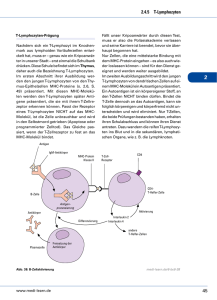
Sevofluran hemmt die DNA-Bindung des Transkriptionsfaktors
Werbung

Aus der Anaesthesiologischen Universitätsklinik der Albert-Ludwigs-Universität Freiburg i. Br. Sevofluran hemmt die DNA-Bindung des Transkriptionsfaktors Aktivator Protein-1 und induziert Apoptose in T-Lymphozyten INAUGURAL-DISSERTATION zur Erlangung des Medizinischen Doktorgrades der Medizinischen Fakultät der Albert-Ludwigs-Universität Freiburg i. Br. vorgelegt 2004 von Patrick Scheiermann geboren in Essen Dekan: Prof. Dr. med. J. Zentner 1. Gutachter: Prof. Dr. med. B. H. J. Pannen 2. Gutachter: Prof. Dr. rer. nat. C. Borner Jahr der Promotion: 2005 Danksagungen: Herzlichen Dank an Prof. Dr. Benedikt Pannen für das spannende Thema dieser Doktorarbeit und die Betreuung sowie Prof. Dr. Christoph Borner für die freundliche Übernahme des Zweitgutachtens. Mein ganz besonderer Dank gilt Dr. Torsten Loop, der mich über die gesamte Zeit äußerst engagiert und zuverlässig betreut hat. Die Zusammenarbeit im Labor hat immer sehr viel Spaß gemacht. Bei Sören Pischke und Tobias Bross möchte ich mich für die ausgezeichnete Zusammenarbeit während der gemeinsamen Laborzeit bedanken. Dr. Matjaz Humar gilt mein Dank für die Unterstützung bei Transfektionen und FACS-Analyse. Ganz besonders bedanken möchte ich mich bei Lotti Egger (Arbeitsgruppe Prof. Dr. C. Borner) für die tatkräftige Unterstützung bei den Arbeiten zur Apoptose. Bei Dr. Philip Hublitz (Arbeitsgruppe Prof. Dr. R. Schüle) möchte ich mich für die Hilfe bei der ELISA-Auswertung bedanken. Für die kritische Bewertung sämtlicher Versuche und Ergebnisse und die äußerst angenehme Zusammenarbeit gilt mein Dank Prof. Dr. Heike Pahl, Niko Andropoulos, Tilo Bächle, David Doviakue, Dr. Alexander Hötzel, Daniel Leitz, Dr. René Schmidt, Armin Welle, Thomas Berger, Christoph Coch, Edith Dörner, Philipp Görttler, Dr. Edgar Kirchner, Dr. Steffen Klippel, Cordula Steimle und Dr. Snezana Temerinac. für Oda, Norbert und Christoph 1 Inhaltsverzeichnis Inhaltsverzeichnis Inhaltsverzeichnis ...........................................................................1 1 Zusammenfassung ...................................................................3 2 Einleitung...................................................................................4 2.1 2.2 2.3 2.4 3 Inhalationsanästhetika.......................................................................... 5 Der Transkriptionsfaktor AP-1 und Lymphozytenfunktion................ 7 Apoptose.............................................................................................. 11 Zielsetzung dieser Arbeit.................................................................... 14 Material und Methoden...........................................................16 3.1 Kultur der Jurkat T-Lymphomzellen .................................................. 16 3.1.1 Material .......................................................................................... 16 3.1.2 Methode ......................................................................................... 16 3.2 Isolation primärer T-Lymphozyten aus peripherem Blut gesunder Spender ................................................................................................ 16 3.2.1 Material .......................................................................................... 16 3.2.2 Methode ......................................................................................... 17 3.3 Versuchsprotokoll ............................................................................... 18 3.3.1 Material .......................................................................................... 18 3.3.2 Methode ......................................................................................... 19 3.4 Nukleäre Extrakte ................................................................................ 20 3.4.1 Material .......................................................................................... 20 3.4.2 Methode ......................................................................................... 21 3.5 „Electrophoretic Mobility Shift Assay” (EMSA) und Kompetitionen ..................................................................................... 22 3.5.1 Material .......................................................................................... 22 3.5.2 Methode ......................................................................................... 25 3.6 Luziferase-Gen-Transfektion.............................................................. 26 3.6.1 Material .......................................................................................... 26 3.6.2 Methode ......................................................................................... 26 3.7 Zytokinbestimmung mittels „Enzyme-Linked Immuno Sorbent Assay“ (ELISA) .................................................................................... 27 3.7.1 Material .......................................................................................... 27 3.7.2 Methode ......................................................................................... 27 3.8 Western Blot ........................................................................................ 28 3.8.1 Material .......................................................................................... 28 3.8.2 Methode ......................................................................................... 29 3.9 „Fluorescence-Activated Cell Sorting” (FACS) für Vollblut ............ 31 3.9.1 Material .......................................................................................... 31 3.9.2 Methode ......................................................................................... 31 3.10 Fluoreszenzmikroskopie ................................................................. 32 3.10.1 Material .......................................................................................... 32 3.10.2 Methode ......................................................................................... 32 3.11 Caspase-Aktivitätsmessung ........................................................... 33 3.11.1 Material .......................................................................................... 33 3.11.2 Methode ......................................................................................... 33 2 Inhaltsverzeichnis 3.12 Statistik............................................................................................. 34 3.12.1 Material .......................................................................................... 34 3.12.2 Methode ......................................................................................... 34 4 Ergebnisse...............................................................................35 4.1 Sevofluran hemmt die PMA/Ionomycin-induzierte DNABindungsaktivität von AP-1 in T-Lymphozyten in vitro.................... 35 4.1.1 Sevofluran hemmt die DNA-Bindungsaktivität von AP-1 ................ 35 4.1.2 Zeitabhängige Hemmung von AP-1 durch Sevofluran ................... 36 4.1.3 Dosisabhängige Hemmung von AP-1 durch Sevofluran ................ 37 4.1.4 Veränderung der DNA-Bindungsaktivität der AP-1-Untereinheiten JunD und c-Jun durch Sevofluran .................................................. 38 4.1.5 Keine Hemmung von AP-1 durch Desfluran und Isofluran ............. 40 4.1.6 Geringer Sevofluran-vermittelter Effekt auf die DNABindungsaktivität von NFAT und NFκB .......................................... 41 4.1.7 Keine Hemmung von SP-1 durch Sevofluran................................. 43 4.1.8 Keine Hemmung von Oct-1 durch Sevofluran ................................ 44 4.2 Sevofluran hemmt die AP-1-abhängige LuziferaseGenexpression in transfizierten Jurkat T-Lymphozyten.................. 45 4.3 Sevofluran hemmt die IL-3-Sekretion in primären Lymphozyten in vitro....................................................................................................... 46 4.4 Sevofluran aktiviert spezifische MAP-Kinasen ................................. 47 4.5 Sevofluran induziert Apoptose .......................................................... 49 4.5.1 Sevofluran induziert morphologische Apoptosemerkmale.............. 49 4.5.2 Sevofluran induziert Annexin V-FITC-Positivität in T-Lymphozyten 50 4.6 Sevofluran induziert Caspase-abhängige Apoptose........................ 51 4.6.1 Nachweis aktiver Caspasen nach Sevofluran-Inkubation in TLymphozyten in vitro ...................................................................... 51 4.6.2 Nachweis aktiver Caspase-3 nach Sevofluran-Inkubation in TLymphozyten in vitro ...................................................................... 53 5 Diskussion...............................................................................55 5.1 5.2 5.3 5.4 5.5 6 Hemmung von AP-1 durch Sevofluran .............................................. 55 Sevofluran und Interleukin-3 .............................................................. 61 Einfluss von Sevofluran auf die MAP-Kinasen ................................. 62 Sevofluran und Apoptose................................................................... 64 Schlussfolgerung ................................................................................ 66 Anhang.....................................................................................68 6.1 6.2 6.3 6.4 6.5 Abkürzungen ....................................................................................... 68 Vektorkarte........................................................................................... 70 Literaturverzeichnis ............................................................................ 71 Wissenschaftliche Leistungen ........................................................... 78 Lebenslauf ........................................................................................... 79 3 Zusammenfassung 1 Zusammenfassung Inhalationsanästhetika verursachen in vitro eine veränderte Sekretion von IL-1β, TNF-α, IL-8 und IFN-γ (32). Der molekulare Mechanismus ist unbekannt. Hypothese dieser Arbeit war, dass Desfluran, Isofluran und Sevofluran über eine Beeinflussung der DNA-Bindungsaktivität von Transkriptionsfaktoren ein verändertes Genexpressionsmuster in primären Lymphozyten in vitro verursachen. Von klinischer Bedeutung ist, dass eine Anästhetika-vermittelte Abnahme immunstimulatorischer Zytokine für eine perioperative Schwächung des Immunsystems verantwortlich sein könnte (10);(52). Die DNA-Bindung der Transkriptionsfaktoren AP-1, NFAT, NFκB, SP-1 und Oct-1 wurde mit PMA und Ionomycin induziert. Im EMSA konnte erstmals eine zeit- und dosisabhängige Hemmung der DNA-Bindungsaktivität des Transkriptionsfaktors AP-1 nach Inkubation mit Sevofluran nachgewiesen werden. Kompetitionsansätze zeigten eine Sevofluran-vermittelte Hemmung der AP-1-Untereinheiten c-Jun und JunD. Eine hemmende Wirkung von Sevofluran auf die Aktivität der AP-1-regulierenden MAP-Kinasen ERK, JNK und p38 konnte im Western Blot nicht nachgewiesen werden. Desfluran und Isofluran beeinflussten die DNA-Bindung von AP-1 nicht. Inkubation mit Sevofluran führte zu geringer Hemmung der DNA-Bindung von NFAT und NFκB und zeigte keine Wirkung auf die DNA-Bindung von SP-1 und Oct-1. Inkubation mit Sevofluran führte im Gegensatz zur Inkubation mit Desfluran zur signifikanten Hemmung der PMA/Ionomycin-induzierten Expression eines in Jurkat T-Lymphozyten transient transfizierten AP-1-abhängigen Luziferase-Reportergenes. Mittels ELISA konnte eine signifikante Sevofluran-vermittelte Hemmung der PMA/Ionomycin-induzierten Sekretion von IL-3 in primären Lymphozyten nachgewiesen werden. Inkubation mit Sevofluran führte zur Aktivierung der p38-MAP-Kinase, die proapoptotische Wirkung haben kann (16). Ebenso induzierte Sevofluran caspase-abhängige Apoptose. Diese Arbeit könnte zum einen dazu beitragen, den molekularen Mechanismus der spezifischen Wirkung von Inhalationsanästhetika auf die lymphozytäre Zytokinsekretion zu klären. Zum anderen scheint es möglich, dass eine Sevofluran-vermittelte Aktivierung der p38-MAP-Kinase zur Induktion caspaseabhängiger Apoptose führt. 4 Einleitung 2 Einleitung In dieser Arbeit sollte untersucht werden, ob Inhalationsanästhetika die DNABindung von Transkriptionsfaktoren beeinflussen. Die Antwort auf diese Frage ist von großer Bedeutung, um spezifische Eigenschaften volatiler Anästhetika untersuchen zu können: So werden Anästhetika unter anderem immunsupprimierende Wirkungen zugeschrieben. Diese Hypothese stützt sich auf Beobachtungen an Patienten (10). Für eine perioperative Immunschwäche können verschiedene Gründe verantwortlich sein (52): Neben der körperlichen Belastung durch den chirurgischen Eingriff könnten Anästhetika die Funktion des Immunsystems beeinflussen. Diese immunmodulatorische Wirkung scheint sehr differenziert und nicht auf einen Angriffspunkt beschränkt zu sein. Volatile und intravenöse Anästhetika unterscheiden sich in ihrer Wirkung auf die Sekretion immunmodulatorischer Zytokine (32). Daher lassen sich die Eigenschaften der Anästhetika immunsupprimierenden Wirkung nicht mit einer zusammenfassen. generalisierten Für intravenöse Anästhetika konnte gezeigt werden, dass sie über eine Hemmung der DNABindung von Transkriptionsfaktoren die zelluläre Immunfunktion beeinträchtigen (37). Sehr unterschiedlich wird der Einfluss von volatilen Anästhetika auf das Immunsystem bewertet. Ein einheitlicher Wirkmechanismus konnte nicht nachgewiesen werden: Während Untersuchungen zeigten, dass in Alveolarmakrophagen der Ratte die Expression der proinflammatorischen Zytokine Interleukin (IL)-8 und Interferon (IFN)-γ unter Narkose mit volatilen Anästhetika zunimmt (32), wird im Gegensatz dazu in peripheren mononukleären Blutzellen (PMBZ) von einem suppressiven Effekt von Inhalationsanästhetika auf die Freisetzung von immunstimulatorischem IL-1β und Tumor Nekrose Faktor (TNF)-α sowie von einer fehlenden Wirkung auf IFN-α berichtet (44). Diese Beobachtungen zeigen zum einen, dass Inhalationsanästhetika je nach Zellart unterschiedliche Wirkungen haben. Andererseits wird deutlich, dass auch innerhalb einer Zellreihe ein sehr spezifisches Genexpressionsmuster vorliegen kann. Daher stellt sich die Frage nach dem molekularen Mechanismus einer Wirkung volatiler Anästhetika auf die Sekretion von Zytokinen. 5 Einleitung Inhalationsanästhetika induzieren Apoptose in Lymphozyten und neutrophilen Granulozyten (13;14;39). Der molekulare Mechanismus ist ungeklärt. Der Transkriptionsfaktor Aktivator Protein-1 (AP-1) sowie die AP-1 regulierende p38-„Mitogen Activated Protein (MAP)-Kinase“ nehmen zentrale Rollen in der Regulation von Zellproliferation und Apoptose ein (16;51;63). Caspasen, eine Gruppe von Serin-Proteasen, sind als Mediatoren der Apoptose für die charakteristische Morphologie apoptotischer Zellen verantwortlich (18). Eine Beteiligung des Transkriptionsfaktors AP-1 und der Caspasen an der Induktion von Apoptose durch volatile Anästhetika wurde bisher nicht untersucht. In dieser Arbeit wurde von der Hypothese ausgegangen, dass Inhalationsanästhetika die Abnahme der lymphozytären Zytokinsekretion durch eine veränderte DNA-Bindung von Transkriptionsfaktoren vermitteln. Dabei sollte auch untersucht werden, ob sich die eingesetzten volatilen Anästhetika in ihrer Wirkung unterscheiden. Zum anderen sollte untersucht werden, ob eine veränderte DNA-Bindungsaktivität des Transkriptionsfaktors AP-1 und aktive Caspasen für die Induktion von Apoptose durch Inhalationsanästhetika mitverantwortlich sind. 2.1 Inhalationsanästhetika In der klinischen Anwendung kommen im wesentlichen drei Inhalationsanästhetika zum Einsatz: Desfluran, Isofluran und Sevofluran. Diese volatilen Anästhetika sind als polyfluorierte Kohlenwasserstoffe auf Diäthylätherbasis lipophil und liegen bei Raumtemperatur (RT) in flüssiger Form vor. Für die klinische Anwendung bedeutet das, dass diese Anästhetika während einer Narkose in einem speziellen Verdampfer vaporisiert werden. Der Siedepunkt von Isofluran liegt bei 48,5°C, der von Sevofluran bei 58,8°C. Desfluran siedet bei 22,8°C, so dass für die Anwendung von Desfluran eine besondere Verdampfertechnologie notwendig ist. Der jeweilige Verdampfer ist über ein Schlauchsystem mit der Frischgaszufuhr verbunden. In diesem System stellt sich ein Gleichgewicht zwischen flüssiger und gasförmiger Phase des Anästhetikums ein. Die Konzentration des Gases oberhalb der Flüssigkeit wird als volumenprozentiger Anteil (Vol%) angegeben. Im klinischen Umgang wird für die Wirkungsstärke der volatilen Anästhetika die spezifische Minimale 6 Einleitung Alveoläre Konzentration (MAC-Wert) verwendet. Der MAC50-Wert eines Inhalationsanästhetikums ist klinisch als die alveoläre Konzentration eines Inhalationsanästhetikums definiert, bei der 50% aller Patienten auf die Hautinzision zu Beginn einer Operation nicht mehr mit Abwehrbewegungen reagieren. Um eine bestimmte Narkosetiefe des Patienten zu erreichen, muss eine Mindestkonzentration des Anästhetikums in der Alveolarluft vorhanden sein, die ein indirektes Maß für den Partialdruck des Anästhetikums im Gehirn darstellt, da im Gleichgewicht Partialdruckgleichheit herrscht. Der MAC-Wert wird in Vol% von 1 Atmosphäre angegeben: je niedriger der MAC-Wert, desto größer die Wirkungsstärke dieses volatilen Anästhetikums. Die MAC-Werte betragen in reinem Sauerstoff für Isofluran 1,28 Vol%, für Desfluran 6-7 Vol% und für Sevofluran 2,05 Vol%. Desfluran und Isofluran unterscheiden sich in ihrer molekularen Struktur durch die Substitution an einem C-Atom, das bei Desfluran ein Fluoratom bindet und bei Isofluran stattdessen ein Chloratom (Abb. 1). Abb. 1: Desfluran, Isofluran und Sevofluran sind polyhalogenierte Diäthylätherverbindungen, die bei RT in flüssiger Form vorliegen und deren Anwendung spezielle Verdampfer erfordert. Desfluran und Isofluran sind stabile Moleküle und werden im Körper nur zu einem geringen Prozentsatz metabolisiert (Desfluran ca. 0,02%, Isofluran ca. 0,2%). Sevofluran unterscheidet sich strukturell: Durch eine komplexere Fluoridierung der Ätherverbindung kommt es zu einer verstärkten hepatischen Metabolisierung (3-5%), in deren Verlauf im Körper anorganisches Fluorid (>10 7 Einleitung µM/l nach 1 MAC-Stunde) anfällt. Desfluran und Sevofluran sind gegenüber Isofluran aufgrund der niedrigeren Blut/Gas-Verteilungskoeffizienten deutlich besser steuerbar, was die Narkoseführung vereinfacht. Sevofluran zeichnet sich gegenüber Desfluran durch einen angenehmeren Geruch aus und reizt die oberen Atemwege deutlich weniger, was es zum bevorzugten Anästhetikum zur inhalativen Narkoseeinleitung in der pädiatrischen Anästhesie macht. 2.2 Der Transkriptionsfaktor AP-1 und Lymphozytenfunktion Der Transkriptionsfaktor AP-1 ist ein Proteindimer, welches aus Untereinheiten mit interagierender, basischer Leuzin-Zipper-Region aufgebaut ist (29). Diese Untereinheiten entstammen folgenden Subfamilien: Die AP-1 Subfamilie zugehörige Untereinheiten Jun c-Jun, JunB, JunD Fos c-Fos, FosB, Fra-1, Fra-2 Maf c-Maf, MafB, MafA, MafG/F/K, Nrl ATF (=CREB) ATF-2, LRF/ATF-3, B-ATF, JDP1, JDP2 Gene dieser Untereinheiten werden durch verschiedene Signaltransduktionswege aktiviert. Dieser Prozess vollzieht sich sehr rasch, so dass der Transkriptionsfaktor AP-1 auch als Teil der „immediate early gene response“ bezeichnet wird (41;48), die eine transkriptionelle Sofortreaktion auf extrazelluläre Signale darstellt (21). Für eine effektive Transaktivierung von AP-1 ist die Dimerisierung essentiell (54). Jun/Fos-Heterodimere und Jun/Jun-Homodimere binden präferenziell an die Sequenz 5’-TGAG/CTCA-3’, die als „12-o-Tetradecanoyl-Phorbol-13-Acetate (TPA) Response Element“ (TRE) bezeichnet wird (29). Damit kommt zum Ausdruck, dass Phorbolesterverbindungen eine DNA-Bindung des Transkriptionsfaktors AP-1 induzieren können (2;35). Jun/ATF-Heterodimere und ATF/ATF-Homodimere binden verstärkt an das „cAMP Response Element“ (CRE), das die Sequenz 5’-TGACGTCA-3’ besitzt (29). Der „Activating Transcription Factor“ (ATF) wird deshalb auch als „cAMP Response Element Binding Protein“ (CREB) bezeichnet. Beide DNA-Sequenzen sind Palindrome, 8 Einleitung d. h. die erste Hälfte der AP-1-Bindestelle (TGAC) stellt die korrespondierenden Basenpaare (bp) der zweiten Hälfte (GTCA) dar. Die AP-1-Dimere unterscheiden sich hinsichtlich Stabilität und transaktivierender Potenz: Jun/Fos-Heterodimere gelten als die AP-1-Komplexe mit der größten Wirkung (49;60), eine Jun/Jun-Homodimerisierung führt zu weitgehendem Verlust der Transaktivation und Instabilität (60). Fos-Untereinheiten bilden im Gegensatz zu ATF keine stabilen Homodimere (24;29). Da Lymphozyten die zentralen Zellen der adaptiven Immunantwort sind und es bei ihrem Funktionsverlust zu schwerwiegenden Defekten der körpereigenen Abwehr kommt, sollen an dieser Stelle die wichtigsten Schritte beschrieben werden, die zur Aktivierung der einzelnen Untereinheiten des Transkriptionsfaktors AP-1 in Lymphozyten führen (21;51): Unter CoStimulation des CD3-Oberflächenantigens (CD3) von T-Lymphozyten führt eine Antigenbindung an den T-Zell-Rezeptor (TCR) über intrinsische Tyrosin-KinaseAktivität (Tyr-K) zu einer Aktivierung der Proteinkinase C (PKC) und der Phospholipase C (PLC). PLC spaltet Phosphatidyl-Ionositol-bis-Phosphat (PIP2) in Diacylglycerol (DAG) und Inositol-tris-Phosphat (IP3). IP3 führt über eine Erhöhung der intrazellulären Kalziumionenkonzentration [Ca2+] zur Aktivierung der Phosphatase Calcineurin, die zum einen direkt den Transkriptionsfaktor Nukleärer Faktor Aktivierter T-Zellen (NFAT), zum anderen möglicherweise auch die c-Jun-NH2-terminale-Kinase (JNK) aktiviert. DAG aktiviert ebenfalls die PKC, die über p21ras eine Kaskade in Gang setzt, die zum einen über die RafKinase (Raf) und die „MAP-Kinase Kinase“ (MEK) die „Extracellular Signal Regulated Protein Kinase“ (ERK), zum anderen über die „MAP-Kinase Kinase Kinase“ (MEKK) und die „Stress Activated Protein-Kinase“ (SEK) direkt die p38MAP-Kinase und JNK aktiviert. Diese MAP-Kinasen-Kaskade wird für die Aktivität der verschiedenen Untereinheiten des AP-1-Komplexes verantwortlich gemacht (56;76). Während diese Mechanismen für die Aktivierung des Transkriptionsfaktors AP-1 ausreichend sind (2), verstärkt eine Co-Stimulation über den CD28-Rezeptor (CD28) und konsekutive Aktivierung von PhosphoInositid-3 (PI3), Phospho-Inositid-3-Kinase (PI3-K) und möglicherweise MEKK die AP-1-abhängige Transkription in Lymphozyten in vitro und in vivo (59). Damit ist die volle Funktion dieser wichtigen Mediatoren für die Immunantwort garantiert. Während in Lymphozyten eine PKC-abhängige Fos-Kinase (Fos-K) 9 Einleitung beschrieben ist, die Fos direkt aktivieren kann (45), wird die Transkription des fos-Genes durch DNA-Bindung vom ERK-abhängigen „Ets-Like Protein-1“ (Elk1) und des „Serum Response Factors“ (SRF) nach Serumzusatz an das „Serum Response Element“ (SRE) induziert. Die Induktion der DNA-Bindung von ATF-2 und c-Jun an die TRE-Sequenz erfolgt über differenzierte Phosphorylierung hauptsächlich via p38-MAP-Kinase (im Falle von ATF-2) und JNK (für c-Jun). Die Chemikalien Phorbol-12-Myristat-13-Azetat (PMA) und Ionomycin können die physiologische, rezeptorvermittelte T-Zell-Aktivierung adäquat ersetzen (38). PMA besitzt eine strukturelle Ähnlichkeit mit DAG. Es kann die PKC direkt aktivieren, welche ihrerseits c-Fos aktiviert, das wiederum einen Teil des AP-1Komplexes darstellt. Das Kalziumionophor Ionomycin stimuliert die JNK über eine Erhöhung von [Ca2+] und führt damit zur Aktivierung von c-Jun, das mit Fos dimerisieren kann (64). AP-1 bindet an spezifische AP-1-Bindestellen (TRE oder CRE) innerhalb der Promotorensequenz, meist in Kombination mit dem Transkriptionsfaktor NFAT (38). AP-1 und NFAT stabilisieren sich durch gemeinsame DNA-Bindung gegenseitig, ein Mechanismus, der ihre transkriptionelle Aktivität deutlich erhöht (Abb. 2). Im IL-3-Promotor liegen die Bindestellen von AP-1 (TGAC/GTCA) und NFAT (GGAAA) (11) nahe beisammen (10 kbp) (38), weshalb das IL-3-Gen AP-1- und NFAT-abhängig transkribiert wird. IL-3 ist ein Zytokin mit stimulatorischer Wirkung auf die hämatopoetischen Stammzellen. Entzug von IL-3 führt abhängig von der Zellart entweder zu einem Wachstums- oder zu einem Differenzierungsstopp (36), kann aber (27)auch Apoptose induzieren (83). Auch die Aktivierung anderer Elemente, die zur DNA-Bindung des Transkriptionsfaktors AP-1 führen, können zur Apoptose führen. Für die JunUntereinheiten von AP-1 sind pro- und antiapoptotische Eigenschaften beschrieben (63), aber auch die Aktivität der p38-MAP-Kinase führte zur charakteristischen Morphologie apoptotischer Zellen (16). 10 Einleitung Abb. 2: Schematischer Ablauf der Aktivierung des Transkriptionsfaktors AP-1: Bindung an den TCR führt über eine intrinsische Tyr-K zur Aktivierung der PLC, welche PIP2 in IP3 und DAG spaltet. IP3 bewirkt über [Ca2+]-Erhöhung eine Aktivierung der Phosphatase Calcineurin, die für die Induktion des Transkriptionsfaktors NFAT nötig ist. Ein möglicher Einfluss Calcineurins auf JNK wird diskutiert. DAG aktiviert die PKC, die entweder c-Fos über die Fos-K ras aktiviert oder über p21 in der Folge JNK aktiviert. Resultat ist eine verstärkte Transkription von c-fos und c-jun, die als c-Fos und c-Jun AP-1 bilden und entweder alleine oder im Komplex mit NFAT an spezifische DNA-Sequenzen (z. B. im IL-3-Promotor) binden. CD28-Co-Stimulation führt über MEKK wahrscheinlich ebenfalls zu JNK-Aktivierung und verstärkter c-jun-Transkription. Eine Stimulation mit PMA und Ionomycin führt unter Umgehung des T-ZellRezeptors direkt zu Aktivierung der PKC und zur [Ca2+]-Erhöhung. 11 Einleitung 2.3 Apoptose Apoptose oder programmierter Zelltod ist ein in der Evolution weitgehend konservierter Prozess, in dessen Verlauf beschädigte, überalterte sowie a- bzw. hyperreaktive Zellen kontrolliert zugrunde gehen (42). Unphysiologische Resistenz gegenüber programmiertem Zelltod führt zu Autoimmunkrankheiten oder Tumoren (23;66), verstärkte Apoptose zu Immunschwäche oder neurodegenerativen Krankheiten (40). Zentrale Mediatoren dieses apoptotischen Zelltodes sind die Caspasen, eine Gruppe von CysteinProteasen, die Proteine nach Aspartat-Resten spalten (18). Obwohl Caspasen auch in gesunden Zellen in gewissem Maße aktiv sind, ist zur vollen Aktivität ein proapoptotischer Stimulus nötig (65). Caspasen werden aufgrund ihrer Funktion in zwei Gruppen unterschieden: Initiator-Caspasen (Caspase-2, Caspase-8, -9, -10 und Caspase-12) sowie Effektor-Caspasen (Caspase-3, -6 und -7) (18). Die Initiator-Caspasen befinden sich am Anfang einer Kette von Signalen, ausgehend von einem proapoptotischen Stimulus. Sie besitzen eine lange (>90 bp) N-terminale Pro-Domäne (auch als „Caspase Recruitment Domain“ oder „Death Effector Domain“ (DED) bezeichnet). Die InitiatorCaspasen liegen in gesunden Zellen als inaktive Monomere vor und werden als Pro-Form durch Adaptoren wie FADD und Apaf-1 dimerisiert (6;17;55). Dieses aktive Initiator-Caspasen-Dimer (18) spaltet dann die in gesunden Zellen bereits dimerisiert vorliegende inaktive Pro-Effektor-Caspase nach einem AspartatRest, was zu einer aktiven Effektor-Caspase (z. B. Caspase-3) und zu einer Verstärkung des proapoptotischen Signals führt. Die Effektor-Caspasen sind verantwortlich für die typische Morphologie apoptotischer Zellen, die aus einem Schrumpfen der Zelle mit Ausstülpung der Plasmamembran besteht und dabei eine Exposition von Phosphatidylserinen auf der Zelloberfläche zeigt (8). Die Zelle degradiert in membranumhüllte „Apoptotic Bodies“, die dann von Makrophagen ohne begleitende Entzündungsreaktion phagozytiert werden (61). Da diese Caspasen-Kaskade unweigerlich zum Zelltod führt, ist eine genaue Regulierung des komplexen Aktivierungsmechanismus erforderlich. Es werden zwei Wege der Caspasen-Aktivierung beschrieben (8): Zum einen führt die Bindung extrazellulärer Liganden der TNF-Superfamilie (TNF-α, FasL/CD95L) Zelloberflächenrezeptoren (33) zur Trimerisierung (TNF-Rezeptor, Fas/CD95). entsprechender Über ihren 12 Einleitung zytoplasmatischen Anteil rekrutieren diese Trimere Adaptorproteine wie FADD oder TRADD. Die sogenannte „Death Domain“ (DD) dieser Adaptorproteine interagiert mit der DED der Initiator-Caspase-8, was zur Dimerisierung und damit zur Aktivierung von Caspase-8 führt. Ein zweiter Mechanismus involviert die Mitochondrien (73) und wird durch die Familie der Bcl-2-Proteine reguliert. Über diesen Weg wird vor allem durch UVund γ-Strahlung, sowie Chemotherapeutika, Mikroorganismen und Entzug von Zytokinen und Wachstumsfaktoren Apoptose induziert. Über weitgehend unbekannte Sensoren kommt es zur Perforation der äußeren Mitochondrienmembran, was zur Freisetzung von Proteinen aus dem mitochondrialen Intermembranenspalt führt (50). Durch Freisetzung von Cytochrom C kommt es zur Oligomerisierung von Apaf-1 und zur Rekrutierung der Initiator-Caspase-9 (1). Dieser große Komplex, der die Wirkung von Caspase-9 erheblich verstärkt, führt zur Dimerisierung und damit zur Aktivierung der Effektor-Caspasen (z. B. Caspase-3) (58). Bcl-2-Proteine verhindern die Perforation der Mitochondrienmembran und damit die Freisetzung proapoptotischer Faktoren wie Cytochrom C ins Zytoplasma (31) (Abb. 3). Der Einfluss der Bcl-2-Proteine auf die Hämatopoese und damit die Funktionalität der Lymphozyten wird durch die Tatsache unterstrichen, dass in transgenen Mäusen eine Überexpression von Bcl-2 zu einer Inhibition der Apoptose (53) mit vermehrtem Überleben von B- und T-Zellen und nachfolgend zum verstärkten Auftreten von Lymphomen führt (71). Das Onkogen bcl-2 führt ebenfalls über eine chromosomale t14/18-Translokation zu follikulären Lymphomen (69). Interessanterweise konnte gezeigt werden, dass volatile Anästhetika wie Isofluran und Sevofluran Apoptose in Lymphozyten induzieren (39). Ebenfalls wurde gezeigt, dass eine berufliche Exposition mit Inhalationsanästhetika zu einer Inhibition der Apoptose in neutrophilen Granulozyten führt (22). Der molekulare Mechanismus blieb jedoch in beiden Fällen weitgehend unklar. Auch der Transkriptionsfaktor AP-1 wird mit der Induktion von Apoptose in Zusammenhang gebracht (20), allerdings scheint die Aktivierung von AP-1 in verschiedenen Zellreihen zu uneinheitlichen Resultaten zu führen (11;54): 13 Einleitung Abb. 3: Schematischer Ablauf der Apoptose: 1) Nach Bindung extrazellulärer Liganden führt eine Trimerisierung von Oberflächenrezeptoren über die Initiator-Caspase-8 zur Aktivierung der Effektor-Caspase-3. 2) Schädigende extrazelluläre Einflüsse bewirken eine Sensoren-vermittelte Freisetzung von Cytochrom C aus dem mitochondrialen Intermembranspalt. Dieser Vorgang kann durch Bcl-2-Proteine verhindert werden. Cytochrom C führt über die Oligomerisierung von Apaf-1 zur Rekrutierung der Initiator-Caspase-9, was in einer Aktivierung der EffektorCaspase-3 resultiert. 14 Einleitung Beschrieben ist sowohl die Induktion von Apoptose durch c-Jun (9), als auch die Notwendigkeit aktiver Jun-Untereinheiten für Zellwachstum und Zelltransformation (64). In lymphoiden Zellreihen konnte nach Entzug von Wachstumsfaktoren und konsekutiver Induktion von Apoptose eine Beteiligung der Protoonkogene c-jun und c-fos nachgewiesen werden (12). Trotz dieser verschiedenen Funktionen ist unbestritten, dass der Transkriptionsfaktor AP-1 und insbesondere die Untereinheit c-Jun eine zentrale regulatorische Rolle hinsichtlich Progression des Zellzyklus und Apoptose einnimmt (63;77). Eine ähnliche Kontroverse besteht hinsichtlich der Beteiligung der MAPKinasen an Proliferation und Apoptose: Da MAP-Kinasen sowohl durch mitogene Stimuli als auch durch schädigende extrazelluläre Einflüsse aktiviert werden (15;76), ist keine verallgemeinernde Aussage zu ihrer Funktion möglich. Eine wichtige Rolle kommt MAP-Kinasen bei der Zelldifferenzierung zu (57), jedoch wurde auch beschrieben, dass MAP-Kinasen Apoptose vermitteln (16). In der vorliegenden Arbeit sollte daher untersucht werden, ob volatile Anästhetika eine veränderte Aktivität der MAP-Kinasen bewirken und welche Konsequenzen sich daraus ergeben. 2.4 Zielsetzung dieser Arbeit Die erste Hypothese dieser Arbeit war, dass volatile Anästhetika über eine Beeinflussung der DNA-Bindung von Transkriptionsfaktoren zu einer veränderten abhängigen Genexpression führen. Daher sollte in dieser Inhalationsanästhetika Arbeit zunächst untersucht Desfluran, Isofluran oder werden, Sevofluran die ob die DNA- Bindungsaktivität der Transkriptionsfaktoren AP-1, NFAT, Nukleärer Faktor κB (NFκB), SV40-Promotor-1 (SP-1) und „Octamer binding Protein-1“ (Oct-1) beeinflussen. In einem zweiten Schritt sollte ein potentieller Effekt der Anästhetika auf die DNA-Bindungsaktivität von Transkriptionsfaktoren hinsichtlich einer eventuellen Zeit- und Dosisabhängigkeit analysiert werden. Für den Fall einer Wirkung einzelner volatiler Anästhetika auf die DNABindungsaktivität unterschiedlicher Transkriptionsfaktoren sollten in einem dritten Schritt die Signaltransduktionswege, die zur Aktivierung dieser 15 Transkriptionsfaktoren führen, auf Einleitung mögliche Beeinflussung durch Inhalationsanästhetika untersucht werden. Eine veränderte DNA-Bindungsaktivität verschiedener Transkriptionsfaktoren könnte so eine Beeinträchtigung der abhängigen Genexpression bewirken. Die zweite Hypothese dieser Arbeit war, dass Inhalationsanästhetika über eine Veränderung der DNA-Bindungsaktivität des Transkriptionsfaktors AP-1 Apoptose induzieren, da AP-1 eine zentrale Rolle in der Regulation von Zellproliferation und Apoptose einnimmt (63). Dabei sollte zunächst untersucht werden, ob eine Inkubation mit Inhalationsanästhetika zur charakteristischen Morphologie apoptotischer Zellen führt (61). Ließe sich Apoptose durch volatile Anästhetika induzieren (39), sollte in einem zweiten Schritt analysiert werden, ob für diese Wirkung der volatilen Anästhetika die Aktivierung der AP-1-regulierenden MAP-Kinasen und aktive Caspasen notwendig sind (16;51). 16 Material und Methoden 3 Material und Methoden 3.1 Kultur der Jurkat T-Lymphomzellen 3.1.1 Material Jurkat T-Lymphomzellen (#ACC 282) DSMZ, D – Braunschweig AIM-V-Medium #12055-091 Gibco-BRL, D – Karlsruhe RPMI-1640-Medium #31870-025 Gibco-BRL, D – Karlsruhe Fetales Kälber-Serum (FCS) #F-7524 Sigma, D – Deisenhofen L-Glutamin #25030-024 Gibco-BRL, D – Karlsruhe Penizillin/Streptomycin DE17-602E Bio Whittaker Europe, B – Verviers 3.1.2 Methode Die Experimente wurden mit Jurkat T-Lymphomzellen durchgeführt. Diese Zellen wurden im Wärmeschrank bei 37°C und 100% Luftfeuchtigkeit unter 21% O2, 5% CO2 und 74% N2 (Frischgas) in RPMI-1640-Medium mit 10% FCS, 1% (=2 mM) L-Glutamin und 1% (=50 µg/ml) Penizillin/Streptomyzin kultiviert. Für die Experimente wurde AIM-V-Medium ohne Zusätze verwandt. 3.2 Isolation primärer T-Lymphozyten aus peripherem Blut gesunder Spender 3.2.1 Material Falcon 50 ml-Reaktionsgefäß Becton & Dickinson, USA – Franklin Lakes, NJ Ficoll-Paque Plus Amersham Pharmacia, D – Freiburg CliniMACS Miltenyi Biotech, D – Bergisch Gladbach Humanes Serum Albumin (HSA) Baxter, D – Unterschleißheim Waschlösung 1 l CliniMACS + 0,5% HSA Sterile Pasteurpipette Brand, D – Wertheim Neubauer Zählkammer Brand, D – Wertheim CD3+-Microbeads Miltenyi Biotech, D – Bergisch 17 Material und Methoden Gladbach MACS-Säule Miltenyi Biotech, D – Bergisch Gladbach 0,4% Trypanblau Sigma, D – Deisenhofen 3.2.2 Methode Die Experimente wurden mit primären Lymphozyten, welche das CD3Oberflächenantigen trugen, aus dem peripheren Blut gesunder Spender durchgeführt. Diese wurden aus Leuko- und Lymphozyten angereicherten Vollblutpräparaten (sogenannten „Buffy Coats“) von der Transfusionsmedizin der Universitätsklinik Freiburg, Abteilung Fraktionierung (Leiterin: Frau PD Dr. G. Zilow) bereitgestellt. Die Isolation dieser CD3+-Lymphozyten erfolgte durch 1,5:1 Volumenschichtung in einem Falcon 50 ml-Reaktionsgefäß auf 15 ml Ficoll-Paque Plus mit anschließender 46-minütiger Dichtezentrifugation bei 1800 rpm. Die sich in der Interphase befindlichen PMBZ wurden mit Hilfe einer sterilen Pasteurpipette abgesaugt und in eine phosphat-gepufferte Waschlösung (CliniMACS) gegeben, um das Ficoll-Paque Plus zu entfernen. Es folgte eine dreimalige Zentrifugation (10 min bei 1600 rpm mit Bremse), um die Zellen zu konzentrieren. Der Überstand wurde jeweils verworfen und die Anzahl sowie die Morphologie der PMBZ in einer NeubauerZählkammer mittels Trypanblau-Färbung ermittelt. Um die T-Lymphozyten mit CD3-Oberflächenantigen isolieren zu können, machte man sich die Tatsache zunutze, dass an Eisen gebundene spezifische Antikörper (sogenannte „CD3+Microbeads“) die Eigenschaft haben, selektiv an das im T-Zellrezeptor integrierte CD3-Oberflächenantigen zu binden. Pro 107 PMBZ wurden 20 µl „CD3+-Microbeads“ sowie 80 µl Waschlösung zugegeben. Das Gemisch wurde 15 min auf Eis inkubiert und über eine Säule mit Magnet filtriert. Durch den Magneten wurden die markierten CD3+ T-Zellen primär in der Säule zurückgehalten und der Durchfluss verworfen. Nach Entfernung der Säule aus dem Magneten wurden die markierten CD3+-Lymphozyten mit Waschlösung eluiert und deren Zahl nochmals mittels Trypanblau-Färbung in der NeubauerZählkammer ermittelt. Die CD3+-Zellen wurden in RPMI-1640-Medium mit 10% FCS, 1% L-Glutamin und 1% Penizillin/Streptomyzin aufgenommen, über Nacht im Brutschrank 18 Material und Methoden inkubiert und am nächsten Tag den Experimenten zugeführt, für die sie in AIMV-Medium ohne Zusätze umgesetzt wurden. 3.3 Versuchsprotokoll 3.3.1 Material 5 ml-Kulturschalen Becton & Dickinson, USA – Franklin Lakes, NJ Falcon 15 ml-Reaktionsgefäß Becton & Dickinson, USA – Franklin Lakes, NJ Desfluran (Suprane) Baxter, D – Unterschleißheim Isofluran (Forene) Abbott, D – Wiesbaden Sevofluran (Sevorane) Abbott, D – Wiesbaden Desfluran-Verdampfer Dräger-Anästhesietechnik, D – Lübeck Isofluran-Verdampfer Dräger-Anästhesietechnik, D – Lübeck Sevofluran-Verdampfer Dräger-Anästhesietechnik, D – Lübeck Gaskonzentrationsmesser PM8050 Dräger- Anästhesietechnik, D – Lübeck Phorbol-12-Myristat-13-Azetat (PMA) Sigma, D – Deisenhofen (50 µg/ml in Ionomycin P1585 DMSO gelöst) I-0634 Sigma, D – Deisenhofen (1,5 µg/µl in DMSO gelöst) Dimethylsulfoxid (DMSO) Roth, D – Karlsruhe 10x PBS-Puffer 1,369 M (80 g) NaCl, 26,8 (2 g) KCl, 14,7 mM (2 g) KH2PO4, 79,7 mM (11,31 g) Na2HPO4 ad 1 l mit ddH2O; auf pH 7,3 eingestellt und autoklaviert; 1x Verdünnung mit ddH2O Natriumchlorid (NaCl) Roth, D – Karlsruhe Kaliumchlorid (KCl) Roth, D – Karlsruhe Kaliumdihydrogenphosphat (KH2PO4) Merck, D – Darmstadt Natriumhydrogenphosphat (Na2HPO4) Merck, D – Darmstadt 19 Material und Methoden 3.3.2 Methode Nach Isolation der CD3+-Zellen wurden diese in Medium aufgenommen und – wie auch die Jurkat T-Zellen – über Nacht im Brutschrank inkubiert. Für das Experiment wurden die Zellen in Kulturschalen in AIM-V-Medium aufgenommen und in einem speziellen Gasschrank inkubiert, in dem dieselben Bedingungen wie im Brutschrank herrschten und an den zusätzlich der jeweilige Verdampfer des volatilen Anästhetikums anzuschließen war (Abb. 4). Abb. 4: Im Gasinkubationsschrank wurden die T-Lymphozyten mit Frischgas inkubiert, dem mittels speziellem Verdampfer Inhalationsanästhetikum zugesetzt wurde. Für die Experimente wurden die klinisch üblichen volatilen Anästhetika Desfluran, Isofluran und Sevofluran verwendet. Die Zellen wurden insgesamt 24 h mit 18 Vol% Desfluran (3 MAC), 5 Vol% Isofluran (4 MAC) oder 8 Vol% (4 MAC) Sevofluran inkubiert. Die auf dem Verdampfer angegebene Konzentration des abgegebenen Gases war nachweislich (Gaskonzentrationsmesser PM8050) gleich der im Brutschrank. 30 min vor Ablauf des Experimentes wurden die Zellen beider Gruppen mit 15 ng/ml Medium PMA und 0,7 µg/ml Medium Ionomycin stimuliert. Danach wurden die Zellsuspensionen geerntet und in 15 ml Falcon-Reaktionsgefäße überführt (Abb. 5). 20 Material und Methoden Abb. 5: Schematischer Ablauf des experimentellen Protokolls T0: Inkubation mit Inhalationsanästhetikum; T1: Zugabe von PMA und Ionomycin; T2: Ende des Versuches Es folgte ein Zentrifugationsschritt (5 min bei 1300 rpm). Der Überstand wurde abgesaugt, das Zentrifugat in 1 ml 1x PBS gewaschen und 1 min bei 13000 rpm zentrifugiert. Der Überstand wurde verworfen und das Zentrifugat der Herstellung von Zellkernproteinextrakten nach modifiziertem Protokoll von Schreiber et al. (62) zugeführt. 3.4 Nukleäre Extrakte 3.4.1 Material Puffer A 10 mM Hepes pH 7,9: 10 mM KCl; 0,1 mM EDTA: 0,1 mM EGTA Puffer C 20 mM Hepes pH 7,9: 0,4 M NaCl; 1 mM EDTA; 1 mM EGTA Hepes-Puffer Roth, D – Karlsruhe EDTA Roth, D – Karlsruhe EGTA Roth, D – Karlsruhe 21 Material und Methoden Aprotinin A-6279 Sigma, D – Deisenhofen Leupeptin L-2023 Sigma, D – Deisenhofen (50 mg/ml in ddH2O gelöst) Natriumfluorid S-1504 Sigma, D – Deisenhofen (1 M in ddH2O gelöst) Phenylmethylsulfonylfluorid Sigma, D – Deisenhofen (100 mM in Isopropanol gelöst) Natriumpyrophosphat-Decahydrat 1,4-Dithiothreitol (DTT) Sigma, D – Deisenhofen (100 mM in S-6422 ddH2O gelöst) #6908.2 Roth, D – Karlsruhe Nonidet P-40 Fluka BioChemika, CH – Buchs (10% in ddH2O) Protein-Assay #500-0006 Bio-Rad, D – München (20% in TrisHCl pH 7,8) Schüttler Ika-Werke, D – Staufen Kühlschrank 4°C Liebherr, D – Biberach a. d. R. Tris-HCl-Puffer Roth, D – Karlsruhe Küvette Greiner bio-one, D – Frickenhausen 3.4.2 Methode Da der aktivierte Transkriptionsfaktor AP-1 in Zellen nur nukleär vorliegt, mussten zur Untersuchung der DNA-Bindungsfähigkeit nukleäre Proteinextrakte gewonnen werden. Die Zellen wurden durch zwei salzreiche Detergenzpuffer (Puffer A und Puffer C) lysiert. Diese Puffer wurden mit den Phosphatase- und Proteinaseinhibitoren Aprotinin (10 µg/ml), Leupeptin (25 µM), Natriumfluorid (10 mM), Phenylmethylsulfonylfluorid (2 mM), Natriumpyrophosphat-Decahydrat (10 mM) und 1,4-Dithiothreitol (1 mM) versetzt. Das Zellzentrifugat aus dem Eppendorfreaktionsgefäß wurde in 400 µl eiskaltem Puffer A resuspendiert und 15 min auf Eis inkubiert. Nach Zugabe von 25 µl Nonidet P-40 (10%) wurde der Ansatz homogenisiert. Es folgte eine Zentrifugation (30 sec bei 13000 rpm). Der Überstand wurde verworfen und das Zentrifugat 5 min bei RT getrocknet, um Flüssigkeitsreste zu eliminieren. Zur Lyse der Kernmembran wurde dem im Reaktionsgefäß verbliebenen Ansatz 50 µl (bei CD3+-Zellen 35 µl) eiskalter Puffer C zugesetzt, wobei eine Resuspension vermieden wurde. Es schloss sich eine viertelstündige 22 Material und Methoden Schüttelung bei 4°C im Vortexrotationsaufsatz an. Nach einem weiteren fünfminütigen Zentrifugationsschritt bei 13000 rpm konnte der Überstand in ein neues Eppendorfreaktionsgefäß überführt und der verbliebene Rest der Zellen verworfen werden. Die Bestimmung der Proteinkonzentration des Lysates erfolgte nach der Bradford-Methode. Hierzu wurde 1 µl der zu untersuchenden Proteinprobe mit 19 µl ddH2O verdünnt und auf 1 ml in einer Küvette mit der Farblösung aufgefüllt. Der enthaltene Farbstoff bindet unspezifisch an Proteine und ändert dabei seine Farbe von rot nach blau. Die dadurch entstehende Änderung der Lichtabsorption kann im Fotometer bestimmt werden. 3.5 „Electrophoretic Mobility Shift Assay” (EMSA) und Kompetitionen 3.5.1 Material AP-1-Oligonukleotid E3201 Promega, D – Heidelberg [1,75 pM/µl bzw. 25 ng/µl] 5’-CGC TTG ATG AGT CAG CCG GAA-3’ 3’-GCG AAC TAC TCA GTC GGC CTT-5’ NFAT-Oligonukleotid NFAT-Oligonukleotid aus dem humanen Promotor des GM-CSF-Gen [1,75 pM/µl bzw. 25 ng/µl] 5’-TTT CTC ATG GAA AGA TGA CAT A-3’ 3’-AAA GAG TAC CTT TCT ACT GTA T-5’ NFκB-Oligonukleotid E3291 Promega, D – Heidelberg [1,75 pM/µl bzw. 25 ng/µl] 5‘-AGT TGA GGG GAC TTT CCC AGG C-3‘ 3‘-TGA ACT CCC CTG AAA GGG TCC G-5‘ SP-1-Oligonukleotid E3231 Promega, D – Heidelberg [1,75 pM/µl bzw. 25 ng/µl] 5‘-d(ATT CGA TCG GGG CGG GGC GAG C)-3‘ 3‘-d(TAA GCT AGC CCC GCC CCG CTC G)-5‘ Oct-1-Oligonukleotid E3241 Promega, D – Heidelberg [1,75 pM/µl bzw. 25 ng/µl] 23 Material und Methoden 5’-TGT CGA ATG CAA ATC ACT AGA A-3’ 3’-ACA GCT TAC GTT TAG TGA TCT T-5’ Magnesiumchlorid (MgCl2) Merck, D – Darmstadt (100 mM in ddH2O) γ-32P-dATP Aktivität: 3000 Ci/mM Amersham Pharmacia, D – Freiburg T4-Polynukleotid Kinase #M0201S New England Biolabs, D – Frankfurt T4-Kinase Puffer #BO2015 New England Biolabs, D – Frankfurt G-25 Microspin Säule #27-5325-01 Amersham Pharmacia, D – Freiburg Heizblock Eppendorf-Netheler-Hinz, D – Hamburg Bovines Serum Albumin (BSA) A-9647 Sigma, D – Deisenhofen (10 mg/ml in Fraktion V 96% proteasefrei ddH2O) Poly[dIdC] Roche Diagnostics GmbH, D – #1219847 Mannheim (50 A260 Units in 2,5 ml ddH2O gelöst) Puffer D+ 20 mM Hepes pH 7,9; 100 mM KCl; 0,5 mM EDTA; 2 mM DTT; 0,1 mM PMSF; 20% Glyzerol; 1% (v/v) NP-40 Glyzerol Fluka BioChemika, CH – Buchs 5x Ficoll+ 20% Ficoll-Paque Plus; 100 mM Hepes pH 7,9; 10 mM DTT; 0,1 mM PMSF; 300 mM KCl 5x Ficoll- 20% Ficoll-Paque Plus; 100 mM Hepes pH 7,9; 10 mM DTT; 0,1 mM PMSF; ad 1 ml mit ddH2O Anti-ATF-1-Kompetitions-AK sc-270 Santa Cruz Biotechnology, D – Heidelberg Anti-ATF-2-Kompetitions-AK sc-187 Santa Cruz Biotechnology, D – Heidelberg Anti-Jun-B-Kompetitions-AK sc-8051 Santa Cruz Biotechnology, D – Heidelberg Anti-Jun-D-Kompetitions-AK sc-74 Santa Cruz Biotechnology, D – Heidelberg Anti-c-Fos-Kompetitions-AK sc-52 Santa Cruz Biotechnology, D – 24 Material und Methoden Heidelberg Anti-c-Jun-Kompetitions-AK sc-44 Santa Cruz Biotechnology, D – Heidelberg Anti-CEBP-Kompetitions-AK sc-61 Santa Cruz Biotechnology, D – Heidelberg Anti-p65-Kompetitions-AK sc-372 Santa Cruz Biotechnology, D – Heidelberg Rotiphorese (30%) 3029.1 Ammoniumperoxiddisulfat (APS) #529K791001 Roth, D – Karlsruhe Merck, D – Darmstadt (10% in ddH2O gelöst) 10x TBE-Puffer 890 mM Tris-HCl; 890 mM Borsäure; 20 mM EDTA pH 8,0; mit ddH2O ad 1 l (für 0,5x TBE verdünnt mit ddH2O) Borsäure TEMED Roth, D – Karlsruhe #T-8133 N,N,N’,N’-Tetramethylethylendiamin, Roth, D – Karlsruhe 2 Glasplatten Thoma, D – Freiburg Abstandhalter und Kamm Thoma, D – Freiburg Polyacrylamidgel 4% 60 ml ddH2O; 10 ml Rotiphorese 30%, 3,5 ml 10x TBE, 400 µl 10% APS; 40 µl TEMED 30% Glyzerol, 70% ddH2O, etwas farbiger Marker Bromphenolblau Bromphenolblau Roth, D – Karlsruhe EMSA-Spannungsquelle Consort, B – Turnhout Gelkammern Thoma, D – Freiburg Whatman-Filterpapier (3 mm) Schleicher & Schüll, D – Dassel Vakuumtrockner Bio-Rad, D – München Plastikfolie (Saran) Roth, D – Karlsruhe X-OMAT-Filme #853 2665 Kodak, USA – Rochester, NY Filmentwicklermaschine Curix 60 Agfa-Gevaert, D – Leverkusen Entwicklerlösungen Agfa-Gevaert, D – Leverkusen Tiefkühlschrank (-80°C) Forma, USA – Marie, OH 25 Material und Methoden 3.5.2 Methode Zunächst musste die Konsensus-Sequenz der AP-1-DNA-Sequenz radioaktiv markiert werden. Hierfür wurde ein AP-1-Oligonukleotid-Mix erstellt. Er bestand aus 37 µl ddH20, 1 µl AP-1-Oligonukleotid, 5 µl T4-Kinasepuffer, 5 µl γ-32PdATP und 2 µl Polynukleotid-Kinase. Dieser wurde 40-50 min bei 37°C inkubiert. Anschließend wurde dieser AP-1-Ansatz über eine mit Resin gefüllte Säule gegeben und die freien Nukleotide durch dreiminütige Zentrifugation bei 3000 rpm von den radioaktiven 32 P-markierten AP-1-Oligonukleotiden getrennt, die 25-30% der Gesamtradioaktivität ausmachten. Das Reaktionsgemisch für die Proteinextrakte bestand aus 20 µg BSA, 2 mg poly[dIdC], 2 µl Puffer D+, 3 µl ddH2O, 4 µl 5x Ficoll+ (für die NFAT-Proben 4 µl 5x Ficoll-), 1 µl 100 mM MgCl2 (nur für AP-1-Proben, sonst 1 µl ddH2O) und 1 µl 32 P-markiertem AP-1- Oligogemisch mit gleichen Mengen an Proteinen (30 µg) in einem Gesamtvolumen von 20 µl. Es folgte eine 25-minütige Inkubation bei RT. Für die Kompetitionsansätze („Supershifts“) wurden zu 3 µl Proteinextrakt 2,5 µl spezifischer Antikörper gegen Untereinheiten des AP-1-Komplexes (ATF-1, ATF-2, Jun-B, Jun-D, c-Fos, c-Jun, CEBP) sowie gegen die p65-Untereinheit des Transkriptionsfaktors NFκB bzw. jeweils 1 µl unmarkiertes NFκB- und AP1-Oligonukleotid zugegeben und der Ansatz 15 min vorinkubiert. Anschließend wurde das oben beschriebene Reaktionsgemisch zugegeben, wobei der Anteil des destillierten Wassers auf 3,5 µl (2,5 µl bei den AP-1-Proben) reduziert wurde. Die 20 µl des Ansatzes wurden nach der Inkubation auf ein vierprozentiges Polyacrylamidgel geladen. 1,5 l 0,5x TBE dienten als Laufpuffer. Die Spannung wurde auf 230 V, die Stromstärke auf 23 mA eingestellt. Die Laufstrecke wurde auf 10 cm festgelegt. Nach Abschluss der Elektrophorese wurde das Gel auf ein Whatman-Filterpapier überführt, die Oberseite mit Folie abgedeckt und im Vakuumtrockner bei 80°C 35 min lang getrocknet. Anschließend wurde das Gelpapier in eine röntgendichte Filmkassette mit Verstärkerfolie gegeben und in der Dunkelkammer ein Film aufgelegt. Die Filmexposition erfolgte bei -80°C. 26 Material und Methoden 3.6 Luziferase-Gen-Transfektion 3.6.1 Material 6-Loch-Platte Greiner bio-one, D – Frickenhausen Sterile Eppendorfreaktionsgefäße Nunc, DK – Roskilde SuperFect Qiagen, D – Hilden #301307 pAP1(PMA)TA-Luc Vector #6056-1 Clontech, USA – Palo Alto, CA (s. Anhang) Gummischaber B. Braun, D – Melsungen Luciferase Assay System E1501 Promega, D – Mannheim Luciferase Reporter Lysepuffer E397A Promega, D – Mannheim Luminometer Microlumat LB96P, EG & G Berthold, D – Bad Wildbad Undurchsichtige 96-Loch-Platte Dynatech, USA – Bridgewater, NJ 3.6.2 Methode Das Plasmid wurde in der Konzentration von 1 µg/µl eingesetzt. 1,5 x 106 zu transfizierende Zellen wurden in 1,5 ml serumfreien RPMI-Medium mit 1% Penizillin/Streptomycin und 1% L-Glutamin in 6-Loch-Platten über Nacht im Brutschrank inkubiert. 150 µl Medium wurden in sterile Reaktionsgefäße vorgelegt. 30 µl SuperFect und 3 µg Plasmid wurden zugegeben und der Ansatz 30 min bei RT belassen. Es wurden nochmals 350 µl Medium je Ansatz zugegeben. Dann wurde der Ansatz vorsichtig gemischt und jeweils tröpfchenweise zu der Zellsuspension gegeben. Nach 4-6 h konnten die nun transient transfizierten Zellen vom Reaktionsgefäßboden abgelöst und in serumfreiem Medium aufgenommen werden. Die Zellen wurden gesammelt, je 1 ml in einer 24-Loch-Platte neu ausplattiert und im Brutschrank 24 h mit dem jeweiligen Inhalationsanästhetikum inkubiert. 9 h später wurden die Zellen mit 15 ng/ml PMA und 0,7 µg/ml Ionomycin stimuliert. Nach 15 h wurden die Zellen wiederum vom Boden losgelöst, in Reaktionsgefäße überführt und 10 min lang bei 3000 rpm zentrifugiert. Der Überstand wurde verworfen und die Zellen in 50 µl 1x Lyse-Puffer resuspendiert. Zum besseren Zellaufschluss wurden die Zellen für kurze Zeit bei -80°C inkubiert. Die Proben wurden anschließend aufgetaut, durchmischt und 5 min bei 13000 rpm zentrifugiert. Nach Vorlage 27 Material und Methoden des Substrates im Luminometer wurden jeweils 20 µl der Probenüberstände in eine waschbare 96-Loch-Platte gegeben und die Änderung der „relative light units“ bestimmt. Zum Abschluss wurde in den gemessenen Proben durch Bradford-Methode der Proteingehalt ermittelt und das Ergebnis normalisiert. 3.7 Zytokinbestimmung mittels „Enzyme-Linked Immuno Sorbent Assay“ (ELISA) 3.7.1 Material Human IL-3 Immunoassay D3000 R & D Systems, D – Wiesbaden Spectra max Plus Lesegerät Molecular Devices Corp. USA – Sunnyvale, CA Computerprogramm Softmax pro Molecular Devices Corp. USA – Sunnyvale, CA 3.7.2 Methode 2,5 x 106 primäre CD3+ T-Lymphozyten aus dem Blut gesunder Spender wurden in 5 ml AIM-V-Medium aufgenommen und im Brutschrank bzw. im Gasschrank inkubiert. Die Inkubation der Zellen erfolgte über einen Zeitraum von 24 h mit 8 Vol% Sevofluran, die Stimulation über 24 h mit 1,5 ng/ml PMA und 0,7 µg/ml Ionomycin. Die Ansätze wurden nach Abschluss des Experimentes 5 min bei 1300 rpm zentrifugiert, die Zellen verworfen und die Überstände asserviert. Die benötigten Reagenzien und IL-3-Standards wurden nach Angaben des Herstellers vorbereitet. 200 µl verdünnter Standards (1000 – 500 – 250 – 125 – 62,5 und 31,25 pg/ml) oder Proben wurden in der beschichteten Platte vorgelegt und 2 h bei RT inkubiert. Anschließend wurde die Flüssigkeit abgesaugt und die Mikrotiterplatte dreimal mit IL-3-Puffer gewaschen. Nach Zugabe von 200 µl „IL-3-conjugate“ wurden die Ansätze wiederum 2 h bei RT inkubiert. Der Waschschritt wurde wiederholt und anschließend 200 µl Substrat-Lösung pro Ansatz zugegeben und der gesamte Ansatz 20 min lichtgeschützt inkubiert. Zur Beendigung der Reaktion wurden 50 µl Stop-Lösung zugegeben. Die Bestimmung erfolgte innerhalb von 30 min bei einer optischen Absorption im Bereich von 450 nm und einer λ-Korrektur bei 540 nm in einem Lesegerät für Mikrotiterplatten. 28 Material und Methoden 3.8 Western Blot 3.8.1 Material Totex-Puffer 20 mM Hepes pH 7,9; 350 mM NaCl; 20% (v/v) Glyzerol; 1% (w/v) Nonidet P-40; 1 mM MgCl2; 0,5 mM EDTA; 0,1 mM EGTA; frisch zugeben: 2 mM DTT; 2 mM PMSF; 15 µg/ml Aprotinin; 0,1% Phosphatase-Inhibitor Cocktail Set II Phosphatase-Inhibitor Cocktail Set II Calbiochem, USA – La Jolla, CA 4x unterer SDS-Puffer 1,5 M Tris-HCl pH 6,8; 0,4% SDS 10% SDS-Polyacrylamidtrenngel 5,2 ml Rotiphorese 30%; 4 ml 4x unterer SDS-Puffer; 6,8 ml ddH2O; 8 µl TEMED; 100 µl APS (10%) Isopropanol Merck, D – Darmstadt 4x oberer SDS-Puffer 0,5 M Tris-HCl pH 8,8; 0,4% SDS SDS-Polyacrylamidsammelgel 1 ml Rotiphorese 30%; 2 ml 4x oberer SDS-Puffer; 5 ml ddH2O; 8 µl TEMED; 80 µl APS (10%) Sodium-Dodecylsulfat (SDS) #4360.1 Roth, D – Karlsruhe 5x-SDS Laufpuffer 125 mM Tris-HCl; 1 M Glyzin; 0,5% SDS; ad 1 l mit ddH2O (1x SDS Laufpuffer verdünnt mit ddH2O) Glyzin Roth, D – Karlsruhe Laemmli-Puffer 1 M Tris-HCl pH 6,8; 1% SDS; 1,5% DTT; 0,1% Bromphenolblau; 50% Glyzerol; ad 20 ml mit ddH2O Präzisions-Marker 161-0372 Methanol Elektrophoresekammer Bio-Rad, D – München Merck, D – Darmstadt 165-3301 Protean III, Bio-Rad, D – München Transfergerät Transblot SD, Bio-Rad, D – München Anodenpuffer I pH 10,4 300 mM Tris-HCl; 10% Methanol; ad 1 l ddH2O Anodenpuffer II pH 10,4 25 mM Tris-HCl; 10% Methanol; ad 1 l 29 Material und Methoden ddH2O Kathodenpuffer pH 9,4 25 mM Tris-HCl; 40 mM 6-Amino-nhexansäure; 10% Methanol; ad 1 l ddH2O 6-Amino-n-hexansäure Roth, D – Karlsruhe Immobilion-Membran #IPVH00010 Millipore Corporation, D – Eschborn 10x TBS-Puffer 100 mM Tris-HCl; pH 8,0; 1,5 M NaCl, ad 1 l ddH2O 1x TBS + Tween (TBST) 10% 10x TBS; 0,1% Tween 20; ad 1l ddH2O Tween 20 Roth, D – Karlsruhe Magermilchpulver Fluka BioChemika, CH – Buchs Assistent-Rotationsschaukel Karl Hecht GmbH, D – Sondheim Phospho-p38-MAPK-AK #9910 Cell Signaling, USA – Beverly, MA Phospho-ERK-AK #9910 Cell Signaling, USA – Beverly, MA Phospho-JNK-AK #9910 Cell Signaling, USA – Beverly, MA p38-MAPK-AK #9212 Cell Signaling, USA – Beverly, MA ERK-AK #9102 Cell Signaling, USA – Beverly, MA JNK-AK #9252 Cell Signaling, USA – Beverly, MA Caspase-3-AK #9662 Cell Signaling, USA – Beverly, MA Cleaved Caspase-3-AK #9661 Cell Signaling, USA – Beverly, MA Anti-rabbit-IgG-HRP-AK #7074 Cell Signaling, USA – Beverly, MA ECLplus-Lösung RPN2132 Amersham Pharmacia, D – Freiburg Pinzetten Aeskulap, D – Tuttlingen Tartan-Plastikfolien 3M, D – Neuss Hyperfilm ECL Amersham Pharmacia, D – Freiburg 3.8.2 Methode Zunächst wurden die Gelkammern mit einem 10% Polyacrylamidtrenngel gefüllt, mit Isopropanol überschichtet und nach Aushärtung mit ddH2O abgewaschen. Nach Polyacrylamidsammelgel oben an. schloss Aus sich den das etwa 1 cm Zellsuspensionen starke wurden gesamtzelluläre Proteinextrakte hergestellt. Die Zellen wurden nach Ende der Gasexpositionszeit in einem Reaktionsgefäß 5 min bei 1300 rpm zentrifugiert, 30 Material und Methoden der Überstand verworfen und die Zellen in 1 ml PBS gewaschen. Nach Überführung des Ansatzes in ein Reaktionsgefäß und einminütiger Zentrifugation bei 13000 rpm wurde das PBS entfernt und das Zentrifugat in 50 µl kaltem Totex-Puffer, der mit Proteinase- und Phosphataseinhibitoren versetzt wurde, resuspendiert und 30 min auf Eis inkubiert. Anschließend wurden die Proben zentrifugiert (10 min bei 13000 rpm und 4°C). Der Überstand wurde asserviert. Der Proteingehalt wurde mittels Bradford-Methode bestimmt, und 50 µg der Proteinproben mit ddH2O auf ein Volumen von 20 µl gebracht. Verdünnungen wurden mit Totex-Puffer durchgeführt. Die Proben wurden 5 min bei 95°C in Laemmli-Puffer denaturiert, kurz bei 13000 rpm zentrifugiert und in die Geltaschen gegeben. Die erste Geltasche enthielt nur 10 µl Präzisionsmarker. Die Elektrophorese erfolgte in 1x SDS-Laufpuffer 30 min bei 30 mA, dann 1 h bei 50 mA sowie konstant 150 mV. Um die aufgetrennten Proteine auf eine Immobilion-PVDF-Membran zu transferieren, wurde diese vorher 10 sec in Methanol, 2 min in ddH2O und 5 min in Anodenpuffer 2 äquilibriert. Die Transfereinrichtung wurde wie folgt aufgebaut: Auf die Anodenseite wurden 4 in Anodenpuffer I gepufferte Filterpapiere gelegt, dann 2 Papiere aus Anodenpuffer II, überschichtet mit der Membran, dem Gel und 6 Papieren aus dem Kathodenpuffer. Die Proteine wurden anschließend mittels eines elektrischen Feldes bei 0,8 mA/cm2 Oberfläche aus dem Gel auf die PVDF-Membran übertragen. Danach wurde die Membran 1 h bei RT auf der Rotationsschaukel in TBST mit 5% Magermilchpulver geschwenkt, wodurch die unspezifischen Proteinbindungen blockiert wurden. Nach dreimaligem Waschen mit TBST wurde die Membran über Nacht bei 4°C auf der Rotationsschaukel mit einem spezifischen ersten Antikörper (1:1000 in TBST + 5% BSA) inkubiert. Nach dreimaligem Waschen mit TBST wurde die Membran dann 1 h bei RT auf der Rotationsschaukel mit dem Anti-rabbit-IgG-HRP-gekoppelten zweiten Antikörper (1:2000 in TBST + 5% Magermilchpulver) inkubiert. Es folgte dreimaliges Waschen mit TBST und anschließende vollständige Benetzung für etwa 30 sec mit ECLplus-Lösung auf der Rotationsschaukel. Die Membran wurde zwischen zwei Plastikfolien in der Dunkelkammer in eine lichtdichte Kassette gegeben und auf einem ECL-empfindlichen Film verschieden lange entwickelt. 31 Material und Methoden 3.9 „Fluorescence-Activated Cell Sorting” (FACS) für Vollblut 3.9.1 Material Durchsichtige 96-Loch-Platte Nunc, DK – Roskilde 10x Annexin-Bindepuffer Becton & Dickinson, USA – Franklin Lakes, NJ (1x Verdünnung in ddH2O) 10x Ammoniumchlorid-Lösung Becton & Dickinson, USA – Franklin Lakes, NJ (1x Verdünnung in ddH2O) Annexin V-FITC Becton & Dickinson, USA – Franklin Lakes, NJ 7-AAD Becton & Dickinson, USA – Franklin Lakes, NJ CD3-PE (Klon SK 7) Becton & Dickinson, USA – Franklin Lakes, NJ Falcon-FACS-Röhrchen Becton & Dickinson, USA – Franklin Lakes, NJ FACSCalibur-Gerät Becton & Dickinson, USA – Franklin Lakes, NJ 3.9.2 Methode Für die Experimente wurden 100 µl heparinisiertes Blut gesunder freiwilliger Spender als Reaktionsansatz in eine 96-Loch-Platte gegeben und 24 h in 8 Vol% Sevofluran-haltiger Atmosphäre (als unbearbeitete Proben galt die entsprechende Zeit im Brutschrank) inkubiert. Nach Ablauf wurden je Ansatz 6 ng/ml (=16 µl) Anti-CD3-Phycoerithrin-Antikörper (CD3-PE) dazugegeben und 30 min lichtgeschützt bei RT inkubiert. Der Ansatz wurde in ein Eppendorfreaktionsgefäß überführt und für 5-10 min jeweils 1 ml 1x Ammoniumchlorid hinzugefügt. Die Proben wurden 5 min bei 2800 rpm zentrifugiert und der Überstand verworfen. Das Zentrifugat wurde in 100 µl 1x Annexin-Bindepuffer resuspendiert und nach Zugabe von jeweils 5 µl Annexin V-FITC 20 min bei RT inkubiert. Annexin V-FITC ist ein Antikörper, der bei der Apoptose spezifische, auf der Membranaußenseite erscheinende Phospholipide (Phosphatidylserin) bindet und so als Apoptosemarker fungiert. Der Antikörper 7-AAD ist ein Marker, der nur dann an DNA binden kann, wenn die Zelle durch 32 Material und Methoden Nekrose geschädigt ist. 5 µl 7-AAD wurden in Falcon-FACS-Röhrchen vorgelegt und lichtgeschützt inkubiert. Die Zellsuspension wurde in je 1 ml 1x Annexin-Bindepuffer aufgenommen und wiederum 5 min bei 2800 rpm zentrifugiert. Der Überstand wurde verworfen, die Zellmasse in 500 µl 1x Annexin-Bindepuffer gelöst und die Messung gestartet. 3.10 Fluoreszenzmikroskopie 3.10.1 Material Staurosporin S4400 Sigma, D – Deisenhofen (1 mg/ml in DMSO gelöst) MG132 PI-102 Biomol GmbH, D – Hamburg (25 mg/ml in DMSO gelöst) Höchst-Farbstoff Propidiumiodid (PI) #33342 Molecular Probes, NL – Leiden Becton & Dickinson, Franklin Lakes, USA – NJ Paraformaldehyd 4% Fluka BioChemika, CH – Buchs Pipes-Lösung Sigma, D – Deisenhofen (0,1 M in ddH2O) Objektträger Langenbrinck, D – Emmendingen Deckgläschen Merck, D – Darmstadt 3.10.2 Methode 5 x 106 Jurkat T-Lymphozyten wurden in 5 ml Kulturmedium ausplattiert, über Nacht im Brutschrank kultiviert und dann 24 h mit 8 Vol% Sevofluran inkubiert. Gleichzeitig wurden zwei Proben 24 h mit 1 µM Staurosporin sowie mit 1 µM MG132 versetzt. Je 100 µl dieser Zellsuspensionen wurden aus den Petrischalen entnommen und in Falcon 15 ml-Reaktionsgefäßen auf Eis gegeben. Die Proben wurden 3 min bei 600 rpm und 4°C zentrifugiert und der Überstand abgesaugt. Als Farbstoffe wurden neben dem Apoptosemarker Annexin V-FITC auch Höchst-Farbstoff verwandt, der Zellkerne anfärbt, sowie Propidiumiodid (PI), das nur bei durch Nekrose geschädigter Membran in der Zelle einen Farbausschlag bewirkt. Je Ansatz wurden nun 50 µl 1x AnnexinBindepuffer zugegeben sowie 0,1% Höchst-Farbstoff, 0,1% Annexin V-FITC 33 Material und Methoden sowie 0,2% PI. Die Ansätze wurden 15 min auf Eis inkubiert und dann 3 min bei 600 rpm und 4°C zentrifugiert. Der Überstand wurde verworfen und das Zentrifugat zur Fixierung in je 20 µl 4% Paraformaldehyd in 0,1 M Pipes-Lösung resuspendiert. Das Zellgemisch wurde auf einen Objektträger gegeben, mit einem Deckgläschen verschlossen und unter fluoreszierendem Licht begutachtet. Aus den verbliebenen Resten der Zellsuspensionen wurden Gesamtzellproteinextrakte mittels Totex-Puffer hergestellt, die in einem Western Blot auf ihren Gehalt an Caspase-3 (Pro-Caspase-3, geschnittene Caspase-3) untersucht wurden. 3.11 Caspase-Aktivitätsmessung 3.11.1 Material Versuchspuffer 100 mM Hepes-KOH pH 7,5; 2 mM DTT Cliniplate-96-Loch-Platte Labsystems, SF – Vantaa Fluorogenes Substrat Acetyl-DEVD-7-Amino-4methylcoumarin; Alexis Corporation, D – Grünberg 3.11.2 Zur Methode Bestimmung der zellulären Caspase-Aktivität wurden 10 µl der Gesamtzellextrakte in Totex-Puffer in 90 µl Versuchspuffer gelöst und in eine schwarze 96-Loch-Platte gegeben. Je 1 µl fluorogenes Substrat wurde zugegeben, um eine Endkonzentration des Substrates von 60 µM zu erhalten. Caspase-3, -6 und -7 spalten das Substrat hinter der DEVD-Sequenz und führen so zu einem Anstieg der Fluoreszenzintensität. Danach wurde die 30minütige Messung im Lesegerät bei 26°C mit Hilfe des Rechners bei einem Exzitationsmaximum von 380 nm und einem Emissionsmaximum von 460 nm gestartet. 34 Material und Methoden 3.12 Statistik 3.12.1 Material SigmaStat Software Paket Jandel Scientific, USA – San Rafael, CA 3.12.2 Methode Luziferase-Reportergenaktivität und IL-3-Bestimmung im ELISA wurden als Box Blot (Median, 25-75% und 95% Konfidenzintervall), Caspase-3-Aktivität als Balkendiagramm dargestellt. Unterschiede zwischen den einzelnen experimentellen Bedingungen wurden mit multiplen paarweisen Vergleichen mittlerer Ränge mit Hilfe des Student-Newman-Keuls-Tests ermittelt. Als statistisch signifikant wurde ein P < 0,05 angenommen. 35 Ergebnisse 4 Ergebnisse 4.1 Sevofluran hemmt die PMA/Ionomycin-induzierte DNABindungsaktivität von AP-1 in T-Lymphozyten in vitro Der Transkriptionsfaktor AP-1 setzt sich aus Proteinkomplexen zusammen, die an spezifische DNA-Abschnitte (TGAC/GTCA) im Promotor binden (29) und so die Transkription abhängiger Gene induzieren. Die DNA-Bindungsaktivität von Transkriptionsfaktoren wie AP-1 kann mittels EMSA untersucht werden. Man macht sich dabei die Tatsache zunutze, dass spezifische Nukleotid-Sequenzen, die an korrespondierende DNA-Abschnitte binden, in einem Polyacrylamidgel verzögert wandern, und bei radioaktiver Markierung charakteristische Bandenmuster liefern. 4.1.1 Sevofluran hemmt die DNA-Bindungsaktivität von AP-1 Die DNA-Bindungsaktivität von AP-1 konnte durch PMA/Ionomycin-Stimulation in T-Lymphozyten in vitro im Vergleich mit unstimulierten Zellen induziert werden (Abb. 6, Spuren 1 und 2). Die Inkubation von T-Lymphozyten über 24 h mit 8 Vol% Sevofluran konnte die Induktion der DNA-Bindungsaktivität von AP1 erheblich hemmen (Abb. 6, Spur 3). Weiterhin zeigte sich, dass eine 24stündige Inkubation von unstimulierten T-Lymphozyten mit 8 Vol% Sevofluran keinen Einfluss auf die DNA-Bindungsaktivität von AP-1 hatte (Abb. 6, Spur 4). Die Hemmung der induzierten DNA-Bindungsaktivität von AP-1 durch 24 h 8 Vol% Sevofluran konnte in Jurkat T-Zellen und in primären CD3+ TLymphozyten aus dem Blut gesunder Spender nachgewiesen werden. JurkatLymphomzellen und primäre Lymphozyten zeigten in den Versuchen identisches Verhalten. Stellvertretend wird daher hier nur ein EMSA mit Extrakten aus Jurkat-Lymphomzellen gezeigt (Abb. 6, Spuren 5-8). 36 Ergebnisse Abb. 6: Der Effekt von Sevofluran auf die DNA-Bindungsaktivität von AP-1 nach 32 PMA/Ionomycin-Stimulation (Autoradiographie P-markierter AP-1-Konsensus+ Sequenzen im EMSA). CD3 T-Lymphozyten aus dem Blut gesunder Spender und Jurkat T-Lymphozyten wurden 23,5 h mit 8 Vol% Sevofluran inkubiert und anschließend 30 min mit 15 ng/ml PMA und 700 ng/ml Ionomycin stimuliert. Gezeigt ist das Ergebnis dreier unabhängiger Experimente. Offenes Dreieck = Position des AP-1-DNA-Komplexes, offener Kreis = unspezifische Radioaktivitätsbindung der Probe, geschlossenes Dreieck = ungebundene Oligonukleotide. 4.1.2 Zeitabhängige Hemmung von AP-1 durch Sevofluran Um die Zeitabhängigkeit der Sevofluran-vermittelten Hemmung der DNABindungsaktivität von AP-1 in stimulierten T-Lymphozyten in vitro zu charakterisieren, wurden die Zellen für 3, 6, 12 und 24 h mit Sevofluran inkubiert. Während bei 3- und 6-stündiger Inkubation mit 8 Vol% Sevofluran keine Hemmung der DNA-Bindungsaktivität von AP-1 erkennbar war (Abb. 7, Spuren 3 und 6), konnte eine Hemmung von AP-1 nach 12 h und nach 24 h 8 Vol% Sevofluran beobachtet werden (Abb. 7, Spuren 9 und 12). 37 Ergebnisse Abb. 7: Die DNA-Bindungsaktivität von AP-1 nach PMA/Ionomycin-Stimulation während Inkubation mit 8 Vol% Sevofluran und verschiedenen 32 P-markierter AP-1-KonsensusExpositionsintervallen (Autoradiographie + Sequenzen im EMSA). CD3 T-Lymphozyten aus dem Blut gesunder Spender wurden 2,5 h, 5,5 h, 11,5 h und 23,5 h mit 8 Vol% Sevofluran inkubiert und anschließend 30 min mit 15 ng/ml PMA und 700 ng/ml Ionomycin stimuliert. Gezeigt ist das Ergebnis dreier unabhängiger Experimente. Offenes Dreieck = Position des AP-1-DNA-Komplexes, offener Kreis = unspezifische Radioaktivitätsbindung der Probe, geschlossenes Dreieck = ungebundene Oligonukleotide. 4.1.3 Dosisabhängige Hemmung von AP-1 durch Sevofluran Um die Dosiskinetik von Sevofluran zu untersuchen, wurden die TLymphozyten mit steigenden Konzentrationen (2, 4, 6 und 8 Vol%) Sevofluran inkubiert. Eine Konzentration von 2 Vol% Sevofluran über einen Zeitraum von 24 h bewirkte bereits eine Hemmung der PMA/Ionomycin-induzierten DNABindungsaktivität von AP-1 in T-Lymphozyten in vitro (Abb. 8, Spur 3). Diese 38 Ergebnisse Hemmung war mit steigender Konzentration (4, 6 und 8 Vol%) von Sevofluran ausgeprägter (Abb. 8, Spuren 6, 9 und 12). Abb. 8: Der Effekt verschiedener Konzentrationen Sevofluran bei 24 h Expositionszeit auf die DNA-Bindungsaktivität von AP-1 nach PMA/Ionomycin32 Stimulation (Autoradiographie P-markierter AP-1-Konsensus-Sequenzen im + EMSA). CD3 T-Lymphozyten aus dem Blut gesunder Spender wurden 23,5 h mit 2, 4, 6 und 8 Vol% Sevofluran inkubiert und anschließend 30 min mit 15 ng/ml PMA und 700 ng/ml Ionomycin stimuliert. Gezeigt ist das Ergebnis dreier unabhängiger Experimente. Offenes Dreieck = Position des AP-1-DNAKomplexes, offener Kreis = unspezifische Radioaktivitätsbindung der Probe, geschlossenes Dreieck = ungebundene Oligonukleotide. 4.1.4 Veränderung der DNA-Bindungsaktivität der AP-1Untereinheiten JunD und c-Jun durch Sevofluran Um die Sevofluran-vermittelte Hemmung der DNA-Bindungsaktivität der verschiedenen AP-1-Untereinheiten zu untersuchen, wurden dem Reaktionsansatz spezifische Antikörper gegen die Untereinheiten des AP-1- 39 Ergebnisse Komplexes zugegeben (Supershift). Je stärker das radioaktive Signal der AP-1Bande durch einen gegen eine bestimmte Untereinheit gerichteten Antikörper abnimmt, um so stärker ist diese Untereinheit an der Bildung des AP-1Komplexes beteiligt. Eine unspezifische Kompetition des Transkriptionsfaktors AP-1 wird durch Zugabe von Oligonukleotiden mit Konsensus-Sequenzen anderer Transkriptionsfaktoren erreicht. Abb. 9: Der Effekt verschiedener spezifischer Kompetitions-Antikörper bzw. verschiedener Oligonukleotide auf die DNA-Bindungsaktivität von AP-1 nach 32 PMA/Ionomycin-Stimulation (Autoradiographie P-markierter AP-1-KonsensusSequenzen im EMSA). 30 min mit 15 ng/ml PMA und 700 ng/ml Ionomycin + stimulierte CD3 T-Lymphozyten aus dem Blut gesunder Spender wurden mit verschiedenen spezifischen Kompetitions-Antikörpern gegen Untereinheiten des AP-1-Komplexes bzw. mit AP-1- und NFκB-Oligonukleotiden vorinkubiert. Offenes Dreieck = Position des AP-1-DNA-Komplexes, offener Kreis = unspezifische Radioaktivitätsbindung der Probe, geschlossenes Dreieck = ungebundene Oligonukleotide. 40 Ergebnisse Der Transkriptionsfaktor AP-1 wurde mit PMA/Ionomycin induziert (Abb. 9, Spur 2). Die Zugabe von Anti-ATF-1- und Anti-ATF-2-Antikörpern (Abb. 9, Spuren 3 und 4) zeigte ebenso wie die Zugabe von Anti-JunB-, Anti-c-Fos-, und AntiCEBP-Antikörpern (Abb. 9, Spuren 5, 7 und 10) keinen Effekt auf die DNABindungskapazität von AP-1. Anti-p65-Antikörper und NFκB-Oligonukleotid hatten auf die DNA-Bindungsaktivität von AP-1 ebenfalls keinen Einfluss (Abb. 9, Spuren 11 und 12). Nach Zugabe eines Anti-JunD-Antikörpers zeigte sich eine zweite starke Bande oberhalb der AP-1-Bande (Abb. 9, Spur 6), die spezifische AP-1-Bande wurde schwächer. Nach Zugabe von Anti-c-JunAntikörper nahm die DNA-Bindungsaktivität von AP-1 in PMA/Ionomycin stimulierten T-Lymphozyten in vitro geringfügig ab, wenig oberhalb der AP-1Bande zeigte sich eine schwache schmale Bande (Abb. 9, Spuren 8 und 9). In der spezifischen Kompetition zeigte sich kein AP-1-spezifisches Signal nach Zugabe nicht-radioaktiv markierten AP-1-Oligonukleotides (Abb. 9, Spur 13). Damit konnte eine Beteiligung der Untereinheiten JunD und c-Jun am PMA/Ionomycin-induzierten AP-1-Komplex nachgewiesen werden. Da sich die DNA-Bindungsaktivität von AP-1 nach Zugabe von Antikörpern gegen ATF, JunB, c-Fos und CEBP nicht änderte, musste davon ausgegangen werden, dass diese Untereinheiten an der Zusammensetzung des PMA/Ionomycininduzierten AP-1-Komplex nicht oder nur zu einem nicht nachweisbaren Anteil beteiligt waren. Ebenso konnte ausgeschlossen werden, dass der Transkriptionsfaktor NFκB an der PMA/Ionomycin-induzierten DNA-Bindung von AP-1 beteiligt war. 4.1.5 Keine Hemmung von AP-1 durch Desfluran und Isofluran Es sollte die Frage beantwortet werden, ob andere volatile Anästhetika ebenfalls in der Lage sind, die DNA-Bindungsaktivität von AP-1 zu hemmen. Im Gegensatz zu Sevofluran konnten weder 18 Vol% Desfluran noch 5 Vol% Isofluran die PMA/Ionomycin-induzierte DNA-Bindungsaktivität von AP-1 in CD3+ T-Lymphozyten in vitro nach 24-stündiger Inkubation hemmen (Abb. 10, Spuren 2 und 3, bzw. Spuren 6 und 7). 41 Ergebnisse Abb. 10: Der Effekt von Desfluran und Isofluran auf die DNA-Bindungsaktivität 32 von AP-1 nach PMA/Ionomycin-Stimulation (Autoradiographie P-markierter + AP-1-Konsensus-Sequenzen im EMSA). CD3 T-Lymphozyten aus dem Blut gesunder Spender wurden 23,5 h mit 18 Vol% Desfluran bzw. 5 Vol% Isofluran inkubiert und anschließend 30 min mit 15 ng/ml PMA und 700 ng/ml Ionomycin stimuliert. Gezeigt ist das Ergebnis dreier unabhängiger Experimente. Offenes Dreieck = Position des AP-1-DNA-Komplexes, offener Kreis = unspezifische Radioaktivitätsbindung der Probe, geschlossenes Dreieck = ungebundene Oligonukleotide. 4.1.6 Geringer Sevofluran-vermittelter Effekt auf die DNABindungsaktivität von NFAT und NFκB Um der Frage nachzugehen, ob neben AP-1 auch andere, für die lymphozytäre Immunantwort relevante Transkriptionsfaktoren durch Sevofluran gehemmt werden, wurden NFAT (38) und NFκB (37) im EMSA untersucht. Die DNABindung beider Transkriptionsfaktoren wurde jeweils durch PMA/Ionomycin 42 Ergebnisse induziert (Abb. 11, Spuren 1 und 2 bzw. 5 und 6). Es konnte nur ein geringer Sevofluran-vermittelter Effekt auf die DNA-Bindungsfähigkeit von NFAT bzw. NFκB nachgewiesen werden (Abb. 11, Spur 3 bzw. 7). Sevofluran ohne PMA/Ionomycin-Stimulation zeigte weder eine Induktion der DNA- Bindungsaktivität von NFAT (Abb. 11, Spur 4), noch von NFκB (Abb. 11, Spur 8). Abb. 11: Der Effekt von 24 h 8 Vol% Sevofluran auf die DNA-Bindungsaktivität 32 von NFκB und NFAT nach PMA/Ionomycin-Stimulation (Autoradiographie P+ markierter Konsensus-Sequenzen im EMSA). CD3 T-Lymphozyten aus dem Blut gesunder Spender wurden 23,5 h mit 8 Vol% Sevofluran inkubiert und anschließend 30 min mit 15 ng/ml PMA und 700 ng/ml Ionomycin stimuliert. Gezeigt ist das Ergebnis dreier unabhängiger Experimente. Offenes Dreieck = Position des NFAT- bzw. NFκB-DNA-Komplexes, offener Kreis = unspezifische Radioaktivitätsbindung der Probe, geschlossenes Dreieck = ungebundene Oligonukleotide. 43 Ergebnisse 4.1.7 Keine Hemmung von SP-1 durch Sevofluran Im Folgenden sollte die Frage beantwortet werden, ob außer den drei besonders wichtigen Transkriptionsfaktoren AP-1, NFAT und NFκB weitere aktivierbare Transkriptionsfaktoren in ihrer DNA-Bindung durch Sevofluran beeinflusst werden. Hierzu wurde durch Stimulation mit PMA/Ionomycin die DNA-Bindung des Transkriptionsfaktors SP-1 induziert (Abb. 12, Spuren 1 und 2). Abb. 12: Der Effekt von 24 h 8 Vol% Sevofluran auf die DNABindungsaktivität von SP-1 nach PMA/Ionomycin-Stimulation 32 (Autoradiographie P-markierter SP-1-Konsensus-Sequenzen im + EMSA). CD3 T-Lymphozyten aus dem Blut gesunder Spender wurden 23,5 h mit 8 Vol% Sevofluran inkubiert und anschließend 30 min mit 15 ng/ml PMA und 700 ng/ml Ionomycin stimuliert. Offenes Dreieck = Position des SP-1-DNA-Komplexes, offener Kreis = unspezifische Radioaktivitätsbindung der Probe, geschlossenes Dreieck = ungebundene Oligonukleotide. 44 Jedoch konnte kein Ergebnisse Sevofluran-vermittelter Effekt auf die DNA- Bindungsaktivität von SP-1 nachgewiesen werden (Abb. 12, Spur 3). 4.1.8 Keine Hemmung von Oct-1 durch Sevofluran Die DNA-Bindungsaktivität des Transkriptionsfaktor Oct-1 wurde nachfolgend im EMSA untersucht. Es war unklar, ob neben induzierbaren Transkriptionsfaktoren wie NFAT, NFκB, AP-1 und SP-1 in CD3+ TLymphozyten auch konstitutiv aktive Transkriptionsfaktoren (Abb. 13, Spur 1) durch Sevofluran gehemmt werden können. Abb. 13: Der Effekt von 24 h 8 Vol% Sevofluran auf die DNABindungsaktivität von Oct-1 nach PMA/Ionomycin-Stimulation 32 (Autoradiographie P-markierter Oct-1-Konsensus-Sequenzen im + EMSA). CD3 T-Lymphozyten aus dem Blut gesunder Spender wurden 23,5 h mit 8 Vol% Sevofluran inkubiert und anschließend 30 min mit 15 ng/ml PMA und 700 ng/ml Ionomycin stimuliert. Offenes Dreieck = Position des Oct-1-DNA-Komplexes, offener Kreis = unspezifische Radioaktivitätsbindung der Probe, geschlossenes Dreieck = ungebundene Oligonukleotide. 45 Ergebnisse Die DNA-Bindungsaktivität von Oct-1 änderte sich nach Stimulation mit PMA/Ionomycin nicht (Abb. 13, Spur 2), eine 24-stündige Inkubation mit 8 Vol% Sevofluran zeigte nur einen geringen Einfluss auf die DNA-Bindungsaktivität von Oct-1 (Abb. 13, Spur 3). 4.2 Sevofluran hemmt die AP-1-abhängige Luziferase- Genexpression in transfizierten Jurkat T-Lymphozyten Ein Vektor mit einem AP-1-abhängigen Luziferasereportergen (s. Anhang) wurde transient in Jurkat T-Zellen transfiziert. Abb. 14: Der Effekt verschiedener volatiler Anästhetika auf die AP-1-vermittelte Reportergenexpression (pAP1(PMA)TA-Luc-Vektor) transient transfizierter Jurkat T-Lymphozyten. Die Zellen wurden während 24-stündiger Inkubation mit 18 Vol% Desfluran oder 8 Vol% Sevofluran mit 15 ng/ml PMA und 700 ng/ml Ionomycin stimuliert. Im Gesamtzelllysat wurde die Luziferase-Aktivität 3 bestimmt (angegeben in RLU x 10 ). *P < 0,05 verglichen mit PMA/Ionomycin in Abwesenheit des volatilen Anästhetikums. Dargestellt ist das Resultat von 6-8 unabhängigen Experimenten. 46 Ergebnisse Das Luziferase-Genprodukt entsteht nach PMA-Stimulation. Wird die DNABindungsaktivität von AP-1 gehemmt, sinkt die in „Relative Lights Units“ (RLU) gemessene Luziferase-Genexpression. Es konnte gezeigt werden, dass sich die Luziferase-Aktivität in transient transfizierten Jurkat T-Lymphozyten in vitro durch Stimulation mit PMA/Ionomycin im Vergleich mit Kontrollzellen induzieren lässt (Abb. 14, 1. und 2. Balken). Es konnte weiterhin gezeigt werden, dass 18 Vol% Desfluran keinen hemmenden Effekt auf die induzierte Luziferase-Aktivität in Jurkat T-Lymphozyten hatte (Abb. 14, 3. Balken). Durch 24-stündige Inkubation mit 8 Vol% Sevofluran wurde die induzierte Luziferase-Aktivität transfizierter und stimulierter T-Lymphozyten in vitro gehemmt (Abb. 14, 5. Balken). Ohne vorausgegangene Induktion mit PMA/Ionomycin hatten weder 24 h 8 Vol% Sevofluran noch 24 h 18 Vol% Desfluran einen Effekt auf die Luziferase-Aktivität transfizierter T-Lymphozyten (Abb. 14, 4. und 6. Balken). 4.3 Sevofluran hemmt die IL-3-Sekretion in primären Lymphozyten in vitro Der Transkriptionsfaktor AP-1 nimmt eine wichtige Funktion in der Regulation von Zellproliferation, Zelltransformation und Zelltod ein (63;64). Wenn sich die DNA-Bindungsaktivität von AP-1 verändert, werden AP-1-abhängig regulierte Gene in entsprechendem Ausmaß transkribiert. Folglich entsteht nach Translation auch eine unterschiedliche Menge an Protein. Als Genprodukt der AP-1-vermittelten Transkription wurde IL-3 (54) mittels ELISA untersucht. Als zentrales Element reguliert IL-3 die zelluläre Transformation und Differenzierung hämatopoetischer Stammzellen (27;36). Primäre CD3+ TLymphozyten aus dem Blut gesunder Spender sezernierten in vitro nach 24stündiger Stimulation mit 15 ng/ml PMA und 700 ng/ml Ionomycin etwa die sechsfache Menge an IL-3 verglichen mit unstimulierten Kontrollzellen (Abb. 15, 1. und 2. Balken). Nach 24-stündiger Inkubation mit 8 Vol% Sevofluran wurde die PMA/Ionomycin-induzierte IL-3-Sekretion in primären T-Lymphozyten aus dem Blut gesunder Spender um etwa ein Drittel reduziert (Abb. 15, 3. Balken). Die 24-stündige Inkubation mit 8 Vol% Sevofluran konnte ohne Zugabe von PMA/Ionomycin die T-lymphozytäre Sekretion von IL-3 nicht induzieren (Abb. 15, 4. Balken). 47 Ergebnisse Abb. 15: Der Effekt von 24 h 8 Vol% Sevofluran auf die IL-3-Sekretion primärer + CD3 T-Lymphozyten in vitro nach PMA/Ionomycin-Stimulation. Die Zellen wurden 24 h mit 15 ng/ml PMA und 700 ng/ml Ionomycin stimuliert und 24 h in 8 Vol% Sevofluran inkubiert. Der Zellüberstand wurde auf den IL-3-Gehalt mittels ELISA untersucht. *P < 0,05 verglichen mit PMA/Ionomycin-Stimulation in Abwesenheit von Sevofluran. Dargestellt ist das Ergebnis dreier unabhängiger Experimente. 4.4 Sevofluran aktiviert spezifische MAP-Kinasen Der Transkriptionsfaktor AP-1 wird durch die zytoplasmatisch vorliegende Kaskade der MAP-Kinasen aktiviert. Insbesondere die JNK sowie die p38-MAPKinase und die ERK kontrollieren durch differenzierte Phosphorylierungsvorgänge die AP-1-Aktivierung (56;57). Diese denaturierten Proteine können in einem SDS-Polyacrylamidgel durch spezifische Antikörper detektiert werden, wenn sie mit einem Peroxidase-gebundenen zweiten Antikörper gekoppelt werden. Nach Detektion mittels eines spezifischen ersten 48 Ergebnisse Antikörpers, der nur die phosphorylierte und damit aktive Variante der MAPKinase erkennt, war eine Aktivierung von ERK in CD3+ T-Lymphozyten durch PMA/Ionomycin nachweisbar (Abb. 16, oberer Teil, Spuren 1 und 2). 24 h 8 Vol% Sevofluran hatte keine Wirkung auf die phosphorylierte ERK (Abb. 16, oberer Teil, Spur 3). Sevofluran induzierte ohne Zugabe von PMA/Ionomycin keine ERK-Phosphorylierung (Abb. 16, oberer Teil, Spur 4). In JurkatLymphomzellen konnte eine Phosphorylierung der JNK nach PMA/IonomycinZugabe nachgewiesen werden (Abb. 16, mittlerer Teil, Spuren 1 und 2). Abb. 16: Western Blot zur Detektion von phosphorylierten MAP-Kinasen in gesamtzellulären Proteinextrakten mit 24 h 8 Vol% Sevofluran inkubierter + CD3 /Jurkat T-Lymphozyten. Peroxidase-Reaktion mittels HRP-konjugiertem 2. Antikörper. Sevofluran hatte auf phosphorylierte JNK keinen Einfluss (Abb. 16, mittlerer Teil, Spur 3). Sevofluran konnte ohne PMA/Ionomycin keine Phosphorylierung von JNK induzieren (Abb. 16, mittlerer Teil, Spur 4). In Jurkat-Lymphomzellen konnte eine Phosphorylierung der p38-MAP-Kinase nach Zugabe von PMA/Ionomycin induziert werden (Abb. 16, unterer Teil, Spuren 1 und 2). Sevofluran zeigte keine Wirkung auf die aktive phosphorylierte p38-MAPKinase (Abb. 16, unterer Teil, Spur 3). Im Unterschied zu den bereits untersuchten Kinasen ERK und JNK bewirkten 24 h 8 Vol% Sevofluran eine Induktion der Phospho-p38-MAP-Kinase (Abb. 16, unterer Teil, Spur 4). Sevofluran konnte somit zum einen die PMA/Ionomycin-induzierte DNA- 49 Ergebnisse Bindungsaktivität von AP-1 hemmen und führte in unbehandelten Zellen zu keiner Induktion von AP-1. Zum anderen zeigte sich in dieser Untersuchung, dass Sevofluran auf die PMA/Ionomycin-aktivierte p38-MAP-Kinase keinen Einfluss hatte, aber zu einer Aktivierung der p38-MAP-Kinase in unbehandelten Zellen führte. Die Aktivität der p38-MAP-Kinase hat nach neuen Erkenntnissen eine wichtige Funktion in der Induktion von Apoptose (51). So wird berichtet, dass die p38-MAP-Kinase verantwortlich ist für die charakteristischen morphologischen Merkmale wie nukleäre Kondensation und Fragmentation sowie Plasmamembranausstülpung (16), die apoptotische Zellen aufweisen. Es sollte daher untersucht werden, ob 24 h 8 Vol% Sevofluran Apoptose induzieren. 4.5 Sevofluran induziert Apoptose 4.5.1 Sevofluran induziert morphologische Apoptosemerkmale Es konnte in vorausgegangenen Untersuchungen gezeigt werden, daß die Inhalationsnarkotika Isofluran und Sevofluran (1,5 mM) Apoptose in peripheren T-Lymphozyten in vitro induzieren (39). In den vorliegenden Experimenten sollte daher zunächst untersucht werden, ob durch Sevofluran in dem gewählten Versuchsaufbau Apoptose in T-Lymphozyten induzierbar ist. Mit Sevofluran inkubierte Jurkat fluoreszenzlichtmikroskopisch auf T-Lymphozyten morphologische wurden Apoptosemerkmale untersucht. Als Kontrolle wurden sowohl unbehandelte als auch mit 1 µM MG132 inkubierte Zellen begutachtet. MG132 gilt durch Proteasom-Inhibition als potenter Apoptose-Induktor. Hoechst-Farbstoff zeigt in Zellen durch spezifische Färbung Zellkernfragmentierung) Zelluntergang eine an. Chromatinverdichtung Annexin Phospholipide V-FITC wie färbt (sogenannte bei apoptotischem Phosphatidylserine auf der Zellmembranaußenseite. Zur Differenzierung zwischen Apoptose und Nekrose wurden die Zellen zusätzlich mit PI gefärbt. Nur bei Nekrose färbt PI den Zellkern an, da die Zellmembran durchlässig geworden ist. In der Einzelzellbeobachtung zeigten unbehandelte Zellen in der Hoechstfärbung runde Zellkerne und waren weder positiv für Annexin V-FITC noch für PI (Abb. 17, obere Reihe). Nach Inkubation mit 1 µM MG132 über 24 h 50 Ergebnisse zeigten die Zellen eine starke Zellkernfragmentierung sowie eine deutliche Annexin-V-FITC-Färbung als Zeichen der Apoptose. In der PI-Färbung ließen sich auch nekrotische Zellen nachweisen (Abb. 17, mittlere Reihe). Sevofluran führte in einigen Zellen ebenfalls zu Zellkernfragmentierung. Es zeigte sich eine Färbung von Phosphatidylserin mit Annexin V-FITC, Nekrose konnte durch die fehlende Anfärbung mit PI ausgeschlossen werden (Abb. 17, untere Reihe). Abb. 17: Einzellzellmikroskopie mit Sevofluran (8 Vol%/24 h) inkubierter Jurkat T-Lymphozyten unter fluoreszierendem Licht. Vergleich mit unbehandelten und mit MG132 (1 µM/24 h) inkubierten Zellen. 4.5.2 Sevofluran induziert Annexin V-FITC-Positivität in TLymphozyten Um diese qualitativen Beobachtungen zu quantifizieren, wurden primäre CD3+ T-Lymphozyten gesunder Spender (Vollblut-Proben) mit Sevofluran inkubiert und in einer FACS-Analyse auf eine Annexin V-FITC- und 7-AAD-Fluoreszenz untersucht. Der Antikörper 7-AAD kann nur dann an DNA binden, wenn die Zellkernmembran durch Nekrose durchlässig geworden ist. In der FACS- 51 Ergebnisse Analyse zeigte sich unter Sevofluran eine etwa dreifache Zunahme der Annexin V-FITC-Positivität gegenüber den unbehandelten Zellen (Abb. 18). Gleichzeitig unterschieden sich unbehandelte Zellen kaum hinsichtlich ihrer 7-AADPositivität von Zellen, die mit Sevofluran inkubiert worden waren. Es konnte somit nachgewiesen werden, dass Sevofluran – wenn auch nur in begrenztem Umfang – signifikant Apoptose induziert,. + Abb. 18: FACS-Analyse primärer CD3 T-Lymphozyten auf 7-AADund Annexin V-FITC-Positivität nach Inkubation mit 24 h 8 Vol% Sevofluran im Vergleich zu Kontrollzellen. T-Lymphozyten wurden nach Markierung ihres CD3-Oberflächenantigens mit 7-AAD und Annexin V-FITC inkubiert. Dargestellt ist das Resultat dreier Experimente. 4.6 Sevofluran induziert Caspase-abhängige Apoptose 4.6.1 Nachweis aktiver Caspasen nach Sevofluran-Inkubation in TLymphozyten in vitro Es stellte sich die Frage nach dem molekularen Mechanismus einer durch Sevofluran induzierten Apoptose. Caspasen gelten als Mediatoren der zellulären Apoptose (18). Mehrere Caspasen liegen zytoplasmatisch in ihrer 52 Ergebnisse inaktiven Form als Pro-Caspasen vor und werden zur Aktivierung an bestimmten Stellen gespalten, wodurch aktive Caspase (sogenanntes „Caspase-Cleavage“) entsteht. Innerhalb der zellulären Caspasen-Kaskade nimmt Caspase-3 eine zentrale Stellung ein, da an dieser Stelle mehrere zelluläre Signaltransduktionswege zusammenlaufen, die in einem apoptotischen Zelluntergang enden (18). Daher sollte nachgewiesen werden, ob nach Sevofluran-Inkubation in T-Lymphozyten in vitro eine veränderte Caspase-3-Aktivität nachweisbar ist. Das Prinzip dieser Methode beruht darauf, dass Acetyl-DEVD-7-Amino-4-methylcoumarin in einem Zelllysat in Gegenwart aktiver Caspase-3, -6 oder -7 gespalten wird (18). Das Spaltprodukt emittiert bei einer bestimmten Wellenlänge fluoreszierendes Licht, welches gemessen wird. Die Intensität des fluoreszierenden Lichtes lässt auf die Menge der aktiven Caspasen in der Probe schließen. Es zeigte sich in diesem „Fluorogenic Assay“, dass aktive Caspasen in unbehandelten Jurkat T-Lymphozyten vorhanden sind (Abb. 19, 1. Balken). 1 µM MG132 über 24 h bewirkte eine 6fache Induktion aktiver Caspasen (Abb. 19, 2. Balken). Abb. 19: „Fluorogenic Assay” zur Messung aktiver Caspasen in Jurkat TLymphozyten in vitro. Induktion aktiver Caspasen mit 1 µM MG132 über 24 h gegenüber unbehandelten Zellen. Vergleich der Caspase-Aktivität nach Inkubation mit 8 Vol% Sevofluran und 18 Vol% Desfluran über 24 h. 53 Ergebnisse Eine 24-stündige Inkubation mit 8 Vol% Sevofluran führte in Jurkat TLymphozyten ebenfalls zu einer Induktion (2,5-fach) aktiver Caspasen (Abb. 19, 3. Balken). Da Sevofluran signifikant, aber in geringem Maß Apoptose induziert, ist entsprechend nur eine schwache Caspase-3-Aktivierung nachweisbar. 18 Vol% Desfluran über 24 h hatten auf die Induktion aktiver Caspasen keinen signifikanten Einfluss (Abb. 19, 4. Balken). 4.6.2 Nachweis aktiver Caspase-3 nach Sevofluran-Inkubation in TLymphozyten in vitro Mittels Western Blot-Technik wurde versucht, Caspase-3 zu detektieren. Caspase-3 liegt zytoplasmatisch als Pro-Caspase-3 mit einem Molekulargewicht von 37 Kilodalton (kD) vor. Bei Aktivierung wird Caspase-3 mit einer Größe von 20 kD abgespalten. Durch sogenanntes „Auto-Cleavage“ eines Fragmentes mit einem Molekulargewicht von 3 kD entsteht eine Caspase3 mit einem Molekulargewicht von 17 kD. Beide Varianten gelten als aktive Caspase-3. Die in der Untersuchung verwendeten spezifischen Antikörper erkannten neben der Pro-Caspase-3 sowohl das 17 kD große Fragment als auch das 20 kD große Caspase-3-Cleavage-Produkt. Neben unbehandelten Zellen wurde als Kontrolle in Jurkat T-Lymphozyten Caspase-Aktivität mit 1 µM Staurosporin und mit 1 µM und 10 µM MG132 induziert. Abb. 20: Anti-Caspase-3-Western Blot aus Totalextrakten mit Sevofluran, + Staurosporin oder MG132 inkubierten CD3 T-Lymphozyten im Vergleich mit unbehandelten Zellen. Peroxidase-Reaktion mittels HRP-konjugiertem 2. Antikörper. Der 1. Antikörper erkennt sowohl Procaspase-3, als auch die geschnittenen Caspase-3-Versionen (Caspase-Cleavage). 54 Ergebnisse Aktive Caspase-3 wurde in Jurkat T-Lymphozyten nach 24-stündiger Inkubation mit 8 Vol% Sevofluran untersucht. In unbehandelten Jurkat T-Lymphozyten ließ sich nur wenig geschnittene (und damit aktive) Caspase-3 nachweisen (Abb. 20, Spur 1). Nach Inkubation mit MG132 oder mit Staurosporin zeigten sich beide aktiven Caspase-3-Formen, aber kaum Pro-Caspase-3. (Abb. 20, Spuren 2-4). Die Inkubation mit 8 Vol% Sevofluran schien eine Caspase-3-Aktivierung zu bewirken, allerdings ist nach wie vor deutlich mehr Pro-Caspase-3 als aktive Caspase-3 nachweisbar (Abb. 20, Spuren 5 und 6). Die Stimulation mit 15 ng/ml PMA und 700 ng/ml Ionomycin über 30 min hatte auf die Caspase-3Aktivität mit Sevofluran inkubierter Zellen keinen Einfluss (Abb. 20, Spuren 7 und 8). 55 Diskussion 5 Diskussion In dieser Arbeit wurde untersucht, wie volatile Anästhetika die DNABindungsaktivität von Transkriptionsfaktoren beeinflussen. Diese Erkenntnisse sind wichtig, um die beobachteten uneinheitlichen Wirkungen von Inhalationsanästhetika zu erklären: Es ist bekannt, dass nach IsofluranInkubation Alveolarmakrophagen verstärkt IL-8 und IFN-γ exprimieren (32). Zum anderen konnte nach Gabe volatiler Anästhetika in peripheren mononukleären Zellen eine Suppression der Expression von IL-1β und TNF-α beobachtet werden (44). Die molekularen Mechanismen, über die diese veränderten Zytokinexpressionsmuster vermittelt wurden, blieben bis jetzt unklar. Die erste Hypothese dieser Arbeit war, dass volatile Anästhetika über eine Beeinflussung der DNA-Bindung der Transkriptionsfaktoren AP-1, NFAT und NFκB, eine Veränderung der abhängigen Genexpressionsmuster bewirken. Inhalationsanästhetika sind nicht nur für ein verändertes Genexpressionmuster verantwortlich (32;44), sondern induzieren auch Apoptose: So konnten im Blut von Patienten unter Isofluran-Narkose vermehrt apoptotische neutrophile Granulozyten und Lymphozyten nachgewiesen werden (13;14). In vitro erhobene Daten zeigen eine Induktion der Apoptose in peripheren Lymphozyten durch Isofluran und Sevofluran (39). Ein möglicher molekularer Mechanismus dieses Phänomens konnte nicht nachgewiesen werden. Daher sollte untersucht werden, welche Gründe für die Induktion von Apoptose durch Inhalationsanästhetika verantwortlich sind. Die Veränderung des DNABindungsverhaltens von AP-1 kann sowohl pro-, als auch antiapoptotische Wirkungen haben (63). Daher war die zweite Hypothese, dass die Veränderung der DNA-Bindung des Transkriptionsfaktors AP-1 und die Aktivierung der AP-1regulierenden MAP-Kinasen an der Induktion von Apoptose durch Inhalationsanästhetika beteiligt sind. 5.1 Hemmung von AP-1 durch Sevofluran Ein wesentliches Ergebnis dieser Arbeit ist die dosis- und zeitabhängige Hemmung der DNA-Bindungsaktivität des Transkriptionsfaktors AP-1 durch Sevofluran. Bei einer langen Inkubationszeit scheint die Sevofluran- 56 Diskussion Konzentration für eine deutliche Hemmung der DNA-Bindungsaktivität von AP-1 offensichtlich unerheblich zu sein, denn bei einer 24-stündigen Inkubation hemmen unterschiedlich hohe Sevofluran-Konzentrationen die DNA- Bindungsaktivität von AP-1 in gleichem Maße. Dies spricht für eine kumulative Wirkung von Sevofluran. Im Einklang mit der Sevofluran-Anreicherung im Medium nahm die DNA-Bindung von AP-1 mit steigender SevofluranKonzentration sukzessive ab. Trotz dieser Proportionalität ist unabhängig von der Konzentration an Sevofluran ab einem bestimmten Punkt die maximale im EMSA nachweisbare Aktivitätshemmung von AP-1 erreicht, so dass eine Dosiserhöhung keinen darüber hinausgehenden Effekt hätte. Bei hohen Sevofluran-Konzentrationen ist bereits nach 12 h eine Abnahme der DNA-Bindungsaktivität von AP-1 zu beobachten, was ein Hinweis darauf ist, dass die spezifischen Wirkungen nach 24 h Sevofluran-Inkubation wahrscheinlich auch nach der Hälfte der Zeit eingetreten wären. Dies stützt die Hypothese eines Punktes der größtmöglichen AP-1-Hemmung. Folge ist, dass für die maximale Hemmung der DNA-Bindung des Transkriptionsfaktors AP-1 eine ganz bestimmte Konzentration an Anästhetikum vorliegen muss. Diese Konzentration wäre um so früher erreicht, je höher die eingesetzte SevofluranKonzentration ist. Als Konsequenz könnte das Versuchsprotokoll in zukünftigen Arbeiten dahingehend modifiziert werden, dass die DNA-Bindung von AP-1 bei maximaler Sevofluran-Konzentration nach deutlich kürzerer Sevofluran- Inkubationszeit als 24 h untersucht wird. Bei langen Expositionszeiten könnte entsprechend die Sevofluran-Konzentration verringert werden. Die Transkriptionsfaktoren NFAT und NFκB wurden – im Gegensatz zu AP-1 – durch Sevofluran nur in einem geringem Ausmaß gehemmt, was zum einen beweist, dass AP-1 gegenüber polyfluorierten Ätherverbindungen sehr empfindlich und deutlich sensibler als NFAT und NFκB zu reagieren scheint. Diese Beobachtung deckt sich mit Angaben zur DNA-Bindungsaktivität von AP1, die sehr sensitiv auf eine Vielzahl heterogener Einflüsse (z. B. Anstieg von cAMP oder [Ca2+], UV-Licht, Wachstumsfaktoren, proinflammatorische Zytokine) auf die Zelle reagiert (21;29). Zum anderen konnte so gezeigt werden, 57 Diskussion dass Sevofluran AP-1-spezifische Wirkungen besitzt, was bisher nicht bekannt war. Die volatilen Anästhetika Desfluran und Isofluran, die ebenfalls im klinischen Alltag eingesetzt werden, zeigten keinen Einfluss auf die DNA-Bindungsaktivität von AP-1. Die Tatsache, dass nur Sevofluran in der Lage war, die DNABindung des Transkriptionsfaktors AP-1 zu hemmen, und Desfluran sowie Isofluran trotz ähnlicher pharmakologischer Eigenschaften keine Hemmung von AP-1 verursachten, war ein weiterer Hinweis für eine differenzierte Wirkung von Sevofluran. Die Spezifität des Sevofluran-vermittelten Effektes auf die DNA-Bindung von AP-1 zeigte sich in der Beobachtung, dass eine Inkubation mit Desfluran im Gegensatz zu Sevofluran nicht zu einem Verlust der Luziferaseaktivität in transfizierten Lymphomzellen führte, und wird durch die Tatsache unterstützt, dass Sevofluran bestimmte pharmakologische Eigenschaften besitzt, die es gegenüber Desfluran und Isofluran abgrenzt: Sevofluran stellt von den untersuchten Inhalationsanästhetika die komplexeste Ätherverbindung dar. Bekannt sind die toxischen Eigenschaften von Äthern, die zu DNA-Schädigung führen und Konsequenzen der chemischen Struktur dieser Verbindungen sind (19). Nach Sevofluran-Narkose entstehen aufgrund verstärkter Metabolisierung (3-5% im Gegensatz zu <0,2% für Desfluran und Isofluran) toxische Stoffwechselprodukte. Neben Kohlenmonoxid (78) (auf das in der vorliegenden Arbeit nicht eingegangen wurde) entsteht zum anderen unter SevofluranNarkose in signifikantem Ausmaß anorganisches Fluorid, da Sevofluran ein sehr instabiler halogenierter Kohlenwasserstoff ist. Toxische Eigenschaften von Fluorid sind seit längerem bekannt (82). Obwohl Fluorid auch in Lymphozyten zyto- und genotoxisch (30) wirkte und in HL-60-Zellen zu Caspase-3Aktivierung und Apoptose führte (3), wurden die in dieser Arbeit verwendeten Lymphozytenkulturen nicht auf ihren Fluoridgehalt untersucht. Desfluran und Isofluran – obwohl ebenfalls halogenierte Kohlenwasserstoffe – sind als Moleküle stabiler als Sevofluran und bilden aus diesem Grund deutlich weniger Fluorid. Die DNA-Bindung der Transkriptionsfaktoren NFAT und NFκB wurde durch Sevofluran in geringem Ausmaß gehemmt. Um eine unspezifische Wirkung 58 Diskussion dieses lipophilen Inhalationsanästhetikums auf diese Transkriptionsfaktoren auszuschließen, wurde die DNA-Bindungsaktivität von SP-1 (induzierbar) und Oct-1 (konstitutiv aktiv) untersucht. Da auch nach 24-stündiger Inkubation mit Sevofluran keine Hemmung der DNA-Bindungsaktivität dieser beiden Transkriptionsfaktoren nachgewiesen werden konnte, spricht dies für einen selektiven Effekt von Sevofluran auf die DNA-Bindung von AP-1. Eine Hemmung von AP-1 und eine fehlende Beeinträchtigung der DNA-Bindung von NFAT und NFκB ist besonders interessant, da diese drei Transkriptionsfaktoren für die „immediate early gene response“ im Rahmen der T-lymphozytär-vermittelten Immunantwort von großer Bedeutung sind. Dabei wird innerhalb kürzester Zeit die Transkription wichtiger Gene der Immunantwort induziert (21;41;48). Der Grund für die starke Wirkung von Sevofluran auf die DNA-Bindung von AP-1 und die vergleichsweise schwache Wirkung auf die DNA-Bindung von NFAT und NFκB liegt möglicherweise in der Eigenschaft der Lymphozyten, auf externe Einflüsse bevorzugt mit der Aktivierung bzw. Hemmung des Transkriptionsfaktors AP-1 zu reagieren. Ein Argument für diese These ist die Heterogenität der Faktoren (z. B. Anstieg von cAMP oder [Ca2+], UV-Licht, Wachstumsfaktoren, proinflammatorische Zytokine), die zu einer Veränderung des DNA-Bindungsverhaltens von AP-1 führt (29). Ebenfalls wird deutlich, dass eine Inkubation mit Sevofluran für eine signifikante Hemmung der aktivierten Transkriptionsfaktoren NFAT und NFκB nicht auszureichen scheint. Dies war unerwartet, da NFAT oft ähnliche Funktionen wie AP-1 besitzt (38) und andererseits auch NFκB auf verschiedene externe Stimuli mit einer Veränderung der DNA-Bindung reagiert (37);(48). Die differenzierte Beeinflussung der DNA-Bindung der untersuchten Transkriptionsfaktoren AP-1, NFAT, NFκB, SP-1 und Oct-1 durch Sevofluran spricht auch für ein selektives Aktivitätsprofil der Lymphozyten. Nicht jeder externe Stimulus führt zur Aktivitätsänderung aller Transkriptionsfaktoren und zeigt, dass die DNA-Bindungsaktivität der Transkriptionsfaktoren je nach Bedarf verändert wird und dabei zunehmen oder abnehmen kann. Bei Untersuchung der an der Sevofluran-vermittelten Hemmung beteiligten Untereinheiten des AP-1-Komplexes stellte sich heraus, dass neben c-Jun vor 59 Diskussion allem JunD unter Inkubation mit Sevofluran eine veränderte DNA-Bindung zeigte, während keine Veränderung des Bindungsverhaltens der AP-1Untereinheiten c-Fos, ATF-1 und ATF-2 sowie CEBP nachweisbar war. Der molekulare Mechanismus der Sevofluran-Wirkung auf die beiden JunUntereinheiten von AP-1 bleibt unklar. Eine Hypothese wäre eine direkte Interaktion von Sevofluran und der basischen Leuzin-Zipper-Region von c-Jun und JunD. Eine Dimerisierung mit Fos-Proteinen und die transaktivierende DNA-Bindung wären so effektiv verhindert. Da Fos-Proteine keine Homodimere bilden können, wäre eine weitgehend funktionslose Fos/JunB- Heterodimerisierung denkbar, da die DNA-Bindung von JunB und der FosUntereinheiten durch Sevofluran-Inkubation unbeeinflusst bleibt. Welche biologische Relevanz könnte die Sevofluran-vermittelte Hemmung der Untereinheiten c-Jun und JunD haben? Die Funktion der Untereinheiten des AP-1-Komplexes scheint sehr unterschiedlich zu sein und je nach Zellart zu variieren (11): c-Jun ist eine der wichtigsten AP-1-Untereinheiten. C-Jun/c-FosHeterodimere haben die höchste transaktivierende Potenz (49), selbst c-JunHomodimere besitzen noch ein hohes Maß an Aktivität (60). Es herrscht Übereinstimmung darüber, dass c-Jun nicht nur eine spezifische Funktion besitzt (63). Phorbolester – wie PMA in der vorliegenden Arbeit – induzieren die DNABindung der c-Jun-Untereinheit, die dadurch die Proliferation hämatopoetischer Stammzellen unterstützt. Dabei konnte auch gezeigt werden, dass die c-Junvermittelte Steigerung der AP-1-Aktivität die Abhängigkeit dieser Zellen von IL-3 vermindert (41). Diese Wachstumsfaktor-ähnliche Eigenschaft wird unterstützt durch die Beobachtung, dass c-Jun über eine Hemmung der Tumorsuppressorgene p53 und p16 einen proliferativen Stimulus darstellen kann (63;64). Somit könnte die Behinderung einer effektiven DNA-Bindung von c-Jun durch Sevofluran zu einer Beeinträchtigung der proliferativen Eigenschaften dieser Untereinheit führen. Durch diesen antiproliferativen Stress könnten proapoptotische Signaltransduktionswege aktiviert werden, was eine mögliche Erklärung für die unter Sevofluran-Inkubation beobachtete Apoptose sein könnte und c-Jun für diesen Fall eine zentrale Rolle zuweist. Andererseits ist die Untereinheit c-Jun des AP-1-Komplexes in der Lage, über 60 Diskussion transkriptionelle Regulierung caspase-abhängige Apoptose in Fibroblasten zu induzieren (9). Da zelluläre Proliferation und Apoptose separat voneinander beobachtet werden konnten, kann davon ausgegangen werden, dass c-Jun neben der Fähigkeit, den Zellzyklus zu regulieren auch davon unabhängige proapoptotische Eigenschaften besitzt. JunD und c-Jun scheinen ähnliche Funktionen zu haben (5), von JunB sind vor allem c-Jun-antagonistische Effekte bekannt (63). JunD gilt im Vergleich mit JunB als stärkerer Partner. Die transaktivierende Fähigkeit von JunD ist aufgrund einer schwächeren Bindung im AP-1-Komplex aber deutlich geringer als die von c-Jun (60). JunD besitzt demnach auch als Homodimer transaktivierende Potenz, welche um ein Vielfaches durch Fos- Heterodimerisierung gesteigert werden kann (60). In hohen Konzentrationen wirkt JunD hemmend auf die AP-1-Aktivität (5). Als möglicher Grund wird eine Verdrängung potenterer Jun-Untereinheiten aus ihren Bindungsstellen angeführt. Neuere Ergebnisse konnten in JunD-/- MEF-Zellen eine p53abhängige Hemmung des Wachstums und morphologische Apoptosemerkmale zeigen (74), was eine antiapoptotische Rolle für JunD bedeuten würde. Unter Sevofluran-Inkubation wurde eine deutliche Hemmung der DNA- Bindungsaktivität von JunD und die Induktion von Apoptose nachgewiesen. Diese Beobachtung könnte darauf hinweisen, dass die Aktivierung von JunD in der vorliegenden Arbeit einen proliferativen Stimulus darstellt. Eine Hemmung der DNA-Bindungsaktivität dieser Untereinheit würde somit einen antiproliferativen Stress darstellen, was möglicherweise zu einem Übergewicht proapoptotischer Stimuli und zur Induktion von Apoptose führt. Eine komplexe Wirkung – wie für c-Jun gezeigt – konnte bisher für JunD nicht nachgewiesen werden. In dieser Arbeit wurde die DNA-Bindung von AP-1 in T-Lymphozyten durch PMA/Ionomycin induziert. Dies könnte einen proliferativen (mitogenen) Stimulus darstellen (41;72). Eine Hemmung des aktivierten Transkriptionsfaktors AP-1 wäre somit als Inhibition einer c-Jun- und JunD-vermittelten Proliferation anzusehen. Diese Proliferation würde auch das Vorhandensein von IL-3 voraussetzen, da dieses Zytokin für Zellwachstum und -differenzierung unerlässlich ist (47). 61 Diskussion 5.2 Sevofluran und Interleukin-3 IL-3 ist als Zytokin für die Entwicklung und Transformation hämatopoetischer Stammzellen von zentraler Bedeutung: Bei vermindertem IL-3-Spiegel kommt es je nach Zellart in vitro zu Apoptose (43) oder ausbleibender Differenzierung (26). In dieser Arbeit konnte gezeigt werden, dass eine Hemmung der DNABindungsaktivität von AP-1 in primären T-Lymphozyten mit einer verminderten IL-3-Sekretion einhergeht. Interessanterweise konnte eine Sevofluran-vermittelte Hemmung der DNABindungsaktivität des Transkriptionsfaktors AP bereits bei niedrigen SevofluranKonzentrationen nachgewiesen werden, womit auch eine Beeinträchtigung der AP-1-abhängigen Zytokinproduktion (IL-3) bei 2 Vol% Sevofluran möglich scheint. Die Transkription des IL-3-Genes nach proinflammatorischer Induktion wird durch verstärkte DNA-Bindung der beiden Transkriptionsfaktoren AP-1 und NFAT reguliert (38;47). NFAT und AP-1 stabilisieren sich durch gemeinsame DNA-Bindung gegenseitig, was ihre transkriptionelle Aktivität deutlich erhöht (38). Sevofluran hemmte in den vorliegenden Experimenten nach Induktion die DNA-Bindungsaktivität von NFAT unwesentlich. Es stellte sich daher die Frage, ob die Abnahme der IL-3-Produktion nach Sevofluran-Inkubation auch durch eine Suppression der DNA-Bindung von NFAT verursacht sein könnte. Die verminderte IL-3-Sekretion spricht für einen deutlichen Aktivitätsverlust der IL-3regulierenden Transkriptionsfaktoren. Ein – wenn auch geringer – Einfluss von NFAT auf diese verminderte IL-3-Sekretion kann nicht ausgeschlossen werden. Da die DNA-Bindung von NFAT durch Sevofluran jedoch kaum beeinflusst wurde, ist davon auszugehen, dass in dieser Arbeit hauptsächlich die Hemmung der DNA-Bindungsaktivität von AP-1 für die geringe IL-3-Sekretion nach Sevofluran-Inkubation verantwortlich war. Von potentieller klinischer Bedeutung ist die Beobachtung, dass bei großen chirurgischen Eingriffen eine perioperative Neutropenie beobachtet wird (7;68), deren Ursachen weitgehend unklar sind. In dieser Arbeit konnte gezeigt werden, dass Sevofluran nach Applikation in hohen Konzentrationen über einen langen Zeitraum mittels Hemmung der DNA-Bindung des Transkriptionsfaktors AP-1 die lymphozytäre IL-3-Sekretion verminderte. Da auch nachgewiesen werden konnte, dass Sevofluran in geringen Konzentrationen die DNA-Bindung von AP-1 hemmt, könnte spekuliert werden, dass in vivo die lymphozytäre IL-3- 62 Diskussion Sekretion während einer langen Sevofluran-Narkose ebenfalls abnimmt. Da IL3 für die Proliferation und Differenzierung hämatopoetischer Stammzellen unerlässlich ist (54), könnte es sich dabei um einen möglichen molekularen Mechanismus dieser perioperativen Schwächung des Immunsystems handeln (37). 5.3 Einfluss von Sevofluran auf die MAP-Kinasen Die Familie der MAP-Kinasen bildet im Zytoplasma von T-Zellen eine Signaltransduktionskaskade, die über differenzierte Phosphorylierung zur Induktion der DNA-Bindung und damit Aktivierung des Transkriptionsfaktors AP-1 führt (15;57). Unklar bleibt ihre Rolle in Bezug auf eine Sevofluranvermittelte Hemmung der AP-1-Untereinheiten c-Jun und JunD. Die Regulation von JunD ist nach wie vor nicht vollständig geklärt. JunD stellt für JNK ein schlechtes Substrat dar (74). Die JNK gilt hingegen als zentrales Element der cJun-Regulation (64;67). Eine Sevofluran-vermittelte Hemmung von JNK konnte auch nach 24-stündiger Inkubation nicht nachgewiesen werden. Auf der anderen Seite konnte Sevofluran die c-Jun-Komponente des AP-1-Komplexes hemmen. Eine mögliche Erklärung hierfür ist, dass in dieser Arbeit nicht untersuchte JNKIsoformen (56;70) in ihrer Aktivität unbeeinflusst blieben. Ein weiterer Grund könnte sein, dass eine Hemmung dieser beiden Jun-Komponenten unabhängig von der Aktivität der regulierenden MAP-Kinase auftritt: Vorstellbar wäre eine direkte Interaktion von Sevofluran und spezifischen Jun-Untereinheiten des AP1-Komplexes und damit ein Verlust der DNA-Bindungsaktivität. Es könnte auch spekuliert werden, (TGAG/CTCA) in dass Sevofluran Wechselwirkung mit tritt der und TRE-Sequenz über einen der DNA unbekannten Mechanismus für die c-Jun- und JunD-Untereinheit des Transkriptionsfaktors AP-1 an dieser Stelle einen kompetitiven Antagonisten darstellt. Dies könnte die Dosis- und Zeitabhängigkeit der Sevofluran-vermittelten Hemmung der DNABindungsaktivität von AP-1 erklären. Sollte die Beendigung der SevofluranInkubation nach einem bestimmten Zeitraum zu einer vollständigen Aufhebung der Hemmung der DNA-Bindung von AP-1 führen, wäre auch das ein weiterer Hinweis auf einen kompetitiven Antagonismus. 63 Diskussion Eine Hemmung der c-Fos-Untereinheit konnte nach Sevofluran-Inkubation nicht beobachtet werden. Somit ist es nicht erstaunlich, dass eine Hemmung der cFos-regulierenden ERK (64) nicht nachgewiesen werden konnte. Inkubation mit Sevofluran zeigte in dieser Arbeit keinen hemmenden Einfluss auf die mit PMA/Ionomycin aktivierten MAP-Kinasen, die für die Regulation der AP-1-Untereinheiten verantwortlich sind. Daher findet sich kein Hinweis, dass die beobachtete Hemmung der DNA-Bindungsaktivität des Transkriptionsfaktors AP-1 nach Sevofluran-Inkubation unter Mitbeteiligung der MAP-Kinasen stattfindet. Interessanterweise zeigte die p38-MAP-Kinase, die hauptsächlich für die Aktivität der ATF-2-Untereinheit des AP-1-Komplexes verantwortlich ist (21;63), nach Sevofluran-Inkubation eine Aktivitätszunahme. Die vier bekannten Isoformen (α-δ) der p38-MAP-Kinase haben nach neuen Erkenntnissen unterschiedliche Funktionen (51). Allen gemeinsam ist ihre Induzierbarkeit durch proinflammatorische Stimuli (67). Unterschiede gibt es jedoch hinsichtlich der Wirkung auf die Aktivierung von AP-1. Innerhalb derselben Zellreihe bewirkt die p38β-Isoform über eine verstärkte c-jun-Transkription eine Zunahme der AP-1-Aktivität, p38γ und p38δ haben auf diese AP-1-Aktivierung keinen oder einen entgegengesetzten Effekt (51). Die Phosphorylierung von c-Jun – ein Vorgang, der ebenfalls zur Aktivierung dieser AP-1-Untereinheit führt (29) – scheint durch die p38β-Isoform gesteigert, durch p38γ- oder p38δ-Isoformen gehemmt zu werden, was von Bedeutung ist, da die Möglichkeit einer c-JunPhosphorylierung durch die p38-MAP-Kinase kontrovers beurteilt wird (25;28;75;79). Der Nachweis einer phosphorylierten p38-MAP-Kinase und der fehlende Nachweis einer AP-1-Aktivierung sprechen daher für eine selektive Induktion der γ- und δ-Isoform dieser Kinase durch Sevofluran. Da die Induktion der DNABindung des Transkriptionsfaktors AP-1 mit PMA/Ionomycin sowohl zu einer Aktivierung der p38-MAP-Kinase als auch zu einer Aktivierung der Untereinheiten c-Jun und JunD des Transkriptionsfaktors AP-1 führt, spricht dies für eine zusätzliche Aktivierung der α- und β-Isoform dieser MAP-Kinase. Um diese Hypothese zu überprüfen, müssten Western Blots mit Antikörpern, die spezifisch die Aktivität der einzelnen p38-Untereinheiten abbilden, 64 Diskussion durchgeführt werden. Da nach Sevofluran-Inkubation eine Hemmung der Untereinheiten c-Jun und JunD, nicht aber der Untereinheit JunB und der p38MAP-Kinase beobachtet wurden, könnte gefolgert werden, dass die im Western Blot detektierte p38-Isoform zu einer selektiven Aktivierung von JunB beiträgt, während eine Hemmung von c-Jun und JunD entweder unabhängig von MAPKinasen oder durch eine nicht-nachgewiesene Isoform der p38-MAP-Kinase vermittelt wurde. Die Aktivierung der p38-MAP-Kinase durch Sevofluran könnte unterschiedliche Folgen haben: Während die β-Isoform Fas-Ligand- und UV-Licht-vermittelte Apoptose abschwächen kann (46) und durch Phosphorylierung die proliferative Aktivität von c-Jun steigern kann (51), wird die p38-MAP-Kinase mit spontaner Apoptose in neutrophilen Granulozyten in Verbindung gebracht (4) und induziert Apoptose nach Entzug von Wachstumsfaktoren (34). Dabei könnte auch das Protoonkogen c-Myc eine entscheidende Rolle spielen (81). Aktive p38-MAPKinase führte demnach zu klassischen morphologischen Apoptosemerkmalen wie Caspase-Aktivierung, nukleärer Kondensation und Fragmentation sowie zur Ausstülpung der Plasmamembran (16). Interessanterweise führt die p38Aktivität sowohl caspase-abhängig wie auch caspase-unabhängig zu diesen morphologischen Veränderungen, was der p38-MAP-Kinase eine frühe Rolle im Mechanismus der Apoptose zuweist (16) und die Frage nach CaspaseBeteiligung bei Sevofluran-induzierter Apoptose aufwarf. 5.4 Sevofluran und Apoptose In dieser Arbeit wurde eine Aktivierung der p38-MAP-Kinase durch Sevofluran nachgewiesen. Gleichzeitig ist bekannt, dass Sevofluran über einen unklaren Mechanismus Apoptose in Lymphozyten induziert (39). Daher sollte die Assoziation zwischen Aktivierung der p38-MAP-Kinase durch Sevofluran einerseits und Induktion von Apoptose andererseits untersucht werden. Nukleäre Kondensation und Fragmentation kann sowohl caspase-abhängig als auch caspase-unabhängig auftreten (16). Nach Sevofluran-Inkubation konnten in den untersuchten Zellen in geringem Ausmaß morphologische ApoptoseKriterien und aktive Caspasen nachgewiesen werden. Charakteristischerweise zeigte sich auch hier eine weitere spezifische Eigenschaft von Sevofluran 65 Diskussion gegenüber Desfluran, da Desfluran-Inkubation zu keiner Caspase-Aktivierung führte. Da der Nachweis der Effektor-Caspase-3 als zentralem Mediator der Sevofluran-induzierten Apoptose gelungen ist, ist es sehr wahrscheinlich, dass Caspasen für die charakteristische Morphologie der beobachteten apoptotischen Zellen (18) verantwortlich sind. Allerdings steht für den definitiven Nachweis ein Versuch mit einem Pan-Caspase-Inhibitor (zVAD.fmk) aus. Ließen sich nach zVAD.fmk-Behandlung keine morphologischen Apoptosemerkmale parallel zu gesteigerter p38-Aktivität nachweisen, könnte damit gezeigt werden, dass die Sevofluran-induzierte und über die p38-MAPKinase-vermittelte Apoptose caspase-abhängig wäre. Wären nach Gabe eines spezifischen p38-MAP-Kinase-Hemmers (SB203580) keine apoptotischen Zellen zu beobachten, wäre auch die zentrale Rolle der p38-MAP-Kinase in der Vermittlung der Sevofluran-induzierten Apoptose bestätigt. Eine direkte Verbindung zwischen AP-1-Hemmung und verminderter IL-3Sekretion sowie caspase-3-abhängiger Apoptose ist nicht bewiesen. Während in Fibroblasten gezeigt werden konnte, dass c-Jun und JunD durch CyclinD1Induktion vor Apoptose geschützt werden (74) und damit möglicherweise reziprok eine Hemmung dieser Jun-Untereinheiten Apoptose induzieren könnte, steht dieser Beweis für Lymphozyten aus. Ein ähnlicher Mechanismus könnte für die Apoptose in Lymphozyten verantwortlich sein, jedoch unterscheidet sich die Funktion der Jun-Untereinheiten je nach Zelltyp (11), so dass hier weitere Untersuchungen (z. B. Microarrays) nötig wären, die den Einfluß von Sevofluran auf die Zellzyklus-regulierenden Proteine klären könnten. IL-3-Entzug führt in hämatopoetischen FDC-P1-Zellen zu Wachstumstopp oder zu Beeinträchtigung des zellulären Wachstums (41;54). Bekannt ist auch, dass IL-3-Entzug zu caspase-abhängiger Apoptose führt (80). Ein Versuchsprotokoll zur Klärung dieser Frage könnte nach Blockade des IL-3-Rezeptors mit spezifischen Antikörpern eine Analyse der Caspase-Aktivität und der Zellmorphologie beinhalten. 66 Diskussion 5.5 Schlussfolgerung In dieser Arbeit konnte gezeigt werden, dass von den klinisch üblichen Inhalationsanästhetika nur Sevofluran die DNA-Bindungsaktivität des Transkriptionsfaktors AP-1 in T-Lymphozyten hemmt. Diese Hemmung von AP1, die sich durch Dosis- und Zeitabhängigkeit charakterisiert, wird durch Desfluran und Isofluran nicht induziert. Als Konsequenz der verminderten DNA-Bindung kommt es zu einer Abnahme der AP-1-abhängigen Sekretion von IL-3. Es findet sich kein Hinweis auf eine Beteiligung der MAP-Kinasen ERK, JNK und p38 an der beobachteten Hemmung der DNA-Bindung von AP-1. Die p38-MAP-Kinase wird durch Inkubation mit Sevofluran induziert. Sehr wahrscheinlich vermittelt die p38-Aktivierung die beobachtete Apoptose nach Sevofluran-Inkubation. In Zellen mit charakteristischer apoptotischer Morphologie werden aktive Caspasen nachgewiesen, so dass davon auszugehen ist, dass Sevofluran caspase-abhängige Apoptose induziert. Als Perspektive für weitergehende Arbeiten könnte möglicherweise der molekulare Mechanismus der Caspase-Aktivierung durch Sevofluran in Betracht gezogen werden. Insbesondere gälte es dabei zu untersuchen, ob die Aktivierung der Caspase-3 über einen Rezeptor vermittelt wird oder ob die Sevofluran-Inkubation über eine Perforation der mitochondrialen Membran zu einer Freisetzung von Cytochrom C aus dem Intermembranspalt führt. Dieser Schritt könnte helfen, um die spezifischen Angriffspunkte von Sevofluran und von möglichen Metaboliten eingehender zu charakterisieren. Über Konsequenzen, die sich aus dieser Arbeit möglicherweise für die klinische Anwendung von Sevofluran ergeben könnten, kann nur spekuliert werden. Die Übertragung der vorliegenden Ergebnisse auf die Situation in vivo ist nicht ohne Einschränkungen zulässig, da eine isolierte Aktivierung und Hemmung spezifischer Transkriptionsfaktoren wie AP-1 unter Narkosebedingungen wahrscheinlich nicht durchführbar wäre. Interessant wäre es, in einer klinischen Studie zu untersuchen, ob sich die in vitro nachgewiesenen Wirkungen von Sevofluran auch in vivo beobachten lassen. Vorstellbar wäre, Blutproben von 67 Diskussion Patienten unter Sevofluran-Narkose auf ihren Gehalt an IL-3 und auf die Menge an apoptotischen Lymphozyten zu untersuchen und beispielsweise mit dem Blut von Patienten einer Kontrollgruppe ohne Narkose oder unter intravenöser Anästhesie zu vergleichen. 68 Anhang 6 Anhang 6.1 Abkürzungen mA Milliampère Abb. Abbildung AK Antikörper AP-1 Aktivator Protein-1 ATF „Activating Transcription Factor” (=CREB) bp, kbp Basenpaare, Kilobasenpaare bzw. beziehungsweise °C Grad Celsius 2+ [Ca ] Kalziumionenkonzentration cAMP zyklisches Adenosinmonophosphat cDNA komplementäre DNA CD3 Oberflächenantigen 3 („Cluster of Differentiation 3“) CD28 Oberflächenantigen 28 („Cluster of Differentiation 28“) CEBP „CCAAT-Enhancer-Binding Protein” (=C/EBP) Ci Curie CO2 Kohlendioxid CRE “cAMP-Response-Element” CREB “CRE-Binding Protein” (=ATF) DAG Diacylglyzerol DNA Desoxyribonukleinsäure (DNS) ELISA “Enzyme-Linked Immuno Sorbent Assay” Elk-1 “ETS-Like Protein” EMSA “Electrophoretic Mobility Shift Assay” ERK “Extracellular Signal Regulated Protein Kinase” ETS “E26-Transformation Specific”-Transkriptionsfaktor FACS “Fluorescence-Activated Cell Sorting” Fos-K Fos-Kinase ng, µg, mg, g Nanogramm, Mikrogramm, Milligramm, Gramm IFN Interferon IL Interleukin 69 IP3 Inositol-trisphosphat JNK c-Jun-NH2-terminale Kinase µl, ml, l Mikroliter, Milliliter, Liter pM, µM, mM, M Picomol(ar), Mikromol(ar), Millimol(ar), Mol(ar) nm, cm Nanometer, Zentimeter MAP-Kinase “Mitogen Activated Protein-Kinase” (MAPK) MEK „Mitogen Activated Protein-Kinase Kinase (MAPKK) MEKK „MAP-Kinase Kinase Kinase (MAPKKK) N2 Stickstoff NFAT Nukleärer Faktor Aktivierter T-Zellen NFκB Nukleärer Faktor κB O2 Sauerstoff Oct-1 „Octamer Binding Protein-1“ PIP2 Phosphatidyl-Inositol-Bisphosphat PI3 Phosphoinositid-3 PI3-K Phosphoinositid-3-Kinase PMA Phorbol-12-Myristat-13-Azetat PMBZ periphere mononukleäre Blutzellen PKC Proteinkinase C PLC Phospholipase C RNA Ribonukleinsäure (RNS) rpm Umdrehungen pro Minute RT Raumtemperatur sec, min, h Sekunde(n), Minute(n), Stunde(n) SEK „Stress Activated Protein-Kinase“ s. (o.) siehe (oben) SP-1 SV40-Promotor-1 TCR T-Zell-Rezeptor TNF-α Tumor Nekrose Faktor-α TPA 12-O-Tetradecanoyl-Phorbol-12-Azetat TRE “TPA-Response-Element” Tyr-K Tyrosin-Kinase u. a. unter anderem mV, V Millivolt, Volt Anhang 70 Vol% Volumenprozent vs. im Vergleich mit z. B. zum Beispiel 6.2 Vektorkarte Abb. 21: der pAP(PMA)-TA-Luc-Vektor enthält ein PMA-abhängiges Luziferasegen und ein AmpicillinResistenzgen. Anhang 71 Anhang 6.3 Literaturverzeichnis 1. Acehan, D., X. Jiang, D.G. Morgan, J.E. Heuser, X. Wang, and C.W. Akey. 2002. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell 9:423-432. 2. Angel, P., M. Imagawa, R. Chiu, B. Stein, R.J. Imbra, H.J. Rahmsdorf, C. Jonat, P. Herrlich, and M. Karin. 1987. Phorbol ester-inducible genes contain a common cis element recognized by a TPAmodulated trans-acting factor. Cell 49:729-739. 3. Anuradha, C.D., S. Kanno, and S. Hirano. 2000. Fluoride induces apoptosis by caspase-3 activation in human leukemia HL-60 cells. Arch. Toxicol. 74:226-230. 4. Aoshiba, K., S. Yasui, M. Hayashi, J. Tamaoki, and A. Nagai. 1999. Role of p38-mitogen-activated protein kinase in spontaneous apoptosis of human neutrophils. J Immunol. 162:1692-1700. 5. Berger, I. and Y. Shaul. 1991. Structure and function of human jun-D. Oncogene 6:561-566. 6. Boatright, K.M., M. Renatus, F.L. Scott, S. Sperandio, H. Shin, I.M. Pedersen, J.E. Ricci, W.A. Edris, D.P. Sutherlin, D.R. Green, and G.S. Salvesen. 2003. A unified model for apical caspase activation. Mol Cell 11:529-541. 7. Borgermann, J., I. Friedrich, S. Flohe, J. Spillner, M. Majetschak, O. Kuss, A. Sablotzki, T. Feldt, J.C. Reidemeister, and F.U. Schade. 2002. Tumor necrosis factor-alpha production in whole blood after cardiopulmonary bypass: downregulation caused by circulating cytokine-inhibitory activities. J Thorac. Cardiovasc. Surg. 124:608617. 8. Borner, C. 2003. The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol Immunol. 39:615-647. 9. Bossy-Wetzel, E., L. Bakiri, and M. Yaniv. 1997. Induction of apoptosis by the transcription factor c-Jun. EMBO J 16:1695-1709. 10. Busoni, P., A. Sarti, M. De Martino, E. Graziani, and S. Santoro. 1988. The effect of general and regional anesthesia on oxygendependent microbicidal mechanisms of polymorphonuclear leukocytes in children. Anesth. Analg 67:453-456. 11. Chinenov, Y. and T.K. Kerppola. 2001. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 20:2438-2452. 12. Colotta, F., N. Polentarutti, M. Sironi, and A. Mantovani. 1992. Expression and involvement of c-fos and c-jun protooncogenes in 72 Anhang programmed cell death induced by growth factor deprivation in lymphoid cell lines. J Biol. Chem 267:18278-18283. 13. Delogu, G., S. Moretti, G. Famularo, A. Antonucci, L. Signore, S. Marcellini, B.L. Lo, and C. De Simone. 2001. Circulating neutrophils exhibit enhanced apoptosis associated with mitochondrial dysfunctions after surgery under general anaesthesia. Acta Anaesthesiol. Scand. 45:87-94. 14. Delogu, G., S. Moretti, G. Famularo, S. Marcellini, G. Santini, A. Antonucci, M. Marandola, and L. Signore. 2001. Mitochondrial perturbations and oxidant stress in lymphocytes from patients undergoing surgery and general anesthesia. Arch. Surg. 136:1190-1196. 15. Dent, P. and S. Grant. 2001. Pharmacologic interruption of the mitogenactivated extracellular-regulated kinase/mitogen-activated protein kinase signal transduction pathway: potential role in promoting cytotoxic drug action. Clin. Cancer Res. 7:775-783. 16. Deschesnes, R.G., J. Huot, K. Valerie, and J. Landry. 2001. Involvement of p38 in apoptosis-associated membrane blebbing and nuclear condensation. Mol Biol Cell 12:1569-1582. 17. Donepudi, M., S.A. Mac, C. Briand, and M.G. Grutter. 2003. Insights into the regulatory mechanism for caspase-8 activation. Mol Cell 11:543-549. 18. Earnshaw, W.C., L.M. Martins, and S.H. Kaufmann. 1999. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68:383-424. 19. El-Zein, R.A., S.Z. Abdel-Rahman, D.L. Morris, and M.S. Legator. 2002. Exposure to ethylene glycol monomethyl ether: clinical and cytogenetic findings. Arch. Environ. Health 57:371-376. 20. Fan, M., M.E. Goodwin, M.J. Birrer, and T.C. Chambers. 2001. The cJun NH(2)-terminal protein kinase/AP-1 pathway is required for efficient apoptosis induced by vinblastine. Cancer Res. 61:44504458. 21. Foletta, V.C., D.H. Segal, and D.R. Cohen. 1998. Transcriptional regulation in the immune system: all roads lead to AP-1. J Leukoc. Biol. 63:139-152. 22. Goto, Y., J. Gallagher, N. Fanning, J. Wang, S. McCusker, P. Redmond, and G. Shorten. 2000. Does chronic occupational exposure to volatile anesthetic agents influence the rate of neutrophil apoptosis? Can. J Anaesth. 47:350-353. 23. Green, D.R. and G.I. Evan. 2002. A matter of life and death. Cancer Cell 1:19-30. 73 Anhang 24. Halazonetis, T.D., K. Georgopoulos, M.E. Greenberg, and P. Leder. 1988. c-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell 55:917-924. 25. Han, J., J.D. Lee, L. Bibbs, and R.J. Ulevitch. 1994. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265:808-811. 26. Hara, T. and A. Miyajima. 1996. Function and signal transduction mediated by the interleukin 3 receptor system in hematopoiesis. Stem Cells 14:605-618. 27. Hasthorpe, S., M. Akinci, and S. Bartelmez. 1988. The kinetics of S phase entry by FMP2.1: effect of IL-3 and GM-CSF receptor expression and ligand affinity. Int. J Cell Cloning 6:30-44. 28. Hu, M.C., Y.P. Wang, A. Mikhail, W.R. Qiu, and T.H. Tan. 1999. Murine p38-delta mitogen-activated protein kinase, a developmentally regulated protein kinase that is activated by stress and proinflammatory cytokines. J Biol Chem. 274:7095-7102. 29. Karin, M., Z. Liu, and E. Zandi. 1997. AP-1 function and regulation. Curr. Opin. Cell Biol. 9:240-246. 30. Kleinsasser, N.H., H. Weissacher, B.C. Wallner, E.R. Kastenbauer, and U.A. Harreus. 2001. [Cytotoxicity and genotoxicity of fluorides in human mucosa and lymphocytes]. Laryngorhinootologie 80:187190. 31. Kluck, R.M., E. Bossy-Wetzel, D.R. Green, and D.D. Newmeyer. 1997. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275:1132-1136. 32. Kotani, N., S. Takahashi, D.I. Sessler, Hashimoto, and A. Matsuki. 1999. expression of proinflammatory macrophages during mechanical 91:187-197. E. Hashiba, T. Kubota, H. Volatile anesthetics augment cytokines in rat alveolar ventilation. Anesthesiology 33. Krammer, P.H. CD95's deadly mission in the immune system. 2000. Nature 407:789-795. 34. Kummer, J.L., P.K. Rao, and K.A. Heidenreich. 1997. Apoptosis induced by withdrawal of trophic factors is mediated by p38 mitogenactivated protein kinase. J Biol Chem. 272:20490-20494. 35. Lee, W., P. Mitchell, and R. Tjian. 1987. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell 49:741-752. 36. London, L. and J.P. McKearn. 1987. Activation and growth of colonystimulating factor-dependent cell lines is cell cycle stage dependent. J Exp Med. 166:1419-1435. 74 Anhang 37. Loop, T., Z. Liu, M. Humar, A. Hoetzel, A. Benzing, H.L. Pahl, K.K. Geiger, and B.H.J. Pannen. 2002. Thiopental inhibits the activation of nuclear factor kappaB. Anesthesiology 96:1202-1213. 38. Macian, F., C. Lopez-Rodriguez, and A. Rao. 2001. Partners in transcription: NFAT and AP-1. Oncogene 20:2476-2489. 39. Matsuoka, H., S. Kurosawa, T. Horinouchi, M. Kato, and Y. Hashimoto. 2001. Inhalation anesthetics induce apoptosis in normal peripheral lymphocytes in vitro. Anesthesiology 95:1467-1472. 40. Mattson, M.P. Apoptosis in neurodegenerative disorders. 2000. Nat. Rev. Mol Cell Biol. 1:120-129. 41. Mayo, M.W., L.S. Steelman, and J.A. McCubrey. 1994. Phorbol esters support the proliferation of a hematopoietic cell line by upregulating c-jun expression. Oncogene 9:1999-2008. 42. Meier, P., A. Finch, and G. Evan. 2000. Apoptosis in development. Nature 407:796-801. 43. Mekori, Y.A., C.K. Oh, and D.D. Metcalfe. 1993. IL-3-dependent murine mast cells undergo apoptosis on removal of IL-3. Prevention of apoptosis by c-kit ligand. J Immunol. 151:3775-3784. 44. Mitsuhata, H., R. Shimizu, and M.M. Yokoyama. 1995. Suppressive effects of volatile anesthetics on cytokine release in human peripheral blood mononuclear cells. Int. J Immunopharmacol. 17:529-534. 45. Nel, A.E., L.K. Taylor, G.P. Kumar, S. Gupta, S.C. Wang, K. Williams, O. Liao, K. Swanson, and G.E. Landreth. 1994. Activation of a novel serine/threonine kinase that phosphorylates c-Fos upon stimulation of T and B lymphocytes via antigen and cytokine receptors. J Immunol. 152:4347-4357. 46. Nemoto, S., J. Xiang, S. Huang, and A. Lin. 1998. Induction of apoptosis by SB202190 through inhibition of p38beta mitogen-activated protein kinase. J Biol Chem. 273:16415-16420. 47. Nimer, S., J. Zhang, H. Avraham, and Y. Miyazaki. 1996. Transcriptional regulation of interleukin-3 expression in megakaryocytes. Blood 88:66-74. 48. Pahl, H.L. 1999. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18:6853-6866. 49. Parra, E., M. Varga, G. Hedlund, T. Kalland, and M. Dohlsten. 1997. Costimulation by B7-1 and LFA-3 targets distinct nuclear factors that bind to the interleukin-2 promoter: B7-1 negatively regulates LFA-3-induced NF-AT DNA binding. Mol Cell Biol. 17:1314-1323. 75 Anhang 50. Patterson, S.D., C.S. Spahr, E. Daugas, S.A. Susin, T. Irinopoulou, C. Koehler, and G. Kroemer. 2000. Mass spectrometric identification of proteins released from mitochondria undergoing permeability transition. Cell Death. Differ. 7:137-144. 51. Pramanik, R., X. Qi, S. Borowicz, D. Choubey, R.M. Schultz, J. Han, and G. Chen. 2003. p38 isoforms have opposite effects on AP-1dependent transcription through regulation of c-Jun. The determinant roles of the isoforms in the p38 MAPK signal specificity. J Biol Chem. 278:4831-4839. 52. Procopio, M.A., A.J. Rassias, J.A. DeLeo, J. Pahl, L. Hildebrandt, and M.P. Yeager. 2001. The in vivo effects of general and epidural anesthesia on human immune function. Anesth. Analg 93:460-5, 4th. 53. Puthalakath, H. and A. Strasser. 2002. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death. Differ. 9:505-512. 54. Redner, R.L., A.W. Lee, G.A. Osawa, and A.W. Nienhuis. 1992. Variable pattern of jun and fos gene expression in different hematopoietic cell lines during interleukin 3-induced entry into the cell cycle. Oncogene 7:43-50. 55. Renatus, M., H.R. Stennicke, F.L. Scott, R.C. Liddington, and G.S. Salvesen. 2001. Dimer formation drives the activation of the cell death protease caspase 9. Proc Natl Acad Sci U S A 98:1425014255. 56. Rincon, M. 2001. MAP-kinase signaling pathways in T cells. Curr. Opin. Immunol. 13:339-345. 57. Rincon, M., R.A. Flavell, and R.J. Davis. 2001. Signal transduction by MAP kinases in T lymphocytes. Oncogene 20:2490-2497. 58. Rodriguez, J. and Y. Lazebnik. 1999. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 13:3179-3184. 59. Rudd, C.E., O. Janssen, Y.C. Cai, A.J. da Silva, M. Raab, and K.V. Prasad. 1994. Two-step TCR zeta/CD3-CD4 and CD28 signaling in T cells: SH2/SH3 domains, protein-tyrosine and lipid kinases. Immunol. Today 15:225-234. 60. Ryseck, R.P. and R. Bravo. 1991. c-JUN, JUN B, and JUN D differ in their binding affinities to AP-1 and CRE consensus sequences: effect of FOS proteins. Oncogene 6:533-542. 61. Savill, J. and V. Fadok. 2000. Corpse clearance defines the meaning of cell death. Nature 407:784-788. 76 Anhang 62. Schreiber, E., P. Matthias, M.M. Muller, and W. Schaffner. 1989. Rapid detection of octamer binding proteins with 'mini-extracts', prepared from a small number of cells. Nucleic Acids Res. 17:6419 63. Shaulian, E. and M. Karin. 2002. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4:E131-E136 64. Shaulian, E. and M. Karin. 2001. AP-1 in cell proliferation and survival. Oncogene 20:2390-2400. 65. Shi, Y. 2002. Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell 9:459-470. 66. Thompson, C.B. 1995. Apoptosis in the pathogenesis and treatment of disease. Science 267:1456-1462. 67. Toone, W.M., B.A. Morgan, and N. Jones. 2001. Redox control of AP-1like factors in yeast and beyond. Oncogene 20:2336-2346. 68. van Sandick, J.W., S.S. Gisbertz, B. ten, I, M.A. Boermeester, van der Pouw Kraan TC, T.A. Out, H. Obertop, and J.J. van Lanschot. 2003. Immune responses and prediction of major infection in patients undergoing transhiatal or transthoracic esophagectomy for cancer. Ann. Surg. 237:35-43. 69. Viardot, A., T.F. Barth, P. Moller, H. Dohner, and M. Bentz. 2003. Cytogenetic evolution of follicular lymphoma. Semin. Cancer Biol. 13:183-190. 70. Waetzig, V. and T. Herdegen. 2003. A single c-Jun N-terminal kinase isoform (JNK3-p54) is an effector in both neuronal differentiation and cell death. J Biol Chem 278:567-572. 71. Wang, J. and C.R. Taylor. 2003. Apoptosis and Cell Cycle-related Genes and Proteins in Classical Hodgkin Lymphoma: Application of Tissue Microarray Technique. Appl. Immunohistochem. Mol Morphol. 11:206-213. 72. Wang, P., P.O. Anderson, S. Chen, K.M. Paulsson, H.O. Sjogren, and S. Li. 2001. Inhibition of the transcription factors AP-1 and NFkappaB in CD4 T cells by peroxisome proliferator-activated receptor gamma ligands. Int. Immunopharmacol. 1:803-812. 73. Wang, X. 2001. The expanding role of mitochondria in apoptosis. Genes Dev. 15:2922-2933. 74. Weitzman, J.B., L. Fiette, K. Matsuo, and M. Yaniv. 2000. JunD protects cells from p53-dependent senescence and apoptosis. Mol Cell 6:1109-1119. 75. Whitmarsh, A.J., S.H. Yang, M.S. Su, A.D. Sharrocks, and R.J. Davis. 1997. Role of p38 and JNK mitogen-activated protein kinases in 77 Anhang the activation of ternary complex factors. Mol Cell Biol 17:23602371. 76. Wilkinson, M.G. and J.B. Millar. 2000. Control of the eukaryotic cell cycle by MAP kinase signaling pathways. FASEB J 14:2147-2157. 77. Wisdom, R., R.S. Johnson, and C. Moore. 1999. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J 18:188-197. 78. Wissing, H., I. Kuhn, U. Warnken, and R. Dudziak. 2001. Carbon monoxide production from desflurane, enflurane, halothane, isoflurane, and sevoflurane with dry soda lime. Anesthesiology 95:1205-1212. 79. Yamagishi, S., M. Yamada, Y. Ishikawa, T. Matsumoto, T. Ikeuchi, and H. Hatanaka. 2001. p38 mitogen-activated protein kinase regulates low potassium-induced c-Jun phosphorylation and apoptosis in cultured cerebellar granule neurons. J Biol Chem. 276:5129-5133. 80. Yoshikawa, H. and K. Tasaka. 2003. Caspase-dependent and independent apoptosis of mast cells induced by withdrawal of IL-3 is prevented by Toll-like receptor 4-mediated lipopolysaccharide stimulation. Eur. J Immunol. 33:2149-2159. 81. Yu, Q., M. He, N.H. Lee, and E.T. Liu. 2002. Identification of Mycmediated death response pathways by microarray analysis. J Biol Chem. 277:13059-13066. 82. Zeiger, E., M.D. Shelby, and K.L. Witt. 1993. Genetic toxicity of fluoride. Environ. Mol. Mutagen. 21:309-318. 83. Zheng, X., A. Karsan, V. Duronio, F. Chu, D.C. Walker, T.R. Bai, and R.R. Schellenberg. 2002. Interleukin-3, but not granulocytemacrophage colony-stimulating factor and interleukin-5, inhibits apoptosis of human basophils through phosphatidylinositol 3kinase: requirement of NF-kappaB-dependent and -independent pathways. Immunology 107:306-315. 78 Anhang 6.4 Wissenschaftliche Leistungen Poster: T. Loop, P. Scheiermann, L. Egger, D. Lachenmeier, D. Doviakue, H. Pahl, K. Geiger, B. Pannen Sevofluran hemmt die Aktivierung des Transkriptionsfaktors ‘Activator Protein-1 (AP-1)’ in humanen T-Lymphozyten in vitro – Abstractband Deutscher Anästhesiekongress – 50. Jahrestagung der Deutschen Gesellschaft für Anästhesiologie und Intensivmedizin, München, 09.04.-12.04.2003 Loop T., Scheiermann P., Doviakue D., Egger L., Schmidt R., Pahl H., Geiger K., Pannen B. Sevoflurane induces Apoptosis and Inhibits ’Activator Protein-1 (AP-1)’ in human T-Lymphocytes in vitro – eur j anaesthesiol 20; suppl 30:A457 Euroanaesthesia 2003, Glasgow, 30.05.-03.06.2003 Publikation: Torsten Loop, M.D., Patrick Scheiermann, David Doviakue, Frank Musshoff, Ph.D., Matjaz Humar, Ph.D., Martin Roesslein, M.D., Alexander Hoetzel, M.D., Rene Schmidt, M.D., Burkhard Madea, M.D., Klaus K. Geiger, M.D., Heike L. Pahl, Ph.D., Benedikt H. J. Pannen, M.D. 2004. Sevoflurane Inhibits Phorbol-Myristate-Acetate-induced Activator Protein1 Activation in Human T Lymphocytes in Vitro: Potential Role of the p38Stress Kinase Pathway. Anesthesiology 101:710-21 Torsten Loop and Patrick Scheiermann contributed equally to this work. 79 Anhang 6.5 Lebenslauf Persönliches: Name: Patrick Alexander Scheiermann Adresse: Moltkestr. 15, D – 79098 Freiburg geboren am: 21.02.1977 in Essen Eltern: Prof. Dr. Norbert Scheiermann Oda Scheiermann, geb. Castrup Ausbildung: Schule: 1983-1987 Cranachschule, Essen 1987/1988 Burggymnasium, Essen 1988-1991 Theodor-Heuss-Gymnasium, Heilbronn 1991-1996 Bischof-Neumann-Schule, Königstein 1993/1994 Austauschschüler an der Norfolk Christian High School, Norfolk, VA., USA Zivildienst: Juli 1996 Abitur 1996/1997 Pflegehelfer in der Chirurgischen Klinik, Kreiskrankenhaus Bad Soden Universität: seit Oktober 1997 Studium der Humanmedizin an der AlbertLudwigs-Universität Freiburg im Breisgau September 1999 Physikum August 2000 1. Staatsexamen 2000/2001 SOKRATES-Austauschstudent an der Université Claude Bernard Lyon 1, Frankreich September 2003 2. Staatsexamen Okt. `03-Aug. `04 Praktisches Jahr in Genf, Gosford und Freiburg November 2004 3. Staatsexamen