Bachelorarbeit - Institut für Theoretische Physik - Goethe
Werbung

Bachelorarbeit
Exakte Diagonalisierung des
Heisenbergmodells: Bestimmung unterer
Schranken für die Grundzustandsenergie
unendlicher Spinsysteme
Tim Herfurth
Institut für Theoretische Physik
Johann Wolfgang Goethe-Universität
Frankfurt am Main
September 2011
Erstprüfer:
Prof. Dr. Roser Valentí
Zweitprüfer:
Prof. Dr. Peter Kopietz
Erklärung
Nach 30 (11) der Prüfungsordnung für den Bachelor- und Masterstudiengang Physik der Johann-Wolfgang-Goethe-Universität Frankfurt am Main
erkläre ich, dass diese Arbeit von mir selbständig und ohne Benutzung anderer als der angegebenen Quellen und Hilfsmittel verfasst wurde. Alle Stellen
der Arbeit, die wörtlich oder sinngemäÿ aus Veröentlichungen oder aus anderen fremden Texten entnommen wurden, sind als solche kenntlich gemacht.
Weiter erkläre ich, dass die Arbeit nicht auch nicht auszugsweise für eine
andere Prüfung verwendet worden ist.
Frankfurt am Main, den 01. September 2011
(Tim Herfurth)
Inhaltsverzeichnis
1
2
3
Einführung
7
1.1
Inhalte und Ziele der Arbeit . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7
1.2
Kapitelgliederung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
8
1.3
Hinweise zur verwendeten Literatur . . . . . . . . . . . . . . . . . . . . . . . .
9
Herleitung und Analyse des Heisenbergmodells
11
2.1
Heitler-London-Näherung
. . . . . . . . . . . . . . . . . . . . . . . . . . . . .
12
2.2
Addition zweier Elektronen-Spins . . . . . . . . . . . . . . . . . . . . . . . . .
17
2.3
Singulett-/Triplett-Zustand und eektiver Hamiltonoperator . . . . . . . . . .
20
Austauschwechselwirkung und Magnetismus
25
3.1
25
3.2
4
Austauschwechselwirkung
. . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.1.1
Wasserstomolekül und direkter Austausch
. . . . . . . . . . . . . . .
25
3.1.2
Superaustausch . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
30
3.1.3
Doppelaustausch
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
32
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
34
Magnetismus
3.2.1
Magnetisches Moment und Magnetisierung
. . . . . . . . . . . . . . .
34
3.2.2
Dia- und Paramagnetismus
. . . . . . . . . . . . . . . . . . . . . . . .
35
3.2.3
Magnetische Ordnung: Ferro- und Antiferromagnetismus . . . . . . . .
38
Exakte Diagonalisierung und untere Schranken der Grundzustandsenergie 47
4.1
Lineare Spin-Kette . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
48
4.2
Addition von Drehimpulsen und Basistransformation . . . . . . . . . . . . . .
52
4.2.1
54
4.3
Basistransformation und Clebsch-Gordan-Koezienten . . . . . . . . .
Untere Schranken der Grundzustandsenergie für unendliche Spinsysteme . . .
57
4.3.1
Methode der unteren Schranke
58
4.3.2
Exakte Diagonalisierung zwei- und dreidimensionaler antiferromagnetischer Spingitter
4.3.3
4.4
. . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
59
Diskussion der Ergebnisse . . . . . . . . . . . . . . . . . . . . . . . . .
80
Ausblick . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
83
Literaturverzeichnis
85
Anhang
87
A.1
Lineare Spinkette: MATHEMATICA-Code . . . . . . . . . . . . . . . . . . . .
87
A.2
Grundzustandsenergie des Doppelkreuzes: MATHEMATICA-Code
88
. . . . . .
5
1 Einführung
1.1 Inhalte und Ziele der Arbeit
Das Thema dieser Bachelorarbeit im Bereich der theoretischen Physik lautet Exakte Diago-
nalisierung des Heisenbergmodells: Bestimmung unterer Schranken für die Grundzustandsenergie unendlicher Spinsysteme . Anders als dieser Titel vielleicht vermuten lässt, leistet diese
Arbeit in weiten Teilen recht allgemeine Abhandlungen zu einem breiten Themenfeld.
Als zentraler Fixpunkt der Arbeit kann das Heisenbergmodell betrachtet werden, das 1928
von Werner Heisenberg und Paul Dirac begründet wurde. Es stellt die Grundlage der formalen
Beschreibung vieler Probleme der Festkörperphysik dar und ist dementsprechend von groÿer
Bedeutung in dieser Disziplin. Rund um dieses Modell bauen sich die Themenfelder dieser
Arbeit auf. Die formale Gestalt des Heisenbergmodells, deren Erläuterung folglich Bestandteil
dieser Arbeit ist, ist die folgende:
HHeis = −J
X
Si · Sj
.
(1.1)
hi,ji
Es kann eine grobe inhaltliche Zweiteilung der Arbeit gemacht werden. Der erste Teil besteht
in der Herleitung und Analyse des Heisenbergmodells. Die Herleitung des Modells ist unabdingbar, da es die Basis für alle folgenden Abhandlungen darstellt und entsprechend begründet
sein sollte. Die Analyse befasst sich mit der Bedeutung des Modells in der Festkörperphysik.
Dabei zeigt sich eine Vielzahl physikalischer Themenfelder, die direkt oder indirekt mit dem
Modell in Zusammenhang stehen. Im Rahmen einer solchen Bachelorarbeit können diese Themengebiete nicht alle vollumfänglich besprochen werden. Eine besondere Stärke bietet das
Modell bezüglich der Erklärung von ferromagnetischem und antiferromagnetischem Verhalten in Isolatoren. Darauf bezugnehmend habe ich deshalb ausführlichere Erläuterungen zum
Themenfeld des Magnetismus vorgenommen. Dies wiederum ist nur durch das Verständnis von
Austauschwechselwirkungen möglich, die ebenfalls einen direkten Bezug zum Heisenbergmodell haben. Auch dieser Aspekt wird umfänglich besprochen. Zusammen mit der Herleitung des
Modells stellen diese beiden Gebiete einen inhaltlich geschlossenen Themenkomplex dar, der
zu einem Gesamtverständnis des Modells und seiner Bedeutung führt. Somit ist es dem Leser
möglich, alle Verwendungen des Modells in einer speziellen Situation auch hinsichtlich eines
gröÿeren Kontext zu bewerten. Wenngleich die Ausführungen diesbezüglich meist allgemeiner
Natur sind und sich selten auf konkrete Systeme beziehen, geben sie dennoch Hinweise auf
die Methoden der konkreten Auswertung des Heisenbergmodells.
Der zweite Teil der Arbeit befasst sich mit einer speziellen Problemstellung, die eine wissenschaftliche Eigenleistung erfordert und den Titel der Arbeit erklärt. Als Grundlage dient auch in diesem Teil das Heisenbergmodell. Beschreibt man mit diesem Modell Systeme
der Gestalt wie sie praktisch vorkommen, dann handelt es sich um zwei- und dreidimensionale Spinsysteme mit quasi unendlicher Ausdehnung. Für solche Systeme existieren allerdings
7
KAPITEL 1. EINFÜHRUNG
keine exakten Lösungen des Heisenbergmodells. Dies ist in der Physik oftmals der Fall und ist
immer mit der Suche nach angemessenen Näherungsmethoden für das jeweilige Problem verbunden. In dem konkreten Fall dieser Arbeit handelt es sich um das Problem der Bestimmung
der Grundzustandsenergie für spezielle, unendliche zwei- und dreidimensionale Spinsysteme,
deren Gitterplätze antiferromagnetisch wechselwirken.
Das hier verwendete Näherungsverfahren besteht in der Bestimmung unterer Schranken für die
gesuchte Grundzustandsenergie und erfolgt über die exakte Auswertung (Diagonalisierung)
kleinerer, endlicher Spinsysteme. Diese Methode wurde in einer Publikation von Rolf Tarrach
und Roser Valentí erstmals vorgestellt [TV90] und in dem selben Zusammenhang benutzt, wie
er in dieser Arbeit auftritt. Somit schlieÿt dieser Teil der Arbeit unmittelbar an die benannte
Publikation an.
Wesentlicher Aufgabenteil war es dabei, allgemeine Verfahren zu entwickeln, die die Diagonalisierung der Spinsysteme gestatten und diese zum Zwecke praktischer Berechnungen in einen
Computeralgorithmus umzusetzen.
1.2 Kapitelgliederung
Es soll nun ein kurzer Abriss der Gliederung der Kapitel und derer Schwerpunkte gegeben
werden.
•
In Kapitel 2 wird die Herleitung des Heisenbergmodells im Rahmen der Heitler-LondonNäherung vorgestellt und das Modell damit begründet. Im Zuge dessen werden einige
Grundlagen der zugehörigen Quantenmechanik, insbesondere des quantenmechanischen
Drehimpulses, eingeführt.
•
Kapitel 3 stellt zunächst einen Zusammenhang des Heisenbergmodells mit dem Begri der Austauschwechselwirkung her. Zudem werden auf allgemeine Weise zusätzliche
Formen der Austauschwechselwirkung erläutert, die keinen direkten Bezug zum Heisenbergmodell haben. Wiederum unmittelbar mit der Austauschwechselwirkung ist der
kollektive Magnetismus in Festkörpern verbunden. Entsprechend erfolgen auch diesbezüglich allgemeine Ausführungen.
•
Kapitel 4 besteht in der Präsentation der von mir durchgeführten Rechnungen. Es wird
zunächst die Methode der Bestimmung unterer Schranken für die Grundzustandsenergie
von Spinsystemen vorgestellt und die theoretischen Grundlagen für die exakte Diagonalisierung solcher Systeme werden geliefert. Es folgt die Beschreibung meiner Vorgehens
bei den Berechnungen. Abschlieÿend werden die Ergebnisse dargelegt, bewertet und
diskutiert sowie ein kurzer Ausblick auf mögliche Erweiterungen der Arbeit geliefert.
8
1.3. HINWEISE ZUR VERWENDETEN LITERATUR
1.3 Hinweise zur verwendeten Literatur
Die von mir verwendete Literatur ist in dem Literaturverzeichnis aufgeführt, das sich den
Kapiteln dieser Arbeit anschlieÿt. Durch entsprechende Markierungen im Text wird auf die
Verwendung eines Literaturteils an zugehöriger Stelle hingewiesen. Es sei aber erwähnt, dass
die meisten der aufgeführten literarischen Quellen als ganz allgemeine Grundlagen dieser
Arbeit dienten und nicht ausschlieÿlich an den ausgezeichneten Stellen Verwendung fanden.
9
2 Herleitung und Analyse des
Heisenbergmodells
Abbildung 2.1: Veranschaulichung des Wasserstomoleküls [Val10]
Wie im vorherigen Teil beschrieben, dient das Heisenbergmodell unter anderem der Erklärung von (anti-)ferromagnetischem und ferrimagnetischem Verhalten von Festkörpern. Mathematisch geschieht dies unter der Berücksichtigung von ausschlieÿlich Elektronenspins auf benachbarten Gitterplätzen. Die Herleitung des entsprechenden Heisenberg-Hamiltonoperators
(1.1) geht deshalb zunächst von der Betrachtung eines Zweielektronen-Systems aus. Gemäÿ
der Situation in einem Festkörper handelt es sich dabei nicht um zwei freie Elektronen, sondern
um an einen Atomkern gebundene Elektronen. Als gröÿtmögliche Vereinfachung betrachtet
man zwei Wasserstoatome auf benachbarten Gitterplätzen. Dies wiederum entspricht auf den
ersten Blick der Betrachtung einer Wasserstomoleküls. Die folgenden Ausführungen werden
aber zeigen, dass dies nur unter Annahme bestimmter Abstände zwischen den Atomkernen
der Fall ist. Nähere Erläuterungen dazu werden an entsprechender Stelle erfolgen.
Als erste Näherung nimmt man für das System unendliche groÿe Kernmassen an, sodass sich
die kinetische Energie des Systems auf diejenige der Elektronen beschränkt. Dann lässt sich
der Hamiltonoperator für das System in der Ortsbasis schreiben als
H=−
~2
(∇2 + ∇22 ) + V (r1 , r2 )
2m 1
.
(2.1)
Die Indizes 1 und 2 kennzeichnen dabei das jeweilige Elektron. Dieselbe Bedeutung als Zuordnung der Elektronen wird auch im Folgenden beibehalten. Tatsächlich handelt es sich bei
Elektronen um ununterscheidbare, quantenmechanische Objekte. Eine klare Zuordnung eines
bestimmten Elektrons an einen bestimmten Ort ist daher nicht möglich. Rechentechnisch ist
es allerdings erforderlich, eine Teilchennummerierung durchzuführen. Diese muss so gehalten
sein, dass eine Vertauschung der Indizes keinen Unterschied hinsichtlich der physikalischen
Bedeutung macht [Nol02].
Der erste Summand in (2.1) stellt die angesprochenen kinetischen Energien der Elektronen
dar. Der zweite Summand ist die zusammengefasste potenzielle Energie des Systems. Sie
11
KAPITEL 2. HERLEITUNG UND ANALYSE DES HEISENBERGMODELLS
hängt von den Ortsvektoren
r1/2
der Elektronen 1 und 2 ab. Sie besteht aus den Coulomb-
Wechselwirkungen der Elektronen mit ihrem Kern, der Elektronen mit dem elektrischen Feld
des jeweils anderen Atomkerns, der Elektronen untereinander (Elektron-Elektron-Wechselwirkung) und der Protonen untereinander. Somit lässt sich schreiben:
V (r1 , r2 ) = −
e2
e2
e2
e2
e2
e2
−
−
−
+
+
4π0 r1A 4π0 r2B
4π0 r1B
4π0 r2A 4π0 r12 4π0 RAB
.
(2.2)
Zum besseren Verständnis sind in Abbildung 2.1 die vorkommenden Vektoren anschaulich
dargestellt. Daraus wird deutlich, dass in der gewählten Notation neben den Elektronen (1
und 2) auch die zugehörigen Kerne durch A beziehungsweise B gekennzeichnet sind.
Die Beschreibung des Systems geschieht nach den Grundlagen der Quantenmechanik durch die
Lösung der Schrödingergleichung. In unserem Fall liegt das Interesse an der Beschreibung des
Grundzustands des Systems. Da der Hamiltonoperator (2.1) keine explizite Zeitabhängigkeit
enthält, handelt es sich um eine zeitunabhängige Schrödingergleichung, die sich im Ortsraum
als folgende Eigenwertgleichung darstellt:
Hψ(r1 , r2 ) = E0 ψ(r1 , r2 )
Hierbei handelt es sich bei
bei
E0
.
(2.3)
ψ(r1 , r2 ) um die Grundzustands-Wellenfunktion im Ortsraum und
um die Grundzustandsenergie des Systems. Die Dierentialgleichung (2.3) ist in dieser
Form nicht exakt lösbar, weshalb Näherungsverfahren zurate gezogen werden müssen. Dies
ist in der Festkörperphysik im Allgemeinen notwendig, da die entsprechende SchrödingerGleichung sich mit der Zahl der beteiligten Teilchen natürlich weiter verkompliziert. Probleme der Festkörperphysik, die typischerweise Systeme von Teilchenzahlen der Gröÿenordnung
1023
behandelt, werden also stets durch verschiedenste, kontextabhängige Methoden der
Näherungsrechnung gelöst.
2.1 Heitler-London-Näherung
Das im Folgenden verwendete Näherungsverfahren geht auf Walter Heitler und Fritz London
zurück. Es wird als Heitler-London-Näherung bezeichnet und wird uns im Laufe dieses Kapitels zur Formulierung des Hamiltonoperators (1.1) des Heisenbergmodells führen.
Der erste Teil der Näherung besteht in der Annahme, dass der Abstand zwischen den Kernen so groÿ ist, dass in (2.2) lediglich die Wechselwirkungsterme der Elektronen mit ihren
Kernen verbleiben. Elektron-Elektron-Wechselwirkung, Proton-Proton-Wechselwirkung und
die Wechselwirkung der Elektronen mit dem jeweils anderen Kern werden zunächst vernachlässigt. Es wird also vorerst die Situation zweier unabhängiger Elektronen angenommen. In
diesem Fall vereinfacht sich der Hamiltonoperator zu
H0 = −
~2 2
e2
~2 2
e2
∇1 −
−
∇2 −
:= H1 + H2
2m
4π0 r1A 2m
4π0 r2B
.
(2.4)
Der zugehörige Hamiltonoperator besteht also aus einer Summe unabhängiger Energieoperatoren. Leicht lässt sich zeigen, dass sich in diesem Fall, die neue Eigenwertgleichung
H 0 ψ(r1 , r2 ) = E0 ψ(r1 , r2 )
12
(2.5)
2.1. HEITLER-LONDON-NÄHERUNG
(2.3) zu zwei entkoppelten, exakt lösbaren, Eigenwertgleichungen umschreiben lässt [CTDL73].
Dies ist auch physikalisch sinnvoll, da wir uns schlieÿlich in einer Näherung unabhängiger
Elektronen benden. Somit ergeben sich die beiden Einteilchen-Schrödingergleichungen
~2 2
e2
−
φA (r1 ) = ε0 φA (r1 )
∇ −
2m 1 4π0 r1A
,
~2 2
e2
−
φB (r2 ) = ε0 φB (r2 )
∇2 −
2m
4π0 r2B
mit den Grundzustands-Wellenfunktionen
ε0 .
φ(r)
(2.6)
(2.7)
und den Einteilchen-Grundzustandsenergien
Im Falle von Wasserstoatomen handelt es sich dabei um bekannte Wellenfunktionen und
Energien.
Bis hierhin war der Spin der Elektronen gänzlich irrelevant, was nicht unbedingt naheliegend
ist, da der Hamiltonoperator des Heisenbergmodells ausschlieÿlich eben deren Spinoperatoren
beinhaltet. Jedoch liegt die Problemstellung tatsächlich nicht in der Beschreibung einzelner
Wasserstoatome, sondern in jener eines Systems zweier Elektronen. Bei Elektronen handelt
es sich um Fermionen, sie tragen einen Spin von 1/2. Folglich gehorchen sie dem Pauli-Prinzip,
welches eine unter Vertauschung der Elektronen antisymmetrische Gesamtwellenfunktion des
Systems fordert. Für zwei ununterscheidbare Fermionen mit Spin s bedeutet dies, dass die
Gesamtwellenfunktion
ψ
die Gleichung
ψ(r1 , s1 ; r2 , s2 ) = −ψ(r2 , s2 ; r1 , s1 )
(2.8)
erfüllen muss [CTDL73, S. 1396f ]. Demzufolge muss die Gesamtwellenfunktion des Systems,
|ψ(r1 , r2 )i ⊗ |χ(s1 , s2 )i aus Ortswellenfunktion ψ und
χ beschrieben werden kann, antisymmetrisch unter Vertauschung der bei-
die basisunabhängig als Tensorprodukt
Spinwellenfunktion
den Elektronenindizes sein. Um dies zu erfüllen, muss entweder die Ortswellenfunktion antisymmetrisch und die Spinwellenfunktion symmetrisch sein oder der genau umgekehrte Fall
vorliegen, sodass in der Ortsdarstellung Gesamtwellenfunktionen der Form
ψ = ψs/a (r1 , r2 )χa/s (s1 , s2 )
(2.9)
physikalisch erlaubt sind. (Der Index s/a weist auf die symmetrische/antisymmetrische Eigenschaft der Funktion hin.) Diese Forderung wird schlussendlich entscheidend für die Begründung des Heisenbergmodells sein. Deshalb werden die möglichen Spinwellenfunktionen an
späterer Stelle näher betrachtet. Zunächst soll aber geklärt werden, was das Pauli-Prinzip für
die Ortswellenfunktion aus (2.5) bedeutet.
Berücksichtigt man nur die Schrödingergleichung (2.5), so wäre
ψ(r1 , r2 ) = φA (r1 )φB (r2 )
(2.10)
die naheliegenste, mögliche Lösung für die Ortswellenfunktion. Physikalisch ist (2.10) allerdings nicht erlaubt, da keine der genannten Symmetriebedingungen erfüllt ist. Da auch Linearkombinationen der eben genannten Wellenfunktion Lösungen der Eigenwertgleichung sind
und aufgrund der Ununterscheidbarkeit der Elektronen die Indizes 1 und 2 vertauschbar sind,
lässt sich folgende symmetrische Ortswellenfunktion konstruieren:
ψs (r1 , r2 ) = φA (r1 )φB (r2 ) + φA (r2 )φB (r1 )
.
(2.11)
13
KAPITEL 2. HERLEITUNG UND ANALYSE DES HEISENBERGMODELLS
(Eine Normierung der Wellenfunktion wird erfolgen.)
Eine antisymmetrische Ortswellenfunktion
ψa (r1 , r2 ) = φA (r1 )φB (r2 ) − φA (r2 )φB (r1 )
(2.12)
lässt sich in diesem Fall durch eine ähnlich intuitive Herangehensweise nden. Mathematisch
sauber lässt sie sich im Allgemeinen durch die sogenannte Slater-Determinate nden [Gri95,
S.210]. Diese Methode lässt sich für beliebig groÿe Systeme verwenden und ist gerade dort
von groÿem Nutzen. Da sie hier nicht weiter von Bedeutung ist, soll sie nicht detaillierter
besprochen werden.
Somit sind die möglichen Ortswellenfunktionen für die Eigenwertgleichung (2.5) des genäherten Problems unabhängiger Elektronen gefunden. Die eigentliche Heitler-London-Näherung besteht nun darin, die Funktionen (2.11) und (2.12) auch als Ortsanteil der Lösungen
der ursprünglichen Eigenwertgleichung (2.3) anzunehmen [AM76]. Augenscheinlich handelt es
sich dabei um eine recht starke Vereinfachung. Die Näherung erweist sich für groÿe ProtonenAbstände als recht plausibel, wird aber für kleinere Abstände schlechter [AM76].
Die gemäÿ (2.9) zu den Ortswellenfunktionen gehörigen antisymmetrischen/symmetrischen
Spinanteile der Gesamtwellenfunktion brauchen hier noch nicht näher untersucht zu werden,
da der Hamiltonoperator (2.1) vom Spin der Elektronen unabhängig ist. Im Allgemeinen
ergeben sich die beiden Gesamtwellenfunktionen, normiert durch
N+ ,
beziehungsweise
N− ,
zu
ψ+ = N+ [φA (r1 )φB (r2 ) + φA (r2 )φB (r1 )] χa (s1 , s2 )
,
(2.13)
ψ− = N− [φA (r1 )φB (r2 ) − φA (r2 )φB (r1 )] χs (s1 , s2 )
.
(2.14)
Leicht ist ersichtlich, dass es sich um orthogonale Funktionen handelt. Es interressiert nun,
welche Energieeigenwerte diese Funktionen für das System liefern. Es ist klar, dass die kleinere
der beiden Energien eine bessere Abschätzung der tatsächlichen Grundzustandsenergie gibt.
Dies wird aus dem Variationsprinzip deutlich [Gri95, S.293]. Demnach gilt für eine beliebige,
normierte Wellenfunktion
ψ:
EGZ ≤ hψ| H |ψi ≡ hHi
.
(2.15)
Ein Beweis dafür lässt sich schnell führen. Eine beliebige, normierte Wellenfunktion
als Linearkombination der orthnormierten, tatsächlichen Eigenfunktionen
ψn
ψ lässt sich
des Hamilton-
operators schreiben.
ψ=
X
cn ψn ,
wobei
Hψn = En ψn
(2.16)
n
Da
ψ
als normiert angenommen wird, gilt:
1 = hψ|ψi =
XX
m
c∗m c∗n hψm |ψn i =
X
n
|cn |2
.
(2.17)
n
Auÿerdem lässt sich der Energieerwartungswert schreiben als
hHi =
*
X
m
14
+
X
XX
X
cm ψm H
cn ψn =
c∗m En cn hψm |ψn i =
En |cn |2
n
m
n
n
.
(2.18)
2.1. HEITLER-LONDON-NÄHERUNG
Per Denition ist aber die Grundzustandsenergie die kleineste der Eigenergien, weshalb
En .
EGZ ≤
Daraus folgt
hHi ≥ EGZ
X
|cn |2 = EGZ
.
(2.19)
n
Die tatsächliche Grundzustandsenergie
EGZ
eines Systems ist also stets kleiner als der En-
ergieerwartungswert für eine Wellenfunktion, die nicht der Grundzustandwellenfunktion entspricht. Das Gleichheitszeichen gilt genau für die exakte Grundzustandswellenfunktion. Die
Energieeigenwerte, die sich aus einer genäherten Wellenfunktion ergeben, bilden somit immer eine obere Schranke für die exakte Grundzustandsenergie. Dies bedeutet auch, dass eine
genäherte Lösung je besser ist desto kleiner ihr Energieerwartungswert ist. Besonders wird
auch die Dierenz der beiden Energieerwartungswerte von Interesse sein.
Es gilt also die Energieerwartungswerte
E± = hHi± = hψ± | H |ψ± i
(2.20)
des Hamiltonoperators (2.1) für die beiden Wellenfunktionen (2.13) und (2.14) auszuwerten.
Hierbei kann der Spinanteil aus genannten Gründen vernachlässigt werden. Zudem sollen die
Spinfunktionen
χa/s
als normiert angenommen werden. Zunächst sind die Normierungskon-
stanten zu bestimmen. Ihr inverses Quadrat entspricht genau
ψs/a ψs/a =
ZZ
|ψs/a (r1 , r2 )|2 dV1 dV2
ZZ
[φA (r1 )φB (r2 ) ± φA (r2 )φB (r1 )]∗ · [φA (r1 )φB (r2 ) ± φA (r2 )φB (r1 )] dV1 dV2
=
.
(2.21)
Da es sich um reelle
φ
handelt, ergibt sich nach Ausmultiplikation
ZZ
Z
2
|ψs/a (r1 , r2 )| dV1 dV2 =
|
Z
±
φ2A dV1
Z
{z
=1
φ2B dV2 +
}
Z
Z
φ2B dV1
{z
}
|
=1
Z
φA (r1 )φB (r1 )dV1
Z
±
φ2A dV2
(2.22)
φA (r2 )φB (r2 )dV2
Z
φA (r2 )φB (r2 )dV2
φB (r1 )φA (r1 )dV1
.
Die ersten beiden Ausdrücke reduzieren sich aufgrund der Normierung der Einteilchen-Wellenfunktionen (hφi |φj i
= δij )
auf 1. Dabei ist eine Unterscheidung zwischen
dV1 und dV2
nicht
nötig, da es sich jeweils um eine Integration über den gesamten Raum handelt. Eine Betrachtung der anderen beiden Summanden zeigt, dass es sich jeweils um Quadrate eines Überlappintegrals handelt. Das Überlappintegral S ist im Allgemeinen durch
Z
Snm =
ψn (r)∗ ψm (r)d3 r
(2.23)
deniert. Den Gesetzmäÿigkeiten der Quantenmechanik zufolge entspricht sein Betragsquadrat
genau der Wahrscheinlichkeit dafür, dass ein Teilchen, das durch
im Orbital
ψm
ψn
beschrieben wird, sich
bendet. Es gibt also ein Maÿ für die räumliche Deckung der beiden Wellen-
funktionen. Im vorliegenden Fall handelt es sich dabei natürlich um den Überlapp von
und
φB :
φA
Z
L=
φA (r1/2 )φB (r1/2 )dV1/2
.
(2.24)
15
KAPITEL 2. HERLEITUNG UND ANALYSE DES HEISENBERGMODELLS
(Das Überlappintegral soll hier
L heiÿen, um einer Verwechslung mit dem Spin vorzubeugen.)
Für das Elektronensystem stellt sich ein groÿer Überlapp als energetisch ungünstig dar. Er
bedeutet eine groÿe räumliche Nähe der Elektronen, was aufgrund der Coulomb-Abstoÿung
mit einer erhöhten potenziellen Energie gleichzusetzen ist. Dieser Aspekt kommt im Zuge der
weiteren Auswertung nochmals zum Tragen.
Zusammengefasst lässt sich (2.21) also ausdrücken durch
ψs/a ψs/a = 1/N 2 = 2(1 ± L2 )
.
(2.25)
Die Auswertung von (2.20) enthält einige Terme und gestaltet sich komplizierter. Es bietet
sich die Denition weiterer Gröÿen an, um die Handhabung bei der Berechnung etwas zu
vereinfachen. Zudem wird die besondere physikalische Bedeutung dieser Gröÿen in Abschnitt
3.1 deutlich werden. Dabei können die Symmetrien des Systems genutzt werden. Man deniert:
ZZ
U=
V 0 (r1 , r2 )|φA (r1 )φB (r2 )|2 dV1 dV2
(2.26)
ZZ
=
ZZ
0
2
V (r1 , r2 )|φA (r2 )φB (r1 )| dV1 dV2
,
V 0 (r1 , r2 )φA (r1 )∗ φA (r2 )φB (r2 )∗ φB (r1 )dV1 dV2
K=
.
(2.27)
Auÿerdem ist es zweckmäÿig den Gesamt-Hamiltonoperator in einen Teil, der unabhängige
Elektronen repräsentiert und einen Teil, der den restlichen Operator darstellt, aufzuteilen.
Der erste Teil ist bereits durch
V
H0
(2.4) gegeben, den verbleibenden Teil von (2.2) nennen wir
0.
H = H 0 + V 0 (r1 , r2 ) = H 0 −
e2
e2
e2
e2
−
+
+
4π0 r1B
4π0 r2A 4π0 r12 4π0 RAB
(2.28)
Somit lässt sich
ψa/s H ψa/s = ψa/s H 0 ψa/s + ψa/s V 0 ψa/s
(2.29)
schreiben. Für den ersten Summanden erhält man
ψa/s H 0 ψa/s =2N 2 ·
ZZ
φA (r1 )∗ φB (r2 )∗ H 0 φA (r1 )φB (r2 )dV1 dV2
|
{z
}
1.
ZZ
2
± 2N ·
φB (r1 )∗ φA (r2 )∗ H 0 φA (r2 )φB (r1 )dV1 dV2
|
{z
}
(2.30)
2.
1. Aufgrund von (2.6)/(2.7) und der Normierung der
ZZ
φ(r)
berechnet man leicht:
φA (r1 )∗ φB (r2 )∗ H 0 φA (r1 )φB (r2 )dV1 dV2 = 2ε0
.
(2.31)
2. Mit der Denition des Überlappintegrals (2.24) ergibt sich:
ZZ
0
ZZ
φB (r1 )φA (r2 )H φA (r2 )φB (r1 )dV1 dV2 =
φB (r1 )φA (r2 )2ε0 φA (r2 )φB (r1 )dV1 dV2
= 2ε0 L2
.
(2.32)
16
2.2. ADDITION ZWEIER ELEKTRONEN-SPINS
Also ist
4ε0 (1 ± L2 )
ψa/s H 0 ψa/s = 4ε0 N 2 (1 ± L2 ) =
= 2ε0
2(1 ± L2 )
.
(2.33)
Prinzipiell analog verläuft die Auswertung des zweiten Summanden.
ZZ
φA (r1 )∗ φB (r2 )V 0 (r1 , r2 )φA (r1 )φB (r2 )dV1 dV2
|
{z
}
1.
ZZ
± 2N 2 ·
φB (r1 )∗ φA (r2 )∗ V 0 (r1 , r2 )φA (r2 )φB (r1 )dV1 dV2
|
{z
}
ψa/s V 0 ψa/s =2N 2 ·
(2.34)
2.
1. Das Integral entspricht genau dem zuvor denierten U.
2. Ein Vergleich zeigt, dass es sich bei dem Integral um K handelt.
Zusammengefasst bedeutet dies, dass
U ±K
2(U ± K)
=
ψa/s V 0 ψa/s = 2N 2 (U ± K) =
2
2(1 ± L )
1 ± L2
.
(2.35)
Damit sind die Energien zu den beiden Gesamtwellenfunktionen bestimmt.
U +K
Hψ+ = 2ε0 +
ψ+
1 + L2
U −K
Hψ− = 2ε0 +
ψ−
1 − L2
⇔
E+ = 2ε0 +
U +K
1 + L2
(2.36)
⇔
E− = 2ε0 +
U −K
1 − L2
(2.37)
Letztlich entscheidend für die Herleitung des Heisenberg-Hamiltonoperators ist die Energiedierenz
J ≡ ∆E = E+ − E− = −2
U L2 − K
1 − L4
(2.38)
zwischen den beiden Eigenenergien.
Es ist damit die Gröÿe
J
deniert worden, die als Kopplungsparameter bezeichnet wird. Sie
ist - wie bereits zu sehen war - von wesentlicher Bedeutung für das Heisenbergmodell und mit
dem quantenmechanischen Eekt der Austauschwechselwirkung verknüpft. Diesem Aspekt ist
ein eigener Abschnitt gewidmet (siehe 3.1), der die zugrundeliegenden Prinzipien eingehender
erläutert.
2.2 Addition zweier Elektronen-Spins
In den bisherigen Ausführungen ist deutlich geworden, dass es in der Heitler-London-Näherung
eine Energieaufspaltung (2.38) zwischen den Gesamtwellenfunktionen mit symmetrischem
und antisymmetrischem räumlichen Anteil gibt. Nach dem Pauli-Prinzip und (2.9) ist dies
äquivalent zu einer Energieaufspaltung zwischen symmetrischer und antisymmetrischer Spinfunktion. Es soll nun genauer untersucht werden, welche Gestalt die möglichen Spinfunktionen für das Problem zweier Wasserstoatome annehmen. Es ist folglich die Kopplung zweier
17
KAPITEL 2. HERLEITUNG UND ANALYSE DES HEISENBERGMODELLS
Elektronen-Spins (s
= 1/2) zu analysieren. Eine Diskussion der Addition beliebig vieler Spins
wird in Abschnitt 4.2 erfolgen.
Beim Spin handelt es sich um einen intrinsischen Drehimpuls quantenmechanischer Objekte.
Er hat keine direkte klassische Entsprechung, da es sich um strukturlose Punktteilchen als
Träger dieses Drehimpulses handelt. Dennoch ist natürlich eine mathematische Beschreibung
des quantenmechanischen Spins möglich. Wie jedem Drehimpuls können auch dem Spin drei
räumliche Komponenten zugeschrieben werden. In der Quantenmechanik entspricht dies den
Sx , Sy
Operatoren
und
Sz .
Sie erfüllen die fundamentalen Vertauschungsrelationen:
[Si , Sj ] = ijk Sk
.
(2.39)
Als weiterer Operator bietet sich das Quadrat des Gesamtspins
S 2 = (Sx +Sy +Sz )2 an. Leicht
lässt sich zeigen, dass die einzelnen Komponenten des Spins mit dem Gesamtspinquadrat
vertauschen.
S 2 , Si = 0 ,
(i = x, y, z)
(2.40)
Dies bedeutet, dass es eine Basis von Zuständen im Hibertraum gibt, die gleichzeitig Eigen-
S2
und einer Komponente Si sind. Es seien nun |smi solche abstrakten Zustände,
s den Gesamtspin und m die z-Komponente des Spins repräsentiert. m kann dabei Werte
zwischen −s und s in ganzzahligen Schritten annehmen. (Für gewöhnlich wird die z-Achse als
zustände zu
wobei
Quantisierungsachse gewählt.) Es gelten dann die Eigenwertgleichungen
S 2 |smi = ~2 s(s + 1) |smi
Sz |smi = ~m |smi
,
(2.41)
.
(2.42)
Für spätere Rechnungen ist es auÿerdem praktisch, die Auf- und Absteigeoperatoren
S±
des
Spins zu denieren.
S± = Sx ± iSy
(2.43)
Nach dieser Denition wirken die Auf-/Absteigeoperatoren nur auf die z-Komponente des
Spins und erfüllen
p
S± |smi = ~ s(s + 1) − m(m ± 1) |sm ± 1i
Zustände mit bereits maximalem
malem
m
m
.
(2.44)
werden vom Aufsteigeoperator und Zustände mit mini-
vom Absteigeoperator vernichtet.
Diese bisherigen Zusammenhänge der Spinalgebra sind für allgemeine Spins
s
gültig und
werden für den allgemeinen Fall auch noch benötigt werden. Nun befassen wir uns aber mit
den Elektronen aus der Heitler-London-Näherung. Da Elektronen als Fermionen einen Spin
von 1/2 tragen, reduziert sich die Situation auf
Der Anschaulichkeit halber soll
m = −1/2
m = 1/2
s = 1/2,
m=±1/2.
als Spin-Up betrachtet und durch
als Spin-Down betrachtet und durch
↓
↑
dargestellt und
dargestellt werden. Ein Zustand aus zwei
Spins kann als Tensorprodukt zweier Spinzustände einzelner Elektronen angegeben werden.
|s1 m1 i ⊗ |s2 m2 i = |s1 m1 s2 m2 i = |m1 m2 i
Im letzten Schritt wurde ausgenutzt, dass
s = 1/2
(2.45)
fest ist und deshalb weggelassen werden
kann. Mit der eben eingeführten Darstellung durch Pfeile ergibt sich eine natürliche Basis
{ei }
aus vier möglichen Spin-Produktzustände für zwei Elektronen:
{|↑↑i , |↑↓i , |↓↑i , |↓↓i}
18
2.2. ADDITION ZWEIER ELEKTRONEN-SPINS
Um Aussagen über den resultierenden Gesamtspin
Sz
s
beziehungsweise dessen z-Komponente
machen zu können, bietet sich die Denition entsprechender Operatoren an.
S ≡ S(1) + S(2)
(2.46)
Sz ≡ Sz(1) + Sz(2)
(2.47)
Die Indizes 1 und 2 sind wieder bezugnehmend auf die beiden Elektronen. Bei Anwendung auf
die Tensorprodukte aus (2.45) wirkt
des gilt auch für
S(1)
und
S(2) .
(1)
Sz
nur auf
(2)
|s1 m1 i und Sz
nur auf
|s2 m2 i. Entsprechen-
Es gilt also
Sz |s1 m1 s2 m2 i = (Sz(1) + Sz(2) ) |s1 m1 i ⊗ |s2 m2 i
= (Sz(1) |s1 m1 i) ⊗ |s2 m2 i + |s1 m1 i (Sz(2) ⊗ |s2 m2 i)
= ~(m1 + m2 ) |s1 m1 s2 m2 i
(2.48)
.
Daraus folgt für die einzelnen Basiszustände:
Sz |↑↑i = ~ |↑↑i
Sz |↑↓i = 0 |↑↓i
(2.49)
Sz |↓↑i = 0 |↓↑i
Sz |↓↓i = −~ |↓↓i
Man erkennt daraus, dass die Spin-Produktzustände Eigenzustände zu
tor
Sz
Sz
sind, der Opera-
also diagonal bezüglich der eingeführten Basis ist. Dies gilt allerdings nicht für den
Gesamtspin-Vektoroperator
S. Unter Verwendung von (2.44) und der Tatsache, dass s = 1/2,
lässt sich
S 2 = (S(1) + S(2) )2 = (S(1) )2 + (S(2) )2 + 2S(1) · S(2)
(1) (2)
(1) (2)
= (S(1) )2 + (S(2) )2 + 2Sz(1) Sz(2) + S+ S− + S− S+
3
(1) (2)
(1) (2)
= ~2 + 2Sz(1) Sz(2) + S+ S− + S− S+
2
(2.50)
schreiben. Bestimmt man nun aus dieser Darstellung die Matrixelemente
(S 2 )ij = ei S 2 ej
,
(2.51)
so ergibt sich folgende Matrixdarstellung des Operators bezüglich der natürlichen Basis:
2
0
S 2 = ~2
0
0
0
1
1
0
0
1
1
0
0
0
0
2
(2.52)
Der Operator ist also augenscheinlich nicht diagonal. Die zu dieser 4×4-Matrix gehörigen
Eigenwerte und (normierten) Eigenzustände lassen sich aber recht einfach bestimmen. Dadurch
ist ein Übergang zu einer Gesamtspin-Basis geschehen, dessen allgemeine Herleitung im Abschnitt 4.2 gezeigt ist. In diesen vier Zuständen
m1 + m2
einer mit
|smi
steht
s
für die Gesamt-z-Komponente. Es ergeben drei Zustände
s=0
m=
mit Gesamtspin s = 1 und
für den Gesamtspin und
[Kop11]:
19
KAPITEL 2. HERLEITUNG UND ANALYSE DES HEISENBERGMODELLS
•
•
s=1:
|1 1i = |↑↑i
1
|1 0i = √ (|↑↓i + |↓↑i)
2
|1 − 1i = |↓↓i
(2.53)
1
|0 0i = √ (|↑↓i − |↓↑i
2
(2.54)
s=0:
s = 1 gehörigen Zustände bezeichnet man als Triplett-Zustände (Triplett, da aus drei
Zuständen bestehend), den Zustand zu s = 0 als Singulett-Zustand.
Die zu
Es ist nun klar, wie die vier möglichen Spinzustände des Zweielektronen-Systems aussehen.
Rückblickend auf die Resultate des vorherigen Abschnitts, ist es notwendig, darunter die symmetrischen/antisymmetrischen Spinzustände zu identizieren. Man erkennt direkt, dass die
Triplett-Zustände die Bedingung für eine symmetrische Funktion erfüllen, also invariant unter
Vertauschung der Elektronenindizes bleiben. Der Singulett-Zustand hingegen ist oensichtlich
ein antisymmetrischer Zustand. Die Spinzustände
χa/s sind also genau die Singulett-/Triplett-
Zustände, abgekürzt mit sing. und trip..
χa (s1 , s2 ) = χsing.
(2.55)
χs (s1 , s2 ) = χtrip.
(2.56)
Das wiederum bedeutet, dass zur Energie
E+
der Singulett-Zustand gehört und zu
E−
die
Triplett-Zustände.
In einer vereinfachten Anschauung können die Triplett-Zustände als eine parallele Ausrichtung
der Spins verstanden werden, während der Singulett-Zustand einer antiparallelen Ausrichtung
entspricht. Entsprechend gehört zu einer symmetrischen Ortswellenfunktion eine antiparallele
Spinanordnung und zu einer antisymmetrischen Ortswellenfunktion eine parallele Spinanordnung.
2.3 Singulett-/Triplett-Zustand und eektiver
Hamiltonoperator
Nun, da bekannt ist, dass für das System 0 und 1 die möglichen Gesamtspins sind und jedes
Elektron den Spin 1/2 trägt, können wir
S2
noch einmal genauer aufschreiben. Die Eigenwerte
für das Quadrat des Gesamtspinoperators sind nach (2.41) 0 für
für
s=1
J
(Singulett) und
2~2
(Triplett). Aus Gründen der Vereinfachung und weil dies auch in weiten Teilen der
Literatur Konvention ist, soll von hieran
in das
s=0
~ = 1
gelten. Tatsächlich wird der Faktor
~2
mit
aus (1.1) gesteckt. Dies sollte im Hinterkopf behalten werden, ändert aber rein gar
nichts an den qualitativen Aussagen der folgenden Erläuterungen. Gemäÿ (2.50) kann man
also schreiben:
20
2.3. SINGULETT-/TRIPLETT-ZUSTAND UND EFFEKTIVER HAMILTONOPERATOR
•
Singulett-Zustand:
S2 = 0 =
•
3
+ 2S(1) · S(2)
2
S(1) · S(2) = −
⇒
3
4
(2.57)
Triplett-Zustand:
S2 = 2 =
3
+ 2S(1) · S(2)
2
S(1) · S(2) =
⇒
1
4
Zuvor hatten wir bereits festgestellt, dass die unterschiedlichen Energieeigenwerte
(2.58)
E+
und
E−
des Systems direkt mit den Singulett- und Triplettzuständen in Zusammenhang stehen. Es
liegt ist nun also zweckmäÿig, einen Hamiltonoperator zu denieren, der die Energiedierenz
∆E = E+ − E− = J
in Abhängigkeit der beiden unterschiedlichen Werte von
S(1) · S(2)
ausdrückt. Dies ist durch den eektiven Hamiltonoperator
1
Hef f = (E+ + 3E− ) − (E+ − Et )S(1) · S(2)
4
(2.59)
gegeben. Er liefert genau die zuvor ermittelten Eigenenergien für die einzelnen Zustände. Um
ihn in einer noch einfacheren Form darzustellen, kann durch eine Verschiebung des Energienullpunktes
von
J
1
4 (E+
+ 3E− ) = 0
gesetzt werden [AM76, S.865]. Durch Einsetzen der Denition
entsteht dann der Spin-Hamiltonoperator
Hspin = −JS(1) S(2)
Hieraus ist ersichtlich, warum
J
.
(2.60)
als Austausch-Kopplungskonstante (Kopplungsparamter)
bezeichnet wird. Die Konsequenz des Hamiltonoperators (2.60) ist nun, dass in Abhängigkeit
des Vorzeichens von
J,
entweder eine parallele (J
> 0)
oder eine antiparallele (J
< 0)
Aus-
richtung der Spins zueinander energetisch begünstigt wird. Dies wiederum ist gleichbedeutend mit ferro- beziehungsweise antiferromagnetischen Stoeigenschaften (Näheres dazu in
3.2). Wie
J
tatsächlich aussieht, hängt natürlich immer von der speziellen Situation, also den
an der Wechselwirkung beteiligten Nachbarn, ab. Es handelt sich dabei nicht um eine magnetische Dipolwechselwirkung wie aus der Herleitung hervorgegangen ist. Deshalb ist nur die
relative Ausrichtung der Spins entscheidend. Im Falle der zwei Wasserstoatome lässt sich
für
J
eine numerische Lösung in Abhängigkeit des angenommenen Kernabstandes nden. Es
zeigt sich dabei, dass
J
bei einem realistischen Kernabstand negativ ist und der Grundzu-
stand des Wasserstomoleküls entsprechend ein Singulett-Zustand ist. Dieses Ergebnis aus der
Näherung wird durch experimentelle Befunde bestätigt, wenngleich es Diskrepanzen bezüglich
der kalkulierten und tatsächlichen Bindungsenergien gibt [HW92, S.68].
Oftmals sind genaue numerische Werte von
J
allerdings nicht von groÿer Bedeutung, da das
Modell unter der Annahme allgemeiner positiver oder negativer Austauschparameter bereits
zu qualitativen Aussagen in der Lage ist. Dies wird sich gerade im Zuge der konkreten Rechnungen am Heisenbergmodell zeigen.
Der Übergang zu eben jenem Modell ist aber noch nicht ganz vollzogen. Der Hamiltonoperator des Heisenbergmodells (1.1) resultiert direkt aus dem Spin-Hamiltonoperator (2.60),
wenn wir nun ein Gittersystem aus Atomen auf
N
Gitterplätzen annehmen. Dabei gehen wir
21
KAPITEL 2. HERLEITUNG UND ANALYSE DES HEISENBERGMODELLS
davon aus, dass ein jedes Atom nur mit den Atomen auf den jeweils nächstbenachbarten Gitterplätzen in Wechselwirkung tritt. Der Hamiltonoperator des Gesamtsystems entspricht dann
genau der Summe über die einzelnen Spin-Hamiltonoperatoren zwischen nächsten Nachbarn.
Dies ist genau der Heisenberg-Hamiltonoperator
HHeis = −
X
JSi · Sj
.
(2.61)
hi,ji
(An der Notation hat sich hier geändert, dass die Indizes an den Spinoperatoren, die den
Gitterplatz kennzeichnen, nun unten stehen, um sie von Potenzen zu unterscheiden.)
Damit ist eine Herleitung des Heisenbergmodells, beziehungsweise seines Hamiltonoperators, erfolgt. Dies ist unter Verwendung zahlreicher Annahmen und Vereinfachungen geschehen,
die allerdings auch gleichbedeutend mit subtileren physikalischen Überlegungen sind. Es ist
klar, dass das Modell keine exakten Ergebnisse liefern kann und davon auszugehen, dass es
nur unter bestimmten Rahmenbedingungen gute Näherungen liefert. Andererseits stellt der
Heisenberg-Hamiltonoperator (2.61) einen eektiven Hamiltonoperator dar, der ganz ohne
quantenmechanische Ortsfunktionen auskommt und lediglich der Betrachtung der Spins bedarf. In Anbetracht der Gröÿe von Systemen der Festkörperphysik wird der groÿe Vorteil eines
solchen eektiven Hamiltonoperators deutlich. Man beachte, dass die Auswertung selbst des
Heisenberg-Hamiltonoperators im Allgemeinen ein schwieriges Unterfangen ist, natürlich in
Abhängigkeit von der Gröÿe des zu untersuchenden Systems. Exakte Lösungen des Modells
für unendlichen Systeme, mit denen man es praktisch zu tun hat, sind im Allgemeinen nur
für eindimensionale Systeme möglich (die Spinkette, berechnet durch Hans Bethe [Bet31]).
In zwei und drei Dimensionen sind nur genäherte Lösungen, zum Beispiel durch Verwendung
von Monte-Carlo-Methoden, zu erzielen. Methoden zur Abschätzung der Grundzustandsenergie unendlicher Systeme sind ein wichtiger Bestandteil dieser Arbeit und Thema des Kapitels
4.
Man kann sich vorstellen, dass ein Modell, das mit weniger Näherungen auskommt, zwar exaktere Lösungen liefert, aber auch weniger praktikabel in seiner Auswertung ist. Nicht zuletzt
deshalb stellt das Heisenbergmodell den Ausgangspunkt tiefergehender Untersuchungen, vor
allem des Magnetismus von Festkörpern, dar.
Verallgemeinerungen des Modells
Bei der Begründung des Hamiltonoperators (2.61)
wurde implizit angenommen, dass zwischen allen Gitternachbarn dieselbe Austauschkopplung
J
besteht. Da sich die Berechnung von
J
aber aus der konkreten Gestalt der wechselwirkenden
Teilchen ergibt, ist ihr Wert für unterschiedliche Bindungspartner im Allgemeinen nicht gleich.
Somit kann eine allgemein gültigere Form des Heisenberg-Hamiltonoperators (2.61) gegeben
werden, indem
J
Jij ersetzt wird. Dadurch ist die Abhängigkeit der Austauschkopplung
i und j ausgewiesen. Zudem kann man auch den Fall betrachten, dass
durch
von den Gitterplätzen
die Kopplungskonstante richtungsabhängig ist, man also von anisotropen Systemen ausgeht.
In diesem Fall schreibt sich der verallgemeinerte Heisenberg-Hamiltonoperator:
Hallg. = −
X <i,j>
22
J x Six Sjx + J y Siy Sjy + J z Siz Sjz
.
(2.62)
2.3. SINGULETT-/TRIPLETT-ZUSTAND UND EFFEKTIVER HAMILTONOPERATOR
Zudem wird für praktische, statistische Berechnungen oftmals ein Übergang zu Modellen
gemacht, in denen die Spins der Gitterplätze sich nur in einer Ebene (XY-Modell) oder nur
entlang einer Achse (Ising-Modell) ausrichten können.
In dieser Arbeit relevant und Grundlage meiner eigenen Berechnungen ist nur der HeisenbergHamiltonoperator isotroper Systeme wie er in (1.1) niedergeschrieben ist.
23
3 Austauschwechselwirkung und
Magnetismus
Das vorausgegangene Kapitel hat eine Begründung des Heisenbergmodells geliefert. Dabei
sind bereits einige Aspekte zur Sprache gekommen, die entweder unmittelbar mit dem Modell
in Zusammenhang stehen oder Teil der in dieser Arbeit behandelten Thematik sind. Dabei sei
beispielsweise an die Theorie von Ferro- und Antiferromagnetismus oder den erwähnten Eekt
der Austauschwechselwirkung gedacht. Diese und andere Aspekte sind für das Verständnis der
Begründung für das Modell, aber auch der Folgerungen aus dem Modell, unabdingbar. Deshalb sollen in diesem Kapitel einige spezielle, weiterführende Themen aufgegrien, analysiert
und diskutiert werden. Diese lassen sich unter den Themen Austauschwechselwirkung und
Magnetismus zusammenfassen.
3.1 Austauschwechselwirkung
Der Heisenberg-Hamiltonoperator (1.1) enthält den Austauschparameter
J.
Welchen Wert
J
in einem System annimmt, hängt von der konkreten Situation ab und ist entscheidend für das
magnetische Verhalten des Systems, wie aus Abschnitt 3.2 hervorgehen wird. Dies ist insbesondere vom Vorzeichen des Wertes abhängig. Die Entstehung des Parameters ist bereits im Zuge
der Herleitung des Heisenbergmodells erläutert worden. Zudem ist bereits erwähnt, dass sich
diese Kopplungskonstante auf dem quantenmechanischen Eekt der Austauschwechselwirkung
begründet, beziehungsweise mit ihr gleichzusetzen ist. Eben diese Austauschwechselwirkung
soll nun diskutiert werden.
3.1.1 Wasserstomolekül und direkter Austausch
Für die Analyse der Austauschwechselwirkung sei noch einmal das Wasserstomolekül aus
der Herleitung des Heisenbergmodells in Kapitel 2 als Verdeutlichung des zugrunde liegenden Prinzips herangezogen. Einer des wesentlichen Punkte der Herleitung war die Forderung
nach antisymmetrischen Gesamtwellenfunktionen für das Zweielektronen-System. Sie hatte
einen rein physikalischen Hintergrund, nämlich den des Pauli-Prinzips. Darauf sei besonders
eindringlich hingewiesen, da es letztlich allein dieses Prinzip der Quantenmechanik ist, das
die Austauschwechselwirkung hervorruft und dazu führt, dass ihr kein klassisches Analogon
zuzuschreiben ist.
Es resultieren daraus die beiden Ortswellenfunktionen
ψs
(2.11) und
ψa
(2.12). Als Folge
25
KAPITEL 3. AUSTAUSCHWECHSELWIRKUNG UND MAGNETISMUS
dessen ist in der gezeigten Rechnung eine Dierenz in den Erwartungswerten der Energien für
die symmetrische und antisymmetrische Ortswellenfunktion entstanden (siehe (2.38)). Diese
Dierenz ist von den Integralen
U
L
(2.24),
K
(2.27) und
U
(2.26) abhängig, wobei
L
und
durch den Überlapp der Wellenfunktionen ungleich null sind. Im Grunde genommen liegt
genau darin eine Denition der Austauschwechselwirkung: Die Austauschwechselwirkung ist
ein quantenmechanischer Eekt, ohne klassisches Analogon, der zu einer Änderung des Energieerwartungswertes eines Systems mehrerer identischer Teilchen führt, wenn ihre Wellenfunktionen überlappen [Mat32]. Die Bedetung dessen wird nun etwas genauer analysiert.
Dazu betrachten wir die zu
J
beitragenden Terme. Zur Erinnerung sind die Integrale noch
einmal aufgeführt:
Z
L=
ZZ
K=
,
ZZ
V 0 (r1 , r2 )|φA (r1 )φB (r2 )|2 dV1 dV2
ZZ
V 0 (r1 , r2 )|φA (r2 )φB (r1 )|2 dV1 dV2
U=
=
φA (r1/2 )φB (r1/2 )dV1/2
,
V 0 (r1 , r2 )φA (r1 )∗ φA (r2 )φB (r2 )∗ φB (r1 )dV1 dV2
Der erste Teil ist das Überlappintegral
L,
.
dessen grundsätzliche Bedeutung bereits geklärt
wurde. Das Überlappintegral kommt nur in geraden Potenzen vor und trägt deshalb selbst
nur positiv bei. Es lohnt sich hierbei die Extremfälle für maximalen und minimalen Überlapp
der Wellenfunktionen in Betracht zu ziehen. Maximal wird das Integral, wenn
φA = φB
ist.
Dies wäre der Fall, wenn beide Elektronen durch dasselbe Orbital beschrieben würden. Also - im Falle der Heitler-London-Näherung - bei unendlich kleinem Kernabstand. Aufgrund
der Normierung der Einteilchen-Wellenfunktionen würde das Überlappintegral eins werden,
wofür
J
nicht mehr deniert wäre, beziehungsweise als divergierend betrachtet werden kann. In
diesem Fall macht eine Denition einer Austauschwechselwirkung allerdings auch keinen Sinn
mehr, da die antisymmetrische Ortsfunktion verschwinden würde, also nur die symmetrische
Ortsfunktion existieren würde. Dies geht auch direkt aus dem Pauli-Prinzip hervor, da demnach Elektronen, die durch dasselbe Orbital beschrieben werden, einen unterschiedlichen Spin
haben müssen. Dies ist gleichbedeutend mit einem Singulett-Zustand und somit auch mit der
symmetrischen Ortswellenfunktion.
Betrachtet man nun den Extremfall zweier Wellenfunktionen, die überhaupt keinen Überlapp haben, so würden sowohl
L als auch K
null werden. Es würde also keine Austauschwech-
selwirkung existieren. Dies ist auch nicht anders zu erwarten, da dies der Situation eines
Systems zweier unabhängiger Elektronen entspricht. Für ein solches System kann es keinen
energetischen Unterschied machen, welche Ausrichtungen die Spins annehmen. Diesen Grenzwert erzielt man durch die Annahme unendlich weit entfernter Atromrümpfe.
Bei den tatsächlich relevanten Problemen handelt es sich natürlich um solche, bei denen eine
Situation zwischen den genannten Extremfällen vorliegt. Entsprechend wird auch für
Situation dazwischen vorliegen. Es kommt dann auf die Integrale
J
und
U
eine
an, welchen Wert
annehmen wird, wie aus (2.38) hervorgeht.
Der nächste zu
26
K
J
J
beitragende Teil ist
U.
Dieser Anteil ist auch klassisch noch interpretier-
3.1. AUSTAUSCHWECHSELWIRKUNG
bar und entspricht letztlich nichts anderem als der Coulombwechselwirkung. Folglich wird
auch als Coulomb-Integral bezeichnet. Da
U
0 eine Summe über die einzelnen Energiebeiträge
V
der Coulombenergien darstellt, lohnt es sich, auch
U
(2.26) als Summe über deren Integrale
U ist dann als
ZZ e2
e2
e2
e2
−
+
+
|φA (r1 )|2 |φB (r2 )|2 dV1 dV2
U=
−
4π0 r1B
4π0 r2A 4π0 r12 4π0 RAB
darzustellen.
zu schreiben. Die von einem Elektron, das durch das Orbital
hende Ladungsdichteverteilung
ρ(r)
φ(r)
(3.1)
beschrieben wird, ausge-
ist gegeben durch
ρ(r) = −e|φ(r)|2
.
(3.2)
Entsprechend stellt das Integral
ZZ
e2
−
|φA (r1 )|2 |φB (r2 )|2 dV1 dV2 =
4π0 r1B
Z
−
e2
ρA (r1 )dV1 ≡ C < 0
4π0 r1B
(3.3)
genau die Coulombsche Wechselwirkungsenergie des Elektrons 1 im Zustand A mit dem Kern
B dar. Vollkommen analog berechnet sich die Coulombenergie des Elektrons 2 im Zustand A
mit Kern B. Aufgrund der Symmetrie des Systems nimmt es den exakt selben Wert
ZZ
−
e2
|φA (r1 )|2 |φB (r2 )|2 dV1 dV2 =
4π0 r2A
Z
−
e2
ρB (r2 )dV2 = C < 0
4π0 r2A
C
an.
(3.4)
Es fehlen nun noch die Coulombenergien, die aus der Elektron-Elektron-Wechselwirkung
(EW W ) und aus der Proton-Proton-Wechselwirkung hervorgehen. Erstere ist gegeben durch
ZZ EW W =
e2
4π0 r12
Es beeinhaltet die Abhängigkeit von
r1
|φA (r1 )|2 |φB (r2 )|2 dV1 dV2 > 0
r2
und
.
(3.5)
und muss deshalb über beide Ortsvariablen
integriert werden. Da die Elektronen sich abstoÿen, stellt diese Wechselwirkung ein repulsives
Potenzial dar und wird stets einen positiven Beitrag liefern.
Das letzte Integral, die Proton-Proton-Wechselwirkung, enthält keine Abhängigkeit von den
Aufenthaltsorten der Elektronen. Bei festem
RAB
erhält man aus diesem ebenfalls repulsiven
Potenzial einfach
ZZ e2
4π0 RAB
|φA (r1 )|2 |φB (r2 )|2 dV1 dV2 =
e2
4π0 RAB
.
(3.6)
Man kann nun einige Feststellung bezüglich des aus aus den Coulomb-Wechselwirkungsenergien resultierenden Vorzeichens von
J
machen. Die Wechselwirkungen der Elektronen mit
dem jeweils anderen Kern stellen ein attraktives Potenzial dar (C
wirkungen groÿ, begünstigen sie nach (2.38) ein negatives
J.
> 0).
Sind diese Wechsel-
Die Elektron-Elektron-Wechsel-
wirkung hat eine gegenteilige Wirkung. Ebenso begünstigt eine groÿe Proton-Proton-Wechselwirkung (3.6) ein positives
J.
Man sieht, dass ihr Beitrag für kleinere Kernabstände wächst.
Für unendlich kleine Kernabstände liegt dann wieder die oben beschriebene Situation vor, die
einen Singulett-Zustand des Systems fordert.
Es verbleibt die Auswertung des Integrals
K
(2.27). Dieses Intgeral hat nun keine klassische
Entsprechung mehr und ist bekanntlich auch erst aufgrund des Pauli-Prinzips entstanden. Es
27
KAPITEL 3. AUSTAUSCHWECHSELWIRKUNG UND MAGNETISMUS
liefert also den phänomenologisch entscheidenden Beitrag zum Austauschparameter
J
und
wird selbst als Austausch-Integral bezeichnet. Deniert man anstelle der Ladungsdichte die
Austauschdichte
%(r) ≡ φA (r)∗ φB (r)
φA
zweier Zustände
und
φB ,
(3.7)
so kann man im Austauschintegral Terme in Analogie zu denen
des Coulombintegrals nden. Wieder ergibt sich eine Summe von Integralen mit den verschiedenen Coulomb-Wechselwirkungen im Integranden, die allerdings in diesem Fall über die
Austauschdichten integriert werden.
ZZ −
K=
e2
e2
e2
e2
−
+
+
4π0 r1B
4π0 r2A 4π0 r12 4π0 RAB
φA (r1 )∗ φA (r2 )φB (r2 )∗ φB (r1 )dV1 dV2
(3.8)
Eine Auswertung des ersten Summanden bringt dann:
ZZ −
e2
φA (r1 )∗ φA (r2 )φB (r2 )∗ φB (r1 )dV1 dV2
4π0 r1B
ZZ e2
−
=
%(r1 )%(r2 )dV1 dV2
4π0 r1B
Z e2
=L
−
%(r1 )dV1
4π0 r1B
(3.9)
≡ LD
Es taucht hierin also wieder das Überlappintegral auf und zudem ein Integral, das als Einelektronen-Austauschintegral gelten kann. Es wird darin der Tatsache Rechnung getragen, dass
auch das Elektron im Zustand B sich mit einer gewissen Wahrscheinlichkeit am Ort
r1
ben-
det und somit zur Wechselwirkung beiträgt.
Der zweite Term stellt aus Symmetriegründen die exakt selbe Einelektronen-Austauschwechselwirkung dar.
ZZ e2
−
4π0 r1B
φA (r1 )∗ φA (r2 )φB (r2 )∗ φB (r1 )dV1 dV2 = LD
(3.10)
Der dritte Term repräsentiert wie schon bei der Coulomb-Wechselwirkung eine Elektron-Elektron-Wechselwirkung, nun aber in Form der Coulombschen Austauschwechselwirkung
ZZ EAW =
e2
4π0 r12
φA (r1 )∗ φA (r2 )φB (r2 )∗ φB (r1 )dV1 dV2
EAW .
(3.11)
Die verbleibende Wechselwirkung der Atomkerne reduziert sich wieder stark und lässt sich
einfach als
ZZ e2
4π0 RAB
φA (r1 )∗ φA (r2 )φB (r2 )∗ φB (r1 )dV1 dV2 = L2 ·
e2
4π0 RAB
(3.12)
schreiben.
Auf recht detaillierte Weise sind damit die einzelnen Bestandteile ermittelt, die den Austauschparameter
J
erzeugen. Welches Vorzeichen
J
im konkreten Fall annimmt, kommt da-
rauf an, welche Werte die einzelnen Terme haben, ist also abhängig von den Partnern der
Wechselwirkung. Es ist bereits bekannt, dass das Pauli-Prinzip für identische Teilchen zu einer Energieaufspaltung zwischen symmetrischen und antisymmetrischen Ortsfunktionen führt.
28
3.1. AUSTAUSCHWECHSELWIRKUNG
Abbildung 3.1: Bindungsenergie des Wasserstomoleküls in Abhängigkeit vom Kernabstand RAB
[Web]
Dies ist direkt mit einer solchen Aufspaltung zwischen den Spinzuständen verbunden. Dies
ist vorallem deshalb bemerkenswert, weil eine direkte Spin-Spin-Wechselwirkung für dieses
Ergebnis nie in Betracht gezogen wurde. Rein elektromagnetische Kräfte führen also zu einer
magnetischen Wechselwirkung. Eine direkte Spin-Spin-Wechselwirkung in Form einer DipolDipol-Wechselwirkung
ED
kann man Abschätzen durch
ED = −
([AM76, S.856];
µB :
2µ0 µ2B
≈ 1, 6 · 10−23 J ≈ 100µeV
4πr3
Bohrsches Magneton; angenommener Abstand
Dies entspricht einer Temperatur von nur etwa 1,2
K.
.
(3.13)
r
von einigen Ångstrøm.)
Es ist klar, dass diese Wechselwirkung
nicht für kooperative magnetische Eekte verantwortlich sein kann. (Bei sehr kleinen Temperaturen ist die Dipol-Dipol-Wechselwirkung natürlich durchaus zu berücksichtigen.) Der
Grund für die groÿe Spin-Spin-Wechselwirkung beruht also letztlich auf den Coulombwechselwirkungen und wird in Form der quantenmechanischen Austauschwechselwirkung vermittelt,
wie sich in den obigen Ausführungen herausgestellt hat.
Gehen wir nocheinmal auf das Bild des Wasserstomoleküls zurück, so ist es genau die
Austauschwechselwirkung, die dafür sorgt, dass ein bindender Zustand existiert, also stabile Wasserstomoleküle bestehen. Die Energien
hängigkeit vom Kernabstand
RAB
E+
(2.36) und
E−
(2.37) lassen sich in Ab-
für das Wasserstomolekül berechnen. Die zugehörigen
Kurven sind in Abbildung 3.1 gezeigt und darin die Kurven für den Singulett-/Triplettzustand
gekennzeichnet. Der Energieverlauf der Kurve für den Singulettzustand hat ein Minimum bei
etwa 0,75 eV, er ist demnach bindend. Der Triplettzustand besitzt kein lokales Minimum und
ist antibindend.
Die Erklärungen zur Austauschwechselwirkung sind gröÿtenteils anhand des Wasserstomoleküls geschehen, das schon Ausgangspunkt der Herleitung des Heisenbergmodells war. Dies
ist allerdings keineswegs als eine beschränkende Bedingung zu bewerten. Die Austauschwechselwirkung tritt bei allen Systemen von identischen Teilchen auf und stellt ein allgemeines
Prinzip dar, wie es hier gezeigt wurde.
29
KAPITEL 3. AUSTAUSCHWECHSELWIRKUNG UND MAGNETISMUS
Alles was bisher bezüglich der Austauschwechselwirkung beschrieben wurde, meint im
Speziellen den sogenannten direkten Austausch. Die Bezeichnung ist naheliegend, weil sich
die magnetische Wechselwirkung als Folge der direkten Coulomb-Wechselwirkung zwischen
den Elektronen der beiden Atomrümpfe ergibt.
Dieser direkte Austausch ist der für die in dieser Arbeit behandelten Probleme und Anwendungen relevante. Es gibt jedoch noch andere Formen der Austauschwechselwirkung, die in der
Festkörpertheorie im Allgemeinen durchaus von Bedeutung sind und deshalb auch Erwähnung
nden sollen.
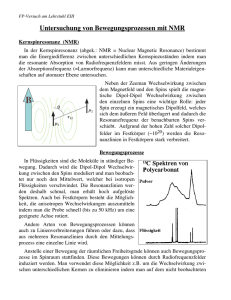
3.1.2 Superaustausch
Die erste der indirekten Austauschwechselwirkungen, die hier benannt werden soll, ist der
sogenannte Superaustausch.
In einer Vielzahl von ionischen Festkörpern existieren magnetische Grundzustände, also feste Ausrichtungen der Ionen-Spins. Dazu zählen einige Oxide und Flouride wie etwa
Manganoxid (MnO) oder Manganourid (MnF2 ), die Antiferromagnete darstellen. Dies ist
auf den ersten Blick dahingehend überraschend, als dass zuvor deutlich geworden ist, dass
ein direkter Austausch nur entsteht, wenn die entsprechenden Atomorbitale überlappen. Zwischen den Mn
2+ -Ionen in den genannten Stoen existiert jedoch kein Überlapp, da sich stets
2− -Ionen zwischen ihnen benden. Es muss also eine langreichweitige Wechselwirkung für
O
den Austausch ursächlich sein. Dies gibt einen Hinweise auf den Namen Super-Austausch.
Der Superaustausch kann also als ein indirekter Austausch zwischen nicht benachbarten magnetischen Ionen verstanden werden, der durch ein
dazwischen liegendes nicht-magnetisches Ion vermittelt wird [Blu08, S. 77]. Schematisch ist die
Anordnung der beteiligten Spins in Abbildung 3.2
gezeigt. Ohne Beschränkung der Allgemeinheit
sind für die Metall-Ionen einzelne, ungepaarte
Elektronen angenommen, während im SauerstoMolekül ein doppelt besetztes p-Orbital vorliegt.
Da es sich nicht um perfekt ionische Bindungen handelt, sind die Elektronen des vermittelnden Ions nicht komplett lokalisiert, sondern können in die benachbarten Orbitale hüpfen (hopping). Die Elektronen sind delokalisiert, was eine
kleinere und somit günstigere kinetische Energie
zur Folge hat.
Abbildung 3.2: Delokalisierte Elektronen in
antiferromagnetischen (a, b und c) und ferromagnetischen (d, e und f) Zuständen [Blu08]
Eine formal-mathematische Betrachtung des
Superaustausches
tisierung
und
erfordert
geschieht
die
über
zweite
Quan-
Störungstheorie
zweiter Ordnung des Hubbard-Modells. In dieser Arbeit möchte ich es bei einer weitgehend
30
3.1. AUSTAUSCHWECHSELWIRKUNG
qualitativen und weniger formalen Erklärung belassen und nur eingeschränkt auf die genannten Methoden zurückgreifen, da der Rahmen dieser Arbeit es nicht erfordert.
Zum besseren Verstädnis sei die Berechnung der Energiekorrektur
En2
zweiter Ordnung der
zeitunabhängigen Störungstheorie aufgeführt. Sie ist gegeben durch
En2 = hn0 |H1 |n1 i =
X hn0 |H1 |k 0 ihk 0 |H1 |n0 i
X |hk 0 |H1 |n0 i|2
=
.
En0 − Ek0
En0 − Ek0
k(6=n)
(3.14)
k(6=n)
Hierbei nummeriert der obere Index jeweils die Ordnung der Korrektur aus der Störungstheorie. Mit 0 im Index sind also die Zustände und Energien des ungestörten Systems gemeint.
Die unteren Indizes
n/k
n-ten/k -ten
meinen den
Eigenzustand des Systems (n
= 0 entspricht
also dem Grundzustand).
Zur Beschreibung des Superaustausches verwendet man den Hamiltonoperator des HubbardModells (siehe zum Beispiel im Buch von Ashcroft und Mermin [AM76]). Er lautet:
H Hubb. = U
X
c†i↑ ci↑ c†i↓ ci↓ − t
i
(σ : Spinrichtung (up oder down).
X
c†iσ cjσ + c†jσ ciσ
.
(3.15)
hiji,σ
i, j :
Gitterplätze.
c, c† :
Vernichtungs-, Erzeugungsoper-
atoren.) Er besteht also aus einem diagonalen Teil, der aus dem Produkt einer Wechselwirkungsenergie
Bei
U
U > 0 und der Anzahl der doppelt besetzten Niveaus der Elektronen besteht.
handelt es sich um die bereits bekannte Abstoÿung durch das Coulombpotential. Dop-
pelt besetzte Orbitale sind also - wie nicht anders zu erwarten - aus elektrostatischer Hinsicht
ungünstig. Der zweite Term ist nicht diagonal und enthält von Null verschiedene Matrixelemente
t
zwischen Zustandspaaren, die sich nur dadurch voneinander unterscheiden, dass ein
einzelnes Elektron ohne Änderung seines Spins von einem gegebenen Gitterplatz auf einen
Nachbarplatz bewegt wurde. Der Parameter
t
repräsentiert also das angesprochene hopping.
Die Energiekorrektur ist im vorliegenden Fall also nichts anderes als zweimal das Quadrat des
Matrixelements
t
geteilt durch die Anregungsenergie
En2 = −
U.
2t2
U
(3.16)
In dem Austauschintegral taucht neben der Korrektur natürlich auch der Teil auf, der die
Elektron-Elektron-Abstoÿung repräsentiert, also der erste Term aus dem Hamiltonoperator
des Hubbard-Modells. Es ist jedoch bereits klar geworden, dass dieser Teil vernachläsigbar
klein ist, wenn die Metall-Ionen weit genug voneinander getrennt sind, was hier angenommen werden kann. Es kann analog zur direkten Austauschwechselwirkung eine eektive Austauschwechselwirkung aufgeschrieben werden.
H = −Jse
X
hiji
Si · Sj
mit
Jse =
−2t2
U
(3.17)
Das virtuelle Hüpfen der Elektronen führt also zu einer aniferromagnetischen HeisenbergAustauschwechselwirkung. Das heiÿt, die Spins sind energetisch günstiger antiparallel ausgerichtet. Dies ist der Superaustausch.
Diese Resultate aus der formellen Analyse lassen sich auf anschauliche Weise gut aus Abbildung 3.2 ersehen. Im oberen Teil ist in (a) der antiferromagnetische Grundzustand (Spins
31
KAPITEL 3. AUSTAUSCHWECHSELWIRKUNG UND MAGNETISMUS
antiparallel) dargestellt. Durch Elektronen-hopping kann dieser mit den angeregten Zuständen (b) und (c) mischen. Die dadurch hervorgerufene Delokalisierung stabilsiert das System.
Der ferromagnetische Grundzustand (d) kann nicht mit angeregten Zuständen mischen. Es
ist leicht zu sehen, dass die Zustände (e) und (f ) gegen das Pauli-Prizip verstoÿen. Dies führt
ebenfalls zu der Erkenntnis, dass der antiferromagnetische Zustand energetisch günstiger ist.
Es ist somit anhand einer Beispielsituation deutlich geworden, wie eine Wechselwirkung zwischen Elektronen(spins) zustande kommt, die nicht direkt wechselwirken, sondern nur indirekt
über einen gemeinsame Bindungspartner. Dies ist genau das Prinzip des Superaustausches und
dient dem qualitativen Verständnis des Phänomens.
Es ist allerdings zu erwähnen, dass natürlich nicht stets die hier angenommenen Geometrien
und Niveaubesetzungen vorliegen. Bisher wurde eine lineare Anordnung der drei Atomrümpfe
und jeweils einfach besetzte Metall-Ionen angenommen. Der Vollständigkeit halber soll kurz
eine weitere Situationen beschrieben werden. Durch die Regeln von Goodenough, Kanamori
und Anderson (GKA-Regeln, siehe [Goo63]) sind Stärke und Art der Austauschwechselwirkung für verschiedene Typen von Systemen bestimmt.
Angenommen sei ebenfalls eine lineare Anordnung aus zwei Metallionen und einem SauerstoIon dazwischen. Allerdings sei nun nur ein Metall-Orbital gefüllt. In diesem Fall (zweite GKARegel) kann ein p-Elektron des Wasserstos in das unbesetzte Metall-Orbital hüpfen. Gemäÿ
1
der zweiten Hundschen Regel
wird es mit paralleler Spinausrichtung eingebaut, während das
andere p-Elektron eine antiferromagnetische Kopplung vorzieht. Insgesamt ergibt sich dadurch
eine parallele Ausrichtung der Spins in den Metall-Ionen, was einer ferromagnetischen Wechselwirkung zwischen den Metall-Ionen gleichkommt.
Zudem sind auch nicht lineare Anordnungen der Atomrümpfe möglich, die wiederum andere
Wechselwirkungen hervorrufen. Die prinzipiellen Ursachen dieser verschiedenen Formen des
Superaustausches sind jedoch stets dieselben.
3.1.3 Doppelaustausch
Die zweite Form der indirekten Austauschwechselwirkung, die hier besprochen werden soll, ist
der Doppelaustausch. Er basiert auf denselben Prinzipien wie der Superaustausch. Es handelt
sich also wiederum um eine Austauschwechselwirkung zwischen zwei Atomen/Ionen, die durch
einen nicht-magnetischen gemeinsamen Bindungspartner vermittelt wird.
Für den Superaustausch wurden jeweils Systeme aus zwei Metall-Ionen (Mangan) und einem
vermittelnden Sauersto-Ion betrachtet. Dabei ist von Metall-Ionen mit gleicher Valenz ausgegangen worden, um die besprochenen Eekte zu beschreiben. Das Verständnis des Doppelaustauschs erfordert nun die Betrachtung von Systemen, deren Metall-Ionen unterschiedliche
Valenzen besitzen. Diese Situation kann praktisch zum Beispiel herbeigeführt werden, in-
3+ -Ionen durch Sr2+ -Ionen ersetzt. In dem entprechenden
dem man in LaMnO3 einige La
La1−x Srx MnO3 wird formal ein Elektron aus einem besetzten Mangan-Orbital entfernt. Auf
1
Die Hundschen Regeln sind fundamentale Regeln der Atomphysik und machen eine Aussage darüber, in
welcher Drehimpulskonguration die Elektronen in den Orbitalen eines Atoms im Grundzustand vorliegen.
Die zweite Hundsche Regel lautet, dass der Gesamtspin der Elektronen in einem Atom den maximal
möglichen Wert annimmt, die Spins also möglichst parallel stehen. Näheres siehe zum Beispiel im Buch
von Haken und Wolf [HW92].
32
3.1. AUSTAUSCHWECHSELWIRKUNG
diese Weise entstehen in dem Material sowohl drei- als auch viervalente Mangan-Ionen, die
weiterhin ein Oxid-Ion als gemeinsamen Nachbarn haben. Das zu dieser Situation gehörige
Orbitalschema ist in Abbildung 3.3 gezeigt.
Dieses Schema lässt das Prinzip des Doppelaustausches gut deutlich werden. Wiederum betrachtet man hopping-Prozesse von Elektronen über
das Oxid-Ion. Aufgrund der gegebenen Orbitalbesetzungen kann das Hüpfen nur vom linken auf
das rechte Mangan-Ion geschehen. Dies kann erfolgen, indem zunächst ein Elektron aus dem
Oxid-Ion auf das Mangan-Ion mit dem unbesetzten Orbital übergeht. Aufgrund der antiferromagnetischen Wechselwirkung zwischen dem
linken Metall-Ion und dem Oxid-Ion und da beim
Hüpfen kein Spin-Flip stattndet, wird dieses
Übergangselektron die selbe Spinrichtung haben
wie die Elektronen des linken Metall-Ions. Der
bereits benannten Hundschen Regel zufolge ist
dieser Übergang aber nur möglich, wenn die Elektronenspins beider Metall-Ionen gleichermaÿen
ausgerichtet sind, wie auch aus der Abbildung
hervorgeht. Völlig analog zur Begründung des Superaustausches gilt auch hier, dass ein Elektronenhüpfen zu einer Minderung der kinetischen
Abbildung 3.3: Der Doppelaustausch erzeugt
eine parallele Anordnung der Elektronenspins
in den Mangan-Ionen. Nur wenn die Spins parallel ausgerichtet sind, ist ein energetisch günstiges hopping möglich (oben). [Blu08]
Energie führt und damit das System energetisch
begünstigt. Schlussendlich führt dies zu einer indirekten Austauschwechselwirkung zwischen den
Mangan-Ionen, dem Doppelaustausch, der eine
parallele Spinausrichtung hervorruft und somit
ferromagnetisch ist.
Es sei darauf hingewiesen, dass im Falle parallel ausgerichteter Spins, im Gegensatz zum
Superaustausch, keine Anregungsenergie (U ) gegen ein Coulombpotenzial geleistet werden
muss. Dies bedeutet, dass die Elektronen leicht zwischen den Metall-Ionen des Gitters hüpfen
können und damit eine gute Leitfähigkeit besteht. Der Doppelaustausch sorgt dadurch für
einen metallischen Charakter der betreenden Materialien. Dies betrit natürlich auch andere Verbindungen als das hier beispielhaft verwendete La1−x Srx MnO3 .
Es existieren darüber hinaus noch andere Formen der indirekten Austauschwechselwirkung,
die mitunter ein sehr materialspezisches Auftreten haben und detailliert betrachtet von
tiefergehenderen elektronischen Eigenschaften der Festkörper abhängen. Die Prinzipien der
direkten und indirekten Austauschwechselwirkung als Phänomen der Quantenmechanik sollten aber durch die bisherigen Ausführungen verständlich geworden sein. Wie ansatzweise
immer wieder zur Sprache gekommen ist, besteht ein direkter Zusammenhang zwischen Austauschwechselwirkung und magnetischem Verhalten von Festkörpern. Der folgende Abschnitt
beleuchtet den Bereich des Magnetismus etwas genauer.
33
KAPITEL 3. AUSTAUSCHWECHSELWIRKUNG UND MAGNETISMUS
3.2 Magnetismus
Es ist im bisherigen Verlauf bereits des Öfteren betont worden, dass Magnetismus im Rahmen
dieser Arbeit eine gewichtige Rolle spielt. Er stellt ein weitreichendes Teilgebiet der Physik
dar und ist insbesondere in der Festkörperphysik von groÿer Bedeutung. Dabei liefert der
Magnetismus eine Vielzahl von unterschiedlichen Aspekten, die hier natürlich nur in einem
geringen Maÿe abgehandelt werden können. Wie sich zeigen wird, steht das Heisenbergmodell in direktem Zusammenhang mit (Anti-)Ferromagnetismus, Formen des kollektiven Magnetismus, die von lokalen magnetischen Momenten erzeugt werden. Sie sollen dementsprechend
behandelt werden. Zudem existieren daneben Formen des itineranten Magnetismus, solchem
der durch Leitungselektronen verursacht wird, der ebenfalls eingeführt werden soll. Auch wenn
hier einige ausgewählte Formen des Magnetismus einzeln vorgestellt werden, sei stets daran
erinnert, dass diese Formen in Materie immer auch gleichzeitig auftreten können und je nach
Substanz speziellerer Natur sind.
3.2.1 Magnetisches Moment und Magnetisierung
Ursächlich für magnetische Felder sind elektrische Ströme (bewegte Ladungen). Betrachtet
man die kleinsten Einheiten von elektrischen Ladungen (Elementarladungen) - zum Beispiel
Elektronen - so wird deutlich, dass ein magnetisches Feld durch einen Translationsimpuls,
einen Bahndrehimpuls
L
und den Elektronenspin
S
erzeugt werden kann. Gebundene Elek-
tronen verfügen über die letzteren beiden Formen des Drehimpulses und ihnen kann der
J = L + S zugeordnet werden. Eine Stromdichteverteilung ~j(~r)
Moment µ
~ , das in seiner allgemeinsten Form deniert ist als
Z
1
µ
~=
d3 r~r × ~j(~r) .
2
Gesamtdrehimpuls
ein magnetisches
erzeugt
(3.18)
Für ein Elektron mit Gesamtdrehimpuls J ist demnach das magnetische Moment:
µ
~ = −gj
Hierin sind
gj = 1 +
der Landé-Faktor und
µB
J
~
.
(3.19)
j(j + 1) + s(s + 1) − l(l + 1)
2j(j + 1)
e~
µB = − 2m
≈ 5, 8 · 10−5 eV/T
e
(3.20)
das Bohrsche Magneton, zwei typ-
ische Kenngröÿen des Magnetismus von Elektronen. Da hier vorerst gebundene Elektronen
behandelt werden, die sich in den Orbitalen ihres Atoms bewegen, handelt es sich um lokale
Momente (itinerante Momente werden später besprochen).
Tatsächlich betrachtet man in Festkörpern natürlich immer eine Vielzahl von Atomen, Ionen
und Molekülen und damit auch eine ebenso groÿe Vielzahl von magnetischen Momenten. Um
eine Aussage über das magnetische Verhalten des Festkörpers als Ganzen machen zu können,
dient die Gröÿe der Magnetisierung
Gesamtmoments
µ
~ =
P
~.
M
Sie ist deniert als der Quotient des magnetischen
~i aller vorhandenen magnetischen Momente
iµ
µi
und des Volumens
der Probe [Stö07].
~
~ = µ
M
V
34
(3.21)
3.2. MAGNETISMUS
Bei einem von auÿen angelegten magnetischen Feld mit der Feldstärke
Induktion
~
B
~
H
ist die magnetische
von der Magnetisierung abhängig, die selber als Ursache eines magnetischen
Feldes gesehen werden kann.
~ = µ0 ( H
~ +M
~)
B
(3.22)
(µ0 : magnetische Feldkonstante)
In Materie mit der relativen Permeabilität
µr
gilt:
~ = µ 0 µr H
~
B
.
(3.23)
Eine wesentliche Eigenschaft eines Materials, die seine magnetische Charakteristik beschreibt,
ist die Reaktion seiner Magnetisierung auf ein äuÿeres Magnetfeld
magnetische Suszeptibilität
~ . Diese Kenngröÿe ist die
H
χm .
χm =
∂M
∂H
(3.24)
In isotropen Medien gilt der Zusammenhang
χm = µ r − 1
.
(3.25)
Aus (3.23) lassen sich damit bereits zwei Klassen magnetischer Substanzen ableiten.
•
paramagnetische Substanzen: 0 <
•
diamagnetische Substanzen: -1<
~,H
~
χm . (M
χm
~,
< 0. (M
parallel)
~
H
antiparallel)
(χm = -1 wird in Supraleitern erreicht.)
Somit sind solche Substanzen klassiziert, die das äuÿere Magnetfeld in ihrem Inneren verstärken (Paramagnetismus) und solche, die es abschwächen (Diamagnetismus). Der Paramagnetismus beinhaltet auch Gruppen, die spontane Magnetisierung zeigen, wie etwa der
Ferromagnetismus.
3.2.2 Dia- und Paramagnetismus
Diamagnetismus
Diamagnetismus tritt als Folge der Lamorpräzession der geladenen Atom-
rumpf-Elektronen in einem äuÿeren Magnetfeld auf. Analog zur Lenzschen Regel der Elektrodynamik sind die dadurch erzeugten Ströme so gerichtet, dass sie das äuÿere Feld abschirmen.
Zur Erklärung sei ein äuÿeres magnetisches Feld in z-Richtung angenommen. Da der Gesamtdrehimpuls eines Elektrons stets eine z-Komponente hat, ist dies gleichbedeutend mit einer
2
Lorentzkraft , die neben der Coulombkraft des Kernes auf das Elektron wirkt. Ähnlich wie
bei einem Kreisel, auf den ein Drehmoment senkrecht zur Drehachse wirkt, kommt es zu einer
Präzession des Drehimpulses um die z-Achse, die als Lamorpräzession bezeichnet wird. Die
zu dieser Präzession gehörige Lamorfrequenz ist gegeben durch
ωL = −
2
e ~
|B|
2me
,
(3.26)
~
Lorentzkraft: F~L = −e(~v × B)
35
KAPITEL 3. AUSTAUSCHWECHSELWIRKUNG UND MAGNETISMUS
wobei
ω
~L
ebenfalls in z-Richtung zeigt. (Eine ausführliche Herleitung ndet sich zum Beispiel
im Vorlessungsskript zu Höhere Experimentalphysik I [Rat11]). Die Präzession erzeugt einen
Ringstrom
I
der Stärke
I = −e
ωL
e2 · B
=−
2π
4πme
.
(3.27)
Dieser wiederum führt zu einem magnetischen Dipolmoment
2
µe = I · A = I · π r 2
3
2
r =
(
3
2
x2 + y 2
.
(3.28)
: mittleres Quadrat des Abstandes des Elektrons vom Kern.)
Durch Aufsummieren aller magnetischen Momente und Ableiten nach dem Magnetfeld (B
µ0 Hz ),
erhält man die magnetische Suszeptibilität des Diamagnetismus:
χD = −
N
≈
µ0 · N · Z · e 2 2 r
6me
steht dabei für die atomare Teilchen-Volumendichte und
.
Z
(3.29)
für die Kernladungszahl.
Der Diamagnetismus tritt seiner Ursache gemäÿ bei allen Substanzen auf. Seine Suszeptibilität liegt jedoch typischerweise in einem Bereich von etwa
10−5 ,
weshalb er leicht von
anderen Formen des Magnetismus dominiert wird. Merklich wird er bei Stoen, die keinen
Paramagnetismus ausbilden. Wie sich im nächsten Teil zeigen wird, betrit dies Stoe, deren
Atome abgeschlossene Schalen haben. Diamagnetismus zeigt sich also zum Beispiel bei Edelgasen und Ionenkristallen der Alkali- und Erdalkalihalogenide. Er ist in einem weiten Bereich
temperaturunabhängig und die resultierende Magnetisierungskurve linear und reversibel.
Paramagnetismus
Die Untersuchung des Diamagnetismus hat es gestattet, einzelne La-
dungsträger (speziell Elektronen) als Ursache von magnetischen Dipolen zu betrachten und
das Verhalten des gesamten Materials als Summe dieses Verhaltens einzelner Dipole aufzufassen. Um sich dem Paramagnetismus zu nähern, ist es erforderlich, sich die elektrische Systematik gesamter Atomhüllen anzusehen (respektive die Elektronenhülle von Molekülen).
Dabei ist vorallem die Verteilung der Drehimpulse (inklusive Spin) von Interesse.
Der Drehimpuls der Elektronen einer Schale lässt sich durch die Quantenzahlen
quantenzahl),
l
(Bahndrehimpulsquantenzahl) und
j
s
(Spin-
(Quantenzahl des Gesamtdrehimpuls-
es) beschreiben. Die Hundschen Regeln geben vor, welche Werte diese Quantenzahlen im
j = 0 nur für
bis auf ein Elektron halbbesetzte Schalen. Ansonsten ist j =
6 0 und die zGesamtdrehimpulses mj kann die Werte −j, −j + 1, . . . , j annehmen. In
Grundzustand für eine bestimmte Elektronenzahl annehmen. Demnach gilt
vollbesetzte oder
Komponente des
einem äuÿeren Magnetfeld, das in z-Richtung zeigt, ergibt sich für das magnetische Moment
eines Atoms aus (3.19) folgender Hamiltonoperator (hier: in Einheiten von
~ = −mj · gj · µB · B
H = −~
µ·B
.
~):
(3.30)
In einem äuÿeren Magnetfeld kommt es also zu einer Energieaufspaltung zwischen den unterschiedlichen Ausrichtungen des Gesamtdrehimpulses, bekannt als Zeemann-Eekt. Dies
36
3.2. MAGNETISMUS
bedeutet, dass sich nach den Gesetzen der statistischen Physik je nach Temperatur unterschiedliche Verteilungen der Ausrichtungen der Drehimpulse ergeben. Eine ungleiche Verteilung der Ausrichtungen wiederum hat eine Netto-Magnetisierung des Festkörpers zur Folge.
N Atomen betrachten, deren
mj = ms = ±1/2 tragen. Be-
Der Einfachheit halber kann man nun zunächst ein System aus
Quantenzahlen die Werte
l = 0, j = s = 1/2
und somit
trachtet man nun die Komponenten des Drehimpulses der einzelnen Atome in Näherung als
unabhängig, so lässt sich eine temperaturabhängige Magnetisierung mithilfe der Boltzmann-
N1 , N2 mit N = N1 + N2
mj = −1/2, dann gilt:
Statistik ausrechnen. Seien
mj = 1/2
beziehungsweise
die Teilchendichten der Atome mit
2µ ·B
N2
− ∆E
− B
= e kB T = e kB T
N1
.
(3.31)
Es folgt, dass die Magnetisierung sich zu
M = µB (N1 − N2 ) = N µB · tanh
µB · B
kB T
berechnet. Für hohe Temperaturen und kleine Magnetfelder, also für
weiter vereinfachen zu
N · µ2B B
kB T
M≈
Für allgemeine Drehimpulse
j 6= 0
(3.32)
µB ·B
kB T
1, lässt sich dies
.
(3.33)
ergibt sich etwas komplizierter im kanonischen Ensemble
(Herleitung zum Beispiel im Skript zur Vorlesung Advanced Solid State Physics II [Val10])
durch Verwendung der statistischen Physik:
M = N gj µB jBj (gj jµB B/(kB T ))
Hierbei sind
Bj (x)
.
3
die Brillouin-Funktionen , die für kleine Argumente
(3.34)
x
durch
1 x
Bj (x) ≈ 1 +
j 3
(3.35)
genähert werden können. Dies führt zu der Magnetisierung
gj2 µ2B j(j + 1)
M =N
B
3kB T
(3.36)
und bringt schlieÿlich die magnetische Suszeptibilität im paramagnetischen Fall.
χP = N
gj2 µ2B j(j + 1)
3kB T
(3.37)
Diese Temperaturabhängigkeit wird als Curie-Gesetz bezeichnet. Mit der materialspezischen
Curie-Konstanten
µ0 ·N ·j(j+1)gj2 µ2B
µ0 N µ2B
(=
für
3kB
kB
C=
χP =
C
T
s = 1/2)
lautet es:
.
Die Suszeptibilität nach dem Curie-Gesetz bewegt sich in Gröÿenordnungen von
(3.38)
10−2 bis 10−3 .
Der Paramagnetismus überwiegt den Diamagnetismus bei gleichzeitigem Vorhandensein also
deutlich.
3
Bj (x) =
2j+1
coth
2j
2j+1
x
2j
−
1
coth
2j
x
2j
,
B 1 (x) = tanh(x)
2
37
KAPITEL 3. AUSTAUSCHWECHSELWIRKUNG UND MAGNETISMUS
Die hier beschriebene Form des Paramagnetismus wird auch als Langevin-Paramagnetismus
bezeichnet und beruht auf einer Vielzahl von Annahmen, beschreibt das Prinzip des Paramagnetismus jedoch erfolgreich. Das Modell geht von einem thermisch isolierten Grundzustand,
dem Nichtvorhandensein der Spin-Bahn-Kopplung und magnetischer Isotropie aus. Zudem
wurde angenommen, dass keine Wechselwirkung zwischen den Teilchen des Festkörpers besteht und somit kein kollektiver Magnetismus existiert. Eben jener soll im nächsten Abschnitt
vorgestellt werden. Zudem sei betont, dass es sich auch hier nur um die Betrachtung lokaler
magnetischer Momente handelt. Ein Paramagnetismus auf Basis von Leitungselektronen wird
ebenfalls Teil dieses Kapitels sein.
3.2.3 Magnetische Ordnung: Ferro- und Antiferromagnetismus
In den vorherigen Betrachtungen zum Magnetismus sind wir stets von untereinander unabhängigen magnetischen Momenten einzelner Elektronen/Atome/Moleküle ausgegangen. In
Abwesenheit eines äuÿeren Feldes würde dies bei jeder beliebigen Temperatur einen thermisch ungeordneten Zustand der magnetischen Momente mit sich ziehen und die mittlere
Magnetisierung eines jeden Atomrumpfes wäre stets Null. Der Abschnitt über Austauschwechselwirkungen hat jedoch gezeigt, dass in kondensierter Materie durchaus gegenseitige Wechselwirkungen der magnetischen Momente bestehen. Dies führt dazu, dass einige Festkörper
unterhalb einer kritischen Temperatur
Tc
einen von Null verschiedenen mittleren Vektor ihrer
einzelnen magnetischen Momente aufweisen. Man bezeichnet solche Festkörper als magnetisch
geordnet. Vorerst liegt das Augenmerk auch hier auf lokalisierten magnetischen Momenten.
Nun können sich die einzelnen, lokalisierten magnetischen Momente in einem magnetischen
Festkörper zu einer eektiv von Null verschiedenen Magnetisierungsdichte für den Festkörper
als Ganzen addieren oder nicht. Tun sie es, so zeigt sich die mikroskopische magnetische Ordnung durch das Vorhandensein einer makroskopischen Magnetisierungsdichte der Probe auch
ohne äuÿeres Feld. Diese Magnetisierungsdichte bezeichnet man als spontane Magnetisierung,
den
geordneten
Zustand
ferromagnetisch
als
[AM76,
S.884 ].
Die Netto-Magnetisierung kommt anschaulich gesprochen
zustande, wenn die einzelnen magnetischen Momente sich
in mindestens einer Ortskomponente parallel anordnen. In
4
Abbildung 3.4
sind schematisch mögliche Anordnungen
der magnetischen Momente für den ferromagnetischen Fall
in einer zweidimensionalen Vereinfachung dargestellt. Aus
dem Hamiltonoperator (1.1) des Heisenbergmodells folgt
direkt, dass für
Abbildung 3.4: mögliche ferromagnetische Anordnungen der magnetischen Momente
J >0
eine parallele Ausrichtung der Elek-
tronenspins energetisch begünstigt ist, also einem ferromagnetischen Festkörper entspricht. Damit ist eine unmittelbare Verbindung zur direkten Austauschwechselwirkung
geschaen, wenn man sich daran erinnert, dass
J
nichts
anderes als eben diese darstellt. Es entscheidet also - wie bereits erwähnt - die Gestalt der
Austauschwechselwirkung über die mögliche Form der magnetischen Ordnung eines Festkör4
Quelle: http://upload.wikimedia.org/wikipedia/commons/c/cc/Ferromagnetic_ordering_illustration.svg
38
3.2. MAGNETISMUS
pers.
In den einfachsten Ferromagneten haben sämtliche lokalen Momente denselben Betrag und
die gleiche mittlere Richtung (obere Reihe in der Abbildung).
Neben der ferromagnetischen Ordnung trit man häuger den Fall, dass sich die einzelnen,
lokalen magnetischen Momente zu einem Gesamtmoment Null addieren und keinerlei spontane
Magnetisierung das Vorhandensein der mikroskopischen Ordnung enthüllt. Solche magnetischen
Zustände
nennt
Sie
kommen
selwirkenden
antiferromagnetisch.
man
zustande,
wenn
Momente,
eine
die
benachbarten,
antiparallele
wech-
Ausrichtung
bevorzugen, sich bei der Addition also aufheben. Folglich
ist dies mit einem negativen Austauschparameter
J
ver-
bunden. Wiederum sind mögliche Spinanordnungen für
5
diesen Fall in Abbildung 3.5
Abbildung 3.5: mögliche antiferromagnetische Anordnungen der magnetischen Momente
gezeigt. Im einfachsten an-
tiferromagnetischen Zustand verteilen sich die lokalen Momente auf zwei ineinanderliegende Untergitter mit identischer Struktur. Innerhalb eines jeden der beiden Untergitter
sind die Beträge und die mittleren Richtungen der magnetischen Momente gleich, die eektiven Momente der beiden Untergitter sind jedoch einander entgegengesetzt gerichtet und
addieren sich zum Gesamtmoment Null. Die obere Reihe der Abbildung entspricht dieser
Situation im eindimensionalen Fall, wobei jedes zweite Momente der Anordnung zum selben
Untergitter gehört. Dies stellt einen einfachen Fall sogenannter bipartiter Gitter dar. In einem
bipartiten Gitter lässt sich jeder Gitterplatz einem Untergitter A oder B so zuordnen, dass er
nur mit Gitterplätzen des jeweils anderen Untergitters wechselwirkt beziehungsweise direkt benachbart ist. Im antiferromagnetischen Fall gilt für solche Gitter, dass die Untergitter für sich
betrachtet als ferromagnetisch geordnet betrachtet werden können und die Magnetisierung
des Gesamtgitters als Dierenz der beiden Magnetisierungen der Untergitter gesehen werden kann. Für bipartite Gitter lassen sich allgemeine Aussagen für den antiferromagnetischen
Grundzustand bezüglich der Gesamtmomente und -spins machen. Dies ist für den nächsten
Teil dieser Arbeit von imenser Bedeutung (siehe zum Beispiel Lieb-Mattis-Theorem).
Während im ferromagnetischen Fall die Grundzustände immer solche sind, in denen die magnetischen Momente alle gleich ausgerichtet sind, also klassisch betrachtet alle Spins up oder
alle down einnehmen, können antiferromagnetische Systeme mehrere Grundzustände haben.
Dazu führe man sich die Anordnungen aus Abbildung
6
3.6
vor Augen. Links ist ein Quadrat aus vier Spins
zu sehen, was ein bipartites Gitter darstellt. Hier ist es
möglich jeweils benachbarte Spins antiparallel auszurichten, wodurch der Grundzustand leicht gefunden ist. Das
Dreieck rechts allerdings lässt dies nicht zu: Betrachtet man
Abbildung 3.6:
frustiertes System;
ertes System.
links:
rechts:
nichtfrustri-
eine Komponente des Spins, stellt man fest, dass es nicht
möglich ist, eine Anordnung zu nden, in der diese Komponente für jeden Spin antiparallel zu beiden Nachbarn ist.
Quelle:http://upload.wikimedia.org/wikipedia/commons/c/c6/Antiferromagnetic_ordering_illustration.
svg
6
http://www.nature.com/nature/journal/v456/n7224/g_tab/456886a_F1.html
5
39
KAPITEL 3. AUSTAUSCHWECHSELWIRKUNG UND MAGNETISMUS
Klassisch betrachtet gibt es nur zwei mögliche Zustände, die keine Grundzustände sind, nämlich alle Spins up oder alle down. Somit existieren sechs Grundzustände. Systeme mit dieser
Eigenschaft bezeichnet man als frustiert. Die Ermittlung von Grundzuständen und Grundzustandsenergien gestaltet sich in ihnen entsprechend kompliziert. Die Rechnungen im nächsten
Kapitel wurden jedoch alle an nicht-frustrierten Systemen vorgenommen.
Es sei erwähnt, dass man die Bezeichnung Ferromagnet auch in einem stärker eingeschränkten Sinne verwendet, wenn man eine Unterscheidung treen will zwischen den zahlreichen
ferromagnetischen Zuständen, die sich ergeben können, wenn die primitive Zelle mehrere,
nicht notwendig identische, magnetische Atomrümpfe enthält. In solchen Fällen benutzt man
das Attribut ferromagnetisch für magnetische Strukturen, in welchen sämtliche magnetische Momente eine positive Komponente in Richtung der spontane Magnetisierung aufweisen.
Festkörper mit spontaner Magnetisierung, welche dieses Kriterium nicht erfüllen, bezeichnet
man als Ferrimagnete.
In einem einfachen Ferrimagneten kann möglicherweise die Austauschkopplung zwischen nächsten Nachbarn eine antiparallele Ausrichtung begünstigen, was bei unterschiedlich starken
magnetischen Momenten der benachbarten Atomrümpfe aber dennnoch eine Netto-Magnetisierung und somit ein restliches, eektives Moment des Festkörpers als Ganzen zur Folge
haben kann.
Heisenbergmodell und Molekularfeldtheorie
Es sollen nun einige quantitative und
formelle Untersuchungen des (Anti-)Ferromagnetismus vorgenommen werden. Zunächst soll
der ferromagnetische Fall besprochen werden, da die Berechnungen zum Antiferromagnetismus
auf darin verwendete Ansätze zurückgreifen. Als Ausgangspunkt dient der Hamiltonoperator
des Heisenbergmodells in einem äuÿeren Magnetfeld
~:
B
H=
X
i
−
X
~ · Si
J Sj + gj µB B
.
(3.39)
j
Es gilt, dass nur über nächste Nachbarn summiert wird (J
Man kann nun ein B-Feld
~≡−
B
X J Sj
~
+B
gµB
6= 0
für
i, j
nächste Nachbarn).
(3.40)
j
so denieren, dass der Hamiltonoperator die selbe Form annimmt wie eine Summe über unabhängige Hamiltonoperatoren (3.30) lokaler magnetischer Momente im externen Magnetfeld:
H = g j µB
X
~ · Si
B
.
(3.41)
i
Um eine exakte Lösung erhalten zu können, dient eine weitere Näherung, die als Molekularfeldtheorie bezeichnet wird. Dabei werden die einzelnen magnetischen Spinmomente als
unabhängig betrachtet und die lokalen Eekte eines einzelnen Moments auf das ihn lokal
umgebende Feld vernachlässigt. Das heiÿt, alle Spins
Si
werden gleichberechtigt und unab-
hängig behandelt und durch ihren Erwartungswert hSi i genähert. Auf diese Weise ersetzt man
eine Vielzahl von komplexen Zwei-Teilchen-Wechselwirkungen durch die Wechselwirkung eines
jeden Spins mit einem gemittelten Feld, das durch den Spinerwartungswert aller anderen Gitterplätze erzeugt wird. Das zusätzliche äuÿere Magnetfeld hat natürlich immer noch Bestand.
40
3.2. MAGNETISMUS
Formell entspricht dies der Substitution
~ef f = −
B
X J · hSj i
~
+B
gµB
,
(3.42)
j
was zu einem eektiven Hamiltonoperator
Hef f = gj µB
X
~ef f · Si
B
(3.43)
i
führt. Die Magnetisierung lässt sich aus dem Erwartungswert der Spins errechnen.
~ = N h~
M
µi = −N gj µB hSi i
(3.44)
(N : Teilchendichte.) Ohne Beschränkung der Allgemeinheit betrachte man nun der Einfachheit halber ein magnetisches Feld
~ = B · ~ez
B
in z-Richtung und Gitterplätze mit
j = s = 1/2.
Aus den Berechnungen zum Paramagnetismus ist bekannt, wie sich der Spin-Erwartungswert
aus dem gegebenen eektiven Hamiltonoperator ermitteln lässt. Dies in die Gleichung für die
Magnetisierung eingesetzt, gibt eine selbst-konsistente Gleichung für die Magnetisierung in
Abhängigkeit vom äuÿeren Magnetfeld und Temperatur.
µB Bef f
M = N µB · tanh
kB T
X
µB
J
= N µB · tanh
B−
Sz
kB T
gj µB j
j
ZJ
µB
B+
M
= N µB · tanh
kB T
N (gj µB )2
(3.45)
In dieser selbst-konsistenten Darstellung ist also die Magentisierung von sich selbst abhängig.
Darin ist
Z
die Zahl nächster Nachbarn eines jeden Gitterplatzes. Es ist sinvoll, eine Mag-
netisierung pro Gitterplatz
m =
M
N zu bestimmen, um die Gröÿe von
N
unabhängig zu
machen. Zudem kann nun das äuÿere magnetische Feld als Null angenommen werden, da
wir an der Temperatur interessiert sind, bei der spontane Magnetisierung ohne Einuss eines
äuÿeren Feldes stattndet.
M
m=
= µB · tanh
N
µB
ZJ
µB A
m ≡ µB · tanh
m
kB T N (gj µB )2
kB T
(3.46)
Eine Lösung dieser ebenfalls selbst-konsistenten Gleichung lässt sich zum Beispiel grasch
nden. Eine solche Lösung zeigt Abbildung 3.7. Es ist zu sehen, dass nicht-triviale Lösungen
m 6= 0 nur durch a < 1 und eine Steigung der Lösungsfunktion
0
ist (f (0) > 1), erfüllt wird. Daraus ergibt sich:
kB Tc =
Damit ist die Temperatur
ZJ
gj2
.
in
m = 0,
die gröÿer als eins
(3.47)
Tc gefunden bei der ein Wechsel von m = 0 zu m 6= 0, beziehungsweise
umgekehrt, stattndet; die kritische Temperatur oder auch Curie-Temperatur. Unterhalb
dieser Temperatur hat ein ferromagnetischer Sto stets eine magnetische Ordnung und somit
eine Magnetisierung, auch ohne äuÿeres Feld. An dieser Stelle ndet also ein Phasenübergang
41
KAPITEL 3. AUSTAUSCHWECHSELWIRKUNG UND MAGNETISMUS
Abbildung 3.7: grasche Lösung der selbst-konsistenten Gleichung für die Magnetisierung (a =
[Val10]
kB T
µ2B A
)
in Form einer spontanen Ordnung statt, der mit einem spontanen Bruch der Rotationsinvarianz verbunden ist; es entsteht eine ausgezeichnete Achse der räumlichen Ordnung (hier die
z-Achse).
Die Betrachtung des Falls für allgemeine
s
gibt für die Curie-Temperatur:
1
kB Tc = ZJ · s(s + 1)
3
.
(3.48)
Interessiert man sich für den Verlauf der Magnetisierung unterhalb und in Nähe der kritischen
Temperatur, so bietet sich eine Taylor-Entwicklung von (3.46). Für kleine Magentisierungen
kann die Reihe nach der dritten Ordnung in
m
abgebrochen werden und es ergibt sich der
folgende Zusammenhang:
s
m(T ) ≈
3µ2B p
Tc − T
Tc2
.
(3.49)
Der kritische Exponent für die Magnetisierung liegt in dieser Näherung also bei 1/2.
Die weitere wichtige Gröÿe im Zusammenhang mit magnetischem Verhalten, die Suszeptibilität, berechnet sich wie gehabt, als Ableitung der Magnetisierung nach dem magnetischen
Feld. Daraus folgt mit der Randbedingung
χCW =
Für
j = s = 1/2
M =0
für
B→∞
das Curie-Weiss-Gesetz.
N µ2B
1
kB T − Tc
(3.50)
ist der Vorfaktor gleich der Curie-Konstanten, also
χCW =
C
T − Tc
.
(3.51)
Der Ferromagnetismus folgt bezüglich seiner temperaturabhängigen Suszeptibilität also einem
ähnlichen Gesetz wie der Langevin-Paramagnetismus lokalisierter, unabhängiger Momente.
Entscheidend ist im Curie-Weiss-Gesetz jedoch die Dierenz zur kritischen Temperatur.
42
3.2. MAGNETISMUS
Die entsprechenden Gröÿen der magnetischen Charakteristik für Antiferromagneten zu ermitteln, gestaltet sich
etwas schwieriger, greift aber letztlich auf die eben verwendeten Methoden zurück. Dazu denke man sich das
zugrundeliegende Gitter mit antiferromagnetischen Austauschkopplungen als bipartites Gitter, das sich durch
zwei Untergitter A und B komplett abdecken lässt. Eine
solche Situation ist für ein zweidimensionales Quadratgitter in Abbildung 3.8 abgebildet. Wie zuvor bereits
beschrieben, kann man beide Untergitter als untereinander
ferromagnetisch wechselwirkend und mit entgegengesetzter
Magnetisierung betrachten. Man benutzt nun wieder die
Abbildung 3.8: bipartites Gitter mit
Untergittern (rot und blau)
Molekularfeldtheorie und deniert die eektiven Magnetfelder
~A
B
und
~B
B
als Gröÿen der Spinerwartungswerte bei-
der Untergitter.
~A = B
~B = B
~−
B
X
iA
Der
zugehörige
X J
J
hSiA i −
hSiB i ≡ λ+ hSiA i + λ− hSiB i
g j µB
gj µB
Hamiltonoperator
(3.52)
iB
stellt
also
eine
Summe
aus
zwei
ferromagnetischen
Hamiltonoperatoren der Untergitter A und B, bekannt aus dem vorherigen Fall, dar.
H = −gj µB
X
~A − gj µB
Si · B
iA
X
~B
Si · B
(3.53)
iB
Völlig analog zur Herleitung des Curie-Weiss-Gesetzes verläuft nun die weitere Berechnung.
Unter der Annahme, dass beide Untergitter die selbe Gestalt haben, muss gelten, dass
~ =M
~ A = −M
~B
M
.
(3.54)
Nach Lösung der selbst-konsistenten Gleichung für die Magnetisierung und anschlieÿender
Taylor-Entwicklung erhält man schlieÿlich die kritische Temperatur
TN
der antiferromag-
netischen Ordnung, die als Néel-Temperatur bezeichnet wird.
TN =
(γ
=
λ+
,
gj2 µ2B N µ0
Γ =
C
(γ − Γ )
2
(3.55)
λ−
)
gj2 µ2B N µ0
Wie gehabt, ist die Magnetisierung der Untergitter nur unterhalb dieser kritischen Temperatur
ungleich Null, bei Temperaturen darüber, besteht keine magnetische Ordnung. Ihre maximale
Magnetisierung haben die Untergitter bei
T = 0.
In diesem klassischen Néel-Zustand liegt
eine optimale antiparallele Ausrichtung benachbarter magnetischer Momente vor. Es sind
genau diese Zustände der antiferromagnetischen Ordnung, die Objekt der noch folgenden
Diagonalisierungs- und Berechnungsmethoden meiner Arbeit sind.
Es sei noch erwähnt, dass die über die Molekularfeldmethode hergeleiteten Ergebnisse, keine
exakten, sondern genäherte sind. Deshalb sind die daraus ermittelten Werte quantitativ nicht
vollständig korrekt, geben aber gute qualitative Ergebnisse, die dem Verständnis der spontanen Magnetisierung dienlich sind.
43
KAPITEL 3. AUSTAUSCHWECHSELWIRKUNG UND MAGNETISMUS
Alle bisher besprochenen Formen des Magnetismus liegen be-
Initeranter Magnetismus
gründet in dem Verhalten von lokalisierten Atomrümpfen und ihrer Elektronen. Es liegt nahe,
dass auch Leitungselektronen, also delokalisierte und aufgrund des Pauli-Prinzips stark korrelierte Elektronen, eine Beitrag zur Suszeptibilität eines Stoes leisten werden. Dies betrit
natürlich besonders Metalle. Es gilt auch hier, dass die Beschreibung des Magnetismus von
Leitungselektronen, auch initernater Magnetismus genannt, unter Betrachtung aller Eekte
äuÿerst komplex ist. Hier sollen grundlegende Konzepte aufgezeigt werden.
Zunächst soll der paramagnetische Anteil des Magnetismus von Leitungselektronen besprochen werden; er wird als Paulischer Paramagnetismus bezeichnet. Es ist dafür hinreichend, im Rahmen der Näherung unabhängiger Elektronen zu arbeiten, die man ausschlieÿlich
als Träger eines magnetischen Spinmoments und nicht als Ladungsträger betrachtet. Dadurch
muss an dieser Stelle die Bedeutung der Bahnbewegung der Elektronen in einem äuÿeren Feld,
die sich sehr kompliziert darstellt, zunächst nicht beachtet werden.
In einem von auÿen angelegten magnetischen Feld
~
H
kann die Feldrichtung als Quantisierungs-
richtung der Spinmomente gesehen werden; sie können also entweder parallel (Volumendichte
N+ )
oder antiparallel (N− ) ausgerichtet sein (N
= N+ + N− ).
Analog zur Herleitung des
Paramagnetismus lokaler Momente, gilt für die Magnetisierung:
M = µB (N− − N+ )
Von Bedeutung ist, wie groÿ die Besetzungen
Niveaudichte
g± ()
eingeführt:
g± ()d
N+
und
.
(3.56)
N−
sind. Zu diesem Zweck wird die
gibt die Anzahl von Elektronen mit parallelem/an-
tiparallelem Spin pro Einheitsvolumen in einem Energiebereich zwischen
Ohne äuÿeres Feld (H
= 0)
und
+ d
liegen beide Spinausrichtungen in gleicher Anzahl vor und ihre
Niveaudichte ist jeweils die Hälfte der gewöhnlichen Zustandsdichte
g().
1
g± () = g()
2
(3.57)
Für Elektronen gilt nach (3.30), dass sich ihre Energie in einem magnetischen Feld
Betrag
µB H
an.
H
um den
vergröÿert beziehungsweise verkleinert. Entsprechend gilt:
1
g± () = g( ∓ µB H)
2
.
(3.58)
Da die aus dem äuÿeren Feld erzeugte Energie selbst bei starken Feldern weit unter der FermiEnergie liegt, kann letztere Funktion mit geringem Fehler entwickelt werden.
1
1
g± () = g() ∓ µB Hg 0 ()
2
2
(3.59)
Die Besetzungszahlen für Fermionen ergeben sich aus dem Integral der Zustandsdichte mul7
tipliziert mit der Fermi-Verteilung
N± =
1
2
Z
f ()
über die Energie.
1
g()f ()d ∓ µB H
2
Z
dg 0 ()f ()
(3.60)
Aus (3.56) folgt direkt
M=
7
W (E) =
exp
1
E−µ
kB T
+1
µ2B H
Z
0
g ()f ()d =
Z
g()
−∂f ()
∂
d
(3.61)
, gibt für unabhängige Fermionen an, mit welcher Wahrscheinlichkeit W sie die Energie
E bei gegebener Temperatur T tragen.
44
µ2B H
3.2. MAGNETISMUS
Am Nullpunkt der Temperatur gilt
−∂f /∂ = δ( − F ) (F :
M = µ2B Hg(F )
Fermienergie), so dass
.
(3.62)
Die Änderungen der Fermifunktion mit der Energie kann auch bei höheren Temperaturen
als vernachlässigbar klein angesehen werden [AM76]. Erst in Bereichen von
104 K
wird diese
Näherung unzulässig. Somit ergibt sich eine weitgehend temperaturunabhängige Suszeptibilität für den Paulischen Paramagnetismus.
χP auli = µ2B g(F )
(3.63)
Diese Suszeptibilität unterscheidet sich von der des Langevin-Paramagnetismus also zum
Einen in ihrer Temperaturunabhängikeit und ist zum Anderen etwa drei Gröÿenordnungen
kleiner. Dies ist darauf zurückzuführen, dass das Pauli-Prinzip der Ausrichtung der Spinmomente sehr viel eektiver entgegenwirkt als thermische Unordnung. Zudem muss man
sagen, dass die gemessenen Suszeptibilitäten des Paulischen Paramagnetismus recht deutliche
Diskrepanzen zu den theoretisch berechneten ausfweisen, was sich durch die Vernachlässigung
der Elektron-Elektron-Wechselwirkung erklären lässt.
Ähnlich den lokalen magnetischen Momenten, erzeugen auch die Leitungselektronen einen
diamagnetischen Beitrag, der ebenfalls auf einer Entsprechung der Lenzschen Regel beruht.
Die Bahnbewegung der Leitungselektronen in einem äuÿeren Magnetfeld erzeugt eine Lorentzkraft auf diese, wodurch sie auf eine spiralförmige Bahn gezwungen werden. Diese kann
man als Ursache für Wirbelströme betrachten, die so gerichtet sind, dass das resultierende
magnetische Moment sich entgegen des äuÿeren Feldes ausbildet. Bei niedrigen Temperaturen
und starken Felder führt diese zu einer komplexen, oszillatorischen Abhängigkeit der Magnetisierung
M (H) [AM76]. Aber auch wenn diese Bedingungen nicht gegeben sind, was in der
Praxis meist der Fall ist, existiert eine von Null verschiedene Magnetisierung aus der Bahnbewegung, der sogenannte Landausche Diamagnetismus. Für freie Elektronen kann man eine
Suszeptibilität dieses Diamagnetismus ermitteln.
1
χLandau = − χP auli
3
(3.64)
Tatsächlich ist es eine bessere Näherung, die Leitungslektronen in einem Metall als in einem
periodischen Potential bendlich anzunehmen. In diesem Fall erweist sich die Analyse ihres
Verhaltens im magnetischen Feld noch komplizierter. Resultat ist aber ebenfalls eine diamagnetische Suszeptibilität gleicher Gröÿenordnung wie die des Paulischen Paramagnetismus.
Praktisch ist die gemessene Suszeptibilität natürlich die Kombination der Beiträge des
Paulischen Paramagnetismus, des Landauschen Diamagnetismus sowie des Larmorschen Diamagnetismus der Rumpfelektronen in abgeschlossenen Elektronenschalen. So zeigen sich Edelmetalle diamagnetisch und die Metalle der ersten drei Hauptgruppen paramagnetisch.
45
4 Exakte Diagonalisierung und untere
Schranken der Grundzustandsenergie
Bis hierhin hat sich meine Arbeit mit der Herleitung des Heisenbergmodells und einigen
weiteren theoretischen Aspekten diesbezüglich befasst. Dieser Teil war sehr allgemein gehalten
und hat die Themen Austauschwechselwirkung und Magnetismus auf grundlegende Art und
Weise behandelt.
Die Eigenleistung und eigentliche Bedeutung dieser Arbeit liegt jedoch in einer bestimmten
Anwendung des Heisenbergmodells auf Modellsituationen. Dabei handelt es sich um die exakte Diagonalisierung des Modells und die Findung unterer Schranken für die Grundzustandsenergien unendlicher zwei- und dreidimensionaler Gitter aus Spin-1/2-Teilchen mit antiferromagnetischer Wechselwirkung. Es soll hierfür in leichter Abwandlung zu (1.1) fortan der
Heisenberg-Hamiltonoperator durch
HHeis = J
X
Si · Sj
(4.1)
hi,ji
J geschehen). In
J > 0. Dies ist schlichtweg eine
gegeben sein (die Denition des Vorzeichens kann über die Denition von
diesem Fall gilt für eine antferromagnetische Wechselwirkung:
Frage der Dention, entspricht aber den gängigen Konventionen, weshalb ich dazu übergehen
möchte.
In diesem Kapitel sollen nun Problemstellung, mathematisch-theoretische Hintergründe, verwendete Methoden und erzielte Resultate vorgestellt und diskutiert werden.
Wie bei jedem Hamiltonoperator, liefern die Eigenwerte des Heisenberg-Hamiltonoperators
die möglichen Energien, die ein System haben kann, das durch den Operator beschrieben wird.
Die Bestimmung dieser Eigenenergien erfordert die Wahl einer geeigneten Basis, wodurch
eine Matrixdarstellung des Hamiltonoperators gefunden werden kann. Die Ermittelung der
Eigenenergien ist dann gleichbedeutend mit der Bestimmung der Eigenwerte der Matrix. Da
dies anhand des exakten Hamiltonoperators ohne weitere Näherungen geschieht, bezeichnet
man dieses Vorgehen als exakte Diagonalisierung. Von Interesse sind hier jeweils die Grundzustandsenergien, also die kleinsten Eigenenergien. Es wird sich zeigen, dass dies an einigen
Stellen zu entscheidenden Vereinfachungen der Berechnung genutzt werden kann. Es ist leicht vorstellbar, dass die Diagonalisierung sich mit zunehmender Gröÿe des zugrundeliegenden Systems verkompliziert. Es wird also darauf ankommen, geeignete Basen und nützliche
Berechnungsmethoden zu nden. Da die durchgeführten Rechnungen zu groÿen Teilen kaum
per Hand durchführbar sind, habe ich mit dem Computeralgebrasystem MATHEMATICA
der Firma Wolfram Research gearbeitet. Die Art der Verwendung des Programms wird an
entsprechender Stelle erläutert.
47
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
Natürlich kann die exakte Diagonalisierung nur an Systemen endlicher Gröÿe durchgeführt
werden (wie sich zeigt, ist die Gröÿe berechenbarer Systeme aus praktischen Gründen stark
beschränkt). Entsprechend ndet die Bestimmung der Grundzustandsenergie für unendliche
Systeme nicht exakt statt, sondern als Abschätzung einer unteren Schranke, welche ihrerseits
exakt ist (mehr dazu im Verlauf des Kapitels).
Einführend soll zunächst eine Kette, also eine eindimensionale Anordnung aus
1/2)
n
Spins (s
=
in Betracht gezogen werden.
4.1 Lineare Spin-Kette
1
2
n-1
n
Abbildung 4.1: Lineare Kette aus n Spin-1/2-Plätzen
Abbildung 4.1 zeigt die hier untersuchte Spin-Kette, bestehend aus
n
Gitterplätzen mit
Spin 1/2. Der Heisenberg-Hamiltonoperator (4.1) schreibt sich für eine solche Kette:
H=J
n−1
X
Sk
· Sk+1
.
(4.2)
k=1
Zu diesem Hamiltonoperator ist natürlich eine Basis zu wählen, die als Quantenzahlen die
Spinwerte der einzelnen Gitterplätze enthält. Dafür benutzt man, wie schon bei der Addition
zweier Spins gesehen, das Tensorprodukt der einzelnen Spins. Da
fest ist, reicht es, nur die z-Komponenten
Basis für eine Kette aus
n
mk
sk = 1/2
für alle Spins
k
der Spins als Quantenzahlen zu benutzen. Die
Spins kann dann als
{|m1 m2 · · · mn i}
geschrieben werden. Dabei können die
mk
die Werte
±1/2
(4.3)
annehmen. Es handelt sich um die
Produktspin-Basis. Es wurde bereits gezeigt, dass auch der Übergang zu einer GesamtspinBasis nützlich sein kann. Dieser Übergang wird an späterer Stelle erfolgen und ist für die
Spin-Kette noch nicht zwingend erforderlich, um einen ersten Einblick in die Berechnugsmethoden zu gewinnen. Es wird allerdings deutlich werden, dass die Verwendung der Produktbasis
bei der Bestimmung der Grundzustandsenergie keine Reduktion auf einen Unterraum der
Zustände zulässt, weshalb die noch berechenbaren Längen der Spinkette stark beschränkt
sind. Um auch gröÿere Systeme noch berechnen zu können, wäre es also nötig, Transformationen in die Basis der Gesamtspins durchzuführen, wie es in den späteren Rechnungen der Fall
ist. Mein tatsächliches Vorgehen war jedoch das hier beschriebene, weshalb ich die Rechnung
auch auf diese Weise vorstellen möchte, um den dokumentarischen Charakter der Arbeit zu
gewährleisten.
Die Basis ist von der Dimension
2n
und der Hamiltonoperator entsprechend eine
2n × 2n -
Matrix. Dies gibt einen Hinweis darauf, wie schnell der Rechenaufwand sich mit zunehmender
48
4.1. LINEARE SPIN-KETTE
Zahl von Spins vergröÿert. Die Ermittelung der Grundzustandsenergie erfordert nun die Diagonalisierung des Operators und somit eine konkrete Matrixdarstellung desselben.
Dafür greifen wir auf Formel (2.50) zurück, wodurch wir das Skalarprodukt zweier Spins
mithilfe der bereits denierten Auf- und Absteigeoperatoren ausdrücken können.
Si
· Sj =
1 (i) (j)
(i) (j)
S+ S− + S− S+ + Sz(i) Sz(j)
2
(4.4)
Die darin auftretenden Spin-Operatoren wirken immer nur gemäÿ ihres eigenen Index auf den
entsprechenden Spin.
(k)
S± |m1 · · · mk · · · mn i = |m1 · · · (mk ± 1) · · · mn i
(k)
S+ |m1 · · · mk = 1/2 · · · mn i = 0
(k)
S− |m1 · · · mk
(4.5)
= −1/2 · · · mn i = 0
Sz(k) |m1 · · · mk · · · mn i = mk |m1 · · · mk · · · mn i
(4.6)
(Hierbei sind alle Spins mit dem Wert 1/2 angenommen. Um etwas Schreibarbeit zu sparen,
gilt zudem
~ = 1,
beziehungsweise kann
J
aus dem Hamiltonoperator in Einheiten von
~2
angenommen werden. Dies soll für alle folgenden Rechnungen vorausgesetzt werden!)
Die Produktbasis ist auÿerdem vollständig und orthogonal.
m1 m2 · · · mn m01 m02 · · · m0n = δm1 ,m01 δm2 ,m02 . . . δmn ,m0n
(4.7)
Mit dieser in den letzten Zeilen geleisteten Vorarbeit ist es prinzipiell einfach, eine Matrixdarstellung des Hamiltonoperators zu nden. Nummeriert man die
Index
i
beziehungsweise
j
durch, so berechnet sich das
Hij = hi| H |ji
(i, j)-te
2n
Basiszustände mit dem
Matrixelement wiefolgt:
.
(4.8)
Diese Matrixelemente sind durch die Kenntnis der Wirkung der in
H
auftretenden Operatoren
vollständig bestimmt. Mithilfe der in MATHEMATICA integrierten Programmiersprache und
bereits implementierten Funktionen lässt sich dieses Verfahren zur Bestimmung des Hamiltonoperators leicht umsetzen. Der entsprechende, kommentierte Code ist in Anhang A.1 abgedruckt. (Ich werde hier noch nicht auf den expliziten Code eingehen, weil dies im Abschnitt
4.3 hinlänglich erfolgt.)
Die Basis wird dabei durch Vektoren repräsentiert, deren
n
Einträge die
mk
widergeben und
entweder 0 (spin-down) oder 1 (spin-up) sind. Durch das Erstellen aller möglichen Permutationen von 0 und 1 auf
n
Stellen, wird die komplette
2n -dimensionale
Basis erzeugt und
S+ ,
(k)
S− und Sz zu implementieren. Für die Auf- und Absteigeoperatoren S± wird zunächst in
zeilenweise in eine Matrix geschrieben. Anschlieÿend ist es notwendig, die Operatoren
jedem Basisverktor der
k -te Eintrag inkrementiert beziehungsweise dekrementiert und die ent-
stehenden Vektoren wieder zeilenweise in eine neue Matrix geschrieben. Die Bestimmung der
Matrixelemente
E
D
(k) (k)
(S± )ij = i S± j
erfolgt nun durch Schleifen, die den
i-ten
Vektor der Basismatrix mit dem
(4.9)
j -ten
Vektor der
inkrementierten/dekrementierten Matrix vergleichen und bei Identität eine 1, andernfalls eine
0 in das Matrixelement
(k)
(S± )ij
schreiben. Weitere Vorfaktoren sind hier nicht notwendig.
Zudem wird auf diese Weise auch die Vernichtung der Zustände berücksichtigt, die bereits
49
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
die maximale beziehungsweise minimale z-Komponente des Spins an
k -ter
Stelle tragen. In
diesen Fällen entstehen in den Vektoren die Einträge 2 oder -1, weshalb ihr Vergleich mit den
Basisvektoren niemals Identität und somit stets das Matrixelement 0 liefert.
Die Implementierung der
(k)
Sz
gestaltet sich sehr einfach, da alle Basisvektoren Eigenvektoren
dieser Operatoren sind. Sie können folglich durch Diagonalmatrizen mit den Eigenwerte 1/2
für spin-up und -1/2 für spin-down dargestellt werden. Um die Einträge auf der Diagonalen
von
(k)
Sz
zu bestimmen, wird per Schleife der
k -te
Eintrag des
j -ten
Basisverktors betrachtet.
(k)
Ist dieser 1, so wird in das j -te Diagonalelement von Sz
1/2 geschrieben, ist er 0, wird -1/2
hineingeschrieben.
Sind die benötigten Operatoren implementiert, kann der Hamiltonoperator gemäÿ (4.2) und
(4.4) als folgende Funktion deniert werden:
H=J
n−1
X
k=1
1 (k) (k+1)
(k) (k+1)
S+ S−
+ S− S+
+ Sz(k) Sz(k+1)
2
.
(4.10)
Der zugrundeliegende Hamiltonoperator ist also eine Summe aus Produkten je zweier der
zuvor bestimmten Matrizen der Spinoperatoren. Die entsprechenden Matrizen-Additionen
und Multiplikationen sind in einem Computeralgebrasystem problemlos auszuwerten. Der
Hamiltonoperator stellt dann eine nicht-diagonale
2n × 2n -Matrix
dar, deren Eigenwerte die
möglichen Energien angeben. Die Bestimmung der Eigenwerte erfolgt ebenfalls computergestützt mithilfe der in MATHEMATICA eingebauten Funktion. Dieser Schritt stellt den
rechenaufwendigsten Teil dar und liefert die Grenze für die Gröÿe der noch berechenbaren
Kettenlänge. Analytische Lösungen sind bereits bei kleinsten Systemen extrem zeitaufwändig.
Bei einer numerischen Bestimmung der Eigenenergien dauerte die Auswertung für eine Kette
aus elf Spins etwa zwei Stunden. Rechnungen für eine Kette aus zwölf Spins habe ich nach
vier Stunden abgebrochen. Sicherlich sind unter Verwendung leistungsfähigerer Rechner und
unter höherem Zeitaufwand auch noch gröÿere Systeme mit dem vorgestellten Algorithmus
auswertbar. Der Rechenaufwand steigt jedoch exponentiell mit der Systemgröÿe, weshalb
Ketten mit etwa
1023
Spins in jedem Fall nicht mehr berechnbar sind. Im Folgenden wer-
den Verfahren aufgezeigt, die unter bestimmten Bedingungen auch zur Berechnung gröÿerer
Systeme befähigen, dennoch ist es natürlich nicht möglich, exakte Lösungen für Systeme, der
in der Praxis relevanten Gröÿe, zu ermitteln. Methoden, die trotzdem eine Abschätzung für
die Grundzustandsenergien unendlicher Systeme ermöglichen, sind im weiteren Verlauf Teil
dieser Arbeit.
Die Diagonalisierung der Spinkette diente vorwiegend dem Verständnis für das grundlegende
Vorgehen, um exakte Lösungen für kleinere Systeme zu erhalten. Dazu zählt auch die Entwicklung eines passenden Algorithmus' für die computergestützte Auswertung. In Abbildung 4.2
sind die Grundzustandsenergien für Spinketten der Länge zwei bis elf grasch dargestellt.
Dazu ist eine lineare Regression der Datenpunkte eingezeichnet. Man erkennt, dass der Verlauf der Grundzustandsenergie prinzipiell linear in der Anzahl der Spins ist. Dies ist auf den
ersten Blick zu erwarten, weil bei jedem hinzukommenden Spin eine weitere Bindung entsteht,
die eine antiferromagnetische Wechselwirkung, also antiparallelen Spin, realisieren kann.
Allerdings sieht man leichte Abweichungen in der Form, dass sich die Energiewerte für gerade
n
stets unter der Regression benden und jene für ungerade
n
darüber. Diese alternierende
Entwicklung ist damit zu begründen, dass nur Ketten mit einer geraden Zahl von Spin-1/2Teilchen einen Gesamtspin von 0 erzeugen können (Begründung siehe im folgenden Abschnitt).
50
4.1. LINEARE SPIN-KETTE
Grundzustandsenergien der Spinkette
2
3
4
5
6
7
8
9
10
11
Grundzustandsenergie E0 in J
0
-0.5
-1
-1
-1.5
-2
-2
-2.5
-3
-3
-3.5
-4
-4
-4.5
-5
-5
2
3
4
5
6
7
8
Zahl der Spins n
9
10
11
Abbildung 4.2: Lineare Kette: Grundzustandsenergie in Abhängigkeit von der Zahl der Gitterplätze
Eine Kette aus einer ungeraden Anzahl von Spin-1/2 kann sich minimal zum Gesamtspin 1/2
addieren. Ein System mit antiferromagnetischer Wechselwirkung bevorzugt stets eine antiparallele Spinausrichtung und damit einen minimalen Netto-Spin. Bei ansonsten selber Struktur
bedeutet das, dass ein System mit geringerem Gesamtspin energetisch begünstigt ist. Genau
dies manifestiert sich im Springen der Energiewerte um die Regressionsgerade. Dabei ist aber
zu beachten, dass dieser Eekt im Vergleich zu dem Energiegewinn bei zusätzlichen Bindungen sehr klein ist, weshalb die lineare Entwicklung der Grundzustandsenergie klar dominiert.
Es wurde erwähnt, dass es von Interesse ist, die Grundzustandsenergien pro Gitterplatz
für ein unendlich ausgedehntes System zu bestimmen. In Abbildung 4.3 sind die Grundzustandsenergien der Ketten aus
n
Spins pro Gitterplatz (E0 (n)/n) gegen die Zahl
n
der Spins
aufgetragen. Dort wird das eben besprochene Alternieren der Energie noch deutlicher. Auÿerdem zeigt sich aber auch, dass die Entwicklung der Energien pro Spin zunehmend kleiner wird.
Auch das ist nicht anders zu erwarten, da der pro Bindung hinzu kommende Erergiebeitrag
etwa konstant ist, während die Grundzustandsenergie des Gesamtsystems sich immer weiter
verringert. Aus diesem Grund ist eine Konvergenz der Energie pro Spin zu erwarten, was aus
der Abbildung auch optisch zu erahnen ist. Eine exakte Lösung für die Grundzustandsenergie
pro Spin wurde bereits 1931 von Hans Bethe gefunden; sie liegt bei
−0, 44135J
[Bet31]. Bei
einer gröÿeren Zahl von berechneten Kettenlängen (viel Rechenzeit!) ist dieser Konvergenzwert sicherlich abschätzbar. Interessanter wird die Abschätzung einer Grundzustandsenergie
allerdings für unendliche Systeme, zu denen keine exakten Lösungen existieren, wie es später
der Fall sein wird.
51
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
Grundzustandsenergien pro Gitterplatz
E0/n in J
2
4
6
8
10
-0.32
-0.32
-0.34
-0.34
-0.36
-0.36
-0.38
-0.38
-0.4
-0.4
-0.42
-0.42
-0.44
-0.44
2
4
6
8
Zahl der Spins n
10
Abbildung 4.3: Lineare Kette: Grundzustandsenergie pro Spin
4.2 Addition von Drehimpulsen und Basistransformation
Für die Berechnungen zur Spinkette wurde mit einer Basis gearbeitet, die aus den Tensorprodukten der Einteilchen-Hilberträume der einzelnen Spins konstruiert wird, die ProduktspinBasis. Bereits im Abschnitt 2.2 zur Addition zweier Elektronenspins ist deutlich geworden, dass es nützlich oder notwendig sein kann, auch eine andere Basis heranzuziehen: die
Gesamtspin-Basis. Dies ist zum Einen naheliegend, da es sich bei Elektronen um ununterscheidbare Teilchen handelt und unter Umständen die Spinwerte eines einzelnen, bestimmten
Elektrons irrelevant sind, aber der Gesamtspin des Systems von Bedeutung ist. Zum Anderen wird die Verwendung einer Gesamtspin-Basis entscheidende Vorteile bei der praktischen Auswertung des Heisenberg-Hamiltonoperators verschiedener Systeme aufweisen. Dabei
interessieren im Allgemeinen die möglichen Gesamtspin-Zustände bei der Kopplung einer beliebigen Anzahl von Spins (hier Spin-1/2).
Deshalb soll nun erläutert werden, wie die orthonormalen Basen eines Systems aus
n Spins,
deren Quantenzahlen die des Gesamtspins enthalten, konstruiert werden können. Für den
Spezialfall zweier Elektronen ist dies bereits geschehen, während im Folgenden der allgemeine
Fall besprochen wird, was auch die Diskussion der Addition von beliebigen Drehimpulsen
beinhaltet.
In Abschnitt 2.2 ist schon einige Vorarbeit geleistet worden. Dort wurden die Eigenschaften
des Einteilchen-Spinoperators
Drehimpulsoperator
J,
S
vorgestellt. Für allgemeine Drehimpulse verwendet man den
der dieselben Eigenschaften besitzt. Da in dieser Arbeit nur Spins als
Drehimpulse relevant sind, wird die Notation mit
52
S
als Drehimpulsoperator beibehalten.
4.2. ADDITION VON DREHIMPULSEN UND BASISTRANSFORMATION
Die Summe zweier Spins
S1
und S2 , also ihr Gesamtspin, ist durch den Operator
S = S1 + S2
(4.11)
gegeben. Dabei handelt es sich ebenfalls um einen Drehimpulsoperator, da er die fundamentale
Kommutatorrelation (2.39) erfüllt [Nol04, S.81]. Auÿerdem ist in (2.48) gezeigt worden, dass
dann auch
Sz
die Tensorprodukt-Zustände
|s1 m1 s2 m2 i
der Einteilchen-Zustände als Eigen-
zustände besitzt. Da aber
S 2 = S12 + S22 + 2S1 · S2
ist, kommutiert
S2
(1)
(4.12)
(2)
Sz und Sz :
h
i
h
i
S 2 , Sz(1) = 2 Sx(1) Sx(2) + Sy(1) Sy(2) , Sz(1)
h
i
h
i
= 2 Sx(1) , Sz(1) Sx(2) + 2 Sy(1) , Sz(1) Sy(2)
h
i
= 2i (S1 × S2 )z = − S 2 , Sz(2)
nicht mit
.
(4.13)
Es gilt andererseits aber:
S 2 , Sz = 0
.
(4.14)
Das bedeutet, dass es einen maximalen Satz kommutierender Observablen
S 2 , Sz , S12 , S22
(4.15)
gibt, die eine gemeinsame, vollständige Basis besitzen. Dabei handelt es sich um die orthonormierte Gesamtspin-Basis
{|s1 s2 ; smi} ,
(−s ≤ m ≤ s)
.
(4.16)
Dieses System aus Observablen und Basiszuständen erfüllt die Eigenwertgleichungen
S 2 |s1 s2 ; smi = s(s + 1) |s1 s2 ; smi
Sz |s1 s2 ; smi = m |s1 s2 ; smi
,
(4.17)
,
2
S1,2
|s1 s2 ; smi = s1,2 (s1,2 + 1) |s1 s2 ; smi
(4.18)
.
(4.19)
Um die Gesamtspin-Basis genau bestimmen zu können, ist es nötig, zu wissen, welche Werte
s
und
m
bei gegebenen
s1
und
s2
annehmen können. Für einen einzelnen Spin
−s ≤ m ≤ s
,
s
gilt, dass
(4.20)
m immer eine Dierenz von 1 liegt.
s einen Gesamtspin
festem s insgesamt 2s + 1 Zustände der
wobei zwischen zwei nebeneinander liegenden Werten von
Es ist nicht verwunderlich, dass derselbe Zusammenhang auch gilt, wenn
repräsentiert. Es folgt daraus auÿerdem, dass bei
|smi existieren. Dies wiederum
|s1 m1 s2 m2 i von der Dimension
Form
bedeutet, dass der Hilbertraum der Produktzustände
d = (2s1 + 1) · (2s2 + 1)
(4.21)
sein muss. Enstprechend besitzt der vollständige Raum der Gesamtspin-Zustände ebenfalls
die Dimension
d.
Es ist somit bekannt, welche Werte
m
bei gegebenem
s
annehmen kann. Diese Kenntnis ist
53
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
aber nur hinreichend, wenn auch bekannt ist, welche Werte
Da es sich bei
S
s
um einen Drehimpulsoperator handelt, muss
bei gegebenen
s≥0
s1 , s2
annimmt.
sein. Auÿerdem ist durch
(2.48) gezeigt worden, dass
m = m1 + m2
Die Bestimmungen der möglichen Werte von
der möglichen
(m1 , m2 )-Kombinationen
s
.
(4.22)
kann unter diesen Annahmen durch Abzählen
bei festen
s1 , s2
geschehen (siehe zum Beispiel im
Buch von W. Nolting [Nol04, S.84 ]). Das Ergebnis ist die Dreiecksungleichung
|s1 − s2 | ≤ s ≤ s1 + s2
.
(4.23)
Die Ungleichung hat ein intuitive, klassische Anschauung: Betrachtet man S1 und S2 als klas-
s = |S1 +S2 | der Summe dieser Vektoren bei antiparalleler
s = |s1 − s2 | und bei paralleler Ausrichtung s = s1 + s2 . Der Quantisierung
s auÿerdem alle Werte in ganzzahligen Schritten dazwischen annehmen. Für die
sische Vektoren, dann ist der Betrag
Ausrichtung
zufolge kann
Dimension der Gesamtspin-Basis gilt dann
sX
1 +s2
(2s + 1) = (2s1 + 1)(2s2 + 1) = d
.
(4.24)
s=|s1 −s2 |
Die geforderte Gleichheit der Dimensionalität beider Basisdarstellungen ist also erfüllt.
Die Benutzung der Gesamtspin-Basis kann - wie erwähnt - vorteilhaft sein. Als die natürliche
Basis eines Systems mehrerer Spins ist jedoch die Produktspin-Basis zu sehen. Der Übergang
von der Produktspin-Basis in die Gesamtspin-Basis geschieht durch unitäre Transformationen.
4.2.1 Basistransformation und Clebsch-Gordan-Koezienten
Die Übergangtransformationen zwischen beiden Basisdarstellungen sollen etwas genauer untersucht werden. Da wir zunächst von festen
s1 , s2
ausgehen, ist folgende Vereinfachung der
Notation möglich und sinnvoll:
|s1 s2 ; smi → |smi ;
Ein Zustand
|s1 m1 s2 m2 i → |m1 m2 i
.
|smi im Gesamtspin-Raum kann durch Zustände |m1 , m1 i aus dem Produktspin-
Raum entwickelt werden.
|smi =
m1 +m
X2 =m
hm1 m2 |smi |m1 m2 i =
m1 +m
X2 =m
C(s1 m1 ; s2 m2 ; sm) |m1 m2 i
(4.25)
m1 ,m2
m1 ,m2
m1 und m2 , die m = m1 + m2
erfüllen. Die Entwicklungskoezienten C(s1 m1 ; s2 m2 ; sm) = hm1 m2 |smi werden als ClebschDie Summe läuft dabei über alle möglichen Kombinationen von
Gordan-Koezienten bezeichnet. Sie sind die Elemente der unitären Transformationsmatrix
T,
die gemäÿ den besprochenen Zusammenhängen vom Rang
Tji = C({s1 m1 s2 m2 }j ; {sm}i );
({s1 m1 s2 m2 }j : Quantenzahlen
{sm}i :
54
Quantenzahlen
(2s1 + 1)(2s2 + 1)
T −1 = T †
sein muss.
(4.26)
s1 , m1 , s2 , m2 des j -ten Basisverktors der Produktspin-Basis.
s, m des i-ten Basisvektors der Gesamtspin-Basis.)
4.2. ADDITION VON DREHIMPULSEN UND BASISTRANSFORMATION
Vollständigkeit und Orthogonalität der beiden Basen führen zu den beiden Orthogonalitätsrelationen der Clebsch-Gordan-Koezienten.
∗
hm1 m2 |smi m1 m2 s0 m0 = δs,s0 δm,m0
,
(4.27)
∗
hm1 m2 |smi m01 m02 sm = δm1 ,m01 δm2 ,m02
.
(4.28)
X
m1 ,m2
X
s,m
Man wählt zudem die Phasen der Zustände
|smi
so, dass die Clebsch-Gordan-Koezienten
stets reell sind.
hm1 m2 |smi = hsm|m1 m2 i
(4.29)
Dies sind einige wesentliche Eigenschaften der Clebsch-Gordan-Koezienten, die aus den
vorherigen Ausführungen hervorgehen beziehungsweise sie bestätigen. In den Berechnungen
zu meiner Arbeit spielen die Clebsch-Gordan-Koezienten eine wichtige Rolle. MATHEMATICA verfügt über eine bereits eingebaute Funktion, die unter Angabe der Quantenzahlen
(s1 , m1 ; s2 , m2 ; s, m)
als Argumente die zugehörigen Clebsch-Gordan-Koezienten ausgibt.
Auÿerdem gibt es zahlreiche Tabellen, in denen die Koezienten für verschiedenste Werte
aufgeführt sind. Deshalb soll nur kurz skizziert werden, wie sie praktisch berechnet werden
können.
s = m = s1 + s2 .
Zunächst betrachte man den Fall maximaler Quantenzahlen, also
wegen (4.22) nur für
m1 = s1
und
m2 = s2
Dies ist
möglich. In diesem Fall besteht (4.25) nur aus
einem Summanden.
|s = s1 + s2 , m = s1 + s2 i = |m1 = s1 , m2 = s2 i
(4.30)
S− auf diesen Zustand lassen sich sämtliche
|s = s1 + s2 , mi und damit die entsprechenden Clebsch-Gordan-Koezienten berech-
Durch sukzessive Anwendung des Absteigeoperators
Zustände
nen. So ist zum Beispiel
S− |s = s1 + s2 , m = s1 + s2 i =
p
2(s1 + s − 2) |s = s1 + s2 , m = s1 + s2 − 1i
.
(4.31)
|s = s1 + s2 , m = s1 + s2 i aus (4.30) bekannt ist, kann |s = s1 + s2 , m = s1 + s2 − 1i leicht
berechnet werden. Auf dieselbe Art erhält man die verbleibenden Zustände für s = s1 + s2 .
Anschlieÿend bestimmt man den Zustand |s = s1 + s2 − 1, m = s1 + s2 − 1i. Für m = m1 +
Da
!
m2 = s1 + s2 − 1
gibt es dann zwei mögliche Kombinationen, also zwei Summanden in (4.25).
Man wählt den allgemeinen Ansatz
|s = s1 + s2 − 1, m = s1 + s2 − 1i = α |m1 = s1 − 1, m2 = s2 i + β |m1 = s1 , m2 = s2 − 1i
(4.32)
mit den Clebsch-Gordan-Koezienten
α
und
β.
Da die Zustände
|smi
orthonormiert sind,
gilt
hs1 + s2 , s1 + s2 − 1|s1 + s2 − 1, s1 + s2 − 1i = 0
Da der Bra-Zustand bereits bekannt ist, liefert dies eine Bestimmungsgleichung für
Zudem müssen die Zustände
|smi normiert sein, wodurch α und β
(4.33)
α
und
β.
dann vollständig bestimmt
sind. Anschlieÿend kann man wieder unter Verwendung der Absteigeoperatoren fortfahren.
Durch wiederholte Benutzung dieser beiden Schritte lassen sich alle Clebsch-Gordan-Koefzienten ermitteln, was bei gröÿeren
s1 , s2 schnell mit einem hohen Arbeitsaufwand verbunden
ist.
55
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
Ferner existieren Rekursionsformeln für die Berechnung sowie explizite, numerische Darstellungen der Clebsch-Gordan-Koezienten [AKHv11].
Es wurde bisher gezeigt, wie sich die Addition zweier Spins mit beliebigen Spinwerten
s1 , s2
vollzieht und welche Darstellungsformen der zugehörigen Zustandsräume bestehen. In dem
Kapitel soll aber auch geklärt werden, wie sich die Addition beliebig vieler Spins vollzieht.
Bedenkt man, dass die Spinwerte
s1 , s2
ihrerseits Gesamtspins einer Addition von Spins sein
können, dann ist klar, dass ein rekursives Vorgehen zielführend ist.
Man betrachte beispielsweise ein System dreier Spins
s1 , s2 , s3
beliebiger Gröÿe, dessen
Zustände in der natürlichen Basis (Produktspin-Basis) die Form
|s1 m1 i ⊗ |s2 m2 i ⊗ |s3 m3 i = |s1 m1 s2 m2 i ⊗ |s3 m3 i
(4.34)
der Tensorprodukte der Einteilchenzustände annehmen. Aus der rechten Seite der ZustandsDarstellung ist ersichtlich, dass dies auch als Tensorprodukt des Zweiteilchenraumes der ersten
beiden Spins mit dem Einteilchenraum des dritten Spins aufgefasst werden kann. Der Zweiteilchenraum wiederum ist gemäÿ der vorherigen Ausführungen in einer Gesamtspin-Basis
darstellbar, sodass das System mittels einer unitären Transformation auch durch Zustände
der Form
|s1 s2 ; s12 m12 i ⊗ |s3 m3 i
darstellbar ist. Dabei handelt es sich bei
bei
m12
s12
(4.35)
um die Quantenzahl zum Spin S12
= S1 + S2
und
um die dazugehörige z-Komponente. Im nächsten Schritt gilt es nun, den Gesamtspin
s12 der ersten beiden Spins mit dem dritten Spin s3 zu koppeln. Dabei entsteht der Gesamtspin
s123 des ganzen Systems als Quantenzahl des Gesamtspinoperators S123 = S12 + S3 . Dies
geschieht wieder, wie beschrieben, durch eine unitäre Transformation unter Verwendung der
Clebsch-Gordan-Koezienten.
T
|s1 , s2 ; s12 m12 ; s3 m3 i −→ |s1 s2 s12 s3 ; s123 m123 i
(4.36)
Die Zustände nach der Transformation (rechts vom Pfeil) bilden die neue Gesamtspin-Basis.
Die Transformationsmatrix ist bestimmt durch
Tji = C({s12 m12 ; s3 m3 }j ; {s123 m123 }i )
wenn wieder die Basiszustände mit
i
beziehungsweise
j
,
(4.37)
durchnummeriert werden.
Es ist zu beachten, dass - im Unterschied zur Addition zweier Spins - hier die
s12
bereits
mehrere verschiedene Werte haben können. Das bedeutet, dass die Unterräume der unter-
s3 gekoppelt werden müssen. Auch ist in einer allgemeineren
s3 ebenfalls einen Gesamtspin darstellen und somit verschiedene
Werte haben. Dann wäre es notwendig, die Basistransformation mit allen Werten von s12 mit
jeweils allen Werten von s3 durchzuführen, was unter Umständen ein kompliziertes Unterschiedlichen
s12
jeweils mit den
Situation vorstellbar, dass die
fangen sein kann. Existierten nun noch endlich viele weitere Spins, so würde man auf die
selbe Art und Weise fortfahren:
s123
mit
s4
zu
s1234
koppeln,
s1234
anschlieÿend an
s5
und so
weiter. In diesem Fall stehen immer mehrere Quantenzahlen in den Produkt- und GesamtspinBasiszuständen, von denen aber immer nur vier bei einer Basistransformation verändert werden (wie gesehen). Allgemein kann man dann für die Addition der Spins
Spin
S12
{|γ; s1 m1 s2 m2 i}j
56
S1
und
S2
zu einem
die Zustände beiden Basen darstellen durch
und
0 0 0
γ ; s1 s2 ; s12 m12
i
.
(4.38)
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
Dabei sind in
γ, γ 0
die jeweils anderen Quantenzahlen in den Produkt-/ Gesamtspin-Ba-
siszuständen zusammengefasst. Die Matrix der Basistransformation hat in diesem allgemeinen
Fall natürlich einen höheren Rang (in Abhängigkeit der Dimension des Unterraums der
γ ) und
ihre Einträge sind gegeben durch
Tji = C({s1 m1 ; s2 m2 }j ; {s12 m12 }i )δγ j ,γ 0i δ(s1 s2 )j ,(s01 s02 )i
.
(4.39)
Das erste Kronecker-Delta trägt der Tatsache Rechnung, dass für vollständige Basisräume die
durch die
γ
aufgespannten Unterräume orthonormal sind, also
0
γ γ = δγ,γ 0
.
(4.40)
Das zweite Kronecker-Delta berücksichtigt den allgemeinen Fall, dass bereits
s1
und
s2
Gesamtspins aus anderen Transformationen sein können und damit keinen festen Wert haben.
Dann folgt aus der Orthonormierung der Basen:
s1 m1 s2 m2 s01 s02 ; s12 m12 ∝ δs1 ,s01 δs2 ,s02
.
(4.41)
Die praktische Bedeutung dieser allgemeinen Transformationsvorschrift wird im Zuge des
Algorithmus der noch folgenden Berechnungen erkenntlich werden.
Durch das zuvor beschriebene Verfahren ist klar, wie die Gesamtspin-Basis eines Systems aus endlichen vielen Spins zu konstruieren ist. Je zwei Einteilchenräumen lassen sich
in Gesamtspinräume transformieren, die dann selber als mehrere Einteilchenräume gröÿerer Spins aufgefasst werden können und somit wieder an andere Einteilchenräume zu einem
Gesamtspinraum gekoppelt werden können. Damit lässt sich die Addition endlich vieler Spins
(oder allgemein: Drehimpulse) auf die rekursive Addition zweier Spins (Drehimpulse) zurückführen. Führt man dabei die Basistransformationen mit allen involvierten Spins durch, so
erhält man am Ende eine Basis, die eine Quantenzahl
S1 + S2 + · · · Sn
s
enthält, die den Gesamtspin S
=
repräsentiert.
Das Beispiel mit den drei Spins hat gezeigt, dass es von der Reihenfolge abhängt, in der man
die einzelnen Spins aneinander koppelt, welche Quantenzahlen am Ende in der GesamtspinBasis stehen. So hätte man beispielsweise erst die Spins
hätte schlieÿlich die Quantenzahl
s13
statt
s12
S1
und
S3
koppeln können und
in der letzten Basis. Welche Reihenfolge der
Spinadditionen am sinnvollsten ist, hängt von dem Problem ab, das gelöst werden soll. Dieses
bestimmt auch darüber, ob es überhaupt nützlich ist, alle vorhandenen Spins bis zum Erhalt
des Gesamtspins zu addieren. Die praktische Bedeutung letzterer Aussagen wird bald deutlich
werden.
4.3 Untere Schranken der Grundzustandsenergie für
unendliche Spinsysteme
In diesem Kapitel haben sich die konkreten Berechnungen bisher auf die Spinkette beschränkt.
Dabei lag das Augenmerk vor allem auf der exakten Berechnung der Grundzustandsenergien
durch Diagonalisierung bei verschiedenen Längen der Kette. Exakte Ergebnisse bezüglich der
57
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
Grundzustandsenergie einer unendlich langen Kette wurden dabei nicht erzielt. Ferner sind
zu den möglichen Zustandsräumen von Systemen mit endlich vielen Spins ausführliche und
allgemeine Erläuterungen erfolgt.
Ziel dieser Arbeit ist es, mittels exakter Diagonalisierung und passenden Berechnungsmethoden, Aussagen über die Grundzustandsenergien verschiedener zwei- und dreidimensionaler Spinsysteme unendlicher Gröÿe (mit antiferromagnetischer Austauschwechselwirkung) zu
treffen. Tatsächlich interessiert natürlich die Grundzustandsenergie pro beteiligten Spin, da
die absolute Grundzustandsenergie mit der Systemgröÿe wächst und im unendlichen Fall divergiert. Selbstverständlich ist es nicht möglich, eine exakte Diagonalisierung für ein unendlich
groÿes Spinsystem vorzunehmen. Die Aussagen über unendliche Systeme können also nur einschränkender Natur sein, wenn sie aus exakten Berechnungen und nicht aus Näherungen oder
numerischen Approximationen hervorgehen.
4.3.1 Methode der unteren Schranke
Erinnern wir uns an den Abschnitt 2.1 über die Heitler-London-Näherung, so haben wir dort
bereits eine der angesprochenen einschränkenden Aussagen über die Grundzustandsenergie
verwendet. Die Rede ist vom Variationsprinzip, das in Formel (2.15) Ausdruck ndet. Mit
diesem Prinzip ist es möglich, für einen bekannten Hamiltonoperator durch Einsetzen einer
Test-Wellenfunktion eine exakte, obere Schranke für die Grundzustandsenergie zu erhalten.
Dabei gilt folglich, dass die Abschätzung umso besser ist je kleiner der erhaltene Energieerwartungswert ist. Die Güte unterschiedlicher Testfunktionen ist somit leicht vergleichbar.
In dieser Arbeit wird eine Methode verwendet, die weniger verbreitet ist und exakte, untere
Schranken für die Grundzustandsenergie liefert. Eingeführt und in selber Weise verwendet
wurde diese Methode in einer Publilation von Roser Valentí und Rolf Tarrach [TV90]. Die
von mir ausgeführten Rechnungen sind Anwendungen der Methode auf Spinsystemen anderer
Geometrie. Die Methode der unteren Schranke soll nun hergeleitet werden.
Dazu betrachte man einen Hamiltonoperator
toren
Hi (i = 1, . . . , n)
H,
der sich in
n
elementare Hamiltonopera-
zerlegen lässt, sodass man schreiben kann:
H=
n
X
Hi
,
(4.42)
i=1
wobei die einzelnen
Hi
im Allgemeinen nicht kommutieren.
[Hi , Hj ] 6= 0, i 6= j
(4.43)
In der Spektraldarstellung lässt sich der Hamiltonoperator dann als
H=
n X
X
i=1
Eis |i, si hi, s|
(4.44)
|i, si hi, s|
(4.45)
s
schreiben, wobei
Ii =
X
s
58
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
Hi sind. Die Unterräume der Hi sind also vollständig.
Der Index i in den Zuständen kennzeichnet, dass er zu dem Hamiltonoperator Hi gehört,
während der Index s über das Spektrum der jeweiligen Hi summiert. Ei0 ist dann die Grundzustandsenergie des Hamiltonoperators Hi und es gilt:
die Identitätsmatrizen im Unterraum der
Ei0 ≤ Ei1 ≤ · · · ≤ Eis ≤ · · · ≤ Ein
Nun sei
.
(4.46)
|ψi eine beliebige, normierte Wellenfunktion. Der Energieerwartungswert des Hamilton-
operators berechnet sich in diesem Zustand unter Ausnutzung der zuvor gemachten Zusammenhänge zu
E = hψ| H |ψi =
n X
X
i=1
Dies gilt für alle Funktionen
E0
von
H
2
Eis | hi, s|ψi | ≥
s
n
X
Ei0
X
2
| hi, s|ψi | =
s
i=1
n
X
Ei0
.
(4.47)
i=1
ψ , also auch für jene, die Eigenfunktion zur Grundzustandsenergie
ist. Daraus folgt dann die Aussage:
E0 ≥
n
X
Ei0
.
(4.48)
i=1
Die Summe der Grundzustandsenergien aller
gie von
H.
Hi
ist kleiner gleich der Grundzustandsener-
Das Gleichheitszeichen gilt nur, wenn die
Hi
untereinander kommutieren. Es
handelt sich unter den gegebenen Annahmen um ein ganz allgemeingültiges, mathematisch
begründetes Theorem.
Die Idee, unter der das Theorem in dieser Arbeit verwendet wird, ist es, aus unendlichen,
periodischen Spingittern Teilstrukturen (Subgitter) zu untersuchen, aus denen das Gesamtgitter aufgebaut werden kann. In solchen Fällen kann der Hamiltonoperator des Gesamtsystems
als eine Summe über unendlich viele Hamiltonoperatoren, die die Teilstrukturen beschreiben,
gesehen werden. Beim Zusammsetzen des Gitters überlappen die einzelnen Teilstrukturen,
die dazugehörigen Hamiltonoperatoren vertauschen also nicht. Dadurch ist eine Situation wie
in (4.42) gegeben. Es gilt auch hier wieder, dass nur die Betrachtung der Grundzustandsenergie pro Gitterplatz sinnvoll ist. Wie dies in der Praxis geschehen kann, wird an entsprechender
Stelle gezeigt.
Es folgt aus dem Theorem der unteren Schranke, dass die benutzen Subgitter umso besser
sind je höhere Grundzustandsenergien pro Gitterplatz sich aus ihnen ergeben. Über das besagte Variationsprinzip lassen sich obere Schranken für die Systeme nden. Bei Vorliegen von
Schranken beiderseits lässt sich die tatsächliche Grundzustandsenergie eines Systems auf eine
gewisse Energiespanne eingrenzen, die möglichst klein werden soll.
Es ist Teil dieser Arbeit, durch Vergleich der unteren Schranken, Aussage darüber zu treen,
nach welcher Systematik die Subgitter am besten zu wählen sind, um möglichst gute Abschätzungen zu erhalten. Grundlegend dafür ist es naheliegenderweise, zunächst die exakten
Grundzustandsenergien der Subgitter zu bestimmen.
4.3.2 Exakte Diagonalisierung zwei- und dreidimensionaler
antiferromagnetischer Spingitter
Um untere Schranken für die Grundzustandsenergie von Spinsystemen zu nden, ist es notwendig, zunächst eine Diagonalisierung der jeweils gewählten Subgitter vorzunehmen. Gemäÿ
59
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
des Theorems der unteren Schranken (4.48) ist es ausreichend, auch von den Subgittern nur
die Grundzustandsenergien zu nden.
Objekte der Berechnungen sind zwei- und dreidimensionale Gitter mit antiferromagnetisch-
n
er Austauschwechselwirkung aus
Spin-1/2-Plätzen. Solche Systeme haben in allen Basis-
darstellungen eine Dimension der Gröÿe
n
berg-Hamiltonoperator eine 2
×
2n ,
entsprechend wird der dazugehörige Heisen-
2n -Mtarix darstellen. Dies führt für gröÿe
n
sehr schnell
zu einem (zu) hohen Rechenaufwand bei der Diagonalisierung. Es ist aber naheliegend, dass
die Eigenräume zur Grundzustandsenergie eines Heisenberg-Spingitters nur einen Unterraum
der Gesamtspin-Basis darstellen. So werden im antiferromagnetischen Spingitter Zustände, die
komplett parallel ausgerichtete Spins repräsentieren, beispielsweise nicht Teil des Grundzustands sein. Es ist also anzunehmen, dass der Heisenberg-Hamiltonoperator gegebenenfalls
blockdiagonal ist. In diesem Fall wäre es zur Bestimmung der Grundzustandsenergie ausreichend, den zugehörigen Block kleinerer Dimension zu nden und diesen zu diagonalisieren.
Damit wäre die Problemstellung auf ein System kleinerer Ordnung reduziert.
Lieb-Mattis-Theorem
Aus einem Theorem, das 1962 von Elliot Lieb und Daniel Mattis
veröentlicht wurde [LM62], folgt, welchen Unterraum der Gesamtspin-Basis der Grundzustand in einem antiferromagnetischen Spingitter einnimmt. Die Gültigkeit des Theorems ist
dabei auf bipartite Gitter beschränkt (bipartite Gitter wurden in Abschnitt 3.2.3 deniert).
Man betrachte wieder ein bipartites Gitter mit den Untergittern A und B. Der maximale Spin
SA des
si(A) .
Systems von Untergitter A ist gegeben durch die Summe seiner einzelnen Spinwerte
SA =
X
si(A)
(4.49)
i(A)
Völlig analog gilt für den maximalen Gesamtspin
SB =
X
SB
si(B)
des Untergitters B:
.
(4.50)
i(B)
Das Theorem von Lieb und Mattis besagt, dass dann bei antiferromagnetischer Wechselwirkung zwischen Spins der Gitter A und B der Gesamtspin
S
des Grundzustandes durch
S = |SA − SB |
gegeben ist. In dieser Arbeit sind die Gitter nur aus Spin-1/2 aufgebaut (si
(4.51)
= 1/2),
sodass
sich in diesem Spezialfall der Gesamtspin des Grundzustandes zu
S = 1/2 · |(n(A) − n(B))|
berechnet, wobei
n(A)
und
n(B)
(4.52)
die Zahl der Spins in den Untergittern A und B sind. Somit
existiert ein bestimmbarer Unterraum, in dem der Grundzustand liegt. Dies ist aber nicht
gleichbedeutend damit, dass alle Zustände dieses Unterraums gleichzeitig den Grundzustand
darstellen.
Das Theorem macht noch allgemeinere Aussagen, die hier nicht von Bedeutung sind. Die von
mir berechneten Gitter sind allesamt antiferromagnetisch und bipartit, sodass der genannte
Spezialfall des Theorems immer gültig ist. Dies ist eine wichtige Voraussetzung um auch für
60
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
etwas gröÿere Untergitter noch eine exakte Diagonalisierung vorzunehmen zu können, da sich
die Dimension des verbleibenden Problems, wie besagt, verkleinert. Dies ist auch der Grund,
weshalb eine Transformation in die Gesamtspin-Basis sinnvoll ist, da nur in dieser Basis der
benötigte Unterraum direkt isolierbar ist.
Es folgt aus dem Theorem indirekt eine weitere Aussage für antiferrmoagnetische, bipartite Gitter, die die Dimension des eektiven Zustandsraums ebenfalls verringert. Demnach
addiert sich der Gesamtspin einer Gruppe von Spins aus Untergitter A, die alle an nur einen
gemeinsamen Gitterplatz aus B koppeln, im Grundzustand zur Summe ihrer einzelnen Spins
(A und B können in dieser Aussage natürlich vertauscht werden). Das bedeutet: Sind
si = 1/2
an nur einen gemeinsamen Gitterplatz aus B gebunden, so kann man diese
n Spins
n Spins
s = 1/2 · n zusammenfassen. Nimmt man an, dass
S und zum System der Hamiltonoperator H gehört, dann gilt
im Grundzustand zu einem Spin der Gröÿe
zu diesen Spins der Gesamtspin
die Aussage unter der Bedingung, dass
2 S ,H = 0
.
(4.53)
Mithilfe der vorgestellten Grundlagen sollen nun verschiedene zwei- beziehungsweise dreidimensionale, unendliche Gitter und deren Grundzustandsenergien pro Gitterplatz abgeschätzt
werden. Dabei soll vorallem untersucht werden, nach welchen Kriterien die Subgitter zu wählen
sind, um möglichst gute untere Schranken zu erhalten. Zu diesem Zweck werden das zweidimensionale Quadrat- und Honeycomb-Gitter sowie das dreidimensionale einfach-kubische
Gitter betrachtet. Dabei werden jeweils ein geschlossenes Subgitter und zwei oene Subgitter
verschiedener Gröÿe berechnet. Was dies im konkreten Einzelfall bedeutet, wird im Folgenden
zu sehen sein. Durch diese gleichbleibende Vorgehensweise entsteht eine Menge von Ergebnissen mit guter Vergleichbarkeit. Aus den Grundzustandsenergien der Subgitter lassen sich
dann, wie zuvor erklärt, untere Schranken für die Grundzustandsenergie pro Spin ermitteln.
Ein Vergleich der Werte für die unterschiedlichen Subgitter lässt erkennen, welche Formen
von Untergittern am besten geeignet sind, untere Schranken zu geben.
Zweidimensionales Quadratgitter und Algorithmus
Abbildung 4.4: Ausschnitt aus einem Quadratgitter
Das erste System, das betrachtet werden soll, ist das Quadratgitter, von dem ein Ausschnitt in Abbildung 4.4 gezeigt ist. Zu diesem Gitter sind die von mir gemachten Berechnungen bereits in der Publikation von Roser Valentí und Rolf Tarrach [TV90] vorgestellt
61
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
worden. Ich habe diese Rechnungen ebenfalls durchgeführt, um ein besseres Verständnis für
die Problemstellung/-lösung entwickeln zu können und um eine Kontrollmöglichkeit für die
von mir erzielten Resultate zu haben, da der verwendete Algorithmus für die weiteren Berechnungen natürlich korrekt sein sollte.
Die erste Rechnung, die ich vorstellen möchte, ist die zum groÿen, oenen Subgitter, dem
3
2
1
4
5
7
6
9
8
Abbildung 4.5: Subgitter des Quadratgitters: Doppelkreuz (1b)
Doppelkreuz. Es ist in Abbildung 4.5 dargestellt. In einem unendlichen Quadratgitter besitzt
jeder Gitterplatz vier Bindungspartner. Das Doppelkreuz bezeichnet man deshalb als oen, da
es getreu seiner Abbildung über sechs Spins verfügt, die nur einen Bindungspartner haben. Ich
stelle es deshalb als erstes vor, weil seine Berechnung sich als eine der komplexesten von mir
durchgeführten darstellt. Dies führt dazu, dass die detaillierte Erläuterung der Vorgehensweise
die wesentlichen Spezialfälle mit einbezieht. Somit wird der verwendete Algorithmus auf eine
Weise vorgestellt, dass die Erläuterung der anderen Rechnungen keine Schritte enthält, die hier
nicht bereits vorkamen. Damit wird die allgemeinste Form der Berechnung also sinnvollerweise
zuerst bekannt gemacht.
Ich möchte dabei so vorgehen, dass ich die allgemeine Bedeutung der einzelnen Schritte
erkläre und parallel aufzeige, wie diese Schritte im Fall des Doppelkreuzes aussehen. Dadurch
können die allgemeinen Ausführungen mittels der beispielhaften Umsetzung besser verständlich
gemacht werden. Meine Implementierung in Form des MATHEMATICA-Codes werde ich an
geeigneten Stellen darlegen. Dabei möchte ich nicht zu sehr auf programmiertechnische Details eingehen, sondern die Prinzipien des Algorithmus' aufzeigen. Der gesamte Code zu dieser
Berechnung ist in Anhang A.2 zu sehen.
Beschreibt man die einzelnen Spins durch
Si ,
wobei der Index
i
die Nummerierung der
Spins gemäÿ der Abbildung wiedergibt, so ergibt sich der Heisenberg-Hamiltonoperator
H1b = J [S1 (S2 + S3 + S4 ) + S6 (S7 + S8 + S9 ) + S5 (S1 + S6 )]
.
(4.54)
Die Auswertung dieses Hamiltonoperators wäre in der natürlichen Basis analog zu der der
Spinkette theoretisch möglich. Allerdings enstünde dabei ein Zustandsraum der Dimension
29 = 512,
der bei der Diagonalisierung schwer zu handhaben ist. Es gilt also die angesproch-
enen Reduktionen des Zustandsraumes für den Grundzustand vorzunehmen.
Es gelte die Notation
Sxy···z = Sx + Sy + · · · + Sz
62
,
(4.55)
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
wobei die Indizes wieder die Spins nummerieren und
sxy···z
die zugehörige Quantenzahl ist.
Aus der Abbildung des Doppelkreuzes lässt sich erkennen, dass
2
2
S234 , H1b = S789
, H1b = 0
gilt. Damit ist bekannt, dass im Grundzustand
(4.56)
s234 = s789 = 3· 12 = 3/2. Der Hamiltonoperator
für das Doppelkreuz lässt sich so schreiben, dass diese Spinwerte direkt darin vorkommen.
H1b = J [S1 S234 + S6 S789 + S5 (S1 + S6 )]
(4.57)
Es ist das ganz allgemeine Vorgehen, den Hamiltonoperator zunächst durch die für den
Grundzustand bekannten Gesamtspin-Operatoren auszudrücken. In diesem Fall hat sich dadurch der Zustandsraum, in dem sich der Grundzustand benden muss, bereits auf die Dimension
42 · 23 = 128
reduziert.
Die entscheidende Reduktion besteht allerdings in der Anwendung des Lieb-Mattis-Theorems. Schat man es, den Hamiltonoperator in einer Basis darzustellen, deren Zustände die
Quantenzahl des Gesamtspins enthält, lässt sich daraus der relevante Block isolieren und aus
diesem wiederum die Grundzustandsenergie erhalten. Dafür muss aber zunächst eine Darstellung des Hamiltonoperators in der Gesamtspin-Basis gefunden werden. Aus dem Abschnitt 4.2
über die Addition von Drehimpulsen sind die Transformationsmatrizen
T
bekannt, die einen
Zustand aus der Gesamtspin-Basis auf den entsprechenden in der Produkspin-Basis abbilden.
Allgemeine Operatoren, dargestellt in der Produktspin-Basis (A), lassen sich dann wiefolgt in
0
eine Gesamtspin-Basisdarstellung (A ) transformieren:
A0 = T −1 AT
Dabei kann
T
.
(4.58)
auch die Hintereinanderausführungen mehrerer Transformationen
Transformationsabfolge eines Vektors
t
ti
sein. Die
a
t
t
t
1
2
3
n
a0 −→
a1 −→
a2 −→
· · · −→
an
(4.59)
lässt sich in der Transformation
T = t1 t2 t3 · · · tn ,
T a0 = an
(4.60)
zusammenfassen.
Um den Grundzustand zu berechnen (nur dann), kann man im letzten Hamiltonoperator
(4.57)
S234
und
S789
als einzelne Spin-3/2 ansehen. Er besteht dann nur aus der Summe von
Produkten aus Spinoperatoren, wie wir es von der Spinkette kennen. Er lässt sich in der natürlichen Basis analog zur Spinkette als Matrix darstellen, wenngleich dafür ein etwas anderer
Algorithmus nötig ist, da er nicht nur Spin-1/2-Operatoren enthält (eine Implementierung
t1 , t2 · · · tn , die
0
Hamiltonoperator H in
dazu wird noch vorgestellt). Unter Kenntnis aller nötigen Transformationen
zu einer Basis führen, die den Gesamtspin enthält, kann man den
dieser Basis darstellen:
H 0 = T −1 HT ,
T = t1 t2 · · · tn
.
(4.61)
Auf diese Weise muss der gesamte Hamiltonoperator transformiert werden, was natürlich mit
einem gewissen Rechenaufwand verbunden ist. Eine nähere Betrachtung zeigt aber, dass es
63
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
möglich ist, den Hamiltonoperator so umzuschreiben, dass er möglichst viele Teile enthält, die
auch in der Gesamtspin-Basis bereits diagonal sind. Im Allgemeinen gilt
2
S1 · S2 = 1/2 S12
− S12 − S22
.
(4.62)
Es lässt sich also jedes Skalarprodukt zweier Spinoperatoren durch die Quadrate der Spinoperatoren und das Quadrat des zugehörigen Gesamtspinoperators ausdrücken. Damit ist
ein solches Skalarprodukt in jeder Basis diagonal, die die Quantenzahlen
s1 , s2 , s12
enthält.
Nun führe man sich auÿerdem vor Augen, wie die Konstruktion der Gesamtspin-Basis erfolgt.
Nach (4.36) werden bei jeder Kopplung zweier Spins die Quantenzahlen
m der z-Komponenten
durch die Quantenzahl des Summenspins und dessen z-Komponente ersetzt. Es entstehen bei
der Kopplung zweier Spins zu einer Gesamtspin-Darstellung also genau die Zustände in denen die rechte Seite von (4.62) unmittelbar diagonal ist (wegen der bekannten Wirkung von
S2
auf einen Zustand, der die Quantenzahl
s
enthält). Es ist somit sinnvoll, im Hamilton-
operator möglichst viele Skalarprodukte durch Quadrate von Spinoperatoren auszudrücken.
Dies ist solange zielführend möglich wie kein Spin in mehreren Summenspins auftritt, da kein
(Gesamt)Spin an einen anderen gekoppelt werden kann, in dem derselbe bereits auftritt. Es
gilt also den Hamiltonoperator durch möglichst viele Quadrate des Spinoperators darzustellen,
ohne solche Dopplungen zu erzeugen. Der Heisenberg-Hamiltonoperator des Doppelkreuzes
nimmt dann die Form an, aus der er berechnet wird.
2
2
2
2
H1b = J/2 S1234
− S12 − S234
+ S6789
− S62 − S789
+ JS5 (S1 − S6 )
(4.63)
(Die Kopplung der Spins 1, 5 und/oder 6 ist hier beispielsweise nicht möglich, da diese Spins
bereits Teil anderer Gesamtspins sind.)
Das Aufstellen der Hamiltonoperatoren dieser Gestalt habe ich jeweils per Hand vorgenommen. Eine Implementierung, die diese Form selbständig aus dem ursprünglichen Hamiltonoperator herleitet ist durchaus umsetzbar; ich habe aus Zeitgründen jedoch darauf verzichtet.
Ist der Hamiltonoperator des Subgitters in seiner endgültigen Form aufgestellt, so gilt
es, zunächst die Transformationen zu nden, die die natürliche Produktspin-Basis in die
Gesamtspin-Basis der gewünschten Form überführen. Dafür wird zunächst die natürliche Basis erstellt. Die darin auftretenden Quantenzahlen sind die
si
und
mi ,
wobei
i
abermals alle
auftretenden Spins des Systems nummeriert. Die Basis enthält also immer doppelt soviele
Quantenzahlen wie die Zahl der Spins im betrachteten Gitter. Während die
si
fest sind, ex-
(−si ≤ mi ≤ si ). Die vollständige
mi . In meinem Code
ist das Erstellen der natürlichen Basis durch die Matrix/Array naturalBasis folgendermaÿen
istieren zu ihnen jeweils
2si + 1 Werte für mi
in der Spanne
Basis besteht nun aus allen vorhandenen Kombinationen der möglichen
realisiert:
names werden die Nummerierungen aller vorkommenden Spins, also beim Doppelkreuz {1,234,5,6,789}, geschrieben. In die Liste spins werden in der zugehörigen Reihenfolge die Spinwerte eingetragen ({1/2,3/2,1/2,1/2,3/2}). Aus diesen Angaben wird
der Variablen numberofspins die Zahl der vorhandenen Spins n zugewiesen. Die Variable
dimension erhält als Wert die Dimension d des zum Spinsystem gehörigen Hilbertraums.
In die Liste
Diese berechnet sich durch
d=
Y
(2si + 1)
i
64
(4.64)
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
und wird auch im Code genau so ermittelt. Mit den bestimmten Gröÿen wird in dem Array
naturalBasis
anschlieÿend die natürliche Basis gespeichert. Es setzt sich aus zwei Teilen
zusammen, wobei der erste Teil eine Liste aus Strings ist, in der die Quantenzahlen in der
Reihenfolge stehen, in der sie in
names
stehen und auch in den Basiszuständen stehen sollen.
Die Liste hat für das Doppelkreuz die Gestalt
{"S1", "m1", "s234", "m234", "S5", "m5",
"S6", "m6", "S789", "m789"}.
Der zweite Teil stellt dann die Matrix dar, die die eigentliche Basis repräsentiert. Dazu werden zunächst alle möglichen Paare
(si , mi ), i = 1 . . . n
erzeugt (in derselben Reihenfolge
wie die zugehörigen Quantenzahlen) und in eine Liste geschrieben, wobei die Paare für jedes
si
wiederum als Liste gespeichert werden. Die eingebaute Funktion
Tuples
erzeugt nun alle
Vektoren, die sich daraus kombinieren lassen, wenn man in jeden Vektor jeweils ein Paar
(si , mi )
für alle
i
schreibt. Damit ist die natürliche Basis in einem Array gespeichert, wobei
jeder einzelne Zustand in einer Liste liegt, die die entsprechenden Werte der Quantenzahlen
enthält. Ein Zustand ist zum Beispiel durch die Liste
1/2, 1/2, -(1/2), 3/2, 1/2}
{1/2, -(1/2), 3/2, -(1/2), 1/2,
gegeben.
Um die einzelnen Transformationsmatrizen zu nden, müssen natürlich die alte und neue
(nach Spinaddition) Basis vorliegen. Das Erstellen einer jeweils neuen Basis durch die Kopplung zweier Spinwerte geschieht über die Funktion
als Argumente die Namen der zu koppelnden Spins
newbasis[oldbasis_, s1_, s2_], die
s1,s2 und die alte Basis (bei der er-
sten Basistransformation immer die natürliche Basis) erwartet. Zunächst wird aus den Namen
der Spins ermittelt, an welchen Stellen die zugehörigen
ten Basis stehen. Anschlieÿend werden diese
s-
m-Quantenzahlen
und
m-Quantenzahlen,
in der al-
die bei der Transformation
verschwinden, in den alten Basiszuständen auf 0 gesetzt. Es bestehen in der alten Basis nun
Zustände, die doppelt vorkommen, da durch das Nullsetzen der Zustandsraum um die Dimension verkleinert wurde, die
s1
und
s2
erzeugen. Mittels einer eingebauten Funktion werden
die doppelten Zustände gelöscht und diese modizierte Basis als
Ausgabe von
newbasis,
cancelledM gespeichert. Die
die neue Basis der gekoppelten Spins, ist dann ebenfalls eine Modi-
zierung der alten Basis. In der Liste, die die Bezeichnungen der Quantenzahlen beinhaltet,
wird per Stringmanipulation
s1
und
s2
m1 durch s12 und m2 durch m12 ersetzt (unter der Annahme, dass
gekoppelt werden, ansonsten analog). Dadurch ist der neue Satz von Quantenzahlen
festgehalten. Es müssen nun noch die Zustände aus
tenzahlen
s12
und
m12
cancelledM
um die Werte der Quan-
erweitert werden, um die komplette Gesamtspin-Basis aufzubauen.
Aus den bisherigen Ausführungen ist bekannt, welche Werte die besagten Quantenzahlen annehmen können. Aus jedem der Zustände, die in
cancelledM liegen, werden (2s1 + 1)(2s2 + 1)
neue Zustände erzeugt, indem jeweils alle Kombinationen der Quantenzahlen-Werte an die
Stelle der beiden 0 gesetzt werden. Damit ist die Basis für den Gesamtspin der gekoppelten
Spins konstruiert; sie hat nötigerweise wieder die Dimension
d
und dieselbe formale Daten-
struktur wie die natürliche Basis.
Konkret bedeutet dies für das Doppelkreuz: Aus dem Heisenberg-Hamiltonoperator (4.63)
sieht man direkt, dass eine Kopplung der Spins
s1234 zu
s6 und s789
tenzahl
von
s1
und
s234
notwendig ist, um die Quan-
erhalten. Ich habe diese Kopplung als erste gewählt, wobei die Kopplung
gleichermaÿen sinnvoll wäre; die Kopplung von
wäre nicht nutzbringend, da in diesem Fall der Spin
s1234
s1
und
s5
beispielsweise
nicht mehr erzeugt werden könnte.
Gemäÿ der Beschreibung enthält die erste transformierte Basis dann die Liste
{"S1 ", "S1234
", "S234 ", "m1234 ", "S5", "m5", "S6", "m6", "S789", "m789"} der Quantenzahlen,
während die eigentlichen Zustände wieder aus Vektoren mit den zehn Einträgen der Werte
65
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
dieser Quantenzahlen bestehen.
Es liegen nun die vollständigen Basen - vor und nach einer Addition zweier Spins - vor.
Die zugehörige Transformationsmatrix ist formal durch (4.39) gegeben und besteht nur aus
Clebsch-Gordan-Koezienten und Nullen. In meiner Implementierung werden die Transformationsmatrizen durch die Funktion
trafo[oldbasis_, s1_, s2_]
bestimmt. Sie wird mit
s1, s2 und der Basis vor der Kopplung als Argumente aufgerufen.
Aus der alten Basis oldbasis erzeugt die Funktion zunächst die neue Basis (durch newbasis;
wie beschrieben) und speichert sie in basis. Anschlieÿend ermittelt trafo die Stellen, an denen die Quantenzahlen m1 , m2 , s1 , S2 in der alten Basis stehen und speichert sie in index1,
index2, indexS1, indexS2. Die Transformationsmatrix ist ein Array der Dimension d × d.
den zu koppelnden Spins
Die in (4.39) enthaltenen Vorschriften für die Matrix werden auf zwei Weisen realisiert. Zum
Einen wird für jeden Eintrag
neuen und der
l-te
(k, l)
durch eine if-Bedingung geprüft, ob der
k -te
Zustand der
Zustand der alten Basis identisch sind, wenn man in ihnen die Quan-
m1
und m2 beziehungsweise s12 und m12 . Sie nden sich genau an den durch index1 und index2
gegebenen Stelle. Gibt diese Abfrage eine Nicht-Identität, so wird das Matrixelement (k, l) au-
tenzahlen löscht, die bei der Transformation ersetzt werden. Dies sind die Stellen von
tomatisch 0. Der in (4.40) und (4.2.1) wiedergegebenen Orthogonalität der Spin-Unterräume
ist dadurch Rechnung getragen. Ist die if-Bedingung erfüllt, so ist das Matrixelement durch
C({s1 m1 ; s2 m2 }k ; {s12 m12 }l ) bestimmt. Dabei handelt es
sich um die Ausgabewerte der von mir denierten Funktion cg[{j1_,m1_},{ j2_,m2_},{
j_,m_}] der Clebsch-Gordan-Koezienten, wobei j1, m1, j2, m2 die Einträge an den Stellen
indexS1, index1, index2, indexS2 des k -ten Vektors der alten Basis und j, m die Einträge
an den Stellen index1, index2 des l-ten Vektors der neuen Basis sind. Diese Funktion habe
ich deniert, weil zwar die Clebsch-Gordan-Koezienten C({s1 m1 ; s2 m2 } ; {s12 m12 }) grundsätzlich durch die eingebaute Funktion ClebschGordan berechnet werden können, diese aber
einen Fehler erzeugt, wenn m1 + m2 6= m12 oder wenn |s1 − s2 | ≤ s12 ≤ s1 + s2 nicht erfüllt
die Clebsch-Gordan-Coezienten
ist. Dass die Clebsch-Gordan-Koezienten in diesem Fall 0 werden, habe ich in der Funktion
cg
in Form einer if-Bedingung als Zusatz zur eingebauten Funktion eingefügt. Die Indizes
und
l
durchlaufen jeweils alle ganzen Zahlen von 1 bis
d.
k
Das Ergebnis ist die gewünschte
Transformationsmatrix zwischen einer gegebenen Basis und der bei Kopplung von
s1
und
s2
resultierenden Basis. Diese Transformationsmatrizen werden nur aus den Angaben der aktuellen Basis und der Spins, die in der neuen Basis addiert werden sollen, erzeugt; sonstige
Angaben sind nicht notwendig.
Es ist für die Berechnung nun erforderlich, nach und nach die Basistransformationen durchzuführen, die zum Einen - ausgehend von der natürlichen Basis - den Gesamtspin aller Spins
im System erzeugen und zum Anderen auf dem Weg dorthin die im Hamiltonoperator auftauchenden anderen Gesamtspins generieren. Zum besseren Verständnis seien die durchgeführten Basistransformationen
ti
aufgeführt, die bei der Berechnung des Doppelkreuzes voll-
zogen wurden.
|s1 m1 s234 m234 s5 m5 s6 m6 s789 m789 i
l t1
|s1 s234 s1234 m1234 s5 m5 s6 m6 s789 m789 i
66
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
l t2
|s1 s234 s1234 m1234 s5 m5 s6 s789 s6789 m6789 i
l t3
|s1 s234 s1234 m1234 s6 s789 s5 s6789 s56789 m56789 i
l t4
|s1 s234 s1234 s6 s789 s5 s6789 s56789 s mi
(Wobei
s ≡ s123456789 , m ≡ m123456789 .)
Welche Spins dabei jeweils gekoppelt wurden, ergibt sich direkt aus dem Vergleich der jeweiligen Basen. Die Matrix
T,
die alle Transformationen zusammenfasst und durch eine einzige
ausdrückt, ist durch das Matrixprodukt
T = t1 t2 t3 t4
gegeben. Im MATHEMATICA-Code
für das Doppelkreuz entsprechen diese Transformationen und das Zusammenfassen zu einer
Gesamttransformation den folgenden Zeilen:
t1
t2
t3
t4
=
=
=
=
trafo[naturalBasis, "S1", "S234"];
trafo[basis, "S6", "S789"];
trafo[basis, "S1234", "S5"];
trafo[basis, "S12345", "S6789"];
T = t1.t2.t3.t4;
In der letzten Basis, nach Durchführung aller Transformationen, sind alle Quantenzahlen
enthalten, deren Operatoren im Hamiltonoperator (4.63) als Quadrate auftauchen. Somit
verbleibt als in der Gesamtspin-Basis nicht-diagonaler Teil beim Doppelkreuz nur der Term
JS5 (S1 − S6 )
.
(4.65)
Natürlich sieht dies im Einzelfall für jedes konkret zu berechnende Problem etwas anders
aus, das Prinzip dahinter bleibt aber stets dasselbe. Es ist nun sehr einfach, den diagonalen
Teil des Hamiltonoperators in der Gesamtspin-Basis zu implementieren. Da sie feste Spins
repräsentieren, haben die Operatoren
2 , S2
S12 , S62 , S234
789
konstante Eigenwerte (gemäÿ (2.41))
und jeder Zustand der Gesamtspin-Basis ist Eigenzustand. Die Operatoren lassen sich dann
d × d-Diagonalmatrizen darstellen, deren Diagonalelemente den Wert si (si + 1) tra= 1, 234, 6, 789). In MATHEMATICA wird das umgesetzt, indem die Operatoren
durch Einheitsmatrizen (IdentityMatrix) der Dimension d (dimension) multipliziert mit
2
dem entsprechenden Eigenwert dargestellt werden. So ist zum Beispiel S1 (s1 = 1/2) durch
3/4*IdentityMatrix[dimension] beschrieben. Auf diese Weise werden alle Spinoperatoren
durch
gen (i
mit festem Spinwert implementiert, deren Operatorquadrate im Hamiltonoperator direkt erscheinen.
Ebenfalls diagonal in der Gesamtspin-Basis, aber ohne konstante Eigenwerte, sind Operatorquadrate von gekoppelten Spins (beim Doppelkreuz:
Funktion
diagonalH[s_,basis_]
2
2
S1234
, S6789
).
Sie werden durch die
implementiert. Diese Funktion erwartet den Spin
Matrixform dargestellt werden soll und die Basis
basis,
s,
der in
in der dies geschehen soll (immer
die letzte Gesamtspin-Basis). Zunächst wird wieder ermittelt, an welcher Stelle der Basisvek-
67
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
toren die entsprechende Quantenzahl
s
steht. Die Matrix ist dann die Diagonalmatrix (einge-
DiagonalMatrix) der Dimension d × d, deren k -tes Diagonalelement durch s(k) (s(k) + 1)
2
s(k) dem Wert der Quantenzahl s im k -ten Basisvektor. S1234
beispielsweise ist somit durch den Ausgabewert von diagonalH[1234,basis] als Matrixdarstellung gegeben. basis stellt damit gemäÿ der Implementierung von trafo immer die
baut;
gegeben ist. Dabei entspricht
letzte Basis dar und somit nach Vollzug aller Transformationen die endgültige GesamtspinBasis.
Um schlussendlich das Energiespektrum des Hamiltonoperators zu erhalten, ist es nötig,
diesen zu vervollständigen, indem man auch die Terme in eine Matrixdarstellung bringt, die
in der Gesamtspin-Basis nicht-diagonal sind. Dazu werden diese zunächst in der natürlichen
Basis dargestellt und anschlieÿend durch
T
transformiert. Die Darstellung in der natürlichen
Basis geschieht prinzipiell genauso wie es bei der Spinkette der Fall war. Nach Formel (4.4)
lassen sich die Skalarprodukte von Spinoperatoren durch die Operatoren
drücken, wobei
(i)
Sz
,
(i)
S+
und
(i)
S−
aus-
i für die am Skalarprodukt beteiligten Spins steht. Diese müssen also zunächst
durch entsprechende Funktionen in die Matrixdarstellung der natürlichen Produktspin-Basis
gebracht werden. Der Operator
(i)
Sz
ist in der natürlichen Basis diagonal und seine Implemen-
tierung entsprechend einfach. Die Funktion
Sz[s_, basis_]
übernimmt das Erstellen dieser
Diagonalmatrix unter Angabe der Basis (praktisch immer die Produkspin-Basis
und des Spins
si ,
dessen
Sz -Operator
nalmatrix mit den Diagonalelementen
Quantenzahl
mi
im
k -ten
naturalBasis)
erstellt werden soll. Die Funktion erzeugt eine Diago-
k,
deren Werte gemäÿ (2.42) gleich den Werten der
Vektor der Produktspin-Basis sind.
Auch die Auf- und Absteigeoperatoren sind analog zu denen der Spinkette implementiert,
allerdings mit dem Unterschied, dass hier der Fall berücksichtigt werden muss, dass im Allgemeinen nicht nur Spin-1/2 auftreten. Sie werden durch die Funktionen
Sup[s_, basis_]
Sdo[s_, basis_] kreiert, wobei die Funktionsargumente die gleiche Bedeutung haben wie schon in Sz. Um die Operatoren korrekt umsetzen zu können, ist die Kenntnis
beziehungsweise
der Wirkung der Auf- und Absteigeoperatoren für allgemeine Spins von Nöten.
S± |s, mi =
p
s(s + 1) − m(m ± 1)|s, m ± 1i
(4.66)
Auÿerdem gilt auch hier, dass Zustände von den Auf-/ Absteigeoperatoren vernichtet wer-
m = s beziehungsweise m = −s gilt. Die Einträge (k, l) der Auf(i)
/Absteigeoperatoren S± werden wiefolgt konstruiert: Es werden stets der k -te und l-te Zustand der Produktspin-Basis verglichen (l, k = 1, 2, . . . , n). Eine if-Bedingung prüft, ob beide
Zustände bis auf die Quantenzahl mi gleich sind (Orthogonalität der Produktspin-Basis) und
ob mi des k -ten Zustands gleich mi +1 (Aufsteigeoperator) beziehungsweise gleich mi −1 (Absteigeoperator) des l-ten Zustands ist. Letztere Bedingung gibt die Inkrementierung/Dekreden, wenn für diese
mentierung der z-Komponente bei Wirkung der Operatoren auf einen Produktspin-Zustand
wieder. Ist eine der beiden Bedingungen nicht erfüllt, muss das Matrixelement 0 sein. Ansons-
p
si (si + 1) − mi (mi ± 1) bestimmt.
(i)
(i)
(i)
Operatoren Sz , S+ und S− erzeugen, habe
ten ist das Matrixelement (k, l) nach (4.66) durch
Nach der Denition der Funktionen, die die
ich
eine weitere Funktion deniert, die unter Verwendung dieser Operatoren das Skalarprodukt
couple[s1_, s2_, basis_] und bezogen auf das Skalarprodukt des letzten Satzes kann man die Argumente s1, s2
als die Nummerierungen i, j der Faktoren Si , Sj betrachten. Das Argument basis übergibt
Si · Sj komplett in Matrixdarstellung bringt. Die Funktion heiÿt
die Basis, in der dies geschehen soll, welche praktisch immer die natürliche Basis ist. Die
68
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
Funktion
couple
übernimmt lediglich die Aufgabe, die Formel (4.4) für gegebene Spins aus-
zuwerten, wobei es sich um eine Summe von Matrixmultiplikationen handelt. Somit kann über
die Funktion das Skalarprodukt jedes beliebigen Paares von Spins in der natürlichen Basis in
einer Matrix dargestellt werden. Die Berechnung des Doppelkreuzes erfordert diese Funktion
nach (4.63) zweimal:
couple[1, 5, naturalBasis]
und
couple[6, 5, naturalBasis].
In
Abhängigkeit von dem konkreten, zu berechnenden Problem kann die Funktion für eine unterschiedliche Zahl von Spin-Paaren notwendig sein.
Da aus genannten Gründen in der Gesamtspin-Basis ausgewertet werden soll, müssen die
Skalarprodukte nun noch entsprechend transformiert werden. Dies erfolgt über die Formel
−1 −1
t eingeführt, die durch t = T −1 = t−1
n · · · t2 t1
deniert ist. Dies ist praktikabel, da MATHEMATICA den Befehl Inverse[T], also die
Berechnung der Inversen von T , so durchführt, dass zunächst T aus den ti berechnet wird
und anschlieÿend invertiert. Dies ist wesentlich rechenaufwendiger als die ti zu invertieren und
(4.58). Ich habe zu diesem Zweck die Variable
anschlieÿend zu multiplizieren.
Mit dem bis hierhin vorgestellten Vorgehen ist es möglich, den gesamten Heisenberg-Hamiltonoperator des Systems als Matrix in der Gesamtspin-Basis darzustellen. Daran anschlieÿend
kann die Diagonalisierung erfolgen. Ich möchte zuvor knapp zusammenfassen, welche Schritte
bis zu diesem Punkt erforderlich waren:
•
Aufstellen des allgemeinen Heisenberg-Hamiltonoperators für das System.
•
Finden von möglichen Spingruppen, die mit dem Hamiltonoperator kommutieren. Diese
haben einen festen Spinwert im Grundzustand. Der Hamiltonoperator kann dann in
Abhängigkeit dieser Gruppen geschrieben werden.
•
Überlegung, welche Skalarprodukte im Hamiltonoperator gekoppelt werden können. Es
sollen soviele Kopplungen wie möglich vorgenommen werden, solange dabei keine Indizes
in Gesamtspins mehrfach auftreten.
•
Ausführen von Basistransformationen, beginnend mit der natürlichen Basis, sodass die
letzte Basis den Gesamtspin und alle im Hamiltonoperator auftauchenden Spins aus
Kopplungen auftreten.
Ist dies erfolgt, kann der Hamiltonoperator in MATHEMATICA in der beschriebenen Form
aufgeschrieben werden. Der Heisenberg-Hamiltonoperator des Doppelkreuzes sieht darin so
aus:
H = J/2 (diagonalH[1234, basis] - 2*(3/4 + 15/4)*IdentityMatrix[dimension]
+ diagonalH[6789, basis]) +
J*t.(couple[1, 5, naturalBasis] + couple[6, 5, naturalBasis]).T;
(Es wird hier zur Berechnung
J = 1 > 0 gesetzt. Dies ist erforderlich, um numerische Auswer-
tungen vornehmen zu können. Zudem ist es üblich, die Energien für das Heisenbergmodell in
Einheiten von
J
anzugeben.)
69
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
Grundsätzlich wäre nun eine Diagonalisierung, also Berechnung der Energieeigenwerte,
möglich. Wie besprochen, empehlt sich aber die Anwendung des Lieb-Mattis-Theorems.
Demzufolge beträgt der Gesamtspin des Doppelkreuzes im Grundzustand 5/2 und es existiert
ein dazugehöriger Block eines unabhängigen Unterraumes im Hamiltonoperator, welchen es
zu ermitteln gilt. Dies gilt natürlich ganz allgemein auch für andere Subgitter. Ich habe dazu
extractTotalSpin[s_, value_] implementiert. Mit dem Argument s wird der
Gesamtspins übergeben (im Doppelkreuz: 123456789), mit dem Argument value
die Funktion
Name des
der Wert des Gesamtspins für den der Block isoliert werden soll. Die Funktion prüft dann in
welchen Vektoren der Gesamtspin-Basis die Quantenzahl
s zum Gesamtspin den übergebenen
Wert hat und erzeugt eine Liste der Vektoren, auf die dies zutrit. Anschlieÿend werden im
Hamiltonoperator die zu diesen Vektoren gehörigen Zeilen und Spalten gestrichen. Als Ausgabewert verbleibt der gewünschte Block, dessen Eigenwerte die Grundzustandsenergie beeinhalten. Führt man die Funktion für das Doppelkreuz mittels
5/2]
aus, so ist das Resultat eine
42 × 42-Matrix.
extractTotalSpin[123456789,
Die Eigenwerte dieser Matrix können
zwar (zumindest numerisch) in einer akzeptablen Zeit berechnet werden lassen, aber es gibt
Überlegungen, die eine weitere Reduktion der Dimensionalität zulassen. Dazu bedenke man,
(2s + 1) verschiedene Werte von m zu jedem Gesamtspin s gehören, also zu jedem s
(2s + 1) Zustände gehören, die sich nur in m unterscheiden. Die Quantenzahl m des Gesamt-
dass
spins kommt aber nicht explizit im Hamiltonoperator vor, weshalb zu jeder Eigenenergie mit
Gesamtspin
s
(2s + 1)-fache Entwartung vorliegen muss. Dementsprechend muss der
s gehört wiederum blockdiagonal sein und Blöcke enthalten,
m zugeordnet werden können. Diese Blöcke ndet man nun ganz analog,
eine
Block, der zu einem bestimmten
die jeweils einem
indem man aus dem vorher ermittelten Block nur die Zeilen und Spalten isoliert, die zu
einem bestimmten, gleichen
sz_, value_, valuez_],
Block für ein gewünschtes
m
extractTotalZ[s_,
extractTotalSpin beinhaltet und zusätzlich den
sz) ausschneidet. Betrachtet man auch hier wieder
gehören. Dies übernimmt die Funktion
die die Funktion
m
(Argument
beispielhaft das Doppelkreuz, so muss eine sechsfache Entartung der Grundzustandsenergie
vorliegen. Dies wiederum bedeutet, dass es sechs
7 × 7-Matrizen
gibt, die dieselben Eigenen-
ergien haben. Es genügt selbstverständlich, nur einen dieser Blöcke auszuwerten, sodass sich
das ganze Problem auf ein siebendimensionales reduziert. Der Block für
über
extractTotalZ[123456789, 123456789, 5/2, 1/2]
m = 1/2
lässt sich
bestimmen. Die Berechnung der
Eigenwerte des Blocks liefert das relevante Ergebnis, die Grundzustandsenergie
E01b
(Mini-
mum des Energiespektrums).
E01b = −2, 96851J
(4.67)
(Die Auswertung der Energieeigenwerte ist bei allen Rechnungen der einzige Schritt, der
numerisch erfolgt.)
Es ist damit das Vorgehen präsentiert worden, durch welches ich die Grundzustandsenergien der verschiedenen Subgitter bestimmt habe. Bei diesem Vorgehen ist ein weiter Teil durch
einen Computeralgorithmus getragen, sodass es nur noch weniger Angaben für die Berechnungen bedarf. Allerdings existieren auch einige Schritte, die per Hand erfolgen müssen,
wie etwa das Aufstellen des Hamiltonoperators in der Form, wie sie vorgestellt wurde oder
auch das entsprechende, sukzessive Transformieren der Basen. Zudem werden einige Funktionen, die zu dem Verfahren gehören, einzeln aufgerufen. An diesen Stellen wäre es durchaus
denkbar, einen allgemeineren Algorithmus zu gestalten. Ich habe mich dabei gedanklich auch
mit einem Algorithmus beschäftigt, der nur durch Angabe der wechselwirkenden Spins die
70
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
gewünschte Grundzustandsenergie ermittelt. Allerdings wäre eine solche Umsetzung natürlich mit einem wesentlich höheren Aufwand verbunden gewesen. Zudem war von vornherein
klar, dass ich eine bestimmte Zahl überschaubarer Gitter berechnen würde. Dies war mit dem
von mir gewählten Verfahren problemlos möglich und insofern wahrscheinlich der für meine
Anforderungen ezientere Weg. Dennoch halte ich die Verallgemeinerung des Algorithmus
für eine interessante Aufgabe und Herausforderung, wenngleich mein Zeitfenster im Rahmen
dieser Arbeit nicht ausreichend war.
Da alle anderen Berechnungen auf demselben Verfahren beruhen, werde ich diese weit
weniger detailliert beschreiben und mich bei der Erläuterung auf diejenigen Punkte beschränken, die für das jeweilige Problem speziell sind. Dabei geht es vorallem um die erwähnten per
Hand vorgenommenen Schritte. Die nächste Berechnung, die vorgestellt werden soll, ist die
2
3
5
1
4
Abbildung 4.6: Subgitter des Quadratgitters: einfaches Kreuz (1a)
zum einfachen, oenen Subgitter des Quadratgitters. Es handelt sich dabei um das einfache
Kreuz (1a) wie es in Abbildung 4.6 dargestellt ist.
Die Ermittelung der Grundzustandsenergie des Kreuzes kann leicht ohne Zuhilfenahme von
Rechnerleistung durchgeführt werden. Für den Grundzustand ist bekannt, dass der Gesamtspin
s12345 = 3/2
sein muss und der Spin
s2345 = 2; dies ergibt sich aus den vorherigen
s1 = 1/2. Der Heisenberg-Hamiltonoperator
Überlegungen dieses Kapitels. Auÿerdem gilt
H1a
des einfachen Kreuzes hat anfänglich die Form
H1a = J/2 (S1 · S2 + S1 · S3 + S1 · S4 + S1 · S5 )
.
(4.68)
Man kann ihn jedoch auch so schreiben, dass er nur aus Operatoren besteht, deren QuantenzahlWerte für den Grundzustand bekannt sind:
2
2
H1a = J/2 S12345
− S12 − S2345
.
(4.69)
Der Hamiltonoperator ist also in der Gesamtspin-Basis komplett diagonal und um die Grundzustandsenergie zu erhalten, reicht es, die bekannten Eigenwerte der Operatorquadrate einzusetzen.
E01a = J/2 (15/4 − 3/4 − 6) = −1, 5J
1
2
4
3
(4.70)
Abbildung 4.7: Subgitter des Quadratgitters: Quadrat (1c)
71
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
Es verbleibt für das Quadratgitter noch die Berechnung zu einem geschlossenen Subgitter,
also einem solchen, dessen Gitterplätze mindestens zwei Bindungen haben. Die einfachste
geschlossene Substruktur des Quadratgitters stellt das Quadrat dar (1c), gezeigt in Abbildung
4.7.
In diesem Gitter ist keine Annahme über den Spinwert von Spingruppen im Grundzustand
zu machen, der Gesamtspin im Grundzustand wird bei
Der Heisenberg-Hamiltonoperator
H1c
s = 0
liegen und es gilt
s1 = 1/2.
schreibt sich, gemäÿ der Spin-Nummerierungen in der
Abbildung,
H1c = J [S1 · (S2 + S4 ) + S2 · S3 + S3 · S4 ]
2
2
= J/2(S124
− S12 − S24
) + J(S2 · S3 + S3 · S4 )
.
(4.71)
Die Gesamtspin-Basis und notwendigen Quantenzahlen erhält man durch folgende Transformationen aus der natürlichen Basis:
|s1 m1 s2 m2 s3 m3 s4 m4 i
l t1
|s1 m1 s2 s4 s24 m24 s3 m3 i
l t2
|s1 s24 s124 m124 s2 s4 s3 m3 i
l t3
|s1 s24 s124 s3 s1243 m1243 s2 s4 i
Das Aufstellen des Hamiltonoperators in MATHEMATICA geschieht wieder durch die Transformation der nicht-diagonalen Teile und die Darstellung der sonstigen Terme durch die
s = s1243 = 0 ist, existiert keine Entartung
des Blocks für s = 0 führt zu einer 2 × 2-Matrix.
vorgestellten Funktionen. Da der Gesamtspin
der Grundzustandsenergie. Das Extrahieren
Die Grundzustandsenergie liegt bei
E01c = −2J
.
(4.72)
Ist die Grundzustandsenergie der Subgitter berechnet, so ist der Groÿteil der Arbeit geleistet. Tatsächlich geht es jedoch um eine Abschätzung der Grundzustandsenergie pro Gitterplatz
im unendlichen, antiferromagnetischen Gitter. Um schlieÿlich diese zu nden, bedarf es noch
weiterer Überlegungen. Die erste wichtige Feststellung ist, dass im unendlichen Quadratgitter
(Abbildung 4.4) zwei Bindungen pro Gitterplatz existieren. Der Ansatz für die Bestimmung
der unteren Schranken war es, die Subgitter als die
Hi
aus der Herleitung in Abschnitt 4.3.1
zu betrachten. Es soll sich also das gesamte, unendliche Gitter aus den Subgittern abdecken
lassen. Diese Abdeckung erfolgt durch das wiederholte Anbringen der Subgitter an allen Gitterplätzen. Für das einfache Quadrat ist diese Situation in Abbildung 4.8 gezeigt. Man sieht
darin in schwarz das erste Kreuz, in grün die Kreuze, die an dessen Gitterplätzen zentriert
werden und in grün-gestrichelt einen Ausschnitt von Kreuzen, die wiederum an deren Gitterplätzen zentriert werden. Da es sich um eine periodische Struktur handelt, reicht der gezeigte
Ausschnitt, um festzustellen, dass bei der Abdeckung des Gesamtgitters an jedem Gitterplatz
vier zusätzliche Bindungen angebracht werden (man sieht es hier am zentralen Gitterplatz).
72
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
Abbildung 4.8: Abdeckung des Quadratgitters durch das einfache Kreuz
Um die Grundzustandsenergie pro Gitterplatz (site)
E0 /site
nach der Abdeckung zu be-
stimmen, muss entsprechend die für das Subgitter bestimmte Grundzustandsenergie mit dem
Vorfaktor (Bindungen pro Gitterplatz/bei der Abdeckung hinzukommende Bindungen) multipliziert werden. Es folgt als untere Schranke für die Grundzustandsenergie pro Gitterplatz im
unendlichen, antiferromagnetischen Quadratgitter unter Verwedung des Kreuzes als Subgitter:
E01a /site = E01a ·
2
= −0, 75J
4
.
(4.73)
Völlig analog geht man für die anderen beiden Subgitter vor und erhält die Vorfaktoren
(1b) und
2
4 (1c). Es ergeben sich als untere Schranken:
E01b /site = E01b ·
2
= −0, 7421J
8
E01c /site = E01c ·
2
= −J
4
,
2
8
(4.74)
.
(4.75)
Die beste Abschätzung bietet also das Doppelkreuz, es folgen das einfache Kreuz und als
schlechteste Abschätzung jene aus dem Quadrat. Eine Diskussion, in welchem Zusammenhang
die Subgitter mit der Qualtität der Abschätzung stehen, wird noch erfolgen.
Zweidimensionales, hexagonales Gitter
Die Rechnungen zum Quadratgitter sind, wie bereits erwähnt, in der Publikation von Roser Valentí und Rolf Tarrach veröentlicht worden [TV90]. Die Rechnungen über das von
mir benutzte Verfahren haben zu denselben Resultaten geführt, was für die Richtigkeit des
Verfahrens spricht. Inhalt meiner Arbeit ist es, zudem für weitere bipartite Gitter untere
Schranken der Grundzustandsenergie pro Gitterplatz zu nden. Dabei habe ich als weiteres
zweidimensionales Gitter, das hexagonale Gitter (auch Bienenwaben- oder Honeycomb-Gitter)
betrachtet. Ein Ausschnitte eines solchen Gitters, das naheliegenderweise aus gleichmäÿigen
Sechsecken aufgebaut ist, ist in Abbildung 4.9 zu sehen. Wieder werden die unteren Schranken
aus zwei oenen und einem geschlossenen Subgitter, aus denen das unendliche Gitter aufgebaut werden kann, ermittelt. Das erste oene Subgitter stellt der Stern (2a) aus Abbildung
4.10 dar. Im Grundzustand gilt hierbei: Der Gesamtspin
s = s1234
hat den Wert 1 und die
73
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
Abbildung 4.9: Ausschnitt aus einem hexagonalen Gitter
1
3
2
4
Abbildung 4.10: einfaches, oenes Subgitter des hexagonalen Gitters (2a)
Spingruppe aus den Spins 1,3 und 4 den Spinwert
s134 = 3/2,
zudem ist
s2 = 1/2.
Der
Heisenberg-Hamiltonoperator ist gegeben durch
H2a = J [S2 · (S1 + S3 + S4 )]
2
2
= J/2(S1234
− S22 − S134
)
(4.76)
.
Wie schon beim einfachen Kreuz, ist es ausreichend, die Eigenwerte der auftretenden Operatorquadrate für den Grundzustand einzusetzen.
E02a = J/2(2 − 3/4 − 15/4) = −1, 25J
1
(4.77)
3
2
4
5
6
Abbildung 4.11: erweitertes, oenes Subgitter des hexagonalen Gitters (2b)
Das zweite, von mir gewählte, oene Subgitter (2b) ist in Abbildung 4.11 gezeigt. Das
s im Grundzustand
= s56 = 1. Auÿerdem
Lieb-Mattis-Theorem lässt folgende Aussagen für den den Gesamtspin
zu:
s = s132456 = 0.
Zudem gilt im Grundzustand desweiteren
s13
gilt natürlich für alle einzelnen Gitterplätze, dass es sich um Spin-1/2-Plätze handelt, also
si = 1/2, (i = 1, 2, . . . 6).
Wieder wird der Heisenberg-Hamiltonoperator durch möglichst
viele bekannte Spinquadrate ausgedrückt.
H2b = J [S2 · (S1 + S3 ) + S4 · (S5 + S6 ) + S2 · S4 )]
2
2
2
2
= J/2(S123
− S22 − S13
+ S456
− S42 − S56
) + JS2 · S4
74
(4.78)
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
Es werden die folgenden Transformationen durchgeführt:
|s13 m13 s2 m2 s4 m4 s56 m56 i
l t1
|s13 s2 s132 m132 s4 m4 s56 m56 i
l t2
|s13 s2 s132 m132 s4 s56 s456 m456 i
l t3
|s13 s2 s132 s4 s56 s456 s132456 m132456 i
Daraus folgt, dass in der gewählten Gesamtspin-Basis nur ein Term nicht-diagonal ist, der über
die Transformationsmatrizen in die Gesamtspin-Basis überführt wird. Es liegen für
s = 0 keine
Entartungen der Grundzustandsenergie vor und die Isolierung des zugehörigen Blocks liefert
eine
2 × 2-Matrix.
Die Auswertung seiner Eigenenergien gibt:
E02b = −2.16421J
.
(4.79)
2
1
3
6
4
5
Abbildung 4.12: geschlossenes Subgitter des hexagonalen Gitters: Hexagon (2b)
Das kleinste geschlossene Gitter des Honeycomb-Gitters ist das Hexagon selber (2c). Es ist
samt Spin-Nummerierungen in Abbildung 4.12 zu sehen. Der Gesamtspin
s im Grundzustand
s = s126435 = 0.
Da es sich um eine
liegt für das System im antiferromagnetischen Fall bei
geschlossene Struktur handelt, sind sonst keine weiteren Annahmen über gekoppelte Spins
machbar. Das Subgitter wird durch den Heisenberg-Hamiltonoperator
H2c = J [S1 · (S6 + S2 ) + S4 · (S5 + S3 ) + S2 · S3 + S5 · S6 )]
2
2
2
2
= J/2(S126
− S26
− S12 + S345
− S35
− S42 ) + J(S2 · S3 + S5 · S6 )
(4.80)
beschrieben. Er enthält zwei nicht-diagonale Terme, wenn man die natürliche Basis wiefolgt
transformiert:
|s1 m1 s2 m2 s3 m3 s4 m4 s5 m5 s6 m6 i
l t1
|s1 m1 s2 s6 s26 m26 s3 m3 s4 m4 s5 m5 i
l t2
75
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
|s2 s6 s1 s26 s126 m126 s3 m3 s4 m4 s5 m5 i
l t3
|s2 s6 s1 s26 s126 m126 s3 s5 s35 m35 s4 m4 i
l t4
|s2 s6 s1 s26 s126 m126 s3 s5 s4 s35 s435 m435 i
l t5
|s2 s6 s1 s26 s126 s435 s126435 m126435 s3 s5 s4 s35 i
Nach Durchführung dieser Transformationen und Bestimmung der zugehörigen Matrizen,
kann der Hamiltonoperator wieder als Matrix in der Gesamtspin-Basis aufgeschrieben werden
(in MATHEMATICA). Extrahiert man den Block zu
s = 0,
so verbleibt ein sechsdimensio-
nales Problem ohne weitere Entartung; die Eigenwerte können bestimmt werden.
E02c = −2.80278J
(4.81)
Es sind somit die Grundzustandsenergien für die drei Subgitter des hexagonalen Gitters
gefunden. Es geht nun wieder darum, jeweils die Grundzustandsenergien pro Gitterplatz zu
nden. Dies geschieht prinzipiell ähnlich, wie es beim Quadratgitter der Fall war. Allerdings ist
ein etwas anderes Vorgehen von Nöten, da das hexagonale Gitter über eine andere Zähligkeit
verfügt. Es ist dadurch nicht sinnvoll möglich, das gesamte Gitter abzudecken, indem an
jedem Gitterplatz ein Subgitter angebracht wird. Man überlegt sich stattdessen, wie durch
Anbringen möglichst weniger Subgitter das gesamte Gitter abgedeckt werden kann. Auÿerdem
betrachtet man nun die Dopplungen der Bindungen beim Abdecken, statt die der Gitterplätze.
Man deckt das komplette Gitter ab und betrachtet anschlieÿend ein einzelnes Hexagon daraus.
Ein solches Hexagon verfügt im Honeycomb-Gitter über sechs innere Bindungen. Durch
Überlappungen der Untergitter kann es dazu kommen, dass in dem Hexagon dann gedoppelte
Bindungen vorkommen, die es wieder rauszurechnen gilt. Baut man das hexagonale Gitter
aus den kleinen, oenen Subgittern (2a) auf, so entstehen diese Dopplungen nicht und jedes
Hexagon hat genau sechs Bindungen. Tut man dasselbe mit dem Subgitter (2b), so zeigen sich
für jedes Hexagon zehn statt sechs Bindungen und bei der Überdeckung durch Sechsecke zeigt
sich, dass in jedem Hexagon alle Bindungen gedoppelt werden, wie sich leicht vorstellen lässt.
Somit resultieren aus der Überdeckung schonmal die Faktoren
1,
6
6
10 und 12 . Da man sich aber
für die Energie pro site und nicht pro Bindung interessiert, muss man zudem berücksichtigen,
dass pro Bindung zwei Gitterplätze existieren; dies erzeugt einen weiteren Vorfaktor
1
2 . Damit
berechnen sich die unteren Schranken für die Grundzustandsenergie pro Gitterplatz zu den
folgenden Werten:
1
· 1 = −0, 625J
2
1 6
E02b /site = E02b · ·
= −0, 64926J
2 10
1 6
E02c /site = E02c · ·
= −0, 7007J
2 12
E02a /site = E02a ·
(4.82)
(4.83)
(4.84)
Es ist auällig, dass die kleinere oene Struktur die beste Schranke liefert. Den schlechtesten
Wert hat hier wiederum das geschlossene Subgitter (Diskussion im Anschluss).
76
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
Dreidimensionales Würfelgitter
Es ist interessant bei der Diskussion der Güte der unteren Schranken auch die Situation
dreidimensionaler Gitter mit einzubeziehen. Aus diesem Grund habe ich die Berechnungen
der unteren Schranke der Grundzustandsenergie auch für Substrukturen des Würfelgitters
durchgeführt. Ein solches Gitter ist die dreidimensionale Erweiterung des Quadratgitters und
setzt sich aus einer periodischen, unendlichen Verknüpfung von Würfeln zusammen. An dem
Vorgehen bei den Berechnungen ändert sich prinzipiell nichts, da die Heisenberg-Hamiltonoperatoren ihre formale Gestalt beibehalten. Wieder werden zwei oene und eine geschlossene
6
5
1
4
7
2
3
Abbildung 4.13: einfaches, oenes Subgitter des Würfelgitters: Stern (3a)
Substruktur betrachtet, die jeweils der dreidimensionalen Erweiterung der im Quadratgitter
verwendeten Strukturen etsprechen. Das kleinste oene Subgitter stellt der Stern (3a), gezeigt
in Abbildung 4.13, dar.
Sein Gesamtspin
s = s1234567
nimmt im Grundzustand den Wert 5/2 an, während die Spin-
s123456 dabei den Wert 3 trägt. Unter diesen Annahmen ist der Heisenberg-Hamiltonoperator H3a des Systems in der Gesamtspin-Basis, analog zum Kreuz, vollständig diagonal:
gruppe
2
2
H3a = JS123456 · S7 = J/2(S1234567
− S123456
− S72 )
Da auch
s7 = 1/2
(4.85)
bekannt ist, folgt nach Einsetzen der Eigenwerte für die Grundzustandsen-
ergie:
E03a = J/2(35/4 − 12 − 3/4) = −2J
.
(4.86)
Es folgt die nächstgröÿere oene Substruktur des Würfelgitters. Auch hier handelt es sich
um die dreidimensionale Erweiterung des Subgitters, das für das Quadratgitter verwendet
wurde ((3b), siehe Abbildung 4.14). Die Berechnung erfolgt völlig analog zu der des Doppelkreuzes, da das Zusammenfassen der Spins
s12345 ≡ s10
und
s9,10,11,12,13 ≡ s90
zum Wert
5/2 für den Grundzustand zur exakt selben Struktur des Heisenberg-Hamiltonoperators führt.
H3b = J [S6 · (S10 + S7 ) + S8 · (S7 + S90 )]
(4.87)
2
2
2
= J/2(S120 6 − S120 − S62 + S89
0 − S8 − S9 ) + JS7 · (S6 + S8 )
Es wurden folglich auch dieselben Transformationen vorgenommen, wobei 10 ,
sprechung von
s234 , s789
s90
die Ent-
des Doppelkreuzes haben. Der Gesamtspin des Systems beträgt
nach dem Lieb-Mattis-Theorem im Grundzustand 9/2 und ist entsprechend zehnfach entartet.
Die Isolierung eines Blocks für
s = 9/2
und einem beliebigen zugehörigen
m
reduizert die
77
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
5
4
1
3
6
2
7
13
9
12
8
10
11
Abbildung 4.14: erweitertes, oenes Subgitter des Würfelgitters: Doppelstern (3b)
Berechnung wieder auf die Auswertung eines siebendimensionalen Problems. Es folgt für die
Grundzustandsenergie:
E03b = −3, 9827J
1
4
(4.88)
2
3
8
5
.
7
6
Abbildung 4.15: geschlossenes Subgitter des Würfelgitters: Würfel (3c)
Folgerichtig wird das geschlossene Subgitter durch einen Würfel repräsentiert ((3c), siehe
Abbildung 4.15). Wie zuvor schon bei den geschlossenen Strukturen, gestaltet sich seine Untersuchung etwas rechenaufwendiger, was darauf zurückzuführen ist, dass die geschlossenen
Strukturen über keine freiliegenden Spins verfügen. Aufgrunddessen können keine Aussagen
über den Grundzustands-Spin von Gruppen gemacht werden und es verbleibt im Hamiltonoperator immer ein groÿer Teil, der in der Gesamtspin-Basis nicht-diagonal ist.
H3c = J[S1 · (S2 + S4 + S8 ) + S6 · (S3 + S5 + S7 )+
S5 · (S4 + S8 ) + S2 · (S3 + S7 ) + S3 · S4 + S7 · S8 ]
2
2
2
2
= J/2(S1248
− S12 − S248
+ S3576
− S62 − S357
)+
(4.89)
J(S5 · S8 + S4 · S5 + S2 · S3 + S2 · S7 + S3 · S4 + S7 · S8 )
Es ergibt sich aufgrund der hohen Zahl von Spins, die im Hamiltonoperator vorkommen, eine
Vielzahl von nötigen transformationen, um in die Gesamtspin-Basis zu gelangen.
|s1 m1 s2 m2 s3 m3 s4 m4 s5 m5 s6 m6 s7 m7 s8 m8 i
l t1
|s1 m1 s2 s4 s24 m24 s3 m3 s5 m5 s6 m6 s7 m7 s8 m8 i
78
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
l t2
|s1 m1 s2 s4 s24 s8 s248 m248 s3 m3 s5 m5 s6 m6 s7 m7 i
l t3
|s2 s4 s24 s8 s1 s248 s1248 m1248 s3 m3 s5 m5 s6 m6 s7 m7 i
l t4
|s2 s4 s24 s8 s1 s248 s1248 m1248 s3 s5 s35 m35 s6 m6 s7 m7 i
l t5
|s2 s4 s24 s8 s1 s248 s1248 m1248 s3 s5 s35 s7 s357 m357 s6 m6 i
l t6
|s2 s4 s24 s8 s1 s248 s1248 m1248 s3 s5 s35 s7 s6 s357 s6357 m6357 i
l t7
|s2 s4 s24 s8 s1 s248 s1248 s6357 s12486357 m12483567 s3 s5 s35 s7 s6 s357 i
Ist dieser Hamiltonoperator in der Gesamtspin-Basis dargestellt, wird abermals der entsprechende Block daraus ermittelt. Es handelt sich dabei um den nicht weiter entarteten Block zu
s = s12486357 = 0, welcher einer 11 × 11-Matrix entspricht. Die Bestimmung der Eigenenergien
gibt:
E03c = −4, 82009J
.
(4.90)
Die Abschätzungen der Grundzustandsenergie pro Gitterplatz für das unendliche Gitter erfolgen wie beim Quadratgitter. Im Würfelgitter existieren drei Bindungen pro Gitterplatz; das
Abzählen der zusätzlichen Bindungen beim Abdecken des Gesamtgitters durch die Subgitter
bestimmt die Nenner der Vorfaktoren. Damit ergeben sich folgende untere Schranken für das
antiferromagnetische Würfelgitter:
E03a /site = E03a ·
3
= −J
6
,
E03b /site = E03b ·
3
= −0, 9957J
12
E03c /site = E03c ·
3
= −1, 8075J
8
(4.91)
,
.
(4.92)
(4.93)
Es sind nun für drei Gittertypen aus jeweils drei Subgittern nach stets derselben Systematik untere Schranken der Grundzustandsenergie errechnet worden. Dieser Teil stellt den
gröÿten/zeitaufwendigsten Part meiner Arbeit dar. Es soll nun die Aussagekraft der Ergebnisse untersucht werden, indem sie verglichen, interpretiert und diskutiert werden. Auÿerdem
sollen die Ergebnisse in Vergleich mit oberen Schranken gesetzt werden, die aus anderen
Methoden bereits ermittelt wurden, um ihre Qualität einschätzen zu können.
79
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
4.3.3 Diskussion der Ergebnisse
Güte der Subgitter
Da es sich stets um untere Schranken handelt, die bestimmt wurden, sind jeweils diejenigen
Werte am besten zu beurteilen, die die gröÿte Grundzustandsenergie pro Gitterplatz liefern.
In der folgenden Tabelle sind die Ergebnisse für die
E0 /site
zusammengefasst:
E0 /site
(a)
(b)
(c)
Quadratgitter
−0, 75J
−0, 625J
−J
−0, 7421J
−0, 6493J
−0, 9957J
−J
−0, 7007J
−1, 8075J
Honeycomb-Gitter
Würfelgitter
Die erste eindeutige Aussage, die der Vergleich der Werte zulässt, ist die, dass die geschlossenen Subgitter (c) jeweils die schlechtesten Abschätzungen geben. Dabei ist die Abweichung zu
den Werten der oenen Subgitter wesentlich gröÿer als die der oenen Subgitter untereinander.
Die geschlossenen Gitter kennzeichnen sich dadurch, dass sie keine Spins beinhalten, die nur
eine Bindung haben, dadurch ist die Zahl der freien Bindungsplätze in ihnen verhältnismäÿig
klein. Das Vorhandensein von einfach gebundenen Spin scheint also ein Qualitätskriterium für
die Subgitter zu sein. Auÿerdem ist auällig, dass alle geschlossenen Gitter im Grundzustand
über einen Gesamtspin von 0 verfügen. Dass dies jedoch nicht hinreichend für eine Aussage
über die Güte der Subgitter ist, zeigt bereits Struktur (2b), die ebenfalls einen Gesamtspin
von 0 im Grundzustand hat, aber eine wesentliche bessere Schranke liefert.
Es scheint also eindeutig, dass komplett geschlossene Subgitter schlechtere Schranken aufweisen
als oene. Es stellt sich nun jedoch die Frage, nach welchen Kriterien sich die Güte oener
Gitter beurteilen lässt; schlieÿlich liefern verschiedene, oene Subgitter allesamt verschiedene
Schranken. Die Betrachtung der Werte für Quadrat- und Würfelgitter, die sich in vieler Hinsicht äquivalent verhalten, könnte vermuten lassen, dass schlicht die Gröÿe der Subgitter ihre
Qualität bestimmt. Auch die Vorteilhaftigkeit von möglichst vielen einfach gebundenen Gitterplätzen ndet sich darin wieder ((1a): vier einfach gebundene Gitterplätze; (1b): sechs einfach
gebundene Gitterplätze). Ein Blick auf die Resultate für das Honeycomb-Gitter widerlegen
diese Vermutungen allerdings. Hier gibt das gröÿere Subgitter (2b) mit der gröÿeren Zahl
freier Gitterplätze die schlechtere Schranke (im Vergleich zu (2a)). Die Überlegung, dass das
Verhältnis von freien zu komplett gebundenen Spins relevant ist, macht zwar für die letzteren
beiden Subgitter Sinn, wird aber durch das Quadratgitter nicht bestätigt. Auf Basis all dieser
Erklärungen scheint also ein Widerspruch zwischen dem Verhalten von (1a), (1b), (3a), (3b)
und (2a), (2b), bezüglich der unteren Schranken der Grundzustandsenergie, zu bestehen.
Zur Klärung dieses Problems betrachte man die Konstruktion der einfachen, oenen Subgitter (a). Sie stellen unter den elementaren Gitter-Bausteinen diejenigen mit dem gröÿten
Grundzustands-Spin dar. Elementar bedeutet dabei, dass sie nur einen Gitterplatz besitzen,
der mehr als eine Bindung hat. Natürlich könnte man die Gesamtgitter auch aus Stücken aufbauen, die nur aus zwei verbundenen Spins bestehen, was auch elementar im hiesigen Sinne
wäre. Die beste untere Schranke liefern aber stets die elementaren Subgitter, die den gröÿten
Gesamtspin im Grundzustand haben [TV90].
80
4.3. UNTERE SCHRANKEN DER GRUNDZUSTANDSENERGIE FÜR UNENDLICHE SPINSYSTEME
Aus Formel (4.48) folgt, dass die Grundzustandsenergie eines Gitters mit dem Hamiltonoperator
H , das sich aus mehreren fundamentalen Gittern mit dem Hamiltonoperator Hi
aufbaut,
höher liegen sollte, als die Summe der Grundzustandsenergien dieser fundmamentalen Gitter.
Dies wiederum bedeutet, dass die Kombinationen aus den fundamentalen Gittern bessere untere Schranken liefern als die fundamentalen Gitter selber. Die bessere Schranke von (1b) im
Vergleich zu (1a) ist eine direkte Folge daraus, denn das Doppelkreuz ist nichts anderes als
das Zusammenfügen zweier einfacher Kreuze an einem Gitterplatz.
Die Subgitter (2a) und (2b) erfüllen diesen Zusammenhang bei genauerem Hinsehen nicht,
denn das Gitter (2b) kann nicht durch einfaches Aneinandersetzen von mehreren Gittern
(2a) erzeugt werden. Die Zusammensetzung von Gitter (2b) kann nur dadurch, erfolgen, dass
zwei Gitter (2a) mit überlappenden Bindungen zusammengesetzt werden. Diese überlappende
Komposition ist keine im vorherigen Sinne beschriebene Kombination zweier fundamentaler
Gitter. Die Kombination aus fundamentalen Gittern bietet nur dann eine bessere Schranke,
wenn die Zahl der Bindungen erhalten bleibt (also Summe der Bindungen der fundamentalen
Gitter=Zahl der Bindungen im kombinierten Gitter). In Abbildung 4.16 ist ein Subgitter
Abbildung 4.16: weiteres Subgitter des Quadratgitters (1d)
(1d) des Quadratgitters eingezeichnet, das sich auf die gleiche Weise aus den fundamentalen Kreuzen zusammensetzt wie (2b) aus (2a). Ich habe auch dafür die untere Schranke der
1d
Grundzustandsenergie ausgerechnet (E0
= −2, 6865J ).
E01d /site = E01d ·
2
= −0, 7664J
7
(4.94)
Auch dieses Subgitter liefert analog zu (2b) eine schlechtere untere Schranke als die anderen
beiden, oenen Gitter. Der vermeindliche Widerspruch diesbezüglich ist also behoben.
Auf Basis der mir vorliegenden Werte für die unteren Schranken und aufgrund theoretischer
Erwägungen ist die Konstruktion guter Subgitter also folgendermaÿen anzugehen: Es wird
zunächst der fundamentale Gitterbaustein mit dem gröÿten Grundzustands-Gesamtspin ermittelt. Die Kombinationen aus diesen Bausteinen, bei denen kein Bindungsüberlapp stattndet, liefern dann noch bessere Schranken. Dies können theoretisch Verknüpfungen aus beliebig vielen fundamentalen Gittern sein, wobei tendenziell die Schranken mit der Anzahl
kombinierter Gitter besser werden. Dabei ist zu beachten, dass es bei der Kombination fun-
Abbildung 4.17: weiteres, kombiniertes Subgitter des Quadratgitters (1e)
damentaler Gitter mehrere Möglichkeiten gibt. Eine weitere Kombination zweier Kreuze für
81
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
das Quadratgitter ist in der besagten Publikation [TV90] ebenfalls berechnet worden ((1e),
siehe Abbildung 4.17). Die untere Schranke für Grundzustandsenergie pro Gitterplatz liegt
auf Basis dieses Subgitters bei
E01e /site = −0, 7391J
.
(4.95)
Dieses Gitter liefert also eine noch bessere Schranke. Verglichen mit dem Doppelkreuz (1b)
entsteht dieses Gitter durch Zusammenfügen zweier Kreuze, wobei zwei Gitterplätze zusammengeführt werden. Dadurch bleibt die Zahl der Bindungen ebenfalls erhalten (acht), aber die
Zahl der Gitterplätze (acht) ist geringer als beim Doppelkreuz (neun). Daraus könnte man
schlieÿen, dass diejenigen Kombinationen zweier fundamentaler Gitter am besten sind, die
das gröÿte Verhältnis von Bindungen zu Gitterplätzen aufweisen (1 und 8/9). Dies deckt sich
auch mit der Situation für das Honeycomb-Gitter. Dort hat (2a) ein solches Verhältnis von 1
und (2b) eines von 5/6.
Die Konstruktion immer besserer Subgitter kann dann natürlich wiederum durch entsprechendes Kombinieren der besten Kompositionen aus den fundamentalen Gittern erfolgen. Somit
wäre eine Art rekursives Vorgehen für die Wahl immer gröÿerer und besserer Subgitter
gegeben, um die Abschätzung durch untere Schranken möglichst gut werden zu lassen.
Ein Vergleich und eine damit einhergehende Interpratation beziehungsweise Diskussion
meiner Ergebnisse sind damit erfolgt. Es soll nun noch untersucht werden, wie gut die gefundenen unteren Schranken, unabhängig vom Vergleich untereinander, einzuschätzen sind.
Vergleich der unteren Schranken mit Ergebnissen aus der Variationsrechnung
Es ist mehrmals erwähnt worden, dass es mittels Variationsrechnung möglich ist, für die von
mir untersuchten (antiferromagnetischen) Systeme obere Schranken für die Grundzustandsenergie pro Gitterplatz zu nden. Je näher die höchste untere Schranke und die niedrigste
obere Schranke beieinander liegen, desto besser ist die tatsächliche Grundzustandsenergie
eingegrenzt.
Zwecks eines Vergleichs sind in der folgenden Tabelle für jeden der drei Gittertypen die beste
1
untere Schranke meiner Berechnungen, eine obere Schranken aus der Variationsrechnung , die
dadurch gegebene Energiespanne und die prozentuale Abweichung aufgeführt.
Quadratgitter
Honeycomb-Gitter
Würfelgitter
untere Schranke
obere Schranke
Spanne
Abweichung
−0, 7421J
−0, 625J
−0, 9957
−0, 6688J
−0, 554
−0, 899J
0, 733J
0, 71J
0, 567J
9, 9%
11, 36%
9, 7%
Die Abweichungen liegen alle in einem Bereich von etwa
etwa
10%.
0, 7J
und prozentual ausgedrückt bei
Es gibt keine besonders signikanten Unterschiede zwischen den einzelnen Gitter-
typen. Die Grundzustandsenergie für die unendlichen, antiferromagnetischen Gitter ist damit
1
aus: [NSJB09] und [Bar72]
82
4.4. AUSBLICK
recht gut eingegrenzt.
Durch verbesserte Methoden sind sicherlich noch bessere Werte für die oberen Schranken
möglich, beziehungsweise liegen gegebenenfalls bereits vor. Was die unteren Schranken anbelangt, sind diese durch die Wahl gröÿerer Subgitter sicherlich auch besser möglich. Jedoch
zeigt sich schon bei meinen Resultaten, dass die Unterschiede zwischen den unterschiedlichen
offenen Strukturen eines Gitters nur sehr gering sind, sodass mit keinen wesentlichen Verbesserungen zu rechnen ist.
4.4 Ausblick
Meine Ergebnisse und der Weg zu diesen wurden ausführlich vorgestellt und diskutiert. Es
soll nun ein knapper Ausblick gegeben werden, welche Erweiterungen sich dieser Arbeit anschlieÿen könnten, um die behandelte Thematik weiter zu vertiefen.
Zum Einen wäre es möglich, den technischen Teil dieser Arbeit zu verbessern. Damit
meine ich die bereits angesprochenene Verallgemeinerung meines Algorithmus. Dieser könnte
so gestaltet werden, dass es nur noch der Angabe der benachbarten Gitterplätze bedarf, um die
Grundzustandsenergie zu erhalten. Eine solchen Implementierung sollte durchaus im Rahmen
des Möglichen liegen, da die notwendigen Schritte für die Berechnung, ausgehend vom zugrundeliegenden Gitter, allesamt bekannt sind. Auch die Anwendung des Lieb-Mattis-Theorems
und die daraus folgende Reduktion auf einen Unterraum für die Grundzustandsenergie könnten in diesen Algorithmus integriert werden.
Da MATHMEATICA über die entsprechenden Elemtente der graschen Benutzereingabe
verfügt, wäre es sogar denkbar, ein Programm zu erstellen, in dem durch das Anordnen
von Kreisen und Bindungen Gitter erstellt werden. Die Implementierung könnte daraus den
zugehörigen Heisenberg-Hamiltonoperator ermitteln und die Grundzustandsenergie berechnen. Eine solche Umsetzung wäre sicherlich ein gröÿeres Projekt.
Die anderen denkbaren Erweiterungen sind eher inhaltlicher Natur. Naheliegend ist es, für
die behandelten Gitter (Quadrat-, Honeycomb- und Würfelgitter) jeweils weitere, gröÿere
Subgitter zu berechnen und daraus die unteren Schranken zu ermitteln. Dies könnte mit
einem, zuvor beschriebenen, verallgemeinerten Algorithmus unkompliziert durchgeführt werden. Auf diese Weise könnten bessere untere Schranken gefunden werden, die zusammen
mit den oberen Schranken der Variationsrechnung eine genauere Abschätzung der Grundzustandsenergie geben würden. Allerdings haben die Ergebnisse der vorangegangenen Abschnitte
gezeigt, dass die zu erwartenden Verbesserungen durch gröÿere Subgitter nur sehr gering sind.
Die Bestimmung von unteren Schranken aus weiteren Subgittern würde zudem die Möglichkeit
geben, die von mir abgeleiteten Regeln für die Güte eines Subgitters zu überprüfen/ bestätigen, da diese Regeln neben theoretischen Erwägungen eine Induktion aus meinen Ergebnissen darstellen. Es sei dabei aber beachtet, dass die Gröÿe der berechenbaren Systeme stark
beschränkt ist.
Eine weitere logische Fortsetzung der Arbeit wäre die Betrachtung anderer bipartiter Gitter
in zwei und drei Dimensionen. Dabei ist beispielsweise an die dreidimensionale Erweiterung
83
KAPITEL 4. EXAKTE DIAGONALISIERUNG UND UNTERE SCHRANKEN DER
GRUNDZUSTANDSENERGIE
des hexagonalen Gitters gedacht. Die Berechnung unteren Schranken nach analogem Vorgehen
würde das Spektrum an Resultaten weiter verbreitern und damit die Aussagekraft der Deutung erhöhen. Die Überlegung, weitere bipartite Gitter in die Berechnungen mit einzubeziehen,
war bereits während der Ausarbeitung dieser Thesis vorhanden. Es hat sich dabei herausgestellt, dass das Finden weiterer, zweidimensionaler, bipartiter Gitterysteme ein schwieriges
Unterfangen darstellt, gerade hinsichtlich periodischer und naturgegebener Gitter.
Da die Methode der unteren Schranken allgemeine Gültigkeit hat, ist es natürlich nicht
zwangsläug notwendig, sich bei den Berechnungen nur auf bipartite Gitter zu beziehen.
Insofern ist es grundsätzlich möglich, dieselben Bestimmungen unterer Schranken für verschiedene nicht-bipartite, frustrierte Systeme durchzuführen (derer gibt es wesentlich mehr).
Solche Systeme weisen ein komplizierteres Verhalten auf und stellen somit sicherlich interessante Untersuchungsobjekte dar. Gerade die Untersuchung der Güte von frustrierten Subgittern und der Vergleich mit dem Verhalten bipartiter Subgitter sollten eine interessante
und anspruchsvolle Ergänzung zu dieser Arbeit aufbieten. Die Betrachtung solcher Systeme
wurde im Rahmen der Arbeit in Erwägung gezogen. Es stellt sich dabei aber ein grundlegendes Problem: Die gute Berechenbarkeit der bipartiten Subgitter beruht auf der Benutzung
des Lieb-Mattis-Theorems und die gesamte Methodik hat seine groÿen Vorzüge in eben dieser
Anwendung des Theorems. Für frustrierte Systeme sind Reduktionen der Dimensionalität für
die Grundzustandsenergie dieser Art nicht möglich; sie sind somit nur mit ungleich höherem
Aufwand auszuwerten.
Eine weitere Möglichkeit wäre es, nicht nur Systeme aus Spin-1/2-Plätzen zu betrachten.
Dies ist deshalb in Erwägung zu ziehen, weil in der Praxix auftretende Gitter nur im seltensten
Fall ausschlieÿlich aus Spin-1/2-Plätzen bestehen. Vielmehr hat man es dort mit Mehrelektronensystemen auf den einzelnen Gitterplätzen zu tun, die einen Gesamtdrehimpuls besitzen,
der die unterschiedlichsten Werte annehmen kann (siehe Hundsche Regeln). Bezüglich des
grundlegenden Vorgehens und des notwendigen Algorithmus' besteht kein Unterschied zu den
von mir untersuchten Spin-1/2-Systemen.
Ich habe nun ein paar mögliche Erweiterungen meiner Arbeit vorgestellt. Es sind sicherlich
noch weitere denkbar, insbesondere wenn man allgemeinere Problemstellungen betrachtet und
beispielsweise den ferromagetischen Fall oder allgemeinere Formen des Heisenbergmodells in
Betracht zieht. Dabei würde man sich aber im Grunde genommen gänzlich anderen Fragestellungen widmen.
Der letztliche Inhalt dieser Arbeit war nicht von Beginn an bis ins Detail vorbestimmt, sondern hat sich durch Entstehen eines Gesamtkonzeptes während der Erstellung dieser Arbeit
entwickelt. Somit sind einige der hier aufgegrienen Erweiterungen bereits Gegenstand meiner
Überlegungen gewesen. Aus Zeitgründen und den gesteckten Rahmen einer Bachelor-Arbeit
ist es erforderlich, sich auf ein begrenztes Feld von Studien zu beschränken. Ich denke, dass
die von mir aufgeführten Ausarbeitungen ein stimmiges und in sich abgeschlossenes Projekt
darstellen.
84
Literaturverzeichnis
[AKHv11] Alex, A. ; Kalus, M. ; Huckleberry, A. ; von Delft, J.:
A numerical
algorithm for the explicit calculation of SU(N) and SL(N,C) Clebsch-Gordan
coecients.
In: Journal of Mathematical Physics 52 (2011), Februar, Nr. 2.
http://dx.doi.org/10.1063/1.3521562. DOI 10.1063/1.3521562
[AM76] Ashcroft, Neil ; Mermin, David: Festkörperphysik. Oldenbourg, 1976
[Bar72] Bartkowski, R. R.: New Variational Method for the Antiferromagnetic Ground
State.
In: Phys. Rev. B
5 (1972), Jun, Nr. 11.
http://dx.doi.org/10.1103/
PhysRevB.5.4536. DOI 10.1103/PhysRevB.5.4536
[Bet31] Bethe, H.: Zur Theorie der Metalle. In: Zeitschrift für Physik A Hadrons and
Nuclei 71 (1931). http://dx.doi.org/10.1007/BF01341708. 10.1007/BF01341708
[Blu08] Blundell, Stephen: Magnetism in Condensed Matter. Oxford University Press,
2008 (Oxford Master Series in Condensed Matter Physics)
[CTDL73] Cohen-Tannoudji, Claude ; Diu, Bernard ; Laloë, Franck: Quantum Mechan-
ics. Bd. 2. Wiley-VCH, 1973
[Goo63] Goodenough, J.B.:
Magnetism and Chemical Bond.
Interscience Publishers,
1963
[Gri95] Griffiths, David J.:
Introduction to Quantum Mechanics. Pearson Education
International, 1995
[HW92] Haken, Hermann ; Wolf, Hans C.: Molekülphysik und Quantenchemie. Springer,
1992
[Kop11] Kopietz, Peter: Vorlesungsskript zur Vorlesung Quantenmechanik II . 2011
[LM62] Lieb, Elliot ; Mattis, Daniel: Ordering Energy Levels of Interacting Spin Systems.
In: J. Math. Phys. 3 (1962), July, Nr. 4. http://dx.doi.org/10.1063/1.1724276. DOI 10.1063/1.1724276
[Mat32] Mattis, Daniel C.:
The Theory of Magnetism Made Simple.
World Scientic,
1932
[Nol02] Nolting, Wolfgang:
Grundkurs Theoretische Physik 7: Viel-Teilchen-Theorie.
Springer, 2002
[Nol04] Nolting, Wolfgang:
Grundkurs Theoretische Physik 5/2: Quantenmechanik -
Methoden und Anwendungen. Springer, 2004
85
LITERATURVERZEICHNIS
[NSJB09] Noorbakhsh, Zahra ; Shahbazi, Farhad ; Jafari, Seyed A. ; Baskaran, Ganapathy:
Variational RVB Wave Function for the Spin-1/2 Heisenberg Model on
Honeycomb Lattice. In: Journal of the Physical Society of Japan 78 (2009), Nr. 5.
http://dx.doi.org/10.1143/JPSJ.78.054701. DOI 10.1143/JPSJ.78.054701
[Rat11] Ratzinger, Ulrich:
Vorlesungsskript zur Vorlesung Höhere Experimentaphysik
I . 2011
[Stö07] Stöcker, Horst (Hrsg.): Taschenbuch der Physik : Formeln, Tabellen, Übersicht-
en. 5., korrigierte Au., Nachdr. Frankfurt am Main : Deutsch, 2007. ISBN
9783817117208 3817117205
[TV90] Tarrach, Rolf ; Valentí, Roser: Exact lower bounds to the ground-state energy
of spin systems: The two-dimensional S=1/2 antiferromagnetic Heisenberg model.
In: Phys. Rev. B 41 (1990), May, Nr. 13. http://dx.doi.org/10.1103/PhysRevB.
41.9611. DOI 10.1103/PhysRevB.41.9611
[Val10] Valentí, Roser: Vorlesungsskript zur Vorlesung Advanced Solid State Theory .
2010
[Web] http://phycomp.technion.ac.il/~riki/H2_molecule.html
86
Anhang
A.1 Lineare Spinkette: MATHEMATICA-Code
h = 1;
J = 1;
In[24]:= H[N_] :=
J*Sum[1/2*(SuperPlus[S][n, N].SuperMinus[S][n + 1, N] +
SuperMinus[S][n, N].SuperPlus[S][n + 1, N]) + (S^z)[n, N].(S^
z)[n + 1, N], {n, 1, N - 1}]
(*hamiltonian in terms of creating-. annihilation- and z-componente \
operator*)
Basismatrix[N_] := Tuples[{1, 0}, N]
(*creates basis matrix by all possible permutations of 1 and 0 where \
1 is spin-up and 0 is spin-down*)
In[27]:= SuperPlus[S][n_, N_] := (
B = ConstantArray[2, {2^N, N}];
a = Basismatrix[N];
For[i = 1, i <= 2^N, i++, a[[i]][[n]] += 1];
For[i = 1, i <= 2^N, i++, B[[i]] = a[[i]]];
Ma = h*Table[
If[ Part[Basismatrix[N], k] == Part[a, j], 1, 0], {k, 1,
2^N}, {j, 1, 2^N}];
Ma)
(*creates creation operator by incrementing basis state quantum \
numbers and comparing to original basis states*)
In[28]:=
SuperMinus[S][n_, N_] := (
B = ConstantArray[2, {2^N, N}];
a = Basismatrix[N];
For[i = 1, i <= 2^N, i++, a[[i]][[n]] -= 1];
For[i = 1, i <= 2^N, i++, B[[i]] = a[[i]]];
Ma = h*Table[
If[ Part[Basismatrix[N], k] == Part[a, j], 1, 0], {k, 1,
2^N}, {j, 1, 2^N}];
Ma)
(*creates annihilation operator by decrementing basis state quantum \
numbers and comparing to original basis states*)
In[29]:=
87
APPENDIX
(S^z)[n_, N_] := (
a = Table[If[Basismatrix[N][[i]][[n]] == 0, -1, 1], {i, 1, 2^N}];
Ma = h/2*DiagonalMatrix[a];
Ma)
(*creates Sz operator by forming a diagonal matrix with m as entries*)
In[30]:= Energien[d_] := N[Eigenvalues[N[H[d]]]];
(*gives numerical evaluation of eigenenergies*)
In[31]:= antiferro = Table[{l, Min[Energien[l]]}, {l, 2, 11}]
(*gives number of spins and according ground state energy in a table*)
Out[31]= {{2, -0.75}, {3, -1.}, {4, -1.61603}, {5, -1.92789}, {6, \
-2.49358}, {7, -2.83624}, {8, -3.37493}, {9, -3.73632}, {10, \
-4.25804}, {11, -4.63209}}
In[32]:= Export[
"Desktop/Bachelor/finding_states/chain/antiferro_chain.csv",
antiferro];
(*exports table into csv format for further manipulations*)
A.2 Grundzustandsenergie des Doppelkreuzes:
MATHEMATICA-Code
h = 1;
J = 1;
(*set h and J to 1 to make numerical evaluations possible*)
names = {1, 234, 5, 6, 789};
(*numbering of the involved spins*)
spins = {1/2, 3/2, 1/2, 1/2, 3/2};
(*according spin values in same order*)
numberofspins = Length[spins];
(*number of spins*)
dimension = Product[2*spins[[k]] + 1, {k, 1, Length[spins]}];
(*dimension of the full hilbert space*)
naturalBasis = {Flatten[
Table[{"S" <> ToString[names[[j]]],
"m" <> ToString[names[[j]]]}, {j, 1, Length[names]}]],
Partition[
Flatten[Tuples[
Table[Table[{spins[[l]], k}, {k, -spins[[l]], spins[[l]]}], {l,
1, Length[spins]}]]], 2*Length[spins]]};
(*creates natural basis of spin system*)
88
A.2. GRUNDZUSTANDSENERGIE DES DOPPELKREUZES: MATHEMATICA-CODE
newbasis[oldbasis_, s1_, s2_] := (
index1 =
Position[oldbasis[[1]], "m" <> StringDrop[s1, 1]][[1]][[1]];
index2 =
Position[oldbasis[[1]], "m" <> StringDrop[s2, 1]][[1]][[1]];
indexS1 = Position[oldbasis[[1]], s1][[1]][[1]];
indexS2 = Position[oldbasis[[1]], s2][[1]][[1]];
cancelledM =
DeleteDuplicates[
Table[ReplacePart[
oldbasis[[2]][[k]], {index1 -> 0, index2 -> 0}], {k, 1,
dimension}]];
{ReplacePart[
oldbasis[[
1]], {(index1) ->
oldbasis[[1]][[indexS1]] <>
StringDrop[oldbasis[[1]][[indexS2]], 1],
index2 ->
oldbasis[[1]][[index1]] <>
StringDrop[oldbasis[[1]][[index2]], 1]}],
Partition[
Flatten[Table[
Table[ReplacePart[
cancelledM[[j]], {index1 -> k, index2 -> l}], {k,
Abs[cancelledM[[j]][[indexS1]] - cancelledM[[j]][[indexS2]]],
cancelledM[[j]][[indexS1]] +
cancelledM[[j]][[indexS2]]}, {l, -k, k}], {j, 1,
Length[cancelledM]}]], 2*numberofspins]}
);
(*creates new basis by coupling s1 and s2*)
cg[{j1_, m1_}, {j2_, m2_}, {j_, m_}] := (
If[m1 + m2 == m && Abs[j1 - j2] <= j <= j1 + j2,
ClebschGordan[{j1, m1}, {j2, m2}, {j, m}], 0]
);
(*Clebsch Gordan coefficients for valid values*)
trafo[oldbasis_, s1_, s2_] := (
basis = newbasis[oldbasis, s1, s2];
index1 =
Position[oldbasis[[1]], "m" <> StringDrop[s1, 1]][[1]][[1]];
index2 =
Position[oldbasis[[1]], "m" <> StringDrop[s2, 1]][[1]][[1]];
indexS1 = Position[oldbasis[[1]], s1][[1]][[1]];
indexS2 = Position[oldbasis[[1]], s2][[1]][[1]];
Table[If[
Delete[oldbasis[[2]][[k]], {{index1}, {index2}}] ==
Delete[basis[[2]][[l]], {{index1}, {index2}}],
cg[{oldbasis[[2]][[k]][[indexS1]],
oldbasis[[2]][[k]][[index1]]}, {oldbasis[[2]][[k]][[indexS2]],
oldbasis[[2]][[k]][[index2]]}, {basis[[2]][[l]][[index1]],
basis[[2]][[l]][[index2]]}], 0], {k, 1, dimension}, {l, 1,
dimension}]
89
APPENDIX
);
(*creates transformation matrix by given s1 s2 and old baswird für is*)
\
Sz[s_, basis_] :=
DiagonalMatrix[
Table[h*basis[[2]][[k]][[
Position[basis[[1]], "m" <> ToString[s]][[1]][[1]]]], {k, 1,
dimension}]];
(*general Sz-operator*)
Sup[s_, basis_] :=
(position = Position[basis[[1]], "m" <> ToString[s]][[1]][[1]];
positionS = Position[basis[[1]], "S" <> ToString[s]][[1]][[1]];
Table[If[
basis[[2]][[k]][[position]] == basis[[2]][[l]][[position]] - 1 &&
ReplacePart[basis[[2]][[k]], position -> 0] ==
ReplacePart[basis[[2]][[l]], position -> 0],
h*Sqrt[(basis[[2]][[k]][[positionS]] basis[[2]][[k]][[position]])*(basis[[2]][[k]][[positionS]] +
basis[[2]][[k]][[position]] + 1)], 0], {k, 1,
dimension}, {l, 1, dimension }]
);
(*S+ operator*)
Sdo[s_, basis_] :=
(position = Position[basis[[1]], "m" <> ToString[s]][[1]][[1]];
positionS = Position[basis[[1]], "S" <> ToString[s]][[1]][[1]];
Table[If[
basis[[2]][[k]][[position]] == basis[[2]][[l]][[position]] + 1 &&
ReplacePart[basis[[2]][[k]], position -> 0] ==
ReplacePart[basis[[2]][[l]], position -> 0],
h*Sqrt[(basis[[2]][[k]][[positionS]] +
basis[[2]][[k]][[position]])*(basis[[2]][[k]][[positionS]] basis[[2]][[k]][[position]] + 1)], 0], {k, 1,
dimension}, {l, 1, dimension }]
);
(*S- operator*)
diagonalH[s_, basis_] := (
position = Position[basis[[1]], "S" <> ToString[s]][[1]][[1]];
DiagonalMatrix[
Table[h^2*
basis[[2]][[k]][[
position]]*(basis[[2]][[k]][[position]] + 1), {k, 1,
dimension}]]
);
(*part which is diagonal in total spin basis*)
couple[s1_, s2_,
basis_] := (1/
2*(Sup[s1, basis].Sdo[s2, basis] +
Sdo[s1, basis].Sup[s2, basis]) + Sz[s1, basis].Sz[s2, basis]);
90
A.2. GRUNDZUSTANDSENERGIE DES DOPPELKREUZES: MATHEMATICA-CODE
(* S1*S2 *)
extractTotalSpin[s_, value_] := (
coloum = Position[basis[[1]], "S" <> ToString[s]][[1]][[1]];
right = Flatten[Position[Transpose[basis[[2]]][[coloum]], value]];
Transpose[H[[right]]][[right]]
);
(*gives sub-hamiltonian for given total spin Ss*)
extractTotalZ[s_, sz_, value_, valuez_] := (
coloum1 = Position[basis[[1]], "m" <> ToString[sz]][[1]][[1]];
right1 =
Flatten[Position[Transpose[basis[[2]]][[coloum1]], valuez]];
coloum2 = Position[basis[[1]], "S" <> ToString[s]][[1]][[1]];
right2 = Flatten[Position[Transpose[basis[[2]]][[coloum2]], value]];
right = Intersection[right1, right2];
Transpose[H[[right]]][[right]]
);
(*gives sub-hamiltonian vor given total spin and given total \
z-componente*)
t1 = trafo[naturalBasis, "S1", "S234"];
t2 = trafo[basis, "S6", "S789"];
t3 = trafo[basis, "S1234", "S5"];
t4 = trafo[basis, "S12345", "S6789"];
(*gives matrices of the single transformations*)
T = t1.t2.t3.t4;
(*total transformation matrix*)
t = Inverse[t4].Inverse[t3].Inverse[t2].Inverse[t1];
(*gives inverse of T*)
H = J/2 (diagonalH[1234, basis] 2*(3/4 + 15/4)*IdentityMatrix[dimension] +
diagonalH[6789, basis]) +
J*t.(couple[1, 5, naturalBasis] + couple[6, 5, naturalBasis]).T;
(*Hamiltonian for ground state energy in total spin basis*)
Eigenvalues[N[extractTotalZ[123456789, 123456789, 5/2, 1/2]]]
(*eigenvalues of block for s=5/2 sz=0. minimum gives ground state \
energy*)
{-2.96851, 1.78947, 1.20711, -1., -0.683182, -0.207107, -0.137775}
91