EulerAlign
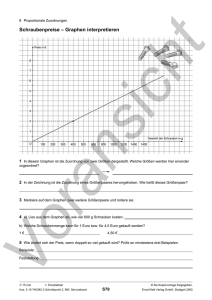
Werbung

EulerAlign
Ausarbeitung zum Seminar „Aktuelle
Themen der Bioinformatik“ von Kai
Scheiffele
Sommersemester 2004
Inhalt:
1. Einleitung
2. Bisherige Ansätze des Multiplen Alignment
2.1 Progressives Alignieren
2.2 Iteratives Alignieren
3. Die Idee von EulerAlign
4. Der Algorithmus von EulerAlign
4.1 Schritt 1: Graphkonstruktion über k-Tupel
4.2 Schritt 2: Transformation des Graphen in einen DAG
4.3 Schritt 3: Pfadsuche
4.4 Schritt 4: Fast-pariwise Alignment
4.5Schritt 5: Endgültiges multiples Alignment
5. Analyse des Algorithmus
6. Komplexität von EulerAlign
7. Auswahl an Simulationsergebnissen
8. Zusammenfassung und Ausblick
Literatur
2
3
3
3
4
4
6
6
7
11
12
13
13
14
15
18
19
1. Einleitung
Beim multiplen Alignment handelt es sich um ein wesentliches und ziemlich
komplexes Problem in der Bioinformatik. Beruhend auf dem paarweisen Alignment, welches
recht schnell und gut berechnet werden kann und Aussagen über die Ähnlichkeit zweier
DNA- oder Protein-Sequenzen ermöglicht, zielt das multiple Alignment darauf ab, aus einer
Vielzahl von Sequenzen die wesentlichen Kernbereiche (i.d.R. konservierte Regionen)
herauszuarbeiten. Im Folgenden wird das Verfahren EulerAlign vorgestellt, ein relativ neuer
Algorithmus mit sehr interessanten Eigenschaften.
Beispiel für die graphische Darstellung eines paarweise Alignment zweier Sequenzen.
Entnommen aus „Dirk Metzler: Alignment und Stammbaumschätzung für biologische Sequenzen“
2. Bisherige Ansätze des Multiplen Alignment
Das multiple Alignieren ein wohlbekanntes Problem in der Bioinformatik, seit langem
Forschungsgegenstand und dementsprechend vielseitig bearbeitet worden. Die Vielzahl
möglicher Lösungsansätze lässt sich allerdings in zwei ausdifferenzierte Hauptansätze
unterteilen:
2.1
Progressives Alignieren
Die Algorithmen welche progressiv alignieren haben gemein, dass sie ein multiples
Alignment dadurch erreichen, dass sie zunächst die beiden ähnlichsten Sequenzen und danach
immer mehr Sequenzen bzw. Cluster von Sequenzen weiter zusammenclustern. Dies wird
solange fortgesetzt, bis alle ursprünglichen Sequenzen zusammengebracht wurden. Doch auch
das progressive Alignieren lässt sich nach unterschiedlichen Methoden klassifizieren. So gibt
es Algorithmen, welche das Clustern über die Ähnlichkeit ganzer Sequenzen betreiben. So
auch z.B. der populärste Vertreter dieser Gangart CLUSTALW, welches wir auch später
gegen EulerAlign antreten lassen werden. Andere Ansätze wiederum versuchen zunächst
3
einmal konservierte Regionen innerhalb der Ausgangssequenzen zu identifizieren und
clustern dann anhand der Ähnlichkeit dieser Sequenzen. Stellvertretend soll hierfür das
Programm GENALIGN genannt sein.
2.2
Iteratives Alignieren
Beim iterativen Alignment wird von einer ursprünglichen Sequenz ausgegangen, welche
dann im Verlauf iterativer Algorithmen nach und nach lokal optimiert wird. Es wird also
immer nur auf einer einzigen Sequenz gearbeitet, wobei die Veränderungen in den einzelnen
iterativen Schritten in der Regel nur Teile der Sequenz anpassen werden und so (iterativ) die
Sequenz der gesuchten immer ähnlicher wird. Beispiele hierfür sind HMMT oder auch
COFFEE.
3. Die Idee von EulerAlign
Die Idee zum Algorithmus von EulerAlign ist ein Stück weit dem Algorithmus des
Programms EULER entliehen [3], welches der Assemblierung von DNA-Sequenzen dient.
Dabei wird das Problem der Assemblierung einfach auf das Problem des Euler-Path reduziert
(es wird ein Weg durch einen Graphen gesucht – wenn es diesen gibt – auf welchem jeder
Kante des Pfades höchstens einmal verwendet wird). Daher stammt auch der Name
EulerAlign, selbst wenn der Algorithmus (wie wir später sehen werden) kaum etwas mit
einem eulerschen Kreis- oder Pfad-Problem zu tun hat.
EulerAlign geht also einen zu EULER entgegen gesetzten Weg und zerschneidet eine
vorhandene Sequenz in (gleich große) Fragmente – anstatt selbige zusammenzusetzen.
Grundlegend ist dabei die Überlegung, dass man eine Teilsequenz bestehend aus k
Nukleotiden/Proteinen in genau zwei k-1 lange Sequenzen zerlegen kann. Dabei überlappen
sich natürlich die ersten bzw. letzten Buchstaben.
4
Entnommen: Folien zum Seminarvortrag
Da es ja EulerAligns Ziel ist einen Graphen zu konstruieren auf welchem dann im
Folgenden operiert wird, werden diese Fragmente einer Sequenz nun in die Kanten und
Knoten eines Graphen uminterpretiert. So kann man die k-1-langen Teilstücke als Knoten
auffassen, welche ja genau eine k-lange Sequenz determinieren – diese entspricht dann der
zwischen ihnen liegenden, gerichteten Kante.
Entnommen: Folien zum Seminarvortrag
Nun hat man bereits alle Elemente, die man zur Konstruktion eines Graphen benötigt
und damit den Background für den folgenden Algorithmus auf dem die Idee und auch die
Stärke von EulerAlign beruhen (Siehe spätere Testergebnisse).
5
4. Der Algorithmus von EulerAlign
Nachdem die Idee des neuen Ansatzes nun erläutert wurde, wird es Zeit sich den
Algorithmus etwas näher im Detail anzusehen. Dazu werden die fünf Schritte von EulerAlign
mal mehr, mal weniger detailliert vorgestellt. Insgesamt gliedert sich der Algorithmus wie
folgt:
(1) Graphkonstruktion über k-Tupel
(2) Transformation des Graphen in einen DAG
(3) Pfadsuche
(4) Fast-pairwise Alignment
(5) Endgültiges multiples Alignment
Nun zu den einzelnen Schritten:
4.1 Schritt 1: Graphkonstruktion über k-Tupel
Über die im vorherigen Abschnitt vorgestellte Idee sind wir nun in der Lage sämtliche
zu alignierenden Sequenzen in k-Tupel zu zerlegen, welche auch sogleich wieder in Kanten
und Knoten eines Graphen uminterpretiert werden. Dabei geht EulerAlign so vor, dass jedes
k-1-Tupel der Sequenzen einzeln betrachtet wird – zunächst schaut man ob es das
entsprechende Tupel bereits gibt. Wenn ja, wird die neue Kante einfach in den schon
vorhandenen Knoten geleitet, ansonsten einfach ein entsprechender Knoten, samt Kante, in
den Graphen eingefügt. Aus dem so entstehenden gerichteten Graph wird sogar automatisch
ein gewichteter Graph indem man den Kanten einfach als Gewicht die Anzahl ihres
Vorkommens mitgibt. Sprich, wenn wir eine Kante einfügen wollen die bereits existiert,
erhöhen wir stattdessen einfach deren Gewicht um „+1“.
…
…
Entnommen: Folien zum Seminarvortrag (Punkte deuten an, dass dies ein möglicher Teilgraph ist – also links
und rechts weiter verlaufen könnte)
6
Wie wir später sehen werden, beruht das schnelle Berechnen einer Konsensus-Sequenz
gegen welche wir alle Eingabesequenzen alignieren wollen darauf, innerhalb des Graphen
einen „längsten Weg“ von einem Sequenzanfang zu einem Sequenzende zu finden. Dafür
reicht es uns aber nicht einen gerichteten gewichteten Graphen zu haben – denn in diesem
können (und werden in der Regel) Kreise sein, welche jeden Algorithmus zum bestimmen
eines längsten Pfades unmöglich machen. Der nächste (und wohl entscheidende) Schritt im
Algorithmus muss nun also darin bestehen den Graphen kreisfrei zu bekommen, sprich, ihn in
einen „Directed acyclic graph“ (DAG) umzuwandeln.
4.2 Schritt 2: Transformation des Graphen in einen DAG
Bei der Eliminierung der Kreise gibt es zwei unterschiedliche Fälle zu beachten. Im
ersten Fall der „Gabelung“ gibt es keine größeren Schwierigkeiten.
Entnommen: Folien zum Seminarvortrag
Nur eine einzige Sequenz beansprucht einen bestimmten Knoten mehrfach und wir
können einfach den entsprechenden Knoten in zwei Knoten aufspalten, wobei jeweils eine der
zuvor eingehenden Kanten einen eigenen Knoten als neues Ziel erhält.
7
Entnommen: Folien zum Seminarvortrag
Je nachdem wie lang die wiederholte Teilsequenz ist, kann es sein, dass der Kreis
dadurch nur kleiner und das Problem nach hinten verlagert wird, durch rekursives
weiterführen dieses Schemas wird über kurz oder lang der Kreis verschwinden.
Ein wenig anders ist der zweite Fall, die „Kreuzung“ gelagert, da man hier zwischen
sicheren und unsicheren Fällen zu unterscheiden hat, sofern der kritische Knoten nicht nur
von einer sondern von mehreren Sequenzen beansprucht wird.
Entnommen: Folien zum Seminarvortrag
Im einfachsten Fall kreuzen sich allerdings wiederum nur zwei Teilsequenzen ein und
derselben Sequenz (wiederum durch rote und grüne Pfeile dargestellt). Dann wird einfach wie
8
bei einer Gabelung verfahren und durch das hinzufügen eines weiteren Knotens der Kreis
entfernt.
Entnommen: Folien zum Seminarvortrag
Kommt nun aber wie erwähnt eine weitere Sequenz (im folgenden durch die gelben
Pfeile symbolisiert) durch den Knoten ergibt sich die genannte Unterteilung in „sichere“ und
„unsichere“ Transformationen, wobei (wie der Name schon preisgibt) die „sichere“ einfacher
zu handhaben ist. Bei der „sicheren“ Transformation verläuft die Teilsequenz der zweiten
Sequenz parallel zu genau einer der beiden Teilsequenzen der ersten Eingabesequenz.
Entnommen: Folien zum Seminarvortrag
Wie bereits mehrfach gesehen erledigt sich das Problem sozusagen von selbst, wenn
einfach durch das Einfügen eines zusätzlichen Knotens das gesamte Gebilde entzerrt wird.
9
Entnommen: Folien zum Seminarvortrag
Worin liegt nun aber die Schwierigkeit, wenn die zweite Sequenz nicht parallel zu
einer der beiden Teilsequenzen der ersten, sondern erst parallel zur einen und dann parallel
zur anderen, verläuft?
Entnommen: Folien zum Seminarvortrag
Wie bereits oben angesprochen wurde, ist die Kantengewichtung für die später
Ermittlung eines längsten Pfades durch den Gesamtgraphen das A und O – denn in den
Kantengewichten spiegelt sich ja sozusagen die Information über die Konserviertheit einer
Teilsequenz wieder. Je höher das Kantengewicht, desto mehr der Eingabesequenzen
beinhalten die Teilsequenz und desto konservierter ist diese folglich.
Im folgenden Bild sieht man nun, dass durch das (gewohnte) hinzufügen eines Knotens der
Kreis zwar entfernt ist (rote und grüne Teilsequenzen laufen nicht mehr durch den gleichen
Knoten v), allerdings nach dem Verlassen der Knoten die grünen und gelben Teilsequenzen
nicht gemeinsam (also parallel) verlaufen, sondern zunächst getrennt sind, was in diesem Fall
10
natürlich zu einem Informationsverlust führt der bei der Pfadsuche (unbemerkt) zu einer
falschen Wahl der längsten Sequenz führen kann (aber auch nicht muss)!
Entnommen: Folien zum Seminarvortrag
Wie wird nun dieser Unsicherheitsfaktor gelöst? Gar nicht, er wird allerdings auf ein
Minimum reduziert. Ganz einfach dadurch, dass zunächst alle anderen Kreise eliminiert
werden und erst zum Schluss die „unsicheren“ Kreuzungen.
Der Graph liegt also nun nach dem Eliminieren aller Kreise als DAG vor und ist damit
bereit für den nächsten wichtigen Schritt, die Pfadsuche.
4.3 Schritt 3: Pfadsuche
Zhang und Waterman gehen auf diesen Punkt zwar nicht näher ein, sondern
verweisen nur darauf, dass man über die Verwendung eines Greedy-Algorithmus einen
genügend richtigen längsten Pfad in linearer Zeit berechnen kann. Da dieser Punkt später bei
der Frage der Komplexität eine nicht ganz unwichtige Rolle spielt, sei hier zumindest ein
Beispiel für einen möglichen Greedy-Algorithmus präsentiert um zu zeigen, dass Linearzeit
für die Berechnung wirklich ausreichend ist (sofern man eine gewisse Fehlertoleranz
bezüglich des wirklich besten Pfades in Kauf nimmt).
Die Pfadsuche gestaltet sich als „längster Pfad“ Problem, da man die Gewichtung der
Kanten als Längen auffassen kann und so mit dem am höchsten gewichteten Pfad auch den
längsten Pfad sucht. Es sei nicht der Anspruch erhoben, dass der gleich vorgestellte GreedyAlgorithmus wirklich der verwendete bzw. der bestmögliche ist. Doch um einen Einblick in
die Funktionsweise eines solch „gierigen“ Algorithmus zu geben sei hier eine Möglichkeit
gezeigt, wie ein potentiell längster Pfad im gegebenen DAG berechnet werden kann.
Die Idee des folgenden Greedy-Algorithmus ist es, wie der Name schon sagt, in jedem
Schritt „gierig“ die Kante aus einer verbleibenden Kantenmenge zu wählen, welche mit hoher
Wahrscheinlichkeit im gesuchten Pfad enthalten sein wird. So schafft es der Algorithmus in
linearer Zeit zu arbeiten, da er jede Kante maximal einmal in eine Ergebnismenge von Kanten
transferieren kann. Nun also zum Algorithmus, wie er aussehen könnte:
11
Erzeuge Kantenmenge E (mit allen in Schritt 1 ermittelten Kanten).
Erzeuge leere Kantenmenge E’.
Solange noch kein Weg durch Graphen gefunden:
Nimm aus E die Kante mit dem höchsten Gewicht und transferiere sie nach E’.
Überprüfe ob Weg durch Graphen mit E’ möglich ist.
Wenn Weg durch Graphen gefunden:
Gib diesen Pfad aus.
Sonst:
Weiter mit erster Schleifenanweisung.
Da die Anzahl der Schleifendurchläufe durch die Anzahl der zuvor ermittelten Kanten
beschränkt ist, läuft der Algorithmus tatsächlich in linearer Laufzeit, sofern innerhalb der
Schleife konstante Zeit benötigt wird.
Und auch dies lässt sich tatsächlich realisieren, indem man einfach jede Kante mit
Marker-Punkten an beiden Seiten ausstattet. Diese werden markiert, wenn die neue Kante
durch weitere Kanten mit einem Anfangs- oder Endpunkt des Graphen in Verbindung treten
kann. Ein Weg durch den Graph ist also dann gefunden, sobald die neu in E’
hinzugenommene Kante an beiden Seiten markiert ist, also einen Anfangspunkt des Graphen
mit einem Endpunkt verbindet.
Da bis zu diesem Punkt nur die jeweils höchstgewichteten Kanten in die
Ergebnissuche einbezogen wurden besteht auch eine sehr gute Chance, dass es sich
tatsächlich um den „längsten Pfad“ handelt.
Auch das der Algorithmus terminiert liegt auf der Hand, denn bei N vielen Sequenzen
gibt es mindestens N viele Verbindungen zwischen globalen Start- und Endknoten und damit
wird früher oder später auf jeden Fall eine solche Verbindung gefunden.
Typisch für einen Greedy-Algorithmus ist, dass er nicht zwangsläufig mit dem besten
Ergebnis terminiert – dies sollte man bei der Anwendung immer in Überlegungen mit
einbeziehen.
4.4 Schritt 4: Fast-pariwise Alignment
Von dem im vorigen Schritt ermittelten längsten Pfad durch den DAG kann man
einfach durch das Entlanghangeln an diesem Pfad eine Konsensus-Sequenz ablesen. Gegen
diese werden nun sämtliche verwendeten Sequenzen paarweise aligniert. EulerAlign
verwendet hierfür einen Algorithmus, welcher ein „banded pairwise alignment“ ermittelt –
das bedeutet, dass der positionelle Unterschied zwischen zwei Zielbuchstaben durch eine
Konstante berücksichtigt wird.
12
4.5 Schritt 5: Endgültiges multiples Alignment
Aus den jeweiligen Alignments wird dann ein endgültiges und globales Alignment
gebildet. Das paarweise alignieren gegen die Konsens-Sequenz (den längsten Weg durch den
Graphen) garantiert hierbei ein möglichst hohes Maß an Übereinstimmung mit dem wirklich
besten globalen Alignment der verwendeten Sequenzen. Den Grund hierfür werden wir im
nächsten Abschnitt verifizieren.
5. Analyse des Algorithmus
Die entscheidende Idee für die mathematische Analyse von EulerAlign ist die folgende:
Es ist zu Zeigen, dass wenn alle Eingabesequenzen von einer Sequenz S0 abgeleitet sind, und
man die Anzahl der verwendeten Eingabesequenzen immer weiter steigert, so wird der
Konsensus-Pfad des EulerAlign-Algorithmus asymptotisch identisch zu S0 werden.
Dabei sind nun die folgenden (bereits verwendeten) Parameter von Interesse:
N
die Anzahl der zu alignierenden Sequenzen,
L
die durchschnittliche Länge der Sequenzen ,
k
die Größe des zugrunde liegenden Tupels, sowie
die Mutationsrate beim Erzeugen der Eingabe.
Des Weiteren definiert man W(e) als das Gewicht einer Kante e, was natürlich von Interesse
ist, da der Algorithmus ja nach einem „längsten Pfad“ sucht und dabei auf genau dieses
Gewichtsmaß zurückgreift.
Es ist nun ein Leichtes sich zu überlegen, dass bei einer Mutationsrate von 0 alle N Sequenzen
identisch zu S0 sind, sowie das Gewicht W(e) = N für jede Kante des „Graphen“ ist. Wenn
nun mutiert wird, werden mehr Kanten mit einem geringeren Gewicht – vornehmlich Kanten
mit einem Gewicht von 1 – im Graphen auftauchen. Mit dem gleichen Maß werden die
entsprechenden Kanten, welche zum Pfad „S0“ gehören an Gewicht verlieren.
Im Graphen mit berücksichtigten Mutationen kann man die Kanten nun sinnvoll in zwei
Klassen (Mengen) unterteilen (welche für die Idee des Beweises essenziell sind): Kanten die
zu S0 gehören, {W(e S0}, sowie die Menge der Kanten, welche nicht zu S0 gehören, {W(e
S0}.
Der Nachweis erfolgt nun über das „Large Deviation Theorem for Binomials“ (L.D.T - siehe
[2]) und basiert auf folgender Idee:
Unter Zuhilfenahme des L.D.T. wird nachgewiesen, dass für N∞ das erwartete Minimum
in der Menge der Gewichte der zu S0 gehörenden Kanten min {W(e S0} größer ist, als das
zu erwartende Maximum in der Menge der Gewichte der nicht zu S0 gehörenden Kanten max
{W(e S0} ist. Dabei ist von Bedeutung, dass man annehmen kann, das die eingestreuten
Mutationen normalverteilt sind – wählt man die Größe der Tupel, also k, entsprechend groß,
13
konvergiert die Anzahl der Tupel welche mutiert sind und öfter als einmal vorkommen sehr
schnell gegen 0 wenn man die Anzahl der Sequenzen, also N, erhöht.
Ist dieser Nachweis erbracht, so kann man sich leicht überlegen, dass der Algorithmus mit
steigender Anzahl an Eingaben auch mit immer größerer Sicherheit S0 als Konsensus-Pfad
ermitteln wird und so ein möglichst gutes Alignment garantiert.
6. Komplexität von EulerAlign
Es sei nun die Komplexität von EulerAlign betrachten. Das Ergebnis wird sein, dass der
Algorithmus sehr effizient und entsprechend praxistauglich ist, was auch schon eine erste
Ahnung rechtfertigt wie das Programm später beim Testlauf gegen das etablierte Programm
CLUSTALW abschneiden wird.
Zunächst zum zeitlichen Aufwand:
(1)
Wie in Schritt 1 gesehen, müssen wir zunächst aus den vorhandenen Sequenzen
einen Graphen bauen und in einen DAG umformen, was allein durch die Anzahl
der Sequenzen N und der Länge L determiniert wird. also beträgt der Aufwand
dafür etwa O(NL).
(2)
bei der Suche nach dem Längsten Pfad kommt uns zugute, dass das
Eingabealphabet endlich und i.d.R. klein ist (4 Nukleotide bzw. 20 Aminosäuren).
Damit hat jeder Knoten auch nur eine durch diese Konstante beschränkte Anzahl
von eingehenden/ausgehenden Kanten – denn von einem Knoten führt eine Kante
zu einem Knoten, bei welchem das „Sequenz-Raster“ nur um eine Position
verschoben ist. Die Anzahl der Kanten (welche ja linear die Komplexität des
Greedy-Algorithmus bestimmen) ist also durch O( Σ NL) also O(NL)
beschränkt.
Dementsprechend ergibt sich eine Gesamtlaufzeit beschränkt durch O(NL).
Zum Speicheraufwand:
Dieser ist auf den Platzbedarf für die Knoten samt ihrer Randdaten beschränkt und
hängt neben der Gesamtzahl der Buchstaben (also auch wieder NL) von dem
gewählten k ab, da dies für den Grad der Zerstückelung der Sequenzen und den damit
verbundenen redundanten Überschneidungshäufigkeiten zusammenhängt.
also O(k NL), was auch sehr effizient ist.
Zusammenfassend lässt sich sagen, dass EulerAlign also sowohl in Bezug auf Rechenzeit,
als auch was den Platzbedarf angeht höchst effizient ist – gerade mal durch einen konstanten
Faktor größer als die Eingabe!
14
7. Auswahl an Simulationsergebnissen
Zum Vergleich mit bestehenden Algorithmen wurde repräsentativ CLUSTALW als
Gegner für EulerAlign ausgewählt und hinsichtlich vier Größen miteinander verglichen. Als
da wären: Sum-of-pairs, Aligning Alignment Score, Identity bezüglich der Qualität der
ermittelten multiplen Alignments, als auch noch die Laufzeit als Maß der effizienten
Geschwindigkeit. Die Ergebnisse folgen nun im Einzelnen: (In den folgenden Ausführungen
steht N für die Anzahl der Sequenzen, L (= 500) für deren Länge und (=5,2%) für die beim
Erzeugen der Daten angelegten Mutationsrate.)
Sum-of-pairs (SP):
Sum-of-pairs ist ein sehr gebräuchliches Scoring-Schema zur Bewertung von
Alignments zwischen equidistanten Sequenzen. Das heißt, die Sequenzen sollten alle
evolutionär „ähnlich weit“ voneinander entfernt liegen. Das genaue Bewertungsschema liegt
zwar nicht vor, doch es ist davon auszugehen, dass andere Bewertungszahlen zu einem
vergleichbaren Ergebnis führen werden. Erwähnt sei darüber hinaus noch, dass der SP-Wert
durch die Anzahl der möglichen Paare normiert ist (also: N(N-1)/2).
800
700
600
SP
500
EA
400
CLW
300
200
100
0
6
10
15
50
100
500
N
Eigene Darstellung. Daten aus [1]
Es wird beim Betrachten des Diagramms schnell klar, dass für gegebene Länge und
Mutationsrate EulerAlign (EA) und CLUSTALW (CLW) bei einer geringen Anzahl N
durchaus vergleichbare Werte abliefern. Je größer die Anzahl der Sequenzen gewählt wird,
desto offensichtlicher zeigt sich, dass EA nahezu konstant im Wert bleibt und nur sehr leicht
ansteigt, CLW allerdings sehr stark in seiner Fehlerzahl (ausgedrückt durch den bestrafenden
Punktewert) ansteigt. Bezüglich Sum-of-pairs macht also eindeutig EA das Rennen.
Aligning Alignment Score (AA):
15
Auch AA ist ein spezifisches Scoring-Schema, welches aber auf der Existenz
phylogenetischer Stammbäume beruht. Kennt man den phylogenetischen Baum, so ist es ein
Leichtes auf diesem basierend eine Fehlerfunktion anzuwenden um die Ergebnisse zwei
verschiedener Verfahren zu vergleichen. Da die für den „EA vs. CLW“-Vergleich
verwendeten Daten zuvor selbst generiert wurden ist auch der phylogenetische Stammbaum
bekannt und AA wird so zu einem sinnvollen Vergleichsverfahren.
700
600
AA
500
400
EA
300
CLW
200
100
0
6
10
15
50
100
500
N
Eigene Darstellung. Daten aus [1]
Bei der Analyse der Ergebnisse zeigt EA bereits bei einer kleinen Anzahl von
Sequenzen seine Stärke gegenüber CLW. Während bei EA die zur Anzahl der Sequenzen
normierte Fehlerzahl nahezu konstant bleibt, bzw. tendenziell leicht ansteigt, werden die
Ergebnisse von CLW deutlich schlechter. Wiederum kann EA CLW insbesondere bei
steigender Zahl von Eingabesequenzen deutlich schlagen.
Identity (ID):
ID ist ein Vergleichsparameter, welcher sich nach folgender Formel errechnet:
ID = (ijk, i<j Iijk) / (ijk, i<j I) x 100%
k wird in dieser Formel als durchlaufende Sequenznummerierung verwendet, i und j
sind Positionen in der k-ten Sequenz. I ist 1, wenn eine Übereinstimmung zwischen dem
ermittelten Alignment und dem wirklichen Alignment (auch hier kommt einem wiederum die
Selbstgenerierung der Daten und somit das Wissen um das wirkliche Alignment zugute).
Damit wird wirklich eine prozentuale Vergleichbarkeit von wirklichem Alignment und
ermittelten Alignment – und damit auch ein Maß zum Vergleich zweier Methoden.
16
120
100
%
80
EA
60
CLW
40
20
0
6
10
15
50
100
500
N
Eigene Darstellung. Daten aus [1]
Da ein Ergebnis von 100% ja eine absolute Übereinstimmung bedeutet, schneidet
natürlich bei diesem Bewertungsschema das Verfahren am besten ab, welches den höchsten
prozentualen Wert vorweisen kann – auch hier hat wiederum EA die Nase vor, insbesondere
wenn es darum geht eine große Anzahl von Sequenzen zu alignieren!
Laufzeit:
Wenn es um das Alignieren kleiner und kurzer Sequenzen geht kann man dies auch
noch sehr genau per Hand erledigen. Die eigentliche Herausforderung besteht (wie eingangs
schon erwähnt) darin, viele (mitunter sehr lange) Sequenzen zu alignieren. Da bisher
verwendete Verfahren und Algorithmen allerdings i.d.R. mit quadratischer Laufzeit zurande
kommen müssen, sind der Praktikabilität bei steigender Sequenzzahl sehr schnell Grenzen
gesetzt. Da nun aber in der Forschung Fragestellungen auftauchen wo es interessant wird
einige Tausend Sequenzen zu alignieren, welche jeweils durchaus einige Millionen
Nukleotide aufweisen können, ist Geschwindigkeit ein sehr wichtiges Gütekriterium. Dabei
kann EulerAlign wirklich mit großen Geschützen aufwarten, wie die folgendes Diagramm
zeigt:
100000
Zeit [sec.]
10000
1000
EA
CLW
100
10
1
6
10
15
50
100
N
Eigene Darstellung. Daten aus [1]
17
500
Die Tests wurden alle auf einem SUN-Rechner mit einer CPU-Geschwindigkeit von
750 MHz durchgeführt, die jeweiligen Laufzeiten für EA und CLW in Sekunden im
Diagramm dargestellt. Für das maximale Testset (500 Sequenzen der Länge 500) benötigt EA
gerade mal etwas über 2 Minuten, CLW hingegen beinahe 4 Stunden (man beachte die
logarithmische Skalierung des Diagramms). Dieses Ergebnis bedarf wohl kaum einer näheren
Bewertung.
8. Zusammenfassung und Ausblick
Zusammenfassend lässt sich sagen, dass es eine revolutionäre Idee ist das Problem des
Alignierens einfach auf ein anderes Problem, in diesem Fall ein Graphtheoretisches Problem,
zu reduzieren. Auf diese Weise ist es gelungen, mit (aus EULER) bekannten Ansätzen nicht
nur bessere Ergebnisse zu produzieren (siehe Auswertung von SOP, AA und ID), sondern
diese sogar in einer signifikant schnelleren Berechnungszeit zu erhalten. Es bleibt offen, ob
das so möglich gewordene Verarbeiten einer solch großen Menge von Eingabesequenzen
wirklich auch zu einem exponentiellen Anstieg des Erkenntnisgewinns führt, doch die
Chancen dazu sind allemal als sehr gut zu bewerten. Es bleibt also abzuwarten wie sich das
Verfahren in der explorativen Praxis bewährt.
Weiterhin offene Probleme seien auch nicht unverschwiegen. So ist die Wahl eines
geeigneten „k“s derzeit noch ein von Hand zu lösendes Problem, welches viel Erfahrung und
Fingerspitzengefühl benötigt. Denn, wählt man k zu groß, so stellt sich nur bei extrem
niedriger Mutationsrate ein zufrieden stellender Konsensus-Pfad ein – andererseits führt ein
zu kleines k nicht zwangsläufig zu eindeutigen Sequenzabschnitten, will heißen: die
Wahrscheinlichkeit Sequenzabschnitte als „gleich“ zu behandeln obwohl sie nichts
miteinander zu tun haben steigt sehr stark an! Schön wäre also ein terminierendes Verfahren,
welches möglichst gut das beste zu verwendende k liefert.
Weiterhin ist bereits in der Beschreibung des Algorithmus das Problem aufgetaucht, dass
das (zwingend notwendige) Eliminieren der Kreise innerhalb des Graphen mitunter zu einem
Informationsverlust führt, welcher im schlimmsten Falle negative Auswirkungen auf das
Ergebnis haben könnte. Es sollten daher auch Veränderungen im Algorithmus angedacht,
getestet und ggf. verifiziert werden, die einen möglichst geringen bzw. nicht existenten
Informationsverlust garantieren. Eine Möglichkeit könnte darin bestehen, bei dem Entzerren
der gesehen „unsicheren Transformationen“ einen neuen Kantentyp zu entwickeln, welcher
dann bei der folgenden Pfadsuche in geeigneter Form mitberücksichtigt wird.
Ein letzter offener Punkt ist der des Auffindens geeigneter Scoring-Verfahren für das
Alighnieren. Dies ist allerdings kein EulerAlign-spezifisches Problem, sondern generell
offener Forschungsgegenstand aller Formen des paarweisen- bzw. multiplen-Alignierens.
18
Literatur:
[1]: Zhang, Y., Waterman, M.S. 2003. An Eulerian Path Approach to Global Multiple
Alignment for DNA Sequences. Journal of Computational Biology. Vol.10, 6, 803-819
[2]: Arratia,R., and Gordon,L. 1989. Tutorial on large deviations fort he binomial distribution.
Bull. Math. Biol. 51, 125-131.
[3]: Pevzner, P.A., Tang, H., Waterman,M.S. 2001. An Eulerian path approach to DNA
fragment assembly. Proc. Natl. Acad. Sci. USA 98, 9748-9753.
19