Bakterien sind die besten Chemiker
Werbung

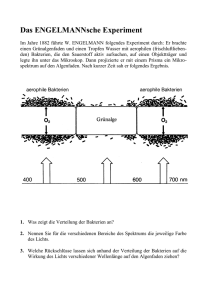
333_359_BIOsp_0406.qxd 346 02.06.2006 10:12 Uhr Seite 346 WISSENSCHAFT Bio-Anorganische Chemie Bakterien sind die besten Chemiker OLIVER EINSLE 1 , GRAZYNA B. SEIFFERT 2 , MARTHA E. SOSA-TORRES 2 , 3 UND PETER M.H. KRONECK 2 1 INSTITUT FÜR MIKROBIOLOGIE UND GENETIK, GEORG-AUGUST-UNIVERSITÄT GÖTTINGEN, 2 FACHBEREICH BIOLOGIE, UNIVERSITÄT KONSTANZ, 3 FACULTAD DE QUÍMICA, UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO “Dear Lord, I fall upon my knees and pray that all my syntheses may cease to be inferior to those conducted by bacteria” ó Dieses Gebet eines Chemikers[1] wird verständlich angesichts der Fülle anorganischer und organischer Moleküle, die von Bakterien meisterlich synthetisiert werden. Sie bestehen in der Regel aus einer einzelnen Zelle, sind von kleiner Gestalt und einfach aufgebaut. Täuschen wir uns nicht, denn Bakterien sind erstaunlich komplexe und faszinierende Lebewesen. Ihre Laboratorien können sie an Extremstandorten aufbauen in Bezug auf Temperatur, Druck, Sauerstoffgehalt und chemischen Stress[2]. Bakterien helfen bei der Verdauung von Gras im Kuhpansen, zusammen mit Flagellaten verwerten sie Holzbestandteile und Stoffwechselprodukte im Termitendarm[3]. „Leben unter tödlichen Bedingungen, bei pH 11 und 300 Gramm Salz pro Liter Wasser“, so lautete die Pressemitteilung des Max-Planck-Instituts in Martinsried über den Anpassungskünstler Natronomonas pharaonis[4]. Bakterien verblüffen die Wissenschaftler stets aufs Neue. Dank geothermischer Strahlung sind sie in 2,4 km Meerestiefe ohne Sonnenlicht zur Photosynthese fähig[5]. In diesen Tiefsee-Oasen gibt es Hydrothermalschlote, die 350° C heißes, an Schwermetallsulfiden reiches Wasser ausstoßen. Ein Paradies für exzentrische Bakterien. Oft müssen Bakterien ihre Nahrung ohne Sauerstoff prozessieren. Ihr Speisezettel zur Energiekonservierung enthält Delikatessen wie Kohlenmonoxid (CO), Methan (CH4), Sulfat (SO42–), Schwefel (S0), Nitrat (NO3–), Arsenat (AsO42–), oder toxische Metallverbindungen[6, 7]. Um das wichtige Element Eisen aufzunehmen, welches je nach Lebensraum in Form schwerlöslicher Eisenoxide oder in Konzentrationen von 0,02 bis 1,0 nM gelöst im Ozean vorliegt, werden Siderophore produziert. Dieses sind Komplexbildner mit zumeist anspruchsvoller Stereochemie und hoher Affinität zum Fe(III)-Ion[8]. Bakterien sind Könner beim Umsatz reaktionsträger Moleküle. Die Reduktion von Lachgas (N2O) zu N2 und H2O läuft problemlos ab bei Raumtemperatur und Normaldruck[9]. Stickstofffixierer spalten die stabile N≡N-Bindung unter Bildung von Ammoniak (NH3), der bioverfügbaren Form des Stickstoffs für Pflanze, Tier und Mensch[10]. Aromatische Kohlenstoffverbindungen, z. B. Benzol, Toluol oder Phenol, werden selbst ohne Sauerstoff zerlegt mittels raffinierter Mechanismen ähnlich zur Birch-Reduktion und der Kolbe-Synthese [11, 12]. Bakterien sind außergewöhnlich geschickte Chemiker, ausgestattet mit einer Vielfalt an neuartigen chemischen Strukturen und Reaktionen. Als repräsentative Beispiele dafür haben wir das Kupferenzym Distickstoffmonoxidreduktase, das Eisen-MolybdänEnzym Nitrogenase und das Multihäm-Enzym Cytochrom c-Nitritreduktase ausgesucht. Dazu stellen wir eine ungewöhnliche Reaktion aus dem Bereich des Aromatenabbaus vor, welche durch das Molybdänenzym Transhydroxylase katalysiert wird. Diese Auswahl soll die fundamentale Bedeutung von Übergangsmetallen und die besondere Rolle des Schwefels für biologische Prozesse und die Evolution des Lebens unterstreichen[6, 13]. Distickstoffmonoxidreduktase (N2OR) Der Umsatz des Treibhausgases N2O (N2O + 2 H+ + 2 e− → N2 + H2O; Eo⬘ = +1,35 V) ist ein wesentlicher Teilschritt der mikrobiellen Denitrifikation. Die N2OR ist ein Head-to-TailHomodimer mit zwei neuartigen Kupferzentren. Jede Untereinheit ist aus zwei Domänen aufgebaut: In der C-terminalen Domäne sitzt das CuA, ein gemischtvalentes Elektronentransfermodul, [Cu1,5+(SCys)2Cu1,5+]. Der CuCu-Abstand liegt, ähnlich wie beim metallischen Kupfer, bei 2,45 Å. Die N-terminale Domäne beherbergt das Katalysezentrum CuZ, einen vierkernigen Kupfersulfidcluster. Dieser findet sich in der Nabe einer siebenblättrigen β-Propeller-Domäne, welche die sieben Histidinliganden für CuZ liefert. Die kürzeste CuA-CuZ-Distanz innerhalb eines Monomers liegt bei 40 Å und ist somit zu groß für einen effizienten Elektronentransfer. Dieses Problem wird elegant gelöst durch die Head-to-Tail-Architektur des Dimers, welche den Abstand auf 10 Å verringert (Abb. 1)[9]. Wo das Gas N2O bindet und wie es umgesetzt wird, ist bisher noch wenig verstanden. Ähnlich wie N2 ist N2O bemerkenswert stabil; es ist aber ein potentes Oxidationsmittel. Seine Affinität zu Metallen ist gering, und es gibt bis heute keine einzige Kristallstruktur eines N2O-Komplexes, nicht einmal für das 1969 von Taube beschriebene Ion [Ru(NH3)5(N2O)]2+. Sowohl CuA als auch CuZ durchlaufen mehrere Redoxzustände; im Enzymtest mit Methylviologen als Elektronendonor muss CuZ in den vollreduzierten Zustand [CuI4(S2–)]2+ überführt werden. N2O könnte dann zwei Kupferzentren überbrücken und gespalten werden[9]. Nitrogenase (N2ase) Der Name Stickstoff des chemischen Elements N rührt von seiner Reaktionsträgheit her. Aber während der großtechnische HaberBosch-Prozess Ammoniak aus N2 und H2 bei hohen Temperaturen und Drücken produziert (über 500° C, 200 bar), erledigen Bakterien dies bei 20° C und 1 bar[10]. Die biologische Stickstofffixierung wird durch die Nitrogenase (N2ase) katalysiert, wobei pro Elektron zwei Moleküle ATP investiert werden: N2 + 8H+ + 8e– + 16 MgATP → 2 NH3 + H2 + 16 MgADP + 16 PO3– 4 Die N2asen lassen sich als Eisen-Molybdän(MoFe)-, Eisen-Vanadium(VFe)-, und Eisen(FeFe)-N2asen klassifizieren. Die am intensivsten untersuchte N2ase ist das MoFeEnzym, dessen Struktur wir bei atomarer Auflösung (1,16 Å) kennen. Es enthält zwei einBIOspektrum | 04.06 | 12. Jahrgang 333_359_BIOsp_0406.qxd 02.06.2006 10:12 Uhr Seite 347 347 zigartige Eisen-Schwefel-Cluster. Der PCluster, ein [8Fe:7S]-Elektronentransferzentrum, reduziert das eigentliche Aktivzentrum der N2ase, den Eisen-Molybdän-Cofaktor (FeMoco). Dieser ist bis heute der größte bekannte biologische Eisen-Schwefel-Cluster (Mo:7Fe:9S:N) und zeigt einen hochsymmetrischen Aufbau mit einem zentralen Stickstoffatom (Abb. 2) [14]. Mit dem Protein ist der FeMoco lediglich über ein Cystein und ein Histidin verbunden; ein Homocitrat bindet über zwei Sauerstoffe an das Molybdän. Die Reaktion der N2ase besteht aus dem komplexen Zusammenspiel des MoFe-Enzyms mit der N2ase-Reduktase, an der ATP gespalten wird. Dadurch werden Elektronen von sehr negativem Potenzial erzeugt, die dann nacheinander an das Aktivzentrum des Enzyms weitergeleitet werden. Erstaunlicherweise ist der molekulare Mechanismus dieser Reaktion noch nicht in allen Einzelheiten aufgeklärt, und selbst die Bindestelle von N2 am Cofaktor bleibt umstritten. Cytochrom c -Nitritreduktase (ccNiR) ˚ Abb. 1: a) Struktur der N2O-Reduktase aus Paracoccus denitrificans, b) CuA und CuZ. ˚ Abb. 2: a) Struktur des Nitrogenase α2β2-Tetramers (grün-blau) im Komplex mit der Reduktase (rosa) aus Azotobacter vinelandii, b) Eisen-Molybdän-Cofaktor, c) P-Cluster. Im biogeochemischen Kreislauf des Stickstoffs nimmt Nitrit (NO2–) eine zentrale Position ein. Je nach Organismus besitzen denitrifizierende Bakterien eine Eisen- oder Kupfer-Nitritreduktase, die Nitrit einelektronisch zu Stickstoffmonoxid (NO) umsetzt. Alternativ wird Nitrit in einem Sechs-Elektronenschritt durch die Cytochrom c-Nitritreduktase (ccNiR) zu Ammoniak reduziert: NO2– + 8H+ + 6e– → NH4+ + 2H2O (Eo⬘ = + 0,340 V). Mögliche Zwischenprodukte der dissimilatorischen Nitratreduktion zu Ammoniak, NO, N2O und Hydroxylamin (NH2OH) werden von ccNiR nicht freigesetzt, sind aber alternative Substrate[15]. Das Enzym ist ein Homodimer, mit fünf über Thioetherbrücken mit dem Protein verknüpfte Hämzentren pro Monomer (Abb. 3)[16]. Die dichte Packung der Hämzentren erlaubt einen effizienten Elektronentransfer und trägt zur hohen katalytischen Aktivität der ccNiR bei. Bisher einmalig ist auch das Aktivzentrum der ccNiR, bei dem im klassischen Häm-Bindemotiv Cys-X-X-CysHis das Histidin durch Lysin ersetzt wird. Die ε-Aminogruppe des Lysins bildet den proximalen Liganden des Eisens; die in trans-Position liegende sechste Koordinationsstelle steht dem Substrat Nitrit zur Verfügung. Die Katalyse wird durch die heterolytische Spaltung einer N-O-Bindung eingeleitet, erleichtert BIOspektrum | 04.06 | 12. Jahrgang ˚ Abb. 3: a) Struktur der Cytochrom c-Nitritreduktase aus Wolinella succinogenes, b) Nitrit koordiniert am Aktivzentrum. durch die Rückbindungswechselwirkung des Nitritstickstoffs mit dem Fe(II)-Hämzentrum. Einen wichtigen Beitrag zur Aktivierung von Nitrit liefert ein Netzwerk von Wasserstoffbrücken, welches die Symmetrie des Nitritmoleküls aufhebt und so eine N-O-Bindung stärkt und die andere schwächt[15]. Das katalytische Hämzentrum ist mit der Proteinoberfläche durch einen Kanal mit einem positiven elektrostatischen Oberflächenpotenzial verbunden, während ein zweiter Kanal mit im wesentlichen negativen elektrostatischen Potenzial auf der gegenüberliegenden Seite hin zur Proteinoberfläche führt. Diese Anordnung gewährleistet einen gerichteten Reaktionsfluss: Aufnahme des Nitritanions und Abgabe des kationischen Produkts NH4+ sind räumlich getrennt, was eine Produktinhibition verhindert und somit zur Optimierung des Enzyms beiträgt[16]. Transhydroxylase (TH) Trihydroxybenzole und ihre Derivate sind weit verbreitete Bausteine aromatischer Naturstoffe und werden durch anaerobe Mikroorganismen abgebaut. Zellextrakte von Pelobacter acidigallici setzen Pyrogallol stöchiometrisch zu Phloroglucin um mithilfe des Cosubstrats 1,2,3,5-Tetrahydroxybenzol. Diese auf dem Papier trivial anmutende Reaktion wird katalysiert von dem Molybdänenzym Transhydroxylase (TH), ein Schlüssel- 333_359_BIOsp_0406.qxd 348 02.06.2006 10:12 Uhr Seite 348 WISSENSCHAFT ˘ Abb. 4: a) Struk- tur der Transhydroxylase aus Pelobacter acidigallici, b) Mo(MGD)2 und [4Fe-4S]-Cluster, c) Transhydroxylierungsreaktion. enzym des fermentativen Abbauwegs von Gallussäure[12]. TH ist ein αβ-Heterodimer und gehört aufgrund der Aminosäuresequenz zur Dimethylsulfoxidreduktase-Familie. Die große α-Untereinheit trägt das Katalysezentrum Mo(MGD)2, wobei das Metall an zwei Moleküle Molybdopterin-Guanin-Dinukleotid über vier Dithiolenschwefelatome koordiniert ist. In der kleineren β-Untereinheit sind drei [4Fe4S]-Cluster verankert, deren Rolle bei der Transhydroxylierungsreaktion noch nicht verstanden ist (Abb. 4). Transhydroxylierungen, bei denen eine Hydroxylgruppe intermolekular zwischen zwei organischen Substraten transferiert wird, sind ein neuartiger Reaktionstyp. Gemäß einem Vorschlag von Janos Rétey (Universität Karlsruhe) wird intermediär ein Diphenylether in der Koordinationssphäre des Molybdäns gebildet. Röntgenstrukturen der TH im Komplex mit Pyrogallol und dem Inhibitor 1,2,4-Trihydroxybenzol begünstigen den Rétey’schen Mechanismus, allerdings bedarf es noch weiterer kinetischer Studien. Molybdän ist eher ein Redox-passiver Zuschauer und leistet nicht wie gewohnt die Sauerstofftransferchemie. Für die wichtige Rolle des Cosubstrats spricht der Befund aus Experimenten mit 18O-markiertem H2O, dass keine der transferierten Hydroxylgruppen dem Wasser entstammt[12]. Bakterien und Biomimetische Chemie Das chemische Leistungsvermögen von Bakterien einschließlich ihrer Anpassungsfähigkeit an extreme Standorte und eine sich rasch verändernde Umwelt muss gesehen werden im Hinblick auf die Evolution des Lebens auf unserer Erde. Packt man das gesamte Geschehen in einen 24-Stunden-Tag, so begannen die Bakterien schon morgens um 5 Uhr ihre Arbeit, der Mensch hingegen erst kurz vor Mitternacht. Er lernt dennoch rasch von der Natur und stellt sich der Herausforderung. Die biomimetische Chemie boomt, und es werden spektakuläre Erfolge erzielt bei der Synthese komplexer organischer Naturstoffe und Metall-abhängiger Katalysatoren. Ein Paradebeispiel ist die Synthese eines Modells des H-Clusters der Eisen-abhängigen Hydrogenase, welche Wasserstoff (H2) sowohl spalten als auch bilden kann. Die Synthese dieses biomimetischen Eisenkomplexes ist ein großer Schritt voran auf dem Weg zu einer effizienten Wasserstoff-Brennzelle, die ohne das teure Platin auskommen könnte[17]. Freuen wir uns also auf weitere Höchstleistungen der Bakterien und sind gespannt, wie diese unser chemisches Wissen und Können voranbringen werden. Danksagung Wir danken Robert Huber, Albrecht Messerschmidt, Norbert Pfennig, Bernhard Schink und Walter Zumft für die Einführung in die faszinierende Welt der Bakterien und Proteinstrukturen, ferner der Deutschen Forschungsgemeinschaft und der Volkswagenstiftung für finanzielle Unterstützung. Literatur [1] Zitat Leslie D. Pettit anlässlich des X. Internationalen Symposiums über Bioanorganische Chemie, Szklarska Poreba, Polen (2005). [2] Groß, M.: Exzentriker des Lebens- Zellen zwischen Hitzeschock und Kältestreß. Spektrum Akademischer Verlag, Heidelberg, Berlin, Oxford, 1997. [3] Brune, A. (2005): Ernährung auf dem Holzweg. Max Planck Forschung 2: 52–57. [4] Falb, M., Pfeiffer, F., Palm, P., Rodewald, K., Hickmannn, V., Tittor, J., Oesterhelt, D. (2005): Living with two extremes: Conclusions from the genome sequence of Natronomonas pharaonis. Genome Research 15: 1336–1343. [5] Beatty, J. T., Overmann, J., Lince, M. T., Manske, A. K., Lang, A. S., Blankenship, R. E., Van Dover, C. L., Martinson, T. A., Plumley, F. G. (2005): An obligately photosynthetic bacterial anaerobe from a deep-sea hydrothermal vent. Proc. Natl. Acad. Sci. (USA) 102: 9306–9310. [6] Kroneck, P. M. H. (2005): The Biogeochemical cycles and the evolution of life. In: Sigel, A., Sigel, H., Sigel, R. K. O. (Hrsg.) Metal Ions in Biological Systems. Marcel Dekker, Basel, 43: 1–7. [7] Oremland, R. S., Stolz, J. F. (2003): The Ecology of Arsenic. Science 300: 939–944. [8] Butler, A. (1998): Acquisition and Utilization of Transition Metal Ions by Marine Organisms. Science 281: 207–210. [9] Zumft, W. G., Kroneck, P. M. H. (2006): Respiratory Transformation of Nitrous Oxide (N2O) to Dinitrogen by Bacteria and Archaea. Adv. Microb. Physiol., in press. [10] Rees, D. C., Tezcan, F. A., Haynes, C. A., Walton, M. Y., Andrade, S. L., Einsle, O., Howard, J. B. (2005): Structural Basis of Nitrogen Fixation. Phil. Trans. R. Soc. A 363: 971– 984. [11] Boll, M., Fuchs, G. (2005): Unusual reactions in anaerobic metabolism of phenolic compounds. Biol. Chem. 386: 989–997. [12] Boll, M., Schink, B., Messerschmidt, A., Kroneck, P. M. H. (2005): Novel bacterial molybdopterin- and tungstopterindependent enzymes: three-dimensional structure, spectroscopy, and reaction mechanism. Biol. Chem. 386: 999–106. [13] Beinert, H. (2000): A tribute to sulfur. Eur. J. Biochem. 267: 5657–5664. [14] Einsle, O., Tezcan, F.A., Andrade, S. L. A., Schmid, B., Yoshida, M., Howard, B. J., Rees, D. C. (2002): Nitrogenase MoFe-Protein at 1.16 Å Resolution: A Central Ligand in the FeMo-Cofactor. Science 297: 1696–1700. [15] Einsle, O., Messerschmidt, A., Huber, R., Kroneck, P. M. H., Neese, F. (2002): Mechanism of the six-electron reduction of nitrite to ammonia by cytochrome c nitrite reductase. J. Am. Chem. Soc. 124: 11737–11745. [16] Einsle, O., Messerschmidt, A., Stach, P., Bourenkov, G. P., Bartunik, H. D., Huber, R., Kroneck, P. M. H. (1999): Structure of cytochrome c nitrite reductase. Nature 400: 476– 480. [17] Tard, C., Liu, X., Ibrahim, S.K., Bruschi, M., De Giola, L., Davies, S.C., Yang, X., Wang, L.S., Sawers, G., Pickett, C.J. (2005): Synthesis of the H-cluster framework of iron-only hydrogenase. Nature 433: 610–613. Korrespondenzadressen: Prof. Dr. Oliver Einsle Institut für Mikrobiologie und Genetik Abt. Molekulare Strukturbiologie Georg-August-Universität Göttingen Justus-von-Liebig-Weg 11 D-37077 Göttingen Tel.: 0551-3914189 Fax: 0551-3914082 [email protected]. Prof. Dr. Peter M. H. Kroneck Fachbereich Biologie Universitätsstr. 10 D-78457 Konstanz Tel.: 07531-882103 Fax: 07531-882966 [email protected] BIOspektrum | 04.06 | 12. Jahrgang 333_359_BIOsp_0406.qxd 02.06.2006 10:12 Uhr AUTOREN Oliver Einsle 1991–1996 Studium der Biologie in Konstanz; 2000 Dissertation an der Universität Konstanz (Peter M. H. Kroneck und Robert Huber, MPI für Biochemie, Martinsried); 2001–2003 Postdoc am California Institute of Technology in Pasadena (Douglas C. Rees); seit 2003 Juniorprofessor an der Universität Göttingen Grazyna B. Seiffert 1998–2003 Studium der Chemie in Wroclaw; seit Oktober 2003 Doktorandin im Labor für Bioanorganische Chemie (Arbeitsgruppe Kroneck) an der Universität Konstanz Martha E. Sosa-Torres 1972–1980 Studium der Chemie in Mexico City; 1983 Dissertation am University College, London (Martin L. Tobe); 1992–1993 Marie-Curie Fellow am King’s College, London (Martin Hughes); seit 1984 Professorin für Anorganische Chemie an der Universidad Nacional Autónoma de México, Mexico City; seit Juli 2005 Sabbatical Jahr an der Universität Konstanz Peter M. H. Kroneck 1962–1967 Studium der Chemie in Basel; 1971 Dissertation an der Universität Konstanz (Peter Hemmerich); 1971–1973 Postdoc an der Utah State University in Logan (Jack. T. Spence); 1980 Habilitation an der Universität Konstanz; seit 1987 Professor für Biochemie, Universität Konstanz; 2002 European Biological Inorganic Chemistry (EUROBIC) Medaille BIOspektrum | 04.06 | 12. Jahrgang Seite 349