Kein Folientitel
Werbung

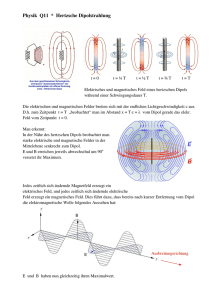
Die Ionische Bindung wird durch das Coulombgesetz beschrieben q1 q 2 r 1 F (r) = 4 πε ε0 ∣r∣2 ∣r∣ Kraft zwischen Ladungen q1 und q2 : r F 2 F 1 q1=-q q2=+q Berechnung der Bindungsenergie r ' =∞ r ' =∞ E ( r) = ∫r ' =r F (r ' ) dr ' = ∫r ' =r q1 q 2 q1 q 2 1 1 dr ' = 2 4 π εε 0 r ' 4 π ε ε0 r Der polare Charakter des Wassers: Ein Molekül mit einem elektrischen Dipol l q- q+ p pH2O 6.17 10 30 Cm Elektrisches Dipolmoment: p =∣q∣ l n p ges = ∑i =1 p i Das Dipolmoment des Wassers ermöglicht die Hydratisierung von Ionen und Proteinen mit polaren Gruppen auf der Oberfläche Auch polare Proteine sind hydratisiert Elektrisches Feld eines Dipols 3u r ( p⋅ur )− p 1 E (r) = 3 4 πε ε0 ∣r∣ gültig nur im größeren Abstand vom Dipol: ∣r∣ ≫ ∣l∣ Elektrisches Feld eines Dipols (gültig nur im größeren Abstand vom Dipol) 3u r ( p⋅ur )− p 1 E (r) = 4 πε ε0 ∣r∣3 Potentielle Energie eines Dipols im elektrischen Feld E E p = − p⋅E Bindungsenergie zwischen zwei Dipolen p1⋅p 2−3(u r⋅p 1)(ur⋅p2 ) 1 E p (r ) = 4 π εε 0 ∣r∣3 p1 p 2 K 1 E p (r ) = 4 π ε ε0 ∣r∣3 K = sin θ 1 sin θ 2 cos(Φ1−Φ2 ) − 2cos θ1 cos θ 2 Induzierte elektrische Dipole sind die Ursache für die van-der-Waals Wechselwirkung p = α ε0 E : Polarisierbarkeit E []=m3 E E p = −∫0 p⋅d E ' = −α ε0∫0 E '⋅d E ' = E = −α ε0 ∫0 1 2 u E⋅u E ∣E '∣dE ' = − α ϵ0∣E∣ 2 2 Quantenmechanische Betrachtung: 3h να E p (r ) = − 4 π2 r 6 h = 6.626 10-34 Js Ein empirisches Potential für die van der Waals Wechselwirkung: Das Lenard-Jones-Potential 6 r0 12 r 0 ELJ (r ) E0 2 r r E/E0 6 E LJ /E 0 4 r=r 0 2 0 -2 0 r/r 0 2 Abstandsabhängigkeit der potentiellen Energie zwischen statischen und dynamischen Ladungsverteilung Ion Ion ~ Dipol induzierter Dipol 1 1 ~ r r Dipol induzierter Dipol ~ 1 1 ~ r2 r 4 ~ 1 1 ~ r4 r4 ~ 1 1 ~ r3 r6 ~ 1 1 ~ r6 r6 ~ 1 1 ~ 6 6 r r *bei Brownscher Molekularbewegung gelten die Ausdrücke in Klammern Wasserstoffbrückenbindungen Eis Flüssiges Wasser Nichtpolare Moleküle gehen keine Wasserstoffbrückenbindungen ein und werden von einer Wasserhülle eingeschlossen. Wasserstoffbrückenbindungen stabilisieren Proteinstrukturen Wasserstoffbrückenbindungen stabilisieren Proteinstrukturen -Helix Paralleles Anti-paralleles -Faltblatt -Faltblatt Empirisches Potential für Wasserstoffbrückenbindung (10-12 Lennard-Jones-Potential) A B E WB (r ) = 12 − 10 r r A, B: Für die jeweilige Wasserstoffbrückenbindung zu bestimmende Konstanten Typische Abstände: F - H ··· F 2.25 Å O - H ··· O 2.90 – 3.00 Å N - H ··· N 2.90 – 3.00 Å Die Polypeptidkette wird durch kovalente Bindungen zusammengehalten Die sechs Atome C-CONH-C liegen in einer Ebene. Die Aminosäurekette ist nur an den CAtomen um die Winkel und drehbar. Empirisches Federpotential E p ,1 (r ) = K 1 (r−r 0 )2 r0 : Gleichgewichtsabstand K1 : Kraftkonstante Bond type r0 (pm) K1 (J mol-1 pm-2) C-C 150,7 132 C=C 133,6 239 C-N 144,9 141 C=N 127,3 239 Empirisches Potential für Bindungswinkel 2 E p ,2 (r ) = K 2 (θ−θ0 ) 0 : Gleichgewichtswinkel K2 : Kraftkonstante Winkel 0 (Grad) K2 (J mol-1 rad-2) C-C-C 112,4 264 C-N-C 121,9 209 Torsionspotential V E p ,3 = [1+cos (n θT −γ)] 2 T: Torsionwinkel, V: Energiebarriere für Rotation, n: Anzahl von Rotationsisomeren, : Phase Beispiel für zwei C-Atome in der Kette X - C – C – X: n = 4, = 0 und V = 16 kJ mol-1 Größenordnungen von Bindungsenergien Kovalente Bindung: Ionische Bindung : 200-500 kJ mol-1 10-30 kJ mol-1 (berechnet für 1mM NaCl in H2O) Wasserstoffbrückenbindung: van-der-Waals Bindung: 10-30 kJ mol-1 1-4 kJ mol-1 Proteindynamik Bewegung von Proteinen ist wesentlich komplizierter als die Dynamik von kleinen Molekülen wie z.B. CO und H2O Normalschwingungen von CO in s-1 Normalschwingungen von H2O Hooksches Gesetz in einer Dimension F Dx F: Kraft (SI-Einheit: N) D: Kraftkonstante (SI-Einheit: N m-1) x: Auslenkung aus der Ruhelage (SI-Einheit: m) Hooksches Gesetz in 9 Dimensionen (für die 9 kartesischen Koordinaten von H2O) F Dx x1 x2 x 3 x4 x x5 , x6 x7 x8 x9 F1 F2 F 3 F4 F F5 , F6 F7 F8 F9 Koordinatenvektor Kraftvektor D1,1 D2,1 ⋮ ⋮ D ⋮ ⋮ ⋮ ⋮ D9,1 D1, 2 D2, 2 ⋮ ⋮ ⋮ ⋮ ⋮ ⋮ D9, 2 ⋯ ⋯ ⋯ ⋯ ⋯ ⋯ D1,9 ⋯ ⋯ ⋯ ⋯ ⋯ ⋯ D2,9 ⋱ ⋮ ⋱ ⋮ ⋱ ⋮ ⋱ ⋮ ⋱ ⋮ ⋱ ⋮ ⋯ ⋯ ⋯ ⋯ ⋯ ⋯ D9,9 Kraftkonstantenmatrix Kraftkonstantenmatrix für H2O 872.4 D= 0.0 0.2 -174.6 0.0 961.4 -70.9 0.0 -54.9 77.7 0.0 -0.1 0.0 0.0 0.1 41.1 0.0 -846.0 -27.0 0.0 870.2 -801.5 0.0 228.5 -6.8 0.0 -14.2 808.3 0.0 -0.1 0.0 0.0 0.0 0.0 0.0 0.1 132.5 0.0 -115.4 81.9 0.0 -24.2 -214.3 0.0 139.6 Kraftkonstantenmatrix für H2O im Hauptachsensystem 0 0 0 0 D= 0 0 176.0 933.8 1016.5 Allgemein: Eigenwerte der Kraftkonstantenmatrix für nicht-lineare Moleküle 3 Eigenwerte gleich Null für Translationen des gesamten Moleküls 3 Eigenwerte gleich Null für Rotationen des Moleküls 3N-6 Eigenwerte ungleich Null für Molekülschwingungen (N : Anzahl der Atome im Molekül)