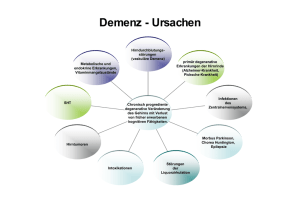
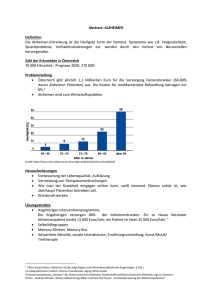
Morbus Alzheimer – Pharmakologische Therapiemöglichkeiten
Werbung

Diplomarbeit Morbus Alzheimer – Pharmakologische Therapiemöglichkeiten heute und in der Zukunft eingereicht von Lars Christian Alexander Haußer zur Erlangung des akademischen Grades Doktor(in) der gesamten Heilkunde (Dr. med. univ.) an der Medizinischen Universität Graz ausgeführt am Institut für Experimentelle und Klinische Pharmakologie unter der Anleitung von Univ.-Prof. Dr. med. univ. Josef Donnerer Graz, 19.10.2016 Eidesstattliche Erklärung Ich erkläre ehrenwörtlich, dass ich die vorliegende Arbeit selbstständig und ohne fremde Hilfe verfasst habe, andere als die angegebenen Quellen nicht verwendet habe und die den benutzten Quellen wörtlich oder inhaltlich entnommenen Stellen als solche kenntlich gemacht habe. Graz, am 19.10.2016 Lars Haußer eh i Danksagungen Zuerst möchte ich mich bei Herrn Univ.-Prof. Donnerer für die freundliche und gute Betreuung bei der Erstellung dieser Diplomarbeit bedanken. Weiter möchte ich mich bei meinen Eltern sowie meiner Schwester für ihre andauernde und unermüdliche Unterstützung bedanken, ohne die dieser Weg nicht möglich gewesen wäre. Zum Schluss möchte ich meinen Freunden, insbesondere Heide Hubertus sowie meiner Verlobten Johanna Grabka für ihre Unterstützung während des Studiums und beim Erstellen dieser Arbeit danken. ii Zusammenfassung Beim Morbus Alzheimer handelt es sich um eine primär degenerative, zerebrale Erkrankung mit progessiver Demenz, deren Prävalenz und Inzidenz ab dem 60. Lebensjahr deutlich zunimmt. Im Hinblick auf die steigende Lebenserwartung in der Bevölkerung sowie des demographischen Wandels ergibt sich daraus die Notwendigkeit wirksamer Therapien, um den Krankheitsverlauf zu beeinflussen. Ziel der Arbeit ist es einen Überblick über die derzeitigen pharmakologischen Therapiemöglichkeiten der Erkrankung zu geben sowie einen Ausblick auf mögliche zukünftige Therapien. Die Arbeit ist das Ergebnis einer Literaturrecherche in der ersten Hälfte des Jahres 2016, vor allem in der Datenbank PubMed. Darüber hinaus wurden Informationen aus Lehrbüchern, Fachinformationen der Hersteller sowie einigen anderen Quellen verwendet. Aktuell werden zur Behandlung der Demenz vom Alzheimer-Typ Wirkstoffe aus der Gruppe der Acetylcholinesterasehemmer sowie der NMDA- Rezeptorantagonist Memantin eingesetzt. Diese Präparate zeigen auf klinischen Messskalen eine kurzzeitige Besserung kognitiver Funktionen, können aber deren weiteren Verlust nicht aufhalten. In der fortgeschrittenen klinischen Erprobung befinden sich derzeit Substanzen vor allem aus dem Bereich der Immuntherapie, der BACE-1 Hemmer, TauAggregationshemmer, Ca2+-Kanalblocker sowie Serotoninrezeptorantagonisten. Einige dieser Substanzen zeigen sowohl in Tierversuchen als auch in den bisherigen klinischen Studien eine Reduzierung der Aβ-Produktion / Ablagerung im Gehirn. Allerdings lässt sich nur bei wenigen Substanzen bisher auch eine Verbesserung der kognitiven Funktion in den klinischen Studien nachweisen, und diese v. a. in einer Subgruppe der Patienten / Patientinnen, die an einer Demenz vom Alzheimer-Typ im leichten oder Prodromalstadium erkrankt sind, und die dann auch nur leicht ausfällt. iii Abstract Alzheimer’s disease is primarily a degenerative cerebral disease that is characterized by progressive dementia. Its prevalence and incidence increases significantly after the age of 60. Considering the increasing life expectancy and the accompanying demographic changes, a strong need for efficient therapies is evident. Aim of this work is to give an overview over currently available pharmacological treatment options, as well as on treatments possibly available in the future. This work is the result of a literature survey conducted during the first half of 2016, where most of the information was retrieved from PubMed. Additional information sources were textbooks, product informations provided by the manufacturers, as well as some additional sources. Currently, members of two substance groups are employed for treatment of Alzheimer’s dementia, namely inhibitors of the enzyme acetyl choline esterase and an antagonist of the NMDA receptor, Memantin. These medications lead to a short-term improvement of cognitive abilities, judged by means of standard clinical tests, however they are not able to stop the progressive cognitive decline characteristic for the disease. Some additional substances are currently in advanced clinical testing. Among them are immunotherapeutics, inhibitors of BACE-1 and inhibitors of tau aggregation, inhibitors of Ca channels, and antagonists of the serotonine receptor. Some of these substances lead to a promising reduction in Aß production and aggregation in the brain, both in animal experiments and in clinical testing. However, only a minority of these new substances leads to an improvement of cognitive abilities in clinical studies, and these effects are small and restricted to patients with early stages of Alzheimer’s disease. iv Inhaltsverzeichnis Eidesstattliche Erklärung...........................................................................................i Danksagungen..........................................................................................................ii Zusammenfassung..................................................................................................iii Abstract....................................................................................................................iv Inhaltsverzeichnis.....................................................................................................v Abkürzungen...........................................................................................................vii Abbildungen.............................................................................................................ix Tabellen....................................................................................................................x 1 Einleitung........................................................................................................1 1.1 Definition...........................................................................................1 1.2 Epidemiologie....................................................................................2 1.3 Ätiologie und Pathogenese...............................................................2 1.4 Risikofaktoren...................................................................................5 1.5 Klinik..................................................................................................5 1.6 Diagnostik..........................................................................................6 1.7 Weitere Neuropsychologische Testverfahren...................................8 1.8 Therapie............................................................................................9 2 Material und Methoden................................................................................13 3 Ergebnisse – Resultate................................................................................14 3.1 Aufbau und Erregungsleitung einer Nervenzelle............................14 3.1.1 3.2 Die Synapse...........................................................................14 3.1.1.1 Elektrische Synapsen.........................................................15 3.1.1.2 Aufbau chemischer Synapsen............................................15 3.1.1.3 Reiz-/Informationsübertragung an chemischen Synapsen.16 Acetylcholinesterase Hemmer........................................................18 3.2.1 Geschichte des Acetylcholin...................................................18 3.2.2 Synthese und Abbau von Acetylcholin...................................19 3.2.3 Acetylcholinesterasen............................................................19 3.2.4 Hemmung der Acetylcholinesterase.......................................20 3.2.5 Acetylcholinesterase-Hemmer bei der Alzheimer-Krankeit....21 3.2.6 Indikation................................................................................22 3.2.7 Übersicht................................................................................23 3.2.8 Wechselwirkungen.................................................................23 v 3.2.9 Nebenwirkungen.....................................................................24 3.2.10 Überdosierung........................................................................25 3.2.11 Donepezil - Allgemeines.........................................................25 3.2.12 Donepezil - Wirkmechanismus...............................................26 3.2.13 Donepezil - Pharmakokinetik..................................................27 3.2.14 Rivastigmin – Allgemeines.....................................................28 3.2.15 Rivastigmin – Wirkmechanismus...........................................28 3.2.16 Rivastigmin - Pharmakokinetik...............................................29 3.2.17 Galantamin – Allgemeines......................................................31 3.2.18 Galantamin - Wirkmechanismus............................................31 3.2.19 Galantamin – Pharmakokinetik..............................................32 3.2.20 Studien....................................................................................34 3.3 NMDA-Rezeptor Antagonist............................................................37 3.3.1 Der Neurotransmitter Glutamat..............................................37 3.3.2 Glutamatrezeptoren................................................................38 3.3.3 NMDA-Rezeptoren.................................................................38 3.3.4 NMDA-Rezeptoren und Alzheimer-Demenz..........................39 3.3.5 Memantin – Allgemeines........................................................41 3.3.6 Memantin – Indikation............................................................41 3.3.7 Memantin - Wirkmechanismus...............................................42 3.3.8 Memantin - Pharmakokinetik..................................................42 3.3.9 Memantin - Wechselwirkungen..............................................43 3.3.10 Memantin - Nebenwirkungen.................................................43 3.3.11 Memantin – Studien................................................................44 3.4 Ausblick...........................................................................................45 3.4.1 Immuntherapie........................................................................45 3.4.1.1 Passive Immuntherapie......................................................46 3.4.1.2 Aktive Immuntherapie.........................................................49 3.4.2 BACE-1 Hemmer....................................................................50 3.4.3 Tau-Aggregations Hemmer....................................................51 3.4.4 Ca2+-Kanalblocker...................................................................52 3.4.5 Serotoninrezeptoragonisten / -antagonisten..........................52 4 Diskussion.....................................................................................................54 5 Literaturverzeichnis.....................................................................................59 vi Abkürzungen 5-HT 5-Hydroxytryptamin Aβ β-Amyloid-Peptid ACh Acetylcholin AChE Acetylcholinesterase ADAS-Cog Alzheimer's Disease Assessment Scale – Cognitive Subscale ADCS Alzheimer's Disease Cooperative Study ADL Activities of Daily Living ADRDA Alzheimer's Disease and Related Disorders Association AMPA α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid AP Aktionspotential ApoE Apolipoprotein E APP Amyloid-Precursor-Protein ARIA Amyloid-related imaging abnormalities AUC Area under the curve BACE β-Site amyloid precursor protein cleaving enzyme BADLS Bristol Activities of Daily Living Score BDNF Brain-derived neurotrophic factor BuChE Butyrylcholinesterase CDR-SB Clinical Dementia Rating sum of boxes CIBIC-plus Clinican's Interview-Based Impression of Change-plus CRP C-reaktives Protein DAD Disability Assessment for Dementia DIAN-TU Dominantly Inherited Alzheimer Network Trial Unit EEG Elektroenzephalographie EOAD Early onset Alzheimer Disease FDA United States Food and Drug Administration GOT Glutamat-Oxalacetat-Transaminase G-Protein Guanosintriphosphat-bindendes Protein ICD International Statistical Classification of Diseases and Related Health Problems INR International Normalized Ratio MMST Mini Mental State Test vii MoCA Montreal Cognitive Assessment nACh- nikotinische Achetylcholin- NFT Neurofibrilläre Tangles NGF Nerve growth factor NINCDS National Institute of Neurological and Communicative Diseases and Stroke NMDA N-Methyl-D-Aspartat NPI Neuropsychiatric Inventory NTB Neuropsychological Test Battery PET Positronen-Emissions-Tomographie sAPPβ soluble amyloid precursor protein beta SIB Severe Impairment Battery SPECT Einzelphoton-Emissionscomputertomographie SSRI Selektive Serotonin-Wiederaufnahme-Hemmer TFDD Test zur Früherkennung von Demenzen mit Depressionsabgrenzung TSH Thyreoidea-stimulierendes Hormon USA Vereinigte Staaten von Amerika y-GT y-Glutamyltransferase ZNS Zentralnervensystem viii Abbildungsverzeichnis Abbildung 1: Schematische Darstellung der Freisetzung von ACh in den ................ synaptischen Spalt gefolgt von der Aktivierung des Rezeptors, .......... dem Abbau durch die AChE und der anschließenden .......................... Rückdiffusion, aus (100)....................................................................17 Abbildung 2: Schematische Darstellung des Abbau von ACh durch die AChE, ........ aus (97)..............................................................................................20 Abbildung 3: Chemische Struktur von Donepezil, aus (99)....................................26 Abbildung 4: Chemische Struktur von Rivastigmin, aus (101)...............................28 Abbildung 5: Chemische Struktur von Galantamin, aus (98)..................................31 Abbildung 6: Chemische Struktur von Memantin, aus (102)..................................41 Abbildung 7: Schematische Darstellung der Entstehung von Aβ bei der .................. Spaltung von APP durch β- und y-Sekretase, modifiziert nach ............ (103)...................................................................................................50 ix Tabellenverzeichnis Tabelle 1: AChE-Hemmer in der Therapie der Demenz vom Alzheimer-Typ (34–36) .................................................................................................................................23 Tabelle 2: Übersicht über die Wirksamkeit der AChE Hemmer aus (51)................35 x 1 Einleitung Unter dem Titel „Über eine eigenartige Erkrankung der Hirnrinde“ veröffentlichte Alois Alzheimer 1907 in der Allgemeinen Zeitschrift für Psychiatrie und psychischgerichtliche Medizin den ersten Fallbericht einer 51-jährigen Patientin, welche an der später nach ihm benannten Erkrankung litt (1). Mit ihren 51-Jahren handelte es sich dabei um eine verhältnismäßig junge Patientin. Heute ist bekannt, dass das Risiko, an einer Demenz vom Alzheimer-Typ zu erkranken, sich mit steigendem Lebensalter erhöht. So liegt bei den 56-70 Jährigen die Prävalenz der Erkrankung bei ca. 1-4% und verdoppelt sich ab diesem Zeitpunkt etwa alle 5 Lebensjahre (2). Die Demenz vom Alzheimer-Typ gewinnt daher sowohl durch die steigende Lebenserwartung, als auch durch die Zunahme des Durchschnittsalters der Bevölkerung, fortlaufend an Bedeutung. Die vorliegende Arbeit gibt im ersten Teil einen Überblick über die derzeit in der Therapie der Erkrankung eingesetzten Medikamente und im zweiten Teil einen Ausblick auf mögliche zukünftige Therapieoptionen. Als Ansatzpunkte zukünftiger Therapien, die auch in der Lage sind den Krankheitsverlauf zu beeinflussen, gelten vor allem die Entwicklung von Antikörpern gegen das β-Amyloid-Peptid (Aβ) sowie β-Sekretase-Inhibitoren. Einige dieser Substanzen befinden sich bereits seit einigen Jahren in verschiedenen Phasen klinischer Studien. 1.1 Definition Bei der Demenz vom Alzheimer-Typ handelt sich um eine primär degenerative zerebrale Erkrankung mit progredienter Demenz (3,4). Es treten für die Erkrankung charakteristische neuropatholgische, pathohistologische und neurochemische Merkmale auf (4,5). Gemäß ICD-10 ist die Demenz selber definiert als „ein Syndrom als Folge einer meist chronischen oder fortschreitenden Krankheit des Gehirns mit Störung vieler höherer kortikaler Funktionen, einschließlich Gedächtnis, Denken, Orientierung, Auffassung, Rechnen, Lernfähigkeit, Sprache und Urteilsvermögen. Das Bewusstsein ist nicht getrübt. Die kognitiven Beeinträchtigungen werden 1 gewöhnlich von Veränderungen der emotionalen Kontrolle, des Sozialverhaltens oder der Motivation begleitet, gelegentlich treten diese auch eher auf.“ (4). 1.2 Epidemiologie Die Demenzen stellen die häufigste Gruppe neurodegenerativer Erkrankungen dar. Von diesen können 60-80% der Untergruppe der Demenz vom Alzheimer-Typ zugeordnet werden. Wobei etwa 50% der an einer Demenz erkrankten Personen ausschließlich Merkmale der Demenz vom Alzheimer-Typ zeigen, bei den restlichen 10-30% zeigen sich sowohl Merkmale einer Demenz vom AlzheimerTyp als auch einer vaskulären Demenz (6,7). Weltweit waren 2014 ca. 40 Millionen Menschen von einer Demenz betroffen, wovon der Großteil der Erkrankten über 60 Jahre alt ist. Bis zum Jahre 2050 rechnen Forscher mit einer Verdoppelung der Krankheitsfälle alle 20 Jahre (8). In der Altersgruppe der über 60-jährigen zeigt sich gegenüber den jüngeren Mitmenschen eine deutliche Zunahme der Prävalenz und Inzidenz von Demenzen (2,6,9,10). So liegt die Wahrscheinlichkeit, eine Demenz zu entwickeln, für Personen die jünger als 50 Jahre sind bei 1:4000, wovon es sich bei etwa 30% um eine Demenz vom Alzheimer-Typ handelt (8). Bei den 56-70-jährigen hingegen beträgt die Prävalenz bereits etwa 1-4% und verdoppelt sich dann alle fünf Lebensjahre (2). Generell ist die Prävalenz bei Frauen höher als bei Männern, in den USA sind zum Beispiel 66% der erkrankten Personen weiblich. Dies ist jedoch vor allem auf eine höhere Lebenserwartung bei Frauen zurückzuführen. Korrigiert man die Werte um die unterschiedliche Lebenserwartung von Männern und Frauen, ergibt sich für beide eine gleich hohe Prävalenz (7). 1.3 Ätiologie und Pathogenese Bis heute ist die Ätiologie und Pathogenese der Erkrankung unklar. Es existieren lediglich verschiedene Hypothesen zur Entstehung. Die AmyloidkaskadenHypothese ist dabei die in den letzten 10-20 Jahren am meisten verfolgte. Diese geht von der Akkumulation des β-Amyloid-Peptids und der Bildung extrazellulärer Amyloid-Plaques als ursächliches pathologisches Ereignis aus. Alle weiteren Prozesse der Erkrankung werden als Folge dieses Ereignisses gewertet (6). 2 Aβ entsteht durch proteolytische Spaltung von APP durch sogenannte β- und γSekretasen. Bei der Spaltung entsteht hauptsächlich eine 40-Aminosäuren lange Form des Aβ (Aβ1-40). In geringerer Konzentration entstehen jedoch auch andere Formen, wovon vor allem die 42-Aminosäuren lange Form (Aβ1-42) zur Aggregation neigt (11,12). Eine Zunahme der Bildung von Aβ, eine Verschiebung des Verhältnisses von Aβ1-40 zu Aβ1-42 oder ein verminderter Abtransport / Abbau von Aβ führen zur Aggregation von Aβ1-42 und damit zur Ausbildung erster diffuser Ablagerungen. Im weiteren Verlauf kommt es vermehrt zur Bildung und Einlagerung von Aβ-Fibrillen sowie zu einer Entzündungsreaktion in deren Umfeld und damit zum Entstehen seniler Plaques. Diese beeinflussen die Phosphorylierung von Tau-Proteinen, welche normalerweise an die Mikrotubuli der Zellen gebunden sind, und die nun ebenfalls aggregieren und die sogenannte Alzheimer-Fibrillen ausbilden. Am Ende kommt es schließlich zu zellulären Funktionsstörungen, die zu einem Mangel an Neurotransmittern und zum Zelltod führen. Hier heraus ergeben sich die für die Demenz vom Alzheimer-Typ typischen Symptome (12–14). Gestützt wird diese Hypothese vorwiegend durch genetische Erkenntnisse, die bei familiär gehäuft auftretenden, früh einsetzenden Demenzen vom Alzheimer-Typ gewonnen werden konnten. In etwa 10% der Fälle von familiär gehäuft auftretenden, früh einsetzenden Demenzen vom Alzheimer-Typ liegt eine autosomal-dominat vererbbare Variante vor. Bei dieser konnten genetische Mutationen an einem der drei Gene APP, Presenilin-1 oder Presenilin-2 nachgewiesen werden (15). Mutationen am Preselin-1 und Preselin-2-Gen, die an der Bildung der γ-Sekretase beteiligt sind, führen zu einer erhöhten Bildung von Aβ1-42, Mutationen im APP-Gen zu einer Angiopathie der Hirngefäße mit abnormaler Ablagerung von Aβ in den kleinen Hirngefäßen (6). Darüber hinaus wurde bereits 1989 nachgewiesen, dass Aβ in Zellkulturen toxisch wirkt. Pieke et. all haben 1995 nachgewiesen, dass die toxische Wirkung von Aβ mit dem Level der Aggregation assoziiert ist (16,17). Ein Kritikpunkt an dieser Theorie ist jedoch, dass es keine Korrelation zwischen der Anzahl / dem Ausmaß an Aβ Plaques im Gehirn und dem Schweregrad einer Demenz gibt (11,17). In den letzten Jahren verlagert sich daher zunehmend die Forschung weg von den festen Aβ-Ablagerungen im Gehirn als Auslöser der Erkrankung hin zu löslichen Aβ-Formen mit niedrigerem Molekulargewicht (11,14). 3 Andere Studien stellen die Theorie auf, dass die gesteigerte APP- und AβProduktion eigentlich einen Kompensationsmechanismus darstellt, um eine anderweitig gestörte Zellfunktion aufrecht zu erhalten (11). Auch eine immunologische Funktion von Aβ konnte in Studien gezeigt werden. So besitzt Aβ in in-vitro Versuchen antimikrobielle Eigenschaften gegen Candia albicans, verschiedene Streptokokken-Spezies und weitere Erreger (18). Eine ältere Theorie geht davon aus, dass die Ursache für eine Demenz vom Alzheimer-Typ in einer primären Degeneration cholinerger Neurone mit nachfolgendem Acetylcholinmangel begründet ist. Bei Untersuchungen an Gewebeproben von Alzheimerpatienten und -patientinnen konnte gezeigt werden, dass bei ihnen die Aktivität der Acetylcholinesterase sowie der Cholinacetyltransferase reduziert ist. Viele der bisher zur Behandlung der Demenz vom Alzheimer-Typ eingesetzten Medikamente basieren auf dieser Theorie (12). Neben Aβ könnte auch das Tau-Protein eine zentrale Rolle in der Pathogenese der Demenz vom Alzheimer-Typ spielen. Das zweite histopathologische Merkmal der Alzheimer-Erkrankung, neben den Aβ-Plaques, sind intrazelluläre Aggregate des Tau-Proteins, die sogenannten Alzheimer-Fibrillen oder Neurofibrillären Tangels (NFT) (6). Beim Tau-Protein handelt es sich um ein Mikrotubuliassoziiertes Protein, welches zum einen an der Organisation und dem Zusammenbau der Mikrotubuli beteiligt ist, zum anderen jedoch auch am axonalen Transport von Zellorganellen, u. a. von Mitochondrien. In Neuronen findet sich Tau vor allem in den Axonen. Bekannt sind einige neurologische Erkrankungen, bei denen Tau eine Rolle spielt. Diese werden auch als Tauopathien bezeichnet. Das normalerweise ungefaltet vorliegende Tau-Protein bildet hyperphosphoryliert Fibrillen und formt NFT. In die entstandenen NFT kann weiteres, normales Tau gebunden werden. Durch die Beeinträchtigung der Funktion der Mikrotubuli sowie des axonalen Transportes kommt es zu einem Verlust von Synapsen und Neuronen (19). Wiederum eine andere Theorie geht davon aus, dass es in Folge eines Fehlens neurotropher Hormone, vor allem von NFG (nerve growth faktor) und BDNF (brain-derived neurotrophic factor), zu einer Abnahme der Wirkung des Acetylcholins kommt (12). 4 1.4 Risikofaktoren Epidemiologisch gesehen ist das zunehmende Lebensalter der Hauptrisikofaktor für das Auftreten einer Demenz vom Alzheimer-Typ. Darüber hinaus sind inzwischen jedoch auch einige weitere, zum Teil beeinflussbare und zum Teil genetisch bedingte Risikofaktoren bekannt. Zum einen gelten alle Risikofaktoren für kardiovaskuläre Erkrankungen auch als Risikofaktor für die Entwicklung einer Demenz vom Alzheimer-Typ. Dazu gehören vor allem Übergewicht, ungesunde Ernährung, Rauchen, körperliche Inaktivität, Bluthochdruck, Diabetes und erhöhte Cholesterinwerte. Des weiteren sind geistige Inaktivität, Depression sowie ein niedriger Bildungsstand als Risikofaktor bekannt (8,20). In Modellen zeigte sich jedoch, dass bei einem Wegfall der sieben wichtigsten, beeinflussbaren Risikofaktoren die Inzidenz der Demenz um lediglich 30% sinken würde (8). Hinzu kommt das Problem, dass in der Praxis zwar eine Reduktion dieser Risikofaktoren möglich sein dürfte, nicht jedoch eine vollständige Elimination. Genetisch gesehen ist das zur Zeit höchste Risiko, an einer sporadischen Form der Alzheimererkrankung zu erkranken, interessanterweise nicht wie bei den familiären, früh einsetzenden Formen auf einem Gen, das an der Bildung und dem Processing von APP beteiligt ist, zu finden, sondern am Apolipoprotein-E-Gen (Apoε). Dieses Gen zeigt einen Polymorphismus, wobei drei Genvarianten auftreten. Die Apoε3-Variante ist hiervon die am weitesten verbreitete. In den USA besitzen etwa 60% der Bevölkerung diese Variante (7). Die Apoε2-Variante scheint protektiv gegen das Auftreten einer Alzheimer-Erkrankung zu sein, wohingegen die Apoε4-Variante mit einem erhöhten Risiko verknüpft ist. So besitzen jene 3% der Bevölkerung, welche homozygot für Apoε4 sind, ein 85%-iges Risiko, an einer Demenz vom Alzheimer-Typ zu erkranken.Für heterozygote Apoε3-/Apoε4-Träger und Trägerinnen, welche etwa 25% der Bevölkerung ausmachen, beträgt das Erkrankungsrisiko immer noch etwa 45-50% (20). 1.5 Klinik Die ersten Anzeichen der Erkrankung sind zum Teil sehr unspezifischer Natur. Zu ihnen zählen Symptome wie Schwindel, Kopfschmerzen und Leistungsabfall. Erstes auffallendes und charakteristisches Symptom ist dann meistens eine Abnahme der Merkfähigkeit für neue Informationen. Die Betroffenen werden 5 vergesslich, verlegen alltägliche Gegenstände und finden sie nicht wieder (z.B. Haustürschlüssel), benötigen plötzlich Einkaufszettel und ähnliches. Darüber hinaus kommt es zu einer Abnahme der örtlichen Orientierung sowie zu Wortfindungsschwierigkeiten, sowohl im Gespräch als auch beim Schreiben. Themenwechsel fallen den Betroffenen schwer. Es treten Störungen im Denk- und Urteilsvermögen auf. Im Verlauf der Erkrankung verlieren die Betroffenen die Fähigkeit, alltägliche Aufgaben, wie z.B. kochen oder sich anzukleiden, auszuführen. Sie sind daher auch nicht mehr in der Lage, ihr Leben und den Haushalt so zu führen, wie sie es gewohnt sind. Neben der Abnahme der kognitiven Leistungsfähigkeit treten jedoch auch nicht kognitive Veränderungen auf, die bei der Therapie und Behandlung berücksichtigt werden müssen. Dazu zählen Symptome wie der soziale Rückzug, Antriebslosigkeit bis hin zur Depression, Unruhe, Schlafstörungen, aber auch Halluzinationen, Aggressionen und Wahnvorstellungen können auftreten. Zu bemerken ist, dass obwohl sich schon früh Störungen in der zeitlichen und örtlichen Orientierung zeigen und es zu einem zunehmenden Verlust der Merkfähigkeit kommt, die Orientierung zur eigenen Person und das Einhalten sozialer Umgangsformen lange erhalten bleibt (2,7). 1.6 Diagnostik Allgemein betrachtet handelt es sich bei der Demenz um eine Beschreibung eines klinisch definierten Syndroms. So handelt es sich gemäß ICD-10 bei einer Demenz um eine Störung höherer kognitiver Funktionen, wie z.B. Sprache, Orientierung, Rechnen oder des Gedächtnises, wobei jedoch keine Bewusstseinsstörung vorliegt und die Sinne an sich intakt sind (4). Grundsätzlich können verschiedene internistische, neurologische oder auch endokrinologische Erkrankungen zur Ausbildung einer Demenz führen, von denen einige gut behandelbar sind und die daher zwingend erkannt werden müssen. Sollte nach Erhebung einer Eigen- und ggf. einer Fremdanamnese der Verdacht auf eine Demenz bestehen, wird empfohlen als erstes die aktuelle kognitive Leistungsfähigkeit mittels einfacher Testverfahren grob zu quantifizieren. In der ärztlichen Praxis wird dafür häufig der sogenannte „Mini Mental State Test“ (MMST) in Kombination mit dem Uhrentest verwendet (21). 6 Dieser Test dient auch als Standard in Therapiestudien, um den Schweregrad einer Demenz einzuschätzen. So entspricht ein Wert von 20-26 Punkten im MMST einer leichten Demenz, ein Wert von 10-19 Punkten einer moderaten bzw. mittelschweren Demenz und ein Wert von < 10 Punkten einer schweren Demenz. Neben dem MMST eignen sich auch der DemTec, der Test zur Früherkennung von Demenzen mit Depressionsabgrenzung (TFDD) sowie der Montreal Cognitive Assessmant Test (MoCA) dazu, den ungefähren Schweregrad einer Demenz zu bestimmten (21). Beim DemTec handelt es sich um einen Test zur Bestimmung der kognitiven Leistungsfähigkeit. Er prüft die Fähigkeiten Merkfähigkeit, Wortflüssigkeit und Zahlen-Umwandeln (22). Der MoCA wurde als Screeninginstrument für Patienten / Patientinnen entwickelt, bei welchen nur eine leichte kognitive Einschränkung vorliegt. Die Anwendung ist, wie auch die des MMST, auch in der Hausarztpraxis nach kurzem Training möglich und benötigt etwa 10 Minuten Zeit (23). Beim TFDD handelt es sich ebenfalls um einen Screeningtest zur Früherkennung von Demenzen. Auch dieser Test lässt sich in der Hausarztpraxis in etwa 10 Minuten durchführen. Im Gegensatz zu den anderen erwähnten Testverfahren enthält dieser Test neben dem Testteil zur Erkennung einer Demenz einen zweiten Testteil, welcher die Abgrenzung einer Demenz von einer Pseudodemenz im Rahmen einer Depression ermöglicht (24). Alle diese Testverfahren besitzen jedoch den Nachteil, dass ihre Sensitivität im Frühstadium einer Demenz begrenzt ist und dass eine ätiologische Zuordnung der Demenz nicht möglich ist (21). Neben der Quantifizierung der kognitiven Leistungsfähigkeit sollte schon früh eine körperliche Untersuchung des Patienten/der Patientin durchgeführt werden, sowie ein psychopathologischer Befund erhoben werden. Hierdurch sollen andere Erkrankungen als Ursache der Demenz ausgeschlossen werden (z.B. Depression, Delir, Suchterkrankungen) und ggf. eine ätiologische Zuordnung der Demenz ermöglicht werden. Zu den weiteren standardmäßig empfohlenen Untersuchungen zählen Blutuntersuchungen, Liquordiagnostik und eine cerebrale Bildgebung (zum Ausschluss z.B. eines Subduralhämatoms, eines Tumors oder eines Normaldruckhydrocephalus sowie der Bestimmung vaskulärer Läsionen). Wichtige Parameter bei der Blutuntersuchung sind Blutbild, Elektrolyte, Blutzucker, TSH, CRP oder Blutsenkung, Leberfunktionswerte (GOT und y-GT), Nierenfunktionswerte (Kreatinin und Harnstoff) sowie die Bestimmung von Vitamin 7 B12 und Folsäure. Hierdurch sollen z.B. Vitaminmangelerkrankungen oder Stoffwechselerkrankungen als Ursache des Demenzsyndroms ausgeschlossen werden. Sollten sich bei diesen Untersuchungen keine Ursache für die Demenz finden lassen, wird eine Liquordiagnostik empfohlen. Im Rahmen dieser werden die notwendigen Parameter zum Ausschluss infektiöser oder entzündlicher Ursachen (z.B. einer Demenz im Rahmen einer Virusenzephalitis) der Demenz bestimmt. Darüber hinaus lässt sich im Liquor von an Alzheimer erkrankten Patienten/Patientinnen schon sehr früh die für diese Krankheit relevanten Parameter Aβ(1-42), Tau und Phosphor-Tau nachweisen. Dies ermöglicht schon früh eine ätiologische Zuordnung der Demenz. Aufgrund der erhöhten Sensitivität und Spezifität bei der Bestimmung aller drei Faktoren wird dies empfohlen. Weitere spezifische Untersuchungen Untersuchungsmethoden wie (SPECT/PET) EEG oder werden in nuklearmedizinische der Leitlinie nur bei bestehender unklarer Ätiologie der Demenz oder zum Ausschluss anderer Verdachtsdiagnosen empfohlen (21). 1.7 Weitere Neuropsychologische Testverfahren Neben den bereits erwähnten Testverfahren zur Quantifizierung der kognitiven Leistungsfähigkeit existieren noch eine Vielzahl weiterer Testverfahren zur Beurteilung der kognitiven Leistungsfähigkeit, des Verhaltens, sowie zur Beurteilung der Möglichkeit, Alltagsaktivitäten auszuüben. Einige dieser Testverfahren, welche auch in klinischen Studien zur Verlaufsbeurteilung verwendet werden, sollen hier noch etwas näher erläutert werden. Einer der am häufigsten in Studien verwendeten Tests zur Quantifizierung der kognitiven Leistungsfähigkeit ist der ADAS-Cog (Alzheimer's Disease Assessment Scale Cognition). Dieser Test besteht aus 11 Untertests und deckt die relevantesten Gebiete, in denen kognitive Einschränkungen im Rahmen einer Demenz auftreten, ab. Er besitzt eine besonders hohe Sensitivität für Veränderungen, die im Verlauf der Erkrankung auftreten, und ist besonders geeignet zur Anwendung bei Patienten / Patientinnen im moderaten Stadium der Erkrankung. Die Durchführung benötigt mit ca. 40 Minuten jedoch relativ viel Zeit, weshalb er vorwiegend in Studien und weniger im klinischen Alltag Verwendung findet (23,25). Da der ADAS-Cog und auch der MMST im frühen Stadium der Demenz vom AlzheimerTyp nicht ausreichend sensitiv ist, um Veränderungen in der kognitiven 8 Leistungsfähigkeit zu erkennen, wurde besonders für diese Gruppe von Patienten / Patientinnen der NTB (Neuropsychological Test Battery) entwickelt. Er besteht aus 9 Untertests zur kognitiven Funktion und benötigt ebenfalls etwa 40 Minuten zur Durchführung (25). Äquivalent zum NTB existiert für Patienten / Patientinnen, die sich im schweren Stadium der Erkrankung befinden, mit der SIB (Severe Impairment Battery) ein besonders geeignetes Testverfahren zur Beurteilung der kognitiven Fähigkeiten. Bei diesem Test werden die noch erhaltenen Fähigkeiten beurteilt (25). Es existieren ebenfalls eine Vielzahl an Tests, die die Fähigkeiten, Aktivitäten des täglichen Lebens (ADL) auszuüben, beurteilen. Die ADCS (Alzheimer's Disease Cooperative Study) hat 45 verschiedene Alltagskompetenzen für die Nutzung in klinischen Studien im Rahmen der Alzheimer-Erkrankung getestet. 27 von diesen Alltagskompetenzen haben sich als geeignet erwiesen, um die Alltagskompetenz von Patienten / Patientinnen zu quantifizieren. Es existieren verschiedene Varianten des ADCSADL, je nach Stadium der Erkrankung (25). Weitere Testverfahren, die in diesem Bereich verwendet werden, sind zum Beispiel die aus 20 Punkten bestehende BADLS (Bristol Activities of Daily Living Scale) (23) sowie die DAD-Skala (Disability Assesment for Dementia). Dieser Test basiert auf einem Interview der Angehörigen / Pflegenden, inwieweit Unterstützung bei der Planung, dem Beginn sowie der Organisation von 10 Tätigkeiten aus den Bereichen Basisfähigkeiten, instrumentelle Fähigkeiten und Freizeitaktivitäten notwendig ist, und wie effektiv die Ausübung ist (25). Zur Beurteilung des Verhaltens eignet sich zum Beispiel das NPI (Neuropsychiatric Inventory), welches typische Verhaltensauffälligkeiten, die im Rahmen einer Demenz auftreten, erfasst (23). Schlussendlich existieren auch noch sehr komplexe Testformen, welche mehrere Bereiche wie kognitive Fähigkeiten, Alltagskompetenzen und Verhalten in einem Test testen. Hierzu zählt zum Beispiel der CIBIC-plus (Clinican's Interview-Based Impression of Changeplus). Der Test besteht aus einem Teil für den Patienten / die Patientin als auch einem für dessen / deren Angehörigen / Pflegenden. Er eignet sich besonders gut, um den Verlauf in verschiedenen Bereichen zu beurteilen (23). 1.8 Therapie Obwohl die Erstbeschreibung der Erkrankung inzwischen über 100 Jahre her ist, gibt es trotz intensiver Forschungsbemühungen bis heute keine Therapie, welche 9 in der Lage ist, das Fortschreiten der Erkrankung aufzuhalten oder zu verlangsamen. Alle derzeitig verfügbaren Therapiekonzepte und Medikamente zielen auf eine vorübergehende Besserung von Alltagskompetenzen und kognitiven Fähigkeiten der Erkrankten sowie der Unterstützung des Patienten / der Patientin und dessen / deren Angehörigen ab. Die Therapie setzt sich zusammen aus einer pharmakologischen Behandlung sowie psychosozialer Intervention, die sowohl dem Betroffenen / der Betroffenen als auch dessen / deren Angehörigen im Umgang mit der Erkrankung helfen soll. Im Rahmen der pharmakologischen Acetylcholinesterase-Hemmer Therapie (AChE-Hemmer) werden Donepezil, vor allem die Rivastigmin und Galantamin sowie der NMDA-Rezeptorantagonist Memantin eingesetzt. Diese wirken positiv auf die Fähigkeit zur Verrichtung von Alltagsaktivitäten, die kognitive Funktion sowie auf den ärztlichen Gesamteindruck (21). Darüberhinaus besteht die pharmakologische Therapie aus der Linderung psychischer Begleitsyndrome wie psychotischen Störungen, Aggression, Agitation und Depressionen. In der Leitlinie der Deutschen Gesellschaft für Neurologie wird die Behandlung leichter und mittelschwerer Demenzen mit einem AChE-Hemmer empfohlen. Die Auswahl des Präparates richtet sich dabei vorwiegend nach dem Neben- bzw. Wechselwirkungsprofil des Medikamentes sowie dessen Verträglichkeit. Außerdem spielen Applikationsart und anfallende Kosten eine Rolle. Da die Wirkung dieser Medikamente von der verabreichten Dosis abhängig ist, wird eine Aufdosierung, abhängig von der Verträglichkeit, bis zur maximalen Tagesdosis empfohlen. Obwohl die AChE-Hemmer zurzeit keine Zulassung zur Behandlung der Demenz vom Alzheimer-Typ im schweren Stadium besitzen, wird ihr Einsatz in der Leitlinie trotzdem empfohlen. Hierbei ist jedoch zu beachten, dass es sich um einen sogenannten Off-Label-Use handelt (21). Zur Behandlung schwerer Formen der Demenz vom Alzheimer-Typ wird primär der NMDA-Rezeptor Antagonist Memantin empfohlen, im moderaten Stadium kann dieser ebenfalls eingesetzt werden, wenn aus irgendwelchen Gründen keine Behandlung mit einem AChE-Hemmer erfolgen kann (21). Im leichten Stadium der Erkrankung konnte keine Besserung bei der Bewältigung von Alltagstätigkeiten und nur geringe Effekte auf die kognitive Funktion durch Memantin nachgewiesen werden, weshalb hier der Einsatz nicht empfohlen wird. Im mittelschweren Stadium konnte zwar ein positiver Effekt gezeigt werden, 10 allerdings zeigte sich bei einer Add-on-Therapie mit Memantin keine Überlegenheit gegenüber einer Einzeltherapie mit Donepezil. Lediglich im schweren Stadium der Alzheimer-Erkrankung ist aus Studien bekannt, dass eine Kombinationstherapie aus Memantin und Donepezil einer Monotherapie mit Memantin oder Donepezil überlegen ist (21). Für die ebenfalls häufig in der Behandlung eingesetzten und beworbenen GinkgoPräparate gibt es laut der Leitlinie keine Empfehlung. Die Studienlage für Ginkgopräparate ist insgesamt sehr heterogen, und ein positiver Effekt im Hinblick auf die Bewältigung von Alltagsaktivitäten existiert lediglich und auch nur fraglich beim Einsatz hoher Dosen (21). Neben der pharmakologischen Therapie ist eine weitere wichtige Säule in der Therapie die psychosoziale Intervention, die im Rahmen dieser Arbeit jedoch nur kurz erwähnt werden soll. Hier kommen Verfahren wie kognitives Training, Ergotherapie, Realitätsorientierung, körperliche Aktivierung, multisensorische Verfahren und vor allem Angehörigenarbeit zum Einsatz. Wie schon erwähnt geht es bei der Therapie der Alzheimer-Erkrankung, neben der Therapie der primären Symptome der Demenz, auch um die Linderung von psychischen Verhaltenssyndromen, die im Laufe der Erkrankung auftreten können. Zur pharmakologischen Therapie von psychotischen Symptomen wie Wahnvorstellungen und Halluzinationen sowie zur Therapie von Agitation und Aggression wird vor allem Risperidon empfohlen. Zur reinen Therapie von Aggression kann darüber hinaus auch Haloperidol verwendet werden. Bei dem Einsatz anderer Präparate aus der Gruppe der Psychopharmaka, wie Aripiprazol, Carbamazepin und Citalopran, handelt es sich wieder um einen sogenannten OffLabel-Use, der aber generell erwogen werden kann. Bei allen Präparaten sollte die Anwendungsdauer so kurz wie möglich sein. Als nicht medikamentöse Behandlungsmethoden von Agitation und Aggression wird rezeptive Musik sowie Aromatherapie empfohlen. Primär sollte versucht werden, Depressionen durch strukturierte Freizeitaktivitäten zu verbessern, und nur bei unzureichender Besserung sollten Antidepressiva eingesetzt werden. Aus der Gruppe der Antidepressiva sollten ausschließlich Präparate ohne anticholinerge Wirkung verwendet werden (21). Zwei weitere oft auftretende Begleitsyndrome, die allerdings in der Regel nicht medikamentös therapiert werden müssen, sind Störungen des Tag-Nacht11 Rhythmus sowie mangelnde Nahrungsaufnahme. Zur Verbesserung des TagNacht-Rhythmus wird eine Strukturierung des Alltages empfohlen. Bei mangelnder Nahrungsaufnahme sollen Betroffene verbale Unterstützung erhalten und eine familiäre Esssituation hergestellt werden (21). 12 2 Material und Methoden Die vorliegende Arbeit verfolgt das Ziel, einen kompakten Überblick über die derzeitigen pharmakologischen Therapieoptionen der Alzheimer-Erkrankung zu geben sowie einen Ausblick auf mögliche zukünftige Therapien, welche sich aktuell in klinischer Erprobung befinden. Bei der durchgeführten Arbeit handelt es sich um eine Literaturrecherche. Die Datenerhebung erfolgte vorwiegend anhand einer systematischen Suche in der Literaturdatenbank PubMed (National Center for Biotechnology Information, U.S. National Library of Medicine, U.S.A.). Ausgehend von Review-Artikeln wurde dabei immer spezifischer in die Thematik weiter recherchiert. Darüber hinaus wurden Informationen aus den Suchmaschinen Google und Google scholar, Lehrbüchern aus den insbesondere Fachinformationen der Pharmakologie, der Arzneimittel-Hersteller, Neurowissenschaften und Genetik, aus dem Bestand der Bibliotheken der Medizinischen Universität Graz sowie der Westfälischen-Wilhelms-Universität Münster entnommen. Informationen über aktuell laufende Studien sind darüber hinaus der Datenbank clinicaltrials.gov entnommen. Die Empfehlungen zur Diagnostik und Therapie im Einleitungsteil der Arbeit sind der aktuellen S3 Leitlinie Demenz der Deutschen Gesellschaft für Neurologie entnommen. 13 3 Ergebnisse – Resultate 3.1 Aufbau und Erregungsleitung einer Nervenzelle Eine Nervenzelle (Neuron) besteht aus einem als Soma oder Perikaryon bezeichneten Zellkörper mit einem relativ großen Zellkern und einer unterschiedlich großen Anzahl an Zellfortsätzen. Diese werden je nach ihrer Funktion als Dendriten oder als Axon bezeichnet (26). Bei den Dendriten handelt sich um meist weit verzweigte Zellfortsätze, die der Aufnahme von Informationen dienen und diese zum Perikaryon leiten. Das Axon hingegen ist ein Zellfortsatz, welcher Informationen vom Perikaryon wegleitet. Es wird auch als Nervenfaser bezeichnet. Im Gegensatz zu den Dendriten ist das Axon weniger stark verzweigt, und jedes Neuron besitzt nur ein Axon. Um eine schnelle Erregungsleitung in dem bis zu einem Meter langen Axon zu gewährleisten, ist dieses von einer von Oligodendrozyten gebildeten Markscheide umhüllt, welche das Axon elektrisch gegen die Umgebung isoliert. Diese Umhüllung wird alle 2-3 mm durch sogenannte Ranvier-Schnürringe unterbrochen (26,27). Meistens lassen sich eine größere Anzahl von Axonen nach ihrer morphologischen Erscheinung zu einer größeren Nervenbahn, einem sogenannten Tractus, zusammenfassen (27). Die Dendriten erhalten ihre Informationen von den Enden der Axone anderer Neuronen. Diese Kontaktstelle, die von Dendrit und Axon gebildet wird, wird als Synapse bezeichnet (26,27). An den Synapsen findet die elektrische oder biochemische Weiterleitung von Signalen statt. 3.1.1 Die Synapse Das Ende eines Axons (Axonterminale) bildet mit der nachfolgenden Zelle eine Synapse aus. Synapsen dienen generell der Reiz-/Informationsweitergabe von einem Neuron zu einem weiteren oder einer anderen Zelle (z.B. Muskelzelle). Das Axonterminale bildet das sogenannte Synapsenendknöpfchen und damit die präsynaptische Seite der Synapse. Die postsynaptische Seite wird von einem Dendrit oder dem Zellkörper der nachfolgenden Zelle gebildet. Ein Reiz wird immer von der präsynaptischen zur postsynaptischen Seite weitergeleitet. 14 Zwischen den Membranen der prä- und postsynaptischen Seite befindet sich ein schmaler Spalt, der als synaptischer Spalt bezeichnet wird (27). Anhand ihrer Funktionsweise lassen sich grundsätzlich zwei Arten von Synapsen unterscheiden. Es existieren zum einen die von Edwin Furshpan und David Potter 1959 nachgewiesenen elektrischen Synapsen, zum anderen die sogenannten chemischen Synapsen. Chemische Synapsen leiten elektrische Impulse nicht direkt, sondern indirekt, durch die Ausschüttung chemischer Botenstoffe, den Neurotransmittern, weiter. Diese chemische Weiterleitung von Nervenimpulsen wurde bereits 1921 von dem Grazer Pharmakologen Otto Loewi nachgewiesen. Im reifen Gehirn des Menschen stellen chemische Synapsen die überwiegende Anzahl der Synapsen dar (28). 3.1.1.1 Elektrische Synapsen Elektrische Synapsen spielen im Rahmen der Demenz vom Alzheimer-Typ und der Therapie dieser Krankheit keine Rolle und werden daher hier nur der Vollständigkeit halber kurz erwähnt. Diese Art der Synapsen dient der direkten Übertragung von Ionenströmen von einer Zelle zur nächsten und wird auch als gap junctions bezeichnet. Im Gegensatz zu den chemischen Synapsen ist der synaptische Spalt sehr schmal (ca. 3nm) und wird von Proteinen (Connexine) durchzogen, die durch die beiden Zellen gebildet werden. Je sechs Connexine der beiden Zellen bilden dabei den als gap junction bezeichneten Ionenkanal. Aufgrund der Tatsache, dass hier direkt Ionenströme weitergeleitet werden, erfolgt die Reizübertragung sehr schnell. Ein wichtiger Unterschied zwischen elektrischen und chemischen Synapsen besteht darin, dass aufgrund der Tatsache, dass die Ionenkanäle bidirektional sind, im Gegensatz zu chemischen Synapsen eine Reizweiterleitung in beide Richtungen erfolgen kann. So kann ein in der Folgezelle ausgelöstes Aktionspotenzial auch wieder in die Ursprungszelle zurück geleitet werden (28). 3.1.1.2 Aufbau chemischer Synapsen Wie bereits erwähnt stellen chemische Synapsen den überwiegenden Anteil der Synapsen im reifen menschlichen Gehirn dar. Diese verwenden zur Informationsübertragung über den mit 20-50nm relativ breiten synpatischen Spalt 15 verschiedene Neurotransmitter. Als Neurotransmitter dienen eine Vielzahl unterschiedlicher chemischer Stoffe aus der Gruppe der Aminosäuren, Amine und Peptide. Im Zusammenhang mit der Therapie bei einer Demenz vom AlzheimerTyp sind aktuell vor allem die beiden Neurotransmitter Acetylcholin (ACh) und Glutamat sowie deren zugehörige Rezeptoren von Interesse. Bei den chemischen Synapsen werden die entsprechenden Neurotransmitter im präsynaptischen Axonterminale synthetisiert und in Vesikeln gespeichert. In der postsynpatischen Membran befinden sich passende Neurotransmitterrezeptoren. Bei diesen Rezeptoren handelt es sich entweder um ligandenabhängige Ionenkanäle oder um G-Protein gekoppelte Rezeptoren (28). 3.1.1.3 Reiz-/Informationsübertragung an chemischen Synapsen Für die Reiz-/Informationsübertragung an chemischen Synapsen ist der Ablauf verschiedener Schritte notwendig. Zuerst muss die Synthese des entsprechenden Neurotransmitters im Neuron/Axonterminale erfolgen. Anschließend muss der synthetisierte Neurotransmitter in Vesikel verpackt und zur präsynaptischen Membran transportiert werden. Dort erfolgt die Freisetzung in den synaptischen Spalt beim Eintreffen eines Aktionspotenzials. Anschließend muss der Neurotransmitter an einen entsprechenden Rezeptor an der postsynaptischen Membran andocken und dort eine elektrische oder chemische Reaktion auslösen. Schlussendlich muss die Entfernung des Neurotransmitters aus dem synaptischen Spalt erfolgen. Die verschiedenen Schritte bieten dabei unterschiedliche Möglichkeiten, den Prozess und damit die Reizübertragung pharmakologisch zu beeinflussen, daher werden die einzelnen Schritte hier etwas genauer betrachtet. Die Synthese kleiner Neurotransmitter (Aminosäuren und Amine) findet direkt im Cytosol des Axonterminale statt. Anschließend werden sie durch spezielle Transporter in Vesikel aufgenommen. Größere Neurotransmitter aus der Gruppe der Proteine werden hingegen im Soma des Neurons produziert, anschließend vom Golgi-Apparat in Vesikel verpackt und zum Axonterminale transportiert. Trifft am Axonterminale ein AP ein, kommt es zur Öffnung spannungsabhängiger Ca 2+Kanäle in der präsynaptischen Membran. Der hier heraus resultierende Ca 2+Einstrom führt zur Verschmelzung der Vesikelmembran mit der präsynaptischen Membran und so zur Freisetzung des Neurotransmitters in den synaptischen Spalt. Bei länger anhaltender Erregung werden „Reserve“-Vesikel vom Zytoskelett 16 als „Nachschub“ in Richtung präsynaptischer Membran transportiert. Nach der Freisetzung des Inhaltes des Vesikels wird seine Membran durch Endocytose zurückgewonnen und kann erneut mit dem Neurotransmitter gefüllt werden. An der postsynaptischen Membran lassen sich zwei Arten von Rezeptoren, an die der Neurotransmitter binden kann, unterscheiden. Zum einen handelt es sich dabei um ligandengesteuerte Ionenkanäle. Sie führen zu einer Rückumwandlung des chemischen Signals in ein elektrisches Signal. Im Gegensatz zu anderen Ionenkanälen sind diese nicht ionenselektiv sondern durchgängig für mehrere Ionen (z.B. Na+, K+, Ca2+). Über ligandengesteuerte Ionenkanäle erfolgt die schnelle synaptische Signalübertragung. Des weiteren existieren G-Protein gekoppelte Rezeptoren. Diese ermöglichen eine Vielzahl an postsynaptischen Aktionen. Nach Bindung des Transmittermoleküles an den Rezeptor aktiviert dieser sogenannte G-Proteine. Die aktivierten G-Proteine können dann ihrerseits entweder Ionenkanäle öffnen oder über die Aktivierung von Proteinen, welche second-messenger synthetisieren, komplexe zelluläre Signalkaskaden auslösen. Abschließend muss der Neurotransmitter wieder aus dem synaptischen Spalt entfernt werden, um eine Übererregung zu vermeiden. Hierfür existieren zwei Möglichkeiten: zum einen die Rückdiffusion durch die präsynaptische Membran, wo dann entweder die Wiederverwendung oder der Abbau des Transmitters erfolgt. Der Vorgang der Diffusion wird dabei häufig durch spezifische aktive Transporter unterstützt. Die zweite Möglichkeit ist die Spaltung / der Abbau des Transmitters direkt im synaptischen Spalt (28). Abbildung 1: Schematische Darstellung der Freisetzung von ACh in den synaptischen Spalt gefolgt von der Aktivierung des Rezeptors, dem Abbau durch die AChE und der anschließenden Rückdiffusion, aus (97) 17 In der Abbildung 1 wird dieser Vorgang zum besseren Verständnis noch einmal schematisch dargestellt. 3.2 Acetylcholinesterase-Hemmer 3.2.1 Geschichte des Acetylcholin Der Neurotransmitter Acetylcholin wurde 1862 erstmals synthetisiert, und einige Jahrzehnte später erfolgte 1913 durch Sir Henry Dale der erste Nachweis von natürlich vorkommendem Acetylcholin. Zu dieser Zeit gab es jedoch noch keine Hinweise darauf, dass Acetylcholin im menschlichen Organismus eine größere Rolle spielen würde, und so nahm das Interesse an diesem Stoff in den folgenden Jahren erst einmal ab. Erst durch einen Versuch des Pharmakologen Otto Loewi von der Universität Graz stieg das Interesse ab 1921 wieder an. Otto Loewi hatte in einem Experiment ein Froschherz inkl. innervierender Nerven präpariert und in eine Nährlösung gelegt. Anschließend hatte er den Vagusnerv des Frosches gereizt und beobachtet, dass die Herzfrequenz abnahm. Nun entnahm er etwas von der das Herz umgebenden Nährlösung und ließ diese über ein zweites, denerviertes Herz laufen. Nachdem auch dieses Herz daraufhin langsamer schlug, vermutete er, dass ein chemischer Stoff an der Übertragung vom Nerven auf das Herz beteiligt ist und nannte diesen „Vagusstoff“ (29). In mehreren weiteren Versuchen konnte nachgewiesen werden, dass es sich bei dem Vagusstoff um einen Cholinester handelt, welcher durch eine Esterase schnell hydrolisiert und damit abgebaut wird. Er kam allerdings nicht auf den Gedanken, dass es sich hierbei um Acetylcholin handeln könnte. Der Nachweis, dass der von Otto Loewi gefundene Vagusstoff und Acetylcholin dieselben Stoffe sind, wurde erst Jahre später von Henry Dale erbracht. Die Untersuchung des Acetylcholins gestaltete sich zu der Zeit auch deshalb schwierig, weil der Stoff schnell wieder abgebaut wurde. Erst in den folgenden Jahren konnte durch Einsatz von Physostigmin, einem Acetylcholinesterase-Hemmer, die physiologische Wirkung von Acetylcholin detaillierter untersucht werden (29). 18 3.2.2 Synthese und Abbau von Acetylcholin Bei Acetylcholin handelt es sich um einen Essigsäureester des Cholins. Die Synthese erfolgt im Cytosol der Axonterminale aus Cholin und Acetyl-CoA durch das Enzym Cholinacetyltransferase. Das für die Synthese notwendige Cholin wird durch spezifische Transportproteine aus der extrazellulären Flüssigkeit aufgenommen, das benötigte Acetyl-CoA stammt aus dem Citratzyklus. Da die Menge des produzierten Acetylcholin von der Menge des vorhandenen Cholins abhängig ist, ist dessen Aufnahme der geschwindigkeitsbestimmende Schritt der Synthese. Nach Freisetzung von Acetylcholin wird dieses durch die Acetylcholinesterase zu Cholin und Acetat hydrolisiert. Der größte Teil des dabei entstehenden Cholins wird dann durch Transporter erneut in das Axonterminale aufgenommen und kann dort wieder zur Synthese von Acetylcholin verwendet werden (30). 3.2.3 Acetylcholinesterasen Acetycholin wird im Acetylcholinesterase, menschlichen aber auch Organismus von der zum einen von der Pseudoacetylcholinesterase (Butyrylcholinesterase) abgebaut. Während die Acetylcholinesterase vorwiegend in Nervenzellen, Muskelzellen und in der Membran von Erythrozyten vorkommt, findet sich die Butyrylcholinesterase (BuChE) vorwiegend in der Leber und im Serum. Im Gegensatz zur AChE handelt es sich bei der BuChE um eine „unspezifische“ Cholinesterase, welche eine Vielzahl an Cholinestern hydrolysiert. Ihr Name ist darauf zurückzuführen, dass sie Butyrylcholin (ein im menschlichen Organismus natürlicherweise nicht vorkommender Cholinester) schneller spaltet als Acetylcholin (31). Bei der AChE handelt es sich um ein für Acetylcholin hoch spezifisches Enzym aus der Gruppe der Serinhydrolasen. Bezeichnend für Serinhydrolasen im Allgemeinen ist das Vorkommen der Aminosäure Serin im katalytischen Zentrum des Enzyms, welches für die Hydrolyse verantwortlich ist. Neben Serin sind an der Bildung der katalytischen Triade bei der AChE noch die Aminosäuren Glutamat und Histidin beteiligt (31). 19 Wie auch aus der Abbildung 2 zu entnehmen ist, kann das aktive Zentrum des Enzyms in zwei Unterzentren unterteilt werden. Zum einen besitzt die AChE ein anionisches Zentrum, zum anderen ein esteratisches Zentrum. Das anionische Zentrum bindet an den positiv geladenen quartären Stickstoff des Cholinanteils von Acetylcholin. Der Essigsäurerest wird dann durch das esteratische Zentrum vom Cholinrest abgespalten. Es entsteht intermediär ein kovalenter Acetyl-EnzymKomplex und freies Cholin. In einem zweiten Schritt findet anschließend eine nucleophile Reaktion von Wasser mit dem gebildeten Acetyl-Enzym-Komplex statt. Hierbei wird die Essigsäure vom Enzym abgespalten und somit das Enzym wieder regeneriert (31,32). 3.2.4 Hemmung der Acetylcholinesterase Aufgrund der Bedeutung des Acetylcholins als Neurotransmitter hat auch die Hemmung der AChE eine große Bedeutung. Wird die AChE gehemmt, kann freigesetztes Acetylcholin nicht mehr abgebaut werden, die Folge ist eine Erhöhung der Acetylcholinkonzentration im synaptischen Spalt und ggf. eine Dauererregung der postsynaptischen Acetylcholinrezeptoren. Ein Überangebot an Acetylcholin führt zu einer Vielzahl an Symptomen, bis hin zur sogenannten cholinergen Krise. Zu den Symptomen zählen: Bradykardie, eine Erhöhung des Muskeltonus im Magen-Darm-Trakt mit Durchfall und Krämpfen, Faszikulationen, Miosis, ein gesteigerter Speichelfluss, erhöhtes Schwitzen, gesteigerte 20 Bronchialsekretion, Bronchokonstriktion, Muskelschwäche und Atemlähmung (20,33). Die Hemmstoffe der AChE können grob in die zwei Gruppen, die der irreversiblen und die der reversiblen Hemmstoffe unterteilt werden. Bei den irreversiblen Hemmstoffen handelt es sich um Phosphorsäureester oder Thiophosphorsäureester. Ihre therapeutische Anwendung beschränkt sich auf die Augenheilkunde, wo sie im Rahmen der Glaukomtherapie Verwendung finden. Ihre Hauptanwendung besitzen sie jedoch als Insektizid und Pestizid sowie, aufgrund der hohen Toxizität vieler Phosphorsäureester, auch als chemischer Kampfstoff („Nervengas“) (31–33). Phosphorsäureester binden analog zum Acetylcholin am esteratischen Zentrum der AChE. Wie auch beim Acetytcholin wird ein Substituent (X) vom Phosphoratom abgespaltet. Es bildet sich ein kovalenter Phosphorsäure-Enzym-Komplex, welcher im Vergleich zum AcetylEnzym-Komplex jedoch stabiler ist. Bis zu diesem Zeitpunkt ist die Hemmung reversibel. Der Phosphorsäurerest kann langsam wieder vom Enzym abgespalten werden und die Acetylcholinesterase ihre Funktion wieder ausüben. Wird allerdings vorher vom Phosphorsäurerest ein weiterer Substituent abgespalten, bildet sich ein irreversibler Phosphorsäure-Enzym-Komplex (31,32). Zu den reversiblen Hemmstoffen zählen die unter anderem ebenfalls als Insektizid eingesetzten Carbaminsäurederivate. Sie finden aber auch vielfach in der Medizin Anwendung, zum Beispiel bei Myasthenia gravis, Glaukom, Lewy-Body-Demenz, Parkinson, postoperativem anticholinergem Syndrom oder verspätetem Erwachen, sowie bei Darm- und Blasenatonie. Im Gegensatz zur Hemmung der AChE mit Phosphorssäureestern wird der Carbamylrest nach ca. 30-40 Minuten wieder vom Enzym abgespalten, und das vorher gehemmte Enzym kann seine Funktion wieder ausüben (31,33). 3.2.5 Acetylcholinesterase-Hemmer bei der Alzheimer-Krankeit Der erste zur Therapie der Demenz vom Alzheimer-Typ zugelassene AChEHemmer war Tacrin (Handelsname: Cognex und Romotal), welcher 1993 auf den Markt kam. Obwohl in Studien sich eine Besserung kognitiver Funktionen zeigte, war sein Nutzen aufgrund starker Nebenwirkungen und hepatotoxischer Wirkung eingeschränkt. Heute sind die drei Acetylcholinesterasehemmer Donepezil, 21 Rivastigmin und Galantamin zur Behandlung der Alzheimer-Krankheit im frühen und mittleren Stadium zugelassen und in Verwendung. 3.2.6 Indikation Alle derzeit auf dem Markt befindlichen AChE-Hemmer zur Therapie der Demenz vom Alzheimer-Typ sind hier lediglich zur symptomatischen Behandlung im frühen und mittleren Stadium (entsprechend einem MMSE bis 10 Punkte) der Erkrankung zugelassen (34–36). In den USA und anderen Ländern hingegen sind AChEHemmer auch für die Behandlung der Demenz vom Alzheimer-Typ im schweren Stadium zugelassen. Die in Deutschland aktuell gültige S3-Leitlinie „Demenz“ der DGPPN und DGN weist ebenfalls darauf hin, dass es eine Evidenz für den Einsatz von AChE-Hemmern auch im schweren Stadium der Erkrankung gibt, und empfiehlt sowohl die Weiterbehandlung als auch den Beginn einer Behandlung mit AChE-Hemmern bei Patienten / Patientinnen im schweren Stadium der Demenz vom Alzheimer-Typ. Sie weist jedoch auch darauf hin, dass es sich hierbei um einen Off-Label-Use handelt, mit den entsprechenden daraus resultierenden Schwierigkeiten (21). 22 3.2.7 Übersicht Tabelle 1: AChE-Hemmer in der Therapie der Demenz vom Alzheimer-Typ (34–36) Wirkstoff Donepezil Rivastigmin Galantamin retard Dosierung 5mg/d, nach einem Monat ist eine Steigerung bis auf 10mg/d möglich 2x 1,5mg/d, nach mind. 2 Wochen ist eine Steigerung auf 2x 3mg/d möglich. Bei guter Verträglichkeit weitere Steigerung bis auf 2x 6mg/d 8mg/d, nach einem Monat ist eine Steigerung auf 16 mg/d. Möglich. Nach einem weiteren Monat ggf. steigern bis auf 24mg/d Einnahme 1x täglich 2x täglich 1x täglich Metabolismus CYP3A4 / 2D6 AChE / BuChE CYP3A4 / 2D6 HWZ ca. 70 – 80 Stunden ca. 1 Stunde ca. 7 Stunden Dosisanpassung bei Nicht erforderlich Niereninsuffizenz Nicht erforderlich (individuelle Verträglichkeit) Schwer: keine Anwendung Dosisanpassung bei Individuelle Leberinsuffizenz Verträglichkeit (leicht bis mittel) Individuelle Verträglichkeit Mittel: Beginn mit 8mg/d alle 2 Tage. Im Verlauf maximal auf 16mg/d steigern Dosisanpassung bei Keine Daten Leberinsuffizenz (schwer) Keine Daten Keine Anwendung Bemerkung Auch als transdermales Pflaster erhältlich (Wechsel alle 24h) Die Tabletten mit sofortiger Wirkstofffreisetzung wurden inzwischen durch retardierte Tabletten ersetzt 3.2.8 Wechselwirkungen Alle AChE-Hemmer besitzen aufgrund ihrer Wirkweise Wechselwirkungen mit anderen Arzneimitteln, die ebenfalls den Acetylcholinstoffwechsel oder die Acetylcholinrezeptoren beeinflussen, sowie mit weiteren Arzneimitteln mit cholinerger oder anti-cholinerger Wirkung. Dies sind zum Beispiel die im Rahmen von Narkosen eingesetzten Muskelrelaxantien, welche die durch Acetylcholin vermittelte neuromuskuläre 23 Übertragung blockieren. Die Wirkung des polarisierenden Muskelrelaxans Succinylcholin wird Muskelrelaxantien dabei (z.B. verstärkt, Pancuronium, die Wirkung Rucoronium depolarisierender und andere) wird abgeschwächt. Hierzu zählen auch β-Blocker, welche eine Wirkung auf die kardiale Erregungsleitung haben. Hier kann die vagotone Wirkung der AChEHemmer verstärkt werden, wodurch eine erhöhte Gefahr für das Auftreten von Bradykardien entsteht (34–38). Bei Donepezil und Galantamin bestehen zusätzlich Wechselwirkungen mit anderen Arzneimitteln, welche als Inhibitoren oder Induktoren an den P450 Isoenzymen wirken, vor allem der Isoenzyme CYP3A4 und CYP2D6. Inhibitoren wie Makrolidantibiotika (z.B. Erythromycin), orale Azol-Antimykotika (z.B. Ketoconazol), Cimetidin und einige weitere führen zu erhöhten Plasmaspiegeln. Enzyminduktoren wie Rifampicin, Carbamazepin und weitere führen zu erniedrigten Plasmaspiegeln. Da die genaue Stärke der Veränderung der Plasmaspiegel für die meisten Präparate unbekannt ist, wird dazu geraten, solche Kombinationen nur mit entsprechender Vorsicht zu verwenden (34,36,37). Rivastigmin hingegen wird nicht über das P450-Isoenzymsystem metabolisiert, so dass hier keine Wechselwirkungen zu erwarten sind (35,38). 3.2.9 Nebenwirkungen Viele häufige Nebenwirkungen, die bei der Einnahme von AChE-Hemmern auftreten, lassen sich durch die Hemmung der AChE im vegetativen Nervensystem erklären. Die Folge der Hemmung ist eine gesteigerte Aktivität des Parasymphathikus. Dies führt unter anderem zu den sehr häufig, vor allem zu Beginn der Therapie und während der Dosissteigerung, auftretenden gastrointestinalen Nebenwirkungen wie Krämpfe, Übelkeit, Erbrechen und Diarrhö. In einem Großteil der Fälle klingen diese Beschwerden jedoch nach kurzer Zeit wieder ab, und eine vorübergehende symptomatische Behandlung der Nebenwirkungen ist ausreichend. Weitere häufige Nebenwirkungen der AChEHemmer sind verminderter Appetit, Halluzinationen, Depression, Synkopen, Schwindel, Tremor, Kopfschmerzen, Somnolenz, Lethargie, Hypertonie, Bradykardien, vermehrtes Schwitzen, Muskelkrämpfe, Müdigkeit, Harninkontinenz, Harnwegsinfekte, Atemwegsinfekte, Ausschlag und Juckreiz (34–36). 24 Bei Patienten / Patientinnen, die mit Reminyl (Galantamin) behandelt worden sind, gab es darüber hinaus vereinzelte Fälle verschiedener schwerer Hauterkrankungen während der Behandlung. Im letzten Jahr sind daher das Stevens-Johnson-Syndrom, die akute generalisierte exanthematische Pustulose und das Erythema multiforme als seltene Nebenwirkungen mit in die Arzneimittelinformation aufgenommen worden. Der Hersteller empfiehlt Patienten / Patientinnen, denen Galantamin verschrieben wird, auf die Gefahr von Hautreaktionen hinzuweisen und das Medikament bei Auftreten sofort abzusetzen, solange ein Zusammenhang der Hautsymptome mit dem Medikament nicht sicher ausgeschlossen werden kann (39). 3.2.10 Überdosierung Bei einer Überdosierung von AChE-Hemmern besteht die Gefahr, eine cholinerge Krise zu entwickeln. Hierbei finden sich als Folge der Zunahme von Acetylcholin im Organismus die Schweißausbrüche, folgenden verstärkter Symptome: Übelkeit, Speichelfluss, Miosis, Erbrechen, Diarrhö, Bronchokonstriktion, Bradykardie, Muskelschwäche ggf. bis zur Atemlähmung, Muskelzuckungen, Kollaps und Krämpfe. Neben unterstützenden Maßnahmen je nach Symptomen bietet sich die Gabe von Atropin als Antidot an (34–37). Die Hersteller empfehlen inital eine Dosis von 12mg Atropin i.v. bei Donepezil, von 0,03mg/kgKG für Rivastigmin und von 0,5-1mg Atropin bei Galantamin. Weitere Dosen werden nach der klinischen Wirksamkeit gegeben (34–36). 3.2.11 Aufgrund Donepezil - Allgemeines der Nebenwirkungen und der dadurch eingeschränkten Anwendungsmöglichkeit der beiden bekannten AChE-Hemmer Neostigmin und Tacrin hat man sich auf die Suche nach besser verträglichen Wirkstoffen gemacht. Dabei wurden verschiedene Indanon-Derivate auf ihre Wirksamkeit getestet. Ein N-Benzylpiperazin zeigte in Tierversuchen eine vielversprechende Wirkung. Nach Austausch der N-Benzylperazin Gruppe durch N-Benzylpiperidin konnte die hemmende Wirkung weiter gesteigert werden. Nach weiteren Modifikationen zeigte sich das Indanon-/Piperidin-Derivat Donepezil-hydrochlorid als 25 erfolgversprechend. Die chemische Struktur von Donepezil wird in Abbildung 3 dargestellt. Abbildung 3: Chemische Struktur von Donepezil, aus (99) 1996 erhielt Donepezil als Acetylcholinesterasehemmer zur Behandlung der leichten und moderaten Form der Alzheimer-Demenz von der FDA die Zulassung. Donepezil unterscheidet sich, neben der deutlich besseren Verträglichkeit, vor allem durch seine sehr hohe Selektivität für die AChE von Tacrine. Darüber hinaus besitzt es mit 70 – 80 Stunden eine deutlich längere Halbwertszeit als andere AChE-Hemmer (40). 3.2.12 Donepezil - Wirkmechanismus Donepezil wirkt als reversibler und hochselektiver Hemmstoff der Acetylcholinesterase, und mit einer deutlich geringeren Wirkung auch als Hemmstoff der Butyrylcholinesterase. Die Wirkung auf die vorwiegend im Zentralnervensystem vorkommende AChE ist dabei etwa 1000mal stärker als auf die vorwiegend peripher vorkommende BuChE (34). Dies führt zu deutlich weniger Nebenwirkungen als sie bei AChE-Hemmern mit geringerer Selektivität, wie zum Beispiel Tacrine, auftreten. Die Enzymhemmung erfolgt vorwiegend durch Bindung von Donepezil ausserhalb des aktiven Zentrums der AChE (nicht-kompetitive Hemmung). Dies führt zu einer Abnahme der Wechselzahl und somit zu einer Abnahme der maximalen Reaktionsgeschwindigkeit der AChE. Zu einem geringeren Anteil bindet Donepezil darüber hinaus auch am aktiven Zentrum der AChE (kompetetive Hemmung) und steht in direkter Konkurrenz zum Acetylcholin. In diesem Fall ist eine Abnahme der Katalysegeschwindigkeit und eine Erhöhung der Michaelis-Konstante die Folge (40,41). 26 3.2.13 Donepezil - Pharmakokinetik Donepezil wird nach oraler Aufnahme rasch resorbiert. Die Resorption ist unabhängig von der Tageszeit und einer evtl. Nahrungsaufnahme (40), trotzdem empfiehlt der Hersteller eine Einnahme kurz vor dem Schlafengehen (34). Nach oraler Einnahme wird die maximale Plasmakonzentration je nach Quelle nach ca. 2,4-4,4 Stunden (40) bzw. 3-4 Stunden (34) erreicht. Unmetabolisiertes Donepezil ist nach der Resorption im Plasma zu 95% an Plasmaproteine gebunden (34). Die durchschnittliche Halbwertzeit des Wirkstoffes beträgt 70 – 80 Stunden und ist damit deutlich länger als die anderer AChE-Hemmer. Sie ist jedoch abhängig vom Alter des Patienten / der Patientin; so beträgt sie bei jungen Patienten / Patientinnen ca. 60 Stunden und bei älteren Patienten / Patientinnen bis zu 104 Stunden. Ein Vorteil der langen Halbwertszeit bestand vor allem, bevor weitere Einnahmeformen (Galantamin retard oder transdermale Rivastigminpflaster) auf den Markt kamen, darin, dass bei Donepezil im Gegensatz zu den anderen AChEHemmern eine einmalige Einnahme am Tag ausreichend war. Drei Wochen nach dem Beginn der regelmäßigen Einnahme einer Einzeldosis von 5mg Donepezil pro Tag wird ein Kumulationsgleichgewicht erreicht, unter dem die Hemmung der AChE 63,4% - 77% beträgt. Nach Erreichen des Kumulationsgleichgewichts gibt es keinen Unterschied der Clearance des Medikaments zwischen jüngeren und älteren Personen mehr, daher ist eine Dosisanpassung für Patienten / Patientinnen im höheren Alter nicht erforderlich. Die Metabolisierung von Donepezil erfolgt in der Leber vorwiegend über das CYP450 Isoenzym CYP3A4 sowie über das Isoenzym CYP2D6. Das Isoenzym CYP2D6 spielt jedoch eine geringere Rolle. Zu den bekannten Metaboliten gehören zum einen das ebenfalls pharmakologisch aktive 6-O-DesmethylDonepezil, zum anderen die Metaboliten Donepezil-cis-N-oxid und 5-ODesmethyl-Donepezil, welche jedoch keine pharmakologische Wirkung besitzen. 11-17% des verabreichten Stoffes wurden in Studien unverändert über die Nieren ausgeschieden und konnten im Urin nachgewiesen werden (34,40). Untersuchungen mit radioaktiv markiertem Donepezil zeigten, dass ein großer Anteil (57%) der verabreichten Radioaktivität im Urin und nur ein geringer Anteil (14%) im Stuhl nachgewiesen werden konnte. Es wird daher angenommen, dass 27 die Ausscheidung von Donepezil und seiner Metaboliten vorwiegend renal erfolgt (34). 3.2.14 Rivastigmin – Allgemeines O N O N H Abbildung 4: Chemische Struktur von Rivastigmin, aus (100) Rivastigmin ist der zweite der drei zugelassenen AChE-Hemmer zur Behandlung der leichten und mittelschweren Form der Demenz vom Alzheimer-Typ. Die Erstzulassung erfolgte 1998 in Europa und zwei Jahre später auch in den USA unter dem Handelsnamen Exelon (42,43). Neben Zubereitungen zur oralen Einnahme sind von Rivastigmin als einzigem AChE-Hemmer derzeit auch transdermale Pflaster zugelassen. Diese haben gegenüber der oralen Einnahme den Vorteil einer geringeren Rate an Nebenwirkungen, insbesondere an Übelkeit und Erbrechen, durch eine niedrigere und langsamer erreichte maximale Plasmakonzentration, sowie eine bessere Compliance der Patienten / Patientinnen (44). 3.2.15 Rivastigmin – Wirkmechanismus Bei Rivastigmin handelt es sich um ein Carbamyl-Derivat, welches zu einer im Prinzip reversiblen Hemmung der AChE und der BuChE führt (35). Da die Hemmung der AChE jedoch deutlich länger anhält, als der Wirkstoff im Plasma nachgewiesen werden kann, wird sie auch als pseudo-irreversible Hemmung bezeichnet. Die Hemmung der im ZNS vorkommenden AChE beträgt bei gesunden Erwachsenen etwa 40%. Die Hemmung der vorwiegend peripher vorkommenden BuChE beträgt ca. 10% (45). Im Liquor von 14 Patienten / Patientinnen, welche an einer Demenz vom Alzheimer-Typ erkrankt waren, konnten keine wesentlichen Unterschiede in der Hemmung der AChE und der 28 BuChE festgestellt werden (35). Rivastigmin besitzt damit eine geringere Selektivität für AChE als Donepezil. Allerdings konnte in Tierversuchen gezeigt werden, dass Rivastigmin die AChE verstärkt in den von Alzheimer besonders betroffenen Hirnregionen, dem Cortex und dem Hippocampus, hemmt. Außerdem hemmt es von den verschiedenen AChE-Isoformen vor allem die globuläre G1Isoform, welche die im Gehirn von Erkrankten, die an einer Demenz vom Alzheimer-Typ leiden, vorherrschende Isoform ist. Die Hemmung der G1-Isoform ist etwa 4-6 stärker als die der G4-Isoform (45). Von der AChE existieren im menschlichen Körper verschiedene Isoformen. Zum einen eine asymmetrische Form und die vier globulären Formen G1-G4. Die Isoform G4 ist die im Zentralnervensystem von gesunden Erwachsenen vorherrschende Form, sie nimmt allerdings mit zunehmendem Alter im Neocortex und Hippocampus ab. Die G1-Isoform hingegen ist zwar im Zentralnervensystem weniger stark vertreten, allerdings bleibt sie auch im Alter konstant. Die Abnahme der G4-Form ist im Gehirn von Patienten / Patientinnen, die an der Alzheimer-Erkrankung erkrankt sind, zusätzlich beschleunigt, so dass es zu einer stärkeren Verschiebung des Verhältnisses der G1- zur G4-Isoform kommt (45). Rivastigmin bindet ähnlich dem Acetylcholin sowohl an das esteratische Zentrum als auch an das anionische Zentrum der AChE (45). Es steht damit in direkter Konkurrenz zum Acetylcholin und führt somit wieder zu einer Abnahme der Katalysegeschwindigkeit. Wie Acetylcholin wird auch Rivastigmin von der AChE hydrolisiert, und es entsteht ein Phenol-Derivat. Im Unterschied zum Acetylcholin, wo der Acetyl-Rest schnell von der AChE abgespalten wird, bleibt der vom Rivastigmin übertragene CarbamylRest deutlich länger an der AChE gebunden und verhindert so eine schnelle Regenerierung des Enzyms. In in-vitro Studien mit Hirngewebe von Ratten zeigte sich, dass es über 24 Stunden bis zur Reaktivierung der carbamylierten AChE dauert (45). 3.2.16 Rivastigmin - Pharmakokinetik Rivastigmin wird nach oraler Aufnahme rasch und fast vollständig resorbiert (35,45). Die maximale Plasmakonzentration wird bereits etwa eine Stunde nach Einnahme erreicht. Die Einnahme zusammen mit einer Mahlzeit verzögert die Resorption und das Erreichen der maximalen Plasmakonzentration um etwa 90 Minuten und verringert die maximale Plasmakonzentration um etwa 30% (35). 29 Durch die verringerte maximale Plasmakonzentration sinkt das Risiko von Nebenwirkungen, insbesondere von gastrointestinalen Nebenwirkungen wie Übelkeit und Erbrechen, weswegen empfohlen wird, Rivastigmin zusammen mit einer Mahlzeit einzunehmen (35,45). Nach der Resorption unterliegt Rivastigmin einem starken First-Pass Effekt, so dass die Bioverfügbarkeit nur bei etwa 36 % (± 13%) liegt. Im Plasma liegt Rivastigmin zu 40% an Plasmaproteine gebunden vor und besitzt eine Halbwertszeit von etwa einer Stunde. Trotz der relativ kurzen Halbwertszeit hemmt Rivastigmin die AChE für neun Stunden, so dass eine zweimalige Einnahme am Tag ausreichend ist (35). Bei Patienten / Patientinnen mit einer Demenz vom Alzheimer-Typ verlängert sich die Hemmung der AChE gegenüber gesunden Menschen auf etwa 12 Stunden. Ein Kumulationsgleichgewicht wird bereits nach Einnahme der zweiten Dosis erreicht (45). Die Metabolisierung erfolgt sowohl peripher als auch zentral vorwiegend über eine durch die Cholinesterasen vermittelte Hydrolyse. Das Cytochrom P450 Isoenzymsystem ist im Gegensatz zum Abbau von Donepezil und Galantamin am Abbau von Rivastigmin nicht oder nur zu einem sehr geringen Teil beteiligt (35,45). Der erste Schritt des Abbaus ist die durch Cholinesterase vermittelte Hydrolyse unter Bildung des decarbamylierten Metaboliten. Darauf folgt entweder eine NDemethylierung, eine Sulfat-Konjugation oder beides. Der erste Metabolit besitzt nur noch eine minimale hemmende Wirkung auf die AChE (etwa 10% des ursprünglichen Medikamentes). Die Ausscheidung erfolgt nach Metabolisierung fast ausschließlich renal, im Stuhl ist weniger als 1% der ursprünglich verabreichten Dosis nachweisbar (35,45). Transdermale Pflaster Wie bereits erwähnt sind von Rivastigmin neben den oralen Zubereitungen seit 2007 auch transdermale Pflaster auf dem Markt. Die Pflaster können für 24 Stunden auf der Haut bleiben und geben in dieser Zeit die Hälfte Ihres Wirkstoffes in die Blutbahn des Patienten ab. Ein Pflaster mit 18mg Rivastigmin setzt etwa 9,5mg in 24 Stunden frei, was aufgrund des nicht vorhandenen First-Pass Effekts einer oralen Einnahme von 12mg/Tag entspricht. Ein wesentlicher Vorteil der transdermalen Gabe gegenüber der oralen Gabe ist die Reduzierung gastrointestinaler Nebenwirkungen, insbesondere Übelkeit und Erbrechen. Dies 30 lässt sich durch eine später erreichte und vor allem deutlich niedrigere maximale Plasmakonzentration erklären. So wird bei der transdermalen Gabe die maximale Plasmakonzentration erst nach 8 Stunden erreicht und ist um 70% niedriger als bei oraler Einnahme (44). 3.2.17 Galantamin – Allgemeines Bei Galantamin handelt es sich um ein ursprünglich aus der Zwiebel des Woronow-Schneeglöckchens (Galanthus woronowii) isoliertes, inzwischen jedoch synthetisch gewonnenes, tertiäres Alkaloid. Neben seiner selektiven, hemmenden Wirkung auf die Acetylcholinesterase wirkt es, als einziges Präparat, auch modulierend auf nikotinische AcetylcholinRezeptoren und verstärkt dadurch die Wirkung des ACh auf diese Rezeptoren (46). 3.2.18 Galantamin - Wirkmechanismus Beim Galantamin lassen sich zwei Wirkmechanismen unterscheiden. Zum einen die Hemmung der Acetylcholinesterase, zum anderen ein modulierender Effekt auf nikotinische Acetylcholinrezeptoren (46). Galantamin konkurriert am aktiven Zentrum der AChE um die Bindung von Acetylcholin. Es bewirkt so eine selektive, kompetitive und reversible Hemmung 31 der AChE (36,46). In in-vitro Versuchen zeigte sich, dass die hemmende Wirkung von Galantamin auf die AChE etwa 53-mal stärker war als auf die BuChE. Dies führt wiederum zu einer besseren Verträglichkeit des Medikaments im Vergleich zu weniger selektiven Hemmstoffen (46). Darüber hinaus wirkt Galantamin modulierend auf nikotinische ACh-Rezeptoren im Gehirn (46). Bei diesen Rezeptoren handelt es sich Transmembranrezeptoren, welche als ligandengesteuerte Ionenkanäle fungieren. Der Rezeptor ist aus vier verschiedenen Untereinheiten aufgebaut (α, β, γ und δ), wobei die Peptidkette der α-Untereinheit die Bindungsstelle für das Acetylcholin darstellt (30). nAChRezeptoren sind unter anderem an der Funktion des Langzeitgedächtnisses beteiligt, eine Abnahme der Rezeptoren führt zu einer Abnahme der Gedächtnisleistung und der Aufmerksamkeit (47,48). Seit längerem ist bekannt, dass es bei an einer Demenz vom Alzheimer-Typ erkrankten Patienten / Patientinnen zu einer Abnahme der nACh-Rezeptoren im Gehirn kommt (46–49), und es konnte gezeigt werden, dass der Verlust dieser Rezeptoren mit der Stärke der kognitiven Beeinträchtigung der Erkrankten korreliert (47). Besonders gut untersucht und pharmakologisch interessant im Rahmen der AlzheimerErkrankung ist die vor allem im Hippocampus und präfrontalen Kortex vorkommende α7-Isoform des Rezeptors (49). Präsynaptische α7-nACh- Rezeptoren sind an der Ausschüttung anderer Neurotransmitter beteiligt, postsynaptische α7-nACh-Rezeptoren lösen über die Bildung von secondmessenger Molekülen unter anderem Signalwege aus, die an der Gedächtnisfunktion beteiligt sind, sowie neuroprotektive Wirkung besitzen (48,49). Galantamin ist in seiner Wirkung nicht selektiv für α7-nACh-Rezeptoren, es wirkt aber auch auf diese. Dieser zweite Wirkmechanismus könnte insbesondere in der Kombination mit dem NMDA-Rezeptorantagonisten Memantin von Interesse sein (50). 3.2.19 Galantamin – Pharmakokinetik Auch Galantamin wird nach oraler Einnahme rasch resorbiert. Die maximale Plasmakonzentration wird nach 0,9 bis 2,0 Stunden erreicht. Die Bioverfügbarkeit des Wirkstoffes ist hoch. Die Einnahme zusammen mit einer Mahlzeit verlangsamt die Resorption deutlich, so dass sich die Zeit bis zum Erreichen der maximalen Plasmakonzentration verdoppelt. Die Plasmabindung ist mit etwa 18 % gering, 32 und die Halbwertszeit mit etwa 7 Stunden kurz (46). Galantamin wird auf mehreren Wegen metabolisiert, wobei der primäre Weg das Cytochrom P450 Isoenzymsystem in der Leber darstellt (36,46), hiervon vor allem die Isoenzyme CYP2D6 und CYP3A4 (36). Zu den Metaboliten von Galantamin zählen Norgalantamin, O-Desmethyl-galantamin, O-Desmethyl-norgalantamin, Epigalantamin und Galantaminone (46). In in-vitro Studien besitzen einige dieser Metaboliten ebenfalls eine hemmende Wirkung auf die AChE, welche jedoch in vivo keine weitere Bedeutung besitzt (36,46). 18-22 % des verabreichten Wirkstoffs wurden in Untersuchungen unverändert über die Niere ausgeschieden und konnten innerhalb von 24 Stunden nach Verabreichung im Urin nachgewiesen werden. Bei Untersuchungen mit 4mg radioaktiv markiertem Galantamin konnte der überwiegende Anteil (90-97%) der verabreichten Radioaktivität innerhalb von 7 Tagen im Urin und nur ein geringer Anteil (2.2%-6.3%) im Stuhl nachgewiesen werden (36). Die Ausscheidung von Galantamin und seinen Metaboliten scheint demnach vorwiegend renal zu erfolgen. Galantamin retardierte Tabletten Neben Tabletten mit sofortiger Wirkstofffreisetzung existieren vom Galantamin auch retardierte Tabletten mit dem Vorteil, dass trotz der kurzen Halbwertszeit des Wirkstoffs eine Einnahme einmal am Tag ausreichend ist. Nach Gabe der retardierten Tabletten wird die maximale Plasmakonzentration nach ca. 4,4 Stunden erreicht und ist um 24 % erniedrigt im Vergleich zur Einnahme nichtretardierter Tabletten. Die AUC ist bioäquivalent (36). 33 3.2.20 Studien J. Grimley Evans et al. veröffentlichten 2004 im International Journal of Neuropsychopharmacology unter dem Titel „Evidence-based pharmacotherapy of Alzheimer's disease“ einen Review, in dem unter anderem die Wirksamkeit der AChE-Hemmer anhand der zu diesem Zeitpunkt vorhandenen Studien evaluiert wurde. In der Auswertung beachtet wurden 32 Placebo-kontrollierte, randomisierte Studien. Eingeschlossen in die Studien wurden Patienten / Patientinnen, die an einer Demenz vom Alzheimer-Typ gemäß der NINCDS-ADRA Kriterien erkrankt waren. Bis auf zwei Studien betrachteten alle dabei lediglich Patienten / Patientinnen, die an einer milden bis moderaten Form der Erkrankung erkrankt waren (das entspricht einem Punktewert von 10 oder 11 bis 24 oder 26 im MMSE). Die Autoren geben hierfür als Gründe an, dass zum einen diese Patientengruppe vermutlich den größten Nutzen von der Therapie habe, zum anderen existieren bei Patienten im schweren Stadium der Erkrankung ethische Schwierigkeiten, die aus der Unmöglichkeit einer ausreichenden Aufklärung bei der Gabe von Medikamenten mit dem Risiko von Nebenwirkungen resultieren. Die allermeisten Studien betrachten die Wirksamkeit des jeweiligen AchE-Hemmers in Bezug auf die weitere Abnahme bzw. Verbesserung der kognitiven Funktion. Der Beobachtungszeitraum ist mit 3-6 Monaten jedoch in den meisten durchgeführten Studien relativ kurz (51). Im Review wurden die Daten aller relevanten Studien mit einem Beobachtungszeitraum von 6 Monaten gepoolt. 34 Tabelle 2: Übersicht über die Wirksamkeit der AChE Hemmer aus (51) Wie der Tabelle 2 zu entnehmen ist, gab es bei Betrachtung des ADAS-Cog einen signifikanten Vorteil für die Therapie mit AChE-Hemmern bei allen drei Präparaten. Bei der Verwendung des MMST als Messinstrument zeigte sich für Donepezil und Rivastigmin ebenfalls ein signifikanter Nutzen. In den mit Galantamin durchgeführten Studien wurde der MMST nicht erhoben. Die DAD-Skala (Dissability Assesment for Dementia scale) wurde in einer Studie mit Donepezil und in zwei Studien mit Galantamin erhoben. Hier fand sich eine signifikante Besserung nur für die Anwendung von Donepezil. Ein ähnliches Ergebnis lieferte die in jeweils einer Studie verwendete NPI (Neuropsychiatric Inventory). Auch hier konnte lediglich für die Gabe von Donepezil eine signifikante Besserung gezeigt werden nicht aber für die Anwendung von Galantamin. Die Autoren betonen, dass sich aus diesen Daten keine Empfehlung für ein bestimmtes Präparat ableiten lasse, da alle Präparate ähnliche Daten liefern und das Studiendesign nicht darauf abzielte, einen Vergleich zu den anderen Präparaten zu liefern. Daten hierfür könnten nur aus direkten Vergleichsstudien kommen. Zwei kleine Vergleichstudien (Bullock et al. (2001) und Passmore et al. (2002)) hätten Donepezil mit Rivastigmin (Bullock et al.) sowie Donepezil mit Galantamin (Passmore et al.) verglichen und waren zu dem Ergebnis gekommen, dass Donepezil den anderen beiden Präparaten im Hinblick auf die Einfachheit der Nutzung und dem Auftreten gastrointestinaler Nebenwirkungen überlegen wäre, 35 jedoch nicht in Bezug auf die kognitive Funktion sowie auf Aktivitäten des täglichen Lebens (ADL) (51). Winblad et al. (2006) führten in Schweden eine Studie mit 248 Patienten / Patientinnen, die an einer Demenz vom Alzheimer-Typ im schweren Stadium litten (MMST 1-10), durch. Ziel war es, die Effektivität von Donepezil bei Patienten / Patientinnen im schweren Stadium der Alzheimer Demenz zu beurteilen, insbesondere in Hinblick auf die Kognition sowie auf Aktivitäten des täglichen Lebens. Von 128 Patienten / Patientinnen in der Verumgruppe beendeten 95 Patienten / Patientinnen den Beobachtungszeitraum von 6 Monaten. Nach 6 Monaten zeigte sich eine statistisch signifikante Verbesserung im SIB (Severe Impairment Battery) und eine geringere Abnahme im ADCS-ADL-severe gegenüber der Placebogruppe (52). Kontrovers diskutiert wurden die Ergebnisse der AD2000 Studie. Hierbei handelte es sich um die erste Studie zur Langzeitwirkung von Donepezil bei der Demenz vom Alzheimer-Typ. Zu Beginn nahmen 565 Patienten / Patientinnen an einer 12 wöchigen „run-in“ Phase teil. Anschließend wurden die 486 Patienten / Patientinnen, die diese Phase abgeschlossen hatten, in eine Verumgruppe und eine Placebogruppe randomisiert. Primäre Endpunkte der Studie waren die Einweisung in eine Pflegeeinrichtung oder der Verlust von zwei Basisfähigkeiten oder von 6 instrumentellen Fähigkeiten auf der BADLS (Bristol Activities of Daily Living Scale). In der Studie zeigte sich, dass es keinen signifikanten Unterschied in der Dauer bis zur Aufnahme in eine Pflegeeinrichtung und keinen signifikanten Unterschied im Hinblick auf den Verlust von instrumentellen Fähigkeiten und der Zunahme von Behinderungen gab. Wie auch in anderen Studien zeigte sich jedoch ein signifikanter Vorteil im Hinblick auf Kognition im MMST. Die Autoren leiten daraus ab, dass Donepezil in der Studie für die Betroffenen keinen relevanten Nutzen bringt (53). Kritikpunkte sind unter anderem die geringe Anzahl an Teilnehmern / Teilnehmerinnen, so dass die statistische Power, um einen signifikanten und relevanten Unterschied zwischen Verum- und Placebogruppe zu erkennen, gering ist. Die Gültigkeit der Ergebnisse der Studie wurde daher in Frage gestellt (54). 36 3.3 NMDA-Rezeptor Antagonist Neben den verschiedenen Wirkstoffen aus der Gruppe der AChE-Hemmer wird auch der NMDA-Rezeptorantagonist Memantin in der pharmakologischen Therapie der Demenz vom Alzheimer-Typ eingesetzt. NMDA-Rezeptoren stellen eine Untergruppe der Glutamat-Rezeptoren im ZNS da. 3.3.1 Der Neurotransmitter Glutamat Die Aminosäure Glutamat zählt wie das Amin Acetylcholin zu den Neurotransmittern, wobei L-Glutamat der wichtigste erregende Neurotransmitter im ZNS ist (30). Bereits um 1950 wurde von Hayashi die Vermutung aufgestellt, dass L-Glutamat ein wichtiger Neurotransmitter im ZNS sein könnte (55). Er hatte in Versuchen an Menschen, Affen und Hunden Glutamat in die graue Substanz des Kortex injiziert und dadurch klonische Krämpfe ausgelöst (55,56). 1959 konnte durch Curtis und seine Kollegen Phillis und Watkins gezeigt werden, dass LGlutamat zur Depolarisation von Neuronen führt. Bis zur allgemeinen Akzeptanz von L-Glutamat als exzitatorischem Neurotransmitter vergingen jedoch noch viele weitere Jahre (57). Glutamat gehört zu den nicht-essentiellen Aminosäuren und kann nach der Aufnahme mit der Nahrung die Blut-Hirn-Schranke nicht überwinden (56). Daher muss Glutamat, um als Neurotransmitter zu wirken, in den Neuronen synthetisiert werden. Die Synthese erfolgt zum einem durch Transaminierung von aus dem Zitratzyklus gewonnenem α-Ketoglutarat, zum anderen wird Glutamat über den Glutamat-Glutamin-Kreislauf wiedergewonnen (30,41). Glutamat wird nach der Freisetzung im synaptischen Spalt durch natriumabhängige Glutamattransporter zu einem kleinen Teil zurück in das Neuron transportiert und zu einem großen Teil in umliegende Gliazellen. In diesen wird mithilfe der Glutaminsynthetase Glutamat und Ammoniak zu Glutamin umgesetzt. Glutamin wird anschließend von den Gliazellen wieder freigesetzt und kann von den Neuronen aufgenommen werden. Durch die mitochondriale Glutaminase wird dort das Glutamin wieder desaminiert, und es entsteht erneut Glutamat, welches in Vesikeln verpackt wieder als Neurotransmitter zur Verfügung steht. Neben seiner Wirkung als erregender Neurotransmitter ist Glutamat auch Ausgangsstoff für die Synthese des hemmenden Neurotransmitters γ-Aminobutyrat (30). 37 3.3.2 Glutamatrezeptoren Glutamatrezeptoren können in metabotrope und ionotrope Glutamatrezeptoren unterteilt werden. Aufgrund ihrer Eigenschaften kann man diese beiden Rezeptorarten in jeweils drei Untergruppen unterteilen (56). Die G-Protein gekoppelten Rezeptoren haben in der Therapie der Demenz vom Alzheimer-Typ zurzeit keine Bedeutung, so dass hier nur die ionotropen Glutamatrezeptoren weiter betracht werden. Diese werden, wie bereits erwähnt, in drei Untergruppen unterteilt, welche aufgrund der Bindung selektiver Agonisten als NMDA-, AMPAund Kainat-Rezeptoren bezeichnet werden (30,58). Ebenfalls zu den ionotropen Glutamatrezeptoren vorkommenden gehören die delta-Rezeptoren. fast ausschließlich an Purkinje-Zellen Diese Rezeptoren binden jedoch kein Glutamat, und ob sie tatsächlich als Ionenkanal fungieren, ist ebenfalls nicht abschließend geklärt, da bisher kein den Kanal öffnender Agonist gefunden wurde. Allerdings zeigten Ady et. al. (2014), dass die Öffnung dieses Rezeptors durch Typ-1-metabotrope Glutamatrezeptoren getriggert werden kann und somit zumindest indirekt durch Glutamat beeinflusst werden kann (59). In der Therapie der Demenz vom Alzheimer-Typ sind jedoch aktuell nur die NMDA-Rezeptoren von Interesse, weshalb hier auf diese noch etwas genauer eingegangen werden soll. 3.3.3 NMDA-Rezeptoren Die NMDA-Rezeptoren, aus der Gruppe der ionotropen Glutamatrezeptoren, besitzen ihren Namen daher, dass sie außer durch Glutamat selektiv auch durch N-Methyl-D-Aspartat geöffnet werden. Sie kommen vorwiegend an der postsynaptischen Membran (58) vor und sind aus je vier Untereinheiten aufgebaut (30), wobei sieben verschiedene Gene für die Untereinheiten kodieren (60). Es existieren also innerhalb der NMDA-Rezeptoren noch einige unterschiedliche Subtypen, wodurch eine höhere Anzahl an Regulationsmöglichkeiten erreicht wird (58). Die verschiedenen ionotropen Glutamatrezeptoren unterscheiden sich in der Geschwindigkeit des Einstroms von Na +-Ionen und Ca2+-Ionen. So erfolgt der Na+Einstrom beim NMDA-Rezeptor langsam und langfristig, beim AMPA-Rezeptortyp jedoch schnell (61). Darüber hinaus erlaubt der aktivierte NMDA-Rezeptor einen höheren Ca2+-Einstrom als andere Rezeptoren (58). Ca2+ wirkt im 38 postsynaptischen Neuron als second-messenger und kann so komplexere Zellfunktionen langfristiger beeinflussen. Solange der Rezeptor nicht aktiviert ist, wird dieser von Mg2+-Ionen blockiert. Diese Blockade ist spannungsabhängig. Die postsynaptische Membran muss also, bevor eine Aktivierung des NMDARezeptors überhaupt möglich ist, vordepolarisiert sein. Dies kann z.B. durch den Einstrom von Na+-Ionen nach Aktivierung der AMPA-Rezeptoren durch Glutamat erfolgen. Eine wichtige Rolle spielen NMDA-Rezeptoren unter anderem im Gedächtnis. Im Tierversuch wurden die NMDA-Rezeptoren von Mäusen so modifiziert, dass diese länger aktiv bleiben. Es zeigte sich, dass die Mäuse mit modifizierten Rezeptoren schneller lernen und sich im Labyrinth besser zurechtfinden. Es wird davon ausgegangen, dass eine geringe Stimulation zu einer Aktivierung postsynaptischer AMPA-Rezeptoren führt, welches zum Auslösen eines einfachen Aktionspotentials im postsynaptischen Neuron führt. Kommt es jedoch zur wiederholten, hochfrequenten oder starken Stimulation, wird das Mg 2+-Ion vom postsynaptischen NMDA-Rezeptor entfernt, und es werden ebenfalls die NMDA-Rezeptoren aktiviert (61). Durch den in der Folge auftretenden Ca 2+-Einstrom kommt es zur Aktivierung der beiden Proteinkinasen Proteinkinase C und der Calcium-Calmodulinabhängigen Proteinkinase II. Diese führen, nach derzeitiger Erkenntnis, über mehrere Wege zu einer erhöhten Sensibilisierung der postsynaptischen Membran für weitere eintreffende Informationen. Zum einen wird durch eine Phosphorylierung der AMPA-Rezeptoren deren Empfindlichkeit erhöht, zum anderen wird die Anzahl an AMPA-Rezeptoren in der postsynaptischen Membran erhöht. Darüber hinaus kommt es zur Bildung neuer dendritischer Dornfortsätze, die neue Synapsen ausbilden können (28). Diese Veränderungen sind länger andauernd, in Tierversuchen bis zu lebenslang, und sind auch unter dem Begriff Langzeit-Potenzierung bekannt (28,61). 3.3.4 NMDA-Rezeptoren und Alzheimer-Demenz Eine Überstimulation oder eine dauerhafte, schwache Erregung der NMDARezeptoren führt zu einem dauerhaften, pathologischen Ca 2+-Einstrom in das Neuron. Diese länger andauernde Ca 2+-Überladung der Nervenzelle führt in der Folge zuerst zu einem Verlust der Funktion der Synapse und schließlich zum Absterben der Nervenzelle. 39 Bei der Demenz vom Alzheimer-Typ kommt es unter anderem ebenfalls zu einer pathologischen Aktivierung von NMDA-Rezeptoren. Memantin als NMDARezeptorantagonist greift an dieser Stelle an. Der genaue Mechanismus, durch den es zur erhöhten Aktivität der NMDA-Rezeptoren im Rahmen der Demenz vom Alzheimer-Typ kommt, ist noch unbekannt. Bekannt ist, dass lösliche Aβ-Formen mit verschiedenen Proteinen interagieren, unter anderem auch mit NMDARezeptoren sowie mit Transportproteinen, die an der Aufnahme / Freisetzung von Glutamat beteiligt sind. Danysz et al. stellen in einem 2012 veröffentlichten Review die aktuellen Erkenntnisse bezüglich der Veränderungen im glutamergen System sowie die Beeinflussung durch Aβ dar (62). Bekannt ist, dass es bei Patienten / Patientinnen, die an einer Demenz vom Alzheimer-Typ erkrankt sind, zu einer Reduzierung bzw. zu veränderten Varianten mit einer reduzierten Funktion der Glutamat-Transporterproteine kommt. Neben Glutamat können jedoch auch andere Stoffe als Agonisten an den Rezeptoren wirken, welche im Rahmen der Alzheimer-Erkrankung ebenfalls erhöht sind. Ein Beispiel ist die Aminosäure Homocystein, welche aufgrund von Veränderungen im Folsäurezyklus erhöht ist, und die unter anderem als Agonist am NMDA Rezeptor wirkt (62). Auf Seiten des Rezeptors ist eine der wichtigsten Veränderungen, die bei chronischen Krankheiten wie der Alzheimer-Erkrankung entstehen können, eine Veränderung des Ruhepotentials der Membran. In Folge der Veränderung des Ruhepotentials kann es zu einer Entfernung des Mg 2+-Ions kommen, welches normalerweise den Rezeptor spannungsabhängig blockiert, dies führt zu einer Sensibilisierung des Rezeptors. Die Sensitivität des Rezeptors kann aber auch durch andere Faktoren erhöht werden. So erhöhen z.B. endogene Polyamine, z.B. Spermine, die Sensitivität von NMDA-Rezeptoren mit NR2B Untereinheit. Aber auch die Abnahme der Anzahl an Rezeptoren, wie sie bei der Demenz vom Alzheimer-Typ vorkommt, führt als Kompensationsmechanismus zu einer Steigerung der Sensitivität der verbleibenden Rezeptoren. Eventuell spielen darüber hinaus auch die Co-Agonisten Glycin und D-Serin eine Rolle im Ausmaß der durch NMDA-Rezeptoren vermittelten Neurotoxizität (62). Studien lassen die Vermutung zu, dass auch Aβ selber einen stimulierenden Einfluss auf den NMDA-Rezeptor sowie, durch eine Hemmung bzw. Umkehr der Glutamat Re-Uptake-Mechanismen, auf die Glutamat-Konzentration im 40 synaptischen Spalt besitzt. Allerdings sind die Versuche hierzu häufig mit relativ hohen Aβ-Konzentrationen durchgeführt worden, so dass eine Aussage über invivo ablaufende Prozesse schwierig ist (62). 3.3.5 Memantin – Allgemeines Abbildung 6: Chemische Struktur von Memantin, aus (102) Memantin ist der einzige derzeit in der Behandlung der Demenz vom AlzheimerTyp zugelassene NMDA-Rezeptorantagonist, und wird zur Therapie im moderaten bis schweren Stadium der Erkrankung eingesetzt (38). Der Gedanke ist, der bei der Alzheimer-Erkrankung auftretenden Daueraktivierung / Sensibilisierung der NMDA-Rezeptoren entgegenzuwirken. Memantin wirkt, ähnlich wie im gesunden Gehirn Mg2+, als stark spannungsabhängiger Antagonist am Rezeptor. Die Folge ist, dass vor allem starke physiologische Signale zu einer Aktivierung des Rezeptors führen (62). 3.3.6 Memantin – Indikation Zugelassen ist Memantin zur Behandlung im moderaten bis schweren Stadium der Demenz vom Alzheimer-Typ (entsprechend im MMST einem Wert von 0-19 Punkten) (63). Allerdings gibt es für Patienten / Patientinnen, die bereits einen AChE-Hemmer erhalten und sich im moderaten Stadium der Demenz vom Alzheimer-Typ befinden, keine überzeugende Evidenz für den zusätzlichen Nutzen von Memantin, so dass in den aktuellen Leitlinien der Einsatz von Memantin bei dieser Patientengruppe nur empfohlen wird, wenn der Einsatz eines AChEHemmers nicht möglich ist bzw. dieser nicht vertragen wird. Eine Add-on 41 Behandlung mit Memantin, zusätzlich zu einem AChE-Hemmer, wird nicht empfohlen. Anders sieht dies bei Patienten / Patientinnen aus, die sich im schweren Stadium der Demenz vom Alzheimer-Typ befinden. Für diese Patienten / Patientinnen ist Memantin das einzige in Europa zugelassene Präparat. Bei diesen Patienten / Patentinnen kann die zusätzliche Gabe von Memantin selbst dann erwogen werden wenn im Rahmen eines Off-Label-Use bereits ein AChE-Hemmer weitergegeben wird (21). 3.3.7 Memantin - Wirkmechanismus Memantin wirkt als nicht-kompetetiver Antagonist am NMDA-Rezeptor. Dabei besitzt es eine hohe Spannungsabhängigkeit und die Fähigkeit, den Rezeptor schnell wieder freizugeben. Es wirkt damit ähnlich dem Mg 2+-Ion, welches im gesunden Gehirn den NMDA-Rezeptor blockiert. Ein pathologischer Ca 2+-Einfluss aufgrund einer Daueraktivierung des Rezeptors / einer erhöhten Sensitivität des Rezeptors und erhöhter Glutamat-Konzentration soll hierdurch verhindert werden. Physiologische, hochfrequente Aktionspotentiale, die normalerweise zu einer Aktivierung des Rezeptors führen, verdrängen jedoch Memantin und können weiterhin zu einer Öffnung des Ionenkanals führen (62,63). Neben diesen Effekten, die zu einer vorübergehenden Verbesserung der kognitiven Fähigkeiten führen, wird auch diskutiert, ob Memantin zusätzlich einen langfristigen neuroprotektiven Effekt bei verschiedenen neuronalen Erkrankungen besitzt (62). 3.3.8 Memantin - Pharmakokinetik Memantin wird nach oraler Aufnahme fast vollständig resorbiert. Die Bioverfügbarkeit liegt bei annähernd 100%. Die Nahrungsaufnahme beeinflusst die Resorption nicht. Die maximale Plasmakonzentration wird nach 3-8 Stunden erreicht. Memantin liegt im Plasma zu etwa 45% an Plasmaproteine gebunden vor (63). Der größte Teil des resorbierten Memantin wird unmetabolisiert wieder ausgeschieden (37). Die Metabolisierung des restlichen Teils erfolgt nicht über das P450-Isoenzymsystem. Zu den bekannten Metaboliten gehören N-3,5-DimethylGludantan, die beiden Isoformen 4- und 6-Hydroxy-Memantin sowie 1-Nitroso-3,5Dimethyl-Adamantan. Keiner dieser Metaboliten besitzt eine pharmakologische Wirkung auf den NMDA-Rezeptor. Die fast vollständig renal erfolgende 42 Ausscheidung wird vermindert durch einen alkalischen Urin, wie er z.B. bei übermäßigem Gebrauch von Säureblockern auftreten kann (63). 3.3.9 Memantin - Wechselwirkungen Obwohl Memantin keine Wirkung auf das P450-Isoenzym System hat und daher Wechselwirkungen aufgrund der Metabolisierung anderer Medikamente unwahrscheinlich sind, treten doch aufgrund der Wirkung am NMDA-Rezeptor sowie der Ausscheidung Wechselwirkungen mit einigen anderen Präparaten auf. So besteht die Gefahr einer Wirkungsverstärkung von Neuroleptika, Anticholinergika, Levodopa, dopaminergen Agonisten sowie anderen NMDARezeptor-Antagonisten wie Amantadin (37,63). Zu erhöhten Plasmaspiegeln kann es ebenfalls bei Kombination mit Wirkstoffen kommen, die das gleiche renale Kationentransportsystem wie Amantadin nutzen. Dies sind zum Beispiel Ranitidin und Nicotin. In Kombination mit dem Diuretikum Hydrochlorothiazid kann es zu einer Abnahme von dessen Plasmaspiegel kommen. Zu Änderungen der Wirksamkeit kommt es ebenfalls bei Baclofen. Hier ist ggf. eine Anpassung der Dosierung notwendig (63). Bei gemeinsamer Anwendung von Memantin und anderen NMDA-Antagonisten wie Ketamin, Amantadin und Dextromethorphan sowie dem Antikonvulsivum Phenytoin besteht die Gefahr des Auftretens pharmakotoxischer Psychosen. Eine Kombination der Präparate sollte daher vermieden werden. Kein kausaler Zusammenhang ist bekannt weshalb es unter Kombination von Memantin mit Warfarin zu einer Erhöhung des INR-Wertes kommen sollte. Trotzdem gibt es Berichte hierüber. Der Hersteller empfiehlt in der Kombination eine engmaschige Kontrolle des INR oder der Prothrombinzeit (63). 3.3.10 Memantin - Nebenwirkungen Insgesamt treten in Studien Nebenwirkungen unter Memantin nicht häufiger auf als in der Placebogruppe. Dabei sind die auftretenden Nebenwirkungen in einem Großteil der Fälle nur leicht bis mittelschwer. Zu den häufigsten, und im Vergleich zur Placebogruppe signifikant erhöhten Nebenwirkungen, zählen Schwindel, Kopfschmerzen, Verstopfung, Schläfrigkeit und erhöhter Blutdruck. Weitere bekannte Nebenwirkungen umfassen Überempfindlichkeitsreaktionen gegen das 43 Arzneimittel, Dyspnoe, erhöhte Leberwerte sowie gelegentlich Verwirrtheit, Halluzinationen, Pilzinfektionen, Störungen des Gangbildes, Herzinsuffizienz, Thrombosen und Erbrechen. In sehr seltenen Fällen kann es darüber hinaus zu Krampfanfällen kommen (63). 3.3.10.1 Memantin – Studien In den vor der Zulassung durchgeführten grundlegenden Monotherapiestudien bei Patienten / Patientinnen im moderaten bis schweren Stadium der Demenz vom Alzheimer-Typ (MMST 3-14 Punkte) zeigte sich im Beobachtungszeitraum von 6 Monaten eine Verbesserung im CIBIC-plus, ADCS-ADLsev sowie in der SIB. Bei Patienten / Patientinnen, die sich im leichten bis moderaten Stadium der Demenz vom Alzheimer-Typ befanden (MMST 10-22 Punkte), zeigte sich ebenfalls eine signifikante Verbesserung im ADAScog und CIBIC-plus nach 24 Wochen. Eine andere Studie zeigte in dieser Patientengruppe, nach einem Beobachtungszeitraum von 24 Wochen, jedoch keinen signifikanten Unterschied zur Placebogruppe (63). Howard et al. veröffentlichten 2012 eine Studie mit 295 Patienten / Patientinnen, welche mindestens in den vergangenen drei Monaten eine Therapie mit Donepezil erhalten hatten und sich im moderaten bis schweren Stadium der Demenz vom Alzheimer-Typ befanden (MMST 5–13 Punkte). Diese wurden in vier Gruppen randomisiert. Eine Gruppe erhielt weiterhin Donepezil, eine weitere nur Placebo, die dritte nur Memantin, und die vierte Donepezil und Memantin. Sowohl für die Patienten / Patientinnen die nur Donepezil erhielten, als auch für diejenigen, welche nur Memantin erhielten, zeigte sich im Vergleich zur Placebogruppe nach 12 Monaten ein höherer Wert im MMST. Im Vergleich zur Abnahme der kognitiven Fähigkeiten und Funktionen, die in allen vier Patientengruppen beobachtet wurde, war die Verbesserung in den Therapiegruppen jedoch relativ gering. Auch die kombinierte Gabe von Memantin und Donepezil zeigte keinen signifikanten Vorteil gegenüber einer Monotherapie (64). 44 3.4 Ausblick Da bis heute, wie bereits erwähnt, lediglich einige Medikamente, die zu einer kurzfristigen Besserung der Symptomatik führen, verfügbar sind, jedoch keine, die den Verlauf der Erkrankung stoppen oder auf lange Zeit verlangsamen können, und wegen der hohen Anzahl an Betroffenen, wird stetig weiter nach neuen Wirkstoffen geforscht. Dabei werden verschiedene Ansätze, wie passive oder aktive Immuntherapie gegen Aβ-Ablagerungen, eine Reduzierung der AβProduktion durch β-Sekretasehemmer, Präparate, die die Aggregation von Tau hemmen, Serotonin-Rezeptorantagonisten und einige weitere besonders verfolgt und befinden sich zur Zeit zum Teil in klinischen Phase III Studien (65). Sehr viele der Wirkstoffe, die es in der Vergangenheit bis in klinische Studien geschafft haben, zeigten in diesen jedoch nicht den gewünschten Effekt. Eine von Cummings et al. im Jahre 2014 veröffentlichte Studie über die im Zeitraum von 2002–2012 in klinischen Studien getesteten Präparate zeigte eine Erfolgsquote von gerade einmal 0,4%, und damit eine der niedrigsten in allen Krankheitsgebieten (66). Auf die derzeit am stärksten verfolgten Therapieansätze, mit dem Ziel den Verlauf der Krankheit zu verändern, soll hier im Folgenden noch etwas genauer eingegangen werden. 3.4.1 Immuntherapie Grundsätzlich kann zwischen einer aktiven und einer passiven Immuntherapie unterschieden werden. Die aktive Immuntherapie besitzt gegenüber der passiven, bei der die Antikörper in regelmäßigen Zeitabständen dem Patienten / der Patientin verabreicht werden, jedoch den Nachteil, dass beim Auftreten von Nebenwirkungen durch die gebildeten Antikörper eine Intervention kaum möglich ist (67). Als pharmakologischer Angriffspunkt im Rahmen der Immuntherapie gilt aktuell vor allem das Aβ. Dabei existieren drei Theorien, wie die Anhäufung von AβPlaques im Gehirn verhindert werden kann: 1) Die Antikörper binden an die existierenden Aβ-Ablagerungen im Gehirn des Patienten. An den Fc-Teil des Antikörpers des so entstandenen Antikörper45 Antigenkomplexes binden die zum mononukleären-phagozytären System gehörenden Mikrogliazellen. Es kommt zur Fc-vermittelten Phagozytose und damit zum Abbau der Aβ-Ablagerungen sowie zur Freisetzung von Entzündungsmediatoren (68,69). 2) In Folge der Bildung von Antikörper-Antigenkomplexen im peripheren Blut gelangt vermehrt Aβ aus dem Gehirn ins Blut, und die Bildung neuer AβAblagerungen wird verhindert. Es kommt zu einer Verschiebung des Aβ Blut-HirnGleichgewichtes und so zu einer vermehrten Entfernung löslicher Aβ-Formen aus dem Gehirn (68). 3) Durch die Antikörper wird zwar nicht die vorhandene Menge an Aβ reduziert, aber die Aggregation zu neuen Plaques verhindert (68). 3.4.1.1 Passive Immuntherapie Erste Hinweise, dass die periphere Gabe von Antikörpern den Verlauf der Erkrankung günstig beeinflussen könnte, veröffentlichten Bard et al. bereits im Jahr 2000 (70). Sie verabreichten PDAPP-Mäusen, welche eine mutierte, mit der Alzheimer-Erkrankung vergesellschaftete APP-Form produzieren, und die viele pathologische Eigenschaften der Alzheimer Erkrankung zeigen, wöchentlich Antikörper gegen Aβ für die Dauer von 6 Monaten. Nach 6 Monaten zeigte sich, dass es unter der Gabe gewisser Antikörper zu einer Reduzierung der Aβ-Plaque Belastung von über 80% gekommen ist im Vergleich zur Kontrollgruppe. In weiteren Studien konnte gezeigt werden, dass nicht nur das neue Entstehen von Aβ-Plaques reduziert wird, sondern dass kleine, bereits vorhandene Plaques sogar abgebaut werden (67,70). Der erfolgreichste Antikörper in diesen Versuchen trug die Bezeichnung 3D6 und wurde anschließend in humanisierter Form unter dem Namen Bapineuzumab in verschiedenen Phase II / Phase III Studien an Patienten / Patientinnen mit einer Alzheimer-Erkrankung getestet (67). Der Antikörper ist gegen das N-terminale Ende von Aβ gerichtet (71). Er bindet sowohl an fibrilläre, oligomere und monomere Aβ-Formen (72). In zwei Phase III Multicenter-, Placebo-kontrollierten Doppelblind-Studien, einer mit Apoε4-positiven Patienten / Patientinnen und einer mit Apoε4-negativen Patienten / Patientinnen, die sich im milden bis moderaten Stadium einer Demenz vom Alzheimer-Typ 46 (MMSE 16-26 Punkte) befanden, konnte nach 78 Wochen kein signifikanter Effekt in den verschiedenen klinischen Skalen zur Messung der kognitiven Funktion (ADAS-cog11, DAD, MMST, u. a.) gegenüber den Placebogruppen nachgewiesen werden. In der Gruppe der Apoε4-positiven Patienten / Patientinnen konnte jedoch eine Abnahme der Aβ-Plaques im PET-Scan nachgewiesen werden (71,72). Aufgrund der fehlenden positiven Auswirkung auf die kognitive Leistung und dem Auftreten von „amyloid-related imaging abnormalities“ (ARIA) mit Erguss oder Ödem, insbesondere bei den Apoε4-positiven Patienten / Patientinnen (15,3%), wurden die Studien 2012 gestoppt (67,72). Auch weitere Antikörper konnten bisher in klinischen Studien nicht überzeugen. Aktuell befinden sich die monoklonalen Antikörper Gantenerumab, Solanezumab und Aducanumab in Phase III Studien (65). Gantenerumab ist im Gegensatz zu Bapineuzumab und Solanezumab kein humanisierter muriner Antikörper, sondern der erste vollständig humane Antikörper gegen die Alzheimer-Erkankung, der sich in klinischer Erprobung befindet (67,73). Gantenerumab bindet selektiv an aggregiertes Aβ, es erfolgt keine Interaktion mit im Plasma vorhandenen löslichen Aβ-Formen. Die Bindung erfolgt zum einen an das N-terminale Ende von Aβ, zum anderen an räumlich angrenzende, zentrale Regionen des Peptides. Die Elimination erfolgt dann am ehesten durch Fcvermittelte Phagozytose durch Mikrogliazellen. In präklinischen Tierversuchen an PS2APP-Mäusen konnte eine Reduzierung der Aβ-Plaques nachgewiesen werden. Auch in der Phase I Studie an 16 Patienten / Patientinnen im leichten bis moderaten Stadium der Demenz vom Alzheimer-Typ (MMST 16-26 Punkte) zeigte sich eine dosisabhängige Reduzierung von Aβ (-15,6% bei 60mg / - 35,7% bei 200mg) im Vergleich zur Placebogruppe. Eine Verbesserung der kognitiven Funktion auf den klinischen Skalen konnte jedoch in dieser kurzen Studie nicht gezeigt werden. Zwei der sechs Patienten / Patientinnen aus der 200mg Verumgruppe (beide Apoε4-Träger / Trägerinnen) zeigten allerdings nach der zweiten bzw. vierten Gabe der Antikörper Auffälligkeiten im Sinne einer ARIA (73). Eine in 2012 gestartete Phase III Studie mit Patienten / Patientinnen, die sich im Prodromalstadium einer Demenz vom Alzheimer-Typ befanden (Einschlusskriterien waren ein MMST von 26 Punkten und höher, ein Wert von 0,5 im „Clinical Dementia Rating“, desweiteren eine verringerte Gedächtnisfunktion im „Free and Cued Selective Reminding Test“ sowie ein positiver Befund im Amyloid 47 PET Scan), wurde Ende 2014 gestoppt, nachdem eine Zwischenanalyse keine signifikante Wirkung der Therapie zeigte (74). In einer post-hoc Analyse wurde jedoch ein dosisabhängiger Effekt auf der ADAScog und im MMST bei Patienten / Patientinnen mit einem besonders schnellen Fortschreiten der Krankheit festgestellt. Es wird diskutiert, ob eventuell die Dosis des verabreichten Antikörpers zu gering war, wobei eine Erhöhung der Dosis auch eine Zunahme an Nebenwirkungen bedeutet. Es wird diskutiert, eventuell neue Studien mit höherer Dosis zu beginnen (75). Aktuell wird Gantenerumab im Rahmen der DIAN-TU Studie sowie in zwei weiteren Phase III Studien zum einen mit Patienten / Patientinnen im leichten Stadium der Alzheimer Demenz untersucht, zum anderen wird die 2014 gestoppte Scarlet RoAD Studie mit Patienten / Patientinnen im Prodromalstadium der Erkrankung als Open-Label-Studie zur Erforschung höherer Wirkstoffdosen weitergeführt (74,76–78). Bei Solanezumab handelt es sich wie bei Bapienezumab um einen humanisierten, murinen Antikörper. Dieser richtet sich jedoch nicht wie die bereits erwähnten Antikörper Bapienuzumab und Gantenerumab gegen fibrilläre Aβ-Ablagerungen im Gehirn, sondern gegen neurotoxische, lösliche Aβ-Formen im Plasma und erhöht damit die Aβ-Clearance, u. a. mit der Folge, dass die Bildung von Aβ-Plaques im Gehirn abnimmt (67,79,80). In den beiden Phase III Studien EXPEDITION-1 /-2 mit 2052 Patienten / Patientinnen, die sich im milden bis moderaten Stadium der Demenz vom Alzheimer-Typ befanden, zeigte sich keine signifikante Verbesserung des Outcomes auf der ADAS-Cog 11 bzw. der ADCS-ADL Skala. In der gepoolten Betrachtung der Subgruppe von Studienteilnehmern / -teilnehmerinnen, die sich im milden Stadium der Alzheimer-Erkrankung befanden, zeigte sich jedoch eine Verlangsamung der Abnahme der kognitiven Fähigkeiten im ADAS-Cog14 und MMST um 34% sowie im ADCS-iADL um 18% (67,79,80). Dieses Ergebnis wäre mit einem den Krankheitsverlauf beeinflussenden Effekt vereinbar (80), allerdings ist die Verbesserung nicht stärker als mit den bisher zugelassenen Medikamenten (67). Mitte 2013 wurde daraufhin eine weitere Phase III Studie mit 2100 Patienten / Patientinnen, die sich im milden Stadium der Erkrankung befanden, gestartet. Die primäre Datenerhebung läuft bis Oktober 2016, so dass mit ersten Ergebnissen zum Ende des Jahres gerechnet wird (80,81). 48 Aducanumab ist ebenfalls ein monoklonaler humaner Antikörper. Dieser Antikörper ist selektiv für Aβ-Oligomere und Fibrillen (67,82,83), wobei er an parenchymale Amyloidablagerungen bevorzugt vor vaskulären Amyloidablagerungen bindet (82). Die Ergebnisse der 2012 gestarteten Phase II Studie PRIME, an der 166 Patienten / Patientinnen im Prodromal- oder milden Stadium der Demenz vom Alzheimer-Typ teilnahmen, zeigte, dass Aducanumab die Blut-Hirn-Schranke überwindet und zu einer dosisabhängigen Abnahme von Aβ führt (67,82,83). Obwohl bereits nach 6 Monaten eine signifikante Abnahme der Aβ-Ablagerungen im Gehirn nachweisbar war, war zu diesem Zeitpunkt keine Verbesserung kognitiver Funktionen nachweisbar. Nach einem Beobachtungszeitraum von einem Jahr zeigte sich jedoch in den Patienten / Patientinnen der Verumgruppe eine Stabilisierung der Abnahme kognitiver Fähigkeiten im CDR-SB und im MMST. Diese Ergebnisse müssen jedoch mit Vorsicht genossen werden, da es sich um eine relative kleine Studie handelt, die vor allem das Ziel hatte, Daten über die Sicherheit, Verträglichkeit und der Abnahme der Aβ-Ablagerungen im Gehirn zu gewinnen (83). 3.4.1.2 Aktive Immuntherapie Ein anderer Ansatz zur Therapie bzw. vor allem zur Prävention der Demenz vom Alzheimer-Typ ist die aktive Immuntherapie. Natürlicherweise in jüngeren Menschen vorkommende Antikörper gegen Aβ deuten darauf hin, dass in jüngeren Jahren eine Immunität gegen die Demenz vom Alzheimer-Typ besteht, welche im höheren Alter mit der generellen Abnahme der Funktion des Immunsystems verloren geht. Wie auch bei Impfungen gegen andere Pathogene könnte es einfacher sein, präventiv gegen die Erkrankung vorzugehen, als zu versuchen, diese nach Entstehung durch eine aktive oder passive Immuntherapie zu heilen. Der erste in klinischen Studien erprobte Impfstoff trug die Bezeichnung AN1792 und bestand aus Aβ1-42 als Immunogen so wie dem starken pro-inflammatorischen Adjuvans QS-21 (84). In einer Phase II Studie entwickelten knapp 20% der Teilnehmer / Teilnehmerinnen Antikörper nach Gabe des Impfstoffes und zeigten signifikant bessere Ergebnisse in einigen Bereichen der NTB (85). Allerdings entwickelten 6% der Teilnehmer / Teilnehmerinnen der Verumgruppe eine Meningoencephalitis, was dazu führte, dass die Studie gestoppt wurde (67,84,85). Vermutet wird, dass die schweren Nebenwirkungen des Impfstoffs durch das 49 Adjuvans QS-21 verursacht worden sind, welches zu einer starken Th1-Zellvermittelten Immunreaktion führte (84). Als Folge der Nebenwirkungen, die in den Studien mit AN1732 auftraten, wurden in der nächsten Generation von Impfstoffen die T-Zell-Epitope vom Aβ-Antigen entfernt (67,84). Aktuell befindet sich kein Impfstoff zur aktiven Immunisierung gegen die Demenz vom Alzheimer-Typ in Phase III Studien (65,86). 3.4.2 BACE-1 Hemmer Wie bereits erwähnt, entsteht Aβ durch die Spaltung von APP mittels β-/γSekretasen. In einem ersten Schritt schneidet die β-Sekretase das APP auf der extrazellulären Seite. Hierbei entsteht zum einen freies sAPPβ, und es verbleibt ein als C99 bezeichneter, transmembranärer Anteil von APP zurück. Die Schnittstelle bildet später das N-terminale Ende von Aβ. In einem weiteren Schritt wird C99 durch eine γ-Sekretase geschnitten. Durch diesen Schritt wird schließlich Aβ gebildet (87). Bei der hauptsächlich im Gehirn vorkommenden β-Sekretase scheint es sich um das „β-site amyloid precursor protein cleaving enzym 1“ (BACE-1) zu handeln. Ein Ansatz zur Therapie der Alzheimer-Demenz ist daher die Hemmung der BACE-1 und somit die Reduktion der Bildung von Aβ. Ein strukturell ähnliches Enzym wird als BACE-2 bezeichnet, scheint aber in-vivo keine Bedeutung bei der Bildung von Aβ zu haben. Allerdings könnte es aufgrund der strukturellen Ähnlichkeit zur BACE-1 bei der Hemmung der BACE-1 eventuell zu Nebenwirkungen kommen, die aus der Hemmung der BACE-2 resultieren (87). 50 In Tierversuchen mit BACE-1 Knockout-Mäusen zeigten sich zuerst keine Auffälligkeiten durch die Inaktivierung von BACE-1, was darauf hoffen ließ, dass keine schweren Nebenwirkungen auftreten würden. Bei genauerer Betrachtung fiel später jedoch auf, dass es zu verschiedenen neurologischen Veränderungen bei den Mäusen kam, so dass zumindest mit dem Auftreten von Nebenwirkungen gerechnet werden muss. In Hinblick auf die Wirksamkeit gegen Demenz vom Alzheimer-Typ zeigte sich nach Kreuzung der BACE-1 Knockout-Mäuse mit APP transgenen Mäusen, dass die Inaktivierung der BACE-1 die Ablagerung von Aβ sowie den Verlust kognitiver Fähigkeiten verhindert (87). Nach anfänglichen Schwierigkeiten bei der Entwicklung von BACE-1 Hemmern, welche eine ausreichende Konzentration im Gehirn erreichen, befinden sich aktuell die beiden BACE-1 Hemmer mit der Bezeichnung „Verubecestat“ („MK8931“) sowie mit der Bezeichnung „AZD-3293“ in Phase III Studien (65,87). Verubecestat zeigte in Phase I Studien eine dosisabhängige Abnahme von Aβ im Liquor. Sowohl in der Phase I Studie als auch in der anschließenden Phase II Studie mit 200 Patienten / Patientinnen zeigten sich keine schweren Nebenwirkungen. Die Phase II Studie wurde daraufhin auf 1960 Teilnehmer / Teilnehmerinnen im leichten bis moderaten Stadium der Demenz vom AlzheimerTyp aufgestockt, und als Phase III Studie weitergeführt (87–89). Darüber hinaus wurde eine weitere Phase III Studie mit Patienten / Patientinnen, die sich im Prodromal-Stadium der Erkrankung befinden, gestartet (87,89). Ebenfalls in Phase III befindet sich der BACE-1 Hemmer „AZD-3293“. Auch dieser BACE-1 Hemmer scheint in den Phase I Studien eine gute Verträglichkeit und eine dosisabhängige Abnahme von Aβ im Liquor zu zeigen. Die genauen Ergebnisse der Studie wurden jedoch bisher nicht veröffentlicht (90,91). 3.4.3 Tau-Aggregations-Hemmer Einen anderen Ansatz versucht die Firma TauRx mit dem Wirkstoff LeukoMethylthioninium. Ausgehend von der Hypothese, dass die pathologische Aggregation von Tau zum Absterben der Neuronen führt, führte sie mit dem TauAggregationshemmer Leuko-Methylthioninium zwei Phase III Studien mit Patienten / Patientinnen im leichten bzw. leichten bis moderaten Stadium der Demenz vom Alzheimer-Typ durch (92). Grundsätzlich gibt es aus Versuchen mit kultivierten Neuronen sowie in Tiermodellen zur Neurodegeneration Hinweise 51 darauf, dass eine Reduzierung der Tau-Menge neuroprotektiv wirkt. Bezogen auf die Demenz vom Alzheimer-Typ konnte anhand von Neuronen aus Tau KnockoutMäusen wie auch bei der Kreuzung von Tau Knockout-Mäusen mit transgenen APP-Mäusen gezeigt werden, dass Tau sowohl in-vivo als auch in-vitro für die toxische Wirkung von Aβ notwendig ist (19). In der durchgeführten Studie mit dem Wirkstoff Leuko-Methylthioninium konnte allerdings kein Nutzen der Therapie bei Patienten / Patientinnen im leichten bis moderaten Stadium der AlzheimerDemenz gezeigt werden (93). 3.4.4 Ca2+-Kanalblocker Wie bereits erwähnt, kommt es im Rahmen der Alzheimer-Demenz zu einem verstärkten Einstrom von Ca2+-Ionen in die Neuronen, mit der Folge, dass die Neuronen geschädigt werden bzw. absterben. In Tierversuchen konnte gezeigt werden, dass die Gabe von Ca 2+-Blockern neuroprotektiv wirkt. In verschiedenen klinischen Studien wurden in der Vergangenheit mehrere Ca 2+-Blocker bei Patienten / Patientinnen, die an einer Demenz litten, untersucht. Die Ergebnisse dieser Studien sind sehr heterogen. In einer direkten Vergleichsstudie zwischen den beiden Ca2+-Blockern Amlodipin und Nilvadipin an Patienten / Patientinnen, die an leichten kognitiven Einschränkungen litten, kam es unter der Therapie mit Nilvadipin zu keiner weiteren Abnahme der kognitiven Fähigkeiten in einem Zeitraum von 20 Monaten. In der Gruppe der Patienten / Patientinnen, die Amlodipin erhielten, hingegen kam es zu einer weiteren Abnahme kognitiver Funktionen. Da die Blutdrucksenkung in beiden Gruppen etwa gleich stark war, scheint der Effekt nicht auf eine Senkung des Blutdruckes zurückzuführen sein (94). Seit 2013 läuft eine Multicenter Studie mit Nilvadipin im Vergleich zu Placebo an einer Gruppe von 500 Probanden / Probandinnen, die an einer Demenz vom Alzheimer-Typ im leichten bis moderaten Stadium leiden. Ergebnisse werden im Laufe des Jahres 2017 erwartet (95). 3.4.5 Serotoninrezeptoragonisten / -antagonisten Ein weiterer Ansatzpunkt in der Entwicklung neuer Therapien gegen die Alzheimer-Demenz sind die Serotoninrezeptoren. In Tierversuchen mit APP/PS1Maus-Modellen zeigte sich, dass es 12-14 Stunden nach Gabe eines SSRI‘s zu 52 einer Reduzierung der Produktion von Aβ um 25% kommt. Auch bei gesunden Probanden / Probandinnen führte die Gabe von Citalopram zu einer reduzierten Produktion und Konzentration von Aβ im Liquor. Eine längere Gabe von SSRI‘s führte in Tierversuchen zu einer Reduzierung der Aβ-Ablagerungen im Gehirn. Auch dieser Effekt konnte an Patienten / Patientinnen nachgewiesen werden. So zeigten sich in PET-Untersuchungen an Patienten / Patientinnen, die die letzten 5 Jahre einen SSRI eingenommen haben, niedrigere kortikale Aβ-Level als in der Kontrollgruppe (96). Die pharmakologische Forschung konzentriert sich dabei aktuell vor allem auf die beiden Untergruppen des Rezeptors 5-HT 4 und 5-HT6. 5-HT4-Rezeptoren beeinflussen die Prozessierung von APP, wobei eine Aktivierung des Rezeptors zur vermehrten Bildung löslichen sAPPβ anstelle von Aβ führt. Hier konnte in Tierversuchen gezeigt werden, dass eine Gabe von 5-HT 4-Agonisten für 2-3 Monate zu einer Abnahme der Aβ-Plaques und zu einer Verminderung des Verlustes kognitiver Fähigkeiten führt (96). Aktuell befindet sich jedoch kein 5-HT 4Agonist in Phase III der klinischen Prüfung (65). Anders siehst dies beim 5-HT6-Rezeptor aus. Hier befinden sich aktuell zwei Präparate (Idalopiridin und AVN-211) in Phase III der klinischen Prüfung (65). Im Gegensatz zum 5-HT4-Rezeptor scheinen beim 5-HT6-Rezeptor jedoch Antagonisten zu einer Besserung der kognitiven Funktion zu führen. So verbesserte die Inaktivierung von 5-HT 6-Rezeptoren in Nagetieren die kognitiven Fähigkeiten in mehreren Verhaltenstests. Zurückzuführen sein dürfte diese kognitive Verbesserung auf eine aus der Hemmung des Rezeptors folgende Stimulation der Freisetzung der Neurotransmitters Glutamat, Acetylcholin und von Katecholaminen. In einer Phase II Studie mit Idalopiridin in Kombination mit Donepezil zeigte sich eine signifikante Verbesserung kognitiver Funktionen im Vergleich zur Placebo- und Donepezil-Gruppe. Andere Studien, die ausschließlich mit 5-HT6-Antagonisten durchgeführt wurden, zeigten jedoch keine signifikante Verbesserung (96). 53 4 Diskussion Bei der Alzheimer-Erkrankung handelt es sich um eine primär degenerative, zerebrale Erkrankung mit progessiver Demenz, deren Prävalenz und Inzidenz ab dem 60. Lebensjahr deutlich zunimmt. Von den an einer Demenz erkrankten Personen leiden etwa 60-80% an einer Demenz vom Alzheimer-Typ oder an einer Mischform. Als Hauptrisikofaktor, an einer Demenz vom Alzheimer-Typ zu erkranken, gilt das Lebensalter, weitere Risikofaktoren sind ein genetisches Risiko sowie ein Risiko für kardiovaskuläre Erkrankungen. Im Hinblick auf eine weiterhin zunehmende Lebenserwartung in der Bevölkerung und dem demographischen Wandel ergibt sich hieraus eine große gesundheits- und gesellschaftspolitische Herausforderung sowie die Notwendigkeit für wirksame, therapeutische Möglichkeiten, um das Auftreten der Erkrankung zu verhindern bzw. das Fortschreiten in einem frühen Stadium zu verlangsamen oder zu stoppen sowie die Lebensqualität der Betroffenen und deren Angehörigen zu erhalten. Bis heute ist die genaue Ätiologie und Pathogenese der Erkrankung unklar. Die am weitesten anerkannte Hypothese geht davon aus, dass es durch Veränderungen in der Produktion oder der Clearance von Aβ zur Bildung unlöslicher Aβ1-42-Ablagerungen im Gehirn kommt, welche in der Folge zu Entzündungsreaktionen in der Umgebung führen und die Phosphorylierung von Tau-Proteinen beeinflussen, wodurch es wiederum zur Bildung intrazellulärer TauAggregate kommt. Am Ende führt dies zu Störungen der Zellfunktionen und dem Absterben von Synapsen / Neuronen. Das fehlende Wissen über die genaue Pathogenese der Erkrankung erschwert jedoch auch das Auffinden pharmakologischer Angriffspunkte und damit die Entwicklung von Therapien gegen die Erkrankung. Aktuell bestehen die pharmakologischen Therapieoptionen je nach Stadium der Erkrankung aus zentral wirksamen Hemmstoffen der Acetylcholinesterase oder einem Antagonisten des NMDA-Rezeptors. Darüber hinaus können lediglich die im Rahmen der Demenz auftretenden psychischen Begleitsymptome wie psychotische Störungen, Aggression, Depression und Agitation pharmakologisch verbessert werden. 54 Die Hemmer der AChE finden ihre Anwendung vorwiegend im frühen bis mittleren Stadium der Erkrankung. Sie sollen durch die Hemmung des Abbaus von ACh dessen Konzentration im synaptischen Spalt erhöhen und damit den Mangel des Neurotransmitters, der aus dem Funktionsverlust / Absterben von Neuronen resultiert, mildern. In durchgeführten Studien mit den verschiedenen AChEHemmern zeigte sich eine signifikante Verbesserung der kognitiven Leistung auf verschiedenen klinischen Messskalen. Die meisten Studien haben allerdings nur einen kurzen Beobachtungszeitraum von 3-6 Monaten. In der Langzeitstudie AD2000, die allerdings unter anderem aufgrund ihrer geringen Teilnehmerzahl kritisiert wird, konnte kein Unterschied bezüglich der Endpunkte Einweisung in eine Pflegeeinrichtung und dem Verlust von instrumentellen Fähigkeiten im Vergleich zur Placebogruppe gezeigt werden. Da es sich bei der Behandlung mit AChE-Hemmern lediglich um eine symptomatische Therapie handelt, die keinen Einfluss auf die Ursache der Erkrankung hat, ist es nicht verwunderlich, dass es unter der Therapie weiterhin zu einer Abnahme der kognitiven Fähigkeiten kommt. Der NMDA-Rezeptorantagonist Memantin wird hingegen vorwiegend im schweren Stadium der Demenz vom Alzheimer-Typ eingesetzt. Dieser soll einer im Rahmen der Erkrankung auftretenden pathologischen Aktivierung von NMDA-Rezeptoren entgegenwirken. Auch hier zeigt sich in Studien eine Verbesserung der kognitiven Leistung, welche jedoch gering ausfällt. Ein Einfluss auf die Ursache der Erkrankung und deren Fortschreiten besitzt Memantin jedoch nicht, so dass auch hier die weitere Abnahme der kognitiven Fähigkeiten nicht verwundert. In den letzten Jahren ist eine Vielzahl an neuen Wirkstoffen sowie Angriffspunkten zur Therapie der Erkrankung präklinisch erforscht worden, und einige haben es in die klinische Prüfung geschafft. Im Rahmen der klinischen Studien scheiterten dann allerdings fast alle Präparate. Aktuell befinden sich vor allem Wirkstoffe aus dem Bereich der aktiven und passiven Immuntherapie, Hemmer der β-Sekretase, Ca2+-Blocker, Präparate mit Wirkung auf die Serotoninrezeptoren sowie Wirkstoffe, die die Aggregation von Tau-Proteinen verhindern sollen, in fortgeschrittener klinischer Prüfung. 55 Bei den Substanzen aus der Gruppe der aktiven sowie passiven Immuntherapie, bei den β-Sekretasehemmern sowie bei den Agonisten am 5-HT 4-Rezeptor kam es zu einer signifikanten Reduzierung der Aβ-Bildung bzw. Ablagerung sowohl im Tierversuch als auch in den klinischen Studien. Betrachtet man die Abnahme der kognitiven Fähigkeiten, so sehen die Ergebnisse, vor allem in den klinischen Studien, jedoch ernüchternder aus. Eine signifikante Reduzierung der Abnahme kognitiver Funktionen konnte in klinischen Studien nur für den 5-HT 6-Antagonisten Idalopiridin (in Kombination mit Donepezil), den Ca 2+-Kanalblocker Nilvadipin sowie für die beiden Antikörper Aducanumab und Solanezumab gezeigt werden. Im Falle von Idalopiridin dürfte die Verbesserung vor allem auf die durch die Hemmung der 5-HT6-Rezeptoren erhöhte Ausschüttung von Neurotransmittern zurückzuführen sein. Allein aufgrund der erhöhten Freisetzung von ACh dürfte eine Besserung der kognitiven Leistung zu erwarten sein. Dieser Mechanismus passt auch dazu, dass die Verbesserung der kognitiven Funktion bei der Gabe von AChE-Hemmern stärker ist, je höher die Dosis ist. Limitierend für eine Erhöhung der Dosis von AChE-Hemmern sind jedoch vor allem die aus der Hemmung der peripheren AChE sowie der Pseudo-AChE resultierenden Nebenwirkungen, so dass hier ein zweiter Wirkweg existieren dürfte, welcher zur Erhöhung der AChKonzentration im synaptischen Spalt führt, bei besserer Verträglichkeit. Eine modifizierende Wirkung auf den Verlauf der Erkrankung ist jedoch unwahrscheinlich. Bei Nilvadipin kam es in einer Vergleichsstudie mit Amlodipin bei Patienten / Patientinnen, die an einer leichten kognitiven Beeinträchtigung litten, zu keiner weiteren Abnahme der kognitiven Funktionen. Allerdings fehlen hier noch die Ergebnisse aktueller Studien zur Wirkung mit einer größeren Gruppe von Patienten / Patientinnen, die an einer Demenz vom Alzheimer-Typ leiden. Da als ein Mechanismus, der zum Verlust der Funktion von Neuronen und schließlich zu deren Absterben führt, ein erhöhter Ca 2+-Einstrom in die Neuronen angenommen wird, welcher im Laufe der Demenz vom Alzheimer-Typ z.B. durch eine Überaktivierung von NMDA-Rezeptoren auftritt, könnte eine entsprechende Blockade von Ca2+-Kanälen tatsächlich neuroprotektive Effekte zeigen. Dies würde 56 allerdings ebenfalls nicht die der Erkrankung zugrunde liegende Ursache beheben. Ausgehend von der Amyloidkaskaden-Hypothese würden Therapien, welche die Bildung / Ablagerung von Aβ beeinflussen, damit auch die Grundlage der Erkrankung bzw. deren Verlauf beeinflussen. Für die Gruppe der BACE-1 Hemmer fehlen hier bisher die Ergebnisse klinischer Studien, inwieweit eine Abnahme der Bildung von Aβ tatsächlich auch zu einer Reduzierung der Abnahme der kognitiven Funktionen führt. Betrachtet man die Immuntherapien, so zeigte sich in den Studien für Solanezumab tatsächlich eine Verlangsamung des Verlustes kognitiver Funktionen, welcher jedoch nicht stärker ausfiel als bei den aktuell eingesetzten Präparaten. Interessant sind die Ergebnisse mit dem Antikörper Aducanumab: hier zeigte sich nach den ersten sechs Monaten eine signifikante Abnahme der AβAblagerungen, und mit einer Latenz von weiteren sechs Monaten auch eine Stabilisierung der Abnahme kognitiver Funktionen. Allerdings handelte es sich hierbei lediglich um eine kleine Phase II Studie, so dass die Ergebnisse der größeren Phase III Studie abzuwarten bleiben. Sollten sich die Ergebnisse bestätigen, wäre dies auch wieder ein neues Indiz für die Gültigkeit der Amyloidkaskaden-Hypothese, welche in letzter Zeit von einigen Wissenschaftlern angezweifelt wurde, da es zwar öfter in Studien zu einer Reduzierung der AβProduktion / Ablagerung kam, jedoch ohne Einfluss auf die kognitive Leistung. In Tierversuchen hingegen zeigte sich häufiger auch ein Effekt auf die kognitiven Fähigkeiten der Tiere. Allerdings werden diese Studien in der Regel an transgenen Mäusen durchgeführt, welche gezielt vermehrt Aβ 1-42 bilden. Sollte im Menschen ein weiterer oder anderer Mechanismus vorhanden sein, würde dieser bei diesen Tieren evtl. gar nicht auftreten. Aktuell gibt es keinen Wirkstoff, der in der Lage ist, den Verlauf der Erkrankung aufzuhalten oder zumindest deutlich zu verlangsamen, so dass weiterhin die Erforschung und Erprobung neuer Wirkstoffe notwendig ist. Da bis heute auch die genauen pathophysiologischen Vorgänge der Erkrankung unbekannt sind, ist auch hier weitere Grundlagenforschung nötig. Die Ergebnisse aus den neuesten Antikörper-Studien können ggf. einen Lichtblick darstellen. Sie zeigen aber auch, dass, sollten sich die Ergebnisse in größer angelegten Studien bestätigen, die 57 Notwendigkeit besteht, sowohl die Bevölkerung als auch die Ärzteschaft weiter für dieses Thema zu sensibilisieren, und dass die Entwicklung einfacher Screeningmaßnahmen nötig ist, um Betroffene in einem möglichst frühen Stadium der Erkrankung zu erkennen, insbesondere vor dem Hintergrund, dass bei beiden Antikörpern ein positiver Effekt auf die kognitiven Fähigkeiten lediglich bei Patienten / Patientinnen, die sich im leichten Stadium oder im Prodromalstadium der Erkrankung befanden, nachgewiesen werden konnte. 58 5 Literaturverzeichnis 1. Stelzmann RA, Norman Schnitzlein H, Reed Murtagh F. An english translation of alzheimer’s 1907 paper, “über eine eigenartige erkankung der hirnrinde”. Clin Anat. 1. Januar 1995;8(6):429–31. 2. Hacke W. Hacke - Neurologie. In: Neurologie. 13. Aufl. Heidelberg: Springer Medizin; 2010. S. 572–6. 3. Alzheimer-Krankheit. In [zitiert 7. März 2016]. Verfügbar unter: http://www.degruyter.com.pschyrembel.han.medunigraz.at/view/kw/4377210? language 4. DIMDI - ICD-10-GM Version 2016 [Internet]. [zitiert 12. März 2016]. Verfügbar unter: http://www.dimdi.de/static/de/klassi/icd-10gm/kodesuche/onlinefassungen/htmlgm2016/block-f00-f09.htm 5. Reuter P. Springer Klinisches Wörterbuch. In: Klinisches Wörterbuch 2007/2008. 1. Aufl. Heidelberg: Springer Medizin; 2007. S. 57–8. 6. Böcker W, Denk H, Heitz P, Höfler G, Kreipe H, Moch, Herausgeber. Denk Pathologie - 5. Auflage. In: Pathologie. 5. Aufl. München: Urban & Fischer; 2012. S. 245–8. 7. Alzheimer’s Association. 2014 Alzheimer’s disease facts and figures. Alzheimers Dement J Alzheimers Assoc. März 2014;10(2):e47–92. 8. Scheltens P, Blennow K, Breteler MMB, de Strooper B, Frisoni GB, Salloway S, u. a. Alzheimer’s disease. The Lancet [Internet]. Februar 2016 [zitiert 12. März 2016]; Verfügbar unter: http://linkinghub.elsevier.com/retrieve/pii/S0140673615011241 9. Österreichischer Demenzbericht 2014 [Internet]. [zitiert 12. März 2016]. Verfügbar unter: http://www.bmg.gv.at/cms/home/attachments/6/4/5/CH1513/CMS1436868155908/dem enzbericht2014.pdf 10. Ziegler U, Doblhammer G. [Prevalence and incidence of dementia in Germany--a study based on data from the public sick funds in 2002]. Gesundheitswesen Bundesverb Ärzte Öffentl Gesundheitsdienstes Ger. Mai 2009;71(5):281–90. 11. Puzzo D, Gulisano W, Arancio O, Palmeri A. The keystone of Alzheimer pathogenesis might be sought in Aβ physiology. Neuroscience. Oktober 2015;307:26–36. 12. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease [Internet]. [zitiert 15. März 2016]. Verfügbar unter: http://www-1sciencedirect1com-1pubmed.han.medunigraz.at/science/article/pii/S0143417915000657 13. Armstrong RA. The pathogenesis of Alzheimer’s disease: a reevaluation of the „amyloid cascade hypothesis“. Int J Alzheimers Dis. 2011;2011:630865. 14. Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. Februar 2007;8(2):101–12. 15. Strachan T, Read AP. Molekulare Humangenetik. In: Molekulare humangenetik. 3. Aufl. München: Elsevier GmbH, Spektrum Akademischer Verlag; 2005. 59 16. Pike C, Walencewicz-Wasserman A, Kosmoski J, Cribbs D, Glabe C, Cotman C. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25-35 region to aggregation and neurotoxicity. J Neurochem. Januar 1995;64(1):253–65. 17. Small DH, Cappai R. Alois Alzheimer and Alzheimer’s disease: a centennial perspective. J Neurochem. November 2006;99(3):708–10. 18. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, u. a. The Alzheimer’s Disease-Associated Amyloid β-Protein Is an Antimicrobial Peptide. Bush AI, Herausgeber. PLoS ONE. 3. März 2010;5(3):e9505. 19. Medina M, Avila J. New perspectives on the role of tau in Alzheimer’s disease. Implications for therapy. Biochem Pharmacol. April 2014;88(4):540–7. 20. McCance KL, Huether SE, Brashers V, Rote NS. Pathophysiology. In: Pathophysiology The Biologic Basis for Disease in Adults and Children. 6. Aufl. Missouri: Mosby Elsevier; 2010. 21. DGN S3 Leitlinie Demenz [Internet]. [zitiert 11. April 2016]. Verfügbar unter: http://www.dgn.org/images/red_leitlinien/LL_2015/PDFs_Download/038013_S3-LLDemenzen-240116.pdf 22. Maercker A. Alterspsychotherapie und klinische Gerontopsychologie. Berlin Heidelberg: Springer; 2015. 23. Sheehan B. Assessment scales in dementia. Ther Adv Neurol Disord. November 2012;5(6):349–58. 24. GmbH DMS. TFDD [Internet]. DocCheck Flexikon. [zitiert 6. Oktober 2016]. Verfügbar unter: http://flexikon.doccheck.com/de/TFDD 25. Robert P, Ferris S, Gauthier S, Ihl R, Winblad B, Tennigkeit F. Review of Alzheimer’s disease scales: is there a need for a new multi-domain scale for therapy evaluation in medical practice? Alzheimers Res Ther. 26. August 2010;2(4):24. 26. Ulfig N. Kurzlehrbuch Histologie. 2. Aufl. Stuttgart: Thieme; 27. Speckman E-J, Hescheler J, Köhling R. Physiologie. 5. Aufl. München: Elsevier GmbH; 28. Bear MF, Connors BW, Paradiso MA. Neurowissenschaften. 3. Aufl. Engel AK, Herausgeber. Heidelberg: Spektrum Akademischer Verlag; 2009. 29. EM T. Henry Dale and the discovery of acetylcholine. - PubMed - NCBI [Internet]. [zitiert 22. April 2016]. Verfügbar unter: http://www-1ncbi-1nlm-1nih-1gov1pubmed.han.medunigraz.at/pubmed/16731499 30. Löffler G, Petrides PE. Biochemie & Pathobiochemie. 7. Aufl. Berlin Heidelberg New York: Springer; 2003. 31. Čolović MB, Krstić DZ, Lazarević-Pašti TD, Bondžić AM, Vasić VM. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr Neuropharmacol. Mai 2013;11(3):315–35. 60 32. Freissmuth M, Böhm S, Offermanns S. Pharmakologie & Toxykologie - Von den molekularen Grundlagen zur Pharmakotherapie. Heidelberg: Springer Medizin; 2012. 33. Mutschler E, Geisslinger G, Kroemer HK, Menzel S, Ruth P. Mutschler Arzneimittelwirkungen. 10. Aufl. Stuttgart: Wissenschaftliche Verlagsgesellschaft; 2013. 34. Fachinfo - Aricept [Internet]. [zitiert 26. Mai 2016]. Verfügbar unter: http://www.fachinfo.de/suche/fi/001940 35. Fachinfo Rivastigmin [Internet]. [zitiert 28. Juni 2016]. Verfügbar unter: http://www.fachinfo.de/pdf/014238#view=FitH&pagemode=none&toolbar=1&statusb ar=0&messages=0&navpanes=0 36. Fachinfo Galantamin [Internet]. [zitiert 28. Juni 2016]. Verfügbar unter: http://www.fachinfo.de/pdf/014662#view=FitH&pagemode=none&toolbar=1&statusb ar=0&messages=0&navpanes=0 37. Ernst Mutschler, Geisslinger G, Kroemer HK, Ruth P, Schäfer-Kortig M. Mutschler Arzneimittelwirkungen. 9. Aufl. Stuttgart: Wissenschaftliche Verlagsgesellschaft; 2008. 38. FachInfo-Service - Exelon [Internet]. [zitiert 14. Juli 2016]. Verfügbar unter: http://www.fachinfo.de/suche/fi/003171 39. Informationsbrief zu Reminyl® [Internet]. [zitiert 6. August 2016]. Verfügbar unter: https://www.bfarm.de/SharedDocs/Risikoinformationen/Pharmakovigilanz/DE/RHB/2 015/inforeminyl.pdf;jsessionid=D598635E1C18A8969EC504D92367CC69.1_cid350? __blob=publicationFile&v=9 40. Shigeta M, Homma A. Donepezil for Alzheimer’s disease: pharmacodynamic, pharmacokinetic, and clinical profiles. - PubMed - NCBI. CNS Drug Rev. Winter 2001;7(4):353–68. 41. Horn F, Moc I, Berghold S, Lindenmeier G. Biochemie des Menschen. 3. Aufl. Stuttgart: Georg Thieme; 2005. 42. European Medicines Agency - Human medicines - Exelon [Internet]. [zitiert 4. Oktober 2016]. Verfügbar unter: http://www.ema.europa.eu/ema/index.jsp? curl=pages/medicines/human/medicines/000169/human_med_000777.jsp&mid=WC0b 01ac058001d125 43. Drug Approval Package: Exelon (Rivastigmine Tartrate) NDA #20-823 [Internet]. [zitiert 4. Oktober 2016]. Verfügbar unter: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2000/20823_Exelon.cfm 44. Sadowsky C, Perez JAD, Bouchard RW, Goodman I, Tekin S. Switching from oral cholinesterase inhibitors to the rivastigmine transdermal patch. CNS Neurosci Ther. 2010;16(1):51–60. 61 45. Polinsky R. Clinical pharmacology of rivastigmine: a new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Clin Ther. August 1998;20(4):634–47. 46. Lilienfeld S. Galantamine--a novel cholinergic drug with a unique dual mode of action for the treatment of patients with Alzheimer’s disease. CNS Drug Rev. Summer 2002;8(2):159–76. 47. Nordberg A. Nicotinic receptor abnormalities of Alzheimer’s disease: therapeutic implications. Biol Psychiatry. 1. Februar 2001;49(3):200–10. 48. Lombardo S, Maskos U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology. September 2015;96:255–62. 49. Terry Jr. AV, Callahan PM, Hernandez CM. Nicotinic ligands as multifunctional agents for the treatment of neuropsychiatric disorders. Biochem Pharmacol. Oktober 2015;97(4):388–98. 50. Nikiforuk A, Potasiewicz A, Kos T, Popik P. The combination of memantine and galantamine improves cognition in rats: The synergistic role of the α7 nicotinic acetylcholine and NMDA receptors. Behav Brain Res. Oktober 2016;313:214–8. 51. Evans JG, Wilcock G, Birks J. Evidence-based pharmacotherapy of Alzheimer’s disease. Int J Neuropsychopharmacol Off Sci J Coll Int Neuropsychopharmacol CINP. September 2004;7(3):351–69. 52. Winblad B, Kilander L, Eriksson S, Minthon L, Båtsman S, Wetterholm A-L, u. a. Donepezil in patients with severe Alzheimer’s disease: double-blind, parallel-group, placebo-controlled study. The Lancet. April 2006;367(9516):1057–65. 53. Courtney C, Farrell D, Gray R, Hills R, Lynch L, Sellwood E, u. a. Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): randomised double-blind trial. Lancet Lond Engl. 26. Juni 2004;363(9427):2105–15. 54. Kaiser T, Florack C, Franz H, Sawicki PT. Abstract: [Donepezil in patients with Alzheimer’s disease--a critical appraisal of the AD2000 study]. Med Klin Munich Ger 1983. 15. März 2005;100(3):157–60. 55. Hayashi T. Effects of Sodium Glutamate on the Nervous System. Keio J Med. 1954;3(4):183–92. 56. Gasparini CF, Griffiths LR. The Biology of the Glutamatergic System and Potential Role in Migraine. Int J Biomed Sci IJBS. März 2013;9(1):1–8. 57. Watkins J. l-glutamate as a central neurotransmitter: looking back. 28. 2000;4:297– 309. 58. Regan MC, Romero-Hernandez A, Furukawa H. A structural biology perspective on NMDA receptor pharmacology and function. Curr Opin Struct Biol. August 2015;33:68–75. 62 59. Ady V, Perroy J, Tricoire L, Piochon C, Dadak S, Chen X, u. a. Type 1 metabotropic glutamate receptors (mGlu1) trigger the gating of GluD2 delta glutamate receptors. EMBO Rep. Januar 2014;15(1):103–9. 60. Mayer ML. Structural biology of glutamate receptor ion channel complexes. Curr Opin Struct Biol. Dezember 2016;41:119–27. 61. Purves WK, Sadava D, Orians GH, Heller HC. Biologie. 7. Aufl. Markl J, Herausgeber. München: Elsevier GmbH; 2006. 62. Danysz W, Parsons CG. Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine--searching for the connections. Br J Pharmacol. September 2012;167(2):324–52. 63. Axura - Suche, Schritt 1 - FachInfo-Service [Internet]. [zitiert 21. August 2016]. Verfügbar unter: http://www.fachinfo.de/suche/fi/006529 64. Howard R, McShane R, Lindesay J, Ritchie C, Baldwin A, Barber R, u. a. Donepezil and Memantine for Moderate-to-Severe Alzheimer’s Disease. N Engl J Med. 8. März 2012;366(10):893–903. 65. Alzheimer-Medikamente: Topthema der Pharmaforschung [Internet]. [zitiert 18. September 2016]. Verfügbar unter: http://www.vfa.de/de/arzneimittelforschung/woran-wir-forschen/alzheimer.html/_3-neue-alzheimer-medikamente-inentwicklung 66. Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6(4):37. 67. Güell-Bosch J, Montoliu-Gaya L, Esquerda-Canals G, Villegas S. Aβ immunotherapy for Alzheimer’s disease: where are we? Neurodegener Dis Manag. Juni 2016;6(3):179– 81. 68. Delrieu J, Ousset PJ, Caillaud C, Vellas B. ‘Clinical trials in Alzheimer’s disease’: immunotherapy approaches: Immunotherapy approaches in Alzheimer’s disease. J Neurochem. Januar 2012;120:186–93. 69. Pezzuetto A, Ulrichs T, Burmester G-R. Taschenatlas der Immunologie. 2. Aufl. Stuttgart - New York: Georg Thieme; 2007. 70. Bard F, Cannon C, Barbour R, Burke R-L, Games D, Grajeda H, u. a. Peripherally administered antibodies against amyloid bold beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 1. August 2000;6(8):916–9. 71. Khorassani F, Hilas O. Bapineuzumab, an investigational agent for Alzheimer’s disease. P T. Februar 2013;38(2):89–91. 72. Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, u. a. Two Phase 3 Trials of Bapineuzumab in Mild-to-Moderate Alzheimer’s Disease. N Engl J Med. 23. Januar 2014;370(4):322–33. 63 73. Nisticò R, Novakovic D, Feligioni M, Scaccianoce S, Piccinin S, Mercuri NB, u. a. Profile of gantenerumab and its potential in the treatment of Alzheimer&amp;#39;s disease. Drug Des Devel Ther. November 2013;1359. 74. Gantenerumab | ALZFORUM [Internet]. [zitiert 20. September 2016]. Verfügbar unter: http://www.alzforum.org/therapeutics/gantenerumab 75. Aducanumab, Solanezumab, Gantenerumab Data Lift Crenezumab, As Well | ALZFORUM [Internet]. [zitiert 20. September 2016]. Verfügbar unter: http://www.alzforum.org/news/conference-coverage/aducanumab-solanezumabgantenerumab-data-lift-crenezumab-well 76. Gantenerumab – SPS - Specialist Pharmacy Service – The first stop for professional medicines advice [Internet]. [zitiert 20. September 2016]. Verfügbar unter: https://www.sps.nhs.uk/medicines/gantenerumab/ 77. A Study of Gantenerumab in Participants With Prodromal Alzheimer’s Disease - Full Text View - ClinicalTrials.gov [Internet]. [zitiert 20. September 2016]. Verfügbar unter: https://clinicaltrials.gov/show/NCT01224106 78. A Study of Gantenerumab in Patients With Mild Alzheimer Disease - Full Text View ClinicalTrials.gov [Internet]. [zitiert 20. September 2016]. Verfügbar unter: https://clinicaltrials.gov/show/NCT02051608 79. Siemers ER, Sundell KL, Carlson C, Case M, Sethuraman G, Liu-Seifert H, u. a. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement. Februar 2016;12(2):110–20. 80. Solanezumab | ALZFORUM [Internet]. [zitiert 20. September 2016]. Verfügbar unter: http://www.alzforum.org/therapeutics/solanezumab 81. Progress of Mild Alzheimer’s Disease in Participants on Solanezumab Versus Placebo Full Text View - ClinicalTrials.gov [Internet]. [zitiert 20. September 2016]. Verfügbar unter: https://clinicaltrials.gov/ct2/show/NCT01900665?term=Solanezumab&rank=1 82. Aducanumab | ALZFORUM [Internet]. [zitiert 20. September 2016]. Verfügbar unter: http://www.alzforum.org/therapeutics/aducanumab 83. Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, u. a. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 31. August 2016;537(7618):50–6. 84. Marciani DJ. A retrospective analysis of the Alzheimer’s disease vaccine progress The critical need for new development strategies. J Neurochem. Juni 2016;137(5):687– 700. 85. Winblad B, Graf A, Riviere M-E, Andreasen N, Ryan J. Active immunotherapy options for Alzheimer’s disease. Alzheimers Res Ther. 2014;6(1):7. 86. ALZFORUM | Therapeutics Active Immunisation [Internet]. [zitiert 20. September 2016]. Verfügbar unter: http://www.alzforum.org/therapeutics/search? fda_statuses=&target_types=&therapy_types%5B%5D=161&conditions=&keywordsentry=&keywords= 64 87. Yan R, Vassar R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. März 2014;13(3):319–29. 88. Merck Presents Findings from Phase 1b Study of Investigational BACE Inhibitor, MK8931, in Patients with Alzheimer’s Disease | Merck Newsroom Home [Internet]. [zitiert 21. September 2016]. Verfügbar unter: http://www.mercknewsroom.com/pressrelease/alzheimers-disease/merck-presents-findings-phase-1b-study-investigationalbace-inhibit 89. Verubecestat | ALZFORUM [Internet]. [zitiert 21. September 2016]. Verfügbar unter: http://www.alzforum.org/therapeutics/verubecestat 90. AZD3293 | ALZFORUM [Internet]. [zitiert 21. September 2016]. Verfügbar unter: http://www.alzforum.org/therapeutics/azd3293 91. AstraZeneca and Lilly announce alliance to develop and commercialise BACE inhibitor AZD3293 for Alzheimer’s disease - AstraZeneca [Internet]. [zitiert 21. September 2016]. Verfügbar unter: https://www.astrazeneca.com/media-centre/pressreleases/2014/astrazeneca-lilly-bace-inhibitor-azd3293-alzheimers-disease16092014.html# 92. LMTM | ALZFORUM [Internet]. [zitiert 24. September 2016]. Verfügbar unter: http://www.alzforum.org/therapeutics/lmtm 93. In First Phase 3 Trial, the Tau Drug LMTM Did Not Work. Period. | ALZFORUM [Internet]. [zitiert 25. September 2016]. Verfügbar unter: http://www.alzforum.org/news/conference-coverage/first-phase-3-trial-tau-drug-lmtmdid-not-work-period 94. Nimmrich V, Eckert A. Calcium channel blockers and dementia: Calcium channels in neurodegenerative disorders. Br J Pharmacol. Juli 2013;169(6):1203–10. 95. Nilvadipine | ALZFORUM [Internet]. [zitiert 25. September 2016]. Verfügbar unter: http://www.alzforum.org/therapeutics/nilvadipine 96. Claeysen S, Bockaert J, Giannoni P. Serotonin: A New Hope in Alzheimer’s Disease? ACS Chem Neurosci. 15. Juli 2015;6(7):940–3. 97. synapse.jpg (1180×724) [Internet]. [zitiert 15. August 2016]. Verfügbar unter: http://daten.didaktikchemie.uni-bayreuth.de/umat/alkaloide/synapse.jpg 98. acetylcholinesterase.png (645×297) [Internet]. [zitiert 15. August 2016]. Verfügbar unter: http://www.atsdr.cdc.gov/csem/cholinesterase/images/acetylcholinesterase.png 99. 2007 F 21:39 14 April. Skeletal formula of donepezil. Created using ACD/ChemSketch 10.0 and Inkscape. [Internet]. 2007 [zitiert 7. Oktober 2016]. Verfügbar unter: https://commons.wikimedia.org/wiki/File:Donepezil_skeletal.svg 100. File:Rivastigmin.svg – Wikimedia Commons [Internet]. [zitiert 7. Oktober 2016]. Verfügbar unter: https://commons.wikimedia.org/wiki/File:Rivastigmin.svg? uselang=de 65 101. Jü. English: Galantamine_Structural_Formulae [Internet]. 2009 [zitiert 7. Oktober 2016]. Verfügbar unter: https://commons.wikimedia.org/wiki/File:Galantamine_Structural_Formulae.png 102. LemiNW. Deutsch: Strukturformel von Memantin [Internet]. 2006 [zitiert 7. Oktober 2016]. Verfügbar unter: https://commons.wikimedia.org/wiki/File:Memantin_Struktur.png?uselang=de 103. Wikipedia I at E. Depiction of en:amyloid precursor protein processing, created by I. Peltan Ipeltan [Internet]. 2006 [zitiert 7. Oktober 2016]. Verfügbar unter: https://commons.wikimedia.org/wiki/File:APP_processing.png 66