MODIFIZIERENDE GENE BEI MUKOVISZIDOSE
Werbung

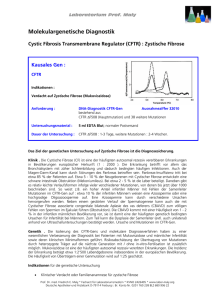
aus Jahresbericht 2006 Modifizierende Gene bei Mukoviszidose Am 2. Juni 2006 hielt Professor Dr. Peter Durie am Dr. von Haunerschen Kinderspital einen Vortrag mit dem Titel „Gene Modifiers and Disease Heterogeneity in Cystic Fibrosis“. Prof. Durie ist Pädiater mit dem Spezialgebiet Kindergastroenterologie und ein international hoch angesehener Wissenschaftler am Forschungsinstitut des Hos­ pital for Sick Children der Universität Toronto, Kanada. Er war wesentlich bei der Identifizierung der Gene für die cystische Fibrose (Mukoviszidose) und das ShwachmanDiamond-Syndrom beteiligt. Für seine Arbeiten wurde er 2003 mit dem renommierten Shwachman Award aus­ gezeichnet. Die cystische Fibrose (CF) ist eine der häufigsten autosomal rezessiv vererbten Erkrankungen der weißen Bevölkerung. In Europa ist 1 von 2.000 bis 3.000 Neugeborenen Das CFTR (Cystic Fibrosis Transmembrane Regulator) Gen codiert für den Chlorid-Kanal in verschiedenen Drüsenzellen. Der Transport für Chlorid und Bikarbonat in das Sekret ist bei Mutationen blockiert. Prof. Dr. med. Sibylle Koletzko betroffen, etwa jeder zwanzigste unter uns ist gesunder Genträger. Über 1.300 verschiedene Mutationen des CFGens (CFTR-Gens) sind inzwischen beschrieben worden. Das betroffe­ne Gen kodiert einen durch cAMP-regulierten Chlorid-Kanal, der sich in verschiedenen Körperzellen be­findet. Unterschieden werden leichtere Mutationen mit noch vorhandener Restaktivität des Chloridkanals, und schwere Mutationen mit einem völligen Ausfall des Chloridtransports. Der gestörte Chlorid-Transport führt in Drüsenzellen zu einem abnorm viskösen Sekret, das der Krankheit im deutschsprachigen Raum den Namen Mukoviszidose gab. Der zähe Schleim führt zu einer Verlegung der Ausführungsgänge in verschiedenen Organen. Durch den Rückstau kommt es zum Gewebsuntergang der Sekret bildenden Zellen. Dieser Prozess beginnt in einigen Organen, z.B. in der Bauchspeicheldrüse, schon im Mutterleib. So besteht bei der Hälfte der Betroffenen bereits zum Zeitpunkt der Geburt eine so stark verminderte Sekretion­ der Verdauungsenzyme, dass eine normale Verdauung besonders von Fett und Eiweiß nicht mehr möglich ist. Trotz normaler Nahrungsaufnahme entwickeln die Kinder bereits in den ersten Lebenswochen eine schwere Gedeihstörung. Andere betroffene Verdauungsorgane sind die Gallenwege mit Gallestau und Leberschädigung, so dass 5 – 10% der betroffenen Kinder bereits in den ersten 10 Lebensjahren eine Leberzirrhose entwickeln. Im Darm kann der zähe Schleim beim ungeborenen Kind einen kompletten Darmverschluss (Mekoniumileus) verursachen. Im späteren Lebensalter kommt es zu schwerster Verstopfung trotz der schlechten Verdauungssituation. In der Lunge führt die Verlegung der kleinen Luftwege durch den zähen Schleim zu einer zunehmenden Zerstörung des Lungengewebes, was bereits im Kindes- oder jungen Erwachsenenalter zum Tode führt. In den Fortpflanzungsorganen verursacht die verminderte Sekretion bei fast allen Männern mit CF eine vollständige Verlegung der Samenstränge mit Zeugungsunfähigkeit. Bei den weiblichen Patientinnen ist die Fruchtbarkeit zum Teil durch Verklebung der Eileiter eingeschränkt. In den Schweißdrüsen führt der gestörte Chloridkanal zu einem sehr salzrei- Dargestellt sind die Hauptorgansysteme, die bei der klassischen Mukoviszidose betroffen sind. Die klinische Ausprägung wird neben der Art der Mutationen von anderen modifizierenden Genen, Umwelteinflüssen und der Therapie mitbestimmt. chen Schweiß. Das macht man sich bei der Diagnosesicherung der Erkrankung durch Messung des Chlorid- und Natriumgehaltes in einer Schweißprobe zu Nutze. Während vor Bekanntwerden des CFTR-Gens die Diagnose einer Mukoviszidose durch die klinischen Symptome am Verdauungstrakt und der Lunge und den erhöhten Chlorid­ gehalt im Schweiß gestellt wurde, wird heute zunehmend auch die direkte Genanalyse mit Nachweis von Mutationen im CFTR-Gen herangezogen. Dieses hat jedoch dazu geführt, dass Menschen identifiziert werden, bei denen das CFTR-Gen auf beiden Chromosomen eine Veränderung, d.h. Mutation, aufweist, die aber bis zum mittleren oder gar höheren Lebensalter noch keine CF-typischen Symptome an der Lunge oder am Verdauungstrakt aufweisen. Betroffen sind vor allem Männer, die wegen Unfruchtbarkeit untersucht werden. Bei ihnen findet man meist die sogenannten leichten Mutationen, bei denen der Chlorid­ kanal noch eine gute Restfunktion hat und dadurch die schweren Ausprägungen der Krankheit verhindert werden. Beim Vergleich der klinischen Ausprägung (Phänotyp) der Erkrankung und den verschiedenen CFTR-Mutationen (Genotyp) hat man für die Bauchspeicheldrüse und die Leber eine gute Übereinstimmung gefunden. Für die Lunge trifft das weniger zu, das heißt auch Patienten mit sogenannten leichten Mutationen können schon früh eine schwere Lungenbeteiligung entwickeln. Für die sehr variable klinische Ausprägung der Mukoviszidose spielen also nicht nur die Mutationen im CFTR-Gen eine Rolle, sondern auch positive und negative Einflussfaktoren aus der Um­welt (z.B. häufige bakterielle Infektionen), und andere Gene, die modifizierend Einfluss nehmen können (sogenannte „gene modifiers“). Mutationen im CFTR-Gen werden in schwere (I, II und III in Rot) und leichtere (IV und V) unterschieden. Liegen bei einem Patienten zwei schwere Mutationen in den beiden Allelen vor, kommt es zur klinischen Ausprägung einer klassischen Mukoviszidose. Liegen zwei leichte Mutationen mit noch Restaktivität im Chloridtransport vor, kann sich die klinische Ausprägung z. B. nur auf die Verlegung der Samenstränge beim Mann beschränken. Diese Erkenntnisse mit Identifizierung der verschiedenen Genmutationen haben dazu geführt, dass bei Patienten, die wegen Entzündungen der Bauchspeicheldrüse oder wegen Infertilität (Unfruchtbarkeit) den Arzt aufsuchen, nach Mutationen im CFTR-Gen gesucht wird. So konnten Mutationen im CFTR-Gen gefunden werden, die nur eine Verlegung des Samenstrangs, aber keine klassische Mukoviszidose verursachen. Untersuchungen des Chloridkanals an der Nasenschleimhaut dieser sonst gesunden Männer weisen in der Tat einen verminderten Transport von Chlorid über die Zellwand auf. Auch ist bei diesen Personen die Chloridkonzentration in Schweiß höher als bei Personen ohne Mutationen, wenngleich häufig noch im Normalbereich. Die Ergebnisse an verschiedenen Populationen mit unterschiedlichem Organbefall haben also gezeigt, dass es sich um ein Kontinuum im Krankheitsspektrum handelt und dass die Unterscheidung zwischen gesund und einer Mukoviszidose durch Erkennung dieser Schwachformen eher schwieriger geworden ist. Die Abbildungen wurden freundlicher Weise von Prof. Dr. Peter Durie für den Artikel zur Verfügung gestellt. Dr. von Haunersches Kinderspital Abteilung für pädiatrische Gastroenterologie und Hepatologie Lindwurmstraße 4 80337 München Tel.: (089) 5160-7854 (oder -2811) Fax: (089) 5160-7898 E-Mail: [email protected] Website: www.hauner.klinikum.uni-muenchen.de Bei den einzelnen Organsystemen spielen Umwelteinflüsse und modifizierende Gene eine unterschiedlich starke Rolle. Während die Manifestation an den mänchlichen Geschlechtsorganen fast ausschließlich von der Art der CFTR-Mutation bestimmt ist, spielen bei den anderen Organen andere Gene (modifier genes) und Umwelteinflüsse eine größere Rolle.