Komplexes aus Corynebacterium glutamicum und Einfluss von
Werbung

Charakterisierung des PyruvatdehydrogenaseKomplexes aus Corynebacterium glutamicum und
Einfluss von Enzymen des anaplerotischen Knotenpunkts auf die Bildung von Valin und
Pantothenat
Dissertation
zur Erlangung des Doktorgrades Dr. rer. nat. der Fakultät für
Naturwissenschaften der Universität Ulm
vorgelegt von
Diana Fiur
aus Heidenheim
Ulm 2004
Die vorliegende Arbeit wurde in der Abteilung Mikrobiologie und Biotechnologie
der Universität Ulm angefertigt:
Amtierender Dekan:
Prof. Dr. Rolf Behm
1. Referent:
Prof. Dr. Bernhard J. Eikmanns
2. Referent:
Prof. Dr. Peter Dürre
Tag der mündlichen Prüfung:
30. Juli 2004
INHALTSVERZEICHNIS
I
Inhaltsverzeichnis
I.
Einleitung ...................................................................................... 1
II. Material und Methoden ................................................................ 9
1.
Bakterienstämme, Plasmide und Oligonukleotide.....................................9
2.
Chemikalien, Geräte, Enzyme ....................................................................13
3.
Nährmedien .................................................................................................14
4.
Kultivierung und Stammhaltung................................................................17
5.
Transformationstechniken .........................................................................18
6.
5.1
Transformation von C. glutamicum mittels Elektroporation ....................18
5.2
Transformation von Corynebakterien .....................................................19
5.3
Konjugativer Transfer von E. coli nach C. glutamicum ...........................19
5.4
Transformation von E. coli......................................................................20
Isolierung und Reinigung von DNA...........................................................20
6.1
Lösungen zur Isolierung von DNA ..........................................................20
6.2
Isolierung von Plasmid-DNA aus E. coli (´Mini-Präp´) ............................21
6.3
Isolierung von Plasmid-DNA aus E. coli (´Midi-Präp´) ............................21
6.4
Isolierung von Plasmid-DNA aus C. glutamicum ....................................22
6.5
Isolierung von chromosomaler DNA aus C. glutamicum ........................23
6.6
Isolierung von DNA-Fragmenten aus Agarosegelen ..............................23
6.7
Reinigung und Konzentrierung von Nukleinsäuren.................................23
Phenolisierung........................................................................................23
Ethanolfällung.........................................................................................24
Isopropanolfällung ..................................................................................24
Butanolfällung.........................................................................................24
Mikrodialyse von DNA ............................................................................24
Reinigung und Konzentrierung von DNA mittels Mikrokonzentratoren ...25
Konzentrations-und Reinheitsbestimmung von Nukleinsäure-Lösungen25
7.
Amplifikation, Rekombination und Analyse von DNA .............................25
7.1
Polymerase-Kettenreaktion (PCR) .........................................................25
INHALTSVERZEICHNIS
II
7.2
Restriktion von DNA ...............................................................................27
7.3
Dephosphorylierung von DNA ................................................................27
7.4
Ligation von DNA-Fragmenten ...............................................................27
7.5
Agarose-Gelelektrophorese....................................................................28
7.6
Sequenzierung .......................................................................................29
7.7
´Southern-Blot´-Hybridisierung ...............................................................29
Digoxigenin-Markierung von DNA-Sonden.............................................29
Auftrennung von DNA und Übertragung auf eine Nylonmembran..........29
Hybridisierung von DNA mit einer Digoxigenin-markierten DNA-Sonde.30
Detektion der hybridisierten Sonde.........................................................30
8.
Isolierung und Analyse von RNA ..............................................................32
8.1
Allgemeine Bemerkungen zur Isolierung von RNA.................................32
8.2
Zellernte zur RNA-Präparation ...............................................................32
8.3
RNA-Präparation mit heißem Phenol .....................................................32
8.4
RNA-Isolierung mit Hilfe des RNeasy-Mini-Kit........................................34
8.5
Radioaktive ´Northern-Blot´-Hybridisierungen ........................................34
Transfer von RNA...................................................................................34
Radioaktive Markierung von Sonden......................................................35
Hybridisierung mit radioaktiven DNA-Sonden ........................................36
9.
8.6
´Primer Extension´-Analyse....................................................................37
8.7
LI-COR-Analyse .....................................................................................37
Bestimmung von Enzymaktivitäten...........................................................38
9.1
Zellaufschluss mit dem RiboLyser ..........................................................38
9.2
Zellaufschluss mittels Universalmischgerät ............................................38
9.3
Proteinbestimmung nach Bradford .........................................................39
9.4
Berechnung von spezifischen Enzymaktivitäten.....................................39
9.5
Bestimmung der Pyruvat-Dehydrogenase-Komplex- und 2-OxoglutaratKomplex-Aktivität....................................................................................40
9.6
Bestimmung der Pyruvat-Decarboxylase-Aktivität..................................40
9.7
Bestimmung der Dihydrolipoamid-Transacetylase-Aktivität....................41
9.8
Bestimmung der Dihydrolipoamid-Dehydrogenase-Aktivität...................42
9.9
Bestimmung der Pyruvat-Kinase ............................................................42
9.10 Bestimmung der PEP-Carboxylase ........................................................43
9.11 Bestimmung der PEP-Carboxykinase ....................................................43
INHALTSVERZEICHNIS
III
9.12 Bestimmung der Pyruvat-Carboxylase ...................................................44
9.13 Bestimmung der Pyruvat-Chinon-Oxidoreduktase .................................44
9.14 Bestimmung der Chloramphenicol-Acetyltransferase-Aktivität ...............45
10. Detektion von Aminosäuren mittels HPLC-Analyse ................................46
10.1 Fermentationen und Probenahme zur Bestimmung von Valin mittels RPHPLC......................................................................................................46
10.2 RP-HPLC zur Quantifizierung von Aminosäuren ....................................46
11. Bestimmung von Pantothenat ...................................................................48
III. Ergebnisse .................................................................................. 50
1.
Analyse des Pyruvat-Dehydrogenase-Komplexes (PDHC) in Corynebacterium glutamicum 13032 ...........................................................................50
1.1
Spezifische Aktivitäten des PDHC im Wachstumsverlauf.......................50
1.2
Charakterisierung der PDHC-Akivität in Zellextrakten von C. glutamicum
13032 .....................................................................................................51
1.3
2.
Bestimmung des Ks-Wertes von Pyruvat für den PDHC.........................53
Etablierung eines Enzymtests zum Nachweis der Pyruvat-Decarboxylase (E1-Untereinheit des PDHC) aus C. glutamicum 13032 ......................56
3.
Isolierung und Untersuchung der Dihydrolipoamid-Transacetylase (E2Untereinheit) aus C. glutamicum 13032 ....................................................59
4.
Transkriptionsanalysen zur Expression der Gene aceE und aceF aus C.
glutamicum 13032.......................................................................................63
4.1
´Northern-Blot´-Experimente der Gene aceE und aceF..........................63
4.2
Promotoraktivitäten der Gene aceE und aceF im Wachstumsverlauf und
auf unterschiedlichen Kohlenstoffquellen ...............................................66
5.
4.3
Transkriptionsstartpunkt-Bestimmung von aceE ....................................68
4.4
Transkriptionsstartpunkt-Bestimmung von aceF ....................................72
Valin- und Pantothenatproduktion mit rekombinanten Stämmen ..........73
5.1
Konstruktion rekombinanter Stämme .....................................................73
5.2
Optimierung der Fermentationsbedingungen .........................................76
5.3
Valinsekretion rekombinanter Stämme...................................................80
5.4
Pantothenatsekretion rekombinanter Stämme .......................................82
INHALTSVERZEICHNIS
6.
IV
Valinproduktion und Wachstumsverhalten von Deletionsmutanten im
Gen aceE .....................................................................................................84
6.1
Nachweis der Deletion des aceE-Gens im Chromosom von
C. gluta-
micum ....................................................................................................85
6.2
Pyruvat-Dehydrogenase und Pyruvat-Decarboxylase-Aktivität in C.glutamicum-Stämmen mit deletiertem aceE-Gen...........................................86
6.3
Wachstumsverhalten und Valinproduktion rekombinanter Valinstämme 87
IV. Diskussion .................................................................................. 90
1.
Charakterisierung des PDHC von C. glutamicum 13032 ........................90
2.
Transkriptionsstudien zu den Genen aceE und aceF in C. glutamicum
13032............................................................................................................94
3.
Valin- und Pantothenatproduktion mit C. glutamicum 13032 .................98
V. Zusammenfassung.................................................................... 106
Summary .................................................................................... 109
VI. Literaturverzeichnis ................................................................. 111
ABKÜRZUNGEN
Abkürzungen
A
Adenin
A.
Azotobacter
α
Alpha
Ac
Acetat
aceE
Pyruvat-Decarboxylase (Gen)
aceF
Dihydrolipoamid-Transacetylase (Gen)
Acetyl-P
Acetylphosphat
(d) ADP
(2´-Deoxy-) Adenosin-5'-diphosphat
AE
Acetat-EDTA-Puffer
A260/280
Absorption bei 260/280 nm
AK
Acetat-Kinase (Protein)
APS
Ammoniumpersulfat
AS
Aminosäure(n)
ATCC
'American Type Culture Collection'
ATP
Adenosin-5'-triphosphat
B.
Bacillus
β
Beta
BHI
'Brain-Heart-Infusion'
BHIS
'Brain-Heart-Infusion'-Sorbitol
bp
Basenpaare
BSA
Rinderserumalbumin
bzw.
beziehungsweise
C
Cytosin, Kohlenstoff
C.
Corynebacterium
°C
Grad Celsius
ca.
circa
CaCl2
Calciumchlorid
CAT
Chloramphenicol-Acetyltransferase
cDNA
komplementäre DNA
cm
Zentimeter
Cm
Chloramphenicol
CMN
Corynebacterium-Mycobacterium-Nocardia
V
ABKÜRZUNGEN
CoA
Coenzym A
CO2
Kohlendioxid
CSPD
Disodium 3-(4-metho xyspiro {1,2-dioxetane-3,2-(5chloro)tricyclo [3.3.1.13,7]decan}-4-yl)phenyl phosphat
CTP
Cytosin-5'-triphosphat
CuSO4
Kupfersulfat
DCPIP
2,6-Dichlorindophenol
DEPC
Diethylpyrocarbonat
Da
Dalton
DHL
Dihydrolipoamid-Transacetylase (Protein)
DIG
Digoxygenin
DMSO
Dimethylsulfoxid
DNA
Deoxyribonukleinsäure
DNase
Deoxyribonuklease
DTNB
Dithionitrobenzoat
DTT
Dithiothreitol
E.
Escherichia, Enterococcus
EDTA
Ethylendiamintetraessigsäure
et al.
'et alii' (und andere)
FAD/FADH2
Flavin-Adenin-Dinukleotid oxidiert/reduziert
FeSO4
Eisensulfat
G
Guanin
g
Gramm; Erdbeschleunigung
GDP
Guanosin-5´-diphosphat
Glk
Glukose
Glukose-6-P
Glukose-6-Phosphat
GmbH
Gesellschaft mit beschränkter Haftung
GTP
Guanosin-5'-triphosphat
h
Stunde(n)
HCl
Salzsäure
HEPES
4-(2-Hydroxylethyl)-1-Piperazinethansulfonsäure
H2O
Wasser
HPLC
´High-performance-liquid-chromatography´
ICL
Isocitrat-Lyase (Protein)
VI
ABKÜRZUNGEN
i. d. R.
in der Regel
IPTG
Isopropyl-1-thio-β-D-galactosid
ilvA
Threonin-Dehydratase (Gen)
ilvBN
Acetohydroxy-Säure-Synthase (Gen)
ilvC
Isomero-Reduktase (Gen)
ilvD
Dihydroxy-Säure-Dehydratase (Gen)
k
Kilo-(103)
K-Acetat
Kaliumacetat
kb
Kilobasen
KCl
Kaliumchlorid
kDa
Kilodalton
KG
Kommanditgesellschaft
KHCO3
Kaliumhydrogencarbonat
K2HPO4
Dikaliumhydrogenphosphat
KH2PO4
Kaliumdihydrogenphosphat
Km
Kanamycin
Km/S
Michaelis-Menten-Konstante
KM
Komplexmedium
KOH
Kalilauge
kV
Kilovolt
l
Liter
λ
Lambda
LB
Luria-Bertani
LDH
Laktat-Dehydrogenase
LiCl
Lithiumchlorid
IDP
Inosin-5´-diphosphat
log
Dekadischer Logarithmus
ln
natürlicher Logarithmus
lpdA
Lipoamid-Dehydrogenase-Gen
µF
Microfarad
µl
Microliter
M
Mol, Molar
m
Milli-(10-3)
mA
Milliampère
VII
ABKÜRZUNGEN
MAE
MOPS-Acetat-EDTA-Puffer
malE
Malat-Enzym (Gen)
MCS
Multiple cloning site
ME
Malat-Enzym
MES
Morpholinoethansulfonsäure
mg
Milligramm
Mg2+
Magnesium
MgCl2
Magnesiumchlorid
MgSO4
Magnesiumsulfat
min
Minute(n)
ml
Milliliter
mm
Millimeter
mM/mmol
Millimolar
MM
Minimalmedium
MnCl2
Manganchlorid
MnSO4
Mangansulfat
mol
Stoffmenge (6,022 x 1023 Teilchen)
MOPS
3-(N-Morpholino)propansulfonsäure
mRNA
'messenger'-RNA
MS
Malat-Synthase (Protein)
mU
Milliunits
N
Amino-
n
Nano-(10-9)
NaAc
Natriumacetat
Na-Citrat
Natrium-Citrat
NaCl
Natriumchlorid
NAD(P)H
Nikotinamidadenindinukleotid (-2'-phosphat), reduziert
NaHCO3
Natriumhydrogencarbonat
NaN3
Natriumazid
NaOH
Natronlauge
ng
Nanogramm
(NH4)2SO4
Ammoniumsulfat
Ni
Nickel
NiCl2
Nickelsulfat
VIII
ABKÜRZUNGEN
nm
Nanometer
NTA
Nitrilotrichloressigsäure
(d)NTPs
(2´-Deoxy) Nukleotide
Ω
Ohm
ODx
Optische Dichte bei einer Wellenlänge von x nm
ODx
Oxalacetat-Decarboxlyase (Protein)
OGDH
2-Oxoglutarat-Dehydrogenase (Protein)
OGDHC
2-Oxoglutaratdehydrogenase-Komplex (Protein)
OPA
ortho-Pthaldialdehyd
ORF
offener Leseraster
p
Pico-(10-12)
P.
Pseudomonas
panB
Keto-Pantoate-Hydroxymethyl-Transferase (Gen)
panC
Pantothenat-Synthetase (Gen)
Pi
anorganisches Phosphat
PAGE
Polyacrylamid-Gelelektrophorese
PC
Phenol-Chloroform
pck
PEP-Carboxykinase (Gen)
PCR
Polymerasekettenreaktion
PCx
Pyruvat-Carboxylase (Protein)
PEG
Polyethylenglykol
PEP
Phosphoenolpyruvat
PEPCk
PEP-Carboxykinase (Protein)
PEPCx
PEP-Carboxylase (Protein)
PEX
´Primer-Extension´
PDC
Pyruvat-Decarboxylase (Protein)
PDH
Pyruvat-Dehydrogenase (Protein)
PDHC
Pyruvat-Dehydrogenase-Komplex (Protein)
pH
Negativer dekadischer Logarithmus der Protonenkonzentration
PK
Pyruvat-Kinase (Protein)
PIPES
Piperazin-N,N'-bis (2-ethansulfonsäure)
pmol
Picomolar
PMSF
Phenylmethylsulfonylfluorid
IX
ABKÜRZUNGEN
ppc
PEP-Carboxylase (Gen)
PTA
Phosphotransacetylase (Protein)
pqo
Pyruvat-Chinon-Oxidoreduktase (Gen)
PQO
Pyruvat-Chinon-Oxidoreduktase (Protein)
pyc
Pyruvat-Carboxylase (Gen)
pyk
Pyruvat-Kinase (Gen)
R
Resistenz
'registered' (eingetragenes Warenzeichen)
RNA
Ribonukleinsäure
RNase
Ribonuklease
RP
´Reversed phase´
rpm
Umdrehungen pro Minute
RT
Raumtemperatur, Reverse Transkriptase
s
Sekunde(n)
S.
Salmonella
SAP
´shrimp alkaline phosphatase´
SDS
Natriumdodecylsulfat
spez.
spezifisch
t
Tonnen
T
Thymin
TAE
Tris-Acetat-EDTA-Puffer
Taq
Abkürzung für die DNA-Polymerase aus Thermus aquaticus
TBE
Tris-Borat-EDTA-Puffer
TE
Tris-EDTA
TEMED
N,N,N',N'-Tetramethyl-ethylendiamin
TM
'trade mark' (Handelsmarke)
Tris
Tris-(hydroxymethyl)-aminomethan
TPP
Thiaminpyrophosphat
TY
´Tryptone Yeast´
µg
Mikrogramm
µm
Mikromolar
U
Einheiten ('units')
ÜN
über Nacht
ÜNK
Übernachtkultur
X
XI
ABKÜRZUNGEN
(d) UTPs
(2´-Deoxy) Uridintriphosphat
UV
Ultraviolett
V
Volt
VADHC
Verzweigtkettiger-Dehydrogenase-Komplex
Val1
C. glutamicum∆ilvA
Val2
C. glutamicum∆ilvA∆panBC
Vol
Volumen
v/v
Volumen pro Volumen
w/v
Gewicht pro Volumen
WT
Wildtyp
Z.
Zymomonas
z. B.
zum Beispiel
ZnSO4
Zinksulfat
Aminosäuren
A
Alanin
M
Methionin
C
Cystein
N
Asparagin
D
Asparaginsäure
P
Prolin
E
Glutaminsäure
Q
Glutamin
F
Phenylalanin
R
Arginin
G
Glycin
S
Serin
H
Histidin
T
Threonin
I
Isoleucin
V
Valin
K
Lysin
W
Tryptophan
L
Leucin
Y
Tyrosin
1
EINLEITUNG
I.
Einleitung
Die Gattung Corynebacterium gehört zusammen mit den Gattungen Mycobacterium und Nocardia zur CMN-Gruppe (Barksdale, 1970) der Actinomyceten (Stackebrandt, 1997). Das Hauptcharakteristikum dieser Gruppe ist der Besitz von die
Zelle umgebenden Mycolsäuren (Minnikin, 1978). Alle zu dieser Gruppe gehörenden Organismen sind nicht-bewegliche und Gram-positive Stäbchen. Die in dieser
Arbeit verwendete Art, Corynebacterium glutamicum, wurde erstmals 1957 von
Kinoshita et al. als unregelmäßig geformtes und nicht sporulierendes Bodenbakterium isoliert, das sich durch einen hohen GC-Gehalt von 53,8 % auszeichnet (Kalinowski et al., 2003). Besondere Bedeutung erlangte C. glutamicum aufgrund seiner Fähigkeit, Aminosäuren mit hohen Erträgen zu produzieren.
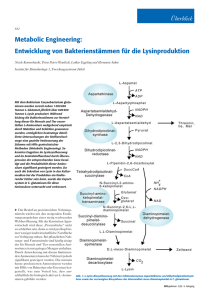
C. glutamicum ist zur Verwertung von Zuckern, Alkoholen und Säuren in der Lage
(Liebl, 1991). In Abbildung 1 ist der Zentralstoffwechsel von C. glutamicum abgebildet. Glukose wird in der Regel über die Glykolyse zu Pyruvat reduziert. Der oxidative Pentosephosphat-Weg kann jedoch alternativ zum Zuckerabbau genutzt
werden. Pyruvat wird anschließend über den Pyruvatdehydrogenase-Komplex
(PDHC) oxidativ zu Acetyl-CoA decarboxyliert, wobei das Acetyl-CoA in den Tricarbonsäure-Zyklus eingeht. Die PDHC ist ein Multienzym-Komplex bestehend
aus
den
drei
Untereinheiten
Pyruvat-Decarboxylase,
Dihydrolipoamid-
Transacetylase und Dihydrolipoamid-Dehydrogenase. Spezifische Aktivitäten
dieses Thiaminpyrophosphat- und Mg2+-abhängigen Komplexes wurden bereits in
diversen C. glutamicum Stämmen nachgewiesen (Shiio et al., 1984, CocaignBousquet und Lindley, 1995 + 1996, Schwinde et al., 2001).
Bei Wachstum auf z. B. Acetat wird dieses über die Enzyme Acetatkinase und
Phosphotransacetylase zu Acetyl-CoA aktiviert (Shiio et al., 1969) und anschließend in den Tricarbonsäure-Zyklus eingeschleust. Weiterhin spielen die Enzyme
Malat-Synthase und Isocitrat-Lyase, zwei Enzyme des Glyoxylat-Zyklus, die von
Reinscheid et al. (1994) charakterisiert wurden, hierbei eine wichtige Rolle. Da
C. glutamicum industriell zur Produktion von Aminosäuren eingesetzt wird, und
hierzu Intermediate aus dem Zentralstoffwechsel abgezogen werden, ist der Glyoxylat-Zyklus als anaplerotische Einheit zum Auffüllen und der Aufrechterhaltung
des Tricarbonsäure-Zyklus unerläßlich.
2
EINLEITUNG
Glukose
Acetat
Acetat
Glukose-6-P
Glykolyse
Glukoneogenese
Phosphoenolpyruvat
CO 2
CO 2
Pyruvat
CO 2
PDHC
Acetyl-P
PTA
Acetyl-CoA
Aspartat
Threonin
Oxalacetat
Malat
MS
Citrat
Acetyl-CoA
Glyoxylat
Lysin
Isoleucin
AK
Valin,
Pantothenat
Isocitrat
ICL
Fumarat
2-Oxoglutarat
Succinat
Glutamat
Succinyl-CoA
CO 2
Abbildung 1: Zentralstoffwechsel von C. glutamicum.
CO 2
EINLEITUNG
3
Ein bedeutender Knotenpunkt im Zentralstoffwechsel von C. glutamicum, insbesondere für die Bereitstellung benötigter Vorläuferprodukte zur Produktion von
Aminosäuren und Vitaminen, ist der PEP-Pyruvat-Oxalacetat-Knotenpunkt, der
auch als anaplerotischer Knotenpunkt bezeichnet wird (Sahm et al., 2000). Er
stellt das Bindeglied zwischen der Glykolyse und dem Tricarbonsäure-Zyklus dar,
ist Startpunkt für die Glukoneogenese und für anaplerotische Reaktionen. Die
wichtigsten Enzyme dieses Knotenpunkts sind die Pyruvat-Kinase (kodiert vom
pyk-Gen), die beiden C3-carboxylierenden Enzyme PEP-Carboxylase (kodiert
vom ppc-Gen) und Pyruvat-Carboxylase (kodiert vom pyc-Gen), sowie die drei C4decarboxylierenden Enzyme PEP-Carboxykinase (kodiert vom pck-Gen), das Malat-Enzym (kodiert vom malE-Gen) und die Oxalacetat-Decarboxylase (kein Gen
bekannt). Der letzte Schritt der Glykolyse, in dem Pyruvat unter ATP-Bildung aus
PEP entsteht, wird von der Pyruvat-Kinase bewerkstelligt (Gubler et al., 1994). Die
PEP-Carboxylase katalysiert die reversible Carboxylierung von PEP zu Oxalacetat. Deletionsmutanten im ppc-Gen haben gezeigt, dass dieses für das Wachstum
nicht essentiell ist (Peters-Wendisch et al., 1993; Gubler et al., 1994), da ein weiteres C3-carboxylierendes Enzym, die Pyruvat-Carboxylase, in C. glutamicum existiert (Peters-Wendisch et al., 1997). Letzteres katalysiert die irreversible Carboxylierung von Pyruvat zu Oxalacetat (Scrutton und Young, 1972) und ist essentiell
für das Wachstum auf Laktat und Pyruvat (Peters-Wendisch et al., 1997). Die beiden genannten Enzyme sind in der Lage, sich bei Wachstum auf Glukose gegenseitig zu ersetzen, wohingegen eine Doppelmutante in den Genen ppc und pyc
nicht mehr zum Wachstum befähigt war (Peters-Wendisch et al., 1998). Das dritte
Enzym, die PEP-Carboxykinase, katalysiert im Allgemeinen die Decarboxylierung
von Oxalacetat zu PEP und ist ATP- oder GTP-abhängig (Utter und Kohlenbrander, 1972), während es in C. glutamicum GTP- oder ITP-abhängig ist (Jetten und
Sinskey, 1993). Die Herstellung einer Deletionsmutante im pck-Gen zeigte, dass
die PEP-Carboxykinase essentiell für das Wachstum auf Aceatat oder Laktat,
nicht jedoch für das Wachstum auf Glukose ist, was auf eine gluconeogenetische
Funktion dieses Enzyms hinwies (Riedel et al., 2001). Es existieren generell zwei
weitere C4-decarboxylierende Enzyme, das Malat-Enzym, das die reversible Decarboxylierung von Malat zu Pyruvat (Fraenkel, 1975) katalysiert und die Oxalacetat-Decarboxylase, die verantwortlich für die Umsetzung von Oxalacetat zu Pyruvat ist (Krampitz und Werkman, 1941). Enzymaktivitäten des Malat-Enzyms konn-
4
EINLEITUNG
ten in verschiedenen C. glutamicum-Stämmen gemessen und das malE-Gen isoliert werden (Cocaign-Bousquet und Lindley, 1995; Cocaign-Bousquet et al., 1996;
Gourdon und Lindley, 1999; Vallino und Stephanopoulos, 1993; Gourdon et al.,
2000). Die Reinigung und Charakterisierung der Oxalacetat-Decarboxylase von C.
glutamicum war bereits möglich, ein dazugehöriges Gen konnte bislang jedoch
nicht isoliert werden (Jetten und Sinskey, 1995). Der PEP-Pyruvat-OxalacetatKnotenpunkt mit allen bedeutenden Enzymen ist in Abbildung 2 dargestellt.
PK
Pyruvat
ODx
Oxaloacetat
Abbildung
2:
Schematische
ME
Darstellung
des
PEP-Pyruvat-Oxalacetat-
Knotenpunkts mit allen involvierten Enzymen.
PK: Pyruvat-Kinase; PEPCx: PEP-Carboxylase; PEPCk: PEP-Carboxykinase;
PCx: Pyruvat-Carboxylase; ODx: Oxalacetat-Decarboxylase; ME: Malat-Enzym;
PQO: Pyruvat-Chinon-Oxidoreduktase; PDHC: Pyruvatdehydrogenase-Komplex
EINLEITUNG
5
Neben der PDHC existiert in C. glutamicum eine Pyruvat-Chinon-Oxidoreduktase,
die Pyruvat direkt zu Acetat umsetzt (M. Schreiner, persönliche Mitteilung). Das für
dieses Enzym kodierende pqo-Gen konnte identifiziert und das Enzym gereinigt
und biochemisch charakterisiert werden, wobei sich mit einem Km-Wert von 30
mM eine sehr geringe Affinität zu Pyruvat zeigte.
C. glutamicum wird als Aminosäureproduzent industriell eingesetzt. Besondere
Bedeutung kommt diesem Organismus bei der Herstellung von Glutamat, von
welchem gegenwärtig 1,5 Millionen t/Jahr produziert werden (Hermann, 2003), zu.
Glutamat wird unter bestimmten induzierenden Bedingungen wie z. B. durch Zugabe von Detergentien wie Tween (Takinami et al., 1965) oder unter Biotinmangel
(Shiio et al., 1962) ins Medium ausgeschieden und kommt in erster Linie als Geschmacksverstärker in der Lebensmittelindustrie zum Einsatz (Kleemann et al.,
1985). Im Gegensatz zu Glutamat findet Lysin, dessen Produktion momentan bei
550 000 t/Jahr liegt, hauptsächlich als Futtermittelzusatz (Leuchtenberger, 1996)
Verwendung.
Eine immer stärker werdende Nachfrage gibt es für die drei verzweigtkettigen Aminosäuren Leucin, Isoleucin und Valin (400-500 t/Jahr). Diese Aminosäuren
kommen als Zusatz zu Futtermittel und Infusionslösungen sowie als Vorstufe für
die chemische Synthese von Herbiziden (Eggeling, 2001; Leuchtenberger, 1996)
zur Anwendung. Ein weiteres biotechnologisch bedeutendes Produkt ist Pantothenat, das mit 4000 t/Jahr zum Großteil chemisch synthetisiert wird (Vandamme,
1992). Pantothenat gehört zu den wasserlöslichen B-Vitaminen (Vitamin B5), ist
der Vorläufer für die Herstellung von Coenzym A und kann von Tieren nicht selbst
synthetisiert werden. Demzufolge liegt der wirtschaftliche Nutzen für Pantothenat
in der pharmazeutischen Industrie und als Zusatz zu Futter- und Nahrungsmitteln.
Die erste Generation von Produktionsstämmen wurde durch ungerichtete Mutagenese hergestellt. Mit dieser Methode konnte im Jahre 1961 eine Homoserinauxotrophe und Lysin-produzierende Mutante erhalten werden (Kitada et al.,
1961). Spätere Verfahren zum Auffinden von Mutanten konzentrierten sich auf
solche Stämme, die auf das Lysin-Analog S-(2-Aminoethyl)-Cystein und Threonin
resistent waren (Sano und Shiio, 1970). Einer dieser Stämme zeichnete sich durch
eine auf Lysin Feedback-resistente Aspartat-Kinase aus, wodurch eine Steigerung
EINLEITUNG
6
der Lysinproduktion möglich war. Die Tatsache, dass die Herstellung von Produktionsstämmen durch ungerichtete Mutagenese unerwünschte Nebeneffekte wie
ein instabiles Produktionsverhalten, eine höhere Sensitivität gegenüber der Temperatur oder des pH-Wertes und eine teilweise kostspielige Supplementation führten dazu, dass in der zweiten Generation von Produktionsstämmen Gene gezielt
in bereits durch ungerichtet Mutagenese hergestellten Stämmen überexprimiert
wurden. Durch gezielte Überexpression der Gene des Lysinbiosynthesewegs in
dem auf Lysin Feedback-resistenten Stamm konnte von Cremer et al. (1991) gezeigt werden, dass neben der Aspartat-Kinase der vom dapA-Gen kodierten Dihydrodipicolinat-Synthase die größte Bedeutung bei der Kontrolle des Flusses in
Richtung Lysin zukommt.
Die beste Möglichkeit, unerwünschte Sekundäreffekte zu vermeiden, geht mit der
Entschlüsselung des Genoms von C. glutamicum einher (Kalinowski et al., 2003),
wodurch nun gezielte genetische Manipulationen zur Steigerung der Aminosäureproduktion durchgeführt werden können. Die gezielte Veränderung von Genen des
Zentralstoffwechsels sollte in dieser Arbeit zur Produktionssteigerung von Valin
und Pantothenat angewandt werden.
Die Gene des Valin- und Pantothenatbiosynthese-Wegs in C. glutamicum konnten
identifiziert und Stämme, die sowohl Valin als auch Pantothenat ins Medium ausscheiden, hergestellt werden (Cordes et al., 1992; Keilhauer et al., 1993; Dusch et
al., 1999; Sahm und Eggeling, 1999; Radmacher et al., 2002; Merkamm et al.,
2003).
Abbildung 3 zeigt sowohl die Biosynthesewege der Aminosäuren Isoleucin und
Valin als auch des Vitamins Pantothenat. Isoleucin wird aus Threonin hergestellt,
wobei der einleitende Schritt dieses Biosynthesewegs von der ThreoninDehydratase katalysiert wird. Bei einer Konzentration von mehr als 5 mM Isoleucin
im Medium (Morbach et al., 1995) wird die Threonin-Dehydratase und alle folgenden Enzyme, die zur Synthese von Isoleucin und Valin notwendig sind, gehemmt.
Die Synthese von Valin erfolgt ausgehend von zwei Molekülen Pyruvat in drei
Schritten durch die von ilvB, N, C und D kodierten Enzyme zum Keto-Isovalerat.
Aus Keto-Isovalerat kann entweder über eine von ilvE kodierte Transaminase B
7
EINLEITUNG
Threonin
Threonin-Dehydratase
(ilvA)
Pyruvat + Pyruvat
Oxobutyrat + Pyruvat
Acetohydroxy-SäureSynthase (ilvB,N)
Acetolaktat
Isomero-Reduktase
(ilvC)
Dihydroxy-Isovalerat
Dihydroxy-SäureDehydratase (ilvD)
Transaminase B
(ilvE)
Keto-Isovalerat
Valin
Aspartat Keto-Pantoatet
AspartatDecarboxylase
(panD)
Keto-PantoatHydroxymethylTransferase (panB)
Isoleucin
Ketopantoat-Reduktase (panE)
β-Alanin Pantothensäure
Pantothenat-Synthetase
(panC)
Pantothenat
Abbildung 3: Biosynthesewege zur Herstellung der Aminosäuren Valin und Isoleucin und des Vitamins Pantothenat.
Valin oder alternativ in drei weiteren Schritten Pantothenat hergestellt werden.
Hierfür wird Keto-Isovalerat in zwei Schritten zu Pantothensäure umgesetzt, die in
einem letzten Schritt mit β-Alanin über eine Pantothenat-Synthetase zu Pantothenat katalysiert wird.
Ein Ziel dieser Arbeit war die Charakterisierung des PDHC von C. glutamicum in
Zellextrakten.
Decarboxylase
Weiterhin
sollten
(E1-Untereinheit,
für
die
kodiert
beiden
von
Untereinheiten
aceE)
und
Pyruvat-
Dihydrolipoamid-
Transacetylase (E2-Untereinheit, kodiert von aceF) der PDHC geeignete Nach-
8
EINLEITUNG
weissysteme etabliert werden und beide Enzyme in Zellextrakten und auf
transkriptioneller Ebene untersucht werden.
Eine weitere Zielsetzung der vorliegenden Arbeit war, ausgehend von bereits bekannten
und
produzierenden
Valin-
und
Pantothenatproduktionsstämmen
(C. glutamicum 13032∆ilvA und C. glutamicum 13032∆ilvA∆panBC), die Enzymaktivitäten des PEP-Pyruvat-Oxalacetat-Knptenpunkts zu variieren und diese
Stämme hinsichtlich ihres Wachtumsverhaltens und der Produktion von Valin und
Pantothenat zu untersuchen.
9
MATERIAL UND METHODEN
II.
Material und Methoden
1.
Bakterienstämme, Plasmide und Oligonukleotide
In Tabelle 1 sind die in dieser Arbeit verwendeten Escherichia- sowie
Corynebacterium- Stämme zusammengetragen; Tabelle 2 zeigt die in dieser
Arbeit verwendeten Plasmide und Tabelle 3 die Oligonukleotide, mit denen gearbeitet wurde.
Tabelle 1: Verwendete Bakterienstämme
Stamm
genetische Eigenschaften
Referenz/
Herkunft
E. coli DH5α
recA1, endA1, hsdR17, sup
E44, relA1, gyrA96, thi-1, lac
Zα
Hanahan, 1985
E. coli S17-1
thi-1, endAR1, hsdR17, sup
E44, pro
Simon et al.,
1983
C. glutamicum 13032
Wildtyp
Abe et al., 1967
C. glutamicum 13032∆aceE
∆aceE
M. Schreiner
C. glutamicum 13032∆ilvA
∆ilvA
C. glutamicum 13032
∆ilvA∆panBC
∆ilvA, ∆panBC
Sahm und Eggeling, 1999
Radmacher et
al., 2002
C. glutamicum 13032
∆ilvA∆panB∆pck
∆ilvA, ∆panBC, ∆pck
diese Arbeit
C. glutamicum 13032
∆ilvA∆panBC∆aceE
∆ilvA, ∆panBC, ∆aceE
diese Arbeit
Escherichia- Stämme
Corynebacterium- Stämme
10
MATERIAL UND METHODEN
C. glutamicum 13032∆ilvA
∆pyc
∆ilvA, ∆pyc
diese Arbeit
C. glutamicum 13032 ilvN/
M13
ilvN resistent gegenüber der
Rückhemmung von Val, Ile,
Leu
V. Elisakova
C. glutamicum 13032 ilvN/
M13 ∆aceE
ilvN resistent gegenüber der
Rückhemmung von Val, Ile,
Leu; ∆aceE
diese Arbeit
C. glutamicum 13032
∆ilvA∆panBC ilvN/M13
∆ilvA, ∆panBC, ilvN resistent
gegenüber der Rückhemmung
von Val, Ile, Leu
V. Elisakova
C. glutamicum 13032
∆ilvA∆panBC
ilvN/M13 ∆aceE
∆ilvA, ∆panBC; ilvN resistent
gegenüber der Rückhemmung
von Val, Ile, Leu; ∆aceE
diese Arbeit
C. glutamicum R127 panC:
pk 18 mob´panC´
panC-Integrationsmutante in
restriktions-negativem Stamm
Sahm und Eggeling, 1999
Tabelle 2: Plasmide
Vektor
Eigenschaften
Referenz
pJC1 ilvBNCD
KmR; trägt Ile-Biosynthesegene
ilvBNCD mit nativen Promotoren
Sahm et al., 1999
pECM3 ilvBNCD
CmR; trägt Ile-Biosynthesegene
ilvBNCD mit nativen Promotoren
Sahm et al., 1999
pEK0
KmR, ori pBL1, ori colE1
Eikmanns et al., 1991a
pEK0-pck
KmR, ori pBL1, ori colE1; Überexpression von pck
Riedel et al., 2001
pEKO-lpdA
KmR, ori pBL1, ori colE1; Überexpression von lpdA
Schwinde et al., 2001
pK19 mobsacB
KmR, oriT (mobilisierbar), oriV
Schäfer et al., 1994
pK19 aceE
KmR, ori pBL1, ori colE1; Vektor zur
Deletion von aceE
M. Schreiner
11
MATERIAL UND METHODEN
pK19 mobsacBpyc
KmR, ori pBL1, ori colE1; Vektor zur
Deletion von pyc
Peters-Wendisch et al.,
1998
pK19 mobsacBpck
KmR, ori pBL1, ori colE1; Vektor zur
Deletion von pck
Riedel et al., 2001
pVWEx1-pyc
KmR, lacIq, Ptac, oripHM1519, oriE,c., pyc
ohne eigenen Promotor
Katsoulidis, Dissertation
pXTpoxBEx
TetR, lacIq, trägt pqo zur Überexpres- B. Bathe, Degussa
sion
pEKEx2
KmR, lacIq, tacP, ori pBL1, ori colE1
Eikmanns et al., 1994
pEKEx2-pyk
KmR, lacIq, tacP, ori pBL1, ori colE1;
trägt pyk-Gen zur Überexpression
diese Arbeit
pMFα-ppc
KmR, trägt ppc-Gen mit nativem Promotor
Eikmanns et al., 1989
pET2
KmR, cat, ori pBL1, ori colE1
Vasicova et al., 1998
pET2-aceE
KmR, cat, ori pBL1, ori colE1
diese Arbeit
Promotortestvektor zur Untersuchung
der aceE-Promotorregion
pET2-aceF
KmR, cat, ori pBL1, ori colE1
diese Arbeit
Promotortestvektor zur Untersuchung
der aceF-Promotorregion
Tabelle 3: Oligonukleotide
´Primer´
DNA-Sequenz
Beschreibung
´Primer´ zur Überexpresssion
over pyk1
CGGGATCCCGAATGGTAGTACCTGTGGC
Hin-´Primer´, BamHI
over pyk2
GGGGTACCCCGCGCTGACACCACGTACA
Rück-´Primer´, KpnI
12
MATERIAL UND METHODEN
´Primer´ zur Klonierung von Promotorfusionen
acE1
ACGCGTCGACCACCAAAAGGACATCAGACC
Hin-´Primer´, SalI
aceE2
CGCGGATCCACACCTCCTGTTGGAATG
Rück-´Primer´,
BamHI
aceF1
ACGCGTCGACGCTCATATTCAATGCGTTCGCG
Hin-´Primer´, SalI
aceF2
CGCGGATCCGCAAGTCGTGTCGTTAAAACGG
Rück-´Primer´,
BamHI
´Primer´ zum Nachweis von Deletionsmutanten
aceE1
ACGCGTCGACCACCAAAAGGACATCAGACC
aceE3
aceE5.3
GGATAGGTGATTGGAAGTTGGGCAAACGAAG
CATGA
CGCGGATCCGCGGGATTTATCTGTCCC
pyc1
GTCGACTCACACATCTTC
pyc2
CACCAGCGAATGGTGCAG
Nachweis der aceEDeletion
Nachweis der aceEDeletion
Nachweis der aceEDeletion
Nachweis der pycDeletion
Nachweis der pycDeletion
IRD800-markierte ´Primer´ für ´Primer´-Extension-Reaktionen
CM4
GAAAATCTCGTCGAAGCTCG
IRD800-markiert
CM5
AAGCTCGGCGGATTTGTC
IRD800-markiert
Sec-Hin2
TGACCATGATTACGCCAAGC
Hin-´Primer´ zur
Sequenzierung von
pEKEx2
sec-REV
CTTCTCTCATCCGCCAAAAC
Rück-´Primer´ zur
Sequenzierung von
pEKEx2
M13 for
GTTTTCCCAGTCACGAC
Universal ´primer´
sec-pyk-kin2
CACAGCGATGGCTCCGAA
´Primer´ zur Sequenzierung des
Mittelstücks von pyk
Sequenzier-´Primer´
Alle ´Primer´ wurden von den Firmen MWG Biotech GmbH (Ebersberg) und Biomers (Ulm) bezogen. Die jeweiligen Schnittstellen sind rot hinterlegt.
13
MATERIAL UND METHODEN
2.
Chemikalien, Geräte, Enzyme
Die eingesetzten Chemikalien wurden von folgenden Firmen bezogen:
Difco Laboratories (Detroit, USA), Geschäftsbereich Fluka, Merck KGaA (Darmstadt), Riedel de Häen AG (Seelze), Serva Feinbiochemica GmbH & Co KG (Heidelberg), Sigma-Aldrich Chemie GmbH (Deisenhofen) (Ausnahmen sind im Text
gesondert erwähnt)
Folgende zum Teil nicht im Text erwähnte Geräte wurden benutzt:
Fotodokumentationsanlage: MWG-Biotech GmbH (Ebersberg)
Photometer:
Ultrospec 3000 (Amersham Pharamacia Biotech
GmbH, Freiburg)
pH-Meter:
WTW pH521 (Wissenschaftlich-Technische Werkstätten, Weilheim)
Waagen:
Sartorius BP 8199 u. BP 2100 (Sartorius AG,
Göttingen)
Zentrifugen:
Centrifuge
5804
R
(Eppendorf-Netheler-Hinz
GmbH, Köln) Heraeus Sepatech Minifuge RF (Heraeus Hoding GmbH, Stuttgart)
Galaxy 14D (VWR International, Darmstadt)
Enzyme:
RNase-freie DNase: Roche Molecular Biochemicals GmbH, Mannheim
Restriktionsenzyme, Taq-Polymerase, T4-DNA-Ligase, SAP ('shrimp alkaline
phosphatase'): MBI-Fermentas GmbH (St. Leon-Rot)
High fidelity PCR Enzyme Mix: MBI-Fermentas GmbH (St. Leon-Rot)
Mul-V- Reverse Transkriptase: MBI-Fermentas GmbH (St. Leon-Rot).
14
MATERIAL UND METHODEN
3.
Nährmedien
Vollmedium 2x TY (Sambrook et al., 2001)
Bacto-Trypton
16
g/l
Hefeextrakt
10
g/l
5
g/l
NaCl
Vollmedium LB (Sambrook et al., 2001)
Bacto-Trypton
Hefeextrakt
NaCl
10
g/l
5
g/l
10
g/l
Zur Herstellung von Platten wurden dem jeweiligen Medium 15 g Agar/l zugesetzt.
SOB-Medium (Sambrook et al., 2001)
Bacto-Trypton
20
g/l
6
g/l
NaCl
0,5
g/l
KCl
0,2
g/l
2
g/l
2,4
g/l
Hefeextrakt
MgCl2 x 6 H2O
MgSO4 x 7 H2O
Die Magnesiumsalze wurden getrennt autoklaviert und danach zugegeben.
CgC-Minimalmedium (modifiziert nach Kase et al., 1972)
(NH4)2SO4
5
g/l
Harnstoff
5
g/l
21
g/l
MOPS
K2HPO4
0,5
g/l
KH2PO4
0,5
g/l
MgSO4
0,25 g/l
CaCl2
0,01 g/l
Spurenelemente-Lösung
1
ml/l
Biotin (200 mg/l)
1
ml/l
15
MATERIAL UND METHODEN
Der pH-Wert wurde für Wachstum auf Glukose auf pH 6,7, für Wachstum auf
Mischsubstrat auf pH 6,5 und für Wachstum auf Säuren mit 1 N NaOH auf pH 6,3
eingestellt. Das Medium wurde bis 90 % des Gesamtvolumens aufgefüllt und
autoklaviert. Biotin, Spurenelemente, Kohlenstoffquellen, sowie gegebenenfalls
Vitamine und Aminosäuren wurden nach dem Autoklavieren zugegeben.
Spurenelemente-Lösung
FeSO4 x 7 H2O
16,4
g/l
MnSO4 x H2O
10
g/l
CuSO4 x 5 H2O
0,2 g/l
ZnSO4 x 7 H2O
1,0 g/l
NiCl2 x 6 H2O
0,02 g/l
Die Lösung wurde mit konzentrierter HCl auf einen pH-Wert von etwa 1 eingestellt.
Kohlenstoffquellen
Glukose
10-40 g/l
K-Acetat
10
g/l
Es wurden 20 %-ige (K-Acetat) bzw. 50 %-ige (Glukose) Stammlösungen hergestellt und sterilfiltriert. Für die Fermentationen zur Untersuchung von Valin und
Pantothenat wurde in der Regel eine Glukose-Endkonzentration von 4 % verwendet. Bei anderen Experimenten lag die Endkonzentration der Substrate bei 1 %.
CGXII-Medium (modifiziert nach Kase et al., 1972)
(NH4)2SO4
20
g/l
5
g/l
42
g/l
K2HPO4
1
g/l
KH2PO4
1
g/l
MgSO4
0,25 g/l
CaCl2
0,01 g/l
Harnstoff
MOPS
Das Medium wurde auf ein Volumen von 750 ml aufgefüllt, auf einen pH von 7
eingestellt und 45 ml in 500 ml Erlenmeyer-Schikanenkolben abgefüllt und au-
16
MATERIAL UND METHODEN
toklaviert. Nach dem Autoklavieren wurden 100 µl Spurenelemente und Biotin zugesetzt, sowie 4,8 ml einer 50 %-igen Glukose-Stammlösung. Das restliche fehlende Volumen wurde auf ein Gesamtvolumen von 60 ml mit H2O aufgefüllt.
Epo-Medium (van der Rest et al., 1999)
Bacto-Trypton
Hefeextrakt
NaCl
10
g/l
5
g/l
10
g/l
Lösen in H2O und autoklavieren
Isonikotinsäurehydrazid
0,4
g/l
Glycin
2,5 g/l
Tween 80
0,1
ml
In 20 ml lösen, sterilfiltrieren und zu 80 ml Vollmedium geben
BHIS-Medium (Liebl et al., 1989)
BHI
Sorbitol
5
g/l
91
g/l
Beide Lösungen wurden getrennt voneinander autoklaviert und dann vereinigt.
LB/BHIS-Platten (Liebl et al., 1989)
BHI
Bacto-Trypton
Hefeextrakt
NaCl
18,5
g/l
5
g/l
2,5 g/l
5 g/l
Sorbitol
91
g/l
Agar
18
g/l
Die Sorbitol-Lösung wurde getrennt angesetzt und autoklaviert und nach dem Autoklavieren zugegeben.
17
MATERIAL UND METHODEN
Saccharose-Platten
LB-Medium
Saccharose
Agar
100
g/l
18
g/l
Medienzusätze
Zusatz
Stammlösung
Arbeitskonzentration
Kanamycin
50 mg/ml
Chloramphenicol
50 mg/ml (in 70% Ethanol)
5 µg/ml
Tetracyclin
10 mg/ml (in 70% Ethanol)
10 µg/ml
Biotin
200 mg/ml
IPTG
100 mM
4.
50 µg/ml
200 µg/ml
1 mM
Kultivierung und Stammhaltung
Die Bakterienstämme wurden in 30 % (v/v) Glycerin (Gesamtvolumen 1 ml) in fest
verschließbaren Gefäßen bei –70°C gelagert. Ausgehend von diesen Stammkulturen wurden Stammplatten (LB-Platten) hergestellt, die als Inokulum für Vorkulturen
dienten. Die Platten für E. coli wurden für 1 Tag bei 37°C inkubiert, die für Corynebacterium für 2 Tage bei 28°C. Diese wurden in der Regel nicht länger als 2
Wochen (Corynebakterien) bzw. nicht länger als 1 Woche (E. coli) verwendet.
Zur Anzucht von E. coli in Flüssigkultur diente LB-Medium. Die Züchtung von
E. coli erfolgte in 5 ml (in Reagenzgläsern) bzw. 50 ml Medium (in 250 ml Erlenmeyerkolben) bei 160 rpm und 37°C. Für Wachstumsversuche mit C. glutamicum
wurde CgC-Medium mit der entsprechenden Kohlenstoffquelle eingesetzt. Für die
Vorkultur wurden die Zellen über Nacht auf 2x TY-Medium gezüchtet (in 500-mlSchikanenkolben bei 28°C, 120 rpm), durch Zentrifugation (4000 rpm, 4°C, 10
min) geerntet und 2x mit 0,9 % NaCl gewaschen. Die gewaschenen Zellen wurden
in 5 ml 0,9 % NaCl resuspendiert und dienten als Inokulum für die Hauptkulturen
auf CgC-Minimalmedium mit der entsprechenden Kohlenstoffquelle. Bei Wachstum auf Glukose lag die optische Dichte bei 600 nm (OD600) zu Beginn bei 0,5, bei
MATERIAL UND METHODEN
18
Wachstum auf Säuren und Mischsubstraten bei 1,0. Die Inkubation erfolgte anschließend bei 28°C und 120 rpm.
5.
Transformationstechniken
5.1
Transformation von C. glutamicum mittels Elektroporation (van der
Rest et al., 1999)
Zur Einbringung von Plasmid-DNA in C. glutamicum wurde eine Elektroporation
mit anschließender Hitzeschockbehandlung durchgeführt. Kompetente Zellen, die
zur Aufnahme von DNA bereit waren, wurden nach folgendem Protokoll hergestellt: 70 ml LB-Medium wurde mit einer Einzelkolonie (frische Platte) inokuliert
und im Schüttelkolben anschließend über Nacht bei 28°C inkubiert. Ausgehend
von dieser Vorkultur wurden 100 ml Epo-Medium in einem 1 l Erlenmeyerkolben
ohne Schikanen zu einer OD600 von 0,3 angeimpft. Diese Kultur wurde dann in
einem zweidimensional schüttelndem Wasserbad (Schüttelfrequenz ca. 1/s) bei
18°C bebrütet - die OD600 entsprach nach 28 h ca. 1. Die Zellen wurden durch
Zentrifugation (5000 rpm, 4°C, 10 min) geerntet und insgesamt viermal mit 50 ml
eiskaltem Glycerin (10 %, v/v) gewaschen. Die Zellen wurden dann in 0,5 ml Glycerin (10 %, v/v) aufgenommen und in 50 µl Aliquots in flüssigem
Stickstoff
schockgefroren. Die Lagerung der kompetenten Zellen erfolgte bei –70°C. Zur
Transformation wurde je ein Aliquot auf Eis aufgetaut, in Elektroporationsküvetten
transferiert und mit 1-5 µl entsalzter Plasmid-DNA versetzt. Nach 15-minütiger Inkubation auf Eis erfolgte die Elektroporation bei 25 µF, 600 Ω und 2,5 kV in einem
Gene-Pulser (Bio-Rad Laboratories GmbH, München). Sofort nach der Elektroporation wurden die Zellen in den Küvetten mit 600 µl BHIS-Medium versetzt und in
einem Wasserbad einem Hitzeschock unterzogen (46°C, 6 min). Anschließend
wurde die Zellsuspension in 1,5 ml- Reaktionsgefäße transferiert und zur Resistenzausprägung für 1 h bei 30°C im Wasserbad inkubiert. Die Zellen wurden zur
Selektion auf LB-Platten mit Kanamycin (15 µg/ml) ausplattiert.
MATERIAL UND METHODEN
5.2
19
Transformation von Corynebakterien (modifiziert nach Liebl et al.,
1989)
Mit einer frischen Kolonie wurden 50-ml 2x TY-Medium angeimpft und über Nacht
inkubiert. Von dieser ÜNK wurden am folgenden Tag 500 ml LB-Medium mit 4
mg/ml Isonikotinsäurehydrazid, 2,5% Glycin und 0,1 % (v/v) Tween 80 (1 l Schikanenkolben) mit 10 ml angeimpft. Die Zellen wuchsen bei 28°C und 120 rpm bis zu
einer End-OD von 0,5. Nach 15-minütiger Kühlung auf Eis wurden die Zellen abzentrifugiert (4500 x g, 4°C, 10 min) und anschließend 2x mit eiskaltem 10%-igem
Glycerin gewaschen. Die Zellen wurden in 1 ml Glycerin aufgenommen und in 100
µl Aliquots bei –70°C gelagert. Zur Elektroporation wurden 100 µl Zellen mit 100 µl
10%-igem kaltem Glycerin verdünnt, mit 1-10 µl Plasmid-DNA versetzt und mit
einem Gene Pulser (Bio-Rad Laboratories GmbH, München) bei 200Ω, 25 µF und
2,5 kV elektroporiert. Anschließend wurden sofort 600 µl BHIS-Medium zugegeben und für 6 min ein Hitzeschock bei 46°C durchgeführt. Die Regeneration erfolgte für 1 h bei 28°C. Zur Selektion wurde der Ansatz auf LB-Platten mit entsprechendem Antibiotikum ausplattiert.
5.3
Konjugativer Transfer von E. coli nach C. glutamicum
Die Konjugation zwischen E. coli und C. glutamicum erfolgte nach einem modifizierten Protokoll von Schäfer et al. (1990). Mit 50 µl einer ÜNK eines rekombinanten E. coli S17-1 Donorstammes wurden 5 ml 2x TY inokuliert. Nachdem die Donorkultur eine OD600 von 1 erreicht hatte, wurde sie für 10 min auf Eis inkubiert.
Der C. glutamicum-Rezipient wurde über Nacht in 70 ml 2x TY gezüchtet (OD600
von 5) und anschließend zur Inaktivierung des Restriktionssystems für 9 min bei
48,5°C inkubiert. Donor und Rezipient wurden im Verhältnis 1:3 gemischt und
durch Zentrifugation sedimentiert. Die Zellen wurden in 150 µl LB-Medium resuspendiert und auf einen Zellulosenitratfilter (25 mm Durchmesser; 0,45 µm Porengröße), der auf einer LB-Platte lag, übertragen. Die Inkubation erfolgte für 20 h
bei 28°C. Danach wurde der Filter in ein Falkon überführt und die Zellen mit 600 µl
LB-Medium abgeschwemmt. Die Zellen wurden kurz sedimentiert und der Ansatz
auf BHIS-Platten mit Nalidixinsäure zum Abtöten von E. coli und Kanamycin zur
Selektion ausplattiert.
MATERIAL UND METHODEN
5.4
20
Transformation von E. coli
Die Transformation von E. coli erfolgte mit Hilfe kaltkompetenter Zellen (Inoue et
al., 1990). Mit 5 ml einer LB-Vorkultur wurden 250 ml SOB-Medium beimpft. Bei
18°C wurde die Kultur bis zum Erreichen einer OD600 von 0,6 geschüttelt (ca. 18 h)
und anschließend auf Eis abgekühlt. Nach der Zellernte durch Zentrifugation
(4000 rpm, 4°C, 10 min) wurde das Zellpellet in 80 ml eiskaltem TB-Puffer gewaschen, 10 min auf Eis gekühlt und erneut abzentrifugiert. Die sedimentierten Zellen wurden anschließend in 20 ml kaltem TB-Puffer aufgenommen, langsam mit
1,5 ml DMSO versetzt und in 200 µl Aliquots in flüssigem Stickstoff schockgefroren. Die Lagerung der kompetenten Zellen erfolgte bei –70°C. Zur Transformation
wurde je ein Aliquot aufgetaut und mit Plasmid-DNA (bis 20 µl) versetzt. Nach Inkubation des Ansatzes auf Eis (40 min) erfolgte ein kurzer Hitzeschock (42°C, 1
min) und eine erneute 10-minütige Inkubation auf Eis. Nach Zugabe von 0,8 ml
LB-Medium wurden die Zellen für 1 h bei 37°C unter Schütteln zur Resistenzausprägung inkubiert und anschließend zur Selektion von Transformanten auf LBPlatten ausplattiert.
TB-Puffer:
10 mM PIPES, pH 6,7
15 mM CaCl2
250 mM KCl
55 mM MnCl2
⇒ PIPES und CaCl2 wurden getrennt von KCl und MnCl2 autoklaviert
6.
Isolierung und Reinigung von DNA
6.1
Lösungen zur Isolierung von DNA
Im folgenden sind die zur Isolierung von DNA verwendeten Lösungen aufgeführt.
TE-Puffer:
10 mM Tris-HCl (pH 8,0)
1 mM EDTA (pH 8,0)
MATERIAL UND METHODEN
21
Lösung A: 50 mM Glucose
25 mM Tris-HCl (pH 8,0)
10 mM EDTA (pH 8,0)
Lösung B: 0,2 N NaOH
1 % SDS
⇒ frisch ansetzen
Lösung C: 3 M K-Acetat
11,5 % Essigsäure
6.2
Isolierung von Plasmid-DNA aus E. coli ('Mini'-Präp, nach Birnboim,
1983, modifiziert)
Die Isolierung von Plasmid-DNA erfolgte nach dem Prinzip der alkalischen Lyse.
Dazu wurden 1,5 ml Zellen einer LB-ÜNK durch kurze Zentrifugation (13000 rpm,
30 s, RT) sedimentiert und in 100 µl Lösung A resuspendiert. Nach Zugabe von
200 µl Lösung B erfolgte die Lyse der Zellen bei gleichzeitiger Denaturierung von
Proteinen und DNA. Durch Zugabe von 150 µl Lösung C ging bei neutralem pH
die Plasmid-DNA wieder in ihre native Form über. Die Fällung der Plasmid-DNA
erfolgte mit Ethanol (siehe 6.7). Nach einmaligem Waschen mit 70 %-igem Ethanol wurde die DNA in 50 µl TE-Puffer gelöst. Die Qualität der Plasmid-DNA war für
Restriktionsanalysen ausreichend.
Um die DNA von RNA-Verunreinigungen zu befreien, wurde der Ansatz mit 1 µl
RNase A (10 mg/ml) versetzt und für 30 min bei 37°C inkubiert. Anschließend
wurde eine Phenolextraktion (sieh 6.7) durchgeführt, um die Plasmid-DNA von
Proteinverunreinigungen zu befreien. Die DNA wurde dann mit Ethanol gefällt und
nach Lufttrocknung in TE-Puffer aufgenommen.
6.3
Isolierung von Plasmid-DNA aus E. coli ('Midi-Präp', Sambrook et al.,
2001)
Große Mengen sauberer Plasmid-DNA aus E. coli wurden aus 50 ml einer LBÜNK isoliert. Das durch Zentrifugation (4000 rpm, 10 min, 4°C) erhaltene Zellpellet
MATERIAL UND METHODEN
22
wurde in 5 ml Lösung A resuspendiert. Anschließend wurden 10 ml Lösung B zugegeben, gemischt und jeweils 10 min auf Eis inkubiert. Nach Zugabe von Lösung
C wurde die Suspension etwa 15x invertiert, um das Präzipitat aufzubrechen,
bevor erneut 10 min auf Eis inkubiert wurde. Nach Abzentrifugieren des flockigen
Präzipitates (20 min, 5000 rpm, 4°C) wurde der Überstand durch 2 Lagen Mullbinden in ein Falkon überführt und die Plasmid-DNA durch Zugabe von 15 ml Isopropanol zum Überstand gefällt und anschließend sedimentiert (5000 rpm, 20 min,
4°C). Das DNA-haltige Pellet wurde in 2 ml H2O gelöst. Zur selektiven und quantitativen Abreicherung der noch zu einem hohen Anteil enthaltenen RNA, wurden 2
ml LiCl-Lösung (5 M LiCl, 50 mM Tris-HCl, pH 7,5) zugegeben. Nach 15-minütiger
Inkubation auf Eis konnte die präzipitierte RNA durch Zentrifugation sedimentiert
werden (5000 rpm, 4°C, 15 min). Der Überstand wurde einer Ethanolfällung unterzogen. Das nach erneuter Zentrifugation erhaltene Pellet wurde in 400 µl TEPuffer gelöst. Zur Entfernung jeglicher RNA-Reste wurde der Ansatz 30 min mit 5
µl RNase A (20 mg/ml) bei 37°C inkubiert. Nach zweimaliger Phenolisierung (siehe 6.7) wurde die Plasmid-DNA mit Ethanol gefällt (siehe 6.7) und in 200 µl TEPuffer aufgenommen.
6.4
Isolierung von Plasmid-DNA aus C. glutamicum
Die Plasmid-Präparation aus C. glutamicum erfolgte in Anlehnung an die Methode,
die für E. coli verwendet wurde (Eikmanns et al., 1994). Es wurden 5 ml einer 2x
TY-ÜNK zentrifugiert (5000 rpm, 5 min, RT), mit 1 ml TE-Puffer gewaschen und
das Zellpellet nach erneuter Zentrifugation in 200 µl Lösung A mit 15 mg Lysozym/ml suspendiert. Der Zellwandabbau durch das Lysozym erfolgte durch Inkubation für 2-3 h bei 37°C. Anschließend wurden 400 µl Lösung B zugegeben.
Nach 5-minütiger Inkubation auf Eis erfolgte die Zugabe von 350 µl eiskalter Lösung C. Nach weiterer 10-minütiger Inkubation auf Eis wurde das Präzipitat durch
Zentrifugation (13000 rpm, 10 min, 4°C) sedimentiert und der Überstand durch
Zugabe von 0,8 Vol Isopropanol (siehe 6.7) in einem frischen Reaktionsgefäß gefällt. Anschließend wurde die Plasmid-DNA abzentrifugiert (13000 rpm, 15 min,
RT) und nach Waschen mit 70 % (v/v) Ethanol in 30 µl TE-Puffer gelöst. Auch hier
wurde optional eine Phenolbehandlung angeschlossen, um eine höhere Reinheit
zu erreichen.
23
MATERIAL UND METHODEN
6.5
Isolierung von chromosomaler DNA aus C. glutamicum
Chromosomale DNA wurde nach der von Eikmanns et al. (1994) beschriebenen
Methode isoliert. Ein Zellpellet aus 5 ml einer ÜNK (2x TY mit 0,5 % (w/v) Glukose) wurde einmal mit TE-Puffer gewaschen und in 1 ml TE-Puffer mit 15 mg Lysozym/ml suspendiert. Nach 3-stündiger Inkubation bei 37°C wurden 3 ml LysisPuffer, 220 µl SDS und 150 µl Proteinase K-Lösung (20 mg/ml) zugegeben und
ÜN bei 37°C inkubiert. Durch anschließende Zugabe von gesättigter NaCl-Lösung
wurden selektiv Proteine präzipitiert. Nach Zentrifugation (20 min, 5000 rpm, RT)
wurde die chromosomale DNA mit kaltem, absolutem Ethanol aus dem Überstand
gefällt (siehe 6.7), mit einer Glasöse gefischt und zum Waschen in 70 %-iges
Ethanol überführt. Nach Lufttrocknung wurde die DNA in 100-200 µl TE-Puffer über Nacht bei 4°C gelöst.
Lysis-Puffer:
10 mM Tris-HCl, pH 8,2
400 mM NaCl
2 mM EDTA
6.6
Isolierung von DNA-Fragmenten aus Agarosegelen
Zur Isolierung von DNA-Fragmenten aus Agarosegelen wurden die gewünschten
DNA-Banden mit einem sauberen Skalpell ausgeschnitten. Die Extraktion der
DNA aus der Agarose erfolgte mit Hilfe des Nucleospin Extract Kit (MachereyNagel GmbH, Düren). Das Verfahren beinhaltet das Aufschmelzen der Agarose
und das Freisetzen der DNA in Gegenwart von Natrium-Jodid. Anschließend erfolgt die Aufreinigung über eine Anionenaustauscher-Säule. Die DNA wurde mit 10
mM Tris (pH 8,0) von der Säule eluiert und konnte direkt weiterverwendet werden.
6.7
Reinigung und Konzentrierung von Nukleinsäuren
Phenolisierung
Die Abtrennung von Proteinen aus DNA-Lösungen erfolgte durch Extraktion mit 1
Vol Phenol-Chloroform (PC) (v/v) (Sambrook et al., 2001).
Das Volumen der
Nukleinsäurelösung wurde mit TE-Puffer (10 mM Tris-HCl, pH 7,6, 1 mM EDTA)
MATERIAL UND METHODEN
24
auf ein Volumen von 400 µl eingestellt und mit 1 Vol PC für 30 s gründlich durchmischt. Nach erfolgter Phasentrennung (Zentrifugation bei 13000 rpm, 5 min, RT)
wurde die obere wässrige Phase in ein neues Reaktionsgefäß überführt. Die Phenolbehandlung wurde so oft wiederholt, bis keine Protein-Interphase mehr erkennbar war. Phenolreste wurden im Anschluß durch einmaliges Ausschütteln mit 1 Vol
Chloroform-Isoamylalkohol (24:1 (v/v)) und erneuter Phasentrennung durch Zentrifugation entfernt. Der DNA-haltige Überstand wurde mit Ethanol gefällt (siehe unten).
Ethanolfällung
Die DNA wurde zu diesem Zweck mit 0,1 Vol 3 M Natriumacetatlösung (pH 5,2)
und 2,5 Vol eiskaltem Ethanol (96% (v/v)) versetzt. Nach Inkubation für 1 h bei
–20°C wurde die präzipitierte DNA zentrifugiert (13000 rpm, 10 min, 4°C) und das
Sediment einmal mit kaltem 70 %-igem Ethanol (v/v) gewaschen. Das Sediment
wurde anschließend luftgetrocknet und danach in einem geeigneten Volumen TEPuffer aufgenommen.
Isopropanolfällung
Die DNA-haltige Lösung wurde mit 0,8 Vol Isopropanol versetzt und sofort zentrifugiert (13000 rpm, 30 min, RT). Anschließend wurde analog zur Ethanolfällung
verfahren.
Butanolfällung (Thomas, 1994)
Im Gegensatz zu Ethanol oder Isopropanol, hat Butanol nicht die Eigenschaft,
Salze zu präzipitieren. Diese Eigenschaft macht man sich zum schnellen und
effektiven Entsalzen von DNA-Lösungen zunutze. Die DNA-Lösung wurde mit 10
Vol 1-Butanol versetzt und für 30 s kräftig durchmischt. Anschließend wurde die
präzipitierte DNA sofort durch Zentrifugation (13000 rpm, 30 min, RT) sedimentiert. Nach einmaligem Waschen mit 70 %-igem Ethanol wurde das DNA-Pellet in
H2O aufgenommen.
Mikrodialyse von DNA
DNA-Lösungen wurden alternativ zur Butanolfällung durch Mikrodialyse gegen
H2O auf sterilen Membranfiltern entsalzt (Marusyk und Sergeant, 1980). Dabei
MATERIAL UND METHODEN
25
wurde die DNA-Lösung auf einen Membranfilter ('VS' Membranfilter, Porengröße
0,025 µm, Millipore GmbH, Eschborn) pipettiert, der auf sterilem H2O schwamm.
Nach 30 min war die Dialyse abgeschlossen und die entsalzte DNA-Lösung wurde
vom Filter in ein Reaktionsgefäß überführt.
Reinigung und Konzentrierung von DNA mittels Mikrokonzentratoren
Kleine Volumina (bis 0,5 ml) von DNA-Lösungen wurden je nach zu reinigender
Fragmentgröße über Microcon-100 oder Microcon-30 Säulchen (Amicon GmbH,
Witten) entsalzt und konzentriert. Diese Methode beruht auf dem Prinzip der Ultrafiltration und erlaubt außerdem eine Abreicherung kleiner, unerwünschter DNAFragmente bis zu einer Größe von 50-100 bp. Die DNA-Lösung wurde dazu auf
die Membran des Konzentrators gegeben und dieser solange zentrifugiert (2500
rpm, RT), bis das Volumen der Lösung auf das gewünschte Maß reduziert war.
Anschließend wurden ca. 500 µl H2O auf die Membran gegeben und erneut zentrifugiert. Durch Wiederholung dieser Schritte konnte eine bessere Entsalzung der
Lösung erreicht werden. Die entsalzte und konzentrierte DNA-Lösung wurde durch
Umdrehen des Konzentrators und Zentrifugation (5000 rpm, 5 min) in einem frischen Reaktionsgefäß gesammelt.
Konzentrations- und Reinheitsbestimmung von Nukleinsäure-Lösungen
Die Konzentrationsbestimmung von Nukleinsäuren in wässrigen Lösungen erfolgte
photometrisch in Quarzküvetten bei einer Wellenlänge von 260 nm. Eine Absorption von 1,0 entsprach dabei 50 µg doppelsträngiger DNA/ml bzw. 40 µg einzelsträngiger RNA. Die Reinheit wurde aus dem Quotienten aus A260 und A280 ermittelt. Reine Nukleinsäure-Lösungen erreichen einen A260/A280 -Quotienten von
1,8 – 2,0.
7.
Amplifikation, Rekombination und Analyse von DNA
7.1
Polymerase-Kettenreaktion (PCR)
Die PCR wurde von Kary B. Mullis entwickelt (Saiki et al., 1985) und von Saiki
(1988) durch Einführen einer hitzestabilen DNA-Polymerase aus Thermus aquaticus (Taq-Polymerase) verbessert. Bei der PCR handelt es sich um eine sehr wir-
26
MATERIAL UND METHODEN
kungsvolle Methode zur Vervielfältigung kurzer Genomabschnitte. Dazu ist es nötig, daß die Sequenz der flankierenden Bereiche bekannt ist. In diesem Fall können zwei Oligonukleotid-´Primer´ synthetisiert werden, die jeweils komplementär
zu einem der beiden 3´-Einzelstrangenden des zu amplifizierenden DNADoppelstranges sind. Im ersten Schritt wird die DNA denaturiert. Im zweiten Schritt
können die ´Primer´ bei einer definierten Annealing-Temperatur an die einzelsträngige DNA entsprechend ihrer Sequenzhomologie hybridisieren. An diesen
kurzen, doppelsträngigen Abschnitten kann die DNA-Polymerase im dritten Schritt
ansetzen, und mit den im Ansatz enthaltenden vier Desoxynucleosidtriphosphaten
die ´Primer´ in 3´-Richtung verlängern. Anschließend wird der Zyklus wiederholt.
Ist n die Anzahl der Zyklen, so können 2n Kopien amplifiziert werden.
Die von MWG oder Biomers bezogenen ´Primer´ wurden in 10 mM Tris-HCl (pH
7,6) gelöst und besaßen eine Endkonzentration von 100 pmol/µl.
Der PCR-Reaktionsansatz enthielt folgende Komponenten:
10x Reaktionspuffer
:
5 µl
DNA
:
ca. 100 ng
25 mM MgCl2
:
2,5 µl
dNTP-Mix (jeweils 2 mM) :
5 µl
10 pmol ´Primer´ A
:
1 µl
10 pmol ´Primer´ B
:
1 µl
Taq-Polymerase (1 U/ µl) :
1 µl
H2O
ad 50 µl
:
Das Standard-PCR- Protokoll ist nachfolgend aufgeführt:
Vorabdenaturierung:
94°C; 5 min
Denaturierung
:
94°C; 30 sec
Annealing
:
55°C; 30 sec
Elongation
:
72°C; 1 min/ 1000 bp
End-Elongation
:
72°C; 10 min
32x
MATERIAL UND METHODEN
27
Nach Ablauf des Programms wurde ein Aliquot jedes Ansatzes auf einem 0,8 %
(w/v) Agarosegel kontrolliert.
7.2
Restriktion von DNA
Plasmid-DNA bzw. chromosomale DNA oder PCR-Produkte wurden mit Restriktionsenzymen nach Angaben des Herstellers verdaut. Analytische Restriktionsverdaus erfolgten im 20 µl–Maßstab, präparative wurden in 50–100 µl Volumina
durchgeführt. Die Restriktionsenzyme sowie die dazugehörigen Puffer wurden von
MBI-Fermentas (St. Leon-Rot) oder New England Biolabs (Schalbach) bezogen.
Wenn möglich, wurden die Ansätze im universalen TangoY-Puffer-System durchgeführt. Die meisten Enzyme sind in diesem Puffer aktiv, was den parallelen Verdau der DNA mit zwei Enzymen ermöglicht. Die Restriktionsenzyme wurden anschließend, wenn möglich, durch Hitze inaktiviert (65–80°C, 10 min). War eines
der eingesetzten Enzyme hitzestabil, erfolgte eine Reinigung mittels MicroconSäulchen oder Nucleospin laut Angaben des Herstellers.
7.3
Dephosphorylierung von DNA-Fragmenten
Um die unerwünschte Religation von geschnittenen Plasmiden zu vermeiden, erfolgte deren Dephosphorylierung mittels 'shrimp alkaline phosphatase' (SAP).
Nach SAP-Behandlung (1-2 U/Reaktion) im 30–50 µl Maßstab (37°C, 30 min)
wurde das Enzym durch Inkubation bei 80°C für 1 h inaktiviert. Die Reinigung des
Ansatzes erfolgte wiederum über Microcon-Säulchen oder Nucleospin.
7.4
Ligation von DNA-Fragmenten
Die Ligation von DNA-Fragmenten erfolgte in 20 µl-Reaktionsansätzen. Es wurden
die dephosphorylierte Plasmid-DNA und das zu klonierende DNA-Fragment ('insert') in einem molaren Verhältnis von ca. 1:5 gemischt und mit 2 µl 10x Ligationspuffer und 1 µl T4-DNA-Ligase versetzt. Nach Inkubation bei 16°C ÜN mit anschließendem Hitzeschritt zum Abstoppen der Reaktion (65°C, 5 min) oder 1,5 h
bei 22°C (mit anschließendem Hitzeschritt für 10 min bei 65°C) wurde der Ligati-
28
MATERIAL UND METHODEN
onsansatz ohne weitere Behandlung in kaltkompetente E. coli–Zellen transformiert.
7.5
Agarose-Gelelektrophorese
Mit Agarosegelen ist es durch Anlegen eines elektrischen Feldes möglich, DNA
nach ihrer Größe aufzutrennen. Die Phosphatgruppen der DNA-Moleküle sind bei
den verwendeten pH-Werten negativ geladen, wodurch die DNA zur Anode wandert. Eine elektrisch neutrale Gelmatrix (Agarose) fungiert hierbei als Sieb, so daß
kleinere DNA-Fragmente schneller wandern als große (Sambrook et al., 2001).
In der Regel wurden 0,8 % (w/v) Agarosegele verwendet. Dazu wurde die Agarose in 1x TAE-Puffer eingewogen und anschließend in der Mikrowelle aufgekocht.
Das Gel wurde nach Abkühlen auf ca. 60°C in die Gelgießvorrichtung gegossen.
Als Laufpuffer wurde ebenfalls 1x TAE-Puffer verwendet. Die Proben wurden mit
1/5 Volumen Ladepuffer versetzt und in die Geltaschen pipettiert. Zur Größenbestimmung wurde eine 1 kb-DNA-Leiter mit aufgetragen. Der Gellauf erfolgte bei
90 V. Anschließend wurde das Agarosegel für 5-30 min in einer EthidiumbromidLösung (1µg/ml) gefärbt und danach in Wasser entfärbt. Die DNA-Banden wurden
dann durch UV-Licht (λ= 312 nm), bei dem das an die DNA angelagerte Ethidiumbromid fluoresziert, sichtbar gemacht und mit einer Fotodokumentationsanlage
fotografiert.
Die hierfür benötigten Puffer sind im folgenden aufgeführt.
50x TAE-Puffer
:
2 M Tris- HCl (pH 8,0)
50 mM EDTA
500 mM Natriumacetat
Ladepuffer
:
0,25 % (w/v) Bromphenolblau
40 % (v/v) Glycerol
MATERIAL UND METHODEN
7.6
29
Sequenzierung
Sequenzierungen von klonierten DNA-Fragmenten wurden von der Firma MWGBiotech GmbH (Ebersberg) durchgeführt. Die notwendige hohe Reinheit der zu
sequenzierenden Proben wurde durch Plasmidpräparation mit dem 'GFX Micro
Plasmid Prep Kit' (Amersham Pharmacia Biotech GmbH, Freiburg), einem Säulenbasierten Anionentauschersystem, erreicht.
7.7
'Southern-Blot'-Hybridisierung (nach Southern, 1975, modifiziert)
Zur Analyse genomischer DNA durch 'Southern-Blot'-Hybridisierung wurde die zu
untersuchende DNA mit geeigneten Restriktionsenzymen verdaut und anschließend gelelektrophoretisch aufgetrennt. Die aufgetrennten DNA-Fragmente wurden
im Gel zunächst denaturiert und anschließend auf eine Nylonmembran übertragen. Digoxigenin-(DIG)-markierte DNA-Sonden wurden mit der auf eine Membran
übertragenen DNA hybridisiert. Die Detektion der hybridisierten Sonden erfolgte
durch Chemilumineszenz.
Digoxigenin-Markierung von DNA-Sonden
Die Markierung der Sonden erfolgte durch Einbau von DIG-dUTPs (Roche Molecular Biochemicals, Mannheim) in einer Standard-PCR. Zu 50 µl PCR-Ansatz
wurde zusätzlich zu den dNTPs 0,5 µl der DIG-dUTP-Lösung hinzugegeben. Mit
Hilfe der sondenspezifischen 'Primer' wurde so das Sondenfragment amplifiziert
und gleichzeitig markiert. Ob der Einbau erfolgreich war, konnte durch Vergleich
der markierten Sonde mit dem jeweiligen unmarkierten Fragment auf einem Agarosegel überprüft werden. DIG-markierte DNA wandert während der Gelelektrophorese immer etwas langsamer und damit weniger weit, als unmarkierte
DNA gleicher Länge.
Auftrennung der DNA und Übertragung auf eine Nylonmembran
Die chromosomale DNA wurde nach einem Restriktionsverdau in einem Agarosegel bei 20 V für 16 h elektrophoretisch aufgetrennt und im Anschluß durch vakuumunterstützte Diffusion mittels eines Vacugene®XL Vakuum Blotting Systems
(Amersham Pharmacia Biotech GmbH, Freiburg) nach einer Methode von Gross
et al. (1988) auf eine positiv geladene Membran (positive Membran Quantum,
30
MATERIAL UND METHODEN
Oncor/Appligene, Heidelberg-Wieblingen) übertragen. Dazu wurde das Gel zuerst
für jeweils 20 min mit Depurinierungslösung, Denaturierungslösung und Neutralisierungslösung und anschließend für 60 min mit Transferlösung überschichtet,
wobei alle 10-15 min die Lösung erneuert wurde. Die Membran wurde kurz zwischen Filterpapier getrocknet und die DNA in einem Stratagene UV Crosslinker
(Stratagene GmbH, Heidelberg) durch UV-Bestrahlung auf der Membran fixiert.
Zur Aufbewahrung der Membran wurde der Blot bei 80°C für etwa 1 h gebacken.
Depurinierungslösung:
0,25 M NaOH
Denaturierungslösung:
1 M NaCl
0,5 M NaOH
Neutralisierungslösung:
1 M Tris-HCl, pH 5,0
2 M NaCl
Transferlösung (20x SSC):
0,3 M Na-Citrat, pH 7,0
3 M NaCl
Hybridisierung der DNA mit einer Digoxigenin-markierten DNA-Sonde
Die Membran wurde zunächst 1 h bei 65°C in 20 ml Prähybridisierungslösung (6x
SSC, 5x Denhardt-Lösung, 0,5 % (w/v) SDS: lösen bei 55°C, dann Zugabe von
400 µl frisch denaturierter Heringssperma-DNA (50 µg/ml)) inkubiert. Die Hybridisierung erfolgte nach Zugabe von 500 ng frisch denaturierter DNA-Sonde im
Hybridisierungsofen ÜN bei 65°C.
Prähybridisierungslösung:
6 x SSC
5 x Denhardt-Lösung
0,5 % (w/v) SDS
50 x Denhardt-Lösung: 1 % (w/v) Ficoll
1 % Polyvinylpyrolidon (w/v)
MATERIAL UND METHODEN
31
1 % (w/v) BSA Fraktion V
⇒ in H2O bei 37°C lösen
Detektion der hybridisierten Sonden
Nicht gebundene DNA-Sonden wurde durch zweimaliges 15-minütiges Waschen
bei RT in 20 ml Waschlösung 1 und anschließendes zweimaliges 20-minütiges
Waschen bei 65°C in 30 ml Waschlösung 2 entfernt. Die folgenden Schritte erfolgten bei RT. Die Membran wurde 1 min in 20 ml Waschpuffer gewaschen. Nach 30minütiger Inkubation in 20 ml Puffer II und kurzem Waschen in Waschpuffer erfolgte die Bindung des Antikörpers durch 30-minütige Inkubation der Membran in 20
ml Puffer II mit Anti-Digoxigenin-AP-Konjugat (150 mU/ml). Das nicht gebundene
Antikörper-Konjugat wurde durch zweimaliges 15-minütiges Waschen in 20 ml
Waschpuffer entfernt. Es folgten 2 min Inkubation in 20 ml Puffer III und 15 min
Inkubation in 10 ml des Chemilumineszenz-Substrates CSPD (2,5 µM in Puffer III).
Überschüssige Feuchtigkeit wurde mit Hilfe von Filterpapier von der Membran entfernt, ohne diese dabei zu trocknen. Die Membran wurde anschließend zwischen
zwei Klarsichtfolien gelegt und 15 min bei 37°C inkubiert. Die Exposition erfolgte 1
h oder länger mit HyperfilmTMECLTM-Film (Amersham Pharmacia Biotech GmbH,
Freiburg).
Waschlösung 1:
2 x SSC
0,1 % (w/v) SDS
Waschlösung 2:
0,1 x SSC
0,1 % (w/v) SDS
Waschpuffer:
Puffer I:
Puffer I mit 0,3 % (w/v) Tween 20
0,1 M Maleinsäure, pH 7,5
0,15 M NaCl
Puffer II:
1 % (w/v) Blocking-Reagenz in Puffer I
⇒ frisch ansetzen und bei 50°C lösen
MATERIAL UND METHODEN
Puffer III:
32
0,1 M Tris-HCl, pH 9,5
0,1 M NaCl
8.
Isolierung und Analyse von RNA
8.1
Allgemeine Bemerkungen zur Isolierung von RNA
Alle wäßrigen Lösungen wurden in DEPC-behandeltem H2O angesetzt. H2O wurde dafür mit 1/1000 Vol DEPC versetzt und für 2 h bei 37°C inkubiert. Anschließend wurde das DEPC durch Autoklavieren inaktiviert. Plastikgefäße und Lösungen wurden grundsätzlich zweimal autoklaviert. Glaspipetten wurden durch
Backen (160°C, 3 h) von RNasen befreit. Nicht autoklavierbare Plastikgefäße wurden durch Waschen mit 0,1 N NaOH und DEPC-behandeltem Wasser von RNasen befreit. Alle kritischen Arbeiten wurden mit Handschuhen durchgeführt. Gestopfte Pipettenspitzen halfen, RNase-Kontamination aus Pipetten zu vermeiden.
8.2
Zellernte zur RNA-Präparation
Wegen der kurzen Halbwertszeit von mRNA in der Zelle - die Halbwertszeit der
mRNA beträgt in manchen Fällen weniger als 30 s (Carpousis et al., 1999) - war
es wichtig, bei der Aufarbeitung der Zellen sehr schnell zu arbeiten. Um den
Stoffwechsel der Zellen zum Stillstand zu bringen, wurde in eine exponentiell
wachsende Kultur von C. glutamicum 1 Vol eiskalter 'Killingbuffer' (Wirkkomponente: Natriumazid) gegossen und die Zellsuspension sofort auf Eiswasser gekühlt.
Nach Zentrifugation in 50 ml Zentrifugenröhrchen (5000 g, 4°C, 10 min) und Dekantieren des Überstands wurde das Zellpellet sofort in flüssigem Stickstoff eingefroren und bis zur Aufarbeitung bei –70°C gelagert.
'Killingbuffer':
20 mM Tris-HCl, pH 8
20 mM NaN3
5 mM MgCl2
MATERIAL UND METHODEN
8.3
33
RNA-Präparation mit heißem Phenol (Schwinde et al., 1993)
1,5 ml saures Phenol (Roth A980.1), 1,5 ml Chloroform-Isoamylalkohol (24:1) und
2,5 ml AE-Puffer wurden in 15 ml Reaktionsgefäßen in einem 60°C Wasserbad
vorgeheizt. 2 ml Schraubdeckelgefäße mit ca. 250 mg 'Glasbeads' (Sigma-Aldrich
Chemie GmbH, Steinheim) wurden mit 0,5 ml saurem Phenol und 0,5 ml chaotropher Salzlösung (RLT-Puffer, RNeasy-Kit, Qiagen GmbH, Hilden) gefüllt und 5
min auf Eis gekühlt. Nach Auftauen der Zellen auf Eis wurden etwa 0,6-1 ml Zellpellet (aus ca. 120 ml Zellen einer OD600 von 3) auf 3 eisgekühlte Schraubdeckelgefäße verteilt. Nach dem Aufschluss der Zellen (3x 45 s, Stufe 6,5) im 'RiboLyser'
(siehe 9.1) wurden die Aufschlussgefäße kurz auf Eis abgekühlt und anschließend
durch Zentrifugation (13000 rpm, 4°C, 5 min) Zelltrümmer und präzipitierte Proteine sedimentiert. Der wässrige Überstand wurde sofort in die Reaktionsgefäße mit
dem auf 60°C vorgeheizten Phenol-Chloroform-Puffer-Gemisch gegeben, und diese im Wasserbad zur ständigen Phasendurchmischung für 10 min geschüttelt.
Nach erneuter Phasentrennung durch Zentrifugation (4000 g, 4°C, 5 min), wurden
die Überstände in neue Reaktionsgefäße überführt und bei RT phenolisiert. Der
Wechsel von Phenolisierung, Überführung in neue Gefäße und Phasentrennung
wurde so oft wiederholt, bis die proteinhaltige Interphase deutlich reduziert war.
Um restliches Phenol zu entfernen, wurde die Probe mit 1 Vol Chloroform/Isoamylalkohol (24:1) versetzt, geschüttelt und wiederholt zentrifugiert. Die
RNA wurde durch Zugabe von 0,1 Vol 3 M NaAc und 2,5 Volumen Ethanol zum
Überstand bei –20°C für mindestens 2 h gefällt. Die anschließend durch Zentrifugation (13000 rpm, 30 min, 4°C) sedimentierte RNA wurde einmal mit 70 %-igem
Ethanol gewaschen und dann luftgetrocknet (20 min) und in 400 µl RNase-freiem
H2O gelöst. Nach Zugabe von 1/10 Vol 10x -DNase-Puffer und 30 µl RNase-freier
DNase (150 U, MBI-Fermentas GmbH, St. Leon-Roth) wurde die DNA durch Inkubation bei 37°C für 20 min abgebaut. Der Verdauansatz wurde danach mit 2 ml
RLT-Puffer (Qiagen, Hilden) und 20 µl ß-Mercaptoethanol versetzt, gemischt und
nach Zugabe von 1,4 ml Ethanol auf die Membran eines Säulchens aus dem
RNeasy-Midi-Kit (Qiagen, Hilden) gegeben. Die weitere Aufarbeitung erfolgte nach
dem Protokoll des Herstellers. Die RNA wurde zweimalig mit je 250 µl RNasefreiem H2O durch Zentrifugation (5000 rpm, 2 min, RT) vom Säulenmaterial eluiert
und in 50 µl-Aliquots in flüssigem Stickstoff schockgefroren und bei –70°C gelagert. Die typische Ausbeute lag bei 600–800 µg RNA. Die Qualität der RNA wurde
MATERIAL UND METHODEN
34
auf einem nativen Agarosegel optisch kontrolliert. In einer RT-PCR wurde die
RNA-Lösung auf das Vorhandensein von mRNA untersucht. In einer PCR wurde
die RNA-Lösung außerdem auf eventuelle Reste an chromosomaler DNA überprüft.
Nach dieser Methode isolierte, sehr saubere RNA wurde für ´Primer Extension´
Experimente eingesetzt.
AE-Puffer: 40 mM Na-Acetat, pH 5,2
1 mM EDTA
8.4
RNA-Isolierung mit Hilfe des Rneasy-Mini-Kit (Quiagen GmbH, Hilden)
Der Rneasy-Mini-Kit eignet sich für die Isolierung von RNA-Molekülen, die größer
als 200 Nukleotide sind. Dazu wurden Zellen wie unter 8.2 beschrieben auf Komplex- und Minimalmedium mit Glukose angezogen und geerntet. Das Pellet wurde
auf Eis aufgetaut, in 350 µl RLT-Puffer resuspendiert und anschließend mittels
RiboLyser (siehe 9.1) (1x 20s, Stufe 6,5; 1x 35s, Stufe 6,5) aufgeschlossen. Das
weitere Vorgehen entsprach den Herstellerangaben.
Die nach dieser Methode isolierte RNA wurde für 'Northern-Blot'-Experimente
eingesetzt.
8.5
Radioaktive 'Northern-Blot'-Hybridisierungen
Transfer von RNA
Um RNA durch 'Northern-Blot'-Hybridisierung zu analysieren wurde diese zunächst nach einer Methode von Alwine et al. (1977) unter denaturierenden Bedingungen in einem Agarosegel aufgetrennt. Dazu wurde ein Formaldehyd-Gel verwendet (20 ml 5x MOPS-Puffer, 63 ml H2O, 0,9 g Agarose: diese Komponenten
wurden aufgekocht und anschließend 17 ml Formaldehyd zugesetzt), das nach
dem Gießen in eine mit 0,1 % SDS gereinigte Kammer für 1-2 h unter dem Abzug
zum Ausdampfen lagerte. Als Laufpuffer diente MOPS. Nach dem Auftragen der
denaturierten Proben lief das Gel für etwa 4 h bei 70-90 V. Die RNA wurde anschließend durch vakuumunterstützte Diffusion mittels eines Vacugene®XL Vaku-
MATERIAL UND METHODEN
35
um Blotting Systems (Amersham Pharmacia Biotech GmbH, Freiburg) nach einer
Methode von Gross et al. (1988) auf eine positiv geladene Membran (positive
Membran Quantum, Oncor/Appligene, Heidelberg-Wieblingen) übertragen. Dazu
wurde das Gel für 2 h mit Transferpuffer überschichtet, wobei die Lösung alle 15
min gewechselt wurde. Die Membran wurde nach dem Transfer zwischen Whatman-Papier getrocknet, im UV-Crosslinker fixiert und für 1 h bei 80°C gebacken.
Transferpuffer:
5x SSC
10 mM NaOH
pH 11,5
5x MOPS:
0,2 M MOPS
0,05 M Na-Acetat
0,01 M EDTA
⇒ Puffer mit autoklaviertem H2O ansetzen und mit NaOH auf
pH 7,0 einstellen
5x RNA-Auftragspuffer: 20 ml Glycerin
400 µl 1 M EDTA
3,8 ml Formamid
1 ml Ethidiumbromid (10 mg/ml)
10x MAE-Puffer
7,2 ml Formaldehydlösung 37%
0,5 % Bromphenolblau
⇒ ad 100 ml H2O
MAE-Puffer:
200 mM Mops
50 mM Na-Acetat
5 mM EDTA
Radioaktive Markierung von Sonden
Die Markierung von DNA-Fragmenten erfolgte nach dem „Random ´Primer´ labeling“ (Feinberg und Vogelstein, 1983) mit dem „Random ´Primers´ DNA Labeling
36
MATERIAL UND METHODEN
System-Kit“ (Invitrogen GmbH, Karlsruhe) nach Herstellerangaben. In der Regel
wurden 25 ng DNA zur Markierung mit [α-32P]dATP eingesetzt. Nicht eingebaute
Nukleotide wurden durch Reinigung über „MicroSpinTM G-25 Säulchen“ (Amersham Pharmacia Biotech GmbH, Freiburg) nach Herstellerangaben entfernt. Für
die Hybridisierung wurden 25 µl markierte Sonde pro Blot eingesetzt.
Hybridisierung mit radioaktiven DNA-Sonden
Auf Nylonmembran fixierte RNA wurde nach einer modifizierten Methode von Du
Pont de Nemours Deutschland GmbH, Dreieich, hybridisiert. Der Blot wurde hierfür angefeuchtet und in ein Hybridisierungsröhrchen überführt. Die Prähybridisierung erfolgte bei 60°C für 2 h. Danach wurde die denaturierte Sonde zugesetzt
und ÜN hybridisiert. Die Membran wurde anschließend in Waschlösung 1 und 2
(siehe 7.7) je 2x für 30 min bei 60°C gewaschen und in Frischhaltefolie eingewickelt. Je nach Stärke des Signals wurde der Blot für 1 h bis mehrere Tage an einer
„BAS-MP Phosphor Imaging“-Platte (Fuji Photo Film Europe GmbH, Düsseldorf)
autoradiographiert. Mit Hilfe des „Bio Imager Fujix BAS 1000“ und dem Programm
„Mac Bas 1000 Image Reader“ wurde das Signal digitalisiert und mit dem „Mac
Bas V 2.31“-Programm ausgewertet.
5x Hybridisierungspuffer:
1% (w/v) Rinderserumalbumin
1% (w/v) Polyvinylpyrolidon (K30)
1% (w/v) Ficoll 400
250 mM Tris-HCl, pH 7,5
0,5% (w/v) Na-Pyrophosphat
⇒ ad 100 ml H2O, pH 7,5; der Puffer wurde
sterilfiltriert und in Aliquots eingefroren
Hybridisierungspuffer:
(für 1 Membran)
4 ml 5x Puffer
12 ml 16,6% Dextransulfat
2 ml 10% SDS
⇒ nach Mischen der Komponenten 15 min auf
65°C erwärmen und anschließend 1,16 g NaCl
zugeben
MATERIAL UND METHODEN
8.6
37
´Primer Extensio´-Analyse
Die Bestimmung der 5´-Enden von mRNA erfolgte nach der Methode der ´Primer
Extension´ basierend auf der Methode von Kellmann et al. (1990). Nach Anlagerung eines spezifischen IRD800 markierten ´Primers´ an der mRNA wurde mit
dem Enzym Reverse Transkriptase eine zu der mRNA komplementäre cDNA hergestellt. Über die Länge der cDNA war es möglich, Rückschlüsse auf das 5´-Ende
der mRNA zu ziehen.
Dazu wurden etwa 100 µg RNA mit Ethanol gefällt (siehe 6.7), an der Luft getrocknet, in 100 µl HP-Puffer resuspendiert und 5 pmol IRD-markierter ´Primer´
zugesetzt. Nach einem Denaturierungschritt für 10 min bei 95°C erfolgte die Primeranlagerung ÜN. Nach Zugabe von 300 µl H2O wurde der Ansatz ethanolgefällt
(siehe 6.7) und mit 70% Ethanol gewaschen. Nach dem Trocknen des Pellets
wurde dieses in 20 µl RTB-Puffer resuspendiert, in ein PCR-Tube überführt und
für 2 min bei 37°C inkubiert. Nach Zugabe von 1 µl Mul-V-Reverse Transkriptase
(MBI Fermentas, St. Leon-Rot) erfolgte die Umschreibung in cDNA für 2 h bei
37°C. Die Reaktion wurde mit 1 µl 0,5 M EDTA gestoppt. Nach einem RNaseVerdau für 30 min bei 37°C wurde der Ansatz mit 150 µl TES-Puffer versetzt und
erneut ethanolgefällt (siehe 6.7). Das Pellet wurde in 3 µl TE-Puffer resuspendiert
und mit 3 µl Stop-Puffer aus dem „SequiTherm EXCELIITM Long-ReadTM Sequencing Kit-LC“ (Epicentre Technologies Corp., Madison, USA) versetzt. Die Analyse
erfolgte auf einem Sequenziergel.
8.7
LI-COR-Analyse
Zur Analyse von ´Primer Extension´-Produkten mit gleichzeitiger Sequenzierung
des entsprechenden Bereichs wurde ein LI-COR 4000L (MWG Biotech AG, Ebersberg) verwendet. Die Sequenzierreaktion wurde mit dem „SequiTherm
EXCELIITM Long-ReadTM Sequencing Kit (Biozym Diagnostik GmbH, Hess, Oldendorf) nach Herstellerangaben durchgeführt. Zur Visualisierung im automatischen
Sequenzierer LI-COR wurden für die Reaktion IRD800 markierte ´Primer´ eingesetzt. Die fertige Sequenzierreaktion konnte dann lichtgeschützt bei –20°C gelagert werden. 1 µl der Reaktion wurde nach 5-minütiger Denaturierung bei 95°C
auf ein 6%-iges (w/v) Polyacrylamidgel (41 cm x 0,25 mm) aufgetragen und ÜN
bei 1500 V, 37 mA, 50 W und 50°C aufgetrennt.
MATERIAL UND METHODEN
38
Polyacrylamidgel: 30 ml SequaGel XR*
7,5 ml SequaGel XR-Pufferlösung*
400 µl DMSO
300 µl APS (10% (w/v) in H2O)
(* National Diagnostic, Hall, England)
9.
Bestimmung von Enzymaktivitäten
9.1
ZellAufschluss mit dem RiboLyser
Zellaufschlüsse von C. glutamicum wurden in der Regel mit dem RiboLyser
(Hybaid GmbH, Heidelberg) durchgeführt. Die Zellen wurden hierbei durch die auf
sie einwirkenden, starken mechanischen Kräfte aufgebrochen.
Zellen aus Kulturen von 50 ml wurden in einem geeigneten Volumen des gewünschten Aufschlusspuffers suspendiert. Die Zellsuspensionen wurden in
Schraubdeckelgefäße, die mit je 250 mg 'Glasbeads' (150–212 µm, Sigma-Aldrich
Chemie GmbH, Steinheim) befüllt waren, zu je 800 µl verteilt. Der Zellaufschluss
erfolgte im RiboLyser bei höchster Stufe (6,5) 4x für jeweils 25 s. Zwischen den
Aufschlüssen wurden die Proben für je 5 min auf Eis gekühlt. Nach dem Aufschluss wurden die 'Glasbeads' und die Zelltrümmer durch Zentrifugation in einer
Mikrozentrifuge (13000 rpm, 4°C, 30 min) sedimentiert.
9.2
Zellaufschluss mittels Universalmischgerät
Alternativ zum Zellaufschluss mit dem RiboLyser wurde ein Universalmischgerät
(Silamat Plus, Vivadent Dental, Ellwangen) benutzt. Dieser ermöglichte einen sehr
guten Zellaufschluss, war jedoch aufgrund der großen Hitzeentwicklung nur für
eine geringe Anzahl an Proben geeignet. Dazu wurden 500 mg ´Glasbeads´ in
Schraubdeckelgefäße gefüllt und 750 µl Zellsuspension dazugegeben. Der Aufschluss erfolgt 3x für 30s, wobei zwischendurch für 10 min auf Eis gekühlt wurde.
Zelltrümmer wurden wiederum abzentrifugiert für 3 min bei 13000 rpm und 4°C.
MATERIAL UND METHODEN
9.3
39
Proteinbestimmung nach Bradford (1976)
Mit der Proteinbestimmung nach Bradford (1976) können Proteinkonzentrationen
quantitativ bei einer Wellenlänge von 595 nm photometrisch bestimmt werden.
Das Funktionsprinzip beruht auf einer Verschiebung des Absorptionsmaximums
des Farbstoffes Coomassie Brilliant Blue 6250, der hauptsächlich mit Argininresten reagiert, von 465 nm auf 595 nm. Durch die Zugabe von Proteinen werden
Aminosäuren an Coomassie Brilliant Blue 6250 gebunden. Dadurch wird die anionische Form des Farbstoffes stabilisiert und eine Verschiebung des Absorptionsmaximums hervorgerufen.
Vor der eigentlichen Proteinbestimmung ist es nötig, eine Kalibriergerade mit BSA
(Bovine Serum Albumine in einer Stammlösung von 1 mg/ml) in einem Konzentrationsbereich von
0-20 µg/ml zu erstellen. Dazu wurden 800 µl BSA-Probe in der
entsprechenden Konzentration mit 200 µl Bradford-Reagenz (0,1 % (w/v) Coomassie Brilliant Blue 6250, 4,8 % (v/v) Ethanol (96 %), 8,6 % (v/v) H3PO4 (86 %))
versehen, gemischt und für 20 min bei RT inkubiert. Anschließend erfolgte die Extinktionsmessung bei einer Wellenlänge von 595 nm. Die Messungen der Proteingehalte der Rohextrakte verliefen ebenfalls nach diesem Prinzip. Der Gehalt der
jeweiligen Proben kann anschließend anhand der Kalibriergeraden ermittelt werden.
9.4
Berechnung von spezifischen Enzymaktivitäten
Die Berechnung spezifischer Enzymaktivitäten erfolgte mit nachfolgender Formel:
spezifische Aktivität [U / mg ] =
∆E / min * Gesamtvolumen
ε * d * Probevolumen * mg Protein
∆E/min
= Extinktionsänderung pro Minute [min-1]
ε
= Extinktionskoeffizient [mmol-1*cm-1]
d
= Schichtdicke der Küvette [cm]
40
MATERIAL UND METHODEN
9.5
Bestimmung
der
Pyruvat-Dehydrogenase-Komplex-
und
2-
Oxoglutarat-Dehydrogenase-Komplex-Aktivität
Der Pyruvat-Dehydrogenase-Komplex (PDHC) katalysiert die oxidative Decarboxylierung von Pyruvat zu Acetyl-CoA unter Bildung von NADH2. Die 2-OxoglutaratDehydrogenase (OGDHC) katalysiert die oxidative Decarboxylierung von 2Oxoglutarat zu Succinyl-CoA, wobei ebenfalls NADH2 entsteht. Die verwendete
Methode zur Bestimmung beider Enzymaktivitäten erfolgte nach Guest und
Creaghan (1972). Der Testansatz enthielt in einem Volumen von 1 ml 100 mM
Tris-HCl, pH 7,2, 1 mM Thiaminpyrophosphat (TPP), 5 mM Pyruvat (1,5 mM 2Oxoglutarat), 3 mM L-Cystein, 10 mM MgCl2, 2 mM NAD und 50 µl Zellextrakt.
Nach Verfolgung des Vorlaufs wurde die Reaktion mit 25 µl einer 8 mM CoALösung gestartet und die Reaktion für 2 min bei 30°C und 340 nm verfolgt (NADHBildung). Der
millimolare Extinktionskoeffizient von NADH beträgt
6,22
mmol-1*cm-1.
Aufschlusspuffer: 100 mM Tris-HCl, pH 7,2
10 mM MgCl2
3 mM L-Cystein
9.6
Bestimmung der Pyruvat-Decarboxylase-Aktivität (E1) (Schwartz und
Reed, 1970)
Die E1-Untereinheit des PDHC katalysiert die Decarboxylierung des Pyruvats unter Entstehung von Hydroxyethyl-TPP:
Pyruvat + TPP
Hydroxyethyl-TPP + CO2
Die Messung der Untereinheit in dieser Form im Komplex ist nicht möglich. Die
Messung der Aktivität erfolgt mit ungebunden vorliegendem Enzym, das dieselbe
Reaktion wie die Pyruvat-Chinon-Oxidoreduktase katalysiert:
Pyruvat + FAD
Acetat + CO2 + FADH2
41
MATERIAL UND METHODEN
Die Aktivitätsbestimmung erfolgt enzymatisch durch Messung der Reduktion des
künstlichen Elektronenakzeptors Kaliumhexacyanoferrat bei 430 nm. Zellen wurden in der logarithmischen Wachstumsphase (OD600=6) geerntet, zweimal mit 50
mM Tris-HCl, pH 7,5 mit 10 % Glycerin gewaschen und in 800 µl desselben Puffers wieder aufgenommen. Der Aufschluss erfolgte im Silamat S5. Der Test enthielt in einem Testansatz von 1 ml 50 mM Tris-HCl (pH 7,5), 10 mM MgCl2, 0,2
mM TPP und 50 µl Rohextrakt. Die Reaktion wurde mit 5 mM Pyruvat gestartet.
Der Test wurde bei 30°C durchgeführt.
9.7
Bestimmung
der
Dihydrolipoamid-Transacetylase-Aktivität
(E2)
(Schwartz und Reed, 1969)
Die Dihydrolipoamid-Transacetylase (DHL) katalysiert die zweite Reaktion des
PDHC. Die Reaktion ist im Folgenden aufgeführt:
Hydroxyethyl-TPP + Lipoamid
6-S-Acetyldihydrolipoamid
+ TPP
S-Acetyldihydrolipoamid + CoA
Acetyl-CoA + Dihydrolipoamid
Der Nachweis der DHL-Aktivität erfolgt enzymatisch nach folgendem Prinzip in
unphysiologischer Richtung:
PTA
Acetylphosphat + CoA
Acetyl-CoA + Pi
Acetyl-CoA + Dihydrolipoamid
6-S-Acetyldihydrolipoamid + CoA
Zur Messung der DHL wurden Zellen aus der exponentiellen Wachstumsphase
verwendet. Als Aufschlusspuffer diente 100 mM Tris-HCl, pH 7,5 (UV-rein) mit
10 % Glycerin. 1 ml Testansatz enthielt 100 mM Tris-HCl, pH 7,5, 10 mM Acetylphosphat, 0,1 mM Li-CoA, 50 µl Dihydrolipoamid (Stammlösung 25 mg/ml Ethanol) und 2 U PTA. Nach Messung des Blindwertes bei 240 nm und 30°C wurde die
MATERIAL UND METHODEN
42
Reaktion mit 50 µl sehr gut verdünntem (1:10) Rohextrakt gestartet und die Reaktion über 2 min verfolgt. Hierbei wird das gebildete S-Acetyldihydrolipoamid gemessen (millimolarer Extinktionskoeffizient 4,4 [mmol-1*cm-1]).
9.8
Bestimmung der Dihydrolipoamid-Dehydrogenase-Aktivität
Die Dihydrolipoamid-Dehydrogenase (E3) katalysiert in den Enzymkomplexen
PDH und OGDH die Oxidation des Dihydrolipoamids unter Bildung von NADH2.
Zur Bestimmung der Enzymaktivität wurde eine Methode nach Guest und Creaghan (1974) angewandt. Der Testansatz enthielt in 1 ml Gesamtvolumen 100 mM
Tris-HCl, pH 7,2, 2 mM NAD+ und 50 µl Zellextrakt in geeigneter Verdünnung. Die
Reaktion wurde durch Zugabe von 25 µl Dihydrolipoamid-Lösung (25 mg/ml Ethanol) gestartet und die Extinktionszunahme des NADH (millimolarer Extinktionskoeffizient 6,22 mmol-1*cm-1) bei 340 nm und 30°C verfolgt.
Der für diesen Enzymtest verwendetet Aufschlusspuffer entspricht dem des
PDHC-Tests (siehe 9.5).
9.9
Bestimmung der Pyruvat-Kinase (Gubler et al., 1994b)
Die Pyruvat-Kinase katalysiert den energiegewinnenden Schritt von PEP zu Pyruvat, das dann nach Decarboxylierung als Acetyl-CoA in den Tricarbonsäure-Zyklus
eingeht. Zum Nachweis wird ein Laktat-Dehydrogenase (LDH)-gekoppelter Test
eingesetzt, der das von der Pyruvat-Kinase gebildete Pyruvat zu Laktat reduziert.
In einem Volumen von 1 ml enthielt der Test 100 mM HEPES (pH 7,0), 100 mM
KCl, 10 mM MgCl2, 0,2 mM NADH, 1,5 mM ADP, 24 U LDH und 50 µl Rohextrakt.
Nach Messung des Blindwerts wurde mit 10 mM PEP gestartet. Die Abnahme von
NADH2 (millimolarer Extinktionskoeffizient: 6,22 mmol-1*cm-1) wird bei 340 nm und
30°C für 2 min verfolgt.
Aufschlusspuffer: 100 mM Tris-HCl, pH 7,6
1 mM DTT
5 mM MgSO4
0,1 mM EDTA
20 mM KCl
MATERIAL UND METHODEN
43
30 % Glycerin
9.10
Bestimmung der PEP-Carboxylase
Die Methode zur Messung der PEP-Carboxylase, die PEP zu Oxalacetat carboxyliert, erfolgt nach einer Methode von Ozaki und Shiio (1969). Die Messung wird bei
30°C durchgeführt, die Abnahme von NADH (millimolarer Extinktionskoeffizient:
6,22 mmol-1*cm-1) wird bei einer Wellenlänge von 340 nm verfolgt. In einem Gesamtvolumen von 1 ml enthielt der Ansatz 100 mM Tris-HCl, pH 8,0, 10 mM
MgCl2, 25 mM NaHCO3, 1 mM DTT, 0,2 mM NADH, 24 U Malat-Dehydrogenase
und 50 µl Rohextrakt. Nach Messung des Blindwertes wurde die Reaktion mit 8
mM PEP gestartet und für 2 min verfolgt.
9.11
Bestimmung der PEP-Carboxykinase
Die Messung der PEP-Carboxykinase beruht auf einer Methode von Bentle und
Lardy (1976). Das Enzym katalysiert die reversible decarboxylierende Reaktion
von Oxalacetat zu PEP. Der Testansatz enthielt in einem Volumen von 1 ml 100
mM HEPES (pH 7,0), 10 mM MnCl2, 100 mM KHCO3, 2 mM Glutathion (reduziert),
0,2 mM NADH, 24 U Malat-Dehydrogenase, 10 mM PEP und 50 µl Rohextrakt in
geeigneter Verdünnung. Die Abnahme von NADH wird bei 340 nm und 30°C verfolgt (millimolarer Extinktionskoeffizient: 6,22 mmol-1*cm-1). Nach Messung des
Blindwertes wird die Reaktion mit 2 mM IDP (alternativ GDP) gestartet. Zur Hemmung der PEP-Carboxylase kann dem Ansatz 250 mM Aspartat zugesetzt werden.
Aufschlusspuffer: 100 mM Tris-HCl, pH 7,0
20 mM KCl
5 mM MgSO4
0,1 mM EDTA
2 mM DTT
30 % Glycerin
44
MATERIAL UND METHODEN
9.12
Bestimmung der Pyruvat-Carboxylase
Die Pyruvat-Carboxylase katalysiert die irreversible Carboxylierung von Pyruvat zu
Oxalacetat unter Verbrauch von ATP. Das gebildete Oxalacetat wurde quantitativ
durch
zugesetzte
Glutamat-Oxalacetat-Transaminase
mit
Glutamat
zu
2-
Oxoglutarat und Aspartat umgesetzt. Das Aspartat konnte über HPLC-Analyse
(siehe 10.2) quantifiziert werden.
Zur Herstellung von Rohextrakten für die Bestimmung der Pyruvat-CarboxylaseAktivität wurden die Zellen in Aufschlusspuffer (100 mM Tris-HCl, pH 7,5, 10 %
Glycerin) aufgenommen. Der Aufschluss erfolgte mit dem RiboLyser (siehe 9.1).
Die Rohextrakte wurden direkt zur Bestimmung der Pyruvat-Carboxylase-Aktivität
eingesetzt oder zur späteren Verwendung in flüssigem Stickstoff eingefroren.
Der Enzymtest basiert auf der Aktivitätsbestimmung von permeabilisierten Zellen
(Peters-Wendisch, 1998). In einem Volumen von 1 ml wurden 100 mM Tris-HCl
(pH 7,5 mit 10 % Glycerin), 10 mM MgCl2, 0,2 mM Pyridoxalphosphat, 10 mM Pyruvat, 4 mM ATP, 25 mM NaHCO3, 2 mM Glutamat und 5 U Glutamat-OxalacetatTransaminase aus Schweineherz zugesetzt. Die Testansätze wurden 1 min bei
30°C vorinkubiert und anschließend die Enzymreaktion mit ca. 400 µg Protein gestartet. Aus dem Reaktionsansatz wurden alle 15 Sekunden Proben entnommen
und die Reaktion durch Zugabe der Probe in 20 % (v/v) eiskalter Perchlorsäure
sofort beendet. Nach Neutralisierung mit 5 M KOH und Zugabe von internem
Standard (100 µm Ornithin) wurden die Proben zentrifugiert und die Überstände
zur Quantifizierung des gebildeten Aspartat durch HPLC-Analyse verwendet.
9.13
Bestimmung der Pyruvat-Chinon-Oxidoreduktase (Cunningham und
Hager, 1971)
Die Pyruvat-Chinon-Oxidoreduktase katalysiert die Decarboxylierung von Pyruvat
zu Acetat. Zellextrakte zur Bestimmung der Pyruvat-Oxidase wurden in der stationären Phase geerntet und zweimal in MES-Puffer, pH 6,0 gewaschen. Der Aufschluss erfolgte mit dem RiboLyser im selben Puffer (siehe 9.1). Die PyruvatChinon-Oxidoreduktase-Aktivität wurde photometrisch bei einer Wellenlänge von
600 nm und 40°C ermittelt, indem die Reduktion des Elektronenakzeptors 2,6Dichloroindophenol (DCPIP) gemessen wurde. Der Testansatz enthielt in 1 ml
Gesamtvolumen 100 mM MES, pH 6,0, 10 mM MgCl2, 0,1 mM TPP, 1% (w/v) Tri-
45
MATERIAL UND METHODEN
ton X-100, 300 µm DCPIP und Rohextrakt. Nach Vorinkubation des Ansatzes für
10 min bei 40°C wurde die Reaktion durch Zugabe von 100 mM Pyruvat gestartet.
Der millimolare Extinktionskoeffizient beträgt 22 mM-1*cm-1.
9.14 Bestimmung der Chloramphenicol-Acetyltransferase-Aktivität (Shaw,
1975)
Nachdem die Zellen auf den gewünschten Medien gezüchtet wurden, wurden sie
in 20 mM Tris-HCl, pH 7,6 gewaschen und in einem geeignete Volumen 1 M TrisHCl, pH 7,6 aufgenommen. Der Aufschluss erfolgte mittels RiboLyser (siehe 9.1).
Die Chloramphenicol-Acetyltransferase katalysiert folgende Reaktion:
Chloramphenicol + 2 Acetyl-CoA
Chloramphenicol-1,3-diacetat + 2
CoA
Das reduzierte Coenzym A (CoA) wird in einer zweiten Reaktion mit 5,5´-Dithiobis2-nitrobenzoesäure (DTNB) weiter umgesetzt:
CoA + DTNB
Dithionitrobenzoyl-CoA + 5-Thio-2-nitrobenzoesäure
Das Reaktionsendprodukt 5-Thio-2-nitrobenzoesäure besitzt einen millimolaren
Extinktionskoeffizienten von 13,6 [mM-1*cm-1] bei 412 nm und läßt sich deshalb bei
dieser Wellenlänge im Photometer nachweisen. Für den Test wurden zunächst
950 µl Reaktionsmischung (4 g DTNB/l, 100 mM Tris-HCl, pH 7,8, 0,2 mM AcetylCoA) in einer Plastikküvette auf 37°C temperiert. Nach Zugabe von 10 µl Zellextrakt einer geeigneten Verdünnung, wurde die Reaktion durch Zugabe von 50 µl
5 mM Chloramphenicol gestartet und die Extinktionszunahme über 1 min photometrisch verfolgt.
46
MATERIAL UND METHODEN
10.
Detektion von Aminosäuren mittels HPLC-Analyse
10.1 Fermentationen und Probenahme zur Bestimmung von Valin mittels
RP-HPLC
Zur Fermentation von Valin-produzierenden Stämmen wurden diese unter entsprechenden Wachstumsbedingungen gezüchtet. Zu bestimmten Zeitpunkten
(i.d.R. 24 und 48) wurde 1 ml Kultur zentrifugiert (13000 rpm, 20 min, RT) und die
Kulturüberstände bis zur weiteren Verwendung bei –20°C eingefroren. Zur Analyse wurden diese Proben mit H2O entsprechend verdünnt, mit 100 µm Ornithin als
internem Standard versetzt und erneut zentrifugiert, bevor die Probe mittels HPLC
untersucht wurde.
10.2 RP-HPLC zur Quantifizierung von Aminosäuren
Die in dieser Arbeit verwendete Methode basiert auf einer Methode von Schuster,
1988 zur Trennung von Aminosäuren nach Vorsäulenderivatisierung zu fluoreszierenden Derivaten und wurde zur Analyse von Valin und Aspartat eingesetzt.
Für die Trennung, Identifizierung und Quantifizierung der Aminosäuren wurde eine
LC 1100 Anlage (Firma Agilent Technologies Deutschland GmbH, Waldbronn) mit
einem Fluoreszenzdetektor (HP G1321A) eingesetzt. Durch Vorsäulenderivatisierung mit ortho-Phtaldialdehyd (OPA) und β-Mercaptoethanol (Pierce, Rockford,
USA) wurden alle primären Aminosäuren zu fluoreszierenden Isoindol-Derivaten
umgesetzt (Ogden und Földi, 1986). Die Derivatisierung erfolgte unter normierten
Bedingungen in einem automatisierten Derivatisierungsprogramm (Tabelle 4), um
identische Aminosäurederivate zu gewährleisten (May und Brown, 1989).
Die Trennung der Aminosäuren erfolgte über ein System bestehend aus Vor- und
Hauptsäule (Vorsäule: C18-Hypersil, ODS-5µ, 40x4 mm, Hauptsäule: C18-Hypersil,
ODS-5µ, 150x3 mm, Chromatographie Service GmbH, Langerwehe) unter folgenden Elutionsbedingungen: 40°C,
Lösungsmittel A (100 mM Natriumacetat, pH
7,2), Lösungsmittel B (Methanol), Durchflußrate von 0,35-0,6 ml/min.
Die Durchmischung der beiden Lösungen wurde wie im Folgenden dargestellt über ein HPLC-Programm gesteuert: 1 min wurde mit 25 % Lösungsmittel B bei
0,35 ml/min eluiert. Anschließend erfolgte in den nächsten 2 min ein linearer
47
MATERIAL UND METHODEN
Tabelle 4: HPLC-Programm zur automatisierten Vorsäulenderivatisierung von
primären Aminosäuren mit OPA.
Operation
´draw´
Volumen [µl] Reagenz
8.5
OPA
Zweck
Aufnahme von Derivatisierungsreagenz
´draw´
2.5
´sample´
´draw´
0.0
´water´
Aufnahme der Probe
Eintauchen der Nadel in demin.
Wasser
´draw´
9.0
OPA
Aufnahme von Derivatisierungsreagenz
´min max. amount in
Durchmischen von Probe und
seat´
OPA
´wait for 1 min´
Reaktion der Aminosäuren mit
OPA zu fluoreszierenden Derivaten
´inject´
5.0
Injektion der Probe auf die Säule
Anstieg des Anteils von Lösungsmittel B auf 45 % und einer Durchflußrate von 0,6
ml/min. Unter Beibehaltung der Durchflußrate wurde für weitere 4 min der Anteil
an Lösungsmittel B linear auf 65 % erhöht. Die folgenden Schritte dienten zur
Spülung und zur Regenration des
Säulenmaterials, indem der Anteil an Lö-
sungsmittel B bis auf 100 % erhöht und die Durchflußrate auf 0,35 ml/min reduziert wurde. Das System wurde mit dieser Durchflußrate für 8,5 min gespült bevor
eine neue Aminosäurebestimmung erfolgen konnte.
Die Detektion erfolgte mit einem Fluoreszenzdetektor bei einer Anregungswellenlänge von 230 nm und einer Emissionswellenlänge von 455 nm. Um die einzelnen
Aminosäuren zu identifizieren, wurden diese einzeln injiziert und eine Regressionsgerade erstellt. Dazu wurde das Programm ´HP-Chemstation for LC Rev.
A.06.01´ von Agilent Technologies verwendet.
48
MATERIAL UND METHODEN
11.
Bestimmung von Pantothenat (Sahm und Eggeling, 1999)
Zur Bestimmung von Pantothenat aus Kulturüberständen, die nach 24 bis 48 h
genommen wurden, wurde der Pantothenat-auxotrophe Stamm C. glutamicum
R127 panC:pK18 mob´panC´ eingesetzt. Vorkulturen dieses Stammes wurden in
50 ml 2x TY-Medium mit 25 µg Kanamycin/ml für 20 h bei 28°C inkubiert. Die Vorkulturen wurden vor Beimpfen des Hauptmediums (CGXII) 2x mit 0,9 % NaCl gewaschen, auf eine Initiations-OD von 0,5 angeimpft und für 28 h bei 28°C inkubiert. Dabei wurden zwei Kolben inokuliert, wobei einer kein Pantothenat enthielt
(End-OD600 von 25), der andere
mit 0,5 mM Pantothenat supplementiert war
(End-OD von 40).
Nach 28 h wurden zu 700 µl Glycerin 1050 µl Kultur gegeben und diese bei –70°C
in flüssigem Stickstoff eingefroren und gelagert.
Anhand des in Tabelle 5 dargestellten Pipettierschemas wurde eine Kalibriergerade erstellt. Dazu wurde eine Stammlösung von 100 µg/l und konzentriertes
CGXII-Medium hergestellt. Jedem Ansatz wurden
100 µl nicht
Pantothenat-
supplementierte Zellen zugesetzt und die Ansätze insgesamt für 48 h bei 180 rpm
geschüttelt. Nach 24 und 48 h wurde die OD600 ermittelt und anhand dieser Werte
die Kalibriergerade angefertigt.
Tabelle 5: Pipettierschema zur Erstellung der Kalibiergerade für die Pantothenatbestimmung.
c Pantothenat
Wasser [µl]
[ng/ml]
Pantothenat-
konzentriertes
stammlösung [µl]
CGXII [ml]
0
1000
0
3
10
900
100
3
20
800
200
3
40
600
400
3
60
400
600
3
80
200
800
3
100
0
1000
3
49
MATERIAL UND METHODEN
Um das zur Pantothenatproduktion nötige β-Alanin in ausreichendem Maße
während der Fermentation zur Verfügung zu stellen, wurde den Stämmen, die auf
die Produktion von Pantothenat untersucht wurden, 20 mM β-Alanin während des
Wachstums zugesetzt. Die Kulturüberstände wurden entsprechend verdünnt, sterilfiltriert und 1 ml in den Test eingesetzt.
Konzentriertes CGXII-Medium:
(NH4)2SO4
5
g/l
Harnstoff
1,25
g/l
MOPS
10,5
g/l
K2HPO4
0,25
g/l
KH2PO4
0,25
g/l
MgSO4
62
mg
CaCl2 (0,1 g/10 ml)
250
µl
Spurenelemente-Lösung
250
µl
Biotin (200 mg/l)
250
µl
Kanamycin (50 mg/ml)
125
µl
Glukose
4%
In 188 ml H2O lösen, auf pH 7,0 einstellen und sterilfiltrieren.
50
ERGEBNISSE
III.
ERGEBNISSE
1.
Analyse des Pyruvat-Dehydrogenase-Komplexes (PDHC) in
Corynebacterium glutamicum 13032
Der Pyruvatdehydrogenase-Komplex in C. glutamicum besteht aus den drei Untereinheiten
Pyruvat-Decarboxylase
(kodiert
von
aceE),
Dihydrolipoamid-
Transacetylase (kodiert von aceF) und Dihydrolipoamid-Dehydrogenase (kodiert
von lpd). Der PDHC katalysiert die zentrale Reaktion der oxidativen Decarboxylierung von Pyruvat zu Acetyl-CoA bei gleichzeitiger Bildung von NADH2. Im folgenden werden die spezifischen Aktivitäten des PDHCs im Wachstumsverlauf, die
Spezifität bezüglich verschiedener Substrate und Kohlenstoffquellen sowie der KsWert für Pyruvat in Zellextrakten untersucht.
1.1
Spezifische Aktivitäten des PDHC im Wachstumsverlauf
Um zu klären, in welcher Wachstumsphase der PDHC die höchste spezifische
Aktivität aufweist, wurde der Wildtyp C. glutamicum 13032 auf Komplexmedium
gezüchtet und pro Stunde 50 ml Zellen zur späteren Messung der spezifischen
Aktivitäten geerntet. Wie aus Abbildung 4 hervorgeht steigen die spezifischen
Aktivitäten bis zur exponentiellen Wachstumsphase an, in der ein Maximum von
0,057 U/mg Protein erreicht wurde. Mit dem Eintreten in die stationäre Phase nach
9 h Wachstum ist die Aktivität auf 31% (0,018 U/mg Protein) des Maximal-Werts
gefallen, nach 24 h verbleibt lediglich eine Restaktivität von 0,007 U/mg Protein,
die 12 % des Maximums entspricht. Aufgrund dieser Ergebnisse wurden alle folgenden Experimente mit Zellextrakten der exponentiellen Wachstumsphase
durchgeführt.
51
ERGEBNISSE
0,05
0,04
OD600
10
0,03
1
0,02
0,01
0,1
spez. PDHC-Aktivität
[U/mg Protein]
0,06
100
0
0
10
20
30
Zeit [h]
Wachstum
PDHC-Aktivität
Abbildung 4: Wachstumsabhängige Untersuchung spezifischer PDHC-Aktivitäten
des Wildtyps C. glutamicum auf Komplexmedium ohne Glukose.
1.2
Charakterisierung
der
PDHC-Aktivität
in
Zellextrakten
von
C. glutamicum 13032
Um die Spezifität des PDHC zu analysieren, wurden aus Zellen der logarithmischen Wachstumsphase nach Wachstum auf Komplexmedium Zellextrakte hergestellt und die Enzymaktivitäten untersucht. Tabelle 6 zeigt, dass bei Durchführung
des Enzymtests unter Standardbedingungen mit allen Reaktionskomponenten eine spezifische Aktivität von 0,11 U/mg Protein erreicht wurde. Wurden die Substrate Pyruvat, CoA oder NAD+ weggelassen, waren keine Aktivitäten mehr bestimmbar, woraus geschlossen werden kann, dass diese Reaktionskomponenten
essentiell sind. In Abwesenheit des Substrates MgCl2 war mit 0,096 U/mg Protein
eine um 13% verringerte Aktivität erkennbar, eine um 52 % verringerte Aktivität
(0,053 U/mg Protein) in Abwesenheit von TPP.
In einer früheren Arbeit (J. Schwinde, Doktorarbeit) konnte bereits ein positiver
Einfluß auf die spezifische Aktivität der PDHC durch Hinzufügen des Reduktionsmittels Cystein zum Reaktionsansatz gezeigt werden. Dieser Effekt konnte in dieser Arbeit durch mehrmalige unabhängige Versuche widerlegt werden. So konnte
ohne Cystein im Ansatz sogar eine um 10 % höhere spezifische Aktivität gemessen werden.
52
ERGEBNISSE
Tabelle 6: Spezifität des PDHC bezüglich der im Reaktionsansatz enthaltenen
Komponenten mit Zellextrakten von auf Komplexmedium gewachsenen Zellen.
Reaktionsansatz
spez. Aktivität [U/mg Protein]
relative Aktivität [%]
vollständig
0,11
100
- MgCl2
0,096
87
- TPP
0,053
48
- Pyruvat
<0,001
<0,01
- CoA
<0,001
<0,01
- NAD
<0,001
<0,01
- Cystein
0,121
110
+
Weitere Hinweise auf die Spezifität des PDHC sollte ein Substrat-Test geben. Neben dem Pyruvatdehydrogenase-Komplex als decarboxylierendem Enzym existiert
in C. glutamicum ein 2-Oxoglutarat-Dehydrogenase-Komplex (OGDHC), der 2Oxoglutarat zu Succinyl-CoA decarboxyliert. Für die Aktivität dieses Enzyms sind,
abgesehen von 2-Oxoglutarat anstelle von Pyruvat als Substrat, die gleichen Reaktionskomponenten erforderlich wie für den PDHC. Um die Spezifität des PDHC
für das Substrat Pyruvat zu beweisen, wurden Zellextrakte aus der exponentiellen
Wachstumsphase untersucht, indem die Substrate Pyruvat und 2-Oxoglutarat getrennt voneinander und in Kombination in den Enzymtest eingesetzt wurden. Dabei wurden spezifische Aktivitäten der Enzyme PDHC und OGDHC auf unterschiedlichen Kohlenstoffquellen untersucht. Die erhaltenen Ergebnisse sind in Tabelle 7 dargestellt. Die höchste spezifische PDHC-Aktivität mit 0,106 U/mg Protein
wurde nach Wachstum auf Komplexmedium ohne Glukose gemessen. Die auf
Komplexmedium mit Glukose bzw. auf Minimalmedium ermittelten Aktivitäten lagen im Bereich von 0,046 U/mg Protein (Komplexmedium mit Glukose) bis 0,057
U/mg Protein (Minimalmedium mit Glukose), was einer prozentualen Aktivität von
43 bis 54 % des Maximalwertes entspricht.
53
ERGEBNISSE
Tabelle 7: Spezifische PDHC- und OGDH-Aktivitäten nach Wachstum auf unterschiedlichen Kohlenstoffquellen mit den Substraten Pyruvat, 2-Oxoglutarat und
beiden in Kombination.
Kohlenstoffquelle
spez. PDHCAktivität
[U/mg Protein]
spez. OGDHCAktivität
[U/mg Protein]
spez. Aktivität mit Pyruvat
und 2-Oxoglutarat
[U/mg Protein]
KM
0,106
0,038
0,162
KM
+ 1 % Glukose
0,046
0,330
0,116
MM
+ 1 % Glukose
0,057
0,034
0,097
MM
+ 1 % Glukose
+ 1 % Acetat
0,048
0,026
0,085
MM + 1 % Acetat
0,049
0,050
0,080
Die spezifischen Aktivitäten der OGDHC verhielten sich auf allen Kohlenstoffquellen nahezu konstant mit Werten von 0,026 bis 0,038 U/mg Protein. Lediglich auf
Minimalmedium mit Acetat war mit 0,05 U/mg Protein die Aktivität leicht erhöht.
Wurden beide Substrate in Kombination eingesetzt entsprach die gemessene Aktivität der Summe aus den Aktivitäten der einzeln zugesetzten Substrate. Daraus
kann geschlossen werden, dass sowohl die Aktivitäten des PDHCs als auch die
des OGDHCs spezifisch für die jeweiligen Substrate sind.
1.3
Bestimmung des Ks-Wertes von Pyruvat für den PDHC
Abgesehen vom anaplerotischen Enzym Pyruvat-Carboxylase, das Oxalacetat aus
Pyruvat bildet, existiert in C. glutamicum ein weiteres Pyruvat-umsetzendes Enzym neben der PDHC, die Pyruvat-Chinon-Oxidoreduktase, die die Umsetzung
von Pyruvat zu Acetat katalysiert. Dieses Enzym wurde bereits gereinigt und biochemisch charakterisiert, eine genaue Funktion im Stoffwechsel kann diesem Enzym jedoch noch nicht zugeschrieben werden (M. Schreiner, persönliche Mitteilung). Da die Pyruvat-Chinon-Oxidoreduktase wahrscheinlich hauptsächlich in der
stationären Phase aktiv ist und der PDHC, wie unter 1.1 gezeigt werden konnte,
ERGEBNISSE
54
seine höchste spezifische Aktivität in der exponentiellen Wachstumsphase aufweist, könnte ein Zusammenhang der beiden Enzyme bezüglich ihrer Funktion im
Zentralstoffwechsel bestehen. Die Bestimmung des Km-Wertes ist nur mit gereinigtem Enzym möglich. Da der PDHC von C. glutamicum sehr geringe spezifische
Aktivitäten besitzt und empfindlich auf diverse Salze (NaCl, KCl, (NH4) reagiert,
war eine Reinigung des Komplexes nicht möglich. Daher wurde die MichaelisMenten-Konstante für das Substrat Pyruvat in Zellextrakten von exponentiell gewachsenen Zellen bestimmt.
Wird bei einer gegebenen Menge Enzym die Konzentration eines Substrates sukzessive erhöht, liegt ab einer gewissen Substratkonzentration das gesamte Enzym
in Form eines Enzym-Substrat-Komplexes vor. Die maximale Reaktionsgeschwindigkeit ist ab diesem Zeitpunkt erreicht. Dieser Zusammenhang kann in Form der
Michaelis-Menten- und Lineweaver-Burk-Darstellung graphisch erfaßt werden.
Die Michaelis-Menten-Gleichung ist im folgenden theoretisch beschrieben:
v=
v max * [ S ]
K m+ [S ]
vmax
= maximale Geschwindigkeit
v
= Reaktionsgeschwindigkeit
[S]
= Substratkonzentration
Km
= Michaelis-Menten-Konstante
Die Michaelis-Menten-Konstante KS ist ein Maß für die Affinität eines Enzyms zu
einem bestimmten Substrat. Bei einem hohen KS-Wert ist erst bei einer relativ hohen Konzentration die halbmaximale Geschwindigkeit erreicht. Ein Enzym wird
bevorzugt jenes Substrat umsetzen, zu dem es eine möglichst kleine Konstante
hat. Die Bestimmung dieses Wertes erfolgt häufig über die Lineweaver-BurkDarstellung. Diese erhält man durch Umformung der Michaelis-Menten-Gleichung
in ihre reziproke Form:
K m + [S ]
Km
1
1
1
=
=
+
*
[S ]
v
v max * [ S ] v max
v max
55
ERGEBNISSE
Um den KS-Wert für Pyruvat für die PDHC zu bestimmen wurde der Test mit unterschiedlichen Endkonzentrationen an Pyruvat im Bereich von 0 bis 5 mM im Test
durchgeführt. In den Abbildungen 5a und 5b sind die entsprechenden Werte sowohl in Michaelis-Menten- als auch in Lineweaver-Burk-Darstellung gezeigt. Aus
Abbildung 5a geht hervor, dass bei einer Endkonzentration von 1,77 mM Pyruvat
im Test zunächst die maximale Aktivität (0,051 U/mg Protein) erreicht ist. Nachdem bis 2,5 mM Pyruvat über 5 Werte die spezifische Aktivität konstant blieb, stieg
sie ab einer Konzentration von 2,75 mM wieder an. Es wurde bis 5 mM Pyruvat im
Test gemessen, da diese Konzentration der im Standardtest eingesetzten Konzentration entsprach. Die maximale Reaktionsgeschwindigkeit läge somit bei 5
mM Pyruvat und 0,074 U/mg Protein. Im Gegensatz zu gereinigtem Protein können im Zellextrakt weitere Faktoren auf die Enzym-Aktivitäten einwirken, weshalb
die Werte lediglich als Hinweis gesehen werden können. Eine konkrete Aussage
darüber, ab welcher Konzentration der Komplex tatsächlich in gesättigter Form
vorliegt, kann nicht getroffen werden. Abbildung 5b zeigt die Ergebnisse in Line-
spez. Aktivität [U/mg Protein]
weaver-Burk-Darstellung. Aus dem Schnittpunkt der Regressionsgeraden mit der
0,08
0,07
0,06
0,05
0,04
0,03
0,02
0,01
0
0
1
2
3
4
5
6
Pyruvat [mM]
Abbildung 5a: Spezifische PDHC-Aktivität in Zellextrakten von C. glutamicum aus
auf Komplexmedium gewachsenen Zellen in Abhängigkeit von der PyruvatKonzentration (0-5 mM) in Michaelis-Menten-Darstellung.
56
ERGEBNISSE
50
y = 17,571x + 9,9011
1/spez. Aktivität [mg/U]
40
30
20
10
0
-1
-0,5
0
0,5
1
1,5
2
-10
1/Pyruvat [1/mM]
Abbildung 5b: Spezifische PDHC-Aktivität in Zellextrakten von C. glutamicum aus
auf Komplexmedium gewachsenen Zellen in Abhängigkeit von der PyruvatKonzentration (0-5 mM) in Lineweaver-Burk-Darstellung.
Ordinate läßt sich die maximale spezifische Aktivität ablesen, aus dem Schnittpunkt mit der Abszisse der KS-Wert für Pyruvat. Der Ordinatenschnittpunkt ergab
eine maximale Aktivität von 0,099 U/mg Protein, der KS-Wert für Pyruvat beträgt
1,74 mM. Dies entsprach in etwa dem in dieser Arbeit zusätzlich ermittelten Wert
von 2,6 mM.
2.
Etablierung eines Enzymtests zum Nachweis der PyruvatDecarboxylase (E1-Untereinheit des PDHC) aus C. glutamicum 13032
Die Pyruvat-Decarboxylase (PDC) wird vom aceE-Gen kodiert, das von Mark
Schreiner isoliert und deletiert wurde. Dabei konnte gezeigt werden, dass eine
Deletion im C. glutamicum Wildtyp zu einem Verlust der Fähigkeit, auf Komplexmedium und Minimalmedium mit Glukose zu wachsen, führte. Dieser Defekt konnte durch Zugabe von 0,5 bis 1 % Acetat auf beiden Medien wieder ausgeglichen
57
ERGEBNISSE
EcoR I
EcoR I
Pst I
orf13
BamH I
aceE
Pst I
BamH I
orf14
2 000
orf15
4000
Abbildung 6: Restriktionskarte der kodierenden Region des aceE-Gens mit dem
stromaufwärts liegenden Gen orf13 und den stromabwärts liegenden Genen orf14
und 15, die in entgegengesetzter Orientierung angeordnet sind. Das Gen aceE
kodiert für die E1-Untereinheit (Pyruvat-Decarboxylase) des PDHC, orf13 und
orf15 kodieren für hypothetische Proteine und orf14 für einen putativen ABCTransporter.
werden. Abbildung 6 zeigt die genetische Organisation des aceE-Gens im Chromosom von C. glutamicum, wobei deutlich wird, dass die kodierenden Gene
der anderen Untereinheiten des PDHC nicht in Nachbarschaft des aceE-Gens liegen.
Die E1-Untereinheit des PDHC katalysiert die Decarboxylierung des Pyruvats unter Entstehung von Hydroxyethyl-TPP:
Pyruvat + TPP
Hydroxyethyl-TPP + CO2
Die Messung der Aktivität dieser Untereinheit war jedoch nur mit nichtkomplexiertem
Enzym,
das
dieselbe
Reaktion
wie
die
Oxidoreduktase katalysiert, möglich:
Pyruvat + FAD
Acetat + CO2 + FADH2
Pyruvat-Chinon-
58
ERGEBNISSE
Tabelle 8: Spezifische PDC-Aktivitäten in Zellextrakten von auf unterschiedlichen
Kohlenstoffquellen kultivierten Zellen.
Kohlenstoffquelle
spez. PDC-Aktivität [mU/mg Protein]
KM
38,8
KM + Glukose
53
MM + Glukose
24
MM + Acetat
<0,001
MM + Glukose + Acetat
<0,001
Die Aktivitätsbestimmung erfolgt enzymatisch durch photometrische Messung der
Reduktion des künstlichen Elektronenakzeptors Kaliumhexacyanoferrat bei 430
nm.
Zur Etablierung des Enzymtests wurden Rohextrakte aus Zellen hergestellt, die
auf Komplexmedium bis zur exponentiellen Wachstumsphase gewachsen waren.
Als Negativkontrolle diente der Stamm C. glutamicum∆aceE, der eine Deletion im
aceE-Gen trägt. Es konnte eine maximale spezifische Aktivität von 38,8 mU/mg
Protein für den Wildtyp C. glutamicum 13032 ermittelt werden, wohingegen für den
Stamm C. glutamicum 13032∆aceE keine Aktivität detektierbar war.
Durch die generell sehr niedrigen Aktivitäten war eine Messung in Korrelation zum
Wachstum schwierig. Erste Versuche weisen allerdings darauf hin, dass auch für
die E1-Untereinheit der PDHC die höchste spezifische Aktivität nach 6 h Wachstum auf Komplexmedium ohne zusätzliche Kohlenstoffquelle mit 27 mU/mg Protein erreicht wird.
Die Untersuchung der spezifischen Aktivitäten auf unterschiedlichen Kohlenstoffquellen (Tabelle 8) zeigte, dass auf Minimalmedium mit Acetat oder Mischsubstrat
gewachsenen Zellen keine Aktivität beobachtet werden konnte. Die auf Minimalmedium mit Glukose ermittelte Aktivität lag bei 24 mU/mg Protein, wohingegen auf
Komplexmedium mit und ohne Glukose die höchsten Aktivitäten mit 53 und 38,8
mU/mg Protein meßbar waren.
Ohne Pyruvat war keine Aktivität detektierbar. Eine Variation der Pyruvatkonzentrationen im Bereich von 0 bis 10 mM zeigte gleichbleibende PDC-Aktivitäten.
59
ERGEBNISSE
Wurde dem Testansatz kein MgCl2 zugesetzt waren im höchsten Fall 12 mU/mg
Protein zu messen, bei Weglassen von TPP maximal 6 mU/mg Protein.
3.
Isolierung
und
Untersuchung
der
Dihydrolipoamid-
Transacetylase (E2-Untereinheit) aus C. glutamicum 13032
Ein weiteres Ziel dieser Arbeit waren Untersuchungen zur Expression des aceFGens, das für die Dihydrolipoamid-Transacetylase (DHL) kodiert. Dieses Gen
konnte durch Sequenz-Vergleiche identifiziert werden und liegt auf dem Chromosom nicht benachbart zu den anderen für den PDHC kodierenden Gene. Das Gen
aceF liegt jedoch benachbart zu zwei Genen der Liponsäurebiosynthese. Da Liponsäure ein kovalent gebundener Bestandteil des PDHC ist, könnte es sich hier
um eine funktionelle Einheit handeln.
XhoI
SalI
SalI
SalI KpnI
SalI
SalI
SalI
KpnI
PvuI
lipB
aceF
1000
PvuI
XhoI XhoI
2000
XhoI
BamHI
lipA
3000
4000
Abbildung 6: Restriktionskarte der kodierenden Region des aceF-Gens mit den
stromabwärts liegenden Genen lipB und lipA. Das aceF-Gen kodiert für die E2Untereinheit (Dihydrolipoamid-Transacetylase) des PDHC, lipB kodiert für eine
Lipoyl-[Acyl-Carrier-Protein]-Protein-N-Lipoyltransferase und lipA für eine LipoylSynthase B.
60
ERGEBNISSE
C.
E.
B.
A.
glutamicum
coli
subtilis
vinelandii
MAFSVEMPELGESVTEGTITQWLKSVGDTVEVDEPLLEVSTDKVDTEIPSPVAGVILEIK
-AIEIKVPDIGADEVE--ITEILVKVGDKVEAEQSLITVEGDKASMEVPSPQAGIVKEIK
------------------------------------------------------------SEIIRVPDIGGDG-E--VIELLVKTGDLIEVEQGLVVLESAKASMEVPSPKAGVVKSVS
60
C.
E.
B.
A.
glutamicum
coli
subtilis
vinelandii
AEEDDTVDVGGVIAIIGDADETPANEAPADEAP-APAEEEEPVKEEPKKEAAPEAPAATG
VSVGDKTQTGALIMIFDSADGAA----DA-----APAQAEE--KKEA-AP--AAAPA-AA
-----------------------------------------------------------VKLGDKLKEGDAIIELEPAAGAAAAPAEAAAVPAAPTQAVD--EAEAPSPGASATPAPAA
120
C.
E.
B.
A.
glutamicum
coli
subtilis
vinelandii
AATDVEMPELGESVTEGTITQWLKAVGDTVEVDEPLLEVSTDKVDTEIPSPVAGTIVEIL
AAKDVNVPDIGSD--EVEVTEILVKVGDKVEAEQSLITVEGDKASMEVPAPFAGTVKEIK
-AFEFKLPDIGEGIHEGEIVKWFVKPNDEVDEDDVLAEVQNDKAVVEIPSPVKGKVLELK
ASQEVRVPDIGSAG-KARVIEVLVKAGDQVQAEQSLIVLESDKASMEIPSPASGVVESVA
180
C.
E.
B.
A.
glutamicum
coli
subtilis
vinelandii
ADEDDTVDVGAVIARIGDANAAAAPAEEEAAPAEEEEPVKEEPKKEAAPEAPAATGAATD
VNVGDKVSTGSLIMVFEVAG-EAGA----AAPAAKQEAA---PA--AAP-AP-AAGVK-E
VEEGTVATVGQTIITFD------------------------------AP------GYE-D
IQLNAEVGTGDLILTLRTTGAQAQP----TAPAAAAAAS---PA--PAPLAPAAAGPQ-E
240
C.
E.
B.
A.
glutamicum
coli
subtilis
vinelandii
VEMPELGESVTEGTITQWLKAVGDTVEVDEPLLEVSTDKVDTEIPSPVAGTIVEILADED
VNVPDIGGD--EVEVTEVMVKVGDKVAAEQSLITVEGDKASMEVPAPFAGVVKEL----LQFK--GSD--------------------------ESDDAKTEA---------------VKVPDIGSAG-KARVIEVLVKAGDQVQAEQSLIVLESDKASMEIPSPAAGVVESV-----
300
C.
E.
B.
A.
glutamicum
coli
subtilis
vinelandii
DTVDVGAVIARIGDANAAAAPAEEEAAPAEEEEPVKEEPKKEEPKKEEPKKEAATTPAAA
-KVNVG---DKVKTGSLIMIFEVEGAAPAAA--PAKQEAAAPAPAAKAEAPAAAPAAKAE
-QVQST---AEAGQ-------DVAKEEQAQE--PAK-------------ATGAGQQDQAE
-AVQLN---AEVGTGDQILTLRVAGAAPSGP--RAR-----GSPGQAAAAPGAAPAPAPV
360
C.
E.
B.
A.
glutamicum
coli
subtilis
vinelandii
SATVSASGDNVPYVTPLVRKLAEKHGVDLNTVTGTGIGGRIRKQDVLAAANG---EAAPA
GKSEFAENDAYVHATPLIRRLAREFGVNLAKVKGTGRKGRILREDVQAYVKEAIKRAEAA
VDP----N-KRVIAMPSVRKYAREKGVDIRKVTGSGNNGRVVKEDIDSFVNGGAQEAAPQ
GAP--SRNGAKVHAGPAVRQLAREFGVELAAINSTGPRGRILKEDVQAYVKAMMQKAKEA
420
C.
E.
B.
A.
glutamicum
coli
subtilis
vinelandii
EAAAPVSAWSTKSVDP-EKAKLRGTTQK-VNRIREITAMKTVEALQISAQLTQLHEVDMT
PAATGGGIPGMLPWPKVDFSKFGEIEEVELGRIQKISGANLSRNWVMIPHVTHFDKTDIT
ETAAPQETAAKPAAAPAPEGEFPETREK-MSGIRKAIAKAMVNSKHTAPHVTLMDEVDVT
PAAGAASGAGIPPIPPVDFAKYGEIEEVPMTRLMQIGATNLHRSWLNVPHVTQFESADIT
480
C.
E.
B.
A.
glutamicum
coli
subtilis
vinelandii
RVAELRKKN-KPAFIEKHGVNLTYLPFFVKAVVEALVSHPNVNASFNAKTKEMTYHSSVN
ELEAFRKQQNEEAAKRKLDVKITPVVFIMKAVAAALEQMPRFNSSLSEDGQRLTLKKYIN
NLVAHRKQF--KQVAADQGIKLTYLPYVVKALTSALKKFPVLNTSIDDKTDEVIQKHYFN
ELEAFRVAQ--KAVAEKAGVKLTVLPLLLKACAYLLKELPDFNSSLAPSGQALIRKKYVH
540
C.
E.
B.
A.
glutamicum
coli
subtilis
vinelandii
LSIAVDTPAGLLTPVIHDAQDLSIPEIAKAIVDLADRSRNNKLKPNDLSGGTFTITNIGS
IGVAVDTPNGLVVPVFKDVNKKGIIELSRELMTISKKARDGKLTAGEMQGGCFTISSIGG
IGIAADTEKGLLVPVVKNADRKSVFEISDEINGLATKAREGKLAPAEMKGASCTITNIGS
IGFAVDTPDGLLVPVIRNVDQKSLLQLAAEAAELAEKARSKKLGADAMQGACFTISSLGH
600
C.
E.
B.
A.
glutamicum
coli
subtilis
vinelandii
EGALSDTPILVPPQAGILGTGAIVKRPVVITEDGIDSIAIRQMVFLPLTYDHQVVDGADA
LGTTHFAPIVNAPEVAILGVSKSAMEPVWNGKE----FVPRLMLPISLSFDHRVIDGADG
AGGQWFTPVINHPEVAILGIGRIAEKAIVRDGE----IVAAPVLALSLSFDHRMIDGATA
IGGTAFTPIVNAPEVAILGVSKASMQPVWDGKA----FQPRLMLPLSLSYDHRVINGAAA
660
C.
E.
B.
A.
glutamicum
coli
subtilis
vinelandii
GRFLTTIKDRLETANFEGDLQLARFITIINNTLS--DIRRLVM-QNALNHIKRLLN--DPQLILMEA
ARFTKRLGDLLA--DIRAILL--
Abbildung 8: Aminosäuresequenz-Vergleich der DHL aus C. glutamicum mit
DHLs von E. coli, B. subtilis und A. vinelandii. Die identischen Bereiche sind rot
hinterlegt. Bereiche zur Bindung des Dihydrolipoamids sind schwarz und eine potentielle E3-Bindedomäne ist grün unterstrichen.
61
ERGEBNISSE
In Abbildung 7 ist eine Restriktionskarte mit dem aceF-Gen und den stromabwärts liegenden Genen lipB und lipA dargestellt. Abbildung 8 zeigt einen Vergleich der Aminosäuresequenz der DHL aus C. glutamicum zu DHLs anderer Organismen.
Die DHL aus C. glutamicum besteht aus 675 Aminosäuren (AS) und hat eine vorhergesagte molekulare Masse von 70,9 kDa. Der AS-Vergleich zeigte Identitäten
zu möglichen E2-Untereinheiten anderer PDHCs und OGDHCs von bis zu 66 %
(C. diphteriae). Abbildung 8 zeigt den AS-Vergleich zu den Organismen B. subtilis (22,4 % Identität) (Hemila et al., 1990), E. coli (31,4 % Identität) (Stephens et
al., 1983) und A. vinelandii (27,6 % Identität) (Hanemaaijer et al., 1988), die
bereits beschrieben und funktionell nachgewiesen sind. Mit Hilfe eines Computerprogrammes (SMART) konnten 3 Domänen von 76 AS für die Bindung von Dihydrolipoamid identifiziert werden (AS 122-197, AS 238-313, AS 4-79). Außerdem
konnte ein Bindungsbereich für die Dihydrolipoamid-Dehydrogenase (E3-BindeDomäne) vorhergesagt werden (AS 371-409).
Die DHL katalysiert die unten aufgeführten Reaktionen:
Hydroxyethyl-TPP + Lipoamid
6-S-Acetyldihydrolipoamid
+ TPP
Acetyldihydrolipoamid + CoA
Acetyl-CoA + Dihydroli-
poamid
Der Nachweis der Enzymaktivität erfolgt enzymatisch nach folgendem Prinzip in
nicht physiologischer Richtung:
PTA
Acetylphosphat + CoA
Acetyl-CoA + Pi
Acetyl-CoA + Dihydrolipoamid
6-S-Acetyldihydrolipoamid
+ CoA
62
ERGEBNISSE
Tabelle 9: Spezifische DHL-Aktivitäten in Zellextrakten von auf unterschiedlichen
Kohlenstoffquellen kultivierten Zellen.
Kohlenstoffquelle
spez. DHL-Aktivität [mU/mg Protein]
KM
233
KM + Glukose
50
MM + Glukose
40
MM + Acetat
33
MM + Glukose + Acetat
39
Um die spezifische Aktivität der DHL zu bestimmen und ein geeignetes Nachweissystem zu etablieren, wurden Zellen aus der exponentiellen Wachstumsphase
(OD600 von 6) nach Wachstum auf Komplexmedium geerntet.
Es konnte eine starke Abhängigkeit der Stabilität des Enzymtests von der CoAKonzentration nachgewiesen werden. Bei einer Konzentration von 0,2-0,4 mM
CoA im Test war keine Aktivität bestimmbar, ebenso beim Einsetzen unverdünnter
Rohextrakte. Eine reproduzierbare Messung von etwa 120 mU/mg Protein war
jedoch in 1:10 verdünnten Rohextrakten aus exponentiell gewachsenen Zellen bei
einer CoA-Konzentration von 0,1 mM im Test möglich.
Mit diesem Testsystem wurden die DHL-Aktivitäten wiederum nach Wachstum auf
unterschiedlichen Kohlenstoffquellen analysiert. Die höchste Aktivität ergab sich
mit 233 mU/mg Protein bei Zellextrakten aus auf Komplexmedium (ohne weitere
Kohlenstoffquelle) gewachsenen Zellen. Auf allen anderen untersuchten Kohlenstoffquellen waren die Aktivitäten im Vergleich zum höchsten Wert (Komplexmedium) mit spezifischen Aktivitäten von 33-50 mU/mg Protein durchgehend niedriger.
ERGEBNISSE
4.
63
Transkriptionsanalysen zur Expression der Gene aceE und
aceF aus C. glutamicum 13032
4.1
´Northern-Blot´-Experimente der Gene aceE und aceF
Es konnte für E. coli gezeigt werden, dass die Gene für die Untereinheiten des
PDHC in einem Operon organisiert sind (Spencer und Guest, 1985). Wie schon
unter III.2 und III.3 dargestellt, liegen die Gene aceE, aceF und lpd in C. glutamicum nicht benachbart auf dem Chromosom. Für das lpd-Gen konnte durch
´Northern-Blot´-Experimente bereits gezeigt werden, dass es monocistronisch
transkribiert wird (Schwinde et al., 2001). Die Frage nach der Genorganisation
sollte nun auch für die Gene aceE und aceF geklärt werden. Zellen von C. glutamicum wurden hierfür auf Komplexmedium und Minimalmedium mit Glukose gezüchtet, in der exponentiellen Wachstumsphase geerntet und die RNA isoliert.
Für die ´Northern-Blot´-Hybridisierungen von aceE wurde ein 579 bp großes
Fragment hergestellt, das durch EcoRI/PstI Verdau aus dem Vektor pdrive-aceE
hervorging und nach radioaktiver Markierung als Sonde eingesetzt wurde. Die Ergebnisse des ´Northern-Blots´ sind in Abbildung 9 dargestellt. Es konnte eine
etwa 2,9-kb-Bande nachgewiesen werden, die auf eine monocistronische Organisation des aceE-Gens hinweist (Strukturgen: 2768 bp). Stromaufwärts und stromabwärts liegen zwei mit orf13 und orf14 bezeichnete Gene, die zum aceE-Gen
eine intergene Region von 206 bp (orf13) und 77 bp (orf14) aufweisen. Da orf14 in
entgegengesetzter Orientierung angeordnet ist und eine Terminationsstruktur am
Ende des aceE-Gens gefunden wurde, entsprach die monocistronische Organisation den Erwartungen. Aus Abbildung 9 wird zudem ersichtlich, dass die Stärke
des Signals mit steigender RNA-Menge zunimmt. Weiterhin ist die Transkriptmenge bei Wachstum auf Komplexmedium deutlich höher als auf Minimalmedium.
Dies weist auf eine Regulation auf RNA-Ebene in Abhängigkeit von der Kohlenstoffquelle hin. Dieses Ergebnis korreliert mit den auf Komplexmedium etwa doppelt so hohen spezifischen Enzymaktivitäten des PDHC (siehe Kapitel III/1.2).
64
ERGEBNISSE
← 2,9 kb
1 2 3
4 5
6 7 8 9 10
Abbildung 9: ´Northern-Blot´-Analyse für das Gen aceE mit RNA aus auf Komplex- und Minimalmedium gewachsenen Zellen. In den Spuren 1-5 ist die Komplexmedium-RNA in steigender Menge (0,7-11,2 µg), in den Spuren 6-10 RNA aus
auf Minimalmedium gewachsenen Zellen derselben Menge aufgetragen.
Für das Gen aceF wurde ebenfalls die genetische Organisation geklärt. Da Versuche zur Deletion dieses Gens bislang erfolglos waren, lag die Vermutung nahe,
dass die Gene aceF und die stromabwärts gelegenen Gene lipB und lipA, die für
Proteine aus der Liponsäurebiosynthese katalysieren, ein Operon bilden, wodurch
beim Deletieren von aceF aufgrund des fehlenden Lipoamids kein Wachstum
mehr möglich ist. Um zu untersuchen, ob diese Gene ein Operon bilden, wurden
´Northern-Blot´-Hybridisierungen mit RNA aus auf Komplexmedium und Minimalmedium gewachsenen Zellen aus der exponentiellen Wachstumsphase mit spezifischen Sonden durchgeführt. Als Sonde diente ein radioaktiv markiertes 617 bpFragment, das aus dem Vektor pEkEx2-aceF durch PstI Verdau hervorging.
65
ERGEBNISSE
← 2 kb
1
2 3 4 5
6 7 8 9 10
Abbildung 10: Northern Blot-Analyse für das Gen aceF mit RNA aus auf Komplex- und Minimalmedium gewachsenen Zellen. In den Spuren 1-5 ist die Komplexmedium-RNA in steigender Menge (0,7-11,2 µg), in den Spuren 6-10 die RNA
aus auf Minimalmedium gewachsenen Zellen derselben Menge aufgetragen.
Da die intergene Region zwischen aceF und dem stromabwärts gelegenen lipBGen lediglich 62 bp beträgt, wurde ein gemeinsames Transkript erwartet. Bei einer
monocistronischen Organisation wäre das zu erwartende Transkript etwa 2 kb
groß (Strukturgen: 2028 bp). Läge das aceF-Gen in einem Operon mit lipB oder
lipB und lipA vor, hätten die erwarteten Transkripte eine Länge von 2,9 kb im ersten bzw. 4 kb im zweiten Fall. Die Ergebnisse des ´Northern-Blots´ sind in Abbildung 10 gezeigt.
Es konnte eine 2-kb-Bande nachgewiesen werden, die auf eine monocistronische
Organisation des Gens hinweist. Die Stärke des Signals nimmt, wie auch schon
für aceE gezeigt werden konnte, mit steigender RNA-Menge zu. Weiterhin ist die
Transkriptmenge auf Komplexmedium deutlich höher als auf Minimalmedium. Dieses Ergebnis korreliert wiederum mit den auf Komplexmedium etwa doppelt so
hohen spezifischen Enzymaktivitäten der PDHC.
66
ERGEBNISSE
4.2
Promotoraktivitäten der Gene aceE und aceF im Wachstumsverlauf
und auf unterschiedlichen Kohlenstoffquellen
Durch die Bestimmung spezifischer PDHC-Aktivitäten im Wachstumsverlauf konnte bereits gezeigt werden, dass die maximale Aktivität in der exponentiellen
Wachstumsphase (OD600 ~ 6 nach Wachstum auf Komplexmedium) erreicht wird
und anschließend auf 12 % des Maximalwerts in der stationären Phase absinkt.
Um eine mögliche Korrelation zu den Promotoraktivitäten zu erhalten, wurden diese ebenfalls wachstumsabhängig für die Gene aceE und aceF untersucht. Der
Stamm zur Untersuchung des aceE-Promotors (C. glutamicum pET2-aceE) trug
ein 391 bp Fragment mit der Promotorregion des Gens auf dem Plasmid pET2
(Promotor-Testvektor
mit
cat-Gen
zur
Messung
der
Chloramphenicol-
Acetyltransferase-Aktivität). Der Stamm C. glutamicum pET2-aceF wurde zur
wachstumsabhängigen Bestimmung der aceF-Promotoraktivitäten herangezogen.
Dieser trug ein 411 bp großes Fragment, das den Promotorbereich des Gens aceF umfaßt. Rohextrakte wurden für diesen Versuch aus auf Komplexmedium gewachsenen Zellen hergestellt, wobei bis zur stationären Phase pro Stunde eine
Probe (50 ml) genommen wurde.
Beide Kulturen zeigten identisches Wachstum und die spezifischen CATAktivitäten nahmen für beide Gene wachstumsabhängig bis zur späten exponentiellen Phase zu, wobei für aceE ein Maximum von 0,54 U/mg Protein, für aceF ein
Maximum von 0,23 U/mg Protein erreicht wurde (Abbildung 11). In der stationären Phase nahm die aceE-Promotoraktivität leicht ab, die aceF-Promotoraktivität
blieb auf dem gleichen Niveau. Die zu Beginn der Experimente gemessenen spezifischen CAT-Aktivitäten sind auf die Vorkulturen zurückzuführen, die noch nahezu die maximale Aktivität besaßen. Vergleichsweise waren die Promotoraktivitäten
des aceE-Gens 2-fach erhöht gegenüber denen des aceF-Gens.
67
ERGEBNISSE
0,6
OD600
0,5
10
0,4
0,3
1
0,2
spez. CATAktivitäten
[U/mg Protein]
100
0,1
0,1
0
5
10
15
20
25
0
30
Zeit [h]
Wachstum C. glutamicum pET2-aceE
Wachstum C. glutamicum pET2-aceF
spez. CAT-Aktivität C. glutamicum pET2-aceF
spez. CAT-Aktivität C. glutamicum pET2-aceE
Abbildung 11: Spezifische CAT-Aktivitäten der Stämme C. glutamicum-pET2aceE und C. glutamicum pET2-aceF im Wachstumsverlauf.
Um die auf unterschiedlichen Kohlenstoffquellen gemessenen spezifischen
PDHC-Aktivitäten in Korrelation zu den CAT-Promotoraktivitäten der Gene aceE
und aceF setzen zu können, wurden die spezifischen CAT-Aktivitäten der Stämme
C. glutamicum pET2-aceE und C. glutamicum pET2-aceF nach Wachstum auf
verschiedenen Kohlenstoffquellen untersucht. Dazu wurden die beiden Stämme
auf Komplexmedium (+/- Glukose) und Minimalmedium (Glukose, Acetat, Glukose-Acetat-Mischsubstrat) bis zur exponentiellen Wachstumsphase gezüchtet und
die spezifischen CAT-Aktivitäten bestimmt. Die Ergebnisse sind in Tabelle 10 zusammengestellt.
Die höchsten CAT-Aktivitäten konnten auf Minimalmedium mit Glukose gemessen
werden mit 0,87 U/mg Protein für aceE, während für die Promotorregion des aceFGens die höchste spezifische CAT-Aktivität auf Komplexmedium mit 0,45 U/mg
Protein ermittelt wurde. Es konnte wiederum auf allen getesteten Kohlenstoffquellen für aceF eine um etwa 50 % verringerte Promotoraktivität gemessen werden.
Deutlich geringere Promotoraktivitäten wurden nach Wachstum auf Komplexmedium mit Glukose mit 0,39 U/mg Protein für das aceE-Gen und 0,16 U/mg Protein
68
ERGEBNISSE
Tabelle 10: Spezifische CAT-Aktivitäten der Stämme C. glutamicum pET2-aceE
und C. glutamicum pET2-aceF auf unterschiedlichen Kohlenstoffquellen nach
Wachstum bis zur exponentiellen Wachstumsphase.
C-Quelle
spez. CAT-Aktivität
[U/mg Protein] des Stammes
C. glutamicum pET2-aceE
spez. CAT-Aktivität
[U/mg Protein] des Stammes
C. glutamicum pET2-aceF
KM
KM + Glukose
0,76
0,39
0,45
0,16
MM + Glukose
0,87
0,44
MM + Acetat
MM + Glukose
+ Acetat
0,44
0,22
0,41
0,23
für das aceF-Gen detektiert. Auf Minimalmedium mit Mischsubstrat oder Acetat
wurden im Vergleich zu den maximal ermittelten Promotoraktivitäten der Gene
aceE und aceF um 50 % erniedrigte spezifische CAT-Aktivitäten bestimmt.
4.3
Transkriptionsstartpunkt-Bestimmung von aceE
Zur Bestimmung der 5´- Enden der RNAs der Gene aceE und aceF wurde die Methode der nicht-radioaktiven ´Primer-Extension´-Analyse (PEX) gewählt. Die hierfür verwendete RNA wurde aus C. glutamicum Stämmen isoliert, die die jeweiligen Promotor-Testvektoren (pET2-aceE bzw. pET2-aceF) trugen. Die Reaktion
wurde mit ungefähr 100 µg RNA durchgeführt. Für die ´Primer-Extension´Reaktion wurden zwei unterschiedliche IRD800-markierte ´Primer´ (CM4 und
CM5) eingesetzt, deren Sequenz zur Vektorsequenz stromabwärts der MCS homolog ist (CM4: Basen 20-39, CM5: Basen 9-26). Diese Primer lagern sich an die
RNA an und werden durch Einsatz der Reversen Transkriptase verlängert bis zum
5´- Ende der Template-RNA. Das Extensionsprodukt kann im Licor-Gel gemeinsam mit einer Sequenzreaktion, für die dieselben Primer verwendet wurden, aufgetrennt und ausgewertet werden.
Bei Durchführung der PEX mit dem Primer CM4 wurden zwei Signale detektiert,
wovon eines stark ausgeprägt war und 121 bp stromaufwärts des Startcodons
ATG lag. Daraus konnte eine Sequenz für die Promotorregion abgeleitet werden,
ERGEBNISSE
69
die der –10-Region des lpd-Gens (TATCCT) entspricht (Abbildung 12). Dieses
Signal konnte mit dem zweiten Primer CM5 nicht bestätigt werden. Dagegen wurde das zweite, 54 bp stromaufwärts des ATG gelegene, Signal mit den Primern
CM4 und CM5 erhalten (Abbildung 13). Für C. glutamicum konnte eine Konsensussequenz für die –10-Region mit der Basenabfolge TA(T/C)AAT festgelegt werden (Patek et al., 2003). Eine mögliche –10-Region für das aceE-Gen mit der Sequenz TATCCT (Abbildung 12) wurde bereits genannt und stimmt in 4 bp mit der
Konsensussequenz überein. Die zweite mögliche –10-Region geht aus Abbildung
13 mit der Sequenz AAGAAT hervor. Diese Sequenz stimmt ebenfalls in 4 Basen
mit der Konsensus-Sequenz für C. glutamicum überein. Ausgehend von diesen
potentiellen –10-Regionen konnten außerdem verlängerte –10- und –35-Regionen
abgeleitet werden. Für das in Abbildung 12 dargestellte Signal lautet die verlängerte –10-Region CGTATCCTTG, die –35-Region 17+/-1 bp stromaufwärts der –
10-Region lautet GTAAAACA. Die Ableitung einer verlängerten –10-Region aus
dem in Abbildung 13 gezeigten Signal ergab die Sequenz CCAAGAATGG, die
Sequenz der –35-Region lautet GGCGTGTC. Untersuchungen zu corynebakteriellen Promotoren führten zu der Festlegung einer Konsensussequenz für die
–35-
Region mit der Sequenz TTGGCA (Patek, 1996). Weiterhin scheinen Guanine 2
bp stromaufwärts und stromabwärts der –10-Region eine wichtige Rolle bei der
Promotorfunktion zu spielen (Vasicova et al., 1999). Beide potentiellen –10Regionen besitzen ein Guanin 2 bp stromabwärts der –10-Region. Die Übereinstimmung mit der Konsensussequenz der -35-Region ist für beide potentiellen
Signale sehr schwach.
70
ERGEBNISSE
A C GT
codogener Strang
C
A
A
C
G
C
A
T
A
G
G
A
A
C
A
A
T
T
A
T
T
G
A
A
T
Abbildung 12: Transkriptionsstartbestimmung (Signal 1) des aceE-Gens mittels
nicht-radioaktiver ´Primer-Extension´-Analyse mit dem IRD800-markierten Primer
CM4; die Reaktion wurde mit 100 µg RNA aus auf Minimalmedium mit Glukose
(C. glutamicum pET2-aceE) gewachsenen Zellen durchgeführt und zusammen mit
einer Sequenzierreaktion auf einem Licor-Gel aufgetrennt.
71
ERGEBNISSE
A C G T
codogener Strang
G
C
T
G
G
T
T
C
T
T
A
C
C
C
T
G
G
C
C
C
T
T
T
G
G
Abbildung 13: Transkriptionsstartbestimmung (Signal 2) des aceE-Gens mittels
nicht-radioaktiver ´Primer-Extension´-Analyse mit dem IRD800-markierten Primer
CM4; die Reaktion wurde mit 100 µg RNA aus auf Minimalmedium mit Glukose
(C. glutamicum pET2-aceE) gewachsenen Zellen durchgeführt und zusammen mit
einer Sequenzierreaktion auf einem Licor-Gel aufgetrennt.
72
ERGEBNISSE
4.4
Transkriptionsstartpunkt-Bestimmung von aceF
Für das aceF-Gen wurden die ´Primer-Extension´-Reaktionen wie bereits für aceE
beschrieben durchgeführt. Es wurde hierfür ein Stamm verwendet, der ebenfalls
das Plasmid pET2-aceF zur Überexpression der Promotorregion trug (C. glutamicum pET2-aceF). Die ´Primer-Extension´-Reaktionen wurden wiederum mit den
Primern CM4 und CM5 durchgeführt.
A C G T
codogener Strang
G
T
G
G
T
G
T
A
G
T
G
T
T
T
A
A
C
T
T
A
G
C
C
A
T
Abbildung 14: Transkriptionsstartbestimmung des aceF-Gens mittels nichtradioaktiver ´Primer-Extension´-Analyse mit dem IRD800-markierten Primer CM4;
die Reaktion wurde mit 100 µg RNA aus auf Minimalmedium mit Glukose
(C. glutamicum pET2-aceF) gewachsenen Zellen durchgeführt und zusammen mit
einer Sequenzierreaktion auf einem Licor-Gel aufgetrennt.
73
ERGEBNISSE
Es konnten bei der Durchführung mit beiden Primern übereinstimmende Signale
detektiert werden (Abbildung 14), woraus sich eine –10-Region mit der Sequenz
TTGAAT ableiten läßt, die in 4 Basen mit der für C. glutamicum festgelegten Konsensussequenz (TA(T/C)AAT) übereinstimmt. Die daraus resultierende verlängerte –10-Region besitzt die Sequenz AATTGAATCG und ein Guanin 2 bp stromabwärts der –10-Region. Für die –35-Region lautet die Sequenz GACCCGAA, die
mit der Konsensussequenz wiederum nur sehr schwach konform geht.
5.
Valin-
und
Pantothenatproduktion
mit
rekombinanten
Stämmen
5.1
Konstruktion rekombinanter Stämme
Aus früheren Arbeiten gab es verschiedene Hinweise darauf, dass die Enzyme
des PEP-Pyruvat-Oxalacetat-Knotenpunkts und deren Veränderung in C. glutamicum einen bedeutenden Einfluss auf die Produktion verschiedener Aminosäuren
haben. So konnte durch Überexpression des Pyruvat-Carboxylase-Gens (pycGen) eine Zunahme der Produktion der Aminosäuren Lysin, Glutamat und Threonin nachgewiesen werden, wohingegen die Deletion zu einer Abnahme der Produktion führte (Peters-Wendisch et al., 2001).
Um den Einfluss der Enzyme, die an der Bildung der Vorstufen PEP, Pyruvat und
Oxalacetat beteiligt sind, zu untersuchen, wurden teils bereits vorhandene Konstrukte zur Überexpression und Deletion bestimmter Gene eingesetzt. Als Grundlage für diese Versuche wurden zwei Ausgangsstämme gewählt, die durch Deletion von drei Genen des Valin-Pantothenat-Biosynthesewegs bereits eine erhöhte
Valinproduktion im Vergleich zum Wildtyp von C. glutamicum aufwiesen. Diese
Stämme werden nachfolgend als Val1 (C. glutamicum 13032∆ilvA) und Val2
(13032∆ilvA∆panBC) bezeichnet. Das Gen ilvA codiert für eine ThreoninDehydratase, die aus Threonin Oxobutyrat und Pyruvat bildet, das anschließend in
mehreren Schritten zu Isoleucin umgesetzt wird. Dieselben Enzyme, die an der
Isoleucinbiosynthese beteiligt sind, sind auch für die Umsetzung von Pyruvat zu
Valin zuständig. Ab einer Konzentration von 5 mM Isoleucin im Medium wird die
Threonin-Dehydratase reprimiert und alle nachfolgenden Gene nicht mehr expri-
74
ERGEBNISSE
miert, was zu einer geringeren Valinausbeute führen würde (Morbach et al., 1995).
Durch die Deletion des ersten Gens im Isoleucinbiosyntheseweg wird die Bildung
von Isoleucin verhindert, wodurch die Hemmung der Expression der Gene des
Isoleucin- und Valinbiosynthesewegs unterdrückt und somit eine höhere Valinsynthese bewirkt wird. Weiterhin steht ein Molekül Pyruvat mehr zur Valinproduktion zur Verfügung. Der Stamm Val1 diente vorwiegend zur Untersuchung des Einflusses auf die Pantothenatproduktion.
Im zweiten Stamm sind zusätzlich zum ilvA-Gen noch die zwei Gene panB und
panC des Pantothenatbiosynthesewegs deletiert. PanB kodiert für eine Ketopantoat-Hydroxymethyl-Transferase, die Ketoisovalerat, das während der Valinbiosynthese entsteht, zu Ketopantoat umsetzt. Das panC-Gen kodiert für eine Pantothenat-Synthetase, die im letzten Schritt des Pantothenatbiosynthesewegs Pantothenat aus Pantoat und β-Alanin synthetisiert. Durch die Deletion dieser Gene
wird die Pantothenatbiosynthese unterbrochen, das anfallende Ketoisovalerat wird
nicht aus dem Valinbiosyntheseweg abgezogen und die Valinproduktion kann gesteigert werden. Außerdem steht weniger bzw. gar kein CoA, das aus Pantothenat
gebildet wird, für den PDHC zur Verfügung. Dieser Stamm wurde zur Untersuchung der Valinsynthese eingesetzt.
Zur Überprüfung der Bedeutung verschiedener Enzyme des anaplerotischen Knotenpunkts auf den Einfluss der Valin- und Pantothenatbiosynthese wurden Konstrukte zur Überexpression des PEP-Carboxylase-Gens (pMFα-ppc), des PyruvatCarboxylase-Gens (pVWEx1-pyc), des PEP-Carboxykinase-Gens (pEK0-pck), des
Pyruvat-Chinon-Oxidoreduktase-Gens
(PXT-poxB-Ex)
und
des
Lipoamid-
Dehydrogenase-Gens (pEK0-lpdA), die bereits vorlagen, in die Stämme Val1 und
Val2 transformiert. Da auch insbesondere der Pyruvat-Kinase eine besondere Bedeutung bei der Beeinflussung des Pyruvat-Pools zugeschrieben wurde, wurde
das Pyruvat-Kinase-Gen (pyk-Gen) in den Vektor pEKEx2 kloniert. Die Überprüfung der Stämme erfolgte durch die Isolierung und entsprechenden Restriktionsverdaus der Plasmide sowie durch Aktivitätstests der einzelnen Enzyme (Tabelle
11). Des weiteren wurde im Stamm Val1 das pyc-Gen und im Stamm Val2 das
pck-Gen deletiert. Beide Stämme wurden mittels PCR auf ihre Richtigkeit kontrolliert. Aus Tabelle 11 geht hervor, dass alle Stämme höhere spezifische Aktivitäten
aufwiesen und somit alle Konstrukte in den Valinstämmen funktionell waren.
75
ERGEBNISSE
Tabelle 11:
spezifischen
Kontrolle der rekombinanten Stämme durch die Bestimmung der
Aktivitäten
der
PEP-Carboxylase
(Val1/2-ppc),
der
PEP-
Carboxykinase (Val1/2-pck), der Pyruvat-Carboxylase (Val1/2-pyc), der PyruvatChinon-Oxidoreduktase (Val1/2-pox), der Pyruvat-Kinase (Val1/2-pyk) und der
Dihydrolipoamid-Dehydrogenase (Val1/2-lpdA).
C. glutamicum Stämme
spez. Aktivität [U/ml]
Val1
Val1-ppc
0,045
0,690
Val2
0,049
Val2-ppc
0,590
Val1
0,039
Val1-pck
0,830
Val2
0,065
Val2-pck
1,010
Val1
1,980
Val1-pyc
6,600
Val2
1,690
Val2-pyc
5,590
Val1
0,037
Val1-pox
0,066
Val2
0,035
Val2-pox
0,046
Val1
0,570
Val1-pyk
1,600
Val2
0,64
Val2-pyk
2,30
Val1
0,11
Val1-lpdA
2,50
Val2
0,25
1,08
Val2-lpdA
ERGEBNISSE
76
Im Stamm Val2, der das Plasmid pEK0-lpdA zur Überexpression des LipoamidDehydrogenase-Gens trägt, wurden zusätzlich die spezifischen PDHC-Aktivitäten
ermittelt. Aus einer früheren Arbeit (J. Schwinde, Doktorarbeit) war bekannt, dass
bei Überexpression dieses Gens sowohl die PDHC- als auch die OGDHC-Aktivität
um etwa 50 % erniedrigt war. Da der PDHC durch seine Funktion zur oxidativen
Decarboxylierung von Pyruvat eine bedeutende Rolle im PEP-Pyruvat-Oxalacetat-Knotenpunkt und zur Vorstufenbereitstellung von Pyruvat spielt, könnte durch
die Erniedrigung der PDHC ein Einfluss auf die Valinsynthese erwirkt werden. Die
Bestimmung der PDHC-Aktivitäten in den Stämmen Val1 und Val2 zeigten bei
Überexpression des lpdA-Gens eine bis zu 20-fach geringere spezifische Aktivität
im Vergleich zu den jeweiligen Ausgangstämmen.
5.2
Optimierung der Fermentationsbedingungen
Um die für die Valinsynthese geeigneten Fermentationsbedingungen einzustellen,
wurde der Einfluss der extern zugesetzten Parameter Isoleucin, Biotin und Pantothenat auf die Valinbildung untersucht. Die Kultivierung der Stämme Val1 und
Val2 erfolgte hierfür in CgC-Medium mit 4% Glukose.
Isoleucin gehört zu den verzweigtkettigen Aminosäuren, die häufig an der Regulation von Stoffwechselenzymen durch Rückhemmungs-Mechanismen beteiligt sind
(Morbach et al., 1995; Eggeling et al., 1987). Es wurde daher untersucht, ob eine
Modifikation der Isoleucinkonzentration im Bereich von 0,25 bis 1,0 mM Isoleucin
Auswirkungen auf das Wachstumsverhalten oder die Valinbildung, z. B. durch Beeinflussung eines oder mehrerer zur Valinproduktion benötigter Synthese-Enzyme,
hat. Bei variierenden Isoleucinkonzentration im Fermentationsexperiment war die
Biotinkonzentration standardmäßig 0,2 mg/l.
Da C. glutamicum nicht zur Biotin-Synthese fähig ist und zwei Biotin-abhängige
Enzyme (Pyruvat-Carboxylase und Acetyl-CoA Carboxylase) besitzt (Jäger et al.,
1996; Peters-Wendisch et al., 1997), die möglicherweise bei Variierung der Biotinkonzentration einen Einfluss auf die Valinproduktion haben, wurden Biotinkonzentrationen im Bereich von 0,1 bis 0,4 mg/l in die Fermentationsexperimente
eingesetzt und die Valinbildung untersucht. Bei variierenden Biotinkonzentrationen
wurde 1 mM Isoleucin eingesetzt.
77
ERGEBNISSE
Es wurden Wachstumskurven aufgenommen und die Valinwerte aus Überständen
von Kulturen nach 48 h mittels HPLC bestimmt. Da sich die Stämme Val1 und
Val2 bezüglich des Wachstumsverhaltens bei verschiedenen Isoleucinkonzentrationen nicht unterschiedlich verhielten, ist in Abbildung 15 exemplarisch das
Wachstumsverhalten für den Stamm Val2 dargestellt. Aus dieser Abbildung geht
hervor, dass eine Reduktion der Isoleucinkonzentration von 1,0 mM , die standardmäßig eingesetzt wurden, auf bis zu 0,25 mM keine negativen Effekte auf das
Wachstum zeigte.
Aus Abbildung 16 wird das Wachstumsverhalten der Stämme Val1 und Val2 bei
variierenden Biotinkonzentrationen ersichtlich. Veränderte Konzentrationen an
Biotin führten, wie aus der Abbildung 16 hervorgeht, zu keinen limitierenden bzw.
verbesserten Wachstumsbedingungen. Die End-OD-Werte des Stammes Val2
sind durchgehend höher, was auch bei weiteren Versuchen bestätigt werden
konnte.
100
OD600
10
1
0,1
0
10
20
30
40
50
60
Zeit [h]
Val2 1.0
Val2 0.75
Val2 0.5
Val2 0.25
Abbildung 15: Wachstum des Stammes Val2 bei Isoleucinkonzentrationen im
Bereich von 0,25-1,0 mM.
78
ERGEBNISSE
100
OD600
10
1
0,1
0
10
Val1 0.2
Val2 0.2
20
30
Zeit [h]
Val1 0.1
Val2 0.1
40
50
60
Val1 0.4
Val2 0.4
Abbildung 16: Wachstum der Stämme Val1 und Val2 bei Biotinkonzentrationen
im Bereich von 0,1-0,4 mg/l.
Tabelle 12 zeigt die ermittelten Valinwerte nach 48 h. Die Werte in Tabelle 12
zeigen keine signifikanten Veränderungen der Valinproduktion bei variierenden
Isoleucinkonzentrationen. Daher wurde in allen folgenden Experimenten eine Isoleucinkonzentration von 1 mM gewählt. Für den Stamm Val1 bzw. Val2 konnte bei
Wachstum auf 0,4 mg Biotin/l eine um 25 % bzw. 30 % verringerte Valinkonzentration vergleichsweise zum Wachstum auf 0,1 mg Biotin/l gezeigt werden. Daher
wurden nachfolgende Versuche mit einer Biotinkonzentration von 0,1 mg/l durchgeführt.
79
ERGEBNISSE
Tabelle 12: Valinbildung nach Wachstum auf unterschiedlichen Isoleucin- bzw.
Biotinkonzentrationen
Stamm
c [mM] nach 48 h
Val1 mit 1,0 mM Isoleucin
8,72
Val1 mit 0,75 mM Isoleucin
9,07
Val1 mit 0,5 mM Isoleucin
8,49
Val1 mit 0,25 mM Isoleucin
8,7
Val2 mit 1,0 mM Isoleucin
8,83
Val2 mit 0,75 mM Isoleucin
8,99
Val2 mit 0,5 mM Isoleucin
8,21
Val2 mit 0,25 mM Isoleucin
7,62
Val1 mit 0,1 mg/l Biotin
8,97
Val1 mit 0,4 mg/l Biotin
6,86
Val2 mit 0,1 mg/l Biotin
8,93
Val2 mit 0,4 mg/l Biotin
6,31
Weiterhin wurden Fermentationsversuche zur Untersuchung des Wachstumsverhaltens und der Valinproduktion bei unterschiedlichen Pantothenatkonzentrationen
durchgeführt. Hierfür wurde wiederum CgC-Medium mit 4% Glukose verwendet,
dem 0,1 mg Biotin/l und 1 mM Isoleucin zugesetzt wurden. Die eingesetzten
Pantothenatkonzentrationen variierten im Bereich von 0,1-5 µM. Eine Limitierung
des Wachstums konnte dabei nicht festgestellt werden, die ermittelten Valinwerte
sind in Tabelle 13 zusammenfassend dargestellt. Daraus geht hervor, dass bei
steigender Pantothenatkonzentration die Konzentration an Valin abnimmt. Bei erhöhter Pantothenatkonzentration kann mehr CoA, das aus Pantothenat synthetisiert wird, gebildet werden. Dadurch steht mehr CoA für die PDHC zur Verfügung,
wodurch möglicherweise mehr Pyruvat zu Acetyl-CoA oxidiert wird und daher ein
verringerter Pyruvat-Pool zur Valinbildung vorhanden ist. Bei weniger Pantothe-
80
ERGEBNISSE
Tabelle 13: Ermittelte Valinwerte nach 24 und 48 h bei Wachstum auf unterschiedlichen Pantothenatkonzentrationen.
Pantothenatkonzentration [mM]
c [Valin] in mM
nach 24 h
c [Valin] in mM
nach 48 h
0,0001
0,0003
0,0007
0,0010
0,0020
0,0050
9,790
10,34
8,400
8,690
5,640
4,420
9,830
10,47
8,570
8,750
5,700
4,480
nat im Medium würde man daher eine gesteigerte Produktion an Valin erwarten,
da weniger Pyruvat zu Acetyl-CoA oxidiert wird und somit ein höherer PyruvatPool zur Valinbildung bereitsteht.
5.3
Valinsekretion rekombinanter C. glutamicum-Stämme
Um die rekombinanten Stämme hinsichtlich ihrer Produktion von Valin zu untersuchen, wurden diese auf CgC-Medium mit den unter 5.2 vorgestellten Bedingungen
auf 4 % Glukose in einer 50 ml ´batch´-Kultur für 48 h fermentiert. Nach 48 h wurden Proben zur Analyse von Valin mittels HPLC entnommen. Aus Abbildung 17
gehen die für die Ausgangs- und rekombinanten Stämme aus mehreren unabhängigen Fermentationen ermittelten Werte hervor.
Wachstumsversuche zeigten
dasselbe Wachstumsverhalten für alle untersuchten Stämme.
Durch die zusätzlichen Deletionen in den Genen panBC ist die gebildete Menge
an Valin im Stamm Val2 mit 9 mM um 22 % höher als in Stamm Val1 (7,01
mM). Die Überexpression der Gene ppc, pyc und pck in den Stämmen Val1 und
Val2 hatten keinen Einfluss auf die Valinproduktion. Der Stamm Val1 zeigte bei
Überexpression des pyk-Gens eine um 100 % erhöhte Valinkonzentration (14,58
mM gegenüber 7,01 mM beim Ausgangsstamm). Dies konnte bei Überexpression
dieses Gens im Stamm Val2 nicht bestätigt werden. Die Überexpression des pqoGens im Stamm Val1 führte bei allen durchgeführten Fermentationen zu einer
Verminderung der Valinmenge um bis zu 53 % (3,03 mM), während im Stamm
81
ERGEBNISSE
18
16
14
Valin [mM]
12
10
8
6
4
2
0
Val1
Val1-ppc
Val1-pyc
Val1-pck
Val1-pyk
Val1-pqo
Val1-lpdA
Val1 delta
pyc
Val2
V al2-ppc
Val2-pyc
V al2-pck
V al2-pyk
Val2-pqo
Val2-lpdA
Val2 delta
pck
12
10
Valin [mM]
8
6
4
2
0
Abbildung 17: Valinbildung der Stämme Val1 und Val2 in mM nach 48 h bei Überexpression der Gene ppc, pck, pyc, pqo, pyk und lpdA, sowie bei Deletion von
pyc und pck.
82
ERGEBNISSE
Val2 die Reduktion des Valinertrages mit 23% (6,9 mM gegenüber 8,93 mM im
Stamm Val2) deutlich geringer ausfiel. In den Stämmen Val1 und Val2 wurde nach
Überexpression des lpdA-Gens kein verändertes Verhalten in der Valinproduktion
detektiert. Weiterhin konnte bei Deletion des pyc-Gens in Val1 eine um 56 % erhöhte Valinkonzentration (12,4 mM) nach 48 h Fermentation gemessen werden,
während die Deletion in Val2 zu keiner Veränderung führte.
Das Wachstumsverhalten aller rekombinanter Stämme entsprach dem der Kontrollstämme.
5.4
Pantothenatsekretion rekombinanter C. glutamicum-Stämme
Zur Untersuchung der Pantothenatproduktion von C. glutamicum 13032 wurden
die im Stamm Val1 hergestellten rekombinanten Stämme herangezogen. Diese
wurden in einer ´batch´-Kultur auf CgC-Medium mit 4 % Glukose fermentiert. Hierbei war es nötig, die Kulturen mit 20 mM β-Alanin, das zur Herstellung von
Pantothenat notwendig ist, zu supplementieren. Generell wird Pantothenat aus
4,5
y = 0,0385x
4
3,5
OD600
3
2,5
2
1,5
1
0,5
0
0
20
40
60
80
100
120
D-Pantothenat [ng/ml]
Abbildung 18: Kalibriergerade im Bereich von 0-100 ng Pantothenat/ml zur Ermittlung der Pantothenatkonzentration aus Kulturüberständen.
83
ERGEBNISSE
einem Molekül Pyruvat und einem Molekül Oxalacetat, aus dem β-Alanin gebildet
wird, synthetisiert.
Das System zur Bestimmung von Pantothenat basiert auf einem biologischen
Testsystem mit einer im panC-Gen nicht mehr funktionstüchtigen Integrationsmutante (Sahm und Eggeling, 1999). Um die gebildeten Mengen Pantothenat zu
bestimmen, war die Erstellung einer Kalibriergerade im Bereich von 0-100 ng Pantothenat/ml notwendig (Abbildung 18). Anhand der abgebildeten Kalibriergeraden
wurden nun alle rekombinanten Stämme untersucht. Dazu wurden nach 24 und 48
h Proben entnommenen und diese in geeigneter Verdünnung und sterilfiltriert in
das biologische Testsystem eingesetzt. Die Messung der optischen Dichte erfolgte
nach 48 h. In Abbildung 19 ist nur die Pantothenatproduktion der Stämme dargestellt, bei denen durch Überexpression bestimmter Gene eine Pantothenatbil-
80
70
Pantothenat [mg/l]
60
50
40
30
20
10
0
Val1
Val1ppc
Val1pck
Val1pqo
Val1-pJC1 Val1-pECM3
ilvBNCD
ilvBNCD
Abbildung 19: Pantothenatproduktion in rekombinanten Stämmen nach 48 h
Fermentation in CgC-Medium mit 4 % Glukose; die Konzentrationen sind in mg
Pantothenat/l angegeben.
ERGEBNISSE
84
dung erwirkt werden konnte. Generell untersucht wurden die Effekte der Überexpression der Gene ppc, pyc, pck, pyk und lpdA im Stamm Val1 sowie der Effekt
Bei Deletion des pyc-Gens. Als Positivkontrolle dienten die Stämme C. glutamicum∆ilvA pJC1ilvBCND und C. glutamicum∆ilvA pECM3ilvBCND, die Plasmide
zur Überexpression der Gene ilvBNCD des Valin-Pantothenat-Biosynthese-Wegs
tragen und sich in der Resistenz der Plasmide unterscheiden. Als direkter Kontrollstamm diente der Stamm Val1.
Aus Abbildung 19 geht hervor, dass drei der getesteten rekombinanten Stämme
eine gesteigerte Pantothenatproduktion aufwiesen. Der Ausgangsstamm Val1 bildete nach 48 h Fermentation kein Pantothenat. Wie sich herausstellte lag die Produktion nach 96 h Fermentation bei 0,7 mg Pantothenat/l. Die Positivkontrollstämme bildeten beide Pantothenat zwischen 50 und 55 mg/l. Eine Pantothenatbildung durch gesteigerte spezifische Aktivitäten von Enzymen des PyruvatOxalacetat-Knotenpunkts konnte bei Überexpression der Gene ppc (9,8 mg/l), pck
(11,4 mg/l) und pqo (15,98 mg/l) erzielt werden. Die Überexpression der Gene
pyk und lpd, sowie die Deletion des pyc-Gens hatten keinen Einfluss auf die Produktion von Pantothenat.
6.
Valinproduktion und Wachstumsverhalten von Deletionsmutanten im Gen aceE
Der PDHC spielt eine bedeutende Rolle in der Pyruvatbereitstellung. Es konnte
bereits gezeigt werden, dass eine durch zufällige Mutagenese hergestellte Mutante mit geringerer Pyruvat-Dehydrogenase-Aktivität eine gesteigerte Lysinproduktion aufwies (Shiio et al., 1984). Des weiteren konnte von M. Schreiner ein Konstrukt zur Deletion des aceE-Gens hergestellt und dieses Gen im C. glutamicum
Wildtyp deletiert werden. Dabei stellt sich heraus, dass die Deletion von aceE in C.
glutamicum zu einem vollständigen Verlust der PDHC-Aktivität führte und dieser
Stamm nicht mehr in der Lage war, auf Glukose als alleiniger Kohlenstoffquelle zu
wachsen (M. Schreiner, persönliche Mitteilung). Da die PDHC einen für die Pyruvatbereitstellung möglicherweise bedeutenden Schritt katalysiert, sollte das aceEGen in bereits Valin-ausscheidenden Stämmen deletiert werden. Als Ausgangs-
85
ERGEBNISSE
stämme zur Untersuchung der Valinproduktion wurden hierfür die Stämme Val2,
C. glutamicum ilvN/M13 und C. glutamicum∆panB ilvN/M13 gewählt. Das Gen ilvN
kodiert in C. glutamicum für eine Acetohydroxy-Säure-Synthase, die durch die
verzweigtkettigen Aminosäuren Leucin, Isoleucin und Valin gehemmt wird (Morbach et al., 2000). Da eine Feedback-Hemmung der Enzyme bzw. Gene des Valinbiosynthesewegs sich negativ auf die Valinproduktion auswirken würde, wurde
der Stamm C. glutamicum ilvN/M13 konstruiert, der im Promotorbereich durch
PCR mutagenisiert wurde und auf die Feedback-Hemmung durch die verzweigtkettigen AS nicht mehr reagiert (V. Elisakova, persönliche Mitteilung).
Die Kontrolle dieser Stämme erfolgte sowohl durch PCR als auch durch
´Southern-Blot´-Experimente (Abbildung 20) und Enzymtests.
6.1
Nachweise
der
Deletion
des
aceE-Gens
im
Chromosom
von
C. glutamicum
Zur Kontrolle aller im Gen aceE deletierten Stämme wurde chromosomale DNA
von diesen wie auch den Referenzstämmen Val2, C. glutamicum ilvN/M13 und
C. glutamicum∆panB ilvN/M13 isoliert, mit SalI verdaut und die DNA über Nacht
auf einem 0,8 %-igen Agarosegel aufgetrennt. Nachdem die DNA auf eine Membran transferiert wurde, erfolgte die Hybridisierung mit einer Digoxigenin-markierten
580 bp großen Sonde. Diese wurde über PCR mit den Primern aceE1 und aceE3
hergestellt und umfasste einen sowohl im C. glutamicum Wildtyp als auch in den
Stämmen mit deletierten aceE-Gen vorhandenen Bereich. Für die Stämme mit
deletiertem aceE-Gen wurde ein um 2077 bp verkürztes Fragment erwartet. Das
Ergebnis des ´Southern-Blots´ ist in Abbildung 20 dargestellt.
Das erwartete Wiltypfragment wies eine Größe von 8270 bp auf, während für die
Deletionsmutanten mit 6193 bp das aceE-Gen erwartungsgemäß um 2077 bp verkürzt war.
86
ERGEBNISSE
1
2
3
4
5
6
8,2 kb
6,2 kb
Abbildung 20: ´Southern-Blot´-Analyse mit chromosomaler DNA von C. glutamicum-Stämmen mit deletiertem aceE-Gen; als Kontrolle dienten die jeweiligen Ausgangsstämme. In den Spuren 1+2 ist die DNA der Stämme C. glutamicum
∆ilvA∆panBC und ∆ilvA∆panBC∆aceE aufgetragen, in den Spuren 3+4 die der
Stämme ilvN/M13 und ilvN/M13∆aceE und in den Spuren 5+6 die der Stämme
∆ilvA∆panB ilvN/M13 und ∆ilvA∆panB ilvN/M13∆aceE.
6.2
Pyruvat-Dehydrogenase-
und
Pyruvat-Decarboxylase-Aktivität
in
C. glutamicum Stämmen mit deletiertem aceE-Gen
Im Wildtyp konnte bereits gezeigt werden, dass die Deletion des aceE-Gens zu
einem vollständigen Verlust der PDHC- und PDC-Aktivität führt (M. Schreiner,
persönliche Mitteilung). Dies sollte nun in allen zur Untersuchung der Valinproduktion hergestellten Stämmen untersucht werden, wobei der Wildtyp als Kontrolle
diente. Dazu wurden Zellen auf Komplexmedium gezüchtet, in der exponentiellen
Wachstumsphase geerntet und nach Aufschluss im RiboLyser auf Aktivität untersucht. Die erhaltenen Werte sind in Tabelle 14 dargestellt.
87
ERGEBNISSE
Tabelle 14 Spezifische Aktivitäten von PDHC und PDC bei Stämmen mit deletiertem aceE-Gen im Vergleich zum Wildtyp nach Wachstum auf Komplexmedium.
Stamm
spez. PDHC-Aktivität
[U/mg]
spez. PDC-Aktivität
[U/mg]
C. glutamicum 13032
C. glutamicum∆aceE
40
<0.01
16.5
<0.01
Val2
38.73
14.62
Val2∆aceE
<0.01
<0.01
C. glutamicum ilvN/M13
46.11
23.1
C. glutamicum ilvN/M13∆aceE
<0.01
<0.01
C. glutamicum∆ilvA
∆panB ilvN/M13
C. glutamicum∆ilvA
∆panB ilvN/M13∆aceE
39.51
20.56
<0.01
<0.01
In allen Ausgangsstämmen konnten PDHC-Aktivitäten im Bereich von 38 bis 46
mU/mg Protein gemessen werden. Alle im aceE-Gen deletierten Stämme zeigten
keine Aktivität mehr. Das gleiche Bild ergibt sich für die spezifischen PDCAktivitäten, die in den Kontrollstämmen eine Aktivität von 14 bis 23 mU/mg Protein aufwiesen, während bei den Deletionsmutanten keine PDC-Aktivität mehr
messbar waren.
6.3
Wachstumsverhalten
und
Valinproduktion
rekombinanter
Valin-
stämme
Alle im aceE Gen deletierten Stämme wurden bezüglich ihres Wachstumsverhaltens und der Valinproduktion untersucht. Vom Wildtyp C. glutamicum ∆aceE war
bekannt, dass dieser ohne Supplementation mit Acetat nicht auf GlukoseMinimalmedium wachsen kann (M. Schreiner, persönliche Mitteilung). Daher wurden alle Stämme mit 1 % Acetat und 1 % Glukose kultiviert. Als Kontrolle der
Wachstumsexperimente wurde einerseits der im aceE-Gen deletierte
Wildtyp-
stamm, andererseits die im aceE-Gen deletierten Valin-ausscheidenden Stämme
mit und ohne Acetat-Supplementation untersucht. Bislang war es, abgesehen
vom Wildtypstamm, lediglich möglich, den Stamm Val2 und Val2∆aceE
88
ERGEBNISSE
100
OD600
Val2 1% Ac
Val2 delta aceE 0% Ac
Val2 delta aceE 1% Ac
10
1
0
5
10
15
20
25
30
Zeit [h]
Abbildung 21: Vergleichende Wachstumsexperimente der Stämme Val2 und
Val2∆aceE auf Minimalmedium mit 1 % Glukose und 1 % Acetat.
bezüglich des Wachstumsverhaltens und der Valinproduktion zu analysieren, da
die beiden Stämme C. glutamicum ilvN/M13∆aceE und C. glutamicum∆ilvA∆panB
ilvN/M13∆aceE nicht zum Wachstum gebracht werden konnten.
Wie aus Abbildung 21 deutlich wird, ist der Stamm Val2 ohne Deletion bis zu
einer OD600 von 23,8 nach
Val2∆aceE
wies
eine
24 h gewachsen.
maximale
OD600
von
Der Stamm
C. glutamicum
11,2
Ohne
auf.
Acetat-
Supplementation war kein Wachstum zu beobachten.
Vom Stamm Val2 und dem Wildtyp C. glutamicum 13032 mit und ohne deletiertem
aceE-Gen wurden mittels HPLC-Analyse die Valinkonzentrationen nach Wachstum auf Minimalmedium mit 1 % Acetat und 1 % Glukose aus Kulturüberständen
nach 24 h untersucht. Aus Tabelle 15 geht hervor, dass in beiden im aceE-Gen
deletierten Stämmen eine Valinbestimmung möglich war. So konnte bei dem Valin-produzierenden Stamm Val2 eine Steigerung um 47 % von 6,9 mM auf 13,1
mM nachgewiesen werden, was auf die OD600 bezogen einer 4-fachen Steigerung
entspricht. Auch beim Wildtyp, der unter normalen Bedingungen kein Valin produ-
89
ERGEBNISSE
Tabelle 15: Messung der Valinkonzentrationen in den Stämmen C. glutamicum
13032, C. glutamicum 13032∆aceE, Val2 und Val2∆aceE.
Stamm
OD600 [24 h]
Valin [mM 24 h]
Valin/OD600
C. glutamicum 13032
C. glutamicum 13032∆aceE
27,0
10,2
0,0
5,4
0,0
0,5
Val2
Val2∆aceE
23,8
10,7
6,9
13,1
0,3
1,2
ziert, konnte nach Deletion von aceE eine Valinkonzentration von 5,4 mM nach 24
Stunden ermittelt werden.
90
DISKUSSION
IV.
Diskussion
1.
Charakterisierung des PDHC von C. glutamicum 13032
Der PDHC stellt in C. glutamicum einen von insgesamt drei Enzyme-Komplexen
dar, die zur oxidativen Decarboxylierung befähigt sind. Neben dem PDHC, der
Pyruvat zu Acetyl-CoA umsetzt, existiert der OGDHC, der 2-Oxoglutarat zu Succinyl-CoA
katalysiert,
und
der
Verzweigtkettige-Aminosäure-Dehydrogenase-
Komplex (VADHC), der die verzweigtkettigen Aminosäuren Leucin, Isoleucin und
Valin zu den entsprechenden Acyl-CoAs katalysiert. Im Allgemeinen besitzen
PDHCs eine molekulare Masse von 0,7 bis 10 MDa, wobei sie in Bakterien in der
Regel durch allosterische Mechanismen oder Endprodukthemmung reguliert werden. In Eukaryoten sind an dieser Regulation zusätzliche Proteine beteiligt (Patel
et al., 1996). Notwendige katalytische und stöchiometrische Cofaktoren für den
Ablauf der PDHC-Reaktion sind TPP, Lipoamid, FAD, CoA und NAD+. Diese
Komponenten waren Hauptbestandteil der enzymatischen Messungen des PDHC
(Guest und Creaghan, 1972), der in C. glutamicum die höchste Aktivität in der exponentiellen Wachstumsphase und bei Wachstum auf Komplexmedium mit 0,106
U/mg Protein aufwies. Um 43 bis 54 % geringere Aktivitäten wurden nach Wachstum auf Minimalmedium mit den Kohlenstoffquellen Glukose, Acetat oder GlukoseAcetat-Mischsubstrat gemessen. Eine Reduktion der PDHC-Aktivität nach Wachstum auf Acetat konnte für den Organismus Pseudomonas aeruginosa gezeigt
werden, wobei die spezifische PDHC-Aktivität im Gegensatz zu derjenigen von C.
glutamicum nach Wachstum auf Glukose-Acetat-Mischsubstrat 6-mal höher war
als auf Acetat als alleiniger Kohlenstoffquelle (Jeyasselan und Guest, 1980a). Da
der PDHC bei Wachstum auf Glukose, nicht aber bei Wachstum auf Acetat notwendig ist, sind die erhöhten Aktivitäten nach Wachstum auf Glukose-AcetatMischsubstrat im Vergleich zu Acetat physiologisch sinnvoll. Für C. glutamicum
waren die ermittelten Aktivitäten nach Wachstum auf Minimalmedium bei allen
Kohlenstoffquellen etwa gleich hoch. Die spezifischen Aktivitäten der PDHC von
C. glutamicum lagen im Bereich von 0,06 bis 0,11 U/mg Protein nach Wachstum
auf Komplexmedium. Im Vergleich zu anderen Organismen sind diese Aktivitäten
relativ gering. Sehr niedrige spezifische Aktivitäten von 0,01 bis 0,039 U/mg Protein wurden für den Organismus Zymomonas mobilis und im mitochondrialen PDHC
des Maises gemessen (Neveling et al., 1998b; Thelen et al., 1998). Sehr hohe
DISKUSSION
91
PDHC-Aktivitäten in Zellextrakten konnte dagegen für den Organismus Azotobacter vinelandii mit 7 U/mg Protein nachgewiesen werden (Bresters et al., 1975),
während die größte Anzahl im Mittelfeld mit 0,05 bis 0,66 U/mg Protein bei den
Organismen P. aeruginosa, B. subtilis, E. coli oder Enterococcus faecalis lag
(Jeyaseelan et al., 1980b; Perham und Lowe, 1988; Visser und Strating, 1982;
Snoep, 1991).
Im Gegensatz zum PDHC von C. glutamicum, der aufgrund sehr geringer spezifischer Aktivitäten und der Empfindlichkeit gegenüber verschiedenen Salzen (NaCl,
NH2SO4 oder KCl) nicht gereinigt werden konnte, war die Reinigung und biochemische Charakterisierung in anderen Organismen wie P. aeruginosa oder Z. mobilis erfolgreich (Jeyaseelan et al., 1980b; Neveling et al., 1998). Eine Empfindlichkeit gegenüber Salzen konnte, abgesehen für den PDHC von C. glutamicum, auch
für den mitochondrialen PDHC des Maises gezeigt werden, der partiell gereinigt
wurde, jedoch bei Konzentrationen von mehr als 50 mM NaCl reduzierte PDHCAktivitäten zeigte (Thelen et al., 1998). Da eine Reinigung des PDHC in C. glutamicum nicht möglich war, wurde der Ks-Wert für Pyruvat in Zellextrakten ermittelt.
Hintergrund für die Ks-Wert-Bestimmung war, dass in C. glutamicum ein als Pyruvat-Chinon-Oxidoreduktase (PQO) bezeichnetes Enzym existiert, dass Pyruvat zu
Acetat umsetzt. Die PQO wurde gereinigt, biochemisch charakterisiert und der KmWert für Pyruvat bestimmt, der mit 30 mM sehr hoch war und eine sehr geringe
Affinität besitzt (M. Schreiner, persönliche Mitteilung). Da es sich bei der PQO
wahrscheinlich um ein in der Stationärphase aktives Protein handelt und der
PDHC im Gegensatz dazu in der exponentiellen Wachstumsphase hoch aktiv ist,
könnte ein physiologischer Zusammenhang der beiden Enzyme bestehen. Die
Bestimmung des Ks-Wertes für Pyruvat in C. glutamicum ergab in zwei unabhängigen Messungen Ks-Werte von 1,88 und 2,6 mM. Damit besitzt der PDHC eine
mehr als 10-fach höhere Affinität für Pyruvat im Vergleich zur PQO, die jedoch
immer noch geringer als die für andere Organismen ermittelten Km-Werte sind. Es
konnten bei anderen Organismen von diversen gereinigten PDHCs Km-Werte für
Pyruvat ermittelt für z. B. E. coli, A. vinelandii oder B. subtilis mit 0,3 mM (Bisswanger, 1980; Bresters et al., 1975; Perham und Lowe, 1988). Etwas höher lag
der Km-Wert mit 0,7 mM für E. faecalis unter anaeroben Bedingungen (Snoep,
1991).
DISKUSSION
92
Für die E1-Untereinheit des PDHC, die vom aceE-Gen kodiert wird, wurde im
Rahmen dieser Arbeit ein Enzymtest zum Nachweis spezifischer Enzymaktivitäten
etabliert. Generell liegt diese Untereinheit des PDHC in Gram-negativen Organismen als Homodimer, in Gram-positiven als Heterodimer vor (Neveling et al.,
1998a). C. glutamicum als Gram-positiver Organismus stellt mit einer homodimeren PDC eine Ausnahme dar. Durch die Bestimmung spezifischer PDC-Aktivitäten
auf unterschiedlichen Kohlenstoffquellen konnten maximale Aktivitäten von 53
mU/mg Protein bei Zellextrakten nach Wachstum auf Komplexmedium (mit und
ohne Glukose) nachgewiesen werden. Die Aktivitäten auf Minimalmedium mit Glukose lagen bei 24 mU/mg Protein, auf Minimalmedium mit Acetat oder GlukoseAcetat-Mischsubstrat waren keine Aktivitäten bestimmbar. In P. aeruginosa wurde
auf Glukose-Acetat-Mischsubstrat eine 3-fach höhere Aktivität der PDC gemessen
als auf Minimalmedium mit Acetat (Jeyaseelan et al., 1980a). Generell konnten für
E1-Untereinheiten anderer Organismen höhere Aktivitäten gemessen werden als
für C. glutamicum. Sehr hohe Aktivitäten der PDC zeigte E. coli K12 mit 27 U/mg
Protein (Bisswanger et al., 1981) gegenüber 1,51 U/mg Protein bei einem anderen
E. coli-Stamm (Guest und Stephens, 1980). Außerdem zeigte dieser Stamm eine
Abhängigkeit der PDC-Aktivität von der Pyruvat-Konzentration mit gereinigtem
Enzym (Guest und Stephens, 1980). Eine Abhängigkeit von Pyruvat in Zellextrakten konnte für C. glutamicum nicht gezeigt werden, da die spezifischen PDCAktivitäten bei Variierung der Pyruvatkonzentration unverändert blieben. Die Messung einer spezifischen PDC-Aktivität in E. faecalis war im Gegensatz zu E. coli
und C. glutamicum nicht möglich (Snoep, 1991).
Eine Besonderheit stellt in C. glutamicum die DHL, die vom aceF-Gen kodiert wird,
dar. Auf Ebene der Aminosäuresequenz (AS) konnte durch Sequenzvergleiche
gezeigt werden, dass dieses Gen sowohl zu E2-Untereinheiten von PDHCs als
auch von OGDHCs anderer Organismen Ähnlichkeiten aufweist. So konnten Identitäten der AS-Sequenz der DHL von C. glutamicum zu E2-Untereinheiten von
PDHCs der Organismen E. coli, B. subtilis und A. vinelandii von 22,4 bis 31,4 %
nachgewiesen werden (Stephens et al., 1983; Hemila et al., 1990; Hanemaaijer et
al., 1988). Der Vergleich der DHL-AS-Sequenz zu E2-Unterheiten der OGDHCs
derselben Organismen zeigte Identitäten von 22,9 bis 25,9 % (Spencer et al.,
1984; Carlsson und Hederstedt, 1989; Westphal und de Kok, 1990).
DISKUSSION
93
Eine Motivsuche in der DHL-AS-Sequenz zeigte das Vorhandensein von drei Lipoamid-Bindedomänen. Diese sind immer am N-Terminus der E2-Untereinheiten
von PDHCs oder OGDHCs lokalisiert. Die E2-Untereinheiten von OGDHCs oder
VADHCs besitzen immer eine Lipoamid-Bindedomäne, während die Anzahl an
Lipoamid-Bindedomänen in PDHCs von eins bis drei variiert (Zhu und Peterskofsky, 1996). Organismen mit einer Bindedomäne sind z. B. Z. mobilis, B.
subtilis oder B. stearothermophilus, wobei Z. mobilis als einziger Vertreter eine
weitere Lipoamid-Bindedomäne in der E1-Untereinheit besitzt (Neveling et al.,
1999; Hemila et al., 1990; Borges et al., 1990). Zwei Lipoamid-Bindedomänen existieren z. B. in P. aeruginosa oder S. faecalis (Rae et al., 1997; Allen und
Perham, 1991). Die Organismen E. coli und A. vinelandii wiesen die höchsten Identitäten zu der AS-Sequenz von C. glutamicum auf und besitzen, wie auch C.
glutamicum, beide drei Lipoamid-Bindedomänen (Stephens et al., 1983; Hanemaaijer et al., 1988). Die Identitäten der Domänen untereinander sind im Allgemeinen sehr hoch, was auch für C. glutamicum gezeigt werden konnte. Zwei der
Domänen stimmen zu 100 % überein. Letztendlich ist der physiologische Sinn dieser unterschiedlichen Anzahl an Lipoamid-Bindedomänen noch nicht geklärt. Für
E. coli konnte ein direkter Zusammenhang der Anzahl an Lipoamid-Bindedomänen
zur Wachstumsrate, die mit steigender Anzahl von eins bis drei Bindedomänen bei
Wachstum auf Glukose ebenfalls stieg, gezeigt werden (Dave et al., 1995). Um die
genaue Funktion der von aceF kodierten E2-Untereinheit aus C. glutamicum für
die Multienzym-Komplexe PDH und OGDH zu klären und Hinweise zu erlangen,
ob das Genprodukt des aceF-Gens als E2-Untereinheit beider Enzym-Komplexe
fungiert, müsste eine Mutante in diesem Gen hergestellt und die Effekte auf die
entsprechenden Enzym-Aktivitäten und das Wachstumsverhalten untersucht werden. Bislang war es jedoch nur möglich, Kolonien zu erlangen, die auch nach
Supplementation mit Acetat und Succinat nicht weiter kultivierbar waren.
Auch für die DHL von C. glutamicum wurde im Rahmen dieser Arbeit ein Enzymtest etabliert und die spezifischen Aktivitäten auf unterschiedlichen Kohlenstoffquellen gemessen. Die höchsten spezifischen Aktivitäten konnten auch hier auf
Komplexmedium ohne zusätzliche Kohlenstoffquelle gezeigt werden. Stark erniedrigt waren die spezifischen Aktivitäten nach Wachstum auf Komplexmedium mit
Glukose und auf Minimalmedium mit Glukose, Acetat oder Glukose-AcetatMischsubstrat. Die maximal ermittelten Aktivitäten für C. glutamicum lagen bei
94
DISKUSSION
0,233 U/mg Protein, was im Vergleich zu den spezifischen Aktivitäten des PDHC
und der PDC detektierten Aktivitäten hoch ist. In anderen Organismen wurden ebenfalls DHL-Aktivitäten untersucht, wobei spezifischen Aktivitäten für E. faecalis
von 0,7 U/mg Protein, für P. aeruginosa von 0,49 U/mg Protein und für E. coli von
6,69 U/mg Protein ermittelt wurden (Snoep, 1991; Jeyaseelan und Guest, 1980b;
Guest und Stephens, 1980). Während für C. glutamicum gleich hohe Aktivitäten
auf Glukose und Glukose-Acetat-Mischsubstrat gezeigt wurden, war bei P. aeruginosa die spezifische Aktivität der DHL nach Wachstum auf Glukose-AcetatMischsubstrat 4-fach erhöht im Vergleich zu den Aktivitäten nach Wachstum auf
Acetat (Jeyaseelan und Guest, 1980a).
Die Etablierung des Enzymtests für die DHL zeigte, dass eine stabile und reproduzierbare Messung nur bei einer Konzentration von 0,1 mM CoA im Test möglich
war. Die Messung erfolgte in nicht-physiologischer Richtung durch photometrische
Bestimmung des durch die DHL gebildeten S-Acetyldihydrolipoamids. Verläuft die
Reaktion in physiologischer Richtung, wird dieses S-Acetyldihydrolipoamid mit
CoA durch die DHL zu Acetyl-CoA und Dihydrolipoamid umgesetzt (Schwartz und
Reed, 1969). Eine mögliche Erklärung für eine instabile Messung bei höheren
CoA-Konzentrationen als 0,1 mM könnte sein, dass sowohl die nichtphysiologische als auch die physiologische Reaktion gleichzeitig stattfinden, wodurch sich die Aktivität in nicht-physiologischer Richtung verringert.
2.
Transkriptionsstudien zu den Genen aceE und aceF von C.
glutamicum 13032
Um Hinweise auf die Transkriptgröße und die Regulation der Gene aceE und aceF
zu gewinnen, wurden ´Northern-Blot´- Experimente, Promotorfusionsstudien und
´Primer-Extension´-Analysen
durchgeführt.
Dabei
konnte
mit
radioaktiven
´Northern-Blot´-Experimenten für das aceE-Gen eine Transkriptgröße von 2,9 kb
und für das aceF-Gen eine Transkriptgröße von 2 kb ermittelt werden, was auf
eine monocistronische Organisation dieser Gene hinweist. Das für die dritte Untereinheit der PDHC kodierende lpdA-Gen konnte bereits in früheren Studien von
Schwinde et al. (2001) auf transkriptioneller Ebene untersucht werden und wird
ebenfalls monocistronisch transkribiert, mit einer Transkriptgröße von 1,45 kb. Die
DISKUSSION
95
Organisation dieser Gene stellt in C. glutamicum eine vollkommen andere dar als
in allen anderen bislang charakterisierten Organismen. Für Gram-positive Bakterien wie B. stearothermophilus und andere Vertreter gilt im Allgemeinen, dass die
kodierenden Gene pdhAα und β, pdhB und lpd geclustert vorliegen (Borges et al.,
1990; Hawkins et al., 1990), wobei das für die dritte Untereinheit der Komplexe
PDH und OGDH kodierende lpd-Gen entweder auf dem Gencluster pdhA-pdhB (z.
B. bei E. coli) oder odhA-odhB (z. B. bei A. vinelandii) lokalisiert ist (Spencer und
Guest, 1985; Westphal und de Kok, 1988). Für E. coli als Gram-negativen Vertreter, bei denen die Gene in der Regel mit aceE, aceF und lpd bezeichnet werden,
konnte ein Transkript von mehr als 7 kb nachgewiesen werden, was auf eine polycistronische Organisation dieser 3 Gene hinwies. Zusätzlich konnte mit der lpdSonde ein kleineres Transkript nachgewiesen werden, dass der Größe des lpdGens entsprach (Spencer und Guest, 1985), woraus geschlossen wurde, dass
dieses Gen für mehrere Enzymkomplexe benötigt wird und daher separat transkribiert werden muss. In A. vinelandii und P. aeruginosa liegt das lpd-Gen nicht im
Cluster mit den für die PDHC oder OGDHC kodierenden Genen vor (Hanemaaijer
et al., 1988; Westphal und de Kok, 1988; Rae et al., 1997). Die für den PDHCkodierenden Gene in C. glutamicum liegen weder geclustert noch in räumlicher
Nähe auf dem Chromosom vor. Z. mobilis ist der einzige bislang beschriebene
Organismus, bei dem die Gene pdhA und pdhB nicht geclustert vorliegen und die
Gene pdhB und lpd durch einen ORF unbekannter Funktion voneinander getrennt
sind (Neveling et al., 1998a). Eine weitere Besonderheit zeigt die Genorganisation
von T. ferrooxidans, bei dem die Gene pdhB und lpd zu einem Gen fusioniert sind
(Powles et al., 1997).
Die Untersuchung von corynebakterieller RNA unter Verwendung von ´NorthernBlot´-Experimenten zeigte bei der Analyse gleicher Mengen RNA aus Komplexund Minimalmedium eine höhere Transkriptmenge der Gene aceE und aceF auf
Komplexmedium. Da diese Ergebnisse mit den spezifischen Enzymaktivitäten des
PDHC auf den entsprechenden Kohlenstoffquellen korrelieren, kann daraus geschlossen werden, dass die Gene aceE und aceF kohlenstoffabhängig reguliert
werden.
Um diese Ergebnisse zu bestätigen, wurden die Promotorregionen beider Gene
durch Messung spezifischer Reportergen-Aktivitäten (CAT) wachstumsabhängig
DISKUSSION
96
und auf unterschiedlichen Kohlenstoffquellen untersucht. Die spezifischen Aktivitäten im Wachstumsverlauf stiegen bis zur späten exponentiellen Phase an, woraus
geschlossen werden kann, dass die Gene aceE und aceF wachstumsabhängig
reguliert werden. Auf unterschiedlichen Kohlenstoffquellen waren die höchsten
Aktivitäten auf Komplexmedium und Minimalmedium mit Glukose detektierbar,
während auf Komplexmedium mit Glukose und Minimalmedium mit Acetat bzw.
Mischsubstrat die spezifischen Aktivitäten geringer waren. Weiterhin waren die
CAT-Aktivitäten, die für den Promotor des aceF-Gens gemessen wurden um den
Faktor zwei geringer als diejenigen, die für den des aceE-Gens ermittelt wurden.
Am besten untersucht ist die Regulation der für den PDHC kodierenden Gene in
E. coli, in dem diese durch einen von pdhR kodierten Regulator kontrolliert werden. Dieser Regulator befindet sich stromaufwärts des aceE-Gens und bildet ein
gemeinsames Operon mit den Genen aceE-aceF-lpd, wobei pdhR zusätzlich einen eigenen Promotor besitzt. Es wurden die Reportergen-Aktivitäten des pdhRPromotors und des Promotors der Gene aceE-aceF nach Wachstum auf Glukose,
Pyruvat und Succinat untersucht. Dabei wurden sehr geringe Aktivitäten für den
Promotor der Gene aceE und aceF allein gemessen, wohingegen ein starker Aktivitätsanstieg beobachtet wurde bei Ermittlung der pdhR-Promotor-Aktivität. Weiterhin konnten auf Pyruvatmedium 3- bis 4-fach höhere Aktivitäten festgestellt
werden als auf Glukose oder Succinat (Quail et al., 1994). Von Kaiser und Sawers
konnte 1994 gezeigt werden, dass der PDHC von E. coli durch Pyruvat induziert
und teilweise durch Glukose, Acetat und Intermediaten des Tricarbonsäure-Zyklus
gehemmt wird. Ein pdhR-analoges Protein konnte bisher in C. glutamicum nicht
identifiziert werden. Weiterhin gibt es Transkriptionsstudien zu den Promotoren
der Gene aceA und aceB (analog zu den Genen aceE und aceF in C. glutamicum)
aus P. aeruginosa. Diese wurden auf Komplexmedium mit den Kohlenstoffquellen
Glukose, Pyruvat, Succinat und Acetat durchgeführt und in den Stämmen P. aeruginosa und E. coli untersucht, wobei in beiden Stämmen Aktivitäten gemessen
werden konnten. Die höchste Aktivität existierte, wie auch für C. glutamicum, auf
Glukosemedium und die geringste auf Acetat. Außerdem waren die für aceB ermittelten Promotor-Aktivitäten, wie es dies auch für C. glutamicum gezeigt wurde,
um 50 % geringer als die des aceA-Gens (Jeremy et al., 1997). Die Promotoraktivitäten sind herunterreguliert durch Acetat, was mit der auf Acetat gemessenen
geringeren PDHC-Aktivität korreliert (Jeyasselan und Guest, 1980). Für Z. mobilis
DISKUSSION
97
konnte hingegen im Gegensatz zu C. glutamicum eine für das pdhB-Gen 6-fach
höhere Promotoraktivität im Vergleich zum pdhA-Gen festgestellt werden (Neveling et al., 1999). Die für C. glutamicum erzielten Ergebnisse deuten auf eine
wachstums- und kohlenstoffabhängige Regulation der Gene aceE und aceF, da
die Promotor-Aktivitäten am höchsten in der späten exponentiellen Wachstumsphase und nach Wachstum auf Glukose sind. Da der PDHC bei Wachstum auf
Glukose eine Rolle spielt, während bei Wachstum auf Acetat die Enzyme AcetatKinase, Phosphotransacetylase und die Glyoxylat-Zyklus-Enzyme Malat-Synthase
und Isocitrat-Lyase eine Rolle spielen, entspricht dieses Ergebnis den Erwartungen.
Untersuchungen der Promotorbereiche der Gene aceE und aceF zeigten zwei potentielle Transkrtiptionsstartpunkte für das aceE-Gen, während für das aceF-Gen
reproduzierbar das gleiche Signal detektiert werden konnte. Für einen der potentiellen Transkriptionsstartpunkte des aceE-Gens (Signal 1) konnte die –10-Region
mit der Sequenz TATCCT abgeleitet werden. Diese Sequenz stimmt mit der für
das lpdA-Gen ermittelten –10-Region überein (Schwinde et al., 2001), konnte jedoch nur mit einem Primer nachgewiesen werden. Die zweite mögliche –10Region, die durch zwei unterschiedliche Primer bestätigt werden konnte, führte zu
der –10-Region AAGAAT (Signal 2). Diese –10-Region konnte bislang für keinen
anderen Promotor in C. glutamicum ermittelt werden. Dasselbe gilt für die für das
aceF-Gen gezeigte –10-Region mit der Sequenz TTGAAT. Die Untersuchung von
288 Promotoren von E. coli (Harley und Reynolds, 1987) und 236 Promotoren von
B. subtilis (Helmann, 1995) führte zu sehr stark konservierten -10- bzw. –35Regionen mit den Sequenzen TATAAT bzw. TTGACA. Durch die Untersuchung
von 33 corynebakteriellen Promotoren in einer früheren Studie (Patek et al., 1996)
und 20 weiteren (Patek et al., 2003) konnte auch für C. glutamicum eine Konsensussequenz für die –10-Region festgelegt werden (TA(T/C)AAT), die in 4 Basen
mit den für aceE und aceF postulierten –10-Regionen übereinstimmt. Sehr gut
untersucht in C. glutamicum ist die Promotorregion des dapA-Gens, das für eine
Dihydropicolinat-Synthase kodiert (Vasicova et al., 1999). Durch das Einfügen von
Mutationen im Promotorbereich und die Messung von Reportergen-Aktivitäten
wurde gezeigt, dass Mutationen in der –35-Region lediglich zu einer schwachen
Reduktion der Aktivität führten, woraus geschlossen wurde, dass diese Region für
DISKUSSION
98
die Transkription in C. glutamicum weniger von Bedeutung für die Erkennung der
RNA-Polymerase zu sein scheint als in anderen Organismen (Vasicova et al.,
1999; Patek et al., 2003). Diese Tatsache erklärt auch die geringe Übereinstimmung der hier ermittelten potentiellen –35-Regionen für die Gene aceE und aceF,
die in maximal zwei Basen mit der Konsensussequenz TTGCCA übereinstimmen
(Patek et al., 2003). Weiterhin konnte in 26 von insgesamt 33 untersuchten
Transkriptionsstartpunkten in C. glutamicum ein Adenin oder Guanin als
Transkriptionsstartpunkt nachgewiesen werden (Patek et al., 1996). Dies stimmt
sowohl mit den für die Gene aceE und aceF als auch für die in z. B. E. coli oder
Milchsäurebakterien ermittelten Transkriptionsstartpunkte überein (Hawley und Mc
Clure, 1983; Van der Vossen et al., 1987).
Die beiden möglichen Transkriptionsstartpunkte für das aceE-Gen weisen darauf
hin, dass ein Signal wahrscheinlich aufgrund von Sekundärstrukturen, die zum
vorzeitigen Abbruch der Reversen Transkriptase geführt haben könnte, zustande
kam. Generell ist es dennoch nicht auszuschließen, dass dieses Gen zwei TS besitzt. Es konnte sowohl für z. B. das dapA-Gen in C. glutamicum (Patek et al.,
1996) als auch für Gene von E. coli und Streptomyces schon mehrere TS nachgewiesen werden (Hawley und Mc Clure, 1983; Strohl, 1992).
Von großer Bedeutung für die RNA-Polymerase in C. glutamicum, besonders bei
schwacher Ausprägung der –35-Region scheinen Guanine 2 bp stromauf- und
stromabwärts der –10-Region zu sein (Vasicova et al., 1999). Bei den hier untersuchten Genen konnte für alle potentiellen –10-Regionen ein Guanin 2 bp downstream der –10-Region gezeigt werden.
3.
Valin- und Pantothenatproduktion mit C. glutamicum 13032
Die industrielle Produktion von Aminosäuren liegt aktuell bei einer jährlichen Gesamtproduktion von 2 Millionen Tonnen und unterliegt einer jährlichen Wachstumsrate von 10 %, wobei die Aminosäuren in verschiedenen Bereichen wie der
Nahrungsmittel- und pharmazeutischen Industrie oder als Futtermittelzusatz zum
Einsatz kommen (Hermann, 2003). Während das Potential von C. glutamicum zur
Glutamat- und Lysinproduktion bereits in den 50-iger Jahren erkannt wurde und
Glutamat seitdem in industriellem Maßstab produziert wird (Kinoshita et al., 1957),
DISKUSSION
99
erlangen heute die verzweigtkettigen Aminosäuren Leucin, Isoleucin und Valin z.
B. als Zusatz zu Infusionslösungen immer größere Bedeutung. Bislang sehr gut
untersucht ist in C. glutamicum die Biosynthese von Isoleucin, das in 10 Schritten
aus Aspartat synthetisiert wird, wobei die letzten 4 Enzyme zusätzlich an der Bildung von Leucin, Valin und Pantothenat beteiligt sind (Umbarger, 1987). Isoleucin
gehört zusammen mit den Aminosäuren Lysin, Threonin und Methionin zur Aspartat-Familie (Eggeling et al., 1987). Da eine Steigerung der Isoleucinakkumulation
durch Änderung von lediglich einer Enzym-Aktivität wie z. B. der HomoserinDehydrogenase nicht möglich war (Eikmanns et al., 1991b), wurden rekombinante
Stämme mit hohen Kopienzahlen unterschiedlicher Synthese-Enzyme hergestellt.
Isoleucinkonzentrationen von 90 mM konnten z. B. durch Einfügen vieler Kopien
eines Feedback-resistenten Homoserin-Dehydrogenase-Gens bei gleichzeitiger
Erhöhung der Kopienzahl des Homoserin-Kinase-Gens und eines Feedbackresistenten Threonin-Dehydratase-Gens erzielt werden (Morbach et al., 1995).
Des weiteren wurde gezeigt, dass eine nicht durch Isoleucin-hemmbare ThreoninDehydratase von E. coli (kodiert vom tdcB-Gen) in C. glutamicum zu einer 50fachen Überproduktion von Isoleucin führte (Guillouet et al., 1999). Die ThreoninDehydratase (kodiert vom ilvA-Gen) leitet ausgehend von Threonin die Isoleucinbiosynthese ein und wird ab einer intrazellulären Isoleucinkonzentration von 2 mM
nahezu vollständig gehemmt (Morbach et al., 1995), was dazu führt, dass alle
nachfolgenden Gene, deren Genprodukte Reaktionen der Isoleucin- und Valinbiosynthese katalysieren, nicht mehr transkribiert. Da diese Rückhemmung negative
Auswirkungen auf die Valinbildung hätte, wurde das ilvA-Gen deletiert. Dadurch
steht außerdem steht ein Molekül Pyruvat zusätzlich zur Valinakkumulation zur
Verfügung. Das durch die Threonin-Dehydratase gebildete Oxobutyrat wird mit
Pyruvat im Isoleucinbiosyntheseweg durch das Enzym Acetohydroxy-SäureSynthase (AHAS) zu Acetohydroxy-Säure umgesetzt. Gleichzeitig katalysiert die
AHAS im Valinbiosyntheseweg die Reaktion von Pyruvat zu Aceto-Laktat. Da die
Affinität der AHAS für Oxobutyrat 2-fach erhöht ist im Vergleich zu der für Pyruvat,
kann der Fluss durch das nicht gebildete Oxobutyrat in der ilvA-Deletionsmutante
zusätzlich in Richtung Valin gelenkt werden (Eggeling et al., 1987). Der C. glutamicum-Stamm mit deletiertem ilvA-Gen ist einer der in dieser Arbeit verwendeten
Stämme (Val1). Außerdem wurde der Stamm Val2, der zusätzliche Deletionen in
den Genen panB und panC des Pantothenatbiosynthesewegs trägt, verwendet
100
DISKUSSION
(Radmacher et al., 2002). Es konnte gezeigt werden, dass dieser Stamm bis zu
20,5 mM Valin nach 48 Stunden akkumuliert. Bei zusätzlicher Überexpression der
Gene ilvBNCD war eine weitere Steigerung von bis zu 92 mM möglich (Radmacher et al., 2002).
In der vorliegenden Arbeit wurden rekombinante Stämme mit veränderten Aktivitäten in den Enzymen des PEP-Pyruvat-Oxalacetat-Knotenpunkts hergestellt, um
den Einfluss auf die Valinbildung zu untersuchen. Bereits im Jahr 1971 wurden
von Kisumi et al. Valin-ausscheidende Stämme des Organismus S. marcescens
beschrieben. Erste Versuche zur Steigerung der Valinsynthese in C. glutamicum
wurden von Burger (persönliche Mitteilung) und Radmacher et al. (2002) unternommen. Eine Deletion im ppc-Gen führte weder zu signifikanten Änderungen des
Pyruvat-Pools noch zu höheren Valin- und Alaninkonzentrationen (Burger, persönliche Mitteilung). Ein weiterer Versuch zur Produktionssteigerung sollte indirekt
durch die Beeinflussung des PDHC bewirkt werden. Von E. coli war bekannt, dass
ein Liponsäuremangel zu einer reduzierten PDHC-Aktivität führt (Yokota et al.,
1994). C. glutamicum besitzt drei Gene der Liponsäure-Biosynthese (lipA, lipB,
lplA), wobei nach Deletion der Gene lipB und lplA keine gesteigerte Valinkonzentration nachgewiesen wurde (Radmacher, persönliche Mitteilung). Eine Deletion im ilvA-Gen (Val1) führte dagegen zu einer Valinausscheidung von 7 mM im
Vergleich zu 1 mM beim Wildtyp (Sahm und Eggeling, 1999).
Bevor die rekombinanten Stämme bezüglich des Wachstums- und Produktionsverhalten untersucht wurden, mussten die Fermentationsbedingungen richtig gewählt werden. Dazu wurden Biotin, Isoleucin und Pantothenat, die zur Supplementation der Stämme Val1 bzw. Val2 zugesetzt werden mussten, im Konzentrationsbereich variiert und das Wachstums- und Produktionsverhalten untersucht.
Eine 2-fache Erhöhung der standardmäßig eingesetzten Biotinkonzentration führte
zu einer um 30 % erniedrigten Valinakkumulation. In C. glutamicum existieren die
zwei
Biotin-abhängigen
Enzyme
Pyruvat-Carboxylase
und
Acetyl-CoA-
Carboxylase (Peters-Wendisch et al., 1997; Jäger et al., 1996). Es konnte nachgewiesen werden, dass bei einer Biotinkonzentration von 100 µg im Vergleich zu 5
µg eine höhere Pyruvat-Carboxylase-Aktivität detektierbar war (Peters-Wendisch
et al., 1997). Daher könnte die geringere Valinproduktion auf eine gesteigerte Py-
DISKUSSION
101
ruvat-Carboxylase-Aktivität, die möglicherweise einen erniedrigten Pyruvat-Pool
zur Folge hat, zurückgeführt werden.
Eine Variation der Isoleucinkonzentration hatte keinen Einfluss auf das Wachstumsverhalten und die Valinsynthese. Um die Aminosäure Isoleucin, die von den
Stämmen Val1 und Val2 nicht selbst synthetisiert werden kann, in ausreichendem
Maße zur Verfügung zu stellen, wurde eine Isoleucinkonzentration von 1 mM in
die Fermentationsversuche eingesetzt. Von Lange et al. (2003) konnte gezeigt
werden, dass bei hohen Valinakkumulationen das Wachstumsverhalten von
C. glutamicum eingeschränkt war. Dies war auf den von den Aminosäuren Isoleucin und Valin gemeinsam genutzten Transporter BrnQ zurückzuführen, der bei
hohen Valinkonzentrationen nicht mehr fähig war, ausreichend Isoleucin in die
Zelle zu transportieren. Da die gebildeten Valinkonzentrationen mit in dieser Arbeit
verwendeten Stämme nicht so hoch waren, dass eine Konkurrenz der beiden Aminosäuren zustande kam, war eine Isoleucinkonzentration von 1 mM ausreichend.
Die Steigerung der Pantothenatkonzentration von 0,1 bis 5 µg Pantothenat im
Fermentationsexperiment führte zu einer Verminderung der Valinkonzentration um
50 %. Da der PDHC ein CoA-abhängiger Enzym-Komplex ist, und der Cofaktor
CoA bei Säugern und E. coli in 4 Schritten aus Pantothenat gebildet wird (Brown,
1959), kann der PDHC durch Variierung der Pantothenatkonzentration indirekt
beeinflusst werden. Durch die Steigerung des Pantothenats konnte mehr CoA gebildet werden, woraus wahrscheinlich eine erhöhte PDHC-Aktivität resultierte, die
das für die Valinbildung nötige Pyruvat zu Acetyl-CoA decarboxylierte. Diese Ergebnisse sprechen dafür, dass der PDHC möglicherweise ein gutes Ziel-Enzym
zur Steigerung der Valinkonzentration sein könnte.
Nachdem die Fermentationsbedingungen festgelegt waren, wurden die rekombinanten Stämme bezüglich des Wachstumsverhalten und der Valinproduktion untersucht werden. Es wurden die Stämme Val1 und Val2 bei Überexpression der
Gene ppc, pck, pyc, pyk, lpdA und pqo untersucht. Des weiteren wurde eine pycDeletionsmutante im Stamm Val1 und eine pck-Deletionsmutante im Stamm Val2
analysiert. Keiner der Stämme zeigte ein verändertes Wachstumsverhalten. Eine
Reduktion des gebildeten Valins um 30 % konnte in den Stämmen Val1 und Val2
bei Überexpression des pqo-Gens gezeigt werden. Weiterhin konnte eine Steige-
DISKUSSION
102
rung um 56 % bei Deletion des pyc-Gens im Stamm Val1 nachgewiesen werden.
Eine um 100 % erhöhte Valinproduktion ließ sich reproduzierbar für den Stamm
Val1 bei Überexpression des pyk-Gens zeigen, die jedoch für den Stamm Val2
nicht bestätigt werden konnte. Zur Bildung von Valin sind 2 Moleküle Pyruvat
notwendig. Eine erhöhte Valinsynthese wurde daher erwartet, wenn eine Änderung der Enzym-Aktivität theoretisch zu einem höheren Pyruvat-Pool führen sollte,
und eine verringerte Valinsynthese bei Änderungen der Enzym-Aktivitäten, die den
Pyruvat-Pool senken sollten. Die Überexpression der Gene pyk, pck und die Deletion im pyc-Gen könnten daher zu einer Steigerung des Pyruvat-Pools führen,
während die Überexpression der Gene pyc, ppc, pqo und die Deletion im pck-Gen
zu einer geringeren Valinsynthese führen sollte. Diese theoretischen Grundlagen
konnten bei Überexpression des pqo- und Deletion des pyc-Gens im Stamm Val1
bestätigt werden. Es konnte in dieser und einer vorangegangene Arbeit (J.
Schwinde, Doktorarbeit) gezeigt werden, dass eine Überexpression des lpdAGens zu einer bis zu 20-fachen Reduktion des PDHC führte. Durch die Überexpression dieses Gens sollte eine Drosselung der PDHC bewirkt werden, womit
theoretisch eine Erhöhung des Pyruvat-Pools und eine Steigerung der Valinkonzentration möglich wäre. Eine Produktionssteigerung konnte bei Überexpression
des lpdA-Gens in den Stämmen Val1 und Val2 jedoch nicht gezeigt werden. Einige der hier untersuchten Gene, die für Enzyme des PEP-Pyruvat-OxalacetatKnotenpunkts kodieren, wurden bereits im Zusammenhang mit der Produktion
anderer Aminosäuren untersucht. Die Herstellung von Integrationsmutante im ppcGen C. glutamicum Stämme nach Wachstum auf Minimalmedium mit Glukose resultierte weder in verändertem Wachstumsverhalten, noch in einer veränderten
Lysinproduktion (Gubler et al., 1994a), während die Inaktivierung des pyk-Gens zu
einer signifikanten Reduktion der Lysinkonzentration bei gleichbleibendem Wachstumsverhalten führte (Peters-Wendisch et al., 1993; Gubler et al., 1994b). Durch
Deletion des pck-Gens konnte eine signifikante Steigerung der Glutamat- und Lysinproduktion, bei Überexpression eine Verminderung der beiden genannten Aminosäuren nachgewiesen werden (Riedel et al., 2001). Die Überexpression des
pyc-Gens hatte eine signifikante Akkumulation der Aminosäuren Glutamat und
Lysin zur Folge, wohingegen die Deletion zu einer Verminderung führte (PetersWendisch et al., 2001). Die hier erhaltenen und früher durchgeführten Untersuchungen der Enzym-Aktivitäten des PEP-Pyruvat-Oxalacetat-Knotenpunkts zei-
DISKUSSION
103
gen, dass der Pyruvat-Pool nicht unbedingt durch die Variierung einer EnzymAktivität verändert werden kann. Eine Erklärung dafür, dass die theoretische Erhöhung des Oxalacetat- und Pyruvat-Pools nicht unbedingt zu einer tatsächlichen
Erhöhung führt, wurde von Petersen et al. (2001) gegeben, der eine Korrelation
zwischen der intrazellulär vorhandenen Menge an Pyruvat zu der von Oxalacetat
zeigte. Eine mögliche Erhöhung des Oxalacetat- bzw. Pyruvat-Pool könnte somit
einen erhöhten ´futile cycle´ zur Folge haben, durch den das gebildete Oxalacetat
bzw. Pyruvat über z. B. glukoneogenetische oder anaplerotische Enzyme wieder
zu Pyruvat bzw. Oxalacetat umgesetzt wird.
Bereits 1984 wurde von Shiio et al. gezeigt, dass durch Reduktion der PDHCAktivität und der wahrscheinlich damit verbundenen Erhöhung des Pyruvat-Pools
eine Steigerung der Lysinproduktion bewirkt werden konnte. Um mögliche Effekte
des PDHC auf die Valinproduktion zu untersuchen, wurde das für die E1-Unterheit
des PDHC kodierende aceE-Gen in unterschiedlichen, Valin-ausscheidenden
Stämmen deletiert und die Effekte auf das Wachstumsverhalten und die Valinproduktion untersucht. Dazu wurden die Stämme Val2, C. glutamicum ilvN/M13 und
C. glutamicum∆panB ilvN/M13 verwendet. Die beiden zuletzt genannten Stämme
sind besonders interessant für die Produktion von Valin aufgrund einer Mutation
im Promotorbereich des ilvN-Gens. Das ilvN-Gen kodiert für eine Untereinheit der
AHAS und wird durch die verzweigtkettigen Aminosäuren Leucin, Isoleucin und
Valin gehemmt. Durch die Mutation im Promotorbereich des ilvN-Gens ist diese
Hemmung aufgehoben. Die Deletion des aceE-Gens war in allen Stämmen möglich und konnte mittels ´Southern-Blots´ bestätigt werden. In allen aceE-deletierten
Stämmen wurde keine PDHC- und PDC-Aktivität mehr gemessen, während die
Ausgangsstämme die entsprechenden Aktivitäten aufwiesen. Durch das bereits im
Wildtyp deletierte aceE-Gen war bekannt (M. Schreiner, persönliche Mitteilung),
dass eine Supplementation mit Acetat zum Wachstum notwendig ist. Dies konnte
in früheren Studien bereits für die Stämme E. coli, P. aeruginosa und S. typhimurium bei Deletion des aceE-Gens gezeigt werden (Henning et al., 1966; Jeyasselan
und Guest, 1980; Langley und Guest, 1974). Durch Acetatsupplementation war
ein Wachstum der Stämme C. glutamicum ilvN/M13 und C. glutamicum∆panB
ilvN/M13 jedoch nicht möglich. Möglicherweise würde eine Supplementation mit
einem anderen Tricarbonsäureintermediat wie Glutamat oder Succinat ein Wachs-
DISKUSSION
tum der Stämme ermöglichen. Die End-OD der Stämme C. glutamicum
104
∆aceE
und Val2∆aceE nach Wachstum auf Glukose-Acetat-Mischsubstrat war erniedrigt
im Vergleich zu den jeweiligen Ausgangsstämmen. Nach einer Fermentationszeit
von 24 Stunden konnte beim Wildtyp C. glutamicum nach Deletion des aceE-Gens
eine Valinakkumulation von 5,4 mM gezeigt werden. Im Gegensatz dazu wurde
beim Wildtyp kein Valin im Medium nachgewiesen. Für den Stamm Val2 konnte
nach Deletion eine Steigerung der Valinkonzentration von 6,9 mM auf 13,1 mM
gezeigt werden. Diese Ergebnisse lassen die Vermutung zu, dass die Stämme C.
glutamicum ilvN/M13 bzw. C. glutamicum∆panB ilvN/M13, die bereits 20 bzw. 100
mM Valin produzieren (V. Elisakova, persönliche Mitteilung), in der Valinsynthese
durch Bereitstellung eines höheren Pyruvat-Pools noch weiter gesteigert werden
könnten. Zukünftige Versuche sollten sich auf die Etablierung des Wachstums
dieser Stämme konzentrieren.
Das Hauptinteresse der Industrie an der Produktion von Vitaminen liegt vorwiegend in der Nutzung als Nahrung- und Futtermittelzusatz sowie als Zusatz zu
pharmazeutischen und kosmetischen Produkten. Die Produktion von Pantothenat
erfolgt größtenteils chemisch (Vandamme, 1992) oder durch Fermentation mit z.B.
E. coli (Moriya et al., 1997). In E. coli erfolgt die Bildung von Pantothenat wie auch
in C. glutamicum in 4 Schritten aus α-Ketoisovalerat und Aspartat (Jackowski et
al., 1996; Sahm und Eggeling, 1999; Dusch et al., 1999; Merkamm et al., 2003).
In dieser Arbeit sollte die Pantothenatproduktion durch veränderte EnzymAktivitäten des PEP-Pyruvat-Oxalacetat-Knotenpunkts optimiert werden. Als Ausgangsstamm diente der im ilvA-Gen deletierte Stamm Val1, der laut Literatur 236
nM Pantothenat nach 24 h Wachstum bildet (Sahm und Eggeling, 1999). Trägt
dieser Stamm das Plasmid pJC1ilvBNCD wurde eine Pantothenatproduktion von
530 nM nach 49 h beobachtet, eine weitere Steigerung auf 4,2 mM war durch zusätzliche Überexpression der Gene panBC auf 4,2 mM möglich (Sahm und Eggeling, 1999). Außerdem konnte gezeigt werden, dass extern zugesetztes β-Alanin,
das zur Pantothenatproduktion notwendig ist, zu einer Produktionssteigerung führte. In dieser Arbeit konnte bei Überexpression der Gene ppc, pck und pqo Pantothenat in Kulturüberständen nachgewiesen werden, während für den Ausgangsstamm Val1 nach 48 Stunden Fermentation kein Pantothenat ermittelt werden
konnte. Überraschend war sowohl die Pantothenatproduktion bei Überexpression
DISKUSSION
105
des pqo-Gens, das zu einer gesteigerten Aktivität der Pyruvat-ChinonOxidoreduktase und damit theoretisch zu einem verringerten Pyruvat-Pool führt,
als auch die Tatsache, dass bei Überexpression des ppc- und pck-Gens, deren
Genprodukte die gegenläufige Reaktion katalysieren, etwa dieselben Mengen
Pantothenat messbar waren. Die letztgenannte Beobachtung könnte wiederum
darin begründet sein, dass das Verhältnis Pyruvat zu Oxalacetat in der Zelle konstant gehalten wird und damit die gleiche Menge an Pantothenat bei Überexpression der Gene ppc und pck gebildet wird (Petersen et al., 2001). Die PyruvatChinon-Oxidoreduktase katalysiert die Umsetzung von Pyruvat zu Acetat und führt
somit zu einer Abnahme des für die Bildung von Pantothenat nötigen PyruvatPools. Theoretisch sollte demnach keine Pantothenatproduktion möglich sein.
Möglich wäre jedoch, dass der Fluss durch den Tricarbonsäure- bzw. GlyoxylatZyklus durch die erhöhte Menge an Acetat gesteigert und das über die PyruvatChinon-Oxidoreduktase gebildete Acetat sofort wieder verstoffwechselt wird.
Erste Erfolge zur Bildung von Pantothenat konnte in dieser Arbeit durch Überexpression von Genen des anaplerotischen Knotenpunkts verzeichnet werden. Um
allerdings eine industrielle Nutzung von C. glutamicum für diesen Zweck anzustreben, müssten noch sehr viele weitere Untersuchungen durchgeführt werden, um
E. coli mit einer 1000-fach höheren Produktionsmenge an Pantothenat als in C.
glutamicum abzulösen oder mit diesem zu konkurrieren.
106
ZUSAMMENFASSUNG
V.
Zusammenfassung
Der PEP-Pyruvat-Oxalacetat-Knotenpunkt in C. glutamicum spielt eine essentielle
Rolle bei der Bereitstellung von Vorläuferprodukten für die Produktion von
Aminosäuren und Vitaminen. Daher ist es von Interesse, die Gene und Genprodukte der daran beteiligten Reaktionen zu charakterisieren und zu verstehen. Ein
Enzym dieses Knotenpunkts, der Pyruvat-Dehydrogenase-Komplex (PDHC), wurde bislang nur wenig untersucht. Dieser Komplex sollte in der vorliegenden Arbeit
auf enzymatischer und transkriptioneller Ebene beschrieben und der Einfluss dieses und anderer Enzyme des anaplerotischen Knotenpunkts bezüglich der Produktion von Valin und Pantothenat analysiert werden.
1.)
Der PDHC zeigte nach Wachstum auf Komplexmedium in der exponentiellen Wachstumsphase mit 106 mU/mg Protein die höchsten spezifischen Aktivitäten. Nach Wachstum auf Minimalmedium mit Glukose, Acetat oder
Glukose-Acetat-Mischsubstrat waren die Aktivitäten um etwa 50 % erniedrigt. Der Ks-Wert für den PDHC für Pyruvat lag bei 2,2 mM.
2.)
Die E2-Untereinheit (Dihydrolipoamid-Transacetylase) des PDHC von C.
glutamicum wird vom aceF-Gen kodiert, das durch Sequenzvergleiche identifiziert wurde. Das abgeleitete Protein besteht aus 675 Aminosäuren mit einer vorhergesagten molekularen Masse von 70,9 kDa. Die AminosäureSequenz der DHL zeigte sowohl Identitäten zu den E2-Untereinheiten des
PDHCs als auch des OGDHCs anderer Organismen mit drei LipoamidBinde-Domänen, wobei die Identität zur E. coli-DHL mit 31,4 % am höchsten war.
3.)
Sowohl für die E2- als auch
für die E1-Untereinheit (Pyruvat-
Decarboxylase), die vom aceE-Gen kodiert wird, wurden Enzymtests etabliert. Die höchsten PDC-Aktivitäten wurden auf Komplexmedium und Komplexmedium mit Glukose detektiert, wohingegen auf Minimalmedium mit
Glukose die spezifische Aktivität um etwa 50 % reduziert, auf Minimalmedium mit Acetat bzw. Glukose-Acetat-Mischsubstrat keine Aktivitäten messbar waren. Während die für die DHL-ermittelte spezifische Aktivität mit 233
ZUSAMMENFASSUNG
107
mU/mg Protein auf Komplexmedium vergleichsweise hoch war, wurden auf
allen anderen getesteten Kohlenstoffquellen mit 33 bis 50 mU/mg Protein
stark verringerte Aktivitäten detektiert.
4.)
Für die Gene der PDC und der DHL wurde eine monocistronische
Transkription nachgewiesen, wobei in beiden Fällen eine höhere Transkriptmenge in auf Komplexmedium gewachsenen Zellen gegenüber Minimalmedium gezeigt wurde. Promotorfusionen mit dem cat-Gen zeigten, dass
die höchsten spezifischen Promotor-Aktivitäten in der späten exponentiellen
Wachstumsphase und auf Komplexmedium bzw. Minimalmedium mit Glukose gemessen werden konnten. Dies weist auf eine wachstums- und
kohlenstoffabhängige Regulation der Gene aceE und aceF hin. Für das
aceE-Gen wurden zwei mögliche Transkriptionsstartpunkte ermittelt, die 54
bzw. 121 bp stromaufwärts des Startcodons liegen. Für das aceF-Gen
konnte ein Transkriptionsstartpunkt 62 bp stromaufwärts des Startcodons
gezeigt werden.
5.)
Die Untersuchung rekombinanter Stämme mit veränderten EnzymAktivitäten des anaplerotischen Knotenpunktes zeigte deutliche Effekte im
Stamm Val1, der 7 mM Valin ausschied. Die Überexpression des PyruvatKinase-Gens und die Deletion des Pyruvat-Carboxylase-Gens führten zu
einem signifikanten Anstieg der Valinkonzentration auf Minimalmedium mit
Glukose auf 14,6 mM bzw. 12,4 mM, während die Überexpression des Pyruvat-Chinon-Oxidoreduktase-Gens zu einer Verringerung der externen Valinkonzentration (3,31 mM) führte. Für den Stamm Val2 (8,9 mM) war der
Effekt bei Überexpression des Pyruvat-Kinase-Gens nicht nachweisbar (8,6
mM) und bei Überexpression des Pyruvat-Chinon-Oxidoreduktase-Gens
(6,9 mM) deutlich geringer. Eine Steigerung der Valinproduktion von 6,9
mM auf 13,1 mM konnte für den Stamm Val2 nach Deletion des aceE-Gens
gezeigt werden. Auch im C. glutamicum Wildtyp, bei dem kein Valin nachweisbar war, waren nach Deletion des aceE-Gens 5,4 mM Valin detektierbar.
ZUSAMMENFASSUNG
6.)
108
Durch Überexpression der Gene ppc, pck und pqo im Stamm Val1 konnten
Pantothenatkonzentrationen im Bereich von 10 bis 16 mg/l gemessen werden, während der Ausgangsstamm Val1 kein Pantothenat ausschied.
SUMMARY
109
Summary
The PEP-pyruvate-oxaloacetate node in C. glutamicum plays an essential role in
the precursor supply for the production of amino acids and vitamins. Thus it is of
interest to characterize and understand the involved reactions. One enzyme of
these node, the pyruvate dehydrogenase complex (PDHC), is not well investigated
yet. In the present work this complex was described on enzymatical and
transcriptional level and the influence of these and other enzymes of the
anaplerotic node with respect to the production of valine and pantothenate were
analyzed.
1.)
The PDHC shows the highest specific activities on complex medium in the
exponential growth phase with 106 mU/mg protein. After growth on minimal
medium with glucose, acetate or glucose-acetate as carbon source the
activities were about 50 % lowered. The Ks-value for the PDHC for pyruvate
was of 2.2 mM.
2.)
The E2-subunit (Dihydrolipoamide transacetylase) of the PDHC of
C. glutamicum is encoded by aceF-gene which was identified by sequence
alignements. The derived protein consists of 675 amino acids with a
predicted molecular mass of 70.9 kDa. The amino acid sequence of the
DHL showed similarities to PDHC´ s as well as OGDHC´ s of other
organisms with three lipoamide-binding sites, whereas the identities were
the highest to the DHL from E. coli with 31.4%.
3.)
For the E2- as well as for the E1-subunit (Pyruvate decarboxylase) which is
encoded by aceE enzyme assays were established. The highest PDCactivities were detected on complex medium and complex medium with
glucose, whereas the specific activities were decreased about 50 % on
minimal medium with glucose and no activity was detectable on minimal
medium with acetate or glucose-acetate as carbon source. The specific
activity meassured for the DHL was relatively high with 233 mU/mg protein
on complex medium, while it was reduced to 33 to 50 mU/mg protein on
other carbon sources.
SUMMARY
4.)
110
For the genes of the PDC and the DHL which are encoded by the genes
aceE and aceF a monocistronic transcription was verified. The amount of
transcript of cells grown on complex medium was in both cases higher
compared to minimal medium. Promoter fusions with the cat-gene showed
the highest promoter-activities in the late exponential growth phase and on
complex and minimal medium with glucose. This indicates a growth- and
carbon source-dependent regulation of the genes aceE and aceF. For aceE
two transcriptional start sites 54 bp and 121 bp upstream of the initiation
codon were detected. For aceF one transcriptional start site 62 bp upstream
of the translational start point was shown.
5.)
The investigation concerning the amino acid production of recombinant
strains with altered enzyme activities of the anaplerotic node showed clear
effects in the strain Val1 which excreted 7 mM valine. The overexpression
of the pyruvate kinase gene and the deletion of the pyruvate carboxylase
gene showed a significant increase on minimal medium with glucose in
valine concentrations with 14.6 mM or 12.4 mM respectively, whereas the
overexpression of the pyruvate quinon oxidoreductase resulted in a
decrease (3.3 mM). The effect on overexpressing the pyruvate kinase gene
(8.62 mM) was not detectable in strain Val2 (8.93 mM) and lowered on
overexpression the pyruvate quinon oxidoreductase gene (6,93 mM). An
increase from 6.9 mM to 13.1 mM was shown for Val2 after deletion of the
aceE gene. For the C. glutamicum wildtype which did not excrete any valine
5.4 mM were detected after the deletion of aceE.
6.)
In strain Val1 pantothenate concentrations in the range from 10 to 16 mg/l
were meassured on overexpression of the genes ppc, pck and pqo, while
the parent strain did not excrete any pantothenate.
LITERATUR
111
VI. Literatur
Abe, S., K.-I. Takayama, S. Kinoshita. 1967. Taxonomical studies on glutamic
acid producing bacteria. J. Gen. Appl. Microbiol. 13: 279-301.
Allen, A. G. und R. N. Perham. 1991. Two lipoyl domains in the dihydrolipoamide
acetyltransferase chain of the pyruvate dehydrogenase multienzyme complex of
Streptococcus faecalis. FEMS Lett. 287: 206-210.
Alwine, J. C., D. J. Kemp und G. R. Stark. 1977. Method for detection of specific
RNAs in agarose gels by transfer to diazobenzyloxymethylpaper and hybridization
with DNA probes. Proc. Natl. Acad. Sci. 74: 5350-5354.
Barksdale, L. 1970. Corynebacterium diphteriae and its relatives. Bacteriological
Reviews 34: 378-422.
Bentle, L. A. und H. A. Lardy. 1976. Interactions of anions and divalent metal
ions with phosphoenolpyruvate carboxykinase. J. Biol. Chem. 251: 2916-2921.
Birnboim, H. C. 1983. A rapid alkaline extraction method for the isolation of
plasmid DNA. Methods Enzymol. 100: 243-255.
Bisswanger, H. 1981. Substrate specificity of the pyruvate dehydrogenase
complex from Escherichia coli. J. Biol. Chem. 256: 815-822.
Borges, A., C. F. Hawkins, L. C. Packmann und R. N. Perham. 1990. Cloning
and sequence analysis of the genes encoding the dihydrolipoamide acetyltransferase and dihydrolipoamide dehydrogenase components of the pyruvate
dehydrogenase multienzyme complex of Bacillus stearothermophilus. Eur. J.
Biochem. 194: 95-102.
112
LITERATUR
Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of
microgram quantities of protein utilizing the principle of protein-dye binding. Anal.
Biochem. 73: 248-254.
Bresters, T. J., R. A. de Abreu, A. de Kok, J. Visser und C. Veeger. 1975. The
pyruvate-dehydrogenase complex from Azotobacter vinelandii. Eur. J. Biochem.
59: 335-345.
Brown, G. M. 1959. The metabolism of pantothenic acid. J. Biol. Chem. 234: 370378.
Carlsson, P. und L. Hederstedt. 1989. Genetic characterization of Bacillus
subtilis
odhA
and
odhB,
encoding
2-oxoglutarate
dehydrogenase
and
dihydrolipoamide transsuccinylase, respectively. J. Bacteriol. 171: 3667-3672.
Carpousis, A. J., N. F. Vanzo und L. C. Raynal. 1999. mRNA degradation– a
tale of poly(A) and multiprotein machines. Trends Genet. 15: 24-28.
Cocaign-Bousquet M. und N. D. Lindley. 1995. Pyruvate overflow and carbon
flux within the central metabolic pathways of Corynebacterium glutamicum during
growth on lactate. Enzyme Microbiol. Technol. 17: 260-267.
Cocaign-Bousquet M., A. Guyonvarch und N. D. Lindley. 1996. Growth rate
dependent modulation of carbon flux through central metabolism and the kinetic
consequences for glucose-limited chemostat cultures of Corynebacterium
glutamicum. Appl. Environ. Microbiol. 62: 429-436.
Cordes, C., B. Möckel, L. Eggeling und H. Sahm. 1992. Cloning, organization
and functional analysis of ilvA, ilvB and ilvC genes from Corynebacterium
glutamicum. Gene 112: 113-116.
Creaghan, I. T. und J. R. Guest. 1972. Amber mutants of the ketoglutarate
dehydrogenase gene of Escherichia coli K 12. J. Gen. Microbiol. 71: 207-220.
113
LITERATUR
Cremer, J., L. Eggeling und H. Sahm. 1991. Control of the lysine biosynthetic
sequence in Corynebacterium glutamicum as analyzed by overexpression of the
individual corresponding genes. Appl. Environ. Microbiol. 57: 1746-1752.
Cunningham, C. C. und L. P. Hager. 1971. Crystalline pyruvate oxidase from
Escherichia coli. 3. Phospholipid as an allosteric effector for the enzyme. J. Biol.
Chem. 246: 1583-1589.
Dave, E., J. R. Guest und M. M. Attwood. 1995. Metabolic engineering in
Escherichia
coli:
lowering
the
lipoyl
domain
content
of
the
pyruvate
dehydrogenase complex adversely affects the growth rate and yield. Microbiol.
141: 1839-1849.
De Kok, A., A. F. Hengeveld, A. Martin und A. H. Westphal. 1998. The pyruvate
dehydrogenase multi-enzyme complex from Gram-negative bacteria. Biochim.
Biophys. Acta 1385: 353-366.
Dusch,
N.,
A.
Pühler
und
J.
Kalinowski.
1999.
Expression
of
the
Corynebacterium glutamicum panD gene encoding L-aspartate-α-decarboxylase
leads to pantothenate overproduction in Escherichia coli. Appl. Environ. Microbiol.
65: 1530-1539.
Eggeling, I., C. Cordes, L. Eggeling und H. Sahm. 1987. Regulation of acetohydroxy acid synthase in Corynebacterium glutamicum during fermentation of αketobutyrate to L-isoleucine. Appl. Microbiol. Biotechnol. 25: 346-351.
Eggeling, L., S. Morbach und H. Sahm. 1997. The fruits of molecular physiology:
engineering
the
L-isoleucine
biosynthesis
pathway
in
Corynebacterium
glutamicum. J. Biotechnol. 56: 167-182.
Eggeling, L. 2001. Amino acids, pp. 281-303. In: C. Ratledge and B. Kristiansen
(ed.), Basic Bio/technology. Cambridge University Press, London, United
Kingdom.
114
LITERATUR
Eikmanns, B. J., M. T. Follettie, M. U. Griot und A. J. Sinskey. 1989. The
phosphoenolpyruvate
carboxylase
gene
of
Corynebacterium
glutamicum:
molecular cloning, nucleotide sequence, and expression. Mol. Gen. Genet. 218:
330-339.
Eikmanns, B. J., E. Kleinertz, W. Liebl und H. Sahm. 1991a. A family of
Corynebacterium glutamicum/ E. coli shuttle vectors for cloning, controlled gene
expression and promoter probing. Gene 102: 93-98.
Eikmanns, B. J., M. Metzger, D. Reinscheid, M. Kircher und H. Sahm. 1991b.
Amplification of three biosynthesis genes in Corynebacterium glutamicum and its
influence on carbon flux in different strains. Appl. Microbiol. Biotechnol. 34: 617622.
Eikmanns, B. J., N. Thum-Schmitz, L. Eggeling, K. U. Ludtke und H. Sahm.
1994. Nucleotide sequence, expression and transcriptional analysis of the
Corynebacterium glutamicum gltA gene encoding citrate synthase. Microbiol. 140:
1817-1828.
Feinberg, A. P. und B. Vogelstein. 1983. A technique for radiolabeling DNA
restriction endonuclease fragments to high specific activity. Anal. Biochem. 132: 613.
Fraenkel, R. 1975. Regulation and physiological functions of malic enzymes. Curr.
Top. Cell. Regul. 9: 157-181.
Gourdon, P. und N. D. Lindley. 1999. Metabolic analysis of glutamate production
by Corynebacterium glutamicum. Metab. Eng. 1: 224-231.
Gourdon, P., M. F. Baucher, N. D. Lindley und A. Guyonvarch. 2000. Cloning
of the malic enzyme gene from Corynebacterium glutamicum and role of the
enzyme in lactate metabolism. Appl. Environ. Microbiol. 66: 2981-2987.
115
LITERATUR
Gross, D. S., K. W. Collins, E. M. Hernandez und W. T. Garrad, 1988. Vacuum
blotting: a simple method for transferring DNA from sequencing gels to nylon
membranes. Gene 74: 347-356.
Gubler, M., M. Jetten, S. H. Lee und A. J. Sinskey. 1994a. Effects of phosphoenol pyruvate carboxylase deficiency on metabolism and lysine production in
Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 40: 857-863.
Gubler, M., M. Jetten, S. H. Lee und A. J. Sinskey. 1994b. Cloning of the
pyruvate kinase gene (pyk) of Corynebacterium glutamicum and site-specific
inactivation of pyk in a lysine-producing Corynebacterium lactofermentum strain.
Appl. Environ. Microbiol. 60: 2494-2500.
Guest, J. R. und I. T. Creaghan. 1972. Lipoamide dehydrogenase mutants of
Escherichia coli K 12. Biochem. J. 130: 8P.
Guest, J. R. und I. T. Creaghan. 1974. Further studies with lipoamide
dehydrogenase mutants of Escherichia coli K 12. J. Gen. Microbiol. 81: 237-245.
Guest, J. R. und P. E. Stephens. 1980. Molecular cloning of the pyruvate
dehydrogenase complex genes of Escherichia coli. J. Gen. Microbiol. 121: 277292.
Guillouet, S., A. A. Rodal, G.-H. An, P. A. Lessard und A. J. Sinskey. 1999.
Expression
of
the
Escherichia
coli
catabolic
threonine
dehydratase
in
Corynebacterium glutamicum and its effect on isoleucine production. Appl. Env.
Microbiol. 65: 3100-3107.
Hanahan, D. 1985. Techniques for transformation of E. coli. In: DNA cloning. A
practical approach. Vol. I (Glover D. M.; Hrsg.), IRL-Pess, Oxford, Washington DC,
USA: S. 109-135.
116
LITERATUR
Hanemaaijer, R., A. Janssen, A. de Kok und C. Veeger. 1988. The
dihydrolipoyltransacetylase component of the pyruvate dehydrogenase complex
from Azotobacter vinelandii. Eur. J. Biochem. 174: 593-599.
Harley, C. B. und R. P. Reynolds. 1987. Analysis of E. coli promoter sequences.
Nucleic acids Res. 5: 2343-2361.
Hawkins, C. F., A. Borges und R. N. Perham. 1990. Cloning and sequence
analysis of the genes encoding the alpha and beta subunits of the E1 component
of
the
pyruvate
dehydrogenase
multienzyme
complex
of
Bacillus
stearothermophilus. Eur. J. Biochem. 191: 337-346.
Hawley, D. K. und W. R. McClure. 1983. Compilation and analysis of Escherichia
coli promoter sequences. Nucleic Acids Res. 11: 2237-3355.
Helmann, J. D. 1985. Compilation and analysis of Bacillus subtilis σA-dependent
promoter sequences: evidence for extended contact between RNA polymerase
and upstream promoter DNA. Nucleic Acids Res. 13: 2351-2360.
Hemila, H., A. Palva, L. Paulin, S. Arvidson und I. Palva. 1990. Secretory S
complex of Bacillus subtilis: sequence analysis and identity to pyruvate
dehydrogenase. J. Bacteriol. 172: 5052-5063.
Henning, U., G. Dennert, R. Hertel und W. S. Shipp. 1966. Translation of the
structural gene of the E. coli pyruvate dehydrogenase complex. Cold Spring
Harbor Symposia on Quantitative Biology 31: 227-234.
Hermann, T. 2003. Industrial production of amino acids by coryneform bacteria. J.
Biotechnol. 104: 155-172.
Inoue, H., H. Nojima und H. Okayama. 1990. High efficiency transformation of
Escherichia coli with plasmids. Gene 96:23-28.
117
LITERATUR
Jackowski S. und C. O. Rock. 1981. Regulation of coenzyme A biosynthesis. J.
Bacteriol. 148: 926-932.
Jäger, W., P. G. Peters-Wendisch, J. Kalinowski und A. Pühler. 1996. A
Corynebacterium glutamicum gene encoding a two-domain protein similar to biotin
carboxylases and biotin-carboxyl-carrier proteins. Arch. Microbiol. 166: 76-82.
Jetten, M. S., M. E. Gubler, S. H. Lee, und A. J. Sinskey. 1994. Structural and
functional analysis of pyruvate kinase from Corynebacterium glutamicum. Appl.
Environ. Microbiol. 60: 2501-2507.
Jetten, M. S. und A. J. Sinskey. 1995. Purification and properties of oxaloacetate
decarboxylase from Corynebacterium glutamicum. Antonie van Leeuwenhoek 67:
221-227.
Jeyaseelan, K. und J. R. Guest. 1980a. Isolation and properties of pyruvate
dehydrogenase complex mutants of Pseudomonas aeruginosa PAO. J. Gen.
Microbiol. 120: 385-392.
Jeyaseelan, K., J. R. Guest und J. Visser. 1980b. The pyruvate dehydrogenase
complex of
Pseudomonas aeruginosa
PAO.
Purification,
properties
and
characterization of mutants. J. Gen. Microbiol. 120: 393-402.
Kaiser, M. und G. Sawers. 1994. Pyruvate formiate-lyase is not essential for
nitrate respiration by Escherichia coli. FEMS Microbiol. Lett. 117: 163-168.
Kalinowski, J. und 26 weitere Autoren. 2003. The complete Corynebacterium
glutamicum ATCC 13032 genome sequence and its impact on the production of Laspartate-derived amino acids and vitamins. J. Biotechnol. 104: 5-25.
Kase, H. und K. Nakayama. 1972. Production of L-threonine by analogueresistant mutants. Agric. Biol. Chem. 36: 1611-1621.
118
LITERATUR
Katsoulidis, E. 2002. Doktorarbeit zum Thema: Isolierung und Charakterisierung
einer
Corynebacterium
glutamicum-Mutante
mit
deregulierter
Pyruvat-
Carboxylase.
Keilhauer, C., L. Eggeling und H. Sahm. 1993. Isoleucine synthesis in
Corynebacterium glutamicum: molecular analysis of the ilvB-ilvN-ilvC operon. J.
Bacteriol. 175: 5595-5603.
Kellmann, J. W., E. Piechersky und B. Piechulla. 1990. Analysis of the diurnal
expression patterns of the tomato chlorophyll a/b binding protein genes. Influence
of light and charcterization of the gene family. Phytobiol. 52: 35-41.
Khyse-Andersen, J. 1984. Electroblotting of multiple gels: a simple apparatus
without buffer tank for rapid transfer of proteins from polyacrylamide to
nitrocellulose. J. Biochem. Biophys. Methods 10: 204-209.
Kinoshita, S., L. Udaka und M. Chimono. 1957. Studies of amino acid
fermentation. In: Production of L-glutamic acid by various microorganisms. J. Gen.
Appl. Microbiol. 3: 193-205.
Kisumi, M., S. Komatsubara und M. I. Sugiura Chibata. 1971. Properties of
isoleucine hydroxamat-resistant mutants of Serratia marcescens. J. Gen.
Microbiol. 69: 291-297.
Kitada, S., K. Nakayama und S. Kinoshita. 1961. Method for producing L-lysine
by fermentation. US Patent 2979439.
Kleemann, A., W. Leuchtenberger, B. Hoppe und H. Tanner. 1985. Amino
acids. Pp. 57-79. In: Ullmann´ s encyclopedia of industrial chemistry. VHC
Verlagsgesellschaft, Weinheim.
Krampitz, L. O. und C. H. Werkman. 1941. The enzymic decarboxylation of
oxaloacetate. Biochem. J. 35: 595-602.
LITERATUR
119
Lange, C., D. Rittmann, V. F. Wendisch, M. Bott und H. Sahm. 2003. Global
expression profiling and physiological characterization of Corynebacterium
glutamicum grown in the presence of L-valine. Appl. Microbiol. Biotechnol. 69:
2521-2532.
Langley, D. und J. R. Guest. 1974. Biochemical and genetic characteristics of
deletion and other mutant strains of Salmonella typhimurium LT2 lacking α-keto
acid dehydrogenase complex activities. J. Gen. Microbio. 82: 319-335.
Leuchtenberger, W. 1996. Amino acids– technical production and use. In
Biotechnology, vol. 6, pp. 465-502. Edited by H.-J.. Rehm, G. Reed, A. Pühler, P.
Stadler & M. Roehr. Weinheim: VCH.
Liebl, W., A. Bayerl, B. Schein, U. Stillner und K. H. Schleifer. 1989. High
efficiency electroporation of intact Corynebacterium glutamicum cells. FEMS
Microbiol. Lett. 65: 299-304.
Liebl, W. 1991. The genus Corynebacterium - nonmedical. In: The procaryotes,
Vol. 2 (Balows, A., Trüper, H. G., Dworkin, M., Harder, W., Schleifer, K. H.; Hrsg.),
Springer, New York, S. 1157-1171.
Marusyk, R. und A. Sergeant. 1980. A simple method for dialysis of smallvolume samples. Anal. Biochem. 105: 403-404.
May, M. E. und L. L. Brown. 1989. Instability of orthophthalaldehyde reagent for
amino acid analysis. Anal. Biochem. 181: 135-139.
Merkmann, M., C. Chassagnole, N. D. Lindley und A. Guyonvarch. 2003.
Ketopantoate reductase activity is only encoded by ilvC in Corynebacterium
glutamicum. J. Biotech. 104: 253-260.
Minnikin, D. E., M. Goodfellow und M. D. Collins. 1978. Lipid composition in the
classification and identification of coryneform and related taxa. In: Bousfield, I. J.
LITERATUR
120
and Calley, A. G. (Ed.), Coryneform bacteria. Academic. Press, London, pp. 85159.
Morbach, S., H. Sahm und L. Eggeling. 1995. Use of feedback-resistant
threonine dehydratase of Corynebacterium glutamicum to increase carbon flux
towards L-isoleucine. Appl. Environ. Microbiol. 61: 4315-4320.
Morbach, S., C. Junger, H. Sahm und L. Eggeling. 2000. Attenuation control of
ilvBNC in Corynebacterium glutamicum: evidence of leader peptide formation
without the presence of a ribosome binding site. J. Biosci. Bioeng. 90: 501-507.
Moriya, T., Y. Hikichi, Y. Moriya und T. Yamaguchi. 1997. Process for
producing D-pantoic acid, D-pantothenic acid or salts thereof. Patent No. WO
97/10340.
Neveling, U., S. Bringer-Meyer und H. Sahm. 1998a. Gene and subunit
organization of bacterial pyruvate dehydrogenase complexes. Biochim. Biophys.
Acta. 1385: 367-372.
Neveling, U., R. Klasen, S. Bringer-Meyer und H. Sahm. 1998b. Purification of
the pyruvate dehydrogenase multienzyme complex of Zymomonas mobilis and
identification and sequence analysis of the corresponding genes. J. Bacteriol. 180:
1540-1548.
Neveling, U., S. Bringer-Meyer und H. Sahm. 1999. Exceptional characteristics
of heterotetrameric (α2β2) E1p of the pyruvate dehydrogenase complex from
Zymomonas mobilis: expression from an own promoter and a lipoyl domain in
E1β. FEMS Microbiol. Lett. 177: 117-121.
Ogden, G. und P. Földi. 1986. Amino acid analysis: an overview of current
methods. LC-GC. 5: 25-40.
Ohnishi, J., S. Mitsuhashi, M. Hayashi, S. Ando, H. Yokoi, K. Ochiai und M.
Ikeda. 2002. A novel methodology employing Corynebacterium glutamicum
LITERATUR
121
genome information to generate a new L-lysine-producing mutant. Appl. Microbiol.
Biotechnol. 58: 217-223.
Ozaki H. und I. Shiio. 1969. Regulation of the TCA and glyoxylate cycles in
Brevibacterium flavum. II. Regulation of phosphoenolpyruvate carboxylase and
pyruvate kinase. J. Biochem. 66: 297-311.
Patek, M., B. J. Eikmanns, J. Patek und H. Sahm. 1996. Promoters from
Corynebacterium glutamicum: cloning, molecular analysis and search for a
consensus motif. Microbiol. 142: 1297-1309.
Patek, M., J. Nesvera, A. Guyonvarch, O. Reyes und G. Leblon. 2003.
Promoters of Corynebacterium glutamicum. J. Biotech. 104: 311-323.
Patel, M. S., T. E. Roche und R. A. Harris. 1996. Alpha-keto acid dehydrogenase
complexes. Basel: Birkhäuser Verlag.
Perham, R. N. und P. N. Lowe. Isolation and properties of the branched-chain 2keto acid and pyruvate dehydrogenase multienzyme complex from B. subtilis.
Meth. Enzymol. 166: 330-342.
Petersen, S., C. Mack, A. A. de Graaf, C. Riedel, B. J. Eikmanns und H. Sahm.
2001. Metabolic consequences of altered phosphoenolpyruvate carboxykinase
activity in Corynebacterium glutamicum reveal anaplerotic regulation mechanisms
in vivo. Metabolic engineering 3: 344-361.
Peters-Wendisch, P. G., B. J. Eikmanns, G. Thierbach, B. Bachmann und H.
Sahm. 1993. Phosphoenolpyruvate carboxylase in Corynebacterium glutamicum
is dispensable for growth and lysine production. FEMS Microbiol. Lett. 112: 269274.
Peters-Wendisch, P. G., V. F. Wendisch, S. Paul, B. J. Eikmanns und H.
Sahm. 1997. Pyruvate carboxylase as an anaplerotic enzyme in Corynebacterium
glutamicum. Microbiol. 143: 1095-1103.
LITERATUR
122
Peters-Wendisch, P. G., C. Kreutzer, J. Kalinowski, M. Patek, H. Sahm und B.
J. Eikmanns. 1998. Pyruvate carboxylase from Corynebacterium glutamicum:
characterization, expression and inactivation of the pyc gene. Microbiol. 144: 915927.
Peters-Wendisch, P. G., B. Schiel, V. F. Wendisch, E. Katsoulidis, B. Möckel,
H. Sahm und B. J. Eikmanns. 2001. Pyruvate carboxylase is a major bottleneck
for glutamate and lysine production by Corynebacterium glutamicum. J. Mol.
Microbiol. Biotechnol. 3: 295-300.
Quail, M. A., D. J. Haydon und J. R. Guest. 1994. The pdhR-aceEF-lpd operon
of Escherichia coli expresses the pyruvate dehydrogenase complex. Mol.
Microbiol. 12: 95-104.
Quail, M. A. und J. R. Guest. 1995. Purification, characterization and mode of
action of PdhR, the transcriptional repressor of the pdhR-aceEF-lpd operon of
Escherichia coli. Mol. Microbiol. 15: 519-529.
Radmacher, E., A. Vaitsikova, U. Burger, K. Krumbach, H. Sahm und L.
Eggeling. 2002. Linking central metabolism with increased pathway flux: L-valine
accumulation by Corynebacterium glutamicum. Appl. Env. Microbiol. 68: 22462250.
Rae, J. L., J. F. Cutfield und I. L. Lamont. 1997. Sequences and expression of
pyruvate dehydrogenase genes from Pseudomonas aeruginosa. J. Bacteriol. 179:
3561-3571.
Reinscheid, D. J., B. J. Eikmanns und H. Sahm. 1994a. Characterization of the
isocitrate lyase gene from Corynebacterium glutamicum and biochemical analysis
of the enzyme. J. Bacteriol. 176: 3474-3483.
Reinscheid, D. J., B. J. Eikmanns und H. Sahm. 1994b. Malate synthase from
Corynebacterium glutamicum: sequence analysis of the gene and biochemical
characterization of the enzyme. Microbiol. 140: 3099-3108.
LITERATUR
123
Riedel, C., D. Rittmann, P. Dangel, B. Möckel, H. Sahm und B. J. Eikmanns.
2001. Characterization, expression, and inactivation of the phosphoenolpyruvate
carboxykinase gene from Corynebacterium glutamicum and significance of the
enzyme for growth and amino acid production. J. Mol. Microbiol. Biotechnol. 3:
573-583.
Sahm H. und L. Eggeling. 1999. D-pantothenate synthesis in Corynebacterium
glutamicum and use of panBC and genes encoding L-valine synthesis for Dpantothenate overproduction. Appl. Env. Microbiol. 65: 1973-1979.
Sahm, H., L. Eggeling und A. A. de Graaf. 2000. Pathway analysis and
metabolic engineering in Corynebacterium glutamicum. Biol. Chem. 381: 899-910.
Saiki, R. K., S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich und N.
Arnheim. 1985. Enzymatic amplification of beta-globin genomic sequences and
restriction site analysis for diagnosis of sickle cell anemia. Science 230: 13501354.
Saiki, R. K., D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi, G. T. Horn, K.
B. Mullis und H. A. Erlich. 1988. Primer-directed enzymatic amplification of DNA
with a thermostable DNA polymerase. Science 239: 487-491:
Sambrook, J., D. W. Russel, N. Irwin und U. A. Janssen. 2001. Molecular
cloning: a laboratory manual. 3rd edition. Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, N. Y.
Sano, K. und I. Shiio. 1970. Microbial production of L-lysine. III. Production by
mutants resistant to S- (2-aminoethyl)- L-cysteine. J. Gen. Appl. Microbiol. 16:
373-391.
Saumweber, H., R. Binder und H. Bisswanger. 1981. Pyruvate dehydrogenase
component of the pyruvate dehydrogenase complex from Escherichia coli K12.
Eur J. Biochem. 114: 407-411.
124
LITERATUR
Schäfer, A., J. Kalinowski, R. Simon, A. H. Seep-Feldhaus und A. Pühler.
1990. High-frequency conjugal plasmid transfer from gram-negative E. coli to
various gram-positive coryneform bacteria. J. Bacteriol. 172: 1663-1666.
Schäfer, A., A. Tauch, W. Jäger, J. Kalinowski, G. Thierbach und A. Pühler.
1994. Small mobilizable multi-purpose cloning vectors derived from the E. coli
plasmids pK18 and pK19: selection of defined deletions in the chromosome of
Corynebacterium glutamicum. Gene 145: 69-73.
Schuster R. 1988. Determination of amino acids in biological, pharmaceutical,
plant and food samples by automated precolumn derivatization and highperformance liquid chromatography. J. of Chromatography 431: 271-284.
Schwinde, J. W., N. Thum-Schmitz, B. J. Eikmanns und H. Sahm. 1993.
Transcriptional analysis of the gap-pgk-tpi-ppc gene cluster of Corynebacterium
glutamicum. J. Bacteriol. 175: 3905-3908.
Schwinde J. 2000. Doktorarbeit zum Thema: Untersuchungen zur Aktivität der 2Oxoglutarat-Dehydrogenase
von
Corynebacterium
glutamicum
und
Charakterisierung des Gens für die Dihydrolipoamid-Dehydrogenase-Untereinheit.
Schwinde, J. W., P. F. Hertz, H. Sahm, B. J. Eikmanns und A. Guyonvarch.
2001. Lipoamide dehydrogenase from Corynebacterium glutamicum: molecular
and physiological analysis of the lpd gene and characterization of the enzyme.
Microbiol. 147: 2223-2231.
Schwartz, E. R. und L. J. Reed. 1969. Alpha-keto acid dehydrogenase
complexes. XII. Effects of acetylation on the activity and structure of the
dihydrolipoyl transacetylase of Escherichia coli. J. Biol. Chem. 244: 6074-6079.
Schwartz, E. R. und L. J. Reed. 1970. Regulation of the activity of the pyruvate
dehydrogenase complex of Eschrichia coli. Biochem. 9: 1434-1439.
125
LITERATUR
Scrutton, M. C. und M. R. Young. 1972. Pyruvate carboxylase. In: Boyer PD
(ed.) The enzymes, Vol. 6, Academic Press, New York, pp. 1-35.
Shaw, W. V. 1975. Chloramphenicol acetyltransferase from chloramphenicolresistant bacteria. Methods. Enzymol. 43: 737-755.
Shiio, I., S. Otsuka und M. Takahashi. 1962. Effect of biotin on the bacterial
formation of glutamic acid. I. Glutamate formation and cellular permeability of
amino acids. J. Biochem. 51: 56-62:
Shiio, I., H. Momose und A. Oyama. 1969. Genetic and biochemical studies on
bacterial formation of L-glutamate. I. Relationship between isocitrate lyase, acetate
kinase, and phosphate acetyltransferase levels and glutamate production in
Brevibacterium flavum. J. Gen. Appl. Microbiol. 15: 27-40.
Shiio, O., Y. Toride und S. Sugimoto. 1984. Production of lysine by pyruvate
dehydrogenase mutants of Brevibacterium flavum. Agric. Biol. Chem. 48: 30913098.
Simon, R., U. Priefer und A. Pühler. 1983. A broad host range mobilization
system for in vivo genetic engineering: transposon mutagenesis in gram-negative
bacteria. Bio/Technology 1: 784-791.
Snoep, J. L., A. H. Westphal, J. A. E. Benen, M. J. Teixeira de Mattos, O. M.
Neijssel und A. de Kok. 1992. Isolation and characterization of the pyruvate
dehydrogenase complex of anaerobically grown Enterococcus faecalis NCTC 775.
Eur. J. Biochem. 203: 245-250.
Southern E. M. 1975. Detection of specific DNA-sequences among DNA
fragments separated by gel electrophoresis. J. Mol. Biol. 98: 503-517.
Spencer, M. E., M. G. Darlison, P. E. Stephens, I. K. Duckenfield und J. R.
Guest.
1984.
Nucleotide
sequence
of
the
sucB
gene
encoding
the
LITERATUR
126
dihydrolipoamide succinyltransferase of Escherichia coli K12 and homology with
the corresponding acetyltransferase. Eur. J. Biochem. 141: 361-374.
Spencer, M. E. und J. R. Guest. 1985. Transcription analysis of the sucAB,
aceEF and lpd genes of Escherichia coli. Mol. Gen. Genet. 200: 145-154.
Stackebrandt, E., F. A. Rainey und N. L. Ward-Rainey. 1997. Proposal for a
new hierarchic classification system. Actinobacteria classic nov. Int. J. Syst.
Bacteriol. 47: 479-491.
Stephens, P. E., M. G. Darlison, H. M. Lewis und J. R. Guest. 1983. The
pyruvate dehydrogenase complex of Escherichia coli K12. Nucleotide sequence
encoding the pyruvate dehydrogenase component. Eur. J. Biochem. 133: 155-162.
Strohl, W. R. 1992. Compilation and analysis of DNA sequences associated with
apparent streptomycete promoters. Nucleic Acids Res. 20: 961-974.
Thelen, J. J., J. A. Miernyk und D. D. Randall. 1998. Partial purification and
characterization of the maize mitochondrial pyruvate dehydrogenase complex.
Plant Physiol. 116: 1443-1450.
Thomas, M. R. 1994. Simple, effective cleanup of DNA ligation reactions prior to
electro-transformation of E. coli. Bio Techniques 16: 988-990.
Umbarger, H. E. 1987. Biosynthesis of the branched-chain amino acids. P: 352367. In F. C. Neidhardt, J. L. Ingraham, K. B. Low, B. Magasanik, M. Schaechter
and H. E. Umbarger (ed.), Escherichia coli and Salmonella typhimurium: cellular
and molecular biology. American Society for Microbiology, Washington, D. C.
Utter, M. F. und H. M. Kohlenbrander. 1972. Formation of oxaloacetate by CO2
fixation on phosphoenolpyruvate. In: The enzymes, Vol VI, Boyer, P. D. (Ed.),
Academic. Press, New York, pp. 117-170.
LITERATUR
127
Vallino, J. J. und G. Stephanopoulos. 1993. Metabolic flux distributions in
Corynebacterium glutamicum during growth and lysine overproduction. Biotechnol.
Bioeng. 41: 633-646.
Vandamme, E. J. 1992. Production of vitamins, coenzymes and related
biochemicals by biotechnological processes. J. Chem. Technol. Biotechnol. 53:
313-327.
Van der Rest, M. E., C. Lange und D. Molenaar. 1999. A heat shock following
electroporation induces highly efficient transformation of Corynebacterium
glutamicum with xenogenic plasmid DNA. Appl. Microbiol. Biotechnol. 52: 541545.
Van der Vossen, J. M., D. van der Lelie und R. Venema. 1987. Isolation and
characterization of Streptcoccus cremoris Wg2-specific promoters. Appl. Environ.
Microbiol. 53: 2452-2457.
Vasicova, P., Z. Abrhamova, J. Nesvera, M. Patek, H. Sahm und B. Eikmanns.
1998. Integrative and autonomously replicating vectors for analysis of promoters in
Corynebacterium glutamicum. Biotechnol. Techniques 12: 743-746.
Vasicova, P., M. Patek, J. Nesvera, H. Sahm und B. Eikmanns. 1999. Analysis
of the Corynebacterium glutamicum dapA promoter. J. Bacteriol. 181: 6188-6191.
Visser, J. und M. Strating. 1982. Pyruvate dehydrogenase complex from
Eschrichia coli. Methods Enzymol. 89: 391-399.
Westphal, A. H. und A. de Kok. 1988. Lipoamide dehydrogenase from
Azotobacter vinelandii. Molecular cloning, organization and sequence analysis of
the gene. Eur J. Biochem. 172: 299-305.
Westphal, A. H. und A. de Kok. 1990. The 2-oxoglutarate dehydrogenase
complex from Azotobacter vinelandii. Eur. J. Biochem. 187: 235-239.
LITERATUR
128
Yokota, A., Y. Terasawa, N. Takaoka, H. Shimizu und F. Tomita. 1994. Pyruvic
acid production by an F1-ATPase-defective mutant of Escherichia coli W1485 lip2.
Biosci. Biotechnol. Biochem. 58: 2164-2167.
Zhu, P.-P. und A. Peterskofsky. 1996. Sequence and organization of genes
encoding enzymes involved in pyruvate metabolism in Mycoplasma capricolum.
Protein Science 5: 1719-1736.
Die vorliegende Arbeit wurde in der Abteilung Mikrobiologie und Biotechnologie
der Universität Ulm angefertigt:
Amtierender Dekan:
Prof. Dr. Rolf Behm
1. Referent:
Prof. Dr. Bernhard J. Eikmanns
2. Referent:
Prof. Dr. Peter Dürre
Tag der mündlichen Prüfung:
30. Juli 2004
Danksagungen
Die vorliegende Arbeit wurde am Institut für Mikrobiologie und Biotechnologie der
Universität Ulm angefertigt.
Mein besonderer Dank gilt Prof. Dr. Bernhard J. Eikmanns für die Möglichkeit an
diesem interessanten Thema zu arbeiten, sein reges Interesse am Fortschreiten
der Arbeit, für seine beneidenswerte Geduld und Hilfsbereitschaft bei der Lösung
kleinerer und größerer Probleme und für die Durchsicht meiner Arbeit.
Für die Übernahme des Korreferats danke ich Prof. Dr. Peter Dürre.
Für das Korrekturlesen dieser Arbeit, viel Unterstützung und Spaß im Labor, in
den Mittagspausen und bei diversen sportlichen und weniger sportlichen
Aktivitäten außerhalb der Universität möchte ich mich ganz besonders bei Annette
Cramer, Heike Gutekunst, Sandra Hujer, Karina Schierling (meiner LieblingsGackerkollegin), Bettina Schiel, Annette Arndt, Ulrike Samen und Steffi Ripka
bedanken.
An dieser Stelle möchte ich auch meiner restliche Arbeitsgruppe
danken, besonders Mark (für eine Menge guter Gespräche über die Wissenschaft
und
andere
Dinge),
Joy
(meiner
Trauzeugin
und
wissenschaftlichen
Leidensgenossin), Stefan, Axel, Gerd, Bastian, Petra und Dieter. Unserer guten
Seele Brigitte Ehrler möchte ich außerdem danken für das fleißige Auffüllen des
Motivase-Glases und nette morgendliche Gespräche. Jiri Hlatko und Sabrina
Lachenmaier danke ich für Ihre Unterstützung während des Praktikums, die
wesentlich zum Gelingen dieser Arbeit beigetragen hat.
Der gesamten Abteilung Mikrobiologie und Biotechnologie danke ich für eine
wahrscheinlich unübertreffbar gute Arbeitsatmosphäre.
Zuletzt möchte ich meiner Familie und ganz besonders meinem Mann Thomas
Fiur danken, der mich nach vielen gescheiterten Versuchen (und kaputten
Zentrifugen) stets geduldig motiviert hat, nicht die Flinte ins Korn zu werfen und
weiterzumachen.
Lebenslauf
24.03.1977
Geburt in Heidenheim
1983-1985
Besuch der Grundschule in Steinheim
1985-1987
Besuch der Grundschule in Schnaitheim
1987-1989
Besuch der Realschule Königsbronn
1989-1996
Besuch des Gymnasiums Oberkochen
06/1996
Allgemeine Hochschulreife
10/1996-05/2001
Biologiestudium an der Universität Ulm
10/96-09/98
Grundstudium der Biologie Vordiplomprüfung in den Fächern
Physik, Chemie, Botanik, Zoologie und Mikrobiologie
10/98-06/00
Hauptstudium der Biologie mit den Studienschwerpunkten
Chemie, Zoologie, Genetik und Mikrobiologie (Hauptfach)
07/00-05/01
Diplomarbeit in der Abteilung Mikrobiologie und Biotechnologie der Universität Ulm zum Thema „Reinigung und Charakterisierung der Acetatkinase aus Bifidobacterium bifidum
MB254“
seit 08/01
Dissertation zum Thema „Charakterisierung des Pyruvatdehydrogenase-Komplexes aus Corynebacterium glutamicum
und Einfluß von Enzymen des anaplerotischen Knotenpunkts
auf die Bildung von Valin und Pantothenat“ in der Abteilung
Mikrobiologie und Biotechnologie der Universität Ulm