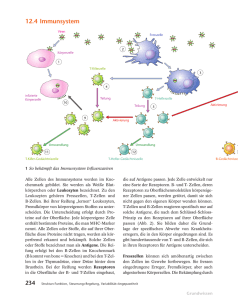
IMMUNOLOGIE SEMINARE
Werbung

IMMUNOLOGIE SEMINARE IMMUNOLOGIE SEMINARE Editor Erna Pap Herausgegeben von Éva Pállinger Edit Buzás András Falus György Nagy Marianna Csilla Holub Sára Tóth László Kőhidai Zsuzsanna Pál Übersetzt von Erna Pap Zoltán Pós Viktor Molnár Ottó Dobozy Fachliche Korrektur Edit Buzás András Falus Sprachliche Korrektur Andreas Christian Reichelt Semmelweis Universität • Budapest, 2012 © Erna Pap, 2012 2 Manuskript vertig gestellt: 31 Mai 2012 ISBN 978-963-9129-83-2 Semmelweis Universität Verantwortlicher für die Gestaltung des Manuskripts: Semmelweis Universität Verantwortlicher Editor: Erna Pap Technischer Redakteur: Adrienn Ádám Länge des Manuskripts: 238 Seiten 3 INHALTSVERZEICHNIS 1. IMMUNOLOGISCHE GRUNDBEGRIFFE. DER AUFBAU DES IMMUNSYSTEMS (ÉVA PÁLLINGER) ..... 10 1.1. 1.2. 1.3. 1.3.1. 1.3.2. 1.3.2.1. 1.3.2.2. 1.3.2.3. 1.3.3. 1.4. 1.5. 1.5.1. 1.5.1.1. 1.6. 1.6.1. 1.6.1.1. 1.6.1.2. 1.6.2. 1.6.3. 1.6.4. 1.6.5. 1.7. 1.7.1. 1.7.1.1. 1.7.1.2. 1.7.1.2.1. 1.7.1.2.2. 1.7.1.3. 1.7.1.4. 1.7.1.5. 1.8. Einführung: die Aufgabe und die Eigenschaften des Immunsystems.............................................. 10 Die angeborene und die erworbene Immunität ............................................................................... 10 Immunologische Grundbegriffe ....................................................................................................... 11 Was erkennt das Immunsystem? .................................................................................................... 11 Durch welche Strukturen werden die Antigene erkannt? ................................................................ 11 Mustererkennungsrezeptoren (PRR) .............................................................................................. 12 T-Zell Rezeptor (TZR) ..................................................................................................................... 12 B-Zell Rezeptor (BZR)..................................................................................................................... 12 Was sind die Folgen der Antigenerkennung? ................................................................................. 12 Lokale Immunantwort ...................................................................................................................... 13 Das Treffen ist die Bedingung der Erkennung und der Antwort. ..................................................... 14 Lymphozyten-Rezirkulation ............................................................................................................. 14 Extravasation .................................................................................................................................. 15 Die Organe des Immunsystems ...................................................................................................... 15 Lymphknoten .................................................................................................................................. 16 Die Zellen der Lymphknoten ........................................................................................................... 16 Wachposten-Lymphknoten (Sentinel) ............................................................................................. 17 Knochenmark .................................................................................................................................. 17 Milz.................................................................................................................................................. 18 Thymus ........................................................................................................................................... 19 Mukosaassozierte lymphatische Gewebe: MALT ........................................................................... 19 Die Zellen des Immunsystems ........................................................................................................ 20 Die Untersuchung der Zellen des Immunsystems ........................................................................... 21 Blutbild und Knochenmarkausstrich Untersuchungen ..................................................................... 21 Zytochemische Reaktionen ............................................................................................................. 22 PAS Reaktion .................................................................................................................................. 22 Nicht-spezifische Esterase Reaktion ............................................................................................... 22 Flusszytometrie ............................................................................................................................... 22 Immunhistochemie .......................................................................................................................... 23 Genetische Untersuchungen ........................................................................................................... 23 Kommunikationen zwischen den Zellen des Immunsystems .......................................................... 23 2. DIE AUF DER ANTIGEN-ANTIKÖRPER WECHSELWIRKUNG BERUHENDEN METHODEN I. (EDIT BUZÁS) ............................................................................................................................................................ 25 2.1. 2.1.1.1. 2.1.2. 2.2. 2.2.1. 2.2.2. 2.2.2.1. 2.2.2.2. 2.2.2.3. 2.2.2.4. 2.2.3. 2.2.4. 2.2.5. 2.2.6. Die Eigenschaften der in der Diagnostik verwendeten Antikörper .................................................. 25 Grundbegriffe .................................................................................................................................. 26 Die Möglichkeiten die Antikörper zu markieren ............................................................................... 27 Methoden ........................................................................................................................................ 28 Flusszytometrie ............................................................................................................................... 28 ELISA (Enzyme Linked Immunosorbent Assay) ............................................................................. 29 Die indirekte ELISA Reaktion .......................................................................................................... 30 Sandwich ELISA ............................................................................................................................. 30 Kompetitive ELISA .......................................................................................................................... 31 Was ist zu beachten, bei der Durchführung von ELISA ? Was kann Probleme verursachen?........ 32 ELFA (Enzyme Linked Immunofluorescent Assay) ......................................................................... 33 ELISPOT ......................................................................................................................................... 33 Immuno blot (Western blot) ............................................................................................................. 34 Auf radioaktiven Markierungen basierende Methoden .................................................................... 36 4 2.2.6.1. 2.2.6.2. 2.2.7. 2.2.7.1. 2.2.7.2. 2.2.7.3. 2.2.7.4. 2.2.7.5. 2.2.7.6. 2.2.8. 2.2.8.1. 2.3. Radioimmunoassay (RIA) ............................................................................................................... 36 Immunoradiometric assay (IRMA) ................................................................................................... 37 Immuncytochemie (Immunhistochemie) .......................................................................................... 38 Direkte Methode .............................................................................................................................. 39 Indirekte Methode ........................................................................................................................... 39 Kontrollen ........................................................................................................................................ 40 PAP Methode (Peroxidase anti-Peroxidase Verfahren) .................................................................. 40 Avidin-Biotin Komplex (ABK) Methode ............................................................................................ 40 Fluorescens-Mikroscopie und Laser konfokale Mikroskopie .......................................................... 40 Lateral flow Tests ............................................................................................................................ 41 Multiplex Immunoassay Systeme .................................................................................................... 42 Welche Immunoassay ist zu verwenden? ....................................................................................... 43 3. AUF ANTIGEN-ANTIKÖRPER INTERAKTION BERUHENDE METHODEN II: DIE IMMUNSEROLOGIE (ANDRÁS FALUS, GYÖRGY NAGY) .............................................................................................................. 45 3.1. 3.2. 3.3. 3.4. 3.5. 3.6. 3.7. 3.8. 3.9. Immunkomplex und Immunpräzipitat .............................................................................................. 45 2D Immundiffusion (2D Einfach- oder 2D Doppeldiffusion) ............................................................. 50 Serumelektrophorese ...................................................................................................................... 51 Immunelektrophorese und Immunfixierung ..................................................................................... 54 Raketenelektrophorese ................................................................................................................... 55 2D Immunelektrophorese ................................................................................................................ 56 Turbidimetrie und Nephelometrie .................................................................................................... 56 Klinische Anwendungen von serologischen Methoden ................................................................... 57 Typen der Agglutination und ihre klinische Anwendungen .............................................................. 58 4. DIE PRAKTISCHE BEZÜGE DES KOMPLEMENTSYSTEMS (EDIT BUZÁS)............................................. 64 4.1. 4.2. 4.3. 4.3.1. 4.3.1.1. 4.3.1.2. 4.3.2. 4.3.3. 4.3.4. 4.3.5. 4.3.6. 4.4. 4.4.1. 4.5. Das Komplementsystem ................................................................................................................. 64 Die Funktionen des Komplementsystems ....................................................................................... 65 Untersuchung der Aktivität des Komplementsystems ..................................................................... 67 Bestimmung den CH50 Wert des Blutserums ................................................................................. 67 Schafserythrozyten Immunoassay .................................................................................................. 68 Liposom Immunoassay ................................................................................................................... 69 Untersuchung der alternativen Komplementaktivation .................................................................... 70 Untersuchung des lektinabhängigen Weges ................................................................................... 70 Komplement Konvertase-Assay (CCA) ........................................................................................... 70 Weitere Verfahren zum Nachweis von Komplementproteinen ........................................................ 71 Sonstige, mit dem Komplementsystem verbundene Verfahren ...................................................... 73 Untersuchung einzelner Komplement-Elemente in verschiedenen pathologischen Zuständen ...... 74 Falldarstell....................................................................................................................................... 74 Aufgaben......................................................................................................................................... 76 5. DIE DURCHFLUSSZYTOMETRIE (ÉVA PÁLLINGER)................................................................................. 77 5.1. 5.2. 5.2.1. 5.2.1.1. 5.2.1.2. 5.2.1.3. 5.3. 5.3.1. 5.3.2. Einleitung ........................................................................................................................................ 77 Das Durchflusszytometer ................................................................................................................ 78 Bildliche Darstellung (data display) ................................................................................................. 81 Histogramm ..................................................................................................................................... 81 Dot plot/Punktwolkendiagramm ...................................................................................................... 82 Kinetische Messungen (time-based collection) ............................................................................... 84 Die Auswertung der FACS-Messdaten ........................................................................................... 84 Grösse und Granularität .................................................................................................................. 84 Fluoreszenz .................................................................................................................................... 85 5 5.4. 5.4.1. 5.4.2. 5.4.3. 5.4.3.1. 5.4.3.2. 5.5. 5.5.1. 5.5.1.1. 5.5.1.2. 5.5.1.3. 5.5.1.4. 5.5.1.5. 5.5.1.6. 5.5.1.7. 5.5.1.8. 5.5.1.9. 5.5.1.10. 5.5.2. 5.5.2.1. 5.5.2.1.1. 5.5.2.1.2. 5.5.2.1.3. 5.5.2.1.4. 5.5.2.2. 5.5.2.2.1. 5.5.2.3. 5.5.2.3.1. 5.5.2.3.2. 5.5.2.4. 5.5.2.4.1. 5.5.2.5. 5.6. 5.7. Probenvorbereitungen für diagnostische Messungen ..................................................................... 88 Herstellung einer Zellsuspension .................................................................................................... 88 Erythrolyse ...................................................................................................................................... 88 Fluoreszenz-markierung ................................................................................................................. 89 Direkte Färbung .............................................................................................................................. 89 Immunphänotypisierung .................................................................................................................. 90 Die Anwendungsgebiete der Durchflusszytometrie ......................................................................... 91 Auf der Methodik beruhende Verteilung .......................................................................................... 91 Proteinnachweisverfahren ............................................................................................................... 91 Untersuchungen der Synthese ........................................................................................................ 92 Enzymaktivität-Messungen ............................................................................................................. 93 Untersuchung der Nukleinsäure ...................................................................................................... 93 Untersuchung der Zellorganellen .................................................................................................... 94 Funktionelle Untersuchungen ......................................................................................................... 94 Bestimmung des relativen Abstandes zwischen Molekülen (FRET) ............................................... 94 Untersuchung löslicher Moleküle (Flow Cytometry Based Fluorescent Bead Immunoassay) ......... 95 Bestimmung der absoluten Zellzahl ................................................................................................ 96 Zellsortierung .................................................................................................................................. 97 Verteilung nach Krankheiten ........................................................................................................... 97 Maligne hämatologische Erkrankungen .......................................................................................... 97 Diagnostik und Differentialdiagnostik .............................................................................................. 97 Diagnostik der Minimal-Resterkrankung ......................................................................................... 98 Nachweis der rezivierenden hämatologischen Erkrankungen ......................................................... 98 Untersuchung der Empfindlichkeit auf die Terapie (MDR = multidrug resistance) .......................... 99 Immundefizienz-Syndrom ............................................................................................................. 100 Phänotyp-Untersuchungen und Nachfolge in der Diagnostik von Immundefekten ....................... 100 Autoimmunerkrankungen .............................................................................................................. 100 Die Diagnostik der Autoimmunerkrankungen ................................................................................ 100 Die Bestimmung der Aktivität von Autoimmunerkrankungen ........................................................ 101 Die Organtransplantation .............................................................................................................. 101 HLA-Assoziation............................................................................................................................ 101 Infektionen .................................................................................................................................... 102 Qualitätskontrolle (quality control) ................................................................................................. 103 Laboratorische Normalbefunde ..................................................................................................... 104 6. IMMUNISIERUNG UND VAKZINATION (EDIT BUZÁS) ............................................................................. 105 6.1. 6.2. 6.3. 6.4. 6.5. Ziel der Immunisierung und ihre praktische Durchführung ............................................................ 105 Die Adjuvantien, ihre Rolle und Gestaltungen ............................................................................... 105 Faktoren, welche die Wirksamkeit der Immunisierung beeinflussen ............................................. 107 Funktionelle Gruppeneinteilung der Antigene und Epitopen ......................................................... 107 Ausführungsmöglichkeiten der Immunisierung ............................................................................. 108 7. VAKZINATION (MARIANNA CSILLA HOLUB)............................................................................................ 109 7.1. 7.1.1. 7.1.1.1. 7.1.2. 7.1.3. 7.2. 7.2.1. 7.2.2. 7.3. Aktive Vakzination ......................................................................................................................... 110 Aktive Vakzination erzeugt starke primäre Antikörper-Antworten ................................................. 110 T-Zell Gedächtnis ist eine der wichtigsten Voraussetzungen einer wirksamen TD B-Zell-Antwort 114 Neben der Antikörper-Produktion kann auch eine zellulären Immunantwort Ziel der Vakzination sein ............................................................................................................................................... 115 Typen der T-Gedächtniszellen ...................................................................................................... 117 Lebensalter-abhängige Impfung-Strategien .................................................................................. 119 Welche Probleme muss die Vakzination in Kleinkindern überwinden? ......................................... 120 Impfung-Strategien für Kleinkinder und Personen in hohem Alter ................................................ 121 Arten von Impfstoffen .................................................................................................................... 122 6 7.4. 7.4.1. 7.4.2. 7.4.3. Neue Wege in der Impfstoffentwicklung ........................................................................................ 123 Verbesserung von gängigen Vakzinen.......................................................................................... 123 Biotechnologische Impfstoffentwicklung........................................................................................ 125 Entwicklung von neuen Vakzinationsverfahren ............................................................................. 129 8. ZELLKULTUREN (SÁRA TÓTH) ................................................................................................................ 130 8.1. 8.2. 8.3. 8.4. 8.5. 8.5.1. 8.5.2. 8.5.3. 8.5.4. 8.6. 8.7. 8.8. Definition der Zell- und Gewebekultivierung, Typen und Formen der Kulturen ............................. 130 Für eine Zellkultur notwendigen Voraussetzungen ....................................................................... 132 Zellzahlveränderungen und unterschiedliche Zellzählungsmethoden ........................................... 134 Separations- und Selektionsverfahren der Zellen ......................................................................... 136 Einige Verwendungsarten der Zellkulturen ................................................................................... 139 Bestimmung der Plating-Effizienz ................................................................................................. 140 Bestimmung der Proliferation und der Vermehrungsfähigkeit der Zellen ...................................... 140 Abmessung der Zytotoxizität ......................................................................................................... 142 Herstellung der monoklonalen Antikörper ..................................................................................... 142 Spezielle Kultivierungsverfahren ................................................................................................... 143 Untersuchungsmöglichkeiten der Zellen in einer Suspensionskultur ............................................ 145 Anwendungsbereiche der Zellkultivierung ..................................................................................... 146 9. DIE HOMING, DIE GEZIELTE MIGRATION VON IMMUNZELLEN. EXTRAVASATION IN ENTZÜNDUNGEN (LÁSZLÓ KŐHIDAI) ...................................................................................................... 148 9.1. 9.2. 9.3. 9.4. 9.5. 9.6. 9.6.1. 9.6.2. 9.7. 9.7.1. 9.7.2. 9.7.3. 9.7.4. 9.7.5. Einführung..................................................................................................................................... 148 Die Hauptformen der Zellmigration ............................................................................................... 148 Die die Chemotaxis auslösenden Moleküle .................................................................................. 149 Chemotaxis Rezeptoren................................................................................................................ 150 Die sich bewegenden Zellen des menschlichen Körpers und ihre Bewegungsformen ................. 152 Zellmotilität und Immunantwort ..................................................................................................... 152 Transendotheliale Migration der Lymphozyten ............................................................................. 153 Migrationswege der dendritischen Zellen ...................................................................................... 154 Die Methoden der Messung der Zellmotilität (Chemotaxis) ........................................................... 156 Reversible Systeme ...................................................................................................................... 157 Irreverzible Systeme ..................................................................................................................... 158 In vivo Techniken .......................................................................................................................... 158 Die neusten Techniken ................................................................................................................. 159 Weitere Methode zur Migrationsanalyse ....................................................................................... 161 10. ANALYSEMETHODEN DER AUTOANTIKÖRPER, HLA-TYPISIERUNG (GYÖRGY NAGY, ZSUZSANNA PÁL) ........................................................................................................................................ 163 10.1. 10.1.1. 10.1.2. 10.1.2.1. 10.1.2.2. 10.1.2.3. 10.1.2.4. 10.1.2.5. 10.2. 10.2.1. 10.2.2. 10.2.2.1. 10.2.2.2. Die Autoantikörper und ihre analytische Methoden ....................................................................... 163 Die Entstehung und allgemeine Eigenschaften der Autoantikörper .............................................. 163 Die wichtigsten Charakteristika von Autoimmunerkrankungen ..................................................... 165 Lupus erythematodes (SLE) ......................................................................................................... 166 ANA (antinukleäre Antikörper) ...................................................................................................... 167 Kryoglobuline ................................................................................................................................ 168 Rheumatoide Arthritis.................................................................................................................... 169 Organspezifische Autoimmunitäten............................................................................................... 170 Die HLA-Typisierung ..................................................................................................................... 172 Die Nomenklatur des HLA-Systems .............................................................................................. 172 Methoden der HLA-Typisierung .................................................................................................... 173 Serologische Methoden: ............................................................................................................... 173 Molekulare Methoden (DNA Sequenzanalyse) ............................................................................. 174 7 10.2.3. 10.2.4. 10.2.5. Gewebe- und Organtransplantation .............................................................................................. 175 HLA Assoziation mit Rheumatoider Arthritis ................................................................................. 177 HLA Assoziation mit Spondylitis ankylosans (SPA, Morbus Bechterew) ....................................... 177 11. ÜBEREMPFINDLICHKEIT TYP I.: ALLERGIE (MARIANNA CSILLA HOLUB) .......................................... 180 11.1. 11.2. 11.3. 11.3.1. 11.3.2. 11.3.2.1. 11.3.2.2. 11.3.3. 11.3.4. 11.3.5. 11.3.5.1. 11.3.5.2. 11.3.5.3. 11.3.6. 11.4. 11.5. 11.5.1. 11.5.2. Die Gruppierung der Überempfindlichkeitsreaktionen ................................................................... 180 Die Entstehung der allergischen Reaktionen ................................................................................ 181 Die Symptome der Allergie ........................................................................................................... 183 Die Type der Allergene ................................................................................................................. 183 Wie entstehen die allergischen Symptome? ................................................................................. 184 Die aus den Mastzellen freigesetzenden Stoffe ............................................................................ 185 Rezeptoren ................................................................................................................................... 185 Allergische Kreuzreaktion ............................................................................................................. 186 Nahrungsmittelintoleranz und Nahrungsmittelallergie ................................................................... 187 Anaphyilaktoide Reaktion / Pseudoallergie ................................................................................... 187 Arzneimittel - ausgelöste Pseudoallergie ...................................................................................... 188 Nahrungsmittel - ausgelöste Pseudoallergie ................................................................................. 188 Physikalische Faktoren – und psychologische Stress-vermittelte Pseudoallergie ........................ 189 Sonnenallergie .............................................................................................................................. 189 Die Diagnose Der Allergie ............................................................................................................. 190 Die Therapie der Allergie .............................................................................................................. 192 Medikamentöse Behandlung ......................................................................................................... 192 Immuntherapie: (Hyposensitisierung/Desensitisierung) ................................................................ 194 12. HYPERSENSITIVITY REACTIONS II: TYPE II-IV HYPERSENSITIVITY REACTIONS (MARIANNA CSILLA HOLUB) ............................................................................................................................................ 196 12.1. 12.1.1. 12.1.1.1. 12.1.1.2. 12.1.1.3. 12.1.2. 12.1.2.1. 12.1.2.2. 12.1.2.3. 12.1.3. 12.1.4. 12.2. 12.2.1. 12.2.2. 12.2.3. 12.2.4. 12.3. 12.3.1. 12.3.2. 12.3.3. 12.3.4. Überempfindlichkeitsreaktion vom Typ II ...................................................................................... 196 Beispiele für eine Typ II Reaktion, die von extrinsischen Antigenen ausgelöst werden ................ 197 Erythroblastosis fetalis (Morbus haemolyticus neonatorum, Immunhämolytische Krankheit bei Feten und Neugeborenen) ............................................................................................................ 197 Typ II Überempfindlichkeit bei der Organtransplantation .............................................................. 197 Medikamenten-induzierten Typ II Reaktionen ............................................................................... 197 Intrinsches Antigen – Beispiele für die von einem Autoantikörper ausgelöste Krankheit .............. 198 Goodpasture-Syndrom (Hauptsächlich wegen den Autoantikörper gegen Kollagen Typ IV in den Nieren) .......................................................................................................................................... 198 Myasthenia gravis (MG) ................................................................................................................ 198 Basedow-Krankheit ....................................................................................................................... 199 Diagnose ....................................................................................................................................... 199 Therapie ........................................................................................................................................ 199 Typ III Überempfindlichkeitsreaktion ............................................................................................. 200 Eine typische Krankheit für den lokalen Typ III ............................................................................. 201 Akute systemische Typ III Hypersensitivitätsreaktion.................................................................... 202 Chronische systemische Typ III Hypersensitivitätsreaktion ........................................................... 202 Therapie ........................................................................................................................................ 202 Überempfindlichkeitsreaktion vom Typ IV oder verzögerter Typ (DTH=delayed type hypersensitivity) ............................................................................................................................ 203 DTH-Hauttests .............................................................................................................................. 203 Kontakthypersensitivität / Kontaktdermatitis.................................................................................. 204 Zöliakie (glutensensitive oder gluteninduzierte Enteropathie) ....................................................... 205 Terapie .......................................................................................................................................... 206 8 13. IMMUNOLOGICAL THERAPIES (GYÖRGY NAGY, ÉVA PÁLLINGER, ZSUZSANNA PÁL) ..................... 207 13.1. 13.2. 13.2.1. 13.2.2. 13.2.2.1. 13.2.2.2. 13.2.2.3. 13.2.2.4. 13.3. 13.3.1. 13.3.2. 13.3.3. 13.3.3.1. 13.3.3.1.1. 13.3.3.1.2. 13.3.3.1.3. 13.3.3.2. 13.3.3.3. 13.3.3.4. 13.3.3.4.1. 13.3.3.4.2. 13.3.3.4.3. 13.4. 13.4.1. 13.4.1.1. 13.4.1.2. 13.4.1.3. 13.4.1.3.1. 13.4.1.3.2. 13.4.1.3.3. 13.4.1.3.4. 13.4.1.3.5. 13.4.1.3.6. 13.4.1.3.7. Grundbegriffe ................................................................................................................................ 207 Gezielte molekulare Therapie (targeted molecular therapy, TMT) ................................................ 208 Biologika oder Biopharmazeutika (biologics) ................................................................................ 208 Targeting ....................................................................................................................................... 208 Targeting mit Nanotechnologie ..................................................................................................... 209 Targeting mit monoklonalen Antikörpern....................................................................................... 209 Targeting mit Peptiden .................................................................................................................. 212 Chemotaktisches targeting (chemotactic drug targeting) .............................................................. 212 Immuntherapie .............................................................................................................................. 212 Aktive Immuntherapie ................................................................................................................... 212 Präventive (prophylaktische) Impfungen ....................................................................................... 213 Therapeutische Impfungen ........................................................................................................... 213 Weiterentwicklung der gängigen Impfungen ................................................................................. 214 Anwendung von Adjuvanzien ........................................................................................................ 214 Unterstützung der Antigenpräsentierung....................................................................................... 215 Verstärkung der Aktivität von T Zellen .......................................................................................... 215 Zytokine in der aktiven Immuntherapie ......................................................................................... 215 Manipulation der Immuntoleranz ................................................................................................... 215 Zelluläre Immuntherapien ............................................................................................................. 216 Induktion der antitumoralen Immunantwort ................................................................................... 216 Anlockung von tumorspezifischen Immunzellen zum Tumor ........................................................ 217 Neutralisierung der tumorvermittelten Immuntoleranz................................................................... 217 Passive Immuntherapie................................................................................................................. 218 Monoklonale Antikörper in der Therapie ....................................................................................... 218 Herstellung der monoklonalen Antikörper ..................................................................................... 218 Risiken und Nebenwirkungen der monoklonalen Antikörper-Therapien ....................................... 220 Therapeutische monoklonale Antikörper in der Rheumatologie .................................................... 220 Der Pathomechanismus der rheumatoiden Arthritis: die Rolle der proinflammatorischen Zytokine ........................................................................................................................................ 221 Die zentrale Rolle des TNFs in der RA ......................................................................................... 221 Gängige Methoden der TNF Blockade .......................................................................................... 222 Nebenwirkungen von TNF-blockierenden Medikamenten............................................................. 222 TNF-Blockade in anderen rheumatologischen Erkrankungen ....................................................... 223 Entzug von B-Zellen mit Hilfe von anti-CD20 monoklonalen Antikörpern...................................... 223 Hemmung der T-Zell-Aktivierung mit CTLA-4 Immunglobulin Fusionsproteinen ........................... 223 14. EINIGE IMMUNOLOGISCHE ANWENDLUNGSMÖGLICHKEITEN DER INFORMATIK (EDIT BUZÁS) .. 226 ABKÜRZUNGEN ................................................................................................................................................ 236 9 1. IMMUNOLOGISCHE GRUNDBEGRIFFE. DER AUFBAU DES IMMUNSYSTEMS (ÉVA PÁLLINGER) 1.1. Einführung: die Aufgabe und die Eigenschaften des Immunsystems Die Aufgabe des Immunsystems ist die Aufrechterhaltung der Identität des Organismus. Das Immunsystem besitzt die Fähigkeit, die eigenen Stoffe von den fremden, und die für den Organismus gefährlichen von den ungefährlichen Stoffen zu unterscheiden. Die Erkennung des Fremdstoffes löst eine Immunantwort aus, die je nach der Natur der Stoffe, den aktuellen Einflüssen aus der Umgebung und den physiologischen Wirkungen entweder als Effektorantwort (Eliminierung des Fremdstoffes), Immuntoleranz oder Ignoranz (immunologische Stummheit) ausgeprägt wird. Die wesentlichsten Eigenschaften des Immunsystems sind: Spezifität, Empfindlichkeit, Selektivität und immunologisches Gedächtnis. 1.2. Die angeborene und die erworbene Immunität Die Funktion des Immunsystems ruht auf zwei Pfeilern: auf der angeborenen und der erworbenen Immunität. Die angeborene (natürliche; nicht-spezifische) Immunantwort ist schnell, sie tritt sofort nach dem Eintreten der Krankheitserreger auf. Sie ist keine antigen-spezifische Reaktion: die Erkennung der Pathogene geschieht durch die Mustererkennungsrezeptoren (PRR), die eine Vielzahl von pathogenassozierten molekularen Mustern (PAMP) erkennen. Die Fremdstoffe und Krankheitserreger phagozytierenden Makrophagen, Granulozyten, die dendritischen Zellen und die natürlichen Killerzellen spielen eine wichtige Rolle bei der Entstehung der angeborenen Immunantwort. Die Funktion des Komplementsystems ist auch bedeutend: die Komponenten des Komplementsystems sind in den körperlichen Flüssigkeiten vorhanden. Der Ablauf der Immunantwort ist unabhängig vom Immunologischen Gedächtnis, da die angeborenen immunologischen Antworten kein Gedächtnis entstehen lassen können. Im Gegensatz dazu, kommt die erworbene (adaptive, spezifische) Immunität erst verzögert zum Zug. Die Antwortreaktion kann erst nach Tagen oder Wochen nach dem Erscheinen der Erreger nachgewiesen werden. Die antigenspezifische Reaktion wird durch spezifische Antigenrezeptoren ausgelöst. Der Ablauf der Antwort wird vom Immunologischen Gedächtnis bestimmt. T und B Lymphozyten sind ihre wesentlichsten zellulären Komponenten . 10 1.3. Immunologische Grundbegriffe Die Immunantwort bedeutet die Erkennung des Fremdstoffes (Zelle / Erreger) und die darauf gegebene Antwortreaktion. Die drei wichtigsten Fragen können so gestellt werden: 1. Was erkennt das Immunsystem? 2. Durch welche Strukturen werden die gefährlichen Strukturen / Antigene erkannt? 3. Was sind die Folgen der Erkennung von Antigenen? 1.3.1. Was erkennt das Immunsystem? Das Immunsystem erkennt Antigene. Irgendwelche Moleküle /Strukturen, die eine Immunantwort auslösen, heißen Antigene. Die Immunogenität ist die Fähigkeit eines Antigens eine Immunantwort auszulösen. Den kleinen Bereich (Molekülabschnitt) des Antigens, der von dem T- oder B-Rezeptor erkannt wird, nennt man Antigendeterminante oder Epitop. Ein einziges Antigen kann mehr Epitope besitzen. Das Mapping der Epitope hat Abbildung 1. Ein einziges Antigen kann mehr Epitope besitzen diagnostische und therapeutische Bedeutung (Abbildung 1.). Man unterscheidet Auto, - Allo – und Xenoantigene. Autoantigen ist ein Antigen, das dem eigenen Körper entstammt, jedoch von dessen Immunsystem nicht als solches erkannt wird. Antikörper können gegen sie produziert werden. Alloantigen ist ein Antigen von der gleichen Spezies aber von einem genetisch unterschiedlichen, anderen Körper, welches eine Immunantwort ( während Transplantation ) hervorruft. Xenoantigen ist ein Antigen einer anderen Spezies, das eine Immunantwort hervorruft. 1.3.2. Durch welche Strukturen werden die Antigene erkannt? Die Antigen-erkennenden Rezeptoren sind in zwei Gruppen aufgeteilt. 1) Rezeptoren, die eine Vielzahl des pathogenassozierten molekularen Mustern – eine allgemeine Struktur auf der Oberfläche der Pathogene - erkennen. Sie sind die Mustererkennungsrezeptoren (PRR). Diese Rezeptoren befinden sich meistens auf den Zellen des nicht-spezifischen Immunsystems. 2) Spezifische Rezeptoren, die die Fähigkeit haben, eine Vielzahl von unterschiedlichen Antigenen zu erkennen. Diese Rezeptoren befinden sich auf der Oberfläche der T- Zellen (TZR) und B-Zellen (BZR). Es ist zu betonen, dass jeder Lymphozyt nur einen Typ von Rezeptor exprimiert, womit er nur ein Epitop des bestimmten Antigens erkennen kann. 11 1.3.2.1. Mustererkennungsrezeptoren (PRR) Die Mustererkennungsrezeptoren erkennen die auf den Pathogenen charakteristischen allgemeinen Strukturen, (PAMP – „pathogen associated molecular pattern” - pathogenassoziertes Molekülmuster) und das veränderte Muster, welches durch den auf die Zelle einwirkenden Stress, umgewandelt wurde. (DAMP - „danger-associated molecular pattern” – gefahrenassoziertes Molekülmuster). Mikrobiale Nukleinsäuren, Peptide und LPS auf der Zellwand der Gram - negativen Pathogene können den pathogenassozierten Strukturen dienen. Intrazelluläre Proteine wie Hitzeschockproteine, HMGB1 (chromatin-associated protein high-mobility group box 1), Proteine der extrazellulären Matrix, Harnsäure, freigesetzte DNS, usw. stellen eingefahreninduziertes Signal (DAMP) dar. Eine Vielzahl von Molekülen können sich als DAMP verhalten, abhängig von der Zusammensetzung der lokalen Zellen. 1.3.2.2. T-Zell Rezeptor (TZR) Die T- Zell Rezeptoren werden auf der Oberfläche der T-Zellen exprimiert, sie erkennen einen Peptidepitop eines Antigens mit großer Spezifität. Die Antigenbearbeitung und die Antigenpräsentierung mit dem MHC Molekül sind Voraussetzungen für die Erkennung durch TZR. Die Zellen, die die Fähigkeit haben Fremdstoffe aufzunehmen, zu bearbeiten und den T-Zellen zu präsentieren, nennt man professionelle antigenpräsentierende Zellen. Die dendritischen Zellen (DZ), die Makrophagen (Mph) und die B Lymphozyten sind die professionellen APZ. Es existieren auch nicht-professionelle APZ, welche die für die Antigenpräsentierung unerlässlichen MHC Moleküle auf ihrer Oberfläche erst nach Zellaktivierung aufweisen. Hierzu gehören die Fibroblasten, einige Epithelzellen (z.B. die Epithelzellen des Thymus und der Schilddrüse), die beta-Zellen der Bauchspeicheldrüse und die Endothelzellen. 1.3.2.3. B-Zell Rezeptor (BZR) Die aus den B Lymphozyten differenzierenden Plasmazellen sezernieren größere Glykoproteine, die Antikörper oder Immunglobulin genannt werden. Die Antikörper sind sowohl im Blut als auch in den biologischen Flüssigkeiten zu finden. Ihre Aufgabe ist es, bakterielle oder virale Antigene zu binden. Der BZR- Komplex besteht aus einem Zelloberfächenimmunglobulin mit jeweils einem Heterodimer (Igα-Igβ). Das Immunglobulin dient als signalerkennender Teil auf den B-Lymphozyten und bindet an das Antigen. Dieses Immunglobulin ist identisch mit einem sezernierten monomeren Immunoglobulin, außer ist es mit einem Heterodimer verbunden. Das Heterodimer kann Signale in das Innere der BZellen weiterleiten. 1.3.3. Was sind die Folgen der Antigenerkennung? Wie es schon im Subkapitel 2. zusammengefasst wurde, reagiert das Immunsystem über zwei Wege: durch die schnell entstehenden natürlichen und durch die langsamer entstehenden spezifischen Immunantworten. 12 Die spezifische Immunantwort unterteilt sich in die so genannte antikörpervermittelte humorale Immunität und die zellgetragene zelluläre Immunität. Die Aktivierung der B Zellen beginnt mit der Erkennung eines Antigens. Darauf folgt die klonale Proliferation, die Entstehung der Zentroblasten, eine Zellteilung mit vielen Mutationen und schließlich die Selektion jener B Zellen, welche Rezeptoren mit großer Affinität besitzen. Danach entstehen die Plasmazellen und die Gedächtniszellen. Treffen T Zellen auf Antigene, kommt es ebenso zur klonalen Proliferation und der Entwicklung von Gedächtniszellen. Durch die Zellaktivierung kommt es zusätzlich zur Sekretion von regulatorischen Proteinen (Cytokine) und es werden die Effektorfunktionen ausgelöst. Es ist zu betonen, dass B und T Zellen so zusagen „eng zusammenarbeiten“: wie auch die dendritischen Zellen, präsentieren auch die B Zellen ihre erkannten, aufgenommenen und abgebauten Antigene den T Zellen durch die MHC-II Expression. Diese Zellen heißen professionelle APZ. Weiters stellen die T aktivierten T Zellen solche Cytokine her, die zur Differenzierung der B Lymphozyten unerlässlich sind. Aus klinischer Sicht, sind die Begriffe der aktiven und passiven Immunität wichtig. Eine aktive Immunität entsteht, wenn Immunogene (Pathogene, Infektion, Schutzimpfungen) in den Körper eintreten. Passive Immunität kann hervorgerufen werden, indem immunologisch bereits kompetente Zellen oder/und Antikörper (Serum) einer immunisierten Person auf einen anderen Körper übertragen werden. 1.4. Lokale Immunantwort Die Erkenntnis der Vielzahl der Immunzellen und ihrer eng miteinander verknüpften Funktionsweise ist bedeutend für die Entwicklung eines medizinischen Modells. Es soll hier durch den Ablauf der lokalen Immunantwort dargestellt werden. Unterschiedliche Erreger und irritierende Stoffe treten den Organismus am Ort der Gewebeverletzung ein. Die Frage ist: wer und wie wird auf diese Fremdstoffe reagiert. In meisten Fällen sind auch Gefäße durch die Verletzung des Gewebes betroffen, oder zumindest erhöht sich ihre Durchlässigkeit. Folglich gelangen Plasma und zelluläre Komponente des Blutes ins Gewebe. Die Extravasation wirkt auf die Funktion sowohl der zellulären als auch der löslichen Komponenten. Die ins Gewebe gelangten Blutplättchen aktivieren sich bereits am Anfang. Ihre Aktivierung fördert die Induktion des Gerinnungssystems. Einerseits entsteht dadurch ein undurchlässiges und unlösliches Fibrinnetz, welches ein mechanisches Hindernis für die Erreger darstellt. Andererseits formt dieses Fibrinnetz, zusammen mit den aktivierten Blutplättchen, den Grund des Thrombus, der dem Verschluss der verletzten Ader dient. Gleichzeitig werden aus den Thrombozyten viele biologisch aktive Stoffe freigesetzt, die auf die benachbarten Zellen wirken, dabei spielen sie eine regulatorische Rolle. Im Blutplasma befinden sich auch die Komponenten des Komplementsystems. Das aktivierende Komplementsystem spielt eine Vielzahl von Rollen. Die Fragmente C3a und C5a üben auf die neutrophilen Granulozyten eine chemotaktische Wirkung aus. C3b opsonisiert die Bakterien und fördert dadurch ihre Eliminierung. Auch dendritischen Zellen, Makrophagen, Mastzellen und 13 Granulozyten besitzen C3b-Rezeptoren, deswegen beeinflussen C3b Fragmente auch die Funktion dieser Zellen. Der membranangreifende Komplex, am Ende des Weges der Komplementaktivierung, hat direkt zytolitische Wirkung. Die ins Gewebe gelangenden neutrophilen Granulozyten haben meistens eine Effektorfunktion. Sie haben die Fähigkeit die Erreger zu fressen. Außerdem bilden sie reaktive Sauerstoffintermediäre (ROI) und proteolytische Enzyme, die nach ihrer Freisetzung die Bakterien extrazellulär zerstören. Dieser Prozess wird jedoch auch von Gewebeverletzungen begleitet. Der Geweberest wird von Makrophagen abtransportiert. Die Makrophagen und die dendritischen Zellen haben die Fähigkeit, die aufgenommenen Stoffe den anderen Zellen zu präsentieren. Mit anderen Wörtern, sind sie professionelle antigenpräsentierende Zellen. Hier treten die Lymphozyten in den Prozess ein, weil sie präsentierte Antigene erkennen können. Die von Makrophagen präsentierten Antigene aktivieren meistens die Th und Tc Zellen, währenddessen aktivieren die von dendritischen Zellen präsentierten Antigene die NKT Zellen. Dadurch werden die natürlichen und spezifischen Immunantworten miteinander verbunden. 1.5. Das Treffen ist die Bedingung der Erkennung und der Antwort. Voraussetzung für die Entwicklung einer spezifischen Immunantwort, ist das Aufeinandertreffen der Lymphozyten mit Antigenen. Deshalb bewachen die T und B Lymphozyten unseren Körper: sie zirkulieren um ihre Ziele zu finden. (T, B Zell Rezirkulation, (Homing)). 1.5.1. Lymphozyten-Rezirkulation Abbildung 2. Lymphozyten-Rezirkulation 14 Während der Lymphozyten-Rezirkulation wandern die naiven Lymphozyten (die, die noch auf kein Antigen getroffen sind) über das Blut von den primären Lymphorganen in die sekundären Lymphorgane/Gewebe ein. Im Falle dass sie auf kein Antigen treffen, treten sie durch die Lymphe wieder in den Blutfluss ein und patrouillieren dort weiter. Die Lymphozyten verlassen den Blutfluss meistens am Ort der „hochendothelialen Venolen“ (HEV), wobei sie dies aber auch wo anders tun könnten. Wenn die Lymphozyten in den sekundären Lymphgeweben auf die für sie geeigneten, spezifischen Antigene treffen, aktivieren sie sich. Durch ihre Aktivierung proliferieren sie sich und sie differenzieren sich zu Effektorzellen. Es ändert sich dadurch das Expressionsmuster der Adhesionsmoleküle an ihrer Zelloberfläche und folglich auch ihre Fähigkeit zu wandern. So ist es ihnen nämlich möglich, unter der Leitung der Chemokine, in das extra-lymphoide Gewebe auszuwandern und direkt am Ort des Angriffes zu wirken (Abbildung 2.). 1.5.1.1. Die Extravasation Wechselwirkung zwischen den Lymphozyten und den Endothelzellen ereignet sich in 4 Schritten. Es wird gewöhnlich „Adhesionskaskade“ genannt. Am Anfang rollen die Lymphozyten auf der Oberfläche des Endothels, wobei sie sich nur an einem Punkt berühren. Selektine leiten den Prozess. Die Bindung wird stärker im dritten und vierten Schritt. Der Prozess wird von Chemokinen, Chemokinrezeptoren und Integrinen gesteuert. Am Ende findet die Transmigration statt, indem die Zellen durch das Endothel hindurch wandern. Die Transmigration wird meistens Abbildung 3. Extravasation von Integrinen reguliert (Abbildung 3.). 1.6. Die Organe des Immunsystems Das Immunsystem – früher Lymphsystem genannt – besteht aus primären (zentralen) und sekundären (peripheren) Lymphorganen. Das Knochenmark und der Thymus gehören zu den zentralen Organen. Die Zellen des Immunsystems entstehen hier und ihre Differenzierung findet hier teilweise statt. Die Umlagerung der Immunglobulingene und der TZR Gene findet hier statt. Mit anderen Wörtern, es entstehen die reifen Klone der B und T Lymphozyten, die spezifische Rezeptoren besitzen und die Fähigkeit haben Antigene zu erkennen. Die sekundären Lymphorgane sind überall im Körper verteilt. Dies ist Sinnvoll, da die Erreger an vielen verschiedenen Stellen in den Körper eintreten können. Die Milz, der Appendix, die Mandeln und 15 die Lymphknoten sind abgrenzt von den übrigen Geweben, im Gegensatz zu den Lymphgewebe der Schleimhaut des Magen-Darm-Trakts, des Luftwegs, des Urogenitalsystems und der Haut. Alle gehören jedoch zu den sekundären Lymphorganen. Diese Organe sind miteinander durch die Lymphgefäße verknüpft. Die Lymphgefäße aus der Peripherie sammeln sich in den Hauptlymphgefässen (truncus lymphaticus dexter und ductus thoracicus), und münden in den Blutfuss. Eigentlich gehören auch die patrouillierenden Lymphozyten und die Antikörper zu den sekundären Lymphorganen. Eine kurze Zusammenfassung: die Rolle der primären Lymphorgane ist die „Herstellung” der Zellen des Immunsystems, währenddessen sichern die sekundären Lymphorgane den Ort des Treffens der Antigene und der Lymphozyten. 1.6.1. Lymphknoten 1.6.1.1. Die Zellen der Lymphknoten Die Lymphknoten sind kleine Strukturen, die sich praktisch überall im Körper befinden. An einigen Orten des Körpers sind sie in größeren Mengen zu finden, z.B. in den Achselhöhlen, in den Lenden, in der submandibularen Regionen und um die Aorta. Die Untersuchung dieser Regionen gehört zur Routine in der physikalischen Untersuchung durch den Arzt. Die Lymphknoten haben zwei wesentliche Aufgaben: 1) Mikroorganismen den und in Körper gelangte ihre Phagozyten entfernen die korpuskuläre Partikel 2) hier findet die Antigenpräsentierung statt. Die Lymphknoten bestehen aus einem äußeren Cortex (Rinde) und einer inneren Medulla (Abbildung 4.). In den anatomisch unterschiedlichen Regionen der Lymphknoten befinden sich unterschiedliche Zellen. Abbildung 4. Lymphknote 16 Die B Lymphozyten und die Makrophagen befinden sich im Cortex. Die B-Zellen wandern in die Lymphknoten durch die HEV ein, wo sie sich in den Folliculi organisieren. Die von Antigenen aktivierten B- Zellen bleiben in den Lymphknoten und beginnen sich zu teilen. Die nicht-stimulierten BZellen gehen in den Kreislauf zurück. Die aktivierten B-Zellen liegen im Zentrum des Folliculus und formen das Keimzentrum (centrum germinativum). Es wird von der marginalen Zone umgeben, in welcher sich naive B-Zellen und wenige T-Zellen befinden. Nach der Blasten-Transformation verlassen die aktivierten B-Zellen den Folliculus und treten in die paracorticalen und medullären Sinusse ein. Aus diesen Zellen entwickeln sich die Antikörper herstellenden Plasmazellen und die B Gedächtniszellen. Die paracorticale und interlobulare Region ist der Treffpunkt der T-Zellen mit dendritischen Zellen. Hier kommen die dendritischen Zellen aus der Peripherie an und präsentieren ihre aufgenommenen Antigene den spezifischen T-Zellen. Die Medulla ist reich in Plasmazellen. Die von Plasmazellen hergestellten Antikörper verlassen die Lymphknoten durch die efferenten Lymphgefäße. 1.6.1.2. Wachposten-Lymphknoten (Sentinel) Da sich Als die ersten Metastasen häufig in den Lymphknoten der Achselhöhlen bilden, kann man seit den 1990-er Jahren eine sogenannte Sentinel-(Wachposten) Lymphknoten-Biopsie durchführen. Das heißt, dass der Fluss der Lymphe von dem Tumor während der Operation beobachtet wird, und man den Lymphknoten, an dem die Lymphe als erstes ankommt – der so genannte Sentinel-Lymphknoten – entfernt. Die pathologische Untersuchung dieses Knoten ergibt das Tumorstadium der Region und erlaubt es so die Region selektiv zu entfernen und mit Bestrahlung zu behandeln. 1.6.2. Knochenmark Abbildung 5. Die Reifung der Immunzellen im Knochenmark. 17 Das rote Knochenmark ist der Ort der Hämatopoese. Die Hämatopoese geht von einer einzelnen hämatopoetischen Stammzelle aus. (Die gemeinsamen hämatopoetischen Stammzellen heißen pluripotente Stammzellen oder aus dem englischen Begriff „hematopietic stem cell” (HSC), aber in einigen Anatomie Bücher werden sie Hämozytoblasten genannt.) Um den Prozess zu vereinfachen, kann man sagen dass sich die hämatopoetischen Stammzellen zu lymphatischen und myeloiden Vorläuferzellen (Progenitorzellen) differenzieren. Die Reifung der myeloiden Zellen und der B Lymphozyten findet im Knochenmark statt, währenddessen wandern die T Progenitorzellen in Thymus ein und differenzieren sich dort. Die endliche Reifung der mononuklearen Phagozyten findet in den peripheren Geweben statt (Abbildung 5.). 1.6.3. Milz Das größte Lymphorgan unseres Körpers liegt in der Bauchhöhle in der Nähe des Magens, im linken Oberbauch unterhalb des Zwerchfells und oberhalb der linken Niere. Die Milz wird von einer bindegewebig bedeckten Kapsel umgeben, von der ein trabekuläres Bindegewebsgerüst in das Parenchym zieht. Das Parenchym besteht aus roter und weißer Pulpa. Die rote Pulpa enthält Blut aus dem die Phagozyten schädliche Partikel entfernen und gealterte rote Erythrozyten abbauen. Die weiße Pulpa ist für die immunologischen Aufgaben verantwortlich. Sie enthält Lymphfollikel, bestehend aus lymphatischem Gewebe mit B-Lymphozyten. Die um die Gefäße angeordneten „periarteriellen lymphatischen Scheiden“ (PALS) mit T-Lymphozyten gehören zur weißen Pulpa. Die B Zellen lagern sich in den Follikeln der weißen Pulpa ab. Die dendritischen Zellen und die größeren, aktivierten Lymphozyten sind die typischen Zellen der um den Follikeln liegenden marginalen Zone (Abbildung 6.). Abbildung 6. Die Milz 18 1.6.4. Thymus Der Thymus des Menschen ist ein zweilappiges Organ im Mediastinum. Seine Größe ist in Erwachsenen geringer, trotzdem funktioniert er unser ganzes Leben. Der Thymus ist der Ort der Differenzierung der T Zellen. Hier lernen die unreifen T Zellen die einzelnen Strukturen zu erkennen. Weiters werden die spezifischen T-Zell Rezeptoren und die CD4, CD8 Moleküle hier exprimiert Die Differenzierung der T Zellen wird von einer großartigen Zellzerstörung begleitet: nur 1-2% der differenzierten Zellen gelangen als T Lymphozyten in die Peripherie. Die Proliferation der aus dem Knochenmark ankommenden T Vorläuferzellen beginnt im Cortex, in dem subkapsulären Bereich. Hier entstehen die CD4- / CD8- doppelt negativen (DN) T Zellen, die die sogenannte positive Selektion durchgehen, während sie zur Medulla wandern. Sie erreichen die cortico-medullare Grenze als CD4+/CD8+ doppelt positive (DP) Zellen. Die sogenannte negative Selektion passiert in der Medulla. Während der Reifung verlieren die Thymozyten entweder ihre CD4, oder ihre CD8 Moleküle und entwickeln sich zu reifen, einfach positiven Th und Tc Zellen (Abbildung 7.). Abbildung 7. Die Selektion der T-Zellen im Thymus. 1.6.5. Mukosaassozierte lymphatische Gewebe: MALT Die Schleimhäute des Mundes, des Respirations – des Darms -und des Geschlechtstrakts haben ihre spezialisierten Immunsysteme, die man unter der Bezeichnung schleimassozierte lymphatische Gewebe zusammenfasst. In den Schleimhäuten des Darms befinden sich die sogenannten Peyer-Plaques, welche lymphatische Follikeln sind. Ihr Epithel besitzt viele intraepitheliale Lymphzyten (IEL), dessen größter Teil sich zu M Zellen entwickelt. Die M Zellen spielen eine besondere Rolle beim Schutz gegen Erreger. Sie nehmen die Antigene aus dem Lumen des Darms auf und befördern sie zu den Immunzellen der Peyer-Plaques. Infolge des Treffens mit den Antigenen, aktivieren sich die naiven und die Gedächtnis B Zellen. Der Prozess setzt sich in den mesenterialen Lymphknoten fort und spezifische Adhesionsmoleküle erscheinen auf der Oberfläche der aktivierten B Zellen (E7). Nach 19 ihrer Wanderung durch den Ductus Thoracicus und das Blut gelangen sie wieder in die Darmmukosa. Mit Hilfe der Adhäsionsmoleküle können sie nun durch die HEV (high endothel venule) Zellen des Darms aus dem Blutfluss auswandern und in der Mukosa ihre Wirkung entfalten (Abbildung 8.). Abbildung 8. Mukosaassozierte lymphatische Gewebe: MALT 1.7. Die Zellen des Immunsystems Die Zusammensetzung der Zellen des Immunsystems entsteht in den primären Immunorganen – im Knochenmark und Thymus –, aber ihre endliche reife Form erhalten sie erst in den peripheren Lymphorganen oder genau am Ort der Immunantwort. Sie werden eher nach ihren funktionellen Eigenschaften als ihrer Morphologie kategorisiert. Eine Verminderte Anzahl oder funktionelle Störungen von Immunzellen führt zu Immundefekten. Die Zellen des Immunsystems entstammen aus einer einfachen Knochenmark-Stammzelle. Diese Zelle hat die Fähigkeit sich zu erneuern und zu differenzieren. Die Differenzierung geht in zwei Richtungen: beide myeloide und lymphoide Linien entstehen. Aus der myeloiden Linie entwickeln sich im Knochenmark granulozyten – erythrozyten – monozyten und – megakaryozyten Kolonie-formende Einheiten. Die lymphoiden Vorläuferzellen differenzieren sich in drei Richtungen. Ein Teil der Zellen bleibt im Knochenmark, aus denen sich die B Zellen entwickeln. Der zweite Teil wandert in den Thymus. Aus diesen Zellen wiederum entwickeln sich die T und NKT Zellen. Aus dem dritten Teil entwickeln sich die NK Zellen. Der genaue Ort dieses Prozesses ist jedoch noch nicht geklärt. Direkte Zell-Zellinteraktionen zwischen den Stromazellen des Knochenmarks und den hämopoetischen Zellen, Cytokine, Wachstumsfaktoren und biogene Amine, spielen eine Rolle bei der Regulation der Hämatopoese. 20 In den letzten Jahren sind zahlreiche neue Gesichtspunkte bezüglich der Hämatopoese aufgetaucht. Es sieht so aus, als ob die strikten Kategorien verschwinden würden. Das könnte bedeuten, dass aus Progenitorenzellen nicht zur eine Zelllinie entsteht, (unidirektionell), sondern es ihnen möglich ist, sich je nach der aktuellen physiologischen Lage multidirektionell zu differenzieren. Neben den morphologischen und zytochemischen Eigenschaften, ist die Identifizierung der hämopoetischen Zellen meistens durch das Expressionsmuster ihrer zytoplazmatischen und Oberflächenproteine möglich. In den 1980 Jahren wurde die Nomenklatur der auf die Leukozyten charakteristischen Differenzierungsmarker/Antigene von einem internationalen Treffen vereinheitlicht. Sie wurde als „Cluster of Differentiation” genannt. (sieh Kapitel 3.) 1.7.1. Die Untersuchung der Zellen des Immunsystems Die Untersuchung der Zellen des Immunsystems besteht aus mehreren Schritten. Die Mengen und die Zusammensetzung der zirkulierenden weißen Blutzellen können durch ein qualitatives oder ein quantitatives Blutbild bestimmt werden. In einigen Fällen können auch Zellen mit pathologischer Morphologie wahrgenommen werden. Knochenmarkaspiration oder Biopsien sind stärker invasive Eingriffe, die in erster Linie durchgeführt werden, um bösartige Tumoren zu beweisen oder auszuschließen. Die genaue Identifizierung der abnormal funktionierenden Zellen ist eine grundlegende Bedingung, um Erkrankungen der Immunzellen zu behandeln und eine Prognose zu erstellen. Zytochemische Untersuchungen, Flusszytometrie und Immunhistochemie werden dazu verwendet. Besonders für den Nachweis der Malignität der Immunzellen und der Prognose bedarf es zytogenetische und molekulargenetische Untersuchungen. 1.7.1.1. Blutbild und Knochenmarkausstrich Untersuchungen Bei der Bewertung eines quantitativen Blutbilds gibt es zwei Gesichtspunkte zu beachten: 1) im Fall fehlender Abweichungen im quantitativen Blutbild, kann man die Gegenwart eines Immundefekts oder einer bösartigen hämatologischen Krankheit noch nicht ausschließen. 2) Im quantitativen Blutbild können Abweichungen auftreten, die nicht die Folgen eines pathologischen Prozesses sind. Z.B. Während einer Schwangerschaft ist eine starke physiologische Leukozytose zu bemerken, bei der kein hämatologisches Krankheitsbild im Hintergrund steht. Bei der Bewertung eines qualitativen Blutbildes gibt es auch zwei ähnliche Gesichtspunkte zu beachten: 1) im Fall fehlender Abweichungen im qualitativen Blutbild, kann man die Gegenwart eines Immundefekts oder einer bösartigen hämatologischen Krankheit noch nicht ausschließen. 2) Abweichungen im qualitativen Blutbild können auftreten, die keine Folge eines pathologischen Prozesses sind. Z.B. Infolge akuten hohen Blutverlustes, oder bei einer Entzündung, erhöht sich die Zellauswanderung aus dem Knochenmark um den Verlust zu kompensieren. Damit gelangen unreife Zellformen in die Peripherie. Bei der Bewertung der Knochenmarkausstriche gibt es drei Gesichtspunkte zu beachten 1. Sind die Zellformen aller Zell-Linien und aller Reifungszustände in der Probe vorhanden? 2. Was ist das Verhältnis der mit einander verglichenen Zell-Linien und Reifeformen. 21 3. Sind abnormale Zellformen in der Probe zu finden? 1.7.1.2. Zytochemische Reaktionen Um die Zell- Linien zu identifizieren, verwendet man häufig zytochemische Reaktionen, da sie auch billig sind. Aus den zahlreichen zytochemischen Reaktionen (Sudan schwarz B Färbung, MPO Nachweis, nicht-spezifische Esterase Reaktion, PAS Reaktion, usw.) beschreiben wir hier zwei Methoden. 1.7.1.2.1. PAS Reaktion Die PAS Reaktion ist nicht spezifisch, weil das Glykogen im Zytoplasma von beiden myeloiden und lymphoiden Zellen nachgewiesen werden kann. Trotzdem können diese Zellen unterschieden werden, da das Zytoplasma der myeloiden Zellen und der reifen Lymphozyten mit PAS homogen gefärbt wird, währenddessen Lymphoblasten eine raue Granularität zeigen. 1.7.1.2.2. Nicht-spezifische Esterase Reaktion Die nicht-spezifische Esterase Reaktion läuft auf Grund von solchen Esterasen Enzymen ab, die sich im Zytoplasma der myeloiden Zellen befinden. Dadurch kann man die myeloiden und die lymphoiden Zellen unterscheiden. Die Funktion der Enzyme im Zytoplasma der Monozyten kann mit Natriumfluorid (NaF) gehemmt werden, nicht aber jene der Granulozyten. Wenn man die Esterase Reaktion mit und ohne NaF durchführt, können die zwei Arten von myeloiden Populationen – Monozyten und Granulozyten – unterschieden werden. 1.7.1.3. Flusszytometrie Flusszytometrie wird für die Immunophenotypisierung der Immunzellen verwendet, wodurch die Stadien der Differenzierung und die Aktivität der Zellen bestimmt werden können. Immunophenotypisierung bedeutet die Identifizierung der Zelloberflächen und der zytoplasmatischen Proteine. Die detaillierte Beschreibung dieser Methode ist im Kapitel 3 zu finden (Abbildung 9.). Abbildung 9. Immunophenotypisierung: die Identifizierung der Zelloberflächen und der zytoplasmatischen Proteine 22 1.7.1.4. Immunhistochemie Immunhistochemische Methoden werden für die Immunophenotypisierung der soliden Gewebe verwendet. 1.7.1.5. Genetische Untersuchungen Die pathologische Funktion der Zellen des Immunsystems kann mit genetischen Methoden untersucht werden. Die funktionellen Störungen der Immunzellen können in den Krankheitsbilden der Immundefekte und in den bösartigen Formen der Zellen ausgeprägt werden. Die molekulargenetischen Diagnostik-Methoden, wie Zytogenetik, FISH, PCR oder die für das Mapping des vollen Genexpressionsmuster fähigen „Chip” Techniken gehören heutzutage zur Routinediagnostik des Immunsystems. (Die detaillierte Beschreibung dieser Techniken sind in den Lehrbüchern der Genetik zu finden.) 1.8. Kommunikationen zwischen den Zellen des Immunsystems Das Immunsystem ist eine funktionelle Einheit, deren Wirken von dem „harmonisierten” Zusammenspiel der überall im Körper anwesenden Immunzellen, Gewebe und Organe abhängt. Dieses Zusammenspiel kann nur durch kontinuierliche Kommunikation verwirklicht werden. Bis jetzt glaubte man, dass Kommunikation zwischen den Zellen nur auf zwei verschiedene Arten passieren kann. Die benachbarten Zellen gehen Wechselwirkungen mit ihren Zelloberflächenstrukturen ein. Das nennt man direkte Zell-Zell Interaktion. (Im Immunsystem ist die Verknüpfung der T Lymphozyten und der APZ ein Beispiel dazu) Die andere Möglichkeit ist die durch von Zellen sekretierte Molekülen vermittelte Signalübertragung. Die sekretierten Moleküle gelangen zu den fernen Zielzellen durch Diffusion, über die Lymphe oder mit dem Blut. (Cytokine stellen eine wesentliche Gruppe der löslichen Signalmoleküle im Immunsystem dar.) Heutzutage ist bereits bekannt, dass auch eine dritte Art von Kommunikation zwischen den Zellen existiert: nämlich, Mikrovesikel vermittelte Signalübertragung. Mikrovesikel sind von den Zellen verschickte membranumgebene Päckchen. Sie können sich an die Rezeptoren der Zielzellen anbinden und dort eine Signalwirkung auslösen, oder sie können ihren Inhalt in die Zielzellen gelangen lassen und die Eigenschaften der Zelle modifizieren. Wenn die Membran der Vesikel mit der Membran der Zielzelle verschmilzt, wird ein neues Membranproteinmuster ausgeprägt, also der „Phenotyp” der Zelle wird verändert. Mikrovesikel sind 30-1000 nm große, von einer Lipiddoppelschicht umgebene Partikel, mit einem auf die Donorzelle charakteristischen Proteinmuster an der Zelloberfläche. Der Inhalt besteht aus Zytoplasma mit Proteinen, Nukleinsäuren und anderen Molekülen. Nach unserer derzeitigen Erkenntnis, haben alle Zellen die Fähigkeit, Mikrovesikel zu verschicken. Dies passiert meistens nach Aktivierung, obwohl es auch in Ruhe vorkommen kann. Mikrovesikel können mit Flusszytometer und Elektronenmikroskopen untersucht werden. 23 Die wichtigsten Eigenschaften der von Mikrovesikeln vermittelten Signalübertragung: 1. Sie wirken mit einem komplexen Proteinmuster, da sie in ihrer Membran verschiedene Proteine tragen. 2. Die Wirkung dauert länger, weil die Proteine membrangebunden sind. (Die Zeitdauer der Wirkung hat eine wichtige regulatorische Rolle.) 3. Solche Stoffe können in Mikrovesikeln von einer Zelle in die anderen gelangen, die unter physiologischen Umständen in der extrazellulären Matrix niemals freigesetzt werden könnten, weil diese Moleküle sofort als Antigene identifiziert wären. (z.B. Nukleinsäuren) Übersetzt von Erna Pap 24 2. DIE AUF DER ANTIGEN-ANTIKÖRPER WECHSELWIRKUNG BERUHENDEN METHODEN I. (EDIT BUZÁS) Eine Reihe von Methoden werden zusammenfassend als Immunassays bezeichnet. Ihr gemeinsames Grundprinzip ist die Erkennung und damit der Nachweis eines Partikels als Antigen durch die Bindung an einen Antikörper. Derzeit beruht der grosse Teil der Untersuchungen in der klinischen Labordiagnostik auf der AntigenAntikörper Wechselwirkung, deswegen werden diese Methoden in diesem Kapitel diskutiert. Die in den Untersuchungen verwendeten Antikörper sind im Handel erhältlich, genau wie die chemischen Reagenzien. Eine Vielzahl von polyklonalen und monoklonalen Antikörpern mit unterschiedlicher Spezifizität stehen zur Verfügung bei den Firmen. 2.1. Die Eigenschaften der in der Diagnostik verwendeten Antikörper Die polyklonalen Antikörper stammen aus dem Serum von immunisierten Tieren; nach der Immunisierung mit speziesfremden Proteinen entstehen grosse Mengen von Antikörpern im Blutserum des immunisierten Organismus. Antikörper Diese reagieren mit unterschiedlichen Konformationsepitopen des Antigens, sie sind polyklonale Antikörper. Die polyklonalen Antikörper werden von vielfaltigen B Zellen/Plasmazellen hergestellt. Die aus dem Serum isolierten Antikörper gehören zu verschiedenen Antikörperklassen. Sie haben verschiedene Affinitätspezifizität, zudem sind sie heterogen bezüglich ihrer Antigenspezifizität, d.h. sie reagieren mit mehreren Antigenen. Nach IgM Typ Antikörper (primäre Immunantwort), einer einzigen Impfung entstehen meistens währenddessen nach wiederholten Impfungen, IgG die dominante Antikörperklasse im Serum ist. Die polyklonalen Antikörper werden meistens als sekundäre Antikörper verwendet. Es ist zweckmässig grössere Tiere (Schwein, Ziege, Kaninchen) für die Herstellung polyklonaler Antikörper zu verwenden, weil grosse Mengen von polyklonalen Antikörpern auf diesem Weg hergestellt werden können. Nur zum Vergleich: vom Kaninchen können 15 ml, von der Maus 0,3 ml, von der Ratte 2 ml, vom Meerschweinchen 5 ml, vom Schaf 200-600 ml, von der Ziege 150-400 ml und vom Pferd 500-7000 ml Serum gewonnen werden. Bei der Auswahl der zu immunisierenden Spezies ist ein weiterer wichtiger Gesichtspunkt der phylogenetische Abstand zwischen dem Antigen und dem immunisierenden Körper. Die Herstellung der polyklonalen Antikörper ist relativ billig und braucht 4-8 Wochen. Das Kaninchen ist die am häufigsten immunisierte Spezies um polyklonale Antikörper herzustellen. Man kann aus ihnen ungefähr 250 mg Antikörper durch Ausbluten gewinnen. Die im Kaninchen hergestellten polyklonalen Antikörper können als sekundäre Antikörper mit monoklonalen Maus-Antikörpern erfolgreich verwendet werden. (Abbildung 2.1.) 25 Abbildung 2.1. Monoklonale und poliklonale Antikörper Die monoklonalen Antikörper werden mit der Hybridom-Technik in Zellkulturen hergestellt, durch somatische Fusion der Tumor- und Plasmazellen. Mit der Fusion entstehen immortalisierte Hybridomzellen, die alle den gleichen monoklonalen Antikörper herstellen. Die Herstellung der monoklonalen Antikörper benötigt im allgemeinen 3-6 Monate. Die im Handel erhältlichen monoklonalen Antikörper gehören zum gleichen Immunglobulin Isotyp. Sie reagieren mit dem gleichen Epitop, das die Firmen in den gewünschten Mengen aus Hybridomzellen herstellen können. Der Vorteil der monoklonalen Antikörper ist nicht nur ihre Herstellung nach Bedarf, sondern auch, dass die hergestellten Antikörper gleich sind, was besonders wichtig ist im Fall der standardisierten klinischen diagnostischen Tests und der Antikörpertherapie. Eine riesige Auswahl von im Handel befindlichen Antikörpern ist unter der Web Adresse http://www.antibodyresource.com/onlinecomp.html zu finden. 2.1.1.1. Grundbegriffe Antigen-Antikörper Bindung: reversibel und beruht auf nicht-kovalenten Wechselwirkungen Affinität: Die Bindungsstärke zwischen einem einzigen Antigenepitop und einer einzigen Bindungsstelle eines Antikörpers, die der Betrag der anziehenden und abstoßenden Kräfte zwischen dem Epitop und dem Antikörper ist. Die Affinität ist die Gleichgewichtskonstante die die AntigenAntikörper Reaktion auszeichnet. Die meisten Antikörper besitzen eine grosse Affinität. Avidität: der Betrag der an mehreren Bindungsstellen gemessenen Bindungsstärken, d.h. die totale Bindungsstärke zwischen multivalenten Antigenen und Antikörpern. (Abbildung 2.2.). 26 Abbildung 2.2. Affinität und Avidität. Spezifizität: die Fähigkeit des Antikörpers nur mit einem einzigen Antigenepitop zu reagieren. Die Antikörper können die verschiedenen Antigene 1) nach ihrer primären Struktur 2) nach ihren Isomerformen und 3) nach ihrer sekundären und tertiären Struktur unterscheiden. Kreuzreaktivität: Ein gegebener Antikörper kann mit mehr als einem Antigen reagieren. Die Ursache kann sein, dass das kreuzreagierende Antigen solche Epitope besitzt, die den Epitopen von anderen Antigenen ähnlich sind. Antikörper Titer: die letzte Verdünnung einer Antikörperlösung, mit der die Antigen-Antikörper Wechselwirkung gemessen werden kann. Sensitivität: sie bezeichnet die Sensitivität des diagnostischen Antikörpers, d.h. wie viel Prozent der Kranken als positiv erkannt werden. Spezifität: sie bezeichnet, wie viel Prozent der Gesunden als negativ (also als gesund) durch den diagnostischen Antikörper erkannt werden. 2.1.2. Die Möglichkeiten die Antikörper zu markieren Um die Lokalisation und/oder die Menge der Antigene festzustellen, muss man die Antigene sichtbar machen. Dazu werden verschiedene Markermoleküle angewendet. (Abbildung 2.3., Tabelle 2.1.und Tabelle 2.2.) Abbildung 2.3. Unterschiedlich markierte Antikörper. 27 Markierung Beispiel Anwendung FITC (fluorescein izotiocianat) PE (Fikoeritrin) Flourokrom Immuncytochemie Rhodamin Texas rot Peroxidase (HRPO) Enzym Alkalische Phosphatase (ALP) ELISA [kolorimetric or chemiluminescent (CLIA)] und ELISPOT, Immunoblot Biotin Biotin Indirekte Immuncytochemie (Detektierung mit Avidin oder Sterptavidin –gekoppeltem Enzym) Goldkolloid Goldkolloid Elektronenmikroskopische Immuncytochemie Radioaktive Markierung 125 I RIA und IRMA Tabelle 2.1. Einige häufige an den Antikörpern gebundene Markermoleküle. Enzym Substrat DAB (diaminobenzidin): braun AEC (aminoetil-karbazol): rot Peroxidase (HRPO) True Blue: blau Luminol: lumineszent NBT (nitroblue tetrazolium): blau Alkalische Phosphatase (ALP) BCIP (bromo-kloro-indoil phosphate): blau Dioxietán Produkte: lumineszent Tabelle 2.2. Einige häufige Enzym-Substrat Systeme 2.2. Methoden 2.2.1. Flusszytometrie Derzeit ein der am häufigsten im klinischen Laborpraktikum verwendeten Immunoassays, der auf Antigen-Antikörper Wechselwirkung beruht. Die detaillierte Beschreibung dieser Methode ist im Kapitel 3. zu finden. 28 2.2.2. ELISA (Enzyme Linked Immunosorbent Assay) ELISA ist der am häufigsten verwendete, auf Antigen-Antikörper Wechselwirkung beruhende nichtradioaktive Immunoassay. Polystiren Platten mit 96 (8x12) Löchern (ELISA plate) werden verwendet. Proteine werden an den Löchern und an der Wand der Löcher der ELISA adsobiert. Die Adsorption wird durch van der Waals Kräfte, hydrophobe- und elektrostatische Wechselwirkungen durchgeführt. Diese Proteine sind indifferent und sind so nicht an der Immunreaktion beteiligt. Es handelt sich um Proteine, die kein Antigen oder keine Antikörper besitzen und deshalb in der ELISA-Reaktion teilnehmen können. Für diesen Zweck verwendet man Krächzen (bovine) Serum Albumin oder Gelatine. Die immunologischen Nachweismethoden können aufgeteilt werden, abhängig davon, mit welchen Verfahren die eigentliche Antigen-Antikörper Reaktion visualisiert wird. Die im ELISA-System verwendeten Antikörper können markiert oder mit einem Enzym und Biotin gekoppelt werden. Für die Markierung der Antikörper wird am häufigsten Horse raddish peroxidase (HRP) oder alkalische Phosphatase (AP) als Enzymmarkierung verwendet. Das Enzym selbst ist nicht sichtbar (z.B. HRP). Es wird dadurch sichtbar, dass es den Elektontransfer zwischen H 2O2 und einem kolorimetrischen Indikator katalysiert. Dadurch verändert sich die Farbe des oxidierten kromogenen Substrats. (TMB, DAB, ABTS) Die Menge des veränderten Kromogenstoffs kann über die optische Densität gemessen werden. Sie ist proportional zur Enzymaktivität. Anstatt kromogener Substrate kann man auch fluorogene Substrate verwenden. Die Biotin Markierung basiert auf der grösseren Affinität zwischen Biotin und Avidin, oder zwischen Biotin und Streptavidin. Die Biotin-Avidin Bindung ist eine der am besten bekannten nicht-kovalenten Protein-Ligand Bindungen. Avidin stammt aus Eiweiß. Es ist ein basisches Glykoprotein dessen Sequenz 30% mit Streptavidin von Streptomyces avidnii gleich ist, aber ihre sekundäre, terziäre und quaternäre Struktur ist fast völlig gleich. Avidin und Streptavidin haben tetramere Struktur, alle Untereinheiten haben die Fähigkeit ein Biotin zu binden. Mehrere Biotin Moleküle können an ein einziges Biotin-markiertes Protein binden, dadurch kann sich das Biotin-gebundene Protein gleichzeitig mit mehreren Avidin Molekülen verkoppeln. (Abbildung 2.4.). Die Avidität des Biotins zu Avidin ist noch größer als die zu Streptavidin. Abbildung 2.4. Biotin Markierung. 29 Im Gegensatz zu Streptavidin, ist Avidin glykolisiert (deswegen kann es auch an Lektine binden), besitzt positive Ladungen (bindet auch an den Zellkern) und ist in der Lage aspezifische Bindungen herzustellen. Im Allgemeinen werden 2-3 parallele Messungen durchgeführt, deren Durchschnitt als Wert genommen wird. Drei Grundtypen der ELISA Methoden sind bekannt: 1. indirekte ELISA 2. Sandwich ELISA 3. kompetitive ELISA 2.2.2.1. Die indirekte ELISA Reaktion Ein bestimmtes Protein (z.B. Virus Antigen) wird an der Oberfläche der Platte adsorbiert. Diesen Schritt nennt man coating. Danach wird die Platte mit einem indifferenten Protein (z.B. bovin serum albumin) blockiert, dazu wird die biologische Probe (Blutserum) zugegeben. Auf die Inkubation folgt das Waschen, danach kommt eine weitere Inkubation mit den gegen die primären Antikörper markierten sekundären Antikörpern. (Die primären Antikörper sind die aus der biologischen Probe nachzuweisenden Antikörper). (Der Marker kann z.B. HRP gekoppelter anti-human Immunglobulin sein.) Nach einem weiteren Waschen werden H2O2 und kromogene in die Löcher eingeführt. Die Farbereaktion wird mit einem Spektrophotometer bei gegebenen Wellenlängen gemessen. Es gibt zweckdienlich eine negative Kontrolle (biologische Probe sicher ohne Antikörper) und eine positive Kontrolle (biologische Probe sicher mit Antikörpern) zu testen. Eine Verdünnungsreihe von Proteinen mit bekannter Konzentration soll auch auf die ELISA Platte als Referenz aufgebracht werden. Dadurch kann eine Kalibrationskurve aufgezeichnet werden, womit die Adsorbenzwerte der unbekannten Proben verglichen werden können. 2.2.2.2. Sandwich ELISA „Capture” Antikörper werden auf der Oberfläche der Platte adsorbiert (diesen Schritt nennt man coating). Nach Blockieren („blocking“) mit indifferenten Proteinen folgt die Inkubation mit dem zum Antikörper passenden Antigen (z.B. Cytokine aus dem Blutserum). Nach dem Waschen kommt die Inkubation mit HRP-markierten Antikörpern, die spezifisch für die selben Antigene sind, aber die mit einem anderen Epitop reagieren. Nach der Zugabe von H 2O2 und Kromogenen wird die Farbereaktion mit einem Spektrophotometer gemessen. (Abbildung 2.5.). In diesem Fall wird die Kalibrationskurve aus den Werten der Verdünnungsreihe der Antigene gezeichnet. (Abbildung 2.6.). 30 Abbildung 2. 5. ELISA Methoden Abbildung 2.6. Kalibrationskurve - ELISA 2.2.2.3. Kompetitive ELISA Im ersten Schritt werden die unmarkierten Antikörper mit den Antigen-enthaltenden biologischen Proben inkubiert. Antigen-Antikörper Komplexe entstehen in den Proben. Diese „präinkubierten” Proben werden auf die Oberfläche der ELISA Platte aufgetragen. Je mehr Antigene in der biologischen Probe vorhanden sind, desto weniger Antikörpermoleküle bleiben frei, um an die auf der ELISA Platte-adsorbierten Antikörper zu binden. Enzym-markierte (HRP) sekundäre Antikörper und kromogene Substrate werden verwendet um die ELISA Reaktion anzuhalten. (Abbildung 2.7.). Der Vorteil der kompetitiven Reaktion ist die Möglichkeit auch kleine Mengen von unmarkierten Antigenen nachzuweisen. 31 Abbildung 2.7. Das Prinzip der kompetitiven ELISA 2.2.2.4. Was ist zu beachten, bei der Durchführung von ELISA ? Was kann Probleme verursachen? 1. Das Waschen der Löcher (genügende Menge der Lösung, genaue Entfernung der Lösung) 2. Austausch der Pipettenspitzen. 3. Parallele Untersuchungen Im Falle einer schwachen oder keiner Farbreaktion, mischen wir zur Kontrolle die enzymmarkierten Antikörper mit der Substrat im 1:1 Verhältnis. Wenn keine Farbreaktion auch in dieser Aufstellung entsteht, können dies die möglichen Ursachen sein: 1. die Substratlösung wurde nicht im Dunkel gelagert, 2. die HRP Lösung ist alt und hat sich teilweise abgebaut. C-Anca P-Anca Anti-Cardiolipin IgG Anti-Cardiolipin IgM ss/DNA ds/DNA Jo-1 Histone Complex AutoimmunDiagnostik Immune Complex Anti-Thyroid Microsomal Anti-Mitochrondrial SSa SSb ANA, SM Sm/RNP Thyroglobulin Anti Thyroglobulin Gliadin IgG, IgA, IgM Androstenedione FSH, HCG, HGH, LH, Estradiol, Estriol Progesterone 17-OH Progesterone Endokrinologie Free-Testosterone Testosterone Cortisol, TSH, T3, T4, T uptake, free-T3, free-T4, TPA Mikrobiologische Untersuchungen Anti-HBsAg ; Anti-HCV; Anti-HIV; Anti-HEV IgM; Anti-HAV IgM, IgG; Anti-tuberculosis IgG; Anti-syphilis; anti-EBV; Anti-Rotavirus AFP, CEA, ferritin Tumordiagnostik HCG, b-HCG, PAP, PSA, Free PSA Tabelle 2.3. Beispiele für ELISA diagnostische Tests. 32 2.2.3. ELFA (Enzyme Linked Immunofluorescent Assay) Ein ultraempfindliches System, das ein fluorogenes Substrat (z.B. 4 Methyl umbilliferyl phosphate (MUP)) verwendet. Da fluoreszierende Moleküle bereits in pikomolaren Mengen detektiert werden können, kann die Empfindlichkeit des Essays mit fluoreszierenden Substraten stark erhöht werden (z.B. der Nachweis der Rotaviren ist hundertmal grösser als mit ELISA). (Abbildung 2.8.). Abbildung 2.8. ELFA (Enzyme Linked Immunofluorescent Assay) 2.2.4. ELISPOT ELISPOT ist ein indirekter Immunoassay, der für den Nachweis für von einzelnen Zellen sekretierten Molekülen (am häufigsten Cytokine) verwendet wird. Die ELISPOT Platte enthält 96 Löcher, die eine spezifische Membran (Nitrocellulose oder PVDF (Polyvinylidenfluorid)) besitzen. Unter sterilen Umständen wird die Membran mit „capture” Antikörpern bedeckt (coating), dann kommt das Blocking mit indifferenten Proteinen (z.B. bovin serum albumin) enthaltendem Puffer. Bekannte Mengen lebender Zellen (aus Lymphozyten 1-300 000 Zellen) werden in die Löcher mit Nährstofflösung o eingelegt, die in CO2 Termostat, auf 37 C für einige Tage kultiviert werden. Danach werden die Zellen ausgewaschen. Ihre sekretierten Moleküle werden durch die Capture Antikörper gebunden. An diese Cytokine binden die markierten Antikörper, die mit bestimmten Substraten unlösbare Komplexe herstellen und damit sichtbar werden. (Abbildung 2.9.). 33 Abbildung 2.9. ELISPOT Nach dem Trocknen können die farbigen Flecken in den Löchern durch Scannen mit einer bildanalysierenden Software ausgewertet werden. ELISPOT ist eine sehr empfindliche Methode, sie bietet die Möglichkeit die positiven Zellen auch in einem 1/300 000 Verhältnis zu bestimmen. 2.2.5. Immuno blot (Western blot) Die relative Menge eines Proteins kann durch einen gegebenen primären Antikörper in den biologischen Proben bestimmt werden. Wir stellen ein Lysat/Homogenisat aus Zellen oder Gewebe her. Der Puffer enthält Protease-Inhibitoren. Die Proben werden auf ein Polyacrylamid Gel aufgetragen und die Proteine werden nach ihrer Grösse mit Gelelektrophorese getrennt. Das eigentliche Blotting ist die Übertragung der Proteine auf einen geeigneten Träger, z.B. Nitrocellulose Membran. Die Lage – die Anordnung – der Proteine bleibt gleich wie sie ursprünglich im Trenngel war. Die freie proteinbindende Oberfläche der Membran wird mit indifferenten Proteinen blockiert. Im nächsten Schritt wird die Membran mit primären Antikörpern inkubiert, diese Antikörper sind fähig das 34 gesuchte Protein zu binden. Dazu werden die sekundären Antikörper zugegeben, welche eigentlich Antikörper-Enzym-Komplexe sind und binden an die primären Antikörper. So wird die Lage sichtbar, an der sich die primären Antikörper gebunden haben. (Abbildung 2.10-11.). Abbildung 2. 10. Gelelektrophorese Abbildung 2.11. Immunoblotting 35 Der am häufigsten verwendete sekundäre Antikörper ist mit HRP gekoppelt. HRP katalysiert die Reaktion, welche mit der Oxydation des Luminols eine 428nm Lichtemission ergibt, welche mit einem lichtempfindlichen Film sichtbar gemacht wird. (Abbildung 2.12.). Die Ergebnisse können auf das Produkt eines housekeeping Gens standardisiert und danach analysiert werden (Beta-Aktin kann einem solchen housekeeping Protein dienen.) Die Ergebnisse informieren über die relative Menge und die Molekularmasse des Proteins in der Probe. Abbildung 2.12. Auswertung mit einem lichtempfindlichen Film. 2.2.6. Auf radioaktiven Markierungen basierende Methoden 2.2.6.1. Radioimmunoassay (RIA) Die Methode ist spezifisch, sehr billig und hat eine grosse Empfindlichkeit. Man kann damit die Konzentration der Antigene, z.B. Hormone messen. Der Nachteil ist, dass man für die Methode spezifisches Equipment (z.B. Gamma Messer) braucht und wegen der Verwendung eines radioaktiven Stoffes, benötigt man eine spezifische Erlaubnis. Des weiteren erfordert die Arbeit besondere Vorsichtsmaßnahmen. Eine Art der Assays ist RAST Test (radioallergosorbent Test). Das Isotop I 125 ist oft an die Aminosäure Tyrosin gebunden. Das radioaktive Antigen wird mit Antikörpern mit einer bekannten Menge gemischt. Danach wird die biologische Probe, mit unbekannten Mengen der zu bestimmenden Antigene, zugegeben. Dadurch tritt das (unmarkierte, kalte) Antigen der Probe mit dem markierten Antigen in Kompetition, um an den Antikörper zu binden. (Abbildung 2.13-14.). Das „S. aureus Protein A (Zysorbin)” ist ein Stoff der Zellwand, der Antikörper binden kann. Dies oder anti-Immunglobulin-Antikörper können die markierten von den unmarkierten Antikörpern trennen. Danach können die markierten Moleküle gemessen werden. 36 Abbildung 2.13. IRMA und RIA Abbildung 2.14. RIA 2.2.6.2. Immunoradiometric assay (IRMA) In diesem System werden monoklonale Antikörper verwendet, die an die innere Oberfläche der polystyren Tuben gebunden sind. Die Proben der Patienten werden mit I 125 -markierten Antikörpern inkubiert. Die unmarkierten, immobilisierten (auf der Wand befestigten) und die in der Lösung vorhandenen markierten Antikörper binden gleichzeitig an die in den Proben vorhandenen Antigene. Da entsteht eigentlich ein Komplex, der auf der soliden Phase angeheftet wird. Die sich nicht bindenden markierten Antikörper werden durch Waschen getrennt. Die gemessene Radioaktivität ist 37 im Verhältnis mit dem Antigengehalt der Probe. Die quantitative Auswertung geschieht mit einer Standardkurve, die mit der Anwendung von Antigenen mit bekannten Mengen gemacht wurde. 2.2.7. Immuncytochemie (Immunhistochemie) Mit diesem Verfahren kann man spezifische Proteine in den Zellen/Geweben mit markierten Antikörpern nachweisen. Schnitte, Blutausstriche, kultivierte angeheftete Zellen und auf Objektträger zentrifugierte Zellsuspensionen (Cytospin Präparate) können untersucht werden. Die Antigen/Antikörper - Bindung kann entweder mit fluoreszierenden Farben, mit Metalkolloiden, mit radioaktiver Markierung oder mit Enzymen sichtbar gemacht werden. Der Antikörper muss die biologische Membran passieren können, um an der die Reaktion in der Zelle oder im Zellkern teilzunehmen. Die Anwendung von Detergenzien (0.25% Triton X-100 oder 0.5% Saponin) kann auch nötig werden, um die Membran durchlässiger zu machen. Wenn Aceton, Methanol oder Ethanol als Fixierungsmittel verwendet wird, ist keine weitere Permeabilisierung nötig. Wenn man Membranantigene nachweisen möchte, ist die Anwendung von Triton zu vermeiden, da Triton die Membran zerstört. Aldehyde, als Fixierungsmittel, stellen inter- und intramolekulare Querverbindungen (Methylenbrücke) in und zwischen bestimmten Strukturproteinen her, die die zellulären/gewebe Antigene maskieren können. Die Wirkung der Maskierung ist von der Dauer und Temperatur der Fixierung, von der Konzentration des Fixierungsmittels und von der Menge der lokalen Proteine abhängig. Um die zellulären/gewebe Antigene zu demaskieren, kann man die Probe mit Proteasen (z.B. mit Trypsin) verdauen oder für eine kurze Zeit im Mikrowellenherd erhitzen. Also werden die gefrorenen oder fixierten Schnitte/Präparate nach Bedarf permeabilisiert. Danach kommt die Blockierung der aspezifischen Bindungsstellen (mit bovine serum albumin, 1% Gelatine oder 10% normales Serum, das aus der gleichen Spezies stammt, wie die sekundären Antikörper). Die Präparate werden zuerst mit primären, danach mit sekundären Antikörpern – mit zwischenzeitlichem Waschen – inkubiert. Es wird empfohlen, die Zellkerne für die HintergrundFärbung auch zu färben (z.B. 0.1-1 μg/ml Hoechst oder DAPI (DNS-Farbe)). Aspezifische Färbung kann infolge der hydrophoben oder elektrostatischen Wechselwirkungen entstehen. Die aspezifische Färbung ist meistens gleichförmig und kann mit Präinkubation mit normalem Serum abgeschwächt werden. Viele Gewebe enthalten endogene Peroxydase-Aktivität. Vor der Anwendung des primären Antikörpers hilft die Präinkubation des Schnittes mit H 2O2 die endogene Peroxidase-Aktivität abzuschaffen. In Bezug auf die endogene alkalische Phosphatase-Aktivität – auch charakteristisch für zahlreiche Gewebe -, kann sie mit Präinkubation mit Levamisole beseitigt werden. Einige Gewebe (z.B. Leber und Nieren) enthalten endogenes Biotin, weswegen in diesen Fällen eine Präinkubation mit nicht-konjugiertem Avidin empfohlen wird (Abbildung 2.15.). 38 Abbildung 2.15. Immuncytochemie (indirekte Methode) 2.2.7.1. Direkte Methode Die Probe wird in einem einzigen Schritt mit markierten (z.B. FITC- markierten) Antikörpern inkubiert. Die Methode ist schnell, kurz und spezifisch, aber wenig empfindlich. 2.2.7.2. Indirekte Methode Ein unmarkierter primärer Antikörper reagiert mit dem Antigen des Gewebes. Danach wird ein markierter sekundärer Antikörper verwendet, der mit dem Immunglobulin der Spezies des primären Antikörpers reagiert. Diese Methode ist empfindlicher. (Das Signal verstärkt sich, da zahlreiche sekundäre Antiköper Moleküle auf den unterschiedlichen Epitopen des primären Antikörpers reagieren können.). Die Methode ist auch billig, weil ein einziger sekundärer Antikörper verwendet werden kann, um an viele unterschiedliche primäre Antikörper anzubinden. In der Immunofluoreszens Markierung wird FITC, Rhodamin oder Texas-Rot für Markierung verwendet, währenddessen Peroxidase oder alkalische Phosphatase in der immunenzymatischen Markierungen üblich ist (Abbildung 2.16.). Abbildung 2.16. Direkte und indirekte Methode, den Antikörper zu markieren 39 2.2.7.3. Kontrollen Positive Kontrollen werden angewendet um zu entscheiden, ob das Protokoll überhaupt funktioniert. Am besten sollte solches Gewebe als Kontrolle verwendet in dem die Reaktion sicher abläuft. Negative Kontrollen werden verwendet, um die Spezifizität des Antikörpers zu bestimmen. Der primäre Antikörper kann ausgelassen werden, indem er mit einem Isotyp Kontroll-Antikörper oder mit normalem Serum der gleichen Spezies ersetzt wird. Wenn die Antikörper neu sind, soll die Reaktion gehemmt werden. Dies kann man auch durch Präinkubation der primären Antikörper mit gereinigtem Antigenen machen. 2.2.7.4. PAP Methode (Peroxidase anti-Peroxidase Verfahren) Diese Methode ist eine indirekte Technik, die aus drei „Schichten” aufgebaut ist: unmarkierte primäre Antikörper, unmarkierte sekundäre Antikörper und ein stabiler Peroxidase- Antiperoxidase Komplex. Die sekundären Antikörper reagieren einerseits mit den primären Antikörpern, andererseits mit der dritten Schicht. Die Empfindlichkeit der Methode ist zirka 100-1000 fach höher, weil die Peroxidase immunologisch und nicht chemisch gebunden ist Deswegen kann man eine viel höhere Primärantikörper Verdünnung verwenden, womit die aspezifischen Bindungen abgeschwächt werden. 2.2.7.5. Avidin-Biotin Komplex (ABK) Methode Es gibt drei „Schichten” auch in dieser Methode. Der ersten „Schicht” entsprechen die unmarkierten primären Antikörper, der zweiten die biotinierten sekundären Antikörper und der dritten der AvidinBiotin-Peroxidase Komplex. 2.2.7.6. Fluorescens-Mikroscopie und Laser konfokale Mikroskopie Ein Fluoreszens-Mikroskop wird gebraucht, um die fluoreszierenden (immuncytochemischen) Proben sichtbar zu machen. Das anregende Licht mit kleinerer Wellenlänge (im Allgemeinen UV, das die Augen schädigen könnte), kommend aus dem optischen System und wird im Fluoreszens-Mikroskop von dem FarbFilter zwischen dem Objektiv und dem Okular absorbiert. So erreicht nur das Licht mit längerer Wellenlänge die Augen oder den farbempfindlichen Film. Damit werden die im Präparat vorhandenen fluoreszierenden Strukturen sichtbar gemacht. Die Fluoreszens kann aus natürlichen Zellkomponenten (Autofluoreszens z.B. aus Vitamin A.) oder aus mit fluorochrom-gefärbten Zellkomponenten stammen. Einige Fluorochrome ändern die Wellenlänge (die Farbe) des emittierten Lichts, abhängig von dem pH Wert oder von der Ca++ Konzentration. Mit Hilfe dieser Fluorochrome können der intrazelluläre pH Wert oder die intrazelluläre Ca++ Konzentration gemessen werden. Das Laser Konfokale Mikroskop ist auch mit einer Fluoreszens-Optik ausgestattet. In diesem Instrument ein dünner Laserstrahl das Präparat ab. Der Fokuspunkt des Laserstrahls kann auch in der Tiefe des Präparats geändert werden. Die aus den Schichten des Präparates gewonnene Information kann im Computer gespeichert und zusammengefasst werden. (Abbildung 2.17.). Auf diese Weise 40 können auch dichtere Präparate untersucht werden, ohne den Verlust der Informationen der dritten Dimension. Das von dem Computer rekonstruierte Bild erscheint auf einem Fernsehmonitor. Mit dieser Methode kann man sich das viel Zeit und Arbeit brauchende Schneiden und die 3DRekonstruktion ersparen. Abbildung 2.17. Das laser konfokale Mikroskop. 2.2.8. Lateral flow Tests Derzeit sind diese Schnelltests weitverbreitet. Sie beruhen auf Immunoassay/Immunkromatographie. Ihre Herstellung ist billig und die Ausführung braucht nur einige Minuten, (im Gegensatz zu den Stunden in den vorgestellten Assays), sie erfordern keine Fachkenntnisse, können zu Hause gemacht werden und es ist einfach, die Ergebnisse zu archivieren. Obwohl die Grundlagentechnologie schon in den 1960er Jahren entwickelt wurde, wurde der erste lateral flow Schwangerschafttest erst in 1988 kommerziell in den Umlauf gebracht. Seitdem sind diese lateral flow Tests in Kliniken, in der Tierheilkunde, in der Agrikultur, in der Lebensmittelindustrie und im Umweltschutz weitverbreitet. Das Prinzip der Methode ist das folgende: der Teststrich wird in die biologische Probe eingetaucht und die Flüssigkeit wandert in dem Teststrich nach dem Prinzip der Kapillarität. Die Probe mischt sich mit gefärbten Mikrogranuli (z.B. Latex), die mit dem für das zu untersuchende (nachzuweisende) Antigen spezifischen Antikörpern bedeckt sind. Die Antigene der Probe binden an die bunten Mikrogranuli und wandern entlang des Teststriches (Abbildung 2.18-19.). An einer bestimmten Position des Teststriches wurde ein anderer Antikörper aufgetragen, der für ein unterschiedliches Epitop des gleichen gegebenen Antigens spezifisch ist. Dort sammeln sich die gefärbten Mikrogranuli, falls das nachzuweisende Antigen (z.B. anti-hCG, LH oder HIV) in der biologischen Probe vorhanden ist. Die 41 weiterwandernden bunten Mikrogranuli sammeln sich noch ein Mal an einer Lage des Teststriches, an der anti-Immunglobulin aufgetragen wurde. Infolgedessen binden alle Mikrogranuli an diese Oberfläche, unabhängig davon, ob irgendein Antigen an die Mikrogranuli gebunden hatte. Diese zweite bunte Linie dient einer inneren Kontrolle des Tests. Abbildung 2.18. Das Prinzip der „lateral flow test“ Methode I. Abbildung 2.19. Das Prinzip der „lateral flow test Methode“ II. Hier folgen einige Beispiele für lateral flow Tests, die kommerziell im Umlauf gebracht sind: H. pylori IgG, H pylori Antigen (aus Stuhl), Schwangerschaftstest, Rotavirus, Adenovirus, Stuhlhemoglobin, Sterptococcus, Morfin, E. coli O157. 2.2.8.1. Multiplex Immunoassay Systeme Die Anwendung der Immunoassays hat sich in jüngster Vergangenheit mehr und mehr verbreitet. 42 RSV, Die Vorteile der multiplex Systeme sind die folgenden: Sie stellen Untersuchungssysteme mit grosser Durchlässigkeit dar. Wenn verwendet, ist nur ein kleineres Volumen der Probe nötig. Sie sind zeit-und kosteneffizient. Mit ihnen hat man die Möglichkeit die Konzentration von mehreren unterschiedlichen Molekülen gleichzeitig zu messen. Sie bieten die Möglichkeit viele unterschiedliche Proteine zu messen. Es gibt solche multiplex Systeme (z.B. Proteome Profiler Arrays) in kommerziellem Umlauf, welche Membranen mit angekoppelten spezifischen capture Antikörpern haben. Diese Systeme ruhen auf dem Prinzip des Nachweises der Chemolumineszenz. Es existieren „96Antibody Array” Systeme, die den parallelen Nachweis unterschiedlichen Molekülen in einer 96 Lücken Mikroplatte ( Mosaic™ von mehreren ELISAs) erlauben (Abbildung 2.20.). In diesen ELISA Systemen können sogar 8-16 Antikörper in jeder Lücke an die Oberfläche in Flecken gebunden werden. Weiterhin ist es auch möglich, eine 384 Lücken Platte mit 25-25 unterschiedlichen capture Antikörpern herzustellen und Untersuchungen auszuführen. Jeder Fleck vertretet unterschiedliche capture Antikörper oder Proteinpopulationen. Auch in diesen Systemen wird das chemolumineszente Signal detektiert und die Auswertung passiert von dem Bild, welches mit der digitalen Kamera gemacht wurde. Abbildung 2.20. Mozaik ELISA Platte 2.3. Welche Immunoassay ist zu verwenden? Die einfachsten und billigsten Untersuchungen sind die, die mit lateral flow Schnelltests gemacht werden können. Wenn der gewünschte Schnelltest (z.B. Schwangerschaftstest) erhältlich ist kann man auch ohne Fachkenntnis auf „ja-nein Typ” Fragen eine Antwort in einigen Minuten bekommen. Wenn die Konzentration der Antigene oder Antikörper im Serum/Blutserum zu bestimmen ist, aber es in kommerziellem Umlauf keinen Schnelltest gibt, ist die ebenfalls relativ schnelle und billige 43 ELISA die Wahl. Es kann nötig sein, die Konzentration der Antikörper (und Antikörperisotypen) in Immundefizienzen, in Entzündungen, in allergischen und Autoimmunkrankheiten zu bestimmen. Außerdem kann man die Immunsuppression durch das Messen der Antikörper verfolgen. Man kann auch ELISA Systeme selbst zusammenstellen. In diesem Fall muss man sich außer der Probe, die Platten, das Antigen, den sekundären Antikörper-Komplex (das Konjugat) und Serum/Plasma als positive und negative Kontrolle besorgen. Die im kommerziellen Umlauf erreichbaren ELISA Kits sind viel kostspieliger als die selbst zusammengestellten. In diesem letzten Fall ist es möglich, standardisierte und optimierte ELISA Reaktionen nach dem Protokoll des Herstellers durchzuführen. Die kommerziellen Kits garantieren das standardisierte und reproduzierbare Messen der routinediagnostischen Testen. Im Fall dass die Empfindlichkeit der ELISA nicht ausreichend ist, ist RIA empfohlen. (z.B. um Hormonkonzentrationen zu bestimmen.) RIA ist eine ziemlich billige Methode, aber wegen der verwendeten radioaktiven Stoffe braucht man spezifisches Equipment (Gamma Messer) und besondere Vorsicht ist geboten. In der Autoimmundiagnostik ist die indirekte Immuncytochemie unverzichtbar, um antinukleäre, gewebespezifische Autoantikörper oder z.B. ANCA (anti-neutrophil, zytoplasmatischer Antikörper) nachzuweisen. Für das Messen der Zellen in Zellsuspensionen, soll man die Flusszytometrie anwenden. In der Routine durch die Identifizierung der Zelloberflächen-Antigene (CD Antigene) können Lymphozytenpopulationen identifiziert werden. Die Methode wird in einem anderen Kapitel beschrieben. Die Western-Blot Methoden werden in der Routindiagnostik nicht verwendet, weil sie zeit-und arbeitsaufwendig sind. Sie dienen die bereits aufgestellte Diagnose weiter zu bestätigen und sie werden auch für die Forschungen verwendet. Das ELFA System wird in der laboratorischen Routinediagnostik verwendet. Es ist eine sensible, standardisierte Technik, die spezifisches Equipment erfordert. Im Vergleich mit den oberen Methoden ist die Verwendung der ELISPOT Untersuchungen relativ wenig verbreitet. Sie werden für einige zellulären immunologischen Untersuchungen angewandt, wenn man die Zahl der (in erster Linie) Zytokin-sezernierenden Zellen bestimmen möchte. Wenn man diese Methode in der Forschung verwendet, bekommt man Ergebnisse über die Immunantwort in Entzündungen, in Tumoren, in allergischen und in Autoimmunkrankheiten. Ein sehr wichtiges Feld der Anwendung der ELISPOT Methode ist die Impfstoffentwicklung, wenn Impfstoffe gegen Viren und Tumoren entwickelt werden. Auch werden die T Zellen mit ELISPOT in der spezifischen Immuntherapie beobachtet. Die multiplex Untersuchungen sind sehr kostspielig, deswegen werden sie meistens nur in der Grundlagenforschung verwendet. Übersetzt von Erna Pap 44 3. AUF ANTIGEN-ANTIKÖRPER INTERAKTION BERUHENDE METHODEN II: DIE IMMUNSEROLOGIE (ANDRÁS FALUS, GYÖRGY NAGY) Unter dem Begriff ”Serologie” verstand man ursprünglich die Analyse des Blutserums, bzw. deren Methodik. Heute wird dieser Begriff etwas ausgedehnter benutzt, insofern dass es auch Methoden hierher geordnet werden welche die Analyse von anderen Körperflüssigkeiten, z.B. Urin, Liquor, usw. ermöglichen. Mit den unterschiedlichen Methoden welche für die Analyse von löslichen Proteinen geeignet sind (ELISA, RIA, Immunoblotting), haben wir uns in dem vorigen Kapitel auseinandergesetzt. In diesem Kapitel fokussieren wir auf Immunkomplexe und Immunpräzipitate. 3.1. Immunkomplex und Immunpräzipitat Die meisten Antigene verfügen über nicht nur ein Epitop (antigene Determinante), sondern eine Vielzahl von ihnen, die meisten von denen nicht uniform, sondern recht unterschiedlich aufgebaut sind. Dazu kommt, dass bestimmte Antikörper (vor allem die pentameren IgM Antikörper) gleichzeitig auch zu mehreren Antigenen binden können, wodurch bei einer Immunreaktion komplizierte Komplexe von Antigenen und Antikörpern, sog. Immunkomplexe entstehen können. Clemens von Pirquet und Béla Schick haben in 1906 als erste beschrieben, dass eine passive intravenöse Immunisierung von Kindern gegen Diphterie mit einem Pferde-anti-Diphterietoxin-Serum als Nebenwirkung innerhalb von 7-14 Tagen zum Auftauchen von extrem großen Mengen von Immunkomplexen in dem Serum der Kinder, eine sog. Immunkomplexkrankheit führen kann. Pirquet und Schick nahmen an, dass man die Symptome darauf zurückführen konnte, dass die immunisierten Kinder gegen das Pferdeserum Antikörper entwickelt haben, welche mit den erkannten körperfremden Pferde-Antigenen verbunden Immunkomplexe gebildet und sich in verschiedenen Organen abgelagert haben. Jahrzehnte später haben Gertmuth und Dixon die Hypothese von Pirquet und Schick zuerst eindeutig beweisen können als sie die Serumkrankheit in Kaninchen untersucht haben. In den Experimenten von Gertmuth und Dixon wurden Kaninchen körperfremde Sera, z.B. Rinde-Serum intravenös verabreicht. Sie haben beobachtet, dass die Konzentration von fremden Antigenen zuerst nur langsam gesunken ist, bis 10-12 Tage nach der Immunisierung ihre Konzentration auf einmal stark abgenommen hat, dass die freien Antigene aus dem Serum fast völlig verschwunden sind. Heute weiß man genau, dass diese 10-12 Tage dem Zeitraum entsprechen, in dem das Immunsystem eines serumkranken Rezipienten beginnt, Antikörper gegen die Fremdantigene zu bilden. Diese bilden schließlich Immunkomplexe welche sich am Ende in verschiedenen Organen, typischerweise in den Nieren, ablagern und die typischen Symptome der Serumkrankheit (siehe später) verursachen. Die Symptome werden milder sobald die Menge der Immunkomplexe abnimmt, aber mit erneuter Antigen-Exposition kehren sie sofort zurück. Durch Messung der Menge des Fremdantigens, des Antikörpers und der Immunkomplexe, können drei Phasen der Serumkrankheit 45 voneinander unterschieden werden: zuerst Antigen Überfluss, dann Äquivalenz und zuletzt Antiköper Überfluss. In der ersten Phase werden kaum Immunkomplexe gebildet da spezifische Antikörper noch praktisch nicht vorhanden sind. In der zweiten Phase werden freie Antikörper und Antigene kaum nachweisbar, weil sie beide fast vollkommen in Immunkomplexe umgewandelt werden. In der dritten Phase werden Immunkomplexe von immer kompliziertere Struktur und immer größerem Molekulargewicht gebildet, jedoch in immer geringerer Konzentration, bis sie eventuell aus dem Kreislauf völlig verschwinden. In der Serumkrankheit ist die gut bekannte proinflammatorische Wirkung der Immunkomplexe, und dadurch die meisten Symptome, vor allem auf ihre Komplementaktivierungskapazität zurückzuführen (das Komplementsystem ist Teil des natürlichen Immunsystems, siehe später in Kapitel 4). Deshalb ist es nicht überraschend, dass in experimentell induzierter Serumkrankheit, parallel zu einer starken Komplementaktivierung, die Menge der freien, noch nicht aktivierten C3 und C4 Komplementproteine massiv abnimmt. Abbildung 3.1. Zusammenhang zwischen der relativen Menge von Antigenen vs. Antikörpern, und die Struktur und Größe der Immunkomplexe Nach den ursprünglichen Beobachtungen von Schick und Pirquet sind die typischen Symptome der Serumkrankheit in Menschen in erster Linie Fieber, Lymphadenopathie, Polyarthritis, Proteinurie und Nesselsucht (Utrikaria). In der Regel erreicht die stets zunehmende Konzentration der Antikörper und die senkende Konzentration der Antigene binnen 7-14 Tage nach Antigenexposition den Äquivalenzpunkt, und demzufolge erscheinen die Symptome auch in diesem Zeitraum. Die in dieser Phase gebildeten Immunkomplexe sind lösliche molekulare Komplexe von Antigenen und Antikörpern. Die absolute Menge dessen hängt von dem stöchiometrischen Verhältnis zwischen Antigen und Antikörper ab. Jederlei Verschiebung dieses chemischen Gleichgewichts aus dem Äquivalenzpunkt, also selbst die kleinste Änderungen in dem Antigen-Antikörper Verhältnis 46 destabilisiert die Immunkomplexe und reduziert ihre Konzentration im Serum (Abbildung 3.1), obwohl die Entstehung und Stabilität des Immunkomplexes natürlich auch von anderen Faktoren beeinflusst wird: dem pH, der Temperatur, der Salzkonzentration, oder der Struktur (Valenz) des Antikörpers (Monomer, Dimer, Pentamer). Falls eine Fällung des Immunkomplexes stattfindet, redet man über die Herausbildung eines Präzipitats, welches auch mit bloßem Auge sichtbar sein kann, und welches nicht mehr wasserlöslich ist. Drittens, die Präzipitat-Bildung von Antigenen deren Größe mit intakten Zellen vergleichbar ist wird als Agglutination bezeichnet. Abbildung 3.2. Agglutination und Präzipitation Die Bindung von Antigenen und Antikörpern selber bleibt aber in allen Fällen eine schnelle und reversible Reaktion, weil sie auf nicht-kovalenten Bindungen, wie den ionischen Bindungen, den van der Waals Bindungen und H-Brücken beruht (Abbildung 3.3.). Viele klinische- und Forschungsmethoden basieren auf Antigen/Antikörper Interaktionen, mit denen wir uns in diesem Kapitel in Detail auseinander setzen werden. Abbildung 3.3. Bindung von Antigenen und Antikörpern Die biologische Wirkung von Immunkomplexen wird streng beeinflusst von einer Reihe von Faktoren, unter anderem von der Affinität mit der sie sich zu Zelloberflächenrezeptoren binden können, (z.B. zu Fc Rezeptoren der Phagozyten), wie schnell sie sich in den verschiedenen Geweben bzw. Organen ablagern (Immunkomplexe mit positiver Ladung 47 lagern sich zum Beispiel mit höherer Wahrscheinlichkeit in den Nierenkörperchen ab), oder der Effizienz womit sie das Komplementsystem aktiveren können. Die Entfernung von Immunkomplexen aus dem Kreislauf erfolgt hauptsächlich mit Hilfe der Komplementrezeptoren der Zelloberfläche von Erythrozyten. Wie wir darauf schon hingedeutet haben, sind die Immunkomplexe in der Lage das Komplementsystem zu aktivieren und demzufolge sind sie häufig auch mit aktivierten Komplementproteinen (C3b, C4b) verbunden. Diese Partikel werden deswegen von den CR1 Komplementrezeptoren der Erythrozyten erkannt und gebunden. Sobald die Erythrozyten später den Leberkreislauf passieren, werden die Komplexe abgegeben und auf die Fc Rezeptoren der leberständigen Makrophagen, den sogenannten Kupffer’sche Zellen, transferiert. Dies geschieht, da der Antikörper-Teil zu FC Rezeptoren mit einer höheren Affinität binden kann als der Komplement-Teil zur CR1. Die übertragenen Immunkomplexe werden von den Kupffer’schen Zellen phagozytiert und vernichtet. Die Erythrozyten bleiben intakt und werden verschont, damit sie erneut in den Kreislauf zurückkehren um Immunkomplexe zu binden und wiederum in die Leber transportieren zu können. Abnormale Mengen von Immunkomplexen können auch in bestimmten Autoimmunerkrankungen diagnostiziert werden, wofür Lupus erythematodes (SLE) ein Paradebeispiel ist. SLE ist eine systemische Autoimmunerkrankung, in der ein breites Repertoire Autoantikörper Viele von vorhanden von ihnen sind Komponente gerichtet des ist. gegen Zellkerns (anti-DNA, ANA), welche unter anderem auch als wichtige diagnostische Merkmale dienen können. erythematodes alle Der Lupus kann praktisch menschlichen Organe angreifen, klinisch jedoch sind die wichtigsten der Befall der Niere und des zentralen Nervensystems (Abbildung 3.4.). Abbildung 3.4. Organe typischerweise betroffen im Systemischen Lupus erythematodes (SLE) Die von diesen Autoantikörpern gebildeten Immunkomplexe häufen sich in den betroffenen Organen massiv an, und spielen damit eine zentrale Rolle in den typischen Symptomen des SLE, welcher dadurch häufig als der Archetyp aller Immunkomplexerkrankungen bezeichnet wird. In Patienten mit SLE kann häufig eine extrem niedrige Konzentration der inaktiven Komplementproteine C3 und C4 nachgewiesen werden, wofür der Grund die kontinuierliche Aktivierung des Komplementsystems durch die Immunkomplexe sein kann. Im Einklang mit dieser Beobachtung sind dagegen die Mengen der gespaltenen, aktiven Komplementproteine (z.B. C4a, ein solubles Proteinfragment) in SLE 48 deutlich höher als normal. In der Tat ist die Serumkonzentration der zirkulierenden Immunkomplexe in den meisten SLE Patienten abnormal hoch, und eine beschleunigte Aktivierung des klassischen und alternativen Komplementweges ist auch typisch für sie. Interessanterweise jedoch, obwohl die Menge der Immunkomplexe mit dem Aktivierungszustand des Komplementsystems in SLE gut korreliert, hat keiner von diesen zwei Markern großen diagnostischen Wert, insofern dass keiner von ihnen eine Verbindung mit der zeitlich stark wechselnden Aktivität der Krankheit selber zeigt. Bezüglich SLE wird angenommen, dass eine genetisch bedingte abnormal niedrige Produktion von den oben erwähnten CR1 Rezeptoren, stark auf dieses Krankheitsbild prädisponiert. Auch der eigentlich seltene, völlige Mangel des Komplementproteins C1q, kann mit nahezu 90 prozentiger Wahrscheinlichkeit zu SLE führen. Weniger selten ist der Mangel an C2 und C4, in dessen Fällen die Chance auf die Entwicklung von SLE im Verlauf des Lebens ca. 33-75 Prozent beträgt. Weil die Immunkomplexe aus dem Kreislauf hauptsächlich durch die auf den leberständigen Makrophagen vorhandenen Fc Rezeptoren eliminiert werden, können genetische Polymorphismen dieser Gene (z.B. FcγRII und FcγRIII) die eine ineffiziente Bindung der Immunkomplexe auf diesen Rezeptoren erzeugen, das Risiko von SLE auch erhöhen (Abbildung 3.5.). Abbildung 3.5. Genetische Polymorphismen welche die Entfernung von Immunkomplexen ändern, und ihr Einfluss auf SLE Es ist auch wichtig zu wissen, dass zusätzlich zu der Exposition auf artfremdes Serum und SLE bzw. manchen gekoppelten Genpolymorphismen, auch bestimmte therapeutische Eingriffe zur Entwicklung einer Serumkrankheit führen können. Zum Beispiel führt die Verabreichung von Beta-LaktamAntibiotika, Sulfonamiden und Thiaziddiuretika, zu einer erhöhten Produktion von Immunkomplexen. Dies kann auch in einer Reihe von anderen immunvermittelten Krankheitsbildern beobachtet werden: z.B. in Kryoglobulinämie, Rheumatoide Arthritis oder leukozytoklastische Angiitis. Die genaue diagnostische Bestimmung der Menge von Immunkomplexen ist stets eine große technische Herausforderung. Obwohl viele solche Methoden existieren, steht zur Zeit keine Methode zur Verfügung, welche gleichzeitig die Konzentration von Immunkomplexen verschiedener Größe und ihre chemische Zusammensetzung bestimmen könnte. Aus dem therapeutischen Gesichtspunkt betrachtet bleibt in Immunkomplexerkrankungen die effizienteste Behandlungsstrategie die Immunsuppression (meistens Steroide). 49 3.2. 2D Immundiffusion (2D Einfach- oder 2D Doppeldiffusion) Abbildung 3.6. 2D Immundiffusion Beide Methoden beruhen auf der Präzipitationsreaktion von Antigenen und spezifischen Antikörpern in einem Agarosegel. Das Ziel ist es, in beiden Fällen die Anwesenheit eines bestimmten Antigens oder Antikörpers in einer flüssigen Probe (z.B. Serum) durch das Erscheinen des Präzipitats im Gel, zu bestätigen bzw. seine Menge semiquantitativ zu bestimmen. 2D Einfachdiffusion (Mancini-Test) Aufgelöste Antikörper werden mit warmen (nicht heißem) flüssigen Agarosegel vermischt. Die Mischung wird auf ein Objektglas gegossen und liegen gelassen bis sie erstarrt. In das harte Gel werden zuerst mit einem Korkbohrer Vertiefungen gebohrt und die zu untersuchenden Lösungen, die möglicherweise das nachzuweisende Antigen beinhalten, in die Vertiefungen pipettiert. Als nächstes wird das Gel 2-3 Tage lang in einer Nasskammer inkubiert, damit das Antigen, falls vorhanden, aus den Vertiefungen in das Gel reindiffundieren kann. Sobald ein Gleichgewicht beim Erreichen des Äquivalenzpunktes entsteht, wird zirkulär um die Vertiefungen auch ein Präzipitat-Ring entstehen. Danach erfolgt die Fixierung: das Gel wird getrocknet und das 50 Präzipitat, falls vorhanden, mit einer aspezifischen Proteinfärbung (z.B. Coomassie) sichtbar gemacht. Je mehr Antigen in der zu analysierenden Probe vorhanden war, desto größer ist der Abstand vom Mittelpunkt des Loches bis zum zirkulären Präzipitat-Ring in dem Gel (Abbildung 3.6.). Selbstverständlich kann die Methode auch umgekehrt benutzt, d.h. zur Analyse von Antikörpern angewandt werden. In diesem Fall werden Antikörper in die Vertiefungen eingetragen, und Antigene bzw. gegen den gesuchten Antikörper gerichtete sekundäre Antikörper zum Gel zugemischt. 2D Doppeldiffusion (Ouchterlony-Test) Bei der 2D Doppeldiffusion werden Antikörper und das nachzuweisende Antigen in zwei separierte Vertiefungen bzw. Kanälchen des selben Agarosegels pipettiert, und dann solange im Gel diffundieren gelassen bis sie im Gel aufeinander treffen. Falls auf diese Weise zwischen den zwei Vertiefungen / Kanälchen im Gel ein Präzipitat entsteht, wird die Präsenz des gesuchten Antigens in der untersuchten Lösung (z.B: Serum) als bewiesen betrachtet. 2D Doppeldiffusion wird auch oft benutzt um den Titer eines Antigens zu bestimmen: dazu werden um eine zentrale, die Antikörper beinhaltende Vertiefung herum mehrere andere Vertiefungen platziert, welche eine Verdünnungsreihe des gesuchten Antigens beinhalten (Abbildung 3.6.). Als Präzipitationstiter wird die größte Verdünnung des Antigens genannt, bei der noch ein sichtbares Präzipitat entstehen kann. 3.3. Serumelektrophorese Die Bestimmung der Gesamtkonzentration aller Serumproteine ist ein wichtiges, täglich ausgeführtes klinisches Routineverfahren. Die Normalwerte der Serumprotein-Konzentration eines gesunden Menschen betragen 60-80 g/l, dessen Mehrheit (35-50g/l) vom Serum-Albumin kommt. Falls sich die Konzentration der Serumproteine unter- oder oberhalb der gesunden Grenzwerte befindet, wird am häufigsten eine Serumelektrophorese gemacht um eine genauere Beschreibung des abnormalen Serumprotein-Profils und die exakte Identifizierung der verantwortlichen Proteinfraktion zu ermöglichen. Die Serumelektrophorese beruht auf Unterschieden in der Ladung und Masse der verschiedenen Serumproteine, welche ihre Auftrennung in einem elektrischen Feld bzw. Gel ermöglichen. Nach der Elektrophorese werden die Proteinfraktionen mit unterschiedlichen Motilität mittels einem geeigneten Farbstoff (z.B. Coomassie Brilliant Blue) gefärbt, dadurch sichtbar gemacht, und basierend auf ihrer Lage im Gel einzeln identifiziert. Im menschlichen Serum ist die am schnellsten wandernde Proteinfraktion die des Thyroxin bindendes Präalbumin, am langsamsten hingegen sind die unterschiedlichen Immunglobuline. In der Serumelektrophorese ist die Präalbumin Fraktion normalerweise kaum oder überhaupt nicht sichtbar. Sie ist auch in dem gezeigten Beispiel (Abbildung 3.7.) nicht zu sehen, weswegen die größten sichtbaren Fraktionen die Fraktion des Albumins, der Globuline (z.B. Coeruloplasmin, -1Antitrypsin, Lipoproteine), der Globuline (z.B. Transferrin, -2- Mikroglobulin) und der Globuline (Immunglobuline) sind. Die Ergebnisse der Serumelektrophorese werden meistens densitometrisch ausgewertet (Abbildung 3.7.). 51 Abbildung 3.7. Serum-elektrophorese Abbildung 3.8. Typische Ergebnisse der Serumelektrophorese in verschiedenen Krankheitsbildern. 52 Abweichungen vom normalen Verhältnis der Serumproteine haben natürlich eine diagnostische Bedeutung (Abbildung 3.8.), z.B. eine verminderte Albumin-Produktion kann auf ein nephrotisches Syndrom, Leberzirrhose, oder chronisch-entzündlichen Darmerkrankungen hinweisen. Erhöhte Mengen von monoklonalen Globulinen können ein Hinweis auf maligne hämatologische Krankheiten bedeuten. Zum Beispiel im Falle von Multiplen Myelomen, wo alle bösartigen Zellen von einer gemeinsamen Vorläuferzelle abstammen, sezernieren alle Krebszellen den selben Antikörper, also identische Immunglobulin Proteine. Aus diesem Grunde werden sowohl die Konzentration der Immunglobuline, als auch die Gesamtkonzentration der Serumproteine erhöht sein, obwohl andere Serumproteine, z.B. Albumin, oft nicht beeinträchtigt sind. Ein anderes Zeichen für Multiples Myelom ist die Blutsenkung, welche bei Patienten mit MM in der Regel auch deutlich höher als normal ist (durchschnittlich über 100 mm/Stunde), wogegen die Konzentration des klassischen Entzündungsmarkers CRP sehr oft in dem gesunden Referenzbereich bleibt. In dieser Situation ist die erhöhte Blutsenkung nicht einer Entzündung zu verdanken, sondern der hohen Mengen von Immunglobulinen, die indirekt auch die Blutsenkung beeinflussen. Deswegen muss man bei gleichzeitiger Diagnose von hoher Blutsenkung und normalen CRP Werten die Möglichkeit eines Multiplen Myleoms immer in Erwägung ziehen. Eine spezielle Variante der Methode ist die 2D Serumelektrophorese. In der 2D Serumelektrophorese werden die Komponente des Serums in zwei Dimensionen, sowohl nach ihrem isoelektrischen Punkt (Dimension Nr. 1) als auch nach ihrer molekularen Größe (Dimension Nr. 2) auf die Oberfläche eines Gels aufgetrennt (Abbildung 3.9.). Dies ermöglicht eine extrem genaue Bestimmung der einzelnen Serumproteine und eine Analyse viel als konventionellen genauere die der Serum- elektrophorese bzw. Western blot. Da aber die Auswertung von 2D Elektrophoresen zurzeit eine große technische Herausforderung darstellt, wird diese Technologie heute noch eher in der Forschung, und nur selten in der klinischen Praxis angewandt. Abbildung 3.9. 2D Elektrophorese 53 3.4. Immunelektrophorese und Immunfixierung Die Immunelektrophorese ist ein weiterer Typ der elektrophoretischen Techniken der Serologie, welche in der Lage ist die verschiedenen Immunglobulinketten voneinander zu unterscheiden und ihre relativen Mengen semiquantitativ zu bestimmen. Sie beginnt mit einer Serumelektrophorese (siehe oben). Danach lässt man aber die aufgetrennten Serumprotein-Fraktionen in dem Gel frei diffundieren, und mit einem polyvalentem, tierischen anti-human Serum, welche alle menschlichen Serumproteine erkennen kann, in der Form einer 2D Immundiffusion (siehe vorher) reagieren. In der Immunelektrophorese reagiert also das zu untersuchende Serum des Patienten mit dem tierischen Serum, anti-human wodurch ein charakteristisches Präzipitat-Muster aller menschlichen Serumproteine erzeugt wird. Die typischen Lage und Form der Präzipitat-Kurven der einzelnen Immunglobulin-Klassen ermöglichen ihre Identifikation und eine mehr oder weniger genaue Bestimmung ihrer relativen Mengen (Abbildung 3.10.). Änderungen in der Form, oder Stärke von einzelnen Kurven mag von diagnostischem Wert sein, und die Methode ermöglicht auch die genaue Identifikation von nicht nur den schweren sondern auch den leichten Immunglobulinketten. Identifizierung von Auch die Antikörpern mit pathologisch erhöhter Expression kann durch starke Verdünnung analysierenden der zu Serumproben erleichtert werden. So werden nämlich die Antikörper langsam im Referenzbereich verschwinden, wogegen abnormal stark exprimierte Antikörper selbst bei starken sichtbar bleiben. Verdünnungen Abbildung 3.10. Immun-elektrophorese Die Immunfixierung ist teilweise ähnlich einer Immunelektrophorese, indem sie auch mit einer Serumelektrophorese beginnt. Danach werden aber die einzelnen Immunglobuline nicht durch eine 2D Immundiffusion, sondern mit Hilfe einer Zellulosemembran, worauf ein polyvalentes tierisches antihuman Serum aufgetragen worden ist, präzipitiert. Diese Membran wird sofort nach der Elektrophorese auf die Oberfläche des Gels gedrückt, wodurch das tierische Serum die Immunglobuline des Patienten an der Stelle, ohne Diffusion, ausfällen lässt (präzipitiert). Im Vergleich mit der Immunelektrophorese ist die Immunfixierung schneller auszuführen, besser reproduzierbar, empfindlicher, einfacher auszuwerten und teilweise auch automatisierbar (Abbildung 3.11). Abbildung 3.11. Serum und Urin Immunfixierung 3.5. Raketenelektrophorese Ähnlich der 2D Einfachdiffusion wird in der Raketenelektrophorese eine Lösung welche möglicherweise die nachzuweisenden Antigene beinhaltet, in eine kleine Vertiefung eines Agarosegels gelegt. Auch diesmal enthält das Agarosegel gegen das Antigen gerichtete Antikörper. Im Gegensatz zu der 2D Einfachdiffusion werden aber hier die Antigene nicht zirkulär, also in alle mögliche Richtungen, frei diffundieren, sondern mit Hilfe einer Elektrophorese nur in eine gewisse Richtung in dem Gel bewegt. Folglich werden auf einer Seite der Vertiefung spitzen- oder raketenförmige Präzipitate entstehen, falls das Antigen in der Tat in der Lösung vorhanden war. Die Entfernung der Spitze der “Raketen“ von dem Mittelpunkt der Vertiefung ist auch hier proportional zu der Menge des Antigens (Abbildung 3.12.). Abbildung 3.12. Raketenelektrophorese 55 3.6. 2D Immunelektrophorese Bei der 2D Immunelektrophorese wird zuerst eine Serumelektrophorese in eine Richtung ausgeführt (Dimension Nr. 1), indem Proteine abhängig von Grösse aufgeteilt werden. In einem zweiten Schritt werden die Komponenten des Serums mit Hilfe eines zweiten Gels (Dimension Nr. 2), welches ein polyvalentes tierisches anti-human Serum beinhaltet, wiederholt, jedoch in eine senkrechte Richtung, erneut aufgelöst. Dies führt zu der Entstehung eines sehr komplizierten Gel-Bildes von einer extrem guten Auflösung, dessen Auswertung aber zu kompliziert ist um Technologie in benutzen können zu 3.13.). der diese Routine (Abbildung Abbildung 3.13. 2D Immunelektrophorese 3.7. Turbidimetrie und Nephelometrie Turbidimetrie und Nephelometrie sind beide klinische Routineverfahren mit dem Ziel die quantitative Messung einer Antigen-Antikörper Interaktion zu ermöglichen. Das Prinzip beider Verfahren ist das die Intensität des Lichtes welches durch eine Kolloidlösung geht, durch die Lichtstreuung der Kolloidpartikel vermindert wird. Die Schwächung der Lichtintensität ist proportional zur Trübheit (Turbidität) der Lösung, welche eigentlich gemessen wird. Im Verlauf beider Verfahren werden in einer wasserklaren Pufferlösung das zu bestimmende Antigen und die damit vermischten spezifischen Antikörper (monoklonal oder polyklonal) Immunkomplexe bilden, die als Kolloidpartikel die Trübheit der Lösung erhöhen. Bei der Turbidimetrie wird die Schwächung eines durchgehenden Lichtstrahls bestimmt. Für die quantitative Analyse wird die Probe mit einer Standardreihe verglichen, welche neben einer stabilen Antikörperkonzentration immer größere Mengen des Antigens beinhaltet bzw. immer stärkere Trübheit vorweist. Dies ermöglicht die Herstellung einer Standardkurve mit dessen Hilfe die Konzentration des Antigens in der Probe bestimmt werden kann. Die Empfindlichkeitsgrenze der Turbidimetrie liegt bei ca. 50 μg/ml. Im Gegensatz zur Turbidimetrie wird in der Nephelometrie nicht die Schwächung eines durchgehenden Lichtstrahls gemessen, sondern die Lichtstreuung in einer Richtung seitlich des eigentlichen Lichtstrahls. Die Empfindlichkeit der konventionellen Turbidimetrie beträgt ungefähr 1 μg/ml. Die Nephelometrie aber kann sowohl gewöhnliches Licht- als auch Laserstrahlen benutzen, wobei die Lasernephelometrie deutlich empfindlicher ist als die konventionelle Methode. Nephelometrie und Turbidimetrie werden häufig benutzt um die Konzentration von Serumproteinen zu bestimmen, u.a. für die quantitative Analyse von Albumin, Immunglobulinen, Transferrin, Lipo- oder Komplementproteine. Die Turbidimetrie wird zusätzlich auch in bestimmten Analysen, welche keine Antigen-Antikörper Interaktion benötigen, angewandt: Zum Beispiel in der Analyse des Wachstums von Bakterien und dadurch der Analyse bakterieller Sensibilität bzw. Resistenz auf bestimmte Antibiotika. Aber auch in der Analyse des Aktivierungszustandes des Gerinnungssystems durch die Bestimmung der Zeit die für die Blutgerinnung benötigt wird. Abbildung 3.14. Turbidimetrie und Nephelometrie 3.8. Klinische Anwendungen von serologischen Methoden Immundiffusion: z.B. für die Identifizierung und Beschreibung von bakteriellen, viralen oder pilzAntigenen - 2D Einfachdiffusion (Mancini-Methode): Vergleichende quantitative Analysen von verschiedenen Antigenen, Serumproteinen (Immunoglobulin Isotypen [IgM, IgG, IgA], Akute-Phase-Proteine, Transferrin, Komplement Komponenten) und Antikörpern von bestimmter Spezifizität in unterschiedlichen Körperflüssigkeiten (Blut, Urin, Speichel usw.) - 2D Doppeldiffusion (Ouchterlony-Methode): Bestimmung des Äquivalenzpunktes in einer bestimmten Antigen-Antikörper Interaktion, Bestimmung der relativen Menge des präzipitierenden Antikörpers (Titer), oder die des Antigens. Elektrophorese: - Immunelektrophorese: Aufteilung der Serumproteine auf einzelne Fraktionen, Diagnose von Abnormitäten der Plasmazellen (z.B. Multyples Myelom, primär Amyloidose) durch den Nachweis von abnormalen Mengen von monoklonalen oder polyklonalen Antikörpern - Raketenelektrophorese: Bestimmung der Konzentration eines Antigens/Antikörpers 57 - 2D Immunelektrophorese: high-throughput, vergleichende qualitative und quantitative Analyse von allen Komponenten von Serum-, Urin-, Zerebrospinalflüssigkeiten. 3.9. Typen der Agglutination und ihre klinische Anwendungen Eine spezifische Wechselwirkung von Partikeln in der Größe von Zellen, und gegen sie gerichtete Antikörper wird als Agglutination bezeichnet. Dieses Phänomen wird in der Diagnostik häufig angewandt um eine spezifische Antigen-Antikörper Interaktion nachzuweisen (Abbildung 3.15.) Abbildung 3.15. Klinischer Nachweis von Agglutination durch geänderte Sedimentation von partikulären Antigenen Agglutination kann von praktisch allen Antikörper-Isotypen erzeugt werden, aber di- oder pentamere Antikörper (z.B. IgM) sind effizienter als monomere Antikörper (z.B. IgG). Ähnlich den klassischen Immunkomplex-Reaktionen benötigt auch die Agglutination ein optimales stöchiometrisches Verhältnis zwischen Antikörper und Antigen. Man redet über direkte Agglutination wenn der Antikörper die Antigene an sich, ohne zusätzliche Hilfe quervernetzen und agglutinieren kann. Abbildung 3.16. Direkte und indirekte Agglutination 58 Direkte Agglutination findet statt z.B. in dem direkten Coombs Test, weil die Antikörper Antigene auf der Zelloberfläche der zur Analyse benutzten Erythrozyten erkennen, und die Agglutination erfolgt durch die Quervernetzung der Erythrozyten durch die Antikörper. (Abbildung 3.17.) Abbildung 3.17. Coombs-test. Verdünnung Nr. 3 in der Verdünnungsreihe gibt den Titer an, also die größte Verdünnung, wobei noch eine Agglutination stattfindet. Die Zellmembran der Erythrozyten beinhaltet eine Vielzahl von bekannten Autoantigenen, von denen die bekanntesten die Agglutinogene A und B sind (Abbildung 3.18.). Abbildung 3.18. Die AB0 Blutgruppenantigene 59 Abhängig von ihrer An- oder Abwesenheit auf Erythrozyten werden die 4 wichtigsten Blutgruppen voneinander unterschieden, d.h. die Blutgruppen A, B, AB und 0 definiert. Die eigentliche Entstehung der Blutgruppen wird von kodominant vererbten Enzymen kontrolliert, welche die Zusammensetzung der Zuckergruppen der Agglutinine, und dadurch die Blutgruppe der jeweiligen Person bestimmen. Die Erkennung der nicht körpereigenen AB0 Blutgruppen-Antigene, also die Immunität gegen nicht kompatiblen Blutgruppen entsteht dadurch, dass viele Bakterien der normalen Darmflora über Antigene ähnlich der AB Antigene verfügen, weswegen alle Personen gegen nicht körpereigene AB Antigene praktisch vom Geburt an immunisiert sind. Zum Beispiel Personen mit Blutgruppe 0 sind immun gegen das A und B Antigen, ihre anti-A und anti-B Antikörper erkennen, agglutinieren und lysieren fremde Erythrozyten welche die entsprechenden Agglutinogene vorweisen (Abbildung 3.19.). Abbildung 3.19. Die AB0 Inkompatibilität. Ein “+“ bedeutet Immunreaktion vom Seiten des Empfängers und eine Agglutination des gegebenen Blutes, “-“ bedeutet keine Immunreaktion. Neben den AB0 Antigenen sind klinisch noch wichtig die Rh Antigene (der Name stammt von Rhesus Affen). Viele Antigene gehören in diese Gruppe, unter ihnen die stärkste Immunogenität dem D Antigen zuzuordnen ist (Abbildung 3.20.). 60 Abbildung 3.20. Das Rh Blutgruppenantigen Im Gegensatz zur AB Immunität, sind die Rh negativen Individuen nicht vom Geburt an immun gegen das D Antigen, sie produzieren nur anti-D Antikörper falls sie irgendwie mit D+ Blut transfundiert und dadurch immunisiert werden. Wenn eine Rh negative Mutter mit einem Rh positives Kind schwanger wird, kann die Mutter während der Geburt, da fetales Blut mit mütterlichem Blut in Kontakt kommt, gegen das D Antigen immunisiert werden. Da der anti-D Antikörper vom Typ IgG ist, ist er plazentagängig, also bei einer erneuten Schwangerschaft mit einem Rh positives Kind können die mütterlichen Antikörper die D Antigene der Erythrozyten des Fetus erkennen, sie agglutinieren, und fetale Erythroblastose verursachen (Abbildung 3.21.). 61 Abbildung 3.21. Die Rh-Inkompatibilität Dies kann prophylaktisch vermieden werden, indem man der gefährdeten Mütter binnen 72 Stunden nach der Geburt, Anti-D Antikörper verabreicht: ein Eingriff der als Rh Prophylaxe bezeichnet wird. In vivo Konsequenzen der Agglutination ABO Inkompatibilität: intravaskuläre Hämolyse (komplementvermittelte Hämolyse) Rh Inkompatibilität: fetale Erythroblastose (Opsonisierung der Erythrozyten, und dann Phagozytose durch Makrophagen) Tabelle 3.1. Die häufigsten Formen der immunvermittelten Erythrozyten-Agglutination . Indirekte Agglutination findet statt, wenn die Antikörper welche das Antigen erkennen selber keine Quervernetzung erzeugen können, nur mit Hilfe eines zweiten Antikörpers, welche den Fc Teil des ersten Antikörpers erkennt, ähnlich wie primäre und sekundäre Antikörper interagieren z.B. in den 62 indirekten immunhistochemischen Verfahren. Des Weiteren handelt es sich um eine passive Agglutination, wenn die zu agglutinierenden Partikel (z.B. Latexperle, Schaf Erythrozyten) das immunogene Antigen in ihrem natürlichen Form nicht vorweisen, das Antigen wird nur künstlich mit ihnen Verbunden, damit eine Agglutination überhaupt stattfinden kann. Passive Agglutination wird in vielen diagnostischen Techniken benutzt um Antigen-Antikörper Interaktion zu visualisieren bzw. quantitativ auszuwerten. Beispielsweise wird der Nachweis des Rheumafaktors (RF, solche Autoantikörper die andere körpereigene Antikörper an ihrem Fc Teil binden können), ein diagnostischer Marker der rheumatoiden Arthritis, Lupus erythematodes, manche Infektionen, usw., oft mit passiver Agglutination ausgeführt. In diesen Tests wird immer der Fc Teil von menschlichen Antikörpern zu Latexperlen, oder mit Hämolysin behandelten Schaf-Erythrozyten (Rose-Waaler Test) gebunden, die danach auf das Serum des Patienten exponiert werden. Falls im Serum des Patienten Rheumafaktoren vorhanden waren, führt dies zu eine sichtbare Agglutination der Latexperle / Erythrozyten. Heute werden Tests dieser Art jedoch immer seltener benutzt, da ELISA, Nephelometrie oder Turbidimetrie ihnen etwas überlegen sind. Als letztes ist es wichtig zu bemerken dass Agglutination von Erythrozyten auch in der völligen Abwesenheit von Antikörpern erfolgen kann (dies ist die nicht-immunvermittelte Hämagglutination), weil bestimmte virale Proteine (Influenza, Mumps) auch in der Lage sind, menschliche Erythrozyten zu verklumpen. Ein nicht-immunvermittelter Mechanismus der Hämagglutination kann aber nur bewiesen werden wenn man zeigen kann, dass antivirale Antikörper das Phänomen hemmen können. Typen der Agglutination, und ihre klinische Anwendungen Direkte Bestimmung der ABO Blutgruppen Agglutination: Widal-Test (Nachweis von Typhuserregern) Indirekte Agglutinat.: Coombs-Test (Nachweis von anti-D Antikörpern) Passive Nachweis des Rheumafaktors Agglutination: Tabelle 3.2. Praktische Anwendungsmöglichkeiten der Agglutination Übersetzt von Zoltán Pós 63 4. DIE PRAKTISCHE BEZÜGE DES KOMPLEMENTSYSTEMS (EDIT BUZÁS) 4.1. Das Komplementsystem Dazu, dass die Einwirkung von Antikörpern zur Lyse von Bakterien oder von anderen Zellen führe, ist nur die Anbindung der Antikörper zu den Zelloberflächenantigenen noch ungenügend, es ist auch irgendeine „ergänzende” (komplementäre) Wirkung des Blutserums (genauer: in ihm befindliche bestimmte Faktoren) notwendig. Die Funktion des Komplementsystems kann am einfachsten durch die Untersuchung der Erythrozyten-Lyse als Einwirkung von Antikörpern nachgewiesen werden. (Abbildung 4.1.) Abbildung 4.1. Komplement-vermittelte Hämolyse Wenn Schafserythrozyten mit polyklonalen Anti-Schafs-Erythrozytenantikörpern inkubiert werden, binden sich Antikörper an die Erythrozytenoberflächen; es erfolgt aber keine Lyse (durch Wirkung der Inkubation findet keine sichtbare Veränderung statt). (Um polyklonale Antikörper zu gewinnen, werden Kaninchen oder Ziegen mit Schafserythrozyten immunisiert; die Antikörper werden aus dem Blutserum des immunisierten Tieres gewonnen.) 64 Wenn zu Schafserythrozyten neben Anti-Schafs-Erythrozytonantikörpern noch humanes Blutserum gegeben wird, entsteht eine gut sichtbare Hämolyse: die Erythrozyten platzen, wodurch Hämoglobin aus den Zellen austritt, und die gebrauchte Pufferlösung anfärbt. Wenn Schafserythrozyten mit Anti-Schafs-Erythrozytenantikörpern und (vorher noch) mit hitzebehandelten (56 o C, für 30 min) Blutserum inkubiert wurden, würde keine Erythrozytenlyse (Hämolyse) stattfinden. Dieses lässt schlussfolgern, dass die sich im Serum befindlichen, für die Zell-Lyse notwendigen Faktoren hitzeempfindlich sind. (Unter solchen Umständen werden keine anderen Proteine im Blutserum inaktiviert.) Während der alltäglichen Gewebezüchtung wird als Ergänzung der Nährmedien fetales Kalb-Serum verwendet; bevor das Serum der Nährlösung zugegeben wird, werden die Komplementproteine im Kalb-Serum durch Hitze inaktiviert. Damit wird in der Zellkultur die durch das Komplementsystem ausgelöste Zell-Lyse und/oder Opsonisierung verhindert. Es ist heute schon bekannt, dass die Bestandteile des Komplementsystems verschiedene, hauptsächlich von Leberzellen produzierte Proteine sind. Ihre Menge beträgt um 12 – 15% der SerumGlobuline; dass heisst, diese Proteine liegen im Serum in sehr grossen Mengen vor. Die Konzentration des C3-Proteins, welches der zentrale Bestandteil des Komplementsystems ist, beträgt 1 g/l. Mitglieder des Komplementsystems sind im Allgemeinen Proenzyme (inaktive Form von proteinspaltenden Enzymen), die im gesunden Organismus funktionell inaktiv vorliegen und nur durch entsprechende Spaltung (Proteolyse) aktiviert werden. Die Aktivierung des Komplementsystems ist ein durch einen entsprechenden Reiz ausgelöster, kettenreaktionsartig ablaufender Vorgang. Die Aktivierung des Komplementsystems führt zur wirksamen Beseitigung von Bakterien, Pilzen oder Viren, bzw. zur Eliminierung von Immunkomplexen. Die einzelnen Elemente des Komplementsystems werden mit grossem C und mit der darauf folgenden Nummer bezeichnet (z.B. C3). Die durch die Spaltung des gegebenen Komplementproteins entstandenen Fragmente werden mit zur Bezeichnung des gegebenen Komplementproteins hinzufügten kleinen a und b bezeichnet. (Durch die Spaltung des C3 Proteins entstandene Fragmente sind C3a und C3b.) 4.2. Die Funktionen des Komplementsystems Heutzutage, ein Jahrhundert nach der Entdeckung des Komplementsystems, ist es eindeutig geworden, dass das Komplementsystem nicht nur in der Eliminierung der Krankheitserreger bzw. der vernichteten Zellen eine Rolle spielt, sondern harmonisiert auch mit den verschiedenen Vorgängen des Immunsystems und gibt Notsignale ab. Damit leistet es einen wichtigen Beitrag für das Aufrechterhalten der Immunhomöstase. Das Komplementsystem kann durch zahlreichen Einwirkungen aktiviert werden. Die Aktivierung des Komplementsystems hat man früher als eine Reihe von linearen, nacheinander kaskadenartig ablaufende Vorgänge gehalten. Nach heutigen Kenntnissen sind die verschiedenen Wege der 65 Komplementaktivierung eher als Netzwerke vorgestellt, in der die Elemente des Komplementsystems sowohl miteinander als auch mit anderen Systemen eng zusammenwirken. Drei Wege der Komplementaktivierung sind bekannt. I. Die Aktivierung des sog. klassischen Weges wird durch den mustererkennenden C1q-Teil des C1 Komplementproteins ausgelöst. Der C1q-Abschnitt eines C1-Proteins bindet sich einerseits an Immunkomplexe (z.B. an IgG- oder an IgM-Kluster), anderseits an Oberflächenkomponenten von Mikroorganismen und von apoptotischen Zellen. (Das C1q kann sich an der Oberfläche der zellulären Elementen sowohl direkt als auch auf indirekte Weise – über den endogenen Mustererkennungsrezeptor – binden.) Durch die Anbindung des C1q entsteht die aktive Konformation des C1-Proteins; dieses spaltet das C4-Protein in zwei Fragmente, in C4a und C4b. Das grössere Fragment, das C4b bindet ein weiteres Komplementprotein, das C2 an. Das derart aktivierte C2 wird von aktivem C1 in zwei Fragmente, in C2a und C2b gespaltet. Das enstandene C2a bleibt auch weiter an C4b gebunden; der enstandene C4b2a-Komplex ist die sog. C3-Konvertase. Die C3-Konvertase bindet ein C3-Komplementprotein an und spaltet es in zwei Fragmente, in C3a und C3b. C3b bleibt an der C3-Konvertase (C4b2a) gebunden; dadurch entsteht ein Heterotrimer, das C4b2a3b, die sog. C5Konvertase. II. Bei der Aktivierung des sog, lektinabhängigen Weges spielen Mannose-bindende Lektine (MBL) und Phykoline (endogene Mustererkennungsrezeptoren, die hauptsächlich Kohlenhydrat-Muster erkennen) die Schlüsselrolle, Am Lektinweg entsteht durch die Spaltung von C4-Komplementprotein das C4b Fragment; von hier an stimmen die Abläufe des lektinabhängigen Weges und des klassischen Weges überein. III. Der sog. alternative Weg ist phylogenetisch der älteste Aktivierungsmechanismus des Komplementsystems. Dabei spielt eine wichtige Rolle die kleingradige aber ständig vorhandene spontane Spaltung von C3-Proteinen im Blutplasma. Dabei entstehen – ohne äussere Einwirkungen – die C3a- und C3b-Fragmente. Wenn das C3b-Proteinfragment an Mikoorganismenoberflächen gebunden wird, wird der alternative Weg aktiviert. Das C3b spaltet mit Hilfe vom im Blutplasma vorhandenen Faktor D, den Faktor B in zwei Fragmente, in Ba und Bb. Der entstandene C3bBbKomplex ist die sog. alternative C3-Konvertase. Diese wird von einem weiteren Faktor, von Properidin (P) stabilisiert. Der stabile Komplex spaltet ein neues C3-Protein; das entstandene C3b-Fragment, bleibt an der C3-Konvertase gebunden: dieser C3bBbP3b-Komplex ist die alternative C5-Konvertase. Alle drei Wege der Komplementaktivierung führen zur Entstehung der C5-Konvertase, die ein proteolytischer Enzymkomplex ist. Der danach folgende Abschnitt, die sog. terminale lytische Phase, ist in allen drei Reaktionswege gleich. Der C3b-Komponent der C5-Konvertase erkennt und bindet das C5-Protein, und spaltet es in zwei Fragmente, in C5a und C5b. C5b verbindet sich mit weiteren Komplementproteinen, mit C6 und C7, und bilden zusammen eine trimere Struktur,(C5b67). Dieser Komplex ist stark hydrophob und kann leicht an der Lipiddoppelschicht der Plasmamembran gebunden werden. Nachdem sich dieser Komplex in der Plasmamembran befestigt hat, bindet an den Trimer ein C8-, später 10-14 C9-Proteine. Der entstandene sog. Membran-angreifende Komplex (MAK) bildet Poren in der Zellmembran. Diese grossen Poren führen zur Lyse der Zelle. 66 In der Komplementkaskade nehmen zahlreiche mustererkennende Moleküle teil. Das C1q bindet an oberflächengebundene IgG-s und IgMs, daneben aber auch an C-reaktive Proteine (CRP) und an mit Pathogenen-assoziierte molekulare Muster (PAMPs). Das Mannose bindende Lektin (MBL) erkennt gewisse Kohlenhydratmuster, Phykoline (1, 2 und 3) sind ebenfalls Kohlenhydratmuster-erkennende Moleküle. Von dem Properidin werden auch PAMPs, und mit Gewebsschädigung-assoziierte molekulare Muster (demage associated molekular pattern, DAMPs) erkannt. Das CRP ist fähig PAMPs und DAMPs an apoptotischen und mikrobiellen Zellen zu erkennen. Das Komplementfaktor-H related Protein (CFHR) befördert monomere CRPs auf nekrotischen Zelloberflächen. Obwohl auch die Oberflächenstrukturen der apoptotischen Zellen von den Mustererkennungsproteinen erkannt werden können, sind gewisse Regulatorproteine in der Lage die Komplementaktivierung zu verhindern. Die opsonierende Wirkung der Komplementproteine trägt zur „stillen” Eliminierung der apoptotischen Zellen bei. 4.3. Untersuchung der Aktivität des Komplementsystems In der Labormedizinischen Praxis kann die Bestimmung der Komplementaktivität vom Blutserum notwendig sein. Es soll aber berücksichtigt werden, dass die Komplementprotein-Konzentrationen im Blutserum einerseits von der Geschwindigkeit deren Synthese, anderseits von der Aktivität des Komplementverbrauches beeinflussen ist. Wie das schon erwähnt war, sind Komplementproteine hitzeempfindlich. Aus diesem Grund dürfen Seren nach Musterabnahme nur für einen minimalen Zeitraum auf Zimmertemperatur sein, vielmehr müssen sie auf Dauer bei -70 °C gelagert werden. 4.3.1. Bestimmung den CH50 Wert des Blutserums Zur Charakrerisierung der klassischen Komplementaktivierung wird am häufigsten der sog.CH50 Wert des untersuchten Blutserums bestimmt. Dieser Wert spiegelt die Komplemenaktivität des Blutserums wieder, und zeigt an, ob und in welchem Maß die mit polyklonalen Antikörpern bedeckten Schafserythrozyten durch das Blutserum lysiert werden. Der CH50 Wert ist der reziproke Wert solcher Serumverdünnung, bei der (unter standardisierten Bedingungen) 50% der mit Antikörpern bedeckten Schafserythrozyten lysiert werden. Nach Vereinbarung enthält 1 CH50 Komplementeinheit diejenige Serummenge, welche zur Lyse von 50% der, mit Antikörpern bedeckten Schafserythrozyten benötigt wird. Serum CH50 In Körperflüssigkeiten C1-Gehalt C3-Gehalt C4-Gehalt Normalwerte von CH50 142-279 CH50 Einheit/mL 1/3, 1/4 vom Vorherigen 16-33 mg/dL Mann: 88-252 mg/dL Frau: 88-206 mg/dL Mann: 12-72 mg/dL Frau: 13- 75 mg/dL Tabelle 4.1. Normalwerte von CH50 67 In den meisten Fallen einer Abweichungen der CH50-Werte wird beobachtet, dass der gemessene Wert höher ist als der normale; dies lässt sich damit erklären, dass die meisten Komplementproteine gleichzeitig auch Akutphasenproteine sind. Der CH50-Wert des Serums sinkt, wenn einige Komplementproteine fehlen oder verbraucht wurden (z.B.bei Komplementdefizienzen und bei bestimmten Vaskulitiden). Ein signifikant niedriger Wert der CH50 kann auch gemessen werden, jedoch lässt sich in einem solchen Fall eine immunologische Erkrankung vermuten (Immunkomplexerkrankung, SLE). Der niedrige CH50 Wert prädisponiert auf ständig wiederkehrenden bakteriellen sinopulmonalen Erkrankungen, durch Neisseria verursachte Meningitis oder Sepsis. Der normale CH50 Wert beweist jedoch nur die Anwesenheit aller Komplementkomponenten, da die Menge einzelner Komplementfaktoren ohne Veränderung der CH50 Aktivität stark gesenkt werden können. Da bei Entzündungen die Synthese der Komplementproteine gesteigert ist, zeigen Normalwerte nicht an, ob eine komplementvermittelte Gewebeschädigung vorliegt. In der Synovialflüssigkeit der betroffenen Gelenke von Patienten, die an verschiedenen entzündlichen Arthritiden (z.B. an rheumatoider Arthritis oder durch Gonokokken verursachten Arthritis) erkrankt sind, werden die Komplementfaktoren lokal verbraucht, dem zu Folge findet man in der Synovialflüssigkeit verringerte CH50 Aktivität, bzw. Komplementproteinkonzentration während im Blutserum der Komplementanteil Normalwerte zeigt. 4.3.1.1. Schafserythrozyten Immunoassay Für die Bestimmung des CH50 Wertes wird das menschliche Komplementsystem durch mit Antikörpern bedeckten Schafserythrozyten aktiviert: mit von einem immunisierten Kaninchen produzierten Anti-Schafserythrozyten Antikörpern, werden Schafserythrozyten behandelt, und die dadurch mit Antikörpern bedeckten Schafserythrozyten werden mit verschiedenen Verdünnungskonzentrationen (sog. Verdünnungsreihe) des zu untersuchenden Blutserums inkubiert. Das Ausmaß der Hämolyse wird bei den unterschiedlichen Serumverdünnungen durch spektrophotometrische Bestimmung des (bei der Zell-Lyse aus den Erythrozyten freigewordenen) Hämoglobins kalkuliert. Der Zusammenhang zwischen der, zu den mit Antikörpern bedeckten Schafserythrozyten gegebenen Serummenge und dem Ausmass der Hämolyse ergibt sich eine sigmoidale Kurve (van Krogh Gleichung). Mit Hilfe von der logarithmischen Transformation wird ein linearer Zusammenhang abgeleitet (Abbildung 4.2.): Y=EM-ERK/ELK wobei E = Extinktion M = Serumprobe RK = Reagenz-Kontrolle (ohne Serum) LK = Kontrolle der Zell-Lyse (Lyse der gesamten Erythrozyten im destillierten Wasser) Yx100 = Menge der lysierten Erythrozyten (in % ausgedrückt) Angesichts dessen, dass die CH50 Kurve keine lineare Steigung zeigt, müssen für die bedeutenden CH50 Abweichungen grosse Mengen Komplementproteine verbraucht werden. 68 Abbildung 4.2. Bestimmung den CH50-Wert des Blutserums 4.3.1.2. Liposom Immunoassay Abbildung 4.3. Die Bestimmung den CH50-Wert des Blutserums mit unterschiedlichen Verfahren 69 In diesem alternativen Verfahren für die Bestimmung des CH50 Wertes des Blutserums werden Liposomen mit DNP (Dinitrophenol)-Hapten bedeckten Oberflächen verwendet. In Hohlräumen der Liposomen ist das Enzym, die Glucose-6-Phosphat-Dehydrogenase (G6PDH), eingeschlossen. Mit Zugabe von Anti-DNP-Antikörpern entstehen an Liposomenoberflächen Antigen-Antikörperkomplexe. Sobald das Komplementsystem aktiviert wird, öffnet sich die Liposomenmembran, und das Enzym G6PDH wird dabei freigesetzt. Durch die Aktivität des Enzyms wird die NAD zu NADH reduziert. Die Menge des gebildeten NADH wird spektrophotometrisch aus der Lichtabsorption bei 340 nm bestimmt. Die Menge des entstandenen NADH ist mit der Komplementaktivität proportional (Abbildung 4.3.). 4.3.2. Untersuchung der alternativen Komplementaktivation 2+ 2+ Wenn man mit Ca -Chelatoren (z.B.EDTA) die Ca -Ionen entzieht, lässt sich der alternative Weg 2+ selektiv untersuchen (Ca -Ionen sind nämlich für den klassischen Weg notwendig). Gleichzeitig 2+ müssen Mg -Ionen unbedingt vorhanden sein, denn sie sind für die Funktionsfähigkeit des Faktors “B” erforderlich. Für die Untersuchung des alternativen Weges können auch (ohne sog. Sensibilisierung, d.h. ohne Antikörper Inkubation) abgewaschene Kaninchenerythrozyten benutzt werden. In diesem System sind die sog. AP50 Werte bestimmbar. 4.3.3. Untersuchung des lektinabhängigen Weges Für Untersuchung des lektinabhängigen Weges soll als erstes verhindert werden, dass die sich im Serum befindenden Mannose-spezifischen Antikörper, den klassischen Weg aktivieren. Geeignet ist dafür z.B. ein C1q-erkennender und -blockierender monoklonaler Antikörper (Mab 2204). Bei Hemmung des klassischen Weges können sowohl der lektinabhängige- als auch der alternative Weg aktiviert werden, jedoch ist die Aktivierung des alternativen Weges nur bei höherer Serumkonzentration (über 1%) möglich, im Gegensatz zum lektinabhängigen Weg, wobei die notwendige Serumkonzentration 0,1-1% betragen soll. Auf dieser Weise sind der lektinabhängige und der alternative Weg unterscheidbar. 4.3.4. Komplement Konvertase-Assay (CCA) Das Komplementsystem kann auch durch Inkubation des Blutes mit gewissen Arzneimitteln, medizinischen Substanzen bzw. mit Materialien der Instrumente aktiviert werden. Der natürliche Regulator der Enzymkaskade (des Komplementsystems) ist die Instabilität der beteiligten Enzyme. Diese Regulatormechanismen können unzureichend werden, wenn Mittel mit komplementaktivierenden Oberflächen verwendet sind. Deshalb – wegen der “Hämokompatibilität” – ist es notwendig die Komplement-Konvertaseaktivität an Arznei-Hilfsmitteloberflächen zu prüfen. Für diese Untersuchung wird chromogenes Substrat verwendet, um kolorometrische Messungen durchzuführen. 70 4.3.5. Weitere Verfahren zum Nachweis von Komplementproteinen 1. Komplementproteine (z.B. C3) können mit der (C3-)ELISA nachgewiesen werden. Für die Untersuchung der in vivo Komplementaktivierung benutzt man selektive Antikörper welche fähig sind entweder die Aktivationsprodukte oder die gebildeten Komplexe zu erkennen. 2. Die Produktion vom C3-Protein bzw. von weiteren Komplementproteinen können mit Hilfe von RT PCR untersucht werden. 3. Weiterhin können die auf den Zelloberflächen abgelagerten (dazu kovalent gebundenen) C3 mit Durchflusszytometrie (FACS) bestimmt werden. 4. Daneben besteht die Möglichkeit zur Untersuchung der Rezeptoren für die unterschiedlichen Komplementproteine (indirekter Weise) durch Bestimmung der entsprechenden mRNAs oder direkt durch die Untersuchung der Rezeptorproteine 5. Für den Immunkomplexnachweis ist die sog. Komplementbindungsreaktion (Komplementkonsumptionstest. KBR) verwendbar. Wenn in der zu untersuchenden Probe Antigen-Antikörperkomplexe (sog.Immunkomplexe) vorhanden sind, binden sie die zur Probe gegebenen Komplementproteine, d.h. bleibt in der Probe kein freies Komplementprotein übrig. Wenn im Komplement-Untersuchungsverfahren oft verwendete mit Antikörpern bedeckte 6. Schafserythrozyten zur Probe gegeben werden, erfolgt Hämolyse in Fall, dem die wenn ursprünglich wenige Immunkomplexe vorhanden waren, freigebliebene komplex so, (nicht dass an gebundene) das Immun- Komplement sich zum mit Antikörpern bedeckten Schafserythrozyten bindet und dadurch deren Lyse verursacht. Sofern im zu untersuchenden Muster bedeutende Mengen von Immun- komplexen vorhanden waren, wurde das gesamte Komplement gebunden, d.h. es bleibt kein freies Komplement mehr übrig, es erfolgt auch keine Hämolyse (Abbildung 4.4.). Abbildung 4.4. Das Prinzip der Komplementbindungsreaktion (Komplementkonsumptionstest) 71 7. Komplementaktivations-ELISA. Es wurde beobachtet, dass zahlreiche Komplementproteine fähig sind, sich an einer entsprechenden Kunststoffoberläche anzukoppeln. Auf dieser Erscheinung basierend wurden ELISA-Systeme entwickelt um die unterchiedlichen Komplementaktivationswege zu untersuchen. Mit Hilfe von IgM bedeckten Oberflächen ist der klassische Aktivierungsweg, mit Hilfe von Lipoplysacchariden (LPS) von Salmonella Typhi behandelten Oberflächen ist der alternative Weg ist untersuchbar. In diesen Fällen wird die Komplementaktivierung entweder mit C9-Neoepitop oder mit Properdin reagierenden monoklonalen Antikörpern nachgewiesen. Die Untersuchung des lektinabhängigen Weges ist einfach mit Mannose vorbehandelten Mikroplattenoberflächen durchzuführen, sofern der klassische Weg mit Anti-C1q Antikörpern blockiert wird (Abbildung 4.5.). Abbildung 4.5. Komplementaktivations-ELISA ® Das sog. Wielisa -Verfahren ist eine Kombination von drei ELISA Systemen, wodurch alle Komplementwege untersucht werden können. ® Im Wielisa -System wird auf entsprechend vorbehandelten Mikroplatten die Aktivierung des Komplementsystems induziert, anschließend werden die terminalen Komplexe detektiert. Die Steuerung der Komplementaktivierung (d.h. die Bestimmung, welcher Komplementweg aktiviert – und dadurch untersucht – werden soll) ist durch die Zusammensetzung der verwendeten Pufferlösung bzw. durch den Verdünnungsgrad des Blutserums 2+ 2+ möglich (Ca - und Mg -Ionen sind für die molekularen Vorgänge des Klassischen- und des Lektinweges erforderlich sowie für den terminalen Abschnitt der Komplementaktivierung; die 2+ 2+ Anwesenheit von Mg -Ionen (neben EDTA, d.h. Fehlen von Ca -Ionen) ist ausreichend für die Aktivierung des alternativen Weges; der klassische- und der lektinabhängige Weg sind bei 1:100 oder bei noch stärkerer Serum-Verdünnung aktiv, der alternative Weg braucht Verdünnungswerte unter ® 1:20). Vorteil des Wielisa -Systems ist, dass keine Schafserythrozyten benötigt werden und alle drei Komplementaktivationswege messbar sind. 72 4.3.6. Sonstige, mit dem Komplementsystem verbundene Verfahren 1. Zum Nachweis von Immunkomplexen mit ELISA-Verfahren werden Anti-C3- oder Anti-C1qAntikörper an ELISA-Mikroplattenoberflächen gebunden. Die angebundene Immunkomplexe werden mit markierten Anti-Immunglobulin-Antikörpern detektiert (Abbildung 4.6.). Abbildung 4.6. Auf ELISA basierende Nachweisemethoden von Immunkomplexen im Blutserum 2. Für die HLA-Typisierung findet der Mikrolymphozytotoxizitäts-Test Verwendung. Dieses Verfahren gehört zu den sog. serologischen Methoden der HLA-Typisierung, welches damals den "Goldstandard" in der HLA-Typisierung bedeutete. Heute werden vielmehr molekularbiologische Methoden zur HLA-Typisierung angewandt. Bei dem Mikrolymphozytotoxitäts-Test werden die sog. Terasaki Platten verwendet. In den Wells (Vertiefungen) dieser Platte werden die (am Ficoll-Gradient) aus dem Blut der zu typisierenden Personen) isolierten Lymphozyten pipettiert. Im Fall der Antigenerkennung bindet der entsprechende HLA-spezifische Antikörper an die Zellen. Die zur Typisierung verwendeten Seren stammen häufig von schwangeren Frauen ab, oder von Personen die mehrmals eine Bluttransfusion erhalten haben. Kaninchenserum kann als Komplement – Quelle benutzt werden, das aktiviert wird, sofern die Antikörper an das HLA-Antigen an Zelloberflächen angebunden sind. Daraufhin, als Resultat der Komplementaktivierung, erfolgt die Zellyse und die Zellmemebran wird für vitale Farbstoffe permeabel. Aufgrund des 73 hineingelangten Farbstoffs, Ethidium-Bromid, oder Tripanblau, ist die bereits erfolgte Zellyse gut detektierbar. 4.4. Untersuchung einzelner Komplement-Elemente verschiedenen pathologischen Zuständen in 1. HANO Typ I.: Die Aktivität des C1-Inhibitors und Antikörpers sind niedrig, C4 niedrig, CH50 niedrig, C3 und C1q liegen im Referenzbereich. 2. AANO-C1-INH (Angioneurotisches Ödem mit Autoimmunursprung): die C1-Inhibitoraktivität ist erniedrigt, Anti-C1-INH-Antikörper positiv, CH50 niedrig, C4 und C1q niedrig, C3 liegt im Referenzbereich. 3. Hämolytisch-urämisches Syndrom (HUS) (mit atypischem Verlauf), im Hintergrund steht die Störung des alternativen Aktivierungswegs. 4.4.1. Falldarstell Die 19-jährige Patientin kommt zur Notdienstaufnahmestelle mit Beschwerden wie leichte Benommenheit, schwere Bauchschmerzen und seit 12 Stunden bestehender Übelkeit mit Erbrechen. Sie berichtet über frühere Bauchschmerzen und über Schwellungen an Händen und Beinen. Man vermutete eine Lebensmittelallergie dabei zu haben. Man vermutete eine Kombination mit einer Lebensmittelallergie. Derartige Beschwerden traten in letzter Zeit jedoch immer häufiger auf. Urtikaria war mit den Symptomen nicht vergesellschaftet. Die Patientin gab an, dass auch ihr Vater ähnliche Beschwerden gehabt haben sollte. Fieber war nicht vorhanden, der Blutdruck beträgt 75/40 mmHg, Puls 120/min. Dieses Krankheitsbild wurde erstmal im Jahre 1888 von Osler beschrieben, als Hereditäres Angioneurotisches Ödem. Es handelt sich dabei um eine Erkrankung mit autosomal dominantem Erbgang und C1-Defizienz. Als Rekurrent-Episoden treten nicht juckende subcutane oder submocosale Ödeme auf. Typische Orte der Ödembildung: Arme, Beine, Oberfuss, Darm, Genitalien, Stamm, Gesicht, Zunge, oder Kehlkopf. Die Häufigkeit der Erkrankung in der Bevölkerung liegt bei 1/50 0000. Die Symptome treten typischerweise im Kindesalter auf, der Gesundheitszustand verschlechtert sich um die Pubertät, die Krankheit besteht bis zum Lebensende. Im Hintergrund der Symptome stehen C1-Inhibitor-Genmutationen (bis heute sind etwa 150 verschiedene Mutationen in HANO Erkrankungen identifiziert). Der C1-Inhibitor hemmt folgende Proteasen: die zwei Untereinheiten vom C1-Komplex (C1r, C1s), gehemmt werden ebenfalls: Kallikrein, Plasmin, der aktivierte Faktor XII (FXIIa) und der aktivierte Faktor XI (FXIa). Obwohl der Typ I. (85% der Fällen) und der Typ II. (15%) der HANO-Krankheiten klinisch voneinander nicht zu unterscheiden sind, stehen im Hintergrund beider Erkrankungen verschiedene Mutationen. Die beim Typ I. mutiert produzierten Proteine werden nicht mit entsprechender Wirksamkeit sezerniert. Aus diesem Grund senkt das C1-Inhibitorniveau sowohl seine Antigenität, als auch seine Aktivität. Im Fall des HANO Typ II. wird das produzierte Protein zwar richtig sezerniert, jedoch ist 74 seine Funktion betroffen. Daher ist zwar die Antigenität des C1-Inhibitors normal, jedoch ist seine Aktivität (Funktion) bedeutend gesenkt. Auf Grund des Mangels an C1-Inhibitor werden während HANO-Anfällen die proteolytischen Kaskaden des Blutplasmas aktiviert, und in erster Linie ist das Bradikinin für die erhöhte Gefässwandpermeabilität verantwortlich. Die Diagnose HANO kommt erst in Verdacht, wenn wiederholt auftretende angioödematöse Krankheitsschübe, in Begleitung mit Bauchschmerzen und ohne Urtikaria, auftreten. Typisch für diese Erkrankung ist der niedrige C4 Antigentiter, jedoch zeigen die C1 und C3 Proteine Normalwerte. Die Therapie besteht aus rekombiniertem C1 der Gabe von intravenösem C1-Inhibitors (500-2000 Units) oder Inhibitor. Wirkungen: Hemmung des Plasmakallikrein, Hemmung von Gerinnungsfaktoren XIIa und XIa sowie C1s, C1r, MASP-1 und MASP-2 und Plasmin wird ebenfalls gehemmt. Abbildung 4.7. Pathomechanismus der HANO-Krankheit und in der Ödembildung beteiligten Faktoren 75 4.5. Aufgaben Beantworten Sie bitte die folgende Fragen 1) Welche CH50 Werte kommen am häufigsten vor: die erhöhten oder die erniedrigten? Was ist die Ursache? Worauf lässt der erniedrigte CH50 Wert hinweisen? 2) Was können wir mit voller Sicherheit aussagen wenn der Gesamtkomplementwert (CH50) im Normberich liegt? 3) Welches Komplementprotein liegt im Serum in höchster Konzentration vor? 1) Worauf weisen die gemeinsam verringerten Werte der C3 und C4 Proteine hin? 2) Worauf weist es hin, wenn der C3 Gehalt sinkt, C4 jedoch im Normbereich liegt? 3) Verringerter CH50 Titer und erniedrigte Konzentration der Komplementproteine sind Typisch für Synovial-Flüssigkeits-Muster bei rheumatoider Arthritis oder Gonokokken Arthritis. In welchem Bereich können die Serumwerte von C3 u.C4 bzw. von der CH50 Wert liegen? Literatur: Farkas H. A herediter angioneuroticus oedema patomechanizmusa és az oedema kiváltásában szerepet játszó provokáló tényezők. (Pathomechanismus der HANO-Krankheit und in der Ödembildung beteiligten Faktoren) Magyar Immunológia 2002;1 (1): 5-11. Zuraw BL. Hereditary Angioedema. New England Journal of Medicine 2008; 359:1027-103. Übersetzt von Ottó Dobozy 76 5. DIE DURCHFLUSSZYTOMETRIE (ÉVA PÁLLINGER) Durch die Entwicklung der laboratorischen Verfahren, die sich aus den Fragestellungen der Proteomik entwickelt haben (wie immunanalytische Verfahren, Massenspektroskopie und monoklonale Antikörperherstellung usw.), haben sich beispiellose Möglichkeiten für die medizinischen Diagnostik eröffnet. Allerdings muss man während der Auswertung von Ergebnisse den Tatsachen ins Auge sehen, weil die Proteinzusammensetzung nicht nur unter den Zelltypen, sondern auch in denselben Zellen von Zeit zu Zeit abweichen kann. Abhängig von dem aktuellen Zustand oder der Umwelt kann sie sich selbst auch ändern. Die biologische Interpretation des detektierten Datesatzes erfordert also molekulare und zellbiologische, sogenannte cytomische (cytomics) Vorkenntnisse. Die Durchflusszytometrie ist eigentlich eine Kombination von Proteomik mit Cytomik, durch die die gleichzeitige Charakterisierung einzelner Zellen nach funktionellen und morphologischen Aspekten möglich ist. Die Durchflusszytometrie gehört zu den Hochdurchsatzverfahren (High throughput screening method = HTS), weil sie während einer sehr kurzen Zeit (wenige Minuten) mehrere tausende Zelle auf multiparametrische Weise detektieren kann. Solche Untersuchungen sind, auf Grund des Paradigmenwechsels auf personalisierte Medizin, nötig geworden. Die HTS –Methoden können die Biomarker-Forschung (die Entdeckung von neuen diagnostischen und prognostischen Parametern) beschleunigen, eine Medikamentenwirkung personalisiert überwachen (individuelle Sensitivität und Leistungsfähigkeit) und eine genauere Zuordnung der molekularen Pathogenese von Krankheiten aufklären. 5.1. Einleitung Die Durchflusszytometrie beschreibt ein laboratorisches Messverfahren, das die quantitative und multiparametrische Analyse von einzelnen Zellen (von physikalischen und/oder chemischen Eigenschaften) in einem außergewöhnlich hohen Tempo erlaubt. Eine fluoreszierende Markierung lässt sich auch verwenden, um die zu analysierende Zelle morphologisch und funktionell zu differenzieren. Die richtig vorbereiteten Zellen werden von einem Laserstrahl beleuchtet und die von dem Laser induzierten optischen Signale werden von dem Gerät detektiert und weiter verarbeitet. Die Anwendung der Durchflusszytomerie ermöglicht die Identifizierung und Separierung (sorting) unterschiedlicher Zellpopulationen aufgrund ihres Immunphänotyps oder auch dem funktionellen Zustand. Seit ihrer in den 40er Jahren begonnenen Geschichte, hat die Durchflusszytometrie signifikante Veränderungen erfahren. Anfangs waren die Instrumente nur einfache Zählgeräte gewesen, die auf dem Prinzip der Messung von Lichtstreuung und von der elektrischen Impedanz funktionierten. Sie 77 wurden dann weiterentwickelt, um die Fluoreszenzsignale auch detektieren zu können. Die Geräte, die schon später routinemäßig angewendet wurden, beinhalteten im Allgemeinen eine einzelne Laserlichtquelle, die 2-6 unterschiedliche Fluoreszenzfarbstoffe (außer der Zellgrösse und Granularität) gleichzeitig differenzieren konnte. Insgesamt waren dies 4-8 gemessene Parameter pro Zelle. Die Durchflusszytometrie war schon damals in der medizinischen Diagnostik weit verbreitet. Hauptsächlich wurde sie für die Immuntypisierung der im Blut zirkulierenden Leukozyten mit 1 oder 2 Farben verwendet. Heutzutage stehen weitere Möglichkeiten zur Verfügung, wie z.B. die auf Mikroperlen basierenden Detektionssysteme für flüssige Moleküle (Cytometric Bead Array =CBA; microsphere-based flow cytometric assay), oder mit der Anwendung von dem FRET-Phänomen (FRET = fluorescence resonance energy transfer) können die intermolekularen Wechselwirkungen (Protein-protein Interaktionen) charakterisiert werden. Mit Antikörpern, die phosphorylierte (modifizierte) Proteine erkennen, lassen sich die intrazellulären Signalübertragungs- und metabolische Wege identifizieren. Die erhöhte Anzahl von gleichzeitig detektierbaren Farben (10-17 Farben in einem Experiment) hat es möglich gemacht, dass die Immunphänotypisierung sogar mit der Detektierung von intrazellulären Zytokinen ergänzt/kombiniert werden kann, d.h. können auch proteomische Untersuchungen auf Zellebene durchgeführt werden. Die Durchflusszytometrie ist dadurch unverzichtbar in dem diagnostischen Arsenal geworden. Neben der morphologischen und funktionellen Charakterisierung der Zelle ist sie auch einzigartig im Bezug auf die Fähigkeit, dass sie gut definierte Zellpopulationen (z. B. mit der Expression von Differentiationsantigenen identifizierte Zellen) mit hoher Geschwindigkeit und Reinheit voneinander separieren kann (Zellseparation, cell sorting). Mit durchflusszytometrischen Verfahren werden folgende Fragestellungen am häuigsten beantwortet: 1. Sind die gesuchten Zellen (z. B. aufgrund ihres Zelloberflächen-Molekül Musters, Immunphänotyp, definierter Population) in dem Untersuchungsmaterial anwesend? 2. Zu welchem Anteil (oder absolute Menge) können die gefragten Zellen nachgewiesen werden? 3. Wie viele unterschiedliche Zellpopulationen (aufgrund z. B. Grösse, Granularität, Immunphänotyp oder funktioneller Zustand) können in der Probe identifiziert werden? 4. In welchem derzeitigen funktionellen Zustand sind die Zellen? 5.2. Das Durchflusszytometer Ein Durchflusszytometer besteht aus 1) dem Flüssigkeitssytem, 2) einem optischen System, 3) elektrischem System, 4) einer Einheit für die Analyse (Computer), 5) Zellsorter (optional). (Abbildung 5.1.) 78 Abbildung 5.1. Genereller Aufbau eines Durchflusszytometers (Ref 1.) Die Detektoren (Photomultiplierröhren) werden mit zunehmendem spektralen Abstand vom Anregungslicht aufsteigend nummeriert (FL1, FL2, FL3 usw.) 1) Von dem Flüssigkeitssystem wird ein Probenstrom in der Messküvette des Leitungssystems in dem Durchflusszytometer erzeugt, in dem die Zellen in einem Flüssigkeitsstrahl aus der Probensuspension einzeln hintereinander („im Gänsemarsch“) durch den Messbereich bewegt werden. Dies erfolgt durch die sog. hydrodynamische Fokussierung, die durch zwei Flüssigkeitsströme, dem Hüll- (sheath fluid) und dem Probenstrom erzielt wird (laminare Strömungsverhältnisse). der Messküvette werden In die nacheinander kommenden, wie in einer Perlenkette aufgereihten Zellen von einem fokussierten beleuchtet. Laserstrahl (Abbildung Flüssigkeitssystem geschlossenes 5.2.) ist System, Das ein da das aufgesaugte Probenmaterial mit der zirkulierenden Flüssigkeit nach der Messung durch einen Schlauch in einem Abfallbehälter (waste container) aufgefangen wird. Abbildung 5.2. Das Funktionsprinzip der Durchflusszelle 2) Das optische System besteht aus einer (oder mehreren) (1) Lichtquelle(n), die für die Beleuchtung der Zellen und Anregung fluoreszierender Farbstoffe geeignet ist (monochromatisches kohärentes Licht: Laser), aus (2) den Laserstrahl fokussierenden Linsen, aus (3) Photomultiplierröhren, die für die Detektion von emittiertem Licht verantwortlich sind und aus (4) einem Spiegelsystem, das das emittierte Licht nach Wellenlängen auftrennt (filtert) und steuert. 3) Die Elektronik konvertiert die detektierten Lichtsignale in digitale Signale für die ComputerVerarbeitung (ADC=Analog-Digital-Umsetzer). 4) Ein Analysesystem (Computer) kümmert sich um die Datensammlung und Speicherung, bildliche Darstellung und die Möglichkeit der Software-beruhenden Verarbeitung. 5) Zellseparator („sorter”) (Die Durchflusszytometrie wird oft als FACS bezeichnet, wobei FACS für "fluorescence activated cell sorting" steht, obwohl noch nicht alle Geräte für die physikalische Trennung der Zellen geeignet sind). Aus technischen Aspekten können die FACS-Instrumente auf zwei Gruppen verteilt werden, abhängig davon, ob sie nach 1) mechanischem oder (2) elektrostatischem Prinzip funktionieren (Abbildung 5.3.). Abbildung 5.3a. Mechanische Separierung der Zellen Die Regel der Trennung kann jeder von dem Gerät detektierender Parameter sein: Zellgrösse, Granularität (FSC und SSC), oder durch identifizierte Fluoreszenzmarkierung Eigenschaften (und irgendeine Kombination von ihnen). Abbildung 5.3b. Elektromechanische Separierung der Zellen 5.2.1. Die Bildliche Darstellung (data display) detektierten Signale können in Histogrammen (Häufigkeit über Intensität) oder für Parameterkombinationen in Punktwolkendiagrammen (dot plot/scatterplot) dargestellt werden. Die an die verschiedenen Fragestellungen angepasste bildliche Darstellung, wird von der zeitlichen Trennung der Messung und der Auswertung ermöglicht, da die Messdaten von allen detektierten Parameter zuerst für jede Zelle einzeln in einen Datenspeicher gespeichert werden, die nach der Messung analysiert werden können. 5.2.1.1. Histogramm Ein einzelner Parameter, im Allgemeinen die Intensität eines Fluoreszenz-Signals, wird in der Funktion der Zellanzahl dargestellt. Mit der Auswertung von Histogrammen kann man über intra- oder extrazelluläre Marker von definierten Zellpopulationen, qualitative bzw. quantitative Informationen erhalten (z. B. die Anwesenheit oder die Stärke der Exprimierung eines Membranproteins). (Abbildung 5.4.) Abbildung 5.4. Histogramm. Auf der y-Achse wird die Zahl von Zellen aufgetragen, die mit einer bestimmten Intensität (entsprechend einer bestimmten Intensitätsstufe oder „Kanäle“) analysiert wurden. Der Vergleich verschiedener Histogramme kann auf spektakulärer Weise durch Überlagerungsdarstellungen (histogram overlay) ermöglicht werden. In diesem Fall werden die MessErgebnisse von den gleichen fluoreszierenden Markierungen unterschiedlicher Proben in dem gleichen Koordinatensystem visualisiert, so dass die Signalintensitätsabweichungen (d.h. die Veränderungen auf Ebene der Protein-Expression) auf den ersten Blick erkannt werden können. (Abbildung 5.5.) Abbildung 5.5. Überlagerungsdarstellung (histogram overlay) Die dreidimensionale Abbildung von „overlay” Histogrammen machen einen ganz kleinen Unterschied auch sichtbar. (Abbildung 5.6.) Abbildung 5.6. 3D-overlay Histogramm 5.2.1.2. Dot plot/Punktwolkendiagramm Im Punktwolkendiagramm können zwei detektierte Parameter (Parameter-Kombination) von denselben einzelnen Zellen dargestellt werden. Durch Auftragung z. B. der Streulichtarten zu einem spezifischen Parameter (ggf. einem Zelloberflächenmarker) auf eine Ordinate und eine Abszisse, können diese Parameter dargestellt und voneinander differenziert werden. 82 Das Bild zeigt die einzelne Zelle als getrennten Punkt. Die Verwendung der Punktwolkendiagramme ermöglicht den gleichzeitigen Nachweis verschiedener Parameter (z. B. Co-Expression) und dadurch können verschiedene Populationen und Untergruppen präziser gefunden werden. (Abbildung 5.7.) Abbildung 5.7. Dot plot Die Differenzierung der schwer zu trennenden Populationen wird mit der Höhenliniendarstellung (contour plot) oder den Dichtediagrammen (density plot) erleichtert, die die spezielle Darstellung von den zwei Parametern sind. (Abbildung 5.8.) Abbildung 5.8. Dot plot, density plot und contour plot 83 5.2.1.3. Kinetische Messungen (time-based collection) Die Zeit, als detektiertes Parameter, kann für kinetische Messungen, wie Enzymaktivität oder für die Charakterisierung der Dynamik eines biologischen Vorganges, wie z.B. die Bindung fluoreszierender Liganden eingesetzt werden. Da die Fluoreszenz-Intensität bei den kinetischen Messungen wesentlich von der verstrichenen Zeit nach der Stimulation beeinflusst wird, wurde die sog. time-window Analyse (oder fixed time flow cytometry) wegen der Vergleichbarkeit eingeführt. Dementsprechend muss die Messung sehr genau, nach der Stimulation oder Behandlung, durchgeführt werden. Insbesondere, wenn man eine schnelle Zellantwort detektiert. Die Dauer der Messungen ist dabei auch definiert. Bei der Darstellung kinetischer Messungen wird die Zeit auf der Ordinatenachse, die FluoreszenzIntensität auf der Abszissenachse repräsentiert. 5.3. Die Auswertung der FACS-Messdaten 5.3.1. Grösse und Granularität Über die Morphologie, Grösse und Granuleninhalt, enthält das auf der Zelloberfläche und auf den Zellkomponenten streuende Licht Informationen. Die Lichtstreuung wird in der Verlängerung der Richtung des Laserstrahls mit einer Fotodiode gemessen. Die in flachem Winkel streuende Lichtstrahlen (Vorwärtsstreulicht, engl. forward scatter [FSC]; FALS = forward angle light scatter) sind mit der Zellgrösse proportional. Das in höheren Winkel streuende Licht, das von den Zellorganellen gestreut wird, wird mit einem Photoelektronenvervielfacher im rechten Winkel zur Achse des Laserstrahls (bei 90° zum einfallenden Licht) (PMT = photomultiplier tube) detektiert und Seitwärtsstreulicht (SS = SSC = side scatter; RALS = right angle light scatter) genannt. Das FSC/SSC-Dotplot (oder Streuungsbild) ist die Abbildung von dem Vorwärtsstreulicht und dem Seitwärtsstreulicht, mit dem man über jede einzelne Zelle Informationen über ihre Zellgröße in der Funktion der Granularität erhält. Bei der Analyse einer FSC/SSC-Dotplot können nicht nur die Zusammensetzung der Zellsuspension (Heterogenität) und die Anteile verschiedener Populationen (Leukozytenpopulationen in dem peripheren Blut) bestimmt werden, sondern auch morphologische Veränderungen der Zelle welche Informationen über ihren funktionellen Zustand geben (z.B, Viabilität, Zelltod, Aktivierung, Zellteilung). Es kann sogar pathologische Vorgänge beweisen, wie zum Beispiel die Präsenz von Blasten im Blut. (Abbildung 5.9.) Abbildung 5.9. Die Bedeutung des Streuungsbildes (FSC/SSC dot plot) in pathologischen Zustände. Gesundes (A) und leukämisches (B) Knochenmark. 84 Punktwolkendiagramme sind auch die Grundlage für weitere Untersuchungen, da sie für die morphologische Definition von Populationen und für das Gating (die Anwendung vordefinierter Regionen zur Auswahl von Zellen in einer erneuten Darstellung) verwendet werden. (Abbildung 5.10.) In dieser Region bzw. dem durch diese Region definierten logischen Gate können die gefragten Zellen fokussiert untersucht werden. Abbildung 5.10. Gating. Die detektierten Partikeln werden entsprechend des „R1 gate“ Bereiches auf dem rechten Bild dargestellt. Die Verteilungsmuster von Zellen können durch Gating aufgrund ihrer Eigenschaften aus verschiedenen Populationen definiert werden. 5.3.2. Fluoreszenz Informationen über Zellen können durch Fluoreszenz-Markierung vervielfältigt werden. Die Fluoreszenz, als Phänomen bedeutet, dass ein Stoff nach Anregung bzw. beim Übergang eines elektronisch angeregten Systems in einen Zustand niedrigerer Energie, Licht emittiert. Die Fluorophore (oder Fluoreszenzfarbstoffe) sind solche Moleküle, die die Fähigkeit haben, Licht einer gegebenen Wellenlänge zu absorbieren und dadurch angeregt werden und nach der Beleuchtung (Exzitation) Licht emittieren (Emission). In der Regel ist das emittierte Licht immer energieärmer als das vorher absorbierte und dementsprechend mit längerer Wellenlänge charakterisiert (Stokessche Regel, s. Biophysik). Viele Fluoreszenzfarbstoffe können eingesetzt werden von denen ein Teil direkt und spezifisch an bestimmte Zellkomponente bzw. Moleküle bindet, andere in Antikörper-gekoppeltem Zustand für Immunfärbung. Die Anzahl von Fluoreszenzfarbstoffen wird sich durch Erweiterung mit eingebauten Laserlichtquellen (die Möglichkeit der Anregung auf mehreren Wellenlängen) und durch Entwicklung von neuen, z.B. aus zwei Fluorophormoleküle zusammengesetzten Tandem-Farbstoffe (TandemKonjugate) ständig steigern. Das Prinzip dieser Konjugate ist es, zunächst den ersten Farbstoff (sog. Donor; z. B. PE, APC) des Tandems durch den entsprechenden Laser anzuregen. Das anschließend emittierte Licht regt wiederum den zweiten Farbstoff (Akzeptor) an, dessen emittiertes Licht dann erst detektiert wird. Diese Konjugate ermöglichen die Erweiterung des Farbstoff-Spektrums und deshalb die Analyse von mehr als vier Farben pro Färbung. (Tabelle 1.) Anregungslicht: Blauer Laser (488 nm) FITC (Fluoresceinisothiocyanat) Alex Fluor 488 PE (Phycoerythrin) PI (Propidium-Iodid) PE-TX red (ECD; Phycoerythrin –Texasrot Tandem) PE-Cy5((Phycoerythrin-Cy5 Tandem) PerCP (Peridinin-chlorophyll-protein complex) PerCP-Cy5.5 (Tandem) PE-Cy7 (Tandem) Anregungslicht: roter Laser (633 nm) APC (Allophycocyanin) APC-Cy7 (Tandem) Anregungslicht: UV/violetter Laser (633 nm) Hoechst 33258 Hoechst 33342 Indo-1 Alexa Fluor 405 Pacific Blue Tabelle 1. Die häufigsten verwendeten Fluoreszenzmoleküle in Durchflusszytometrie Bei den diagnostischen Messungen ist das Ziel hauptsächlich, die Anwesenheit und die Menge (Anteil) von verschiedenen Zellpopulationen nachzuweisen, oder seltener den funktionellen Zustand zu charakterisieren. Im Allgemeinen wird dies durch Immunphänotypisierung, d. h. durch zelloberflächen-Markierung oder intrazelluläre Markierung, die auf der Verwendung von Fluorophorkonjugierten monoklonalen Antikörpern, derer Spezifizität auf den Antigen-Antikörper Reaktionsprinzip beruht, durchgeführt. 86 Bei der Anwendung eines Fluorophors werden die detektierten Signale in einem Histogramm zusammengefasst. Aufgrund der Fluoreszenz-Intensität kann abgelesen werden, ob an die gefragten Zellen ein zugegebener Antikörper gebunden hat. Dies liefert wichtige biologische Informationen und ob die Zellen ein bestimmtes Antigen exprimieren (ja oder nein Antwort; z.B. Differenzierungs- oder Aktivierungsmarkern). Die nummerische Auswertung (Berechnung von Flächeninhalten unter Kurven) reflektiert den prozentualen Anteil (%) von positiven (das gefragte Protein exprimierenden) und negativen (nicht-exprimierenden) Populationen. Das Intensitätsmaximum hängt von der Menge gebundener Fluoreszenzfarbstoffe und indirekterweise von der Menge gebundener Antikörper ab. Das bedeutet, dass je höher das detektierte Fluoreszenzsignal ist, desto mehr gefragte Antigene werden von den Zellen exprimiert. Durch die Anwendung von Kalibrierungsperlen mit bekannter Menge von Fluoreszenzstoffen, kann die Proteinexprimierung quantifiziert werden. (Abbildung 5.11.) Abbildung 5.11. Absolut Quantifizierung mit einer Kalibrierungskurve. Da die richtige Identifizierung bestimmter Zellgruppen nur durch die Messung mehrerer Parameter (mehrere Marker: Protein-Expressionsmuster) erfolgen kann, wird die einfarbige Markierung immer öfter durch die parallele Verwendung von Fluoreszenzmolekülen welche unterschiedliche Wellenlängen emittierenden, ersetzt. Bei der mehrfarbigen Markierung werden die detektierten Fluoreszenzsignale in einem Punktwolkendiagramm dargestellt. Auf den Punktwolkendiagrammen können, ähnlich zu den Histogrammen, die Anwesenheit der Exprimierung (ja oder nein Antwort) und der prozentuale Anteil an Zellen, die auf die gefragten Antigen positiv sind, sichtbar gemacht werden. Mit der Bestimmung der Fluoreszenzintensität wird das Expressionsniveau und dadurch die CoExpression aufgeklärt (Abbildung 5.12.). Da die Fluorophoren kein Licht einer diskreten Wellenlänge, sondern in einem breiten Band emittieren, muss man mit spektralen Überlappungen rechnen. Solche Überlappungen der Farbstoffe müssen nach dem Messvorgang durch Kompensation korrigiert werden. 87 Abbildung 5.12. Immunphenotypisierung. Neben der Co-Expression muss man mit der Stärke der Expression einer bestimmten Marker rechnen. 5.4. Probenvorbereitungen für diagnostische Messungen 5.4.1. Herstellung einer Zellsuspension Da mit einem Durchflusszytometer in einem Flüssigkeitsstrahl vereinzelte Partikel gemessen werden, muss man im Allgemeinen aus jeder biologischen Probe die eingesetzt werden soll, eine Suspension gewinnen. (natürlich nicht aus Zellen, die sich schon in einer Suspension befinden, wie z. B. im peripheren Blut, dem Knochenmark-Aspirat, Liquor, Urin). Bei einem soliden Gewebe ist der erste Schritt die Vorbereitung der Zellen die in Suspension gebracht werden sollen (Vereinzelung). Dafür sind viele Verfahren bekannt; von der mechanischen Separierung bis zur enzymatischen Verdauung. Es hängt von der Sensitivität der Zellen, der Struktur des Gewebes usw. ab, welches Verfahren angewandt wird. Während der Isolation muss man jedoch berücksichtigen, dass die gewonnenen Zellen in einem verhältnismäßig intakten Zustand zu bewahren sind. (Vor allem ist Vorsicht geboten, die Epitope der Zelloberflächenproteine nicht zu zerstören und die Struktur, evtl. die korrekte Funktion der Zellen, zu bewahren.) 5.4.2. Erythrolyse Bei der Vorbereitung der Proben aus dem peripheren Blut oder aus Knochenmark-Aspirat es ist erforderlich, die in der grössten Menge vorkommenden Blutkörperchen durch Lyse zu eliminieren. 88 Zahlreiche Verfahren sind bekannt: Behandlung mit einer hypotonischen Lösung oder mit Ammoniumchlorid. 5.4.3. Fluoreszenz-markierung Die Markierung mit einem Fluoreszenzmolekül kann auf zwei technisch möglichen Weisen erfolgen: 1) direkte Färbung: wenn das fluoreszierende Molekül an einer Zellorganelle oder an einem Molekül spezifisch bindet, oder 2) Immunphänotypisierung: wenn eine Struktur (ein Antigen) mit einem Fluoreszenzmolekül-gekoppelten Antikörper detektiert wird. 5.4.3.1. Direkte Färbung In der klinischen Praxis ist das meist verbreitete direkte Verfahren die Propidiumiodid (PI)-Markierung des DNA-Gehaltes. PI wirkt wie Ethidiumbromid als Nukleinsäureinterkalator und es kann stöchiometrisch (1PI pro 5 Basenpaaren) an die doppelsträngigen DNA und RNA Moleküle binden. Auf diese Weise (nach RNase-Behandlung) kann der DNA-Gehalt quantitativ bestimmt werden und dadurch eine Zellzyklusanalyse durchgeführt werden (Abbildung 5.13.). Abbildung 5.13. Zellzyklusanalyze mit Propidiumiodide Durch die direkte Markierung kann der funktionelle Zustand der Zellen auch in vielen Fällen charakterisiert werden, wie z.B. die Enzym-Aktivität mit fluoreszierenden Substratmolekülen, die Apoptose durch die Detektierung von veränderten Lipidzusammensetzungen der Plasmamembran, oder die ROI (reaktive Sauerstoff Arten) Produktion von Granulozyten welche mit der Hilfe von auf Sauerstoffradikale empfindlichen Fluorophoren bestimmt werden kann. 89 5.4.3.2. Immunphänotypisierung Bei der Färbung der Zelle mit Fluorophor konjugierten Antikörpern ist die Spezifizität durch die Antigen-Antikörper Reaktion gesichert. Aufgrund der Eigenschaften der Fluorophore und durch die Spezifizität gebenden monoklonalen (selten polyklonale) Antikörper, können direkte und indirekte Techniken unterschieden werden. Im Fall der direkten Technik wird ein Fluorophor an den gegen das Antigen spezifischen (primäre) Antikörper, gebunden. Bei der indirekten Technik wird der primäre Antikörper nicht markiert, weswegen die Anwendung eines weiteren, markierten (sekundären) Antikörpers erforderlich ist. Dieser richtet sich gegen den ersten Antikörper, um die Bindung zu visualisieren. Eine spezielle Möglichkeit ist die Verwendung eines mit Biotin-verbundenen primären Antikörpers wobei ein Fluorophor-konjugiertes Streptavidin für die Sichtbarmachung zugegeben wird. (Abbildung 5.14.) Abbildung 5.14. Direkte und Biotin/Streptavidin vermittelte Markierung. Beide Anwendungen haben Vorteile: bei dem direkten Verfahren gibt es eine geringere Wahrscheinlichkeit für unspezifische Bindungen, aber das Signal ist stärker bei der indirekten Färbung, da an einem antigenspezifischen Antikörper mehrere sekundäre gleichzeitig binden können. In der klinischen Praxis trifft man am öftesten auf die Immunphänotypisierung, bei der die Zellen aufgrund des Proteinmusters auf den Zelloberflächen und auch dem innerhalb der Zellen, identifiziert und charakterisiert werden. Die während Untersuchungen der weißen Blutzellen entdeckten Antigene werden anlässlich der „Internationalen Arbeitsgemeinschaften“ seit den 1980er Jahren (International Workshops on Human Leukocyte Differentiation Antigens) verglichen und kategorisiert. Die eingeordneten Antigene bekommen wegen der leichteren Identifizierung eine Nummer, die mit der Abkürzung für „Cluster of Differentiation” gekoppelt wird. Der neunte Workshop wurde in Barcelona 2010 veranstaltet, nachdem die Anzahl von derzeitig identifizierten und eingeordneten Antigenen 363 beträgt (CD363=S1PR1 [sphingosine-1-phosphate receptor 1]). (Tabelle 2.) 90 Myeloidische Zelle Stammzelle-Marker Granulozyt Monozyt Megakariozyt Lymphozyten Erytrozyt TZelle B- Zelle CD34 intrazell. MPO CD14 CD41 Glycophorin A CD2 CD19 CD117 CD13 HLA-DR CD61 CD71 CD3 CD20 CD133 CD33 CD4 CD42a CD4 CD22 CD15 CD13 CD33 CD8 HLA-DR CD11b CD33 CD13 CD5 CD79a CD7 (CD10) CD16 Tabelle 2. Die wichtigste CD Markern 5.5. Die Anwendungsgebiete der Durchflusszytometrie Im Folgenden werden die Anwendungsgebiete nach zwei Annäherungen präsentiert: 1) auf der Methodik beruhende Verteilung und 2) klinische Anwendungen. 5.5.1. Auf der Methodik beruhende Verteilung (bei der aufgereihten Verteilung wurde keinen Anspruch auf Vollständigkeit erhoben.) 5.5.1.1. Proteinnachweisverfahren Mit der FACS-Methodik ist es also möglich, Proteine direkt oder indirekt nachzuweisen. Die spezifische Detektierung beruht auf Antigen-Antikörper-Bindungsprinzip, weil die Proteine (als Antigene) mit Fluoreszenzstoff markierten Antikörpern markiert werden (Immunphänotypisierung). Ein unspezifischer Nachweis lässt sich mit der in situ Detektierung von Amino- und Thiol-Gruppen erzielen, die nur über die Anwesenheit, Menge und Lokalisierung von Proteinen informativ ist, aber nicht über die Zusammensetzung. Die beiden Verfahren sind auch für die Charakterisierung von Rezeptor-Ligand-Wechselwirkungen geignet bzw. die Thiol-reaktiven Stoffe sind verwendbar für die Feststellung der Konformationsveränderungen der Proteine. Spezielle Möglichkeiten wurden durch die Entdeckung des grün fluoreszierenden Proteins (Abkürzung GFP; engl. green fluorescent protein) erreicht, das sich auch bei der Überprüfung des Transfektionswirkungsgrades bewährt hat. Bei der Transfektion (das Einbringen von Fremd-DNA oder RNA in eukaryotische Zellen) wird das GFP-kodierende gfp Gen direkt vor dem Stop-Kodon gefragter Proteine eingebaut und auf diese Weise wird das transkribierte Gen mit dem gfp-Gen zusammen exprimiert (GFP-Fusionsproteine) und erscheint als fluoreszierendes Protein in der Zelle. Die Anwesenheit der Fluoreszenz beweist eine erfolgreiche Expression gefragter Proteine. Wird die gfpcDNA nach einem Promotor-Bereich eingebaut, wird das GFP immer sichtbar, sobald der Promotor aktiviert wird und dadurch die Steuerung der GenExpression untersucht werden kann. Die Co- 91 Transfektion mit GFP-Reporterplasmid hat noch einen weiteren Vorteil, nämlich dass die erfolgreich transfektierten Zellen sortiert werden können. Für die Diagnosestellung oder der prognostischen Beurteilung von einigen pathologischen Zuständen muss das Zelloberflächen-Expressionsniveau bestimmter Proteine aufgeklärt werden. In bestimmten Fällen es ist genug, wenn man die Unterschiede zwischen den pathologischen und gesunden Zellen findet, wie z.B. bei dem Leukozyten-Adhäsionsdefekt Typ I. (LAD-I; Mutationen im ITGB2-Gen (21q22.3), das für das Beta-2-Integrin/CD18 kodiert, das mit CD11a bildet LFA-1) eine verringerte Expression von CD11b und CD18, bei dem Typ II. (extrem selten) das Sialyl-Lewis XBlutgruppenantigen ist nicht auf den neutrophile Granulozyten nachgewiesen werden können. Bei der Spondylitis ankylosans (Bechterewsche Krankheit) im Gegenteil, wurde eine enge Assoziation mit der Präsenz von HLA-B27 gefunden. Es sind auch klinische Zustände bekannt, in denen die absolute Menge eines Proteins maßgebend ist, z.B. in Sepsis hat die quantitative Bestimmung von HLA-DR auf den Monozyten und CD64 (FcγRI) auf den Granulozyten eine Bedeutung in der Beurteilung des Verlaufs. Bei der quantitativen Bestimmung werden fluoreszierende Kalibrierungsperlen bekannter Menge benutzt. 5.5.1.2. Untersuchungen der Synthese Die fluoreszierenden Bausteine (z.B. Nukleotide, Monosaccharide, Lipidsäure, usw.) oder in den Zellen physiologischerweise fehlende Stoffe können für die Beobachtung der Bildung neuer Moleküle verwendet werden. Dazu gehört z. B. die Inkorporierung von Timidin-Analoga (wie BrdU = Bromdesoxyuridin, EdU = 5-ethynyl-2´-deoxyuridin) mit denen eine Zellzyklusanalyse durchgeführt werden kann. (Abbildung 5.15.) Abbildung 5.15 Darstellung des Zellzyklus mittels BrdU-Färbung. Vor (blau) und nach (grün) der Stimulation. In dem vordefinierten Lymphozyten-Bereich (R1 Gate) werden die CD4 und CD8 positiven Zellen dargestellt. Die Histogramme (unten) zeigen die BrdU-Inkorporierung von den entsprechenden positiven Zellen (in R3 und R2 weitergeführt). 92 5.5.1.3. Enzymaktivität-Messungen In lebenden Zellen, kann die Enzymaktivität mit fluoreszierenden Substratmolekülen in situ durchgeführt werden. Wenn man mehrere Parameter gleichzeitig in Betracht zieht, kann mit der Identifizierung einer bestimmten Zellpopulation, die Enzymaktivität Zelltyp-spezifisch bestimmt werden. Heutzutage stehen solche Fluoreszenzproben zur Verfügung für Glykosidase, Phosphatase, Peptidase, Protease und Oxidase. Diese Messungen ermöglichen die umfassende Charakterisierung der Zellfunktion (metabolische Wege, Signalübertragung, Aktivierung usw.) (Abbildung 5.16). Abbildung 5.16. Ein Bespiel für eine Enzymaktivität-detektierende Fluoreszent-Probe: Ein Karboxyderivat von Fluoreszein, das Karboxy-H2DCFDA. Damit lassen sich die reaktiven Sauerstoffradikale quantitativ messen. (http://probes.invitrogen.com/media/pis/mp36103.pdf) 5.5.1.4. Untersuchung der Nukleinsäure Der DNA- und RNA-Gehalt wird mit fluoreszierenden Nukleinsäure-Farbstoffen (anlässlich des Zellzyklus oder Apoptose) bestimmt. Die nicht-permeablen Farbstoffe können auch die Viabilität (Lebensfähigkeit; s. 3.19.) kennzeichnen, weil sie nur in den sterbenden Zellen ins Zytoplasma gelangen können. In der Diagnostik wird die Bestimmung des DNA-Gehaltes am häufigsten für die Schätzung der Teilungsfähigkeit von Tumorzellen oder ihrer Zelltodrate nach der Behandlung angewandt. Die Retikulozyten lassen sich aufgrund ihres RNA-Gehaltes im Blut identifizieren. Der 93 Nachweis der Nukleinsäuren hat eine mikrobiologische Bedeutung, einerseits weil die Kenntnisse über die Viabilität von Erregern auf die Prognose hinweist, anderseits weil der Tod der infizierten Zellen (durch Apoptose oder Nekrose) auch detektiert werden kann. 5.5.1.5. Untersuchung der Zellorganellen Viele unterschiedliche, für Durchflusszytometrie adaptierte und spezifische Nachweisverfahren von Zellorganellen sind bekannt, z. B. wird mit Fluoreszenzmarkierung die in vivo Umgestaltung der Zytoskelette untersucht und mit fluoreszierenden Lipid-Analoga wird die Arbeitsweise von Plasmamembran- und zytoplasmatischen Membransystemen beobachtet. Mit Fluoreszenzstoffmarkierten Fettsäuren können auch die Signalübertragungswege und Zellaktivierung untersucht werden. 5.5.1.6. Funktionelle Untersuchungen Wegen der Vielfältigkeit der funktionellen Untersuchungen werden hier die Möglichkeiten nur aufgereiht. Die detailierten Erklärungen unterschiedlicher Methoden können in der Fachliteratur gefunden werden. Gezielte Lebensvorgänge: Viabilität Zellteilung/Zellzyklus Apoptose (Kaspase-Aktivität, Messung des mitochondrialen Membranpotentials, TUNEL = Terminal DeoxynucleotideTransferase dUTP Nick End Labeling Method) Proteinsynthese (z.B. Zytokineproduktion) Phagozytose/Endozytose Signalübertragung (z.b. fluoreszierende Fettsäure, anti-Phosphoprotein Antikörpern, usw.) zytoplasmatische Ionenkonzentration (ionenempfindliche fluoreszierende Farbstoffe) Funktionsfähigkeit der Transportproteine (mittels fluoreszierender Substrate z.B. MultidrugRezistenz = MDR) Reaktive Sauerstoffradikale (ROI) und Stickstoff-monoxide (NO) 2+ 2+ 2+ Messung von Metallionen (Ca , Mg , Zn Indikatoren) pH- Messung Messung des Membranpotentials Kinetische Messungen (z.B. zeitliche Bestimmung der Enzymaktivität) Bestimmung des Redoxzustandes (Quantifizierung der Thiol-Gruppen und DisulfideBindungen) 5.5.1.7. Bestimmung des relativen Abstandes zwischen Molekülen (FRET) Um den Abstand zwischen Moleküle oder zwischen intramolekularen Domains zu messen, werden zwei Fluoreszenzstoffe gleichzeitig verwendet. Im Rahmen des Förster-Resonanzenergietransfers (FRET) wird die Energie eines angeregten Farbstoffs, auch Donor (für die Wellenlänge des anregenden Lichtes empfindlich) genannt, auf einen zweiten Farbstoff, auch Akzeptor (für die 94 Wellenlänge des emittierten Lichtes des Donors empfindlich) genannt, übertragen. Wenn die zwei Fluoreszenzstoffe nahe genug zu einander liegen und der Donor angeregt wird, wird das Fluoreszenzlicht des Akzeptors erscheinen. Wenn die zwei Moleküle voneinander entfernt sind, kann nur dem Donor entsprechendes Licht detektiert werden. (Abbildung 5.17.) Abbildung 5.17. Prinzip der durchflußzytometrischen Messung von Rezeptorinteraktion mittels FRET. 5.5.1.8. Untersuchung löslicher Moleküle (Flow Cytometry Based Fluorescent Bead Immunoassay) An Moleküle (Proteine, Oligonukleotide, Lipide oder KGGohlenhydrate), die auf die Oberfläche synthetischer Mikroperlen gebunden werden, können verschiedene lösliche Analyte spezifisch binden. Aufgrund der Grösse der Mikroperlen und der Detektionsantikörper mit Fluoreszenzfarbstoffen unterschiedlicher Farben, können die gefragten löslichen Antigene quantifiziert werden. Der Vorteil des Verfahren ist, dass man mehrere Analyte gleichzeitig aus sehr geringen Mengen Probenmaterials, quantitativ messen kann (mehr als 10 Zytokine aus weniger als 25µl Serum). Die Empfindlichkeit ist ähnlich zu dem ultrasensitiven ELISA Verfahren (in pg/ml Bereich). (Abbildung 5.18) Abbildung 5.18a. Grundsätze der Mikroperle-Methoden 95 Abbildung 5.18b. Ein Beispiel für ein fluoreszierendes Bead-Immunoassay und eine Multiparameteranwendungen (englisch: Multiplexing). 5.5.1.9. Bestimmung der absoluten Zellzahl Durch Anwendung von Referenzperlen (als innere Kontrolle) aus Polystiren in einer bekannten Konzentration, kann die absolute Zellzahl bestimmt werden. (Abbildung 5.19.) 96 Abbildung 5.19. Die Verwendung der Referenzpartikel ermöglicht eine volumenbezogene Zählung (Absolutzählung). In diesem Experiment wurden die apoptotischen und lebenden Zellen zusätzlich getrennt (FL1: annexinV-FITC, FL2: propidium-iodide). 5.5.1.10. Zellsortierung Die Sortierung mit dem Durchflusszytometer hat viele Vorteile im Vergleich zu anderen Verfahren: Die Trennung von Zellpopulationen kann mit der Berücksichtigung von mehreren (multiparametrischen) Kriterien erfolgen. (FS/SS; Fluoreszenz). Die Geschwindigkeit der Separierung ist sehr hoch: mehr als 100000 Zelle können pro Sekunden sortiert werden und zugleich ist der Wirkungsgrad (Reinheit) höher als 95%. Die modernen Geräte können schon eine sichere Sortierung solcher Zellen durchführen, die in einer nur sehr geringe Menge vorhanden sind (<0.1% pro Untersuchungsmaterial): („rare event sorting”). Diese Möglichkeit hat eine sehr grosse Bedeutung z.B. in der Nachsorge einer Minimal-Resterkrankung (minimal residual disease) oder in der Isolierung von für die autologe Stammzelletransplantation nötigen Stammzellen oder Vorläuferzellen. Die Sortierung kann unter sterilen Umständen durchgeführt werden und die Zellen können auf diese Weise weiter gezüchtet oder klonal vervielfältigt werden. 5.5.2. 5.5.2.1. Verteilung nach Krankheiten Maligne hämatologische Erkrankungen 5.5.2.1.1. Diagnostik und Differentialdiagnostik Die Aspekte der Durchflusszytometrie für die Diagnose und Differentialdiagnose von malignen hämatologischen Erkrankungen, beruhen auf der Feststellung der zellulären Herkunft und der Reifungsstadien, die durch die Messung der Zelloberflächen- und zytoplasmatischen Antigene (CD Markern) bestimmt werden. Da die Mehrheit der Oberflächen-Antigene in mehreren Stadien einer Zellreihe, ggf. in mehreren Zellreihen exprimiert werden, sollen für diagnostische Zwecke fast immer multiparametrische Messungen durchgeführt werden. Die Methodik der Verfahren, die eine entsprechende diagnostische und differentialdiagnostische Aussagekraft haben, soll möglicherweise auch kostengünstig sein, und unter den verschiedenen Laboratorien vergleichbare Ergebnisse erzielen. Sie werden in Konsens- Protokollen zusammengefasst. Mehrere Typen von Konsens-Protokollen werden benutzt: Das Ziel des sog. „Grundpanel” ist die Bestätigung von festgestellten Diagnosen, die mit dem klinischen Bild und zytochemischen genetischen ergänzt und mit ggf. Untersuchungen werden. Immuntypisierung Für die werden relative wenige Oberflächenund zytoplasmatische Marker benötigt. Hauptsächlich die Trennung der myeloiden und lymphoiden Stämme und die Untersuchung der Expression der grundsätzlichen prognostischen dabei Marker wichtig. CD10=CALLA) ist (z.B. (Abbildung Abbildung 5.20. Diagnostisches Workflow für die LeukämieDiagnostik 5.20.). Die ergänzten Konsens-Protokolle wurden für die Feststellung einer richtigen Stadiumeinteilung (staging) ausgearbeitet. Bei der Immuntypisierung sind die für Zellreihe spezifische Differenzierungsmarker und andere Antigene, die für die Reifungsstadien bestimmter Zelltypen charakteristische, oder wichtige Prognose-Faktoren sind, wichtig. Wegen der besseren Kenntnisse über den Immunphänotyp des malignes Klons, wird die Durchflusszytometrie neben der Diagnosestellung für die Nachsorge von bösartigen hämatologischen Erkrankungen, auch für die Erhebung der therapeutischen Wirksamkeit und der frühen Entdeckung von Rezidiven angewendet. 5.5.2.1.2. Diagnostik der Minimal-Resterkrankung Die Minimal-Resterkrankung (minimal residual disease) ist ein trotz der Therapie verbliebener Zustand ohne klinische Symptome. Der Vorteil der Durchflusszytometrie ist ihre Geschwindigkeit und die sehr grosse Anzahl untersuchter Zellen (500.000 – 1.000.000 / Untersuchung). Ihr Nachteil ist die geringere Empfindlichkeit im Vergleich zu genetischen Verfahren. 5.5.2.1.3. Nachweis der rezivierenden hämatologischen Erkrankungen Obwohl die molekularbiologischen Methoden für den Nachweis eines Rezidivs einer Leukämie wichtigere Rollen spielen, ist die multiparametrische Immunphänotypisierung vernachlässigen. Ihre Vor- und Nachteile darin sind die gleichen wie vorher. 98 nicht zu 5.5.2.1.4. Untersuchung der Empfindlichkeit auf die Terapie (MDR = multidrug resistance) Es gibt mehrere Möglichkeiten, wenn man die Wirksamkeit einer Behandlung beurteilen möchte. Folgendes kann man untersuchen: 1) die Menge von malignen Zellen, auf denen die Diagnose beruht und die Proportion der bösartigen und gesunden Zellen (durch Immuntypisierung), 2) die Präsenz der mit Medikamenten- Resistenz zusammen- hängenden proteine Membran- (durch Immun- typisierung), oder 3) die Beurteilung der Pump- funktion von Proteinen, die für den Resistenz verant- Abbildung 5.21a. MDR Membranprotein Immuntypisierung. wortlich sind. (Abbildung 5.21.) Abbildung 5.21b-c. Funktionelle Charakterisierung eines MDR-Proteins. 99 5.5.2.2. Immundefizienz-Syndrom Die Immundefekte die sind durch quantitative und/oder funktionelle Störungen einer oder mehrerer Zelltypen des Immunsystems entwickelt haben, sind durch eine vorübergehende oder irreversible Schwächung der Abwehrfunktion gekennzeichnet. Je nach ihrem Entstehen gibt es angeborene und erworbene Formen. Die Symptome werden von morphologischen oder/und funktionellen Veränderungen der Immunzellen verursacht. Als Folgen treten Infektionskrankheiten häufiger auf, oder sie verlaufen erschwert. Die Ziele der Untersuchung des Immunsystems: 1. Diagnosestellung (Nachweis der Immunkrankheit) 2. Charakterisierung der Erkrankung 3. Der Verlauf der Krankheitsaktivität 4. Die Monitorisierung der therapeutischen Antwort 5.5.2.2.1. Phänotyp-Untersuchungen und Nachfolge in der Diagnostik von Immundefekten Die Bestimmung der relativen Anteile von zirkulierenden Lymphozyten-Subpopulationen (T- bzw. B-Zelle, CD4:CD8 Anteil, usw.) Die Charakterisierung Anwesenheit der von zirkulierenden Gedächtniszellen, Lymphozyten-Subpopulationen Identifizierung von aktivierten (z.B. Lymphozyten, Anwesenheit der Antigen-spezifischen Lymphozyten, usw.) Verlaufsmonitorisierung einer HIV-Infektion (die absolute Anzahl CD4-positiver Zellen bei HIV Infizierten hat eine therapeutische Bedeutung. Die antivirale Behandlung von einem, an einer HIV-Infektion leidenden Patienen, muss nach den WHO-Richtwerten (World Health Organisation) von 2010, dann beginnen, wenn die absolute Menge der CD4+ Lymphozyten auf unter 350 Zelle/mm3 abgefallen ist.) Die Funktionstests der Phagozyten Die Messung der ROI (reaktive Sauerstoff-Radikalen)-Produktion Die Untersuchung der Fähigkeit der Zytokine-Produktion Die Untersuchung der zytotoxischen Aktivität von natürlichen Killerzellen (NK-Zellen) usw. 5.5.2.3. Autoimmunerkrankungen Die Durchflusszytometrie kann für die Untersuchung pathologischer Autoimmunität, die durch eine gegen die körpereigenen Antigene gerichtete und zur Zellschädigung leitende Immunantwort gekennzeichnet ist, mehrfach angewendet werden. Durch die Präsenz zirkulierender Autoantikörper, der Anzahl und des funktionellen Zustandes von den Zellen, die an dem Autoimmunprozess teilnehmen und auch durch deren Aktivität, kann der Verlauf beurteilt und monitorisiert werden. 5.5.2.3.1. Die Diagnostik der Autoimmunerkrankungen Eine mögliche Methode den zirkulierenden Autoantikörper nachzuweisen, ist die Detektierung mit Hilfe von Mikroperlen. Die Autoantikörper in dem Serum von dem Patienten werden auf die Oberfläche von mit Autoantigen-bedeckten fluoreszierenden Kunststoff-Mikroperlen, 100 gebunden und mit auf unterschiedlichen Wellenlängen emittierenden Fluorophor konjugierten anti-human IgG-Antikörpern, sichtbar gemacht. Da die fluoreszierenden Partikel je nach Fluoreszenzintensität unterschieden werden, ist die Messung mehrerer Autoantikörpern gleichzeitig möglich (pl. dsDNA, SS-A, SS-B, Sm, Sm/RNP, Scl70, Jo-1, usw.). Die Vorteile sind 1) ihre Geschwindigkeit (es ist nicht nötig, auf mehrere Patientenproben zu warten, wie bei den ELISA-Systemen), 2) die notwendige Menge der Proben ist gering (~25-30 µl) und 3) es gibt die Möglichkeit, mehrere Autoantikörper simultan (bis hin mehr als 10) nachzuweisen. Eine spezielle Möglichkeit, die zirkulierenden Antikörper zu untersuchen ist es, die Antikörper aus dem Patientenserum auf die aus gesunden Individuellen isolierte Zellen zu binden und sie dann mit Fluorophor konjugierten anti-human Immunglobulinen zu visualisieren. In Autoimmunthrombozytopenie (ITP) wird dieses Verfahren für den Nachweis von Autoantikörpern, die auf den Thrombozyten spezifisch gebunden sind, verwendet. 5.5.2.3.2. Die Bestimmung der Aktivität von Autoimmunerkrankungen Die Bestimmung von bestimmten Zellpopulationen (Anteil, Absolutwerte und funktioneller Zustand), die in einem Autoimmunprozess beteiligt sind (wie regulatorische T-Zellen, Th17-Lymphozyten, usw.), kann bei der Beurteilung der Prognose helfen. Es muss aber betont werden, dass die Charakterisierung von Lymphozyt-Subpopulationen im Blut nicht immer die typische Lokalisierung einer Krankheit reflektiert. Deswegen ist es sinnvoll, die Zellzusammensetzung der Gelenkflüssigkeit bei rheumatischer Arthritis und von Liquorproben bei Multiple Sklerose zu untersuchen. Heutzutage beginnt man die Bedeutung von den aus aktivierten und sterbenden Zellen freigesetzten Partikeln, die sogenannten Mikrovesikeln, zu erkennen. (Durch ihre Oberflächenproteine kann man auf ihren Ursprung rückschließen) Mehrere Publikationen deuten darauf hin, dass sich ihre Menge in aktiven Autoimmunprozessen erhöht, und ihre Präsenz auch als Biomarker verwendet werden kann. Das meist verbreitete Untersuchungsverfahren dafür ist auch die Durchflusszytometrie. 5.5.2.4. Die Organtransplantation Eine grundsätzliche Voraussetzung einer erfolgreichen Organtransplantation ist die Verhinderung einer immunologischen Abstoßungsreaktion (Rejektion). Die immunsuppressive Behandlung macht den Patienten für Infektionen empfindlich. Mit Hilfe der Durchflusszytometrie können die immunologischen Abwehrreaktionen der transplantierten Patienten auf mehreren Stufen beurteilt werden: 1) die alloreaktiven Antikörper können nachgewiesen und klassifiziert werden. Auch können Donor-Rezipient Paare, mit erhöhtem Risiko für eine Abstoßungsreaktion, identifiziert werden; 2) die Komponenten der zellulären Immunantwort (Rejektion versus Infektion: Differenzialdiagnose) können aufgeklärt werden; 3) der Erfolg der Transplantation durch eine TZR-Analyse kann bestimmt werden. 5.5.2.4.1. HLA-Assoziation Da eine enge Assoziation zwischen einigen Erkrankungen und HLA-Haplotypen bekannt ist, hat die Bestimmung der Oberflächen-Expression von HLA-Haplotypen eine klinische Bedeutung. In der alltäglichen Praxis ist die FACS-Analyse der HLA-B27 Expression auf den Lymphozyten am meisten 101 verbreitet. Sie wird hauptsächlich bei der familiären Vermehrung der Bechterewschen Krankheit (Spondylitis ankylosans) verwendet. 5.5.2.5. Infektionen Die Methoden, die auf dem Nachweis von Nukleinsäure beruhen, bilden heute einen wichtigen Teil der alltäglichen mikrobiologischen Diagnostik. Neben ihren Vorteilen (sie beschleunigen die Feststellung der Diagnose, erhöhen die Spezifizität und ermöglichen die Identifizierung jener Pathogene, die gar nicht oder nur ganz schwierig vermehrt werden, wie z.B. Mycobacterium tuberculosis, Chlamydia trachomatis) darf man nicht vergessen, dass einerseits der Nachweis der Nukleinsäure nicht gleichzusetzen mit der Anwesenheit der replikationsfähigen Keime ist, anderseits ermöglichen diese Verfahren keine rasche Orientierung über das Immunsystem des Wirtes! Gerade wegen des Ersatzes von diesen Funktionen ist die Durchflusszytometrie ein nützliches Werkzeug der mikrobiologischen Diagnosestellung geworden. Der grösste Vorteil von Durchflusszytometrie ist im Vergleich mit anderen auf Nukleisäure-nachweis beruhenden Techniken die individuelle, auf der Stufe der Zelle erfolgte Detektierung. Wegen ihrer Schnelligkeit (die Probe kann 2 Stunden nach dem Empfang Ergebnisse liefern) hat sie große Bedeutung für den Nachweis von langsam wachsenden Mikroben, wie Mycobakterien und Pilzen. Mit der entsprechenden Methode (Probenvorbereitung) werden die Erregern genau identifiziert, womit sie besonders erfolgreich ist in der Diagnostik von gemischten Infektionen. Nicht zuletzt kann man weitere Informationen über das Immunsystem des Wirtes (durch den Nachweis spezifischer Antikörpern oder durch die Detektierung einer zytotoxische Immunantwort) und von der anti-mikrobiellen Behandlung erhalten. Anwendungsmöglichkeiten der Durchflusszytometrie: Identifizierung der Erreger: 1. Heterogenitätsuntersuchungen (Lichtstreuung; spezifische Fluorophore, fluoreszierende Gram Färbung) 2. Monoklonale Antikörper in der mikrobiologischen Diagnostik: Immunphänotypisierung 3. Die Untersuchung der Wechselwirkung zwischen den Mikroorganismen und dem Wirt 4. Identifizierung mit Hilfe von Fluoreszenzfarbstoff-konjugierten Oligonukleotiden bzw. mit dem auf die Nukleinsäure bindende Fluorophoren. Die Untersuchung des physiologischen Zustandes von Mikroorganismen 1. Viabilitätsuntersuchungen 2. Funktionelle Untersuchung der Mikroben (Messung des Membranpotentials oder der Enzymaktivität usw.) Serologische Untersuchungen Empfindlichkeitstestung für Antibiotiken Separierung von Mikroorganismen (Sortierung) Beurteilung des Immunstatus 102 Absolute Indikationen Relative Indikationen Diagnostik und Differentialdiagnostik von malignen Nachfolge hämatologischen Erkrankungen Autoimmunerkrankungen MDR-Testung Nachfolge von Infektionszustände Nachfolge einer HIV-Infektion Nachweis von HLA-Assoziation Vorbereitung und Nachfolge einer Transplantation Tumorprognostik (DNA-Gehalt/Zellzyklus) Nachfolge der Immundefekten Bestimmung der Retikulozyt-Anzahl PNH Diagnostik (CD55, CD59 Expression; der Aktivität von Untersuchung der Thrombozyt-Funktion paroxysmalen nächtlichen Hämoglobinurie; PIG-AGen, das kodiert für das Enzym N- Acetylglukosaminyltransferase, welches für die Bildung sog. GPI-Anker verantwortlich ist) Tabelle 3. Die klinische Indikationsbereiche der Durchflusszytometrie 5.6. Qualitätskontrolle (quality control) Die in der Durchflusszytometrie erforderliche Qualitätskontrolle muss auf der Überprüfung der Arbeitsvorgänge von nicht nur dem Gerät sondern auch den Proben beruhen. Zusätzlich zu den Kontrollen während bzw. über den Arbeitsablauf, gibt es auch solche Kontrollmaßnahmen, die die Instrument betreffen und mit täglicher oder monatlicher Regelmäßigkeit durchgeführt werden müssen. Die Ergebnisse geben Informationen über die Empfindlichkeit und Stabilität des Gerätes. Für den klinischen Aspekt ist es am wichtigsten die Probenvorbereitungen zu validieren. Während der Immunphänotypisierung kann das Immunglobulinmolekül auch mit ihrem Fc-Teil auf die Zellen binden, wenn sich Fc-Rezeptoren auf der Oberfläche der untersuchten Zellen (Granulozyten, NK-Zelle usw.) befinden. Um diese unspezifische Bindungen nachzuweisen, werden IsotypKontrollantikörper benutzt. Der Isotyp-Kontrollantikörper ist für ein irrelevantes Antigen (im Allgemeinen ein Antigen aus einer anderen Tierart) spezifisch, das in den untersuchten Zellen sicherlich nicht exprimiert wird. Deshalb kann er nur durch seinen Fc-Teil binden. In einem Parallelansatz kann ein Isotyp-Kontrollantikörper zur Kontrolle neben dem spezifischen Antikörper, der gegen die zu analysierenden Antigene gerichtet ist, verwendet werden. In identischer Konzentration zeigen sie die gleiche Affinität zu Fc-Rezeptoren, wie die spezifischen Antikörper des gleichen Isotyps. Damit er kann als Maß für die unspezifische Bindung und Eigenfluoreszenz der Zellen dienen (Hintergrundsignal). (Abbildung 5.23.) 103 Abbildung 5.22. Isotyp-Kontrollantikörper Kennzeichnung 5.7. Laboratorische Normalbefunde Die Normalbereiche von zirkulierenden Lymphozyt-Populationen der Erwachsenen wird in Tabelle 5.4. dargestellt. x1000 /mikroliter % CD19+ B Zelle CD3+/CD4+ Th Zelle CD8+/CD4+ Tc Zelle CD4 : CD8 Ratio CD3/CD56+ NK Zelle 0,78-2,24 0,080.49 0,49-1,64 0,17-0,88 0,95,0 0,08-0,69 53-83 5-21 30-59 10-40 Lympho cyt CD3+ Zelle 1,143,38 T 5-32 Tabelle 5.4. Referenz Übersetzt von Viktor Molnár Literatur Rothe G: Technische und methodische Grundlagen der Durchflusszytometrie. In: Sack U, Tárnok A, Rothe G (Hrsg): Zelluläre Diagnostik. Grundlagen, Methoden und klinische Anwendungen der Durchflusszytometrie. Basel, Karger, 2007, pp 27–70 (Ref1) 104 6. IMMUNISIERUNG UND VAKZINATION (EDIT BUZÁS) 6.1. Ziel der Immunisierung und ihre praktische Durchführung Unter Immunisierung versteht sich eine - durch beabsichtigtes Einbringen eines bestimmten Antigens ausgelöste - Immunantwort. Mit der Immunisierung können bestimmte Ziele erreicht werden: a.) In verschiedenen Immunoassays verwendet man als sekundäre Antikörper sog. polyklonale Antikörper, welche durch Immunisierung von Versuchstieren mit artfremden Antigenen gewonnen wurden. Ebenso können polyklonale Antikörper hergestellt werden, indem man Kaninchen oder Schweine mit Schafserythrozyten immunisiert. Die entstandenen Anti-Schafs-Erythrozyten-Antikörper finden weitläufige Verwendung für Untersuchung des Komplementsystems (siehe Kapitel 4.). b.) Die Immunisierung ist auch für die Herstellung monoklonaler Antikörper der grundlegende Schritt. Dafür werden vorwiegend Mäuse verwendet. Wenn im Blutserum des immunisierten Organismus der antigenspezifische Antikörper in großen Mengen (hohen Titer) vorhanden ist, werden aus der Milz der immunisierten Maus Plasmazellen isoliert. Durch somatische (Zell)fusion von den auf diese Weise gewonnenen Plasmazellen, mit Zellen einer Maus-Lymphom-Zellinie (Sp2/0), entstehen die monoklonalen Antikörper produzierenden sog. Hybridom-Zellen (siehe Kapitel 7.). c.) Bei Nagetieren, die genetische Anfälligkeit für Autoimmunerkrankungen zeigen (in erster Linie sind es Mäuse und Ratten), können durch Immunisierung Autoimmunkrankheiten induziert werden. Diese Tierversuchsmodelle geben uns die Möglichkeit den Pathomechanismus der gegebenen Erkrankung unter standardisierten und kontrollierten Bedingungen zu untersuchen. Auf diese Weise werden verschiedene Arthritis-Modelle (z.B. adjuvante Arthritis – AA, Kollagen induzierte Arthritis – CIA, Proteoglykan induzierte Arthritis – PGIA, etc.) konstruiert. Für Sclerosis Multiplex (SM) ist z.B. ein oft untersuchtes Mausmodell die experimentelle allergische Encephalomyelitis (EAE). d.) In der alltäglichen ärztlichen Praxis sind die häufigsten und wichtigsten Immunisierungen die Schutzimpfungen (die sog. Vakzination). Ein sehr wichtiger Anwendungsbereich der Immunisierung ist die Vakzine-Herstellung, besonders in seiner präklinischen Tierversuchsphase. 6.2. Die Adjuvantien, ihre Rolle und Gestaltungen Adjuvantien (wirkungssteigernde Substanzen) die zusammen mit Antigenen verwendet werden, begünstigen durch ihre kostimulatorischen Wirkung die Ausbildung der Immunantwort. Die in der Praxis häufig verwendeten Adjuvantien sind Gemische verschiedener Substanzen. Wenn zur Immunisierung neben dem Antigen auch Adjuvantien verwendet werden, wirken sie auf zwei unterschiedliche Arten und Weisen. Erstens, Adjuvantien erstellen oft ein Antigen-Depot, aus 105 dem das Antigen langsamer, kontinuierlicher und gleichmäßiger freigesetzt wird. Damit steigt die Wahrscheinlichkeit, dass die Antigenmoleküle auf die mit spezifischen Antigenrezeptoren ausgestatteten Lymphozyten treffen. Anderseits, Adjuvantien, welche die Liganden für Musterund/oder Notsignalerkennungsrezeptoren (PAMP oder DAMP Rezeptoren) der natürlichen Immunität enthalten, steigern die Wirksamkeit der Antigenpräsentation (durch Steigerung der Expression von MHC- und kostimulatorischen Molekülen in antigenpräsentierenden Zellen). Das sog. inkomplette Freundsche-Adjuvans (Incomplete Freund Adjuvant, IFA) ist eine Wasser-in-Öl Emulsion (außer Wasser und Öl beinhaltet es keine andere Substanzen, wie z.B. abgetötete Mykobakterien). Am häufigsten verwendet man jedoch das Komplette Freundsche Adjuvans (Complete Freund Adjuvant, CFA). Dieses ist ebenfalls eine Wasser-in-Öl Emulsion, welche aber auch abgetötete und getrocknete Mykobakterien enthält (im Allg. Mycobakterium tuberculosis). Die Mykobakterien in dem CFA induzieren die Kostimulationsmoleküle der Makrophagen extrem effizient: So stark, dass das CFA beim Mensch als Adjuvans wegen dessen ausgeprägten Immunogenität gar nicht verwendet werden darf. In der klinischen Praxis, also der menschlichen Immunisierung, gehören zu den am häufigsten verwendeten Adjuvantien andere Adjuvantien. Zum Beispiel das Aluminium-Phosphat und das Aluminium-Hydroxid (Alum). Diese Adjuvantien stellen einerseits ein Antigen-Depot dar, anderseits steigern sie die Antigenaufnahme der Makrophagen. Zu weiteren Adjuvantien gehören noch die Liposomen und das ISCOM (immunostimulating complex). Die ISCOM sind kleine Partikel von 40 nm Durchmesser, welche beim Vermischen im entsprechenden Verhältnis von Cholesterinen, Phospholipiden und Saponinen, spontan entstehen. Durch ihre Wirkung gelangen die Antigene unmittelbar ins Zytosol, wodurch T-Zellen induziert werden. Abbildung 6.1. Die wichtigsten Arten von Adjuvantien und ihr Wirkungsmechanismus Mit diesen zur Immunisierung verwendeten Adjuvantien ist es möglich verschiedene Rezeptoren der antigenpräsentierenden Zellen gezielt zu erreichen. Alle einzelnen Zelltypen die Antigene präsentieren, besitzen einen für sie spezifischen Rezeptorbestand. Durch intrazelluläre Kommunikation – sog. crosstalk – beeinflussen sich die Signalwege der einzelnen Rezeptoren gegenseitig. Die Agonisten der Mustererkennungsrezeptoren sind wichtige Komponenten der Adjuvantien. Diese Substanzen sind in der 106 Lage mit gezielter Einwirkung auf die plasmamembranständigen und/oder endosomalen Mustererkennungsrezeptoren die TH1- und/oder die TH2-Immunantwort zu begünstigen. Im Zytosol befindliche NOD-like Rezeptoren (NLR) und die RIG-like Helikasen (RLHs) sind fähig die intrazellulären bakteriellen und viralen Signale zu erkennen. Über die Wirkung solcher Agonisten stehen noch wenige Kenntnisse zur Verfügung; bestimmte Agonisten (wie z.B. der MDP) werden jedoch in der Veterinärmedizin bereits mit Erfolg verwendet. Die C-Typ Lektin-Rezeptoren (CLR) und die Scavenger Rezeptoren steigern die Antigen-bindung und Präsentation, auf diese Weise können sie zur Immuntoleranz vermeidenden stimulierenden Wirkungen beitragen. 6.3. Faktoren, welche die Wirksamkeit der Immunisierung beeinflussen Die Wirksamkeit der Immunisierung kann auf zahlreiche Weise gesteigert werden: 1. Mit Verwendung von Adjuvantien. 2. Es wird ein, vom Wirtsorganismus (aus welchem das Antigen stammt) phylogenetisch weit entferntes Lebewesen immunisiert, wobei eine niedrigere Immuntoleranz, gegenüber dem zur Immunisierung verwendeten Antigen zu erwarten ist. 3. Der Wirkungsgrad der Immunisierung wird gesteigert, indem die Immunisierung aufgrund der strukturellen Eigenschaften des Antigens (z.B. aufgrund der zu erwartenden MHC-Bindung) mit den prädiktierten, „stärksten” (immundominanten) Epitopen des Antigens durchgeführt wird. 4. Die optimale Dosis des Antigens wird bestimmt und verwendet. 5. Die optimale Art der Immunisierung wird ausgewählt. 6.4. Funktionelle Gruppeneinteilung der Antigene und Epitopen Die T-Zellen erkennen kurze, lineare Peptid-Epitope. Demgegenüber sind die von den Antikörpern erkannten B-Zellen-Epitope überwiegend dreidimensionale Strukturen (sog. Konformationsepitope). Diese Epitope gehen durch Denaturierung wegen Veränderung der Raumstruktur verloren. Nur ein kleiner Teil der B-Zell-Epitopen sind lineare Peptid-Epitope. Nach Denaturierung von proteinartigen Antigenen können solche Antikörper entstehen, die nur denaturierte, lineare Peptid-Sequenzen (des Antigens) erkennen können, die sich mit nativen Antigenmolekülen nicht verbinden. Es können aber auch solche Antikörper produziert werden, welche sowohl im nativen als auch im denaturierten Protein eine lineare Peptid-Sequenz erkennen. Durch Enzymwirkung entstandene Strukturveränderung des Antigens, kann es zur Ausbildung von neuen Epitopen kommen. Enzyme, wie z.B. Proteasen oder Glykosidasen, rufen NeuepitopStrukturen hervor. Ein komplexes Antigen löst eine polyklonale Immunantwort aus, verschiedenartigen Antikörpern mit unterschiedlicher Epitopen-Spezifität führt. 107 welche zur Bildung 6.5. Ausführungsmöglichkeiten der Immunisierung Intravenöse Impfung Mit der intravenösen Impfung gelangt das Antigen primär direkt in die Milz, sekundär in den Lymphknoten. Bei ölhaltigen Emulsionen besteht unmittelbare Emboliegefahr. Ferner wird kein Antigen-Depot gebildet. Erhöhtes Risiko besteht dabei für die Ausbildung der Immuntoleranz oder einer Anaphylaxie. Deshalb ist die intravenöse Impfung nur mit Liposomen oder mit Alum geeignet. Häufiger wird die intravenöse Impfung für booster (Auffrischimpfung), welche kein Adjuvans enthält, verwendet. Dabei werden Gedächtniszellen aktiviert; es braucht keine (oder kaum) Kostimulation. Intradermale Impfung Die intradermale Injektion als Impfung wird häufig verwendet. Dabei gelangt das Antigen langsam in die Blutbahn, anderseits aber werden die Antigene (Impfstoffe) von den Langerhans Zellen der Haut schnell aufgenommen und in die regionalen Lymphknoten transportiert. Ein Nachteil der intradermalen Impfungen ist die häufige Geschwürbildung an der Impfstelle (Ulzeration). Subkutane Impfung Ihr Vorteil ist die leichte Durchführbarkeit; wahrscheinlich wird aus diesem Grund wird dieser Weg am häufigsten zur Immunisierung gewählt. Auf dieser Weise ist es möglich gleichzeitig größere Mengen vom Impfstoff zu verabreichen. Der Impfstoff (das Antigen) wird langsam resorbiert (in erster Linie entlang der Lymphbahnen); die Resorptionsgeschwindigkeit hängt sehr stark von der Blutversorgung des Impfortes ab. In gewissen Fällen soll man in Betracht ziehen, dass der Impfstoff im Unterhautsraum (subcutanen Raum) auch „abwandern” kann. Bei anamnestisch bereits bestehender großer Anaphilaxiegefahr ist unbedingt dieser Immunisierungsweg erforderlich. Die intramuskuläre Impfung Impfungen dieser Art sichern eine schnelle Antigenresorbtion in den Blut- und Lymphbahnen, die Resorption hängt aber von der Menge des Antigens (Impfstoffes) ab. Das ist der richtige Weg zur Impfung von Molekülen mit kleineren Molekulargewicht und/oder mit potenziell irritierender Wirkung; jedoch werden größere Moleküle wahrscheinlich eher durch die Lymphbahnen entlang der Faszien abtransportiert. Es können zwar größere Mengen des Impfstoffes auf diese Weise verabreicht werden, ein Nachteil ist aber die verschleppende Resorption entlang der Faszien, als dessen Folge Entzündungen und Nervenlesionen auftreten können. Bedeutenden Nachteil stellt die nicht monitorisierbare (untersuchbare) Stelle der Impfung dar. Die intraperitoneale Impfung Diese Art Impfung findet bei Nagetieren am häufigsten Verwendung. Dabei gelangt das Antigen über Lymphbahnen schnell in die Lymphknoten. Es können größere Mengen von Impfstoffen und verschiedenartigen Adjuvantien gegeben werden. Bei Auffrischimpfungen (booster) besteht große Gefahr, wegen der raschen Resorption des Impfstoffes in die Blutbahn und die dadurch hervorgerufene Anaphylaxie. Übersetzt von Ottó Dobozy 108 7. VAKZINATION (MARIANNA CSILLA HOLUB) Vakzination kann entweder mit therapeutischem oder mit präventivem Ziel ausgeführt werden. Als Therapie wird der Impfstoff nach der Infektion bzw. Erkrankung verabreicht: nach Infektion - passive Immunisierung In der passiven Immunisierung beinhaltet der Impfstoff präformierte Antikörper (IgG) gegen ein bestimmtes Antigen, und gibt dadurch passiv, (fast) ohne Beteiligung des Immunsystems, den Patienten einen fertiggestellten Schutz. Antikörper für solche Impfstoffe werden aus einem, mit dem fraglichen Antigen oder Pathogen hyperimmunisierten Tier, oder aus Menschen nach Antigenexposition, gewonnen. Passive Immunisierung wird meistens in akut lebensbedrohlichen Situationen angewandt, wo es keine Zeit oder Möglichkeit gibt, dass der Patient durch aktive Immunisierung selber eine Immunität aufbaut. Vorteile: - Verleiht einen schnellen Schutz gegen das Antigen Nachteile: - Der Schutz ist nur vorübergehend, ein immunologisches Gedächtnis wird nicht entstehen (die Halbwertszeit von IgG ist ca. 7-23 Tage im menschlichen Körper) - Eine Elimination bzw. Neutralisierung des Antikörpers kann auch schneller erfolgen, - nicht-menschliche Antikörper können Hypersensitivität oder eine art-spezifische Immunantwort gegen tierischen IgG Epitope erzeugen, Indikation: - Rabies - Tetanus - Botulismus - Schlangengifte verschiedenster Art - Hepatitis-A (als Schutzmaßnahme für Neugeborene und immunsupprimierte Personen welche in der Umgebung von mit Hepatitis A-infizierten Patienten leben) nach Erkrankung - Vakzination mit antigen-beladenen dendritischen Zellen: bei manchen chronischen Krankheitsbildern, z.B. manche experimentelle Tumorvakzine - adoptiver Zell-Transfer: aus dem fraglichen Patienten selbst, bzw. aus einem histokompatiblen Donor werden antigenspezifische immunkompetente Zellen selektiert bzw. gentechnologisch hergestellt, (Knochenmark, Tyhmus oder Blut-Lymphozyten) und in den Körper des Patienten zurück-, bzw. eingeführt. z.B. bei Kindern mit angeborenen Immundefizienzen, manche experimentelle Tumorvakzine. 109 Als Präventivmaßnahme wird der Impfstoff noch vor einer wahrscheinlichen, zukünftigen Infektion angewandt: aktive Vakzination In der aktiven Immunisierung beinhaltet der Impfstoff das Antigen, wogegen die Immunisierung wirken soll. Dadurch wird aktiv, also mit der aktiven Beteiligung des Immunsystems der geimpften Person, Immunität aufgebaut. Aktive Immunisierung wirkt nur in immunkompetenten Personen, welche nicht akut von dem fraglichen Antigen bedroht sind. Das Ziel ist es den Anteil der spezifischen antigenerkennenden Zellen zu erhöhen und ein immunologisches Gedächtnis gegen das Antigen zu erzeugen. Ideale Eigenschaften eines wirksamen Impfstoffes Sicher Hat keine seriösen Nebenwirkungen, das Risiko von potentiell erzeugten Erkrankungen oder Tod ist minimal Protektiv Gibt sicheren Schutz gegen Infektion mit dem aktiven, lebenden Krankheitserreger Stabil Wirkt über mehrere Jahre oder Jahrzehnte hinaus Ruft die Produktion von neutralisierenden Antikörpern hervor Manche Erreger (z.B. Poliovirus) infizieren solche Zellen, (z.B. Neuronen), welche unersetzbar sind. Neutralisierende Antikörper können die Infektion und Zerstörung von solchen Zellen verhindern. Induziert eine protektive Antwort von T-Zellen Bestimmte Pathogene (vor allem die intrazellulär aktiven Erreger) können mit Hilfe einer zellvermittelten Immunantwort besser eliminiert werden. Praktische Überlegungen Biologische Stabilität, einfache Verabreichung, billige Herstellung Tabelle 7.1. Ideale Eigenschaften eines wirksamen Impfstoffes 7.1. Aktive Vakzination 7.1.1. Aktive Vakzination erzeugt starke primäre AntikörperAntworten Zur Entstehung einer Immunantwort bzw. bis zum Beginn der Antikörperproduktion, wird nach einer Antigenexposition oder Vakzination, ein relativ langer Zeitraum benötigt (ungefähr 5-10 Tage). In dieser, sog. primären Immunantwort werden nur vergleichsweise geringe Mengen von typischerweise IgM-Typ Antikörpern gebildet. Die Affinität dieser Antikörper bleibt relativ niedrig, weil sie von schnell reagierenden Plasmazellen in den Primär-Foci der Lymphknoten produziert werden. Da diese BZellen sich außerhalb der Keimzentren befinden, sind sie nicht in der Lage die Affinität ihrer Antikörper durch gezielte Mutationen und Selektion (Affinitätsreifung) zu steigern. Mit der Zeit wird aber ein Teil der aktivierten B-Zellen in das Keimzentrum der Sekundärfollikel einwandern. Hier gehen sie durch den Prozess der Affinitätsreifung, wodurch der Anteil der 110 hochaffinen Antikörper im Serum der auf das Antigen exponierten / geimpften Person, schrittweise aber deutlich erhöht wird. Weil die meisten von diesen B-Zellen in den Keimzentren auch den IgKlassenwechsel ausführen, werden mit der Zeit nicht nur mehr hochaffine Antikörper, sondern auch mehr Antikörper vom nicht-IgM-Typ (IgG, IgA, usw) gebildet. Letztlich wird sich eine kleine Minderheit der auf diese Weise entstandenen B-Zellen, in B-Gedächtniszellen umwandeln, welche bei erneuter Exposition auf das Antigen oder bei einer Auffrischimpfung die erneute Immunantwort auslösen. Bei einer sekundären aus den solchen, Antwort oben sog. werden genannten Gründen hochaffine Antikörper, meistens vom Typ IgG, viel schneller (innerhalb von 1-3 Tagen) und in deutlich höheren Mengen sezerniert, als nach der ersten Antigenexposition (Abbildungen 7.1 und 7.2). Abbildung 7.1. Typischer Ablauf einer primären und sekundären humoralen Immunantwort Abbildung 7.2. Aufbau, Aktivierung und Verstärkung des humoralen Gedächtnisses durch widerholte Infektionen. 111 Latenz nach der Antigenexposition Stärke der Antwort Isotyp der Antikörper Affinität der Antikörper Variabilität in der Spezifizität der Ak. Primäre humorale Antwort 5-10 Tage Sekundäre humorale Antwort 1-3 Tage schwächer IgM >> IgG stärker vor allem IgG (dank der Klassenwechsel, in manchen Fällen auch IgA, IgE) durchschnittlich hoch (dank der Affinitätsreifung) relativ niedrig (dank der Affinitätsreifung) durchschnittlich niedrig relativ groß Tabelle 7.2. Vergleich einer primären und sekundären humoralen Antwort. Antigene, die für eine aktive Vakzination angewandt werden können, müssen einer Vielzahl von Kriterien entsprechen. Als erstes müssen sie solche Antigene des gezielten Pathogenes sein, die im menschlichen Körper nicht vorkommen. Also klassische Fremdantigene, wogegen eine Immunantwort keine Kreuzreaktionen gegen menschliche Autoantigene hervorrufen könnte. Zweitens ist es wichtig, dass Antigene nicht nur eine Vielzahl von Epitopen vorweisen, sondern auch dass für eine aktive Vakzination auch sog. protektive Epitope unter ihnen vorkommen. Protektive Epitope sind nicht nur spezifisch für das das fragliche Pathogen, sondern sie sind auch stark immunogen. Sie werden als protektiv bezeichnet, weil durch ihre Erkennung alleine, unabhängig von anderen Epitopen des Antigens ist das Immunsystem in der Lage, einen vollen Schutz gegen eine produktive Infektion zu sichern. Solche Epitope können sowohl zu den T-abhängigen (TD, T-dependent) als auch T-unabhängigen (TI, T-independent) Antigenen gehören. Also wird für eine B-Zell-Antwort gegen sie T-Zell Hilfe benötigt (TD) oder nicht (TI). Erzeugung einer T-Zell Antwort durch die Vakzination ist selbstverständlich kritischer bei TD Antigenen, schadet aber nie, oder kann wichtig sein bei TI Antigenen auch, z.B. im Falle von TI Antigenen von intrazellulären Pathogenen, welche letzten Endes nur mit Hilfe von aktivierten Th und/oder Tc Zellen eliminiert werden können. Letztlich ist es extrem wichtig, dass die Induktion der Immunantwort stark genug sein muss, um ein langfristiges spezifisches Gedächtnis zu erzeugen, ohne das der eigentliche Sinn der aktiven Immunisierung nicht gesichert wäre. Antikörper, die durch die Erkennung und Bindung des Krankheitserregers seine weitere Verbreitung im Körper vollständig hemmen können, werden als neutralisierende Antikörper bezeichnet. Die Erzeugung von neutralisierenden Antikörpern ist selbstverständlich das wichtigste Kriterium für allerlei Vakzinationen. Neutralisierende Antikörper müssen fähig sein die wichtigsten pathogenen Mechanismen des Erregers zu blockieren. Zum Beispiel im Falle von Viren, müssen sie selbst die Bindung des Virus zu den Virusrezeptoren der Zielzellen vorbeugen können. Andererseits gibt es unter den neutralisierenden Antikörpern auch solche, welche die effektive Bekämpfung des Krankheitserregers durch andere Mechanismen ermöglichen: Zum Beispiel indem sie - die Entfernung des Pathogenes oder der infizierten körpereigenen Zellen unterstützen (z.B. durch FcR-vermittelte Phagozytose des Antikörper-gebundenen Erregers) 112 - die Replikation des Erregers in dem Wirtsorganismus oder Wirtszellen verhindern (z.B. durch Modifizierung von inter- oder intrazellulären Signalübermittlungswegen) - die Freisetzung des Erregers aus den Wirtszellen, bzw. den interzellulären Transfer des Erregers und so seine weitere Verbreitung unterdrücken (z.B. durch Modifizierung der räumlichen Anordnung von viralen Hüllenproteinen in der Membran). Abbildung 7.3. Effekte von Antikörpern auf virusinfizierten Zellen Die infolge der ersten Antigenexposition entstehende primäre Immunantwort führt schleunigst zur Bildung von spezifischen Antikörpern im Blutserum. Die Abwesenheit von bestimmten Antikörpern im Serum nennt man generell Seronegativität, hingegen die Präsenz von bestimmten Antikörpern gegen ein Antigen, Seropositivität. Bei allen Vakzinationsverfahren ist es selbstverständlich ein wichtiges Kriterium, dass das Verfahren in der Lage sein muss, die Produktion von Antikörpern hervorzurufen. Mit andern Worten, dass die geimpfte Person durch die Vakzination vom ursprünglichen, seronegativen Status ins seropositive konvertiert wird. Diese Änderung in dem immunologischen Status des Patienten wird als Serokonversion bezeichnet, weil das Gegenteil davon, also ein mögliches Verschwinden der Antikörper Seroreversion heißt. Ein wirksamer Impfstoff sichert aber nicht nur die Produktion von Antikörpern, sondern sorgt auch dafür, dass die Antikörper in solchen Mengen sezerniert werden, dass die geimpfte Person einen aktiven Schutz vor dem Antigen erhält. Das ist die Seroprotektion. Die exakte Menge der Antikörper benötigt für einen kompletten Schutz vor dem Antigen ist abhängig vom Erreger bzw. Impfstoff selbst, 113 z.B. bei Hepatitis A beträgt sie 20 mlU/ml Antikörper; bei Hepatitis B 10 mlU/ml Antikörper, usw. (mIU/ml = milli-international units per milliliter) 7.1.1.1. T-Zell Gedächtnis ist eine der wichtigsten Voraussetzungen einer wirksamen TD B-Zell-Antwort Künstlich auslösbare T-unabhängige B-Zell-Antwort In der Auslösung einer konventionellen, T-abhängigen B-Zell-Antwort spielt die T-B Zell Wechselwirkung, und insbesondere die CD40-CD40L Interaktion eine Schlüsselrolle. Abbildung 7.4. Die CD40-CD40L Interaktion Eine mögliche Alternative der Vakzin-Entwicklung für T-abhängige Antigene (also die überwiegende Mehrheit aller Antigene) ist es solche Vakzine zu entwickeln, welche selbst für T-abhängige Antigene eine T-unabhängige B-Zell-Antwort auslösen, und damit den Immunisierungsprozess stark vereinfachen bzw. abkürzen. Dies wird erreicht indem man T-Zellen durch den Impfstoff absichtlich nicht aktiviert, sondern ihre Anwesenheit den B-Zellen nur vortäuscht. Dazu werden aktivierende antiCD40 Antikörper zum Impfstoff gemischt, welche die CD40 Moleküle auf B-Zellen genauso aktivieren können, wie ein CD40L Molekül einer aktivierten T Zelle. Auf diese Weise kann man auch Impfstoffe herstellen, die nutzlos wären, da ihre Schlüsselantigene T-Zellen einfach nicht effizient genug aktivieren könnten. Zweitens, da der relativ lange Prozess der T-Zell-Aktivierung mit dieser Strategie aus dem Spiel ist, kann man auf diese Weise auch schneller eine Antikörperproduktion durch Vakzination hervorrufen. Nachteile der Strategie sind die Gefahr einer unabsichtlichen polyklonalen BZell-Antwort, und im extremen Fällen Autoimmunität oder Beschädigung des T-Zell-Gedächtnis. 114 7.1.2. Neben der Antikörper-Produktion kann auch eine zellulären Immunantwort Ziel der Vakzination sein Um die durch Vakzination erzeugte T Zell Antworten zu verstehen, müssen wir uns zuerst mit den drei Phasen der T-Zell-Antwort auseinandersetzen. Diese sind: eine klonale Expansion von reaktiven T Zellen in der Präsenz des Antigens nach Beseitigung des Antigens kommt es zu einer Kontraktion der Immunantwort und letztlich, parallel zu der letzteren Phase, beginnt der Aufbau des immunologischen Gedächtnis An dieser Stelle ist es wichtig zu sehen, dass es einige Unterschiede zwischen den Reaktionen der CD8+ Zellen und der CD4+ in den oben genannten drei Phasen gibt. Abbildung 7.5. Antivirale CD8+ und CD4+ T-Zell-Antwort. Im Falle der CD8+ T Zellen laufen die drei Phasen der T-Zell-Antwort folgendermaßen ab: Abbildung 7.6. CD8+ T-Zell-Antwort auf eine Infektion. 115 1. Nach einer Infektion bzw. Impfung erfolgt die Aktivierung der naiven CD8+ T Zellen mit Hilfe von dendritischen Zellen. Die Aktivierung von naiven CD8+ T Zellen läuft meistens schneller ab als die der naiven CD4+ T Zellen, (manchmal innerhalb von 2 Stunden nach Antigenexposition, wogegen CD4+ T Zellen mindestens 6 Stunden brauchen), benötigt niedrigere Antigen-Konzentrationen und ist weniger abhängig von Kostimulation. 2. In der Phase der Expansion entsteht die primäre T-Zell-Antwort. Nur 24 Stunden nach der Antigenexposition sind die antigenspezifischen CD8+ T Zellen in der aktiven Proliferation (im Falle der CD4+ T Zellen findet dies schon wieder viel langsamer statt). Funktionell gesehen ist die durch klonale Expansion entstandene Zellmenge recht heterogen: die meisten Zellen sind IFN- produzierende oder TNF- sezernierende zytotoxische T Zellen. 3. In der darauf folgenden Phase der Kontraktion stirbt dann ungefähr 90-95 % aller primär aktivierten CD8+ T-Zellen durch Apoptose ab. 4. Die Zellen welche der Apoptose entgehen, bilden das Repertoir der frühen primären CD8+ T Gedächtniszellen. Die Anzahl der Gedächtniszellen hängt von der Anzahl der ursprünglichen Effektorzellen ab; je mehr Effektorzellen entstanden sind, desto mehr Gedächtniszellen werden gebildet: meistens ist die Anzahl der Gedächtniszellen ungefähr 5-10% der Effektorzellen. Auf ihrer Zelloberfläche exprimieren sie noch bestimmte CD Marker, die auch noch auf den naiven CD8+ T Zellen zu sehen waren. Im Gegensatz zu naiven Zellen sezernieren sie aber schon mehrere Zytokine wie, z.B. IFN- und TNF- gleichzeitig. 5. CD8+ T Gedächtniszellen, welche die Kontraktionsphase überleben, und neben IFN- und TNF- auch IL-2-t sezernieren, haben die Kapazität sich zu erneuern, und sie bilden die Mehrheit der langfristige CD8+ T Gedächtniszellen. Ihre Fähigkeit zur Selbsterneuerung gibt die Erklärung darauf, warum am meisten antigenspezifischen CD8+ T Gedächtniszellen 2-3 Tage nach der Beseitigung des Antigens im menschlichen Körper zu finden sind. Der Unterschied in der Menge der antigenspezifischen, reagierenden T Zellen in sekundären vs. primären Immunantworten ist generell ähnlich zum Unterschied der Antikörper unter den selben Umständen. Nach einer erneuten Antigenexposition, also in einer sekundären Immunantwort nimmt die Menge der reaktiven T Effektorzellen zu, und dasselbe gilt für die T Gedächtniszellen. Nachdem also bei einer gewöhnlichen Impfung mit der ersten Impfung eine Grundimmunisierung stattgefunden hat, werden durch die wiederholten Impfungen, die sog. Auffrischimpfungen (manchmal als Boosterimpfung bezeichnet), eigentlich sekundäre Immunantworten ausgelöst. In diesen Antworten werden die Phasen der Kontraktion der T-Zell-Antwort immer länger, und nach jeder neuen Antigenexposition entstehen neue T-Gedächtniszellen in immer größere Mengen. Andererseits interessant ist auch die Tatsache, dass nach jeder solchen neuen Antigenexposition, ein immer kleinerer Anteil der neulich gebildeten Gedächtniszellen in der Lage sein wird, IL-2 zu produzieren und sich selber zu erneuern. Weil nur sie sich aber, im Gegensatz zu den primären Gedächtniszellen, effektiv erneuern können, werden sie über die Jahre - früher oder später - die Mehrheit der überlebenden Gedächtniszellen und dadurch die Hauptkomponente des Gedächtnis bilden. 116 Abbildung 7.7. Kinetik von primären und sekundären CD8+ T Zell-Antworten. 7.1.3. Typen der T-Gedächtniszellen Aus den in der primären Immunantwort aktivierten T Zellen differenzieren sich zwei Typen von TGedächtniszellen, welche voneinander durch ihre funktionellen Eigenschaften und ihre Oberflächenmarker, wie z.B. Homing-Rezeptoren relativ einfach unterschieden werden können. TCM: Zentrale T-Gedächtniszellen Sowohl CD4+, als auch CD8+ T Zellen können sich zu TCM entwickeln Ihre Oberflächenmarker ermöglichen ihre Migration zu und zwischen sekundären Lymphorganen (z.B. die, auf naiven T Zellen auch exprimierten CCR7 und CD62L/L-Selektin) – weswegen TCM Zellen meistens in den Lymphknoten, Tonsillen, Milz und in der Blutzirkulation zu finden sind. Sie sind empfindlicher für das Antigen als naive T Zellen Benötigen auch weniger Kostimulation für ihre Aktivierung, als naive T Zellen. Nach Aktivierung exprimieren sie CD40L stärker als naive T Zellen (dieses Feedback ist ein wichtiges Signal für die APZ, z.B. B-Zellen oder dendritische Zellen). Nach Aktivierung sezernieren sie IL-2, wodurch sie autokrin ihre eigene Teilung fördern und letzten Endes ihre Selbsterneuerung ermöglichen können. Infolge einer Antigen-Aktivierung führen sie oft assymetrische Teilungen aus: ungefähr die Hälfte der Tochterzellen wandelt sich in TEM (oder durch TEM in T-Effektorzellen) um, die andere Hälfte bleibt aber eine TCM Zelle, damit das Gedächtnis gegen das jeweilige Antigen erhalten bleibt. TEM: Effektor T-Gedächtniszellen Sowohl CD4+, als auch CD8+ T Zellen können sich zu TEM entwickeln Im Gegensatz zu TCM Zellen exprimieren sie weder CCR7 noch CD62L/L-Selektin, dafür aber solche Chemokinrezeptoren und Integrin-Typ Adhesionsmoleküle, welche ihre schnelle Auswanderung in die Antigen-exponierten, entzündeten Geweben ermöglichen. Aus diesem 117 Grund sind sie in sekundären Lymphorganen (bis auf die Milz) kaum, jedoch in anderen Geweben wie in den Lungen, Leber, Darmtrakt oder im Blut in relativ hohen Anzahl nachweisbar. Sie reagieren auf einen Antigenstimulus extrem schnell, und wandeln sich in vollständig aktivierte Effektrozellen binnen Stunden um, welche je nach Funktion der ursprünglichen TEM Zelle (CD4+, oder CD8+ TEM) IFN- / IL-4 produzierende Th Effektorzellen, oder Perforin/Granzym positive Tc Effektorzellen sein können. Abbildung 7.8. Zentrale T Gedächtniszellen (TCM) und Effektor T Gedächtniszellen (TEM). Da die Entwicklung des T-Zell-Gedächtnis vor allem von den Umständen der primären Antigenexposition abhängt und danach relativ schwer zu beeinflussen ist, werden die schon einmal aktivierten T-Zellen die Phasen der Expansion, Kontraktion und den Aufbau der Gedächtniszellen quasi vorprogrammiert ausführen. Die Anzahl der Gedächtniszellen bleibt über eine längere Zeit, jedoch mindestens über Monate, erhalten. Dies ist auch ziemlich unabhängig von äußeren Einwirkungen. Aus diesem Grund dürfen die vom STIKO (vom Ständigen Impfkommission) empfohlene Zeiträume im Impfkalender, die der Arzt zwischen den einzelnen Impfungen eines jeweiligen Impfungsprotokolls einhalten soll, nur als Minimum interpretiert werden. Eine unabsichtliche Verlängerung von diesen Pausen (z.B. die Person reist im Mitte des Protokolls unerwartet ab, wird krank, und kann nicht rechtzeitig geimpft werden), wird auf die Effektivität des Impfungsprogramms normalerweise keine relevante Einwirkung haben. Dies aus dem einfachen Grund, da die Mengen der langzeitigen T Gedächtniszellen in der Regel von solchen Unterbrechungen bzw. Abweichungen vom Standardprogramm nicht beeinflusst werden. Mit anderen Worten, selbst wenn die empfohlenen Pausen zwischen wiederholten Impfungen aus jeglichen, nicht voraussehbaren Gründen verlängert werden müssen, gibt es keinen Grund das Impfungsprogramm umzustrukturieren oder neu zu starten. Selbstverständlich ist das Gegenteil, also eine Verkürzung des empfohlenen Zeitraums zwischen zwei Impfungen definitiv nicht zu empfehlen, weil in diesem Fall das Immunsystem nicht genügend Zeit hat, die Antwort abzuschließen, was die Effektivität der kommenden Vakzination deutlich kompromittieren würde. 118 Weiters ist auch Fieber generell eine Kontraindikation bei Impfungen. Bei solchen akuten, nicht banalen Infektionen bzw. Krankheitsbildern, wo hohes Fieber (es gibt unterschiedliche Empfehlungen diesbezüglich, das Robert Koch Institut empfehlt >+38.5C) gemessen wird, darf die Impfung nur nach einer kompletten Heilung des Patienten verabreicht werden. Dies kann man dadurch erklären, dass Fieber bei vielen Impfungen eine mögliche Nebenwirkung ist, und zusätzliche Erhöhung eines bereits vorhandenen Fiebers durch die Impfung nicht sinnvoll ist bzw. eine unnötige Belastung der Patienten darstellt. Hinzu kommt auch, dass Fieber verursacht durch eine Infektion, kann eines der wichtigeren Symptome einer schiefgelaufenen Impfung, d.h. Fieber als Nebenwirkung der Vakzination, für den Arzt praktisch unsichtbar machen kann. Letztlich gibt es auch einige Publikationen in der Literatur die besagen, dass akute Entzündungen die Umwandlung von aktivierten naiven CD8+ T Zellen in CD8+ T-Gedächtniszellen beeinflussen können. Bei akuten Infektionen spielen die inflammatorischen Zytokine eine wichtige Rolle in der Unterstützung der Aktivierung, Reifung, und Antigenpräsentierung der dendritischen Zellen (DC). Die meisten DC-aktivierten CD8+ T Zellen, welche in der Phase der Expansion entstanden sind, werden in der Phase der Kontraktion verschwinden, damit die überlebenden T Zellen praktisch nur aus T-Gedächtniszellen bestehen, und die T-Effektorzellen nur als eine stets schrumpfende Minderheit der überlebenden Zellen zurück bleiben. Falls aber in einer geimpften Person, parallel zu der Impfung auch eine schwere Infektion oder starke Entzündung vorhanden ist, werden wegen der starken, ständigen Aktivierung die T-Effektorzellen in einer höheren Anzahl am Leben bleiben, als normal. Sie teilen sich sehr intensiv, wodurch die, von dem Vakzin induzierten, T-Gedächtniszellen die Minderheit in der T-Zell-Fraktion bilden werden, und nicht umgekehrt. Dazu kommt, dass die homöostatischen Zytokine, welche für das Überleben der TGedächtniszellen kritisch sind, während einer starken Infektion, im Vergleich mit inflammatorischen Zytokinen, in nur relativ geringen Mengen produziert werden. In so einem Zytokin-Millieu, also im Millieu einer aktiven Infektion können deswegen die Gedächtniszellen nicht stabilisiert werden. Sie erleiden verschieden funktionelle Schäden und letzten Endes sterben viele unter ihnen durch Apoptose ab. Dies kann selbstverständlich ernsthafte Konsequenzen in Bezug auf die langfristige Wirksamkeit des Vakzins haben. 7.2. Lebensalter-abhängige Impfung-Strategien Sowohl Kleinkinder, als auch Personen im hohen Alter (über 65 Jahren) geben typischerweise schwache Immunantworten auf neue Fremdantigene. In ähnlicher Weise reagieren sie nur schwach und ineffizient auf Impfungen. Aus diesem Grunde ist es sehr wichtig, dass sehr junge und sehr alte Personen spezielle Impfungen erhalten, welche eine starke Produktion von neutralisierenden Antikörpern auch in diesen gefährdeten Lebensalter-Kategorien ermöglichen. Im Säuglingsalter stellt das Knochenmark noch extrem große Mengen von naiven B-Zellen unterschiedlicher Spezifizität her. Mit der Zeit wird aber ein immer größerer Anteil des hämatopoetisch aktiven Knochenmarks mit Fettzellen ersetzt, wodurch die Bildung von neuen B-Zellen mit zunehmendem Alter immer geringer wird. Die Menge der homöostatischen Zytokine, welche B-Zellen 119 am Leben erhalten ist auch limitiert. Deshalb werden mit zunehmendem Alter die bereits vorhandenen B-Gedächtniszellen und reifen Plasmazellen im Kampf um homöostatische Überlebenssignale auch immer weniger Platz den neugebildeten B-Zellen übrig lassen. Dadurch verringern sie ihre Halbwertszeit und Überlebenschancen. Dies ist einer der Gründe, warum die Kapazität auf neue BZell-Antworten mit zunehmendem Alter stets geringer wird. Da die Halbwertszeit von IgG im Blutserum nur 21-30 Tage beträgt, wird der Schutz der Kleinkinder durch die mütterlichen Antikörper nach der Geburt bzw. nach dem Abstillen rapid verringert. Die transplazentalen bzw. durch die Muttermilch transferierten mütterliche Antikörper werden innerhalb von Monaten praktisch verschwinden. Das eigene B-Zell-Repertoir dagegen ist aber noch nicht immer schon reif genug um allerlei Infektionen effizient bekämpfen zu können. Typische Infektionen in Kleinkindern, die häufig kritische Situationen auslösen, sind Hib, Rotavirus, oder Salmonella, welche bei Erwachsenen praktisch nie oder nur in extremen Fällen fatale Auswirkungen haben können. In der Evolution der Säugetiere bzw. Menschen mussten also Jungtiere oder kleine Kinder, über eine längere Zeit nach der Geburt, mit einem noch nicht vollkommen reifen Immunsystem zum Überleben auskommen. Durch die Aufnahme von mütterlichen IgG und IgA Antikörpern über die Muttermilch sind sie nur passiv gegen eine Vielzahl von Krankheitserregern immunisiert. Dies kann aber aus der Perspektive der Vakzination ernsthafte Probleme verursachen. In älteren Personen ist die Situation letzten Endes ähnlich. Natürlich nicht wegen des unreifen Status, sondern auf Grund der Alterung des Körpers und seinem Immunsystem: Die Rückbildung des Thymus bzw. des Knochenmarks, und die Degeneration des T- und B-Zell Gedächtnisses. Deswegen werden Infektionen im hohen Alter manchmal erneut deutliche Herausforderungen darstellen, welchen aber das Immunsystem des Patienten schon ein oder mehrmals in seinem Leben schon erfolgreich entgegnet ist. Die Symptome werden stärker, Heilung erfolgt langsamer, und die Chance auf fatale Komplikationen ist höher, wie z.B. das im Falle von Influenza-infizierten alten Personen häufig zu sehen ist. Letzten Endes führen aber diese Ursachen auch zu ineffizienten Antworten in der Vakzination. 7.2.1. Welche Probleme muss die Vakzination in Kleinkindern überwinden? Eine erfolgreiche Vakzination in Kleinkindern ist technisch schwer, aber natürlich nicht unmöglich zu erreichen. Aus mehreren Gründen: Erstens, in den sekundären Lymphorganen der Kleinkinder gibt es noch sehr wenig oder kaum Marginal Zonen- (MZ-) B-Zellen, welche auf Polysacharid-artige TI– Antigene (T-unabhängige Antigene) reagieren und durch Umwandlung in Plasmazellen gegen sie Antikörper erzeugen könnten. Aus diesem Grund sind Impfungen welche TI-Antworten benötigen (z.B. Hib Vakzin) in jungen Kindern ineffizient. Zweitens werden Kostimulationsmoleküle kaum, oder nur in suboptimalen Mengen auf den B-Zellen von Kleinkindern exprimiert, weswegen auch die dendritische Zelle – T-Zelle – B-Zelle Wechselwirkung bei TD-Antigenen (T-abhängige Antigene) weniger wirksam sein wird. Dies beeinflusst wie die meisten Peptidantigene präsentiert werden und verringert dadurch die Wirksamkeit vieler Peptidvakzine (z.B. Masern- / Morbilli-Vakzin). 120 Drittens, weil die follikulären dendritischen Zellen auch noch unreif sind und in den Keimzentren der Lymphknoten die Affinitätsreifung, somatische Hypermutation und Isotyp-Klassenwechsel anders ablaufen als bei Erwachsenen. Die aktivierten B-Zellen reifen meistens zu Gedächtniszellen und nur seltener zu Plasmazellen, wodurch die Antikörperproduktion nach Antigenexposition ziemlich begrenzt bleibt. Viertens werden in kleinen Kindern nach Antigenexposition viel weniger Plasmazellen aus den sekundären Lymphorganen in das Knochenmark wandern. Auch ihre Chancen aufs langfristige Überleben im Zytokin-Millieu eines jungen Knochenmarks sind deutlich geringer. Also ist in Kleinkindern die Umwandlung von mit hochaffinen BZR ausgestatteten B-Zellen in entsprechenden Plasmazellen, aus den oben genannten Gründen, limitiert. Eine langfristige Immunität könnte jedoch immer noch, durch B-Zellen mit niedrigaffinen BZR, bzw. ihren Gedächtniszellen gesichert werden. Interessanterweise aber, aus Gründen die bis heute noch nicht vollkommen geklärt worden sind, dies ist nicht der Fall. Gedächtniszellen welche in Kleinkindern erzeugt werden haben eine begrenzte Lebensdauer, und bleiben nie ein Leben lang erhalten. Zum Beispiel in Erwachsenen, welche als Säuglinge eine Hepatitis B-Infektion überstanden haben, kann man HepB-spezifischen BGedächtniszellen bzw. Schutz gegen das Virus nicht mehr nachweisen. 7.2.2. Impfung-Strategien für Kleinkinder und Personen in hohem Alter Verbesserte Aktivierung von naiven B-Zellen: neue Adjuvantien sind in der Lage durch spezielle Immunomodulation die Erreichbarkeit des Vakzins zu erhöhen: z.B. in dem neuesten Hib Vakzin wird nicht mehr das reine Hib Kapselpolysaccharid als Impfstoff benutzt, sondern es wird zu einem Dyphterietoxoid Protein als Träger gekoppelt, und der dabei entstandene sog. Konjugat-Impfstoff wird angewandt. Beschleunigung der Wanderung von naiven B-Zellen in die sekundären Lymphorgane, und der Aufbau von reifen Keimzentren: mit höherer Antigendosis, oder mit Hilfe eines VakzinDepots (z.B. in der Fluval-P Vakzin wird das Antigen in ein Aluminium-Phosphat Gel eingebettet, welches das Antigen nur langsam, über eine längere Zeit gleichmäßig verteilt, freisetzt), oder damit, dass die Grundimmunisierung selbst auch aus mehreren Impfungen besteht, welche dafür sorgen, dass das Antigen kontinuierlich im Körper erhalten bleibt. Nur später, in weiteren unabhängigen Schritten erfolgen die Auffrischimpfungen. Beispielsweise in der Diphtherie-Keuchhusten-Wundstarrkrampf-Kombinationsimpfungen wird die Grund- immunisierung selbst aus drei Schnitten bestehen (I/a, I/b, I/c) die für Kinder im Alter von 2, 3 und 4 Monaten verabreicht werden müssen. Verstärkung der Aktivierung der naiven B-Zellen: dies wird erreicht mit höheren Antigendosen und mit wirksameren Adjuvantien. Unterstützung der Differenzierung von Plasmazellen: dies kann man in erster Linie mit besseren Adjuvantien erreichen. 121 Stabilisierung des Gedächtnis für mehrere Jahrzehnte: durch Auffrischimpfungen, also wiederholte Exposition auf niedrige Dosen des Antigens: z.B. die Tetanusimpfung muss alle 10 Jahre wiederholt werden. 7.3. Arten von Impfstoffen Impfstoffe werden auf die unterschiedlichsten Arten und Weisen klassifiziert. Eine mögliche, praktische Einteilung ist wie folgt: Ganzpartikelimpfstoffe/Vollvakzine: Attenuierter Lebendimpfstoff Totimpfstoff / inaktivierter Impfstoff Lebender rekombinanter Impfstoff Untereinheitimpfstoffe/Spaltvakzine: Proteinimpstoffe syntetische Peptidimpstoffe DNA (RNA) - Vakzine Gegen den selben Erreger werden oft mehrere Impfstoffe von den unterschiedlichsten Typen entwickelt. Eines der besten Beispiele für diese Tatsache war vielleicht die große Auswahl von Influenzavakzine, welche alle gegen das 2009-2010 H1N1 Influenzavirus entwickelt und für die klinische Anwendung irgendwo in der Welt zugelassen worden sind. Ganzpartikelimpfstoff mit inaktivierten (zerstörten) Viren Spaltvakzin mit durch Detergentien aufgelösten und inaktivierten Partikeln Spaltvakzin bestehend aus, von inaktivierten Viren gereinigten Hemagglutinin und Neuraminidase (Oberflächenantigene des Virus) Ganzpartikelimpfs toffe mit attenuierter Lebendimpfstoff, kälte-adaptierte Virusstämme, also mit nicht pathogenischen ganzen lebenden Viren Baxter (EMEA), 8 Hersteller in China, Novartis (USA) Medimmune (USA) Omninvest + AlPhosphat Adjuvans (Ungarn), CSL (Australien, USA) Novartis + M59 Adjuvans (EMEA) Microgen (Russland Cantacuzino (Rumänien) Sanofi Pasteur (USA), Green Cross (Korea), GSK+ASO3 Adjuvans (EMEA, Kanada) Tabelle 7.3. Arten der zugelassenen monovalenten Impfstoffe gegen das 2009-2010 Influenza (Quelle: WHO). 122 7.4. Neue Wege in der Impfstoffentwicklung 1. Weiterentwicklung von gängigen Vakzinen 2. Entwicklung von neuen Vakzinen 3. Experimentelle Methoden biotechnologische Entwicklung von Vakzinen Die wichtigsten Ziele in der weiteren Verbesserung der heutigen Vakzine sind immer die Steigerung der Immunogenität, und die Verminderung der Toxizität der Impfung. Um die Immunogenität der Impfung zu steigern muss nicht nur das bestmögliche Epitop, sondern auch ein geeignetes Adjuvans, und der wirksamste Weg der Verabreichung des Impfstoffes ausgewählt werden. Um dagegen die Toxizität möglichst minimalisieren zu können, also die Sicherheit des Impfstoffes soweit wie möglich zu erhöhen, muss gesichert werden dass das angewandte Adjuvans die kritischen Eigenschaften einer protektiven Immunantwort nicht kompromittiert (z.B. Th1/Th2 Gleichgewicht, idealer Isotyp der produzierten Antikörper, usw.) der Impfstoff durch Ähnlichkeit zu menschlichen Antigenen keine Autoimmunreaktionen auslöst (Molekulare Mimikry) der Impfstoff in einer reinen Form bereitgestellt werden kann (keine Kontamination mit aktiven Erregern, Bestandteile von anderen Mikroorganismen, Prionen, toxische Derivate des Impfstoffes oder Chemikalien angewandt in der industriellen Prozessieren, usw.), der Impfstoff möglichst keine stabilen oder auf gar keinen Fall vererbbaren Änderungen im Genom des Patienten hervorruft (dies gilt in erster Linie für DNA und RNA-Vakzine). 7.4.1. Verbesserung von gängigen Vakzinen Im Falle der attenuierten Lebendimpfstoffe wird Immunität gegen ein, im Großen und Ganzen intaktes, lebendes Pathogen erzeugt, dessen Virulenz aber künstlich vermindert worden ist. Dieses attenuierte Pathogen kann entweder eine modifizierte, weniger virulente Form, oder ein eng verwandter nicht oder weniger virulenter Stamm oder Variante des eigentlichen Pathogenes sein. So oder so, erzeugt das attenuierte Pathogen zwar Immunität gegen das eigentliche Pathogen durch die Ähnlichkeit ihrer Antigene (also durch Kreuzreaktion), es verfügt aber über keine nennenswerte Virulenzfaktoren, weswegen es relativ harmlos ist, es in den Körper eines naiven Menschen einzuführen. Zum Beispiel zur Impfung gegen Influenza werden oft solche Influenzaviren benutzt, die über eine längere Zeit nicht in menschlichen Zellen, sondern in den Zellen eines anderen Tieres kultiviert worden sind (oft in Affenzellen). Im Laufe der Kultivierung in einem fremden Wirtsorganismus adaptiert sich das Virus mit einer Reihe von Mutationen an die neue Umgebung. Dabei entstehen, unter anderem, auch solche Viren, welche die Zellen des ursprünglichen Wirtes immer noch infizieren können, sich in dieser alten Umgebung aber nicht mehr vermehren können. Diese Variante wird als Impfstoff benutzt, wodurch eine limitierte Infektion und eine volle Immunantwort gegen das ursprüngliche pathogene Virus ausgelöst wird, ohne aber eine produktive, also potentiell gefährliche Infektion zu riskieren. (Abbildung 7.9.) 123 Abbildung 7.9. Attenuierung von menschlichen Viren durch Kultivierung in tierischen Zellen. Attenuierung kann auch durch künstliche Mutagenese werden. oder Zum Gendeletion Beispiel im erreicht Falle von Influenzaviren ist der häufigste Virusstamm von Jahr zu Jahr anders. Deswegen werden die saisonalen Influenzaimpfungen jedes Jahr in der Industrie durch Mutagenese so verändert, dass sie gegen die, im jeweiligen Jahr am wahrscheinlichsten auftauchende neue Varianten, Immunität sichern. Natürlich müssen solche Verfahren strengstens überwacht werden, damit das attenuierte, mit der Impfung verabreichte Virus weder durch neue Mutationen noch durch Rekombination, seine Virulenz zurückgewinnen kann. Ein gutes Beispiel für attenuierte Vakzine, deren Weiterentwicklung ein dringendes Problem ist, ist das BCG Vakzin. Der gegen Tuberkulose (Mycobacterium tuberculosis) eingesetzte Impfstoff beinhaltet ein durch gezielte Züchtung Bakterium, das erzeugtes Bacillus verwandtes Calmette-Guérin (BCG). Dies ist ein Mycobacterium bovis Stamm, dessen Genom mehrere Gendeletionen beinhaltet und deswegen stark attenuiert ist, aber wegen gemeinsamen Antigenen eine Kreuzreaktion gegen das M. Abbildung 7.10. Entwicklung von inaktivierten, nicht pathogenen Mutanten eines Erregers. 124 tuberculosis auslöst. In vielen Ländern ist zwar die Bevölkerung virtuell vollständig mit BCG geimpft worden, dennoch kehrt Tuberkulose in den letzten Jahrzehnten fast überall in der Welt zurück. Heute steht schon fest, dass trotz dem anfänglichen Erfolg, die Wirksamkeit der Impfung über die Jahrzehnte ständig nachgelassen hat. Heute schon gibt sie keinen vollen Schutz mehr gegen TBC. Andererseits ist sie immer noch effizient genug gegen die schwersten und lebensbedrohlichen Formen von TBC: der Miliartuberkulose und der tuberkulösen Meningitis. Wegen der verminderten Wirksamkeit haben mehrere Länder (z.B. Deutschland) die BCG-Impfung aus dem Impfkalender gestrichen, weil andere (z.B. Ungarn) die Impfung immer noch anwenden. Eine vergleichende Genomanalyse der BCG Stämme der Vakzin und der heute häufigsten M. tuberculosis hat bewiesen, dass durch die ständige künstliche Kultivierung der BCG, Mutationen entstanden sind. Durch diese Mutationen ist der BCG Impfstoff in dem menschlichen Wirtsorganismus nicht mehr so aktiv wie seine ursprünglichen Formen vor Jahrzehnten, als die Impfung eingeführt worden ist. Dies ist der Grund, warum die heutigen BCG Vakzine das Immunsystem nicht mehr so wirksam aktivieren, wie vor einigen Jahrzehnten. Im Falle der Untereinheitimpfstoffe beinhaltet das Vakzin nur ein Teil des Krankheitserregers, ein Molekül oder ein molekulares Fragment welches das protektive Epitop trägt. Zum Beispiel beinhalten manche Influenza Vakzine nur gereinigtes Hemagglutinin und Neuraminidase Proteine. Also nur die wichtigsten Oberflächenantigen des Virus und alle anderen Komponente des Virus werden während der Herstellung des Vakzins entfernt. Für die Weiterentwicklung der Untereinheitimpfstoffe ist das beste Beispiel vielleicht die neueste Generation des Hib (Haemophilus influenzae B) Vakzins für Kleinkinder, welche Hib-verursachte Meningitis und Epiglottitis vorbeugen. Die ursprüngliche Form des Vakzins beinhaltete nur gereinigtes H. influenzae Kapsel-polysaccharid. Es hat sich aber herausgestellt, dass dieses Vakzin in der Praxis nicht immunogen genug war. In der heute benutzten Form des Anti-Hib Vakzins ist deswegen das Kapselpolysaccharid zu einem Carrier-Protein gebunden. Da dieser Konjugatimpfstoff sowohl aus einem TI Polysacharid-, als auch aus einem TD Peptidantigen besteht, ist das Vakzin in der Lage beide (TD und TI) Antigenantworten auszulösen. Das Carrier-Protein PRP-D (Polyribosil-RibitolPhosphat-Diphtherietoxoid) regt spezifische T-Zellen an, die durch Ihre Zytokinsekretion so ein Zytokin-Milieu kreieren, welches auch die Aktivierung von B-Zellen unterstützt. Diese erkennen nicht das Protein, sondern die Zuckerkomponente des Impfstoffs. (Das PRP-D Toxoid ist ein Exotoxin, dessen Toxizität durch Formaldehydbehandlung eliminiert wurde, ohne dabei seine Immunogenität zu verringern). 7.4.2. Biotechnologische Impfstoffentwicklung Rekombinate Proteinvakzine werden hergestellt indem Gene von bestimmten immunogenen Proteinen eines Krankheitserregers kloniert, in eine Zellkultur (oft Hefezellen) transgenenetisch eingeführt, und exprimiert werden. Auf diese Weise wird zum Beispiel das heutige Vakzin gegen Hepatitis B industriell hergestellt. Dazu werden Hefezellen verwendet, welche ein Kapsidprotein des Virus in industriellen Mengen sekretieren. Neulich wird aber neben Bakterien, Hefe und ähnliche Einzeller, oder menschliche Zellen auch mit pflanzlichen Zellen experimentiert. Dies verspricht neue Wege der Verabreichung von Impfstoffen: Falls essbare Pflanzen auch Proteine von Krankheitserreger transgenisch exprimieren könnten, wäre es möglich, Vakzine einfach als Essen zu verabreichen. Beispielsweise wurde das Choleratoxin B-t schon erfolgreich transgenetisch in Kartoffeln eingeführt und exprimiert. In Mausexperimenten konnte so sowohl lokal, als auch systemisch die Bildung von Antikörpern hervorgerufen werden. Bei der neuesten Generation des Vakzins wird das Ziel der verbesserten Immunogenität häufig durch eine vorsichtige Selektion des besten Epitops bzw. Adjuvans erreicht. Vor allem bei experimentellen synthetischen Peptidvakzinen liegt der Schwerpunkt in der Identifizierung oder Erzeugung von den bestmöglichen T- und B-Zell-Epitopen. Herstellung von solchen Vakzinen kann entweder mittels konventioneller Laborstrategien oder durch die sogenannte in silico (durch Computer-basierte) Epitop-Analyse erreicht werden. Konventionelle (wet lab) Strategie: Der Krankheitserreger Komponente werden konventioneller bzw. mit seine Hilfe von Labortechnologie vermehrt oder einzeln kloniert, überlappende Peptide dieser Antigenproteine werden einzeln synthetisiert, und letztlich wird ihre Immunogenität einzeln ausgewertet um die besten Epitope für das Vakzin zu finden. Reverse (in silico) Strategie: Die Analyse geht nicht, wie gewöhnlich, von dem Pathogen oder ihren Komponenten aus, sondern aus seinem Genom, also der Sequenz welche diese Eigenschaften bestimmt (reverse Strategie). Die genomische Sequenz bioinformatischen Techniken wird mit analysiert (Computer-basierende, also “in silico“ Analyse), um die bestmöglichen Epitope zu finden, ohne dabei alle Komponente des Pathogens manuell in einem Labor analysieren zu müssen. Nur die Antigene oder Epitope, welche in der ComputerAnalyse am vielversprechendsten waren, werden kloniert oder synthetisiert, und letztlich wird ihre Immunogenität ausgewertet um die besten Epitope für das Vakzin zu finden. Abbildung 7.11. Die Entwicklung von einem Vakzin gegen Malaria. 126 Weil wir uns mit der Epitop-Prädiktion in einem anderen Kapitel im Detail beschäftigen, werden wir sie hier nur kurz, am Beispiel der Entwicklung eines zukünftigen Malaria-Vakzins erklären. Populationsgenetische Untersuchungen haben ergeben, dass eine allelische Variante des MHCI, nämlich HLA-B53, stark mit einem genetisch bedingten Schutz vor Malariainfektion assoziiert ist. Als antigenpräsentierendes Protein bindet das MHCI Variante HLA-B53 solche Nonapeptide, in denen in der zweiten Aminosäure-Position ein Prolin steht. Da die genomische Sequenz und dadurch alle Proteine des Erregers schon seit Jahren bekannt sind, kann man aus der genomischen Bibliothek auch eine Peptidbibliothek von Malaria erzeugen. Aus so einem Katalog aller möglichen Peptidfragmente der Proteine von Malaria, kann man auch alle Nonapeptide mit einem Prolin als zweite Aminosäure auswählen. In der Entwicklung eines zukünftigen Malaria-Vakzins werden diese Peptide künstlich synthetisiert, und es wird getestet, ob sie 1) tatsächlich zum HLA-B53 binden können 2) wenn ja, dann ob sie in mit Malaria infizierten Personen eine T-Zell-Antwort auslösen können 3) wenn ja, ob sie protektiv wirken und als Vakzin anwendbar sind. Dieses Vakzin sollte theoretisch effizient wirken, aber natürlich nur in solchen Personen, welche HLA-B53 tragen. Aus diesem Grunde muss man ein Vakzin herstellen, welche nicht nur aus diesem Peptid, sondern aus einer Mischung von Peptiden besteht, welche die häufigsten MHC-Varianten binden können, und dadurch der Mehrheit der Population Schutz vor Malaria verleihen. Eine alternative Lösung für dieses Problem wäre die sogenannte Epitop Affinität-Steigerung (epitopenhancement). Dies kann man mit zwei möglichen Strategien erreichen: MHC Affinität-Steigerung: Von den häufigsten Allelvarianten der MHC Proteine ist es meistens bekannt, Peptide welcher Länge sie bevorzugt binden, bzw. was für welche Aminosäure(n) und welcher Abschnitt genau in der gebundenen Peptidsequenz am wichtigsten für eine starke Bindung ist. Mit Hilfe dieser Information ist es möglich die Peptide eines Vakzins so zu modifizieren, dass sie an dieser kritischen Stelle(n) die Aminosäure(n) mit höchster Affinität zum häufigsten MHC Moleküle beinhalten, während die immunogenen Aminosäuren, also diejenigen, die nicht zum MHC, sondern vor allem zum TZR binden, unverändert bleiben. Mit anderen Worten, durch gezielte Manipulation der Peptidsequenz ist es möglich die Bindung zum MHC zu verstärken ohne dabei die Bindung zum TZR zu vermindern. TZR Affinität-Steigerung: Bei vielen Tumorvakzinen ist es ein Ziel die Anzahl der T Zellen, welche die Tumor-Epitope erkennen, auf ein mögliches Maximum zu steigern. Durch Manipulation der Peptidsequenz ist es machbar, dass bestimmte TZRs und dadurch bestimmte T Zell Populationen aktiviert werden, während andere gar nicht. Oder dass z.B nur. hochaffine T-Zell-Klone, nicht aber niedrigaffine Klonen induziert werden. Auf ähnlicher Weise werden bei manchen neuen experimentellen Virusvakzinen solche Peptide angewandt, welche gegen nicht nur einen, sondern gleichzeitig mehreren Stämmen des Virus protektiv wirken. Sie beinhalten ein bioinformatisch bestimmtes und synthetisch hergestelltes Peptid (ein sog. Peptidchimäre), welches in keinem der natürlichen Virusvarianten vorkommt, jedoch durch seine überlappenden Teile gegen eine Vielzahl der natürlichen Virusvarianten eine Immunantwort auslöst. Bezüglich der synthetischen Peptid-Vakzine muss letztlich auch bemerkt werden, dass reine Peptide von dieser Größe meistens kaum immunogen sind. Ihre Aufnahme durch APZs und die Reifung der APZ muss künstlich, durch die Impfung unterstützt werden. Bei Peptid-Vakzinen werden hierzu oft ISCOMs (immune stimulatory complex) benutzt, welche Liposom-ähnliche Lipidkomplexe sind. Sie dienen nicht nur als Carrier für die Peptide, sondern durch Membranfusion mit der APZ sorgen sie auch für ihre intrazelluläre Aufnahme und gleichzeitig üben sie eine immunstimulierende Wirkung auf die APZ aus. Ein weiterer Vorteil der ISCOMs in der Virusvakzination ist, dass mittels ISCOMs gelieferte exogen zugegebene Peptide wegen der Membranfusion in dem intrazellulären Raum der APZ gelangen. Deswegen werden sie als endogene Peptide bearbeitet, und in erster Linie auf MHCI präsentiert, So können antivirale CD8+ T Zellen stark induzieren werden. Das DNA–Vakzin ist eine durch Genmanipulation hergestellte rekombinante DNA-Sequenz. In der einfachsten Form besteht ein DNA-Vakzin aus einem Vektor mit einem eingebauten Gen, welches für das für die Vakzinierung verwendete Antigen kodiert. Der Vektor sorgt dafür, dass das Gen in die Zellen des Patienten eingeführt und exprimiert wird, wodurch eine Immunantwort gegen das Antigen aufgebaut werden kann. Auf ähnlicher Weise werden in manchen experimentellen Vakzinierungsversuchen auch Immunisierungen gegen Gen-Bibliotheken, also praktisch alle Gene eines Erregers, versucht. Nackte DNA Vakzine, in denen das Vektor einfach ein bakterielles Plasmid ist, sind in einer reinen Form relativ einfach herstellbar, weswegen sie in Vergleich mit anderen Vakzin-Arten als sehr sicher gelten. Dazu kommt, dass ein Plasmid, als bakterielle, also unmethylierte CpG DNA, durch die Aktivierung von TLR9 auch immunstimulatorisch auf das Immunsystem wirkt. Obwohl diese Technologie für menschliche Behandlung noch nicht zugelassen ist, ist sie definitiv vielversprechend und wirkt oft gut in Tiermodellen, wo das Plasmid häufig mit einer sog. “gene gun“ in den Körper eingeführt wird. Dies ist ein Kanonen-ähnliches Instrument welches mit Hilfe von komprimiertem Heliumgas mikroskopische Gold oder Platinpartikel in die Haut, und dadurch auch in die unterhalb gelegenen Muskelzellen schießt. Die Partikel tragen das Plasmidvektor auf ihrer Oberfläche, wodurch der Vektor in die Zellkerne von Haut- und Muskelzellen gleichzeitig eindringt. Dennoch ist ihre Effizienz unter in vivo Umständen relativ niedrig, weswegen intensive Forschung getrieben wird um andere mögliche, z.B. virale Vektoren auszuprobieren. Mit den Arten, Eigenschaften, Vor- und Nachteilen, und Anwendungsbereichen der unterschiedlichen gentechnologischen Vektoren werden wir uns im Detail im Fach Genetik beschäftigen. Abbildung 7.12. DNA-Vakzin. 128 7.4.3. Entwicklung von neuen Vakzinationsverfahren Für allerlei Vakzinationen stellt die gezielte Verabreichung des Antigens eine Herausforderung von kritischer Bedeutung dar. In experimentellen Vakzinen wird deswegen häufig die Immunisierung mit dendritischen Zellen: ausprobiert. Theoretisch soll sie einer der besten Wege sein, um ein Antigen sicher und gezielt in die sekundären Lymphorgane einzuführen. Diese Technologie beruht auf körpereigenen Monozyten, welche aus dem Blut der zu impfenden Person einfach isoliert werden und mit Hilfe von geeigneten Zytokinen in vitro in dendritische Zellen (DC) umgewandelt werden können (siehe Kapitel über Zellkultur). Solche DC sind dann relativ einfach mit Antigenen aufzuladen und in den Körper zurückzuführen. Mit Antigenen geladene DC lösen schnell antigenspezifische T-Zell-Antworten aus, und weil sie ohne Entzündung, also ohne eine parallel erscheinende Welle von inflammatorischen Zytokinen in den Lymphknoten auftauchen, werden sie relative wenig T-Effektor-, dagegen viel T-Gedächtniszellen erzeugen. Weil die Technologie für einfache Impfungen heute noch relativ umständlich und kostspielig ist, wird die Vakzination mit DC in erster Linie für Tumorvakzine entwickelt. Auch da bei diesen Krankheitsbildern die Erzeugung einer starken T-Zell-Antwort absolut essentiell ist. Übersetzt von Zoltán Pós 129 8. ZELLKULTUREN (SÁRA TÓTH) 8.1. Definition der Zell- und Gewebekultivierung, Typen und Formen der Kulturen Bei der Zell- und Gewebekultivierung werden die tierischen oder pflanzlichen Gewebemuster in vitro (d.h. in Reagenzglas in Laboratoriumsumständen) entweder zur Erhaltung oder zur Zellvermehrung gezüchtet. Im weiteren fassen wir die Informationen für die tierischen/humanen Gewebeproben zusammen. Für die Kultivierung vorgesehenen Gewebemuster können entweder durch eine Biopsie, durch eine Operation oder unmittelbar nach dem Tod entnommen werden. Abhängig von der Grösse und Beschaffenheit der, aus dem Körper entnommenen Gewebeprobe, sind drei Hauptsorten der Kultivierung unterschieden: Organkultur, Gewebekultur und die Zellkultur Unter Organkultur versteht man im wahren Sinne des Wortes keine eigentliche Züchtung des Organs, sondern es wird lediglich das Überleben der organbildenden Zellen gesichert, wodurch das gegebene Organ und dessen Funktionen aufrechterhalten wird. Im Grunde genommen werden die Organe zwischen der Organentnahme bis zur Transplantation (einige Stunden dauernde Zeitspanne) in einer Organkultur (Organaufrechterhaltung) aufbehalten. Für die Herstellung einer Organkultur verwendet man am häufigsten entsprechend kleine, embryonale Organe, wobei auch eine begrenzte Zellvermehrung und -Differenzierung stattfinden kann. Im Fall der Herstellung einer Gewebekultur wird vom Körper eine entsprechend kleine Gewebeprobe entfernt, und ihre ursprüngliche histologische Struktur behaltend wird das Gewebestück unter künstlichen Umständen erhalten. Während der Kultivierung vermehren sich die Zellen der Probe jedoch nur in begrenzter Anzahl. Am häufigsten verwendetes Kultivierungsverfahren ist die sog. Zellkultur, wobei Zellen eines aus dem Körper entfernten (gesundes oder tumoriges) Gewebestückes (meistens mit Hilfe bestimmter proteolytischer Enzyme) isoliert werden. Die derartig gewonnenen, miteinander nicht in der gewöhnlichen Gewebearchitektur vorhandenen Zellen bleiben erhalten und vermehren sich sogar. Die drei Sorten der Zellkulturen sind die primäre Zellkultur, der Zellstamm und die Zellinie. Eine primäre Kultur ist der eigentliche Beginn jeglicher Kulturen, wobei die Zellen aus direkt aus dem Körper entnommenen Gewebe stammen: die Zellen des Gewebestückes werden so voneinander und auch von der extrazellulären Matrix isoliert, dass nur ein einziger Zelltyp für das Kultivierungsverfahren übrig bleibt. Wenn, die in vitro Umstände entsprechend sind, werden die Zellen nicht nur überleben, sondern beginnen sich auch zu vermehren, und für den Zelltyp charakteristische physiologische Parameter zeigen. 130 In dem Fall, indem die Zellen sich vermehren und weiterhin züchtbar sind, entstehen für uns zwei Möglichkeiten: Entweder es entsteht ein sog. Zellstamm, der nur für begrenzte Zeit (einige Wochen, Maximum einige Monate) aufrechterhalten werden kann, aber alle physiologischen Parameter, dem Zelltyp entsprechend, zeigt. Die Chromosomenzahl der Zellen bleibt der Art entsprechend auch normal. Oder es entsteht eine sog. Zellinie, die aus sog. transformierten Zellen besteht. Diese Zellen sind am häufigsten aneuploide, d.h. ihre Chromosomenzahlen weichen von arttypischen Chromosomenzahlen ab, werden aber unsterblich, sozusagen für unbegrenzte Zeit (Jahren, Jahrzehnten lang) haltbar. Sie zeigen weder die arttypischen physiologischen, noch die Gewebeeigenschaften, oder es sind nicht alle Eigenschaften/Funktionen vorhanden. Die Zelltransformation kann spontan während der in vitro Kultivierung entstehen, oder schon vor der Entnahme im Gewebe vorhanden gewesen sein (z.B. bei Tumorgeweben). Wahrscheinlich ist die bekannteste humane Zellinie die HeLa-Zellinie. Diese Kultur wurde von Gebärmutterkrebs (cervicales Adenocarcinom) Frau Henrietta Lachs in USA im Jahre 1951 entnommen, und wird seit dem kultiviert, und diese Zellinie ist bis heute erhalten geblieben! Die Zellen der Zellstämme und Zellinien werden in einem speziellen Nährmedium, welches 10% krioprotektive Substanz (in allg. Dimethylsulphoxide /DMSO/ oder Glyzerin) enthält, gefroren und in flüssigen N2, bei -196 °C in sog. Zellbänken langzeitig erhalten. Grosse internationale Zellbänke (www.atcc.org und www.hpacultures.org.uk) geben uns die Möglichkeit, bestimmte Untersuchungen (wie z.B. eine Arzneimitteltestung) in unterschiedlichen Zeitpunkten und geographischen Orten an denselben biologischen Objekten durchführen zu können. Dieses bewirkt eine ausserordentliche Reproduzierbarkeit und Vergleichbarkeit der Versuchsergebnisse. Die Zellkulturen – Abhängig von den Ausgangsproben – sind entweder monolayer (einschichtige) Zellkulturen, oder Suspensionskulturen. In einer sog. monolayer (einschichtige, anhaftende) Zellkultur haften die Zellen an eine bestimmte Unterlage (an Boden der Kulivierungsschale), breiten sich aus und bilden so eine einzige Zellschicht. Die Zellen benötigen enge Zell-Matrix und oft auch enge Zell-Zell Beziehung, d.h, man soll für Kultivierung haftungsfähiger Zellen einen entsprechenden Nährboden und Matrix anbieten. Eine Suspensionszellkultur ist für Zellkulturen, die aus hämatopoetischen Zellen stammen charakteristisch; diese Zellen benötigen während der Kultivierung keine Matrix, Zell-Zell Kontakte sind nicht charakteristisch. Seltener, jedoch möglich ist auch die stark transformierten Zellen in Suspensionskulturen zu erhalten, da diese Zellen für die normalen Zellen charakteristische Fähigkeiten wie die sog. Kontakthemmung (d.h. die Vermehrungsfähigkeit der Zellen ist von Zelldichte abhängig) oft schon früher verloren haben. In einer monolayer Zellkultur (Zellkultur angehafteten Zellen) werden die Zellen durch Vermehrung früher oder später einander erreichen, so kommt eine Zelle mit Zahlreichen anderen Zellen in Berührung, und wegen dieses Kontaktes wird die Vermehrung der normalen (nicht transformierten) Zellen gehemmt. Sonstige Funktionen werden 131 jedoch nicht geschädigt. Für transformierte Zellen ist die Kontakthemmung nicht typisch; sie teilen sich weiter, wachsen aufeinander zu und gestalten damit eine mehrreihige Zellkultur, im extremsten Fall können sie sich sogar auch in einer Zellsuspension weiter vermehren. 8.2. Für eine Zellkultur notwendigen Voraussetzungen Für die Zellkulturen (für das Aufrechterhalten ihrer Funktionen und um die Zellvermehrungsfähigkeiten zu sichern) müssen die im lebenden Organismus vorhandenen Zustände ziemlich genau nachgeahmt werden. Physikalische Parameter (z.B. Temperatur, osmotischer Druck), chemische Parameter (z.B. pH, CO2-Konzentration, entsprechende Konzentration der Nährstoffe usw.) sowie die biologischen Parameter (z.B. extrazellulärer Matrix) müssen dem lebendigen Organismus entsprechende Werte erreichen oder annähern. Gelöste Substanzen Zell – Zell- Zell – MatrixWechselwirkungen Wechselwirkungen Physikalische Parameter Nährmedium Zellwechselwirkungen: Kultivierungsoberfläche z.B. Temperatur Blutserum Homologe =:Zell-Densität Kunststoff, pH (Indikator + Puffer) Hormone Heterologe = gemeinsame Kultivierung (coculture) Glas, Membrane, Oxygen-/KohlendioxidKonzentration Metaboliten und Produkte Mikroperlen Wasserdampfgehalt Autokrine und Parakrine Wirkung Überzug (coat) z.B. Osmolarität Kollagen, Statische oder dynamische Kultur Wachstumsfaktoren Haftungsfaktoren Stoffwechselrate Gelatine, feeder layer (nährende Zellschicht) Matrixgel, Biomatrix Tabelle 8.1. Notwendige Umstände der Zellkulturen Für Kultivierung tierischer Zellen braucht man irgendwelche flüssigen oder seltener (in erster Linie im Fall der Kultivierung der hämatopoetischen Zellen) halb-flüssige (semi-soliden) Mittel, sog. Kultivierungsmedium (Nährmedium). Diese sind synthetische Medien, d.h. ist ihre Zusammensetzung bekannt, und sie beinhalten die notwendigen Salze, Aminosäuren, Kohlenhydrate, Nukleotiden und Vitamine. Das energiesichernde Kohlenhydrat ist am häufigsten Glukose, seltener benutzt man Fruktose oder Galaktose. Der durchschnittliche Zuckergehalt beträgt vom 1,0 g/l bis 4,5 g/l. Höheren Zuckergehalt benötigen die sich schnell teilenden embryonalen und die Tumorzellen, oder die Glukose schnell verbrauchenden (metabolisierende) glykolytisch sehr aktiven Zellen. Die Nährmedien beinhalten auch einen Puffer (z.B. Phosphat oder HEPES) und einen Indikator (am häufigsten Phenolrot), welche ermöglichen sowohl die Verminderung der, wegen der Stoffwechselvorgänge erfolgenden pH-Veränderungen, als auch die Nachfolgerungen einer pHÄnderung. Für die meisten Zellkulturen beträgt der optimale pH-Wert 6,8 - 7,2. 132 Weil die Zellen für normale Funktionen auch bestimmte Proteine (wie z.B. Wachstumsfaktoren, usw.) und Lipide benötigen, müssen auch diese Substanzen gesichert werden, am häufigsten durch Zugabe von tierischem Blutserum. Für die meisten menschlichen und tierischen Zellen entspricht das 5 - 10%ige fetale Kalbserum (FKS, fetal calb serum, FCS) enthaltende Nährmedien. Es wird kein humanes Blutserum benutzt, weil einerseits dessen Zugänglichkeit begrenzt ist, anderseits dessen Sicherheitsrisiko (Infektionsgefahr) grösser ist. Weiterhin ist der Antikörpergehalt des fetalen Blutserums sehr gering und mit Hitzebehandlung ist auch die Aktivierung des Komplementsystems leicht eliminierbar. Das Blutserum ist kein synthetisches Zuschlagsmaterial, seine genaue Zusammensetzung ändert sich von Charge zu Charge, deshalb können sich auch die Konzentrationen der sich im Blutserum befindlichen wachstumsfördernden und wachstumshemmenden Faktoren stark unterscheiden. (Zweckmässigerweise werden die Proben der einzelnen Chargen an den verwendeten Zellkulturen vorgetestet.) Obwohl das Serum von kontrollierten, für die Abnahme gezüchteten Herden gewonnen wird, wo das Vermeiden der mikrobiellen Kontamination (Bakterien, Mycoplasmen, Viren, Prionen, Pilze) strengstes kontrolliert ist, wird für die sehr empfindlichen Zellkulturen Blutserum aus Südamerika (wo BSE noch nicht vorgekommen war) empfohlen. Wegen dieser eigenartigen Problematik werden zur Zeit, immer intensiver, synthetische, mit bekannter Zusammensetzung Serumersatzmittel benutzt. Ihre breite Verwendung ist jedoch wegen des hohen Preises begrenzt. In vielen Fällen wird sog. konditioniertes Medium verwendet, das jedoch nicht anders ist als ein von anderen Zellkultur weggenommenes, „gebrauchtes“ Nährmedium, in dem die gezüchteten Zellen verschiedene, sogar chemisch noch nicht definierte Substanzen abgegeben haben. Dieses wird als Ergänzung zum frischen synthetischen Nährmedium der Zellkulturen von gleicher oder von anderer Zellart gegeben, um eine sehr erfolgreiche Kultur zu erhalten .Während der Kultivierung verbrauchen die Zellen die zur Verfügung stehenden Nährstoffe, und geben gleichzeitig ihre Stoffwechselprodukte in das Nährmedium ab. Die Nährflüssigkeit wird dadurch nach 2-3 Tagen verbraucht und muss entfernt und mit frischer ersetzt werden (Nährmediumsaustausch). Die Oben genannten Umstände entsprechen den Bedürfnissen auch der prokariotischen Bakterien sehr gut, weshalb eine bakterielle Kontamination (Infektion) unbedingt verhindert werden soll. Zu einer erfolgreichen Kultivierung sind strenggenommene eine sterile Umgebung (durch sterile Kabinen, sog. Laminäre Boxen gesicherter Arbeitsplatz) und sterile Lösungen und Instrumente unbedingt erforderlich. Bezüglich der physikalischen Parameter sind für die meisten Zellarten 37 °C und 5% CO 2-Gehalt bei 80 – 90 % Luftfeuchtigkeit die besten Voraussetzungen. Dieses kann durch die sog. CO 2-Thermostate (Blutschranke) gesichert werden. Die O2-Konzentration ist kein limitierender Parameter, denn die atmosphärische O2-Konzentration ist den meisten Zellen vollkommen ausreichend. Gleichzeitig aber, ist die im Körper vorkommende (d.h. für die Zellen notwendige) ca. 5% CO 2-Konzentration ein Vielflaches der für die Atmosphäre typischen Konzentration (d.h. 0,035%). In bestimmten Fällen z.B. bei den in der Molekularbiologie vorkommenden Transfektionsverfahren (Ca-Präzipitation) ist aber eine niedrigere CO2-Konzentration erforderlich. Für das Bebrüten der Zellen werden heute schon einweg-Kunststoff-Gefässe, sog. Petri-Schalen und verschiedene Flaschen benutzt. Ihre Oberflächenstruktur und Fassungsvermögen (im Fall der 133 monolayer Zellkulturen) können abhängig der Versuchsanordnung unterschiedlich sein, von ganz kleinen von 200—800 μl Volumen bis mehrere Liter betragene sog. „Zellfabriken“. Im Handel angebotene Gefässe können von der Fabrik derart oberflächenbehandelt werden, dass die meisten Zelltypen für ihre Haftung eine günstige adhäsive Oberfläche besitzen. Bestimmte Zellen werden jedoch beim Vorhandensein einer entsprechenden extrazellulären Matrix empfindlich reagieren. Aus diesem Grund bietet der Handel mit verschiedenen Matrix Komponenten (z.B. mit Kollagen, Laminin, Ornithin, usw.) beschichtete Kultivierungsschalen an. In gut proliferierenden adhesiven (monolayer) Zellkulturen kann die Schalenoberfläche limitierend wirken, da diese einfach verbraucht wird. Deshalb müssen die Zellkulturen durch Aufteilung der Zellkultur, die sog. Subkultivierung (Passagieren), auf neue Oberflächen übertragen werden. Dabei müssen die Zellen enzymatisch (z.B. mit Trypsin oder mit Trypsin-EDTA) von der Gefässoberfläche entfernt werden. In einigen primären Zellkulturen die nicht aus einem embryonalen sondern aus einem erwachsenen Organismus stammen, kann deren extrazellulärer Matrix, die von den Zellen produziert wird, reich an Kollagen sein. In diesem Fall benutzt man für die Subkultivierung der Zellen solche Enzymgemische, welche Kollagenase und/oder evtl. DNA-se enthalten. Die letzte ist wichtig für den Abbau evtl. aus zugrundegegangenen Zellen freigewordene DNA. Es sind schnellwachsende Zellkulturen, deren Zellen aus Tumorgewebe stammen; für ihre Passage ist eine 0,02%-ige EDTA Lösung, d.h. eine 2+ einfache Anbindung der Ca -Ionen genügend. Wenn durch die enzymatische Behandlung der Kulturen die Zellen sich von der Oberfläche ablösen, – wir überzeugen uns davon durch mikroskopische Untersuchung –, werden die Verdauungsprozesse abgestellt. Dieses kann mittels spezifischer aber teuer Trypsin-Inhibitor oder durch die preisgünstigere Variante, zum gegebenen Zelltyp verwendbare, protein- (d,h. Blutserum-)haltigen Nährmedium erreicht werden. Die dadurch entstehene Zellsuspension wird abzentrifugiert, und durch das Abgiessen des Überstandes werden nicht nur die überschüssigen Enzyme, sondern auch die Bruchstücke der Zellen entfernt. Wenn man nun zu denen, sich im Pelett befindlichen Zellen, neues Medium zugibt, kann man eine neue Kultur anlegen. Für empfindlichstere Zellen oder wenn das Ziel der Untersuchung die, in kleinen Mengen vorkommenden Zelloberflächenmoleküle sind, können vom Handel angebotene verschiedene, nicht enzymatische Systeme benutzt werden. 8.3. Zellzahlveränderungen und unterschiedliche Zellzählungsmethoden Bei verschiedenen Untersuchungsanordungen müssen Zellen mit gleicher Anzahl in Parallelkulturen verwendet werden, nur dadurch wird gesichert statistisch zuverlässige, auswertbare und verlässlicherweise vergleichbare Ergebnisse zu erhalten. Dazu muss die Zellzahl der gegebenen Zellkulturen bestimmt werden. Für die Zellzählung wird eine Zellsuspension benötigt (entweder sind die Zellen von Anfang an in einer Zellsuspension kultiviert, oder im Fall einer monolayer Zellkultur sollen - während der Subkultivierung - in Suspension gebracht werden). 134 Üblicherweise geschieht die Zellzählung m.H. von sog. Hämozytometer (Zählkammer nach Bürker). Auf Grund, der mit einem Hämoztometer bestimmten Zellzahl ist die Anzahl der Zellen in 1 ml Suspension kalkulierbar (Abbildung 8.1.). Neuerdings werden auch verschiedene Zählautomaten verwendet. Abbildung 8.1. Anwendung der Zählkammer nach Bürker Aus der Sicht des Kultivierungsverfahrens ist die Anzahl der lebendigen Zellen viel wichtiger, als die Gesamtzellzahl der Kultur, d.h. die Bestimmung der Anzahl der lebendigen Zellen spielt eine ausserordentlich wichtige Rolle. Am häufigsten wird für die Bestimmung der Anzahl von lebendigen Zellen die sog. Trypanblau Exclusions(Verdrängungs)methode benutzt. Die Zellen werden mit 0.04 %iger Trypanblau-Lösung gefärbt (Zellsuspension/Farbstoffproportion ist 1:10, d.h. für die Zellsuspension bedeutet es eine 10-malige Verdünnung), wobei nur die abgestorbenen Zellen 135 angefärbt werden. Lebendige Zellen lassen kein Farbstoff durch ihre Plasmamembran. (Im solchen Fall wird in der zur Bestimmung der Gesamtzellzahl benutzten Formel noch der Faktor 10x – die Verdünnung der Zellsuspension von Farbstofflösung – als Multiplikator aufgenommen. Die Anzahl der lebendigen Zellen in 1ml Zellsuspension = die Anzahl im Hämozytometer über mit drei 2 Linien begrenzten Quadrant (deren Oberfläche 1 mm beträgt) betreffende mit Trypanblau nicht 4 angefärbten Zellen (keine blaue Zellen) x10 x10. Die Zellzählung mit Hämozytometer ist zeitaufwendig, braucht Erfahrung, jedoch lässt grösseren Raum für die individuelle Beurteilung – im Fall z.B. wenn der Zelltyp zur Klumpung neigt, was ausgewertet werden soll und ob die einzelnen Zellen von kleineren Zellgruppen separiert werden können. Zugleich arbeiten aber die Zellzählerautomaten (entweder anhand der elektrischen Leitfähigkeit oder Lichtstreuung) im streng begrenzten Aussenmassenbereich; jede, von der Grösse abweichende kleinere oder grössere Zellen, z.B. in der Anaphase sich befindende, elongierte oder durch Mutation zu aneuploid oder polyploid gewordene Zelle, sowie durch Zellaggregation entstandenen Zellgruppen werden von der Zählung ausgeschlossen, dadurch könnte eine heterogene Zellpopulation als homogene behandelt werden. Andererseits besteht bei den Automaten keine Möglichkeit für die Beobachtung der für den gegebenen Zelltyp charakteristischen morphologischen Veränderungen/Abweichungen. Zweckmässigerweise soll die Art der Zellzählung nach Erfordernissen des Laboratoriums, nach Erfahrung des Personals, nach den verwendeten Zelltypen (z.B. die Bestimmung der Anzahl der Zellen in einem Suspensionskultur von hämatopoetischen Zellen ist besser automatisierbar), und entsprechend der Versuchsrichtungen der Untersuchungen ausgewählt werden. 8.4. Separations- und Selektionsverfahren der Zellen Öfters ist die Ausgangskultur (primäre Zellkultur) noch heterogen, sie besteht aus unterschiedlichen Zellen, da das Ursprungsgewebe verschiedene Zelltypen enthält; für die Untersuchung kann jedoch nur ein bestimmter Typ von Zellen benötigt werden. Für Herstellung von einer homogenen Zellsuspension, die nur einen einzigen Zelltyp beinhaltet, verwendet man entweder spezifische Antikörper gegen den auf den gegebenen Zelltyp charakteristischen Zelloberflächenmarker, oder die verschiedenen Zelltypen können nach deren Grösse m.H. durch Dichtegradientenzentrifugation separiert werden. In diesem Fall wird für die Herstellung des Dichtegradientes FICOLL (ein Gemisch von neutralen, ® wasser- löslichen, verzweigten Polysacchariden mit grosser Molekularmasse) verwendet (Abbildung 8.2.). Abbildung 8.2. Mit Hilfe von Dichtegradientenzentrifugation erfolgte Leukozytenseparation 136 Heutzutage wird von den auf Antikörperbildung beruhenden Zellseparationsverfahren am häufigsten das negative oder positive Selektionsverfahren m.H. von MACS-Mikroperlen benutzt (Abbildung 8.3.). Dem zu Folge werden Antikörper gegen charakteristische (d.h. in anderen Zellen nicht vorhandenen) Oberflächenantigenen auf den zu selektionierenden Zelltyp an Oberflächen von magnetische Nanoteilchen (Mikroperle) gebunden. Wenn die Zellsuspension über eine bestimmte Zeit lang mit den magnetisierten (magnetmarkierten) Antikörpermolekülen inkubiert wird, werden die Magnetteilchen durch auf ihren Oberflächen befindlichen Antikörpermolekülen zu den entsprechenden Zellen, die den Oberflächenmarker tragen, gebunden. Auf dieser Weise werden die oberflächenmarkertragenden Zellen magnetisch markiert, und die anderen Zellen, die die gegebenen Oberflächenmarker nicht tragen, werden nicht markiert. Die magnetische Oberflächenmarkierung der Zellen (d.h. die Anbindung der magnetisierten Antikörpermoleküle an den Oberflächenantigenen) verändert weder die Zellstruktur, noch die Zellfunktion oder die Aktivität der Zelle. Abbildung 8.3. Prinzip der magnetischen Selektion der Zellen Die Zellsuspension, die aus einer Mischung von unmarkierten und markierten Zellen besteht, wird in einer Säule (d.h. in einem Rohr) in einen sog. MACS Magnetseparator gestellt. Durch das im Gerät herrschende magnetischen Kraftfeld werden die magnetisch markierten Zellen in der Säule zurückgehalten, jedoch die unmarkierten Zellen nicht, die durch die Säule abfliessen und verlassen das Gerät. 137 Bei dem sog. negativen Selektionsverfahren finden, die vom magnetischen Kraftfeld zurückgehaltenen Zellen, kein Gebrauch mehr, nur die in der Durchflussfraktion vorhandenen unmarkierten Zellen. Auf dieser Art wurde die Probe (d.h. die Zellsuspension) von den unerwünschten, die Untersuchung störenden Zellen gereinigt. Im sog. positiven Selektionsverfahren werden die jenige Zellen benötigt, die im Magnetseparator zurückgehalten worden sind (d.h. welche markiert sind). Die durchfliessende Fraktion wird abgegossen, die Säule wird vom Separator entfernt, ausserhalb des magnetischen Kraftfeldes fliessen die magnetisch markierten Zellen durch die Säule; diese Zellfraktion kann für weitere Untersuchungen aufgefangen und benutzt werden. Im Fall der Untersuchung von Knockenmark- oder peripheren Blutzellen ist für die Separation des gewünschten Zelltypes auch ein anderes, ebenso ein antikörperverbrauchendes, auf der Rosettenbildung basierendes Verfahren geeignet (Abbildung 8.4.). Es wird ein spezifischer tetramer Antikörperkomplex verwendet (z.B. ® RosettaSep ) der Kreuzverbindungen zwischen den unerwünschten Zellen und Erythrozyten bildet. Diese kreuzverbundenen Zellgruppen sind als „Rosetten“ (daher der Name des Verfahrens) im Lichtmikroskop erkennbar. Der nächste Schritt ist die Dichtegradientenzentrifugation. Dabei werden sich die Rosetten am Boden der Zentrifugenröhre als Pellett absetzen, im Gegensatz dazu werden sich die unmarkierten, sich nicht in Rosetten befindenden Zellen, entsprechend deren Dichte, in dem oberen Teil der Zentrifugenröhre, in der Flüssigkeit (in der Supernatant) befinden. Aus dieser Position können die Zellen für weitere Untersuchung gesammelt werden. Auf ähnlicher Weise funktioniert das auch markierte Antikörper benutzende FACS-Verfahren, welches für die Separation der gewünschten Zellen geeignet ist (siehe separates Kapitel). Abbildung 8.4. Zellseparationsverfahren auf Grund der Rosettenbildung 138 Von den Leukozyten ist die selektive Vermehrung der verschiedenen Lymphozyten auch möglich. Da die Lymphozyten im peripheren Blut, sich in der G 0-Phase befinden, können mit entsprechenden Induktionsmitteln (z.B. mit ConA oder mit LPS) entweder die T- oder die B-Lymphozyten zur Lymphoblasten umgewandelt werden (sog. Blast-Transformation), d.h. von G0 zur G1-Phase, und so zur Vermehrung gebracht werden (Tabelle 8.2.); damit ist es möglich deren Anhäufung zu erreichen. Mitogene Substanz Zielzelle Concanaralin A (ConA) Phytohämagglutinin (PHA) Kermesbeeren (Pokeweed, PWM) T-Zellen Lypopolysaccharide (LPS) B-Zellen T- und B-Zellen Tabelle 2. Mitogene Substanze Dieses Verfahren sogar zu der, durch ein Antigen ausgelöste T-Zell Proliferation sich ähnelt, ist aber aspezifisch, in diesem Fall werden alle T- und/oder B-Zellen das Blast-Stadium erreichen, nicht nur die antigenspezifische. Zur Routine Chromosomenuntersuchungen wird auch die lymphoblastische Transformation durch pflanzliche Lektine (Phytohämagglutinin, PHA) erreicht, denn durch eine einfache Blutabnahme vom peripheren Blut nach 48–72 Stunden dauernde Kultivierung viele teilende Lymphoblasten gewonnen werden können. Zur Karyotypisierung notwendige Menge den Methaphasenzellen stehen schnell zur Verfügung. Für die Separation von Zellpopulationen mit Knochenmarksursprung, steht ein einfaches Verfahren zur Verfügung, wobei die Adhesionsunterschiede der einzelnen Zellpopulationen ausgenutzt werden. Die aus dem Knochenmark frisch isolierte Monozyten/Makraphagenpopulation haftet relativ rasch an den Boden des Kultivierungsgefässes, bis die Lymphozyten und Granulozyten sich gar nicht anhaften. Nach einigen Stunden Inkubation wird das Nährmedium abgegossen, so dass auch die nicht anheftenden Zellen entfernt werden und die angereicherte Monozyten/Makrophagenfraktion weiter kultiviert werden kann. 8.5. Einige Verwendungsarten der Zellkulturen Die Bedeutung des Zellkulturverfahrens liegt vor allem darin, dass mit seiner Hilfe ein Teil von Tierversuchen, bzw. solche Versuche die aus ethischen, moralischen oder juristischen Gründen an Menschen nicht ausführbar sind, in Laborverhältnissen ersetzt werden können. Diese Versuche sind genau planbar und reproduzierbar. Das Zellkulturverfahren bietet Möglichkeiten für Untersuchungen der meisten Funktion von zahlreichen Zell- und Gewebearten, die von der komplizierten Wechselwirkung des Körpers entrissen wurden, sogar spezielle Lebensfunktionen differnzierter Zellen können auch studiert werden. Gleichzeitig aber kann die Tatsache, dass die kultivierten Zellen aus dem Körper isoliert sind, ein Hindernis des in vitro Zuchtverfahrens darstellen; die Funktion von Zellen 139 und ihr Verhalten im Organismus sind noch von zahlreichen anderen Zelltypen beeinflusst, weshalb die erhaltenen Daten nicht auf die in vivo-Zustände direkt bezogen werden können. Dies auszuschliessen, werden immer häufiger die sog. Kokultivierungssysteme verwendet, in denen verschiedene Zelltypen in gemeinsamer Nährlösung gezüchtet werden, so dass die einzelnen Zelltypen voneinander mit Hilfe von Membran-Insert getrennt sind. Auf dieser Weise sind die durch die löslichen Molekülen vermittelten Wechselwirkungen zwischen den einzelnen Zellarten nachweisbar und nachvollziehbar. Weitere Überlegungen fordert die Tatsache, dass besonders während einer längeren Kultivierungszeit von Zellinien, bedeutende Anhäufung von Mutationen, und Selektionen auftreten können. Heute kann man behaupten, dass die Zell-Linien unter in vitro Umständen an einem spontanen Evolutionsvorgang teilnehmen. Folglich bedeutet die Anwendung der gleichen Zellinie nicht unbedingt ein StandardSystem. Bekanntlich besitzen Zellen breitbasig verwedeter Zellinien eine für die Zellkultur typische sog. modale-Chromosomenanzahl, die von der Chromosomenzahl der gegebenen Art auch abweichen kann. Diese Zahl beträgt im Fall der CHO-Zellinie 20 (jedoch ist diefür den chinesischen Hamster charakteristische Chromosomenzahl: 2n=22), bzw. bei der Hela-Zellen 56 (welche von der normalen humanen Chromosomenzahl 2n=46 weit enfernt liegt). Deshalb schenkt man heutzutage erhöhte Aufmerksamkeit den primären Zellkulturen und Zellstämmen. Heute lagern und vertreiben die Zellbanken immer mehr solche Zellen oder Zellstämme, die mit ihren Eigenschaften annähernd bei der Norm liegen. 8.5.1. Bestimmung der Plating-Effizienz Die Plating-Effizienz (PE) ist diejenige Indexzahl, die die Wirksamkeit der Explantierung bestimmt. Diese Zahl gibt an, wieviel Prozent der explantierten Zellen in der Zellkultur Zellkolonien zustande bringen werden können (anders ausgedrückt: wieviele Zellkolonien aus einer einzelnen Zelle entspringen); PE = Anzahl der Zellkolonien/Anzahl der explantierten Zellen X 100. Mit Hilfe der Empfindlichkeit dieses Verfahrens kann man den Nährstoffbedarf unterschiedlicher Zelltypen bestimmen, bzw. ist es so auch möglich herauszufinden, welche Serum-Chargen sich für die Kultur am besten eignen oder auch die Zytotoxizität zu bestimmen. 8.5.2. Bestimmung der Proliferation und der Vermehrungsfähigkeit der Zellen Die Proliferationsfähigkeit von Zellen wird vor allem mit den zugegebenen verschiedenen Wachstumsfaktoren / Zytokinen gemessen, bzw. üblicherweise, in den Versuchssystemen wegen unterschiedlichen Testsubstanzen bestimmt. Für die Bestimmung der Proliferation, bzw. dem Gegenteil: der Zytotoxizität, stehen zahlreiche Methoden zur Verfügung. Im sog. direkten Verfahren mit irgendwelchem – bereits früher erwähnten – Verfahren (z.B. mit Hämozytometer oder mit Hilfe von automatischen Zellzählern) kann die Zellzahl (Zellzahländerung in Abhängigkeit der Zeit) unmittelbar bestimmt werden. 140 Mit dem sog. indirekten Verfahrenwerden irgendwelche, sich mit der Zellzahl (mit Änderung der Zellzahl) proportional ändernde Parameter (z.B. der Einbau eines radioaktives Isotopes, oder die metabolische Aktivitätsänderung eines Enzyms der Zelle) bestimmt. Vom indirekten Verfahren ist die älteste und zugleich die bekannteste Methode das Thymidin3 3 Inkorporations-Assay. In dieser Methode wird mit Tritium ( H) markiertes Thymidin ( H-Thymidin) verwendet. Das markierte Thymidin wird während der DNA-Replikation in den neu synthetisierten DNA-Strang eingebaut. So ist das Mass der Inkorporation mit der Anzahl der DNA Replikationen und damit auch mit der Zahl der Zellteilungen proportional. Mit Hilfe von einem Szintillations-β-Zähler wird die Radioaktivität das Lysatum von behandelten und unbehandelten Zellen gemessen, und daraus kann auf die Auswirkung der Testsubstanz auf die Zellproliferation geschlossen werden. In letzter Zeit bestrebt man immer öfters anstatt von radioaktiven Isotopen, viel mehr nicht-radioaktive Substanzen zu verwenden. Aus diesem Grund sind mehrere solche Proliferations-Assays ausgearbeitet worden, welche auf anderen Grundprinzipien basiert sind. Eine von diesen ist das auf 5Brom-Desoxyuridin (BrdU)-Inkorporation basierte Verfahren. Der BrdU ist ein (nicht strahlendes) Thymin-Analog, welcher bei der DNA-Replikation in die neu synthetisierten Ketten (als Basenpaare von Adenin) anstatt Thymin-Basen eingebaut wird. Darauffolgend können - mit Anwendung Anti-BrdUAntikörper (ohne Zellyse) mit einer immunzytochemischen Reaktion - die Zellen, welche während der Behandlung BrdU eingebaut haben (das heisst, solche Zellen, welche während dieser Zeit in Teilung – in der S-Phase des Zellzykluses – waren) nachgewiesen werden. Dementsprechend kann die Proportion der BrdU-beinhaltenden (markierten) und der nicht BrdU-haltigen (unmarkierten) Zellen, und somit auch die Wirkung einer Testsubstanz auf der Proliferationsrate bestimmt werden. Neuerdings wird ein mit einem Fluorochrom (5-Ethynilil -2 Desoxyuridin) markiertes Nucleosid verwendet und somit braucht man keine Antikörper für die unmittelbare Verfolgung der Zellproliferationsänderungen. Andere grosse Gruppen der indirekten Proliferations-Assays messen die Aktivitätsänderungen der mitochondrialen Enzyme. Bei dem sog. MTT-Assay wandelt sich in lebendigen Zellen das wasserlösliche MTT (ein Tetrasolium-Salz) durch die Wirkung von mitochondrialen Enzymen (Dehydrogenasen) in unlösbare braun-lila Formasan um. Diese Substanz wird erst solubilisiert, dann kann die Formasan Konzentration bei 570 nm photometrisch bestimmt werden. Die FormasanKonzentration ist mit der Anzahl lebendiger Zellen proportional. Es ist empfohlen, Zellzahlbestimmungen die auf metabolischen Veränderungen beruhen, mit einem direkten oder auf Basisanalogen beruhende indirektem Verfahren zu kontrollieren, denn es könnte vorkommen, dass die Testsubstanz unmittelbar auf mitochondiale Enzyme wirkend, die FormasanMenge senkt, ohne dass sich die Zellzahl dabei verringern würde. Gleichzeitig soll jedoch betont werden, dass die Zellzahl auch deshalb sinken könnte, weil die Testsubstanz einfach toxisch war. In diesem Fall ist die Zellzahl-Verringerung nicht als Resultat der Proliferationsratenrückganges anzusehen, sondern als Folge von Zelluntergang. 141 8.5.3. Abmessung der Zytotoxizität Obwohl die Zellproliferation und der Zelluntergang zusammengehörende Erscheinungen sind, während des Assays – jedoch vom Versuchszweck und Testsubstanz abhängig – sind, haben entweder die Anzahl lebendigen Zellen oder sogar die Absterberate von Zellen eine Bedeutung. In einen Teil des oben erwähnten Zellproliferationsverfahrens kann ausgehend von einer bekannten Zellzahl gleichzeitig auf die proliferierenden (d.h. auf die lebendigen) Zellen und auch auf den Zelluntergang (z.B. die plating Effizienz oder der MTT Assays) geschlossen werden. Ein anderer Bereich der Zytotoxizitätsbestimmung beruht auf der Untersuchung der Plasmamembranintegrität. Eine solche Methode ist, die bei der Zellzahlbestimmung früher erwähnte Trypanbau-Färbung, wobei nur die Zellen mit nicht intakten Zellmembranen (d.h. Zellen die nicht mehr lebendig sind) den Farbstoff aufnehmen, oder die bei der Durchflusszytometrie (FACS) besprochene Propidium-Jodid-Färbung, wobei der Farbstoff ebenfalls nur durch Zellmembranen von abgestorbenen Zellen in das Zellinnere gelangen kann. Mit anderen Verfahren zur Zytotoxizitätsbestimmung können Membrantransporte solcher Substanzen untersucht werden, die innerhalb der Zelle nicht aktiv sind, jedoch aus den Zellen aktiv befördert werden oder umgekehrt. Solche Substanzen sind einerseits die Laktose-Dehydrogenase, anderseits auch für die lebendigen oder für die abgestorbenen Zellen charakteristischen Proteasen. Im Falle der anhaftungsfähigen Zellen gehören zu den neuesten Methoden Messungen,, welche auf dem Prinzip der elektrischen Impedanz basieren (z.B. ECIS. siehe im Kapitel, Migration der Zellen), wobei durch die Erhöhung der abgestorbenen, nicht mehr anhaftungsfähigen Zellen sich die elektrische Impedanz verringert, d.h. eher die Kinetik wird als Endergebnis des Zellunterganges untersucht. 8.5.4. Herstellung der monoklonalen Antikörper Abbildung 8.5. Selektionsverfahren der Hybridomzellen 142 Eine hervorragende Bedeutung des Anwendungsbereichs der Zellsuspensionskulturen hat die Herstellung von sog. monoklonalen Antikörpern. In diesem Verfahren werden erst von der Milz mit entsprechendem Antigen immunisierte Tiere antikörperproduzierende B-Zellen isoliert, diese werden mit Hilfe von einem die Zellmembran destabilisierendem Faktor (z.B. Poliethylen-Glykol oder SendaiVirus) mit unbegrenzt teilungsfähigen Myelomzellen (B-Zellinie) fusioniert. Diese Myelomzellen produzieren in sich keine Antikörper gegen das Antigen, und es fehlt von denen entweder die HGPRT (Hypoxanthyn-Guanin-Phosphorybosil-Transferase) oder das Enzym TK (Thymidin-Kinase). Ein Teil der auf diese Weise entstandenen Zellen hat die Fähigkeit zur unbegrenzten Zellvermehrung bzw. die Fähigkeit Antikörper zu produzieren (Hybiddom). Diese Hybridom-Zellen, werden durch das sog. HAT-Selektionsverfahren (Abbildung 8.5.) von den nicht hydbrid Zellen separiert und anschliessend zur Vermehrung (Mitose) angeregt. Während der Selektionsmethode liegen die Zellen in Hypoxantin-Aminopterin-Thymidin (HAT) Medium. Im Selektionsmedium sterben die Partnerzellen der Hybridisierung (d.h. die nicht hybridisierten Zellen) ab, weil: 1. Die aus der Milz isolierten intakten B-Zellen, als primäre Zellkultur nur zu einer begrenzten Anzahl an Teilungen (Replikationen) fähig sind; 2. In mutierten Zellen mit Myelomursprung hemmt das Aminopterin den klassischen Weg der DNASynthese. bzw. funktioniert der alternative DNA-Syntese-Weg auch nicht wegen Mangel an TK oder HGPRT. Auf diese Weise werden im HAT-Medium nur die Hybridomzellen überleben, weil die Heterozygoten, und deswegen sowohl auf HGPRT, als auch auf das TK betreffend sind, und so fähig sind für die alternative DNA-Synthese somit auch für Zellprolieration. Von diesen Hybridom-Zellen können mit Zell-Klonierung und darauffolgend mit einem ELISAVerfahren die sog. monospezifischen antikörperproduzierenden Zellen ausgewählt werden, d.h. diese Zellen, nur auf eine einzige Bindestelle des Antigens (Epitop) spezifische Antikörper herstellen. Anschliessend werden die isolierten Zellen im Bioreaktor massenhaft zur Vermehrung angeregt, was zweckmässig ist für die Herstellung von grösseren Mengen von monoklonen Antikörpern. 8.6. Spezielle Kultivierungsverfahren Am häufigsten wird die übliche Petri- oder die einfache Kultivierungsschale für Laboranalyse-oder Versuchszwecke verwendet. Es kommen jedoch oft Fälle vor, wobei von einzelnen Zelltypen grössere Mengen benötigt werden. Für solche Fälle – abhängig vom Zelltyp (angehaftete oder suspensions Zellkultur) – entwickelte man verschiedene finanziell relevante Laborgeräte, mit denen bei Zugabe von verhältnissmässig geringen Mengen Serum, Nährmedium und sonstigen Adjuvanten mehrere Millionen von Zellen gleichzeitig erhalten werden können. Im Fall der Suspensionszellkulturen verwendet man z.B. die "Spinner Flasche", bei adherenten (anhaftenden, monolayer) Zellkulturen verwendet man die rotierende Flasche (roller bottle). Im ersten Fall befindet sich in dem Flascheninneren ein Dreharm, der in die Zellsuspension hineinreichend, ständig und gleichmässig Zellen und Nährmedium umrührt so, dass für jede einzelne Zelle die gleiche und gleichmässige Nährstoffzufuhr gesichert ist. 143 Im zweiten Fall sind die Zellen in grossflächigen zylindrischen Gefässen an der Innenseite angehaftet, unddie Zellen wachsen unter einer dünnen Nährmediumsschicht. Im Gerät wird durch die gleichmässige Drehbewegungen des Kultivierungsgefässes die gleichmässige und notwendige Menge der Nährmediumszufuhr ermöglicht, wodurch das Austrocknen der Zellen verhindert wird. In beiden Fällen der Zellvermehrungen ergeben sich günstige Zell/Nährlösungsverhältnisse. Ein spezielles Verfahren ist für die Zellvermehrung (scale up) im Fall der adhesiven (ahgehafteten) ® Zellkulturen, wenn die Zellen auf den Oberflächen von Mikroperlen (Cytodex ) in einer "Spinner Flasche" kultiviert sind. Zum Nährmedium werden Mikroperlen-Partikel zugegeben, wodurch ein extreme Oberflächenvergrösserung erreicht wird. Die Mikroperlen-Partikel werden bald von Zellen besiedelt. Durch Strömung des Nährmediums können die auf die Mikroperlen angesiedelten Zellen gleichmässig Nährstoffe aufnehmen und Stoffe abgeben. Auf ähnlichem Prinzip basieren die Techniken der statischen und dynamischen Zellfabriken. In diesen ist auch der Atemgasaustausch gesichert. Bioreaktoren werden für die Herstellung grosser Mengen von monoklonalen Antikörpern, bzw. für die durch Vermehrung von genetisch modifizierten Eukariotenzellen rekombinant produzierte Proteine (z.B. Insulin, Wachstumsfaktoren) benutzt. Im medizinischen Heilverfahren könnten die Leberzell-Reaktoren eine bedeutende Rolle spielen, diese dynamischen Kulturen sind bis zur Ankunft des Donororgans fähig das Blut des Patienten zu detoxieren / entgiften, und damit sein Leben zu verlängern. Die Methylzellulose enthaltenden sog. semisoliden (halbflüssige) Medien verhindern das Anheften von Zellen am Gefässboden zusätzlich aber sind die Nährstoffe gesichert. Weil diese Medien die Knochenmarksverhältnisse imitieren; wird dieses Verfahren hauptsächlich für die Kultivierung hämatopoetischer Stammzellen angewandt. Ausgehend von Hämatopoetischen Stammzellen in einem mit Zytokinen angereicherten semisoliden Medium erscheinen die differenzierenden Hämatopoetische Zellen in einer stark bestimmten Reihenfolge. Zuerst erscheinen die erythroiden Progenitorzellen, dann der CFU-GEMM (Granulozyten, Erythrozyten, Monozyten, Makrophagen, d.h. die multipotenten) Progenitorkolonien, letztlich die Unipotenten, nur einen einzigen Zelltyp beinhaltenden Kolonien (z.B.Makrophagen oder Monozyten). Bei Kultivierung von embryonalen Stammzellen benötigt man ein anderes spezielles Verfahren. In diesem Fall werden die Zellen auf eine andere Zellschicht, welche durch vorherige Behandlung mit Mitomycin C (MM) oder durch Bestrahlung in seinem Wachstum gehemmt ist, zerstreut. Die auf diese Weise konstruierte sog. nährende Zellschicht oder feeder layer produziert Faktoren (Zytokine, Wachstumsfaktoren) die für die Erhaltung des undifferenzierten pluripotenten Zustandes der Stammzellen nötig ist, ohne dasdie zur Vermehrung erwünschten Stammzellen (von den nährenden Zellen) überwuchert werden. Grösstes Problem dieses Verfahrens bedeutete, dass sowohl die Maus als auch die humanen Embryonalen Stammzellen auf Maus-Fibroblast feeder gezüchtet wurden. Das daraus folgende Risiko der artfremden Kontamination ergab das grösste Hinderniss den therapeutischen Einsatz von Humanen Embryonalen Stammzellen. Aus diesem Grund verwendet man heute anstatt von aus Maus stammenden nährenden Zellschichten (feeder layer) synthetische extrazelluläre Matrix mit genau definierter Zusammensetzung. 144 8.7. Untersuchungsmöglichkeiten der Zellen in einer Suspensionskultur Als Anschluss der Subkultivierung teilt man die Zellvermehrung in einer Zellkultur auf mehrere Abschnitte auf (Abbildung 8.6.). Nach Abfertigung der Zellkultur ist die Kultur in der sog.lag-(oder verzögerte)-Phase; die Zellen regenerieren sich, wachsen, dann beginnen sie sich zu teilen, ihre Anzahl wächst nur langsam. In der darauffolgender sog. log-Phase, die Abbildung 8.6. Wachstumsphasen einer Zellkultur (Abschnitte der Zellzahlsänderungen) Zellen vermehren sich intensiv, ihre Zahl wächst exponentiell. In der (log Phase) folgenden Plateauphase sinkt der Nährstoffgehalt (und im Falle von adhesiven Zellen auch der freie Oberfläche) immer mehr, die Zellvermehrung wird verlangsamt, schliesslich hört sie auf, die Zellzahl bleibt für kürzere Zeit konstant. Wenn in diesem Zustand das Nährmedium nicht gewechselt wird oder die Zellen nicht in mehreren Kulturschalen subkultiviert werden, dann beginnen in der letzten, sog. Untergangsphase die Zellen abzusterben, die Zellzahl sinkt stetig. Am besten geeigneter Zeitpunkt zur Untersuchung der gezüchteten Zellen, ist die intensive Wachstumsphase, die log Phase Die adherenten (angehafteten) Zellen werden nach irgendwelcher zytologischen Färbung bzw. nach einer immunzytochemischen Reaktion im Lichtmikroskop in situ untersucht. Für Untersuchung der lebendigen adherenten Zellkulturen verwendet man eine Invers-Lichtmikroskop (Abbildung 8.7.) bzw. eine Videomikroskop. Abbildung 8.7. Prinzip des Inversmikroskopes 145 Zellen der Suspensionszellkulturen müssen erst mit entsprechender Technik auf einen Objektträger gebracht werden, um untersuchbar zu sein. Eine solche Methode ist z.B. die Herstellung eines Ausstrichpräparates, eines Dicktropfpräparates und das Zytozentrifugieren. Die Herstellung des Ausstrichs (Abbildung 8.8.) geht aus einer hochzelligen Zellsuspension, ein kleines Tröpfchen dieser Suspension wird auf den Objektträger gelegt, welche mit der Kante eines anderen Objektträgers auf eine einzige Zellschicht erweitert wird. Im Fall des Dicktropfpräparates ist die Ausgangszellsuspension dünn (deren Zellzahl ist niedrig), aus dieser Suspension wird ein kleines Volumen auf einen Objektträger pipettiert. Während dem Zytozentrifugieren wird, abhängig von der Zelldichte der Ausgangssuspension, eine Muster mit kleinerem oder grösserem Volumen in spezielle L-Förmige Trichter gebracht, woraus alle Zellen durch die Zentrifugalkraft auf einen Oberflächenteil mit 0.5 cm Durchmesser von einem dahinter befestigten Objektträger aufgefangen werden. Mit jedem des Obigen Verfahrens können Objektträger mit der Zellen, die auf gewünschten den Verfahren gebracht wurden, nach trocknen und fixieren als Präparate im Mikroskop untersucht werden. Abbildung 8.8. Herstellung eines Ausstrichpräparates aus einer Zellsuspension 8.8. Anwendungsbereiche der Zellkultivierung Zellen von Zellkulturen können auf verschiedenster Weise untersucht werden (Tabelle 8.3.). Von den üblichen licht- oder elektronenmikroskopischen bis zu den modernen molekularbiologischen (wie z.B. Genexpressionsuntersuchungen) Techniken. Die Zellkultivierungsverfahren ermöglichen die Untersuchungen sowohl mit den (embryronalen, erwachsenen und/oder induzierten) pluripotenten Stammzellen als auch mit differenzierten Zellen. Mit Hilfe von in Zellkulturen in grossen Menge hergestellten hoffnungsvolle normalen oder Aussichten in gerade der genetisch gewöhnlichen menschlichen Erkrankungen. 146 eröffneten modifizierten/korrigierten Zellen, oder verschiedenartigen Gentherapie von Umweltwechselwirkungen Infektionen (Viren, Bakterien, Parasiten) Toxikologie Immunologie Karzinogenese Biotransformation der Xenobiotica Genetik Transformation Zellfusion Zellzyklus Zellbiologie Zell - Zell und Zell - Matrix Wechselwirkungen Genexpression Zellproliferation Differentierung Zellmigration, -Invasion Intrazelluläre Aktivität Replikaton, Transkription RNS Metabolismus Proteinbiosynthese Intermediäres Stoffwechsel Biotechnologie/Tissue engineering Zytokine/Wachstumsfaktoren, Hormone, Antikörperproduktion Künstliche Gewebe Tabelle 8.3. Anwendungsmöglichkeiten von der Zellkultivierung Übersetzt von Ottó Dobozy 147 9. DIE HOMING, DIE GEZIELTE MIGRATION VON IMMUNZELLEN. EXTRAVASATION IN ENTZÜNDUNGEN (LÁSZLÓ KŐHIDAI) 9.1. Einführung Die Fähigkeit der Zellen zu wandern ist ein grundlegender Prozess. In den frühen Entwicklungsphasen der Phylogenese entwickelte sich schon diese Fähigkeit, da es für die primitiven einzelligen Organismen immer ein bedeutender Vorteil war, die schädlichen (toxischen) Stoffe von den günstigen Nahrstoffen zu unterscheiden. Zellmigration ist eine der grundlegenden zellphysiologischen Prozesse, indem sie solche physiologischen Prozesse, wie die Befruchtung oder die Angiogenese, beeinflusst. Zellmigration ist auch an pathologischen Prozessen beteiligt, wie der Entstehung von Entzündungen, von Metastase der Tumoren oder der Entstehung der Atherosklerose. Die Migration ist der Fokus einiger Therapien, in denen die Arzneimittel das Ziel selektiv erreichen sollen. 9.2. Die Hauptformen der Zellmigration Die Migration der Zellen kann nach verschiedenen Ansichten gruppiert werden. Nach einem häufig verwendeten Prinzip unterscheidet man (i) Kinesen, bei denen die Richtung der Bewegung von der Konzentration der die Bewegung auslösenden Moleküle nicht abhängt, so dass die Bewegung random ist und (ii) Taxis, bei denen der Konzentrationsgradient bedeutend ist und die Richtung der Bewegung vektoriell ist (Abbildung 9.1.) Abbildung 9.1. Die Hauptformen der Zellbewegungen Bei den Kinesen spricht man auch über die Geschwindigkeit der Bewegung (Ortokinese), über die Frequenz (Klinokinese) und über die ad hoc Richtung (Chemokinese). 148 Lösliche Moleküle aus der Umgebung lösen Chemotaxis aus, der Lockstoffgradient an der Zelloberfläche drückt sich als Haptotaxis aus. Die aus den abgestorbenen Zellen freigesetzten Stoffe induzieren Nekrotaxis (Abbildung 9.2.). Abbildung 9.2. Die von chemischen Stoffen ausgelöste Motilität der Zellen 9.3. Die die Chemotaxis auslösenden Moleküle Diese Stoffe werden nach ihrer Wirkung in zwei Hauptgruppen unterteilt. (i) Chemoattractante Stoffe beeinflussen Stoffkonzentrationsgradienten, die die Zellen Fortbewegungsrichtung wandern in der der Zellen durch Richtung des Konzentrationsgradienten. (ii) Chemorepellente Stoffe verursachen die Bewegung in die umgekehrte Richtung, in Richtung des niedrigeren Konzentrationsgradienten. Als Chemokine fungieren unterschiedliche Substanzgruppen. Stark chemoattractante oder repellente Stoffe können ebenso einfache Ionen und relativ kleine Moleküle (z.B. Aminosäuren) als auch relativ grosse Moleküle (z.B. Chemokine, syntetische Artzneimittel) sein. In der Biologie, Physiologie, Pathophysiologie und Immunologie existieren so genannte professionelle chemoattractante Moleküle (z.B. formil-Met-Leu-Phe, Chemokine, Komplement 5a). 149 Andererseits können weitere Moleküle (z.B. Hormone), deren primäre Aufgabe keine ChemotaxisAuslösung ist, auch als Chemoattractante / Chemorepellente fungieren. Die Klassifizierung der vier Subgruppen der Chemokine beruht auf der Lokalisation der die Peptidstruktur beeinflussenden Disulfidbrücken und auf dem Abstand zwischen den zentralen Cistein Aminosäuren. Aus dem Grund der Funktion sind die Moleküle der CX3C Gruppe sehr bedeutend. Die Mitglieder dieser Gruppe können sich durch die hydrophobe Domäne des C-Endes in der Membran verankern, wodurch sie praktisch Haptotaxis auslösende Liganden werden (Abbildung 9.3.) Abbildung 9.3. Die Grundlage der Gruppierung der Chemokine nach ihrer Struktur 9.4. Chemotaxis Rezeptoren Während der Evolution wurden verschiedene Ligand-Rezeptor Interaktionen für die Auslösung der Chemotaxis selektioniert. In Prokaryonten dienen solche Transmembranproteine den chemotaxis Rezeptoren, die relativ einfache Moleküle (z.B. einfache Saccharide, Aminosären und ihre Dimere, Trimere) binden können (z.B. Asparaginsäure-Tar Rezeptor). Ihre Struktur wird von vier Domänen ausgebildet, deren Funktion unterschiedlich ist: (i) extrazelluläre, ligandbindende Region, (ii) die sogenannte „coiled coil” Domäne, (iii) die Methylgruppe-bindende Region, die für Rezeptoraktivierung 150 und Signalübertragung verantwortlich ist und schließlich (iv) die zytoplasmatische Domäne, die durch die Phosphorilierung der chemotaktischen Proteine eine signalisierende Funktion besitzt. Es ist zu betonen, dass die Dimerisierung der Rezeptoren bereits in diesen früh entwickelten Organismen auftritt und eine höhere Aktivität aufweisen. In der Membran der Zellen der höheren Lebewesen (ebenso der Menschen als auch der Tiere) befinden sich auch die chemotaxis Rezeptoren, die die Fähigkeit haben, die kurzen Formil-Methionin (formil-Met-Leu-Phe) besitzenden, während einer Infektionen freigesetzten bakteriellen Peptide zu binden. Diese Rezeptoren gehören schon zur Familie der 7TM Rezeptoren, und extrazelluläre glykosilierte Sequenzen spielen eine Rolle bei der Bindung der Liganden. Der das C5a Peptid bindende Rezeptor ist genauso ein 7TM Rezeptor. Die Signalübertragung dieser zwei chemotaxis Rezeptoren ist an ein trimeres G-Protein gekoppelt. Weitere intrazelluläre Signalübertragungsnetzwerke? sind in der Signalvermittlung beteiligt (z.B.. PLC2+ PIP2-IP3-Ca ; Ras/Raf-MEK1/MEK2-ERK1/ERK2; MEKK-MEK3-p38-MAPK) Die Chemokinrezeptoren gehören auch zur Familie der 7TM Rezeptoren. Hier bindet auch der Ligand an das extrazelluläre N-Ende des Rezeptors. Die intrazelluläre Signalisierung(Signalübertragung) wird von der Interaktion des trimeren G-Proteins und des Rezeptors ausgelöst. Eine DRY Sequenz liegt auf der 2ten Schleife des Rezeptors, diese Region ist für die Interaktion verantwortlich. Das C-Ende des Rezeptors aktiviert das G-Protein. Die Phosphorilierung dieser Region erlaubt die Bindung an Beta-Arrestin, wodurch weitere Signale generiert werden (Abbildung 9.4.). Ausser diesen Interaktionen ist bei mehreren Chemokinen (z.B. IL8) beschrieben, dass auch die GAG Moleküle auf der extrazellulären Oberfläche der Membran, in der Nachbarschaft der Chemokinrezeptoren eine wichtige Rolle bei der Stabilisierung des LigandRezeptor Komplexes spielen. M. O'Hayre, C.L. Salanga,T.M. Handel, and S.J. Allen. (2008) Biochemical J. 409:635-49 Abbildung 9.4. Signalübertragungsmechanismen von Chemokinrezeptoren 151 9.5. Die sich bewegenden Zellen des menschlichen Körpers und ihre Bewegungsformen Viele Arten von Zellen sind Zielzellen der Chemokine in höheren Lebewesen. Aus physiologischen und klinischen Gründen sind die neutrophilen Granulozyten, die Monozyten, die Lymphozyten, die eosinophilen Granulozyten und die Endothelzellen der Gefässe die wichtigsten. In der so genannten mesenchymalen Form der Migrationsmechanismen in höheren Lebewesen wird die Polarisation der Zellen von den Integrinen und der Aktivität der extrazellulär wirkenden Proteasen determiniert. Die klassische amöboide Migration unterscheidet sich darin, dass der Abbau der extrazellulären Umgebung keine wichtige Rolle spielt. In den letzten Jahren wurde eine neue Bewegungsform beschrieben, die so genannte kollektive Migration. In dieser Form wandern Zellgruppen zusammen, die durch Zelladhäsionsstrukturen miteinander gekoppelt sind. Diese Migration ist während der Morphogenese der embryonalen Gewebe und bei dem Wachsen einigen Tumoren zu finden (Abbildung 9.5.). Abbildung 9.5. Bewegungsformen der menschlichen Zellen 9.6. Zellmotilität und Immunantwort Die Komplexität des Proczsses der Migration und ihrer zelluläre Regulation kann durch die Migration der Lymphozyten und dendritischen Zellen (DZ) erfolgreich untersucht werden. Auf diese Weise kann der Weg der DZ – Progenitor - unreife DZ – reife antigenpräsentierende DZ aus dem Knochenmark durch die nicht-lymphoiden Gewebe zu den Immunorganen aufgezeichnet 152 werden Abbildung 9.6. zeigt den Prozess, dessen bedeutender Teil die Migration und Zirkulation zwischen den Organen ist. Abbildung 9.6. Die Migration der Lymphozyten und der dendritischen Zellen 9.6.1. Transendotheliale Migration der Lymphozyten Der oben dargestellte Prozess zeigt ein nuanciertes Bild in den Räumen der Gewebe. Das Endothel der Gefässe ist die wichtigste Oberfläche für die Migration aus immunologischer/pathologischer Gesichtsweise. Die gerichtete Migration (Homing, Entzündungsprozesse) von vielen Zellpopulationen erschieht? (entstand/erschien?) durch diese riesige Oberfläche. Die Zellpopulationen verlassen das Kapillarsystem oder gelangen zurück, – Prozesse die von der Expression von bestimmten Membranadhäsionsproteinen und löslichen Faktoren (Chemokine, bakteriell Peptide) induziert und gerichtet werden. Die Entstehung der Phasen wird in den postkapillären Venulen (?) während der Transmigration der Lymphozyten von den hier erklärten Mechanismen ermöglicht Abbildung 9.7: 1. Rolling - Selektin-Sialomucin Verbindung (oder Integrin alfa4beta1 und alfa4beta7) 2. Aktivierung – die Chemokine des Endothels binden an die Chemokinrezeptoren der Lymphozyten, wodurch die Lymphozyten aktiviert werden. Die Chemokine befinden sich auf den Proteoglykanen des Heparansulphat. 3. Adhäsion – als Folge der Aktivierung anscheinen Integrine mit erhöhter Affinität und Avidität auf der Oberfläche der Lymphozyten. Sie binden an ihre Liganden (ICAM, VCAM usw) auf den Endothelzellen. 153 4. Diapedesis – die Bindung mit den junktionalen Adhäsionsmolekülen (JAM, PECAM-1=CD31) ergibt die Öffnung der Breschen (vllt. Zellzwischenräume) zwischen den Endothelzellen und ermöglicht die Transmigration der Lymphozyten. Die Migration der Lymphozyten ist von Adhäsionsmolekülen, Chemokinen und Chemokinrezeptoren stark reguliert. Abbildung 9.7. Die transendotheliale Migration der Lymphozyten Das Homing der naiven T Zellen und ihre Rezirkulierung zwischen den sekundären Immunorganen wird von der Expression des Integrins alfa4beta7-t (Mucosa) und L-Selektins (Lymphknoten) ermöglicht. Die Migration der aktivierten T Zellen zum Ort der Entzündung schließt die Bindung von mehreren Ligand-Rezeptor Paaren ein. Selektin-Sialomucin, Integrin alfa4beta1-VCAM-1, Integrin alfa4beta1-CS-1 und CD44-Hialuronsäure. Im Falle der T Gedächtniszellen ist die „homing signature” ein spezifisches Adhäsions-und Chemokinrezeptorprofil, welches ihnen die Fähigkeit zu einer selektiven und gewebespezifischen Migration verleiht. Die naiven B Zellen ko-exprimieren L-Selektin und alfa5beta7 Integrin, die es ihnen erlauben in die Mucosa und in die periphären Lymphknoten einzutreten. Die im germinalen Zentrum der PeyerPlaques abläufenden Reaktionen rufen die Expression des alfa4beta7 Integrins der B Gedächtniszellen hervor. Diese Zellen differenzieren sich zu IgA sekretierenden Plasmazellen. Der grosse Teil der B Gedächtniszellen stammen aus den Lymphknoten und sie differenzieren sich zu IgG sekretierenden Plasmazellen. Diese Zellen exprimieren CXCR4, alfa4beta1 Integrin und LFA-1, die für das Homing ins Knochenmark nötig sind. Im Knochenmark entwickeln sich diese Zellen zu langlebigen Plasmazellen. 9.6.2. Migrationswege der dendritischen Zellen Die das Knochenmark verlassenden dendritischen Zellen erreichen ihre bestimmten Zielorgane / Zielgewebe durch den Blutkreislauf. Der Thymus, die Lymphknoten, die Milz und die Haut sind von der höchsten Wichtigkeit. Während plasmazytoide dendritische Zellen in allen vier Zielorganen zu finden sind, kommen konventionelle DZ Zellen im Thymus und in der Milz auch vor. In der Haut und in 154 den Lymphknoten treten dazu präkursor DZ Zellen auch auf. Unterschiedliche Rezeptor-Ligand Bindungen sichern die Ankunft der DZ Zellen in die jeweiligen Zielorgane. Im Falle der Lymphknoten und des Thymus sind die VLA4-VCAM1 und die PSLG-1 – CD62 (E/P) Verbindungen von grosser Bedeutung. In den Follikuli der Milz ist die CXCR5-CXCL13 Verbindung, in den periarteriolen Räumen der weissen Pulpa ist die CCR7-CCL19/CCL21 Interaktion charakteristisch. Zahlreiche Interaktionen wurden in der Haut und in den Entzündungsreaktionen beschrieben, in den PSLG-1 – CD62 (E/P), CXCR1-Fraktalkin, CCR6-CCL20 und CCR2-CCL2 typisch sind. Schöne Beispiele sind im Falle der Lymphknoten und der Haut für die molekulare Regulation der Zellmigration zu finden (Abbildung 9.8.). Abbildung 9.8. Die Migration der dendritischen Zellen in den Lymphknoten. Im Falle der Zellmigration ist in Menschen die Haut ein sehr interessantes „System”. Die in den oberen Epithelschichten, nah zur Körperoberfläche patrouillierenden DZ Zellen können relativ schnell zu den zentralen Komponenten des lymphatischen Systems gelangen. Damit wird es möglich diese Zellen für experimentelle und therapeutische Ziele zu verwenden. Die sich im Epithelgewebe befindenden DZ Zellen können einige aus den von Mikroben angegriffenen Zellen freigesetzten Stoffe (TNFa, IL-1b, PAMP) detektieren. Diese Moleküle wirken auf die DZ Zellen als Migrationssignale. Die DZ Zellen bauen ihre E-Cadherin Verbindungen ab, wodurch sie sich mobilisieren lassen. Der nächste Schritt der Migration ist der Abbau der basalen Membran, oder der 155 dazu assoziierten Komponenten der extrazellulären Matrix (EZM). Dieser Prozess wird von Matrixmetalloproteinasen (MMP2, MMP9) durchgeführt. Gleichzeitig erscheinen Chemokinrezeptoren (CXCR4, CCR7, CCR8) auf der frontalen Oberfläche der Zellen. Sie können bestimmte Chemokine (CXCL12, CCL21, CCL1) in sehr kleinen Konzentrationen wahrnehmen, das hilft in der Wahl der Richtung der Migration. Die DZ Zellen adhärieren sich an das Endothel der afferenten Lymphgefässe im Raum des Bindegewebes unter der Haut. In diesem Prozess sind die CCR7-CCL21, VLA-4 – VCAM-1, LFA-1-ICAM-1 und Sphingosin 1-Phosphat Signale und der promiskuide Chemokinrezeptor (D6) des Endothels beteiligt. Der letzte Schritt ist die Internalisierung der DZ Zellen in die Lymphgefässe, wo das Signal CCL21 der Wanderung hilft. 9.7. Die Methoden der Messung der Zellmotilität (Chemotaxis) Die Messung der Zellmotilität ermöglicht die Analyse von vielen immunologischen und zellphysiologischen Zuständen. (i) Verschiedene Zellpopulationen können (? getrennt gemessen / unterschieden) werden, (ii) die Migrationsaktivität von Zellgruppen können mit Hilfe von Referenzliganden (z.B. fMLF, IL-8) bestimmt werden, (iii) die chemotaktischen Wirkungen von einigen Körperflüssigkeiten oder von aus ihnen isolierte Stoffe können analysiert werden und schließlich (iv) die Wirkungsmechanismen von neuen, aus pharmakologischen Gründen wichtige Stoffe oder späteren Arzneimittelmolekülen können untersucht werden. Die Wahl der geeigneten Modelzellen und Untersuchungstechniken braucht besondere Sorgfalt. Abbildung 9.9. zeigt zwei verschiedene Systeme bei der Messung der Chemotaxis. Die Methoden werden abhängig davon verwendet, ob sich die Zellen in 3 oder 2 Dimensionen bewegen. Im reversiblen System sind die Zellen in freiem Raum, jedoch in einem filter-getrennten System ist die Chance minimal, dass die Zellen zurückwandern würden. In diesen Systemen entscheidet der Kon- zentrationsgradient, wie lange die Untersuchung dauert. Wenn der Konzentration-gradient ausgeglichen wird, misst man keine Chemotaxis, aber chemokinetische Aktivität. Abbildung 9.9. Hauptformen Zellmigration zu messen Die Aktivität der chemotaktischen Zellen und die Induzierbarkeit der Chemotaxis können durch mehreren Methoden gemessen werden. Die Menge der erhaltenen Informationen steht nicht im Verhältnis mit der Zuverlässigkeit der Methoden. Einerseits erlaubt die Dunn-Kammer z.B. – eine der die meisten Informationen gebenden Methoden – die Messung von relativ wenigen Zellen. 156 6 Andererseits ergeben die Rezeptorbindungs-Untersuchungen nur eine Angabe, obwohl 10 Zellen gemessen werden. Die besten Methoden sind jene, die mehr Parameter untersuchen, und zahlreiche Methoden werden endlich analysiert. Unten folgen nur einige der vielen Anwendungstechniken. 9.7.1. Reversible Systeme Die zu diesen Gruppe gehörenden Methoden ermöglichen die Untersuchung der Zellen, die sich mit amöboiden Bewegung oder mit Zilien/Geisseln bewegen. Die Technik mit dem am längste (24 Stunden) fortbestehenden Konzentrationsgradient wird DunnKammer genannt (Abbildung 9.10.). Das Bild zeigt den Durchschnitt des Systems, mit zwei konzentrischen ringartigen Kam- mern. Die Migration der Zellen erfolgt unter der Abdeckungsplatte, auf dem Bereich zwischen den Kammern. Der Konzentrations- gradient entsteht auf diesem dünnen Flüssigkeitsfilm, welcher kontinuierliche, Bewegung die vektorielle der Zellen Abbildung 9.10. Prinzip der Dunn-Kammer aufrechterhaltet. Eine seit langem verwendete Technik ist die Agarplatten-Methode, die die Bewegung auf einer Oberfläche misst (Abbildung 9.11.). Zusätzlich gibt es viele weitere Chemotaxis Assays. Die Agarplatten-Methode ist ähnlich dem zelluläre Assay, dessen Grundprinzip ähnlich der immunologischen Technik der Messung von Antigen-Antikörper Interaktionen ist. Die qualitativen Eigenschaften der Methode können mit der Messung der Abstände zwischen dA und dK in quantitative umgewandelt werden. Abbildung 9.11. Messung der Zellmigration mit Agarplatte Technik 157 9.7.2. Irreverzible Systeme Der Boyden-Kammer (Abbildung 9.12.) ist die am häufigsten verbreitete Technik, um Chemotaxis zu messen. Die Technik wurde 1962 von Stephen Boyden beschrieben. Ihr Prinzip ist ein zwei-Kammer System, wo die Kammern mit einem Filter voneinander getrennt sind. Der Filter ist nur für jene Zellen durchlässig, die sich mit aktiver Bewegung bewegen. Die Zellen werden in die obere Kammer eingegeben, die untersuchende Probe ist in der unteren Kammer. Während der Inkubationszeit wandern die Zellen durch die Poren des Filters entlang des Konzentrationsgradienten in die untere Kammer. Die Migration der Zellen kann durch die Messung der Zellzahl ausgewertet werden, oder durch den Nachweis von einigen spezifischen Enzymen oder durch die Bestimmung der ATP Moleküle in den Zellen. Der Nachteil der Methode ist, dass eine Kammer nur einem Messpunkt entspricht, also viele Kammern müssen gleichzeitig verwendet werden. Um dieses Problem zu lösen, wurde eine Form von 96-Well-Multititerplatte auch entwickelt, wodurch viele Abbildung 9.12. Struktur und Prinzip der Boyden- Kammer – Rechts: wandernde Zellen durch die Poren des Filters parallele Experimenten gleichzeitig durchgeführt werden können. 9.7.3. In vivo Techniken Wachstumsfaktor Konzentration in Matrigel Konzentration in GFR- Matrigel EGF 0.5-1.3 ng/ml < 0.5 ng/ml bFGF < 0.1-0.2 pg/ml n.d. NGF < 0.2 ng/ml < 0.2 ng/ml PDGF 5-48 pg/ml < 5 pg/ml IGF-1 11-24 ng/ml 5 ng/ml TGF-b 1.7-4.7 ng/ml 1.7 ng/ml GFR – Spiegel vermindert; n.d. – kann nicht detektiert werden Tabelle 9.1. Der Inhalt der Wachstumsfaktoren in Matrigel Präparaten Wenn ein lebendes Tier verwendet wird um die Experimenten durchzuführen, wird oft ein aus dem Tumorgewebe gewonnenes EZM (extrazelluläre Matrix) Konzentrat, das Matrigel, verwendet. Matrigel ist ein Netzwerk, das eine künstliche Umgebung für die wandernden Zellen darstellt. Dieses Material wird in den Scheiben-Assays verwendet, wenn man mit Filtern bedeckte Scheiben in ein Versuchstier implantiert. Darin enthalten ist Matrigel und der zu untersuchende Stoff. Nach einigen Tagen nimmt 158 man die Scheiben heraus. Die Anzahl der eingewanderten Zellen und das Verhältnis der unterschiedlichen Zellpopulationen können dann bestimmt werden. Tabelle 9.1. zeigt die Grenzen der Verwendbarkeit der Matrigel-EZM Mischung. Die Mischung besitzt einige Wachstumsfaktoren in viel grösseren Konzentrationen als physiologisch benötigt wäre, und das kann in einigen Experimenten auch oft ein Nachteil sein. Eine Lösung ist die Einführung von solchen Matrigel Systemen, die niedrigere Mengen von Wachstumsfaktoren enthalten. (GFR = growth factor reduced) Eine weitere geistreiche Methode gehört zu den in vivo Assays, die Sephadex Perlen in den intraperitonealen Raum des Versuchstiers bringt. Die Perlen lösen eine starke Zellmigration aus. Sie ziehen die Zellen in den Raum an. Verschiedene Stoffe können an die Perlen gebunden werden, wodurch.ihre Chemotaxis-induzierende Wirkung studiert werden kann. Die Auswertung erfolgt nach 648 Stunden durch das Absaugen der peritonealen Flüssigkeit. Die Wundheilung oder ‘wound healing’ Assays werden auch zusammen mit diesen Techniken diskutiert. Die aus Epithelzellen geformten konfluenten Schichten werden beschädigt. Dann wird beobachtet, wie schnell sich die zwei verletzten Oberflächen annähern. Eine verlässliche Methode Wunden herzustellen ist die Anwendung von einer Platte mit 24-48-96 Spitzen, die sich unter gut kontrolliertem Druck bewegen und die Epitheloberfläche beschädigen. 9.7.4. Die neusten Techniken Abbildung 9.13. Prinzip der ECIS Technik Das Prinzip der auf der Impedanz liegenden Messungen wurde von dem Nobelpreis Gewinner für Physik, Ivar Giaver, beschrieben. Diese Messungen können bei dem ersten Schritt der Chemotaxis, 159 der Zelladhäsion verwendet werden und auch für die Untersuchung der Migration selbst. Nach dem Prinzip adhärieren sich die Zellen auf der Elektrode die in den Stromkreis des Wechselstroms eingeschaltet wird. Da die Zellen gute Isolationsmittel sind, ergeben sie steigende Ohm Resistenz (R) und Impedanz (Z) Werte. Die Induzierbarkeit und die Hemmbarkeit der Zellen sich zu adhärieren können erfolgreich gemessen werden. Der Wert der Methode ist dass die Prozesse mehrere Tage in real-time – in wirklicher Zeit – verfolgt werden können (Abbildung 9.13.). Eine weitere Methode welche das Prinzip der Boyden-Kammer und der Impedanz-Messung (xCELLigence – Roche) vereint, wurde auch entwickelt. Der Filter ist mit Elektroden zwischen den zwei Kammern bedeckt, also die wandernden Zellen generieren ein elektrisches Signal und die addierten Signale stellen die Impedanz her. Die steigende Impedanz ist proportional mit der Anzahl der positiven (der wandernden) Zellen. Elektroden können auch der pünktlicheren und genaueren Messung der Wundheilung dienen. Diese Methode ist die so weit am objektivsten ausgearbeitete Technik um die Wundheilung zu studieren. Die sogenannte intravitale Mikroskopie kann auch für die Untersuchung der Zellmigration verwendet werden. Lebende Tiere oder grössere Organe können ohne Fixierungsmittel analysiert werden. Die Kamera funktioniert mit einer extrem schnellen Verschlusszeit, damit vaskuläre Prozesse, wie die Adhärenz der Zellen zur Gefässwand und Transmigration, monitorisiert werden können. „traction force microscopy” (TFM) Diese Technik ist die empfindlichste Methode um sich bewegende Zellen zu untersuchen. (Abbildung 9.14.). Ihr grundsätzliches Prinzip beruht auf den entstehenden Kräften zwischen den sich bewegenden Zellen und der unterliegenden Oberfläche bei den fokalen Kontakten während der amöboiden Bewegung. Die Kräfte sind mit Hilfe von Mikronadeln messbar. Die Grösse der Kraft ist in nN (nanoNewton). Abbildung 9.14. Kräfte bei der Anwendung der „traction force microscopy” 160 9.7.5. Weitere Methode zur Migrationsanalyse Die Untersuchung der Zellmigration hat nicht nur mit der Untersuchung der sich bewegenden Zellen zu tun, aber bedeutet auch der molekuläre Nachweis der Rezeptoren, der Liganden und der Expression ihrer Gene. Die gesuchte RNA wird erst mit ihrem markierten, antisens- Paar hybridisiert, nachdem eine nur die einsträngige RNA Moleküle verdauende RN-ase verwendet wird. Die zweisträngigen markierten Moleküle können getrennt werden. Es existieren viele Chemokin/Chemokinrezeptor Kits. Übersetzt von Erna Pap Literatur Alvarez D, Vollmann EH, von Andrian UH. Mechanisms and consequences of dendritic cell migration. Immunity. 2008 Sep 19;29(3): 325-42. Boyden S. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. The Journal of Experimental Medicine 1962 Mar 1;115: 453-66. Chu Y, Dietert RR. The chicken macrophage response to carbohydrate-based irritants: temporal changes in peritoneal cell populations. Developmental and Comparative Immunology 1968 Winter; 12(1): 109-19. Dembo M, YL Wang. Stresses at the cell-to-substrate interface during locomotion of fibroblasts. Biophysical Journal 1999 Apr; 76(4): 2307-16. Frevert CV, Wong VA, Goodman RB, Goodwin R, Martin TR. Rapid fluorescence-based measurement of neutrophil migration in vitro. Journal of Immunological Methods 1998 Apr 1;213(1): 41–52. Hadjout N, Xinyun Y, Knecht D, Lynes M. Automated real-time meausrements of leukocyte chemotaxis. Journal of Immunological Methods 2007 Mar 30;320(1-2): 70-80. Kőhidai L, Lemberkovits É, Csaba G. Molecule dependent chemotactic responses of Tetrahymena pyriformis elicited by volatile oils. Acta Protozoologica 1995;34: 181-5. Kragh M, Hjarnaa P-JV, Bramm E, Kristjansen PEG, Rygaard J, Binderup L. In vivo chamber angiogenesis assay: an optimized Matrigel plug assay for fast assessment of anti-angiogenic activity. International Journal of Oncology 2003 Feb;22(2): 305-11. Leick V, Helle J. A quantitative assay for ciliate chemotaxis. Analytical Biochemistry 1983 Dec;135(2): 466-9. Neils CM, Tyree Z, Finlayson B, Folch A. Combinatorial Mixing of Microfluidic Streams. Lab on a Chip 2004 Aug; 4(4): 342-50. Nelson RD, Quie PG, Simmons RL. Chemotaxis under agarose: a new and simple method for measuring chemotaxis and spontaneous migration of human polymorphonuclear leukocytes and monocytes. Journal of Immunology 1975 Dec;115(6): 1650-56. 161 O'Hayre M, Salanga CL, Handel TM, Allen SJ. Chemokines and cancer: migration, intracellular signalling and intercellular communication in the microenvironment. The Biochemical Journal 2008 Feb 1;409(3): 635-49. Pals ST, de Gorter DJJ, Spaargaren M. Lymphoma dissemination: the other face of lymphocyte homing. Blood 2007 Nov 1;110(9): 3102-11. Repesh LA. A new in vitro assay for quantitating tumor cell invasion. Invasion & Metastasis. 1989;9(3): 192-208. Yarrow JC, Perlman ZE, Westwood NJ, Mitchison TJ. A high-throughput cell migration assay using scratch wound healing, a comparison of image-based readout methods. BMC Biotechnology 2004 Sep 9;4: 21. Zantl R, Rädler U, Horn E. Chemotaxis in µ-channels. Imaging and Microscopy 2006 Mar; 8(1): 30-2. Zicha D, Dunn GA, Brown AF. A new direct-viewing chemotaxis chamber. Journal of Cell Science 1991 Aug;99 (Pt 4): 769-75. Zinn K, DiMaio D, Maniatis T. Identification of two distinct regulatory regions adjacent to the human beta-interferon gene. Cell. 1983 Oct;34(3): 865-79. http://dendritic-cells-research.com/Dendritic%20Cells.html 162 10. ANALYSEMETHODEN DER AUTOANTIKÖRPER, HLATYPISIERUNG (GYÖRGY NAGY, ZSUZSANNA PÁL) 10.1. Die Autoantikörper und ihre analytische Methoden 10.1.1. Die Entstehung und allgemeine Eigenschaften der Autoantikörper Die Vielfalt der Antikörper der B-Zellen verleiht dem menschlichen Körper einen wirksamen Schutz vor den unzähligen Pathogenen welche unseren Organismus alltäglich angreifen. Im Verlauf der wiederholten Aktivierung von B-Zellen entstehen, dank der Affinitätsreifung, Antikörper mit immer höher Bindungskraft und, dank des Isotypenwechsel, Antikörper vom Typ IgG, IgA, IgE, usw., die verschiedene immunologische Funktionen ausüben können. Andererseits ist ein wirksamer Schutz vor diesen Pathogenen auch mit gewissen Risiken verbunden, weil die Prozesse, welche die Vielfalt der Antikörper herstellen, manchmal auch zur Bildung von potentiell gefährlichen Autoantikörpern führen können. Autoantikörper kommen in allen Menschen vor. Eine wahre Autoimmunerkrankung entsteht jedoch nur in ca. 5% der menschlichen Population. Die Entstehung von solchen Krankheiten ist ein langer Prozess mit vielen, unter anderen genetischen, hormonellen, epigenetischen und umweltbeeinflussten Faktoren, die alle die Pathogenese beeinflussen können. Die Autoantikörper selber können dementsprechend in zwei größeren Kategorien eingeteilt werden: in den natürlichen, also in allen gesunden Menschen vorkommenden, und in den pathologischen Autoantikörper, die für Autoimmunerkrankungen typisch sind. Natürliche Autoantikörper sind vor allem von den B1 Zellen (CD5+ B Zellen) sezernierte polyreaktive Antikörper, welche in dem menschlichen Serum in einer relativ hohen Konzentration vorkommen, und vor allem gegen vitale, konservierte Antigene gerichtet sind. Sie bauen den immunologischen Homunculus auf, schützen aktiv körpereigene Strukturen, aber nehmen auch an dem primären Schutz teil, auch in der ersten Linie der Verteidigung gegen manche Pathogene. In den meisten Fällen sind sie Antikörper vom Typ IgM oder IgA, und sind nur selten eine somatische Hypermutation durchgegangen. Meistens werden sie von den B-1a Zellen gebildet, welche aus der embryonalen Leber nach der Geburt in das Peritoneum und in die Pleura einwandern. Hier haben sie einen langsamen Turnover, vermehren sich sehr langsam, nur damit die abgestorbenen B-1a Zellen kontinuierlich ersetzt bleiben, und ihre Antikörperproduktion ein Leben lang aufrechterhalten bleibt. Pathologische Autoantikörper hingegen werden vorwiegend von B2 Zellen gebildet, und eine Reihe von Beweisen existieren, die besagen dass sie meistens lokal, an oder nahe zur Stelle der Autoimmunreaktion gebildet werden. In der Rheumatoiden Arthritis erscheinen in der synovialen Membran, in Myasthenia gravis im Thymus, autoantikörper-produzierende ektopische Keimzentren, 163 also solche atypischen Keimzentren die außerhalb ihrer regulären Lage, also außerhalb der sekundären Lymphorgane entstehen. Ähnlicher Weise, in der Multiplen Sklerose kann man z.B. intrathecal IgG Produktion in der Rückenmarksflüssigkeit nachweisen. Pathologische Autoantikörper haben eine hohe Affinität, sind entweder vom IgG, IgA oder IgM Typ, und können auch eine hohe Konzentration im Serum erreichen. Generell kann man sagen, dass zwischen dem Beginn der Produktion der ersten Autoantikörper, und der Beschädigung eines jeweiligen Zielorgans in einer Autoimmunerkrankung, immer eine gewisse Zeit vergeht. Mit anderen Worten, um die klinisch nachweisbaren Symptome zu erreichen, muss ein bedeutender Teil der Zellen des Zielorgans schon beschädigt oder vernichtet worden sein (z.B. müssen in Diabetes mellitus die Anzahl der b-Zellen der Bauchspeicheldrüse schon deutlich vermindert sein). Aus diesen Gründen sind Autoantikörper im Serum oft früher nachweisbar als sich die ersten Symptome der Krankheit manifestieren könnten. Demzufolge kann der Nachweis von pathologischen Autoantikörpern im Serum nicht nur als diagnostischer, sondern auch als prognostischer Marker benutzt werden. Hierzu kommt, dass man eine Vielzahl von ihnen auch in der Einschätzung der Effizienz einer bestimmten möglichen Therapie benutzen kann. Sie können also auch als prädiktive Marker angewandt werden, und viele von ihnen sind aussagekräftig was die Effizienz einer gewählten Therapie betrifft. Pathologische Autoantikörper können durch viele unabhängige Mechanismen entstehen. Infektionen können ihre Produktion durch mehrere Wege auslösen: 1. Durch die molekulare Mimikry, also Ähnlichkeiten zwischen Antigenen des Pathogens auf die das Immunsystem reagiert und Antigenen des menschlichen Körpers, welche durch Kreuzreaktion eine Autoimmunerkrankung erzeugen. Ein Beispiel dafür ist die, nach Infektion mit Campylobacter jejuni entstehende Polyneuropathie, das Guillain-Barre Syndrom. 2. Infektionen können zu einer verstärkten MHCII Expression führen, oder einer erheblich gesteigerten Expression von kostimulatorischen Molekülen. Dies kann dann dazu führen, dass auch körpereigene Antigene (die zwar von der derselben APZ wie die Pathogen-Antigene präsentiert werden, aber normalerweise toleriert werden) eine Immunantwort induzieren. 3. Manche Erreger, z.B. das Epstein-Barr Virus können eine polyklonale Aktivierung von BZellen, oder manche Superantigene eine polyklonale Induktion von T-Zellen erzeugen. Durch die folglich starke Proliferation der Zellen können sich unter ihnen auch autoreaktive Klone befinden, welche von der peripheren Toleranz nicht mehr kontrolliert werden können, und eine Autoimmunität hervorrufen. Neben Infektionen können Autoimmunerkrankungen auch von verschiedenen Fehlern in der Regulation der Immunhomeostase erzeugt werden: 1. Pathologische Abnormitäten der Antigenprozessierung können Autoimmunerkrankungen erzeugen, zum Beispiel im Falle des Morbus bechterews (siehe später). 2. Myasthenia gravis ist eine Autoimmunerkrankung in der die neuromuskuläre Endplatte beeinträchtigt ist, indem Antikörper gegen verschiedene Komponenten der quergestreiften Muskelzellen gebildet werden, welche mit der neuromuskulären Transmission interferieren, und 164 dadurch die für die Krankheit typische Muskelschwäche erzeugen. In dieser Krankheit spielt eine Dysfunktion des Thymus eine wichtige Rolle. Die Mehrheit der jungen Patienten weist eine Thymus Hyperplasie vor, und 8-10% aller Patienten leiden an einem Thymom. In den Patienten mit Thymus Hyperplasie wird ein Autoantikörper gegen den nikotinergen AChR induziert. In den ThymomPatienten versagen die Selektionsmechanismen der T-Zellen, weswegen hohe Mengen von autoreaktiven T-Zellen mit unterschiedlicher Spezifizität dem Thymus entkommen, und die Peripherie erreichen. 3. Genetisch bedingte Fehler in der Differenzierung von T-Zellen im Thymus können auch zur Produktion von Autoantikörpern führen: eine rezessive Mutation des AIRE Gens bedingt z.B. das APECED Syndrom. 4. Eine ungenügende Effizienz der Entfernung von apoptotischen Zellen kann zur Freilassung von großen Mengen von intrazellulären Antigenen führen, wie zum Beispiel im SLE, wo z.B. anti-dsDNA Autoantikörper gegen sie gebildet werden. 5. Therapeutische Anwendung von manchen Medikamenten, wie die des Procainamids oder Hydralazins können durch die Beeinflussung der Genexpression der DNA Methylasen wirken. Sie beeinflussen letzten Endes die DNA-Methylierung, üben epigenetische Einwirkungen auf Gene welche in dem Pathomechanismus der Krankheit eine Rolle spielen aus, und rufen so eine Autoantikörperproduktion hervor. 6. Die Freilassung von Antigenen aus den für das Immunsystem normalerweise unzugänglichen Bereichen des Körpers, also den Zonen des sog. Immunprivilegs, kann auch die Sekretion von Autoantikörpern fördern, wie es im Falle der sympathischen Ophtalmopathie z.B. zu sehen ist. 7. Auch funktionelle Abnormitäten in den regulatorischen T-Zellen können die Autoantiköperproduktion fördern. Mutation des typischerweise in Treg Zellen exprimierten Foxp3 Gens ist stark gekoppelt zum IPEX Syndrom. 8. Eine Mutation des AID Gens, das in der Entwicklung des B-Zell-Repertoirs unumgänglich ist, verursacht nicht nur Hyper-IgM Syndrom in AID knockout Mäusen, sondern steigert auch ihre Empfindlichkeit für Autoimmunerkrankungen. 10.1.2. Die wichtigsten Charakteristika von Autoimmunerkrankungen Autoimmunerkrankungen sind in systemische und organspezifische Kategorien einzuteilen. Von den systemischen Autoimmunkrankheiten sind die häufigsten das Sjögren Syndrom, die Rheumatoide Arthritis, und SLE, in denen eine Autoimmunität gegen eine Reihe von herkömmlichen häufigen körpereigenen Antigenen entwickelt wird. Bei den organspezifischen Autoimmunitäten gibt es dagegen eine Immunantwort gegen spezifische, auf bestimmte Organe beschränkte Antigene, wie z.B. anti-TSHR Antikörper, welche die Schilddrüse, oder anti-nikotinerg AChR Antikörper, welche Funktionen der quergestreiften Muskelzellen beschädigen. Diese zwei Antikörper zeigen auch, wie Autoantikörper die Zielorgane beeinflussen können. Die anti-AChR Antikörper interferieren mit der Signalübermittlung des nACh-Rezeptors, die anti-TSHR Antikörper können die TSH Rezeptoren überstimulieren, und durch unkontrolliertes Wachstum eine Hyperthyreose und Morbus Basedow verursachen. Drittens, in der Hashimoto 165 Thyreoiditis fördern gegen die Schilddrüsen gerichtete Antikörper die immunvermittelte Zerstörung der Schilddrüse und erzeugen eine Hypothyreose. Abhängig vom Isotyp der sezernierten Autoantikörper kann der Mechanismus der Erzeugung der Gewebeschäden auch unterschiedlich sein. In der MG sind die produzierten Anti-AchR Antikörper meistens IgG1 und IgG3 Typ Antikörper, welche neben der induzierten Internalisierung der Rezeptoren auch Komplementaktivierung und ADCC auslösen können. In einem anderen Subtyp der MG, in dem anti-MuSK Antikörper typisch sind, sind die Antikörper vor allem vom Typ IgG4, und infolge einer posttranslationellen Modifizierung sind sie nicht in der Lage das Komplement zu aktivieren. In diesem Fall sind die Gewebeschäden wahrscheinlich von einem durch AK beschleunigten Abbau von MuSK verursacht. Generell muss man sehen dass in den systemischen Autoimmunerkrankungen eine Vielzahl von Autoantikörpern gleichzeitig vorhanden sein können, wogegen Autoantikörper auch infolge von Infektionen gebildet werden können, die aber später meistens keinerlei klinischen Konsequenzen haben werden. Wenn ein Verdacht auf eine Autoimmunerkrankung in der klinischen Praxis besteht, wird am öftesten ein Nachweis der in der jeweiligen Krankheit am häufigsten vorkommenden Autoantikörper verlangt. Trotzdem ist die Präsenz von diesen Antikörpern an sich noch kein spezifischer Beweis für eine gewisse Autoimmunerkrankung, also dürfen die Autoantikörper-Werte nur zusammen mit dem klinischen Bild interpretiert werden. Auf den nächsten Seiten werden wir anhand von zwei Beispielen von systemischen Autoimmunerkrankungen (SLE und RA) die verschiedenen Methoden der klinischen Analyse von Autoantikörpern erklären. Von den organspezifischen Autoimmunitäten und ihre Autoantikörper werden wir uns nur mit der Myasthenia gravis im Detail auseinandersetzen. 10.1.2.1. Lupus erythematodes (SLE) Der Lupus erythematodes ist typischerweise die Erkrankung von jungen, 20-50 Jahre alten Frauen. Obwohl Autoimmunkrankheiten generell öfter in Frauen als Männern vorkommen, ist der SLE das Paradebeispiel für diesen Unterschied, da das Verhältnis von kranken Frauen vs. Männern bei SLE ca. 9:1 beträgt. Die Prävalenz in Frauen ist zurzeit 1:800-1000, Tendenz steigend. Der SLE ist der Archetyp von komplexen Autoimmunkrankheiten. Je nach beeinträchtigtem Organ können die Symptome recht variabel sein: z.B. Arthritis, Pleuritis (Brustfellentzündung) und Pleuraerguss, Herzrhythmusstörung, Fieber, Ermüdung, Schwäche, Schmetterlingserythem am Gesicht, Nephritis, Anämie, Raynaud-Syndrom, neuronale Probleme, Vaskulitis, usw. Eine Vielzahl von Autoantikörpern werden gebildet, wie z.B. antinukleärer Antikörper (ANA), Rheumafaktor (RF), anti-C1q (Komplement), anti-Erythrozyten Antikörper, anti-doppelsträngige DNA Antikörper (anti-ds-DNA), und oft sind Kryoglobuline auch zu sehen, also aus Immunglobulinen bestehende Komplexe, die bei niedrigeren Temperaturen im Serum Präzipitate bilden. 166 Abbildung 10.1. Zusammenhang zwischen Aktivität von SLE und Konzentration der Serum anti-DNA Antikörper bzw. Komplement-Aktivierung 10.1.2.2. ANA (antinukleäre Antikörper) ANA ist ein Sammelbegriff von Antikörpern die verschiedene Zellkern-Komponente erkennen. ANAs tauchen in vielerlei autoimmunen und infektiösen Krankheiten auf. Zum Beispiel in SLE sind ANA Antikörper vom Typ anti-Chromatin, anti-dsDNA, anti-Histon, anti-Sm (=anti-Smith, gegen ein snRNP), in der Sklerodermie anti-U1RNP (auch gegen ein snRNP), anti-Scl-70 (gegen Topoisomerase 1), antiCentromer, im Sjögren Syndrom die SS-A und SS-B Antikörper, in Polymyositis die anti-Jo1 (HistidintRNA Synthetase) Antikörper typisch. Diese Antikörper werden normalerweise mit Immunhistochemie nachgewiesen, und danach wird ihr Titer genauer, mit ELISA bestimmt. Ein screening test für ANAs ist die Analyse von ANA-erzeugte Immunfluoreszenz an der HEP2 Zellinie (ein menschliches epidermoides Karzinom). Das verdünnte, zu analysierende Serum wird zu den Hep2 Zellen gegeben, und dann, falls ANA vorhanden ist, wird ihre Bindung nachgewiesen bzw. ihr Bindungsmuster im Zellkernen wird mit Fluoreszenz-markiertem anti-human IgG, mittels indirekter Immunhistochemie analysiert. Bei solchen Analysen wird als diagnostisches Endergebnis sowohl die angewandte Verdünnung, als auch das Muster der Antikörperbindung im Zellkern (nukleolär, granular, homogen, peripher), angegeben. Falls möglich, muss man jedoch auch bestimmen und angeben, gegen welche Kernkomponente genau die ANAs gerichtet sind. Diese sind alles wichtige Informationen, weil z.B. unter 1:40 Serumverdünnung eine Positivität auch bei gesunden Menschen vollkommen normal ist. Der Labortest ist eigentlich so eingestellt worden, dass es bei einer Verdünnung von 1:160 in 5% der gesunden Population ein positives Resultat gibt. Mit anderen Worten, die Spezifizität des Tests beträgt 95%, d.h. 5% ist der erwartete Anteil der falsch positiven. Falsch positiv ist eine Testperson ohne klinischen Symptome, aber mit granulärer Zellkernfärbung (nur dieses Muster kann in gesunden Menschen vorkommen). Pathologische, z.B. homogene Kernfärbung geben Patienten mit anti-dsDNA Antikörper in SLE, periphere Kernfärbung ist typisch z.B. bei anti-Histon Antikörpern in SLE und Sklerodermie, nukleoläre Kernfärbung ist charakteristisch im Falle von anti-Topoisomerase Antikörpern in Sklerodermie, 167 granuläre Kernfärbung kann man bei z.B. anti-RNPs sehen (SLE, Sklerodermie, Mischkollagenose, Sjögren), obwohl sie auch in gesunden Personen, nach bestimmten viralen oder anderen Infektionen, vorkommen können. Indirekte Immunfluoreszenz wird auch angewandt wenn man Immunkomplexe in beeinträchtigten Organen (Niere oder Haut), in entsprechenden Gewebe-Biopsien, nachweisen möchte. Wenn der Screening-test ein positives Ergebnis zeigt, dann folgen spezifische Testverfahren um festzustellen, um welchen Antikörper unter dem Sammelbegriff ANA es sich in dem jeweiligen Fall eigentlich handelt. Dies zu bestimmen ist durch spezifische ELISAs, Immundiffusion, oder Western blot möglich. Da wir diese Techniken schon in anderen Kapiteln besprochen haben, werden wir in diesem Bereich hier nicht in weitere Details gehen. Bestimmung der Präsenz und Titer von Autoantikörpern ist nicht nur diagnostisch, sondern es ist auch prognostisch und prädiktiv wichtig. Zum Beispiel wenn in SLE der Titer von anti-dsDNA Antikörpern sinkt, und parallel dazu die Serumkomplement-Konzentration steigt, dann geht die Krankheit in eine Remission, bzw. die Therapie wirkt gut. Die Präsenz von bestimmten anderen Autoantikörpern z.B. die der anti-Chromatin AKs, ist häufiger in Lupus Nephritis, also kann prognostisch auf eine Nieren-Beschädigung in SLE hindeuten. Unter den Autoantikörpern mit prognostischem Wert in SLE können z.B. die anti-Ro (=SSA) Antikörper, die 10 Jahre vor dem Ausbruch der Krankheit schon detektierbar sind, erwähnt werden, die anti-dsDNA Antikörper, die 2.5 Jahre und letztlich die anti-Sm Antikörper, die einige Monate vor den ersten Symptome im Blut erscheinen. 10.1.2.3. Kryoglobuline Kryoglobuline sind Serum Immunglobuline welche bei Temperaturen unter +37°C ein Präzipitat bilden, aber erneut aufgewärmt gehen sie schon wieder in die Lösung. An dieser Stelle ist es wichtig klarzumachen, dass Kryoglobuline zwar in der Autoimmunität oft vorkommen, sie sind aber nicht spezifisch für Autoimmunerkrankungen, weil sie auch in anderen Krankheitsbildern, wie z.B. in manchen malignen hämatologischen Krankheiten und auch in der Hepatitis C Infektion ziemlich typisch sind. Sie zeigen mehrere Untergruppen: Typ I: isolierte monoklonale Immunglobuline (wie monoklonales IgG oder IgM, selten andere Isotypen), Typ II: monoklonales IgM welches als Rheumafaktor polyklonale IgG Antikörper an ihren Fc Teil bindet, und Typ III: ähnlich wie Typ II, bloß das IgM Rheumafaktor polyklonal ist. Der Nachweis von Kryoglobulinen war früher eine umständliche und lange Prozedur, in der zuerst Serum bei +37°C isoliert, und danach 2-7 Tage lang bei +4°C gelagert worden ist. Falls ein Präzipitat in der Lösung zu sehen war, wurden die Proben zentrifugiert, nach Entfernung des Überstands mit physiologischer Salzlösung vermischt und wieder auf +37°C erwärmt. Wenn das Präzipitat wieder löslich geworden ist, wurde es als ein Kryoglobulin identifiziert. Heute werden Kryoglobuline mit Immunelektrophorese oder Immunfixierung nachgewiesen, welche nicht nur den Isotyp der Kryoglobuline verraten, sondern auch viel schneller auszuführen sind, als die klassische Methode. 168 10.1.2.4. Rheumatoide Arthritis Die Rheumatoide Arthitis ist eine autoimmun Entzündung welche eine Destruktion der knorpel- und synovialen Membranen hervorruft. Ihre Häufigkeit beträgt 1:100 in der nord-amerikanischen und europäischen Population. Die Krankheit ist systemisch, und sie hat auch extra-aurikulare Manifestationen, welche vor allem in den Personen zu beobachten sind, die für die Krankheit typische Autoantikörper produzieren. Systemische Symptome können z.B. Perikarditis, Lungenfibrose, trockene Augen, und Polyneuropathie sein. ANAs sind oft vorhanden, und auch anti-zyklisches citrulliniertes Peptid (anti-CCP) Antikörper sind häufig. Neulich hat man in RA auch Antikörper gegen citrulliniertes Vimentin und citrullinierte alpha-Enolase entdeckt, mit anderen Worten, die citrullinierten Proteine bzw. Autoantikörper gegen sie scheinen in der Pathogenese von immer grösserer Bedeutung zu sein. Citrullin ist eine proteinogene Aminosäure, Citrullinierung, also das Erscheinen von Citrullin in Proteinen kann jedoch nur infolge einer posttranslationellen Modifizierung entstehen, weil das Genom keine Citrullin-spezifische DNA oder RNA Codons beinhaltet. Citrullin entsteht deshalb aus Arginin Aminosäuren die bereits in einer Peptidkette eingebaut sind: der Prozess wird von einem Enzym namens Peptidyl-Arginil-Deiminase (PAD) katalysiert. Abbildung 10.2. Die Biosynthese von Citrullin Viele Tierexperimente haben gezeigt, dass citrullinierte Antigene Arthritis verursachen können, obwohl der genaue Mechanismus dahinter noch unbekannt ist. Interessanterweise hat man auch bewiesen, dass das Rauchen die Citrullinierung generell verstärkt, und weil es auch bekannt ist, dass Rauchen ein wichtiger Risikofaktor bei RA ist, es ist möglich dass die zwei miteinander kausativ gekoppelt sind. Die anti-CCP Antikörper sind in der RA vor allem diagnostisch wichtig, weil ihr Niveau nicht mit der Aktivität der Krankheit korreliert. Die anti-CCP Antikörper kommen in 65% der Patienten vor (Sensitivität des Markers ist 65%) und ungefähr 95% der anti-CCP positiven Personen haben tatsächlich RA (Spezifizität des Markers beträgt 95%). Anti-CCP Antikörper werden in der Regel mit ELISA nachgewiesen. 169 Der Rheumafaktor (RF) ist auch ein typischer Autoantikörper in RA. Dies ist ein Antikörper welcher gegen andere Immunglobuline produziert wird. Ungefähr 80% der RA Patienten sind Rheumafaktor positiv, obwohl seine Serumkonzentration in anderen Autoimmunitäten und nach manchen Infektionen auch erhöht wird. Dementsprechend weisen ungefähr 10% der gesunden Erwachsenen auch detektierbare Mengen von Rheumafaktoren vor. In den RA Patienten prognostiziert ein höherer Spiegel von Rheumafaktoren immer einen destruktiveren Ablauf der Krankheit. Abbildung 10.3. Der Rheumafaktor Der RF wird normalerweise mit zwei Methoden nachgewiesen (es gibt eine Vielzahl von solchen Methoden, wie Z.B. ELISA, aber in der klinischen Praxis sind diese zwei am weitesten verbreitet): 1. Latex Agglutinationstest: Die zu analysierende Serumprobe wird mit zu menschlichen IgG gekoppelten Latex Mikroperlen vermischt. Wenn RF in der Serumprobe ist, quervernetzen sich die Perlen durch die IgG und es bildet sich dadurch ein sichtbares Präzipitat. Dieser Test ist für Screening geeignet. Ähnlich war früher der sog. Rose-Waaler Test, in dem anstatt Latexperlen Schaf Erythrozyten benutzt wurden, aber dieser Test ist heute aus praktischen Gründen nicht mehr so populär, als er einmal war. 2. Nephelometrie: Sie freut sich einer etwas höheren Popularität als der Latex Agglutinationstest, weil sie etwas sensitiver ist. In der Nephelometrie wird zur Serumprobe eine sehr hohe Konzentration von menschlichen IgG gegeben. Falls RF vorhanden ist, entstehen kleine Immunkomplexe im Serum, welche die Lichtstreuung der Probe erhöhen und auf dieser Weise mit einem Laser-Nephelometer nachweisbar sind. Die Menge der Komplexe ist direkt proportional zur Stärke der Lichtstreuung. Genauso wie in dem SLE können der Rheumafaktor und die anti-CCP Antikörper auch in der RA viele Jahre vor den ersten Symptomen erscheinen. Wenn die zwei Antikörper gleichzeitig ausgewertet werden, kann der Arzt einen ausgezeichnet sensitiven und spezifischen prognostischer Test benutzen. 10.1.2.5. Organspezifische Autoimmunitäten Organspezifische Autoimmunerkrankungen sind unter anderem: autoimmun Thyreoiditis, Pemphigus vulgaris, autoimmun Myasthenia gravis, usw. In der Myasthenia gravis zeigen mehr als 80% aller Patienten anti-AchR Antikörper. Diese Antikörper werden meistens mit einem Radio-Immunassay (RIA) nachgewiesen. In solchen Testverfahren wird zur Serumprobe des Patienten ein radioaktiv markiertes Antigen, meistens 125 I alpha-Bungarotoxin-markiertes AchR gegeben. Wenn AchR-spezifische Antikörper im Serum sind, 170 bilden sie Immunkomplexe mit dem markierten AchR. Diese Immunkomplexe werden dann mit der Zugabe von Anti-human IgG quervernetzt und präzipitiert. Das Präzipitat wird durch Zentrifugierung von der Lösung getrennt, und seine Radioaktivität, welche direkt proportional zur Konzentration der Autoantikörper ist, wird mit einer Gamma-Zähler gemessen. Im Falle der, für die Hashimoto thyreoditis charakteristischen anti-TSHR Antikörper wird eher das Radio Rezeptor Assay (RRA) benutzt. RRA ist ein kompetitives Assay, in dem bekannte Mengen von radioaktiv markierten TSH Molekülen in eine Kompetition treten mit einer unbekannten Menge von Anti-TSHR Autoantikörpern des Patienten. Im Test versuchen diese zwei Moleküle kompetititv zu einer begrenzten Menge von Bindungsstellen an TSH Rezeptoren zu binden. Je mehr Anti-TSHR Autoantikörpern vorhanden sind im Serum des Patienten, desto weniger radioaktiv markierte TSH Moleküle könne zu den Rezeptoren binden, und vice versa. Die Stärke der Radioaktivität des Präzipitats wird also hier, im Gegensatz zu RIA, umgekehrt proportional zur Menge des nachzuweisenden Autoantikörpers sein. Abbildung 10.4. Das Radio Rezeptor Assay (RRA) Neben diesen Testverfahren wird neulich auch sehr viel mit Antigen-Arrays experimentiert um die ganze Komplexität der autoimmunen humoralen Immunantwort erfassen zu können. Diese Technologie beruht auf einer katalogisierten Sammlung verschiedener Autoantigene, welche von Autoantikörpern in Autoimmunerkrankungen oft erkannt werden. Diese Antigene werden rekombinant hergestellt, und in einer bestimmten räumlichen Ordnung, in Reihen und Kolumnen, zu einer speziellen Glas- oder Kunststoff-Oberfläche gebunden (Antigenarray). Nachdem die aspezifische Proteinbindung zur Oberfläche blockiert wird, wird das Serum des Patienten auf das Array aufgetragen. Nach einer gewissen Inkubationszeit werden die Autoantikörper des Patienten, falls vorhanden, zu ihrem erkannten Autoantigen binden. Diese Antikörper werden in dem nächsten Schritt 171 mit Fluoreszenz-markiertem anti-human IgG nachgewiesen, und mit Hilfe eines speziellen ArrayScanners, basierend auf ihrer räumlichen Position und emittierten Lichtintensität, auf dem Array identifiziert bzw. ihre relative Menge gemessen. Der Vorteil dieser Technologie ist die Vielzahl von Autoantikörpern die gleichzeitig analysiert werden können. Auf diese Weise könnten auf einem Array praktisch alle bekannten Autoantikörper aller Autoimmunkrankheiten in ein und demselben Test ausgewertet werden. Neben diesen klassischen Antigenarrays gibt es auch solche in Entwicklung, mit denen die komplementaktivierende Wirkung von Autoantikörpern auch bestimmt werden kann, also ihre Funktionen auch beschrieben werden können. 10.2. Die HLA-Typisierung Die menschlichen HLA Gene die für die MHC Proteine kodieren, befinden sich auf dem kurzen Arm des menschlichen Chromosoms 6. Die Aufgabe der von ihnen kodierten MHC Proteine ist die Regulation der Erkennung von körpereigenen und fremden Antigenen. Die MHC Proteine haben zwei wichtigere Untergruppen. Die MHC I Proteine werden von den HLA A, B und C Genen kodiert. Sie werden auf der Oberfläche von allen kernhaltigen Zellen exprimiert, wirken nur im Komplex mit einem zusätzlichen Protein namens beta2-Mikroglobulin, welches von Chromosom 15 kodiert ist, und präsentieren endogene Antigene für CD8+ Tcit Zellen. Die MHC II Proteine werden dagegen von fünf HLA Gengruppen kodiert, von denen die letzten zwei nicht so bedeutend sind: HLA DP, DQ, DR, und DM, DO. MHC II Proteine erscheinen vor allem auf antigenpräsentierenden Zellen, wirken in Form eines alpha-beta Heterodimers, und präsentieren exogene Antigene für CD4+ Th Zellen. 10.2.1. Die Nomenklatur des HLA-Systems Die Nomenklatur des HLA Systems, also die diagnostische Beschreibung der einzelnen, von Person zu Person meist unterschiedlichen HLA Typen musste mit der Zeit kontinuierlich geändert werden, weil mit den neueren Methoden eine immer genauere Identifizierung der HLA Typen ermöglicht wurde. Vor zwei Jahrzehnten waren für die HLA Typisierung nur serologische Methoden erhältlich, mit der Entwicklung neuer Technologien benutzt man heute aber eher molekulargenetische Untersuchungsmethoden. Die Nomenklatur: HLA-A: HLA-A Gen Lokus HLA-A1: Serologisch definiertes Antigen. Serologische Methoden ermöglichen nur eine begrenzte Auflösung. Dies wurde mit der Verbreitung der Techniken der Molekulargenetik erheblich verbessert. HLA-A1*: Der “*” markiert Ergebnisse von Typisierungs-Analysen welche mit einer molekulargenetischen Untersuchung ausgeführt wurden. HLA-A1*0101 – 4 Nummern Auflösung: Beschreibt solche allelische Unterschiede in der kodierenden Region der DNA-Sequenz des HLA-Gens, die zu Unterschieden in der Proteinsequenz führen 172 HLA-A1*010101 – 6 Nummern Auflösung: Beschreibt solche allelische Unterschiede in der kodierenden Region des HLA-Gens, welche aber wegen der Codon-Degeneration die Proteinsequenz nicht beeinflussen können HLA-A1*01010101 – 8 Nummern Auflösung: Sequenz-Unterschiede in den intronischen oder anderen, nicht-kodierenden Regionen des HLA-Gens 10.2.2. Methoden der HLA-Typisierung 10.2.2.1. Serologische Methoden: Mikrozytotoxizitätstest: dieser Test beruht auf einer komplementvermittelten Zytotoxizität, welche spezifisch gegen die individuellen MHC Varianten eines jeweiligen Patienten ausgelöst wird. Er wird benutzt für die MHC I Typisierung (HLA-A, -B und -C). Im Laufe des Tests werden zuerst aus dem Blut des Patienten durch Ficoll-Zentrifugierung Lymphozyten gewonnen. Kleine Aliquote von diesem Isolat werden dann auf eine 96-Well Platte aufgetragen. Auf der Platte werden alle Wells mit einem anderen Antikörper inkubiert, die die verschiedenen HLA Serotypen spezifisch erkennen. Im nächsten Schritt wird zu den Wells Kaninchenserum mit aktivem Komplement-Inhalt gegeben. Die Komplementproteine zerstören die Zellen in den Wells, wo eine Antikörperbindung stattgefunden hat. Diese Lyse wird dann mit Exclusionsaassays wie z.B. Trypanblau sichtbar gemacht und so der HLA-Serotyp des Patienten bestimmt. Trypanbalu ist ein Farbstoff der die Plasmamembran nicht durchdringen kann. Abbildung 10.5. Mikrozytotoxizitätstest Er gelangt also nicht in intakte Zellen, sondern nur nach deren Lyse Der HLA Genotyp/Serotyp wird hier also anhand der Position der Wells, d.h. die Spezifizität der Antikörper welche eine Lyse erzeugt haben und Zellfärbung mit Trypan blau ermöglichten, bestimmt. Vorteile des Tests sind die niedrigen Kosten und dass kein großer Laborhintergrund benötigt wird. Seine Nachteile sind 173 die relativ großen Mengen an Lymphozyten, die zum Test gebraucht werden, und dass es fürs Labor manchmal schwer ist gegen alle seltene HLA-Serotypen spezifische Antikörper zu finden. Gemischte Lymphozytenkultur (Mixed Lymphocyte Culture, MLC): dieser Test beruht auf einer T-Zell-vermittelten Erkennung von individuellen MHC Varianten eines jeweiligen Patienten. Er wird benutzt für MHC II Typisierung (vor allem HLA-DR und -DQ), und wird typischerweise als Standardverfahren vor Organtransplantationen ausgeführt. In diesem Fall werden Lymphozyten des Spenders mit Lymphozyten des Empfängers vermischt, und in eine Zellkultur gebracht, wo sie typischerweise 48 Stunden lang kultiviert werden. Falls eine MHC Inkompatibilität vorliegt, werden T Zellen von den fremden MHCs angeregt, was man durch ihre Teilung (mit Hilfe von 3HThymidin Inkorporation) oder Cytokin-Sekretion (mit IFNgamma ELISA) nachweisen kann. Da in diesem Fall (two-way MLC) nicht klar wird, welche Zellen, d.h. ob die Zellen des Spenders oder des Empfängers reagiert haben, also ob es nach Transplantation eine Graft vs Host oder Host vs Graft Reaktion zu erwarten ist, werden manchmal die Zellen des einen Partners mit Bestrahlung vor dem Test inaktiviert (one-way MLC). Abbildung 10.6. Gemischte Lymphozytenkultur für HLA-Typisierung Weiter, wenn der Test zur HLA-Typisierung benutzt wird (Abbildung 10.6.), werden kleine Aliquote von Lymphozyten des zu testenden Patienten in den Wells einer 96-Well Platte mit Lymphozyten von bekanntem HLA-Typ z.B. HLA-Dw-1, 2, usw. vermischt, und dann wird die Aktivierung gemessen. Dies ermöglicht die Bestimmung von speziellen HLA Typen, welche in der Diagnostik immer in als „HLADw…“ Typen angegeben werden. 10.2.2.2. Molekulare Methoden (DNA Sequenzanalyse) Diese Techniken tauchten am Ende der ’80-er mit dem RestriktionsfragmentlängenpolymorphismusAnalyse (RFLP) auf, wurden erleichtert in den 90-ern mit der Einführung der Polymerase- 174 kettenreaktion (PCR), und noch später mit der Verbreitung der modernen einfachen Sequenzierungsanalysen. RFLP: Praktisch fast alle genetische Polymorphismen, und so auch die der HLA Gene können mit RFLP typisiert werden. In der RFLP werden Unterschiede in den Längen der, mit sequenzspezifischen Restriktionsendonukleasen (REs) verdauten DNA, verglichen. REs erkennen und spalten bei typischerweise 4-8 Basenpaaren lange DNA Sequenzen. Falls die Erkennungssequenz durch eine andere allelische Variante geändert wird, findet die Spaltung nicht statt, und die Länge der DNA Fragment wird geändert. Diese Unterschiede können mit verschiedenen Gelelektrophorese-gekoppelten Techniken nachgewiesen und zur HLA Typisierung benutzt werden. PCR SSOP (PCR Sequenzspezifische-Oligonukleotide-Sonde) HLA-Gensegmente des Patienten werden mit PCR vermehrt, durch die Primer mit Biotin gekoppelt, und denaturiert. Danach werden sie auf eine Membran aufgetragen, die in einer bestimmten räumlichen Ordnung (Reihen und Kolumnen) alle analysierten HLA-Typen in der Form von einzelsträngigen DNA-Sonden beinhaltet. Die denaturierte, einzelsträngige Biotin-markierte DNA des Patienten wird mit der einzelsträngigen DNA-Sonde der Membran hybridisiert. Die Positionen der spezifischen Bindungen an der Membran werden dann mit der Zugabe von einem Avidin-HRP Enzym Komplexes und letztlich mit der Zugabe eines HRP Substrats sichtbar gemacht. So wird ein sichtbares Produkt an den Stellen der spezifischen Bindungen entstehen, welche den HLAGenotyp des Patienten angeben. SSP (Sequenzspezifische Primers): Diese Methode bestimmt den HLA-Genotyp mit sequenzspezifischen PCR Primers, ein PCR Produkt von bestimmter Länge wird erzeugt, falls ein bestimmtes HLA Allel in der DNA des Patienten vorhanden ist. Meistens werden mehrere PCR Reaktionen mit jeweils mehreren PCR Primerpaaren gleichzeitig angewandt(Multiplex PCR). Die Länge der erhaltenen Produkte wird am Ende der PCR mit Hilfe von Gelelektrophorese bestimmt, und so der Genotyp anhand der Länge der PCR Produkte bestimmt. SBT (Sequence Based Typing): Die genomische DNA des Patienten wird mit PCR und nachfolgender Sequenzierung von Base zu Base vollständig abgelesen, und der Genotyp mit Hilfe von online DNA-Sequenzdatenbanken bestimmt. 10.2.3. Gewebe- und Organtransplantation Die Transplantation ist bei vielerlei Krankheiten, wie malignen hämatologischen Krankheiten, vielen fortgeschrittenen Nierenerkrankungen, schweren Kardiomyopathien usw. eine, meistens zwar für den Arzt technisch herausfordernde, aber reale Möglichkeit, und oft die letzte therapeutische Chance für die Patienten. Heute sind schon die technischen Möglichkeiten für eine Transplantation der z.B. Hornhaut, Lunge, Herz, Bauchspeicheldrüse, Dünndarm, Knochenmark, Haut, Nieren, usw. gegeben. Die ideale Situation bei Transplantationen ist der Fall in dem der Patient (Empfänger) ein Organ von einem Spender mit völlig identischen HLA-Allelen erhalten kann. Dies ist der Fall wenn eine isogene Transplantation zwischen eineiigen Zwillingen, oder seltener, wenn eine allogene Transplantation 175 zwischen HLA-identisches Geschwister ausgeführt werden kann. Solche glücklichen Konstellationen sind aber extrem selten. Abbildung 10.7. Vielfalt der klassischen HLA Moleküle an der Oberfläche einer einzelnen Antigenpresentierende Zelle (APZ), und der genetische Hintergrund dieser Variabilität Meistens gibt es klare individuelle Unterschiede zwischen den MHC Proteinen des Spenders und des Empfängers die Transplantationszwischenfälle, sprich eine Abstoßung, auslösen können. Bei den alltäglichen allogenen Transplantationen wird es also immer ein wichtiges Ziel bleiben solche Spender zu finden, dessen HLA-Allelkombination am nahesten zu der des Empfängers liegt. Ein begrenztes Mismatch ist generell akzeptabel, falls es zu keiner großen Reduktion der Halbwertszeit des Organs / Überleben des Patienten führt, obwohl der Arzt in extrem schweren Fällen auch erhöhte Risiken eingehen mag. Da HLA Genotypen als Haplotyp vererbt werden, ist es empfehlenswert, nach einem passenden Spender zuerst in der Verwandtschaft des Empfängers zu suchen. Wenn nämlich z.B. zwischen zwei Geschwistern ein HLA-Genotyp übereinstimmt, ist die Chance wegen der Haplotype, dass auch die anderen HLA-Genotypen übereinstimmen, in der selben Familie relativ hoch. Letztlich muss man auch sehen, dass die relative Wichtigkeit der Übereinstimmung von MHC I und II Proteine und anderen, z.B. Blutgruppenantigenen bei Abstoßungsreaktionen von Organ zu Organ unterschiedlich ist. Bei Nierentransplantationen zum Beispiel sind sowohl MHC I als auch MHC II mehr oder weniger genauso wichtig, aber HLA-DR Mismatches sind extrem kritisch. Was die andere Seite der Transplantationszwischenfälle, also statt Abstoßung die Graft versus Host Reaktionen (GVHD) angeht, ist hier die Knochenmarktransplantation bei malignen hämatologischen Krankheiten eine spezielle Kategorie. Die Übereinstimmung der Blutgruppenantigene ist in diesem Fall nicht so wichtig, weil nach einer Knochenmark-Transplantation die roten Blutkörperchen des Empfängers auch ersetzt werden. Andererseits aber, weil die Blutgruppenantigene neben den Erythrozyten, zwar sehr schwach, aber auch von den Endothelzellen der Kapillaren exprimiert sind(!), 176 ist eine Graft versus Host Reaktion selbst hier nicht vollkommen auszuschließen. Wie bei der Abstoßung, gilt auch bei der GVHD dass je grösser die Unterschiede in den HLA Genen sind, desto höher sind die Chancen der Komplikationen. 10.2.4. HLA Assoziation mit Rheumatoider Arthritis HLA Typisierung ist eine Analyse die nicht nur bei Transplantationsreaktionen, sondern auch bei vielen Autoimmunitäten empfehlenswert ist auszuführen. Der Grund für diese Tatsache liegt daran, dass es schon seit langer Zeit bekannt ist, dass die Mehrheit dieser Krankheiten genetisch bedingt sind und (unter anderem) auch mit bestimmten HLA-Allelen gekoppelt ist. HLA-Assoziation ist auch in der RA stark: ungefähr 30% aller Patienten tragen einen bestimmten, nämlich den HLADRB1 Serotyp. Heute ist dies die stärkste bekannte genetische Assoziation in der RA, obwohl in der Ära der genomweiten Assoziationsstudien (genome wide asszociation study, GWAS) man es erwarten kann, dass innerhalb einer kurzen Zeit immer mehr Gene bekannt werden, die auch in dieser Krankheit von Bedeutung sind. Es ist auch wichtig zu sehen, dass nicht alle, zum HLA-DRB1 Serotyp gehörenden Genotypen / Allele zur Krankheit mit der selben Wahrscheinlichkeit assoziiert sind. Ein weiterer interessanter Punkt ist, dass in dem HLA-DRB1 Serotyp, in der dritten hypervariablen Region der DRβ1 Kette, die Aminosäuren Nr. 70-74 (QKRAA oder QRRAA) zwischen den HLA-DRB1*01 und HLADRB1*04 Allelen völlig übereinstimmen. Da die zwei Allele auf RA mit ähnlicher Stärke prädisponieren, geht man davon aus, dass dies den Aminosäuren Nr. 70-74 zu verdanken ist, die deswegen als “shared epitopes” bezeichnet werden. Weitere, mit der RA gekoppelte Genpolymorphismen sind z.B. Polymorphismen des protein tirosine phosphatase non-receptor 22 (PTPN22, eine Komponente der T-Zell-Rezeptor Signalübermittlung) oder das cytotoxic T-lymphocyte antigen 4 (CTLA-4, ein negativer Kostimulator-Rezeptor auf aktivierten T Zellen). Generell könnte man sagen, dass Polymorphismen von solchen und ähnlichen Immungenen meistens nicht nur mit einer, sondern einer Vielzahl von Autoimmunerkrankungen assoziiert sind, und sehr oft treten sie mit Umweltfaktoren auch in Interaktion. Zum Beispiel wird die prädisponierende Wirkung von HLADRB1 auf anti-CCP positive RA vom Rauchen deutlich gesteigert. 10.2.5. HLA Assoziation mit Spondylitis ankylosans (SPA, Morbus Bechterew) Morbus Bechterew ist eine Autoimmunkrankheit die mit starken Entzündungen in den Wirbelsäulengelenken charakterisiert ist. Die chronischen Entzündungen führen in der Regel zu einer langsamen und schmerzhaften Verschmelzung der Wirbel, eingeschränkter Bewegungsfreiheit und sogar Brüchen in der Wirbelsäule. Typische radiologische Befunde sind Verknöcherungen des Wirbelkörperbandapparates, die am Ende die für die Krankheit typische Bambusrohrform der Wirbelsäule erzeugen. Im Gegensatz zu vielen anderen Autoimmunerkrankungen kommen in der SPA Autoantikörper nicht vor. Die SPA zeigt familiäre Häufung mit starker HLA-Assoziation (mit HLA-B27). In der Tat, ungefähr 90% der Weißen mit SPA sind HLA-B27 positiv. Die Prävalenz der Krankheit ist ca. 0.25-0.5%. Sie betrifft meistens Männer unter 40 Jahren, und ist in Männern durchschnittlich siebenmal häufiger als in Frauen. Die Patienten werden meistens mit Taillenschmerz diagnostiziert, 177 der nach längerer Inaktivität, Erholung oder morgens schlimmer wird, aber mit Bewegung sich etwas verbessert. Die Verkalkung der Sehnen und Gelenke ist ein langer Prozess, findet über mehrere Jahrzehnte hinaus statt, und ist mit typischen zusätzlichen Symptomen gekoppelt, wie Sehnen- und Gelenkschmerz, Aorteninsuffizienz oder Iritis (Entzündung der Regenbogenhaut). Die Frage warum HLA-B27 auf die Krankheit prädisponiert, wurde in Tiermodellen analysiert, und es stellte sich heraus, dass HLA-B27 transgene Tiere, welche das menschliche HLA-B27 Allel exprimiert haben, mit der Zeit oft auch SPA-ahnliche Symptome zeigten. Wenn die andere Komponente der MHCI, also B2 Mikroglobulin gentechnologisch entnommen wurde, haben die Tiere die Symptome mit unveränderter Häufigkeit gezeigt, und in den HLA-B27 positiven Tiermodellen ist die Krankheit selbst in der vollen Abwesenheit von CD8+ T Zellen aufgetreten. Diese Ergebnisse deuten darauf hin, dass SPA nicht von CD8+ T Zellen, bzw. nicht durch Antigenpräsentierung des aktiven HLA-B27 Moleküls abhängig ist. Die wahrscheinlichste Erklärung für diese Ergebnisse ist die Hypothese, dass in den SPA Patienten in der Proteinsynthese der HLA-B27 Moleküle die Faltung der schweren Kette falsch ausgeführt wird. Die Synthese der schweren Kette und ihre Verkopplung mit dem B2 Mikroglobulin findet im ER statt, noch vor seinem Transport zur Zelloberfläche. Man nimmt an dass die abnormale Faltung des Proteins inkorrekt positionierte Disulfidbrücken, Kopplung zu einem Chaperon namens BiP, und eine Akkumulation des HLA-B27 Proteins im ER erzeugt. Diese Akkumulation wird erst unter dem Einfluss von Cytokinen, die die Expression von MHC Porteinen verstärken, extrem kritisch und erzeugt eine Stressreaktion des ERs (sog. ”unfolded protein response”), welche die Zellfunktionen beschädigt. Zweitens, die betroffene Proteine können wahrscheinlich auch MHC-I Dimere bilden (HC-B27 Dimere), und diese vollkommen abnormale Strukturen erzeugen eine inkorrekte Aktivierung von Th und NK Zellen (Abbildung 7.8). Der Prozess ist deswegen nicht von den CD8+, sondern von CD4+ TZellen und NK-zellen vermittelt. Abbildung 10.8. Der wahrscheinliche immunologische Pathomechanismus des Morbus bechterews 178 Wie wir es vorher schon erwähnt haben, sind verschiedene HLA Allele mit mehreren Autoimmunerkrankungen gekoppelt. Es gibt HLA Allele die mit Typ-1-Diabetes mellitus, RA, Morbus bechterew, Myasthenia gravis, oder Narkolepsie assoziiert sind. Die Narkolepsie ist eine neuronale Erkrankung mit unkontrollierbaren, plötzlichen Schlafanfällen und REM-Phasen. Die Patienten leiden an Schlaflähmung, also Paralyse nach dem Erwachen, Kataplexie, d.h. von Stress induziertem Tonusverlust und hypnagogen Halluzinationen (in der REM-Phase beginnenden, entweder im Schlaf oder an der Grenze des Schlafes und Erwachens auftauchende extrem lebensnahe Halluzinationen) Der Hintergrund der Krankheit ist unbekannt, obwohl in narkoleptischen Patienten man schon AntiOrexin Antikörper gefunden hat, welche, zusammen mit der HLA-Assoziation eine immunologische Pathogenese verdächtigen. Ein weiteres Beispiel ist Myasthenia gravis, wo die jugendlichen antiAChR positiven Fälle sich stark von anderen, z.B. anti-MuSK positiven Fällen unterscheiden; eine Tatsache, welche auf die Existenz von unterschiedlichen Pathomechanismen hindeutet. Übersetzt von Zoltán Pós 179 11. ÜBEREMPFINDLICHKEIT TYP I.: ALLERGIE (MARIANNA CSILLA HOLUB) 11.1. Die Gruppierung der Überempfindlichkeitsreaktionen Gell und Coombs haben nach dem Mechanismus, den entstehenden Effektormolekülen, den beteiligten Zellen und der Zeit der Entwicklung des Prozesses die Hypersensitivitätsreaktionen in zwei Gruppen aufgeteilt. Da diese Aufteilung meistens in der heutigen klinischen Anwendung verwendet wird, übernehmen wir sie auch in diesem Handzettel. Sobald jedoch mehr über die Ursachen und die Mechanismen der Überempfindlichkeitsreaktionen bekannt wird, können sich diese Kategorien auch noch ändern. So wird zum Beispiel in der englischen Literatur – aber nicht in der amerikanischen – häufig eine zusätzliche fünfte Klasse (Gruppe) verwendet. Diese fünfte Klasse entspricht dem Typ II. Die untenliegende Tabelle ist die Zusammenfassung der vier Überempfindlichkeitsreaktionen nach der Aufteilung von Gell und Coombs. Typ Reaktion I. Allergie - Anaphylaxie IgE –vermittelte Antigen-Antikörper Reaktion II. cytotoxisch An den Zelloberfläche-Antigene (extrinsic) oder Gewebsantigene (intrinsic) spezifisch gebundene Ak (IgG) III. Immunkomplex Zirkulierende Antigen-Antikörper Komplexe lagern in den Gewebe ein IV. Zellvermittelt - verzögerter Typ T-Zelle Antwort auf die Antigene Tabelle 11.1. Die 4 Typen von Überempfindlichkeitsreaktionen Nach der Allergenexposition entstehen die Symptome am schnellsten im Falle einer Typ I Überempfindlichkeitsreaktion. Zeitlich gesehen folgen Typ II und dann Typ III. In einer Typ IV Überempfindlichkeitsreaktion vergeht am meisten Zeit, bis es zum Erscheinen von Symptomen kommt. Man nennt die Typ I. Reaktion eine Sofortreaktion, die Typ II.Reaktion eine späte Überempfindlichkeitsreaktion. Die Schwierigkeit der klinischen Symptome verhält sich im umgekehrten Sinne zur Nummerierung der Type. So können die Symptome einer Sofortreaktion in einigen Minuten zum Tod führen. Der Arzt hat sehr wenig Zeit diese Symptome zu behandeln. 180 11.2. Die Entstehung der allergischen Reaktionen Die wichtigste biologische Wirkung des Allergens ist es, eine IgE-vermittelte Th2 Typ Immunantwort auszulösen. Dadurch wird im Allgemeinen eine Entzündungsreaktion verursacht. 1. Sensitisierung (in genetisch prädisponierten Personen) Die für das Allergen charakteristischsten Parameter sind die folgenden: Sehr kleine Mengen lösen schon die Reaktion aus (z.B. ist 1µg/Jahr aus Ambrosia Pollen genug) Nicht prozessiertes, intaktes Molekül Soluble Moleküle im Allgemeinen mit kleinem Molgewicht, die in ausgetrockneten Formen sehr stabil werden können. Solche ist die allergene Komponente des Kots der Milben, das Der p1 mit 15kD Gewicht. Die Schleimhaut ist am häufigsten der Ort des Eintrittes der Allergene in den Körper oder der Ort der Allergenexposition (z.B. respiratorisches Epithel), an dem die ausgetrockneten Antigene wieder löslich werden. Es existieren Allergene, die die Möglichkeit eines Eintrittes durch ihrer enzymatische Aktivität steigen. Das Der p1 Allergen des Kots der Milben besitzt eine Komponente mit Cistein-Serin Protease Aktivität. Das Ergebnis ist die Spaltung der Membranproteine (Okkludin, Klaudin) der tight junctions, wodurch die Membranpermeabilität der Bronchien steigt und so die Allergene einfacher in die subepitheliale Zone eintreten können. Abbildung 11.1. Allergische Reaktion – Sensitisierung Die Allergene können nicht nur ihren einfacheren Eintritt in den Körper fördern, sondern gleichzeitig auch die Antigenaufnahme, die Antigenpräsentierung und die T-Zell Aktivierung. Das Der p1 Allergen wirkt auf die PAR-2 Rezeptoren (protease activated receptor 2) der Epithelzellen durch seine proteolytische Aktivität und löst die Sezernierung der proinflammatorischen Cytokine (IL-6, IL-8, GMCSF) aus. Diese Mikroumgebung erleichtert die Entstehung der Entzündungsprozesse. Der Kot der Milben besitzt neben den Allergenen solche Moleküle, die als Adjuvantien dienen und die Antigenaufnahme der dendritischen Zellen fördern. Ein solches Molekül ist die bakterielle DNA, das 181 Endotoxin. Überwiegt das an TLR-4 gebundene Endotoxin, fördert es eine Th2 Typ Antwort, überwiegt die an das TLR-9 gebundene hypometilierte DNA, wird eine Th1 Typ Antwort gefördert. Zusammenfassend kann das Allergen seine Aufnahme von dendritischen oder von irgendwelchen antigenpräsentierenden Zellen (APZ) begünstigen. Das von APZ und B Zellen aufgenommene und prozessierte Allergen wird den geeigneten Th2 Zellen präsentiert und von TZR erkannt. Es führt zur Herstellung der IL-4 und IL-13 Cytokine. IL-4 und IL-13 fördern den Ig- Klasswechsel in der Umgebung der auf das Allergen spezifischen B Zellen: IgE Antikörper werden hergestellt. IgE kann an seinen hochaffinen Rezeptor (FcRI) binden, der auf der Oberfläche der Mastzellen, Basophilen und dendritischen Zellen zu finden ist. IgE kann auch an seinen weniger affinen Rezeptor (FcRII) binden, der sich auf B Zellen befindet. Zur autokrinen kostimulierenden Wirkung aktiviert dieser Rezeptor die weitere Produktion der IgE. Das „Der p1“ wirkt auf die Epithelzellen, wodurch es eine direkte Rolle bei der Aktivierung der B und T Zellen spielt. Durch seine proteolytische Aktivität spaltet es CD23 auf den B Zellen und CD25 auf den T Zellen. Einige Allergene – wie „Der p1“ – haben die Fähigkeit die IgE unabhängige IL-4, IL-13 Herstellung in Mastzellen oder in basophilen Granulozyten auszulösen. 2. Die Sofortreaktion Im Körper befinden sich allergenspezifische IgE B – Gedächtniszellen und T- Gedächtniszellen (nötig für die Kostimulierung). Diese werden aktiviert, wenn das Allergen wiedermal im Körper vorkommt und ihre Aktivierung führt zur Herstellung von IgE in großen Mengen. Die Sofortreaktion selbst wird durch die Kreuzbindung der Antigene, die an die schon auf den Mastzellen vorhandenen IgE binden, ausgelöst. Als Folge der Degranulation werden die vorgeformten (Histamin und Leukotrien) Mediatoren und die neu synthetisierten (Cytokine und Chemokine) Mediatoren aus den Mastzellen freigesetzt. Diese Mediatoren wirken vor Ort auf das herumliegenden Gewebe. Die wichtigsten Wirkungen der Mediatoren: Steigerung der Gefäßpermeabilität Kontraktion der glatten Muskulatur der Bronchien Steigerung der Schleimsekretion der Atemwege Anziehung der inflammatorischen Zellen zum Ort der Allergenexposition 3. Die späte Reaktion Die späte Reaktion entwickelt sich einige (2-24) Stunden nach dem Eintritt des Allergens. Einerseits bedeutet die Mastzellantwort die Aktivierung des Lipoxigenase, Cyklooxigenase Wegs und die Veränderung der Genexpression. Diese führen zur Freisetzung und Herstellung von Lipidmediatoren und neuen Th2 Cytokinen, die für die Aufrechterhaltung des Prozesses verantwortlich sind. 182 Andererseits kann das aus den allergenspezifischen Th Zellen und Mastzellen freisetzende IL-5 Cytokin die Eosinophilen aktivieren und eine Eosinophilie verursachen. Die eosinophilen Granulozyten stellen weitere proinflammatorische Mediatoren (Leukotriene, kationische Proteine, eosinophile Peroxidase, eosinophile Neurotoxine) und weitere IL-13 und IL-5 her. Abbildung 11.2. Die Effektorphase der allergischen Reaktion 11.3. Die Symptome der Allergie 11.3.1. Die Type der Allergene Der Typ des Allergen und sein Eintritt in den Körper bestimmen den Ablauf der allergischen Antwort. Die unterliegende Tabelle ist eine Zusammenfassung. HÄUFIGE DER ORT DES ALLERGENE EINTRITTES Kontaktallergie Kontaktallergene: Haut (Urticaria) Insektenstich, Latex Nahrungsmittelallergi Erdnuss, Mund e Fische, Muschel, Milk, Ei SYNDROM Allergische Rhinitis - Pollen, Heuschnupfen Kot der Milben Schleimhaut: Nase, Augen, Bronchien Allergisches Asthma Tierhaar, Pollen, Einatmung Kot der Milben 183 ANTWORT Steigerung des Blutstroms, der Durchlässigkeit der Gefäße Brechdurchfall Diarrhö, Juckreiz Urtikaria selten Anaphylaxie Ödem und Juckreiz der Schleimhaut der Nase Schwellung, Reizung und Schleimbildung in den Nasenwegen Allergische Konjunktivitis Verengung der Atemwege, gesteigerte Schleimsekretion, Entzündung der Atemwege Husten, Dyspnea Systemische Anaphylaxie Medikamente, Erdnuss, Tiergift Intravenös, oder direkt Systemische Steigerung des durch die Schleimhaut Blutstroms, der Durchlässigkeit des Mundes der Gefäße, Trachealer Verschluss, Kreislaufkollaps, Koma, Tod Tabelle 11.2. Die Type der Allergene 11.3.2. Wie entstehen die allergischen Symptome? Die Wirkung der Degranulation der Mastzellen hängt von der Gewebe-Umgebung ab: Im Gastrointestinaltrakt verursachen die erhöhte Sekretion von Verdauungsenzymen und die Peristaltika Brechdurchfall, Diarrhö, Krämpfe. In den Atemwegen verursachen die Einschnürung und die erhöhte Schleimsekretion das Husten, die pfeifende Atmung, beengende Schmerzen in der Brust und keuchende Atmung. Der gesteigerte Blutstrom in den Gefäßen und die erhöhte Durchlässigkeit verursachen Ödeme und Entzündung und der Antigentransport in die Lymphknoten wird wirksamer. Urticaria und Angiödeme sind gute Beispiele für die Veränderungen in den Geweben Beide entstehen auf die gleiche Art und Weise: rote juckende Flecken erscheinen temporär. Ihre Ränder sind ohne scharfe Grenzen und heben sich aus ihrer Umgebung ab. Das Urticaria ist das Ödem der Dermis, während ein Angiödem das Ödem der Subkutangewebe ist. Abbildung 11.3. Entstehung der Urticaria. Als Folge der ins Blut gelangenen Allergene entwickelt sich eine Anaphylaxie, die zwei oder mehrere Organsysteme betrifft. Die Permeabilität der Kapillärien wird erhöht, ein Ödém entsteht, die Gewebe (z.B die Zunge) schwellen, der Blutdruck sinkt und die Sauerstoffversorgung der Gewebe nimmt ab, bis es zum anaphylaxischer Schock mit Bewusstlossigkeit kommt. Als Folge der Kontraktion der glatten 184 Muskulatur in den Bronchien tritt schwere und keuchende Atmung auf. Wegen der Kontraktion des Darms können diese Symptome Durchfall und Diarrhö verursachen. 11.3.2.1. Die aus den Mastzellen freigesetzenden Stoffe Enzyme z.B. Tryptasen, Kinasen: das Bindegewebe wird reorganisiert wegen des Abbaus der Bronchus-erweiternden Peptide entsteht ein Bronchospasmus. Toxische Mediatoren z.B. Histamin: die Permeabilität der Kapillaren wird erhöht, steigt die glatte Muskelkontraktion, verursacht Juckreiz in der Haut z.B Heparin: hemmt die Blutgerinnung. Cytokine z.B IL-4, IL-13: stimulieren Th2 z.B. IL-5: stimuliert die eosinophilen Granulozyten z.B. TNF alfa: wirkend auf das Endothelium und viele Immunzellen, es löst Entzündungen aus. Chemokine z.B. CCL3: zieht Monozyte, Makrophage und Neutrophyle an. Lipidmediatoren z.B. Leukotriene: steigern die glatte Muskelkontraktion, die Permeabilität der Kapillaren, die Schleimsekretion z.B. PAF (platelet activating factor): zieht Leukozyte an, aktiviert die Neutrophilen, die Eosinophilen und die Blutplättchen. 11.3.2.2. Rezeptoren Die während der allergischen Reaktionen freigesetzten Stoffe werden von ihren Rezeptoren wahrgenommen. Die Wirkung wird durch die Rezeptoren ausgelöst und oft verstärken sie den Effekt zusätzlich untereinander. Histamin Rezeptoren sind in der Haut, im gastrointestinalen Trakt, im Herz, im Kapillarsystem, in den glatten Muskeln der Lunge, auf den Zellen des Knochenmarks, auf den Zellen der Milz, auf den peripheren Sinnesneuronen zu finden. Bei den allergischen Reaktionen spielen drei Typen von Histamin Rezeptoren eine Rolle, deren wichtigste Wirkungen die folgenden sind: - Histamin durch H1 Rezeptor verursacht: Steigerung der Permeabilität der Kapillaren, Vasodilatation, Tachykardie, die Kontraktion der Bronchien und des Verdauungssystems. - Histamin durch H2 Rezeptor verursacht: Steigerung der Permeabilität der Kapillaren, stärkere Sekretion der Magensäure, stärkere Schleimsekretion des Atemsystems. - Histamin durch H4 Rezeptoren: Juckreiz der Haut, die Rekrutierung der Eosinophilen und der Mastzellen. (Die H3 Rezeptoren sind im zentralen Nervensystem lokalisiert, sie spielen keine Rolle bei den allergischen Reaktionen.) 185 Die Lipidmediatoren (Prostaglandin und Leukotrien) wirken auf ihre Rezeptoren, die sich in der Haut, in der glatten Muskulatur der Lungen und im Kapillarsystem befinden. Die schwerste Folge dieser Wirkung ist die Kontraktion der glatten Muskulatur der Lungen. Weiterhin sind die Lipidmediatoren – neben Histamin – an der Entstehung der Urticaria beteiligt. 11.3.3. Allergische Kreuzreaktion Das Immunsystem gibt eine gleiche Antwort auf verschiedene, aber nach ihrer molekularen Struktur ähnliche Antigene. Diese Antwort ist nämlich eine allergische Reaktion. Die häufigsten Kreuzreaktionen entwickeln sich zwischen Latex und Obst, beziehungsweise zwischen Pollen und Obst. Einige Beispiele: Pollen der Buche – Apfel, Birne, Pfirsich, Nuss, Walnuss, Erdnuss, Sellerie Pollen der Gräser – Orange, Wassermelone, Tomate, Erdnuss Latex – Banane, Avokado, Kiwi, Maroni, Papaya, Feige. 70% der Patienten mit Nahrungsmittelallergien weisen ein orales allergisches Syndrom wegen einer Kreuzreaktion auf. Abbildung 11.4. Beispiel auf die Kreuzreaktion zwischen Nahrungsmittelallergenen und anderen Allergenen. In primären Nahrungsmittelallergien ist das sensitisierende (sensibilisierende) und das die allergische Reaktion auslösende Allergen gleich. Das Allergen gelangt durch den Mund in den Körper, es sensitisiert als Nahrungsmittelallergen und später, bei einer weiteren Exposition löst es eine orale / systemische anaphylaktische Reaktion aus. In sekundären Nahrungsmittelallergien ist das sensitisierende und das die allergische Reaktion auslösende Allergen unterschiedlich. Das sensitisierende Allergen ist kein Nahrungsmittel, es gelangt 186 in den Körper durch Inhalation oder durch die Haut. Gleichzeitig löst ein strukturell ähnliches, aber nicht-sensitisierendes Nahrungsmittelallergen durch den Mund mit einer Kreuzreaktion die allergische Reaktion aus. Oft ist Latex solch ein Allergen, welches eine sekundäre Nahrungsmittelallergie auslöst. Latex ist in dem natürlichen Gummi vorhanden, ist eine Komponente der Arzthandschuhe und findet sich oft im Arbeitsumfeld von Personen die im Gesundheitswesen tätig sind. Das Allergen des Latex ist eine Kitin-bindende hevein Domäne, anwesend im Kitinase Enzym der Avokados z.B. Solche, evolutionär konservative, Allergie-verursachende Moleküle heißen Panallergene. 11.3.4. Nahrungsmittelintoleranz und Nahrungsmittelallergie Nahrungsmittel lösen oft allergische Reaktionen aus, wie auch die Nahrungsmittelintoleranz, welche ähnliche Symptome aufweisen kann. Jedoch ist Nahrungsmittelintoleranz nicht gleichzusetzen mit Nahrungsmittelallergie. 1. Überempfindlichkeit; wenn das Immunsystem eine Rolle bei der Reaktion spielt: - Die echten Nahrungsmittel- Überempfindlichkeitsreaktionen sind meistens mit IgE-vermittelter Mastzelldegranulation gekoppelt. Die Nahrungsmittelallergie Typ I. kann primär oder sekundär sein. (schon früher diskutiert) - Jedoch existiert auch eine nicht IgE, sondern T-Zell-vermittelte Nahrungsmittel- Überempfindlichkeitsreaktion (glutensensitive Enteropathie / Zöliakie). (Siehe später im Kapitel über die Typ IV. Überempfindlichkeitsreaktionen!) 2. Intoleranz; wenn Immunsystem keine Rolle bei der Reaktion spielt: - Die nicht Immunsystem-vermittelte Nahrungsmittel- Überempfindlichkeit kann eine sogenannte Pseudoallergie sein. Diese Pseudoallergie löst eine von Mastzelldegranulation verursachte anaphylaktoide Reaktion aus. - Nahrungsmittelintoleranz kann auch ohne Mastzelldegranulation entstehen. Ein Beispiel ist dafür die Milchempfindlichkeit. Während die Milchallergie im Säuglingsalter entsteht und die Ursache eine abnormale, von dem Protein (Kasein) der Krächzenmilch ausgelöste Reaktion ist, ist die Laktose die Ursache der Milchempfindlichkeit, deren Abbau entlang des Alters langsamer wird oder vollständig verloren geht. Im Hintergrund steht ein Polymorphismus in der regulatorischen Region des Gens des Enzyms Laktase. 11.3.5. Anaphyilaktoide Reaktion / Pseudoallergie In Pseudoallergien ist das Immunsystem in der Behandlung der Symptome eigentlich nicht beteiligt. Der die Reaktion auslösende Stoff kann durch IgE-unabhängige Wege Mastzell – und Basophilendegranulation oder Membranzerstörung initieren, infolgedessen Histamin freigesetzt wird. Daher wird die Reaktion von dem Antigen bei dem ersten Treffen ausgelöst. Die unterliegende Tabelle ist eine Zusammenfassung der häufigsten Stoffe, die anaphylaktische oder anaphylaktoide Reaktion auslösen. 187 Stoffe für Anaphylaktoide direkte Mastzelldegranulation/ Pseudoallergie Anaphylaxische/ allergische/ IgEvermittelte Stoffe Nahrungsmittel (Nuss, Meeresfrüchte) Arzneimittel (z.B. NSAID, Aspirin) Arzneimittel (z.B. beta-laktam Antibiotika) CT/MR Kontrastmittel Insektenstich (Bienen, Wespe, Ameise) Ernährung mit hohem Histamingehalt (Rotwein) Latex Physikalische Faktoren (Bewegung, Warm, Kalt) Bakterieller Abbau des Histidins (Fischvergiftung – schlecht gelagerter Fisch mit hohem Histidingehalt) Tabelle 11.3. Der häufigsten Stoffe die Anaphylaxische oder anaphylaktoide Reaktion auslösen 11.3.5.1. Arzneimittel - ausgelöste Pseudoallergie Die CT und MRI Untersuchungen stellen ein hohes Risiko im klinischen Praktikum dar. Das intravenös eingegebene Kontrastmittel verursacht bei 10% der Patienten eine anaphylaktoide Reaktion. Der Mechanismus ist noch nicht bekannt. Es wird vermutet, dass die ionische Zusammensetzung, die Osmotoxizität und Chemotoxizität die Stabilität der Zellmembranen – auch der Mastzellmembran – beeinflussen. Selten können auch nicht-steroidale entzündungshemmende Medikamente (z.B. Ibuprofen) und Acetylsalicylsäuren (z.B. Aspirin) eine anaphylaktoide Reaktion verursachen. Nach Vermutungen steht eine Enzymhemmung im molekulären Hintergrund: diese Medikamente hemmen die Prostaglandinsynthese durch Hemmung des COX-1 Enzyms und verschieben den ArachidonsäureMetabolismus in Richtung der Lipoxigenase-vermittelten Leukotriensynthese. Demzufolge vermindert sich die Menge des antiinflammatorischen Prostaglandins (PGE2) und die proinflammatorische Leukotriensynthese (LT-A4, -B4,-C4, -D4) steigt. Synthetische Nahrungsmittelfarbstoffe, die solchen Medikamenten chemisch ähnlich sind (z.B. Tartrazin/E102), können die gleiche Wirkung hervorrufen. 11.3.5.2. Nahrungsmittel - ausgelöste Pseudoallergie Pseudoallergien können durch Nahrungsmittel mir hohem Histamingehalt ausgelöst werden. Unter normalen Umständen wird Histamin im Körper über Aminooxidasen abgebaut. Die Diaminooxidase (DAO) spielt die größte Rolle bei dem Abbau des in der Ernährung enthaltenden Histamins. DAO wird großteils im Darm exprimiert. Das Enzym ist fähig Histamin in der extrazellulären Matrix zu „neutralisieren“ indem es die transepitheliale Absorption des Histamins hemmt. Bei den Personen, deren DAO-Aktivität niedriger ist oder die kein DAO Enzym exprimieren, gelangt Histamin zusammen mit der Ernährung in den Kreislauf, wo es die gleiche Wirkung ausübt, wie das endogen freigesetzte Histamin. Die verminderte Aktivität oder die ungenügende Menge der DAO kann von einem angeborenem Enzymmangel herrühren, oder es kann das Ergebnis einer gastrointestinalen Krankheit (z.B. Morbus Crohn) sein. In der Crohn Krankheit stellen die Enterozyten wenig DAO her. DAO blockierende Medikamente (z.B. cefalosporin Antibiotika) oder Ernährungen (z.B. Rotwein selbst) können selten für die Histamin-abbauende Fähigkeit verantwortlich gemacht werden. Die Histaminempfindlichkeit, verursacht durch einen genetisch bedingten funktionellen DAO Mangel, ist eigentlich eine metabolische Krankheit. Es wurde nachgewiesen, dass bestimmte SNP (single 188 nucleotide polymorphism) im Gen der DAO mit der Ausprägung einiger gastrointestinalen Krankheiten korrelieren. (Das in den Kreislauf gelangte und von den Zellen aufgenommene Histamin wird über ein anderes intrazelluläres Enzym, die Histamin-N-Metiltransferase (HNMT) abgebaut. HNMT ist hauptsächlich in der Leber zu finden) Einige Nahrungsmittel erreichen solch einen hohen Histamingehalt, dass es trotz normaler DAO Aktivität in gesunden Personen zu Vergiftungen kommt. Das häufigste Beispiel dafür ist die Fischvergiftung. Eine Fischvergiftung soll man nicht mit der echten Allergie gegen Fischproteine und Meeresfrüchte verwechseln, obwohl die klinischen Symptome in beiden Fällen ähnlich sind. Im Falle einer Fischvergiftung wird Histidin in Fischen mit hohen Histidingehalt über vermehrte Bakterien dekarboxiliert, wodurch Histamin entsteht. (Das schon entstandene Histamin kann später nicht mehr entfernt werden, weder mit kochen noch mit einfrieren oder mit Räuchern.) Einige Nahrungsmittel mit wenig Histamin (z.B. Zitruspflanzen, Erdbeere) können auch eine anaphylaktoide Reaktion auslösen. Die Membran der Mastzellen wird irgendwie im Duodenum destabilisiert, was die gleichzeitige Degranulation der Mastzellen verursacht. Der Mechanismus ist noch nicht bekannt. Es ist sehr wichtig zu betonen, dass die Patienten die eine Histaminempfindlichkeit aufweisen, mit keinen CT und MRI Kontrastmittel behandelt werden dürfen. Im Falle mangelnder Alternativen, dürfen diese Mittel nur nach geeigneter Antihistamin Behandlung verwendet werden. 11.3.5.3. Physikalische Faktoren – und psychologische Stress-vermittelte Pseudoallergie Physikalische Faktoren – Bewegung, Wärme, Kälte – und Stress können Pseudoallergien verursachen. In allen Fällen steht eine Mastzelldegranulation im Hinntergrund, obwohl der genaue Mechanismus noch nicht bekannt ist. Die Symptome werden niemals so schwerwiegend wie im Fall der durch Arzneimittel oder Nahrungsmittel ausgelösten Reaktionen sein, in denen die Faktoren durch die Schleimhaut in den Körper eintreten. In solchen Fällen kommt es meistens nur zu Urticaria. Die so genannte cholinergische Urticaria wird durch Bewegung ausgelöst und es steht in Beziehung mit der Thermoregulation des Körpers. Die Bezeichnung beruht auf der Erfahrung, dass eine Acethylcholin-Injektion bei Betroffenen die Urtikaria auch auslösen kann. Zur gleichen Zeit ist man sich nicht bewusst, welche Rolle Acethylcholin bei der Entstehung der Symptome spielt. Kälteallergie kann durch jeglichen Kontakt mit einem kalten physikalischen Medium ausgelöst werden: kaltes Wasser, Klimatisierung, kalte Nahrungsmittel / Getränke, Schwimmen im kalten Wasser und kalter Schweiß. Wiedermal ist es unklar, warum die Membran der Mastzellen sich destabilisiert. Wärmeallergie wird durch Kontakt mit einem Medium über 43C ausgelöst, der Mechanismus ist auch nicht verstanden. Die wegen psychologischem Stress verursachte Pseudoallergie ist sehr selten. 11.3.6. Sonnenallergie 189 Obwohl eine Sonnenallergie von physikalischen Faktoren ausgelöst wird, tritt eine IgE-vermittelte Mastzelldegarnulation auf. Die so genannten Fotoallergene gehören zum breiten Spektrum (290-800 nm). Die Type der Sonnenallergie werden nach den Wellenlängen unterschieden, abhängig davon, welche Wellenlänge die Reaktion hervorruft. 11.4. Die Diagnose Der Allergie Die diagnostischen Tests weisen die durch die Mastzelldegranulation freigesetzten Stoffe und die für das Allergen spezifischen IgE Antikörper nach. Die IgE unabhängige Pseudoallergie kann durch das Messen des Histaminspiegels und das gleichzeitige Messen des DAO Spiegels klassifiziert werden. 1. Allergie Tests Die am häufigsten in der heutigen klinischen Praxis verwendeten Allergie – Tests: Das Messen des Triptase- Spiegels im Serum Das Messen des Histamin Spiegels im Urin (nicht im Serum, weil die Halbwertszeit des Histamins im Serum viel kürzer ist als im Urin.) 2. Hautprobe/ Prick Test: das Antigen wird intradermal eingebracht, danach wird der Grad der Mastzelldegranulation in vivo in der Haut der sensibilisierten Person gemessen. Früher wurde das Antigen mit einer Injektionsnadel eingetragen, heute existieren auch andere Formen. Z.B. der so genannte Morrow-Brown Nadel, usw. http://www.lincolndiagnostics.com/ Neben den Antigenen werden PBS als negativ Kontrolle und Lösungen mit bekannten Histaminkonzentrationen als positive Kontrolle verwendet. Ein gewöhnlicher Prick Test enthält 20 verschiedene allgemeine aufgelöste Allergene (regionale Pollen, Katzen- und Hundehaare, Milbe Mischung, Schimmelpilz, Milch, Ei, Feder). Außerdem existieren es auch spezifische Tests. (http://www.frank-diagn.hu/main.php?mi=allergen&kod=LOFA ) 15 Minuten nach der Auftragung wird die Reaktion abgelesen, während dieser Zeit führt die Mastzelldegranulation zur Entstehung einer lokalen Schwellung. Der Test ist positiv, wenn der Durchmesser der Schwellung größer als 3 mm ist. Die Tabelle zeigt die Beziehung zwischen dem Durchmesser und seiner Darstellung. +++ = gleicher Durchmesser wie bei der positiven Kontrolle ++++ = größerer Durchmesser als bei der positiven Kontrolle ++ = Durchmesser ist 2/3 der positiven Kontrolle + = Durchmesser ist 1/3 der positiven Kontrolle Es existieren einige Allergien, die nur mit dieser Methode nachgewiesen werden können. (z.B. Sonnenallergie). Außerdem geben die Reaktion die IgE unabhängige anaphylaktoide Stoffe auch. Die Anwesenheit der allergenspezifischen IgE Antikörper kann mit mehreren Methoden aus dem Serum des Patienten nachgewiesen werden. 3. RAST (RadioAllergoSorbent Test): das Messen der allergenspezifischen IgE in vitro 190 Die Blutprobe des Patienten wird mit unterschiedlichen Allergenen in Reaktion gebracht: die Allergene sind dabei zu einem speziellen Teststäbchen gebunden. - Der Stab mit Allergenen wird ins Serum des Patienten eingetaucht. Falls vorhanden, binden die Allergene an die spezifischen IgE Moleküle während der Inkubation. - Die nichtspezifischen Antikörper werden ausgewaschen. - Der Stab wird in einer Lösung inkubiert, die radioaktiv markierte anti-IgE sekundäre Antikörper enthält. - Die nicht spezifisch bindenden Antikörper werden ausgewaschen. - Die Radioaktivität des Teststäbchen wird mit einem Geigerzähler gemessen, um die radioaktive Strahlung verschiedener Allergen-IgE Komplexe bestimmen zu können. Die quantitative Menge von allergenspezifischen IgE-Antikörpern im Blut korreliert mit der Radioaktivität (RAST 1-6, der kleinere Wert entspricht einer schwächeren Reaktion. So bezeichnet z.B Stufe 6 eine starke Allergie.) Der Vorteil dieser Methode im Vergleich mit dem Prick Test ist, dass sie nicht unangenehm für den Patienten ist und dass gleichzeitig die Empfindlichkeit gegen viele Antigene untersucht werden kann. Die Arzneimittelallergien werden am häufigsten mit dieser Methode nachgewiesen. 4. Laterale Immunkromatographie: das Messen der allergenspezifischen IgE in vitro Die Methode wurde schon im Kapitel 2. beschrieben. In diesem Fall ist das Antigen das lösliche Detektormolekül, das an das spezifischen IgE des Patienten bindet, wodurch sie als Komplex wandern. Ein zweiter, an eine Membran gebundener IgE spezifischer Antikörper, fängt den Komplex ein wodurch es zu einer Farbreaktion (in Form eines Striches) an der Stelle der Membran kommt. 5. Der allergenspezifische sandwich immunoassay ist die modifizierte Form des RAST Tests. Die Methode wurde schon im Kapitel 2. beschrieben. In diesem Fall sind die zu untersuchenden Allergene als Antigene auf eine solide Oberfläche gebunden, an die die im Serum des Patienten vorkommendenen IgE Antikörper binden. Diese werden mit der schon bekannten Methode mit Sekundärantikörpern detektiert. Der Sekundärantikörper ist Enzym-gekoppelt und gibt eine Farbreaktion. 6. Allergen microarray: Rekombinante Allergene können durch molekulare Klonierung hergestellt werden. Sie enthalten die für ein Antigen jeweils Epitope. Diese können dann in multi-allergenen Testsystemen verwendet werden, wie das „allergen microarray“. Die solubilisierten Allergenepitope werden an ein Glas gebunden, werden mit den Serum IgE des Patienten inkubiert, und werden schließlich mit fluorescensmarkierten anti-IgE detektiert. Nach dem Ablesen der Fluorescensintensität, werden die Angaben mit einer Software ausgewertet. Der Vorteil ist, dass das „Allergenprofil“ des Patienten mit einer einzigen Untersuchung bestimmt werden kann, und auch die Stärke der Reaktion nachgewiesen werden kann, d.h welche Epitope die stärkste Antwort auslösen. 191 Abbildung11.5. Allergenprofil mit Microarray 7. Immuno RCA (rolling circle DNA amplification): Die IgE im Serum des Patienten binden an die solide Oberfläche gebundenen Antigene. Die IgE werden mit einem Oligonukleotidstrang-konjugierten Antikörper erkannt. Eine komplementäre, zirkuläre DNA wird dem Oligonukleotidstrang zugegeben und mit DNA Polymerase und Nukleotiden läuft eine DNA –Amplikikation ab, eine PCR Reaktion eventuell. Als Ergebnis entsteht eine Wiederholung von mehreren hundert kontinuierlichen Strängen, denen am Ende der Reaktion fluorescensmarkierte komplementäre Proben zugegeben werden. Diese werden detektiert. 11.5. Die Therapie der Allergie Der Patient kann oligo- oder polisensitisiert werden, d.h. dass er gegen einzelne / einige oder gegen viele verschiedene Allergene empfindlich ist. Bei dem oligosensitisierten Patienten kann die Vermeidung des Allergens, die allergenspezifische oder die Antikörper-Immuntherapie erfolgreich sein. Bei dem polisensitisierten Patienten ist die Medikamentöse Therapie gezielt, um die Symptome abzuschaffen. 11.5.1. Medikamentöse Behandlung Die Medikamentöse Behandlung hemmt die Mastzelldegranulation, die Wirkung der schon freigesetzten Stoffe und dreht die schon entwickelten Symptome zurück. Abhängig davon, wie schwerwiegend die Symptome sind, sollen die Patienten mit unterschiedlichen Medikamenten nach dem geeigneten Protokoll behandelt werden. Adrenalin: der einzige antagonistische Wirkstoff, der die dem Histamin entgegengesetzten Prozesse auslöst und die Symptome dadurch abschafft. Bei einer anaphylaktischen Reaktion ist Adrenalin das erste zu verabreichende Medikament. - Als alfa-Rezeptor Agonist kontrahiert es die Gefäße, vermindert das Ödem, hemmt den Juckreiz. 192 - Durch Beta-Rezeptoren entspannt es die glatte Muskulatur, erweitert es die Atemwege, beendet die gastrointestinalen Koliken, steigert die Kraft der Herzmuskelkontraktion, das Herz schlägt schneller, erhöht sich der Blutdruck, vermindert sich die Histamin-Permeabilität der Zellmembran, wird die Freisetzung des Histamins und des Leukotriens gehemmt, wird die inflammatorische Antwort vermindert. Da Adrenalin nicht die Wirkung des sich schon im Körper befindlichen und an Zellen gebundenen Histamins hemmen kann, muss Adrenalin solange verabreicht werden, bis die Wirkung des Histamins nachlässt. Bei sehr niedrigem Blutdruck oder falls der Zustand des Kranken sich verschlimmert, muss Adrenalin intravenös jede 10-15 Minuten injektiert werden. Antihistaminika: pharmakologische Antagonisten, die an Histaminrezeptoren binden und so die Bindung von Histamin an seine Rezeptoren hemmen, wodurch die durch Histamin ausgelösten Prozesse gehemmt werden. Antihistaminika sind Arzneimittel, die die Wirkung von Histamin aufheben. Die schon entwickelten Histamin-vermittelten Reaktionen laufen weiter ab da sie durch Antihistaminika nicht beendet werden. Deswegen sollen Antihistaminika (Histamin-Antagoniste) bei den anaphylaktischen Reaktionen nach Adrenalin gegeben werden. Glukokortikoide: allgemeine immunsuppressive und entzündungshemmende Stoffe (unabhängig von der Ursache der Entzündung.) Sie hemmen die Synthese der inflammatorischen Cytokine: von IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8 , IFN-γ. Sie hemmen die Chemotaxis, die Adhesion, die Wanderung aus den Gefäßen in die Gewebe, die Phagozytose der Leukozyten und der Entzündungszellen. Der molekulare Hintergrund ihrer Wirkung: 1. Hemmung der Gene der inflammatorischen Cytokine 2. Induktion der Expression und Sekretion des Annexin-1 (Lipokortin-1) in den Zellen des natürlichen Immunsystems. Annexin wirkt auf autokrine und parakrine Weise. Es hemmt die Synthese der Eikosanoide durch die Hemmung der Phospholipase -A2 (PLA2), d.h. dass die Prostaglandin und Leukotriensynthese aufgehoben wird. Kalcium und Natrium-Kromoglykate: sie stabilisieren die Membran der Mastzellen und hemmen dadurch die Degranulation. Leukotrien-Antagonisten: sie sind Lipoxigenase- Leukotriensynthese vermindert wird. 193 hemmende Stoffe, wodurch die Abbildung 11.6. Die Therapie der Allergie mit Arzneimitteln. (Die Wirkungen des Adrenalins sind nicht hier dargestellt, sieh den Text!.) 11.5.2. Immuntherapie: (Hyposensitisierung/Desensitisierung) allergenspezifische Immuntherapie: sie ist indiziert, wenn der Patient gegen nur einige Antigene empfindlich ist. Bei dieser Therapie werden dem Patienten verdünnte Antigen(suspensionen) subkutan injektiert. Über 12 Wochen wird bei jeder nachfolgenden Impfung die Allergenkonzentration gestiegert. Danach wird die höchste Allergendosis als aufrechterhaltende Behandlung jeden Monat über Jahre (manchmal 3Jahre) dem Patienten gegeben. Die Therapie soll immer im Krankenhaus durchgeführt werden, wegen der Gefahr der Entstehung einer Anaphylaxie. Während der Therapie verändert sich die zelluläre und die humorale Immunantwort und Toleranz entwickelt sich. Es werden verdünnte Allergenmengen injektiert und keine hohen Konzentrationen welche eine stärke/stärkere Reaktion hervorrufen. Dendritische Zellen, die nun auf Allergenepitope treffen, reifen auf Grund des mangelnden Entzündungssignals nur unvollständig. Durch die Steigerung der Dose verändert sich das Verhältnis der T Zellen. Die kostimulierenden Moleküle der unvollständig reifen dendritischen Zellen treten in eine tolerogene Wechselwirkung mit den T Zellen der Lymphknoten. Infolgedessen steigt die Anzahl der funktionellen Treg Zellen (Tr1) – die in der Peripherie entstehen und IL-10 und TGF-beta sekretieren. (Die Anzahl der Th2 vermindert). Außerdem, auf die steigende Antigendose steigt die IL-10 Sekretion der Monozyten, der Makrophagen und der B Zellen. Die vorstehenden Cytokine hemmen die Effektor-T-Zellen, die Entzündungsreaktionen, aber verschieben den Immunglobulin-Klasswechsel in Richtung der IgA und IgG. Diese Ig Antikörper kompetieren um die Allergenbindung, wodurch die Entstehung der 194 IgE.vermittelten Sofortreaktion vermindert wird, also die sofort Histaminreaktion und die spätere eosinophilreaktion. Es gibt klinische Versuche um eine sublinguale Therapie zu entwickeln in der das Allergen oral eingenommen wird. Die Methode ist nachgewiesenermaßen sicherer als die subkutane Methode, da sie keine anaphylaktische Reaktion auslöst. Aber die Wirksamkeit ist kleiner in Erwachsenen als im Fall der subkutanen Methode. In Milchallergie bei Säuglingen ist die Methode erfolgreich. In der Mehrheit der Kinder „wächst“ die Nahrungsmittelallergien bis zum Alter von 7 Jahren aus, da die kontinuierliche Anwesenheit des Allergens in kleinen Dosen die Entstehung der natürlichen Toleranz vorantreibt. Das Immunsystem der Säuglinge ist unreif, deswegen hilft die zugegebene Milch in steigenden Konzentrationen der Entstehung der natürlichen Immunität, gleich mit der Entstehung der Immuntoleranz durch subkatane Methode. Abbildung 11.7. Allergenspezifische Desensitizierung. Prophylaktische Vakzination: Die Immuntherapie der Zukunft. In den Säuglingen kann IgG spezifische Antikörpersynthese mit hypoallergenen Allergenepitopen hervorgerufen werden, wodurch ist es möglich ist, die Sensitizierung und die IgE spezifische Reaktion vorzubeugen. Auf diese Weise bleibt der IgE Spiegel auch in der Anwesenheit des natürlichen Antigens immer niedrig. Antikörper Therapie: Behandlung mit monoklonalen IgE-spezifischen Antikörper: Der Antikörper bindet an die zirkulierenden IgE Moleküle oder an die in Allergien vorkommenden Cytokine, und die Entstehung der allergischen Reaktion wird dadurch gehemmt. Anti IgE wird nur bei den schwierigsten, auf Kortikosteron nicht reagierenden allergischen Asthmatikern angewendet. Die anti-Cytokin Therapie ist momentan in Untersuchungsstadien. Übersetzt von Erna Pap 195 12. HYPERSENSITIVITY REACTIONS II: TYPE II-IV HYPERSENSITIVITY REACTIONS (MARIANNA CSILLA HOLUB) Die Überempfindlichkeitsreaktionen vom Typ II und III beruhen ebenfalls auf dem Antikörper-Antigen Bindungsprinzip. Die Immunkomplexe, die von Antikörpern und Antigenen gebildet werden, entstehen jedoch auf unterschiedlicher Weise während der beiden Prozesse. Bei einer Typ II Reaktion binden die Antikörper an Antigene, die auf der Oberfläche der Zelle (oder des Gewebes) fixiert/gekoppelt sind. In einer Typ III Reaktion ist das Antigen löslich und die Immunkomplexe zirkulieren unabhängig von den Zellen, in freiem Zustand im Kreislauf. 12.1. Überempfindlichkeitsreaktion vom Typ II Die Immunkomplexe werden von Zelloberflächen- oder Gewebsantigenen bzw. die auf ihnen spezifisch gebunden IgG Antikörpern gebildet. Die Antigene können in extrinsische und intrinsische unterteilt werden. Die in den Körper von außen eintretenden, fremden, aber an eigenen Zellen bindenden Antigene werden als extrinsisch klassifiziert. Andere sind die intrinsischen Antigene, die auf der Oberfläche der Zellen erscheinen. Die Bindung von Antikörpern leitet unterschiedliche Mechanismen ein: Die Antigen-exprimierenden, mit Antikörpern bedeckten Zellen werden empfindlich gegenüber der Komplement-vermittelten Lyse, gegen Antikörper-vermittelter zellulärer Zytotoxizität (ADCC=antibody-dependent cellular cytotoxicity) und/oder gegenüber der Opsonisierung mit Antikörpern folgenden Phagozytose. Diese Effektormechanismen führen zum Untergang der Zellen. Die auf den Zellen exprimierten Antigene, die mit Antikörpern bedeckt sind, lösen eine starke Phagozytose aus, indem lytische Enzyme von den Phagozyten freigesetzt werden. Infolge dessen wird nicht nur die gegebene Zelle geschädigt, sondern auch die anderen in ihrer Umgebung. Es kommt also zu einer Gewebsschädigung. In bestimmten Fällen kann der Antikörper die Funktion der antigen-exprimierenden Zellen hemmen oder stimulieren (zB. bei Myasthenia gravis [MG] verhindern die den Acetylcholinrezeptor blockierenden Autoantikörper die Bindung von Acetylcholin an seinem Rezeptor). Dabei kann auch eine Gewebsschädigung entstehen. (bei MG führt die Zerstörung der postsynaptischen Endplatte zum Verlust Depolarisierungsversagen kommt. 196 von AchR-en, wodurch es zu einem 12.1.1. Beispiele für eine Typ II Reaktion, die von extrinsischen Antigenen ausgelöst werden 12.1.1.1. Erythroblastosis fetalis (Morbus haemolyticus neonatorum, Immunhämolytische Krankheit bei Feten und Neugeborenen) Pathologischer Zustand bei Neugeborenen, für den die Auflösung von Erythrozyten (Hämolyse) verantwortlich ist. Obwohl normalerweise keine fötalen Zellen in den mütterlichen Kreislauf während der Schwangerschaft übertreten sollen, kommt es während der Geburt meistens zur Vermischung von kindlichem und mütterlichem Blut. Dabei können jene Erythrozyten in den mütterlichen Kreislauf eintreten, auf denen vom Vater erworbene Antigene liegen, weswegen sie von der Mutter als körperfremd erkannt werden. Gegen sie werden von dem Körper der Mutter Antikörper hergestellt, die durch die Plazenta in den fötalen Kreislauf übertreten. Die Antikörper binden dann an die Rh-positiven Erythrozyten des Kindes, die dadurch zur Hämolyse kommen. Bei der ersten Schwangerschaft haben die produzierten Antikörper keine Folgen, sie können nur die Entwicklung weiterer Kinder beeinflussen. (Ein ähnlicher Vorgang läuft ab, wenn die Schwangeren eine Transfusion mit körperfremden Stoffen bekommen. Die Antikörper, die gegen die natürlich gebildeten Blutgruppenantigene gerichtet sind, oder die vorher gebildet wurden, können nach einer Transfusion auch das erste Kind schädigen. Die häufigste Ursache der immunhämolytischen Krankheit bei Feten ist die Rh-Unverträglichkeit zwischen dem Fetus und der Mutter. Die Rh-Blutgruppe wird von den D-Antigen kodierendem RHD Gen und dem RHCE Gen zusammen bestimmt. Die häufigste ist DCE (D+) unter den Caucasoiden (Europiden). Die Rh-negativen haben einen cde-Genotyp im Allgemeinen. Die Unverträglichkeiten von AB0-Antigenen oder Kell-Antigenen (CD238) können die seltene Ursache einer fötalen Hämolyse sein. In diesen Fällen sind die Symptome viel milder, und sie verschlechtern sich bei den weiteren Schwangerschaften. Der Ikterus (Gelbsucht) bei den Neugeborenen, bei dem sie als Symptom für Hämolyse erscheinen, entsteht wegen der Vermehrung/Einlagerung/erhöhter Konzentration von Bilirubin. Die Gallenausscheidung der Leber kann nicht mit dem vermehrten Anfall des Bilirubins Schritt halten, und eine Gelbfärbung von Haut und der Sklera des Auges wird verursacht. (Die höhere Konzentration des Bilirubins kann Gehirnschädigung und Taubheit verursachen.) Die Fototerapie, die bei diesen Neugeborenen angewendet wird, fördert den Abbau des Bilirubins. 12.1.1.2. Typ II Überempfindlichkeit bei der Organtransplantation Wenn eine Übereinstimmung von AB0-Blutgruppen zwischen Spender und Empfänger vorliegt, kommt es zu einer sog. Transfusionsreaktion. Die hyperakute Abstoßung wird von allogenem HLA-Typ des Spenders, als Antigen ausgelöst. 12.1.1.3. Medikamenten-induzierten Typ II Reaktionen Bei den Medikamenten-induzierten Typ II Reaktionen wird die Schädigung zirkulierender Zellen von den Medikamentenmoleküle verursacht, die auf den korpuskularen Elemente des Blutes gebunden sind. 197 - Hämolytische Medikament, Anämie: das auf das den Blutkörperchen fixiert ist, ist in der Regel ein Penizillin-Derivat (seltener Tetrazyklin oder Erythromycin) - Thrombozytopenie: das Medikament bindet an Blutplättchen, meistens ein Kinin-Derivat. - Agranulozytose: sie kann mit Anlagerung eines Pirazolon-Derivat (zB. Aminophenazon, Noramino- phenazon) in Zusammenhang liegen. (Abbildung 12.1) Abbildung 12.1. Beispiele für den Medikamenten-induzierten Typ II Reaktionen 12.1.2. Intrinsches Antigen – Beispiele für die von einem Autoantikörper ausgelöste Krankheit 12.1.2.1. Goodpasture-Syndrom (Hauptsächlich wegen den Autoantikörper gegen Kollagen Typ IV in den Nieren) Hier binden die Antikörper an das sog. Goodpasture-Antigen, das die nicht-kollagene Domäne (von alpha3-Ketten) des Kollagen IV der Basalmembran ist. Da es in den Nierenglomeruli und in den Lungenalveolen exprimiert ist, werden diese Organe beschädigt. 12.1.2.2. Myasthenia gravis (MG) Bei Myasthenia gravis (MG) wird ein hemmender Antikörper an den Acetylcholinrezeptoren (AchR) der postsynaptischen Membran der neuromuskulären Endplatte binden, wodurch die Erregungsleitung aussetzt. Da die Krankheit in den meisten Fällen in Verbindung mit Thymusneoplasien oder – hyperplasie vorkommt, vermutet man, dass die Entstehung der Autoantikörper auf die überflüssige Anwesenheit eines ähnlichen (homologes) Proteins zurückgeführt werden kann, welches nur in tumorösen Geweben exprimiert wird. Die dagegen sensibilisierten, tumorspezifischen helfer T-Zellen können die Entstehung von hochaffinen, IgG-produzierenden Plasmazellen fördern. Diese Vermutung ist auch davon unterstützt, dass eine spontane Produktion von anti-Acetylcholinrezeptor Antikörpern auch unter den in vitro kultivierten Zellen, die aus dem während der Therapie entfernten Thymus gewonnen wurden, nachgewiesen werden kann. Unter den Neugeborenen der MG-Mütter können mit einer Wahrscheinlichkeit von 1:8, vorübergehende (2-3 wochenlang dauernde) Symptome erscheinen, die auf das Übertreten von Autoantikörpern durch die Plazenta hinweist. 12.1.2.3. Basedow-Krankheit Ein gegen die TSH-Rezeptoren gerichteter, stimulierender Autoantikörper kann die Wirkung von TSH vortäuschen, und dadurch die Schilddrüse zu einer überflüssigen Aktivität zwingen, die zu einer Hyperthyreose führt. Die stimulierenden Antikörper können während der Schwangerschaft über die Plazenta von der Mutter auf den Embryo übertragen werden, und eine neonatale Hyperthyreose des Neugeborenen verursachen. Deswegen ist es besonders wichtig, die Hormone- und den Autoantikörperspiegel von an dieser Krankheit leidenden Müttern zu überwachen. 12.1.3. 1. Der Diagnose Nachweis von Antikörpern, die die Neugeborenen/Organempfänger/transfundierte Zellen bedecken Person/mit (im Körper Medikament von den behandelter Patient/Autoimmunpatient) Agglutinationstest: - direkter Coombs-Test (DAT= direct antiglobulin test): Die mit Antikörpernbedeckten Zellen agglutinieren nach der Reaktion mit Coombs-Reagenz, das aus einer Ziege stammt, und antihuman IgG6/IgM/C3 enthält. - Mikrosäule-Test Die mit Antikörpern bedeckten Zellen werden auf einer Matrix mit hoher Dichte geschichtet. Durch die Matrix werden sie zentrifugiert und sie werden darunter an den anti-human IgG6/IgM/C3 Antikörper binden. Bei dem nächsten Zentrifugationsschritt können nur die nichtagglutinierten Zellen das unterliegende Gel passieren. Werden die Zellen mit Antikörpern bedeckt, agglutinieren sie, und bleiben auf dem oberen Teil der Säule stecken (s. MikrosäuleTest Folien). 2. Nachweis von im Serum frei zirkulierenden Antikörpern (im Körper von Schwangeren/Patienten vor Organtransplantation/Patienten vor medikamentösen Therapien) - indirekte Coombs-Test: Zum Serum, das die Antikörper enthält, wird zuerst das Blut von Rh-positiven Patienten/Spendern, dann in einem zweiten Schritt das Coombs-Reagenz zugegeben. Eine Agglutination weist auf die Präsenz des Antikörpers hin. 12.1.4. Therapie Das primäre Ziel ist es, die Bindung von Antikörpern an Antigene zu verhindern. Hiermit wird mit zwei Beispielen illustriert, welche immunologischen Eingriffe die Symptome beseitigen können. 1. Bei Rh-Inkompatibilität wird anti-D IgG (Rh-Immunglobuline) als Prophylaxe in einer intramuskulären Injektion der Mutter gegeben, innerhalb 72 Stunden nach der Geburt des ersten Kindes oder nach einem spontanen oder künstlichen Abortus. In der entsprechenden Phase der nächsten Schwangerschaft muss die Prophylaxe erneut durchgeführt werden. Die fötalen Blutkörperchen (Bk), die in die mütterliche Milz gelangen, werden dadurch mit dem künstlich zugeführten Antikörper bedeckt. Sie werden befördern: 199 - die Entfernung der fötalen Bk-en durch Phagozytose, so können sie nicht mit den mütterlichen B-Zellen in Kontakt treten, die für D Antigen spezifisch sind. - wenn sie mütterliche trotzdem B-Zellen auf spezifische treffen, sind die Epitope von den zugeführten Antikörpern bedeckt, sodass sie für die mütterlichen Antikörper nicht zugänglich sind. - wenn die mütterlichen B-Zellen trotzdem auf diese Epitope treffen, hemmen die gegebenen Antikörper, die auf der Oberfläche der Bk-en liegen, die Teilung der B-Zellen durch das sog. Antikörper Feedback Phänomen. (Abbildung 12.2) Abbildung 12.2. Negative Kostimulation der B-Zellen 2. Bei jenen Krankheiten, die von stimulierenden und hemmenden Autoantikörpern verursacht werden, können die Ursachen bisher gar nicht oder nur zum Teil behoben werden. Bei Patienten mit Myasthenia gravis, in der ein hemmender Autoantikörper vorkommt, kann man die Behebung der Ursache in bestimmten Fällen durch die Entfernung der Thymus erreichen. In anderen Fällen können die Symptome durch die Erhöhung der Halbwertzeit von endogenen Acetylcholinesterasen (AchE) durch die Gabe von Cholinesterase-Inhibitoren behandelt werden. Bei einem schweren Aufflammen der Krankheit können die auslösenden anti-AchR Antikörper durch Plasmapherese entfernt oder durch die intravenöse Gabe eines hemmenden Antikörpers inhibiert werden. Natürlich können auch andere, nicht-immunologische medikamentöse Therapien eingesetzt werden, um die Symptomen zu vermindern. 12.2. Typ III Überempfindlichkeitsreaktion Die Antigene des Immunkomplexes können exogener (wie z. B. aus Medikamenten, oder aus einem Erreger stammende) oder endogener (wie z. B. die DNA in dem Lupus erythematodes) Herkunft sein. Die Menge der Antigene übertrifft in der Regel die Menge der spezifischen Antikörper. Die Antikörper mit niedriger Affinität bevorzugen die Entstehung einer Immunkomplexkrankheit: es kommt die mangelnde Eliminierung zum Vorschein, und es begünstigt sie in der Kreislauf zu bleiben und somit die Neigung zur Bildung eines Komplexes. Die Größe der entstehenden Immunkomplexe kann auch die Bildung der Krankheit beeinflussen. Die größeren Antikörper werden von den Phagozyten mit höherer Leistungsfähigkeit einverleibt und aus dem Kreislauf entfernen als die kleineren. Gewebszerstörende Immunkomplexe: sie entsehen im Blut, dann treten sie aus dem Kreislauf aus, und setzen sich in den Wänden der Gefäße fest. Die kationische Antigene enthaltenden Immunkomplexe binden an der negative-geladenen Basallamina der Gefäßwände und der Nierenglomeruli. Die Ausdehnung der Ablagerung hängt nicht nur von der Größe der Immunkomplexe (je kleiner sie sind, desto 200 länger bleiben sie in dem Kreislauf, und desto wahrscheinlicher lagern sie sich ab), sondern auch von ihrer Zusammensetzung ab. Dabei ist aber nicht das Antigen, sondern der Antikörper entscheidend. Der typischste Antikörper, der eine Gewebsschädigung verursacht, ist IgG, manchmal IgM, und äußerst selten lassen sich IgA in den Komplexen nachweisen. sie können auch in situ, an dem Ort der Antigenlokalisierung entstehen. Ein inflammatorischer Prozess wird von den abgelagerten Immunkomplexen induziert. Von den Immunkomplexen wird das Komplementsystem aktiviert (die Chemotaxis und die Rekrutierung von Enzündungszelle erhöht sich dadurch) und die neutrophilen und eosinophilen Granulozyten (durch FcγRIII), bzw. die Thrombozyten werden angelockt. Die vasoaktiven Amine, die von basophilen Granulozyten und den Thrombozyten freigesetzt werden, erhöhen die Gefäßpermeabilität. Die zu den abgelagerten Immunkomplexen wandernden neutrophilen Granulozyten schädigen durch frustrierte Phagozytose 1 die Zellen und das Gewebe, die in der Umgebung der Ablagerung liegen. Der Mechanismus der Gewebsschädigung ist unabhängig von dem Ort der Ablagerung. Die Konsequenzen der Gewebsschädigung hängen jedoch von dem Ort der Ablagerung ab. Nach der Häufigkeit des Auftretens sind meistens die Haut, seltener die Nieren, das gastrointestinale System und die Gelenke von den Ablagerungen betroffen. Dadurch kann in den Gefäßwänden eine Vaskulitis, in der Basalmembran der Nierenglomerulen ein Glomerulonephritis und in der synovialen Membran der Gelenke eine Arthritis entstehen. Beispiele für die Krankheiten: Arthus-Reaktion: sie entsteht in der Haut, typischerweise wegen der Komplexe mit IgG. Schönlein-Henoch purpura (Kapillarblutung)- die Haut und Nieren sind betroffen, Immunkomplexe mit IgA Nach den Infektionskrankheiten, wenn der Erreger nicht eliminiert wird. zB. poststreptokokkale Glomerulonephritis oder Endocarditis. SLE: chronische, systemische Autoimmunkrankheit, bei der die Nieren, die Gelenke, die Haut, die Kapillaren des Herzens, und die serösen Oberflächen betroffen sein können. Akute Serumkrankheit: systemische Vasculitis, Glomerulonephritis und Arthritis mit IgGenthaltenden Immunkomplexen. 12.2.1. Eine typische Krankheit für den lokalen Typ III Farmerlunge – es ist häufig unter den Menschen, die landwirtschaftliche Arbeit haben, und täglich mit Futtermitteln (Heu für Rindernahrung) in Kontakt kommen, weswegen es so genannt wird. Das Antigen stammt typischerweise aus einem Bakterium, das in verschimmeltem Heu lebt (Thermoactinomyces), und durch Einatmung in den Körper gelangt. Trotz der Typ I Reaktion können die Antigene in einer großen Menge in der Umwelt des Patienten gefunden werden. Die Symptome der Krankheit: (lokale) Entzündungssymptome, in diesem Fall die Entzündung der Lunge (Pneumonitis). 1 Neutrophile sind "frustriert", wenn sie zu große Partikel aufnehmen müssen. In diesem Fall entlassen sie ihre Granuleninhalt (voll mit Proteasen und Sauerstoffradikalen) einfach nach außen, um evtl. die Errerger zu zerstören. Dabei verursachen sie aber auch Schäden an Nachbarzellen. 201 Zur immunologischen Diagnose werden die Tests für gegen das Bakterium gerichtete Antikörper, bzw. bakterielle Antigene verwendet: Immundiffusion: Prezipitationstest, Komplementfixation, Latexagglutination. 12.2.2. Akute systemische Typ III Hypersensitivitätsreaktion Serumkrankheit: Das Antigen kann ein Medikament, wie zB. Penicillin sein. Die Symptome entstehen 7-10 Tagen nach der Antigenexposition: Vasculitis, Glomerulonephritis, Arthritis. Die Feststellung der Diagnose läuft, wie oben (s. lokale Typ III). 12.2.3. Chronische systemische Typ III Hypersensitivitätsreaktion SLE: Das Antigen ist ein endogen nukleares Antigen. Da die Krankheit selbst im Rahmen von vorigen Praktiken erwähnt wurde, kann man die Symptome und die Diagnostik dort finden. In der Pathogenese der Typ I, II und III Überempfindlichkeitsreaktionen spielen Antikörper ebenso eine Rolle. In vielen Fällen können sogar die gleichen Antigene die Ursache sein (zB. Penicillin). Es ist wichtig, die wichtigsten Unterschiede in Tabelle 12.1. zusammenzufassen: Tabelle 12.1. Typ I-III Überempfindlichkeitsreaktionen 12.2.4. Therapie Bei den lokalen und systemischen Erkrankungen ist die Vermeidung auslösender Stoffe am zweckmäßigsten. Neben der medikamentösen Hemmung der Entzündung, kann man in den schweren Fällen die Plasmapherese, als ein Verfahren für die Entfernung zirkulierender Antikörper, verwenden. In chronischen Zuständen (wie zB. SLE) können durch biologische Therapiemaßnahmen B-Zellen oder andere, in der Kostimulation teilnehmende Zielmoleküle (z. B. CD40L, s. entsprechende Folien), mit gegen sie gerichteten blockierenden Antikörpern, inaktiviert werden. Dadurch lassen sich die 202 Anzahl der B-Zellen, und unter diesen die für die Autoantikörperproduktion verantwortlichen B-Zelle reduzieren. Im Rahmen von frühen oder präklinischen Studien werden solche Therapien untersucht, bei denen die Entwicklung einer Toleranz gegen bekannte Antigene erzielt wird. 12.3. Überempfindlichkeitsreaktion vom Typ IV oder verzögerter Typ (DTH=delayed type hypersensitivity) Weil das Auftreten der Symptome mehr als 12 Stunden oder sogar Wochen benötigen kann, bezeichnet man diese Reaktion auch als Überempfindlichkeitsreaktion vom verzögerten Typ. Das Antigen bindet nicht an den Antikörper, sondern es löst eine zelluläre Immunantwort aus: die Reaktion entwickelt sich mit der Mitwirkung von CD4+, CD8+ T-Zellen, Makrophagen und basophilen Granulozyten. In der Sensibilisierung spielen die CD4+ Th1-Zellen durch ihre Antigenerkennung eine wichtige Rolle. Am Ort der Antigen-Exposition werden unterschiedliche Zytokine (wie IFN-gamma, IL2, TNF-beta, TNF-alfa, GM-CSF, IL-3) freigesetzt, die die Rekrutierung und Aktivierung von Makrophagen und von zytotoxischen T-Zellen auslösen, und infolgedessen eine lokale Gewebszerstörung verursachen. Die untere Tabelle fasst die Eigenschaften von den 3 Unterklassen der Überempfindlichkeitsreaktion Typ IV zusammen. Syndrom verzögerter Typ der Überempfindlichkeit (DTH in engerem Sinne) ANTIGEN Folgen Proteine: -lokales Hautödem Insektenproteine -Erythem (Hautrötung) mycobakterielle Proteine (Tuberculin, Lepromin) -Induration -zelluläre Infiltration -Dermatitis Kontakthypersensitivität -Haptene: lokale Hautreaktion: Pentadeka-katechol (Gift von Giftsumach), Paraphenylenediamine -Erythem -kleine Metallionen: -intraepitheliale Knötchen -zelluläre Infiltration -Blasen Ni, Cr Zöliakie (glutensensitive oder gluteninduzierte Enteropathie) Bestandteile von Gluten in vielen Getreidesorten vorkommende Klebereiweiß, (bei Weizen: Gliadin) -Zottenatrophie in der Dünndarmschleimhaut -Verdauungstörung -Gedeihstörung Tabelle 12.2. Unterklassen der Überempfindlichkeitsreaktion Typ IV 12.3.1. DTH-Hauttests Die DTH-Hauttests werden in der klinischen Praxis verwendet, um die schon abgelaufene und aktuelle Infektionen bzw. die von uns zugeführten Antigene nachzuweisen. Wenn der Körper schon früher auf den Erreger getroffen ist, löst er durch das T-Zellen Gedächtnis eine intensive Hautreaktion auf 203 bestimmte Antigene des Erregers aus. (Mit diesem Untersuchungsverfahren kann man nicht genau entscheiden, ob das Individuum/der Patient zur Zeit an der Krankheit leidet oder schon überstanden hat und deshalb eine Immunität besitzt.) Der Stoff, der in dem Test benutzt wird, kann z. B. Tuberculin (für Nachweis von TB; Mantoux-Test, Lepromin (für Lepra), Histoplasmin (für Histoplasma capsulatum, hauptsächlich bei Lungeninfektionen), oder Candidin (für Candida-Infektion) zum Nachweis sein. Bei den Untersuchungen wird der Extrakt aus dem nicht-pathogenen Mycobakterium tuberculosis Erreger in die Haut (intracutan) injiziert. Der Extrakt kann OT (=old tuberculin), das ein Konzentrat aus dem in vitro Kulturmedium in dem M. tuberculosis gezüchtet wurde; oder häufiger PPD (purified protein derivate), die ebenso aus dem Medium präzipitiert wurden, sein. Die Immunzellen bilden eine Schwellung an dem Ort der Injektion, die Ausdehnung der begleitenden Entzündung hängt davon ab, ob die jeweilige Person zur Zeit gesund ist oder früher an TB gelitten hat. Die entzündliche Veränderung erreicht ihre maximale Ausdehnung später und wird deshalb erst nach 3-5 Tagen abgelesen und ausgewertet. Wenn bei jemandem die Ansteckung früher erfolgt hat, ist der Körper empfindlicher, und zeigt eine intensivere Reaktion auf den Erreger, aus dem der Extrakt hergestellt wurde (T-Zelle Gedächtnis). Auf der Haut des Gesunden überschreitet die Induration (verhärtete und erhöhte Hautveränderung) nicht den Durchmesser von 5 mm . Bei Betroffenen können es aber mehr als 10 mm sein. Die Rötung allein zählt nichts. Ein Beispiel für die diagnostische Anwendung: (Mantoux): Tuberculin-Test Die Tuberculose- Infektion (in Ungarn auch für die Kontrolle der BCG-Vakzinierung) kann damit nachgewiesen werden. Tabelle 12.3. Tuberculin / Mantoux Test (Abbildung 12.3) 12.3.2. Kontakthypersensitivität / Kontaktdermatitis Bei der Kontakthypersensitivität / Kontaktdermatitis binden Haptene (solche kleine Molekülen, die selbst keine Immunantwort auslösen können) an die eigenen Proteine. Die häufigsten Stoffe die Dermatitis auslösen: Metalle, zB. zum Gerben von Leder benutztes Kobalt, Nickel in Kleinod, pflanzliche Öle, zB. Urushiol oder Pentadeka-katechol (Gift von Giftsumach) 204 synthetische Stoffe, zB. Paraphenylene-diamine (nicht zu vermischen mit purified protein derivate, die auch PPD abgekürzt wird) für höhere Farbintensität in den kosmetischen Produkten, Kleidungen und Pelzwaren. Die dendritischen Zellen nehmen sie auf, und präsentieren sie den CD4+ oder CD8+ T-Zellen, die als haptenspezifische effektor Zellen aus den Lymphknoten in die Haut wandern. Bei der nächsten Exposition werden die Hapten-Peptide den lokalen, in der Haut liegenden T-Zellen präsentiert, und induzieren die Aktivierung der T-Zellen und ihre lokalen effektorischen Wirkungen. Ein Beispiel für die Diagnostik: epikutane Hauttest (Patch-Test) für Kontaktdermatitis Bei diesem Verfahren wird ein Pflaster, das mit den häufigsten Allergenen eingetränkt wurde, auf die Haut des Rückens geklebt und 48 Stunden später wieder entfernt. Das Ergebnis der Reaktion wird 96 Stunden (4.-6. Tagen) später abgelesen. In den am häufigsten verwendeten diagnostischen Standardreihen werden die häufigsten Stoffe (33 Allergene) getestet. Es gibt auch spezifische Pflastern für z.B. die Materialen, die in der zahnärztlichen Praxis verwendet werden (15 basis + 8 zusätzliche Allergene), oder ein anderes welches für Nachweis einer Metallallergie (die komplette Reihe besteht aus 24 Allergene) geeignet ist. Es gibt noch weitere Reihen, die Proben aus Medikamenten, Lebensmittelzusatzstoffe, Reinigungsmittel, Farbstoffe und Duftstoffe beinhalten. Die kommerziellen Standardreihen werden auch als finn chamber (Finnkammern) zum Verkauf zugelassen. Beim Ablesen sind die Rötung und die örtliche Schwellung auch in Betracht zu ziehen. 12.3.3. Zöliakie (glutensensitive oder gluteninduzierte Enteropathie) Für die Entstehung der Überempfindlichkeit ist ein aus 33 Aminosäuren bestehendes Peptid verantwortlich, das aus dem Gluten stammt. Das Gluten (Klebereiweiss) befindet sich in den Samen von bestimmten Getreidearten (Weizen, Gerste, Hafer, Roggen), und diese Eiweißmischung kann man im Weizen auf Gliadine (Prolaminfraktion) und Glutenine (Glutelinfraktion) unterteilen. Während der Verdauung wird das Gliadine in ein Peptid aus 33 Aminosäuren gespalten. Dieser Gliadinabschnitt kann an ein Enzym, die Gewebstransglutaminase (tTG= tissue transglutaminase) binden. Die im Peptid vorhandene Aminosäure Glutamin wird vom der tTG aus Glutaminsäure gebildet, und die Veränderung verleiht dem Peptid eine negative Ladung. Dieser Charakter verstärkt die Fähigkeit, an bestimmten HLA-Typen zu binden, die auf den antigenpräsentierenden Zellen der Lamina propria exprimiert werden und so den T-Zellen präsentiert werden. Bei den Individuen, die einen bestimmten Haplotyp (konkreterweise DQ2 oder DQ8, kodiert von HLA-A1 und -B1) haben, werden die glutenreaktiven T-Zellen aktiviert, produzieren Zytokine wie z. B. IFN-gamma, und leiten einen entzündlichen Vorgang im Darm ein. Während des Vorganges beginnen die Plasmazellen, die sich in der Umgebung des entzündetes Darmes befinden, Antikörper und Autoantikörper zu produzieren. Bei einem Verdacht einer Zöliakie werden zuerst serologische Untersuchungen durchgeführt, aber zur Diagnose sind weitere diagnostische Maßnahmen erforderlich, wie Dünndarmbiopsie histologischen Feststellung der Zottenatrophie mit begleitender Entzündung. Zur Diagnose von Zöliakie verwendete serologische, spezifische Antikörper Nachweisverfahren Die Tests zielen auf den Nachweis der IgA und IgG Antikörper aus dem Serum: 205 zur anti-deaminiertes Gliadinpeptid AK (a-DGP) Der Nachweis von AK, die während der Immunantwort auf Gliadin entstehen, ist wichtig, weil die Erkrankung dadurch von anderen Zottenatrophie-Krankheiten, die von der Gluteneinnahme unabhängig sind, differenziert werden kann. anti-Endomysium AK (EMA) Das Endomysium ist ein Bindegewebsprotein, das die feinen Muskelfasern des Dünndarmes umhüllt. Der Körper stellt anti-Endomysium-AK als Antwort auf die ständige Zerstörung des Darmes her. (Den Nachweis kann man auch in den Dünndarmbiopsie-Proben durchführen.) anti-Transglutaminase AK (a-tTG) Die (eigene) Transglutaminase bildet mit dem (fremden) Gliadinpeptid eine netzähnliche Struktur, und ein auf diese Weise entstehendes Autoantigen wird sehr stabil. Die anti-tTG und anti-EMA Antikörper weisen auf ähnlicher Weise auf eine Gewebszerstörung hin. Die Konzentration der Antikörper im Blut ist proportional mit dem Schweregrad der Krankheit, und man kann den Ablauf gut verfolgen. Die IgA-Tests sind viel spezifischer (IgA wird von den lokalen Plasmazellen produziert), weshalb die IgG-Messung mit der IgA-Messung zu ergänzen ist. 12.3.4. Terapie Die wirksamste Therapie ist, wie auch bisher, die Vermeidung des auslösenden Stoffes: der Kontakt mit bestimmten Metallen ist zu minimalisieren und eine glutenfreie Ernährung sollte eingehalten werden. Neben der entzündungsstillenden medikamentösen Therapie können noch die immunologische Therapie, wie z.B. die Hemmung der T-Zell- Antwort mit Immunsuppressiva (z. B. Cyclosporin) eingesetzt werden. Daneben werden jedes Jahr neue Wirkstoffe für die Hemmung der TZell-Antwort entwickelt, die zB. durch das Blockieren des B7 Kostimulationsmoleküls oder des IL-2Rezeptors wirken. Das neueste Ziel ist die Induktion der Toleranz von pathologischen T-Zellen, aber zur Zeit sind noch keine überzeugenden klinischen Ergebnisse im Zusammenhang damit bekannt. Übersetzt von Viktor Molnár 206 13. IMMUNTHERAPIE (GYÖRGY NAGY, ÉVA PÁLLINGER, ZSUZSANNA PÁL) Die Hauptziele der Medizin sind immer die Heilung oder Vorbeugung von Krankheiten, und falls möglich, nicht nur die Beseitigung der Symptome, sondern auch der Ursachen der jeweiligen Krankheit. Im Laufe der mehrere Jahrtausende langen Geschichte der europäischen Medizin wurde eine unglaubliche Vielzahl von Behandlungsstrategien entwickelt, dessen wichtigstes Prinzip aber immer die Doktrin des „primum nil nocere”, also „zuerst einmal nicht schaden“ war. Obwohl sämtliche pharmazeutische Forschung in erster Linie die Entwicklung von effektiven aber gleichzeitig auch nebenwirkungsfreien Medikamenten erzielt, verfügt selbst die heutige Medizin über solchen idealen Medikamente nicht, oder kaum. Manchmal sieht es so aus, als wären Wirkung und Nebenwirkung voneinander untrennbar, selbst wenn der Traum eines jeden Arztes eindeutig eine Welt ohne jeglichen Nebenwirkungen ist. Diese Situation scheint sich langsam zu ändern. Die explosionsartige Entwicklung der molekularen Medizin in den letzten Jahrzehnten, und vor allem das Humangenomprojekt ermöglichten eine vorher unvorstellbar tiefe Einsicht in die molekularen Pathomechanismen der verschiedenen Krankheiten, und enthüllten eine fast unverständliche Vielzahl von möglichen therapeutischen Zielen, welche zusammen die Entwicklung einer neuen Generation von gezielten molekularen Therapien ermöglichten. Als nächstes führten die moderne molekulare Genetik und die schnelle Analyse des genetischen Fingerabdrucks die ersten individualisierten Beschreibung des menschlichen Genoms Therapien ein. Schließlich beschleunigte die neben der pharmazeutischen Forschung auch die Genehmigung von neuen Medikamenten und vereinfachte die Verfolgung ihrer wahren Effizienz in der klinischen Praxis. 13.1. Grundbegriffe Immuntherapie Die Immuntherapien sind solche Therapien, die die Funktionen des körpereigenen Immunsystems ausnutzen um durch gezielte Beeinflussung von immunologischen Mechanismen ein therapeutisches Ziel zu erreichen. Sie modifizieren eine Immunantwort oder bestimmte Immunfunktionen (Normalisierung, Stimulation, Suppression, usw.) damit das Immunsystem eine Krankheit besser oder effizient genug bekämpfen kann. Modifizierfaktoren von biologischen Antworten (biological response modifiers, BRMs) BRMs sind vom menschlichen Organismus produzierte natürliche Moleküle, die unter physiologischen Umständen nur in extrem niedrigen Konzentrationen hergestellt werden, jedoch einen starken biologischen Einfluss auf die Immunantwort haben, und die für Zwecke einer Immuntherapie relativ einfach auszunutzen sind. In den Immuntherapien induzieren BRMs die Antwortreaktionen des 207 Immunsystems auf verschiedene immunologische Herausforderungen. Ihre therapeutische Anwendung wird dadurch ermöglicht, dass sie mit den modernen Labortechniken in industriellen Mengen, rein, reproduzierbar, und sicher herstellbar geworden sind. 13.2. Gezielte molekulare Therapie (targeted molecular therapy, TMT) Eine gezielte molekulare Therapie ist in der Regel gegen solche körpereigenen Strukturen und Mechanismen gerichtet, die nur unter pathologischen Umständen erscheinen oder aktiviert werden, also im physiologischen Zustand im Körper nicht vorhanden sind. Im Gegensatz zu der klassischen pharmazeutischen Therapie, erzielt die TMT durch diese Strategie ausschließlich die kranken Zielzellen (targets) zu beeinflussen ohne dabei die gesunden Zellen zu beschädigen oder gefährden. Dies ermöglicht eine starke Einschränkung des Spektrums der Nebenwirkungen und gleichzeitig auch die Steigerung der therapeutischen Effizienz. Der Begriff TMT wird meistens in Krebstherapien verwendet, wo TMTs typischerweise essentielle molekulare Regulationswege der Tumorentwicklung angreifen, wogegen die klassische Chemotherapie alle sich intensiv teilenden Zellen des Patienten beschädigt. Die wichtigsten Instrumente der heute in der Praxis erhältlichen, d.h. für klinische Zwecke genehmigten TMTs können in zwei Untergruppen geordnet werden: 1) kleine Moleküle (small molecules) und 2) monoklonale Antikörper. Die kleinen Moleküle (<500 Dalton) binden mit großer Affinität zu Proteinen, Nukleinsäuren oder Polysacchariden der Zielzellen und verändern dadurch ihre Aktivität oder Funktion. Ihr kleines Molekulargewicht ermöglicht eine schnelle Penetranz durch die Plasmamembran, sie können also ihre Wirkung auch intrazellulär ausüben. In den nachfolgenden Kapiteln beschäftigen wir uns zuerst mit diesen Therapien. Mit den monoklonalen Antikörpern werden wir uns später, in Kapitel 13.4.1.1. detailliert auseinandersetzten. 13.2.1. Biologika oder Biopharmazeutika (biologics) Biologika sind solche therapeutische Agenzien, die aus einer biologischen Quelle stammen (z.B. mit Hilfe von rekombinanter DNA Technologie / Gentechnologie / Genmanipulation). Heute sind sie (noch) meistens Makromoleküle, vor allem Proteine. Wegen ihren vielen Vorteilen sind Biologika die wichtigsten Wirkungsmittel der neuen TMTs. 13.2.2. Targeting Die Mehrheit der heutzutage angewendeten Medikamente erreicht die Stelle ihrer Wirkungszone im Körper durch die Blutzirkulation. Dies führt dazu, dass sie in relativ hohen Dosen verabreicht werden müssen, weil sie im Blut stark verdünnt werden. Zweitens wirken sie nicht ausschließlich nur an der Stelle der erzielten pharmazeutischen Aktivität , da sie durch die Blutzirkulation in vielen Körperteilen 208 auch deponiert werden die mit der Wirkung des Medikaments nichts zu tun haben. Aus diesen Gründen ist der Anspruch auf einen spezifischen, gezielten Medikamententransfer (Targeting) schon seit langem entstanden. Dem Prinzip nach sollten diese Medikamente irgendwie so modifiziert werden, dass ihre Bindung zu den Zielzellen spezifischer wird, mit anderen Worten ihre Wirkung auf die Zielzellen limitiert wird. Auf diese Weise könnte man die für die therapeutische Wirkung benötigten Dosen verringern, die Gewebe-Verteilung der Medikamente optimieren und auch die Nebenwirkungen drastisch vermindern. 13.2.2.1. Targeting mit Nanotechnologie Die Nanotechnologie spezialisiert sich auf Technologien mit denen Aufbau und Modifikation von Strukturen in der Größe von einzelnen Atomen bis hin zu der Größenordnung von 100 nm erreichbar ist. Die darauf bauende Nanomedizin fokussiert sich auf zwei Bereiche: auf die Entwicklung von Nanomedikamenten und die der neuen medizinischen Bildverarbeitungstechniken. Das große Versprechen der Nanomedizin ist die Verbesserung der Effizienz der heutigen Medikamente durch Optimierung ihrer Aufnahme, Freilassung im Körper, und Verteilung in dem Organismus durch intelligente Änderung der Struktur der heutigen Medikamente in extrem kleine Maßstäbe. Diese verbesserten Molekularstrukturen sollen viele Probleme, die heutige Medikamente noch plagen, wie z.B. die oft begrenzte Löslichkeit, die fragliche Stabilität, usw. beseitigen und früher als kompliziert und umständlich geltende Verabreichungswege ermöglichen (Inhalation oder intranasale Verabreichung) oder auch eine gewisse Individualisierung der Therapie irgendwie machbar machen. Die wichtigsten Gemeinsamkeiten aller Nanomedikamente sind die Multimodularität und die Multifunktionalität. Multimodularität bedeutet, dass die kleinste voll funktionelle molekulare Einheit der Nanomedikamente selbst auch aus vielen kleineren Modulen besteht, welche funktionell unterschiedliche Aufgaben erfüllen. Das für die eigentliche therapeutische Funktion verantwortliche Medikament-Molekül wird z.B. von einer Transporter-Einheit getragen, oder in ihrem Inneren versteckt. Zum Transporter werden dann weitere Module gekoppelt, die unabhängig voneinander für die Verbesserung der Absorption, Metabolismus, Verteilung und Targeting des Medikaments verantwortlich sind. Zusätzlich sind manche experimentelle Nanomedikamente auch mit Markierungsmodulen verkoppelt (z.B. Fluoreszenz), damit ihre Aktivität im Körper besser verfolgt werden kann. Von der Nanotechnologie wird in erster Linie in der Therapie der neuropsychiatrischen Erkrankungen viel erwartet, weil sie durch die Blut-Hirn-Schranke eine bessere Absorption ermöglichen, und dadurch ein schon seit langer Zeit vorhandenes kritisches Problem lösen sollen. 13.2.2.2. Targeting mit monoklonalen Antikörpern Die mit Medikamentenmolekülen, Radioisotopen, oder Toxinen gekoppelten monoklonalen Antikörper können auch als “Zielfinder“ (homing device) fürs Targeting verwertet werden. 209 Abbildung 13.1. Arten der Immuntherapie mit monoklonalen Antikörpern Sie verbessern die therapeutische Wirksamkeit der Medikamente und minimalisieren den Schaden der gesunden körperlichen Zellen ähnlich zu den Nanomedikamenten. Ihre möglichen Nebenwirkungen sind meistens mit denen der zum Antikörper gebundenen Wirkstoffe identisch, obwohl der Antikörper-Teil selber unter Umständen auch seine eigenen Nebenwirkungen haben kann (siehe später). Die Klassifizierung der Therapien in denen monoklonale Antikörper als Lieferanten verwendet werden, beruht auf den zu transportierten Wirkstoffen. Dementsprechend bezeichnet man die auf mit Radioisotopen konjugierten Antikörpern basierenden Therapien als Radioimmuntherapien (z.B. Ibritumomab tiuxetan, Tositumomab), die mit chemotherapeutischen Agenzien gekoppelten Antikörper als Chemoimmutherapeutika (ist noch in der experimentellen Entwicklungsphase), die mit bakteriellen (Diphtherietoxin, Pseudomonas Exotoxin) oder pflanzlichen (Rizin A, Saporin) Toxinen gekoppelten Antikörper als Immuntoxintherapien (z.B. Gemtuzumab ozogamicin, BL22). Einige für die Anwendungsmöglichkeiten von solchen wie folgt: Beispiele Antikörpern sind Abbildung 13.2. Nackte monoklonale Antikörper. 210 Nicht alle monoklonale Antikörper tragen Konjugate. Die sog nackte Antikörper werden meistens zur Neutralisierung eines Ligands oder Blockade seines Rezeptors verwendet. Abbildung 13.3. Mono-klonale Antikörper mit Radionuklide Radioisotope chemisch werden zu den entweder Antikörpern gebunden, also mit ihnen direkt verkoppelt verabreicht (einstufige Therapie), oder nach erst der Verabreichung des Antikörpers ins Blut geführt und mit Hilfe eines Biotin-Avidin Systems Antikörpern, bzw. gebunden Die (zweistufige zweite flexibler, Strategie da es zu den Zielzellen Therapie). ist etwas unterschiedliche Isotopen mit dem selben Antikörper Abbildung 13.4. Monoklonale Enzym-Prodrug Therapie anwendbar macht, und theoretisch ermöglicht sie auch eine stärkere lokale Dose zu erreichen, ist aber auch aspezifischer, weil Biotin auch an nicht-gezielten körperlichen Zellen vorkommen kann. In diesem Fall sind die Antikörper mit einem (meistens nicht-menschlichen) Enzym verkoppelt. Das Enzym ist in der Lage ein von den Antikörpern unabhängig, in einer zweiten Runde der Therapie verabreichtes, inaktives und deswegen für gesunde körperliche Zellen harmloses Prodrug umzuwandeln. Da die Umwandlung des Prodrugs in ein aktives (z.B. toxisches Medikament, wie ein Zytostatikum) nur an den Zielzellen (z.B. Tumorzellen) stattfindet, wird die Therapie die Zielzellen gezielt und effizient, für andere körperliche Zellen jedoch besonders schonend, und dadurch fast ohne Nebenwirkungen beeinflussen. Abbildung 13.5. Liposom-Therapie ergänzt mit monoklonalen Antikörpern Liposomen sind relativ leicht mit monoklonalen Antikörpern zu verbinden. Weil sie praktisch alle Formen von therapeutischen Agenzien verschlossen, von dem Rest des Körpers separiert transportieren, und nur den Zielzellen abgeben können, ist diese Strategie besonders vielversprechend. Liposomen sind oft PEG-modifiziert um ihre Halbwertszeit zu verlängern. 13.2.2.3. Targeting mit Peptiden Da die Zielpunkte der meisten Medikamente sich im Zytoplasma oder Zellkern befinden, ist/spielt der Transfer des Wirkstoffes in den intrazellulären Raum eine Schlüsselfrage/Schlüsselrolle in allen gezielten Therapien. Die Entdeckung der sog. zell-penetrierenden Peptiden (cell penetrating peptides = CPPs), kleine Peptide wie das z.B. Protein Tat des HIV Virus, haben diese Frage/dieses Thema stark beeinflusst. Solche Peptide erlauben bzw. sichern eine stark verbesserte Penetranz für Medikamente oder Markierungsmoleküle durch die sonst nur sehr selektiv durchdringbaren Plasmamembranen. Die medizinische Benutzung der CCPs hat heutzutage nur noch zwei wichtigere Bereiche: die medizinische Bildverarbeitung und die molekulare Radiotherapie, obwohl neulich/seit kurzem immer mehr auch mit gewebespezifischen CPP-Konstruktionen experimentiert wird (z.B: gewebespezifisch aktivierbare CPPs, usw.). 13.2.2.4. Chemotaktisches targeting (chemotactic drug targeting) Eine spezielle Form der gezielten immunologischen Therapien ist CDT, bei der die zu behandelnden Zellen zu den Medikamentenmolekülen geleitet werden, und nicht umgekehrt. Die CDT Konjugate bestehen aus mehreren Teilen: 1) aus einem carrier, welcher die zelluläre Aufnahme und Internalisierung des Medikaments bestimmt 2) einem chemotaktisch aktiven Ligand, welcher die Zielzelle spezifisch, durch seine chemotaktische Wirkung zur Aufnahme des Medikaments anlockt, 3) dem Mediment-Molekül selbst, und 4) einem spacer, welcher das Medikament zum carrier bindet und seine Abspaltung vom carrier nach der Aufnahme, innerhalb der Zelle ermöglicht. 13.3. Immuntherapie Die Immuntherapien werden auch nach mehreren Eigenschaften kategorisiert. Je nachdem, ob die Immuntherapie auch eine aktive Beteiligung des Immunsystems des Patienten benötigt oder nicht, werden die aktiven und passiven Immuntherapien voneinander unterschieden. Zweitens, von der Richtung des Eingriffs abhängig redet man über Immunstimulation, Immunsuppression oder Immunmodulation. Es ist wichtig zu sehen dass diese zwei möglichen Gruppierungen nicht einander hierarchisch unter- oder übergeordnet sind, d.h. alle ihrer möglichen Kombinationen sind vorstellbar (aktive Suppression, passive Immunmodulation, usw.). 13.3.1. Aktive Immuntherapie Die aktiven Immuntherapien sind solche medizinischen Einwirkungen, welche dem Immunsystem des Patienten die benötigten Einsatzbefehle erteilen, gegen eine gewisse Krankheit aufzutreten oder 212 einen gewissen Erreger zu beseitigen, damit der Organismus selber das Problem behandeln kann. Eine aktive Immuntherapie gegen ein Zielmolekül kann mittels Induktion der Immunantwort (z.B. Impfung), Verstärkung einer bereits laufenden Immunantwort (z.B. durch Verstärkung der Antigenpräsentierung), oder sogar durch die Beeinflussung der eingebauten regulatorischen Mechanismen der Immunantwort (z.B. Aufheben der Toleranz gegen ein Tumorantigen) erzielt werden. Aktive Immuntherapie wird am häufigsten in akuten und chronischen infektiösen Erkrankungen, und in manchen nicht-infektiösen chronischen Krankheitsbildern wie Krebs und Autoimmunität verwendet. 13.3.2. Präventive (prophylaktische) Impfungen Gegen potentiell lebensbedrohliche akute Infektionen werden meistens präventive Impfungen eingesetzt, welche getötete oder attenuierte (in ihrer Virulenz künstlich beschränkte) Krankheitserreger beinhalten. Sie sind bei solchen Infektionen von Bedeutung, gegen die ein langfristiges Gedächtnis aufgebaut werden kann (z.B. Pocken, Tollwut, Typhus, Cholera, HBV, Diphtherie, Tetanus, usw.). Präventive Impfungen werden in einen gesunden Organismus, noch vor seiner Exposition auf den wahren Erreger eingeführt, um den klassischen, potentiell tödlichen Verlauf der Infektion vorzubeugen, den Verlauf dem Patienten zu erleichtern, oder die typischen Schäden und Risiken einzudämmen. (Eine wichtige Ausnahme dieser Definition ist der Fall der Tollwut, dessen Behandlung unter gewissen Umständen auch auf einer prophylaktischen Impfung beruht, welche aber nach der Exposition verabreicht werden kann). Aktive Immunisierung in oder gegen chronische Krankheitsbilder bzw. Infektionen ist in der Regel weniger effizient. In diesem Bereich fokussiert sich die heutige Forschung vor allem auf solche Krebskrankheiten, hinter denen eine Virusinfektion als instrumenteller Faktor bekannt ist: z.B. Leberzellkarzinom (HBV, HCV), Gebärmutterhalskrebs (HPV), Burkitt- und Hodgkin Lymphom (EBV), T-Zell-Leukämie und Lymphomen (HTLV-1), usw. Diese, gegen Krebsentwicklung eingesetzten prophylaktischen Impfungen (cancer preventive vaccines) müssen jedoch klar von den therapeutischen Impfungen getrennt werden, die gegen einen bereits vorhandenen Krebs angewandt werden (cancer treatment vaccines). 13.3.3. Therapeutische Impfungen Das Ziel der Entwicklung von therapeutischen Impfungen ist die Verbesserung der gängigen Therapien gegen manche langsam ablaufende chronische Krankheiten, wie bestimmte Krebsarten, Autoimmunkrankheiten, AIDS, Tuberkulose, Malaria, usw. Therapeutische Impfungen mobilisieren die zelluläre oder humorale Immunantwort des Patienten, damit die Schwierigkeit der Krankheit etwas vermindert wird, oder in dem extremen Fall, die Krankheit vollständig geheilt wird. Diese therapeutischen Impfungen liefern oft die selben Antigene oder Epitope dem kranken Organismus, gegen die man in einer passiven Immunisierung monoklonale Antikörper verabreichen würde. Für klinische Zwecke genehmigte therapeutische Impfungen gibt es viele, ein Beispiel dafür ist in dem Fall der Multiplen Sklerose zu sehen. Die in Schüben verlaufende Multiple Sklerose kann höchst 213 effektiv durch einen Impfstoff unter dem kommerziellen Namen Copaxone beeinflusst werden. Der Impfstoff beinhaltet ein synthetisches Copolimer namens Glatiramerazetat. das sich dem MyelinBasischen Protein ähnelt, also dem Autoantigen, welches in der MS von dem Immunsystem angegriffenen wird. Obwohl der Wirkungsmechanismus vom Glatiramerazetat unbekannt ist, wird angenommen, dass es seine Wirkung wenigstens teilweise durch die Verschiebung des Th1-Th2 Gleichgewichts in die Th2 bzw. eine immunsuppressive Richtung ausübt. Auf diese Weise werden anti-inflammatorische Mechanismen und für das Glatiramerazetat spezifische Treg Zellen aktiviert. 13.3.3.1. Weiterentwicklung der gängigen Impfungen 13.3.3.1.1. Anwendung von Adjuvanzien Eine Schlüsselfrage der Impfstoff-Entwicklung ist immer, wie stark und wie lang die von den Antigenen ausgelöste Immunantwort sein wird. Ideal wäre selbstverständlich eine lebenslange Immunität zu erzeugen, aber das ist bei vielen Erregern und Antigenen unmöglich. Die zur Immunisierung angewandten natürlichen oder rekombinanten Antigene weisen meistens eine relativ niedrige Immunogenität vor. Deshalb müssen sie im Impfstoff nicht in ihrer reinen Form, sondern zusammen mit Adjuvanzien, d.h. mit Molekülen welche ihre Immunogenität steigern, eingesetzt werden. Die Wirkungsmechanismen der Adjuvanzien sind vielseitig, aber sie wirken typischerweise aspezifisch, also von den Eigenschaften des Antigens des Impfstoffes unabhängig. Zum Beispiel sorgen viele von ihnen für eine gleichmäßige und langsame Freilassung des immunisierenden Antigens aus dem Impfstoff über die Zeit (Deponierungseffekt), wodurch die Antigenpräsentierung länger läuft, mehr T Zellen aktiviert werden können, und die Immunantwort auch stabiler bleibt. Solche Adjuvanzien sind z.B. die Aluminiumhydroxyd oder Aluminiumphosphat Gels, obwohl Wasser-Öl Emulsionen zu diesem Zweck auch oft eingesetzt werden. Andere Adjuvanzien wirken durch Stimulierung des natürlichen Immunsystems durch PRRs, Zum Beispiel Aktivierung des TLR2 und TLR4 durch Adjuvanzien steigert die Sekretion von immunaktivierenden inflammatorischen Zytokinen wie TNF, verstärkt die Expression von iNOS, und beschleunigt die Migration mancher APCs (DCs und Makrophage): sie aktivieren also viele Verteidigungsmechanismen gegen infektiöse Krankheisterreger. In den Tumortherapien erhöht die Sekretion von TNF die Permeabilität der Kapillaren des Tumors für die Chemotherapie, und verstärkt dadurch ihre Wirkung. Eine höhere iNOS Expression führt zur Freisetzung von NO in hohen Konzentrationen, welche Apoptose induzieren kann selbst in solchen Tumorzellen die schon Resistenz gegen die Chemotherapie entwickelt haben. Schnellere Wanderung von DC und Makrophagen in und aus dem Stroma von Tumoren verbessert die Präsentierung von Tumorantigenen für CD8+ T Zellen. In der Tat gibt es klinische Beweise dafür, dass eine parenterale Behandlung mit TLR2 und TLR4 das Überleben von Patienten verlängert, selbst wenn sie auf die konventionelle Chemotherapie nicht mehr antworten können, oder einen Rückfall gehabt haben. Teilweise führten diese Kenntnisse zu den ersten Versuchen Nanomedikamente mit TLR4 oder TLR7 Agonisten-Gehalt zu entwickeln. Laut den zurzeit erhältlichen Daten erzeugen diese Strategien eine starke und, in manchen Krankheiten und glücklichen Fällen, sogar lebenslange Immunität. 214 13.3.3.1.2. Unterstützung der Antigenpräsentierung Die Stärke der antitumoralen Impfstoffe kann auch durch die Unterstützung der APC in der Ausübung ihrer Präsentierungsfunktionen verbessert werden. Hierfür gibt es mehrere Möglichkeiten, wie die Beschleunigung der Teilung der Vorläuferzellen (IL-3, GM-CSF), Beschleunigung der Differenzierung und Reifung der APCs mit ausgewählten Zytokinen (GM-CSF), Aktivierung der Expression von kostimulatorischen Molekülen, Interferenz mit negativen Kostimulatoren (z.B. Interferenz mit CTLA-4), oder Verbesserung der Antigen-Aufnahme der APCs (z.B. ex vivo Auffüllung von DCs mit synthetischen Antigen-Peptiden, oder sogar ex vivo Fusion von DCs mit Tumorzellen). 13.3.3.1.3. Verstärkung der Aktivität von T Zellen Die gegen bestimmte Fremdantigene oder Tumorassoziierte Antigene (TAA) spezifischen T Zellen können zum Zwecke der Impfung, in erster Linie durch eine verbesserte Aktivierung des TZRs auch stärker aktiviert werden. Dies kann am einfachsten durch eine Verbesserung der Prozedur der Antigen-Auswahl erreicht werden. Optimierung der TZR Aktivierung durch verbesserte Antigenpeptide ist jedoch keine leichte Aufgabe, da ein essentiell wichtiger Teil der Verteidigungsmechanismen von vielen Krankheitserregern und Tumoren ihre relativ starke Ähnlichkeit zu körpereigenen Antigenen ist. Für die Verbesserung der TZR-Aktivierung durch intelligent ausgewählte Peptide wurde deshalb in den letzten Jahren eine ganz neue Wissenschaft, das sog. Epitop-Engineering etabliert. 13.3.3.2. Zytokine in der aktiven Immuntherapie Wegen ihrer starken und vollkommen natürlichen Wirkung auf die Immunantwort sind Zytokine ideale BRMs für die Behandlung von einer Vielzahl von Krankheiten in denen das Immunsystem eine wichtige Rolle spielt. Die Wirkungen von therapeutisch eingesetzten Zytokinen können sehr vielseitig sein: z.B. in Tumortherapien mögen sie sowohl die antitumorale Antwort verstärken, als auch direkt an den Tumorzellen wirken. Zum Beispiel IFN alpha, eines der am besten bekannten immuntherapeutischen Zytokine, erhöht die MHC Expression sowohl an DC als auch an Tumorzellen, unterstützt die Killer-Aktivität von Tcit und NK Zellen, hebt die Toleranz gegen Tumoren auf und treibt Tumorzellen in die Apoptose. Interleukin-2 (IL-2), ein natürlicher Wachstumsfaktor von T Zellen, kann in hohen Dosen T Zellen nicht nur zur Teilung anregen sondern sie auch überstimulieren, eine massive Freisetzung von vielen T-Zell-Zytokinen erzeugen (cytokine storm), extrem starke akute Entzündungen und Abstoßung von selbst etablierten Tumoren oder volle Regression ihrer Metastasen hervorrufen. Andere, in der Tumortherapie noch nicht etablierte aber vielversprechende Zytokine sind TNF und TRAIL, welche auch direkte zytotoxische Wirkungen auf Tumorzellen haben. 13.3.3.3. Manipulation der Immuntoleranz Die Immuntoleranz ist eine kontrollierte, absichtliche Unfähigkeit des Immunsystems auf bestimmte Antigene zu reagieren. Die Toleranz ist vor allem gegen körpereigene Strukturen sinnvoll und dementsprechend wird sie als eine wichtige natürliche und vollkommen physiologische Funktion des Immunsystems ausgeübt. Interessanterweise wird aber Toleranz auch gegen bestimmte, nichtkörpereigene aber harmlose fremde Antigene, z.B. Antigene der Kommensalen in dem Darmtrakt 215 entwickelt. Toleranz wird nur problematisch, wenn gefährliche Viren, Bakterien, Parasiten und andere Krankheitserreger, Schritte machen um eine Toleranz gegen sich selbst aktiv hervorzurufen. Ein Paradebeispiel dafür ist das Mycobacterium leprae, oder wenn die Toleranz gegen körpereigene Antige in den Autoimmunitäten ausfällt. Therapeutisch induzierte Toleranz wurde zuerst in der Transplantationsmedizin eingeführt um die allogen transplantierten Organen vor der Abstoßung zu schützen. Heute wird aber diese Option auch bei vielen Autoimmunkrankheiten und Hypersensitivitätsreaktionen eingesetzt. Erzeugung der Toleranz ist durch mehrere Wege möglich: 1) immunsuppressive Steroide (z.B. Prednisolon), 2) Interferenz mit der DNA-Replikation und Zellteilung der T Zellen (Azathioprin) 3) Interferenz mit der Signalübermittlung in T Zellen (Tacrolymus) 4) durch Vernichtung der aktivierten T-Zellen mit monoklonalen Antikörpern (z.B. anti-CD3, anti-CD25), 5) die noch experimentelle Strategie der Hemmung der Tcit und NK Zellen durch die Verabreichung von solublen MHCI Molekülen oder 6) die gezielte Induktion von Treg Zellen. 13.3.3.4. Zelluläre Immuntherapien Das ständig verbesserte Verständnis der Mechanismen der antitumoralen Immunantworten führte zur Entstehung von solchen Immuntherapien, welche die mit dem Tumor interagierenden Immunzellen direkt, in situ zu beeinflussen versuchen. Je nach Wirkungsmechanismus können diese Therapien in drei Gruppen eingeteilt werden: 1) Induktion der antitumoralen Immunantwort 2) Anlockung von tumorspezifischen Immunzellen an den Ort des Tumors 3) Neutralisierung der tumorvermittelten Immuntoleranz. 13.3.3.4.1. Induktion der antitumoralen Immunantwort In den zellulären Tumorimmuntherapien sind der adoptive Zell-Transfer (ACT) und die DC Impfung alternative Strategien mit dem selben Ziel, nämlich der Erzeugung einer starken Immunantwort mit Hilfe von manipulierten Immunzellen. Beim ACT werden T Zellen, meistens tumor-infiltrierende T Zellen (TILs) aus Krebsläsionen des Patienten isoliert, und außerhalb des Körpers, unter künstlichen Umständen, in speziellen Zellkulturen (ex vivo) aktiviert bzw. manipuliert. Die am öftesten benutzte Strategie ist die künstliche Aktivierung von T-zellen mit Exposition auf Tumorantigene und gleichzeitiger IL-2 Behandlung. Eine andere Strategie ist die Entnahme von T Zellen und ihre gentechnologische Transfektion mit Vektoren, welche die Expression von tumorspezifischen TZRs forcieren. In beiden Fällen werden die ex vivo aktivierten, manipulierten und vermehrten, also gegen den Tumor so stark wie möglich angeregten T Zellen am Ende des Prozesses dem Patienten in extrem großen Mengen zurückgegeben (adoptiver Zell-Transfer). In der DC Impfung werden aus dem peripheren Blut des Patienten Monozyten isoliert und mit einem speziellen Protokoll ex vivo, meistens mit Hilfe unter anderem, von IL-4 und GM-CSF zu DC differenziert. Parallel zur Entnahme des Blutes werden auch Tumorzellen aus dem Patienten isoliert, lysiert und die DC mit ihren Antigenen und manchen DC Reifungsfaktoren zusammen so lange inkubiert, bis die DC mit den Tumorantigenen aufgefüllt und vollständig reif sind. Die DC werden dann 216 dem Patienten zurückgegeben um eine tumorspezifische T-Zell-vermittelte Immunantwort zu erzeugen. Sowohl ACT als auch DC Impfung sind wirksame therapeutische Möglichkeiten, jedoch sind beide noch in der experimentellen Phase und noch extrem kompliziert in einem regulären Krankenhaus auszuführen. 13.3.3.4.2. Anlockung von tumorspezifischen Immunzellen zum Tumor Zu diesem Zweck werden in erster Linie solche TLR Agonisten benutzt, die eine starke Entzündung an der Stelle des Tumors erzeugen und dadurch eine so starke Infiltration von Immunzellen jeglicher Arten erzeugen, dass sie am Ende die tumorerzeugte Toleranz durchbrechen und den Tumor abstoßen. Ein Beispiel für diese Strategie ist der TLR7 Agonist namens Imiquimod, welcher einfach in der Form einer Creme auf manchen Hauttumoren wie Basalzellkarzinomen aufgetragen wird, und sie rasch, parallel zur Entwicklung einer starken Entzündung, abstoßen lässt. Eine andere Strategie ist die der Anwendung von bispezifischen Antikörpern. Diese entstehen durch enzymatische Spaltung und Neukopplung, oder durch gentechnologisch erzeugte Antikörper-Chimäre, deren zwei Fab Teile zwei verschiedene Spezifitäten vorweisen (!). Der eine Fab Teil bindet einen Marker der Tumorzellen, während der andere einen Marker der anzulockenden Immun-zelle, z.B. CD3 der T Zellen erkennt. Diese Konstruktion verbindet Tumorzellen also mit Immunzellen um die Erkennung und Vernichtung Krebszellen der zu Abbildung 13.6. Bispezifische monoklonale Antikörper begünstigen. 13.3.3.4.3. Neutralisierung der tumorvermittelten Immuntoleranz Therapeutische Strategien dieser Art können die systemische Immunsuppresion oder lokale Immuntoleranz durch das Ausschalten von T-Zell-hemmenden Mechanismen neutralisieren (antiCTLA-4, anti-PD-L1). Eine andere Möglichkeit ist die Sichtbarkeit der Tumorzellen für die Immunzellen durch die forcierte Expression von bestimmten, stark immunogenen Antigenen (manche TAAs), oder immunstimulierenden Zytokinen (GM-CSF, IL-2, usw) zu steigern. 217 13.4. Passive Immuntherapie In einer passiven Immuntherapie wird ein immuntherapeutisches Ziel entweder ohne, oder nur mit begrenzt aktiver Beteiligung des Immunsystems des Patienten erreicht. Dies bedeutet in erster Linie die Verabreichung von Medikamenten, zum Beispiel monoklonalen Antikörpern, die dem Patienten einen fertiggestellten Schutz liefern oder eine therapeutisch nützliche Immunantwort mechanistisch anregen. 13.4.1. Monoklonale Antikörper in der Therapie Die monoklonalen Antikörper wirken genauso wie die Antikörper des Patienten selbst. Sie können entweder „nackte“, also nicht-modifizierte, nicht-gekoppelte, quasi-native Antikörper, oder chemisch bzw. gentechnologisch mit irgendeinem Molekül gekoppelte modifizierte Antikörper sein. Die gekoppelten oder modifizierten Antikörper können Toxine, Radioisotope (typischerweise starke gamma oder beta-Emitter), Enzyme, Medikamente, immunmodulatorische Moleküle wie z.B. Zytokine tragen, oder zwei unabhängige Bindungsstellen mit unterschiedlicher Spezifizität vorweisen (bispezifische Antikörper, siehe vorher, Kapitel 13.3.3.4.2). 13.4.1.1. Herstellung der monoklonalen Antikörper Die industrielle Herstellung von monoklonalen Antikörpern Die Erzeugung von monoklonalen Antikörpern wurde erst in den ‘70ern, durch die Entwicklung der Hybridom-Technik möglich gemacht. Die Technik ist den Forschern Georges J.F. Köhler und César Milstein zu verdanken, wofür sie beide in 1984 den Nobelpreis in Medizin erhielten. Diese Technologie ermöglichte zum ersten Mal die Produktion von wortwörtlich unbegrenzten Mengen von Antikörpern mit einer einzigen, bestimmten Spezifizität, und wird seitdem mit großem Erfolg benutzt für sowohl diagnostische als auch therapeutisch Zwecke. Zur Erstellung von monoklonalen Antikörpern wird zuerst ein Tier, meist eine Maus mit dem fraglichen menschlichen Antigen, wogegen die monoklonalen Antikörper benötigt werden, intravenös immunisiert. Aus dieser Maus werden dann die durch Immunisierung aktivierten, milz-ständigen BZellen entnommen und in vitro kultiviert. Im nächsten Schritt wird zu diesen B-Zellen eine MyelomZelllinie gegeben, und die zwei werden miteinander vermischt. Danach werden die Zellen in dieser gemischten Kultur miteinander zu zufälligen Fusionen gebracht. Die Zellfusion wird meistens mit Polyethylenglykol (PEG) begünstigt, obwohl heute weitere, alternative Methoden, wie die der elektromagnetischen Fusion, usw. auch immer öfter benutzt werden. PEG ist ein starker Dipol, der die räumliche Orientierung der Wassermoleküle um die Plasmamembran der Zellen herum verändert, und dadurch eine Fusion zweier PMs ermöglicht. Da die Fusionen rein zufällig ablaufen, wird der Anteil der eigentlich gewünschten B-Zell - Myelomzell Fusionszellen, der sog. Hybridomen, sehr gering, nur ca. 1-2%. Aus diesem Grund müssen die unerwünschten Zellen (nicht-fusionierte B-Zellen und nicht-fusionierte Myelomzellen), vor den weiteren Schritten des Prozesses durch Selektion eliminiert werden. Die Selektion wird mittels eines speziellen Zellkulturmediums, des sog. HAT-Mediums (Hypoxanthin218 Aminopterin-Thymidin) ausgeführt. Das HAT Medium blockiert durch seinen Aminopterin Inhalt den klassischen Weg des Folsäure-Metabolismus und dadurch die DNA-Synthese. Andererseits aber, durch die Zugabe von Hypoxanthin und Thymidin, schafft es auch alle Voraussetzungen für einen alternativen Weg der DNA Synthese (sog. salvage pathway), welcher aber das Enzym ThymidinKinase (TK) benötigt. Da die nicht fusionierten Myelomzellen für genau diesen alternativen Stoffwechselweg Mangelmutanten sind (TK -/-), sterben sie in diesem Medium rasch ab. Die nicht fusionierten B-Zellen sterben zwar auch, aber etwas später ab, weil sie wie alle gesunden Zellen, eine begrenzte Lebensdauer haben. Sie werden also einige Wochen in der Kultur nicht überleben können. Mit anderen Worten können eine HAT-Selektion nur die immortalisierten und gleichzeitig auch mit einer aktiven Thymidin-Kinase ausgestattete Zellen, also die Hybridomzellen, eine längere Zeit überstehen. Diese Hybridomzellen können dank der Myelomzellen-Gene als jede Zellinie, unbegrenzt weiter kultiviert und in industriellen Mengen vermehrt werden. Andererseits, dank ihrer B-Zellen-Gene, werden die Hybridomzellen auch die Antikörper des B-Zell-Fusionspartners kontinuierlich sezernieren. Diese Antikörper werden die selbe Spezifizität und Isotyp vorweisen, wie die ursprüngliche, mit dem Antigen in der Maus immunisierte B-Zelle. Diese Zellen werden dann mit weiteren Techniken weiter selektiert um die besten von ihnen, d.h. die Antikörper mit der besten Affinität zum Antigen zu finden, und massenweise vermehrt. Dies ist möglich 1) in konventionellen in vitro Kultur der Hybridomzellen 2) in einer Massenkultur mit Hilfe eines Bioreaktors (auch Fermenter genannt) oder 3) durch Implantation der Hybridomzellen in eine andere Maus. In den ersten zwei Fällen werden die monoklonalen Antikörper vom Überstand der Kulturen, im dritten aus der peritonealen Flüssigkeit der Maus zurückgewonnen. Die Rolle der Gentechnologie in der Herstellung von monoklonalen Antikörpern Mit gentechnologischen Methoden ist es gelungen, aus monoklonalen Maus-Antikörpern solche Antikörper zu erzeugen, die in vielen Eigenschaften schon fast menschlich sind. Diese Antikörper sind entweder in allen Bereichen des Antikörpers bis auf die variablen Domänen, (Chimära-Antikörper) oder in allen Bereichen des Antikörpers bis auf die CDR Regionen der variablen Domänen, (humanisierter Antikörper) mit menschlichen Sequenzen ersetzt. Neulich wird auch mit solchen Technologien experimentiert, die vollkommen menschliche Antikörper erzeugen könnten. Im Vergleich mit Maus Antikörpern monoklonalen werden gentechnologisch diese veränderten Antikörper vom menschlichen Immunsystem viel seltener als fremd erkannt. Abbildung 13.7. Klassifizierung und Nomenklatur der monoklonalen Antikörper nach Herkunft und Ähnlichkeit zu menschlichen Antikörpern 219 Dies ist ein riesiger Vorteil weil sie deswegen viel weniger Komplikationen erzeugen und im Körper des Patienten auch nur langsam abgebaut werden. 13.4.1.2. Risiken und Nebenwirkungen der monoklonalen Antikörper-Therapien Die erste Generation der damals noch vollständig Maus monoklonalen Antikörper hatte noch viele praktische Probleme welche ihre industrielle Produktion und routinemäßige Anwendung erheblich komplizierte. Diese Maus Antikörper haben nämlich in den menschlichen Patienten oft starke Immunreaktionen induziert, welche das Leben der Patienten durch eine akute Hypersensitivität vom Typ I und einem davon hervorgerufenen anaphylaktischen Schock bedrohte. Zweitens haben diese Immunreaktionen auch die langfristige Wirksamkeit der der monoklonalen Antikörper gefährdet, weil gegen sie relativ schnell neutralisierende menschliche anti-Maus Antikörper (HAMA) und ein immunologisches Gedächtnis entstanden sind. Um diese Nebenwirkungen und inhärenten Schwierigkeiten der Therapie in den Griff zu bekommen, hat man einerseits versucht alle, für die therapeutische Funktion unnötigen Teile dieser Antikörper mit menschlichen Sequenzen zu ersetzen: so entstanden die ersten Chimäre und humanisierten Antikörper. Anderseits hat man, wenn möglich, die Maus-Segmente sogar völlig entfernt, wie es bei den therapeutischen monoklonalen Antikörper-Fragmenten (Fab2 Dimere, Fab Monomere, scFv) der Fall ist. Die kleine Molekülgröße der Antikörperfragmente hatte auch einen zusätzlichen Vorteil: sie verbesserte Penetranz auch von die diesen Antikörpern in die Gewebe, welche sehr wichtig ist in der Therapie von soliden Tumoren. Dies kommt jedoch zu einem Preis: mit der verminderten Größe kommt nämlich auch eine schnellere Elimination, welche die Halbwertszeit der Antikörper therapeutische und ihre Effektivität vermindert. Abbildung 13.8. Klassifizierung Nomenklatur der monoklonalen Antikörper nach Herkunft und Ähnlichkeit zu menschlichen Antikörpern 13.4.1.3. Therapeutische monoklonale Antikörper in der Rheumatologie Monoklonale Antikörper werden neben der Onkologie auch in anderen Bereichen der Immunotherapie, wie zum Beispiel in der Immuntherapie der Autoimmunerkrankungen oder der Rheumatologie mit Erfolg benutzt. In der Rheumatologie ist das Zytokin TNF einer der wichtigsten Zielpunkte der monoklonalen Antikörper, da er als proinflammatorisches Zytokin in den meisten rheumatischen Entzündungen eine kritische Rolle spielt, und durch mehrere Mechanismen in der Lage ist, Entzündungen des synovialen Raumes zu induzieren oder verstärken. 13.4.1.3.1. Der Pathomechanismus der rheumatoiden Arthritis: die Rolle der proinflammatorischen Zytokine Rheumatoide Arthritis (RA) ist eine systemische Autoimmunerkrankung, die sich vor allem mit dem typischen Gelenkschmerz und Schwellungen der Finger und Zehengelenke manifestiert. Die Patienten beschweren sich zuerst oft über eine Morgensteifigkeit der Hände, dessen Dauer mit der Aktivität der Krankheit eine positive Korrelation zeigt. Diese Symptome erscheinen meistens symmetrisch, und können sich individuell, mit der Zeit, zusätzlich auf die Handgelenken, Ellbogen und Knie ausbreiten. In den betroffenen Gelenken entstehen chronische Entzündungen, und die dadurch verstärkte Osteoklasten-Aktivität führt zu typischen Erosionen der Gelenkstruktur, die in Röntgen- oder MRAnalysen leicht erkennbar sind. Interessanterweise bleibt die Wirbelsäule fast immer verschont, bis auf das Atlantoaxialgelenk, wo das entzündete, proliferierende Gewebe eine Rückenmarkkompression verursachen kann. In der RA werden eine Vielzahl von Entzündungsmediatoren freigesetzt: zu den charakteristisch sezernierten proinflammatorischen Zytokinen gehören IL-1, IL-6, IL-17, IL-18, IL-33 und TNF, und die in der RA typischen anti-inflammatorischen Zytokine sind IL-10 und IL-35. In den aktiven Stadien der RA können meistens erhöhte Blutsenkung und hohe CRP Werte im Serum gemessen werden. Typisch sind auch Autoantikörper wie z.B. Rheumafaktoren und anti-zyklische citrullinierte Peptide (anti-CCP: siehe in Kapitel 10 im Detail). Letzten Endes führen diese starken chronischen Entzündungsprozesse zur Destruktion und begrenzter Funktionalität der Gelenke: die RA ist eine der häufigsten Ursachen der begrenzten Arbeitsfähigkeit in den europäischen Ländern. Ungefähr 60-70 Prozent der Patienten sind Frauen, deren ersten Symptome erscheinen meistens im Alter von 30-40 Jahren. Rauchen ist ein wichtiger Risikofaktor. 13.4.1.3.2. Die zentrale Rolle des TNFs in der RA Die Hauptfunktion von TNF ist der Schutz des menschlichen Körpers vor Krankheitserregern durch die Aktivierung von lokalen und systemischen akuten Entzündungsreaktionen. Der Name TNF (Tumornekrosefaktor) hat eine lange und komplizierte Geschichte hinter sich. In 1890 ist es dem Chirurgen William B. Coley in New York zuerst aufgefallen, dass in schweren, inoperablen Krebspatienten manchmal erstaunliche Heilungen stattgefunden haben, oft gleich nachdem der krebskranke Patient eine schwere bakterielle oder virale Infektion überstanden hat. Er fing deshalb an, manche seiner Krebspatienten absichtlich mit solchen Erregern zu infizieren. Coley hat festgestellt, dass die antitumorale Wirkung dieser akuten Infektionen in bestimmten, seltenen Fällen auch so, künstlich induzierbar war. Heute weiß man, dass die von schweren Infektionen induzierten akuten Entzündungen, unter anderem durch die Freilassung von TNF, tatsächlich manchmal die chronische Entzündung des Tumorstromas und die dadurch erzeugte Immuntoleranz durchbrechen, und eine Abstoßung des Tumors erzeugen können. Erst in 1985 ist es aber Bruce A. Beutler klar geworden, dass das hormon-artige Protein namens Cachexin, das die Abmagerung der Tumorpatienten (Kachexie) erzeugte, und der Schock-induzierende Entzündungsmediator namens TNF, das ein und 221 selbe Molekül sind, und so erhielt der Name TNF endlich seinen Platz im Kanon der Zytokinen. Für seine Arbeit hat Beutler in 2011 den Nobel Preis in Medizin erhalten. Neben Infektionen und Krebs ist TNF auch in vielen autoimmun-inflammatorischen Krankheiten, wie z.B. in der RA überexprimiert. Nichts desto trotz blieb für die Therapie von RA TNF als ein möglicher Zielpunkt für eine lange Zeit unbekannt, und selbst bis zum Anfang der ‘90er blieben die Steroide, die nichtsteroidalen Antiphlogistika und die Zytostatika die wichtigsten Mittel in den Händen der Mediziner. Die ersten Versuche mit einer völlig neuen Methode, die eine Blockade von Zytokinen erzielte um die Entzündung zu verringern, haben erst in den ’80ern stattgefunden. Marc Feldmann und Sir Ravinder Maini haben beobachtet, dass die aus RA-Patienten entfernten synovialen Gewebeproben in einer Zellkultur noch tagelang große Mengen von TNF und IL-1 freigesetzt haben. Ähnlich dem TNF ist IL-1 auch ein Entzündungsmediator, welcher zu den typischen Erosionen der RA auch einen wichtigen Beitrag leistet. Die Gruppe hat sowohl anti-TNF, als auch anti-IL-1 Antikörper ausprobiert um die Symptome zu behandeln, und es stellte sich heraus, dass in der Zellkultur die effektivsten die antiTNF Antikörper waren, die sogar auch die Sekretion von IL-1 indirekt hemmen konnten. Später haben auch klinische Untersuchungen die ausgezeichnete therapeutische Aktivität der anti-TNF Antikörper in der RA erwiesen, und heute ist die anti-TNF Therapie weltweit etabliert in der Behandlung der RA. Feldmann und Maini erhielten für ihre Arbeit den Albert Lasker Preis in 2003. 13.4.1.3.3. Gängige Methoden der TNF Blockade Die wichtigsten, für die klinische Anwendung genehmigten, TNF-blockierende Medikamente sind 1: der humanisierte Maus anti-TNF monoklonale Antikörper Infliximab (für i.v. Verabreichung) 2: der erste vollkommen humane monoklonale Antikörper, der Anti-TNF monoklonale Antikörper Adalimumab (s.c. Verabreichung) 3: das PEG-konjugierte monoklonale Fab Antikörper-Fragment Certolizumab pegol (s.c.), 4: der vollkommen menschliche monoklonale Antikörper Golimumab (s.c.), 5: und der als Fusionsprotein eines löslichen TNF-Rezeptors und eines Immunglobulin Fc-Teils synthetisierter Etanercept (s.c.) In Europa erhalten jährlich Zehntausenden RA-Patienten anti-TNF Medikamente, die Therapie wird aber von der Gesundheitskassen oft nur dann unterstützt, wenn klassische Behandlungen (z.B. Methotrexat) sich als nicht genügend wirksam erstellt haben. 13.4.1.3.4. Nebenwirkungen von TNF-blockierenden Medikamenten Die wichtigste Nebenwirkung der TNF-Blockade ist eine erhöhte Anfälligkeit für praktisch allerlei bakterielle, virale, und Pilzinfektionen, von denen viele zu den opportunistischen Infektionen gehören. Die häufigsten sind Infektionen der oberen Atemwege, wobei die wichtigste Komplikation die Tuberkulose ist, vor allem ihre extrapulmonale Form. Das Risiko der Tuberkulose ist soweit erhöht, dass alle mit anti-TNF Medikamenten behandelten RA Patienten kontinuierlich unter pulmonologischer Kontrolle stehen müssen. Es wird angenommen, dass die TNF Blockade vor allem durch die indirekte Hemmung der IFN Expression das Risiko von TBC steigert. Das von aktivierten Th-Zellen freigesetzte IFN ist von kritischer Bedeutung in der Aktivierung der TBC-infizierten Makrophagen und in dem Aufbau von Granulomen, also letzten Endes in der Isolierung und erfolgreichen Beseitigung des Erregers. Bei vielen, mit anti-TNFs behandelten RA Patienten ist ohne diese Funktionen die Verteidigung des Körpers gegen Tuberkulose grundlegend kompromittiert worden. 222 13.4.1.3.5. TNF-Blockade in anderen rheumatologischen Erkrankungen Weil TNF neben RA auch in der Pathogenese von vielen rheumatologischen Krankheiten eine Schlüsselrolle spielt, wird die TNF-Blockade auch in anderen Bereichen der Rheumatologie in der täglichen Routine angewandt. Beste Beispiele für solche Krankheiten sind vielleicht die Behandlung der SPA (Spondylitis ankylosans, für eine detaillierte Beschreibung siehe Kapitel 10.2.5), und die der juvenilen Arthritis. 13.4.1.3.6. Entzug von B-Zellen mit Hilfe von anti-CD20 monoklonalen Antikörpern B-Lymphozyten spielen auch wichtige Rollen in der Pathogenese von RA. Neben ihrer AutoantikörperProduktion, wie die des Rheumafaktors oder der anti-CCP Antikörper, präsentieren sie auch die von ihnen erkannten Autoantigene für T-Zellen, und sezernieren Zytokine, welche die Krankheit verstärken. In der RA sind Rheumafaktoren und anti-CCP Antikörper beide prognostische Marker, aber ihre Konzentration zeigt keine Korrelation mit der stets schwankenden Aktivität der Krankheit selber. Entzug der B-Zellen durch Antikörper, welche ihre charakteristischen CD20 Marker erkennen, ist eine ausgezeichnete therapeutische Strategie in RA. Der intravenös verabreichte Maus-Mensch Chimäre monoklonale anti-CD20 Antikörper Rituximab verursacht eine selektive Suppression der BZellen durch ihre komplement- oder ADCC-vermittelte Lyse. Nach einer Behandlung dauert es einige Monate, bis CD20+ B-Zellen erneut in relevanten Mengen im Körper auftauchen, weswegen die Behandlung in der Regel nur zweimal im Jahr durchgeführt werden muss. Da CD20 auf fast allen BZellen, jedoch nicht auf Plasmazellen vorhanden ist, sollte die Therapie komischerweise die autoantikörper-produzierenden B-Zellen eigentlich nicht einmal eliminieren können. Dennoch wurde nachgewiesen, dass die Ritumximab-Therapie die Konzentration der Autoantikörper stark verringert, ohne dabei die meisten physiologischen Antikörper zu beeinflussen. Man vermutet, und es gibt einige Beweise dafür, dass die Erklärung für diesen Widerspruch einfach eine abnormal hohe Expression des CD20 auf autoreaktiven Plasmazellen in RA ist, welche sie für die Therapie viel empfindlicher machen als normale Plasmazellen es sind. Was die Nebenwirkungen angeht, macht die RituximabBehandlung die Patienten auch deutlich empfindlicher für manche Infektionen. 13.4.1.3.7. Hemmung der T-Zell-Aktivierung mit CTLA-4 Immunglobulin Fusionsproteinen T-Lymphozyten sind auch gute therapeutische Zielpunkte in der Immuntherapie der RA. Zu diesem Zweck wird ein Fusionsprotein bestehend aus CTLA-4 und dem Fc-Teil eines Immunglobulins angewandt (Abatacept). Der Zugabe des Ig Fc Teils ist benötigt, damit das integrale PlasmamembranProtein CTLA-4 auch als von der Membran separiertes lösliches Serumprotein, also intravenös verabreichtes Medikament in einer Lösung stabil bleibt. Da der Fc Teil an sich unerwünschte EffektorFunktionen (z.B. Komplement-Aktivierung) auslösen könnte, wurden diese Funktionen in der zweiten Generation der Abatacept-Therapie durch gezielte Mutationen inaktiviert. Der Prinzip der Therapie ist relativ einfach: das Fusionsprotein bindet mit seiner CTLA-4 Untereinheit zum B7 Molekül der antigenpräsentierenden Zellen. Dank der Tatsache, dass CTLA-4 eine höhere Affinität für B7 vorweist als CD28 auf T Zellen, verringert das Fusionsprotein kompetitiv die Kostimulation und so die erfolgreiche Aktivierung der T-Zellen (Abbildung 13.9-10.). Abatacept ist zugelassen für die Therapie von RA, wird als Infusion verabreicht, und wegen ihrer stark suppressiven 223 Wirkung auf T-Zellen erzeugt sie auch eine erhöhte Empfindlichkeit für verschiedene Infektionen in den behandelten Patienten. Abbildung 13.9. Prinzip der Abatacept-Therapie 224 Abbildung 13.10. Das Abatacept Fusionsprotein Übersetzt von Zoltán Pós 225 14. EINIGE IMMUNOLOGISCHE ANWENDLUNGSMÖGLICHKEITEN DER INFORMATIK (EDIT BUZÁS) Dieses Praktikum soll hauptsächlich einen Überblick über das Themaverschaffen, und ein Konzept versucht über die bioinformatischen und web-Applikationen mit der Hilfe von konkreten Beispielen vorzustellen. Ein wichtiger Ausgangspunkt (Startseite) ist die Homepage von National Center for Biotechnology Information (NCBI, www.ncbi.nlm.nih.gov), die nach der Erkennung der wissenschaftlichen Bedeutung von computerisierter Datenverarbeitung in der Medizin erstellt wurde. Seit 1988 funktionierte sie als die Abteilung National Library of Medicine (NLM), in den Rahmen von National Institutes of Health (NIH), die von der US-Regierung unterstützt wird. Das NCBI unter anderen etabliert Datenbanken, sorgt für das Aufrechterhalten und koordiniert die Zugangsberechtigung für bestimmte Daten und Software. Durch die standardisierten Datenformen werden die Speicherung und der Informationsaustausch zwischen den Benutzern ermöglicht. Auf der Startseite von NCBI soll man oben im Klappfenster „Search“ die PubMed Datenbasis, die eine Datenbank von publizierten biomedizinischen Artikeln ist, auswählen. Ins unterliegende breite Fenster tippen wir den gefragte Ausdruck oder Ausdrücke, nach denen wir in der Datenbank suchen möchten. Sucht man z. B. nach dem Wort „immunity“, findet man zirka 300.000 Übereinstimmungen. Wenn man den Titel des ersten Artikels anklickt, befindet sich auf der geöffneten neuen Seite eine Zusammenfassung (das Abstract), und ein auf der oberen rechten Seite liegendes Piktogramm (icon) leitet zu dem Volltext weiter (kostenlos erreichbar sind z. B. „Free PubMed article in PubMed Central” oder „Royal Society Publishing fulltext available” usw), dann kann man ihn herunterladen. Natürlich können nicht alle Artikel auf diese Weise erreicht werden, sondern nur die Abstrakte (Zusammenfassungen) der Herausgabe können in vielen Fällen gelesen werden. 226 Wenn man sich hauptsächlich über die immunologischen Zeitschriften informieren möchte, steht eine Liste hier zur Verfügung. Klickt man den Name der Zeitschrift an, gelangtman direkt auf die Homepage des Journals. Es ist sogar auch möglich, die sog. Meta-Datenbanken aufzusuchen, die selbst Sammlungenanderer Datenbanken sind. Ein Beispiel hierfür, im Rahmen derimmunologischen Thematik, ist die BioMed Central Datenbank, in der man z. B. auf dbMinor (Minor Histocompatibility Knowledge Database) oder Defensins Knowledgebase spezialisierte Wissensdatenbanken findet. Auf diesen Links können viele Informationen (auf überraschender Weise) über die Defensine und über minor Histokompatibilitätsantigene gefunden werden; hier kann die jährlichen Ausdehnung der Fachliteratur verfolgt werden. Zahlreiche Allergen-Datenbanken können auch über das Internet erreicht werden. Nach Registrierung kann man einen eigenen Login-Name und Password erhalten. Suchen wir die vorliegenden Informationen über Derp1 Allergen auf! Ein sehr wichtiges Modellsystem sind die genetisch modifizierten Organismen für die in vivo immunologischen Experimente, in den ein oder gleichzeitig mehrere Gene ausgeschaltet oder überexprimiert werden, damit die Bedeutung bestimmter Moleküle beurteilt werden kann. Der häufigste verwendete Modellorganismus ist die Maus in der Immunologie. Auf dieser URL befindet sich eine Datenbank der genetisch modifizierten Mausstämme, in dem z.B. die beta-2-mikroglobulin knock-out (KO) Mäuse ausgewählt werden können. Das beta-2-mikroglobulin Protein bindet üblicherweise an den alpha-Ketten von MHC I oder CD1 Molekülen. Der genetische Mangel verursacht den Fehler des funktionsfähigen MHC I, was zum Fehlen der CD4-CD8+ zytotoxischen TZellen führt. 227 Die Datenbank für kommerzielle Antikörper kann man in der Praxis sehr gut anwenden: z. B. http://www.antibodyresource.com/. Wenn man auf den Link klickt, erreicht man nicht nur die Angebote und die Homepage der Hersteller, die in ihren Katalogen unterschiedliche Antikörper in nichtkonjugierten oder z. B. mit Enzym gekoppelten Formen anbieten, sondern auch solche Hersteller, die sog. Custom Antibody Serviceleistung zur Verfügung stellen. Diese Firmen stellen solche Antikörper nach Bedarf der Besteller her, gegen deren spezifischen Zielantigene keine Antikörper auf dem Markt zur Verfügung stehen. Mit der Benutzung der bioinformatischen Werkzeuge zur Epitop-Prediktion werden die für gegebenes Protein spezifischen Sequenzen ausgewählt, die voraussichtlich die stärkste Antigenität zeigen. Aufgrund dieser Informationen wird ein synthetisches Peptidhapten hergestellt, mit dem Versuchstiere immunisiert werden und mit dessen Hilfe poly- oder monoklonale Antikörper produziert werden. Es ist auch möglich, das Antigen als rekombinantes Protein zur Immunisierung zu exprimieren, bzw. DNA-Immunisierung zur Antikörperherstellung durchzuführen. Fast 200 ähnliche, je nach Bedarf Antikörper herstellende (custom antibody service) Firmen bieten ihre Serviceleistungen für den Benutzer an. Wenn man nicht aus dem Anwendungsbereich von „Custom antibody suppliers” wählt, sondern man auf „Catalog antibody suppliers” klickt, wirdwieder ein umfangreiches Angebot zur Verfügung stehen. Unter diesen bietet z. B. die Firma Abcam mehr als 40.000 Artikeln an, die nach Themen auf der Homepage der Firma gruppiert werden. Nicht nur einzelne primäre und sekundäre Antikörper und entsprechende Isotypekontrollen, sondern auch z. B. custom antibody arrays stehen hier zur Verfügung. Im Fall von dieser letzten Möglichkeit wird eine Matrix von gewählten Antikörpern gegen unterschiedliche Antigene hergestellt. Es ist aber auch möglich, Probematerial einzuschicken (z.B.: aus >1x10 Zellen, die >100 g Proteinextrakt bedeutet, 6 oder >10 mg Gewebsprobenmaterial, oder >20 l Serum oder >100 g extrahierte Proteinlösung) mittels dem die Firma ein Antikörperarray erstellt. In so einem Fall, wirkt die Firma als reinen Dienstleister mit. Die IMGT (THE INTERNATIONAL IMMUNOGENETICS INFORMATION SYSTEM®) wurde von Marie-Paule Lefranc in 1989 vorgeführt, um die Immunglobuline, T-Zelle Rezeptoren, MHC-Molekülen 228 in ein informatisches System zu integrierten. Die Datenbanken, Webquellen und online-Dienste können einen vielfältigen Bedarf decken. In der Datenbank für Immunglobulin-Strukturen kann man die variablen Regionen (CDR-Regionen) von leichten Ketten bestimmter monoklonaler Antikörper untersuchen, wie z.B. bei den 17B, 3D6 oder 9E genannten Antikörpern können die Aminosäuresequenzen von CDR1, CDR2 und CDR3 Regionen mit bunten Buchstaben gezeichnet werden. Die in Klammern stehenden Nummern, z.B. (27-38) zeigen die Ausdehnung bestimmter CDR-Regionen. Diese CDR1-Region enthält maximal 11 Aminosäuren, die zwischen den 27. und 38. Positionen liegen. Die Sequenz kann z.B. ESVSSD sein und besteht so aus 6 Aminosäuren. Auf dieser Seite kann man unten sehen, dass dieser Bereich auch länger sein kann. Auf einer anderen Seite werden die Antikörper-Moleküle als perlenschnurförmiges (Collier-de-Perles) 2D-Modell dargestellt, in dem die Aminosäuren, die zu den CDR-Regionen gehören, als bunte Perlen markiert werden. Wird die Länge der Aminosäure-Kette von z.B. CDR3-Regionen verändert, kann das Modell durch das Klicken auf den Button DRAW wiedergezeichnet werden. Die Datenbank Immunepitope (IEDB) enthält experimentell verifizierte und charakterisierte Epitope der B- und T-Zellen und die Ergebnisse der Bindungs- und Ligand Elution-Experimente mit MHC. Hier werden nicht nur die molekularen Strukturen von den Zellen der adaptiven Immunantwort zusammengefasst, sondern auch die experimentellen Umstände, unter denen die Daten gewonnen wurden. In dieser Datenbank finden sich die Epitope des Immunsystems des Menschen, nichtmenschlichen Primaten, Nagetieren, Schweinen, Katzen und noch anderen. Während der letzten vier 229 Jahre wurden 180.978 Experimente nach der publizierten Literatur grundlegend analysiert, die ~ 99 % von öffentlichen Daten bedeutet. Daneben wurden 129.186 experimentale Daten, die direkt von den Forschern eingeschickt wurden, auch verarbeitet. Etliche Anwendungen zu Prädiktion von T-und B-Zelle Epitopen und für die Analyse von Epitopen sind von der IEBD zur Verfügung gestellt. Im Fall von den letzten ist es möglich, die Populationshäufigkeit zu extrahieren, eine Analyse von Sequenzkonservation oder Clusteranalyse von denEpitopen durchzuführen. Auf der Seite von MBIM (Microbiology and Immunology resources) können unter anderen die Informationen über CD-Antigene (cluster of differentiation) von CD1 bis CD247 finden. Über die MetaDatenbank von University of Pittsburgh und UPMC können auch viele andere immunologische Datenbanken und Prädiktions-Servern, wie z.B. EVALLER, die die potenziellen AllergenEigenschaften eines Proteinantigens vorhersagen können, oder IIDB (The Innate Immune Database) erreicht werden. In den folgenden konkreten Beispielen wird die Untersuchung einer potentiellen Antigenität illustriert. Wenn wir uns dafür interessieren, welche Antigene als potenzielle Autoantigene bei Rheumatoider Arthritis in Frage kommen können, können die publizierte Daten auf der PubMed Webseite durchsucht werden. Wenn Sie im Suchfenster „candidate autoantigens early rheumatoid arthritis” eingeben, steht der folgende gefundene Artikel frei zur Verfügung. Candidate autoantigens identified by mass spectrometry in early rheumatoid arthritis are chaperones and citrullinated glycolytic enzymes.Goëb V, Thomas-L'Otellier M, Daveau R, Charlionet R, Fardellone P, Le Loët X, Tron F, Gilbert D, Vittecoq O. Arthritis Res Ther. 2009;11(2):R38. Epub 2009 Mar 10. (link) Klicken wir auf den Titel, erreichen wir den Abstrakt des Artikels, in dem die in der frühen Rheumatoid Arthritis durch Massenspektroskopie identifizierte Autoantigene zusammengefasst werden, unter den das alpha-Enolase Enzym inbegriffen ist. Brauchen wir mehrere Informationen darüber, wählen wir den durch die NCBI-Seite erreichbaren Link der Protein Datenbank neben dem Suchfenster. Wenn wir „alpha-Enolase“ eintragen, werden wireine ziemlich grosse Menge an Treffernbekommen. Unter diesen soll man die menschliche alpha-Enolase (species: Homo sapiens) auswählen. Auf der 230 geöffneten Seite befindet sich eine standardisierte Bezeichnung vom alpha-Enolase Protein mit der Anzahl der beteiligten Aminosäure (366), und grundlegenden Publikationen über das Molekül (Titel, Autor, Zeitschrift, Jahr, Band, Seite und Link). Außerdem kann man auch die verfügbaren Informationen über die Domän-Struktur des Proteins (wie die Lokalisation von z.B.) und schließlich die Reinfolge der Aminosäuren finden. Wenn wir wissen möchten, wie das Protein konserviert ist, um ähnliche mikrobielle Sequenzen zu finden, die ebenfalls eine Autoimmunreaktion mit alpha-Enolase, auf Grund molekularen Mimikris auslösen, können wir sie dort finden. Um die Frage zu testen, bleiben wir noch auf der Startseite von NCBI, und wählen wir unter Säule „Popular resources” rechts denLink von BLAST. BLAST ist ein Acronym aus den ersten Buchstaben von „Basic Local Alignment Search Tool”, und ermöglicht die Suche und die Ausrichtung der Sequenzen von Nukleotiden oder Aminosäuren. Auf der Seite BLAST soll man „Protein blast“ unter der „Basic Blast“ Option heraussuchen, dann soll man in das geöffnete Fenster „Enter query sequence” unsere Aminosäuresequenz aus der früheren Seite kopieren. Die Sequenz wird 60 Aminosäuren pro Zeile dargestellt, die am Anfang der Zeile liegende Nummern stört nicht, also kann man die Sequenz mit ihnen zusammen übernehmen. Klicken wir auf den Button Blast, wird die Analyse gestartet. Nach kurzer Zeit erscheinen die Suchergebnissen in der Mitte des Bildschirmes, in einem Rahmen befindet sich die graphische Darstellung. Die große Anzahl der untereinander liegenden roten durchgezogenen Linien weisen darauf hin, dass sehr viele komplette Übereinstimmungen vorliegen, also die Aminosäuresequenz von alpha-Enolase während der Evolution stark konserviert geblieben ist. Man kann nach ähnlichen Sequenzen in der Hoffnung darauf suchen, dass eine ähnliche Aminosäureabfolge in den Infektionserregern vorkommen kann, die eine potentielle Rolle mittels eines molekularen Mimikris in der Entstehung der Autoimmunprozesse spielen können. Da wir mit der Sequenz von alpha-Enolase eine Blast-Suche gestartet haben, können wir es unter die ähnlichen Sequenzen überprüfen. In diesem Fall haben wir eine grosse Anzahl von 100% Übereinstimmungen gefunden, unter denen es aber gar keine prokaryotischen Sequenzen gibt. Wenn man die Immunantwort gegen alpha-Enolase untersuchen möchte, und es steht kein gereinigtes oder rekombinantes Protein zur Verfügung, kann die T-Zelle-Antwort auch mit synthetischen Peptiden 231 untersucht werden. Welche Peptidsequenzen muss untersucht werden? Dies kann beantwortet werden, indem wir auf die Seite CBS (Center of Biological Sequence Analysis, Denmark) gehen. Wählen wir die Prediction Server Funktion aus, und führen wir eine Epitop-Vorhersage z.B. mit NetMHCII durch. Hier kann die Sequenz ins Fenster eingeschrieben werden, bestimmte HLA-Allele ausgewählt werden und die Länge von dem Peptid-Epitop eingestellt werden. Der Algorithmus betrachtet die möglichen Peptide nacheinander, und sagt die Affinität der Bindung zur HLA voraus (strong binder, weakbinder). Der NetMHCII 2.2 Server prädiktiert die Bindung des Peptids zu den HLA-DR, HLA-DQ, HLA-DP Molekülen, durch eine Methode, dieauf dem Prinzip der künstlichen neuronalen Netzwerke beruht. Die Peptidbindung kann für 14 HLA-DR Allele (damit werden die 9 bekannten sog. HLA-DR Supertypen abgedeckt), und für 6 HLA-DQ und 6 HLA-DP Allele modellieren werden. Die Prediktionswerte werden in nM IC50 (bei mittlere inhibitorischer Konzentration wird eine halbmaximale Inhibition beobachtet) angegeben, und Peptide werden neben einer hierarchischen Zuordnung mit starker oder schwacher Bindung (strong binder, SB vagyweakbinder, WB) gekennzeichnet. Kopieren/Einfügen wir die ganze Aminosäuresequenz von alpha-Enolase aus der NCBI-Seite ins Fenster von NetMHCII 2.2, dann wählen wir das HLA-DRB1*0404 Allel unter den möglichen aus, das mit der Rheumatoiden Arthritis stark assoziert ist. Klicken wir auf die kleine Box neben dem „sort by affinity” Befehl, und schicken wir die Aufgabe zu dem CBS Server mit dem Button „submit“. Die Ergebnisse der Vorhersage werden in Form einer Liste angezeigt, in der die aus 15 Aminosäuren bestehenden, also zum MHCII-Molekül bindenden Peptide je nach ihrer Affinität geordnet sind. Es ist zu betonen, dass der Algorithmus insgesamt 5 stark bindende (SB) Peptide inmitten des alphaEnolase Enzyms identifiziert. Unter diesen steht auf dem ersten Platz das an der 43. Position mit EVILPVPAFNVINGG anfangendePeptid, das am stärksten zu dem bestimmten HLA vorraussichtlich binden kann. Das Detail „core epitope” von diesem ist VPAFNVING, das für die Erkennung unerlässlich ist. Die Aminosäuren, die anden N oder C terminalen Bereichen neben dem „core epitope” liegen, die sog. flanking regions, sind nicht essentiell für die Erkennung einer Peptidsequenz. 232 Also wir haben das EVILPVPAFNVINGG Peptid gewählt. Auf Ähnliche Art und Weise kann eine Vorhersage von der MHC-Bindung im Fall von anderen Allelen, wie z.B. HLA-DRB1*0101, durchgeführt werden, und der gleiche Peptid-Treffer, als der am stärksten bindende Abschnitt des alpha-Enolase Enzyms wird auch enthalten sein. Die nächste Frage ist, was man im Besitz dieser Information noch weiteres machen kann? Man kann z. B. die Seite einer Ansammlung besuchen, auf dem die Firmen, die synthetische Peptide auf Anfrage herstellen, erhältlich sind. Neben dem synthetischen Peptid aus Alpha-Enolase kann man auch Kontroll-Peptide bestellen, die z.B. die gleiche Länge, die gleiche Aminosäure- Zusammensetzung besitzen, aber zufällige Sequenzen von Aminosäuren enthalten oder Peptide, die von anderen irrelevanten Proteinen stammen. Die hergestellten Proteine können in vitro getestet werden: wenn die Leukozyten, die aus dem Blut von Kranken oder Kontroll-Individuen isoliert wurden, mit dem Peptid behandelt werden, ist die Frage, ob es in den Kranken spezifische T-ZellenAktivierung bzw. die Sekretion spezifischer Zytokine induziert. In diesem Fall wird das Antigen von den Blutzellen der Kranken (z.B. von den Monozyten, von dendritischen Zellen) präsentiert. Eine Alternative ist, wenn man ein MHC-Tetramer (ein Komplex aus vier MHC-Molekülen, z.B. HLA DRB1*0101) von einem Hersteller bestellt, der eine solche Konstruktion erzeugt, in dem ein gegebenes Peptid in den peptidbindenden Teil des MHC-Moleküls enthalten ist, und es auch mit einem Fluoreszenzfarbstoff markiert. Die T-Zellen, die zum Tetramer binden, können einfach in dem peripheren Blut identifiziert werden, wenn z.B. das Tetramer mit einem roten, der für die T-Zellen spezifischen Antikörper (z.B. anti-CD3ε) mit einem grünen Farbstoff markiert ist. Wenn wir uns nicht für von der T-Zelle sondern von der B-Zelle vermittelten Antwort interessieren, dann können wir eine Vorhersage eines B-Zell Epitopes (Link) durchführen. Diese Prädiktion beruht auf einem Konsensus zahlreicher Sequenzanalysen, in denen die Antigenität, die hydrophoben und hydrophilen Eigenschaften, die Flexibilität bestimmter molekularer Regionen, die sekundäre Struktur wie β-Faltblatt-Struktur und die physiko-chemischen Parametern MHC-Tetramer ebenso in Betracht gezogen werden. Man kennt zwei Typen von B- (invitrogen.com) Zelle Epitopen: lineare und Konformationsepitope. Wenn die 3DStruktur des Proteins bekannt ist, ist es auch möglich, das Epitop durch z.B. Oberfläche-Exposition zu identifizieren. In diesem Fall kann die 3D-Struktur des möglichen Epitops dargestellt werden. Die Fertigstellung der Vorhersage von linearen Epitopen wird als Service für den Kunden von der oben genannten Firma angeboten. Die den prädiktierten Epitopen entsprechenden synthetischen Peptide bzw. ihre konjugierten Formen mit einem indifferenten Protein (wie KLH=keyhole limpet hemocyanin; aus der Hämolymphe der Großen Kalifornischen Schlüssellochnapfschnecke gewonnen) oder mit Biotin werden für Immunisierung benutzt, die die Untersuchung der B-Zellen Reaktivität mit einem einfachen ELISA System ermöglicht. 233 Zur weiteren ist es auch möglich, die Funktionen des Immunsystems (z.B. Virusinfektion oder AIDS) mit einem Bioinformatik-Werkzeug zu simulieren. Auf der von Immunogrid kann man unter Simulationen wählen, die verschiedene Vorgänge (z.B. Virus, Tumor, Atherosclerose) beschreiben und modellieren. Unter den Virusinfektionen kann die HIV und die EBV Infektionen simuliert werden. Das Tumor Simulation Modell enthält die Beschreibung des Wachstums und des Brustkrebses. Wenn wir z.B. ein deskriptives Modell wählen, und wir klicken auf die „lymph node WITHOUT infection” Option, dann auf „view results”, können wir die Veränderungen der Lymphknoten verfolgen. Die unterschiedlichen Panele zeigen, wie sich die Anzahlen von dendritischen Zellen, B-Zellen und THelfer-Zellen während des Ablaufs ändern. Das AG stellt die erforderliche Zeit für die Zuführung eines löslichen Antigens bzw. für die Aufladung von dendritischen Zellen mit einem Antigen dar. Wenn das Antigen die Lymphknoten erreicht, erhöht sich die Dichte von Lymphozyten wegen der Einströmung von B- und T-Zellen. Die Mehrheit der Antigene wird von dendritischen Zellen und B-Zellen aufgenommen, das aus dem plötzlichen Anstieg der Anzahl von antigenpräsentierenden Zellen auch sichtbar ist. Die dendritischen Zellen induzieren die rasche Proliferation der T-Helfer-Zellen (gestrichelte Linie auf der Abbildung TH), inzwischen tritt der Anstieg der B-Zellen verzögert auf (gestrichelte Linie auf der Abbildung B), weil sie von der Kostimulation abhängig sind. Solange das Immunsystem neue Antigene detektiert, bleibt es aktiv: die gesamte Anzahl von Lymphozyten wächst monoton fünffach und die Zelle teilen sich, um den verfügbaren Pool aus antigenspezifischen T-Zellen zu verstärken (lange gestrichelte Linie auf der Abbildungen B und TH). Im Rahmen des Immunologie-Seminars und –Vorlesungen wurden zahlreiche Krankheiten erwähnt (wie z.B. CGD: chronic granulomatous disease, asthma bronchiale, APECED-Syndrom: Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy), deren Beschreibung und Zusammenfassung von ihren genetischen Hintergründen sich in der OMIM Datenbank (OMIM: Online Mendelian 234 Inheritance in Man) befindet. Es leitet auf die in dem zweiten Semester gelehrter Genetik und Genomik weiter. Wählen wir auf der NCBI Seite rechts das „popular resources” Link. Hier kann man nicht nur nach Krankheiten suchen, sondern man kann darüber auch Information erhalten, mit welchen menschlichen Krankheiten ein gegebenes Protein bzw. sein kodierendes Gen assoziiert ist. Wenn wir bei unserem Bespiel bleiben, schreiben wir ins Suchfenster alpha-Enolase. Unter vielen anderen Informationen können wir hier lesen, dass alpha-Enolase als ein mögliches Autoantigen von Hashimoto-Thyreoiditis sein kann, aber man hat noch keine Assoziation von Polymorphismen mit den Autoimmunkrankheiten erforscht. Übersetzt von Viktor Molnár 235 ABKÜRZUNGEN 7TM: Rezeptoren – Membranrezeptorfamilie aus Sebenpfad Transmembrandomainen AChR: Acetylcholin-Rezeptor ACT: Adoptiver Zell-Transfer (adoptive cell transfer) ADCC: Antikörperabhängige zellvermittelte Zytotoxizität AID: Aktivierungsinduzierte Cytidin-Deaminase AIRE: Autoimmune Regulator ANA: Antinukleärer Antikörper APC: Antigenpräsentierende Zelle (antigen presenting cell) APECED: Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy BALT: Bronchienassozierte lymphatische Gewebe (bronchus-associated lymphoid tissue) BRM: Modifizierfaktoren von biologischen Antworten (biological response modifiers) BZR: B –Zell-Rezeptor CCL19: Makrophag-inflammatorisches Protein -3-beta (MIP-3-beta) CCL2: Monozyt-chemotaktisches Protein -1 (MCP-1) CCL20: Makrophag-inflammatorisches Protein -3 (MIP-3-alpha) CCL21: Sekundäres lymphatisches Gewebe Chemokin (SLC) CCP: Zyklisches Citrulliniertes Peptid CD: Cluster of Differentiation CD: Cluster of differentiation CD: Cluster of Differentiation CD62: Selektin CPPs: Zell-penetrierende Peptide (cell penetrating peptides) CTLA-4: Cytotoxic T-lymphocyte antigen 4 CXCL12: Stromal cell-derived factor-1 (SDF) CXCL13: B Lymphozyte chemoattractant (BLC) DAMP: Danger associated molecular pattern - DC: Dendritische Zelle (dendritic cell) EALT: Augenassozierte lymphatische Gewebe (eye-associated lymphoid tissues): Conjunctiva [CALT], Lacrimal Duct [LDALT] associated lymphoid tissues) ELISA: Enzyme Linked Immunosorbent Assay ER: Endoplasmatisches Retikulum ERK: Extrazelluläre-signalregulierte Kinase 236 fMLF/fMLP: Formil Met-Leu-Phe GAG: Glucose amino glycane GALT: Darmassozierte lymphatische Gewebe (gut-associated lymphoid tissue ) HAMA: menschliche anti-Maus Antikörper(human anti-mouse antibodies) HLA: Humanes Leukozytenantigen ICAM: Interzelluläres Adhäsionsmolekül (CD54) Ig: Immunoglobulin IL: Interleukin IL-1b: Interleukin 1 beta iNOS: induzierbare Stickstoffmonoxid-Synthase (inducible nitric oxide synthase) IP3: Inozitol tri-phosphate IPEX: Immune dysfunction, Polyendocrinopathy, and Enteropathy, X-linked JAM: Junctionales Adhäsionsmolekül LALT: Larynx-associated lymphoid tissue LFA-1: Lymphozyt-funktionsassoziertes Antigen (CD11a/CD18) MAC: Membrane attack complex MALT: Darmassozierte lymphatische Gewebe (mucosa-associated lymphoid tissue ) MAPK: Mitogen-aktivierte Protein-Kinase MEK: MAP Kinase Kinase MEK3: MAP Kinase Kinase3 MEKK: MAP Kinase Kinase Kinase MG: Myasthenia gravis MTT: 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide MuSK: Muskelspezifische Rezeptor-Tyrosinkinase MV: Mikrovezikel NALT: Nose-associated lymphoid tissue NO: Stickstoffmonoxid (nitric oxide) PAMP: Pathogen-associated molecular patterns PAMP: Pathogen-assoziertes molekuläres Muster PD-L1: Programmed death ligand 1 PECAM-1: Platelet endothelial cell adhesion molecule (CD31) PEG: Polyethylenglykol PIP2: Phosphatidylinositol 4,5-bisphosphate PLC: Phospholipase C PRR: Pattern recognition receptors - Mustererkennungsrezeptoren PSLG-1: P-Selektin Glycoprotein Ligand-1 (CD162) RA: Rheumatoide Arthritis 237 RA: Rheumatoide Arthritis RF: Rheumafaktor RFLP: Restriktionsfragmentlängenpolymorphismus ROI: Reaktiver Sauerstoffsintermediär SALT: Skin-associated lymphoid tissue SLE: (Systemischer) Lupus erythematodes SPA: Spondylitis ankylosans, Morbus Bechterew SPA: Spondylitis ankylosans (Morbus Bechterew) SSOP: sequenzspezifische Oligonukleotid-Probe SSP: Sequenzspezifische Primer TAA: Tumorassoziierte Antigene TCR: T-Zell-Rezeptor TIL: Tumor-infiltrierende T Zelle TLR: Toll-like Rezeptor TMT: Gezielte molekulare Therapie (targeted molecular therapy) TNF: Tumornekrosefaktor TNFa: Tumor necrosis factor TSH: Thyreoidea-stimulierendes Hormon VALT: Vascular-associated lymphoid tissue VCAM: Vascular cell adhesion molecule (CD106) VLA4: Very late antigen-4 = integrin alpha4beta1 (CD49d/CD29) 238