittenen Punkt. potentiometrische ner (25) Vitamin B 12 bemerkenswert ist. mit B 12 allgemeinen gelingt Natronlauge erhalten, Kobalts am Vitamin B 12- Eine Reduktion des stärkeren Reduktionsmittel (28) beobachteten, im zur mit konzentrierter des Gegenwart auch in zweiwertigen Stufe auch durch Lauge die zweiwertige und Stufe erreicht. tracht der (26) R. Yamada nach auch mit in Salzsäure oder nach H.Diel Platinoxid. an einwertige Stufe des Kobalts Ammonchloridlösung oder Reduktion besten bewährt hat. nach Die ver¬ stehen verschiedene Metho¬ wobei sich letzteres in dieser Arbeit werden, erreicht einwertigen Stufe, zur planaren Corrin-Ligandensystem Zink in Essigsäure oder in mit u.a. zwei- wie auch zur dem aus Für die Reduktion des B drängt wird. den sowohl Kobalt ohne dass dadurch das nucleophile Reaktivität. (33) zu ca. 5 bestimmt wurde. Schrauzer nimmt an, - 26 - dass B 12 ten soll, mit der konjugaten Saure, die Behandlung welches mit der gelost, mit der eine Der anfänglich farblose, Die nach Phase, ein in ungefähr vier der sich das Ueberdruck Reaktionsgefass eindringt. Hand der Versuche, in Metha¬ Hahns im ohne dass verschlossenen wer¬ vorübergehenden nun da violette, Luft un¬ Scheide¬ im in das Einspritzen des elektrophilen Reagens durch den zweiphasigen Reduktion ermöglicht es, die nucleophile Reak¬ sofort in Abwesenheit rot, wobei überschüssigem Reduktionsmittel von zu Co(I)-cobester-Komplex enthaltende Aether- braungelbe ölige Tropfchen ausfallen. diese um ein an verfolgen. gelbbraune UV/VIS-spektroskopischen Vergleich sich dabei in sogleich ausgeglichen einer dunkelgrün. Die werden, setzt Konkurrenzreaktion mit Sauerstoff ausgeschlossen. Methanol-Wasser(l:l)-Gemisch es Edukt Spatere bei denen durch Ausschütteln der oxidierten Aether-Hexan-Phase zeigten durch dass Oeffnen des Phase wird nach Natriumborhydrid gelost hat, kann, Farbanderung der Losung genau Hexan-Losung einem obere Luftkontakt wird die grüne, Bei re¬ Scheidetrichter im dass der Ueberdruck so Minuten Schuttein Durch eine Co(I)-cobesters tion des langsam und Jodo- Chloro-cyano- Natriumborhydridzugabe leicht abgelassen ist, Polyathylenstopfen wird Diese Methode der sehr nur unpolaren 1:1 Gemisches Aether-Hexan wieder durch kurzes immer trichter Natriumborhydrid Nach der Wasserstoffentwicklung ein, Rotfarbung erwies orangerote Aquo-cyano-Komplex bleibt auch beim Schuttein den tere in Co(m)-cobester-Kom- fur die Reduktion Ausgangsprodukt eines Scheidetrichter muss. Metho¬ spezielles Reduktions¬ gleichen Menge Wasser verdünnt und unteren, wasserigen Phase. starke ein Bei der Scheffoldschen Methode wird das ungefähr doppelten Menge uberschichtet. der Als Gut verwendbar fur die Reduktion sind auch cyano-Co(HI)-cobester. nol einwertiges Ko¬ hiesigen Institut bei Versuchen am Letzteres wird mit Dicyano-Derivat. duziert. dass an, experimentelle dieser Arbeit fur die Reduktion der in er Wasser-Methanol in zugängliche Aquo-cyano-Co(m)-cobester-perchlorat gunstiger als sich das leicht das (34) R.Scheffold übernommen werden konnte. plexe erforderten besondere Co(m)-l,7,7,12,12-pentamethylcorrine der Reihe der verfahren, Hypothese gibt seiner mit Diazomethan und hohe Reaktivität der formal Corrin-Komplexe So entwickelte Köbalt-Wasserstoffbindung enthal¬ verestert werde. Luftempfindlichkeit Extreme B 12 von Bildung des Methylderivats balt enthaltenden den. eine Gleichgewicht steht. Als Beweis im diese Saure bei der unter - Substanz isoliert mit authentischem mit wurde, Material, zweiwertiges Kobalt enthaltendes Cobester-Derivat 27 - handelt. Im dazu wird nach Gegensatz durch Luft sofort zu Lösung zeigt ähnliches Spektrum, der grünen von UV/VIS im ein jedoch cobester einen Rf-Wert oder mehrere Edukts als können. Abspaltung von dass das spricht, man Cyanid dass sein, % etwa 75 nur Carboxylate reagieren eine des bei der Reduktion des teilweise mit Komplexen. violetten zu Aquo-cyano-cobester- Auf Grund der Löslichkeit in dass der in der Aether-Hexan-Phase sei. apolarem Medium vorliegende grüne Ob axial ein Wassermolekül ligandiert ist, offen bleiben. Luftempfindlichkeit der Co(I)-cobester auch Da wegen der extrem bei der gefüllten "Drybox" Charakterisierung Der in diesem nicht isoliert werden auf ein Um die UV/VIS-Spektrum Versuch verwendete UV-Zelle im oben beschriebenen nen, dass Luftoxidationsprodukt der grünen Aether-Hexan-Phase mehr enthält. annehmen, mit Stickstoff ziert. Dicyano-Co(HI)- wird angenommen, Aether-Hexan Phase extrahiert werden abgespaltenen Cyanidionen Co(I)-cobester-Komplex ungeladen muss in die Die in diesem Gemisch unlöslichen Cyanogruppe keine unter den Stand¬ bei denen der Methylestergruppen unter basischen Reduktionsbedingungen verseift Co(I)-cobester Komplex Für die könnte läuft, nicht Dies wird auch der Grund dafür den durch die Reduktion kations jenem fast identisch mit Dünnschichtchromatogramm im 0,4 zeigt, von Dicyano-Co(HI)-cobester- Kaliumcyanid von ardbedingungen (Silicagel/Benzol-Methylacetat 1:4), worden sind. in Wasser Aether-Hexan-Lösung abgetrennte verunreinigtes das durch Zusatz Da dieses Produkt wird. (35)B12 K. Bernhauer Hydroxo- (resp. Aquo-) Co(HI)-cobalamin oxidiert. Die nach der Reduktion violette - Zweiphasensystem Konzentration der grünen mit wurde in der bestimmen Zelle mit Luft des erhaltenen zu kön¬ oxidiert, Dicyano-Co(III)-cobesters Aus der Extinktion konnte auf die Konzentration der cobester-LÖsung sich Natriumborhydrid redu¬ Co(I)-cobester Lösung Kaliumcyanid zugegeben und das Spektrum in der man in Aether-Hexan beschränken. Chloro-cyano-Co(ni)-cobester wurde nach der Aufnahme des Spektrums in der aufgenommen. konnte, musste Dicyano-Co(m)- und daraus auf jene des reduzierten Produkts geschlossen wer¬ den. Das ( e = UV/VIS-Spektrum 39'000), spricht jenem 394 nm ( des B 12 charakteristische l = des Co(I)-cobesters 38'000), 465 nm (vgl. Figur 10). ( t = mit Absorptionsmaxima 3'50O) Nach So ordnet nm G.Schrauzer Spektrum der Co(I)-corrin-Derivate schiebung der Hauptbanden zustande. und 576 er durch eine die ( £ = (16) bei 282 4'300) nm ent¬ kommt das hypsochrome Ver¬ «.-Bande der Absorption 28 - bei 394 nm und die langwelligen - g* -Bande jener bei 282 Banden oberhalb 400 nm Die viel weniger zu. intensiven, danach d-d Metallorbital- oder entsprechen nm Metall-Ligand Chargetransfer-Uebergängen. 4H 104 l UV/VIS-Spektrum 300 von Co(I)-cobester 400 1:1 in Aether-Hexan 500 600 Figur 10 Neben der schon oben beschriebenen Oxidation mit Luft wurde auch die Reaktivität des Co(I)-cobesters gegenüber Säure geprüft. Einspritzen Beim von wässeriger Perchlorsäure blieb die grüne Aether-Hexan-Phase unverändert, wäh¬ rend die ser zugegebene wurde E. Säure eine ein Sind die Alkylierungsreaktionen wurden, in der über die Reaktion der reduzierten Der in dieser Arbeit durch am spektralen system. Dabei B 12 , die im In die¬ von auch am beschrieben, Co(I)so ist Verbindungen mit Halogenen nichts veröffent¬ Umsetzung mit Jod erhaltene Folgeprodukte und chemischen Einfluss der axialen war Folgenden Literatur verschiedentlich cobester und die daraus entwickelten den bildete. Co(n)-cobester-Derivat nachgewiesen. Dijodo-Co(III)-cobyrinsäure-heptamethylester cobester untersucht licht. gelbe, wässerige Phase untere, UV/VIS-spektroskopisch experimentellem Vorteil, brachten Liganden dass Dijodo-Co(III)- neue am Kenntnisse über Kobalt-cor r in - durch die axiale Ligandie- 29 - ungeladene, Jodidionen von rung Der Cobester-Komplexe stabil gespritzten konzentrierten Jodlosung spontan. Die und die hielt ses braungrune Färbung verschwanden klare, eine cobesters mit Jod den che darstellt, zeigten, einen Additionskomplex die erste Oxidationsstufe des um gegenüber einstufig mer Die hier beobachtete den in sich, es zweistufige Alkylierungsreaktionen Dijodo-Co(III)-cobesters mit halbem Rf-Wert auf. stanz um cobester ein handelt, der Chromatogramm einem Benzol-Hexan an noch ein Formel von Co(m)-cobester-Denvate in Benzol ist etwas nach 604 Komplexes bathochrom auf -Bande drei v- (vgl. Figur 21, Feinsprit der gegenüber Fall an maxima fur die einen der modifiziert. 91). S. nm und 562 in Das blauviolettes Nebenprodukt sich bei dieser Sub¬ es und Dijodo-Co(rn)Nach Di30do-Co(III)-cobester aus jenen der bisher untersuchten nm jenen des verschoben UV/VIS-Spektrum in wird axiales Jodid durch Alkohol verdrangt, des Jodo-cyano-Co(in)- sind, bei 358 nm, sowohl ß -Banden und «.- Benzol vollkommen verschieden. treten 371 nm im Ge¬ und 393 nm Methanol als auch Wie beim was auch m Jodo-cyanodiesem in Orangefarbung der Losung festgestellt werden kann. Die Absorptions- der alkoholischen Losung bei 316 Bildung eines einem nm und 469 nm Dialkoholo-Co(rn)-cobester-Kations, Co(n)-cobester Komplex (vgl. konnte bei im¬ mit Jod trat neben dem Wahrend die überlappende Absorptionen ist von jenem Co(HI)-cobester dar, Jodo-cyano-Co(HI)-cobester entspricht. Silicagel kristallisierte mit da jene Silicagel-Dunn- Im Dicyano-Co(III)- Dijodo-Co(III)-cobesters erwartungsgemass gegenüber biet der den Jodo- um Besonderheit eine in rotvioletten Nadelchen. in UV/VIS-Spektrum Das Dijodo-Co(ni)- Co(I)-cobesters Oxidation des Spatere Versuche ergaben, dass Conproportionierungsprodukt das er¬ spatere Versu¬ wie einwertigen Edukts, schichtchromatogramm des Rohprodukts der Umsetzung braunroten Fleck des man 45). S. Zweielektronen-Oxidation verlaufen. einer einer ein¬ Trübung und Jodzugabe, des weinroten (vgl. werden braungrunen Zwischenprodukt handelt Co(II)-cobester. Jod stellt bei weiterer mit auftretende vorerst kann durch kurze Reduktion mit Natriumtmosulfat Di]odo-Co(HI)-Komplex übergeführt Bei dem wurden, Losung. Bei Joduberschuss wird die Losung braun. Die¬ weinrote das vermutlich Produkt, erhalten waren. geloste grüne Co(I)-cobester reagierte Aether-Hexan in kristalline Laboratoriumsbedingungen welche unter - S. 33 ). Ein ahnliches sprechen jedoch mcht sondern vielmehr fur Phänomen dieser Art spateren Versuch nochmals beobachtet werden (vgl. S. 33). 30 - Das in des NMR-Spektrum - Di]odo-Co(III)-cobesters Deuterochloroform oder in Deuterobenzol ist weit weniger gut strukturiert und auf gelost als die der bis¬ her untersuchten Co(in)-cobester-Komplexe (vgl. Figur 11). Da nur fur die Me¬ thylestergruppen scharfe Signale auftreten, und die zwischen 6,0 und 7,0 ppm erwartete Resonanz des de vorerst vermutet, Verbindung NMR-Spektrum 10-Stellung vollständig fehlt, Protons der vinylischen die von sei wur¬ partiell paramagnetisch. Dijodo-Co(III)-cobester in Deuterobenzol i 1 6 5 1 • 1 3 4 1 1 2 1 • r~ ppm Figur 11 Dieser Paramagnetisms konnte theoretisch Teil der ein unreinigt. lerweise ration sen dreiwertigen oder die vor, ein Teil der vorliegen, so bei dem die 42) dagegen netismus 1) noch Temperatur Der weisen einer 70 C Ein bei strukturiert, Veränderung. Ursachen haben: partiell mit in seinen einer sechs, erhöht sein. Ein highspin Konfigu¬ in auf einen ganz der wurde, ergab jedoch Kristalle bei 77° die¬ dann entsprechender Ver¬ keine sichtba¬ Grund fur die verwaschene Struktur ist nicht bekannt. Suszeptibilitat ver¬ norma¬ aufgenommenes NMR-Spektrum sollte 45 hegt Co(n)-cobester ist durch Temperaturerhöhung mehr Kerne "paramagnetischer" um Entweder highspin-Elektronenkonfiguration dreiwertigen Kobaltionen mussten bei sungen der magnetischen S. zwei einer lowspin angeordneten 3d-Elektronen Zustand übergehen. such, in NMR-Spektrum untersuchte Probe im Wurde noch wemger re Kobaltionen K und 293° K Mes¬ (vgl. schwachen, temperaturinduzierten Paramag- hm. Der bisher einzige bekannte hexafluor id -Anion. highspin Kobalt(III)-Komplex ist das Kobalt(ni)- 31 - - Anderseits haben weitere NMR-Versuche paramagnetischem Jodo-Co(II)-cobester lediglich ser abflacht und etwas Effekt nicht der beim bei welcher die verbreitert, ergeben, dass Zugabe 20 von % Signale des Dicyano-Co(III)-cobesters sonst aber ohne Einfluss bleibt. Dijodo-Co(m)-cobester beobachteten Da die¬ Störung entspricht, Methylestergruppen gar nicht, die Signale der naher beim Kobalt liegenden Protonen jedoch mehr verbreitert sind, wird netisms fur unwahrscheinlich NMR-Spektrum die von auch externer Paramag¬ gehalten (vgl. Figur 12). Dicyano-Co(III)-cobester + 25% Jodo-Co(II)-cobester m Deuterobenzol C„D TMS »*iSf>Hw^*« 8 7 6 3 2 ppm Figur 12 Im Massenspektrum des die aus den axialen jodid (142) ten in Dijodo-Co(III)-cobesters Liganden gebildeten temperaturabhangiger Intensität auf. Acetyl (Masse 43) verglichen temperatur das treten von erwartungsgemass Jod (127) und Methyl- Mit dem als Standard gewähl¬ erreicht ihre Intensität bei 203 Ionenquellen- Maximum. Durch Versetzen serigem Massenzahlen einer Kahumcyamd, Co(III)-cobester. benzohschen erhielt Dieser man in Dijodo-Co(III)-cobester Losung 98%-iger Ligandaustausch konnte folgt werden. Dabei wurde, um eine einphasige Ausbeute kristallinen auch im Reaktion mit Dicyano- UV/VIS-Spektrum zu haben, wäs¬ ver¬ benzolische Blausaurelosung zugetropft. Die entstehende Jodwasserstoffsaure neutralisierte man mit festem inert war. Die Kahumcarbonat, Bildung des das gegenüber den Dicyano-Co(III)-cobesters Co(III)-cobester-Derivaten konnte dabei an der zuneh- 32 - Extinktion menden der y -Bande des - Produkts 372 bei abgeschätzt nm wer¬ den. Darstellung des Dijodo-Co(HI)-cobesters schon kurz erwähnt, Wie bei der Dicyano-Co(m)-cobester kann dieser mit che conproportionieren. Glei¬ überraschend Komplexe wurden benzolischer Lösungen dieser Mengen äquimolarer, zusam¬ mengegeben und unter Stickstoff bei RT gerührt. Die langsam ablaufende Kreu¬ zungsreaktion lässt sich ses nach im zwanzig nach zwölf Stunden jene der Edukte, ungefähr gleich intensive der braunrote des Dicyano-Komplexes (Rf-Wert 0,15), der violette des rend der Dünnschichtchromatogramm gut verfolgen. Zeigt die¬ Minuten noch drei dritte, blauviolette (Rf-Wert 0,4) Silicagel gereinigt, konnte diese blauviolette Substanz siert werden. Dire Daten, wie chromatographisches Verhalten des bekannten den sind ein um Gemisch Zusammenhang Dicyano-Co(m)-cobester mit ist Kobalt(II)-Komplexe (kristalliner Die axialen des nur sehr und schwach, wäh¬ Chromatographie an Benzol-Hexan kristalli¬ Mikroanalyse Dass beiden bemerkenswert, gleiche Conproportionierungsprodukt aus es mit dass jenen sich bei dem Con- Ligandenisomeren kann nicht Jodo-Co(ü)-cobester sind NMR-Spektren und dünnschicht- identisch. der noch ganz Durch mit Ausnahme der Cyano-Jodo-Co(in)-cobester handelt, In diesem F. IR- und Jodo-cyano-Co(HI)-cobesters proportionierungsprodukt um UV/VIS-, so Dijodo- (Rf-Wert 0,6) nur dominiert. Flecken, oder ausgeschlossen werden. aus einem Gemisch unter analogen von Bedingungen das langsam entsteht. Cobyrinsäur e-heptamethylester s Jodo-Co(II)-cobyrinsäure-heptamethylester) Jodogruppen im Co(ni)-Cobester-Komplex werden auch durch Silberionen leicht abgespalten. einer benzolischen Bei Zusatz von Silberperchlorat-Monohydrat Dijodo-Co(in)-cobester Lösung schlug fort nach orange um, und ein feiner trum der abdekantierten Niederschlag zu die rotviolette Farbe fiel aus. Die im so¬ UV/VIS-Spek- Lösung auftretenden Banden bei 350 nm, 494 nm und - 525 lassen durch nm So ist das - Vergleich auf das Diaquo-Co(HI)-cobester-Kation schliessen. (14) publizierte Spektrum K. Bernhauer von 33 cobinamid-Derivats in Wasser gegenüber dem obigen verschoben. Weitere besprochen. Nach Entfernen des Eigenschaften teln mit Wasser wurde das dieses um 316 und 469 nm vates, UV/VTS-Spektrum sondern vielmehr dieses jenem findet nach in (37) Lösung gelbe eine Parallele bei ent¬ mit Banden bei (25) H.Diehl von Cobester-Produkt einerseits auf 40° Diaquo-Co(III)-cobinamid-Deri- eines schon 1952 bei getrocknet. gelösten Produkts in Benzol beschriebenen jenem des Vitamin Diaquo-Co(in)-cobester-Komplexes F.Wagner hypsochrom werden auf Seite 52 Rotationsverdampfer am zweiwertiges Kobalt enthaltenden Cobalaminderivates, Die Reduktion des nm überschüssigen Silberperchlorats durch Ausschüt¬ Lösungsmittel gleicht nicht mehr dem nm 2-6 Cobester-Komplexes fernt und der Rückstand am Hochvakuum bei RT 18 Stunden Das Diaquo-Co(m)- des nur beim Trocknen am Cobinamid-Komplexen. Silicagel B 12 . Vakuum Da das und Alox Dünnschicht¬ platten mehrere rötliche und gelbbraune, verschmierte Flecken zeigte und ander¬ seits nicht direkt kristallisiert werden Bestimmung Das konnte, wurde auf eine mikroanalytische verzichtet. NMR-Spektrum des Trocknungsprodukts in Deuterobenzol unterstützt die Annahme der formalen Der durch die Zweiwertigkeit des Kobalts in diesem Cobester-Komplex. ungerade Anzahl d-Elektronen des formal zweiwertigen Kobalts bedingte Paramagnetismus verbreitert oder verschiebt alle Signale derart, dass einzig noch die Methylestergruppen Die den, xes (vgl. Die 35) S. Absorptionsmaxima die von 66%-iger des nm Diehl für die Die Banden des gegenüber denen des wer¬ Co(II)-cobester-KompleJodo-Co(II)- Ausbeute kristalliner UV/VIS-Spektrums des Co(II)-cobester-Komple- als auch in Feinsprit mit jenen des auf kleine Verschiebungen der Bande bei 315 Lösung des erhalten wurde. stimmen sowohl in Benzol in Wasser bis in Hügel erkenntlich sind. experimentell dadurch bestätigt einer benzolischen wässerigem Kaliumjodid mit als breiter des Kobalts konnte dass beim Schütteln cobester xes Zweiwertigkeit von 4-6 nm gut überein. Die Extinktion dagegen liegt beim Cobester-Derivat entsprechende Bande des B 12 Co(II)-cobester-Komplexes sind (16) zugeordneten Banden um 25 % höher als angegebene. wie Diaquo-Co(ni)-Kations hypsochrom der nach G.Schrauzer Vitamin B 12 jene des Co(I)-cobesters verschoben. Beim Vergleich zeigt sich, dass sowohl die 34 - ot-Bande bei 469 als nm zwischen den plexes auch die - bei y-Bande entsprechenden des Co(I)- 318 Co(II)-cobester-Kom- des nm Diaquo-Co(m)-Komplexes und des liegen. (vgl. 16) S. dieses Massenspektrum Das ligandiertes Wasser oder einen Co(II)-cobester-Derivats gibt anderen einen Anhaltspunkt, da Liganden noch fur weder fur axial Perchloratgegemon Massenzahlen eindeutig temperaturabhangige fehlen. Da auch das konnte IR-Spektrum keinen Hinweis fur sich bei der es Co(II)-cobester vorliegenden Verbindung Absaugen am mit Dijodo-Co(m)-cobester Vakuum wahrend 18 Stunden bei Aquo-Co(II)-cobester-perchlorat In Ermangelung eindeutig nicht als auch in identifiziert werden. Dieser ungeladenen Hydroxo- (innert rasch in der Reaktion Sil- von Benzol und anschliessen¬ RT, wurde jedoch eher man bisher amorphe, zur Methanol in Stunden) Minuten bis festem Zustand gelost gegen Luftoxidation langsam (innert Tagen), er nicht kristal- Verfugung stehenden Daten ist sowohl Komplex wasserfreiem Benzol oder abs. jedoch Saure kann der auf Grund der In wasserigem Methanol wird stabil. von Mikroanalyse einer gibt, erwarten. Co(II)-cobester-Komplex lisierbare Perchloratamon den Auf Grund der Hersteilungsweise, handeln. berperchlorat-Monohydrat dem ein um zum in Gegenwart Diaquo-Co(in)-cobester- Komplex oxidiert. Dieser cobester in sehr leicht Versuch, erwähnt, dass es sich ein um dieses zu dass unter erwähnte Reaktion des gewissen Umstanden Dijodo-Co(HI)- Cobester-Komplexe übergehen. Umsetzung der grünen Co(I)-cobester Losung mit Jod tritt, Bei der ester auch die früher die zweiwertige Stufe in oben sucht, wie Alkohol zeigen, grün braunes einen Produkt Zwischenprodukt auf, Jodo-Co(II)-cobester Komplex durch einelektronische von dem vermutet wie wurde, handelt. Deshalb wurde Reduktion aus ver¬ Dijodo-Co(III)-cob- erhalten. Vorversuche Reduktion von mit Wasserstoff und Aquocobalamm zu B 12 Dijodo-Co(m)-cobester Losung ergaben tionsgemisches. braungrun und ten Edukt noch Nach zeigte einen circa im zwei Platinoxid, verwendet eine Stunden die H.Diehl hatte, an einer (25) schon zur benzolischen langsame Farbanderung des Reak¬ war die an der Luft Silicagel Dunnschichtchromatogramm langsamer laufenden grünen Fleck. bestandige Losung neben dem rotviolet¬ 35 - Wesentlich bessere Ausbeuten erreichte Umsatz jedoch man rasch ester-Lösung bar nicht nur das wenig Edukt, Bei der triumthiosulfat abgetrennten ten noch %. Diese %, schein¬ Na- vom deshalb neben 59 man wahrscheinlich auf der Säule % rückgebil¬ Auf umgesetztes Material berechnet beträgt Versuche in schönen an Desaktivierung des neutralem Aluminiumoxid liefer¬ aus ist speziell darauf Methylenchlorid-Hexan kugeliges oder Benzol-Hexan Material. hinzuweisen, dass sungen und bei RT kristallisiert werden muss, Ein Forcieren der nicht aus Bei stark Co(II)-cobester noch nicht trüben erfolgen. Jodo-Co(n)-cobester die schichtchromatogramm Mutterlauge erkennt man von Im Cobester-Komplexen wird nicht farblos sondern braunrot. einen erhöhten Anteil an Lö¬ Zur durch Stehen¬ man Methylacetat-Hexan-Lösung kristallinen in Produkt ausölt. während drei Wochen unter Calciumchlorid-Verschluss. den anderen untersuchten ergaben übersättigten Kristallisation darf erst nach etwa 48 Stunden angeimpften, aus diesem Cobester- da ansonst das röntgenographischen Untersuchung geeignete Kristalle erhielt lassen einer konnte grünschwarzen Nädelchen kristallisiert werden. geringerer Ausbeute ein unschönes, ester. des Benzol-Methylacetat 1:1 eluierte Jodo-Co(II)-cobester Kristallisationsversuche zu Na- ungünstigere Resultate. Methylacetat-Hexan satz wässerigen Silicagel an Nebenreaktion konnte auch durch nicht verhindert werden. Der mit Komplex n sich die benzolische Cob- längerer Reaktionszeit Reduktionsprodukts erhielt detes, Dijodo-Co(IH)-cobester-Edukt. 70 auch bei Chromatographie grünem Jodo-Co(II)-cobester noch 17 Silicagels 0,1 Reduktion mit einer braungrün, und nach fünf Minuten zeigte das Dünnschichtchro- noch umgesetzt wurde. die Ausbeute grüner Substanz und einen viel rascheren an durch Beim Schütteln mit dieser färbte triumthiosulfatlösung. matogramm - Jodo- Gegen¬ beim Im Dünn- Dijodo-Co(III)-cob- Dies konnte jedoch auch durch Kristallisation unter Stickstoff nicht verhin¬ dert werden. Dass es sich bei dem ziertes Derivat mit formal in Benzol deutlich. Komplex ist vorliegenden grünen Cobester-Komplex um zweiwertigem Kobalt handelt, NMR-Spektrum zeigt ein Wie schon bei dem auf Seite 33 besprochenen auch hier nur ein breites Signal ein redu¬ Co(ü)-cobester- zwischen 3 und 4 ppm erkennbar. - NMR-Spektrum 36 - Jodo-Co(II)-cobester von in Deuterobenzol TMS 3 2 ppm Figur 13 Viel schwieriger Art der axialen zu welches neben der in den Mikroanalyse Auskunft Bei dem reduzierten 127) Nicht axiale war die Liganden geben sollte, etwas nur Produkt sind allein die Intensitäten des ist auf Grund des oder Stickstoff sich, gewichts durch die zum von Verhältnis Massenspektrums, dass das Produkt einer die zweite axiale im zum Jods Standard (Mas¬ Acetyl geringer. Hydrierung der Natriumthiosulfat-Reduktion identisch sen über Methyljodids (Masse 142) und des (Masse 43) tigung, Wertigkeit des Kobalts temperaturabhängigen Fragmenten jenem des Dijodo-Co(III)-cobesters sehr ähnlich. se entscheiden als die formale Liganden. Das Massenspektrum der schwarzgrünen Kristalle, Ligandstelle gebrückte, wie mindesten in frei von ist, kann auch Dimere Mikroanalyse möglich. in einem mit einer Der jenem Sauerstoff Kobalt-Kobalt in Benzol mit ungeladenen Komplex oder durch Wasser besetzt sein. Lösung, wegen des 1115 ausschliessen. sondern auch unter Berücksich¬ Dijodo-Co(m)-cobester Bindung las¬ gefundenen Molekular¬ Entscheid zwischen Struktur 1) und 2) wird 37 - - OH2 U^j U^J 53,66 6,32 5,06 10,90 4,81 % 52,82 6,40 4,99 10,74 4,74 % Analyse A 53,42 6,39 5,14 10,83 4,89 % Analyse B 53,48 6,32 5,15 10,99 4,81 % 1) C52H73CoJN4014 MG 2) 1163 = C52H75CoJN4015 MG 1181 = Es wird daraus vorliegt. (vgl. S. geschlossen, dass der fünffach koordinierte Jodo-Co(II)-cobester Ob bei der Reduktion des Dijodo-Co(III)-cobesters 17) Jodligand abgespalten wird, kann erst durch der eine obere, labilere Rontgenanalyse ent¬ schieden werden. koordinierte, Fünffach wie sie m der Cobalamin- und Cobaloxim-Reihe bisher unbekannt auch mit anderen die formal zweiwertiges Kobalt enthaltende meisten, wie Ligandsystemen stellen Von diesen sind überdies Seltenheit dar. Dichlor-bis(2-dimethylaminoaethyl)oxid-kobalt(n) (vgl. I) das trigonal-bipyramidal eine Komplexe, sind, koordiniert und deshalb nicht mit Jodo-Co(II)-cobester ver¬ gleichbar. Durch Rontgenstrukturanalyse gesicherte, tetragonal-pyramidale Koor¬ dination zeigen a) zwei P. Aldermann Komplexe: (62): Nitroso-bis(dimethyldithiocarbamato)-kobalt(II) (vgl. II) b) M. Gerloch (36): Dichloro-(pyridin-2-aldehyd-2'-pyridyl-hydrazon)-kobalt(II) (vgl. HI) Fur das Grund tur Kaliumpentacyanokobaltat(H) von ESR- und geschlossen. in Losung wird Elektronenspektren auf Im Kristall liegt eine von H. Gray (63) auf tetragonal-pyramidale Struk¬ die dimere Form vor. - 38 - N«*f*° Cl ii i*y CHs h3c/ Cl to III Das UV/VIS-Spektrum tionsmaxima bei zeigt nur plexes. so erhält und 468 eine Löst nm Absorption cobester rechnet). Jodo-Co(II)-cobesters 323 nm, 435 nm, geringe Aehnlichkeit man man des grünen 299 nm, mit eine gelbe Lösung, mit deren 1163) resp. Absorp- in Benzol mit und 603 in Feinsprit UV/VIS-Spektrum jenem des bekannten und Extinktion übereinstimmt. = nm nm (vgl. Figur 14) jenem des anderen Co(II)-cobester-Kom- jedoch Jodo-Co(II)-cobester (vgl. Figur 15) (MG 492 (Die oder abs. Methanol, mit Banden bei 316 nm Co(II)-cobester-Komplexes Extinktionen wurden auf "Aquo"-Co(II)-cobester-"perchlorat" (MG = in Jodo-Co(II)1155) be¬ 39 - UV/VIS-Spektren von - Jodo-Co(II)-cobester £-104 /^S\ 2- \ I i in Benzol V ] ^ 700 600 500 400 300 nm Figur 14 —i 300 r- 1 1 400 500 600 Figur 15 Das IR-Spektrum des jodo-Co(III)-cobesters schieden. im fünffach Es Jodo-Co(n)-cobesters im ist daher zu Gebiet der vermuten, in Nujol differiert. gegenüber jenem des Dinur dass die Geometrie des Corrin hgandierten Komplex nicht wesentlich jodo-Co(III)-cobesters ist C=N; C=C-Streckschwingungen von wenig ver¬ -rr-Systems jener des oktahedralen Di- 40 - Da der kristallin und gensatz sowohl festen Zustand im analysenrein erhaltene Jodo-Co(II)-cobester wie auch in im Ge¬ Kobalt enthaltenden Cobaloximen oder zweiwertiges benzolischer Losung gegen Luftoxidation Tage stabil ist, wurde versucht, weitere Beweise fur die Zweiwertigkeit über des Vitamin B 12 zu - Kobalts m diesem Komplex zu erbringen. So wurden Untersuchungen mit fol¬ genden Methoden durchgeführt. a) magnetische Suszeptibilitat Co(III)-cobyrinsaure-heptamethylester-Komplexen b) Oxidation ad a) In nachfolgender Anmerkung zu wird die Theorie und Messung der magnetischen Suszeptibilitat besprochen. Anmerkung Die einer magnetische Suszeptibilitat Substanz. Man unterscheidet magnetisches Verhalten, wobei ist ein Audruck des magnetischen Moments paramagnetisches, diamagnetisches auf letztes in und ferro- dieser Arbeit nicht eingegangen wird. Paramagnetische Teilchen, die in Folge ungepaarter Elektronen ein eigenes magnetisches Moment haben, werden m ein Magnetfeld hineingezogen. Ihre mag¬ netische Suszeptibilitat ist positiv und nimmt mit steigender Temperatur gemäss dem Curie-Gesetz ab. Dieser Paramagnetismus ist sowohl durch den Spin als auch durch die Bahnbewegung der ungepaarten Elektronen bedingt. Diamagnetische Teilchen, die alle Elektronen in abgeschlossenen Schalen haben, werden aus einem Magnetfeld hinausgestossen. Ihre magnetische Suszep¬ tibilitat ist negativ und temperaturunabhangig. Findet man nun eine negative magnetische Suszeptibilitat, die temperatur abhangig ist, so sind zwei Falle zu unterscheiden: a) Der absolute Betrag steigt mit steigender Temperatur, d.h. cher, dem Curie-Gesetz folgender Paramagnetismus vor. b) Der absolute Betrag sinkt mit steigender Temperatur. Dies es liegt ein schwa¬ bedeutet, dass ein folgende Begrün¬ kleiner Paramagnetismus induziert wird. Dieser Effekt konnte dung haben: bei einem Uebergangsmetall mit gerader Anzahl d-Elektronen in lowAnordnung ein unbesetztes d-Orbital nur wenig hoher als das oberste be¬ setzte, so konnte schon eine kleine Aktivierungsenergie ein Elektron in das leere Orbital promovieren. In einem solchen Fall nähme die Besetzungswahrscheinlichkeit des leeren Orbitals mit steigender Temperatur zu. Liegt spm Molekül,z.B. ein Metallkomplex,neben einem paramagnetischen diamagnetische Teilchen z.B. Kohlenstoff-, Stickstoff Halogenatome, so wird in den allermeisten Fallen der Paramagnetismus Enthalt ein Metallzentralatom noch oder auch - 41 - - überwiegen, da dieser bei RT im allgemeinen circa 1000 mal stärker als der Diamagnetismus eines Teilchens ist. So sollte auch beim Jodo-Co(II)-cobester der diamagnetische Anteil des Cobesters mit C52H73N4O14 und des Jods einen Paramagnetisms des zweiwertigen Kobalts nicht überdecken können. In dieser Arbeit wurde die magnetische Suszeptibilität von Kristallen nach der Gouy-Methode bestimmt. Man misst dabei die Gewichtsänderung der Probe beim Einbringen in ein magnetisches Feld. Die Auswertung der Resultate erfolgt indirekt durch Vergleich mit dem für eine Referenzsubstanz, Misst man in diesen Versuchen Quecksilber, erhaltenen Wert. Probe und Eichsubstanz unter Messröhrchen mit der Probe direkt gleichen Bedingungen und im gleichen gleichem Füllvolumen, so lässt sich die Massensuszeptibilität aus den zwei Messungen berechnen: mo X I • go I m 0 X = Massensuszeptibilität X = Massensuszeptibilität der Eichsubstanz für Quecksilber 'go der Probe „ 2 27>10 = - ' - 13 m = eingewogene Menge Eichsubstanz m = eingewogene Menge Iq = Gewichtsänderung der Eichsubstanz I = Gewichtsänderung der Dimension von X: Wird im Probe Probe c.g.s.-System gerechnet, oder mit dem e.g.s.u./Mol Durch 6 c.g.s.u./g Zusatz: für die Molsusz. Multiplikation der Massensuszeptibilität man die Molsuszeptibilität Xm. so wird X dimensionslos für die Grammsusz. resp. angegeben. mit dem Molekulargewicht der Pro¬ be erhält Xm = Xg ' Moleew- Um den für die Berechnung des magnetischen Moments notwendigen, genauen Be¬ erhalten, müssen die diamagnetischen Anteile aller Ligandatome summiert und zu dem unkorrigierten Wert von Xm addiert werden. Aus der kor¬ rigierten Molsuszeptibilität und der Messtemperatur lässt sich dann das magne¬ trag von X zu tische Moment u (Einheit: Bohrsches [B.M] Magneton) = der Probe berechnen: 2,84\/xk0rr diamagnetischen Substanzen jedoch, deren Molsuszeptibilität einen negativen hat, kann das magnetische Moment nicht nach obiger Formel ausgerechnet und in Bohrschen Magnetonen ausgedrückt werden. Man gibt deshalb in solchen Fällen die nicht korrigierte Molsuszeptibilität an, und bei allfälliger Temperatur¬ abhängigkeit der Werte die Messtemperatur. Bei Wert 42 - a. - Magnetische Suszeptibihtat Die untersuchten Co(HI)-cobester nach der Proben, Jodo-Co(II)-cobester Dicyano-Co(HI)-cobester und Gouy-Methode 77°K m' 293°K m' Dijodo-Co(HI)-cobester 77°K m' Xm, 77°K Dicyano-Co(in)-cobester (Alle Werte Die Messungen wurden Festkorper-Physik Auffallend und unerwartet in standig Widerspruch magnetisms Das zu dem tat(II) Phänomen gleiche Substanz einer gelost ramagnetismus man = - = - und eine mit einer ein eine in 10"4 6,11 10"4 4,70-10"4 2,77-10~4 - = Institut fur am ist der fur den kristal¬ Das Resultat scheint voll- Benzol beobachteten Para¬ A.Adamson von Kobalt(II)-Komplex, von zweier -6,0- Kahumcyanid, Molsuszeptibilitat magnetisches Moment am 10 . so von von genstrukturanalyse man +1,24«10 ebenfalls man jedoch einen _3 1,72 B.M., im Kristall einem fest. was diamagnedie gleiche deutlichen Pa- Umgerechnet einem er¬ ungepaarten von Adams on zurückgeführt. Dadurch können die ungepaar¬ Kobaltzentralatome Cyanogruppe an Kobalt(II) entspricht. miteinander Elektronenpaarung bewirkt. Ob bei diesen eine Misst stellt (41) Kahumpentacyanokobal- dem Diamagnetismus der festen Substanz wird Dimerenbildung rekt oder über 293°K durchgeführt. NMR-Spektrum im lowspin-Anordnung einer ten Elektronen 6,03 293°K A. Menth Dr. jedoch wurde 1951 wasserigem Der unerwartete was - 10"4 obigen Zusammenstellung Molsuszeptibilitat in daraus Elektron und auf = 5,06 Im kristallinen Zustand ist diese Substanz beobachtet. mit und stehen. zu ebenfalls fünffach ligandierten halt von - Jodo-Co(II)-cobester gefundenen Diamagnetismus. im 77°K e.g.s.u./Mol) der ETH an der in = 293°K m' linen Vergleich Dijodo- zum gemessen. Jodo -Co(n) -cobester tisch und wurden jeweils bei in miteinander verbunden entschieden werden. Wechselwirkung kommen, Dimeren sind, zwei Metallatome di¬ konnte durch Ront- 43 - Jodo-Co(II)-cobesters Im Falle des stenschen Gründen unwahrscheinlich. 2, ca (gemessen 5 A gebildet meren am scheint eine Metall-Metall solche Kobalt-Kobalt Eine Dikobaltoktacarbonyl (59)) So beschreibt werden. - kann Tnphenylphosphin-Co(II)-cobaloxim-Dimeres, Form isoliert werden konnte. L. Smith Brücke eine mindestens von Koordination des noch Cobalamin ähnlicheren Kobalts Versuche haben ! dass gezeigt, fünffach andere Art der Di- resp. eine von L^lj fur ein Bindung bei Cobaloximdi- diamagnetisches bisher nicht (43) dem die an Bei der pyramidalen Polymerenbildung formulieren. Die ungepaarKobaltzentralatome konnten über gemeinsam gebundenes Jod gepaart werden. Q / baltzentren ware als von in tan. I Der aus dem ungefähr 5 A mit Smith o um etwa 3 A kurzer dargestellten 1,4-Dicobalamm-bu- Ueber die wirklichen Verhaltnisse im Jodo-Co(n)- cobester Kristall kann auch erst die Rontgenstrukturana- ' lyse Aufschluss geben. Die dass bei höherer Temperatur der Tatsache, negative Betrag der Suszeptibilitat etwas einen ganz schwachen Paramagnetimus hin, der ist, grosser nach dem Curie-Gesetz steigender Temperatur abnimmt. mit Die kehrte der Resultate der Messungen Temperaturabhangigkeit. Suszeptibilitat (vgl. S. hedralen Jodid kleiner. mit dem magnetismus 40). Ausgehend vom ist dieser Anmerkung angedeuteten sind beim (vgl. -Orbitale des zeigen ist der Mechanismus axialen der t» - umge¬ zu Para- erklaren Kobalts im Liganden okta- durch (44)). Kobalts werden durch t\ eine negative Betrag temperaturinduzierte Ersatz der axialen f. Cotton leeren Orbitalen des dadass der Abstand grosser wird. Temperatur Niveau-Schema der 3d-Orbitale des Folge,zur liegenden, Dijodo-Co(III)-cobester Möglicherweise der Einflüsse denkbar Die besetzten t„ am Bei höherer Dicyano-Co(in)-cobester zwei hoher in Folge,zur a) reiner dem Cob- den Kovalenzradien berechnete Abstand der beiden Kb- I deutet auf in Dimerenbüdung ist. I I / aus von cir¬ ligandierten Jodo-Co(II)-cobester lasst sich ten Elektronen zweier ein jedoch Methylengruppen notig vier im (42) G.Schrauzer das ester jedoch Bindung von -rr-Wechselwirkung Liganden stabilisiert. den e*-Orbitalen am Das mit hat Kobalt 44 - b) Die besetzten t„ -Orbitale des tiefer besetzten - Kobalts werden durch liegenden, Dadurch wird die 5p-Orbitalen Aufspaltung A des axialen der d-Orbitale mit -Wechselwirkung 2g Liganden destabihsiert. Kobalt kleiner. am 2g 7 Î / A' "<, t l2g ' 2g==afi= =**H=t, 2g Co Co Halogenidionen beeinflussen die Uebergangsmetalle der in der normalen Oxidationsstufe ser Liganden Im balt so eines t„ der in spektrochemischen Dijodo-Co(HI)-cobester klein werden, -Elektrons in gemäss dass ein e Serie eine geringe -Orbital und b), was zum ersten grossen Periode auch zur in der Stellung die¬ Ausdruck kommt. Aufspaltung A konnte die schon Fall der d-Orbitale Temperaturerhöhung Induktion von zur Ko¬ am Promotion Paramagnetismus ge¬ nügt. Im Gegensatz dazu cobesters ist die erwartungsgemass temperaturunabhangig. Zusammenfassend ist Dijodo-Co(III)- und Die Messresultate cobalamm Suszeptibihtat des diamagnetischen Dicyano-Co(m)- von X festzustellen, Jodo-Co(II)-cobester, sind mit dem = von dass alle drei in kristalliner H.Diehl Proben, Dicyano-Co(III)-, Form diamagnetisch (45) angegebenen -1,59-10" e.g.s.u./Mol vergleichbar. Die Wert fur sind. Aquo- temperaturab- hangigen Differenzen der Resultate liegen ausserhalb der Messfehlergrenzen und müssen durch die Struktur der Proben bedingt sein. 45 - Oxidation b. Liegt sollte so Co(III)-cobyrinsaure-heptamethylester-Komplexen zu Jodo-Co(n)-cobester dem grünen in wirklich zweiwertiges durch quantitative Oxidation desselben man (25) H.Diehl plex erhalten. konnte B 12 Kahumhexacyanoferrat quantitativ Jodo-Co(II)-cobester naheliegend es - mit eine einer in einen Co(III)-cobester-Kom- durch oxidimetrische Titration mit Aquocobalamin überfuhren. einphasige Oxidation benzolischen Kobalt vor, in Jodlosung Benzol zum Da jedoch beim angestrebt wurde, war Dijodo-Co(III)-cobester zu oxidieren. In einem Zugabe 92 in von praparativen Ansatz erhielt halben einem % Ausbeute. cobesters mit In Spektrum des Edukts Jodo-Co(II)-cobester aus anderen Versuch wurde die Oxidation des einem benzolischen einer man und die Jodlosung im nm (vgl. Figur 21, Kreuzungspunkte übergehen und das Jod erkannte auf, nur man Beersche am 90). wenn zwei Gesetz Nach Verbindungen den isobestischen Punkten. Absorptionsbanden des Dijodo-Co(HI)-cobesters mit überschüssigem Additionsverbindung setzung Wert von von graphisch 0,5 gegenüber 0,6 produkts vor, in der letzterem so das bildeten man Komplex von wieder schlecht abtrennen. von und am unverbrauchtem Abweichen nahezu konstant. Liegen lasst kleine Kristallisation des Schuttein mit wasseriger Dies Jodadditionskomplex analog ist. h I i i braune konnte auch bei der Um¬ nm beobachtet werden. Dijodo-Co(III)-cobester. ein nm Dijodo-Co(III)-cobester Durch kurzes Nebenprodukt bei 300 Joduberschuss bleibt solches bei der Mutterlauge. sung erhalt dass mit nm treten solche Dijodo-Co(HI)-cobester gebildete, Absorption mit starker Co(I)-cobester von Jod und (46) eine 474 Gleichzeitig blieb die Extinktion Kurven der Die 414 nm, ohne Nebenreaktion ineinander erfüllt ist. Das Vorliegen der von 336 nm, A. Weissberger der Extinktion bei 300 Ansteigen Das jeder Jodzugabe aufgenommenen bildeten nach S. Jodo-Co(II)- UV/VIS-Spektrum verfolgt. Kurvenschar mit fünf isobestischen Punkten bei 283 nm, und 633 durch Molaquivalent Jod kristallinen Dijodo-Co(III)-cobester Mit einem Rf- sich chromato¬ Mengen dieses Neben¬ Dijodo-Co(III)-cobesters Natriumthiosulfat Lö¬ scheint dem sie darauf aus hinzudeuten, Jodid und Jod ge¬ 46 - Im Zusammenhang Resultaten obigen mit - geplant, war plexen polarographische Messungen durchzuführen, als auch hatten. nommen tonitril G.Schrauzer (40) Löslichkeitsversuche (Fluka) zeigten jedoch trums, dass vermutlich durch Spuren den. Aussagen Um aber sichere Dijodo-Co(m)-cobester von von machen Alkohol Jodoliganden ausgetauscht können, zu sollten die Messungen unzersetzten, definierten Produkt ausgeführt werden. Da nem sungsmitteln, in denen axiale Messungen möglich sind, in reinstem Ace- Farbänderung und des UV/VTS-Spek- Hand der an (39) sowohl O. Müller Cobaloxim-Derivaten unter¬ Cobalamin- resp. an diesen Cobester-Kom- an wie sie nur wur¬ an ei¬ polaren Lö¬ in Jodogruppen abgespalten werden, polarographische auf musste Untersuchungen mit dieser Methode verzich¬ tet werden. In Anbetracht der Stabilität des Oxidationsempfindlichkeit des Jodo-Co(n)-cobesters an wurde die Zustand und in Lösung untersucht. in festem Die schwarzgrünen Kristalle werden Vitamin B 12 der Luft und bei Tageslicht über längere Zeit nicht zersetzt. Eine benzolische Lösung bleibt unter gleichen Bedingungen mindestens vier (UV/VIS-Spektrum! ). Tage lang unverändert In (innert Tagen) nol oder Aethanol erhält Lösung, trum nen einen man eine gelbbraune Co(n)-cobester Komplex Jodo-Co(ü)-cobester-Kristalle hält man enthält. Versetzt oxidiert. die gemäss man dagegen In Metha¬ UV/VTS-Spekdie dunkelgrü¬ Tetrachlorkohlenstoff, mit absolutem sofort eine violette Lösung. Chloroform und Methylacetat, Schwefelkohlenstoff wird die Substanz langsam Dies deutet darauf hin, so er¬ dass dabei das Lö¬ sungsmittel mit dem Substrat gramm der Lösung keinen Jodo-Co(II)-cobester mehr. Das violette Hauptprodukt mit einem Rf-Wert licagel von abtrennen und mutung, dass handelt, wird einerseits es 0,6 aus reagiert. liess So zeigt auch ein Dünnschichtchromato- sich durch Dickschichtchromatographie Tetrachlorkohlenstoff-Hexan kristallisieren. sich bei diesem Produkt um den an Si- Die Ver¬ Chloro-jodo-Co(HI)-cobester durch das NMR- anderseits durch das Massenspektrum gestützt. Das NMR-Spektrum lässt eindeutig auf einen Co(m)-cobester-Komplex schliessen. teten 142) Im Massenspektrum treten bei 200 lonenquellentemperatur die erwar¬ Spaltprodukte Methylchlorid (Massenzahl 50) und Methyljodid (Massenzahl mit einer Intensität wählten von 430 Acetyl (Massenzahl 43) stoff eingeschlossen % auf. resp. Da 120 jedoch % gegenüber dem als Standard ge¬ diese Kristalle Tetrachlorkohlen¬ haben, und bis anhin die Kristallisation aus einem anderen - Losungsmittel nicht gelang, 47 - ist die vermutete Struktur nicht durch eine Mikro¬ analyse gesichert. Die Reaktivität lichen Charakter Chlorradikals gegenüber Tetrachlorkohlenstoff lasst Jodo-Co(n)-cobesters des aus das Cumol elektrophile Chloratom hingen reagiert wohl beide Die auf radikalahn- einen Abspaltung dem Tetrachlorkohlenstoff entspricht dem Verhalten nucleophilen Radikals. Das Kobalt wird an schliessen. eine zur unter formaler Abgabe dreiwertigen Stufe oxidiert. eines eines eines Elektrons und p-Xylol Mit Jodo-Co(II)-cobester Losung nicht, benzolische ob¬ stabile Radikale bilden. Cl Cl Cl Cl Cl Cl Cl Cl / Com/ I Zum Beweis ablauft, dafür, wurde versucht merisation Gegensatz Styrol von zu einem dass die Reaktion mit Tetrachlorkohlenstoff radikalisch mit dem entstehenden zu initiieren. Ein Blindversuch ohne Trichlormethylradikal entsprechender Vorversuch Tetrachlorkohlenstoff, die eine zeigte, erwartete Poly¬ im Po¬ lymerenbildung. L. Haines chlorkohlenstoff lymerisation nomeren (47), der die Reaktion untersuchte, von Fasst man vor in mit Tetra¬ analoge Radikalbildung weiteren Versuchen die Resultate der verschiedenen im Mo¬ Jodo-Co(n)-cobesters mit geprüft werden. Versuchsgruppen zusammen, allem auf Grund der Oxidationsversuche die Annahme Kobalts durch Po¬ Die Verwendbarkeit dieses fur den Radikainachweis bei der Reaktion des scheint wertigen Dimangandecacarbonyl eine Methylmethacrylat nachweisen. Tetrachlorkohlenstoff soll so von konnte dabei eines zwei¬ Jodo-Co(n)-cobester gerechtfertigt. Ergänzende Versuche, 48 - ein - in festem und Elektronenspinresonanz-Spektrum gelöstem Zustand und die Messung der magnetischen Suszeptibilität einer benzolischen Lösung Röntgenstrukturanalyse G. sind sowie eine vorgesehen. AIkyl-Co(III)-cobyrinsäure-heptamethylester-Komplexe Kobalt-Alkylbindung Die für eine natürliche chanismus der Coenzym B 12 Arbeit oximen beim schon beabsichtigt, in Coenzym B 12, Dunkelkammer einer trieben wurde. Das In Alkylcobalaminen durchgeführt, V/60 speziellen Fällen, Zeit eine neuartig Taschenlampe diente, wenn das mit es Co(I)-cobesters sammeln. zu Alkylcobal- Untersuchungen der Alle Versuche wurden als Lichtquelle die aber mit eine nur in 60 V be¬ Experiment erforderte, wurde Rotlicht verwendet. dessen Cobalamin-Analogon (vgl. F.Wagner (8)), schiedentlich beschrieben worden ist zu. Erfahrungen und den auch die wobei W Birne Methyl-Co(HI)-cobester-Derivat, setzen des bekannten absolut nur Kobalt-Alkyl Bindung den dargestellten Alkyl-cobester-Kbmplexe. drei Meter Höhe montierte 220 für kurze nicht Cobester-Modell am festgestellte Lichtempfindlichkeit erschwerte in dieser Arbeit deshalb war abhängigen Reaktionen eine zentrale Bedeutung war über die Stabilität und die Reaktivität der Die B 12 Coenzym sondern vermutlich kommt ihr auch in dem Me¬ Verbindung, vorliegenden In der im mit einem schon ver¬ konnte durch Um¬ elektrophilen Alkylierungsreagens leicht erhalten werden. Beim die Einspritzen auch bei weiterer Entstehen eines ben, von Methyljodid Lösung sofort rot, wobei im Methyljodidzugabe grüne Aether-Hexan-Phase wurde zur Umsetzung bestehen blieb. Diese mit Jod, Trübung die Trübung wird dem positiv geladenen Methyl-Co(III)-cobester-Komplexes zugeschrie¬ der mit einem Wasser-Methanol 1:1-Gemisch ausgezogen werden konnte. Das Ausbleiben eines Jodoliganden am Farbumschlages bei dieser Operation macht einen axialen Methylierungsprodukt einheitliches Gegenion zu geben, von H.Gschwend (48) als unwahrscheinlich. wurde der Natriumperchlorat-Lösung geschüttelt. ten in die Gegensatz Um dem Komplex ein Wasser-Methanol-Auszug mit einer Dieses Anion hatte sich schon bei Arbei¬ Gegenion positiv geladener Metallkomplexe be- 49 - wahrt. Methylenchloridextraktion erhielt Durch Zustand und ziegelrot Struktur in Losung dunkelgelb Dunnschichtchromatogramm mit an erhalten. zu unteren hin, dass (vgl. pfindlichkeit ein in an Flecken, und neutralem und er¬ gute eine die auch schon bei dem positiv worden ist, gela¬ deutet partieller Austausch des ein durch Fremdionen stattfindet. Aus der Lichtem- gewanderten Flecken den Dunnschichtchromatogrammen kann Austausch der Methylgruppe ausgeschlossen werden. Anfanglich wurde das Aluminiumoxid, te sich als das war Rohprodukt. Methyljodids mieden werden kann. konnten bisher zur Das aminen Methylenchlorid extrahierte Produkt stellte sich Es aus Beide das heraus, amorphe Material zeigt mit jenen des in weitgehend wie und durch keine nm und 460 nm. Methylgruppe Die grosse resp. kann die Alkyl-Komplex derart erniedrigen, dass vergleichbar wird. Neben nm ver¬ rohe, G.Schrauzer sie (16) Alkylcobal- zwei Ab¬ Aehnlichkeit dieser Spektren des B 12 ist bemerkenswert! Elektronendichte mit jener ordnete Corrinoid-Komplexen die ot-Bande der Absorption jener bei 306 dem spezielle Me¬ mit den am hohen und dadurch die Elektronegativitat der Stickstoffatome des rivat das auch bei den Methylenchlorid das Co(n)-cobester-Komplexes Der induktive Effekt der vor gebracht werden. (in Wasser) beobachtete, typische UV/VIS-Spektrum sorptions maxima bei 305 zeig¬ dass durch sorg¬ violetten Phase chromatographierte Losungsmittelgemisch keinem Kristallisation eine Es UV/VTS-spektroskopisch Verunreinigung des Produkts Produkte, basischem an chromatographiert. Abtrennen der bei der Reduktion entstehenden Einspritzen des thode mit das sich dafür als geeignet erwies, dass das mit Chloroform eluierte Produkt aber, nicht reiner faltiges im vorlaufig die gelingt nicht, es Co(II)-cobester gemacht und dem 18) Liganden S. auch wie auf dem Adsorbens wahrscheinlich der festem in da Hinweise auf angenommen, Sihcagel Beobachtung, Diese Aquo-cyano-Co(III)- darauf das wird Benzol-Methylacetat 1:4 als mobile Phase, scheinen meist mehrere verschmierte denen Produkt, ein Fur dieses Liganden fehlen. basischem Aluminiumoxid Trennung man war. Methyl-Co(III)-cobesters-Derivates eines den zweiten axialen Im - in bei 460 im den nm Kobalt er- Cornnsystems Co(n)-cobester-Dealkylligandierten und die »--Bande zu. Methylenchlorid ist als Lösungsmittel fur Methyl-Co(HI)-cobester-perchlorat-Komplexes Wasserl:! geeignet (vgl. Figur 23, S. 98). auch ein ein In Alkohol UV/VIS-Spektrum Gemisch von (Methanol des Monoglym- oder Feinsprit) 50 - zeigt das Spektrum bei 480 dung auch (20) E.Winnacker von Bei Aufnahme des die Schulter bei 330 nm, eine schwach erkennbar ist. ganz nur zeigt dieses Benzol in bisher unerklärte Diese bei durch die Bil¬ bedingt vermutlich Schulter, eine Methyl-alkoholo-Co(HI)-cobester-Kbmplexes. eines trums nm - Spek¬ Methylenchlond in Erscheinung wurde Methyl-Co(III)-l,2,2, 7,7,12,12-hepta- einem methylcornn-Kbmplex beobachtet. Das NMR-Spektrum jenen anderer in Chloroform ist Co(HI)-cobester-Denvate Stellung dagegen erscheint bei 6,7 ppm, nach tieferem Feld entspricht. (Vitamin 12) ebenfalls um entspricht wohl der Effekt ser B chemischen Serie zu Donatoreffekts der 0,2 und 4 ppm Proton der 10- vinylische Verschiebung zu 0,3 um (15) H. Hill von Vergleich im jenem des ppm ist das CyanocobalDie¬ ppm nach tieferem Feld verschoben. Stellung der Liganden gemäss der erwartenden einer Beobachtungen Nach Vinylproton-Signal des Methylcobalamins amins Das gegenüber dem ebenfalls positiv ge¬ was Aquo-cyano-Co(m)-cobester-Komplex ladenen Gebiet zwischen 1 im ahnlich. ist aber Tendenz, Methylgruppe überraschend. Die in in der spektro- Anbetracht des induktiven Protonen der axialen Methyl¬ gruppe dagegen sind stark abgeschirmt und erscheinen als Singlett bei -0,15 ppm rel. TMS. Diese zu nach hohem Feld konnte nicht sondern auch auf die sterische system, pe mit Verschiebung nur auf das Corrin- Wechselwirkung der axialen Methylgrup¬ Methylestergruppen zurückzuführen sein. Das entsprechende Signal Methyl-aquo-Co(in)-l, 2,2,7,7,12,12-heptamethylcorrin-perchlorat tritt bei im 0,78ppm auf. Wahrend bei der Umsetzung an kristallinem, musste Co(III)-cobester-Komplexes bezügliche Information ein nur Signal bei 6,71 kyliert werden, Coenzym Im ppm wobei Gegensatz zu Methyljodid ein erhalten. mit eine werden, aus hohe Ausbeute stereospezifische Reaktivität des Da dieses dem NMR-Spektrum sowohl fur den axialen eine dies¬ Methylli¬ Singlett bei -0,15 ppm als auch fur das Vinylproton zeigt, wird auch der wird, Ligandstelle am Co(I)-cobester stereospezifisch dass der Alkylligand in nur al- Analogie zum (20) Me- Kobalt besetzt. diesen Resultaten erhielt des Methyljodid die vorliegenden Fall des amorphen Methyl- im versucht angenommen B 12 die obere thylierungsversuchen len zu scharfes ein B 12 einheitlichem Material fur Co(I)-cobalamins spricht, ganden von E.Winnacker bei 15-Cyano-Co(I)-l,2,2,7,7,12,12-heptamethylcorrins lichtempfindliches Gemisch, das im NMR-Spektrum Methylgruppen entsprechende Signale bei 0,20 und 0,25 ppm im zwei mit axia¬ Verhältnis 51 - 2:1 zeigte. Molekulteil. satz mit dem Chlor Ligand kein Die Intensität der zu. jeweils relativ spaltung von zur S. zu Massenzahl konnte, wurde ganden den in 16). zum 50 steigt dass geladenen Komplexe (wie Methylcobalamin die vom beim an Chlorid) Chlor oder (49) einem % bei tritt die Ab¬ als Kon¬ kombinieren, 3 % jener der Mas¬ was Komplexbildungskonstante Cyanocobalamin zum Bindung Verschiedene Autoren (vgl. un¬ eine chromatographische Alkylcobalammen zum Er Reini¬ den dass beim gibt an, Methyl-cyanocobalamin 10 ist und mal begründet dies Einfuhrung amomscher Liganden Bildung ungeladener, kristalliner Alkyl-Co(III)- unter Jodmethylgruppe Diese Be¬ Bildungstendenz dieser Dicyano-Derivat Die als untere axiale Li¬ einzufuhren. der schnellen Austauschbarkeit Liganden untersucht. unteren cobester-Komplexe gelingt jedoch dann, Spaltung an hat bei verschiedenen einer der auf 64 eher kristallisiert werden Cyanid der geringen trans zierten die In¬ steigender Tempera¬ % bei 205 50 bei 210 Masse Chlorid oder Azid, Nitrit, thermodynamischen Transeffekt. oder Gegen¬ bestimmt werden. mit dem Co-C Im jedoch Acetyl (Masse 43) Temperatur mit ungeladener Komplex ein H. Hill kleiner als jene Cyanmethyl- 15 Bei höherer derartiger Liganden (wie Azid oder Cyanid), zu von Methyl-Co(III)-cobester-perchlorat-Komplex gegenüber erreicht Vergleich geeigneten Aquo-cyano-Co(HI)-cob- mühungen scheiterten aber entweder Transeffekt vereinigt hat. %. Dadurch kann der Anteil des durch die axiale Methyl¬ versucht, gung verhinderte. Liganden axialen dass sich der bedeutet, Dies sondern nimmt mit 43. Massenzahl gebildeten Methylchlor ids Annahme, eines jener des Standards Maximum, Bei dem 34 43 und bei 255 In der Kombination dem Perchloratamon ester-Perchlorat betragt die Intensität der gruppe Grignard- mit abgespaltene Methylgruppe erscheint bei Gerust-Methylgruppen, die ebenfalls kurrenzreaktion auf. se Alkylierung Co(III)-cobester-Derivaten Verhältnis im und 240 180 zwischen 240 aus den bisher untersuchten zu tensität der Masse 50 tur Die thermisch Methylchlorid (Masse 50) (vgl. als axiale Möglichkeit der nicht beobachtete bisher einem 205 durch nur Massenspektrum des Methyl-Co(m)-cobester-perchlorat-Kömplexes zeigt Das mit konnte erhalten werden. Reagentien eine Alkylprodukt Einheitliches - wenn die axiale ersetzt wird F. (vgl. Wagner (8)) Kobalt-Kohlenstoffbindung in S. Methylgruppe durch eine 53). haben sich mit der lichtindu¬ Alkylcobalammen und im Coen- 52 - zym B 12 befasst. jedoch nur in hchkeit der - Wahrend diese Reaktion bei den naturlichen polaren Losungsmitteln untersucht werden Cobester-Komplexe zusätzliche Versuche wurde auch früheren Arbeiten kann, Alkyl-Derivaten erlaubt die Wie Benzol. in Los- schon in dieser zwischen aeroben und anaeroben Be¬ in dingungen unterschieden. Belichtung des Methyl-Co(m)-cobester-perchlorat-Komplexes Bei Monoglym-Wasser 1:1 Gemisch mit Banden bei 345 nm, 490 Co(III)-cobester-Denvat Spektrum Setzt mit man dem die )f <* - und ß hypsochrom nm in dem in UV/VIS-Spektrum S. 98 Zugabe 491 von und 518 nm Monoglym-Wasser 1:1 in 524 -Banden bei Ebenfalls Spektrum liegen. Durch bei 348 nm, Belichtungsprodukt -Bande bei 342 ein (vgl. Figur 23, nm Tetramethylammoniumhydroxid-Losung zu, in man ), das fur em Trifluoressig- Diaquo-Co(in)-cobester-Komplex zugeordnete dem Absorptionsmaxima dem 510 und nm charakteristisch ist. entsteht daraus das saure der Luft erhalt an erhalt so und 507 nm Vergleich im man nm einen ein (vgl. (14)). Tropfen 0,1 n UV/VIS-Spektrum, bathochrom und die nm dem mit Saure erhaltenen zu Monoglym-Wasser 1:1 wurde fur das Belichtungs¬ produkt des Methyl-Co(in)-cobester-perchlorat-Komplexes der pK*-Wert durch Mikrotitration zu 6,2 ca. bestimmt. dingungen gemäss UV/VIS-Spektrum Form das Diaquocobinamid Nach aerober Wasser in Belichtung absolutem Methanol oder 495 den bei 336 nm, 512 nem (in Benzol), nm nm in Bei der warten. 0 und der basischen H. Hill (49) gibt fur an. nm Benzol UV/VIS-Spektren zeigen die (in Methanol) resp. 340 nm, 495 fuhrt beiden Fallen in 499 nm und 525 zu einer in Ban¬ und nm Co(III)-cobester-Komplexe sprechen. Zugabe nach 350 nm, von sofortigen ei¬ Ver¬ nm. Photolyse des Methyl-Co(III)-cobester-perchlorat-Komplexes unter Bedingungen Eine dreimal Stunde dem analog dem ist am unter diesen Umstanden ein aus Alkylcobal- Co(II)-cobester-Komplex Hochvakuum entgaste und bei 0,005 zu Torr wahrend er¬ ei¬ Tageslicht ausgesetzte benzolische Losung des Methyl-Co(III)- cobester-Komplexes zeigte chenden Banden bei 314 dieses 6, von Be¬ Methyl-Co(III)-cobester-perchlorat-Komplex oder Coenzym B 12 gebildeten B 12 aminen sauren Erwartungen. pK-Wert absolutem Tropfen Trifluoressigsaure anaeroben einen Belichtung unter neutralen Gemisch der ein Wert den von und 512 die fur schiebung der Banden ner dieser vorliegt, entspricht Da bei der Spektrums nm dann auch die und 475 mit jenem des nm. Edukts einem Co(II)-cobester-Komplex entspre¬ Die schon oben erwähnte Aehnlichkeit geht aus Figur 16 deutlich hervor. 53 - UV/VIS-Spektrum von - Methyl-Co(III)-cobester-Komplex tungsprodukt H in anaeroben Belich¬ resp. Benzol t-104 Methy l-Co(ni) -cobester Komplex - 500 400 300 nm Figur 16 jedoch B 12 Wahrend ist die stabil Wasser rasch in erhaltene benzolische (vgl. aerober auch S. 33). Lichtspaltung durch Luft oxidiert tion des ebenfalls Kobalt(III) Aquo-Co(ni)-cobalamin zu Co(n)-cobester-Losung an oxidiert Dies deutet darauf hin, Benzol entstehende Co(ü)-cobester-Komplex in sondern dass wird, dass der vermutlich auch bei möglicherweise Folgeprodukte gebildeten Methylradikals wird, der Luft über Stunden nicht direkt einer mit Sauerstoff fur die Oxidation Reak¬ zum verantwortlich sind. Der fur Austauschversuche (vgl. S. 60) benotigte Trideuteromethyl-Co(III)- cobester-Komplex wurde analog der undeutenerten Verbindung methyljodid Co(I)-cobester dargestellt. und glym-Wasser 1:1 UV/VIS-spektroskopisch Das aus amorphe Produkt mit dem Trideutero- war Mono- in Methyl-Co(III)-cobester-per- chlorat-Komplex identisch. Das IR-Spektrum des deutenerten Produktes unterscheidet des undeutenerten Spektrum fehlt nur in C-D-Streckschwingung bei der deutenerten Methylgruppe bei -0,15 Im der bei 2100 sich cm von und jenem im NMR- Verbindung lediglich das Signal der axialen ppm. Massenspektrum erreicht die Intensität des Trideuteromethylchlonds (Massenzahl 53) das Maximum. bei 210 mit 10 In demselben % relativ zum Spektrum betragt Standard Acetyl (Massenzahl 43) die Intensität der Massenzahl 50 54 - (Methylchlorid) nur 2 % (rel. 43) - während Aquo-cyano-Co(m)-cobester-perchlorat beim Um gewisse Spezialfragen der Reaktivität substituierter genden 3 die Co(in)-cobester-Derivate Jodmethyl- und Temperatur gleicher bei sie % (rel. 43) ausmacht. der axialen an untersuchen zu Methylgruppe können, wurden im Fol¬ Cyanmethyl-Co(III)-cobester-Komplexe dargestellt. CH2I CH„I„ ?J— / Co1 / / Co111/ I Diese ebenfalls stark Co(I)-cobesters Unterschied lichtempfindlichen Methylenjodid mit Dies deutet darauf mit dass in diesen beiden Fällen hin, einem Methanol-Wasser-Gemisch da dabei vermutlich ein labiler in diesen beiden Fällen das zur Trockene Das durch nes axialer schlug mit dem Rf-Wert =0,4. mit an aus rotes 0,3. Durch in Aether- die Farbe nach hellrot um, am Daher wurde Rotationsverdampfer mit aus Produkt mit einem Rf-Wert Zusätzlich beobachtete Nädelchen; von 0,6 und ein brau¬ ein rotviolettes Nebenpro¬ worden ist. mg Das rote Produkt kristallisier¬ misslangen wie auch Von den roten Kristallen wird angenommen, analyse bestätigt man beim braunen Methylenchlorid-Hexan Jodmethyl-jodo-Co(HI)-cobester handelt, im Benzol-Methylacetat 1:4 als mobile Silicagel-Dickschichtchromatographie konnten 120 Benzol-Hexan in feinen züglichen Versuche Struktur: ungeladene, Methylenjodid erhaltene Rohprodukt zeigte Silicagel rote und 70 mg braune Substanz isoliert werden. te trüb sondern Jodoligand verdrängt wird. Reaktionsgemisch direkt Alkylierung hauptsächlich ein dukt mit Rf Im hierbei eingedampft. Dünnschichtchromatogramm Phase Einspritzen des Reagens nicht war Komplexe gebildet werden. Beim Ausschütteln der Aether-Hexan- Hexan lösliche Phasen Jodacetonitril synthetisiert werden. Darstellung des Methyl-Co(HI)-cobester-Komplexes zur die Aether-Hexan-Phase nach dem klar. resp. Produkte konnten durch Umsetzen des dass es aus alle diesbe¬ Benzol-Hexan. sich dabei um den obwohl diese Struktur durch keine Folgende Punkte jedoch sprechen für Mikro¬ eine solche - - Der Rf-Wert spricht fur einen - Der Komplex enthalt gemäss - Der Komplex ist - Die Struktur der 55 - ungeladenen Komplex. Mikroanalyse pro Mol ungefähr 1,9 Jodatome. lichtempfindlich. Photolyseprodukte. Belichtung einer benzolischen Losung der Substanz unter aeroben Bedingungen entsteht Dijodo-Co(III)-cobester in einer UV/VIS-spektroskopischen Ausbeute von 95% (vgl. Figur 24, S. 104). Wird die gleiche Reaktion unter Argon durchgeführt, so entsteht zu 75 % gemäss UV/VIS-Spektrum und Dunnschichtchromatogramm Jodo-Co(n)-cobester. Nach erfolglosen Kristallisationsversuchen an dem Rohprodukt konnten nach einer Chromatographie an Silicagel noch 25 % kristalliner Jodo-Co(II)-cobester isoliert werden, welche UV/VIS-spektroskopisch und dunnschichtchromatographisch mit analysenrei¬ Bei der Material identisch nem - waren. Das Massenspektrum Im Massenspektrum treten nur Jod (Massenzahl 127) und Methyljodid (Mas¬ senzahl 142) als temperaturabhangige Fragmente auf. Erwartungsgemass ist diesem Spektrum die relative Intensität m von 142 gegenüber 127 grosser als Di]odo-Co(III)-cobesters. Bei der angenommenen Jodmethyl-jodoStruktur kann Methyljodid im Massenspektrum nicht nur durch Kombination ei¬ sondern auch durch Abstraktion nes axialen Jodids mit einer Methylgruppe, eines Wasserstoffs durch die Jodmethylgruppe gebildet werden. jenem des in Die UV/VIS-, IR- und NMR-spektroskopischen sten Cobester-Derivaten wenig aufschlussreich. mit Absorptionsmaxima die anderen 20'000 (bei 350 nm, 468 Alkyl-Co(in)-cobester-Komplexe eine 294 bei 294 nm, Einfluss des Jods tertiären ser nm und 530 maximale zeigt nm mei¬ Benzol in auch wie Extinktion von gegenüber jenem des Methyl-Co(III)-cobester-Komplexes Methylgruppen zugeordnet werden. der bei den nur nm). tieferem Feld verschoben. tons wie UV/VIS-Spektrum NMR-Spektrum ist das Signal der axialen Methylenprotonen durch den Im heit Daten sind Das 10-Stellung Arbeit bei einem bei Es kommt dadurch des Cobesters ppm. die Nahe des liegen und zu In Deuterochloroform 6,30 in kann nicht mehr erscheint das Signal Dies stellt den tiefsten Wert ungeladenen Komplex in nach Singletthaufens der mit Sicher¬ des dar, Vmylpro- der die¬ in diesem Losungsmittel beobachtet wurde. Die Struktur der braunen Substanz konnte bisher werden. Da nicht kristallisiert werden zichtet. Die konnte, thyl-jodo-Co(III)-cobester und im eindeutig geklart eine Mikroanalyse ver¬ spektroskopischen Unterschiede gegenüber dem sogenannten Jodmesind viel geringer als auf Grund des unterschiedlichen Färbung auf der Dunnschicht-Platte sen- nicht wurde auf IR-Spektrum sind die beiden zu Verhaltens und der erwarten ware. Verbindungen sehr ähnlich. Im Mas¬ Im NMR- - 56 - Spektrum zeigt das Signal des Vinylprotons eine Jodmethyl-jodo-Kömplex unerwartet grosse im Vergleich rel. zu Deuterobenzol " Jodmethyl-jodo" braune Substanz Eine ähnlich unterschiedliche tren der beiden Substanzen in 17 sogenannten Lösungsmittelabhängigkeit. Stellung des Vinylprotons ppm zum TMS in in Deuterochloroform 6,35 6,30 6,40 6,65 Lösungsmittelabhängigkeit zeigen Benzol, Methylenchlorid die UV/VIS-Spek¬ und Methanol (vgl. Figur 19). - UV/VIS-Spektren des sog. Jodmethyl-jodo-Co(in)-cobesters und der braunen Substanz <.104 in Benzol -'"""\^K"~^ "Jodmethyl-jodo" x---X braune Substanz \ 300 500 400 600 Figur 17 £-104 in Methylenchlorid "Jodmethyl-jodo" 2 1 - braune Substanz - 300 400 500 Figur 18 600 nm 57 - - in abs. Methanol e-io* "Jodmethyl-jodo" /~\ braune Substanz * \ ^ö<r 400 300 Figur 19 Das UV/VIS-Spektrum einer an der braunen Substanz deutet auf die der Luft belichteten benzolischen Bildung von apolarem Lösungsmittel grosse spektroskopische stanz mit dem sog. schen Daten und der lassen eine strukturelle Verbindungen vermuten. Auf Herstellungsmethode wären Die in Aehnlichkeit der braunen Sub¬ Jodmethyl-jodo-Co(ni)-cobester Verwandtschaft der beiden Lösung Dijodo-Co(HI)-cobester. u.a. Grund der folgende spektroskopi¬ zwei Strukturvor- schläge möglich. ZW V2 A) B) I du CH2I i Struktur A scheidet auf Grund der Resultate griff re Co(I)-cobesters auf ein Jod des zu widersprechen scheint, Methylenjodids gebildet Jodo- und dann der untere Die Unterschiede (43) (vgl. S. 43) aus. könnte durch werden. nucleophilen An¬ So würde zunächst der obe¬ Jodmethyl-Ligand gebunden. gegenüber Dünnschichtchromatogramm L.Smith aufgestellten Hypothese der stereoselektiven Reakti¬ Struktur B, welche der oben vität des von dem sog. wie auch im Jodmethyl-jodo-Co(in)-cobester NMR-Spektrum im in Chloroform und im 58 - UV/VIS-Spektrum bilität des Jodo-Liganden als den beiden isomeren Ligand Komplexen erklaren lassen. der oberen Koordinationsstelle labi¬ an stand oder in sie m dass möglich, es Benzol gelost den wahrend spektroskopischen Daten, die Auf Grund dieser gerecht werden, scheint stellt, konnten sich durch die unterschiedliche Sta¬ möglicherweise gegenüber polaren Losungsmitteln "empfindlicher" der unteren. an sen in dass dieser So ist anzunehmen, ler und Methylenchlorid in - die braune Substanz Hypothe¬ den in ist festem Zu¬ Jodo-jodmethyl-Co(m)cobester (Struktur B) dar¬ polareren Losungsmitteln als Jodmethyl-Co(m)-cobester- jodid vorliegen wurde. Jodmethyl-jodo-Co(ni)-cobesters Um die Struktur des sogenannten chern und über die Reaktivität der de deren Reduzierbarkeit Jodmethylgruppe Methylliganden zum Aufschluss untersucht. Da zu si¬ erhalten, zu wur¬ bei allen jedoch diesbezüglichen Reaktionen die Produkte nicht mehr lichtempfindlich waren, wird angenommen, dass bei den untersuchten Kohlenstoff-Bindung unumgänglich Bedingungen die Spaltung der Kobalt- So entsteht auch bei der ist. Methyl-Co(in)-cobester-perchlorat-Komplex Methanol grüner empfindlich Spektrum Co(I)-cobester, ist und das sche Messungen dass schon die Addition von 315 und 468 nm G.Schrauzer einem von von absolutem in nicht mehr licht¬ Co(II)-cobester-Komplex entsprechende UV/VIS- einem mit Banden bei Luftoxidationsprodukt dessen Behandlung Natriumborhydrid mit zeigt. nm (40) an Elektron zeigten polarographi- Ebenso Methylcobaloximen zur Spaltung in Acetonitril, der Kobalt-Kohlenstoff- Bindung fuhrt. Alle folgenden Reduktionsversuche wurden mit einigen Jodmethyl-]odo-Co(in)-cobesters durchgeführt, qualitativ durch UV/VIS-Spektrum oder so dass Milligrammen des sog. die Reaktionsprodukte Dunnschichtchromatographie nur identifiziert werden konnten. - Natriumborhydrid Mit oder Calciumhydrid oder Natriumamalgam, methanohscher Losung erhielt noch einem erkennbaren, trum bei 470 - Mit man schwachen ein lichtunempfindliches Adsorptionsmaximum alle in Produkt mit im nur UV/VIS-Spek- nm. Zink und Ammonchlorid zol entstand gemäss 490 nm und 520 nm in Methanol oder mit UV/VIS-Spektrum ein mit Silberperchlorat Absorptionsbanden Diaquo-Co(in)-cobester-Komplex. bei in ca. Ben¬ 350 nm, 59 - - Mit Co(I)-cobester ne, nicht - in Aether-Hexan als Reduktionsmittel erhielt lichtempfindliche Lösung, deren UV/VIS-Spektrum rid mit Maxima bei 325 nm, 480 430 nm, nm und 635 eine man grü¬ Methylenchlo¬ in jenem des Jodo-Co(n)- nm cobesters ähnlich ist. Konnte auch unter keiner der obigen thyl-gruppe zum Methylliganden gefunden werden, Anhaltspunkte dafür, werden Bedingungen dass das Jodid der so Methylenchlorid violettes mit folgende Versuch Methylengruppe durch Cyanid gruppe bei 2135 Lösung des substituiert Jodmethyl-jodo-Co(III)-cobesters sog. im Dunkeln erhielt wässerigem Kaliumcyanid Produkt, das cm" unten beschriebenen im IR-Spektrum neben der Bande noch ein ganz schwaches Jodids entstandenen tritt bei 160 Cyanmethyl-Co(III)-cobester-Derivat (Massenzahl 41) Cyanid-Behandlungsprodukt, lenchlorid bei 330 nm, ches Material 395 nm, Isolierung Die Darstellung des jenigen des sog. spritzen dessen nm tritt die 590 nm und genauere liegen, weder Da in Charakterisierung als Jodacetonitril sofort mit dem jedoch Methy¬ ein einheitli¬ werden konn¬ chromatographiert Cyanmethyl-jodo-Co(in)-cobesters reagierte Im Bande Massenspektrum. Darin unmöglich. konnte Jodmethyl-jodo-Co(m)-cobesters durchgeführt im Dunkeln gleiche UV/VIS-Absorptionsbanden und ein dunkel¬ Cyanid- zeigt. mit grosser Intensität auf. noch kristallisiert noch darstellte, erwiesen sich 460 man axialen in eines durch Substitution des auch das Cyanmethylliganden spricht Acetonitril einer Signal bei 2200 cm" jedoch sehr viel intensiver auf. Für das Vorliegen te, gibt der kann, ohne dass dabei die Kobalt-Kohlenstoffbindung gespalten wird. Durch Vibrieren einer dieses Reduktion der Jodme- eine analog der¬ werden. Beim Ein¬ Co(I)-cobester unter Bildung einer klaren dunkelroten Lösung. Das direkt durch Entfernen der Lö¬ sungsmittel und des überschüssigen Reagens liche Produkt erwies sich im Methylacetat (Rf = 0,5) erhaltene, lichtempfind¬ 1:4 als mobile Phase als Gemisch eines weinroten und drei bräunlichen Nebenprodukten an Cyanmethyl-jodo-Co(in)-cobester UV/VIS-Spektrum jodo-Co(ni)-cobesters um in Benzol ist 20 - 40 nm (Rf = mit Benzol- Hauptproduktes 0,8, 0,2, 0,1). Silicagel konnte aus Nach zwei¬ Benzol-Hexan der kristallin erhalten werden. Fall ist die angenommene Struktur durch eine Das Vakuum Silicagel-Dünnschichtchromatogramm maliger Dickschichtchromatographie weinrote am In diesem Mikroanalyse gesichert. gegenüber jenem des sog. bathochrom verschoben und Jodmethyl- zeigt Banden 60 - bei 303 nm, bei 303 nm, de, 457 375 nra, nm betragt Chloroform erkennt Im NMR-Spektrum des Cyanmethyl-jodo-Co(III)-cobesters 0,68 in ppm ein Signal, Im Massenspektrum mit stark 371 nm, licagel handelt H. einer einer 557 sich es zur Methylgruppe Seit einiger eine cm . Deuterobenzol integriert, dem Methylengruppe aus den axialen Ligan¬ (Massenzahl 41) und Methyljodid (Massen¬ man um erst bei ein ihren Kulminationspunkt. Cyanmethyl-jodo-Co(IH)-cob- Produkt, dessen UV/VTS-Absorptionsbanden liegen. ein steigender Temperatur 207° benzolischen Losung des nm Wahrend die der Mas¬ Intensität auf. Gemäss Dunnschichtchromatogramm Gemisch aus ca. 80 % einer an Si- blauvioletten und grünen Substanz. Versuche der in erwartungsgemass die Acetonitril der Luft erhalt bei 321 nm, % Protonen Kobalt koordinierten axialen jene der Masse 142 Beim Belichten an zwei das Maximum erreicht und bei absinkt, zeigt esters das fur ca. temperaturabhangiger 41 schon bei 170 rasch 20 treten gebildeten Fragmente 142) am man werden können. zugeordnet se die Nitrilbande bei 2200 IR-Spektrum vermutlich die Protonen der zahl Die Extinktion der intensivsten Ban¬ nm. etwa 20'000. Im erscheint bei den und 557 - in Cobyrinsaure-heptamethylester-Komplexen Zeit wurde Methylenprotonen zentrale Deprotonierung der axialen, kobaltgebundenen in vermutet, dass der Labilität resp. C-,-Stellung des 5'-Desoxyadenosylrests der Aciditàt CoenzymBl2 im Bedeutung bei den enzymatischen Reaktionen dieses Moleküls zu¬ kommt. Um 1964 fand K. Bernhauer cobalamin mit Zink und Essigsaure kylierung mit (9), in dass das durch Reduktion Dimethylsulfat erhaltene Methylcobalamm gruppe tritiert war. R. Keese (10), der tauschreaktionen der Protonen der axialen Abhängigkeit des mit maximal 15 von Cyano- tritiertem Wasser und anschliessende Al- am an der axialen Methyl¬ hiesigen Laboratorium die Aus¬ Methylgruppe am Methylcobalamm in pH-Werts der Losung untersuchte, erhielt schwankende Resultate % Austausch. 61 - - In Anbetracht dieser beiden Informationen wurde fur die ein neues Die vorliegende Arbeit Konzept entworfen. B 12 Rontgenstrukturanalyse des Coenzyms hatte fur den Bindungswinkel Co-C-C130 D.Hodgkin (4) durch C,,-Methylenkohlenstoff am des 5'-Des- oxyadenosylrestes einen Autoren auf partiell ionischen Charakter der Kobalt-Kohlenstoffbindung ge¬ einen Wert von Aus diesem Wert geliefert. war von den schlossen worden. Der grosse Winkel von 130° spricht den beiden betreffenden Orbitalen in am fur erhöhten s-Charakter C-,-Kohlenstoff und erhöhten (33-50 %) p-Charakter denjenigen Cet-Orbitalen, die Wasserstoffatome binden. Die Tendenz des balt-Bindungsorbitals deutet darauf men am C5,-Kbhlenstoffatom dass die hin, grosserem in Ko¬ s-Charakter anzuneh¬ Bindungselektronen der Kobalt-Kohlenstoff-Bin- dung mehr beim Kohlenstoff lokalisiert sind. Bei steht einer eine diesem Winkel C-.-H Bindung C sp2 entsprechenden Hybridisierung senkrecht 120° Oc- zur am C^-Kohlenstoff Kobalt-Kohlenstoff-Bindung. H C 180° (Cr sp L^lJ Auf der Annahme, dass der grosse Winkel eine Folge der elektronischen Eigen¬ schaften der direkt beteiligten Bindungsorbitale und mcht der sterischen Verhalt¬ ist, nisse wurde Gelingt es in ester-Komplexes ren, so konnte folgende Hypothese aufgebaut. die zweite axiale Koordinationsstelle des einen ein Liganden Proton der mit Methylgruppe labilisiert werden, da Carbanion durch Delokalisation stabilisiert wurde. anions über ein t„ -Orbital am Methyl-Co(III)-cob- guten Ruckbindungseigenschaften einzufuh¬ Kobalt und das den wurde den Austausch der Protonen an der das entstehende Diese Delokalisation des Carb- tt-System des transaxialen Methylgruppe beschleunigen. Ligan¬ 62 - In Anbetracht der nenkonfiguration schwache 6 lenmonoxid, transständigen Methylgruppe solcher Reaktivität (1965) ter 150 Atmosphären eine intermediäre Neben Koh- befähigt schien, wurden von von uns geltenden Stickstoff noch als inert Volpin (61), der M. Aethylmagnesiumbromid mit Stickstoff Ammoniak nachweisen konnte. lineare Elektro¬ Zeit dieser Versuche bekannter Uebergangsmetallchloriden von für die allem zur später erschienen die Arbeiten Erst nach Behandlung zu auch dem damals Eigenschaften vor -lowspin die durch eine Liganden geeignet, das auf Grund verschiedener zugesprochen. dass und der d -Bindung gebunden starke Rückbindungsfähigkeit zeigen. Metallcarbonyl-Komplexe solche scheinen jene Kobalt am - Diese un¬ Versuche, Stickstoffixierung angenommen wurde, zeigen, Uebergangsmetallkomplexe mit hochreduziertem Zentralatom zu solcher Reaktion geeignet sind. Um die axiale Koordination dieser schwach zu erzwingen, komprimierte ren. man Unter diesem Druck Hess rat-Komplex gelöst nukleophilen Liganden sie auf einen Arbeitsdruck man von stehen. In einem weiteren Ansatz je Ein plex Zersetzung und positives Resultat her bekannten NMR-Spektrum wäre am Auftreten des vom aufgearbeitet, auf Austausch im geprüft. Methyl-Co(HI)-cobester-Kbm- Signals der Protonen der axialen Methylgruppe bei 0,16 ppm, welches beim Edukt Hand der im wur¬ Nach durch¬ Essigsäure zugesetzt. schnittlich drei Tagen wurden die Proben in der Dunkelkammer auf Kobalt Atmosphä¬ jeweils Trideutero-Co(III)-cobester-perchlo- in Methanol bei RT den Natriumacetat oder Natriumacetat und UV/VIS-Spektrum 1000 am Integration fehlt, zu erkennen. des erwarteten 30 mg deuteriertes Material zur Eine quantitative Signals erfolgen. Verfügung standen, Da Bestimmung jedoch hätten aus sollte pro Versuch an nur apparativen Grün- - den Austauschquoten nur von 63 Minimum 25 - % mit Sicherheit festgestellt werden können. Leider war Massenspektrum würde es zur zu erlauben, methylchlorid), Zeit dieser Versuche die (vgl. erkennen noch nicht bekannt 51). S. (Trideutero- erweisen. Zusammenstellung der Resultate (vgl. Tabelle 2 und 3, S. 110/111) Dieser in einem Versuch festgestellte Austausch konnte nicht reproduziert werden. entsprechenden Versuch wart von mäss diese Bei den Ansätzen in Tage bei RT in Luftkontakt. wie alle anderen untersuchten Proben Gegenwart ungeklärt von Ge¬ un- Natriumacetat zeigte das NMR- ist auch die andere in den Proben beobachtete Veränderung. ein starkes Signal bei Signal, das beim analogen in dem einem Versuch für Singlett Arbeit von obige R. Abeles ca. zwei im anderen (50) mit abgeschlossen zym B 12 1,2-Propandiol wird Tritium in den für war, folgenden Ergebnissen: C-j-Stellung des 5'-Desoxyadenosylliganden nicht radioaktivem jedoch von Dieses ppm auf. erschien, integriert ungefähr elf Proto¬ Verunreinigung sprechen. Versuchsserie überträgt bei der enzymatischen Umlagerung von +0,06 bei Versuch mit Kohlenmonoxid nicht Dies könnte für eine externe Nachdem die NMR-Spektren der behandelten Bei zwei mit Stickstoff in Gegenwart Natriumacetat behandelten Ansätzen trat ein rung In dem ppm. Bisher nen. im Dunkeln auch nach Ausschütteln der Probe mit Wasser Spektrum 1,24 war nur die Probe nach der Stickstoffbehandlung in Gegen¬ Natriumacetat noch zwei UV/VIS-Spektrum zersetzt. war be¬ zu mit tritiertem Al- dass mit einer Ausnahme kein Austausch beobachtet wurde. zeigt, im Diese Methode 52 und 51 auch kleinere Austauschraten mit Sicherheit Experimentell günstig dürften sich auch Versuche stimmen. Liganden axiale Hand der Intensitäten der Massenzahlen 53 an kyl-Co(m)-cobester-Kbmplexen Die Möglichkeit zu am erschien 1966 Tritiertes Propionaldehyd Coenzym mit in B 12. Cg,-Stellung eine 1,2-Propandiol Tritium auf die Bei der Umlage- tritiertem Coen¬ gebildeten Propionaldehyd eingebaut. - Der Firma F. 64 - Hoffmann-La Roche & Co AG zügige Ueberlassung grösserer Mengen Vitamin Die Durchführung der Austausch-Versuche danke ich für die gross- B 12. unter hohen Drucken wurde durch das freundliche Wohlwollen des technisch-chemischen Laboratoriums an der ETH unter dem Vorstand licht. von Herrn Prof. Dr. A. Guy er ermög¬ 65 - - EXPERIMENTELLER Folgenden nachstehender Herrn Prof. Herren resp. TEIL ihren Mitarbeitern danke ich für die Bestimmung physikalischer Daten: Dr. W. Simon für die UV/VIS-Spektren, aufgenommen Modell 14 für die UV 137 Für die laufenden ein Perkin-Elmer Spektrophotograph verwendet. Angaben bedeuten: Max: Adsorptionsminimum, Min: Cary Spektrophotometer Charakterisierungen. Untersuchungen wurde Die auf Schrägstrich: Extinktion. Adsorptionsmaximum, Seh: Schulter. (Wellenlängen in Zahlen nach dem nm) IR-Spektren, Perkin-Elmer Gitter-Spektrophotograph 257. Die w Intensitätsabschätzungen bedeuten: schwach (Werte in s stark, m mittel, ). cm NMR-Spektren, aufgenommen auf Varian A 60 Spektrometer. Die chemischen Standard s = von Singlett, Verschiebungen beziehen Tetramethylsilan (TMS) S m = (vgl. Lösungsmittel W. Simon pK*-Werte, J. Seibl Herrn W. Manser für die durchgeführt im angege¬ (55)). bestimmt in einem ' (vgl. für die Massenspektren, spektrograph 0 ppm. nach der thermoelektrischen Methode 1,2-Dimethoxyaethan Herrn PD Dr. sich auf den Multiplets Molekulargewichtsbestimmungen, benen = AEI MS 2/H 1 : 1 Gemisch Wasser- W.Simon (56 und 57)). aufgenommen auf Massen- oder Hitachi RMU/6A. Mikroanalysen. 1) Fluka, 1,2-Dimethoxyaethan (Monoglym) purum, destilliert über Natriumhydrid. 66 - Ganz speziell körperphysik an ich Herrn Dr. danke (Vorstand der ETH der Resultate. Schmelzpunkte wurden : im Vakuum vom (vgl. Laboratorium für Fest¬ G.Busch) für die Hilfe bei der von M. Hub Apparatur nach Dr. Messun¬ Interpretation Gouy auf einer er au¬ (58)). Tottoli im offenen korrigiert. Säulenchromatogramme wurde, Merck, Kieselgel Korngrösse 0,05 - 0,2 mm wo nichts anderes vermerkt ist, verwendet und mit Benzol einge¬ Für die Schüttsäule wurde ein Verhältnis Durchmesser schlämmt. 1 sowie für die in einer Röhrchen gemessen und sind nicht Für die Dr. erfolgten nach der Methode kompensierenden Waage tomatisch Die Die Messungen A. Menth Herr Prof. magnetischen Suszeptibilität gen der - zu Höhe von 10 angestrebt. Die für die stellt mit Dünnschichtchromatographie verwendeten Platten wurden herge¬ Merck, Kieselgel viert während circa vier wahrt. Als G für Dünnschichtchromatographie nach Stahl, Stunden bei 140 und ein bis fünf Standard-Lauf mittel diente ein Gemisch akti¬ Tage bei RT aufbe¬ Benzol-Methylac etat 1 : 4. Kaliumcyanid-Silicagel-Dünnschichtplatten Silicagel-Dünnschichtplatten obiger Qualität wurden gesättigten methanolischen dem heissen Föhn Kaliumcyanidlösung besprüht getrocknet. vor Gebrauch mit einer und zwei Minuten mit . 67 - - CN B 12 Vit. / » / Co I CN 1,3 nol kristallines g (gereinigt 10 -4 wurden mit 100 ml Metha¬ vollständig (51)) am ungefähr 20 gekocht. mit 200 ml Wasser 200 ml extrahiert. zu dampfer erhielt noch analog wie oben lenstoffextrakt, gelöst und an Silicagel (75 g), der 90-fachen Menge Methylacetat 2:3 gelöst, geimpft eluierten 990 mg mit 100 ml Hexan in Benzol bei 373 (£ kristallisierten aus nm = 28'900), erhalten wurden zugesetzt, wurden in 100 ml Ben¬ einem Vorversuch Nach gelassen. und nach nm = 29'700). je so dass insgesamt 925 mg kristalliner (Ausbeute = 89 %). S. Das 66) Produkt nur Charakterisierung gelangten zwei 27 mg ( abge- Mutterlauge e in Benzol bei Dicyano-Co(HI)-cobester zeigte auf einer Kaliumcyanid- einen Fleck Charakterisierung). Zur an¬ 36 Stun¬ nochmaligem Stehenlas¬ Von den 55 mg 5 ml Benzol und 15 ml Hexan weitere Silicagel-Dünnschichtplatte (vgl. der aus Kaliumcyanid mit 1500 ml Benzol- während 48 Stunden im Kühlschrank konnten 898 mg violette Nädelchen nutscht werden 373 Kristallen und 36 Stunden im Dunklen verschlossen stehen den wurden zweimal je 30 ml Hexan sen mit wenig Benzol in dem 2 g festes Dicyano-Co(in)-cobester versetzt, rückflussiert und vereinigt, beigemischt worden waren, chromatographiert wurden. Die zol Rotationsver¬ Methylenchlorid¬ 260 mg Tetrachlorkoh¬ Man erhielt dabei weitere aufgearbeitet. die mit den schon erhaltenen 800 mg zuerst mit Schwefelsäure ge¬ unter reinem Stickstoff Tagen ver¬ einen mit drei Portio¬ am 800 mg Tetrachlorkohlenstoff- und 320 mg einmal während zwei an durch Zugabe Methylenchlorid, je Nach Entfernen der Lösungsmittel Letzterer wurde in 30 ml Methanol und 3 ml konz. extrakt. löst, man neutralisiert, Die Lösung, was Kaliumcyanid dunkelviolett geworden, wurde Tetrachlorkohlenstoff und anschliessend mit nen von Nach dem Ein¬ Rückfluss einem dunkleren Rot bewirkte. 700 mg festem circa von Lösung karminrote, Die ml wurde dünnt und unter Rühren mit festem Natriumbicarbonat Farbumschlag nach einer kalten löste. wurde vier Tage unter sauerstoffreiem Lösung auf Mol) Zugabe erst nach nach L. Meites Rotationsverdampfer am engen • Schwefelsäure in 50 Methanol Schwefelsäure circa 1 molare Stickstoff (9,6 wobei sich die Substanz versetzt, 10 ml konz. Vitamin B 12 Proben: (vgl. Fussnote am Schluss 68 - - Aus dem oben beschriebenen Ansatz wie Probe A: folgt umkristallisiert: 200 mg kristallinem Dicyano-Co(in)-cobester 20 ml Benzol wurde mit 20 ml Hexan versetzt, angeimpft und Eine Lösung von den bei RT verschlossen stehen gelassen. Nach Zugabe in 24 Stun¬ weiteren von 15 ml Hexan und noch 24 Stunden bei RT und 6 Stunden im Kühlschrank konnten 198 mg Kristalle abfiltriert werden. lassen, Stehen weiteren analogen Umkristallisationen dieses 190 mg Kristalle, und RT) (zwei Tage die nach dem Trocknen Charakterisierung gelangten ( zur Nach zwei Materials erhielt bei man 0,003 tin Benzol bei 373 Torr nm = 29'500). Aus einem Probe B: (t Analyse = 29'000) C H Co N ber. 59,55 6,76 5,41 7,72 gef. 59,50 6,79 5,33 7,73 136 Probe B: Smp. nm Probe B C54H73CoNg014 nach der Umkristallisation aus Benzol-Hexan Vorversuch, in Benzol bei 373 - 138° (unzersetzt gemäss UV/VTS-Spektrum: denintensitäten) UV/VIS (in EtOH + nm Max/53'000-260 nm Min/5'900-278 nm Max/11'200 289 nm Min/ 5'900-314 nm Max/9'500-331 nm Min/ 4'800 354 nm Seh/14*500-369 nm Max/30'800-410nm Min/ 2'100 419 nm Max/ 2'400-442 nm Min/ 5*200 545 nm Max/ 8*600-562 nm Min/ 7'600-584nmMax/10*900 * 1*400-514nm (vgl. Figur 6, Probe B Ban¬ Probe B: 213 UV/VIS (in Benzol) IR 0,1 o/oo Kaliumcyanid) rel. * Seh/ 15) S. 291 nm Min/ 5*800-316 nm Max/ 9*800-327 nm Sch/7*300-337 nm Min/5'500 359 nm Sch/14'400-373 nm Max/29'800-414 nm Min/2'900-424 nm Max/3'400 450 nm Min/ nm nmMax/9'200-567 nm Min/8'000 591 nm Max/12'000 (in Chloroform) 1*800-520 Probe A Seh/ (vgl. Figur 3, 2990 m, 2950 m, 2125 w, 1365 m, 1350 m, 1150 m, 1105 m, 1730 s, Probe B einmal umkristallisiert Smp. : 153-156° (vgl. Lit. 5*100-552 (5)). aus S. 1580 m, 1005 12) 1500 m, 1435 m, 1400 m, m. Methylenchlorid-Diisopropyläther- 69 - 0,97 -1,64 (Singletthaufen (5 1,64-3,00 (m, (2,07 3,00 - 4,06 (m, (5 5,67 (s * bei s 12) S. 0,99, 1,13, 1,17, 1,36, 1,49, Total H*) 20 ca. (vgl. Figur 4, Probe A (in Deuterobenzol) NMR 100 MC - 2,20 s, s Total ca. 3,24 zwischen s ) H*) 34 3,46) und H) Total 25 H) 1 internen Standard bezogen auf den hier als Intensitäten 25 Protonen zu gewählten Abschnitt zwischen 3,00 und 4,06ppm (vgl. S. 13). (in Deuterochloroform) NMR 60 MC 0,81 -1,69 (Singletthaufen (5 Total 1,69 -3,17 (m, (2,19 3,17 4,06 (m, - 5,60 (s * 4 s, 2,23 s) zwischen s Total 3,62 und ca. 34 3,81) Intensitäten bezogen auf den hier als Vergleich der Intensitäten der bei 180° 27 = 60 % bei 210° 27 = 37 % von sche aus Werte, da = - und 43 (CHgCO) aus Ansatz; einem speziellen ( l in Benzol bei 373 nm kristallisiert 0,003 = Torr ge¬ 28*800) 10"4 e.g.s.u./Mol (diamagnetisch) 2,77 früheren Ansätzen: Kristallisation entfernen ist. ren 293°K) (HCN) 13). S. Benzol-Hexan und 24 Stunden RT trocknet) & Massen 27 (vgl. 43 aus XM (77°K ppm 25 Protonen zu (base peak) 43 von magnetische Suszeptibilität (Kristalle Bei H) internen Standard gewählten Abschnitt zwischen 3,17 und 4,06 Erfahrungen H*) Total 25 H) 1 MS Probe B 13) S. 1,20, 1,27, 1,37, 1,52, 1,57) H*) 20 ca. (vgl. Figur 5, Probe A bei s aus Methylenchlorid-Hexan ergab Methylenchlorid Chromatographie mit Chloroform offenbar an nur sehr schwer wurde der aus Mikroanalyse ca. 65 den Kristallen %. Auf Dicyano-Co(HI)-cobester cyanid-Silicagel-Dünnschichtplatten zeigte dasselbe Material zu normalen Sili- zersetzt. nur Fleck. Analysenreines fal¬ Sillcagel ohne festes Kaliumcyanid und Eluie- verringerten die Ausbeute auf cagel-Dünnschichtplatten die Material auf unbehandelter Silicagel-Platte: Auf Kalium- einen violetten 70 - \ / I L_ _LJ \ I i rötlich /co+/ /Co / " CIO I I CN CN Dicyano-Co(III)-cobester (9,2- 100 mg kristalliner ' Methylenchlorid vibriert. Saure am rend 24 Stunden 30%-iger und mit 10 ml lenchlorid-Hexan 10 -4 Mol) wurden Perchlorsaure drei Blausaure Scheidetrichter die orange, am Hausvakuum in 20 ml Minuten 14 Torr von organische Phase 20 ml Wasser und filtrierte durch Watte. mit von der Entfernen des Rotationsverdampfer und Trocknen des Ruckstandes am Lo¬ HV wah¬ ergaben 103 mg (96 %) amorphes Aquo-cyano-Co(IÎI)-cobester- Da das perchlorat. im man ab, wusch sungsmittels gelost Absaugen der entstandenen Nach bei RT trennte Produkt weder aus dem wurde auf kristallisierte, Gemisch Benzol-Hexan noch eine Mikroanalyse Methy- verzichtet. spektroskopischen Charakterisierung gelangte das Rohprodukt. Zur UV/VIS (in Benzol) (vgl. Figur 7, 275 nm Max/12'000-292 356 nm Max/22'700-407 nm 514 nm Mm/ nm nm 7'500-521 19) S. Mm/6'600-326 nm Max/8'900-331 nm Sch/5'700-427 nm Min/3'550-490 nm Min/13'800 Max/ 9'000 Max/7'700 (in Chloroform) 3400 w (breit), 1350 m, NMR (in 1,82 2,93 In - - - 2135 w, 2950 m, 1148 m, 1130 m, Deuterochloroform 1,11 1) violett OH, CN IR - + 1018 1 1730 s, 2,93 (m, (2,33 diesem s s, 1579 m, 1495 m, 1438 m, Tropfen Trifluoressigsaure) 1,82 (Signalhaufen, (1,26 s), 4,29 (m, (6 1605 w, m 2,38 s) zwischen Total ca. Total ca. 27 3,63 und 3,82) 24 H*) H*) Total 25 H) Versuch wurde ausschliesslich Methylenchlorid verwendet, Gebrauch durch basisches Aluminiumoxid filtriert worden war. das vor 71 - - 6,48 (si H) * Intensitäten bezogen auf den hier als internen Standard gewählten Abschnitt zwischen 2,93 und 4,29ppm (vgl. MS Vergleich der Intensitäten der Massen 27 (HCN), 43 bei 210° 27 bei 225° 27 (base peak) 50 255° 345 % von 43 43 3 % von = 208 % von 43 = 9 % von 43 = (base peak) 50 bei = 27 = 3 % von 43 50 = 34 % von 43 25 Protonen zu S. 13). (CHgCO) (CHgCl) und 50 (base peak) Dunnschichtchromatographisches Verhalten: Das Produkt zeigte auf einer Silicagel-Platte mehrere orange und rötliche ineinander fliessende Flecken. pH-Abhängigkeit des UV/VIS-Spektrums l • 104 UV/VIS-Spektren 300 in Monoglym-Wasser 1:1 500 400 600 nm Figur 20 A 406 X- Aquo-cyano-Co(III)-cobester-perchlorat (3, misch B Monoglym 1) 5 • 10 _7 Mol) in 10 ml Ge- -Wasser 1:1. 216 nm Max/49'000-265 nm Min/10'300-273 nm Max/11'700-291 nm Min/7'300 324 nm Seh/H'700-355 nm Max/28'100-404 nm Seh/ 5'000-423 nm Min/3'400 498 nm Max/ nm Min/ nm Max/ 9'000 Min/7'500 nach Zusatz 9'000-513 von 3 Tropfen 0,1 n 8'500-528 Tetramethylammoniumhydroxid 220 nm Max/43'600-265 nm Min/10'500-275 nm Max/11'000-294 nm 360 nm Max/20'500-438 nm Min/ nm Max/ Sch/8'400 1) Fluka, 1,2-Dimethoxyaethan, vor 3'100-524 Gebrauch über 8'400-547 Natriumhydrid destilliert. 72 - Durch trum Zugabe von 1 Tropfen konz. - erhielt Perchlorsaure man wieder Spek¬ wobei die Extinktionen wegen der Verdünnung etwas kleiner waren. A, nm Max/50'000-264 323 nm Seh 513 nm Min/ 214 Auf Grund Grenze des /ll'600-354 8'600-526 nm Min/11'000-273 nm Max/12'200-292 nm Max/27'300-404 nm Seh Max/ nm Mikrotitration einer in um Mm/7'800 nm Max/9'100 9'000 Monoglym-Wasser 1:1 liegende pK*-Wert Messbereichs / 5'000-498 zu > 11 kann der abgeschätzt der an werden. Cl CN >/Lj à«y CN CN 100 mg kristalliner Dicyano-Co(HI)-cobester (9,2 10 • _5 Mol) wurden 15 ml in Methylenchlond gelost und mit 10 ml 30%-iger Perchlorsaure drei Minuten vib¬ Nach riert. der entstandenen Blausaure Absaugen wurde die orange Phase Methylenchlond im trennt und zweimal mit je 20 ml Wasser 15%-iger Vakuum am Scheidetrichter gewaschen. Durch thylenchlorids Nach Abtrennen am organische Phase Rotationsverdampfer wurden die 107 mg Rohprodukt Tetrachlorkohlenstoff aufgenommen chlorid beigemischt worden war, und an 10 g abge¬ 50 ml Phase und Entfernen des Me- der wasserigen von der Saure Schuttein mit wasseriger Natriumchlorid Losung färbte sich die rasch weinrot. 14 Torr bei RT von von Sihcagel, denen 1 chromatographiert Nach . in wemg g festes einer Natrium- Vorfraktion von 5 mg violettem Produkt mit 200 ml Chloroform konnten mit weiteren 500 ml Chlo¬ roform 76 mg weinroter Methylenchlond gelost, Vorversuch weitere angeimpft 20 ml Hexan Chloro-cyano-Co(III)-cobester mit 60 ml Hexan versetzt und mit liess zu verschlossen stehen. man und hess 24 Stunden Plattchen abfiltriert werden konnten Zur Charakterisierung gelangte knstalhsierte und 24 Stunden bei Benzol bei 370 nm = 22'500) 1) Merck, Kieselgel 0,05 - aus 0,2 (60%. £ eine 0,003 einem mm stehen, m Kristallen einem gab man worauf 62 mg dunkelrote aus Torr und RT aus Nach drei Tagen Benzol bei 371 dreimal In 10 ml eluiert werden. nm = 22*200). Methylenchlorid-Hexan getrocknete Probe ( um- tin analogen Ansatz. / eingeschlammt mit Tetrachlorkohlenstoff. 73 - (zusätzlich Analyse - Tage unter gleichen Bedingungen getrocknet) zwei C H Cl Co N ber. 57,94 6,70 3,23 5,37 6,38 gef. 57,88 6,77 2,89 5,11 6,30 C53H73C1CoN5°14 § § Zweite Chloranalyse 3,07 einer Probe aus aus einem Vorversuch gleicher Reinheit, (in Methylenchlorid) Mol. Gew. berechnet : 1098 gefunden : 1111 Smp. 137 UV/VIS IR 30°C bei = 3,589 mg/g 140° (unzersetzt gemäss UV/VIS-Spektrum; - (in Benzol) (vgl. Figur 8, S. Lsm Bandenintensitäten) rel. 22) 306 nm Min/7'700-316 Seh/ 8'300-332 nm Sch/10'000-370 nm Max/22'500 405 nm Min/3'100-420 nmMax/ 3'600-439 nm Min/ nm Max/ 544 nm Min/9'000-565 nm 2'200-531 9*500 Max/10'800 nm (in Chloroform) 2120 w, 2940 m, 1200 NMR 1726 s, 1578 m, 1496 m, 1431 m, breit, 1160 m, 1150 m, 1100 m, 1000 1360 m, 1350 m, m (in Deuterochloroform) 1,10-1,97 (Singletthaufen, (6 Total ca. 1,97 3,00 - - 5,95 (s * 3,00 (m, (2,25 4,47 (m, (6 1 s 24 s, s bei 1,25, 1,31, 1,42, 1,47, 1,64, 1,88) H*) 2,30 s) zwischen Total 3,62 H*) 26 ca. und 3,78) H) Total 25 H) Intensitäten bezogen auf den gewählten Abschnitt MS c hier als internen Standard zwischen 3,00 und 4,47 ppm (vgl. Vergleich der Intensitäten der Massen 27 (HCN), 43 bei 175° 27 bei 187° 27 % 43 und 1'700 (base peak) 11 % von 43 (base peak) = 250 % von 43 und 290 = 87 % von 43 50 50 210 = = von 25 zu (CHgCO) % % von von Protonen 15). S. 50 50 und 50 (CHgCl) 74 - bei bei 195° 200° 27 = 28 % von 43 50 = 67 % von 43 27 = 5 % von 43 50 = 23 % von 43 - (base peak) und 43 % von 50 (base peak) und 22 % von 50 Dunnschichtchromatographisches Verhalten: Analysenreines Material zeigte auf Silicagelplatte folgendes Bild: einer __^>? / / ' ; —; rot CN 1 I violett I / /Co / / Co I I CN CN 150 mg kristalliner 20 ml Dicyano-Co(m)-cobester (1,38 Methylenchlorid gelost Im Scheidetrichter chlorsaure vibriert. mit je wasseriger Rotationsverdampfer entfernt. Die 180 Losungsmittel Dunnschichtchromatogramm drei Flecken zeigten, blauviolett Rf wenig Benzol in fraktion 4 b) : 1 von gelost 21 mg und eluiert worden waren, Animpfen Stehen lassen 40 ml Hexan im an 0,5 (stark), 14 g erhielt c) rotviolett Rf man die mit 280 ml mit 560 ml Jodo-cyano-Co(in)-cobester. mit Kristallen a) 30%-iger die aus Dunkeln wahrend 48 einem Stunden, Per- mg = so¬ wur¬ Rohprodukt, 0,3 (mittel) Nach = 0,6 wurden einer Vor- Benzol-Methylacetat Benzol-Methylacetat Aus mit Farbe braunrot Rf Silicagel chromatographiert. Dijodo-Co(III)-cobester, 138 mg blauvioletten Hexan nach = in Nach dem Abtrennen der wasserigen Phase die (schwach), wurden Methylenchlondphase de das am Mol) 10 ml Kaliumjodidlosung geschüttelt, wobei fort nach blauviolett umschlug. im -4 Saure abgetrennt und zweimal der von 10 Minuten mit zwei 20 ml Wasser gewaschen wurde die orangerote 1%-iger, 10 ml und wahrend • 7 : 3 20 ml Benzol und 50 ml Vorversuch und verschlossen wobei nach 24 Stunden nochmals zugegeben wurden, kristallisierten 129 mg schwarzblaue Tetraeder. 75 - Da die 10 mg Mutterlauge ( Ausbeute 78 % xan - nicht mehr kristallisiert werden t in Benzol bei 375 nm 16'100). = umkristallisierte Probe hatte eine Extinktion gelangte rung 0,003 Torr einmal eine getrocknete Eine von konnten, betrug einmal 17'800. aus Zur Benzol-He¬ Charakterisie¬ Benzol-Hexan umkristallisierte und drei aus ( Probe t in Benzol bei 375 nm 18'100) = die Tage bei einem aus anderen Ansatz. Analyse (Probe zusätzlich 48 Stunden über verlust von C53H73CoJN5°14 MG 1'190 = 145 Smp. - UV/VIS H J N ber. 53,49 6,18 10,66 5,89 gef. 53,40 6,14 10,50 5,76 147 (unzersetzt gemäss UV/VIS-Spektrum; (in Benzol) (vgl. Figur 9, Gewichts¬ Sch/15'500-342 nm Min/8'500-375 nm 459 nm Min/ nm Max/8'500-575 nm 1'600-561 rel. Bandenintensitäten) 24) S. nm Max/18'100-424 Min/ 8'300-596 Sch/3'000 nm nmMax/9'400 Methanol) absolutem (Aufnahme einen was C 304 UV/VIS (in IR PoOn getrocknet, 1,93 % bewirkte) auf Perkin-Elmer 273 nm Max/12'600-291 400 nm Seh/ 5'300-424 Spektrophotograph nm Min/8'200-323 nm nm Min/3'800-490 nm UV 137) (vgl. Figur 9, Sch/13'600-352 Max/ 8'800-516 nm S. nm Sch/7'800 (in Chloroform) NMR 2950 m, 2130 w, 1730 s, 1392 w, 1353 m, 1150 m, 1608 w, 1580 m, 1105 m, 1005 1497 m, 1440 m, 1478 w, m (in Deuterobenzol) 0,85 - 1,78 (Singletthaufen, (5 Total ca. 1,78 3,03 - - 3,03 (m, (2,13 4,18 (m, 5 s 20 s, s bei 1,02, 1,20, 1,27, 1,48, 1,70) H*) 2,26 s) zwischen Total ca. 3,28 und 27 3,48, H*) Total 25 H) 6,21 (si H) * Intensitäten gewählten bezogen auf den hier als internen Standard Abschnitt zwischen 3,03 und 4,8 ppm (vgl. S. zu 25 Protonen 13). 24) Max/26'400 76 - der Intensitäten der Vergleich MS und 142 bei 185° bei - Massen 27 (HCN), 43 (CHgCO), 127 (J) (CH3J) 205° 27 = 9 % von 142 127 = 35 % von 142 142 = 600 % von 43 27 = 49 % von 142 127 = 50 % von 142 142 = 55 % von 43 (base peak) Dünnschichtchromatographisches Verhalten: Analysenreines Material zeigte auf rötlich Bild: > \ i Silicagel-Platte folgendes —* °n T T einer violett blauviolett Cl /to / / Co1 / » I CN Von einer Methanol 1 : 1 Lösung wurden von 0,5 3 mg ml Chloro-cyano-Co(HI)-cobester (2,7 • Mol) 10 hydrophobisierte UV-Zelle gegeben, durch deren schliessender Gummischlauch gesteckt worden konnte die Zelle luftdicht verschlossen werden. Aether-Hexan 1 cher : 1 Gemisch Oeffnung war. Flussäure ein am Rand dicht ab¬ Mit einer Schlauchklemme Durch den Schlauch und 50 mg Natriumborhydrid zu, gab man 3 ml worauf unter schwa¬ Wasserstoffentwicklung die Reduktion ablief. Durch kurzes Schütteln und Druckausgleich durch die Schlauchklemme se in 5 ml Wasser- in eine mit verdünnter grün und die untere violett gefärbt. le unter Ausnützung nach drei Schlauchklemme UV/VIS-Spektrums war die Minuten die obere Pha¬ Letztere wurde durch Umkehren der des Ueberdrucks durch den Schlauch dass sie bei der Aufnahme des verschlossener war so weit nicht mehr störte. Wasserstoffentwicklung Zel¬ abgelassen, nur Bei fest noch gering. 77 - - Nach der Aufnahme des violette Phase noch vollständig herausgesogen. takt rotlich gewordenen Losung wurden 2 mg festes UV/VIS-Spektrum konnte auf die Konzentration der nm den. Um wurde Losung stimmte uberein. im zweiter ein sung und Gebiet zwischen 400 Aether-Hexan 1: 1) Max/39'000-345 282 nm 459 nm Min/ 576 nm Max/ des nm 1 : 1 geschüttelt. demjenigen! mit 3,5 Gemisch auf Minuten Absorptionen grossere authenti¬ von zu wer¬ erhalten, Chloro-cyano-Co(m)-cobester 1 ml von ml Das Co(I)-cobester Losung geschlossen Gemisch Lo¬ durchgeführt. Co(I)-cobesters (vgl. Figur 10, Min/10'800-394 nmMax/ 3'450-465 untere, Vergleich der Extinktion bei Durch nm Versuch, ausgehend ml Aether-Hexan 1 :1 2,5 UV/VIS (in 600 - die Nach Auffüllen der beim Luftkon¬ und drei Kaliumcyamd zugesetzt dieser violetten Injektionsspritze einer Monoglym Wasser mit Dicyano-Co(m)-cobester schem 373 Spektrums wurde mit 3'500-478 nm Max/38'000-439 nm Seh / 28) S. 3'400-504 nm Sch/4'100 nm Min/1'800 4'300 CN I /Co / -/co / I N I Dicyano-Co(in)-cobester (1,84 200 mg kristalliner 20 ml zwei Methylenchlond gelost Minuten vibriert. Nach bei RT wurde die orange re abgetrennt, dampfer zur lost und in Absaugen Methylenchlond-Phase einen 500 ml Scheidetrichter Hexan sowie 30 ml Wasser vorgelegt heissen Glasstab mit war, verschlossen. lief, musste gewaschen und mit einem einer in von wurde ca. flachen NS 29 starker Anfang schwach rot, Vakuum von der Sau¬ Rotationsver¬ 1,3 g Natrium- Polystopfen, färbte sich durch jedoch wel¬ gemacht worden Wasserstoffentwicklung ausgeglichen werden. Die obere Phase, die am am 30 ml Methanol ge¬ Stelle etwas dunner Da die Reduktion unter in dem je 150 ml Aether und Zugabe Nach am in jeweils nach kurzem Schuttein der Ueberdruck Oeffnen des Hahns farblos war, an wurden Scheidetrichter im gegeben, waren. borhydrid wurde der Scheidetrichter cher Mol) Perchlorsaure wahrend Der Ruckstand wurde eingedampft. -4 der entstandenen Blausaure zweimal mit je 20 ml Wasser Trockene einem 30%-iger und mit 15 ml 10 • ver¬ sorgfaltiges vor mit der Reduktion fortschrei- 78 - Als nach dunkelgrün. tender Reaktion - Minuten die Wasserstoffent¬ sieben ca. wicklung nachgelassen hatte, nutzung des Ueberdrucks Scheidetrichter abfliessen dunkelgrüne Aether-Hexan-Phase spritzte In die Polystopfen bereiteten -4 Mol) 1,5 in schwand nach sungsmittel Rohprodukt grun Rf mit 30 ml Benzol =0,6 (stark), einem Chromatogramm eluiert, welche nem in hen gelassen. 1 gelost, angeimpft violette Nadelchen Rf vier = Aus der Mutterlauge kristallisierten braun- und konnten Dijodo-Co(HI)-cobester mg blauviolette Sub¬ dunnschichtchromatographisch Drjodo-Co(HI)-cobester Der versetzt, mit Kristallen im Benzol bei 359 nm Benzol-Hexan weitere = aus ei¬ Dunkeln ste¬ Kühlschrank konnten 128 mg tin aus c) 0,0 (sehr schwach). und 48 Stunden bei RT verschlossen abgenutscht werden ( das Flecken: Menge Silicagel (18 g) 80 ml Hexan im ver¬ weinrote Lo¬ gelost zeigten die 155 mg 138 mg braunroter Nach weiteren 24 Stunden (3,4- Trübung klare, 0,3 (schwach), = identisch waren. mit vor¬ Scheidetrichter, spulte In wenig Benzol UV/VIS-spektroskopisch 15 ml Benzol Vorversuch : eine Benzol-Methylacetat 3:2 wurden 17 Mit 250 ml durch den nun Rundkolben und entfernte die Lo¬ einen d) braungrun Jodo-cyano-Co(III)-cobester wurde in den man der 90-fachen an man erhielt man blauviolett Rf Benzol-Methylacetat 4 eluiert werden. stanz b) 0,1 (sehr schwach), = mit 600 ml mit und Silicagel Dunnschichtchromatogramm im braunrot Rf Aufar¬ 85 mg frisch sublimiertem Jod von Rotationsverdampfer. am (Die gelassen. Die sofort auftretende braungrune ein. vollständiger Jodzugabe, Reaktionsgemisch Aus Losung eine ml Aether Nach kurzem Umschwenken öffnete sung. a) nun besprochen. ) dieser Losung wird spater beitung 10 im unter Aus¬ violette untere Phase wurde die (59 %) 26'500). 10 mg (5 %) glei¬ cher Reinheit. Charakterisierung gelangte Zur sierte und bei 358 Analyse nm zwei = dreimal eine Benzol-Hexan umkristalli- aus Tage bei 0,003 Torr und RT getrocknete Probe ( 26'300) (zusatzlich aus einem £. in Benzol früheren Ansatz. noch 24 Stunden ui.ter denselben Bedingungen getrocknet) C H Co J N C52H73CoJ2N4°14 ber* 48'38 5'70 4>57 19>66 4>34 1291 gef. 48,49 5,65 4,39 19,52 4,39 MG = 79 - Mol. (in Benzol) Gew. berechnet: 1291 gefunden: 1338 158 Smp. - - 160 bei 25°C c 6,291 mg/g = (unzersetzt gemäss UV/VTS-Spektrum; Lsm Bandenintensitäten) rel. UV/VIS (in Benzol) 301 nm Min/9'600-358 um Max/26'300-371 um Sch/25'700-393 um 428 nm Min/3'900-492 nm Max/ 8-200-538 nm Min/ nmMax/ 584 nm Min/6'500-604 nm Max/ 7'600 6'700-562 Sch/17'800 7'600 UV/VIS (in Feinsprit) IR IR 206 nm Min/47'300-216 nm Max/54'000-246 nm Min/12'200-267 nm 282 nm Min/16'600-316 nm Max/20'800-341 nm Sch/15'400-388 nm 470 nmMax/ Min/ 4'800 9'800 (in Nujol) 1735 s, 1610 w, 1220 m, 1160 m, 1100 (in NMR Kaliumbromid 1572 m, 1732 s, 1200 m, 1160 m, 1610 1100 (in Deuterobenzol) 2,94 * - - 1417 w, 1295 w, (breit) w, 1570 m, 1490 m, (vgl. Figur 13, Total ca. S. 36) 17 H*) 2,94 (unstrukturierter Signalhaufen, 3,71 (m, (5 zwischen s 1431 m, gewählten Abschnitt 3,27 Total ca. 3,47) und 31 Total 25 zwischen 2,94 3,71 und ppm Vergleich der Intensitäten der Massen 43 (CH,CO), bei 200° 208° 127 = 23 % von 142 142 = 40 % von 43 127 = 36 % von 142 1180 % von 43 142 = magnetische Suszeptibilität 1249 w, 1292 w, m H*) H) Intensitäten bezogen auf den hier als internen Standard bei 1251 w, Pille) 2950 m, 1,62 1490 m, 1540 w, m 0,84-1,62 (Signalhaufen, MS Max/20'500 (Kristalle, aus (vgl. 127 zu S. (J) 25 Protonen 15). und 142 (base peak) aus einem speziellen Ansatz kristallisiert Benzol-Hexan und 24 Stunden bei RT und Torr (CH„J) getrocknet. ( £ in Benzol bei 358 nm = 0,003 25'800). - (77°K) X„ M XM (293°K) 80 - 10" e.g.s.u./Mol (diamagnetisch) =-4,70-10" e.g.s.u./Mol (diamagnetisch) = 6.11 - • Dunnschichtchromatographisches Verhalten: Analysenreines Material zeigte auf einer o 0 I 1 I 1 1 i i ausgeschüttelt, violetten Losung wurde Die violette Phase_aus_dem_Reduktions_gemisch mit zwei cyanid Material wurde Bedingungen aufgehoben Schwefelsaure partiell konz. Methylenchlorid Methylenchlorid Material, welches zu im Nachveresterung 40 ml je Zusatz Kahum- von ähnlich war, Dunnschichtchromatogramm und durch zu Nach dem Rotationsverdampfer am nach Dicyano-Co(III)-cobester UV/VIS-spektroskopisch unter den normalen ses Portionen wodurch die wasserige Phase fast farblos wurde. Man erhielt 45 mg violettes dem 1 braunrot Trocknen über Natriumsulfat wurde das entfernt. ! i i braungrun Aufarbeiten der wässerigen^ Silicagel-Platte folgendes Bild. mit jedoch nicht lief. Die¬ Methanol und Dicyano-Co(III)-cobester aufgearbeitet. CN I >lkj iLy CN I 44 mg kristalliner Benzol blasen. ' Dijodo-Co(III)-cobester (3,42 10 _5 Mol) wurden in 20 ml gelost und wahrend 90 Minuten mit sauerstoffreiem Stickstoff durchge¬ (Darstellung vgl. (51)). laren wasserigen 1) Merck, • Benzol Nach dem Einspritzen Kaliumcyamd Losung (1,5- 10 (kristallisierbar). 4 Mol) von 0,25 rührte ml einer man 0,6 60 Minuten moun- 81 - ter wobei sich die Farbe Stickstoff, Rotationsverdampfer Benzol vom aufgenommen und Lösungsmittel befreite Rohprodukt wurde durch Chromatographie mit Dicyano-Co(HI)-cobester violette Animpfen Hexan nach weinrot nach violett änderte. von sigen Kaliumcyanid abgetrennt. Der te, - kristallisierte mit authentischem 29'400) wie auch in wenig vom überschüs¬ 5 ml Benzol und 10 ml aus (98,5 %) von waren UV/VIS-spektroskopisch ( UV/VIS (in Benzol) (Aufnahme Silicagel Material innerhalb Dicyano-Co(m)-cobester mit reinem 3 g am 100 ml Methylacetat vollständig eluier- quantitativ. Die erhaltenen 36,6 mg Kristalle chromatographisch an Das 40 Stunden fast sowohl dünnschicht- t in Benzol bei 371 nm identisch. auf Perkin-Elmer Spektrophotograph UV 137) 294 nm Min/ 6'000-316 nm Max/ 358 nm Sch/14'200-373 nm Max/29'400-415 nmMin/2'600-426 nmMax/3'100 455 nmMin/ nm Seh/ 594 nm 1'600-521 9'900-328 4'900-555 I / + Sch/7'800-339 Min/5'600 nmMax/9'000-570nm Min/7'800 /Co / / I I I CN mg kristalliner Dicyano-Co(m)-cobester (2- CN kristalliner nm I Co I 21,8 nm Max/H'200 CN /Co / = Dijodo-Co(III)-cobester (2# lutem Benzol ' 10 _5 Mol) gelöst und verschlossen bei RT 10 Mol) und 25,8 mg wurden zusammen in 20 ml abso¬ gerührt. An Hand von Silicagel- Dünnschichtchromatogrammen konnte das Fortschreiten der Reaktion gut verfolgt werden. sive Zeigte dieses nach 20 Minuten Reaktionszeit drei ungefähr gleich Flecken a) braunrot Rf 0,4 (Produkt), c) violett Rf =0,6 (Dijodo-Co(IH)-cobester), b) =0,15 (Dicyano-Co(m)-cobester), tionsabbruch nach zwölf Stunden die c) im Verhältnis zu Rotationsverdampfer b) auf circa 5 ml wurde Benzol-Methylacetat 4:1 1) Merck, Benzol von Nach dem an 4 mg wurden mit (kristallisierbar). blau-violett Rf so waren 5 g = bei Reak¬ Edukten entsprechenden Flecken sehr viel schwächer. Nach einer braunroten Vorfraktion 100 ml den inten¬ a) und Einengen der Lösung am Silicagel chromatographiert. Dijodo-Co(IH)-cobester 550 ml eluiert mit Benzol-Methylacetat 7:3 50 mg 82 - blauviolette erhalten. In 20 ml Animpfen mit Substanz setzt liess man nach Nach verschlossen stehen. ten 39 mg schwarzviolette Zugabe 0,003 24 Stunden bei mung Zur aus einem 375 nm Jodo-cyano-Co(III)-cobester = und im Analyse, Schmelzpunkt- UV/VIS-Spektrum 17*100). Reinheit. getrocknet umknstallisierte und 48 Stunden bei ver¬ Vorversuch 12 Stunden 10 ml Hexan und weiteren 12 Stunden konn¬ isoliert und als (6 %) gleicher Torr und RT auch fur das wie von tin Benzol bei kristallisierten weitere 3 mg trum charakterisiert. Benzol gelost und mit 60 ml Hexan Kristallen Kristalle (82 %, identifiziert werden - wurde IR-, und eine Mutterlauge Produkt wurde und NMR- MS-Spek- Molekulargewichtsbestimzweimal Torr und RT 0,003 Aus der Dieses aus Benzol/Hexan getrocknete Probe des oben beschriebenen Ansatzes verwendet. Analyse C H Co J N C53H73CoJN5014 ber. 53,49 6,18 4,96 10,67 5,89 1190 gef. 52,46 5,93 5,06 9,77 5,55 MG = (in Benzol) Mol. Gew. berechnet: 1190 gefunden: 1145 Smp. 146 - bei (unzersetzt 148 25°C c = 604; 7, mg/g UV/VIS-Spektrum; gemäss Lsm rel. Bandemntensitaten) UV/VIS (in Benzol) IR 303 nm 458 nm Sch/15'000-342 Min/ 2'200-560 Min/8'700-375 nm nmMax/8'500-575 nm nm Max/17'600-426 nm Sch/3'400 Min/81 '000-597 nmMax/9'100 (in Chloroform) NMR 2970 m, 2950 m, 2130 w, 1730 s, 1390 m, 1332 m, 1105 m, 1008 1610 w, 1580 m, 1497 m, 1438 m, m (in Deuterobenzol) 0,85 - 1,78 (Singletthaufen (5 Total ca. 1, 78-3,03 (m, (2,15 3,03 - 4,18 (m, 6,20 (s * 1 5 s 25 s, s bei 1,02, 1,20, 1,27, 1,48, 1,70) H*) 2,26 s) zwischen Total 3,28 ca. und 31 3,48, H*) Total 25 H) H) Intensitäten bezogen auf den hier als gewählten Abschnitt zwischen 3,03 internen Standard und 4,18ppm (vgl. zu S. 25 Protonen 15). - MS Vergleich der bei 175° Massen 27 = 112 % von 142 127 = 27 % von 142 142 = 73 % von 43 27 = 70 % von 142 127 = 27 % von 142 142 = 430 % von 43 220° Dunnschichtchromatographisches Die - 27 (HCN), (J) 127 Verhalten: mikroanalytisch untersuchte Probe zeigte gleiche Bild (CHgCO), 43 (CH3J) und 142 bei Intensitäten der 83 wie Jodo-cyano-Co(III)-cobester (vgl. auf S. Silicagel-Platte einer das 76). I /Co111/ / Co11/ I I I I Dijodo-Co(ffl)-cobester (7,0 90 mg kristalliner Benzol und nach gelost losung fünf Zugabe von die am im Rotationsverdampfer braun Rf 0,0 (schwach), Mit eluiert werden. stallen 6 ml aus 0,1 in Die organische wurden vier wenig Benzol Die 110 mg Flecken grün Rf zeigten, = a) 1) Methylacetat, abgetrennt Rohprodukt, braunrot Rf =0,6 0,2 (stark), d) braungrun aufgenommen und an Rf = 10 g Silicagel chro- 210 ml Benzol-Methylacetat 4:1 wurden 15 mg Substanz eluiert, Methylacetat 1: 1 Nach Entfernen des Methylacetat einem 30 ml in Natnumthiosulfat- n Phase wurde Losungsmittel befreit. (schwach), c) 4 auf, Vorversuch ge verschlossen bei RT filtriert. Mol) Dijodo-Co(III)-cobester dunnschzchtchromatographisch Mit 420 ml Benzol in = wurden matographiert. welche mit man vom Dunnschichtchromatogramm (mittel), b) 10 lang geschüttelt. Die anfangs braunrote Losung verfärbte Minuten sich wahrend der Reduktion braungrun. und • 40 g Eis mit 15 ml vor im an. konnten 56 mg grüner Losungsmittels versetzte mit Die nur Dunkeln stehen am identisch waren. Jodo-Co(II)-cobester Rotationsverdampfer nahm 25 ml Hexan und impfte mit Kri¬ schwach trübe gelassen, Losung wurde wodurch 45 mg Gebrauch zweimal durch Kahumcarbonat zwei (55 %) (Merck p.a.) Ta¬ grun- 84 - Aus den 10 mg die gleiche Ein Benzol bei 323 Reinheit wie das Probe A: Probe Torr und RT zweimal B: aus £ aus ergaben, die nm 26'200), = ( Analyse Tage bei nm = früheren Ansätzen: aus Benzol-Pentan und und Benzol-Pentan Benzol bei 323 in 13 mg Kristalle Benzol bei 358 Proben zwei getrocknet kristallisiert und drei ( in Animpfen 20'700). = 4,5 %. zweimal umkristallisiert 0,003 £ 21'400). = umgesetztes Edukt berechnet werden. Diese be¬ Charakterisierung gelangten Zur ( Edukt hatten können die Ausbeuten auch auf tragen dann 65 % resp. nm nm Benzol-Hexan nach aus Drjodo-Co(III)-cobester eluierten 15 mg Da die tin Benzol bei 323 Mutterlauge kristallisierten (3,8 %) ( 3 mg weitere ( Nadelchen erhalten wurden schwarzer - einmal nm 21'600). = Methylac etat-Hexan aus RT und Tage bei zwei Benzol bei 323 t in 0,003 Torr 21'300). C H Co J N C52H73CoJN4014 ber. 53,66 6,32 5,06 10,90 4,81 Probe A gef. 53,42 6,39 5,31 10,83 4,89 Probe B* gef. 53,48 6,32 5,15 10,99 4,81 * (zusatzlich Mol. Gew. Smp. 24 h über getrocknet: 1,38% Gewichtsverlust) (in Benzol) berechnet: 1163 gefunden: 1115 Probe B 189 - UV/VIS (in Benzol) (Probe A) bei 25°C c = 191°C (gemäss UV/VIS-Spektrum (vgl. Figur 14, Probe B S. Max/20'500-309 8,875 mg/g unter Lsm Zersetzung) 39) 279 nm Min/14'200-299 nm 365 nm Seh/ 9'600-409 nm Min/ 7'000-435 nmMax/ 8'500-482 nm Mm/ 7'500 492 nmMax/ 7'600-558 nm Mm/ 2'300-603 nmMax/ 2'900-638 nm Seh/ 2'400 UV/VIS (in Feinsprit) IR Po^s um¬ getrocknet (vgl. Figur 15, Probe B nm S. 216 nm Max/40'500-246 nm Min/8400-266 nm 316 nm Max/25'000-382 nm Min/5400-415 nm (Nujol) Min/19'800-323 1610 w, 1595 w, 1560 m, 1530 w, Max/21'600 39) Max/16'800-279 Seh/ 6'800-469 Probe B 1730 s, nm 1200 m, 1150 m nm Min/11'800 nm Max/10'700 85 - IR (Kaliumbromid Pille) NMR 2940 m, 1730 s, 1350 m, 1200 m, Probe A 1610 w, Probe A 142 bei 180° (vgl. Figur 13, 127 = 142 = 127 142 31 % von 142 168 % von 43 = 36 % von 142 = 220 % von 43 magnetische Suszeptibihtat 0,003 destens unter und =- 5,06 = 6,03 - aus Tage an allem die aus 127 (J) speziellen Ansatz, kristal¬ getrocknet) Torr nm einem und 48 Stunden RT Methylacetat-Hexan aus ( t in Benzol bei 21'200) = 10"4 e.g.s.u./Mol (diamagnetisch) • 10"4 e.g.s.u./Mol (diamagnetisch) in kristalliner Form oder der Luft stabil (vgl. keine Vorteile Saulenchromatographie mitteln durchzufuhren. platten (CHgCO), Massen 43 • Stickstoffatmosphare vor 3,9 früheren Ansätzen Produkt ist vier 36) S. und (base peak) (Kristalle 323 Das 1430 m, (base peak) lisiert Erfahrungen 3,0 Hügel zwischen 1480 m, m Vergleich der Intensitäten der 203° XM (77°K) XM (293°K) 1530 m, (CH3J) und bei 1000 1100 m, Probe A paramagnetisch;breiter MS 1560 m, 1592 m, 1158 m, (in Deuterobenzol) - Analysenreines braunviolettes © o grün gelost 46). brachte, mit in bei abs. Wahrend die ist es Benzol über min¬ Kristallisation ratsam, diese Operation entgasten, sauerstoffreien Losungs¬ Material zeigte auf aufgetragen und laufen gelassen lerweise kein oxidiertes S. Silicagel-Dunnschicht- Laboratoriumsbedingungen Produkt norma¬ (Dijodo-Co(III)-cobester). 86 - Eine che alten solche Nebenreaktion wurde o \ Wo¬ Katalytische Hydrierung von - 13 mg I M I grün braunviolett Di]odo-Co(III)-cobester (1 at) Katalysator, / .. I In früheren Versuchen wurde bei RT mit Wasserstoff / 1 / V\ die eine o / \ 1 Edukt, einigen Fallen auf mehr als in Silicagel-Platten beobachtet. © war - über Dijodo-Co(III)-cobester einem 10 ml abs. benzolischer Losung in reduziert Platinoxid-Katalysator Benzol). Nach einer anfänglich braunrote Losung braungrun geworden (20 mg Stunde Reaktionszeit und zeigte un UV/VIS- Spektrum die charakteristischen Jodo-Co(n)-cobester Absorptionsbanden bei 304 nm, 324 nm, 369 nm (Seh), 440 nm, 491 nm, 604 nm und 638 nm (Seh) in den entsprechenden Intensitäten. Im beim Dunnschichtchromatogramm jedoch zeigte Rohprodukt Flecken (Rf = der 0,4) diese Substanz an Stelle des Natriumthiosulfat-Reduktion beobachteten schwachen braunen einen intensiveren orangen (Rf = 0,5). 1/ v grün stark Durch anschliessende ; orange mittel braunviolett schwach Saulenchromatographie an Silicagel konnte dieses orange Ne¬ benprodukt nicht abgetrennt werden, und die Kristallisation des thylacetat 1:4 eluierten grünen Produkts gelang nicht oder Kristalhsierbares an Silicagel, waren. dem ca. Eluat erhielt 20 man % pulverisiertes erst nach nur mit Benzol-Me- sehr schlecht. zweimaliger Chromatographie Natriumthiosulfat beigemischt worden 87 - - I Z_W ZÇo^/ »/Co"/ I I 20 mg kristalliner Dijodo-Co(m)-cobester (1,6- 10 -4 Mol) wurden in 25 ml Benzol gelost und mit einer Losung von 40 mg Silberperchlorat-Monohydrat -4 Mol) in 5 ml Benzol versetzt, wobei die braunrote Farbe sofort nach (1,8- 10 orange umschlug. schlag mit einer tionen Wasser Nach in 15 ml ' Methylenchlorid die organische Phase. mit 20 ml Wasser bei 350 nm, am Rotationsverdampfer trum noch unverändert. te Substanz hatte Banden bei 320 0,1 494 Diaquo-Co(in)-cobester-Komplex entsprach. tels Losung vollständig. Dreimalige brachte den Mit 30 ml SüberjodidniederSchuttein mit drei Por¬ orangefarbigen Komplex Perchlorsaure geschüttelt und n gewaschen zeigte die Methylenchlorid Phase Absorptionsmaxima trum mit abgetrennt werden; 20 ml entfärbte die benzolische zu Extraktion mit je wieder Minute Schuttein konnte der einer G4 Glasfilternutsche 0,005 Die bei jedoch nm bei 40 C gelost und 525 nm, Nach dem ein UV/VIS-Spek- welches UV/VIS-Spek- Torr und RT wahrend 18 Stunden welches fur das einem Absaugen des Losungsmit¬ die Substanz gemäss Methylenchlorid in und 470 nm, war nm ein getrockne¬ UV/VTS-Spektrum Vorliegen mit Co(II)-cobester- eines Komplexes sprach (vgl. S. 32). Da das Produkt weder chromatographiert (vgl. Verhalten auf oben Dunnschichtplatte) erhaltene, amorphe wurde auf eine noch kristallisiert werden Material MG nm Charakterisierung. konnte, gelangte das Aus demselben Grund mikroanalytische Bestimmung verzichtet. UV/VIS (in Benzol) (Extinktionen 316 zur = berechnet auf Aquo-Co(II)-cobester-perchlorat 1150) Max/25'400-385 nm Min/5'200-414 nm Sch/6'900-469 Mm/11'600-315 nm Max/12'000 UV/VIS (in Feinsprit) IR 245 nm Min/7'100-266 nm Max/16'700-281 nm 382 nm Min/3'100-417 nm Seh/ nmMax/10'000 6'200-469 nm Max/23'000 (in Chloroform) 3680 w, 3400 w, 1390 m, 1352 m, 1) Methylenchlorid, 2950 m, 1152 m, vor 1730 s, 1105 m, 1600 w, 1570 m, 1050 m, 1008 1490 m, 1438 m, (Merck) filtriert. m Gebrauch durch basisches Alox NMR (m Deuterochloroform) Hügel breiter paramagnetisch zwischen bei 160° bei 205u 50 = 5 bei 240° 50 = 9 50 = % 6 von 43 % von 43 % von 43 Dunnschichtchromatographisches Das charakterisierte 4,0 (CHgCO) Vergleich der Intensitäten der Massen 43 MS und 3,4 und 50 (CHgCl) (base peak) Verhalten: Material zeigte auf einer Silicagel-Platte folgendes Bild: \ / orange . 18 mg des durch rötlich Behandlung von Di3odo-Co(III)-cobester rat-Monohydrat erhaltener Co(n)-cobester-Komplex wurden lost. Mit 30 ml 0,1 schüttelt, verfärbte Braungrun. Nach Kaliumjodid Losung n sich das im im in Rotationsverdampfer Dijodo-Co(III)-cobester in entfernt. Das nebst grünem ein (66 %) und 10 ml eine ( £ in Benzol bei 323 = am Jodo-Co(n)-cobester identisch. Sihcagel-Dunnschichtchromatogramm Startpunkt nur noch Die 10 mg Dijodo-Co(III)-cobester. einer mit 30 ml Nach drei 20'400 nebst wel¬ nicht laufende Methylacetat gelost, nm filtriert, Rohprodukt, schwache, direkt kristallisiert werden. angeimpft, abfiltriert mit authentischem ten gemäss schmutziges Jodo-Co(II)-cobester analysenreinem Material). Dunnschichtchromatographisch nigung 30 ml Benzol ge¬ Dunkeln verschlossen Stehenlassen wurden 12 mg kristalliner cobester von am Verunreinigung zeigte, konnte, Hexan versetzt und RT der benzolischen Phase Silicagel Dunnschichtchromatogramm noch etwas braunroten braune in und 10 g Eis wahrend 5 Minuten ge¬ Abtrennen der wasserigen Losung wurde durch Watte und das Losungsmittel ches Orange mit Süberpercnlo- Tagen bei Jodo-Co(II)- gegenüber 21'300 war das Produkt Mutterlauge enthiel¬ braunen Verunrei¬ UV/VIS (in Benzol) (Aufnahme Max/19'800-310 auf Perkin-Elmer Min/19'000-323 300 nm 413 nm Min/ 6*500-442 nmMax/ 608 nm Max/ 3*200-645 nm nm 8*100-488 Seh/ Spektrophotograph 137) UV nm Max/20'400-368 nm Sch/8'900 nm Seh/ nm Min/2'000 7*100-518 2*100 I I präparativ 46,5 Benzol gelöst ' laren benzolischen 56 mg Unter Rühren Jodlösung (total Nach Entfernen des weiter. Rohprodukt, roten Fleck die im zeigten, authentischem aus 2 tropfte 10" • und Dijodo-Co(III)-cobester abfiltriert werden. UV/VIS-spektroskopisch ( am und reiner (Darstel- am und rührte zehn Minuten Dunklen konnten Dijodo-Co(III)-cobester auf Animpfen Min/9'500-360 500 nm Max/8*500-542nm Min/ 608 nm Max/7'700 (91 %) violet¬ nm der = 26*500) Mutterlauge, Substanz, die mit reinem Dijo- welche gemäss knapp hinter dem (1,5 %) dünnschichtchromato¬ kristallisiert werden. Max/26'500-369 (kristallisierbar). mit Nach 24 Stun¬ 7 mg Perkin-Elmer Spektrophotograph UV nm nm 46, einen braun¬ nur dünnschichtchromatographisch war eine braune mo¬ Rotationsverdampfer konnten die konnten weitere 1 mg 302 Benzol zu 2,72-10 ml einer direkt kristallisiert werden. Das Produkt durch UV/VIS (in Benzol) (Aufnahme 1) Merck, Jod) £ in Benzol bei 360 lief, verunreinigt war, graphisch wurden in 40 ml 20 ml Hexan durch do-Co(III)-cobester (£= 26*300) identisch. Aus Produkt Mol) Silicagel Dünnschichtchromatogramm 15 ml Benzol Dünnschichtchromatogramm 7,35 man Mol Lösungsmittels den verschlossen Stehen lassen bei RT te Nädelchen -5 und während 30 Minuten mit sauerstoffreiem Stickstoff (51)) gespült. lung vgl. Jodo-Co(II)-cobester (4,0-10 mg kristalliner 7*000-566 nm Sch/26'100-432 nmMax/ 8*100-588 137) nm Min/4*000 nm Min/6'800 90 - - I n / / / L I I I I ni / UV/VIS-spektroskopisch 406 zol 1) Tropfenweise setzte Losung zu man je in 0,01 (= 0,395 Aequivalent), 414 nm, 474 nm und 633 ml nm. Dies deutet darauf Jodo-Co(II)-cobester vorlag £ wurden cm 2,72-10"'J einer in 10 ml absolutem Ben- Schichtdicke) gegeben. molaren benzolischen Jod- jeder Zugabe das wobei nach liefen durch isobestische nm. Die nach Jodlosung (0,79 Aequivalent) aufgenommene bei 300 (1 UV-Zelle eine aufgenommen wurde. Diese Kurven 336 nm, Mol) Jodo-Co(II)-cobester (3,47-10 jf gelost und 4 ml davon einer UV/VIS-Spektrum 283 nm, Punkte bei Zugabe von total 0,02 ml Kurve zeigte die kleinste Extinktion hin, dass nach dieser Zugabe der vorgelegte verbraucht war, und freies Jod neben Dijodo-Co(III)-cobester (vgl. Figur 22). .104 300 400 600 500 700 Figur 21 a) vorgelegter Jodo-Co(II)-cobester b) nach Zugabe von total 0,01 ml in Benzol Jodlosung (0,395 Aequivalent) c) " " " " 0,02 ml " (0,79 d) " " " " 0,025 ml " (0,977 Aequivalent) 1) Merck, Benzol (kristalhsierbar). Aequivalent) nm 91 - UV/VIS (in Benzol) Werte der Min/10'300 (9'600) 301 nm 495 nmMax/ 8'200 585 nm Min/ 6'800 in Benzol bei zum Max/25'500 (26'300) - 359 nm (8'200) - 540 nm Min/ 7'200 ( 6'700) (6'500) - 604 nm Max/ 7'500 ( 7'600) Vergleich (aus Zugabe c) analysenreinen Dijodo-Co(III)-cobesters Extinktion des In Klammer: Figur 22 Kurve - von ^3ÖtT einem anderen benzolischer Min/4'600 (3'900) - 428 nm - 531 nmMax/8'100 (7'600) Ansatz) Dijodo-Co(m)-cobester Jod-Lösung. "40T loT ~e5ö" Figur 22 a) Dijodo-Co(m)-cobester in Benzol b) nach Zugabe von total 0,1 Mol-Aequivalent Jod c) nach Zugabe von total 0,2 Mol-Aequivalent Jod Cl /coV i i 100 mg dunkelgrüner, den unter Stickstoff man zu kristalliner 80 ml absolutem Tetrachlorkohlenstoff eine dunkelrote Lösung erhielt. stoff bei RT wurde das und der Rückstand in 1) Merck, Jodo-Co(II)-cobester (8,6 • 10" Mol) wur¬ gegeben, wodurch Nach 15 Minuten Stehen lassen unter Stick¬ Lösungsmittel am Rotationsverdampfer bei 40 C entfernt wenig Tetrachlorkohlenstoff aufgenommen. Da das Silicagel Tetrachlorkohlenstoff p.a. - 92 - Dunnschichtchromatogramm nebst schwach sung zwei auf eine einer 20 rote im Nadelchen aus, Diese a) Das : Hypothese stutzt sich auf fur NMR-Spektrum spricht Von S. 97) einer gel¬ man Mit nach Entfernen des MethylLosungs¬ In 30 ml absolutem Produkt. Hexan versetzt und wahrend 48 Stunden gelassen kristallisierten 65 mg blau¬ nm folgende einen Chloro-jodo-Co(III)-cobesters eines Benzol bei 335 in noch wurde die Lo¬ Chromatographien. mg dunkelrotes fur die die Struktur i 0,1), separat weggekratzt werden. mit 150 ml gelost, (54 %, und 0,6) dem Produkt laufenden Vorfraktion ab¬ vor Dunkeln verschlossen stehen genommen wird 1 durch Cehte filtriert erhielt Tetrachlorkohlenstoff bei RT 1 Benzol-Methylacetat Rotationsverdampfer 82 am 0,8 = = Silicagel Dickschichtplatte (Herstellung vgl. cm die blaurote Substanz eluiert und mittels x weinroten, unmittelbar konnte getrennt, acetat Nebenprodukte zeigte (Rf 20 cm Hauptprodukt (Rf blauroten einem rötliche und mit aufgetragen ben und - = an¬ 19'500). Informationen: zwei diamagnetischen Co(III)-cobester-Köm- plex. b) Die im stark temperaturabhangigen Massenspektrum auftretenden, Methylchlond (Massenzahl 50) und Methyljodid (Massenzahl 142) Chlorid und Jodid als axiale Da bis anhin eine Liganden Hexan umkristallisierte und fünf zur Charakterisierung ( Die stoff hin. Umkristalhsation weder thylac etat-Hexan erfolgreich war, gelangte Tage bei Benzol bei i in eingeschlossen bruchstucken im Dies bis zu + nm in = 0,003 H noch aus Me- Tetrachlorkohlenstoff- Torr getrocknete Probe 19'800). den etwa 160 C Analyse C52H73ClCoJN4014 CCl4 einmal aus eine 335 Benzol-Hexan Kristallen Tetrachlorkohlen¬ wird durch das Auftreten Massenspektrum C52H73ClCoJN404 aus RT und Mikroanalyse lasst vermuten, dass ist. Fragmente deuten auf von Tetrachlorkohlenstoff- unterstutzt. Cl J N ber. 52,07 6,14 2,96 10,59 4,67 ber. 47,04 5,51 13,10 9,38 4,14 gef. 47,12 5,68 15,18 6,21 4,23 1) Merck, Tetrachlorkohlenstoff p.a. 93 - UV/VIS (in Benzol) (Aufnahme IR Min/14'900-335 298 nm 432 nmMax/ auf Perkin-Elmer nm 3*300-451 - nm Max/19'800-3ö5 Min/ 3'100-531 UV 137) Sch/15'800-419 nm Min/3'200 nm Seh/ nm Max/9'500 7'600-555 (in Chloroform) NMR 1608 w, 2950 m, 1730 s, 1152 m, 1103 m, 1008 1579 m, 1497 m, - 1,83 3,20 * 19 - - 3,20 (m, Total 4,70 (m, (4 Intensitäten ca. s 1,08, 1,22, 1,28, 1,36, 1,58, 1,72) H*) 29 3,28 auf den hier bezogen 3,46) und Total bei bei bei 200° 207° 240° H) 25 als internen Standard und 4,70ppm (vgl. Vergleich der Intensitäten der Massen 43 (CH„CO), und 142 zu S. 25 Protonen 15) (CKLC1), 50 127 (J) (CH3J) 127 == 30 % von 142 50 == 340 % von 142 und 430 142 == 120 % von 43 127 == 31 % von 142 142 und 105 50 == 350 % von 142 == 30 % von 43 127 == 32 % von 142 50 == 18 % von 142 und 4 142 == 27 % von % % von 43 % von 43 von 43 43 CH CN Co 1163 m, H*) zwischen s bei gewählten Abschnitt zwischen 3,20 I 1353 m, m 1,83 (Singletthaufen, (6 Total ca. MS 1436 m, (in Deuterobenzol) 0,84 / Spektrophotograph nm / / I Co V C104" I CN 134 mg kristalliner Methylenchlorid gelost Dicyano-Co(III)-cobester (1,24-10 und analog den cobesters beschriebenen Verfahren (vgl. bei der S. 76) -4 Darstellung mit 12 ml Mol) des wurden in 15 ml Dijodo-Co(III)- 30%-iger Perchlorsau- 94 - re - behandelt und in je 130 ml Aether und Hexan und je 30 ml Wasser und Me¬ Natriumborhydrid thanol mit rationen mussten bei stark Nach Abtrennen der (3,4-10 _3 Mol) im Scheidetrichter gedämpftem Licht (vgl. S. 96) violetten Phase wurden unteren, in die reduziert. dunkelgrüne Aether-Hexan Lösung sofort ziegelrot und trüb wurde. 0,4 Phase durchgeführt ml zwei wurden Minuten vereinigt und geschüttelt. 110 mg man (76 %) ( t in jeweils 0,003 Monoglym-Wasser 1:1 Gemisch bei 304 nm = 20'800) '. Methylenchlorid-Hexan, Methylenchlorid- aus Benzol-Hexan und spektroskopischen Charakterisierung gelangte Torr und RT Methylacetat-Hexan eine Tage bei zwei getrocknete Probe. 1 242 nm Min/8'400-263 nm 371 nm Sch/8'400-406 nm : 1 ' Gemisch) Max/20'200-278 Min/ (vgl. Figur 23, nm nmMax/ 5'000-461 S. Min/12'300-304 98) nm Max/20'800 8'700 Methylenchlorid)2* UV/VIS (in 263 nm Max/22'800-278 nm Min/12'200-304 nm Max/24'400-359 376 nm Max/ nm Min/ nm Max/ 9'100-408 5*500-456 nm Min/8'500 9'800 (in Chloroform) 2950 m, 1100 1) 2) Rotationsverdampfer am amorphes Material ergaben, wurde auf eine Mikroanalyse verzichtet. nur UV/VIS (in Monoglym-Wasser IR Lösungsmittels Methylenchlorid-Diisopropylaether, Zur je 40 ml Methylen¬ Extraktion mit lichtempfindlicher Methyl-Co(ni)-cobester-perchlorat-Kbmplex Da Kristallisationsversuche Pentan, je 30 ml, zu 15%-iger Natriumperchlorat-Lösung mit 20 ml dreimalige Durch chlorid und anschliessendes Entfernen des erhielt 1 : vollständig entfärbte. Die dunkelgelben Wasser-Metha¬ die organische Phase nol-Auszüge wodurch die erst 30 Sekunden unter Luftausschluss und dann mit drei Portionen Wasser Methanol 1 was werden. Methyljodid eingespritzt, schüttelte Man Alle folgenden Ope¬ Fluka 1730 s, 1615 w, 1598 w, 1570 m, 1490 m, 1438 m, 1349 m, s Methyljodid puriss. (stabilisiert Extinktion berechnet mit Mol. Gew. perchlorat). mit 1168. Silberwolle). (Methyl-"aquo"-Co(m)-cobester- 95 - - (in Deuterochloroform) NMR - 0,15 (s, 0,72 - 3 H) 1,94 (Singletthaufen (5 Total 1,94- 3,14 (m, (2,35 3,14 - 4,59 (m, (6 2,44 s) s, zwischen s bei s 1,03, 1,24, 1,44, 1,64, 1,80) H*) 24 ca. Total 3,59 und ca. H*) 27 3,79) Total H) 25 6,71 (s, 1H) * Intensitäten bezogen auf den hier als gewählten Abschnitt zwischen 3,14 4,59 und bei 205° 50 = 15 % von 43 (base peak) bei 240° 50 = 64 % von 43 (base peak) Erfahrungen aus (vgl. zu 15). S. und 50 25 Protonen (CHgCl) früheren Versuchen Chromatographie produkte. ppm (CH3CO) Massen 43 Vergleich der Intensitäten der MS internen Standard Silicagel oder Alox lieferte hauptsächlich Zersetzungs¬ an Produkt auf So zeigte das oben charakterisierte Silicagel-Dunn- einer schichtplatte folgendes Bild: •\>i>,)) orange Bei setzt, Chromatographie aber nur zu ca. 50 an % o rötlich basischem Alumimumoxid eluiert werden. methyl-Co(III)-cobester-perchlorat-Komplex, Material liess sich aber pisch nicht reiner. entscheidend So zeigte ein ist, (In S. 96). Das auch nicht kristallisieren und diesem bei der kann das (Vgl. Darstellung Zusammenhang ist darauf Reduktion die untere Phase UV/VIS-spektrosko- hinzuweisen, Bild: dass es vollständig abzutrennen: ) Dunnschichtchromatogramm der beiden Proben (Woellm) folgendes unzer- Trideutero- chromatographierte so war Produkt von auf basischem Alox - a) 96 - \\/ I, rötlich violett chromatographierter Methyl-Co(III)-cobester-perchlorat-Komplex mcht b) chromatographierter Tndeutero-methyl-Co(III)-cobester-perchlorat-Komplex Lichtempfindlichkeit der Alkyl-Co(ni)-cobester-Komplexe Unter den 60 W Birne erforderte, wurde Bei drei Meter Hohe und mit in alle untersuchten ren Transport erfolgte bei nur ca. Komplexe über Stunden stabil. Wenn zusätzlich am eine rote Taschenlampe 60 V gespiesen, es das V/ wa¬ Experiment verwendet. Tageslicht wurden die Reaktionsgefasse oder die gefüll¬ Zellen mit Alufolie gut umwickelt. ten erhellt durch normale 220 Arbeitsbedingungen: Dunkelkammer, montiert Die Aufnahme der IR- und NMR-Spektren gedämpftem Tageslicht, wobei die Substanzen gemäss UV/VIS-Spek- trum nicht zersetzt wurden. Fur die Bedingungen der Lichtspaltung die und Folgeprodukte vgl. folgende Seiten. CDq OH„ /c° y CIO. •/c° V cio4" I CN 88 mg in amorphes Aquo-cyano-Co(III)-cobester-perchlorat (8-10 30 ml Methanol und Hexan gelost analog dem benen Verfahren mit den und nach bei der fort Herstellung Natriumborhydrid im in die -4 Mol) 30 ml Wasser und je des Licht Mol) (dargestellt wurden 50 ml Aether Dijodo-Co(III)-cobesters Scheidetrichter reduziert. Operationen mussten bei stark gedämpftem Tndeuteromethyljodid (7,9-10 den Zugabe von beschrie¬ Alle folgen¬ durchgeführt werden. 0,05 nach F. Cotton (52, 53)) dunkelgrüne Aether-Hexan Phase eingespritzt, wodurch die Losung ziegelrot und trüb wurde. Analog dem undeutenerten ml wur¬ so¬ Methyl-cobester-Kom- 97 - plex aufgearbeitet (vgl. S. Tetrachlorkohlenstoff gelost phiert wurden. einer Nach den mit 150 ml 94) erhielt te, gelangte = 38 mg 21*600) 2) welche ' wenig in chromatogra- 0,003 wur¬ lichtempfindlicher Trideuteromethyl- eluiert (44 %, Da auch dieser . 24 Stunden bei eine Rohprodukt, violetten Vorfraktion mit Tetrachlorkohlenstoff Methylenchlorid nm 50 mg 10 g basischem Aluminiumoxid an Co(in)-cobester-perchlorat-Komplex Gemisch bei 304 man - £ Monoglym-Wasser 1 in : 1 nicht kristallisiert werden konn¬ Torr und RT getrocknete Probe zur spek¬ troskopischen Charakterisierung. UV/VIS (in Monoglym-Wasser 242 nm Min/9'100-262 nm 373 nm Seh/8'800-406 nm UV/VIS (in Monoglym-Wasser 1 : 1 Gemisch)2' Max/21'600-279 Min/ 1 : 1 4'700-461 nm Min/13'300-304 nmMax/ Gemisch belichtet nm Max/21'600 9'000 + 1 Tropfen konz. Per- chlorsaure) IR 270 nm Sch/8'300-317 nm Min/7'400-348 nm Max/28'000 390 nm Min/ 3'800-402 nmMax/4'100-423 nm Min/2'800-491 nm Max/ 504 nm Min/ 9'000-519 nmMax/9'900 Max/18'500-309 9'600 (in Chloroform) NMR 2950 m, 2100 w, 1730 s, 1350 m, 1100 s, 1050 1615 w, 1570 m, 1600 m, 1490 m, 1438 m, m (in Deuterochloroform) Vinylproton Ein MS nm der Vergleich 53 m Stellung 10, axialen der Intensitäten der Massen 43 (CHjCO), bei 50 -0,15ppm (CHgCl) fehlt. und (CDgCl) bei 210° 53 = 50 bei 225° 53 bei 6,68 (s) Methylgruppe entsprechendes Signal 250° 10 % = 2 = 1 50 = 53 50 von 43 % von 43 % von 43 6 % von 43 = 3 % von 43 = 34 % von 43 (base peak) 1) Woellm, basisches Aluminiumoxid, emgeschlammt mit 100 ml MethylacetatMethanol 1: 1 und anschliessend mit 100 ml Methylacetat und dann mit 100 ml Tetrachlorkohlenstoff gespult. 2) Extinktion berechnet mit Mol. Gew. 1171. 98 - CH3 I / Co - *1—> CIO" T/ aerob 435 y- amorpher Methyl-Co(m)-cobester-perchlorat-Komplex (3,7-10 und in 10 ml wurden unter Lichtausschluss eingewogen misch Eine mit dieser gelöst. Schichtdicke) röhren, die gemäss hielt man Monoglym-Wasser Lösung gefüllte, verschlossene UV-Zelle (1 10 von UV/VIS-Spektrum cm montiert waren. ein Gemisch zwischen aquo-Co(III)-cobester-Komplex säure zugegeben 1 Mol) 1-Ge- : cm während einer Minute zwischen zwei 40 W Fluoreszenz- Abstand im _7 vermuten Dem Diaquo-Co(III)- wurde ein Hess, Belichtungsprodukt, und das Hydroxo- Tropfen konz. Perchlor¬ (vgl. Figur 23). Figur 23 a) Methyl-Co(III)-cobester-perchlorat-Komplex b) gleiches c) Material nach der " " " in " " nach konz. Gab man dem niumhydroxydlösung Belichtungsprodukt zu, so erhielt UV/VIS (in Monoglym-Wasser Da ein Gemisch vorliegt, 254 nm Min-262 495 nm Max-518nm Max um 1 : einen man Zusatz von 1 Tropfen Perchlorsäure Tropfen 0,1 n Tetramethylammo- das unten tabellierte Spektrum. 1-Gemisch belichtet) kann die Max-299 Monoglym-Wasser 1 :1 Belichtung nm Extinktion nicht angegeben werden. Min-346 nm Max-380 nm Min-411 nm Max 99 - UV/VIS (in Monoglym-Wasser (Extinktion chlorsaure) 225 nm 317 nm 413 nm 1 : 1 - Gemisch belichtet Min/11'400-270 nm Min/ 5'700-348 nmMax/27'800-388 nm Min/ 1'900-491 nmMax/ nm Max/41'500-254 nm UV/VIS (in Monoglym-Wasser 1 : 1 9'200-503 nm 388 nm Durch 6,2 eine Min/ 2'900-413 Mikrotitration / CIO." 4,0 wurden 8'700-518 nmMax/9'500 + 1 Tropfen 0,1 Mol. Gew. Sch/15'400-297 nm Min/8'000-342 nm nm Seh/ nmMax/9'800-524 nm 1 : wurde n Tetra- 1170) Max/19'200 Seh/ pK*-Wert ein 9'500 von ca. mg in anaerob amorpher Methyl-Co(III)-cobester-per chlor at-Komplex 5 ml absolutem Benzol gegeben und mit dicht umwickelt und Kolben man nmMax/3'500 Mm/ nm 3'800-507 Sch/6'600 Min/ berechnet mit Monoglym-Wasser 1 in nm 3'100-401 erhalten. /Co ben Max/33'800-264 Per- 1271) Max/16'000-309 Gemisch belichtet methylammomumhydroxyd) (Extinktion 213 1 Tropfen konz. + berechnet mit Mol. Gew. am Hochvakuum einer von Nach neuem. evakuiert. auftauen, Dabei war 0,25 erkennen. nur eine in der eine Mol) Mit Alufolie licht¬ zu wurde der entgasen, hess Kaltemischung Stunde dem schwache Farbveranderung der verdünnt absolutem Benzol (3,5-10~ 50 ml Rundkol¬ einen und dreimaliger Wiederholung dieses Zyklus wurde die davon, welche ml in Um die Losung kühlte wieder Losung unter Vakuum bei geschlossenem Hahn setzt. gelost, Isopropanol-Trockeneis-Mischung gekühlt, (0,01 Torr) bei geschlossenem Hahn evakuierte Dunkeln Schliffstuck mit Hahn verschlossen. einem in im nicht das Tageslicht ausge¬ dunkelgelben Losung luftempfindlich waren, zeigten einem Co(II)-cobester-Komplex entsprechende UV/VIS-Spektrum. UV/VIS (in Benzol) (vgl. Figur 16, 314 nm 1) Merck, Max/~ Benzol (Aufnahme S. auf Perkin-Elmer Spektrophotograph UV 53) 23'000-412 nm (kristalhsierbar) zu mit 3 ml Min/~ 5'900-475 nm Max/~ 11'000 137) - / / /Ç° /C° / / Co CH2I I CN Dicyano-Co(m)-cobester (1,83-10 200 mg kristalliner cobesters beschriebenen Verfahren behandelt und in Mol) (vgl. 77) S. mit 12 ml Natriumborhydrid in nen wurden bei stark nen der unteren Phase wurden Perchlorsau- im (vgl. Licht gedämpftem 0,2 ml folgenden Operatio¬ S. 96) durchgeführt. Methylenjodid 1) (2,5-10 -3 Nach Abtren- Mol) die grü¬ in Aether-Hexan-Phase eingespritzt, wobei die Farbe sofort nach weinrot schlug. Nach 30 Sekunden schuttein unter Luftausschluss goss sung einen in fernte die Kolben, spulte Losungsmittel (stark), Rf b) 0,0 = cm : 4 und Zur Trennung chromatographiert. loste die man Nach Die beiden Entfernen des 120 mg rotbraunen gabe von Hauptprodukte, i in Animpfen RT ( l mit Dunkeln im Benzol bei 294 nm = Aus der 10 mg Mutterlauge konnten weitere den in Benzol bei 294 nm = mit 0,6 = d) braun Losung auf eine Benzol-Methyl am eluiert und Rotationsverdampfer in Kristallen stehen. 15 ml aus Nach Benzol, einem einer Zu¬ 51 mg rote Nadelchen ab- 20100). 5 mg (2 %) kristallisiert wer¬ 19'800). 1) Fluka, Methylenjodid (purum), gereinigt durch Destillation über Silberwolle 67-69°/ll Torr. 2) Merck, Kieselgel PF254 fur prap. Schichtchromatographie, 40 g und 65 ml ser pro Platte. - die rotbraune und die Methylacetat mit Losungsmittels nach 24 Stunden verschlossen bei (21 %, rotbraun Rf 0,25 (stark), aufgetragen und 40 ml Hexan und weiteren 24 Stunden konnten filtnert werden = Jodmethyl-jodo-Co(III)-cobester versetzte mit 50 ml Hexan und hess Vorversuch Rf wurde die benzolische 2) braungelbe Substanz, wurden separat weggekratzt, durch Celite filtriert. a) Flecken, c) braungelb =0,4 (schwach), Silicagel Dickschichtplatte cm vier über¬ von gelost, zeigten die 250 mg wenig Benzol in Dunnschichtchromatogramm violett Rf die klare Lo¬ Rotationsverdampfer. Am Hochvakuum am 0,05 (schwach). - 20 x acetat 1 im man um¬ den Scheidetrichter mit 50 ml Benzol nach und ent¬ schüssigem Methylenjodid befreit Rohprodukt 20 ml Dijodo-Co(III)- 30%-iger Alle Scheidetrichter reduziert. mit 20 wurden je 150 ml Aether und Hexan und je 30 ml Methanol und Was¬ ser ne -4 bei der Darstellung des analog den und Methylenchlorid gelost re - CH,I I CN I 100 bei Was¬ 101 - braungelbe Substanz, die Die 70 mg noch schwache eine - im Verunreinigung zeigten, Dunnschichtchromatogramm konnten nicht kristallisiert werden. Zur Charakterisierung des Jodmethyl-jodo-Co(III)-cobesters gelangte mal aus 0,003 umkristalhsierte und Methylacetat-Hexan ( Probe getrocknete Torr fc zwei Benzol bei 294 in nur eine zwei¬ Tage bei RT und nm = 20'000) aus einem an¬ deren Ansatz. mikroanalytischen Werte Die über Phosphorpentoxid CJ-„H_(-CoJ„N401 berechneten Werten uberein. . C H Co ber. 48,78 5,79 4,52 19,45 4,29 gef. 47,75 5,53 6,00 18,48 4,14 Analyse C53H75CoJ2N4014 1305 Mol. Gew. Smp. 185 - UV/VIS (in 294 468 189° (Zersetzung Benzol Max/20'000-326 nm Max/ 7*100-492 in Min/16'200-350 nm Min/ 6'700-530 nm Max/18'000-423 nm Max/ Spektrophotograph Gebrauch über bas. Alox 255 nm Min/17'200-268 nm Max/19'200-274 nm 320 nm Min/16'100-348 nm Max/19'200-413 nm 492 nm Min/ 6'500-519 nm Max/ absolutem Sch/5'700-480 nm Min/5'500 7'800 UV 137) filtriert) (vgl. Figur 18, (vgl. Figur 19, S. Max/19'200 Min/ nm Max/ 5'100-462 Max/20'200-388 nm Min/4'100 (in Chloroform) NMR 2950 m, 1730 s, 1165 m, 1105 m, 1602 w, 1005 1573 m, 1490 m, 1438 m, 1390 w, 1352 m, m (in Deuterobenzol) 0,65 - 1,57 (Singletthaufen Total 1,57 - ca. 2,96 (m, (2,18 21 s, (5 s bei 1,03, 1,24, 1,28, 1,37, 1,48) H) 2,26 s) 7'100 57) nm Total ca. 30 56) Min/18'700-283 Max/10'400 nm S. nm 6'800 Max/16'800-284 Min/12'300-328 nm 421 Methanol N 56) S. nm Perkin-Elmer Methylenchlorid (vor 263 IR auf J UV/VIS-Spektrum) gemäss (vgl. Figur 17, nm UV/VIS (Aufnahme in stimmten auch nach zusätzlichem Trocknen wahrend 48 Stunden nicht mit den auf die Summenformel H) 102 - 2,96 - 4,33 (m, (6 s 3,28 zwischen - 3,48) und 25 Total H) 6,35 (s,l H) NMR (m Deuterochloroform) (6 1,09-1,86 (Singletthaufen Total 1,86 3,39 - - 3,39 (m, (2,30 s) 4,75 (m, (5 s s bei 1,51, 1,62, 1,73) 1,18, 1,26, 1,40, H) 21 ca. Total 29 ca. 3,65 zwischen H) und 3,76) 25 Total H) 6,30 (s,l H) MS Vergleich der 142 (CHgJ) bei 200° Intensitäten der 127 142 56 % =1110 % = Die von 142 von 43 (CHgCO), 127 (J) und (base peak) 5 Minuten spater gleiche Temperatur, in Massen 43 127 = 41 % 142 = 222 % von 142 von 43 (base peak) Charakterisierung der amorphen braungelben Substanz erfolgte diesem Ansatz erhaltenen und 12 Stunden bei RT und 0,003 an der Torr getrockneten Probe. Da die Substanz Mikroanalyse UV/VIS (alle Spektren UV 137 in in m nicht kristallin erhalten werden wurden auf (vgl. Figur 17, 304 nm Max/17'800-321 422 nm Min/ 6'000-456 Methylenchlorid (vor S. nm Min/14'500-296 nm 357 nm Max/13'000-393 nm 452 nm eine Spektrophotographen 56) Min/17'400-332 6'300-493 Sch/17'200-318 Min/ 7'300-420 nm nm Max/17'700-375 Min/ Alox 5'700-538 nm Sch/13'600 nmMax/ 6'600 filtriert) (vgl. Figur 18, S. (vgl. Figur 19, Max/16'400-279 Seh/ 6'100-468 nm S. Max/17'800-345 nm Min/12'700 nm Max/ nm Min/ 9'100-438 8'400 57) Min/11'800-326 nmMax/ 8'100 56) nm Max/9'000 absolutem Methanol nm Perkin-Elmer Gebrauch durch bas. nm nm einem nmMax/ 278 420 wurde auf aufgenommen) Benzol 263 konnte, verzichtet. nm Max/16'700-388 nm Min/5'500 103 - IR - (in Chloroform) NMR 2950 m, 2920 m, 1730 s, 1170 m, 1105 m, 1008 1600 w, 1490 m, 1570 m, 1350 m, 1439 m, m (in Deuterobenzol) Vinylproton in Stellung 10, 6,40 (s) NMR (in Deuterochloroform) Vinylproton MS Stellung 10, 6,65 (s) in 142 bei bei 225° 250° 127 = 38 % von 142 142 = 390 % von 43 127 = 31 % von 142 142 = 150 % von 43 127 = 65 % von 142 142 = 42 % von 43 (J) und (base peak) (base peak) I CH,I I Co 127 (CH3J) bei 185° / (CHgCO), Vergleich der Intensitäten der Massen 43 I Ü* / aerob I / /Co | I I 470 y im Dunkeln und in 10 ml Benzol füllte und verschlossene UV-Zelle nute unter den bei der Mol) Jodmethyl-jodo-Co(ni)-cobester (3,7-10 kristalliner abgewogen (1 cm gelöst. Schichtdicke) wurden mit dieser Lösung ge¬ Eine wurde während einer Mi¬ Belichtung des Methyl-Co(HI)-cobester-perchlorat-Komplexes angegebenen Bedingungen (vgl. S. 98) belichtet. Ohne Schütteln färbte sich die Lösung rasch rotviolett. authentischem Das UV/VIS-Spektrum Dijodo-Co(III)-cobester Extinktionen bei 359 nm berechnete war mit reinem 1) Merck, (kristallisierbar) Benzol am man eine davon entsprach demjenigen Auf Grund des Umsetzung Rotationsverdampfer chromatographisch Belichtungsprodukt das in Benzol. Dijodo-Co(III)-cobester zu vom 95 von Vergleichs der %. Dünnschicht- Lösungsmittel befreite identisch. 104 - UV/VIS (m belichtet) Benzol Min/9'400-359 301 nm 495 nmMax/7'500-540 605 nm - (vgl. Figur 24) nm nm Max/24'900-368 Min/ 6'100-563 nm Sch/24'300-430 nm Min/3'900 nm Max/ nm Min/6'100 7*000-585 Max/6'900 UV/VIS-Spektrum von sog. Jodmethyl-jodo-Co(m)-cobester hchtungsprodukt " in Benzol Jodmethyl -îodo -Co(III) -cobester aerobes " Behchtungsprodukt 500 400 300 aerobemBe- resp. 600 Figur 24 W /Co /- Im anaerob folgenden gedämpftem Licht /Co"/ Versuch mussten alle 42 mg kristalliner 90 ml Benzol 1) den Sauerstoff Operationen vollständig zu entfernen, bei der anaeroben der evakuierte Kolben mit 75 W Lampe aus 20 Argon 2) wurde die Losung bei stark _5 bei in Distanz wurden in Um demselben 100 ml S 99) entgast Am Schluss wurde Argon gefüllt, verschlossen und wahrend cm Mol) RT gespult. Belichtung des Methyl-Co(III)-cobester- perchlorat-Komplexes angegebenen Methode (vgl einer Belichtung Jodmethyl-]odo-Co(III)-cobester (3,2-10 gelost und wahrend 45 Minuten mit Kolben nach der der vor durchgeführt werden. belichtet, 1) Merck, Benzol (kristallisierbar) 2) Wasserstoffwerk Luzern, Argon (reinst) zwei Stunden mit wobei mit dem kalten Föhn gekühlt 105 - wurde. Bei der - Belichtung färbte sich die anfangs nach Entfernen des Lösungsmittels rote Lösung braungrün. Die Rotations verdampf er am erhaltenen 40 mg Rohprodukt enthielten gemäss Dünnschichtchromatogramm und UV/VIS-Spektrum ungefähr 80 % Jodo-Co(n)-cobester. zu Methylacetat-Hexan erfolglos waren, Silicagel chromatographiert. acetat 7 250 ml : 3, zwei mit 30 ml Hexan versetzt und gelöst, werden, 21'300) mit % entsprach. UV/VIS-spektroskopisch ( und (Aufnahme Max/20'400-311 auf Perkin-Elmer Min/19'800-323 302 nm 412 nm Min/ 6'900-439 nmMax/ 608 nm Max/ 3'000-644 nm CN man In 10 ml konnten nach angeimpft, Kristalle - mit Jodo-Co(n)dünnschicht- waren £ in Benzol bei 323 nm = nm Seh/ 8'500-490 Spektrophotograph UV 137) nm nm Max/21'300-367 Seh/ nm 7'600-564 Sch/9'700 nmMin/2'400 2'500 CH„CN I / »/C° / I CN 200 mg kristalliner Methylenchlorid gelöst Dicyano-Co(m)-cobester (1,83-10 und analog den S. 77) mit 15 ml behandelt und in je 150 ml Aether und Hexan und je Natriumborhydrid ser mit nen wurden bei stark im Scheidetrichter gedämpftem Licht 2) Phase wurden 0,5 ml Jodacetonitril ' reduziert. durchgeführt. (7-10 -3 1) Methylacetat, vor Gebrauch zweimal durch 2) Dargestellt nach J. Scholl (54). Gereinigt Natriumthiosulfat bei 68-69°/*0 Torr. -4 Darstellung bei der (vgl. cobesters beschriebenen Verfahren re eluierte RT 10 mg kristalliner Die 5 g an analysenreinem Material identisch. UV/VIS (in Benzol) /C° 25 was aus Benzol-Methyl Jodo-Co(II)-cobester. 24 mg grünen Tagen verschlossen Stehenlassen bei chromatographisch wenig Benzol gelöst und Nach einer Vorfraktion mit 150 ml Benzol-Methylacetat 1:1 cobester isoliert I wurde in Kristallisationsversuche Dijodo-Co(m)-cobester enthielt, welche 9 mg Methylacetat jedoch Da Mol) Mol) des wurden in 15 ml Dijodo-Co(m)- 30%-iger Perchlorsäu¬ 50 ml Methanol und Was¬ Alle folgenden Operatio¬ Nach Abtrennen der unteren in die grüne Aether-Hexan- Kaliumcarbonat p.a. filtriert. durch Destillation über festem 106 - eingespritzt, Phase - wobei die Farbe sofort nach weinrot Nach 30 Se¬ umschlug. kunden Schütteln unter Luftausschluss wurde die klare Reaktionslösung mit 50 ml gespült, Benzol in einen Rundkolben dampfer acetonitrüs a) d) = braun Rf = 0,0 - Dickschichtplatte (Herstellung mit 4 : b) 0,8 (schwach), 0,2 (schwach). in = Das weinrote zeigte vier Flecken, Dieses 20 eine cm x hen man aus Zur nm = ( Zwei £ in Benzol bei 304 Benzol-Hexan weitere am vom mg Material im eine braunrote Verunreini¬ lieferte 70 mg Animpfen Tage bei RT weggekratzt, mit Produkt, Kristallen aus ei¬ Dunkeln verschlossen ste¬ Tage bei RT und Gewichtsverlust 1204 162 = 21'000). (3,5 %) eine 0,003 zweimal Torr Aus der Mutterlauge erhielt violette Nädelchen aus gleicher Reinheit. Benzol-Hexan umkristalli¬ getrocknete Probe ( 24 Stunden bei RT über von t in Benzol bei - P,0- getrocknet, was einen 1,34 % bewirkte) C H Co N J ber. 53,86 6,28 4,90 5,82 10,55 gef. 53,63 6,27 5,04 5,78 10,57 C54H75CoJN5014 Smp. nm 20-700). Analyse (Probe zusätzlich = 8 mg Charakterisierung gelangte sierte und drei MG % etwa 10 Silicagel- cm Rotationsverdampfer am 0,2 wur¬ gelassen, konnten 48 mg (21,5%) kristalliner Cyanmethyl-jodo-Co(III)-cobester isoliert werden 304 zu = Benzol-Methyl- wurde Hauptprodukt 10 ml Benzol und 20 ml Hexan nach aus Vorversuch kristallisierte. nem noch 20 und mit zweites, analoges Dickschichtchromatogramm Ein gung. immer orange Rf Trennung der Substanzen Zur durch Celite filtriert und Dünnschichtchromatogramm c) 0,5 (stark), Lösungsmittel befreit. In wenig Benzol gelöst zeigten die 110 welches Rotationsver¬ am wenig Benzol aufgenom¬ 100) aufgetragen siehe S. chromatographiert. Methylacetat eluiert, weinrot Rf Rohprodukts auf de die benzolische Lösung des acetat 1 Rohprodukt Dünnschichtchromatogramm untersucht. braunrot Rf (schwach), Lösungsmittel und das Absaugen des zurückgebliebenen, überschüssigen Jod- Hochvakuum wurde das am und im men Nach entfernt. 165° (Zersetzung gemäss UV/VIS-Spektrum) UV/VIS (in Benzol) 303 nm Max/20'700-347 nm Min/12'700-275 nm Max/14'000-425 nm Min/4'800 457 nm Max/ nm Min/ nm Max/ nm Sch/6'200 5'000-487 4'400-557 8'200-605 107 - IR - (in Chloroform) 2950 m, 2200 w, 1350 m, 1150 1730 s, 1490 m, 1571 m, 1605 w, 1070 1100 m, (breit), s 1436 m, 1390 w, m (in Deuterobenzol) NMR Vinylproton in Stellung 10, 6,25 (s) (CHgCN), Vergleich der Intensitäten der Massen 41 MS bei bei bei (CHgCO), 43 127 (J) (CH3J) und 142 170° 205° 207° 41 (base peak) = 70 % von 142 142 = 116 % von 43 41 (base peak) 127 = 142 = = % 168 48 % von 142 161 % von 43 = 5 % von 127 = 42 % von 142 142 = 53 % von 43 41 % 295 = 127 43, von 43, von % 20 43, von 380 % 225 % 127 und 250 % 127 und 105 % von von 127 und 8 % von von von 142 142 142 (base peak) Austauschversuche Alle Operationen mussten bei stark 109/110 Die Resultate sind auf Seite gedämpftem Licht durchgeführt werden. tabelliert. Versuche unter Stickstoff a) Methanol in 25 mg (2,2-10 -5 amorpher Trideuteromethyl-Co(in)-cobester-perchlorat-Komplex Mol) glas gefüllt. wurden in 3 ml Methanol Mit einer verschlossen, stellte Schliffkapillaren, man dem mit 250 at Stickstoff Autoklaven mit einem das 2) 1) gelöst und welche in ein 15 ml nicht in die Schliffreagenz- Lösung eintauchte, Gefäss in einen 100 ml Hochdruckautoklaven. gespült worden war, Kompressor auf 1000 at 1) Fluka, absolutes Methanol (puriss.). 2) Sauerstoff & Wasserstoff Werk Luzern, Nach- wurde der Stickstoffdruck im gebracht. Stickstoff Nach 60 Stunden Stehen nachgereinigt. 108 - bei RT wahrend deren, um - Auslaufen der Losung ein zu vermeiden, nur gelegent¬ lich und schwach geschüttelt werden konnte, wurde der Ueberdruck sorgfaltig ab¬ geblasen, und Losungsmittel bei RT das getrocknet, Tetramethylsilan) Gebiet im gruppe wurde loste und Cobester, wie Rotationsverdampfer entfernt. Am am den Ruckstand 3,72 ppm verwendet. kein Signal tion ausgeschlossen werden konnte. Das hess keine b) ppm, molare Losung wurde form wie Der Ruckstand wurde 50 ml Wasser befreit. in in gewaschen und (3.10 ' -4 eine gelost, zeigte Bei das auf Grund 1,24 hess, das 0,06 a) Versuch am trat Die an Rotationsverdampfer wieder ein neues Integration grossen getrocknet und in jenem des auf eine tausch hinweisendes Gemäss Signal von bei vom -0,15 ppm in Edukts Ein weiteres war auch in Methylenchlond duzierbarkeit dieses Resultats zu 1) Merck, (wasserfrei) Natriumacetat p. a. folgende Un¬ noch zwei Dieses mit der Signal, neues Ein auf Signal bei einen Aus¬ diesem Spektrum nicht In Methanol mit Natriumacetat prüfen, Losungsmittel wenig Deuterochloro¬ Verunreinigung schlies- Acetationen zugeschrieben. UV/VIS-Spektrum dem Versuch unzersetzt. externe aufgear¬ zweimal mit je Signal auf, welches verglichen konnte bisher nicht zugeordnet werden. Ueberresten Natriumacetat mit Stickstoff behandelt und Methylenchlorid aufgenommen, NMR-Spektrum gegenüber ppm seiner ppm wurde erkennen. Austauschreak¬ der behandelten Probe Integration der sieben Methylestergruppen 11 Protonen entsprach. sen bei NMR-Spektrum im 3 ml absolutem Methanol ge- in Mol) zugesetzt. Am Hochvakuum bei RT 3 Stunden terschiede. Methylester-Signal Bedingungen UV/VIS-Spektrum Cobester-Edukt wurden 1) 0,1 Methyl¬ Natriumacetat mit und 24 mg Natriumacetat beitet. axialen Veränderung gegenüber dem Edukt erkennen. 25 mg deuteriertes lost, einer Als interner Standard das höchste dass unter diesen so (ohne speziellen die Integration Die mit Stickstoff behandelte Probe zeigte -0,15 Methanol in im Ausgangsmatenals. spateren Versuchen Hochvakuum wenig Deuterochloroform chemical shift der Protonen mit jenem des auch bei den bei in verglich das NMR-Spektrum -0,15 ppm,dem von am man war die Substanz nach wurden, weitere zu um Versuche die Repro- durchgeführt. 109 - Ein analoger Ansatz wurde 70 Stunden doch nach dem den das NMR-Spektrum -0,15 bei Austausch der Deuteronen durch Protonen Spektrum In Methylenchlond zeigte in in ppm, unter wurde, arbeitete Deuterochloroform traten das ungefähr 1,5 nur ähnliches nicht identifizierte Protonen Die stehen so gelassene einen UV/VIS-Spektrum c) 25 mg acetat und tionszeit (1,5-10 -4 Mol) 1,24 in einen UV/VIS-Spektrum in liess, wurde versetzt. wurde getrocknet. Die wie im keine wesentliche -0,15 Methylenchlond eine 98 in Ein auf jedoch einen NMR-Spektrum Veränderung ppm. Gemäss Essigsaure %. (1,5-10 gepufferte Losung in pH-Wert einen Versuch Da weder Austausch erwartete sen. 1) Merck, Eisessig zeigte diesem Fall ppm auf. _4 war von a) durchgeführten Stickstoffbehandlung, betrug, Hochvakuum eine vollständig. und Essigsaure je 0,05 molar und hatte roforra das fur kennen Letztere Probe nach dem Aufarbeiten Ausgangsmaterial Wahrend Dunkeln stehen gelas¬ welches und mit 12 mg Natriumacetat gelost 170 Stunden am mit 48 mg Natrium- Tndeuteromethyl-Co(in)-cobester-perchlorat-Komplex 1) UV/VTS- der erste Ansatz: Wiederum wie ppm, ppm fehlte Natriumacetat Nach der gemäss Versuch bei RT 0,06 und jenes bei zeigte einem Im beide Proben unzersetzt. mit absolutem Methanol Essigsaure im Austausch hinweisendes Signal bei waren Methanol in -0,15 Probe Chloroform gegenüber dem auch kein auf bei entsprach, Austausch deutendes Signal bei in NMR-Spektrum Signal Methylgrup¬ entsprach. 1000 at Stickstoff belassen. die andere Hàlfte sofort auf. man ein % axialen Integration Veränderung. gelost und Hälfte der Losung nach dem Entgasen erst 48 Stunden sen je¬ Trideuteromethyl-Co(III)-cobester- 6 ml absolutem Methanol angesetzt und 120 Stunden 65 circa zu einer dessen das Produkt keine dritten Versuch wurden 40 mg einem perchlorat-Kömplex acetat aufgearbeitet, Deuterochloroform das Protonen in gehalten; sondern noch 48 Stun¬ gelassen. Nach analogem Aufarbeiten zeigte entsprechende Signal Cobester am pe der Luft stehen an 1000 at Stickstoff unter nicht sofort Druckausgleich Dunkeln im - eine das Signal wurden Mol) an bei -0,15 ppm Natrium¬ wobei die Reak¬ und 2 Stunden in Deuterochlo- zeigte, noch das Veränderung gegenüber dem Edukt Austauschreaktion unter diesen 3 ml ungefähr 4,7. b) aufgearbeitet NMR-Spektrum in und 9 mg er¬ Bedingungen ausgeschlos¬ 110 - - Versuche unter Kohlenmonoxid <,D In einer zweiten Serie wurde Bei verwendet. gleicher Versuchsanordnung analog aufgearbeiteten Proben ein auf einen Austausch hinweisendes nem Versuch das delten Proben neue waren Stickstoff Kohlenmonoxid wie bei den Experimenten jenen Fälle in keinem der drei bei Signal 0,06 bei Signal mit von mit Stick¬ angegebenen Reaktionszeiten zeigten die NMR-Spek- stoff und den in der Tabelle tren der Stelle an ppm auf. -0,15 ppm. (a, b oder Ebenso trat in kei¬ UV/VIS-Spektren Die c) der behan¬ der Edukte identisch. Tabelle 2 Zusammenstellung der Resultate der Austauschversuche Co(m)-cobester-perchlorat-Komplex (TDM-cob. ) Edukt in 3 ml 24 mg NaOAc MeOH at N, 60" 1000 at N„ 48h at at N2 + N„ -» TDM-cob. in 6 ml 48 mg NaOAc MeOH + 48 Austausch keine Luftkontakt 120" 1000 NMR-Spektrum bei 60" 1000 70" 1000 24 mg NaOAc 50 mg. Veränderungen im bedingungen 25 mg TDM-cob. Trideuteromethyl- unter Stickstoff. Reaktions- Zusätze mit Luftkontakt 0,06 ppm 1,25 ppm -0,15 ppm 1,25 ppm 0,06 ppm 1,25 ppm keine 25 mg 12 mg NaOAc TDM-cob. in 3 ml 9 mg HOAc MeOH 1) Techn. ehem. serstoff. 170" 1000 at N„ keine Laboratorium, ETH, Kohlenoxid, Hergestellt durch kat. Spaltung von über 99 % rein/Restgas Ameisensäure. Was¬ - Ill Tabelle - 3 Zusammenstellung der Resultate der Austauschversuche Co(m)-cobester-perchlorat-Komplex (TDM-cob.) Edukt 25 mg TDM-cob. Reaktions- Zusätze bedingungen 24 mg NaOAc 60 1000 at CO 72 1000 at CO Trideuteromethyl- mit unter Kbhlenmonoxid. Veränderungen NMR-Spektrum im bei keine 1,24 ppm in 3 ml MeOH , „ 12 .. mg ,.. NaOAc 9 mg HOAc , h j keine Austausch - 112 - ZUSAMMENFASSUNG Ausgehend dellsubstanz in wurden die sucht, vom Dicyano-Co(in)-cobyrinsäure-heptamethylester, 89%-iger Ausbeute aus Vitamin B 12 der als Darstellung und Eigenschaften axial substituierter Komplexe wobei man sich vor allem auf die Alkyl- und Unter balt enthaltender der Luft und in benzolischer Lösung cobyrinsäure-heptamethylester unter¬ Halogenid-ligandierten De¬ rivate konzentrierte. an anderen konnte dabei ein Mo¬ hergestellt werden konnte, formal kristallin erhalten werden. zweiwertiges stabiler Ko¬ Jodo-Co(II)- 113 - - LITERATURVERZEICHNIS E.L.Smith, 2) D.Hodgkin, J.Pickworth, I.G. White, H.Barker, 4) D.Hodgkin, 5) A. Eschenmoser, 7) G.Schrauzer, 8) F.Wagner, 9) K.Bernhauer, Roy. Keese, 11) R. Bonnet, 12) IUPAC, 13) A.Fischli, 14) K. 15) H.Hill, 16) G.Schrauzer, 17) Coord. Chem. Information bulletin J.Pratt, Naturwissenschaften C.Jörgensen, 20) E.L.Winnacker, 21) R.Firth, Theoret. Prog. H.Hill, Acta 393 23) A.Todd, 24) H.Hill, 25) H.Diehl, 26) R.Yamada, J. Soc. 33_5, 443 (1962) 2859(1965) 53, 459(1966) (1967) 73 (1962) Comm. 400 hiesigen Institut am R.Thorp, J.Williams, (1967) Ann. S.Glauser, N.Y. Acad. Sei. 88, (1960) Nature 174, 1168(1954) J. J.Williams, R.Murie, Dolphin, Chem. Z. 7, 328(1967) J.Pratt, Cannon, J.Pratt, 2, 109 Inorg. Chem. 4, D. Irvine, P.George, J. unveröffentlichte Arbeiten Chem. D. Rev. Chim. P.Day, 19) Biochem. F.Wagner, J.Williams, Chem. 112; 586(1964) 26_ (1966) P.Renz, Bernhauer, Sei. hiesigen Laboratorium (1967) ETH Diss. Acad. N.Y. am (1966) 405 (1963) 573 63, Rev. 79_, 836(1967) (1964) 3056 97, Bern. Ann. Pesaro, D.Bormann, Angew. Chem. 3_5, Rev. of Biochem. F.Wagner, 18) 27) Chem. J.Kohnle, M. (1965) 306 M.Pesaro, unveröffentlichte Arbeiten 10) 22) ^88, A.Wick, A. Eschenmoser, Ann. R. P.Day, A Soc. 480(1960) (1961) 937 E.Bertele, R.Scheffold, Proc. E.Winnacker, Trueblood, K. N. J.Biol.Chem. 235, 192, Nature P.G.Lenhert, A.Fischli, J.Feiner, (1948) VIII J.H.Robertson, H.Weissbach, R.Smith, H.Gschwend, 43_, J. 178, 325(1955) Nature 3) 6) Biochem. L.F.Parker, 1) Biochim. Iowa State Coll. Biophys. Acta A.Johnson, Proc. Theoret. J. Sei. Biol. 3, 26, 555(1951) 93_, 196(1964) Chem. Soc. 423 311 (1963) (1962) 114 - 28) O.Schindler, 29) H.Hill, 30) G.Wilkinson, H.Griffith, 31) G.Wilkinson, A.Mays, 32) G.Schrauzer, W. Hieber, 34) R. Scheffold, 35) K. Bernhauer, Chem. J.Williams, J.Pratt, 33) 34, 1356(1951) Acta Chim. Helv. - Chem. 98, 3324(1965) Ber. 7b, 322(1952) Naturforschg. Z. 2757(1959) 203, 1167(1964) Nature Windgassen, J. W.Hübel, 197(1964) Ind. Soc. Chem. J. private Mitteilung O.Müller, Angew. Chem. F.Wagner, 75, 1145 (1963) 36) M.Gerloch, Chem. J. 37) F.Wagner, 38) K. 39) T. L. 40) G.Schrauzer, 41) A.Adamson, 42) G.Schrauzer, 43) L.Smith, Bogard, Am. J. Chem. Chem. H.Diehl, 47) L.Haines, 48) H.Gschwend, 49) G.Hayward, Z. Diss. R.Sealock, Haar, Nature N.Y. Acad. 112. chemistry, p. 604(1962) Iowa State Coll. J. 6485 Soc. 50) R.Abeles, P.Frey, L.Meites, Anal. 52) F.Cotton, J.Fasnacht, 53) R.Kuhn, J. Biol. Chem. 157, A 65 (1931) 699(1967) 215, J.Pratt, 51) 20, Chem. 984 J.Williams, N.Vanston, (1965) 241_, 2732 (1966) (1948) J. Chem. Soc. 4138(1959) 61^, 117(1958) 54) J. 55) W.Simon, C.Tomlinson, 56) W.Simon, Helv. 57) W.Simon, D. Wegmann, Chem. Sei. (1964) ETH H.Hill, J. Chem. Scholl, Ann. Advanced inorganic Phys. Chem. A.Poe, Ann. (1966) (1951) (1951) 19 26, A. Weissberger, 3738 (1964) Vander 46) 88, (1966) 602 A.W.Johnson, G.Wilkinson, R. (1964) 521 hiesigen Institut am 5710 73, Soc. 99, Ber. 339, Z. J. Am. Chem. Soc. Windgassen, J. 565 F.Cotton, Biochem. E.Irion, unveröffentlichte Arbeiten L.Mervyn, 45) (1966) 1317 private Mitteilung Bernhauer, 44) (A) Soc. Ber. Chim. 29, 2416 (1896) 14_, 301(1960) 4^, 1835(1958) Chimia Acta Helv. Chim. Acta 45, 962 (1962) Sei. - 58) 59) G.Gardner Summer, 732 60) Z. G.Natterer, M.Huber, P.Löliger, Diss. 61) M.Volpin, 62) P.Aldermann, 63) H.Gray, H. 115 - Angew. Klug, Math. & Phys. L.Alexander, 13_, 92(1962) Acta Cryst. 17 (1964) ETH V.Shur, (1967) Nature J. Chem. J.Alexander, Soc. 209, 668 J.Am. 1236 (1966) (1962) Chem. Soc. 89, 3356 (1967) LEBENSLAUF Am 22. Januar 1940 wurde ich in Basel geboren. der Primarschule trat ich in das Humanistische Frühjahr 1959 die Maturitätsprüfung Typus begann ich das Zürich, schloss. A. an der welches ich Ende 1963 mit dem am Organisch-Chemischen Eschenmoser, A in Basel ablegte. Im ein, gleichen Besuch wo Jahr Eidgenössischen Technischen Hochschule Nach einem durch Militärdienst Juni 1964 Dr. Chemiestudium vierjährigem Gymnasium ich im in Nach Diplom als Ingenieur-Chemiker ab- bedingten Unterbruch arbeitete ich seit Institut im Laboratorium unter dessen von Herrn Prof. Leitung ich neben anderem die vorlie¬ gende Dissertation ausführte. Herbst 1967 Lucius Werthemann