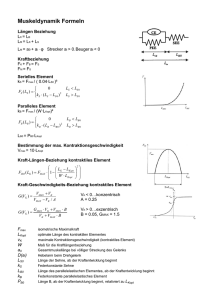
Die Reaktionsgeschwindigkeit
Werbung

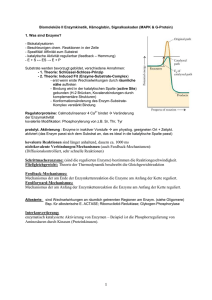
Die Reaktionsgeschwindigkeit Die grundlegende Größe, mit der in der Kinetik gearbeitet wird, ist die Reaktionsgeschwindigkeit. Sie gibt an, wie viele Teilchen pro Zeit in einer chemischen Reaktion umgesetzt werden. Diese Geschwindigkeit hängt dabei von vielen Faktoren ab. Je nach zugrunde liegendem Modell gibt es unterschiedliche Möglichkeiten, die Reaktionsgeschwindigkeit zu betrachten. Ein wichtiger Faktor, der zu berücksichtigen ist, ist die Konzentration der vorliegenden Stoffe. Je mehr Teilchen in einem Volumen vorliegen, desto mehr Kollisionen werden pro Zeiteinheit vorkommen. Da eine Reaktion aber nur stattfinden kann, wenn zwei Teilchen miteinander kollidieren, steigt die Reaktionsgeschwindigkeit mit der Konzentration der Edukte. Wenn eine Reaktion folgenden Typs vorliegt so gilt für die Hinreaktion das Geschwindigkeitsgesetz wobei vR die Reaktionsgeschwindigkeit, ( − ∆cA) die Abnahme der Konzentration des Stoffes A und ∆t die verstrichene Zeit ist. Diese Reaktionsgeschwindigkeit ist die Durchschnittsgeschwindigkeit der Reaktion, da einzelne Moleküle unterschiedliche lange Zeitintervalle benötigen, bevor sie in ein Reaktionsereignis eintreten. Da die Abnahme der Edukte der Zunahme der Produkte entsprechen muss, gilt außerdem Dieses stark vereinfachte Modell bedarf noch einiger Verfeinerungen bezüglich: • • • • • • • • der Aktivität, also die effektive Konzentration der Menge der Edukte im Verhältnis zu der Menge der Produkte und gegebenenfalls des Lösungsmittels der Temperatur der Stoßenergie der Anwesenheit von Katalysatoren der Reaktionen mit auftretenden Gasen vom Partialdruck der Ausrichtung großer Reaktionspartner (Enzyme, Katalysatoroberfläche) beim Zusammenstoß des Zerteilungsgrades Die Reaktionsordnung Für jede Reaktion lässt sich eine Reaktionsgleichung formulieren, die beschreibt, wie viele Edukt-Teilchen miteinander reagieren, um eine bestimmte Anzahl Produkt-Teilchen zu bilden. Läge etwa folgende Reaktionsgleichung vor würde dies bedeuten, dass zwei Teilchen A mit einem Teilchen B, einem Teilchen C und einem Teilchen D kollidieren müssten, um das Produkt E zu bilden. Jedoch ist die Wahrscheinlichkeit, dass fünf Teilchen gleichzeitig und zudem mit ausreichender Energie kollidieren, äußerst gering. Viel wahrscheinlicher ist, dass zunächst zwei oder drei Teilchen zusammen treffen, ein Zwischenprodukt bilden, dieses Zwischenprodukt kollidiert dann mit weiteren Teilchen gegebenenfalls unter Bildung weiterer Zwischenprodukte und zuletzt bildet sich das Produkt E, hier ein Beispiel: Die Zerlegung der Gesamtreaktion in einzelne Schritte, in Elementarreaktionen, und deren Untersuchung zeigt, wie die Reaktion genau abläuft. Experimentell oder auf der Grundlage modellhafter Annahmen kann ermittelt werden, wie die Reaktionsgeschwindigkeiten der Elementarreaktionen von den jeweiligen Konzentrationen der Komponenten A, B, C und D abhängen. Die Abhängigkeit der Reaktionsgeschwindigkeit vom Exponenten mit dem die Konzentration eines bestimmten Reaktanden in das Geschwindigkeitsgesetz eingeht, wird als Reaktionsordnung in Bezug auf diesen Reaktanden bezeichnet. Die Gesamtordnung einer Reaktion ist die Summe der Reaktionsordnungen aller an ihr beteiligten Reaktanden. Die drei häufigsten, weil wahrscheinlichsten, Reaktionsordnungen Reaktio sind: Reaktionen nullter Ordnung Derartige Reaktionen sind unabhängig von der der Konzentration der Reaktanden, hier ist die Reaktionsgeschwindigkeit also konstant. Dies kann z. B. der Fall sein, wenn es sich um lichtabhängige Reaktionen handelt (dann ist der Faktor k von der Lichtintensität abhängig) wobei v – Reaktionsgeschwindigkeit CA(t) – Konzentration des Stoffes A zum Zeitpunkt t t – Zeit k – Geschwindigkeitskoeffizient Beispiele sind photochemische Reaktionen oder katalytische Reaktionen. Integration beider Seiten ergibt: Zu Beginn der Reaktion (t = 0 ) ist die Konzentration des Reaktanten [A]0. Dementsprechend gilt für die Integrationskonstante C = [A]0 Somit resultiert die integrierte Form des Zeitgesetzes: Diee Auftragung von [A] gegen t liefert eine Gerade mit der Steigung -k. Reaktionen erster Ordnung Hier handelt es sich um katalytische oder radioaktive Zerfallsprozesse. Die Reaktionsgeschwindigkeit ist nur von der Konzentration des zerfallenden Stoffes abhängig. integriert mit [A]t – Konzentration von A zur Zeit t [A]0 – Anfangskonzentration von A Umformung des Geschwindigkeitsgesetzes ergibt Gleichung (2) stellt die differentielle Form des Zeitgesetzes dar Durch Integration dieser Gleichung und Bestimmung der Integrationskonstanten C erhält man das entsprechende Zeitgesetz in integrierter Form. Integration der Gleichung (2) gibt: Die Integrationskonstante C kann anhand der Integrationsgrenzen bestimmt werden. Zu Beginn der Reaktion (t = 0) ist [A]0 die Konzentration von A. Für ln[A]0 ergibt sich: Deshalb: Es folgt damit: Eine Auftragung von ln[A] oder ln[A] / [A]0 gegen die Zeit t ergibt eine Gerade mit der Steigung -k. Bei einer Reaktion erster Ordnung sollten die entsprechenden Messpunkte bis zu einem Reaktionsumsatz von 80-90% auf dieser Geraden liegen. Gleichung (6) kann auch wie folgt formuliert werden: Dies bedeutet, dass die Konzentration von A exponentiell mit der Zeit abnimmt. Die Geschwindigkeitskonstante k läßt sich auch aus der Halbwertszeit t1/2 ermitteln. Unter der Halbwertszeit versteht man die Zeit, in der die Hälfte der ursprünglichen Konzentration des Reaktanten A verbraucht ist ==> [A] = 1/2 [A]0 . Es gilt entsprechend Gleichung (6):: Reaktionen zweiter Ordnung In diesem Falle reagieren zwei Edukte zu einem oder mehreren Produkten. Die Reaktionsgeschwindigkeit ist abhängig von den Konzentrationen der Ausgangsstoffe. oder bei nur einem Stoff Trennung der Variablen und Integration ergeben: Für die Konzentration von A zu Beginn der Reaktion t = 0 wird A0 gesetzt. Die Integrationskonstante C hat folglich den Wert 1 / [A]0. Somit ergibt sich die integrierte Form des Zeitgesetzes mit Eine Auftragung von 1 / [A] gegen die gegen die Zeit t ergibt gibt eine Gerade mit der Steigung k. Bei einer Reaktion zweiter Ordnung (Konzentrationen der Reaktionspartner identisch) sollten die entsprechenden Messpunkte bis zu einem Reaktionsumsatz von 50% auf dieser Geraden liegen. Sofern [A]0 and [B]0 unterschiedlich sind, führt man zweckmäßig die Umsatzvariable x ein. Für Gleichung (1) ergibt sich: wobei [A]0 - x = [A], [B]0 - x = [B] und x die Abnahme von A und B darstellt. Nach Trennung der Variablen und Partialbruchzerlegung läßt sich Gleichung (5) integrieren wobei C die Integrationskonstante darstellt. Unter der Voraussetzung dass x = 0, sofern t = 0 wird der Wert für C erhalten und Gleichung (6) wird zu Eine Auftragung von gegen t ergibt eine Gerade mit der Steigung ([A]0 - [B]0) k. Gleichung (8) kann auch, wie folgt formuliert werden Allerdings gibt es einen Sonderfall, bei dem einer der Reaktanden in einem sehr hohen Überschuss vorliegt, so dass die Konzentrationsänderung über die Zeit der Reaktion verschwindend gering ist. Das ist zum Beispiel der Fall, wenn Wasser sowohl Reaktionspartner als auch das Lösungsmittel darstellt (z. B. bei einer Esterhydrolyse). In diesem Fall folgt die Reaktionsgeschwindigkeit den Gesetzmäßigkeiten einer Reaktion erster Ordnung. Da es sich aber trotzdem um eine bimolekulare Reaktion handelt, spricht man von Reaktionen pseudoerster Ordnung (oder scheinbar erster Ordnung). Ein weiterer Sonderfall ergibt sich, wenn experimentell eine Reaktion 0. Ordnung gemessen wird (obwohl eine Reaktion höherer Ordnung vorliegt). Dann spricht man von einer Reaktion scheinbar nullter Ordnung. Dies kann insbesondere bei katalytischen Prozessen der Fall sein (wie z. B. der Enzymkatalyse oder der katalytischen Hydrierung von Ethen) wenn der limitierende Faktor die Anzahl „Plätze“ am Katalysator sind und nicht wie oft die Moleküle zusammenprallen (da dann die direkte Elementarreaktion so gut wie keine Rolle mehr spielt). Gleichgewichtsreaktionen Viele Reaktionen sind Gleichgewichtsreaktionen bei denen das gebildete Produkt wieder zum Edukt zurück reagieren kann. Die Änderung der Konzentration A ist in diesem Fall bestimmt durch die Differenzialgleichung mit den Reaktionsparametern kab der Reaktion und kba der Rückreaktion . Es ergibt sich daraus das folgendes Geschwindigkeitsgesetz: Geschwindigkeitsgesetz Zusammenfassung Differentielles Integriertes Linearer Reaktionsordnun Zeitgesetz Plot Zeitgesetz g Steigung Einheiten der der Geschwindigkeitskonstant Geraden e 0 - d[A] / dt = [A] = [A]0 [A] k kt gegen t -k mol · L-11 · s-1 1 - d[A] / dt = [A] = [A]0 e ln[A] - kt k [A] gegen t -k s-1 2 - d[A] / dt = 1 / [A] = 1 / 1 / [A] k [A]2 [A]0 + kt gegen t k L · mol-11 · s-1 Temperaturabhängigkeit der Reaktionsgeschwindigkeit RGT-Regel (Reaktionsgeschwindigkeit sgeschwindigkeit-Temperatur-Regel): Regel): Wird die Temperatur für eine chemische Reaktion um 10 K erhöht, dann erhöht sich die Reaktionsgeschwindigkeit um das 2- bis 4-fache fache (für eine Reaktion mit einer Aktivierungsenergie um die 50 kJ/mol!) Mathematisch und physikalisch sikalisch wird dies mit dem Ansatz von Arrhenius begründet. Nach diesem kann die Temperaturabhängigkeit der Reaktionsgeschwindigkeit mit einer Exponentialfunktion beschrieben en werden. Wie oben schon beschrieben lautet die Bestimmungsgleichung für eine Reaktion zweiter Ordnung: Die Temperaturabhängigkeit ist in dem so genannten Geschwindigkeitskoeffizienten enthalten, denn es gilt hier die Arrhenius-Gleichung. Arrhenius . Der Geschwindigkeitskoeffizient k ist nur dann eine Konstante, solange die Temperatur nicht verändert wird und kann dann auch als Geschwindigkeitskonstante bezeichnet werden. we Überblick über die wichtigsten Formeln der Reaktionskinetik Berechnung der MomentanMomentan Berechnung der Reaktionstyp geschwindigkeit v Halbwertszeit 0. Ordnung 1. Ordnung 2. Ordnung • 0. Ordnung: • 1. Ordnung: • 2. Ordnung: In allen Fällen erhält man k aus der Steigung der entstehenden Graphen. Je mehr Messpunkte Mes man hat, desto genauer wird das Ergebnis. Arrhenius Arrhenius-Gleichung Es ist eine bekannte Tatsache, dass die Geschwindigkeit einer chemischen Reaktion im allgemeinen mit steigender Temperatur zunimmt. Die quantitative Beziehung zwischen Reaktionsgeschwindigkeit und Temperatur wird durch die Arrhenius Arrhenius-Gleichung beschrieben: Svante Arrhenius Ea = Aktivierungsenergie R = 8.314 J/mol·K T = absolute Temperatur in Kelvin A = präexponentieller Faktor oder Häufigkeitsfaktor A = p · Z, wobei Z die Stoßzahl ist und p einen sterischen Faktor darstellt. Z erweist sich als nur schwach abhängig von der Reaktionstemperatur. Somit stellt der Häufigkeitsfaktor für die entsprechende Reaktion eine Konstante dar. Die Arrhenius-Gleichung basiert auf der Stoßtheorie, wonach die Reaktanten sowohl in der korrekten Orientierung als auch mit der nötigen kinetischen Energie zusammenstoßen müssen, damit sie in die Produkte umgewandelt werden. Logarithmiert man beide Seiten der Gleichung (1), so erhält man Die Auftragung von lnk gegen 1/T liefert eine Gerade mit der allgemeinen Gleichung y = -mx + b, wobei x = 1/T y = lnk m = - Ea / R b = lnA Die Aktivierungsenergie Ea lässt sich aus der Steigung m der Geraden ermitteln: Ea = -m · R Die Aktivierungsenergie wird in Veröffentlichungen auf eine Kommastelle angegeben. Bei der Angabe der lnA-Werte sind 2 Dezimalen statthaft. Bestimmung von Ea Um die Aktivierungsenergie möglichst exakt zu bestimmen sollte das entsprechende Experiment bei mindestens drei verschiedenen Reaktionstemperaturen durchgeführt werden. Die Temperaturintervalle sollten mindestens 5°C betragen. "Zweipunktegleichung" Rein rechnerisch läßt sich die Aktivierungsenergie einer gegebenen Reaktion anhand der experimentellen Daten bestimmen, die man bei zwei verschiedenen Reaktionstemperaturen gewonnen hat. Es gilt bei T1: bei T2: Subtraktion der Gleichung (4) von Gleichung (3) führt zu Unformen obiger Gleichung und Auflösen nach Ea gibt Enzymkinetik Michaelis-Menten Menten-Theorie Die Michaelis-Menten-Theorie legt den Grundstein für die Enzymkinetik. Hier wurde das theoretische Rüstzeug erarbeitet, Enzyme nicht nur hinsichtlich hinsichtlic ihrer Aktivität zu charakterisieren, sondern auch die Stoffmenge (Konzentration) zu finden, welche eine den Gegebenheiten angepasste Umwandlung ermöglicht. ermöglicht Im Allgemeinen sind Enzyme in der Lage, schwankende Substrat-Konzentrationen Konzentrationen auszugleichen, d. h. ein Fließgleichgewicht („steady state“) dadurch einzustellen, dass sie ihre Tätigkeit dem Angebot anpassen. Im Gegensatz zur Kinetik chemischer Reaktionen gibt es in der Enzymkinetik das Phänomen der Sättigung:: bei sehr hohen Substratkonzentrationen kann die Umsatzgeschwindigkeit v nicht weiter gesteigert werden d. h. es wird ein Wert Vmax erreicht. Theoretische Begründung Als Biokatalysatoren bilden Enzyme E mit ihrem Substrat S einen Komplex ES (Enzym(Enzym Substrat-Komplex), Komplex), aus dem heraus sich die Reaktion zum Produkt P vollzieht: k1 und k'1 sind die Geschwindigkeitskonstanten für die Assoziation Assoziation von E und S bzw. die Dissoziation des Enzym-Substrat-Komplexes Komplexes ES. k2 und k'2 sind die entsprechenden Konstanten für die Reaktion zum Produkt bzw. die Rückreaktion zum Substrat. Diese Rückreaktion findet unter den Bedingungen der Enzymkinetik, d. d h. unmittelbar nach Mischung der Komponenten E und S, noch nicht statt. statt. Ferner wird die Umwandlung von ES zu EP (und nicht die spontanee Freisetzung von P) gemessen, so dass die folgende Vereinfachung gerechtfertigt ist: Hierin ist k2 ein Maß der maximalen Reaktionsgeschwindigkeit bei Substratsättigung ( Vmax), auch Wechselzahl,, molekulare Aktivität, „turnover number“ oder kcat genannt (kcat = Vmax / [E0]). Die Michaeliskonstante,, die Substratkonzentration, die bei der Halbsättigung vorliegt (die Umsatzgeschwindigkeit also v = Vmax/2 beträgt), ergibt sich zu (Michaelis-Menten-Fall, Fall, gegeben wenn k2<< k-1) oder allgemeiner zu Situation [Zerfallswege von ES/Bildungsweg von ES] für den Fall, dass k2 (Briggs-Haldane-Situation gegenüber k1 nicht vernachlässigt werden kann) Sättigungsverlauf: eine Hyperbel zwischen Km und Vmax Sättigungshyperbel Km entspricht 1/2 Vmax igungsfunktion eines “Michaelis-Menten “Michaelis Menten Enzyms” lässt sich unter Verwendung der Die Sättigungsfunktion Parameter Km und Vmax wie folgt formulieren: formulieren Dies ist die Michaelis-Menten Menten Beziehung, Beziehung d. h. die Gleichung einer Hyperbel mit den folgenden in der Abbildung gezeigten Eigenschaften: • Ihre Asymptoten entsprechen den Werten Vmax (realer Ast) und Km (imaginärer Ast, gestrichelt); • • • Gleicht die Substratkonzentration [S] dem Km-Wert, so liegt die Hälfte des ursprünglich vorhandenen Enzyms [Eo] in Form des Enzym-Substrat-Komplexes [ES] vor, die andere Hälfte ist frei [E]. Da die Sättigung asymptotisch angenähert wird, sind hierzu Substratkonzentrationen erforderlich, die mehr als dem zehnfachen Km-Wert entsprechen. Im Umkehrschluss gilt: Hat man für ein Enzym eine Sättigungshyperbel gemessen, d. h. die Umsatzgeschwindigkeit v als Funktion der Substratkonzentration [S] bestimmt, so lassen sich daraus Vmax (die Aktivität) und Km (die reziproke Affinität) ableiten. Ein relativ neues, einfaches und doch präzises Verfahren zu diesem Zweck ist die direktlineare Auftragung (Enzymkinetik). Inhibitoren, darunter wichtige Medikamente und Gifte, ändern die Eigenschaften von Enzymen und können mit den hier eingeführten Methoden näher charakterisiert und in ihrer Wirkungsweise besser verstanden werden: • • • „kompetitive“ Inhibitoren erhöhen den Km-Wert, verändern Vmax jedoch nicht „unkompetitive“ Inhibitoren erhöhen ebenfalls den Km-Wert, senken zudem aber Vmax „nichtkompetitive“ Inhibitoren senken Vmax, verändern den Km-Wert aber nicht Enzymkinetik Die Enzymkinetik ist ein Teilgebiet der biophysikalischen Chemie. Sie beschreibt, wie schnell enzymkatalysierte chemische Reaktionen verlaufen. Die Enzymkinetik findet breite Anwendung in Biologie und Medizin, da auch biologische Substrate (Reaktionspartner) – darunter solche, die im Menschen auftreten – untersucht werden. Ein Hauptziel der Enzymkinetik ist die Beschreibung der Konzentrationsabhängigkeit der Reaktionsgeschwindigkeit mit geeigneten Formeln, sowie die Bestimmung der dazugehörigen Parameter für ein bestimmtes Protein. Da Enzyme dazu dienen, Reaktionen zu beschleunigen und zu lenken, ist die enzymkinetische Analyse zum Verständnis von Enzymfunktionen unerlässlich. Die Bindungsreaktion kann nach dem Massenwirkungsgesetz beschrieben werden, sie wird charakterisiert durch Dissoziationskonstante Kd = k − 1 / k + 1. Dabei ist Kd diejenige Konzentration des Substrates, bei der (in Abwesenheit der Umsetzung zu Produkt) die Hälfte aller Enzymmoleküle Substrat gebunden haben. Sie ist umgekehrt proportional zur Affinität des Enzymes für das Substrat. Die Umsetzung des Enzym-Substrat-Komplexes zu Enzym und Produkt wird bestimmt durch eine Geschwindigkeitskonstante 1. Ordnung, kcat. Die tatsächliche Geschwindigkeit der enzymatischen Reaktion ergibt sich zu v = kcat * [ES]. Allerdings vermindert die Zerfallsreaktion die Konzentration von [ES], es ist deshalb erforderlich, diese statt mit Kd mit zu berechnen. Dann ergibt sich[1] Dies ist die Gleichung einer Hyperbel, sie nähert sich für unendlich hohe Substratkonzentrationen, wenn alle Enzymmoleküle Substrat gebunden haben, einem Grenzwert an. Die Konzentration bei der die Reaktionsgeschwindigkeit gerade die Hälfte des Maximalwertes erreicht, ist die MichaelisKonstante Km. Zur Bestimmung von Vmax und Km aus Meßreihen von v und [S] dienen computergestützte Verfahren wie die nichtlineare Regressionsanalyse (Simplex- oder Levenberg-Marquardt-Verfahren). Graphische Extrapolationsverfahren (Linearisierungen) wie etwa die doppelt-reziproke Auftragung nach Lineweaver und Burk sollten dafür nicht verwendet werden, da sie zu ungenau sind. Sie eignet sich jedoch sehr gut zur Präsentation der Ergebnisse enzymkinetischer Versuche, weil das menschliche Auge Abweichungen von einer Gerade leichter erkennen kann als die von einer Kurve. Direkt-lineare Auftragung Wenig bekannt ist die Tatsache, dass sich enzymkinetische enzymkinetische Parameter bequem und präzise direkt aus einer Sättigungshyperbel gemäß der Abbildung herleiten lassen („direkt-lineare („direkt Auftragung“ auch „Cornish-Bowden Bowden-Diagramm“ Diagramm“ genannt). In dieser Hyperbel ist die enzymatische Umsatzgeschwindigkeit v (Y-Achse) (Y als Funktion der Substratkonzentration [S] dargestellt. Für die direkt-lineare lineare Auftragung überträgt man die Anfangsgeschwindigkeiten des enzymatischen Umsatzes direkt in das v-[S]-Diagramm. v Die [S]-Werte sind vor Versuchsbeginn bekannt (eingestellte Substratkonzentrationen); Substratkonzentrationen); während der Versuchsreihe ist dann der Ordinatenwert für v (die Anfangsgeschwindigkeit) nachzutragen. Im Gegensatz zu normalen Auftragungen werden die Messwerte also nicht als Punkte dargestellt, sondern in Form von Linien, die sich im günstigsten stigsten Fall in einem gemeinsamen Punkt schneiden, aus dessen Koordinatenwerten Km und Vmax folgen. Die katalytische Effizienz folgt übrigens aus der Steigung der Tangente an den Ursprung: Vmax/Km; daraus ergibt sich kcat/Km. Die Fehlerbehandlung wird im direkt-linearen direkt linearen Plot weitgehend vereinfacht: die Linien im zweiten Quadranten werden mehr oder weniger um einen Punkt streuen. Mittelwertsbildung gibt dann die wahrscheinlichen Werte für die Parameter Km und Vmax. Bei Inspektion der Streubreite der Messpunkte können Ausreißer leicht leic identifiziert und sogenannte Mediane abgelesen werden. Linearisierungsverfahren Linearisierungsverfahren wurden in der Vergangenheit sehr sehr häufig für die schnelle grafische Bestimmung der wichtigen Kinetikparameter Km und Vmax verwendet. Sie sind zwar einprägsam und verbreitet, führen jedoch zu einer teils erheblichen Verfälschung des Ergebnisses durch Messfehler und sind zur Fehlerbetrachtung Fehlerbetrachtung mehr oder weniger ungeeignet. Mittlerweile hat die Ermittlung der Michaelis-Menten-Parameter Michaelis Parameter durch nichtlineare Regression stark an Bedeutung gewonnen, die zu deutlich genaueren Ergebnissen führt. Lineweaver-Burke-Diagramm Lineweaver-Burk-Diagramm Diagramm (doppelt-reziproke (doppelt Auftragung) 1/v als Funktion von 1/[S]. zur Datenrepräsentation meist verwendet, zur Auswertung jedoch am wenigsten verlässlich. Kleine Fehler in v ergeben bei kleinen [S]-Werten [S] Werten eine große Abweichung in 1/v, bei großen [S]-Werten ist diese eher zu vernachlässigen. Eadie-Hofstee-Diagramm Das Eadie-Hofstee-Diagramm nimmt eine Mittelstellung ein. Hierbei wird v über (v/[S]) aufgetragen. Eine Umformung der Michaelis-Menten-Gleichung ergibt die folgende Gleichung: Aus dem Diagramm lassen sich vmax als Y-Achsenabschnitt und Km als negative Steigung der Regressionsgeraden ableiten. Der Fehler wächst mit v/[S]. Da v bei beiden Koordinaten eingeht, konvergieren alle Abweichungen zum Ursprung. Scatchard-Diagramm Das Scatchard-Diagramm v/[S] als Funktion von v. gleicht dem vorigen (Achsen sind vertauscht) und wird zumeist zur Repräsentation von Bindungsmessungen (anstelle enzymkinetischer Daten) angewendet. Scatchard- und EadieHofstee-Diagramme gelten als die besten Werkzeuge zur Diagnose kooperativer Phänomene. Im Falle negativer Kooperativität oder nicht-identischer, isolierter Bindungsplätze entsteht ein konkaver Verlauf mit linearem Endast. Die Steigungen entsprechen hier den Affinitäten (Kd beziehungsweise Km) und die Gesamtzahl der Bindungsplätze (aktiven Zentren) ist aus dem Schnittpunkt mit der x-Achse abzulesen. Hill-Diagramm Graphische Methode, vorrangig für die Bestimmung eines kooperativen Bindungsvorganges für ein Protein (Enzym). Gebunden werden Substrate S (bzw. Liganden L) Die Graphik setzt Kenntnis von Vmax (beziehungsweise n) voraus. Die Auftragung von log v/(Vmax-v) als Funktion von log [S] beziehungsweise log r/(n-r) als Funktion von log [L] ist eine Gerade der Steigung 1 (nH = 1), wenn die Bindungsplätze voneinander unabhängig sind. Bei kooperativen Systemen kann die Steigung am Nullpunkt (der Hill-Koeffizient nH) theoretisch der Zahl der Untereinheiten (n) gleichen, wird aber praktisch darunter bleiben. Falls die individuellen Km-(Kd-)Werte nicht durch Tangenten an die 45°- Ausläufer der Kurve extrapolierbar sind, ist nH ein Ersatzmaß der Kooperativität einzusetzen; nur für nH = 1 gleicht der Nulldurchgang ln Km (beziehungsweise ln Kd). Für Hämoglobin (n = 4), das als klassisches, hochkooperatives Protein gilt, wurde nH zu 2,8-2,9 bestimmt. Inhibitoren Viele Therapeutika und Gifte sind Hemmstoffe (Inhibitoren) von Enzymen. Aus diesem Grunde ist der Aufklärung des Wirkungsmechanismus immer eine besondere Bedeutung zugekommen. Die Nomenklatur der Hemmtypen wurde von W.W. Cleland 1963 auf eine systematische Grundlage gestellt, leider werden in vielen Lehrbüchern immer noch Begriffe abweichend verwendet. Hier sollte allerdings beachtet werden, dass sich klassische Analysen auf reversibel bindende Stoffe beschränken. Irreversible Bindung einer Substanz an ein Enzym führt zur Inaktivierung, nicht zur Hemmung. Abgeleitet aus der Michaelis-Menten-Gleichung (v = Vmax x [S] / (Km + [S]) stellt sich die allgemeine Inhibitionsgleichung wie folgt dar: Danach kann das Verhältnis des Ki-Wertes -Wertes (Dissoziationskonstante des Komplexes EI) und des Kii-Wertes Wertes (Dissoziationskonstante des Komplexes EIS) zur Ableitung des Inhibitionstyps dienen: Kompetitiv Inhibitor und Substrat schließen sich gegenseitig von der Bindung an das Enzym aus. Dies bedeutet jedoch nicht notwendigerweise, notwendigerweise, dass der Inhibitor an der gleichen Bindungsstelle bindet wie das Substrat. Auch wenn die Bindung von Substrat bzw. Inhibitor zu Konformationsänderung im Enzym führen, welche die Bindungsstelle für den jeweils anderen blockieren, ist die Hemmung kompetitiv. kompetitiv. Wenn Substrat und Inhibitor allerdings die gleiche Bindungsstelle haben, dann ist der Hemmtyp notwendig kompetitiv. Bei der Kompetitiven Hemmung kann der Inhibitor durch Substrat aus dem Enzym verdrängt werden, Vmax ändert sich also nicht. Allerdings wird für jede gewünschte Geschwindigkeit eine höhere [S] benötigt, die scheinbare Km wird also mit steigender [I] höher. Im Lineweaver-Burk-Diagramm führt dies zu einer Schar von Geraden, die einen gemeinsamen Schnittpunkt auf der y-Achse (1 / Vmax) haben. Unkompetitiv Der Inhibitor bindet nicht an das freie Enzym, sondern an den ES-Komplex. Höhere Konzentrationen des Substrates können daher den Hemmstoff nicht vom Enzym verdrängen, sondern führen zu vermehrter Bindung. Umgekehrt vermindert Bindung des Hemmstoffes die Konzentration von ES, nach dem Prinzip von Le Chatelier muss sich also zusätzliches ES aus E und S bilden: Die scheinbare Km vermindert sich, die Affinität des Enzymes für das Substrat steigt mit steigender [I]. Gleichzeitig nimmt natürlich Vmax ab. Im Lineweaver-BurkDiagramm finden wir eine Schar paralleler Geraden. Nicht-kompetitiv Der Inhibitor kann sowohl an E als auch an ES binden. Im einfachsten Fall ist dabei Ki = Kii, d.h. das die Substratbindung die Affinität des Enzymes für den Inhibitor nicht verändert, etwa durch Konformationsänderung. Dann folgt natürlich auch, dass die Bindung des Inhibitors die Affinität des Enzymes für das Substrat nicht ändert und Ks = Kss. Wegen des Zusammenhangs zwischen Ks und Km ändert die Bindung von Inhibitor also auch nicht Km. Es lässt sich nun zeigen (durch Substitution und Eliminierung aus den Definitionen von Ki,Kii,Ks und Kss), dass Ki / Kii = Ks / Kss. Wenn also Ki < Kii, dann folgt Ks < Kss und die scheinbare Km steigt mit [I]. Falls andererseits Ki > Kii, dann folgt Ks > Kss und die scheinbare Km sinkt mit steigendem [I]. Die nicht-kompetitive Hemmung führt im Lineweaver-Burk-Diagramm zu einer Schar von Geraden mit gemeinsamen Schnittpunkt links von der y-Achse, der Schnittpunkt liegt auf der x-Achse wenn Ki = Kii, er liegt über der x-Achse falls Ki < Kii und unter der x-Achse falls Ki > Kii.