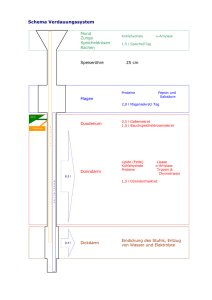
Regulatorische Protein-Protein-Interaktionen in der plastidären
Werbung

Regulatorische Protein-Protein-Interaktionen in der plastidären Transkription Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften der Fakultät für Biologie und Biotechnologie an der Internationalen Graduiertenschule Biowissenschaften der Ruhr-Universität Bochum angefertigt in der Arbeitsgruppe Pflanzliche Zellphysiologie und Molekularbiologie vorgelegt von Andrea Kolpack aus Gelsenkirchen Bochum Februar, 2010 Erstgutachter: Prof. Dr. Gerhard Link Zweitgutachter: Prof. Dr. Ulrich Kück Regulatory Protein-Protein Interactions in the Plastid Transcription Dissertation To obtain the degree Doctor Rerum Naturalium (Dr. rer. nat.) at the Faculty of Biology and Biotechnology, Ruhr-University Bochum International Graduate School Biosciences Ruhr-University Bochum Laboratory of Plant Cell Physiology and Molecular Biology submitted by Andrea Kolpack from Gelsenkirchen, Germany Bochum February 2010 accepted on the recommendation of Prof. Dr. Gerhard Link, first supervisor Prof. Dr. Ulrich Kück, second supervisor für Thorsten Danksagung Mein besonderer Dank gilt meinem Doktorvater Herrn Prof. Dr. G. Link für die Überlassung des sehr interessanten und vielschichtigen Forschungsthemas. Die wertvollen Diskussionen und sein fachlicher Rat haben maßgeblich zum Gelingen der Arbeit beigetragen. Bei Herrn Prof. Dr. U. Kück möchte ich mich für die freundliche Übernahme des Korreferats und sein stetiges Interesse an der Arbeit bedanken. Ganz herzlich möchte ich mich bei allen Mitgliedern der Arbeitsgruppe „Pflanzliche Zellphysiologie und Molekularbiologie“: Frau Brigitte Link, Dr. Jennifer Schweer, Dipl.-Biol. Hacer Türkeri, Dr. Heike Loschelder, Dipl.-Biol. Sylvia Bock, Ena Husic und Thomas Pieta für das familiäre Arbeitsklima und die gute Zusammenarbeit bedanken. Besonders möchte ich mich bei Frau Brigitte Link für ihre exzellente technische Arbeit und die schöne Zeit im Labor bedanken. Meiner Familie und meinen Freunden danke ich für ihr langjähriges Interesse, Verständnis und ihre liebevolle Unterstützung. Mein herzlichster Dank gilt Thorsten und Agnes, die mich beide durch die Höhen und Tiefen dieser Arbeit begleitet haben. Inhaltsverzeichnis Inhaltsverzeichnis: Abkürzungsverzeichnis V Abbildungsverzeichnis IX Genbezeichnungen X 1.Einleitung 1 1.1 Der Plastid 1 Ursprung der Plastiden 1 Differenzierung der Plastiden 1 Das Plastom 3 1.2 Vergleich der pro- und eukaryotischen Transkriptionsinitiation 3 1.3 Die plastidäre Transkription 5 DNA-abhängige RNA-Polymerase NEP 5 DNA-abhängige RNA-Polymerase PEP 6 1.4 Sigmafaktoren 8 1.5 Regulation der Aktivität von Sigmafaktoren 10 1.6 Interaktionen zwischen Pflanzen und Phytopathogenen 13 1.7 Protein-Protein-Interaktionen 15 2. Zielsetzung 17 3. Material und Methoden 19 3.1 Material 19 3.1.1 Chemikalien und Enzyme 19 3.1.2 Nährmedien 20 3.1.2.1 Nährmedien für die E. coli-Kultivierung 20 3.1.2.2 Nährmedien für die Anzucht von A. thaliana 20 3.1.3 Linien von A. thaliana 21 I Inhaltsverzeichnis 3.1.4 Bakterienstämme von E. coli 21 3.1.5 Oligonukleotide 22 3.1.6 Plasmide 24 3.1.7 Internetadressen 30 3.2 30 Methoden 3.2.1 Anzucht von A. thaliana 30 3.2.2 Isolierung von Nukleinsäuren 31 3.2.2.1 Isolierung genomischer DNA 31 3.2.2.2 Isolierung von Plasmid-DNA 31 Plasmid-DNA-„Easy“-Präparation 31 Plasmid-DNA-Midipräparation 31 3.2.2.3 Isolierung von RNA 31 3.2.3 Amplifizierung von Nukleinsäuren 31 3.2.3.1 Reverse Transkription 31 3.2.3.2 Polymerase-Kettenreaktion 32 3.2.3.3 ortsgerichtete in vitro Mutagenese 32 3.2.3.4 DNA-Sequenzierung 32 3.2.4 Elution von DNA-Fragmenten 33 3.2.5 Markierung von Nukleinsäuren 33 3.2.5.1 radioaktive Endmarikierung von DNA 33 3.2.5.2 nicht-radioaktive DIG-Markierung von RNA 33 3.2.6 „Northern“-Transfer von RNA 33 3.2.7 RNA/RNA-Hybridisierung 33 3.2.8 Expression rekombinanter Proteine 34 II Inhaltsverzeichnis 3.2.8.1 Expression mittels des pMAL-Systems 34 3.2.8.2 Expression mittels des pGEX-Systems 34 3.2.9 Gelelektrophorese von Proteinen 35 3.2.10 „Western“-Transfer von Proteinen 35 3.2.11 Immunodetektion von Proteinen 35 3.2.12 Protein Overlay Blot 36 3.2.13 Färbung von Proteinen 36 3.2.14 Bestimmung der Proteinkonzentration 36 3.2.15 Nachweis von DNA-Protein-Interaktionen 36 3.2.16 Circulardichroismus (CD) –Spektroskopie 37 4. Ergebnisse 38 4.1 Überexpression und Funktionsanalyse des AtSIG1-Proteins 38 4.2 Analyse der Protein-Bindungsfunktion von AtSIG1 mittels ausgewählter Mutanten-Konstrukte 43 4.3 Expression und Reinigung der Proteine AtSIB1 und AtSIB2 48 4.4. Interaktionsstudien mit dem AtSIB1- und AtSIB2-Proteinen 50 4.5 Funktionsanalysen der Interaktion zwischen den AtSIG1- und den beiden AtSIB-Proteinen 54 4.6 Bioinformatische Analyse der Sigma-Bindeproteine 58 4.7 Analyse der plastidären Genexpression des sig1-Gens in einer sig1-1 Einzel-Knockout-Linie 64 Analyse der Phänotypen von sig1 und sig6 Einzel-KnockoutLinien und von den Doppel-Knockout-Linien sig1xsig6 und sig6xsig1 unter unterschiedlichen Licht-Bedingungen 68 4.8 5. Diskussion 5.1 Funktionsanalysen des rekombinanten AtSIG1-Proteins und ausgewählter Konstrukte 71 71 III Inhaltsverzeichnis 5.2 Charakterisierung der Sigma-Bindeproteine aus A. thaliana 75 Analyse der physischen Protein-Protein-Interaktion von AtSIG1und beiden AtSIB-Proteinen 76 Analyse der funktionellen Protein-Protein-Interaktion von AtSIG1und beiden AtSIB-Proteinen 79 5.3 5.4 In silico Analyse der Sigma-Bindeprotein AtSIB1 und AtSIB2 84 Einfluss des sig1-Gens in einer sig1-1 Einzel-Knockout-Linie auf die plastidäre Transkription und phänotypische Effekte in Einzel- und Doppelmutanten durch Licht 87 Ausblick 90 6. Zusammenfassung 92 7. Summary 93 8. Literatur 95 9. Anhang 115 10. Kongressberichte 117 11. Lebenslauf 118 12. Erklärung 119 IV Abkürzungen Abkürzungen: A Adenosin bzw. Ampere Abb. Abbildung Abk. Abkürzung A. tumefaciens Agrobacterium tumefaciens amp Ampicillin A. bidest/Aqua bidest zweimal destilliertes Wasser Aqua dest. destilliertes Wasser APS Ammoniumpersulfat AS Aminosäure A. thaliana Arabidopsis thaliana ATP Adenosintriphosphat AtSIG1-6 Sigmafaktor 1 bis 6 aus A. thaliana B. subtilis Bacillus subtilis BLRP Blue-Light-Responsive-Promoter bp Basenpaare BCIP 5-Bromo-4-chloro-3-indoyl-β-D-phosphat BPB Bromphenolblau BSA Rinder-Serumalbumin („bovine serum albumin“) bspw. beispielsweise bzw. beziehungsweise °C Grad Celsius cam Chloramphenicol cDNA „copy“ DNA Ci Curie CK2 Proteinkinase CK2 cpCK2α chloroplastidäre CK2α CTAB Cethyltrimethylammoniumbromid C-terminal carboxyterminal Da Dalton d.h. das heißt dH2O destilliertes Wasser DNA Desoxyribonukleinsäure V Abkürzungen DNase Desoxyribonuklease dNTP Desoxynukleosidtriphosphat (dATP, dCTP, dGTP, dTTP) DTT Dithiothreitol E. coli Escherichia coli EDTA Ethylendiaminotetraessigsäure et al. und andere (lat.: „et alii“) g Gramm bzw. Gravitationskonstante; Erdbeschleunigung (9,81 m/s2) ggf. gegebenenfalls GST Glutathion-S-Transferase h Stunde HR-Antwort hypersensitive response IPTG Isopropyl-β,D-thiogalaktopyranosid IR Regionen mit gegenläufigen Sequenzwiederholungen („inverted repeats“) kb Kilobasenpaare kDa Kilodalton kV Kilovolt M Molar µ micro min Minute mol Stoffmengeneinheit mRNA Boten („messenger”)-RNA MT Mutante NBT Nitroblue Tetrazolium NCBI National Center for Biotechnology Information NEP kernkodierte, phagenähnliche RNA-Polymerase („nuclear-encoded RNA-Polymerase“) N.tabacum Nicotiana tabacum nt Nukleotid(e) N-terminal aminoterminal OD optische Dichte VI Abkürzungen O. sativa Oryza sativa PAA Polyacrylamid PAGE Polyacrylamid-Gelelektrophorese PAMPs pathogen (or microbe)-associated molecular patterns PCR Polymerase-Kettenreaktion („polymerase chain reaction”) PEND „Plastid Envelope DNA-binding Protein“ PEP plastidenkodierte RNA-Polymerase („plastidencoded RNA Polymerase”) pH Logarithmus der H3O+-Ionenkonzentration in mol/l ppGpp 5‘-Diphosphat-Guanosin-3‘-Diphosphat PR-Proteine pathogenesis related Proteine PRRs Pattern recognition receptors P. syringae Pseudomonas syringae PTI-Antwort PAMP-triggered immunity PTK plastidäre Transkriptionskinase RBP RNA-Bindeprotein RNA Ribonukleinsäure RNase Ribonuklease ROS reactive oxygen species rpm Umdrehungen pro Minute („revolts per minute“) rRNA ribosomale RNA RT Raumtemperatur RubisCO Ribulose-1,5-bisphosphat-Carboxylase/Oxygenase s Sekunde SAR systemic aquired resistance S. alba Sinapis alba SASIG σ-Faktor von S.alba SDS Natriumdodecylsulfat σ-Faktor Sigmafaktor SIG Sigmafaktor SLF σ-ähnlicher-Faktor („sigma-like-factor“) Tab. Tabelle VII Abkürzungen TAC Membran-gebundener PEP-Polymerase Komplex TBS „Tris Buffered Saline“ TBP TATA-Bindeprotein TCA Trichloressigsäure TEMED NNN’N’-Tetramethylethylendiamin TFII Transkriptionsfaktor der RNA-Polymerase II TIC translocon at the inner membrane of chloroplasts TOC translocon at the outer membrane of chloroplasts TP Transit-Peptid Tris Tris(hydroxymethyl)aminoethan TritonX-100 t-Octylphenoxypolyethoxyethanol tRNA Transfer-RNA U Unit (Enzymeinheit) u.a. unter anderem UKB unkonservierter Bereich UV ultraviolettes Licht V Volt v/v Volumen/Volumen („volume per volume“) w/v Gewicht/Volumen („weight per volume“) WT Wildtyp XCFF Xylencyanolblau X-Gal 5-Brom-4-chloro-3-indolyl-β-galaktopyranosid z.B. zum Beispiel Die Aminosäuren wurden entsprechend den Empfehlungen der IUPAC-IUPKomission für biochemische Nomenklatur mit jeweils einem Buchstaben abgekürzt. VIII Abbildungsverzeichnis Abbildungsverzeichnis: Abb. 1.1 Abb. 1.2 Abb. 1.3 Abb. 1.4 Abb. 1.5 Modell zur Plastiden-Differenzierung PEP-Formen und ihre spezifischen Promotorelemente Sigmafaktoren in Prokaryoten und A. thaliana Regulation der Sigmafaktoren durch Anti-Sigmafaktoren Modell zur pflanzlichen Pathogen-Abwehrantwort Abb. 4.1.1 Expression und Reinigung des rekombinanten AtSIG1-Proteins Bindungsstudien des AtSIG1-Proteins mittels EMSA Deletions- und Punktmutanten des AtSIG1-Proteins CD-Spektren der AtSIG1-Punktmutanten im Vergleich zum AtSIG1-Protein Funktionsstudien der AtSIG1-Mutanten mittels EMSA Expression und Reinigung der Proteine AtSIB1 und AtSIB2 Schematische Darstellung der AtSIG1-Proteinkonstrukte Interaktionsstudien zwischen AtSIG1-Konstrukten und beiden AtSIB-Proteinen mittels Protein-Overlay-Blots Sequenzdarstellung der verwendeten plastidären Promotoren EMSA-Studien mit den rekombinanten AtSIG1- und AtSIBProteinen Sequenzalignment mit Sekundärstrukturanalyse der AtSIBProteine Analyse der Sequenz und der Phylogenie von VQ-Motiv enthaltenden Proteinen aus verschiedenen Pflanzenarten Merkmale der sig1-Knockout-Linie Northern Blot Analysen der sig1 Knockout Linie im Vergleich zum Wildtyp Homozygotie-Nachweis der Doppelmutanten Phänotypen der sig1 und sig6 Einzel- und DoppelKnockout-Mutanten im Vergleich zum Wildtyp Col0 unter verschiedenen Lichtbedingungen Abb. 4.1.2 Abb. 4.2.1 Abb. 4.2.2 Abb. 4.2.3 Abb. 4.3.1 Abb. 4.4.1 Abb. 4.4.2 Abb. 4.5.1 Abb. 4.5.2 Abb. 4.6.1 Abb. 4.6.2 Abb. 4.7.1 Abb. 4.7.2 Abb. 4.8.1 Abb. 4.8.2 Abb. 5.1 Abb. 5.2 Abb. 5.3 Abb. 5.4 Abb. 5.5 Sequenzvergleich des HTH-Sequenzmotives der Subregion 4.2 Modell zur AtSIB-/AtSIG1-Interaktion Modell zu den Funktionen der AtSIB-Proteine im PEPKomplex Einordnung des SIB1-Proteins in den pflanzliche Pathogen-Abwehr Darstellung des SIB-spezifischen VQ-Motives 2 7 9 12 14 39 42 44 45 47 49 51 53 55 57 60 63 65 67 68 69 75 79 81 83 86 IX Genbezeichnungen Genbezeichnungen : atpB Gen für die β-Untereinheit der ATP-Synthase atpE Gen für die ε-Untereinheit der ATP-Synthase psaA Gen für die Untereinheit A des Photosystems I psbA Gen für das D1-Protein des Photosystems II rbcL Gen für die große Untereinheit der RubisCO rpoA Gen für die α- Untereinheit der PEP rpoB Gen für die β-Untereinheit der PEP rpoC1 Gen für die β´-Untereinheit der PEP rpoC2 Gen für die β´´-Untereinheit der PEP rpoTm Gen für die mitochondrial-lokalisierte NEP rpoTp Gen für die plastiden-lokalisierte NEP rpoTmp Gen für die NEP, die in Plastiden und Mitochondrien lokalisiert ist rps16 Gen für das ribosomale Protein S16 trnQ Gen für die tRNA für die Aminosäure Glutamin trnV Gen für die tRNA für die Aminosäure Valin Atsig1-6 Gene für die Sigmafaktor 1-6 aus Arabidopsis thaliana Atsib1 Gen für das Sigma-Bindeprotein 1 aus Arabidopsis thaliana Atsib2 Gen für das Sigma-Bindeprotein 2 aus Arabidopsis thaliana Sasig1 Gen für den Sigmafaktor 1 aus Sinapis alba X Einleitung 1. Einleitung 1.1 Der Plastid: Ursprung, Differenzierung und Plastom Pflanzenzellen enthalten drei gentragende Kompartimente: den Zellkern, Mitochondrien und Plastiden. Letztere sind charakteristisch für pflanzliche Zellen und haben entscheidende Funktionen für das Leben auf der Erde. Ursprung der Plastiden Der Ursprung der Plastiden wird durch die Endosymbiontenhypothese erklärt (Palmer, 2003). Nach dieser – weithin anerkannten – Vorstellung nahm eine bereits kompartimentierte Eukaryotenzelle ein photosynthetisches Bakterium (höchstwahrscheinlich ein Cyanobakterium) durch Endozytose auf (Kuroiwa et al., 2008). Nachdem das Bakterium integriert und nicht aufgelöst wurde, bildeten die beiden Zellen zunächst eine Symbiose. Im Laufe der Zeit wurden bakterielle Gene in den eukaryotischen Kern oder in die Mitochondrien verlagert oder gingen verloren (Martin und Müller, 1998; McFadden, 2001). Die transferierten Gene stehen unter der Kontrolle des eukaryotischen (Kern-)Transkriptionssystems und dadurch hat das ursprünglich „freie“ Bakterium seine vollständige Autonomie verloren und ist zu einem semiautonomen Organell geworden (Reski, 2009). Im „einfachsten“ Fall sind Plastiden von einer Doppel-Membran umgeben, welches auf eine einmalige Endozytose hindeutet. Die betreffende Organismusgruppe – Archaeplastida - basiert wahrscheinlich auf einem gemeinsamen Vorfahr und umfasst die Landpflanzen, Grünalgen (Viridiplantae), Rotalgen und eine kleine Gruppe von Protisten (Glaucophytes) (Adl et al., 2005; Lang et al., 2008). Andere Plastiden, die durch drei (Euglenophyta, einige Dinoflagellaten) oder sogar vier Membranen (Haptophyta, Cryptophyta, Chlorarachnophyta) umhüllt werden, stammen offenbar aus einer zweiten Endosymbiose (Zimmer et al., 2007). Differenzierung der Plastiden Die Plastiden sind eine Gruppe mit großer Formen- und Funktionsvielfalt darunter z.B. Chloroplasten, Etioplasten, Leukoplasten und Chromoplasten. Die Größe kann 1 Einleitung zwischen 2 und 20 µm variieren. Alle Formen gehen aus den Proplastiden oder, durch entsprechende Reize, aus einer anderen reifen Plastidenform hervor. Eine de novo Entstehung von Plastiden ist nicht möglich, obwohl sie über ein eigenes Genom verfügen. Der Proplastid kommt in embryonalen Pflanzenzellen vor und ist eine Vorstufe, die z.B. bei der Eizellbildung nach Degeneration vorhandener, reifer Plastiden entstehen kann. Die Etioplasten entwickeln sich im Dunkeln und sind chlorophyllfrei; unter Lichteinwirkung entstehen aus ihnen Chloroplasten. Chloroplasten enthalten als Orte der Photosynthese neben Chlorophyll auch die anderen Komponenten des Photosyntheseapparates: den Calvin-Zyklus und viele andere Syntheseschritte. Zur Speicherung dienen die Leukoplasten, die sich in Proteinoplasten (Proteinspeicherung), Elaioplasten (Lipidspeicherung) und Amyloplasten (Stärkespeicherung) gliedern und meist in Wurzeln oder Früchten vorkommen. Chromoplasten vermitteln durch einen hohen Gehalt an Carotinoiden eine gelbe bis rötliche Färbung. Dieser Typ kommt nur in Pflanzen mit farbigen Blütenblättern vor. Jede Zelle enthält nur eine Plastidenform. (Kirk und Tilney- Basset, 1978; Reski, 1998; zur Übersicht Abb.1.1). Abb. 1.1: Modell zur Plastiden-Differenzierung Alle Plastiden entwickeln sich aus der Vorstufe des Proplastiden. Jeder reife Plastidentyp kann aus einer anderen Plastidenform differenzieren. Die Pfeile zeigen die Differenzierungsrichtungen an. Genauere Erläuterungen sind im Text gegeben. (nach Kull, 1993; modifiziert) 2 Einleitung Das Plastom Plastiden haben im Laufe der Organellen-Evolution zwar Gene an den Nucleus verloren, besitzen aber trotzdem noch ein eigenes Genom – das Plastom – sowie ein eigenes Genexpressionssystem. Früher wurde das Plastom bakterienähnlich als zirkuläres DNA-Molekül beschrieben (Hermann et al., 1975; Bendich, 1987). Neuere Ergebnisse lassen eher auf eine überwiegend lineare und konzentrierte Form der DNA schließen, die mit der inneren Membran des Plastiden durch PEND („Plastid Envelope DNA-binding Protein“) und andere Proteine verbunden ist (Sato et al. 2003; Wycliffe et al., 2005). Charakteristische Sequenzwiederholungen („inverted repeats“) teilen bei vielen Landpflanzen das Gesamtmolekül in eine kleine und große Einzelkopienregion (Sugiura, 1995), jedoch sind auch Abweichungen von dieser Struktur z.B. bei den Leguminosen gefunden worden (Link, 1996; Stern et al., 1997). Pro Organell können 10 bis 300 Kopien des Plastoms vorliegen (Bendich, 1987; Sugiura, 1992). Das Plastom hat eine Größe zwischen 120 und 160 kbp und enthält zwischen 120 und 135 Gene, welche für Teile des Genexpressions- und Photosynthese-Apparates kodieren (Barbrook et al., 2006). In den Plastiden liegen aber vermutlich 2100 (Arabidopsis) bis 4800 (Reis) Proteine vor (Richly and Leister, 2004), die kernkodiert sind. Diese Genprodukte werden im Cytosol als Vorläuferproteine synthetisiert, über N-terminale Transit-Peptide und die Translokasekomplexe TIC und TOC (translocon at the inner/outer membrane of chloroplasts) in die Plastiden transportiert und dort weiter prozessiert (Jarvis und Soll, 2002). 1.2 Vergleich der pro- und eukaryotischen Transkriptionsinitation Unterschiede zwischen der pro- und eukaryotischen Transkription sind sowohl in der Anzahl der DNA-abhängigen RNA-Polymerasen als auch in der Struktur dieser Komplexe sichtbar. Während Prokaryoten nur über eine einzige DNA-abhängige RNA-Polymerase verfügen, welche die Transkription aller RNA-Arten durchführt, besitzen Eukaryoten im Zellkern mindestens drei verschiedene Isoformen dieses zentralen Enzyms. 3 Einleitung Die bakterielle RNA-Polymerase wird aus den Untereinheiten α2, ω, β und β’ gebildet, welche die Elongation und Termination als „core“-Komplex ausführen. Die Initiation benötigt ein zusätzliches Protein - den Sigmafaktor, welcher konservierte Sequenzmotive - Promotorelemente - spezifisch erkennt und dadurch die promotorspezifische Transkription einleitet. Nach der Promotor-Erkennung durch den holo-Komplex – bestehend aus Sigmafaktor und core-Komplex – wird der DNADoppelstrang aufgeschmolzen und die ersten Nukleotide transkribiert. Wenn der Komplex in die Elongationsphase überwechselt, löst sich der Sigmafaktor vom Polymerase-Komplex und das core setzt die Transkription bis zum Ende fort. Die bakteriellen Promotorelemente liegen im 5‘-Bereich des Gens und zwar als -35Region („TTGACA“) und -10-Region („TATAAT“) stromaufwärts des Transkriptionsstarts (+1) (Murakami und Darst, 2003; Browning und Busby, 2004). Die eukaryotische Transkription im Zellkern führen drei DNA-abhängige RNAPolymerasen durch, wobei jede für die Transkription einer RNA-Art verantwortlich ist. Alle bestehen aus 10 Untereinheiten, die sich in zwei große (140 und 180-220 kDa) und acht kleine (16 bis 40 kDa) Einheiten gliedern. Die RNA-Polymerase I bildet die Vorläufer-rRNAs und Polymerase III hauptsächlich „kleine“ RNAs wie tRNAs oder URNAs. RNA-Polymerase II synthetisiert die Vorläufer-mRNA vor dem SplicingProzess (Kuhlemeier, 1992; Schwechheimer et al., 1998). Für die spezifische Transkriptions-Initiation benötigen diese RNA-Polymerasen neben konservierten Sequenzmotiven auch Polymerase-spezifische Transkriptionsfaktoren. Die Initiation benötigt fünf generelle Faktoren (TFIIB, -D, -E, -F und -H), die jeweils eine Funktion wie das Aufwinden der DNA im Promotor-Bereich vermitteln (Boeger et al., 2005) und für die Elongation freigesetzt werden (Martinez, 2002). Die eukaryotische Transkription wird durch Promotoren eingeleitet, für die meist eine typische AT-reiche Sequenz charakteristisch ist: die TATA-Box. Diese liegt etwa 25 bis 30 bp stromaufwärts des Transkriptionsstartpunktes (+1). Nach Anlagerung des TATA-Bindeproteins und weiterer Transkriptionsfaktoren bildet sich dort der Initiatiatorkomplex aus, zu dem dann die RNA-Polymerase „rekrutiert“ wird. Weitere cis-Elemente regulieren und modulieren die Transkription durch Wechselwirkungen mit DNA-Bindeproteinen positiv (enhancer) oder negativ (silencer) (Roeder, 1996). 4 Einleitung 1.3 Die plastidäre Transkription Die Transkription in den Plastiden ist komplexer als ihre ursprüngliche bakterielle Herkunft erwarten lässt, da sich zwei DNA-abhängige RNA-Polymerasen diese Aufgabe teilen. In Cyanobakterien erfolgt die Transkription trotz ihres größeren Genoms wie bei allen Bakterien durch einen einzigen Protein-Komplex. Ihre „domestizierten“ Nachkommen verfügen neben einem „bakterienähnlichen“ Multiprotein-Komplex noch über ein „phagenähnliches“ Enzym bestehend aus lediglich einem einzelnen Polypeptid. Die Gliederung der plastidären Gene in Klassen basiert auf ihren Promotoreigenschaften: Klasse I Gene werden nur durch den Multiprotein-Komplex transkribiert, Klasse III Gene ausschließlich durch das Einzelpolypeptid-Enzym. Die Gen-Klasse II weist Promotoren beider Polymerasen auf und eine Transkription ist durch beide möglich (nach Hajdukiewicz et al., 1997). DNA-abhängige RNA-Polymerase NEP Die „phagenähnliche“ RNA-Polymerase wird auch als NEP (nuclear encoded polymerase) bezeichnet (Maliga, 1998). Sie besteht aus einer einzigen katalytischen Untereinheit, ist kernkodiert und mit Bakteriophagen-RNA-Polymerase wie T3, T7 oder SP6 verwandt. Für Mitochondrien-Gene ist eine Transkription durch eine kernkodierte, phagenähnliche RNA-Polymerase schon länger bekannt (Masters et al., 1987). Insgesamt sind in Arabidopsis drei Gene für diese Polymerasen (bezeichnet als RpoT-Gene) nachgewiesen worden (Hedtke et al., 2002), deren Lokalisation durch GFP-Importstudien ermittelt wurde. Diese RNA-Polymerasen werden in die Mitochondrien (RpoTm), die Plastiden (RpoTp) und in beide Organellen (RpoTmp) transportiert (Hedtke et al., 1997; Hedtke et al., 2000; Kobayashi et al., 2001a; Kobayashi et al., 2001b; Hedtke et al., 2002; Kobayashi et al., 2002). NEP-typische Promotoren vom Typ I verfügen mindestens über eine sogenannte YRTA-Box (Typ Ia), die durch eine vorgelagerte GAA-Box (Typ Ib) verstärkt werden kann (Kapoor und Sugiura, 1999; Liere et al., 2004; SwiateckaHagenbruch et al., 2007; zur Übersicht siehe Shiina et al., 2005). „Nonconsensus“NEP-Promotoren (Typ II) weisen hingegen keine konservierten Elemente auf (Kapoor et al., 1997; Sriraman et al., 1998). Die höchste Aktivität der NEP liegt in den Proplastiden vor; in ausdifferenzierten Plastiden spielt hingegen die Multiprotein- 5 Einleitung RNA-Polymerase eine zunehmend größere Rolle (Emanuel et al., 2004, Emanuel et al., 2006). DNA-abhängige RNA-Polymerase PEP Die aus den Untereinheiten α2, β, β’ und β’‘ aufgebaute Multiprotein-RNAPolymerase wird als PEP (plastid encoded polymerase) bezeichnet (Maliga, 1998: zur Übersicht Abb.1.2). Diese Untereinheiten formen das „core“ des Enzyms und ihre rpo-Gene (rpoA, rpoB, rpoC1 und rpoC2) werden durch die NEP-Polymerase in den Plastiden transkribiert (Hu und Bogorad, 1990; Hu et al., 1991; Hess und Börner, 1999; Ishizaki et al., 2005). Die spezifische Initiation der Transkription PEPabhängiger Plastidengene wird durch einen kernkodierten, transient-gebundenen Sigmafaktor vermittelt (Murakami und Darst, 2003; Pfalz et al., 2006). Eine Isolierung der PEP ist in zwei verschiedenen Formen möglich: als lösliches Enzym ohne DNA-gebunden zu sein oder als unlöslicher DNA-Protein-Komplex TAC („transcriptionally active chromosome“: Transkriptionsaktives-Chromosom). Das TAC besteht aus DNA, RNA und ungefähr 100 Proteinen, deren Funktionen erst teilweise bekannt sind (Hallick et al., 1976; Briat et al., 1979; Reiss und Link, 1985; Bülow et al., 1987; Igloi und Kössel, 1992; Krause und Krupinska 2000; Pfalz et al., 2006). Die „lösliche“ PEP konnte in zwei Formen aus Senf-Keimlingen isoliert werden (PEPA und PEP-B). Beide unterscheiden sich in ihren biochemischen Eigenschaften in vitro. PEP-B kommt überwiegend in Etioplasten vor und ist ein kleinerer Komplex von 420 kDa, der wie bakterielle Polymerasen durch das Antibiotikum Rifampicin gehemmt wird. In Chloroplasten überwiegt die A-Form, die ein Komplex aus 13 bis 15 Untereinheiten von etwa 750 kDa ist und unempfindlich gegen Rifampicin ist. PEP-A konnte in vitro mittels Phosphorylierung teilweise in die B-Form umgewandelt werden (Pfannschmidt und Link, 1994 und 1997; Pfannschmidt et al., 2000; Ogrzewalla et al., 2002). Die PEP-assozierten Proteine der A-Form sind alle kernkodiert und erfüllen unterschiedliche Aufgaben. Hierzu zählen ein RNABindeprotein (RBP), ein Protein für RNA-Prozessierung, eine Fe-SuperoxidaseDismutase und ein Annexin-ähnliches Protein für die „Entgiftung“ reaktiver Nebenprodukte der Photosynthese sowie die plastidäre Transkriptionskinase PTK (Pfannschmidt et al., 2000; Ogrzewalla et al., 2002; Link, 2003). PEP-Promotoren besitzen ebenso wie die Promotoren vieler prokaryotischer Gene eine -35- und -10-Regionen, welche für die plastidäre Transkription in vitro wie in vivo 6 Einleitung notwendig sind (Link, 1994; Eisermann et al., 1990; Allison und Maliga, 1995; Hess und Börner, 1999). Allerdings zeigt sich eine gewisse Flexibilität: Beispiele dafür sind die „fehlende“ (nicht-konservierte) -10-Region im atpB-Gen von Spinat oder die ebenso fehlende typische -35-Region im rps16-Gen von Senf (Gruissem und Zurawski, 1985; Neuhaus et al., 1989). Andererseits kommt – für Plastiden-DNA eher unerwartet – auch ein sogenanntes TATA-ähnliches Motiv vor. Dieses kann offenbar ein „fehlendes“ Element kompensieren wie beim rps16-Gen oder die Bindungseigenschaften eines „vollständigen“ Promotors verstärken wie beim psbAGen (Link und Langridge, 1984; nach Eisermann et al., 1990). Abb. 1.2: PEP-Formen und ihre spezifischen Promotorelemente Modell der plastidären DNA-abhängigen RNA-Polymerase PEP-Formen A und B. Die basale Form B ist in Etioplasten verbreitet und weist keine der mit PEP-A extraassozierten Proteine auf. Neben dem holo-Enzym bestehend aus den Untereinheiten α, β, β', β'' und dem Sigmafaktor (σ) sind bei der A-Form ein RNA-Bindeprotein (RBP), ein putatives RNA-Prozessierungsprotein (RNA-ProP), ein Annexin-ähnlichesProtein (AlP), eine Fe-Superoxidase-Dismutase (Fe-SOD) und die plastidäre Transkriptionskinase (PTK) mit dem Komplex assoziert. Die kernkodierten Proteine sind gelb dargestellt und die plastidenkodierten Untereinheiten in hellgrün. Der PEPPromotor besteht aus einer -35 und -10-Region. (Zeichnung nach Schweer, Dissertation 2009, modifiziert; PEP-A-/PEP-B-Formen: nach Originaldaten Pfannschmidt et al., 2000; Loschelder et al., 2004). 7 Einleitung 1.4 Sigmafaktoren Sigmafaktoren sind essentiell für die spezifische Promotor-Bindung der RNAPolymerase in Bakterien und Plastiden. Die Faktoren erkennen die typischen prokaryotischen Promotorelemente (Konsensus-Sequenzen): die -35-Region („TTGACA“) und die -10-Region („TATAAT“). Bald nach der Entdeckung des ersten Sigmafaktors überhaupt (Burgess et al., 1969) wurden sehr schnell weitere prokaryotische Transkriptionsfaktoren identifiziert und klassifiziert: mindestens 14 verschiedene Klassen von Sigmafaktoren werden heute unterschieden (Wösten, 1998), basierend auf der Molekülmasse und den spezifischen Promotor-Erkennungssequenzen (Helmann und Chamberlin, 1998; Wösten, 1998). Diese Einordnung wurde durch eine phylogenetische Einteilung der Sigmafaktoren in vier Gruppen ergänzt. Sigmafaktoren der Gruppe I sind essentielle Hauptfaktoren mit einer hohen Sequenzhomologie zu Sigma70. Sigma70 ist der häufigste und am besten charakterisierte „primäre“ Sigmafaktor aus E. coli, der die Transkription der meisten „Haushalts“- und Zellwachstumsgene koordiniert. Die Gruppe II wird aus Sigma70-ähnlichen Proteinen gebildet, die aber nicht essentiell sind und deren Verlust kompensiert werden kann. Typische Stress-Sigmafaktoren wie der Hitzeschockfaktor Sigma32 bilden die Gruppe III und sind eher entfernt mit Sigma70 homolog. Die Synthese der Stressfaktoren erfolgt durch spezifische Signale wie einem Temperaturanstieg bei Sigma32 und führt zur Transkription spezifischer Regulons mit Sigma-spezifischen Promotoren. Die ECF Sigmafaktoren (extracytoplasmatic function) bilden Gruppe IV, die sehr verzweigt und groß ist. ECF Sigmafaktoren werden durch Signale im Periplasma aktiviert (Lonetto et al., 1992; Raivo und Silhavy, 2001; Helmann, 2002; Paget und Helmann, 2003). Der strukturelle Aufbau von Sigmafaktoren besteht nach Sequenzvergleichen aus vier konservierten Regionen, die unterschiedliche Funktionen vermitteln. Die Regionen 2 und 4 weisen die höchste Konservierung auf und vermitteln die core- und DNA-Bindung (Region 2.1), die Erkennung der Promotorelemente (2.4 für die -10Region und 4.2 für die -35-Region) und das Aufschmelzen des DNA-Doppelstranges (Region 2.3). Die Konformation der Region 2 wird durch Region 1.2 stabilisiert. Die Region 1.1 ist nur in prokaryotischen Sigmafaktoren der Gruppen I und II sehr konserviert und ist für die Grundfunktion wohl unnötig (Lonetto et al., 1992; Gruber und Gross, 2003; Zenkin et al., 2007). 8 Einleitung Abb. 1.3: Sigmafaktoren in Prokaryoten und A. thaliana In der Abbildung ist die Struktur von Sigmafaktoren in Prokaryoten und Pflanzen gezeigt. Alle Sigmafaktoren weisen vier konservierte Regionen (1-4) auf, die spezifische Aufgaben in der Transkriptionsinitiation erfüllen. Die Unterschiede zwischen prokaryotischen und pflanzlichen Sigmafaktoren sind die fehlende Region 1.1 und das Vorhandensein eines N-terminalen Transit-Peptides (TP) sowie einer nicht-konservierten Region vor Region 1.2 bei den Pflanzen. In den Grundfunktionen stimmen die bakteriellen und plastidären Sigmafaktoren überein. (Weitere Details im Text). (A: nach Browning und Bushy (2004), modifiziert; B: nach Loschelder, Dissertation 2007) Erste Hinweise auf pflanzliche Sigmafaktoren ergaben sich aus immunologischen Untersuchungen mit Antikörpern gegen E. coli Sigmafaktoren (Lerbs et al., 1988; Troxler et al., 1994) und etwa gleichzeitig wurden bereits die ersten Sigmafaktoren 9 Einleitung aus Senfplastiden isoliert (SLF67, SLF52 und SLF29, Bülow und Link, 1988; Tiller et al., 1991). Trotz prinzipieller Ähnlichkeiten bestehen auch Unterschiede zwischen bakteriellen und pflanzlichen Sigmafaktoren. Hierzu gehöht das regelmäßige Fehlen der Region 1.1 bei Pflanzen, das N-terminal vorhandene plastidäre Transit-Peptid bei den Vorläufern sowie ein – ebenfalls N-terminaler – hoch-variabler Bereich (UKB, nicht-konservierter Bereich) pflanzlicher Sigmafaktoren (Hakimi et al., 2000; Vergleich der Abb. 1.3). In vielen mono- und dikotylen Pflanzen sind weitere Sigmafaktoren identifiziert worden. Der Modellorganismus A. thaliana besitzt sechs dieser Proteine, die als AtSIG1-6 bezeichnet werden (Tanaka et al., 1997; Isono et al., 1997; Kanamura et al., 1999; Fujiwara et al., 2000). Sie werden in die Gruppen I und II der Sigmafaktoren eingeordnet und stellen eine monophyletische Gruppe dar, die offenbar durch Duplikation aus einem oder mehreren Ur-Genen hervorgegangen sind (Shiina et al., 2005; Lysenko, 2006). Die meisten Sigmafaktoren erfüllen spezifische Funktionen und Aufgaben in der plastidären Transkription. So vermittelt z.B. SIG2 die Transkription essentieller Gene der Protein- und Tetrapyrrolsynthese und SIG5 fungiert als Anti-Stressfaktor, der z.B. unter Trocken- oder Redoxstress-Bedingungen sehr aktiv ist (Yao et al., 2003; Tsunoyama et al., 2004; Onda et al., 2008). Der Faktor SIG1 ist am häufigsten und kommt womöglich als „primärer“ Sigmafaktor am ehesten in Frage, zumal bisher keine Daten für eine (zusätzliche) Spezialfunktion für diesen Faktor vorliegen (Privat et al., 2003; Nagashima et al., 2004). 1.5 Regulation der Aktivität von Sigmafaktoren Sigmafaktoren sind Regulatoren, die über ihre Bindungseigenschaften an den Promotor und die core RNA-Polymerase die Effizienz der Transkription bestimmen (Campbell et al., 2009). Wie die Regulation der plastidären Sigmafaktoren selbst erfolgt, ist aber erst in Anfängen untersucht. Reversible Phosphorylierung und Dephosphorylierung ist eine sehr verbreitete posttranslationale Modifikation und als Regulationsmechanismus der Transkription und Translation eingehend belegt. Die Aktivität der plastidären Sigmafaktoren ist abhängig von ihrem Phosphorylierungsgrad, der durch eine PEP-assoziierte Ser/ThrProteinkinase, die plastidäre Transkriptionskinase (PTK), verändert werden kann. In 10 Einleitung Chloroplasten sind die Sigmafaktoren gering phosphoryliert und nur transient mit der core RNA Polymerase verbunden, wodurch der Sigmafaktor schnell abdiffundiert und die Transkription ungehindert in die Elongationsphase überwechselt. Bei stark phosphorylierten Sigmafaktoren wie in Etioplasten liegt eine starke Bindung an das core-Enzym vor und damit ist ein Wechsel in die Elongationsphase nicht möglich (Tiller und Link, 1993a, b). Prokaryotische Sigmafaktoren weisen ein großes Spektrum an Regulationsmöglichkeiten auf. Einerseits zählen dazu Effektor-Moleküle wie das ppGpp (5‘-Diphosphat-Guanosin-3‘-Diphosphat), welches die Promotor-Spezifität des core-Enzym durch Bindung modifiziert. Die Identifikation von homologen, plastidären ppGpp-Synthetasen (RelA und SpoT) in Arabidopsis (van der Biezen et al., 2000), Chlamydomonas reinhardtii (Kasai et al., 2002) und Tabak (Givens et al., 2004) zusammen mit einer Zunahme der ppGpp-Menge unter verschiedenen Stressbedingungen lassen eine Funktion in Plastiden vermuteten. ppGpp hemmt in vitro die PEP-Aktivität; ein spezifischer Nachweis der ppGpp-Funktion steht noch aus (Cashel et al., 1996; Toulokhonov et al., 2001; Givens et al., 2004; Takahashi et al., 2004). Ein weiterer Regulationsmechanismus zur Kontrolle der Sigmafaktor-Aktivität betrifft die Anti-Sigmafaktoren, die in Prokaryoten sehr verbreitet sind. Anti-Sigmafaktoren erkennen spezifisch ihren Sigmafaktor, der durch die Bindung konformations- und funktionsmäßig modifiziert wird. Diese reversible Interaktion verhindert die Bindung an die core-RNA-Polymerase und somit das Erkennen der spezifischen Promotoren (Hughes und Mathee, 1998, zur Übersicht Abb.1.3). Der Anti-Sigmafaktor selbst kann wiederum durch Anti-Anti-Sigmafaktoren deaktiviert werden, das durch Modifikationen oder Export aus der Zelle erfolgen kann. Zwei Beispiele für die unterschiedlichen Aufgaben der Anti-Sigmafaktoren in Prokaryoten seien hier kurz vorgestellt: der Anti-Sigmafaktor SpoIIAB in der Sporulation von Bacillus subtilis und der Anti-Sigmafaktor AsiA bei der T4Bakteriophagen-Infektion von E. coli. 11 Einleitung Abb. 1.4: Regulation der Sigmafaktoren durch Anti-Sigmafaktoren Die obere Abbildung zeigt die Auswirkungen der reversiblen Interaktion zwischen Sigmafaktoren (σ) und ihren spezifischen Anti-Sigmafaktoren. Diese Regulation ist in Bakterien sehr verbreitet und eine Wieder-Aktivierung des Sigmafaktors ist durch Bindung eines Anti-Anti-Sigmafaktors an den Anti-Sigmafaktor möglich. Genauere Erläuterungen und Beispiele für Regulationen sind im Text gegeben. (nach Hughes und Mathee, 1998; verändert und vereinfacht) Die Sporulation bei Bacillus subtilis wird durch Stresssignale wie Nährstoffmangel in Form einer Kaskade bestehend aus vier verschiedenen Sigmafaktoren (σF, σE, σK und σG) aktiviert. Unter optimalen Lebensbedingungen sind die Sigmafaktoren σF und σG an den Anti-Sigmafaktor SpoIIAB gebunden und liegen inaktiv vor. Eine Aktivierung dieser Sigmafaktoren erfolgt, wenn der Komplex aus Sigmafaktor und Anti-Sigmafaktor SpoIIAB durch den Anti-Anti-Sigmafaktor SpoIIAA gebunden und der Sigmafaktor freigesetzt wird. Die Sigmafaktoren σF und σG sind dann aktiv und können die Transkription für ihre spezifischen Operons vermitteln. Eine andere „Strategie“ wird beim Anti-Sigmafaktor AsiA sichtbar, der spezifisch den E. coli Sigma70 an Region 4.2 im holo-Komplex während einer Infektion mit dem Bakteriophagen T4 bindet: die AsiA-Bindung hemmt nicht nur die Funktion von Sigma70, sondern verändert auch die Struktur der Region 4. Die konservierte Region 4 vermittelt die Erkennung der -35-Region durch ein Helix-Turn-Helix-Strukturmotiv, 12 Einleitung welches durch die AsiA-Bindung zu einer durchgängigen Helix modifiziert wird. Die AsiA-gebundene holo-RNA-Polymerase wird für die Transkription von T4-Genen nachfolgend durch das T4-kodierte Bindeprotein MotA rekrutiert (zur Übersicht Campbell et al., 2009; Colland et al., 1998; Severinova et al., 1998; Baxter et al., 2006). Die Funktionen der Anti-Sigmafaktoren sind vielfältig und regulieren in den Prokaryoten die Aktivität ihrer spezifischen Sigmafaktoren je nach Bedarf der Zelle und der Umgebung. Gleichzeitig stellen sie selbst ebenfalls auch Angriffspunkte für Regulationen dar wie oben für SpoIIAB bzw. AsiA ausgeführt. Als erste potentielle Kandidaten für eine Rolle als pflanzliche Anti-Sigmafaktoren können das SigmaBindeprotein 1 und das „Genprodukt T3K9.5“ gelten, die AsiA-ähnlich Region 4 des SIG1 von Arabidopsis binden können (Morikawa et al., 2002). Ein konkreter Nachweis funktioneller Konsequenzen dieser Interaktionen fehlte für beide pflanzlichen „Anti-Sigmafaktoren“ jedoch bisher. 1.6 Interaktionen zwischen Pflanzen und Phytopathogenen Neben Herbivoren müssen sich Pflanzen auch gegen pflanzliche Pathogene wie Agrobacterium tumefaciens oder Pseudomonas syringae verteidigen. P. syringae infiziert die Pflanzenzellen durch die Injektion von Effektor-Proteinen wie AvrPtoB. Die Funktion der Effektor-Proteine ist die Unterdrückung der pflanzlichen Immunantwort, um eine fortgesetzte Vermehrung der Bakterien in den Apoplasten der Pflanzenzelle zu ermöglichen. Vermutlich werden durch diese Proteine auch Nährstoffe in den Apoplasten freigesetzt, welche P. syringae für die Vermehrung benötigt (Alfano und Collmer, 1996; Mudgett, 2005). Für die Unterdrückung der pflanzlichen Antwort inaktivieren die Effektor-Proteine entscheidende Enzyme der pflanzlichen Abwehr-Kaskade oder erhöhen die Expression der Phytohormone Abscisinsäure und Ethylen, welche die Antagonisten der Salizylsäure-vermittelten Antwort und damit des „freiwilligen Zelltodes“ sind (Munkvold und Martin, 2009). Die Abwehr der Pflanzenzellen beginnt mit der Erkennung von pathogenspezifischen, kleinen Molekülen, die als PAMPs (pathogen (or microbe)-associated molecular patterns) bezeichnet werden, durch spezifische Rezeptoren (Pattern recognition receptors: PRRs). Dies löst die PTI-Antwort (PAMP-triggered immunity) 13 Einleitung mit einer nachfolgenden MAPK-Kaskade und einer Aktivierung spezifischer Transkriptionsfaktoren aus (Jones und Dangl, 2006; Chisholm et al., 2006). Durch die Transkriptionsfaktoren findet eine erhöhte Expression von anti-mikrobiellen Komponenten wie ROS (reactive oxygen species), PR-Proteine (pathogenesis related) wie Glucanasen und Phytoalexinen statt (Gómez-Gómez et al., 1999). Das Ende der pflanzlichen Abwehr-Antwort stellt die HR-Antwort (hypersensitive response) dar, welche eine Art programmierter Zelltod der infizierten Pflanzenzelle ist und dem Pathogen seine Wachstumsgrundlage nimmt (zur Übersicht Abb.1.5). Eine Infektion benachbarter Pflanzenzellen wird durch die systemisch erworbene Resistenz (systemic aquired resistance, SAR) verhindert, da durch die PTI-Antwort die Abwehr-Kaskade in allen Pflanzenzellen aktiviert wurde (Pitzschke et al., 2009). Abb. 1.5: Modell zur pflanzlichen Pathogen-Abwehrantwort Das Schema spiegelt die ersten Reaktionsschritte der pflanzlichen PathogenAbwehrantwort dar, welche mit Erkennung der Pathogen-spezifischen PAMPs beginnt und über eine MAPK-Kaskade in der Expression von Abwehr-Proteinen endet. Diese leiten die weiteren Schritte der pflanzlichen Abwehr ein, die mit der HRAntwort („programmierter Zelltod“) endet. (Erläuterungen siehe Text). (nach Pitzschke et al., 2009, modifiziert). 14 Einleitung 1.7 Protein-Protein-Interaktionen Protein-Protein-Interaktionen – vor allem in Protein-Komplexen - sind für fast alle biologischen Stoffwechselwege und Funktionen verantwortlich und kommen in Pround Eukaryoten vor. Dabei können (rein) physische und funktionelle Interaktionen unterschieden werden. Physische Protein-Protein-Interaktionen müssen weder stabil noch direkt sein. Das bedeutet, dass diese Interaktionen nur transient und/oder indirekt sein können. Eine indirekte Protein-Protein-Interaktion ist denkbar, wenn zwei Proteine ein gemeinsames Substrat haben oder wenn zwei Komponenten eines MultiProteinkomplexes nur durch die Existenz einer weiteren gehaltenen werden. Eine Aussage über die Aufgaben und die Funktionen solcher Protein-Protein-Interaktion wird durch physische Protein-Protein-Interaktionen nicht getroffen. Trotzdem bilden diese Interaktionen entscheidende Grundlagen für die Beschreibung von Signalketten, Protein-Netzwerken und den Aufbau von Übersichtskarten „globaler“ Protein-Protein-Interaktionen eines Systems, welches als Interactomics bezeichnet wird. Zur Bildung der Netzwerk-Übersichten bzw. Interaktome wird auf ProteinDatenbanken und spezifische Vorhersage-Programme wie STRING für Netzwerke zurückgegriffen (Alberts, 1998; Jensen et al., 2009). Für unterschiedliche Organismen wurden schon theoretische Karten von allen Protein-Protein- Interaktionen auf Basis der sequenzierten Genome berechnet. Beispiele dafür sind Arabidopsis, Saccharomyces cerevisiae, Caenorhabditis elegans und das HumanGenom (Geisler-Lee et al., 2007; Uetz et al., 2000; Giot et al., 2003; Li et al, 2004; Miller et al., 2005; Rual et al., 2005; Gandhi et al., 2006). Die Möglichkeit, statt ganzer Organismen nur Netzwerke komplexer Stoffwechselwege oder für Organellen wie Chloroplasten berechnen zu lassen, besteht ebenfalls. In die Vorhersagen werden aber oft auch hypothetische Proteine ohne nachgewiesene Funktionen einbezogen, deren Expression und Funktion experimentell noch bewiesen werden muss (Baginsky und Gruissem, 2004). Eine funktionelle Protein-Protein-Interaktion bezeichnet Interaktionen, die neben einer physischen Interaktion auch zu einer funktionellen Veränderung führen. Dazu zählen posttranslationale Modifikationen, welche Änderungen der Aktivität oder der Konformation mindestens eines der interagierenden Proteine verursachen. Dies bildet die Grundlage der meisten Signal- und Stoffwechselwege aller zellulären 15 Einleitung Prozesse. Beispiele stellen die MAP-Kinase-Signalkette sowie die PathogenAbwehrmechanismen dar (Pitzschke et al., 2009). Die Interaktionen der Sigmafaktoren sind ebenfalls Komponenten eines regulatorischen Netzwerkes von Protein-Protein-Interaktionen. Die Regulation der Sigma-Aktivität beginnt mit der Translation der Vorläufer-Proteine und wird durch modifizierende Protein-Protein-Interaktionen an den „reifen“ Proteinen ergänzt. Dabei interagieren die Sigmafaktoren in den Plastiden mindestens mit der core-RNAPolymerase und modifizieren diesen Komplex im Hinblick auf eine spezifische Transkriptionsinitiation. Die Regulation und Modulation der Sigmafaktor-Aktivität durch Protein-Protein-Interaktion in Plastiden ist dagegen weitgehend unbekannt. Die vorliegende Arbeit soll eine Basis für eine systematische Analyse der ProteinProtein-Interaktionen schaffen, indem die Protein-Protein-Interaktionen von dem Sigmafaktor AtSIG1 und seinen spezifischen Interaktoren genauer analysiert werden. 16 Zielsetzung 2. Zielsetzung In A. thaliana wurden sechs Sigmafaktoren identifiziert, die gemeinsam mit der plastidären PEP-RNA-Polymerase an der Transkription der Plastiden-Gene beteiligt sind. Spezifische Gen- und Proteinfunktionen sind für die Sigmafaktoren 2 bis 6 schon recht gut bekannt. Dies wird beispielsweise an der besonders eingehend untersuchten entwicklungs- und genspezifischen Rolle des Sigmafaktors 6, die in der Arbeitsgruppe umfassend charakterisiert wurde und sein Einfluss auf die plastidäre Transkription weiterhin untersucht wird (Loschelder et al., 2006; Schweer et al., 2006; Schweer et al., 2010). Eine solche Charakterisierung fehlt bisher noch für den Sigmafaktor 1 (AtSIG1), der zwar relativ häufig (abundant) ist und deshalb sogar als möglicher „Hauptsigmafaktor“ interpretiert wurde (Shiina et al., 2005), aber in Mutantenlinien keinen oder nur wenig sichtbaren Phänotypen hinterlässt (Privat et al., 2003). Das Interesse gerade an diesem Faktor ist auch deshalb groß, weil es (nur hier) vor einigen Jahren gelungen war, zwei Interaktionsproteine SIB1 und SIB2 (sigma factor binding proteins 1 und 2) zu identifizieren; allerdings blieb die genaue Rolle dieser beiden Interaktoren bisher unbekannt (Morikawa et al., 2002). Eine experimentelle Beantwortung dieser Teilfragen könnte mithelfen, ein neues Verständnis der plastidären Gen-Regulation durch ("Anti")Sigma-Interaktionsproteine zu erzielen. Die Gene (AT2G41180 für AtSIB2 und AT3G56710 für AtSIB1) der Interaktoren sind kernlokalisert und die Genprodukte werden wie auch die Sigmafaktoren posttranslational in die Plastiden transportiert (Morikawa et al., 2002). An diese Ergebnisse von Vorarbeiten knüpft nun die vorliegende Arbeit direkt an: Es geht um regulatorische Protein-Protein-Interaktionen in der plastidären Transkription. Im Fokus der Arbeit steht vor allem die Interaktion zwischen dem AtSIG1 und den beiden AtSIB-Proteinen. Unter Beachtung der folgenden Aspekte: • Hat das rekombinante AtSIG1-Protein ein ähnliches Promotorspektrum wie das bereits untersuchte homologe SaSIG1-Protein Sinapis alba (Homann und Link, 2003) 17 Zielsetzung • Wie wirken sich Mutationen einzelner Aminosäuren in funktions-wichtigen Regionen des AtSIG1-Proteins auf die Funktion als Sigmafaktor aus? • Worin besteht die Funktion der Interaktion aus AtSIB- und AtSIG1-Proteinen und welche direkte Folge hat diese Interaktion auf die Promotor-Bindung? Wie äußert sich die Regulation des AtSIG1 durch die AtSIB-Interaktion? • Die Sequenzen der AtSIB-Gene bzw. -Proteine liegen inzwischen komplett vor: Gibt es spezifische Sequenzmotive, die für die Interaktion mit AtSIG1 verantwortlich sein könnten? Ist das Motiv konserviert? Wie ist die Verbreitung dieses Sequenzmotives und gibt es SIB-ähnliche Proteine? • Bisherige Ergebnisse sprechen für eine Rolle der konservierten Region 4 von AtSIG1 bei der Interaktion mit SIB1/SIB2. Haben auch andere Region(en) einen Einfluss? • Wie äußert sich das Fehlen des AtSIG1-Proteins in einer sig1-KnockoutPflanze und einer sig6xsig1-Doppel-Knockout-Pflanzen? Gibt es Unterschiede in der Ausprägung des Phänotyps in verschiedenen Entwicklungsphasen und Anzuchtbedingungen? 18 Material und Methoden 3. Material und Methoden 3.1 Material 3.1.1 Chemikalien und Enzyme Hersteller: Material: Amersham: positiv geladene Nylonmembran Hybond™-N+ Biomol: Dialyse-Membranen Typ 8 und 20, X-Gal [5-Brom-4-Chlor-3-Indolyl-βD-Galaktosid] BioRad: Precission Protein Standards All Blue Epicentre: E. coli RNA-Polymerase “core” (bestehend aus α2ββ’) und E. coli RNA-Polymerase “holo” (bestehend aus α2ββ’ und σ70 im Überschuss), jeweils 1U/µl GE Healthcare: Bulk GST Purification Modules, Anti-GST-Antikörper Gerbu: Bis-Tris [Bis-(2-Hydoxyethyl)amino-tris (hydroxymethyl) –methan], Isopropyl-β,D-thiogalactopyranosid, Nitroblue Tetrazolium, Chloramphenicol, Sulfadiazin Hartmann Analytics: [γ32P-]ATP Invitrogen: DnaseI, Lambda-DNA Machery & Nitrocellulose-Membran, NucleoSpinPlasmid, NucleoBondPlasmid, Nagel: GSH-Sepharose Merck: Proteinase K New England T4-Polynukleotidkinase, Anti-MBP-Antikörper, pMAL™ Protein Fusion Biolabs: and Purification System Promega: Restriktionsendonukleasen, T4-DNA-Ligase, Dithiothreitol, GoTaq®HotStart-Polymerase, pGEM®-T Easy Vector Systems, 19 Material und Methoden Magnesiumchlorid, AMV Reverse Transkriptase, Recombinant RNasin® Ribonuclease Inhibitor, Pfu-Turbo-Polymerase, TSAP, Sp6/T7-/T3-RNA-Polymerase, RNA Marker Qiagen: QIAquick Gel Extraction Kit, DNeasy Plant Mini Kit, PCR Purification Kit Roche: Lysozym, Anti-Digoxigenin-AP, Blockierungsreagenz, CDP-Star, DIGUTP, Random Primed DNA Labeling Kit Roth: Rotiphorese 40 und 30 (Acrylamid/Bisacrylamid in 40% bzw. 30% (w/v), Desoxynukleosidtriphosphate, Random Primer, BCIP, Magermilchpulver, Roti®-Phenol, SDS, TEMED, Tween20, NLauroylsarkosin Serva: Agarose, Bromphenolblau Sigma: Ammoniumpersulfat, Ampicillin, β-Mercaptoethanol, BSA, RNase A, sekundäre Antikörper mit alkaliner Phosphatase, Ethidiumbromid QuickChange® Site-Directed Mutagenesis Kit Stratagene: Alle nicht gesondert aufgeführten Standard-Chemikalien wurden in jeweils benötigten Labor-Reinheitsgraden von ausgewiesenen Herstellerfirmen bezogen. 3.1.2 Nährmedien 3.1.2.1 Nährmedien für die E. coli-Kultivierung LB-Medium: 1% Pepton, 0,5% Hefe-Extrakt, 0,5 % NaCl LBamp-Medium: LB-Medium enthält 50 mg/l Ampicillin LBamp+cam-Medium: LB-Medium enthält jeweils 50 mg/l Ampicillin und Chloramphenicol Agar-Platten: 15 mg/ml Bacto-Agar im entsprechenden Nährmedium 3.1.2.2 Nährmedien für die Anzucht von A. thaliana MS-Medium: je 1 % Makroelemente, 1 % Mikroelemente, Eisen-Quelle, Saccharose, mit einem pH von 5,6-5,7 20 Material und Methoden MS-Medium mit 0,5 % (w/v) Gelrite und Zusatz von MS-Platten: Vitaminen MSsulf-Platten: 3.1.3 MS-Platten mit 11,25 µg/ml Sulfadiazin Linien von A. thaliana Pflanze Arabidopsis thaliana L Heynh. Cv Columbia Arabidopsis thaliana sig6 Mutante Arabidopsis thaliana sig1 Mutante Genotyp Referenz NASC - European Arabidopsis Stock Centre T-DNA Insertion in Exon 6 Linie 242G06 des AtSig6 Gens GABI-Kat, MPI Köln (AT2G36990) Rosso et al., 2003 T-DNA Insertion in Exon 8 Linie 758B02 des AtSig1 Gens GABI-Kat, MPI Köln (AT1G64860) Rosso et al., 2003 T-DNA Insertionen in den Arabidopsis thaliana Genen Atsig1 sig1xsig6 Mutante (AT1G64860) und Atsig6 Kreuzung der sig1 und sig6 Mutante diese Arbeit (AT2G36990) T-DNA Insertionen in den Arabidopsis thaliana Genen Atsig6 sig6xsig1 Mutante (AT2G36990) und Atsig1 Kreuzung der sig6 und sig1 Mutante (AT1G64860) 3.1.4 diese Arbeit Bakterienstämme von E. coli DH5α: E. coli K12 deoR, endA1, gyrA96, usdR17 (rk- mk-), recA1, supA1, supE44, thi-1, Δ(lacZYA-argFV169), Φ80lacZ ΔM15, F- (Stratagene) BL21-CodonPlus-RIL: E. coli B F-, ompT, hsdS(rB- mB-), dcm+, Tetr, gal, endA, Hte [argU ileY leuW Camr] (Statagene) 21 Material und Methoden XL1blue: E. coli K12 recA1, endA1, gyrA96, thi-1, hsdR17, supE44, relA1, lac [(F- proAB lacIqZ ΔM15 Tn10(TetR)] (Stratagene) 3.1.5 Oligonukleotide Die aufgeführten Oligonukleotide wurden durch die Firma Eurofins MWG Synthesis GmbH synthetisiert. Oligonukleotide für die Homozygotie-Prüfung: Sig6-HO1 5‘ CCACTCGCCTATTGTTGGTT 3‘ Sig6-HO2 5‘ GGAGAGGAGGCAGTTTGATG 3‘ SIG1-HO1 5‘ GTTTCCATGTGTCTCTCCTC 3‘ SIG1-HO2 5‘ CTCACCCTCTCTCTCGATAA 3‘ AtSIG1-Oligonukleotide AtSIG1-TP-F 5‘ GAATTCGAATTCGCTTCTACTGAGAAGCCATGGCTA 3‘ AtSIG1-TP-R 5‘ CTGCAGGGATCCTCAATTCTTAAGGATCATTG 3‘ SIG1AS81-215-R 5‘ CTGCAGGGATCCACCAGATTTGATTTTC 3‘ SIG1AS257-340-F 5‘ GAATTCGAATTCCATCTAGCTAGAGAGAAG 3‘ SIG1AS257-340-R 5‘ CTGCAG GGATCCTAGGTGAGTAGGTAACCT 3‘ SIG1AS358-413-F 5‘ GAATTCGAATTCGGAATCACACCCTCCATTGAT 3‘ SIG1AS358-413-R 5‘ CTGCAGGGATCCAGTATCCGCAATGTAACT 3‘ SIG1AS438-502-F 5‘ GAATTCGAATTCGCAACACTCGGAGAACGG 3‘ 321Q/H: 5‘ TATATTGGTGGATTCGACACGGTGTCTCAAGAGCACTAG3‘ 5‘ CTAGTGCTCTTGAGACACCGTGTCGAATCCACCAATATA3‘ 458C/S: 5‘ GGTCTAGACAAAGAAAGTCTCACATGGGAAGACATTAG 3‘ 5‘ GCTAATGTCTTCCCATGTGAGACTTTCTTTGTCTAGACC 3‘ 460T/A: 5’GTCTAGACAAAGAATGTCTCGCATGGGAAGACATTAGTAAACG 3‘ 5‘ CGTTTACTAATGTCTTCCCATGCGAGACATTCTTTGTCTAGAC 3‘ 22 Material und Methoden 463D/A: 5’GACAAAGAATGTCTCACATGGGAAGCAATTAGTAAACGGATAGG 3‘ 5‘ CCTATCCGTTTACTAATTGCTTCCCATGTGAGACATTCTTTGTC 3‘ 465S/A: 5‘ GTCTCACATGGGAAGACATTGCTAAACGGATAGGATTATCGAGAG 3‘ 5‘ CTCTCGATAATCCTATCCGTTTAGCAATGTCTTCCCATGTGAGAC 3‘ 467R/A: 5’ GGGAAGACATTAGTAAAGCGATAGGATTATCGAGAGAGAGGG 3’ 5’ CCCTCTCTCTCGATAATCCTATCGCTTTACTAATGTCTTCCC 3’ 471S/A: 5’ GTAAACGGATAGGATTAGCGAGAGAGAGGGTGAG 3’ 5’ CTCACCCTCTCTCTCGCTAATCCTATCCGTTTAC 3’ 476R/H: 5‘ TCGAGAGAGAGGGTGCACCAGGTAGGGCTTGTGGCAC 3‘ 5‘ GTGCCACAAGCCCTACCTGGTGCACCCTCTCTCTCGA 3‘ AtSIB1-Oligonukleotide AtSIB1-F 5‘ GAATTCGAATTCATGGAGTCATCATCGTCGACT 3‘ AtSIB1-R 5‘ CTGCAGCTCGAGTCACATAGAATCGATGCTTCC 3‘ AtSIB2-Oligonukleotide AtSIB2-F 5‘ GAATTCGAATTCATGGATCAGTCATCATCAACGTTGC 3‘ AtSIB2-R 5‘ CTGCAGCTCGAGTCAGAGAGAACCAATGCTTCC 3‘ Promotor-Oligonukleotide: rbcL-1 5‘ GTACTGCAGGTACCGGACCAATGATTTG 3‘ rbcL-2 5‘ GAGAAGCTTAAGATATCAGTATCCTTGGT 3‘ rps16-F 5‘ GTCGTACGTTGCTTTCTACC 3‘ rps16-R 5‘ CGGGTAAGTAAGGTAGAACC 3‘ Vektor-Oligonukleotide: pBS: T3: 5’ AAT TAA CCC TCA CTA AAG GG 3’ T7: 5’ TAA TAC GAC TCA CTA TAG GGC 3’ 23 Material und Methoden pGEM: pMAL: pGEX: 3.1.6 T7: 5‘ TAA TAC GAC TCA CTA TAG GG 3‘ Sp6: 5‘ ATT CTA TAG TGT CAC CTA AAT 3’ Primer1: 5’ CTA ACC ATC TTC AGG AGG CGC GCA 3’ Primer2: 3’ GGA GGA TAG AGC GGG CTA ATT CGA 5’ 5’-Primer: 5’ GGG CTG GCA AGC CAC GTT TGG TG 3’ 3’-Primer: 5’ CCG GGA GCT GCA TGT GTC AGA GG 3’ Plasmide Plasmide ohne Angabe einer Bezugsquelle wurden im Rahmen dieser Arbeit selbst erstellt. pBSIIKS (+), pBSIISK (+): Klonierungsvektor, der die „Blau/Weiss“-Selektion und die in vitro-Transkription klonierter DNA (mittels T3- und T7-RNA-Polymerase) ermöglicht (Stratagene). pGEM-T-easy: Klonierungsvektor, der eine vereinfachte Klonierung von amplifzierten Fragmenten mit „Blau/Weiss“-Selektion und Taq-Polymerase in vitro-Transkription klonierter DNA (mittels T7- und Sp6-RNA-Polymerase) ermöglicht (Promega). pMAL-c4x: Vektor zur heterologen Überexpression von Proteinen mit N-terminal-fusioniertem Maltosebindeprotein in E. coli (New England Biolabs). pGEX-6P-1: Vektor zur heterologen Überexpression von Proteinen mit N-terminalen GSTFusionsproteinen (GE Healthcare/ Amersham Biosciences). pBS/Sig1-TP: Aus cDNA wurde das sig1-Gen aus A. thaliana ohne Transit-Peptid mittels PCR (Oligonukleotid-Paar AtSIG1-TP) amplifiziert und unter Verwendung der Restriktionsschnittstellen EcoRI und BamHI in den Vektor pBSIIKS kloniert. 24 Material und Methoden pMAL/Sig1-TP: Das Fragment aus dem Klon pBS/Sig1-TP wurde mittels der Restriktionsschnittstellen EcoRI und BamHI in den Vektor pMAL-c4x kloniert. pBS/Sig1AS81-215: Aus dem Plasmid pBS/Sig1-TP wurde unter Verwendung des Primerpaares AtSIG1TP-F und SIG1AS81-215-R die konservierte Region 1 des sig1-Gens von A. thaliana amplifiziert und mittels EcoRI und BamHI in den Vektor pBSIIKS kloniert. pMAL/Sig1AS81-215: Das Fragment aus dem Klon pBS/Sig1AS81-215 wurde mittels der Restriktionsschnittstellen EcoRI und BamHI in den Vektor pMAL-c4x kloniert. pBS/Sig1AS257-340: Aus dem Plasmid pBS/Sig1-TP wurde unter Verwendung des Primerpaares SIG1AS257-340 die konservierte Region 2 des sig1-Gens von A. thaliana amplifiziert und mittels EcoRI und BamHI in den Vektor pBSIIKS kloniert. pMAL/Sig1AS257-340: Das Fragment aus dem Klon pBS/Sig1AS257-340 wurde mittels der Restriktionsschnittstellen EcoRI und BamHI in den Vektor pMAL-c4x kloniert. pBS/Sig1AS358-413: Aus dem Plasmid pBS/Sig1-TP wurde unter Verwendung des Primerpaares SIG1AS358-413 die konservierte Region 3 des sig1-Gens von A. thaliana amplifiziert und mittels EcoRI und BamHI in den Vektor pBSIIKS kloniert. pMAL/Sig1AS358-413: Das Fragment aus dem Klon pBS/Sig1AS358-413 wurde mittels der Restriktionsschnittstellen EcoRI und BamHI in den Vektor pMAL-c4x kloniert. 25 Material und Methoden pBS/Sig1AS438-502: Aus dem Plasmid pBS/Sig1-TP wurde unter Verwendung des Primerpaares SIG1AS438-502-F und AtSIG1-TP-R die konservierte Region 4 des sig1-Gens von A. thaliana amplifiziert und mittels EcoRI und BamHI in den Vektor pBSIIKS kloniert. pMAL/Sig1AS438-502: Das Fragment aus dem Klon pBS/Sig1AS438-502 wurde mittels der Restriktionsschnittstellen EcoRI und BamHI in den Vektor pMAL-c4x kloniert. pBS/Sig1AS81-413: Aus dem Plasmid pBS/Sig1-TP wurden unter Verwendung des Primerpaares AtSIG1-TP-F und SIG1-AS358-413-R die konservierten Regionen 1 bis 3 des sig1Gens von A. thaliana amplifiziert und mittels EcoRI und BamHI in den Vektor pBSIIKS kloniert. pMAL/ Sig1AS81-413: Das Fragment aus dem Klon pBS/Sig1AS81-413 wurde mittels der Restriktionsschnittstellen EcoRI und BamHI in den Vektor pMAL-c4x kloniert. pBS/321Q/H: Der Klon pBS/Sig1-TP wurde als Grundlage für eine ortsgerichtete Mutagenese unter Verwendung des Primerpaares 321Q/H verwendet. Die Mutation führt an der Aminosäureposition 321 zu einem Austausch der Aminosäure Glutamin gegen Histidin. pMAL/321Q/H: Ausgehend von Klon pBS/321Q/H wurde das mutierte Sig1-Gen mittels EcoRI und BamHI in den Vektor pMAL-c4x kloniert. pBS/458C/S: Ausgehend von Klon pBS/Sig1-TP wurde mittels ortsgerichteter Mutagenese (Primerpaar 458C/S) die Aminosäure Cystein an Position 458 gegen ein Serin ersetzt. 26 Material und Methoden pMAL/458C/S: Das mutierte Sig1-Gen aus Klon pBS/458C/S wurde mittels EcoRI und BamHI in den Vektor pMAL-c4x kloniert. pMAL/460T/A: Im sig1-Gen wurde mittels ortsgerichteter Mutagenese (Primerpaar 460T/A) die Aminosäure Threonin an Position 460 gegen ein Alanin ersetzt. Das mutierte Fragment wurde über EcoRI und BamHI in den Vektor pMAL-c4x kloniert (Husic, Bachelorarbeit 2009). pMAL/463D/A: Im sig1-Gen wurde mittels ortsgerichteter Mutagenese (Primerpaar 463D/A) die Aminosäure Asparaginsäure an Position 463 gegen ein Alanin ersetzt. Das mutierte Fragment wurde über EcoRI und BamHI in den Vektor pMAL-c4x kloniert (Husic, Bachelorarbeit 2009). pMAL/465S/A: Im sig1-Gen wurde mittels ortsgerichteter Mutagenese (Primerpaar 465S/A) die Aminosäure Serin an Position 465 gegen ein Alanin ersetzt. Das mutierte Fragment wurde über EcoRI und BamHI in den Vektor pMAL-c4x kloniert (Husic, Bachelorarbeit 2009). pMAL/467R/A: Im sig1-Gen wurde mittels ortsgerichteter Mutagenese (Primerpaar 467R/A) die Aminosäure Arginin an Position 467 gegen ein Alanin ersetzt. Das mutierte Fragment wurde über EcoRI und BamHI in den Vektor pMAL-c4x kloniert (Husic, Bachelorarbeit 2009). pBS/471S/A: Ausgehend von Klon pBS/Sig1-TP wurde mittels ortsgerichteter Mutagenese (Primerpaar 471S/A) die Aminosäure Serin an Position 471 gegen ein Alanin ersetzt (Husic, Bachelorarbeit 2009). 27 Material und Methoden pMAL/471S/A: Das mutierte sig1-Gen aus Klon pBS/471S/A wurde mittels EcoRI und BamHI in den Vektor pMAL-c4x kloniert. pBS/476R/H: Ausgehend von Klon pBS/Sig1-TP wurde mittels ortsgerichteter Mutagenese (Primerpaar 476R/H) die Aminosäure Arginin an Position 476 gegen ein Histidin ersetzt. pMAL/476R/H: Das mutierte sig1-Gen aus Klon pBS/476R/H wurde mittels EcoRI und BamHI in den Vektor pMAL-c4x kloniert (Husic, Bachelorarbeit 2009). pBS/Sib1: Das sib1-Gen aus A. thaliana wurde mittels PCR (Oligonukleotid-Paar AtSIB1) aus cDNA amplifiziert und unter Verwendung der Restriktionsschnittstellen EcoRI und XhoI in den Vektor pBSIIKS kloniert. pGEX/Sib1: Das Fragment aus dem Klon pBS/Sib1 wurde mittels der Restriktionsschnittstellen EcoRI und XhoI in den Vektor pGEX-6P-1 kloniert. pBS/Sib2: Das Gen T3K9.5 bzw. sib2 aus A. thaliana wurde mittels PCR (Oligonukleotid-Paar AtSIB2) aus cDNA amplifiziert und unter Verwendung der Restriktionsschnittstellen EcoRI und XhoI in den Vektor pBSIIKS kloniert. pGEX/Sib2: Das Fragment aus dem Klon pBS/Sib2 wurde mittels der Restriktionsschnittstellen EcoRI und XhoI in den Vektor pGEX-6P-1 kloniert. pGEM/rbcL: Vektor pGEM mit einem 1 kb großen Insert, das den rbcL-Promotor trägt (Hanaoka et al., 2003). 28 Material und Methoden pGEM/NrbcL: Vektor pGEM mit einem Teil der cDNA des rbcL-Gens, wird als Northern Blot-Sonde verwendet (Kollektion der Arbeitsgruppe). pGEM/atpB: Vektor pGEM mit einem Insert entsprechend der mRNA des atpB-Gens, wird als Northern Blot-Sonde verwendet (Schweer et al., 2006). pGEM/atpE: Das Plasmid pGEM trägt ein Insert, das als Northern Blot-Sonde gegen die mRNA von atpE eingesetzt werden kann (Schweer et al., 2006). pGEM/trnV: Das Plasmid pGEM trägt ein Insert, das als Northern Blot-Sonde gegen die mRNA von trnV eingesetzt werden kann (Schweer et al., 2006). pSPT-452a: Das enthaltene Insert kann als Northern Blot-Sonde gegen das Transkript des psbAGens genutzt werden (Liere et al., 1995). pBS/Hinf120: Das enthaltene Insert enthält den Promotor des psbA-Gens. pBS/psaA: Das Plasmid trägt den psaA-Promotor (Summer et al., 2000). pBS/rpoB: Das 1,2 kb große Insert weist den NEP-Promotor des rpoB-Gens auf (Homann, Dissertation, 2002). pBS/trnV: Das enthaltene Insert trägt den Promotor des trnV-Gens (GAC) (Hanaoka et al., 2003). 29 Material und Methoden pUC13/trnQ: Das enthaltene Insert trägt den trnQ-Promotorbereich (Neuhaus, 1989). pUCE1.45: Das Plasmid trägt den Promotor und Teile des trnK-Gens (Homann und Link, 2003). pGEM/rps16: Das Insert von 298 bp weist den stromaufwärts liegenden Promotor des rps16-Gens auf (Neuhaus et al, 1989 und Homann und Link, 2003). 3.1.7 Internetadressen NCBI http://www.ncbi.nlm.nih.gov BLAST http://www.ncbi.nlm.nih.gov/blast/Blast.cgi CLUSTLW2 http://www.ebi.ac.uk/Tools/clustalw2/index.html ExPASy tools http://www.expasy.ch/tools Phytozome v4.1 http://www.phytozome.net/ PSIPRED http://bioinf.cs.ucl.ac.uk/psipred/psiform.html Restrictionmapper http://www.restrictionmapper.org/ Primerfox http://www.primerfox.com/ Pfam 24.0 http://pfam.sanger.ac.uk/ 3.2 Methoden Nachfolgend sind die in dieser Arbeit verwendeten Methoden dargestellt. Die nicht gesondert aufgeführten Standardmethoden wurden nach Sambrook et al. (2001), Ausubel et al. (1995) oder nach Herstellerangaben durchgeführt. 3.2.1 Anzucht von A. thaliana Für die Anzucht von A. thaliana Pflanzen – sowohl Wildtyp als auch Mutanten – wurden Samen unter sterilen Bedingungen sterilisiert und anschließend auf entsprechende MS-Platten aufgetragen. Die Sterilisation erfolgte durch Inkubationen 30 Material und Methoden in 70% (v/v) Ethanol und 5% (w/v) Natriumhypochlorit mit anschließenden Waschschritten in destilliertem Wasser. Für das Ausplattieren der Samen wurden diese mit 0,1% (w/v) Agarose vermischt und auf MS-Platten verteilt. Nach 2 Tagen in Dunkelheit bei 4°C wurden die Platten mit den Samen im Phytotron unter verschiedenen Lichtbedingungen kultiviert. 3.2.2 Isolierung von Nukleinsäuren 3.2.2.1 Isolierung genomischer DNA Die Isolierung von Gesamt-DNA aus A. thaliana-Pflanzen erfolgte mit dem DNeasy Plant Mini Kit (Qiagen, Hilden), welches auf der Methode von Doyle und Doyle (1987) basiert und damit auch das Detergenz CTAB zur Isolierung nutzt. 3.2.2.2 Isolierung von Plasmid-DNA Plasmid-DNA-„Easy“-Präparation Nach Berghammer und Auer (1993) wurde eine Methode zur Plasmid-DNAIsolierung verwendet, die zeitlich schnell ist, aber zu keiner hochreinen Plasmid-DNA führt. Plasmid-DNA-Midipräparation Plasmid-DNA-Präparationen für reinere und höher konzentrierte DNA wurden nach Birnboim und Doly (1979) oder mittels des NucleoSpin®- bzw. des Nucleo-Bond-Kits (Macherey & Nagel) durchgeführt. 3.2.2.3 Isolierung von RNA Die Isolierung von pflanzlicher Gesamt-RNA wurde nach Chomczynski und Sacchi (1987) durchgeführt. 3.2.3 Amplifizierung von Nukleinsäuren 3.2.3.1 Reverse Transkription Die Reverse Transkription wurde mittels des Enzyms AMV-Reverse-Transkriptase (Avian Myeloblastosis Virus Reverse Transcriptase, Promega) durchgeführt, indem 31 Material und Methoden isolierte RNA (siehe 3.2.2.3) mit Hilfe von Oligonukleotiden in cDNA umgeschrieben wurde. Die synthetisierte cDNA konnte direkt in eine PCR-Reaktion (siehe 3.2.3.2) als Matrize eingesetzt werden, um eine ausreichende Menge an DNA für weitere Schritte zu amplifizieren. 3.2.3.2 Polymerase-Kettenreaktion (PCR) Die Polymerase-Kettenreaktion – kurz PCR (engl.: Polymerase Chain Reaction) – ist eine Methode, um beliebige DNA-Fragmente in vitro amplifizieren zu können (modifiziert nach Mullies und Faloona, 1987). Für eine entsprechende Reaktion in 50 µl wird die zu amplifizierende DNA als Matrize mit Desoxynukleotiden, spezifischen Oligonukleotiden und 1 U Polymerase (GoTagHotStart®- bzw. PfuPolymerase, Promega) unter spezifischen Reaktionsbedingungen vermischt und durch eine Abfolge von Denaturierungs-, Annealing- und Elongationsschritten amplifiziert. Anschließend können die Ansätze analysiert und weiterverwendet werden. 3.2.3.3 ortsgerichtete in vitro Mutagenese Die Erstellung von spezifischen Punktmutationen erfolgte mittels ortsgerichteter in vitro Mutagenese durch das „QuickChange® Site-directed Mutagenesis Kit“ der Firma Stratagene. Es wurden nach Herstellerangaben entsprechende Oligonukleotide mit den spezifischen Mutationen synthetisiert und die PCR-Reaktion mit anschließendem DpnI-Verdau der Matrizen-DNA durchgeführt. Nach erfolgreicher Transformation in den E. coli-Stamm XL1Blue wurden Plasmide von Einzelkolonien isoliert und durch DNA-Sequenzierung überprüft. 3.2.3.4 DNA-Sequenzierung Alle für diese Arbeit notwendigen Sequenzierungen wurden am Lehrstuhl für molekulare Neurobiochemie (Ruhr-Universität Bochum, Fakultät für Chemie) unter Verwendung eines Kapillarelektrophoresegerät „3130cl Genetic Analyser“ (Applied Biosystems) ausgeführt (nach Sanger et al., 1977). Die Auswertung der detektierten Signale erfolgte mittels des zugehörigen Programmes „Sequence Scanner 1.0“ (Applied Biosystems). 32 Material und Methoden 3.2.4 Elution von DNA-Fragmenten Die Elution von DNA-Fragmenten wie beispielsweise PCR-Produkten erfolgte mittels „QIAquick Gel Extraktion Kit“ (Qiagen). Dafür wurden DNA-Fragmente in einem präparativen Gel elektrophoretisch aufgetrennt, die gewünschte Bande eluiert und die DNA-Extraktion nach Herstellerangaben durchgeführt. 3.2.5 Markierung von Nukleinsäuren 3.2.5.1 radioaktive Endmarkierung von DNA Die radioaktive Endmarkierung von DNA diente der Markierung von PromotorFragmenten für EMSA-Studien (siehe 3.2.15). Nach Rao (1994) wurde die T4Polynukleotidkinase dazu verwendet die γ-ständigen Phosphatgruppen des γ-32PATPs auf die dephosphorylierten 5‘-OH-Gruppen von Promotor-Fragmenten zu übertragen. Eine Abtrennung der ungebundenen Nukleotide erfolgte mittels illustra MicroSpinTMG-50 Columns (GE Healthcare), welche das Säulenmaterial Sephadex verwenden. 3.2.5.2 nicht-radioaktive DIG-Markierung von RNA Die Markierung von RNA für Northern Blot-Analysen erfolgte mittels Digoxigenin-11UTP durch in vitro-Transkription nach den Angaben des Herstellers für DIG (Roche Diagnostics). 3.2.6 „Northern“-Transfer von RNA Proben von Gesamt-RNA wurden in 1,5%igen Agarosegelen unter denaturierenden Bedingungen elektrophoretisch aufgetrennt und im Anschluss durch Kapillarkräfte auf positiv geladene Nylonmembranen übertragen. 3.2.7 RNA/RNA-Hybridisierung Für die RNA/RNA-Hybridisierung wurden DIG-markierte RNA-Fragmente verwendet (siehe 3.2.5.2), die mit Membranen ü. N. bei 68°C hybridisiert wurden. Die anschließende Detektion mittels Chemilumineszenz erfolgte nach Herstellerangaben (Roche Diagnostics) unter Verwendung von Anti-Digoxigenin-Alkalische- Phosphatase-Konjugat mit seinem Substrat CDP-Star. 33 Material und Methoden 3.2.8 Expression rekombinanter Proteine 3.2.8.1 Expression mittels des pMAL-Systems Das pMAL-System (New England Biolabs) basiert auf dem Vektor pMAL-c4x, der es ermöglicht einklonierte Genfragmente als MBP-Fusionsproteine in E. coli zu synthetisieren. Die vorliegenden Klone benötigten zur Überexpression den E. coliStamm BL2-CodonPlus-RIL (Stratagene), der bevorzugte tRNA-Codons aus Eukaryoten trägt und damit schwierige eukaryotische Gene synthetisieren kann. Für die Überexpression wurden 1l LB-Medium mit Ampicillin und Chloramphenicol (je 50 µg/ml) 2%tig mit einer Übernachtkultur angeimpft und anschließend bis zu einer OD590 von 0,5-0,7 bei 37°C kultiviert. Die Überexpression wurde mit IPTG (1 mM Endkonzentration) induziert und nach 4 h durch die Zellernte beendet. Die Zellen wurden in Säulenpuffer (nach Herstellerangaben: 200 mM Tris/HCl, 200 mM NaCl, 1 mM EDTA, 1 mM DTT) resuspendiert und mittels Ultraschall aufgeschlossen. Nach Entfernung der Zelltrümmer durch Zentrifugation (4000 x g, 30 min, 4°C) wurden die Fusionsproteine mit einer äquilibrierten Amylose-Matrix gebunden und konnten durch Maltose-haltigen Säulenpuffer (10 mM) eluiert werden. 3.2.8.2 Expression mittels des pGEX-Systems Die Überexpression von Proteinen mittels des pGEX-Systems (GE Healthcare/ Amersham Biosciences) erfolgte durch die Klonierung des entsprechenden GenFragments in den Vektor pGEX-6P-1. Die Expression erfolgt als GST-Fusionsprotein im E. coli-Stamm BL21-CodonPlus-RIL (Stratagene). Für eine Expression wurden 250 ml LB-Medium mit Ampicillin und Chloramphenicol (je 50 µg/ml) 1%tig mit einer Übernachtkultur angeimpft und bis zu einer OD590 von 0,5 bei 30°C inkubiert. Die Expression wurde mit IPTG (0,5 mM Endkonzentration) induziert und nach 2 h Kultivierung durch Ernte der Zellen gestoppt. Die Resuspendierung der Zellen erfolgte in 1 x PBS-Puffer (10 mM Na2HPO4, 1,8 mM KH2PO4, 2,7 mM KCl, 140 mM NaCl, 5 mM DTT, pH 7,3) und nach einer Inkubation mit Lysozym (1 mg/ml Endkonzentration) wurden die Zellen durch Ultraschall aufgebrochen. Nach einer Inkubation mit DNaseI und einer Zentrifugation (4000 x g, 30 min, 4°C) wurde der Überstand auf eine äquilibrierte GSH-Sepharose-Matrix (Macherey & Nagel) gegeben. Das gebundene Fusionsprotein konnte durch GSH-haltigen Puffer (10 mM GSH, 50 mM Tris/HCl pH 8,0) eluiert werden. 34 Material und Methoden 3.2.9 Gelelektrophorese von Proteinen Die Auftrennung von Proteinen nach ihrem Molekulargewicht erfolgte mittels durchgängigen 8% - 10% Polyacrylamid-Bis-Tris-Gelen oder in NuPAGE Bis-Tris Fertiggelen (Invitrogen). Die Proben für die Elektrophorese wurden jeweils mit Probenpuffer (50 mM Bis-Tris/HCl pH 6,6, 2 mM EDTA, 50 mM DTT, 2 % SDS, 7 % Glycerin und 0,02% BPB) vermischt und für 5 Minuten bei 70°C denaturiert. Die Gele können aufgrund ihrer Zusammensetzung (Polyacrylamid und Puffer (1,25 M BisTris, pH 6,6)) als native sowie auch als denaturierende Gele genutzt werden. In der vorliegenden Arbeit wurden nur denaturierende Gele dieses Systems verwendet und daher wurde ein MOPS-SDS-haltiger Laufpuffer (50 mM MOPS, 50 mM Tris, 0,1 % SDS, 1 mM EDTA, pH 7,0) eingesetzt. 3.2.10 „Western“-Transfer von Proteinen Nach der Methode von Towbin et al. (1992) wurden die Proteine nach der Gelelektrophorese in Transferpuffer (20 mM Tris, 150 mM Glycin, 0,04 % SDS, 20 % Methanol) durch eine Halbtrocken-Blotkammer (Roth) auf reine Nitrocellulose transferiert. Der Protein-Transfer fand in 20 min bei 300 mA und 4°C statt und wurde mittels einer reversiblen Ponceau-S-Färbung (1% Ponceau, 5 % Essigsäure) überprüft. Anschließend erfolgte eine Immunodetektion (siehe 3.2.11) 3.2.11 Immunodetektion von Proteinen Eine Immunodetektion ermöglicht es auf Nitrocellulose-Membran transferierte Proteine spezifisch nachzuweisen. Nach einem Western-Transfer (siehe 3.2.10) wurde die Membran für 1 h mit 2% (w/v) Magermilchpulver in 1 x TBS (50 mM Tris/HCl pH 8,0, 150 mM NaCl) bzw. mit 5% (w/v) Magermilchpulver in 1 x PBS-T (10 mM Na2HPO4, 1,8 mM KH2PO4, 2,7 mM KCl, 140 mM NaCl, 0,3% Tween 20, pH 7,3) blockiert. Anschließend wurde für eine weitere Stunde der erste und proteinspezifische Antikörper (Anti-MBP- bzw. Anti-GST-Antikörper) nach Herstellerangaben (New England Biolabs bzw. GE Healthcare) in der entsprechenden Lösung und Verdünnung mit der Membran inkubiert. Die nachfolgenden Waschschritte und Inkubation mit dem zugehörigen, sekundären Antikörper wurden ebenfalls nach Herstellerangaben (Sigma) durchgeführt. Die Detektion erfolgte durch die Spaltung der Substrate NBT (0,33 mg/ml) und BCIP (0,165 mg/ml) durch AlkalischePeroxidase, welche Teil des sekundären Antikörpers ist. 35 Material und Methoden 3.2.12 Protein Overlay Blot Für die Untersuchung von Protein-Protein-Interaktionen wurde eine Variante der PullDown-Analyse verwendet: der Protein Overlay Blot (modifiziert nach Jelich-Ottmann et al., 2001). Dafür wurde das auf Interaktion zu untersuchende Protein in Gelen aufgetrennt und dann auf Nitrocellulose transferiert (siehe 3.2.10). Diese Membran wurde mittels Magermilchpulver-Lösung (je nach folgendem Antikörper) geblockt und anschließend ü. N. mit einem potentiellen Interaktionsprotein (200 µg) inkubiert. Nach den nötigen Waschschritten erfolgte eine Immunodetektion gegen das verwendete Interaktionsprotein (siehe 3.2.11). Wenn eine Interaktion zwischen den Proteinen stattgefunden hat, wurde das transferierte Protein mittels eines indirekten Nachweises durch einen anderen Antikörper detektiert. 3.2.13 Färbung von Proteinen Die Färbung von Proteinen erfolgte entweder direkt im Protein-Gel mittels Coomassie-Blau (Merril et al., 1983) oder durch eine reversible Ponceau-S-Färbung (1% Ponceau, 5 % Essigsäure nach Salinovich und Montelaro, 1986) bei auf Nitrocellulose transferierten Proteinen. 3.2.14 Bestimmung der Proteinkonzentration Die Bestimmung der Proteinkonzentration in Proben wurde nach der von Bradford (1976) beschriebenen Methode durchgeführt. Als Proteinstandard diente Rinderserumalbumin (BSA). 3.2.15 Nachweis von DNA-Protein-Interaktionen Zum Nachweis von DNA-Protein-Interaktionen wurde der EMSA („electrophoretic mobility shift assay“) verwendet. Diese Technik nutzt die Tatsache aus, dass bei einer elektrophoretischen Auftrennung freie DNA-Fragmente schneller durch ein Gel laufen als Protein-DNA-Komplexe (Fried und Crothers, 1981; Garner und Revzin, 1981). Eine Detektion der freien DNA-Fragmente und der Protein-DNA-Komplexe war aufgrund der radioaktiven Markierung der DNA-Fragmente (siehe 3.2.5.1) möglich. Ein Reaktionsansatz bestand aus 2,5 ng radioaktiv-markiertem Promotorfragment, 700 ng E. coli core-RNA-Polymerase (1 U) mit 1 µg poly[dIdC] im entsprechenden Reaktionspuffer (30 mM Tris/HCl pH 7, 0,5 mM β-Mercaptoethanol, 0,5 mM EDTA, 36 Material und Methoden 5% (v/v) Glycerin) und 50 ng rekombinantem AtSIG1-MBP-Protein. Für Interaktionsstudien wurden meist 150 ng Protein eines potentiellen Interaktors (AtSIB1-/AtSIB2-GST-Protein) zugefügt. Nach einer Inkubation von 15 min bei 25°C wurden die Ansätze auf 4%ige native Polyacrylamid-Gele aufgetragen, aufgetrennt und getrocknet. Die Auswertung der Signale erfolgte mit dem FLA3000-PhosphoImager und anschließender Analyse mit dem Programm AIDA v4.04. 3.2.16 Circulardichroismus (CD) –Spektroskopie Die CD-Spektroskopie ist eine wichtige Methode zur Analyse der Konformation von Proteinen. Für eine entsprechende Analyse erfolgte jeweils die Aufnahme von 10 Spektren im Bereich von 190 bis 320 nm. Als Leerprobe diente der verwendete Messpuffer (25 mM Natrium-Phosphat-Puffer, pH 8.0), in dem ebenfalls die Proteine für die Messung gelöst vorlagen. Die Messung selbst wurde bei 20°C in einem JASCO715-Spektropolarimeter (am Lehrstuhl Biochemie der Pflanzen) durchgeführt. Die Auswertung der erhaltenen Daten erfolgte mit dem zum Spektropolarimeter gehörigen Programm Spectra Analysis. 37 Ergebnisse 4. Ergebnisse 4.1 Überexpression und Funktionsanalyse des AtSIG1-Proteins Um die Interaktion des AtSIG1-Proteins mit den beiden AtSIB-Proteinen zu untersuchen, mussten diese Proteine in ausreichender Menge, hinreichend rein und funktionsfähig vorliegen. Da diese Voraussetzungen am ehesten von geeigneten rekombinanten Proteinen erfüllt werden, wurden zunächst verschiedene System geprüft und dann für die Herstellung des AtSIG1-Proteins das pMAL-System (New England Biolab) ausgewählt. Dieses System führt zur Expression von rekombinanten Proteinen in Fusion mit dem Maltose-Bindungs-Protein MBP. Dies hat mindestens drei Vorteile: 1) Affinitätschromatographie-Reinigung mittels Amylose-Matrix 2) Abspaltung des MBP-Anteils möglich 3) MBP verleiht vielen Fusionsproteinen lösliche „native“ Eigenschaften (Fox et al., 2003) Für eine Expression wurde aus cDNA das sig1-Gen aus A. thaliana mit Genspezifischen Oligonukleotiden (siehe 3.1.5) ohne Transit-Peptid mittels PCR amplifiziert, in den Vektor pBSIIKS (Stratagene) kloniert und in den Überexpressionsvektor pMAL umkloniert (siehe Abb. 4.1.1A). Die Expression selbst erfolgte im E. coli-Stamm BL21-CodonPlus-RIL, der ein Plasmid mit trn-Genen enthält und darüber die Synthese von schwierigen eukaryotischen Genen ermöglicht (Stratagene). 38 Ergebnisse Abb. 4.1.1: Expression und Reinigung des rekombinanten AtSIG1-Proteins A) Schematische Darstellung des AtSIG1-Proteins. Die konservierten Regionen 1 bis 4 (Lonetto et al., 1992, Paget und Helmann, 2003) sind ebenso wie der unkonservierte Bereich (UKB) sowie N-terminal das Fusionsprotein MBP eingezeichnet. B) Überexpression und Reinigung des AtSIG1-Proteins. Das gezeigte Coomassiegefärbte Proteingel stellt zunächst die Expression vor und nach der Induktion mit 1 mM IPTG dar (Spuren 1 und 2). Danach folgen die Proteinmuster der Aufreinigungsschritte im pMAL-System (New England Biolabs): vor und nach der Affinitätschromatographie an der Amylose-Matrix (Durchlauf- (d) und Waschschritt (w) in den Spuren 5 und 6). Die Muster nach Zentrifugation des Gesamtextraktes nach Trennung in Überstand (ü, Spur 3) und Sediment (p, Spur 4) sind ebenfalls gezeigt. Der Anstieg der Elution vom AtSIG1-MBP-Fusionsprotein ist sichtbar und basiert auf einen Maltosegradienten (Spuren 7 bis 12). Weitere Fraktionen wurden hier nicht gezeigt. Ein Duplikat des Proteingels wurde mittels Western-Technik auf eine Nitrocellulose-Membran transferiert und eine immunologische Detektion mit dem AntiMBP-Antikörper durchgeführt. Ein Ausschnitt ist unter dem Coomassie-gefärbten Gel 39 Ergebnisse gezeigt (spezifischen Nachweis des Fusionsproteins in allen Spuren; siehe Material und Methoden, Kapitel 3.2.11). Abb.4.1.1B zeigt exemplarische Proteindaten auf dem Weg vom bakteriellen Rohextrakt zum gereinigten Protein. Die einzelnen Schritte der Aufreinigung bis zur Elution des AtSIG1-Proteins sind jeweils mit Hilfe einer Proteinfärbung und eines immunologischem Nachweises festgehalten. Zu Beginn wurde eine Überexpressionskultur mit 1 mM IPTG für 4 Stunden induziert, die Induktion und damit die Synthese des Proteins ist im gefärbten Gel deutlich zu erkennen (Vergleich der Spuren 1 und 2, oben). Die optimale Bedingung für eine Expression von bis zu 5 mg nativem und löslichem Protein (pro Liter Bakterienkultur) ist bei einer Temperatur von 25°C. Die Reinigung des AtSIG1-Proteins erfolgte mittels Affinitätschromatographie über eine Amylose-Matrix (New England Biolabs; Spuren 2-12, oben). Zur Kontrolle und Detektion des exprimierten, rekombinanten Proteins diente der Anti-MBP-Antikörper (New England Biolabs), der den N-terminalen Fusionsanteil – das MBP-Protein – spezifisch erkennt (siehe Abb.4.1.1.B, unten). Um das rekombinante AtSIG1-Fusionsprotein auf enzymatische Sigma-Aktivität zu testen, wurden DNA-Bindungsstudien durchgeführt. Die Grundfunktionen von Sigmafaktoren in Bakterien und Pflanzen sind die Vermittlung der Promotor-Bindung als Teil des RNA-Polymerase-Komplexes und die anschließende Initiation der Transkription (Burgess et al., 1969, Campbell et al., 2009). Eine Überprüfung der Promotor-Bindung ist durch EMSA-Experimente möglich (Fried und Crothers, 1981; Garner und Revzin, 1981). Diese in publizierten Vorarbeiten der Arbeitsgruppe verwendete Methode (Link, 1984, Tiller et al., 1991; Tiller und Link, 1993, Homann und Link, 2003) ermöglicht eine relativ schnelle und einfache Abschätzung der Sigma-Aktivität. Sie basiert auf der Bildung eines „heterologen“ holo-EnzymPromotor-Komplexes aus den folgenden drei Komponenten: - E. coli core RNA-Polymerase (Epicentre) - Promotor-haltiges DNA-Fragment - zu prüfender Sigmafaktor. Als Promotor diente der starke psbA-Promotor als Bestandteil eines klonierten DNAFragments (routinemäßig aus Sinapis alba). Die Sequenz ist identisch mit der des psbA-Promotors aus A. thaliana (Liere et al., 1995). Die jeweiligen EMSA40 Ergebnisse Reaktionsansätze wurden über ein natives Polyacrylamid-Gel aufgetrennt, das Gel getrocknet und die radioaktiven Signale mittels eines Phospho-Imagers (FLA-3000) detektiert. Zu den Ansätzen zählen auch Kontrollen wie das allein aufgetragene psbA-Promotorfragment (siehe Abb. 4.1.2, Spur 1), das gewissermaßen die Position des ungebundenen Basissignals (ohne Proteinbindung) angibt. Eine unspezifische DNA-Bindung durch das core-Enzym allein ohne Sigmafaktor wird durch die Verwendung von 1 µg poly[dIdC] in den Ansätzen verhindert (siehe Spur 2), und eine Promotor-Bindung ohne core-Enzym kann nicht durch das AtSIG1-Protein vermittelt werden (Spur 4). Der MBP-Fusionsproteinanteil ohne den Sigmaanteil hat zusammen mit dem core-Enzym auch keine Promotor-bindenden Eigenschaften (Spur 3). Als Positiv-Kontrolle dient ein holo-RNA-Polymerase-Komplex (Epicentre) von E. coli, der aus dem core-Enzym und dem bakteriellen HauptstandardSigmafaktor σ70 besteht und ein starkes, spezifisches Bindungssignal mit dem psbAPromotor zeigt (Spur 5). Die Bindung des heteromeren Komplexes bestehend aus AtSIG1-Protein, E. coli core-Enzym und den psbA-Promotor (Spur 6) ist verglichen mit derjenigen des E. coli holo-Enzyms eher schwach, aber dennoch spezifisch wie Kompetitionsexperimente mit nicht-markierten Promotorfragmenten zeigen (Spuren 7-9). Beispielsweise wurde nicht-markierter rpoB-Promotor im Überschuss (100 ng) eingesetzt, welcher nur spezifisch durch die NEP-Polymerase (siehe Einleitung, Kapitel 1.3) erkannt und gebunden wird. Das entsprechende Bindungssignal wird durch diesen nicht-markierten Promotor gar nicht oder nur sehr schwach negativ beeinflusst (vergleiche Spuren 7 und 8). Folglich wurde der nicht-markierte Promotor nicht durch den AtSIG1-core-Komplex gebunden und ist kein wirksamer Kompetitor. 41 Ergebnisse Abb. 4.1.2: Bindungsstudien des AtSIG1-Proteins mittels EMSA Der mit 32P-markierte Promotor wurde für EMSA-Studien eingesetzt, um die Promotor-Bindefähigkeit des AtSIG1-Proteins zu analysieren. Die Negativ-Kontrollen bestanden aus dem radioaktiv-markierten Promotor allein (Spur 1) und in Anwesenheit des core-Enzyms ohne Sigmafaktor (Spur 2). Die fehlende Promotorbindung des Fusionsproteins MBP wurde in Anwesenheit des core-Enzyms (Spur 3) gezeigt. Die erfolgreiche Promotorbindung konnte nicht durch das AtSIG1Protein allein (Spur 4), sondern nur als AtSIG1-core-Komplex erreicht werden (Spur 6). Als Positiv-Kontrolle wurde die holo-RNA-Polymerase aus E. coli zusammen mit dem radioaktiv-markierten psbA-Promotor eingesetzt (Spur 5). Die Spuren 8 und 9 zeigen Kompetition mit nicht-markiertem psbA-Promotor-Fragment und die Spuren 7 und 8 zeigen keine Kompetition mit dem nicht-markierten NEP-spezifischen rpoBPromotor. Die dargestellten Signale basieren auf einem nativen Polyacrylamid-Gel, das getrocknet mittels eines Phospho-Imagers (FLA-3000) analysiert wurde. Abk.: B: Bindungssignal, F: freie Sonde. Hingegen hat die Verwendung von nicht-markiertem psbA-Promotor (100 ng) als Kompetitor eine dramatisch „schwächende“ Wirkung auf das radioaktive Bindungssignal (vergleiche Spuren 8 und 9). Der AtSIG1-core-Komplex konnte in diesem Fall sowohl den radioaktiv-markierten als auch den nicht-markierten Promotor binden. Da statistisch gesehen beide Promotoren einen Teil der Komplexe binden, reduziert sich die Signalstärke und beweist die Wirksamkeit des psbAPromotors als Kompetitor und verifiziert gleichzeitig auch die Spezifität der gezeigten DNA-Protein-Interaktion. 42 Ergebnisse Nachdem so durch Expression, Reinigung und DNA-Bindungsanalyse für das rekombinante AtSIG1-Protein sichergestellt war, dass von „nativen“ SigmafaktorEigenschaften ausgegangen werden kann, konnten weiterführende Analysen zur Charakterisierung von AtSIG1 und seiner Protein-Protein-Interaktionsfähigkeit folgen. 4.2 Analyse der Protein-Bindungsfunktion von AtSIG1 mittels ausgewählter Mutanten-Konstrukte Das klonierte Expressionskonstrukt AtSIG1 (siehe 4.1) diente als Ausgangsmaterial für die Herstellung von Punktmutanten mittels ortsgerichteter Mutagenese (siehe Material und Methoden, Kapitel 3.2.3.3). Die ausgewählten Stellen lagen größtenteils in der konservierten Region 4.2 des Sigmafaktors, welche die Erkennung der -35Promotorregion vermittelt (Paget und Helmann, 2003). Diese Region enthält ein sogenanntes HTH-Sequenzmotiv (Helmann und Chamberlin, 1988) und wurde als potentielle Interaktionsstelle der beiden AtSIB-Proteine von Morikawa et al. (2002) vermutet. Basierend auf diesen Ergebnissen und konzeptionellen Vorgaben von Vorarbeiten wurden im Rahmen der vorliegenden Dissertation Sequenzvergleiche (siehe vollständige Abb. 9.1 im Anhang) mit den anderen fünf Sigmafaktoren von A. thaliana und SIG1 aus S. alba (SaSIG1) durchgeführt. Es wurden Aminosäuren in AtSIG1 ausgewählt, die in den anderen Faktoren von A. thaliana an diesen Positionen nicht vorkommen, aber in SaSIG1 aus S. alba konserviert sind: Positionen 460, 463, 465, 467 und 471. Ebenfalls einbezogen wurden die Positionen 321 und 476, weil diese beiden Aminosäuren (Glutamin bzw. Arginin) sich in SaSIG1 als wichtig für die Sigma-Aktivität erwiesen hatten (Homann und Link, 2003). Darüber hinaus wurde auch die Position 458 ausgewählt, da dieser Cystein-Rest offenbar an einer Dimer-Bildung des rekombinanten AtSIG1-Proteins unter schwach reduzierenden Bedingungen beteiligt ist (Daten nicht gezeigt). Dies könnte bedeuten, dass die Sigma-Aktivität des AtSIG1-Proteins durch Änderungen der Redoxbedingungen moduliert wird. Als generelle Negativ-Kontrolle für diese Punktmutationen diente ein Verkürzungskonstrukt, dem die gesamte konservierte Region 4 C-terminal fehlte; dieses wird als SIG1AS81-413 bezeichnet. 43 Ergebnisse Abb. 4.2.1: Deletions- und Punktmutanten des AtSIG1-Proteins A) Schematische Darstellung der Mutanten des AtSIG1-Proteins. Die jeweils erstellten Punktmutationen geben die ursprüngliche und die ersetzte Aminosäure an. Die Region 4 wurde aufgrund der Akkumulation der Punktmutationen in diesem Bereich vergrößert dargestellt. Das Deletionskonstrukt SIG1AS81-413 wurde um die konservierte Region 4 verkürzt und ist dementsprechend dargestellt. Alle gezeigten Konstrukte werden in Fusion mit dem MBP-Protein synthetisiert. B) Nachweis der gereinigten, rekombinanten AtSIG1-Mutanten-Proteine sowie des „Wildtyp“-Proteins. Es werden ein Coomassie-gefärbtes Proteingel und ein entsprechender Immunoblot gegen das MBP-Fusionsprotein gezeigt. Das AtSIG1-TP (Spur 9): Ausgangsprotein ohne Mutation. Die Bezeichnungen der Mutanten-Proteine (oben) ergeben sich aus denjenigen in A. Gleiche Volumina (nicht Proteinmengen) der affinitätschromatographisch gereinigten Präparate wurden eingesetzt. 44 Ergebnisse Die Abb. 4.2.1A zeigt eine schematische Abbildung aller Konstrukte, die – wiederum in Fusion mit dem MBP-Protein – bakteriell überexprimiert und durch MaltoseAffinitätschromatographie gereinigt wurden (vergleiche Kapitel 4.1). Die Konstrukte 458 und 471 wurden speziell für diese Arbeit angefertigt, während die Herstellung der übrigen Punktmutanten Bestandteil einer Laborkooperation und eigenständig angefertigten Bachelor-Arbeit (E. Husic, 2009) waren. Die erfolgreich gereinigten Mutanten-Proteine sind in Abb. 4.2.1B im Coomassie-gefärbten Proteingel (oben) und nach Immunodetektion gegen das Fusionsprotein MBP (unten) gezeigt. Die weitere Analyse dieser Mutanten sollte sowohl einen Einblick in die Konformation als auch in die Funktion dieser Konstrukte liefern. Für die Konformations-Analyse wurde die Circulardichroismus-Spektroskopie (CD-Spektroskopie) ausgewählt. Diese Technik nutzt die Tatsache, dass optisch aktive Moleküle zirkular polarisiertes Licht absorbieren können und liefert ein CD-Spektrum. Abb. 4.2.2: CD-Spektren der AtSIG1-Punktmutanten im Vergleich zum AtSIG1Protein Die gezeigten CD-Spektren wurden mittels des JASCO715-Spektropolarimeter am Lehrstuhl für Biochemie der Pflanzen im Bereich von 190 bis 320 nm aufgenommen. Jedes Spektrum basiert auf jeweils 10 Analysen, die gemittelt wurden. Als Leerprobe diente der verwendete Messpuffer mit Phosphat. Als Vergleichs-„Standard“ für die Auswertung der Punktmutationen diente das nicht-mutierte AtSIG1-Protein. Die Mutanten-Bezeichnungen sind wie in Abb. 4.2.1 angegeben. Für die erhaltene Abb. 4.2.2 wurden für jedes Konstrukt jeweils 10 einzelne Spektren-Messungen mit dem JASCO715-Spektropolarimeter am Lehrstuhl Biochemie der Pflanzen durchgeführt. Diese Spektren wurden im Bereich von 190 bis 45 Ergebnisse 320 nm aufgenommen. Als Vergleichskontrollen wurden der Phosphat-Messpuffer allein als Leerprobe und das AtSIG1-Ausgangsprotein als Vergleichsprobe zu den Punktmutanten verwendet. Die durchgeführte Analyse zeigte, dass die Punktmutanten insgesamt mit dem nicht-mutierten AtSIG1-Protein eine relativ hohe Vergleichbarkeit und Ähnlichkeit in den Spektren zeigten. Die höchsten Ähnlichkeiten zeigten dabei die Mutationen der Aminosäuren 467 und 463. Mit etwas Abstand folgten die Punktmutationen der Aminosäuren 460, 321 und 458. Den größten Abstand zum Wildtyp zeigten die Mutationen der Aminosäuren an den Positionen 471, 476 und 465. Diese Kurven legen nahe, dass sich die Konformationen der zuletzt genannten Aminosäuren am meisten vom ursprünglichen AtSIG1-Protein unterscheiden. Da die CD-Spektren aber noch nichts über die Funktionsfähigkeit dieser Mutanten aussagen, sollten in vitro Studien mittels EMSA zur Klärung dieser Frage beitragen. Dafür wurden wie bereits in Kapitel 4.1 beschrieben, Reaktionsansätze mit den Sigma-Mutanten-Proteinen zusammen mit dem radioaktiv-markierten psbA- Promotor-Fragment und E. coli core-RNA-Polymerase inkubiert, auf ein natives Polyacrylamid-Gel aufgetragen, getrocknet und mittels eines Phospho-Imagers detektiert. Die erhaltenen Bindungsmuster wurden mit Hilfe des Programms AIDA v4.04 quantifiziert. Dabei diente das DNA-Bindungssignal mit dem AtSIG1-Protein im Komplett-Ansatz als Vergleichswert (100%). Nach dieser Methode wurden sowohl die Punktmutanten als auch das Verkürzungskonstrukt SIG1AS81-413 in Hinblick auf DNA-Bindungsfähigkeit analysiert (mindestens drei unabhängige Werte verschiedener EMSA-Studien). Die erhaltenen Werte der Bindungsfähigkeit wurden statistisch ausgewertet, die Standardabweichung errechnet und werden durch Säulendiagramme in Abb. 4.2.3 repräsentiert. Das Verkürzungskonstrukt SIG1AS81413 zeigte im Vergleich zum AtSIG1-Protein in den Bindungsstudien eine etwas höhere Fähigkeit an die Promotor-DNA zu binden, was aber durch die ermittelte Standardabweichung relativiert wird. Der Verlust der DNA-Bindungsfähigkeit der meisten Punktmutanten im Vergleich zum AtSIG1-Protein liegt zwischen 10 bis 30%, d.h. diese Mutanten weisen noch eine Bindungsfähigkeit von mindestens 70% auf. Die zwei auffälligsten Mutationen liegen an den Positionen 471 und 476, die zu einem Verlust an DNA-Bindungsfähigkeit um 85% bzw. 65% führen, d.h. die entsprechenden Mutanten weisen nur noch sehr geringe Sigma-Aktivität auf. Zusammenfassend zeigten die Analysen ein sehr unterschiedliches Bild von den 46 Ergebnisse untersuchten Konstrukten, da der völlige Verlust der konservierten Region 4 eher keine Auswirkungen auf die Bindungsfähigkeit des AtSIG1-Proteins an den psbAPromotor hat, aber einzelne Punktmutationen wie an Position 471 und 476 zu gravierenden Verlusten in der DNA-Bindungsfähigkeit führen können. Es ist darauf hinzuweisen, dass die EMSA-Daten ausschließlich die DNA-Bindungsfähigkeit betreffen und es bleibt zu klären, inwieweit diese Aminosäuren einen Einfluss auf die Art der regulatorischen Protein-Protein-Interaktionen von AtSIG1 haben. Abb. 4.2.3: Funktionsstudien der AtSIG1-Mutanten mittels EMSA Die gezeigten Säulen basieren auf ausgewerteten EMSA-Studien, die mittels des Programms AIDA v4.04 ausgewertet wurden. Als Vergleichswert und Kontrolle diente das AtSIG1-Protein für welches eine DNA-Bindungsfähigkeit mit 100 % angesetzt wurde. Die dargestellten Werte wurden aus mindestens drei verschiedenen EMSAStudien ermittelt, statistisch ausgewertet und nach Bestimmung der Standardabweichung mit Excel erstellt. Die Mutanten-Bezeichnungen decken sich mit denjenigen in Abb. 4.2.1 und 4.2.2. 47 Ergebnisse 4.3 Expression und Reinigung der Proteine AtSIB1 und AtSIB2 Die Untersuchung der Interaktion zwischen AtSIG1- und den beiden AtSIB-Proteinen sollte nicht nur die Interaktion umfassen, sondern auch ein globaleres Bild der Transkriptionsregulation in den Plastiden ermöglichen. Die Erforschung der Funktion der Sigmafaktoren ist für sich allein genommen hinsichtlich spezifischer Regulation und Modulation der Sigma-Aktivität jedes einzelnen Faktors bereits recht weit fortgeschritten (siehe z.B. Schweer et al., 2010). Meine Arbeit soll als Teil dieser Untersuchungen die Regulation durch Protein-Protein-Wechselwirkungen am Beispiel von AtSIG1 und seinen beiden Interaktoren AtSIB1 und AtSIB2 beleuchten. Die beiden Interaktoren des AtSIG1-Proteins – AtSIB1 (AT3G56710) und AtSIB1„verwandt“ (wird nachfolgend als AtSIB2 bezeichnet; AT2G41180) – wurden durch Hefe-Zwei-Hybrid-Analysen identifiziert (Morikawa et al., 2002) und mittels Pulldown-Analysen verifiziert. In diesen Analysen wurde nur die konservierte Region 4 des AtSIG1-Proteins verwendet, welche spezifisch mit beiden AtSIB-Proteinen interagiert. Die Funktion dieser Interaktionen ist aber ungeklärt geblieben und Morikawa et al. (2002) vermuteten einen Inhibitor, der Ähnlichkeit zu den aus Bakterien gut untersuchten Anti-Sigmafaktoren hat (Hughes und Mathee, 1998). Um die Interaktion und die Funktion der beiden Interaktoren zu beleuchten, müssen nicht nur das AtSIG1-Protein (siehe 4.1) in ausreichender Menge und Qualitiät vorliegen, sondern auch beide AtSIB-Proteine in dieser Form. Deshalb wurden die beiden Proteine rekombinant in E. coli unter Verwendung des pGEX-Systems (GE Healthcare) synthetisiert. Das System ermöglicht – ähnlich dem pMAL-System – die Expression von rekombinanten Proteinen in N-terminaler Fusion mit dem Protein Glutathion-S-Transferase (Abk.: GST). Die Expression basiert auf der cDNA eines sib-Gens (AT3G56710 bzw. AT2G41180) aus A. thaliana, welches durch Genspezifische Oligonukleotide (siehe 3.1.5) mittels PCR amplifiziert, in den Vektor pBSIIKS (Stratagene) kloniert und in den ausgewählten Überexpressionsvektor pGEX-6P-1 umkloniert wurde (GE Healthcare). Die Synthese der Proteine erfolgte im E. coli-Stamm BL21-CodonPlus-RIL (Stratagene), der auf die Herstellung von codonmäßig „schwierigen“ Genen ausgerichtet ist. 48 Ergebnisse Abb.4.3.1: Expression und Reinigung der Proteine AtSIB1 und AtSIB2 Die beiden gezeigten, Coomassie-gefärbten Proteingele stellen die Expressionen der AtSIB2- und AtSIB1-Protein (links bzw. rechts) vor und nach der Induktion mit 0,5 mM IPTG dar (Spuren 1 und 2 für AtSIB2 bzw. Spuren 8 und 9 für AtSIB1). Es folgen jeweils die Schritte der Aufreinigung (nach Macherey und Nagel): vor der GSHSepharose-Matrix Pellet (Spur 3 für AtSIB2 bzw. Spur 10 für AtSIB1) und Überstand (Spur 4 bzw. 11 für AtSIB2 bzw. AtSIB1) einer Zentrifugation, Durchlauf- (Spur 5 für AtSIB2 bzw. Spur 12 für AtSIB1) und Waschschritte (Spur 6 für AtSIB2 bzw. Spur 13 für AtSIB1). Für AtSIB2 ist nur der Elutionspeak des Proteins (Spur 7) gezeigt, der aber dem Anstieg der Elution des AtSIB1-Proteins (Spuren 14-20) ähnelt. Duplikate der Proteingele wurden mittels der Western-Methode auf eine NitrocelluloseMembran transferiert und anschließend eine immunologische Detektion mit dem AntiGST-Antikörper (GE Healthcare) durchgeführt. Ausschnitte der entsprechenden Detektionen sind unter den Coomassie-gefärbten Gelen gezeigt. Die Fusionsproteine wurden bei ca. 45 kDa (siehe Pfeile) in den entsprechenden Spuren sichtbar und spiegeln die Intensität der Coomassie-gefärbten Banden dieser Größe wieder. Der Weg zu den gereinigten Proteinen über die Expression unter Darstellung der einzelnen Aufreinigungsschritte zeigt Abb. 4.3.1 in Form eines Proteingels und eines immunologischen Nachweises für die AtSIB-Proteine. Zu Beginn wurden Überexpressionskulturen mit 0,5 mM IPTG für 2 Stunden induziert, die Induktion und damit die Synthese der Proteine sind im gefärbten Gel zu erkennen (siehe Spuren 8 und 9 für AtSIB1 und Spuren 1 und 2 für AtSIB2). Die optimalen ExpressionsBedingungen zur Gewinnung von löslichem und nativem Protein lagen für beide 49 Ergebnisse AtSIB-Proteine bei einer Temperatur von 30°C. Über Affinitätschromatograpie an äquilibrierten GSH-Sepharose-Matrices (GE Healthcare und Macherey & Nagel) wurden die AtSIB-Proteine isoliert, die einzelnen Schritte sind exemplarisch für die AtSIB-Proteine in Abb. 4.3.1 gezeigt. Eine Kontrolle der isolierten Proteine erfolgte durch den Anti-GST-Antikörper (GE Healthcare), der das N-terminal fusionierte GSTProtein spezifisch detektiert (siehe auch Abb.4.3.1). Die rekombinanten AtSIB1- und AtSIB2-Proteine liegen in ausreichender Menge und Qualität vor und können in weiteren Analysen auf ihre Interaktionsfähigkeit und Funktionsfähigkeit untersucht werden. 4.4 Interaktionsstudien mit den AtSIB1- und AtSIB2-Proteinen Nachdem die beiden Proteine AtSIB1 und AtSIB2 in ausreichender Menge und Qualität vorlagen, waren die Voraussetzungen für Funktionsuntersuchungen dieser Proteine gegeben. Aus den Arbeiten von Morikawa et al. (2002) war bekannt, dass beide AtSIB-Proteine mit der konservierten Region 4 des AtSIG1-Proteine interagieren. Es blieb in jener Arbeit aber ungeklärt, ob dies der einzige Interaktionsbereich des Sigmafaktors für die Bindung an AtSIB-Proteine ist. Deshalb wurden für die Analyse der Interaktionen zwischen ATSIG1- und AtSIB-Proteinen verschiedene Konstrukte des AtSIG1-Proteins ausgewählt. Zu den Konstrukten zählten das AtSIG1-Protein ohne Transit-Peptid, aber auch Verkürzungen, die nur den konservierten Regionen 1 bis 4 des AtSIG1-Proteins entsprechen. Abb. 4.4.1 zeigt die erstellten Verkürzungen schematisch. Für die Herstellung der Verkürzungskonstrukte wurde das Volllängen-Konstrukt aus Kapitel 4.1 verwendet, indem es als Matrize in einer PCR eingesetzt wurde. Die Fragmente wurden durch spezifische Oligonukleotide amplifiziert und anschließend in die Vektoren pBSIIKS (Stratagene) und pMAL (New England Biolabs) kloniert. Die Peptide wurden im E. coli-Stamm BL21-CodonPlus-RIL (Stratagene) mit einem MBP-Fusionsanteil synthetisiert. Die anschließende Reinigung der Fusionsproteine erfolgte äquivalent zu den anderen Reinigungen des pMAL-Systems (New England Biolabs) nach den Angaben des Herstellers (siehe auch Kapitel 4.1, Abb.4.1.1). Die isolierten Proteine 50 Ergebnisse konnten in ausreichender Menge und Qualität gewonnen werden, wie in Abb. 4.4.2A im Coomassie-gefärbten Proteingel gezeigt. Als Vergleich sind auch Proben des kompletten AtSIG1-Fusionsproteins und die Fusionsproteine MBP bzw. GST allein aufgetragen. Abb.4.4.1: Schematische Darstellung der AtSIG1-Proteinkonstrukte Zur Untersuchung der Interaktionsfähigkeit der AtSIG1- und AtSIB-Proteine wurde die Pull-down-Variante Protein-Overlay-Blot (nach Jelich-Ottmann et al., 2001) ausgewählt. Bei dieser Technik wurden Proteine mittels eines Proteingels elektrophoretisch aufgetrennt, auf Nitrocellulosemembran mittels eines WesternTransfers geblottet und diese Membran mit Magermilch-Lösung (2 % (w/v)) abgeblockt. Anschließend wurde die so vorbereitete Membran mit einem potentiellen Interaktor (200 µg) über Nacht bei 4°C in einem Bindungspuffer zusammen inkubiert. Nach drei Waschschritten erfolgte eine spezifische Immunodetektion gegen das Fusionsprotein des möglichen Interaktors. Für die in Abb. 4.4.2 dargestellten Ergebnisse wurden Proteingele mit den Proteinen AtSIG1, SIG1AS81-215, SIG1AS257-340, SIG1AS358-413 und SIG1AS438-502 für die Interaktion und den GST- und MBP-Proteinen als Kontrollen elektrophoretisch aufgetrennt. Ein Duplikat dieser Gele wurde Coomassie-gefärbt und als Kontrolle für Vergleichszwecke gezeigt (siehe Abb. 4.4.2). In allen Spuren wurden gleichwertige 51 Ergebnisse Proteinkonzentrationen (pro Spur 250 ng) nach einer Proteinbestimmung nach Bradford (1976) aufgetragen. Um nicht-spezifische Kreuzreaktionen zwischen den aufgetrennten Proteinen mit dem Primär-Antikörper gegen das GST-Protein (GE Healthcare) ausschließen zu können, wurde ein Blot nur in Bindungspuffer inkubiert und immunologisch detektiert. Eine spezifische Bindung war nur bei dem aufgetrennten Kontrollprotein GST (Spur 14) durch den primären Antikörper möglich. Als Sekundärantikörper wurde ein Anti-Ziege-IgG-Alkalische Phosphatase-Konjugat verwendet, welches eine sichtbare Farbreaktion durch BCIP und NBT auslöst (siehe Spur 14). Die Immunodetektion hat bei MBP- und seinen Fusionsproteinen keine sichtbare Farbreaktionen hervorgerufen (vergleiche Spuren 8 bis 13). Als zusätzliche Kontrolle wurde auch eine Inkubation mit BSA (200 µg) in Bindungspuffer durchgeführt, das zu einem mit 4.5.2A äquivalenten Ergebnis führte (Daten nicht gezeigt). In der Abb.4.5.2C wurde die geblockte Membran mit dem AtSIB1-Protein inkubiert. Die nachfolgende Immunodetektion zeigt Farbreaktionen bei dem Kontrollprotein GST, aber auch bei den Proteinen AtSIG1-TP, SIG1AS81-215, SIG1AS257-340, SIG1AS358-413 und SIG1AS438-502 (Spuren 15 bis 19 und 21). Diese Proteine wurden indirekt durch die Bindung mit dem AtSIB1-Protein angezeigt. Folglich bindet das AtSIB1-Protein nicht ausschließlich die konservierte Region 4 des AtSIG1-Proteins, sondern auch die ausgewählten Regionen von AS 81 bis 215, AS 257 bis 340, AS 358 bis 413 und AS 438 bis 502 des Sigmafaktors. Eine Bindung des AtSIB1-Proteins an das MBP-Protein kann ausgeschlossen werden, da dieses Protein nicht indirekt nachgewiesen wurde (Spur 20). Die Ergebnisse für das AtSIB2Protein sind in Abb. 4.4.2D gezeigt und stellen ein mit AtSIB1 vergleichbares Ergebnis dar. Die gezeigten Nachweise wurden durch mindestens fünfmalige Wiederholung verifiziert und durch revers durchgeführte Ansätze (Daten nicht gezeigt, fünf Wiederholungen) bestätigt. Die durchgeführten Interaktionsstudien zeigten eine Bindung der beiden AtSIBProteine mit allen ausgewählten AtSIG1-Konstrukten. Die Bindung an die konservierte Region 4 des Sigmafaktors durch die Proteine AtSIB1 und AtSIB2 konnte bestätigt werden. Es wurde auch eine Bindung an die Bereiche des AtSIG1 von AS 81 bis 215 entsprechend der konservierten Region 1, von AS 257 bis 340 entsprechend der konservierten Region 2 und von AS 358 bis 413 entsprechend der konservierten Region 3 durch die beiden AtSIB-Proteine gezeigt. 52 Ergebnisse Abb. 4.4.2: Interaktionsstudien zwischen AtSIG1-Konstrukten und beiden AtSIB-Proteinen mittels Protein-Overlay-Blots Unter A) ist ein Coomassie-gefärbtes Proteingel mit den aufgetrennten AtSIG1-MBPKonstrukten gezeigt: AtSIG1-TP (Spur 1), SIG1AS81-215 (Spur 2), SIG1AS257-340 (Spur 3), SIG1AS358-413 (Spur 4) und SIG1AS438-502 (Spur 5). Als Kontrollproteine wurden das MBP- und GST-Protein aufgetragen. (pro Spur 250 ng Protein) Für B) bis D) wurden Duplikate des Proteingels mittels Western-Transfer auf Nitrocellulosemembranen geblottet und geblockt. Zum Ausschluss von Kreuzreaktionen wurde ein Kontroll-Blot durchgeführt, der ü. N. nur mit Bindungspuffer bei 4°C inkubiert wurde (siehe B)). Die Interaktionsstudien wurden mit C) und D) durchgeführt, da die geblockten Membranen mit AtSIB1-Protein (C)) bzw. mit AtSIB2-Protein (D)) inkubiert wurden (jeweils 200 µg). Die Membranen wurden anschließend auf Immunodetektionen gegen das GST-Protein hin ausgewertet. Im Kontrollblot (B)) wurde nur das Kontrollprotein GST nachgewiesen. In den Blots C) und D) wurden durch die Proteine AtSIB1 bzw. AtSIB2 indirekt alle AtSIG1- 53 Ergebnisse Proteinkonstrukte detektiert. Das MBP-Protein, mit dem alle AtSIG1-Konstrukte fusioniert sind, wurde in keinem Blot nachgewiesen. Die gezeigten Ergebnisse wurden mindestens durch 5 Wiederholungen verifiziert und durch revers durchgeführte Experimente bestätigt. 4.5 Funktionsanalysen der Interaktion zwischen den AtSIG1- und den beiden AtSIB-Proteinen Das Protein AtSIG1 liegt in ausreichender Menge und Qualität vor und seine funktionelle Aktivität wurde durch DNA-Bindungsstudien nachgewiesen (siehe Kapitel 4.1). Eine Interaktion der rekombinanten AtSIB-Proteine konnte auch mit dem AtSIG1-Protein und Verkürzungsproteinen gezeigt werden (siehe Kapitel 4.4). Die Protein-Overlay-Blot-Technik ist eine Methode zum Nachweis der physischen Interaktion, liefert jedoch keine funktionellen Informationen. Für diesen Aspekt wurde daher wie zur Analyse der Grundfunktion – Promotor-Bindung und TranskriptionsInitiation (Burgess et al., 1969; Campbell et al., 2009) – der EMSA ausgewählt. Die Methode erlaubt nicht nur die schnelle Überprüfung der Promotor-Bindung, sondern auch die quantitative Auswertung der Intensität des Bindungssignals durch das Programm AIDA v4.04. Da die Promotorregionen von plastidären Genen teilweise aus sehr unterschiedlichen Sequenzelementen bestehen, sollte es möglich sein, innerhalb einer Promotorkollektion Präferenzen für den Sigmafaktor AtSIG1 allein und in Kombination mit AtSIB-Protein(en) vergleichend zu erfassen. Eine solche Präferenz kann auf die Transkription einer bestimmten Genklasse durch das AtSIG1-Protein hindeuten, welche durch die Interaktion mit einem AtSIB-Protein verändert werden kann. Abb. 4.5.1 stellt die Promotorelemente der ausgewählten Promotoren schematisch dar. Zu diesen Promotoren zählen der unter Kapitel 4.1 beschriebene psbA-Promotor, der aus S. alba stammt aber mit dem A. thaliana-Promotor identisch ist (Link, 1984; Tiller et al., 1991; Tiller und Link, 1993a,b; Homann und Link, 2003). Die Promotorelemente im psbA-Promotor sind die aus E. coli bekannten Regionen -35 und -10, welche durch ein „TATA-Box-ähnliches“ Motiv sowie eine „extended“-10Region verbunden sind. Über eine hohe Homologie im Konsensus-Bereich verfügen auch die S. alba-Promotoren trnK (Neuhaus und Link, 1987), trnQ (Neuhaus et al., 54 Ergebnisse 1989) und psaA (Summer et al., 2000), die ebenfalls verwendet wurden. Darüber hinaus wurden speziell im Rahmen der vorliegenden Arbeit die Promotoren der Gene rbcL und trnV (GAC) aus A. thaliana nach der Publikation von Hanaoka et al. (2003) isoliert, welche die konservierten Promotorelemente der -35- und -10-Region enthalten. Schließlich wurde auch der rps16-Promotor aus A. thaliana isoliert, der – wie sein Pendant aus S. alba (Neuhaus et al, 1989; Homann und Link, 2003) – eine -10-Region und ein „TATA-Box-ähnliches“ Sequenzmotiv besitzt. Abb. 4.5.1: Sequenzdarstellung der verwendeten plastidären Promotoren In der obigen Abbildung sind die plastidären Promotoren: psbA (Link, 1984), trnK (Neuhaus und Link, 1987), trnQ (Neuhaus et al., 1989), psaA (Summer et al., 2000), rbcL und trnV (GAC) (Hanaoka et al., 2003) und rps16 (Neuhaus et al., 1989). Die Promotorelemente -10-, -35-Regionen, die TATA-Box-ähnlichen Sequenzmotive und das „extented“-10-Motiv sind farblich in den Sequenzen gekennzeichnet. Die prokaryotische Konsensussequenz (E. coli σ70-Promotoren; Murakami und Darst, 2003, Browning und Busby, 2004) ist im oberen Bereich angegeben. Bei der Funktionsanalyse durch die Bindung der AtSIB-Proteine an das AtSIG1Protein wurde zu einem Standard-Reaktionsansatz entweder das AtSIB1- oder das 55 Ergebnisse AtSIB2-Protein zugefügt. Ein Standard-Reaktionsansatz bestand aus isolierter coreRNA-Polymerase aus E. coli (Epicentre), einem radioaktiv markierten PromotorFragment, AtSIG1-Protein und PolyIdC zur Unterdrückung unspezifischer Bindungen. Die Auftrennung der Ansätze fand in nativen Polyacrylamid-Gelen statt, die getrocknet und deren radioaktive Signale mit Hilfe des Phospho-Imagers FLA-3000 und dem Programm AIDA v4.04 ausgewertet wurden. Die in Abb. 4.5.2 gezeigten Ergebnisse basieren auf mindestens drei unabhängigen Experimenten und reproduzierten Werten. Für jeden Promotor wurden Kontrollen durchgeführt, welche die Spezifität der Bindungssignale bestätigen sollen. Dazu zählen die allein aufgetragenen radioaktivmarkierten Promotor-Fragmente (siehe Spuren 1 für alle Promotoren), die auch als Vergleiche für mögliche Signal-Verschiebungen dienen. Die Bindung an den Promotor kann weder durch das AtSIG1-Protein (Spuren 2) noch das core-Enzym (Spuren 3) allein vermittelt werden. Die Bildung des holo-Komplexes - bestehend aus core und AtSIG1 – führt zu einer Signalverschiebung und zeigt eine Bindung – wenn auch eine relativ schwache – an den Promotor (Spuren 7). Die beiden Fusionsanteile – MBP und GST – können diese Bindung zusammen mit dem core-Enzym nicht vermitteln (Spuren 5 und 6). Starke und spezifische Signale vermittelt der E. coli holo-RNA-Polymerase-Komplex (Epicentre), welcher aus dem core-Enzym und dem Standard-Sigmafaktor σ70 besteht (Spuren 4). Eine wichtige Negativkontrolle umfassten Reaktionsansätze mit Sonde, core und Sigma-Bindeprotein AtSIB1 oder AtSIB2 allein in Abwesenheit von AtSIG1 (Spuren 11 und 9). Die Reaktionsansätze aus core-, AtSIB2 oder AtSIB1 sowie zusätzlich AtSIG1 (Spuren 8 bzw. 10) führen dagegen zu einer – unerwartet starken – spezifischen Signalverschiebung (vergleiche das relativ schwache Bindungssignal in Spur 7, d.h., ohne AtSIB1 oder AtSIB2). Ebenfalls zu einem Promotor-Bindungssignal führt eine Interaktion des verkürzten AtSIG1-Protein (AS81-413, Kapitel 4.2) mit AtSIB2 in core-Anwesenheit (Daten nicht gezeigt). Somit ist die konservierte Region 4 nicht allein das Ziel der funktionellen Bindung durch die beiden Interaktoren AtSIB1 und AtSIB2. 56 Ergebnisse Abb. 4.5.2: EMSA-Studien mit den rekombinanten AtSIG1- und AtSIB-Proteinen Die Promotor-Fragmente:psbA (Link, 1984), trnK (Neuhaus und Link, 1987), trnQ (Neuhaus et al., 1989), psaA (Summer et al., 2000), rbcL, trnV (GAC) (Hanaoka et al., ) und rps16 (Neuhaus et al., 1989) wurden radioaktiv markiert und in GelBindungstests eingesetzt. Als Negativ-Kontrollen wurden die Promotoren (Spuren 1), das AtSIG1-Protein (Spuren 2) und das core-Enzym (Spuren 3) allein eingesetzt. Die Fusionsproteine GST und MBP (Spuren 5 und 6) konnten genauso wie die beiden 57 Ergebnisse AtSIB-Proteine (Spuren 9 und 11) zusammen mit dem core-Enzym keine PromotorBindung vermitteln. Als Positiv-Kontrolle wurde die E. coli holo-RNA-Polymerase (Spuren 4) verwendet worden. Die Signale der AtSIG1-core-Komplexe sind in den Spuren 7 gezeigt, welche mit den Interaktionsansätzen aus AtSIG1/AtSIB2- (Spuren 8) und AtSIG1/AtSIB1-Interaktionen (Spuren 10) verglichen werden sollen. Die dargestellten Signale basieren auf einem nativen Polyacrylamid-Gel, das getrocknet mittels eines Phospho-Imagers (FLA-3000) analysiert und durch mindestens drei unabhängige Durchführungen reproduziert wurde. Abk.: b: Bindungssignal, f: freie Sonde. Im weiteren Verlauf dieser Untersuchungen wurden vergleichende EMSAs mit verschiedenen Promotoren durchgeführt. Die Promotoren der Gene psbA, rbcL und trnK wurden mit beiden Bindeproteine getestet, diejenigen von trnV (GAC), trnQ und rps16 nur mit AtSIB2. In allen Fällen führten Interaktionen zwischen AtSIG1- und den AtSIB-Proteinen in Gel-Bindungsstests zu einer erhöhten Bindung an den eingesetzten Promotor. Zusammengefasst lässt sich feststellen, dass AtSIG1 durch einen AtSIB-Bindungspartner extrem aktiviert zu werden scheint. Die Bindungseffizienz des AtSIG1-Proteins ist im Vergleich zu dem SIB-gebundenen AtSIG1-Protein als schwach zu betrachten. 4.6 Bioinformatische Analyse der Sigma-Bindeproteine Nach der Betrachtung des AtSIG1-Proteins und seiner funktionellen Veränderungen durch die Interaktion mit seinen spezifischen Bindeproteine AtSIB1 und AtSIB2, sollten sowohl die Struktur als auch die Verbreitung der beiden Interaktoren mittels in silico Analysen genauer beschrieben und identifiziert werden. Beide Proteine werden im Kern nur durch ein Exon kodiert und nach Expression über ein plastidäres Transit-Peptid in die Plastiden transportiert. Dies wurde zumindest für AtSIB1 über Chloroplastenimport-Analysen mit GFP-Fusionsproteinen nachgewiesen (Morikawa et al., 2002). Die Homologie der Proteine zueinander liegt bei 56%. Diese Homologie basiert größtenteils auf einem konservierten Sequenzmotiv, welches auch in einem Sequenzvergleich der beiden Proteine gezeigt und hervorgehoben ist (siehe auch Abb. 4.6.1). 58 Ergebnisse Das Sequenzmotiv wurde durch Bateman (pers. Beobachtungen) im Pfam-Eintrag PF05678 der Pfam-Datenbank des Welcome Trust Sanger Institute erstmals genauer beschrieben und charakterisiert (Finn et al., 2008). Das Motiv wurde nach den beiden hochkonservierten Aminosäuren Valin und Glutamin als VQ-Motiv bezeichnet und ist durch die Abfolge folgender Aminosäuren: FXhVQChTG (X: jede Aminosäure, h: hydrophobe Aminosäure) charakterisiert. Es kommt in A. thaliana mindestens in 34 Proteinen vor und ist auch in anderen Pflanzen weit verbreitet. In nicht-pflanzlichen Organismen wurde noch kein Protein mit VQ-Motiv identifiziert (Bateman, Andreasson et al., 2005). Eine Funktion des VQ-Motives konnte bisher nicht gezeigt werden, da die bisher analysierten Proteine keine gemeinsamen Funktionen aufweisen wie bspw. MKS1 und ein Calmodulin-Bindeprotein (Andreasson et al, 2005). Da aber bisher unbekannt ist, ob andere Pflanzen Proteine enthalten, die den SIBProteinen in Bezug auf ihre Funktion als Sigma-Bindeproteine ähnlich sind, wurde zu Beginn eine Sekundärstruktur-Analyse mit beiden Proteinen durchgeführt, um weitere auffällige Sequenzen identifizieren zu können. Dafür wurde das VorhersageProgramm PSI-PRED v2.6 (Jones, 1999) verwendet, welches für das VQ-Motiv eine Helixstruktur vorhersagt. Es wurde auch eine Faltblatt-Struktur vor dem VQ-Motiv in beiden Proteinsequenzen erwartet. Die Abb. 4.6.1 zeigt den Sequenzvergleich zusammen mit der vorhergesagten Sekundärstruktur der jeweiligen Proteinsequenz im direkten Vergleich. Die hier vorgestellte bioinformatische Sekundärstrukturanalyse identifizierte eine zweite konservierte Sequenz mit einem charakteristischen Sequenzmotiv in beiden AtSIB-Proteinen. Dieses Motiv wurde als Grundlage verwendet, um mit Hilfe von Datenbankanalysen nach SIB-ähnlichen Proteinen auch in anderen Organismen zu suchen. Das Ziel war es zu ermitteln, ob das Vorkommen von SIB-Proteinen auf A. thaliana beschränkt oder eher weiter verbreitet ist. Der in silico Ansatz begann mit der Auswahl der Daten aus den Datenbanken NCBI (National Center for Biotechnology Information) und Phytozyme v4.1 des Joint Genome Institute, welche zusammen eine große Vielfalt von vollständig sequenzierten Genomen frei zugänglich machen. 59 Ergebnisse Abb. 4.6.1: Sequenzalignment mit Sekundärstrukturanalyse der AtSIB-Proteine Die Abbildung zeigt ein Sequenzalignment basierend auf den Algorithmen des BLAST-Programms (Altschul et al., 1997 und 2005) der beiden AtSIB-Proteine (AtSIB1: AT3G56710 und AtSIB2: AT2G41180). Die mittlere Zeile gibt die übereinstimmenden Aminosäuren beider Proteine an. Mit jeder Proteinsequenz wurde eine Analyse der Sekundärstruktur mit dem Programm PSI-PRED v2.6 (Jones, 1999) durchgeführt. Die Vorhersage der Sekundärstruktur gliedert sich in coil-, Faltblatt- und Helix-Strukturen, welche durch Strich, Pfeil und Tonne dargestellt werden. Das VQ-Sequenzmotiv ist durch einen roten Kasten hervorgehoben und durch einen blauen Kasten die konservierte Sequenz stromaufwärts des VQ-Motives. Das allgemeine VQ-Motiv ist durch die Sequenz FXhVQChTG (X: jede Aminosäure, h: hydrophobe Aminosäure) charakterisiert. Für die Sequenzvergleiche wurde das Programm BLAST verwendet, das mit der Matrix BLOSUM62 arbeitet. Es wurde kein Protein mit den Sequenzmotiven in Hefen, Pilzen oder Algen identifiziert, aber ein weiteres SIB-ähnliches Protein in A. thaliana. (nachfolgend als „AtSIB3“ bezeichnet). Alle identifizierten Proteine stammen aus mono- und dikotylen Pflanzen und sind in Tabelle 4.6.1 dargestellt. Insgesamt handelt es sich um 25 Proteine aus 11 Pflanzen, die alle eher klein mit einer Länge 60 Ergebnisse von 120 bis 220 Aminosäuren sind und bisher keine bekannte Funktion haben. Eine Untersuchung der Proteine auf plastidäre Transit-Peptide erfolgte mit dem Programm ChloroP (Emanuelsson et al., 1999) und führte zu einem eher gemischten Ergebnis. Nach diesen Daten haben alle Pflanzen mindestens ein Protein mit einem „typischen“ plastidären Transit-Peptid. AtSIB3, das SIB-ähnliche Protein aus A. thaliana verfügt allerdings (im Unterschied zu AtSIB1 und AtSIB2) nicht über ein solches. Dabei sollte jedoch berücksichtigt werden, dass inzwischen generell zunehmend auch „nichtorthodoxe“ Transitpeptide für plastidenorientierte Proteinvorläufer beschrieben wurden (Emanuelsson et al., 2000). Tabelle 4.6.1: Bioinformatisch identifizierte SIB-ähnliche Proteine 61 Ergebnisse Die Sequenz des VQ-Motivs nach der Pfam-Datenbank (Eintrag PF05678) wird wie folgt beschrieben: FXhVQChTG (X: jede Aminosäure, h: hydrophobe Aminosäure) und wurde mit den identifizierten Proteinen verglichen, um eine mögliche Untergruppe der Familie von VQ-tragenden Proteinen erkennen zu können. Diese Gruppe wird dann auch durch das stromaufwärts des VQ-Motiv liegende Sequenzmotiv charakterisiert. Dafür wurde mit dem Programm CLUSTALW2 (Larkin et al., 2007) ein Vergleich aller identifizierten Proteinsequenzen durchgeführt. Ein Ausschnitt aus diesem Vergleich zeigt die beiden Motive (siehe Abb. 4.6.2A) in allen Sequenzen unter Angabe der Motivsequenzen. Die Sequenz für das Prä-VQ-Motiv lautet KhXYI und das SIB-VQ-Motiv hat die Aminosäureabfolge von FREALVQELTG. Der Sequenzvergleich ist auch die Grundlage für einen phylogentischen Stammbaum, basierend auf dem Neighbor-Joining Algorithmus, der die Ähnlichkeiten der identifizierten Proteine zueinander bildlich darstellt (siehe Abb. 4.6.2B). Es zeigt sich, dass häufig zwei identifizierte Proteine der gleichen Pflanze eine große Ähnlichkeit zueinander haben, vergleichbar der Situation der beiden AtSIB-Proteine aus A. thaliana (siehe Abb. 4.6.1 und 4.6.2B). Insgesamt hat die bioinformatische Analyse der AtSIB-Proteine gezeigt, dass in anderen Pflanzen auch Proteine mit einer hohen Sequenzähnlichkeit vorkommen. Es konnten 27 Proteine aus 11 Pflanzen identifiziert werden, die gemeinsam über die beiden Sequenzmotive „KhXYI“ und „FREALVQELTG“ mit den wahrscheinlichen Sekundärstrukturen eines Faltblatt und einer Helix verfügen. Die Schlussfolgerung könnte somit sein, dass SIB-Proteine als Interaktionspartner von pflanzlichen Sigmafaktor(en) eine universelle Rolle bei pflanzlichen Organismen spielen. 62 Ergebnisse Abb. 4.6.2: Analyse der Sequenz und der Phylogenie von VQ-Motiv enthaltenden Proteinen aus verschiedenen Pflanzenarten A) Es ist ein Ausschnitt des Sequenzvergleichs von VQ-Motiv enthaltenen Proteinen gezeigt. Der Vergleich wurde durch das Programm CLUSTALW2 (Larkin et al., 2007) erstellt und wird durch die vorhergesagte Sekundärstruktur der Sequenzen (Programm PSI-Prediction v2.6) vervollständigt. Das bekannte VQ-Motiv (FXhVQChTG, X: jede Aminosäure, h: hydrophobe Aminosäure) wird im Vergleich zum SIB-VQ-Motiv (FREALVQELTG) gezeigt, welches durch das stromaufwärts liegende Motiv (KhXYI) vervollständigt wird. 63 Ergebnisse B) Darstellung eines phylogenetischen Stammbaums, der auf dem Sequenzvergleich aus A) basiert und mit dem Neighbor-Joining Algorithmus berechnet wurde. Die eingezeichneten Kreise verdeutlichen die nahe verwandten VQ-Proteine innerhalb einer Spezies. 4.7 Analyse der plastidären Genexpression des sig1-Gens in einer sig1-1 Einzel-Knockout-Linie Der Sigmafaktor 1 in ausgewachsenen Exemplaren der Modellpflanze A. thaliana (Rosettenstadien) einer der häufigsten Vertreter der Sigmafamilie. In jungen Stadien der Keimlingsentwicklung ist er hingegen wenig abundant und kann erst nach sieben Tagen in der Pflanze nachgewiesen werden (Tsunoyama et al., 2004). Dieser Faktor verfügt bisher als einziger über bekannte Interaktoren (SIB1 + SIB2), die eine regulatorische Funktion auf diesen Faktor haben. Morikawa et al. (2002) vermuteten eine Funktion als Anti-Sigmafaktor, welche in Bakterien weit verbreitet sind und dort die bakterielle Transkription regulieren können (Hughes und Mathee, 1998). Auf diesen grundlegenden Befunden wurde im Rahmen der vorliegenden Arbeit aufgebaut. Zur Charakterisierung der Interaktionen zwischen AtSIG1 und beiden AtSIB-Proteinen wurde eine sig1 Knockout-Pflanze ausgewählt, um den Einfluss von Sigmafaktor 1 und seinen putativen Regulatoren auf die plastidäre Genexpression zu prüfen. Zu diesem Zweck wurde die geeignet erscheinende Knockout-Linie 758B02 aus der Sammlung der GABI-Kat-Linien des Max-Planck-Institus für Züchtungsforschung ausgewählt (Rosso et al., 2003). Diese Linie weist eine T-DNAInsertion 2524 Nukleotide stromabwärts des Startcodons in Exon 8 des sig1-Gens (At1G64860) auf. Die T-DNA verhindert aufgrund ihrer Größe eine Transkription des sig1-Gens. Die Lokalisation der T-DNA erfolgte durch die Sequenzierung der Begrenzungsbereiche (rechte und linke Begrenzungen: RB: right border; LB: left border) der T-DNA. Die Abbildung 4.7.1A stellt die sig1-Genstruktur genauer dar und verdeutlicht die Einbaustelle der T-DNA ebenfalls. Da zu Beginn meiner Arbeit schon eine stabile und homozygote Linie aus dem Saatgut der GABI-Kat-Sammlung identifiziert worden war (Loschelder, persönliche Mitteilung), begann meine Arbeit mit der Vermehrung des Saatgutes für die 64 Ergebnisse weiterführenden Arbeiten. Die homozygote Linie wies keinen sichtbaren Phänotyp auf, d.h. sie entspricht vom äußeren Erscheinungsbild dem Wildtyp wie in Abbildung 4.7.1B gezeigt. Basierend auf dieser Beobachtung wurden auch die nachfolgenden Generationen auf ihre Homozygotie analysiert. Dies erfolgte durch genomische PCR, wie in Abbildung 4.7.1C beispielhaft dargestellt ist. Dafür wird mit spezifischen sig1Oligonukleotiden (siehe 3.1.5) versucht, ein DNA-Fragment des Sigmafaktors zu amplifizieren. Abb. 4.7.1.: Merkmale der sig1-Knockout-Linie A) Schematische Darstellung der Exon-Intron-Struktur des Atsig1-Gens mit gekennzeichneter T-DNA-Insertionsstelle (2524 Nukleotide stromabwärts des Startcodons in Exon 8). Das sul-Gen in der T-DNA stellt die Resistenz gegen Sulfadiazin dar. Dem Schema sind auch die abgeleitete mRNA und Protein zu entnehmen. [TP: Transit-Peptid, UR: unkonservierte Region, 1-4: konservierte Region 1-4] B) Phänotyp des Wildtyps Col0 im Vergleich zur sig1-Knockout-Mutante nach Kultivierung unter Kurztagbedingungen für 3 Tage. 65 Ergebnisse C) Beispielhafter Homozygotie-Nachweis mittels genomischer PCR. Durch Sig1spezifische Oligonukleotide wird bei keiner T-DNA-Insertion ein Bereich von 859 Nukleotiden (siehe A), rot markiertes Amplifikat) amplifiziert. Eine entsprechende Amplifikation eines 800 bp Produktes war nur beim Wildtyp möglich. Die T-DNA-Insertion in der Knockout-Linie hat höchstwahrscheinlich eine nur ineffiziente (oder völlig fehlende) Amplifikation des Fragmentes aufgrund der Vergrößerung des Fragmentes zur Folge. Somit ist die verwendete Mutanten-Linie trotz Abwesenheit eines sichtbaren Phänotyps homozygot, und es folgte eine Analyse der plastidären Transkripte, um Effekte basierend auf den Verlust des Sig1Genproduktes zu erkennen. Dafür wurden das Saatgut unter Standard-Bedingungen im Kurztag (8 h Licht, 60 μmol m2s-1, 24°C, 16 h Dunkelheit bei 20°C) angezogen. Veränderungen der plastidären Transkripte in der sig1 Knockout-Pflanze im Vergleich zum Wildtyp liefern Informationen über die Funktion des SIG1-Proteins und damit auch über seine Interaktoren. Es wird aber auch unser Gesamtbild von der Regulation der plastidären Transkription durch die Sigmafaktoren vervollständigt. Dies sollte auf Ebene der RNA mit ausgewählten plastidären Zielgenen geschehen. Dafür wurden Pflanzen in einer Spanne von 7 bis 28 Tagen angezogen, geerntet, die RNA aufgearbeitet und anschließend Northern Blot Analysen durchgeführt. Die ausgewählten Zielgene entsprachen den Genklassen I-II, welche zusammen mit der Genklasse III durch (NEP transkribierte Gene) die funktionellen Gengruppen des Plastoms darstellen (zur Übersicht siehe Einleitung und Shiina et al., 2005). Die Gene der Klasse I werden durch die PEP transkribiert und durch die Transkripte von psbA und rbcL sowie durch das Vorläufer-Transkript des trnV-Gens repräsentiert. Für alle diese Transkripte konnten keine Unterschiede zwischen der sig1-Mutante und dem Wildtyp beobachtet werden. Folglich zeigt die Abbildung 4.7.2 auch keine Unterschiede in der Signalstärke bei den dargestellten Transkripten trotz gleicher RNA-Mengen. Die jeweils eingesetzte RNA-Menge ist anhand der 25S rRNA-Banden gezeigt, die aus den Ethidiumbromid-gefärbten RNA-Gelen stammen und die Vergleichbarkeit der Signale verdeutlichen. Die Genklasse II transkribiert sowohl NEP als auch PEP und wurde durch die Transkripte des atpB- und atpE-Gens repräsentiert. Auch diese Expressionsmuster weisen keine Unterschiede zwischen der sig1-Mutante und dem Wildtyp im direkten Vergleich auf wie in der Abbildung 4.7.2 gezeigt. Da der sig1-Knockout-Mutante ein sichtbarer Phänotyp auch auf 66 Ergebnisse molekularer Ebene unter Standard-Bedingungen fehlt, sind gravierende und generelle Folgen des Verlust des sig1-Genproduktes auszuschließen. Abb. 4.7.2: Northern Blot Analysen der sig1 Knockout Linie im Vergleich zum Wildtyp Die isolierte Gesamt-RNA aus Keimlingen unterschiedlichen Alters (7, 10, 14 & 28 Tagen) wurde aufgetrennt (1 µg pro Spur), geblottet und mit DIG-markierten Sonden hybridisiert. Die Sonden umfassten die plastidären Genklassen I & II und die Ergebnisse sind entsprechend auf der linken Seite (unter Angabe der Klasse und der verwendeten Sonde) gekennzeichnet. Die 25S rRNA-Bande spiegelt die Verwendung gleicher RNA-Mengen wider. Der obere Bereich gibt das Alter und die Unterscheidung zwischen Wildtyp (WT) und der sig1 Knockout-Mutante (MT) an; auf der rechten Seite werden die Größen der Transkripte in kb (nach dem Promega-RNAMarker) gezeigt. Die zugrundeliegenden Hybridisierungen wurden mindestens dreimal mit RNA aus unterschiedlichen Isolierungen wiederholt. 67 Ergebnisse 4.8 Analyse der Phänotypen von sig1 und sig6 Einzel-Knockout-Linien und von den Doppel-Knockout-Linien sig1xsig6 und sig6xsig1 unter unterschiedlichen Licht-Bedingungen Da das Fehlen des sig1-Gens zu keinen gravierenden Folgen in der Knockout-Linie führt, wurden Kreuzungen mit der bereits charakterisierten sig6-Knockout-Linie durchgeführt. Die sig6-Knockout-Linie hat einen stark ausgeprägten visuellen und molekularen Phänotyp und ist in unserer Arbeitsgruppe eingehend untersucht (Loschelder et al., 2006; Schweer et al., 2006). Die erfolgreiche Kreuzung wurde durch genomische PCR überprüft. Eine Darstellung der entsprechenden Gele ist in Abb. 4.8.1 gezeigt. Abb. 4.8.1: Homozygotie-Nachweis der Doppelmutanten Der Nachweis des sig1- und sig6-Gens erfolgte durch genomische PCR mittels Sigma-spezifischer Oligonukleotide. Die amplifizierten Genbereiche haben eine Größe von 859 nt (sig1) bzw. von 873 nt (sig6). Die fehlenden Amplifikate korrelieren mit der inserierten T-DNA in den entsprechenden Genbereichen. Für Analysen unter erhöhten Stress-Bedingungen wurden die vorhandenen Einzelund Doppel-Knockout-Linien wie auch der Wildtyp sterilisiert und unter verschiedenen Lichtbedingungen für 7 und 14 Tage kultiviert. Die Lichtbedingungen waren der Kurztag (8 h Licht, 60 μmol m2s-1, 24°C, 16 h Dunkelheit bei 20°C), der Langtag (16 h Licht, 60 μmol m2s-1, 24°C, 8 h Dunkelheit bei 20°C) und Dauerlicht (24 h Licht, 60 µmol m2s-1, 25°C). 68 Ergebnisse Abb. 4.8.2: Phänotypen der sig1 und sig6 Einzel- und Doppel-KnockoutMutanten im Vergleich zum Wildtyp Col0 unter verschiedenen Lichtbedingungen Es wurden die Lichtbedingungen Kurztag (Abk.: KT, 8 h Licht, 60 μmol m2s-1, 24°C, 16 h Dunkelheit bei 20°C), Langtag (Abk.: LT, 16 h Licht, 60 μmol m2s-1, 24°C, 8 h Dunkelheit bei 20°C) und Dauerlicht (Abk.: Dauer, 24 h Licht, 60 µmol m2s-1, 25°C) 69 Ergebnisse verwendet um Keimlinge für 7 und 14 Tagen auf MS-Platten zu kultivieren. Der Wildtyp Col0 und die sig1 Mutante verfügen unter allen 3 Lichtbedingungen über einen grünen und kräftigen Phänotyp. Die sig6 Mutante und beide Doppelmutanten weisen weiße Kotyledonen auf, denen im Kurztag grüne Blätter folgen und im Langtag und Dauerlicht weiße bis schwach grüne Blätter folgen. In Abb. 4.8.2 sind die Phänotypen gezeigt, die die Linien unter diesen Bedingungen zeigten. Anhand dieser Abbildungen ist erkennbar, dass unter den KurztagBedingungen die sig1-Knockout-Linie einen mit dem Wildtyp Col0 vergleichbaren Phänotyp aufweist, der ohne Unterschiede grüne Kotyledonen und Blätter hat. Diese Beobachtung bleibt auch für die beiden anderen Lichtbedingungen unter allen gezeigten Stadien der Keimlinge bestehen. Die sig6-Knockout-Linie weist unter Kurztag-Bedingungen einen typischen Phänotyp mit weißen Kotyledonen auf. Der Phänotyp wird unter Langtag- und Dauerlicht-Bedingungen noch stärker, indem die Folgeblätter hellgrün bis weiß sind und damit den Kotyledonen ähneln. Die beiden Doppel-Knockout-Linien sig1xsig6 und sig6xsig1 gleichen in ihren Phänotypen beide der sig6-Knockout-Linie unter allen Lichtbedingungen. Folglich führten auch keine Veränderungen in den Belichtungsbedingungen zu einem veränderten Phänotyp der Einzel- und Doppel-Knockout-Linien von sig1. 70 Diskussion 5. Diskussion Das Ziel dieser Arbeit war die funktionelle Charakterisierung des SIG1-Proteins und seiner Interaktionsproteine SIB1 und SIB2 aus A. thaliana. Im Modellorganismus A. thaliana sind sechs Sigmafaktoren bekannt, die überwiegend in internationaler Kooperation schon sehr umfassend funktionell charakterisiert werden konnten. Eine Ausnahme bildet das – eher häufige – AtSIG1-Protein, über welches zum Beginn der vorliegenden Arbeit kaum funktionelle Informationen vorhanden waren. Dies ist umso bemerkenswerter, als es schon seit mehreren Jahren bekannt ist, dass dieser Faktor zwei Interaktionspartner, AtSIB1 und AtSIB2, besitzt (Morikawa et al., 2002). Auch die Interaktionen zwischen AtSIG1 und den AtSIB-Proteinen waren in der Zwischenzeit nicht weiter spezifisch und funktionell charakterisiert worden. Um dieses Informationsdefizit zu reduzieren, wurden rekombinante Proteine synthetisiert und deren funktionelle Bindungsspezifität mittels in vitro Studien mit Verkürzungskonstrukten und Punktmutanten vom AtSIG1 charakterisiert. Mit Hilfe einer sig1-knockout-Linie von A. thaliana wurden komplementär zum in vitro Ansatz die Auswirkung des Genverlustes auf die plastidären Transkripte untersucht. 5.1 Funktionsanalysen des rekombinanten AtSIG1-Proteins und ausgewählter Konstrukte Für eine Funktionsanalyse vom AtSIG1 wurde das Protein in ausreichender Qualität und Quantität benötigt. Diese Aufgabe wurde gelöst, indem das Gen als cDNA isoliert, in E. coli transformiert und das Protein durch Überexpression, Zellextraktion und Affinitätschromatographie gewonnen wurde. Die Funktionsfähigkeit der rekombinanten AtSIG1-Proteine wurde mittels Gel-Bindungstest – kurz EMSA – geprüft. Diese Methode nutzt die Tatsache aus, dass Protein-DNA-Komplexe in nativer Gelelektrophorese langsamer laufen als ungebundene DNA (Link, 1984; Tiller et al., 1991; Tiller und Link, 1993a,b; Homann und Link, 2003). Sigmafaktoren bilden 71 Diskussion sowohl in Prokaryoten als auch in Plastiden zusammen mit der core RNAPolymerase das holo-Enzym, welches selektiv an Promotoren binden kann und eine spezifische Initiation möglich macht. Isolierte Sigmafaktoren außerhalb des Polymerase-Komplexes binden nicht an die Promotor-DNA, dieses wird erst durch eine Konformationsänderung des Faktors als Teil des holo-Enzym-Komplexes möglich (Dombroski et al., 1992; Campbell et al., 2009). Vorarbeiten der Arbeitsgruppe hatten gezeigt, dass authentische, biochemisch gereinigte, plastidäre Sigmafaktoren aus S. alba zusammen mit der core-RNAPolymerase aus E. coli zu einem „gemischten“ holo-Enzym zusammen treten können (Homann und Link, 2003). Dieses Verhalten wurde auch in den hier durchgeführten EMSA-Studien ausgenutzt, indem gereinigte E. coli core-RNA-Polymerase mit rekombinantem Sigmafaktoren und einem radioaktiv-markierten Promotor inkubiert wurden. Nach der Auftrennung konnten gebildete Protein-DNA-Komplexe über eine Signalverschiebung im Vergleich zur ungebundenen Sonde identifiziert werden. Die Spezifität der DNA-Bindung wurde durch Positiv- und Negativ-Kontrollen abgesichert und durch die Verwendung des nicht-selektiven doppelsträngigen „Kompetitors“ poly[dIdC] erhöht, in dessen Gegenwart die Wirkung etwaiger unspezifisch bindender Proteine minimiert werden kann (Tiller und Link, 1993b). Für das rekombinante AtSIG1-Protein konnte eine EMSA-Signalverschiebung gezeigt werden, welche die Fähigkeit der Bindung an das core-Enzym nachweist. Die Spezifität der Promotorbindung wurde durch Kompetitionsexperimente belegt: die Anwesenheit von nicht-radioaktiv-markierten psbA-Promotorfragmenten im Überschuss führte zu einer starken bis vollständigen Reduzierung des radioaktiven Bindungssignals. Diese Reduktion basiert auf der Konkurrenz der markierten bzw. nicht-markierten Fragmente mit dem gleichen Promotor um vorhandene holoEnzyme. Das Bindungssignal wurde dagegen nicht in seiner Intensität geschwächt, wenn der rpoB-Promotor als nicht-markiertes „Wettbewerber“-Fragment eingesetzt wurde. Die fehlende Kompetitionswirkung ist in diesem Fall darauf zurückzuführen, dass der rpoB-(NEP)-Promotor vom Sigma-core-Komplex nicht erkannt und nicht benutzt wird. Der Vergleich der Intensität der Signalverschiebung zwischen der E. coli holo-RNA-Polymerase mit dem Haupt-Sigmafaktor SIGMA70 und dem heterologen holo-Enzym aus E. coli core-RNA-Polymerase und rekombinantem AtSIG1-Protein zeigt eine deutlich geringere Bindungseigenschaft im letzten Fall. In 72 Diskussion den publizierten Vorarbeiten von Homann und Link (2003) war mit rekombinantem SIG1 aus S. alba eine mit der E. coli holo RNA-Polymerase vergleichbare Intensität des Verschiebungssignals sichtbar. Dieser graduelle Unterschied könnte darauf zurückzuführen sein, dass das in der vorliegenden Arbeit verwendete E. coli holoEnzym (Epicentre) einen höheren Sigma-Anteil als das früher verwendete, im Labor selbst hergestellte Präparat, aufweist und somit aktiver ist. Für die weiterführende Analyse der Bindungseffizienz des AtSIG1-Proteins wurden ausgewählte Punktmutanten durch ortsgerichtete Mutagenese in den konservierten Regionen 2 und 4 erstellt und analysiert. Die Auswahl basierte auf publizierten Vorarbeiten von Homann und Link (2003), sowie eigenen Sequenzvergleichen zwischen den SIG1-Proteinen aus A. thaliana und S. alba. Die ausgewählten Aminosäuren befanden sich an den Positionen 321, 458, 460, 463, 465, 467, 471 und 476 (Abb.5.1 und schematisch Abb. 4.2.1). Die Konstrukte wurden äquivalent zum „Wildtyp“-AtSIG1-Protein rekombinant synthetisiert, isoliert und zu Beginn mittels CD-Spektroskopie untersucht. Die Methode der CirculardichroismusSpektroskopie basiert auf der Absorption von zirkular polarisiertem Licht durch optisch aktive Moleküle wie Peptide und Proteine. Diese Absorption kann durch das Spektrometer gemessen und computergestützt quantifiziert werden; im Ergebnis werden Informationen über die Konformation der analysierten Proteine geliefert. Die gemessenen Spektren der AtSIG1-Konstrukte waren sich alle sehr ähnlich, was darauf hindeutet, dass die eingefügten Mutationen keine gravierenden strukturellen Veränderungen bewirkten. Die größten Abweichungen zeigten die Spektren der Proteine mit Mutationen der Aminosäuren 471, 476 oder 465. Die CD-Spektroskopie konnte nur Aussagen über die Konformation der analysierten Konstrukte liefern, aber keine Informationen über Funktionsveränderungen der Proteine durch die mutierten Aminosäuren. Um diesen Aspekt zu untersuchen, wurden Gel-Bindungstests wie schon zur Funktionsprüfung des AtSIG1-Proteins durchgeführt. Als Kontrolle wurde unter anderem das Verkürzungskonstrukt SIG1AS81-413 eingesetzt, dem die konservierte Region 4 fehlt. Der Funktionsvergleich der Proteine erfolgte durch die Quantifizierung der Intensität ihrer Bindungssignale im Verhältnis zum AtSIG1-Protein. Das verkürzte Kontrollprotein SIG1AS81-413 zeigte eine mit dem AtSIG1-Protein vergleichbare Bindung an den im Testansatz vorhandenen psbA-Promotor. Dies wird durch die 73 Diskussion Promotorstruktur des psbA-Gens erklärt, welche neben den -35- und -10-Regionen unter anderem ein extended -10 Motiv aufweist. Das Motiv vermittelte im C-terminal verkürzten Sigmafaktor SIGMA70 eine spezifische Promotorerkennung und Transkriptionsinitiation. Ein bakterieller Sigmafaktor kann „normalerweise“ ohne die konservierte Region 4.2 nicht mehr einen typischen bakteriellen Promotor aus -35und -10-Region erkennen und so die Transkription einleiten. Bei Vorhandensein eines extended -10-Motiv ist eine Transkription jedoch auch mit dem verkürzten Sigmafaktor möglich (Kumar et al., 1993). Die meisten AtSIG1-Punktmutanten zeigten auch nur einen geringen Verlust an DNA-Bindungsfähigkeit von 10% bis 30%. Zusammenfassend haben die Aminosäuren der Positionen 321, 458, 460, 463, 465 und 467 keine spezifische Funktion bei der Erkennung und Bindung des psbAPromotors, der sehr starke Promotorelemente aufweist. Für die Mutation der Aminosäure 321 deckt sich diese Beobachtung mit den Ergebnissen aus Vorarbeiten. Damals wurde für das homologe Glutamin im SaSIG1-Protein eine Beteiligung an der Bindung der -10-Region vermutet, da diese im bakteriellen SIGMA70 gezeigt wurde (Waldburger et al., 1990). Das mutierte SaSIG1-Protein zeigte eine vergleichbare Bindung an das core-Enzym und den psbA-Promotor und wies nur eine bessere Bindung an eine Promotormutante auf (Homann und Link, 2003). Auch hier zeigte die Umwandlung eines Glutamins in ein Histidin unter Standardbedingungen keine auffälligen Veränderungen der Bindung. Nachdem in Beobachtungen eine Dimer-Bildung des AtSIG1-Proteins und der Verkürzung SIG1AS438-502 unter schwach reduzierenden Bedingungen in Proteingelen beobachtet wurde, sollte die funktionelle Relevanz dieser Beobachtung durch die Mutation der Aminosäure Cystein an Position 358 in ein Serin geprüft werden. Auch diese Mutation führte in den Gel-Bindungstests zu kaum veränderten Bindungseigenschaften des Sigmafaktors und die beobachtete Dimerisierung des Faktors ist wohl eher artifiziell als funktionell. Die Aminosäuren der Positionen 460, 463, 465, 467, 471 und 476 liegen in der Region und bilden das Helix-Turn-HelixMotiv, welches die Bindung an die -35-Promotorregion vermittelt. Durch die Bedeutung des Sequenzmotives waren sehr gravierende Änderungen der Bindungseigenschaften zu erwarten, was sich nur bei den Aminosäuren 471 und 476 mit Reduzierungen von 85% bzw. 65% der Bindungseffizienz zeigte. 74 Diskussion Abb. 5.1: Sequenzvergleich des HTH-Sequenzmotives der Subregion 4.2 Darstellung des Helix-Turn-Helix-Motiv der Region 4.2 aus dem E. coli SIGMA70 (EcSIG70), des SIG1 aus S. alba und des SIG1 aus A. thaliana. Die Helices werden durch die ersten acht und die letzen neun Aminosäuren gebildet, welche durch drei Aminosäuren (Turn) getrennt werden. Insgesamt ist die Bindungseffizienz des AtSIG1-Proteins in vitro an den psbAPromotor als eher schwach zu bezeichnen, wenn man die Effizienz mit dem SaSIG1Protein und dem E. coli holo-Enzym vergleicht. Eine Erhöhung der Effizienz konnte auch nicht durch Punktmutationen an einzelnen Aminosäuren beobachtet werden. Die Mutationen zeigten nur für zwei Aminosäuren an den Positionen 471 und 476, welche im wichtigen Helix-Turn-Helix-Strukturmotiv liegen, eine verringerte Effizienz an Promotoren zu binden. Eine Erklärung für diese geringe Effizienz könnten fehlende posttranslationale Modifikationen darstellen oder das Fehlen eines spezifischen Aktivators sein. Potentielle Regulatoren des AtSIG1, welche die Eigenschaften des AtSIG1-Proteins verändern könnten, sind seine spezifischen Bindeproteine, AtSIB1 und AtSIB2. 5.2 Charakterisierung der Sigma-Bindeproteine aus A. thaliana Das AtSIG1-Protein ist der bisher einzige pflanzliche Sigmafaktor, für den spezifische Interaktoren identifiziert wurden. Die beiden identifizierten Genprodukte (AT3G56710 für AtSIB1 und AT2G41180 für AtSIB2) binden die konservierte Region 4 des AtSIG1-Proteins in vitro (Morikawa et al., 2002). Beide Gene sind kernkodiert und 75 Diskussion bestehen nur aus einem Exon. Die Genprodukte sind relativ klein und verfügen über N-terminale Transit-Peptide, über welche ihr Transport wie auch der Transport der pflanzlichen Sigmafaktoren in die Plastiden vermittelt wird. Ein DNA-Bindemotiv wurde nicht nachgewiesen. Die funktionelle Bedeutung der Bindung durch die AtSIBProteine wurde bisher nicht gezeigt. Es wurde aber vermutet, dass es sich um AntiSigmafaktoren handelt, die in Bakterien sehr verbreitet sind (Morikawa et al., 2002). Bakterielle Anti-Sigmafaktoren modulieren und regulieren durch reversible Bindung ihres spezifischen Sigmafaktors dessen Funktion. In Prokaryoten sind Anti-Sigmafaktoren sehr verbreitet als Inhibitoren von Stressund ECF-Sigmafaktoren, welche durch bestimmte Einflüsse induziert und aktiviert werden. Der erste identifizierte Anti-Sigmafaktor AsiA moduliert die Bindungseigenschaften des E. coli Hauptsigmafaktors SIGMA70 (Colland et al., 1998; Severinova et al., 1998; Baxter et al., 2006; Campbell et al., 2009). AsiA bindet bei einer Infektion mit dem Bakteriophagen T4 den SIGMA70, verhindert die weitere Transkription der bakteriellen Gene und vermittelt mit dem Protein MotA zusammen die Transkription von Phagen-Genen. Die AsiA-Interaktion findet mit der Region 4 des SIGMA70-Protein statt und verhindert die Erkennung der -35-Region der Promotoren (Helmann, 1999). Auf struktureller Ebene wurde gezeigt, dass die AsiABindung das Helix-Turn-Helix-Motiv der Region 4.2 in eine einzelne Helixstruktur umwandelt (Campbell et al., 2009). SIGMA70 kann aber auch durch das Polypeptid Rsd gehemmt werden, das auch die Region 4 bindet aber die Assoziierung mit dem core-Komplex verhindert (Campbell et al., 2009). Basierend auf diesen – jeweils neuen – Befunden an prokaryotischen Transkriptionssystemen sind die AtSIBProteine wahrscheinlich Anti-Sigmafaktoren. Analyse der physischen Protein-Protein-Interaktion von AtSIG1- und beiden AtSIB-Proteinen Die Zielsetzung der vorliegenden Arbeit war die Charakterisierung des AtSIG1Proteins und seiner beiden Interaktoren AtSIB1 und AtSIB2. Die Art der Regulation und damit der Einfluss auf die plastidäre Transkription durch diese Interaktionen sind nicht bekannt. Als Interaktionsstelle ist bisher nur die konservierte Region 4 des AtSIG1-Proteins bekannt. Ein Ausschluss einer anderen Interaktionsstelle war aber 76 Diskussion basierend auf den publizierten Ergebnissen nicht möglich (Morikawa et al., 2002). Um diesen Aspekt zu klären wurden pull down Interaktionsstudien mit rekombinanten Proteinen durchgeführt. Neben den Volllängenproteinen AtSIB1, AtSIB2 und AtSIG1 wurden von AtSIG1 auch Verkürzungen aufgereinigt, welche den vier konservierten Regionen des Sigmafaktors entsprechen. Die ausgewählte Variante der pull down-Experimente war der Protein Overlay Blot, für den Proteine gelelektrophoretisch aufgetrennt und anschließend auf Nitrocellulose-Membranen transferiert wurden. Die Membran wurde dann mit Magermilchpulver-Lösung abgeblockt und mit dem potentiellen Interaktor der aufgetrennten Proteine inkubiert. Nach der Inkubation wurde das ungebundene Protein abgewaschen und ein immunologischer Nachweis gegen den potentiellen Interaktor durchgeführt. Bei einer erfolgreichen Interaktion wurde das aufgetrennte Protein indirekt durch seinen spezifischen Interaktor nachgewiesen. Diese Reaktion musste auch in der umgekehrten Durchführung zu dem gleichen Ergebnis führen. Ein negativer Nachweis bedeutete dagegen keine Interaktion zwischen den aufgetrennten Proteinen und dem getesteten Interaktor. Alle rekombinanten Proteine, die auf ihre Interaktionsfähigkeit getestet wurden, waren N-terminal mit dem Fusionsprotein MBP bei den Sigmafaktoren (MBP-System, NEB) bzw. mit dem Fusionsprotein GST bei den Sigmabindeproteinen (GST-System, GE Healthcare) verbunden. Eine Verwendung nicht-fusionierter Proteine war nicht möglich, da es bisher für diese Proteine keinen spezifischen Antikörper gibt. Antikörper, die spezifisch die Fusionsproteine erkennen, waren von den Herstellern problemlos zu erhalten. Für die Analyse der Interaktionen wurden das rekombinante AtSIG1-Protein wie auch die vier Verkürzungsproteine SIG1AS81-213, SIG1AS257340, SIG1AS358-413 und SIG1AS438-502 elektrophoretisch in Proteingelen aufgetrennt. Zusätzlich wurden auch die Kontrollproteine GST und MBP gelelektrophoretisch aufgetrennt, um die Spezifität der Interaktionen (MBP) und eine Erfolgskontrolle der Detektion (GST) nachzuweisen. Ein für Vergleichszwecke Coomassie-gefärbtes Proteingel zeigte, dass alle Proteine in ausreichender Qualität und Quantität vorlagen. Duplikate dieser Gele wurden für die Interaktionstests angefertigt und die Nitrocellulose-Membranen wurden nach dem „Absättigen“ in Magermilchpulver-Lösung entweder nur in Bindungspuffer, oder in Gegenwart von AtSIB1- oder AtSIB2-Protein allein in Bindungspuffer inkubiert. Diese Inkubationen 77 Diskussion ergaben jeweils nur ein Signal an der Position des isolierten „Kontrollproteins“ GST und schlossen damit unspezifische Kreuzreaktionen des Antikörpers aus. Diese Kontrolle unterstreicht die Spezifität der detektierten Interaktionen in den anschließenden eigentlichen Bindungsstudien. Die Immunodetektion nach der AtSIB1-Inkubation wies indirekt neben dem AtSIG1-Protein und der Verkürzung SIG1AS438-502 auch die anderen Verkürzungen SIG1AS81-213, SIG1AS257-340 und SIG1AS358-413 nach. Folglich bindet das AtSIB1-Protein neben der konservierten Region 4 des AtSIG1-Proteins auch die anderen konservierten Regionen 1 bis 3. Ein vergleichbares Ergebnis erbrachte auch die Detektion der AtSIB2-Interaktion, die ebenfalls indirekt alle AtSIG1-Konstrukte nachwies. Auch die AtSIB2/AtSIG1-Interaktion findet wahrscheinlich nicht nur zwischen einer einzelnen Bindungsstelle statt. Daraus entwickelt sich die Frage nach der Art der Interaktionen zwischen den Bindungspartnern. Es bestehen dafür zwei Möglichkeiten: einerseits kann ein AtSIB-Protein allein das AtSIG1-Protein an verschiedenen Stellen binden oder ein einzelnes AtSIG1-Protein kann durch mehrere AtSIB-Proteine an verschiedenen Stellen gebunden werden (siehe Abb. 5.2). Die Interaktionsstudien mittels Protein Overlay Blot wiesen mehrere Bindungsstellen der beiden AtSIB-Proteine an das AtSIG1-Protein nach. Um detailliertere Aussagen zur Protein-Protein-Interaktion zu treffen, müssten die Mutanten-Studien weiter verfeinert werden, z.B. unter Einbeziehung einzelner Aminosäuren des VQ-Motives wie auch des Prä-VQ-Motives. Die Untersuchung solcher Punktmutanten benötigt wahrscheinlich auch noch höher auflösende Methoden wie Surface Plasmon Resonance (SPR), welche Rückschlüsse auf die Assoziation und Dissoziation der Interaktoren wie auch die Bindungskonstanten ermöglicht. Die genaue Kenntnis dieser physischen Protein-Protein-Interaktionen könnte sogar die Bildung und Berechnung kompletter pflanzlicher „Interaktome“ erleichtern, in denen aufgrund der oben skizzierten Datenbankanalysen zahlreiche VQ-Proteine vorkommen. Eine „klassische“ Anti-Sigmafaktor-ähnliche Funktion der AtSIB-Proteine ist durch das Vorliegen mehrerer Bindungsstellen unwahrscheinlicher geworden, kann aber ohne verfeinerte Funktionsnachweise auch nicht ausgeschlossen werden. 78 Diskussion Abb. 5.2: Modell zur AtSIB-/AtSIG1-Interaktion Die Abbildung zeigt die möglichen Arten der Interaktionen zwischen AtSIB-Proteinen und AtSIG1. Da beide AtSIB-Proteine prinzipiell alle Bereiche des AtSIG1-Proteins binden können, sind mehrere unterschiedliche Bindungssituationen vorstellbar: so kann eines der SIB-Proteine allein binden (B) oder es kann (an verschiedenen Stellen) zur Bindung mehr als eines SIB-Proteins kommen (A). Die klassische Antisigma-Funktion ist eine Inhibierung des Sigmafaktors. Zwar zeigen die EMSADaten (Abb. 4.5.2) eine dramatische Steigerung (keine Schwächung) der DNABindungsaktivität. Gerade dieser Effekt könnte sich negativ auswirken durch „Einfrieren“ des Bindungskomplexes und Hemmung der nachfolgenden Transkription (Tiller und Link, 1993b) Analyse der funktionellen Protein-Protein-Interaktion von AtSIG1- und beiden AtSIB-Proteinen Zur Klärung der Funktion der Interaktionen zwischen AtSIG1 und beiden AtSIBs wurden wiederum Gel-Bindungstests ausgewählt, die mögliche Veränderungen in der Effizienz der Promotorbindung und damit Aktivitätsänderungen des AtSIG1-Proteins wiedergeben können. Die Bindungsfähigkeit des AtSIG1-Proteins als Teil des rekonstruierten holo-Enzyms an den psbA-Promotor ist eher gering. Auch die Bindung an andere Promotoren wie rbcL, psaA, trnK, trnV, trnQ und rps16 war eher schwach (siehe Ergebnisse, Kapitel 4.5 , Abb. 4.5.2). Bei einer Funktion der beiden 79 Diskussion AtSIB-Proteine als „klassische“ Anti-Sigmafaktoren müsste diese geringe Bindungsfähigkeit an die unterschiedlichen Promotoren vollständig gehemmt werden (Campbell et al., 2009). Der psbA-Promotor ist ein starker Promotor, der neben den bakteriellen Promotorelementen wie die -35- und -10-Region noch ein extended -10-Motiv und ein TATA-ähnliches Bindemotiv aufweist (Link und Langridge, 1984; Eisermann et al., 1990). Trotzdem vermittelte das AtSIG1-Protein zusammen mit dem core-Enzym nur eine schwache Bindung an den Promotor. Dies änderte sich gravierend nach der Zugabe des AtSIB1- oder AtSIB2-Proteins, welches in beiden Fällen zu einer starken Zunahme der Promotorbindung führte. Die Zunahme wird durch eine vollständige Verschiebung der radioaktiv-markierten psbA-Promotorfragmente gezeigt. Folglich erhöhen beide AtSIB-Proteine die Bindungsfähigkeit des AtSIG1-Proteins an den psbA-Promotor. Eine Bindung der AtSIB-Proteine an den Promotor kann ausgeschlossen werden, da keines dieser beiden Proteine zusammen mit dem coreEnzym allein ein Bindungssignal vermitteln konnte. Es stellt sich die Frage, wie es bei schwächeren Promotoren aussieht, die nur die -35 und -10-Region wie rbcL, trnK, trnV und trnQ oder wie rps16 ein TATA-ähnliches Motiv zusammen mit einer -10-Region aufweisen. Die beiden AtSIB-Proteine könnten unterschiedliche Promotor-Präferenzen bei den AtSIG1-Proteinen auslösen und dadurch eine spezifische Regulation der Aktivität des AtSIG1-Proteins bewirken. Für die Promotoren rbcL und trnK erhöhten die beiden AtSIB-Proteine ebenso wie am psbA-Promotor die Bindungseffizienz des AtSIG1-Proteins, ohne selbst eine Promotorbindung vermitteln zu können. Für die Promotoren trnV, trnQ, psaA und rps16 wurde die erhöhte Bindungsfähigkeit daher nur noch für das AtSIB2-Protein getestet und nachgewiesen. Eine Änderung der Promotor-Präferenz durch die SIBProteine war über die gesamte Auswahl verwendeter Promotoren in vitro nicht gegeben, welches aber in planta anders sein kann. Beide AtSIB-Proteine erhöhen durch ihre Interaktion mit AtSIG1 seine Fähigkeit, an Promotoren zu binden. Die Funktion dieser erhöhten Promotorbindung kann einerseits eine Blockierung der Promotoren darstellen, andererseits aber die Aktivierung des AtSIG1-Proteins sein. Die letztere Funktion ist aber wahrscheinlich, da die AtSIB-Proteine (ohne AtSIG1) keine DNA-bindenden Eigenschaften besitzen. 80 Diskussion Für eine Aktivator-Funktion spricht auch die Fähigkeit der AtSIB-Proteine, den gesamten AtSIG1 spezifisch zu binden und nicht einzelne, strikt definierte Bereiche. Abb. 5.3: Modell zu den Funktionen der AtSIB-Proteine im PEP-Komplex Die Abbildung zeigt drei hypothetische Möglichkeiten wie AtSIB-Proteine die Aktivierung des AtSIG1-Proteins und das Verstärken der Promotorbindung bewirken könnten. Dazu zählen eine bestehende und ausschließliche Interaktion mit dem AtSIG1-Protein (A); eine Funktion als „Mediator“, der AtSIG1 mit dem core-Enzym reversibel stärker bindet (B); eine beschleunigte Dissoziation des AtSIB-Proteins vom PEP-Komplex und nachfolgende erneute Bindung (= erhöhter Bindungs-Turnover). Die Konsequenzen dieser möglichen Mechanismen auf nachfolgende Schritte (Initiation, Elongation) sind nicht vorhersagbar aufgrund der erhaltenen Ergebnisse. Anti-Sigmafaktoren binden eher einzelne Regionen mit einer essentiellen Funktion für den Sigmafaktor (Campbell et al., 2009). Andererseits kann eine Aktivierung des Sigmafaktor negative Konsequenzen für die Transkription haben, falls es dadurch zu einer (ir)reversiblen Konformationsänderung des gesamten holo-Enzym-Komplexes („Einfrieren“) kommt (Tiller und Link, 1993b). Die Möglichkeiten der Aktivierung sind vielfältig und werden erst durch Analysen der Zeitpunkte und der Ereignisse der Assoziation und Dissoziation der Interaktoren geklärt werden können. Dazu zählen Chaperon-ähnliche Funktionen wie die Faltung des Sigmafaktors durch eines oder mehrere Bindeproteine, da auch nicht das Verhältnis der Interaktion bekannt ist. Die 81 Diskussion AtSIG1-Aktivierung könnte auch darauf basieren, dass die beiden Bindeproteine den Sigmafaktor vielleicht um sich selbst falten und damit aktivieren. Eine Funktion als „Mediator“ zwischen AtSIG1 und dem core-Enzym wäre eine weitere mögliche Art der Interaktion, wobei es Teil des PEP-Komplexes oder vor der Transkriptionsinitiation auch abdiffundieren könnte. Beweise für jede dieser Thesen müssten noch erbracht werden. Dies würde letztlich in den Bereich der (Röntgen-) Strukturanalysen führen, der ausdrücklich (noch) nicht Gegenstand der vorliegenden Arbeit war. Die AtSIB-Proteine fungieren also eher als Aktivatoren des AtSIG1-Proteins, soweit dies die Promotorbindung betrifft. Für das Atsib1-Gen wurde durch Microarray- und QRT-PCR-Analysen eine erhöhte Transkriptmenge nach einer Behandlung der Pflanzen mit Salizylsäure festgestellt (Narusaka et al., 2008). Salizylsäure ist charakteristisch in der Pathogen-Abwehr von Pflanzen und aktiviert die systemisch erworbene Resistenz (SAR) der gesamten Pflanze. SAR ist notwendig für eine erfolgreiche pflanzliche Verteidigung gegen Pathogene und verhindert eine Verbreitung der Pathogene. Eine kausale Beteiligung des AtSIB1-Proteins am Salizylsäure-Signalweg konnte nicht gezeigt werden, obwohl in sib1- Überexpressions-Linien die Transkription von ROS-Genen hochreguliert war. Die Regulation der Atsib1-Genexpression ist charakteristisch für biotische Stressbedingungen. Die Promotorstrukturen des Gens bestehen aus einer DNABindestelle für WRKY Transkriptionsfaktoren, mehreren ACGT Sequenzen und TGABoxen (Narusaka et al., 2008). Bei Infektionen mit Pseudomonas syringae wurde ebenfalls eine induzierte Hochregulation des sib1-Transkripts gemessen. Eine etwas stärkere Resistenz zeigten sib1-Überexpressions-Linien, jedoch ohne Unterschiede zum Wildtyp in den Signalwegen der Abwehr (Xie et al., 2010) Eine Einordnung des SIB1-Proteins in das Signal-Netzwerk der Pathogen-Abwehr lässt eine Rolle bei den pathogenesis related-Proteinen (PR-Proteine) vermuten. Vor dieser gesteigerten SIB1-Expression fand die unspezifische PTI-Antwort der Pflanzenzelle bestehend aus einer Erkennung von Molekülen des Pathogens und einer MAPK-Kaskade und der Aktivierung von spezifischen Transkriptionsfaktoren statt. Diese Transkriptionsfaktoren vermitteln neben der Expression der PR-Proteine auch die Bildung von mikrobiell-hemmenden Komponenten wie ROS (reactive oxygen species) und Phytoalexinen (Gómez-Gómez et al., 1999). Zu den PR-Proteinen 82 Diskussion zählen Glucanasen aber auch Proteine der verschiedenen Signalwege wie der Salizylsäure-vermittelten-Antwort. SIB1 könnte als Teil der PR-Proteine synthetisiert werden, da in den Überexpressionslinien auch eine Erhöhung der ROSKomponenten nachgewiesen wurde. Es bleibt zu klären, ob SIB1 weitere Funktionen während der Pathogen-Abwehr als Teil eines Signalnetzwerkes im Cytosol und in den Plastiden innehat. Die Aktivierung der plastidären Transkription durch AtSIG1Bindung könnte weiteren funktionellen Protein-Protein-Interaktionen vorangegangen sein (Zur Übersicht Abb.5.4). Abb. 5.4: Einordnung des SIB1-Proteins in den pflanzliche Pathogen-Abwehr Die obere Abbildung ordnet das SIB1-Protein als Teil der PR-Proteine ein, die nach der PTI-Antwort der Pflanzenzelle synthetisiert werden und die spezifische PathogenAbwehr einleiten. Eine funktionelle Protein-Protein-Interaktion des SIB1-Proteins mit dem Sigmafaktor SIG1 führt zu dessen Aktivierung und damit zu plastidärer ExtraTranskription in der „Notsituation“ der Pflanze in Antwort auf das Phytopathogen. Die große Bedeutung der Chloroplasten für die Pathogen-Abwehr und die Veränderungen der plastidären Transkription ist ausführlich dokumentiert (Roberts und Paul, 2006; Jones et al., 2006; Thilmony et al., 2006). Die physische Bindung des SIB1-Proteins an SIG1 aktiviert den Sigmafaktor funktionell und damit 83 Diskussion möglicherweise die spezifische Transkription von Plastiden-Genen für (dringend benötigte) Proteine während der Pathogen-Abwehr. So gesehen konnte das SIG1Protein als Stressfaktor aufgefasst werden, der durch eine Interaktion seiner spezifischen Bindeproteine SIB1 und SIB2 aktiviert wird. SIG1 wird bei einer Pathogen-Infektion durch SIB1 aktiviert; es bleibt aber zu klären, unter welchen Stressbedingungen eine Expression von SIB2 und eine nachfolgende SIG1Aktivierung stattfinden. In silico Analyse der Sigma-Bindeprotein AtSIB1 und AtSIB2 Die Zunahme der sequenzierten Genome und abgeleiteten Proteine in Datenbanken wie z.B. PFAM führte einerseits zu einem verbesserten Zugriff auf die gespeicherten Proteinsequenzen, ermöglichte aber auch die Identifizierung kleiner und noch unbekannter Sequenzmotive. Dazu zählte auch das VQ-Sequenzmotiv, dessen Funktion unbekannt ist und zu deren Trägern auch die AtSIB-Proteine gehören. Zur Familie der VQ-Proteine gehören viele unterschiedliche Proteine häufig ohne bekannte Funktionen und bisher auch nur Proteine aus mono- und dikotylen Pflanzen (Datenbank-Eintrag von PFAM: PF05678). Daraus lässt sich schließen, dass die AtSIB-Proteine eukaryotische Regulatorproteine sind, die sowohl eine hemmende Wirkung (Anti-Sigma-ähnlich) als auch eine Aktivator-Funktion haben können. Dies ist auch ein erster Hinweis zur Klärung der schwachen Promotorbindung des AtSIG1-Proteins in vitro. Das MKS1-Protein zählt ebenfalls zu den VQ-Proteinen und ist Teil einer Signalkette in der Pathogen-Abwehr von Pflanzen. In diesem Signalweg wird das MKS1-Protein als Substrat der Kinase MAP4 phosphoryliert und ist ein Bindungspartner der Transkriptionsfaktoren WRKY25 und WRKY33, die ihrerseits durch die MAP4-Kinase phosphoryliert werden. Es liegt somit nahe, dass auch das MKS1-Protein regulatorische Aufgaben besitzt (Andreasson et al., 2005). Die Sequenz des VQ-Motivs ist im PFAM-Eintrag 05678 wie folgt beschrieben: FXhVQChTG. Dabei entsprechen das X jeder möglichen Aminosäure und das h einer hydrophoben Aminosäure. Die Sequenz der beiden AtSIB-Proteine weicht von diesem allgemeinen Motiv leicht ab: FRELVQELTG. In diesem Motiv kommen zwei 84 Diskussion Aminosäuren anstelle der X-Aminosäure vor und auch das konservierte Cystein liegt nicht vor und wurde durch Glutaminsäure ersetzt. Neben dem VQ-Motiv sind auch weitere Sequenzübereinstimmungen erkennbar. Ein recht gut konservierter Bereich, etwa 20 Aminosäuren umfassend, liegt vor dem modifizierten VQ-Motiv. Eine Struktur wurde für das Motiv bisher noch nicht beschrieben. Um diese Frage zu klären, wählte ich als Vorhersage-Programm für Sekundär-Strukturen PSI-PRED v2.6 (Jones, 1999) aus. Mit diesem Programm kann die wahrscheinliche SekundärStruktur einer Proteinsequenz berechnet werden. Die Berechnungen wurden mit den Proteinsequenzen der beiden AtSIB-Proteine durchgeführt und für das VQ-Motiv der beiden Proteine wurde eine Helix-Struktur vorhergesagt. Aber auch für die Bereiche, die vor dem Motiv liegen und in den AtSIB-Proteinen übereinstimmen, wurden zwei Faltblatt-Strukturen vorhergesagt. Bisher wurden keine weiteren SIB-Proteine publiziert und es blieb durch DatenbankRecherchen zu klären, ob die beiden einzigen bekannten SIB-Proteine eine Besonderheit des – dann gerade nicht repräsentativen – Modellorganismus A. thaliana sind oder ob doch Hinweise für ihre weitere Verbreitung vorhanden sind. Ein allgemeines Vorkommen der SIB-Proteine würde für eine wichtige Funktion in der Regulation der plastidären Transkription und damit auch für die Bedeutung dieser Proteine und ihres Sequenzmotives sprechen. Für die Klärung des Aspektes war das VQ-Motiv alleine eher von Nachteil, da es sehr verbreitet vor allem bei nichtplastidären Proteinen ist (Andreasson et al., 2005). Doch die Vorhersage der Sekundärstruktur der beiden AtSIB-Proteine lieferte neben dem leicht veränderten VQ-Motiv auch einen weiteren konservierten Sequenzbereich mit auffälliger Sekundärstruktur, der für die bioinformatische Suche nach ähnlichen – vielleicht auch funktionell-äquivalenten – Proteinen dienen konnte. Zumindest würden Proteine mit allen hier gefundenen Sequenzen und Strukturen eine Unterfamilie der VQ-MotivFamilie bilden. Nach einer Vorauswahl wurde die Datenbank-Suche auf die NCBI(National Center of Biological Information)-Kollektion und die Phytozyme-Datenbank v4.1 des Joint Genome Institute eingegrenzt, welche eine große Auswahl an freizugänglichen Genomen lieferten. Die Sequenzvergleiche der SIB-Proteine wurden mit BLAST durchgeführt und manuell überprüft. Alle ausgewählten Sequenzen weisen neben dem VQ-Motiv davor auch die beiden Faltblatt-Strukturen auf. Basierend auf diesen Bedingungen konnten aus 11 Pflanzen insgesamt 25 85 Diskussion Proteine identifiziert werden, einschließlich eines weiteren Proteins aus A. thaliana. Die gefundenen Pflanzenspezies sind sehr unterschiedlich und zählen zu den Monound Dikotylen. Auch die Anzahl der identifizierten Proteine pro Art variiert stark. Alle ausgewählten Proteine weisen alle drei „Sequenz-Motive“ auf und sind eher klein (100-220 Aminosäuren). Die Frage der Expression und Lokalisation kann nicht vollständig beantwortet werden. Einige Proteine werden nur als hypothetische Proteine durch die Datenbanken beschrieben und der Nachweis der Expression muss noch experimentell erbracht werden. Dies gilt auch für die Lokalisation der identifizierten Proteine, für die teilweise ein N-terminales plastidäres Transit-Peptid mittels ChloroP erwartet wird, aber noch verifiziert werden muss. Für das dritte A. thaliana-Protein wurde eine Cytosol- oder Kern-Lokalisation vorhergesagt und damit eine SIB-Funktion in den Plastiden wahrscheinlich ausgeschlossen. Eine Sequenzähnlichkeit der gefundenen Proteine ist aber gegeben und diese könnten eine Untergruppe der VQ-Proteinfamilie repräsentieren. Um ein genaueres Motiv der Untergruppe angeben zu können, wurde ein multipler Sequenzvergleich mit allen Proteinsequenzen durchgeführt. Das Ergebnis führte zu einem Prä-VQ-Motiv, das durch eine Faltblatt-Struktur charakterisiert ist und durch „KhXYI“ beschrieben wird. Das h steht für eine hydrophobe Aminosäure und das X für eine beliebige Aminosäure. Das SIB-spezifische VQ-Motiv wird durch eine Helix beschrieben und besteht aus der Aminosäurefolge: „FREALVQELTG“ (siehe Abb. 5.5). Abb. 5.5: Darstellung des SIB-spezifischen VQ-Motives Die Abbildung zeigt die Sequenzbereiche der beiden AtSIB-Proteine, welche die SIBspezifischen Sequenzmotive enthalten. Im Prä-VQ-Bereich ist die Sequenz KhXYI konserviert und das VQ-Motiv wurde für Unterfamilie der SIB-spezifischen VQProteine in FREALVQELTG spezifiziert. Beide Motive basieren auf insgesamt 25 Proteinen aus 11 verschiedenen Pflanzen und enthalten dieses Sequenzmotiv. (h: hydrophobe Aminosäure, X: jede Aminosäure) 86 Diskussion Der multiple Sequenzvergleich bildete aber auch die Grundlage für einen phylogenetischen Stammbaum, der auf dem Neighbor-Joining-Algorithmus beruhte. In dem Baum ist auffällig, dass meist zwei Proteine einer Pflanzenart wie bei den SIB-Proteinen aus Arabidopsis sehr ähnlich zueinander sind. Diese könnten durch Gen-Duplikationen während der Evolution entstanden sein. Es gilt aber zu beachten, dass dieser phylogenetischer Stammbaum nur auf bioinformatischen Daten beruht und noch experimentell verifiziert werden muss. Zusammenfassend konnten mittels der bioinformatischen Analysen SIB-ähnliche Proteine gefunden werden und somit ist ein globales Vorkommen von SIB-Proteinen auch in Plastiden sehr wahrscheinlich. Die in silico Daten bilden die Grundlage für weitere experimentelle Analysen, wozu auch die Funktion des VQ-Motives zählt. Die Charakterisierung der AtSIB-Proteine zeigt, dass diese Proteine zu der VQProteinfamilie gehören und wahrscheinlich eine Unterfamilie mit einem weiteren Sequenzmotiv bilden. Beide Proteine binden nicht nur spezifisch die konservierte Region 4 des AtSIG1-Proteins sondern binden den gesamten Faktor, was gegen eine Funktion als „klassische“ Anti-Sigmafaktoren spricht. Beide Bindeproteine erhöhen die Fähigkeit des „schwachen“ AtSIG1-Proteins an ganz unterschiedlichen Plastiden-Promotoren in vitro. Die Expression und damit die Induzierung der AtSIBFunktion erfolgt unter Stressbedingungen, welche für AtSIB1 die PathogenInfektionen darstellen. 5.3 Einfluss des sig1-Gens in einer sig1-1 Einzel-Knockout-Linie auf die plastidäre Transkription und phänotypische Effekte in Einzel- und Doppelmutanten durch Licht Um die Funktion des AtSIG1-Proteins in der plastidären Transkription in vivo untersuchen zu können, bot es sich an auf eine sig1 Knockout-Linie von A. thaliana zurückzugreifen. Die Mutanten-Linie sig1-1-Knockout-Linie (758B02) lag in der 87 Diskussion Sammlung der Arbeitsgruppe vor und wurde zur Beginn meiner Arbeit erneut positiv auf Homozygotie durch genomische PCR-Analysen geprüft. Im Gegensatz zu der sig6-2 Knockout-Linie, die einen auffälligen Phänotyp mit weißen Kotyledonen zeigt (Loschelder et al., 2006), entspricht das äußere Erscheinungsbild der sig1-1-Linie dem Wildtyp. Um dennoch Unterschiede zum Wildtyp, herausarbeiten zu können, wurde auch der molekulare Phänotyp, soweit er sich im plastidären Transkriptmuster äußert, in die Untersuchungen einbezogen. Veränderungen im plastidären Transkript-Spektrum (Erhöhung oder Reduktion der Transkriptmenge bzw. Veränderungen in der Größe von Transkripten) können transkriptionell, aber auch post-translational bedingt sein. Daher wurden weitere Überlegungen wie folgt einbezogen: die plastidären Gene gliedern sich in drei Klassen, nach der transkribierenden RNA-Polymerase. Für meine Analyse wählte ich nur Gene der Klasse I und II, da beide Genklassen durch die PEP-RNA-Polymerase abgelesen werden. Die Klasse I Gene werden ausschließlich durch die PEP abgelesen und werden in der vorliegenden Arbeit durch die Gene psbA, rbcL und trnV repräsentiert. Die Gene der Klasse II können von der NEP und der PEP abgelesen werden und werden durch die Gene atpB und atpE repräsentiert, die zusammen ein Operon bilden (Schweer et al., 2006). Die Gene psbA, rbcL und trnV können als Haushaltsgene bezeichnet werden, da diese im Chloroplasten durchgängig gebraucht und deshalb transkribiert werden. Als Hinweis auf transkriptionelle (Sigma-) Regulationen kann somit gelten, wenn ein PEP-abhängiges, nicht aber NEP-abhängiges Gen in der Mutante veränderte RNA-Level ergibt. Das SIG1-Protein wurde bisher als genereller Faktor beschrieben, der die Transkription der Haushaltsgene vermittelt (Privat et al., 2003). Für 20 Tage alte sig1-Mutanten aus O. sativa wurden reduzierte Transkripte für beide Photosysteme nachgewiesen, aber diese Mutanten zeigen auch einen hellgrünen Phänotyp in diesen Stadien im Gegensatz zu meiner Mutante (Tozawa et al., 2007). Der visuelle Phänotyp der sig1-1 Knockout-Linie veränderte sich auch nicht durch andere Lichtbedingungen als Kurztag. Unter Langtag und Dauerlicht blieb der visuelle Phänotyp der sig1-1 Knockout-Linie vergleichbar mit dem Wildtyp (siehe Kapitel 4.8, Abb.4.8.1). Es blieb zu klären, ob die sig1-1 Knockout-Pflanze trotz eines kräftig grünen Phänotyps entsprechende Änderungen auf Transkriptebene aufweist. Dafür wurden 88 Diskussion Northern Blot-Analysen durchgeführt, für welche die oben aufgeführten Gene als Sonden dienten. Die Ergebnisse der Analysen erbrachten aber vergleichbare Transkriptmengen im Wildtyp und der sig1-1-Knockout-Pflanze für die geprüften Gene (siehe Kapitel 4.7, Abb.4.7.2). Daraus lässt sich schließen, dass diese Gene nicht ausschließlich oder gar nicht durch das SIG1-Protein erkannt werden. Folglich kann einerseits ein anderer Sigmafaktor der A. thaliana-Familie die Aufgaben des SIG1-Proteins übernommen haben oder SIG1 ist nicht an der Transkription der ausgewählten Gene beteiligt. Die SIG1-Funktion als genereller Faktor wird durch die gezeigten Ergebnisse etwas unwahrscheinlicher, da das Fehlen eines solchen Sigmafaktors zu einem auffälligen molekularen Phänotyp und sehr veränderten Transkriptmengen führen sollte. Ein spezifischer Inhibitor für diesen Sigmafaktor wäre ebenfalls unwahrscheinlich. Auswirkungen des sig1-Genverlusts auf plastidärer Transkriptebene könnten noch breiter durch Microarray-Analysen z.B. aller Plastidengene geklärt werden. Um zu klären, ob das Fehlen des sig1-Gens durch einen anderen Sigmafaktor kompensiert wird, wurde die sig6-2-Knockout-Linie ausgewählt. Die sig6-2-KnockoutLinie hat einen ausgeprägten visuellen und molekularen Phänotyp, welcher die entwicklungs- und genspezifische Rolle des SIG6 in der plastidären Transkription wiederspiegelt (Loschelder et al., 2006; Schweer et al., 2006). Diese Untersuchungen zeigten, dass der Verlust des sig6-Gens in frühen Stadien (4 Tage) zu reduzierten Transkriptmengen der Gene psbA, rbcL und trnV führte (Schweer, Dissertation). Vermittelt aber SIG1 die Transkription dieser Gene in den späteren Stadien, könnte das SIG6-Protein deren Transkription bei Verlust des sig1-Gens weiterhin übernehmen. Um diese Möglichkeit zu prüfen, wurden durch Kreuzungen Doppel-Knockout-Linien der beiden Gene sig1 und sig6 angefertigt. Beide Linien müssten einen verstärkten visuellen Phänotyp der sig6-2-Knockout-Linie zeigen, wenn SIG1 eine Funktion als genereller Faktor hat und durch SIG6 ersetzt wird. Da der visuelle Phänotyp der Doppel-Knockout-Linien aber unter allen Anzuchtbedingungen (Kurztag, Langtag und Dauerlicht) der sig6-Knockout-Linie glich und keine weitere Verstärkung dieses Phäns beobachtet wurde, ist die Funktion des SIG1-Proteins als genereller Faktor eher unwahrscheinlich. Basierend auf den Daten von Onda et al. (2008), die eine Induktion des sig1-Transkripts bei dunkeladaptierten Pflanzen durch Belichtung mit Rot- (660 nm) und Blaulicht (470 nm) 89 Diskussion gemessen haben, erscheint es wahrscheinlicher, dass SIG1 eher einen StressFaktor darstellt. Einem generellen „primären“ Sigmafaktor wie bei Prokaryoten kommt wahrscheinlich der (hier nicht untersuchte) Plastidenfaktor SIG2 nahe, der eine entscheidende Rolle in der Transkription von Photosynthese- und ProteinsyntheseGenen über einen längeren Zeitraum zu spielen scheint (Kanamaru und Tanaka, 2004). 5.4 Ausblick Die Ergebnisse meiner Arbeit zeigen, dass die AtSIB-Proteine nicht nur physisch mit AtSIG1 interagieren, sondern auch zu funktionellen Änderungen führen. Diese Untersuchungen liefern somit die Grundlage für detailliertere Untersuchungen der SIG1-abhängigen Transkription in vitro und in planta. Die Ergebnisse der vorliegenden Arbeit sprechen dafür, dass AtSIG1 wahrscheinlich ein Stressfaktor ist, der durch Bindung seiner spezifischen Interaktoren funktionell aktiviert wird. Diese Aktivierung erhält die PEP-vermittelte Transkription auf einem aktiven Zustand und damit die Genexpressions-Homeostasis im Plastiden selbst unter „schwierigen“ Bedingungen. Durch den gewählten Ansatz war eine Klärung der genauen Bindungsdeterminanten leider nicht möglich. Zur Erklärung der Bindungsdeterminanten sind Analysen der beiden bioinformatisch-identifizierten SIBSequenzmotive auf ihre genauen Eigenschaften und Funktionen in der Interaktion nötig. Weitere Fortschritte können z.B. durch die Surface plasmon resonance (SPR) Technik zusammen mit dem Einsatz von rekombinanten Punkt- und Deletionsmutanten aller Interaktoren erwartet werden, da diese Methode die Bestimmung der Assoziation und Dissoziation wie auch der Bindungskonstante ermöglicht. Selbst eine Analyse von multiplen Interaktionen wie dem PEP-Komplex ist möglich (Lalonde et al., 2008). Die in silico identifizierten und „homologen“ Sigma-Bindeproteine in unterschiedlichen Pflanzenspezies sollten durch experimentelle Funktionsansätze verifiziert werden, 90 Diskussion um eine Allgemeingültigkeit für die regulatorischen Eigenschaften der SIB-Proteine und ihrer spezifischen Sequenzmotive zu zeigen. Die putative Funktion des AtSIB1-Proteins als Teil der Pathogen-Abwehr sollte ebenfalls näher überprüft werden, um Interaktionspunkte des Proteins zu identifizieren und eine mögliche Signalkaskade der plastidären Pathogen-Abwehr nachvollziehen zu können. Eine Grundlage sollte dabei die sig1-1 Knockout-Pflanze bilden, da eine Pathogen-Infektion in dieser Umgebung wahrscheinlich zu Änderungen auf Proteinebene führen wird. 91 Zusammenfassung 6. Zusammenfassung Plastiden sind gentragende Kompartimente von Pflanzenzellen und verfügen über eigene Transkriptionsenzyme. Die Haupt-RNA-Polymerase der Chloroplasten (PEP) ist ein Multiproteinkomplex mit katalytischem Kern („core“) sowie Sigmafaktor(en) für die spezifische Initiation der Transkription ganz ähnlich wie bei Bakterien. In Arabidopsis sind sechs dieser Sigmafaktoren bekannt, die als Regulatoren die Aktivität der PEP je nach „Bedarf“ der Zelle steuern. Wenngleich diese „prokaryotischen“ Transkriptionsregulatoren alle kernkodiert sind, so werden sie dennoch in die Plastiden transportiert und scheinen ausschließlich hier ihre Wirkung zu entfalten. Die vorliegende Arbeit gilt regulatorischen Protein-Protein-Interaktionen in der plastidären Transkription am Beispiel der Interaktion zwischen dem Sigmafaktor SIG1 und SIB1 bzw. SIB2 – zwei als spezifische SIG1-Interaktionspartner identifizierte und ebenfalls kernkodierte Chloroplastenproteine. Als Arbeitshypothese wurde geprüft, inwieweit die beiden Bindeproteine – in Analogie zu bakteriellen Systemen – über Anti-Sigma-ähnliche Eigenschaften verfügen und die Funktion von SIG1 regulatorisch beeinflussen (Morikawa et al., 2002). Durchgeführte EMSAFunktionsanalysen mit SIG1 allein (ohne SIB1/SIB2) zeigten eine zwar spezifische, aber (im Vergleich zu anderen Sigmafaktoren) nur schwache Bindung selbst an „starken“ Promotoren wie z.B. denjenigen für psbA. Dies erschien für SIG1 – als einen in der Pflanze relativ abundanten und möglicherweise „generellen“ Faktor – eher unwahrscheinlich. Die Bindung eines regulatorischen Interaktors war eine Möglichkeit, den eher „schwachen“ Sigmafaktor in eine stärkere Form zu konvertieren. Tatsächlich erbrachten EMSA-Analysen von SIG1 in Gegenwart von SIB1 bzw. SIB2 eine dramatische Steigerung Bindungsstellen zeigten, der dass Promotor-Bindungsaktivität. beide SIB-Proteine fähig Die Analysen sind, neben der der konservierten SIG1-Region 4 auch die konservierten Regionen 1 bis 3 spezifisch zu binden. Ein Einfluss auf die Promotor-Präferenz von SIG1 durch SIB1 bzw. SIB2 92 Zusammenfassung wurde hingegen innerhalb des breiten Spektrums ausgewählter Promotoren nicht sichtbar. Das Fehlen eines ausgeprägten Phänotyps der sig1-Knockout-Linie (A. thaliana) schließt eine einfache „An/Aus“-Anti-Sigmafaktor-ähnliche Funktion der beiden SIBProteine wahrscheinlich aus, da der Verlust eines abundanten und „primären“ Sigmafaktors eine visuell starke Veränderung zeigen würde. Möglich sind allerdings kompensatorische Mechanismen, die das Fehlen dieses Faktors ausgleichen. Sib1/sib2-Knockout-Linien existieren bisher nicht, dennoch kann die erhöhte Fähigkeit des SIG1-Proteins in Anwesenheit von SIB-Proteinen effizienter an Promotoren zu binden durch neuere, publizierte Daten aus der Phytopathologie erklärt werden, wonach SIB-Proteine Teil der pflanzlichen Pathogenantwort zu sein scheinen (Narusaka et al., 2008). Danach kann das SIG1-Protein auch als ein AntiStressfaktor gesehen werden, der durch Interaktion mit seinen spezifischen Bindeproteinen SIB1 und SIB2 aktiviert wird und mithilft, die plastidäre Genexpression unter diesen Bedingungen aufrechtzuerhalten. In silico Analysen zeigten, dass auch in anderen Pflanzen SIB-ähnliche Proteine vorkommen. Basierend auf den identifizierten Sequenzen konnten zwei SIBspezifische Sequenzmotive ermittelt werden. Die Expression und die Funktion dieser „homologen“ Proteine müssen zwar experimentell noch bewiesen werden, dennoch deuten diese bioinformatischen Daten schon jetzt auf ein verbreitetes Vorkommen von Sigmafaktor-Bindeproteinen hin. 93 Summary 7. Summary Plastids are one of the gene containing compartments of plant cells and have their own set of transcription enzymes. The main RNA polymerase of chloroplasts (PEP) is a multi enzyme complex with a catalytic core and sigma factor(s) that confer the specific transcription initiation as in bacteria. In Arabidopsis six sigma factors are known that regulate PEP activity as “required” by the plant cell. All of these “prokaryotic” transcription regulators are nucleus encoded and subsequently transported into the plastids, i.e. their sole sites of action. In this work regulatory protein-protein interactions in plastid gene transcription were addressed, with a focus on the interaction between sigma factor SIG1 and SIB1 or SIB2. Both have been identified as specific interactors of SIG1 and are nucleus encoded chloroplast proteins. As a working hypothesis it was tested if both binding proteins – in analogy to prokaryotic systems – have “anti sigma”-like characteristics and can specifically modulate the SIG1 activity (Morikawa et al., 2002). Functional EMSA studies with SIG1 alone (without SIB1/SIB2) showed a specific, but weak binding (in comparison with other sigma factors) even at “strong” promoters such as that one for psbA, too. Weak promoter binding was considered unlikely for an abundant and potential by general sigma factor in plants. Binding of regulatory interactor is one possibility to convert a “weak” sigma factor into a more active conformation. In fact, a dramatic increase in promoter binding activity was measured in EMSA experiments for SIG1 in the presence of SIB1 or SIB2. Analysis of the SIG1 binding sites showed that both SIB proteins are able to bind the conserved sigma region 4 as well as the other conserved regions 1 to 3, respectively. A set of plastid promoters widely differing in architecture was used to test for a promoter preference, indicating no obvious changes in the absence or presence of SIB1 or SIB2. Unlikely, a simple on and off function of the anti sigma-like proteins SIB1 and SIB2 is given fact, because sig1 knockout lines from A. thaliana have no distinct phenotypes. Possible explanations are compensatory mechanisms that may help, adjust gene 94 Summary expression despite, the missing sigma factor. No knockout lines of sib1 or sib2 are available to date, but the increasing SIG1 ability to bind specific promoters in presence of SIB1 or SIB2 is in accordance with new data in phytopathology, suggesting that SIB proteins might be involved in plant pathogenic response (Narusaka et al., 2008). These data imply that SIG1 could be viewed as an anti stress factor that is activated by specific interaction with its binding proteins SIB1 and SIB2, thus providing for relatively undisturbed plastid gene expression under these conditions. In silico analysis identified SIB-like proteins in other plants. The sequences of these proteins form the basis of two SIB specific sequence motifs. Expression and function of the homologous proteins awaits to be experimentally confirmed, but in any case the bioinformatics data indicate a broad distribution of sigma factor binding proteins such as SIB1 and SIB2. 95 Literatur 8. Literatur Adl, SM, Simpson, AGB, Farmer, MA, Andersen, RA, Anderson, OR, Barta, JR, Bowser, SS, Brugerolle, G, Fensome, RA, Fredericq, S, et al., (2005). The new higher level classification of eukaryotes with emphasis on the taxonomy of protists. J Eukaryot Microbiol 52, 339–451. Alberts, B (1998). The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell 92, 291-294. Alfano, JR und Collmer, A (1996). Bacterial pathogens in plants: life up against the wall. Plant Cell 8, 1683-1698. Allison, LA und Maliga, P (1995). Light-responsive and transcription-enhancing elements regulate the plastid psbD core promoter. EMBO J 14, 3721-30. Altschul, SF, Madden, TL, Schäffer, AA, Zhang, J, Zhang, Z, Miller, W und Lipman DJ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389-4402. Altschul, SF, Wootton, JC, Gertzm EM, Agarwala, R, Morgulis, A, Schäffer, AA und Yu, YK (2005). Protein database searches using compositionally adjusted substitution matrices. FEBS J 272, 5101-5109. Andreasson, E, Jenkins, T, Brodersen, P, Thorgrimsen, S, Petersen, NHT, Zhu, S, Qiu, JL, Micheelsen, P, Rocher, A, Petersen, M, Newman, A et al. (2005). The MAP kinase substrate MKS1 is a regulator of plant defense responses. EMBO J 24, 2579.2589. Ausubel, FM, Breut, R, Kingston, RE, Moore, DD, Seidmann, JG, Smith, JA, Strutl, K (1995). Current protocols in molecular biology. John Wiley & Sons, New York. 95 Literatur Baginsky, S und Gruissem, W (2004). Chloroplast proteomics: potentials and challenges. Journal of Experimental Biology 55, 1213-1220. Barbrook, AC, Howe, CJ und Purton, S (2006). Why are plastid genomes retained in non-photosynthetic organisms? Trends Plant Sci. 11, 101-108. Baxter, K, Lee, J, Minakhin, L, Severinov, K und Hinton, DM (2006). Mutational analysis of sigma 70 region 4 needed for appropriation by the bacteriophage T4 transcription factors AsiA and MotA. J Mol Biol 363, 931-944. Bendich AJ (1987). Why do chloroplasts and mitochondria contain so many copies of their genome? Bioessays 6, 279-82. Berghammer, H und Auer, B (1993). „Easypreps“: fast and easy plasmid minipreparation for analysis of recombinant clones in E. coli. Biotechniques 14, 524528. Birnboim, HC und Doly, J 1979. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Research 7, 1513-1523. Boeger, H, Bushnell, DA, Davis, R, Griesenbeck, J, Lorch, Y, Strattan, JS, Westover, KD und Kornberg, RD (2005). Structural basis of eukaryotic gene transcription. FEBS Letters 579, 899-903. Bradford, MM (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248-254. Briat, JF, Laulhere, JP und Mache, R (1979). Transcription activity of a DNA protein complex isolated from spinach plastids. Eur J Biochem 98, 285-292. Browning, DF und Busby, SJW (2004). The regulation of bacterial transcription initiation. Nat Rev Microbiol 2, 57-65. 96 Literatur Bülow, S, Reiss, T und Link, G (1987). DNA-binding proteins of the transcriptionally active chromosome from mustard (Sinapis alba, L.) chloroplasts. Curr Genet 12, 157159. Bülow, S und Link, G (1988). Sigma-like activity from mustard (Sinapis alba L.) chloroplasts conferring DNA-binding and transcription specificity to E. coli core RNA polymerase. Plant Mol. Biol. 10, 349-357. Burgess, RR, Travers, AA, Dunn, JJ und Bautz, EKF (1969). Factor stimulating transcription by RNA polymerase. Nature 221, 43-46. Campbell, EA, Westblade, LF und Darst, SE (2009). Regulation of bacterial RNA polymerase σ factor activity: a structural perspective. Curr Opin Microbiol 11(2), 121127. Cashel, M, Gentry, DM, Hernandez, VJ und Vinella, D (1996). The stringent response. In: Neidhardt FC (ed) Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology, ASM Press, Washington D.C., S. 1458-1496. Chisholm, ST, Coaker, G, Day, B und Staskawicz, BJ (2006). Host-microbe interactions: shaping the evolution of the plant immune response. Cell 124, 803-814. Chomczynski, P und Sacchi, N (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162, 156-159. Colland, F, Orsini, G, Brody, E, Bue, H und Kolb, A (1998). The bacteriophage T4 AsiA protein: a molecular switch for sigma 70-dependent promoters. Molecular Microbiology 27, 819-829. Dombroski, AJ, Walter, WA, Record, MT, Jr., Siegele, DA und Gross, CA (1992). Polypeptides containing highly conserved regions of transcription initiation factor σ 70 exhibit specificity of binding to promoter DNA. Cell 70, 501-512. 97 Literatur Doyle, JJ und Doyle JL (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11. Eisermann, A, Tiller, K und Link, G (1990). In vitro transcription and DNA binding characteristics of chloroplast und etioplast extracts from mustard (Sinapis alba) indicate differential usage of the psbA promoter. EMBO J 9, 3981-3987. Emanuel, C, Weihe, A, Graner, A, Hess, WR und Börner, T (2004). Chloroplast development affects expression of phage-type RNA polymerases in barley leaves. Plant J 38, 460-472. Emanuel, C, von Groll, U, Muller, M, Borner, T, Weihe, A (2006). Developmentand tissue-specific expression of the RpoT gene family of Arabidopsis encoding mitchondrial and plastid polymerases. Planta 223, 998-1009. Emanuelsson, O, Nielsen, H und von Heijne, G (1999). ChloroP, a neural networkbased method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci 8, 978- 984. Emanuelsson, O, Nielsen, H, Brunak, S und von Heijne, G (2000). Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300, 1005-1016. Finn, RD, Tate, J, Mistry, J, Coggill, PC, Sammut, JS, Hotz, HR, Ceric, G, Forslund, K, Eddy, SR, Sonnhammer, EL und Bateman, A (2008). The Pfam protein families database. Nucleic Acid Res 36: D281-D288. Fox, JD und Waugh, DS (2003). Maltose-binding protein as a solubility enhancer. Methods Mol Biol 205, 99-117. Fried, M und Crothers, DM (1981). Equilibria and kinetics of lac repressor-operator interaction by polyacrylamid gel electrophoresis. Nucleic Acids Res 9, 6505-6525. 98 Literatur Fujiwara, M, Nagashima, A, Kanamaru, K, Tanaka, K, und Takahashi, H (2000). Three new nuclear genes, sigD, sigE und sigF, encoding putative plastid RNA polymerase σ factors in Arabidopsis thaliana. FEBS Lett 481, 47-52. Gandhi, TK, Zhong, J, Mathivanan, S, Karthick, L, Chandrika, KN, Mohan, SS, Sharma, S, Pinkert, S, Nagaraju, S, Periaswamy, B, et al. (2006). Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 38, 285-293. Garner, MM und Revzin, A (1981). A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res 9, 3047-3060. Geisler-Lee, J, O’Toole, N, Ammar, R, Provart NJ, Millar, AH und Geisler, M (2007). A predicted interactome for arabidopsis. Plant Physiol. 145, 317-329. Giot, L, Bader, JS, Brouwer, C, Chaudhuri, A, Kuang, B, Li, Y, Hao, YL, Ooi, CE, Godwin, B, Vitols, E, et al. (2003). A protein interaction map of Drosophila melanogaster. Science 302, 1727-1736. Givens, RM, Lin, MH, Taylor, DJ, Mechold, U, Berry, JO und Hernandez, VJ (2004). Inducible expression, enzymatic activity, and origin of higher plant homologous of bacterial RelA/SpoT stress proteins in Nicotiana tabacum. J Biol Chem 279, 7495-7504. Gómez-Gómez, L, Felix, G und Boller, T (1999). A single locus determines sensitivity to bacterial flagellin in Arabidopsis thaliana. Plant J 18, 277-284. Gruber, TM und Gross, CA (2003). Multiple sigma subunits and the partitioning of bacterial transcription space. Annu Rev Microbiol 57, 441-466. Gruissem, W und Zurawski, G (1985). Analysis of promoter regions for the spinach chloroplast rbcL, atpB und psbA genes. EMBO J 4, 3375-3383. 99 Literatur Hajdukiewicz, PTJ, Allison, LA und Maliga, P (1997). The two RNA polymerases encoded by the nuclear and the plastid compartments transcribe distinct groups of genes in tobacco plastids. EMBO J 16, 4041-4048. Hakimi, MA, Privat, I, Valay, JG und Lerbs-Mache, S (2000). Evolutionary conservation of C-terminal domains of primary sigma70- type transcription factors between plants and bacteria. J Biol Chem 275, 9215-9221. Hallick, RB, Lipper, C, Richards, OC und Rutter, WJ (1976). Isolation of a transcriptionally active chromosome from chloroplasts of Euglena gracilis. Biochemistry 15, 3039-3045. Hanaoka, M, Kanamaru, K, Takahashi, H und Tanaka, K (2003). Molecular genetic analysis of chloroplast gene promoters dependent on SIG2, a nucleus-encoded sigma factor for the plastid-encoded RNA polymerase, in Arabidopsis thaliana. Nucleic Acids Res 31, 7090-7098. Hedtke, B, Börner, T und Weihe, A (1997). Mitochondrial and chloroplast phagetype RNA polymerases in Arabidopsis. Science 277, 809-811. Hedtke, B, Börner, T und Weihe, A (2000). One RNA polymerase serving two genomes. EMBO Rep. 1, 435-440. Hedtke B, Legen J, Weihe A, Herrmann RG und Börner T (2002). Six active phage-type RNA polymerase genes in Nicotiana tabacum. Plant J 30, 625-637. Helmann, JD und Chamberlin, MJ (1988). Structure and function of bacterial sigma factors. Annu Rev Biochem 57, 839-72. Helmann, JD (1999). Anti-sigma factors. Current Opinion in Microbiology 2, 135-141. Helmann, JD (2002). The extracytoplasmic function (ECF) sigma factors. Adv Microb Physiol 46, 47-110. 100 Literatur Herrmann RG, Bohnert HJ, Kowallik KV und Schmitt JM (1975). Size, conformation and purity of chloroplast DNA of some higher plants. Biochim Biophys Acta 378, 305–317. Hess, WR und Börner, T (1999). Organellar RNA polymerases of higher plants. Int Rev Cytol 190, 1-59. Homann, A (2002). Molekulargenetische Analyse der plastidären Transkription: Sigmafaktoren und ihre Gene. Dissertation, Ruhr-Universität Bochum. Homann, A und Link, G (2003). DNA-binding and transcription characteristics of three cloned sigma factors from mustard (Sinapis alba L.) suggest overlapping and distinct roles in plastid gene expression. Eur J Biochem 270, 1288-1300. Hu, J und Bogorad, L (1990). Maize chloroplast RNA polymerase: The 180-, 120-, und 38-kilodalton polypeptides are encoded in chloroplast genes. Proc Natl Acad Sci USA 87, 1531-1535. Hu, J, Troxler, RF und Bogorad, L (1991). Maize chloroplast RNA polymerase: The 78- kilodalton polypeptide is encoded by the plastid rpoC1 gene. Nucleic Acids Res. 19, 3431- 3434. Hughes, KT und Mathee, K (1998). The anti-sigma factors. Annu Rev Microbiol 52, 231-86. Husic, E (2009). Versuche zur Interaktion eines pflanzlichen Sigma-Faktors, AtSIG1, mit seinen plastidären Bindungspartner AtSIB-rP. Bachelorarbeit, Ruhr-Universität Bochum. Igloi, GL und Kössel, H (1992). The transcriptional apparatus of chloroplasts. Crit Rev Plant Sci 10, 525-558. 101 Literatur Ishizaki, Y, Tsunoyama, Y, Hatano, K, Ando, K, Shinmyo, A, Kobori, M, Takeba, G, Nakahira, Y und Shiina, T (2005). A nuclaer-encoded sigma factor, Arabidopsis SIG6, recognizes sigma-70 type chloroplast promoters and regulates early chloroplast development in cotyledons. Plant J. 42, 133-144. Isono, K, Niwa, Y, Satoh, K und Kobayashi, H (1997). Evidence for transcriptional regulation of plastid photosynthesis genes in Arabidopsis thaliana roots. Plant Physiol. 114, 623-630. Jarvis P und Soll J (2002). Toc, tic, and chloroplast protein import. Biochim Biophys Acta 1590 (1-3), 177-189. Jelich-Ottmann, C, Weiler, EW und Oecking, C (2001). Binding of regulatory 14-33 proteins to the C terminus of the plant plasma membrane H+-ATPase involves part of its autoinhibitory region. J Biol Chem 276, 39852-39857. Jensen, LJ, Kuhn, M, Stark, M, Chaffron, S, Creevey, C, Muller, J, Doerks, T, Julien, P, Roth, A, Simonovic, M, Bork, P und von Mering, C (2009). STRING 8 a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 37, D412-D417. Jones, DT (1999). Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292, 195-202. Jones, AME, Thomas, V, Bennett, MH, Mansfield, J und Grant, M (2006). Modifications to the Arabidopsis Defense Proteome Occur Prior to Significant Transcriptional Change in Response to Inoculation with Pseudomonas syringae. Plant Physiology 142, 1603-1620. Jones, JD und Dangl, JL (2006). The plant immune system. Nature 444, 323-239. Kanamura, K, Fujiwara, M, Seki, M, Katagiri, T, Nakamura, M, Mochizuki, N, Nagatani, A, Shinozaki, K, Tanaka, K und Takahashi, H (1999). Plastidic RNA polymerase σ factors in Arabidopsis. Plant Cell Physiol 40, 832-842. 102 Literatur Kanamaru, K und Tanaka, K (2004). Roles of chloroplast RNA polymerase sigma factors in chloroplast development and stress response in higher plants. Biosci Biotechnol Biochem 68(11), 2215-23. Kapoor, S, Suzuki, JY und Sugiura, M (1997). Identification und functional significance of a new class of non-consensus-type plastid promoters. Plant J 11, 327338. Kapoor, S und Sugiura, M (1999). Identification of two essential sequence elements in the nonconsensus type II PatpB-290 plastid promoter by using plastid transcription extracts from cultured tobacco BY-2 cells. Plant Cell 11, 1799-1810. Kasai, K, Usami, S, Yamada, T, Endo, Y, Ochi, K und Tozawa, Y (2002). A RelASpoT homolog (Cr-RSH) identified in Chlamydomonas reinhardtii generates stringent factor in vivo and localizes to chloroplasts in vitro. Nucleic Acids Res 30, 4985-4992. Kirk, JTO und Tilney-Basset, RAE (1978). Proplastids, etioplasts, amyloplasts, chromoplasts and other plastids. Ed 2. Amsterdam: Elsevier/North-Holland Biomedical Publishing, S. 219–241 Kobayashi, Y, Dokiya, Y, Sugita, M (2001a). Dual targeting of phage-type RNA polymerase to both mitochondria and plastids is due to alternative translation initiation in single transcripts. Biochem Biophys Res Commun 289, 1106-1113. Kobayashi, Y, Dokiya, Y, Sugiura, M, Niwa, Y, Sugita, M (2001b). Genomic organization and organic-specific expression of a nuclear gene encoding phage-type RNA polymerase in Nicotiana sylvestris. Gene 279, 33-40. Kobayashi, Y, Dokiya, Y, Kumazawa, Y, Sugita, M (2002). Non-AUG translation initiation of mRNA encoding plastid-targeted phage-type RNA polymerase in Nicotiana sylvestris. Biochem Biophys Res Commun 299, 57-61. Krause, K und Krupinska, K (2000). Molecular and functional properties of highly purified transcriptionally active chromosomes from spinach chloroplasts. Physiol Plant 109, 188-195. 103 Literatur Kuhlemeier, C (1992). Transcriptional and post-transcriptional regulation of gene expression in plants. Plant Mol Biol 19, 1-14. Kumar, A, Malloch, RA, Fujita, N, Smillie, DA, Ishihama, A und Hayward, RS (1993). The minus 35-recognition region of Escherichia coli sigma 70 is inessential for initiation of transcription at an "extended minus 10" promoter. J Mol Biol 232, 406418. Kuroiwa, T, Misumi, O, Nishida, K, Yagisawa, F, Yoshida, Y, Fujiwara, T und Kuroiwa, H (2008). Vesicle, mitochondrial, and plastid division machineries with emphysis on dynamin and electron-dense rings. Int. Rev. Cell Mol. Biol. 271, 97–152. Lalonde, S, Ehrhardt, DW, Loqué, D Chen, J, Rhee, SY und Frommer, WB (2008). Molecular and cellular approaches fort he detection of protein-protein interactions: latest techniques and current limitations. The Plant Journal 53, 610-635. Lang, D, Zimmer, AD, Rensing, SA und Reski, R (2008). Exploring plant biodiversity: the Physcomitrella genome and beyond. Trends Plant Sci 13, 542–549. Larkin, MA, Blackshields, G, Brown, NP, Chenna, R, McGettigan, PA, McWilliam, H, Valentin, F, Wallace, IM, Wilm, A, Lopez, R, Thompson, JD, Gibson, TJ und Higgins, DG (2007). ClustalW and ClustalX version 2. Bioinformatics 23(21), 2947-2948. Lerbs, S, Bräutigam, E und Mache, R (1988). DNA-dependent RNA polymerase of spinach chloroplasts: Characterization of sigma-like and alpha-like polypeptides. Mol Gen Genet 211, 459-464. Li, S, Armstrong, CM, Bertin, N, Ge, H, Milstein, S, Boxem, M, Vidalain, P-O, Han, JJ, Chesnau, A, Hao, T, et al. (2004). A map of the interactome network of the metazoan C. elegans. Science 303, 540-543. 104 Literatur Liere, K, Kestermann, M, Müller, U und Link, G (1995). Identification and characterization of the Arabidopsis thaliana chloroplast DNA region containing the genes psbA, trnH and rps19'. Curr Genet 28, 128-130. Liere, K, Kaden, D, Maliga, P, Börner, T (2004). Overexpression of phage-type RNA polymerase RpoTp in tobacco demonstrates its role in chloroplast transcription by recognizing a distinct promoter type. Nucleic Acids Res 32, 1159-1165. Link, G (1984). DNA sequence requirements for the accurate transcription of a protein-coding plastid gene in a plastid in vitro system from mustard (Sinapis alba L.). EMBO J 3, 1697-1704. Link, G und Langridge, U (1984). Structure of the chloroplast gene for the precursor of the Mr32,000 photosystem II protein from mustard (Sinapis alba L.). Nucleic Acids Res 12, 945- 958. Link, G (1994). Plastid differentiation: organelle promoters and transcription factors. In plant promoters and transcription factors – Results and problems in cell differentiation, Vol. 20. L. Nover, ed. (Berlin Heidelberg: Springer-Verlag), pp. 65-85. Link, G (1996). Green life: control of chloroplast gene transcription. BioEssays 18, 465-472. Link, G (2003). Redox Regulation of Chloroplast Transcription. Antioxidants & Redox Signaling 5, 79-87. Lonetto, M, Gribskov, M und Gross, CA (1992). The σ70 family: Sequence conservation and evolutionary relationships. J Bacteriol 174, 3843-3849. Loschelder, H, Homann, A, Ogrzewalla, K und Link, G (2004). Proteomics-based sequence analysis of plant gene expression - the chloroplast transcription apparatus. Phytochemistry 65, 1785-1793. 105 Literatur Loschelder, H, Schweer, J, Link, B und Link, G (2006). Dual temporal role of plastid sigma factor 6 in Arabidopsis development. Plant Physiol 142, 642-650. Loschelder, H (2007). Funktionsanalyse der Transkription in den Plastiden: Zur differentiellen Rolle von Mitgliedern der Sigmafaktor Familie aus Arabidopsis thaliana. Dissertation, Ruhr-Universität Bochum. Lysenko, EA (2006). Analysis of the Evolution of the Family of the Sig Genes Encoding Plant Sigma Factors. Russian Journal of Plant Physiology 53, 605-614. Maliga P (1998). Two plastid RNA polymerases of higher plants: an evolving story. Trends Plant Sci 3, 4-6. Martin W und Müller M 1998. The hydrogen hypothesis for the first eukaryote. Nature 392, 37-41 Martinez, E. (2002). Multi-protein complexes in eukaryotic gene transcription. Plant Mol Biol 50, 925-947. Masters, BS, Stohl, LL und Clayton, DA (1987). Yeast mitochondrial RNA polymerase is homologous to those encoded by bacteriophages T3 and T7. Cell 51, 89-99. McFadden, JI (2001). Chloroplast origin and intergration. Plant Physiology 125, 5053. Merill, CR, Goldman, D und van Keuren, MR (1983). Silver staining methods for polyacrylamide gel electrophoresis. Methods in Enzymology, Grossmann, L. und Moldeve, K., Eds., New York, Academic Press, 230-239. Miller, JP, Lo, RS, Ben-Hur, A, Desmarais, C, Stagljar, I, Noble, WS und Fields, S (2005). Large-scale identification of yeast integral membrane protein interactions. Proc Natl Acad Sci USA 102, 12123-12128. 106 Literatur Morikawa, K, Shiina, T, Murakami, S und Toyoshima, Y (2002). Novel nuclearencoded proteins interacting with a plastid sigma factor, Sig1, in Arabidopsis thaliana. FEBS letters 514, 300-304. Mudgett, MB (2005). New insights to the function of phytopathogenic bacterial type III effectors in plants. Annual Review of Plant Biology 56, 509-531. Mullies, KB und Faloona, FA (1987). Specific synthesis of DNA in vitro via a polymerase-catalysed chain reaction. Methods Enzymol 155, 335-350. Munkvold, KR und Martin, GB (2009). Advances in experimental methods for the elucidation of Pseudomonas syringae effector function with a focus on AvrPtoB. Molecular Plant Pathology 10(6), 777-793. Murakami, KS und Darst, SA (2003). Bacterial RNA polymerases: the wholo story. Current Opinion in Structural Biology 13, 31-39. Nagashima, A, Hanaoka, M, Motohashi, R, Seki, M, Shinozaki, K, Kanamaru, K, Takahashi, H und Tanaka, K (2004). DNA microarray analysis of plastid gene expression in an Arabidopsis mutant deficient in a plastid transcription factor sigma, SIG2. Biosci Biotechnol Biochem 68, 694–704. Narusaka, M, Kawai, K, Izawa, N, Seki, M, Shinozaki, K, Seo, S, Kobayashi, M, Shiraishi, T und Narusaka, Y (2008). Gene coding for SigA-binding protein from Arabidopsis appears to be transcriptionally up-regulated by salicylic acid and NPR1dependent mechanisms. J Gen Plant Pathol 74, 345-354. Neuhaus, H und Link, G (1987). Nucleotide sequence and transcriptional organization of the chloroplast tRNALys(UUU) gene from mustard (Sinapis alba). Curr. Genet. 11, 251-257. Neuhaus, H, Scholz, A und Link, G (1989). Structure and expression of a split chloroplast gene from mustard (Sinapis alba): Ribosomal protein gene rps16 reveals unusual transcriptional features und complex RNA maturation. Curr Genet 15, 63-70. 107 Literatur Ogrzewalla, K, Piotrowski, M, Reinbothe, S und Link, G (2002). The plastid transcription kinase from mustard (Sinapis alba L.). A nuclear-encoded CK2-type chloroplast enzyme with redox-sensitive function. Eur J Biochem 269, 3329-3337. Onda, Y, Yagi, Y, Saito, Y, Takenaka, N und Toyoshima, Y (2008). Light induction of Arabidopsis SIG1 and SIG5 transcripts in mature leaves: differential roles of cryptochrome 1 and cryptochrome 2 and dual function of SIG5 in the recognition of plastid promoters. Plant J 55(6), 968-978. Paget, MSB und Helmann, JD (2003). The σ70 family of sigma factors. Genome Biology 4, 203. Palmer, JD (2003). The symbiontic birth and spread of plastids: how many times and whodunit? J. Phycol. 39, 4-11. Pfalz, J, Liere, K, Kandlbinder, A, Dietz, KJ und Oelmuller, R (2006). pTAC2, -6 and -12 are components of the transcriptionally active plastid chromosome that are required for plastid gene expression. Plant Cell 18, 176-97. Pfannschmidt, T und Link, G (1994). Separation of two classes of plastid DNAdependent RNA polymerases that are differentially expressed in mustard (Sinapis alba L.) seedlings. Plant Mol Biol 25, 69-81. Pfannschmidt, T und Link, G (1997). The A und B forms of plastid DNA-dependent RNA polymerase from mustard (Sinapis alba L.) transcribe the same genes in a different developmental context. Mol Gen Genet 257, 35-44. Pfannschmidt, T, Ogrzewalla, K, Baginsky, S, Sickmann, A, Meyer, HE und Link, G (2000). The multisubunit chloroplast RNA polymerase A from mustard (Sinapis alba L.): Integration of a prokaryotic core into a larger complex with organellespecific functions. Eur J Biochem 267, 253-261. Pitzschke, A, Schikora, A und Hirt, H (2009). MAPK cascade signaling networks in plant defence. Current Opinion in Plant Biology 12, 421-426. 108 Literatur Privat, I, Hakimi, MA, Buhot, L, Favory, J-J und Lerbs-Mache, S (2003). Characterization of Arabidopsis plastid sigma-like transcription factors SIG1, SIG2 and SIG3. Plant Mol Biol 51, 385-399. Rao, VB (1994). Direct sequencing of polymerase chain reaction-amplified DNA. Anal Biochem 216, 1-14. Raivio, TL und Silhavy, TJ (2001). Periplasmic stress and ECF sigma factors. Annu Rev Microbiol 55, 591-624. Reiss, T und Link, G (1985). Characterization of transcriptionally active DNA protein complexes from chloroplasts and etioplasts of mustard (Sinapis alba L.). Eur J Biochem 148, 207-212. Reski, R (1998). Development, genetics and molecular biology of mosses. Botan. Acta 111, 1–15. Reski, R (2009). Challenges to our current view on chloroplasts. Biol Chem 390, 731–738. Richly, E und Leister, D 2004. An improved prediction of chloroplast proteins reveals diversities and commonalities in the chloroplast proteomes of Arabidopsis and rice. Gene 329, 11–16. Roberts, MR und Paul, ND (2006). Seduced by the dark side: integrating molecular and ecological perspectives on the influence of light on plant defence response against pests and pathogens. New Phytologist 170, 677-699. Roeder, RG (1996). The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem Sci 21, 327-335. Rosso, MG, Li, Y, Strizhov, N, Reiss, B, Dekker, K und Weisshaar B (2003). An Arabidopsis thaliana T-DNA mutagenized population (GABI-Kat) for flanking sequence tag-based reverse genetics. Plant Mol Biol 53, 247-259. 109 Literatur Rual, JF, Venkatesan, K, Hao, T, Hirozane-Kishikawa, T, Dricot, A, Li, N, Berriz, GF, Gibbons, FD, Dreze, M, Ayivi-Guedehoussou, N, et al., (2005). Towards a proteome-scale map of the human protein-protein interaction network. Nature 437, 1173-1178. Salinovich, O und Montelaro, RC (1986). Reversible staining and peptide mapping of proteins transferred to nitrocellulose after separation by sodium dodecylsulfatepolyacrylamide gel electrophoresis. Anal Biochem 156, 341-347. Sambrook, J, Fritsch, EF, Maniatis, T (2001). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Press, Cold Spring Harbor, NY Ed.2. Sanger, F., Nicklen, S. und Coulson, A.R. (1977). DNA sequencing with chain termination inhibitors. Proc Natl Acad Sci USA 74, 5463-5467 Sato, N, Terasawa, K, Miyajima, K, Kabeya. Y. (2003). Organization, developmental dynamics and evolution of plastid nucleoids. International Review of Cytology 232, 217–262. Schwechheimer, C, Smith, C, Bevan MW (1998). The activities of acidic and glutamine-rich transcriptional activation domains in plant cells: design of modular transcription factors for high level expression. Plant Mol Biol 36, 195-204. Schweer, J, Loschelder, H und Link, G (2006) A promoter switch that can rescue a plant sigma factor mutant. FEBS Lett 580, 6617-6622. Schweer, J (2009). Rolle kernkodierter Transkriptionsfaktoren bei der Steuerung der plastidären Genexpression in A. thaliana: Konstruktion und molekular-genetische Funktionsanalysen von Mutantenlinien. Dissertation, Ruhr-Universität Bochum. Schweer, J, Tuerkeri, H, Link, B und Link, G (2010). AtSIG6, a plastid sigma factor from Arabidopsis, reveals functional impact of cpCK2 phosphorylation. Plant Journal accepted. 110 Literatur Severinova, E, Severinova, K und Darst, SE (1998). Inhibition of Escherichia coli RNA polymerase by bacteriophage T4 AsiA. J Mol Biol 279, 9-18. Shiina, T, Tsunoyama, Y, Nakahira, Y und Khan, MS (2005). Plastid RNA polymerases, promoters, and transcription regulators in higher plants. Int Rev Cytol 244, 1-68. Sriraman, P, Silhavy, D und Maliga, P (1998). Transcription from heterologous rRNA operon promoters in chloroplasts reveals requirement for specific activating factors. Plant Physiol 117, 1495-1499. Stern, DB, Higgs, DC und Yang, J (1997). Transcription and translation in chloroplasts. Trends Plant Sci. 2, 308-315 Sugiura M (1992). The chloroplast genome. Plant Mol Biol 19,149-168. Sugiura, M (1995). The chloroplast genome. Essays Biochem 30, 49-57 Summer, H, Pfannschmidt, T und Link, G (2000). Transcripts and sequence elements suggest differential promoter usage within the ycf3-psaAB gene cluster on mustard (Sinapis alba L.) chloroplast DNA. Curr. Genet. 37, 45-52. Swiatecka-Hagenbruch, M, Liere, K und Börner, T (2007). High diversity of plastidial promoters in Arabidopsis thaliana. Mol Gen Genomics 277, 725-734. Takahashi, K, Kasai, K und Ochi, K (2004). Identification oft he bacterial alarmone guanosine 5‘-diphosphate 3‘-diphosphate (ppGpp) in plants. Proc Natl Acad Sci USA 101, 4320-4324. Tanaka, K, Tozawa, Y, Mochizuki, N, Shinozaki, K, Nagatani, A, Waskasa, K und Takahashi, H (1997). Characterization of three cDNA species encoding plastid RNA polymerase sigma factors in Arabidopsis thaliana: Evidence for the sigma factor heterogeneity in higher plant plastids. FEBS Lett. 413, 309-313. 111 Literatur Thilmony, R, Underwood, W und He, SY (2006). Genome-wide transcriptional analysis of the Arabidopsis thaliana interaction with the plant pathogen Pseudomonoas syringae pv. tomato DC3000 and the human pathogen Escherichia coli O157:H7. The Plant Journal 46, 34-53. Tiller, K, Eisermann, A und Link, G (1991). The chloroplast transcription apparatus from mustard (Sinapis alba L.)--Evidence for three different transcription factors which resemble bacterial σ factors. Eur J Biochem 198, 93-99. Tiller, K und Link, G (1993a). Sigma-like transcription factors from mustard (Sinapis alba L.) etioplast are similar in size to, but functionally distinct from, their chloroplast counterparts. Plant Mol Biol 21, 503-513. Tiller, K und Link, G (1993b). Phosphorylation and dephosphorylation affect functional characteristics of chloroplast and etioplast transcription systems from mustard (Sinapis alba L.). EMBO J 12, 1745-1753. Tozawa, Y, Teraishi, M, Sasaki, T, Sonoike, K, Nishiyama, Y, Itaya, M, Miyao, A und Hirochika, H (2007). The plastid sigma factor SIG1 maintains Photosystem I activity via regulated expression of the psaA Operon in rice chloroplasts. Plant J 52, 124-132. Toulokhonov, II, Shulgina, I und Hernandez, VJ (2001). Binding of the transcription effector ppGpp to Escherichia coli RNA polymerase is allosteric, modular, and occurs near the N terminus of the beta’-subunit. J Biol Chem 276, 1220-1225. Towbin, H, Staehelin, T, Gordon, J (1992). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. 1979. Biotechnology 24, 145-149. Troxler, RF, Zhang, F, Hu, J und Bogorad, L (1994). Evidence that σ factors are components of chloroplast RNA polymerase. Plant Physiol 104, 753-759. 112 Literatur Tsunoyama, Y, Ishizaki, Y, Morikawa, K, Kobori, M, Nakahira, Y, Takeba, G, Toyoshima, Y und Shiina, T (2004). Blue light-induced transcription of the plastidencoded psbD gene is mediated by a nuclear-encoded transcription initiation factor, AtSig5. Proc Natl Acad Sci USA 101, 3304-3309. Uetz, P, Giot, L, Cagney, G, Mansfield, TA, Judson, RS, Knight, JR, Lockshon, D, Narayan, V, Srinivasan, M, Pochart, P et al. (2000). A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisisae. Nature 403, 623-627. van der Biezen, EA, Sun, J, Coleman, MJ, Bibb, MJ und Jones, JD (2000). Arabidopsis RelA(SpoT homologs implicate (p)ppGpp in plant signaling. Proc Natl Acad Sci USA 97, 3747-3752. Waldburger, C, Gardella, T, Wong, R und Susskind, MM (1990). Changes in conserved region 2 of Escherichia coli σ 70 affecting promoter recognition. J Mol Biol 215, 267-276. Wösten, MM (1998). Eubacterial sigma-factors. FEMS Microbiol. Rev. 22, 127-150. Wycliffe, P, Sitbon, F, Wernersson, J, Ezcurra, I, Ellerstrom, M, Rask, L (2005). Continuous expression in tobacco leaves of a Brassica napus PEND homologue blocks differentiation of plastids and development of palisade cells. The Plant Journal 44, 1–15. Xie, YD, Li,W, G, D, Dong, J, Zhang, Q, Ren, D, Peng, M und Xia, Y (2010). The Arabidopsis gene SIGMA FACTOR-BINDING PROTEIN 1 plays a role in the salicylate- and jasmonate-mediated defense response. Plant Cell Environ, accepted. Yao, J, Roy-Chowdhury, S, Allison, LA (2003). AtSig5 is an essential nucleusencoded Arabidopsis σ-Like factor. Plant Physiol 132, 739-747. Zenkin, N, Kulbachinskiy, A, Yuzenkova, Y, Mustaev, A, Bass, I, Severinov, K und Brodolin, K (2007). Region 1.2 of the RNA polymerase sigma subunit controls recognition of the -10 promoter element. EMBO J 26(4), 955-964. 113 Literatur Zimmer, A, Lang, D, Richardt, S, Frank, W, Reski, R, und Rensing, SA (2007). Dating the early evolution of plants: detection and molecular clock analyses of orthologs. Mol Genet Genomics 278, 393–402. 114 Anhang 9. Anhang Abb. 9.1 : Sequenzvergleich der sechs Sigmafaktoren (AtSIG1-AtSIG6) aus Arabidopsis thaliana und dem Sigmafaktor SaSIG1 aus Sinapis alba Sterne, Punkte und Doppelpunkte geben den Grad der Homolgie der Proteine untereinander an. (Programm: CLUSTAL W 2.0.12) SaSIG1 AtSIG1 AtSIG4 AtSIG2 AtSIG6 AtSIG3 AtSIG5 MATTAVIGLNAGKRLLSSSFYYSDVTEKFLSVNDLCSSQYHTRSGITAKKTS------SS MATAAVIGLNTGKRLLSSSFYHSDVTEKFLSVNDHCSSQYHIAS--TKSGIT------AK --------------------------------------------MATTIPTT------AT MSSCLLPQFKCPPDSFSIHFRTSFCAPKHNKGSVFFQPQCAVSTSPALLTSM------LD ------MEATRNLVSSSPSFQTKTHLKSSYSSPSSVVMLHDQTTTPVVNSRH------LN MASFNSFPIPKQIVGSSSSSSSSTSRPRILVRSSLTSSMTSTNSMLVFVHPHPLIKHWLS -MGVVSISSSAARSPLGLGNDLLTHRSSLKKPSIVAFKADDSSNSALIIPPREQVLIPAE 54 52 10 54 48 60 59 SaSIG1 AtSIG1 AtSIG4 AtSIG2 AtSIG6 AtSIG3 AtSIG5 SSSNYSPSFPSSSRQTQSAKALKES---VASSVQPWLPPGSDQELEE------------KASNYSPSFPSSNRHTQSAKALKESV-DVASTEKPWLPNGTDKELEEE-----------ATMCPSPPVPTISPLLRTTHQCQPS----PSLSSPFSIKLSTALVCG------------VAKLRLPSFDTDSDSLISDRQWTYTRPDGPSTEAKYLEALASETLLTSDEAVVVAAAAEA SLSRHFPASVLSQEPREESRPLSHALRDDRTSQLTLERRQFDELVSSREDE--------LLRSDQTSFPIFVRIPMTSVVATRWSFLSSVKEESRIYQNDSLKACGCASVSPYTAQNNV KHNEKKREVTRRKPCKTPKKPLSLEHNSAPSCSLGVDYNEAAARLESIYKLS-------- 98 99 53 114 99 120 111 SaSIG1 AtSIG1 AtSIG4 AtSIG2 AtSIG6 AtSIG3 AtSIG5 ------------DD---IDHSVEALLLLQRSML-EKQWNLSFEKTR-----------------------CYDDDDLISHSVEAILLLQKSML-EKSWNLSFEKAVSSEY---------------------------DTTVDRVVDSSVMIK-PEKWGIQSEKRR-------------VALARAAVKVAKDATLFKNSNNTNLLTSSTADK-RSKWDQFTEKERAGILGHLAVSDNGI ----------KFEQQLLHSTGLWNLLISPLTSE-TKLPAVVSPLADAELCDVVALAQKAL YVELKDPKENIGVGSAERSYSSRSMLQYNLLAKNLLALEETFVALDSVRMERDIMLQMGK -----------PATTTLVEDDVEDGGSKAKVSRRRKRKESGEEKKVVVRN---------. 128 138 80 173 148 180 150 SaSIG1 AtSIG1 AtSIG4 AtSIG2 AtSIG6 AtSIG3 AtSIG5 -----------------------------------KKVP-VTCSGISARQRRIGAKKKTN ---------------------------PGKGTIRKKKIPVITCSGISARQRRIGAKKKTN ---------------------------------------------KRRRRRRVGYERLEP VSDKITASASNKESIGDLESEKQEEVELLEEQPSVSLAVRSTRQTERKARRAKGLEKTAS SASKQAALLVDDTEANPSDNIKDSLSTSSSMSLPEKGNIVRSKRQLERRAKNRRAPKSND LGAAELFKTCLSRYRGSSITSCLSDTTELVDTTPNQQVFVSSRRKVKKKARRSSVTAENG ------------------------------NVKKEKRMSLDKRIALKRNVQEKPVVASVD : 152 171 95 233 208 240 180 SaSIG1 AtSIG1 AtSIG4 AtSIG2 AtSIG6 AtSIG3 AtSIG5 ---VKAVSEVNFQNNLVKGYVKG----------VISDHVLSHAEVVRLSKKIKSGLRLDE MTHVKAVSDVSSGK-QVRGYVKG----------VISEDVLSHVEVVRLSKKIKSGLRLDD --EEEENAGVEAEAETISVPVVG----------ASRSGFLSRLEEVQLCLYLKEGAKLEN GIPSVKTGSSPKKKRLVAQEVDHNDPLRYLRMTTSSSKLLTVREEHELSAGIQDLLKLER VDDEGYVPQKTSAKKKYKQGADNDDALQLFLWGPETKQLLTAKEEAELISHIQHLLKLEK DQSSLPIGLRTTWNNIDVPRVRRPP-------KYRKKRERISRNETEMSTGVKIVADMER KKVTKRQQEEEKIERLVRDYSASNDIVSLDWKKMKIPPVLSSTEHAWLFKLMQPMKALLE : : :: : 199 220 143 293 268 293 240 SaSIG1 AtSIG1 AtSIG4 AtSIG2 AtSIG6 AtSIG3 AtSIG5 HKSRLTDRLGCEPSDEQLAMSLKISRAELQAWLMECHLAREKLAMSNVRLVMSIAQRYDHKSRLKDRLGCEPSDEQLAVSLKISRAELQAWLMECHLAREKLAMSNVRLVMSIAQRYDLGTSVEE---NEMVSVLLASGRGKKKRSANEILCRRKEAREKITRCYRRLVVSIATGYQLQTELTERSGRQPTFAQWASAAGVDQKSLRQRIHHGTLCKDKMIKSNIRLVISIAKNYQVKTKLESQNGCEPTIGEWAEAMGISSPVLKSDIHRGRSSREKLITANLRLVVHIAKQYQIRTQLEEESGKVASLSCWAAAAGMNEKLLMRNLHYGWYCRDELVKSTRSLVLFLARNYRVKDVLQKSLGREPREAEIAGEINMTAGEVKKKIEIGRAARNKLIKHNLRLVLFVMNKYFH : . * : .:::: **: : * 258 279 199 352 327 352 300 115 Anhang SaSIG1 AtSIG1 AtSIG4 AtSIG2 AtSIG6 AtSIG3 AtSIG5 --NMGAEMSDLVQGGLIGLLRGIEKFDSSKGFRISTYVYWWIRQGVSRALVDNSRTLRLP --NLGAEMSDLVQGGLIGLLRGIEKFDSSKGFRISTYVYWWIRQGVSRALVDNSRTLRLP --GKGLNLQDLIQEGSIGLLRGAERFDPDRGYKLSTYVYWWIKQAILRAIAHKSRLVKLP --GAGMNLQDLVQEGCRGLVRGAEKFDATKGFKFSTYAHWWIKQAVRKSLSDQSRMIRLP --NRGLNFQDLLQEGSMGLMKSVEKFKPQSGCRFATYAYWWIRQSIRKSIFQNSRTIRLP --GLGIAHEDLIQAGYVGVLQGAERFDHTRGYKFSTYVQYWIRKSMSTMVSRHARGVHIP EFTNGPKFQDLCQAGMRGLITAIDRFEPKRKFRLSTYGLFWIRHAIIRSMTTSN-FTRVP * .** * * *:: . ::*. :::** :**::.: : ::* 316 337 257 410 385 410 359 SaSIG1 AtSIG1 AtSIG4 AtSIG2 AtSIG6 AtSIG3 AtSIG5 THLHERLGLIRNAKLRLQ-EKG-ITPSIDRIAESLNMSQKKVRNATEAVSKIFSLDRDAF THLHERLGLIRNAKLRLQ-EKG-ITPSIDRIAESLNMSQKKVRNATEAVSKVFSLDRDAF GSMWELTAKVAEASNVLT-RKLRRQPSCEEIAEHLNLNVSAVRLAVERSRSPVSLDRVAS FHMVEATYRVKEARKQLY-SETGKHPKNEEIAEATGLSMKRLMAVLLSPKPPRSLDQKIG ENVYMLLGKVSEARKTCV-QEGNYRPSKEELAGHVGVSTEKLDKLLYNTRTPLSMQQPIW SSIIRTINHIQKARKTLKTSHGIKYAADEEIAKLTGHSVKKIRAANQCLKVVGSIDKKVG FGLESVRVEIYKAKTELWFEMG-RPPTEDEVVERLKITPERYREVLRAAKPVLSLNSKHS : : :* . :.:. . . *:: 374 395 316 469 444 470 418 SaSIG1 AtSIG1 AtSIG4 AtSIG2 AtSIG6 AtSIG3 AtSIG5 PSLNGLPGETHHSYIADNRLENNPWHGYDALALKDEVSKLISVTLGEREREIIRLYYGLD PSLNGLPGETHHSYIADTRLENNPWHGYDDLALKEEVSKLISATLGEREKEIIRLYYGLD Q-----NGRMTLQEIVRGPDETRPEEMVKREHMKHEIEQLLGS-LTARESRVLGLYFGLN MN-----QNLKPSEVIADPEAVTSEDILIKEFMRQDLDKVLDS-LGTREKQVIRWRFGME SD-----QDTTFQEITPDSGIETPTMSVGKQLMRNHVRNLLNV-LSPKERRIIKLRFGID DC-----FTTKFLEFTPDTTMESPEEAVMRQSARRDIHDLLEG-LEPREKQVMVLRYGLQ VT------QEEFINGITDVDGVGANNSRQPALLRLALDDVLDS-LKPKESLVIRQRYGLD . : : .:: * :* :: :*:: 434 455 370 523 498 524 471 SaSIG1 AtSIG1 AtSIG4 AtSIG2 AtSIG6 AtSIG3 AtSIG5 KEC-LTWEDISQRIGLSRERVRQVGLVALEKLKHAARKRKMEAMILKNKEC-LTWEDISKRIGLSRERVRQVGLVALEKLKHAARKRKMEAMILKNGETPMSFEEIGKSLKLSRERVRQINGIALKKLRNVHNVNDLKIYYSSSE DGRMKTLQEIGEMMGVSRERVRQIESSAFRKLKNKKRNNHLQQYLVAQS GGKQRSLSEIGEIYGLSKERVRQLESRALYRLKQNMNSHGLHAYADLLV DYRPKSLEEIGKLLKVSKEWIRKIERRAMAKLRDQPNAEDLRYYLNQ-GKGDRTLGEIAGNLNISREMVRKHEVKALMKLKHQARVDYLRQYIV--: :*. :*:* :*: *: :*:. . :. 481 502 419 572 547 571 517 116 Kongressberichte Kongressberichte: J Schweer, H Loschelder, H Türkeri, B Link, A Kolpack und G Link (2007). A Promoter Switch that can rescue a Plant Sigma Factor Mutant. FEBS Advanced Lecture Course “Origin and Evolution of Mitochondria and Chloroplasts”, Acquafredda di Maratea, Italien H Türkeri, H Loschelder, A Kolpack, J Schweer, B Link und G Link (2007). Redox Regulation of Chloroplast Transcription: The Plastid Transcription Kinase. GermanJapanese Workshop „Evolutionary and Functional Aspects of Redox Regulation in Photosynthetic Organisms“, Konstanz H Loschelder, J Schweer, H Türkeri, A Kolpack, B Link und G Link (2007). Dual temporal Role of Plastid Sigma Factor 6 in Arabidopsis Development. 17. Tagung der Gesellschaft für Entwicklungsbiologie, Marburg A Kolpack, H Türkeri, H Loschelder, J Schweer und G Link (2007). Developmentally regulated plastid transcription in plants: Interactions partners and signalling mechanisms. 17. Tagung der Gesellschaft für Entwicklungsbiologie, Marburg A Kolpack, H Türkeri, J Schweer, H Loschelder und G Link (2007). Regulation of Chloroplast Transcription. Interne Tagung SFB 480, Velen H Türkeri, J Schweer, A Kolpack, H Loschelder, B Link und G Link (2008). Analyse zur Funktion der plastidären Transkriptionskinase (PTK) bei der Regulation der Genexpression in Arabidopsis thaliana. 21. Tagung „Molekularbiologie der Pflanzen“, Dabringhausen H Türkeri, J Schweer, A Kolpack, H Loschelder und G Link (2008). Redox Regulation of Chloroplast Transcription: The Plastid Transcription Kinase. Lucca (Barga), Italien J Schweer, H Türkeri, A Kolpack, H Loschelder und G Link (2008). Multi-facetted controls in chloroplasts gene regulation: impact of multiple sigma factors and their master regulator. FESPB 2008 Congress, XVI Congress of the Federation of the European Societies of the Plant Biology, Tampere, Finnland A Kolpack, H Türkeri, J Schweer und G Link (2009). Protein-Protein Interaktionen in der plastidären Genexpression: Sigma Faktoren und ihre Interaktoren. 22. Tagung „Molekularbiologie der Pflanzen“, Dabringhausen 117 Lebenslauf LEBENSLAUF Persönliches Andrea Kolpack geboren am 22.09.1981 in Gelsenkirchen ledig Eltern: Renate Elfriede Herta Robbauer, geb. Eisenschmidt, ehemals Kolpack Frank Rudolf Wilhelm Kolpack Schulausbildung Aug. 1988 – Juli 1992 Grundschule an der Wannerstrasse in Gelsenkirchen Aug. 1992 – Juli 2001 Grillo-Gymnasium in Gelsenkirchen Abschluss: Abitur Hochschulbildung Okt. 2001 - Okt. 2006 Studium der Biologie an der Ruhr-Universität Bochum Abschluss: Diplom Diplomarbeit in der Arbeitsgruppe „Pflanzliche Zellphysiologie und Molekularbiologie“ zum Thema: “Versuche zur in vitro Funktionsanalyse rekombinanter Sigma-Faktoren und der rekombinanten Transkriptionskinase CK2a aus Arabidopsis thaliana“ Seit Feb. 2007 Anfertigung der Dissertation in der Arbeitsgruppe „Pflanzliche Zellphysiologie und Molekularbiologie“ zum Thema: „Regulatorische Protein-Protein-Interaktionen in der plastidären Transkription“ 118 ERKLÄRUNG Hiermit erkläre ich, dass ich die Arbeit selbständig verfasst und bei keiner anderen Fakultät eingereicht und dass ich keine anderen als die angegebenen Hilfsmittel verwendet habe. Es handelt sich bei der heute von mir eingereichten Dissertation um sechs in Wort und Bild völlig übereinstimmende Exemplare. Weiterhin erkläre ich, dass digitale Abbildungen nur die originalen Daten enthalten und in keinem Fall inhaltsverändernde Bildbearbeitung vorgenommen wurde. Bochum, den _____________________________________ (Unterschrift)