Kollagenosen - Rheuma
Werbung

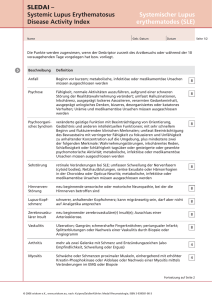
Kollagenosen Der Begriff „Kollagenosen“ wurde Anfang des 20. Jahrhunderts für Krankheiten eingeführt, die nicht organspezifisch waren, sondern viele Organe gleichzeitig betrafen und daher als Erkrankungen des Bindegewebes eingeordnet wurden. Heute wird vielfach die Bezeichnung „Autoimmunkrankheiten des Bindegewebes“ (autoimmune connective tissue diseases) vorgezogen, die jedoch ebenfalls nicht klar definiert ist. Alle Erkrankungen dieser Gruppe gehen mit einer Dysregulation des Immunsystems einher. Diese äußert sich im Auftreten autoreaktiver T-Lymphozyten, die entweder direkt oder durch Stimulation der Bildung von Autoantikörpern durch B-Lymphozyten eine Gewebeschädigung herbeiführen können. Die Autoantikörper sind überwiegend gegen Kernbestandteile (Nukleinsäuren, Kernproteine, Enzyme des Nukleinsäurestoffwechsels) und zu einem kleineren Anteil gegen zytoplasmatische Proteine (in Ribosomen, Mitochondrien) gerichtet. Die Ursachen der Immunstörung sind vielfältig und im einzelnen nicht genau bekannt. Eine Rolle spielen genetische Faktoren polygen vererbte Prädisposition zur Erkrankung, familiäre Häufung (beim LE Konkordanz bei eineiigen Zwillingen etwa 25-50%), starke Gynäkotropie (offenbar infolge einer Beeinflussung des Immunsystems durch Geschlechtshormone), besondere HLA-Klasse-II-Typen wie DR2 oder DR3, über die die Antigenpräsentation an T-Helfer-Zellen erfolgt, Mutationen in Genen, die die B-Zell- oder T-Zell-Aktivierung beeinflussen (beim LE z.B. nachgewiesen für den T-Zell-Rezeptor und TNF-), hereditäre Komplement-Defizienzen (dadurch gestörte Elimination von Eigen- und Fremdantigenen, gestörte Clearance von Immunkomplexen und von apoptotischen Zellen) sowie erworbene Faktoren virale, bakterielle oder parasitäre Antigene, die zu Kreuzreaktionen mit spezifischen körpereigenen Antigenen führen (z.B. interagieren Picornaviren zur Produktion ihrer RNA mit Histidyl-tRNA-Synthetase und könnten für Induktion der gegen dieses Enzym gerichteten Anti-Jo-1-Antikörper bei Dermatomyositis verantwortlich sein; Epitop des Scl-70-Antigens ist dem retroviralen p30gag-Protein ähnlich; Onchocerca volvolus verfügt über Homolog des Calcium-bindenden Enzyms Calreticulin, gegen das SSA-Antikörper gerichtet sind), virale oder bakterielle Antigene, die das Immunsystem unspezifisch stimulieren (wie z.B. Epstein-Barr-Virus), UV-Licht (v.a. UVB führt zur Aggravation von LE und Dermatomyositis; mögliche Mechanismen sind u.a. Untergang von Keratinozyten bei starker UV-Exposition, Transfer des SSA-Antigens vom Zellkern zur Zellmembran, DNA-Veränderungen), Geschlechtshormone (betroffen sind v.a. Frauen zwischen Menarche und Menopause; v.a. beim LE Verschlechterung bei Schwangerschaft und exogener Östrogengabe, teilweise auch zyklusabhängige Verschlechterung; Östrogene fördern humorale Immunreaktionen durch TH2-Differenzierung von T-Lymphozyten, Androgene dagegen eine TH1-Differenzierung; beim LE wurde erhöhte Cytokin-Produktion durch TH2-Zellen unter Östrogen-Einfluss nachgewiesen, u.a. IL-10; auch Prolaktion fördert humorale Immunantworten), Medikamente (beim LE Auslösung u.a. durch aromatische Amine wie Procainamid und Hydralazin, durch Isoniazid, Chlorpromazin, Phenytoin, Minocyclin, Thiazide, βBlocker, Captopril, Allopurinol, Piroxicam etc.; nach Absetzen nicht immer Abheilung; bei Dermatomyositis ist v.a. Auslösung durch D-Penicillamin, nichtsteroidale Antiphlogistika und Lipidsenker beschrieben), Tumoren (paraneoplastisches Auftreten der Dermatomyositis). Im Unterschied zu gewöhnlichen Immunreaktionen kann ein Autoantigen nicht eliminiert werden. Dies führt zur Perpetuierung der Immunantwort und zur Ausweitung auf andere, assoziierte Autoantigene („Epitope spreading“). Trotz der vielfältigen Ursachen äußern sich die Störungen in reproduzierbaren Krankheitsbildern, die zwar im Einzelfall gewisse Übergänge zeigen, jedoch in der Regel gut voneinander abzugrenzen sind. Die Klassifikation dieser Krankheitsbilder als „Kollagenosen“ ist willkürlich und differiert daher von Lehrbuch zu Lehrbuch. Traditionell werden vor allem der Lupus erythematosus, die Dermatomyositis und die Sklerodermie als „Kollagenosen“ bezeichnet. Weitere Krankheiten, die dieser Gruppe zugerechnet werden, sind das Sjögren-Syndrom („Sicca-Syndrom“; Minderproduktion von Tränen, Speichel etc. infolge einer Zerstörung exokriner Drüsen; an der Haut v.a. Xerose, Vaskulitis, anuläre Erytheme; oft SSA- und SSB-Antikörper, Kryoglobuline; Lippenbiopsie zeigt lymphozytäre Infiltration kleiner Speicheldrüsen; oft Zeichen eines SLE; Komplikation: extranodale Non-Hodgkin-Lymphome), das Sharp-Syndrom („mixed connective tissue disease“; zur Diagnose werden U1RNP-Antikörper gefordert; wahrscheinlich nur Sammelbegriff für ungewöhnliche Verlaufsformen anderer Kollagenosen; gleichzeitiges Auftreten von Sklerodermieund LE-typischen Hautveränderungen, Raynaud-Phänomen, Arthritis, Myalgien; im weiteren Verlauf oft Einordnung als Sklerodermie oder SLE möglich), die rezidivierende Polychrondritis (durch Antikörper gegen Typ-II-Kollagen, das in Knorpelgewebe zu finden ist; rezidivierende Erysipel-artige Rötung und Schwellung der Ohren unter Aussparung der Ohrläppchen, im weiteren Verlauf narbige Schrumpfung der Ohren; auch Befall des Nasenknorpels mit Entwicklung einer „Sattelnase“ sowie thorakal betonte Arthritis durch Gelenkbefall; oft kombiniert mit anderen Kollagenosen; histopathologisch perichondriales Infiltrat mit Dominanz von Neutrophilen, später Plasmazellen), das Still-Syndrom (Fieber und Arthritis, auch Lymphadenopathie, Splenomegalie, Serositis, Iridocyclitis; juveniler Typ mit Beginn im Kindesalter wird vom Erwachsenen-Typ unterschieden; für Diagnose wird Dauer für > 3 Monate und Befall von > 3 Gelenken gefordert; Rheumafaktor ist negativ; Hautveränderungen sind häufig und können der Arthritis vorausgehen; initial in ca. 25% der Fälle stammbetontes Exanthem; ferner persistierende erythematöse Plaques, die histopathologisch ein interstitielles, oft um Ausführungsgänge ekkriner Drüsen betontes Infiltrat mit Dominanz von Neutrophilen aufweisen, enstprechend der rheumatoiden neutrophilen Dermatose), die rheumatoide Arthritis (symmetrische Polyarthritis meist mit Beginn an Fingergrund- und -mittelgelenken, oft Fieberschübe, Skleritis; Rheumafaktoren = Antikörper, meist vom IgM-Typ, gegen IgG gerichtet, sind in ca. 80% nachweisbar, aber nicht spezifisch; Hautveränderungen sind häufig, haben unterschiedliche Spezifität und umfassen u.a. Rheumaknoten (bei 20% der Patienten; hohe Spezifität; gehen selten der Arthritis voraus; v.a. an Extremitätenstreckseiten im Bereich größerer Gelenke; histopathologisch große palisadenförmige Granulome aus einem Ring von Epitheloidzellen um zentrale fibrinreiche Nekrosezone), Rheumatoide neutrophile Dermatose (asymptomatische, meist mehrere Wochen persistierende erythematöse Plaques; histopathologisch dichtes - - - - - Neutrophilen-reiches Infiltrat in der gesamten Dermis, keine Granulome, keine Vaskulitis), Interstitielle granulomatöse Dermatitis mit Arthritis (auch bei SLE, StillSyndrom und anderen immunologischen Grundkrankheiten; klinisch strangförmige, lineäre Indurationen oder erythematöse Plaques; histopathologisch anfangs interstitielles Infiltrat mit Dominanz von Neutrophilen, später zunehmend Histiozyten mit Bildung von palisadenartigen Granulomen sowie von Churg-Strauss Granulomen, die zentral degeneriertes Kollagen zeigen, das von Histiozyten, Neutrophieln und Eosinophilen umgeben wird), Leukozytoklastische Vaskulitis (Immunkomplex-Vaskulitis; unspezifisch; recht typisch für die rheumatoide Arthritis sind jedoch kleine, meist rasch abheilende Infarkte an den Fingerspitzen), Pyoderma gangraenosum (unspezifisch; auch bei M. Crohn, Colitis ulcerosa, SLE, hämatologischen Erkrankungen; Beginn als Follikulitis, mit übersteigerter Entzündungsreaktion, zur Akkumulation von massenhaft neutrophilen Granulozyten und nachfolgender Ulceration führend). Erythema nodosum (unspezifisch; meist akut auftretende, durckschmerzhafte, subkutane, erythematöse, manchmal hämorrhagische Knoten; histopathologisch septal betonte Pannikulitis; meist in Assoziation mit Streptokokkeninfektionen, Yersiniose, Tuberkulose, Sarkoidose), Sweet-Syndrom (unspezifisch; plötzlich auftretende, sukkulente erythematöse Papeln und Plaques; histopathologisch gekennzeichnet durch knotiges bis diffuses Infiltrat mit Dominanz neutrophiler Granulozyten; meist nach fieberhaften Infekten sowie bei Lymphomen und Leukämien, auch bei anderen Autoimmunkrankheiten wie M. Crohn, Sjögren-Syndrom, SLE). Andere Krankheiten, die auf einer Dysregulation des Immunsystems beruhen und mit Autoantikörpern einhergehen können, aber gewöhnlich nicht als „Kollagenose“ apostrophiert werden, sind z.B. der Lichen planus, der M. Behçet, die Wegener’sche Granulomatose und die Graft-versus-host-disease. Zu den „klassischen“ Kollagenosen wird die progressive Sklerodermie gerechnet, gekennzeichnet durch die Kombination von Entzündung, Gefäßveränderungen und Sklerose. Ätiologie und Pathogenese sind unbekannt; neben einer genetischen Prädisposition könnten auch Umwelttoxine eine Rolle spielen (Parallelen zu Erkrankungen, die wahrscheinlich durch aromatische Amine ausgelöst werden: Eosinophilie-Myalgie-Syndrom infolge exogener LTryptophan-Zufuhr und „Toxic-oil-Syndrom“ durch Einnahme anilinvergällten Rapsöls; in beiden Fällen Eosinophilie, Myalgie, evtl. Lungenödem und Pleuritis, juckendes makulopapulöses Exanthem, sklerodermiforme Hautveränderungen ohne Beteiligung des Gesichts und ohne Raynaud-Phänomen). Eine primär immunologische Ursache wird durch Ähnlichkeit mit der chronischen Graft-vs.-Host-Reaktion nahegelegt. Im Rahmen der Immunreaktion gebildete Zytokine könnten sowohl für Gefäßveränderungen als auch für die gesteigerte Kollagensynthese durch Fibroblasten verantwortlich sein. T-Lymphozyten können die Kollagensynthese u.a. durch Interleukin-4 (TH2-Zellen) und transforming-growth-factor (TGF-) stimulieren; auch vermehrte Freisetzung von TGF- bei Hypoxie. Histopathologisch findet sich nur in Frühstadien ein interstitielles lymphoplasmazelluläres Infiltrat; im weiteren Verlauf zunehmende Verdichtung des Kollagens mit Verbreiterung der Dermis. Klinisch äußert sich die Entzündung in einem Ödem mit meist nur leichter Rötung (fast nur an Fingern), die Gefäßveränderungen in einem Raynaud-Phänomen (Spasmen der Fingerarterien unter Kälteeinfluss mit schmerzhafter Weißverfärbung und anschließender Zyanose; bei fast allen Patienten; oft anderen Manifestationen um Jahre vorausgehend), bleibenden Gefäßstenosen und ischämischen Ulcera (v.a. an den Fingerkuppen, mit eingesunkenen Narben abheilend, durch Sklerosierung mitverursacht), Kapillarerweiterungen und -verlust am Nagelfalz sowie Teleangiektasien (v.a. an Lippen, Gesicht, Händen), und die Sklerosierung in einer Sklerodaktylie mit zunehmender Bewegungseinschränkung (von den Fingern auf Hände und distale Unterarmabschnitte übergehend), einer Mikrostomie mit radiären Furchen („Tabaksbeutelmund“), einer Amimie und einer Verkürzung und Fibrosierung des Zungenbändchens. Weitere häufige Hautveränderungen sind eine Calcinosis cutis, und eine in druckexponierten Bereichen akzentuierte Hyperpigmentierung. Neben der Haut sind auch der Gastrointestinaltrakt (u.a. Schluckstörungen und Refluxösophagitis durch Ösophagusbeteiligung, Obstipation durch reduzierte Motilität und Peristaltik), der Respirationstrakt (fibrosierende Alveolitis mit Belastungsdyspnoe und pulmonaler Hypertension), die Nieren (Sklerose der Nierenarteriolen mit Untergang des Nierenparenchyms, renale Hypertonie), das Herz, Muskulatur, Skelettsystem und Nervensystem betroffen. Anhand von Hautveränderungen, Beteiligung innerer Organe und Laborparametern werden Varianten der progressiven Sklerodermie unterschieden, die eine zunehmend schlechtere Prognose aufweisen: Typ I (Hautbefall nur der Akren bis zum Hand- bzw. Sprunggelenk; oft Antizentromeren-Antikörper = ACA), Typ II (Hautveränderungen auch weiter proximal an Extremitäten und im Gesicht; oft ACA) und Typ III (oft initial Allgemeinsymptome und erst später Raynaud-Phänomen, stark ausgeprägte Nagelfalz-Läsionen, Reiben der Sehnenscheiden an Extremitäten, oft Topoisomerase I-Antikörper = Scl70-Ak; häufig rasche Progredienz; 5-Jahres-Überlebensrate 70%). Eine klare Abgrenzung der Varianten ist allerdings nicht möglich. Eine besondere Manifestationsform der limitierten progressiven Sklerodermie (Typ I und II) mit meist milder Ösophagusbeteiligung ist das CREST-Syndrom (Calcinosis, Raynaud, Esophagus, Sklerodactyly, Teleangiectases). Zu den Differenzialdiagnosen der progressiven Sklerodermie zählen u.a.: Morphaea (= zirkumskripte Sklerodermie; fraglich Auslösung durch Borrelien bei entsprechender genetischer Prädisposition; histopathologisch gleiche Stadien wie progressive Sklerodermie; Therapie v.a. mit UVA1 oder PUVA; unterschieden werden der plaqueförmige Typ (Beginn mit rundlicher Rötung, dann durch Sklerosierung zentrale Weißverfärbung und Verhärtung mit erythematösem Randsaum; Abheilung mit atrophischen, hyperpigmentierten, leicht eingesunkenen Herden, die nicht selten ohne Anamnese entzündlicher oder sklerotischer Vorstadien beobachtet und auch als Atrophodermia Pasini-Pierini bezeichnet werden; in akuten Stadien oft gutes Ansprechen auf Penicillin), der lineäre Typ (meist im Kindesalter; längliche Läsionen, oft mit Fasziitis und nachfolgender, tief greifender narbiger Schrumpfung; meist an Extremitäten, aber auch am Kopf unter dem Bilde der „Sclerodermia en coup de sarbe“ oder der „Hemiatrophia faciei“), der kleinmakulöse Typ (Morphaea guttata; runde, oft derb infiltrierte Herde; wegen oberflächlicher Sklerose oft sehr weiß; Typ III der progressiven Sklerodermie kann mit ähnlichen Herden beginnen), - - - - - - - der generalisierte Typ (pansklerotische Morphaea, multiple, symmetrisch verteilte Herde am gesamten Körper, Einschränkungen der Beweglichkeit und der Atmung), die Morphaea profunda (Entzündung und Sklerosierung v.a. in Subkutis), die eosinophile Fasziitis (durch Befall der Fasziien tiefe Induration, v.a. an Extremitäten unter Aussparung von Händen und Füßen, oft frühzeitig Beugekontrakturen), der Lichen sclerosus et atrophicus (wahrscheinlich keine einheitliche Erkrankung und nur zum Teil dem Spektrum der Morphaea zugehörig; klinisch weißliche, oft bizarr konfigurierte, atrophische Plaques, meist im Genitalbereich und am Stamm; histopathologisch atrophische Epidermis mit Lymphozyten in der Basalzellschicht, subepidermale Sklerose und bandförmiges lymphoplasmazelluläres Infiltrat im Bereich des superfiziellen Gefäßplexus, am Stamm häufig kombiniert mit typischen histopathologischen Zeichen der Morphaea), Sklerodermie-artige Veränderungen durch exogene chemische Faktoren (u.a. LTryptophan beim Eosinophile-Myalgie-Syndrom, Vinylchlorid-Monomere, organische Lösungsmittel wie Xylol und Benzol, Pestizide wie DDT, Chemotherapeutika wie Bleomycin und Cisplatin, Silikate), Sklerodermie-artige Veränderungen durch physikalische Faktoren (Sklerosierung durch chronische Vibrationstraumata, Morphaeae-artige Veränderungen nach Radiatio), Chronische Graft-versus-Host-Disease (oft ausgedehnter Organbefall; SiccaSymptomatik; Erosionen und Narben der Mundschleimhaut; an der Haut lichenoide Papeln, retikuläre Hyperpigmentierungen, Sklerodermie-ähnliche Läsionen; histopathologisch oft Assoziation von lichenoider Interface-Dermatitis und Sklerose im Stratum reticulare), Porphyria cutanea tarda (angeborene oder durch Leberschädigung akquirierte Defizienz der Uroporphyrinogen-Decarboxylase in Hepatozyten; vermehrter Nachweis von Uroporphyrinen im Harn; neben erhöhter Verletzlichkeit der Haut mit Ausbildung verkrusteter Erosionen und Milien, Blasen, Hyperpigmentierung, Hypertrichose des Gesichts sowie Zeichen chronischer Lichtschädigung ist auch massive Sklerosierung möglich; histopathologisch PAS-positives Material in Gefäßwänden), Scleredema Buschke (rasch fortschreitende Induration mit leichter Rötung, v.a. im Nacken und am oberen Rücken; oft assoziiert mit Diabetes mellitus, auch nach Infekten; histopathologisch massive Verdickung der Dermis ohne Vermehrung von Fibroblasten und Muzinablagerungen zwischen Kollagenfaserbündeln), Skleromyxödem (generalisierte Verlaufsform des Lichen myxoedematosus; meist bei monoklonaler Gammapathie; großflächig konfluierende Papeln v.a. an Streckseiten der Extremitäten, oberem Rumpf und Gesicht; histopathologisch Fibrose mit starker Vermehrung von Fibroblasten und Muzinablagerungen). Die Therapie der progressiven Sklerodermie ist schwierig. Steroide und Immunsuppressiva (v.a. Cyclosporin, Methotrexat, Cyclophosphamid) haben einen nur geringen Effekt. Antifibrotische Substanzen (insbesondere -Interferon, das Wirkung von TGF- auf Fibroblasten hemmt) werden erprobt. Zur Therapie des Raynaud-Syndroms werden Vasodilatatoren eingesetzt (v.a. Nifedipin, cave: bei Fingerkuppennekrosen evtl. Durchblutungsverschlechterung durch Blutdrucksenkung). Das Prostazyklin-Derivat Iloprost hat eine protektive Wirkung auf Endothelzellen (u.a. durch Hemmung der Plättchenaktivierung, Reduktion endothelialer Adhäsionsmoleküle wie ICAM-1 und VCAM- 1) und hemmt TGF--Wirkung auf Fibroblasten (anhaltende Besserung des RaynaudPhänomens nach 5tägiger Infusionstherapie zu Beginn der kalten Jahreszeit, gute Wirkung bei Fingerkuppennekrosen). Wichtig sind Allgemeinmaßnahmen (Schutz vor Kälte, Bewegungsübungen) sowie eine an den Symptomen orientierte Therapie (z.B. Protonenpumpen-Hemmern wie Omeprazol bei Reflux-Ösophagitis). Die Dermatomyositis ist charakterisiert durch die Assoziation einer Polymyositis (lymphozytäre Myositis mit symmetrischer Myopathie v.a. der Schulter- und Beckengürtelmuskulatur, oft schmerzhaft, Sicherung durch EMG und Biopsie des M. triceps brachii, Ultraschall, Erhöhung von CK und Aldolase im Serum) mit charakteristsichen Hautveränderungen. Beide Komponenten können anfangs (teilweise auch dauerhaft) isoliert vorhanden sein. Evtl. Kombination mit anderen Autoimmundermatosen (v.a. progressive Sklerodermie) und Befall weiterer Organe (in bis zu 40% Reizleitungsstörungen und Tachyarrhythmien infolge meist milder Beteiligung des Herzmuskels, seltener Dysphagie, interstitielle Pneumonitis etc.). Evtl. Nachweis von Autoantikörpern (u.a. Anti-Jo-1, gerichtet gegen Histidyl-tRNA-Synthetase, bei ca. 20% der Patienten, Indikator für Lungenbeteiligung). Altersgipfel in der Kindheit (juvenile Form) und im mittleren bis höheren Erwachsenenalter (adulte Form; in 10 bis 50% Assoziation mit Karzinom). Die Hautveränderungen sind v.a. Folge einer Interface-Dermatitis, d.h. einer Entzündung mit Schwerpunkt im Bereich der dermoepidermalen Junktion (vakuoläre Veränderungen an der Junktion; einzelne nekrotische Keratinozyten v.a. im Stratum basale, teilweise in die obere Dermis abtropfend; Abtropfen von Pigment aus Epidermis in obere Dermis mit nachfolgender Speicherung in Melanophagen), die durch T-Lymphozyten hervorgerufen wird. Die Epidermis ist meist atrophisch, die Basalmembran verdickt, im Stratum reticulare findet sich vermehrt Muzin. Im Unterschied zum diskoiden Lupus erythematosus, der ansonsten die gleichen Veränderungen zeigt, geht die Dermatomyositis meist mit einem nur spärliches Lymphozyteninfiltrat in der oberen Dermis einher. Klinische Zeichen der Dermatomyositis sind ein livides Erythem in lichtexponierten Arealen, v.a. periokulär (kombiniert mit Lidödem) und an den Streckseiten der Extremitäten (unter Bevorzugung der Ellenbogen-Streckseiten und der Streckseiten der Fingergelenke), weißliche, zentral eingesunkene Papeln über den Streckseiten der Fingergelenke („Gottron’sche Papeln“) sowie Teleangiektasien und Hyperkeratosen am Nagelfalz. Oft besteht Juckreiz. Bei längerer Bestandsdauer geht das Erythem in eine Poikilodermie (Atrophie, Teleangiektasien und Pigmentverschiebungen als Folge der chronischen InterfaceDermatitis) über. Die Therapie der Dermatomyositis besteht in einer initial hoch dosierten Prednisongabe (etwa 1 mg/kg KG mit allmählicher Dosisreduktion über Monate), evtl. in Kombination mit Methotrexat oder Azathioprin. Meist wird so innerhalb von 2 bis 3 Jahren eine vollständige Abheilung erreicht. Bei Karzinom-assoziierten Fällen führt eine erfolgreiche Therapie des Karzinoms meist zur Besserung. Der Lupus erythematosus ist eine Systemkrankheit mit vielen unterschiedlichen Manifestationen. Die Verschiedenartigkeit des klinischen Bildes und des Verlaufs ist auf Unterschiede in Ätiologie und Pathogenese all jener Manifestationen zurückzuführen, die dem Spektrum des Lupus erythematosus zugerechnet werden. Die häufige Kombination dieser Manifestationen spricht jedoch für große pathogenetische Gemeinsamkeiten und war für ihre Zusammenfassung unter einem Namen verantwortlich. Trotz der Verschiedenartigkeit der Manifestationen ist der Lupus erythematosus letztlich ein charakteristisches Krankheitsbild, das sich von anderen Autoimmunkrankheiten in der Regel gut abgrenzen lässt. Der Lupus erythematosus geht in schweren Fällen mit Fieber und Abgeschlagenheit einher und betrifft u.a. Haut, Schleimhäute (v.a. Erosionen der Mundschleimhaut), Gelenke (symmetrische Polyarthritis, v.a. Finger-, Hand-, und Kniegelenke), Muskeln (Myalgien), Nervensystem (periphere Neuropathie, Infarkte im Gehirn, Kopfschmerzen, epileptische Anfälle, Psychosen), Nieren (Nephritis unterschiedlichen Schweregrades; histopathologisch werden u.a. eine fokale proliferative Lupus-Nephritis, eine diffuse proliferative Nephritis und eine membranöse Nephritis mit starker Verdickung der Basalmembranen der Glomeruli unterschieden; zu den Symptome zählen Proteinurie, Hämaturie, nephrotisches Syndrom; renale Hypertension ist Hinweis auf schlechte Prognose), Herz (Perikarditis, auch Myokarditis mit Reizleitungsstörungen, verruköse Endokarditis = Libman-Sacks-Endokarditis, Arteriitis der Koronararterien mit Infarkt), Lungen (Pleuritis, Lupus-Pneumonitis), Gastrointestinaltrakt (Überkeit, Anorexie, krampfhafte Schmerzen bei Infarkt und Darmperforation infolge einer Arteriitis der Mesenterialarterien, Aszites), Milz (Splenomegalie), Lymphknoten (Lymphadenopathie), Augen (Konjunktivitis, Episkleritis), Speicheldrüsen (Parotitis, oft assoziiert mit Sjögren-Syndrom). Vom American College of Rheumatology wurden 11 Kriterien für die Diagnose eines systemischen Lupus erythematosus (SLE) propagiert, von denen vier erfüllt sein sollten (1. Schmetterlingserythem; 2. diskoide kutane LE-Herde; 3. Photosensitivität; 4. orale Ulcera; 5. Arthritis; 6. Serositis, d.h. Pleuritis oder Perikarditis; 7. Nierenbeteiligung; 8. neurologische Veränderungen in Form von Psychose oder epileptischen Anfällen; 9. hämatologische Veränderungen in Form von hämolytischer Anämie, Leukopenie, Lymphopenie, Thrombozytopenie; 10. immunologische Veränderungen, u.a. anti-DNA-Ak, Anti-SmAntikörper, falsch pos. unspezifische Lues-Serologie; 11. Antinukleäre Antikörper). Diese Kriterien wurden von Rheumatologen v.a. mit dem Ziel entwickelt, den LE von anderen Erkrankungen mit Gelenkbeschwerden abzugrenzen. Tatsächlich ist die Diagnose jedoch bereits anhand einer typischen Hautläsion zu stellen, denn der Lupus erythematosus ist immer eine systemische Erkrankung. Die Unterscheidung zwischen systemischem und nichtsystemischem LE ist willkürlich, sachlich nicht gerechtfertigt und für die Patientenbetreuung nicht hilfreich. In schweren Fällen mit ausgedehnten Manifestationen an Haut und/oder anderen Organen ist stets ein Staging mit anschließender adäquater Therapie erforderlich, unabhängig davon, ob vier der propagierten Kriterien erfüllt sind oder nicht. Der Lupus erythematosus nimmt meist einen milden Verlauf mit nur geringen morphologischen und funktionellen Veränderungen. An inneren Organen sind diese oft nicht nachweisbar. An der Haut werden dagegen schon geringfügige Veränderungen erfasst, so dass umschriebene Hautläsionen oft die einzige Manifestation eines Lupus erythematosus sind. Diese Läsionen werden im wesentlichen durch autoreaktive T-Lymphozyten verursacht und manifestieren sich meist in Form einer Interface-Dermatitis. T-Lymphozyten führen dabei entweder direkt (zytotoxische T-Zellen, CD8+) oder durch Freisetzung von Zytokinen wie Interferon und TNF- (T-Helfer-Zellen, CD4+; NK-Zellen) zu einer Schädigung der Junktionszone und zum Untergang (Apoptose) von Keratinozyten. Ein gleichartiger Mechanismus liegt auch anderen T-Zell-vermittelten Immunreaktionen zugrunde (z.B. Graftversus-Host-Disease, Lichen planus, Erythema exsudativum multiforme, lichenoide Arzneimittelreaktionen). Autoantikörper spielen in der Pathogenese kutaner LE-Herde nur eine untergeordnete Rolle. Im Serum finden sich Antinukleäre Antikörper (als sensibelster Screening-Test) bei limitiertem Hautbefall in Form des dermoepidermalen LE nur in etwa 25% und beim rein dermalen LE tumidus in etwa 10%. An der dermoepidermalen Junktion lassen sich beim dermoepidermalen LE immunfluoreszenzmikroskopisch in der Regel Niederschläge von IgG, IgM, IgA und/oder Komplement (C3) nachweisen, wenn Läsionen in belichteter Haut biopsiert werden (in frühen Läsionen jedoch oft negativ; zudem sehr unspezifisch, da Niederschläge auch in lichtgeschädigter Haut gesunder Personen und bei anderen Krankheiten wie Rosazea vorkommen können). Beim Lupus erythematosus tumidus, bei dem die Junktionszone intakt ist, sind junktionale Niederschläge von Immunglobulinen und/oder Komplement nur in etwa 10% der Fälle nachweisbar. Erst bei stärkerer Krankheitsaktivität (klinisch erkennbar an ausgedehnten Hautveränderungen bzw. Symptomen vonseiten anderer Organe) ist die direkte Immunfluoreszenz auch in nicht-läsionaler Haut positiv (beim SLE in lichtexponierter Haut in etwa 80%; in nicht lichtexponierter Haut seltener, aber hinweisend auf hohe Krankheitsaktivität, einschl. Nierenbeteiligung). Erst bei stärkerer Krankheitheitsaktivität sind Autoantikörper im Serum regelmäßig nachweisbar, insbesondere Antinukleäre Antikörper (ANA; sehr sensibel, aber wenig spezifisch; bei SLE in 95% nachweisbar; auch bei anderen Autoimmunkrankheiten, chronischen Infektionen etc.; durch manche Medikamente induzierbar, z.B. Procainamid, Hydralazin, Diphenylhydantoin, Isoniazid; bei gesunden Personen in 5% nachweisbar, v.a. bei alten Personen), Anti-DNS-Antikörper (nur Antikörper gegen Doppelstrang-DNS sind aussagekräftig; mäßig sensibel, dafür recht spezifisch für LE; bei SLE in 50 bis 70% nachweisbar; Konzentration korreliert mit Krankheitsaktivität, v.a. bezüglich Nierenbeteiligung), Anti-Histon-Antikörper (mäßig sensibel, mäßig spezifisch; bei SLE in etwa 50% nachweisbar, aber auch bei rheumatoiden Arthriitis und anderen Krankheiten; bei medikamenteninduziertem LE meist positiv), Anti-Sm-Antikörper (gerichtet gegen kleine nukleäre Ribonucleoproteine, = snRNP; wenig sensibel, aber recht spezifisch; bei SLE in ca. 30% nachweisbar; korrelieren mit Nierenbeteiligung), Anti-nRNP-Antikörper (gerichtet gegen snRDPs; wenig sensibel, wenig spezifisch; ursprüngliche Korrelation mit mixed connective tissue disease ließ sich nicht bestätigen; Vorkommen bei verschiedenen Kollagenosen, bei SLE in ca. 10% nachweisbar); SSA-Antikörper (= Ro-Antikörper; gerichtet gegen kleine cytoplasmatische Ribonucleoproteine, = scRNP, u.a. Calreticulin, ein auch bei pathogenen Keimen nachweisbares 46 kDA-Antigen in endoplasmatischen Retikulum, das u.a. nach UVExposition an die Zelloberfläche transloziert wird; mäßig sensibel, mäßig spezifisch; ursprünglich beim Sjögren-Syndrom beschrieben, aber auch bei anderen Kollagenosen vorkommend; beim SLE in ca. 25% nachweisbar, auch bei ANA-negativen Fällen; Korrelation mit besonderen LE-Manifestationen, u.a. starker Photosensitivität, subakut-kutanem LE, neonatalem LE und LE bei hereditärem C2- und C4Komplementmangel), - SSB-Antikörper (= La-Antikörper; gerichtet gegen scRNP; oft gemeinsam mit SSAAntikörpern auftretend, aber weniger sensibel, bei SLE in ca. 10% nachweisbar), Anti-C1q-Antikörper (nicht sensibel, aber sehr spezifisch für Lupus-Nephritis), Anti-Neutrophilen-Cytoplasmatische Antikörper (= ANCA; wenig sensibel, wenig spezifisch; bei SLE in ca. 20% nachweisbar; v.a. perinukleäre ANCA, = pANCA; im Gegensatz zur Wegener’schen Granulomatose keine zytoplasmatischen ANCA, = cANCA), Rheumafaktoren (Anti-IgG-Antikörper; wenig sensibel, wenig spezifisch; bei SLE in ca. 30% nachweisbar); Anti-Phospholipid-Antikörper (heterogene Gruppe von Antikörpern gegen Phosphodiestergruppen in DNS und Phospholipiden wie Phosphatidylcholin, Phosphatidylserin und Cardiolipin; erstmals bei der Lues beschrieben und noch heute im VDRL-Test zur Lues-Diagnostik genutzt, später Beschreibung als „lupus anticoagulant“; wenig sensibel, wenig spezifisch; bei SLE in ca. 30% nachweisbar, aber auch bei anderen Autoimmunkrankheiten und ohne assoziierte Autoimmunkrankheit; klinisch relevant infolge einer Beeinflussung der Gerinnung, die zu erhöhter Thromboseneigung führt und sich an der Haut in Thrombophlebitiden, Ulcera crurum, Gangrän und Livedo racemosa äußern kann; auch Migräne und zunächst passagere Sprachstörungen etc. infolge zentralnervöser Thrombosen; Detektion u.a. durch verlängerte PTT infolge reduzierter Umwandlung von Prothrombin in Thrombin durch Phospholipid-haltigen Prothrombinaseaktivatorkomplex). Das Auftreten so zahlreicher Autoantikörper ist auf eine Dysregulation des Immunsystems zurückzuführen (herabgesetzte Funktion von zytotoxischen T-Zellen und TH1-Zellen bei Überwiegen einer TH2-Immunantwort; TH2-Zytokine wie IL10 stimulieren AntikörperProduktion durch B-Zellen; ferner reduzierte Hemmung von B-Zellen durch antiidiotypische Antikörper; Antigen-Spreading durch Zellschädigung bei erhöhtem Aktivitätsniveau von BZellen). Die meisten Antikörper sind lediglich Epiphänomene. Für besondere Manifestationen des LE scheinen sie jedoch direkt verantwortlich zu sein (u.a. liegt dem neonatalen LE ein diaplazentarer Übergang von SSA-Antikörpern von der Mutter auf das Kind zugrunde; antiDNS-Antikörper lösen im Mausmodell eine Nephritis aus; zirkulierende Antigen-AntikörperKomplexe können zu einer leukozytoklastischen Vaskulitis führen; anti-PhospholipidAntikörper führen zu Thromben und Ulzerationen). Die Hautveränderungen des Lupus erythematosus werden in spezifische und unspezifische Läsionen eingeteilt. Letztere entsprechen teilweise besonderen Stadien der spezifischen Veränderungen (z.B. Pigmentanomalien, Urticae), teilweise sind sie Ausdruck einer Kombination mit anderen, verwandten Krankheitsbildern (z.B. Rheumaknoten, Sklerodaktylie). Zu den unspezifischen Hautveränderungen zählen (mit Angabe der ungefähren Häufigkeit bei SLE) Teleangiektasien (45-65%; auffällig v.a. am Nagelfalz), leukozytoklastische Vaskulitis (10-20%; Immunkomplex-Vaskulitis, Purpura v.a. an Unterschenkeln), Thrombophlebitis (5-10%), Raynaud-Phänomen (15-20%), Livedo racemosa (im englischen Sprachraum als Livedo reticularis bezeichnet; 10%; meist verursacht durch Anti-Phospholipid-Antikörper, die zu Thromben und Gefäßverschlüssen führen; auch polyklonale Kryoglobulinämie kann verantwortlich sein; klinisch minder durchblutete weißliche Hautareale, umgeben von bizarr konfigurierten lividen Rötungen, die durch Hyperämie bei sauerstoffarmem Blut bedingt sind; v.a. an Beinen und Gesäß; bei gleichzeitigem Vorliegen zentralnervöser - - - - - Komplikationen infolge von Gefäßverschlüssen sind Kriterien für Diagnose des Sneddon-Syndroms erfüllt), die Atrophie blanche (<5%; schmerzhaft, Weißverfärbung mit randständiger livider Rötung infolge einer Minderdurchblutung, oft ulzerierend, fast nur an Unterschenkeln), der Morbus Degos (= maligne atrophische Papulose; <5%; klinisch gekennzeichnet durch im Durchmesser ca. 5 mm große Papeln mit eingesunkenem, weißlichem Zentrum und erythematösem Rand an Stamm und Extremitäten; histopathologisch oft typisches Bild des dermoepidermalen LE, zentral teilweise Nekrosezone aufgrund eines Gefäßverschlusses nachweisbar; bei vielen Patienten gleichartige Veränderungen im Gastrointestinaltrakt, die infolge Perforation und Peritonitis oft zum Tode führen; auch andere Manifestationen eines SLE), chronische Ulcera der Haut (5%), Erosionen der Mundschleimhaut (7%), Gangrän (<5%, schwarze Verfärbung, v.a. Finger, Zehen), Perniones (<5%; livide Maculae oder Papeln an den Akren nach Kälteexposition; histopathologisch superfizielles und tiefes perivaskuläres Lymphozyteninfiltrat mit interstitieller Muzinvermehrung, vom dermalen LE nicht abzugrenzen; bei Entwicklung von Hyperkeratosen und anderen Zeichen des chronisch-diskoiden LE wird vom Chilblain-Lupus gesprochen; Beziehungen zwischen Kälte-induiziertem Chilblain-Lupus und Perniones sind unklar); Anetodermie (<5%; umschriebener Verlust elastischer Fasern, der sich klinisch in weißlichen, leicht erhabenen und leicht eindrückbaren Papeln und Plaques manifestiert; ursächlich sind evtl. Mikrothromben infolge von Anti-PhospholipidAntikörpern), Rheumaknoten (5-10%), Pyoderma gangraenosum (<5%), Sklerodaktylie (10%), Calcinosis cutis (<5%), Urticae (7-15%; oft mit hämorrhagischer Note) Pigmentveränderungen (10%). Die spezifischen Veränderungen des kutanen Lupus erythematosus weisen ein charakteristisches klinisches Bild auf, nämlich Erytheme, die bevorzugt in lichtexponierten Arealen auftreten (v.a. Nasenrücken und Wangen; Ohren; Hals und Nacken; DecolletéBereich; Finger, dort im Unterschied zur Dermatomyositis nicht über, sondern zwischen Interphalangealgelenken), häufig mit Hyperkeratosen einhergehen und meist mit atrophischen, hyper- oder hyperpigmentierten Patches abheilen. Histopathologisch findet sich meist eine Interface-Dermatitis (vakuoläre Veränderungen an der Junktion, unscharfe Junktionszone, einzelne nekrotische Keratinozyten, Melanophagen im Stratum papillare) in Assoziation mit einem fast rein lymphozytären perivaskulären Infiltrat und interstitiellen Muzinablagerungen. Das histopathologische Bild entspricht damit dem der Dermatomyositis (siehe dort); allerdings ist das Entzündungsinfiltrat in der Regel stärker ausgeprägt und betrifft nicht nur die obere, sondern die gesamte Dermis. Die Epidermis ist jedoch nicht immer betroffen, und die Entzündung kann auch die Subkutis erfassen. Zudem variiert das histopathologische Bild in Abhängigkeit von Bestandsdauer und Akuität der Veränderungen: bei akuten Läsionen gehören dem Infiltrat auch neutrophile (viel seltener eosinophile) Granulozyten an. Weitere Zeichen der Akuität sind ein Ödem und Erythrozytenextravasate. Bei lange bestehenden Läsionen sieht man meist eine ausgeprägte Atrophie der Epidermis (Ausnahme: hypertropher LE, bei dem es infolge anhaltenden Kratzens bzw. Reibens zu einer reaktiven Epidermishyperplasie kommt) und der Dermis (klinisch sind die Herde dann oft eingesunken), die Basalmembran ist verdickt, im Stratum papillare finden sich zahlreiche Melanophagen (= „Pigmentinkontinenz“, verantwortlich für die klinisch wahrnehmbaren Hyper- und Hypopigmentierungen; in Assoziation mit Epidermisatrophie und ektatischen Gefäßen Bild der Poikilodermie), und es findet sich oft eine follikulär betonte Hyperkeratose (wegen der starken sensiblen Innervation der Follikel führt Kratzen an den follikulären Hyperkeratosen zu Schmerzen, = „Tapeziernagelphänomen“). Der Verlauf des kutanen LE ist in der Regel chronisch-rezidivierend: über Jahre hinweg treten (oft an den gleichen Stellen, v.a. nach UV-Exposition) immer wieder Entzündungsschübe auf, die sich über Wochen bis Monate zurückbilden, meist einen atrophischen, hyper- oder hypopigmentierten Patch hinterlassend. Nur selten heilt die Erkrankung nach einem einzigen oder wenigen Schüben spontan ab. Die spezifischen Hautveränderungen des LE wurden nach unterschiedlichen Gesichtspunkten eingeteilt. Eine Klassifikation anhand betroffener anatomischer Strukturen wurde 1940 von Irgang vorgenommen. Dieser unterschied zwischen dem dermoepidermalen LE (= klassischer diskoider Lupus erythematosus): häufigste Manifestationsform; klinisch charakterisiert durch meist scheibenförmige, scharf begrenzte, teilweise konfluierende Plaques, die – abhängig von Stadium und Akuität – alle typischen Charakteristika des kutanen LE aufweisen können; bei Befall des behaarten Kopfes Alopezie (anfangs mit Rötung und follikulären Hyperkeratosen einhergehend; Abheilung mit Verlust der Haarfollikel und permanenter Alopezie unter dem unspezifischen Bild der „Pseudopelade Brocq“); dermalen LE (Lupus erythematosus tumidus): keine Epidermisbeteiligung oder nur minimale Hinweise darauf, wie zum Beispiel einige Melanophagen im Stratum papillare; histopathologisch gekennzeichnet durch ein perivaskuläres Lymphozyteninfiltrat in der gesamten Dermis mit interstitieller Muzinvermehrung; ANA nur in 10% nachweisbar, direkte Immunfluoreszenz nur in 10% positiv; klinisch rötliche Papeln ohne Veränderungen der Oberfläche, häufig anuläre Läsionen, keine Hyperkeratosen, keine Pigmentverschiebung; wegen fehlender Epidermisbeteiligung Abheilung der Läsionen mit klinischer Restitutio ad integrum, d.h. ohne Atrophie und Hyper- bzw. Hypopigmentierung; Varianten des LE tumidus wurden unter verschiedenen Namen beschrieben (wobei wegen fehlender Epidermisbeteiligung Zugehörigkeit zum Spektrum des LE meist nicht erkannt wurde): lymphocytic infiltration Jessner-Kanof (rötliche Papeln und Plaques, meist im Gesicht; durch UV-Licht provozierbar, meist rasche Besserung durch Hydroxychloroquin); retikuläre erythematöse Muzinose (= REM-Syndrom; klinisch anuläre und retikuläre Rötungen im Decolleté-Bereich und am oberen Rücken; durch UVLicht provozierbar, meist rasche Besserung durch Hydroxychloroquin); tiefes gyriertes Erythem (= tiefer Typ des Erythema anulare centrifugum; meist wenige anuläre Herde); Lupus erythematosus gyratus repens (sich rasch ausdehnende, zentral wieder abheilende anuläre Herde am gesamten Integument; rasch wechselnde Konfiguration der Effloreszenzen; die ursprüngliche Beschreibung des Erythema anulare centrifugum 1916 durch Darier betraf einen solchen Fall); subkutanen LE (Lupus profundus): in zwei Dritteln der Fälle assoziiert mit Veränderungen an der dermoepidermalen Junktion und in der Dermis; histopathologisch lymphozytäres Infiltrat v.a. in den Läppchen der Subkutis, oft lymphozytärer „Kernstaub“ durch Zerfall von Lymphozyten, lymphozytäre Vaskulitis, subkutane Nekrosen; klinisch tiefe, unscharf begrenzte rötlich-bräunliche Knoten, nicht selten ulzerierend und mit eingesunkenen Narben abheilend; v.a. im Gesicht, an Oberarmen und Oberschenkeln, am behaarten Kopf, am oberen Stamm und am Gesäß; ANA fast immer nachweisbar; direkte Immunfluoreszenz in etwa einem Drittel der Fälle positiv). Eine Klassifikation anhand der Ausdehnung der Hautveränderungen erfolgte schon 1872 durch Kaposi, nämlich eine Unterscheidung zwischen dem limitierten kutanen LE (von Kaposi als diskoider Lupus erythematosus bezeichnet): meist nur wenige „scheibenförmige Flecken“, keine Beeinträchtigung des Allgemeinzustandes; disseminierten kutanen LE (von Kaposi als Lupus erythematosus discretus et aggregatus bezeichnet): zahlreiche kleine Flecken, oft in Gruppen stehend, meist assoziiert mit schweren, z.T. lebensbedrohlichen Allgemeinerscheinungen. Zahlreiche kleine, meist sich sehr rasch vermehrende Effloreszenzen deuten auf eine hohe Krankheitsaktivität hin und sind häufig mit Manifestationen an inneren Organen und einer schlechten Prognose assoziiert. Eine Klassifikation anhand des Verlaufes der Hautveränderungen wurde 1981 von Gilliam und Sontheimer vorgenommen. Sie unterschieden zwischen dem chronisch-kutanen Lupus erythematosus: diskoider Lupus erythematosus, hypertropher LE, Lupus profundus; nur in 5-10% Entwicklung eines SLE; subakut-kutanen Lupus erythematosus (= SCLE): in ca. 15% Entwicklung eines SLE; starke Photosensitivität; SSA-Antikörper positiv; histopathologisch meist relativ geringes, oberflächliches Entzündungsinfiltrat, jedoch keine grundsätzlichen Unterschiede zum chronisch-diskoiden LE; klinisch sind zentrale Gesichtspartien meist ausgespart bei bevorzugtem Befall von seitlichen Gesichtspartien, Hals, Decolleté-Bereich und Streckseiten der Arme; unterschieden werden ein anulärer Typ (erythematöser, indurierter Randsaum, meist ohne Epidermisbeteiligung) und ein papulosquamöser Typ (= „psoriasiformer Typ“); als eigenständige Variante des subakut-kutanen LE gilt der neonatale Lupus erythematosus (ausgelöst durch transplazentaren Übergang von SSA-Antikörpern von der Mutter auf das Kind; der Säugling entwickelt SCLE-typische Veränderungen, jedoch bevorzugt im Gesicht, v.a. periokulär; starke Photosensitivität; oft AV-Block; weitere Komplikationen sind Kardiomyopathie, Thrombozytopenie, hepatobiliäre Störungen mit Hyperbilirubinämie; Abheilung innerhalb von Monaten, allerdings oft mit irreversiblen Schädigungen); akuten kutanen Lupus erythematosus: plötzliches Auftreten der Hautveränderungen, meist nach UV-Exposition; Rückbildung oft mit Hyper- oder Hypopigmentierungen; meist erythematöse Maculae oder flache, ödematöse Plaques ohne Schuppung; typische Manifestationen sind das Schmetterlingserythem (Rötung an Nasenrücken und Wangen; meist rasches Auftreten nach UV-Exposition und spätere Rückbildung), exanthematische Hautveränderungen („morbilliform“) und bullöse Hautveränderungen (bullöser Lupus erythematosus; histopathologisch einhergehend mit Ansammlungen neutrophiler Granulozyten in dermalen Papillen mit nachfolgender subepidermaler Blasenbildung, ähnlich der Dermatitis herpetiformis und der linearen IgA-Dermatose; wahrscheinlich induziert durch Autoantikörper gegen Antigene der dermoepidermalen Junktion, v.a. Typ-VII-Kollagen; in der direkten Immunfluoreszenz sind meist IgG, IgA, IgM und C3 an der Junktion nachweisbar); - Erythema-exsudativum-multiforme-artige Hautveränderungen (auch als Rowell-Syndrom bezeichnet; ursprünglich in Verbindung mit ANA, SSBAntikörpern und positivem Rheumafaktor beschrieben; diese Laborparameter sind jedoch nur inkonstant nachweisbar); Der akute kutane LE weist in der Regel auf das Vorliegen eines SLE hin (beim bullösen LE fast immer). Er wurde teilweise als Folge einer Vaskulitis bezeichnet, tatsächlich liegen jedoch keine Vaskulitiszeichen vor; mit Ausnahme des bullösen LE (bei dem Autoantikörper ein Dermatitis herpetiformis-ähnliches Bild induzieren) entsprechen die Veränderungen histopathologisch denen des diskoiden LE (oft mit zusätzliches Zeichen der Akuität: Beteiligung neutrophiler Granulozyten am Infiltrat, Ödem, Erythrozytenextravasate). Die verschiedenen Manifestationen des LE bilden ein Spektrum mit fließenden Übergängen; jede Klassifikation bleibt daher ungenau. Die Einteilung der Hautveränderungen anhand betroffener anatomischer Strukturen hat den Vorteil einer morphologischen Charakterisierung der Einzeleffloreszenzen (und der absehbaren Konsequenzen wie Narbenbildung, Pigmentalterationen, Restitutio ad integrum). Die Einteilung anhand von Ausdehnung oder Akuitätsgrad ermöglicht Rückschlüsse auf die Gefährlichkeit der Erkrankung: disseminierte und akut auftretende Läsionen sind Hinweise auf eine hohe Krankheitsaktivität und sollten zur einer genauen Untersuchung auf (klinisch oft noch nicht manifeste) Komplikationen vonseiten innerer Organe Anlass geben. In der Therapie des Lupus erythematosus wird im Unterschied zur Dermatomyositis und einigen bullösen Autoimmundermatosen eine zurückhaltende Strategie bevorzugt, d.h. eine möglichst niedrig dosierte Behandlung, die den klinischen Erfordernissen angepasst ist (anstelle einer initial massiven Unterdrückung des Immunsystems): bei wenigen Hautläsionen Lokaltherapie mit Corticosteroiden, ferner Lichtschutz; in therapierefraktären Fällen, bei größerer Zahl von Hautveränderungen und/oder bei extrakutanen Manifestationen (v.a. Arthritis) Gabe von Antimalariamitteln (z.B. 200400 mg Hydroxychloroquin/die; u.a. verminderte Prostaglandin-Synthese durch Hemmung der UV-induzierten Aktivität von Phospholipase A und C; Hemmung der Freisetzung von Proteasen aus Lysosomen; reduzierte Wirksamkeit bei Rauchern evtl. wegen Hemmung der Aufnahme in Lysosomen; wegen seltener Komplikation einer irrevesiblen Retinopathie möglichst intermittierende Gabe, v.a. in Sommermonaten, und regelmäßige opthalmologische Kontrollen; auch Neuropathie, Leukopenie etc. möglich), initial evtl. kombiniert mit Steroiden; alternativ können Retinoide versucht werden (anfangs z.B. 50 mg Etretinat oder Acitretin/die; cave: Teratogenität); bei fehlender Besserung durch Antimalariamittel und akutem Verlauf (subakut-kutaner LE, akuter kutaner LE, bullöser LE) DADPS, evtl. kombiniert mit Steroiden, bei schwererem Verlauf Corticosteroide (initial z.B. 60-120 mg Prednison/die) in Kombination mit Immunsuppressiva (z.B. Azathioprin, Cyclophosphamid), therapeutische Alternativen umfassen u.a. Thalidomid (v.a. diskoider LE), Cyclosporin-A; in schweren Fällen hochdosierte Pulstherapie mit Steroiden oder Immunsuppressiva, Photopherese, gezielte Therapie besonderer Manifestationen, z.B. ASS bei erhöhtem Thromboserisiko durch Anti-Phospholipid-Antikörper.