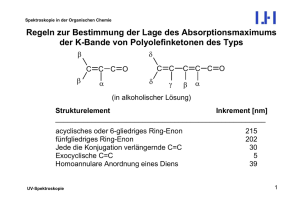
Dynamische Prozesse – Chemischer Austausch
Werbung

Spektroskopie in der Organischen Chemie Dynamische Prozesse – Chemischer Austausch Weil die Messfrequenz der Kernresonanz (100 MHz bis 1 GHz, also ca. 107 bis 1010 s-1) in einem Frequenzbereich liegt, in dem auch viele Molekülbewegungen (Moleküldynamik) stattfinden, können Veränderungen in der Geschwindigkeitskonstanten von Molekularbewegungen Einfluss auf das Aussehen von NMR-Signalen haben. Am einfachsten kann man Geschwindigkeitskonstanten durch Temperaturveränderung verändern, sodass man zu ihrer Beobachtung die NMR-Spektren bei variabler Temperatur aufnimmt (Dynamische NMR, DNMR, VT NMR). Das heißt, man misst Serien von Spektren bei jeweils verschiedenen Temperaturen; „variabel“ heißt nicht, dass die Temperatur während einer Messung verändert wird, sondern von Messung zu Messung. Es gibt zwei verschiedene Arten von dynamischen Prozessen: (a) Intramolekulare Dynamik: z.B. Konformationsumwandlungen beweglicher Moleküle (b) Intermolekulare Dynamik: Austauschprozesse. Hier können neben der Temperatur- auch Konzentrationsänderungen einzelner Komponenten das Aussehen der Spektren verändern. NMR-Spektroskopie 1 Spektroskopie in der Organischen Chemie Intramolekulare Dynamik: Konformationsumwandlungen Die Effekte der Temperaturvariation seien am einfachst möglichen Beispiel erläutert, einem Zwei-Spinsystem, bei dem die beiden Spins (Protonen) durch einen konformativen Umwandlungsprozess ihre Plätze tauschen. Es sei erwähnt, dass es sich hierbei um „gehinderte Rotationen“ handeln muss, bei denen die zwischen den beiden Konformations-Grundzuständen liegende Energiebarriere relativ hoch sein muss. Ungehinderte Rotationen, wie z.B. solche um C-C-Einfachbindungen in Alkanketten werden durch den NMR-Resonanzbereich nicht mehr erfasst. Das hier dargestellte Beispiel sei die Rotation um die C-N-Bindung in N,NDimethylbenzamid, die wegen ihres Doppelbindungsanteils gehindert ist. O CH3 C Ph NMR-Spektroskopie N CH3 2 Spektroskopie in der Organischen Chemie Es sind grundsätzlich drei Temperaturbereiche zu unterscheiden: (a) Tiefftemperaturbereich (im Bsp.: 193 K): Die Molekülbewegung ist sehr langsam (relativ zur NMR-Zeitskala), und man sieht die Signale der beiden austauschenden, in unterschiedlichen chemischen Umgebungen befindlichen Methylgruppen einzeln. Man sagt im Laborjargon, die „Rotation sei eingefroren“, obwohl sie das nicht notwendigerweise sein muss. Zustand 1 Zustand 2 NMR-Spektroskopie 3 Spektroskopie in der Organischen Chemie (b) Koaleszenzbereich: Hier werden die Signale der austauschenden Spezies bei zunehmender Temperatur und damit zunehmender Austauschgeschwindigkeit verbreitert, wandern aufeinander zu, verschmelzen bei der Koaleszenztemperatur Tc und bilden schließlich ein Durchschnittssignal, das sich bei weiterer Temperaturerhöhung wieder verschmälert. T < Tc NMR-Spektroskopie T = Tc = 285 K im Bsp. T > Tc 4 Spektroskopie in der Organischen Chemie (c) Hochtemperaturbereich (im Bsp.: 343 K): Hier ist die Molekülbewegung sehr schnell, und man erhält nur noch ein Durchschnittssignal mit natürlicher Linienbreite, also ohne koaleszenzbedingte Verbreiterung. NMR-Spektroskopie 5 Spektroskopie in der Organischen Chemie Annahme: reversible Umwandlung zwischen A und B mit Kinetik 1. Ordnung: ka kb A → B und A ← B -> temperaturabhängiges Gleichgewicht: nb/na = exp (-ΔG/(RT)) = K na und nb: relative Populationen (na+nb = 1) Durch Spektrensimulation können für jede Temperatur T die Geschwindigkeitskonstanten k erhalten werden. Für die Geschwindigkeitskonstante k gilt die Eyring-Gleichung: k = χ (kBT)/(h) exp (-ΔG≠/(RT)) kB = Boltzmann-Konstante, NA = Avogadrozahl, χ wird hier gleich 1 gesetzt ΔG≠ = RT (23.76 – ln(k/T)) ln(k/T) = 23.76 – (ΔH≠/R)*1/T + (ΔS≠/R) Auftragung von ln(k/T) gegen 1/T -> Steigung = – (ΔH≠/R) und Achsenabschnitt = (23.76 + (ΔS≠/R)) Somit erhält man also die Freie Enthalpie und die Entropie der Aktivierung. NMR-Spektroskopie 6 Spektroskopie in der Organischen Chemie Basierend auf diesem Verfahren sind die Aktivierungsparameter T-unabhängig. Die Temperaturabhängikeit von K kann bei Bedarf basierend auf der van’t Hoff Gleichung ermittelt werden. Alternativ zur Eyring-Gleichung können die Daten basierend auf der Arrhenius-Gleichung analysiert werden (s. Günther). Aus dem Bereich des langsamen Austausches erhält man den Abstand der Resonanzfrequenzen Δν = νa-νb und die natürliche Linienbreite. Dabei macht man die nicht immer korrekte Annahme, dass diese T-unabhängig sind. Diese Auswertungen erfordern eine exakte experimentelle Temperaturbestimmung und genaue Anpassung, und das auch in Temperaturbereichen, die weiter weg von der Koaleszenztemperatur liegen, d.h. wo die Simulationen ohnehin weniger präzise sind (laut Prof. Duddeck). Aus diesem Grunde und weil ΔS≠ bei Konformationsumwandlungen meist nicht sehr groß ist, verzichtet man oft auf die Berechnung der Temperaturabhängigkeit und begnügt sich mit einer Näherungsformel, nach der man die Höhe der Rotationsbarriere bei der Koaleszenztemperatur (ΔGc≠) abschätzen kann. Dazu benötigt man nur die möglichst exakte Koaleszenztemperatur Tc und den Abstand der beiden Linien Δν im Tieftemperaturbereich (langsamer Austausch). NMR-Spektroskopie 7 Spektroskopie in der Organischen Chemie kc = π · Δν ≈ 2.22 · Δν Reaktionsgeschwindigkeit am Koaleszenzpunkt 2 oder bei J-Kopplung kc ≈ 2.22 · (Δν ) 2 + 6 J ( AB) 2 und ΔGc≠ ≈ R Tc [22.96 + ln (Tc/Δν)] [J/mol] (s. Günther) bzw. ΔGc≠ ≈ -4.58 · 10-3 · Tc [10.32 + log (Tc/kc)] [kcal/mol] ΔGc≠ ≈ -1.91 · 10-2 · Tc [10.32 + log (Tc/kc)] [kJ/mol] Bei einem gekoppelten AX-System mit Δν/J(AX) >> 10 ist der Faktor „6J(AX)2“ gegenüber „Δν2“ kaum noch von Bedeutung für die Größe von ΔGc≠. Im Falle des N,N-Dimethylformamids ist Δν = 653 Hz und Tc = 285 K. Damit: kc = 1450 s-1 und ΔGc≠ ≈ 14.4 kcal/mol (60.2 kJ/mol). NMR-Spektroskopie 8 Spektroskopie in der Organischen Chemie Man erkennt aus der Näherungsformel, dass es zwei bestimmende Einflüsse für die Koaleszenztemperatur Tc gibt, die Energiebarriere ΔG≠ selbst (also die Geschwindigkeitskonstante kc), aber auch der Abstand der chemischen Verschiebung der austauschenden Spezies Δν. Letzteres erklärt, warum in Molekülen, die mehrere Paare austauschender Kerne aufweisen, unterschiedliche Koaleszenztemperaturen Tc existieren können, obwohl natürlich für alle die gleiche Geschwindigkeitskonstante kc gilt. Ein Beispiel dafür sind die temperaturabhängigen (VT = variable temperature) 13 C-NMR-Spektren von cis-1,2-Dimethylcyclohexan. Hier wird die Ringinversion (Racemisierung) eingefroren, und innerhalb mehrerer Kohlenstoffpaare (C-1 und C-2, C-3 und C-6, C-4 und C-5, C-7 und C-8,) tauschen die Kohlenstoffatome ihre Positionen aus. 7 CH3 5 4 NMR-Spektroskopie 6 3 1 2 CH3 8 4 5 3 8 CH 3 6 2 1 7 CH3 9 Spektroskopie in der Organischen Chemie NMR-Spektroskopie 10 Spektroskopie in der Organischen Chemie Man erkennt sehr schön, dass die Koaleszenztemperatur deutlich vom Abstand der Linien der austauschenden Kerne bei tiefer Temperatur abhängt; vgl. z. B. die Signale bei δ = 35.05 (Δδ = 2) und 16.25 (Δδ = 8.5). NMR-Spektroskopie 11 Spektroskopie in der Organischen Chemie VT-1H-NMR eines Methyl-octachlortrypticens, links: experimentell, rechts: simuliert. Cl H Cl H C H Man erkennt das Einfrieren der Methylrotation. Bei -720C ist aus dem A3-Spinsystem ein A2X-Spinsystem mit einem Triplett (Int. 1) und einem Dublett (Int. 2) geworden (kc = 2.5 s-1). Die Austauschgeschwindigkeit bei Tc (etwa -400C) ist 156 s-1. Bei -720C ist sie aber immer noch 2.5 s-1! Von völligem „Einfrieren“ kann also keine Rede sein. NMR-Spektroskopie 12 Spektroskopie in der Organischen Chemie Intermolekulare Dynamik: Protonenaustausch Der wichtigste Prozess des intermolekularen Austauschs ist der Wechsel von Protonen von einem sauren Molekül zu nächsten, wie er z.B. in Alkoholen, Carbonsäuren und Aminen existiert. Auch dies ist in der 1H-NMR-Spektroskopie beobachtbar und führt bei Raumtemperatur i.a. zu einer Verbreiterung der entsprechenden OH- und NH-Signale, wobei eventuelle Aufspaltungen durch skalare Kopplungen meist nicht mehr erkennbar sind, weder an diesen Signalen, noch an denen der Kopplungspartner. Es gibt jedoch Lösungsmittel, die OH-Gruppen so stark komplexieren und damit von der weiteren Umgebung abschirmen können, dass die OH-Protonen eine deutlich verlängerte Verweilzeit am Sauerstoffatom aufweisen. Sie dürfen jedoch selbst nicht protisch sein! In solchen Fällen ist eine Kopplungsaufspaltung, wie man sie zwischen C-H-Protonen kennt, beobachtbar. Ein typisches Beispiel für ein solches Lösungsmittel ist Hexadeutero-Dimethylsulfoxid (CD3-SO-CD3). Auch einen intermolekularen Austausch kann man durch Temperaturerniedrigung verlangsamen: NMR-Spektroskopie 13 Spektroskopie in der Organischen Chemie 1 H-NMR-Spektrum von Methanol (CH3-OH) bei verschiedenen Temperaturen: 283 K 208 K Quartett OH 263 K NMR-Spektroskopie Dublett CH3 310 K 14 Spektroskopie in der Organischen Chemie Befinden sich in der Messlösung saure Protonen aus unterschiedlichen chemischen Umgebungen, beobachtet man i.a. nur ein einziges gemeinsames Durchschnittssignal. Dies kann dazu dienen, OH- bzw. NH-Signale zu identifizieren. Handelt es sich bei dem Substrat um einen Alkohol, kann man ein OH-Signal bei δ ≈ 4 erwarten. (OH- und NH-Signale sind stark abhängig von äußeren Einflüssen wie Konzentration, Temperatur, Säurespuren u.a.) Wenn es viele C-H-Signale gibt, mag es schwierig sein, das OH-Signal eindeutig zu identifizieren. Gibt man jedoch eine geringe Menge einer Säure (δ ≈ 12) hinzu, erhält man ein Durchschnittssignal von Alkohol- und Säure-Proton, also irgendwo zwischen δ = 4-12, je nach den relativen molaren Mengen. Durchschnittssignal bei δ = 6.0 Alkoholsignal bei δ = 4.0 Carbonsäuresignal bei δ = 12.0 (0.33 molar bzgl. Alkohol) Δδ = 8.0 10 NMR-Spektroskopie 5 0 ppm 15 Spektroskopie in der Organischen Chemie Intermolekularer Austausch kann auch zwischen Molekülen stattfinden und durch VT-NMR beobachtet werden. Als Beispiel sei der Ligandenaustausch von Phenylseleniden in Dirhodiumkomplex-Addukten genannt: R Se = Ph-Se CH2-R Sea + [Seb Rh - Rh Se] Rh - Rh = O R Seb + [Sea Rh - Rh Se] O O O Rh Rh O O O R O R Das Molverhältnis beträgt 5:1, sodass zwei Selenidmoleküle komplexiert werden, während drei weitere als freie Liganden vorliegen und um die Komplexierungsstellen konkurrieren. Die folgenden 1H-NMR-Spektren zeigen links die ortho-Protonen der Se-PhGruppen und rechts das Signal der Se-CH2-Protonen. NMR-Spektroskopie 16 Spektroskopie in der Organischen Chemie Se H Bei Raumtemperatur sieht man nur Durchschnittssignale, während bei tiefen Temperaturen die Signale der Se-Ligand-Spezies einzeln sichtbar werden. Se CH2 Signal aus Addukt Se-Rh-Rh-Se Signal des freien Se Ph-Se-CH2-R = Se NMR-Spektroskopie 17 Spektroskopie in der Organischen Chemie Ein anderes wichtiges Beispiel für intermolekularen Austausch ist die Bildung kinetisch labiler Komplexe zwischen Lanthaniden-Verschiebungsreagenzien (lanthanide shift reagent, LSR) und Lewis-basischen Ligandenmoleklülen. Dies kann zur Vereinfachung von überlagernden 1H-Signalen und zur Strukturbestimmung des Liganden ( Lanthaniden-Verschiebungsreagenzien). Wenn das LSR selbst chiral ist, kann es auch zur chiralen Erkennung genutzt werden. Ob man zwei getrennte Signale für den freien und gebundenen Zustand sieht oder ein koalesziertes breites oder schmales Signal hängt nicht nur von der Termperatur ab sondern auch von der Dissoziationskonstante Kd bzw. den Geschwindigkeitskonstanten kon und koff (kA und kB im Bild unten) der Komplexbildung und dem Konzentrationsverhältnis der Bindungspartner ab. NMR-Spektroskopie 18 Spektroskopie in der Organischen Chemie Beispiel: Protein-Ligandenwechselwirkungen (Protein-Protein, Protein-kleine organische Moleküle, Protein-DNA etc.) hochaffiner Ligand (Kd ca. nM): 2 getrennte Signale, Intensität der Linien des freien und gebundenen Zustands hängt vom jeweiligen Anteil von freiem und gebundenem Protein ab (Überschuss an Ligand – nur noch Linien für gebundenen Zustand sichtbar) -> slow exchange Ligand mittlerer Affinität (Kd ca. µM): 1 breites – sehr breits koalesziertes Signal (‚Mischsignal’) -> intermediate exchange Ligand niederer Affinität (Kd ca. mM): 1 schmales Mischsignal, Lage gewichtet entsprechend den Anteilen des freien und gebundenen Zustands -> fast exchange -> Bestimmung von Kd möglich, wenn Spektren in Anwesenheit schrittweise steigender Ligandenkonzentrationen gemessen werden NMR-Spektroskopie 19 Spektroskopie in der Organischen Chemie Aus: I. A. Kleckner & M.P. Foster An introduction to NMR-based approaches for measuring protein dynamics, Biochim. Biophys. Acta (2010), doi:10.1016/j.bbapap.2010.10.012 NMR-Spektroskopie 20 Spektroskopie in der Organischen Chemie Allgemein können dynamische Informationen aus einer Vielzahl von NMRParametern erhalten werden, zu T1- und T2- Relaxationszeiten und NOE (im Bild unten als NSR bezeichnet) siehe auch IM3. Aus: I. A. Kleckner & M.P. Foster An introduction to NMR-based approaches for measuring protein dynamics, Biochim. Biophys. Acta (2010), doi:10.1016/j.bbapap.2010.10.012 NMR-Spektroskopie 21 Spektroskopie in der Organischen Chemie Lanthaniden-Verschiebungsreagenzien (Lanthanide Shift Reagents, LSR) Im Jahre 1969 entdeckte Hinckley, dass gewisse Lanthanidionen die Halbwertsbreiten der 1H-NMR-Signale von Ligandenmolekülen nicht sehr stark verbreitern, obwohl sie paramagnetisch sind. Dadurch blieb es möglich, die Signale einschließlich ihrer Feinstruktur in Anwesenheit solcher Ionen zu beobachten. Allerdings werden die Signale mehr oder weniger stark verschoben. Dies eröffnete zwei wichtige Anwendungsgebiete: (a) Stark überlappende Signale konnten „auseinander gezogen“ werden; zur damaligen Zeit waren Signalüberlagerungen ein ernstes Problem, weil noch keine hohen Magnetfelder zur Verfügung standen. (b) Die Signalverschiebungen konnten in erster Näherung nach der Dipoltheorie interpretiert werden (siehe unten) und erlaubten damit Aussagen zur Struktur der Ligandenmoleküle. Beide Aspekte sind heute nicht mehr allzu aktuell, jedoch ist der letztere keineswegs überholt, sondern bietet manchmal verblüffend einfache Problemlösungen an. (& Inspiration für neue Experimente -> z.B. Analyse von Protein-Liganden NMR-Spektroskopie 1 Spektroskopie in der Organischen Chemie WW mit PREs). Die Anwendung chiraler LSR zur Enantiomerenerkennung ist dagegen auch heute noch von großer Bedeutung (siehe später). Theoretischer Hintergrund LSR sind Komplexe von Ln3+-Ionen (i.a. Ln = Eu, Yb und gelegentlich Pr) mit drei β-Dionaten (siehe unten). Sie sind harte Lewissäuren und besitzen noch eine freie Ligandenstelle, an die Lewis-basische Liganden gebunden werden können. Diese LSR-Ligand-Komplexe (LSR···L) sind kinetisch instabil, d.h. sie haben – relativ zur NMR-Zeitskala – eine kurze Lebensdauer und zeigen schnellen Ligandenaustausch. LSR + L LSR···L Damit erscheinen im NMR-Spektrum die Ligandensignale nicht zweimal (freies L und komplexiertes L), sondern man erhält nur ein Durchschnittssignal, dessen Position im Spektrum vom Unterschied der chemischen Verschiebung (Δ = δkomplex - δfrei) sowie von der relativen molaren Zusammensetzung der beiden Komponenten LSR und L in der Lösung abhängt. NMR-Spektroskopie 2 Spektroskopie in der Organischen Chemie Δ ist die paramagnetische Signalverschiebung, die sich aus zwei Komponenten zusammensetzt, dem dipolaren und dem Kontaktterm: Δ = Δdipolar + Δkontakt Der dipolare oder Pseudo-Kontaktterm beschreibt die magnetisch-dipolare Wechselwirkung von Lanthanidion und Ligandenkernen durch den Raum. Sie ist die Wechselwirkung, die zur Strukturermittlung der Ligandenmoleküle genutzt werden kann (siehe unten). Der zweite Term Δkontakt steht für mögliche Spin-Delokalisationen innerhalb des Komplexes und wirkt durch die Bindungen. Die durch diese Wechselwirkung verursachten Signalverschiebungen sind nur schwer quantitativ zu erfassen und sollten – wenn möglich – unterdrückt bzw. vermieden werden. Dipolarer oder Pseudo-Kontaktterm Man kann davon ausgehen, dass man das Lanthanidion als einen magnetischen Punktdipol auffassen kann, der ein axial-symmetrisches dipolares Magnetfeld erzeugt (Anisotropie). Damit wird es möglich, seine Feldwirkung gemäß der McConnel-Robertson-Gleichung zu beschreiben: NMR-Spektroskopie 3 Spektroskopie in der Organischen Chemie z Kern i des Liganden θi r Ln3+ € y x z NMR-Spektroskopie 2 3 cos θi − 1 Δdipolar(i) = K ri3 Die z-Richtung ist die Hauptmagnetisierungsachse, die durch die Kernverbindungslinie des Ln3+-Ions mit dem direkt gebundenen Atom des Liganden, z.B. O in (H9C4) 2O---Eu(fod)3 (mod. aus Hesse, Meier, Zeeh & s.u.) definiert ist. K ist ein Kalibrierungsfaktor, mit dem man die berechneten und die experimentellen Werte verknüpfen kann und der für alle i Kerne eines Ligandenkomplexes gleich ist. Das axial-symmetrische Feld erzeugt einen Doppelkegel. Beim Wechsel von der Innen- zur Außenseite des Kegels kehrt sich das Vorzeichen der Signalverschiebung um; auf dem Kegelmantel selbst ist sie Null. 4 Spektroskopie in der Organischen Chemie Der Öffnungswinkel des Kegels kann damit leicht berechnet werden: 2 Eu3+ + Pr3+ - - + 3 cos θ i − 1 = 0 und damit: θi = 54.70. Kerne innerhalb der Kegelmäntel werden durch Eu3+ und Yb3+ entschirmt [δ-Wert vergrößert; (+)] aber durch Pr3+ abgeschirmt [δ-Wert verkleinert; (-)]. Außerhalb der Kegelmäntel ist es umgekehrt. Liegt der Kern gerade auf einem Kegelmantel, wird sein Signal nicht verschoben. In den meisten Fällen liegen die 1H- oder 13C-Kerne eines Liganden innerhalb der Kegelmäntel. Häufig ist es sogar so, dass θi < 400 ist. Dann kann man relative Entfernungen der Kerne i von Ln3+ in recht guter Näherung und halbquantitativ nach der Formel abschätzen: Δ ≈ K · 1/r-3 NMR-Spektroskopie 5 Spektroskopie in der Organischen Chemie 90-MHz-1H-NMR-Spektrum von 30 mg 1-Hexanol; unten mit 50 mg Eu(dpm)3 NMR-Spektroskopie 6 Spektroskopie in der Organischen Chemie Der Kontakt-Anteil an Δ sollte so klein wie möglich sein, da er die Interpretation nach dem Dipolmodell verfälscht. Letzteres ist zum Glück für 1H-Signale nur dann der Fall, wenn das Proton nur sehr wenige Bindungen vom Ln3+-Ion entfernt ist, also z.B. das α-Proton eines Alkohols. In diesem Fall sollte man keine auch nur halbquantitative Auswertung durchführen. Bei 13C ist dieses Problem leider erheblich gravierender. Dies sei am Beispiel von Pyridin (komplexierendes Atom: N) mit Eu(dpm)3 dargestellt: γ 13 β N Δ(1H) α C Δ(13C) Differenz % Kontakt berechn. Δ(13C) exptl. berechn. exptl. α β 31.02 31.10 90.0 67.8 22.2 25 10.68 10.59 -0.9 24.33 -25.2 ca. 100 γ 9.71 9.33 30.2 18.7 11.5 38 1 H zeigt kaum Kontaktanteil, wohl aber 13C! Aus diesem Grunde ist es bei 13C besser, Yb-Salze als LSR zu verwenden. Dann ist oft nur C-α betroffen. NMR-Spektroskopie 7 Spektroskopie in der Organischen Chemie Struktur der LSR Das Metallion eines LSR ist ein Ln3+-Ion mit seinen ungepaarten f-Elektronen. Mit der Ausnahme von Lanthan (f0; nicht paramagnetisch) kann man prinzipiell jedes Lanthanid einsetzen. Es hat sich aber in der Praxis gezeigt, dass Europium und Ytterbium, gelegentlich auch Praseodym (wegen der Umkehrung bei der Signalverschiebungsrichtung) ausreichen. In anderem Zusammenhang ist auch Gadolinium (Gd3+) interessant. Bei ihm ist gerade jedes f-Orbital einfach besetzt, sodass sich die Anisotropien gegenseitig kompensieren. Ein Gadolinium-LSR verschiebt deshalb die Signale nicht; vielmehr gibt es hier eine Entfernungsabhängigkeit der Signalverbreiterung (r-3), die gelegentlich für Strukturuntersuchungen genutzt wurde. Gd3+-Salze werden als Kontrastmittel in der Kernspin-Tomographie eingesetzt. (& solvent PREs) Das Lösungsmittel für die oben gezeigten LSR-Experimente muss unpolar sein, damit es nicht die Ligandenstelle blockiert. Lösungsmittel wie Methanol-d4, Dimethylsulfoxid-d6, Aceton-d6 usw. scheiden aus. Das gängige Lösungsmittel ist CDCl3. Wasser ist der „Tod“ des LSR, weil es sich kaum wieder entfernen lässt. Man erkennt Wasser daran, dass das LSR deutlich gelb gefärbt ist. NMR-Spektroskopie 8 Spektroskopie in der Organischen Chemie Durch die Einengung auf unpolare Lösungsmittel ergibt sich das Problem, ein lösliches Ln3+-Salz zu erhalten. Dies ist möglich, wenn das LSR drei Liganden des Anions eines β-Diketons enthält, wobei diese zur Löslichkeitsverbesserung größere Alkyl- oder Perfluoralkylreste tragen: O 1 R O O R2 1 R - O R2 Die am häufigsten auftretenden Dionate sind: dpm: dipivaloylmethanato R1 = R2 = tert-Butyl fod: 1,1,1,2,2,3,3-heptafluor-7,7-dimethyl-4,6-octandionato R1 = tert-Butyl; R2 = CF2CF2CF3 fod-Komplexe sind besser löslich als dpm-Komplexe und auch stärker Lewissauer, was zu größeren Signalverschiebungen führen kann. Es gibt auch deuterierte LSR, bei denen zur Vermeidung von eigenen 1HSignalen alle Protonen durch Deuterium ersetzt sind. NMR-Spektroskopie 9 Spektroskopie in der Organischen Chemie Funktionelle Gruppen der Substrate Nach dem HSAB-Prinzip binden vorzugsweise stark Lewis-basische Liganden an das Lanthanidion. Die Größe von Δδ hängt dabei von den Komplexbildungskonstanten ab. Von Seiten des Liganden spielen zwei Effekte eine Rolle: (a) Basizität des Substrats, z.B. -NH2 > -OH > C=O > -COOR > -CN > -S(b) Sterische Effekte Sterische Abschirmung kann die Wirksamkeit eines Atoms oder einer funktionellen Gruppe stark schwächen oder gar ganz aufheben. Bei Verbindungen mit mehr als einer funktionellen Gruppe muss auf die dominierende Komplexierungsstelle im Liganden geachtet werden, wobei beide Argumente – Basizität und sterische Abschirmung – abgewogen werden müssen. Als quantitatives Maß für Δδ wurde der sog. G-Wert vorgeschlagen. Er entspricht der Signalverschiebung in ppm für ein äquimolares Verhältnis von LSR und L (1:1). NMR-Spektroskopie 10 Spektroskopie in der Organischen Chemie Einige typische G-Werte: RCH2NH2 RCH2OH RCH2NH2 RCH2OH RCOOCH3 RCH2CN RCH2SR´ G-Wert 150 100 35 25 14 5 1 Diese G-Werte sind allerdings nur von begrenztem Wert, weil sie voraussetzen, dass die Signalverschiebungen bis hin zu einem Molverhältnis von 1:1 linear bleiben. Dies ist aber keineswegs der Fall, was u.a. daran liegt, dass die Kinetik nicht so einfach ist wie eingangs beschrieben: LSR + L LSR···L Vielmehr gibt es auch andere Spezies, wie LSR···L2 , LSR2··L oder (LSR···L)2. Dadurch wird die Kinetik sehr viel komplexer, und die Abhängigkeit der Signalverschiebung Δδ vom Molverhältnis LSR/L hat einen deutlich anderen Verlauf. Man stellt fest, dass der Anstieg nur bis zu LSR/L ≈ 0.3 linear bleibt. Darüber hinaus flacht sie deutlich ab, sodass es bei LSR-Experimenten ratsam ist, diese Grenze nicht zu überschreiten. NMR-Spektroskopie 11 Spektroskopie in der Organischen Chemie G 1 2 LSR/L Der schwarze Kurvenverlauf stellt die „ideale“ Abhängigkeit dar, die rote die reale: Der Anstieg beginnt meist mit einer kleinen Verzögerung, was an Spuren von Wasser oder anderen polaren Verunreinigungen liegen kann. Dann folgt ein Abschnitt mit linearem Anstieg bis LSR/L ≈ 0.3 bis 0.5, gefolgt von einer Abflachung; Die reale Kurve kann die theoretische aber auch bei LSR/L > 1 überschreiten. NMR-Spektroskopie 12 Spektroskopie in der Organischen Chemie Anwendungen (a) Verbesserung der Dispersion NMR Signale Ein gutes Beispiel war bereits vorher gezeigt worden, 1-Hexanol. Auf der folgenden Seite sind die 1H-NMR-Spektren von trans-4-tert-Butylcyclohexanol in Gegenwart steigender Mengen Eu(dpm)3 gezeigt: OH 3 4 2 1 Man erkennt sehr gut die vollständige Separierung der einzelnen Signale im obersten Spektrum; die Signale der tert-Butylgruppe sind jeweils rechts zu sehen und nur mit 1/10 ihrer wahren Intensität dargestellt. Es ist anzumerken, dass solche Spektrenverbesserungen angesichts der heute verfügbaren viel höheren Magnetfelder sowie moderner Messmethoden zur Separierung überlagerter Signale mehr oder weniger obsolet geworden sind. NMR-Spektroskopie 13 Spektroskopie in der Organischen Chemie OH 3 4 2 NMR-Spektroskopie 1 14 Spektroskopie in der Organischen Chemie (b) Konfigurationsbestimmung Die Abbildung zeigt die 13C-Signalverschiebungen von Isoborneol aufgetragen gegen die jeweiligen LSR : Isoborneol-Verhältnisse. (Für C-2 gilt die rechte, für alle anderen die linke ppm-Skala.) In der Strukturdarstellung sind die G-Werte angezeigt. Man erkennt an den geminalen Methyl-C´s (10.0 vs. 4.2), dass das diastereomere Borneol (OH endo) nicht vorliegen kann. NMR-Spektroskopie 15 Spektroskopie in der Organischen Chemie Heute würde man solche Konfigurationsbestimmungen anders durchführen (z.B. durch NOE-Messungen), aber dennoch kann die Rückbesinnung auf LSRExperimente wegen ihrer Einfachheit durchaus angezeigt sein: 16.2 24.0 21.2 12.8 12.8 7.7 E N OH HO N Z Die 13C-Signalverschiebungen wurden für die beiden diastereomeren Butanonoxime bestimmt (Komplexierungsstelle OH). Vergleicht man paarweise die GWerte für die Kohlenstoffe, ist die Konfigurationszuordnung offensichtlich. Bei Konformationsbestimmungen (‚Strukturbestimmungen’) sollte man nie vergessen, dass es nicht garantiert ist, dass die bevorzugte Konformation eines Ligandenmoleküls im LSR-Komplex die gleiche ist wie im freien Liganden. Es ist daher von solchen Experimenten abzuraten, zumal sie mit hohem Mess- und Interpretations-aufwand verbunden sind und es heute einfachere Methoden gibt. (c) Chirale LSR = CLSR -> chirale Erkennung s.u. NMR-Spektroskopie 16 Spektroskopie in der Organischen Chemie NMR-Methoden zur chiralen Erkennung Mit Ausnahme der CD-Spektroskopie (CD = Circulardichroismus) sind alle spektroskopischen Methoden – auch die NMR – achiral, d.h. sie können nicht zwischen den Enantiomeren einer zu untersuchenden Verbindung (Substrat) unterscheiden. Dennoch kann man sie zur chiralen Erkennung heranziehen, wenn man bei der Messung einen chiralen Hilfsstoff (Auxiliar) in enantiomerenreiner Form verwendet. Dieses Auxiliar soll mit dem Substrat in irgendeiner Weise interagieren, sodass aus den Substrat-Enantiomeren Diastereomere werden, deren physikalische Eigenschaften, also auch spektroskopische, sich unterscheiden. Man wird dann statt identischer Signale für die Enantiomeren unterschiedliche Signale für die Diastereomeren erhalten. Man nennt dies Dispersion, genauer: diastereomere Dispersion. Für die Verwendung in der NMR-Spektroskopie gibt es zwei grundsätzlich unterschiedliche Typen von Auxiliaren: NMR-Spektroskopie 1 Spektroskopie in der Organischen Chemie (1) Chirale Derivatisierungsreagenzien; chiral derivatizing agents (CDA) Bei diesen Reagenzien werden die enantiomeren Substratmoleküle durch chemische Reaktion (Ausbildung kovalenter Bindungen; gelegentlich auch Salzbildung) in Diastereomere überführt. (2) Chirale Solvatisierungsreagenzien; chiral solvating agents (CSA) Hier werden die enantiomeren Substratmoleküle in eine chirale Umgebung gebracht, ohne dass eine kovalente Bindung geknüpft wird. Dies kann geschehen durch (a) Messung in einem chiralen Lösungsmittel (b) Adduktbildung des Substrats an einen chiralen, enantiomerenreinen Komplex. Beide Methoden werden heute vielfältig verwandt und stellen neben der Chromatographie an chiralem Trägermaterial die verbreitetsten und einfachsten Methoden zur chiralen Erkennung, d.h. der Enantiomerendifferenzierung, dar. NMR-Spektroskopie 2 Spektroskopie in der Organischen Chemie (1) Chirale Derivatisierungsreagenzien (CDA) Die Methode kann natürlich nur angewendet werden, wenn die zu untersuchenden Substrate über eine genügend reaktive funktionelle Gruppe verfügen. Es bieten sich Alkohole, Amine, Carbonsäuren etc. an. Das Substrat, dessen Enantiomerenreinheit geprüft werden soll, wird – oft im NMR-Röhrchen - mit einem enantiomerenreinen Auxiliar vermischt, sodass Diastereomere entstehen. Typische Auxiliare sind: OCH3 Ph C H3C O O H3C O CH3 O Cl O C COOH Se CH3 CH3 P CF3 COOH H3C Ph C H NCO H N C O H3C (R)-(+)-(α)-Methoxyα-trifluormethyl- Anderson-Shapiro- (1,S,4R)-(-)-ω- (R)-(α)-Methylphenylessigsäure Reagenz Camphansäure benzylisocyanat (Mosher-Säure) NMR-Spektroskopie Ph Selon Silks III 3 Spektroskopie in der Organischen Chemie Diese Auxiliare und noch eine ganze Reihe ähnlicher Verbindungen sind vor allem für Alkohole und Amine geeignet, die Säuren werden als Säurechloride eingesetzt. Zum Teil sind sie auch als solche Verbindungen käuflich zu erwerben. Das mit Abstand wichtigste Reagenz aus dieser Reihe ist die Mosher-Säure. Sie liefert leicht detektierbare NMR-Signale der gebildeten diastereomeren Ester bzw. Amide; vor allem 1H von OCH3, 19F von CF3, beides Singulettsignale von hoher Intensität und Empfindlichkeit. Im Falle des Anderson-Shapiro-Reagenzes bietet sich das Signal des ebenfalls recht empfindlichen 31P-Kerns an. Für Carbonsäuren gibt ein sehr interessantes selenhaltiges Reagenz (Selon), bei dem man das 77Se-NMR-Signal misst. Dieser Kern zeichnet sich durch besonders große Dispersionen aus; er erkennt ein Chiralitätszentrum auch noch in großer Entfernung. NMR-Spektroskopie 4 Spektroskopie in der Organischen Chemie 7 Bindungen CH3 * O Se C N O * HC 3 Enantiomer 1 Enantiomer 2 77Se-NMR-Signal *Ph Das 77Se-NMR-Signal ist trotz der großen Entfernung zwischen dem Selenatom und dem Chiralitätszentrum in der Säurekette (*) eindeutig dispergiert. NMR-Spektroskopie 5 Spektroskopie in der Organischen Chemie O Ph H H Se C N O H3 C Ph O Ph D H Se C N O H3 C Ph O Ph D D Se C N O H3 C NMR-Spektroskopie Ph Noch eindrucksvoller ist, dass der 77Se-Kern in der Lage ist, sogar die beiden diastereomeren α-Monodeuterophenylessigsäuren (Mitte) zu unterscheiden. Man beachte auch die deutlichen Isotopenverschiebungen der 77Se-NMRSignale. 6 Spektroskopie in der Organischen Chemie (2) Chirale Solvatationsreagenzien (CSA) Das Grundprinzip der Wechselwirkung zwischen Substratmolekül und Auxiliar ist hier nicht die Ausbildung einer kovalenten Bindung. Vielmehr wird hier die Chiralität der Solvathülle bei chiralen solvatisierenden Molekülen ausgenutzt. Allerdings muss diese Wechselwirkung stark genug sein, um eine erkennbare Diastereomerie zu erzeugen. (a) Chirale Lösungsmittel Verwendet man ein Lösungsmittel, muss man darauf achten, dass beide Moleküle (Substrat und Lösungsmittel) über funktionelle Gruppen verfügen, die sich stark genug anziehen, um einen ausreichend starken, also langlebigen, Lösungsmittelkomplex zu erzeugen. Der erste, der dieses Prinzip verwendet hat, war Pirkle, der für die chirale Diskriminierung von Aminen in der Messlösung enantiomerenreine Alkohole (Pirkles Alkohole) beigefügt hat, bzw. umgekehrt enantiomerenreine Amine für Alkohole: NMR-Spektroskopie 7 Spektroskopie in der Organischen Chemie CF3 C CF3 CF3 H C OH C H OH OH CF3 C H CF3 H NH2 C H NH2 Diese sind in der Lage, über starke Wasserstoffbrückenbindungen zu assoziieren, wobei aber die aromatischen Gruppen zur Ausbildung sekundärer Wechselwirkungen benötigt werden. Nur dadurch wird die gegenseitige Beeinflussung groß genug, um sichtbare Dispersionen zu erzeugen. NMR-Spektroskopie 8 Spektroskopie in der Organischen Chemie (b) Chirale Lanthaniden-Verschiebungsreagenzien (CLSR) CLSR spielen auch heute noch eine wichtige Rolle bei der chiralen Erkennung von Liganden, also bei der Feststellung, ob eine chirale Verbindung als Racemat oder nichtracemisches Gemisch vorliegt. Die CLSR produzieren aus den Enantiomeren diastereomere Komplexe, sodass enantiotope Kerne diastereotop und damit anisochron werden. Eine Integration liefert dann das relative Verhältnis der diastereotopen Signalpaare als Abbild des Verhältnisses der enantiomeren Ligandenmoleküle. CLSR tragen die Chiralität in den Dionate-Resten, die i.a. vom D-Campher abgeleitet sind. Typische Vertreter sind tfc (3-trifluoracetylcamphorato) oder hfm (3-heptafluorpropylhydroxymethylen) R - R = CF3 oder CF2CF2CF3 O O NMR-Spektroskopie 9 Spektroskopie in der Organischen Chemie Eine interessante Anwendung ist die, wo durch Überführung enantiotoper Protonen in diastereotope eine Rotationsbarriere durch DNMR beobachtbar wird: H3C Ph-CH2 N O=N H3C O=N N H3C Ph-CH2 H3C In diesem Beispiel wurde Atropisomerie bei N-Nitrosoanilinen vermutet. Die Koaleszenz der Methylprotonen kann jedoch ohne ein CLSR (Komplexierung an NNO) nicht beobachtet werden, weil sie enantiotop sind. Erst im diastereomeren CLSR-Komplex werden sie anisochron (δ = 3.26 und 3.08). NMR-Spektroskopie 10 Spektroskopie in der Organischen Chemie (c) Chirale Dirhodium-Komplexe CLSR funktionieren nur, wenn das Substrat eine deutlich Lewis-basische Funktionalität besitzt (z.B. -OH, NH2, COOR, C=O), weil sie selbst starke LewisSäuren sind. Ist dies nicht der Fall, versagt die Methode. Hier bietet sich eine alternative Methode an, bei der die Metallatome (Rhodium) selbst schwache Säuren sind (weich): R O O R O O Rh Rh O O O R O OCH3 R= C Ph CF3 R Es handelt sich um einen enantiomerenreinen Dirhodium-Komplex mit vier Mosher-Säure-Resten. NMR-Spektroskopie 11 Spektroskopie in der Organischen Chemie Ähnlich wie bei den LSR bilden sie mit den Liganden (L) Addukte. Hier sind allerdings 1:1- und 2:1-Addukte möglich, je nach stöchiometrischen Verhältnissen. L [Rh-Rh] + L (1:1-adduct) -L L [Rh-Rh] (2:1-adduct) L [Rh-Rh] +L Bei Raumtemperatur sind die Addukte i.a. kinetisch nicht stabil, d.h. es erfolgt schneller Austausch – ähnlich wie bei den LSR –, und man beobachtet nur Durchschnittssignale. NMR-Spektroskopie 12 Spektroskopie in der Organischen Chemie Als Beispiel sei die Dispersion eines nichtracemischen Phosphinsulfids gezeigt, beobachtet an den 1H- und 31P-NMR-Signalen: S H3 C O P Ph (H3C)3C NMR-Spektroskopie 13