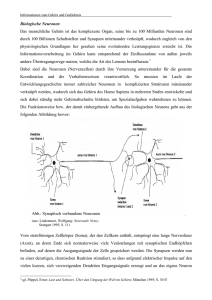
fortschritte in der hirnforschung
Werbung

FORTSCHRITTE IN DER HIRNFORSCHUNG Ausgabe 2007 Die Neuroethik entwickelt sich Essay von Steven E. Hyman, MD FORTSCHRITTE IN DER HIRNFORSCHUNG Die Neuroethik entwickelt sich Essay von Steven E. Hyman, MD Ausgabe 2007 THE EUROPEAN DANA ALLIANCE FOR THE BRAIN EXECUTIVE COMMITTEE William Safire, Chairman Edward F. Rover, President Colin Blakemore, PhD, ScD, FRS, Vice Chairman Pierre J. Magistretti, MD, PhD, Vice Chairman Carlos Belmonte, MD, PhD Anders Björklund, MD, PhD Joël Bockaert, PhD Albert Gjedde, MD, FRSC Sten Grillner, MD, PhD Malgorzata Kossut, MSc, PhD Richard Morris, Dphil, FRSE, FRS Dominique Poulain, MD, DSc Wolf Singer, MD, PhD Piergiorgio Strata, MD, PhD Eva Syková, MD, PhD, DSc Executive Committee Barbara E. Gill, Executive Director Die European Dana Alliance for the Brain (EDAB) ist ein Zusammenschluss von 186 führenden Wissenschafterinnen und Wissenschaftern aus 27 Ländern. Zu ihren Mitgliedern zählen fünf Nobelpreisträger. Die EDAB hat sich zum Ziel gesetzt, die Gesellschaft auf die Bedeutung der Gehirnforschung aufmerksam zu machen. Die Organisation wurde 1997 gegründet und versteht sich als Schnittstelle zwischen der medizinischen Laborarbeit, der Forschung sowie der breiten Öffentlichkeit. Für weitere Informationen: The European Dana Alliance for the Brain Dr Béatrice Roth, PhD Centre de Neurosciences Psychiatriques Site de Cery 1008 Prilly / Lausanne E-mail : [email protected] Deckel: Jennifer Suehs FORTSCHRITTE IN DER HIRNFORSCHUNG Ausgabe 2007 Die Neuroethik entwickelt sich 5 11 Einleitung von David C. Van Essen, PhD Fünf Jahre Neuroethik: Das Gebiet entwickelt sich weiter von Steven E. Hyman, MD Fortschritte in der Hirnforschung im Jahr 2006 19 In der Kindheit auftretende Störungen 27 Bewegungsstörungen und andere Störungen der Motorik 35 Schädigungen des Nervensystems 43 Neuroethik 51 Neuroimmunologische Erkrankungen 61 Schmerz 69 Psychiatrische Erkrankungen, Verhaltensstörungen und Suchtkrankheiten 79 Störungen der Sinnes- und Körperfunktion 87 Stammzellen und Neurogenese 97 Denken und Erinnern 105 Referenzen 117 Stelle Dir eine Welt vor... Einleitung von David C. Van Essen, PhD Präsident der Society for Neuroscience D er vorliegende Jahresbericht fasst mehr als 100 Forschungsbefunde zusammen und illustriert damit den Beitrag der neurowissenschaftlichen Forschung zum besseren Verständnis und auch zu Diagnose und Behandlung vieler verheerender Krankheiten und Störungen des Nervensystems. Jedes der zehn Kapitel stellt Entdeckungen in den Mittelpunkt, die sich entweder auf eine bestimmte Krankheitsform oder auf ein übergreifendes Thema, etwa Neuroethik, beziehen. Die einzelnen Befunde („Kleinode der Neurowissenschaft“) und auch die allgemeinen Themen, die aus dem Bericht als Ganzem hervorgehen, bilden einen wesentlichen Teil der grossen Herausforderung, das menschliche Gehirn in Gesundheit und Krankheit zu verstehen. Das menschliche Gehirn ist eine erstaunlich komplizierte Struktur, die Informationen verarbeitet und sämtliche Aspekte unseres Verhaltens steuert. Der verworrene Nervenschaltkreis des Gehirns mit Milliarden von Neuronen und Trillionen von Synapsen ist weitaus komplizierter als jedes andere Organsystem des Körpers. Diese Komplexität zeigt sich auf vielen Ebenen. Auf der molekularen und zellulären Ebene etwa kommen höchst choreographierte molekulare Signale zum Einsatz, um Informationen von einer Zelle zur anderen zu übermitteln und die Stärke dieser Signale im Laufe der Entwicklung und des Lernens zu regulieren. Auf der Systemebene ist eine Symphonie von koordinierten neuralen Aktivitätsmustern involviert, die tausende von verschiedenen Hirnstrukturen umfassen, die ihrerseits über zehntausende von anatomischen Bahnen miteinander kommunizieren. Hinzu kommt ein hohes Mass an individueller Variabilität von Hirnstruktur und -funktion bei verschiedenen Personen, woraus die enorme Vielfalt unserer individuellen Persönlichkeiten und intellektuellen Fähigkeiten resultiert. Angesichts dieser erstaunlichen Komplexität – sie übertrifft jene eines Spaceshuttles oder 5 eines Supercomputers bei weitem – überrascht es wohl nicht, dass im Nervensystem ungezählte Formen von Fehlfunktion möglich sind. Tatsächlich wurden bereits über 1000 Störungen und Krankheiten des Nervensystems genau bestimmt, und die Liste wird immer länger. Von den meistverbreiteten Beschwerden – Alzheimersche Krankheit, Schizophrenie, Hirnschlag und Lernstörungen – ist ein grosser Teil der Bevölkerung betroffen; sie auferlegen der Gesellschaft eine grosse Last, sowohl was die ökonomischen Auswirkungen als auch was Verzweiflung und menschliches Leid anbelangt. Ohne entscheidende Fortschritte bezüglich Vorbeugung und Behandlung von Krankheiten des Nervensystems wird diese Bürde aufgrund der steigenden Lebenserwartung der Menschen weiter zunehmen. Zur Beschleunigung des Fortschritts müssen wir die Krankheitsmechanismen und die normalen Mechanismen von Hirnfunktion und Hirnplastizität, also der Anpassungsfähigkeit, wesentlich besser verstehen. Entsprechende Fortschritte, dazu gehören auch die im vorliegenden Bericht dargestellten, werden uns in die Lage versetzen, die Fähigkeit des Nervensystems, sich selbst zu regenerieren, zu reparieren und mit einer Schädigung umzugehen, besser zu nutzen und zu fördern. Die Errungenschaften der Hirnforschung im Jahr 2006 haben zu eigenen Themenbereichen geführt. Einer davon betrifft Fortschritte in der Charakterisierung jener genetischen Faktoren, die zu den unterschiedlichsten neurologischen und psychiatrischen Krankheiten beitragen. Hierhin gehört etwa die Klärung, welche Rolle spezifische Gene bei der familiären Parkinsonschen Krankheit spielen, oder auch die Bestimmung vieler mit Angst zusammenhängender Gene in einem Mausmodell 1-3. 6 Eine weitere potente Strategie besteht darin, unser Wissen über Gene mit anderen experimentellen Ansätzen, etwa Neuroimaging, zu kombinieren. Ein erstaunliches Beispiel im vorliegenden Bericht betrifft den Einsatz der Magnetresonanz-Tomographie zur Charakterisierung von (sowohl strukturellen als auch funktionellen) Hirnanomalien bei Personen, die zwar Träger einer bestimmten, bei gewalttätigem Verhalten vorkommenden genetischen Variante sind, aber keine psychiatrische Krankengeschichte haben 4. Auch die Entdeckung bestimmter struktureller Hirnanomalien bei der Aufmerksamkeitsdefizit-Hyperaktivitätsstörung (ADHS) 5 sowie funktioneller Abweichungen bei Autismus 6 belegen die Leistungsfähigkeit des Neuroimaging. Einleitung Die Erforschung degenerativer Erkrankungen des Nervensystems – dazu gehören die Parkinsonsche, die Huntingtonsche und die Alzheimersche Krankheit sowie die Amyotrophe Lateralsklerose (ALS) – steht in vielen Laboratorien weiterhin im Mittelpunkt. Im Bereich der Zellbiologie verstehen wir heute besser, weshalb es gewissen Proteinen nicht gelingt, sich zu richtig funktionierenden molekularen Konfigurationen zu falten, und weshalb es zu einer Degeneration kommt, wenn die Mechanismen zur Bewältigung falsch gefalteter Proteine versagen. Ein weiterer Ansatz basiert auf interventionellen Strategien, bei denen Nervenzellen durch geeignete Therapien vor Schädigung und Zerstörung geschützt werden 7. Andere Forschungsarbeiten untersuchen die Rolle des Immunsystems in Bezug auf das Gehirn. Normalerweise ist das „Zusammenspiel“ gut, doch wiesen im vergangenen Jahr viele neue Befunde auf die verheerenden Auswirkungen hin, zu denen es kommen kann, wenn das Immunsystem zu einem Angriff auf das Gehirn provoziert wird. Hierher gehören etwa entzündliche Reaktionen, welche die Schädigung der Neuronen noch verstärken, die durch degenerative Prozesse des Nervensystems infolge der Alzheimerschen, der Parkinsonschen, der Huntingtonschen Krankheit oder der ALS ausgelöst wurden. Diesbezügliche Erkenntnisse haben zu neuen therapeutischen Verfahren geführt: Um die Schädigung der Neuronen bei all diesen degenerativen Erkrankungen des Nervensystems zu reduzieren, werden entzündungshemmende Medikamente eingesetzt. Die Aussicht einer Nervenersatztherapie ist für viele Neurowissenschafter und Neurowissenschafterinnen so etwas wie ein heiliger Gral; dies vor allem deshalb, weil die Neurogenese bei Adulten in vielen anderen Spezies verbreitet ist. Bei Autoimmunkrankheiten wie Multipler Sklerose scheint die Attacke des Immunsystems in einem direkten Angriff auf Gliazellen zu bestehen. Neue Ergebnisse im vergangenen Jahr erbrachten wichtige Erkenntnisse zur Identität jener Schlüsselproteine, die den Immunangriff vermitteln, und zur Bestimmung eines Antikörper-Biomarkers, dank dem sich bestimmte Behandlungen bei Autoimmunkrankheiten besser steuern lassen 8, 9. Im menschlichen Gehirn sind zerstörte Neuronen insofern unersetzbar als die Geburt neuer Neuronen (Neurogenese) bei Erwachsenen nur in bestimmten Hirnregionen vorkommt. Die Aussicht einer Nervenersatztherapie ist für viele Neurowissenschafter und Neurowissenschafterinnen so etwas wie ein heiliger Gral; dies vor allem deshalb, weil die 7 Neurogenese bei Adulten in vielen anderen Spezies verbreitet ist. Haarzellen in der Innenohrschnecke sind diesbezüglich ganz besonders interessant; zum einen sind sie Teil eines relativ einfachen Nervenschaltkreises, zum andern ist Hörverlust eine sehr häufige und behindernde Funktionsstörung. Neuere Befunde in der Charakterisierung jener Gene, welche die Proliferation von Haarzellen steuern, lassen weitere Fortschritte erhoffen 10. Weitere intensive Bemühungen gelten sowohl der Frage, wodurch Neurogenese im Hippokampus und in anderen Hirnregionen, in denen sie vorkommt, gesteuert wird als auch der Verwendung von Stammzellen, die eine Neurogenese in weiteren Hirnregionen und im Rückenmark fördern sollen 11, 12. Dass Fortschritte in Diagnose und Behandlung von Hirnkrankheiten auch zu besonderen Herausforderungen mit ethischen und gesundheitspolitischen Implikationen führen können, ist ein allumfassendes Thema. Mitglieder der Dana Alliance for Brain Initiatives trugen entscheidend zur Ausgestaltung dieses neu entstehenden Gebiets der Neuroethik bei. Steven Hymans Aufsatz zur Neuroethik gibt einen gut durchdachten Überblick über die Geschichte dieses neuen Gebiets und die Probleme, mit denen es zu ringen hat. Auf letztere konzentriert sich insbesondere das Kapitel Neuroethik im Hauptteil des vorliegenden Berichts, in dem eine Reihe von Fragen und Kontroversen beleuchtet werden, darunter auch Themen im Zusammenhang mit der Intimsphäre des Gehirns, dem Gehirn ohne Bewusstsein und den Implikationen von Nervenprothesen. Wie geht es mit der Hirnforschung weiter? Drei eindeutige Entwicklungen werden das Gebiet auch in Zukunft stark beeinflussen. Wie geht es mit der Hirnforschung weiter? Drei eindeutige Entwicklungen werden das Gebiet auch in Zukunft stark beeinflussen: 8 Methoden ermöglichen Entdeckungen. Die meisten hier beschriebenen Entdeckungen wären vor einem Jahrzehnt nur schon deshalb nicht möglich gewesen, weil entscheidende experimentelle Methoden, etwa die funktionelle Magnetresonanz-Tomographie und die High-Throughput Gensequenzierung, entweder gar nicht vorhanden oder nur unzulänglich waren. Methodenorientierte Fachpersonen im akademischen Bereich und in der Privatwirtschaft erarbeiten laufend eine Vielfalt von Methoden zur Beschaffung und Analyse von Informationen über das Gehirn. Um diese Fortschritte weiter zu beschleunigen, muss in diese Projekte nachhaltig investiert werden. Einleitung Vom Labor zum Krankenbett zum Labor. Der gebräuchliche Ausdruck „vom Labor zum Krankenbett“ bezeichnet die Umsetzung von Entdeckungen der neurowissenschaftlichen Grundlagen- und Translationsforschung in eine verbesserte medizinische Betreuung. Heute erkennt man, dass auch der Informationsfluss in die entgegengesetzte Richtung, also vom klinischen Bereich zurück zur neurowissenschaftlichen Grundlagenforschung, sehr wichtig ist. Die Erforschung von Krankheiten und Krankheitsmechanismen vermittelt Forschenden in der Neurowissenschaft oft Erkenntnisse über die grundlegenden Mechanismen der Hirnfunktion und -entwicklung. Am Jahrestreffen 2006 der Society for Neuroscience berichteten beispielsweise alle vier Vorsitzenden in ihrem Vortrag, dass ihre eigene Forschungsarbeit von den bidirektionalen Interaktionen zwischen neurowissenschaftlicher Grundlagenforschung und klinischer Neurowissenschaft profitiert hatte. Bei mir persönlich hat diese Perspektive eine starke Resonanz und widerspiegelt neuere Veränderungen meines eigenen Forschungsprogramms. Noch vor wenigen Jahren war ich ein „reiner“ neurowissenschaftlicher Grundlagenforscher, doch gegenwärtig richtet sich die Forschungstätigkeit in meinem Laboratorium hauptsächlich auf bestimmte neurologische oder psychiatrische Krankheiten; mittels neuer Methoden analysieren wir die Struktur und Funktion der Grosshirnrinde. Am Jahrestreffen der Society for Neuroscience sind Krankheiten das Thema der Forschung, das am raschesten wächst, ein Hinweis darauf, dass sich die Forschungstätigkeit der Neurowissenschaft insgesamt zunehmend mit Krankheiten befasst. Eine Informationsflut. Die im vorliegenden Bericht beschriebenen Studien repräsentieren nur die Spitze eines grossen und rasch anwachsenden Eisbergs von Informationen, welche die neurowissenschaftliche Gemeinschaft Jahr für Jahr hervorbringt. Nur ein Bruchteil der potentiell wertvollen Informationen wird publiziert oder in Datenbanken zugänglich gemacht. Ausserdem ist selbst das in Onlinezeitschriften und Datenbanken zugängliche Material nicht so leicht und effektiv auffindbar, wie man es sich wünschen würde. Dies wird sich voraussichtlich im kommenden Jahrzehnt drastisch ändern, da Verbesserungen der Informationstechnologie neue Möglichkeiten eröffnen sollen, von denen wir heute nur träumen können; dann werden wissenschaftlich und klinisch tätige Fachpersonen sowie die Öffentlichkeit rasch, zuverlässig und bequem eine Fülle von Informationen über das Nervensystem abrufen können. 9 Fünf Jahre Neuroethik: Das Gebiet entwickelt sich weiter von Steven E. Hyman, MD G erade weil das Gehirn für unser Menschsein und unsere wertvollsten Fähigkeiten von zentraler Bedeutung ist, sind Hirnkrankheiten besonders verheerend und stehen im Mittelpunkt intensiver Forschungsbemühungen. Indem sich die Wissenschaft intensiv darum bemüht, die verheerendsten Krankheiten der Menschheit zu bekämpfen, gewinnt sie auch wichtige Erkenntnisse zum menschlichen Denken, Fühlen und Verhalten in Gesundheit und Krankheit. Allerdings werfen neue Methoden, dank denen wir die Tätigkeit des menschlichen Gehirns beobachten und seine Funktion beeinflussen können, auch entscheidende ethische und politische Fragen auf. Fortschritte in der Bildgebung des Gehirns könnten es dereinst ermöglichen, in jenen Fällen eine objektive Diagnose zu stellen, bei denen wir uns heute ausschliesslich auf klinische Beobachtung stützen. Fortschritte in Genetik und Molekularbiologie lassen hoffen, dass es möglicherweise binnen einem Jahrzehnt gelingen wird, Behandlungen zu entwickeln, um die Progredienz degenerativer Erkrankungen des Nervensystems, etwa der Alzheimerschen und der Parkinsonschen Krankheit, aufzuhalten. Neben der Genetik macht auch die kognitive und soziale Neurowissenschaft Fortschritte; diese deuten auf neue Methoden hin, um bei Schizophrenie- und Autismuskranken die volle Gesundheit und Funktionsfähigkeit wiederherzustellen. Fortschritte an der Schnittstelle von Neurowissenschaft und Technik lassen eine Zeit erahnen, in der Interaktionen zwischen Gehirn und Computer dazu führen werden, dass von einer Lähmung Betroffene eine beachtliche Bewegungsfähigkeit wiedererlangen werden. Vorläufige Versuche mit tiefer Hirnstimulation lassen darauf schliessen, dass ein besseres Verständnis der Hirnschaltkreise 11 zu wirksameren Behandlungen von Depression, Angstkrankheiten und anderen Erkrankungen, welche Emotion und Kognition betreffen, führen könnte. Fortschritte im Kampf gegen Hirnkrankheiten sind in unserer Gesellschaft zu Recht ein zentrales Ziel. Da das Gehirn sehr kompliziert ist und sich beispielsweise die höchsten Ebenen der menschlichen Kognition nicht leicht in Tiermodellen untersuchen lassen, müssen diese Fortschritte mühsam errungen werden. Wirksamer gegen Krankheiten vorzugehen und herauszufinden, wie wir die intelligentesten jungen Menschen für diese Aufgabe begeistern und ihnen das notwendige Rüstzeug geben können, sind allerdings nicht die einzigen Herausforderungen, vor denen wir stehen. Dies ist der zweite von der Dana Alliance herausgegebene Bericht über Fortschritte in der Hirnforschung, der sich mit Neuroethik befasst (der erste erschien im Jahr 2003). Mitglieder dieser Organisation haben sich in Artikeln und Vorträgen intensiv mit den ethischen Herausforderungen auseinandergesetzt, die aus der Hirnwissenschaft hervorgehen. Bedenken, die sich aus der Erforschung des Gehirns, des Verhaltens und des geistigen Lebens ergeben, waren verschiedentlich bereits früher, in oft kleinen Gruppierungen von Fachpersonen aus der Wissenschaft, Ethik und anderen Bereichen ansatzweise behandelt worden. Ein weit reichendes und dauerhaftes Engagement für diese Fragen, die unter der Bezeichnung „Neuroethik“ zusammengefasst wurden, kristallisierte sich an einer von der Dana Foundation in San Francisco gesponserten Konferenz mit dem Titel „Neuroethik – das Gebiet abstecken“ („Neuroethics: Mapping the Field“) heraus. Seit diesem Meeting im Mai 2002 liess eine wachsende Zahl von Treffen, Artikeln und Büchern ein dynamisches interdisziplinäres Gebiet entstehen, zu dem eine vielseitige Gemeinschaft ihre Beiträge leistete; sie umfasst unter anderem Fachpersonen aus Wissenschaft, Philosophie, Medizin, Jurisprudenz, Soziologie, Politikwissenschaft sowie politisch Verantwortliche. Angesichts dieses wachsenden Interesses kam im Mai 2006 eine Gruppe in Asilomar, Kalifornien, zusammen und beschloss die Gründung einer Neuroethischen Gesellschaft (www.neuroethicssociety.org). 12 Die Teilnehmenden der Asilomar-Konferenz waren der Ansicht, eine Gesellschaft würde zur Bildung einer dauerhaften Gesprächsplattform beitragen, so dass Wissenschafter und Wissenschafterinnen aus ganz Mit ihrem Bemühen, Aufmerksamkeit auf diesen Bereich zu lenken, ist diese im Entstehen begriffene Gesellschaft keineswegs allein. Die American Association for the Advancement of Science behandelte an ihren Treffen regelmässig neuroethische Themen. Die Society for Neuroscience bot auf ihrer jährlichen Konferenz seit 2003 eine Vorlesung zu Neuroethik an und hielt drei Symposien über Neuroethik ab, unter anderem eines im Oktober 2006, das sich auf ein weites Spektrum internationaler Fragen konzentrierte; es ging um Forschende, die Freiwillige und Gemeinschaften in ärmeren Ländern ausbilden, oder auch darum, dass Regierungen von reicheren Ländern die ethischen Richtlinien für die Hirnforschung ausarbeiten. Im laufenden Jahr werden die Cognitive Neuroscience Society und die Association for Psychological Science Symposien zur Neuroethik organisieren. Die Wellcome Trust Bioethics Summer School befasst sich seit zwei Jahren mit Neuroethik. Die American Academy of Arts and Sciences hat Anfang 2007 ein Symposium über den Einsatz von Neuroimaging zur Lügendetektion durchgeführt. Trotz dieses Wachstums nehmen Publikationsmöglichkeiten erst jetzt zu. Neue interdisziplinäre Arbeitsbereiche stossen bei Veröffentlichungen aus zwei Gründen oft auf Schwierigkeiten. Erstens passt ihre Arbeit möglicherweise nicht genau zum Bereich von bestehenden Disziplinen, welche wissenschaftliche Zeitschriften herausgeben. Zweitens kann es für einen Arbeitsbereich, dessen Erfolg auf bedeutenden Beiträgen aus mehreren Disziplinen beruht, schwierig sein, an das relevante Quellenmaterial heranzukommen. Fachleute aus Neurowissenschaft, Rechtswissenschaft und Philosophie wissen oft nicht, wo sie nach Artikeln suchen sollen, die sich mit Problemen zur Neuroethik befassen. Fünf Jahre Neuroethik: Das Gebiet entwickelt sich weiter unterschiedlichen Fachbereichen in interdisziplinären Arbeitsgebieten interagieren können. Aufgrund der wachsenden Erfahrung und Vertiefung wird es ihnen hoffentlich möglich sein, grundlegende Probleme anzugehen. Meiner Ansicht nach ist es der besondere Status des Gehirns, der die Neuroethik zu einem derart zentralen Anliegen macht. Es sind daher verschiedene Bemühungen im Gang, um neue Publikationsmöglichkeiten zu schaffen. Die Neuroethics Society traf mit dem American Journal of Bioethics eine Vereinbarung, um spezielle Nummern (American Journal of Bioethics – Neuroscience) herauszugeben, die diesem Arbeitsbereich gewidmet sind, und auch das Journal of Cognitive Neuroscience erweitert gegenwärtig seinen Publikationsbereich. 13 Jetzt, im Anfangsstadium der Neuroethik wird immer wieder die Frage aufgeworfen, weshalb denn überhaupt ein neuer Arbeitsbereich nötig sei, warum das grössere Zelt der Bioethik nicht ausreiche? Selbstverständlich sind viele der Wissenschafter und Wissenschafterinnen, die sich mit Neuroethik befassen, fest in bestehenden bioethischen Aktivitäten verwurzelt, und dies soll auch so bleiben. Viele Anliegen der Neuroethik sind auch für die Bioethik von zentraler Bedeutung, z. B. Einverständniserklärungen von Menschen mit einer kognitiven Beeinträchtigung oder Patientenverfügungen. Hirnforschung erfordert jedoch mehr. Meiner Ansicht nach ist es der besondere Status des Gehirns, der die Neuroethik zu einem derart zentralen Anliegen macht; eine eingehende Beachtung der Implikationen der Hirnforschung führt uns weit über die gewohnten Grenzen der Bioethik hinaus. Die Möglichkeit, mittels Bildgebung des Gehirns neuere Erlebnisse einer Person zu rekonstruieren oder ihre Glaubwürdigkeit zu überprüfen, wirft nicht nur herkömmliche bioethische Fragen zur Intimsphäre auf, sondern betrifft auch weitere Berufsgruppen – unter anderem Fachleute für Strafverfolgung und nationale Sicherheit – die in Diskussionen zur Bioethik oft nicht vertreten sind. Was die Neuroethik noch einzigartiger macht, ist die Erkenntnis, dass das Gehirn nicht nur Objekt ethischer Untersuchungen ist sondern auch die Basis unserer ethischen Grundsätze. Analog dazu geben Bestrebungen, die Hirntätigkeit zu beeinflussen oder zu steuern, zu ethischen Fragen Anlass, die sich nur teilweise mit jenen decken, die aus der Beeinflussung der Herz- oder Nierentätigkeit hervorgehen. Dies beruht darauf, dass es bei der Steuerung der Hirntätigkeit um den Kern unseres Selbst und unserer Autonomie geht. Was die Neuroethik noch einzigartiger macht, ist die Erkenntnis, dass das Gehirn nicht nur Objekt ethischer Untersuchungen ist sondern auch die Basis unserer ethischen Grundsätze. 14 Dieser letzte Punkt wird allerdings von jenen bestritten, die glauben, ethische Grundsätze beruhten auf einem Naturgesetz oder einer göttlichen Vorgabe. In einem Land wie den USA, in dem Religion wichtig ist, bleibt diese Diskussion lebendig und bedeutsam. Nun da wir anfangen, neuronale Grundlagen der sozialen Interaktion – dazu gehören auch Belange Fünf Jahre Neuroethik: Das Gebiet entwickelt sich weiter wie Vorurteil und Vertrauen – zu verstehen, und sich Möglichkeiten ergeben, solche Interaktionen (z. B. durch pharmakologische oder elektrische Stimulation) zu beeinflussen, werden grundlegende Fragen nach der Herkunft unserer unterschiedlichen ethischen Systeme aufgeworfen. Sind sie von zeitlosen, rationalen Prinzipien abgeleitet? Handelt es sich um Zufallsprodukte eines sich entwickelnden Gehirns? Oder beides? Um mit dem wissenschaftlichen Fortschritt möglichst sinnvoll umgehen zu können, braucht es die Neuroethik. 15 Fortschritte in der Hirnforschung im Jahr 2006 17 In der Kindheit auftretende Störungen Hirnanomalien bei Autismus 20 Neue Erkenntnisse zur ADHS 23 Zerebrale Lähmung: Der Einfluss von Infektionen 26 19 E ine der bedeutsamen Studien des Jahres 2006, welche die Kindheit betrafen, galt der Lokalisierung spezifischer Hirnbereiche, die zum vergrösserten Hirnvolumen bei der Autismusspektrumstörung beitragen. Die Forschung gewann auch neue Einsichten in gewisse neuroanatomische und biochemische Veränderungen, die für die kognitiven Schwierigkeiten verantwortlich zu sein scheinen, unter denen Kinder mit einer Aufmerksamkeitsdefizit-Hyperaktivitätsstörung leiden; ausserdem ergaben sich weitere Hinweise auf den Einfluss von Virusinfektionen auf Entwicklungsstörungen, wie zerebrale Lähmung. Hirnanomalien bei Autismus Bei der Autismusspektrumstörung (Autism spectrum disorder, ASD), einer Gruppierung, die Autismus und ähnliche Störungen umfasst, handelt es sich um eine tief greifende Entwicklungsstörung, die sich hauptsächlich in einer stark verminderten Interaktions- und Kommunikationsfähigkeit manifestiert. Niemand weiss genau, was zu ASD führt, doch wurden viele neurologische Anomalien nachgewiesen, die möglicherweise zu den für ASD typischen sozialen und kognitiven Defiziten beitragen. Abweichende Aktivitäten in bestimmten Hirnregionen wurden mit ASD in Verbindung gebracht. Laut einem von Marco Iacoboni, einem Neurowissenschafter an der University of California, Los Angeles, geleiteten Forschungsteam etwa, war bei Kindern mit ASD ein bestimmter Teil des Gehirns, der so genannte inferiore frontale Gyrus, merklich weniger aktiv während der Durchführung bestimmter Aufgaben, die mit sozialer Interaktion zusammenhingen 1. In einer Studie, die in Nature Neuroscience publiziert wurde, untersuchte das Team von Iacoboni mittels funktioneller Magnetresonanz-Tomographie die Nervenaktivität von zehn hochbegabten Kindern mit ASD und zehn sich normal entwickelnden Kindern, während sie emotionale Gesichtsausdrücke beobachteten und nachahmten. Das Ausmass der verminderten Aktivität korrelierte mit dem Schweregrad ihrer Symptome. 20 Vermutlich gehört der inferiore frontale Gyrus zum so genannten Spiegelneuronsystem (mirror neuron system), das für die Wahrnehmung und den Ausdruck von Emotionen bedeutsam ist und das Empfinden von Empathie ermöglicht. Der Befund deutet auf ein fehlerhaftes Spiegelneuronsystem als mögliche Grundlage der bei Autismus beobachteten sozialen Defizite hin. Man weiss zwar nicht, welche Bedeutung der Kortexdicke auf der Ebene einzelner Zellen zukommt, doch vermuten die Forschenden, sie könnte das Ausmass der „Arborisation“ angeben, also der Verästelung von Verbindungen zwischen Hirnzellen. Im Verlauf der normalen Entwicklung des Gehirns findet eine massive Überproduktion von Zellen und Verbindungen mit anderen Hirnzellen (Synapsen) statt. Darauf folgt ein kompetitiver Abbau oder ein „Zurückschneiden“ (pruning) von Neuronen und ihren Verbindungen. Die Gruppe nimmt an, dieser Abbau führe zu einer Verdünnung des Kortex. In der Kindheit auftretende Störungen Auch Abweichungen der Hirngrösse wurden mit ASD in Verbindung gebracht. Eine im American Journal of Psychiatry veröffentlichte Untersuchung einer Forschungsgruppe, die von Antonio Hardan, einem Psychiater der Stanford University, geleitet wurde, verglich die Grösse des Kortex (der Hirnrinde) von 17 Kindern mit Autismus mit jener von 14 Kindern ohne diese Krankheit 2. Die Kortexdicke ist ein empfindliches Mass der normalen Hirnentwicklung. Die Analyse der Aufnahmen des Gehirns zeigte, dass die Kortexdicke in den Schläfenlappen und Scheitellappen der autistischen Kinder erhöht war. Die Forschenden nehmen an, diese anatomischen Unterschiede seien für das grössere Gehirn bei ASD mitverantwortlich. Aufgrund dieses Befundes werden die Forschenden untersuchen, was die Verringerung der Kortexdicke normalerweise steuert – dazu gehören auch genetische Einflüsse. Sie haben vor, die verschiedenen an diesem Prozess beteiligten Gene zu untersuchen und hoffen, einen Hinweis zu erhalten, der verstehen lässt, wodurch die dickeren Hirnstrukturen beim Autismus verursacht werden; dies könnte zu neuen Behandlungen führen. Einer in Archives of General Psychiatry 3 veröffentlichten Studie zufolge scheint auch eine vergrösserte Amygdala zum grösseren Gehirn bei Autismus beizutragen. Die Amygdala ist ein Teil des Gehirns, der für die sozio-emotionale Funktion bedeutsam ist. In dieser von Stephen Dager von der University of Washington geleiteten Untersuchung wurde mittels Magnetresonanz-Tomographie die Grösse der Amygdala bei 45 Kindern mit ASD im Alter von drei bis vier Jahren gemessen. Dabei entdeckten die Forschenden einen Zusammenhang zwischen einer vergrösserten Amygdala – insbesondere der rechten Seite – und der 21 Untersuchung des Autismus Im Rahmen einer gemeinsam mit Dennis Shaw durchgeführten Studie zur Messung der Grösse der Amygdala bei Kindern mit einer Autismusspektrumstörung prüft der Forscher Stephen Dager (im Vordergrund) ein Hirnscan. Ausprägung der Symptome bei diesen Kindern. Als sie dieselben Kinder etwa drei Jahre später untersuchten, zeigte sich, dass die sprachlichen und sozialen Fähigkeiten bei den Kindern mit einer stärkeren Vergrösserung der rechten Amygdala schwächer entwickelt waren. Diese Ergebnisse zeigen einen klaren Zusammenhang zwischen Amygdala-Anomalien und den bei Autismus festgestellten Verhaltensstörungen; sie deuten überdies darauf hin, dass die Grösse der rechten Amygdala zur Vorhersage des klinischen Verlaufs der Krankheit dienen könnte. 22 Forschende desselben Laboratoriums berichteten in einem neueren Artikel in Neurology, die bei autistischen Kindern im Vergleich zu Kindern mit einer Entwicklungsverzögerung festgestellten Behinderungen könnten einer verstärkten „transversalen Relaxation“ von Hirnzellen (graue Hirnsubstanz) zugeschrieben werden 4. Die mittels Magnetresonanz-Tomographie (magnetic resonance imaging, MRI) festgestellte transversale Die Forschenden verglichen 60 autistische Kinder mit 16, die an einer Entwicklungsverzögerung litten, und 10, die sich normal entwickelten. Alle Kinder waren zwischen zwei und vier Jahre alt. Es zeigte sich, dass die Zellen der sich normal entwickelnden Kinder im Vergleich zu den autistischen Kindern bedeutend dichter angeordnet waren. Kinder mit einer Entwicklungsverzögerung lagen, was die Dichte anbelangt, zwischen den sich normal entwickelnden und den autistischen Kindern. Autismus bleibt eine mysteriöse Krankheit. Diese Bildgebungs-Studien deuten auf Abweichungen in der Entwicklung der neuronalen Strukturen hin, die möglicherweise bereits zu Beginn der Schwangerschaft auftreten. In der Kindheit auftretende Störungen Relaxation gibt an, wie dicht Hirnzellen angeordnet sind. Dies wird durch die Wasserverdrängung im Gehirn gemessen, eine Methode, die über den Zeitverlauf der Hirnreifung Aufschluss gibt. Neue Erkenntnisse zur ADHS Bei der Aufmerksamkeitsdefizit-Hyperaktivitätsstörung (ADHS) handelt es sich um eine Entwicklungsstörung, deren Auswirkungen den schulischen und beruflichen Erfolg verringern und das Risiko von Depression, Drogenmissbrauch und unfallbedingten Verletzungen, möglicherweise sogar mit Todesfolge erhöhen. Charakteristisch für ADHS sind Ruhelosigkeit und Ablenkbarkeit, die vermutlich auf der Unfähigkeit beruhen, gewisse Impulse innerhalb des Gehirns zu unterdrücken. Die Wissenschaft hatte schon lange vermutet, ADHS beruhe auf einem Mangel an Dopamin. Neueres Beweismaterial belegt dies. Mit Medikamenten, welche die Verfügbarkeit des hemmenden Neurotransmitters Dopamin im Gehirn erhöhen, lässt sich ADHS in den meisten Fällen erfolgreich behandeln. Die Wissenschaft hatte schon lange vermutet, ADHS beruhe auf einem Mangel an Dopamin. Neueres Beweismaterial belegt dies und sieht in Defekten der „Dopamin-Transporter“ die Hauptursache: Die Transporter nehmen zu viel Dopamin auf, bevor dieses von einer Hirnzelle zur nächsten weitergegeben werden kann. Ein von Donald Gilbert, einem Kinderneurologen am Cincinnati Children’s Hospital Medical Center, geleitetes Forschungsteam untersuchte bei 16 Kindern und Jugendlichen mit ADHS die Bewegungshemmung durch den motorischen Kortex des Gehirns bevor und nachdem sie Medikamente erhielten, welche die Verfügbarkeit von Dopamin im Gehirn steigerten 5. 23 Drücken! Nicht drücken! Wechseln! Hirnaktivität bei ADHS Kinder mit einer Aufmerksamkeitsdefizit-Hyperaktivitätsstörung zeigten eine reduzierte Hirnaktivität im medialen präfrontalen Kortex, wenn sie eine motorische Reaktion vermeiden sollten (links) und in frontotemporalen Hirnregionen, wenn sie von einer Aufgabe zu einer anderen wechseln sollten (rechts). Die daraus resultierenden grösseren Mengen von Dopamin hemmten bei allen getesteten Kindern die Aktivität des motorischen Kortex, doch war die Wirkung des Medikaments bei jenen Kindern wesentlich grösser, welche die genetische Variante DAT1 aufwiesen, die normalerweise eine zu hohe Aktivität des Dopamintransporters verursacht. Dies führt zu einem Mangel an hemmendem Dopamin. Diese Untersuchung bringt ADHS mit genetisch veränderten Dopamintransportern in Verbindung. Katya Rubia vom Institute of Psychiatry am King’s College in London und ihre Mitarbeitenden fanden in einem ähnlich angeordneten Versuch bei 19 Knaben mit ADHS, deren Krankheit nie mit Medikamenten behandelt worden war, einen vergleichbaren Mangel in den für motorische Hemmung und sprunghaftes Verhalten verantwortlichen Hirnregionen 6. Dies, so betonen sie, ist deshalb bedeutsam, weil die bisherigen Untersuchungen an Kindern erfolgt waren, die bereits Medikamente gegen ADHS erhalten hatten, was die Resultate beeinflusst haben könnte. 24 Mittels Magnetresonanz-Tomographie stellte Rubias Team bei diesen „medikamentös unbelasteten” Kindern und Jugendlichen mit ADHS fest, Wie bei Autismus wurde auch bei ADHS die Kortexdicke als Indikator für die Hirnentwicklung untersucht. Wie bei Autismus wurde auch bei ADHS die Kortexdicke als Indikator für die Hirnentwicklung untersucht. Unter der Leitung von Philip Shaw massen Forschende des National Institute of Mental Health die Kortexdicke bei einer Gruppe von 166 Kindern mit ADHS 7. Etwa alle zwei Jahre machten sie Magnetresonanz-Aufnahmen von diesen Kindern und verglichen sie mit Scans von gesunden Kindern. In der Kindheit auftretende Störungen dass ihre Hirnaktivität sowohl bei Aufgaben vermindert war, die eine motorische Hemmung erforderten als auch bei solchen, die einen Wechsel von einer Aufgabe zur nächsten verlangten (was kognitive Flexibilität voraussetzt). Der Befund lässt darauf schliessen, dass die mangelnde Aktivität dieser Patientengruppe nicht mit einer Langzeitbehandlung durch Stimulantien zusammenhängt. Während beiden Aufgaben war eine mangelnde Aktivität in präfrontalen Hirnregionen offensichtlich, ebenso in temporalen und parietalen Regionen, die man bis anhin nicht mit ADHS in Verbindung gebracht hatte. Die Analyse dieser Bilder ergab, dass der Kortex von Kindern mit ADHS in jenen Hirnbereichen, die für die Steuerung der Aufmerksamkeit wichtig sind, dünner war. Spätere Scans dieser Kinder zeigten, dass bei jenen mit schlechteren klinischen Ergebnissen der Kortex im vorderen Bereich des Gehirns besonders dünn war, und zwar in der Nähe einer Region, welche Aspekte der Aufmerksamkeit, z. B. das Unterdrücken unangemessener Verhaltensweisen steuert. Zudem fand sich bei Kindern mit ADHS, deren klinische Ergebnisse besser waren, ein charakteristisches Muster von kortikalen Veränderungen im rechten parietalen Kortex, der einige der grundlegendsten Aspekte der Aufmerksamkeit steuert. An dieser Stelle erreichte der Kortex bis zur späten Adoleszenz dieselbe Dicke wie bei gesunden Kindern. Bei Kindern mit schlechten klinischen Resultaten fand diese „Normalisierung“ jedoch nicht statt. Die Ergebnisse waren unabhängig davon, ob die Kinder Medikamente gegen ADHS erhielten. Diese Arbeit vermittelt ein sehr detailliertes Bild des Kortex von Kindern mit ADHS und hebt den Forschenden zufolge Veränderungen des Gehirns hervor, welche die Besserung dieser Krankheit widerspiegeln oder sogar bewirken könnten. 25 Wie beim Autismus geben auch bei ADHS Studien mittels Bildgebenden Verfahren Hinweise darauf, welche Hirnfunktionen schief laufen. Untersuchungen dieser Art können sowohl diagnostisch als auch therapeutisch von Nutzen sein. Zerebrale Lähmung: Der Einfluss von Infektionen Im Jahr 2006 wurden weitere Beweise für den Einfluss von Infektionen auf Entwicklungsstörungen, wie z. B. die zerebrale Lähmung, erbracht. Bei der zerebralen Lähmung handelt es sich um eine bleibende und oft schwerwiegende Hirnkrankheit, die bereits in der frühen Kindheit nachweisbar ist und eine Fehlsteuerung von Körperbewegungen oder Körperhaltungen bewirkt. Die Ursachen der zerebralen Lähmung sind weitgehend unbekannt, und zurzeit kann man ihr nicht vorbeugen. Catherine Gibsons Team von der University of Adelaide, Australien, berichtete in einem Artikel im British Medical Journal von Untersuchungen über einen möglichen Zusammenhang von Virusinfektionen und zerebraler Lähmung 8. Die Forschenden suchten in getrockneten Blutproben von Neugeborenen, von denen 443 an der Krankheit litten und 883 nicht, nach Herpesviren, einer Gruppe von Viren, zu der auch jene gehören, die für Windpocken und Fieberbläschen verantwortlich sind. Die Ergebnisse zeigten, dass Babys mit zerebraler Lähmung den Herpesviren im Verlauf der Schwangerschaft ihrer Mütter wesentlich öfter ausgesetzt waren als die anderen Babys – ein Hinweis darauf, dass diese Viren während der Schwangerschaft zur Entstehung der Krankheit beitragen könnten. Diese Untersuchungen müssen noch an weiteren Populationen bestätigt werden. 26 Bewegungsstörungen und andere Störungen der Motorik Falsch gefaltete Proteine: Freunde oder Feinde? 28 Entzündungen und Parkinsonsche Krankheit 30 Genetik der Parkinsonschen Krankheit 32 Überwachung und Behandlung der Huntingtonschen Krankheit 32 27 A uf dem weiten Weg von der Grundlagenforschung bis zu neuen Therapien zur Behandlung von Krankheiten, die mit der Bewegung des Menschen zusammenhängen, gab es im Jahr 2006 Fortschritte. Laborstudien über Proteinfaltung, Entzündungen, Wachstumsfaktoren und Genetik zeigten neue Möglichkeiten zur Überwachung und Behandlung solcher Krankheiten auf. Einige Therapien werden zurzeit an Tieren und Menschen getestet. Falsch gefaltete Proteine: Freunde oder Feinde? Was ein Protein im Körper macht, wird durch seine Form bestimmt. Zellen bilden Proteine, die aus langen Ketten von Untereinheiten, so genannten Aminosäuren, bestehen und sich zu dreidimensionalen Gebilden aufreihen und falten. Fehlerhaft gefaltete Proteine interagieren mit anderen Proteinen. Es kommt auch vor, dass sich falsch gefaltete Proteine aneinander festmachen und Klumpen bilden, so genannte Inklusionen, die im Gehirn von Menschen mit neurologischen Krankheiten häufig vorkommen. Alpha-Synuclein ist eine Hauptkomponente jener Inklusionen (der so genannten Lewy-Körper), die man typischerweise in Hirnzellen von Personen findet, die an der Parkinsonschen Krankheit leiden, einer Krankheit die Starre, Zittern und verlangsamte Bewegung verursacht. Lewy-Körper kommen auch bei einer ähnlichen Form der Demenz vor; man spricht hier von einer Lewy-Körper-Demenz. Inklusionen mit sehr viel AlphaSynuclein finden sich auch bei der Multiplen Systematrophie, die mit der Parkinsonschen Krankheit Ähnlichkeiten aufweist und Sprach-, Gleichgewichts- und Koordinationsstörungen hervorrufen kann. Zwei neuere Untersuchungen von Thomas Südhof und Mitarbeitenden (über die in Cell berichtet wurde) sowie von Tracey Dickson und Mitarbeitenden (über die in Experimental Neurology berichtet wurde) lassen darauf schliessen, dass Alpha-Synuclein normalerweise die Aufgabe hat, Nervenzellen vor Schädigungen zu schützen 1, 2. Demnach scheint ein normales Niveau von richtig gefaltetem Alpha-Synuclein Zellen zu schützen, während Überproduktion, Fehlfaltung und Anhäufung des Proteins mit Krankheiten verbunden sind. Wie ist das zu erklären? 28 Zu dieser Frage gibt es zwar kontroverse Ansichten, doch geht man allgemein davon aus, die Fehlfaltung und Anhäufung des Proteins trage zum Zelltod bei; der Vorgang selbst bleibt jedoch unklar. Möglicherweise Ausgehend von der Hypothese, wonach Inklusionen zur Schädigung einer Zelle beitragen, werden zurzeit Therapien entwickelt, welche Anhäufungen und Inklusionen verhindern sollen. Den umgekehrten Ansatz wählte ein Team unter der Leitung von David Housman und Aleksey Kazantsev, die ihre Ergebnisse in Proceedings of the National Academy of Sciences veröffentlichten 5. Sie gingen davon aus, die Anhäufung der falsch gefalteten Proteine könnte ein Vorgang sein, durch den sich eine Zelle gegen die Verteidigt sich die Zelle? Möglicherweise um sich selbst vor den schädigenden Auswirkungen falsch gefalteter Proteine bei degenerativen Erkrankungen des Nervensystems, etwa der Parkinsonschen Krankheit, zu schützen, produziert eine gestresste Zelle (im Zentrum) mehr Alpha-Synuclein, ein Hirnprotein. Bewegungsstörungen und andere Störungen der Motorik können falsch gefaltete Proteine ihre normale Aufgabe nicht erfüllen; ausserdem scheinen sie auch andere Funktionen der Zelle zu stören. Eine von Richard Morimoto geleitete Studie, über die in Science berichtet wurde, lässt darauf schliessen, dass ein Übermass an falsch gefalteten Proteinen das System der „Qualitätskontrolle“ der Zelle beeinträchtigen kann, was zur Fehlfaltung weiterer Proteine führt 3. Eine andere Untersuchung, die Susan Lindquist und Mitarbeitende in Science publizierten, deutet darauf hin, dass ein Übermass an Alpha-Synuclein die Bewegung von Proteinen innerhalb von Zellen beeinträchtigt 4. 29 schädliche Wirkung von falsch gefalteten Proteinen schütze; demnach würden Inklusionen die Zellen schützen und nicht schädigen. Sie machten Versuche mit einer B2 genannten Substanz, welche die Bildung von Inklusionen fördert, und stellten fest, dass sie das Ausmass der Zellschädigung in Zell-Modellen der Huntingtonschen und der Parkinsonschen Krankheit tatsächlich verminderte. In einem Kommentar, der in Experimental Neurology erschien, bot Mark Cookson eine Erklärung für die offensichtlich paradoxen Wirkungen von Alpha-Synuclein 6. Seiner Ansicht nach schützen normale, mässige Konzentrationen von Alpha-Synuclein Nervenzellen. Sobald eine Zelle gestresst wird, versucht sie sich vor einer Schädigung zu schützen, indem sie mehr Alpha-Synuclein produziert. Alpha-Synuclein beginnt kleine Anhäufungen zu bilden, welche die normale Zelltätigkeit stören. Wenn es gelingt, diese kleineren Anhäufungen zu Inklusionen zusammenzufassen, werden sie daran gehindert, die Zelle zu schädigen. Ein besseres Verständnis der Funktion falsch gefalteter Proteine bei degenerativen Erkrankungen des Nervensystems wird die Entwicklung neuer Medikamente begünstigen, die diese Schädigung verhindern. Entzündungen und Parkinsonsche Krankheit Bei der Parkinsonschen Krankheit kommt es bei einer bestimmten Population von Nervenzellen zu einem vorzeitigen Zelltod. Es stellt sich die Frage: warum? Möglicherweise spielen Entzündungen, also die Ansammlung reaktiver Zellen, eine Rolle. James Bower und seine Mitarbeitenden am Mayo Clinic College of Medicine verglichen die Krankengeschichten von 196 Parkinsonkranken mit denen von 196 entsprechenden Kontrollpersonen. Ihre in Neurology publizierte Studie ergab, dass die späteren Parkinson-Kranken häufiger an Asthma, allergischer Rhinitis oder Heuschnupfen gelitten hatten als die Kontrollpersonen 7. 30 Dieser Befund lässt darauf schliessen, dass bei gewissen Personen Immunreaktionen auftreten, die sowohl an Allergien als auch an der Parkinsonschen Krankheit beteiligt sind. Dazu passt auch die Entdeckung von Bowers Gruppe, dass Medikamente, die eine Entzündung verhindern, z. B. nichtsteroide entzündungshemmende Medikamente (nonsteroidal anti-inflammatory drugs, NSAIDs), einen Schutz bilden können – so dass die Parkinsonsche Krankheit möglicherweise bei Personen, die NSAIDs verwenden, mit geringerer Wahrscheinlichkeit auftritt. Insgesamt lassen diese Befunde einen Zusammenhang von Entzündungen und Bei gewissen Personen treten Immunreaktionen auf, die sowohl an Allergien als auch an der Parkinsonschen Krankheit beteiligt sind. Eine von Miguel Hernán geleitete Gruppe publizierte in Neurology eine ähnliche Studie 8. Sie hatte entdeckt, dass die Wahrscheinlichkeit, eine Parkinsonsche Krankheit zu entwickeln, bei Männern, die andere NSAIDs als Aspirin (z. B. Ibuprofen) verwendeten, um 20% niedriger war als bei Personen, die keine solchen Medikamente verwendeten; bei Frauen, die NSAIDs verwendeten, war sie hingegen um 20% höher. Der unerwartete Geschlechtsunterschied bestätigt die Ergebnisse einiger anderer Studien, denen zufolge die Risikofaktoren für die Parkinsonsche Krankheit bei Männern und Frauen unterschiedlich waren. Eine andere Studie ergab, dass ein Antibiotikum, das seit den 1970er Jahren zur Behandlung von Akne eingesetzt wurde, Entzündungen hemmt und Neuronen schützt. Raymond Swanson und Mitarbeitende am University of California and Veterans Affairs Medical Center in San Francisco untersuchten an im Laboratorium gezüchteten Nervenzellkulturen, wie das Antibiotikum Minocyclin Neuronen schützt 9. In ihrer in Proceedings of the National Academy of Sciences veröffentlichten Studie wiesen sie nach, dass Minocyclin PARP-1 hemmt, ein Protein, das als Reaktion auf eine DNA-Schädigung Entzündungen und den Zelltod begünstigt. Sie schlossen daraus, dass die entzündungshemmende und neuroprotektive Wirkung von Minocyclin auf der Hemmung von PARP-1 beruhen könnte. Dass Minocyclin, Entzündungen hemmt und Neuronen schützt, könnte klinisch genutzt werden; entsprechende Untersuchungen an Tiermodellen der Parkinsonschen Krankheit, der Huntingtonschen Krankheit und der Amyotrophen Lateralsklerose (ALS oder Lou Gehrig-Krankheit) führten denn auch zu viel versprechenden Ergebnissen. Die in Neurology veröffentlichten Resultate einer klinischen Pilotstudie sprechen für die Durchführung weiterer klinischer Minocyclin-Studien bei der Parkinsonschen Krankheit 10. Auch im Zusammenhang mit der Huntingtonschen Krankheit und mit ALS werden klinische Studien mit Minocyclin vorbereitet. Bewegungsstörungen und andere Störungen der Motorik Parkinsonscher Krankheit erkennen, doch braucht es weitere Studien, um bestimmen zu können, welcher Art diese Beziehung ist. Das Verständnis dieses Zusammenhangs könnte wichtige neue Erkenntnisse zum Krankheitsprozess ermöglichen und auf neue Behandlungsstrategien hinweisen. 31 Genetik der Parkinsonschen Krankheit Die familiäre Form der Parkinsonschen Krankheit macht ca. 10% aller Krankheitsfälle aus, und es sind mindestens fünf Genmutationen bekannt, die an der ererbten Form dieser Krankheit mitwirken. Die Untersuchung dieser Gene hat zu Erkenntnissen über den Krankheitsprozess geführt, die allen Parkinson-Kranken zugute kommen könnten. Zwei Studien, die in Nature veröffentlicht wurden, befassten sich mit der Beziehung zwischen zwei verschiedenen Genen, die mit der ererbten Parkinsonschen Krankheit in Verbindung gebracht werden 11, 12. Es zeigte sich, dass die Gene „Parkin“ und „PINK1“ zusammenwirken, um die Tätigkeit der Mitochondrien, der Kraftwerke der Zelle, aufrechtzuerhalten. Diese und weitere Studien bestätigen die seit langem gehegte Ansicht, Funktionsstörungen der Mitochondrien könnten zur Parkinsonschen Krankheit beitragen. Ein Zusammenhang von „Parkin“- und „PINK1“-Mutationen und der Parkinsonschen Krankheit wurde erstmals in Bezug auf Personen beschrieben, bei denen beide Kopien des „Parkin“-Gens oder des „PINK1“-Gens fehlerhaft waren. Obwohl auch Personen mit einer einzigen fehlerhaften Kopie diese an ihre Kinder weitergeben können, war die klinische Relevanz unklar. Zwei in Archives of Neurology und eine in Movement Disorders veröffentlichte Studien zeigten nun, dass sich auch eine einzige fehlerhafte Kopie auf die Entwicklung der Parkinsonschen Krankheit auswirken kann 13-15. Bei Personen mit einer einzigen fehlerhaften Kopie von „PINK1“ war das Risiko, an Parkinson zu erkranken höher als bei ihren Verwandten mit zwei normalen Kopien dieses Gens. Bei Personen mit einer einzigen fehlerhaften Kopie von „PINK1“ war das Risiko, an Parkinson zu erkranken höher als bei ihren Verwandten mit zwei normalen Kopien dieses Gens. Und Personen mit einer einzigen fehlerhaften Kopie von „Parkin“ waren beim Ausbruch der Parkinsonschen Krankheit jünger als die meisten Betroffenen, einschliesslich ihrer eigenen Verwandten mit zwei normalen Kopien. Da viel öfter nur eine Kopie eines Gens fehlerhaft ist und nicht beide Kopien, könnten mehr Personen von diesen Mutationen betroffen sein als bisher angenommen wurde. Überwachung und Behandlung der Huntingtonschen Krankheit 32 Bei der Huntingtonschen Krankheit handelt es sich um eine genetische Erkrankung, die gewöhnlich bei Erwachsenen im Alter von 40 bis 50 Jahren Die Wahrscheinlichkeit das Krankheitsgen zu erben, beträgt für jedes Kind eines an Huntington erkrankten Elternteils 50%; mithilfe eines Testes lässt sich heute mit hoher Treffsicherheit feststellen, ob eine bestimmte Person es tatsächlich geerbt hat. Viele Risikopersonen ziehen es jedoch vor, sich nicht testen zu lassen, denn es gibt keine Heilbehandlung, keine Mittel zur Prävention und nur wenige wirksame Symptombehandlungen. Sowohl die Progression der Krankheit als auch die Wirksamkeit allfälliger Behandlungen lassen sich möglicherweise durch eine Überwachung der immunen „Mikrogliazellen“ verfolgen. Diese Zellen tragen vermutlich dadurch zur Krankheit bei, dass sie aktiviert werden und Entzündungen fördernde Substanzen freisetzen. Eine Forschungsgruppe unter Paola Piccini zeigte mittels Positronen-Emissions-Tomographie, dass das Ausmass der Mikroglia-Aktivierung mit der Schwere der Huntingtonschen Krankheit korreliert. Dieser in Neurology publizierte Befund bestätigt eine Beteiligung der Mikroglia an dieser Krankheit 16. Der Befund könnte auch für andere degenerative Erkrankungen des Nervensystems gelten. Mithilfe des Wachstumsfaktors GDNF (glial-line derived neurotrophic factor) lässt sich die Huntingtonsche Krankheit möglicherweise behandeln. Er vermag Nervenzellen zu schützen und fördert sogar deren Regeneration. Ein grosser klinischer GDNF-Versuch mit Menschen war zwar früher einmal abgebrochen worden, doch gab es im Jahr 2006 kleinere Studien mit unterschiedlichen Resultaten, in denen GDNF als Behandlungsmöglichkeit der Parkinsonschen Krankheit überprüft wurde 17-19. Die Verwendung von GDNF zur Behandlung der Huntingtonschen Krankheit wurde auch an einem Mausmodell getestet; die Studie ist in Proceedings of the National Academy of Sciences erschienen 20. Forschende unter der Leitung von Jeffrey Kordower brachten GDNF mithilfe eines Virus ins Gehirn von Mäusen und erzielten dadurch eine Besserung des Verhaltens, weniger zerstörte Nervenzellen und weniger Inklusionskörper. Weitere Studien sind notwendig um zu entscheiden, ob GDNF als wirksame Behandlung der Huntingtonschen Krankheit beim Menschen in Frage kommt. Obwohl wir zurzeit über keine Therapien verfügen, die auf den der Huntingtonschen Krankheit zugrunde liegenden Krankheitsprozess gerichtet Bewegungsstörungen und andere Störungen der Motorik ausbricht. Charakteristisch sind progressive unwillkürliche Bewegungen, emotionale Störungen und ein Abbau der intellektuellen Fähigkeiten. 33 sind, lässt sich die Lebensqualität dieser Kranken durch Medikamente welche die Symptome lindern verbessern. Eine klinische Studie mit einer solchen Behandlung wurde in Neurology vorgestellt 21. Die Studie dauerte zwölf Wochen und ergab, dass Kranke, die mit dem Medikament Tetrabenazin behandelt wurden, deutlich weniger unwillkürliche Bewegungen ausführten als Kranke, die ein Placebo erhielten. 34 Schädigungen des Nervensystems Gedanken nutzbar machen 36 Instandsetzung des Rückenmarks 37 Hirnschlagforschung 39 Hirntumoren 40 35 B ei der Erforschung von Schädigungen des Zentralnervensystems (ZNS) stellt sich allgemein die Frage, was die Grundlagenforschung zur Entwicklung von Therapien beitragen kann. Bezüglich aller entscheidenden ZNS-Schädigungen – Rückenmarkverletzung, Hirnschlag und Hirntumoren – fehlen Behandlungen, was zu einem grossen Teil an der Komplexität der zugrunde liegenden Prozesse liegt. Wenn sich die Forschung vor allem darauf konzentriert, die Prozesse des Zelltodes, der Nervenregeneration und der Tumorentstehung aufzudecken, hat sie stets das Ziel vor Augen, dieses Wissen in molekular ausgerichtete Behandlungen umzusetzen, um dadurch die Schädigung des Nervensystems zu verhindern oder zu beheben. Gedanken nutzbar machen Zu den am meisten Aufsehen erregenden Schlagzeilen dieses Jahres gehört jene über einen gelähmten Mann, der mittels seiner Gedanken einen Computer bediente. Dieser Fortschritt ist der Höhepunkt jahrzehntelanger Grundlagenforschung, die jenem Hirnzentrum galt, das für die motorische Steuerung verantwortlich ist (dies wird im Kapitel Neuroethik, S. 45 weiter diskutiert). Eine Pilotstudie über diesen einen Patienten – John Donoghue von der Brown University und ein in Harvard stationiertes Team von Mitarbeitenden berichteten darüber in Nature – bestätigte das Konzept, wonach eine Hirn-Computer Schnittstelle die Nervenaktivität des primären motorischen Kortex einer Person aufzeichnen und in spezifische Aktionen auf externen Geräten übertragen kann 1. 36 Der in dieser Studie vorgestellte Mann, der infolge einer drei Jahre zuvor erlittenen Rückenmarkverletzung vom Hals abwärts gelähmt war, konnte Email-Nachrichten öffnen, einen Fernseh- und einen Lichtschalter betätigen, eine Handprothese öffnen und schliessen sowie rudimentäre Tätigkeiten mit einem aus mehreren Gelenken bestehenden künstlichen Arm ausführen. Die hier vorgestellte Arbeit ist ein erster Schritt in Richtung auf gedankenbetriebene Roboter, die als Hilfsmittel dienen könnten, um Personen, welche infolge einer Schädigung des Zentralnervensystems gelähmt sind, eine gewisse Unabhängigkeit zu ermöglichen. Die Autoren weisen deutlich darauf hin, dass die Technik weiter entwickelt werden muss, bevor eine praktische Anwendung ausserhalb einer Forschungsanordnung ins Auge gefasst werden kann. Die vielfältigen Aspekte von Rückenmarksverletzungen erfordern entsprechend unterschiedliche Behandlungsansätze, und die Forschung beginnt erst jetzt, verschiedene therapeutische Strategien in Tierversuchen zu kombinieren. Das Bemühen, Axonen – jene Nervenfasern, welche Signale von einer Zelle zur andern leiten – zum Nachwachsen zu veranlassen, ist weiterhin mit grundlegenden Schwierigkeiten verbunden. Unter anderem gilt es herauszufinden, wie man beschädigte Nervenfasern veranlassen kann, in die richtige Richtung zu wachsen und sich mit den richtigen Zielstrukturen zu verbinden, so dass die neuronale Kommunikation wiederhergestellt wird. Zu den Problemen, die diese Schwierigkeiten noch vergrössern, gehören die Lücke, die entsteht, wenn das Rückenmark durch einen Riss oder eine Quetschung verletzt wird; die Entstehung einer undurchdringbaren „GliaNarbe“ am Ort der Verletzung; das Vorhandensein von hemmenden Molekülen in der Narbe und im Rückenmark, die ein Nachwachsen von Axonen (Kommunikationskabeln) verhindern; sowie die komplizierte Dynamik von richtungsweisenden Axonen. Die Forschung konzentriert sich darauf, Substanzen zu identifizieren und zu testen, welche diesen körpereigenen Inhibitoren des Axon-Wachstums entgegenwirken könnten. Schädigungen des Nervensystems Instandsetzung des Rückenmarks Eine Substanz, die zurzeit getestet wird, ist Chondroitinase ABC, ein natürlich vorkommendes bakterielles Enzym, das früheren Forschungsarbeiten zufolge die Bildung inhibitorischer Moleküle, so genannter Proteoglykane, in der Glia-Narbe verhindert. James Massey und Mitarbeitende an der University of Louisville berichteten im Journal of Neuroscience, die Injektion von Chondroitinase ins Stammhirn von Ratten mit einer zervikalen Rückenmarkverletzung habe das Aussprossen von Nerven am Ort der Verletzung gefördert; dies bestätigt frühere Berichte 2. Forschende an der Johns Hopkins University und an der University of Michigan unter der Leitung von Ronald Schnaar berichteten in Proceedings of the National Academy of Sciences ebenfalls, Chondroitinase ABC habe in einem Tiermodell der Rückenmarkverletzung das Wachstum von Axonen induziert. Ausserdem entdeckten sie ein zweites bakterielles Enzym, Sialidase, welches das Wachstum von Axonen bei Ratten mit Nervenverletzungen zu verdoppeln scheint 3. Forschende tragen nicht nur dazu bei, die angeborenen Inhibitoren des Axon-Wachstums zu überwinden, sondern arbeiten auch daran, jene 37 grundlegenden biologischen Vorgänge zu ermitteln, die Axonen zum Wachsen und Eingehen richtiger Verbindungen veranlassen. Drei Forschungsgruppen berichteten im Jahr 2006 über erste Ergebnisse auf diesem Gebiet. Forschende arbeiten daran, jene grundlegenden biologischen Vorgänge zu ermitteln, die Axonen zum Wachsen und Eingehen richtiger Verbindungen veranlassen. Yuqin Yin und Larry Benowitz vom Children’s Hospital Boston berichteten in Nature Neuroscience, sie hätten einen natürlich vorkommenden Wachstumsfaktor, Oncomodulin, entdeckt, der bei Ratten mit einer Verletzung des Sehnervs die Regeneration der Nerven um das fünf- bis siebenfache verstärkte 4. Aus dem Laboratorium von Samuel Pfaff im Salk Institute stammt einem Bericht in Neuron zufolge der Nachweis, dass ein Wachstumsfaktor anderer Art, der Fibroblast-Wachstumsfaktor, wachsende Axonen dazu bringt, sich wieder mit den richtigen Zielzellen in den Muskeln zu verbinden 5. Und Forschende unter der Leitung von Paul Forscher in Yale schrieben in Nature Cell Biology, sie hätten neue Funktionen des molekularen „Motor“-Proteins Myosin-II entdeckt, das an der Spitze von Axonen die Steuerung des Nervenwachstums unterstützt 6. Diese Berichte geben weiteren Aufschluss darüber, wie Vorgänge, die an der Entwicklung des Nervensystems beteiligt sind, zur Regeneration von Nerven nach einer Verletzung genutzt werden könnten. 38 Andernorts, an der Case Western Reserve University, verwendete eine Forschungsgruppe unter der Leitung von Jerry Silver Chondroitinase ABC zusammen mit einer „neuralen Brücke“, um in einem Rattenmodell das Nachwachsen von Axonen über eine Rückenmarkverletzung hinweg zu erleichtern. Als erstes transplantierte das Team ein Segment des Ischiasnervs des Tieres in die durch die Verletzung entstandene Lücke. Dieses Transplantat bildete eine Brücke, über die neu sprossende Axonen wachsen konnten. Dann wurde mittels einer implantierten Pumpe eine konstante Dosis des Enzyms Chondroitinase ABC zugeführt, was das Aussprossen förderte und eine weitere Narbenbildung am Ort der Verletzung verhinderte. Bei diesen Ratten kam es zu einer entscheidenden Verbesserung der Mobilität – dies im Vergleich zu Ratten, welche zwar derselben Prozedur unterzogen wurden, jedoch statt Chondroitinase ABC eine inaktive Kochsalzlösung erhielten. Bei letzteren zeigte sich weder ein neues Wachstum von Axonen noch eine verbesserte Mobilität. Die Ergebnisse erschienen im Journal of Neuroscience 7. Diese ersten Berichte über Tiermodelle von Rückenmarkverletzungen helfen mit, mögliche Verfahren zu bestimmen, die eines Tages bei Menschen Anwendung finden könnten. Schädigungen des Nervensystems Unter der Leitung von Douglas Kerr verfolgten Forschende an der Johns Hopkins University einen ähnlichen Ansatz. Sie transplantierten Motoneuronen in Tiere mit einer Rückenmarkverletzung und behandelten dann dieses Gebiet mit einem Cocktail von Substanzen, der jene Signale neutralisieren sollte, die das Wachstum von Axonen verhindern. Schliesslich infundierten sie einen Nervenwachstumsfaktor, der Axone dazu veranlasst, die richtigen Verbindungen einzugehen. Auf diese Weise, so ein Bericht in Annals of Neurology, erreichten sie eine teilweise Wiederherstellung der Funktionsfähigkeit der gelähmten Tiere 8. Hirnschlagforschung Dank Medikamenten zur Senkung der beiden hauptsächlichsten Risikofaktoren – Bluthochdruck und Cholesterin – traten seit ein paar Jahrzehnten jedes Jahr drastisch weniger neue Hirnschläge auf. In den Fällen, in denen der Hirnschlag infolge eines Blutgerinnsels erfolgt (ischämischer Hirnschlag), hilft ein Gewebe-Plasminogen-Aktivator (tissue plasminogen activator, tPA) – vorausgesetzt er wird innert dreier Stunden nach Eintritt des Schlaganfalls verabreicht – das Gerinnsel aufzulösen und den Schaden möglicherweise auf ein Minimum zu begrenzen. Allerdings wird tPA heute viel zu selten zur Akutbehandlung eingesetzt; dies liegt zum Teil daran, dass nur wenige der dafür in Frage kommenden Kranken innerhalb der erforderlichen drei Stunden nach Einsetzen der ersten Symptome in eine Spezialstation für Schlaganfall gelangen. Daten eines den ganzen Bundesstaat umfassenden Hirnschlag-Registers in Minnesota ergaben, dass nur 2% der Kranken mit einem Blutgerinnsel tPA erhielten. Von den Kranken, die kein tPA erhielten, waren 41% erst nach dem dreistündigen therapeutischen Fenster im Spital angelangt, und weitere 38% konnten nicht genau angeben, wann die Symptome eingesetzt hatten. Mathew Reeves von der Michigan State University leitete diese Minnesota-Register-Studie, über die in Neurology berichtet wurde 9. Diese Untersuchungen unterstreichen die Notwendigkeit, Therapien zu entwickeln, welche selbst dann wirksam zur Verbesserung der Hirnfunktion und zur Genesung beitragen, wenn sie nicht innerhalb von drei 39 Stunden nach dem Einsetzen eines ischämischen Hirnschlags durchgeführt werden. In erster Linie geht es weiterhin darum, Gerinnsel auflösende Mittel mit einem grösseren Zeitfenster zu entwickeln. In diese Richtung weisen Resultate einer klinischen Studie mit einem neuroprotektiven Medikament zur Eindämmung der unmittelbar nach einem ischämischen Hirnschlag auftretenden Hirnschädigungen. Bemühungen um die Entwicklung neuroprotektiver Medikamente gab es zwar schon seit zwei Jahrzehnten, doch ist das Präparat NXY-059 das erste Medikament, das gemäss den neuen Expertenrichtlinien zur Förderung der klinischen Hirnschlagforschung entwickelt wurde. Wenn das Medikament innert sechs Stunden nach einem ischämischen Schlaganfall verabreicht wurde, war eine Behinderung 90 Tage nach dem Hirnschlag seltener. Warren Wasiewski von der Western Infirmary in Glasgow, Schottland, der Versuchsleiter dieser Multisite-Studie, über die das New England Journal of Medicine berichtete, hielt jedoch fest, dass bezüglich der neurologischen Funktion keine Verbesserungen beobachtet wurden 10. In erster Linie geht es also weiterhin darum, Gerinnsel auflösende Mittel mit einem grösseren Zeitfenster zu entwickeln. Hirntumoren Gliome, tödliche Hirntumoren, sind therapieresistent, und im Allgemeinen sterben die Betroffenen innerhalb von zwei Jahren nach der Diagnose. Zu Entstehung, Prävention und Behandlung dieser Tumoren, gibt es erst wenige wissenschaftliche Anhaltspunkte. Die im Rahmen der neurowissenschaftlichen Grundlagenforschung vorgenommenen Untersuchungen konzentrierten sich weitgehend auf die Beziehung von Stammzellen und Tumorzellen und schlossen sich an frühere Forschungsarbeiten an mit der Frage, ob Stammzellen Substanzen bilden, die das Krebswachstum fördern. Jeremy Rich und Mitarbeitende an der Duke University beschrieben in Cancer Research eine spezifische Art von Gliomzellen, die sie wegen ihrer gemeinsamen Merkmale mit normalen Stammzellen als „stammzellartige Gliom-Krebszelle“ bezeichneten 11. 40 Die Forschenden untersuchten, wodurch diese Art von Gliomzellen das Tumorwachstum verstärkt. Sie entdeckten, dass diese Zellen grosse Mengen einer natürlichen Substanz, des so genannten vaskulären endothelialen Wachstumsfaktors (vascular endothelial growth factor, VEGF), Aufgrund von wissenschaftlichen Arbeiten, die unter der Leitung von Howard Fine am National Institute of Neurological Disorders and Stroke sowie am National Cancer Institute durchgeführt und in Cancer Cell vorgestellt wurden, geht man inzwischen davon aus, dass ein als StammzellFaktor (stem cell factor, SCF) bezeichneter Wachstumsfaktor ganz entscheidend zum Tumorwachstum beiträgt 12. Wie VEGF scheint auch SCF die Progression von Krebs dadurch anzukurbeln, dass es für eine lokale Umgebung sorgt, welche die Bildung von Blutgefässen anregt. Eine wichtige therapeutische Strategie besteht darin, herauszufinden, wie man das Wachstum von Blutgefässen rund um einen Tumor hemmen und so die Tumoren bezüglich Blut und Sauerstoff aushungern kann. Schädigungen des Nervensystems produzieren; dieser unterstützt die Bildung von Blutgefässen, welche Sauerstoff und Nährstoffe zu den Gliomzellen bringen und so deren Wachstum und Wuchern fördern. Gegenwärtig wird auch ein allfälliger Einsatz von Stammzellen zur Behandlung von Gliomen untersucht. Ein von Arturo Alvarez-Buylla geleitetes Team an der University of California, San Francisco, schrieb in Neuron, ein Signalmolekül, das die Entwicklung von Hirnzellen bei Erwachsenen reguliert, verursache, wenn es übermässig angeregt werde, bei Mäusen ein invasives tumorartiges Wachstum; werde diese Stimulation aufgehoben, so bilde sich der Tumor zurück 13. Demnach könnte die Blockierung der Signalwege eine mögliche Behandlungsstrategie zur Verhinderung von bösartigen Gliomen darstellen. 41 Neuroethik Placebos in klinischen Versuchen 44 Intimsphäre des Gehirns 44 Neu entstehende Technologien und das menschliche Gehirn 45 Abstufungen nicht bewusster Zustände 48 43 M it der Gründung der Gesellschaft für Neuroethik im Jahr 2006 hat das Gebiet der Neuroethik eine klarere Gestalt angenommen. Die von hervorragenden Mitgliedern aus den Bereichen Wissenschaft, Jurisprudenz und Ethik gegründete Gesellschaft betreibt eine Webseite, www.neuroethicssociety.org sowie zwei „Partner-Publikationen“, das American Journal of Bioethics und das Journal of Cognitive Neuroscience. In vier Hauptbereichen der Neuroethik kam es dieses Jahr zu entscheidenden (natürlich von grossen Diskussionen begleiteten) Fortschritten. Es ging dabei um Interventionen bei Affekt- und Verhaltensstörungen, die Intimsphäre des Gehirns, die Auswirkungen neu aufkommender Technologien, sowie subtile Veränderungen im Verständnis von nicht bewussten Hirnzuständen, etwa dem dauerhaften vegetativen Zustand. Placebos in klinischen Versuchen Dass etwa in klinischen Versuchen Placebos zum Einsatz kommen, führt zu ethischen Bedenken. Eine im British Journal of Psychiatry veröffentlichte Studie von Sumant Khanna, in der etwa zwölf Dutzend manisch Kranke anstelle einer Behandlung mit dem gebräuchlichen antipsychotisch wirkenden Medikament Risperidon ein Placebo erhielten, löste kürzlich eine Debatte aus 1. Wie Ganapati Mudur im British Medical Journal festhielt, gab es Ärzte, welche die Gültigkeit der von den Versuchsteilnehmenden abgegebene Einverständniserklärung bezweifelten 2. Solche Versuche werfen die Frage auf, ob Gemütskranke überhaupt in der Lage sind, eine Einverständniserklärung abzugeben. Intimsphäre des Gehirns Die Bemühung um zunehmend ausgeklügelte bildgebende Verfahren stellt alte Ansichten über den Geist, z. B. die Unantastbarkeit der unausgesprochenen Gedanken eines Menschen, in Frage. Unternehmerische Forschende haben Lügendetektoren entwickelt, die auf funktioneller Magnetresonanz-Tomographie (functional magnetic resonance imaging, fMRI) basieren und angeblich eine grössere Treffgenauigkeit bieten als die herkömmlichen Polygraphen, welche die Reaktionen des sympathischen Nervensystems messen. 44 In einer fMRI-Studie, bei der die Abklärung einer Schiesserei in einem Spital simuliert wurde, konnten Feroze Mohamed und Mitarbeitende acht Hirnregionen bestimmen, die während des Lügens signifikant aktiver Neuroethik waren als in einer neutralen Situation und zwei Regionen, die während einer wahrheitsgemässen Aussage aktiver waren als in einer neutralen Situation. Sie veröffentlichten ihre Arbeit in Radiology 3. Zurzeit halten sich die meisten Forschenden der Neurowissenschaft mit ihren Ansichten zurück, aber ein Leitartikel in Nature fordert die neurowissenschaftliche Gemeinschaft dringend auf, Bedenken laut und deutlich zu äussern und sich damit auf eine lange öffentliche Diskussion sowohl über die ethischen Implikationen dieser Technologien als auch über die grundsätzliche Bedeutung der Intimsphäre einzulassen 4. Feroze Mohamed und Mitarbeitende konnten acht Hirnregionen bestimmen, die während des Lügens signifikant aktiver waren als in einer neutralen Situation und zwei Regionen, die während einer wahrheitsgemässen Aussage aktiver waren als in einer neutralen Situation. Dank einer neuen Technik zur Prüfung von Neuroimaging-Daten, der so genannten Mustererkennung, kann die Wissenschaft recht genaue Aussagen darüber machen, was eine Person betrachtet, noch bevor dies der Person selbst bewusst ist. Zwar könnte diese Möglichkeit die beunruhigende Vorstellung des Gedankenlesens heraufbeschwören, doch gelten laut einem Leitartikel in Nature Neuroscience für die Bemühungen, mittels Mustererkennung Lügen aufzudecken, dieselben Einschränkungen wie für den herkömmlichen Polygraphen – dass etwa emotionale Antworten das Signal verrauschen. Die Relevanz von Mustererkennungstechniken dürfte eher im Bereich der Grundlagenforschung liegen, denn sie wird nicht nur erkennen lassen, „wo“ Informationsverarbeitung im Gehirn stattfindet sondern auch „wie“ 5. Interessant ist auch der Versuch, biologische Marker zu identifizieren, etwa Hirnanomalien oder spezifische Genmutationen, die auf eine Neigung zu Gewalttätigkeit schliessen lassen. Eine durchdachte Übersichtsarbeit von Nigel Eastman und Colin Campbell in Nature Reviews Neuroscience wirft die Frage auf, ob solche Marker als Kausalität im juristischen Wortsinn angesehen werden können und, falls ja, ob eine Person mit einem oder mehreren solchen Biomarkern zu Recht vorsorglich interniert werden darf, um die Öffentlichkeit vor einem künftigen Schaden zu schützen 6. Neu entstehende Technologien und das menschliche Gehirn Eine einzigartige klinische Studie, bei der Gehirn und Computer zusammenwirken, betrifft BrainGate, eine von Matt Nagle benutzte, massentwickelte 45 Arm- und Handprothese – die Titelgeschichte der Ausgabe von Nature am 13. Juli 2006 (siehe auch das Kapitel Schädigungen des Nervensystems, S. 36). Nagle, der infolge einer verletzungsbedingten Rückenmarksdurchtrennung an Tetraplegie leidet, betätigt das Gerät nur mit der Kraft seiner Gedanken – d. h. durch Hirnsignale, welche seine Absicht verkörpern, den Arm auszustrecken, seine Hand zu öffnen und zu schliessen, usw. Die von Nagles bewusstem Gehirn ausgesandten Bewegungssignale werden von einer in den motorischen Kortex implantierten, 96 Elektroden umfassenden Schaltmatrix aufgenommen, übersetzt und weitergeleitet, und steuern so die Bewegung der Prothese. Im Gegensatz zu verschiedenen anders gearteten technischen Hilfsmitteln für Kranke mit einer multiplen Lähmung – eines wird z. B. durch elektrische Aktivität auf der Kopfhaut gesteuert, ein anderes basiert auf Augenbewegungen – erfordert diese neuromotorische Prothese weder ein monatelanges Training noch die volle Aufmerksamkeit des Benützers. Wie Leigh Hochberg und seine Mitarbeitenden in Nature berichten, kann Nagle ein Gespräch führen, während er fingierte Emails öffnet oder die Robothand bzw. den Arm bewegt 7. Wenn es gelingt, die Geschwindigkeit der Informationsverarbeitung und damit die Leistungsfähigkeit künftiger neuromotorischer Prothesen zu steigern, werden sich neue Fragen stellen: Wem sollen sie zugute kommen und nach welchen Kriterien (therapeutisch, finanziell, vielleicht sogar psychosozial) erfolgt die Auswahl? Stephen Scott weist in derselben Ausgabe darauf hin, dass die Verwendung solcher Prothesen aufgrund von im Gehirn bereits bestehenden Informations-Rückkoppelungs-Schaltkreisen die Organisation der Hirnsignale subtil verändern könnte, so dass sich Gehirn und von Menschenhand angefertigte Geräte immer besser vereinen – eine hoffnungsvolle Perspektive für Menschen mit einer Lähmung 8. 46 Andere technologische Fortschritte im Bereich der Bildgebung des Gehirns führen zu Bedenken bezüglich „zufälliger Befunde“, unerwarteter Hinweise auf eine mögliche Krankheit, die im Verlauf der Erforschung eines nicht damit zusammenhängenden Themas entdeckt werden. Berichte über zufällige Befunde häufen sich. Eine Arbeitsgruppe, der etwa 50 Fachleute aus den Bereichen medizinische Bildgebung, biomedizinische Ethik und Rechtswissenschaft angehörten, behandelte in Science die Frage, ob Forschende verpflichtet seien, den an ihren Studien Teilnehmenden solche zufälligen Befunde mitzuteilen und, falls ja, unter welchen Umständen 9. Neuroethik Die Antworten sind alles andere als selbstverständlich. Einige der möglicherweise entdeckten Krankheiten, können sehr schwerwiegend sein, und falls in einer Versuchsanordnung häufig falsch positive Resultate vorkommen, erhielte man erst durch eine zweite, diesmal von einem diagnostischen Radiologen ausgewertete Aufnahme ein sicheres Resultat. Angenommen zufällige Befunde würden auf einem Forschungsscan sichtbar: Hat die Versuchsperson ein Recht darauf, diese zu erfahren, oder ein Recht, sie „nicht“ zu erfahren, oder beides? Die Arbeitsgruppe fordert alle Forschenden, die mit Bildgebung des Gehirns arbeiten, dringend dazu auf, die Entscheidung über den Umgang mit zufälligen Befunden im Voraus zu treffen. Das entsprechende Protokoll sollte im Rahmen der Einverständniserklärung (informed consent) figurieren. Natürlich kann es sein, dass künftige Erfahrungen mit zufälligen Befunden zu neuen Empfehlungen führen, doch wird als Richtlinie weiterhin gelten, dass es darum geht, die wissenschaftliche Integrität und das öffentliche Vertrauen sicherzustellen. Abstufungen nicht bewusster Zustände Im Jahr 2006 lenkten Forschungsarbeiten die Aufmerksamkeit auf ungewöhnliche Patienten mit schweren Hirnverletzungen. Im Journal of Clinical Investigation beschrieben Henning Voss, Nicholas Schiff und weitere Forschende aus New York, New Jersey und Neuseeland die spontane Genesung eines Mannes, der sich nach einem Autounfall 19 Jahre lang in einem minimal bewussten Zustand befunden hatte und unfähig war, sich zu bewegen oder zu sprechen 10. Sein Zustand hatte sich im Laufe der Jahre schrittweise gebessert; dennoch waren die Wiedererlangung des Bewusstseins, der fliessenden Sprache, der Kognition und der Bewegungsfähigkeit von drei der vier Extremitäten beispiellos. Die Forschenden untersuchten sein Gehirn mittels eines nicht-invasiven bildgebenden Verfahrens, der Diffusions-Tensor-Magnetresonanz-Tomographie. Sie fanden Hinweise auf ein Nachwachsen von Axonen, welche die Bildung neuer Verbindungen im Gehirn ermöglichten. Ein zweiter im selben Artikel beschriebener Patient, der (ebenfalls infolge eines Autounfalls) mehr als ein Jahr in einem dauerhaften vegetativen Zustand und weitere vier Jahre in einem minimal bewussten Zustand zugebracht hatte, zeigte zwar weder eine vergleichbare klinische Besserung noch ein Nachwachsen von Axonen, doch könnte dies noch geschehen. Die Autoren fordern weitere Bildgebungs-Studien mittels Diffusions-Tensor-Bildgebung und Positronen-Emissions-Tomographie, die bereits kurz 47 Patientin Gesunde Versuchspersonen Tennis Spielen fMRI-Scans Räumliche Vorstellung Hirnaktivität bei vegetativem Zustand Als Reaktion auf gesprochene Aufforderungen, sie solle sich vorstellen, Tennis zu spielen oder durch ihr Haus zu gehen, reagierte eine Patientin in einem vegetativen Zustand mit Aktivitäten in denselben Hirnregionen wie gesunde Versuchspersonen. nach einer Hirnverletzung einsetzen sollten und weitere Einblicke in das hoffnungsvolle Phänomen einer langfristigen Neuverschaltung des Gehirns vermitteln könnten. Auch in einer von Adrian Owen geleiteten Forschungsarbeit, die in Science erschienen ist, kam es zu einer viel versprechenden Beobachtung bezüglich der Hirnaktivität einer Patientin in einem dauerhaften vegetativen Zustand (persistent vegetative state, PVS) 11. Bei einer teilnahmslos wirkenden jungen Frau, die nach einem Autounfall fünf Monate in einem dauerhaften vegetativen Zustand verbracht hatte, machte man ein fMRIScan des Gehirns; es belegte eindeutig, dass sie verschiedene komplizierte kognitive Aufgaben ganz normal zu bewältigen vermochte. 48 Als Reaktion auf gesprochene Sätze, selbst auf solche, die doppeldeutig klingende Wörter enthielten („The ,creak‘ came from a beam in the ,ceiling“. „Das ,Knacken‘ stammte von einem ,Träger‘ in der ,Decke“) entsprach die Aktivität in den Sprachzentren ihres Gehirns jener von gesunden Neuroethik Versuchspersonen. Darüber hinaus reagierte sie auf die gesprochene Aufforderung, sich vorzustellen, sie würde Tennis spielen oder durch die Zimmer ihres Hauses gehen, mit einer normalen Hirnaktivität. Diese Befunde sind besonders beeindruckend, da sie sehr präzise anzeigen, dass vielen Aspekten dessen, was wir gemeinhin als Bewusstsein bezeichnen, eine Hirnaktivität zugrunde liegt: Intention, Körpergefühl, einem (vorgestellten) sich bewegenden Gegenstand nachblicken, und die Erinnerung an eine (vorgestellte) bekannte Umgebung. Bereits früher hatte die Neurowissenschaft angenommen, Kranke in einem dauerhaften vegetativen Zustand verfügten über „Inseln“ von erhalten gebliebenen Funktionen, die mittels herkömmlicher klinischer Methoden nicht aufgespürt werden können. Die geistige Mobilität dieser immobilen Kranken scheint zu bestätigen, dass solche „Inseln“, wie vermutet, vorkommen können, und lässt hoffen, dass irgendeine Art von Fenster zu diesen Schlupfwinkeln von nicht bewussten Zuständen gefunden wird. Diese Befunde sind besonders beeindruckend, da sie sehr präzise anzeigen, dass vielen Aspekten dessen, was wir gemeinhin als Bewusstsein bezeichnen, eine Hirnaktivität zugrunde liegt. Da es nur bei einer winzig kleinen Zahl von Kranken zu einer Besserung kam, stellt sich die Frage, ob man bei allen Kranken versuchen soll, mittels kostspieliger experimenteller Eingriffe (z. B. tiefer Hirnstimulation oder transmagnetischer Stimulation) eine gewisse Kommunikationsfähigkeit wiederherzustellen. Das Bestreben der gegenwärtigen Forschung herauszufinden, ob und unter welchen Umständen solche Interventionen wirksam sein werden, könnte mithelfen, jene Kranken zu eruieren, die wohl am ehesten davon profitieren. 49 Neuroimmunologische Erkrankungen Multiple Sklerose 52 Ziel eines Autoimmun-Angriffs 53 Die Immunreaktion unter Kontrolle halten 54 Das Immunsystem und die Alzheimersche Krankheit 55 Neuropathischer Schmerz 58 Plastizität 58 Depressionen können Entzündungen hervorrufen 59 51 D ie Beziehung zwischen dem Immunsystem des Menschen und seinem Gehirn ist oft getrübt; das Gehirn ist ein „immunprivilegiertes“ Gebiet, in dem nur eine Art von Immunzellen, die so genannte Mikroglia, vorkommt. Bakterien, Viren und Toxine können ins Gehirn eindringen, indem sie die Blut-Hirn-Schranke durchbrechen, jene überaus dichte Schicht von Zellen in der Wand von Blutgefässen, welche den Übertritt von Substanzen aus dem Blut ins Gehirn kontrolliert. Sobald dies geschieht, strömen Immunzellen ins Gehirn, um die Eindringlinge zurückzuschlagen. Manchmal jedoch verwechseln Immunzellen irrtümlicherweise normales Hirngewebe mit Eindringlingen und greifen es an. Ein Beispiel dafür ist die Multiple Sklerose, bei der Immunzellen ausrasten und das unentbehrliche, isolierende Myelin angreifen, das die Axonen in Gehirn und Zentralnervensystem umgibt. Das Immunsystem setzt auch einen Angriff auf die Amyloid-Proteine in Gang, welche sich im Gehirn von Alzheimer-Kranken bilden; dieser Angriff ist so aggressiv, dass er eine für Neuronen schädliche Entzündung hervorruft. Bei der Parkinsonschen Krankheit gibt es möglicherweise einen ähnlichen Vorgang (siehe Bewegungsstörungen und andere Störungen der Motorik, S. 30). Worauf es beruht, dass bei Multipler Sklerose gewisse Immunzellen so umgewandelt werden, dass sie Myelin angreifen, wurde 2006 entdeckt und zählt zu den bedeutendsten Fortschritten auf dem Gebiet der Neuroimmunologie dieses Jahres. Weitere Forschungsarbeiten untersuchten, wie das Immunsystem eine durch die Alzheimersche Krankheit hervorgerufene Degeneration verhindern oder sogar rückgängig machen könnte. Multiple Sklerose 52 Bei Multipler Sklerose führen die wiederholten Angriffe des Immunsystems zu Lücken in der Myelinhülle von Nervenzellaxonen und damit zu einer Unterbrechung der neuralen Signalübermittlung. Diese Unterbrechung ruft eine Reihe von Symptomen hervor. Bis vor kurzem nahm man an, solche Angriffe auf das Myelin gingen von fehlerhaften ImmunT-Helferzellen (so genannten TH1-Zellen) aus, welche das Immunsystem normalerweise über in die Zelle eingedrungene Bakterien oder Viren informieren. 2005 haben Forschende jedoch herausgefunden, dass eine andere T-Helferzelle, TH17, als Auslöser eines Autoimmunangriffs auf Myelin eine entscheidende Rolle spielt. Eine unter der Leitung von Neuroimmunologische Erkrankungen Estelle Bettelli durchgeführte und in Nature veröffentlichte Studie von Forschenden an der Harvard Medical School in Boston ergab, dass TH17Zellen entstehen, wenn unreife T-Zellen der Kombination von zwei anderen Molekülen ausgesetzt sind 1. Eines dieser Moleküle ist ein signalgebendes Protein, das als transformierender Wachstumsfaktor Beta (transforming growth factor-beta, TGF-beta) bezeichnet wird. Das andere ist ein entzündungsförderndes Immunmolekül, das so genannte Interleukin-6 (IL-6), das von T-Zellen freigesetzt wird. IL-6 defiziente Mäuse hatten keine TH17-Zellen und entwickelten auch keine Mäuseversion der Multiplen Sklerose. Yoichiro Iwakura und Harumichi Ishigame entdeckten zudem, dass das Molekül Interleukin-23 (IL-23), ein Wachstumsfaktor, unreife T-Zellen in TH17-Zellen umwandelt 2. Ihre im Journal of Clinical Investigation veröffentlichte Arbeit zeigte, dass sie im Tiermodell die Entwicklung der Multiplen Sklerose sowie einer weiteren Auoimmunerkrankung, der entzündlichen Darmerkrankung (inflammatory bowel disease), durch die Blockierung von IL-23 entscheidend unterdrücken konnten. Zusammengefasst lassen diese beiden Studien darauf schliessen, dass Therapien, welche die Umwandlung von Immun-T-Zellen in TH17-Zellen blockieren, zumindest bei einigen Autoimmunerkrankungen, einschliesslich der Multiplen Sklerose, wirksam sein könnten. Ziel eines Autoimmun-Angriffs Bei einer weiteren entzündlichen Erkrankung, der Neuromyelitis optica, greift das Immunsystem das Myelin um den Sehnerv an, was zu teilweiser oder völliger Blindheit führt. Zwar wird Neuromyelitis optica manchmal irrtümlich für eine erste Manifestation von Multipler Sklerose gehalten, doch hat man kürzlich entdeckt, dass ein NMO-IgG genannter Antikörper, der im Falle von Neuromyelitis optica irrtümlicherweise das Myelin angreift, bei Kranken mit Multipler Sklerose nicht vorkommt. Dieser Befund lässt darauf schliessen, dass es sich bei Neuromyelitis optica um eine eigenständige Krankheit handelt. Forschungsarbeiten deuten auch daraufhin, dass NMO-IgG zur transversen Myelitis beiträgt, einer Krankheit, bei der das Immunsystem das Myelin um Axonen im Rückenmark angreift und Bewegungsstörungen oder Lähmungen verursacht. Brian Weinshenker und Mitarbeitende an der Mayo Clinic berichteten in Annals of Neurology, ca. 40% der Kranken mit einer weit reichenden transversen Myelitis seien positiv auf NMO-IgG 53 getestet worden, und bei mehr als der Hälfte dieser positiv getesteten sei es binnen Jahresfrist zu einem Rückfall gekommen 3. Personen ohne diesen Antikörper im Blut hatten keinen Rückfall. Vor dieser Entdeckung konnte man nicht feststellen, bei welchen dieser Kranken, einschliesslich jener mit transverser Myelitis, das Risiko eines neuerlichen Autoimmunangriffs auf das Rückenmark bestand. Durch den Nachweis dieses Biomarkers lassen sich nun die Rückfallgefährdeten identifizieren, und man kann den Einsatz von immunsuppressiven Behandlungen erwägen. Aber wogegen richtet sich der Angriff des NMO-IgGAutoimmun-Antikörpers? Unter der Leitung von Vanda Lennon entdeckten Forschende an der Mayo Clinic im Jahr 2006, dass NMO-IgG fälschlicherweise auf Aquaporin-4 abzielt, ein kürzlich entdecktes Protein im Zentralnervensystem, dank dem Wasser in Zellen ein- und ausströmen kann 4. Aquaporin-4 wird im Gehirn vor allem durch sternförmige Zellen, so genannte Astrozyten gebildet, welche die Blut-Hirn-Schranke verstärken, schädliche Substanzen im Blut beseitigen und davon abhalten ins Gehirn zu gelangen. Hohe Konzentrationen von Aquaporin-4 kommen im Sehnerv, im Rückenmark und in bestimmten Teilen des Hirnstamms vor – lauter Ziele, die das Immunsystem von Kranken mit Neuromyelitis optica angreift. Dies deutet darauf hin, dass an diesen Orten möglicherweise NMO-IgG aus Blutgefässen durchsickert, dort auf Aquaporin-4 stösst und es angreift. Jedenfalls ist die Erkenntnis, dass NMO-IgG einen zuverlässigen Marker für diese Krankheit darstellt, für die Diagnose der Neuromyelitis optica ein bedeutender Fortschritt. Die Immunreaktion unter Kontrolle halten Eine unkontrollierte Immunreaktion im Gehirn kann Multiple Sklerose, Lupus und andere Erkrankungen verursachen – wodurch aber gerät das Immunsystem ausser Kontrolle? Eine unkontrollierte Immunreaktion im Gehirn kann Multiple Sklerose, Lupus und andere Erkrankungen verursachen – wodurch aber gerät das Immunsystem ausser Kontrolle? 54 Chemokine sind Signalträger zwischen Zellen und regulieren den Einsatz der Leukozyten genannten Immunzellen. Ein von Richard M. Ransohoff geleitetes Team berichtete in Nature Neuroscience, um die Immunreaktion im Gehirn unter Kontrolle zu halten, seien Fraktalkine, eine ungewöhnliche c e b d f CX3CR1+/– CX3CR1–/– Neuroimmunologische Erkrankungen a Schwierigkeiten mit der Kontrolle im Immunsystem Mäuse, denen das Gen für das Fraktalkin-Rezeptorprotein fehlte (unten) ein Protein, das auf Entzündungszellen des Gehirns vorkommt, entfalten mit der Zeit eine grössere Aktivität der Mikroglia (von links nach rechts), was im Mäusemodell menschlicher Krankheiten eine grössere Schädigung von Neuronen bewirkt. Variante von Chemokinen, unentbehrlich 5. Durch die Freisetzung von Fraktalkinen verhindern Immunzellen des Gehirns (also die Mikroglia), dass andere an einer Immunreaktion beteiligte Zellen überreagieren. Ransohoff entdeckte, dass Mäuse, denen das Gen für Fraktalkine fehlte, zwar normal aussahen, jedoch während der heftigen Entzündungsreaktionen in Mausmodellen menschlicher Erkrankungen, einschliesslich der Parkinsonschen Krankheit und der Amyotrophen Lateralsklerose (LouGehrig-Krankheit), wesentlich grösseren Schaden nahmen. Das Immunsystem und die Alzheimersche Krankheit Das Immunsystem hält die Beta-Amyloid-Partikel, die sich im Gehirn von Alzheimerkranken anhäufen, für fremde Eindringlinge, die zerstört werden müssen. Daraus resultiert eine Entzündung, welche die Krankheit sicher verschlimmert, sie möglicherweise sogar verursacht. Ein klinischer Versuch mit einem therapeutischen Impfstoff zur Dezimierung dieser Plaques musste im Jahr 2002 abgebrochen werden, da die Immunreaktion bei einigen Teilnehmenden eine schwere Hirnhautentzündung hervorrief. 55 Aber laut Michal Schwartz und Mitarbeitenden am Weizmann Institute of Science in Israel lassen sich die immunen T-Zellen, die diese Entzündung hervorriefen, dennoch wirksam und gefahrlos als Verbündete im Kampf gegen diese Plaques einsetzen. Die Forschenden beschrieben in Proceedings of the National Academy of Sciences, sie hätten Mäuse, die zur Entwicklung von Amyloiden Plaques gezüchtet wurden, durch die Gabe von Glatiramer-Acetat (GA), einem zur Behandlung der Multiplen Sklerose verwendeten Immunsystem-Modulator (Handelsname „Copaxon“), therapeutisch immunisiert. Diese Therapie, welche T-Zellen anregt, reduzierte die Plaque-Belastung der Mäuse und förderte das Wachstum von Zellen im Hippokampus, was zu einer Verbesserung von Gedächtnis und Lernfähigkeit führte 6. Die Forschenden führen die Wirkung dieser Therapie darauf zurück, dass Mikrogliazellen angeregt wurden, ein als Insulinartiger Wachstumsfaktor-1 (insulin-like growth factor-1, IGF-1) bezeichnetes Hormon zu exprimieren und nicht das destruktive Zytokin, den so genannten Tumor-Nekrose-Faktor-Alpha (TNF-alpha), der Entzündungen auslöst. Mittels einer derartigen Feinabstimmung der Immunreaktion könnten solche Therapien, wie die Autoren glauben, einen Angriff auf das Beta-Amyloid im Gehirn anregen, ohne eine destruktive Entzündung auszulösen. Schwartz wies ausserdem nach, dass ins Gehirn von Mäusen injizierte immune T-Zellen in gewissen Gebieten zur Neubildung von Neuronen beitragen, und zwar auch im Hippokampus, der durch die Alzheimersche Krankheit stark geschädigt wird. Sie und ihr Team verglichen die Gehirne von zwei Mäusegruppen in einer anregenden Umgebung voller Spielobjekte und neuartiger Gegenstände: normale Mäuse und Mäuse mit einem schweren kombinierten Immundefekt (severe combined immune deficiency, SCID), der bewirkte, dass die Tiere praktisch keine T-Zellen hatten. Bei den normalen Mäusen kam es zu einer kräftigen Neurogenese im Hippokampus, wo das Kurzzeitgedächtnis entsteht; bei SCID-Mäusen ohne T-Zellen war dies praktisch nicht der Fall. 56 Im Zusammenhang mit der Alzheimerschen Krankheit gab es 2006 einen weiteren, mit der Unterdrückung von Entzündungen einhergehenden, viel versprechenden Forschungsansatz. Im Rahmen einer sechsmonatigen von Edward Tobinick geleiteten Pilotstudie behandelten Forschende der University of California, Los Angeles, 15 Kranke mit einer mässigen bis schweren Alzheimerschen Krankheit mit Etanercept; diese Therapie wirkt Die wöchentlichen Injektionen führten bei den Teilnehmenden zu einer wesentlichen Verbesserung der mentalen Funktion. Die Studie bekräftigt die Theorie, wonach die Entzündung selbst entscheidend zur Demenz von Alzheimer-Kranken beiträgt und sich der mentale Abbau durch die Entzündungshemmung verlangsamen oder gar verhindern lässt. Eine Forschungsgruppe der Case Western Reserve University hält das Bestreben, die Alzheimersche Krankheit durch die Beseitigung der Plaques im Gehirn zu heilen, für grundsätzlich falsch. Forschende an der Case Western Reserve University stellten allerdings die weit verbreitete Hypothese in Frage, die für die Alzheimersche Krankheit charakteristischen amyloiden Plaques und die von ihnen ausgelöste Entzündung seien die Ursache der Erkrankung. Sie vermuten vielmehr, die Symptome rührten von oxidativem Stress, der erhöhten Produktion von Oxidantien her, welche Neuronen zerstöre. In einem Bericht in Current Alzheimer Research behaupten Hyoung-gon Lee, Mark Smith, George Perry und Mitarbeitende, die Plaques seien der Versuch des Gehirns, den oxidativen Stress zu mindern 8. Ihrer Ansicht nach ist das Bestreben, die Alzheimersche Krankheit durch die Eliminierung der Plaques im Gehirn zu heilen, völlig falsch. Sie halten es für sinnvoller, die Ursachen von oxidativem Stress zu identifizieren und ihren Einfluss zu vermindern. Neuroimmunologische Erkrankungen TNF-Alpha entgegen und hat sich bei der Unterdrückung arthritischer Entzündungen als wirksam erwiesen 7. Inzwischen entdeckten Forschende der Northwestern University Medical School unter der Leitung von Abdelhak Belmadani, dass Chemokine nicht nur Signale aussenden und Leukozyten regulieren, sondern ebenso die Migration von neuralen Vorläuferzellen an alle entzündeten Stellen im Gehirn regulieren – auch bei der sehr ausgedehnten Entzündung, die durch die Alzheimersche Krankheit verursacht wird. Sobald eine Entzündung Nervenzellen schädigt, aktivieren Astrozyten Chemokine; diese lenken dann adulte neurale Vorläuferzellen zur von der Entzündung betroffenen Stelle. Wie die Forschenden in Journal of Neuroscience schreiben, könnte dieser Befund zur Entwicklung von Medikamenten gegen Hirnschädigung führen, indem sie die Migration von neuralen Vorläuferzellen an die verletzte Stelle fördern, wo diese sich zu neuen Neuronen entwickeln könnten 9. Es wäre denkbar, dass dies zur Wiederherstellung eines durch Alzheimersche Krankheit geschädigten Hippokampus beitragen könnte. 57 Neuropathischer Schmerz Andere Forschungsarbeiten liessen eine Verbindung von Mikroglia und neuropathischem Schmerz erkennen, einer chronischen und oft qualvollen Erkrankung, bei der ein Schmerz noch lange fortdauert, nachdem die Verletzung, die Infektion oder das Toxin, das ihn ausgelöst hatte, beseitigt wurde (vgl. auch das Kapitel Schmerz, S. 62). Neuropathischer Schmerz kann als Folge einer Verletzung von „peripheren“ Nerven (jenen ausserhalb von Gehirn und Rückenmark) entstehen. Zu dieser abnormen Schmerzreaktion kommt es, wenn Mikrogliazellen, die einzigen in Gehirn und Rückenmark vorhandenen Immunzellen, aktiviert werden und einen aus dem Hirn stammenden neurotrophen Faktor freisetzen, der die Schmerzsignale zwischen Mikroglia und Neuronen verstärkt – dies die Ansicht eines von Michael Salter geleiteten Forschungsteams der University of Toronto. Dieser Vorgang bringt die normale Schmerzbekämpfung zum Erliegen, so dass Neuronen selbst ohne das Vorhandensein irgendwelcher schmerzhaften Stimuli überempfindlich sind. Die Forschenden stellten ihre Ergebnisse im European Journal of Physiology vor 10. Diese Forschungsarbeit lässt darauf schliessen, dass die Signal gebenden Komponenten der Mikrogliazellen viel versprechende therapeutische Angriffspunkte zur Linderung von chronischen peripheren Nervenschmerzen darstellen könnten. Andernorts, am Columbia University Medical Center, beantragten Forschende ein Patent zur Entwicklung von Medikamenten, die ein als Protein Kinase G (PKG) bekanntes Enzym ausschalten und dadurch chronischen Schmerz hemmen sollen. Wie sie in Neuroscience berichteten, führt PKG zu einer neuronalen Hypererregbarkeit, die bewirkt, dass andauernd Schmersignale erzeugt werden 11. Sie beobachteten, dass der Schmerz aufhörte, sobald das PKG ausgeschaltet wurde; das Enzym eignet sich somit ideal als Angriffspunkt für Medikamente. Plastizität 58 Solange das Gehirn jung ist, reagiert es mit einer kräftigen Neuverkabelung auf Verletzungen. Eine solche Neuverkabelung, die so genannte Plastizität, wird mit zunehmendem Alter langsamer; nun haben Forschende an der Harvard Medical School entdeckt, dass möglicherweise ein Protein des Immunsystems, der so genannte Paired-immunoglobulinlike receptor-B (PirB), im Verlauf der Zeit die Plastizität hemmt und Neuroimmunologische Erkrankungen Mikroglia vermittelt Schmerz Nach einer Verletzung peripherer Nerven wird die Mikroglia im dorsalen Horn des Rückenmarks aktiviert; dadurch werden Neuronen überempfindlich, und es kommt selbst dann zur Empfindung von Schmerz, wenn kein schmerzhafter Reiz vorhanden ist. dadurch Hirnverbindungen stabiler macht. Ein von Josh Syken geleitetes Team publizierte diese Ergebnisse in Science 12. Sie beobachteten, dass PirB-deprivierte Mäuse während ihres ganzen Lebens über eine grössere Fähigkeit zur Neuverkabelung verfügten. Würde man einen Weg finden, um PirB zu reduzieren, könnten Verbindungen zwischen Neuronen, die durch Rückenmarkverletzungen, Hirnschlag oder andere Traumen geschädigt wurden, möglicherweise leichter wiederhergestellt werden. Depressionen können Entzündungen hervorrufen Forschende der Emory University School of Medicine in Atlanta entdeckten, dass das Immunsystem von depressiven Männern, die in ihrer Jugend unter Stress gelitten hatten, bei Stress übertriebene Entzündungsreaktionen aufweist, was zur schlechten Prognose bei Entzündungskrankheiten beitragen könnte. Stresserlebnisse führen gewöhnlich zu einer erhöhten Produktion von Interleukin-6 (IL-6) und diese fördert Entzündungen. Unter der Leitung von Andrew H. Miller und Christine Heim unterzogen die Forschenden 28 Männer – bei der Hälfte von ihnen waren eine Major Depression und Stress in der Jugend diagnostiziert worden – einem Test. Nachdem sie 59 Aufgaben ausgeführt hatten, die Stress erhöhen, z. B. rechnen und öffentlich sprechen, wurden Blutproben entnommen und auf das Vorhandensein von IL-6 getestet. Wie die Forschenden im American Journal of Psychiatry berichteten, nahm IL-6 zwar bei allen Teilnehmenden zu, doch stiegen die Konzentrationen bei der depressiven Gruppe fast doppelt so stark an 13. Die Untersuchung liefert erste Hinweise auf einen Zusammenhang von Major Depression, jugendlichem Stress und Gesundheitszustand. 60 Schmerz Hauptschalter für chronische Schmerzen identifiziert 62 Die Erwartung von Schmerz kann ebenso schlimm sein, wie der Schmerz selber 63 Schmerzbekämpfung dank Schädlingsbekämpfung 64 „Emotionale Ansteckung“ bei Schmerz 65 Die Art eines Placebos wirkt sich aus 66 61 S chmerz stellt für Ärzte und Ärztinnen sowie für die Allgemeinheit gleichermassen ein enormes Problem dar; laut der gemeinnützigen Organisation Partners Against Pain belaufen sich der daraus resultierende Produktivitätsverlust und die Kosten im Gesundheitswesen auf jährlich insgesamt fast 100 Milliarden Dollars. Weltweit gelang es der Schmerzforschung im Jahr 2006, besser zu verstehen, was akuten und chronischen Schmerzen zugrunde liegt und wie man sie lindern kann. Eine Studie entdeckte einen „Hauptschalter“ für die Entwicklung von neuropathischen Schmerzen – eine chronische Art von Schmerzen, die sich grundlegend von akuten, verletzungsbedingten Schmerzen unterscheidet. Eine zweite ergab, dass es schlimmer sein kann, Schmerzen zu erwarten als sie tatsächlich zu erleiden. Auf der Suche nach einem Schädlingsbekämpfungsmittel stiessen Forschende auf einen Enzymhemmer, der möglicherweise Entzündungsschmerzen lindert und zugleich das mit Medikamenten wie Rofecoxib (Vioxx) verbundene Herzinfarktrisiko verringert. Kanadische Forschende stellten fest, dass Mäuse Empathie entwickeln und schmerzempfindlich werden, wenn sie beobachten, dass eine andere Maus Schmerzen hat. Ausserdem erkannten Forschende, dass die Art eines Placebos und die Umstände, unter denen es verabreicht wird, den Placeboeffekt bezüglich Schmerzempfindung steigern können. Hauptschalter für chronische Schmerzen identifiziert Charakteristisch für neuropathischen Schmerz, der von einer Verletzung „peripherer“ Nerven ausserhalb von Gehirn und Rückenmark herrührt, ist die Empfindung von chronischen einschiessenden oder brennenden Schmerzen. Auf die Behandlung mit Opiaten, den stärksten Schmerzmitteln, zu denen etwa Morphium, Codein und Oxycodon (mit dem Markennamen Oxycontin) zählen, spricht dieser Schmerztyp nur schlecht an. 62 In der Zeitschrift Neuron berichteten Forschende der Harvard Medical School, sie hätten einen „Hauptschalter“ für die Entwicklung neuropathischer Schmerzen gefunden 1. Dieser Schalter, das Runx1-Gen, wird nur in sensorischen Nervenzellen, den nozizeptiven Zellen, exprimiert, die mit Schmerzempfindung zu tun haben. Via Ionenkanäle, besondere Poren in der Membran von Nervenzellen, übersetzen diese Zellen schmerzhafte Stimuli in Nervensignale. Schmerz Die von Qiufu Ma geleitete Gruppe setzte „Knockout“-Mäuse (deren Runx1-Gen entfernt worden war) thermischen, mechanischen, entzündlichen und neuropathischen Reizen aus und nahm die Dauer, während der die Tiere daraufhin ihre Pfote entweder anhoben oder leckten, als Mass für die Schmerzreaktion. Die Runx1-defizienten Mäuse reagierten auf den mechanischen Schmerzreiz, nicht jedoch auf schmerzhafte thermische, neuropathische oder entzündliche Reize. Die Entwicklung ihrer Schmerzrezeptorzellen war beeinträchtigt, und die Ionenkanäle, die bekanntlich zur Empfindung von thermischen und neuropathischen Schmerzen nötig sind, waren nichtexistent. Wie die Forschenden festhielten, könnte dieser Befund weit reichende Implikationen für die Entwicklung neuer, besser wirksamer Behandlungsstrategien bei neuropathischem Schmerz haben, die möglicherweise darauf beruhen, dass man die Expression des Runx1-Gens bei Kranken mit chronischen Schmerzen ausschaltet. Die Erwartung von Schmerz kann ebenso schlimm sein, wie der Schmerz selber Während sie in der Arztpraxis auf eine Injektion warten oder einem schmerzhaften medizinischen Eingriff entgegensehen, denken manche Leute: Wenn es nur schon vorbei wäre. Egal, wie weh es tut!“ Die Wissenschaft hat heute möglicherweise eine Erklärung dafür: Für manche Menschen ist es ebenso schlimm, einen Schmerz zu erwarten, wie ihn tatsächlich zu erleiden. Ein Forschungsteam an der Emory University School of Medicine verwendete Hirnscans, um die Biologie der Furcht zu untersuchen; dabei stellte sich heraus, dass beinahe ein Drittel der Versuchspersonen, die bereit waren, sich einem Elektroschock auszusetzen, einen stärkeren Schock wählte, wenn dadurch die Wartezeit abgekürzt wurde und sie nicht auf einen schwächeren warten mussten. Die Freiwilligen wurden in einem Magnetresonanz-Tomographie-Gerät platziert und erhielten am Fuss eine Serie von 96 unterschiedlich starken Elektroschocks. Die meisten Freiwilligen zogen es vor, einem stärkeren Elektroschock ausgesetzt zu werden, wenn dafür die Wartezeit vor dem Schock kürzer war. Die von Gregory Berns und Mitarbeitenden in Science veröffentlichten Ergebnisse lassen erkennen, dass die meisten Versuchspersonen während 63 des Wartens den Schock fürchteten 2. Jene, die keinen Aufschub ertrugen und einen unverzüglichen und schmerzhafteren Schock wählten, wurden als „extrem Furchtsame“ eingestuft, während die „mässig Furchtsamen“ mit der Aussicht auf einen leichteren Schock einen Aufschub ertrugen. Die Magnetresonanz-Scans zeigten, dass Teile der „Schmerzmatrix“ des Gehirns – ein Netzwerk von Hirnregionen, die auf schädliche Stimuli, einschliesslich Schmerz, reagieren -aktiv wurden, noch bevor den Versuchspersonen ein Schock verabreicht wurde. Aufgrund von Reaktionen anderer, für Angst und Angstgefühle zuständiger Hirnregionen unterschieden sich die mässig und die extrem Furchtsamen vor dem Schock nicht. Die Ergebnisse zeigen, dass die Schmerzzentren des Gehirns der Zeit, bis ein Ereignis eintritt, umso mehr Aufmerksamkeit schenken, je mehr jemand dieses Ereignis fürchtet. Die Schmerzzentren des Gehirns schenken der Zeit, bis ein Ereignis eintritt, umso mehr Aufmerksamkeit, je mehr jemand dieses Ereignis fürchtet. Man weiss zwar noch nicht, welcher Zusammenhang zwischen diesen Präferenzen und dem Umgang mit bekanntlich unangenehmen Ereignissen, etwa einem mit einem schmerzhaften Eingriff verbundenen Arztbesuch, besteht, doch könnten die neurobiologischen Grundlagen der Furcht dereinst gewisse Anhaltspunkte für ein besseres Schmerzmanagement liefern. Schmerzbekämpfung dank Schädlingsbekämpfung Nachdem das verbreitete Schmerzmittel Vioxx (Rofecoxib) wegen Sicherheitsbedenken aus dem Handel genommen wurde, stiessen Forschende der University of California, Davis, möglicherweise auf einen unbedenklicheren Weg, Menschen mit Arthritis oder anderen Entzündungskrankheiten die nötige Schmerzlinderung zu gewährleisten. Die Forschenden hatten ursprünglich ein ganz anderes Ziel: Sie suchten ein biologisches Schädlingsbekämpfungsmittel, um die Entwicklung von Insektenlarven zu beeinflussen. Im Verlauf ihrer Studie entdeckten sie jedoch ein neues menschliches Enzym, das indirekt die Produktion der mit Schmerz und Entzündung zusammenhängenden COX2-Proteine hemmt. 64 Eine kombinierte Anwendung der beiden Therapien könnte Entzündungsschmerzen lindern und die Nebenwirkungen der zur Schmerzlinderung verwendeten Medikamente verringern. Tests an Nagetieren ergaben, dass Schmerz der Enzymblocker ebenso wirksam war wie niedrige Dosen von Rofecoxib und einem anderen COX2-Hemmer, Celecoxib (Celebrex), ohne jedoch jene chemischen Veränderungen im Blut zu bewirken, die im Rahmen einer früheren Studie mit schweren kardiovaskulären Komplikationen, einschliesslich Herzinfarkt, in Zusammenhang gebracht worden waren; dieser Befund hatte dazu geführt, dass Vioxx aus dem Handel gezogen wurde. Diese neue Untersuchung wurde in Proceedings of the National Academy of Sciences publiziert 3. Eine Kombination dieser beiden Arten von COX2-Hemmern könnte die Konzentration der für eine wirksame Behandlung von Entzündungen notwendigen COX2-Inhibitoren entscheidend verringern, meinten die Forschenden. Diese Kombination führt offensichtlich zu chemischen Veränderungen des Blutes, die der Entwicklung von Blutgerinnseln, einem Hauptfaktor des Herzinfarkts, entgegenwirken. Eine derartige Kombinationstherapie könnte mithelfen, das Dilemma zu lösen, ob zur Behandlung von Entzündungsschmerzen wirksame COX-2-Inhibitoren zum Einsatz kommen sollen. „Emotionale Ansteckung“ bei Schmerz Bereits früher hat die Schmerzforschung nachgewiesen, dass Erfahrungen der Jugendzeit und gewisse soziale Faktoren chronische Schmerzen verschlimmern können. In Deutschland hatte man z. B. festgestellt, dass durch soziale Faktoren hervorgerufene Veränderungen der Hirnfunktion zu verstärkten Schmerzempfindungen führen können 4. Eine andere Studie hatte ergeben, dass in der Jugendzeit erlittene Schmerzen einen Einfluss darauf haben können, wie Schmerz im Erwachsenenalter empfunden wird 5. Eine neue Untersuchung, über die Jeffrey Mogil und Mitarbeitende in Science berichteten, zeigt nun, dass die Schmerzreaktion einer Maus verstärkt wird, wenn eine andere Maus da ist, die ebenfalls Schmerzen hat; dies deutet darauf hin, dass bei Schmerz Empathie eine Rolle spielt 6. Mittels eines Essigsäure-Writhing-Tests, der leichte Magenschmerzen vortäuscht, konnten die Forschenden am McGill University’s Pain Genetics Laboratory bei untereinander vertrauten Mäusen, eine bestimmte Art von Empathie, die so genannte „emotionale Ansteckung“ nachweisen; diese besteht darin, dass eine Maus den emotionalen Zustand einer anderen erkennt und sich ihm anpasst. Die Forschenden stellten fest, dass eine Maus empfindlicher auf Essigsäure reagierte, wenn sie gleichzeitig sah, dass eine andere Maus unter einem schmerzhaften Hitzestimulus litt. 65 Für die Interaktion unter ihresgleichen sind Mäuse auf Pheromone (chemische Substanzen, welche Signale zwischen Mitgliedern derselben Spezies übermitteln) angewiesen. Selbst wenn die Forschenden den Geruchsinn, die Sicht sowie das Gehör der Nagetiere blockierten, wussten diese um den Schmerz der andern; die Kommunikation der Schmerzreaktion war offenbar auf andere Weise erfolgt. Da der Umgang mit chronischen Schmerzen stark von sozialen Interaktionen abhängig ist, könnte McGills Befund für die Schmerzforschung bei Menschen bedeutsam sein. Ausgehend von den am Mausmodell gefundenen Ergebnissen könnte man sowohl die an der Schmerzempfindung beteiligten Hirnmechanismen von Menschen als auch den Einfluss sozialer Faktoren auf das Schmerzmanagement untersuchen. Die Art eines Placebos wirkt sich aus Vor über 50 Jahren beschrieb Henry K. Beecher, ein Anästhesiologe in Harvard, erstmals die medizinische Bedeutung von Placebos. Als Placeboeffekt bezeichnet man das Phänomen, dass sich die Symptome von Kranken infolge einer an sich unwirksamen Behandlung bessern, weil die Betroffenen diese Wirkung erwarten oder an sie glauben. Der Placeboeffekt lässt sich durch die Art des verabreichten Placebos und durch die Umstände, unter denen es abgegeben wird, beeinflussen. In zwei Untersuchungen, die im British Medical Journal und im Journal of Neuroscience veröffentlicht wurden, wies ein von Ted Kaptchuk geleitetes Forschungsteam am Harvard Medical School’s Osher Institute nach, dass sich der Placeboeffekt durch die Art des verabreichten Placebos und durch die Umstände, unter denen es abgegeben wird, beeinflussen lässt 7, 8. Um festzustellen, welche Placebo-Behandlung wirksamer war, führte Kaptchuks Gruppe in der ersten Studie an 135 Kranken mit starken Armschmerzen eine Schein-Akkupunktur durch, während weitere 135 eine inaktive Pille bekamen. Keine der Behandlungen übertraf die andere. In einer späteren Studie wurde bei der Hälfte einer jeden Gruppe die ursprüngliche Placebo-Behandlung fortgesetzt, während die andere Hälfte eine wirkliche Behandlung erhielt. 66 Die Personen, die einer Schein-Akkupunktur unterzogen wurden, berichteten von einer grösseren Schmerzlinderung, als jene, die unwirksame Schmerz Pillen einnahmen. Laut Kaptchuk deutet dieser Befund darauf hin, dass der grössere Placeboeffekt der Schein-Akkupunktur darauf beruhen könnte, dass die Behandlung bei diesem „Ritual“ mit einem Instrument erfolgt – eine mögliche Erklärung, der er und seine Mitarbeitenden nun weiter nachgehen. In dem Testdurchgang, in welchem einige Personen eine wirkliche Behandlung erhielten, bestimmten die Forschenden ausserdem mittels funktioneller Magnetresonanz-Tomographie, welche Netzwerke des Gehirns durch die Schein-Akkupunktur aktiviert wurden. Ausgehend von früheren Studien, denen zufolge Bahnen im präfrontalen Kortex, im Striatum und im Hirnstamm an der Verarbeitung von Placebos mitwirken, entdeckten sie, dass bestimmte Hirnbereiche besonders eng mit dem Placeboaffekt verknüpft sind. Einer davon ist der anteriore insulare Kortex, der Körperempfindungen, einschliesslich Schmerz, aktiviert. Diese Studien bestätigen frühere Hinweise darauf, dass der so genannte Placeboeffekt auf veränderten Hirnfunktionen beruht. 67 Psychiatrische Erkrankungen, Verhaltensstörungen und Suchtkrankheiten Schizophrenie 70 Gewalt und Aggression 73 Angsterkrankungen 74 Depression 74 Suizid bei Teenagern 76 Kokainsucht 77 69 W ie schon im Jahr 2005 konzentrierte sich die Erforschung psychischer Krankheiten auch 2006 auf den Einfluss von Genen bei psychiatrischen Erkrankungen und auf die Interaktion dieser Gene mit Umweltfaktoren. Allerdings richtete sich die Erforschung der klinischen und genetischen Behandlung dieser Erkrankungen 2006 auf einen neuen Fokus. Die Schizophrenieforschung verglich die klinische Wirksamkeit neuerer Neuroleptika mit der ihrer Vorgänger. Genetische Studien zur Depression konzentrierten sich auf mögliche Prädiktoren für die therapeutische Wirksamkeit von Antidepressiva; auf einen allfälligen Zusammenhang einer Behandlung mit Antidepressiva und Suizid; sowie auf die Frage, inwiefern die Behandlung depressiver Mütter einen Einfluss darauf hat, ob ihre Kinder depressive Symptome entwickeln bzw. als depressiv diagnostiziert werden. Schizophrenie An Schizophrenie Erkrankte wurden lange Zeit hauptsächlich mit Neuroleptika behandelt. Leider haben viele herkömmliche Medikamente eine Menge unangenehmer Nebenwirkungen, die mit der Unterdrückung des Neurotransmitters Dopamin zusammenhängen. Deshalb verschreibt man heute in der Psychiatrie oft Neuroleptika der zweiten Generation, d. h. „atypische“ Neuroleptika, da die Wahrscheinlichkeit, dass sie die Dopaminübertragung in von der Krankheit nicht direkt betroffenen Hirngebieten hemmen, kleiner ist. Aber ist diese neue Klasse therapeutischer Wirkstoffe wirksamer, und wird sie von den Kranken besser ertragen als die Medikamente der ersten Generation? Für die meisten trifft dies nicht zu, so die Arbeiten von Jeffrey Lieberman und Mitarbeitenden in den Jahren 2005 und 2006. Eine im Jahr 2005 publizierte Forschungsarbeit ergab für Neuroleptika der ersten und zweiten Generation keine unterschiedliche Wirksamkeit 1. Hinsichtlich der Verträglichkeit wurde Olanzapin, ein Medikament der zweiten Generation, von den Kranken zwar etwas seltener eigenmächtig abgesetzt als andere Medikamente, aber es führte zu unerfreulichen Gewichtszunahmen und hatte Nebenwirkungen auf den Stoffwechsel. 70 Liebermans Gruppe setzte ihre Arbeit im Jahr 2006 fort und publizierte im American Journal of Psychiatry zwei Aufsätze, welche die Behandlung mit Neuroleptika detaillierter beleuchteten. Es zeigte sich, dass chronisch Schizophreniekranke die Behandlung mit Olanzapin und Risperidon eher fortsetzten als mit anderen atypischen Neuroleptika 2. Dementsprechend untersuchten die Forschenden bei Kranken, die auf atypische Neuroleptika überhaupt nicht angesprochen hatten, die Wirksamkeit von Clozapin, einem Medikament mit starken Nebenwirkungen, das bei therapieresistenten Kranken als „letzter Ausweg“ eingesetzt wird. Sie stellten fest, dass diese auf Clozapin besser ansprachen als auf ein zweites atypisches Neuroleptikum 3. Psychiatrische Erkrankungen, Verhaltensstörungen und Suchtkrankheiten Unterschiedliche antipsychotische Medikamente Der Forscher Jeffrey Lieberman verglich die Wirksamkeit von Neuroleptika der ersten und zweiten Generation und stellte fest, dass die jüngere Generation im Grossen und Ganzen weniger wirksam ist. Ganz unabhängig davon untersuchten auch Peter Jones und sein Forschungsteam in Cambridge die Wirksamkeit von Neuroleptika der zweiten Generation zur Behandlung von chronischer Schizophrenie. Die am Versuch Teilnehmenden erhielten zufällig ein Neuroleptikum der ersten oder der zweiten Generation und wurden ein Jahr lang durch einen Arzt/eine Ärztin beurteilt, der/die nicht wusste, wem welches Medikament zugeteilt worden war. Erfasst und verglichen wurden Symptome, Nebenwirkungen und die Lebensqualität. Jones Team hatte eigentlich erwartet, dass die atypischen 71 Medikamente wirksamer wären als ihre Vorgänger, doch das Gegenteil war der Fall. Kranke, welche Medikamente der ersten Generation einnahmen, sprachen auf die Behandlung besser an und erzielten auf Lebensqualität-Skalen eine höhere Punktzahl 4. Im Zusammenhang mit der Forschungsarbeit von Lieberman rief dieser Befund in der Psychiatrie eine gewisse Konsternation hervor. Insgesamt deuten diese Ergebnisse darauf hin, dass Versuche mit atypischen Neuroleptika im Allgemeinen vor allem dann erfolgen sollten, wenn Kranke auf Neuroleptika der ersten Generation resistent sind. Die Erforschung der schwer fassbaren Ursachen der Schizophrenie konzentriert sich weiterhin auf den Einfluss dopaminerger Neuronen. Die Erforschung der schwer fassbaren Ursachen der Schizophrenie konzentriert sich weiterhin auf den Einfluss dopaminerger Neuronen. Erste Arbeiten brachten eine exzessive Dopaminübertragung mit den Verhaltensauffälligkeiten der Krankheit in Verbindung. Aber Michael O’Donovan, Michael Owen und weitere Mitarbeitende untersuchten die abweichende Funktion von Hirnzellen, der so genannten Glia, als einen weiteren möglichen Wegbereiter der Krankheit; sie stützten sich dabei auf frühere, mittels Autopsie und Neuroimaging gewonnene Hinweise auf strukturelle sowie das Volumen betreffende Unterschiede der weissen Substanz des Gehirns (den neuralen Verbindungen) bei Schizophreniekranken und gesunden Kontrollpersonen. Gliazellen produzieren in Interaktion mit Neuronen das Myelin, einen fetthaltigen Isolator, der die Übertragung elektrischer Signale von einer Hirnzelle zur nächsten begünstigt. Die in Proceedings of the National Academy of Sciences veröffentlichten Resultate der Gruppe besagen, dass Personen mit Varianten des Gens OLIG2, das die Bildung von Myelin reguliert, für Schizophrenie anfällig sind. Dies lässt erwarten, dass aus der weiteren Erforschung jener Gene, welche den Einfluss der Glia auf die Myelinproduktion steuern, wichtige Einblicke in den komplizierten Vorgang der Schizophrenie hervorgehen könnten 5. Gewalt und Aggression 72 Seit Jahrhunderten bemüht sich die Wissenschaft genau zu bestimmen, was der menschlichen Gewalt und Aggression zugrunde liegt. Während der Einfluss der sozialen Umwelt eingehend erforscht wurde, erwies sich Am besten gesichert ist bisher der genetische Zusammenhang von gewalttätigem Verhalten und Monoamin Oxidase A (MAOA), einem Enzym, das direkt am metabolischen Abbau des Neurotransmitters Serotonin beteiligt ist. In einer Arbeit, die in Proceedings of the National Academy of Sciences veröffentlicht wurde, berichteten Andreas Meyer-Lindenberg und Mitarbeitende von den Ergebnissen einer Studie, in welcher der Einfluss von MAOA mittels voxelbasierter Morphometrie, einer Computergestützten neuroanatomischen Methode zur Messung der unterschiedlichen Konzentrationen im Hirngewebe, und funktioneller Magnetresonanz-Tomographie untersucht wurde. Sie entdeckten, dass das Gehirn von Personen, die zwar nie an einer gewalttätigen psychiatrischen Störung gelitten hatten, bei denen aber eine Genvariante vorkam, welche die Expression des MAOA-Enzyms verminderte, im Vergleich zu Personen mit einer höheren MAOA-Expression deutliche strukturelle und funktionelle Unterschiede aufwies. Bei Personen mit einer verminderten MAOA-Expression war das Volumen der grauen Substanz im cingulären Gyrus, in der Amygdala und im anterioren cingulären Kortex reduziert; ausserdem kam es bei ihnen zu einer Zusammenhang eines Gens mit Gewalt Die Gruppenergebnisse zweier Arten von Magnetresonanz-Scans zeigen auf, dass das Volumen und die Aktivität des so genannten anterioren cingulären Kortex bei Personen mit einer Genvariation, die mit Aggression verbunden ist, reduziert sind (in dunklem grau dargestellt). Dieser Bereich ist an der Steuerung von emotionalen und aggressiven Reaktionen beteiligt. Psychiatrische Erkrankungen, Verhaltensstörungen und Suchtkrankheiten die Untersuchung der zugrunde liegenden genetischen Komponenten als schwieriger und widersprüchlicher. 73 stärkeren Aktivierung der Amygdala und der limbischen Regionen – Bereiche, die an der Verarbeitung von Emotionen beteiligt sind – wenn sie wütende und ängstliche Gesichtern voneinander unterscheiden sollten. Die Untersuchung machte auch eine Geschlechtskomponente sichtbar: Während einer emotionalen Gedächtnisaufgabe war die Aktivität von Amygdala und Hippocampus bei männlichen Teilnehmenden stärker erhöht als bei weiblichen. Obwohl viele Faktoren zu gewalttätigem Verhalten beitragen, deutet der Befund darauf hin, dass insbesondere bei Männern mit dieser besonderen Genvariante, eine mögliche biologische Disposition zu impulsiver Gewalttätigkeit besteht 6. Angsterkrankungen Auch die Erforschung von angstbedingten Erkrankungen konzentrierte sich im Jahr 2006 auf Gene. Anhand von Mausmodellen identifizierte Carrolee Barlows Gruppe am Salk Institute 17 Gene, deren Expressionsmuster mit typischen Symptomen von Angsterkrankungen verbunden waren. Als mögliche Ursachen für angstbedingte Erkrankungen diskutieren sie in ihrem in Nature publizierten Aufsatz auch zwei Gene, die in den oxidativen Stressmetabolismus involviert sind, also in die vermehrte Produktion von Oxidantien, die zur Degeneration von Neuronen führt. Barlows Team transferierte diese Gene mittels Viren in Zellen und stellte fest, dass die übermässige Expression der Gene bei Mäusen ängstliches Verhalten verstärkte 7. Auf ähnliche Art und Weise untersuchte eine von David Goldman geleitete Gruppe Gene, die mit einer ganz bestimmten Angsterkrankung, der Zwangskrankheit (obsessive compulsive disorder, OCD) zusammenhängen. Die Gruppe stellte fest, dass HTT, ein Serotonin-Transporter-Gen, an OCD beteiligt ist. In einem Aufsatz, der im American Journal of Human Genetics veröffentlicht wurde, diskutieren sie den Befund, dass HTTLPR über drei und nicht, wie bisher angenommen, über zwei Genvarianten verfügt. Multiple Genotyp-Methoden deckten eine bisher unbekannte Genvariante auf, die zu einem neuen Verständnis der Neurobiologie der OCD führen könnte 8. Bisher konnte man die Grundlagen dieser Verhaltensauffälligkeiten nur schwer erkennen. Möglicherweise werden diese neueren genetischen Ansätze zu eindeutigeren Informationen führen. Depression 74 Tiefe Hirnstimulation als mögliche Behandlung von Personen, deren Depression auf herkömmliche Medikamente nicht anspricht, stösst Daneben gab es unter anderem Untersuchungen von genetischen Faktoren, welche die Reaktion auf Behandlungen mit herkömmlichen Antidepressiva beeinflussen könnten. Forschende unter der Leitung von Francis McMahon suchten nach genetischen Grundlagen für die individuell unterschiedlichen Resultate von Behandlungen mit Antidepressiva. Sie untersuchten die DNA von 1953 Kranken mit einer Major Depression, die mit dem gebräuchlichen Antidepressivum Citalopram behandelt wurden, und wiesen einen eindeutigen Zusammenhang zwischen einer erfolgreichen Behandlung und der für die Serotoninaufnahme verantwortlichen Genvariante A, HTR2A, nach. Die Ergebnisse dieser Studie sind im American Journal of Human Genetics dargestellt 10. Des Weiteren stellten sie fest, dass diese A-Variante bei weissen Kranken sechsmal häufiger vorkam als bei afroamerikanischen Kranken, die entsprechend schlechter auf eine Behandlung mit Citalopram ansprachen. Der Befund liefert ein überzeugendes Argument dafür, dass dieses Gen die Wirksamkeit von Antidepressiva beeinflusst, und könnte zur Erklärung von rassenbedingt unterschiedlichen Reaktionen auf Behandlungen mit Antidepressiva beitragen. Eine dreimonatige erfolgreiche medikamentöse antidepressive Behandlung von Müttern führte dazu, dass bei ihren Kindern depressive und andere Symptome sowie die entsprechende Diagnose seltener wurden. Andernorts veranschaulichten Myrna Weissman und Mitarbeitende ein interessantes Phänomen bezüglich Depression bei Kindern. Dass Kinder depressiver Eltern mit hoher Wahrscheinlichkeit selbst auch eine depressive Krankheit entwickeln, war längst bekannt. Weissmans Gruppe berichtete nun im Journal of the American Medical Association, eine dreimonatige erfolgreiche medikamentöse antidepressive Behandlung der Mütter hätte dazu geführt, dass bei ihren Kindern depressive und andere Symptome sowie die entsprechende Diagnose seltener wurden. Umgekehrt litten Kinder von Müttern, deren Depression fortdauerte, unter vermehrten Symptomen. Dies lässt darauf schliessen, dass auch Umweltfaktoren die Psychopathologie dieser risikoreichen Gruppe von Kindern beeinflussen können 11. Psychiatrische Erkrankungen, Verhaltensstörungen und Suchtkrankheiten weiterhin auf Interesse. Ob diese Behandlung bei einer weiter gefassten Gruppe von Kranken wirksam ist, könnten weitergehende Studien von Helen Mayberg und Mitarbeitenden an der Emory University zeigen 9. 75 Suizid bei Teenagern Forschende unter der Leitung von Mark Olfson untersuchten einen anderen Aspekt der Behandlung mit Antidepressiva: ihre Beziehung zu Suizidversuchen und vollendeten Suiziden bei Erwachsenen und Kindern. Die in Archives of General Psychiatry veröffentlichten Ergebnisse ihrer Fallstudie mit entsprechender Kontrollgruppe zeigten, dass bei Erwachsenen kein Zusammenhang zwischen der Behandlung mit Antidepressiva und Suizidversuchen oder vollendeten Suiziden bestand; bei Kindern und Jugendlichen hingegen war der Zusammenhang sowohl mit Suizidversuchen als auch mit vollendeten Suiziden signifikant. Der Befund legt zumindest nahe, dass die medikamentöse Behandlung von jüngeren Kranken durch Fachpersonen im Spital und durch Eltern gut überwacht werden muss 12. Das Augenmerk der Forschungsgruppe von Eric Nestler galt der Neurobiologie von stressbedingten Depression. In einer in Nature Neuroscience publizierten Studie wurden Mäuse dem Stress andauernder sozialer Niederlagen, einem häufigen Vorläufer depressiver Erkrankungen, ausgesetzt und anschliessend einer Dauerbehandlung mit dem Antidepressivum Imipramin unterzogen. Nestlers Gruppe stellte fest, dass dieser durch Niederlagen hervorgerufene Stress zu einer Abnahme des neurotrophen Proteins BDNF (brain-derived neurotrophic factor) im Hippokampus führte und zu einer vermehrten Modifikation bestimmter mit der Gentranskription assoziierter Proteine, der so genannten Histon Methylierung. Das Antidepressivum machte diesen Vorgang rückgängig, ebenso eine Infusion von BDNF selbst. Der Befund deutet darauf hin, dass Histon Methylierung und die damit zusammenhängenden neurobiologischen Vorgänge ein neues Interessengebiet für die Therapie von Depressionen eröffnen könnten 13. 76 Inzwischen entdeckten Michel Lazdunski und seine Mitarbeitenden ein anderes Interessengebiet für die künftige Entwicklung von Antidepressiva. Lazdunskis Gruppe berichtete in Nature Neuroscience, sie hätten entdeckt, dass bei Mäusen ein „background“ Kaliumkanal, TREK-1, der durch Serotonin reguliert wird, an der Resistenz gegen Depressionen beteiligt war. Tiere ohne den TREK-1-Kanal waren unter Stress depressionsresistent – ein Hinweis darauf, dass dieser Kanal als Angriffspunkt für neue medikamentöse Behandlungen der Depression in Frage kommt 14. Liegt dem heftigen Verlangen nach Drogen eine erhöhte Freisetzung von Dopamin zugrunde? Forschende unter der Leitung von Nora Volkow untersuchten eingehend, woran es liegt, dass von Drogen ausgelöste Reize bei ehemals Süchtigen dauerhafte konditionierte Reaktionen hervorrufen können; sie kamen zum Schluss die Antwort auf obige Frage könnte Ja lauten. Frühere, mittels Bildgebung des Gehirns durchgeführte Studien brachten diese Reaktionen mit einer Aktivierung bestimmter limbischer Strukturen in Verbindung. Mithilfe der Positronen-Emissions-Tomographie konnte Volkows Gruppe bei ehemals Kokainsüchtigen, denen ein Video zum Thema Kokain gezeigt wurde, eine konditionierte Dopaminfreisetzung im dorsalen Striatum nachweisen. Diese im Journal of Neuroscience publizierten Resultate lassen darauf schliessen, dass Therapien, welche die Zunahme von Dopamin einschränken, eine Suchtbehandlung unterstützen könnten 15. Schliesslich zeigten mit funktioneller Bildgebung des Gehirns durchgeführte Studien auf, dass verschiedene Hirnregionen, unter anderem der präfrontale Kortex und die Amygdala, durch mit Drogen assoziierte Reize aktiviert werden. Diese Regionen stehen mit einer anderen Region des Gehirns, dem ventralen tegmentalen Bereich (ventral tegmental area, VTA) in Verbindung. Veränderungen der Synapsen im VTA könnten der Forschung Hinweise auf die neurobiologischen Ursachen von Drogensucht, Entzug und Rückfall geben. Mu-Ming Poo und Mitarbeitende untersuchten die dopaminergen Neuronen im VTA von Ratten nach einem Kokainentzug und berichteten darüber in Nature Neuroscience. Sie fanden in jenen Zellen eine Erhöhung des neurotrophen Faktors BDNF. Möglicherweise werden die dopaminergen Zellen des VTA während eines Entzugs durch diese verstärkte Expression von BDNF stimuliert und setzen eine Kette von Ereignissen in Gang, die dazu führen, dass ein heftiges Verlangen nach der Droge einsetzt, sobald jemand mit Erinnerungen an den früheren Drogengebrauch konfrontiert wird 16. Psychiatrische Erkrankungen, Verhaltensstörungen und Suchtkrankheiten Kokainsucht 77 Störungen der Sinnesund Körperfunktion Das Gehör: Regeneration von Haarzellen bei Säugetieren 80 Weshalb Gesichter vertraut sind 81 Pheromon-Erkennung 82 REM-Schlaf-Schaltkreise 83 Zirkadiane Uhren und Ernährung 83 Rezeptor für sauren Geschmack identifiziert 85 Stammzellen und Sehvermögen 86 79 D ank der Spitzentechnologie hat die Wissenschaft heute mehr denn je Einblick in die innere Tätigkeit des Gehirns, was zu einem besseren Verständnis der komplizierten Beziehung zwischen dem Gehirn, den Sinnen und den Körperfunktionen führt. Das riesige Gebiet reicht von spezialisierten Genom-Techniken zur Erforschung des Mechanismus des REMSchlafs (rapid-eye-movement) über Studien zum Einfluss zirkadianer Uhren auf die Ernährung bis hin zur Entdeckung, dass ein Teils des Gehirns nur eine einzige Aufgabe hat: Gesichter zu erkennen. Andere Untersuchungen des Jahres 2006 brachten neue Informationen in Bezug auf das Gehör, den Geruch- und Geschmacksinn sowie das Sehvermögen; dadurch wurden alte Fragen beantwortet und neue aufgeworfen. Das Gehör: Regeneration von Haarzellen bei Säugetieren Die zentrale Hörstörung, eine zurzeit irreversible Krankheit, ist in den USA die häufigste Form der Taubheit. Verursacht wird sie durch eine Schädigung von spezialisierten Haarzellen des Innenohrs aufgrund von Alter, lauten Geräuschen oder Nebenwirkungen gewisser Medikamente. Wissenschafter und Wissenschafterinnen, die sich für die Entwicklung neuer Therapien bei gewissen Formen des Hörverlusts interessierten, bekamen 2006 grossen Auftrieb, da Forschungsergebnisse darauf schliessen lassen, dass diese für das Gehör unerlässlichen, spezialisierten Haarzellen möglicherweise regenerieren können. Zwar können beschädigte sensorische Zellen im Innenohr der Cochlea, von Vögeln und anderen niedrigen Wirbeltieren regenerieren, doch sind Zellen in der Cochlea von Säugetieren, einschliesslich des Menschen, dazu nicht in der Lage. Die Erforschung neuer Therapien der zentralen Hörstörung hatte ihr Augenmerk schon lange auf Haarzellen gerichtet. Neil Segil, Andy Groves und Mitarbeitende am House Ear Institute in Los Angeles berichteten in Nature über ihre Entdeckung, dass das Gen p27Kip1 die Zellteilung im Innenohr verhindert 1. 80 Die Forschenden untersuchten sensorische Zellen von Mäusen in Zellkulturen. Dabei stellten sie fest, dass dieses Gen bei neugeborenen Mäusen ausgeschaltet war und sich die Bindegewebszellen stark vermehren und zu Haarzellen differenzieren konnten. Untersuchungen an Kulturen von zwei Wochen alten Mäusen ergaben hingegen, dass dieses Gen, kurz p27, nun angeschaltet war und die Zellteilung stoppte. Jedoch konnten Zellen von zwei Wochen alten Mäusen, denen das Gen p27 aufgrund einer genetischen Manipulation fehlte, Haarzellen hervorbringen; dies deutet darauf hin, dass es möglich sein könnte, die Hörfähigkeit wiederherzustellen, wenn es gelingt, dieses Gen auszuschalten und so das Wachstum von Haarzellen im Innenohr anzuregen. Störungen der Sinnes- und Körperfunktion Hören, Haar Das mittels eines Hochleistungs-Scanner-Elektronenmikroskops erhaltene Bild einer Haarzelle zeigt Haarbündel, die auf der Zelloberfläche hervorstehen. Forschende stellten bei Mäusen fest, dass durch Stilllegung eines Gens das Wachstum von Haarzellen ermöglicht wird, wodurch das Gehör wieder hergestellt werden könnte. Im Hinblick auf die Entwicklung neuer Therapien zur Heilung der Taubheit von Menschen ist die Arbeit viel versprechend. Obwohl dieser Vorgang nur in Mäusekulturen nachgewiesen wurde, ist die Arbeit auch im Hinblick auf die Entwicklung neuer Therapien zur Heilung der Taubheit von Menschen viel versprechend. Weshalb Gesichter vertraut sind In der anhaltenden Diskussion der Neurowissenschaften, ob das Gehirn in spezialisierte Bereiche mit einer je spezifischen Aufgabe unterteilt ist, führt eine in Science veröffentlichte Studie zu neuen Einsichten 2. Forschende der Harvard Medical School und der Universität Bremen in Deutschland identifizierten im visuellen Kortex eine Region, in der fast alle Neuronen für eine einzige Aufgabe spezialisiert sind: die Wahrnehmung von Gesichtern. Mittels funktioneller Magnetresonanz-Tomographie bestimmte ein von Margaret Livingstone geleitetes Team im Kortex von Makak-Affen drei Regionen, die für das Erkennen von Gesichtern bedeutsam sein sollen. Den Affen wurden 96 Abbildungen von Gesichtern, Körpern, Früchten, Spielsachen, Händen und wirren Gittermustern gezeigt. Dabei wurden die neuronalen Impulse der einzelnen Zellen in der grössten dieser drei 81 Hirnregionen registriert. Man stellte fest, dass beinahe alle Neuronen (97%), die überhaupt auf visuelle Reize ansprachen, 50-mal häufiger auf Gesichter reagierten als auf die anderen visuellen Reize. Die einzigen anderen Bilder, die bei diesen Neuronen eine Reaktion auslösten, waren runde Objekte, deren Form Gesichtern ähnlich war. Menschen verfügen möglicherweise über spezialisierte Zellen, die ausschliesslich dem Erkennen von Gesichtern dienen. Die grosse physiologische Ähnlichkeit des Gehirns von Makaken und Menschen lässt darauf schliessen, dass Menschen über ebensolche spezialisierte Zellen verfügen, die ausschliesslich dem Erkennen von Gesichtern dienen. Die Verwendung der Bildgebung, um den Vorgang der Gesichtererkennung zu analysieren, könnte somit zu neueren Methoden führen, um Lügen zu detektieren (siehe auch Neuroethik, S. 45). Pheromon-Erkennung Bei vielen Tieren ist das Gehirn so aufgebaut, dass es Pheromone erkennt, chemische Signale, die dazu dienen, Partner anzulocken. Dem Gehirn des Menschen scheint diese Fähigkeit zu fehlen – ein wissenschaftliches Rätsel angesichts neuerer Befunde, denen zufolge Menschen auf Pheromone reagieren können. Es wäre allerdings möglich, dass das olfaktorische Epithel, das über Neuronen verfügt, welche gewöhnliche Gerüche erkennen, auch Pheromone aufspürt. Forschende am Fred Hutchinson Cancer Research Center in Seattle untersuchten eine allfällige Beteiligung des Epithels an der Pheromon-Erkennung bei Mäusen. In ihrer in Nature veröffentlichten Arbeit identifizierten Stephen Liberles und Linda Buck einen Satz von olfaktorischen Rezeptoren in der Nase von Mäusen, die so genannten TAA-Rezeptoren (trace amine-associated receptors, TAARs) 3. Diese Rezeptoren sind anders als jene, die Gerüche wahrnehmen, und frühere Studien deuteten darauf hin, dass einige von ihnen durch Substanzen aktiviert werden, die im sehr pheromonhaltigen Urin von Mäusen vorkommen. In ihrer Studie fanden Liberles und Buck heraus, dass Isoamylamin, eine der Substanzen, die von TAARs erkannt werden, als ein Pheromon fungiert, das die Geschlechtsreife weiblicher Mäuse beschleunigt. 82 Zusammen mit anderen Befunden lässt dies darauf schliessen, dass TAARs eine weitere Methode der Pheromon-Erkennung bieten. Da die Gene, die REM-Schlaf-Schaltkreise Vor über 50 Jahren entdeckte die Wissenschaft den mit Träumen assoziierten REM-Schlaf (rapid-eye-movement sleep). Wie das Gehirn vom REM- in den Non-REM-Schlaf wechselt, ist aber immer noch unklar. Forschende der Harvard Medical School haben 2006 Ergebnisse vorgelegt, die auf ein neues Erklärungsmodell für den Vorgang, wie das Gehirn den Übergang in und aus dem REM-Schlaf steuert, schliessen lassen. Die Studie von Clifford Saper und Mitarbeitenden, über die in Nature berichtet wurde, untersucht ausserdem, was die Traumphasen und den während des REM-Schlafs auftretenden Verlust des Muskeltonus aktiviert 4. Frühere Modelle basierten auf Interaktionen zwischen cholinergen Neuronen, die während des REM-Schlafs aktiv sind, und monoaminergen Neuronen, die während des REM-Schlafs inaktiv sind. Das vom Harvard-Team entwickelte Modell, der so genannte „Flip-Flop-Wechsel“ zeigte jedoch, dass eine Deaktivierung dieser beiden Neuronen-Arten den REM-Schlaf nur wenig beeinflusste. Störungen der Sinnes- und Körperfunktion TAARs kodieren, auch bei Fischen und Menschen vorkommen, könnte es sein, dass Menschen Pheromone mithilfe dieser Rezeptoren erkennen. Stattdessen fanden sie heraus, dass eine wechselseitige Interaktion zwischen Neuronen zur Freisetzung des chemischen Botenstoffs GammaAminobutyric-Säure führt; dieser ist im Gehirn weit verbreitet, bindet an Neuronen und vermindert deren Aktivität. Probleme bei der Steuerung dieser Interaktion könnten eine Reihe rätselhafter Schlafstörungen erklären, etwa die REM-Verhaltensstörung, bei der die Betroffenen ihre Träume ausleben, und hypnagoge Halluzinationen, bei denen sie im Wachzustand traumartige Halluzinationen erleben. Bei den bisherigen Ansätzen, Schlaf anstossende Medikamente zu entwickeln, wurden die unterschiedlichen Schlafstadien im Allgemeinen nicht berücksichtigt. Das Verständnis der Steuerung dieser Stadien könnte zu besseren Therapien von Schlafstörungen führen. Zirkadiane Uhren und Ernährung Die Wissenschaft weiss seit langem, dass Tiere, denen nur während ihrer normalen Schlafzyklen Zugang zu Nahrung gewährt wird, ihre Schlafmuster und biologische Funktionen so ändern, dass sie dann wach und aktiv sind, wenn Nahrung zur Verfügung steht. Zwei voneinander unabhängige 83 Kortikosteroid Wachheit, Nahrungsaufnahme Ausschüttung Kortex Hypothalamus CRH 0,4 mm seitlich zur Mittellinie Orexin MCH PVH Schlaf LHA GABA VLPO dSPZ Thermoregulation Glutamat TRH MPO vSPZ DMH VMH SCN ARC Hunger Gefühl Leptin Ghrelin Essen und schlafen um zu überleben Der dorsomediale Nukleus des Hypothalamus (DMH) koordiniert unter anderem die Zyklen von Schlaf und Wachheit, der Nahrungsaufnahme und der Körpertemperatur. Die Forschung hat ergeben, dass diese Region die biologische Uhr des Gehirns, den suprachiasmatischen Nukleus (SCN), über die zeitliche Steuerung des Futterangebots ausser Kraft setzen kann. Studien, die im Jahr 2006 von Teams der Harvard Medical School und dem University of Texas Southwestern Medical Center veröffentlicht wurden, erklären vielleicht, was die Tiere zu dieser Umstellung befähigt. Forschende stellten fest, dass der DMH die biologische Uhr des Gehirns ausser Kraft setzen und eine neue Uhrzeit etablieren kann, um die Verfügbarkeit von Futter auszunützen. 84 Das eine Team – es wurde von Saper in Harvard geleitet – konzentrierte sich auf den dorsomedialen Nukleus des Hypothalamus oder DMH, eine Hirnregion, die mit Teilen des Gehirns kommuniziert, die unter anderem an Vorgängen mitwirken, welche die Ernährung, den Energieverbrauch, die Regulierung von Wachheit und Schlafen sowie die Körpertemperatur betreffen. Laut einem Bericht in Nature Neuroscience stellten die Forschenden fest, dass der DMH die biologische Uhr des Gehirns ausser Kraft setzen und eine neue Uhrzeit etablieren kann, um die Verfügbarkeit von Futter auszunützen 5. Die Ergebnisse dieser beiden Studien setzen der künftigen Erforschung von zirkadianen Uhren und Ernährung neue Ziele und lassen erwarten, dass sich diese Arbeit eines Tages auch auf die Behandlung von Fettleibigkeit auswirken könnte. Rezeptor für sauren Geschmack identifiziert Der Mensch schmeckt Nahrungsmittel mithilfe von Geschmacksknospen auf der Zunge, die fünf spezifische Geschmäcke erkennen: bitter, süss, salzig, sauer und umami (den Geschmack von Natriumglutamat). Störungen der Sinnes- und Körperfunktion Die zweite Studie wurde von Masashi Yanagisawa und Mitarbeitenden in Proceedings of the National Academy of Sciences publiziert; sie zeigt, dass DMH-Neuronen über eine Uhr verfügen, die normalerweise keine Funktion hat, sich aber, wenn die Verfügbarkeit der Nahrung eingeschränkt ist, der Zeit anpasst, in der Nahrung zur Verfügung steht. Auf diese Weise kann die DMH ihre eigenen zirkadianen Rhythmen etablieren, welche die biologische Uhr des Gehirns ausser Kraft setzen 6. Im Jahr 2006 identifizierten zwei voneinander unabhängige Forschungsgruppen in ihren Studien ein Protein, dank dem Menschen und gewisse Tiere saure Geschmäcke wahrnehmen können, unter anderem auch Aromen, die vor dem Verzehr verdorbener oder unreifer Nahrung warnen. Die Forschenden berichteten in Nature und Proceedings of the National Academy of Sciences über ihre Arbeiten und gaben dem Protein die Bezeichnung PKD2L1. Es kommt in einigen Geschmacksknospen vor, allerdings nicht in jenen, die süsse, bittere und umami Geschmäcke erkennen 7, 8. Eine von Charles Zuker geleitete Gruppe an der University of California, San Diego, die ihre Arbeit in Nature vorstellte, veränderte Mäuse gentechnisch so, dass ihnen PKD2L1 fehlte. Diese Mäuse reagierten zwar auf andere Geschmäcke, nicht aber, wenn man ihnen Zitronensäure, Essig oder andere saure Geschmäcke gab. Inzwischen wies ein von Hiroaki Matsunami geleitetes Team an der Duke University in Proceedings of the National Academy of Sciences darauf hin, dass diese Befunde möglicherweise zu einem besseren Verständnis der sensorischen Informationsverarbeitung des Gehirns führen und eines Tages von der verarbeitenden Industrie zur Veränderung des Geschmacks von Nahrungsmitteln genutzt werden könnten. 85 Stammzellen und Sehvermögen Die Erforschung des Sehvermögens erbrachte Hinweise darauf, dass embryonale Stammzellen zur Behandlung der altersbedingten Makuladegeneration in Frage kommen könnten. Diese Krankheit, die häufigste Erblindungsursache bei über 65jährigen, beruht auf einer Schädigung der Retina und der in ihrem Zentrum gelegenen Makula, die ein detailliertes, zentrales Sehen ermöglicht. Retinale Pigmentepithelzellen, die den Grund der Retina auskleiden, können bei einigen Arten der Makuladegeneration beschädigt sein. In einer Studie, über die in Cloning and Stem Cells berichtet wurde, transformierte ein von Raymond Lund geleitetes Team der Oregon Health and Science University embryonale Stammzellen in solche retinale Pigmentepithelzellen und injizierte diese anschliessend in die erkrankten Augen von Ratten 9. Etwa sechs Wochen später überprüften die Forschenden die Sehkraft der Ratten und stellten fest, dass jene, welche die transformierten Stammzellen erhalten hatten, einen grossen Teil ihres Sehvermögens bewahrt hatten, wohingegen die unbehandelten Ratten beinahe blind waren. Die Forschenden überprüften die Sehkraft der Ratten und stellten fest, dass jene, welche die transformierten Stammzellen erhalten hatten, einen grossen Teil ihres Sehvermögens bewahrt hatten, wohingegen die unbehandelten Ratten beinahe blind waren. In einer ähnlichen Studie transplantierten Forschende unter der Leitung von Robin Ali am University College London Photorezeptoren, also die empfangenden Zellen der Retina 10. Insgesamt ergeben diese Studien, dass es möglich sein könnte, Stammzellen und andere primitive Zellen zur Behandlung der Makuladegeneration einzusetzen. Diese Ergebnisse sind zwar viel versprechend, doch weisen die Forschenden warnend darauf hin, dass die Augenkrankheit der Ratten nicht mit der Makuladegeneration des Menschen gleichzusetzen ist; daher sind weitere Studien nötig um zu bestimmen, ob Stammzellen auch zur Behandlung der menschlichen Form dieser Krankheit verwendet werden können. 86 Stammzellen und Neurogenese Neurogenese im Kortex 88 Eine natürliche Reaktion auf Verletzungen 90 Neurogenese und Epilepsie 92 Antidepressiva verstärken Neurogenese in einem bestimmten Stadium 93 Krankheitsproteine und die Entwicklung von Hirnzellen 93 Stützzellen werden krebsartig 95 Das Protein Notch aktiviert Stammzellen 95 87 B ekanntlich dauert die Neubildung von Neuronen, die so genannte Neurogenese, in einigen Hirnregionen während des ganzen Lebens an. Aufgrund der Forschung zeigt sich immer deutlicher, dass dieser Vorgang zu den Selbstheilungsmethoden des Gehirns zählt und therapeutisch genutzt werden könnte; eine abnormale Neurogenese trägt möglicherweise zu gewissen Krankheiten bei und könnte neue Zugänge zur Therapie eröffnen. Auch die Verwendung von unreifen und wandlungsfähigen Zellen, so genannten Stammzellen, ist eine viel versprechende Behandlungsmöglichkeit. Im Jahr 2006 gelang es der Forschung, die Wege, über die sich Stammzellen in Neuronen verwandeln, ein Stück weit zu enträtseln. Aber können Stammzellen ganz bestimmte Aufgaben im Gehirn übernehmen? Neurogenese im Kortex Seit 1998 weiss man, dass im Hippokampus des erwachsenen menschlichen Gehirns Neurogenese stattfindet. Weniger klar ist, ob auch in anderen Gebieten neue Neuronen gebildet werden und ob die erstaunliche Anpassungsfähigkeit des Gehirns, seine Plastizität, auf dem Umbau bestehender Zellen oder der Bildung von neuen beruht. Eine innovative Methode, um das Alter von Hirnzellen zu bestimmen, besteht in der Zuhilfenahme von Radiokarbon (14C), das im Verlauf der überirdischen Nukleartests in den 1950er Jahren in riesigen Mengen ausgestossen wurde und seither messbar abgenommen hat, da es über die Erdatmosphäre in die DNA von Pflanzen, Tieren und Menschen gelangt ist. Im Jahr 2005 stellte ein von Jonas Frisen geleitetes Team am Karolinska Institute in Stockholm aufgrund der 14C-Datierung fest, dass die 14C-Konzentrationen im Kortex von erwachsenen Menschen mit jenen übereinstimmten, die zur Zeit ihrer Geburt in der Atmosphäre bestanden hatten – ein Hinweis darauf, dass im Verlaufe des Lebens, wenn überhaupt, dann nur wenige kortikale Neuronen gebildet wurden. 88 Frisen und seine Mitarbeitenden taten sich mit verschiedenen anderen Laboratorien für eine weiter ausgedehnte Studie zusammen, über die in Proceedings of the National Academy of Sciences berichtet wurde 1. Die Forschenden arbeiteten mit Hirngewebe, das aus der Autopsie von sieben zwischen 1933 und 1973 geborenen Personen stammte, und erfassten Neuronen aus allen Lappen des Kortex. Die Forschenden vermuten, dass Neurogenese im Hippokampus zwar bei gewissen Arten des Gedächtnisses eine Rolle spielt, dass aber kognitive Funktionen, etwa lernen und analysieren, von kortikalen Zellen ausgeübt werden, die von Geburt an vorhanden sind, und dass die Stabilität des Kortex wichtiger ist als seine Plastizität. Kortikale Neuronen gehören möglicherweise zu den ersten Zellen, die gebildet werden, wenn der menschliche Embryo Gestalt annimmt. Stammzellen und Neurogenese Wiederum entsprachen die 14C-Konzentrationen bei jeder Person den bei ihrer Geburt vorkommenden Werten – ein überzeugender Beweis dafür, dass sich die Neurogenese im Kortex auf die Zeit beschränkt, in der sich das Gehirn herausbildet. Der Kortex ist der Sitz von „höheren“ Funktionen wie Denken und Analyse und gilt als jener Teil des Gehirns, der den Menschen von anderen Arten unterscheidet. Eine in Nature Neuroscience veröffentlichte Studie macht deutlich, dass kortikale Neuronen möglicherweise zu den ersten Zellen gehören, die gebildet werden, wenn der menschliche Embryo Gestalt annimmt. Unter der Leitung von Colin Blakemore von der University of Oxford und Pasko Rakic von der Yale University bestimmten Forschende eine besondere Population von Neuronen, die sich in den ersten paar Wochen der Schwangerschaft herausbilden. Diese Vorläuferzellen entstehen in jenem Gebiet, das sich zum Kortex entwickelt, und sind vor anderen Neuronen vorhanden, die tiefer im Gehirn gebildet werden und dann im Laufe der Hirnentwicklung an Stellen im Kortex wandern. Die Vorläufer bilden auch eine andere Art von Marker-Proteinen – ein Hinweis darauf, dass es sich bei ihnen um eine einzigartige Zellpopulation handelt. Der Befund ist ein Hinweis darauf, dass die ersten Zellen des spezifisch „menschlichen“ Gehirns bereits in einem sehr frühen Stadium der embryonalen Entwicklung vorhanden sind, was für die Entwicklung des normalen Gehirns und möglicherweise auch für viele kognitive Störungen von Bedeutung ist 2. So deuten etwa neuere Erkenntnisse über den Autismus darauf hin, dass bei dieser Krankheit Abweichungen der kortikalen Entwicklung vorliegen (siehe In der Kindheit auftretende Störungen, S. 21). Die Verwendung von Stammzellen zu Therapiezwecken hängt davon ab, ob sie sich zu spezifischen Zelltypen entwickeln können, die dann genau 89 jene Funktion übernehmen, die zur Heilung einer bestimmten Krankheit nötig ist. Laut einem Bericht in Nature Neuroscience ist diese Plastizität oder „Pluripotenz“ begrenzt 3. Unter der Leitung von Sally Temple vom Albany Medical College in New York fanden Forschende heraus, dass die zeitliche Koordinierung, welche die kortikale Entwicklung bestimmt, in den Vorläuferzellen kodiert ist, aus denen diese Neuronen entstehen – sie wird also nicht von Faktoren aus der Umgebung der Neuronen signalisiert. Der Kortex entwickelt sich in Schichten, und die Neuronen einer jeden Schicht werden gemäss einem vorhersehbaren Zeitplan gebildet. Die Forschenden stellten fest, dass Neuronen aus neuralen Vorläuferzellen, die Mäusen entnommen und isoliert in Kulturen aufgezogen wurden, in derselben Reihenfolge in Erscheinung traten, wie dies im Gehirn des Embryos der Fall gewesen wäre. Mit jedem Stadium büssten die Zellen etwas von ihrer Plastizität ein. Indem die Forschenden ein Foxg1 genanntes Gen, das für die Entwicklung des Kortex nötig ist, reduzierten, konnten sie den zeitlichen Ablauf bei Neuronen der mittleren Gestation zurücksetzen, nicht aber den der späten Gestation. Der Befund hat wichtige Implikationen für die Stammzelltherapie, da er darauf hindeutet, dass die Abfolge der Entwicklung von allem Anfang an programmiert ist und die Zellen nur während eines kurzen Zeitfensters von ihrem „Schicksal“ abgelenkt werden können. Eine natürliche Reaktion auf Verletzungen Viele Tierstudien haben aufgezeigt, dass das adulte Gehirn auf Verletzungen oder einen „Insult“ mit einem akuten Anstieg der Neurogenese reagieren kann – eine Fähigkeit, die zur Behandlung von Verletzungen infolge eines Traumas oder eines Hirnschlags genutzt werden könnte, wenn man die genauen Schritte verstehen würde. Einem Bericht im Journal of Neuroscience zufolge stellten T. Yamashita und Mitarbeitende fest, dass eine Gruppe von neuralen Stammzellen, die normalerweise nur olfaktorische Zellen bildet, nach einem Hirnschlag in der Lage ist, im Striatum, wo die Schädigung erfolgt war, neue Neuronen hervorzubringen 4. Diese neuen Neuronen waren fähig, Verbindungen mit angrenzenden Zellen des Striatum einzugehen. Der Befund hat Implikationen für die Behandlung des Hirnschlags und anderer neurologischer Erkrankungen. 90 David Greenberg und seine Forschungsgruppe vom Buck Institute for Age Research wollten herausfinden, welche Auswirkungen Neurogenese auf Neurogenese könnte auch als natürliche Reaktion auf Rückenmarkverletzungen vorkommen und zu therapeutischen Zwecken genutzt werden. Stammzellen und Neurogenese die Genesung nach einem Hirnschlag beim Menschen hat und suchten in Hirnbiopsien aus Hirnschlag-induzierten Hirnläsionen, nach neu gebildeten Neuronen. Wie sie in Proceedings of the National Academy of Sciences berichteten, gab es in den Gebieten rund um die verletzte Stelle molekulare Marker von neu gebildeten Neuronen – insbesondere in der Nähe von Blutgefässen, die Wachstumsfaktoren bilden, welche die Teilung und das Wachstum von Neuronen während der Neurogenese fördern 5. Der Befund lässt darauf schliessen, dass Neurogenese in einem gewissen Ausmass stattfindet und dass es therapeutisch sinnvoll sein könnte, diesen Vorgang medikamentös zu unterstützen. Neurogenese könnte auch als natürliche Reaktion auf Rückenmarkverletzungen vorkommen und zu therapeutischen Zwecken genutzt werden. Im Journal of Neuroscience berichteten Michael Tuszynski, Fred Gage und weitere Mitarbeitende vom Salk Institute, nach einer experimentell induzierten Rückenmarkverletzung sei die Zahl der neu geteilten Zellen Neurogenese und Rückenmarkverletzung Ein von Fred Gage (oben) und Michael Tuszynski – beide vom Salk Institute – geleitetes Team stellte fest, dass sich nach einer Rückenmarkverletzung neue Neuronen entwickeln. Dieser Vorgang könnte zu neuen Therapien führen. 91 bei erwachsenen Rhesusaffen um mehr als das 80fache erhöht 6. Sieben Monate nach der Verletzung hatten sich viele dieser Zellen zu unterschiedlichen Arten von Helferzellen entwickelt; einige bildeten Myelin, eine für die Axonen von verletzten Neuronen äusserst wichtige fetthaltige Isolierung. Mit dieser Arbeit wurde nachgewiesen, dass eine gewisse Neurogenese zur Wiederherstellung nach Rückenmarkverletzungen stattfindet; sie könnte durch entsprechende Therapien gefördert werden. Neurogenese und Epilepsie Einigen Tierstudien zufolge regen epileptische Anfälle Neurogenese an. Allerdings ergab eine Studie, die in einer Spezialausgabe der Zeitschrift Hippocampus vorgestellt wurde, dass die neu gebildeten Zellen nicht zu Ersatzneuronen heranwachsen, sondern zu Glia (zu Zellen, die „Hilfs“ Funktionen wahrnehmen, indem sie z. B. Myelin produzieren, aber keine Signale übermitteln) 7. Jack Parent und sein Team vom University of Michigan Medical Center fanden bei Ratten nach chemisch induzierten Krampfanfällen eine deutliche Vermehrung von Hirnzellen; dies wurde während der auf den Eingriff folgenden zweiwöchigen Beobachtungszeit durch eine Substanz angezeigt, die an in Teilung begriffenen Zellen bindet. Weshalb reagiert das Gehirn auf unterschiedliche Verletzungen mit der Produktion unterschiedlicher Zellen? Da sich diese Zellen, im Gegensatz zu jenen neugeborenen Neuronen, die nach einer hirnschlagbedingten Verletzung entstehen, zu Glia entwickelten, wirft diese Studie eine wichtige Frage auf: Weshalb reagiert das Gehirn auf unterschiedliche Verletzungen mit der Produktion unterschiedlicher Zellen? Weitere Forschungsarbeiten sollen über Neurogenese zu Reparaturzwecken Aufschluss geben und möglicherweise Wege zu neuen Behandlungen der Epilepsie weisen. 92 Ein anderes von Parent geleitetes Team fand heraus, dass die nach einem Krampfanfall einsetzende Neurogenese selbst zum Problem beitragen könnte. Bei Menschen mit Temporallappenepilepsie und auch in experimentellen Modellen, weist z. B. ein Teil des Hippokampus, die so genannte Körnerschicht des Gyrus dentatus, häufig Abweichungen auf. Die Forschenden berichteten in Annals of Neurology, dass bei Ratten mit länger dauernden Krämpfen Vorläuferzellen aus dieser Schicht abwandern und sich abnormal entwickeln 8. Obwohl also Neurogenese in gewissen Bereichen des Hippokampus während des ganzen Lebens andauert, wird Antidepressiva verstärken Neurogenese in einem bestimmten Stadium Antidepressiva steigern vermutlich die Neurogenese im Hippokampus. Allerdings dauert es bei den heute verfügbaren Medikamenten drei bis vier Wochen, bis sich die Stimmung von Kranken mit einer Depression bessert, und rund ein Drittel der Kranken spricht auf die Behandlung überhaupt nicht an. Viele Studien deuten darauf hin, dass die Wirkung von Medikamenten wie Fluoxetin (Prozac) letztlich auf der Förderung von Neurogenese beruht; ein besseres Verständnis der involvierten Schritte könnte zur Entwicklung von Medikamenten führen, welche Neurogenese direkter stimulieren. Stammzellen und Neurogenese die Migration der Neuronen – so die Vermutung der Forschenden – durch Krämpfe gestört, was zu einer fehlerhaften Integration neu gebildeter Zellen und möglicherweise zu erneuten Krämpfen führt. In Proceedings of the National Academy of Sciences berichteten Forschende der Cold Spring Harbor Laboratories, sie hätten einen Stamm von „Reporter“-Mäusen mit einem blau fluoreszierenden Protein im Kern jener Zellen entwickelt, die aus neuralen Vorläuferzellen stammten 9. Das Team testete die „blauen“ Neuronen auf verschiedene Markerproteine und brachte sie dann mit Fluoxetin (Prozac) in Kontakt; so stellten sie sicher, dass ausschliesslich Stammzellen in einem ganz bestimmten Entwicklungsstadium Ziel des Medikaments waren. Die Entdeckung von unmittelbareren Wegen zur Vermehrung dieser Zellpopulation könnte zu rascher einsetzenden und wirksameren Therapien der Depression führen. Krankheitsproteine und die Entwicklung von Hirnzellen Die krankmachende Wirkung des Prion-Proteins ist bestens bekannt: Falsch gefaltet ist es für Enzephalopathien verantwortlich, etwa Rinderwahn und sein menschliches Äquivalent, die Creutzfeldt-Jakob-Krankheit. Studien der letzten Jahre ergaben jedoch, dass Prion-Proteine nicht definitionsgemäss abnorm sind, sondern sich durch eine Auffaltung und Neufaltung in eine krankmachende Form verwandeln; ob es zu einer Krankheit kommt, hängt von der Menge der falsch gefalteten Prionen ab. Über den normalen Beitrag der Prion-Proteine zur Hirntätigkeit weiss man weniger. Jeffrey Macklis von der Harvard University, Susan Lindquist vom Massachusetts Institute of Technology und Mitarbeitende wiesen nach, dass Prion-Proteine in jenen Bereichen des Gehirns, in denen Neurogenese 93 Über den Rinderwahn hinaus Forschende haben mehr über die normale Aufgabe des Prionproteins herausgefunden, das vor allem für seine Rolle bei Krankheiten bekannt ist. Hier sieht man das normale Protein in den Nuklei von sich entwickelnden Neuronen (weisse Streifen). Die molekulare SpulenStruktur des normalen Moleküls ist im Hintergrund sichtbar. stattfindet, reichlich vorkommen. Ihre Studie, die in Proceedings of the National Academy of Sciences veröffentlicht wurde, zeigte einen engen Zusammenhang zwischen der Menge von Prion-Proteinen und der Zahl der Vorläuferzellen, die zu Neuronen differenzierten; bei experimentellen Mäusen mit einer Überproduktion des Proteins gab es mehr sich vermehrende Hirnzellen als bei normalen oder Knockout-Mäusen 10. Weitere Studien werden über die Aufgabe, die dieses Protein im normalen Hirn erfüllt, Aufschluss geben und möglicherweise zu neuen Methoden führen, um Prion-Erkrankungen zu verhindern oder zu heilen. Wie Stem Cells Development berichtete, wiesen Forschende unter der Leitung von Kiminobu Sugaya an der University of Central Florida nach, dass übermässige Mengen des amyloiden Vorläuferproteins, wie sie z. B. bei der Alzheimerschen Krankheit vorkommen, die Produktion neuer Neuronen hemmen könnten, indem sie Stammzellen dazu verleiten, sich zu Astrozyten zu entwickeln 11. 94 Wenn in Kulturen gezüchtete menschliche neurale Stammzellen mit diesem Protein stimuliert wurden, differenzierten sie häufiger zu Astrozyten, wohingegen die Hemmung des Proteins mithilfe eines Antikörpers diese Differenzierung verhinderte. Bei transgenen, Beta-Amyloid produzierenden Mäusen entwickelten sich transplantierte menschliche Stammzellen zu Glia und nicht zu Neuronen. Stützzellen werden krebsartig Derselbe Vorgang, der darüber bestimmt, aus welchen neuralen Stammzellen Neuronen und aus welchen Stützzellen hervorgehen, könnte eine weitere, verhängnisvollere Beobachtung erklären: Stammzellen können sich auch zu Tumoren zu entwickeln. Einem Bericht in Neuron zufolge, identifizierte ein von Arturo Alvarez-Buylla an der University of California in San Francisco geleitetes Team eine Gruppe neuraler Stammzellen, die Träger eines Rezeptors für einen Wachstumsfaktor waren 12. Die Infusion dieses Wachstumsfaktors führte dazu, dass diese Zellen exzessiv wuchsen und gewisse Charakteristika von Tumoren entwickelten (siehe auch Schädigungen des Nervensystems, S. 40). Stammzellen und Neurogenese Die Ergebnisse deuten darauf hin, dass hohe Konzentration des amyloiden Vorläuferproteins die Selbstheilungsbemühungen des Gehirns dadurch vereiteln könnten, dass sie den Kurs von Zellen ändern, die sonst zu Ersatzneuronen herangewachsen wären; diese Funktion gilt es besser zu verstehen, bevor wir Stammzellen dereinst als Therapie der Alzheimerschen Krankheit und weiterer Erkrankungen einsetzen können. Derselbe Vorgang, der darüber bestimmt, aus welchen neuralen Stammzellen Neuronen und aus welchen Stützzellen hervorgehen, könnte eine weitere, verhängnisvollere Beobachtung erklären: Stammzellen können sich auch zu Tumoren zu entwickeln. Inzwischen berichteten Patricia Casaccia-Bonnefil von der Robert Wood Johnson Medical School und ihre Mitarbeitenden im Journal of Neuroscience, Glia könnte infolge fehlender Apoptose, also des programmierten Zelltods, krebsartig werden 13. Die Forschenden untersuchten KnockoutMäuse denen das Gen p53, das die Apoptose in Gang setzt, fehlte. Das Fehlen von p53 hatte nicht automatisch Krebs zur Folge. Wenn man jedoch die Mäuse einem experimentellen, Krebs verursachenden Stimulus aussetzte, kam es zu drastischen, dem Krebs entsprechenden Veränderungen der neuralen Stammzellen – beispielsweise teilten sie sich rascher und differenzierten nicht vollständig. Das Protein Notch aktiviert Stammzellen Das eigentliche Ziel der Stammzelltherapie besteht darin, „endogene“ Stammzellen zu aktivieren – also jene, die im Körper der Kranken selbst vorhanden sind. In Nature berichtete Ronald McKay vom National Institute of Neurological Disorders and Stroke über ein Modell der StammzellExpansion, das zur Verwirklichung dieses Ziels beitragen könnte 14. 95 Die Aktivierung des Rezeptors Notch induziert eine Folge von Ereignissen, die das Überleben von neuralen Stammzellen fördern. Wenn man adulte Ratten mit einem Molekül behandelte, das an den Notch-Rezeptor bindet, vermehrte sich die Zahl der Vorläuferzellen, und nach einer Schädigung durch einen experimentellen Hirnschlag verbesserte sich ihre Bewegungsfähigkeit. Diese Arbeit weist auf eine Methode der StammzellExpansion sowohl in Kulturen als auch in einem mit Transplantaten behandelten Wirtstier hin, dank der Stammzellen sogar erneut angeschaltet werden könnten. Das Forschungsgebiet der Stammzellen floriert, aber die oben vorgestellten Studien zeigen, wie viel immer noch nicht wissen. Die Entwicklung von Stammzellen so zu lenken, dass aus ihnen bestimmte Zelltypen (Nervenzellen oder Glia) werden und nicht etwa Krebszellen, bleibt auch weiterhin die grosse Herausforderung. 96 Denken und Erinnern Alzheimersche Krankheit 98 Wer das Vollbild der Alzheimerschen Krankheit entwickelt 100 Eine Ursache der frontotemporalen Demenz 101 Das normale Gedächtnis – ein grosser Schritt vorwärts 102 97 D ie Erforschung des Denkens und Erinnerns führte 2006 zu uneinheitlichen Resultaten: in manchen Bereichen gab es bemerkenswerte Entdeckungen, in anderen zeigte sich, dass es notwendig ist, eine Pause einzulegen und die Ansätze neu zu überdenken. Alzheimersche Krankheit Kennzeichnend für die Alzheimersche Krankheit ist das Vorhandensein von Beta-Amyloid Plaques im Gehirn der Kranken. Seit etwa einem Jahrzehnt konzentrierten sich viele wissenschaftliche Arbeiten auf diese physiologischen Manifestationen der Krankheit; dahinter stand die Absicht, die Bildung von Plaques womöglich zu verhindern oder diese zu beseitigen, um dadurch die Auswirkungen der Krankheit auf das Verhalten zu mildern. Doch kamen 2006 verschiedene Gruppen aufgrund ihrer Ergebnisse zum Schluss, bei den Plaques selbst handle es sich nicht um die eigentliche Ursache der Krankheit. Die Beta-Amyloid Plaques sind Ansammlungen eines kleinen Peptids, das von einem grösseren Protein, dem so genannten Amyloid-Vorläufer-Protein, abgetrennt und in den Bereich zwischen Neuronen freigesetzt wird. Frühere Arbeiten mit Mäusen, die das menschliche Amyloid-VorläuferProtein exprimieren, zeigten, dass Verhaltensauffälligkeiten, etwa Defizite im räumlichen Gedächtnis, lange vor dem zu Tage treten von Plaques bestanden. Somit sind entweder nicht die Proteinfragmente das Problem, oder aber die Neuronen werden durch kleinere, nicht wie Plaques aussehende Ansammlungen beschädigt. Eine einzige Plaque besteht aus hunderten oder tausenden von Proteinfragmenten, doch Sylvain Lesné von der University of Minnesota Medical School in Minneapolis und seine Mitarbeitenden entdeckten, dass zu der Zeit, als das Gedächtnis der Tiere nachzulassen begann, kleine Ansammlungen von nur gerade zwölf Fragmenten vorhanden waren. 98 Mehr noch, als die Forschenden diese kleinen, dem Gehirn von verstorbenen Tieren entnommenen Klumpen reinigten und ins Gehirn von gesunden Tieren injizierten, verloren diese die Fähigkeit, sich die räumliche Gestalt eines Labyrinths einzuprägen. Diese Forschungsarbeit wurde in Nature vorgestellt 1. Schuld daran schien bei diesen Tieren ein anderes kleines Fragment des Amyloid-Vorläufer-Proteins zu sein, das so genannte C-31. Die Forschenden folgern daraus, dass die zwischen den Neuronen liegenden Plaques möglicherweise für den Beginn der Einbussen verantwortlich sind, dass aber C-31 den vernichtenden Schlag versetzt, indem es ins Innere der Nervenzellen gelangt. Beide Forschungsgruppen vermuten, Medikamente, welche entweder die Bildung der kleinen Klumpen oder die Freisetzung von C-31 verhindern, könnten dazu beitragen, beim Menschen das Ausmass der durch die Alzheimersche Krankheit hervorgerufenen Symptome und Schädigung zu begrenzen. Denken und Erinnern Etwas Ähnliches berichteten Forschende des Buck Institute for Age Research in Novato, Kalifornien, in Proceedings of the National Academy of Sciences; wenn die genetisch veränderten Mäuse eine Proteinvariante exprimierten, aus der das Beta-Amyloid nicht freigesetzt werden kann, fehlten ihnen zwar die für die Alzheimersche Krankheit typischen Plaques, aber es kam dennoch zu Gedächtnisstörungen 2. Auch Forschungsarbeiten an Menschen stellen heute die Bedeutung der Beta-Amyloid Plaques als eine Ursache der Krankheitssymptome bei Alzheimer in Frage. Die Wissenschaft weiss seit Jahrzehnten, dass die Krankheit nicht bei allen Personen mit Plaques ausbricht. Um herauszufinden, wie verbreitet Plaques im Gehirn von gesunden Erwachsenen sind, untersuchten Forschende unter der Leitung von David Bennett vom Rush Alzheimer’s Disease Center an der Universität Chicago mehr als 2000 gesunde Erwachsene in zwei verschiedenen Gemeinden. Ihre Ergebnisse veröffentlichten sie in Neurology 3. Die Studienteilnehmenden werden jedes Jahr neuropsychologisch getestet; dadurch soll sichergestellt werden, dass sie bei ihrem Ableben nicht an Demenz leiden. Von den 134 Teilnehmenden, die bisher gestorben sind und ihr Gehirn für eine Obduktion zur Verfügung stellten, wiesen zwei Plaques im Neokortex des Gehirns auf, was den aktuellen Kriterien der Pathologie zufolge besagt, dass sie mit grosser Wahrscheinlichkeit an der Alzheimerschen Krankheit litten; bei weiteren 48 wurden Plaques in den limbischen Regionen des Gehirns festgestellt, was einem mittleren Risiko entspricht. Der einzige Unterschied, den Bennett und seine Mitarbeitenden bezüglich der mentalen Funktion dieser 50 Kranken im Vergleich mit den übrigen 84 Teilnehmenden ohne nachweisbare Plaques fanden, betraf eine leichte Abnahme des episodischen oder ereignisorientierten Gedächtnisses. 99 Aufgrund dieser Daten kam Bennetts Team zu zwei Schlussfolgerungen. Erstens weisen sie darauf hin, dass bereits ein geringfügiges Nachlassen des episodischen Gedächtnisses den Beginn einer Alzheimerschen Krankheit anzeigen kann. Zweitens verfügen Menschen generell über mehr Neuronen als sie im Alltag brauchen – die Forschenden sprechen von einer „neurologischen Reserve“. Deshalb können in vielen Fällen beträchtliche neuronale Schädigungen und eine für Alzheimer typische Pathologie vorliegen, ohne dass es zu einem drastischen Gedächtnisverlust oder einer Demenz kommt. Woran es liegt, dass einige Kranke trotz Plaques gesund bleiben, während die Krankheit bei anderen, welche dieselbe Neuropathologie aufweisen, ausbricht, ist eine Schlüsselfrage. Woran es liegt, dass einige Kranke trotz Plaques gesund bleiben, während die Krankheit bei anderen, welche dieselbe Neuropathologie aufweisen, ausbricht, ist eine Schlüsselfrage, die heute für viele Forschende im Mittelpunkt steht. Wer das Vollbild der Alzheimerschen Krankheit entwickelt Ähnliche Forschungsarbeiten geben Aufschluss darüber, weshalb nicht alle älteren Erwachsenen mit Gedächtnisstörungen das Vollbild der Alzheimerschen Krankheit entwickeln. Es gibt keinen anerkannten Test um vorauszusagen, bei welchen Kranken der Zustand stabil bleibt und bei welchen er sich weiter verschlimmern wird. Ein solches Wissen könnte die ärztliche Beratung von Kranken und ihren Familien verbessern und dazu beitragen, das weitere Vorgehen in jenen Fällen zu planen, bei denen die Alzheimersche Krankheit aller Wahrscheinlichkeit nach ausbrechen wird. Zwei Studien, über die 2006 berichtet wurde, stellen bedeutende Schritte in diese Richtung dar. In der einen Studie beobachteten Forschende des New York State Psychiatric Institute und der Columbia University in New York, unter der Leitung von Matthias Tabert 63 gesunde Erwachsene und 148 Kranke mit einer leichten kognitiven Beeinträchtigung, einem Zwischenstadium zwischen normaler Gedächtnisfunktion und Demenz. Wie die Forschenden in Archives of General Psychiatry berichten, litten nach drei Jahren 34 der Kranken mit einer leichten kognitiven Beeinträchtigung an der Alzheimerschen Krankheit 4. 100 Dabei war das Risiko einer Verschlechterung für jene Kranken mit einer leichten kognitiven Beeinträchtigung, die ausschliesslich Gedächtnisdefizite Denken und Erinnern aufwiesen, relativ gering; nur bei zwei von 20 Kranken dieser Gruppe entwickelte sich die Alzheimersche Krankheit. Im Gegensatz dazu, entwickelte sich im selben Zeitraum bei der Hälfte jener 64 Kranken, die von Anfang an ausser Gedächtnisproblemen auch andere kognitive Defizite aufwiesen, die Alzheimersche Krankheit. Aufgrund neuropsychologischer Tests könnte man somit bei Kranken mit einer leichten kognitiven Beeinträchtigung diese beiden Situationen unterscheiden und vorhersagen, wer mit grösster Wahrscheinlichkeit mit weiteren Problemen rechnen muss. Unterdessen gelang es Forschenden der University of California in Los Angeles, anhand körperlicher Merkmale, zu bestimmen, welche Kranken mit einer leichten kognitiven Beeinträchtigung besonders gefährdet waren, die Alzheimersche Krankheit zu entwickeln 5. Mittels hochauflösender Magnetresonanz-Tomographie stellten sie einem Bericht in Archives of Neurology zufolge fest, dass Kranke, deren Hippokampus ein kleineres Volumen aufwies, mehr gefährdet waren, die Alzheimersche Krankheit zu entwickeln als solche mit einem grösseren Volumen. Ausserdem wiesen Kranke, bei denen sich später das Vollbild der Krankheit entwickelte, zu Beginn der Studie eine stärkere Atrophie in bestimmten Regionen des Hippokampus auf als Kranke, deren Zustand stabil blieb. Falls es gelingen sollte, Therapien zu entwickeln, die den Ausbruch der Alzheimerschen Krankheit verhindern oder hinausschieben, wäre es äusserst wichtig, diese „Prä-Alzheimer“-Fälle zu identifizieren. Eine Ursache der frontotemporalen Demenz Zwar handelt es sich bei der Alzheimerschen Krankheit um jene Art von Demenz, über die am meisten gesprochen wird, doch ist sie keineswegs die einzige. Die frontotemporale Demenz ist bei unter 65jährigen die zweithäufigste Art von Demenz. Kranke mit dieser Art Demenz weisen Verhaltensauffälligkeiten sowie Persönlichkeitsveränderungen und eine Enthemmung auf. Im Allgemeinen bleibt jedoch ihr Gedächtnis intakt. Die frontotemporale Demenz hat eine starke genetische Komponente, und man weiss bereits, dass einige Formen dieser Krankheit durch Mutationen des Mikrotubuli-assoziierten Proteins Tau verursacht werden. Bei Kranken ohne Mutationen des Tau-Gens war die Krankheitsursache bisher jedoch unbekannt. Im Jahr 2006 entdeckten zwei Forschungsgruppen bei diesen Kranken Mutationen des Gens für einen Wachstumsfaktor, das so genannte Progranulin. 101 Dieses Gen wird in vielen verschiedenen Neuronen der Hirnrinde und in Mikrogliazellen, den Immunzellen des Gehirns, exprimiert. In zwei Studien, über die in Nature berichtet wurde, stellten Forschende die Hypothese auf, Progranulin sei für das Überleben von Neuronen wichtig und der Verlust einer einzigen Kopie des Progranulin-Gens reiche aus, um eine Nervendegeneration zu verursachen 6, 7. In Tiermodellen scheint Progranulin die Expression anderer Wachstumsfaktoren zu induzieren, die möglicherweise zum Überleben von Zellen beitragen. Die Identifizierung der Mutation, die dieser Art der frontotemporalen Demenz zugrunde liegt, eröffnet Möglichkeiten, Therapien für diese Kranken zu entwickeln. Das normale Gedächtnis – ein grosser Schritt vorwärts Die Wissenschaft vermutet seit langem, die Speicherung von Erinnerungen erfolge über Veränderungen der Stärke der synaptischen Verbindungen zwischen Neuronen. Sobald also eine Erinnerung gespeichert wird, würde die Stärke der Synapse und damit ihre Kommunikationsfähigkeit mit ihrem Nachbarn wachsen. Diesen Vorgang nennt man Langzeitpotenzierung (long-term potentiation, LTP). Heute liegen drei Studien mit entscheidenden Belegen dafür vor, dass LTP tatsächlich die neurale Grundlage für den Aufbau des Gedächtnisses darstellt. Forschende richteten ihr Augenmerk auf drei Kriterien: Die Hemmung von LTP durch chemische Inhibitoren müsste Lernen verhindern; das Erlernen einer bestimmten Aufgabe oder Information müsste in der dafür zuständigen Hirnregion LTP hervorrufen; und nach erfolgtem Lernen müsste das Auslöschen der LTP durch chemische Substanzen zu einer Amnesie führen und das erlernte Verhalten beseitigen. Frühere Forschungsarbeiten hatten nachgewiesen, dass das erste Kriterium erfüllt wurde. 102 In einem der Experimente des Jahres 2006 versetzten Jonathan Whitlock und Mitarbeitende am Howard Hughes Medical Institute und am Massachusetts Institute of Technology Ratten jedes Mal, wenn sie den dunklen Bereich ihres Käfigs aufsuchten, einen leichten elektrischen Schock, so dass sie lernten, diesen Ort zu meiden. Wie die Gruppe in Science berichtete, kam es, während die Tiere diese räumliche Information lernten, zu einer LTP im Hippokampus, der bei Nagern für räumliches Lernen zuständig ist 8. Denken und Erinnern Presynaptisches Neuron Neurotransmitter Lernen Rezeptoren Postsynaptisches Neuron Ionen Protein PKM-Zeta Danke für das Gedächtnis Forschende stellten fest, dass die Langzeitpotenzierung, welche dem Gedächtnis zugrunde liegt, vom Protein PKM-Zeta abhängt. Wenn dieses Protein gehemmt wird, vergessen Ratten ein gelerntes Verhalten. Agnès Gruart von der Universidad Pablo de Olavide in Sevilla, Spanien, berichtete im Journal of Neuroscience von ähnlichen Resultaten 9. Ihr Team stellte fest, dass Lernen im Hippokampus von Mäusen eine LTP induzierte, und dass Substanzen, welche die neurale Übermittlung verhinderten, sowohl das Lernen als auch die Entwicklung von LTP hemmten. Eine von Eva Pastalkova am SUNY Downstate Medical Center in Brooklyn, New York, geleitete Gruppe führte diese Idee in ihrer Studie einen Schritt weiter und berichtete darüber in derselben Ausgabe von Science. Sie wiesen nach, dass Tiere das erlernte Verhalten vergassen, wenn die LTP chemisch rückgängig gemacht wurde 10. Die Behandlung verunmöglichte jedoch nicht jegliche synaptische Übertragung und verhinderte auch nicht späteres Lernen. Diese Studien liefern wichtige Hinweise darauf, dass die langjährige Hypothese über den Aufbau von Erinnerungen, wahrscheinlich richtig ist. 103 Referenzen Einleitung 1 Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA, Golbe LI, Mark MH, Racette BA, Perlmutter JS, Parsian A, Guttman M, Nicholson G, Xu G, Wilk JB, Saint-Hilaire MH, DeStefano AL, Prakash R, Williamson S, Suchowersky O, Labelle N, Growdon JH, Singer C, Watts RL, Goldwurm S, Pezzoli G, Baker KB, Pramstaller PP, Burn DJ, Chinnery PF, Sherman S, Vieregge P, Litvan I, Gillis T, MacDonald ME, Myers RH, and Gusella JF. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease. Archives of Neurology. 2006 63(6):826-832. 2 Hedrich K, Hagenah J, Djarmati A, Hiller A, Lohnau T, Lasek K, Grunewald A, Hilker R, Steinlechner S, Boston H, Kock N, Schneider-Gold C, Kress W, Siebner H, Binkofski F, Lencer R, Munchau A, and Klein C. Clinical spectrum of homozygous and heterozygous PINK1 mutation in a large family with Parkinson disease. Archives of Neurology. 2006 63(6):833-838. 3 Hovatta I, Tennant RS, Helton R, Marr RA, Singer O, Redwine JM, Ellison JA, Schadt EE, Verma IM, Lockhart DJ, and Barlow C. Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature. 2005 438(7068):662-666. 4 Meyer-Lindenberg A, Buckholtz JW, Kolachana B, R Hariri A, Pezawas L, Blasi G, Wabnitz A, Honea R, Verchinski B, Callicott JH, Egan M, Mattay V, and Weinberger DR. Neural mechanisms of genetic risk for impulsivity and violence in humans. Proceedings of the National Academy of Sciences USA. 2006 103(16):6269-6274. 5 Shaw P, Lerch J, Greenstein D, Sharp W, Clasen L, Evans A, Giedd J, Castellanos FX, and Rapoport J. Longitudinal mapping of cortical thickness and clinical outcome in children and adolescents with attention-deficit/hyperactivity disorder. Archives of General Psychiatry. 2006 63(5):540-549. 6 Dapretto M, Davies MS, Pfeifer JH, Scott AA, Sigman M, Bookheimer SY, and Iacoboni M. Understanding emotions in others: Mirror neuron dysfunction in children with autism spectrum disorders. Nature Neuroscience. 2006 9(1):28-30. 7 McBride JL, Ramaswamy S, Gasmi M, Bartus RT, Herzog CD, Brandon EP, Zhou L, Pitzer MR, Berry-Kravis EM, and Kordower JH. Viral delivery of glial cell line-derived neurotrophic factor improves behavior and protects striatal neurons in a mouse model of Huntington’s disease. Proceedings of the National Academy of Sciences USA. 2006 103(24):9345-9350. 8 Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, and Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006 441(7090):235-238. 9 Weinshenker BG, Wingerchuk DM, Vukusic S, Pittock SJ, and Lennon VA. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Annals of Neurology. 2006 59(3):566-569. 10 White, PM, Doetzlhofer A, Lee YS, Groves AK, and Segil N. Mammalian cochlear supporting cells can divide and trans-differentiate into hair cells. Nature. 2006 441(7096):984-987. 11 Steele AD, Emsley JG, Ozdinler PH, Lindquist S, and Macklis JD. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proceedings of the National Academy of Sciences USA. 2006 103(9):3416-3421. 105 12 Yamashita T, Ninomiya M, Hernandez Acosta P, Garcia-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, and Sawamoto K. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. Journal of Neuroscience. 2006 26(24):6627-6636. In der Kindheit auftretende Störungen 1 Dapretto M, Davies MS, Pfeifer JH, Scott AA, Sigman M, Bookheimer SY, and Iacoboni M. Understanding emotions in others: Mirror neuron dysfunction in children with autism spectrum disorders. Nature Neuroscience. 2006 9(1):28-30. 2 Hardan AY, Muddasani S, Vemulapalli M, Keshavan MS, and Minshew NJ. An MRI Study of Increased Cortical Thickness in Autism. American Journal of Psychiatry. 2006 163(7):1290-1292. 3 Munson J, Dawson G, Abbott R, Faja S, Webb SJ, Friedman SD, Shaw D, Artru A, and Dager SR. Amygdala volume and behavioral development in autism. Archives of General Psychiatry. 2006 63(6):686-693. 4 Petropoulos H, Friedman SD, Shaw DWW, Artru AA, Dawson G, and Dager SR. Gray matter abnormalities in autism spectrum disorder revealed by T2 relaxation. Neurology. 2006 67(4):632-636. 5 Gilbert DL, Wang Z, Sallee FR, Ridel KR, Merhar S, Zhang J, Lipps TD, White C, Badreldin N, and Wassermann EM. Dopamine transporter genotype influences the physiological response to medication in ADHD. Brain. 2006 129(Pt. 8):2038-2046. 6 Smith AB, Taylor E, Brammer M, Toone B, and Rubia K. Task-specific hypoactivation in prefrontal and temporoparietal brain regions during motor inhibition and task switching in medicationnaive children and adolescents with attention deficit hyperactivity disorder. American Journal of Psychiatry. 2006 163(6):1044-1051. 7 Shaw P, Lerch J, Greenstein D, Sharp W, Clasen L, Evans A, Giedd J, Castellanos FX, and Rapoport J. Longitudinal mapping of cortical thickness and clinical outcome in children and adolescents with attentiondeficit/hyperactivity disorder. Archives of General Psychiatry. 2006 63(5):540-549. 8 Gibson CS, MacLennan AH, Goldwater PN, Haan EA, Priest K, and Dekker GA. Neurotropic viruses and cerebral palsy: Population based casecontrol study. British Medical Journal. 2006 332(7533):76-80. Bewegungsstörungen und andere Störungen der Motorik 106 1 Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, and Südhof TC. Alpha-synuclein cooperates with CSP-alpha in preventing neurodegeneration. Cell. 2005 123(3):383-396. 2 Quilty MC, King AE, Gai WP, Pountney DL, West AK, Vickers JC, and Dickson TC. Alphasynuclein is upregulated in neurones in response to chronic oxidative stress and is associated with neuroprotection. Experimental Neurology. 2006 199(2):249-256. 3 Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, and Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006 311(5766):1471-1474. 4 Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, and Lindquist S. Alpha-synuclein blocks ER-golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006 313(5785):324-328. Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT, McLean PJ, Young AB, Housman DE, Kazantsev AG. Pharmacological promotion of inclusion formation: A therapeutic approach for Huntington’s and Parkinson’s diseases. Proceedings of the National Academy of Sciences USA. 2006 103(11):4246-4251. 6 Cookson, MR. Hero vs. antihero: The multiple roles of alpha-synuclein in neurodegeneration. Experimental Neurology. 2006 199(2):238-242. 7 Bower JH, Maraganore DM, Peterson BJ, Ahlskog JE, and Rocca WA. Immunologic diseases, anti-inflammatory drugs, and Parkinson disease: A case-control study. Neurology. 2006 67(3):494-496. 8 Hernán MA, Logroscino G, and García Rodriguez LA. Nonsteroidal antiinflammatory drugs and the incidence of Parkinson disease. Neurology. 2006 66(7):1097-1099. 9 Alano CC, Kauppinen TM, Valls AV, and Swanson RA. Minocycline inhibits poly (ADP-ribose) polymerase-1 at nanomolar concentrations. Proceedings of the National Academy of Sciences USA. 2006 103(25):9685-9690. 10 The NINDS NET-PARKINSON’S DISEASE Investigators. A randomized, double-blind, futility clinical trail of creatine and minocycline in early Parkinson disease Neurology. 2006 66(5):664-671. 11 Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, and Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006 441(7097):1157-1161. 12 Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, and Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006 441(7097):1162-1166. 13 Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA, Golbe LI, Mark MH, Racette BA, Perlmutter JS, Parsian A, Guttman M, Nicholson G, Xu G, Wilk JB, Saint-Hilaire MH, DeStefano AL, Prakash R, Williamson S, Suchowersky O, Labelle N, Growdon JH, Singer C, Watts RL, Goldwurm S, Pezzoli G, Baker KB, Pramstaller PP, Burn DJ, Chinnery PF, Sherman S, Vieregge P, Litvan I, Gillis T, MacDonald ME, Myers RH, and Gusella JF. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease. Archives of Neurology. 2006 63(6):826-832. 14 Hedrich K, Hagenah J, Djarmati A, Hiller A, Lohnau T, Lasek K, Grunewald A, Hilker R, Steinlechner S, Boston H, Kock N, Schneider-Gold C, Kress W, Siebner H, Binkofski F, Lencer R, Munchau A, and Klein C. Clinical spectrum of homozygous and heterozygous PINK1 mutation in a large family with Parkinson disease. Archives of Neurology. 2006 63(6):833-838. 15 Zadikoff C, Rogaeva E, Djarmati A, Sato C, Salehi-Rad S, St George- Hyslop P, Klein C, and Lang AE. Homozygous and heterozygous PINK1 mutations: Considerations for diagnosis and care of Parkinson’s disease patients. Movement Disorders 2006 21(6): 875-879. 16 Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, Brooks DJ, and Piccini P. Microglial activation correlates with severity in Huntington’s disease. A clinical and PET study. Neurology. 2006 66(11):1638-1643. 17 Chebrolu H, Slevin JT, Gash DA, Gerhardt GA, Young B, Given CA, and Smith CD. MRI volumetric and intensity analysis of the cerebellum in Parkinson’s disease patients infused with glialderived neurotrophic factor (GDNF). Experimental Neurology. 2006 198(2):450-456. 18 Lang AE, Gill S, Patel NK, Lozano A, Nutt JG, Penn R, Brooks DJ, Hotton G, Moro E, Heywood P, Brodsky MA, Burchiel K, Kelly P, Dalvi A, Scott B, Stacy M, Turner D, Wooten VG, Elias WJ, Laws ER, Dhawan V, Stoessl AJ, Matcham J, Coffey RJ, and Traub M. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Annals of Neurology. 2006 59(3):459-466. Referenzen 5 107 19 Slevin JT, Gash DM, Smith CD, Gerhardt GA, Kryscio R, Chebrolu H, Walton A, Wagner R, andYoung AB. Unilateral intraputaminal glial cell linederived neurotrophic factor in patients with Parkinson disease: Response to 1 year each of treatment and withdrawal. Neurosurgical Focus. 2006 20(5):E1-E7. 20 McBride JL, Ramaswamy S, Gasmi M, Bartus RT, Herzog CD, Brandon EP, Zhou L, Pitzer MR, Berry-Kravis EM, and Kordower JH. Viral delivery of glial cell line-derived neurotrophic factor improves behavior and protects striatal neurons in a mouse model of Huntington’s disease. Proceedings of the National Academy of Sciences USA. 2006 103(24):9345-9350. 21 Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: A randomized controlled trial. Neurology. 2006 66(3):366-372. Schädigungen des Nervensystems 108 1 Hochberg LR, Serruva MD, Friehs GM, Mukand JA, Saleh M, Caplan AH, Branner A, Chen D, Penn RD, and Donoghue JP. Neuronal ensemble control of prosthetic devices by a human with tetraplegia. Nature. 2006 442(7099):164-171. 2 Massey JM, Hubscher CH, Wagoner MR, Decker JA, Amps J, Silver J, and Onifer SM. Chondroitinase ABC digestion of the perineuronal net promotes functional collateral sprouting in the cuneate nucleus after cervical spinal cord injury. Journal of Neuroscience. 2006 26(16):4406-4414. 3 Yang LJ, Lorenzini I, Vajn K, Mountney A, Schramm P, Schnaar RL. Sialidase enhances spinal axon outgrowth in vivo. Proceedings of the Nacional Academy of Sciences USA. 2006 103(29):11057-11062. 4 Yin Y, Henzl MT, Lorber B, Nakazawa T, Thomas TT, Jiang F, Langer R, and Benowitz LI. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nature Neuroscience. 2006 9(6):843-852. 5 Shirasaki R, Lewcock JW, Lettieri K, and Pfaff SL. FGF as a target-derived chemoattractant for developing motor axons genetically programmed by the LIM code. Neuron. 2006 15(50):841-853. 6 Medeiros NA, Burnette DT, and Forscher P. Myosin II functions in actinbundle turnover in neuronal growth cones. Nature Cell Biology. 2006 8(3):215-226. 7 Houle JD, Tom VJ, Mayes D, Wagoner G, Phillips N, and Silver J. Combining an autologous peripheral nervous system “bridge” and matriz modification by chondroitinase allows robust, functional regeneration beyond a hemisection lesion of the adult rat spinal cord. Journal of Neuroscience. 2006 12(28):7405-7415. 8 Deshpande DM, Kim YS, Martinez T, Carmen J, Dike S, Shats I, Rubin LL, Drummond J, Krishnan C, Hoke A, Maragakis N, Shefner J, Rothstein JD, and Kerr DA. Recovery from paralysis in adult rats using embryonic ítem cells. Annals of Neurology. 2006 60(1):32-44. 9 Deng YZ, Reeves MJ, Jacobs BS, Birbeck GL, Kothari RU, Hickenbottom SL, Mullard AJ, Wehner S, Maddox K, Majid A and Coverdell P, for the National Acute Stroke Registry Michigan Prototype Investigators. IV tissue plasminogen activator use in acute stroke: Experience from a statewide registry. Neurology. 2006 66(3):306-312. 10 Lees KR, Zivin JA, Ashwood T, Davalos A, Davis SM, Diener HC, Grotta J, Lyden P, Shuaib A, Hardemark HG, and Wasiewski WW for the Stroke-Acute Ischemic NXY Treatment (SAINT I) Trial Investigators. NXY-059 for acute ischemic stroke. New England Journal of Medicine. 2006 354(6):588-600. Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, Shi Q, McLendon RE, Bigner DD, and Rich JN. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. American Association for Cancer Research. 2006 66(16):784-788. 12 Sun L, Hui AM, Su Q, Vortmeyer A, Kotliarov Y, Pastorino S, Passaniti A, Menon J, Walling J, Bailey R, Rosenblum M, Mikkelsen T, and Fine HA. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell. 2006 9(4):287-300. 13 Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones- Hinojosa A, VandenBerg S, and Alvarez-Buylla A. PDGRR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006 51(2):187-199. Referenzen 11 Neuroethik 1 Khanna S, Vieta E, Lyons B, Grossman F, Eerdekens M, and Kramer M. Risperidone in the treatment of acute mania: A double-blind, placebo-controlled study. British Journal of Psychiatry. 2005 187(3):229-234. 2 Mudur G. Indian study sparks debate on the use of placebo in psychiatry trials. British Medical Journal. 2006 332(754):566. 3 Mohamed FB, Faro SH, Gordon NJ, Platek SM, Ahmad H, and Williams JM. Brain mapping of deception and truth telling about an ecologically valid situation: Functional MR imaging and polygraph investigation – initial experience. Radiology. 2006 238(2):679-688. 4 Editorial. Neuroethics needed. Nature. 2006 441(7096):907. 5 Editorial. What’s on your mind? Nature Neuroscience. 2006 9(8):981. 6 Eastman N and Campbell C. Neuroscience and legal determination of criminal responsibility. Nature Reviews Neuroscience. 2006 7(4):311-318. 7 Hochberg LR, Serruya MD, Friehs GM, Mukand JA, Saleh M, Caplan AH, Branner A, Chen D, Penn, RD, and Donoghue JP. Neuronal ensemble control of prosthetic devices by a human with tetraplegia. Nature. 2006 442(7099):164-171. 8 Scott S. Converting thoughts into action. Nature. 2006 442(7099):141-142. 9 Illes J, Kirschen MP, Edwards E, Stanford LR, Bandettini P, Cho MK, Ford PJ, Glover GH, Kulynych J, Macklin R, Michael DB, Wolf SM, and members of the Working Group on Incidental Findings in Brain Research. Incidental findings in brain imaging research. Science. 2006 311(5762):783-784. 10 Voss HU, Ulug AM, Dyke JP, Watts R, Kobylarz EJ, McCandliss BD, Heier LA, Beattie BJ, Hamacher KA, Vallabhajosula S, Goldsmith SJ, Ballon D, Giacino JT, and Schiff ND. Possible axonal regrowth in late recovery from the minimally conscious state. The Journal of Clinical Investigation. 2006 116(7):2005-2011. 11 Owen AM, Coleman MR, Boly M, Davis MH, Laureys S, and Pickard JD. Detecting awareness in the vegetative state. Science. 2006 313(5792):1402. Neuroimmunologische Erkrankungen 1 Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, and Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006 441(7090):235-238. 109 2 Iwakura Y and Ishigame H. The IL-23/IL-17 axis in inflammation. Journal of Clinical Investigation. 2006 116(5):1218-1222. 3 Weinshenker BG, Wingerchuk DM, Vukusic S, Pittock SJ, and Lennon VA. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Annals of Neurology. 2006 59(3):566-569. 4 Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, and Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. Journal of Experimental Medicine. 2005 202(4):473-477. 5 Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, and Ransohoff RM. Control of microglia neurotoxicity by the fractalkine receptor. Nature Neuroscience. 2006 9(7):917-924. 6 Butkovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, and Schwartz M. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proceedings of the National Academy of Sciences USA. 2006 103(31):11784-11789. 7 Tobinick E, Gross H, Weinberger A, and Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: A six-month pilot study. Online publication in Medscape General Medicine. 2006: http://www.medscape.com/viewarticle/529176. 8 Lee H, Zhu X, Nunomur A, Perry G, and Smith MA. Amyloid beta: The alternate hypothesis. Current Alzheimer Research. 2006 3(1):75-80. 9 Belmadani A, Tran PB, Ren D, and Miller RJ. Chemokines regulate the migration of neural progenitors to sites of inflammation. Journal of Neuroscience. 2006 26(12):3182-3191. 10 Trang T, Beggs S, and Salter MW. Purinoceptors in microglia and neuropathic pain. European Journal of Physiology. 2006 452(5):645-652. 11 Sung YJ, Chiu DT, and Ambron RT. Activation and retrograde transport of protein kinase G in rat nociceptive neurons after nerve injury and inflammation. Neuroscience. 2006 141(2):697-709. 12 Syken J, GrandPre T, Kanold PO, and Shatz CJ. PirB restricts oculardominance plasticity in visual cortex. Science. 2006 313(5794):1795-1800. 13 Pace TWW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH, and Heim CM. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. American Journal of Psychiatry. 2006 163(9):1630-1633. Schmerz 110 1 Chen CL, Broom DC, Liu Y, de Nooij JC, Li Z, Cen C, Samad OA, Jessell TM, Woolf CJ, and Ma Q. Runx1 determines nociceptive sensory neuron phenotype and is required for thermal and neuropathic pain. Neuron. 2006 49(3):365-377. 2 Berns GS, Chappelo J, Cekic M, Zink CF, Pagnoni G, and Martin-Skurski ME. Neurobiological substrates of dread. Science. 2006 312(5774):754-758. 3 Schmelzer, KR, Inceoglu B, Kubala L, Kim I-H, Jinks SL, Eiserich JP, and Hammock BD. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proceedings of the National Academy of Sciences USA. 2006 103(37): 13640-13645. 4 Carl T. Hall, “Social factors may deepen chronic pain”, San Francisco Chronicle, November 4, 2002. Sternberg WF, Scorr L, Smith LD, Ridgway CG, and Stout M. Long-term effects of neonatal surgery on adulthood pain behavior. Pain. 2005 13(3):347-353. 6 Langford DJ, Crager SE, Zarrar S, Smith SB, Sotocinal SG, Levenstadt JS, Chanda ML, Levitin DJ, and Mogil JS. Social modulation of pain as evidence for empathy in mice. Science. 2006 312(5782):1967-1970. 7 Kaptchuk TJ, Stason WB, Davis RB, Legedza A, Schnyer RN, Kerr CE, Stone DA, Nam BH, Kirsch I, and Goldman RH. Sham device v. inert pill: Randomized controlled trial of two placebo treatments. British Medical Journal. 2006 332(7538):391-397. 8 Kong J, Gollub RJ, Rosman IL, Webb JM, Vangel MG, Kirsch I, and Kaptchuk TJ. Brain activity associated with expectancy-enhanced placebo analgesia, as measured by functional magnetic resonance imaging. The Journal of Neuroscience. 2006 26(2):381-388. Referenzen 5 Psychiatrische Erkrankungen, Verhaltensstörungen und Suchtkrankheiten 1 CLieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, and Hsiao JK for the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New England Journal of Medicine. 2005 353(12):1209-1223. 2 Stroup TS, Lieberman JA, McEvoy JP, Swartz MS, Davis SM, Rosenheck RA, Perkins DO, Keefe RS, Davis CE, Severe J, and Hsiao JK for the CATIE Investigators. Effectiveness of olanzapine, quetiapine, risperidone, and ziprasidone in patients with chronic schizophrenia following discontinuation of a previous atypical antipsychotic. American Journal of Psychiatry. 2006 163(4):611-622. 3 McEvoy JP, Lieberman JA, Stroup TS, Davis SM, Meltzer HY, Rosenheck RA, Swartz MS, Perkins DO, Keefe RS, Davis CE, Severe J, and Hsiao JK for the CATIE Investigators. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Archives of General Psychiatry. 2006 63(10):1079-1087. 4 Jones PB, Barnes TRE, Davies L, Dunn G, Lloyd H, Hayhurst KP, Murria RM, Markwick A, and Lewis SW. Randomized controlled trial of the effect on quality of life of second- vs firstgeneration antipsychotic drugs in schizophrenia. Archives of General Psychiatry. 2006 63(10):1079-1087. 5 Georgieva L, Moskvina V, Peirce T, Norton N, Bray NJ, Jones L, Holmans P, Macgregor S, Zammit S, Wilkinson J, Williams H, Nikolov I, Williams N, Ivanov D, Davis KL, Haroutunian V, Buxbaum JD, Craddock N, Kirov G, Owen MJ, and O’Donovan MC. Convergent evidence that oligodendrocyte lineage transcription factor 2 (OLIG2) and interacting genes influence susceptibility to schizophrenia. Proceedings of the National Academy of Sciences USA. 2006 103(33): 12469-12474. 6 Meyer-Lindenberg A, Buckholtz JW, Kolachana B, R Hariri A, Pezawas L, Blasi G, Wabnitz A, Honea R, Verchinski B, Callicott JH, Egan M, Mattay V, Weinberger DR. Neural mechanisms of genetic risk for impulsivity and violence in humans. Proceedings of the National Academy of Sciences USA. 2006 103(16):6269-6274. 7 Hovatta I, Tennant RS, Helton R, Marr RA, Singer O, Redwine JM, Ellison JA, Schadt EE, Verma IM, Lockhart DJ, and Barlow C. Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature. 2005 438(7068):662-666. 111 8 Hu XZ, Lipsky RH, Zhu G, Akhtar LA, Taubman J, Greenberg BD, Xu K, Arnold PD, Richter MA, Kennedy JL, Murphy DL, and Goldman D. Serotonin transporter promoter gain-of-function genotypes are linked to obsessive-compulsive disorder. American Journal of Human Genetics. 2006 78(5):815-826. 9 Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, Schwalb JM, and Kennedy SH. Deep brain stimulation for treatmentresistant depression. Neuron. 2005 45(5):651-660. 10 McMahon FJ, Buervenich S, Charney D, Lipsky R, Rush AJ, Wilson AF, Sorant AJ, Papanicolaou GJ, Laje G, Fava M, Trivedi MH, Wisniewski SR, and Manji H. Variation in the gene encoding the serotonin 2a receptor is associated with outcome of antidepressant treatment. American Journal of Human Genetics. 2006 78(5):804-814. 11 Weissman MM, Pilowsky DJ, Wickramaratne PJ, Talati A, Wisniewski SR, Fava M, Hughes CW, Garber J, Malloy E, King CA, Cerda G, Sood AB, Alpert JE, Trivedi MH, and Rush AJ for the STAR*D-Child Team. Remissions in maternal depression and child psychopathology: A STAR*Dchild report. JAMA. 2006 295(12):1389-1398. 12 Olfson M, Marcus SC, and Shaffer D. Antidepressant drug therapy and suicide in severely depressed children and adults: A case-control study. Archives of General Psychiatry. 2006 63(8):865-872. 13 Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, and Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depresión and antidepressant action. Nature Neuroscience. 2006 9(4):519-525. 14 Heurteaux C, Lucas G, Guy N, El Yacoubi M, Thuemmler S, Peng XD, Noble F, Blondeau N, Widmann C, Borsotto M, Gobbi G, Vaugeois J-M, Debonnel G, and Lazdunski M. Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nature Neuroscience. 2006 9(9):1134-1141. 15 Pu L, Liu Q, and Poo M. BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nature Neuroscience. 2006 9(5):605-607. 16 Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Childress AR, Jayne M, Ma Y, and Wong C. Cocaine cues and dopamine in dorsal striatum: Mechanism of craving in cocaine addiction. Journal of Neuroscience. 2006 26(24):6583-6588. Störungen der Sinnes- und Körperfunktion 112 1 White PM, Doetzlhofer A, Lee YS, Groves AK, and Segil N. Mammalian cochlear supporting cells can divide and trans-differentiate into hair cells. Nature. 2006 441(7096):984-987. 2 Tsao DY, Freiwald WA, Tootell RB, and Livingstone MS. A cortical region consisting entirely of face-selective cells. Science. 2006 311(5761):670-674. 3 Liberles SD and Buck LB. A second class of chemosensory receptors in the olfactory epithelium. Nature. 2006 442(7103):645-650. 4 Lu J, Sherman D, Devor M, and Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006 441(7093):589-594. 5 Gooley JJ, Schomer A, and Saper CB. The dorsomedial hypothalamic nucleus is critical for the expression of food-entrainable circadian rhythms. Nature Neuroscience. 2006 9(3):398-407. 6 Mieda M, Williams SC, Richardson JA, Tanaka K, and Yanagisawa M. The dorsomedial hypothalamic nucleus as a putative food-entrainable circadian pacemaker. Proceedings of the National Academy of Sciences USA. 2006 103(32):12150-12155. Huang AL, Chen X, Hoon MA, Chandrashekar J, Guo W, Trankner D, Ryba NJ, and Zuker CS. The cells and logic for mammalian sour taste detection. Nature. 2006 442(7105):934-938. 8 Ishimaru Y, Inada H, Kubota M, Zhuang H, Tominaga M, and Matsunami H. Transient receptor potential family members PKD1L3 and PKD2L1 form a candidate sour taste receptor. Proceedings of the National Academy of Sciences USA. 2006 103(33):12569-12574. 9 Lund RD, Wang S, Klimanskaya I, Holmes T, Ramos-Kelsey R, Lu B, Girman S, Bischoff N, Sauve Y, and Lanza R. Human embryonic stem cellderived cells rescue visual function in dystrophic RCS rats. Cloning and Stem Cells. 2006 8(3):189-199. 10 MacLaren RE, Pearson RA, MacNeil A, Douglas RH, Salt TE, Akimoto M, Swaroop A, Sowden JC, and Ali RR. Retinal repair by transplantation of photoreceptors precursors. Nature. 2006 444(7116):203-207. Referenzen 7 Stammzellen und Neurogenese 1 Bhardwaj RD, Curtis MA, Spalding KL, Buchholz BA, Fink D, Björk- Eriksson T, Nordborg C, Gage FH, Druid H, Eriksson PS, and Frisen J. Neocortical neurogenesis in humans is restricted to development. Proceedings of the National Academy of Sciences USA. 2006 103(33): 12564-12568. 2 Bystron I, Rakic P, Molnar Z, and Blakemore C. The first neurons of the human cerebral cortex. Nature Neuroscience. 2006 9(7):880-886. 3 Shen Q, Wang Y, Dimos JT, Fasano CA, Phoenix TN, Lemischka IR, Ivanova NB, Stifani S, Morrisey EE, and Temple S. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nature Neuroscience. 2006 9(6):743-751. 4 Yamashita T, Ninomiya M, Hernandez Acosta P, Garcia-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, and Sawamoto K. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. Journal of Neuroscience. 2006 26(24):6627-6636. 5 Jin K, Wang X, Xie L, Mao XO Mao, Shu W, Wang Y, Shen J, Mao Y, Banwait S, Greenberg DA. Evidence for stroke-induced neurogenesis in the human brain. Proceedings of the National Academy of Sciences USA. 2006 103(35):13198-131202. 6 Yang H, Lu P, McKay HM, Bernot T, Keirstead H, Steward O, Gage FH, Edgerton VR, and Tuszynski MH. Endogenous neurogenesis replaces oligodendrocytes and astrocytes after primate spinal cord injury. Journal of Neuroscience. 2006 26(8):2157-2166. 7 Parent JM, von dem Bussche N, and Lowenstein DH. Prolonged seizures recruit caudal subventricular zone glial progenitors into the injured hippocampus. Hippocampus. 2006 16(3):321-328. 8 Parent JM, Elliott RC, Pleasure SJ, Barbaro NM, and Lowenstein DH. Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Annals of Neurology. 2006 59(1):81-91. 9 Encinas JM, Vaahtokari A, Enikolopov G. Fluoxetine targets early progenitor cells in the adult brain. Proceedings of the National Academy of Sciences USA. 2006 103(21):8233-8238. 10 Steele AD, Emsley JG, Ozdinler PH, Lindquist S, and Macklis JD. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proceedings of the National Academy of Sciences USA. 2006 103(9):3416-3421. 11 Kwak YD, Brannen CL, Qu T, Kim HM, Dong X, Soba P, Majumdar A, Kaplan A, Beyreuther K, and Sugaya K. Amyloid precursor protein regulates differentiation of human neural stem cells. Stem Cells Development. 2006 15(3):381-389. 113 12 Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, VandenBerg S, and Alvarez-Buylla A. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006 51(2):187-199. 13 Gil-Perotin S, Marin-Husstege M, Li J, Soriano-Navarro M, Zindy F, Roussel MF, Garcia-Verdugo JM, and Casaccia-Bonnefil P. Loss of p53 induces changes in the behavior of subventricular zone cells: Implication for the genesis of glial tumors. Journal of Neuroscience. 2006 26(4):1107-1116. 14 Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK, Kittappa R, and McKay RD. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006 442(7104):823-826. Denken und Erinnern 114 1 Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, and Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006 440(7082):352-357. 2 Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, Greenberg DA, and Bredesen DE. Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proceedings of the National Academy of Sciences USA. 2006 103(18):7130-7135. 3 Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, and Wilson RS. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006 66(12):1837-1844. 4 Tabert MH, Manly JJ, Liu X, Pelton GH, Rosenblum S, Jacobs M, Zamora D,Goodkind M, Bell K, Stern Y, and Devanand DP. Neuropsychological prediction of conversion to Alzheimer disease in patients with mild cognitive impairment. Archives of General Psychiatry. 2006 63(8):916-924. 5 Apostolova LG, Dutton RA, Dinov ID, Hayashi KM, Toga AW, Cummings JL, and Thompson PM. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps. Archives of Neurology. 2006 63(5):693-699. 6 Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, and Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006 442(7105):916-919. 7 Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, and Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006 442(7105):920-924. 8 Whitlock JR, Heyman AJ, Shuler MG, and Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006 313(5790): 1093-1097. 9 Gruart A, Munoz MD, and Delgado-Garcia JM. Involvement of the CA3-CA1 synapse in the acquisition of associative learning in behaving mice. Journal of Neuroscience. 2006 26(4):10771087. 10 Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006 313(5790):1141-1144. Abbildungen / Fotos S. 5: S. 11: S. 22: S. 24: S. 29: S. 47: S. 55: S. 59: S. 71: S. 73: S. 81: S. 84: S. 91: S. 94: S. 103: Photograph courtesy of David C. Van Essen Photograph courtesy of Steven E. Hyman Photographs courtesy of Stephen Dager Image courtesy of Katya Rubia Image courtesy of Mark Cookson Image courtesy of Adrian Owen Image courtesy of Richard Ransohoff Image courtesy of Simon Beggs Photograph courtesy of Jeffrey Lieberman Image courtesy of Andreas Meyer-Lindenberg and Joshua Buckholtz, NIMH/IRP Image courtesy of House Ear Institute Illustration by Benjamin Reece Photograph courtesy of Fred Gage Image courtesy of Jeffrey Macklis Illustration by Benjamin Reece 115 Stelle Dir eine Welt vor . . . … in der Krankheiten wie Alzheimer, Parkinson, Lou Gehrig (ALS) sowie Retinitis pigmentosa und andere Ursachen von Erblindung jeweils in einem frühen Stadium erkannt und umgehend mit Medikamenten behandelt werden, die eine Verschlimmerung, noch vor dem Auftreten schwerwiegender Schädigungen verhindern. … in der die genetischen Bahnen und die umweltbedingten Auslöser, die Menschen für Geisteskrankheiten disponieren, bekannt sind, so dass entsprechende diagnostische Tests und zielgerichtete Therapien – einschliesslich Medikamente, Beratung und vorbeugende Eingriffe – in grossem Umfang zur Verfügung stehen und umfassend angewendet werden. … in der neue Erkenntnisse über die Entwicklung des Gehirns dazu verwendet werden, die entscheidenden Vorteile des Lernens in den ersten Lebensjahren zu fördern und mit dem Altern zusammenhängende Krankheiten zu bekämpfen. … in der Rückenmarksverletzungen nicht länger zu lebenslänglichen Lähmungen führen, da das Nervensystem dazu gebracht werden kann, Nervenschaltkreise neu zu gestalten und die Bewegung der Muskeln wieder herzustellen. … in der Drogenabhängigkeit und Alkoholismus das Leben von Menschen nicht länger im Griff haben, da leicht zugängliche Behandlungen jene Veränderungen im Gehirn beeinflussen können, die für das Absetzen von Abhängigkeit erzeugenden Substanzen verantwortlich sind, aber auch Sucht und Verlangen hervorrufen können. … 118 in der das tägliche Leben der Menschen nicht mehr von depressiven Episoden oder Angstattacken beeinträchtigt wird, da wirksamere Medikamente zur Behandlung dieser Krankheiten verfügbar werden. Es mag zwar vielen unrealistisch und utopisch vorkommen, aber wir dürfen festhalten, dass wir gegenwärtig in einer ausserordentlich aufregenden Zeit der Geschichte der Neurowissenschaft leben. Die im vergangenen Jahrzehnt erfolgten Fortschritte in der Forschung haben uns weiter gebracht als wir gehofft hatten. Wir verstehen die grundlegenden Mechanismen der Hirntätigkeit wesentlich besser und sind nun an dem Punkt angelangt, an dem wir diese Erkenntnisse für therapeutische Zwecke fruchtbar machen können. Wir haben bereits angefangen, Strategien, neue Techniken und Behandlungsformen zur Bekämpfung einer ganzen Reihe neurologischer Krankheiten und Störungen zu entwickeln. Indem wir Therapieziele festlegen und unser Wissen anwenden, werden wir wirksame Behandlungen und in einigen Fällen wohl auch Heilmethoden entwickeln. Bei allem, was wir in letzter Zeit im Bereich der Neurowissenschaft gelernt haben, erkennen wir immer deutlicher, wie vieles wir nicht wissen. Dadurch wird es immer dringlicher, dass wir die Grundlagenforschung vorantreiben, die sich mit der weiterreichenden Frage, wie lebende Organismen überhaupt funktionieren, befasst. Dies wird dazu beitragen, jene komplexen Fragestellungen anzugehen, welche zu wissenschaftlichen Entdeckungen führen. Die koordinierte Arbeit von Tausenden, die in den verschiedenen Bereichen der Grundlagenforschung und der klinischen Forschung wissenschaftlich tätig sind, hat uns eine grosse Menge an Informationen gebracht; sie umfassen so unterschiedliche Gebiete wie die Strukturanalyse von Molekülen, die gezielte Entwicklung neuer Pharmaka, die Genomforschung, bildgebende Untersuchungen des Gehirns, kognitive Neurowissenschaft und klinische Studien. Dieses ganze Wissen Um unsere Aufgabe erfolgreich zu erfüllen, sind wir auch auf das Vertrauen der Öffentlichkeit angewiesen. Forschende und Laien müssen daher aus den neuen Erkenntnissen der Hirnforschung entstehenden ethischen und sozialen Konsequenzen gemeinsam erörtern. Die Dana Alliance for Brain Initiatives und die European Dana Alliance for the Brain ist eine Gemeinschaft von Neurowissenschaftlern und Neurowissenschaftlerinnen, die sich hochgesteckte Ziele gesetzt haben ; dies zeigte sich bereits im Jahre 1992, als in Cold Spring Harbor, New York, ein Forschungsplan aufgestellt wurde und dann im Jahre 1997, als die neu gebildete europäische Gruppe sich auf ihre eigenen Zielsetzungen verpflichtete. Beide Gruppen sind gegenwärtig daran, ihre konkreten Zielvorstellungen so anzupassen, dass sie die erreichten Fortschritte optimal ausnützen können. Wir stecken uns auch neue Ziele, die uns den Weg zu bald Erreichbarem weisen, und stellen langfristige Pläne auf. Indem wir uns ausmalen, welche positiven Auswirkungen diese neue Ära der Neurowissenschaft voraussichtlich haben wird, beschleunigen wir die auf das Erreichen unserer Ziele ausgerichteten Entwicklungen. Die Ziele Die verheerenden Auswirkungen der Alzheimer-Krankheit bekämpfen. Bei der Alzheimer-Krankheit kommt es zur Ansammlung eines Proteinfragments von Amyloid, welches die Nervenzellen schädigt. Der Mechanismus dieser Ansammlung wurde inzwischen in Tierversuchen biochemisch genetisch untersucht. Aufgrund dieser Tiermodelle werden gegenwärtig therapeutische Substanzen und ein möglicherweise wirksamer Impfstoff entwickelt, die die Anhäufung dieser schädlichen Substanz verhindern oder ihren Abbau beschleunigen sollen. Diese neuen Therapien, die schon bald an Menschen erprobt werden können, wecken die begründete Hoffnung, dass der Krankheitsverlauf wirkungsvoll behandelt werden kann. Stelle Dir eine Welt vor ... können wir nun breit zur Behandlung neurologischer Krankheiten und Störungen einsetzen. Diese wissenschaftliche Arbeit werden wir auch weiterhin nicht nur individuell und ausgerichtet auf die das eigene spezifische Interessengebiet weiterführen, sondern gemeinsam mit Kollegen aller wissenschaftlichen Bereiche nach Möglichkeiten der interdisziplinären Zusammenarbeit suchen. Die optimale Behandlung der ParkinsonKrankheit herausfinden. Medikamente, die auf die Dopaminbahnen des Gehirns einwirken, wurden erfolgreich zur Behandlung der motorischen Störungen der Parkinson-Krankheit eingesetzt. Leider verliert sich dieser therapeutische Effekt bei vielen Patienten nach 5 bis 10 Jahren. Nun werden neue Medikamente entwickelt ; sie sollen die Wirkung der auf Dopamin beruhenden Behandlungen verlängern und den für die Krankheit verantwortlichen selektiven Untergang von Nervenzellen verzögern. Patienten, die auf die medikamentöse Behandlung nicht ansprechen, könnten von chirurgischen Methoden, etwa der tiefen Hirnstimulation, profitieren. Dank neueren Formen der Bildgebung des Gehirns lässt sich feststellen, ob diese Behandlungsformen tatsächlich Nervenzellen vor dem Untergang bewahren und die normalen Schaltkreise wieder herstellen können. Das Auftreten von Hirnschlag reduzieren und die Therapie des Hirnschlags verbessern. Herzkrankheiten und Hirnschlag treten beträchtlich seltener auf, wenn Leute aufhören zu rauchen, auf einen tiefen Cholesterinspiegel achten, durch Diät und sportliche Betätigung ihr normales Gewicht beibehalten und wenn ein vorhandener Diabetes 119 diagnostiziert und behandelt wird. Wenn ein Hirnschlag aufgetreten ist, können die rasche Erhebung des Befunds und sofortige Behandlung eine erstaunliche Verbesserung mit weniger Folgeerscheinungen bewirken. Neue Behandlungsmethoden, um die akuten Auswirkungen eines Schlaganfalls auf Hirnzellen weiter zu reduzieren, sind im Entwicklungsstadium. Weitere Verbesserungen erwarten wir von neuen Rehabilitationsverfahren, die auf der neuen Erkenntnis von Reorganisationsvorgängen im Gehirn nach Schädigungen beruhen. Neue, wirkungsvolle Ansätze zur Vorbeugung und Behandlung der Multiplen Sklerose finden. Heute stehen uns erstmals Medikamente zur Verfügung, die erlauben, den Verlauf dieser Krankheit zu beeinflussen. Neue Medikamente, die die Immunreaktion des Körpers verändern, werden Anzahl und Intensität der Schübe der Multiplen Sklerose weiter vermindern. Ausserdem werden wir neue Methoden anwenden, um die langfristige Progression aufzuhalten, die durch den Untergang von Nervenfasern verursacht wird. Erfolgreichere Behandlungen von Gemütskrankheiten entwickeln wie Depression, Schizophrenie, Zwangserkrankung und manisch-depressive Erkrankung. Zwar wurden im letzten Jahrzehnt die für diese Krankheiten verantwortlichen Gene noch nicht gefunden, doch dürfte die Sequenzierung des menschlichen Genoms einige an diesen Krankheiten beteiligte Gene aufdecken. Neue bildgebende Verfahren gepaart mit Erkenntnissen über die Aktivitäten dieser Gene im Gehirn werden erkennen lassen, was bei diesen Erkrankungen des Gemüts und des Denkens in den einzelnen Hirnschaltkreisen schief läuft. Dies wird die Grundlage für eine bessere Diagnose, für eine wirksamere Anwendung der heute zur Verfügung stehenden Medikamente und für die Entwicklung völlig neuartiger therapeutischer Substanzen bilden. Bessere Behandlungen bei Hirntumoren entwickeln. Viele Arten von Hirntumoren, vor allem die bösartigen und solche, die durch Ableger einer Krebserkrankung ausserhalb des Gehirns zustande kommen, lassen sich nur schwer behandeln. Bildgebende Verfahren, die Behandlung mit fokussierter Bestrahlung, verschiedene Methoden, um Medikamente in den Tumor zu bringen, und die Bestimmung von genetischen Markern, die zur Diagnose beitragen werden, bilden die Grundlage zur Entwicklung innovativer Therapien. Die genetischen und neurobiologischen Ursachen der Epilepsie aufdecken und die Behandlung verbessern. Das Verständnis der genetischen Grundlagen der Epilepsie und der neuralen Vorgänge, die zu Anfällen führen, wird präventive Diagnosen und zielgerichtete Therapien ermöglichen. Die Fortschritte der elektronischen und chirurgischen Therapien lassen wirkungsvolle Behandlungs120 möglichkeiten erwarten. Die Erholung nach traumatischen Hirn- und Rückenmarksverletzungen verbessern. Wir sind dabei, Behandlungsmöglichkeiten zu erproben, die unmittelbar nach einer Verletzung den Umfang des verletzten Gewebes verringern sollen. Andere Wirkstoffe zielen darauf ab, die Schaltkreise der Nervenfasern wiederherzustellen. Techniken zur Förderung der Zellregeneration im Gehirn, um die abgestorbenen und beschädigten Nervenzellen zu ersetzen, werden ausgehend von Tiermodellen schon bald auch an Menschen klinisch erprobt werden. Gegenwärtig werden elektronische Prothesen entwickelt, die die Mikrochip-Technik verwenden, um Nervenschaltkreise zu steuern und dadurch die Bewegungsaktivität gelähmter Gliedmassen wieder zu ermöglichen. Die Ursachen der Abhängigkeit auf der Ebene des Gehirns behandeln. Forschende konnten jene Nervenschaltkreise im Gehirn bestimmen, die an der Abhängigkeit aller gängigen Mittel beteiligt sind, und haben die wichtigsten Rezeptoren für diese Wirkstoffe geklont. Neue bildgebenden Verfahren, werden die neurobiologischen Mechanismen feststellen lassen, die ein normales Gehirn in ein abhängiges Gehirn verwandeln, und die Entwicklung von Therapien ermöglichen, um diese Veränderung entweder rückgängig zu machen oder zu kompensieren. Die Hirnmechanismen verstehen, die der Reaktion auf Stress, Angst und Depression zugrunde liegen. Geistige Gesundheit ist eine Vorbedingung für eine gute Lebensqualität. Stress, Angst und Depression schaden nicht nur dem Leben der davon betroffenen Personen, sie können auch verheerende Auswirkungen auf die Gesellschaft haben. Wenn es uns gelingt, die Stressreaktion des Organismus sowie die an Angst und Depression beteiligten Hirnschaltkreise besser zu verstehen, werden wir wirksamere präventive Massnahmen entwickeln können und auch bessere Behandlungsverfahren, um ihre Auswirkungen zu lindern. Die Strategie Die Entdeckungen der Erforschung des Genoms ausnützen. Die vollständige Sequenz aller Gene, des menschlichen Genoms wird schon bald zur Verfügung stehen. Dies bedeutet, dass wir im Verlauf der nächsten 10 bis 15 Jahre in der Lage sein werden, für jeden Bereich des Gehirns und für jedes Lebensstadium – vom frühen embryonalen Leben an, über die Kindheit, die Adoleszenz bis zum Erwachsenenalter – zu bestimmen, welche Gene aktiv sind. Wir werden feststellen können, welche Gene bei verschiedensten neurologischen und psychiatrischen Krankheiten verändert sind, so dass ihre Proteinprodukte entweder ganz fehlen oder auf eine abnorme Weise funktionieren. Dank dieser Methode ist es bereits möglich, die genetische Grundlage von Krankheiten wie Huntington, spinozerebelläre Ataxie, Muskeldystrophie und fragiles X-Syndrom zu bestimmen. Stelle Dir eine Welt vor ... Neue Methoden für den Umgang mit Schmerzen entwickeln. Der Schmerz muss heute in der Medizin nicht mehr einfach hingenommen werden. Die Erforschung der Ursachen von Schmerzen sowie der Nervenaktivität, die für ihn verantwortlich ist, wird den Neurowissenschaftlern Mittel in die Hand geben, um wirksamere und zielgerichtete Therapien zur Schmerzbekämpfung zu entwickeln. Insgesamt verspricht die Entdeckung von Genen und ihre Anwendung zur klinischen Diagnose die Neurologie und Psychiatrie grundlegend zu verändern und stellt eine der grössten Herausforderungen der Neurowissenschaft dar. Zum Glück verfügen wir über Mikroarrays oder „Gen-Chips“, die diese Entwicklungen sehr beschleunigen und uns sowohl für die Diagnose als auch für die Entwicklung neuer Therapien wirkungsvolle Mittel in die Hand geben. Unser Wissen über die Entwicklung des Gehirns anwenden. Von der Empfängnis bis zum Tod durchläuft das Gehirn ganz bestimmte Entwicklungsstadien mit jeweils unterschiedlicher Anfälligkeit für Schädigungen und Fähigkeit zur Entwicklung, Tendenzen, die entweder gefördert oder gehemmt werden können. Um die Behandlung von Entwicklungsstörungen wie Autismus sowie Aufmerksamkeits- und Lernstörungen zu verbessern, wird die Neurowissenschaft eine detailliertere Darstellung der Hirnentwicklung erarbeiten. Da gewisse Probleme der Hirnentwicklung mit anderen Entwicklungsphasen 121 wie der Adoleszenz oder dem Altern zusammenhängen, wird uns das Verständnis der Veränderungen des Gehirns im Verlauf dieser Perioden neue Therapien ermöglichen. Das riesige Potential der Plastizität des Gehirns ausnutzen. Wenn wir die Neuroplastizität – die Fähigkeit des Gehirns sich selbst wiederherzustellen und anzupassen – ausnutzen, kann die Neurowissenschaft Behandlungen von degenerativen neurologischen Erkrankungen fördern und Möglichkeiten zur Verbesserung von gesunden und kranken Hirnfunktionen bereitstellen. In den kommenden zehn Jahren werden Zellen therapeutisch ersetzt werden und die Förderung der Neubildung von Zellen wird zu neuen Behandlungen von Hirnschlag, Rückenmarksverletzungen und der Parkinson Krankheit führen. Unser Verständnis des spezifisch Menschlichen vergrössern. Wie funktioniert das Gehirn ? Die Neurowissenschaft ist nun so weit, dass sie die entscheidenden Fragen nicht nur stellt, sondern auch anfängt sie zu beantworten. Welche Mechanismen und grundlegenden Nervenschaltkreise ermöglichen es uns, Erinnerungen zu speichern, aufmerksam zu sein, unsere Emotionen wahrzunehmen und auszudrücken, Entscheidungen zu treffen, Sprache zu gebrauchen und kreativ zu sein ? Die Bemühungen, eine „einheitliche Feldtheorie“ des Gehirns zu entwickeln, werden grosse Möglichkeiten eröffnen, das menschliche Potential zu maximieren. Die Methoden Zellen ersetzen. Ausgewachsene Nervenzelle können sich nicht replizieren, um die durch eine Krankheit oder eine Verletzung verloren gegangenen Zellen zu ersetzen. Methoden, die sich die Fähigkeit der Nerven122 stammzellen (den Vorläufern von Nervenzel- len) zunutze machen, sich zu neuen Nervenzellen zu differenzieren, werden die Behandlung neurologischer Erkrankungen möglicherweise revolutionieren. Die Verpflanzung von Nervenstammzellen, die bisher an Tiermodellen durchgeführt wird, wird schon bald das Stadium von klinischen Studien an Menschen erreichen. Wie die Entwicklung dieser Zellen gesteuert werden kann, wie sie an den richtigen Ort gebracht und veranlasst werden können, die geeigneten Verbindungen zu bilden, sind aktuelle Themen der Forschung. Reparaturmechanismen von Nervenzellen. Dank der dem Nervensystem innewohnenden Fähigkeit der Wiederherstellung – in gewissen Fällen werden neue Nervenzellen regeneriert, in andern die Verkabelung wiederhergestellt – hat das Gehirn die Möglichkeit, sich selbst „wieder in Ordnung zu bringen“. Wenn es uns gelingt, diese Prozesse zu fördern, dürfen wir hoffen, Patienten mit Rükkenmarks- oder Kopfverletzungen heilen zu können. Verfahren, um die Degeneration des Nervensystems aufzuhalten oder ihr vorzubeugen. Viele Erkrankungen wie Parkinson, Alzheimer, Huntington und ALS sind die Folge einer Degeneration spezifischer NervenzellPopulationen in bestimmten Hirnbereichen. Die heutigen Behandlungsmethoden beeinflussen zwar die Symptome einer Krankheit wie Parkinson, nicht aber den fortschreitenden Untergang der Nervenzellen. Techniken, die auf unseren Kenntnissen der Mechanismen des Zelltods aufbauen, werden vermutlich zu Methoden führen, die die Degeneration von Nervenzellen verhindern und damit ein Fortschreiten der Krankheit aufhalten können. Verfahren, um die Expression von Genen im Gehirn zu verändern. Es ist möglich, die Wirkung bestimmter Gene im Gehirn von Versuchstieren entweder zu verstärken oder zen Chemie erlauben es Forschenden, neue Pharmaka in einem nie zuvor gekannten Ausmass hervorzubringen, von welchen viele in der klinischen Anwendung von beträchtlichem Nutzen sein könnten. Die Entwicklung neuer, rascher Screening-Verfahren, die auf „Gen-Chips“ und anderen hochentwickelten Techniken beruhen, werden in gewissen Fällen das Zeitintervall zwischen der Entdeckung einer neuen Substanz und ihrer klinischen Erprobung von mehreren Jahren auf einige Monate reduzieren. Stelle Dir eine Welt vor ... zu blockieren. Mutierte Gene von Menschen, die neurologische Krankheiten wie Huntington und ALS verursachen, werden bei Versuchstieren eingesetzt, um die Entwicklung neuer Therapien zur Prävention der Neurodegeneration voranzutreiben. Solche Techniken haben uns bereits wertvolle Informationen über normale Vorgänge wie die Entwicklung des Gehirns, Lernen und die Bildung neuer Erinnerungen vermittelt. Diese Techniken bieten uns die Möglichkeit, normale und abnorme Hirnprozesse wesentlich intensiver als je zuvor zu untersuchen; sie werden wohl mit der Zeit auch klinisch zur Behandlung verschiedener Hirnkrankheiten angewendet werden. Verbesserte bildgebende Verfahren. Die Abbildungen sowohl der Hirnstrukturen wie auch der Hirnfunktionen wurden stark verbessert. Dank der Entwicklung von Verfahren, die Hirnfunktionen ebenso rasch und genau abbilden wie sie stattfinden, sind „Echtzeit“-Abbildungen von Hirnfunktionen möglich geworden. Diese Techniken erlauben es den Forschenden genau zu verfolgen, welche Teile des Gehirns am Denken, Lernen und Erleben von Emotionen beteiligt sind. Elektronische Hilfsmittel als Ersatz für nicht funktionstüchtige Hirnbahnen. Mit der Zeit wird es wohl möglich sein, verletzte Hirnbahnen zu umgehen. Wir hoffen, dass die Verwendung von Multielektroden-Implantaten und Mikro-Computer-Vorrichtungen – welche die Aktivität im Gehirn aufzeichnen und in Signale übersetzen, die ans Rückenmark, an die motorischen Nerven oder direkt an die Muskeln weitergeleitet werden – uns so weit bringen wird, dass Verletzte auf die Wiederherstellung ihrer Funktionstüchtigkeit hoffen dürfen. Neuartige Methoden um Heilmittel zu entdecken. Fortschritte der strukturellen Biologie, der Genomforschung und der rechnergestüt- 123 Members of EDAB AGID Yves Hôpital de la Salpêtrière, Paris, France AGUZZI Adriano University of Zurich, Switzerland CHERNISHEVA Marina University of St Petersburg, Russia ANTUNES João Lobo University of Lisbon, Portugal CHVATAL Alexandr Institute of Experimental Medicine ASCR, Prague, Czech Republic AUNIS Dominque INSERM Strasbourg, France CLARAC François CNRS, Marseille, France AVENDAÑO Carlos University of Madrid, Spain CLEMENTI Francesco University of Milan, Italy ANDERSEN Per University of Oslo, Norway BADDELEY Alan University of York, UK BARDE Yves-Alain University of Basel, Switzerland BELMONTE Carlos Instituto de Neurosciencias, Alicante, Spain. BENABID Alim-Louis INSERM and Joseph Fourier University of Grenoble, France BEN-ARI Yehezkel INSERM-INMED, Marseille, France COLLINGRIDGE Graham University of Bristol, UK; President of the British Neuroscience Association CUÉNOD Michel University of Lausanne, Switzerland CULIC Milka University of Belgrade, Yugoslavia DAVIES Kay University of Oxford, UK DEHAENE Stanislas INSERM, Paris, France BERGER Michael University of Vienna, Austria DELGADO-GARCIA José Maria Universidad Pablo de Olavide, Seville, Spain; President of the Spanish Neuroscience Society BERLUCCHI Giovanni Università degli Studi di Verona, Italy DICHGANS Johannes University of Tübingen, Germany BERNARDI Giorgio University Tor Vergata-Roma, Italy DOLAN Ray University College, London, UK BENFENATI Fabio University of Genova, Italy BERTHOZ Alain Collège de France, Paris, France BEYREUTHER Konrad University of Heidelberg, Germany BJÖRKLUND Anders Lund University, Sweden BLAKEMORE Colin Medical Research Council, UK BOCKAERT Joel CNRS, Montpellier, France BORBÉLY Alexander University of Zurich, Switzerland DUDAI Yadin Weizmann Institute of Science, Rehovot, Israel ELEKES Károly Hungarian Academy of Sciences, Tihany, Hungary; President of the Hungarian Neuroscience Society ESEN Ferhan Osmangazi University, Eskisehir, Turkey EYSEL Ulf Ruhr-Universität Bochum, Germany BRANDT Thomas University of Munich, Germany BRUNDIN Patrik Lund University, Sweden FERRUS Alberto Instituto Cajal, Madrid, Spain BUDKA Herbert University of Vienna, Austria FIESCHI Cesare University of Rome, Italy BUREŠ Jan Academy of Sciences, Czech Republic FOSTER Russell University of Oxford, UK BYSTRON Irina University of St Petersburg, Russia FRACKOWIAK Richard University College London, UK CARLSSON Arvid University of Gothenburg, Sweden FREUND Hans-Joachim University of Düsseldorf, Germany CATTANEO Elena University of Milan, Italy FREUND Tamás University of Budapest, Hungary CHANGEUX Jean-Pierre Institut Pasteur, Paris, France FRITSCHY Jean-Marc University of Zurich, Switzerland GARCIA-SEGURA Luis Instituto Cajal, Madrid, Spain KERSCHBAUM Hubert University of Salzburg, Austria GISPEN Willem University of Utrecht, The Netherlands KETTENMANN Helmut Max-Delbrück-Centre for Molecular Medicine, Berlin, Germany GJEDDE Albert Aarhus University Hospital, Denmark KORTE Martin Technical University Braunschweig, Germany GLOWINSKI Jacques Collège de France, Paris, France KOSSUT Malgorzata Nencki Institute of Experimental Biology, Warsaw, Poland GREENFIELD Susan The Royal Institution of Great Britain, London, UK KOUVELAS Elias University of Patras, Greece GRIGOREV Igor Institute of Experimental Medicine, St Petersburg, Russia GRILLNER Sten Karolinska Institute, Stockholm, Sweden KRISHTAL Oleg Bogomoletz Institute of Physiology, Kiev, Ukraine LANDIS Theodor University Hospital Geneva, Switzerland LANNFELT Lars University of Uppsala, Sweden HARI Riitta Helsinki University of Technology, Espoo, Finland LAURITZEN Martin University of Copenhagen, Denmark HARIRI Nuran University of Ege, Izmir, Turkey; President of the Turkish Neuroscience Society LERMA Juan Instituto de Neurociencias, CSIC-UMH, Alicante, Spain HERMANN Anton University of Salzburg, Austria LEVELT Willem Max-Planck-Institute for Psycholinguistics, Nijmegen, The Netherlands HERSCHKOWITZ Norbert University of Bern, Switzerland HIRSCH Etienne Hôpital de la Salpêtrière, Paris, France HOLSBOER Florian Max-Planck-Institute of Psychiatry, Munich, Germany HOLZER Peter University of Graz, Austria HUXLEY Sir Andrew University of Cambridge, UK INNOCENTI Giorgio Karolinska Institute, Stockholm, Sweden IVERSEN Leslie University of Oxford, UK IVERSEN Susan University of Oxford, UK JACK Julian University of Oxford, UK JEANNEROD Marc Institut des Sciences Cognitives, Bron, France LEVI-MONTALCINI Rita EBRI, Rome, Italy LIMA Deolinda University of Porto, Portugal LOPEZ-BARNEO José University of Seville, Spain MAGISTRETTI Pierre University of Lausanne, Switzerland MALACH Rafael Weizmann Institute of Science, Rehovot, Israel MARIN Oscar Universidad Miguel HernandezCSIC, Spain MATTHEWS Paul University of Oxford, UK MEHLER Jacques SISSA, Trieste, Italy MELAMED Eldad Tel Aviv University, Israel MONYER Hannah University Hospital of Neurology, Heidelberg, Germany JOHANSSON Barbro Lund University, Sweden MORRIS Richard University of Edinburgh, Scotland; President, Federation of European Neuroscience Societies KACZMAREK Leszek Nencki Institute of Experimental Biology, Warsaw, Poland. MOSER Edvard Norwegian University of Science and Technology, Trondheim, Norway KASTE Markku University of Helsinki, Finland KATO Ann Centre Médical Universitaire, Geneva, Switzerland NALECZ Katarzyna Nencki Institute of Experimental Biology, Warsaw, Poland KENNARD Christopher Imperial College School of Medicine, London, UK NEHER Erwin Max-Planck-Institute for Biophysical Chemistry, Göttingen, Germany NIETO-SAMPEDRO Manuel Instituto Cajal, Madrid, Spain SINGER Wolf Max-Planck-Institute for Brain Research, Frankfurt, Germany NOZDRACHEV Alexander State University of St Petersburg, Russia SMITH David University of Oxford, UK OERTEL Wolfgang Philipps-University, Marburg, Germany STEWART Michael The Open University, UK OLESEN Jes Glostrup Hospital, Copenhagen, Denmark; Chairman, European Brain Council ORBAN Guy Catholic University of Leuven, Belgium PÁRDUCZ Árpád Hungarian Academy of Sciences, Szeged, Hungary PEKER Gonul University of Ege Medical School, Izmir, Turkey; President, Turkish Neuroscience Society PETIT Christine Institut Pasteur & Collège de France, Paris, France POCHET Roland Université Libre de Bruxelles, Belgium POEWE Werner Universitätsklinik für Neurologie, Innsbruck, Austria POULAIN Dominique Université Victor Segalen, Bordeaux, France; President, French Neuroscience Society PROCHIANTZ Alain CNRS and Ecole Normale Supérieure, Paris, France PYZA Elzbieta Jagiellonian University, Krakow, Poland SPERK Günther University of Innsbruck, Austria STOERIG Petra Heinrich-Heine University, Düsseldorf, Germany STRATA Pierogiorgio University of Turin, Italy SYKOVA Eva Institute of Experimental Medicine ASCR, Prague, Czech Republic THOENEN Hans Max-Planck-Institute for Psychiatry, Martinsried, Germany TOLDI József University of Szeged, Hungary TOLOSA Eduardo University of Barcelona, Spain TSAGARELI Merab Beritashvili Institute of Physiology, Tblisi, Republic of Georgia VETULANI Jerzy Institute of Pharmacology, Krakow, Poland VIZI Sylvester Hungarian Academy of Sciences, Budapest, Hungary WALTON John Lord of Detchant University of Oxford, UK WINKLER Hans Austrian Academy of Sciences, Innsbruck, Austria RAFF Martin University College London, UK RAISMAN Geoffrey Institute of Neurology, University College London, UK RIBEIRO Joaquim Alexandre University of Lisbon, Portugal ZEKI Semir University College London, UK ZILLES Karl Heinrich-Heine-University, Düsseldorf, Germany RIZZOLATTI Giacomo University of Parma, Italy ROSE Steven The Open University, UK ROTHWELL Dame Nancy University of Manchester, UK RUTTER Sir Michael King’s College London, UK BAW Term Members AZOUZ Rony Ben-Gurion University of the Negev, Israel BATTAGLINI Paolo University of Trieste, Italy SAKMANN Bert Max-Planck-Institute for Medical Research, Heidelberg, Germany SCHWAB Martin University of Zurich, Switzerland CASTRO LOPES José University of Porto, Portugal CLARKE Stephanie University of Lausanne, Switzerland; President, Swiss Society of Neuroscience SEGAL Menahem Weizmann Institute of Science, Rehovot, Israel DEXTER David Imperial College, London, UK SEGEV Idan Hebrew University, Jerusalem, Israel DE ZEEUW Chris Department of Neuroscience, Erasmus MC, Rotterdam, The Netherlands SHALLICE Tim University College London, UK DIETRICHS Espen University of Oslo, Norway GRAUER Ettie Israel Institute of Biological Research, Israel HAGOORT Peter F.C. Donders Centre for Cognitive Neuroimaging, The Netherlands LYTHGOE Mark University College London, UK MALVA João University of Coimbra, Portugal MOHORKO Nina University of Ljubljana, Slovenia MOLDOVAN Mihai The Panum Institute, University of Copenhagen, Denmark NALEPA Irena Polish Academy of Sciences, Warsaw, Poland HUCHO Ferdinand Freie Universität Berlin, Germany; President, European Society for Neurochemistry JOËLS Marian University of Amsterdam, The Netherlands; President, The Dutch Neurofederation KHECHINASHVILI Simon Beritsashvili Institute of Physiology, Tblisi, Republic of Georgia; President, Georgian Neuroscience Association KOSTOVIC Ivica Institute for Brain Research, Zagreb, Croatia; President, Croatian Society for Neuroscience REPOVS Grega Washington University, St Louis, USA MENDLEWICZ Julien ULB Erasme Hospital, Brussels, Belgium; President, European College of Neuropsycopharmacology SKALIORA Irini Biomedical Research Foundation of the Academy of Athens, Greece PITKÄNEN Asla University of Kuopio, Finland; FENS Secretary General STAMATAKIS Antonis University of Athens, Greece STOOP Ron University of Lausanne, Switzerland PRZEWLOCKI Ryszard Polish Academy of Sciences, Krakow, Poland; President, Polish Neuroscience Society ZAGREAN Ana-Maria Carol Davila University of Medicine and Pharmacy, Bucharest, Romania ROTSHENKER Shlomo The Hebrew University of Jerusalem; President, Israel Society of Neuroscience ZAGRODZKA Jolanta Nencki Institute of Experimental Biology, Warsaw, Poland SAGVOLDEN Terje University of Oslo, Norway; President, Norwegian Neuroscience Society Federation of European Neuroscience Societies Presidents BÄHR Mathias University Hospital Göttingen, Germany; President, German Neuroscience Society BARTH Friedrich G. Austrian Academy of Sciences, Austria; President, Austrian Neuroscience Society STENBERG Tarja Institute of Biomedicine/ Physiology Biomedicum, Helsinki, Finland; President, Finnish Brain Research Society STYLIANOPOULOU Fotini University of Athens, Greece; President, Hellenic Society for Neuroscience SYKA Josef Academy of Sciences, Prague, Czech Republic; President, Czech Neuroscience Society ZAGREAN Leon Carol Davila University of Medicine, Bucharest, Romania; President, National Neuroscience Society of Romania BOER Gerard Netherlands Institute for Brain Research, The Netherlands; President, Dutch Neurofederation BRESJANAC Marja Institute of Pathophysiology, Ljubljana, Slovenia; President, Slovenian Neuroscience Association (SINAPSA) DE OLIVEIRA Catarina Resende University of Coimbra, Portugal; President, Portuguese Society for Neuroscience DE SCHUTTER Erik University of Antwerp, Belgium; President, Belgian Society for Neuroscience DI CHIARA Gaetano University of Cagliari, Italy; President, Italian Society for Neuroscience (SINS) FRANDSEN Aase Copenhagen University Hospital, Denmark; President, Danish Society for Neuroscience January 2007 A Dana Alliance for the Brain Inc Publication prepared by EDAB, the European subsidiary of DABI Gedruckt in der Schweiz 6.2007