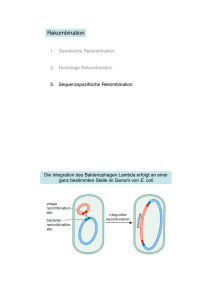
Mutagenese des nuo-Operons von Escherichia coli
Werbung

Mutagenese des nuo-Operons von Escherichia coli INAUGURALDISSERTATION zur Erlangung der Doktorwürde der Fakultät für Chemie, Pharmazie und Geowissenschaften der Albert-Ludwigs-Universität Freiburg im Breisgau vorgelegt von Diplom-Biologin Mareike Uhlmann Freiburg im Breisgau, April 2005 Vorsitzender des Promotionsausschusses: Prof. Dr. Georg E. Schulz Referent: Prof. Dr. Thorsten Friedrich Korreferent: Prof. Dr. Peter Gräber Tag der Bekanntgabe des Prüfungsergebnisses: 07.Juli 2005 Wesentliche Teile dieser Arbeit wurden in folgendem Artikel veröffentlicht: Mareike Uhlmann und Thorsten Friedrich (2005) The EPR signals assigned to Fe/S cluster N1c of the Escherichia coli NADH:ubiquinone oxidoreductase (complex I) derive from cluster N1a. Biochemistry 44: 1653-1658 Das Schönste, was wir entdecken können, ist das Geheimnisvolle. Albert Einstein Inhaltsverzeichnis T 1 Zusammenfassung 4 2 Einleitung 6 2.1 Das aerobe Elektronentransportsystem von Escherichia coli 6 2.2 Die NADH:Ubichinon Oxidoreduktase 7 2.3 Die NADH:Ubichinon Oxidoreduktase aus E. coli 9 2.3.1 Das nuo-Operon 9 2.3.2 Der isolierte Komplex I 11 2.4 Mutagenese im nuo-Operon 15 2.4.1 Die extrachromosomale Klonierung 15 2.4.2 λ-Red vermittelte Rekombination in E. coli 17 2.4.3 Selektion auf Rekombination 19 2.5 Thema der Arbeit 21 3 Material und Methoden 22 3.1 Medien und Lösungen 22 3.2 Mikroorganismen, Vektoren und Oligonukleotide 25 3.3 Mikrobiologische Methoden 28 3.3.1 Anzucht von E. coli 28 3.3.2 Herstellung von Dauerkulturen 29 3.3.3 Herstellung kompetenter Zellen 29 3.4 Molekularbiologische Methoden 30 3.4.1 Analytische Plasmidisolierung mittels „Boiling lysis“ 30 3.4.2 Analytische Plasmidisolierung mittels alkalischer Lyse 30 3.4.3 Präparative Plasmidisolierung 31 3.4.4 Präparation genomischer DNA 31 3.4.5 Restriktion und Modifikation von DNA 31 3.4.6 Agarosegelelektrophorese 32 -1- 3.4.7 Isolation und Aufreinigung von DNA-Fragmenten 32 3.4.8 Polymerase-Kettenreaktion 33 3.4.9 Einführung von Punktmutationen mittels ortsgerichteter Mutagenese 34 3.4.10 Weitere molekularbiologische Methoden 34 3.5 Proteinchemische Methoden 34 3.5.1 Isolierung der Plasmamembranen 34 3.5.2 Isolierung des Komplex I aus dem Stamm ANN0221/pBADnuo 35 3.5.3 Isolierung des Komplex I über Affinitäts-Chromatographie an StrepTactin 36 3.5.4 Aufreinigung des NADH-Dehydrogenase Fragmentes über eine AffinitätsChromatographie an StrepTactin 37 3.5.5 Verschiedene analytische Methoden 37 4 Ergebnisse 42 4.1 Klonierung des extrachromosomalen Expressionssystems pBADnuo 42 4.2 Produktion von Komplex I vom Plasmid pBADnuo 47 4.2.1 Wachstumskurven von nuo/ndh-Doppeldeletionsstämmen mit pBADnuo 47 4.2.2 Atmungsaktivität des E. coli Stammes ANN0221/pBADnuo 51 4.2.3 Assemblierung des Komplex I in ANN0221/pBADnuo 53 4.2.4 Isolierung des Komplex I aus dem Stamm ANN0221/pBADnuo 54 4.3 Das Subklonsystem des Plasmids pBADnuo 56 4.3.1 Vektor für die Subklonierung des Plasmids pBADnuo 58 4.3.2 Klonierung der pBADnuo-Subklone und der kleinen Subklone 61 4.4 Einführung einer StrepTag II-Sequenz in pBADnuo 63 4.4.1 Modifizierung von NuoB 63 4.4.2 Modifizierung von NuoF 68 4.4.3 Modifizierung von NuoCD 71 4.5 λ-Red vermittelte Rekombination zur Mutagenese im nuo-Operon 80 4.5.1 Deletionsmutanten in pBADnuo 80 4.5.2 Punktmutationen in nuoM 92 4.6 Mutagenese an einem Fe/S-Bindemotiv auf NuoG 102 4.6.1 ESR-spektroskopische Charakterisierung des NADH-Dehydrogenase Fragmentes im Cytoplasma 105 Isolierung des NADH-Dehydrogenase Fragmentes aus den NuoG-Mutanten 109 4.6.2 -2- 4.6.3 ESR-spektroskopische Charakterisierung des aufgereinigten NADHDehydrogenase Fragmentes 111 4.6.4 ESR-spektroskopische Charakterisierung des NADH-Dehydrogenase Fragmentes in aerob und anaerob gezogenen Zellen 113 5 Diskussion 115 5.1 Das extrachromosomale Expressionssystem pBADnuo 115 5.2 Nutzung des Plasmids pBADnuo zur Mutagenese 117 5.3 Vergleich der λ-Red vermittelten Rekombination mit der Klonierung 118 5.4 Kombination von Klonierung und Rekombination 121 5.5 Isolierung des Komplex I über eine Affinitäts-Chromatographie 122 5.6 Das zusätzliche Fe/S-Bindemotiv auf NuoG 123 6 Literatur 127 -3- Zusammenfassung 1. Zusammenfassung Die protonenpumpende NADH:Ubichinon Oxidoreduktase, die auch als respiratorischer Komplex I bezeichnet wird, koppelt den Transfer von zwei Elektronen von NADH auf ein Chinon mit der Translokation von vier Protonen über die Membran. Der Elektronenweg in dem Enzymkomplex verläuft über ein Flavin Mononukleotid und bis zu neun Eisen-Schwefel (Fe/S)-Zentren. Da die Struktur des Komplexes auf molekularer Ebene bislang nicht bekannt ist, ist die Elektronenübertragung und ihre Kopplung mit der Protonentranslokation noch nicht verstanden. Der Komplex I setzt sich modular aus einem NADH-Dehydrogenase Fragment, einem verbindenden Fragment und einem Membranfragment zusammen. Der Komplex I aus dem Bakterium Escherichia coli besteht aus 13 verschiedenen Untereinheiten, NuoA bis NuoN, die von den Genen des 15 kbp großen nuo-Operons (von NADH:Ubichinon Oxidoreduktase) kodiert werden. Die Gene nuoC und nuoD sind in E. coli zu einem Gen fusioniert. In dieser Arbeit sollte ein System entwickelt werden, mit dem beliebige Manipulationen am nuo-Operon vorgenommen werden können. In einem ersten Schritt sollte das Operon in einen Expressionvektor kloniert werden, um später große Mengen des mutierten Komplexes zu erhalten. Dann sollten Subklone des Operons erstellt werden, die in einer PCR-basierten Mutagenese eingesetzt werden sollten. Die Subklone sollten das gesamte nuo-Operon abdecken und nach Mutation in den Expressionsvektor rückkloniert werden können. Dieses System sollte eingesetzt werden, um den Komplex mit Affinitätspeptiden für eine schnelle Aufreinigung zu modifizieren, um die Assemblierung des Komplexes durch Deletion einzelner Gene zu untersuchen und um Bindestellen für Kofaktoren durch stellengerichtete Mutagenese zu charakterisieren. Die Gene nuoA und nuoB wurden aus dem Chromosom mit PCR-Methoden amplifiziert und in ein Plasmid ligiert, so dass vor nuoA eine neue Restriktionsschnittstelle für einen Promotor geschaffen wurde. Das nuo-Operon wurde aus dem Chromosom geschnitten und in ein Cosmid ligiert. Aus dem Cosmid wurden die Gene nuoCD bis nuoG herausgeschnitten und in das Plasmid hinter nuoB eingefügt. Aus dem Expressionsvektor pBAD33 wurde der Arabinose-Promotor ausgeschnitten und in die neu geschaffene Schnittstelle vor nuoA kloniert. Die restlichen Gene des nuo-Operons wurden sukzessiv aus dem Cosmid eingefügt. Dieser Expressionsvektor wurde pBADnuo -4- genannt. Von pBADnuo wurde ein Zusammenfassung vollassemblierter und enzymatisch aktiver Komplex I produziert. Der Gehalt in der Membran war etwa drei- bis vierfach höher als in dem Ausgangsstamm. Dies ist das erste Beispiel für eine extrachromosomale Produktion von Komplex I. Um die nuo-Gene des Plasmids pBADnuo einer Mutagenese zugänglich zu machen, wurden Subklone der Gene nuoA bis nuoL sowie für einen Teil von nuoM (nuoM´) erhalten. Eine direkte Rückklonierung in pBADnuo war aber nur für die Gene nuoA bis nuoE´ möglich, da die benötigten Zwischenvektoren nicht hergestellt werden konnten. Durch Etablierung der λ−Red vermittelten Rekombination war es jedoch möglich, auch Mutationen aus nuoE bis nuoM´ in pBADnuo einzufügen. Mit diesem System wurden die Untereinheiten NuoB, NuoC und NuoF jeweils C-terminal mit einem StrepTag II-Peptid modifiziert. Die Modifizierung an NuoF verhinderte die Assemblierung des Komplex I. Die Modifizierung an NuoB führte zu einem fragmentierten Komplex I, dem die Untereinheiten NuoA, NuoJ und NuoK sowie die Fe/S-Zentren N2 und N4 fehlten. Die StrepTag II-Sequenz wurde so in das Gen nuoCD eingeführt, dass ein Stoppkodon für nuoC sowie ein neues Startkodon für nuoD entstand. Der so modifizierte Komplex wurde über eine Affinitäts-Chromatographie mit äußerst geringen Ausbeuten präpariert. Durch die Deletion der Gene nuoCD, nuoM, nuoG und nuoE bis F von dem Plasmid pBADnuo wurde Komplex I nicht mehr assembliert. Allerdings war im Cytosol des Stammes BL21(DE3)/pBADnuo/∆nuoCD das assemblierte NADH-Dehydrogenase Fragment nachzuweisen. Die übrigen Mutanten wurden bislang nicht auf das Vorhandensein dieses NADH-Dehydrogenase Fragmentes untersucht. Die Untereinheit NuoG trägt ein zusätzliches Bindemotiv für ein Fe/S-Zentrum. Da in der Präparation des Komplexes ein neues ESR-Signal gefunden wurde, wurde dieses dem Motiv auf NuoG zugeordnet und als „N1c“ bezeichnet. Mittels ortsgerichteter Mutagenese wurden die Cysteine des Bindemotivs gegen Alanine ausgetauscht. Die ESR-spektroskopische Charakterisierung der mutierten Proteine zeigte, dass das ESR-Signal von dem zweikernigen Zentrum N1a auf NuoE stammt. Da die Mutationen die Assemblierung störten, bindet das zusätzliche Bindemotiv wahrscheinlich ein bislang nicht nachgewiesenes Fe/S-Zentrum, das als N7 bezeichnet wird. -5- Einleitung 2. Einleitung 2.1 Das aerobe Elektronentransportsystem von Escherichia coli Das Gram-negative, fakultativ anaerobe Enterobakterium Escherichia coli, das im Darm von Säugetieren lebt, deckt Elektronentransportsysteme. seinen Diese Energiebedarf bestehen aus über aerobe und anaerobe membrangebundenen primären Dehydrogenasen und terminalen Reduktasen bzw. Oxidasen, die bei aerobem Wachstum durch Ubichinon und bei anaeroben Wachstumsbedingungen durch Menachinon oder im Fall der Nitrat-Atmung durch Dimethylmenachinon miteinander verbunden sind (Unden, 1988; Wissenbach et al., 1990). Die von diesen Komplexen katalysierten Redoxreaktionen sind mit der Bildung eines elektrochemischen Protonengradienten gekoppelt, der für die ATPSynthese und andere energieverbrauchende Prozesse genutzt wird (Mitchell, 1961; Ingledew und Poole, 1984; Neijssel und Teixeira de Mattos, 1994; Rich, 2003). Das aerobe Elektronentransportsystem von E. coli umfasst im Wesentlichen drei energiewandelnde Enzymkomplexe, die NADH:Ubichinon Oxidoreduktase (NADH- Dehydrogenase Typ I; NDH-I) und zwei terminale Cytochrom-Oxidasen bo und bd, sowie eine alternative, nicht-energiewandelnde NADH-Dehydrogenase (NADH-Dehydrogenase Typ II; NDH-II) (Yagi et al., 1998; Abb. 2-1). Die Cytochrom bd-Oxidase besteht aus zwei Polypeptidketten und besitzt zwei Hämgruppen vom b-Typ und eine vom d-Typ. Sie transloziert pro Elektron ein Proton über die Membran (Puustinen und Wikström, 1999). Die aus vier Polypeptidketten bestehende Cytochrom bo3-Oxidase enthält je eine Hämgruppe vom b-Typ und vom o-Typ, sowie zwei Kupferionen. Dieser Komplex transloziert zwei Protonen pro übertragenem Elektron über die Membran, so dass die Energiekonservierung doppelt so hoch wie bei der Cytochrom bd-Oxidase ist (Anraku und Gennis, 1987; Puustinen et al., 1991). Die Cytochrom bo3-Oxidase wird unter aeroben Bedingungen, die Cytochrom bdOxidase unter mikroaeroben Bedingungen exprimiert (Cotter et al., 1990; Iuchi et al., 1990). -6- Einleitung Abb. 2-1: Schematische Darstellung des aeroben Elektonentransportsystems in E. coli Die energiewandelnden Enzymkomplexe mit ihrer Energiebilanz sind grau dargestellt. NDH-I bezeichnet die NADH:Ubichinon Oxidoreduktase, NDH-II die alternative NADHDehydrogenase, bo3-Komplex die Cytochrom bo3-Oxidase und bd-Komplex die Cytochrom bd-Oxidase. Die Pfeile zeigen die Richtung des Elektronenflusses, Q steht für den Elektronenüberträger Ubichinon. Beide NADH-Dehydrogenasen übertragen Elektronen von NADH auf Ubichinon. Im Gegensatz zu der NDH-I ist die Reaktion der NDH-II nicht mit einer Protonentranslokation gekoppelt. Die NDH-II weist eine zehnfach geringere Affinität zum natürlichen Substrat NADH und eine etwa tausendfach geringere Affinität zum künstlichen Substrat Desamino (d)-NADH auf. Dies wird zur Unterscheidung der beiden Dehydrogenasen in enzymatischen Tests genutzt (Matsushita et al., 1987). So stammt die NADH-Oxidaseaktivität von beiden Enzymen, während die d-NADH-Oxidaseaktivität nahezu vollständig von der NDH-I stammt. Der Besitz verschiedener NADH-Dehydrogenasen und terminaler Oxidasen ermöglicht E. coli, sich an verschiedene Wachstumsbedingungen anzupassen. 2.2 Die NADH:Ubichinon Oxidoreduktase Die protonenpumpende NADH:Ubichinon Oxidoreduktase, die auch als respiratorischer Komplex I oder NDH-I bezeichnet wird, koppelt den Transfer von zwei Elektronen des NADH auf ein Chinon mit der Translokation von vier Protonen über die Membran nach -7- Einleitung folgender Reaktionsgleichung (Walker, 1992; Friedrich, 2001; Magnitsky et al., 2002; Brandt et al., 2003; Yagi et al., 2003): NADH + Q + 5H+n NAD+ + QH2 + 4H+p P PB B P P B B P PB B Hierbei steht Q für Ubichinon und H+n und H+p für Protonen auf der negativen inneren bzw. P PB B P PB B positiven äußeren Seite der Membran. Durch diese Protonentranslokation trägt die NADH:Ubichinon Oxidoreduktase zum Aufbau der protonenmotorischen Kraft bei, die essentiell für energieverbrauchende Prozesse ist. Homologe Formen des Komplexes kommen in der Cytoplasmamembran vieler Bakterien und in der inneren Mitochondrienmembran der meisten Eukaryonten vor. Während der mitochondriale Komplex I von Eukaryonten aus bis zu 46 Untereinheiten aufgebaut ist (Carroll et al., 2002), besteht der bakterielle Komplex I im Allgemeinen aus 14 verschiedenen Untereinheiten, die homolog zu Untereinheiten des mitochondrialen Komplexes sind. Sieben dieser 14 Untereinheiten sind periphere Proteine, darunter alle Untereinheiten, die die bislang nachgewiesenen Redoxgruppen des Komplexes, ein Flavin Mononukleotid (FMN) und bis zu neun Eisen-Schwefel (Fe/S)-Zentren beinhalten. Bei den restlichen sieben Untereinheiten handelt es sich um hydrophobe Proteine, die laut Sekundärstrukturvorhersage mit 54 α-Helices in der Membran verankert und in bislang unbekannter Weise an der Protonentranslokation beteiligt sind (Weiss et al., 1991; Walker, 1992; Friedrich et al., 1995; Brandt, 1997). UV/vis-spektroskopische Untersuchungen des Komplexes ergaben Hinweise auf die Existenz eines weiteren Kofaktors in den membranständigen Untereinheiten, dessen chemische Struktur noch unbekannt ist (Friedrich, 1998; Schulte et al., 1998 und 1999; Friedrich et al., 2000). Der mitochondriale Komplex I besitzt eine molekulare Masse von nahezu 1 MDa, während die des bakteriellen Komplexes etwa 550 kDa beträgt. Von allen Atmungskettenkomplexen ist der Komplex I aufgrund seiner Größe und komplizierten Zusammensetzung der Einzige, dessen Struktur noch nicht auf molekularer Ebene gelöst wurde. Bisherige Strukturuntersuchungen konzentrierten sich auf elektronenmikroskopische Aufnahmen von Komplex I-Einzelpartikeln und zweidimensionalen Membran-Kristallen des Komplexes aus Rinderherz (Sazanov und Walker, 2000; Grigorieff 1998), dem filamentösen Pilz Neurospora -8- Einleitung crassa (Leonard et al., 1987; Hofhaus et al., 1991; Guenebaut et al., 1997 und 1998a), der aeroben Hefe Yarrowia lipolytica (Djafarzadeh et al., 2000) und den Bakterien E. coli (Guenebaut et al., 1998b) und Aquifex aeolicus (Peng et al., 2003). Die Untersuchungen an dem Komplex aus verschiedenen Organismen zeigten eine L-förmige Struktur mit einem Membranarm und einem peripheren Arm, der in die mitochondriale Matrix bzw. das bakterielle Cytoplasma ragt. Der isolierte E. coli Komplex I kann je nach Ionenstärke des Mediums zwei unterschiedliche Konformationen annehmen: In Puffern hoher Ionenstärke bildet er die oben beschriebene L-förmige Struktur, während in Puffern niedriger Ionenstärke eine so genannte Hufeisen-Form vorliegt (Böttcher et al., 2002; Abb. 2-2). Wie mittels analytischer Ultrazentrifugation gezeigt wurde, gehen beide Formen reversibel ineinander über. In der Hufeisen-Form, von der man auf Grund ihrer enzymatischen Aktivität annimmt, dass sie den nativen Zustand des Komplexes darstellt, sind peripherer Arm und Membranarm parallel zueinander angeordnet, in der L-Form senkrecht zueinander. Abb. 2-2: Schematische Darstellung der Konformationen des Komplex I aus E. coli. (a) zeigt die L-Form, die bei hohen Ionenstärken vorliegt und (b) die Hufeisenform, die bei niedrigen Ionenstärken vorliegt. Die Modelle wurden aus elektronenmikroskopischen Aufnahmen an Einzelpartikeln berechnet (Böttcher et al., 2002). 2.3 Die NADH:Ubichinon Oxidoreduktase aus E. coli 2.3.1 Das nuo-Operon Der Komplex I aus E. coli besteht aus 13 verschiedenen Untereinheiten. Die für diese Untereinheiten kodierenden Gene sind im sogenannten nuo-Operon organisiert (von -9- Einleitung NADH:Ubichinon Oxidoreduktase) und werden mit nuoA-N bezeichnet (Weidner et al., 1993; Abb. 2-3). Das Operon hat eine Größe von 15 kbp und befindet sich auf min 51 des E. coli Chromosoms (Weidner et al., 1993). In E. coli liegen die Gene nuoC und nuoD fusioniert vor (Braun et al., 1998; Friedrich, 1998). Diese Fusion findet sich ebenfalls in Buchnera aphidicola, Yersinia pestis, Pseudomonas aeruginosa und Salmonella typhimurium (Clarke et al., 1997; http://www.scripps.edu/mem/biochem/CI/images/operons.gif). Die Homologen von nuoC und D sind in der Formiat-Hydrogenlyase von E. coli ebenfalls zu einem Gen hycE fusioniert, welches für die große Untereinheit der Hydrogenase kodiert (Sauter et al., 1992; Videira und Azevedo, 1994; Friedrich und Weiss, 1997). Die Anordnung der Gene im nuoOperon ist konserviert und findet sich ebenfalls in den homologen Gen-Loci von Paracoccus denitrificans, Rhodobacter capsulatus, Thermus thermophilus und Synechocystis spec. (Friedrich et al., 1995; Friedrich und Weiss, 1997). Die Promotorregion des nuo-Operons besteht aus einer 560 bp langen stromaufwärts gelegenen Region. Die Expression des Operons wird durch O2 und Nitrat über die Regulatoren ArcA, NarL, FNR und andere Faktoren, welche C4-Dicarboyxlate beeinhalten, reguliert (Bongaerts et al., 1995). Bei einem anaeroben Wachstum reprimiert ArcA die Expression der nuo-Gene, durch Nitrat wird sie über NarL stimuliert. FNR reprimiert die Expression der alternativen NADH-Dehydrogenase (Bongaerts et al., 1995). Abb. 2-3: Modell des nuo-Operons. Die Gene, die für die peripheren Proteine kodieren, sind blau dargestellt, die die für die membranständigen Gene kodieren gelb. Das nuo-Operon ist schematisch mit den aus der Primärstruktur vorhergesagten Bindemotiven für Redoxgruppen und Substrate gezeigt. Der E. coli Komplex I wird aus der Cytoplasmamembran durch Detergenzextraktion und zwei Ionenaustausch-Chromatographien sowie einer Größenausschluss-Chromatographie bei pH 6 isoliert (Leif, 1995; Spehr et al., 1999; Stolpe, 2003). Je mehr Verunreinigungen die - 10 - Einleitung Präparation enthält, desto mehr dieser Schritte sind nötig, wobei sich bei jeder weiteren Aufreinigung die Ausbeute verringert. Die Ausbeute der bisherigen Präparation beträgt 2-5 % (Spehr et al., 1999; Stolpe, 2003). Um eine möglichst hohe Ausbeute und große Reinheit der Präparation zu erreichen, ist die Affinitäts-Chromatographie durch kleine Peptide (Tags) die Methode der Wahl (Bailon et al., 2000). Die beiden gebräuchlichsten Systeme sind der HisTag und der StrepTag. Im Fall des StrepTags wird gewöhnlich das StrepTag II-System verwendet (Voss und Skerra, 1997), das die natürliche Bindung zwischen Avidin und Biotin nachahmt, die eine der höchsten Bindungskonstanten in der Natur aufweist. Avidin ist neben Lysozym und Conalbumin ein Bestandteil des antibakteriellen Systems des Hühnereis, wobei die antibakterielle Wirkung darin besteht, der Umgebung Biotin zu entziehen, wobei bis zu vier Moleküle Biotin an das aus vier Untereinheiten bestehende Glykoprotein gebunden werden. Das StrepTag II-System nutzt das dem Avidin homologe bakterielle Streptavidin in Form des trägergebundenen Liganden StrepTactin. An das aufzureinigende Protein wird mit Hilfe molekularbiologischer Methoden der so genannte StrepTag angebracht, eine kurze Peptidsequenz von acht Aminosäuren, die die Eigenschaften des Biotins imitiert. Im Fall des His-Tags wird eine Sequenz von vier bis zehn Histidinen eingeführt, über die das Protein an eine Nickel-beladene Matrix gebunden wird. Gewöhnlich wird vor dem Tag eine kurze Sequenz von meist zwei weiteren Aminosäuren eingebracht, die als Bindeglied (Linker) bezeichnet wird und als Abstandshalter zwischen Tag und Protein dient, um eine Exposition des Tags sicherzustellen. Es wird berichtet, dass der Komplex I aus Y. lipolytica und aus T. thermophilus über einen His-Tag C-terminal zu NuoC effizient über eine AffinitätsChromatographie aufgereinigt wurde (Kashani-Poor et al., 2001; T. Ohnishi, persönliche Mitteilung). Aufgrund der Homologie der NADH:Ubichinon Oxidoreduktasen ist zu vermuten, dass auch der E. coli Komplex I nach einer Tag-Modifizierung dieser Untereinheit durch eine Affinitäts-Chromatographie aufzureinigen wäre. Allerdings liegt NuoC im E. coli Komplex I nicht als einzelne Untereinheit sondern fusioniert mit NuoD vor. 2.3.2 Der isolierte Komplex I Bei Anheben des pH-Wertes auf 7.5 und Detergenzwechsel zu Triton X-100 zerfällt der isolierte Komplex in drei Fragmente, das NADH-Dehydrogenase Fragment, das verbindende Fragment und das Membranfragment (Leif et al., 1995; Abb. 2-4). - 11 - Einleitung Abb. 2-4: Schematische Darstellung von Komplex I aus E. coli und seiner Fragmente, in die der Komplex I bei Anheben des pH-Wertes und Detergenzwechsel zu Triton X-100 zerfällt. Gelb dargestellt ist das NADH-Dehydrogenase Fragment, orange das verbindende Fragment und blau das Membranfragment. Die Elektronen fliessen vermutlich von NADH über das FMN und die Fe/S-Zentren N1a, N1b, N3, N4 und N5 des NADH-Dehydrogenase Fragmentes zu den Fe/S-Zentren N6a, N6b und N2 des verbindenden Fragmentes, wo die Reduktion des Ubichinons (Q) zu Ubichinol (QH2) stattfindet. Die vermutlich an diesen Prozess gekoppelten Protonentranslokationen sind mit gepunkteten Pfeilen dargestellt. Das lösliche NADH-Dehydrogenase Fragment ist der Elektroneneingang des Komplexes und besteht aus den Untereinheiten NuoE, F und G (Braun et al., 1998; Leif et al., 1995; Friedrich und Scheide, 2000). Es enthält die NADH-Bindestelle, das FMN und die ESRspektroskopisch nachweisbaren Fe/S-Zentren N1b, N1c, N3 und N4 (Sled et al., 1993; Leif et al., 1995; Braun et al., 1998; Friedrich, 1998; Bungert et al., 1999; Tab. 2-1) und weist eine Masse von 160 kDa auf. Ein weiteres konserviertes Motiv für ein zweikerniges Fe/S-Zentrum befindet sich auf NuoE (Yano et al., 1994; Braun et al., 1998). Dieses wurde dem Zentrum N1a zugeordnet. Das Zentrum N1a zeigt ein pH-abhängiges Mittenpotential im Bereich von -410 bis -370 mV (Ohnishi, 1998; Zu et al., 2002). Es wurde angenommen, dass das Mittenpotential des Zentrums N1a in E. coli so negativ ist, dass es in der NADH-reduzierten Probe oxidiert vorlag und somit nicht nachgewiesen wurde (Sled et al., 1993). - 12 - Einleitung Tab. 2-1: Lokalisation der in der Präparation von E. coli nachgewiesenen Fe/S-Zentren Fe/S-Zentrum N1a** N1b N1c** N2 N3 N4 N5* N6a/N6b N7*** Typ Untereinheit 2Fe/2S 2Fe/2S 2 Fe/2S 4Fe/4S 4Fe/4S 4Fe/4S 4Fe/4S 2x 4Fe/4S 4Fe/4S NuoE NuoG NuoG NuoB NuoF NuoG NuoG NuoI NuoG Fragment des Komplexes NADH-Dehydrogenase Fragment NADH-Dehydrogenase Fragment nicht existent Verbindendes Fragment NADH-Dehydrogenase Fragment NADH-Dehydrogenase Fragment NADH-Dehydrogenase Fragment Verbindendes Fragment NADH-Dehydrogenase Fragment * Wurde bislang in dem E. coli Komplex I nicht nachgewiesen. ** Siehe diese Arbeit. *** Bislang noch nicht nachgewiesen, vermutlich von einem zusätzlichen Bindemotiv auf NuoG ligiertes Fe/S-Zentrum, siehe diese Arbeit. Ein weiteres Bindemotiv für ein Fe/S-Zentrum befindet sich auf der Untereinheit NuoG (Abb. 2-5) Dieses zusätzliche Motiv wurde bislang außer bei E. coli nur bei Salmonella thyphimurium, T. thermophilus, A. aeolicus und wenigen anderen Bakterien gefunden (Finel, 1998; Scheide et al., 2002). E. A. T. P. R. N. H. coli aeolicus thermophilus denitrificans capsulatus crassa sapiens 221 * * * * 270 WDMQFAPSICQQCSIGCNISPGERYGELRRIENRY-NGTVNHYFLCDRGRF WLLEKGRTVCNLCPVGCEIQIEYGVGDWRSKRKVYRTKPTDELNICAKGFF WEMEETPTTCALCPVGCGITADTRSGELLRIRARE-VPEVNEIWICDAGRF WELTKTESIDVMDALGSSIRIDTKGREVMRILPRN-HDGVNEEWISDKTRF WELVKTESIDVMDALGSNIRIDAKGREVKRIVPRN-HDGVNEEWLSDKGRF WELKKTESIDVLDGLGSNIRVDTRGLEVMRILPRL-NDEVNEEWINDKTRF WETRKTESIDVMDAVGSNIVVSTRTGEVMRILPRM-HEDINEEWISDKTRF Abb. 2-5: Sequenzvergleich der NuoG-Homologen von E. coli, A. aeolicus, T. thermophilus, P. denitrificans, R. capsulatus, N. crassa und H. sapiens. Die Nummerierung bezieht sich auf die E. coli Sequenz. Die Cysteine des zusätzlichen Fe/S-Zentrum Bindemotivs sind rot gedruckt und ihre Position mit einem Stern markiert. - 13 - Einleitung ESR-Spektren der Präparation des E. coli Komplex I zeigen ein ESR-Signal eines zweikernigen Fe/S-Zentrums mit gx,y,z = 1.92, 1.94 und 2.00. Aufgrund seines Mittenpotentials von –240 mV, das keine pH-Abhängigkeit zeigt, wurde dieses Signal nicht dem Zentrum N1a, sondern einem neuartigen Zentrum, N1c, zugeordnet (Sled et al., 1993; Leif et al., 1995). Es wurde sowohl in den cytoplasmatischen Membranen von E. coli, dem isolierten Komplex I und dem isolierten NADH-Dehydrogenase Fragment nachgewiesen. Das Signal war in den Spektren der cytoplasmatischen Membran eines E. coli Komplex I Überproduzenten deutlich verstärkt (Spehr et al, 1999). Dieses Signal wurde daher dem zusätzlichen Bindemotiv auf NuoG zugeordnet (Sled et al., 1993; Leif et al., 1995; Braun et al., 1998; Bungert et al., 1999). In anderen Enzymen wie der E. coli Formiat Dehydrogenase H (Boyington et al., 1997), der periplasmatischen Nitrat Reduktase von Desulfovibrio desulfuricans (Dias et al., 1999) oder im Fall der Ferredoxin-Oxidoreduktasen von Pyrococcus furiosus (Hu et al., 1999; Chan et al., 1995) ligiert ein Bindemotiv mit ähnlichen Cystein-Abständen allerdings ein vierkerniges Fe/S-Zentrum. Es wurde diskutiert, ob das als N1c bezeichnete Fe/S-Zentrum möglicherweise zwischen einem zweikernigen und einem vierkernigen Zustand wechselt, je nachdem ob Sauerstoff vorhanden ist oder ein reduzierter Zustand des Zentrums vorliegt (Nakamaru-Ogiso et al., 2002). Eine solche Umwandlung eines [4Fe-4S]-Zentrums wurde z.B. für den Transkriptionsfaktor FNR und der Biotinsynthase gezeigt (Lazazzera et al., 1996; Bates et al., 2000, Khoroshilova et al., 1997; Kiley und Beinert, 1998; Duin et al., 1997; Ugulava et al., 2000; Ollagnier-De Choudens et al., 2000). Zusätzlich zu dem dem Fe/S-Zentrum N1c zugeordneten Bindemotiv befindet sich auf NuoG ein weiteres Bindemotiv für ein vierkerniges Fe/S-Zentrum. Bislang war umstritten, ob dieses als N5 bezeichnetes Zentrum einen tatsächlichen Kofaktor des Komplex I darstellt, da seine Konzentration in Präparationen des Komplexes sehr gering ist (Ohnishi, 1979 und 1998; Beinert und Albracht, 1982). N5 wurde in dem überproduzierten Homolog von NuoG aus P. denitrificans nachgeweisen (Yano et al., 1995; Ohnishi, 1998). Vermutlich ist es in stöchiometrischen Mengen in Komplex I enthalten, aber ein Großteil des N5 Spins könnte im S = 3/2 Grundzustand vorliegen, was zu einer Unterschätzung seiner Konzentration gemessen an den Spins in der g = 2 Region führen würde (Yano et al., 1995; Ohnishi, 1998). Das amphipathische, verbindende Fragment ist nur in detergenzhaltigen Puffern löslich und besteht aus den Untereinheiten NuoB, CD und I und hat eine molekulare Masse von 124 kDa - 14 - Einleitung (Leif et al., 1995; Rasmussen et al., 2001). Es verbindet das NADH-Dehydrogenase Fragment mit dem Membranfragment und enthält die Fe/S-Zentren N2, N6a und N6b (Rasmussen et al., 2001; Tab. 2-1). Das Membranfragment besteht aus den sieben hydrophoben Untereinheiten NuoA, NuoH und NuoJ-N mit einer molekularen Masse von 241 kDa. Es enthält weder ein konserviertes Motiv zur Bindung eines Kofaktors, noch wurde ein Kofaktor in dieser Untereinheit nachgewiesen. Es ist möglicherweise an der Chinonbindung und der Protonentranslokation beteiligt (Weiss et al., 1991; Walker, 1992; Friedrich et al., 1995; Brandt, 1997). Die Ubichinon-Bindestelle wurde bislang aufgrund der Hydrophobiezität des Substrates in dem Membranfragment vermutet. NuoH wurde durch Photoaffinitätsmarkierungen und durch Mutagenesestudien von menschlicher mitochondrialer DNA als Ubichinon- und Rotenon-bindendes Protein vorgeschlagen (Earley und Ragan, 1984; Earley et al., 1987; Degli Esposti et al., 1994; Majander et al., 1996; Lunardi et al., 1998). Kürzlich wurde postuliert, dass sich die Ubichinon-Bindestelle an der Kontaktfläche zwischen peripherem Arm und Membranarm befindet und die Untereinheiten NuoH und NuoD an der Bindung beteiligt sind (Darrouzet et al., 1998; Dupuis et al., 2001). Weiterhin wurde zur Lokalisierung der Ubichinonbindestelle ein photoaktivierbares Azido-Ubichinon-Derivat, 3-Azido-2-methyl-5-methoxy[3H]-6-decyl-1,4-benzochinon, verwendet, das nach Bestrahlung mit UV-Licht kovalent an NuoM gebunden war (Gong et al., 2003). 2.4 Mutagenese im nuo-Operon 2.4.1 Die extrachromosomale Klonierung Eine gängige Technik zur Untersuchung von Struktur-Funktionsbeziehungen von Enzymen ist eine Mutagenesestudie. Solche Mutationen in das nuo-Operon im Genom von E. coli einzubringen, ist allerdings sehr aufwendig (Spehr et al., 1999). Einfacher ist es, die entsprechenden Gene extrachromosomal auf einem Plasmid zu mutagenisieren. Voraussetzung hierfür ist, dass entweder das gesamte Operon oder ein Teil davon in einen Vektor kloniert wurde. Ein Expressionsvektor, der das gesamte nuo-Operon enthält, ermöglicht direkt die Expression mutierter Gene. Erfolgt die Expression in einem Stamm, in dem das nuo-Operon aus dem Genom deletiert wurde, so produzieren die Zellen nur den gezielt veränderten Komplex I. Eine Alternative stellt die Komplementation dar, d.h. es wird nur ein Teil des Operons im Genom deletiert und dieser Teil wird auf einem Plasmid in die - 15 - Einleitung Zellen eingebracht. Nach Expression der auf dem Plasmid mutierten Gene erfolgt in der Regel der Einbau der Untereinheiten zu dem Gesamtkomplex. Allerdings ist eine solche Komplementation nicht in jedem Fall möglich, da die Assemblierung des Komplexes bei getrennter Expression der Gene von dem Chromosom und von einem Plasmid nicht unbedingt stattfinden muss. Außerdem können unterschiedliche Transkriptstabilitäten die Menge des produzierten Proteins beeinflussen. Daher ist es von Vorteil, das gesamte Operon auf einem extrachromosomalen Expressionssystem zur Verfügung zu haben. Die Klonierung des nuo-Operons ist aufgrund seiner Größe von 15 kbp nicht trivial. Zwar wurde auch das atp-Operon der ATP-Synthase kloniert, die mit 535 kDa eine mit dem Komplex I vergleichbare Masse besitzt, jedoch besteht die ATPase aus 8 Untereinheiten, die in einer Stöchiometrie von a1:b2:c10-14:α3:β3:γ1:δ1:ε1 vorkommen (Noji und Yoshida, 2001). B B B B B B B B B B B B B B B B Dadurch hat das aus den Genen atpIBEFHAGDC bestehende Operon eine Größe von nur 7 kbp (Gunsalus et al., 1982; Kasimoglu et al. 1996). Klonierungsvektoren besitzen im Allgemeinen eine Klonierungskapazität von etwa fünf bis zehn kbp. Es gibt nur vereinzelte Berichte, dass größere DNA-Fragmente in Expressionsvektoren kloniert wurden. Die hohe Ringspannung destabilisiert das Plasmid und erschwert eine Expression der klonierten Gene und eine Replikation des Plasmids. In der Literatur werden die höchsten Kapazitäten mit bis zu 12 kbp für den ZAP Express® Vektor, Fa. Stratagene, angegeben. Mit so genannten P P Lambda-Austauschvektoren können neun bis 23 kbp kloniert werden, bei der Verwendung von Cosmiden bis zu 42 kbp, jedoch ist eine Expression der Gene von diesen Systemen nicht möglich (Sambrook und Russel, 2001). Um bei der Klonierung des nuo-Operons das Problem der hohen Ringspannung zu vermeiden, müsste das Operon in mehreren kleinen Schritten kloniert werden. Als Vektor würde sich der pBAD33 eignen, der durch einen ArabinosePromotor sehr gut regulierbar ist. Er enthält den PBAD-Promotor des araBAD-Operons und das B B araC-Gen, das für den negativen und positiven Regulator dieses Promotors kodiert (Guzman et al., 1995). Die wichtigsten Eigenschaften dieses Plasmids sind eine sehr geringe Hintergrundexpression im nicht-induzierten Zustand, eine schnelle und hohe Expression in Gegenwart von L(+)-Arabinose sowie die Möglichkeit zur Modulation der Expression über die Konzentration des Induktors Arabinose. - 16 - Einleitung 2.4.2 λ-Red vermittelte Rekombination in E. coli Mit dem Zusammenspiel von Restriktionsenzymen und einer Ligase entsteht eine Vielzahl von Möglichkeiten zur Modifikation von DNA, die unter dem Begriff der Klonierung zusammengefasst werden. Mittels Klonierung können z.B. einzelne Gene in einen Vektor integriert werden, Deletionen oder Insertionen eingeführt oder einzelne DNA-Bereiche ausgetauscht werden. Voraussetzung ist allerdings, dass geeignete Erkennungssequenzen für Restriktionsenzyme vorhanden sind. Oftmals müssen diese palindromischen Bereiche erst durch eine Mutagenese eingeführt werden, während in anderen Fällen das zu häufige Vorkommen einer Erkennungssequenz zu einer unerwünschten Fragmentierung der DNA führt. Dadurch sind vor allem der Modifikation des Chromosoms Grenzen gesetzt. Zur Modifikation von DNA wird bei der Hefe Saccharomyces cerevisiae häufig anstelle der Klonierung die Rekombination genutzt. Bringt man durch Transformation lineare DNAFragmente in die Zellen ein, so werden diese in das Genom eingebaut, wenn sie zu der chromosomalen Sequenz homologe Bereiche aufweisen. Dies geschieht mit dem sehr effizienten Rekombinasesystem von S. cerevisiae über einen Doppelstrangbruch im Genom und anschließende Ligation mit dem homologen Einzelstrang (Oliver et al., 1998). Auf diese Art und Weise können synthetische Oligonukleotide genutzt werden, um über die Rekombination jedes gewünschte DNA-Fragment in das Genom von S. cerevisiae einzuführen. Die Nutzung der Rekombination zur Einführung von linearen DNA-Fragmenten in das Genom von E. coli ist durch das Rekombinasesystem von E. coli begrenzt (Abb. 2-6), das einen Homologiebereich von mehreren tausend Basen zwischen Fragment und Zielsequenz benötigt, dabei wenig effizient ist und vor allen Dingen nicht geeignet zur Rekombination linearer DNA ist, da diese durch die intrazelluläre RecBCD-Exonuklease abgebaut wird (Lorenz und Wackernagel, 1994). Bei einer Deletion von recBCD wird lineare DNA zwar nicht abgebaut, jedoch findet keine Rekombination mehr statt, Plasmide werden nur noch in geringem Maß repliziert und das Zellwachstum ist gestört (Cosloy und Oishi, 1973). Ermöglicht wird die Rekombination linearer DNA, wenn statt des E. coli eigenen Rekombinasesystems das λ-Red System des Phagen λ genutzt wird (Murphy, 1998). Dieses besteht aus den drei Rekombinase-Untereinheiten Gam, Exo und Beta. λ-Gam inhibiert die Nukleaseaktivität von RecBCD, so dass lineare DNA in E. coli transformiert werden kann, ohne dass sie abgebaut wird. λ-Exo führt einen Doppelstrangbruch in die DNA ein und - 17 - Einleitung erzeugt 3´-Überhänge, die als Substrat für die Rekombination benötigt werden. λ-Beta ist ein Einzelstrang-bindendes Protein, das die Renaturierung von komplementären DNAEinzelsträngen fördert und so einen Strangaustausch vermitteln kann (Ellis et al., 2001). Die λ-Red vermittelte Rekombination ist im Vergleich zu dem RecBCD-System nicht nur wesentlich effizienter, es wird außerdem ein wesentlich kleinerer homologer Bereich für den Austausch benötigt. Schon ab einer Länge von 20 bp findet eine Rekombination statt, eine genügend hohe Effizienz ist ab 36 bp oder mehr gegeben (Datsenko und Wanner, 2000). Abb. 2-6: Das Rekombinasesystem von E. coli. RecBCD bindet an ein freies Ende der doppelsträngigen DNA und entwindet unter ATP-Verbrauch die Helix. Es entstehen zwei einzelsträngige Schleifen, in denen RecBCD vier bis sechs Nukleotide vor dem 3´-Ende einer sogenannten Chi-Sequenz schneidet. So entsteht ein offener Einzelstrang, an den RecA zur Stabilisierung bindet und an dem die homologe Rekombination stattfindet (nach: http://gsbs.gs.uth.tmc.edu/courses/experimental_genetics/fig_9.jpg). Durch die λ-Red Rekombinase sind alle Möglichkeiten der Modifikation von DNA in E. coli gegeben, die auch durch Klonierung erreicht werden können, allerdings vollkommen sequenzunabhängig. Oftmals kann daher das „Rekombineering“ die Methode der Wahl z.B. - 18 - Einleitung zur Deletion eines Gens oder für das Einführen von Mutationen sein, wobei vor allem die genomische Manipulation im Vergleich zur Klonierung wesentlich einfacher ist. 2.4.3 Selektion auf Rekombination Erfolgt eine Rekombination ohne Selektion über eine Antibiotikaresistenz, so trägt sowohl das Ausgangsplasmid als auch das rekombinierte Plasmid die gleiche Resistenz und eine Unterscheidung der Zellen ist nicht möglich. In diesem Fall müssen entweder sehr viele Kolonien mittels Plasmidpräparation und anschließender Restriktionsanalyse oder über eine Polymerase-Kettenreaktion (PCR) durchsucht werden. Alternativ bieten sogenannte Suizidsysteme die Möglichkeit, rekombinierte von nicht-rekombinierten Plasmiden zu unterscheiden. Ein Beispiel für ein solches System ist das nptI-sacR-sacB-System (Ried und Collmer, 1987). Das sacB-Gen aus Bacillus subtilis kodiert für die Levansucrase, ein Enzym, das Saccharose in Levan umbaut. Levan akkumuliert in den Zellen und führt zu einer Lyse (Gay et al., 1985). Gram-negative Bakterien, die dieses Gen zusammen mit seinem Regulator sacR enthalten, können nicht auf Saccharose-haltigen Medien wachsen. In diesem Fall spricht man von einem negativen Selektionsmarker. Bei Verwendung der nptI-sacR-sacB-Kassette benutzt man zusätzlich als positiven Selektionsmarker das nptI-Gen, das für eine Neophosphotransferase kodiert und eine Kanamycin-Resistenz vermittelt. Wird diese Kassette in ein E. coli Plasmid eingeführt, können Zellen auf Kanamycin-enthaltendem Medium auf die Insertion der Kassette überprüft werden. Das Medium darf dabei keine Saccharose enthalten, da die Zellen ansonsten lysiert werden. Die Kassette kann über Rekombination in den Bereich insertiert werden, in den eine Mutation eingeführt werden soll. Die Mutation wird wiederum über Rekombination derart in das Plasmid eingeführt, dass die nptI-sacR-sacBKassette vollständig von dem Bereich ersetzt wird, in dem die Mutation liegt. Bei Anzucht auf Medium mit Saccharose können nur Zellen überleben, die die Kassette nicht mehr enthalten und bei denen die Rekombination erfolgreich war. Soll statt der Klonierung die Rekombination genutzt werden, um ein DNA-Fragment in einen Vektor einzuführen, so steht die Möglichkeit der Antibiotika-Selektion ebenfalls nicht zur Verfügung. Bei der Klonierung wird der Vektor mit Restriktionsenzymen linearisiert und wird bei nicht erfolgter Ligation als lineare DNA in E. coli vom RecBCD-Rekombinasesystem abgebaut (Lorenz und Wackernagel, 1994). Nur geschlossene - 19 - Einleitung Plasmide werden in E. coli propagiert und produzieren das die Resistenz vermittelnde Protein. Zellen, die kein erfolgreich ligiertes Plasmid enthalten, können auf Antibiotika-haltigen Medien nicht wachsen. Im Fall der Rekombination wird der Ursprungsvektor in seiner superspiralisierten Form in die Zellen transformiert. Es erfolgt kein Abbau des Plasmids und die Zellen besitzen die Plasmid-kodierte Antibiotika-Resistenz, egal ob ein Gen durch Rekombination in das Plasmid eingeführt wurde oder nicht. Hier kann zwischen rekombinanten und nicht-rekombinanten Zellen mittels einer Blau-Weiß-Selektion unterschieden werden. Hierzu wird ein Plasmid benötigt, das in seiner multiplen Klonierungsstelle (multiple cloning site, MCS), einem DNA-Bereich mit vielen Erkennungssequenzen für Restriktionsenzyme, das Gen für das N-terminale α-Fragment der β-Galaktosidase, bezeichnet mit lacZ´, enthält. β-Galaktosidase setzt normalerweise Galaktose zu Fructose und Glucose um. Andere Substrate wie 5-Bromo-4-chloro-3-indolyl[beta]-D-galaktopyranosid (X-Gal), das farblos ist, werden zu einem farbigen Produkt umgesetzt. Produziert der Bakterienstamm das chromosomal kodierte C-terminale ωFragment der β-Galaktosidase, das nur zusammen mit dem α-Fragment katalytische Aktivität hat, so erfolgt die sogenannte α-Komplementation. Wird in die MCS des verwendeten Vektors ein DNA-Fragment insertiert, so wird die für das α-Fragment kodierende Sequenz unterbrochen und es wird keine funktionelle β-Galaktosidase mehr produziert. X-Gal wird nicht mehr zu einem farbigen Produkt umgesetzt und solche Plasmide enthaltende Bakterienkolonien bleiben farblos. Kolonien, die Plasmide ohne Insert enthalten, sind weiterhin zur Produktion von β-Galaktosidase fähig und setzen X-Gal um, wodurch sie eine blaue Färbung annehmen. Blaue Kolonien scheiden als Rekombinanten aus, wodurch die Anzahl zu untersuchender Kolonien wesentlich verringert wird. Plasmide, die in ihrer MCS für lacZ´ kodieren und somit für eine Blau-Weiß-Selektion geeignet sind, werden im Normalfall nur zur Klonierung und nicht zur Expression genutzt. Der Vektor pETBlue-1 (Invitrogen) ermöglicht nicht nur die Blau-Weiß-Selektion, sondern auch eine auf den T7 lac-Promotor basierende Expression. Selektion und Expression verlaufen getrennt voneinander, da die Expression des insertierten Gens und von lacZ´ über zwei Promotoren in verschiedene Richtungen verläuft (s. Anhang). - 20 - Einleitung 2.5 Thema der Arbeit Bislang existiert kein extrachromosomales Expressionssystem für das nuo-Operon. Um Mutagenesestudien zu erleichtern, sollte das gesamte nuo-Operon in den Expressionsvektor pBAD33 kloniert werden. Zur Verringerung der Ringspannung war eine Klonierung in mehreren Klonierungsschritten geplant. Um die einzelnen nuo-Gene einer Mutagenese zugänglich zu machen, sollte ein Subklonsystem erstellt werden, in dem Fragmente des Operons in Klonierungsvektoren zur Verfügung stehen, die bei einer maximalen Größe von 6 kbp für eine PCR-basierte Mutagenese geeignet sind. In die Subklone sollten verschiedene Mutationen in nuo-Gene eingeführt und diese anschließend zur Expression der mutierten Gene in das Expressionsplasmid rückkloniert werden. Es sollten Deletionen einzelner Gene bzw. Gengruppen, verschiedene Tag-Sequenzen und der Austausch einzelner Aminosäuren in das nuo-Operon eingeführt werden. Da die genetische Manipulation über Mutagenese und Klonierung nicht immer zugänglich ist, sollte die sequenzunabhängige Methode der λ-Red vermittelten Rekombination als Ergänzung zum Subklonsystem etabliert werden. . - 21 - Material und Methoden 3. Material und Methoden 3.1 Medien und Lösungen LB-Medium Pepton (Invitrogen) 1 % (w/v) NaCl (Roth) 1 % (w/v) Hefeextrakt (AppliChem) 0.5 % (w/v) Für feste Nährböden: Agar (AppliChem) 2 % (w/v) LBAmp LB-Medium + Ampicillin (AppliChem) 100 mg/l LBKan LB-Medium + Kanamycin (Merck) LBCam LB-Medium + Chloramphenicol (Fluka) 30-60 mg/l feste Medien 20 mg/l flüssige Medien 170 mg/l LBTet LB-Medium + Tetracyclin (AppliChem) SOC-Medium Pepton (Invitrogen) 2 % (w/v) Hefeextrakt (AppliChem) TB-Medium 50 mg/l 0.5% (w/v) NaCl (Roth) 10 mM KCl (Merck) 2.5 mM MgSO4.7H2O (Fluka) 10 mM MgCl2.6H2O (Riedel-de Haën) 10 mM Glucose (Serva) 20 mM Pepton (Invitrogen) 1.2 % (w/v) Hefeextrakt (AppliChem) 2.4 % (w/v) Glycerin (aus Institutsbeständen) 0.2 % (v/v) KH2PO4 (Merck) 17 mM K2HPO4 (Aldrich) 72 mM - 22 - Material und Methoden M9-Medium Na2HPO4 (Merck) 0.6 % (w/v) KH2PO4 (Merck) 0.3 % (w/v) NaCl (Roth) 0.05 % (w/v) NH4Cl (Riedel-de Haën) CaCl2 (Fluka) 0.1 mM MgSO4 (Fluka) 10 mM Casein (Merck) 0.1 % (w/v) Tryptophan (Merck) M46-Medium 0.01 % (w/v) Mannitol (Serva) 30 mM Mannitol (Serva) 30 mM Natriumacetat (Riedel-de Haën) Casein (Difco) L-Methionin (Sigma) 0.2 mM Uracil (Fluka) 0.2 mM L-Prolin (Sigma) 0.15 mM 1 mM L-Histidin-HCl (Merck) 0.15 mM L-Isoleucin (Fluka) 0.3 mM L-Valin (Merck) 0.3 mM L-Tryptophan (Aldrich) 0.2 mM Thiamin (Sigma) 1 µM Pepton (Invitrogen) 1 % (w/v) NaCl (Roth) 1 % (w/v) Hefeextrakt (AppliChem) DMSO (Roth) STET-Puffer 4 mM 0.1 % (w/v) Adenin-HCl (Aldrich) TSS 0.1 % (w/v) 0.5 % (w/v) 5 % (w/v) MgSO4 (Fluka) 20 mM Tris-HCl, pH 8.0 (Merck) 50 mM EDTA (Roth) 50 mM Saccharose (Merck) 8 % (w/v) Triton X-100 (Serva) 5 % (w/v) - 23 - Material und Methoden Puffer P1 (Qiagen) Tris-HCl, pH 8.0 (Merck) 50 mM EDTA (Roth) 10 mM 100 µg/ml RNAse (Qiagen) Puffer P2 (Qiagen) NaOH (Roth) 0.2 M SDS (Serva) Puffer P3 (Qiagen) 1 % (w/v) Kaliumacetat, pH 5.5 (Merck) Tris-Borat, pH 8.3 (Merck) Biuret-Reagenz 1 mM NaOH (Roth) 0.2 M Na-K-Tartrat (Merck) 5 g/l Cu2SO4 (Merck) 3 g/l KI (Fluka) 5 g/l B B B SDS (USB) 8 % (w/v) Glycerin (aus Institutsbeständen) Tris-Acetat, pH 6.8 (Merck) EDTA (Roth) Blot-Puffer 1M Bromphenolblau (Merck) 0.3 % (w/v) Tris-HCl, pH 7.5 (Merck) 25 mM 192 mM SDS (USB) 0.05 % (w/v) Methanol (aus Institutsbeständen) 20 % (v/v) NaCl (Roth) 0.8 % (w/v) KCl (Merck) 0.02 % (w/v) Na2HPO4 (Merck) 0.144 % (w/v) KH2PO4 (Merck) 0.024 % (w/v) B B B PBST 20 % (w/v) 2.5 mM Glycin (Roth) PBS 89 mM EDTA (Roth) B SDS-Probenpuffer 3M B B B B B PBS + Tween 20 (Acros) - 24 - 0.01 % (w/v) Material und Methoden TBE Tris-Borat, pH 8.3 (Merck) 89 mM EDTA (Roth) 1 mM SDS-Kammerpuffer Glycin (Roth) 14 % (w/v) Tris-Acetat, pH 8.3 (Merck) 3 % (w/v) SDS (Serva) 0.5 % (w/v) 3.2 Mikroorganismen, Vektoren und Oligonukleotide Tab. 3-1: Verwendete E. coli Stämme Stamm Genotyp Quelle DH5α supE 44 ∆lacU169 (Φ80lacZ∆M15) hsdR17 (Hanahan et al., 1983) recA1 endA1 gyrA96 thi-1 re1A1 BL21(DE3) BF- ompT hsdS (rB- mB-) gal dcm P P B PB P B (Studier et al., 1986) P PB RosettaBlue(DE3) endA1 hsdR17 (rK12-mK12+) supE44 thi-1 T P P Novagen P P recA1 gyrA96 relA1 lacF' [proA+B+ P P P P lacIqZ∆M15::Tn10(tetR)] (DE3)pRARE (CmR) P P RR1∆M15 P P Fhsd 320 (rB- mB-) recA+ ara-14 pro A2 lac B PB P B PB P P P T (Bolivar et al., 1977) 1qZ, ∆M15 gal K2 rspL20strR xyl-5 mtl-1 supE P P P P 44 LambdaP P AN387 (Wallace und Young, 1977) ANN023 AN387, ∆nuoB Spehr et al., 1996 ANN0221 AN387, ndh::tet; nuoB::nptI (Böck, 1996) ANN0222 AN387, ndh::tet; nuoB::nptI-sacB-sacR (Böck, 1996) ANN0921 AN387, ndh::tet; nuoI::nptI (Böck, 1996) ANN0922 AN387, ndh::tet; nuoI::nptI-sacB-sacR (Böck, 1996) ANN003 AN387, 5´nuo::lacI-aadA+-T7-Promotor (Spehr et al., 1999) P P - 25 - Material und Methoden Tab. 3-2: Verwendete Vektoren Vektor Quelle pET11-a AGS, Heidelberg pET24-a Novagen, Darmstadt pUC18 Biolab, Schwalbach pBAD33 Guzman et al., 1995 pETBlue-1 Novagen, Darmstadt pKD46 Datsenko und Wanner, 2000 pCP20 Datsenko und Wanner, 2000 pKD3 Datsenko und Wanner, 2000 pAR1219 Davanloo et al., 1984 pBAD33-nuoB Flemming, 2003 pASK-IBA3 IBA, Göttingen Die Vektorkarten von pET11-a/nuoB-G/NuoFC, pBADnuo, pBAD33, pET24-a, pET11-a und pKD46 sind im Anhang gezeigt. Tab. 3-3: Verwendete Oligonukleotide. Die Oligonukleotide wurden entweder von MWG Biotech (Ebersberg) oder von der Hermann GbR (Freiburg) synthetisiert. Oligonukleotid Sequenz N1c-1Pvuf 5´-CAGTTTGCGCCGTCGATCGCCCAGCAATGTTCCATCGGC-3´ N1c-1Pvur 5´-GCCGATGGAACATTGCTGGGCGATCGACGGCGCAAACTG-3´ N1c2 5´-GCATCTGCCAGCAAGCTTCCATCGGCTGTAAC-3´ N1c2r 5´-GTTACAGCCGATGGAAGCTTGCTGGCAGATGC-3´ N1c3 5´-CAATGTTCCATCGGCGCCAACATCAGCC-3´ N1c3r 5´-GGCTGATGTTGGCGCCGATGGAACATTG-3´ N1c4 5´-CACTACTTCCTCGCGGACCGTGGTCGTTTCG-3´ N1c4r 5´-CGAAACGACCACGGTCCGCGAGGAAGTAGTG-3´ pET11M-NdeI 5´-AAGATCCATATGTTACTACCCTGGC-3´ pET11M-NheI 5´-GTCATGCTAGCTTACGGCCTTGTAGTAG-3´ pET11CD-NdeI 5´-CCATTTCATATGGTGAACAATATGACCG-3´ - 26 - Material und Methoden Tab. 3-3; Fortsetzung LambdaCD1-rev 5´-TCTTCGCTTATTCCTAAATCTATTTCGCGAAGCTTACTGCGT GTAGGCTGGAGCTGCTTC-3´ LambdaCD1-for 5´-AGATAAACAATCAGGTCAGACACCAGGCTGCCGCGGATCG CATATGAATATCCTCCTTAG-3´ IBA-M-for 5´-GAATAAGGTCTCCAATGTTACTACCCTGGCTAATATTAATT C-3´ IBA-M-rev 5´-TATTGTGGTCTCGGCGCTCGGCCTTGTTAGTAGTAACGGAA TTAAC-3´ Lambda-M-L 5´-GCCTTCGCGGTGCATCCGACACTACCCCCAGTCCGAAGAGG TGTAGGCTGGAGCTGCTTC -3´ Lambda-M-N 5´-CCACTGCTGGATATTGCCAATCGCGGAGTGCGAGGTATCCC ATATGAATATCCTCCTTAG-3´ StrepC-for 5´-CTCGCCGTTTGAGCTGACCAAAGCCAAACAGGATCTGGAGG CTAGCTGGAGCCACCCGCAGTTCGAG-3´ StrepC-rev 5´-TCATCCCCCACTCTTCCGGTTTGAAGGTCAGGGCTTCCATTT ACTTCTCGAACTGCGGGTGGCTCCAGC-3´ 6TagCSD-for 5´-CTCGCCGTTTGAGCTGACCAAAGCCAAACAGGATCTGGAGG CTGCAGCTGCTGCTGCTAG-3´ 6TagCSD-rev 5´-GGCTTCCATTTACTTCTCGAACTGCGGGTGGCTCCAGCTAGC AGCAGCAGCTGCAGC-3´ SeqB-Cterm 5´-GCGCGAACGCAAGCGGTG-3´ SeqC-Cterm 5´-CGGCGATCCGCGGCAGC-3´ pUC25-for 5´-CCCAATACGCAAACCGCTCGAGCCTCTCCCCGCGCGTTGG -3´ pUC25-rev 5´-CCAACGCGCGGGGAGAGGCTCGAGCGGTTTGCGTATTGGG -3´ 24Mred-for 5´-AAATAATTTTGTTTAACTTTAAGAAGGAGATATACATATGT TACTACCCTGGCTAATATTAATTCCC-3´ 24Mred-rev 5´-CGGATCCGCGACCCATTTGCTGTCCACCAGTCATGCTAGCTT ACGGCCTTGTAGTAGTAACGG-3´ Leu244Ile-for 5’-CCGCTGCATGGCTGGATCCCGGATGCGCACTC-3’ Leu244Ile-rev 5’-GAGTGCGCATCCGGGATCCAGCCATGCAGCGG-3’ - 27 - Material und Methoden Tab. 3-3; Fortsetzung Asp246Asn-for 5’-GTGGTTCCGCTCCATGGCTGGCTGCCGAATGCGCACTCCCA G-3’ Asp246Asn-rev 5’-CTGGGAGTGCGCATTCGGCAGCCAGCCATGGAGCGGAACC AC-3’ -EFG-CD 5´-GGGCAGTATCGATTTTGTTATGTCAGATGTGGACCGCTAAG TGTAGGCTGGAGCTGCTTC-3´ -EFG-H 5´-AGTTCCGGTGATATCCAACTCATTGTTGTGCCTCCTTGAGCA TATGAATATCCTCCTTAG-3´ -G-for 5´-TAACCGAATTTCGATTAACGCTCAGTCTCTGACTGAGAAAG TGTAGGCTGGAGCTGCTTC-3´ -EF-rev 5´-GCCATTAGCATGCTTCCAGTTTTCTCAGTCAGAGACTGAGC ATATGAATATCCTCCTTAG-3´ 3.3 Mikrobiologische Methoden 3.3.1 Anzucht von E. coli Einzelne Kolonien einer Platte wurden mit Hilfe eines sterilen Zahnstochers in 4 ml bzw. 50 ml LB-Medium mit den entsprechenden Antibiotika überführt und für 16 Std. bei 37°C bzw. im Fall temperatursensitiver Plasmide bei 30°C geschüttelt. Diese Übernachtkulturen wurden zur Anzucht von E. coli in größerem Maßstab oder zur Plasmidpräparation verwendet. E. coli Zellen wurden je nach benötigter Zellmenge im Litermaßstab oder in 10 l LB-Medium im Glasfermenter angezogen. Als Zusätze wurden dem Medium 0.01 g/l Eisen-AmmoniumCitrat, 0.05 g/l Riboflavin und 0.02 g/l Natriumsulfid zugegeben. Die Zellen wurden aerob bei 37°C angezogen. Bei Bedarf wurde die Expression bei einer optischen Dichte bei 600 nm (OD600) von 0.8 in einer 1 l-Kultur bzw. bei einer OD600 von 1.2 bei einer Zucht im 10 l Fermenter induziert. Die Zellen wurden etwa zwei weitere Stunden bis zum Erreichen der stationären Phase angezogen, abzentrifugiert (9´000 Upm, Rotor GS-3, 30 min, 4°C, RC-5 - 28 - Material und Methoden Centrifuge, Sorvall), mit 50 mM 2-(N-Morpholino)-ethansulfonsäure (MES)/50 mM NaCl, pH 6.0 gewaschen und das Zellsediment bis zur Verwendung bei -80°C aufbewahrt. 3.3.2 Herstellung von Dauerkulturen Zu 500 µl sterilem 80 % Glycerin wurde 300 µl Bakterienkultur gegeben, gemischt und sofort in flüssigem Stickstoff schockgefroren. Die Dauerkulturen wurden bei -80°C aufbewahrt. 3.3.3 Herstellung kompetenter Zellen Herstellung elektrokompetenter Zellen: LB-Medium wurde im Verhältnis 1:100 (v/v) mit einer E. coli Übernachtkultur angeimpft. Bei einer OD600 von 0.5 bis 0.6 wurden die Zellen im Anzuchtkolben 15 min auf Eis gekühlt und anschließend für 10 min bei 5´000 Upm abzentrifugiert (4°C, Zentrifuge 5804R, Eppendorf). Das Sediment wurde in dem Ausgangsvolumen eiskaltem sterilem H2O resuspendiert und anschließend erneut abzentrifugiert. Dieser Schritt wurde zweimal wiederholt. Das Sediment der letzten Zentrifugation wurde in dem 1.5 fachen Volumen eiskaltem 15 % (v/v) Glycerin resuspendiert und anschließend bis zur Verwendung bei -80°C gelagert. Wurden die Zellen sofort zur Transformation eingesetzt wurden sie in H2O statt Glycerin resuspendiert. Transformation durch Elektroporation: 0.1 bis 2.5 µl Plasmid-DNA wurden mit 90 µl elektrokompetenten Zellen vermischt, in eine Elektroporationsküvette (Eppendorf) mit einem Elektrodenabstand von 1 mm gegeben und im Elektroporator (Eppendorf, Electroporator 2510) für 5 ms einem Puls von 1´700 V ausgesetzt. Anschließend wurde sofort 1 ml SOC-Medium zugegeben und die Bakteriensuspension für 1 Std. bei 650 Upm und 30°C bzw. 37°C zur Ausbildung der Antibiotika-Resistenzen geschüttelt. Die Zellen wurden für 4 min bei 4´000 Upm abzentrifugiert (Eppendorf, Zentrifuge 5415 D), in 100 µl SOC-Medium resuspendiert und auf LB-Agarplatten mit dem entsprechenden Antibiotikum ausplattiert. - 29 - Material und Methoden Transformation nach der TSS-Methode: U E. coli Zellen wurden aus einer frischen Übernachtkultur angeimpft und bis zu einer OD600 B B von 0.3 bis 0.4 bei 37°C angezogen. 1 ml der Zellsuspension wurde bei 3´000 Upm für 10 min zentrifugiert (Zentrifuge 5804R, Eppendorf) und das Sediment in einem Zehntel des Ausgangsvolumen eiskaltem TSS-Puffer resuspendiert. 1-10 µl DNA wurden mit der Zellsuspension gemischt und 30 min auf Eis inkubiert. Die Suspension wurde für 45 sec bei 42°C inkubiert und anschließend für 2 min auf Eis abgekühlt. Nach Zugabe von 900 µl SOCoder LB-Medium mit 20 mM Glucose wurde für 1 Std. bei 37°C und 650 Upm geschüttelt. Die Zellen wurden 4 min bei 4´000 Upm abzentrifugiert (Zentrifuge 5804R, Eppendorf) und das resuspendierte Sediment auf Platten mit dem entsprechenden Antibiotikum ausplattiert. 3.4 Molekularbiologische Methoden 3.4.1 Analytische Plasmidisolierung mittels „Boiling lysis“ Zur analytischen Plasmidisolierung nach der „Boiling lysis“-Methode (Holmes und Quigley, 1980) wurden 1.5 ml einer E. coli Übernachtkultur für 4 min bei 4´000 Upm abzentrifugiert (Zentrifuge 5804R, Eppendorf). Das Sediment wurde in 500 µl eiskaltem STET-Puffer resuspendiert und nach Zugabe von 20 µl Lysozym (50 mg/ml, Fluka) wurden die Proben zuerst 15 min auf Eis inkubiert und anschließend für 2.5 min in einem kochenden Wasserbad erhitzt. Die Probe wurde 5 min auf Eis abgekühlt, die denaturierte genomische DNA 10 min bei 14´000 Upm (Zentrifuge 5804R, Eppendorf) abzentrifugiert und mit einem sterilen Zahnstocher aus dem Reaktionsgefäß entfernt. Die verbleibende Plasmid-DNA wurde durch Zugabe von 500 µl Isopropanol, eine 20 minütige Inkubation bei -20°C und anschließende Zentrifugation bei 14´000 Upm (15 min, 4°C, Zentrifuge 5804R, Eppendorf) ausgefällt. Das DNA-Sediment wurde an der Luft getrocknet und anschließend in 40 µl H2O durch Schütteln B B bei 50°C und 1´150 Upm gelöst. 3.4.2 Analytische Plasmidisolierung mittels alkalischer Lyse 2 ml einer E. coli Übernachtkultur wurden 4 min bei 4´000 Upm abzentrifugiert (Zentrifuge 5804R, Eppendorf). Das Sediment wurde in 250 µl P1-Puffer resuspendiert und zur Lyse der - 30 - Material und Methoden Zellen 250 µl P2-Puffer zugegeben. Nach 4 min Inkubation bei Raumtemperatur wurde die alkalische Suspension mit 250 µl P3-Puffer neutralisiert und 15 bis 30 min bei 4°C inkubiert. Danach wurde 10 min bei 14´000 Upm (Zentrifuge 5804R, Eppendorf) zentrifugiert und der Überstand in ein neues Reaktionsgefäß überführt. Nach Zugabe von 750 µl Isopropanol und 20 min Inkubation bei -20°C wurde die Plasmid-DNA 15 min bei 14´000 Upm (Zentrifuge 5804R, Eppendorf) abzentrifugiert, an Luft getrocknet und durch Schütteln bei 50°C und 1´150 Upm in 40 µl H20 resuspendiert. B B 3.4.3 Präparative Plasmidisolierung Plasmide wurden aus E. coli im präparativen Maßstab mit dem QIAGEN Midi Kit (Qiagen, Hilden) nach Herstellervorschrift isoliert. Hierfür wurden für high copy Plasmide (über 200 Plasmide pro Zelle) 50 ml, für low copy Plasmide (weniger als 50 Plasmide pro Zelle) 100 bis 200 ml einer Übernachtkultur eingesetzt. Bei Plasmiden, die für die Sequenzierung bestimmt waren, wurde die Suspension nach der Isopropanolfällung für nur 5 min auf Eis inkubiert. 3.4.4 Präparation genomischer DNA 3 ml einer E. coli Übernachtkultur wurden 3 min bei 3´000 Upm abzentrifugiert (Zentrifuge 5804R, Eppendorf) und das Sediment in 200 µl Lysis-Puffer resuspendiert. Nach 10 min Inkubation bei Raumtemperatur wurde die Suspension mit 66 µl 5 M NaCl gemischt. Das Gemisch wurde 10 min bei 17´000 *g bei 4°C zentrifugiert (Zentrifuge 5804R, Eppendorf), der Überstand mit 266 µl Chloroform versetzt und durch fünfzigmaliges vorsichtiges Umdrehen des Reaktionsgefäßes gemischt. Nach 3 min Zentrifugation bei 17´000 *g (Zentrifuge 5804R, Eppendorf) wurde die wässrige Phase mit 266 µl Isopropanol gemischt, für 10 min bei -20°C inkubiert und die ausgefällte genomische DNA 15 min bei 25´000 *g abzentrifugiert (Zentrifuge 5804R, Eppendorf). Das Sediment wurde zweimal mit 70 % (v/v) Ethanol gewaschen, getrocknet und in 30 µl H2O gelöst (Chen und Kuo, 1993). B B 3.4.5 Restriktion und Modifikation von DNA Analytische Restriktionsansätze wurden in 20 µl durchgeführt. Für je 1 µg DNA wurde 1 bis 10 U Restriktionsenzym (MBI Fermentas, St. Leon-Rot) in dem jeweiligen Restriktionspuffer - 31 - Material und Methoden eingesetzt und nach Herstellervorschriften verdaut. Das Volumen eines präparativen Restriktionsansatzes betrug 20 bis 60 µl. DNA-Überhänge wurden mit alkalischer Phosphatase (CIAP, MBI Fermentas, St. Leon-Rot) nach Herstellervorschriften dephosphoryliert und mit T4 Polynukleotid Kinase (MBI Fermentas, St. Leon-Rot) phosphoryliert. Für Ligationen wurde T4 Ligase eingesetzt (MBI Fermentas, St. Leon-Rot). In einem 20 µl Ligationsansatz wurden 50 ng Vektor im molaren Verhältnis von 1:3 bis 1:5 mit dem zu ligierenden DNA-Fragment gemischt und mit 5 U Ligase im entsprechenden Puffer über Nacht bei 4°C bzw. für 4 Std. bei Raumtemperatur inkubiert. 3.4.6 Agarosegelelektrophorese Zur Auftrennung von DNA-Fragmenten in einem Größenbereich von 0.6 bis 8 kbp wurden 0.8 % (w/v) Agarosegele in 0.5x TBE mit 500 µg/l Ethidiumbromid verwendet. Die Fragmente wurden bei einer Spannung von 2 bis 8 V/cm in 0.5x TBE voneinander getrennt und durch die UV-Fluoreszenz des DNA-Ethidiumbromid-Komplexes bei 254 nm nachgewiesen. Die Größen der DNA-Fragmente konnten durch gleichzeitige Auftrennung von DNA-Fragmenten bekannter Größe abgeschätzt werden. 3.4.7 Isolation und Aufreinigung von DNA-Fragmenten Zur Aufreinigung einzelner DNA-Fragmente wurde die DNA auf einem Agarosegel aufgetrennt, das aufzureinigende Fragment mit einem Skalpell aus dem Gel ausgeschnitten und mit dem Novagen SpinPrep Gel DNA Kit (Novagen, Schwalbach) nach Herstellervorschriften aufgereinigt. PCR-Ansätze wurden mittels des Novagen PCR Clean-up Kit (Novagen, Schwalbach) nach Herstellervorschrift aufgereinigt. Kleine DNA-Fragmente bzw. Plasmide wurden durch Zugabe des 0.7-fachen Volumens Isopropanol und eines Zehntel Volumens 3 M Natriumacetat, pH 5.2 für 20 min bei -20°C ausgefällt und 15 min bei 14´000 Upm (Zentrifuge 5804R, Eppendorf) abzentrifugiert. Nach Trocknen des Sediments an Luft wurde die DNA durch Schütteln bei 1´400 Upm und 50°C in H2O gelöst. Große B B DNA-Fragmente bzw. Plasmide wurden analog mit dem 2.5 fachen Volumen Ethanol statt Isopropanol ausgefällt. - 32 - Material und Methoden 3.4.8 Polymerase-Kettenreaktion Die Polymerase-Kettenreaktion (PCR) wurde in 50 µl Ansätzen in dünnwandigen Reaktionsgefäßen (Biozyme) in einem GeneAmp PCR-System 2400 (Perkin Elmer) oder in einem Mastercycler (Eppendorf Thermocycler) durchgeführt. In einem PCR-Ansatz wurden 25 bis 150 ng Vorlage-DNA, je 10 bis 100 pmol der beiden Oligonukleotide (Primer), 1 µl 10 mM dNTPs, 5 µl 10x PCR-Puffer (MBI Fermentas, St. Leon-Rot) und 2.5 U Long PCR Enzyme Mix (MBI Fermentas, St. Leon-Rot) eingesetzt. Nach einem ersten Denaturierungsschritt bei 94°C für 2 bis 5 min wurden 25 bis 30 Zyklen mit den drei Phasen Denaturierung, Anlagerung der Primer (Annealing) und Elongation durchlaufen. Die DNAStränge wurden bei 94°C für 20 bis 30 sec denaturiert und bei einer Temperatur von 5°C unterhalb der Schmelztemperatur der Primer für 30 sec zusammengelagert. Die Elongation erfolgte bei 68°C, wobei pro kb PCR-Produkt eine Elongationszeit von 60 sec gewählt wurde. Mit dem letzten Elongationsschritt von 7 bis 10 min sollten nicht komplett synthetisierte DNA-Stränge verlängert werden. Abschließend wurden die Ansätze bis zur weiteren Benutzung auf 4°C gekühlt. Gegebenenfalls wurde die Vorlage-DNA mit dem Restriktionsenzym DpnI, das das nicht-methylierte PCR-Produkt nicht abbaut, für 1 Std. bei 37°C verdaut. Sollten DNA-Fragmente mit einer Länge von mehr als 7 kbp amplifiziert werden, wurden zunächst zehn Zyklen mit den drei oben beschriebenen Phasen durchlaufen, gefolgt von 17 weiteren Zyklen, in denen die Elongation jeweils um 10 bis 20 sec verlängert wurde. Bei nicht erfolgreicher Amplifikation wurde die Reaktion erneut mit einem Zusatz von 4 % (v/v) DMSO in dem Ansatz durchgeführt. Gegebenenfalls wurde die MgCl2-Konzentration im Ansatz von 12.5 bis 30 mM variiert. Eine analytische PCR wurde sowohl an ganzen Zellen als auch an aufgereinigten Plasmiden durchgeführt. Es wurde 2.5 U Taq-Polymerase (MBI Fermentas, St. Leon-Rot) zur Amplifikation eingesetzt. Bei der Ganzzell-PCR wurde die DNA im ersten Schritt bei 94°C für 10 min denaturiert, alle darauf folgenden Schritten wurden analog zum allgemeinen PCRProtokoll durchgeführt. - 33 - Material und Methoden 3.4.9 Einführung von Punktmutationen mittels ortsgerichteter Mutagenese Zur Einführung von Punktmutationen wurde das QuikChange® II Site-Directed Mutagenesis Kit (Stratagene) nach Herstellerangaben verwendet. Alternativ wurde eine Mutagenese-PCR mit dem Expand Long Template PCR System (Roche) durchgeführt, die Template-DNA mit dem Restriktionsenzym DpnI verdaut und das PCR-Produkt nach Aufreinigung mittels Elektroporation in elektrokompetente E. coli Zellen transformiert. Um die mutierten Plasmide von den Wildtyp-Plasmiden zu unterscheiden, wurden die Mutagenese-Primer so gewählt, dass durch die Mutagenese eine Restriktionsschnittstelle eingefügt oder entfernt wurde. 3.4.10 Weitere molekularbiologische Methoden Plasmid-DNA wurde von der Firma GATC Biotech AG (Konstanz) sequenziert. Zur Klonierung in ein Cosmid und zur Transformation in E. coli wurden große Fragmente mit dem Expand cloning Kit (Roche) nach Herstellerangaben in Phagen verpackt. 3.5 Proteinchemische Methoden 3.5.1 Isolierung der Plasmamembranen Zur Isolation von Plasmamembranen wurden E. coli Zellen im 2.5fachen Volumen 50 mM MES/NaOH, pH 6.0, 50 mM NaCl unter Zusatz von 200 µl 0.1 M Phenylmethylsulfonylfluorid (PMSF, Sigma), einer Spatelspitze DNase (Boehringer) und einer Spatelspitze Lysozym (Fluka) in einem Teflon-in-Glas-Homogenisator homogenisiert. Dabei entstandener Schaum wurde durch Zugabe von möglichst wenig Antischaum-Emulsion (Antifoam 204, Sigma) entfernt. Alle weiteren Schritte erfolgten bei 4°C. Die Zellen wurden in einem Durchgang in der „French Press“ (SML Aminco) bei 110 MPa aufgeschlossen. Zelltrümmer und nicht aufgeschlossene Zellen wurden durch eine 20minütige Zentrifugation bei 36´000 *g (16´000 Upm, Rotor 8.24, RC-5 Superspeed Refrigerated Centrifuge, Sorvall) abgetrennt. Durch eine weitere Zentrifugation für 1 Std. bei 250´000 *g (50´000 Upm, Rotor 60Ti, L8-70M Ultrazentrifuge, Beckman) wurden die Membranen sedimentiert. Das Membransediment wurde in 1 ml/g 50 mM MES/NaOH, pH 6.0 resuspendiert und gegebenenfalls bei -80 °C bis zur Verwendung gelagert. - 34 - Material und Methoden 3.5.2 Isolierung des Komplex I aus dem Stamm ANN0221/pBADnuo Der E. coli Doppeldeletionsstamm ANN0221 enthält weder Komplex I noch die alternative NADH Dehydrogenase, da das ndh-Gen durch Insertion einer Tetracyclin-Resistenzkassette und das nuoB-Gen durch eine Kanamycin-Resistenzkassette zerstört wurden (Böck, 1996). In diesem Stamm wurden die nuo-Gene auf dem Plasmid pBADnuo eingebracht und von dem Plasmid exprimiert. Dieser Stamm wurde in einem 10 l- Fermenter angezogen und die Zellen bis zu ihrer Verwendung bei -80°C gelagert. Für eine Präparation des Komplex I aus diesem Stamm wurden 60 bis 70 g Zellen (Feuchtgewicht) in 250 ml 50 mM MES/NaOH, pH 6.0 unter Zusatz von 200 µl PMSF (0.1 M in Isopropanol, Sigma), 0.5 g Lysozym (Fluka) und einer Spatelspitze DNase (Boehringer) resuspendiert und wie oben beschrieben die Plasmamembranen hergestellt. Die Membranen wurden in einen Teflon-in-Glas-Homogenisator überführt und ihr Gewicht bestimmt. Zur Extraktion von Komplex I aus den Membranen wurde je g Zellen 1 ml 50 mM MES/NaOH, 50 mM NaCl, 5 mM MgCl2, pH 6.0 (A*-Puffer), 4 µl 0.1 M PMSF und soviel B B einer 20 % (w/v) n-Dodecyl-β-D-maltopyranosidlösung (DDM, Glycon) zugegeben, wie zur Einstellung einer Endkonzentration von 3 % Detergenz nötig war. Die Suspension wurde vorsichtig homogenisiert und für 10 min inkubiert. Nach Zentrifugation bei 250´000 *g (50´000 Upm, Rotor 60Ti, L8-70M Ultrazentrifuge, Beckman) für 20 min wurde der Überstand mit 7 ml/min auf eine 80 ml Anionentauscher-Chromatographie Source15 Q (26 mm x 400 mm; Pharmacia Biotech) aufgetragen, die mit 50 mM MES/NaOH, 50 mM NaCl, 0.1 % (w/v) DDM, pH 6.0 (ADM-Puffer) äquilibriert worden war. Gebundene Proteine wurden B B mit einem linearen 700 ml Gradienten von 50 mM NaCl bis 350 mM NaCl in 50 mM MES/NaOH, 0.1 % (w/v) DDM, pH 6.0 bei einer Flussgeschwindigkeit von 5 ml/min eluiert. Die Fraktionen mit NADH/Ferricyanid Reduktaseaktivität, einer artefiziellen Aktivität des peripheren Arms des Komplexes, wurden vereinigt. Die Proteine wurden durch Zugabe einer 50 % (w/v) Polyethylenglycol 4´000-Lösung in ADM-Puffer (PEG, Boehringer) bei einer B B PEG-Endkonzentration von 9 % unter 10 min rühren auf Eis ausgefällt. Das ausgefällte Protein wurde für 5 min bei 12´000 *g (10´000 Upm, Rotor 8.24, RC-5 Superspeed Refrigerated Centrifuge, Sorvall) abzentrifugiert, das Sediment zweimal mit H2O gewaschen B B und in 1 ml A*DM-Puffer unter Rühren resuspendiert. Die so aufkonzentrierte Proteinlösung B B wurde auf eine 450 ml Größenausschluss-Chromatographiesäule (26 mm x 850 mm Sephacryl S300, Amersham Biosciences) aufgetragen, die mit A*DM-Puffer äquilibriert worden war. Die B - 35 - B Material und Methoden Proteine wurden in A*DM-Puffer mit einer Flussgeschwindigkeit von 20 ml/Std. bei einer Fraktionsgröße von 1.75 ml eluiert. Die Fraktionen mit NADH/Ferricyanid Reduktaseaktivität wurden vereinigt und bei -80°C gelagert. 3.5.3 Isolierung des Komplex I über Affinitäts-Chromatographie an StrepTactin Die DNA-Sequenz, die für das StrepTag II kodiert, wurde in pBADnuo jeweils an die Gene nuoB, nuoC und nuoF angefügt, so dass die entsprechenden Proteine C-terminal mit dem Affinitätspeptid versehen waren. Das so modifizierte Plasmid wurde in den Doppeldeletionsstamm ANN0221 transformiert und die nuo-Gene von pBADnuo exprimiert. Die Zellen wurden in 1 l-Kolben bzw. im 10 l-Fermenter angezogen und bis zu ihrer Verwendung bei -80°C gelagert. Aus 30 g Zellen wurden wie oben beschrieben Plasmamembranen hergestellt. Die Membranen wurden in 1 ml 50 mM MES/NaOH, 50 mM NaCl, pH 6.0 pro g Membran mit 50 µl PMSF (0.1 M in Isopropanol) resuspendiert. Pro ml Lösung wurde zur Extraktion der Membranproteine 0.175 ml 20 % (w/v) DDM zugesetzt. Nach 10 min Inkubation wurde 20 min bei 250´000*g (50´000 Upm, Rotor 60Ti, L8-70M Ultrazentrifuge, Beckman) zentrifugiert und der Überstand mit 6 ml/min auf eine 20 ml Source15 Q (50 mm x 75 mm; Pharmacia Biotech) aufgetragen, die mit ADM-Puffer äquilibriert worden war. Die Proteine wurden mit einem linearen 160 ml Gradienten von 150 mM NaCl bis 350 mM NaCl in 50 mM MES/NaOH, 0.1 % (w/v) DDM, pH 6.0 eluiert. Fraktionen mit NADH/Ferricyanid Reduktaseaktivität wurden vereinigt und auf eine 20 ml StrepTactin-Sepharosesäule (50 mm x 75 mm; Pharmacia Biotech) aufgetragen. Die Säule wurde mit ADM-Puffer gespült, bis die Absorbanz bei 280 nm auf das Grundniveau zurückgegangen war. Die gebundenen Proteine wurden mit 2.5 mM D-Desthiobiotin (Sigma) in ADM-Puffer eluiert. Fraktionen mit NADH/Ferricyanid Reduktaseaktivität wurden vereinigt und bis zur Verwendung bei -80°C gelagert. Das Säulenmaterial wurde mit 1 mM 4-Hydroxylbenzen-2-carboxylsäure (HABA, IBA) in ADM-Puffer regeneriert. - 36 - Material und Methoden 3.5.4 Aufreinigung des NADH-Dehydrogenase Fragmentes über eine AffinitätsChromatographie an StrepTactin 20 g Zellen des E. coli Stammes BL21(DE3)/pET11-a/nuoB-G/NuoFC wurden in 60 ml B B 50 mM MES/NaOH, 50 mM NaCl, pH 6.0, 20 µg Avidin, 10 µg/ml DNaseI (Boehringer), mit einer Tablette Proteaseinhibitor (Complete, Boehringer) in einem Teflon-in-GlasHomogenisator resuspendiert. Die Zellen wurden mit einem einmaligen Durchgang in der „French Press“ (SLM Aminco) bei 110 MPa aufgeschlossen und das Cytoplasma durch Ultrazentrifugation für 1 Std. bei 250´000*g (50´000 Upm, Rotor 60Ti, L8-70M Ultrazentrifuge, Beckman) erhalten. Das Cytoplasma wurde auf eine 50 ml DEAESepharosesäule (28 x 100 mm, Pharmacia) aufgetragen, die mit 50 mM MES/NaOH, 50 mM NaCl, pH 6.0 äquilibriert worden war. Die gebundenen Proteine wurden in einem linearen 400 ml Gradienten von 50 mM NaCl bis 400 mM NaCl in 50 mM MES/NaOH, pH 6.0 eluiert. Fraktionen mit NADH/Ferricyanid Reduktaseaktivität wurden vereinigt und der pH entweder mit 0.1 N oder 1 N NaOH unter Rühren auf Eis auf 7.0 eingestellt. Die Probe wurde auf eine 5 ml StrepTactin-Sepharose-Säule (7 x 65 mm, IBA) aufgetragen, die mit 100 mM Tris/HCl, 1 mM EDTA, pH 7.0 äquilibriert worden war. Das NADH-Dehydrogenase Fragment wurde mit 2.5 mM D-Desthiobiotin (Sigma) in 100 mM Tris/HCl, 1 mM EDTA, pH 7.0 eluiert. Die Fraktionen mit NADH/Ferricyanid Reduktaseaktivität wurden vereinigt, durch Ultrafiltration (Centricon 100, Amicon) aufkonzentriert und bei -80°C gelagert. Das Säulenmaterial wurde mit 1 mM HABA regeneriert. 3.5.5 Verschiedene analytische Methoden Proteinbestimmungen: U Die Proteinkonzentration wurde nach der Biuret-Methode (Beisenherz et al., 1953) bestimmt. Hierzu wurde 10 µl bis 50 µl Probe mit 1 ml 6% Trichloressigsäure versetzt und die denaturierten Proteine für 1 min bei 13´200 Upm (Zentrifuge 5415D, Eppendorf) abzentrifugiert. Der Überstand wurde verworfen und das Sediment in 1 ml Biuret-Reagenz unter Schütteln und bei 1´400 Upm bei Raumtemperatur für 30 min resuspendiert. Die Absorption der Proben bei 546 nm wurde gemessen. Nach Entfärbung der Proben durch Zugabe von einigen Körnchen KCN wurde die Absorption bei 546 nm erneut vermessen und als Hintergrund, bedingt durch die Trübung durch Lipide, abgezogen. Aus der so bestimmten - 37 - Material und Methoden Absorbanz wurde die Proteinmenge anhand einer zuvor aufgenommenen Eichgerade mit verschiedenen Mengen Rinderserumalbumin (Sigma) als Standard abgelesen. Die Konzentration des isolierten Komplex I wurde durch Messung der Absorbanz der Probe bei 278 nm (Ultrospec 1000; Pharmacia Biotech) bestimmt. Der Extinktionskoeffizient von Komplex I bei 278 nm (ε = 760 mM-1cm-1) wurde aus der von der DNA abgeleiteten P P P P Aminosäuresequenz mit Inkrementen für Tyrosin, Tryptophan und Cystin berechnet (Gill und von Hippel, 1989). Die Proteinkonzentration wurde aus der gemessenen Absorbanz nach dem Lambert-Beer´schen Gesetz berechnet. Zuckergradientenzentrifugation: U Die Assemblierung des Komplex I in den einzelnen E. coli Stämmen wurde mittels Zuckerdichtegradienten-Zentrifugation untersucht. Dazu wurden die Membranproteine der isolierten Cytoplasmamembranen mit DDM in einer Endkonzentration von 3 % (w/v) solubilisiert und nicht lösliche Bestandteile durch Zentrifugation bei 20´000*g (Kühlzentrifuge Sorvall Instruments RC-5, Rotor A 8.24, 16´000 Upm, 4°C) abgetrennt. Die Suspension wurde auf einen Zuckergradienten von 5-25 % (w/v) Saccharose in ADM-Puffer B B aufgetragen und 16 Std. bei 180´000 *g zentrifugiert (Ultrazentrifuge Beckman L8-70M, Rotor Ti 70, 36´000 Upm, 4°C). Die Gradienten wurden in 600 µl Portionen fraktioniert und deren NADH/Ferricyanid Reduktaseaktivität bestimmt. Da nur Komplex I in einem Molekulargewichtsbereich über 200 kDa diese Aktivität aufweist, weist ein Aktivitätsgipfel im letzten Drittel des Gradienten auf einen vollständig assemblierten Komplex I hin. Bestimmung der NADH/Ferricyanid Reduktaseaktivität: U Zu 1 ml 50 mM MES/NaOH, 50 mM NaCl, 1 mM K3[Fe(CN)6], pH 6.0 wurden 0.2 mM B B B B NADH gegeben. Nach Zugabe von 5 bis 20 µl Probe wurde die zeitliche Änderung der Absorbanz bei 410 nm bei Raumtemperatur gemessen (Ultrospec 1000; Pharmacia Biotech). Der molare Extinktionskoeffizient von K3[Fe(CN)6] beträgt ε = 1 mM-1cm-1 (Friedrich et al., B B B B P P P P 1989). Bestimmung der Atmungsaktivität: U Die (d-)NADH-Oxidase Aktivität der E. coli Plasmamembranen wurde durch Messung des Sauerstoffverbrauchs mit einer Clark-Sauerstoffelektrode bestimmt. Als Probenpuffer wurde 50 mM MES/NaOH, pH 6.0 verwendet, das Gesamtvolumen in der Probenkammer betrug - 38 - Material und Methoden 1 ml. Die Aktivität wurde bei 30°C gemessen. Zur Eichung der Elektrode wurde der Stromfluss in O2-gesättigtem Puffer und nach Reduktion des O2 durch Zugabe eines Überschusses Natriumdithionit gemessen. Die gemessene Differenz entspricht nach der Löslichkeit von O2 in H2O bei 30°C 237 µM O2. Es wurden 5 bis 20 µl Membransuspension eingesetzt. Die Reaktion wurde durch Zugabe von 10 µl einer 0.1 M NADH (Gerbu) bzw. d-NADH (Sigma) Lösung gestartet und der Verlauf der Sauerstoffabnahme mit einem Schreiber registriert. SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE): Es wurden diskontinuierliche Laemmli-Gele mit 3 %igem Sammel- und 16 %igem Trenngel verwendet (Laemmli, 1970). Je nach Proteinmenge wurden 5 bis 30 µl Probe mit 5 bis 10 µl Probenpuffer versetzt und 30 min bei Raumtemperatur geschüttelt. Die Proteine wurden bei einer Stromstärke von 30 mA aufgetrennt und die Proteinbanden durch Färbung in Coomassie-Blau-Lösung und anschließende Entfärbung in 30 % (v/v) Ethanol, 10 % (v/v) Essigsäure sichtbar gemacht. Silberfärbung von SDS-Gelen: SDS-Gele wurden zur Silberfärbung für mindestens 1 Std. in 10 % Essigsäure und 30 % Ethanol fixiert. Die Gele wurden dreimal mit 30 % Ethanol für 15 min gewaschen, bevor sie in 200 mg/l Na2S2O3.6 H2O für 1 min sensibilisiert wurden. Nach Waschen mit destilliertem H2O für zweimal 30 sec wurden die Gele mit 2 g/l AgNO3, 0.0125 % (v/v) Formaldehyd für 25 min imprägniert. Anschließend wurde zweimal für 20 sec mit destilliertem H2O gewaschen und mit 60 g/l Na2CO3, 200 mg/l Na2S2O3.6 H2O, 0.05 % (v/v) Formaldehyd bis zur deutlichen Färbung der Proteinbanden entwickelt. Die Reaktion wurde durch Zugabe konzentrierter Essigsäure gestoppt. Western Blot-Analyse: Nach ihrer gelelektrophoretischen Auftrennung wurden einzelne Proteine über eine Western Blot-Analyse nachgewiesen. Dazu wurden die Proteine in einer Tank-Blot-Apparatur in BlotPuffer bei 4°C und 400 mA je nach Größe des Proteins für 1 bis 2 Std. aus dem Polyacrylamid-Gel auf Cellulosenitrat-Membranen (Schleicher & Schüll) transferiert (Towbin et al., 1979). Alle weiteren Inkubationsschritte erfolgten bei Raumtemperatur auf einem Kippschüttler. Die Membranen wurden für 1 Std. in PBS mit 3 % (w/v) Magermilchpulver (Glücksklee) geschüttelt, um freie Bindeplätze der Membran abzudecken und anschließend - 39 - Material und Methoden 5 min mit PBST gewaschen. Die Membran wurde mit Antikörpern einer Verdünnung von 1:500 in PBST für 2 Std. inkubiert. Anschließend wurde zweimal für 5 min mit PBST gewaschen, um überschüssige Antikörper zu entfernen und für 1 Std. in PBST mit 1:5´000 verdünnter Protein A-gekoppelter Meerrettich-Peroxidase (Bio-Rad) inkubiert. Daraufhin wurden die Membranen je zweimal für 5 min erst mit PBS, anschließend mit 25 mM NaPhosphat, pH 7.2 gewaschen. Die gebundenen Zweit-Antikörper wurden durch Inkubation mit 25 mM Na-Phosphat, pH 7.2, 0,05 % (w/v) 3.3´-Diaminobenzidin (Sigma), 0.03 % (w/v) NiSO4, 0.03 % (w/v) CoCl2 und 0.1 % H202 sichtbar gemacht (De Blas und Cerwinski, 1983). B B B B B B B B ESR-Spektroskopie: U ESR-Spektren wurden in Zusammenarbeit mit Prof. Dr. Thorsten Friedrich aufgenommen. Proben wurden zur Messung auf 300 µl aufkonzentriert. Das aufgereinigte NADHDehydrogenase Fragment wurde in Gegenwart von Redoxmediatoren mit Dithionit reduziert. Cytoplasma-Proben wurden mit 20 mM NADH (Endkonzentration) reduziert. Zum Vergleich wurde ein Aliquot des Cytoplasmas mit dem gleichen Volumen Puffer verdünnt. Für die Differenzspektroskopie wurde das ESR-Spektrum dieser Probe von dem der reduzierten Probe abgezogen. Die Proben wurden zunächst in einer 1:5 (v/v) Isopentan/Methylcyclohexan-Kältemischung bei 150 K und anschließend in flüssigem Stickstoff gefroren und bis zur Messung bei -80°C gelagert. Tieftemperatur-ESR-Messungen wurden mit einem Bruker EMX 1/6 Spektrometer durchgeführt. Die Probentemperatur wurde mit einem ESR-9 Helium-Kryostaten (Oxford Instruments) kontrolliert. Das Magnetfeld wurde mit einem Bruker Standard geeicht. Die Mikrowellenfrequenz betrug 9.461 GHz, die Modulationsamplitude 6 G, die Zeitkonstante 164 ms und die Aufnahmegeschwindigkeit 17.9 mT/min. Permeabilisierung ganzer Zellen: U 4 g Zellen wurden mit 100 ml 50 mM MES, 50 mM KCl, pH 6.0 gewaschen und das Sediment in 50 ml 50 mM MES, 50 mM KCl, pH 6.0 resuspendiert. Die Suspension wurde 18 Std. bei Raumtemperatur leicht gerührt. Nach 10 min Zentrifugation bei 10´000 Upm (Zentrifuge 5804R, Eppendorf) wurde das Sediment in 10 ml 50 mM MES, 50 mM KCl, pH 6.0 resuspendiert, mit 300 µl Toluol versetzt und 30 min bei 4°C gerührt. Nach erneuter Zentrifugation für 10 min bei 10´000 Upm (Zentrifuge 5804R, Eppendorf) wurde das Sediment wiederum in 10 ml 50 mM MES, 50 mM KCl, pH 6.0 resuspendiert, 300 µl Toluol zugegeben und 30 min bei 4°C gerührt. Daraufhin wurden die Zellen dreimal mit je 50 ml - 40 - Material und Methoden 50 mM MES, 50 mM KCl, pH 6.0 wie oben beschrieben gewaschen, anschließend in 5 ml 50 mM MES, 50 mM KCl, pH 6.0 resuspendiert und bei -80°C gelagert. - 41 - Ergebnisse 4. Ergebnisse 4.1 Klonierung des extrachromosomalen Expressionssystems pBADnuo Die chromosomale DNA des Komplex I Überproduktionsstammes ANN003/pAR1219 (Spehr et al., 1999) wurde isoliert und daraus das nuo-Operon mit dem Restriktionsenzym ScaI herausgeschnitten. Die DNA-Fragmente wurden in einem Agarosegel aufgetrennt und ein 27´866 bp großes Fragment, welches das nuo-Operon enthält, durch Gelelution aufgereinigt. Es wurde in das Cosmid III (Roche) ligiert und mittels einer Phagenverpackung (Expand cloning kit, Roche) in E. coli eingeführt. Das Konstrukt wird im Folgenden als ScaCos bezeichnet und enthält das nuo-Operon, zwei T7-Promotoren sowie eine Ampicillin- und Spectinomycin-Resistenz. Es ist aber aufgrund der falschen Lage der Promotoren nicht zur Expression des nuo-Operons geeignet. Daher sollte das nuo-Operon von dem Cosmid ScaCos in das Plasmid pBAD33, das einen Arabinose-Promotor enthält, umkloniert werden. Zunächst wurde ein Konstrukt kloniert, in dem beide T7-Promotoren gegen einen Arabinose-Promotor aus dem Plasmid pBAD33 ausgetauscht wurden. Um den Beginn des nuo-Operons optimal zum Promotor auszurichten, wurde er mittels PCR amplifiziert und dabei stromaufwärts vor nuoA eine NsiI-Schnittstelle eingefügt (Abb. 4-1). Das PCR-Produkt, das die Gene nuoA und nuoB enthält, wurde mit den Restriktionsenzymen NsiI und SmaI geschnitten und in den mit SmaI verdauten Vektor pUC18 kloniert. Dieses Konstrukt wurde mit pUCPCR-AB bezeichnet. Aus dem Cosmid ScaCos wurden weitere nuo-Gene (nuoC-F und der Beginn von nuoG, bezeichnet als nuoG´) mit SmaI herausgeschnitten und in pUCPCR-AB eingefügt. In das resultierende Konstrukt pABS1 wurde der Arabinose-Promotor aus pBAD33 unter Verwendung der Restriktionsenzyme NsiI und PstI eingefügt. Das entstandene Konstrukt wurde mit pPromABS bezeichnet und enthält den Arabinose-Promotor und den Beginn des nuo-Operons bis zum Gen nuoG´ (Abb. 4-1). - 42 - Ergebnisse Abb. 4-1: Klonierung des Plasmids pPromABS, das den Arabinose-Promotor des Plasmids pBAD33 und den Beginn des nuo-Operons mit den Genen nuoA-nuoG´ enthält. Ausgangspunkt ist das Konstrukt ScaCos, das das mit ScaI einklonierte nuo-Operon enthält. Die folgenden Klonierungsschritte dienten zur korrekten Positionierung des ArabinosePromotors stromaufwärts zum nuo-Operon. Die restlichen Gene des nuo-Operons wurden in weiteren Klonierungsschritten eingefügt. Der Arabinose-Regulator und -Promotor und der im Leseraster gelegene Beginn des nuo-Operons wurden aus pPromABS mit den Restriktionsenzymen NsiI und BglII herausgeschnitten und in das mit NsiI und BamHI geschnittene Plasmid pBAD33 umkloniert, so dass der Promotorbereich von pBAD33 gegen den Promotor mit dem Beginn des nuo-Operons - 43 - Ergebnisse ausgetauscht wurde (Abb. 4-2). Dieses Konstrukt wurde als pBADProm bezeichnet Die fehlenden nuo-Gene wurden aus dem Cosmid ScaCos eingefügt. Hierfür wurde pBADProm mit PstI geschnitten, die überhängenden Enden enzymatisch durch das Klenow-Fragment geglättet und anschließend mit SgfI geschnitten. Das Konstrukt ScaCos wurde mit EcoO109I geschnitten, mit dem Klenow-Fragment geglättet und anschließend mit SgfI verdaut. Das resultierende 12´706 bp große Fragment wurde mit dem 8´572 bp großen DNA-Fragment aus pBADProm ligiert (Abb. 4-2). Abb. 4-2: Klonierung des Plasmids pBADnuo, ausgehend von den Plasmiden pBAD33 und pPromABS, das den Arabinose-Promotor im Leseraster des Starts des nuo-Operons enthält. Nach Austausch des Promotorbereichs aus pBAD33 gegen den aus pPromABS wurden die restlichen nuo-Gene von dem Cosmid ScaCos in das Plasmid pBADProm eingefügt. Der Ligationsansatz der DNA-Fragmente pBADProm/PstI(blunt)+SgfI und ScaCos/EcoO109I(blunt)+SgfI wurde in die E. coli Stämme RR1∆M15 und BL21(DE3) transformiert. Aus den Transformanden wurde mit einer Mini-Präparation die Plasmid-DNA aufgereinigt und mit einer Restriktionsanalyse untersucht. Es zeigte sich, dass die PlasmidDNA von fast allen Restriktionsenzymen auch bei geringen Enzymkonzentrationen zersetzt wurde. Bei der Auftrennung der Restriktionsansätze auf einem Agarosegel wurde bei verschiedenen Ansätzen nur ein Gemisch von DNA-Fragmenten aller Größen detektiert. Nur - 44 - Ergebnisse bei dem Restriktionsenzym KpnI wurde in mehreren Plasmid-Präparationen das erwartete Restriktionsmuster des Plasmids pBADnuo festgestellt (Abb. 4-3). Abb. 4-3: Restriktionsanalyse mit KpnI nach Ligation der DNA-Fragmente pBADProm/PstI(blunt)+SgfI und ScaCos/EcoO109I(blunt)+SgfI zur Klonierung des Plasmids pBADnuo. Im Fall der gelungenen Klonierung wurden Fragmentgrößen von 12´342 bp und 8´936 bp erwartet (Spur 1, mit Pfeilen gekennzeichnet). Als Standard wurde pUC-Mix 4 (MBI Fermentas, St. Leon-Rot) verwendet (pUC4). Da nur bei der Restriktionsanalyse mit dem Enzym KpnI die erwarteten Fragmente erhalten wurden, bei Verwendung anderer Restriktionsenzyme die Plasmid-DNA abgebaut wurde, ist zu vermuten, dass dies aus der hohen Ringspannung des 21´278 bp großen Plasmids resultiert oder aber Verunreinigungen der Präparation sich störend bei einer Restriktion der Plasmide auswirken. Die Plasmide, die bei dem Schnitt mit KpnI das erwartete Restriktionsmuster zeigten, wurden nach verschiedenen Methoden im Mini- und auch im Midi-Maßstab präpariert, um möglicherweise störende Einflüsse von Verunreinigungen der Mini-Präparation auszuschliessen. Die Plasmid-DNA dieser Präparationen wurden durch alle verwendeten Restriktionsenzyme außer KpnI abgebaut. Hieraus wurde geschlossen, dass die Ringspannung der Plasmid-DNA der Grund für den unspezifischen Abbau ist. Das Plasmid pBADnuo wurde in E. coli DH5α transformiert, aus den Transformanden die Plasmide präpariert und mittels Restriktionsanalyse untersucht. Es wurde eine Kolonie erhalten, deren Plasmide mit verschiedenen Restriktionsenzymen das jeweils erwartete Restriktionsmuster des Plasmids pBADnuo zeigten (Tab. 4-1, Abb. 4-4). Gegenüber den - 45 - Ergebnisse vorher genutzten B-Stämmen BL21(DE3) und RR1∆M15 zeigt sich der K12-Stamm DH5α zur Vermeidung hoher Ringspannungen geeignet. Tab. 4-1: Erwartete Fragmentgrößen des Plasmids pBADnuo nach Restriktionsanalyse mit verschiedenen Restriktionsenzymen. Restriktionsenzym Fragmentgrößen [bp] NcoI 14´036 5´903 1´339 HindIII 10´938 6´549 2´710 1´081 EcoRV 7´331 5´643 5´320 1´573 1´411 PstI 17´229 4´049 KpnI 12´342 8´936 NheI 18´993 2´285 - 46 - Ergebnisse Abb. 4-4: Restriktionsanalyse der Transformanden des Plasmids pBADnuo in E. coli DH5α. Das jeweils verwendete Restriktionsenzym ist angegeben. Die erwarteten Fragmentgrößen finden sich in Tab. 4-1. Als Standard wurde pUC-Mix 4 (pUC4; MBI Fermentas, St. LeonRot) aufgetragen. 4.2 Produktion von Komplex I vom Plasmid pBADnuo Das Plasmid pBADnuo soll hauptsächlich zu Mutagenesestudien des E. coli Komplex I eingesetzt werden. Zu diesem Zweck muss die Expression der Gene des nuo-Operons, die korrekte und vollständige Assemblierung des Komplexes und eine möglichst große Proteinproduktion gewährleistet sein. Weiterhin sollte der Stamm, in dem das Plasmid exprimiert wird, keine chromosomale Kopie des nuo-Operons enthalten. Um dies zu überprüfen, wurden von den jeweiligen Transformanden Wachstumskurven in Mangelmedien aufgenommen, die Atmungsaktivität der Plasmamembranen gemessen und die Stabilität des Komplexes nach Detergenzextraktion in einem Saccharosegradienten überprüft. 4.2.1 Wachstumskurven von nuo/ndh-Doppeldeletionsstämmen mit pBADnuo Um bei der Expression des nuo-Operons vom Plasmid pBADnuo eine Hintergrundaktivität der chromosomalen nuo-Kopie auszuschließen, sollten E. coli Stämme verwendet werden, die keinen Komplex I produzieren. Es wurden Stämme aus Arbeitsgruppenbeständen benutzt, bei denen in nuoB (ANN0221, ANN0222) bzw. nuoI (ANN0921, ANN0922) eine KanamycinResistenzkassette insertiert worden war. Durch diese Insertion wird die Transkription stromabwärts gelegener Gene verhindert und der Komplex I assembliert nicht (Böck, 1996). - 47 - Ergebnisse Alle vier Stämme tragen außerdem eine Tetracyclin-Resistenzkassette in dem Gen ndh, das für die alternative NADH-Dehydrogenase kodiert und ebenfalls nicht transkribiert wird (Böck, 1996). Das Plasmid pBADnuo wurde in diese vier Doppeldeletionsstämme transformiert und von den Transformanden Wachstumskurven in Mangelmedium aufgenommen. Es wurde erwartet, dass die Doppeldeletionsstämme ohne membranständige NADH-Dehydrogenasen in Mangelmedium kaum Wachstum zeigen, während Deletionsstämme mit dem Plasmid pBADnuo bei korrekter Produktion des Komplexes auch in Mangelmedium wie der Wildtyp AN387 wachsen sollten (Abb. 4-5). Abb. 4-5: Anzucht von zehn Kolonien des E. coli Stammes AN387 auf LB-Medium (links) und M46/Mannitol-Medium (rechts). Die Doppeldeletionsstämme ANN0221, ANN0222, ANN0921 und ANN0922, die aus acht Jahre alten Stammkulturen angeimpft wurden, wurden zunächst daraufhin überprüft, ob sie auf M46-Mangelmedium mit 20 mM Mannitol tatsächlich ein verlangsamtes Wachstum zeigten. Die Stämme wurden auf LB-Medium vereinzelt und jeweils 10 Kolonien auf M46/Mannitol-Medium überimpft. Von diesen Kolonien wurde je eine augewählt, die auf Mangelmedium schlechtes und auf LB-Medium gutes Wachstum zeigte (Abb. 4-6). Als Vergleich wurde der Wildtyp-E. coli-Stamm AN387 (Abb. 4-5) sowie der Komplex IÜberproduktionsstamm ANN003/pAR1219 (Abb. 4-7) ebenfalls auf Mangelmedium und LBMedium gezogen. Im Fall des Überproduktionsstammes wurde dem Medium IPTG zur Induktion der Komplex I-Produktion zugesetzt (Abb. 4-7). Auf den Platten mit Kolonien der Stämme AN387 und ANN003/pAR1219 war deutlich zu sehen, dass sich das Wachstum der Zellen auf Mangelmedium nur wenig von dem auf LB-Medium unterscheidet (Abb. 4-5, 4-7). - 48 - Ergebnisse Abb. 4-6: Anzucht einzelner Kolonien der E. coli Stämme ANN0221 (links oben), ANN0222 (rechts oben), ANN0921 (links unten) und ANN0922 (rechts unten) auf (a) LB-Medium und (b) M46/Mannitol-Mangelmedium. Abb. 4-7: Wachstum von zehn Kolonien des E. coli-Stammes ANN003/pAR1219 auf LBMedium mit 0.1 % (w/v) IPTG (links) und M46/Mannitol-Mangelmedium (rechts). Alle Kolonien der Doppeldeletionsstämme ANN0221, ANN0222 und ANN0922 zeigten auf Mangelmedium auch nach 60 Std. Inkubation bei 37°C nahezu kein Wachstum, während sie auf LB-Medium normal wuchsen. Allerdings zeigten nur vier Kolonien des Stammes ANN0921 ein verlangsamtes Wachstum, während die übrigen Kolonien vergleichbar schnell wie auf LB-Medium wuchsen (Abb. 4-6). Von den Kolonien, die auf M46/MannitolMangelmedium ein geringes und auf LB-Medium ein gutes Wachstum zeigten, wurde jeweils eine Übernachtkultur angesetzt. Diese Kulturen wurden zur Herstellung von Dauerkulturen - 49 - Ergebnisse sowie elektrokompetenter Zellen verwendet. Das Plasmid pBADnuo wurde durch Elektroporation in diese Zellen transformiert. Die resultierenden Stämme ANN0221/pBADnuo, ANN0222/pBADnuo, ANN0921/pBADnuo, ANN0922/pBADnuo wurden 1:100 in M9-Mangelmedium mit 20 mM Mannitol angeimpft. Die Expression der nuo-Gene wurde mit 0.2 % (w/v) L(+)-Arabinose induziert und Wachstumskurven der einzelnen Stämme aufgenommen (Abb. 4-8). Zum Vergleich wurden Wachstumskurven der nicht transformierten Doppeldeletionsstämme aufgenommen (Abb.4-8). Abb. 4-8: Wachstumskurven der E. coli-Stämme (a) ANN0221 ( ), ANN0222 ( ), ANN0921( ), ANN0922 ( ) sowie (b) ANN0221/pBADnuo( ), ANN0222/pBADnuo ( ), ANN0921/pBADnuo( ) und ANN0922/pBADnuo ( ) in M9-Medium mit 0.2 % (w/v) L(+)-Arabinose. - 50 - Ergebnisse Nur der Doppeldeletionsstamm ANN0221 wuchs unterschiedlich schnell, je nachdem, ob er das Plasmid pBADnuo enthielt oder nicht (Abb. 4-8). Die übrigen Stämme zeigten ohne die Expression der nuo-Gene ein gutes Wachstum in Mangelmedium (Abb. 4-8). Es ist anzunehmen, dass diese Deletionsmutanten im Lauf ihrer achtjährigen Lagerung mit WildtypZellen verunreinigt wurden. Daher wurde der Stamm ANN0221/pBADnuo für weitere Arbeiten ausgewählt. 4.2.2 Atmungsaktivität des E. coli Stammes ANN0221/pBADnuo Der E. coli-Stamm ANN0221/pBADnuo wurde in 1 l LB-Medium angezogen und die Expression der nuo-Gene durch Zugabe von 0.2 % (w/v) L(+)-Arabinose induziert. Aus 2.5 g Zellen wurden die Membranen präpariert und in 200 µl 50 mM MES, 50 mM NaCl, pH 6.0 resuspendiert. Analog dazu wurden die Membranen des Stammes ANN0221 präpariert und von beiden Membranen die d-NADH-Oxidase Aktivität mit einer Sauerstoffelektrode gemessen. Die Membranen des Stammes ANN0221 zeigten -wie erwartet- nach Zugabe von NADH keinen Sauerstoffverbrauch (Abb. 4-9). Wurde in die gleiche Messung Membranen des E. coli Wildtyp-Stammes AN387 zugegeben, wurde eine starke Abnahme der Sauerstoffkonzentration beobachtet (Abb. 4-9). Abb. 4-9: Sauerstoffverbrauch von Cytoplasma-Membranen des E. coli Stammes ANN0221 nach Zugabe von NADH und Sauerstoffverbrauch von Membranen des E. coli Wildtyps AN387 in der gleichen Messung. Die Sauerstoffkonzentration wurde mit einer ClarkeElektrode bei 37°C bestimmt. Die Messung enthielt 10 µg Membranprotein des Stamms ANN0221 und 43 µg Membranprotein des Wildtyp-Stammes. - 51 - Ergebnisse Die Membranen des Stammes ANN0221/pBADnuo zeigten sowohl mit NADH als auch mit d-NADH als Substrat eine deutliche und vergleichbare Atmungsaktivität (Abb. 4-10). Die Aktivität betrug mit NADH als Substrat 0.23 µmol O2/min.mg, B B P P mit d-NADH 0.25 µmol O2/min.mg (Tab. 4-2). Diese Ergebnisse zeigen, dass beide Dehydrogenase B B P P Aktivitäten von dem auf pBADnuo kodierten Komplex I stammen und nicht von genomisch kodiertem Komplex I oder der alternativen NADH-Dehydrogenase aus E. coli. Die im Vergleich zum Wildtyp etwas 3.5fach erhöhte d-NADH-Oxidase Aktivität (Tab. 4-2) zeigt, dass der Komplex I in dem Stamm ANN0221/pBADnuo etwa 3- bis 4-fach überproduziert wird. Die NADH-Oxidase Aktivität der Membranen des Stammes ANN0221/pBADnuo ist mit der des Wildtyps AN387 vergleichbar. Im Stamm ANN0221/pBADnuo stammt diese Aktivität aber vollständig von Komplex I, während sie im Wildtyp AN387 zum größten Teil von der alternativen NADH-Dehydrogenase stammt. Dies ist an der wesentlich geringeren d-NADH-Oxidase Aktivität des Wildtyp-Stammes ersichtlich, die ausschließlich von Komplex I hervorgerufen wird. Abb. 4-10: Sauerstoffverbrauch der Cytoplasma-Membranen des Stammes ANN0221/pBADnuo gemessen mit einer Clark-Sauerstoffelektrode bei Zugabe von d-NADH (a) und NADH (b). Die Sauerstoffkonzentration wurde bei 37°C bestimmt. Beide Messungen enthielten 10 µg Membranprotein. - 52 - Ergebnisse Tab. 4-2: (d-)NADH-Oxidase Aktivitäten der Cytoplasmamembranen der E. coli Stämme ANN387 und ANN0221/pBADnuo. Die Werte für den Stamm AN387 stammen von Friedrich et al. (1994). E. coli Stamm NADH-Oxidase Aktivität d-NADH-Oxidase Aktivität [µmol O2/min.mg] B B P P AN387 (Wildtyp) 0.26 0.07 ANN0221/pBADnuo 0.23 0.25 4.2.3 Assemblierung des Komplex I in ANN0221/pBADnuo Um die Assemblierung und Stabilität des in dem Stamm ANN0221/pBADnuo gebildeten Komplex I zu untersuchen, wurden die Membranproteine der Cytoplasmamembran mit 3 % DDM (w/v) solubilisiert. Der Extrakt wurde auf Zuckerdichtegradienten von 5 bis 20 % (w/v) Saccharose aufgetragen und 16 Std. bei 180´000 g zentrifugiert. Die Gradienten wurden fraktioniert und die NADH/Ferricyanid Reduktaseaktivität der einzelnen Fraktionen vermessen. Als Vergleich diente ein Extrakt der Cytoplasmamembranen des Stammes ANN0221 (Abb. 4-11). Der Extrakt der Membranen des Stammes ANN0221/pBADnuo zeigte einen deutlichen Gipfel der NADH/Ferricyanid Reduktaseaktivität im zweiten Drittel des Gradienten. Dies entspricht der Position des Komplex I aus Wildtyp (Leif et al., 1994). Dieser Gipfel ist im Extrakt des Stammes ANN0221 nicht nachzuweisen. Der Aktivitätsgipfel im vorderen Teil des Gradienten rührt von der alternativen NADH Dehydrogenase her. - 53 - Ergebnisse Abb. 4-11: Zuckergradienten-Zentrifugation der Detergenzextrakte der CytoplasmaMembranen der Stämme ANN0221 ( ) und ANN0221/pADnuo ( ). Gezeigt ist die NADH/Ferricyanid Reduktaseaktivität der einzelnen Fraktionen. Die Gradienten wurden mit je 5 mg Protein beladen. 4.2.4 Isolierung des Komplex I aus dem Stamm ANN0221/pBADnuo 67 g Zellen ANN0221/pBADnuo wurden in der „French-press“ aufgeschlossen und 10 g Cytoplasmamembranen durch differentielle Zentrifugation erhalten. Die Membranproteine wurden mit 3 % DDM solubilisiert, nicht gelöste Bestandteile abzentrifugiert und der Extrakt auf eine Anionenaustausch-Chromatographie an Source 15Q aufgetragen. Fraktionen mit NADH/Ferricyanid Reduktaseaktivität wurden vereinigt, mit PEG 4´000 ausgefällt, in einem geringen Volumen ADM-Puffer gelöst und durch Größenausschluss-Chromatographie auf Sephacryl S300 weiter aufgereinigt. Fraktionen mit NADH/Ferricyanid Reduktaseaktivität wurden vereinigt und die Reinheit der Präparation über eine SDS-PAGE überprüft. Nach der Größenausschlusschromatographie waren nur wenige Verunreinigungen in der Präparation vorhanden (Abb. 4-12), so dass für den Nachweis, dass der vollständige Komplex aufgereinigt wurde, kein weiterer chromatographischer Schritt notwendig war. - 54 - Ergebnisse Abb. 4-12: Isolierung des Komplex I aus dem Stamm ANN0221/pBADnuo über eine Anionenaustausch-Chromatographie an Source 15Q (a) und GrößenausschlussChromatographie auf Sephacryl S300 (b). Die durchgezogene Kurve zeigt die Absorbanz bei 280 nm, die Punkt-Strich Linie den Verlauf der NADH/Ferricyanid Reduktaseaktivität und die gestrichelte Linie die Lage des NaCl-Gradienten. In (c) ist die SDS-PAGE zur Auftrennung der Proben der einzelnen Reinigungsschritte gezeigt. M: Membranen (5 µl Auftrag; 52 mg/ml); E: Detergenz-Extrakt (5 µl Auftrag; 27 mg/ml); SQ: Gipfelfraktion nach Anionenaustausch-Chromatographie (40 µl Auftrag; 2.6 mg/ml); KI: aufgereinigter Komplex I aus dem Stamm ANN003/pAR1219 nach der Standard-Präparation (15 µl Auftrag; 1 mg/ml); Sep: Gipfelfraktionen nach Größenausschluss-Chromatographie (je 40 µl Auftrag; vereinigte Fraktionen 0.7 mg/ml). - 55 - Ergebnisse Tab. 4-3: Isolierung von Komplex I aus E. coli ANN0221/pBADnuo. Es wurden 67 g Zellen (Feuchtgewicht) eingesetzt. Präparationsschritt Volumen Proteinmenge [ml] [mg] NADH/Ferricyanid Ausbeute Reduktaseaktivität [%] gesamt spezifisch [µmol/min] [µmol/(min•mg)] Membranen 64 3328 3962 1.2 100 Detergenzextrakt 58 1566 3550 2.3 90 AnionenaustauschChromatographie 22 57 461 8.1 12 GrössenausschlussChromatographie 14 10 347 34.7 9 Bei einer typischen Präparation des Komplex I aus dem Überproduzenten ANN003/pAR1219 wurden aus 55 g Zellen mit den gleichen Reinigungsschritten 25 mg Protein einer spezifischen NADH/Ferricyanid Reduktaseaktivität von 48 µmol/(min•mg) isoliert (Stolpe und Friedrich, 2004). Dies zeigt, dass die hier erhaltene Präparation nicht so sauber ist, wie die aus dem Stamm ANN003/pAR1219. Bezieht man die Präparationen auf die gleiche Ausgangsmenge Zellen, wurde etwa ein Drittel des Proteins im Vergleich zu der Präparation aus ANN003/pAR1219 gewonnen. 4.3 Das Subklonsystem des Plasmids pBADnuo Das Plasmid pBADnuo sollte für Mutagenese-Studien eingesetzt werden, wobei seine Größe von gut 21 kbp ein Problem für die Mutagenese mittels PCR und nachfolgende Sequenzierungen darstellt. Um zu verhindern, dass die Polymerase Fehler einbaut und dass die Elongation verfrüht abgebrochen wird, sollte eine Mutagenese-PCR an kleineren Plasmiden durchgeführt werden. Hierzu wurde das Operon durch singuläre Restriktionsschnittstellen in drei Subklone aufgeteilt: pBAD-A, pBAD-B und pBAD-C (Abb. 4-13). Da die Subklone immer noch zwischen 5.9 kbp und 10.3 kbp groß sind, sollten sie in zwei bis drei noch kleinere Subklone aufgeteilt werden. Hierbei wurden Schnittstellen verwendet, die zwar nicht in pBADnuo, aber in den Subklonen pBAD-A, -B und -C singulär - 56 - Ergebnisse vorkommen (Abb. 4-13). So sollten die nuo-Gene in den kleinen Subklonen modifiziert werden können und diese anschließend über den größeren Subklon in das Plasmid pBADnuo zurückkloniert werden, von dem aus dann die Expression der mutierten Gene stattfindet (Abb. 4-13). Abb. 4-13: Schema des Subklonsystems des nuo-Operons. Ausgehend vom Plasmid pBADnuo sollten drei Subklone pBAD-A, pBAD-B und pBAD-C erstellt werden, von denen jeweils zwei oder drei kleinere Subklone kloniert werden sollten. Die in den Plasmiden enthaltenen nuo-Gene und die benötigten, jeweils singulären Schnittstellen sind angegeben. - 57 - Ergebnisse 4.3.1 Vektor für die Subklonierung des Plasmids pBADnuo Für die Subklone mussten Vektoren verwendet werden, in denen die singulären Erkennungssequenzen in pBADnuo ebenfalls singulär nur in der MCS vorkommen. Für die Subklone pBAD-A, pBAD-B und pBAD-C musste ein Vektor mit Sequenzen für die Restriktionsenzyme SacI, XhoI, ApaI und NdeI zur Verfügung stehen. Für die kleineren Subklone wurden weiterhin Erkennungssequenzen für die Enzyme EcoRI, BglII, KpnI und EcoRV benötigt. Aus dem Klonierungsvektor pUC18 wurde zunächst mittels PCR ein Vektor hergestellt, in den 34 zusätzliche Basenpaare eingeführt wurden (Abb. 4-14). Das resultierende Konstrukt wird im Folgenden als pUC20 bezeichnet. DpnI PruI ApaI ↓ ↓ ↓ 5´-AAC AAT TTG CTC AGC GAT CGC GTA CGG GCC CTA AGG ACC G-3´ ↑ MboI ↑ SgfI ↑ Eco0109I Abb. 4-14: In pUC18 eingefügte Sequenz zur Herstellung des Vektors pUC20. Pfeile kennzeichnen die Positionen von Erkennungssequenzen verschiedener Restriktionsenzyme. Um weitere Restriktionsschnittstellen in den Vektor pUC20 einzufügen, wurde pUC20 mittels PCR weiter mutagenisiert. Die Erkennungssequenzen für die Restriktionsenzyme SmaI, EcoRV, RsrII und NruI (Abb. 4-15) wurden mittels überhängenden Primern (pUC25-for und pUC25-rev) zwischen der MCS und der NdeI-Schnittstelle von pUC20 eingeführt. Das resultierende Plasmid wird im Folgenden als pUC24 bezeichnet. RsrII EcoRV SmaI NruI ↓ ↓ ↓ ↓ 5´-CGG TCC GAT ATC CCC GGG GGA CTT TCG CGA-3´ Abb. 4-15: Zur Herstellung des Vektors pUC24 in den Vektor pUC20 eingeführte Sequenz. Die Pfeile geben die Positionen der Erkennungssequenzen verschiedener Restriktionsenzyme an. - 58 - Ergebnisse Nach der Mutagenese-PCR wurde der Ursprungsvektor mit DpnI verdaut. Das unmethylierte PCR-Produkt, das nicht fragmentiert wurde, wurde in E. coli RR1∆M15 transformiert. Aus einzelnen Kolonien wurden die Plasmide präpariert und mittels Restriktionsanalyse charakterisiert (Abb. 4-16). Abb. 4-16: Restriktionsanalyse des Vektors pUC24 mit EcoRV+ScaI (Spur 1) bzw. RsrII+ScaI (Spur 2). Die erwarteten Fragmentgrößen betrugen 1´969 bp und 784 bp (EcoRV+ScaI) bzw. 1´962 bp und 791 bp (RsrII+ScaI). Als Standard wurde in Spur 3 pUCMix 4 (MBI Fermentas, St. Leon-Rot) aufgetragen. In den Vektor pUC24 wurde eine weitere Restriktionsschnittstelle für XhoI mittels einer Mutagenese-PCR mit den Primern pUC25/2-rev und pUC25/2-for eingefügt, die hinter der NruI-Schnittstelle für die Sequenz 5´-CTC GAG-3´ kodiert. Das so hergestellte Plasmid pUC25 wurde mittels Restriktionsanalyse verifiziert (Tab. 4-4, Abb. 4-17). - 59 - Ergebnisse Tab. 4-4: Erwartete Fragmentgrößen nach Restriktionsverdau des Plasmids pUC25 mit verschiedenen Enzymen. Restriktionsenzyme Fragmentgrößen [bp] RsrII + ScaI 1´968 791 EcoRV + ScaI 1´975 784 SmaI + ScaI 1´981 778 NruI + ScaI 1´991 768 XhoI + ScaI 1´515 1´244 Abb. 4-17: Restriktionsanalyse des Plasmids pUC25 mit RsrII und ScaI (Spur 2), EcoRV und ScaI (Spur 3), SmaI und ScaI (Spur 4), NruI und ScaI (Spur 5) und XhoI und ScaI (Spur 6). Als Standard wurde pUC-Mix 4 eingesetzt (Spur 1 und 7; MBI Fermentas, St. Leon-Rot). - 60 - Ergebnisse 4.3.2 Klonierung der pBADnuo-Subklone und der kleinen Subklone Um Subklone des Plasmids pBADnuo herzustellen, wurde es mit den in Tab. 4-5 aufgeführten Restriktionsenzymen verdaut und die entstandenen DNA-Fragmente mittels Gelelution aufgereinigt. Der Vektor pUC25 wurde mit den gleichen Restriktionsenzymen geschnitten. Die Fragmente von pBADnuo wurden mit dem entsprechend geschnittenen und aufgereinigten Vektor ligiert und in E. coli RR1∆M15 transformiert. Die zur Herstellung der Fragmente benutzten Restriktionsenzyme wurden ebenfalls zur Analyse der Klone verwendet. Tab. 4-5: Subklone des Plasmids pBADnuo. Aufgeführt sind die verwendeten Restriktionsenzyme, die in den entsprechenden Fragmenten enthaltenen nuo-Gene, die Größe der Fragmente und die Gesamtgröße des resultierenden Subklons. Subklon Restriktionsenzyme Enthaltene nuo- Fragmentgröße Größe des Plasmids Gene [bp] [bp] pBAD-A SacI + XhoI nuoA-nuoE´ 3´401 5´942 pBAD-B XhoI + ApaI nuoE´-nuoL´ 7´417 9´928 pBAD-C ApaI + NdeI nuoL´-nuoN 5´091 7´547 pBAD-A1 SacI + EcoRI nuoA-nuoCD´ 1´806 4´563 pBAD-A2 EcoRI + XhoI nuoCD´-nuoE´ 1´595 4´138 pBAD-B1 XhoI + EcoRI nuoE´-nuoF´ 1´100 3´643 pBAD-B2 EcoRI + KpnI nuoF´-nuoI´ 4´441 7´184 pBAD-B3 KpnI + ApaI nuoI´-nuoL´ 1´876 4´619 pBAD-B4 XhoI + KpnI nuoE´-nuoI´ 5´541 8´068 pBAD-C1 ApaI + EcoRV nuoL´-nuoM´ 2´199 4´746 pBAD-C2 EcoRV + NdeI nuoM´-nuoN 2´892 5´560 Von den geplanten Subklonen wurden in dieser Arbeit die Plasmide pBAD-A, pBAD-A1, pBAD-A2, pBAD-B2, pBAD-B3, pBAD-B4 und pBAD-C1 hergestellt (Abb. 4-18). Nicht kloniert wurden die Plasmide pBAD-B, pBAD-B1, pBAD-C und pBAD-C2. Auch Versuche, die Inserts, d.h., die in den Vektor zu inserierenden DNA-Fragmente, in einen anderen Vektor - 61 - Ergebnisse und unter Verwendung anderer Restriktionsenzyme mit gleicher Erkennungssequenz zu klonieren, waren nicht erfolgreich. Abb. 4-18: Restriktionsanalyse der Plasmide pBAD-A (Spur 1), pBAD-A1 (Spur 2), pBAD-A2 (Spur 3), pBAD-B2 (Spur 5), pBAD-B3 (Spur 6), pBAD-B4 (Spur 7) und pBAD-C1 (Spur 9). Die Plasmide wurden jeweils mit den Enzymen verdaut, mit denen sie hergestellt wurden (Tab. 4-5). Als Standard wurde pUC-Mix 4 (Spur 4 und 10) bzw. λ-DNA/EcoRI+HindIII (Spur 8; beide MBI Fermentas, St. Leon-Rot) eingesetzt. Mit dem in dieser Arbeit etablierten Subklonsystem sind die Bereiche nuoA-nuoM´ des nuoOperons abgedeckt und stehen für Mutagenesen zur Verfügung. Eine direkte Rückklonierung mutierter Gene ist nur mit den kleinen Subklonen pBAD-A1 und pBAD-A2 über das Plasmid pBAD-A möglich. Für die Rückklonierung von Mutationen in den kleinen Subklonen pBADB2, pBAD-B3, pBAD-B4 und pBAD-C1 in pBADnuo fehlen bislang die benötigten Subklone pBAD-B und pBAD-C. Da die Schnittstellen der nuo-Fragmente in den kleinen Subklonen der Plasmide pBAD-B und pBAD-C in pBADnuo nicht singulär vorliegen, können diese Fragmente nicht direkt in pBADnuo rückkloniert werden. Die kleinen Subklone von pBAD-B und pBAD-C sind zur Mutagenese geeignet, die Mutationen müssen aber auf anderem Weg in pBADnuo transferiert werden. Eine Alternative zur Klonierung ist die λ-Red vermittelte Rekombination (Datsenko und Wanner, 2000), bei der Mutationen mit beliebigen Restriktionsenzymen aus den kleinen Subklonen herausgeschnitten und in pBADnuo gegen den Wildtyp-Bereich ausgetauscht werden können. Das Subklonsystem ist für die Mutagenese dieser Bereiche nicht geeignet. Die λ-Red vermittelte Rekombination zur Einführung von Punktmutationen, Deletionen oder Insertionen in pBADnuo kann in E. coli Zellen durchgeführt werden, die außer dem Zielplasmid pBADnuo das Plasmid pKD46 enthalten (s. Anhang). pKD46 kodiert für die - 62 - Ergebnisse λ-Red Rekombinase, deren Expression über einen Arabinosepromotor durch die Induktion mit L-Arabinose erfolgt. pBADnuo und pKD46 werden durch Kotransformation in E. coli eingeführt und bei 30°C in LB-Medium mit den Antibiotika Chloramphenicol und Ampicillin gemeinsam propagiert. pBADnuo kodiert dabei für eine Acetyltransferase, die die Chloramphenicolresistenz vermittelt, pKD46 für β-Lactamase, die Ampicillinresistenz vermittelt. Das zu rekombinierende DNA-Fragment, das neben dem auszutauschenden Bereich flankierende homologe Enden mit einer Länge von mindestens 36 bp aufweist, wird durch Elektroporation in die E. coli Zellen mit den beiden Plasmiden transformiert. Das Plasmid pKD46 enthält einen temperatursensitiven Replikationsursprung, so dass bei 37°C keine weitere Replikation des Plasmids stattfindet. Werden die Zellen nach der Rekombination bei 37°C ohne Ampicillin angezogen, wird nur pBADnuo weiterhin repliziert. 4.4 Einführung einer StrepTag II-Sequenz in pBADnuo 4.4.1 Modifizierung von NuoB Bei den in diesem Kapitel beschriebenen Arbeiten wurde Frau Julia Hinderberger im Rahmen ihrer Diplomarbeit (2004) angeleitet. In den Subklon pBAD-A wurde am 5´-Ende von nuoB die StrepTag II-Sequenz eingefügt. Dazu wurde zunächst über eine PCR nuoB und nuoC amplifiziert, wobei der Primer am 3´-Ende von nuoB und am 5´-Ende von nuoCD jeweils für das Affinitätspeptid kodierte. Die PCR-Produkte wurden in einer weiteren PCR eingesetzt, um aus beiden Fragmenten ein DNA-Fragment, das die StrepTag II-Sequenz (Abb. 4-19) enthält, zu bilden (Abb. 4-20). StrepTag II-Sequenz mit Linker: 5´-gct agc tgg agc cac ccg cag ttc gaa aaa-3´ Ala Ser Trp Ser His Pro Gln Phe Glu Lys Abb. 4-19: Nukleotid- und Aminosäuresequenz des StrepTag II mit zwei vorangefügten Aminosäuren als sogenannter „Linker“ (Alanin und Serin), die für einen Abstand zwischen Tag und Protein sorgen sollen. - 63 - Ergebnisse P1 a) P3 3´ 5´ 3´ 5´ nuoB nuoCD P2 P4 PCR 5´ 3´ 3´ 5´ b) 5´ 5´ 3´ 3´ P1 P4 PCR 5´ 3´ nuoB Tag-Sequenz nuoCD Abb. 4-20: PCR zur Einführung eines StrepTag II in NuoB, der Abschnitt des nuo-Operons ist schwarz, die Tagsequenz rot dargestellt. Die verwendeten Primer sind mit P1-4 bezeichnet. a) Es wurden je ein Außen- (P1 und P4) und ein Innenprimer (P2 und P3) zur Amplifizierung der Fragmente nuoA-B bzw. nuoCD-E´ eingesetzt. Die Außenprimer sind in den Bereichen nuoA (P1) und nuoE´ (P4) komplementär zum nuo-Operon und enthalten Restriktionsschnittstellen für die Enzyme SacI (P1) und XhoI (P4). Die Innenprimer enthalten neben komplementären nuo-Sequenzen die vollständige Tagsequenz. b) Durch Annealing der Produkte des ersten Schrittes in der überlappenden Tagsequenz wurde die Vorlage für den zweiten PCR-Schritt gebildet. Nach Restriktion mit SacI und XhoI wurde das Fragment gegen das entsprechende native Fragment im Vektor pBADnuo ausgetauscht. - 64 - Ergebnisse Das in pBAD-A enthaltene Fragment des nuo-Operons aus nuoA-nuoE´ mit der StrepTag IISequenz am 3´-Ende von nuoB wurde in das Plasmid pBADnuo kloniert. Das resultierende Plasmid, das im Folgenden als pBADnuo/NuoBc bezeichnet wird, wurde in den Komplex IDeletionsstamm ANN023 transformiert. Der Stamm wurde angezogen und die Expression der nuo-Gene durch Zugabe von 0.2 % (w/v) L(+)-Arabinose induziert. Frau Hinderberger hat in ihrer Arbeit gezeigt, dass der Komplex I in diesem Stamm assembliert wird und mit dem Detergenz Dodecylmaltosid extrahiert werden kann (Hinderberger, 2004). Aus den Zellen wurden die Plasmamembranen präpariert und der Komplex I über eine AntionenaustauschChromatographie an DEAE-Sepharose und eine Affinitäts-Chromatographie an StrepTactinSepharose aufgereinigt. Es zeigte sich, dass der Komplex I zwar an das AffinitätsChromatographie-Material gebunden und eluiert wurde, jedoch enthielt die Präparation nicht alle Untereinheiten (Abb. 4-21, 4-22). Die Banden des Elutionsgipfels wurden anhand ihrer scheinbaren molekularen Massen den Untereinheiten NuoB, CD, E, F, G, H, I, L, M und N zugeordnet. Die hydrophoben Untereinheiten NuoA, NuoJ und NuoK konnten nicht nachgewiesen werden. Auffällig ist, dass die Intensitätsverhältnisse der Banden der einzelnen Untereinheiten nicht übereinstimmen. So ist z.B. die Intensität der NuoG zugeordneten Bande wesentlich geringer als die der Bande von NuoI (Abb. 4-22). Dies deutet darauf hin, dass die Untereinheiten dieser Präparation nicht in einem stöchiometrischen Verhältnis vorliegen. Abb. 4-21: Elutionsprofil der Affinitäts-Chromatographie an StrepTactin. (-) kennzeichnet die Absorbanz bei 280 nm in willkürlichen Einheiten, (+) die NADH/Ferricyanid Reduktaseaktivität der Fraktionen ist mit einem „+“ gekennzeichnet. Der Pfeil markiert den Beginn der Elution mit 2.5 mM Desthiobiotin. - 65 - Ergebnisse A B C D E F G H I J 100 70 50 a) 25 20 10 b) 4 7 1 2 3 5 6 8 9 Abb. 4-22: SDS-PAGE der mit Affinitäts-Chromatographie gereinigten Komplex I Präparation, (a) Färbung mit Coomassie Brilliant Blue und (b) Silberfärbung. Es wurden jeweils 40 µl Probe aufgetragen. (A) 0-5 ml des Waschschritts, (B) 5-20 ml des Waschschritts, (C) Fraktion 8 des Eluats (31.4 ml), (D) Fraktion 10 des Eluats (33 ml), (E) Frei, (F) Fraktion 14 des Eluats (36.2 ml), (G) Fraktion 16 des Eluats (37.8 ml), (H) Fraktion 18 des Eluats (39.4 ml), (I) Fraktion 20 des Eluats (41 ml) und (J) Standard. Die molekulare Masse einzelner Banden in a) ist in kDa angegeben. Die Banden in den Spuren D und F bis I wurden aufgrund ihrer scheinbaren molekularen Massen den Untereinheiten NuoG (1), NuoCD (2), NuoN und M (3), NuoF (4), NuoL (5), NuoH (6), NuoB (7), NuoI (8) und NuoE (9) zugeordnet. Die vierkernigen Fe/S-Zentren N2 (gz = 2.054 und gx,y = 1.922) und N4 (gz = 2.100, gx = 1.862) wurden in ESR-Spektren der Präparation nicht nachgewiesen (Abb. 4-23). Wie aus der Präparationstabelle ersichtlich (Tab. 4-6), wurde weniger als 10 % der von der DEAE-Säule eluierten Aktivität nach Affinitäts-Chromatographie wiedergewonnen. Somit ist die Modifizierung des C-terminalen Endes von NuoB mit dem StrepTag II-Affinitätspeptid nicht geeignet, um den Komplex I aufzureinigen. - 66 - Ergebnisse Abb. 4-23: ESR-Spektren von Komplex I-Präparationen aus dem Stamm ANN003/pAR1219 nach konventioneller Chromatographie (a) und dem Stamm ANN0221/pBADnuo/NuoBC nach Affinitäts-Chromatographie (b). Die Spektren wurden bei 13 K und 10 mW Mikrowellenleistung aufgenommen. Die Mikrowellenfrequenz betrug 9.44 GHz, die Modulationsamplitude 6 mT, die Zeitkonstante 0.064 s und die Aufnahmegeschwindigkeit 17.9 mT/min. Die bei 40 K und 2 mW Mikrowellenleistung aufgenommenen Spektren sahen vergleichbar aus (Daten nicht gezeigt). B B Tab. 4-6: Isolierung von Komplex I aus E. coli ANN0221/pBADnuo/BC. Es wurden 28 g Zellen (Feuchtgewicht) eingesetzt. B Präparationsschritt B Volumen Proteinmenge NADH/Ferricyanid Ausbeute [ml] [mg] Reduktaseaktivität [%] gesamt spezifisch [µmol/min] [µmol/(min•mg] Membranen 18 1500 2250 1.5 100 Detergenzextrakt 58 692 1800 2.6 80 Anionenaustauschchromatographie 54 104 1080 10.4 48 0.27 3.8 89 24 4 AffinitätsChromatographie - 67 - Ergebnisse 4.4.2 Modifizierung von NuoF Aus vorangegangenen Arbeiten war im Arbeitskreis ein Plasmid vorhanden, in dem die Gene nuoB-nuoG in das Plasmid pET11-a eingefügt und am 3´-Ende von nuoF die StrepTag IISequenz insertiert worden war. Von diesem als pET11-a/nuoB-G/NuoFC bezeichneten B B Plasmid wurden im Stamm BL21(DE3) die nuo-Gene exprimiert. Es wurde gezeigt, dass das NADH-Dehydrogenase Fragment aus den Untereinheiten NuoE, F und G aus diesem Stamm an StrepTactin-Sepharose bindet und dass die Modifizierung von NuoF weder die Assemblierung noch die enzymatische Aktivität des Fragmentes stört (Bungert et al., 1999). Der Bereich mit der StrepTag II-Sequenz an nuoF wurde aus dem Plasmid pET11-a/nuoB-G/NuoFC mit DraIII herausgeschnitten. Das 2´692 bp große Fragment wurde B B durch Gelelution aufgereinigt und in den Stamm DH5α/pBADnuo+pKD46 durch Elektroporation transformiert. Die Zellen waren zuvor bei 30°C mit 0.2 % (w/v) L(+)Arabinose angezogen worden, um die Expression der λ-Red Rekombinase zu induzieren. Nach der Transformation wurden die Zellen in 4 ml LB-Medium für 16 Std. bei 37°C angezogen und anschließend die Plasmide mittels Mini-Präparation isoliert. Die Restriktionsanalyse mit NheI, das eine Sequenz in dem Bereich, der die StrepTag II-Sequenz mit nuoF verbindet, erkennt, zeigt ein Gemisch der Plasmide pBADnuo und dem nach Rekombination resultierenden Plasmid pBADnuo/NuoFC (Tab. 4-7, Abb. 4-24). B B Tab. 4-7: Erwartete Fragmentgrößen der Plasmide pBADnuo und pBADnuo/NuoFC nach Restriktionsanalyse mit NheI. B Restriktionsenzym NheI pBADnuo pBADnuo/NuoFC [bp] [bp] 18´993 14´098 2´285 4´925 B 2´285 - 68 - B Ergebnisse Abb. 4-24: Restriktionsanalyse des Rekombinationsansatzes der StrepTag II-Sequenz in pBADnuo mit NheI (Spur 1). Als Standard wurde λ-DNA/EcoRI+HindIII (Spur 2; MBI Fermentas, St. Leon-Rot) aufgetragen. Um die Plasmide pBADnuo und pBADnuo/NuoFC zu trennen, wurde die Präparation 1:100 B B mit destilliertem Wasser verdünnt und in E. coli DH5α transformiert. Ein Fünftel des Transformationsansatzes wurde auf LB-Medium mit Chloramphenicol ausplattiert. Einzelne Kolonien wurden für 16 Std. bei 17°C angezogen, die Plasmide präpariert und mit NheI und ScaI geschnitten. Zwei Klone zeigten das erwartete Fragmentmuster mit Größen von 13´103, 4´925, 2´285 und 995 bp (Abb. 4-25). Abb. 4-25: Restriktionsanalyse des Rekombinationsansatzes des DraIII-Fragmentes des Plasmids pET11-a/nuoB-G/NuoFC in pBADnuo (Spur 1). Es wurde mit dem Enzym NheI B B geschnitten. Spur 2 zeigt den Standard λ-DNA/EcoRI+HindIII (MBI Fermentas, St. LeonRot) aufgetragen. - 69 - Ergebnisse Eine Sequenzierung der Klone mit dem erwarteten Restriktionsmuster zeigte die korrekte Rekombination der StrepTag II-Sequenz in pBADnuo. Das Plasmid pBADnuo/NuoFC wurde in den Doppeldeletionsstamm ANN0221 transformiert und unter Induktion mit 0.2 % (w/v) L(+)-Arabinose angezogen. Kurz vor der stationären Phase wurden die Zellen geerntet. Aus 4.3 g Zellen wurden 550 mg Membranen präpariert, die mit 500 µl 50 mM MES, pH 6.0 resuspendiert und in der Sauerstoffelektrode vermessen wurden. Mit d-NADH als Substrat wurde kein Sauerstoffverbrauch nachgewiesen (Daten nicht gezeigt). Die Membranen wurden mit DDM solubilisiert, auf einen Zuckergradienten von 5 % - 20 % Saccharose aufgetragen und für 16 Std. bei 180´000 *g zentrifugiert. Die Gradienten wurden in 2 ml Portionen fraktioniert und die NADH/Ferricyanid Reduktaseaktivität der einzelnen Fraktionen bestimmt (Abb. 4-26). Abb. 4-26: Zuckergradienten-Dichtezentrifugation des Detergenzextraktes der cytoplasmatischen Membranen des Stammes ANN0221/pBADnuo/NuoFC. Gezeigt ist die NADH/Ferricyanid Reduktaseaktivität der Fraktionen des Gradienten. Der Gradient wurde mit 5 mg Protein beladen. Der Gradient zeigte zwei Aktivitätsgipfel. Ein Gipfel entsprach der Position der alternativen NADH-Dehydrogenase in den Fraktionen 3 und 4. Der zweite Aktivitätsgipfel entspricht der Position des NADH-Dehydrogenase Fragmentes. In den Fraktionen 10 bis 12, in denen der Komplex I normalerweise sedimentiert, wurde keine Aktivität gemessen. Dies zeigt, dass in dem Stamm ANN0221/pBADnuo/NuoFC der Komplex I nicht assembliert wird, was vermutlich durch das Affinitätspeptid an NuoF verhindert wird. - 70 - Ergebnisse 4.4.3 Modifizierung von NuoCD Aus Arbeiten mit Y. lipolytica und T. thermophilus war bekannt, dass eine Modifizierung des Komplex I mit einem Affinitätspeptid C-terminal an NuoC zu einer intakten Präparation des Komplexes über eine Affinitäts-Chromatographie führt (Kashani-Poor et al., 2001; T. Ohnishi, persönliche Mitteilung). Es ist jedoch nicht ohne weiteres möglich, die StrepTag II-Sequenz an dieser Position in den E. coli Komplex I einzuführen, da in E. coli die Gene nuoC und D zu einem Gen nuoCD fusioniert sind. Zuerst müssen die Gene voneinander getrennt werden. Mittels Sequenzvergleichen mit Organismen, in denen nuoC und nuoD als getrennte Gene vorliegen, wurde ein potentielles Startkodon für nuoD in E. coli identifiziert. Vor dieses Startkodon sollte über eine PCR die für den StrepTag II kodierende Sequenz mit einem stromaufwärts dazu gelegenen Stoppkodon für nuoC eingeführt werden (Abb. 4-27). Eine Ribosomenbindestelle für nuoD sollte an dem 3´-Ende der StrepTag II-Sequenz eingeführt werden. Um die Sequenz einer „klassischen“ Ribosomenbindestelle ähnlicher zu machen, wurde die ursprüngliche Sequenz GAA AAA in GAG AAG umgewandelt, ohne dass die Aminosäuresequenz beeinflusst wurde (Abb. 4-19, 4-27). Die Primer StrepC-for und StrepC-rev für die Amplifikation der StrepTag II-Sequenz wurden so synthetisiert, dass sie jeweils für 34 bp bzw. 33 bp der StrepTag II-Sequenz kodieren, wobei sie 30 zueinander komplementäre bp aufweisen, so dass in diesem Bereich eine Anlagerung der Primer aneinander stattfindet (Abb. 4-27). Außerdem kodierten die Primer jeweils für 40 bp der am 3´-Ende von nuoC bzw. am 5´-Ende von nuoD gelegenen Sequenz, an denen über Rekombination die Tag-Sequenz in das Konstrukt pBADnuo eingeführt werden sollte. 5´-GCT AGC TGG AGC CAC CCG CAG TTC GAG AAG-3´ Ala Ser Linker Trp Ser His Pro Gln Phe Glu Lys StrepTag II-Sequenz Abb. 4-27: Nukleotid- und Aminosäuresequenz des StrepTag II. Zur Amplifikation der Sequenz mit flankierenden Enden, die homolog zum 3´-Ende von nuoC bzw. 5´-Ende von nuoD sind, sollten Primer verwendet werden, die sich ohne Matrizen-DNA aneinander anlagern (in der Nukleotid-Sequenz unterstrichen markiert). In der Aminosäuresequenz ist jeweils die Linkersequenz und die StrepTag II-Sequenz unterstrichen dargestellt. Um den StrepTag II in den entsprechenden Bereich innerhalb von nuoCD in das Konstrukt pBADnuo einzubringen, wurde ein 113 bp langes DNA-Fragment eingesetzt, das jeweils 40 - 71 - Ergebnisse bp von nuoC bzw. nuoD enthält, welche die StrepTag II-Sequenz flankieren. Hinter der TagSequenz befindet sich ein Stoppkodon für nuoC sowie ein Startkodon für nuoD. Dieses Fragment sollte mittels PCR hergestellt werden. Es wurden Primer genutzt, die sich jeweils in einem Bereich von 23 bp überlappen. Durch diese Überlappung lagern sich die Primern aneinander an und eine Elongation erfolgt jeweils komplementär zum Gegenprimer. In diesem Fall wurde keine Matrizen-DNA für die PCR benötigt, sie wurde als so genannte „Template-lose PCR“ durchgeführt (Abb. 4-28). Das PCR-Produkt sollte anschließend über eine Rekombination in pBADnuo eingebracht werden, das resultierende Konstrukt in einen Komplex I-defizienten Stamm transformiert und aus diesem nach Anzucht Komplex I präpariert werden. Abb. 4-28: Schema zur Herstellung eines DNA-Fragmentes zur Rekombination eines StrepTag II zwischen nuoC und nuoD im Konstrukt pBADnuo. Das Fragment sollte mittels einer Templat-losen PCR mit Primern hergestellt werden, die jeweils 40 bp von nuoC bzw. nuoD sowie einen Teil der StrepTag II-Sequenz beinhalten und aneinander annealen, so dass keine Matrizen-DNA benötigt wird. TAA bezeichnet das neu einzufügende Stoppkodon von nuoC. In einer PCR mit den Primern StrepC-for (69 bp) und StrepC-rev (67 bp) wurde ein 113 bp großes Fragment amplifiziert, das das 3´-Ende von nuoC, eine 6 bp lange verbindende Sequenz, die StrepTag II-Sequenz, ein Stoppkodon und das 5´-Ende von nuoD enthält. Dieses PCR-Produkt (Abb. 4-29) wird im Folgenden als nuoC-StrepTag II-nuoD bezeichnet. Neben dem 113 bp großen PCR-Produkt wurde eine weitere, schwach angefärbte Bande in dem Agarosegel nachgewiesen, die aufgrund ihrer Größe vermutlich von einem Dimer des PCRProduktes gebildet wird (Abb. 4-29). - 72 - Ergebnisse Abb. 4-29: Gelelektrophoretische Auftrennung der PCR-Produkte zur Amplifizierung von nuoC-StrepTag II-nuoD in einem 2 %igen Agarosegel (Spur 1). Als Standard wurde pUCMix 8 (pUC8; MBI Fermentas, St. Leon-Rot) aufgetragen. Der Pfeil kennzeichnet das (Haupt)-PCR-Produkt. Das Fragment nuoC-StrepTag II-nuoD wurde in den E. coli Stamm DH5α mit den Plasmiden pBADnuo und pKD46 transformiert und bei 37°C 1 Std. unter Schütteln inkubiert. Der Ansatz wurde in 4 ml LB-Medium für 16 Std. bei 37°C gezogen. Die Plasmide wurden mittels Mini-Präparation aus der Kultur isoliert. Nach einer 1:100-Verdünnung wurden sie zur Vereinzelung erneut in E. coli DH5α transformiert. Nach Ausplattieren der Zellen auf LBMedium mit Chloramphenicol wurden die Zellen bei 37°C über Nacht inkubiert. Von den so erhaltenen Kolonien wurden 12 in je 4 ml LB-Medium über Nacht angezogen, die Plasmide mittels Mini-Präparation aufgereinigt, und mit NheI geschnitten (Abb. 4-30). Das Plasmid pBADnuo ergibt zwei Fragmente von 18´993 und 2´285 bp. Das nach Rekombination erwartete Plasmid pBADnuo/nuoC-StrepTag II-nuoD sollte durch das Enzym NheI in drei Fragmente von 17´159, 2´285 und 1´867 bp gespalten werden. Der korrekte Einbau der StrepTag II-Sequenz und der optimierten Ribosomenbindestelle wurde durch eine Sequenzierung bestätigt (s. Anhang). Abb. 4-30: Restriktionsanalyse des Rekombinationsansatzes des PCR-Produktes nuoCStrepTag II-nuoD in pBADnuo. Es wurde mit dem Restriktionsenzym NheI geschnitten (Spur 1). Als Standard wurde pUC-Mix 4 (Spur 2; MBI Fermentas, St. Leon-Rot) aufgetragen. - 73 - Ergebnisse Um den Komplex I, der von dem Plasmid pBADnuo/nuoC-StrepTag II-nuoD exprimiert wird, zu isolieren, wurde das Plasmid in E. coli ANN0221 transformiert. Der Stamm ANN0221/pBADnuo/nuoC-StrepTag II-nuoD wurde wie beschrieben in einem 10 l-Maßstab angezogen. Aus 26.3 g Zellen wurden 7.3 g Cytoplasmamembranen erhaltenen. Die Membranproteine wurden durch Zugabe von DDM extrahiert und mittels einer Anionenaustausch-Chromatographie auf einer 40 ml DEAE-Säule von dem Überschuß Detergenz befreit. Durch eine Stoßelution mit 50 mM MES/NaOH, 320 mM NaCl, 0.1 % (w/v) DDM, pH 6.0 wurden die Proteine von der Säule gewaschen und mit PEG 4.000 gefällt, abzentrifugiert und in 1 ml 50 mM MES/NaOH, 50 mM NaCl, 0.1 % (w/v) DDM, pH 7.0 resuspendiert. Die resuspendierte Lösung wurde auf eine 8 ml StrepTactin-Sepharosesäule aufgetragen, die Säule mit 50 mM MES/NaOH, 50 mM NaCl, 0.1 % (w/v) DDM, pH 7.0 gespült, bis die Absorbanz bei 280 nm auf die Grundlinie zurückgegangen war. Gebundene Proteine wurden mit 2.5 mM Desthiobiothin im gleichen Puffer eluiert. Es wurde nur ein sehr kleiner Elutionsgipfel detektiert (Abb. 4-31). Die Fraktionen dieses Gipfels wurden auf ein SDS-Gel aufgetragen, die einzelnen Proteine gelelektrophoretisch voneinander getrennt und durch Silberfärbung sichtbar gemacht (Abb. 4-31). - 74 - Ergebnisse Abb. 4-31: Reinigung des Komplex I aus dem Stamm ANN0221/pBADnuo/nuoC-StrepTag II-nuoD. Elutionsprofil der IonenaustauschChromatographie an DEAE-Sepharose (a), der Affinitäts-Chromatographie an StrepTactinSepharose (b) und Proteinzusammensetzung der Gipfelfraktionen der Absorbanz bei 280 nm nach SDS-PAGE (c). Die SDS-PAGE zeigt weiterhin die Proteinzusammensetzung der vereinigten Fraktionen der Ionenaustausch-Chromatographie (IEC) und des Durchflusses der StrepTactin-Säule (D). Die Pfeile markieren den Beginn der Stoßelution mit 320 mM NaCl in (a) und 2.5 mM D-Desthiobiotin in (b). - 75 - Ergebnisse Aus dem E. coli Stamm ANN0221/pBADnuo/nuoC-StrepTag II-nuoD kann der Komplex I über Affinitäts-Chromatographie an StrepTactin-Sepharose aufgereinigt werden. Die Präparation enthält Banden, die anhand ihrer scheinbaren molekularen Massen Untereinheiten des Komplexes entsprechen. Die Ausbeute der Präparation ist allerdings so gering, dass die Proteinkonzentration und die NADH/Ferricyanid Reduktaseaktivität nicht bestimmt werden konnte. Im Vergleich zu der Standardpräparation des Komplex I aus dem Stamm ANN0221/pBADnuo waren sowohl die Ausbeuten als auch die NADH/Ferricyanid Reduktaseaktivität der einzelnen Schritte bis zur Affinitäts-Chromatographie miteinander vergleichbar (Tab. 4-3, Tab. 4-8). Die geringe Ausbeute nach Affinitäts-Chromatographie liegt somit nicht an einer größeren Instabilität des Komplexes durch die Modifizierung mit dem StrepTag II, sondern daran, dass das Affinitätspeptid keine ausreichende Bindung zu dem Chromatographie-Material ausbildet. Tab. 4-8: Isolierung von Komplex I aus 26.3 g (Feuchtgewicht) E. coli ANN0221/pBADnuo nuoC-StrepTag II-nuoD. Aufgrund der geringen Ausbeute der Affinität-Chromatographie war es nicht möglich, die Proteinmenge und die NADH/Ferricyanid Reduktaseaktivität dieses Schrittes zu bestimmen. Präparationsschritt Volumen Proteinmenge NADH/Ferricyanid Ausbeute [ml] [mg] Reduktaseaktivität [%] gesamt spezifisch [µmol/min] [µmol/(min•mg] Membranen 64 506 1280 2.5 100 Detergenzextrakt 62 242 1240 5.1 97 AnionenaustauschChromatographie AffinitätsChromatographie 35 32 420 13 33 3 - - - - Da der Komplex I aus dem E. coli Stamm ANN0221/pBADnuo nuoC-StrepTag II-nuoD assembliert und prinzipiell über eine Anionenaustausch-Chromatographie gereinigt werden kann, wurde versucht, die Wechselwirkung des Affinitätspeptids C-terminal an NuoC mit dem StrepTactin-Material zu verbessern, indem der verbindende Bereich zwischen NuoC und - 76 - Ergebnisse dem Affinitätspeptid von einem Alanin und einem Serin auf sechs Alanine und ein Serin verlängert wurde. Zur Einfügung weiterer Nukleotide in den verbindenden Bereich des Plasmids pBADnuo/nuoC-StrepTag II-nuoD wurde es als Matrize für eine weitere PCR mit den Primern 6TagCSD-for und –rev verwendet. Die Primer kodierten jeweils für eine Hälfte der StrepTag II-Sequenz und enthielten die Kodons für fünf zusätzliche Alanine in dem verbindenden Bereich. Die PCR wurde wie oben für das Fragment nuoC-StrepTag II-nuoD beschrieben durchgeführt. Das resultierende PCR-Produkt (Abb. 4-32) wird im Folgenden als nuoC-6 StrepTagII-nuoD bezeichnet. Abb. 4-32: Gelelektrophoretische Auftrennung der PCR-Produkte zur Amplifizierung von nuoC/6 StrepTag II/nuoD (Spur 1). Als Standard wurde pUC-Mix 8 (Spur 2; MBI Fermentas, St. Leon-Rot) aufgetragen. Das (Haupt)-PCR-Produkt ist mit einem Pfeil gekennzeichnet. Das PCR-Produkt nuoC/6 StrepTag II/nuoD wurde in elektrokompetente DH5α/pBADnuo+pKD46 transformiert. Die Rekombinase wurde durch Induktion mit 0.2 % (w/v) L(+)-Arabinose exprimiert. Nach der Transformation wurden die Zellen bei 37°C für 16 Std. angezogen, die Plasmide durch eine Mini-Präparation isoliert und mit einer Restriktionsanalyse charakterisiert. Durch den verlängerten verbindenden Bereich wurde eine Erkennungssequenz für das Restriktionenzym PstI eingefügt. In Tab. 4-9 sind die erwarteten Fragmentgrößen der verschiedenen Restriktionen aufgeführt. Das Restriktionsmuster ist in Abb. 4-33 gezeigt. - 77 - Ergebnisse Tab. 4-9: Erwartete Fragmentgrößen der Plasmide pBADnuo/nuoC-StrepTag II-nuoD und pBADnuo/nuoC-6 StrepTag II-nuoD nach Restriktion mit verschiedenen Enzymen. Restriktions- pBADnuo/nuoC-StrepTag II-nuoD pBADnuo/nuoC-6 StrepTag II- enzym [bp] nuoD [bp] PstI 17´229 9´700 4´049 7´600 4´049 PstI + XbaI 11´492 9´701 5´700 5´700 4´100 4´100 1´839 KpnI 12´342 12´342 8´984 8´936 Abb. 4-33: Restriktionsanalyse des Rekombinationsansatzes des PCR-Produktes nuoC-6 StrepTag II-nuoD in pBADnuo/nuoC-StrepTag II-nuoD mit den Enzymen PstI (Spur 1), PstI+XbaI (Spur 2) und KpnI (Spur 3). Als Standard wurde λ–DNA/EcoRI+HindIII (Spur 4; MBI Fermentas, St. Leon-Rot) verwendet. Wie die Restriktionsanalyse zeigte, pBADnuo/nuoC-StrepTag II-nuoD und wurde ein Gemisch der beiden pBADnuo/nuoC-6 StrepTag II-nuoD Plasmide präpariert (Abb. 4-33). Zur Trennung der Plasmide wurde die Probe 1:100 verdünnt und in E. coli DH5α transformiert. Von den auf LB-Medium mit Chloramphenicol gewachsenen Kolonien - 78 - Ergebnisse wurden 24 Übernachtkulturen angesetzt, daraus mit einer Mini-Präparation die Plasmide isoliert und diese mit einer Restriktionsanalyse charakterisiert (Abb. 4-34). Das erwartete Fragmentmuster zeigt Tabelle 4-9. Abb. 4-34: Restriktionsanalyse der Transformation des Plasmidgemisches pBADnuo/nuoCStrepTag II-nuoD und pBADnuo/nuoC-6 StrepTag II-nuoD in E. coli DH5α. Die Plasmide wurden mit dem Enzym PstI (Spur 1 und 2) bzw. mit PstI und XbaI (Spur 3 und 4) geschnitten. In den Spuren 1 und 3 ist das Fragmentmuster des Plasmids pBADnuo/nuoCStrepTag II-nuoD, in den Spuren 2 und 4 das des Plasmids pBADnuo/nuoC-6 StrepTag IInuoD zu finden. Als Standard wurde λ-DNA/EcoRI+HindIII (Spur 5; MBI Fermentas, St. Leon-Rot) aufgetragen. Zwei Klone zeigten das erwartete Fragmentmuster der Plasmide. Die Sequenz wurde mit einer DNA-Sequenzierung bestätigt (s. Anhang). Aus E. coli DH5α/pBADnuo/nuoC6 StrepTag II-nuoD wurde, wie oben für den Stamm ANN0221/pBADnuo/nuoC-StrepTag IInuoD beschrieben, der Komplex I über die Präparation der Cytoplasmamembranen, eine Detergenzextraktion, eine Anionenaustausch-Chromatographie an DEAE-Sepharose und eine Affinitäts-Chromatographie an StrepTactin Komplex I isoliert. Die Ausbeuten der einzelnen Schritte und die enzymatischen Aktivitäten unterschieden sich nur unwesentlich von der Präparation aus dem Stamm ANN0221/pBADnuo/nuoC-StrepTag II-nuoD (Tab. 4-8, Daten nicht gezeigt). Das Elutionsprofil der Affinitäts-Chromatographie an StrepTactin unterschied sich nicht von dem Profil der Aufreinigung von Komplex I mit dem um vier Alanine kürzeren verbindenden Bereich (Abb. 4-35). Somit scheint die Wechselwirkung des Affinitätspeptids mit dem Säulenmaterial nicht Grund für die fehlende Bindung des Enzyms an das Material zu sein. - 79 - Ergebnisse Abb. 4-35: Reinigung des Komplex I aus dem Stamm DH5α/pBADnuo/nuoC-6 StrepTag IInuoD. Elutionsprofil der Affinitäts-Chromatographie an StrepTactin-Sepharose. Aufgetragen wurde das Eluat einer Anionenaustausch-Chromatographie an DEAE-Sepharose, das Elutionsprofil der Anionenaustausch-Chromatographie entsprach dem der Aufreinigung von Komplex I aus dem Stamm ANN0221/pBADnuo/nuoC-6 StrepTag II-nuoD. Der Pfeil kennzeichnet den Beginn der Elution mit D-Desthiobiotin. 4.5 λ-Red vermittelte Rekombination zur Mutagenese im nuo-Operon 4.5.1 Deletionsmutationen in pBADnuo Um Deletionen in pBADnuo mittels λ-Red vermittelter Rekombination einzuführen, wurde der zu deletierende Bereich zunächst durch ein Resistenzgen ersetzt. An dieses Resistenzgen wurde über eine PCR jeweils am 3´- und 5´-Ende ein Bereich von 40 bp eingefügt, der homolog zu der Sequenz war, in die das Resistenzgen insertiert werden sollte (entsprechend 4-36; Datsenko und Wanner, 2000). Nach der Selektion positiver Klone mittels Antibiotikaresistenz wurde das Resistenzgen durch die FLP-Rekombinase über flankierende Sequenzen, die als FRT-Sequenzen (FLP recognition target) bezeichnet werden, entfernt. Die U U U U U U Eliminierung der Antibiotikaresistenz hinterlässt eine 82 bis 85 bp lange Sequenz, die sogenannte „Narbe“, in dem deletierten Bereich. Diese Sequenz enthält in allen sechs Leserastern Stoppkodons, eine idealisierte Ribosomenbindestelle und Startkodons, damit die Expression stromabwärts gelegener Gene nicht gestört wird (Datsenko und Wanner, 2000). - 80 - Ergebnisse . Abb. 4-36: Deletion eines Gens durch die λ-Red vermittelte Rekombination nach Datsenko und Wanner (2000). H1 und H2 bezeichnen homologe Bereiche zur Zielsequenz, P1 und P2 die Sequenzen der Primer, die sich an das Resistenzgen anlagern. FRT bedeutet „FLP recognition target“ und bezeichnet eine Sequenz, die von der FLP-Rekombinase erkannt wird. U U U U U U Deletion von nuoCD: U Es ist bekannt, dass zur Überproduktion des NADH-Dehydrogenase Fragmentes auch die Gene nuoB und nuoCD, deren Produkte nicht Teil des Fragmentes sind, überexprimiert werden müssen (Braun et al., 1998). Um zu überprüfen, welches Protein für die Assemblierung des Fragmentes benötigt wird, sollte nuoCD im Plasmid pBADnuo deletiert werden. Das Gen nuoCD wurde durch einen Austausch gegen eine KanamycinResistenzkassette deletiert. Die Kanamycin-Resistenzkassette wurde durch eine PCR am Plasmid pKD4 mit den Primern LambdaCD1-rev und LambdaCD1-for, die flankierend homologe Enden zu nuoCD enthielten, amplifiziert. Die Template-DNA wurde nach der PCR mit KpnI geschnitten und das 1´547 bp große PCR-Produkt aufgereinigt. Das die λ-Red Rekombinase kodierende Plasmid pKD46 wurde in E. coli DH5α mit dem Zielplasmid der Rekombination, pBADnuo, transformiert. Nach der Transformation wurden die Zellen auf LB-Medium mit Ampicillin und Chloramphenicol angezogen, um die Propagation sowohl von pBADnuo als auch von pKD46 zu gewährleisten. Die Transformanden wurden bei 30°C mit 0.2 % (w/v) L(+)-Arabinose angezogen, um die Expression der Rekombinase zu induzieren. Die Zellen wurden elektrokompetent gemacht und mit 250 ng des PCR-Produktes - 81 - Ergebnisse transformiert. Diese Transformanden wurden auf LB-Medium mit Chloramphenicol und Kanamycin ausplattiert, um auf Klone zu selektionieren, bei denen die KanamycinResistenzkassette in das die Chloramphenicol-Resistenz tragende Plasmid pBADnuo insertiert worden war. Die Plasmid-DNA von neun Kolonien wurde über das SpinPrep PCR purification Kit (Novagen) aufgereinigt. Die E. coli Stämme BL21(DE3), DH5α und RR1∆M15 wurden mit diesem Plasmid transformiert. Von dem Stamm BL21(DE3) wurde ein Klon isoliert, der nach Restriktionsanalyse das erwartete Fragmentmuster für das Plasmid pBADnuo/nuoCD::nptI aufwies (Abb. 4-37). Die nach Restriktionsanalyse mit den Enzymen XbaI, NdeI, PstI, BglII und SalI erwarteten Fragmentmuster der Plasmide pBADnuo und pBADnuo/nuoCD::nptI sind in Tab. 4-10 aufgeführt. Tab. 4-10: Erwartete Fragmentgrößen der Plasmide pBADnuo und pBADnuo/nuoCD::nptI nach Restriktionsverdau mit verschiedenen Enzymen. Restriktionsenzym XbaI pBADnuo pBADnuo/nuoCD::nptI [bp] [bp] 21´278 18´309 1´393 1´263 NdeI 21´278 12´879 8´086 PstI 17´229 9´342 4´049 7´574 4´049 BglII 9´939 9´939 9´181 7´509 2´158 2´158 1´359 SalI 19´303 - 82 - 20´965 Ergebnisse Abb. 4-37: Restriktionsanalyse des Plasmids pBADnuo/nuoCD::nptI. Als Enzym eingesetzt wurde XbaI (Spur 1), NdeI (Spur 2), PstI (Spur 3), BglII (Spur 5) und SalI (Spur 6). In Spur 4 wurde als Standard pUC-Mix 4 (MBI Fermentas, St. Leon-Rot) aufgetragen. Der Stamm E. coli BL21(DE3)/pBADnuo/nuoCD::nptI wurde in 50 ml LB-Medium angezogen und seine Plasmide mittels einer Midi-Präparation isoliert. Die KanamycinResistenzkassette wurde aus diesen Plasmiden mit der FLP-Rekombinase entfernt. Flankierend zu der Kanamycin-Resistenzkassette liegen die Erkennungssequenzen der Rekombinase, an denen die Kassette ausgeschnitten wird. Die FLP-Rekombinase ist auf dem Plasmid pCP20 kodiert. In den Stamm BL21(DE3)/pBADnuo/nuoCD::nptI wurde das Plasmid pCP20 über eine Elektroporation transformiert. Nach der Transformation wurden die Zellen bei 30°C auf LB-Agar mit Chloramphenicol und Ampicillin angezogen. Einzelne Kolonien wurden zunächst für 16 Std. bei 37°C in Medium mit Chloramphenicol und Kanamycin und dann für 10 Std. bei 43°C zur Expression der FLP-Rekombinase in LBMedium mit Chloramphenicol angezogen. Aus Einzelkolonien wurden Plasmide präpariert und durch Restriktionsanalysen mit RsrII und NcoI charakterisiert (Abb. 4-38). Nach Entfernung der Kanamycin-Resistenzkassette aus pBADnuo sollte der Schnitt mit RsrII zu einer Linearisierung eines 19´572 bp großen DNA-Fragmentes führen (Abb. 4-38, Spur 2). Bei nicht erfolgter Rekombination sollten zwei Fragmente mit Größen von 14´496 und 6´469 bp entstehen. Die Restriktion des Plasmids pBADnuo/∆nuoCD mit NcoI sollte drei Fragmente mit den Größen 12´330, 5´903 und 1´339 bp ergeben (Abb. 4-38, Spur 3). Aus dem nicht rekombinierten Plasmid pBADnuo würden bei Restriktion mit NcoI vier Fragmente mit 8´306, 5´903, 5´417 und 1´339 bp entstehen. - 83 - Ergebnisse Abb. 4-38: Restriktionsanalyse von pBADnuo/∆nuoCD. In Spur 1 ist der Restriktionsverdau der Probe mit NcoI, in Spur 2 der Restriktionsverdau mit RsrII und in Spur 3 der DNAStandard λ-DNA/EcoRI+HindIII (MBI Fermentas, St. Leon-Rot) aufgetragen. Das Plasmid mit dem erwarteten Fragmentmuster nach RsrII- und NcoI-Verdau wurde mit einer Midi-Präparation isoliert und sequenziert. Die Sequenzierung zeigte, dass nuoCD aus dem Plasmid pBADnuo deletiert wurde und dass durch die Rekombination eine „Narbe“ von 85 bp zurückblieb, die eine idealisierte Ribosomenbindestelle, sowie Startkodons in allen drei Leserastern enthält (Datsenko und Wanner, 2000). Die Deletionsmutante pBADnuo/∆nuoCD wurde weiterhin von Frau Saskia Kissel verwendet, um den Effekt der Deletion von nuoCD auf die Assemblierung von Komplex I bei Expression von pBADnuo/∆nuoCD zu untersuchen (Kissel, 2003). Das Plasmid pBADnuo/∆nuoCD wurde durch Elektroporation in E. coli BL21(DE3) transformiert, die Zellen angezogen und nach Induktion mit 0.2% (w/v) L-(+)-Arabinose nach 3 Std. Wachstum bei einer OD600 von B B 0.8 geerntet. Die Bakterien wurden durch einen einmaligen Durchgang in einer „French Press“ aufgeschlossen. Das Cytosol wurde als Überstand einer Ultrazentrifugation erhalten und durch Ultrafiltration etwa 10-fach aufkonzentriert. Es wurde eine NADH-reduzierte und eine Luftsauerstoff-oxidierte Probe angesetzt. Die Differenz dieser Spektren ist in Abb. 4-39 gezeigt. Es sind deutlich die Signale der Fe/S-Zentren des NADH-Dehydrogenase Fragments zu erkennen. Dies zeigt, dass NuoCD nicht essentiell für die Assemblierung dieses Fragmentes ist. - 84 - Ergebnisse Abb. 4-39: ESR (reduziert-minus-oxidiert) Differenz Spektrum des Cytosols des Stammes BL21(DE3)/pBADnuo/∆nuoCD. Das Spektrum wurde bei 40 K und 50 mW Mikrowellenleistung aufgenommen. Im Bereich zwischen 310 und 330 mT sind noch Reste des Mn2+-Signals zu erkennen, was auf eine gering unterschiedliche Proteinkonzentration der beiden Proben zurückzuführen ist. Die Proteinkonzentration betrug etwa 50 mg/ml. P P Deletion von nuoM: U Mit Hilfe eines radioaktiv-markierten und photoaktivierbaren Ubichinon-Analog wurde NuoM selektiv markiert (Gong et al., 2004). Um die Rolle von NuoM bei der UbichinonBindung zu untersuchen, sollte das Gen nuoM im Plasmid pBADnuo mutiert werden. Für die Komplementation mit dem mutierten Gen sollte nuoM zunächst aus pBADnuo deletiert werden. Außerdem sollte die Assemblierung des Komplexes in der Deletionsmutante pBADnuo/∆nuoM untersucht werden. Zunächst wurde in pBADnuo nuoM durch eine Kanamycin-Resistenzkassette ersetzt. Hierfür wurde die Resistenzkassette auf dem Plasmid pKD4 mit den Primern Lambda-M-L und Lambda-M-N amplifiziert, die flankierend für je 40 Basen kodierten, die homolog zu den stromauf- und stromabwärts zu nuoM gelegenen Bereichen sind. Das PCR-Produkt mit einer Größe von 1´547 bp wurde mit einer Ethanolfällung gereinigt und durch Elektroporation in Zellen transformiert, die das Zielplasmid pBADnuo sowie das die λ-Red Rekombinase kodierende Plasmid pKD46 enthielten. Diese Zellen waren mit 0.2 % L(+)-Arabinose bei 30°C angezogen worden. Die Transformanden wurden auf LB-Medium mit Chloramphenicol - 85 - Ergebnisse und Kanamycin ausplattiert, um Zellen zu selektionieren, bei denen die KanamycinResistenzkassette in das die Chloramphenicol-Resistenz tragende Plasmid pBADnuo insertiert worden war. Kolonien wurden für 16 Std. in LB-Medium angezogen, ihre Plasmide mit einer Mini-Präparation isoliert und durch Restriktion mit NdeI analysiert. Das Plasmid pBADnuo/nuoM::nptI sollte zwei Fragmente der Größen 18´860 und 2´369 bp ergeben, während bei nicht erfolgter Rekombination nur ein Fragment von 21´278 bp entstehen sollte. Alle untersuchten Klone zeigten das Restriktionsmuster von pBADnuo/nuoM::nptI (Abb. 4-40). Abb. 4-40: Restriktionsanalyse von vier verschiedenen Präparationen des Plasmids pBADnuo/nuoM::nptI mit dem Enzym NdeI (Spur 1 bis 4). Als Standard aufgetragen wurde pUC-Mix 4 (Spur 5; MBI Fermentas, St- Leon-Rot). Zellen mit dem Plasmid pBADnuo/nuoM::nptI wurden elektrokompetent gemacht und mit dem Plasmid pCP20, das für die FLP-Rekombinase kodiert, transformiert. Die Zellen wurden bei 30°C auf LB-Medium mit Ampicillin und Chloramphenicol angezogen, um die Propagation beider Plasmide zu gewährleisten. Anschließend wurde zur Induktion der Expression der FLP-Rekombinase für 16 Std. bei 37°C inkubiert. Aus den Kulturen wurden die Plasmide isoliert und mit einer Restriktionsanalyse untersucht. Tab. 4-11 zeigt die zur Analyse verwendeten Restriktionsenzyme und die erwarteten Fragmentgrößen. - 86 - Ergebnisse Tab. 4-11: Erwartete Fragmentgrößen der Plasmide pBADnuo/∆nuoM pBADnuo/nuoM::nptI nach Restriktionsverdau mit verschiedenen Enzymen. Restriktionsenzym PstI pBADnuo/∆nuoM pBADnuo/nuoM::nptI [bp] [bp] 17´229 17´229 2´607 2´881 und 1´119 XbaI 12´037 12´037 7´799 7´799 1´393 BglII 9´181 9´181 8´487 6´944 2´158 2´946 2´158 Die Zellen einer Kolonie, die das erwartete Fragmentmuster zeigte (Abb. 4-41), wurden angezogen und deren Plasmide mit einer Midi-Präparation isoliert. Durch eine Sequenzierung wurde die Deletion von nuoM in pBADnuo bestätigt. Abb. 4-41: Restriktionsanalyse des Plasmids pBADnuo/∆nuoM. Als Standard wurde pUCMix 4 verwendet (Spur 4; MBI Fermentas, St. Leon-Rot). Als Restriktionsenzym wurde PstI (Spur 1), XbaI (Spur 2) und BglII (Spur 3) eingesetzt. - 87 - Ergebnisse Deletion von nuoE bis nuoG: U Das NADH-Dehydrogenase Fragment ist homolog zu der Familie der NAD-abhängigen [NiFe]-Hydrogenasen (Friedrich und Scheide, 2000). Um zu überprüfen, ob eine dieser Hydrogenasen in der Lage ist, das NADH-Dehydrogenase Fragment im Komplex I funktionell zu ersetzen, sollte die Gengruppe nuoE bis G aus dem Plasmid pBADnuo deletiert werden. Das Plasmid pBADnuo/∆nuoEFG sollte zusammen mit einem Plasmid in einem Komplex I Deletionsstamm exprimiert werden, dass die zu nuoE, F und G homologen Hydrogenasegene enthält (Schmitz et al., 2002). Die Gengruppe nuoE-G sollte durch eine Kanamycinresistenzkassette ersetzt werden, die am Plasmid pKD4 amplifiziert wurde. Dafür wurden die Primer –EFG-CD und –EFG-H verwendet, die sich jeweils an das 5´- bzw. 3´-Ende der Kanamycinkassette anlagern sollen und außerdem homolog zu je 40 bp der Gene nuoCD und nuoH waren. Die Gengruppe nuoEG konnte jedoch in mehreren Versuchen mit veränderten Bedingungen nicht durch die Kanamycin-Resistenzkassette ersetzt werden. Vermutlich ist der zu ersetzende Bereich mit 4´613 bp zu groß, um die Rekombination zu ermöglichen. Da das große Plasmid pBADnuo wahrscheinlich stark verdrillt vorliegt, können sich die homologen Enden der 1´467 bp großen Kanamycinkassette nicht an die stromauf- und abwärts von nuoE-G gelegenen Bereiche anlagern. Daher wurde versucht, die Gene nuoE und F sowie nuoG einzeln mittels der λ-Red vermittelten Rekombination zu deletieren und in einem dieser Konstrukte den jeweils anderen Bereich zu entfernen. Im Fall von nuoE-F würden 1´334 bp deletiert werden, bei nuoG 2´732 bp. Die Kanamycin-Resistenzkassette wurde mit jeweils homologen Enden zu den stromaufwärts und stromabwärts zu nuoEF bzw. nuoG gelegenen Bereichen amplifiziert. Zur Deletion von nuoEF wurden dabei die Primer –EFG-CD und –EF-rev verwendet, zur Deletion von nuoG die Primer –G-for und –EFG-H. Die PCR-Produkte wurden aufgereinigt und anschließend in elektrokompetente DH5α/pBADnuo+pKD46 transformiert, die zuvor bei 30°C mit 0.2 % (w/v) L(+)-Arabinose angezogen worden waren. Die Zellen wurden auf LB-Agarplatten mit Chloramphenicol und Kanamycin ausgestrichen. Auf diesem Medium sollten nur Zellen wachsen, die das Chloramphenicol-resistente Plasmid pBADnuo mit der insertierten Kanamycin-Resistenzkassette enthalten. Kolonien wurden in jeweils 4 ml LB-Medium für 16 Std. bei 37°C angezogen. Nach Mini-Präparation wurden die Plasmide durch Restriktionsverdau mit XbaI analysiert. Das Plasmid pBADnuo sollte in ein lineares Fragment - 88 - Ergebnisse von 21´278 bp geschnitten werden, während das Plasmid pBADnuo/nuoEF::nptI in drei Fragmente von 16´415, 3´093 und 1´393 bp gespalten werden sollte. Nahezu alle Klone zeigten das erwartete Fragmentmuster (Abb. 4-42). Abb. 4-42: Restriktionsanalyse des Plasmids pBADnuo/nuoEF::nptI mit dem Enzym XbaI (Spur 1 und 2). Als Standard wurde λ-DNA/EcoRI+HindIII (Spur 3; MBI Fermentas, St. Leon-Rot) aufgetragen. Die Insertion der Kanamycin-Resistenzkassette in das Plasmid pBADnuo anstelle der Gene nuoE und F wurde durch eine Sequenzierung bestätigt (s. Anhang). Die Resistenzkassette wurde entfernt, indem das Plasmid pBADnuo/nuoEF::nptI mit dem Plasmid pCP20 in E. coli DH5α kotransformiert wurde. Die Expression der FLP-Rekombinase wurde durch Anzucht der Zellen bei 42°C induziert. Die Plasmide wurden aus den Zellen mit einer MiniPräparation isoliert und mit dem Restriktionsenzym XbaI analysiert (Tab. 4-12, Abb. 4-43). Tab. 4-12: Fragmentgrößen der Plasmide pBADnuo, pBADnuo/nuoEF::nptI pBADnuo/∆nuoEF nach Verdau mit dem Restriktionsenzym XbaI. pBADnuo pBADnuo/nuoEF::nptI pBADnuo/∆nuoEF [bp] [bp] [bp] 21´641 16´415 16´415 3´093 3´093 1´393 - 89 - und Ergebnisse Abb. 4-43: Restriktionsanalyse der Plasmide pBADnuo (Spur 1), pBADnuo/nuoEF::nptI (Spur 2) und pBADnuo/∆nuoEF (Spur 3) mit dem Enzym XbaI. Als Standard wurde λ-DNA/EcoRI+HindIII verwendet (Spur 4; MBI Fermentas, St. Leon-Rot). Die Insertion der Kanamycin-Resistenzkassette in nuoG wurde über einen Restriktionsverdau mit HindIII, PstI und NcoI analysiert (Abb. 4-44). Die erwarteten Fragmentgrößen zeigt Tab. 4-13. Tab. 4-13: Erwartete Fragmentgrößen der Plasmide pBADnuo und pBADnuo/nuoG::nptI bei Restriktionsanalyse mit verschiedenen Enzymen. Restriktionsenzym HindIII pBADnuo pBADnuo/nuoG::nptI [bp] [bp] 10´938 10´775 6´549 6´549 2´710 2´710 1´081 PstI 17´229 11´278 4´049 4´707 4´049 NcoI 14´036 9´121 5´903 5´903 1´339 3´671 1´339 - 90 - Ergebnisse Abb. 4-44: Restriktionsanalyse des Plasmids pBADnuo/nuoG::nptI mit den Restriktionsenzymen HindIII (Spur 1), PstI (Spur 2) und NcoI (Spur 3). Als Standard wurde λ-DNA/EcoRI+HindIII (Spur 4; MBI Fermentas, St. Leon-Rot) verwendet. Nach Transformation des Plasmids in elektrokompetente DH5α/pBADnuo/nuoG::nptI und anschließende Hitzeinduktion der Expression der FLP-Rekombinase bei 42°C wurden die Plasmide aus einzelnen Kolonien durch Mini-Präparation isoliert und mit XhoI analysiert (Tab. 4-14; Abb. 4-45). Tab. 4-14: Erwartete Fragmentgrößen bei Restriktionsanalyse der Plasmide pBADnuo, pBADnuo/∆nuoG und pBADnuo/nuoG::nptI mit dem Enzym XbaI. pBADnuo pBADnuo/∆nuoG pBADnuo/nuoG::nptI [bp] [bp] [bp] 21´278 13´674 13´674 4´967 4´967 1´393 - 91 - Ergebnisse Abb. 4-45: Restriktionsanalyse der Plasmide pBADnuo/nuoG::nptI (Spur 1), pBADnuo/∆nuoG (Spur 2) und pBADnuo (Spur 3) mit XbaI. Als Standard wurde λDNA/EcoRI+HindIII (Spur 4; MBI Fermentas, St. Leon-Rot) aufgetragen. Die korrekte Sequenz von pBADnuo/∆nuoG wurde durch eine Sequenzierung bestätigt (s. Anhang). Es wurde noch nicht versucht, in diesem Plasmid eine Deletion der Gene nuoE und F bzw. eine Deletion von nuoG im Plasmid pBADnuo/∆nuoEF durchzuführen. 4.5.2 Punktmutationen in nuoM Die Untereinheit NuoM wurde mit einem radioaktiv markierten, photoaktivierbaren AzidoUbichinon-Derivat (3-Azido-2-methyl-5-methoxy[3H]-6-decyl-1,4-benzochinon) markiert und der Bereich des Proteins, an den das Derivat gebunden hatte, durch Sequenzierung tryptischer Fragmente identifiziert (Gong et al., 2003). Anhand von elektrochemischinduzierten FT-IR Differenzspektren in Gegenwart und Abwesenheit von Ubichinon wurde außerdem vermutet, dass Lysin(e) und Aspartat(e) oder Glutamat(e) an der Chinon-Bindung beteiligt sind (P. Hellwig, mündliche Mitteilung). Ein Sequenzvergleich ergab, dass in dem mit dem Chinon-Derivat markierten Bereich von NuoM ein Leucin und ein Aspartat an den Positionen 244 bzw. 246 konserviert sind (Abb. 4-46). Das Leucin sollte zu Isoleucin und das Aspartat zu einem Asparagin in dem Plasmid pBADnuo mutiert werden. Der Komplex sollte in den Mutanten produziert und charakterisiert werden. - 92 - Ergebnisse E. S. S. Y. R. R. S. M. 221 * * 255 YLLMLGFFIAFAVKMPVVPLHGWLPDAHSQAPTAG YLLMLGFFIAFAVKMPVVPLHGWLPDAHSQAPTAG YLLMLGFFIAFAVKMPVVPLHGWLPDAHSQAPTAG YLLMLGFFIAFAVKMPVVPLHGWLPDAHSQAPTAG TLLFLAFFASFAVKMPMWPVHTWLPDAHVQAPTAG QILWWAIFIAFAVKIPIIPFHTWLPDAHVQAPTSG SWLFFAFLVAFAVKVPMVPVHTWLPDAHVQAPTAG KALFLGFMFAFAIKAPLWPFHRWLPDAAVESTPAT coli flexneria typhimurium pestis capsulatus conorii meliloti tuberculosis Abb. 4-46: Sequenzvergleich der NuoM-Homologen von E. coli, S. flexneria (Shigella flexneria), S. typhimurium (Salmonella typhimurium), Y. pestis (Yersinia pestis), R. capsulatus (Rhodobacter capsulatus), R. conorii (Rickettsia conorii), S. meliloti (Sinorhizobium meliloti) und M. tuberculosis (Mycobacterium tuberculosis). Die Nummerierung bezieht sich auf die E. coli-Sequenz. Das konservierte Leucin an Position 244 und Aspartat an Position 246, die an der Ubichinonbindung beteiligt sein könnten, sind rot gedruckt und mit einem Stern markiert. Die Punktmutationen Leu244Ile und Asp246Asn wurden über eine Mutagenese-PCR im P P P P Subklon pBAD-C1 in nuoM eingeführt. Für die Mutation Leu244Ile wurde das Kodon CTG P P gegen das Kodon ATC ausgetauscht, so dass eine Schnittstelle für das Enzym BamHI entstand. Für die Mutation Asp246Asn wurde das Kodon GAT gegen das Kodon AAT P P ausgetauscht, allerdings musste eine weitere Base im Primer ausgetauscht werden, so dass eine Erkennungssequenz für das Enzym NcoI entstand. Der Subklon pBAD-C1 wurde jeweils als Template-DNA zur Mutagenese-PCR eingesetzt. Für das Plasmid pBAD-C1/nuoM(Leu244Ile) wurden die Primer Leu244Ile-for und -rev, für P P das Plasmid pBAD-C1/nuoM(Asp246Asn) die Primer Asp246Asn-for und -rev eingesetzt. Die P P PCR-Produkte wurden aufgereinigt und in E. coli DH5α transformiert. Einzelne Kolonien wurden über Nacht angezogen, die Plasmide über eine Mini-Präparation isoliert und durch Restriktionsanalyse auf den Basenaustausch überprüft. Bei der Restriktionsanalyse der Mutation pBAD-C1/nuoM(Leu244Ile) mit BamHI sollten Fragmente der Größe 4´230, 363 und P P 153 bp entstehen, während im Fall des Plasmids pBAD-C1 zwei Fragmente von 4´593 und 153 bp entstehen sollten. Bei drei Klonen wurde das Fragmentmuster des Plasmids pBADC1/nuoM(Leu244Ile) nachgewiesen (Abb. 4-47). Das 153 bp große Fragment ist aufgrund P P seiner geringen Länge nicht zu sehen. - 93 - Ergebnisse Abb. 4-47: Restriktionsanalyse der Plasmide pBAD-C1 (Spur 1) und pBAD-C1/nuoM(Leu244Ile) (Spur 3) nach Behandlung mit BamHI. Als Standard wurde in Spur 2 λ-DNA/EcoRI+HindIII (MBI Fermentas, St. Leon-Rot) aufgetragen. P P Für die Mutation pBAD-C1/nuoM(Asp246Asn) wurde nach Restriktion mit NcoI ein P P Fragmentmuster aus drei Fragmenten der Größen 2´791, 1´339 und 616 bp erwartet. Das Plasmid pBAD-C1 sollte entsprechend zwei Fragmente von 3´407 und 1´339 bp ergeben. Es wurden drei Klone identifiziert, die das erwartete Restriktionsmuster des Plasmids pBAD-C1/nuoM(Leu244Ile) zeigten (Abb. 4-48). P P Abb. 4-48: Restriktionsanalyse des Plasmids pBAD-C1/nuoM(Leu244Ile) (Spur 1) und pBAD-C1 (Spur 3) nach Behandlung mit NcoI. Als Standard wurde λ-DNA/EcoRI+HindIII (Spur 2; MBI Fermentas, St. Leon-Rot) aufgetragen. P P Die jeweiligen Mutationen wurden durch DNA-Sequenzierung bestätigt (s. Anhang). Aus den gewünschten Plasmiden wurde ein HincII-Fragment mit der jeweiligen Mutation erzeugt, das - 94 - Ergebnisse in das Plasmid pBADnuo rekombiniert werden sollte (Abb. 4-49). Um die Punktmutationen über die λ-Red Rekombinase in pBADnuo einzuführen, wurden lineare DNA-Fragmente mit den entsprechenden Mutationen in E. coli Zellen transformiert, die die Ziel-DNA sowie ein λ-Red Rekombinase exprimierendes Plasmid trugen (Abb. 4-49). Es wurde nicht auf eine Antibiotika-Resistenz selektioniert, weil die auf dem Plasmid kodierte Resistenz durch Einführen der Mutation nicht verändert wurde. Abb. 4-49: Allgemeines Schema zur Einführung einer Mutation in das Genom oder in ein Plasmid mittels λ-Red Rekombinase. Zellen, die die Ziel-DNA, in die die Mutation eingeführt werden soll, und das Plasmid pKD46 enthalten, das für die Rekombinase kodiert, werden bei 30°C angezogen. Nach Transformation mit einem linearen DNA-Fragment (grün dargestellt), das die betreffende Mutation enthält und flankierend dazu einen mindestens 36 bp großen homologen Bereich aufweist, in den die Mutation eingeführt werden soll, wird durch L(+)-Arabinose die Produktion der λ-Red Rekombinase induziert, die die Rekombination katalysiert. Durch Anzucht bei 37°C geht das Plasmid pKD46 aufgrund seines temperatursensitiven Replikationsursprungs verloren. - 95 - Ergebnisse Tab. 4-15: Erwartete Fragmentgrößen bei Restriktion der Plasmide pBADnuo und pBADnuo/nuoM(Mutation) mit verschiedenen Enzymen. Restriktionsenzym pBADnuo Mutation pBADnuo/nuoM(Mutation) [bp] BamHI [bp] Leu244Ile 7´893 7´893 P P 7´231 7´231 4´395 4´395 1´518 1´155 153 363 88 153 88 NcoI 14´036 Asp 246 P Asn 14´036 P 5´903 5´287 1´339 1´339 616 Die Restriktionsanalyse des Plasmids pBADnuo/nuoM zeigte, dass keiner der Klone das erwartete Fragmentierungsmuster (Tab. 4-15) der Mutation nuoM(Leu244Ile) trug (Abb. 4-50). P P Anscheinend war die Rekombination nicht effizient genug. Trotz Variation verschiedener Parameter der Rekombinationsansätze, wobei vor allem verschiedene Konzentrationen des mutierten Fragmentes eingesetzt wurden, gelang es nicht, die Mutation, die zu dem Austausch Leu244Ile in NuoM führt, in pBADnuo einzuführen. In dem Rekombinationsansatz, der zu der P P Mutation nuoM(Asp246Asn) führen sollte, wurde jeweils ein Gemisch der Plasmide pBADnuo P P und pBADnuo/nuoM(Asp246Asn) isoliert (Abb. 4-50). Auch nach mehrfacher Verdünnung der P Plasmide und P Retransformation in E. coli pBADnuo/nuoM(Asp246Asn) nicht vereinzelt werden. P P - 96 - DH5α konnte die Mutante Ergebnisse Abb. 4-50: Restriktionsanalyse der Rekombinationsansätze zur Erzeugung der Mutationen Leu244Ile (Spur 1) und Asp245Asn (Spur 3) auf NuoM. Die isolierten Plasmide wurden mit BamHI (Spur 1) und NcoI (Spur 3 und 4) geschnitten. Spur 1 zeigt das Fragmentmuster von pBADnuo und Spur 3 ein Gemisch der Plasmide pBADnuo/nuoM(Asp245Asn) und pBADnuo. Spur 4 zeigt das Fragmentmuster des Plasmids pBADnuo. Als Standard wurde λ-DNA/EcoRI+HindIII (Spur 2) und pUC-Mix 4 (Spur 5; beide MBI Fermentas, St. LeonRot) aufgetragen. P P P P P P Daraufhin wurde versucht, die Mutationen mittels λ-Red vermittelter Rekombination in ein Plasmid einzubringen, in dem nuoL und nuoM durch eine nptI-sacR-sacB-Kassette ersetzt worden war (pBADnuo/nuoLM::nptI-sacB, von Thomas Pohl aus dem Arbeitskreis zur Verfügung gestellt). Zellen mit diesem Plasmid können nur auf Medium ohne Saccharose wachsen und können über ihre Kanamycin-Resistenz selektioniert werden. Die Gene nuoL und nuoM wurden an den Plasmiden pBAD-C1/nuoM(Leu244Ile) P P und pBAD-C1/nuoM(Asp246Asn) amplifiziert und mit pBADnuo/nuoLM::nptI-sacB rekombiniert, P P indem die aufgereinigten PCR-Produkte in DH5α/pBADnuo/nuoLM::nptI-sacB+pKD46 transformiert wurden, die zuvor in Medium mit 0.2 % (w/v) L(+)-Arabinose angezogen und anschließend elektrokompetent gemacht wurden. Die Zellen wurden auf LB-Medium mit Saccharose und Chloramphenicol angezogen, um auf den Verlust der nptI-sacR-sacBKassette zu selektionieren. Zwar wuchsen einige Kolonien auf diesem Medium; eine MiniPräparation zeigte allerdings, dass sie keine Plasmide enthielten. Da diese Zellen resistent gegenüber Chloramphenicol waren, wurde angenommen, dass die ChloramphenicolResistenzkassette in das Genom an einer unbekannten Stelle integriert wurde. Da es nicht gelang, die gewünschten Mutationen in NuoM in das Plasmid pBADnuo zu rekombinieren, wurde in einem nächsten Schritt versucht, nuoM in einen Expressionsvektor einzubringen und damit die Expression aller nuo-Gene von dem Plasmid pBADnuo/∆nuoM zu komplementieren. Diese Methode hätte den Vorteil, dass alle gewünschten Mutationen in nuoM direkt in dieses Konstrukt eingebracht werden könnten. - 97 - Ergebnisse Zunächst wurde nuoM in den Vektor pET11-a kloniert. Hierzu wurde das Gen in einer PCR mit den Primern pET11M-NdeI und pET11M-NheI so amplifiziert, dass es von den Erkennungssequenzen der Restriktionsenzyme NdeI und NheI flankiert wurde. Das PCRProdukt und der Vektor pET11-a wurden mit diesen Enzymen geschnitten und ligiert. Nach Transformation des Ligationsansatzes in E. coli wurden auf Ampicillin-haltigen LBAgarplatten allerdings keine Kolonien gefunden. Mehrere Versuche mit Variationen bei der PCR-Amplifizierung, der Aufreinigung der Restriktionsansätze und des Ligationsansatzes führten nicht zu der Klonierung von nuoM in pET11-a. Der Versuch, das Gen nach Behandlung mit NheI und NdeI in den Vektor pET24-a zu klonieren, scheiterte ebenfalls. pET24-a unterscheidet sich von pET11-a durch die komplementäre Lage der MCS, in diesem Konstrukt würde das Gen in der falschen Orientierung vorliegen und nicht exprimiert werden. So kann ausgeschlossen werden, dass eine mögliche basale Produktion von NuoM, das nach Sekundärstrukturvorhersagen zehn transmembrane Helices besitzt (Fearnley und Walker, 1992; Weidner et al., 1993), letal für die Zellen ist. In Kontrollexperimenten wurde die Aktivität der Restriktionsenzyme nachgewiesen. Warum die Klonierung erfolglos blieb, ist unklar. In einem weiteren Versuch sollte nuoM mit der λ-Red vermittelten Rekombination in das Plasmid pET24-a eingebracht werden, das eine Kanamycin-Resistenz besitzt. Da das Plasmid pKD46 eine Ampicillin-Resistenz vermittelt, wird durch Verwendung beider Antibiotika die gleichzeitige Propagation beider Plasmide sichergestellt. Das Gen nuoM wurde über eine PCR mit den Primern 24Mred-for und 24Mred-rev amplifiziert, die jeweils homolog zu 20 Basen des 3´- und 5´-Endes von nuoM sind und flankierend jeweils 30 Basen der MCS des Vektors enthielten. Als Matrize wurde das Plasmid pBADnuo verwendet. Das PCR-Produkt mit einer Größe von 1´376 bp wurde durch Gelelektrophorese nachgewiesen (Abb. 4-51). - 98 - Ergebnisse Abb. 4-51: Gelelektrophoretische Auftrennung des PCR-Ansatzes zur Amplifizierung von nuoM (Spur 1). Es wurde eine Größe von 1´376 bp erwartet. Als Standard wurde λ-DNA/EcoRI+HindIII (Spur 2; MBI Fermentas, St. Leon-Rot) verwendet. Die Plasmide pKD46 und pET-24a wurden durch Elektroporation in E. coli DH5α kotransformiert. Die Zellen wurden bei 30 °C 1 Std. geschüttelt und eine Übernachtkultur in 50 ml LB-Medium mit Ampicillin und Kanamycin bei 30 °C angesetzt. Die Expression der Rekombinase wurde mit 0.2 % (w/v) L(+)-Arabinose induziert, die Zellen elektrokompetent gemacht und auf Eis gelagert. Danach wurde das amplifizierte PCR-Produkt durch Elektroporation in die elektrokompetenten Zellen transformiert und der Ansatz 1 Std. bei 30°C geschüttelt. Die Zellen wurden auf LB-Medium mit Kanamycin ausplattiert und über Nacht bei 37 °C inkubiert, um die weitere Replikation von pKD46 zu unterbinden. Eine Restriktionsanalyse der isolierten Plasmide einer Übernachtkultur zeigte, dass nur der leere Vektor isoliert worden war. Es wurde angenommen, dass die erfolgreiche Rekombination zu selten stattfand und somit die Anzahl positiver Klone zu gering war. Um zwischen Zellen, die das Plasmid ohne Insert und das rekombinierte Plasmid mit nuoM beinhalten, zu unterscheiden, sollte daher der Vektor pETBlue-1 (s. Anhang) mit einer Blau-Weiß-Selektion verwendet werden. Das Gen nuoM wurde mit den Primern pETBlueM2-for und pETBlueM2-rev amplifiziert, die jeweils für 40 Nukleotide kodierten, die homolog zur MCS von pETBlue-1 waren und den Start- bzw. den Stopp-Bereich von nuoM enthielten. Als Matrize diente das Plasmid pBADnuo. Das PCR-Produkt mit einer Größe von 1´584 bp, das im Folgenden als BlueM bezeichnet wird, wurde durch Gelelektrophorese nachgewiesen (Abb. 4-52). - 99 - Ergebnisse Abb. 4-52: Gelelektrophoretische Auftrennung des PCR-Ansatzes zur Amplifizierung von nuoM mit zum Vektor pETBlue-1 homologen Enden (Spur 1). Es wurde eine Größe von 1´584 bp erwartet. Als Standard verwendet wurde λ-DNA/EcoRI+HindIII (Spur 2; MBI Fermentas, St. Leon-Rot). Das Plasmid pKD46 wurde durch Elektroporation in E. coli DH5α transformiert. Die Zellen wurden bei 30°C für 1 Std. geschüttelt, in 50 ml LB-Medium mit Ampicillin übergeimpft und für 16 Std. angezogen. Die Zellen wurden elektrokompetent gemacht und die Expression der Rekombinase mit 0.2 % (w/v) L(+)-Arabinose induziert. In diese Zellen wurde das Plasmid pETBlue-1 und das aufgereinigte PCR-Produkt BlueM transformiert. Der Ansatz wurde nach Schütteln für 1 Std. bei 37 °C abzentrifugiert, die Zellen in 100 µl SOC-Medium resuspendiert und nach Zugabe von 10 µl 0.1 M IPTG auf LB-Medium mit Ampicillin und XGal ausplattiert. Die Zellen wurden bei 37 °C über Nacht inkubiert und anschließend bei 4°C gelagert, um die Farbreaktion zu fördern. Da sowohl der leere Vektor als auch das rekombinante Plasmid für eine Ampicillin-Resistenz kodieren, wurde die Zellsuspension vor dem Ausplattieren 1:20 mit LB-Medium verdünnt, um Einzelkolonien auf den Platten zu erhalten. Auf der Schale wuchsen neben einigen blauen auch eine große Anzahl weißer Kolonien, die das rekombinierte Plasmid enthalten sollten (Abb. 4-53). - 100 - Ergebnisse Abb. 4-53: LB-Ampicillin-Schale mit X-Gal nach Ausstrich der Transformation des PCRProduktes BlueM in E. coli DH5α/pETBlue-1+pKD46. Von 12 weißen Kolonien wurden Übernachtkulturen mit Ampicillin angesetzt, die Plasmide präpariert und mit Eco31I und XbaI+ScaI geschnitten (Abb. 4-54). Die erwarteten Fragmente sind in Tab. 4-16 aufgeführt. Tab. 4-16: Erwartete Fragmentgrößen bei Restriktion der Plasmide pETBlue-1 und pETBlue-1/nuoM mit verschiedenen Enzymen. Restriktionsenzym Eco31I XbaI + ScaI pETBlue-1 pETBlue-1/nuoM [bp] [bp] 2´385 3´911 1´091 1´095 1´995 3´525 1´481 1´421 - 101 - Ergebnisse Abb. 4-54: Restriktionsanalyse mit den Enzymen Eco31I (Spur 1) und XbaI + ScaI (Spur 3) der Rekombinationsansätze des PCR-Produktes BlueM in das Plasmid pETBlue-1. Als Standard wurde λ-DNA/EcoRI+HindIII (Spur 2; MBI Fermentas, St. Leon-Rot) aufgetragen. Aus allen Kulturen wurde das Plasmid pETBlue-1 isoliert, obwohl die ausgesuchten Kolonien weiß gewesen waren. Diese Kolonien besaßen einen Durchmesser von über 1 mm, so dass eine fehlende Anfärbung trotz einer aktiven β-Galaktosidase nicht auf einer zu geringen Größe der Kolonien beruht. Trotz mehrfacher Versuche mit abgeänderten Bedingungen konnten nur noch blaue Kolonien gefunden werden. Die Methode der Blau-weiß-Selektion war aus nicht bekannten Gründen für die Selektion von rekombinanten Zellen nicht geeignet. 4.6 Mutagenese an einem Fe/S-Bindemotiv auf NuoG Die vier konservierten Cysteine auf NuoG, die ein zusätzliches Bindemotiv für ein Fe/SZentrum ausbilden, wurden mittels einer PCR-basierenden Mutagenese zu Alaninen mutagenisiert. Hierbei wurden Primer eingesetzt, in denen jeweils ein oder mehrere Basen gegenüber der Template-Sequenz ausgetauscht waren, wodurch die beabsichtigte Mutation eingeführt wurde. In einer PCR wurden die Primer mit den veränderten Basen verlängert. Die Template-DNA wurde durch das Restriktionsenzym DpnI abgebaut, das nur methylierte DNA spaltet und nicht das unmethylierte PCR-Produkt (Abb. 4-55). Das zirkuläre, aber nicht ligierte PCR-Produkt wurde in E. coli Zellen transformiert, die das Plasmid ligierten und replizierten. - 102 - Ergebnisse Abb. 4-55: Schematische Darstellung einer ortsgerichteten Punktmutation mittels PCR auf einem Plasmid. Die Primer mit der gewünschten Mutation (durch ein x dargestellt) lagern sich an das Template an (1) und werden verlängert, ohne ligiert zu werden (2). Das Plasmid mit der Mutation liegt aber zirkulär vor. Nach Abbau des Ausgangsplasmids (4) wird das PCR-Produkt in E. coli-Zellen transformiert, wo es ligiert und propagiert wird (3) (nach dem Manual des QuikChange® II Site-Directed Mutagenesis Kit; Stratagene). Als Template wurde der Subklon pBAD-B2 eingesetzt, der die Gengruppe nuoF´-nuoI´enthält und 7´184 bp groß ist. Das Plasmid pET11-a/nuoB-G/NuoFC, in das letztendlich die B B Mutationen eingeführt werden sollten, war mit 13´059 bp zu groß für eine Mutagenese-PCR. Als Primer wurden für die Mutante pBAD-B2/NuoG(Cys230Ala) die Oligonukleotide N1cP P Pvuf und N1c-Pvur eingesetzt, für die Mutante pBAD-B2/NuoG(Cys233Ala) die P P Oligonukleotide N1c2 und N1c2r, für die Mutante pBAD-B2/NuoG(Cys237Ala) die P P Oligonukleotide N1c3 und N1c3r und für die Mutante pBAD-B2/NuoG(Cys265Ala) die P P Oligonukleotide N1c4 und N1c4r. Nach der PCR wurde das Plasmid pBAD-B2 durch DpnI fragmentiert und die amplifizierten Plasmide in E. coli transformiert. Nach der Transformation wurde der Ansatz auf LB-Medium mit Ampicillin ausgestrichen. Jeweils sechs der Kolonien wurden für 16 Std. bei 37°C in LB-Medium mit Ampicillin angezogen, aus diesen Übernachtkulturen die Plasmide isoliert und die erwünschten Klone durch Restriktionsanalyse identifiziert. Aus diesen Klonen wurde jeweils ein DraIII-Fragment mit der betreffenden Mutation herausgeschnitten. Aus dem Plasmid pET11-a/nuoB-G/NuoFC B B wurde ebenfalls das 10´367 bp große DraIII-Fragment isoliert und aufgereinigt. Dieses Fragment wurde jeweils mit dem 2´692 bp großen DraIII-Fragment der mutierten Plasmide ligiert und in E. coli RR1∆15 transformiert und ausplattiert. Die Transformanden wurden für - 103 - Ergebnisse 16 Std. bei 37°C angezogen, aus diesen Übernachtkulturen die Plasmide isoliert und durch Restriktionsanalyse charakterisiert (Abb. 4-56). Die Primer der Mutagenese-PCR enthielten im Bereich der Mutation eine singuläre Erkennungssequenz für ein Restriktionsenzym. Tab. 4-17 zeigt die erwarteten Fragmentgrößen des Plasmids pET11-a/nuoB-G/NuoFC mit und ohne Mutation. Tab. 4-17: Erwartete Fragmentgrößen des Plasmids pET11-a/nuoB-G/NuoFC und der Plasmide mit den entsprechenden Mutationen auf NuoG nach Restriktionsanalyse mit verschiedenen Enzymen. Mutation Cys230Ala Restriktions- pET11-a/nuoB-G/ pET11-a/nuoB-G/ enzym NuoFC NuoFC(Mutation) [bp] [bp] 5´764 5´764 5´635 3´211 844 2´424 708 844 108 708 PvuI 108 Cys233Ala HindIII 6´069 6´069 3´169 2´710 2´710 2´540 1´111 1´111 629 Cys237Ala EheI 6´894 5´392 4´153 4´153 1´182 1´565 695 1´182 114 695 21 114 21 Cys 265 Ala RsrII 13´059 9´209 3´850 - 104 - Ergebnisse Abb. 4-56: Restriktionsanalyse der Plasmide pET11-a/nuoB-G/NuoFC(Cys230Ala) (Spur 5) mit PvuI, pET11-a/nuoB-G/NuoFC(Cys233Ala) (Spur 2) mit dem Restriktionsenzym HindIII, von pET11-a/nuoB-G/NuoFC(Cys237Ala) (Spur 3) mit EheI und 265 pET11-a/nuoB-G/NuoFC(Cys Ala) (Spur 4) mit RsrII. Als Standard wurde pUC-Mix 4 (Spur 1 und 6; MBI Fermentas, St. Leon-Rot) aufgetragen. Sämtliche Mutationen wurden durch Sequenzierung bestätigt und die mutierten Plasmide in den Stamm BL21(DE3) transformiert. In diesem Stamm wurden die nuo-Gene durch IPTGZugabe exprimiert. 4.6.1 ESR-spektroskopische Charakterisierung des NADH-Dehydrogenase Fragmentes im Cytoplasma Durch die Überexpression von nuoB-G wird das NADH-Dehydrogenase Fragment mit dem StrepTag II C-terminal an NuoF vollständig im Cytoplasma assembliert (Bungert et al., 1999). Für die korrekte Assemblierung werden die Untereinheiten NuoB und NuoCD benötigt, diese sind aber in der Präparation des Fragmentes nicht nachweisbar (Braun et al., 1998). In einer Western-Blot-Analyse des Cytosols des Stammes BL21(DE3)/pET11-a/nuoB-G/NuoFC mit spezifischen Antikörpern gegen NuoB und NuoCD wurden beide Untereinheiten nachgewiesen (Abb. 4-57). Beide Proteine wurden in der Kontrolle, dem Überproduktionsstamm ANN003/pAR1219, einer Komplex I-Präparation, ebenfalls eindeutig nachgewiesen. Im Cytosol des Überproduzenten ANN003/pAR1219 wurde ebenfalls eine geringe Menge der Untereinheit NuoB detektiert (Abb. 4-57 a). Dies könnte von einem Abbau des Komplex I oder einem Assemblierungsintermediat stammen. NuoCD wurde nicht erkannt (Abb. 4-57 b). - 105 - Ergebnisse Abb. 4-57: Western-Blot-Analyse des Cytosols der Stämme BL21(DE3)/pET11-a/nuoB-G/NuoFC (Spur 1 und 4) und ANN003/pAR1219 (Spur 2 und 6) sowie von aufgereinigtem Komplex I (Spur 3 und 5) mit Antikörpern gegen NuoB (a) und NuoCD (b). Die Positionen der Untereinheiten sind mit einem Pfeil gekennzeichnet. B Das Cytoplasma des B Stammes BL21(DE3)/pET11-a/nuoB-G/NuoFC B B wurde nach Ultrazentrifugation der aufgebrochenen Zellen gewonnen und aliquotiert. Ein 300 µl Aliquot wurde mit 5 µl 1 M NADH versetzt, ein anderes mit dem gleichen Volumen Puffer. Beide Proben wurden 5 min auf Eis inkubiert, in ESR-Röhrchen überführt und tiefgefroren. Von beiden Proben wurden ESR-Spektren bei 40 K und 10 mW Mikrowellenleistung aufgenommen. Das Spektrum der Probe, die mit Puffer verdünnt worden war, wurde von dem Spektrum der mit NADH-reduzierten Probe abgezogen. Das Differenz-Spektrum zeigt die Signale der paramagnetischen Spezies des Cytoplasmas, die mit NADH reduzierbar sind (Abb. 4-58). Das Differenz-Spektrum zeigt alle Signale der zweikernigen Fe/S-Zentren des NADH-Dehydrogenase Fragmentes, die auch in der Präparation beschrieben wurden (Braun et al., 1998). Die Signale des Zentrums N1b bei g2,⊥ = 2.03, 1.94 und die Signale bei B B gx, y, z = 1.92, 1.94, 2.00, die dem Zentrum N1c zugeordnet wurden, sind zu erkennen (Abb. B B 4-58). Das Spektrum bei 13 K zeigt außerdem die vierkernigen Fe/S-Zentren N2 und N4, die jedoch so stark von Signalen anderer Fe/S-Zentren überlagert sind, dass sie nicht ausgewertet werden können (Daten nicht gezeigt). - 106 - Ergebnisse Abb. 4-58: ESR (NADH-reduziert minus Luft-oxidiert) Differenz-Spektrum des Cytoplasmas aus BL21(DE3)/pET11-a/nuoB-G/NuoFc. Die Einzelspektren wurden bei 40 K und 10 mW Mikrowellenleistung aufgenommen. Die übrigen Messparameter waren: Mikrowellenfrequenz, 9.44 GHz; Modulationsamplitude, 6 mT; Zeitkonstante, 0.064 s und Aufnahmegeschwindigkeit 17.9 mT/min. Die Signale sind einzelnen Fe/S-Zentren zugeordnet. Die Spektren des Cytoplasmas der Mutantenstämme wurden in gleicher Weise erhalten. Die Differenzspektren des Cytoplasmas aus den Mutanten Cys233Ala und Cys237Ala zeigten nur die Signale des Zentrums N1c (Abb. 4-59 c, d). Die Signale des Zentrums N1b waren nicht nachweisbar. Die Differenzspektren des Cytoplasmas der Mutanten Cys230Ala und Cys265Ala zeigten ein sehr kleines Signal des Zentrums N1c, deren Amplituden weniger als 5 % der des Signals im Wildtyp Cytoplasma betrugen (Abb. 4-59 d, e). Die Signale des Fe/S-Zentrums N1b war in keiner Mutante nachweisbar (Abb. 4-59 d, e). - 107 - Ergebnisse Abb. 4-59: ESR-(NADH-reduziert minus Luft-oxidiert)-Differenzspektren des Cytoplasmas des Stammes BL21(DE3)/pET11a/nuoB-G/NuoFc (a) und der NuoG-Mutantenstämme Cys230Ala (b), Cys233Ala (c), Cys237Ala (d) und Cys265Ala (e). Die Einzelspektren wurden bei 40 K und 10 mW Mikrowellenleistung aufgenommen. Die übrigen Messparameter betrugen: Mikrowellenfrequenz von 9.44 GHz, Modulationsamplitude von 6 mT, Zeitkonstante von 0.064 s und Aufnahmegeschwindigkeit von 17.9 mT/min. Die Signale sind einzelnen Fe/SZentren zugeordnet. - 108 - Ergebnisse 4.6.2 Isolierung des NADH-Dehydrogenase Fragmentes aus den NuoG-Mutanten Um sicherzustellen, dass die ESR-Differenzspektren des Cytoplasmas der Mutanten die Signale der Fe/S-Zentren des NADH-Dehydrogenase Fragmentes widerspiegeln, wurde das Fragment aus dem Cytosol mittels Anionenaustausch-Chromatographie und AffinitätsChromatographie aufgereinigt. Das Enzym eluierte von der Anionenaustausch- Chromatographie bei 160 mM NaCl. Bei der Affinitäts-Chromatographie eluierte das Protein in einem einzigen Peak. In Abb. 4-60 sind die Elutionsprofile der Präparation aus der Mutante Cys233Ala gezeigt. Die Präparationen des NADH-Dehydrogenase Fragmentes aus den Mutanten Cys237Ala und Cys265Ala zeigten ähnliche Elutionsprofile. Abb. 4-60: Isolierung des NADH-Dehydrogenase Fragmentes aus dem Stamm BL21(DE3)/pET11-a/nuoB-G/NuoFc/NuoG(Cys233Ala) über eine Chromatographie an DEAE-Sepharose (a) und Chromatographie an StrepTactin-Sepharose (b). Die durchgezogene Kurve zeigt die Absorbanz bei 280 nm, die Punkt-Strich Linie den Verlauf der NADH/Ferricyanid Reduktaseaktivität und die gestrichelte Linie die Lage des NaClGradienten. In (c) ist die Polypetidzusammensetzung der gekennzeichneten Fraktionen der Affinitäts-Chromatographie nach SDS-PAGE gezeigt. Der Pfeil markiert den Beginn der Elution mit D-Desthiobiotin-Puffer. - 109 - Ergebnisse Aus 10 g Zellen (Feuchtgewicht) des Stammes BL21(DE3)/pET11-a/nuoB-G/NuoFc wurden 5 mg NADH-Dehydrogenase Fragment erhalten. Aus 50 g Zellen der NuoG-Mutanten wurden nur 0.5 bis 1.5 mg erhalten. Die Präparationen aus den NuoG-Mutanten Cys233Ala, Cys237Ala und Cys265Ala bestand aus den Untereinheiten NuoE, F und G des Fragmentes (Fig. 4-58 a, b, c). Jedoch tendierten Präparationen aus der NuoG-Mutante Cys237Ala zu einem Verlust von NuoG, wenn der pH-Wert vor dem Auftrag der Probe auf die AffinitätsChromatographiesäule mit 1 M statt mit 0.1 M NaOH auf pH 7.0 eingestellt wurde (siehe Material und Methoden). Abhängig von der Einstellung des pH-Werts enthielt die Präparation aus dieser Mutante entweder das komplett assemblierte NADH-Dehydrogenase Fragment oder einen aus den Untereinheit NuoE und NuoF bestehenden Subkomplex (Abb. 4-61 d). Das NADH-Dehydrogenase Fragment konnte aus der NuoG-Mutante Cys230Ala nicht isoliert werden, da es vermutlich nicht assembliert wurde. Dies steht in Einklang damit, dass im ESRDifferenz-Spektrum des Cytosols dieser Mutante kein Fe/S-Zentrum des Fragmentes detektiert wurde (Abb. 4-61 b). Aus dieser Mutante wurde nur die Untereinheit NuoF, die mit dem StrepTag modifiziert ist, aufgereinigt (Abb. 4-61 e). Abb. 4-61: SDS-PAGE der Präparationen des NADH-Dehydrogenase Fragmentes aus den Stämmen BL21(DE3)/pET11-a/nuoB-G/NuoFc (a) und den NuoG-Mutanten Cys265Ala (b), Cys237Ala (c, d) und Cys230Ala (e). Das NADH-Dehydrogenase Fragment der Mutante Cys237Ala wurde entweder unter milden Bedingungen isoliert (c) oder vor der AffinitätsChromatographie mit 1 M NaOH titriert (d). Die Positionen der Untereinheiten NuoE, NuoF und NuoG sind angezeigt. Die zusätzliche Bande mit einer apparenten molekularen Masse von 18 kDa in (c) stammt von einem proteolytischen Fragment von NuoG (Bungert et al., 1999). - 110 - Ergebnisse 4.6.3 ESR-spektroskopische Charakterisierung des aufgereinigten NADH- Dehydrogenase Fragmentes Die Präparationen wurden mit einem Überschuß Dithionit in Gegenwart von Redoxmediatoren reduziert und bei 40 K und 2 mW Mikrowellenleistung vermessen. Die ESR-Spektren des isolierten NADH-Dehydrogenase Fragmentes unterschieden sich nicht signifikant von den Differenzspektren des Cytosols der Mutanten (Abb. 4-59 und 4-62). Die ESR-Spektren des Fragmentes der NuoG-Mutanten Cys233Ala, Cys237Ala und Cys265Ala zeigten die Signale, die dem Zentrum N1c zugeschrieben wurden (Abb. 4-62). Signale des Zentrums N1b äußerst geringer Intensität waren ebenfalls nachzuweisen (Abb. 4-62). Die Signale der Fe/S-Zentren N3 und N4 wurden in den Präparationen nicht detektiert (Daten nicht gezeigt). Abb. 4-62: ESR-Spektren der Präparationen des NADH-Dehydrogenase Fragmentes aus den Stämmen BL21(DE3)/pET11-a/nuoB-G/NuoFc (a) und aus den NuoG-Mutanten Cys233Ala (b), Cys237Ala (c) und Cys265Ala (d). Die Spektren wurden bei 40 K und 2 mW aufgenommen. Die Proben wurden mit einem Überschuss an Dithionit in der Gegenwart von Redoxmediatoren reduziert. Das mit einem Stern markierte Radikal-Signal stammt von den Redoxmediatoren. Die Signale sind einzelnen Fe/S-Zentren zugeordnet. Die übrigen Messparameter waren: 9.44 GHz Mikrowellenfrequenz, 6 mT Modulationsamplitude, 0.064 s Zeitkonstante, Aufnahmegeschwindigkeit 17.9 mT/min. - 111 - Ergebnisse Mutationen in dem zusätzlichen Bindemotiv auf NuoG hatten einen Einfluss auf die ESRSpektren aller Fe/S-Zentren, bis auf die Signale des Zentrums N1c. Das ESR-Spektrum des Subkomplexes aus den Untereinheiten NuoE und NuoF aus der Mutante zeigte ebenfalls die ESR-Signale des Zentrums N1c (Abb. 4-63). Daher kann das Signal nicht von einem auf der Untereinheit NuoG liegenden Fe/S-Zentrum stammen. Die Zuordnung des Signals „N1c“ zu dem zusätzlichen Bindemotiv auf NuoG ist somit falsch. Wie aus der Präparation vor allem der Mutanten Cys237Ala und Cys230Ala ersichtlich ist, scheint die Mutagenese dieses Bindemotives die Assemblierung des NADH-Dehydrogenase Fragmentes zu stören. Es ist somit wahrscheinlich, dass dieses Bindemotiv die Liganden für ein Fe/S-Zentrum zur Verfügung stellt, welches bislang nicht ESR-spektroskopisch nachgewiesen wurde. Abb. 4-63: ESR-Spektren der Präparationen des NADH-Dehydrogenase Fragmentes aus den Stämmen BL21(DE3)/pET11-a/nuoB-G/NuoFc (a) und dem NuoEF-Subkomplex aus der NuoG-Mutante Cys237Ala (b). Die Spektren wurden bei 40 K und 2 mW aufgenommen. Die Proben wurden mit einem Überschuss an Dithionit in der Gegenwart von Redoxmediatoren reduziert. Das mit einem Stern markierte Signal wird von den Redoxmediatoren verursacht. Die Signale sind einzelnen Fe/S-Zentren zugeordnet. Die übrigen Messbedingungen waren: 9.44 GHz Mikrowellenfrequenz, 6 mT Modulationsamplitude, 0.064 s Zeitkonstante, Aufnahmegeschwindigkeit 17.9 mT/min. - 112 - Ergebnisse 4.6.4 ESR-spektroskopische Charakterisierung des NADH-Dehydrogenase Fragmentes in aerob und anaerob gezogenen Zellen Es war vorgeschlagen worden, dass das zusätzliche Motiv auf NuoG ein Fe/S-Zentrum bindet, das als „molekularer Schalter“ für die Anpassung an aerobes und anaerobes Wachstum dient, indem es von einem zweikernigen in einem vierkernigen Zustand wechselt (Nakamuru-Ogiso et al., 2000). Um diese Annahme zu überprüfen, wurde der Stamm BL21(DE3)/pET11-a/nuoB-G/NuoFC einmal aerob unter starker Luftzufuhr und einmal anaerob unter Stickstoffbegasung angezogen. Die unterschiedlichen Anzuchtbedingungen spiegelten sich in der Färbung der Zellen wider (Abb. 4-64) Abb. 4-64: Suspensionen der Zucht von E. coli BL21(DE3)/pET11-a/nuoB-G/NuoFC unter aeroben (O2) und anaeroben (N2) Bedingungen. Die Zellen wurden mit Toluol permeabilisiert und aliquotiert. Ein Aliquot wurde mit NADH reduziert, das andere mit dem gleichen Volumen Puffer verdünnt. Von den vier Proben wurden ESR-Spektren bei 40 K und 10 mW aufgenommen. Abb. 4-65 zeigt die (NADHreduziert minus wie-isoliert) Differenzspektren der aeroben und anaeroben Zucht. Es konnten keine signifikanten Unterschiede zwischen den Spektren nachgewiesen werden, so dass keine Aussage zu der Theorie des „molekularen Schalters“ gemacht werden kann. Die zusätzlichen Signale in den Differenz-Spektrum der aerob gezogenen Probe stammen von Mangan, was damit zu erklären ist, dass anscheinend die oxidierte Probe zu stark verdünnt wurde. - 113 - Ergebnisse Abb. 4-65: ESR (NADH-reduziert minus wie-isoliert)-Differenz-Spektrum permeabilisierter ganzer Zellen von E. coli BL21(DE3)/pET11-a/nuoB-G/NuoFc einer aeroben (a) und einer anaeroben (b) Zucht. Die Einzelspektren wurden bei 40 K und 10 mW Mikrowellenleistung aufgenommen. Die übrigen Messbedingungen waren: 9.44 GHz Mikrowellenfrequenz, 6 mT Modulationsamplitude, 0.064 s Zeitkonstante und 17.9 mT/min Aufnahmegeschwindigkeit. - 114 - Diskussion 5. Diskussion 5.1 Das extrachromosomale Expressionssystem pBADnuo Das Plasmid pBADnuo ist das erste extrachromosomale Expressionsystem sämtlicher Komplex I-Gene. Ein solches System wurde in der Literatur bislang nicht beschrieben. Es ist zur homologen Überproduktion des Komplexes in E. coli geeignet. Es wurde gezeigt, dass eine Produktion der 13 Untereinheiten NuoA-N bei Expression der zugehörigen nuo-Gene vom Plasmid pBADnuo stattfindet (Abb. 4-12) und dass ein enzymatisch aktiver Komplex I aus allen Untereinheiten NuoA-N assembliert wird. Dies wurde durch ZuckergradientenZentrifugation (Abb. 4-11), Messung der Atmungsaktivität (Abb. 4-10) und Wachstumskurven (Abb. 4-8) belegt. Der einzige Bericht in der Literatur über eine extrachromosomale Expression und Produktion von mehreren Komplex I-Genen beschreibt die Überproduktion des NADH-Dehydrogenase Fragmentes durch Expression der Gengruppe nuoB-G in E. coli (Braun et al., 1998; Bungert et al., 1999). Einzelne Untereinheiten des Komplex I aus Paracoccus denitrificans wurden heterolog in E. coli exprimiert und die einzelnen Untereinheiten spektroskopisch charakterisiert (Yano et al., 1994 a und b; Yano et al., 1995; Yano et al., 1996; Yano et al., 1996; Yagi et al., 1998; Yagi und Matsuno-Yagi, 2003). Allerdings wichen die spektroskopischen Eigenschaften der Kofaktoren in den überproduzierten einzelnen Untereinheiten stark von denen der Kofaktoren im Gesamtkomplex ab. Mit dieser Methode wurden die Liganden einzelner Kofaktoren identifiziert, die Rolle dieser Kofaktoren für den Mechanismus des Komplexes blieb aber unklar. Bislang wurde Komplex I durch verstärkte Expression der chromosomalen Gene nuoA-N überproduziert. Dies gelang durch den Austausch des nuo-Promotors gegen den induzierbaren T4-Phagen Promotor (Spehr et al., 1999). Die Ausbeuten der Präparationen des Komplexes aus ANN0221/pBADnuo waren etwa ein Drittel der Ausbeuten des chromosomalen Überproduzenten ANN003/pAR1219. Das pBADnuo-System stellt somit eine Alternative zur Überproduktion von Komplex I im Stamm ANN003/pAR1219 dar. Die Induktion der Expression mit L(+)-Arabinose ist wesentlich preisgünstiger als die mit IPTG, die beim Überproduzenten ANN003/pAR1219 benötigt wird. Gemessen an der NADH-Oxidase Aktivität wird der Komplex in beiden Stämmen etwa vier- bis fünffach überproduziert (Tab. 4-2; Spehr et al., 1999). Dies ist eine geringe Überproduktionsrate, z.B. im Vergleich zu - 115 - Diskussion der 20-fachen Überproduktion der ATP-Synthase in E. coli (von Meyenburg et al., 1984)). Die vergleichbare Produktion des Komplexes bei chromosomaler und Plasmid-gerichteter Überproduktion zeigt, dass die geringe Überproduktion keine Folge von noch unbekannten Regulationsfaktoren auf dem Chromosom ist. Eine mögliche Ursache könnte die fehlerhafte bzw. unzureichende Assemblierung des Komplexes sein. Durch die gleichzeitige Expression von Chaperonen könnte die Proteinproduktion gesteigert werden. Dieser Ansatz wird derzeit in unserem Arbeitskreis verfolgt. Weiterhin sollte untersucht werden, inwiefern sich die Anzucht der Zellen in Mangelmedium auf die Proteinproduktion auswirkt. Es wurde gezeigt, dass der Arabinosepromotor in reichhaltigem Medium eine drei- bis vierfach geringere Expression als in Mangelmedium aufweist (Guzman et al., 1995). Dies könnte aus einer geringeren Kopienzahl in reichhaltigem Medium oder aus der Gegenwart von KatabolitRepressoren oder Inhibitoren resultieren. Dieser Effekt wurde auch bei anderen Promotoren, wie z.B. dem tac-Promotor, beobachtet (Guzman et al., 1995). Als Expressionsvektor wurde das Plasmid pBAD33 ausgewählt (Guzman et al., 1995). Die Expression über den Arabinose-Promotor wird durch die Konzentration von L(+)-Arabinose sehr gut reguliert. Der Vektor zeigt im Vergleich zu den auf dem T7-Promotor basierten Plasmiden kaum Hintergrundexpression, da die Expression durch Glucose vollständig reprimiert wird (Guzman et al., 1995). Dadurch ist es möglich, die Expression des nuoOperons durch Zugabe von Arabinose bzw. Glucose zu dem Medium gezielt an- und auszuschalten. Die Regulation der Expression über die Arabinosekonzentration verhindert auch eine zu starke Expression des nuo-Operons, was für die Zelle unter Umständen letal sein könnte. Der Promotor sollte außerdem nicht zu stark sein, da eine Protein-Überproduktion von mehr als 30 % des Gesamtproteins der Zelle zu einer Zerstörung der mRNA und der Ribosomen sowie zu einer Akkumulation von Hitzeschockproteinen führen kann (Dong et al., 1995). Bei der Konstruktion des 21 kbp großen Plasmids pBADnuo ergaben sich mehrere Probleme. Zum einen gingen sehr große DNA-Fragmente bis 12 kbp bei der Aufreinigung durch Gelelution oder über eine PCR-Aufreinigungsmethode meist vollständig verloren, da sie so stark an die Silica-Säulen banden, dass sie nicht eluierten. Daher wurde auf eine präparative Trennung der Fragmente eines Restriktionsansatzes verzichtet und die Fragmente nur über eine Ethanolfällung aufgereinigt. Dadurch kam es zwar mitunter zu einer Ligation falscher Fragmente mit dem Vektor, die Ausbeute an gewünschtem Produkt war aber dennoch höher - 116 - Diskussion als bei einer Ligation mit einem aufgereinigten Fragment geringer Konzentration. Die großen Konstrukte konnten nur über eine Elektroporation in E. coli transformiert werden, da bei einer chemischen Transformation die Konstrukte vermehrt fragmentieren. Ein weiterer wichtiger Faktor ist die Verwendung des E. coli Stammes DH5α zur Propagation sehr großer Plasmide. Die E. coli Stämme BL21(DE3) und RR1∆M15 waren dazu nicht geeignet, was sich in einer Fragmentierung der Plasmid-Konstrukte zeigte. DH5α scheint als E. coli K12-Derivat gegenüber den B-Typ Derivaten BL21(DE3) und RR1∆M15 für eine stabile Propagation großer Konstrukte besser geeignet zu sein. Nach der Vermehrung des Plasmids pBADnuo in DH5α war es stabil genug, um auch in anderen Stämmen erfolgreich repliziert zu werden. 5.2 Nutzung des Plasmids pBADnuo zur Mutagenese Eine weitere Anwendung des pBADnuo-Expressionssystemes stellt die Mutagenese der nuoGene dar. Eine Mutagenese des chromosomalen nuo-Operons ist sehr umständlich und aufwendig, da Mutationen stets unmarkiert eingeführt werden müssen, um die Expression der stromabwärts gelegenen nuo-Gene nicht zu beeinflussen. Als Kompromiss stellte sich ein Ansatz heraus, mit dem auf der Untereinheit NuoB eine Reihe von Mutationen eingeführt wurden (Flemming et al., 2003). Das Gen nuoB wurde in das Plasmid pBAD33 kloniert und dort mutagenisiert. Dieses Plasmid wurde in einen Stamm transformiert, in dem nuoB chromosomal deletiert worden war. In diesem Ansatz wurde NuoB von dem Plasmid produziert, die restlichen Komplex I-Untereinheiten von dem Chromosom. Diese in transKomplementation ermöglichte die Produktion des vollständig assemblierten Komplexes, der die auf dem Plasmid kodierten Mutationen in NuoB trug (Flemming et al., 2003). Um prinzipiell Mutationen einzelner Untereinheiten auf einem Plasmid einzuführen, wird für die in trans-Komplementation ein E. coli Stamm mit der entsprechenden genomischen Deletion benötigt. Außerdem muss das Gen der betreffenden Untereinheit in ein Expressionsplasmid kloniert werden. Es ist aber nicht bekannt, ob eine in trans-Komplementation mit jedem Gen des nuo-Operons möglich ist, da die Deletion die Expression stromabwärts gelegener Bereiche des Operons stören könnte oder die Stabilität der mRNA beeinträchtigen könnte. Eine sinnvolle Alternative ist deshalb die Expression des gesamten nuo-Operons von einem Plasmid, in dem Mutationen eingeführt werden können. Mit einer Größe von über 21 kbp ist das Plasmid pBADnuo allerdings viel zu groß für eine direkte Mutagenese. PCR-basierte Mutagenesemethoden beschränken sich auf Plasmide mit einer Größe bis etwa 6 kbp, da - 117 - Diskussion oberhalb dieser Größe die Syntheseleistung der Polymerase zu gering ist und außerdem die Fehlerrate stark ansteigt, so dass das PCR-Produkt weitere unerkannte Mutationen tragen könnte. Solche Mutationen können im besten Fall zum Austausch einer Aminosäure führen, schlimmstenfalls kann es zur Einführung eines Stoppkodons oder zur Verschiebung des Leserasters kommen. Deshalb sollten kleinere Plasmide mutagenisiert werden, die nur Teile des nuo-Operons enthalten. Um nicht für jede Mutagenese ein neues Konstrukt zu klonieren, wurde ein sogenanntes Subklon-System des nuo-Operons aufgestellt. Dieses sollte aus drei Subklonen pBAD-A, pBAD-B und pBAD-C bestehen, die wiederum in noch kleinere Subklone aufgeteilt werden sollten. Diese kleinen Subklone sollten durch PCR-Methoden mutagenisiert werden und anschließend in einen großen Subklon und dann in das Plasmid pBADnuo zurückkloniert werden. Mit dieser Methode wurde z.B. eine StrepTag II-Sequenz am 3´-Ende von nuoB eingeführt (Kap. 4.4.1). In dieser Arbeit wurde der Grundstein des Subklonsystems gelegt, da nur die über den Subklon pBAD-A abgedeckten Gene nuoA-nuoE´ zugänglich sind. Die Subklone pBAD-B und pBAD-C konnten bislang nicht hergestellt werden. Die Größe der zu klonierenden nuo-Bereiche erschwert die Herstellung dieser Subklone. Eine Möglichkeit, diese Subklone herzustellen, wäre, neue singuläre Restriktionssequenzen in pBADnuo einzufügen. Aufgrund der Größe des Plasmids ist eine PCR an pBADnuo allerdings nicht möglich. Solche Sequenzen könnten über die λ-Red vermittelte Rekombination in pBADnuo eingeführt werden. 5.3 Vergleich der λ -Red vermittelten Rekombination mit der Klonierung Da die Subklone pBAD-B und pBAD-C bislang nicht zur Verfügung stehen, wurde nach Alternativen gesucht, Mutationen von den kleinen Subklonen in pBADnuo einzubringen. Klonierungsmethoden sind dabei nicht möglich, da keine weiteren singulären Schnittstellen vorhanden sind, die für die Rückklonierung genutzt werden können. Eine Möglichkeit stellt die Rekombination dar, die bislang in E. coli nur wenig genutzt wird, da das Rekombinasesystem von E. coli für eine genetische Manipulation nicht geeignet ist. Zum einen wird zur Rekombination ein homologer Bereich von mehreren tausend bp benötigt, zum anderen ist die Rekombination linearer DNA nicht möglich, da die RecBCD-Nuklease lineare DNA abbaut. Wird aber in E. coli die λ-Red Rekombinase aus dem Phagen λ exprimiert, so - 118 - Diskussion verhindert diese den Abbau linearer DNA durch Inhibition der RecBCD-Nuklease. Die Rekombination mit der λ-Red Rekombinase benötigt einen homologen Bereich von nur etwa 36 bp. Mit der λ-Red Rekombinationsmethode können lineare DNA-Fragmente in genomische oder Plasmid-DNA eingebaut werden, wenn sie flankierende Bereiche von mindestens 36 bp besitzen, die der genomischen oder der Plasmidsequenz entsprechen. Mit der Methode der λ-Red vermittelten Rekombination wurde versucht, Deletionen, Insertionen und Punktmutationen in Plasmide einzuführen. Punktmutationen auf nuoM wurden in den kleinen Subklon pBAD-C1 eingeführt (Abb. 4-47, 4-48), konnten aber nicht in pBADnuo zurückkloniert werden, da der große Subklon pBAD-B nicht vorhanden war (Kap. 4.3.2). Es wurde deshalb versucht, ein Fragment von pBAD-C1, das die gewünschte Mutation trug, in pBADnuo zu rekombinieren (Kap. 4.5.2). Dieser Versuch scheiterte zum einen daran, dass stets nur ein Gemisch beider Plasmide erzeugt wurde, zum anderen an der fehlenden Selektion auf das rekombinante Plasmid. Eine Selektion über das sacR-sacB-Suizidsystem lieferte nur die Rekombination des Fragmentes in das Genom. Diese Methode müsste an die gewünschte Anwendung angepasst werden. Ohne eine Selektionsmöglichkeit müssen nach der Rekombination eine große Anzahl Klone analysiert werden. Dies muss bei Punktmutationen durch Restriktionsanalyse erfolgen, eine PCRbasierte Analyse wie im Fall von Insertionen oder Deletionen ist nicht möglich. Dies zeigt, dass die λ-Red vermittelte Rekombination nicht unbedingt für die Einführung von Punktmutationen geeignet ist. Es wäre wahrscheinlich sinnvoller, das mutierte Gen in ein Expressionsplasmid Deletionsstamm zu einzubringen und exprimieren. die Untereinheit Allerdings ist es in einem entsprechenden nicht trivial, chromosomale Deletionsmutanten von E. coli mit den gebräuchlichen Methoden herzustellen. Hier ist das λ-Red Rekombinationssystem eine effektive Methode zur Deletion einzelner Gene und Gengruppen. Die Deletionen können dabei sowohl auf dem Chromosom als auch auf dem Plasmid pBADnuo eingeführt werden. Mittels der λ-Red vermittelten Rekombination wurden verschiedene Deletionen in das Plasmid pBADnuo eingefügt: pBADnuo/∆nuoB (Kissel, 2003), pBADnuo/∆nuoCD (Abb. 4-38), pBADnuo/∆nuoEF (Abb. 4-43), pBADnuo/∆nuoG (Abb. 4-45) und pBADnuo/∆nuoM (Abb. 4-41). Die deletierten Bereiche waren zwischen 662 bp (nuoB) und 2´732 bp (nuoG) groß. Die Deletion des 4´613 bp großen Genbereiches nuoE-nuoG gelang nicht. Nach der Rekombination wurden zwar resistente Kolonien gefunden, die aber keine Plasmide - 119 - Diskussion enthielten. Es ist möglich, dass die Resistenzkassette in das Chromosom statt in das Plasmid pBADnuo rekombiniert wurde. Es wurde berichtet, dass mit dieser Methode Deletionen von bis zu 5´178 bp möglich sind (Datsenko und Wanner, 2000). Diese Deletionen wurden allerdings ausschließlich ins Genom eingeführt. Vermutlich ist die starke Ringspannung des über 21 kbp großen Plasmids pBADnuo für die Rekombination eines großen Bereiches hinderlich. Der 4.6 kbp große Bereich nuoEFG sollte durch die 1.6 kbp große Kanamycinresistenzkassette ersetzt werden. Zur Anlagerung der Resistenzkassette an die homologen Bereiche müsste das Plasmid stark verdrillt werden, daher ist es vermutlich einfacher, die Resistenz in das Genom einzubauen. Um diesen Bereich dennoch zu deletieren, könnte in die Plasmide pBADnuo/∆nuoEF eine Deletion von nuoG bzw. in pBADnuo/∆nuoG eine Deletion von nuoE und nuoF eingeführt werden. Durch die λ-Red Rekombinase wurde die StrepTag II-Sequenz mit einem verbindenden Bereich, der für zwei bzw. sieben Aminosäuren kodiert, in das Plasmid pBADnuo eingebracht. Hierbei wurde sowohl eine schon innerhalb einer Gengruppe des nuo-Operons vorhandene Tag-Sequenz genutzt als auch eine Tag-Sequenz neu eingeführt. Im ersten Fall wurde ein Fragment aus dem Expressionsplasmid pET11-a/nuoB-G/NuoFC, in dem die B B StrepTag II-Sequenz am 3´-Ende von nuoF angefügt wurde (Bungert et al., 1999), in pBADnuo rekombiniert. Da es keine positive Selektion gab, wurden sehr viele Klone analysiert. Etwa 3 % der Klone enthielten ein Plasmidgemisch, aus dem das gewünschte Konstrukt durch Retransformation nach Verdünnung des Plasmidgemisches vereinzelt werden konnte. Auch die Neusynthese der StrepTag II-Sequenz an nuoC in pBADnuo über die Rekombination eines PCR-Produktes war sehr effizient. Dabei wurde eine so genannte „Template-lose“ PCR eingesetzt (Abb. 4-28), in der die Oligonukleotide aneinander angelagert und verlängert werden, anstatt an einer Matrizen-DNA verlängert zu werden. Diese PCR war gut geeignet, das Fragment für die Rekombination zu synthetisieren. Allerdings ist die Größe des PCR-Produktes durch die Länge der Primer eingeschränkt, da eine Synthese längerer Oligonukleotide unsicher ist und zur Ausbildung von Fehlbasen führen kann, die Rekombination behindern können. Die Insertion von 30 bp war problemlos möglich (Abb. 4-30). Das Gen nuoM konnte weder mit einer Klonierung noch einer Rekombination in ein Plasmid eingeführt werden. Die Rekombination scheiterte vor allem an einer geeigneten Selektionsmöglichkeit. Eine klassische Blau-Weiß-Selektion führte ebenfalls nicht zum - 120 - Diskussion gewünschten Ergebnis. Die Selektion wurde auch dadurch erschwert, dass das Plasmid pKD46 die gleiche Resistenz wie das Zielplasmid besitzt. Möglicherweise wurde pKD46 in den weißen Kolonien bei 37°C weiterhin repliziert. Es hatte sich gezeigt, dass zum vollständigen Verlust der Propagation dieses Plasmids eine Temperatur von 42°C über einen längeren Zeitraum nötig war, wenn Zellen auf die auf diesem Plasmid kodierte Ampicillinresistenz angewiesen waren. Es wäre sinnvoll ein Rekombinase-kodierendes Plasmid mit einer anderen Resistenz zu konstruieren. Dieses Plasmid sollte auch einen anderen Replikationsursprung haben, da einige Plasmide aufgrund der nicht-kompatiblen Replikationsursprünge nicht mit Hilfe von pKD46 rekombiniert werden können. Allgemein zeigte sich bei der Insertion von DNA in pBADnuo, dass eine Rekombination erleichtert wird, wenn das Zielplasmid und das zu rekombinierende DNA-Fragment in E. coliZellen mit dem Plasmid pKD46 kotransformiert werden. Wird das zu rekombinierende DNAFragment in Zellen transformiert, die die Plasmide pKD46 und pBADnuo enthalten, so erfolgt gleichzeitig mit der Induktion der Expression der Rekombinase auch eine Expression der nuoGene, da beide unter der Kontrolle des Arabinose-Promotors stehen. Rekombinations-Ansätze in Zellen, in denen vor der Transformation des DNA-Fragmentes eine gemeinsame Propagation von pBADnuo und pKD46 stattfand, führten zu keinem Erfolg. Dabei war es unerheblich, ob beide Plasmide kotransformiert wurden oder ob ein Plasmid in Zellen transformiert wurde, die schon das andere Plasmid enthielten. 5.4 Kombination von Klonierung und Rekombination Das in dieser Arbeit erstellte Subklonsystem ist sehr gut geeignet, um Mutationen in Komplex I einzuführen. Allerdings ist nur der Genbereich nuoA-nuoE´ zugänglich, da zwei der drei großen Subklone nicht erhalten wurden. Alle weiteren Gene sind auf den kleinen Subklonen zugänglich und können mit der λ-Red vermittelten Rekombination in pBADnuo eingebracht werden. Diese Methode hat sich bei Insertionen bewährt, während Punktmutationen deutlich schwerer zu rekombinieren und selektionieren sind. Diese Mutanten können besser über eine in trans-Komplementation erzeugt werden. Die hierfür benötigten E. coli-Stämme, in denen die nuo-Gene auf dem Chromosom deletiert wurden, können durch Rekombination hergestellt werden. Allerdings enthielten diese Zellen nur wenig Protein, was die spätere Protein-chemische Charakterisierung des Komplexes erschweren - 121 - Diskussion würde. Deshalb sollte getestet werden, ob statt der chromosomalen Deletion nicht auch eine Deletion in dem Plasmid pBADnuo verwendet werden kann. Beide Plasmide pBADnuo/∆nuoX und z.B. pET11-a/nuoX - müssten in einem Komplex I-defizienten Stamm exprimiert werden. Es ist klar, dass Klonierung und Rekombination zwei Methoden sind, die sich nicht ausschließen, sondern gegenseitig ergänzen. Ihre Kombination kann zur Herstellung rekombinierender DNA-Fragmente auch ohne singuläre Restriktionsschnittstellen genutzt werden. 5.5 Isolierung des Komplex I über eine Affinitäts-Chromatographie Es ist bislang nicht möglich, den E. coli Komplex I so mit einem Affinitätspeptid zu versehen, dass er über eine Affinitäts-Chromatographie aufgereinigt werden kann. Nur das NADHDehydrogenase Fragment wird mit Hilfe eines StrepTag II aufgereinigt. Es zeigte sich, dass nur eine C-terminale Modifizierung einer Untereinheit mit dem Affinitätspeptid sinnvoll ist, da das Fragment sonst nicht assembliert (Bungert et al., 1999). Zur Aufreinigung des Komplex I wurden die Untereinheiten NuoB, NuoC und NuoF C-terminal mit dem StrepTag II modifiziert, allerdings ohne Erfolg. Da das NADHDehydrogenase Fragment mit dem Affinitätspeptid an NuoF assembliert und in einer enzymatisch aktiven Form über eine Affinitäts-Chromatographie isoliert werden kann (Bungert et al., 1999), scheint diese Stelle im Komplex I von anderen Untereinheiten abgeschirmt zu sein. Dies wird dadurch bestätigt, dass das Affinitätspeptid die Assemblierung des Komplexes stört, wie durch Zuckergradientenzentrifugation gezeigt wurde (Abb. 4-26). Die Untereinheit NuoB wurde mit der StrepTag II-Sequenz modifiziert, da sie hydrophil ist. Hydrophobe Untereinheiten sind zur Modifizierung ungeeignet, da sie bei der Präparation durch Detergenzmoleküle abgeschirmt werden würden. Auch diese Modifizierung störte die Assemblierung des Komplex I. Die Untereinheiten NuoA, J und K des Komplexes konnten in der Präparation nicht nachgewiesen werden (Abb. 4-22), ebenso die Fe/S-Zentren N2 und N4 (Abb. 4-23). Es ist jedoch nicht auszuschliessen, dass der Komplex in der Membran vollständig assembliert vorlag, aber durch das Affinitätspeptid destabilisiert wurde, so dass er bei der Detergenzextraktion desintegrierte. Die C-terminale Modifizierung von NuoC führte zwar zu einem funktionellen Komplex I (Abb. 4-31), jedoch wurde der Komplex nur sehr schwach von dem StrepTactin-Material gebunden (Abb. 4-31). Die Bindung konnte auch - 122 - Diskussion nicht durch eine Verlängerung des verbindenden Bereiches, die das Affinitätspeptid exponieren sollte, verbessert werden. Da nur geringste Mengen des Komplexes über diese Methode aufgereinigt wurden, stellt sie keine Alternative zur Standardaufreinigung von Komplex I dar. Es wurde gezeigt, dass der Komplex I aus Y. lipolytica über eine Modifizierung mit einem His-Tag C-terminal an dem Homolog von NuoC möglich ist (Kashani-Poor et al., 2001). Warum eine entsprechende Modifizierung mit dem homologen Enzym aus E. coli offensichtlich nicht zu dem gewünschten Erfolg führt, ist nicht verstanden. Es ist aber nicht auszuschliessen, dass die schwache Bindung eine Folge des pH-Wertes des Puffers ist. Die Affinitäts-Chromatographie erfolgte bei pH 6.0, da der Komplex bei diesem pH-Wert sein Stabilitätsoptimum hat (Leif et al., 1995). Bei diesem pH-Wert zeigt das StrepTactin aber nur eine geringe Affinität zu dem StrepTag II (Voss und Skerra, 1997). In weiterführenden Arbeiten sollte versucht werden, den Komplex kurz vor der AffinitätsChromatographie auf pH 7.5 zu titrieren, schnell auf die Säule in pH 7.5-Puffer aufzutragen und mit dem üblichen pH 6.0-Puffer von der Säule zu eluieren. Die Versuche zur Modifizierung von NuoC zeigen, dass die Trennung der Untereinheit NuoCD nicht zu einer Änderung der Eigenschaften des Komplexes führt (Abb. 4-31). Dies zeigt auch, dass der am 3´-Ende der StrepTag II-Sequenz gelegene Bereich tatsächlich als Ribosomenbindestelle für nuoD dient. Stromabwärts vom Gen nuoCD wurde in dem Subklon pBAD-A die Sequenz für den His- und den StrepTag II eingeführt (Hinderberger, 2004). Nach Rekombination bzw. Rückklonierung in pBADnuo kann überprüft werden, ob das Affinitätspeptid an dieser Position an das entsprechende Affinitätsmaterial bindet. 5.6 Das zusätzliche Fe/S-Bindemotiv auf NuoG Die ESR-Signale bei gx, y, z = 1.92, 1.94 und 2.00, die in den cytoplasmatischen Membranen von E. coli und in Präparationen des E. coli Komplex I gemessen wurden, wurden einem neuartigen zweikernigen Fe/S-Zentrum zugeordnet, das mit N1c bezeichnet wurde (Sled et al., 1993; Leif et al., 1995; Braun et al., 1998). Zwar erinnern die spektralen Eigenschaften dieses Zentrums an die des zweikernigen Fe/S-Zentrums N1a aus anderen Organismen (Ohnishi, 1998), doch zeigten sie nicht die Eigenschaften dieses Zentrums, nämlich sein sehr niedriges und pH-abhängiges Mittenpotential (Sled et al., 1993; Leif et al., 1995). Somit wurden diese ESR-Signale einem neuartigen Zentrum N1c zugeordnet, welches auf NuoG lokalisiert sein sollte, da diese Untereinheit ein zusätzliches Bindemotiv für ein Fe/S-Zentrum - 123 - Diskussion enthält. In dieser Arbeit wurde gezeigt, dass diese ESR-Signale von dem Zentrum N1a stammen und dass es kein Zentrum N1c gibt. Die Mutagenese der vier Cysteine des zusätzlichen Bindemotivs auf NuoG führten zu einem nahezu vollständigen Verlust aller Fe/S-Zentren dieser Untereinheit, während die Signale, die N1c zugeordnet wurden, nachweisbar waren (Abb. 4-59, 4-62). Diese Signale wurden auch in dem NuoEFSubkomplex aus der Mutante Cys237Ala detektiert (Abb. 4-63). Dies zeigt, dass diese Signale nicht von einem Fe/S-Zentrum auf der Untereinheit NuoG stammen können. Das einzige Bindemotiv für ein zweikerniges Fe/S-Zentrum in dem Subkomplex NuoEF befindet sich auf NuoE und bindet das Zentrum N1a (Yano et al., 1994). Dies bedeutet, dass die dem Zentrum N1c zugeordneten ESR-Signale von dem Zentrum N1a der Untereinheit NuoE stammen. Die überproduzierte und aufgereinigte Untereinheit NuoE von E. coli enthält ein zweikerniges Fe/S-Zentrum mit g-Werten und einem Mittenpotential, die identisch zu denen des als N1c bezeichneten Zentrum sind (Zu et al., 2002). Es war allerdings nicht klar, ob das Zentrum N1a auf der einzelnen Untereinheit NuoE die gleichen spektralen und thermodynamischen Eigenschaften wie im NADH-Dehydrogenase Fragment oder im Komplex I zeigen würde. Es ist bekannt, dass ESR-Spektren von Fe/S-Zentren in isolierten Untereinheiten sich erheblich von denen des gleichen Zentrums im assemblierten Komplex unterscheiden können (Yano et al., 1996). Zusätzlich wurde gezeigt, dass das Mittenpotential des Zentrums N1a aus E. coli etwa 100 mV positiver ist als das des Zentrums in anderen Organismen und dass seine pHAbhängigkeit stark von der Ionenstärke des Puffers beeinflusst wird (Zu et al., 2002). Bei Salzkonzentrationen größer als 100 mM zeigt das Mittenpotential von N1a keine pHAbhängigkeit, während es in einem Puffer mit 10 mM NaCl, im pH-Bereich von 3.5 bis 9.5 zwischen -348 mV und -246 mV variiert (Zu et al., 2002). Das Fe/S-Zentrum N1a in Komplex I von E. coli zeigte im pH-Bereich von 6.0 bis 8.2 keine pH-Abhängigkeit, da der Komplex in einem Puffer mit 300 mM NaCl vermessen wurde. Dies führte zu seiner falschen Zuordnung als N1c (Sled et al., 1993; Leif et al., 1995). Die Mutagenese des zusätzlichen Bindemotivs auf der Untereinheit NuoG lässt vermuten, dass die betreffenden Cysteine tatsächlich an der Bindung eines Fe/S-Zentrums beteiligt sind, da sie einen Einfluss auf die Assemblierung des NADH-Dehydrogenase Fragmentes hat. Vor allem in den Mutanten Cys230Ala und Cys265Ala wurde durch ESR-Differenzspektroskopie ein deutlich verringerter Anteil des NADH-Dehydrogenase Fragments gezeigt (Abb. 4-62). Das NADH-Dehydrogenase Fragment der Mutante Cys237Ala zeigte eine verringerte - 124 - Diskussion Stabilität, da es zu einem Subkomplex aus den Untereinheiten NuoE und F zerfällt (Abb. 4-63). Ein ähnlicher Subkomplex wurde durch Überexpression von NuoE und F in P. denitrificans erhalten (Yano et al., 1997). Das Fe/S-Zentrum, das an das zusätzliche Motiv auf NuoG gebunden ist, muss weitergehend charakterisiert werden. Es wurde diskutiert, ob dieses Zentrum ein molekularer Schalter zwischen aeroben und anaerobem Wachstum sein könnte (Nakamuro-Ogiso et al., 2002). In dieser Arbeit wurden keine Hinweise für diese Funktion gefunden (Abb. 4-65). Weiterhin wird diskutiert, dass dieses Zentrum mit der Fähigkeit des Komplexes zur Reaktion mit Menachinon besteht (Finel, 1998). Beide Vorschläge beschreiben verwandte Phänomene (Unden und Bongaerts, 1997). Es wurde vorgeschlagen, dass das zusätzliche Bindemotiv ein vierkerniges Fe/S-Zentrum ligiert. Ähnliche Bindemotive wurden in anderen Enzymen wie z.B. der E. coli Formiat Dehydrogenase gefunden, wo sie immer ein vierkerniges Fe/S-Zentrum binden (NakamuruOgiso et al., 2002). Die Überexpression eines kurzen Fragmentes von NuoG, das das betreffende Motiv enthält, führte zur Produktion eines Peptids, das ein vierkerniges Zentrum enthält (Nakamuru-Ogiso et al., 2002). Dies zeigt, dass das Motiv auf NuoG in E. coli Komplex I ebenfalls ein vierkerniges Zentrum binden könnte. Es ist möglich, dass dieses Zentrum bislang noch nicht ESR-spektroskopisch nachgewiesen wurde, weil es entweder ein sehr niedriges Mittenpotential besitzt oder einen Spin-Grundzustand höher als s=½. Ein wichtiges Folgeexperiment ist deshalb die ESR-spektroskopische Untersuchung eines elektrochemisch reduzierten Komplex I. Da die Namensgebung „N1c“ auf der Annahme beruhte, dass es sich hierbei um ein zweikerniges Fe/S-Zentrum handelt, sollte der Name des Zentrums auf der Untereinheit NuoG geändert werden. Zweikernige Fe/S-Zentren werden mit N1 und einem Buchstaben in der Reihenfolge der Entdeckung (N1a, N1b, darauffolgend N1c) bezeichnet. Vierkernige Fe/S-Zentren werden von N2 aufsteigend nach ihrer chronologischen Entdeckung nummeriert. Das neue Zentrum auf NuoG wird folglich nun als N7 benannt werden. Als nächster Schritt wäre zu untersuchen, ob sich die Mutagenese der betreffenden Cysteine im Gesamtkomplex ebenfalls auf die Assemblierung auswirkt. Sollte der Komplex in diesen Mutanten assemblieren, könnte er ESR-spektroskopisch untersucht werden. Zur Herstellung der Mutanten gibt es sowohl die Möglichkeit der Klonierung als auch der Rekombination. Da die Cysteine über eine Mutagenese-PCR im Subklon pBAD-B2 gegen Alanine ausgetauscht wurden, könnten die Mutationen in pBADnuo umkloniert werden. Das dafür benötigte - 125 - Diskussion Plasmid pBAD-B steht aber dafür nicht zur Verfügung. Da die zur Herstellung von pBAD-B2 verwendeten Restriktionsschnittstellen im Konstrukt pBADnuo nicht singulär vorliegen, kann keine direkte Rückklonierung erfolgen. Deshalb wäre es sinnvoll, die Mutationen des Bindemotivs auf NuoG über eine Rekombination in pBADnuo einzuführen. Hierfür können die Mutationen mit DraIII aus dem ursprünglichen Klon herausgeschnitten und dieses Fragment zur Rekombination in pBADnuo eingesetzt werden. Da keine Selektionsmöglichkeit über eine unterschiedliche Antibiotikaresistenz besteht, wäre es empfehlenswert, zuerst den betreffenden Bereich in pBADnuo gegen eine nptI-sacR-sacBKassette auszutauschen, um nach der Rekombination dieser Kassette gegen das die Mutationen tragende Fragment auf Saccharose-haltigem Medium zu selektieren. - 126 - ________________________________________________ ______ Literatur 6. Literatur Anraku, Y., und Gennis, R.B. (1987) The aerobic respiratory chain of Escherichia coli. Trends Biochem. Sci. 12, 262-266. Bailon, P., Ehrlich, G.K., Fung, W.-J., und Berthold, W. (2000) Affinity chromatography – Methods and protocols. Humana press, Totowa. Bates, D.M., Popescu, C.V., Khoroshilova, N., Vogt, K., Beinert, H., Münck, E., und Kiley, P.J. (2000) Substitution of leucine 28 with histidine in the Escherichia coli transcription factor FNR results in increased stability of the [4Fe-4S](2+) cluster to oxygen. J. Biol. Chem. 275, 6234-6240. Beinert H., und Albracht S.P.(1982) New insights, ideas and unanswered questions concerning iron-sulfur clusters in mitochondria. Biochim. Biophys. Acta. 683, 245-77. Beisenherz, G., Balze, H.J., Bücher, T., Czok, R., Garbade, K.H., Meyer-Ahrendt, E., und Pfeiderer, G. (1953) Phosphofructo-Aldolase, Phosphoglyceraldehyd-Dehydrogenase, Milchsäure-Dehydrogenase, Glycerophosphat-Dehydrogenase und Pyruvat-Dehydrogenase aus Kaninchenmuskulatur in einem Arbeitsgang. Z. Naturforsch. 8b, 555-577. Bongaerts, J., Zoske, S., Weidner, U., und Unden, G. (1995) Transcriptional regulation of the proton translocating NADH dehydrogenase genes (nuoA-N) of Escherichia coli by electron acceptors, electron donors and gene regulators. Mol. Microbiol. 16, 521-534. Böttcher, B., Scheide, D., Hesterberg, M., Nagel-Steger, L., und Friedrich, T. (2002) A novel, enzymatically active conformation of the Escherichia coli NADH:ubiquinone oxidoreductase (complex I) J. Biol. Chem. 277, 17970-17977. Boyington, J.C., Gladyshev, V.N., Khangulov, S.V., Stadtman, T.C., und Sun, P.D. (1997) Crystal structure of formate dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science 275, 1305-1308. Brandt, U. (1997) Proton-translocation by membrane-bound NADH:ubiquinoneoxidoreductase (complex I) through redox-gated ligand conduction. Biochim. Biophys. Acta 1318, 79-91. Brandt, U., Kerscher, S., Dröse, S., Zwicker, K., und Zickermann, V. (2003) Proton pumping by NADH:ubiquinone oxidoreductase. A redox driven conformational change mechanism? FEBS Lett. 545, 9-17. Braun, M., Bungert, S., und Friedrich, T. (1998) Characterization of the overproduced NADH dehydrogenase fragment of the NADH:ubiquinone oxidoreductase (complex I) from Escherichia coli. Biochemistry 37, 1861-1867. Bungert, S., Krafft, B., Schlesinger, R., und Friedrich, T. (1999) One-step purification of the NADH dehydrogenase fragment of the Escherichia coli complex I by means of Strep-tag affinity chromatography. FEBS Lett. 460, 207-211. - 127 - ________________________________________________ ______ Literatur Carroll, J., Shannon, R.J., Fearnley, I.M., Walker, J.E., und Hirst, J. (2002) Definition of the nuclear encoded protein composition of bovine heart mitochondrial complex I. Identification of two new subunits. J. Biol. Chem. 277, 50311-50317. Chan, E.K., Mukund, S., Kletzin, A., Adams, M.W.W., und Rees, D.C. (1995) Structure of a hyperthermophilic tungstopterin enzyme, aldehyde ferredoxin oxidoreductase. Science 267, 1463-1469. Chen, W., und Kuo, T. (1993) A simple method for preparation of gram-negative bacterial genomic DNA. Nucl. Acids Res. 21, 2260. Clark, M.A., Baumann, L., und Baumann, P. (1997) Buchnera aphidicola (Aphid endosymbiont) contains genes encoding enzymes of histidine biosynthesis. Curr. Microbiol. 35, 122-123. Cosloy, S.D., und Oishi, M. (1973) Genetic transformation in Escherichia coli K12. Proc. Natl. Acad. Sci. USA 70, 84-87. Cotter, P.A., Chepuri, V., Gennis, R.B., und Gunsalus, R.P. (1990) Cytochrom o (cyoABCDE) and d (cydAB) oxidase gene expression in Escherichia coli is regulated by oxigen, pH and fnr gene product. J. Bacteriol. 172, 6333-6338. Darrouzet, E., Issartel, J.P., Lunardi, J., und Dupuis, A. (1998) The 49-kDa subunit of NADH-ubiquinone oxidoreductase (Complex I) is involved in the binding of piericidn and rotenone, two quinone-related inhibitors. FEBS Lett. 431, 34-38. Datsenko, K.A., und Wanner, B.L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640-6645. Davanloo P., Rosenberg A.H., Dunn J.J., und Studier F.W. (1984) Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 81, 2035-2039. De Blas, A.L., und Cherwinski, H.M. (1983) Detection of antigens on nitrocellulose paper immunoblots with monoclonal antibodies. Anal. Biochem. 133, 214-219. Degli Esposti, M., Carelli, V., Ghelli, A., Ratta, M., Crimi, M., Sangiorgi, S., Montagna, P., Lenaz, G., Luagresi, E., und Cortelli, P. (1994) Functional alterations of the mitochondrially encodes ND4 subunit associated with Leber´s hereditary optic neuropathy. FEBS Lett. 352, 375-379. Dias, J.M., Than, M.E., Humm, A., Huber, R., Bourenkov, G.P., Bartunik, H.D., Bursakov, S, Calvete, J., Caldeira, J., Carneiro, C., Moura, J.J.G., Moura, I., und Romão, M.J. (1999) Crystal structure of the first dissimilatory nitrate reductase at 1.9 A solved by MAD methods. Structure 7, 65-79. Djafarzadeh, R., Kerscher, S., Zwicker, K., Radermacher, M., Lindahl, M., Schägger, H., und Brandt, U. (2000) Biophysical and structural characterization of proton-translocating NADHdehydrogenase (complex I) from the strictly aerobic yeast Yarrowia lipolytica. Biochim. Biophys. Acta 1459, 230-238. - 128 - ________________________________________________ ______ Literatur Dong H., Nilsson, L., und Kurland, C.G. (1995) Gratuituos overexpression of genes in Escherichia coli leads to growth inhibition and ribosome destruction. J. Bacteriol. 177, 14971504. Duin, E.C., Lafferty, M.E., Crouse, B.R., Allen, R.M., Sanyal, I., Flint, D.H., und Johnson, M.K. (1997) [2Fe-2S] to [4Fe-4S] cluster conversion in Escherichia coli biotin synthase. Biochemistry 36, 11811-11820. Dupuis, A., Prieur, I., und Lunardi, J. (2001) Toward a characterization of the connecting module of complex I. J. Bioenerg. Biomembr. 33, 159-168. Earley, F.G.P., und Ragan, C.I. (1984) Photoaffinity labelling of mitochondrial NADH dehydrogenase with arylazidoamorphigenin, an analogue of rotenone. Biochem. J. 224, 525534. Earley, F.G.P, Patel, S.D., Ragan, C.I., und Attardi, G. (1987) Photolabelling of a mitochondrially encoded subunit of NADH dehydrogenase with [3H]dihydrorotenone. FEBS Lett. 219, 108-112. Ellis, H.M., Yu, D., DiTizio, T., und Court, D.L. (2001) High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligo-nucleotides. Proc. Natl. Acad. Sci. USA 98, 6742-6746. Fearnley, I.M., und Walker, J.E. (1992). Conservation of sequences of subunits of mitochondrial complex I and their relationships with other proteins. Biochim. Biophys. Acta 1140, 105-134. Finel, M. (1998) Organization and evolution of structural elements within complex I. Biochim. Biophys. Acta 1364, 112-125. Flemming, D., Hellwig, P., and Friedrich, T. (2003) Involvement of tyrosines 114 and 139 of subunit NuoB in the proton pathway around cluster N2 in Escherichia coli NADH:ubiquinone oxidoreductase. J. Biol. Chem. 278, 3055-3062. Flemming, D., Schlitt, A., Spehr, V., Bischof, T., and Friedrich, T. (2003) Iron-sulfur cluster N2 of the Escherichia coli NADH:ubiquinone oxidoreductase (complex I) is located on subunit NuoB. J. Biol. Chem. 278, 47602-47609. Flemming, D. (2005) Die Bedeutung des Eisen-Schwefel-Zentrums N2 für den Mechanismus der NADH:Ubichinon Oxidoreduktase. Inauguraldissertation, Albert-Ludwigs-Universität Freiburg. Friedrich, T., Hofhaus, G., Ise, W., Nehls, U., Schmitz, B., und Weiss, H. (1989) A small isoform of NADH:ubiquinone oxidoreductase (complex I) without mitochondrially encoded subunits is made in chloramphenicol-treated Neurospora crassa. Eur. J. Biochem. 180, 173180. Friedrich, T., Steinmüller, K., und Weiss, H. (1995) The proton-pumping respiratory complex I of bacteria and mitochondria and its homologue in chloroplasts. FEBS Lett. 367, 107-111. - 129 - ________________________________________________ ______ Literatur Friedrich, T., und Weiss, H. (1997) Modular evolution of the respiratory NADH:ubiquinone oxidoreductase and the origin of its modules. J. Theor. Biol. 187, 529-541. Friedrich, T. (1998) The NADH:ubiquinone oxidoreductase (complex I) from Escherichia coli. Biochim. Biophys. Acta 1364, 134-146. Friedrich, T., Brors, B., Hellwig, P., Kintscher, L., Scheide, D., Schulte, U., Rasmussen, T., Mäntele, W., und Weiss, H. (2000) Characterization of two novel redox groups in the respiratory NADH:ubiquinone oxidoreductase (complex I). Biochim. Biophys. Acta 1459, 305-310. Friedrich, T., und Scheide, D. (2000) The respiratory complex I of bacteria, archaea and eukarya and its module common with membrane-bound multisubunit hydrogenases. FEBS Lett. 479, 1-5. Friedrich, T. (2001) Complex I: A chimaera of a redox and conformation-driven proton pump? J. Bioenerg. Biomembr. 33, 169-177. Gay, P., Le Coq, D., Steinmetz, M., Bekelman, T., und Kado, C.I. (1985) Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J. Bacteriol. 164, 918-921. Gill, C., und von Hippel, P.H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319-326. Gong, X., Xie, T., Yu, L., Hesterberg, M., Scheide, D., Friedrich, T., und Yu, C.A. (2003) The ubiquinone-binding site in NADH:ubiquinone oxidoreductase from Escherichia coli. J. Biol. Chem. 278, 25731-25737. Grigorieff, N. (1998) Three-dimensional structure of bovine NADH:ubiquinone oxidoreductase (complex I) at 22 A in ice. J. Mol. Biol. 277, 1033-1046. Guenebaut, V., Vincentelli, R., Mills, D., und Weiss, H. (1997) Three-dimensional structure of NADH-dehydrogenase from Neurospora crassa by electron microscopy and conical tilt reconstruction. J. Mol. Biol. 276, 409-418. Guenebaut, V., Schlitt, A., Weiss, H., Leonard, K., und Friedrich, T. (1998) Consistent structure between bacterial and mitochondrial NADH:ubiquinone oxidoreductase (complex I). J. Mol. Biol. 276, 105-112. Gunsalus, R.P., Brusilow, W.S.A., und Simoni, R.D. (1982) Gene order and gene-polypeptide relationship of the proton-translocating ATPase operon (unc) of Escherichia coli. Proc. Natl. Acad. Sci. USA 79, 320-324. Guzman, L.-M., Belin, D., Carson, M.J., und Beckwith, J. (1995) Tight regulation, modulation, and high-level expression by vectors containing the Arabinose PBAD Promotor. J. Bacteriol. 177, 4121-30. Hatefi, Y. (1985) The mitochondrial electron transport and oxidative phosphorylation system. Ann. Rev. Biochem. 54, 1015-1069. - 130 - ________________________________________________ ______ Literatur Hinderberger, J. (2004) Modifizierung der Escherichia coli NADH:Ubichinon Oxidoreduktase zur Aufreinigung mittels Affinitätschromatographie. Diplomarbeit, AlbertLudwigs-Universität Freiburg. Hofhaus, G., Weiss, H., und Leonard, K. (1991) Electron microscopic analysis of the peripheral and membrane parts of mitochondrial NADH dehydrogenase (complex I). J. Mol. Biol. 221, 1027-1043. Holmes, D.S., und Quigley, M. (1981) A rapid boiling method for the preparation of bacterial plasmids. Anal. Biochem. 114, 193. Hu, Y., Faham, S., Roy, R., Adams, M.W.W., und Rees, D.C. (1999) Formaldehyde ferredoxin oxidoreductase from Pyrococcus furiosus: the 1.85 A resolution crystal structure and its mechanistic implications. J. Mol. Biol. 286, 899-914. Ingledew, W.J., und Poole, R.K. (1984) The respiratory chains of Escherichia coli. Microbiol. Rev. 48, 222-271. Iuchi, S., Chepuri, V., Fu, H.A., Gennis, R.B., und Lin, W.C.C. (1990) Requirements for terminal cytochromes in generation of the aerobic signal of the arc regulatory system in Escherichia coli: study utilizing deletions and lac fusions of cyo and cyd. J. Bacteriol. 172, 6020-6025. Jones, H.M., Brajkovich, C.M., und Gunsalus, R.P. (1983) In vivo 5´ terminus and length of the mRNA for the proton translocating ATPase (unc) operon of Escherichia coli. J. Bacteriol. 155, 1279-1287. Kashani-Poor, N., Kerscher, S., Zickermann, V., und Brandt, U. (2001) Efficient large scale purification of his-tagged proton translocating NADH:ubiquinone oxidoreductase (complex I) from the strictly aerobic yeast Yarrowia lipolytica. Biochim. Biophys. Acta. 1504, 363-370. Kasimoglu, E., Park, S.J., Malek, J., Tseng, C.P., und Gunsalus, R.P. (1996) Transcriptional regulation of the proton-translocating ATPase (atpIBEFHAGDC) operon of Escherichia coli: control by cell growth rate. J. Bacteriol. 178, 5563-5567. Kiley, P.J., und Beinert, H. (1998) Oxygen sensing by the global regulator, FNR: the role of the iron-sulfur cluster. FEMS Microbiol. Rev. 22, 341-352. Kissel, S. (2003) Über die Assemblierung des NADH-Dehydrogenase Moduls der NADH:Ubichinon Oxidoreduktase aus Escherichia coli. Diplomarbeit, Albert-LudwigsUniversität Freiburg. Koroshilova, N., Popescu, C., Münck, E., Beinert, H., und Kiley, P.J. (1997) Iron-sulfur cluster disassembly in the FNR protein of Escherichia coli by O2: [4Fe-4S] to [2Fe-2S] conversion with loss of biological activity. Proc. Natl. Acad. Sci. USA 94, 6087-6092. Laemmli, U.K. (1970) Cleavage if structural proteins during the assembly of the bacteriophage T4. Nature 227, 17566-17575. - 131 - ________________________________________________ ______ Literatur Lazazzera, B.A., Beinert, H., Khoroshilova, N., Kennedy, M.C., und Kiley, P.J. (1996) DNA binding and dimerization of the Fe-S-containing FNR protein from Escherichia coli are regulated by oxygen. J. Biol. Chem. 271, 2762-2768. Leif, H., Sled, V., Ohnishi, T., Weiss, H., und Friedrich, T. (1995) Isolation and characterization of the proton-translocating NADH:ubiquinone oxidoreductase from Escherichia coli. Eur. J. Biochem. 230, 538-548. Leonard, K., Haiker, H., und Weiss, H. (1987) Three-dimensional structure of NADH: ubiquinone reductase (complex I) from Neurospora mitochondria determined by electron microscopy of membrane crystals. J. Mol. Biol. 194, 277-286. Lorenz, M.G., und Wackernagel, W. (1994) Bacterial gene transfer by natural genetic transformation in the environment. Microbiol. Rev. 58, 563-602. Lunardi, J., Darrouzet, E., Dupuis, A., und Issartel, J.P. (1998) The nuoM arg368his mutation in NADH:ubiquinone oxidoreductase from Rhodobacter capsulatus: a model for the human nd4-11778 mtDNA mutation associated with Leber´s hereditary optic neuropathy. Biochim. Biophys. Acta 1407, 114-124. Magnitsky, S., Toulokhonova, L., Yano, T., Sled, V., Hagerhall, C., Grivennikova, V.G., Burbaev, D.S., Vinogradov, A.D., und Ohnishi, T. (2002) EPR characterization of ubisemiquinones and iron-sulfur cluster N2, central components of the energy coupling in the NADH-ubiquinone oxidoreductase (complex I) in situ. J. Bioenerg. Biomembr. 34, 193-208. Majander, A., Finel, M., Savontaus, M.L., Nikoskelaninen, E., und Wikström, M. (1996) Catalytic activity of complex I in cell lines that posess replacement mutations in the ND genes in Leber´s hereditary optic neuropathy. Eur. J. Biochem. 239, 201-207. Matsushita, K., Ohnishi, T., und Kaback, H.R. (1987) NADH-Ubiquinone oxidoreductases of Escherichia coli aerobic respiratory chain. Biochemistry 26, 7732-7737. Mitchell, P. (1961) Coupling of phosphorylation to electron and hydrogen transfer by a chemiosmotic type of mechanism. Nature 191, 144-148. Murphy, K.C. (1998) Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol. 180, 2063-2071. Nakamaru-Ogiso, E., Yano, T., Ohnishi, T., und Yagi, T. (2002) Characterization of the ironsulfur cluster coordinated by a cysteine binding motif (CXXCXXXCX27C) in the Nqo3 subunit in the proton-translocating NADH-ubiquinone oxidoreductase (NDH-1) of Thermus thermophilus HB-8. J. Biol. Chem. 277, 1680-1688. Nakamaru-Ogiso, E., Yano, T., Yagi, T., und Ohnishi, T. (2005) Characterization of the ironsulfur cluster N7 (N1c) in the subunit NuoG of the proton-translocating NADH-quinone oxidoreductase from Escherichia coli. J. Biol. Chem. 280, 301-307. Neijssel, O.M., und Taxeira de Mattos, M.J. (1994) The energetics of bacterial growth: a reassesment. Mol. Microbiol. 13, 172-182. - 132 - ________________________________________________ ______ Literatur Noji, H., und Yoshida, M. (2001) The rotary machine in the cell, ATP synthase. J. Biol. Chem. 276, 1665-1668. Ollagnier-De Choudens, S., Sanakis, Y., Hewitson, K.S., Roach, P., Baldwin, J.E., Munck, E., und Fontecave, M. (2000) Iron-sulfur center of biotin synthase and lipoate synthase. Biochemistry 39, 4165-4173. Ohnishi, T. (1979) Mitochondrial Iron-Sulfur Flavodehydrogenases. pp. 2-80 in “Membrane Proteins in Energy Transduction” (Ed. Roderich Capaldi), M. Dekker Inc., New York. Ohnishi, T. (1998) Iron-sulfur clusters/semiquinones in complex I. Biochim. Biophys. Acta 1354, 186-206. Oliver, S.G., Winson, M.K., Kell, D.B., und Baganz, F. (1998) Systematic functional analysis of the yeast genome. Trends Biotechnol. 16, 373-378. Peng, G., Fritzsch, G., Zickermann, V., Schägger, H., Mäntele, R., Lottspeich, F., Bostina, M., Radermacher, M., Huber, R., Stetter, K.O., und Michel, H. (2003) Isolation, characterization and electron microscopic single particle analysis of the NADH:ubiquinone oxidoreductase (complex I) from the hyperthermophilic eubacterium Aquifex aeolicus. Biochemistry 42, 3032-3039. Porter, A.C.G., Brusilow, W.S.A., und Simoni, R.D. (1983) Promotor for the unc operon of Escherichia coli. J. Bacteriol. 155, 1271-1278. Puustinen, A., Finel, M., Haltia, T., Gennis, R.B., und Wikström, M. (1991) Properties of the two terminal oxidases of Escherichia coli. Biochemistry 30, 3936-3942. Puustinen, A., und Wikström, M. (1999) Proton-exit from the heme-copper oxidase of Escherichia coli. Proc. Natl. Acad. Sci. USA 96, 35-37. Rasmussen, T., Scheide,D., Brors, B., Kintscher, L., Weiss, H., und Friedrich, T. (2001) Identification of two tetranuclear FeS clusters on the ferredoxin-type subunit of NADH:ubiquinone oxidoreductase (complex I). Biochemistry 40, 6124-6131. Rich, P. (2003) Chemiosmotic coupling: The cost of living. Nature 421, 583. Ried, J.L., und Collmer, A. (1987) A nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker-exchange-eviction mutagenesis. Gene 57, 239-246. Sambrook, J., und Russel, R.W. (2001) Molecular cloning: A laboratory manual, 3rd ed. Cold Spring, NY: Cold Spring Harbor Laboratory Press. Sauter, M., Böhm, R., und Böck, A. (1992) Mutational analysis of the operon (hyc) determining hydrogenase 3 formation in Escherichia coli. Mol. Microbiol. 6, 1523-1532. Sazanov, L.A., und Walker, J.E. (2000) Cryo-electron crystallography of two sub-complexes of bovine complex I reveals the relationship between the membrane and peripheral arms. J. Mol. Biol. 392, 455-464. - 133 - ________________________________________________ ______ Literatur Scheide, D., Huber, R., und Friedrich, T. (2002) The proton-pumping NADH:ubiquinone oxidoreductase (complex I) of Aquifex aeolicus. FEBS Lett. 512, 80-84. Schmitz, O, Boison, G., Salzmann, H., Bothe, H., Schulz, K., Wang, S.K., und Happe, T. (2002) HoxE--a subunit specific for the pentameric bidirectional hydrogenase complex (HoxEFUYH) of cyanobacteria. Biochim Biophys Acta. 1554, 66-74. Schulte, U., Abelmann, A., Amling, N., Brors, B., Friedrich, T., Kintscher, L., Rasmussen, T., und Weiss, H (1998) Search for novel redox groups in mitochondrial NADH:ubiquinone oxidoreductase (complex I) by diode array UV/VIS spectroscopy. BioFactors 8, 177-186. Schulte, U., Haupt, A., Abelmann, A., Fecke, W., Brors, B., Rasmussen, T., Friedrich, T., und Weiss, H. (1999) A reductase/isomerase subunit of mitochondrial NADH:ubiquinone oxidoreductase (complex I) carries an NADPH and is involved in the biogenesis of the complex. J. Mol. Biol. 292, 569-580. Sled, V., Friedrich, T., Leif, H., Weiss, H., Meinhardt, S., Fukumori, Y., Calhoun, M., Gennis, R., und Ohnishi, T. (1993) Bacterial NADH-quinone oxidoreductases: iron-sulfur clusters and related problems. J. Bioenerg. Biomembr. 25, 347-356. Spehr, V., Schlitt, A., Scheide, D., Guenebaut, V., und Friedrich, T. (1999) Overexpression of the Escherichia coli nuo-Operon and isolation of the overproduced NADH:ubiquinone oxidoreductase (complex I). Biochemistry 38, 16261-16267. Stolpe, S., und Friedrich, T. (2004) The Escherichia coli NADH:Ubiquinone Oxidoreductase (complex I) is a primary proton-pump but may be capable of secondary sodium antiport. J. Biol. Chem. 279, 18377-18383. Towbin, H., Staehlin, T., und Gordon, J. (1979) Electrophoretic transfer of proteins from polyacrylamid gels to nitrocellulose sheets: procedure and some application. Proc. Natl. Acad. Sci. USA 76, 4350-4353. Ugulava, N.B., Gibney, B.R., und Jarrett, J.T. (2000) Iron-sulfur cluster interconversions in biotin synthase: dissociation and reassociation of iron during conversion of [2Fe-2S] to [4Fe4S] clusters. Biochemistry 39, 5206-5214. Unden, G. (1988) Differential roles for menaquinone and dimethylmenaquinone in anaerobic electron transport of E. coli and their fnr-independent expression. Arch. Microbiol. 164, 8190. Unden, G., und Bongaerts, J. (1997) Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim. Biophys. Acta 1320, 217-234. Videira, A., und Azevedo, J.E. (1994) Two nuclear-coded subunits of mitochondrial complex I are similar to different domains of a bacterial formate hydrogenlyase subunit. Int. J. Biochem. 26, 1391-1393. von Meyenburg, K., Jørgensen, B.B., van Deurs, B. (1984) Physiological and morphological effects of overproduction of membrane-bound ATP synthase in Escherichia coli K-12. EMBO J. 3,1791–1797. - 134 - ________________________________________________ ______ Literatur Voss, S., und Skerra, A. (1997) Mutagenesis of a flexible loop in streptavidin leads to higher affinity for the Strep-Tag II peptide and improved performance in recombinant protein purification. Protein. Eng. 10, 975-982. Walker. J.E. (1992) The NADH:ubiquinone oxidoreductase (complex I) of respiratory chains. Quart. Rev. Biophys. 25, 253-324. Weidner, U., Geier, S., Ptock, A., Friedrich, T., Leif, H., und Weiss, H. (1993) The gene locus of the proton-translocating NADH:ubiquinone oxidoreductase in Escherichia coli. Organization of the 14 genes and relationship between the derived proteins and subunits of mitochondrial complex I. J. Mol. Biol. 233, 109-122. Weiss, H., Friedrich, T., Hofhaus, G., und Preis, D. (1991) The respiratory-chain NADH dehydrogenase (complex I) of mitochondria. Eur. J. Biochem. 197, 563-576. Wissenbach, U., Kröger, A., und Unden, G. (1990) The specific functions of menaquinone and demethylmenaquinone in aerobic respiration with fumarate, dimethylsulfoxide, trimethylamine N-oxide and nitrate by Escherichia coli. Arch. Microbiol. 154, 60-66. Yagi, T., Yano, T., Di Bernardo, S., und Matsuno-Yagi, A. (1998) Procaryotic complex I (NDH-1), an overview. Biochim. Biophys. Acta 1364, 125-133. Yagi, T., und Matsuno-Yagi, A. (2003) The proton-translocating NADH-quinone oxidoreductase in the respiratory chain: the secret unlocked. Biochemistry 42, 2266-2274. Yano, T., Sled, V., Ohnishi, T., und Yagi, T. (1994 a) Identification of amino acid residues associated with the [2Fe-2S] cluster of the 25 kDa (NQO2) subunit of the proton-translocating NADH-quinone oxidoreductase of Paracoccus denitificans. FEBS Lett. 345, 160-164. Yano, T., Sled, V., Ohnishi, T., und Yagi, T. (1994 b) Expression of the 25-kilodalton ironsulfur subunit of the energy-transducing NADH-ubiquinone oxidoreductase of Paracoccus denitrificans. Biochemistry 33, 494-499. Yano T., Yagi T., Sled V.D., und Ohnishi T. (1995) Expression and characterization of the 66-kilodalton (NQO3) iron-sulfur subunit of the proton-translocating NADH-quinone oxidoreductase of Paracoccus denitrificans. J. Biol. Chem. 270, 18264-18270. Yano, T., Sled, V., Ohnishi, T., und Yagi, T. (1996) Expression and characterization of the flavoprotein subcomplex composed of 50-kDa (NQO1) and 25 kDa (NQO2) subunits of the proton-translocating NADH-ubiquinone oxidoreductase of Paracoccus denitrificans. J. Biol. Chem. 271, 5907-5913. Zu, Y., Di Benardo, S., Yagi, T., und Hirst, J. (2002) Redox properties of the [2Fe-2S] center in the 24 kDa (NQO2) subunit of the NADH:ubiquinone oxidoreductase (complex I). Biochemistry 41, 10056-10069. - 135 - Anhang Anhang Anhang 1: Vektorkarte des Plasmids pETBlue-1. Ap bezeichnet dasAmpicillin-Resistenz. Gezeigt sind die Replikationsursprünge (f1 origin, pUC or), der tet-Promotor, T7-Promotor, T7-Terminator, rrnB-Terminator, die Ribosomenbindestelle (T7 RBS) sowie das lacZ-Gen Anhang 2: Vektorkarte des Plasmids pBAD33. Die Lage des araC-Regulators und araC– Promotors ist eingezeichnet, außerdem die Position der singulären Schnittstellen der Restriktionsenzyme NdeI, SacI, ApaI und XhoI. Cam bezeichnet das ChloramphenicolResistenzgen. - 136 - Anhang Anhang 3: Vektorkarte des Plasmids pET11-a/nuoB-G/NuoFc. nuo bezeichnet die Position der einzelnen Komplex I-Untereinheiten und die Pfeile geben die Transkriptionsrichtung an. Amp bezeichnet die Lage des Ampicillin-Resistenzgens. Außerdem ist die Modifizierung der Untereinheit NuoF durch den Strep-Tag II gezeigt. Anhang 3: Vektorkarte des Plasmidkonstruktes pBADnuo. nuo bezeichnet die Lage der einzelnen Komplex I-Untereinheiten nuoA-N, die Pfeile geben die Transkriptionsrichtung an. Cam steht für das Chloramphenicol-Resistenzgen. Die Lage des araC-Regulators und araC– Promotors ist eingezeichnet, außerdem die Position der singulären Schnittstellen der Restriktionsenzyme NdeI, SacI, ApaI und XhoI. - 137 - Anhang Anhang 4: Vektorkarte des Plasmids pET24-a.Gezeigt sind die Replikationsursprünge (ori, f1 origin), das Kanamycin-Resistenzgen (Kann), der T7-Promotor und das lacI-Gen (LacI). Die in der multiple cloning site (MCS) liegenden Restriktionsschnittstellen NheI und NdeI sind markiert. Im Plasmid pET11-a liegt die MCS in umgekehrter Orientierung, der Rest des Plasmids ist identisch. Anhang 5: Vektorkarte des Plasmids pKD46. Amp bezeichnet das Ampicillin-Resistenzgen und araC den Arabinosepromotor. Gezeigt sind die Gene gamma, beta und exo, die für die λRed Rekombinase kodieren. - 138 - Anhang Anhang 6: Ergebnis der Sequenzierung von pBADnuo/nuoC-StrepTag II-nuoD pBADcgnnnnnnnnnnnnnnnnnnnnaCAAGccaACAGGATCTGGAGGCTAGCTGGAGCCAC CCGCAGTTCGAGAAGtaaAATGGAAGCCCTGACCTTCAAACCGGAAGAGTGGGGG ATGAAGCGCGGCACCGAAAACGAGGACTTCATGTTCCTCAACCTCGGTCCGAACC ACCCGTCGGCGCACGGGGCTTTCCGTATCGTTTTGCAACTCGATGGCGAAGAGAT TGTCGACTGCGTACCAGACATCGGTTACCACCACCGTGGTGCGGAGAAAATGGGC GAACGCCAGTCCTGGCACAGCTACATTCCGTATACTGACCGTATCGAATACCTCG GCGGCTGCGTTAACGAAATGCCTTACGTGCTGGCGGTAGAGAAACTGGCCGGGA TCACCGTGCCGGATCGCGTTAACGTCATTCGCGTTATGCTCTCCGAACTGTTCCGC ATCAACAGTCACCTGCTGTATATCTCGACCTTTATTCAGGACGTCGGCGCAATGA CGCCAGTGTTCTTCGCCTTTACCGATCGTCAGAAAATTTACGATCTGGTGGAAGC AATCACTGGTTTCCGTATGCACCCGGCGTGGTTCCGTATTGGCGGCGTAGCGCAC GACCTGCCGCGCGGCTGGGATCGCCTGCTGCGTGAGTTCCTCGACTGGATGCCGA AACGTCTGGCGTCTTACGAGAAAGCGGCGCTGCAAAACACCAttCTGaannnnnAGG GCGTTGCCGCCTATGGCGCGaa Anhang 7: Ergebnis der Sequenzierung von pBADnuo/nuoC-6 StrepTag II-nuoD ACGagaAAGCGGCGCTGCAAAACgactgacAAGccaACAGGATCTGGAGGCTGCAGCT GCTGCTGCTAGCTGGAGCCACCCGCAGTTCGAGAAGTAAAATGGAAGCCCTGAC CTTCAAACCGGAAGAGTGGGGGATGAAGCGCGGCACCGAAAACGAGGACTTCAT GTTCCTCAACCTCGGTCCGAACCACCCGTCGGCGCACGGGGCTTTCCGTATCGTTT TGCAACTCGATGGCGAAGAGATTGTCGACTGCGTACCAGACATCGGTTACCACCA CCGTGGTGCGGAGAAAATGGGCGAACGCCAGTCCTGGCACAGCTACATTCCGTAT ACTGACCGTATCGAATACCTCGGCGGCTGCGTTAACGAAATGCCTTACGTGCTGG CGGTAGAGAAACTGGCCGGGATCACCGTGCCGGATCGCGTTAACGTCATTCGCGT TATGCTCTCCGAACTGTTCCGCATCAACAGTCACCTGCTGTATATCTCGACCTTTA TTCAGGACGTCGGCGCAATGACGCCAGTGTTCTTCGCCTTTACCGATCGTCAGAA AATTTACGATCTGGTGGAAGCAATCACTGGTTTCCGTATGCACCCGGCGTGGTTC CGTATTGGCGGCGTAGCGCACGACCTGCCGCGCGGCTGggATCGCCTGCTGCGTG AGTTCCTCGACTGGATGCCGAAACGTCTGGCGTCTTACCATTCTgaaaggTCGtTCc Anhang 8: Ergebnis der Sequenzierung von pBADnuo/∆nuoEF CGATTTTGTTATGTCAGATGTGGACCGCTAAGTGTAGGCTGGAGCTGCTTCGAAG TTCCTATACTTTCTAGAGAATAGGAACTTCGGAATAGGAACTAAGGAGGATATTC ATATGCTCAGTCTCTGACTGAGAAAACTGGAAGCATGCTAATGGCTACAATTCAT GTAGACGGCAAAGAATACGAGGTCAACGGAGCGGACAACCTGCTGGAAGCTTGT CTGTCTCTGGGCCTTGATATTCCTTACTTTTGCTGGCATCCGGCGCTGGGAAGTGT CGGTGCTTGCCGCCAGTGTGCGGTGAAGCAATACCAAAACGCGGAAGACACGCG TGGTCGCCTGGTGATGTCCTGTATGACACCGGCTTCCGATGGCACCTTTATTTCCA TTGACGACGAAGAAGCGAAACAGTTCCGTGAAAGCGTGGTCGAGTGGTTGATGA CCAACCACCCGCACGACTGTCCGGTATGTGAAGAGGGCGGTAACTGCCATCTTCat gATATGACTGTGATGACCGGACACAGCTTCCGTCGCTACCGTTTCACCAAACGTA CCCACCGTAATCAGGATTTGGGGCCATTCATCTCTCACGAAATGAACCGCTGCAT CGCCTGCTACCGCTGTGTGCGTTACTACAAAGATTACGCTGACGGTACAGATCTG GGCGTTTAcGGTGCGCACGACaACGTCTACTtCGGTCGCCCGGAagacgGCACGCTgg - 139 - Anhang Anhang 9: Ergebnis der Sequenzierung von pBADnuo/∆nuoG aCTCAttgtTGTGCCTCCTTGAGCATATGAATATCCTCCTTAGTTCCTATTCCGAAGTT CCTATTCTCTAGAAAGTATAGGAACTTCGAAGCAGCTCCAGCCTACACTTTCTCA GTCAGAGACTGAGCGTTAATCGAAATTCGGTTACCAGCGCTCTTTCAGCAGGTTC GGCTGAATCCCATTAATCAAATGGGTATTGCTGAACGGCTGTTTGATTCCCGCCTC AAATTCTTCGCGGAAATATTTGATGGCGCTCTGTAACGGCTCCACTGCACCAGGT GCGTGGGCACAGAAAGTTTTACCCGGGCCTAAGAATCGACACAGTTGCTCAAGTG TTTCGATATCGCCCGGCTGACCTTCACCACGCTCCAGCGCACGCAGAATTTTCAC GCTCCACGGCAGACCGTCGCGGCACGGCGTACACCAGCCGCAGGACTCACGGGC GAAAAACTCTTCCAGGTTACGCACCAGCGACACCATGTTGATCTCATGGTCAACC GCCATCGCCAGCGCCGTACCCAGACGGCTGCCCGCTTTACCGATACTTTCGAATT CCATCGGCAGATCAAGGTGCGCTTCGGTCAGGAAGTCAGTCCCCGCGCCGCCTGG CTGCCAGGCTTTAAATTTCagnnnATCACGCATACCACCGGCGTAATCTTCGaggAtcT CGCGTGcg Anhang 10: Ergebnis der Sequenzierung von pBAD-C1/nuoM(Leu244Ile) P P gacttacctataAATAGGCGTATCACGAGGCCCTTTCGTCTCGCGCGTTTCGGTGATGAC GGTGAAAACCTCTGACACATGCAGCTCCCGGAGACGGTCACAGCTTGTCTGTAAG CGGATGCCGGGAGCAGACAAGCCCGTCAGGGCGCGTCAGCGGGTGTTGGCGGGT GTCGGGGCTGGCTTAACTATGCGGCATCAGAGCAGATTGTACTGAGAGTGCACCA TATGCGGTGTGAAATACCGCACAGATGCGTAAGGAGAAAATACCGCATCAGGCG CCATTCGCCATTCAGGCTGCTCGCGAGTCCCCCGGGATATCGGTCTGGGCGAAGG CCATCCACGCACCGTAGAAGATGCCGATAACACCCAGCCACATAGCAATTGGCG CGAACTCTGCCGACGCGTTCGGGAACAGCGGCAGGGAGAAACGCAGCAAACCGT AAGCGGCAGTTTTCAGCAAGATCCCCGCGAGGTCAACGGAACCGGCGGTCGGAG CCTGGGAGTGCGCATCCGGGATCCAGCCATGCAGCGGAACCACCGGCATTTTGAC TGCGAAGGCGATGAAGAAGCCCAGCATTAACAGGTATTCCACACCACTGGACAT TGGCGTATTCAGCAGCTCTTCATAGTTGAAGGTCCAGACGCCGGTCGCATTGTAG TGAACAAAAACCAGCGCCAGGATGGCAATCAACATCACCAGGCCACTCGCCTGG GTGTAAATGAAGAACTTGGTTGCCGCCGTGATACGCGTTTTACCGTCAGaggCTTT ATGCCCCCACAGTGCGATCAGGAAGTACATCGGCACCAGCATCATTTCCCAGAAG aaGaaGAACAGGAACATGTCGaTGGCAAGgaacAcgccga Anhang 11: Ergebnis der Sequenzierung von pBAD-C1/ nuoM(Asp246Asn) P P AGGCGTATCACGAGGCCCTTTCGTCTCGCGCGTTTCGGTGATGACGGTGAAAACC TCTGACACATGCAGCTCCCGGAGACGGTCACAGCTTGTCTGTAAGCGGATGCCGG GAGCAGACAAGCCCGTCAGGGCGCGTCAGCGGGTGTTGGCGGGTGTCGGGGCTG GCTTAACTATGCGGCATCAGAGCAGATTGTACTGAGAGTGCACCATATGCGGTGT GAAATACCGCACAGATGCGTAAGGAGAAAATACCGCATCAGGCGCCATTCGCCA TTCAGGCTGCTCGCGAGTCCCCCGGGATATCGGTCTGGGCGAAGGCCATCCACGC ACCGTAGAAGATGCCGATAACACCCAGCCACATAGCAATTGGCGCGAACTCTGC CGACGCGTTCGGGAACAGCGGCAGGGAGAAACGCAGCAAACCGTAAGCGGCAGT TTTCAGCAAGATCCCCGCGAGGTCAACGGAACCGGCGGTCGGAGCCTGGGAGTG CGCATTCGGCAGCCAGCCATGGAGCGGAACCACCGGCATTTTGACTGCGAAGGC GATGAAGAAGCCCAGCATTAACAGGTATTCCACACCACTGGACATTGGCGTATTC AGCAGCTCTTCATAGTTGAAGGTCCAGACGCCGGTCGCATTGTAGTGAACAAAAA - 140 - Anhang CCAGCGCCAGGATGGCAATCAACATCACCAGGCCACTCGCCTGGGTGTAAATGA AGAACTTGGTTGCCGCCGTGATACGCGTTTTACCGTCAGAggCTTTATGCCCCCAC AGTGCGATCAGGAAGTACATCGGCACCAGCATCATTTCCCAGaagaaGAagaacaggaA CATGTCGATggcanngaacAC - 141 - Danksagung Ich danke Prof. Dr. Thorsten Friedrich für die Bereitstellung des interessanten Themas und seine unermüdliche Hilfestellung bei allen Fragen. Er hat es immer geschafft, mich aufzumuntern, zu ermutigen und mit seinem Enthusiasmus zu motivieren. Mit den Mitgliedern des Arbeitskreises habe ich nicht nur viele schöne Stunden im Institut verbracht, sondern auch außerhalb des Labors. Es hat mir mit den „Friedrichs“ viel Spaß gemacht, und ich werde diese Zeit sehr vermissen. Ich hoffe, einige dieser Freundschaften mit in meine Zukunft zu nehmen. Für die Erledigung so vieler Kleinigkeiten gilt mein Dank Franz Butz, Helga Lay und Edith Fitzke. Ich danke außerdem meinen Diplomanden und Mitarbeiterpraktikanten, von denen viele mir die Arbeit erleichtert haben. Ich danke den Leuten, die mir (auch in schlechten Zeiten) gezeigt haben, wie schön es ist, gute Freunde zu haben. Mein besonderer Dank gilt dabei Christiane Schäfer und Stefan Tacke, Saskia Kissel, Stefan Stolpe, Markus Kohlstädt, Annette, Thom und Marlon Bunk, Kathrin Lauckner und Andreas Kranzusch, außerdem meinen Freunden aus Mönchengladbach, Christian Bönsch, Andrea Peper und Stephy Foltin. Alle anderen seien sich sicher, dass ich sie nicht vergessen habe (und nur der Platz nicht ausreicht). Dirk Flemming möchte ich trotz allem danken, dass ich einige schöne Stunden mit ihm erlebt habe. Michael Ochsenkühn danke ich dafür, ein Stück des Weges neben mir zu laufen. Den Pferden Farina, Chamba und Ohio und einer Horde von „Pferdeleuten“ verdanke ich etliche Stunden der Zerstreuung vom Laboralltag… Meine Familie hat mir nicht nur das Studium ermöglicht, ich verdanke ihr wesentlich mehr; meiner Mutter, dass sie den Ehrgeiz in mir gefördert hat, meinem Vater, dass er mir früh die Augen für die wunderbaren kleinen Dinge dieser Welt geöffnet hat, meinem Bruder danke ich dafür, dass er der beste Mensch der Welt ist und meinen Omas dafür, dass sie für mich da waren. …und letztendlich danke ich dem Tuniberg, auf dem ich im Sonnenschein diese Zeilen schrieb… Das Leben ist schön! - 142 -