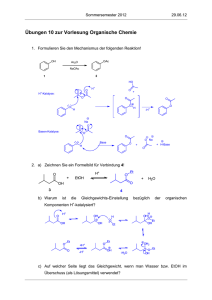
Synthese und Umwandlung von Funktionellen Gruppen
Werbung

Synthese und Umwandlung von Funktionellen Gruppen Bisherige Vorlesungen in OC • OC1: Einführung in OC, Bindungsverhältnisse, elementare Stereochemie, Substanzklassen in der OC, elementare Reaktionen dieser Substanzklassen, einfache Reaktionsmechanismen • OC2: detaillierte systematische Betrachtung aller Reaktionsmechanismen in der OC. OC2 geht der Frage nach: welche Produkte entstehen, wenn man mit einer bestimmten Ausgangssubstanz eine SNReraktion, eine Eliminierung, eine Oxidation usw. macht? R OH R R R Hal R SN R SH R NH2 R COOH Br2 O R R S R R R NH R R R Ad E R OOCR R R HBr Br R HOH H RCOOOH R R H Br R R R Br OH R R O 2 Hier in der OC4 • Behandeln wir die Frage: mit welchen Reaktionen kann man ein vorgegebenes Strukturelement aufbauen? • OC4 orientiert sich an der täglichen Praxis des synthetisch arbeitenden Chemikers. R' O R R' OH El Ox R' O CC R R R R' O Ad • Voraussetzung für diese Vorgehensweise: beherrschen aller Reaktionsmechanismen, kennen von Reaktivitäten, Kompatibiltäten • Je mehr Reaktionen man kennt, um so besser!!! Diese Reaktionen sollen Sie in der OC4 lernen. Reaktionen, die Sie schon aus der OC2 kennen, werden unter Verweis auf das OC2-Skript nur kurz erwähnt (sind aber 3 trotzdem klausurrelevant). Inhalt Übersicht über funktionelle Gruppen Halogene aus Alkanen durch radikalische Halogenierung aus Alkenen durch Wohl-Ziegler-Bromierung aus Alkenen durch Halolactonisierung aus Alkenen durch Addition von HX aus Alkenen durch Addition von X2 aus Alkenen und Alkinen durch Hydroborierung/Halogenierung aus Alkoholen durch SNi aus Alkoholen durch Appel-Reaktion und Mitsunobu-Reaktion aus Halogenen und Tosylaten durch Finkelsteijn-Reaktion aus Carbonsäuren durch Hunsdiecker-Reaktion aus Carbonylverbindungen durch α-Halogenierung aus Aromaten durch Kernhalogenierung aus Aromaten durch Chloralkylierung aus Diazoverbindungen durch Sandmeyer-Reaktion und durch Schiemann-Reaktion Alkene aus Dreifachbindungen durch Lindlar-Hydrierung aus Propargylalkoholen durch LAH-Reduktion aus Dreifachbindungen durch Hydroborierung, Hydrostannylierung aus Doppelbindungen durch Isomerisierung aus Aldehyden/Ketonen durch Wittig-Reaktion aus Aldehyden/Ketonen durch Horner-Emmons-Reaktion 4 Alkene aus Aldehyden/Ketonen durch Still-Gennari-Reaktion aus Aldehyden/Ketonen durch Julia-Olefinierung und Julia-Lythgoe-Olefinierung aus Aldehyden/Ketonen durch Stevens-Reaktion aus Aldehyden/Ketonen durch Shapiro-Reaktion aus Aldehyden/Ketonen durch Tebbe-Reaktion aus Aldehyden/Ketonen durch McMurry-Reaktion aus Bis-Alkenen durch Olefin-Metathese aus Aldehyden/Ketonen durch Silylenoletherbildung aus Alkoholen durch Eliminierung aus Halogeniden durch Eliminerung aus Epoxiden durch Eliminerung/Reduktion aus Aromaten durch Birch-Hückel-Reduktion aus Alkenen und Dienen durch Diels-Alder-Reaktion Alkine aus Olefinen durch Eliminerung aus Aldehyden/Ketonen durch Seyferth-Reaktion bzw. Bestmann-Variante aus Aldehyden durch Corey-Fuchs-Reaktion aus Aldehyden/Ketonen durch Addition von Li-CH2X aus Halogeniden/Tosylaten und Acetyliden durch SN aus Alkenon durch Eschenmoser-Fragmentierung aus Aldehyden/Ketonen durch Reppe-Reaktion aus Acetyliden durch Isomerisierung aus Halogeniden durch Glaser-Kupplung 5 Alkohole aus Halogeniden durch Hydrolyse aus Estern durch Hydrolyse aus Epoxiden durch Reduktion aus Olefinen durch Addition von Wasser aus Olefinen durch Hydroborierung/Oxidation aus Aldehyden/Ketonen durch Reduktion aus Aldehyden/Ketonen/Carbonsäurederivaten durch AdNC=O aus Epoxiden durch nucleophile Substitution aus Carbonsäurederivaten durch Reduktion aus Ethern durch Etherspaltung aus Aminen durch Diazotierung/Verkochung aus Alkoholen durch Mitsunobu-Reaktion 1,2-Diol aus Olefinen durch cis-Hydroxylierung aus Olefinen durch Epoxidierung/Hydrolyse aus Epoxiden durch Hydrolyse aus 1,2-Diketon/2-Ketoaldehyd/2-Ketocarbonsäurederivat durch Reduktion aus α-Hydroxyketon/α-Hydroxyaldehyd/α-Hydroxycarbonsäurederivat durch Reduktion aus Aldehyden/Ketonen durch Pnakol-Kupplung 1,3-Diol aus 1,3-Diketonen durch Reduktion aus Aldolen durch Reduktion aus α,β-Epoxyketonen durch Reduktion aus α,β-ungesättigten Ketonen durch Oxa-Michael-Addition/Reduktion 6 Ether/Epoxid/Oxetan aus Alkohol und Halogenid (Williamson-Ether-Synthese) aus Ester durch Reduktion aus Acetal durch Eliminierung von Alkohol aus Olefin durch Prileschaev-Reaktion aus α,β-ungesättigtem Keton durch Scheffer-Weitz-Reaktion aus Aldehyd/Keton durch Darzens-Glycidester-Synthese aus Aldehyden/Ketonen und Schwefel-Yliden Aldehyd aus prim. Alkohol durch Oxidation aus prim. Halogenid durch Oxidation aus Carbonsäurederivat durch Reduktion aus Acetal/Halbacetal durch Hydrolyse aus Dithioacetal druch Hydrolyse aus gem. Dihalogenid durch Hydrolyse aus Aromat durch Formylierungs-Reaktion aus Olefin durch Ozonolyse aus Vinyl-Allyl-Ether durch Claisen-Umlagerung aus 1,2-Diol durch Glycolspaltung aus Imin/Oxim/Hydrazon/Semicarbazon durch Hydrolyse aus prim. Nitroverbindung durch Nef-Reaktion Halbacetal/Acetal/Dithioacetal aus Aldehyd/Keton und Alkohol aus Lacton durch Reduktion aus Aldehyd/Keton durch Ethandithiol/1,1-Propanditiol 7 Keton aus sec. Alkohol durch Oxidation aus Imin/Oxim/Hydrazon/Semicarbazon durch Hydrolyse aus sec. Nitroverbindung durch Nef-Reaktion aus Vinyl-Allyl-Ether durch Claisen-Umlagerung aus gem. Dihalogenid durch Hydrolyse aus Halbacetal/Acetal/Dithioacetal durch Hydrolyse aus Imin/Oxim/Hydrazon/Semicarbazon durch Hydrolyse aus Carbonsäurederivat durch Nucleophile Substitution aus Aromat durch Acylierung α,β-ungesättigtes Keton/Aldehyd aus α-Halogenketon/Aldehyd durch Eliminierung aus β-Halogenketon/Aldehyd durch Eliminierung aus Aldehyden/Ketonen durch Aldol-Kondensation aus α-Pheylseleno-Aldehyden/Ketonen durch Eliminierung aus Alkenen durch Oxidation in Allylstellung 2-Hydroxy-Aldehyd/Keton aus Aldehyden/Ketonen durch Corey-Seebach-Reaktion aus α-Halogen-Aldehyden/Ketonen durch Hydrolyse aus α,β-Epoxy-Aldehyden/Ketonen durch nucleophile Substitution aus Aldehyden durch Benzoinkondensation aus Estern durch Acyloinkondensation aus Enolaten durch Davis-Oxidation aus Enolaten durch Oxidation mit MoOPH aus Silylenolethern durch Hassner-Rubottom-Oxidation 8 3-Hydroxy-Aldehyd/Keton aus Aldehyden/Ketonen durch Aldol-Reaktion aus α,β-Epoxy-Aldehyden/Ketonen durch Reduktion Carbonsäure aus Carbonsäurederivaten durch Hydrolyse aus prim. Alkohol durch Oxidation aus Keton durch Baeyer-Villiger-Oxidation/Hydrolyse aus metallogenischer Verbindung und CO2 Carbonsäurehalogenid aus Carbonsäure und Thionylchlorig/Oxalylchlorid/Phosphortribromid/… Carbonsäureanhydrid aus Carbonsäure und P4O10 aus Carbonsäuresalz und Carbonsäurehalogenid aus Carbonsäure und Carbonsäureanhydrid Ester/Lacton aus Carbonsäure und Alkohol durch Versterung aus Estern durch Umesterung aus Keton durch Baeyer-Villiger-Oxidation aus aromatischem Keton durch Dakin-Reaktion aus ω-Hydroxycarbonsäure durch Macrolactonisierung 9 Amid/Lactam aus Carbonsäure und Amin aus Carbonsäurehalogenid und Amin aus Oxim durch Beckmann-Umlagerung aus ω-Aminocarbonsäure durch Macrolactamisierung aus Carbonsäurehalogenid und Methoxymethylamin (Æ Weinreb-Amid) aus Carbonsäuren und Isonitrilen durch Ugi-Reaktion α-Halogencarbonsäure/carbonsäureester aus Carbonsäuren durch Hell-Vollhard-Zelinski-Reaktion aus α-Aminosäuren durch Diazotierung/Halogenierung aus Carbonsäureesterenolat durch Halogenierung α-Hydroxycarbonsäuren aus α-Halogencarbonsäuren durch Hydrolyse aus α-Aminosäuren durch Diazotierung/Hydrolyse aus Cyanhydrinen durch Hydrolyse aus Carbonsäureesterenolat durch Davis-Oxidation aus Carbonsäureesterenolat durch Oxidation mit MoOPH aus Silylketenacetal durch Rubottom-Oxidation α-Aminocarbonsäure aus α-Halogencarbonsäure durch Aminolyse aus Aldehyden, KCN und NH3 durch Strecker-Synthese aus Aldehyden durch Erlenmeyer-Azlacton-Synthese aus Bislactimethern durch Schöllkopf-Reaktion α,β-ungesättigte Carbonsäure aus α-Halogencarbonsäure durch Michaelis-Arbuzov-Reaktion/Horner-Emmons-Reaktion 10 Nitril aus Halogenid durch SN-Reaktion aus Carbonsäureamid durch Wasserabspaltung aus Oxim durch Wasserabspaltung Isonitril aus Formamid durch Wasserabspaltung (Ugi-Isonitril-Synthese) aus Halogenid und AgCN Amin aus Ammoniak und Halogenid durch Alkylierung aus Carbonsäureamid durch Reduktion aus Imin durch Reduktion aus Nitril durch Reduktion aus Aldehyd/Keton, Formaldehyd, Amin durch Mannich-Reaktion aus Halogenid durch Gabriel-Synthese aus Aldehyd und Amin durch reduktive Aminierung (Eschweiler-Clark-Reaktion) Hydroxylamin aus Aminen durch Oxidation aus Hydroxylamin durch Alkylierung Imin/Schiffsche Base aus Aldehyd/Keton und Amin Oxim aus Aldehyd/keton und Hydroxylamin aus Alkanen durch radikalische Nitrosierung aus CH-aziden Verbindungen durch Nitrosierung/Tautomerie 11 Hydrazon aus Aldehyd/Keton und Hydrazin Diazoniumionen aus aromatischen Aminen durch Diazotierung Diazoverbindungen aus α-Aminocarbonylverbindungen durch Diazotierung aus N-Alkyl-N-Nitroso-Harnstoff durch Umlagerung/Elimnierung Azoverbindungen aus Diazoniumionen durch Azokupplung Thiol/Thioether aus Halogenid durch SN Sulfoxid/Sulfon aus Thioether durch Oxidation aus Sulfoxid durch Alkylierung aus Sulfon durch Alylierung aus Sulfonylchlorid durch Alkylierung Sulfonsäure aus Thiol durch Oxidation aus Aromaten durch Sulfonierung aus Aromaten durch Sulfochlorierung/Hydrolyse Phosphonat aus Halogenid und Michaelis-Arbuzov-Reaktion Phosphat aus Halogenid und Phosphorsäuresalz 12 Literatur • „Reaktionsmechanismen“, R. Brückner, Spektrum-Verlag, 3. Aufl. 2004, 70 €, d a s Lehrbuch zu Reaktionen in der Organischen Chemie • „Advanced Organic Chemistry“ Teil A + Teil B , F. A. Carey/ R. J. Sundberg, Springer-Verlag, 5. Aufl. 2007, ≈ 100 € sehr gutes Lehrbuch für Organische Chemie (keine Stoffchemie) • „Organic Chemistry“, J. Clayden/ N. Greeves/ S. Warren, Oxford University Press, 2. Aufl. 2012, ≈ 80 €, sehr gutes Lehrbuch für Organische Chemie; wird es ab WS 2013/14 auch auf deutsch geben. • „Advanced Organic Chemistry“, J. March/ M. B. Smith, Wiley, 6. Aufl. 2007, ≈ 80 €, hervorragend für Fortgeschrittene und als Nachschlagewerk • „Classics in Total Synthesis I“, K. C. Nicolaou, E. J. Soerensen, WileyVCH, 1996, ≈ 60 €, hervorragendes weiterführendes Werk zur Naturstoffsynthese • „Classics in Total Synthesis II“, K. C. Nicolaou, S. A. Snyder, Wiley-VCH, 2003, ≈ 60 €, hervorragendes weiterführendes Werk zur 13 Naturstoffsynthese • „Classics in Total Synthesis III“, K. C. Nicolaou, J. S. Chen, Wiley-VCH, 2011, ≈ 60 €, hervorragendes weiterführendes Werk zur Naturstoffsynthese • „Strategic Applications of Named Reactions in Organic Synthesis“, L. Kürti, B. Czacko, Elsevier-Verlag, 84 €, didaktisch hervorragendes Buch zu Namensreaktionen • „Comprehensive Organic Transformations“, R. C. Larock, Wiley-VCH, 2. Aufl. 1999, ≈ 160 €, Nachschlagewerk („Chemical Abstaracts“ für den Schreibtisch mit 15000 Reaktionen und mit 32000 Literaturstellen) • „Protective Groups in Organic Synthesis“, T. W. Geene, P. Wuts, WileyVCH, 4. Aufl. 2005, ≈ 80 €, Nachschlagewerk zu Schutzgruppen • „Protecting Groups in Organic Chemistry“, P. Kochinsky, Thieme-Verlag, 3. Aufl. 2006, ≈ 60 €, Nachschlagewerk zu Schutzgruppen • „Lehrbuch der Organischen Chemie“, H. Beyer, W. Walter, W. Francke, Hirzel-Verlag, 24. Aufl. 2005, ≈ 80 €, d a s Lehrbuch zu Stoffchemie 14 1. Übersicht über funktionelle Gruppen • Halogene R R1 1 R R2 X R2 X X R1 X R X 1 X R2 3 R X = F, Cl, Br, I • Alkene R • R2 1 R R2 2 R R1 R3 R4 1 R R3 Alkine R H R1 R2 15 • Alkohole, Diole, Triole,… R • R1 R2 R2 R1 OH R 3 HO H R1 2 R HO H R2 HO H H OH Ether, Epoxide, Oxetane R • OH HO H R1 HO H 1 R1 O O R R1 R2 2 O R2 R3 Aldehyde, Ketone, α,β−ungesättigter Aldehyd/Keton, α-subst. Aldehyd/Keton, β-subst. Aldehyd/Keton O R H R O 1 R X O R H R 1 X R R 2 O H X R2 R1 O R O O 2 X H R1 X = F, Cl, Br, I, OH, OR3, SH, SR4, NH2, NR25, Aryl, Alkyl,... O R1 16 • Thioalkohole, Thioether, Thiirane, Thietane, Thioaldehyde, Thioketone R1 HS H R SH R1 • R2 SH 1 R 3 R S 2 R R1 R2 R1 R2 S R3 S S R R2 S R1 H R2 Amine, Hydroxylamine, Hydrazine, Azide R NH2 R N H OH 1 R N H R N H R2 NH2 1 R N R3 R N R2 N N 17 • Halbacetal, Acetal, Dithioacetal, N,O-Acetal, Enolether, Enolester, Enamine, Enamide R3O OH R1 R3O OR4 R2 R1 R2 OR3 R • 1 R 2 R3S SR4 O R1 R2 R1 O R3O NHR4 R2 R1 R 1 O H N NHR3 R3 R2 2 R R1 R2 R3 Imine, Oxime, Hydrazone, Semicarbazone N 1 R R3 2 R N 1 R OH 2 R N 1 R NH2 2 R N R1 H N NH2 O R2 18 • Carbonsäuren, Carbonsäurederivate O R O R1 OH R R2 O O R1 X O R2 X =(F), Cl, Br,( I) O R1 O O N R2 R C N R3 • Diazoverbindungen, Azoverbindungen, Diazoniumsalze R • H C N N R1 N R2 R N N N Nitroso-Verbindungen, Nitroverbindungen, Nitrate R N O R O N O R O N O O 19 • Sulfoxide, Sulfone, Sulfensäuren, Sulfinsäuren, Sulfonsäuren O S R1 R2 • R S OH R O S O O S R OH OH Phosphine, Phosphite, Phosphonate, R1 • O O S R1 R2 R2 P R3 OR2 P R1O OR3 R1 O P OR3 OR2 Kohlensäurederivate O HO O O OH 1 R O 2 OR RO O X X =(F), Cl, Br,( I) 1 R 2 O R R1 O N N R4 R3 20 R2 • Wichtige Bindungslängen [pm] C (sp3) C (sp2) C (sp) H 109 108 107 C (sp3) 153 151 147 C-C: 148 C=C: 132 C-C: 143 C=C: 131 C (sp2) C-C: 138 C≡C: 118 C (sp) F 140 134 127 Cl 179 173 163 Br 197 188 179 I 216 210 199 O C-O: 143 C-O: 134 C=C: 121 C=O: 116 N C-N: 147 C-N: 138 C=N: 128 C≡N: 114 S 182 175 C-S: 168 C=S: 167 Wenn keine konkrete Bindung angegeben ist, handelt es sich immer um Einfachbindungen. 21 • Wichtige Bindungsenergien Bindung Bindungsenergie [KJ/mol] Bindung Bindungsenergie [KJ/mol] C-H 410 S-H 340 C-C 350 Cl-Cl 243 C=C 620 Br-Br 192 C≡C 830 I-I 151 C-F 480 O-O 130-210 C-Cl 330 N-N 160 C-Br 275 N=N 420 C-I 220 N≡N 950 C-O 370 P=O 575 C=O 740 Si-C 290 C-N 300 Si-H 320 C=N 600 Si-O 370 C≡N 850 Si-F 580 C-S 250 Si-Cl 391 O-H 460 Si-Br 310 N-H 390 Si-I 230 22 2. Synthese von Halogeniden 2.1. aus Alkanen durch radikalische Halogenierung • OC2 Folien 6-21 • Radikalkettenmechanismus • Bindungsenergien entscheidend für Reaktivität und Selektivität • Radikalische Fluorierung sehr stark exotherm (explosiv) • Radikalische Chlorierung stark exotherm, wenig selektiv • Radikalische Bromierung exotherm, gut kontrollierbar, selektiv • Radikalische Iodierung endotherm, läuft nicht ab 23 Radikalische Halogenierungen spielen zur Synthese von Grundchemikalien in der Industrie eine große Rolle. Beispiel 1) Synthese von Methylchlorid, Methylenchlorid, Chloroform, Tetrachlorkohlenstoff CH4 + Cl2 CH3Cl + CH2Cl2 + CHCl3 + CCl4 + Cl2 + CH4 CH3· 2 Cl· CH3Cl 37% CH2Cl2 41% CHCl3 19% CCl4 – HCl 3% Beispiel 2) n-Propylchlorid und iso-Propylchlorid Cl2 Cl Cl + 300 °C 48% 52% 24 2.2. aus Alkenen durch Wohl-Ziegler-Bromierung O N – I Br – Br O H N O + H 2 Br · Br Br O 1 2 R R Br · R 1 2 R Br—Br R 1 R · H H H H Br • OC2 Folie 22 • NBS sorgt für geringe Konzentration an elementarem Brom • bei geringen Br2-Konzentrationen ist allylische Substitution kinetisch bevorzugt • funktionert auch in Benzylstellung • Nebenreaktionen: Doppelbindungsverschiebung, cis-transIsomerisierung, Mehrfachbromierung 25 2 Beispiel 1) Teilschritt aus einer Synthese von Ginkgolid B H O Br 10 °C / 7 h O O H O H tBu O O H 48% H O OMe O O O H tBu H OMe Br NBS / CCl4 O O H OMe HO O H O H 24% OMe H O H HO O H tBu O Ginkgolid B H tBu O H E. J. Corey et al., J. Am. Chem. Soc. 110, 649-651 (1988) E. J. Corey et al., Chem. Soc. Rev. 17, 111-133 (1988) 26 Beispiel 2) Synthese von Octulosonsäurederivaten MEMO H NBS / CCl4 O O COOMe O COOMe O Br MEMO H O O OH H Octulosonsäure O HO COOH HO spielt eine Rolle beim Überleben von Helicobacter pylorii im Magen J. Tadanier et al., Carbohydr. Res. 201, 185-207 (1990) 27 2.3. aus Alkenen/Alkinen durch Addition von HX • OC2 Folien 97, 98 • HX kann nach ionischem oder radikalischem Mechanismus an Doppelbindungen addiert werden • radikalische Addition verläuft nach Anti-Markownikow HBr R Br R O2 • radikalische Addition in Gegenwart von Luftsauerstoff oder Radikalstarter • ionische Addition verläuft nach Markownikow Br HBr R • N2 R ionische Addition unter Ausschluss von Luftsauerstoff! 28 Beispiel 1) Teilschritt aus einer Synthese von Cycloviracin B1 HBr Br (Ph-COO)2 Hexan, 0° C OH HO HO HO O HO OH O O O HO OH O O OMe O O O HO OH O OH O OH OH OH O OH O OH A. Fürstner et al., J. Am. Chem. Soc. 125, 13132-13142 (2003) OH OH 29 Beispiel 2) Teilschritt aus einer Synthese von Streptomyceten-“Abgasen“ HBr Br O2 O O muffiger Geruch von Pilzkulturen S. Schulz et al., J. Nat. Prod. 67, 395-401 (2004) 30 2.4. aus Alkenen durch Addition von X2 • OC2 Folie 90 • X2 kann ionisch an Doppelbindung addiert werden • ionische Halogenierungen verlaufen über ein Halogeniumion, wenn ein intermediär auftretendes Carbeniumion nicht stabilisiert werden kann • ionische Halogenierungen laufen über ein einges Ionenpaar, wenn ein intermediär auftretendes Carbeniumion durch geeignete Substituenten stabilisiert werden kann 1 R R 2 Br Br R 1 Br + R2 – Br Br R R 1 2 Br 31 • X2 kann radikalisch an Doppelbindungen addiert werden • ionische und radikalische Addition zeigen unterschiedlichen stereochemischen Verlauf 1 R 2 R Br · Br R 1 R · 2 Br Br Br R R 1 2 Br 32 Beispiel 1) Ausschnitt aus einer Synthese von N-Methyl-Maysenin Br Br2 OH OH Br Cl MeO N N-Methyl-Maysenin O O MeO N H OH O E. J. Corey et al., J. Am. Chem. Soc. 100, 29162918 (1978) 33 Beispiel 2) Isolierbares Bromoniumion Br2 Br + Br – R. S. Brown et al. J. Am. Chem. Soc. 107, 4504-4508 (1985) 34 2.5. aus Alkenen durch Halolactonisierung und verwandten Reaktionen O I I OH I HO I O O I + OH – + –H – HO O + NaHCO3 O O NaHCO3 –H I + I I O O • OC2 Folien 91-94 • Halolactonisierungen führen zur Ringbildung mit eindeutiger Stereochemie • Analoge Reaktion: Haloalkoxylierung, Halohydroxylierung 35 Beispiel 1) Teilschritt einer Synthese von Giberellin A3 HO HO H I OH I2 / NaHCO3 / H2O HO • HO H COOMe COOH Me THF / CH2Cl2 / RT • Me O O O COOMe OH H HO Me OH H O COOH E. J. Corey et al., J. Am. Chem. Soc. 100, 8031-8034 und 8034-8037 (1978) 36 Beispiel 2) Teilschritt einer Synthese Perhydrohistionicotoxin Br NBS N O OH N CH2Cl2 / 0 °C O O 83% Perhydrohistionicotoxin N H H OH neurotoxisch G. E. Keck et al., J. Org. Chem. 47, 3590-3591 (1982) 37 2.6. aus Alkenen und Alkinen durch Hydroborierung - Halogenierung • OC2 Folien 100-102 • Hydroborierung von Alkenen ergibt Trialkylborane • geeignete Hydroborierungsreagenzien: Boran, Thexylboran, 9-BBN • Bromierung ergibt in einer Radikalreaktion α-Brom-boran • Das α-Brom-boran wird von HBr in Bromalkan umgewandelt. R R BH3 R R B R B R – HBr R + HBr Br + Br2 Br Br R Br 38 • Hydroborierung-Halogenierung von Alkinen ist die beste Methode zur Herstellung von Vinylbromiden und Vinyliodiden • geeignete Hydroborierungsreagenzien: Disiamylboran, Catecholboran • Hydroborierung führt zunächst zum Trialkenylboran (AntiMarkownikow) • Trialkenylborane können mit elementarem Halogen in Gegenwart von Base zu den entsprechenden Alkylhalogeniden umgesetzt werden • Man muss zwei Mechanismen unterscheiden 1) Zugabe von Catecholboran – Zugabe von Halogen – Zugabe von NaOMe Æ führt zur Konfigurationsumkehr im Vergleich zum Vinylboran 2) Zugabe von Catecholboran – Zugabe von NaOMe – Zugabe von Halogen Æ führt zu Retention der Konfiguration im Vergleich zum Vinylboran 39 Zugabe von Catecholboran – Zugabe von Halogen – Zugabe von NaOMe O B H O O R Br2 B O R O Br B O R Br O H R Br ” B O Me O Br H –” Me O O O H R Br B H O Br Br H R B H O Br O – B OMe O R Br – Br” H H 40 Zugabe von Catecholboran – Zugabe von NaOMe – Zugabe von Halogen Br Br O B H O O R – ” Me O O B O R R ” B O O Me – Br” R O – Br B OMe O 41 Beispiel 1) Teilschritt aus einer Synthese von Dihydroxyeicosatetraensäuremethylester 1) O B H TBDMSO O TBDMSO MeOOC MeOOC OTBDMS I OTBDMS 2) NaOH 3) I2 OH COOMe OH K. C. Nicolaou et al., Angew. Chem. 101, 621-623 (1989) 42 Beispiel 2) Teilschritt aus einer Synthese von Bombykol O 1) O B H Br 2) Br2 3) NaOMe OH Bombykol: Sexuallockstoff des Seidenspinners Bombyx mori A. Suzuki et al., Tetrahedron 39, 3271-3276 (1983) 43 2.7. aus Alkoholen durch SN- und SNi-Reaktion • OC2 Folie 61 • Reagenzien: Thionylchlorid SOCl2, Phosphortribromid PBr3, Phosphortrichlorid PCl3, HCl, HBr, HI • Nachteil: Edukte und Produkte müssen säurestabil sein! • In Gegenwart von Pyridin verläuft die Reaktion mit Thionylchlorid unter Inversion der Konfiguration an einem secundären C O O R1 H Cl S Cl R2 + N O 1 – N H “ ” Cl R1 –” O H “ S Cl O Cl 1 2 R R R2 R O S Cl 2 R Cl Inversion weil SN2 44 • Ohne Pyridin verläuft die Reaktion unter Retention der Konfiguration an einem secundären C –” O H “ S Cl O Cl 1 2 R R O O R1 H Cl S Cl R2 O 1 R S “ O R1 – HCl O S R Cl 2 O ” Cl R2 Cl SNi 1 R 2 R • mit PCl3 und PBr3 bilden sich zunächst Phosphite, die dann durch Chlorid oder Bromid substituiert werden. • analoges gilt für die Reaktionen mit HCl, HBr und HI. • Nebenreaktionen: Umlagerungen, Eliminierungen 45 Beispiel 1) Teilschritt aus einer Synthese von Hybocarpon OMe OMe SOCl2 OH MeO Cl MeO OMe OMe OH O O HO HO OH HO O O OH HO O OH Hybocarpon Pflanzenwachstumsregulator aus Flechten C. L. L. Chai et al., J. Org. Chem. 71, 992-1001 (2006) 46 Beispiel 2) Teilschritt aus einer Synthese von Mispyrinsäure OTBDPS OTBDPS PBr3 / Py OH Br COOH HOOC Mispyrinsäure DNA-Polymerase-Inhibitor aus Mischocarpus pyriformis K. Mori et al. Org. Biomol. Chem. 2, 2236-2244 (2004) 47 2.8. aus Alkoholen durch Appel-Reaktion • OC2 Folien 33-44 • Reagenzien: PPh3/CCl4 oder PPh3/CBr4 oder PPh3/CI4 • Reaktion verläuft streng nach SN2 am secundären C-Atom X CX3 Ph3P O H 1 ” ” CX3 2 R R “ Ph3P X “ Ph3P R R2 1 R R 1 2 R1 PPh3 O O R1 O O “ ” X Ph3P X R 2 R2 X R1 R2 48 • Appel-Reaktion ist ein Spezialfall der Mukaiyama-Redoxkondensation PPh3 / X-Y OH R1 R2 X R1 – Ph3P=Y R2 O X-Y = N Hal Hal–Hal PhS–SPh PhSe–SePh O X 1 R 2 R Hal Hal = 1 R R 2 R1 SePh SPh R2 R1 R2 R1 R2 49 Beispiel 1) Teilschritt aus einer Synthese von Lentiginosin OBn PPh3 / CBr4 OBn Br OH NEt3 OBn OBn OBn OBn CH2Cl2, 0 °C 89% HO H HO N Y. H. Jung et al., Org. Lett. 8, 4101-4104 (2006). 50 Beispiel 2) Teilschritt aus einer Synthese von Dictyostatin OTBDMS TBDMSO PPh3 / I2 OH OTBDMS TBDMSO I ImH OH HO O O OH OH P. V. Ramachandran et al., Org. Lett. 9, 157-160 (2007). 51 2.9. aus Halogeniden durch Finkelstein-Reaktion • OC2 Übung 3 Aufgabe 4c • Umwandlung von Halogeniden ineinander, meist Chloride in Bromide oder Iodide und Bromide in Iodide • verwandte Reaktion: Umwandlung von Tosylaten in Halogenide durch Alkalimetallhalogenide oder Tetrabutylammoniumhalogenide • Es handelt sich um Gleichgewichtsreaktionen • GG wird durch Reagenzüberschuss auf die Produktseite verschoben NaI /Aceton Cl R1 R2 R1 nBu4NCl /THF OTs R1 – NaCl I R2 – nBu4NOTs R2 Cl R1 R2 52 Beispiel 1) Teilschritt aus einer Synthese von Brevetoxin B O O Br NaI / 18-K-6 O O O Aceton / RT O I O HO H O H O O H O H H H O H O H HO O H H O H H H O H O H H O K. C. Nicolaou et al., J. Am. Chem. Soc. 117, 10227-10264 (1995) 53 Beispiel 2) Teilschritt aus einer Synthese von Östron OH I 1) TsCl / Py 2) NaI / Aceton O H H H HO K. P. C. Vollhardt et al., J. Am. Chem. Soc. 102, 5253-5261 (1980) 54 2.10. aus Carbonsäuren durch HunsdieckerReaktion • Umsetzung von Carbonsäuren mit HgO/Br2 oder Ag2O/Br2 • Radikalischer Mechanismus + Ag2O O R OH – H2O + Br2 O R OAg – AgBr O R O Br R Br – CO2 • Ausbeuten selten über 60% 55 Beispiel: Teilschritt aus einer Synthese von Exaltolid MeOOC MeOOC COOH HgO / Br2 Br O O Exaltolid Geschlechtsspezifischer Riechstoff V. R. Mamdapur et al., J. Ind. Inst. Sci. 81, 299-312 (2001). 56 2.11. aus Carbonylverbindungen durch αHalogenierung • geeignete Halogene: Cl2, Br2, I2, NBS/CCl4, SO2Cl2 • Man kann die Reaktion säurekatalysiert durchführen H+ O R 1 R HH 2 O R1 H Br — Br O R1 2 R – HBr R2 H Br • Die säurekatalysierte Reaktion verläuft unter thermodynamischer Kontrolle, d.h. über das stabilste Enol! • Die säurekatalysierte Reaktion kann man nach der Monohalogenierung anhalten 57 • Man kann die Reaktion auch im Basischen durchführen. Dann verläuft die Reaktion über das Enolat. • Reaktion im Basischen verläuft unter kinetischer Kontrolle, d.h. über das Enolat, das sich am schnellsten bildet! O – O + OH 1 R R1 R2 HH ” Br — Br R2 – H2O – + OH O R1 – H2O ” R Br O 1 R – Br– Br — Br – Br – H Br O R 2 R2 1 R2 BrBr • Reaktion im Basischen kann man nicht nach der Monobromierung anhalten • Steuerung der Selektivität durch Verwendung von Silylenolethern, Silylketenacetalen oder Enaminen 58 Beispiel 1) Teilschritt aus einer Synthese von Velloziolon NIS OTMS O O I HO H B. B. Snider et al., J. Am. Chem. Soc. 57, 4883-4888 (1992) 59 Beispiel 2) Teilschritt aus einer Synthese von Oxosilphiperfolen O OTMS OEt NaI / mCPBA I OEt TMS O H C. Sha et al., J. Org. Chem. 63, 2699-2704 (1998) 60 • verwandte Reaktion: Hell-Vollhard-Zelinski-Reaktion P / Br2 O R R OH Br2 R Br Br OH R Br Br O O R HBr O OH O R OH Br + O R Br 61 • verwandte Reaktion: Haloform-Reaktion • OC2 Folie 161 O R H ” O—H O O NaOH / Br2 Br HH ” O Br R H Br Br — OH O Br R O H + ” Br Br Br BrBr O + O O NaOH / Br2 O OH R BrBr R NaOH / Br2 Br — OH ” O—H Br R O R R HH Br — OH ” + CHBr3 +H R OH 62 2.12. aus Aromaten durch Kernhalogenierung • OC2 Folien 124-126 • zur Einführung von Cl, Br, I • Reaktivität: Cl > Br > I • Elektrophile aromatische Substitution KKK (Kälte Katalysator Kern) • Geeignete Katalysatoren: FeX3, AlX3, generell: Lewis-Säure • reaktive Aromaten benötigen zur Halogenierung keinen Katalysator • direkte Fluorierung schwierig • direkte Iodierung geht nur bei Phenolderivaten gut 63 Beispiel: Teilschritt aus einer Synthese von Durocarmycin SA ICl / THF / 60 °C H2N BnO I H2N NO2 BnO MeOOC OMe H N O NO2 OMe N O N H OMe K. Hiroya et al., Org. Lett. 6, 2953-2956 (2004) 64 2.13. aus Aromaten durch Sandmeyer-Reaktion • OC2 Folie 145 • zur Einführung von Cl, Br, CN in Aromaten • gelegentlich wird auch die Einführung von I und NO2 als SandmeyerReaktion bezeichnet • Radikalmechanismus induziert durch CuX (X = Cl, Br, CN, BF4) Ar–Cu–X “ Ar N N – Y + Cu+X– Ar · Ar N N · – Cu2+X–Y– – N2 + Cu2+X–Y– Ar X – Cu+Y– X Ar Cu Y • Nebenprodukte: Biaryle, Phenole, Azoverbindungen 65 Beispiel: Teilschritt aus einer Synthese von Dichroanal B OH OMe NH2 CHO 1) NaNO2 / HBr / 0 °C 2) CuBr / H2O / 80 °C OH OMe Br CHO OH OMe OHC A. K. Baerjee et al., Org. Lett. 5, 3931-3933 (2003) 66 2.14. aus Aromaten durch Schiemann-Reaktion • OC2 Folie 145 • zur Einführung von F in Aromaten • die Reaktion wird mit ArN2+BF4- durchgeführt, das aus ArN2+Cl- durch NaBF4 oder HBF4 ausgefällt wird. • Neuere Variante: Diazotierung in Gegenwart von Py-HF • Reaktion verläuft nach einem SNAr1-Mechanismus über ein Phenylkation. 67 Beispiel: Teilschritt aus einer Synthese von 2-Fluorapomorphin MeO OPiv MeO NH2 OPiv 1) HPF6 / tBuONO 2) N H N N“ BF4– Bn HO F OH H N Bn F Fluorapomorphin H N Potentielles Medikament gegen Parkinson H3C M. Begtrup et al., Eur. J. Org. Chem. 2005, 4428-4433 68 2.14. aus Aromaten durch Haloalkylierung • OC2 Folie 137 • Umsetzung von Aromaten mit Formaldehyd oder höheren Aldehyden in Gegenwart von HCl (Blanc-Reaktion) oder HBr • elektrophile aromatische Substitution gefolgt von SN1 H H O H H+ H H O “ OH OH – H+ “ H “ OH2 + H+ X “ -H2O – +X 69 Beispiel: Teilschritt einer Synthese von Nakijichinon A MeO OMe (CH2O)n MeO OMe MeO OMe HBr / HOAc MeO OMe Br H O NaI / Aceton MeO OMe MeO OMe N COOH H OH O I H. Waldmann et al., J. Am. Chem. Soc. 123, 11586-11593 (2001) 70 3. Synthese von Alkenen 3.1. aus Alkinen durch Lindlar-Hydrierung • Lindlar-Paladium = Pd auf CaCO3 oder auf Kohle vergiftet mit Pb(OAc)2 oder mit BaSO4 oder mit Chinolin • vergiftetes Pd ist weniger reaktiv als unvergiftetes Pd • endständige Alkine werden zum endständigen Alken hydriert • innere Alkine werden stereospezifisch zum cis-Alken hydriert • Wasserstoffaufnahme muss kontrolliert werden! Reaktion nach Aufnahme von einem Äquivalent Wasserstoff abbrechen! Wasserstoffmenge 2 eq Unvergiftetes Pd 1 eq Lindlar-Pd Reaktion abbrechen! Zeit 71 Beispiel: Teilschritt einer Synthese von Hybridalacton O H O OTBDMS H H O O Et H H2 / Pd / CaCO3 Chinolin O H OTBDMS H H O O O Et H O H O H H Et O H E. J. Corey et al., J. Am. Chem. Soc. 106, 2375-2377 (1984). 72 3.2. aus Propargylalkoholen durch WhitingReaktion • Propargylalkohole werden durch LiAlH4 oder Red-Al bei RT oder darunter stereospezifisch zu trans-Allylalkoholen reduziert. • Reaktion geht schlecht bei isolierten Dreifachbindungen (T>>100 °C) H ” H Al H H R O H H ” H Al H H R – H2 O O Al ” O O H – ” O R H + R O Al ” O O + H / H2O O H R H 73 Beispiel: Teilschritt einer Synthese von Bengamid E OH O OH LiAlH4 OH S OBn Et2O / Rückfluss OH OH O O OBn N N OH OMe M. Hanaoka et al., J. Org. Chem. 60, 5910-5918 (1995). 74 • Verwandte Reaktion: Birch-Hückel-artige Reduktion von Alkinen OH O ( )9 N BOC OTBDMS OH Li / EtNH2 – 78 °C ( )9 HO OTBDMS NH2 R. Bittmann et al., J. Org. Chem. 68, 7046-7050 (2003). 75 3.3. aus Alkenen durch Isomerisierung der Doppelbindung • Isomerisierung der Doppelbindungsgeometrie: cis-trans-Isomerisierung • cis-trans-Isomerisierungen können bei katalytischen Hydrierungen ablaufen (merkt man aber oft nicht! Warum?) • cis-trans-Isomerisierungen können durch Bestrahlen mit UV-Licht bewirkt werden • cis-trans-Isomerisierungen können durch I2 katalysiert werden • cis-trans-Isomerisierung führt zum stabilieren trans-Isomer • Isomerisierung der Doppelbindungsposition • kann durch Hydrierkatalysatoren ohne Zusatz von Wasserstoff bewirkt werden 76 Beispiel 1) Isomerisierung von Allylethern (Schutzgruppe für Alkohole) und Allylestern (Schutzgruppe für Säuren) O O H H NaH Molekül O Molekül OH K2Cr2O7 H2SO4 / H2O Br Aceton O O HO Pd / C / MeOH Molekül O HO Molekül O O PPTS / H2O HO Molekül OH 77 Beispiel 2) Teilschritt einer Synthese von Anhydromarasmon O O O O H H Ir (CO)Cl (PPh3)2 O O O H O H Ethanol / Wasser H 70 °C / 6 h O O O O H H C. Wallner et al., Eur. J. Org. Chem. 2003, 3060-3064. 78 3.4. aus Aldehyden/Ketonen durch Wittig-Reaktion • OC2 Folien 194, 195 • Aldehyde/Ketone reagieren mit Phosphor-Yliden in einer thermischen konzertierten [2+2]-Cycloaddition zu Oxaphosphetanen, die dann in einer [2+2]-Cycloreversion zu Alken und Triphenylphosphinoxid zerfallen • Stabilisierte Ylide ergeben trans-Alkene • Destabilisierte Ylide ergeben cis-Alkene • Nicht-stabilisierte Ylide ergeben Mischungen aus cis-und trans-Alkenen Ph Ph “ P R ” › Ph P Ph Ph R Ph [2+2] H O Ph Ph P Ph O R H H R' R' R R' O PPh3 79 Beispiel 1) Teilschritt einer Synthese von Mycolacton “ Ph3P ” COOEt Me O PGO PGO Me H Toluol / 110 °C COOEt PGO PGO H 12 h / 84% OH OH O O OH O O Y. Kishi et al., Org. Lett. 6, 4901-4904 (2004) OH OH 80 Beispiel 2) Teilschritt einer Synthese von Cryptophycin 3 “ Ph3P O Ph TBDMSO COOtBu ” H Ph COOtBu TBDMSO H H O Ph O O O O H N N H Cl O OMe M. E. Maier et al., Eur. J. Org. Chem. 2005, 317-325. 81 3.5. aus Aldehyden durch Horner-EmmonsReaktion • OC2 Folie 196 • Aldehyde reagieren mit deprotonierten Phosphonaten zu trans-Alkenen • Mechanismus: Addition an Carbonylgruppe – Ringschluss zum Oxaphosphetan – konzertierter Zerfall des Oxaphosphetans zu Phosphorsäureester (wasserlöslich!) und Alken O MeO MeO O 1 R R2 ” P ” HO 1 R H O P H R2 R1 2 R + HO OMe OMe R1 O P H R2 ” OMe OMe ” O P OMe O OMe 82 Beispiel 1) Teilschritt einer Synthese von Isodominsäure O MeO COOMe MeO P ” COOMe COOMe Me OHC MeOOC COOMe N BOC COOMe N BOC COOH HOOC N H COOH J. Clayden et al., J. Am. Chem. Soc. 127, 2412-2413 (2005) 83 Beispiel 2) Teilschritt einer Synthese von Kuhistaferon O O O O Et3N / LiBr O O O O P O THF / 60 °C MeO OMe O 80 % O H O O O OH K. Shishido et al., Tetrahedron Lett. 46, 1269-1271 (2005) 84 3.6. aus Aldehyden durch Still-Gennari-Reaktion und Ando-Reaktion • Aldehyde reagieren mit deprotonierten Phosphonaten zu cis-Alkenen • Mechanismus: vermutlich analog zur Horner Emmons-Reaktion • veränderte elektronische Eigenschaften am P bewirken vermutlich den umgekehrten stereochemischen Verlauf Horner-Emmons CH3 O O CH3 O P COOMe HR LDA, LHMDS, NaH, KOtBu Still-Gennari CH2 CF3 CF3 Ando O O O CH2 O P COOMe HR KHMDS / 18-K-6 P O O COOMe HR KHMDS / 18-K-6 85 Beispiel: Teilschritt einer Synthese von Discodermolid OCH2CF3 P OCH2CF3 MeO O O O O O NaH / THF ” MeO O O O O – 10 °C O OCH2CF3 P OCH2CF3 O H OTBDMS OPMB OTBDMS MeO H O O O O OTBDMS OPMB OTBDMS HO O O OH OH O OH O NH2 I. Paterson et al., Org. Lett. 6, 4933-4936 (2004) 86 3.7. aus Aldehyden/Ketonen durch PetersonOlefinierung • OC2 Folie 193 • Trialkylsilylsubstituierte Carbanionen [R3Si-CH-Z] können mit Aldehyden oder Ketonen zu Alkenen umgesetzt werden („Silyl-WittigReaktion“ wegen Analogie zu Wittig). Z = elektronenziehende Gruppe - O Me3Si Base Z O ” Me3Si Z H R O ” Me3Si Z H H H Z H Z R H R H H H R R Me3Si Z H H H Rotation R ” Z Me3Si H ” Me3Si Z H H O ” Z H Z H R O ” H ” Me3Si O R H Me3Si O H R 87 Beispiel 1) Teilschritt einer Synthese von Buflavin O iPrO SiMe3 N SiMe3 CHO O CsF / DMF iPrO 110 °C MeO MeO MeO N H H N MeO V. Sneckius et al., Tetrahedron Lett. 39, 1325-1326 (1998). 88 Beispiel 2) Teilschritt einer Synthese von Lancifolol Me3Si COOEt TBDMSO TBDMSO O TBDMSO COOEt N” Li+ EtOOC 93 % 7% HO H. Monti et al., Tetrahedron Lett. 42, 6125-1328 (2001). 89 3.8. aus Aldehyden/Ketonen durch BamfordStevens- und Shapiro-Reaktion • Bei der Bamford-Stevens-Reaktion werden Aldehyde oder Ketone mit mindestens einem α-H-Atom in Alkene überführt. • Zunächst wird aus der Carbonylverbindung das Tosylhydrazon hergestellt. Das Tosylhydrazon kann basisch unter protischen Bedingungen(NaOEt/EtOH oder KOtBu/tBuOH) gespalten werden. O H S NH NH2 O O N N 2 R1 R ” N N 2 R 1 R cat. H+ 1 ” R R2 +H H H R + H H “ N N LM R H “ R2 1 H H H Ts R2 1 H H protisches “ N N – Ts– Ts ” O – N2 1 R H H R2 -H+ 1 R R2 90 • Man kann das Tosylhydrazon auch unter aprotischen Bedingungen (LDA/THF, NaH/Dioxan usw.) spalten. O H S NH NH2 O O N N 2 R1 R R cat. H – Ts– R ” H H N ” N N Ts 2 R 1 R H H H H aprotisches “ N N 1 Ts R2 1 + ” LM R R2 2 1 – N2 R R 1 R2 H H 91 • Verwendet man als Base 2 Äquivalente MeLi, nBuLi, secBuLi, tBuLi oder eine andere Li-organische Verbindung, dann läuft die ShapiroReaktion ab. O H S NH NH2 O O N N 2 R 1 R Li N R 1 Li N R N N ” H H R R H H H aprotisches Li LM 1 Ts R2 1 R 2 – Ts 2 cat. H+ – Ts ” – N2 R R 1 2 H2O 1 R R2 H 92 Beispiel 1) Teilschritt einer Synthese von Ibogamin COOEt H 1) Ts-NH-NH2 COOEt COOEt 2) Na / Ethylenglycol H O N N H H J. W. Huffman et al., J. Org. Chem. 50, 1460-1464 (1985) 93 Beispiel 2) Teilschritt einer Synthese von Phytocassan D O OTBDMS H 1) Ts–NH-NH2 PPTS / THF H OTBDMS H TBDMSO H 2) LDA / THF H TBDMSO H O H O H HO H K. Mori et al. Eur. J. Org. Chem. 2000, 4079-4091. 94 3.9. aus Alkoholen durch Wasserabspaltung • Prinzip: Überführung von OH-Gruppe in bessere Abgangsgruppe (Wasser, anorganischer Ester, organischer Ester ). • Säurekatalysierte Abspaltung von Wasser verläuft nach E1 • es bildet sich das stabilste Alken (thermodynamische Kontrolle) • Nach Überführen der OH-Gruppe in Tosylat, Mesylat, Triflat erfolgt Eliminierung im Basischen nach E2 • Für säureempfindliche Substanzen kann man Wasser unter sehr milden Bedingungen mit Burgess-Reagenz oder mit Martin-Reagenz abspalten O “ Et3N S N COOMe O ” Burgess Ph F3C CF3 Ph O S Ph F3C CF3 O Ph Martin 95 Burgess-Reagenz O O O “ Et3N S N COOMe O ” H ” ” “ Et3N S N COOMe H ” O O “ O “ Et3N S N COOMe ” O O – H+ O ” O S N COOMe O HH Martin-Reagenz H Ph F3C CF3 Ph O S ∆ Ph F3C CF3 O Ph – Ph F3C CF3 F3C CF3 Ph Ph O Ph S “ O – H+ Ph F3C CF3 Ph O Ph S O O ” Ph ∆ – “S F3C CF3 Ph O ” Ph O H H 96 Beispiel 1) Teilschritt einer Synthese von Polygodial O COOMe COOMe HO COOMe COOMe F3C S Cl O DMAP / CH2Cl2 H H RT / 86% CHO CHO Frasshemmer für Insekten H K. Mori et al., Tetrahedron 42, 273-281 (1986) 97 Beispiel 2) Teilschritt einer Synthese von Herbolid E 1) Tf2O / Py CH2Cl2 / – 15 °C OH O 2) Li2CO3 / DMA O 60 °C / 30 min. O O O O OH HO O O J. R. Pedro et al., Tetrahedron Lett. 33, 5253-5256 (1992). 98 Beispiel 3) Teilschritt einer Synthese von Ikarugamycin H N O HO H O H Burgess-Reagenz H H N H COOMe COOMe N H H O C6H6 / 80 °C O OMe MeO H O O OMe MeO H O H N H H N O H N H H H O OH L. Paquette et al., J. Am. Chem. Soc. 112, 9284-9292 (1990). 99 Beispiel 4) Teilschritt einer Synthese von Forskolin O O OTBDPS O O Martin-Reagenz H H H OTBDPS OH Toluol / 60 °C HO H O O HO H OAc OH P. Weltzel et al., Tetrahedron 51, 2947-2952 (1995). 100 3.10. aus vicinalen Diolen durch Corey-WinterReaktion • 1,2-Diole werden mit Thiophosgen und Base umgesetzt. Das cyclische Thiocarbonat wird mit PPh3 oder P(OMe)3 reduktiv gespalten. • cis-Diole ergeben cis-Alkene, trans-Diole ergeben trans-Alkene H O H S “ Cl Cl – CO2 S ” O Cl Cl H O H (MeO)3P O ” O S O P(OMe)3 “ O O – HCl S Cl O S O – HCl H O S O O P(OMe)3 O 101 Beispiel: Teilschritt einer Synthese von trans-Cycloocten S 1) 1) mCPBA OH 2) NaOH OH Cl Cl 2) P(OMe)3 H H E. J. Corey et al., J. Am. Chem. Soc. 87, 934-935 (1965). 102 3.11. aus Halogeniden durch Eliminierung von HX • OC2 Folien 76-80 • Oft überführt man OH-Gruppen in situ in das Halogenid und eliminiert im Basischen (mit POCl3) • Eliminierung verläuft nach E2 und liefert bevorzugt das SaytzeffProdukt (höher substituerte Doppelbindung) • Es kann nicht ganz augeschlossen werden, dass die Eliminierung bereits auf der Stufe des Phosphorsäureesters stattfindet (vgl. 3.9). 103 Beispiel: Teilschritt einer Synthese von Anisatin OAc OAc POCl3 / Py HO O 100 °C O HO O O O O OH HO HO O O H. Niwa et al. J. Org. Chem. 51, 1015-1018 (1986). 104 3.12. aus Aminen durch Hofmann- und CopeEliminierung • OC2 Folien 65-86 • Hofmann-Eliminierung: Amin wird in quartäres Ammoniumsalz überführt („erschöpfende Methylierung“), das dann mit einer Base (NaOH, KOH, AgOH) zusammen erhitzt wird. Man erhält bevorzugt das weniger substituierte Alken (Hoffmann-Produkt) • Cope-Eliminierung: ein tertiäres Amin wird durch H2O2, mCPBA oder DMDO (Dimethyldioxiran, Acetonperoxid, Adam-Reagenz) in das Aminoxid überführt und erhitzt. Das Aminoxid fungiert als Base, die sich selbst eliminiert. 105 Beispiel 1) Teilschritt einer Synthese von Picrasin MeO MeO 1) MeI O O 2) NaOH NMe2 MeO O O H H O O D. S. Watt et al., Tetrahedron Lett., 30, 5989-5992 (1989). 106 Beispiel 2) Teilschritt einer Synthese von Ervitsin H H N NMe2 H H N H O N 1) mCPBA 2) Toluol / 110 °C COOMe H H N H O COOMe N H N H O J. Bosch et al., J. Am. Chem. Soc. 115, 5340-5341 (1993). 107 3.13. aus Estern durch Esterpyrolyse • OC2 Folie 86 • Esterpyrolysen sind syn-Eliminierungen • Esterpyrolysen finden häufig erst ab 400 °C in der Gasphase statt, gelegentlich auch schon viel früher! • Wenn mehrere geeignete H-Atome zur Verfügung stehen, dann erhält man oft Produktgemische. Besser: Estergruppen an Ringsystemen Beispiel: Teilschritt aus Syntheseversuchen von Azadirachtin O O O COOMe OH OH O O O O AcO MeOOC OH H O H O O 0,1 Torr, 165 °C COOMe OH OH O O 97% O S. V. Ley et al., Tetrahedron 47, 9231-9246 (1991) AcO MeOOC OH H O H O O 108 • Verwandte Reaktion: Chugaeff-Reaktion; verläuft bereits ab ca. 150 °C • OC2 Folie 86 Beispiel: Teilschritt aus einer Synthese von Dihydroclerodin H O H H O H 1) NaH / CS2 / MeI HO O O H O H O H 2) 220 °C / 48 h O O H O O H O OAc OAc A. De Groot, J. Org. Chem. 64, 9178-9188 (1999). 109 • Verwandte Reaktion: Sulfoxid-Pyrolyse (bei 100-150°C) und Selenoxid-Pyrolyse (bereits ab -40 °C!) H O HH O HH H S“ S O Ph Ph + H O HH O Se S Ph Ph HH H Ph + HH ” OH H S HH – H2O Ph O Ph OH HH ” OH “ Se S O S O S H HH – H2O Se S Ph O O Se Ph Ph Se OH 110 Beispiel: Teilschritt einer Synthese einer Teilstruktur von Azadirachtin TBDMSO S O O H Ph 1) mCPBA / CH2Cl2 TBDMSO O 2) Toluol / 110 °C O H S. V. Ley et al., Tetrahedron Lett. 28, 221-224 (1987) 111 3.14. aus Alkenen/Alkinen durch Diels-AlderReaktion • OC2 Folien 115-120 • Es reagieren bevorzugt elektronenreiche Diene mit elektronenarmen Dienophilen; Diels-Alder-Reaktion mit normalem Elektronenbedarf. • Die Reaktion verläuft über einen 6-gliedrigen wannenförmigen Übergangszustand, der durch secundäre Orbitalwechselwirkungen (Regel von Alder) stabilisiert wird. • Diels-Alder-Reaktionen werden durch Lewis-Säuren, durch hohen Druck und durch Ultraschall beschleunigt und sind sowohl inter- als auch intramolekular durchführbar. Regioselektivität D D Z Z Z D D D D Z Z Z 112 • gute Diene OMe MeO O • gute Dienophile O O H COOR O CN NO2 OR COOR O O O O O 113 Beispiel 1) Teilschritt einer Synthese von Caribenol A MeOOC O O COOMe Toluol / BHT TBDMSO TBDMSO 120 °C / 24 h / 92 % H O O OH H H Z. Yang et al., J. Am. Chem. Soc. 2010, 132, 13608-13609. 114 Beispiel 2) Teilschritt einer Synthese von Herbindol B Ts Ts N H OMe N OMe OMe OMe O CH2Cl2 / 0 °C H O H N M. A. Kerr et al., Org. Lett., 7, 1215-1218 (2005). 115 3.15. aus Alkenen durch Alder-En-Reaktion • Alken mit allylischem Wasserstoff wird mit einem Enophil (Alken, Aldehyd, Keton, Imin, Thioketon) bei erhöhter Temperatur umgesetzt. • Die Reaktion verläuft über einen 6-gliedrigen wannenförmigen Übergangszustand. • Alder-En-Reaktionen werden durch Lewis-Säuren, durch hohen Druck und durch Ultraschall beschleunigt. • Alder-En-Reaktionen sind sowohl intermolekular als auch intramolekular durchführbar; die intramolekulare Variante verläuft besser. H H H O H O 116 Beispiel: Teilschritt einer Synthese von α-Himachalen H 1) HCl H H OH 2) 150 °C MeO H H OMe H H A. Shrikrishna et al., Tetrahedron Lett., 7, 6867-6870 (2004). 117 3.16. aus Allyl-Vinyl-Ethern durch ClaisenUmlagerung • Umlagerungen von Allyl-Vinyl-Ethern oder Allyl-Phenyl-Ethern bezeichnet man als Claisen-Umlagerung • Die Reaktion verläuft über einen 6-gliedrigen Übergangszustand. • O O O O H HO Es sind zahlreiche Varianten dieser Reaktion entwickelt worden: Johnson-Claisen-Umlagerung, Ireland-Claisen-Umlagerung, Kazmaier-Claisen-Umlagerung 118 Beispiel: Teilschritt einer Synthese von Kuehneromycin B O 150 °C O H O H CHO CHO H C. Wallner et al., Synthesis 2004, 685-688. 119 3.17. aus Alkenen/Alkinen durch CopeUmlagerung • 1,5-Diene reagieren thermisch zu isomeren 1,5-Dienen über einen 6gliedrigen Übergangszustand R R • Oxy-Cope-Umlagerung: R = OH HO • Anionische Oxy-Cope-Umlagerung: R = O ” O O HO ” O _ H2O O 120 Beispiel: Teilschritt einer Synthese von Bilosespen A O O O KH / 18-K-6 OH OMe O THF / 60 °C 97% O H O O OMe COOH H H C. Liao et al., Org. Lett. 5, 4741-4743 (2003). 121 Mechanismus der Cope-Umlagerung bei der Synthese von Bilosespen A 15 O 13 16 17 11 10 16 17 9 9 OMe 7 11 10 12 OH 8 8 O 13 2 12 3 O 9 5 8 O 4 O 4 2 3 1 O 2 3 11 16 O 1 6 10 6 5 4 17 = OMe 7 O 1 OH 6 5 14 15 14 15 12 H 14 7 O O OMe 13 Vorgehensweise bei komplizierten Umlagerungen • Atome durchnumerieren • Bindungen verschieben (eigentliche Umlagerung durchführen) • Gerüst in übliche Darstellung umformen 122 3.18. aus Aromaten durch Birch-HückelReduktion • Birch: Na in fl. NH3 + Aromat + tert.Butanol (Eintopf) • Hückel: Na in fl. NH3 + Aromat; wenn Reaktion fertig, dann vorsichtig ges. NH4Cl-Lösung zugeben (2 Schritte) • Varianten: Li + Amine oder auch Li + HMPT • Aromaten mit elektronenziehenden Substituenten ergeben andere Diene als Aromaten mit elektronenschiebenden Substituenten (Regioselektivität). Z Z Na / NH3 fl. – 37 °C D D Na / NH3 fl. – 37 °C 123 Beispiel: Teilschritt einer Synthese von Aeruginosin 298-A Li / NH3 fl. COOH NH2 MeO COOH EtOH NH2 MeO HO O HO N OH H H O H NH N O N H CH2OH J. Bonjoch et al., Chem. Eur. J. 7, 3446-3462 (2001) N H NH2 124 4. Synthese von Alkinen 4.1. aus Vinylhalogeniden durch Eliminierung von HX • Aus Vinylhalogeniden kann durch starke Basen (Alkoholate, DBN, DBU, LDA usw.) HX eliminiert werden. • E2-Eliminierung: H und Halogen trans eliminieren schnell, H und Halogen cis eliminieren langsam. • Reaktion muss nicht unbedingt vom Vinylhalogenid ausgehen, man kann auch von geminalen oder von vicinalen Dihalogeniden ausgehen (man benötigt dann 2 oder 3 Äquivalente Base; wann benötigt man 3 Äquivalente Base?) • Aus vicinalen oder geminalen Dihalogeniden kann sich auch das Allen bilden, das dann aber zum thermodynamisch stabileren Alkin isomerisiert. 125 Beispiel: Teilschritt einer Synthese von Prostacyclin-Analoga COOtBu COOtBu O O NaOH H H Br H HO HO H nBu4NHSO4 cat. Et2O / Toluol 50 °C H H HO H HO M. Shibasaki et al. J. Org. Chem. 53, 1227-1231 (1988). 126 4.2. aus Aldehyden durch Seyferth-Reaktion • Im Prinzip eine Variante der Horner-Emmons-Reaktion zur Synthese von Alkinen ” O O O ” “N N P MeO MeO H nBu Li O “ N N MeO P MeO 2 ” R H O H MeO P ” R MeO N“ N ” O MeO P O MeO N • N ” R – (MeO)2PO2– R H H R ” N“ – N2 R H H N Nachteil: Instabilität vom Phosphonat; muss immer erst hergestellt werden (umständlich). 127 • besser: Variante von Bestmann: käufliches Reagenz, mildere Reaktionsbedingungen O O ” MeO P MeO N“ N ” K2CO3 / MeOH MeO ” O O ” OMe MeO P MeO N“ N O – O “ N P N MeO MeO 2 ” OMe 128 Beispiel: Teilschritt einer Synthese von Formamycin O PivO TESO O OHC O nC5H11 H HO HO ” “ (MeO)2P–CH–N2 PivO H TESO O O KOtBu /THF nC5H11 H –78 °C O O O O O OH O O HO OMe W. R. Roush et al., Org. Lett. 3, 453-456 (2001). 129 4.3. aus Aldehyden durch Corey-Fuchs-Reaktion • Der Aldehyd wird mit PPh3/CBr4 mit oder ohne Zn umgesetzt. • Im entstandenen Dibromolefin wird mit nBuLi ein Br durch ein Li ausgetauscht und HBr eliminiert. Ph Ph “ P Br Ph Ph Br Br P Ph Ph Br Br Ph Ph P Br Br Ph “ Br ” Br P Br Ph Ph Br Ph Br Ph Br Br O P Ph Ph R ” Br Br Ph Br P Ph Ph Li R Br R Br H3O+ H H Br Wittig R H nBu–Li Li R Li–nBu Br H 130 Beispiel: Teilschritt einer Synthese von Spicigerolid O TBDMSO O O H H 1) PPh3 / CBr4 O O H OTBDMS 2) nBuLi TBDMSO O H H H OTBDMS 3) DMF O O O TBDMSO H H OTBDMS H OAc OAc O O OAc OAc J. A. Marco et al., Tetrahedron Lett. 44, 539-541 (2003). 131 4.4. aus terminalen Alkinen durch Reppe-Reaktion • Das terminale Alkin wird durch eine geeignete Base deprotoniert. • Das in situ erzeugte Acetylid-Anion wird mit Elektrophilen umgesetzt. O Base 1 R H 1 R ” R2 H H R 1 OH R2 wässerige Aufarbeitung • Mit Acetylen kann die Reaktion nur auf einer Seite oder auf beiden Seiten durchgeführt werden. • Acetylide kann man auch einfach mit Alkylierungsmitteln umsetzen und erhält dann interne Acetylene. Base 1 R H R1 ” R2 R – Br– • Br 1 R2 Als Alkylierungsmittel können auch Epoxide eingesetzt werden. 132 Beispiel: Teilschritt einer Synthese von Breynolid OTBDPS OTBDPS tBuLi / THF / –78 °C H H O H O OSEM O OMEM OMe HO H OMEM OMe OH H H O OSEM O H H O S HO HO O O HO D. R. Williams et al., J. Am. Chem. Soc. 112, 4552-4554 (1990). 133 • Weitere Reppe-Reaktionen H EtOH H O Et 130 - 150 °C H H H Ni-Kat. Ni-Kat. 134 4.5. aus α,β-ungesättigten Ketonen durch Eschenmoser-Tanabe-Fragmentierung • Eschenmoser-Variante: Das α,β-ungesättigte Keton wird mittels SchefferWeitz-Reaktion mit H2O2 und NaOH in das α,β-Epoxyketon überführt. Dieses wird mit Tosylhydrazin ins Tosylhydrazon umgewandelt und anschließende mit Base (NaOH, NaOEt, Pyridin usw.) fragmentiert. • Die Reaktion eignet sich vor allem zur Herstellung von mittleren und großen Ringen aus bicyclischen α,β-ungesättigte Ketonen. O H2O2 O N Ts–NH–NH2 O NaOH N N H N Ts ” O–H O + cat. H Ts – Ts– ” O O O 135 • Fehr-Ohlof-Büchi-Variante: Das α,β-ungesättigte Keton wird mittels Tosylhydrazin in das Tosylhydrazon überführt. Dieses wird mit NBS und nBuOH gerührt. Vorteil: a) man muss kein Epoxid herstellen b) Reaktion ist nach 3 min. fertig. O Ts–NH–NH2 N H N Ts NBS N N Ts H O O – H+ R Ts H O R – H+ cat. H+ N Br N R H2O RO OR O 136 Beispiel: Teilschritt einer Synthese von Exaltolid O 1) Ts–NH–NH2 H2 / Pd / C 2) NBS (CH2)14 (CH2)10 (CH2)10 3) sec.BuOH mCPBA (CH2)14 O O O O CH2Cl2 NaHCO3 G. Ohloff et al., Helv. Chim. Acta. 62, 2655-2660 (1979). 137 4.6. aus terminalen Alkinen durch GlaserKupplung • Glaser-Kupplung: Ein terminales Acetylen wird mit wässerigem NH3 und CuCl in schwerlösliches Cu-Acetylid überführt, das an der Luft langsam zum Diacetylen abreagiert. • Eglinton-Variante: Cu(OAc)2 / MeOH / Pyridin / Luft erlaubt homogene Reaktionsführung und damit schnellere Reaktion und bessere Ausbeuten. • Hay-Variante: CuCl / MeOH / TMEDA / reiner Sauerstoff ergibt noch schnellere Reaktion und fast quantitative Ausbeuten. N + Py R H R + – PyH ” + CuCl Cu N R N Cu R N O2 R R – Cu2+ 138 • Verwandte Reaktion: Cadiot-Chodkiewitz-Kupplung 1) CuCl / NH2OH / EtNH2 1 R MeOH / N2 H 2) • 2 R 1 R 2 R Br erlaubt die Synthese von unsymmetrischen Diinen 139 Beispiel: Teilschritt einer Synthese von Endiandrinsäure A SPh H COOMe Ph Ph Cu(OAc)2 / Py / MeOH SPh MeOOC H Ph H H H H H COOMe H K. C. Nicolaou et al., J. Am. Chem. Soc. 104, 5555-5562 (1990). 140 5. Synthese von Alkoholen 5.1. aus Alkylhalogeniden durch Hydrolyse • OC2 Folien 30-56 • Hydrolyse von Alkylhalogeniden wird meist unter basischen Bedingungen durchgeführt (K2CO3 / H2O / Lösungsvermittler) • Reaktion verläuft nach SN1 oder SN2 • Vorsicht bei zusätzlichen hydrolysierbaren Gruppen im Molekül: Ester, Anhydride, Säurehalogenide, Silylether, Enolether, Acetale usw. • Manchmal gelingen Hydrolysen unter neutralen Bedingungen 141 Beispiel: Teilschritt einer Synthese einer Modellverbindung für Azadirachtin H2O / Aceton Br O HO CF3COOAg O 60 °C / 60 h / 98% S. V. Ley et al., Tetrahedron Lett. 31, 431-432 (1990). 142 5.2. aus Epoxiden durch Reduktion • Mit LiAlH4, Red-Al, LiEt3BH wird das Epoxid von der sterisch weniger gehinderten Seite angegriffen (SN2-artiger Verlauf) • Mit NaBH3CN / BF3 wird das Epoxid von der sterisch gehinderten Seite angegriffen (SN1-artiger Verlauf) • Mit DIBALH hängt die Regioselektivität von den Substituenten ab. H ” “ Li H Al H H O ” “ Na H Al O H H ” “ H B CN Na H O Li O “ Et ” H B Et Et Al H 143 Beispiel 1) Teilschritt einer Synthese von Alliacol HO H O LiEt3BH THF / RT O HO H O OH 83% O O OH O K. D. Moeller et al., J. Am. Chem. Soc. 126, 9106-9111 (2004). 144 Beispiel 2) Teilschritt einer Synthese von Disorazol C Na(MeOCH2CH2O)2AlH2 O OH OH OH O N O OMe O O O OMe N O A. I. Meyers et al., J. Org. Chem. 66, 6037-6045 (2001). 145 5.3. aus Epoxiden durch Addition von CNucleophilen • Mechanismus: SN2, Epoxid wird immer von der sterisch leichter zugänglichen Seite her angegriffen • Geeignete C-Nucleophile: Grignard-Reagenzien, Li-organische Verbindungen, Dialkylcuprate, Diarylcuprate Mg R R Br Li LDA R R Z O Z= R O OR NO2 MgBr CN Li2CuCl4 SO2R nBuLi R Br (RCHZ)2CuLi Z Li2CuCl4 R Li (RCH2)2CuLi 146 Beispiel: Teilschritt einer Synthese von Obtusenin H O H OBn MeMgBr Et2O / RT Br H OH H OBn Cl O M. T. Crimmins et al., J. Am. Chem. Soc. 125, 7592-7595 (2003). 147 5.4. aus Aldehyden/Ketonen durch Reduktion • OC2 Folien 176-180 • geeignete Reduktionsmittel: Li+AlH4−, Na+BH4−, Zn2+(BH4− )2, Li+Et3BH−, Na+(MeOCH2CH2O)2AlH2− (Red-Al), Na+BH3CN−, Li+BH3CN−, AlH3, Li+Al(OR)nH4-n−, DIBALH, Na+BH4-n(OAc)n−, BH3, Sia2BH (Secundär-IsoAmyl-Boran), 9-BBN, L-, N-, K-Selectrid, LS-Selectrid • Aldehyde in Gegenwart von Ketonen: NaBH4/tiefe Temperatur, NaBH(OAc)3, BH3-tBuNH2, LiAl(OtBu)3H, LiBH4 • Ketone in Gegenwart von Aldehyden: NaBH4/CeCl3 (Luche-Reduktion) • α,β-ungesättigte Aldehyde/Ketone zu Allylalkoholen: NaBH4/CeCl3 (Luche-Reduktion) • sterisch wenig gehinderte Ketone in Gegenwart von sterisch gehinderten Ketonen: L-Selectrid • sterisch gehindertes Keton in Gegenwart von sterisch weniger gehindertem Keton: MAD/DIBALH 148 Beispiel 1) Teilschritt einer Synthese von Brefeldin A O HO K-Selctrid CN OTBDMS H CN OTBDMS OH O O HO H ” B H M “ M-Selectrid J. Nokami et al., Tetrahedron Lett. 32, 2409-2412 (1991). 149 Beispiel 2) Teilschritt einer Synthese von Saragossasäure MeOOC MeOOC HO MeOOC MeOOC O O O LiBH4 / THF MeOOC HO – 78 °C MeOOC O H O OH O O OH OAc HOOC HOOC HO O O COOH S. F. Martin et al., J. Org. Chem. 67, 4200-4208 (2002). 150 Beispiel 3) Teilschritt einer Synthese von Cheimonophyllon E O H OBz NaBH4 HO H OBz CeCl3 · 7H2O MeOH / – 20 °C HO OH H H O O K. Tadano et al., J. Org. Chem. 67, 6690-6698 (2002). 151 5.5. aus Aldehyden/Ketonen durch Addition von CNucleophilen • OC2 Folie 215 • Grignard-Reagenzien R-MgX R Mg O R OH Br 1) H O 1) R1 H R 2) H3O+ 1) MgBr 2) H3O+ O R2 R1 R 2 R OH + 1 R H R 2) H3O R1 H OH • Nebenreaktionen mit Grignard-Reagenzien: Grignard-Kupplung und 152 Grignard-Reduktion • Grignard-Reaktionen verlaufen meist über einen 6-gliedrigen Übergangszustand, an dem 2 Grignard-Reagenzien beteiligt sind. • selten treten 4-gliedrige Übergangszustände auf • in Einzelfällen liegt ein Radikalmechanismus vor • Schlenck-Gleichgewicht: Br R Mg Mg Br R MgBr2 Br R Mg + Br R Mg R Mg R Mg Br R 153 • häufigster Mechanismus: 1 R Mg R1 Mg Br O O Mg RH Br R1 Br Mg H R 1 Br H3O+ R H O H R Br Mg R Br 1 O O Mg RH R1 Mg R1 Br H R Mg R 1 1 R H3O+ Br 154 • Li-organische Verbindungen reagieren ebenfalls mit Aldehyden und Ketonen, jedoch über einen 4-gliedrigen oder über einen offenen Übergangszustand (hängt vom Lösungsmittel ab!) O 3 2 R R1 R R 1 Li O Li 3 R R 2 L O R3 R1 R2 R1 Li Chelat- ” “ Li L O R1 3 R Li 2 R H3O + 1 R O R3 H R2 O 2 R3 R Ligand • wichtiger Unterschied: bei α,β-ungesättigten Carbonylverbindungen machen Li-organische Verbindungen fast ausschließlich 1,2-Addition, Grignard-Reagenzien dagegen sowohl 1,2- als auch 1,4-Addition 155 (Produktgemisch!) • Zn-organische Verbindungen reagieren ohne Zusatz von Lewis-Säure nur mit Aldehyden, mit Zusatz von Lewis-Säure auch mit Ketonen (Selektivität!) R1 R MgI ZnCl2 R ZnX O ZnCl H 2 R Mg Zn R I R H O ZnX ZnI R 1 Li R Li 2 R + H3O ZnBr2 R ZnBr HO H R1 • R2 Zn-organische Verbindungen können Ester, Amide, Nitril-Gruppen, Sulfoxide, Sulfone, Amine, Dreifachbindungen usw. enthalten, mit denen 156 sie nicht reagieren. • verwandte Reaktion: Aldol-Reaktion • OC2 Folien 188-190 1 O R O LDA 1 R R1 2 HH N R O Li R H H R O R2 H O H Li R2 ” Li HO O H3O+ R2 R R1 H H OH O R2 R 1 RH 157 • verwandte Reaktion: Reformatzki-Reaktion • OC2 Folie 187 OR O Br O Br O O Zn R Zn BrZn R O Zn O Br RO H O O ZnBr O R1 H R2 R1 O R O R O Zn Br R2 ZnBr O 1 R R2 H3O O OR + OH O R1 R2 OR 158 • verwandte Reaktion: Blaise-Reaktion (ergibt β-Ketocarbonsäureester) OR O Br O Br O O Zn R Zn BrZn O R Zn O Br RO H O ZnBr O R 1 CN H R1 N R O Zn Br R O ZnBr N R1 H3O+ O OR O R 1 O OR 159 • verwandte Reaktion: Cyanhydrin-Bildung KCN / H+ O R1 1 TMSCN R TMSCN O H R2 TMSO CN R1 2 R R R1 R2 O HO CN R2 TMSO CN R LDA H Aldehyd TMSO CN ” “ R Li umgepolter Aldehyd Umpolung nach Hünig 160 Beispiel 1) Teilschritt einer Synthese von Cyclindricin C BnO BOC N C6H13 BnO C6H13 O H HO MgBr BOC N H OH N C6H13 O C. Kobayashi et al., Tetrahedron Lett. 45, 5921-5924 (2004). 161 Beispiel 2) Teilschritt einer Synthese von Barbasconsäure O CHO H Li O O O OO H H OH O O OO O H H O O COOH H. Hagiwara et al., J. Org. Chem. 70, 2250-2255 (2005). 162 Beispiel 3) Teilschritt einer Synthese von Macrolactin A HO H CHO H ZnBr H OTIPS OTIPS O O O O OH O O HO HO J. P. Marino et al., J. Am. Chem. Soc. 124, 1664-1668 (2002). 163 5.6. aus Carbonsäurederivaten durch Reduktion • OC2 Folie 214 • Reduktion von Carbonsäuren: LiAlH4, LiBH4, Red-Al, LiEt3BH, NaBH4 + AlCl3 • Reduktion von Carbonsäurehalogeniden: LiAlH4, NaBH4, LiEt3BH, DIBALH, 9-BBN • Reduktion von Estern: Na/NH3fl., BH3, LiBH4, LiEt3BH, AlH3, DIBALH, LiAlH4, • Reduktion von Anhydriden: LiAlH4 • Die Reduktionen laufen prinzipiell zweistufig: 1. Reduktion führt zum Aldehyd (SNC=O); 2. Reduktion führt nach wässeriger Aufarbeitung zum primären Alkohol (AdNC=O) • Vorsicht: Reduktion von Carbonsäureamiden liefert in der Regel Amine! 164 Beispiel 1) Teilschritt einer Synthese von Mniopetal E MOMO O O MOMO MOMO O O MOMO LiAlH4 O OH OEt HO HO O OH O CHO H H K. Tadano et al., J. Org. Chem. 65, 8595-8607 (2000). 165 Beispiel 2) Teilschritt einer Synthese von Brefeldin A H2 / Pd / C DIBALH OTBDMS OTBDMS COOEt OTBDMS OH COOEt TsCl / Py H OTBDMS Li OTBDMS H OTs H OH O O HO H Y. G. Suh et al., J. Org. Chem. 67, 4127-4137 (2002). 166 5.7. aus Carbonsäurederivaten durch Addition von C-Nucleophilen • OC2 Folie 215 • wichtigste C-Nucleophile hierfür: Grignard-Reagenzien, Li-organische Verbindungen • geeignete Carbonsäurederivate: Anhydride, Säurehalogenide, Ester • Mechanismus: SN2t gefolgt von AdNC=O H COOR 2 1 R MgX OH H R1 R1 R2 COOR 2 1 R R2 MgX O R O C O R 2 1 R MgX 1 R OH R1 R1 OH R1 R1 167 Beispiel 1) Teilschritt einer Synthese von Acutumin O MeO MeO HO MeMgI / THF OMe 85 % MeO OMe OMe OMe MeO OH Cl O N O OMe Me Acutumin bewirkt die Wiederherstellung von Erinnerungen bei Gedächtnisverlust OMe S. L. Castle et al., Org. Lett. 7, 1089-1092 (2005). 168 Beispiel 2) Teilschritt einer Synthese von Calcitriol H O MeO TBDMSO MeMgI / Et2O H HO 0 °C / 88 % H TBDMSO H H H HO H HO OH H. Maehr et al., Eur. J. Org. Chem. 2004, 1703-1713. 169 5.8. aus Estern durch Hydrolyse oder Alkoholyse • OC2 Folie 210 • Hydrolyse von Estern liefert Alkohole und Carbonsäuren • Mechanismus: Nucleophile Substitution am Carbonyl-C SN2t • Hydrolyse kann säurekatalysiert oder basenkatalysiert durchgeführt werden O 1 R + O R +H 2 R1 + +H O 1 R O H • “ H O H R2 O“ H O H R O O H 2 H 1 R O O H“ H “ H O – R-OH R1 O H O + –H R2 1 H 2 R O O H R O – H+ 1 R O Säurekatalysierte Hydrolysen sind Gleichgewichtsreaktionen! H 170 • Basenkatalysierte Hydrolysen sind keine Gleichgewichtsreaktionen! Der letzte Schritt ist irreversibel! • Deshalb: Hydrolysen von Carbonsäurederivaten immer basenkatalysiert durchführen! ” O H O R 1 O R O ” 2 1 R + H3O+ wässerige HO 2 – R –O 2 O R – ” O H O R1 O H – H2O O R1 O ” O R1 O H Aufarbeitung 171 Beispiel: Teilschritt einer Synthese von Mniopetal F HO O OMenthyl O H O2N COOH O PNBO OMenthyl O OTBDPS OTBDPS H H DCC / DMAP / CH2Cl2 H RT / 3h / 98% 1) TBAF PNBO O OMenthyl O CHO H 2) DMSO / Py-SO3 H K2CO3 / MeOH HO O OMenthyl O CHO H RT / 1h / 72% J. Jauch et al., Eur. J. Org. Chem. 2001, 473-476. H 172 5.9. aus Alkoholen durch Mitsunobu-Reaktion • Primäre und secundäre Alkohole werden durch Nucleophile mit azidem H in Gegenwart von Diethylazodicarboxylat und Triphenylphosphin in die entsprechenden Substitutionsprodukte überführt • Diethylazodicarboxylat (DEAD) EtOOC-N=N-COOEt dient zum Aktivieren von PPh3 • Carbonsäuren als Nucleophile: aus secundären Alkoholen entstehen zunächst Ester, die nach Hydrolyse in die invertierten Alkohole überführt werden • Mechanismus: bei sekundären Alkoholen immer SN2 • Als Nucleophile sind ebenfalls geeignet: Alkohole, Phenole, Thiole, Thiophenole, Stickstoffwasserstoffsäure HN3, Metallhalogenide 173 H O O EtO PPh3 EtO N N COOEt R 2 R 1 O R 2 R O R “ Ph O P Ph Ph OH– R2 O O Ph Ph EtO “ P Ph N N H OEt O R1 1 R O O 1 R1 Ph N N P“ OEt Ph Ph O ” ” O H EtO N N H O ” OEt O R EtOOC H N N – H COOEt O R 2 R ” O – R2 “ Ph O P Ph Ph Ph3P=O R1 2 O H R 174 Beispiel: Teilschritt einer Synthese von Fasicularin 1) O2N O C6H13 Cbz N OH O PPh3 / DEAD 2) NaOH / THF N COOH C6H13 Cbz N OH C6H13 NCS C. Kibayashi et al., J. Am. Chem. Soc.. 122, 4583-4592 (2000). 175 5.10. aus Alkenen durch Addition von Wasser • OC2 Folien 97, 103 • Säurekatalysierte Addition von Wasser an Doppelbindungen entspricht der Umkehrung der E1-Eliminierung (Prinzip der mikroskopischen Reversibilität) • Als Säuren werden üblicherweise H2SO4, HClO4 oder HNO3 eingesetzt (wenig nucleophiles Anion) • Reaktion geht nur bei Molekülen ohne säureempfindliche Gruppen • Man erhält Addition nach Markownikow (stabilstes Carbeniumion) • Gelegentlich beobachtet man Umlagerungen (Wagner-MeerweinUmlagerung) von Alkylgruppen, Arylgruppen oder Hydrid unter Stabilisierung vom ersten gebildeten Carbeniumion • Weitere Methode: Oxymerkurierung mit Hg(OAc)2 in nassem Lösungsmittel mit anschließender Reduktion mit NaBH4 • Oxymerkurierung/Reduktion ergibt ausschließlich MarkownikowAddition von Wasser an Alkene 176 OAc R1 2 R Hg R3 OAc 4 R OAc OAc Hg “ Hg R1 2 R 3 R R4 1 R 2 R “ H 3 R R4 Merkuriniumion H ” H B H H OAc R1 R2 HO R1 R2 HO Hg 3 R R2 R 4 R HO H 1 3 R R4 + R R2 HO H Hg R R 4 R R2 – Hg • HO R1 R2 3 • Hg 1 R3 4 R • Hg H – H+ H H 1 O HO • R21 Hg 3 R R 4 HO R 3 R R4 R3 4 R 177 Beispiel: 1) Teilschritt einer Synthese von Sclerophytin A H H HO O 1) Hg(OAc)2 THF / H2O H H H H H H O 2) NaBH4 O HO OH H H O H H H H HO O OH vermutete Struktur O HO H H OH richtige Struktur L. E. Overman et al., J. Am. Chem. Soc.. 123, 9033-9044 (2001). 178 Beispiel: 2) Teilschritt einer Synthese von Lineatin 1) Hg(OPiv)2 / THF H2O O OH O O O O 2) NaBH4 Aggregationspheromon von Trypodendron lineatum J. D. White et al., J. Am. Chem. Soc.. 104, 5486-5489 (1982). 179 5.11. aus Alkenen durch Hydroborierung/Oxidation • OC2 Folien 100-102 • Die Sequenz Hydroborierung/Oxidation führt zur Anti-MarkownikowAddition von Wasser an unsymmetrisch substituierte Doppelbindungen (warum?) • Hydroborierungsreagenzien: 9-BBN, Catecholboran, Thexylboran, Disiamylboran, BH3•SMe2 • Oxidation: NaOH / H2O2 / EtOH / THF • Aus 9-BBN entsteht Cyclooctan-1,5-diol und Borsäure. Welche Nebenprodukte treten bei Catecholboran, Thexylboran und Disiamylboran auf? 180 ” 9-BBN R ” R – – OH O R O OH ” R B O B HO ” R ” O B R HO O OH R O O H ” B R THF / RT O OH O B” O O O H B O OH – OH– O O B O ” O H+ / H2O – OH– R OH R O O B O HO + B(OH)3 + OH 181 Beispiel: 1) Teilschritt einer Synthese von Huperzin A N TBDMSO OMe OTBDMS HO H 1) BH3 • SMe2 N 2) NMO CH3 N TBDMSO OMe OTBDMS OMe NH2 J. Mann et al., Org. Biommol. Chem. 2007, 301-306. 182 Beispiel: 2) Teilschritt einer Synthese von Reveromycin A O H OTBDMS O BnO OH 1) BH3 • THF O 2) NaOH / H2O2 Me BnO H H OTBDMS O Me O O O HOOC Me H COOH Me H Me COOH O Me OH M. A. Rizzacasa et al., Org. Lett. 6, 3001-3004 (2004). 183 5.12. aus Aminen durch Diazotierung/Verkochung • Diazotierung von prim. aliphatischen Aminen mit NaNO2/H2SO4 verd. mit anschließender Verkochung geht nur gut bei Aminosäuren • Alle anderen primären Amine ergeben Produktmischungen (warum?) NH2 O O H NaNO2 H2SO4 verd. N N “ H O“ O O H – N2 H O H O OH OH O • Doppelte Inversion = Retention! 184 Beispiel: Teilschritt aus der Synthese von Indolactam V NH2 O O H OH NaNO2 OH O H2SO4 verd. Me H N N OH O N H T. P. Kogan et al., Tetrahedron 46, 6623-6632 (1990). 185 5.13. aus Ethern durch Etherspaltung • Ether werden oft als Schutzgruppe für Alkohole verwendet. Zur Abspaltung der Schutzgruppe muss der Ether gespalten werden. • Methylether: HI (nur für einfache Ether ohne weitere funktionelle Gruppen), BBr3 mit und ohne Dimethylsulfid, BF3 + Ethanthiol, Trimethylsilyliodid • Mechanismus: Aktivierung des Ether-Sauerstoffs durch Elektrophil, Spaltung der Alkyl-O-Bindung durch SN1 (Benzylether, Allylether, tert.Butylether) oder SN2 (Methylether u. a.) R O Br ” Br B Br O R “ CH3 BBr3 CH3 – CH3Br R O Br Br B O R “ CH3 + H 2O BBr2 – B(OH)3 R O Br ” H 186 • Benzylether: Benzylether können selektiv in Gegenwart von anderen Ethern durch katalytische Hydrierung (H2/Pd/C) gespalten werden (wann kann man diese Methode nicht anwenden?). R • H2 / Pd / C O R + OH CH3 p-Methoxybenzylether: p-Methoxybenzylether können selektiv in Gegenwart von anderen Ethern durch Oxidation mit DDQ in feuchtem Dichlormethan gespalten werden. NC CN O O R O CH3 Cl Cl O Hydridübertragung ! HH O R O O HO H O R “ CH3 CH3 H O H – H+ H O R OH + O H CH3 187 • Silylether: Silylether können selektiv in Gegenwart von anderen Ethern durch Tetrabutylammoniumfluorid TBAF gespalten werden. R O R Si R-OTMS • O Si R R-OTES O R Si R-OTIPS O Si R R-OTBDMS O Si R-OTBDPS Wichtig: Abspaltung der Silylgruppe ist kein SN2-Mechanismus, sondern ein Additions-Eliminierungsmechanismus (Oktettaufweitung am Si!) R O F Si ” R O Si F ” R O ” H2O R OH – TMS-F 188 Beispiel: 1) Teilschritt aus der Synthese von Jatropholon A OH OH Me H Bn O Me BF3 • OEt2 C2H5SH H H HO H NaOAc OH Me Me H H O H Amos B. Smith III et al., J. Am. Chem. Soc. 46, 3040-3048 (1986). 189 Beispiel: 2) Teilschritt aus der Synthese von Syncarpinsäure O O MeO BBr3 / CH2Cl2 OMe –80 °C bis RT OMe 90%% HO HO OH OH O O D. Hartmann et al., bisher noch nicht publiziert 190 Beispiel: 3) Teilschritt aus der Synthese von Kuehneromycin A OH DDQ / CH2Cl2 / H2O O O O 0 °C bis RT 100 % OH O O O CHO H H U. Reiser et al., Synlett 2001, 90-92. 191 6. Synthese von Diolen 6.1. aus Alkenen durch cis-Dihydroxylierung • OC2 Folie 159 • cis-Dihydroxylierung von Alkenen wird am besten mit OsO4 in Gegenwart von Wasser (Aceton/Wasser oder tBuOH/Wasser oder THF/Wasser) durchgeführt. Dabei bildet sich ein Bis-osmatester, der in situ zum Diol hydrolysiert wird. • Der elektrophile Angriff von OsO4 an die Doppelbindung erfolgt immer von der sterisch besser zugänglichen Seite. ” O 2“ O Os O ” O R1 + 2H2O – H2OsO4 • R1 R1 O O “ O Os O ” R1 O O Os O O OH OH Wichtig: Os(VIII) wird dabei in Os(VI) überführt. 192 • Nachteil: OsO4 hat bei RT bereits hohen Dampfdruck (Flüssigkeit!), ist sehr giftig und deshalb auch sehr teuer! • Ausweg: OsO4 wird nur in katalytischen Mengen eingesetzt und mit Hilfe eines Cooxidationsmittels immer wieder aus H2OsO4 regeneriert. • Geeignete Cooxidationsmittel: H2O2, tBu-OOH, NMO, K3[Fe(CN)6], früher auch noch NaOCl (Nachteil davon?) • Weitere Verbesserung: Statt flüssigem OsO4 wird gleich festes K2OsO4 eingesetzt. • Weiteres geeignetes Reagenz zur cis-Hydroxylierung: KMnO4 im Alkalischen! Im Sauren und Neutralen werden die erzeugten Diole von KMnO4 oxidativ gespalten. Mechanismus der cis-Hydroxylierung mit KMnO4 analog zum Mechanismus mit OsO4. • Die Reaktion von KMnO4 mit Doppelbindungen ist die sogenannte Baeyer-Probe für Alkene. Das Entstehen von Braunstein zeigt vorhandene Doppelbindungen an. 193 Beispiel: Teilschritt aus der Synthese von Mniopetal E O OMenthyl O CHO OsO4 cat. / NMO HO HO O CHO H H H OMenthyl O Aceton / Wasser / 0 °C H 1 Woche / 69 % 80 % TFA / H2O HO HO O OH O CHO H RT / 4 h / 100 % H J. Jauch et al., Synlett 2001, 87-89. 194 6.2. aus Alkenen durch Epoxidierung/Hydrolyse • OC2 Folie 95 • Alkene lassen sich in trans-1,2-Diole überführen, indem man sie epoxidiert und in situ gleich hydrolysiert. • Reagenzien dazu: Ameisensäure/H2O2, Essigsäure/H2O2 oder Trifluoressigsäure/H2O2. • Zunächst bildet sich die entsprechende Persäure, die mit dem Olefin eine Prileshaev-Reaktion eingeht. • Das Epoxid wird stereoselektiv säurekatalysiert zum trans-Diol hydrolysiert. H2O2 O R R1 O OH R H “O + H+ 1 R H 2 R R2 O – H+ H O OOH HO R 1 R1 R2 R2 OH 195 • Verwandte Reaktionen: „Prevost – trocken“ und „Prevost – nass“ (Woodward-Modifikation der Prevost-Reaktion). • „Prevost – trocken“ führt zu trans-1,2-Diol. O R1 R1 I2 R2 “ I O ” R2 O R1 I R2 O O – I– R1 O R2 O “ O O ” R1 O NaOMe R1 OH R2 O MeOH R2 OH O • „Prevost – nass“ (stöchiometrische Menge Wasser! WoodwardModifikation der Prevost-Reaktion) führt zum cis-1,2-Diol. R1 O 2 O R “ H O H R1 O R2 O H O O R 1 R2 O OH OH– R 1 R 2 OH OH 196 Beispiel: Teilschritt aus der Synthese von Dihydrofastigilin C H O I2 / AcOAg / H2O AcOH / 60 °C O OAc O 12 h / 90% O OAc H O O OH H O O O O O P. T. Lansbury et al., Tetrahedron 43, 5583-5592 (1987). 197 6.3. aus 1,n-Dicarbonylverbindungen, aus 1,nHydroxycarbonylverbindungen und aus Lactonen • geeignete Reduktionsmittel: LiAlH4, NaBH4, Zn(BH4)2, LiEt3BH, Na(MeOCH2CH2O)2AlH2 (Red-Al), NaBH3CN, LiBH3CN, AlH3, LiAl(OR)nH4-n, DIBALH, NaBH4-n(OAc)n, BH3, Sia2BH, 9-BBN, L-, N-, K- Selectrid, katalytische Hydrierung mit Raney-Ni und anderen Katalysatoren, O R2 R1 OH red R 1 O O 2 R R R2 1 OH OH O R1 O red red R2 R 1 R O red OH OH 2 R 1 OH R 2 O O red HO OH 198 Beispiel: 1) Teilschritt aus der Synthese von Ipomeamaron Raney-Ni / H2 O O OH OH Weinsäure / NaBr O H O O T. Sugimura et al., Tetrahedron 50, 11647-11658 (1994). 199 Beispiel: 2) Teilschritt aus der Synthese von Schmetterlingspheromonen O O NaBH4 / MeOH O O Rückfluss O OH OH O OH O O Pheromonkomponente von Idea leuconoe S. Schulz et al., Eur. J. Org. Chem. 2002, 3884-3892. 200 6.4. aus α,β-Epoxyketonen durch Reduktion • Reduktion von α,β-Epoxyketonen mit Samarium(II)iodid in Methanol ergeben β-Hydroxyketone, die mit anderen Reduktionsmitteln (vgl. 6.3.) zu Diolen reduziert werden können. O R O 2+ + Sm 1 2 R + Sm2+ 3+ – Sm R1 – Sm 3+ OH O ” R2 O O R1 ” R1 O ” + +H • R2 O • R2 R1 OH R2 OH H2O / H+ OH O R 1 2 R OH OH red R1 R2 201 Beispiel: Teilschritt aus der Synthese von Maitotoxin TBDPSO TBDPSO H BnO O H H OBn O H BnO H OBn H O O O H SmI2 MeOH / 0 °C BnO O H H OBn OBn O H BnO OH H O O Me4N+ BH(OAc)3– H 10 min. / 96% H O H OBn OBn H O H OBn OBn TBDPSO H BnO O H H OBn OBn O H BnO H O H OH H O OH H OBn OBn K. C. Nicolaou et al., J. Am. Chem. Soc. 130, 7466-7476 (2008). 202 6.5. aus Aldehyden und Ketonen durch PinakolKupplung • Behandelt man Aldehyde oder Ketone mit Na, Mg, Al oder anderen Metallen, die als Einelektronendonatoren fungieren können, dann erhält man in guten Ausbeuten symmetrische 1,2-Diole (Pinakole). • Gekreuzte Pinakol-Kupplungen sind selten und erfordern spezielle Methoden. • Intramolekulare Reaktionen führen zu cyclischen Diolen. R1 O R H + 2 • + Na ” – Na HO R1 2 R + 1 O R • R2 dim. ” O R2 1 R 2 O” 1 R R R2 1 R OH 203 Beispiel: Teilschritt aus der Synthese von Taxol O O H OBn OH OBn TiCl3 / DME H O HO O Zn-Cu-Leg. O O O O O 23 % O O O O Ph Ph N H HO AcO O OH O O HO AcO O BzO K. C. Nicolaou et al., Nature 367, 630-634 (1994). •Neues Beispiel, weil eigentlich McMurryReaktion!!! 204 7. Synthese von Ethern und Epoxiden 7.1. durch Williamson-Ethersynthese • Alkoholate oder Phenolate werden mit Alkylierungsmitteln R-X (X = Cl, Br, I, OTs, OMs, OTf usw.) zu Dialkylethern oder Aryl-Alkylethern umgesetzt (warum kann man keine Diarylether synthetisieren?) • Mechanismus: Meist SN2 mit Inversion der Konfiguration am Alkylierungsmittel, selten SN1. • Intramolekulare Williamson-Ethersynthesen führen zu cyclischen Ethern. Ringschlusstendenz in Abhängigkeit von der Ringgröße beachten! 1 R R 1 OH Cl R Base R1 OH Base O 2 R 2 O Cl R2 R2 R1 1 R OH R2 R1 Cl Base O 205 2 R Beispiel: 1) Teilschritt aus der Synthese von Asimicin O O O OH O H OMe 1) TsCl / Py O 2) K2CO3 / MeOH 91 % O C10H21 O H O C10H21 HO OH O O O O HO T. R. Hoye et al., Tetrahedron Lett. 36, 1981-1984 (1995). 206 Beispiel: 2) Teilschritt aus der Synthese von Asimicin O O O O OMs OH HO O Pyridin / Rückfluss O 3 h / 81 % O C10H21 HO C10H21 HO OH O O O O HO E. Keinan et al., J. Org. Chem. 65, 6035-6051 (2000). 207 7.2. aus Estern oder Lactonen durch Reduktion • Geeignete Reagenzien: Cl3SiH + BF3•OEt2, Et3SiH + BF3•OEt2, LiAlH4 + BF3•OEt2, NaBH4 + BF3•OEt2; statt BF3•OEt2 kann auch TiCl4 oder Trifluoressigsäure verwendet werden. • Mechanismus: die Lewis-Säure aktiviert die Ester-CarbonylGruppe und das Silan überträgt ein Hydrid-Ion. O R1 O R – BF3OSiEt3 • BF3 2 – R1 EtBF 3 ” HO Si Et “ BF3 O Et 2 O R 2 R R1 O 1 R2 1 R OEt R O – Et3Si+ Et H Et Si H H Si Et Et Et Et H Si Et H 2 “ Et R R2 1 R O O H H – Et3Si+ ” BF3 Et ” Et Si “ BF3 Et O R2 1 R O H Lactone ergeben bei dieser Reaktion cyclische Ether. 208 Beispiel: 1) Teilschritt aus der Synthese von Boswelliasäureethylether Et3SiH / BF3OEt2 O H O HOOC H RT / 6 h / 90% H O HOOC H J. Bergmann et al., Eur. J. Org. Chem. 2003, 4752-4756. 209 Beispiel: 2) Teilschritt aus der Synthese von Sesaminon O HO BnO HO O BnO Et3SiH / BF3OEt2 O O O O O O CH2Cl2 / -78 °C / 64% O O HO O O O O O O O H. Yoda et al., Synlett 2001, 400-402. 210 7.3. durch Prileshaev-Reaktion • OC2 Folie 95 • Elektronenreiche Doppelbindungen werden durch Persäuren (meist in Gegenwart von NaHCO3 als Puffer) in einer konzertierten Reaktion in Epoxide Überführt. • Käufliche Reagentien: mCPBA, MMPP, • Reagenzien, die man in situ herstellen muss: HCOOOH, CH3COOOH, CF3COOOH, CH3CNHOOH, DMDO • Mechanismus: 1 R RCOOOH R2 • 1 R H O R2 O 1 R O R – RCOOH O R2 Addition des Peroxosauerstoffs erfolgt stereospezifisch cis. 211 Beispiel: 1) Teilschritt aus der Synthese von Epothilon A S OH N DMDO CH2Cl2 / 2 h O O - 35 ° C / 48 % OH O O S OH N O O OH O D. Schinzer et al., Angew. Chem. 109, 543-544 (1997). 212 Beispiel: 2) Teilschritt aus der Synthese von Erythronolid A PMPh O O PMPh O OH mCPBA O C O O O O OH O C O HO OH OH O O OH OH R. W. Hoffmann et al., Chem. Ber. 127, 2519-2526 (1994). 213 7.4. aus α,β-ungesättigten Ketonen durch Scheffer-Weitz-Reaktion • OC2 Folie 107 • Elektronenarme Doppelbindungen werden durch NaOH / H2O2 glatt epoxidiert. • Statt NaOH / H2O2 können auch DMDO oder TBHP / DBU verwendet werden. • Geeignete Substrate: α,β-ungesättigte Ester, α,β-ungesättigte Aldehyde und Ketone, α,β-ungesättigte Sulfoxide, Sulfone und Sulfonsäuren, Nitroalkene • Mechanismus: ” O R1 R2 H O O H O R 1 O O ” 2 R O – OH– R1 O R2 214 Beispiel: Teilschritt aus der Synthese von Manumycin O H N O Ac TBDP / DMDO O CH2Cl2 / RT / 3 d OH Ac Warum entsteht das Epoxid trans zur Methylgruppe? OH 78% O H N H N O OH • • O H N O HO W. Adam et al., Synlett 2002, 510-511. 215 7.5. durch Darzens-Glycidestersynthese • OC2 Folie 197 • α-Halogencarbonsäureester reagieren in Gegenwart von Basen (NaOR, NaNH 2 oder anderen starken Basen) mit Aldehyden oder Ketonen zu sogenannten Glycidestern (α,β-Epoxy-Ester). • Mechanismus ” O O H 2 O R R2 1 R ” – HO R Cl O R3 – Cl– O R O 1 O R 2 O ” R H O R1 O R3 Cl 2 O • R 3 O R2 Cl R2 1 Statt α-Halogencarbonsäureestern kann man auch einsetzen: O R1 H N Cl R3 R2 R1 H R1 CN Cl H Cl O R1 S R2 H Cl O R1 O 2 R H Cl N R3 R2 216 Beispiel: Teilschritt aus der Synthese von Epiasarenin 1) LDA / THF – 20 °C O Br O 2) O O O O CHO O O O O O O O O H H O P. G. Steel et al., Org. Lett. 4, 1159-1162 (2002). 217 7.6. durch Corey-Chaykovsky-Reaktion • Trimethylsulfoniumhalogenide reagieren in Gegenwart von Basen mit Aldehyden oder Ketonen zu Epoxiden. • Reaktion funktioniert auch mit α,β-ungesättigten Aldehyden und Ketonen. O H3C “ S CH2 H H3C ” H – H2 H3C “ ” S CH2 R1 H3C R2 “ S H3C H3C O ” CH2 1 R 2 R O – Me2S • R2 R1 Reaktion funktioniert analog mit (aber nicht mit α,β-ungesättigten Carbonylverbindungen) H3C O H3C I ” “ S CH2 H 218 Beispiel: 1) Teilschritt aus der Synthese von Periplanon B O O Me3S+ I– OTBDMS OTBDMS O nBuLi / THF – 5 °C / 30 min. O 75% O O O K. Mori et al., Tetrahedron 43, 2689-2699 (1987). 219 Beispiel: 2) Teilschritt aus der Synthese von Phyllanthocin H BnO H O O H Me2SOCH2 O O DMSO / THF BnO H O O O O 0 °C / 1 h 88% H MeOOC H O O O O Ph O A. B. Smith et al., J. Am. Chem. Soc. 113, 2071-2092 (1991). 220 8. Synthese von Aldehyden und Ketonen 8.1. aus Alkoholen durch Oxidation • Problem bei der Oxidation von primären Alkoholen zu Aldehyden: Aldehyde können zur Carbonsäure weiteroxidiert werden. • Um Aldehyde zu erhalten, kann man nur solche Oxidationsmittel verwenden, die Aldehyde nicht oder nur sehr langsam weiteroxidieren. Die Oxidation von sekundären Alkoholen zu Ketonen bereitet üblicherweise keine Probleme. • Geeignete Oxidationsmittel: CrO3/Pyridin (Collins-Oxidation) PCC, PDC, TEMPO + Cooxidationsmittel, DMSO/(COCl)2 (SwernOxidation), DMSO/Py-SO3 (Parikh-Doering-Oxidation), TPAP (LeyOxidaton), Dess-Martin-Reagens, und unzählige Variationen dieser Oxidationsmittel, Oppenauer-Oxidation, MnO2. “ N H ” O O Cr O PCC Cl “ N H ” 2 PDC O O Cr O ” O 221 N O • TEMPO “ N ” O “ ” O Ru O O TPAP AcO OAc OAc I O O Dess-Martin-Reagens • Wichtig bei der Anwendung all dieser Oxidationsmittel zur Synthese von Aldehyden: auf Wasserausschluss achten! (Ausnahme: TEMPO). Deshalb setzt man oft gemahlenes getrocknetes Molsieb (3 Å oder 4 Å) zu. • Genereller Mechanismus: der primäre Alkohol bindet an das Oxidationsmittel und über einen cyclischen Übergangszustand wird von der CH2-Gruppe ein H+ und vom Alkoholsauerstoff ein Elektronenpaar entfernt. H M: Heteroatom in hoher X X H HH Oxidationsstufe +N. +N M +N2 M R X: basisches Atom. R O O 222 • Oxidation mit PDC (analog PCC, Collins-Reagens und andere Chrom(VI)-Verbindungen) • OC2 Folie 151 H HH R O H O O O Cr O Cr O O H H O R ” H O O “ O Cr O Cr H O O O R H R O O O O Cr O Cr H O O O “ H O HH H HH O R – H2O HH H ” O O Cr O Cr O O O O O Cr O Cr O O H O H O 223 • Oxidation mit TPAP (Ley-Oxidation) ” R O H O “ ” O Ru O O H R H O “ ” O Ru O – HRuO3 O O R H • TPAP ist sehr teuer. Deshalb wird es nur in katalytischen Mengen eingesetzt und durch ein geeignetes Cooxidationsmittel immer wieder regeneriert (ähnlich wie OsO4 bei cis-Dihydroxylierungen). • Geeignete Cooxidationsmittel für TPAP-Oxidationen: O2 oder NMO. O2 RuO4– HRuO3 – H2O 224 • TEMPO-Oxidation TEMPO ist ein stabiles Radikal (Gefrierschrank -30 °C), das durch ein geeignetes Cooxidationsmittel aktiviert wird. H OAc OAc N O • – I – 2 AcO R + AcO– – AcOH I + O O “ N “N O ” O OAc – R I H H O R OAc O R • + H N OH häufig eingesetzte Cooxidationsmittel für TEMPO-Oxidationen: NaOCl / NaHCO3 / KBr oder Ph-I(OAc)2 (wenn der zu oxidierende Alkohol eine oder mehrere Doppelbindungen enthält). 225 • Dess-Martion-Oxidation Das Dess-Martin-Reagens wird aus o-Iodbenzoesäure, Oxon (Mischung aus KHSO5, KHSO4 und K2SO4) und Acetanydrid hergestellt. AcO HO O I O Oxon I OH OAc I OAc O Ac2O / AcOH O O o-Iodbenzoesäure O Ioxoxybenzoesäure IBX Dess-Martin-Reagens R O – AcO O AcO I O O – R O H I H H OAc O O O ” CH3 + –H O H H O O OAc O AcO O R O HH AcO “I AcO OAc I OAc O OAc O I CH3 O – AcOH – AcO O + R – H 226 • Swern-Oxidation (OC2 Folie 153) Oxalylchlorid wird in CH2Cl2 gelöst und auf −60 °C bis −78 °C abgekühlt. Dann gibt man DMSO zu, dann den zu oxidierenden Alkohol und dann Triethylamin und lässt auftauen. Statt Oxalylchlorid kann auch Trifluoressigsäureanhydrid verwendet werden. S O O O Cl Cl ” O O S O Cl Cl “ O O “ S O – – Cl O O Cl S O Cl – + Cl Cl HH –H H H O O S O Cl + O R R H O H H H “ S H O R R “ Cl S HH – CO2 – CO – – Cl S – ” H O – CO2 – CO – Cl– + Et3N H H H S “ H –H H H O H + R – – Cl O R H Cl S O R 227 • Parikh-Doering-Reaktion Bei der Parikh-Doering-Oxidation wird DMSO / Py-SO3 /NEt3 in einem geeigneten Lösungsmittel verwendet. Reaktion verläuft im Gegensatz zur Swern-Oxidation bei Raumtemperatur. HH R • OH DMSO / Py-SO3 NEt3 / RT H R O Pfitzner-Moffatt-Oxidation Bei der Pfitzner-Moffatt-Oxidation wird DMSO / DCC /NEt3 in einem geeigneten Lösungsmittel verwendet. Reaktion verläuft im Gegensatz zur Swern-Oxidation bei Raumtemperatur. HH R OH DMSO / DCC NEt3 / RT H R O 228 • Oppenauer-Oxidation (OC2 Folie 154) Bei der Oppenauer-Oxidation werden primäre Alkohole mit Aceton und Aluminiumisopropylat in Aceton oder Benzol als Lösungsmittel zu Aldehyden oxidiert. • Reaktion funktioniert auch mit sekundären Alkoholen. Diese werden sehr viel schneller oxidiert als primäre Alkohole (Methode zur Oxidation von sekundären Alkoholen in Gegenwart von primären Alkoholen, ohne diese zu schützen) O iPrO HH iPrO Al OiPr iPrO iPrO iPrO ” Al iPrO R ” Al O “ H O OiPr Al “ O iPrO iPrO H O R O “ O Al OiPr iPrO ” iPrO – iPrO– + R H 229 Beispiel: 1) Teilschritt aus der Synthese von Mniopetal E OH O PDC / CH2Cl2 OTBDMS H OTBDMS H OTBDMS MS 4 Å / RT / 3h 67% OH O TEMPO / PhI(OAc)2 OTBDMS RT / 90 min. 98% HO HO O OH O CHO H H J. Jauch et al., Synlett 2001, 87-89. 230 Beispiel: 2) Teilschritt aus der Synthese von Erythronolid A PMPh O O O O OH TPAP / NMO CH2Cl2 / MS 3 Å 0 °C / 30 min. 79% O C PMPh O O O O O H O C O HO OH OH O O OH Swern-Oxidation, PCC, ParikhDoering, Corey-Kim-Oxidation und Dess-Martin-Oxidation führten alle zur Zersetzung des Ausgangsmaterials. OH R. W. Hoffmann et al., Chem. Ber. 127, 2519-2526 (1994) 231 Beispiel: 3) Teilschritt aus der Synthese von Tautomycin OH OBz DMSO / (COCl)2 O CH2Cl2 / NEt3 OPMB OBz OPMB H – 78 °C / 69% O O HO O OH O O OMe OH O H H O O O R. W. Armstrong et al., J. Org. Chem. 61, 3106-3116 (1996). 232 Beispiel: 4) Teilschritt aus der Synthese von Epothilon A S S Dess-Martin-Reagens H N HO OTBDMS N O CH2Cl2 / RT / 1h OTBDMS 78% S OH N O O OH O D. Schinzer et al., Angew. Chem. 109, 543-544 (1997). 233 Beispiel: 5) Teilschritt aus der Synthese von Lycodolin Me HO KH / Benzophenon N O Toluol / 110 °C Me HO N O 45% H OH O Me HO N H O C. H. Heathcock et al., J. Am. Chem. Soc. 104, 1054-1068 (1982). 234 8.2. aus Halogeniden durch Oxidation • Kornblum-Oxidation: Primäre und secundäre Alkylhalogenide und Benzylhalogenide können mit DMSO in Gegenwart von Na2CO3 oder NaHCO3 zu Aldehyden und Ketonen oxidiert werden. Analog reagieren Tosylate. O S R X H HH R – –X S O “ H H Na2CO3 + –H HH R O ”H H “ S O – S R H • Reaktivität: Tosylate > Iodide > Bromide > Chloride. Gelegentlich werden wenig reaktive Chloride zuvor mit Silbertosylat in die Tosylate überführt. • Bei α-Halogenketonen oder α-Halogenestern tritt kein Schwefel-Ylid als Intermediat auf. Ein α-H des Ketons wird direkt deprotoniert.235 • Verwandte Reaktion: Sommelet-Oxidation N N Ar N N “ Ar X N N N H2O N – NH3 HH Ar NH2 – CH2O + NH3 H H O NH H – H2O H HO H H – NH3 H H O Ar + H+ Ar NH2 + H2O NH2+ H “ Ar NH2 • Wichtig: Bei der Oxidation des Benzylamins findet eine Hydridübertragung auf das Methyleniminiumion statt. Der erste Teilschritt (Bildung des Amins aus dem Halogenid) heißt Delepine-Reaktion. • Verwandt mit der Sommelet-Oxidation: Kröhnke-Reaktion = Oxidation von Benzylhalogeniden mit p-Nitroso-N,N-Dimethylanilin, 236 Pyridin und Wasser. Beispiel: Teilschritt aus der Synthese von Solidago-Alkohol Br CHO 1) DMSO / AgBF4 100 °C / 10 h OBz 2) Et3N 7 RT / 30 min OBz 60% O OH H. S. Liu et al., Synthesis 2004, 271-275. 237 8.3. aus Carbonsäurederivaten durch Reduktion • Carbonsäurederivate werden üblicherweise zu Alkoholen reduziert. Unter speziellen Bedingungen bleibt die Reduktion auf der Aldehydstufe stehen, bis die Reaktion wässerig aufgearbeitet wird. • Carbonsäurechloride können mit schwachen Reduktionsmitteln wie NaBH4 oder mit sterisch stark gehinderten Reduktionsmitteln LiAlH(OtBu)3 bei tiefen Temperaturen zu Aldehyden reduziert werden. NaBH4 O R Cl • Cl – 78 °C O R H2O Cl H R LiAlH(OtBu)3 O R – 78 °C O ” BH3 ” Al(OtBu)3 Cl H O R H2O H O R H Klassische Methode: Reduktion von Carbonsäurechloriden mit Lindlar-Pd / H2 zu Aldehyden (Rosenmund-von Braun-Reduktion) 238 • Carbonsäureester können mit DIBALH bei tiefen Temperaturen in nicht-koordinierenden Lösungsmitteln (CH2Cl2, Toluol, Hexan) zu Aldehyden reduziert werden. iBu2AlH O R • OR' – 78 °C AliBu2 O R H2O OR' H O R H Tertiäre Amide und Weinreb-Amide können mit DIBALH ebenfalls bei tiefen Temperaturen zu Aldehyden reduziert werden. iBu2AlH O 1 R 2 NR R iBu2AlH O R – 78 °C N R OMe – 78 °C AliBu2 O R 1 2 H2 O NR R H O ” AliBu2 O N “ Me R H R O H R H2O O R H 239 • Carbonsäuren können mit Thexylbromboran selektiv zum Aldehyd reduziert werden. Br Br B H O R Br R O B O H B O O H Br B R – H2 O Br B H O OH– R H 240 Beispiel: 1) Teilschritt aus der Synthese von (-)-α-Thujon O O O OEt O DIBALH Et2O / – 78 °C O H O 81% O W. Oppolzer et al., Helv. Chim. Acta 80, 623-639 (1997). 241 Beispiel: 2) Teilschritt aus der Synthese von Camptothecin COOMe MOMO CONEt2 DIBALH CHO MOMO THF / – 78 °C CONEt2 100% N O N O HO O M. A. Ciufolini et al., Angew. Chem. 108, 1789-1791 (1996). 242 8.4. aus Acetalen durch Hydrolyse • OC2 Folien 167, 168 • Acetale werden prinzipiell sauer hydrolysiert (THF/Wasser/Säure). • Basisch können Acetale nicht angegriffen werden! Acetale = basenstabile Carbonylschutzgruppen! • Mechanismus: Umkehrung der Acetalbildung! RO OR + H+ H“ RO OR R1 R2 R1 – H+ RO R • 1 OH 2 R – ROH R2 + +H H RO“ R 1 R R R 2 “H O 1 R H H RO OH R1 2 “H RO OH – ROH 1 O 2 R R2 – H+ O R1 R2 Alle Schritte sind reversibel! Das Gleichgewicht wird nur durch die Menge an Wasser verschoben! 243 Beispiel: 1) Teilschritt aus der Synthese von Swainsonin O H OH O N HO HCl halbkonz. H OH HO THF / RT 12 h N 96% H. Pearson et al., J. Org. Chem. 61, 7217-7221 (1997). Beispiel: 2) Teilschritt aus der Synthese von Mniopetal E HO HO O O O CHO 80 % TFA / H2O HO HO O CHO H H H OH O RT / 4 h / 100 % J. Jauch et al., Synlett 2001, 87-89. H 244 8.5. aus Dithioacetalen durch Hydrolyse • Geeignete Reagenzien: H+ / H2O / THF oder HgCl2 / H2O / THF / BF3⋅OEt2 oder PhI(OAc)2 / MeOH / H2O. S Hg2+ S R1 S R2 “ + S Hg “S R2 R 1 R “ H O R 1 R S Hg 1 H + R2 O – H+ 2 1 S R1 R AcO 2 – AcO – “ H O R1 “ S I S R1 R2 Ph S O H 1 2 R R “S S I R1 R 2 Ph H O – H+ H Ph AcO I S O H R1 R2 O + 2 OAc OAc –H R H R2 R AcO S + O – H+ I Ph Hg 1 R R 2 245 Beispiel: 1) Teilschritt aus der Synthese von Maytansin Cl MeO N O OTBDMS Cl HgCl2 / CaCO3 S MeO OCONH2 MeO N O OTBDMS OCONH2 CH3CN / H2O / RT O 12h S MeO O N Cl MeO N O O O O H O HO MeO N H O E. J. Corey et al., J. Am. Chem. Soc. 102, 6613-6615 (1980). 246 Beispiel: 2) Teilschritt aus der Synthese von Roflamycoin OH OBn S S OH PhI(OOCCF3)2 MeOH / H2O MeO O OBn RT / 5 min. / 81% O OH O O OH OH OH OH OH OH OH OH S. D. Rychnovsky et al., J. Am. Chem. Soc. 102, 2621-2622 (1994). 247 8.6. aus Aromaten durch Formylierungs- und Acylierungsreaktionen • Vilsmeier-Formylierung OC2 Folie 138 O P Cl O H ” O Cl P Cl O Cl Cl Cl N H – Cl– N “ Cl – PO2Cl2 “ N H H H H H O H + Cl “ N H O H H N O P Cl O Cl Cl H N – N “ Cl “ H Cl N“ – O P Cl O Cl + –H H N – Cl– O – Me2NH2+ H 248 • Gattermann-Formylierung HCl (g) H N H “ N H H “ N H N H H ZnCl2 +H + H“ N H H + H2O O – NH4+ H • Varianten: Gattermann-Adams-Formylierung: verwendet Zn(CN)2 (leichter zu handhaben als HCN und ZnCl2). • Gattermann-Koch-Formylierung: verwendet CO / HCl / AlCl3. Dieses Gemisch verhält sich wie Ameisensäurechlorid HCOCl. Wichtig: Ameisensäurechlorid ist nur unterhalb von −60 °C stabil, bei dieser Temperatur aber nicht reaktiv genug. Bei höheren Temperaturen zerfällt es in CO und HCl. 249 • Reimer-Tiemann-Reaktion • OC2 Folie 139 ” Cl Cl Cl O H H O + H2O / H • ” Cl Cl ” Cl Cl H Cl Cl O – Cl– – + OH ” O Cl O ” H H O Cl O – Cl – ” H Cl ” Cl O H OH O H Reaktion geht nur bei aktivierten Aromaten! 250 • Weitere Formylierungsreaktionen: Cl OMe + AlCl3 O – – AlCl4 Cl Cl O “ H + OH– Cl – Cl – OMe H OH O – MeOH • H Duff-Reaktion H + (CH2)6N4 H / H2O H H N“ H H H2O NH2 H H H H N“ H – CH3NH2 “ NH2 H O H 251 Beispiel: 1) Teilschritt aus der Synthese von Normalindin OBn OBn 10 eq. POCl3 N Bn DMF / 45 °C / 95% H H N O N Bn H N N F. A. Davis et al., J. Org. Chem. 71, 8761-8766 (2006). 252 Beispiel: 2) Teilschritt aus der Synthese von Illudin C O 2,5 eq. POBr3 TESO 3,0 eq. DMF H Br CH2Cl2 / 72 h / RT 64% O OH R. L. Funk et al., Org. Lett. 3, 2611-2613 (2001). 253 Beispiel: 3) Teilschritt aus der Synthese von Podocarpin-Derivaten OH Br CHCl3 / NaOH / Et3N 60 °C / 71% OH O H Br OH O H C. L. Zhang et al., Chin. Chem. Lett. 17, 163-164 (2006). 254 8.7. aus Alkenen durch Ozonolyse • Mit Ozon können Doppelbindungen gespalten werden. Je nach Aufarbeitung kann man Alkohole, Aldehyde und/oder Ketone oder Carbonsäuren erhalten. • Reduktive Aufarbeitung mit einem geeigneten Reduktionsmittel wie NaBH4, LiAlH4 oder Zn / HOAc führt zu Alkoholen. • Reduktive Aufarbeitung mit Dimethylsulfid oder Triphenylphosphin ergibt Aldehyde und/oder Ketone. • Oxidative Aufarbeitung mit H2O2 ergibt Carbonsäuren und/oder Ketone. LiAlH4 R R1 R3 2 4 R R O3 Me2S R3 R1 2 OH HO R1 2 R4 R3 O O R R4 H2O2 R1 R3 R2 = R4 HO LM / 78 °C =H O O OH 255 • Mechanismus in CH2Cl2: 1,3-Diploare Cycloaddition gefolgt von 1,3Diploarer Cycloreversion gefolgt von 1,3-Diploarer Cycloaddition gefolgt von Aufarbeitung. O “ ” O O O 1 O R O 1 O 3 2 R R ” O R 4 R1 R2 O“ R3 R4 R1 R2 3 R R R2 R4 O O O 1 R R1 R2 O R R3 R4 2 H ” O R 3 R1 4 R ” R1 R2 O O 2 H– R2 O“ O R2 O H O R3 3 Homologe! R4 R3 R4 O O – OH– R1 R2 R3 R4 H OH H OH R1 R3 R4 O O ” O O R3 R4 256 R1 2 R R3 4 R O ” R1 2 R S O O O 1 R H O H+ 3 R H O O R 1 OH O H • H2O2 1 R H H “ O O R3 R4 S O “ R H 1 R O O HO H O R3 O • R 1 OH O R1 – DMSO + H2O2 3 O H H O R2 R3 R4 3 R H O O H O HO H O O R OH 3 – H2O R3 H R3 HO • Mechanismus in MeOH: 1,3-Diploare Cycloaddition gefolgt von Peroxyacetalbildung! (MeOH fängt Carbonyloxide ab!). 257 Beispiel: 1) Teilschritt aus der Synthese von Roflamycoin OTBDMS OTBDMS O3 / MeOH MeO O OBn -78 °C / 90 % H O MeO O O OBn OH O O OH OH OH OH OH OH OH OH S. D. Rychnovsky et al., J. Am. Chem. Soc. 102, 2621-2622 (1994). 258 Beispiel: 2) Teilschritt aus der Synthese von Damsin O O3 / MeOH / CH2Cl2 O – 86 °C / (MeO)3P O O COOMe COOMe H O O O F. A. Davis et al., J. Am. Chem. Soc. 98, 3379-3380 (1976). 259 8.8. aus 1,2-Diolen durch Glycolspaltung • 1,2-Diole (Glycole) können mit verschiedenen Reagenzien zu Aldehyden oder Ketonen gespalten werden. • Criegee-Spaltung: Glycole werden mit Pb(OAc)4 unter wasserfreien Bedingungen zu Aldehyden oder Ketonen gespalten. HO R1 OAc AcO Pb OAc AcO OH 4 3R 2 R – AcO R ” AcO AcO Pb HO 1 R 2 R AcO OAc Pb O O –H 1 • R1 R 2 4 3R R – Pb(OAc)2 R OAc AcO Pb O H “ 4 3R R HO R “ –H O “ AcO OAc 1 O 2 R 4 3R R – AcO” AcO OAc Pb H O O “ 1 R 2 R R 3R O 2 R 3 R 4 R Als Lösungsmittel verwendet man üblicherweise Eisessig. Für R2 = R4 = H erhält man Aldehyde (keine Weiteroxidation!) 260 4 • Malaprade-Spaltung: Glycole werden mit Periodsäure HIO4 (H5IO6) in wässeriger Lösung zu Aldehyden oder Ketonen gespalten. In der Praxis wird diese Reaktion oft mit NaIO4 unter sauren Bedingungen durchgeführt. HO 1 R R – H2O • HO 4 3R R1 R O “ OH I O H O O 1 R “ 2 R I “ H O 1 R 2 R O H “ 4 3R R O HO I O H “ –H O O 2 R 3R R O “ 1 R O 2 R +H “ 4 3R H“ O O H HO I O H O O R R 1 2 R 4 3R R OH I –H R1 “ O 4 HO +H OH O 4 – H 3R R O O HO I O ” O O I O OH OH 2 O O O O 4 R R2 R3 – HIO3 R1 O R2 R3 R4 Auch wenn NaIO4 in wässeriger Lösung eingesetzt wird, spricht man von Malaprade-Spaltung 261 • Verwandte Reaktionen: Bei der Lemieux-Johnson-Oxidation werden C=C-Doppelbindungen mit NaIO4/OsO4 oxidativ gespalten. Dabei wird die Doppelbindung durch katalytische Mengen OsO4 zunächst in das cis-Diol überführt, das dann mit NaIO4 oxidativ gespalten wird. Das Os(VI)-Intermediat wird durch NaIO4 wieder zu OsO4 oxidiert. OsO4 1 R • 2 R HO R1 OH NaIO4 R2 R1 CHO + R2 CHO Bei der Lemieux-von Rudloff-Oxidation ist OsO4 durch KMnO4 ersetzt. KMnO4 1 R 2 R HO R1 OH R2 NaIO4 R1 CHO + R2 CHO 262 Beispiel: 1) Synthese von D-Glycerinaldehyd-acetonid MeO OMe OH OH HO OH OH OH OH O NaIO4 O O O O SnCl2 cat. DME, Rückfl. OH O NaHCO3 / H2O CHO CH2Cl2 J. D. Bryant et al., Org. Synth. 72, 6-10 (1995) Beispiel: 2) Synthese von L-Glycerinaldehyd-acetonid HO OH O O H2 / Pd / C HO OH NaIO4 / NaOH O O OH O O pTsOH cat. H DMF / RT HO O OH O O RT O H H HO HO OMe OH CHO C. Hubschwerlen et al., Org. Synth. 72, 1-4 (1995) 263 Beispiel: 3) Teilschritt aus der Synthese von Preclavulon A H H O OH Pb(OAc)4 MeOH / -5 °C / 1h 72% H H COOMe CHO H COOH H E. J. Corey et al., Tetrahedron Lett. 29, 995-998 (1988). 264 Beispiel: 4) Teilschritt aus der Synthese von Premisakinolid A OMe O OMe 1) OsO4 cat. O NMO OTBDMS OPMB Dioxan / H2O 2) NaIO4 O H OTBDMS OPMB 72% M. J. Krische et al., Org. Lett. 17, 4686-4689 (2015). 265 8.9. aus Vinyl-Allyl-Ethern durch ClaisenUmlagerung • Allyl-Vinyl-Ether sind aus Aldehyden oder Ketonen und Allylalkoholen leicht zugänglich. R3 O OH R3 R3 R H R 2 cat. H 3 R OO + R – H2O • O cat. H+ 1 1 H 3 – R OH R2 R1 H 2 R Erhitzt man Allyl-Vinyl-Ether, dann findet eine sogenannte sigmatrope Umlagerung statt (griech. tropos = betreffend, einwirkend), bei der 3 Elektronenpaare in einem 6-gliedrigen Übergangszustand verschoben werden. Es bildet sich ein γ,δungesättigter Aldehyd oder ein γ,δ-ungesättigtes Keton. O R3 1 R H R 2 O Δ R3 H R1 R2 266 Beispiel: Teilschritt aus der Synthese von Garsubellin A O O O O 200 °C O O O NaOAc O 96% O OMOM HO O OMOM O O O O M. Shibasaki et al., J. Am. Chem. Soc. 127, 14200-14201 (2005). 267 8.10. aus Iminen, Oximen und Hydrazonen durch Hydrolyse • Imine, Oxime und Hydrazone lassen sich säurekatalysiert zu Aldehyden bzw. Ketonen hydrolysieren. N 1 OH 2 R R R R O H O H “ OH H H N R H H OH N “ 1 +H “ 2 1 R 2 “H O – NH2OH R1 R2 H N OH H –H R1 R2 “O H H “ N OH + H“ R1 R2 O H O – H“ R1 R2 • Wichtig: alle Schritte sind Gleichgewichtsreaktionen. Durch große Mengen Wasser wird das Gleichgewicht auf die Seite der Carbonylverbindung verschoben. • Analog verlaufen die Hydrolysen von Iminen und Hydrazonen. 268 Beispiel: Derivatisierung von 3-Oxo-Tirucallensäure zur Isolierung und Reinigung HOOC HOOC O H2N H O NH2 NH H H2N pTsOH cat. H NH N O + andere Substanzen H kristallisiert HOOC HOOC Reinigung durch Umkristallisieren H H2N NH O N H H2 O HCl H H. Safayhi et al., Mol. Pharm. 60, 267-273 (2001). O H 269 8.11. aus Nitroverbindungen durch Nef-Reaktion • Substituierte Nitroalkane werden mit Basen deprotoniert und dann sofort wieder angesäuert. Die erhaltene tautomere Form (Aci-NitroForm) wird im stark sauren zum Aldehyd oder Keton hydrolysiert. • Substanz muss säurestabil sein. • Klassische Anwendung in der Zuckerchemie (Emil Fischer). 1 R H 2 R – H+ – H+ OH– O N“ O” H O R 1 R2 R1 O H N R2 O ” + H+ O ” N“ O” H O R1 H2SO4 R1 halbkonz. R2 O H N R2 O H +H + H O O H N“ O” R1 H O 1 H “ O H – NOH N H 2 R O H H R O H “O N H R2 O ” H“ O R1 R2 O R1 R2 2 NOH N2O + H2O 270 Beispiel: Teilschritt aus der Synthese von Isocomen NO2 OTMS SnCl4 NO2 O O 1) NaOH 2) H2SO4 H2O O L. A. Paquette et al., J. Am. Chem. Soc. 103, 1835-1838 (1981). 271 8.12. aus Carbonsäurederivaten durch Nucleophile Substitution • Carbonsäurederivate können nur durch solche nucleophilen Reagenzien in Ketone überführt werden, die selbst nicht mit Ketonen reagieren. • Organocadmium-Reagenzien R 1 Mg Br • R R • MgBr CdCl2 R 1 Cd R R 1 Organozink-Reagenzien 1 Mg Br • 1 R 1 O R 1 MgBr ZnCl2 2 Cl Br R 1 CuCl Li 2 R 1 R O 2 R 1 R ZnCl O Cl Lithiumdialkylcuprate Li O 2 R 1 R 2 1 R Cu R 1 + Li O Cl R R 2 1 O Cl – Cl O Cl – Cl – ” O R ” 2 R 1 R – R 2 R 1 ” O Cl – Cl – R 2 R Lithiumenolate lassen sich analog mit Säurechloriden (und auch mit Estern) zu 1,3-Diketonen umsetzen. 272 1 Beispiel: Teilschritt aus der Synthese von Centrolobin MeO MeOOC COOMe MgBr O MeO Cl O CuBr / LiBr THF / RT / 98% H MeO O H OH T. Nakata et al., Tetrahedron Lett. 49, 6462-6465 (2008). 273 8.13. aus Carbonsäurederivaten durch ClaisenKondensation Bei der Claisen-Kondensation werden Carbonsäureester in αPosition deprotoniert und üblicherweise mit sich selbst umgesetzt. • O O R' O R O R ” O H HH O O R HH R O H H H R' R ” O O R H H H R' O R' R OO R' R' O – R O” R' ” R' O O” O O HH R' R H+ Aufarb. O R' O R H H H R' • Als Base verwendet man üblicherweise das Alkoholat, das formal auch im Erster vorkommt. Andere Alkoholate würden teilweise zur Umesterung führen. • Man kann als Base auch LDA (oder vergleichbar starke Basen) 274 oder NaH verwenden. • Man kann auch gekreuzte Claisen-Kondensationen mit zwei verschiedenen Estern durchführen. Um zu vermeiden, dass sich Produktgemische bilden, darf einer der beiden Ester keine α-HAtome besitzen. Der andere Ester wird dann mit LDA o.ä. irreversibel deprotoniert. O N O R' O 1 ” O R H HH O O R R OO R2 R3 R R O 2 3 R R HR 1 R O 1 O 2 O R1 ” R 3 R R H R' R' O – R O” R ” O O” O R1 O 2 3 R R R1 R + H Aufarb. O 1 R O 2 3 R 1 R R HR • Die Reaktion von Esterenolaten mit Aldehyden oder Ketonen βHydroxyestern bzw. die Reaktion von Ketonenolaten mit Estern zu 1,3-Diketonen bezeichnet man allgemein als Claisen-Reaktion. • Intramolekulare Esterkondensationen werden als DieckmannKondensationen bezeichnet. 275 Beispiel: 1) Teilschritt aus der Synthese von Violapyron C O 1) LDA / THF / -78 °C COOtBu O tBuOOC 2) MeO OH O O O O J. S. Lee et al., Eur. J. Org. Chem. 2014, 4472-4476. O 276 Beispiel: 2) Teilschritt aus der Synthese von Fredericamycin O O 1) iPr2NH / EtOH RT / 3 h CH2 EtOOC HO 2) HOAc EtO N EtO COOEt O CH2 HO N COOEt O MeO O HO O HO O N H D. L. Boger et al., J. Am. Chem. Soc. 117, 11839-11849 (1995). 277 8.14. durch Pinacol-Umlagerung • Klassisch können 1,2-Diole nach Behandlung mit Schwefelsäure oder anderen Brønsted-Säuren zu Ketonen umlagern. HO OH H + H “ HO OH “ - H2O Pinakol OH “ O H O + -H Pinakolon • Als Zwischenprodukte oder Nebenprodukte können Epoxide auftreten. • Bei unsymmetrischen Diolen bildet sich zunächst das stabilste Carbeniumion. Die verbleibenden Reste am tertiären Alkohol wandern um so leichter, je nucleophiler sie sind, weil sie mit ihrem Elektronenpaar wandern (Anionotrope Umlagerung oder Nucleophile Umlagerung). 278 • Statt Brønsted-Säuren kann man auch Lewis-Säuren verwenden. Oft ändert sich dann das erhaltene Produkt. O OH BF3-OEt2 70% HClO4 OH CH2Cl2 – 20 °C 0 °C • Man kann die Reaktion auch unter basischen Bedingungen durchführen, wenn eine der beiden OH-Gruppen selektiv in eine gute Abgangsgruppe überführt werden kann. TfO • O H2O O H |B” O Verwandte Reaktion: Semipinakol-Umlagerung H2N OH NO+ N “ N OH “ - N2 OH “ O H O + -H 279 Beispiel: Teilschritt aus der Synthese von Hopeanol MeO MeO OMe OH OH OMe CHO H3PO4 MeO MeO MeO OMe OMe MeO OMe OMe HO OH COOMe O O HO HO OH S. A. Snyder et al., Angew. Chem. 124, 4156-4160 (2012). 280 • Verwandte Reaktion: Umlagerung von Epoxiden Epoxide lassen sich in Gegenwart von BF3⋅Et2O oder MgBr2 oder anderen Lewis-Säuren zu Ketonen umlagern. Die Umlagerung verläuft über das stabilere Carbeniumion. Üblicherweise wandert der nucleophilste Rest. O 1 R 2 R BF3 3 R R4 R 2 R “ ” O BF3 1 R R2 1 ” BF3 O“ 3 R 4 R 4 R R 3 1 R – BF3 R2 R 1 R 2 “ ” O BF3 3 R R 4 O 4 R 3 R 281 8.15. aus α,β-ungesättigten Ketonen durch Reduktion • Doppelbindungen in α,β-ungesättigten Carbonylverbindungen können durch katalytische Hydrierung zu Carbonylverbindungen reduziert werden. • Geeignete Katalysatoren: Raney-Ni, Pd/C, Nickelborid Ni2B, NiCl2 NaBH4 Ni2B MeOH / H2O • Doppelbindungen in α,β-ungesättigten Carbonylverbindungen können ionisch durch Hydrid-übertragende Reagenzien reduziert werden. • Triethylsilan/Stryker-Osborn-Reagens und Variationen davon sind besonders effektiv in der 1,4-Reduktion von α,β-ungesättigten Carbonylverbindungen. Cu(OAc)2 PPh3 Ph2SiH2 (Ph3PCuH)6 282 Beispiel: 1) Teilschritt aus der Synthese von Ingwer-Keton O O Ni2B / H2 HO RT / 2h 98% HO V. Kovalenko et al., Z. Naturforsch. B69, 885-888 (2014). Beispiel: 2) Teilschritt aus der Synthese von Isomigrastatin Ph Ph P MeO O H N P O HO O O H O N Ph Ph OBz O O Cu H Tol. / RT / 59% O H N O MeO O HO OBz O O MeO O HO O O A. Fürstner et al., Chem. Eur. J. 19, 7370-7383 (2013). 283 8.16. aus α,β-ungesättigten Aldehyden und Ketonen durch Michael-Addition • Bei Michael-Additionen (Konjugierte Addition, 1,4-Addition) werden C-Nucleophile an das β-C-Atom der α,β-ungesättigten Carbonylverbindung addiert. Das intermediär gebildete Enolat wird entweder bei der wässerigen Aufarbeitung protoniert oder mit einem anderen Elektrophil abgefangen. • Als C-Nucleophile eignen sich deprotonierte β-Ketoester, deprotonierte Malonsäureester, deprotonierte Nitroalkane und auch Cyanid, jeweils üblicherweise unter protischen Bedingungen. • Unter aprotischen Bedingungen können auch Enolate von Ketonen und von Estern eingesetzt werden. Unter Lewis-Säure-Katalyse können auch Silylenolether und Silylketenacetale eingesetzt werden. H 1 O R1 Z R2 ” Z 2 Z1 R1 H Z2 O ” R2 H + Z1 R1 H Z2 O H Z1 R2 R1 H Z2 O R2 284 • Wichtige C-Nucleophile sind Cu-haltige metallorganische Reagenzien mit unterschiedlicher Zusammensetzung. Man unterscheidet Dialkyl-Lithium-Cuprate R2CuLi, Dialkyl-LithiumCyano-Cuprate R2CuCNLi2, Dialkyl-Cyano-Zink-Cuprate R2CuCNZn. • Die genannten Cuprate übertragen immer nur einen Rest R. Bei wertvollen Resten R geht einer verloren. Deshalb setzt man oft gemischte Cuprate ein, die neben dem wertvollen Rest R noch einen sogenannten Dummy-Substituenten enthalten, der nicht übertragen wird. Häufig als Dummy-Substituent verwendet wird der 2Thiophenyl-Rest. • Cuprat-Reagenzien sind weiche Nucleophile (HASAB; wegen hoher Polarisierbarkeit von Cu) und deshalb nur schwach basisch. Weiche Nucleophile reagieren mit α,β-ungesättigten Carbonylverbindungen bevorzugt am β-C-Atom (weicher als das Carbonyl-C) untert 1,4Addition. Harte Nucleophile (Organolithiumverbindungen, GrignardReagenzien) reagieren bevorzugt mit dem Carbonyl-C (1,2-Addition). 285 Beispiel: 1) Teilschritt aus der Synthese von Longiborneol O LiHMDS / THF -78 °C, 1h COOMe O 0 °C, 3h 94% COOMe O OH M. Ihara et al., J. Org. Chem. 65, 4112-4119 (2000). 286 Beispiel: 2) Teilschritt aus der Synthese von Fredericamycin CH3 1) LDA / -78 °C 30 Sekunden (!!!) EtOOC EtO N COOEt 2) Cyclopentenon 20 Sekunden (!!!) 3) HOAc O HO O CH2 EtOOC EtO N COOEt O MeO O HO O HO O N H D. L. Boger et al., J. Am. Chem. Soc. 117, 11839-11849 (1995). 287 Beispiel: 3) Teilschritt aus der Synthese von Aplykurodionon-1 * H O O MgBr H H H O TMSCl / HMPA O CuI / LiCl / -78 °C O H * H O THF / 96% C. C. Li et al., Org. Lett. 16, 4380-4383 (2014). 288 8.17. aus Alkinen durch Hydratisierung • Die Hydratisierung von Alkinen verläuft entsprechende der Oxymerkurierung von Alkenen. • Verschiedene Übergangsmetallverbindungen (Cu2+, Hg2+, Ag+ usw.) katalysieren die Markownikow-Addition von Wasser an Alkine. • Terminale Alkine ergeben Methylketone, während interne Alkine Mischungen von Ketonen ergeben. Hg R 1 2+ 1 2 R R1 O H + Hg 2 R 2 R R Hg2+ 1 + H+ R 2+ – Hg H O H 2 R H O H R H “ O H 1 Hg+ – H+ R2 HH R 1 R 2 O 289 Beispiel: Teilschritt aus der Synthese von Lycoran HOOC O pTsOH•2H2O NO2 O Toluol / 110 °C 60-70% O O O COOH O O NO2 H N Z. H. Shao et al., Org. Lett. 16, 3044-3047 (2014). 290 8.18. durch Decarboxylierung von β-Ketocarbonsäuren • β-Ketocarbonsäuren decarboxylieren üblicherweise sehr leicht beim Erhitzen oder auch schon bei RT. • Sterische Hinderung oder konformative Starrheit kann die Decarboxylierung verlangsamen oder ganz unmöglich machen. • Unter neutralen Bedingungen verläuft die Reaktion über einen 6gliedrigen Übergangszustand. O R O 1 O O H O O R1 R2 • H H O H O – CO2 R1 R2 R 1 R2 R2 Im alkalischen verläuft die Reaktion über einen offenen Übergangszustand. O O R1 O 2 R O ” – CO2 ” H 1 R R2 O H+ 1 R R2 291 • Bicyclo[2.2.1]heptan-2-on-2-carbonsäure kann thermisch bis 200 °C nicht decarboxyliert werden. ~90 ° O O H O – CO2 O H O H • Grund (klassisch): Bredt´sche Regel: In Bicyclen mit mindestens einer Einerbrücke (Bicyclo[X.Y.1], X,Y>1) geht vom Brückenkopfatom keine Doppelbindung aus. • Grund (modern): Die in der Bredt´schen Regel erwähnte Doppelbindung würde aus zwei p-Orbitalen bestehen, die annähernd senkrecht aufeinander stünden. Senkrecht aufeinander stehende Orbitale können aber keine Bindung bilden! • Wenn die Summe der beiden größten Brücken X+Y ≥ 8, dann gilt die Bredt´sche Regel nicht mehr. Für X+Y=8 kann man das System als Derivat von trans-Cycloocten auffassen. Trans-Cycloocten ist stabil! Wie wird es hergestellt? • Generell können auch substituierte Malonsäuren, α-Nitro-carbon292 säuren, α-Sulfonylcarbonsäuren u.ä. decarboxyliert werden. • Verwandte Reaktion: Krapcho-Decarboxylierung. • β-Ketocarbonsäureester werden in DMSO mit NaCN auf 150-200 °C erhitzt. Dabei bildet sich ein Alkylnitril, CO2 und ein Keton. Statt NaCN kann man auch NaCl, NaBr, LiCl oder LiBr oder auch LiI verwenden. Die Reaktion verläuft in trockenem DMSO, kann aber auch in nassem DMSO (1-10 eq. H2O) durchgeführt werden. O O 1 R O R 2 CH2R3 ” CN O R H H 1 2 R O 1 R R2 • Die Reaktion funktioniert auch bei Malonsäureestern, α-Cyanocarbonsäureestern, α-Nitrocarbon-säureestern, α-Sulfonylcarbonsäureestern u. ä. Verbindungen (allgemein: Z-C-COOH). • Methylester werden leichter decarboxyliert als Ethylester. Wichtig: die Reaktion funktioniert auch mit tert.Butylestern! 293 • Verwandte Reaktion: Carroll-Reaktion • Die Carroll-Reaktion ist eine Kombination von Claisen-Umlagerung und Decarboxylierung von β-Ketocarbonsäuren. Manchmal wird sie auch als decarboxylative Claisen-Umlagerung oder Kimel-CopeUmlagerung bezeichnet. 1 4 R R 5 RR 2 O O O 100 200 °C oder: 2 LDA – 78 °C R3 OH O O R5 4 R R R 2 5 R R3 1 R R2 R4 5 R3 • O O R4 R H O4 R R1 3 OH – CO2 O 1 R R2 5 R R3 1 R R2 Wichtig: die Allylgruppe des Esters reagiert unter Allylverschiebung! 294 Beispiel: 1) Teilschritt aus der Synthese von Paspalin 1 H H 60 °C HOOC – CO2 O O O O O O H N H H H H O A. B. Smith III et al., J. Am. Chem. Soc. 111, 5761-5768 (1989). 295 Beispiel: 2) Teilschritt aus der Synthese von Vetiselinen COOMe MeOOC DMSO / H2O (3 eq.) NaCl (1 eq.) / 150 °C O 67% COOMe H O H J. R. Porter et al., J. Org. Chem. 51, 5450-55452 (1986). 296 8.19. durch Riley-Oxidation Durch Riley-Oxidation mit SeO2 kann eine CH2-Gruppe in α-Position zu einem Keton zum Keton oxidiert werden. Riley-Oxidationen funktionieren auch in Allylposition (→Allylalkohol oder Enon). • H O R2 R1 R R1 HH 2 R 1 R R Se O + H2O R 2 1 R H Se O HO O – H2O 1 H 2 O O O Se O O O 1 R O O H R2 Se Se OH O R2 1 – H2SeO H HO R2 R O 297 Beispiel: Teilschritt aus der Synthese von Bruceolin E O O SeO2 / Dioxan N COOEt 100 °C / 16h 95% O O N COOEt O N H J. C. Badenock et al., Tetrahedron Lett. 52, 6772-6774 (2011). 298 8.20. durch Tiffeneau-Demjanov-Umlagerung • Bei der Tiffeneau-Demjanov-Umlagerung (Ringerweiterung) läuft eigentlich eine intramolekulare Semipinakol-Umlagerung ab. • Ein primäres α-Hydroxy-amin wird mit HNO2 diazotiert, wobei N2 abgespalten wird und das entstandene primäre Carbeniumion durch Umlagerung einer C-C-Bindung stabilisiert wird. N “N NH2 OH OH NaNO2 / HCl (CH2)n “ OH – N2 (CH2)n H O “ (CH2)n (CH2)n O – H+ (CH2)n 299 Beispiel: Teilschritt aus der Synthese von Homospectinomycin Cbz HO N N NaNO2 / AcOH Cbz O HO O H O H2 N HO H2O / 40 min. 33% Cbz HO N HO N HO HO HO N O H O + Cbz O HO N N N Cbz O HO Cbz Cbz O H O + Cbz O HO O O H O O E. Fritzen et al., J. Antibiot. 42, 1445-1451 (1988). 300 8.21. durch Friedel-Crafts-Acylierung von Aromaten • Benzol oder elektronenreichere Aromaten lassen sich durch FriedelCrafts-Acylierung in Aryl-Aryl-Ketone oder in Aryl-Alkyl-Ketone überführen. Geeignete Acyclierungsmittel sind Säurehalogenide (Reaktivität I > Br > Cl > F), Anhydride, Carbonsäuren, Ketene und auch Ester. • Als „Katalysator“ verwendet man Lewis-Säuren wie z.B. AlCl3, FeCl3, ZnCl2, TiCl4 usw., aber auch H2SO4, H3PO4 oder Polyphosphorsäure. Man benötigt mehr als 1 Äquivalent Katalysator, da die Lewis-Säure bis zur Hydrolyse der Reaktionsmischung am Carbonyl-Sauerstoff koordiniert bleibt. Ausnahme: Sc(OTf)3 kann in katalytischen Mengen verwendet werden. • Mehrfachacylierungen treten nicht auf, da die eingeführte Acylgruppe den Aromaten elektronenarm macht und dadurch desaktiviert. • Dirigierende Effekte bereits vorhandener Substituenten müssen berücksichtigt werden. 301 • Mechanismus: AlCl3 O 1 R ” AlCl3 O“ Cl 1 R “ O ” AlCl3 R1 Cl R2 R2 R wässerige Aufarbeitung 1 “ ” O AlCl4 1 R Cl ” AlCl3 1 R O H Cl H “ 1 R H – Cl– R2 – CO R1“ AlCl4” ” AlCl ” AlCl3 O“ H “ R1 O“ H – H+ R2 σ-Komplex O H R 3 2 • Zwischen Edukt und σ-Komplex sowie zwischen σ-Komplex und Produkt tritt jeweils noch ein π-Komplex auf (nicht gezeichnet). • Wenn sich stabilisierte Carbeniumionen ausbilden können, dann decarbonylieren die Acyliumionen und es bilden sich auch 302 Alkylierungsprodukte (Nebenreaktion). • Phenole werden erst am O acyliert und reagieren dann in einer Fries-Umlagerung zu den C-acylierten Produkten. “O O O OH R O R O AlCl3 Cl ” AlCl3 O ” Cl3Al O R “ R – HCl ” Cl3Al SN2t ” Cl3Al O “ O O “ O HO AlCl2 O“ R – HCl R wässerige ” OH O R Aufarbeitung • Man braucht ebenfalls > 1 eq. Lewissäure. Ob die Reaktion intraoder intermolekular abläuft, ist nicht vollständig geklärt. • Meist entsteht das p-Produkt als Hauptprodukt. 303 Beispiel: Teilschritt aus der Synthese von Lacinilen O OMe O OMe O 1) Zn / HCl konz. 7h Rückfluss HOOC AlCl3 / Nitrobenzol 0 - 5 °C / 85% OMe O 2) TFAA / 0 °C O 10 min. / 72% OMe HO O K. Krohn et al., J. Org. Chem. 63, 4140-4142 (1998). 304 • Verwandte Reaktion: Houben-Hoesch-Reaktion. Bei der Houben-Hoesch-Reaktion werden sehr reaktionsfähige Aromaten (Resorcin-, Phloroglucin- und Pyrogallol-Derivate u.a.) nur monoacyliert, während solche Aromaten bei Friedel-CraftsAcylierungen auch mehrfach acyliert werden können. • Man setzt sehr reaktive Aromaten mit Nitrilen in Gegenwart von ZnCl2 und gasförmiger HCl zu monoacylierten Aromaten um. R N H+ “ N H R Cl ” N H R Cl EDG N EDG H HR “ H EDG – H+ N H R ZnCl2 – ZnCl3 R “ N H ” H+ / H2O EDG O R 305 8.22. Thioaldehyde/Thioketone aus Aldehyden/Ketonen und Lawesson-Reagens • Die Addition von H2S an Aldehyde und Ketone führt unter Wasserabspaltung zu Thioaldehyden und Thioketonen, die extrem leicht flüchtig sind und ganz übel riechen (1888/89: Evakuierung der Umgebung des Chemischen Instituts der Uni Freiburg nach einem Versuch, Thioaceton herzustellen). • Wichtig: Thioaldehyde und Thioketone sind relativ instabil und trimerisieren üblicherweise. H2S O R 1 R 2 HO SH R + cat. H 1 1 R 2 H2S S HS SH + cat. H R 1 R 2 – H2S R1 R2 2 RR S 1 R 2 R S S 1 R R2 306 • Statt H2S kann man auch P4S10 verwenden. Mit P4S10 kann man auch Iminne, Oxime und Hydrazone in Thioketone überführen. • Wichtigstes Reagenz zur Umwandlung von Carbonyl- in Thiocarbonylgruppen ist das Lawesson-Reagenz, das aus Anisol und P4O10 hergestellt wird. S P S S S P S S S MeO P S P S S S S MeO P P S S MeO P S „P-S-Ylen“ MeO OMe S “ S P S ” „P-S-Ylid“ 307 • Beim Erhitzen zerfällt es in zwei Hälften, die als Phosphor-SchwefelYlide aufgefasst werden können. • Der Mechanismus der Reaktion verläuft vermutlich analog zur WittigReaktion konzertiert über eine [2+2]-Cycloaddition. R1 R1 MeO P S R2 S MeO O O P S S R1 O MeO R2 R2 S P S • Thioaldehyde und Thioketone mit α-H-Atomen liegen sehr oft in der Thioenolform vor. • Das Lawesson-Reagenz reagiert auch mit anderen Carbonylverbindungen wie z.B. Amiden und Estern. 308 Beispiel: Synthese von Thiosteroiden O H S S P MeO H O P S O H OMe H S S H H Touol / Rückfluss / 12 h / 60 % O H H S S MeO P O P S H OMe S H O H H H H Touol / Rückfluss / 12 h / 60 % J. Sarek et al., Tetrahedron 64, 3736-3743 (2008). O H H H S H 309 9. Synthese von Halbacetalen, Acetalen und Dithioacetalen 9.1. aus Aldehyden und Ketonen und Alkoholen • Regel: offenkettige Halbacetale sind instabil! Ausnahmen??? R3–OH O R 1 2 R R • 1 1 R R3–OH O 2 R R3O OH R 2 R3O OH R 1 R 2 1 2 R , R = Alkyl, Aryl, H R3 = Alkyl, (Aryl) 1 2 R , R = Elektronenziehende Reste R3 = Alkyl, (Aryl) Cyclische Halbacetale (werden oft auch als Lactole bezeichnet) sind stabil, wenn sie fünf oder sechs Ringglieder aufweisen. Typisches Beispiel: Glucose (Gleichgewicht mit der offenkettigen Form). 310 R 1 1 R • H O O O R1 O R 2 R H O O 1 R2 OH R2 OH R2 In Gegenwart von Basen sind alle Halbacetale instabil und zerfallen in ihre Bestandteile (Gleichgewichtsreaktion). ” 3 ROO H R1 1 R O R2 O O OH R1 R2 H 2 R ” R 1 + O O R3O” R2 OH (CH2)n (CH2)n n = 2, 3 n = 2, 3 311 • Mit 2 Equivalenten Alkohol reagieren Aldehyde und Ketone zu Acetalen. Alle Schritte sind Gleichgewichtsreaktionen, die durch Entfernen von Wasser auf die Seite des Acetals verschoben werden. O 1 R “H O + +H 2 R R1 + ROH R2 RO “ 1 R RO“ R1 R1 + ROH R2 – H+ OH 2 R H H“ RO OH – H2O R2 H RO R 1 “ OR R 2 RO OH R1 R2 – H+ + +H RO OR R1 R2 • Zur Herstellung von Ethylacetalen setzt man Aldehyde und Ketone häufig mit cat. Mengen H+ in Ethanol um und setzt Orthoester (z.B. Ameisensäureorthoethylester oder Essigsäureorthoethylester) zu. Die Orthoester wirken als wasserentziehende Mittel und setzen dabei zusätzlich noch Ethanol frei. • Als Säure verwendet man H2SO4, pTsOH oder Camphersulfonsäure, aber auch schwache Säuren wie PPTS, NH4NO3 oder auch schwache Lewis-Säuren. 312 Beispiel: 1) Synthese von stabilen offenkettigen Halbacetalen O HO F3C CF3 O HO O F3C THF / RT / 24 h / 66% O CF3 H. Lateef et al., J. Fluorine Chem. 104, 167-171 (2000). Beispiel: 2) Teilschritt eines Versuchs zur Totalsynthese von PPAPs O H OEt OEt OEt EtOH / HC(OEt)3 H NH4NO3 10 min. / 80 °C 74% O 1) BH3 2) Ph–OH 3) Cl2CH–OCH3 Et3COK 4) NaOH / H2O2 E. Feidt, unveröffentlicht O OEt O OH Hyperforin O 313 Beispiel: 3) Synthese eines stabilen cyclischen Halbacetals BH H2O2 NaOH PCC HO OH CH2Cl2 HO O O OH P. H. Singh et al., J. Mol. Struct. 685, 139-145 (2004). 314 9.2. aus Lactonen durch Reduktion • Analog zu der Reduktion von Estern zu Aldehyden lassen sich Lactone zu Lactolen (= cyclische Halbacetale) reduzieren. • Weil cyclische Halbacetale nur stabil sind, wenn sich 5- oder 6gliedrige Ringe ausbilden können, funktioniert diese Reaktion auch nur bei 5- und 6-gliedrigen Lactonen. • Geeignete Reduktionsmittel: DIBALH bei tiefen Temperaturen, LiAlH(OEt)3 bei tiefen Temperaturen. iBu O H Al O iBu O iBu O O H Al iBu O O H iBu Al iBu H2O / NaOH OH H O oder H2O / HCl iBu Al iBu O H2O / NaOH H oder H2O / HCl O OH H 315 Beispiel: Teilschritt aus der Synthese Crispatenin O O DIBALH / Toluol O – 70 °C / 95% 40% AcO OAc OAc OH O OH 40% OH CHO 20% Abwehrstoff der Alge Elysia crispata gegen Fraßfeinde. J. L. Perrain et al., J. Org. Chem. 72, 3770-3775 (2007). 316 9.3. aus Aldehyden und Ketonen und Thiolen • Thiole reagieren mit Aldhyden und Ketonen zu Thioacetalen. Üblicherweise wird die Reaktion in Gegenwart von Lewis-Säuren zur Aktivierung durchgeführt. Aber auch mit Toluolsulfonsäuren als Katalysator entstehen Thioacetale. • Mit 1,2-Dithiolen entstehen 1,3-Dithiolane (5-gliedrige cyclische Thioacetale). • Mit 1,3-Ditiolen entstehen 1,3-Dithiane (6-gliedrige cyclische Thioacetale). O R1 ” “ BF3 O BF3 R2 R1 “ – F3BO– R1 S R3 R2 3 R ” H “ F3B O S R3 S H – H+ R2 3 R S H ” F3B O S R3 H “ R3 S S R3 R1 R2 R3 S S R3 – H+ R1 R2 317 HS O R1 R2 O R1 1S 2 SH BF3-OEt2 S3 R1R2 BF3-OEt2 HS R2 SH 1 S 2 S3 R1R2 1,3-Dithiolan, olan deutet in der HeterocyclenNomenklatur auf einen vollständig gesättigten 5Ring hin. 1,3-Dithian, an deutet in der HeterocyclenNomenklatur auf einen vollständig gesättigten 6Ring hin. • Thioacetale werden üblicherweise als Schutzgruppen für Aldehyde und Ketone verwendet, die in Gegenwart von Acetalen mit HgCl2/H2O gespalten werden können (Acetale und Thioacetale sind zueinander orthogonale Schutzgruppen). • Thioacetale von Aldehyden können umgepolt werden (pKS = 31-37) und fungieren dann als Acyl-Anion-Äquivalent (Corey-SeebachUmpolung) 318 Beispiel: Teilschritt aus der Synthese von Amphidinolid T3 S I S / tBuLi S O O NaHCO3 / CH3I S THF / HMPT OBn – 78 °C / 83% OBn CH3CN / H2O / RT 16 h / 95% O HO O O H O O OBn O G. Zhao et al., J. Org. Chem. 71, 4625-4635 (2006). 319 • Verwandte Reaktion: Smith-Tietze-Linchpin-Reaktion (Linchpin = Angelpunkt, Aufhängung, Drehpunkt). • Ein 2-Trialkylsilyl-1,3-Dithian wird deprotoniert (Umpolung!) und dann mit einem Epoxid umgesetzt. Nach spontaner Umlagerung der Silylgruppe von C nach O (Brook-Umlagerung) bildet sich wieder ein deprotoniertes Dithian, das mit einem weiteren Epoxid (oder einem anderen Elektrophil) umgesetzt werden kann. O S S Li–nBu R1 S ” S R3Si H R3Si S R3Si O R1 OSiR3 R2 2 R ” S S O Brook-Umlagerung (C-Si → O-Si) ” 1) H3O+ 1 R S ” R3Si O R1 O Linchpin S ” S S S R1 O R2 R1 HO OH 2) TBAF OSiR3 3) HgCl 2 320 9.4. aus Ortho-Estern durch nucleophile Substitution • Ortho-Ester können durch Gringnard-Reagenzien in Acetale überführt werden. Statt Grignard-Reagenzien können auch Lithiumorganische Reagenzien oder Lithiumenolate in Gegenwart einer Lewis-Säure verwendet werden. R1 • O O O LA LA “ O 1 R O O R1 “ O O R2–MgX R1 R2 O O Wenn keine zusätzlichen Lewis-Säuren zugesetzt werden, dann fungiert das Metall der metallorganischen Verbindung als LewisSäure. 321 Beispiel: Teilschritt aus der Synthese von Lonomycin A O O O O O 1) TiCl4 / DIPEA N CH2Cl2 / – 78 °C O 2) Bn O O O O N Bn O O OMe OMe HOOC A O OH OMe H B O O C D H O HO E H O F OMe OMe OH D. A. Evans et al., J. Am. Chem. Soc. 117, 3448-3467 (1995). 322