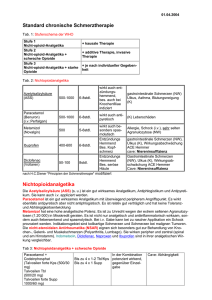
Volltext herunterladen
Werbung

Bachelorarbeit Grundlagen eines aus pharmakologischer Sicht qualifizierten Schmerzmanagements mit Bezugnahme auf besondere Bedürfnisse von Patientinnen und Patienten aus pädiatrischen und geriatrischen Fachbereichen eingereicht von Julia Došen zur Erlangung des akademischen Grades Bachelor of Science (BSc) an der Medizinischen Universität Graz Auenbruggerplatz 2 8036 Graz ausgeführt am Institut für experimentelle und klinische Pharmakologie Universitätsplatz 4/I 8010 Graz unter der Anleitung von Betreuerin Ao.Univ.-Prof. Dr.med.univ. Ulrike Holzer Graz, 02. August 2016 „Ich erkläre ehrenwörtlich, dass ich die vorliegende Arbeit selbstständig und ohne fremde Hilfe verfasst habe, andere als die angegebenen Quellen nicht verwendet und die den benutzten Quellen wörtlich oder inhaltlich entnommenen Stellen als solche kenntlich gemacht habe. Graz, am 18. Juli 2016 Julia Došen, eh“ 1 Einleitung ……………………………………………………………………………….. 1 2 Definitionen und Begriffserklärungen zum Thema Schmerz ………………….. 2 2.1 Nahezu alle Menschen wissen, wie es sich anfühlt Schmerzen zu haben, doch wie wird Schmerz definiert und was ist seine Funktion? …............. 2 2.2 Kategorisierungen des Schmerzes …………………………………………...... 3 2.2.1 Die vier verschiedenen Schmerztypen ………………………….............. 4 2.3 Wie entstehen Schmerzen und welche körperlichen Strukturen sind an der Schmerzverarbeitung beteiligt? …………………………………………... 5 2.3.1 Funktionsweise von Nozizeptoren ………………………………………... 6 2.3.2 Schmerzimpulsverarbeitung und Schmerzbahn ……………………….. 7 3 Welche Arten von Schmerzmedikamenten gibt es? …………………………... 10 3.1 Pharmakologischer Wirkmechanismus und häufig verwendete Substanzen von Nicht – Opioidanalgetika ……………………………………………. 10 3.1.1 Saure antipyretische-antiphlogistische Analgetika oder NSAIDs ….. 11 3.1.2 Selektive Cyclooxygenase 2 – Hemmer (Coxibe) ……………………… 15 3.1.3 Nicht – saure antipyretische Analgetika ………………………………… 16 3.1.4 Analgetika ohne antipyretisch-antiphlogistische Wirkung ……….…. 18 3.2 Pharmakologischer Wirkmechanismus und häufig verwendete Substanzen von Opioidanalgetika ……………………………………………………... 20 3.2.1 Einteilung der Opioidanalgetika nach pharmakologischen Aspekten 22 3.2.2 Einteilung der Opioidanalgetika nach WHO – Stufenschema ……. 26 3.2.3 Schwach wirksame Opioide ……………………………………………. 27 3.2.4 Stark wirksame Opioide ………………………………………………… 28 3.3 Unerwünschte Opioidwirkungen und Gefahren von Überdosierungen .. 32 4 Spezialanforderungen in der pharmakologischen Schmerztherapie ………. 35 4.1 WHO – Stufenschema ……………………………………………………………… 37 4.2 Bezugnahme auf spezielle Bedürfnisse bestimmter Patientengruppen .. 39 4.2.1 Besonderheiten bei der Schmerztherapie von Patientinnen und Patienten aus dem Fachbereich der Geriatrie ………………………………….. 39 4.2.2 Besonderheiten bei der Schmerztherapie von Patientinnen und Patienten aus dem Fachbereich der Pädiatrie ………………………………….. 42 5 Schlussfolgerungen und Ausblick ……………………………………………….. 45 Literaturverzeichnis …………………………………………………………………….. 47 Abbildungsverzeichnis ………………………………………………………………… 49 Zusammenfassung Schmerz ist ein wichtiges Symptom für krankhafte Prozesse und der Hauptgrund, weshalb Patientinnen und Patienten ärztliche Hilfe in Anspruch nehmen. Die pharmakologische Schmerztherapie ist bei der Therapie von Schmerzzuständen stets eine bedeutende Grundlage. Hauptsächlich kommen dabei zwei Substanzklassen von Medikamenten zum Einsatz: Nicht-Opioidanalgetika und Opioidanalgetika. Die Kenntnis des genauen Wirkungs- und Nebenwirkungsprofils der einzelnen Medikamente ist für ein qualifiziertes Schmerzmanagement besonders wichtig. Darüber hinaus kann nur auf dieser Basis für den individuellen Patienten oder die individuelle Patientin die richtige medikamentöse Schmerztherapie ausgewählt werden. Patientinnen und Patienten aus dem Bereich der Geriatrie und Pädiatrie scheinen diesbezüglich aufgrund des Vorliegens von relativ geringen Forschungsergebnissen über die pharmakologische Schmerztherapie für diese Gruppen stark benachteiligt zu sein. Abstract Pain is an important sign of various diseases or pathological processes and the main reason why patients seek medical aid. Pharmacological pain therapy plays a major role in the treatment of pain conditions. For this purpose, the following two different substance classes are most commonly used: Non-opioid analgesics and opioid analgesics. The knowledge of the precise effects and side effects of each individual medication is of particular significance for a qualified pain management, since this is the only way to find the right drug treatment for each individual pain therapy plan for every single patient. Patients from the fields of paediatrics and geriatrics appear to be disadvantaged in terms of pharmacological pain therapy as relatively few research results for these groups of patients are available. 1 Einleitung Im Bereich der Pflege und Medizin ist Schmerz ein Symptom auf Seiten der Patientinnen und Patienten, mit dem alle Berufsgruppen des Gesundheitssektors sehr häufig konfrontiert sind. Die pharmakologische Schmerztherapie stellt seit jeher einen wichtigen Grundpfeiler in der Behandlung von Schmerzzuständen dar. Aus diesem Grund habe ich mich entschieden, diese Thematik in meiner Bachelorarbeit genauer zu beleuchten und mich mit den Grundlagen der aus pharmakologischer Sicht möglichen Therapieformen des Schmerzes auseinanderzusetzen. Dabei interessierte mich besonders die Frage, welche Patientengruppen in Bezug auf die Schmerztherapie besondere Bedürfnisse aufweisen. Meine Forschungsfragen lauteten daher: Was sind die physiologischen Grundlagen für die Schmerzentstehung, wo setzen pharmakologische Schmerzmedikamente zur Schmerzreduktion an? Welche pharmakologischen Schmerzmedikamente werden verwendet, wie wirken diese Substanzen? Welche besonderen Bedürfnisse haben Patientinnen und Patienten aus dem Fachbereich der Pädiatrie und Geriatrie in Bezug auf eine qualifizierte Schmerztherapie? Die vorliegende Bachelorarbeit stellt eine Beantwortung dieser Fragestellungen in Form einer wissenschaftlichen Literaturrecherche dar. In Kapitel zwei und drei wird ein Überblick über die Grundlagen der pharmakologischen Schmerztherapie aufgezeigt. Darauf folgend werden in Kapitel vier die speziellen Anforderungen in der pharmakologischen Schmerztherapie beschrieben. In Kapitel fünf wird die gesamte Thematik abschließend diskutiert und ein Ausblick gegeben. Die angegebenen, beispielhaften Medikamentennamen in dieser Arbeit wurden der Internet-Seite der AGES Medizinmarktaufsicht des Bundesministeriums für Sicherheit im Gesundheitswesen entnommen (Quelle: AGES, Wirthumer-Hoche, 2016). 1 2 Definitionen und Begriffserklärungen zum Thema Schmerz Der Schmerz steht unter den Gründen, weshalb ärztliche Hilfe in Anspruch genommen wird, an erster Stelle und auch die meisten Krankheiten äußern sich auf körperlicher Ebene primär über das Symptom Schmerz. Einerseits ist Schmerz auf diese Art ein wichtiger Hinweisgeber für unser Befinden, andererseits empfinden wir seine Bürde oft als unerträglich, wenn er anhält, obwohl wir seine Hinweisfunktion nicht mehr benötigen. Größtenteils sind alle Bereiche unseres menschlichen Körpers dazu fähig, auf Schmerzreize zu reagieren. Es gibt allerdings auch Organe, wie Gehirn und Leber, welche dazu nicht in der Lage sind (vgl. Mutschler et.al. 2013, S. 194). 2.1 Nahezu alle Menschen wissen, wie es sich anfühlt Schmerzen zu haben, doch wie wird Schmerz definiert und was ist seine Funktion? Eine mögliche Definition von Schmerz nach Mutschler lautet: „Schmerz ist eine unangenehme Sinneswahrnehmung und entsteht dann, wenn mechanische, thermische, chemische oder elektrische Reize einen Schwellenwert (Schmerzschwelle) überschreiten und dadurch (meist) zu einer Gewebeschädigung mit einer Freisetzung von Schmerzmediatoren sowie konsekutiv zur Bildung von (afferenten) Schmerzimpulsen führen.“ (Mutschler et.al. 2013, S. 194) Die Hauptfunktion des Schmerzes ist ein Warnsignal des Körpers vor möglichen Vorgängen, die das von Schmerzen betroffene Gewebe schädigen könnten. Nur durch eine Registrierung und Weiterleitung dieser Vorgänge an das Gehirn besteht die Option schadhafte Prozesse wahrnehmen und aufhalten zu können. Auf diese Weise kann durch eine adäquate Schmerzwahrnehmung in den meisten Fällen eine irreversible Zerstörung körperlicher Strukturen verhindert werden (vgl. Zeilhofer, Sittl 2007, S. 78). Allerdings kann auch eine andauernde Schmerzempfindung aufgrund von verschiedensten, teilweise medizinisch noch unvollständig erforschten, krankhaften Prozessen ausgelöst werden, welche keinen direkten Nutzen als Hinweisfaktor für ein Problem des Körpers mehr darstellt. Gerade in diesem Hinblick besteht eine Notwendigkeit für ein qualifiziertes Schmerzmanagement auf pharmakologischer Ebene (vgl. Höllt, Allgaier 2009, S. 207). 2 2.2 Kategorisierungen des Schmerzes Aufgrund der im vorherigen Absatz aufgezeigten Problematik ist eine Unterscheidung zwischen einem akuten Schmerz, der nur so lange andauert wie der schädigende Einfluss auf das Gewebe stattfindet, und einem chronischen Schmerz, der auch vorhanden bleibt, obwohl seine Warnfunktion vom Organismus nicht mehr benötigt wird, sinnvoll. Es gibt viele Ursachen, die dazu führen, dass aus akuten Schmerzen chronische Schmerzen werden. Diese dauern dann entweder über mehrere Monate an oder treten immer wieder in regelmäßigen Abständen auf. Um an dieser Stelle nur ein Beispiel von vielen zu nennen, könnte eine ununterbrochene Reizung der Schmerzrezeptoren durch ein Tumorgeschehen der Auslöser dafür sein. Allerdings sind die genauen Mechanismen dieses Überganges und der Manifestation des chronischen Schmerzes nur zum Teil wissenschaftlich erforscht. Dies erschwert eine qualifizierte Therapie von betroffenen Patienten und Patientinnen (vgl. Höllt, Allgaier 2009, S. 208). Neben einer groben Unterscheidung zwischen akutem und chronischem Schmerzgeschehen kann auch eine Unterteilung in verschiedene Schmerzqualitäten vorgenommen werden. Dabei ist der somatische Schmerz derjenige, der ein Schmerzgeschehen bezeichnet, welches in der Haut, den Muskeln, den Gelenken, den Knochen oder dem Bindegewebe stattfindet. Der sogenannte viszerale Schmerz hat die Bedeutung des Eingeweideschmerzes und bezeichnet einen solchen Schmerz, der bei Dehnung der Bauchorgane oder bei Krämpfen der glatten Muskulatur dieser Organe entsteht. Eine solche Schmerzempfindung kann aber auch aus einer Mangeldurchblutung derselben Organe mit eventueller zusätzlicher Entzündung resultieren. Darüber hinaus kann eine Gegenüberstellung von einem Oberflächenschmerz und einem Tiefenschmerz durchgeführt werden. Ein typisches Beispiel für den Oberflächenschmerz wäre ein Nadelstich, dieser ist örtlich klar wahrnehmbar und von hellem Charakter, außerdem hält er nur kurz an. Der Tiefenschmerz wäre typischerweise der Kopfschmerz, der einen dumpfen Charakter besitzt und örtlich schwer zu begrenzen ist, oft strahlt er in den Umkreis der betroffenen Region aus. Teilweise entstehen auch zusätzliche Symptome, die durch das vegetative Nervensystem ausgelöst werden, und es kann dadurch beispielsweise zu Schweißausbrüchen, Übelkeit oder Blutdruckabfall kommen (vgl. Mutschler et.al. 2013, S. 200-201). 3 2.2.1 Die vier verschiedenen Schmerztypen 1. Physiologischer Nozizeptorschmerz Die durch direkte Reizung der Schmerzrezeptoren ausgelöste Schmerzempfindung entsteht bei Einwirkung von Druck, schädlichen chemischen Substanzen oder Hitze auf den menschlichen Körper. Dieser für unser Überleben wichtige Schmerztyp dient mit seiner Warnfunktion der Auslösung eines Schutzreflexes zur sofortigen Beendigung der schädigenden Einwirkung (vgl. Mutschler et.al. 2013, S. 195). 2. Pathophysiologischer Nozizeptorschmerz Dieser Schmerztyp wird mit dem Entzündungsschmerz gleichgesetzt und als Folge von Entzündungsgeschehen oder Gewebeschädigungen empfunden. Eine Schmerzempfindung in einem solchen Zusammenhang tritt auch in Ruhe auf. Zusätzlich kann sich eine verstärkte Schmerzempfindung entwickeln, welche in der Fachsprache als Hyperalgesie bezeichnet wird. Oder aber es können sogar Schmerzen durch vormals nicht schmerzhafte Stimulierungen ausgelöst werden. Dieser Zustand wird fachlich korrekt Allodynie genannt (vgl. Mutschler et.al. 2013, S. 195). 3. Neuropathischer Schmerz Dieser Schmerztyp basiert auf einer Nervenschädigung, welche durch unterschiedliche Ursachen ausgelöst werden kann. Hierzu zählen beispielsweise Bandscheibenvorfälle, Amputationen, Entzündungsgeschehen oder eine Erkrankung an Diabetes Mellitus. Typischer Weise sind neuropathische Schmerzen äußerst unangenehm. Durch eine Veränderung im Schmerzleitungssystem können die geschädigten Nerven teilweise eine Überempfindlichkeit auf ansonsten nicht als schmerzhaft empfundene Reize, wie Berührung oder Kälte auslösen. Zusätzlich können neuropathische Schmerzen auch mit Ausfallserscheinungen einhergehen. Auch Phantomschmerzen werden zu dieser Gruppe gezählt (vgl. Mutschler et.al. 2013, S. 195). 4. Funktioneller Schmerz Laut Höllt kann auch ein vierter Schmerztyp definiert werden, der aus einer nicht der Norm entsprechenden körperlichen Reaktion im Zentralnervensystem auf Schmerzimpulse resultiert. Dieser Art Schmerzen liegt keine organische Ursache zu Grunde (vgl. Höllt, Allgaier 2009, S. 208). 4 2.3 Wie entstehen Schmerzen und welche körperlichen Strukturen sind an der Schmerzverarbeitung beteiligt? Ein wichtiger Schlüsselbegriff der Fachsprache im Zusammenhang mit der körperlichen Schmerzverarbeitung ist die Nozizeption, deren Definition lautet wie folgt: „Die Auslösung, Weiterleitung und zentrale Verarbeitung der Schmerzimpulse wird als Nozizeption bezeichnet.“ (Mutschler et.al. 2013, S. 194) Die Schmerzrezeptoren werden in der Fachsprache als Nozizeptoren bezeichnet und laut Höllt folgendermaßen beschrieben: „Bei den Schmerzrezeptoren (Nozizeptoren) handelt es sich um freie Nervenendigungen. Sie reagieren auf mechanische, thermische und chemische Reize. Zahlreiche Nozizeptoren reagieren auf alle Arten von Reizen und werden deshalb als polymodal bezeichnet. Andere sind nur mechanisch oder mechanisch und thermisch erregbar. “ (Höllt, Allgaier 2009, S. 208) Genauer bezeichnet handelt es sich bei diesen freien Nervenendigungen einerseits um die der sogenannten sensorischen C-Fasern, die einen dumpfen und schlecht lokalisierbaren Schmerzcharakter empfinden lassen. Andererseits handelt es sich dabei auch um die freien sensorischen Nervenendigungen der schnell leitenden Aδ-Fasern, durch welche ein heller Schmerzcharakter empfunden wird (vgl. Höllt, Allgaier 2009, S. 209). Abbildung 1: Schmerzverarbeitung (Quelle: Pirman, 2016) 5 Bei einer Schädigung des menschlichen Körpergewebes oder einer Dysfunktionalität im Stoffwechsel eines bestimmten Gewebes wird in den meisten Fällen eine Schmerzempfindung ausgelöst. Diese Schmerzempfindung kommt auch dadurch zu Stande, dass die von der Schädigung betroffenen Zellen des Gewebes bestimmte Stoffe an ihre Umgebung abgeben oder produzieren, auf welche die Nozizeptoren reagieren und ansprechen. Solche Stoffe oder chemische Substanzen werden als Schmerzmediatoren bezeichnet (vgl. Mutschler et.al. 2013, S. 196). 2.3.1 Funktionsweise von Nozizeptoren Die Nozizeptoren erfüllen die wichtige Aufgabe der Schmerzwahrnehmung und Weiterleitung aus der Peripherie. Diese langsam leitenden Nervenfasern übertragen die gewonnene Information oder die erfahrenen Reize in die zentralen Schaltstellen des menschlichen Körpers. Problematisch bei dem System der körperlichen Schmerzverarbeitung kann es werden, wenn eine Dauerüberlastung mit Reizen stattfindet und eine überproportionale Wahrnehmung von Schmerzen ausgelöst wird. Daraus resultiert die Gefahr einer Chronifizierung des Schmerzgeschehens, das therapeutisch meist einen weitaus komplizierteren Behandlungsaufwand nötig macht, als ein akutes Schmerzgeschehen. Diese Tatsache muss bei der Therapie von Schmerzen und Schmerzerkrankungen stets berücksichtigt werden (vgl. Zeilhofer, Sittl 2007, S. 78). Die TRP-Kanal-Gruppe („transient receptor potential“) stellt eine typische Vertreterklasse von Sensoren dar, die dazu dienen können, eine Nozizeption zu vermitteln. Capsaicin ist die in Chili-Pfefferoni enthaltene Substanz, welche beispielsweise über den Vanilloid-Rezeptor ihre Wirkung vermittelt. Dieser zur TRP-Gruppe gehörende Rezeptor (TRPVR1) ist ein ligandengesteuerter Ionenkanal, welcher Hitze- und Säurereize leiten kann. Andere Rezeptoren dieser Gruppe können nozizeptive Hitze- und Kälteempfindungen registrieren. Ein Einstrom von Natrium- und Kalziumionen in die Zelle als Folge der TRP-Kanal-Aktivierung führt zu einer Zellmembran-Depolarisation, die sich aktivierend auf spannungsabhängige Natriumkanäle auswirkt. Die Konsequenz daraus ist die Entstehung eines Aktionspotentials, welches den grundlegenden Mechanismus hinter der Arbeitsweise der Nozizeptoren bildet. Zusätzlich zu dieser auf chemisch-physikalischer Basis ablaufenden Wirkungsweise des schmerzverarbeitenden Systems, besteht auch die Möglichkeit, dieses System auf 6 körperlicher Ebene noch additiv zu verstärken. Dies geschieht durch sogenannte algogene oder schmerzversursachende Substanzen. Diese werden, wie im vorherigen Absatz beschriebenen, unter dem Überbegriff der Schmerzmediatoren zusammengefasst. Von Zerstörung betroffene Zellen geben sowohl Kaliumionen, als auch Adenosintriphosphat in ihre Umgebung ab. Dies führt zu einem veränderten pH-Wert, dessen Absinken sich auch auf die Funktion der Nozizeptoren förderlich auswirkt. Bei der Aktivierung des Immunsystems im Rahmen von Entzündungsreaktionen kommt es zu einer Zellinvasion in das betroffene Gewebe und dadurch auch zur Ausschüttung von Stoffen, die sich aktivierend auf die Nozizeptoren auswirken. Als wichtige Vertreter dieser Substanzen wären besonders das Kinin Bradykinin und die Prostaglandine hervorzuheben. Es kommt in diesem Zusammenhang aber auch zu einer Freisetzung von Zytokinen und Wachstumsfaktoren. Bradykinin und Prostaglandin E2 führen beispielsweise über Wechselwirkung mit bestimmten Rezeptoren durch Proteinkinasen zu einer Phosphorylierung des Vanilloid-Rezeptors (TRPVR1) und machen diesen dadurch stärker empfindlich für die Reize, auf die der Rezeptor normalerweise reagiert. Genau an dieser Stelle setzten die häufig eingesetzten Nicht-Opioidanalgetika an, indem sie eine Hemmung der Prostaglandin-Bildung auslösen und auf dies Weise eine abnorme Schmerzempfindung verhindern (vgl. Höllt, Allgaier 2009, S. 208-209). Im peripheren Nervensystem spielt auch das Hormon Serotonin eine Rolle bei der Empfindung von Schmerzreizen, hier führt Serotonin im Gegenteil zum Zentralnervensystem zu einer verstärkten Schmerzempfindung (vgl. Mutschler et.al. 2013, S. 196). 2.3.2 Schmerzimpulsverarbeitung und Schmerzbahn Generell wird jeder Schmerzreiz aus der Peripherie des Körpers primär ins Rückenmark geleitet, wo der Reiz im Hinterhorn des Rückenmarks über ein oder zwei Nervenzellen auf die Gegenseite umgeleitet wird. Die Schmerzimpulsweiterleitung erfolgt dann auf der Gegenseite über den sogenannten Tractus spinothalamicus lateralis vom Rückenmark zum Gehirn. Im Gehirn erfolgt eine weitere Umleitung im Thalamus, wobei die letztliche Registrierung und Verarbeitung der Schmerzreize zum größten Teil im Gyrus postcentralis sattfindet. Dabei handelt es sich um das Areal der Großhirnrinde, welches für die Verarbeitung von sensorischen Reizen zuständig ist. In Abbil- 7 dung 2 ist dieses Schmerzleitungssystem des menschlichen Körpers sehr anschaulich, visuell dargestellt. Für emotionale und vegetative Auswirkungen von Schmerzerlebnissen sind limbisches System und Hypothalamus verantwortlich. Das konkrete Bewusstwerden der Schmerzempfindung und zusätzlich die Beurteilung der Stärke und des Entstehungsortes von Schmerzen bewerkstelligt, wie bereits erwähnt, der Bereich des Gyrus postcentralis in der Großhirnrinde (vgl. Mutschler et.al. 2013, S. 198-199). Abbildung 2: Schmerzleitungssystem (Quelle: Pirman, 2016) Der Mechanismus, der hinter einer Fortleitung von Nerven- und Schmerzimpulsen steht, basiert auf der Freisetzung und Wirkung bestimmter chemischer Substanzen. Transmittersubstanzen, die bei der Weiterleitung des Nervenimpulses im Rahmen einer Schmerzwahrnehmung eine Rolle spielen, wären beispielsweise Substanz P und Glutamat. Diese führen zu einer Aktivierung von bestimmten Rezeptoren im Hinterhorn des Rückenmarks. Typischerweise wäre hier der sogenannte NMDA-Rezeptor zu nennen, welcher durch eine Blockade mit Magnesium unter normalen Bedingungen verschlossen bleibt. Die Nervenzellen im Rückenmark können allerdings durch die Wirkung von Glutamat und Substanz P teilweise depolarisiert werden, wodurch in Folge 8 die Blockade am NMDA-Rezeptor durch Magnesium aufgehoben wird und eine vollständige Depolarisation mit Weiterleitung des Schmerzreizes bis ins Gehirn stattfinden kann (vgl. Mutschler et.al. 2013, S. 198-199). Auf der Strecke der Weiterleitung von Schmerzimpulsen gibt es mehrere Abschnitte, an welchen eine Hemmung des Impulses stattfinden kann. An den Annahmestellen im Zentralnervensystem, welche die Impulse von Nozizeptoren aus der Peripherie empfangen, befinden sich auch Rezeptoren für Opioide, Noradrenalin, Serotonin und Gamma-Aminobuttersäure (GABA). Bei Aktivierung dieser präsynaptischen Rezeptoren im Rückenmark durch den entsprechenden Stoff kann eine Blockade in der Reizweiterleitung stattfinden. In der Substantia gelatinosa, einem Abschnitt im Hinterhorn des Rückenmarks, befinden sich Zwischenneurone, welche Opioide enthalten, über deren Freisetzung sie ebenfalls zu einer Hemmung der Impulsweiterleitung zum Gehirn befähigt sind. Dies bewerkstelligen die Zwischenneurone, indem sie sogenannte Enkephaline und Dynorphine freisetzen, welche zu einer Aufhebung der Reizleitung zwischen Rückenmark und Thalamus führen. Enkephaline beispielweise gehören zu den Opioidpeptiden und sind opioidartige Stoffe, die körpereigen hergestellt werden können und als Neurotransmitter fungieren. Absteigende Nervenzellen aus dem sogenannten Nucleus raphe magnus im Gehirn, welche Serotonin enthalten, sind in der Lage, dieses auszuschütten und dadurch eine Freisetzung der körpereigenen Opioide aus den Zwischenneuronen in der Subsatntia gelatinosa des Rückenmarks auszulösen. Ein weiterer Ausgangspunkt, der durch Nervenzellen gekennzeichnet ist, die Opioide enthalten, ist das sogenannte periaquäduktale Grau oder zentrales Höhlengrau, die Substantia grisea centralis. Diese Ansammlung von Nervenzellkörpern im Gehirn ist wiederum in der Lage, die stimulierende Wirkung auf den eben beschrieben Nucleus raphe magnus auszuüben. Zusätzlich befindet sich auch noch im sogenannten Locus coeruleus im Gehirn ein zur Blockade befähigtes Zellareal von Nervenzellen, welche Noradrenalin enthalten und dieses vom Gehirn absteigend abgeben können. Die Freisetzung der jeweiligen verschiedenen Neurotransmitter übt eine entsprechende hemmende Wirkung auf die Weiterleitung von Schmerzimpulsen aus (vgl. Höllt, Allgaier 2009, S. 209). 9 3 Welche Arten von Schmerzmedikamenten gibt es? Schmerzmedikamente werden in der medizinischen Fachsprache als Analgetika bezeichnet. „Analgetika sind Substanzen, die die Schmerzempfindung verringern bzw. unterdrücken, ohne eine allgemein-narkotische Wirkung zu besitzen.“ (Mutschler et.al. 2013, S.194) Nach aktuellen wissenschaftlichen Kriterien und pharmakologischen Standards werden diese in zwei große Gruppen unterteilt: 1. Nicht-Opioidanalgetika Nicht-opioidartige Analgetika können Opioidrezeptoren nicht beeinflussen und greifen auf eine andere Weise in den menschlichen Stoffwechsel ein, um das Schmerzerleben zu vermindern. Zu dieser Gruppe von Analgetika zählen verschiedene Substanzen, die im nächsten Kapitel 3.1 genauer beschrieben werden (vgl. Höllt, Allgaier 2009, S. 221). 2. Opioidanalgetika Opioidartige Analgetika sind in der Lage, eine Wirkung auf Opioidrezeptoren auszuüben, die zu einer Schmerzlinderung führt. Über die Bindung an diese Rezeptoren werden im Körper verschieden Wirkungen ausgelöst, diese werden in Kapitel 3.2 aufgezeigt (vgl. Zeilhofer, Sittl 2010, S. 70). 3.1 Pharmakologischer Wirkmechanismus und häufig verwendete Substanzen von Nicht-Opioidanalgetika Die meisten Schmerzmittel aus der Gruppe der Nicht-Opioidanalgetika hemmen mehr oder weniger stark das Enzym Cyclooxygenase (COX) und entfalten hauptsächlich dadurch ihre schmerzlindernde Wirkung. Diese Art der Schmerzmedikation wird am häufigsten verwendet, sie zeichnet sich zusätzlich durch eine fiebersenkende und von der Dosierung abhängige, entzündungshemmende Wirkungsweise aus (vgl. Höllt, Allgaier 2009, S. 223). 10 Nicht-Opioidanalgetika können des Weiteren in drei Gruppen unterteilt werden: 1. Saure antipyretisch-antiphlogistische Analgetika Diese Schmerzmittel haben aus chemischer Sicht mit Ausnahme der selektiven Cyclooxygenase 2-Hemmer sauren Charakter und wirken, abgesehen von ihrem analgetischen Einfluss, antipyretisch (fiebersenkend) und antiphlogistisch (entzündungshemmend). Sie werden gerne auch, aus der englischen Fachsprache abgeleitet, als NSAIDs (non-steroidal anti-inflammatory drugs) bezeichnet. 2. Nicht-saure antipyretische Analgetika Dabei handelt es sich um Medikamente, die chemisch gesehen keine Säuren sind und neben ihrer analgetischen Wirkung auch eine antipyretische (fiebersenkende) Wirkung aufweisen. 3. Analgetika ohne antipyretisch-antiphlogistische Wirkung Die dritte Gruppe der Nicht- Opioidanalgetika umfasst solche Medikamente, die rein analgetisch wirken und keine zusätzlichen antipyretischen (fiebersenkenden) oder antiphlogistischen (entzündungshemmenden) Eigenschaften besitzen (vgl. Höllt, Allgaier 2009, S. 221-222). 3.1.1 Saure antipyretische-antiphlogistische Analgetika oder NSAIDs Die erste Gruppe der Nicht-Opioidanalgetika zeichnet sich zusätzlich zu ihrer schmerzlindernden auch durch eine fiebersenkende und in hoher Dosierung durch eine ausgeprägte und therapeutisch gut nutzbare, entzündungshemmende Wirkung aus. Daher werden diese Medikamente auch gerne als nichtsteroidale Antiphlogistika oder NSAIDs (non-steroidal anti-inflammatory drugs) bezeichnet. Die Wirkung dieser Medikamentengruppe beruht auf einer Hemmung der Cyclooxygenasen-vermittelten Spaltung der ungesättigten Fettsäure, Arachidonsäure, in die Vorstufen bestimmter stoffwechselaktiver Stoffe, wie etwa der Prostaglandine und Thromboxane. Prostaglandine zählen zu den Schmerzmediatoren, deren Rolle bei der Schmerzentstehung bereits in Kapitel 2.3.1 beschrieben wurde. Durch eine Blockade eines bestimmten Enzyms, der Cyclooxygenase (COX), wird die Bildung der Prostaglandine gehemmt 11 und ihre schmerzfördernde Auswirkung auf den Organismus wird somit eingeschränkt oder bei hohen Dosierungen fast gänzlich unterbunden (vgl. Mutschler et.al. 2013, S.205). Es existieren zwei Arten von Isoenzymen des Enzyms Cyclooxygenase (COX), die Cyclooxygenase 1 (COX 1) und die Cyclooxygenase 2 (COX 2). Der wichtige Unterschied dieser beiden Enzymformen liegt darin, dass die COX 1 fortlaufend im menschlichen Organismus gebildet wird, wohingegen die COX 2 überwiegend erst durch bestimmte äußere Reize in höherem Maß aktiviert wird. Diese Aktivierung der COX 2 geschieht besonders im Rahmen von entzündlichen Prozessen, Schmerzgeschehen oder Schädigungen eines Gewebes. Wobei COX 1 im Gegenteil dazu hauptsächlich zur normalen, physiologisch notwendigen Bildung von Prostaglandinen führt, die für wichtige Stoffwechselvorgänge des Körpers benötigt werden. Beispiele dafür wären die Hemmung der Säureproduktion im Magen durch Prostaglandine oder eine Wirkung auf die Funktion von Thrombozyten bei der Blutgerinnung. Aufgrund dieser Tatsache hat die Hemmung der COX 2 eine größere therapeutische Bedeutung in Bezug auf die ungewollten Erscheinungsformen krankhafter Prozesse und deren gewünschte Beseitigung. Denn gerade die Verstärkung der Bildung von COX 2 bei Entzündungen oder Schmerzen soll ja durch die pharmakologische Intervention verhindert werden und somit auch im weiteren Sinne eine übermäßige Produktion von Prostaglandinen. Allerdings sind die Wirkungsweisen von Prostaglandinen im menschlichen Körper außerordentlich vielfältig, so dass sich deren Blockade in Bezug auf erwünschte und unerwünschte Wirkungen relativ die Waage hält und eine nebenwirkungsfreie Anwendung dieser Medikamente unmöglich macht (vgl. Mutschler et.al. 2013, S.206). Aus einer Unterdrückung der vermehrten Prostaglandin-Bildung resultiert keine direkte Schmerzausschaltung im menschlichen Organismus. Die Schmerzreduktion basiert auf einer indirekten Wirkung, da die Prostaglandine dazu führen, dass Nozizeptoren für schmerzauslösende Reize empfindlicher werden. Besonders Prostaglandin E2 und Prostaglandin I2 (Prostacyclin) erniedrigen die zur Auslösung eines Aktionspotentials nötige Schwelle. In diesem Zusammenhang spielen auch weitere Schmerzmediatoren, wie Bradykinin, Histamin oder Serotonin eine große Rolle, welche als auslösende Substanzen für die Reizweiterleitung fungieren (vgl. Höllt, Allgaier 2013, S. 212-213). Die Indikation zur Gabe von nichtsteroidalen Antiphlogistika stellt sich bei Schmerzgeschehen und Entzündungen vielfältigen Ursprungs. Im Besonderen bei akuten und 12 chronischen Krankheitsbildern der Arthritis, postoperativen Schmerzen, Schmerzen bei Tumorerkrankungen, während der Menstruation, bei Migräne und generell beim Auftreten von Fieber. Eine spezielle Indikation stellt auch ein nach der Geburt nicht verschlossener Ductus arteriosus Botalli bei Früh- beziehungsweise Neugeborenen dar, denn die Prostaglandine PGE2 und PGI2 spielen einer Rolle bei der Offenhaltung dieser Verbindung zwischen Aorta und Lungenarterie vor der Geburt. Eine medikamentöse Therapie mit Substanzen, welche die Prostaglandin-Bildung blockieren, wirkt sich förderlich auf einen Verschluss aus (vgl. Mutschler et.al. 2013, S.206). Acetylsalicylsäure (bspw. Medikamentennamen Aspirin®, Aspro®), Ibuprofen (bspw. Medikamentennamen Aktren®, Brufen®, Dolormin®), Diclofenac (bspw. Medikamentennamen Deflamat®, Arthrotec®, Diclobene®) und Naproxen (bspw. Medikamentenname Naprobene®, Proxen®) stellen die gängigsten Vertreter der in diesem Kapitel beschriebenen Substanzgruppe dar. Neben ihrem sauren Charakter besitzen diese Medikamente die Fähigkeit gut an Plasmaproteine zu binden (vgl. Höllt, Allgaier 2013, S. 209). Die Acetylsalicylsäure ist unter den NSAIDs die einzige Substanz, welche die COX Enzyme irreversibel hemmt und aus diesem Grund auch zu einer Verhinderung der Aggregation von Thrombozyten führt (vgl. Zeilhofer, Sittl 2010, S. 73). „Bereits durch eine einmalige Gabe einer subanalgetischen Dosis (50-100mg) wird infolge der Hemmung der COX die Synthese von Thromboxan A2 in den Thrombozyten vermindert und damit die Thrombozytenaggregation gehemmt. Dieser Effekt hält mehrere Tage an, weil die kernlosen Thrombozyten nicht zur Proteinsynthese fähig sind und damit keine COX nachsynthetisieren können. “ (Höllt, Allgaier 2013, S. 213) Dies erklärt ihre Anwendung zur Prophylaxe von Herzinfarkt und Schlaganfall. Die im Vergleich zu den anderen Vertretern der NSAIDs starken Nebenwirkungen der Acetylsalicylsäure auf den Magen-Darm-Trakt, lassen sie für eine langfristige, effektive analgetische Therapie zunehmend als ungeeignet erscheinen (vgl. Zeilhofer, Sittl 2010, S. 73). Historisch betrachtet ist das Medikament Aspirin® bereits in der Antike in Form der Weidenrinde (Weide heißt auf lateinisch salix) zur Behandlung von Schmerzen und Fieber verwendet worden. Nach der Isolierung des Salicins und der Oxidation des Salicylalkohols zur Salicylsäure im neunzehnten Jahrhundert, gelang schließlich die Synthese der Reinform von Acetylsalicylsäure im Jahre 1899. Zu diesem Zeitpunkt wurde 13 das immer noch gängige Aspirin® von der Pharmafirma Bayer erstmals auf den Markt gebracht (vgl. Höllt, Allgaier 2013, S. 211). NSAIDs als Substanzgruppe sind generell Medikamente mit einem nicht zu unterschätzenden, starken Nebenwirkungsprofil, welches gerade aufgrund ihrer weit verbreiteten Anwendung als problematisch zu betrachten ist. Unter den leichteren Nebenwirkungen wären Übelkeiten, Sodbrennen und Erbrechen zu nennen, welche oft auftreten können. Die typischen, bei diesen Medikamenten auftretenden Komplikationen können die Entstehungen von Geschwüren des Magen- und Darmtraktes beinhalten oder zu einer Verschlimmerung von einer diesbezüglich bereits bestehenden Symptomatik bei Einnahme dieser Medikamente führen. Bei Personen, die zu allergischen Reaktionen neigen, können teils gravierende körperliche Reaktionen, wie Asthmaanfälle („Aspirin-Asthma“) oder Hautausschläge bei der Einnahme von Acetylsalicylsäure auftreten. Das in Bezug auf den Abbau von Aspirin entstehende Salicylat kommt in der Niere mit der Harnsäure in Interaktion und die beiden Stoffe konkurrieren um die Ausscheidungskapazität des Organs. Daher führen empfohlene Mengen zur Behandlung von Schmerzzuständen zu einer verminderten Ausscheidung von Harnsäure. Bei der Einnahme der empfohlenen, höheren Mengen zur Behandlung von rheumatisch bedingten Schmerzzuständen tritt wiederum eine erhöhte Harnsäure-Ausscheidung auf und es kann auch zu unerwünschten Wirkungen auf das zentrale Nervensystem kommen, die sich etwa mit Schwindel, Ohrensausen, Hör- und Sehstörungen, Übelkeit oder Erbrechen äußern. Ein langfristiger und hochdosierter Gebrauch von NSAIDs kann zur irreversiblen Schädigung der Nierenfunktion führen (vgl. Höllt, Allgaier 2013, S. 214-215). Intoxikationen fallen primär durch eine Hyperventilation auf und äußern sich in der Folge durch Entgleisungen des Säure-Basen Haushaltes im menschlichen Organismus. Eine Hämodialyse kann in schwerwiegenden Fällen lebensrettendes Mittel der Wahl sein, bereits zehn Gramm Acetylsalicylsäure können zu einem tödlichen Ausgang führen. Ibuprofen und Diclofenac werden nach oraler Einnahme überwiegend im Dünndarm resorbiert, sie haben im Vergleich mit der bereits im Magen resorbierten Acetylsalicylsäure einen verzögerten Eintritt der Wirkung. Diclofenac fällt zusätzlich auch durch eine relativ niedrige Bioverfügbarkeit bei oraler Einnahme auf, die von Mensch zu Mensch sehr stark variieren kann und trotzdem treten oft bei längerfristiger Anwendung die typischen, starken Nebenwirkungen auf. In Hinsicht auf mögliche 14 Überdosierungen wäre bei Ibuprofen der Vorteil zu nennen, dass dieses Medikament sich bei hoher und mehrfacher Einnahme nicht im Organismus anhäuft und dadurch die Gefahr von lebensgefährlichen Intoxikationen eingeschränkt ist (vgl. Höllt, Allgaier 2013, S. 215). 3.1.2 Selektive Cyclooxygenase 2-Hemmer (Coxibe) Der größte Teil der nichtsteroidalen Antiphlogistika hemmt beide Enzymformen, sowohl COX 1, als auch COX 2. Es wurden allerdings aufgrund der Bestrebung, die durch die Hemmung der COX 1 typischen Nebenwirkungen zu verhindern, selektive COX 2Inhibitoren entwickelt, die nicht gänzlich den gewünschten Effekt der Nebenwirkungsfreiheit erzielt haben. COX 2-selektive NSAIDs oder auch COXIBE genannt, haben komplexere Nebenwirkungen als COX 1-Hemmer, diese sind dennoch nicht unbedeutend. COX 2 spielt beispielweise eine Rolle bei Wundheilungsprozessen und ebenfalls bei der Entwicklung von Tumorzellen, daher ergeben sich auch Anwendungsgebiete dieser Medikamente in der Tumor-Therapie (vgl. Mutschler et.al. 2013, S.207). Coxibe sind gleich gut antiphlogistisch und analgetisch wirksam wie NSAIDs, allerdings haben sie den Nachteil eines relativ verzögerten Wirkungseintritts bei oraler Verabreichung im Vergleich zu den herkömmlichen nichtsteroidalen Antiphlogistika. Der Konzentrations-Anstieg der Substanzen im Plasma dauert länger, daher sind sie zur Anwendung bei akuter Schmerzsymptomatik ungeeignet. Hervorzuheben sind allerdings die geringen Nebenwirkungen im Gastrointestinaltrakt, in Bezug auf Pseudoallergien, wie Anlagetika-induziertes Asthma, sowie betreffend einer negativen Beeinflussung der Blutgerinnung dieser Medikamente. Nebenwirkungen auf die Nieren sind hingegen nicht zu vernachlässigen und mit den unerwünschten Wirkungen von NSAIDs gleich zu setzen, wobei sie sich hauptsächlich in einem Anstieg des Blutdrucks durch verminderte Ausscheidung von Wasser und Natrium äußern (vgl. Zeilhofer, Sittl 2010, S. 73). Laut Mutschler zählen zu den häufigsten spezifischen Nebenwirkungen dieser Medikamentengruppe Atemwegs-Infektionen, Diarrhö, Dyspepsie, Beschwerden im oberen Bauch und Kopfschmerzen. Negativ aufgefallen sind die Coxibe durch die Substanz Rofecoxib, welche bereits vom Markt genommen wurde. Es konnte nachgewiesen 15 werden, dass die Einnahme das Risiko für Herz-Kreislauf-Erkrankungen erhöhte. Generell ist die Dosierung von herkömmlichen NSAIDs und Coxiben so niedrig wie möglich und die Verabreichung über eine möglichst kurze Zeitspanne vorzunehmen, da eine lange Anwendung ein erhöhtes Risiko für das Auftreten von Problemen im HerzKreislauf Bereich vermuten lässt. Während der Schwangerschaft dürfen diese Medikamente ebenfalls nicht angewendet werden. Die hauptsächlich zur Anwendung kommenden Substanzen der selektiven COX 2-Hemmer sind Celecoxib (bspw. Medikamentenname Celebrex®), Etoricoxib (bspw. Medikamentenname Arcoxia®) und Parecoxib (bspw. Medikamentenname Dynastat®). Celecoxib wird teilweise zur Behandlung von Darmkrebs und Darmpolypen eingesetzt, weil das Enzym COX 2 bei diesen Erkrankungen eine entscheidende Rolle zu spielen scheint. Neben Celecoxib wird auch Etoricoxib zur Behandlung von Arthrose und Arthritis eingesetzt. Parecoxib kommt bei der kurzzeitigen Behandlung von Schmerzen nach operativen Eingriffen zur Anwendung (vgl. Mutschler et.al. 2013, S.213-214). 3.1.3 Nicht-saure antipyretische Analgetika Die exakte Art der schmerzstillenden Wirkung dieser Medikamentengruppe auf körperlicher Ebene ist nicht so genau untersucht und verstanden wie die Wirkungsweise der NSAIDs. Ihr Angriffspunkt befindet sich eher im zentralen Nervensystem, da sie leicht die Blut-Hirn- Schranke passieren können. Dies führt im Rückenmark und Gehirn zur Blockade einer übermäßigen Prostaglandin-Bildung und lässt die für die eher peripher wirkenden NSAIDs typischen Nebenwirkungen äußerst gering ausfallen. Weil die Produktion von Prostaglandinen im peripheren Körpergewebe nicht beeinflusst wird, kann durch diese Medikamente auch keine entzündungshemmende Wirkung erzielt, sehr wohl aber eine fiebersenkende Wirkung erreicht werden (vgl. Mutschler et.al. 2013, S.215). Die antipyretische Wirkung sowohl von sauren, als auch von nicht – sauren antipyretischen Analgetika kommt aufgrund einer Blockade des Enzyms Cyclooxygenase 2 (COX2) zu Stande. Fieber entsteht durch eine komplexe körperliche Reaktion auf so genannte exogene Pyrogene, die durch Viren oder Bakterien in den Körper gelangen können. Die Folge ist eine vermehrte Bildung von COX2 und dadurch auch ein höherer 16 Spiegel von Prostaglandin E2 (PGE2), der über cAMP eine Veränderung des Sollwertes der Körperkerntemperatur im vorderen Hypothalamus bewirkt. Eine Verhinderung der Bildung von PGE2 kann eine Antipyrese durch Analgetika zu Stande bringen (vgl. Höllt, Allgaier 2013, S. 213). Bei der Gegenüberstellung der sauren und nicht-sauren antipyretischen Analgetika führen die nicht-sauren zu einer weitaus geringeren Hemmung des Enzyms Cyclooxygenase, als die sauren Substanzen. Für einen therapeutischen Effekt in einem von Entzündungserscheinungen betroffenen, peripheren Gewebe reichen Medikamente dieser Gruppe daher nicht aus, außerdem befindet sich die Stelle ihrer Wirkung auch hauptsächlich im zentralen Nervensystem. Die Blockade der COX 2 im Rückenmark und Gehirn genügt aber, um Schmerzen zu unterdrücken. Teilweise haben die beschriebenen Substanzen auch eine zusätzliche Wirkung auf den TRPA1-Kanal in Nervenzellen des Rückenmarks. Die Bedeutung dieser Kanäle in den Zellmembranen für die Weiterleitung von Informationen wurde auch in Kapitel 2.3.1 dargelegt (vgl. Höllt, Allgaier 2013, S. 212-213). Unter den häufig angewendeten Substanzen von nicht-sauren antipyretischen Analgetika wären vor allem das Paracetamol (bspw. Medikamentennamen ben-u-ron®, Mexalen®, Dolomo®) und das Metamizol (bspw. Medikamentenname Novalgin®) zu nennen. Phenazon (bspw. Medikamentenname Coffo-Selt-Brausetabletten®) und Propyphenazon (bspw. Medikamentenname Adolomed®) sind bezüglich ihrer Wirkungsweise dem Metamizol sehr ähnlich (vgl. Mutschler et.al. 2013, S.215-217). Bei der Behandlung von Schmerzen und Fieber mit Paracetamol muss trotz des geringeren Nebenwirkungsprofils im Vergleich zu den NSAIDs besonderes Augenmerk hinsichtlich der potentiell schädigenden Wirkung dieses Medikamentes auf die Leber gelegt werden und auf die geringe therapeutische Breite dieses Medikamentes hingewiesen werden. Es wird hauptsächlich in der Leber verstoffwechselt und durch eine Kopplung mit den Verbindungen Glucuronid und Sulfat zur Ausscheidung über die Nieren befähigt. Ein sehr geringer Anteil von drei Prozent wird ohne Veränderung der chemischen Struktur des Stoffes renal aus dem Körper ausgeschieden. Für die chemische Veränderung der Substanz in der Leber müssen allerdings genügend funktionsfähige Einheiten von Leberzellen zur Glucuronidierung und Sulfatierung vorhanden sein. Dies kann im Falle einer Überdosierung oder einer eingeschränkten Leberfunktion verhindert werden. Als Folge davon entstehen giftige Stoffwechselprodukte des 17 Paracetamols im Organ und führen zu dessen Untergang. Mutschler empfiehlt für Erwachsene ohne beeinträchtigte Leberfunktion eine Einzeldosis von 500-1000 mg und für Kinder eine Einzeldosis von bis zu 10mg/kg. Die Tageshöchstdosis von Paracetamol darf bei Erwachsenen 4000 mg und bei Kindern 50mg/kg nicht überschreiten. Das bei nicht ausreichender Kapazität der Leber zum Abbau des Medikaments bedeutendste, giftige Stoffwechselprodukt ist das N-Acetylchinonimin. Die Zwischenprodukte dieser für die Leber toxischen Stoffe binden an Proteine der Leberzellen und führen zu deren Nekrotisierung „Dosen über 10g (entsprechend dem 2,5-fachen der Tageshöchstdosis) führen dementsprechend unbehandelt zu schweren, meist tödlichen Leberzellnekrosen. Bei Patienten mit vorgeschädigter Leber (z.B. durch Alkohol) können bereits Dosierungen von 4-6 g oder darunter problematisch sein.“ (Mutschler et.al. 2013, S.216) Metamizol ist ein hoch wirksames Analgetikum mit zusätzlicher antipyretischer und spasmolytischer Wirkung. Als seltene starke Nebenwirkung dieses Medikamentes muss die Agranulozytose genannt werden, welche zum Tode führen kann. Zusätzlich sind auch bei schneller intravenöser Gabe in manchen Fällen kritische Schockzustände bei PatientInnen aufgetreten, daher darf dieses Medikament intravenös nur kurzzeitig angewendet werden (vgl. Zeilhofer, Sittl 2010, S. 74). Die Daten über die Gefährlichkeit der Nebenwirkungen von Metamizol variieren stark innerhalb von Europa, bei längerer Anwendung muss in jedem Fall eine Überprüfung des Blutbildes stattfinden. Weniger gravierende unerwünschte Wirkungen können sich in Veränderungen von Schleimhäuten, Hautreaktionen und Blutdruckabfall äußern (vgl. Mutschler et.al. 2013, S.216). 3.1.4 Analgetika ohne antipyretisch-antiphlogistische Wirkung Die dritte Gruppe der Nicht-Opioidanalgetika entfaltet ihre Wirkung ebenso, wie die im vorherigen Kapitel beschriebenen Substanzen, im zentralen Nervensystem. Eine Beeinflussung für Rezeptoren, die auf Opioide ansprechen, findet dabei nicht statt und es kommt auch zu keinem Gewöhnungseffekt. Höllt zählt fünf Substanzklassen zu dieser Gruppe von Analgetika, darunter Flupirtin, Ziconotid, welches eine hundert- bis tausendfach stärkere Wirkung in Bezug auf die Schmerzstillung hat, als das Opioida18 nalgetikum Morphin, Ketamin, Capsaicin und Cannabinoide. Das sind alles sehr unterschiedliche Substanzen, welche eben nur unter den Gesichtspunkten der groben Eigenschaften, weder antipyretisch noch antiphlogistisch wirksam zu sein, in einer Gruppe zusammengefasst werden können. Bei Ketamin handelt es sich um eine besonders stark schmerzlindernde Substanz, welche vor allem in Notfällen zur raschen Schmerzreduktion verwendet werden kann. Capsaicin stammt aus Chili-Pfefferoni und wirkt über die in Kapitel 2.3.1 bereits dargestellten TRPVR1-Kanäle lokal schmerzstillend, indem es nach einer primären Aktivierung nachfolgend zu einer Aufhebung der Sensibilität dieser Schmerzrezeptoren führt. Tetrahydrocannabinol (THC oder Dronabinol) ist die wirksame, schmerzlindernde Substanz der Cannabinoide, Medikamente mit diesem Wirkstoff sind eingeschränkt bei neuropathisch bedingten Schmerzen zugelassen (vgl. Höllt, Allgaier 2013, S. 218). Flupirtin (bspw. Medikamentenname Katadolon®) wirkt neben seinem analgetischen Effekt auch relaxierend auf die Muskulatur und bewerkstelligt dies über die Beeinflussung von Kaliumkanälen in Nervenzellen, allerdings ist der genaue Wirkmechanismus dieses Medikamentes noch unbekannt. Es wird in der Leber verstoffwechselt und über die Niere ausgeschieden, daher ist Vorsicht geboten bei der gleichzeitigen Anwendung mit anderen Medikamenten, welche ebenfalls in der Leber verstoffwechselt werden, wie etwa das typische Beispiel Paracetamol. Ferner ist bei diesem Medikament besonders auf Wechselwirkungen mit anderen Substanzen zu achten, durch seinen Angriffspunkt im zentralen Nervensystem interagiert es besonders mit solchen Stoffen, die ebenfalls an dieser Stelle ihre Wirkung entfalten. Zu den unerwünschten Wirkungen von Flupirtin zählen neben den typischen zentral bedingten Störungen, wie Müdigkeit und Schwindel, auch solche des Magen-Darmtraktes wie Übelkeit, Durchfall oder Verstopfung. Ziconotid (bspw. Medikamentenname Prialt®) ist ein nach Vorbild des Giftes einer Meeresschnecke künstlich hergestelltes Medikament zur Injektion in den Liquorraum bei therapieresistenten, chronischen Schmerzzuständen. Das Medikament entfaltet seine Wirkung über die Hemmung bestimmter Kalziumkanäle von Nervenzellen. Da es eher selten zur Anwendung kommt, fehlen ausreichend Daten zur genauen Beurteilung der Risiken bezüglich Nebenwirkungen und Interaktionen mit anderen Medikamenten. Eine gleichzeitige Anwendung mit intrathekalen Chemotherapeutika ist jedenfalls unzulässig und es sollte nur von Ärzten mit ausreichend Erfahrung angewendet werden. Zu den häufigen, unerwünschten Wirkungen zählen hauptsächlich zentralnervöse Symptome, wie Schwindel, Übelkeit, Nystagmus, abnormales 19 Gangbild, abnorme Schläfrigkeit, Störungen der Gedächtnisleistung und Kopfschmerzen, um nur einige von vielen zu nennen. Flupirtin und Ziconotid sind in Deutschland zugelassen, in Österreich jedoch nicht, daher beziehen sich die angegebenen Medikamentennamen auf den Handelsraum Deutschland (vgl. Mutschler et.al. 2013, S.216). 3.2 Pharmakologischer Wirkmechanismus und häufig verwendete Substanzen von Opioidanalgetika Opioidanalgetika sind sehr potente Schmerzmittel, die ihre Wirkung über die Besetzung sogenannter Opioidrezeptoren entfalten. Hauptsächlich spielen in dieser Hinsicht die µ-Opioidrezeptoren eine Rolle. Aufgrund der Gefahr eines körperlichen und geistigen Gewöhnungseffektes unterliegt die Abgabe dieser Medikamentengruppe großteils strengen gesetzlichen Regelungen, die im Suchtmittelgesetz schriftlich verankert sind. Dennoch stellen diese Medikamente eine wichtige Therapieoption in der Schmerzbehandlung dar und sind bei fachgerechter Anwendung gut zu handhaben (vgl. Zeilhofer, Sittl 2010, S. 70). Die Verwendung von derartigen Schmerzmitteln geht aus geschichtlicher Sicht sehr weit in das Altertum zurück und wurde bereits einige Jahrtausende vor Christus beschrieben. Die therapeutische Behandlung basierte bereits in frühen Zeiten auf Extrakten aus Schlafmohn (Papaver Somniferum), dessen getrockneter Pflanzenkapsel-Saft als Opium bezeichnet wird. Opium enthält bis zu zwölf Prozent Morphin und bis zu ein Prozent Codein, darüber hinaus auch geringfügig Thebain und andere Stoffe, wie Papaverin oder Noscapin. Aus dem Opium gewonnene Arzneimittel werden als Opiate bezeichnet, darunter werden sowohl die natürlich vorkommenden, therapeutisch genutzten Inhaltsstoffe des Opiums, sowie auch die daraus zu gewissen Teilen künstlich hergestellten Arzneien gezählt. Grob zusammenfassend ausgedrückt also alle chemischen Verbindungen, die strukturell eine Ähnlichkeit mit Morphin aufweisen. Die Bezeichnung Opioide stellt eine zum Großteil immer anwendbare und aktuell gültige Begriffsdefinition dar. Die Stoff-Gruppe der Opioide beinhaltet sämtliche Substanzarten, die sowohl körpereigen als auch körperfremd über eine Wechselwirkung mit sogenannten Opioidrezeptoren Wirkungen des Morphins hervorrufen (vgl. Höllt, Allgaier 2013, S. 218-219). 20 „Opioidrezeptoren kommen in unterschiedlicher Dichte sowohl prä- als auch postsynaptisch im Zentralnervensystem und peripher vor. In besonders großer Anzahl werden sie im limbischen System, Thalamus, Hypothalamus und Striatum, sowie in der Formatio reticularis und der Substantia gelatinosa des Rückenmarks gefunden.“ (Mutschler et.al. 2013, S.218) Es gibt drei unterschiedliche Arten von Opioidrezeptoren: µ-Opioidrezeptoren, δ-Opioidrezeptoren, К-Opioidrezeptoren und zusätzlich noch einige Untertypen. Wobei die µ-Rezeptoren für die klassischen Opioidwirkungen verantwortlich sind, auf welche im folgenden Text genauer eigegangen wird. Die δ-Rezeptoren können neben einer schmerzstillenden Wirkung auch emotionale Verstimmungen und Halluzinationen auslösen. Die К-Opioidrezeptoren führen abgesehen von der Schmerzreduktion ebenfalls zu Sedierung und Pupillenverengung (vgl. Mutschler et.al. 2013, S.218). Laut Höllt sind allerdings die möglicherweise als Nebenwirkung auftretenden emotionalen Verstimmungen eher auf die Aktivierung der К-Opioidrezeptoren zurückzuführen, wobei die δOpioidrezeptoren gleich den µ-Opioidrezeptoren eine Atemdepression auslösen können. Grundsätzlich sind aber alle drei Rezeptortypen für die Schmerzstillende Wirkung verantwortlich und lösen ebenfalls jeweils eine Blockade der zu Weiterleitung des Speisebreis nötigen autonomen Beweglichkeit des Magens bzw. Darmes aus (vgl. Höllt, Allgaier 2013, S. 220-221). In ihrer Funktionsweise gleichen sich die drei Opioidrezeptor-Typen dahingehend, dass sie zu den G-Protein-gekoppelten Rezeptoren zählen und generell erst aufgrund einer möglichen Hemmung ihrer Aktivierung durch den Antagonisten der Opioid-Wirkungen, dem sogenannten Naloxon, als echte Opioidrezeptoren gelten (vgl. Höllt, Allgaier 2013, S. 219-220). Die Verhinderung der Wahrnehmung von Schmerzreizen kommt auf zellulärer Ebene durch eine Beeinflussung der Aktivität von Ionenkanälen zu Stande, diese wird durch das Binden der Opioide an die entsprechenden Rezeptoren vermittelt. Detailliert sind hier die Calciumkanäle und Kaliumkanäle betroffen. Die Blockade bestimmter Enzyme, der sogenannten Adenylatcyclasen, führt zu einer Erniedrigung eines Signalstoffes, dem cyclischen Adenosinmonophosphat (cAMP) und in Folge zu einer erniedrigten Öffnung von Calciumkanälen und einer erhöhten Öffnung von Kaliumkanälen. Da diese Rezeptoren für Opioide im gesamten Körper verteilt sind, ergibt sich ein relativ vielschichtiges Spektrum an Wirkungen, welche nach deren Lokalisation in Wirkungen 21 bezüglich des zentralen Nervensystems und Wirkungen bezüglich des peripheren Nervensystems unterteilt werden (vgl. Mutschler et.al. 2013, S.218): 1. Zentrale Wirkungen von Opioidanalgetika „Analgesie: Opioide unterdrücken auf spinaler Ebene nozizeptive Impulse, aktivieren das endogene schmerzhemmende System und verändern im limbischen System das Schmerzerlebnis. (Die Schmerzen werden nicht mehr als so unangenehm und bedrohlich empfunden.)“ (Mutschler et.al. 2013, S.218) Weitere zentrale Wirkungen von Opioiden sind Sedierung, Angstlösung, positive wie negative Auswirkungen auf die emotionale Befindlichkeit (Euphorie bzw. Dysphorie), Blockade des Atem- und Hustenzentrums im zentralen Nervensystem, Pupillenverengung (Miosis), Erhöhung des Widerstandes der Skelettmuskulatur und Erhöhung der Konzentration von ADH (Antidiuretisches Hormon). Darüber hinaus können Opioide zentral eine Aktivierung des Brechzentrums im Gehirn bewirken, diese geht in Folge dann oft in eine Hemmung des Brechzentrums über (vgl. Mutschler et.al. 2013, S. 218). 2. Periphere Wirkungen von Opioidanalgetika Auch im peripheren Nervensystem bewirken Opioide eine Hemmung der Schmerzempfindung. Darüber hinaus führen diese Medikamente zu einer Verzögerung der Entleerung des Magens, zu Verstopfung, einer erschwerten Abgabe von Gallenflüssigkeit und Harn aufgrund einer Erhöhung der Spannung der Schließmuskulatur dieser Organe, zu einer verminderten Wandspannung der Blutgefäße und somit der Gefahr eines stark erniedrigten Blutdruckes und sie können aufgrund einer Erhöhung der Ausschüttung von Histamin zu allergischen Reaktionen führen (vgl. Mutschler et.al. 2013, S.219). 3.2.1 Einteilung der Opioidanalgetika nach pharmakologischen Aspekten Grundsätzlich kann bei Opioiden eine Unterscheidung bezüglich der Geschwindigkeit ihrer Rezeptorbindung sowie der Stärke und Dauer ihrer Wirkung getroffen werden. Darüber hinaus wird aus pharmakologischer Sicht eine Einteilung nach ihrem Bindungsverhalten an den Opioidrezeptoren neben ihrer durch die Rezeptorbindung hervorgerufenen Wirkstärke, der sogenannten intrinsischen Aktivität, vorgenommen. 22 Demzufolge sind vier unterschiedliche Typen dieser Substanzgruppe zu nennen, wobei die reinen Opioidrezeptor-Agonisten hauptsächlich über die Bindung an µ-Opioidrezeptoren ihre Wirkung erreichen (vgl. Mutschler et.al. 2013, S. 219): 1. Reine Opioidrezeptor-Agonisten Opioidanalgetika sind großteils reine Opioidrezeptor-Agonisten. Der Prototyp der Opioide, das Morphin, fällt in diese Gruppe und wird aufgrund seiner relativ schwierigen chemischen Herstellung heute noch immer meist direkt aus dem Opium extrahiert, wobei Morphin als Einzelsubstanz auch als erstes Opioid aus dem Opium chemisch abgetrennt wurde (vgl. Zeilhofer, Sittl 2010, S. 70). Morphin entfaltet seine Wirkung hauptsächlich über die Bindung an die µ-Opioidrezeptoren, durch deren Aktivierung, die Schmerzreizleitung gehemmt wird. Eine geringe Abänderung im molekularen Aufbau der opioidartigen Substanzen kann dazu führen, dass sie auf verschiedene Opioidrezeptoren stärker beziehungsweise schwächer wirken. Dies wurde genutzt, um Opioidanalgetika zu synthetisieren, die bestimmte unerwünschte Wirkungen von Opioiden umgehen (vgl. Mutschler et.al. 2013, S. 219-220). Diacetylmorphin oder Heroin ist ebenfalls ein typischer Agonist an Opioidrezeptoren mit einer niedrigeren Neigung an µ-Rezeptoren zu binden als Morphin. Durch die fettlösliche Eigenschaft dieser Verbindung kann diese vom Blut rasch in das Gehirn übertreten. Im zentralen Nervensystem findet eine chemische Umwandlung des Heroins statt, die Bindung führt an den µ-Rezeptoren zu einer höheren Wirkstärke, die das von Süchtigen gewünschte Hochgefühl bei intravenöser Anwendung auslöst und daher das hohe Suchtpotenzial dieser Substanz verdeutlicht. Hydromorphon ist eine chemisch abgewandelte Substanz von Morphin mit einer etwas stärkeren Wirkung als die Ausgangssubstanz und aufgrund seiner chemischen Eigenschaften einer Nieren-schonenden Abbaudynamik im Körper. Codein ist ein Opioidrezeptor-Agonist mit schwächerer Wirkung als Morphin an den µ-Rezeptoren. Bei Personen mit gewissen genetischen Veränderungen in der Art und Weise, wie diese Substanz verstoffwechselt wird, besitzt Codein nahezu keine schmerzstillende Wirkung. Codein und Dihydrocodein werden eher gerne als Medikamente zur Hustenstillung eingesetzt (vgl. Höllt, Allgaier 2013, S. 224-225). 23 Substanzen, welche eine viel höhere Wirkstärke als Morphin in Bezug auf Schmerzreduktion aufweisen, wären die hauptsächlich intensivmedizinisch verwendeten und parenteral verabreichten Opioide Fentanyl, Sufentanil, Remifentanil und Alfentanil. Sie besitzen auf chemischer Ebene alle vier lipophilen Charakter, was zu einer raschen Verteilung im menschlichen Körper führt. Remifentanil hat neben der potentesten Wirkung gegen Schmerzen allerdings auch die kürzeste Wirkdauer. Weitere Substanzen, die in diese erste Gruppe der reinen Opioidrezeptor-Agonisten fallen wären auch noch Levomethadon, welches länger als andere Opioide braucht, um wieder aus dem Körper ausgeschieden zu werden, daher eine Eignung zur Entzugstherapie aufweist und ebenfalls als Analgetikum angewendet wird (vgl. Zeilhofer, Sittl 2010, S. 71). Darüber hinaus sind auch die Substanzen Pethidin, als erstes künstlich hergestelltes Opioid und dessen abgeänderte Form, das Loperamid, zu nennen. Pethidin hat neben einer geringeren Wirkung gegen Schmerzen als Morphin aufgrund einer schwächeren Bindung an die µ-Opioidrezeptoren auch die ungünstige Eigenschaft, sich bei langfristiger Anwendung im Körper anzuhäufen, daher ist nur eine kurzfristige Medikation sinnvoll. Loperamid hingegen hat eine sehr hohe Bindungsstärke an die µ-Opioidrezeptoren, die sogar zweihundertmal stärker ist als das Bindungsverhalten von Morphin. Aufgrund seiner chemischen Eigenschaften erreicht dieses Opioid allerdings nur geringe Konzentrationen im Körper und kann auch nicht in das Gehirn übertreten, daher wird es hauptsächlich zur Beeinflussung der Opioidrezeptoren im Magen-Darm-Bereich verwendet und als Medikament gegen Durchfall eingesetzt (vgl. Höllt, Allgaier 2013, S. 225-226). 2. Gemischte Opioidrezeptor-Agonisten und Opioidrezeptor-Antagonisten Diese künstlich hergestellten Opioide wurden mit dem Hintergedanken, eine Verhinderung des Substanzmissbrauches zu gewährleisten, erzeugt. Sie wirken über eine Bindung an К-Opioidrezeptoren und blockieren als Antagonisten eine Aktivierung der µ-Opioidrezeptoren, die auch eine Rolle für die Suchtentstehung spielen. Als Opioide, die in diese zweite Gruppe fallen, wären nur Pentazocin und Nalbuphin zu nennen. Pentazocin konnte sich aufgrund geringer Vorteile gegenüber Morphin in der Anwendung am Markt nicht durchsetzen. Nalbuphin hingegen weist eine zehnfach höhere 24 antagonistische Wirkung auf die µ-Opioidrezeptoren auf als Pentazocin und löst deshalb bei missbräuchlicher Verwendung durch Suchtkranke Symptome des Entzugs aus (vgl. Mutschler et.al. 2013, S. 220). Während Pentazocin in Bezug auf die Auslösung von Halluzinationen und Zuständen der Dysphorie negativ aufgefallen ist, hat sich Nalbuphin im Bereich der Geburtshilfe bewährt, da es wegen seiner antagonistischen Wirkung auf die µ-Opioidrezeptoren eine geringere Gefahr für die Entstehung einer Atemdepression beim Neugeborenen mit sich bringt. Darüber hinaus ist die Verwendung von Nalbuphin nicht gesetzlich reglementiert (vgl. Höllt, Allgaier 2013, S. 227). 3. Opioidrezeptor Partialagonisten Partielle Opioidrezeptor-Antagonisten wurden ebenfalls mit dem Hintergedanken hergestellt, eine Verhinderung des Missbrauches zu erreichen. Die Substanz, die in diese Gruppe fällt, wäre das Buprenorphin. Es weist eine sehr starke Rezeptorbindung auf, allerdings führt es im Vergleich zu Morphin zu keiner vollständigen Besetzung der µOpioidrezeptoren (vgl. Zeilhofer, Sittl 2010, S. 71). Dies ändert sich auch durch eine Steigerung der Dosis von Buprenorphin nicht und wirkt sich besonders positiv in Bezug auf die Nebenwirkungen von Opioiden aus, die dadurch vermindert werden können, weil keine vollständige Rezeptoraktivierung stattfinden kann (Ceiling-Effekt). Allerdings können Symptome eines Entzugs auftreten, wenn nach einer längerfristigen Anwendung eines reinen µ-Opioidrezeptor-Agonisten auf einen Partialagonisten umgestellt wird (vgl. Mutschler et.al. 2013, S. 220). 4. Reine Opioidrezeptor-Antagonisten Hier wären die Substanzen Naloxon und Naltrexon zu nennen, welche beide reine Antagonisten an Opioidrezeptoren sind, sie ähneln in ihrer chemischen Struktur den Opioiden und können so an die Opioidrezeptoren binden ohne eine Wirkung hervorzurufen. Dadurch konkurrieren sie mit anderen opioidartigen Stoffen um die RezeptorBindungsstellen und hemmen auf diese Weise indirekt deren Wirkstärke, weshalb sie bei Vergiftungen oder bei zu hohen Dosierungen von Opioiden zur Anwendung kommen. Naloxon hat die Eigenschaft, an alle Arten von Opioidrezeptoren zu binden, in kleinen Dosen vorrangig an die µ-Opioidrezeptoren, in größeren Dosen auch an alle anderen. Bei suchtkranken Personen führt die Verabreichung von Naloxon zu einer akuten Entzugssymptomatik, daher ist hier Vorsicht geboten und eine Anwendung mit 25 Bedacht durchzuführen. Generell wird Naloxon relativ schnell wieder aus dem Körper eliminiert, daher muss es bei Überdosierungen von Opioiden oft hintereinander intravenös verabreicht werden. Naltrexon hat eine viel längere Wirkdauer als Naloxon und zwar ein bis zwei Tage, daher eignet es sich für eine langfristige Entzugstherapie und kann oral eingenommen werden. Vorsicht ist bei Patienten mit vorgeschädigter Leber geboten, da die Substanz giftig für die Leberzellen ist (vgl. Höllt, Allgaier 2013, S. 226227). 3.2.2 Einteilung der Opioidanalgetika nach WHO-Stufenschema Des Weiteren können Opioidanalgetika, abgesehen von ihren pharmakologischen Aspekten, nach dem WHO-Stufenschema grob in zwei Gruppen eingeteilt werden: 1. Schwach wirksame Opioide Diese Medikamentengruppe weist eine geringere Vergiftungs- und Suchtgefahr auf, als die Gruppe der stark wirksamen Opioide, da trotz Dosissteigerung keine Zunahme der Wirkung eintritt (Ceiling-Effekt) (vgl. Zeilhofer, Sittl 2010, S. 70). Schwach wirksame Opioide werden laut Weltgesundheitsorganisation (WHO) in die Gruppe zwei des in Kapitel 4.1 beschriebenen Stufenschemas der Schmerztherapie eingeteilt (vgl. Mutschler et.al. 2013, S. 225). 2. Stark wirksame Opioide Alle Medikamente aus dieser Gruppe unterliegen dem Suchtmittelgesetz und sind aufgrund ihres relativ hohen Gefahrenpotentials nur für eine fachgerechte Anwendung vorgesehen. Stark wirksame Opioide werden laut Weltgesundheitsorganisation (WHO) in die Gruppe drei des in Kapitel 4.1 beschriebenen Stufenschemas der Schmerztherapie eingeteilt (vgl. Mutschler et.al. 2013, S. 226). „Ärztliche Behandlung, Verschreibung und Abgabe § 8. Suchtmittelhaltige Arzneimittel dürfen nur nach den Erkenntnissen und Erfahrungen der medizinischen, zahnmedizinischen oder veterinärmedizinischen Wissenschaft, insbesondere auch für Schmerz- sowie für Entzugs- und Substitutionsbehand- 26 lungen, verschrieben, abgegeben oder im Rahmen einer ärztlichen, zahnärztlichen oder tierärztlichen Behandlung am oder im menschlichen oder tierischen Körper unmittelbar zur Anwendung gebracht werden.“ (Bundeskanzleramt: SuchtmittelgesetzSMG, 2016, § 8) 3.2.3 Schwach wirksame Opioide Tramadol (bspw. Medikamentennamen Adamon®, Lanalget®, Tradolan®) ist ein schwaches Opioid, das seine Wirkung über die µ-Opioidrezeptoren entfaltet und gleichzeitig auch einen Einfluss auf den Serotonin- und Noradrenalin Haushalt nimmt, da es zu einer verminderten Rückaufnahme von freigesetzten Molekülen dieser Stoffe führt. Daher ist Tramadol bei Epilepsie kontraindiziert, da es das Risiko von Krampfanfällen erhöht, insbesondere in der Kombination mit anderen Medikamenten, die ähnlich in den Hirnstoffwechsel eingreifen. Insgesamt verteilt sich Tramadol im Körper relativ gut, es kann auch einen Übertritt in das zentrale Nervensystem oder die Plazenta bewerkstelligen und braucht in etwa sechs Stunden, um wieder aus dem Körper entfernt zu werden. Je nach Veranlagung können bei der Verabreichung von Tramadol eher die unerwünschten Wirkungen überwiegen oder die Schmerzlinderung ungenügend sein, da die Stoffwechselprozesse zur Verarbeitung dieses Wirkstoffes aufgrund von genetischen Unterschieden anscheinend bei verschiedenen Menschen mit unterschiedlichen Geschwindigkeiten ablaufen können. Dies beruht darauf, dass die aktive, analgetisch wirksame Form von Tramadol erst durch chemische Umwandlungsprozesse im Körper entsteht. Zusätzlich ist auch zu beachten, dass Tramadol bei schweren Schmerzzuständen keine ausreichende Wirkung entfaltet und bei Langzeitgabe immer in Darreichungsformen mit verzögerter Wirkstofffreisetzung verabreicht werden soll (vgl. Cascorbi 2013, S. 27-29). Tilidin als nächste Substanz der WHO-Gruppe zwei ist pharmakologisch nahezu inaktiv und wird erst durch bestimmte Stoffwechselprozesse im Organismus zum aktiven Wirkstoff umgewandelt. Diese Stoffwechselprozesse finden in der Leber statt. In der Anwendung ist Tilidin hauptsächlich in Kombination mit dem Opioidantagonisten Naloxon erhältlich, diese Kombination wurde entwickelt, um den Missbrauch dieser Substanz für Suchtzwecke einzudämmen. Aus diesem Grund ist eine Anwendung von Tilidin/Naloxon Kombinationspräparaten bei suchtkranken Patienten kontraindiziert, da 27 bei Gabe dieser Substanz starke Entzugs-Symptome ausgelöst werden können. Darüber hinaus macht eine Gabe bei Leberfunktionsstörungen wenig Sinn. In Österreich ist Tilidin nicht erhältlich (vgl. Cascorbi 2013, S. 27-29). Codein (bspw. Medikamentenname Codipertussin®) und das Dihydrocodein (bspw. Medikamentenname Codidol®) mit einer dreifach stärkeren Wirkung, gehören ebenfalls zu den schwachen Opioiden der WHO-Stufe zwei. Meistens wird Codein als Kombinationspräparat mit anderen Medikamenten, speziell auch mit nicht-opioidartigen Schmerzmitteln, angewandt. Allerdings hat Codein aufgrund seiner unberechenbaren Wirkungsentfaltung, die wegen genetischer Prädispositionen von Patient zu Patient sehr stark variieren kann, zunehmend an Bedeutung in der Anwendung als Schmerzmedikament verloren (vgl. Mutschler et.al. 2013, S. 225). 3.2.4 Stark wirksame Opioide Morphin (bspw. Medikamentenname Vendal®), der Opioid Prototyp mit der mengenmäßig höchsten Datenlage bezüglich Anwendungserfahrung und Wirkungsweise, ist das Paradebeispiel für die zur Behandlung von sehr starken Schmerzzuständen indizierten stark wirksamen Opioide. Morphin ist für die verschiedensten Darreichungsformen erhältlich und kann in der Menge und der Anwendungsart an den jeweiligen Patienten beziehungsweise die jeweilige Situation angepasst werden (vgl. Cascorbi 2013, S. 36). Es entfaltet eine andauernde Wirkung von ungefähr vier Stunden, diese kann bei Darreichungsformen mit verzögerter Wirkstofffreisetzung auf zwölf bis sogar vierundzwanzig Stunden verlängert werden. Bei vorsichtiger Anwendung kann es auch bei Leberfunktionsstörungen verabreicht werden und es eignet sich gut zur kombinierten Gabe mit anderen nicht-opioidartigen Schmerzmitteln (vgl. Beubler 2012, S. 8283). Zu beachten ist, dass Morphin nach oraler Aufnahme relativ niedrige Blutspiegel aufweist, da dieser Stoff bei der ersten Leberpassage stark dezimiert wird (First-PassEffekt). Daher wird bei der Verabreichung in Tablettenform eine dreifach höhere Dosis benötigt als bei intravenöser Gabe von Morphin (vgl. Mutschler et.al. 2013, S. 222). Hydromorphon (bspw. Medikamentenname Hydal®) hat eine in etwa sieben Mal höhere Wirkung gegen Schmerzen als Morphin, es ist aber ansonsten ähnlich dem Morphin, allerdings bei Patienten mit eingeschränkter Nierenfunktion empfehlenswerter und besonders geeignet für eine subkutane Verabreichung. Es wird nach nur wenigen 28 Stunden wieder zur Hälfte aus dem Körper ausgeschieden und eignet sich nicht für die Therapie von chronischen Schmerzzuständen (vgl. Cascorbi 2013, S. 43). Oxycodon (bspw. Medikamentenname Oxycodon Accord®) ist ein stark wirksames Opioid, das ein wenig stärker schmerzreduzierend wirkt als Morphin und hauptsächlich in Form von Tabletten mit verzögerter Wirkstofffreisetzung angewendet wird (Retard), es besitzt keinen hohen First-Pass Metabolismus und ist auch in Kombination mit Naloxon, dem typischen Opioid-Antagonisten, erhältlich. Die antagonisierende Wirkung des Naloxons wird dabei gezielt ausgenutzt, um bei oraler Gabe die typische OpioidNebenwirkung der Obstipation im Darm zu verringern. (vgl. Mutschler et.al. 2013, S. 225). Buprenorphin (bspw. Medikamentennamen Astec®, TTS: Bupacal®, Bupensan®) ist das typische Beispiel für einen partiellen Opioidrezeptor-Agonisten. Gleichzeitig handelt es sich bei dieser Substanz um ein stark wirksames Opioid mit einem Schutzmechanismus gegen die gefürchtete Opioid-Nebenwirkung der Atemdepression. Für die Langzeittherapie bei chronischen Schmerzzuständen gibt es auch die Darreichungsform von Buprenorphin als sogenanntes transdermal therapeutisches System (TTS) oder umgangssprachlich als Schmerzpflaster bezeichnet. Buprenorphin ist daher ein in der Anwendung besonders sicheres Opioid, das allerdings aufgrund der hohen Substanzverluste nach der ersten Leberpassage bei oraler Gabe unbrauchbar ist, jedoch bei anderen Darreichungsformen immerhin eine Wirkung von bis zu acht Stunden erreicht (vgl. Cascorbi 2013, S. 56-57). Fentanyl (bspw. Medikamentennamen Breakyl®, Effentora®, TTS: Durogesic®) ist ein sehr stark wirksames Opioid, das intravenös verabreicht sehr gut steuerbar ist und rasch seine Wirkung im Organismus entfaltet, daher ist es prädestiniert für die Anwendung bei akuten Schmerzen und im operativen Bereich. Es existieren allerdings auch vielfältige andere Darreichungsformen für die Langzeitanwendung von Fentanyl, insbesondere TTS, also Schmerzpflaster. Die Gefahr einer Atemdepression ist bei einer Anwendung von Fentanyl stets im Auge zu behalten, dies trifft in besonders hohem Maß auf die Anwendung in der Anästhesie zu (vgl. Beubler 2012, S. 75-76). Die Wirkstärke von Fentanyl in Bezug auf die Schmerzreduktion ist immerhin hundertmal so hoch wie die von Morphin, daraus lässt sich die Gefahr einer Überdosierung 29 ableiten. Diese ist bei der Langzeitanwendung in Form von TTS oder oralen Darreichungsformen allerdings nur bei geänderten äußeren Umständen wie Fieber oder vermehrter Hautdurchblutung (Sauna) oder mehrmaliger Gabe hintereinander relevant. Ansonsten bleibt diese Substanz im Blut relativ lange in gleicher Konzentration bestehen und wird nach Beendigung der Zufuhr nach ungefähr siebzehn Stunden zur Hälfte aus dem Organismus ausgeschieden (vgl. Cascorbi 2013, S. 51-52). Piritramid (bspw. Medikamentenname Dipidolor®), Remifentanil (bspw. Medikamentenname Ultiva®) und Sufentanil (bspw. Medikamentenname Sufenta®) wären an dieser Stelle noch als Beispiele für starke wirksame Opioide zu nennen, welche im Rahmen von operativen Eingriffen zur intravenösen Anwendung kommen. Piritramid wirkt um ein Vielfaches (siebzig Prozent) stärker als Morphin über einen Zeitraum von circa sechs Stunden und wird gerne nach Operationen eingesetzt. Remifentanil fällt durch seine zweihundertfach höhere Fähigkeit zur Schmerzreduktion in der Gegenüberstellung mit Morphin und seinen extrem schnellen Wirkungs-Eintritt beziehungsweise Wirkungs-Austritt auf. Aus diesem Grund kann Remifentanil niemals zur langfristigen Schmerztherapie eingesetzt werden, da seine potente Wirkung viel zu schnell wieder nachlässt, es ist aber während Operationen das Opioid, welches am besten anästhesiologisch zu steuern ist. Sufentanil findet hier Erwähnung, da es die höchste Potenz aller Opioide hat, Schmerzen zu lindern, diese ist bis zu tausend Mal höher als die Schmerzreduktion von Morphin. Es wird ebenfalls relativ schnell im Stoffwechsel verwertet (vgl. Mutschler et.al. 2013, S. 225-226). Eine Übersicht über die grundlegenden in der medikamentösen Schmerztherapie angewendeten Opioide und deren beispielhafte Medikamentennamen veranschaulicht die Tabelle auf der nachfolgenden Seite 31. Die erste Substanz auf der Liste, Tilidin, ist in Österreich nicht erhältlich, wird aber in Deutschland angewendet. Das Kombinationspräparat von Tilidin mit Naloxon heißt in Deutschland beispielsweise Valoron® und enthält acht Prozent Naloxon. Es kann ohne gesetzliche Einschränkungen durch das Betäubungsmittelgesetz verschrieben werden und ist die am stärksten wirksamste Substanz der Opioidanalgetika, die in Deutschland noch unreglementiert abgegeben werden darf. 30 Alle anderen unter Ausnahme von Tilidin in der folgenden Tabelle angeführten Analgetika, sind auch in Österreich erhältlich und fallen unter das Suchtmittelgesetzt: Abbildung 3: Grundlagen medikamentöser Schmerztherapie – Opioide (Quelle: Seifert, 2016) 31 3.3 Unerwünschte Opioid-Wirkungen und Gefahren von Überdosierungen Die typischen, auch unerwünschten, zentralen und peripheren Wirkungen der Opioidanalgetika wurden bereits im Überblick in Kapitel 3.2 dieser Arbeit dargestellt. In der therapeutischen Anwendung stellen vor allem die Übelkeit mit Brechreiz und die Verstopfung die größte Herausforderung für Patienten und Therapeuten dar. Aufgrund der Toleranzentwicklung des Brechzentrums im Gehirn stellt sich die Problematik der Übelkeit nur zu Beginn einer Opioidtherapie dar. Ein Medikament gegen Erbrechen sollte vorbeugend in den ersten zwei Wochen zusätzlich eingenommen werden. Die Verstopfung (Obstipation) zählt zu den mengenmäßig dominantesten Nebenwirkungen mit hohen Anforderungen an die Therapie (vgl. Cascorbi 2013, S. 39). „Die Obstipation unterliegt keiner Toleranzentwicklung und muss deshalb während der gesamten Opioidtherapie konsequent mit Laxanzien (Lactulose) behandelt werden.“ (Cascorbi 2013, S. 39) Laut Cascorbi konnte in klinischen Studien keine Unterscheidung bezüglich der Wirkstärke von WHO-zwei- und WHO-drei-Stufen-Opioiden dargelegt werden, lediglich bezüglich des Auftretens von Nebenwirkungen konnte eine unterscheidende Gegenüberstellung dieser beiden Substanzklassen aufgezeigt werden. In diesem Zusammenhang treten Übelkeit und Erbrechen, Verstopfung, Juckreiz und Dämpfung des zentralen Nervensystems mit erhöhtem Schlafbedürfnis bei den WHO-Medikamenten der Gruppe zwei seltener auf, als bei den Medikamenten der dritten Gruppe, wobei die restlichen, typischen Opioide-Nebenwirkungen keine Zuordnung zu einer der beiden Gruppen aufweisen (vgl. Cascorbi 2013, S. 26). Bei der Anwendung von Opioid-Analgetika kommt es außerdem rasch zu einer erhöhten Empfindlichkeit der Rezeptoren gegenüber Schmerzreizen, dies muss bei einer Schmerztherapie mit Opioiden berücksichtigt werden. Bestenfalls wird dem Mechanismus entgegengewirkt, das kann in Form von gleichzeitiger Gabe von nicht-opioidartigen Schmerzmedikamenten oder anderen Medikamenten erfolgen. Der Vorgang wird wie folgt bezeichnet (vgl. Cascorbi 2013, S. 25): „Opioid-induzierte Hyperalgesie Opioide können auch zu einer Schmerzverstärkung führen, welche auf einer Aktivierung pronozizeptiver Systeme beruht.“ (Cascorbi 2013, S. 25) 32 Die Kombination mehrerer Medikamente, die eine Auswirkung auf den Serotoninspiegel im Gehirn haben, kann in Kombination mit Opioiden ebenfalls problematisch werden und zu dem sogenannten Serotoninsyndrom führen. Dieses Syndrom äußert sich durch eine Reihe verschiedener Symptome, wie etwa Krampfanfällen oder Schüttelfrost, die alle mit einem erhöhten Serotoninspiegel einhergehen (vgl. Beubler 2012, S. 62). Medikamente, die sich hemmend auf die Freisetzung des Neurotransmitters Acetylcholin auswirken, sind in der Lage bei gleichzeitiger Anwendung mit Opioiden, auch deren anticholinerge Nebenwirkungen zu verstärken. Zu dieser Gruppe von pharmakologischen Substanzen zählen sowohl bestimmte Antidepressiva, als auch andere Arzneistoffe aus dem psychiatrischen Bereich oder Medikamente, die bei einer Parkinson-Erkrankung zum Einsatz kommen. Darüber hinaus kommt es bei Opioidgabe in Kombination mit Substanzen aus der Gruppe der sogenannten MAO-Hemmer zu einer Verstärkung der Neurotoxizität dieser Medikamente. Insbesondere in Zusammenhang mit dem Wirkstoff Pethidin konnten gravierende Komplikationen beobachtet werden (vgl. Cascorbi 2013, S. 41): „Bei Vorbehandlung von Patienten mit bestimmten Antidepressiva (MAO-Hemmstoffen) innerhalb der letzten 14 Tage vor der Opioidanwendung sind lebensbedrohende Wechselwirkungen auf Zentralnervensystem, Atmungs- und Kreislauffunktion mit Pethidin beobachtet worden.“ (Cascorbi 2013, S. 41) Trotz der relativ vielschichtigen, typischen Nebenwirkungen von Opioiden muss dennoch darauf hingewiesen werden, dass sie als Medikamentengruppe ein äußerst geringes schädigendes Potential für den menschlichen Organismus aufweisen (vgl. Diener et al. 2008, S. 369). „Es gibt bis heute keinen Anhalt für eine Organtoxizität von Opioiden. Dies ist sicher der bedeutendste Vorteil der Opioide.“ (Diener et al. 2008, S. 369) Die gefürchtete Nebenwirkung der Atemdepression ist bei der Schmerztherapie mit Opioidanalgetika, wenn diese fachgerecht angewendet werden, relativ gering. Zu beachten ist dabei die langsame Erhöhung der Menge an Opioiden, welche verabreicht wird. Das vorsichtige Herantasten an die richtige Dosis für den individuellen Patienten, 33 besonders bei solchen Patienten, die noch nicht in Kontakt mit Opioiden gekommen sind, ist dabei besonders wichtig. Zu beachten ist auch, dass das Schmerzerleben in Wechselwirkung mit der Atmungsblockade steht, was bedeutet, dass eine Atemdepression leichter entsteht, wenn der Patient von seinen Schmerzen befreit wurde, oder anders ausgedrückt, dass der Schmerzreiz in gewisser Weise einen Schutz vor einer Atemdepression bietet. Daher sollte ab dem Moment der Beseitigung der Schmerzursache eine Opioidgabe nicht mehr fortgesetzt werden. Gefährlich in Bezug auf das Auftreten einer Atemdepression ist immer ein schnelles Übertreten einer hohen Opioid-Dosis in das Gehirn. Notfallmedikament der Wahl ist der am Ende von Kapitel 3.2.1 beschriebene Opioid-Antagonist Naloxon (vgl. Beubler 2012, S. 59-60). Bei gut eingestellter Unterdrückung der Schmerzempfindung durch Opioide sind Nebenwirkungen im Zentralnervensystem geringer, starke Komplikationen bei der Anwendung lassen auf eine geringe Opioid-Sensitivität im Gehirn gegenüber der angewendeten Substanz schließen und haben nichts mit der Dosierung zu tun. Die aufgrund der peripheren Wirkungen von Opioid-Analgetika ausgelösten Nebenwirkungen hingegen stehen in Zusammenhang mit der Dosierung und der verabreichten Substanz. Diese können durch einen Medikamentenwechsel (Opioidrotation) oder ein Herabsetzen der verabreichten Dosis verbessert werden (vgl. Diener et al. 2008, S. 355). 34 4 Spezialanforderungen in der pharmakologischen Schmerztherapie Grundsätzlich steht für die pharmakologische Schmerztherapie von verschiedensten, leichten Schmerzzuständen, die nicht über einen längeren Zeitraum andauern, die breite Palette der gesamten Nicht-Opioidanalgetika zur Verfügung. Bei der Anwendung dieser Medikamente ist das Vermeiden von sogenannten Kombinationspräparaten zu beachten, da diese bei häufiger Anwendung trotz nicht existentem therapeutischen Zusatznutzen mit dem Auftreten schwerer Nierenschäden korrelieren. Leider werden sie dennoch häufig angewendet und sind rezeptfrei erhältlich, was fälschlicher Weise zu ihrer Verharmlosung beiträgt. Eine dauerhafte Anwendung von NSAIDs stellt einen fragwürdigen Aspekt dar, denn sie hat schädigende Auswirkungen auf die verschiedenen Organsysteme zur Folge, dabei sind neben möglichen Leber- und Nierenschäden vor Allem Probleme im Magen-Darm-Bereich häufig (vgl. Höllt, Allgaier 2013, S. 229). Opioid-Analgetika stehen für die Behandlung von starken Schmerzzuständen, wie etwa bei einem Herzinfarkt oder nach operativen Eingriffen, aber auch bei traumatisch bedingten Schmerzen zur Verfügung. Darüber hinaus finden sie Einsatz in der Onkologie und bei chronischen Schmerzzuständen, die nicht auf eine Tumorerkrankung zurückzuführen sind. Eine Eignung von Opioid-Analgetika zur Behandlung von neuropathischen Schmerzen konnte ebenfalls klinisch nachgewiesen werden. Eine Anwendung von Opioid -Analgetika bei chronischen Schmerzpatienten ist in Fachkreisen weiterhin ein strittiger Punkt (vgl. Diener et al. 2008, S. 356). „Eine international anerkannte Definition persistierender oder chronischer Schmerzen besteht nicht. Wenn Schmerzen länger als ein halbes Jahr bestehen oder einen Monat länger als der zu erwartende Genesungszeitraum, wird im Allgemeinen von einem chronischen Schmerzzustand gesprochen.“ (Nikolaus 2013, S. 209) Opioide sollten zur Langzeittherapie nur dann in Frage kommen, wenn der Grad der Schmerzen mit anderen Medikamenten nicht unter Kontrolle zu bringen ist, die betreffenden Schmerzen grundsätzlich opioidsensitiv sind und sowohl die Nebenwirkungen, als auch die Gefahr einer Zunahme des chronischen Zustandes akzeptabel sind (vgl. Diener et al. 2008, S. 356). 35 „Nach der S3-Leitlinie zur Langzeitanwendung von Opioiden bei nichttumorbedingten Schmerzen LONTS (Reineke u. Sorgatz 2009) sollte eine Opioidanwendung nicht zwangsläufig lebenslang durchgeführt werden. Der Versuch einer Dosisverringerung bzw. ein Auslassversuch sollte nach 3–6 Monaten eingeplant werden, insbesondere dann, wenn eine Verbesserung der Grundkrankheit oder die positive Wirkung anderer Behandlungen es sinnvoll erscheinen lassen. Diese Empfehlungen sind sicherlich auch auf viele andere Substanzgruppen, die in der Schmerztherapie verwendet werden, zu übertragen.“ (Strumpf 2013, S.114) Die Auswahl des richtigen Medikamentes für die Schmerztherapie ist immer eine Individualentscheidung, bei der sämtliche Gegenanzeigen in Bezug auf die Anwendung bei dem jeweiligen Patienten genau durchdacht werden müssen und auch psychologische Aspekte beachtet werden sollten. Die Bedeutsamkeit von Analgetika in der Schmerztherapie ist hoch, sollte aber nicht überbewertet werden (vgl. Strumpf 2013, S. 107). Das Vorgehen bei jeder qualifizierten Schmerztherapie folgt gewissen Grundprinzipien, die sich folgendermaßen darstellen: „Ein unzureichend wirksames Analgetikum ist, falls möglich, höher zu dosieren oder durch ein potenteres zu ersetzen.“ (Diener et al. 2008, S. 338) Wenn ein eingenommenes Schmerzmittel keine zufriedenstellende Wirkung erzielt, ist es nicht weiter anzuwenden, die Wirkung des eingenommenen Schmerzmittels ist mittels geeigneter Messinstrumente (unterschiedliche Schmerzskalen) nach Angabe des Patienten zu ermitteln, schriftlich festzuhalten und in die Entscheidung über Dosierung und Auswahl des Medikamentes miteinzubeziehen (vgl. Diener et al. 2008, S. 339). „Ohne Prüfung der Wirksamkeit ist die Verabreichung von Analgetika ein Behandlungsfehler.“ (Diener et al. 2008, S. 339) Gerade bei chronifizierten Schmerzzuständen oder in der Tumor-Schmerztherapie kann auch eine prophylaktische Schmerzmittel-Gabe zum Einsatz kommen, um Schmerzspitzen zu vermeiden, außerdem sind Analgetika mit verzögerter Wirkstofffreisetzung oder langwirksamer Biokinetik zu präferieren. Eine Schmerzlinderung sollte jeweils immer mit der niedrigsten möglichen Dosierung herbeigeführt werden 36 und bei gleichwertiger Schmerzreduktion ist dem Mittel mit dem geringeren Nebenwirkungsprofil bzw. Schadenspotential der Vorzug zu geben. Die auftretenden unerwünschten Wirkungen während einer Schmerztherapie sind stets schriftlich festzuhalten und therapeutisch zu berücksichtigen. Der Therapeut muss sich aktiv beim Patienten nach Nebenwirkungen erkundigen. Eine qualifizierte Überwachung des Therapieverlaufs ist dringend nötig, genau wie die Begründung für ein Fortbestehen der Therapie in regelmäßigen Abständen zu hinterfragen ist (vgl. Diener et al. 2008, S. 339-341) „Bei der medikamentösen Therapie chronischer Dauerschmerzen werden Analgetika und Nichtanalgetika nach einem festen Zeitschema eingesetzt, das sich an der pharmakologischen Wirkungsdauer des verwendeten Präparates orientiert.“ (Strumpf 2013, S.109) Der Behandlungsplan orientiert sich in den meisten Fällen an dem im folgenden Kapitel beschriebenen WHO-Stufenschema (vgl. Höllt, Allgaier 2013, S. 229). 4.1 WHO-Stufenschema Das Stufenschema der WHO (World Health Organisation) geht bereits auf das Jahr 1986 zurück und stellt einen Leitfaden für die Vorgehensweise bei der Behandlung von chronisch persistierenden Schmerzen dar, es ist in Abbildung 4 auf Seite 37 visuell dargestellt (vgl. Höllt, Allgaier 2013, S. 229). Grundsätzlich wurde dieses Stufenschema zu Behandlung von tumorbedingten Schmerzen entwickelt und ist mittlerweile weitläufig als Behandlungsschema für chronische Schmerzen anerkannt. Wenn eine Schmerzlinderung auf einer jeweiligen Stufe nicht mehr ausreichend ist, dann wird jeweils die nächst höhere Stufe als Behandlungsoption gewählt. Zusätzlich besteht auch die Möglichkeit, eine niedrige Stufe auszulassen, wenn die Schmerzintensität zu hoch eingeschätzt wird um damit einen Therapieerfolg zu erzielen (vgl. Zeilhofer, Sittl 2010, S. 76-77). Stufe zwei und drei kann mit Medikamenten unterstützt werden, die die schmerzlindernde Wirkung des ausgewählten Schmerzmittels unterstützen (sogenannte Koanalgetika oder Adjuvantien). Dabei handelt es sich beispielsweise um bestimmte Antidepressiva oder Kortikosteroide (Vgl. Beubler 2012, S.10). 37 Abbildung 4: WHO-Stufenschema zur Pharmakotherapie von Tumorschmerzen (Quelle: Stackelberg, 2016) „Dem WHO-Stufenplan ist zwar zu verdanken, dass sich die Schmerztherapie und vor allem die Verschreibung von Opioiden weltweit gebessert hat, seine kritiklose Anwendung trägt aber in den seltensten Fällen den Bedürfnissen der Schmerzpatienten Rechnung.“ (Beubler 2012, S. 48) In vielen Fällen, gerade im klinischen Bereich, ist die erste Stufe von Beginn an schon zu niedrig um Schmerzfreiheit zu erreichen, dies betrifft besonders bei Schmerzen nach operativen Eingriffen oder traumatischen Ereignissen zu. Hier macht eine Anwendung des Stufenplans der WHO in umgekehrter Reihenfolge eher Sinn. Das gleiche gilt für Patienten mit chronischen Schmerzzuständen, die bereits bei der ersten Vorstellung so große Schmerzen haben, dass eine Anwendung der Stufe-eins-Medikamente keine Erleichterung verschaffen würde. Oft würde ein Therapieversuch mit zu schwachen Medikamenten mehr körperlichen Schaden als Nutzen anrichten, dies betrifft im Besonderen die Nicht-Opioidanalgetika aufgrund ihres starken Nebenwirkungsprofils (vgl. Beubler 2012, S. 48). 38 4.2 Bezugnahme auf spezielle Bedürfnisse bestimmter Patientengruppen Kinder haben mindestens das gleiche Schmerzerleben wie Erwachsene und sind dennoch in Hinsicht auf eine adäquate Schmerztherapie untertherapiert (vgl. Greffe et al. 2014, S. 1022). Beubler spricht von Kindern als einer besonders benachteiligten Patientengruppe in Hinblick auf die Therapie mit Analgetika (vgl. Beubler 2012, S. 110). „Auch die Annahme, dass Kinder Schmerzen schneller vergessen als Erwachsene ist falsch. Kinder, ja selbst Früh und Neugeborene, zeigen Reaktionen der Angst und der Abwehr, wenn ärztliche und pflegerische Maßnahmen vorgenommen werden, die zuvor häufiger mit Schmerzreizen verbunden waren. Kinder haben genauso ein Recht auf eine effiziente Schmerztherapie wie Erwachsene und die Vorstellung, dass Analgetika hinsichtlich ihrer pharmakokinetischen und pharmakodynamischen Wirkung beim Kind anders zu bewerten sind, muss korrigiert werden.“ (Beubler 2012, S.110) Ebenso verhält es sich mit der Schmerztherapie von alten Menschen. Aus wissenschaftlich erhobenen Daten geht hervor, dass bis an die Hälfte der Menschen, die ein Lebensalter jenseits von 65 Jahren erreicht haben, an beeinträchtigenden Schmerzen leiden. Dies impliziert immer eine gewisse Verleitung, diese Problematiken aufgrund des häufigen Vorkommens zu verharmlosen (vgl. Beubler 2012, S. 110) 4.2.1 Besonderheiten bei der Schmerztherapie von Patientinnen und Patienten aus dem Fachbereich der Geriatrie Laut Schäfers sind immerhin 25% der über 65-jährigen von chronischen Schmerzleiden betroffen, dennoch gibt es wenig klinische Studien zu dieser Thematik und geriatrische Patientinnen und Patienten sind hinsichtlich einer adäquaten Schmerztherapie untertherapiert. Erschwerend für eine effiziente Therapie gegen Schmerzen kommt die im Alter häufig auftretende Multimorbidität hinzu (vgl. Schäfers 2008, S. 314). Wehling spricht sogar von einem generellen Auftreten von Schmerzen bei über 70-jährigen von bis zu 70% (vgl. Wehling 2013, S. 191). Im Zusammenhang mit Analgetika im Alter ist besonders die Veränderung auf organischer Ebene bezüglich der Ausscheidungsmechanismen von Leber und Niere zu beachten. Diese wirken sich dahingehend aus, dass höhere Medikamenten-Plasmaspiegel über einen längeren Zeitraum bestehen bleiben. Zusätzlich besteht häufig auch 39 eine verminderte Flüssigkeitsaufnahme und meist die Einnahme von mehreren Medikamenten zur gleichen Zeit (vgl. Schäfers 2008, S. 316). „Generell sind bei älteren Menschen kleinere Anfangsdosierungen zu wählen, die Dosisanpassung soll langsamer erfolgen als bei jungen Menschen (start low and go slow!). Beim Einsatz von Retardformen kann das Dosierungsintervall nach Bedarf verlängert werden.“ (Beubler 2012, S. 121) Bei der Messung von Schmerzen im geriatrischen Schmerzassessment sind die Schmerzmessinstrumente an den kognitiven und körperlichen Zustand des jeweiligen Menschen anzupassen, wenn nötig muss auch die Körpersprache, das Verhalten und die Mimik gedeutet werden (vgl. Beubler 2012, S. 120). Im deutschsprachigen Raum stehen beispielsweise Messinstrumente für die Fremdbeurteilung von Menschen mit Demenz zur Verfügung, wie die BESD-Skala (Beurteilung des Schmerzes bei Demenz) oder die BISAD-Skala (Beobachtungsinstrument für das Schmerzassessment bei alten Menschen mit Demenz) (vgl. Lukas, Drebenstedt 2014, S. 420). In Hinblick auf die Beurteilung des Gefahrenpotentials von Medikamentengruppen, welche im geriatrischen Fachbereich zur Schmerztherapie eingesetzt werden, ist wohl gerade der Einsatz der häufig verkauften NSAIDs sehr kritisch zu bewerten. Dies betrifft vor Allem die Anwendung dieser Medikamente über einen längeren Zeitraum. Ältere Menschen haben ein erhöhtes Risiko für das Auftreten der typischen Nebenwirkungen dieser Substanzgruppe, die besonders das Verdauungssystem und die Funktion der Nieren betreffen, außerdem fördern NSAIDs bei alten Menschen die Entstehung von Bewusstseinsstörungen (Delir) und anderen Beschwerden des Gehirnstoffwechsels (vgl. Wehling 2013, S. 196-197). „Aufgrund ihrer fehlenden Organtoxizität können passende Opioide für ältere Menschen häufig deutlich günstiger sein.“ (Lukas, Drebenstedt 2014, S. 421) Wenn Opioide nicht intravenös verabreicht werden, beinhalten sie sehr niedriges Gefahrenpotential zum Missbrauch, dies ist ein weiterer Grund, welcher der relativ zurückhaltenden Anwendung dieser Substanzgruppe bei älteren Menschen entgegen zu halten ist (vgl. Nikolaus 2013, S. 213). Problematisch kann bei Opioidanwendungen in der Geriatrie zu geringes Trinken in Kombination mit verminderter Funktion der Nieren 40 werden, dies betrifft im Besonderen die Substanz Morphin, da sich bestimmte Stoffwechselprodukte dieser Substanz im Gehirn anreichern können. Laut Beubler sind Hydromorphon und Oxycodon besser geeignet und diesbezüglich unbedenklich. Zu Beginn der Therapie tritt die Nebenwirkung der Sedierung bei der Opioid-Therapie von alten Menschen häufiger auf als sonst, diese lässt aber in kurzer Zeit wieder nach und sollte keinen Grund für einen Therapieabbruch darstellen (vgl. Beubler 2012, S. 122123). Multimorbidität und Polypharmazie sind zwei typische Faktoren des Alters, die miteinander in Verbindung stehen, und der Grund dafür sind, dass sogenannte unerwünschte Arzneimittelwirkungen (UAW) oder Nebenwirkungen auf der ganzen Welt im Alter weit häufiger vorkommen, als in jungen Jahren (vgl. Mühlberger, Sieber 2014, S. 245). Die Amerikanische Gesellschaft für Geriatrie (American Geriatric Society, AGS) hat bereits in den neunziger Jahren eine Liste mit Medikamenten zusammengestellt, die eine mögliche Gefährdung für Patientinnen und Patienten aus dem Fachbereich der Geriatrie darstellen. Dieser im Jahre 2012 letztmals aktualisierten und von der AGS veröffentlichten, sogenannten Beers-Liste, entspricht das deutschsprachige Äquivalent der sogenannten PRISCUS-Liste (www.priscus.net). Folgende Analgetika scheinen auf diesen Listen auf (vgl. Mühlberger, Sieber 2014, S. 251): Bestimmte NSAIDs und selektive COX-2-Inhibitoren (Indometacin, Acemetacin, Ketoprofen, Piroxicam, Meloxicam, Phenylbutazon, Etoricoxib) sind wegen Flüssigkeitsretention, Verschlimmerung einer Herzinsuffizienz und der Gefahr von Geschwüren im Magen-Darm-Trakt zu vermeiden. Die PRISCUS-Liste nennt Paracetamol und schwache Opioide wie Tramadol als Alternative (vgl. Mühlberger, Sieber 2014, S. 257). Unter den Opioiden ist lediglich das Pethidin auf der Negativliste anzutreffen, da die Verabreichung eine erhöhte Gefahr sowohl für Stürze aufgrund von orthostatischen Beschwerden, als auch für das Auftreten eines Delirs mit Nebenwirkungen des Zentralnervensystems und kognitiver Leistungsabnahme beinhaltet. Laut PRISCUS-Liste sind Paracetamol und Opioide mit geringerem Risiko für die Delir-Entstehung (Morphin, Tilidin/Naloxon, Oxycodon, Buprenorphin, Hydromorphon) die passende Alternativtherapie (vgl. Mühlberger, Sieber 2014, S. 254). 41 4.2.2 Besonderheiten bei der Schmerztherapie von Patientinnen und Patienten aus dem Fachbereich Pädiatrie Leider wurde in der Medizin über einen langen Zeitraum davon ausgegangen, dass Kinder, besonders Säuglinge, ein geringeres Schmerzempfinden als Erwachsene haben würden, was zu einer inadäquaten Schmerztherapie mit zahlreichen Leidtragenden führte (vgl. Beubler 2012, S. 110). In der Tat ist die Erfassung des Schmerzes bei sehr jungen Kindern schwierig, daher müssen geeignete Messinstrumente für die Schmerzintensität zum Einsatz kommen. Frühgeborene, die knapp an der Überlebensgrenze (24. Schwangerschaftswoche) zur Welt kommen, empfinden bereits Schmerzen, wobei eine Sensibilisierung auf Schmerzreize im zentralen Nervensystem im Erwachsenenalter nachweisbar ist. Es ist sogar anzunehmen, dass derart früh geborene Kinder ein erhöhtes Schmerzempfinden aufweisen. Der sogenannte Sedierungsbogen nach Hartwig kann zur Schmerzbeurteilung bei beatmeten früh- und neugeborenen Kindern herangezogen werden. Die kindliche Unbehagens- und Schmerzskala (KUSS) kann hervorragend für die Schmerzmessung bis zum vierten Lebensjahr verwendet werden. Ab dem fünften Lebensjahr können dann bereits Gesichterskalen zur Selbsteinschätzung der Schmerzsituation beim Kind zum Einsatz kommen, wobei bei Kindern mit besonderen Bedürfnissen individuelle Konzepte von den betreuenden Personen zu erarbeiten sind (vgl. Zernikow 2008, S. 286). Eine besondere Herausforderung stellt die Schmerztherapie bei Früh- und Neugeborenen in jedem Fall für das behandelnde Team von Fachkräften dar, sie basiert auf Entscheidungen, die bei jeder einzelnen Behandlung stets wieder individuell auf das betreffende Kind bezogen, getroffen werden müssen (vgl. Zernikow 2008, S. 288): Für die Abschätzung der richtigen Entscheidungen ist ein großer Erfahrungsschatz nötig. Die Qualitätssicherungsgruppe der Gesellschaft für Pädiatrische Onkologie und Hämatologie arbeitet beispielsweise daran, geeignete und gültige Messinstrumente für Schmerzen festzulegen, um krebskranken Kindern die bestmögliche Schmerztherapie zukommen zu lassen (vgl. Beubler 2012, S. 110). Paracetamol wird für Kinder in der Fachliteratur wiederholt als sicherstes Analgetikum postuliert, dies betrifft besonders die Neugeborenenperiode, da die für die Leber so 42 giftigen Stoffwechselprodukte in diesem Lebensalter noch nicht gebildet werden können. Bei älteren Kindern ist stets auf die genaue, dem Gewicht des Kindes entsprechende, Dosierung zu achten. Der Wirkstoff Metamizol ist aufgrund der potenziellen, schweren Nebenwirkungen der Agranulozytose und des Kreislaufschocks bei Kindern nur sehr kritisch zu betrachten, Beubler empfiehlt Metamizol nur in Sonderfällen und dann gemeinsam mit Paracetamol (vgl. Beubler 2012, S.112). Acetylsalicylsäure kann bei Kindern, die noch nicht in der Pubertät angelangt sind, in Kombination mit einer viralen Infektion, die schwere Komplikation des sogenannten Reye-Syndroms auslösen. Diese Nebenwirkung ist zwar selten, beeinträchtigt aber mit einem tödlichen Ausgang in fünfundzwanzig Prozent aller Fälle in äußerst gravierender Form Gehirn und Leber. Daher darf Acetylsalicylsäure bei Viruserkrankungen von Kindern unterhalb der Pubertätsgrenze niemals zum Einsatz kommen (vgl. Höllt, Allgaier 2013, S. 214). Laut Seyberth tritt das Reye-Syndrom in Zusammenhang mit Acetylsalicylsäure zwar selten, aber immerhin bis zu einem Alter von achtzehn Jahren auf (vgl. Seyberth, Schwab 2010, S.460). Nur bei Neugeborenen ist die Geschwindigkeit bestimmter Abbauprozesse im Stoffwechsel noch langsamer, daher sind die Plasmaspiegel der verabreichten Substanzen höher und die Wirkung stärker, dies spielt aber nur bis zum dritten Lebensmonat eine Rolle, denn dann errechnet sich die richtige Dosis nur streng nach dem Körpergewicht des Kindes (vgl. Beubler 2012, S. 111). Laut Wilhelm ist Paracetamol, welches allerdings eine nur sehr schwache analgetische Wirksamkeit aufweist, ab der Geburt und auch bei Frühgeborenen zugelassen. NSAIDs sind für Kinder ab dem sechsten Lebensmonat zugelassen und Metamizol darf ab dem dritten Lebensmonat angewendet werden (vgl. Wilhelm, Kreiter 2015, S. 68 - 69). Morphin als im klinischen Bereich am häufigsten angewandtes Opioid, ist für alle Altersklassen zugelassen, Codein ab dem zwölften Lebensjahr und Tramadol ab dem ersten Lebensjahr (vgl. Wilhelm, Kreiter 2015, S. 69-70). Intravenöse Opioidgaben müssen bei Kindern, die jünger als sechs Monate sind, stets pulsoxymetrisch überwacht werden. Häufig kommt im Bereich der Pädiatrie das schwache Opioid Tramadol zum Einsatz, starke Opioide eher nach der Neugeborenenperiode (vgl. Zernikow 2008, S. 190-192). Am besten untersucht in Hinblick auf die Verwendung bei Neugeborenen sind Morphin und Fentanyl, Pethidin ist hingegen 43 wegen der Anhäufung im kindlichen Organismus und der Auslösung von Nebenwirkungen grundsätzlich ungeeignet (vgl. Zernikow 2008, S. 288). „Bei extrem unreifen Frühgeborenen werden starke Opioide als Einzeldosen verabreicht, da die Wirkdauer starken Schwankungen unterlegen ist und bei Anwendung einer Dauerinfusion nach Meinung vieler Neonatologen das Risiko einer nekrotisierenden Enterokolitis weiter ansteigt.“ (Zernikow 2008, S. 288) Laut Greffe wurde die intravenöse Gabe von Opioiden gut untersucht und hat sich in der Pädiatrie bewährt (vgl. Greffe et al. 2014, S. 1024). Greffe nennt ebenfalls Paracetamol als häufig und bei richtiger Dosierung sicher anzuwendendes Medikament in der Pädiatrie, die Gefahren der Überdosierung ergeben sich hiernach eher aus den rezeptfrei erhältlichen Kombinationspräparaten und deren unvernünftigem Gebrauch (vgl. Greffe et al. 2014, S. 1022). Zur Schmerztherapie vor kleinen lokalen Eingriffen in der Kinderheilkunde sollten Emla-Pflaster und Emla-Gel zum Einsatz kommen, diese enthalten ungefährliche Mittel zur lokalen Betäubung der Haut. Zur Behandlung von entzündlichen Erkrankungen bei Kindern sind NSAIDs geeignet, allerdings darf bei Ausbleiben des Therapieerfolges keinesfalls eine Dosissteigerung vorgenommen werden, sondern die Grenzen der Höchstmengen sind strikt einzuhalten. Eine additive Gabe eines schwachen oder sogar starken Opioids ist in diesem Fall sinnvoll und angezeigt. Beubler empfiehlt die Gabe von NSAIDs in Kombination mit Magenschutz aufgrund des vorhandenen Risikos für die Entstehung eines Geschwürs im Verdauungstrakt (vgl. Beubler 2012, S.111-113). Als gut geeignetes Kurzzeitmedikament in der akuten Schmerztherapie bei Kindern ist Ibuprofen zu nennen, welches eine weitaus größere therapeutische Breite besitzt als das Paracetamol, bei dessen Verabreichung nie auf die potentielle Giftigkeit für die Leber vergessen werden darf (vgl. Zernikow 2008, S. 289) „Immer müssen mögliche Nebenwirkungen adjuvanter Schmerzmittel mit Eltern und Kind nicht zuletzt aus forensischen Gründen dezidiert besprochen werden. Die Inhalte des Gesprächs sind zu dokumentieren.“ (Zernikow 2008, S. 292) 44 5 Schlussfolgerungen und Ausblick Schmerzen weisen nicht nur sehr oft auf ein Krankheitsgeschehen hin, sondern zählen auch zu den häufigsten Beweggründen auf Patientenseite für die Konsultation des ärztlichen Berufsstandes. Schmerz wird daher als wichtiges Symptom zur Orientierung bei der Suche nach Krankheitsursachen begriffen. Zusätzlich nimmt in logischer Konsequenz auch die Therapie von Schmerzen einen besonders großen Raum im medizinischen Sektor ein (vgl. Mutschler et.al. 2013, S. 225-226). Die recht häufig ohne ärztliche Überwachung stattfindende Einnahme von Nicht-Opioidanalgetika ist auf Seiten der von Schmerzen betroffenen Patientinnen und Patienten oft auch die Ausweichstrategie für eine misslungene Analgetika Therapie auf ärztlicher Seite. In der Folge nehmen Verschlimmerungen des Krankheitszustandes aber mehr zu als ab. Diesbezüglich ist der Bedarf an zusätzlicher Aufklärung für Betroffene gegeben, denn Gefahren für die Gesundheit werden bei der Anwendung unterschätzt (vgl. Diener et al. 2008, S. 355). „Das Leberversagen ist sehr selten (0,5–1/100 0000 Userjahre). Aber 5 % bis 10 % aller medikamenteninduzierten Fälle eines akuten Leberversagens sind auf NSIAD zurückzuführen. Die Sterblichkeit beträgt bis zu 25 %.“ (Diener et al. 2008, S. 353) Die Einnahme von traditionellen NSAIDs ist der Hauptgrund für das Auftreten des akuten Nierenversagens und beträgt immerhin sechsunddreißig Prozent (vgl. Diener et al. 2008, S. 352). „Ein hochgradiges Risiko eines Nierenversagens besteht für alle NSAR bei Hypovolämie jeder Genese (z.B. perioperativ durch Nachblutung oder bei Dehydratation durch Mangelernährung) sowie bei Patienten mit Herzinsuffizienz. Daher müssen Patienten nach Eingriffen mit auch nur möglichem höherem Blut- oder Volumenverlust von jeder prophylaktischen Gabe eines tNSAR oder Coxibe ausgeschlossen werden, bis ein komplikationsloser Verlauf der Operation sichergestellt ist.“ (Diener et al. 2008, S. 352) Zusätzlich kommt hinzu, dass bestimmte Gruppen von Patientinnen und Patienten in Bezug auf eine adäquate Schmerztherapie untertherapiert sind: 45 Ältere Menschen wenden am meisten Analgetika an und dennoch gibt es recht wenige Forschungsergebnisse über die Risiken bei der Anwendung solcher Medikamente in dieser Altersgruppe. Aufgrund von altersbedingten physiologischen Unterschieden ist eine Übertragung von Erkenntnissen aus klinischen Forschungen an jüngeren Menschen problematisch. Optimaler Weise sollte im geriatrischen Bereich eine multimodale Schmerztherapie zum Einsatz kommen, welche die besonderen Bedürfnisse dieser immer größer werdenden Gruppe von Patientinnen und Patienten berücksichtigt (vgl. Lukas, Drebenstedt 2014, S. 421). „Schätzungen gehen davon aus, dass 45-80% aller älteren Schmerzpatienten inadäquat behandelt werden.“ (Lukas, Drebenstedt 2014, S. 421) Die Multimorbidität im Alter ist die Hauptursache für die problematische Polypharmazie, die mit den so häufig auftretenden Komplikationen und unerwünschten Arzneimittelwirkungen im geriatrischen Bereich in Zusammenhang steht (vgl. Siegmund, May (2010, S. 462). Auch Kinder sind in Bezug auf eine qualifizierte Schmerztherapie untertherapiert. Dabei kommt erschwerend hinzu, dass nur eine eingeschränkte Auswahl an Medikamenten zur Verfügung steht (vgl. Seyberth, Schwab 2010, S.460). Im pädiatrischen Fachbereich sind immerhin achtzig Prozent der gesamten Medikamente, die für Erwachsene zum Einsatz kommen, nicht zugelassen, darunter fallen auch viele Analgetika. Dies ist auf Vorkommnisse von schweren Komplikationen aufgrund von für Kinder ungeeigneten Substanzen oder Darreichungsformen zurückzuführen (vgl. Seyberth, Schwab 2010, S.456). „Tatsächlich sind Kinder hinsichtlich Schmerztherapie eine besonders benachteiligte Gruppe. Eine Reihe von mythischen Glaubenssätzen verhindert nach wie vor, dass Kinder die medikamentöse Schmerztherapie erhalten, die sie benötigen.“ (Beubler 2012, S. 110) Die klinische Forschung im Bereich der kindlichen Schmerztherapie ist nicht ausreichend, um auf genügend gesicherte Forschungsergebnisse zurückgreifen zu können. Im Besonderen betroffen ist der Bereich der Neonatologie, aber generell besteht für die pädiatrische Schmerztherapie ein Ergänzungs-Bedarf an klinischen Studien (vgl. Zernikow et. al 2015, S. 158). 46 Literaturverzeichnis Beubler, E (2012): Kompendium der medikamentösen Schmerztherapie. Wirkungen, Nebenwirkungen und Kombinationsmöglichkeiten. 5. Auflage. Springer Medizin Verlag: Wien, S. 6123. Bundeskanzleramt: Rechtsinformationssystem (RIS), Fassung vom 05.07.2016: Bundesgesetz über Suchtgifte, psychotrope Stoffe und Drogenausgangsstoffe (Suchtmittelgesetz – SMG) (2016) https://www.ris.bka.gv.at/GeltendeFassung.wxe?Abfrage=Bundesnormen&Gesetzesnummer=10011040 (abgerufen am 05.07.2016) Cascorbi, I (2013): Analgetika, in: Cascorbi, I, Sorge, J, Strumpf, M (Hrsg.): MedikamentenPocket Schmerztherapie. Springer Medizin Verlag: Berlin Heidelberg, S. 3-67. Diener, H, C, Maier, C, Kindler, D, Gerwig, M (2008): Medikamentöse Schmerztherapie, in: Diener, H, C, Maier, C (Hrsg.): Die Schmerztherapie. Interdisziplinäre Diagnose- und Behandlungsstrategien. 3., überarbeitete Auflage. Elsevier Urban & Fischer: München · Jena, S. 338391. Greffe, B, S, Galinkin, J, L, King, N, A (2014): Pain Management & Pediatric Palliative & Endof-Life Care, in: Hay, W, W, Deterding, R, R, Levin, M, J, Abzug, M, J (Hrsg.): Current Diagnosis & Treatment: Pediatrics, Twenty-Second Edition. International Edition. McGraw-Hill Education: New York, S. 10222-1031. Höllt, V, Allgaier, C (2009): Analgetika, in: Aktories, K, Förstermann, U, Hofmann, F, Starke, K (Hrsg.): Allgemeine und spezielle Pharmakologie und Toxikologie. 10., überarbeitete Auflage. Elsevier GmbH, Urban&Fischer Verlag: München, S. 219-243. Höllt, V, Allgaier, C (2013): Analgetika, in: Aktories, K, Förstermann, U, Hofmann, F, Starke, K (Hrsg.): Allgemeine und spezielle Pharmakologie und Toxikologie. 11., überarbeitete Auflage. Elsevier GmbH, Urban&Fischer Verlag: München, S. 207-231. Lukas, A, Drebenstedt, C (2014): Schmerzen, in: Pantel, J, Schröder, J, Bollheimer, C, Sieber, C, Kruse, A (Hrsg.): Praxishandbuch Altersmedizin. Geriatrie – Gerontopsychiatrie – Gerontologie. 1. Auflage. Verlag W. Kohlhammer: Stuttgart, S. 415-424. Mutschler, E, Geisslinger, G, Kroemer, H, K, Menzel, S, Ruth, P (2013): Mutschler Arzneimittelwirkungen. Pharmakologie. Klinische Pharmakologie. Toxikologie. 10., vollständig überarbeitete und erweiterte Auflage. Wissenschaftliche Verlagsgesellschaft: Stuttgart, S. 194-234. Mühlberg, W, Sieber, C (2014): Iatrogene Schäden durch Polypharmazie im Alter, in: Pantel, J, Schröder, J, Bollheimer, C, Sieber, C, Kruse, A (Hrsg.): Praxishandbuch Altersmedizin. Geriatrie – Gerontopsychiatrie – Gerontologie. 1. Auflage. Verlag W. Kohlhammer: Stuttgart, S. 245-260. 47 Nikolaus, T (2013): Persistierender Schmerz, in: Zeyfang, A, Hagg-Grün, U, Nikolaus, T (Hrsg.): Basiswissen Medizin des Alterns und des alten Menschen. 2., überarbeitete Auflage. Springer Medizin Verlag: Berlin Heidelberg, S. 207-220. Schäfers, M (2008): Schmerztherapie im Alter, in: Diener, H, C, Maier, C (Hrsg.): Die Schmerztherapie. Interdisziplinäre Diagnose- und Behandlungsstrategien. 3., überarbeitete Auflage. Elsevier Urban & Fischer: München · Jena, S. 314-318. Seyberth, H, W, Schwab, M (2010): Besonderheiten der Arzneimitteltherapie im Kindesalter, in: Lemmer, B, Brune, K (Hrsg.): Pharmakotherapie. Klinische Pharmakologie. 14. Auflage. Springer Medizin Verlag: Heidelberg, S. 453-459. Siegmund, W, May, K (2010): Arzneitherapie im Alter, in: Lemmer, B, Brune, K (Hrsg.): Pharmakotherapie. Klinische Pharmakologie. 14. Auflage. Springer Medizin Verlag: Heidelberg, S. 482-489. Strumpf, M (2013): Probleme der medikamentösen Schmerztherapie, in: Cascorbi, I, Sorge, J, Strumpf, M (Hrsg.): Medikamenten-Pocket Schmerztherapie. Springer Medizin Verlag: Berlin Heidelberg, S. 106-118. Wehling, M, Burkhardt, H, Schwarz, S, Fröhlich, L, Wedding, U (2013): Spezielle Aspekte bezogen auf Organsysteme nach geriatrisch klinischer Bedeutung, in: Wehling, M, Burkhardt, H (Hrsg.): Arzneitherapie für Ältere. 3., vollständig überarbeitete und aktualisierte Auflage. Springer Medizin Verlag: Berlin Heidelberg, S. 191-208. Wilhelm, I, Kreiter, B (2015): Schmerz und Sedation, in: Schöni, M, H, Simonetti, G, D, Aebi, C (Hrsg.): Berner Datenbuch Pädiatrie. 8., vollständig überarbeitete Auflage. Verlag Hans Huber: Bern, S. 65-71. Zeilhofer, H, U, Sittl, R (2007): Therapie mit Analgetika und Lokalanästhetika, in: Lemmer, B, Brune, K (Hrsg.): Pharmakotherapie. Klinische Pharmakologie. 13., überarbeitete und aktualisierte Auflage. Springer Medizin Verlag: Heidelberg, S. 78-88. Zeilhofer, H, U, Sittl, R (2010): Therapie mit Analgetika und Lokalanästhetika, in: Lemmer, B, Brune, K (Hrsg.): Pharmakotherapie. Klinische Pharmakologie. 14. Auflage. Springer Medizin Verlag: Heidelberg, S. 69-77. Zernikow, B (2008): Schmerztherapie bei Kindern, in: Diener, H, C, Maier, C (Hrsg.): Die Schmerztherapie. Interdisziplinäre Diagnose- und Behandlungsstrategien. 3., überarbeitete Auflage. Elsevier Urban & Fischer: München · Jena, S. 286-294. Zernikow, B, Hünseler, C, Roth, B, Michel, E (2015): Klinisch-pharmakologische Grundlagen der Schmerztherapie, in: Zernikow, B (Hrsg.): Schmerztherapie bei Kindern, Jugendlichen und jungen Erwachsenen. 5. Auflage. Springer-Verlag: Berlin Heidelberg, S. 105-158. 48 Abbildungsverzeichnis Abbildung 1 (entnommen aus): Pirman, T, TAD Pharma GmbH (2016) http://www.tad.de/de/gesundheit/schmerz/schmerz/schmerzverarbeitung/ (abgerufen am 04. 07.2016) Abbildung 2 (entnommen aus): Pirman, T, TAD Pharma GmbH (2016) http://www.tad.de/de/gesundheit/schmerz/schmerz/schmerzverarbeitung/ (abgerufen am 04.07.2016) Abbildung 3 (entnommen aus): Seifert, V, der – allgemeinarzt.com (2016) http://www.allgemeinarzt-online.de/a/allgemeine-palliativversorgung-grundlagen-medikament%C3%B6ser-schmerztherapie-1563192 (abgerufen am 04. 07. 2016) Abbildung 4 (entnommen aus): Stackelberg, H, Deutsches Grünes Kreuz (2016) http://www.forum-schmerz.de/schmerz-infos/krebsschmerzen/therapie/who-stufenschema.html (abgerufen am 04.07. 2016) 49