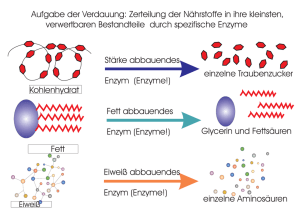
Katalytische Strategien 9
Werbung

9. Katalytische Strategien 9.0.1 Einige grundlegende katalytische Mechanismen sind vielen Enzymen gemeinsam Die enzymatische Katalyse beginnt mit der Substratbindung. Die Bindungsenergie, die dabei als freie Enthalpie freigesetzt wird, bestimmt die Substratspezifität und erhöht die katalytische Effizienz. Die Wechselwirkungen zwischen Enzym und Substrat stabilisieren auch den Übergangszustand und verringern die Aktivierungsenergie. Ausserdem kann die Bindungsenergie Strukturveränderungen im Enzym oder Substrat unterstützen und so die Katalyse erleichtern, was man als induced fit bezeichnet. Mechanismen der Katalyse: 1. Kovalente Katalyse: Das aktive Zentrum einer reaktiven Gruppe enthält eine starke nucleophile Gruppe, die temporär kovalent verändert wird. Bsp: Chymotropsin (9.1.2) 2. Allgemeine Säure-Base-Katalyse: Ein anderes Molekül als Wasser besitzt die Funktion eines Protonendonors oder -akzeptors. Bsp: Histidinrest, der als basischer Katalysator bei Chymotropsin, Nucleophilie eines Serinrestes verstärkt (9.1.2) 3. Metallionenkatalyse: Ein Metallion kann als elektrophiler Katlysator wirken, indem es die negative Ladung eines Zwischenproduktes der Reaktion stabilisiert, oder eine nucleophile Gruppe erzeugen, indem es die Acidität eines benachbarten Moleküls erhöht. Bsp: Carboanhydrase (9.2.2) Das Metallion kann an das Sustrat binden und dadurch die Bindungsenergie erhöhen. Bsp: NMP-Kinase (9.4.3) 4. Katalyse durch Annäherung: Substrate binden ans Enzym und werden so zusammengebracht. Bsp: NMP-Kinase (9.4.3) 9.1 Proteasen ermöglichen eine schwer durchführbare Reaktion Proteasen spalten Proteine durch eine Hydrolysereaktion, die Addition eines Wassermoleküls an eine Peptidbindung. Ohne Katalysator beträgt die Halbwertszeit dafür zehn bis 1000 Jahre. Peptidbindungen müssen aber bei einigen biochemischen Reaktionen innerhalb von Millisekunden gespalten werden. Für die kinetische Stabilität von Peptidbindungen ist ihre Resonanzstruktur verantwortlich. Das Kohlenstoffatom der Carbonylgruppe ist wegen dem Doppelbindungscharakter zwischen dem Kohlenstoff- und Stickstoffatom weniger elektrophil. Ein Enzym, das eine Spaltung bewirken soll, muss also einen nucleophilen Angriff auf eine normalerweise nicht reaktive Carbonylgruppe ermöglichen. 9.1.1 Chymotrypsin besitzt einen hochreaktiven Serinrest Ein Enzym, das am Abbau von Proteinen beteiligt ist, ist Chymotrypsin. Es spaltet Peptidbindungen selektiv auf der carboxyterminalen Seite von grossen hydrophoben Aminosäuren, z.B. Tryptophan, Tyrosin, Phenylalanin, Methionin. Eine stark nucleophile Gruppe des Enzyms greift die Carbonylgruppe des Substrats an und wird während der Katalyse kurzzeitig kovalent mit dem Substrat verknüpft –> Kovalente Modifikation. Beim katalytischen Mechanismus von Chymotrypsin spielt ein ungewöhnlich reaktiver Serinrest (Serin 195) eine Rolle. 9.1.2 Die Chymotrypsinreaktion erfolgt in zwei Schritten, die über ein kovalent gebundenes Zwischenprodukt miteinander verknüpft sind Um die Kinetik der Enzymreaktion leicht messen zu können, lässt man das Enzym mit einem Substratanaloga reagieren, das ein farbiges Produkt hervorbringt. Bei Chymotrypsin ist ein solches chromogenes Substrat N-Acetyl-L-phenylalanin-pnitrophenylester und eines der Produkte, die bei der Chymotrypsinspaltung entstehen ist das gelbe p-Nitrophenolat. Zu Beginn der Reaktion zeigt sich eine Phase des schnellen Anstiegs (burst). Sobald das Gleichgewicht erreicht ist, entsteht das Produkt langsamer. Dies deutet darauf hin, dass die Hydrolyse in zwei Schritten erfolgt, von denen der erste rascher erfolgt als der zweite. Die Bildung von p-Nitrophenolat erfolgt also nach Zugabe des Substrats mit grosser Geschwindigkeit, sobald das Acyl-Enzym-Zwischenprodukt entstanden ist, während das Enzym für ein “Zurücksetzen“ durch die Hydrolyse des Acyl-Enzym-Zwischenproduktes länger braucht. 9.1.3 Serin ist Teil einer katalytischen Triade mit Histidin und Aspartat Chymotrypsin ist etwa kugelförmig und besteht aus drei Polypeptidketten, die durch Disulfidbrücken miteinander verbunden sind. Das aktive Zentrum welches Serin 195 enthält, liegt in einer Spalte an der Oberfläche des Enzyms. Die Seitenkette vom Serin ist über eine Wasserstoffbrücke mit dem Imidazolring von Histidin 57 verbunden, dessen NH-Gruppe wiederum über eine Wasserstoffbrücke mit der Carboxylatgruppe von Aspartat 102 verknüpft ist. Diese Anordnung bezeichnet man als katalytische Triade. Der Histidinrest positioniert die Serinseitenkette und polarisiert deren Hydroxylgruppe. Durch den Abzug des Protons von der Hydroxylgruppe entsteht ein Alkoxidion, das stärker nucleophil ist als ein Alkohol. Der Aspartatrest stützt die Orientierung des Histidins und macht es durch elektrostatische Effekte zu einem besseren Protonenakzeptor. Mechanismus für die Hydrolyse von Peptiden (Abb. 9.8): Nach der Substratbindung erfolgt ein nucleophiler Angriff der Hydroxylgruppe von Serin 195 auf das Carbonylkohlenstoff des Substrats. Die Umgebung des Kohlenstoffatoms verändert sich von einer trigonal planaren zu einer tetraedrischen Struktur. Dieses in sich instabile tetraedrische Zwischenprodukt erhält formal eine negative Ladung am Sauerstoffatom der Carbonylgruppe, die durch NH-Gruppen eines Proteinbereiches, den man als Oxyaniontasche bezeichnet, stabilisiert wird. Das Zwischenprodukt zerfällt anschliessend und es entsteht das Acyl-Enzym. Ein Proton wird vom positiv geladenen Histidin auf die bei der Spaltung der Peptidbindung entstehende Aminogruppe übertragen, welche sich so ungehindert vom Enzym lösen kann und durch ein Wassermolekül ersetzt wird. Die nun folgende Hydrolyse der Estergruppe des Acyl-Enzyms entspricht im Prinzip der vorangegangenen Reaktion. Der Histidinrest wirkt nun als allgemeiner Säurekatalysator und bildet ein tetraedrisches Zwischenprodukt, bei dessen Zerfall sich das Carbonsäureprodukt bildet. Schliesslich ist das Enzym nach Freisetzung des Carbonsäureprodukts wieder bereit für einen neuen Katalysezyklus. Eine tiefe, verhältnismässig hydrophobe Tasche (S1-Tasche), in welche die langen, ungeladenen Seitenketten von Aminosäureresten wie Phenylalnin und Tryptophan hineinpassen, erklärt die Bevorzugung von Peptidbindungen direkt hinter solchen Aminosäureresten. Die Bindung einer geeigneten Seitenkette in diese Tasche positioniert die daran anschliessende Peptidbindung im aktiven Zentrum für die Spaltung. Andere Proteasen zeigen noch komplexere Spezifitätsmuster mit zusätzlichen Taschen. 9.1.4 Katalytische Triaden kommen auch bei anderen hydrolytischen Enzymen vor Einige davon, wie beispielsweise Trypsin und Elastase, sind zu Chymotrypsin homolg. Sie zeigen dieselben Reaktionsmechanismen, unterscheiden sich jedoch in der Substratspezifität. Weitere Vertreter der Chymotrypsinfamilie sind eine Gruppe von Proteinen, die bei der Blutgerinnung mitwirken und eine Reihe von Proteasen bei Bakterien und Viren. Auch andere Enzyme, die nicht zu Chymotrypsin homolog sind, enthalten sehr ähnlche aktive Zentren mit einer katalytischen Triade, z.B. Subtilisin, eine Bakterienprotease, oder Carboxypeptidase II aus Weizen. Dieselbe Strategie für die Hydrolyse von Peptid- und ähnlichen Bindungen ist also im Lauf der Evolution mindestens dreimal entstanden und ist somit ein Beispiel für konvergente Evolution. 9.1.5 Die katalytische Triade wurde mithilfe ortsspezifischer Mutagenese genau untersucht Mit ortsspezifischer Mutagenese konnte der jeweilige Beitrag der einzelnen Aminosäuren zur katalytischen Leistungsfähigkeit des Enzyms bestimmt werden. Der Austausch von Serin oder Histidin gegen Alanin verringerte den Wert für k kat auf weniger als ein Millionstel des normalen Wertes. Die Umwandlung von Aspartat hatte einen geringeren Effekt. 9.1.6 Cystein-, Aspartat- und Metalloproteasen sind weitere wichtige Klassen von peptidspaltenden Enzymen Die Strategie der Cysteinproteasen stimmt grösstenteils mit der von der Chymotrypsinfamilie überein. Bei ihnen übernimmt ein Cysteinrest, der von einem Histidinrest aktiviert wird, die Funktion der nucleophilen Gruppe, die ähnlich dem Serinrest bei den Serinproteasen die Peptidbindung angreift. Bsp: Papain aus der Papayafrucht; Cathepsine, im Immunsystem von Säugern; Caspasen, Enzyme bei der Apoptose Bei den Aspartatproteasen ist das wichtigste Merkmal des aktiven Zentrums ein Paar von Asparaginsäureresten, die zusammen einem Wassermolekül ermöglichen eine Peptidbindung anzugreifen. Der eine Asparaginsäurerest verschiebt in seiner deprotonierten Form das Gleichgewicht des angreifenden Wassermoleküls zur Deprotonierung und der andere polarisiert in seiner protonierten Form die Peptidcarbonylgruppe und erhöht so deren Empfindlichkeit für einen Angriff. Zu dieser Enzymklasse gehört Renin, das bei der Blutdruckregulierung beteiligt ist, und das Verdauungsenzym Pepsin. Das aktive Zentrum der Metalloproteasen enthält ein gebundenes Metallion. Dies ist fast immer ein Zinkion, das ein Wassermolekül aktiviert, welches dann als nucleophile Gruppe die Peptidcarbonylgruppe angreift. Bsp: Thermolysin, ein bakterielles Enym; Carboxypeptidase, ein Verdauungsenzym. Bei jeder dieser drei Strategien wird Wasser oder eine andere nucleophile Gruppe aktiviert, die Peptidcarbonylgruppe polarisiert und die anschliessende Stabilisierung eines tetraedrischen Zwischenprodukts ermöglicht. 9.1.7 Proteaseinhibitoren sind wichtige Medikamente Chemische Verbindungen, welche die Aktivität von Proteasen blockieren oder verändern, zeigen starke biologische Auswirkungen. Die meisten natürlichen Proteaseinhibitoren besitzen ähnliche Strukturen wie die Peptidsubstrate der Enzyme, die sie hemmen. Mehrere wichtige Medikamente sind Proteaseinhibitoren z.B. Captopril, Inhibitor der Metalloprotease ACE für die Regulierung des Blutdrucks; Crixivan, Inhibitor der HIV-Protease für die Behandlung von AIDS. Spezifität ist bei diesen als Medikamente verwendeten Proteaseinhibitoren sehr wichtig. 9.2 Carboanhydrasen machen eine schnelle Reaktion noch schneller Kohlendioxid ist eines der hauptsächlichen Endprodukte im aeroben Metabolismus. Bei komplexen Organismen wird Kohlendioxid ins Blut freigesetzt und zum Ausatmen in die Lungen transportiert. Im Blut reagiert Kohlendioxid mit Wasser, wobei sich Kohlensäure bildet, die durch Dissoziation eines Protons zu einem Hydrogencarbonation wird. Diese Hydratisierung erfolgt auch ohne Katalysator bei mittlerer Geschwindigkeit. Die Carboanhydrasen sind als Katalysatoren aber notwendig, da die CO2Hydratisierung und die Kohlensäure-Dehyratisierung häufig an schnelle Reaktionen gekoppelt sind. Die aktivsten dieser Enzyme, z.B. die menschliche Carboanhydrase II, hydratisieren CO2 mit einer Geschwindigkeit von bis zu kkat=10^6s^-1. 9.2.1 Carboanhydrasen enthalten ein gebundenes Zinkion, das für die katalytische Aktivität essenziell ist Ein Drittel aller bekannten Proteine enthalten entweder gebundene Metallionen oder benötigen solche Ionen für ihre Aktivität. Die chemische Reaktivität von Metallionen ist bedingt durch ihre positive Ladung, durch ihre Fähigkeit zur Bildung von relativ starken, aber kinetisch instabilen Bindungen und manchmal noch durch verschiedene stabile Oxidationszustände. Zink kommt in biologischen Systemen nur in der Oxidationsstufe +2 vor. Bei der Carboanhydrase ist ein Zinkatom immer an mindestens vier Liganden gebunden, an Imidazolringe von drei Histidinresten und an ein Wassermolekül. 9.2.2 Bei der Katalyse kommt es zur Aktivierung eines Wassermoleküls durch Zink Die Bindung eines Wassermoleküls an ein positiv geladenes zinkhaltiges Zentrum verringert den pKs-Wert des Wassermoleküls von 15,7 auf 7. Aufgrund des erniedrigten pKs-Wertes bildet sich eine gewisse Menge an Hydroxidionen. Ein zinkgebundenes Hydroxidion ist ausreichend nucleophil, sodass es Kohlendioxid viel besser angreifen kann als Wasser. Mechanismus der Kohlendioxidhydratisierung (Abb. 9.25): Zink ermöglicht die Freisetzung eines Protons aus dem Wassermolekül, sodass ein Hydroxidion entsteht. Das CO2-Substrat bindet an das aktive Zentrum des Enzyms und wird so positioniert, dass es mit dem Hydroxidion reagiert. Das Hydroxidion wandelt das Kohlendioxidmolekül in ein Hydrogencarbonation um. Dessen Freisetzung und die Bindung eines neuen Wassermoleküls regeneriert das katalytische Zentrum. Die Bindung eines Wassermoleküls an Zink begünstigt also die Bildung eines Übergangszustandes. 9.2.3 Ein Protonen-Shuttle ermöglicht die schnelle Regeneration der aktiven Enzymform Am Ende einer Kohlendioxidhydratisierung muss das zinkgebundene Wassermolekül ein Proton abgeben, damit wieder die aktive Form des Enzyms entsteht. Die Geschwindigkeit der Rückreaktion, das heisst die Protonierung des zinkgebundenen Hydroxidions, wird begrenzt durch die Geschwindigkeit der Protonendiffusion. Protonen diffundieren sehr schnell mit Geschwindigkeitskonstanten zweiter Ordnung von etwa10^11 M^-1s^-1 (k-1). Die Geschwindigkeitskonstante der Rückreaktion muss also geringer sein als dieser Wert. Da die Gleichgewichtskonstante bei pKs=7 K=k1/k-1=10^-7 ist, muss k1 geringer oder gleich 10^4s^-1 sein. Wenn jedoch Kohlendioxid mit einer Geschwindigkeit von 10^6s^-1 hydratisiert wird, muss dieser Schritt des Mechanismus mindestens genauso schnell erfolgen. Lösung: Für die höchsten Geschwindigkeiten der Kohlendioxidhdratisierung muss ein Puffer vorhanden sein. Der Puffer kann Protonen binden oder freisetzen. Der Vorteil besteht darin, dass die Konzentration von Protonen und Hydroxidionen bei neutralem pH-Wert auf jeweils 10^-7 begrenzt ist, während die Konzentration von Pufferkomponenten viel grösser sein kann. Die molekularen Komponenten zahlreicher Puffer sind aber zu gross, um in das aktive Zentrum der Carboanhydrase gelangen zu können. Bei den Carboanhydrasen gibt einen Protonen-Shuttle, dessen Hauptkomponente, das Histidin 64, Protonen vom zinkgebundenen Wassermolekül an die Proteinoberfläche und dann weiter in den Puffer überträgt. 9.2.4 Durch konvergente Evolution sind bei verschiedenen Carboanhydrasen aktive Zentren auf der Basis von Zink entstanden Carboanhydrasen, die zu menschlichen Enzymen homolog sind und die man als Carboanhydrasen bezeichnet, kommen bei Tieren, einigen Bakterien und Algen häufig vor. -Carboanhydrasen sind bei höheren Pflanzen und zahlreichen Bakterienspezies zu finden. Die dritte Familie, die -Carboanhydrasen, hat man zuerst bei Methanosarcina thermophila von den Archaea entdeckt. Eine konvergente Evolution hat also mindestens dreimal Carboanhydrasen hervorgebracht, die auf koordinierten Zinkionen basieren und deren katalytische Aktivität mit den zinkgebundenen Wassermolekülen verknüpft ist. 9.3 Restriktionsenzyme führen hochspezifische Spaltungsreaktionen an DNA aus Bakterien und Archaea haben Mechanismen entwickelt, um sich selbst vor viralen Infektionen zu schützen. Restriktionsendonucleasen (Restriktionsenzyme) erkennen in der viralen DNA bestimmte Basensequenzen (Erkennungssequenzen oder –stellen) und spalten die DNA an definierten Positionen. Die Typ-IIRestriktionsenzyme schneiden die DNA innerhalb ihrer Erkennungssequenz, andere Arten an etwas von den Erkennungssequenzen entfernten Positionen. Restriktionsendonucleasen müssen hochspezifisch sein, um nur DNA-Moleküle zu schneiden, die Erkennungssequenzen enthalten, und die Wirts-DNA überhaupt nicht anzugreifen. Die Wirts-DNA wird durch andere Enzyme geschützt, die man als Methylasen bezeichnet. Sie methylieren Adeninbasen in den Erkennungssequenzen der Wirtszelle. Die Enzympaare, Restriktionsendonuclease und Methylase, bezeichnet man als Restriktions-Modifikations-Systeme. 9.3.1 Die Spaltung erfolgt über eine in-line-Verdrängung des 3’Sauerstoffatoms am Phosphor durch magnesiumaktiviertes Wasser Die Restriktionsendonucleasen katalysieren die Hydrolyse des Phosphodiesterrückgrats der DNA. Dabei wird spezifisch die Bindung zwischen dem 3’-Sauerstoffatom und dem Phosphoratom gespalten. Die Produkte dieser Reaktion sind DNA-Stränge mit einer freien 3’-Hydroxylgruppe und einer 5’-Phosphorylgruppe. Diese Reaktion erfolgt über einen nucleophilen Angriff am Phosphoratom. Zwei Mechanismen bieten sich dafür an: Mechanismus 1 mithilfe einer starken nucleophilen Gruppe (Nu) über ein kovalent gebundenes Zwischenprodukt oder Mechanismus 2 in einer direkten Hydrolyse (Abb. S.270/p 246). Jeder Mechanismus geht von einer anderen nucleophilen Gruppe aus, die das Phosphoratom angreift. In beiden Fällen handelt es sich jedoch um eine in-line-Verdrängung. Die ankommende nucleophile Gruppe greift das Phosphoratom an, sodass ein fünffachkoordinierter Übergangszustand in Form einer zweifachen trigonalen Bipyramide mit dem Phosphoratom in der Mitte, der ankommenden nucleophilen Gruppe an einer der Pyramidenspitzen und die zu erstzende Gruppe an der anderen Spitze entsteht. Da beim ersten Mechanismus am Phosphor zwei Verdrängungsreaktionen stattfinden, würde sich die stereochemische Konfiguration am Phophoratom zweimal hintereinander umkehren, sodass dieselbe Konfiguration erhalten bliebe. Beim zweiten Mechanismus würde sich die Konfiguration jedes Mal bei einer Verdrängungsreaktion umkehren. Die Analyse zeigte, dass sich die stereochemische Konfiguration am Phosphoratom bei der Spaltung nur einmal umkehrt, was für einen direkten Angriff von Wasser spricht. 9.3.2 Restriktionsenzyme benötigen für die katalytische Aktivität Magnesium Restriktionsendonucleasen erfordern für ihre Aktivität Magnesium oder ein anderes bivalentes Kation. Das Magnesiumion ist von sechs Liganden gebunden: drei Wassermolekülen, zwei Carboxylate der Aspartatreste des Enzyms und ein Sauersroffatom an der Schnittstelle. Untersuchungen haben gezeigt, dass ein zweites Magnesiumion an einer benachbarten Position vorhanden sein muss, damit die Restriktionsenzyme die DNA schneiden können. 9.3.3 Der vollständige katalytische Apparat bildet sich nur mit Komplexen aus passenden DNA-Molekülen und sichert so die Spezifität Die Erkennungssequenzen der meisten Restriktionsendonucleasen sind umgekehrte Wiederholungen (inverted repeats). Diese Anordnung gibt der dreidimensionalen Struktur der Erkennungsstelle eine zweizählige Rotationssymmetrie. Die Restriktionsenzyme zeigen eine entsprechende Symmetrie, um die Erkennung zu ermöglichen. Das Enzym umgibt die DNA in Form einer festen Klammer. Zwischen dem Enzym und einer passenden DNA-Sequenz kommt es zu charakteristischen Wechselwirkungen. Innerhalb der 5’-GATATC-3’-Sequenz treten die G- und die A-Base am 5’-Ende eines jeden Stranges zusammen mit ihren Watson-Crick-Bindungspartnern über Wasserstoffbrücken zu Aminosäureestern, die sich in zwei Protonenschleifen befinden, in direkten Kontakt mit dem Enzym. Dabei ragt bei jeder Untereinheit des Enzyms eine der Schleifen aus der Oberfläche heraus. Das Auffälligste an diesem Komplex ist jedoch die Verformung der DNA, die in der Mitte deutlich geknickt wird. Die mittleren beiden TA-Basenpaare treten nicht mit dem Enzym in Kontakt, sind aber offenbar für den Knick erforderlich, weil sie sich leicht verformen lassen. Die Störung der DNA-Struktur an dieser Stelle wirkt sich gravierend auf die Spezifität der enzymatischen Reaktion aus. Bei der EcoRV-Endonuclease hat man festgestellt, dass sich das Enzym bei Abwesenheit von Magnesium mit annähernd derselben Affinität an alle DNASequenzen bindet, passende oder nicht. Die nicht passende DNA wird aber kaum verformt, was wichtige Auswirkungen auf die Katalyse hat. Keine Phosphorylgruppe gelangt in ausreichende Nähe zu den Aspartatresten des aktiven Zentrums, sodass sich die Magnesiumbindungsstelle nur unvollständig bildet. Die Verformung des Substrats und die anschliessende Bindung des Magnseiumions verbessern die katalytische Spezifität der EcoRV-Endonucleasen um den Faktor 1'000'000, trotz der geringen Unterschiede in Bezug auf die Substratbindung. Bei der Bindung an das Enzym wird die DNA so verformt, dass zusätzliche Kontaktstellen zwischen dem Enzym und dem Substrat entstehen; das erhöht die Bindungsenergie. Jedoch wird dieser Gewinn durch den Energieaufwand beim Verformen der DNA aus der entspannten Konformation wieder ausgeglichen. Also besteht kein grosser Unterschied zwischen den Bindungsaffinitäten zu spezifischer und unspezifischer DNA. Allerdings erhalten durch die Verformung des spezifischen Komplexes die Bindungsstellen für das Magnesiumion ihre vollständige Struktur. Enzyme können also die verfügbare Bindungsenergie nutzen, um Substrate zu verformen und für die chemische Umwandlung vorzubereiten. Wechselwirkungen innerhalb des Komplexes mit einem verformten Substrat stabilisieren den Übergangszustand, der zur Hydrolyse der DNA führt. Wenn sich an der Aminogruppe des Adeninnucleotids am 5’-Ende der Erkennungssequenz eine Methylgruppe befindet, verhindert diese die Bildung einer Wasserstoffbrücke und in Folge sind andere Wechselwirkungen zwischen dem Enzym und dem DNA-Substrat unterbrochen; die für die Spaltung erforderliche Verformung findet nicht statt. Also lässt sich durch die DNA-Verformung erklären, wie die Methylierung die Katalyse blockiert und die DNA der Wirtszelle schützt. 9.3.4 Typ-II-Restriktionsenzyme besitzen einen übereinstimmenden katalytischen Core-Bereich und sind wahrscheinlich durch horizontalen Gentransfer miteinander verwandt Typ-II-Restriktionsenzyme kommen vor allem bei Archaea und Eubakterien vor. Die gemeinsame Core-Struktur umfasst -Stränge, die die Aspatatreste enthalten, welche die Bindungsstellen für Magnesiumionen bilden. Bakterien haben möglicherweise von anderen Spezies Gene, die solche Enzyme codieren, durch horizontalen Gentransfer übernommen. Die Sequenzen von EcoRI und RsrI stimmen beispielsweise in einem Bereich von 266 Aminosäuren zu 50% überein, allerdings sind diese Bakterienspezies nicht eng miteinander verwandt, was darauf hindeutet, dass die beiden Spezies das Gen für das Enzym nach ihrer Arttrennung aus demselben Ursprung erhalten haben. Bei Restriktion-Modifikations-Systemen hat offenbar der Schutz vor Virusinfektionen den horizontalen Gentransfer begünstigt. 9.4 Nucleosidmonophosphat-Kinasen katalysieren den Austausch von Phosphorylgruppen ohne vorhergehende Hydrolyse Nucleosidmonophosphat-Kinasen (NMP-Kinasen) katalysieren die Übertragung der endständigen Phosphorylgruppe von einem Nucleosidtriphosphat (NTP), normalerweise ATP, auf die Phosphorylgruppe eines Nucleosidmonophosphats. Dabei müssen sie die Konkurrenzreaktion, Übertragung der Phosphorylgruppe auf ein Wassermolekül verhindern, was mithilfe des induced fit-Mechanismus möglich ist. Ein Metallion verstärkt zusätzlich die Wechselwirkungen zwischen Enzym und Substrat, wobei das Metall hier mit dem Substrat einen Komplex bildet. 9.4.1 NMP-Kinasen sind eine Familie von Enzymen, die P-Schleifen-Strukturen enthalten NMP-Kinasen enthalten eine konservierte NTP-Bindungsdomäne, welche aus einem zentralen -Faltblatt, das an beiden Seiten von -Helices umgeben ist, besteht. Zwischen dem ersten -Strang und der ersten Helix befindet sich die sogenannte PSchleife (Gly-X-X-X-X-Gly-Lys), welche mit den Phosphorylgruppen des gebundenen Nucleotids in Wechselwirkung tritt. 9.4.2 Komplexe von Nucleosidtriphosphaten mit Magnesium (oder Mangan) sind die eigentlichen Substrate für grundsätzlich alle NTP-abhängigen Enzyme NMP-Kinasen sind ohne bivalente Metallionen, wie beispielsweise Magnesium oder Mangan, grundsätzlich inaktiv. Der Metallion-Nucleotid-Komplex ist das eigentliche Substrat der Enzyme. Die Bindung des Metallions an das Nucleotid erhöht die Bindungsenergie. Erstens neutralisiert das Magnesiumion einen Teil der negativen Ladung der Polyphosphatkette und verringert so unspezifische ionische Wechselwirkungen zwischen dem Enzym und der Polyphosphatgruppe des Nucleotids. Zweitens halten die Wechselwirkungen zwischen Magnesiumion und Sauerstoffatom der Phosphorylgruppe das Nucleotid in einer genau definierten Konformation fest. Ein Magnesiumion ist normalerweise in einer oktaedrischen Struktur von sechs Gruppen umgeben, zwei Sauerstoffatomen und Wassermolekülen. Drittens liefert das Magnesiumion zusätzliche Kontaktstellen für eine Wechselwirkung zwischen dem ATP-Magnesium-Komplex und dem Enzym. Manchmal binden Seitenketten des Enzyms direkt an das Magnesiumion. In anderen Fällen interagiert das Enzym über Wasserstoffbrücken der Wassermoleküle, die an das Ion angelagert sind, indirekt mit dem Magnesioumion. 9.4.3 Die Bindung von ATP induziert starke Konformationsänderungen Die Strukturveränderungen im Kinasemolekül bei der Substratbindung sind ein klassisches Beispiel für einen induced fit (Abb. 9.51). Die P-Schleife schliesst sich über dem oberen Ende der Polyphosphatkette. Diese Bewegung ermöglich eine Abwärtsbewegung der obersten Domäne des Enzyms, sodass diese über dem gebundenen Nucleotid eine Art Deckel bildet. Wenn das ATP-Molekül auf diese Weise gebunden ist, positioniert sich seine -Phosphorylgruppe in der Nähe der Bindungsstelle des zweiten Substrats NMP. Die Bindung des zweiten Substrats NMP führt zu zusätzlichen Konformationsänderungen. Beide Veränderungen stellen sicher, dass nur dann eine katalytisch aktive Konformation entsteht, wenn sowohl Donor als auch Akzeptor gebunden sind, um die hier sinnlose Übertragung der Phosphorylgruppe auf Wasser zu verhindern. Das Enzym bindet seine beiden Substrate in grosser Nähe zueinander und in der korrekten Orientierung, sodass der Übergangszustand stabilisiert wird, der schliesslich zur Übertragung führt -> Katalyse durch Annäherung. 9.4.4 P-Schleife-NTPase-Domänen sind in zahlreichen wichtigen Proteinen vorhanden Ähnliche und fast immer homologe Domänen wie bei den NMP-Kinasen kommen bei vielen biochemisch essenziellen Proteinen vor. Bsp: ATP-Synthase, molekulare Motorproteine (Myosin), signalübertragende Proteine (Transducin), Proteine für die Translation von mRNA in Proteine, DNA-und RNA-entwindende Helikasen. Die weite Verbreitung von P-Schleife-NTPase-Domänen lässt sich wahrscheinlich am besten dadurch erklären, dass sie durch Bindung grundlegende Konformationsänderungen ausführen können.