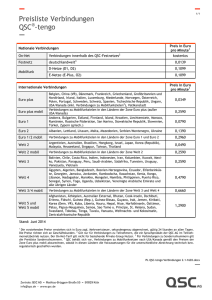
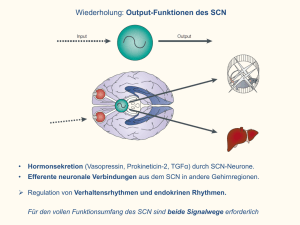
Darstellung, Struktur und Eigenschaften neuer Übergangsmetall
Werbung

Darstellung, Struktur und Eigenschaften neuer Übergangsmetall-Koordinationspolymere Diplomarbeit im Fachbereich Chemie der Christian-Albrechts-Universität Kiel vorgelegt von Mario Wriedt Kiel, Mai 2008 Darstellung, Struktur und Eigenschaften neuer Übergangsmetall-Koordinationspolymere Diplomarbeit im Fachbereich Chemie der Christian-Albrechts-Universität Kiel vorgelegt von Mario Wriedt Kiel, Mai 2008 Meiner Mama gewidmet, der wunderbarsten Person, die ich je kannte. Kiel, Mai 2008 Danksagung Die vorliegende Arbeit wurde in der Zeit von November 2007 bis Mai 2008 im Institut für Anorganische Chemie der Christian-Albrechts-Universität zu Kiel angefertigt. An dieser Stelle möchte ich all denen danken, die mir bei der Durchführung und Fertigstellung dieser Arbeit hilfreich zur Seite gestanden haben. Insbesondere möchte ich Herrn PD Dr. Christian Näther dafür danken, dass er mir das Thema dieser Arbeit zur weitgehend selbständigen Bearbeitung überlassen und mich durch seine stete Diskussionsbereitschaft mit vielseitigen Denkanstößen bereichert hat. Christian, vor allem möchte ich mich bei Dir für die aufopferungsvolle Unterstützung während meines gesamten Studiums bedanken. Frau Inke Jeß bin ich zu besonderem Dank für ihre sehr gute und enge Zusammenarbeit verpflichtet – vielen Dank, Inke, für Deine uneingeschränkte und stetige Hilfsbereitschaft. Herr Prof. Dr. Wolfgang Bensch war so freundlich, mir die unbegrenzte Nutzung seiner experimentellen Ausstattung zu gestatten, vielen Dank dafür. Frau Uschi Cornelissen und Frau Stephanie Pehlke aus der Spektroskopie möchte ich für die freundliche Unterstützung sowie Frau Maren Rasmussen und Herrn Henning Lühmann für die Magnetmessungen danken. Der Studienstiftung des Deutschen Volkes gebührt mein besonderer Dank für die finanzielle Förderung. An dieser Stelle möchte ich auch Dir, Oma, und Dir, Paps, für die monetäre Unterstützung während meines Studiums danken. Des Weiteren war Herr Dr. Wolfgang Gräber so freundlich, mich während meiner gesamten Studienzeit als Wissenschaftliche Hilfskraft zu beschäftigen. Wolfgang, die Zusammenarbeit mit Dir hat mir immer sehr viel Spaß gemacht. Auch meinem Kommilitonen Jan Boeckmann gebührt mein Dank – auf Dich, Jan, konnte ich mich immer verlassen. Zuletzt möchte ich der wichtigsten Person in meinem Leben danken. Liebe Anja, vielen Dank für Deine unendliche Geduld als Lektorin und für Deine Hilfsbereitschaft in allen Lebenslagen. Inhaltsverzeichnis 1 Einleitung 1.1 Motivation zur Untersuchung von Koordinationspolymeren ...................................... 1 1.2 Einteilung und Übersicht über Koordinationspolymere .............................................. 7 1.3 Thermische Abbaureaktionen .................................................................................... 10 1.4 Auswahl geeigneter Liganden ................................................................................... 11 1.5 Stand der Forschung .................................................................................................. 14 1.5.1 Ausgangspunkt: CuX-Pyrazin-Koordinationspolymere (X = Cl, Br, I) ............. 14 1.5.2 CuX-4,4’-Bipyridin-Koordinationspolymere (X = Cl, Br, I) ............................. 15 1.5.3 CuX-2-Methyl-/2-Chlorpyrazin-Koordinationspolymere (X = Cl, Br, I) .......... 17 1.5.4 Kontrolle der thermischen Reaktivität durch Mischkristallbildung ................... 18 1.5.5 Einfluss der Kinetik thermischer Abbaureaktionen auf die Produktbildung...... 19 1.5.6 Thermische Abbaureaktionen zur Herstellung von Koordinationspolymeren mit kooperativen magnetischen Eigenschaften .................................................. 20 1.6 2 Zielsetzung und Übersicht über die Arbeit ................................................................ 22 Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin 2.1 Einleitung ................................................................................................................... 24 2.2 Kristallstrukturen der Verbindungen ......................................................................... 25 2.2.1 [ZnCl2(2,5-Dimethylpyrazin)]n .......................................................................... 25 2.2.2 (ZnCl2)2(2,5-Dimethylpyrazin)3 ......................................................................... 26 2.2.3 [ZnBr2(2,5-Dimethylpyrazin)]n – Modifikation I ............................................... 28 2.2.4 [ZnBr2(2,5-Dimethylpyrazin)]n – Modifikation II ............................................. 28 2.2.5 (ZnBr2)2(2,5-Dimethylpyrazin)3 ......................................................................... 30 2.2.6 [ZnBr2(2,5-Dimethylpyrazin)]2·2,5-Dimethylpyrazin........................................ 30 2.2.7 [ZnI2(2,5-Dimethylpyrazin)]n ............................................................................. 32 2.2.8 ZnI2(2,5-Dimethylpyrazin) ................................................................................. 32 2.3 Thermoanalytische Untersuchungen der Verbindungen ............................................ 34 2.4 Synthetische Aspekte ................................................................................................. 40 2.5 Vergleich von ZnX2(Pyrazin) und ZnX2(2,5-Dimethylpyrazin) (X = Cl, Br, I)........ 41 3 Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden 3.1 Einleitung ................................................................................................................... 43 3.2 Verbindungen auf Basis von Übergangsmetall-Thiocyanaten und Pyrazin .............. 45 3.2.1 3.2.1.1 [Ni(SCN)2(Pyrazin)2]n ................................................................................. 45 3.2.1.2 [M(SCN)2(Pyrazin)]n (M = Fe, Co, Ni) ...................................................... 46 3.2.2 Thermoanalytische Untersuchungen der Verbindungen .................................... 48 3.2.3 Magnetische Eigenschaften der Verbindungen .................................................. 51 3.3 Verbindungen auf Basis von Übergangsmetall-Cyanaten und Pyrazin ..................... 54 3.4 Verbindungen auf Basis von Übergangsmetall-Thiocyanaten und Pyrimidin .......... 56 3.4.1 4 Kristallstrukturen der Verbindungen .................................................................. 45 Kristallstrukturen der Verbindungen .................................................................. 56 3.4.1.1 [Mn(SCN)2(Pyrimidin)2]n ........................................................................... 56 3.4.1.2 M(SCN)2(Pyrimidin)4 (M = Co, Ni) ........................................................... 58 3.4.2 Thermoanalytische Untersuchungen der Verbindungen .................................... 59 3.4.3 Magnetische Eigenschaften der Verbindungen .................................................. 60 Verbindungen auf Basis von Übergangsmetall-Halogeniden und 2,2'sowie 4,4'-Dipyridyldisulfid 4.1 Einleitung ................................................................................................................... 62 4.2 Kristallstrukturen der Verbindungen ......................................................................... 62 4.2.1 ZnX2(2,2'-Dipyridylsulfid) (X = Cl, Br, I) ......................................................... 62 4.2.2 ZnBr2(2,2'-Dipyridyldisulfid) ............................................................................. 64 4.2.3 Cu6X6S4(4-Pyridiniumthiolat)4 (X = Cl, Br) ...................................................... 65 5 Zusammenfassung und Ausblick ................................................................................... 68 6 Experimenteller Teil 6.1 Darstellung der Verbindungen ................................................................................... 72 6.1.1 Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin.... 72 6.1.2 Verbindungen auf Basis von Übergangsmetall-Thiocyanaten und Pyrazin ....... 76 6.1.3 Verbindungen auf Basis von Übergangsmetall-Cyanaten und Pyrazin ............. 79 6.1.4 Verbindungen auf Basis von Übergangsmetall-Thiocyanaten und Pyrimidin ... 79 6.1.5 Verbindungen auf Basis von Zink(II)halogeniden und 2,2'-Dipyridylsulfid ..... 80 6.1.6 Verbindungen auf Basis von Zink(II)halogeniden und 2,2'-Dipyridyldisulfid .. 81 6.1.7 Verbindungen auf Basis von Kupfer(I)halogeniden und 4-Pyridiniumthiolat ... 81 6.2 7 8 Methoden ................................................................................................................... 82 6.2.1 Röntgen-Einkristallstrukturanalyse .................................................................... 82 6.2.2 Röntgen-Pulverdiffraktometrie........................................................................... 84 6.2.3 Raster-Elektronenmikroskopie und Röntgenfluoreszenzanalyse ....................... 85 6.2.4 Thermogravimetrie gekoppelt mit der Massenspektroskopie und Differenzthermoanalyse...................................................................................... 86 6.2.5 Dynamische Differenzkalorimetrie .................................................................... 88 6.2.6 Weitere verwendete Geräte und Software .......................................................... 89 6.3 Verwendete Chemikalien ........................................................................................... 90 6.4 Einkristallstrukturdaten .............................................................................................. 91 Publikationen und Konferenzbeiträge 7.1 Publikationen ........................................................................................................... 113 7.2 Konferenzbeiträge .................................................................................................... 115 Literatur ......................................................................................................................... 116 1. Einleitung -1- 1 Einleitung 1.1 Motivation zur Untersuchung von Koordinationspolymeren Die Synthese neuer Verbindungen, die bestimmte physikalische Eigenschaften oder eine definierte Reaktivität aufzeigen, gehört sowohl an den Universitäten als auch in der Industrie seit jeher zu den Hauptaufgaben in der chemischen Forschung. Die meisten modernen Technologien sind weitgehend von der Entdeckung neuartiger Materialien mit neuen oder verbesserten Eigenschaften abhängig. Einen großen Anteil daran bilden Festkörperverbindungen. Diese stellen die Grundlage für viele wichtige industriell genutzte Bereiche dar. Jedoch ist die Synthese neuer Verbindungen auch heute noch überwiegend dem Zufall überlassen. Größtenteils werden neue Materialien systematisch auf die Existenz bestimmter Eigenschaften hin untersucht, wobei die Auswahl der Verbindungsklasse z.T. auf Erfahrungen basiert. Neben dieser empirischen Vorgehensweise wurden in den letzten Jahren verstärkt Untersuchungen mit dem Ziel durchgeführt, zu einem rationaleren Design neuer Festkörperverbindungen zu gelangen. Hierbei gibt es allerdings eine Vielzahl an Problemen zu überwinden, deren Lösung höchstwahrscheinlich einige Zeit in Anspruch nehmen wird. Die Eigenschaften kristalliner Verbindungen sind neben der chemischen Zusammensetzung auch von der dreidimensionalen Anordnung der Kristallbausteine, d.h. der Kristallstruktur, abhängig. Diese Tatsache ist der Ausgangspunkt für Überlegungen hinsichtlich eines rationaleren Festkörperdesigns. Als einfaches Beispiel hierfür können die Kohlenstoffmodifikationen von Graphit[1] und Diamant[2] herangezogen werden: Im Graphit ist jedes Kohlenstoffatom sp2-hybridisiert und bildet σ-Bindungen zu seinen drei Nachbaratomen aus (Abb. 1.1: links). Das vierte Elektron ist an einer π-Bindung beteiligt und ist dafür verantwortlich, dass das Graphit zu einem elektrischen Leiter wird. Es besitzt eine Schichtstruktur aus kondensierten C6-Ringen mit einer Kantenlänge von 1.4210 Å. Die Schichten sind um 2.46 Å gegeneinander verschoben, so dass es über und unter der Mitte jedes Sechsecks sowie unter und über jedem zweiten Kohlenstoffatom, zu einer Überlappung zwischen der unteren und der oberen Schicht kommt. Dieses Muster wird mehrfach wiederholt, so dass sich auf diese Weise eine Schichtstruktur bildet. Da die Wechselwirkung zwischen den Schichten sehr schwach ist, stellt Graphit ein gutes Schmiermittel dar. Im Diamant sind die Kohlenstoffatome dagegen sp3-hybridisiert und in einem Abstand von 1.5445 Å tetraedrisch jeweils von vier weiteren Kohlenstoffatomen umgeben (Abb. 1.1: rechts). Diese Verknüpfung erzeugt ein dreidimensionales Netzwerk. Aufgrund dessen ist 1. Einleitung -2- Diamant eines der härtesten Materialien.[3] Da keine Elektronen für eine elektrische Leitung zur Verfügung stehen, ist Diamant ein Isolator. Abb. 1.1: Kristallstruktur von Graphit (links) und Diamant (rechts). Besonders interessante physikalische Eigenschaften (Tab. 1.1) treten nur dann auf, wenn eine bestimmte Kristallstruktur vorliegt. Werden z.B. bestimmte kristalline Festkörperverbindungen mit Licht bestrahlt, so emittieren diese elektromagnetische Strahlung, die neben der ursprünglich eingestrahlten Frequenz auch Licht der doppelten Frequenz enthält.[4-6] Diese sogenannten "Frequenzverdoppler" sind daher u.a. in den Materialwissenschaften von großem Interesse. Alle Kristallstrukturen können grob in zwei Klassen unterteilt werden: Diejenigen, die mit, und diejenigen, die ohne Inversionszentrum kristallisieren. Untersuchungen haben ergeben, dass der Effekt der Frequenzverdopplung nur in Verbindungen mit nicht-zentrosymmetrischen Strukturen auftreten kann.[7] Ähnlich verhalten sich z.B. pyro- oder piezoelektrische Verbindungen.[8] Für ein rationaleres Design von Festkörperstrukturen ist es daher zunächst notwendig, derartige Struktur-Eigenschafts-Beziehungen systematisch zu untersuchen. Tab. 1.1: Zusammenfassung der wichtigsten Eigenschaften von Festkörperverbindungen. Mechanische Eigenschaften Härte Spaltbarkeit Elektrische Eigenschaften Piezo- und Pyroelektrizität Elektrische Leitfähigkeit Optische Eigenschaften Optische Aktivität Nichtlineare optische Eigenschaften Magnetische Eigenschaften Kooperative magnetische Phänomene Wichtigstes Kriterium für ein rationaleres Design von Festkörperverbindungen ist demnach die Kenntnis über die Zusammenhänge zwischen der Kristallstruktur und den physikalischen Eigenschaften oder der chemischen Reaktivität eines Stoffes. Sind diese bekannt, kann unter 1. Einleitung -3- Berücksichtigung der chemischen Zusammensetzung versucht werden, gezielt Verbindungen mit ähnlichen Kristallstrukturen zu erzeugen, so dass die gewünschte Eigenschaft hervorgerufen wird. Solche Überlegungen werfen die Fragestellung auf, wie eine bestimmte Kristallstruktur gezielt erzeugt werden kann. Dieses Problem wurde bereits 1988 in einer provokativen Art und Weise formuliert: "One of the continuing scandals in the physical sciences today is that it remains in general impossible to predict the structure of even the simpliest crystalline solids from a knowledge of their chemical composition".[9] Aufgrund der geschilderten Überlegungen wäre die Beantwortung dieser Frage jedoch von großer wissenschaftlicher und technologischer Bedeutung. Die Kristallstruktur wird zum größten Teil von den Wechselwirkungen zwischen den einzelnen Bausteinen bestimmt. Daher ist es für die Schaffung eines rationalen Strukturdesigns unbedingt notwendig, diese zu analysieren und einzuordnen. Die auftretenden Wechselwirkungen können daher unterschiedlich klassifiziert werden (Tab. 1.2). Tab. 1.2: Einteilung von Wechselwirkungen zwischen Kristallbausteinen. Einteilung von Wechselwirkungen stark schwach bindend nicht-bindend attraktiv repulsiv intramolekular intermolekular van der Waals Coulomb nicht direktional direktional Die Abstandsabhängigkeit und Direktionalität stellen die weitaus wichtigsten Kriterien für ein rationales Strukturdesign dar. Für einen gezielten Aufbau von Kristallstrukturen können besonders gerichtete Wechselwirkungen, z.B. Wasserstoffbrückenbindungen oder Metall-Ligand-Wechselwirkungen, herangezogen werden. Das menschliche Wissen über die Wechselwirkungen dieser Kräfte ist jedoch bislang noch sehr begrenzt, so dass wir von einem gezielten "Kristalldesign" noch weit entfernt sind. Dennoch ergeben sich aus derartigen Überlegungen vielversprechende Ansätze. 1. Einleitung -4- In den letzten Jahren hat sich für ein rationaleres "Materialdesign" der Begriff des "Crystal Engineering" etabliert. Dies bedeutet, aufbauend auf einem umfassenden Verständnis der Wechselwirkungen in Kristallen, die gezielte Erzeugung ganz bestimmter Kristallstrukturen mit definierten Eigenschaften.[10-12] Jedoch ist zu berücksichtigen, dass sich der Begriff des "Crystal Engineering" von einer Vorhersage von Kristallstrukturen unterscheidet. Die Vorhersage von Kristallstrukturen bedeutet die exakte Vorhersage der Struktur in Bezug auf genaue Informationen über die Gitterparameter, die Symmetrie und die Raumgruppe. Ein "Crystal Engineering" oder "Strukturdesign" ist bei weitem nicht so restriktiv und umfasst eher die Vorhersage von Netzwerktopologien. Dazu zählt die ungefähre Verknüpfung der Bausteine, was auch Aspekte der Kristallsymmetrie, z.B. die Zentrosymmetrie oder Nicht-Zentrosymmetrie von Strukturen mit einschließt. Hierbei muss jedoch auch die häufig vorkommende Polymorphie chemischer Verbindungen berücksichtigt werden. Dieses Phänomen beschreibt das Vorkommen chemischer Verbindungen in unterschiedlichen kristallinen Modifikationen. Es hat seinen Ursprung darin, dass das komplexe Zusammenspiel der Wechselwirkungen in Kristallen dazu führt, dass die Energieunterschiede der unterschiedlichen Modifikationen für unterschiedliche Anordnungen der Bausteine in einem Kristall nur gering sein können.[11,13,14] Die physikalischen und chemischen Eigenschaften der auftretenden Modifikationen sind dabei unterschiedlich. Deshalb kommt diesem Phänomen beim "Crystal Engineering" eine besondere Bedeutung zu. Es können unterschiedliche Festkörpereigenschaften bei sonst gleicher chemischer Zusammensetzung, jedoch unterschiedlicher Kristallstruktur, miteinander verglichen werden. Solche Aspekte sind besonders in der Arzneistoffforschung von außerordentlichem Interesse.[15-18] Für die Analyse der in kristallinen Verbindungen auftretenden Wechselwirkungen stehen prinzipiell verschiedene Möglichkeiten zur Verfügung. Sie können einerseits mit modernen quantenchemischen Verfahren theoretisch näher untersucht werden, was sich in Abhängigkeit des zu lösenden Problems sowie der angestrebten Genauigkeit immer noch als extrem schwierig und langwierig gestaltet. Anderseits können Kristallstrukturen mit Hilfe moderner Datenbanken und entsprechenden Auswerteroutinen systematisch analysiert werden.[19-25] Zudem eignen sich besonders polymorphe Modifikationen für eine Analyse intermolekularer Wechselwirkungen in Kristallen,[26] da bei diesen – wie oben beschrieben – die strukturellen Unterschiede ausschließlich aus Unterschieden in der Packung der Moleküle resultieren. 1. Einleitung -5- Die erlangten Erkenntnisse über die in Kristallen auftretenden Wechselwirkungen können nun für einen gezielten Aufbau von Kristallstrukturen verwendet werden. Es empfiehlt sich hierbei, zunächst Festkörperverbindungen zu untersuchen, deren Struktur zum überwiegenden Teil von gerichteten (anisotropen) Wechselwirkungen bestimmt wird. Da deren Struktur oftmals durch ungerichtete (isotrope) Wechselwirkungen bestimmt wird, sind klassische anorganische Festkörperverbindungen für ein rationales Kristalldesign weit weniger geeignet. Nicht einmal ansatzweise kann für diese Verbindungen die Kristallstruktur vorherbestimmt werden. Dies ist insofern nachteilig, da viele dieser Verbindungen, u.a. wegen ihrer hoch vernetzten Strukturen, sehr interessante magnetische oder optische Eigenschaften zeigen. Ein Analogon zu den dreidimensional vernetzten Festkörperstrukturen stellen sogenannte koordinationspolymere Verbindungen dar. Sie bestehen, wie klassische Koordinationsverbindungen auch, aus einem von Liganden umgebenen Zentralatom. Im Gegensatz zu den "diskreten" Koordinationsverbindungen (Abb. 1.2: links) sind in Koordinationspolymeren (Abb. 1.2: rechts) jedoch benachbarte Zentralatome zusätzlich durch mehrzähnige Liganden miteinander verbrückt. Abb. 1.2: Schematische Darstellung der strukturellen Unterschiede zwischen "diskreten" Koordinationsverbindungen (links) und Koordinationspolymeren (rechts). In Abhängigkeit der Zusammensetzung dieser Verbindungen sowie der koordinativen Eigenschaften der Liganden und der Metallkationen, können ein-, zwei- oder dreidimensionale Koordinationsnetzwerke zumindest teilweise gezielt aufgebaut werden. Überdies handelt es sich bei Koordinationspolymeren um "molekular" aufgebaute Verbindungen mit gerichteten Metall-Ligand-Wechselwirkungen. Aus diesem Grund können die Zusammensetzung und die Struktur meist wesentlich effektiver kontrolliert werden, als dies bei rein anorganischen Verbindungen der Fall ist. 1. Einleitung -6- Die Vielfalt koordinationspolymerer Verbindungen ist groß. Werden beispielsweise zur Darstellung von Koordinationspolymeren paramagnetische Übergangsmetallkationen als Zentralatome verwendet, und ist der verbrückende Ligand nicht zu groß gewählt, wie u.a. in Aziden[27] (Abb. 1.3: links) oder Thiocyanaten[28] (Abb. 1.3: rechts), kann im Prinzip das Auftreten kooperativer magnetischer Eigenschaften wie z.B. Ferro- oder Antiferromagnetismus erwartet werden.[29,30] Darüber hinaus kann man in den meisten Fällen aufgrund einfacher Überlegungen, beispielsweise hinsichtlich der Ladung und Stöchiometrie der beteiligten Bausteine, die Zusammensetzung von Koordinationsverbindungen relativ einfach einstellen. Abb. 1.3: Kristallstrukturen von Poly[bis-(µ2-Azido)-(µ2-Pyrazin-N,N')-Mangan(II)],[27] ferromagnetisch (links) und Poly[bis-(µ2-Thiocyanato-N,S)-(µ2-Pyrazin-N,N')-Mangan(II)],[28] antiferromagnetisch (rechts). Vom Standpunkt des "Crystal Engineering" betrachtet, besitzen Koordinationspolymere weitere wichtige Vorteile. Im Gegensatz zu den bereits erwähnten klassischen anorganischen Festkörper- oder den "diskreten" Koordinationsverbindungen (Abb. 1.2: links), kann die Festkörperstruktur koordinationspolymerer Verbindungen (Abb. 1.2: rechts) besser kontrolliert werden. Dies liegt daran, dass die Packung, z.B. von "diskreten" Komplexen, im Kristall zum größten Teil von isotropen, d.h. nicht-gerichteten, van-der-Waals-Wechselwirkungen dominiert wird. Dadurch ist diese praktisch nicht mehr zu kontrollieren. Werden jedoch die verbrückenden Liganden in koordinationspolymeren Verbindungen geeignet gewählt, wird die relative Orientierung benachbarter Komplexe festgelegt. Die Struktur, oder zumindest die Topologie der auftretenden Koordinations-Netzwerke, und damit auch deren Eigenschaften, können somit weitaus effektiver vorherbestimmt werden.[31] Demnach eignen sich koordinationspolymere Verbindungen hervorragend, um den Wirkungsgrad des Konzepts des "Crystal Engineering" näher zu untersuchen. Diese Untersuchung ist daher ein Hauptbestandteil der vorliegenden Diplomarbeit, in der neue Koordinationspolymere von Übergangsmetallen dargestellt, strukturell charakterisiert sowie hinsichtlich ihrer physikalischen Eigenschaften umfangreich untersucht werden. 1. Einleitung 1.2 -7- Einteilung und Übersicht über Koordinationspolymere Aufgrund der bereits erwähnten Eigenschaften von Koordinationspolymeren hinsichtlich des Designs von Strukturen mit interessanten physikalischen Eigenschaften, wurde an der Thematik dieser Verbindungsklasse besonders in der letzten Zeit mit großem Interesse geforscht. Dies wird u.a. an dem drastischen Anstieg der Arbeiten, die sich mit Koordinationspolymeren beschäftigen, deutlich.[27,32-80] Im Folgenden sollen daher nur anhand von Beispielen einige ausgewählte strukturelle Merkmale vorgestellt werden. Koordinationspolymere Verbindungen können unterschiedlich klassifiziert werden. Eine erste Einteilung kann basierend auf der Dimensionalität der auftretenden Koordinationsnetzwerke erfolgen.[79] Bestimmt wird diese hierbei überwiegend von den Koordinationseigenschaften der Liganden, von der Natur der Metallkationen sowie vom Verhältnis zwischen Metall und Ligand. Um die Dimensionalität zu einem eindeutigen Kriterium werden zu lassen, muss zunächst jedoch festgelegt werden, auf welche Art von Wechselwirkungen diese Einteilung beruht. Werden beispielsweise neben reinen Metall-Ligand-Wechselwirkungen auch Wasserstoffbrückenbindungen zwischen den Liganden berücksichtigt, so ergibt sich daraus eine veränderte Dimensionalität der Netzwerke. Bei ausschließlicher Betrachtung von Metall-Ligand-Wechselwirkungen finden sich in der Literatur viele Strukturen eindimensionaler Koordinationspolymere, welche überwiegend aus zick-zack-förmigen[36,48,64] oder helixartigen Ketten[49,57,75] aufgebaut sind. Helixartige Strukturen sind deshalb von besonderer Bedeutung, weil diese chiral sind. Derartige Verbindungen kristallisieren oft in chiralen Raumgruppen, sind polar und verfügen z.B. über die Fähigkeit zur Frequenzverdopplung. Ein weiteres eindimensionales Strukturelement, das in koordinationspolymeren Verbindungen immer wieder auftritt, sind Leiterstrukturen.[44,46,61,78] Im Vergleich zu den Erstgenannten findet sich in diesen Verbindungen meist eine veränderte Stöchiometrie, da sich das Verhältnis von Metall zu Ligand ändert. Auch Strukturen zweidimensionaler Koordinationspolymere finden sich vielfach in der Literatur.[27,39,50,52,56,63,67,68] Sie entstehen in der Regel, wenn lineare "Spacerliganden" verwendet werden. Die Mehrheit dieser Strukturen ist aus quadratischen Gittern aufgebaut, jedoch treten in Abhängigkeit der Liganden auch andersartige Koordinationsnetzwerke auf. Diese Netzwerke enthalten Poren, deren Durchmesser von der Größe und der Topologie der Liganden abhängt. Kleine Poren werden mit Cyanoliganden[67,68] und größere mit z.B. Pyrazin oder 4,4‘-Bipyridin[27,39,50,52,63] (Abb. 1.4) gebildet. Außerdem sind zweidimensionale Kristallstrukturen zusätzlich als potenzielle Wirt-Einschlussverbindungen von großem Interesse. 1. Einleitung -8- Abb. 1.4: Zweidimensionales großporiges Gitter von Poly[bis(µ2-4,4'-Bipyridyl-N,N')-di-(µ-Isothiocyanato-N)Cobalt(II)]-Diethylether,[63] übersichtshalber ohne Wasserstoffatome und Diethylether dargestellt. Als übergeordnetes Ziel im Rahmen des "Crystal Engineering" wird allerdings der Aufbau dreidimensionaler Netzwerke verfolgt. In diesen können nur noch wenige Architekturen auftreten. Zu den Bekanntesten zählen hier die diamantartigen (Abb. 1.5: links) sowie die kubischen (Abb. 1.5: rechts) Netzwerke.[35,47,51,53,59,69] Abb. 1.5: Diamantartiges Netzwerk von Poly[[NMe4][CuPt(CN)4]],[51] übersichthalber sind nur die Metallzentren dargestellt (links) und oktaedrisches Netzwerk von Poly[[Ag(Pyrazin)3](SbF6)],[47] übersichtshalber ohne Wasserstoffatome und SbF6 dargestellt (rechts). In den diamantartigen Koordinationsnetzwerken sind normalerweise die Metallzentren tetraedrisch von Liganden umgeben.[35,51,53] Vergleichend hierzu sind in kubischen Netzwerken die Metallzentren oktaedrisch von den Liganden umgeben. Neben diesen einfachen existiert eine Vielzahl anderer Netzwerke, beispielsweise die sogenannten interpenetrierenden Netzwerke (Durchdringungsnetzwerke) oder Netzwerke, in denen Schichten durch Liganden verknüpft werden (Pillaring).[79] Die bisher erwähnten Koordinationspolymere bestehen aus Übergangsmetallzentren, welche über organische Anionen sowie über neutrale organische Liganden verknüpft sind. Bei einer weiteren Art von Koordinationspolymeren kommen neben organischen auch typisch anorganische Baueinheiten vor. Diese Verbindungen werden beispielsweise. als "anorganisch-orga- 1. Einleitung -9- nische Hybridstrukturen" bezeichnet und haben in der letzten Zeit zunehmend an Bedeutung gewonnen.[32,33] Die Besonderheit dieser "Hybridstrukturen" besteht in ihrer strukturellen Vielfalt. Hierbei werden die Strukturen von rein anorganischen Festkörperverbindungen, und damit ihren verbundenen physikalischen Eigenschaften, durch den Einbau geeigneter organischer Liganden in weiten Teilen modifiziert. Zu diesen "anorganisch-organischen Hybridverbindungen" zählen auch Koordinationspolymere auf Basis von Kupfer(I)halogeniden und -pseudohalogeniden sowie N-Donorliganden. In diesen Verbindungen treten typische CuX-Substrukturen (X = Cl, Br, I, SCN, CN) wie z.B. Einfach- und Doppelketten auf (Abb. 1.6).[42,58,60,65,70,73,74,80-84] Abb. 1.6: Bespiele für verschiedene CuX-Substrukturen in CuX-Koordinationspolymeren (X = Cl, Br, I) mit NDonorliganden. Durch geeignete organische Liganden können diese CuX-Substrukturen zu mehrdimensionalen Koordinationspolymeren verknüpft werden (Abb. 1.7). Wird beispielsweise Pyridin als ein organischer Ligand verwendet, der nur über eine einzige Koordinationsstelle verfügt, werden eindimensionale Strukturen erhalten (Abb. 1.7: links).[85] Werden jedoch bifunktionelle, linear verbrückende Liganden eingesetzt, wie z.B. Pyrazin, so werden die einzelnen CuXSubstrukturen gezielt zu zweidimensionalen Netzwerken verknüpft (Abb. 1.7: rechts).[58] Abb. 1.7: Eindimensionale Ketten von Poly[bis(µ3-Chloro)-Pyridin-Kupfer(I)][85] (links) und zweidimensionale Struktur von Poly[bis(µ3-Chloro)-(µ2-Pyrazin)-di-Kupfer(I)][58] (rechts). 1. Einleitung - 10 - Im Rahmen des "Crystal Engineering" kann die Art der auftretenden CuX-Substrukturen jedoch bislang nur bedingt, z.B. durch die Reaktionsbedingungen wie Temperatur, Reaktionszeit, Lösungsmittel oder die Stöchiometrie der Reaktanden, vorherbestimmt werden. Sind Kupfer(I)halogenid und Ligand zu gleichen Teilen enthalten, so werden diese im Folgenden als 1:1-Verbindungen bezeichnet. Entsprechend setzen sich 2:1-Verbindungen aus einem CuX zu Ligand Verhältnis von 2:1 zusammen. Bei den 1:1-Verbindungen finden sich in der Regel kettenartige CuX-Substrukturen, während leiterartige CuX-Substrukturen überwiegend in 2:1Koordinationspolymeren auftreten. 1.3 Thermische Abbaureaktionen Ein wesentlicher Aspekt in der vorliegenden Diplomarbeit bildet die Tatsache, dass ligandenreichere, z.B. 1:1-Verbindungen, durch thermische Abbaureaktionen in ligandenärmere, z.B. 2:1-Verbindungen, überführt werden können – eine Methode, die sich daher zur Darstellung von neuen Koordinationspolymeren eignet. Deren Darstellung beschränkte sich bisher jedoch auf Reaktionen in Lösung (Abb. 1.8). Häufig resultieren daraus entweder Gemische verschiedener Phasen oder bestimmte, beispielsweise thermodynamisch metastabile Verbindungen, können nicht dargestellt werden. MXL4 ±L MXL3 ±L MXL2 ±L MXL Abb. 1.8: Reaktionsgleichgewicht eines Koordinationspolymers in Lösung (M = Metall, X = Anion, L = Ligand). Einen alternativen Ansatz für die Synthese derartiger Verbindungen bieten thermische Abbaureaktionen geeigneter Vorläuferverbindungen (Precursoren), wie beispielsweise Solvate oder ligandenreiche Verbindungen. Diese werden erhitzt, so dass eine stufenweise Abgabe eines Teils der Liganden unter Bildung ligandenärmerer Verbindungen erfolgt (Abb. 1.9). Die Reaktionsgleichgewichte werden hierbei in Richtung der ligandenärmeren Verbindungen verschoben, so dass diese in quantitativer Ausbeute erhalten werden. 1. Einleitung - 11 - Masseverlust MXL4 (1:4) MXL3 (1:3) MXL2 (1:2) MXL (1:1) (Metall : Ligand) Temperatur Abb. 1.9: Idealisierte TG-Kurve einer Thermischen Abbaureaktion. Ein ligandenreiches Koordinationspolymer wird in mehreren Teilschritten zu einem ligandenarmen abgebaut. Diese Methode fand zum einen bisher keine Beachtung und zum anderen fehlen systematische Untersuchungen zum Mechanismus sowie dem Auftreten intermediärer Zwischenphasen. In aktuellen Forschungen der Gruppe Näther et al.[86-93] zeigt sich jedoch, dass koordinationspolymere Verbindungen beim gezielten thermischen Abbau neue Koordinationspolymere als Intermediate bilden, die meist phasenrein und in 100%-iger Ausbeute abgefangen werden können. Deren Forschungsergebnisse belegen auch, dass thermodynamisch metastabile Phasen und unterschiedliche polymorphe Modifikationen zugänglich sind, und dass die Produktbildung von der Kinetik der Reaktionen abhängt. 1.4 Auswahl geeigneter Liganden Das Ligandensystem koordinationspolymerer Verbindungen muss bestimmte Voraussetzungen erfüllen, wenn eine gewisse Strukturkontrolle oder das Auftreten kooperativer magnetischer Eigenschaften angestrebt wird. Im Folgenden werden daher einige der Gründe aufgeführt, die zu der Auswahl der in den vorliegenden Untersuchungen verwendeten Liganden (Abb. 1.12) geführt haben. Wichtig ist zunächst, dass die Liganden über mehrere potentielle Koordinationsstellen verfügen, damit zumindest zwei Metallkationen zu einer polymeren Verbindung verknüpft werden können. Zudem sollten die Koordinationsstellen im Ligand möglichst weit voneinander entfernt sein, um "diskrete" chelatartige Komplexe zu vermeiden. Darüber hinaus ist es von besonderer Bedeutung, dass das Ligandensystem über möglichst wenig konformationelle Freiheitsgrade verfügt, da sonst die resultierende Struktur lediglich begrenzt kontrolliert werden kann. Ferner sollte bei der Betrachtung der koordinativen Eigenschaften des Ligandensystems berücksichtigt werden, dass für die Metallkationen nur wenige Möglichkeiten vorhanden sein 1. Einleitung - 12 - sollten, sich relativ zum Liganden anzuordnen. Es reicht also nicht aus, nur die Koordinationssphäre der Metallzentren zu betrachten. Die am besten geeigneten Liganden werden die sein, bei denen eine eindeutige Vorzugsorientierung der Metalle zu den Liganden auftritt. Dies soll am Beispiel von N-Donorliganden verdeutlicht werden. Es ist möglich, neben aromatischen auch aliphatische Amine als Liganden zu verwenden. Hierbei stellt sich die Frage, welche dieser Liganden im Rahmen des "Crystal Engineering" für eine verbesserte Strukturkontrolle geeigneter sind. Derartige Untersuchungen können relativ einfach mit Hilfe von Datenbankanalysen in der "Cambridge Structure Data Base" (CSD) durchgeführt werden. Hierzu wird in der CSD nach Strukturen gesucht, die neben Übergangsmetallkationen auch Pyrazin als aromatischen oder Dimethylamin als aliphatischen Liganden enthalten. Im direkten Vergleich wird anschließend die Orientierung der Metallkationen relativ zum Liganden analysiert.[94,95] Die Lage der Metallkationen relativ zum Liganden lässt sich hierbei mit Hilfe von Polarkoordinaten beschreiben, die durch die drei folgenden Parameter charakterisiert sind: Dem Abstand zwischen dem Metallkation und dem Stickstoffatom des N-Donorliganden r(N-M), dem Auslenkungswinkel des Metallkations aus der CN-C-Ebene φ sowie dem Winkel senkrecht zu dieser Ebene θ (Abb. 1.10). Abb. 1.10: Beschreibung der Lage von Metallkationen relativ zum Stickstoffliganden durch die drei Parameter r(N-M), θ und φ. Erwartungsgemäß ordnen sich die Metallkationen im Falle des sp2-hybridisierten Stickstoffatoms des Pyrazins in Richtung des N-N-Vektors sowie innerhalb der Molekülebene (Abb. 1.11: links) an. Es resultiert eine eher schmale Verteilung. Im Gegensatz hierzu wird im Falle des sp3-hybridisierten Stickstoffatoms des Dimethylamins eine wesentlich breitere Verteilung gefunden (Abb. 1.11: rechts). 1. Einleitung - 13 - Abb. 1.11: Punktdiagramm, das die Lage von Metallkationen relativ zu Pyrazin (rechts) oder Dimethylamin (links) zeigt, übersichtshalber sind einige Wasserstoffatome nicht dargestellt. Aus diesen Untersuchungen geht hervor, dass aromatische N-Donorliganden für einen gezielten Aufbau von mehrdimensionalen Koordinationspolymeren weitaus geeigneter sind als sp3-hybridisierte aliphatische Aminliganden. Bei letzteren kann die Struktur nicht eindeutig kontrolliert werden, so dass deshalb in der vorliegenden Diplomarbeit ausschließlich die in Abb. 1.12 dargestellten aromatischen Diamine verwendet werden. N S N S N S S 4,4'-Dipyridyldisulfid N 2,2'-Dipyridyldisulfid N N N H3 C N CH 3 N N Pyrazin 2,5-Dimethylpyrazin Pyrimidin Abb. 1.12: Strukturformeln ausgewählter aromatischer Diamine und deren Derivate. Da es sich bei diesen aromatischen N-Donorliganden um neutrale Moleküle handelt, wird zur Ladungskompensation der Metallkationen ein Gegenion benötigt. Dieses sollte ähnliche Voraussetzungen wie die N-Donorliganden erfüllen. Es muss sich daher um Anionen handeln, die verbrückend und nicht chelatisierend wirken. Außerdem sollten diese Gegenionen über nur wenig konformationelle Freiheitsgrade verfügen und nur wenige Möglichkeiten für eine Anordnung der Metallkationen relativ zum Ligandensystem vorhanden sein. Sollen z.B. gezielt Verbindungen mit kooperativen magnetischen Eigenschaften dargestellt werden, sollten neben paramagnetischen Metallzentren sehr kleine verbrückende Anionen, wie das Azid-[27] oder das Thiocyanatanion,[28] verwendet werden. 1. Einleitung 1.5 - 14 - Stand der Forschung Im Folgenden wird ein kurzer Überblick über die Entwicklung der Forschung des Arbeitskreises Näther et al.[28,86-93,96-112] im Bereich der Synthese neuer koordinationspolymerer Verbindungen, deren strukturellen Charakterisierung und deren thermischen Reaktivität gegeben. Diese Ergebnisse sind der Ausgangspunkt für die im Rahmen dieser Diplomarbeit bearbeitete Aufgabenstellung. 1.5.1 Ausgangspunkt: CuX-Pyrazin-Koordinationspolymere (X = Cl, Br, I) Wird Kupfer(I)chlorid mit Pyrazin umgesetzt, werden zwei Verbindungen gebildet.[103] Die Struktur der ligandenreichen 1:1-Verbindung Poly[CuCl(µ2-Pyrazin-N,N‘)] besteht aus "zickzack"-förmigen CuCl-Einzelketten, die über µ-N,N‘-Koordination zu Schichten verknüpft sind (Abb. 1.13: links). In der ligandenarmen 2:1-Verbindung Poly[(CuCl)2(µ2-Pyrazin-N,N‘)] werden CuCl-Doppelketten gefunden, die ebenfalls durch die Pyrazin-Liganden zu Schichten verknüpft sind (Abb. 1.13: rechts). Abb. 1.13: Kristallstrukturen von Poly[CuCl(µ2-Pyrazin-N,N‘)] (links) und Poly[(CuCl)2(µ2-Pyrazin-N,N‘)] (rechts). Die Kristallstrukturen der 1:1-Verbindung mit CuBr sowie der 2:1-Verbindung mit CuBr und CuI wurden später ebenso charakterisiert und zeigen die gleiche Topologie wie die Koordinationsnetzwerke der 1:1- und 2:1-Verbindungen mit CuCl. Jede dieser Verbindungen lassen sich leicht direkt aus der Reaktion der Kupfer(I)halogenide mit Pyrazin in verschiedenen Lösungsmitteln herstellen, wobei sich die 1:1-Verbindungen mit CuCl und CuBr in großen Mengen und sehr rein synthetisieren lassen. Im Gegensatz dazu lassen sich in Lösung die entsprechenden 2:1-Verbindungen nur als Mischung der 1:1- und 2:1-Verbindung herstellen. Mit CuI allerdings wird die ligandenarme 2:1-Verbindung in Lösung phasenrein erhalten, wobei die 1:1-Verbindung nicht auftritt. 1. Einleitung - 15 - Bei einer thermogravimetrischen Analyse der 1:1-Verbindung mit CuCl werden zwei Massestufen in der TG-Kurve beobachtet (Abb. 1.14: links). Hierbei stimmt der experimentelle Masseverlust jeder Stufe mit dem überein, der für den Verlust eines halben Pyrazin-Liganden berechnet wird. Der Rückstand nach der ersten Stufe kann durch Röntgen-Pulveruntersuchungen als 2:1-Verbindung und nach der zweiten Stufe als CuCl identifiziert werden (Abb. 1.14: rechts). Temperaturabhängige Röntgen-Pulveruntersuchungen belegen dabei, dass sich die 1:1-über die 2:1-Verbindung zu CuCl zersetzt. endo Δm = -20.7 % TG Tp=273°C relative Intensität Δm und ΔT in belibiege Einheiten Δm = -20.4 % gemessen DTA Tp=176°C berechnet 50 100 150 200 Temperatur / °C 250 300 10 15 20 25 30 2-Theta / ° 35 40 45 Abb. 1.14: DTA-TG Kurve der 1:1-Verbindung Poly[CuCl(µ2-Pyrazin-N,N‘)] (links), experimentelles Pulverdiffraktogramm für den Rückstand nach der ersten TG-Stufe (rechts oben) und berechnetes Pulverdiffraktogramm der 2:1-Verbindung Poly[(CuCl)2(µ2-Pyrazin-N,N‘)] (rechts unten). In weiteren Untersuchungen wurde nachgewiesen, dass die 1:1-Verbindung mit CuBr gleich reagiert, wobei die 2:1-Verbindung mit CuI direkt zum Kupfer(I)iodid zerfällt. Durch diese thermische Abbaureaktion konnten zum ersten Mal die ligandenarmen 2:1-Verbindungen mit CuCl und CuBr phasenrein hergestellt werden. Dies belegt, dass neue CuX-Koordinationspolymere durch thermische Abbaureaktionen hergestellt werden können. Davon ausgehend wurden die Darstellung, die Struktur und die thermischen Eigenschaften von CuX-Koordinationspolymeren auf Basis verschiedener Liganden systematisch untersucht. 1.5.2 CuX-4,4’-Bipyridin-Koordinationspolymere (X = Cl, Br, I) Zur Überprüfung, ob die bisher erlangten Ergebnisse nicht nur auf die CuX-Pyrazin-Koordinationspolymere beschränkt sind, wurden weitere Untersuchungen von Koordinationspolymeren auf Basis von CuX und dem linearen Liganden 4,4‘-Bipyridin durchgeführt.[102] Es galt 1. Einleitung - 16 - herauszufinden, ob eine Beziehung zwischen der Struktur dieser Verbindungen und ihrem thermischen Verhalten besteht. Die Strukturen der ligandenarmen 2:1-Verbindungen Poly[(CuX)2(µ2-4,4’-Bipyridin-N,N‘)] weisen die gleiche Netzwerktopologie wie die entsprechenden 2:1-Verbindungen mit Pyrazin auf: CuX-Doppelketten, die durch die 4,4’-Bipyridin-Liganden zu Schichten verknüpft sind (Abb. 1.15: rechts). Dem gegenüber zeigen die ligandenreichen 1:1-Verbindungen Poly[CuX(µ2-4,4’-Bipyridin-N,N‘)] eine Netzwerktopologie sowie CuX-Substruktur, die sich im Vergleich zu den entsprechenden 1:1-Verbindungen mit Pyrazin völlig unterscheidet: (CuI)2Dimere, die durch die 4,4’-Bipyridin-Liganden zu pseudo hexagonalen Schichten verknüpft sind (Abb. 1.15: links). Abb. 1.15: Kristallstrukturen von Poly[CuX(µ2-4,4’-Bipyridin-N,N‘)] (links) und Poly[(CuX)2(µ2-4,4’-Bipyridin-N,N‘)] (rechts). Das thermische Verhalten der ligandenreichen 1:1-Verbindungen mit 4,4‘-Bipyridin entspricht dem der Verbindungen mit Pyrazin als Liganden. Es wurde herausgefunden, dass die 1:1-Verbindungen beim Aufheizen quantitativ in ligandenarme 2:1-Verbindungen umwandeln, bevor sie zum Kupfer(I)halogenid zerfallen. Die Ergebnisse belegen eindeutig, dass diese Methode zur Herstellung von CuX-Verbindungen allgemein anwendbar und nicht nur auf Pyrazin-Koordinationspolymere beschränkt ist. Überdies ist kein einfacher Zusammenhang zwischen der Struktur der Edukte bzw. Produkte und ihrem thermischen Verhalten erkennbar. 1. Einleitung 1.5.3 - 17 - CuX-2-Methyl-/2-Chlorpyrazin-Koordinationspolymere (X = Cl, Br, I) In den bisherigen Untersuchungen wurde gezeigt, dass die thermische Reaktivität der CuXKoordinationspolymere auf Basis von 4,4‘-Bipyridin und Pyrazin identisch ist. Beim Aufheizen wandeln die topologisch unterschiedlichen ligandenreichen Verbindungen quantitativ in die topologisch gleichen ligandenarmen Verbindungen um. Aus diesem Grund wurden weitere Untersuchungen mit ligandenreichen Verbindungen, die eine identische Netzwerktopologie aufweisen, durchgeführt. Als geeigneter Ligand in diesem Zusammenhang wurde das 2Methylpyrazin gewählt.[104] Wie erwartet, ist die Topologie der 1:1-Verbindungen mit CuCl und CuBr (Abb. 1.16: links) sowie der 2:1-Verbindungen mit CuBr und CuI (Abb. 1.16: rechts) identisch zu denen in den Verbindungen mit Pyrazin als Ligand (Abb. 1.13). Einzige Ausnahme stellt die Orientierung der Methylgruppe dar. Ebenso verhält sich die thermische Reaktivität dieser Verbindungen ähnlich: Die 1:1-Verbindungen wandeln sich beim Aufheizen quantitativ in die entsprechenden ligandenarmen 2:1-Verbindungen um, bevor sie zum Kupfer(I)halogenid zerfallen. Abb. 1.16: Kristallstrukturen von Poly[CuX(µ2-2-Methylpyrazin-N,N‘)] (links) und Poly[(CuX)2(µ2-2Methylpyrazin-N,N‘)] (rechts). In weiteren Studien wurden die Eigenschaften von Koordinationspolymeren auf Basis von 2Chlorpyrazin untersucht.[105] Hier wurden gleiche Kristallstrukturen erwartet, da die van-derWaals-Radien der Methylgruppe und des Chloridanions vergleichbar sind. In manchen Fällen werden sogar isotype Verbindungen, d.h. Verbindungen gleichen Strukturtyps mit sehr ähnlichen Gitterparametern, beobachtet. Dieses Verhalten wird als "Chlor-Methyl-Austauschregel" bezeichnet. Wie erwartet, sind die Strukturen der ligandenreichen 1:1-Verbindungen mit CuCl und CuBr topologisch mit denen des 2-Methylpyrazins identisch, jedoch nicht isotyp. Einzige Ausnahme ist, dass die relative Orientierung der Chloridanionen nicht alterniert (Abb. 1.17: links). 1. Einleitung - 18 - Wider Erwarten ist jedoch die Kristallstruktur der CuI-Verbindung mit 2-Chlorpyrazin vollkommen unterschiedlich: CuI-Doppelketten werden nicht durch den 2-Chlorpyrazin-Liganden verbrückt (Abb. 1.17: Mitte). Abb. 1.17: Kristallstrukturen von Poly[CuX(µ2-2-Chlorpyrazin-N,N‘)] (X = Cl, Br) (links), Catena[CuI(2Chlorpyrazin-N)] (Mitte) und Poly[CuI(µ2-2-Chlorpyrazin-N,N‘)] (rechts), übersichtshalber ohne Wasserstoffatome dargestellt. Des Weiteren ist die thermische Reaktivität von Poly[CuX(µ2-2-Chlorpyrazin-N,N‘)] (X = Cl, Br) im Vergleich zu den 2-Methylpyrazin-Koordinationspolymeren verschieden. Beim Aufheizen zerfallen die ligandenreichen 1:1-Koordinationspolymere mit 2-Chlorpyrazin direkt zum Kuper(I)halogenid, ohne eine ligandenarme Phase zu bilden. Im Gegensatz dazu bildet Catena[CuI(2-Chlorpyrazin-N)] beim Aufheizen eine ligandenarme 2:1-Verbindung (Abb. 1.17: rechts). 1.5.4 Kontrolle der thermischen Reaktivität durch Mischkristallbildung Die bisherigen Ergebnisse aus den CuX-2-Methyl- und CuX-2-Chlorpyrazin-Koordinationspolymeren (X = Cl, Br, I) zeigen, dass kein Zusammenhang zwischen ihrer Struktur und ihrer thermischen Reaktivität besteht, da die 2-Methylpyrazin-Koordinationspolymere beim thermischen Abbau im Vergleich zu denen mit 2-Chlorpyrazin eine ligandenarme Phase bilden. Aufgrund ihrer Netzwerktopologie und der unterschiedlichen thermischen Reaktivität wurde versucht, letztere durch Mischkristallbildung zu beeinflussen.[101] Bei der Reaktion von CuCl mit äquimolaren Mengen von 2-Chlor- und 2-Methylpyrazin entstehen Mischkristalle, die zur Hälfte aus den beiden Liganden bestehen und isotyp zu Poly[CuCl(µ2-2-Chlorpyrazin-N,N‘)] sind. Thermogravimetrische Untersuchungen in Kopplung mit der Massenspektroskopie belegen, dass beim Aufheizen eine ligandenarme intermediäre 2:1-Verbindung gebildet wird, die 1. Einleitung - 19 - nur 2-Chlorpyrazin enthält. Darüber hinaus kann im Vergleich zu den Mischkristallen der thermische Abbau einer Mischung aus Poly[CuCl(µ2-2-Chlorpyrazin-N,N‘)] und Poly[CuCl(µ2-2-Methylpyrazin-N,N‘)] als eine Summe zweier verschiedener thermischer Reaktionen beschrieben werden (Abb. 1.18). Diese ersten Experimente beweisen, dass die thermischen Eigenschaften derartiger Verbindungen durch Mischkristallbildung beeinflussbar sind. Abb. 1.18: TG-Kurven von Poly[CuCl(µ2-2-Methylpyrazin-N,N‘)] (1), Poly[CuCl(µ2-2-Chlorpyrazin-N,N‘)] (2), Poly[CuCl(µ2-2-Methylpyrazin-N,N‘)0.5(2-Chlorpyrazin)0.5 (3) sowie eine 1:1-Mischung von Poly[CuCl(µ2-2Methylpyrazin-N,N‘)] und Poly[CuCl(µ2-2-Methylpyrazin-N,N‘)] (4). 1.5.5 Einfluss der Kinetik thermischer Abbaureaktionen auf die Produktbildung Aus den Untersuchungen über die CuX-2-Methyl- und CuX-2-Chlorpyrazin-Koordinationspolymere (X = Cl, Br, I) geht hervor, dass die thermische Reaktivität zweier topologisch gleicher Verbindungen vollkommen unterschiedlich sein und durch Mischkristallbildung beeinflusst werden kann. In diesem Zusammenhang ist zu klären, von welchen Parametern das Auftreten verschiedener intermediärer Phasen während einer thermischen Abbaureaktion abhängt. Es wird angenommen, dass die Kinetik der Reaktion eine wesentliche Rolle spielt. Dies kann durch heizratenabhängige TG-Messungen untersucht werden. Diese Annahme wurde durch folgendes Beispiel überprüft: In der Reaktion von CuCl mit 2-Ethylpyrazin können die 1:1- und 2:1-Verbindungen hergestellt werden. Beim Aufheizen der ligandenreichen 1:1-Verbindung mit einer kleinen Heizrate werden zwei Massestufen beobachtet, in der die erste Stufe der Reaktion der ligandenarmen 2:1-Verbindung und die zweite Stufe dem CuCl zuzuordnen ist. Wird die Heizrate schrittweise erhöht, verringert sich der Wert der ersten Massestufe auf einen Wert, der der Bildung einer 3:2-Verbindung entspricht (Abb. 1.19). Die Strukturen aller drei Verbindungen konnten durch Einkristallstrukturanalyse charakterisiert werden.[91] 1. Einleitung - 20 - Abb. 1.19: Heizratenabhängige TG-Messungen für die ligandenreiche 1:1-Verbindung Poly[CuCl(µ2-2Ethylpyrazin-N-N‘)]. 1.5.6 Thermische Abbaureaktionen zur Herstellung von Koordinationspolymeren mit kooperativen magnetischen Eigenschaften Aus den bisherigen Untersuchungen geht hervor, dass neue ligandenarme Koordinationspolymere auf Basis von aromatischen Diaminen und Kupfer(I)halogeniden durch thermische Abbaureaktionen hergestellt werden können. Im Folgenden ist zu klären, ob diese Methode allgemein anwendbar ist und dadurch auch zur Herstellung anderer Koordinationspolymere genutzt werden kann. Als besonders interessant im Bereich der kooperativen magnetischen Eigenschaften gelten Koordinationspolymere auf Basis von paramagnetischen Übergangsmetallen und kleinen Anionen, wie Thiocyanaten. Aus diesem Grund wurden Koordinationspolymere auf Basis von Mangan(II)thiocyanat und Pyrazin hergestellt.[28] Auch hier konnte eine ligandenreiche (Abb. 1.20: links) und eine ligandenarme Verbindung (Abb. 1.20: rechts) hergestellt werden. Abb. 1.20: Kristallstrukturen der ligandenreichen 1:2-Verbindung Poly[Mn(SCN)2(µ2-Pyrazin-N,N‘)2] (links) und der ligandenarmen 1:1-Verbindung Poly[Mn(SCN)2(µ2-Pyrazin-N,N‘)] (rechts). 1. Einleitung - 21 - In der ligandenreichen Verbindung sind die Metallzentren nur durch die Pyrazin-Liganden zu Schichten verknüpft, während in der ligandenarmen Verbindung die Metallzentren zusätzlich durch die Thiocyanatanionen über µ-N,S-Koordination zu Schichten verknüpft sind. Letzteres ist eine wichtige Voraussetzung für das Auftreten kooperativer magnetischer Austauschwechselwirkungen. Zum Nachweis der in der ligandenarmen Verbindung auftretenden magnetischen Austauschwechselwirkungen, wird diese in größeren Mengen phasenrein benötigt. Jedoch wird sie nur als Gemisch der ligandenarmen und der ligandenreichen Verbindung erhalten. Jedoch ist es möglich, die ligandenreiche Verbindung in großen Mengen phasenrein herzustellen. Daraufhin wurde, basierend auf den Ergebnissen der CuX-Verbindungen, das thermische Verhalten der ligandenreichen Verbindung untersucht. Wie erwartet, lassen sich zwei Massestufen beobachten, von denen die erste als die ligandenarme Verbindung Poly[Mn(SCN)2(µ2-Pyrazin-N,N‘)] und die zweite als Mn(SCN)2 identifiziert werden kann. Die ligandenarme Verbindung wird dadurch in großen Mengen und phasenrein hergestellt. Aus magnetischen Untersuchungen geht hervor, dass die ligandenreiche Verbindung einen "normalen" Curie-Weiss-Paramagnetismus zeigt (Abb. 1.21: links) und die ligandenarme Verbindung einen antiferromagnetischen Ordnungspunkt besitzt (Abb. 1.21: rechts). Abb. 1.21: Magnetische Suszeptibilität χ als Funktion der Temperatur für die Verbindung Poly[Mn(SCN)2(µ2Pyrazin-N,N‘)2] (links) und Poly[Mn(SCN)2(µ2-Pyrazin-N,N‘)] (rechts). Folglich belegen die o.g. Untersuchungen, dass ein breites Feld neuer Koordinationspolymere durch thermische Abbaureaktionen geeigneter ligandenreicher Precursoren dargestellt werden kann, welche interessante physikalische Eigenschaften, wie z.B. kooperative magnetische Eigenschaften, besitzen können. 1. Einleitung 1.6 - 22 - Zielsetzung und Übersicht über die Arbeit Wie bereits erläutert, eignen sich insbesondere Koordinationspolymere für den gezielten Aufbau von Kristallstrukturen. Aus diesem Grund sind derartige Verbindungen Schwerpunkt der vorliegenden Diplomarbeit, die sich mit der Darstellung, der strukturellen Charakterisierung und der umfassenden Untersuchung der physikalischen Eigenschaften neuer Koordinationspolymere auf Basis von N-Donorliganden und Übergangsmetallen beschäftigt. Im Rahmen des "Crystal Engineering" wird versucht, mit einer gezielten Auswahl von Liganden und Anionen – deren Kriterien im Abschnitt 1.4 ausführlich erklärt wurden – den Aufbau von Strukturen zu kontrollieren oder zumindest stark zu beeinflussen. Hierbei wird angenommen, die Netzwerktopologie innerhalb gewisser Grenzen vorherbestimmen zu können. Darüber hinaus gilt es vorrangig herauszufinden, – basierend auf vorherigen Arbeiten über die thermischen Eigenschaften von Koordinationspolymeren auf Basis von Kupfer(I)halogeniden und N-Donorliganden – ob die Methode des thermischen Abbaus ligandenreicher Precursoren auch zur Darstellung ligandenarmer thermodynamisch stabiler und metastabiler Verbindungen sowie polymorpher Modifikationen anderer Systeme geeignet ist. Zu Beginn dieser Diplomarbeit erfolgt eine Übersicht über die Untersuchungen zur Darstellung, zur Struktur und zu den Eigenschaften von Koordinationspolymeren auf Basis aromatischer N-Donorliganden und Zink(II)halogeniden. Hierzu wurden von der Gruppe Näther et. al. bereits einige Vorarbeiten geleistet.[86,96-100,113-117] Aufgrund dessen war eine ausführliche Einarbeitung und Vertiefung in die Methoden der modernen Festkörperanalytik möglich. Hierzu zählen insbesondere die Röntgen-Pulverdiffraktometrie, die Röntgen-Einkristallstrukturanalyse und die Energiedispersive Röntgenfluoreszenzspektroskopie sowie die thermischen Analyseverfahren, der Differenzthermoanalyse und der Thermogravimetrie in Kopplung mit der Massenspektroskopie sowie der dynamischen Differenzkalorimetrie. Basierend auf den vorangegangenen Ergebnissen und Erfahrungen werden im zweiten Teil Untersuchungen zur Darstellung, zur Struktur und zu den Eigenschaften von Koordinationspolymeren auf Basis von N-Donorliganden und paramagnetischen Übergangsmetallen vorgestellt. Diese stellen potenzielle Verbindungen mit interessanten magnetischen Eigenschaften dar, die in der Literatur nur unvollständig dokumentiert sind. Hierzu zählen beispielsweise Koordinationspolymere auf Basis von aromatischen N-Donorliganden und ÜbergangsmetallThiocyanaten, die z.T. kooperative magnetische Eigenschaften aufweisen.[28,89,106,118-126] Da es sich bei Thiocyanaten und Cyanaten um isovalenzelektronische und strukturell gleichartige 1. Einleitung - 23 - Ionen handelt, stellt sich die Frage, ob Koordinationspolymere auch auf Basis von Übergangsmetall-Cyanaten ähnliche magnetische, thermische und strukturelle Eigenschaften aufweisen. Hierzu finden sich in der Literatur nur wenige Beispiele.[119,122] Daher werden im Rahmen dieser Diplomarbeit neben den Übergangsmetall-Thiocyanaten auch die -Cyanate untersucht. Hierzu zählt insbesondere die Untersuchung der thermischen Eigenschaften von neuen und literaturbekannten ligandenreichen Koordinationsverbindungen auf Basis paramagnetischer Übergangmetall-Thiocyanaten und -Cyanaten sowie die magnetische Charakterisierung ihrer ligandenarmen thermischen Abbauprodukte. Zum Abschluss dieser Diplomarbeit werden einige Verbindungen auf Basis derivatisierter NDonorliganden und Übergangsmetalle vorgestellt, die sich nicht in die ursprüngliche Fragestellung einordnen lassen oder für weitere Untersuchungen nicht phasenrein dargestellt werden konnten. Diese wurden aus Liganden hergestellt, die sich im Verlauf der Reaktionen zersetzten und sich somit als ungeeignet zur Darstellung von Koordinationspolymeren erwiesen haben. 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 24 - 2 Verbindungen auf Basis von Zink(II)halogeniden und 2,5Dimethylpyrazin 2.1 Einleitung In den bislang beschriebenen Untersuchungen wurde gezeigt, dass ligandenarme Koordinationspolymere auf Basis von Kupfer(I)halogeniden oder auch Pseudohalogeniden und N-Donorliganden einfach durch den thermischen Abbau geeigneter ligandenreicher Precursor-Verbindungen dargestellt werden können. Die entstehenden Intermediate sind sehr rein und lassen sich nahezu immer in 100 %iger Ausbeute herstellen. Außerdem können in einigen Fällen thermodynamisch metastabile Verbindungen und polymorphe Modifikationen erhalten werden. Aus diesem Grund ist der thermische Abbau eine alternative Methode zur Herstellung derartiger Verbindungen in Lösung – besonders in den Fällen, in denen die Produkte lediglich als Mischungen oder überhaupt nicht dargestellt werden können. Ausgehend von diesen Ergebnissen wurde geprüft, ob der thermische Abbau nur für die Entdeckung und Darstellung von Kupfer(I)halogenid-Koordinationsverbindungen genutzt werden kann oder ob diese Methode auch zur Darstellung anderer Übergangsmetall-Koordinationsverbindungen verwendet werden kann. Hierzu wurden Zink(II)halogenid-Koordinationspolymere ausgewählt. Im Gegensatz zu den koordinationspolymeren Verbindungen auf Basis von Kupfer(I)halogeniden, in denen das Kupfer(I)kation ausschließlich tetraedrisch koordiniert ist, ist in diesen Verbindungen das Zink(II)kation tetraedrisch,[127-130] oktaedrisch[131-134] oder auch pentaedrisch[135-138] koordiniert. Diese strukturelle Vielfalt ist eine wichtige Voraussetzung für die Darstellung neuer ligandenarmer Verbindungen durch thermische Abbaureaktionen. Daher handelt es sich bei Zink(II)halogenid-Koordinationspolymeren um potenzielle Verbindungen für die Darstellung neuer ligandenarmer Verbindungen. Dies wurde bereits in einigen Untersuchungen am Beispiel von Zink(II)halogeniden und N-Donorliganden als Ligand, wie z.B. Pyrazin[115], Pyrimidin[100,117], Pyridazin[97,99,113] und 2-Chlorpyrazin[114] nachgewiesen. Im ersten Teil dieser Diplomarbeit wird geprüft, ob sich ligandenreiche Koordinationspolymere auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin ebenfalls zur Darstellung ligandenarmer Verbindungen eignen. Im folgenden Kapitel steht daher die Darstellung und die Charakterisierung derartiger Koordinationspolymere im Vordergrund. Hierzu zählen die Untersuchung der thermischen Eigenschaften sowie die Darstellung neuer thermodynamisch stabiler und meta- 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 25 - stabiler Zink(II)halogenid-Koordinationsverbindungen und polymorpher Modifikationen. Insgesamt wurden die folgenden acht Verbindungen dargestellt und charakterisiert:[139] ¾ [ZnCl2(2,5-Dimethylpyrazin)]n (1) IUPAC: Catena-(µ2-2,5-Dimethylpyrazin-N,N')-Dichlorido-Zink(II) ¾ (ZnCl2)2(2,5-Dimethylpyrazin)3 (2) IUPAC: Bis(dichlorido)bis(2,5-Dimethylpyrazin-N)(µ2-2,5-Dimethylpyrazin-N,N')di-Zink(II) ¾ [ZnBr2(2,5-Dimethylpyrazin)]n (3I + 3II) IUPAC: Catena-(µ2-2,5-Dimethylpyrazin-N,N')-dibromido-Zink(II) ¾ (ZnBr2)2(2,5-Dimethylpyrazin)3 (4) IUPAC: Bis(dibromido)bis(2,5-Dimethylpyrazin-N)(µ2-2,5-Dimethylpyrazin-N,N')di-Zink(II) ¾ [ZnBr2(2,5-Dimethylpyrazin)]2·2,5-Dimethylpyrazin (5) IUPAC: [Dibromido-bis(2,5-Dimethylpyrazin-N)-Zink(II)]·2,5-Dimethylpyrazin ¾ [ZnI2(2,5-Dimethylpyrazin)]n (6) IUPAC: Catena-(µ2-2,5-Dimethylpyrazine-N,N')-diiodido-Zink(II) ¾ ZnI2(2,5-Dimethylpyrazin)2 (7) IUPAC: Diioido-bis(2,5-dimethylpyrazine-N)-Zink(II) Zunächst werden die Kristallstrukturen der o.g. Verbindungen beschrieben. Anschließend erfolgt eine ausführliche Vorstellung deren thermischen Eigenschaften und ein Vergleich der Strukturen und Eigenschaften der o.g. Verbindungen mit denen aus Zn(II)halogeniden und Pyrazin als Ligand. 2.2 Kristallstrukturen der Verbindungen 2.2.1 [ZnCl2(2,5-Dimethylpyrazin)]n Die 1:1-Chlorid-Verbindung Catena-(µ2-2,5-Dimethylpyrazin-N,N')-Dichlorido-Zink(II) (1) kristallisiert in der orthorhombischen Raumgruppe Pnma mit vier Formeleinheiten in der Elementarzelle (Tab. 2.1). Die Zink(II)kationen sowie die beiden kristallografisch unabhängigen Chloridanionen befinden sich auf einer Spiegelebene und die 2,5-Dimethylpyrazin-Liganden sind auf einem Inversionszentrum angeordnet. Die Zink(II)kationen sind in der Kristallstruktur jeweils von zwei kristallografisch unabhängigen Chloridanionen und zwei symmetrieäquivalenten 2,5-Dimethylpyrazin-Liganden verzerrt tetraedrisch koordiniert (Abb. 2.1: links). Die Zn-N- 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 26 - Abstände betragen 2.1027(15) Å und die Bindungswinkel um die Zink(II)kationen liegen zwischen 97.00(8) und 117.83(4)° (Tab. 2.2). Es werden ZnCl2-Monomere als ZnX2-Substruktur gefunden, welche durch die 2,5-Dimethylpyrazin-Liganden über µ-N,N'-Koordination zu linearen Ketten in Richtung der kristallografischen a-Achse verbunden sind (Abb. 2.1: rechts). Abb. 2.1: Kristallstruktur der 1:1-Chlorid-Verbindung 1 mit Blick auf die Koordinationssphäre des Zink(II)kations mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden (links) und Blick auf die Kristallstruktur in Richtung der kristallografischen a-Achse (rechts). Symmetrie-Operationen: A = -x + 2, -y + 1, -z + 1; B = x, -y + ½, z. 2.2.2 (ZnCl2)2(2,5-Dimethylpyrazin)3 Die 2:3-Chlorid-Verbindung Bis(dichlorido)bis(2,5-Dimethylpyrazin-N)(µ2-2,5-Dimethylpyrazin-N,N')di-Zink(II) (2) kristallisiert in der triklinen Raumgruppe P-1 mit einer Formeleinheit in der Elementarzelle und allen Atomen in allgemeiner Lage (Tab. 2.1). In der Kristallstruktur werden diskrete [(ZnCl2)2(2,5-Dimethylpyrazin)3]-Einheiten gefunden, die auf einem Inversionszentrum lokalisiert sind. Jedes der beiden Zink(II)kationen ist von zwei Chloridanionen sowie einem Stickstoffatom eines terminalen und einem Stickstoffatom eines überbrückenden 2,5-Dimethylpyrazin-Liganden koordiniert. Somit werden in der Kristallstruktur die beiden Zink(II)kationen durch einen Liganden über µ-N-N'-Koordination zu einem diskreten Komplex verbrückt (Abb. 2.2: links). Die Zn-N-Abstände liegen zwischen 2.085(4) und 2.091(4) Å und die Bindungswinkel um die Zink(II)kationen betragen zwischen 104.56(12) und 108.53(12)° (Tab. 2.2). In der Kristallstruktur sind die diskreten Komplexe über intermolekulare C-H···ClWasserstoffbrückenbindungen (∠(C2-H2···Cl2) = 169.753(17)°, d(H2···Cl2) = 2.8391(4) Å) zu Ketten verknüpft (Abb. 2.2: rechts). 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 27 - Abb. 2.2: Kristallstruktur der 2:3-Chlorid-Verbindung 2 mit Blick auf die Koordinationssphäre der Zink(II)kationen mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden (links) und Blick auf die Kristallstruktur in Richtung der kristallografischen a-Achse (rechts). Intermolekulare Wasserstoffbrückenbindungen sind als gestrichelte Linien gezeigt. Symmetrie-Operation: A = -x + 1, -y + 2, -z. Tab. 2.1: Ausgewählte Einkristallstrukturdaten der 1:1- 1 und 2:3-Chlorid-Verbindung 2. Verbindung Kristallsystem Raumgruppe a/Å b/Å c/Å α /° β /° γ /° Z V / Å3 R1 1 orthorhombisch Pnma 6.0621(4) 10.2156(8) 13.9518(8) 90 90 90 4 864.01(10) 0.0241 2 triklin P-1 7.2149(10) 9.1905(11) 10.1410(13) 79.973(15)° 70.138(15)° 70.264(15)° 1 593.94(13) 0.0496 Tab. 2.2: Ausgewählte Bindungslängen [Å] und -winkel [°]der 1:1- 1 und 2:3-Chlorid-Verbindung 2. Zn(1)-N(1) N(1)-Zn(1)-N(1B) N(1)-Zn(1)-Cl(1) Cl(1)-Zn(1)-Cl(2) Zn(1)-N(1) Zn(1)-N(11) N(1)-Zn(1)-N(11) N(1)-Zn(1)-Cl(1) N(11)-Zn(1)-Cl(1) Verbindung 1 2.1027(15) Zn(1)-Cl(1) 97.00(8) Zn(1)-Cl(2) 117.83(4) N(1)-Zn(1)-Cl(2) 113.80(2) Verbindung 2 2.085(4) Zn(1)-Cl(1) 2.091(4) Zn(1)-Cl(2) 113.05(16) N(1)-Zn(1)-Cl(2) 108.53(12) N(11)-Zn(1)-Cl(2) 105.86(12) Cl(1)-Zn(1)-Cl(2) 2.2129(6) 2.2312(6) 103.99(4) 2.2148(14) 2.2358(13) 104.56(12) 105.64(12) 119.41(5) 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin 2.2.3 - 28 - [ZnBr2(2,5-Dimethylpyrazin)]n – Modifikation I Die polymorphe Modifikation 3I der 1:1-Bromid-Verbindung Catena-(µ2-2,5-DimethylpyrazinN,N')-dibromido-Zink(II) kristallisiert in der orthorhombischen Raumgruppe Pnma mit vier Formeleinheiten in der Elementarzelle und ist isotyp zu Verbindung 1 (Abb. 2.3 und Tab. 2.3). Die Zn-N-Abstände betragen 2.110(4) Å und die Bindungswinkel um die Zink(II)kationen liegen zwischen 97.5(2) und 118.20(11)° (Tab. 2.4). Abb. 2.3: Kristallstruktur der 1:1-Bromid-Verbindung 3I mit Blick auf die Koordinationssphäre des Zink(II)kations mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden. Symmetrie-Operationen: A = -x, -y + 1, -z + 1; B = x, -y + ½, z. 2.2.4 [ZnBr2(2,5-Dimethylpyrazin)]n – Modifikation II Die polymorphe Modifikation 3II der 1:1-Bromid-Verbindung Catena-(µ2-2,5-DimethylpyrazinN,N')-dibromido-Zink(II) kristallisiert in der orthorhombischen Raumgruppe Pnma mit vier Formeleinheiten in der Elementarzelle (Tab. 2.3). Die Modifikationen 3I und 3II sind topologisch identisch. In der asymmetrischen Einheit der Modifikation 3II sind die Zink(II)kationen sowie ein kristallografisch unabhängiges Chloridanion auf einer C2-Achse und der 2,5-Dimethylpyrazin-Ligand auf einem Inversionszentrum angeordnet. Die Zink(II)kationen sind in der Kristallstruktur jeweils von zwei symmetrieäquivalenten Chloridanionen und zwei symmetrieäquivalenten 2,5-Dimethylpyrazin-Liganden verzerrt tetraedrisch koordiniert (Abb. 2.4: links). Der Zn-N-Abstand beträgt 2.121(2) Å und die Bindungswinkel um die Zink(II)kationen liegen zwischen 93.57(13) und 119.86(7)° (Tab. 2.4). Es werden ZnCl2-Monomere als ZnX2-Substruktur gefunden, die durch die 2,5-Dimethylpyrazin-Liganden über µ-N,N'-Koordination zu linearen Ketten in Richtung der kristallografischen c-Achse verknüpft sind (Abb. 2.4: rechts). 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 29 - Abb. 2.4: Kristallstruktur der 1:1-Bromid-Verbindung 3II mit Blick auf die Koordinationssphäre des Zink(II)kations mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden (links) und Blick auf die Kristallstruktur in Richtung der kristallografischen b-Achse (rechts). Symmetrie-Operationen: A = -x + 1, -y, -z + 1; B = -x + 1, y, -z + ½. Tab. 2.3: Ausgewählte Einkristallstrukturdaten der 1:1-Bromid-Verbindungen 3I und 3II. Verbindung Kristallsystem Raumgruppe a/Å b/Å c/Å α /° β /° γ /° Z V / Å3 R1 3I orthorhombisch Pnma 6.2698(6) 10.3167(7) 14.3842(9) 90 90 90 4 930.42(12) 0.0431 3II monoklin C2/c 16.1453(18) 6.4913(6) 9.8755(10) 90 115.096(11) 90 4 937.29(17) 0.0271 Tab. 2.4: Ausgewählte Bindungslängen [Å] und -winkel [°]der 1:1-Bromid-Verbindungen 3I und 3II. Br(1)-Zn(1) Br(2)-Zn(1) N(1B)-Zn(1)-N(1) N(1)-Zn(1)-Br(1) Zn(1)-N(1) N(1B)-Zn(1)-N(1) N(1)-Zn(1)-Br(1) Verbindung 3I 2.3510(10) Zn(1)-N(1) 2.3711(10) N(1)-Zn(1)-Br(2) 97.5(2) Br(1)-Zn(1)-Br(2) 118.20(11) Verbindung 3II 2.121(2) Zn(1)-Br(1) 93.57(13) N(1)-Zn(1)-Br(1B) 105.32(6) Br(1)-Zn(1)-Br(1B) 2.110(4) 105.01(10) 111.05(4) 2.3560(5) 119.86(7) 112.38(3) 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin 2.2.5 - 30 - (ZnBr2)2(2,5-Dimethylpyrazin)3 Die 2:3-Bromid-Verbindung Bis(dibromido)bis(2,5-Dimethylpyrazin-N)(µ2-2,5-Dimethylpyrazin-N,N')di-Zink(II) (4) kristallisiert in der triklinen Raumgruppe P-1 mit einer Formeleinheit in der Elementarzelle und ist isotyp zu Verbindung 2 (Abb. 2.5 und Tab. 2.5). Die Zn-N-Abstände liegen zwischen 2.082(5) und 2.085(5) Å, die Bindungswinkel um die Zink(II)kationen betragen zwischen 106.79(15) und 119.37(4)° (Tab. 2.6). Im Vergleich zur isotypen 2:3-Chlorid-Verbindung 2, sind auch hier in der Kristallstruktur die diskreten Komplexe über intermolekulare CH···Br-Wasserstoffbrückenbindungen (∠(C2-H2···Cl2) = 172.433(15)°, d(H2···Cl2) = 2.9427(5) Å) zu Ketten miteinander verknüpft. Abb. 2.5: Kristallstruktur der 2:3-Bromid-Verbindung 4 mit Blick auf die Koordinationssphäre des Zink(II)kations mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden. Symmetrie-Operationen: A = -x + 1, -y + 2, -z. 2.2.6 [ZnBr2(2,5-Dimethylpyrazin)]2·2,5-Dimethylpyrazin Die ligandenreiche 1:3-Bromid-Verbindung [Dibromido-bis(2,5-Dimethylpyrazin-N)-Zink(II)] ·2,5-Dimethylpyrazin (5) kristallisiert in der monoklinen Raumgruppe C2/c mit vier Formeleinheiten in der Elementarzelle (Tab. 2.5). In der asymmetrischen Einheit sind die Zink(II)kationen sowie ein kristallografisch unabhängiges Bromidanion auf einer C2-Achse angeordnet. Der nicht am Zink(II)kation koordinierte 2,5-Dimethylpyrazin-Ligand befindet sich auf einem Inversionszentrum. Das kristallografisch unabhängige Bromidanion ist über zwei Positionen fehlgeordnet und wurde mit einem Split-Modell sowie Besetzungsfaktoren von jeweils 0.5 verfeinert. In der Kristallstruktur liegen diskrete [(ZnBr2)(2,5-Dimethylpyrazin)2]·(2,5-Dimethylpyrazin)-Einheiten vor, in denen die Zink(II)kationen jeweils von zwei symmetrieäquivalenten Bromidanionen und zwei symmetrieäquivalenten 2,5-Dimethylpyrazin-Liganden verzerrt tetraedrisch koordiniert sind. Der freie 2,5-Dimethylpyrazin-Ligand ist dabei nicht mit dem Zink(II)kation verknüpft (Abb. 2.6: links). Die Zn-N-Abstände betragen 2.066(3) Å und die Bindungswinkel um die Zink(II)kationen liegen zwischen 104.26(11) und 115.0(6)° (Tab. 2.6). In der Kristallstruktur sind die diskreten Komplexe in gewellten Ketten angeordnet (Abb. 2.6: rechts). 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 31 - Abb. 2.6: Kristallstruktur der 1:3-Bromid-Verbindung 5 mit Blick auf die Koordinationssphäre des Zink(II)kations mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden (links) und Blick auf die Kristallstruktur in Richtung der Diagonalen der kristallografischen a/b-Achse (rechts). Symmetrie-Operationen: A = -x, y, -z + ½; B = -x, -y + 2, -z. Tab. 2.5: Ausgewählte Einkristallstrukturdaten der 2:3- 4 und 1:3-Bromid-Verbindung 5. Verbindung Kristallsystem Raumgruppe a/Å b/Å c/Å α /° β /° γ /° Z V / Å3 R1 4 triklin P-1 7.2840(8) 9.4775(16) 10.1762(11) 81.004(13) 72.489(12) 71.306(12) 1 633.21(14) 0.0548 5 monoklin C2/c 8.5956(9) 11.4237(7) 23.1531(16) 90 100.174(10) 90 4 2237.7(3) 0.0622 Tab. 2.6: Ausgewählte Bindungslängen [Å] und -winkel [°]der 2:3- 4 und 1:3-Bromid-Verbindung 5. Zn(1)-N(1) Zn(1)-N(11) N(1)-Zn(1)-N(11) N(1)-Zn(1)-Br(1) N(11)-Zn(1)-Br(1) Zn(1)-N(2) N(2)-Zn(1)-N(2A) N(2)-Zn(1)-Br(1') N(2A)-Zn(1)-Br(1') N(2A)-Zn(1)-Br(1A') Br(1')-Zn(1)-Br(1A') N(2)-Zn(1)-Br(1) Verbindung 4 2.082(5) Zn(1)-Br(1) 2.088(5) Zn(1)-Br(2) 113.6(2) N(1)-Zn(1)-Br(2) 107.88(15) N(11)-Zn(1)-Br(2) 106.79(15) Br(1)-Zn(1)-Br(2) Verbindung 5 2.066(3) Zn(1)-Br(1') 114.43(15) Zn(1)-Br(1) 111.9(3) Br(1')-Zn(1)-Br(1) 106.33(18) Br(1A')-Zn(1)-Br(1) 111.9(3) N(2)-Zn(1)-Br(1A) 105.6(9) Br(1A')-Zn(1)-Br(1A) 104.95(19) Br(1)-Zn(1)-Br(1A) 2.3534(10) 2.3801(10) 105.42(15) 103.98(15) 119.37(4) 2.323(4) 2.391(3) 9.9(4) 115.0(6) 104.26(11) 9.9(4) 124.5(4) 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin 2.2.7 - 32 - [ZnI2(2,5-Dimethylpyrazin)]n Die 1:1-Iodid-Verbindung Catena-(µ2-2,5-Dimethylpyrazin-N,N')-diiodido-Zink(II) (6) kristallisiert in der monoklinen Raumgruppe C2/c mit vier Formeleinheiten in der Elementarzelle und ist isotyp zu Verbindung 3II (Abb. 2.7 und Tab. 2.7). Die Zn-N-Abstände betragen 2.132(2) Å und die Bindungswinkel um die Zink(II)kationen liegen zwischen 93.65(12) und 120.32(7)° (Tab. 2.8). Abb. 2.7: Kristallstruktur der 1:1-Iodid-Verbindung 6 mit Blick auf die Koordinationssphäre des Zink(II)kations mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden. Symmetrie-Operationen: A = -x + 1, -y, -z + 1; B = -x + 1, y, -z + ½. 2.2.8 ZnI2(2,5-Dimethylpyrazin) Die ligandenreiche 1:2-Iodid-Verbindung Diioido-bis(2,5-dimethylpyrazin-N)-Zink(II) (7) kristallisiert in der monoklinen Raumgruppe P21/c mit vier Formeleinheiten in der Elementarzelle und allen Atomen in allgemeiner Lage (Tab. 2.7). In der Kristallstruktur finden sich diskrete Komplexe, in denen die Zink(II)kationen jeweils von zwei kristallografisch unabhängigen Iodidanionen und zwei Stickstoffatomen der 2,5-Dimethylpyrazin-Liganden verzerrt tetraedrisch koordiniert sind (Abb. 2.8: links). Die Zn-N-Abstände betragen zwischen 2.092(3) und 2.100(3) Å und die Bindungswinkel um die Zink(II)kationen liegen zwischen 101.81(12) und 116.517(19)° (Tab. 2.8). In der Kristallstruktur werden die diskreten Komplexe durch schwache intermolekulare C-H···I-Wasserstoffbrückenbindungen zu einem zweidimensionalen Netzwerk verknüpft (Abb. 2.8: rechts und Tab. 2.9). 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 33 - Abb. 2.8: Kristallstruktur der 1:2-Iodid-Verbindung 7 mit Blick auf die Koordinationssphäre des Zink(II)kations mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden (links) und Blick auf die Kristallstruktur in Richtung der kristallografischen b-Achse (rechts). Intermolekulare Wasserstoffbrückenbindungen sind als gestrichelte Linien gezeigt. Tab. 2.7: Ausgewählte Einkristallstrukturdaten der 1:1- 6 und 1:2-Iodid-Verbindung 7. Verbindung Kristallsystem Raumgruppe a/Å b/Å c/Å α /° β /° γ /° Z V / Å3 R1 4 monoklin C2/c 16.6286(13) 6.8351(4) 10.0199(8) 90 113.947(9) 90 4 1040.81(13) 0.0208 5 monoklin P21/c 12.2908(8) 8.2723(7) 17.2054(13) 90 101.266(8) 90 4 1715.6(2) 0.0300 Tab. 2.8: Ausgewählte Bindungslängen [Å] und -winkel [°] der 1:1- 6 und 1:2-Iodid-Verbindung 7. Zn(1)-N(1) N(1B)-Zn(1)-N(1) N(1)-Zn(1)-I(1) Zn(1)-N(1) Zn(1)-N(11) N(1)-Zn(1)-N(11) N(1)-Zn(1)-I(1) N(11)-Zn(1)-I(1) Verbindung 6 2.132(2) Zn(1)-I(1) 93.65(12) N(1)-Zn(1)-I(1B) 105.91(6) I(1)-Zn(1)-I(1B) Verbindung 7 2.092(3) Zn(1)-I(1) 2.100(3) Zn(1)-I(2) 101.81(12) N(1)-Zn(1)-I(2) 110.42(9) N(11)-Zn(1)-I(2) 103.69(9) I(1)-Zn(1)-I(2) 2.5568(4) 120.32(7) 110.60(2) 2.5600(5) 2.5744(5) 105.85(9) 117.59(9) 116.517(19) Tab. 2.9: Längen und Winkel der intermolekularen Wasserstoffbrückenbindungen der 1:2-Iodid-Verbindung 7. C14-H14···I1 C15-H15···I2 C16-H16···I2 distance (H···I) / Å 3.3873(2) 3.8614(3) 3.5735(2) angel (C-H···I) / deg 139.687(4) 116.753(4) 153.832(5) 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin 2.3 - 34 - Thermoanalytische Untersuchungen der Verbindungen Beim Aufheizen der ligandenreichen 2:3-Chlorid-Verbindung 2 in einer Thermowaage auf 500 °C werden zwei gut aufgelöste Massestufen in der TG-Kurve beobachtet, welche mit endothermen Signalen in der DTA-Kurve verbunden sind. Aus der MS-Trend-Scan-Kurve wird ersichtlich, dass nur der Ligand (m/z = 108) während dieser beiden Stufen emittiert wird (Abb. 2.9: links). Der experimentelle Masseverlust der ersten Stufe i.H.v. 18.1 % stimmt exakt mit dem berechneten Verlust für einen Drittel des Liganden (1 mol; 18.1 %) überein. Der Masseverlust für die zweite Stufe von 18.7 % stimmt nahezu mit dem berechneten Verlust für einen Drittel des Liganden (1 mol; 18.1 %) überein. Deshalb ist davon auszugehen, dass in der ersten Stufe eine 1:1-Verbindung und in der zweiten Stufe eine ligandenarme 2:1-Verbindung als Intermediat gebildet wird. Bei weiterem Aufheizen werden die restlichen Liganden emittiert, so dass ZnCl2 entsteht, welches bei weiterem Aufheizen verdampft (Abb. 2.9: links). Der thermische Abbau der 1:1-Chlorid-Verbindung 1 erfolgt zum Verlauf des thermischen Abbaus von Verbindung 2 DTG Δm = 18.1 % Δm = 18.7 % endo TG TP = 290°C TP = 309°C TP = 98°C DTA m/z = 108 50 DTG Δm = 21.9 % endo Δm = 21.9 % TP = 301°C TP = 264°C TG m/z = 108 MID 100 150 200 250 300 350 400 450 500 Temperatur / °C ΔT, Δm, (dm/dT) und Ionenstrom in beliebigen Einheiten ΔT, Δm, (dm/dT) und Ionenstrom in beliebigen Einheiten ab der zweiten Stufe äquivalent (Abb. 2.9: rechts). DTA MID 150 200 250 300 350 400 450 Temperatur / °C Abb. 2.9: DTA-, TG-, DTG- und MS-Trend-Scan-Kurve (simultane Messung) für die 2:3-Chlorid-Verbindung 2 (links) und 1:1-Chlorid-Verbindung 1 (rechts). Gegeben sind die Masseverluste (%) und Peak-Temperaturen TP (°C); Heizrate: 4 K/min; dynamische Stickstoffatmosphäre (75 mL/min); Al2O3-Tiegel. Beim Aufheizen der ligandenreichen 1:3- Bromid-Verbindung 5 wird ein ähnliches Verhalten wie bei der 2:3-Chlorid-Verbindung 2 beobachtet. Der Masseverlust i.H.v. 36.8 % in der ersten 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 35 - Stufe stimmt ungefähr mit dem berechneten Verlust für zwei Drittel der Liganden (2 mol; 39.4 %) überein, so dass die Bildung einer 1:1-Verbindung als Intermediat anzunehmen ist. Bemerkenswerterweise ist diese erste TG-Stufe mit zwei endothermen Signalen in der DTA-Kurve verbunden, was auf die Bildung eines weiteren Intermediates hindeutet (Abb. 2.10: links). Heizratenabhängige Messungen zeigen jedoch keine weitere TG-Stufe (vgl. B in Abb. 2.12). Der experimentelle Masseverlust i.H.v. 10 %, der in der zweiten TG-Stufe zu beobachten ist, stimmt annähernd mit dem erwarteten Verlust für einen Sechstel der Liganden (0.5 mol; 9.8 %) überein, was auf die Bildung einer neuen ligandenarmen 2:1-Verbindung der Zusammensetzung (ZnBr2)2(2,5-Dimethylpyrazin) schließen lässt. Bei weiterem Aufheizen werden die restlichen Liganden emittiert, so dass ZnBr2 entsteht, das anschließend verdampft (Abb. 2.10: links). Der thermische Abbau der polymorphen 1:1-Modifikationen 3I (Abb. 2.10: Mitte) und 3II (Abb. 2.10: rechts) erfolgt zum Verlauf des thermischen Abbaus von Verbindung 5 ab der zweiten Stufe äquivalent. endo Δm = 10.0 % TP = 106°C TP = 65°C TP = 341°C TG TP = 285°C DTA m/z = 108 50 100 150 200 MID 250 Temperatur / °C 300 350 400 150 DTG Δm = 16.5 % endo Δm = 5.2 % Δm = 7.3 % TP = 266°C TP = 337°C TG TP = 305°C m/z = 108 200 DTA MID 250 300 Temperatur / °C 350 400 ΔT, Δm, (dm/dT) und Ionenstrom in beliebigen Einheiten Δm = 36.8 % ΔT, Δm, (dm/dT) und Ionenstrom in beliebigen Einheiten ΔT, Δm, (dm/dT) und Ionenstrom in beliebigen Einheiten DTG DTG Δm = 13.6 % endo Δm = 10.2 % TP = 290°C TG TP = 339°C DTA MID m/z = 108 150 200 250 300 350 400 Temperatur / °C Abb. 2.10: DTA-, TG-, DTG- und MS-Trend-Scan-Kurve (simultane Messung) für die 1:3- Bromid-Verbindung 5 (links) sowie der polymorphen 1:1-Modifikationen 3I (Mitte) und 3II (rechts). Gegeben sind die Masseverluste (%) und Peak-Temperaturen TP (°C); Heizrate: 4 K/min; dynamische Stickstoffatmosphäre (75 mL/min); Al2O3-Tiegel. Beim Aufheizen der ligandenreichen 1:2-Iodid-Verbindung 7 werden drei gut aufgelöste Massestufen in der TG-Kurve beobachtet. Der Masseverlust i.H.v. 20.2 % in der ersten Stufe stimmt exakt mit dem berechneten Verlust für die Hälfte der Liganden (1 mol; 20.2 %) überein. Der Masseverlust i.H.v. 13.5 % in der zweiten Stufe stimmt ebenfalls exakt mit dem erwarteten Verlust für einen Drittel der Liganden (0.67 mol; 13.5 %) überein. Daher ist anzunehmen, dass in der ersten 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 36 - TG-Stufe eine 1:1-Verbindung gebildet wird, die bei weiterem Aufheizen in eine neue ligandenarme 3:1-Verbindung der Zusammensetzung (ZnI2)3(2,5-Dimethylpyrazin) zerfällt. Bei weiterem DTG Δm = 20.2 % Δm = 13.5 % endo Δm = 7.0 % TG TP = 301°C TP = 109°C TP = 265°C m/z = 108 50 100 150 200 DTA MID 250 300 350 400 ΔT, Δm, (dm/dT) und Ionenstrom in beliebigen Einheiten ΔT, Δm, (dm/dT) und Ionenstrom in beliebigen Einheiten Aufheizen wandelt sich die 3:1-Verbindung in ZnI2 um. DTG Δm = 16.5 % endo Δm = 8.6 % TG TP = 271°C TP = 297°C DTA MID m/z = 108 150 200 Temperatur / °C 250 300 350 400 Temperatur / °C Abb. 2.11: DTA-, TG-, DTG- und MS-Trend-Scan-Kurve (simultane Messung) für die 1:2-Iodid-Verbindung 7 (links) und 1:1-Iodid-Verbindung 6 (rechts). Gegeben sind die Masseverluste (%) und Peak-Temperaturen TP (°C); Heizrate: 4 K/min; dynamische Stickstoffatmosphäre (75 mL/min); Al2O3-Tiegel. Die simultanen DTA-TG-MS-Messungen geben Hinweise darauf, dass die ligandenreichen Verbindungen 2, 5 und 7 in der ersten TG-Stufe in die 1:1-Verbindungen und in der zweiten Stufe in neue ligandenarme 2:1- oder 3:1-Intermediate zerfallen. Durch die Durchführung weiterer TGMessungen und Isolation der entsprechenden Rückstände nach der ersten bzw. zweiten Stufe mit Hilfe von Abbruchexperimenten, galt es, diese Annahmen zu überprüfen. Durch anschließende Röntgen-Pulveruntersuchungen und Elementaranalyse sollten die Rückstände charakterisiert werden. Da jedoch die TG-Stufen nicht immer gut aufgelöst sind, kann sich die Isolierung solcher Intermediate als schwierig erweisen. Zur Überprüfung, ob die Auflösung der einzelnen TGStufen verbessert werden kann oder ob evtl. noch unbekannte Intermediate sichtbar werden, wurden für alle Verbindungen heizratenabhängige Messungen mit Heizraten von 1, 4, 8 und 16 K/min durchgeführt (Abb. 2.12). Daraus wurde ersichtlich, dass sich besonders für Verbindung 1 bei einer Heizrate von 1 K/min die Auflösung verbessert (vgl. D in Abb. 2.12). Für alle Abbruchexperimente wurde eine Heizrate von 4 K/min gewählt. 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin (A) - 37 - (D) Relativer Masseverlust Relativer Masseverlust (B) (C) 50 100 150 200 250 Temperatur / °C 300 350 400 (E) (F) 150 200 250 300 350 400 Temperatur / °C Abb. 2.12: Heizratenabhängige Messungen für die ligandenreichen Verbindungen 2 (A), 5 (B) und 7 (C) sowie der ligandenarmen Verbindungen 1 (D), 3I (E) und 6 (F); dynamische Stickstoffatmosphäre (75 mL/min); Al2O3-Tiegel). Das experimentelle Röntgen-Pulverdiffraktogramm des Rückstandes nach der ersten TG-Stufe der ligandenreichen 2:3-Chlorid-Verbindung 2 stimmt exakt mit dem aus Einkristallstrukturdaten berechneten Diffraktogramm der 1:1-Verbindung 1 überein (vgl. A mit B in Abb. 2.13). Zudem weisen die Resultate der Elementaranalyse eine gute Übereinstimmung mit der berechneten Zusammensetzung für die 1:1-Verbindung auf (berechnet/gefunden: C 29.48/29.74, H 3.30/3.19, N 11.46/11.63). Daraus geht hervor, dass Verbindung 1 in dieser thermischen Abbaureaktion sehr rein gebildet wird. Das Pulverdiffraktogramm des Intermediates, isoliert nach der zweiten TGStufe, unterscheidet sich von dem der 1:1-Verbindung 1 (vgl. C mit A in Abb. 2.13). Eine genaue Analyse zeigt, dass weiterhin einige Reflexe der 1:1-Verbindung 1 und ZnCl2 vorhanden sind. Trotz mehrerer Abbruchexperimente bei unterschiedlichen Temperaturen nach der zweiten TG-Stufe gelingt es nicht, dieses Intermediat phasenrein darzustellen. Letztlich stimmen nur die Ergebnisse der Elementaranalyse mit der berechneten Zusammensetzung für eine ligandenarme 2:1-Chlorid-Verbindung gut überein (berechnet/gefunden: C 18.93/18.43, H 2.12/2.25, N 7.36/7.26). 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 38 - Relative Intensität (A) (B) (C) 5 10 15 20 25 30 35 40 2 Theta / ° Abb. 2.13: Experimentelles Röntgen-Pulverdiffraktogramm des nach der ersten (A) und zweiten (C) TG-Stufe isolierten Rückstandes der 2:3-Chlorid-Verbindung 2 und berechnetes Röntgen-Pulverdiffraktogramm der 1:1-ChloridVerbindung 1 (B); Cu-Kα-Strahlung. Aus dem Vergleich des Pulverdiffraktogramms des Rückstandes, isoliert nach der ersten TGStufe, in der thermischen Abbaureaktion der ligandenreichen 1:3-Bromid-Verbindung 5 mit denen der aus Einkristallstrukturdaten berechneten Diffraktogramme für die polymorphen 1:1-Modifikationen 3I und 3II wird deutlich, dass das Intermediat als Mischung beider 1:1-Modifikationen erhalten wird (vgl. A mit B und C in Abb. 2.14). Die Elementaranalyse des Intermediates im Vergleich mit der berechneten Zusammensetzung für eine 1:1-Verbindung bestätigt diese Annahme (berechnet/gefunden: C 21.62/21.81, H 2.42/2.24, N 8.40/8.23). Das Pulverdiffraktogramm des Rückstandes, isoliert nach der zweiten TG-Stufe, deutet auf die Bildung einer neuen ligandenarmen 2:1-Verbindung hin. Jedoch sind immer noch Reflexe der 1:1-Verbindungen und ZnBr2 vorhanden (vgl. D in Abb. 2.14). Trotz mehrerer Abbruchexperimente bei unterschiedlichen Temperaturen nach der zweiten Stufe ist es nicht möglich, dieses Intermediat phasenrein darzustellen. Letztlich stimmen auch hier lediglich die Ergebnisse der Elementaranalyse mit der berechneten Zusammensetzung für eine ligandenarme 2:1-Bromid-Verbindung überein (berechnet/gefunden: C 12.90/12.33, H 1.44/1.53, 5.02/5.59). Des Weiteren geben die Pulverdiffraktogramme Hinweise darauf, dass die ligandenarmen 2:1Chlorid- und Bromid-Verbindung isotyp sind (vgl. C in Abb. 2.13 mit D in Abb. 2.14). 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 39 - Relative Intensität (A) (B) (C) (D) 5 10 15 20 25 30 35 40 2 Theta / ° Abb. 2.14: Experimentelles Röntgen-Pulverdiffraktogramm des nach der ersten (A) und zweiten (D) TG-Stufe isolierten Rückstandes der 1:3-Bromid-Verbindung 5 und berechnetes Röntgen-Pulverdiffraktogramm der 1:1-Modifikationen 3I (B) und 3II (C); Cu-Kα-Strahlung. Das Pulverdiffraktogramm des Rückstandes, isoliert nach der ersten TG-Stufe in der thermischen Abbaureaktion der ligandenreichen 1:2-Iodid-Verbindung 7, ist in exakter Übereinstimmung mit dem aus Einkristallstrukturdaten berechneten Diffraktogramm für die 1:1-Iodid-Verbindung 6 (vgl. A mit B in Abb. 2.15). Weiterhin stimmen die Ergebnisse der Elementaranalyse annähernd mit der berechneten Zusammensetzung für die 1:1-Verbindung überein (berechnet/gefunden: C 16.86/16.95, H 1.89/1.86, N 6.56/6.70). Auffällig ist, dass das Pulverdiffraktogramm des Rückstandes, isoliert nach der zweiten TG-Stufe, dem nach der ersten Stufe gleicht. Weitere Abbruchexperimente bei höheren Temperaturen zeigen, dass jeweils lediglich Reflexe der 1:1-Verbindung 6 vorliegen. Dieses ungewöhnliche thermische Verhalten lässt sich bisher nicht erklä- Relative Intensität ren. (A) (B) (C) 5 10 15 20 25 30 35 40 2 Theta / ° Abb. 2.15: Experimentelles Röntgen-Pulverdiffraktogramm des nach der ersten (A) und zweiten (C) TG-Stufe isolierten Rückstandes der 1:2-Iodid-Verbindung 7 und berechnetes Röntgen-Pulverdiffraktogramm der 1:1-IodidVerbindung 6 (B); Cu-Kα-Strahlung. 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin 2.4 - 40 - Synthetische Aspekte Die Bildung und Stabilität dieser Verbindungen wurde durch Lösungsvermittelte Umwandlungsexperimente untersucht. Hierzu wurde ein Überschuss von Zink(II)halogenid und Ligand in verschieden stöchiometrischen Verhältnisse in Methanol gemischt. Der resultierende Niederschlag wurde für drei Tage gerührt, um die thermodynamisch stabilste Verbindung bei Raumtemperatur zu erhalten. Anschließend erfolgte eine Untersuchung der Produkte durch Röntgen-Pulverdiffraktometrie (Tab. 2.10). Tab. 2.10: Ergebnisse der Reihenversuche in Methanol als Funktion der molaren Verhältnisse zwischen Zn(II)halogenid und 2,5-Dimethylpyrazin. ZnX2 / Ligand X = Cl X = Br X=I 1:6 1:4 7 (1:2) 1:3 1:2 2:3 1:1 3:2 1 (1:1) 3I (1:1) 6 (1:1) 2:1 4:1 Diese Experimente zeigen deutlich, dass nur die Verbindungen 1, 3I, 6 und 7 stabil sind und in Lösung dargestellt werden können. Dabei ist zu bemerken, dass die 1:2-Iodid-Verbindung 7 nicht durch bloßes Mischen der Reaktanden in der durch die Zusammensetzung vorgegebenen Stöchiometrie dargestellt werden kann. Die Reaktion von ZnI2 und Ligand im molaren Verhältnis 1:2 resultiert in der Bildung der 1:1-Verbindung 6, wohingegen im Chlorid- und BromidSystem nur die 1:1-Verbindungen unabhängig von den stöchiometrischen Verhältnissen der Edukte gebildet werden. Des Weiteren können die 2:3-Chlorid-Verbindung 2 sowie die 1:3Bromid-Verbindung 5 nur in einer lösungsmittelfreien Reaktion phasenrein dargestellt werden. Dies trifft auf die 2:3-Bromid-Verbindung 4 weder mit noch ohne Lösungsmittel zu. Zur Überprüfung, welche der beiden polymorphen 1:1-Bromid-Modifikation 3I und 3II die thermodynamisch stabilere Verbindung bei Raumtemperatur ist, wurden äquivalente Mengen beider Formen zusammen in Methanol gerührt. Nach drei Tagen war nur noch die Modifikation 3I vorhanden, was beweist, dass 3I die stabile bei Raumtemperatur und 3II die metastabile Modifikation ist (Abb. 2.16). Zusätzliche DSC-Untersuchungen in Kombination mit der RöntgenPulverdiffraktometrie zeigen außerdem keine Umwandlungen zwischen den beiden Modifikationen. Daher ist nicht feststellbar, ob sich die Modifikationen enantiotrop oder monotrop verhalten (Abb. 2.17). 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 41 - Relative Intensität (A) (B) (C) 5 10 15 20 25 30 35 40 2 Theta / ° Abb. 2.16: Experimentelles Röntgen-Pulverdiffraktogramm einer Mischung der polymorphen 1:1-Bromid-Modifikation 3I und 3II vor (A) und nach (B) drei Tagen Rühren in Methanol sowie berechnetes Röntgen-Pulverdiffraktogramm der Modifikation 3I (C); Cu-Kα-Strahlung. Die Pfeile zeigen die Reflexlagen der Modifikation 3II an. TP = 358 °C TP = 225 °C dQ/dT endo TP = 350 °C TP = 225 °C 50 100 150 200 250 300 350 400 Temperatur / °C Abb. 2.17: DSC-Kurve der polymorphen Modifikationen 3I (oben) und 3II (unten). Gegeben sind Peak-Temperaturen TP (°C); Heizrate: 3 K/min; dynamische Stickstoffatmosphäre; Al-Tiegel. 2.5 Vergleich von ZnX2(Pyrazin) und ZnX2(2,5-Dimethylpyrazin) (X = Cl, Br, I) Für die entsprechenden Koordinationspolymere auf Basis von Zink(II)halogeniden und Pyrazin als Ligand sind bereits einige ligandenreiche und ligandenarme Verbindungen literaturbekannt.[115,140-142] In den Kristallstrukturen der ligandenreichen 1:2-Verbindungen mit ZnCl2 und ZnBr2 ist jedes Zink(II)kation von zwei Halogenidanionen und vier Pyrazin-Liganden oktaedrisch koordiniert. Des Weiteren sind die Zink(II)kationen durch die Pyrazin-Liganden zu Schich- 2. Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin - 42 - ten verknüpft. Dagegen sind in allen 2,5-Dimethylpyrazin-Verbindungen die Zink(II)kationen durch zwei Halogenidanionen und zwei Liganden tetraedrisch koordiniert. Für die ligandenreichen Verbindungen führt dies zur Bildung von Oligomeren in den Verbindungen 2 und 4 und diskreten Einheiten in Verbindung 5 und 7. Es ist anzunehmen, dass die sterisch anspruchsvolleren Methylgruppen, benachbart zu den Stickstoffatomen, für dieses abweichende Verhalten verantwortlich sind. Zudem konnte ungewöhnlicherweise eine ligandenreiche 1:2-Verbindung mit ZnI2 nur mit 2,5-Dimethylpyrazin als Ligand hergestellt werden. Für dieses außergewöhnliche Verhalten gibt es bisher keine Erklärung. Demgegenüber werden in nahezu allen Kristallstrukturen der ligandenreichen 1:1-Verbindungen – mit Ausnahme der Kristallstruktur der 1:1-Chlorid-Pyrazin-Verbindung, in der die Metallzentren oktaedrisch koordiniert sind – topologisch identische Netzwerke gefunden, in denen die Zink(II)halogenid-Einheiten durch die Pyrazin- oder 2,5-Dimethylpyrazin-Liganden zu Ketten verknüpft sind. In der 1:1-Chlorid-Pyrazin-Verbindung sind die Zink(II)kationen jedoch durch kantenverknüpfte Chloridanionen zu Ketten verbunden, die darüber hinaus durch die N-Donorliganden zu Schichten verknüpft sind.[142] Des Weiteren ist die thermische Reaktivität aller ligandenreichen Verbindungen mit Pyrazin oder 2,5-Dimethylpyrazin identisch. Beim Aufheizen wandeln sie sich in zwei Stufen in die ligandenarmen 2:1-Verbindungen um, wobei die 1:1-Verbindungen als Intermediate gebildet werden. Diese Ergebnisse verdeutlichen unzweifelhaft, dass es keinen einfachen Zusammenhang zwischen deren Kristallstrukturen und thermischen Eigenschaften gibt. 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 43 - 3 Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden 3.1 Einleitung Eines der wichtigsten Ziele im Bereich der Forschung an Koordinationspolymeren ist die Synthese neuer Verbindungen mit interessanten physikalischen Eigenschaften.[27,69,143-146] Für die Untersuchung derartiger Verbindungen ist es unabdingbar, diese phasenrein und in größeren Mengen herzustellen. Da Koordinationspolymere jedoch meist in Lösung hergestellt werden, kommt es mitunter zur Bildung von Mischungen verschiedener Phasen. Dabei ist es teilweise unmöglich, eine thermodynamisch metastabile Verbindung überhaupt in Lösung darzustellen. Im vorangegangenen Kapitel sowie in weiteren Arbeiten[28,86,87,89,91,92,100,104,114,117,147-150] wurde bereits gezeigt, dass ligandenreiche Koordinationspolymere auf Basis von Kupfer(I)- und Zink(II)halogeniden sowie aromatische N-Donorliganden durch einen kontrollierten thermischen Abbau in ligandenärmere Koordinationspolymere überführt werden können. Je nach dem untersuchten System gelingt es in einigen Fällen sogar, das ligandenärmere Intermediat in großen Ausbeuten und hoher Reinheit zu isolieren. Aus diesem Grund ist die Methode des kontrollierten thermischen Abbaus ein alternativer Weg zur Darstellung neuer Koordinationspolymere (s. Kapitel 1.3). Daraus ergibt sich die Frage, ob diese Methode auch zur Darstellung von Verbindungen anderer Systeme geeignet ist. Da ein thermischer Abbau von Koordinationspolymeren auf Basis von aromatischen N-Donorliganden und Übergangsmetallen mit kleinen Anionen, z.B. Thiocyanaten, Cyanaten oder Cyaniden, in vielen Fällen zu neuen Verbindungen mit einem höheren Grad der Vernetzung führt, stehen derartige Untersuchungen hier im Vordergrund. Bei Anwesenheit von paramagnetischen Übergangsmetallen sind auf diese Weise mitunter kooperative magnetische Phänomene zu erwarten. In diesem Zusammenhang sind die bereits literaturbekannten ligandenreichen Verbindungen [Mn(SCN)2(Pyrazin)2]n,[28] [Fe(SCN)2(Pyrazin)2]n[151] und [Co(SCN)2(Pyrazin)2]n[144] als potenzielle Precursorverbindungen von hohem Interesse. In vorherigen Untersuchungen wurde nachgewiesen, dass die ligandenreiche Verbindung [Mn(SCN)2(Pyrazin)2]n, die einen "normalen" Curie-Weiss-Paramagnetismus zeigt, durch einen kontrollierten thermischen Abbau in die ligandenarme Verbindung [Mn(SCN)2(Pyrazin)]n, die beim Abkühlen einen antiferromagnetischen Ordnungspunkt aufweist, quantitativ überführt werden kann (s. Kapitel 1.56).[28] 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 44 - Der zweite Teil dieser Diplomarbeit beschäftigt sich daher mit Untersuchungen, ob sich weitere Koordinationspolymere auch auf Basis anderer paramagnetischer Übergangsmetalle herstellen lassen. Hierzu werden neben den Übergangsmetall-Thiocyanaten auch die -Cyanate und zusätzlich zu Pyrazin auch Pyrimidin als Ligand untersucht. Im folgenden Kapitel steht daher die Darstellung und Charakterisierung derartiger Koordinationspolymere im Vordergrund. Hierzu zählen insbesondere die Untersuchung der thermischen Eigenschaften von neuen und literaturbekannten ligandenreichen Koordinationsverbindungen auf Basis paramagnetischer Übergangmetalle sowie die magnetische Charakterisierung der ligandenreichen sowie evtl. auftretender ligandenarmen thermischen Abbauprodukte. Insgesamt wurden die folgenden zwei literaturbekannten und sieben neuen Verbindungen dargestellt und charakterisiert: ¾ [Fe(SCN)2(Pyrazin)2]n[151] IUPAC: Poly[bis(thiocyanato-N)-bis(µ2-Pyrazin-N,N')-Eisen(II)] ¾ [Fe(SCN)2(Pyrazin)]n IUPAC: Poly[bis(µ2-thiocyanato-N,S)-bis(µ2-Pyrazin-N,N')-Eisen(II)] ¾ [Co(SCN)2(Pyrazin)2]n[144] IUPAC: Poly[bis(thiocyanato-N)-bis(µ2-Pyrazin-N,N')-Cobalt(II)] ¾ [Co(SCN)2(Pyrazin)]n IUPAC: Poly[bis(µ2-thiocyanato-N,S)-bis(µ2-Pyrazin-N,N')-Cobalt(II)] ¾ [Ni(SCN)2(Pyrazin)2]n IUPAC: Poly[bis(thiocyanato-N)-bis(µ2-Pyrazin-N,N')-Nickel(II)] ¾ [Ni(SCN)2(Pyrazin)]n IUPAC: Poly[bis(µ2-thiocyanato-N,S)-bis(µ2-Pyrazin-N,N')-Nickel(II)] ¾ [Co(OCN)2(Pyrazin)2]n IUPAC: Poly[bis(cyanato-N)-bis(µ2-Pyrazin-N,N')-Cobalt(II)] ¾ [Mn(SCN)2(Pyrimidin)2]n IUPAC: Poly[bis(thiocyanato-N)-bis(µ2-Pyrimidin-N,N')-Mangan(II)] ¾ Co(SCN)2(Pyrimidin)4 IUPAC: Tetrakis(Pyrimidin-κN)-bis(thiocyanato-κN)-Cobalt(II) ¾ Ni(SCN)2(Pyrimidin)4 IUPAC: Tetrakis(Pyrimidin-κN)-bis(thiocyanato-κN)-Nickel(II) Zunächst werden die Kristallstrukturen der o.g. Verbindungen beschrieben. Anschließend erfolgt eine ausführliche Vorstellung deren thermischer und magnetischer Eigenschaften. 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 45 - 3.2 Verbindungen auf Basis von Übergangsmetall-Thiocyanaten und Pyrazin 3.2.1 Kristallstrukturen der Verbindungen 3.2.1.1 Die [Ni(SCN)2(Pyrazin)2]n ligandenreiche 1:2-Nickel-Verbindung Poly[bis(thiocyanato-N)-bis(µ2-Pyrazin-N,N')- Nickel(II)] kristallisiert in der zentrosymmetrischen monoklinen Raumgruppe C2/m mit zwei Formeleinheiten in der Elementarzelle (Tab. 3.1: links) und ist isotyp zu den schon literaturbekannten Verbindungen [M(SCN)2(Pyrazin)2]n (M = Mn,[28] Fe,[151] Co[144]) Die asymmetrische Einheit besteht aus einem Nickel(II)kation, das die Position 2/m einnimmt, einem Thiocyanatanion, das auf einer kristallografischen Spiegelebene lokalisiert ist sowie einem PyrazinLiganden, der auf einem Inversionszentrum angeordnet ist. In der Kristallstruktur sind die Nickel(II)kationen von vier Pyrazin-Liganden sowie von zwei Thiocyanatanionen geringfügig verzerrt oktaedrisch umgeben (Abb. 3.1: links). Die Ni-N-Bindungslänge zu den Stickstoffatomen der negativ geladenen Thiocyanatanionen beträgt 2.033(2) Å und ist signifikant kürzer als die Bindung zu den Stickstoffatomen der Pyrazin-Liganden mit 2.1622(13) Å. Die Bindungswinkel um das Nickel(II)kation liegen zwischen 88.27(7) und 91.73(7)° sowie 180° (Tab. 3.1: rechts). Die Thiocyanatanionen agieren nicht als verbrückender Ligand, sondern sind lediglich über das Stickstoffatom an das Nickel(II)kation koordiniert. Zudem werden die Metallzentren durch die Pyrazin-Liganden über µ-N,N'-Koordination zu Schichten entlang der kristallografischen cAchse verbrückt (Abb. 3.1: rechts). Abb. 3.1: Kristallstruktur der 1:2-Nickel-Verbindung mit Blick auf die Koordinationssphäre des Nickel(II)kations mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden (links) sowie Blick auf die Struktur in Richtung der kristallografischen c-Achse (rechts). Symmetrie-Operationen: A = -x, -y, -z + 1; B = -x, y, -z + 1; C = x, -y, z; D = -x + ½, -y + ½, -z + 1. 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 46 - Tab. 3.1: Ausgewählte Einkristallstrukturdaten (links), Bindungslängen (Å) und -winkel (°) (rechts) von [Ni(SCN)2(Pyrazin)2]n. Kristallsystem Raumgruppe a/Å b/Å c/Å α /° β /° γ /° Z V / Å3 R1 3.2.1.2 monoklin C2/m 9.9283(16) 10.2127(19) 7.2079(12) 90 118.498(10) 90 2 642.29(19) 0.0277 Ni(1)-N(1) Ni(1)-N(3) S(1)-C(3) C(3)-N(3) N(1A)-Ni(1)-N(1C) N(3)-Ni(1)-N(1) N(3)-Ni(1)-N(1A) N(1)-Ni(1)-N(1C) N(3A)-Ni(1)-N(3) N(1A)-Ni(1)-N(1) N(1C)-Ni(1)-N(1B) 2.1622(13) 2.033(2) 1.624(3) 1.163(4) 88.27(7) 89.66(6) 90.34(6) 91.73(7) 180.0 180.0 180.00(8) [M(SCN)2(Pyrazin)]n (M = Fe, Co, Ni) Die ligandenarmen 1:1-Verbindungen Poly[bis(µ2-thiocyanato-N,S)-bis(µ2-Pyrazin-N,N')-Eisen(II)], Poly[bis(µ2-thiocyanato-N,S)-bis(µ2-Pyrazin-N,N')-Cobalt(II)] und Poly[bis(µ2-thiocyanato-N,S)-bis(µ2-Pyrazin-N,N')-Nickel(II)] kristallisieren in der zentrosymmetrischen monoklinen Raumgruppe C2/m mit zwei Formeleinheiten in der Elementarzelle (Tab. 3.2). Die EisenVerbindung ist mit der literaturbekannten Mangan-Verbindung[28] und die Cobalt-Verbindung mit der Nickel-Verbindung isotyp. Die asymmetrische Einheit beinhaltet ein Metall(II)kation und einen Pyrazin-Liganden, die beide die Position 2/m besetzen sowie ein Thiocyanatanion, das auf einer Spiegelebene liegt. Der Pyrazin-Ligand ist bei der Cobalt- und der Nickel-Verbindung über zwei Orientierungen fehlgeordnet und die Besetzung jeweils symmetriebedingt auf 0.5 festgelegt. Zur Überprüfung der Wahl der richtigen Raumgruppe wurde die Symmetrie reduziert und in den Raumgruppen C2 und Cm verfeinert. Die Fehlordnung blieb allerdings erhalten und es ließen sich keine Anzeichen von Überstruktur-Reflexen erkennen. In der Kristallstruktur sind die Metall(II)kationen jeweils von zwei Stickstoffatomen der PyrazinLiganden sowie jeweils von zwei Schwefel- und zwei Stickstoffatomen der Thiocyanatanionen geringfügig verzerrt oktaedrisch koordiniert (Abb. 3.2). Im Gegensatz zu den ligandenreichen Verbindungen werden hier die Metall(II)kationen über eine µ2-N-S-Koordination von den Thiocyanatanionen zu Ketten verbrückt, die zusätzlich durch die Pyrazin-Liganden zu Schichten verknüpft werden (Abb. 3.3). Wie bei den ligandenreicheren Verbindungen [M(SCN)2(Pyrazin)2]n (M = Mn,[28] Fe,[151] Co,[144] Ni[s.o.]), sind auch hier die M-N-Bindungslängen zu den Stickstoffatomen der negativ geladenen Thiocyanatanionen mit Fe-NCS = 2.073(3), Co-NCS = 2.056(3) bzw. Ni-NCS = 2.031(3) Å kleiner als die Bindungslängen zu den Stickstoffatomen der Pyrazin-Liganden mit Fe-NPyz = 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 47 - 2.257(3), Co-NPyz = 2.174(3) bzw. Ni-NPyz = 2.129(3) Å. Die M-S-Bindungslängen betragen FeS = 2.6377(9), Co-S = 2.5713(9) bzw. Ni-S = 2.5215(9) Å und die Bindungswinkel um die Metall(II)kationen liegen bei der Eisen-Verbindung zwischen 87.49(9) und 92.51(9)° sowie 180°, bei der Cobalt-Verbindung zwischen 86.64(7) und 93.36(7)° sowie 180° und bei der Nickel-Verbindung zwischen 86.31(9) und 93.69(9)° sowie 180°. Im direkten Vergleich der 1:1-Mangan,[28] Eisen-, Cobalt- und Nickel-Verbindungen ist ein deutlicher Trend der M-NCS- und M-NPyzBindungslängenverkürzung von Mangan zu Nickel sowie eine geringfügige Änderung des NCSM-NCS-Winkels und der intra- und intermolekularen Metall-Metall-Abstände erkennbar (Tab. 3.3 und Tab. 3.4). Diese Beobachtung resultiert aus den sich von Mangan zu Nickel verkleinernden Ionenradien (Mn = 0.97, Fe = 0.92, Co = 0.90 und Ni = 0.84 Å).[159] Abb. 3.2: Kristallstrukturen der 1:1-Verbindungen [M(SCN)2(Pyrazin)]n mit M = Fe (links), M = Co (Mitte) und M = Ni (rechts) mit Blick auf die Koordinationssphäre der Metall(II)kationen mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden. Symmetrie-Operationen: A = -x, -y + 1, -z; B = -x, -y + 1, -z + 1; C = x, y, z + 1 für die Eisen-Verbindung; A = -x + 1, -y + 1, -z + 2; B = x, y, z - 1; C = -x + 1, -y + 1, -z + 1 für die Cobalt-Verbindung und A = -x, -y, -z; B = x, y, z-1; C = -x, -y, -z + 1 für die Nickel-Verbindung. Abb. 3.3: Kristallstruktur der 1:1-Verbindungen [M(SCN)2(Pyrazin)]n (M = Fe, Co, Ni) mit Blick auf die Struktur; übersichtshalber ohne Fehlordnung abgebildet. 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 48 - Tab. 3.2: Ausgewählte Einkristallstrukturdaten von [M(SCN)2(Pyrazin)]n (M = Mn,[28] Fe, Co, Ni). 1:1-Verbindung Kristallsystem Raumgruppe a/Å b/Å c/Å β /° Z V / Å3 R1 Mn monoklin C2/m 10.6165(17) 7.4554(14) 5.7646(7) 94.368(16) 2 454.94(13) 0.0333 Fe monoklin C2/m 10.5311(11) 7.3224(10) 5.6957(6) 94.588(12) 2 437.80(9) 0.0325 Co monoklin C2/m 11.4570(18) 7.1608(8) 5.6630(9) 104.892(18) 2 448.99(11) 0.0338 Ni monoklin C2/m 11.4697(12) 7.0560(6) 5.6086(5) 105.127(11) 2 438.18(7) 0.0296 Tab. 3.3: Ausgewählte Bindungslängen [Å] und -winkel [°] von [M(SCN)2(Pyrazin)]n (M = Mn,[28] Fe, Co, Ni). 1:1-Verbindung M(1)-N(1) M(1)-N(2B) M(1)-S(1) S(1)-C(2) C(3)-N(2) N(2C)-M(1)-S(1) N(2B)-M(1)-S(1) N(1)-M(1)-S(1) N(2B)-M(1)-N(1) N(2B)-M(1)-N(1A) N(2B)-M(1)-N(2C) S(1)-M(1)-S(1A) N(1A)-M(1)-N(1) N(2)-C(2)-S(1) Mn 2.327(2) 2.126(2) 2.7071(8) 1.639(3) 1.159(4) 87.58(7) 92.42(7) 90.0 90.0 90.0 180.0 180.0 180.00(10) 179.8(3) Fe 2.257(3) 2.073(3) 2.6377(9) 1.642(3) 1.165(5) 87.49(9) 92.51(9) 90.0 90.0 90.0 180.0 180.0 180.0 178.9(3) Co 2.183(3) 2.063(3) 2.5746(10) 1.642(4) 1.157(5) 86.67(9) 93.33(9) 90.0 90.0 90.0 180.0 180.0 180.0 180.0(3) Ni 2.129(3) 2.031(3) 2.5215(9) 1.645(4) 1.160(5) 86.31(9) 93.69(9) 90.0 90.0 90.0 180.0 180.0 180.00(18) 180.00(5) Tab. 3.4: Intra- und intermolekulare Metall-Metall-Abstände [Å] von [M(SCN)2(Pyrazin)]n (M = Mn,[28] Fe, Co, Ni). 1:1-Verbindung M-SCN-M (intra) M-Pyrazin-M (intra) M-M (inter) 3.2.2 Mn 5.765 7.455 6.486 Fe 5.696 7.622 6.413 Co 5.663 7.161 6.755 Ni 5.609 7.056 6.733 Thermoanalytische Untersuchungen der Verbindungen Beim Aufheizen aller drei ligandenreichen 1:2-Verbindungen [M(SCN)2(Pyrazin)2]n (M = Fe, Co, Ni) zeigt sich ein ähnliches thermisches Verhalten. Es lassen sich zwei gut aufgelöste Massestufen in der TG-Kurve beobachten, die mit einem endothermen Signal in der DTA-Kurve verbunden sind. Aus der MS-Trend-Scan-Kurve wird ersichtlich, dass ausschließlich der Ligand (m/z = 80) während dieser beiden Stufen emittiert wird (Abb. 3.4). Die experimentellen Masseverluste stimmen annähernd mit dem überein, der für den Verlust jeweils der Hälfte der Liganden berechnet wird (Tab. 3.5). 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 49 - Tab. 3.5: Vergleich der experimentellen mit den berechneten Masseverlusten für den Verlust eines Mols Pyrazin für die Verbindungen [M(SCN)2(Pyrazin)2]n (M = Fe, Co, Ni). 1:2-Verbindung Δm für 1. Stufe Δm für 2. Stufe Δm berechnet (-1 mol Pyrazin) Fe 23.6 23.4 24.1 Co 23.2 23.5 23.9 Ni[a] 21.1 21.2 23.9 [a] Die etwas größere Abweichung lässt sich bisher nicht erklären. Deshalb ist davon auszugehen, dass in der ersten Stufe eine ligandenarme 1:1-Verbindung der Zusammensetzung [M(SCN)2(Pyrazin)]n (M = Fe, Co, Ni) als Intermediat gebildet wird. Bei weiterem Aufheizen werden die restlichen Liganden emittiert, so dass in der zweiten Stufe M(SCN)2 (M = Fe, Co, Ni) entsteht, welches sich bei weiterem Aufheizen zersetzt. endo Δm = 23.4 % TG TP = 258°C TP = 318°C DTA MID m/z = 80 50 100 150 200 250 300 350 400 450 500 Temperatur / °C Δm = 23.2 % endo Δm = 23.5 % TP = 239°C TP = 306°C TG TP = 335°C DTA MID m/z = 80 50 100 150 200 250 300 350 400 450 500 Temperatur / °C DTG ΔT, Δm, (dm/dT) und Ionenstrom in beliebigen Einheiten Δm = 23.6 % DTG ΔT, Δm, (dm/dT) und Ionenstrom in beliebigen Einheiten ΔT, Δm, (dm/dT) und Ionenstrom in beliebigen Einheiten DTG Δm = 21.1 % endo Δm = 21.2 % TP = 267°C TP = 327°C TG TP = 397°C DTA MID m/z = 80 50 100 150 200 250 300 350 400 450 500 Temperatur / °C Abb. 3.4: DTA-, TG-, DTG- und MS-Trend-Scan-Kurve (simultane Messung) für die ligandenreichen 1:2Verbindungen [M(SCN)2(Pyrazin)2]n mit M = Fe (links), M = Co (Mitte) und M = Ni (rechts). Gegeben sind die Masseverluste (%) und Peak-Temperaturen TP (°C); Heizrate: 4 K/min; dynamische Stickstoffatmosphäre (75 mL/min); Al2O3-Tiegel. Berechnungen zufolge entstehen nach der ersten Massestufe die ligandenarmen 1:1-Verbindungen. Um diese Annahme zu überprüfen, wurden weitere TG-Experimente durchgeführt, in denen die Rückstände nach der ersten Massestufe isoliert und durch Elementaranalyse und RöntgenPulverbeugung untersucht wurden. Die experimentell ermittelten Ergebnisse der Elementaranalyse stimmen gut mit der berechneten Zusammensetzung der ligandenarmen 1:1-Verbindungen überein (Tab. 3.6). 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 50 - Tab. 3.6: Vergleich der experimentellen Ergebnisse der Elementaranalyse mit denen der berechneten Zusammensetzung der 1:1-Verbindungen [M(SCN)2(Pyrazin)]n (M = Fe, Co, Ni). Ctheo Cexp Ntheo Nexp Stheo Sexp Htheo Hexp Fe / % Co / % 28.59 28.24 28.39 28.06 22.22 21.95 22.48 22.08 25.44 25.13 25.54 25.26 1.60 1.58 1.54 1.52 Ni / % 28.27 28.47 21.98 21.57 25.16 24.92 1.58 1.53 Des Weiteren stimmen die experimentellen Röntgen-Pulverdiffraktogramme der Rückstände nach der ersten TG-Stufe der ligandenreichen 1:2-Verbindungen exakt mit den aus Einkristallstrukturdaten berechneten Diffraktogrammen der 1:1-Verbindungen überein. Aufgrund der beim Eisen auftretenden Fluoreszenz bei Verwendung von Cu-Kα-Strahlung, kann der Rückstand der Verbindung [Fe(SCN)2(Pyrazin)]n nicht mittels Röntgen-Pulveruntersuchungen charakterisiert werden. Relative Intensität (A) (B) (C) (D) 10 15 20 25 30 35 40 45 2 Theta / ° Abb. 3.5: Experimentelle Röntgen-Pulverdiffraktogramme der nach der ersten TG-Stufe isolierten Rückstände der 1:2-Verbindungen [M(SCN)2(Pyrazin)2]n mit M = Co (A) und M = Ni (C) sowie die berechneten Röntgen-Pulverdiffraktogramme der 1:1-Verbindungen [M(SCN)2(Pyrazin)]n mit M = Co (B) und M = Ni (D); Cu-Kα-Strahlung. 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden 3.2.3 - 51 - Magnetische Eigenschaften der Verbindungen Zur Untersuchung der magnetischen Eigenschaften war die Herstellung der Verbindungen in größeren Mengen erforderlich. Die Darstellung der ligandenreichen 1:2-Verbindungen und der ligandenarmen 1:1-Nickel-Verbindung erfolgte unproblematisch durch Reaktion stöchiometrischer Mengen der Edukte in Lösung. Die ligandenarmen Eisen- und Cobalt-Verbindungen in Lösung konnten allerdings nur als Mischungen beider Phasen hergestellt werden, so dass deren Darstellung durch den thermischen Abbau ihrer ligandenreichen Precursoren entstand. Die Auswertung der magnetischen Eigenschaften der Verbindungen geschah auf Grundlage des Curie-Weiss-Gesetzes. Die Daten wurden für den Core-Diamagnetismus korrigiert. 1 exp 3·R·C·M N NA · µB 2.828 · √C M = Molgewicht = 1, wenn C in mol-1 µB = 9.2740·10-21 A m2 N = Anzahl magnetischer Atome R = 8.3145·107 J K-1 mol-1 C = Curie-Konstante in cm3 K mol-1 NA = 6.0221·1023 mol-1 µeff = effektives magnetisches Moment in µB θ = Weiss'sche Konstante in K χM = molare magnetische Suszeptibilität in cm3 mol-1 Das Auftreten von θ ≠ 0 deutet auf eine Wechselwirkung zwischen den benachbarten magnetischen Momenten hin. Anhand des Vorzeichens der Weiss'schen Konstante lässt sich die Art der in der Substanz vorherrschenden magnetischen Wechselwirkungen empirisch charakterisieren. Bei θ < 0 handelt es sich um überwiegend antiferromagnetische Wechselwirkungen zwischen den benachbarten Spin-Momenten, wohingegen θ > 0 auf eine ferromagnetische Kopplung hindeutet. Der Betrag von θ gibt die Stärke der Wechselwirkungen an. Für die ligandenreiche 1:2-Verbindung [Fe(SCN)2(Pyrazin)2]n findet sich in Übereinstimmung mit der Literatur[151] bei einer Neel-Temperatur von 8.5 K ein antiferromagnetischer Ordnungspunkt, der durch einen indirekten Austausch über die Pyrimidin-Liganden zustande kommt (Abb. 3.6). Auch der Wert der Weiss'schen Konstante i.H.v. -6.68 K deutet auf ein antiferromagnetisches Austauschphänomen hin (Tab. 3.7). Die Abweichung des experimentellen effektiven mag- 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 52 - netischen Moments von 5.25 µB vom berechneten high-spin-Wert von 4.9 µB für ausschließlichen "spin-only" Paramagnetismus ist auf die Spin-Bahn-Kopplung zurückzuführen (Tab. 3.7). Für die ligandenreichen 1:2-Verbindungen [M(SCN)2(Pyrazin)2]n (M = Co, Ni) ist in Übereinstimmung mit der Literatur[143,144] ein rein paramagnetisches Verhalten zu finden (Abb. 3.6). Der Abfall in der χΜT-Kurve in Richtung kleinerer Temperaturen deutet zudem auf antiferromagnetische Wechselwirkungen zwischen den benachbarten Spin-Momenten hin (Abb. 3.6: rechts). Während für die 1:2-Cobalt-Verbindung der Wert der Weiss'schen Konstante von -15.12 K dieses Verhalten bestätigt, weicht für die 1:2-Nickel-Verbindung θ mit 1.04 K nicht signifikant ab (Tab. 3.7). Für die 1:2-Nickel-Verbindung stimmt das experimentelle effektive magnetische Moment von 2.77 µB mit dem berechneten Wert von 2.83 µB für ausschließlichen "spin-only" Paramagnetismus annähernd überein. Die Abweichung der 1:2-Cobalt-Verbindung von 4.76 µB von dem berechneten high-spin-Wert von 3.87 µB ist auf die Spin-Bahn-Kopplung zurückzuführen (Tab. 3.7). 320 0.36 3.5 16 14 0.32 280 3.0 χM / mol cm -3 12 0.28 10 -1 240 2.5 8 6 -3 0.20 3 χM / mol cm χM / cm mol 0 10 20 30 40 2.0 50 Temperatur / K 3 -1 200 χM*T / cm K mol -1 0.24 160 1.5 -1 0.16 0.12 120 0.08 80 1.0 0.04 0.5 40 0.00 0.0 0 0 50 100 150 200 Temperatur / K 250 300 0 50 100 150 200 Temperatur / K 250 300 0 50 100 150 200 250 300 Temperatur / K Abb. 3.6: Magnetische Suszeptibilität als Funktion der Temperatur (links), reziproke molare magnetische Suszeptibilität als Funktion der Temperatur (Mitte) und Produkt der magnetischen Suszeptibilität und Temperatur als Funktion der Temperatur (rechts) für die ligandenreichen 1:2-Verbindungen [M(SCN)2(Pyrazin)2]n mit M = Fe (■), M = Co (●) und M = Ni (▲). 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 53 - Tab. 3.7: Ergebnisse der Auswertung nach dem Curie-Weiss-Gesetz für die ligandenreichen 1:2-Verbindungen [M(SCN)2(Pyrazin)2]n (M = Mn,[28] Fe, Co, Ni). 1:2-Verbindung C / cm3 K mol-1 θ/K µeff (exp) / µB µeff (theo) / µB antiferromag. Ordnungspkt. / K Auswertungsbereich / K Mn 4.51 -1.72 6.00 5.92 10-300 Fe 3.44 -6.68 5.25 4.90 8.5 30-300 Co 2.84 -15.12 4.76 3.87 25-300 Ni 0.96 1.04 2.77 2.83 4-300 Da bei den ligandenarmen 1:1-Verbindungen [M(SCN)2(Pyrazin)2]n (M = Co, Ni, Fe) die Metall(II)kationen über die Thiocyanatanionen verknüpft sind, sind kooperative magnetische Phänomene bei tieferen Temperaturen zu erwarten. Diese Vermutung bestätigt sich jedoch nicht (Abb. 3.7). Im direkten Vergleich der 1:1-Eisen-Verbindung mit der 1:2-Verbindung ist kein antiferromagnetischer Ordnungspunkt mehr vorhanden. Stattdessen lassen sich ferromagnetische Wechselwirkungen beobachten, was durch den Anstieg in der χΜT-Kurve der Eisen-Verbindung in Richtung kleinerer Temperaturen (Abb. 3.7: rechts) und durch den Wert der Weiss'schen Konstante i.H.v. 5.42 K bestätigt wird (Tab. 3.8). Hinsichtlich der 1:1-Cobalt-Verbindung finden sich in Übereinstimmung mit der 1:2-Verbindung weiterhin antiferromagnetische Wechselwirkungen, deren Auftreten durch den Wert der Weiss'schen Konstante i.H.v. -8.84 K bestätigt wird (Tab. 3.8). Nach einem ersten Abfall der χΜT-Kurve in Richtung kleinerer Temperaturen lässt sich jedoch ab 30 K ein starker Anstieg beobachten, der bisher nicht erklärbar und deshalb durch weitere Messungen aufzuklären ist (Abb. 3.7: rechts). Bei der 1:1-Nickel-Verbindung ändert sich im Vergleich zur 1:2-Verbindung die Art der Wechselwirkung zwischen den benachbarten Spin-Momenten. Es finden sich ferromagnetische Wechselwirkungen, was durch den Anstieg in der χΜT-Kurve der Nickel-Verbindung in Richtung kleinerer Temperaturen (Abb. 3.7: rechts) und durch den Wert der Weiss'schen Konstante i.H.v. 15.59 K bestätigt wird (Tab. 3.8). Zusammenfassend lässt sich bislang nicht erklären, weshalb nur die ligandenarme 1:1-ManganVerbindung[28] einen antiferromagnetischen Ordnungspunkt aufweist. So zeigt z.B. die topologisch identische ligandenarme 1:1-Verbindung [Co(SCN)2(Dimethylpyrazin-N,N'-dioxid)]n ebenfalls ein antiferromagnetisches Austauschphänomen bei tiefen Temperaturen.[152] Da alle 1:1-Verbindungen [M(SCN)2 (Pyrazin)]n (M = Mn, Fe, Co, Ni) bis auf geringfügige Unter- 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 54 - schiede in den Bindungslängen und -winkeln sowie hinsichtlich der intra- und intermolekularen Metall-Metall-Abstände ähnlich sind, können strukturelle Ursachen hierfür ausgeschlossen werden (Tab. 3.3 und Tab. 3.4). Gleiches gilt für die ligandenreichen 1:2-Verbindungen. Auch hier lässt sich nur bei der 1:2-Eisen-Verbindung ein antiferromagnetischer Ordnungspunkt beobachten. Die Ursachen dieses Phänomens müssen im Vergleich zu den entsprechenden ligandenreichen Mangan-, Cobalt- und Nickel-Verbindungen in künftigen Arbeiten aufgeklärt werden. 6.0 280 1.1 260 1.0 5.5 240 0.9 5.0 220 0.8 4.5 200 0.7 χM / cm mol -1 0.6 3.5 -1 3 0.4 3 140 0.5 4.0 χM*T / cm K mol 160 χmol / mol cm -3 -1 180 3.0 120 100 0.3 80 0.2 60 2.5 2.0 1.5 40 0.1 20 1.0 0.0 0 0 50 100 150 200 250 300 0 50 100 150 200 250 300 0 50 100 150 200 250 300 Temperatur / K Temperatur / K Temperatur / K Abb. 3.7: Magnetische Suszeptibilität als Funktion der Temperatur (links), reziproke molare magnetische Suszeptibilität als Funktion der Temperatur (Mitte) und Produkt der magnetischen Suszeptibilität und Temperatur als Funktion der Temperatur (rechts) für die ligandenarmen 1:1-Verbindungen [M(SCN)2(Pyrazin)]n mit M = Fe (■), M = Co (●) und M = Ni (▲). Tab. 3.8: Ergebnisse der Auswertung nach dem Curie-Weiss-Gesetz für die ligandenarmen 1:1-Verbindungen [M(SCN)2(Pyrazin)]n (M = Mn,[28] Fe, Co, Ni). 1:1-Verbindung C / cm3 K mol-1 θ/K µeff (exp) / µB µeff (theo) / µB antiferromag. Ordnungspkt. / K Auswertungsbereich / K 3.3 Mn 4.36 -69.55 5.90 5.92 26.0 60-300 Fe 3.67 5.42 5.41 4.90 40-300 Co 3.05 -8.84 4.94 3.87 55-300 Ni 1.05 15.59 2.90 2.83 85-300 Verbindungen auf Basis von Übergangsmetall-Cyanaten und Pyrazin Koordinationspolymere auf Basis von Übergangsmetall-Cyanaten sind in der Literatur nur unvollständig beschrieben.[119,122] Aufgrund der außerordentlichen Instabilität derartiger Verbindungen, gelang es lediglich, eine ligandenreiche Verbindung der Zusammensetzung 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 55 - [Co(OCN)2(Pyrazin)2]n darzustellen. Da diese Verbindung zur weiteren Charakterisierung nicht phasenrein dargestellt werden konnte, wird nur deren Struktur im folgenden Abschnitt beschrieben. Die ligandenreiche 1:2-Cobalt-Verbindung Poly[bis(cyanato-N)-bis(µ2-Pyrazin-N,N')-Cobalt(II)] kristallisiert in der zentrosymmetrischen monoklinen Raumgruppe C2/c mit acht Formeleinheiten in der Elementarzelle und allen Atomen in allgemeiner Lage (Tab. 3.9). Das kristallografisch unabhängige Cyanatanion ist über zwei Orientierungen fehlgeordnet und wurde mit einem SplitModell sowie Besetzungsfaktoren von 0.75 sowie 0.25 verfeinert. In der Kristallstruktur sind die Cobalt(II)kationen von vier Pyrazin-Liganden sowie von zwei Cyanatanionen geringfügig verzerrt oktaedrisch umgeben (Abb. 3.8: links). Die Co-N-Bindungslänge zu den Stickstoffatomen der negativ geladenen Thiocyanatanionen beträgt 2.040(4)-2.059(3) Å und ist signifikant kürzer als die Bindung zu den Stickstoffatomen der Pyrazin-Liganden mit 2.191(3)-2.200(3) Å. Die Bindungswinkel um das Cobalt(II)kation liegen zwischen 89.22(12) und 90.71(10)° sowie 179.58(11)° (Tab. 3.9: rechts). Die Cyanatanionen agieren nicht als verbrückender Ligand, sondern sind lediglich über das Stickstoff-Atom an das Cobalt(II)kation end-on koordiniert. Zudem werden die Metallzentren durch die Pyrazin-Liganden über µ-N,N'-Koordination zu Schichten entlang der kristallografischen a-Achse verbrückt (Abb. 3.8: rechts). Die entsprechenden Mangan,[28] Eisen-,[151] Cobalt-[144] und Nickel-Thiocyanat-Verbindungen besitzen eine identische Netzwerktopologie. Abb. 3.8: Kristallstruktur der 1:2-Cobalt-Verbindung mit Blick auf die Koordinationssphäre des Cobalt(II)kations mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden (links) und Blick auf die Kristallstruktur in Richtung der kristallografischen a-Achse (rechts); übersichtshalber ohne Fehlordnung abgebildet. Symmetrie-Operationen: A = x, -y + 1, z - ½; B = x, -y + 2, z + ½. 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 56 - Tab. 3.9: Ausgewählte Einkristallstrukturdaten (links), Bindungslängen (Å) und -winkel (°) (rechts) von [Co(OCN)2(Pyrazin)2]n. Kristallsystem Raumgruppe a/Å b/Å c/Å α /° β /° γ /° Z V / Å3 R1 monoklin C2/c 25.5712(17) 10.1230(8) 10.1863(7) 90 104.763(8) 90 8 2549.8(3) 0.0507 Co(1)-N(1) Co(1)-N(11) Co(1)-N(21) Co(1)-N(31) N(21)-C(21) C(21)-O(21) N(11)-Co(1)-N(1) N(31)-Co(1)-N(1) N(21)-Co(1)-N(1) C(21)-N(21)-Co(1) N(21)-C(21)-O(21) 2.200(3) 2.191(3) 2.040(4) 2.059(3) 1.151(8) 1.206(9) 178.54(11) 89.50(12) 90.57(12) 153.4(5) 177.1(9) 3.4 Verbindungen auf Basis von Übergangsmetall-Thiocyanaten und Pyrimidin 3.4.1 Kristallstrukturen der Verbindungen 3.4.1.1 [Mn(SCN)2(Pyrimidin)2]n Die 1:2-Mangan-Verbindung Poly[bis(thiocyanato-N)-bis(µ2-Pyrimidin-N,N')-Mangan(II)] kristallisiert in der zentrosymmetrischen orthorhombischen Raumgruppe Cmcm mit vier Formeleinheiten in der Elementarzelle (Tab. 3.10: links) und ist isotyp zur entsprechenden Cobalt-Verbindung.[144] Die asymmetrische Einheit beinhaltet ein, die Position 2/m besetzendes, Mangan(II)kation, ein, auf einer Spiegelebene liegendes, Thiocyanatanion und einen Pyrimidin-Liganden, der auf einer, durch C1 und C3 verlaufenden, C2-Achse liegt. In der Kristallstruktur sind die Mangan(II)kationen von vier Pyrimidin-Liganden sowie von zwei Thiocyanatanionen geringfügig verzerrt oktaedrisch umgeben (Abb. 3.9: links). Die Mn-N-Bindungslänge zu den Stickstoffatomen der negativ geladenen Thiocyanatanionen beträgt 2.1324(17) Å und ist signifikant kürzer als die Bindung zu den Stickstoffatomen der Pyrimidin-Liganden mit 2.3497(11) Å. Die Bindungswinkel um das Mangan(II)kation liegen zwischen 88.20(5) und 91.80(6)° sowie 180° (Tab. 3.10: rechts). Die Thiocyanatanionen agieren nicht als verbrückender Ligand, sondern sind lediglich über das Stickstoffatom an das Mangan(II)kation end-on koordiniert. Zudem werden die Metallzentren durch die Pyrimidin-Liganden über µ-N,N'-Koordination zu Schichten parallel zur kristallografischen b-Achse verbrückt, in denen die Pyrimidin-Liganden senkrecht zur Schichtebene angeordnet sind (Abb. 3.9: rechts). 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 57 - Abb. 3.9: Kristallstruktur der 1:2-Mangan-Verbindung mit Blick auf die Koordinationssphäre des Mangan(II)kations mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden (links) und Packungsbild (rechts). Symmetrie-Operationen: A = -x, -y + 1, -z; B = x, -y + 1, -z; C = -x, y, z; D = -x + ½, y, -z + ½. Tab. 3.10: Ausgewählte Einkristallstrukturdaten (links), Bindungslängen (Å) und -winkel (°) (rechts) von [Mn(SCN)2(Pyrimidin)2]n. Kristallsystem Raumgruppe a/Å b/Å c/Å α /° β /° γ /° Z V / Å3 R1 orthorhombisch Cmca 9.7693(9) 15.6505(10) 8.6220(6) 90 90 90 4 1318.26(17) 0.0276 Mn(1)-N(1) Mn(1)-N(11) S(1)-C(11) C(11)-N(11) N(1)-Mn(1)-N(1C) N(11)-Mn(1)-N(1B) N(11)-Mn(1)-N(1) N(1)-Mn(1)-N(1B) N(1A)-Mn(1)-N(1) C(11)-N(11)-Mn(1) N(11)-C(11)-S(1) 2.3497(11) 2.1324(17) 1.620(2) 1.173(3) 88.20(5) 89.73(4) 90.27(4) 91.80(6) 180.00(5) 143.91(16) 179.51(19) Aus Pulver-Untersuchungen geht hervor, dass die entsprechende 1:2-Nickel-Verbindung isotyp Relative Intensität zu den o.g. Verbindungen ist (Abb. 3.10). Jedoch fehlen die Einkristallstrukturdaten bisher. 5 10 15 20 25 30 35 40 45 2 Theta / ° Abb. 3.10: Experimentelles Röntgen-Pulverdiffraktogramm der 1:2-Nickel-Verbindung [Ni(SCN)2(Pyrimidin)2]n (oben) und berechnetes Röntgen-Pulverdiffraktogramm der 1:2-Cobalt-Verbindung (unten); Cu-Kα-Strahlung. 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden 3.4.1.2 - 58 - M(SCN)2(Pyrimidin)4 (M = Co, Ni) Die ligandenreichen 1:4-Verbindungen Tetrakis(Pyrimidin-κN)-bis(thiocyanato-κN)-Cobalt(II) und Tetrakis(Pyrimidin-κN)-bis(thiocyanato-κN)-Nickel(II) sind isotyp und kristallisieren in der zentrosymmetrischen monoklinen Raumgruppe P21/c mit vier Formeleinheiten in der Elementarzelle (Tab. 3.11: links). Alle Atome befinden sich in allgemeiner Lage. In den Kristallstrukturen sind die Metall(II)kationen von vier Pyrimidin-Liganden sowie von zwei Thiocyanatanionen geringfügig verzerrt oktaedrisch umgeben (Abb. 3.11). Im Vergleich zu den 1:2-Verbindungen M(SCN)2(Pyrimidin)2 (M = Co,[144] Ni[s.o.]) ist die Kristallstruktur zwar aus den gleichen Einheiten aufgebaut, jedoch werden diese nicht durch die Liganden verknüpft, wodurch diskrete Komplexe gefunden werden. Wie in den 1:2-Verbindungen sind hier die M-N-Bindungslängen zu den Stickstoffatomen der negativ geladenen Thiocyanatanionen mit Co-NCS = 2.060(2)2.072(2) bzw. Ni-NCS = 2.0424(19)- 2.0498(18) Å kleiner als die Bindungslängen zu den Stickstoffatomen der Pyrimidin-Liganden mit Co-NPym = 2.185(2)-2.2172(19) bzw. Ni-NPym = 2.1292(18)- 2.1547(18) Å. Die Bindungswinkel um die Metall(II)kationen liegen bei der CobaltVerbindung zwischen 87.84(8) und 92.74(7)° sowie zwischen 177.57(7) und 178.56(7)° und bei der Nickel-Verbindung zwischen 88.24(7) und 92.41(7)° sowie zwischen 177.62(7) und 178.79(7)° (Tab. 3.12). Im direkten Vergleich dieser beiden isotypen Verbindungen ist ein geringfügiger Trend der M-NCS- und M-NPym-Bindungslängenverkürzung von der Cobalt- zur Nickel-Verbindung sowie eine geringfügige Änderung des NCS-M-NCS-Winkels erkennbar (Tab. 3.12). Abb. 3.11: Kristallstrukturen der 1:4-Verbindungen Co(SCN)2(Pyrimidin)4 (links) und Ni(SCN)2(Pyrimidin) (rechts) mit Blick auf die Koordinationssphäre der Metall(II)kationen mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden. 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 59 - Tab. 3.11: Ausgewählte Einkristallstrukturdaten von M(SCN)2(Pyrimidin)4 (M = Co, Ni). 1:4-Verbindung Kristallsystem Raumgruppe a/Å b/Å c/Å β /° Z V / Å3 R1 Co monoklin P21/c 8.9454(5) 12.4819(9) 20.1588(12) 98.785(7) 4 2224.4(2) 0.0339 Ni monoklin P21/c 8.9152(6) 12.4948(8) 20.0048(16) 98.630(9) 4 2203.2(3) 0.0367 Tab. 3.12: Ausgewählte Bindungslängen [Å] und -winkel [°] von M(SCN)2(Pyrimidin)4 (M = Co, Ni). 1:4-Verbindung M(1)-N(1) M(1)-N(11) M(1)-N(21) M(1)-N(31) M(1)-N(51) M(1)-N(61) S(1)-C(51) S(2)-C(61) C(51)-N(51) C(61)-N(61) N(21)-M(1)-N(1) N(31)-M(1)-N(11) N(51)-M(1)-N(61) N(61)-M(1)-N(1) N(51)-M(1)-N(1) N(31)-M(1)-N(1) N(51)-C(51)-S(1) C(51)-N(51)-M(1) 3.4.2 Co 2.198(2) 2.2172(19) 2.186(2) 2.185(2) 2.060(2) 2.072(2) 1.627(2) 1.629(2) 1.165(3) 1.165(3) 177.57(7) 178.56(7) 177.99(8) 87.84(8) 90.70(8) 92.74(7) 179.5(2) 175.8(2) Ni 2.1401(18) 2.1547(18) 2.1292(18) 2.1296(19) 2.0424(19) 2.0498(18) 1.632(2) 1.631(2) 1.160(3) 1.165(3) 177.62(7) 178.79(7) 178.34(8) 88.24(7) 90.68(7) 92.41(7) 179.5(2) 175.87(19) Thermoanalytische Untersuchungen der Verbindungen Da die ligandenreichen 1:4-Verbindungen zur weiteren Charakterisierung nicht phasenrein dargestellt werden konnten, werden im folgenden Abschnitt lediglich die thermischen Eigenschaften der 1:2-Mangan-Verbindung beschrieben. Weitere thermoanalytische Untersuchungen der Verbindungen [M(SCN)2(Pyrimidin)2]n (M = Mn, Co, Ni) mit Charakterisierung deren thermischer Abbauprodukte folgen in künftigen Arbeiten. Beim Aufheizen der 1:2-Verbindung Mn(SCN)2(Pyrimidin)2 werden drei gut aufgelöste Massestufen in der TG-Kurve beobachtet, die mit endothermen Signalen in der DTA-Kurve verbunden sind. Aus der MS-Trend-Scan-Kurve wird ersichtlich, dass ausschließlich der Ligand (m/z = 80) 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 60 - während dieser Stufen emittiert wird (Abb. 3.12). Der experimentelle Masseverlust der ersten Stufe i.H.v. 22.4 % stimmt annähernd mit dem berechneten Verlust für die Hälfte der Liganden (1 mol; 24.2 %) überein. Der Masseverlust für die zweite Stufe i.H.v. 8.7 % stimmt nahezu mit dem berechneten Verlust für einen Sechstel der Liganden (1 mol; 8.1 %) überein. Deshalb ist davon auszugehen, dass in der ersten Stufe eine 1:1-Verbindung der Zusammensetzung Mn(SCN)2(Pyrimidin) und in der zweiten Stufe eine ligandenarme 3:2-Verbindung als Intermediat gebildet wird. Bei weiterem Aufheizen werden die restlichen Liganden emittiert, so dass Mn(SCN)2 entsteht, welches sich bei weiterem Aufheizen zersetzt. ΔT, Δm, (dm/dT) und Ionenstrom in beliebigen Einheiten DTG Δm = 22.4 % endo Δm = 8.7 % Δm = 16.7 % TP = 403°C TP = 263°C TP = 223°C TP = 196°C m/z = 80 50 TG DTA MID 100 150 200 250 300 350 400 450 500 Temperatur / °C Abb. 3.12: DTA-, TG-, DTG- und MS-Trend-Scan-Kurve (simultane Messung) für die ligandenreiche 1:4-Verbindung Mn(SCN)2(Pyrimidin)4. Gegeben sind die Masseverluste (%) und Peak-Temperaturen TP (°C); Heizrate: 4 K/min; dynamische Stickstoffatmosphäre (75 mL/min); Al2O3-Tiegel. 3.4.3 Magnetische Eigenschaften der Verbindungen Im folgenden Abschnitt werden ebenfalls lediglich die magnetischen Eigenschaften der 1:2Mangan-Verbindung beschrieben. Weitere magnetische Untersuchungen der Verbindungen [M(SCN)2(Pyrimidin)2]n (M = Mn, Co, Ni) und deren thermischer Abbauprodukte werden Thema künftiger Arbeiten sein. Die Auswertung erfolgte analog zu der in Abschnitt 3.2.3 beschriebenen. Für die 1:2-Verbindung [Mn(SCN)2(Pyrimidin)2]n ist bis zu einer Temperatur von 10 K ein rein paramagnetisches Verhalten zu beobachten. Unterhalb dieser Temperatur ist ein Maximum bei 3 K in der χM-1-Kurve erkennbar, das auf einen antiferromagnetischen Ordnungspunkt hinweist, allerdings nicht charakteristisch für einen solchen ist (Abb. 3.13: Mitte). Die Ursache des unge- 3. Verbindungen auf Basis von Übergangsmetall-(Thio)cyanaten und N-Donorliganden - 61 - wöhnlichen Verlaufs der χM-1-Kurve ist durch unterschiedliche Messstrategien in künftigen Arbeiten aufzuklären. Der Abfall in der χΜT-Kurve in Richtung kleinerer Temperaturen deutet auf antiferromagnetische Wechselwirkungen zwischen den benachbarten Spin-Momenten hin (Abb. 3.13: rechts). Der Wert der Weiss'schen Konstante i.H.v. -3.63 K bestätigt diese Annahme. Weiterhin stimmt das experimentelle effektive magnetische Moment von 5.93 µB mit dem für ausschließlichen "spin-only" Paramagnetismus berechneten high-spin-Wert von 5.92 µB nahezu überein (Tab. 3.13). 0.40 70 0.40 3.1 0.38 -3 0.37 χM / mol cm 3 χM / cm mol 4.0 3.0 60 -1 0.35 4.5 3.2 0.39 0.36 -1 0.30 2.9 3.5 2.8 50 0.35 2 3 4 5 6 7 40 8 Temperatur / K 3.0 2.5 1 2 3 4 5 6 7 8 Temperatur / K -1 3 1 2.6 3 -1 0.33 χM*T / cm K mol -3 χM / mol cm 0.34 0.25 χM / cm mol -1 2.7 0.20 2.5 30 2.0 0.15 20 1.5 0.10 10 0.05 0.00 1.0 0.5 0 0 50 100 150 200 250 300 0 50 Temperatur / K 100 150 200 Temperatur / K 250 300 0 50 100 150 200 250 300 Temperatur / K Abb. 3.13: Magnetische Suszeptibilität als Funktion der Temperatur (links), reziproke molare magnetische Suszeptibilität als Funktion der Temperatur (Mitte) und Produkt der magnetischen Suszeptibilität und Temperatur als Funktion der Temperatur (rechts) für [Mn(SCN)2(Pyrimidin)2]n. Tab. 3.13: Ergebnisse der Auswertung nach dem Curie-Weiss-Gesetz für [Mn(SCN)2(Pyrimidin)]n. C / cm3 K mol-1 θ/K µeff (exp) / µB µeff (theo) / µB antiferromag. Ordnungspkt. / K Auswertungsbereich / K 4.40 -3.63 5.93 5.92 10-300 4. Verbindungen auf Basis von Übergangsmetall-Halogeniden und 2,2'- sowie 4,4'-Dipyridyldisulfid - 62 - 4 Verbindungen auf Basis von Übergangsmetall-Halogeniden und 2,2'- sowie 4,4'-Dipyridyldisulfid 4.1 Einleitung Die in diesem Kapitel vorgestellten Verbindungen auf Basis von Übergangsmetall-Halogeniden mit 2,2'- und 4,4'-Dipyridyldisulfid wurden im Rahmen der vorliegenden Diplomarbeit dargestellt und strukturell charakterisiert. Die Liganden 2,2'- und 4,4'-Dipyridyldisulfid erwiesen sich jedoch aus nachfolgenden Gründen als ungeeignet zur Darstellung von Koordinationspolymeren: Bei der Umsetzung von ZnX2 (X = Cl, Br, I) mit 2,2'-Dipyridyldisulfid unter solvothermalen Synthesebedingungen wurde deutlich, dass die Disulfid-Gruppe im 2,2'-Dipyridyldisulfid in-situ unter S-S-Bindungsbruch zum 2,2'-Dipyridylsulfid reagiert und ZnX2(2,2'-Dipyridylsulfid)Chelatkomplexe bildet.[109-111] Ferner ist bei der Umsetzung von ZnBr2 mit 2,2'-Dipyridyldisulfid die Bildung des entsprechenden ZnBr2(2,2'-Dipyridyldisulfid)-Chelatkomplexes zu beobachten.[112] Des Weiteren fiel bei der Umsetzung von CuX (X = Cl, Br) mit 4,4'-Dipyridyldisulfid unter solvothermalen Synthesebedingungen auf, dass in diesem Fall die Disulfid-Bindung ebenfalls thermisch labil ist und auf die Bildung von diskreten Komplexen, bestehend aus Cu6X6S4Clustern und 4-Pyridiniumthiolat-Liganden, hinausläuft.[107,108,153] Zusammenfassend betrachtet, sind die Liganden 2,2'-und 4,4'-Dipyridyldisulfid aufgrund ihrer Labilität nicht zur Darstellung neuer Koordinationspolymere durch thermische Abbaureaktionen geeignet. Außerdem bilden sie bei der Umsetzung mit Übergangsmetall-Halogeniden bevorzugt Chelat- bzw. diskrete Komplexe, die sich nur bedingt in die ursprüngliche Fragestellung einordnen lassen. Da diese Verbindungen zur weiteren Charakterisierung nicht phasenrein dargestellt werden konnten, werden nur deren Strukturen in diesem Kapitel beschrieben. 4.2 Kristallstrukturen der Verbindungen 4.2.1 ZnX2(2,2'-Dipyridylsulfid) (X = Cl, Br, I) Die Verbindungen Dichlorido(di-2-pyridyl sulfid-κ2N,N‘)zink(II)[109] (1) (Abb. 4.1) und Dibromido(di-2-pyridylsulfid-κ2N,N‘)zink(II)[111] (2) (Abb. 4.2) kristallisieren monoklin in der zentrosymmetrischen Raumgruppe P21/c mit vier Formeleinheiten in der Elementarzelle (Tab. 4.1). In der asymmetrischen Einheit befinden sich alle Atome in allgemeiner Lage. 4. Verbindungen auf Basis von Übergangsmetall-Halogeniden und 2,2'- sowie 4,4'-Dipyridyldisulfid - 63 - Die Verbindung Diiodido(di-2-pyridylsulfid-κ2N,N‘)zink(II)[110] (3) (Abb. 4.3) kristallisiert orthorhombisch in der zentrosymmetrischen Raumgruppe Pnma mit vier Formeleinheiten in der Elementarzelle (Tab. 4.1). In der asymmetrischen Einheit befinden sich die Zink(II)kationen, Schwefelatome und die Iodidanionen auf einer kristallografischen Spiegelebene. Alle drei Verbindungen sind isostrukturell, jedoch nicht isotyp. Das Zink(II)kation ist jeweils verzerrt tetraedrisch von zwei Halogenidanionen und zwei Pyridyl-N-Atomen koordiniert. Hieraus resultiert die Bildung eines sechsgliedrigen Chelat-Ringes in einer Boot-Konfiguration, wobei die Sulfidgruppe – wie üblich bei dieser Art von Komplexen – nicht an der Zink-Koordination teilnimmt. Allgemein ist 2,2'-Dipyridylsulfid aufgrund seiner konformativen Flexibilität als zweizähniger Chelat-Ligand bekannt.[154-156] In 1 betragen die Zn-X- bzw. Zn-N-Bindungslängen 2.2192(8)-2.2261(8) bzw. 2.057(2)-2.061(2) Å, in 2 2.058(3)-2.055(3) bzw. 2.3527(5)-2.3504(6) Å sowie in 3 2.063(3) bzw. 2.5447(6) Å. Die N-Zn-X Bindungswinkel liegen in 1 im Bereich zwischen 107.42(7) und 115.66(7)°, in 2 zwischen 107.43(8) und 115.35(8)° sowie in 3 zwischen 108.33(8) und 113.67(7)° (Tab. 4.2). Tab. 4.1: Ausgewählte Einkristallstrukturdaten von ZnX2(2,2'-Dipyridylsulfid) (X = Cl, Br, I). Verbindung Kristallsystem Raumgruppe a/Å b/Å c/Å α /° β /° γ /° Z V / Å3 R1 1 monoklin P21/c 12.1944(12) 7.6404(4) 14.2572(15) 90 110.426(12) 90 4 1244.82(19) 3.56 2 monoklin P21/c 11.0385(8) 8.9627(5) 13.1572(11) 90 91.663(9) 90 4 1301.16(16) 3.30 3 orthorhombisch Pnma 13.9418(8) 10.9742(10) 9.1913(6) 90 90 90 4 1406.27(18) 2.86 Tab. 4.2: Ausgewählte Bindungslängen (Å) und -winkel (°) von ZnX2(2,2'-Dipyridylsulfid) (X = Cl, Br, I). Verbindung Zn1-N1 Zn1-N11 Zn1-X1 Zn1-X2 N1-Zn1-X1 N11-Zn1-X1 N1-Zn1-X2 N11-Zn1-X2 Diederwinkel 1 2.057(2) 2.061(2) 2.2192(8) 2.2261(8) 109.32(7) 115.66(7) 108.68(7) 107.42(7) 50.4(1) 2 2.058(3) 2.055(3) 2.3527(5) 2.3504(6) 108.56(8) 107.43(8) 109.76(8) 115.35(8) 52.7(1) 3 2.063(3) 2.5447(6) 113.67(7) 108.33(8) 60.1(1) 4. Verbindungen auf Basis von Übergangsmetall-Halogeniden und 2,2'- sowie 4,4'-Dipyridyldisulfid - 64 - Abb. 4.1: Kristallstruktur von Dichlorido(di-2-pyridylsulfid-κ2N,N‘)zink(II) (1) mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden. Abb. 4.2: Kristallstruktur von Dibromido(di-2-pyridylsulfid-κ2N,N‘)zink(II) (2) mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden. Abb. 4.3: Kristallstruktur von Diiodido(di-2-pyridylsulfid-κ2N,N‘)zink(II) (3) mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden. Symmetrie-Operation: A = x, -y + ½, z. 4.2.2 ZnBr2(2,2'-Dipyridyldisulfid) Die Verbindung Dibromido(di-2-pyridyldisulfid-κ2N,N‘)zink(II)[112] (Abb. 4.4) ist isotyp zu der entsprechenden Chlorid-Verbindung.[157] Sie kristallisiert triklin in der zentrosymmetrischen Raumgruppe P-1 mit zwei Formeleinheiten in der Elementarzelle (Tab. 4.3: links). Die asymmetrische Einheit besteht aus einem 2-Pyridylsulfid-Liganden sowie dem ZnBr2 in allgemeiner Position. Das Zink(II)kation ist in allen drei Verbindungen verzerrt tetraedrisch von zwei Bro- 4. Verbindungen auf Basis von Übergangsmetall-Halogeniden und 2,2'- sowie 4,4'-Dipyridyldisulfid - 65 - midanionen und zwei Pyridyl-N-Atomen koordiniert. Hieraus resultiert die Bildung eines siebengliedrigen Chelat-Ringes, wobei die Disulfidgruppe – wie üblich bei dieser Art von Komplexen – nicht an der Zink-Koordination teilnimmt. Die Zn-Br- bzw. Zn-N-Bindungslängen betragen 2.091(5)-2.042(5) bzw. 2.3897(10)-2.3664(10) Å und die N-Zn-Br Bindungswinkel liegen im Bereich zwischen 100.99(15) und 112.77(15)° (Tab. 4.3: rechts). Die S-S-Bindungslänge (2.050(3) Å) ist etwas länger als die im freien Liganden (2.016 Å).[158] Tab. 4.3: Ausgewählte Einkristallstrukturdaten (links), Bindungslängen (Å) und -winkel (°) (rechts) von ZnBr2(2,2'Dipyridyldisulfid). Kristallsystem Raumgruppe a/Å b/Å c/Å α /° β /° γ /° Z V / Å3 R1 triklin P-1 7.7610(8) 8.2962(8) 12.3576(13) 95.488(12) 107.161(12) 112.950(11) 2 679.70(12) 4.57 Zn1-N1 Zn1-N11 Zn1-Br1 Zn1-Br2 S1-S2 N1-Zn1-Br1 N11-Zn1-Br1 N1-Zn1-Br2 N11-Zn1-Br2 N11-Zn1-N1 Br2-Zn1-Br1 2.091(5) 2.042(5) 2.3897(10) 2.3664(10) 2.050(3) 100.99(15) 103.35(15) 103.61(14) 112.77(15) 117.2(2) 119.06(4) Abb. 4.4: Kristallstruktur von Dibromido(di-2-pyridyldisulfid-κ2N,N‘)zink(II) mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden. 4.2.3 Cu6X6S4(4-Pyridiniumthiolat)4 (X = Cl, Br) Die Verbindungen Hexachloridotetrakis(µ3-4-pyridiniumthiolato-κ3S:S:S)hexakupfer(I)[108,153] (1) (Abb. 4.5) und Hexabromidotetrakis(µ3-4-pyridiniumthiolato-κ3S:S:S)hexakupfer(I)[107] (2) (Abb. 4.6) sind isotyp und kristallisieren tetragonal in der zentrosymmetrischen Raumgruppe I41/a mit vier Formeleinheiten in der Elementarzelle (Tab. 4.4). Die asymmetrische Einheit be- 4. Verbindungen auf Basis von Übergangsmetall-Halogeniden und 2,2'- sowie 4,4'-Dipyridyldisulfid - 66 - steht aus zwei Kupfer(I)kationen, einem Bromidanion und einem 4-Pyridiniumthiolat-Liganden in allgemeiner Position sowie aus einem Bromidanion, das auf einer C2-Achse angeordnet ist. Da sich zudem ein Kupfer(I)kation neben der C2-Achse befindet, ist dieses symmetriebedingt ungeordnet. Das zweite unabhängige Kupfer(I)kation ist über zwei Positionen mit Besetzungsfaktoren von 0.6 und 0.4 in allgemeiner Position ungeordnet. Im Allgemeinen besteht die Kristallstruktur aus hexanuklearen Cu6Cl6S4-Clustern, die auf einer vierzähligen Drehinversionsachse angeordnet sind. Die Kupfer(I)kationen bilden ein stark verzerrtes Oktaeder, in dem jedes Kupfer(I)kation mit einem Bromidanion verbunden ist. Während zwei dieser Bromidanionen als terminale Liganden agieren, verknüpfen die übrigen von ihnen die Cu6Cl6S4-Cluster über µ3-Koordination. Die Cu2Br2-Einheiten sind auf einem Inversionszentrum angeordnet und die Sulfidanionen sind jeweils über µ3-Koordination mit drei Kupfer(I)kationen verknüpft. Tab. 4.4: Ausgewählte Einkristallstrukturdaten von Cu6X6S4(4-Pyridiniumthiolat)4 (X = Cl, Br). Verbindung Kristallsystem Raumgruppe a/Å b/Å c/Å α /° β /° γ /° Z V / Å3 R1 1 tetragonal I41/a 15.0563(9) 15.0563(9) 13.0852(9) 90 90 90 4 2966.3(3) 4.93 2 tetragonal I41/a 15.3161(9) 15.3161(9) 13.2602(8) 90 90 90 4 3110.6(3) 3.24 4. Verbindungen auf Basis von Übergangsmetall-Halogeniden und 2,2'- sowie 4,4'-Dipyridyldisulfid - 67 - Abb. 4.5: Kristallstruktur von Hexachloridotetrakis(µ3-4-pyridiniumthiolato-κ3S:S:S)hexakupfer(I) mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden. Symmetrie-Operationen: A = 1 - x, 1½ - y, z; B = 1¼ - y, ¼ + x, 1¼ - z; C = -¼ + y, 1¼ - x, 1¼ - z. Abb. 4.6: Kristallstruktur von Hexabromidotetrakis(µ3-4-pyridiniumthiolato-κ3S:S:S)hexakupfer(I) mit Nummerierung und 50 % Wahrscheinlichkeitsellipsoiden. Symmetrie-Operationen: A = 1 - x, 1½ - y, z; B = 1¼ - y, ¼ + x, 1¼ - z; C = -¼ + y, 1¼ - x, 1¼ - z. 5. Zusammenfassung und Ausblick - 68 - 5 Zusammenfassung und Ausblick Im Rahmen dieser Diplomarbeit standen die Darstellung, die strukturelle Charakterisierung sowie die Untersuchung der physikalischen Eigenschaften neuer Übergangsmetall-Koordinationspolymere im Vordergrund. Daher sollten zum einen, im Sinne des "Crystal Engineering", ein rationaleres Design von Festkörperstrukturen mit dem Ziel entwickelt werden, Struktur-Eigenschaftbeziehungen aufzuzeigen. Basierend auf vorherige Arbeiten über die thermischen Eigenschaften von Koordinationspolymeren auf Basis von Kupfer(I)halogeniden und N-Donorliganden, galt es darüber hinaus vorrangig herauszufinden, ob die Methode des thermischen Abbaus ligandenreicher Precursoren auch zur Darstellung ligandenarmer thermodynamisch stabiler und metastabiler Verbindungen sowie polymorpher Modifikationen anderer Systeme geeignet ist. Hierzu wurden zahlreiche neue Verbindungen mit interessanten Eigenschaften dargestellt und untersucht. Da allerdings in den einzelnen Themenbereichen noch einige Fragen unbeantwortet blieben, müssen diese in künftigen Untersuchungen bearbeitet werden. Der erste Teil dieser Ausarbeitung beschäftigt sich mit der Darstellung und Charakterisierung von Koordinationspolymeren auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin als Ligand. Insgesamt wurden acht neue Verbindungen synthetisiert und charakterisiert. Die Reaktion von ZnX2 (X = Cl, Br, I) und 2,5-Dimethylpyrazin in Methanol führte zur Bildung der ligandenarmen Verbindungen [ZnX2(2,5-Dimethylpyrazin)]n (X = Cl 1, Br 3I und 3II, I 6) sowie der ligandenreichen Verbindung ZnI2(2,5-Dimethylpyrazin)2 7. Weitere ligandenreiche Verbindungen der Zusammensetzung (ZnX2)2(2,5-Dimethylpyrazin)3 (X = Cl 2, Br 4) und [ZnBr2(2,5Dimethylpyrazin)]·2,5-Dimethylpyrazin 5 konnten in einer lösungsmittelfreien Reaktion dargestellt werden. In der Kristallstruktur aller Verbindungen sind die Zink(II)kationen jeweils von zwei Halogenidanionen und zwei 2,5-Dimethylpyrazin-Liganden verzerrt tetraedrisch koordiniert. In den 1:1-Verbindungen 1 und 6 sowie in den polymorphen 1:1-Modifikationen 3I und 3II sind die Zink(II)kationen außerdem durch die Liganden zu Ketten verknüpft. Dagegen ist die Kristallstruktur der 2:3-Verbindungen 2 und 4 aus oligomeren diskreten [(ZnX2)2(2,5-Dimethylpyrazin)3]-Einheiten (X= Cl, Br) aufgebaut, in der jedes Zink(II)kation jeweils von zwei Halogenidanionen sowie von einem verbrückenden und terminalen Liganden koordiniert ist. Die Struktur der ligandenreichen 1:2- 7 und 1:3-Verbindung 5 besteht aus diskreten monomeren [ZnX2(2,5-Dimethylpyrazin)2]-Einheiten (X = Br, I). Bei dem kontrollierten thermischen Abbau der ligandenreichen Verbindungen 2, 5, und 7 lassen sich verschiedene Massestufen beobachten. Während in der ersten Stufe die ligandenarmen 1:1-Verbindungen 1 und 6 in quantitativen Ausbeuten sowie 3I und 3II als Mischung gebildet werden, entstehen in der zweiten Stufe neue li- 5. Zusammenfassung und Ausblick - 69 - gandenarme 2:1-Verbindungen. Bei weiterem Aufheizen werden die restlichen Liganden emittiert, so dass ZnX2 (X = Cl, Br, I) entsteht, welches bei weiterem Aufheizen verdampft. Reaktionen verschiedenster stöchiometrischer Zusammensetzungen der Edukte in Methanol zeigen, dass unabhängig von den molaren Verhältnissen von ZnX2 (X = Cl, Br) und Ligand lediglich die 1:1Verbindungen 1 und 3I gebildet werden, die daher die thermodynamisch stabilsten Verbindungen bei Raumtemperatur darstellen. Für ZnI2 wird bei Verhältnissen zwischen ZnI2 und Ligand größer als 1:3, die 1:2-Verbindung 7 erhalten, wohingegen bei kleineren Verhältnissen stets die 1:1-Verbindung 6 entsteht. Der Schwerpunkt des zweiten Teils dieser Arbeit lag auf der Darstellung und Charakterisierung von Koordinationspolymeren auf Basis paramagnetischer Übergangsmetalle mit kleinen Anionen und N-Donorliganden. Da ein thermischer Abbau derartiger Verbindungen in vielen Fällen zu neuen Verbindungen mit einem höheren Grad der Vernetzung führt, sind auf diese Weise mitunter kooperative magnetische Phänomene zu erwarten. Daher wurden in diesem Zusammenhang Thiocyanate und Cyanate als kleine Anionen sowie Pyrazin und Pyrimidin als Ligand untersucht. Insgesamt wurden sieben neue Verbindungen synthetisiert und charakterisiert. Die Reaktion von M(SCN)2 (M = Fe, Co, Ni) mit Pyrazin in Wasser führte zur Bildung der ligandenreichen 1:2-Verbindungen [M(SCN)2(Pyrazin)2]n (M = Fe, Co, Ni) sowie der ligandenarmen 1:1Verbindung [Ni(SCN)2(Pyrazin)]n. Die ligandenarmen 1:1-Verbindungen [M(SCN)2(Pyrazin)]n (M = Fe, Co) ließen sich nur durch den thermischen Abbau ihrer ligandenreichen Precursoren darstellen. In der Kristallstruktur der ligandenreichen 1:2-Verbindungen sind die Metall(II)kationen von vier Pyrazin-Liganden sowie von zwei Thiocyanatanionen geringfügig verzerrt oktaedrisch umgeben. Die Thiocyanatanionen agieren nicht als verbrückender Ligand, sondern sind lediglich über das Stickstoffatom an das Metall(II)kation koordiniert. Zudem werden die Metallzentren durch die Pyrazin-Liganden über µ-N,N'-Koordination zu Schichten verknüpft. In der Kristallstruktur der ligandenarmen 1:1-Verbindungen sind die Metall(II)kationen jeweils von zwei Stickstoffatomen der Pyrazin-Liganden sowie von zwei Schwefel- und zwei Stickstoffatomen der Thiocyanatanionen geringfügig verzerrt oktaedrisch koordiniert. Im Gegensatz zu den ligandenreichen Verbindungen werden hier die Metall(II)kationen über eine µ2-N-S-Koordination von den Thiocyanatanionen zu Ketten verbrückt, die zusätzlich durch die Pyrazin-Liganden zu Schichten verknüpft werden. Bei dem kontrollierten thermischen Abbau der ligandenreichen 1:2-Verbindungen lassen sich zwei Massestufen beobachten. In der ersten Stufe bilden sich die ligandenarmen 1:1-Verbindungen in quantitativen Ausbeuten. Bei weiterem Aufheizen werden die restlichen Liganden emittiert, so dass M(SCN)2 (M = Fe, Co, Ni) entsteht, welches sich 5. Zusammenfassung und Ausblick - 70 - bei weiterem Aufheizen zersetzt. Bei der Untersuchung der magnetischen Eigenschaften findet sich für die ligandenreiche 1:2-Verbindung [Fe(SCN)2(Pyrazin)2]n bei einer Neel-Temperatur von 8.5 K ein antiferromagnetischer Ordnungspunkt, während sich bei den ligandenreichen 1:2Verbindungen [M(SCN)2(Pyrazin)2]n (M = Co, Ni) ein rein paramagnetisches Verhalten beobachten lässt. Obwohl bei ihnen am ehesten kooperative magnetische Phänomene erwartet werden, weisen die ligandenarmen 1:1-Verbindungen [M(SCN)2(Pyrazin)]n (M = Fe, Co, Ni) erstaunlicherweise ebenfalls nur ein rein paramagnetisches Verhalten auf. Im direkten Vergleich mit der literarturbekannten topologisch identischen 1:1-Verbindung [Mn(SCN)2(Pyrazin)]n, die einen antiferromagnetischen Ordnungspunkt bei 26 K aufweist, ist dieses magnetische Verhalten bisher nicht erklärbar und deshalb Thema künftiger Untersuchungen. Die Reaktion von Co(OCN)2 mit Pyrazin in Wasser führte zur Bildung der ligandenreichen 1:2Verbindung [Co(SCN)2(Pyrazin)2]n, die eine identische Netzwerktopologie mit den entsprechenden ligandenreichen Thiocyanat-Verbindungen aufweist und deshalb eine Precursorverbindung für eine ligandenarme Verbindung mit interessanten magnetischen Eigenschaften darstellt. Für weitere Untersuchungen konnte diese Verbindung jedoch nicht phasenrein dargestellt werden. Ziel künftiger Arbeiten wird daher die phasenreine Darstellung weiterer Cyanate sein. Die Reaktion von M(SCN)2 (M = Mn, Co, Ni) mit Pyrimidin in Wasser führte zur Bildung der ligandenreichen 1:2-Verbindungen [M(SCN)2(Pyrimidin)2]n (M = Mn, Co, Ni). Weitere ligandenreichere 1:4-Verbindungen der Zusammensetzung Mn(SCN)2(Pyrimidin)4 (M = Co, Ni) konnten in einer lösungsmittelfreien Reaktion dargestellt werden. Die Kristallstruktur der 1:2Verbindungen weist eine identische Netzwerktopologie mit den entsprechenden Pyrazin-Verbindungen auf. Dagegen sind die Kristallstrukturen der 1:4-Verbindungen zwar aus den gleichen Einheiten aufgebaut, jedoch werden diese nicht durch die Liganden verknüpft, so dass diskrete Komplexe zu finden sind. Alle diese Verbindungen stellen potenzielle ligandenreiche Precursoren für den thermischen Abbau dar, dessen ligandenarme Abbauprodukte in folgenden Arbeiten charakterisiert werden sollen. Zusammenfassend wurde im Rahmen dieser Untersuchungen gezeigt, dass sich durch einen kontrollierten thermischen Abbau neue Koordinationspolymere darstellen lassen, die z.T. in Lösung nicht, oder nur als Gemische verschiedener Phasen, dargestellt werden können. Durch lediglich ein Experiment – ausgehend von den ligandenreichsten Verbindungen – können so weitgehend alle ligandenarmen Verbindungen eines Systems entdeckt werden. Ein Ziel dieser Untersuchungen war die Aufklärung der Zusammenhänge zwischen den Strukturen der während des thermischen Abbaus entstehenden Intermediate und deren ligandenreichen Precursoren sowie 5. Zusammenfassung und Ausblick - 71 - zwischen den Strukturen und der thermischen Reaktivität bzw. Stabilität der Ausgangsverbindungen. Außerdem galt es zu prüfen, inwiefern die magnetischen Eigenschaften von den Strukturen der ligandenreichen Precursoren und deren ligandenarmen Intermediate abhängig sind. Die hier vorgestellten Untersuchungen – z.T. im direkten Vergleich mit vorherigen Arbeiten – haben jedoch ergeben, dass kein einfacher Zusammenhang zwischen diesen Parametern besteht. Folglich ist ein rationaleres Design von Koordinationspolymeren mit definierten Strukturen und Eigenschaften komplizierter als dies häufig in der Literatur dargestellt wird. Es ist daher notwendig, weitere dieser Verbindungen darzustellen und deren thermische sowie magnetische Eigenschaften zu charakterisieren. Demgemäß werden in künftigen Arbeiten Untersuchungen an Koordinationspolymeren auf Basis von weiteren Übergangsmetall-Halogeniden und derivatisierten Pyrazin-Liganden folgen. 6. Experimenteller Teil - 72 - 6 Experimenteller Teil 6.1 Darstellung der Verbindungen 6.1.1 Verbindungen auf Basis von Zink(II)halogeniden und 2,5-Dimethylpyrazin [ZnCl2(2,5-Dimethylpyrazin)]n: Kristallines Pulver dieser Verbindung wurde durch Reaktion von 272.6 mg ZnCl2 (2.0 mmol) und 216.3 mg (2.0 mmol) 2,5-Dimethylpyrazin in 1 mL Methanol unter Rühren bei RT erhalten. Nach drei Tagen wurde der weiß gefärbte, mikrokristalline Rückstand über einem Porzellanfiltertiegel (A2) abgesaugt und mit Diethylether gewaschen. Die Phasenreinheit wurde durch Röntgen-Pulverdiffraktometrie (Abb. 6.1) und Elementaranalyse überprüft. Ausbeute: 411.5 mg (84.2 %). Elementaranalyse für C6H8Cl2N2Zn (244.41) berechnet: C 29.48, H 3.30, N 11.46; gefunden: C 29.41, H 3.29, N 10.86. IR (KBr): = 3127 (w), 3040 (m), 2929 (w), 1827 (w), 1624 (w), 1508 (s), 1451 (m), 1381 (w), 1346 (s), 1273 (w), 1165 (m), 1078 (s), 1037 (m), 992 (w), 903 (w), 754 (w), 527 (w), 446 (m) cm-1. Einkristalle dieser Verbindung wurden durch Reaktion von 68.1 mg (0.5 mmol) ZnCl2 und 108.1 mg (1.0 mmol) 2,5Dimethylpyrazin in 2 mL Methanol unter solvothermalen Bedingungen bei 100 °C innerhalb von Relative Intensität einem Tag erhalten. 5 10 15 20 25 30 35 40 2 Theta / ° Abb. 6.1: Experimentelles (oben) und berechnetes (unten) Röntgen-Pulverdiffraktogramm der Verbindung [ZnCl2(2,5-Dimethylpyrazin)]n; Cu-Kα-Strahlung. (ZnCl2)2(2,5-Dimethylpyrazin)3: Kristallines Pulver dieser Verbindung wurde durch Reaktion von 136.3 mg ZnCl2 (1.0 mmol) in 648.9 mg (6.0 mmol) 2,5-Dimethylpyrazin ohne Lösungsmittel und ohne Rühren bei RT erhalten. Nach einer Woche wurde der weiß gefärbte, mikrokristalline Rückstand über einem Porzellanfiltertiegel (A2) abgesaugt und mit Diethylether gewaschen. Die Phasenreinheit wurde durch Röntgen-Pulverdiffraktometrie (Abb. 6.2) und Ele- 6. Experimenteller Teil - 73 - mentaranalyse überprüft. Elementaranalyse für C18H24Cl4N6Zn2 (596.97) berechnet: C 36.21, H 4.05, N 14.08; gefunden: C 36.50, H 4.00, N 14.13. IR (KBr): = 3105 (w), 3042 (m), 2954 (w), 2918 (w), 2859 (w), 1982 (w), 1830(w), 1635 (w), 1497 (s), 1445 (s), 1382 (m), 1335 (s), 1268 (w), 1233 (w), 1158 (s), 1077 (s), 1060 (s), 1039 (m), 979 (m), 894 (w), 751 (w), 453 (s), 425 (s) cm-1. Einkristalle dieser Verbindung, in Form von farblosen Blöcken, wurden durch Reaktion von 68.2 mg (0.5 mmol) ZnCl2 in 108.1 mg (1.0 mmol) 2,5-Dimethylpyrazin ohne Lö- Relative Intensität sungsmittel und Rühren bei RT innerhalb von drei Tagen erhalten. 5 10 15 20 25 30 35 40 2 Theta / ° Abb. 6.2: Experimentelles (oben) und berechnetes (unten) Röntgen-Pulverdiffraktogramm der Verbindung (ZnCl2)2(2,5-Dimethylpyrazin)3; Cu-Kα-Strahlung. [ZnBr2(2,5-Dimethylpyrazin)]n: Kristallines Pulver der Modifikation 3I wurde durch Reaktion von 450.4 mg ZnBr2 (2.0 mmol) und 216.3 mg (2.0 mmol) 2,5-Dimethylpyrazin in 1 mL Methanol unter Rühren bei RT erhalten. Nach drei Tagen wurde der weiß gefärbte, mikrokristalline Rückstand über einem Porzellanfiltertiegel (A2) abgesaugt und mit Diethylether gewaschen. Die Phasenreinheit wurde durch Röntgen-Pulverdiffraktometrie (Abb. 6.3: links) und Elementaranalyse überprüft. Ausbeute: 542.7 mg (81.4 %). Elementaranalyse für C6H8Br2N2Zn (333.33) berechnet: C 21.62, H 2.42, N 8.40; gefunden: C 21.59, H 2.32, N 7.78. IR (KBr): = 3124 (w), 3035 (m), 2926 (w), 1816 (w), 1615 (w), 1506 (s), 1445 (m), 1389 (m), 1343 (s), 1272 (m), 1164 (s), 1078 (s), 1035 (m), 987 (m), 897 (w), 753 (w), 525 (w), 425 (m) cm-1. Kristallines Pulver der Modifikation 3II wurde ohne Rühren wie 3I hergestellt. Nach Animpfen mit Einkristallen der Modifikation 3II und anschließendem Verdunsten des Lösungsmittels kristallisierte die Modifikation 3II phasenrein aus (Abb. 6.3: rechts). Elementaranalyse C6H8Br2N2Zn (333.33) berechnet: C 21.62, H 2.42, N 8.40; gefunden: C 21.35, H 2.20, N 8.59. IR (KBr): = 3126 (w), 3039 (m), 1818 (w), 1616 (w), 1500 (s), 1438 (m), 1384 (m), 1339 (s), 1270 (m), 1163 (s), 1080 (s), 1033 (m), 973 (w), 893 (w), 750 (w), 528 (w), 432 (s) cm-1. Einkristalle beider Verbindungen wurden durch Reaktion von 112.6 mg (0.5 mmol) ZnBr2 und 54.1 mg (0.5 mmol) 2,5-Dimethyl- 6. Experimenteller Teil - 74 - pyrazin in 2 mL Methanol ohne zu rühren im Rollrandglas bei RT innerhalb von drei Tagen erhalten. Hierbei kristallisierten beide polymorphe Modifikationen zusammen aus: Modifikation 3I 5 Relative Intensität Relative Intensität in Plättchen- und Modifikation 3II in Stäbchen-Form. 10 15 20 25 30 35 40 5 10 15 20 25 30 35 40 2 Theta / ° 2 Theta / ° Abb. 6.3: Experimentelles (links: oben) und berechnetes (links: unten) Röntgen-Pulverdiffraktogramm der Modifikation 3I sowie experimentelles (rechts: oben) und berechnetes (rechts: unten) Röntgen-Pulverdiffraktogramm der Modifikation 3II der Verbindung [ZnBr2(2,5-Dimethylpyrazin)]n; Cu-Kα-Strahlung. (ZnBr2)2(2,5-Dimethylpyrazin)3: Einkristalle dieser Verbindung in Form von farblosen Blöcken wurden durch Reaktion von 28.1 mg (0.125 mmol) ZnBr2 in 108.1 mg (1.0 mmol) 2,5Dimethylpyrazin ohne Lösungsmittel und Rühren bei RT innerhalb von drei Tagen erhalten. Kristallines Pulver konnte nicht phasenrein dargestellt werden. ZnBr2(2,5-Dimethylpyrazin)3: Kristallines Pulver und Einkristalle dieser Verbindung wurden durch Reaktion von 450.4 mg (2.0 mmol) ZnBr2 in 1297.7 mg (12.0 mmol) 2,5-Dimethylpyrazin ohne Lösungsmittel und ohne Rühren bei RT innerhalb von eienr Woche erhalten. Die Phasenreinheit wurde durch Röntgen-Pulverdiffraktometrie (Abb. 6.4) und Elementaranalyse überprüft. Elementaranalyse für C18H24Br2N6Zn (549.62) berechnet: C 39.34, H 4.40, N 15.29; gefunden: C 39.35, H 4.38, N 15.25. IR (KBr): = 3039 (w), 2921 (w), 2363 (w), 1819 (w), 1635 (w), 1499 (s), 1446 (m), 1383 (m), 1337 (s), 1270 (m), 1163 (s), 1081 (s), 1039 (m), 972 (w), 891 (w), 751 (w), 669 (w), 508 (w), 430 (m) cm-1. - 75 - Relative Intensität 6. Experimenteller Teil 5 10 15 20 25 30 35 40 2 Theta / ° Abb. 6.4: Experimentelles (oben) und berechnetes (unten) Röntgen-Pulverdiffraktogramm der Verbindung ZnBr2(2,5-Dimethylpyrazin)3; Cu-Kα-Strahlung. [ZnI2(2,5-Dimethylpyrazin)]n: Kristallines Pulver dieser Verbindung wurde durch Reaktion von 1276.7 mg ZnI2 (4.0 mmol) und 432.6 mg (4.0 mmol) 2,5-Dimethylpyrazin in 1 mL Methanol unter Rühren bei RT erhalten. Nach drei Tagen wurde der leicht gelb gefärbte, mikrokristalline Rückstand über einem Porzellanfiltertiegel (A2) abgesaugt und mit Diethylether gewaschen. Die Phasenreinheit wurde durch Röntgen-Pulverdiffraktometrie (Abb. 6.5) und Elementaranalyse überprüft. Ausbeute: 542.7 mg (67.9 %). Elementaranalyse für C6H8I2N2Zn (427.31) berechnet: C 16.86, H 1.89, N 6.56; gefunden: C 16.82, H 1.83, N 6.54. IR (KBr): = 3118 (w), 3031 (m), 1813 (w), 1497 (s), 1438 (m), 1378 (w), 1337 (m), 1268 (m), 1159 (m), 1077 (s), 1030 (m), 891 (w), 750 (w), 526 (w), 433 (m) cm-1. Einkristalle dieser Verbindung wurden durch Reaktion von 159.6 mg (0.5 mmol) ZnI2 und 54.1 mg (0.5 mmol) 2,5-Dimethylpyrazin in 2 mL Relative Intensität Methanol ohne zu rühren im Rollrandglas bei RT innerhalb von drei Tagen erhalten. 5 10 15 20 25 30 35 40 2 Theta / ° Abb. 6.5: Experimentelles (oben) und berechnetes (unten) Röntgen-Pulverdiffraktogramm der Verbindung [ZnI2(2,5-Dimethylpyrazin)]n; Cu-Kα-Strahlung. ZnI2(2,5-Dimethylpyrazin)2: Kristallines Pulver dieser Verbindung wurde durch Reaktion von 1276.7 mg ZnI2 (4.0 mmol) und 2595.4 mg (24.0 mmol) 2,5-Dimethylpyrazin in 1 mL Methanol 6. Experimenteller Teil - 76 - unter Rühren bei RT erhalten. Nach zwei Tagen wurde der leicht gelb gefärbte, mikrokristalline Rückstand über einem Porzellanfiltertiegel (A2) abgesaugt und mit Diethylether gewaschen. Die Phasenreinheit wurde durch Röntgen-Pulverdiffraktometrie und Elementaranalyse überprüft. Ausbeute: 542.7 mg (66.0 %). Elementaranalyse für C12H16I2N4Zn (535.46) berechnet: C 26.92, H 3.01, N 10.46; gefunden: C 27.04, H 3.04, N 10.90. IR (KBr): = 2911 (w), 1804 (w), 1628 (w), 1501 (s), 1436 (m), 1384 (m), 1333 (s), 1268 (w), 1239 (w), 1162 (m), 1062 (s), 1035 (m), 971 (w), 883 (w), 749 (w), 672 (w), 429 (s) cm-1. Einkristalle dieser Verbindung, in Form von farblosen Blöcken, wurden durch Reaktion von 75.8 mg (0.25 mmol) ZnCl2 in 108.1 mg Relative Intensität (1.0 mmol) 2,5-Dimethylpyrazin bei RT innerhalb von einem Tag erhalten. 5 10 15 20 25 30 35 40 2 Theta / ° Abb. 6.6: Experimentelles (oben) und berechnetes (unten) Röntgen-Pulverdiffraktogramm der Verbindung ZnI2(2,5-Dimethylpyrazin)2; Cu-Kα-Strahlung. 6.1.2 Verbindungen auf Basis von Übergangsmetall-Thiocyanaten und Pyrazin [Fe(SCN)2(Pyrazin)2]n:[151] Kristallines Pulver dieser Verbindung wurde durch Reaktion von 278.0 mg (1.0 mmol) FeSO4·7H2O, 194.4 mg (2.0 mmol) KSCN und 160.2 mg (2 mmol) Pyrazin in 5 mL Wasser unter Rühren bei RT erhalten. Nach drei Tagen wurde der rotbraun gefärbte, mikrokristalline Rückstand über einem Porzellanfiltertiegel (A2) abgesaugt und mit Ethanol und Diethylether gewaschen. Die Phasenreinheit wurde durch Röntgen-Pulverdiffraktometrie (Abb. 6.7) und Elementaranalyse überprüft. Ausbeute: 292.6 mg (88.1 %). Elementaranalyse für C10H8FeN6S2 (332.19) berechnet: C 36.16, H 2.43, N 25.30, S 19.31; gefunden: C 35.82, H 2.33, N 24.89, S 20.02. IR (KBr): = 3449 (m), 3132 (w), 3048 (w), 2359 (w), 2056 (s), 1636 (w), 1486 (w), 1416 (m), 1161 (w), 1112 (w), 1052 (m), 798 (w), 456 (m) cm-1. - 77 - Relative Intensität 6. Experimenteller Teil 10 15 20 25 30 35 40 45 50 2 Theta / ° Abb. 6.7: Experimentelles (oben) und berechnetes (unten) Röntgen-Pulverdiffraktogramm der Verbindung [Fe(SCN)2(Pyrazin)2]n; Cu-Kα-Strahlung. [Fe(SCN)2(Pyrazin)]n: Einkristalle dieser Verbindung, in Form von orangebraunen Stäbchen, wurden durch Reaktion von 139.0 mg (0.5 mmol) FeSO4·7H2O, 97.2 mg (1.0 mmol) KSCN und 10.0 mg (0.125 mmol) Pyrazin in 3 mL Wasser unter solvothermalen Bedingungen bei 120 °C innerhalb von vier Tagen erhalten. Kristallines Pulver konnte nicht phasenrein dargestellt werden. [Co(SCN)2(Pyrazin)2]n:[144] Kristallines Pulver dieser Verbindung wurde durch Reaktion von 237.9 mg (1.0 mmol) CoCl2·6H2O, 194.4 mg (2.0 mmol) KSCN und 160.2 mg (2 mmol) Pyrazin in 5 mL Wasser unter Rühren bei RT erhalten. Nach drei Tagen wurde der rosa gefärbte, mikrokristalline Rückstand über einem Porzellanfiltertiegel (A2) abgesaugt und mit Ethanol und Diethylether gewaschen. Die Phasenreinheit wurde durch Röntgen-Pulverdiffraktometrie (Abb. 6.8) und Elementaranalyse überprüft. Ausbeute: 289.1 mg (86.2 %). Elementaranalyse für C10H8CoN6S2 (335.28) berechnet: C 35.82, H 2.41, N 25.07, S 19.13; gefunden: C 35.53, H 2.33, N 24.92, S 19.18. IR (KBr): = 3444 (m), 3118 (w), 3053 (w), 2065 (s), 1633 (w), 1490 (w), Relative Intensität 1415 (m), 1163 (m), 1127 (m), 1055 (m), 969 (w), 800 (m), 465 (m) cm-1. 10 15 20 25 30 35 40 45 50 2 Theta / ° Abb. 6.8: Experimentelles (oben) und berechnetes (unten) Röntgen-Pulverdiffraktogramm der Verbindung [Co(SCN)2(Pyrazin)2]n; Cu-Kα-Strahlung. 6. Experimenteller Teil - 78 - [Co(SCN)2(Pyrazin)]n: Einkristalle dieser Verbindung, in Form von orangefarbenen Stäbchen, wurden durch Reaktion von 43.8 mg (0.25 mmol) Co(SCN)2 und 5.0 mg (0.0625 mmol) Pyrazin in 400 µL Ethanol unter solvothermalen Bedingungen bei 120 °C innerhalb von fünf Tagen erhalten. Kristallines Pulver konnte nicht phasenrein dargestellt werden. [Ni(SCN)2(Pyrazin)2]n: Kristallines Pulver dieser Verbindung wurde durch Reaktion von 237.7 mg (1.0 mmol) NiCl2·6H2O, 194.4 mg (2.0 mmol) KSCN und 160.2 mg (2 mmol) Pyrazin in 5 mL Wasser unter Rühren bei RT erhalten. Nach drei Tagen wurde der blau gefärbte, mikrokristalline Rückstand über einem Porzellanfiltertiegel (A2) abgesaugt und mit Ethanol und Diethylether gewaschen. Die Phasenreinheit wurde durch Röntgen-Pulverdiffraktometrie (Abb. 6.9) und Elementaranalyse überprüft. Ausbeute: 292.6 mg (88.1 %). Elementaranalyse für C10H8NiN6S2 (335.04) berechnet: C 35.85, H 2.41, N 25.08, S 19.14; gefunden: C 35.63, H 2.26, N 24.73, S 19.33. IR (KBr): = 3441 (m), 3123 (w), 3060 (w), 2083 (s), 1629 (w), 1491 (w), 1416 (m), 1165 (m), 1126 (m), 1058 (m), 974 (w), 803 (m), 476 (m) cm-1. Einkristalle dieser Verbindung, in Form von blauen Stäbchen, wurden durch Reaktion von 43.7 mg (0.25 mmol) Ni(SCN)2 und 40.1 mg (0.5 mmol) Pyrazin in 3 mL Wasser unter solvothermalen Bedingungen Relative Intensität bei 120 °C innerhalb von einem Tag erhalten. 10 15 20 25 30 35 40 45 50 2 Theta / ° Abb. 6.9: Experimentelles (oben) und berechnetes (unten) Röntgen-Pulverdiffraktogramm der Verbindung [Ni(SCN)2(Pyrazin)2]n; Cu-Kα-Strahlung. [Ni(SCN)2(Pyrazin)]n: Kristallines Pulver dieser Verbindung wurde durch Reaktion von 174.9 mg (1.0 mmol) Ni(SCN)2 und 80.1 mg (1 mmol) Pyrazin in 5 mL Wasser unter Rühren bei RT erhalten. Nach sieben Tagen wurde der grün gefärbte, mikrokristalline Rückstand über einem Porzellanfiltertiegel (A2) abgesaugt und mit Ethanol und Diethylether gewaschen. Die Phasenreinheit wurde durch Röntgen-Pulverdiffraktometrie und Elementaranalyse überprüft. Ausbeute: 292.6 mg (88.1 %). Elementaranalyse für C6H4N4NiS2 (254.95) berechnet: C 28.27, H 1.58, N 6. Experimenteller Teil - 79 - 21.98, S 25.16; gefunden: C 28.47, H 1.53, N 21.51, S 24.68. IR (KBr): = 3461 (m), 3094 (w), 2123 (s), 1632 (w), 1490 (w), 1419 (m), 1166 (w), 1124 (w), 1059 (m), 806 (m), 479 (m) cm-1. Einkristalle dieser Verbindung, in Form von grünen Stäbchen, wurden durch Reaktion von 43.7 mg (0.25 mmol) Ni(SCN)2 und 5.0 mg (0.0625 mmol) Pyrazin in 3 mL Methanol unter sol- Relative Intensität vothermalen Bedingungen bei 120 °C innerhalb von fünf Tagen erhalten. 10 15 20 25 30 35 40 45 50 2 Theta / ° Abb. 6.10: Experimentelles (oben) und berechnetes (unten) Röntgen-Pulverdiffraktogramm der Verbindung [Ni(SCN)2(Pyrazin)]n; Cu-Kα-Strahlung. 6.1.3 Verbindungen auf Basis von Übergangsmetall-Cyanaten und Pyrazin [Co(OCN)2(Pyrazin)2]n: Einkristalle dieser Verbindung, in Form von gelben Blöcken, wurden durch Reaktion von 145.5 mg (0.5 mmol) Co(NO3)2·6H2O, 81.1 mg (1.0 mmol) KOCN und 40.1 mg Pyrazin (0.5 mmol) in einem Gemisch aus 0.5 mL Wasser und 0.5 mL Methanol unter solvothermalen Bedingungen bei 120 °C innerhalb von drei Tagen erhalten. Kristallines Pulver konnte nicht phasenrein dargestellt werden. 6.1.4 Verbindungen auf Basis von Übergangsmetall-Thiocyanaten und Pyrimidin [Mn(SCN)2(Pyrimidin)2]n: Kristallines Pulver dieser Verbindung wurde durch Reaktion von 323.6 mg (2.0 mmol) MnCl2·2H2O, 388.8 mg (4 mmol) KSCN und 320.4 mg (4 mmol) Pyrimidin in 1.5 mL Wasser unter Rühren bei RT erhalten. Nach drei Tagen wurde der weiß gefärbte, mikrokristalline Rückstand über einem Porzellanfiltertiegel (A2) abgesaugt und mit Ethanol und Diethylether gewaschen. Die Phasenreinheit wurde durch Röntgen-Pulverdiffraktometrie und Elementaranalyse überprüft. Ausbeute: 292.6 mg (88.1 %). Elementaranalyse für C10H8MnN6S2 6. Experimenteller Teil - 80 - (331.28) berechnet: C 36.26, H 2.43, N 25.37, S 19.36; gefunden: C 35.95, H 2.29, N 24.73, S 19.46. Einkristalle dieser Verbindung, in Form von farblosen Stäbchen, wurden durch Reaktion von 40.5 mg (0.25 mmol) MnCl2·2H2O, 48.6 mg (0.5 mmol) KSCN und 20.0 mg (0.25 mmol) Pyrimidin in 250 µL Wasser unter solvothermalen Bedingungen bei 120 °C innerhalb von drei Relative Intensität Tagen erhalten. 5 10 15 20 25 30 35 40 45 2 Theta / ° Abb. 6.11: Experimentelles (oben) und berechnetes (unten) Röntgen-Pulverdiffraktogramm der Verbindung [Mn(SCN)2(Pyrimidin)2]n; Cu-Kα-Strahlung. Co(SCN)2(Pyrimidin)4: Einkristalle dieser Verbindung, in Form von rosafarbenen Blöcken, wurden durch Reaktion von 43.8 mg (0.25 mmol) Co(SCN)2 in 120.2 mg (1.5 mmol) Pyrimidin ohne Lösungsmittel und Rühren bei RT innerhalb von drei Tagen erhalten. Kristallines Pulver konnte nicht phasenrein dargestellt werden. Ni(SCN)2(Pyrimidin)4: Einkristalle dieser Verbindung, in Form von blauen Blöcken, wurden durch Reaktion von 43.7 mg (0.25 mmol) Ni(SCN)2 in 120.2 mg (1.5 mmol) Pyrimidin ohne Lösungsmittel und Rühren bei RT innerhalb von drei Tagen erhalten. Kristallines Pulver konnte nicht phasenrein dargestellt werden. 6.1.5 Verbindungen auf Basis von Zink(II)halogeniden und 2,2'-Dipyridylsulfid ZnCl2(2,2'-Dipyridylsulfid): Einkristalle dieser Verbindung wurden durch Reaktion von 4.3 mg (0.031 mmol) ZnCl2 und 27.5 mg (0.125 mmol) 2,2'-Dipyridyldisulfid in 3 mL Methanol unter solvothermalen Bedingungen bei 110 °C innerhalb von drei Tagen erhalten. Kristallines Pulver konnte nicht phasenrein dargestellt werden. 6. Experimenteller Teil - 81 - ZnBr2(2,2'-Dipyridylsulfid): Einkristalle dieser Verbindung wurden durch Reaktion von 7.0 mg (0.031 mmol) ZnBr2 und 27.5 mg (0.125 mmol) 2,2'-Dipyridyldisulfid in 3 mL Methanol unter solvothermalen Bedingungen bei 110 °C innerhalb von drei Tagen erhalten. Kristallines Pulver konnte nicht phasenrein dargestellt werden. ZnI2(2,2'-Dipyridylsulfid): Einkristalle dieser Verbindung wurden durch Reaktion von 39.9 mg (0.125 mmol) ZnI2 und 27.5 mg (0.125 mmol) 2,2'-Dipyridyldisulfid in 3 mL Methanol unter solvothermalen Bedingungen bei 110 °C innerhalb von vier Tagen erhalten. Kristallines Pulver konnte nicht phasenrein dargestellt werden. 6.1.6 Verbindungen auf Basis von Zink(II)halogeniden und 2,2'-Dipyridyldisulfid ZnBr2(2,2'-Dipyridyldisulfid): Einkristalle dieser Verbindung wurden durch Reaktion von 28.2 mg (0.125 mmol) ZnBr2 und 27.5 mg (0.125 mmol) 2,2'-Dipyridyldisulfid in 3 mL Methanol unter solvothermalen Bedingungen bei 110 °C innerhalb von vier Tagen erhalten. Kristallines Pulver konnte nicht phasenrein dargestellt werden. 6.1.7 Verbindungen auf Basis von Kupfer(I)halogeniden und 4-Pyridiniumthiolat Cu6Cl6S4(4-Pyridiniumthiolat)4: Einkristalle dieser Verbindung wurden durch Reaktion von 12.4 mg (0.125 mmol) CuCl und 27.5 mg (0.125 mmol) 4,4'-Dipyridyldisulfid in 1 mL Ethanol unter solvothermalen Bedingungen bei 120 °C innerhalb von drei Tagen erhalten. Kristallines Pulver konnte nicht phasenrein dargestellt werden. Cu6Br6S4(4-Pyridiniumthiolat)4: Einkristalle dieser Verbindung wurden durch Reaktion von 17.0 mg (0.125 mmol) CuBr und 27.5 mg (0.125 mmol) 4,4'-Dipyridyldisulfid in 1 mL Ethanol unter solvothermalen Bedingungen bei 120 °C innerhalb von drei Tagen erhalten. Kristallines Pulver konnte nicht phasenrein dargestellt werden. 6. Experimenteller Teil 6.2 - 82 - Methoden Die im Rahmen dieser Diplomarbeit erarbeiteten Ergebnisse wären ohne die Zuhilfenahme moderner festkörperanalytischer Methoden nicht zustande gekommen. Zum besseren Verständnis werden deshalb nachfolgend die wichtigsten Grundlagen dieser Methoden vorgestellt. 6.2.1 Röntgen-Einkristallstrukturanalyse Die Röntgen-Einkristallstrukturanalyse zählt zu den wichtigsten Methoden der Strukturaufklärung kristalliner Verbindungen. Die exakte räumliche Anordnung aller Bausteine, z.B. Atome, Ionen oder Moleküle, in kristallinen chemischen Verbindungen wird hierbei mit Hilfe von Röntgenstrahlen bestimmt. Durch die Strukturbestimmung können überdies Pulverdiffraktogramme berechnet werden, die zur Identifizierung und Reinheitsbestimmung kristalliner Verbindungen unabdingbar sind. Die Durchführung einer Röntgen-Einkristallstrukturanalyse besteht aus mehreren, aufeinanderfolgenden Schritten: ¾ Kristallauswahl ¾ vorbereitende Messung mit anschließender Indizierung der Beugungsreflexe ¾ Bestimmung des vorläufigen Kristallsystems, des Bravais-Typs sowie der Elementarzellabmessungen ¾ Datensammlung durch Intensitätsmessungen der Beugungsreflexe ¾ Bestimmung der Raumgruppe und Lauesymmetrie ¾ Strukturlösung durch Bestimmung der Atomlagen in der Elementarzelle ¾ Strukturverfeinerung ¾ Auswertung: Gütefaktoren, Abstände, Winkel Nach Auswahl eines geeigneten Einkristalls unter dem Mikroskop erfolgt die Messung mit einem Diffraktometer. Grundlage hierfür bildet die Röntgenbeugung, die vereinfacht als Reflektion an sogenannten Netzebenen des Kristalls aufgefasst werden kann. Die Netzebenen bezeichnen hierbei solche Ebenen, die durch Gitterpunkte im Raumgitter des Kristalls verlaufen. Die Beugungsbedingung wird durch die Bragg'sche Gleichung beschrieben (Abb. 6.12). 6. Experimenteller Teil - 83 - Abb. 6.12: Beugung von Röntgenstrahlung an Netzebenen gemäß der Bragg'schen Gleichung. Nur wenn diese Gleichung erfüllt wird, kann mit einem Beugungsmaximum gerechnet werden. Im ersten Schritt der Messung wird nach Reflexen gesucht, die anschließend indiziert werden, um das vorläufige Kristallsystem, den Bravais-Typ sowie die Elementarzellparameter zu bestimmen. Je nach Art der auftretenden Symmetrieelemente können alle Kristalle insgesamt sieben Kristallsystemen zugeordnet werden, die selbst sieben Formen von primitiven Elementarzellen entsprechen. Zusätzlich gibt es sieben verschiedene Typen zentrierter Elementarzellen, was zusammen mit den sieben primitiven Elementarzellen, insgesamt vierzehn verschiedene Formen von Elementarzellen, die sogenannten Bravais-Typen, ergibt. Nach Abschluss der vorbereitenden Messungen und Messung der Intensitäten aller notwendigen Reflexe, werden zunächst die Laue-Symmetrie und die Raumgruppe anhand der Analyse von beobachteten systematischen Auslöschungen bestimmt. Diese Auslöschungen werden von Translations-Symmetrieelementen wie z.B. Schraubenachsen, Gleitspiegelebenen oder Zentrierungen verursacht. In der anschließenden Strukturlösung werden den gemessenen Reflexen Phasen zugeordnet, woraus die Berechnung einer Elektronendichtekarte erfolgt. Den dabei erhaltenen Elektronendichtemaxima werden schrittweise Atome zugeordnet und so ein vorläufiges Strukturmodell erstellt, welches in der nachfolgenden Strukturverfeinerung noch optimiert wird. Die Güte der Verfeinerung wird letztendlich durch mehrere Gütefaktoren angegeben. Das weitaus wichtigste Kriterium ist jedoch, dass das erhaltene Strukturmodell chemisch sinnvoll ist und mit den Erwartungen des Experimentators weitgehend übereinstimmt. Ist ein zufriedenstellendes Strukturmodell gefunden worden, erfolgt die Auswertung der gewonnenen Daten. Bindungslängen und Winkel werden berechnet und geeignete Abbildungen erstellt. 6. Experimenteller Teil - 84 - Verwendetes Gerät und Analyse-Software: Alle Einkristalle wurden auf einem "STOE IPDS-1" der Firma STOE & Cie, Darmstadt, Deutschland, gemessen. Zur Kühlung während der Messung wurde die Tieftemperatureinrichtung "Cryosteam Cooler" der Firma Oxford-Instruments, Oxon, England, verwendet. Die Auswertung erfolgte mit dem Programmpaket "STOE" Ver. 2.89. Zur Analyse der gesamten Daten wurde das Programmpaket "Shelxtl" Ver. 5.1 der Firma Bruker AXS, Karlsruhe, Deutschland, verwendet. 6.2.2 Röntgen-Pulverdiffraktometrie Die Röntgen-Pulverdiffraktometrie zählt zu den wichtigsten destruktiven analytischen Methoden in der Festkörperchemie, die zur Identifizierung und Reinheitsbestimmung kristalliner Verbindungen eingesetzt wird. Neben einer quantitativen Phasenanalyse können mit dieser Methode auch Informationen über Teilchengröße und Kristallinität erlangt werden. Unter bestimmten Voraussetzungen können sogar vollständige Strukturbestimmungen durchgeführt werden. Bei der Röntgen-Pulverdiffraktometrie wird die Intensität der Beugungsreflexe als Funktion des Beugungswinkels (2-Theta) gemessen. Es resultiert ein Pulverdiffraktogramm, das durch folgende Parameter charakterisiert ist: ¾ Die Reflexlage charakterisiert die Abstände der Netzebenen im Kristall, aus denen die Gitterparameter berechnet werden können. ¾ Die Reflexintensität hängt von der Art und Anordnung der Atome in der Elementarzelle ab, wobei Abweichungen in der Intensität aufgrund einer Vorzugsorientierung (Textur) der Kristallite auftreten können. ¾ Die Reflexbreite und die Reflexform enthalten Informationen über die Teilchengröße sowie "Stress" und "Strain" der Pulverpartikel. Die Identifizierung unbekannter kristalliner Pulver kann durch Vergleich mit bekannten Pulverdiffraktogrammen erfolgen. Eine Vielzahl davon findet sich in der PDF-Datenbank (Powder Diffraction File). Sollten keine passenden Diffraktogramme vorliegen, können diese aus Einkristallstrukturdaten berechnet werden. Diese können z.B. aus Datenbanken wie der "Cambridge Structural Database" (CSD) für organische oder der "Inorganic Crystal Structure Database" (ICSD) für rein anorganische Verbindungen entnommen werden. 6. Experimenteller Teil - 85 - Verwendetes Gerät und Analyse-Software: Alle Pulverdiffraktogramme wurden auf einem "STOE STADI-P-Gerät" der Firma STOE & Cie, Darmstadt, Deutschland, gemessen. Als Steuer- und Analysesoftware wurde das Programmpaket "STOE WinXPOW" Ver. 2.11, der Firma STOE & Cie, Darmstadt, Deutschland, verwendet. 6.2.3 Raster-Elektronenmikroskopie und Röntgenfluoreszenzanalyse Die Raster-Elektronenmikroskopie (REM) ist eine destruktive Methode, um die Morphologie und Topographie kleiner Partikel, wie z.B. Kristalle, zu untersuchen. Die Untersuchung erfolgt durch das Abrastern einer Probe mit einem Elektronenstrahl im Hochvakuum oder, bei vakuumempfindlichen Verbindungen, bei annäherndem Normaldruck im ESEM-Modus. Durch die Wechselwirkung der Elektronen des Elektronenstrahls mit der Probe wird eine Abbildung der Objektoberfläche mit einer hohen Schärfentiefe erzeugt. Der maximale theoretische Vergrößerungsfaktor liegt bei ca. 1 : 1.000.000, was einer Auflösung von bis zu 2 Å entspricht. Zur qualitativen und halbquantitativen Charakterisierung der Elementzusammensetzung kleinster Probenbereiche ist das REM häufig mit einem EDX-Gerät gekoppelt (EDX = Energiedispersive Röntgenfluoreszenz). Dieses analysiert die charakteristische Röntgenstrahlung, die entsteht, wenn ein Elektron des Elektronenstrahls im Atom der Probe ein kernnahes Elektron aus seiner Position schlägt. Diese Lücke wird sofort von einem energiereicheren Elektron aus einem höheren Orbital aufgefüllt. Die dabei freiwerdende Energie wird in Form eines Röntgenquants frei (Röntgenfluoreszenz). Die dadurch entstehende Röntgenstrahlung ist charakteristisch für den Übergang und somit für das Element. Es resultiert ein Spektrum, in dem die Intensität der Röntgenquanten als Funktion der Energie dargestellt ist, so dass man qualitative und quantitative Informationen über die in der Probe vorliegenden Elemente erhält. Nachteilig ist, dass der Elektronenstrahl nicht sehr tief in die Probe eindringt und somit lediglich Informationen über bestimmte Oberflächenbereiche erhalten werden. Des Weiteren ist eine quantitative Analyse sehr leichter Elemente, wie z.B. Kohlenstoff oder Stickstoff, extrem ungenau. Im Rahmen der vorliegenden Diplomarbeit wurde diese Methode daher nur zur Überprüfung der Elementarzusammensetzung von Einkristallen angewandt, bevor diese am Einkristalldiffraktometer gemessen wurden. Verwendete Geräte und Analyse-Software: Für die EDX- und die elektronenmikroskopischen Aufnahmen wurde ein PHILIPS EDAX/XL30-ESEM-Gerät der Firma Royal Philips Electronics, Redhill, England, benutzt. 6. Experimenteller Teil 6.2.4 - 86 - Thermogravimetrie gekoppelt mit der Massenspektroskopie und Differenzthermoanalyse Abb. 5.6.13: Schematische Abbildung einer Thermogravimetrie-Apparatur gekoppelt mit der Massenspektroskopie (links) und Differenzthermoanalyse (rechts). Die Thermogravimetrie (TG) ist eine Methode zur Messung der Gewichtsänderung einer Probe während eines vorgegebenen Temperatur-Zeit-Programms in einer möglichst definierten Atmosphäre (Abb. 5.6.13: links). Derartige thermogravimetrische Untersuchungen sind nur dann möglich, wenn die Probe Materie mit der Umgebung austauscht, wie dies z.B. bei der thermischen Zersetzung von Solvaten der Fall ist. Bei jedem Masseverlust erscheint eine Massestufe, die auf eine Probenreaktion hindeutet. Durch den Vergleich experimenteller Masseverluste mit dem für die entsprechende Reaktion berechneten Masseverlust können erste Hinweise auf die Reaktion gewonnen werden. Zudem kann zur besseren Darstellung die differenzierte thermogravimetrische Kurve (DTG-Kurve) berechnet werden, die zur besseren Ermittlung des Prozessanfangs bzw. -endes der Reaktion dient. Da aus einer TG-Messung nur der Masseverlust bestimmt wird, werden keine weiteren Informationen über die Art der abgespaltenen Produkte erhalten. Hierfür kann die Thermowaage mit einem Massenspektrometer (MS) gekoppelt werden (Abb. 5.6.13: links). Dabei kann zwischen zwei verschiedenen Messmethoden unterschieden werden: ¾ Analog-Scan: Bei einem Analog-Scan werden alle Massen innerhalb eines bestimmten Bereichs als Funktion der Zeit oder Temperatur detektiert. Dabei erhält man Informationen über die bei der thermischen Zersetzung freiwerdenden Stoffe. ¾ MID-Scan (Multiple Ion Detection): Sind die bei einer Reaktion freiwerdenden Stoffe bekannt, wird ein sogenannter MID-Scan oder MS-Trend-Scan durchgeführt. Hier werden definierte Massen als Funktion der Zeit oder Temperatur detektiert. 6. Experimenteller Teil - 87 - Durch die Thermogravimetrie können, wie bereits beschrieben, nur Reaktionen untersucht werden, in denen die Probe Materie mit der Umgebung austauscht, jedoch können keinerlei Informationen über die Richtung des Wärmeaustausches ermittelt werden. Um zu überprüfen, ob es sich bei der thermischen Zersetzung um exo- oder endotherme Prozesse handelt, wird die Thermogravimetrie daher häufig mit der Differenzthermoanalyse (DTA) kombiniert (Abb. 5.6.13: rechts). Dies ist eine Methode, bei der die Temperaturdifferenz zwischen einer Probe und einer Vergleichsprobe gemessen wird, während die Probe einem vorgegebenen Temperatur-Zeit-Programm unterworfen wird. Wie bei allen o.g. thermischen Analysemethoden, haben bei der Messung vielerlei Faktoren Einfluss auf den Reaktionsablauf: ¾ Temperatur-Zeit-Programm ¾ Probeneigenschaften: Struktur der Probensubstanz ¾ Atmosphäre im Probenraum: Gasdruck und Gasart ¾ Wärmeleitung: Temperaturinhomogenitäten in der Probe ¾ Auftriebs- und Strömungseffekte Verwendete Geräte und Analysesoftware: Alle DTA-TG-MS-Messungen wurden auf einer Thermowaage "STA-409CD" der Firma Netzsch, Selb, Deutschland, durchgeführt, an das ein Quadrupol-Massenspektrometer "QMA400" der Firma Balzers über eine Skimmerkopplung angekoppelt ist. Als Analysesoftware für TG-DTA-Auswertungen wurde das Programm "Proteus", Ver. 4.8.0, der Firma Netzsch, Selb, Deutschland, und für MS-Auswertungen das Programm "Quadstar 422" Ver. 6.03, der Firma Balzers, verwendet. 6. Experimenteller Teil 6.2.5 - 88 - Dynamische Differenzkalorimetrie Abb. 5.6.14: Schematische Abbildung eines Wärmestrom-Differenzkalorimeters (links) und eines Leistungskompensations-Kalorimeters (rechts). Die Dynamische Differenzkalorimetrie (DDK) ist eine Methode zur Messung auftretender Wärmestromdifferenzen zwischen einer Probe und einer Vergleichsprobe, während diese einem vorgegebenen Temperatur-Zeit-Programm unterworfen werden. Im Gegensatz zur DTA besteht die Möglichkeit, die Reaktions-, die Umwandlungs-, die Zersetzungs-, bzw. die Schmelzenthalpie zu bestimmen. Generell werden zwei verschiedene Messmethoden unterschieden: ¾ Bei der Wärmestrom-Differenzkalorimetrie ist das eigentliche Messsignal, wie bei der DTA, eine Temperaturdifferenz, die sich jedoch proportional zum übertragenen Wärmestrom verhält (Abb. 5.6.14: links). ¾ Bei der Leistungskompensations-Kalorimetrie werden die Probe sowie die Referenzprobe mit unterschiedlichen Öfen beheizt, um die Temperatur beim Auftreten eines exo- oder endothermen Vorgangs konstant zu halten. Der zum Beheizen notwendige Strom wird gemessen, so dass es sich um eine echte Leistungsmessung handelt (Abb. 5.6.14: rechts). Verwendete Geräte und Analysesoftware: Alle DDK-Messungen wurden mit einem Wärmestrom-Differenzkalorimeter "DSC-204" der Firma Netzsch, Selb, Deutschland, durchgeführt. Als Analysesoftware wurde das Programm "Proteus", Ver. 4.8.0, der Fa. Netzsch, Selb, Deutschland, verwendet. 6. Experimenteller Teil 6.2.6 - 89 - Weitere verwendete Geräte und Software ¾ Infrarotuntersuchungen: ATI Mattson Genesis Series FTIR Spektrometer, Steuersoftware: "Winfirst", der Firma ATI Mattson. ¾ Magnetische Untersuchungen: PPMS der Firma Quantum Design, San Diego, USA. ¾ Textverarbeitung: Microsoft Word 2007, Ver. 12 SP1, Microsoft, Redmond, USA. ¾ Tabellenkalkulation: Microsoft Excel 2007, Ver. 12 SP1, Microsoft, Redmond, USA. ¾ Grafikerstellung: Microcal Origin, Ver. 7.5G, Microcal OriginLab, Northampton, USA. ¾ Vektorgrafik: CorelDRAW X3, Ver. 13, Corel Corporation, San Francisco, USA. ¾ Strukturformeln: ChemDraw Ultra, Ver. 10, CambridgeSoft, Cambridge, USA. ¾ Kristallstrukturen: Diamond, Ver. 2.1e, Crystal Impact, Bonn, Deutschland. 6. Experimenteller Teil 6.3 - 90 - Verwendete Chemikalien Verbindung Hersteller Reinheit / % 4,4'-Bipyridyldisulfid Alfa Aesar >98 2,2'-Bipyridyldisulfid Alfa Aesar >98 Pyrazin Alfa Aesar >98 Pyrimidin Alfa Aesar >99 2,5-Dimethylpyrazin Alfa Aesar >99 KOCN Merck >98 KSCN Merck >99 CuCl Fluka >97 CuBr Riedel-de Haën >97 ZnCl2 Alfa Aesar >99.99 ZnBr2 Alfa Aesar >99.9 ZnI2 Alfa Aesar >98 Co(NO3)·6H2O Merck >99 CoCl2·6H2O Merck >97 NiCl2·6H2O Merck >97 MnCl2·2H2O Merck >97 Ni(SCN)2 Alfa Aesar >98 Co(SCN)2 Alfa Aesar >98 6. Experimenteller Teil 6.4 - 91 - Einkristallstrukturdaten [ZnCl2(2,5-Dimethylpyrazin)]n Table 1. Crystal data and structure refinement for [ZnCl2(2,5-dimethylpyrazine)]n. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C6H8Cl2N2Zn 244.41 170(2) K 0.71073 Å orthorhombic Pnma a = 6.0621(4) Å α= 90°. b = 10.2156(8) Å β= 90°. c = 13.9518(8) Å γ = 90°. 864.01(10) Å3 4 1.879 Mg/m3 3.393 mm-1 488 0.22 x 0.04 x 0.16 mm3 2.47 to 28.01°. -8<=h<=7, -13<=k<=13, -18<=l<=18 9309 1092 [R(int) = 0.0350] 99.5 % Full-matrix least-squares on F2 1092 / 0 / 57 1.072 R1 = 0.0241, wR2 = 0.0641 R1 = 0.0258, wR2 = 0.0648 0.017(2) 0.544 and -0.562 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 28.01° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were allowed to rotate but not to tip. A numerical absorption correction was performed min/max transmission: (0.521/0.869). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Zn(1) Cl(1) Cl(2) N(1) C(1) C(4) C(5) x 6081(1) 3210(1) 5170(1) 8362(2) 8862(2) 9500(3) 7707(3) y 2500 2500 2500 4042(1) 4729(2) 4313(2) 4439(2) z 5100(1) 4120(1) 6649(1) 4977(1) 4177(1) 5779(1) 3259(1) U(eq) 9(1) 17(1) 18(1) 10(1) 10(1) 11(1) 17(1) 6. Experimenteller Teil - 92 - (ZnCl2)2(2,5-Dimethylpyrazin)3 Table 1. Crystal data and structure refinement for (ZnCl2)2(2,5-dimethylpyrazine)3. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C18H24Cl4N6Zn2 596.97 170(2) K 0.71073 Å triclinic P-1 a = 7.2149(10) Å α= 79.973(15)°. b = 9.1905(11) Å β= 70.138(15)°. c = 10.1410(13) Å γ = 70.264(15)°. 593.94(13) Å3 1 1.669 Mg/m3 2.487 mm-1 302 0.12 x 0.07 x 0.09 mm3 2.36 to 27.97°. -9<=h<=9, -11<=k<=11, -13<=l<=13 5213 2740 [R(int) = 0.0529] 95.6 % Full-matrix least-squares on F2 2740 / 0 / 140 1.074 R1 = 0.0496, wR2 = 0.1277 R1 = 0.0719, wR2 = 0.1369 0.038(5) 0.890 and -1.195 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 27.97° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were allowed to rotate but not to tip. A numerical absorption correction was performed min/max transmission: (0.75362/0.831). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Zn(1) Cl(1) Cl(2) N(1) N(2) C(1) C(2) C(3) C(4) C(5) C(6) N(11) C(11) C(14) C(15) x 8244(1) 11364(2) 7591(2) 7618(6) 7307(7) 7681(8) 7548(8) 7192(7) 7374(8) 7844(10) 6829(9) 6220(6) 4150(7) 7055(7) 3189(8) y 8535(1) 7598(1) 10576(1) 6801(5) 4492(5) 5391(6) 4265(6) 5907(6) 7050(6) 5128(7) 6229(7) 9250(5) 9742(6) 9512(6) 9379(6) z 2076(1) 596(1) 3265(1) 3625(4) 5775(5) 3358(5) 4452(6) 6041(5) 4942(5) 1910(6) 7512(5) 861(4) 1409(5) -529(5) 2935(5) U(eq) 15(1) 20(1) 19(1) 15(1) 24(1) 19(1) 24(1) 19(1) 18(1) 29(1) 26(1) 15(1) 14(1) 16(1) 20(1) 6. Experimenteller Teil - 93 - [ZnBr2(2,5-Dimethylpyrazin)]n – Modifikation I Table 1. Crystal data and structure refinement for [ZnBr2(2,5-dimethylpyrazine)]n. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C6H8Br2N2Zn 333.33 170(2) K 0.71073 Å orthorhombic Pnma a = 6.2698(6) Å α= 90°. b = 10.3167(7) Å β= 90°. c = 14.3842(9) Å γ = 90°. 930.42(12) Å3 4 2.380 Mg/m3 11.166 mm-1 632 0.11 x 0.03 x 0.08 mm3 2.43 to 28.08°. -8<=h<=8, -13<=k<=13, -18<=l<=16 7814 1164 [R(int) = 0.1052] 97.3 % Full-matrix least-squares on F2 1164 / 0 / 57 1.063 R1 = 0.0431, wR2 = 0.1078 R1 = 0.0524, wR2 = 0.1145 0.0073(13) 1.006 and -1.795 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 28.08° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were allowed to rotate but not to tip. A numerical absorption correction was performed min/max transmission: (0.361/0.722). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Br(1) Br(2) Zn(1) N(1) C(4) C(1) C(5) x 6711(1) 4896(1) 3771(1) 1564(6) 527(7) 1027(7) 2034(9) y 2500 2500 2500 4038(4) 4323(4) 4720(4) 4408(5) z 5957(1) 3369(1) 4943(1) 5041(3) 4257(3) 5809(3) 6726(3) U(eq) 20(1) 22(1) 12(1) 13(1) 14(1) 13(1) 22(1) 6. Experimenteller Teil - 94 - [ZnBr2(2,5-Dimethylpyrazin)]n – Modifikation II Table 1. Crystal data and structure refinement for [ZnBr2(2,5-dimethylpyrazine)]n. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C6H8Br2N2Zn 333.33 170(2) K 0.71073 Å monoclinic C2/c a = 16.1453(18) Å α= 90°. b = 6.4913(6) Å β= 115.096(11)°. c = 9.8755(10) Å γ = 90°. 937.29(17) Å3 4 2.362 Mg/m3 11.084 mm-1 632 0.19 x 0.06 x 0.11 mm3 2.79 to 28.04°. -21<=h<=21, -8<=k<=8, -13<=l<=13 4836 1134 [R(int) = 0.0378] 99.5 % Full-matrix least-squares on F2 1134 / 0 / 53 0.999 R1 = 0.0271, wR2 = 0.0672 R1 = 0.0343, wR2 = 0.0707 0.0037(4) 0.840 and -0.755 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 28.04° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were allowed to rotate but not to tip. A numerical absorption correction was performed min/max transmission: (0.249/0.521). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Zn(1) Br(1) N(1) C(1) C(4) C(5) x 5000 6293(1) 4927(2) 4192(2) 5718(2) 3296(2) y 3822(1) 5842(1) 1585(4) 418(4) 1167(4) 835(5) z 2500 3912(1) 4011(2) 3796(3) 5198(3) 2517(3) U(eq) 12(1) 22(1) 13(1) 13(1) 13(1) 18(1) 6. Experimenteller Teil - 95 - (ZnBr2)2(2,5-Dimethylpyrazin)3 Table 1. Crystal data and structure refinement for (ZnBr2)2(2,5-dimethylpyrazine)3. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C18H24Br4N6Zn2 774.81 170(2) K 0.71073 Å triclinic P-1 a = 7.2840(8) Å b = 9.4775(16) Å c = 10.1762(11) Å 633.21(14) Å3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 28.02° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole α= 81.004(13)°. β= 72.489(12)°. γ = 71.306(12)°. 1 2.032 Mg/m3 8.222 mm-1 374 0.13 x 0.09 x0.06 mm3 2.27 to 28.02°. -9<=h<=9, -12<=k<=12, -13<=l<=13 5880 2991 [R(int) = 0.0548] 97.7 % Full-matrix least-squares on F2 2991 / 0 / 140 1.078 R1 = 0.0525, wR2 = 0.1449 R1 = 0.0655, wR2 = 0.1526 0.020(3) 1.706 and -1.335 e.Å-3 Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were allowed to rotate but not to tip. A numerical absorption correction was performed min/max transmission: (0.403/0.621). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Zn(1) Br(1) Br(2) N(1) N(2) C(1) C(2) C(3) C(4) C(5) C(6) N(11) C(11) C(14) C(15) x 8228(1) 11435(1) 7633(1) 7599(8) 7326(10) 7560(10) 7461(12) 7312(10) 7489(10) 7577(13) 7085(13) 6221(8) 4221(10) 6996(9) 3336(10) y 8532(1) 7518(1) 10669(1) 6887(6) 4663(7) 5542(7) 4433(8) 6015(8) 7114(7) 5279(9) 6319(8) 9247(6) 9718(7) 9531(7) 9346(8) z 2088(1) 611(1) 3285(1) 3618(6) 5752(7) 3367(7) 4454(8) 5993(7) 4916(7) 1944(8) 7441(7) 860(5) 1371(6) -505(6) 2873(7) U(eq) 12(1) 18(1) 16(1) 14(1) 25(1) 18(1) 26(2) 18(1) 16(1) 27(2) 27(2) 12(1) 14(1) 13(1) 19(1) 6. Experimenteller Teil - 96 - [ZnBr2(2,5-Dimethylpyrazin)]2·2,5-Dimethylpyrazin Table 1. Crystal data and structure refinement for [ZnBr2(2,5-dimethylpyrazine)2]·2,5-dimethylpyrazine. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C18H24Br2N6Zn 549.62 170(2) K 0.71073 Å monoclinic C2/c a = 8.5956(9) Å α= 90°. b = 11.4237(7) Å β= 100.174(10)°. c = 23.1531(16) Å γ = 90°. 2237.7(3) Å3 4 1.631 Mg/m3 4.683 mm-1 1096 0.13 x 0.10 x 0.06 mm3 3.00 to 26.93°. -10<=h<=10, -13<=k<=14, -28<=l<=28 7691 2368 [R(int) = 0.0622] 98.3 % Full-matrix least-squares on F2 2368 / 2 / 137 0.995 R1 = 0.0425, wR2 = 0.0992 R1 = 0.0707, wR2 = 0.1122 0.0026(5) 0.483 and -0.699 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 26.93° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were allowed to rotate but not to tip. A numerical absorption correction was performed min/max transmission: (0.571/0.751). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Zn(1) Br(1) Br(1') N(1) N(2) C(1) C(2) C(3) C(4) C(5) C(6) N(12) C(11) C(12) C(15) x 0 2370(8) 2026(15) 1395(4) 641(3) 2368(4) 1974(4) -334(4) 87(4) 3857(5) -1797(4) 1140(3) -1309(4) -169(4) -2768(4) y 9201(1) 10176(6) 10431(12) 7277(3) 8222(2) 8040(3) 8501(3) 7436(3) 6990(3) 8370(4) 7067(4) 10690(3) 9636(3) 10314(3) 9241(4) z 2500 2947(1) 2934(2) 803(1) 1829(1) 1129(2) 1635(2) 1512(1) 1005(1) 913(2) 1719(2) 351(1) 192(1) 533(2) 412(2) U(eq) 42(1) 79(1) 92(2) 31(1) 24(1) 30(1) 30(1) 23(1) 28(1) 48(1) 36(1) 29(1) 24(1) 27(1) 32(1) 6. Experimenteller Teil - 97 - [ZnI2(2,5-Dimethylpyrazin)]n Table 1. Crystal data and structure refinement for [ZnI2(2,5-dimethylpyrazine)]n. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C6H8I2N2Zn 427.31 170(2) K 0.71073 Å monoclinic C2/c a = 16.6286(13) Å b = 6.8351(4) Å c = 10.0199(8) Å 1040.81(13) Å3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 27.99° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole α= 90°. β= 113.947(9)°. γ = 90°. 4 2.727 Mg/m3 8.237 mm-1 776 0.13 x 0.03 x 0.09 mm3 2.68 to 27.99°. -21<=h<=21, -8<=k<=9, -13<=l<=13 5219 1240 [R(int) = 0.0300] 99.3 % Full-matrix least-squares on F2 1240 / 0 / 53 1.051 R1 = 0.0208, wR2 = 0.0513 R1 = 0.0249, wR2 = 0.0524 0.0024(2) 0.654 and -0.864 e.Å-3 Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were allowed to rotate but not to tip. A numerical absorption correction was performed min/max transmission: (0.411/0.785). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Zn(1) I(1) N(1) C(1) C(4) C(5) x 5000 6339(1) 4938(2) 4230(2) 5686(2) 3371(2) y 3613(1) 5743(1) 1479(4) 364(4) 1119(4) 730(4) z 2500 3929(1) 4007(2) 3816(3) 5173(3) 2563(3) U(eq) 12(1) 21(1) 11(1) 12(1) 13(1) 18(1) 6. Experimenteller Teil - 98 - ZnI2(2,5-Dimethylpyrazin)2 Table 1. Crystal data and structure refinement for ZnI2(2,5-dimethylpyrazine)2. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C12H16I2N4Zn 535.46 170(2) K 0.71073 Å monoclinic P21/c a = 12.2908(8) Å α= 90°. b = 8.2723(7) Å β= 101.266(8)°. c = 17.2054(13) Å γ = 90°. 1715.6(2) Å3 4 2.073 Mg/m3 5.024 mm-1 1008 0.13 x 0.09 x 0.06 mm3 2.41 to 27.10°. -15<=h<=15, -10<=k<=10, -22<=l<=22 12810 3671 [R(int) = 0.0480] 96.8 % Full-matrix least-squares on F2 3671 / 0 / 173 0.979 R1 = 0.0300, wR2 = 0.0738 R1 = 0.0394, wR2 = 0.0773 0.0023(2) 0.745 and -0.846 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 27.10° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were allowed to rotate but not to tip. A numerical absorption correction was performed min/max transmission: (0.582/0.735). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Zn(1) I(1) I(2) N(1) N(2) C(1) C(2) C(3) C(4) C(5) C(6) N(11) N(12) C(11) C(12) C(13) C(14) C(15) C(16) x 2651(1) 4155(1) 1119(1) 3357(3) 4417(3) 2827(3) 3383(4) 4949(3) 4411(3) 1657(4) 6129(4) 2157(3) 1553(3) 1450(3) 1155(4) 2287(4) 2587(4) 1017(4) 2773(5) y 5866(1) 7935(1) 6813(1) 3818(4) 1325(4) 2417(5) 1195(5) 2718(5) 3955(4) 2152(5) 2879(5) 4964(4) 3544(5) 5724(5) 4952(5) 2817(5) 3549(5) 7351(6) 1226(6) z 5324(1) 5197(1) 6048(1) 5937(2) 6886(2) 6004(2) 6483(2) 6807(2) 6339(2) 5578(3) 7235(3) 4168(2) 2681(2) 3590(2) 2848(2) 3259(2) 4000(2) 3735(3) 3091(3) U(eq) 18(1) 27(1) 28(1) 18(1) 26(1) 20(1) 24(1) 20(1) 19(1) 32(1) 28(1) 20(1) 33(1) 22(1) 30(1) 27(1) 23(1) 34(1) 39(1) 6. Experimenteller Teil - 99 - [Fe(SCN)2(Pyrazin)]n Table 1. Crystal data and structure refinement for [Fe(SCN)2(pyrazine)]n. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C6H4FeN4S2 252.10 293(2) K 0.71073 Å monoclinic C2/m a = 10.5311(11) Å b = 7.3224(10) Å c = 5.6957(6) Å 437.80(9) Å3 2 1.912 Mg/m3 2.149 mm-1 252 0.12 x 0.09 x 0.08 mm3 3.39 to 28.09°. -13<=h<=12, -9<=k<=9, -7<=l<=7 1973 573 [R(int) = 0.0322] 99.0 % Full-matrix least-squares on F2 573 / 0 / 38 1.008 R1 = 0.0325, wR2 = 0.0823 R1 = 0.0406, wR2 = 0.0869 0.064(7) 0.403 and -0.438 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 28.09° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole α= 90°. β= 94.588(12)°. γ = 90°. Comments: All non-hydrogen atoms were refined using anisotropic displacement parameters. The C-H hydrogen atoms were located in difference map but were positioned with idealized geometry and were refined using a riding model. A numerical absorption correction was performed min/max transmission: (0.6955/0.7670). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Fe(1) S(1) C(3) N(3) N(1) C(1) x 0 2135(1) 1549(3) 1113(3) 0 469(2) y 5000 5000 5000 5000 1918(4) 947(3) z 0 2757(1) 5342(6) 7159(6) 0 1863(4) U(eq) 19(1) 26(1) 21(1) 27(1) 22(1) 27(1) 6. Experimenteller Teil - 100 - [Co(SCN)2(Pyrazin)]n Table 1. Crystal data and structure refinement for [Co(SCN)2(pyrazine)]n. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C6H4CoN4S2 255.18 293(2) K 0.71073 Å monoclinic C2/m a = 11.4570(18) Å b = 7.1608(8) Å c = 5.6630(9) Å 448.99(11) Å3 2 1.888 Mg/m3 2.327 mm-1 254 0.12 x 0.06 x 0.09 mm3 3.39 to 27.92°. -15<=h<=15, -9<=k<=9, -7<=l<=7 2153 576 [R(int) = 0.0711] 99.0 % Full-matrix least-squares on F2 576 / 0 / 47 1.013 R1 = 0.0338, wR2 = 0.0777 R1 = 0.0429, wR2 = 0.0806 0.009(4) 0.366 and -0.538 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 27.92° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole α= 90°. β= 104.892(18)°. γ = 90°. Comments: All non-hydrogen atoms were refined using anisotropic displacement parameters. The C-H hydrogen atoms were located in difference map but were positioned with idealized geometry and were refined using a riding model. The pyrazine ligand is disordered in two orientations and was refined using a split model. The disordering remains constant in space group C2 or Cm and there are no hints for super structure reflections. A numerical absorption correction was performed min/max transmission: (0.6706/0.7435). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Co(1) N(1) C(1) C(1') S(1) C(3) N(2) x 5000 5000 4957(6) 4008(4) 6974(1) 6447(3) 6076(3) y 5000 1951(4) 969(8) 969(7) 5000 5000 5000 z 5000 5000 2969(10) 5027(9) 8430(2) 10866(6) 12584(6) U(eq) 14(1) 19(1) 28(1) 20(1) 23(1) 16(1) 21(1) 6. Experimenteller Teil - 101 - [Ni(SCN)2(Pyrazin)2]n Table 1. Crystal data and structure refinement [Ni(SCN)2(pyrazine)2]n. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C10H8N6NiS2 335.05 293(2) K 0.71073 Å monoclinic C2/m a = 9.9283(16) Å α= 90°. b = 10.2127(19) Å β= 118.498(10)°. c = 7.2079(12) Å γ = 90°. 642.29(19) Å3 2 1.732 Mg/m3 1.828 mm-1 340 0.12 x 0.10 x 0.08 mm3 3.07 to 30.02°. -13<=h<=1, -14<=k<=14, -9<=l<=10 2165 991 [R(int) = 0.0477] 99.9 % Full-matrix least-squares on F2 991 / 0 / 51 1.003 R1 = 0.0277, wR2 = 0.0695 R1 = 0.0412, wR2 = 0.0733 0.0009(16) 0.445 and -0.466 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 30.02° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were refined using a riding model. A numerical absorption correction was not performed. Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Ni(1) S(1) C(3) N(3) N(1) C(1) C(2) x 0 3501(1) 2070(3) 1077(3) 1537(2) 2936(2) 1107(2) y 0 0 0 0 1520(1) 1721(2) 2310(2) z 5000 12320(1) 9938(4) 8209(4) 5060(2) 6652(3) 3412(3) U(eq) 19(1) 48(1) 25(1) 28(1) 23(1) 33(1) 33(1) 6. Experimenteller Teil - 102 - [Ni(SCN)2(Pyrazin)]n Table 1. Crystal data and structure refinement for [Ni(SCN)2(pyrazine)]n. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C6H4N4NiS2 254.96 293(2) K 0.71073 Å monoclinic C2/m a = 11.4697(12) Å b = 7.0560(6) Å c = 5.6086(5) Å 438.18(7) Å3 2 1.932 Mg/m3 2.640 mm-1 256 0.13 x 0.10 x 0.07 mm3 3.42 to 27.98°. -15<=h<=15, -9<=k<=9, -7<=l<=7 2468 436 [R(int) = 0.0435] 76.5 % Full-matrix least-squares on F2 436 / 0 / 47 1.058 R1 = 0.0296, wR2 = 0.0759 R1 = 0.0335, wR2 = 0.0780 0.034(6) 0.412 and -0.454 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 27.98° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole α= 90°. β= 105.127(11)°. γ = 90°. Comments: The crystal investigated was nonmerohedral twinned. Both individuals were indexed and integrated separately. Addition weak reflections were observed leading to a doubled unit cell with a = 13.7870, b = 7.0580, C = 11.2210, β = 126.900 and space group C2/c. However refinement in C2/c or Cc also leads to a disordering of the pyrazine ligand which is in Cc in a general position. All non-hydrogen atoms were refined using anisotropic displacement parameters. The C-H hydrogen atoms were located in difference map but were positioned with idealized geometry and were refined using a riding model. Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Ni(1) N(1) C(1) C(1') S(1) C(3) N(2) x 0 0 -48(6) -988(5) 1940(1) 1429(3) 1059(3) y 0 -3018(5) -4016(7) -4006(7) 0 0 0 z 0 0 -2051(10) 55(10) 3390(2) 5871(7) 7609(6) U(eq) 15(1) 19(1) 26(1) 21(1) 23(1) 18(1) 21(1) 6. Experimenteller Teil - 103 - [Co(OCN)2(Pyrazin)2]n Table 1. Crystal data and structure refinement for [Co(OCN)2(pyrazine)2]n. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C10H8CoN6O2 303.15 170(2) K 0.71073 Å monoclinic C2/c a = 25.5712(17) Å α= 90°. b = 10.1230(8) Å β= 104.763(8)°. c = 10.1863(7) Å γ = 90°. 2549.8(3) Å3 8 1.579 Mg/m3 1.353 mm-1 1224 0.14 x 0.26 x 0.07 mm3 2.85 to 27.10°. -32<=h<=32, -12<=k<=12, -13<=l<=13 11369 2684 [R(int) = 0.0385] 95.5 % Full-matrix least-squares on F2 2684 / 0 / 199 1.080 R1 = 0.0507, wR2 = 0.1377 R1 = 0.0642, wR2 = 0.1447 0.0000(7) 0.709 and -1.183 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 27.10° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were refined using a riding model. A numerical absorption correction was performed min/max transmission: (0.6268/0.8097). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Co(1) N(1) C(1) C(2) N(2) C(3) C(4) N(11) C(11) C(12) N(12) C(13) C(14) N(21) C(21) O(21) O(21') C(21') N(31) C(31) O(31) C(31') O(31') x 6293(1) 6284(1) 6619(2) 6620(2) 6283(1) 5945(2) 5941(2) 6314(1) 6705(2) 6701(2) 6309(1) 5922(2) 5924(2) 5468(1) 5047(3) 4603(3) 4627(6) 5054(8) 7125(1) 7539(3) 7963(3) 7530(9) 8018(11) y 7506(1) 5944(3) 5983(3) 5014(3) 3980(3) 3936(4) 4920(3) 9093(3) 9152(3) 10102(3) 11005(3) 10958(4) 10000(4) 7557(3) 7083(8) 6596(9) 7504(16) 7581(16) 7442(3) 7287(7) 7136(8) 7620(19) 7720(20) z 6350(1) 7846(3) 9072(4) 10044(3) 9778(3) 8538(3) 7578(3) 4903(3) 4247(4) 3261(4) 2930(3) 3588(4) 4576(4) 5817(3) 5444(8) 5110(10) 4188(18) 4980(20) 6873(3) 7699(8) 8512(7) 7340(20) 7880(30) U(eq) 13(1) 16(1) 22(1) 23(1) 17(1) 23(1) 23(1) 16(1) 23(1) 22(1) 17(1) 26(1) 26(1) 24(1) 35(2) 109(4) 53(5) 23(4) 22(1) 25(2) 74(3) 30(6) 100(8) 6. Experimenteller Teil - 104 - [Mn(SCN)2(Pyrimidin)2]n Table 1. Crystal data and structure refinement for Mn(SCN)2(pyrimidine)2. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C10H8MnN6S2 331.28 170(2) K 0.71073 Å orthorhombic Cmca a = 9.7693(9) Å α= 90°. b = 15.6505(10) Å β= 90°. c = 8.6220(6) Å γ = 90°. 1318.26(17) Å3 4 1.669 Mg/m3 1.311 mm-1 668 0.3 x 0.1 x 0.1 mm3 2.60 to 27.98°. -12<=h<=12, -20<=k<=19, -11<=l<=11 5654 844 [R(int) = 0.0431] 99.9 % Full-matrix least-squares on F2 844 / 0 / 52 1.108 R1 = 0.0276, wR2 = 0.0654 R1 = 0.0314, wR2 = 0.0666 0.0084(12) 0.372 and -0.249 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 27.98° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and refined using a riding model. Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Mn(1) N(1) C(1) C(3) C(4) S(1) C(11) N(11) x 0 1674(1) 2500 2500 1722(1) 0 0 0 y 5000 5708(1) 5321(1) 7028(1) 6570(1) 3565(1) 3794(1) 3966(1) z 0 1477(1) 2500 2500 1453(2) 4768(1) 2936(2) 1611(2) U(eq) 13(1) 17(1) 18(1) 19(1) 18(1) 28(1) 16(1) 21(1) 6. Experimenteller Teil - 105 - Co(SCN)2(Pyrimidin)4 Table 1. Crystal data and structure refinement for Co(SCN)2(pyrimidine)4 Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C18H16CoN10S2 495.46 200(2) K 0.71073 Å monoclinic P21/c a = 8.9454(5) Å α= 90°. b = 12.4819(9) Å β= 98.785(7)°. c = 20.1588(12) Å γ = 90°. 2224.4(2) Å3 4 1.479 Mg/m3 0.987 mm-1 1012 0.2 x 0.15 x 0.15 mm3 2.82 to 26.02°. -10<=h<=11, -15<=k<=15, -24<=l<=24 13647 4156 [R(int) = 0.0439] 95.2 % Full-matrix least-squares on F2 4156 / 0 / 281 0.972 R1 = 0.0339, wR2 = 0.0809 R1 = 0.0512, wR2 = 0.0879 0.0080(9) 0.399 and -0.289 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 26.02° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and refined using a riding model. A numerical absorption correction was performed min/max transmission: (0.7810/0.9262). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Co(1) N(1) C(1) N(2) C(2) C(3) C(4) N(11) C(11) N(12) C(12) C(13) C(14) N(21) C(21) N(22) C(22) C(23) C(24) N(31) C(31) N(32) C(32) C(33) C(34) S(1) C(51) N(51) S(2) C(61) N(61) x 4232(1) 3183(2) 3991(3) 3549(3) 2180(4) 1263(3) 1817(3) 6148(2) 5964(3) 7087(3) 8479(3) 8773(3) 7564(3) 5364(2) 5972(3) 6926(3) 7262(4) 6639(4) 5690(3) 2348(2) 1728(3) 537(3) -85(3) 472(4) 1726(4) 6798(1) 5946(3) 5334(3) 1815(1) 2589(3) 3143(3) y 2571(1) 1954(2) 1336(2) 999(2) 1312(2) 1940(2) 2241(2) 3153(1) 3642(2) 4050(2) 3985(2) 3496(2) 3073(2) 3194(2) 4174(2) 4576(2) 3934(3) 2926(3) 2571(2) 2034(2) 2657(2) 2416(2) 1457(3) 749(3) 1062(2) -790(1) 334(2) 1136(2) 5799(1) 4754(2) 4012(2) z 3527(1) 2545(1) 2188(1) 1562(1) 1271(1) 1594(1) 2243(1) 3046(1) 2450(1) 2152(1) 2500(2) 3118(2) 3368(1) 4488(1) 4524(1) 5034(1) 5564(1) 5596(1) 5034(1) 4018(1) 4441(1) 4734(1) 4582(2) 4157(1) 3887(1) 4134(1) 3911(1) 3746(1) 2586(1) 2984(1) 3274(1) U(eq) 22(1) 25(1) 31(1) 41(1) 40(1) 40(1) 34(1) 23(1) 29(1) 39(1) 39(1) 38(1) 32(1) 26(1) 29(1) 38(1) 45(1) 45(1) 35(1) 29(1) 32(1) 43(1) 44(1) 47(1) 41(1) 43(1) 27(1) 33(1) 38(1) 24(1) 30(1) 6. Experimenteller Teil - 106 - Ni(SCN)2(Pyrimidin)4 Table 1. Crystal data and structure refinement for Ni(SCN)2(pyrimidine)4. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C18H16N10NiS2 495.24 200(2) K 0.71073 Å monoclinic P21/c a = 8.9152(6) Å α= 90°. b = 12.4948(8) Å β= 98.630(9)°. c = 20.0048(16) Å γ = 90°. 2203.2(3) Å3 4 1.493 Mg/m3 1.098 mm-1 1016 0.3 x 0.2 x 0.2 mm3 2.83 to 28.08°. -11<=h<=10, -16<=k<=16, -25<=l<=26 15101 5112 [R(int) = 0.0485] 95.3 % Full-matrix least-squares on F2 5112 / 0 / 281 1.043 R1 = 0.0367, wR2 = 0.0917 R1 = 0.0520, wR2 = 0.0983 0.0079(10) 0.667 and -0.594 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 28.08° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and refined using a riding model. Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Ni(1) N(1) C(1) N(2) C(2) C(3) C(4) N(11) C(11) N(12) C(12) C(13) C(14) N(21) C(21) N(22) C(22) C(23) C(24) N(31) C(31) N(32) C(32) C(33) C(34) S(1) C(51) N(51) S(2) C(61) N(61) x 4230(1) 3207(2) 4019(3) 3580(3) 2206(3) 1280(3) 1836(3) 6103(2) 5919(3) 7052(3) 8450(3) 8747(3) 7531(3) 5335(2) 5939(3) 6894(3) 7228(3) 6604(4) 5655(3) 2380(2) 1757(3) 565(3) -49(3) 504(3) 1765(3) 6804(1) 5940(3) 5326(2) 1795(1) 2591(2) 3153(2) y 2570(1) 1966(1) 1345(2) 1003(2) 1312(2) 1941(2) 2249(2) 3129(1) 3636(2) 4047(2) 3961(2) 3450(2) 3029(2) 3177(1) 4158(2) 4566(2) 3922(2) 2915(2) 2555(2) 2048(2) 2674(2) 2429(2) 1462(2) 762(2) 1072(2) -772(1) 352(2) 1149(1) 5782(1) 4740(2) 3997(1) z 3525(1) 2563(1) 2203(1) 1575(1) 1282(1) 1608(1) 2259(1) 3053(1) 2459(1) 2159(1) 2503(1) 3115(1) 3369(1) 4466(1) 4501(1) 5017(1) 5550(1) 5582(1) 5017(1) 4004(1) 4428(1) 4731(1) 4579(1) 4152(1) 3874(1) 4134(1) 3909(1) 3744(1) 2595(1) 2991(1) 3279(1) U(eq) 21(1) 24(1) 30(1) 40(1) 39(1) 38(1) 32(1) 22(1) 27(1) 37(1) 38(1) 36(1) 31(1) 26(1) 29(1) 37(1) 43(1) 44(1) 34(1) 28(1) 32(1) 41(1) 43(1) 46(1) 40(1) 42(1) 26(1) 31(1) 36(1) 23(1) 28(1) 6. Experimenteller Teil - 107 - ZnCl2(2,2'-Dipyridylsulfid) Table 1. Crystal data and structure refinement for ZnCl2(2,2'-dipyridylsulfide). Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C10H8Cl2N2SZn 324.51 170(2) K 0.71073 Å monoclinic P21/c a = 12.1944(12) Å α= 90°. b = 7.6404(4) Å β= 110.426(12)°. c = 14.2572(15) Å γ = 90°. 1244.82(19) Å3 4 1.732 Mg/m3 2.541 mm-1 648 0.13 x 0.09 x 0.07 mm3 2.95 to 28.14°. -16<=h<=15, -8<=k<=10, -15<=l<=18 7272 2939 [R(int) = 0.0358] 96.5 % Full-matrix least-squares on F2 2939 / 0 / 146 1.002 R1 = 0.0356, wR2 = 0.0885 R1 = 0.0515, wR2 = 0.0959 0.0121(14) 0.470 and -0.600 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 28.14° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and refined using a riding model. A numerical absorption correction was performed min/max transmission: (0.5358/0.7520). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Zn(1) Cl(1) Cl(2) N(11) C(11) C(12) C(13) C(14) C(15) N(1) C(1) C(2) C(3) C(4) C(5) S(1) x 7512(1) 8503(1) 7151(1) 5975(2) 5996(2) 4973(3) 3906(3) 3878(3) 4925(2) 8325(2) 8192(2) 8775(2) 9546(2) 9679(2) 9039(2) 7353(1) y 3898(1) 1410(1) 5439(1) 3765(3) 3642(4) 3575(4) 3682(4) 3870(4) 3910(4) 5499(3) 5231(4) 6216(4) 7517(4) 7808(4) 6795(4) 3421(1) z 2194(1) 2369(1) 793(1) 2488(2) 3433(2) 3656(3) 2884(3) 1910(3) 1739(2) 3397(2) 4286(2) 5128(2) 5049(2) 4139(3) 3328(2) 4443(1) U(eq) 21(1) 28(1) 31(1) 22(1) 23(1) 30(1) 35(1) 33(1) 27(1) 20(1) 21(1) 25(1) 29(1) 29(1) 23(1) 30(1) 6. Experimenteller Teil - 108 - ZnBr2(2,2'-Dipyridylsulfid) Table 1. Crystal data and structure refinement for ZnBr2(2,2'-dipyridylsulfide). Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C10H8Br2N2SZn 413.43 170(2) K 0.71073 Å monoclinic P21/c a = 11.0385(8) Å α= 90°. b = 8.9627(5) Å β= 91.663(9)°. c = 13.1572(11) Å γ = 90°. 1301.16(16) Å3 4 2.111 Mg/m3 8.162 mm-1 792 0.14 x 0.10 x 0.07 mm3 2.75 to 28.04°. -14<=h<=14, -11<=k<=11, -17<=l<=17 14781 3105 [R(int) = 0.0411] 98.5 % Full-matrix least-squares on F2 3105 / 0 / 146 1.029 R1 = 0.0330, wR2 = 0.0799 R1 = 0.0458, wR2 = 0.0853 0.0076(6) 1.153 and -1.155 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 28.04° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and refined using a riding model. A numerical absorption correction was performed min/max transmission: (0.2849/0.3938). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Zn(1) Br(1) Br(2) N(1) C(1) C(2) C(3) C(4) C(5) S(1) N(11) C(11) C(12) C(13) C(14) C(15) x 7460(1) 7200(1) 7628(1) 8929(3) 8968(3) 9955(4) 10912(4) 10854(4) 9853(3) 7789(1) 6206(3) 6435(3) 5613(3) 4528(4) 4303(4) 5160(3) y 8474(1) 10885(1) 8209(1) 7499(3) 6003(4) 5287(4) 6142(5) 7682(5) 8322(4) 4873(1) 7109(3) 5653(3) 4683(4) 5268(4) 6768(5) 7667(4) z 5091(1) 5757(1) 3323(1) 5825(2) 5939(2) 6405(3) 6789(3) 6702(3) 6215(3) 5409(1) 5746(2) 5880(2) 6314(3) 6640(3) 6535(3) 6085(3) U(eq) 18(1) 33(1) 36(1) 17(1) 19(1) 26(1) 31(1) 32(1) 25(1) 26(1) 17(1) 18(1) 25(1) 29(1) 31(1) 24(1) 6. Experimenteller Teil - 109 - ZnI2(2,2'-Dipyridylsulfid) Table 1. Crystal data and structure refinement for ZnI2(2,2'-dipyridylsulfide). Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C10H8I2N2SZn 507.41 170(2) K 0.71073 Å orthorhombic Pnma a = 13.9418(8) Å α= 90°. b = 10.9742(10) Å β= 90°. c = 9.1913(6) Å γ = 90°. 1406.27(18) Å3 4 2.397 Mg/m3 6.261 mm-1 936 0.15 x 0.10 x 0.07 mm3 2.65 to 28.06°. -18<=h<=16, -14<=k<=14, -12<=l<=12 11604 1784 [R(int) = 0.0291] 99.0 % Full-matrix least-squares on F2 1784 / 0 / 80 1.025 R1 = 0.0286, wR2 = 0.0702 R1 = 0.0357, wR2 = 0.0739 0.0034(4) 0.895 and -0.864 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 28.06° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and refined using a riding model. A numerical absorption correction was performed min/max transmission: (0.3521/0.4636). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Zn(1) I(1) I(2) N(1) C(1) C(2) C(3) C(4) C(5) S(1) x 5141(1) 3318(1) 5876(1) 5775(2) 5819(2) 6253(2) 6670(3) 6652(3) 6195(3) 5244(1) y 2500 2500 2500 3873(2) 3762(3) 4633(3) 5630(3) 5741(3) 4844(3) 2500 z 1438(1) 1575(1) -1099(1) 2632(3) 4090(4) 4967(4) 4311(5) 2819(5) 2005(4) 4926(1) U(eq) 21(1) 36(1) 38(1) 22(1) 24(1) 33(1) 41(1) 42(1) 32(1) 30(1) 6. Experimenteller Teil - 110 - ZnBr2(2,2'-Dipyridyldisulfid) Table 1. Crystal data and structure refinement for ZnBr2(2,2'-dipyridyldisulfide). Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C10H8Br2N2S2Zn 445.49 170(2) K 0.71073 Å triclinic P-1 a = 7.7610(8) Å α= 95.488(12)°. b = 8.2962(8) Å β= 107.161(12)°. c = 12.3576(13) Å γ = 112.950(11)°. 679.70(12) Å3 2 2.177 Mg/m3 7.969 mm-1 428 0.09 x 0.12 x 0.07 mm3 2.89 to 26.02°. -9<=h<=9, -10<=k<=10, -15<=l<=15 6076 2609 [R(int) = 0.0535] 97.4 % Full-matrix least-squares on F2 2609 / 0 / 155 1.058 R1 = 0.0457, wR2 = 0.1143 R1 = 0.0579, wR2 = 0.1201 0.0071(16) 1.310 and -1.428 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 26.02° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were refined using a riding model. A numerical absorption correction was performed min/max transmission: (0.27617/ 0.40172). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Br(1) Br(2) Zn(1) S(1) N(1) C(2) C(5) C(1) C(3) C(4) S(2) N(11) C(15) C(11) C(14) C(13) C(12) x 9936(1) 3820(1) 7046(1) 7276(3) 7378(8) 7679(9) 7566(10) 7414(9) 7933(10) 7888(10) 5117(2) 7411(7) 8390(9) 6512(9) 8464(10) 7523(10) 6535(10) y 7573(1) 4841(1) 5058(1) 2365(2) 5577(7) 5176(10) 7263(9) 4556(9) 6913(10) 7975(10) 1119(2) 2888(7) 2991(9) 1280(8) 1511(10) -148(9) -270(9) z 2302(1) 1297(1) 2290(1) 4464(2) 4048(5) 5987(6) 4404(5) 4828(5) 6347(6) 5545(7) 2831(2) 1671(5) 928(6) 1897(5) 359(6) 587(6) 1385(6) U(eq) 19(1) 20(1) 14(1) 22(1) 17(1) 22(1) 19(1) 17(1) 25(2) 26(2) 21(1) 15(1) 18(1) 16(1) 24(2) 23(1) 23(1) 6. Experimenteller Teil - 111 - Cu6Cl6S4(4-Pyridiniumthiolat)4 Table 1. Crystal data and structure refinement for Cu6Cl6S4(4-pyridiniumthiolato)4. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C20H20Cl6Cu6N4S4 1038.58 170(2) K 0.71073 Å Tetragonal I41/a a = 15.0563(9) Å α= 90°. b = 15.0563(9) Å β= 90°. c = 13.0852(9) Å γ = 90°. 2966.3(3) Å3 4 2.326 Mg/m3 5.070 mm-1 2032 0.12 x 0.11 x 0.08 mm3 3.40 to 28.06°. -19<=h<=19, -19<=k<=12, -17<=l<=15 7864 1783 [R(int) = 0.0480] 98.7 % Numerical 0.672 and 0.553 Full-matrix least-squares on F2 1783 / 0 / 106 1.042 R1 = 0.0493, wR2 = 0.1283 R1 = 0.0646, wR2 = 0.1382 0.0012(4) 2.593 and -0.692 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 28.06° Absorption correction Max. and min. transmission Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were refined using a riding model. A numerical absorption correction was performed min/max transmission: (0.4733/0.5172). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Cu(1) Cu(1') Cu(2) Cl(1) Cl(2) S(1) C(1) C(2) N(1) C(3) C(4) C(5) x 4532(2) 4782(2) 4933(7) 4014(1) 5000 3822(1) 3074(3) 3198(3) 1870(3) 2576(4) 1737(4) 2327(3) y 5775(2) 5778(3) 7690(5) 4412(1) 7500 6960(1) 6748(3) 6055(3) 6474(4) 5919(4) 7137(5) 7284(4) z 5997(2) 5726(3) 4562(1) 5708(1) 2868(1) 5384(1) 4396(3) 3693(4) 2840(3) 2931(4) 3518(5) 4290(4) U(eq) 35(1) 29(1) 73(2) 34(1) 27(1) 19(1) 20(1) 31(1) 41(1) 40(1) 47(2) 39(1) 6. Experimenteller Teil - 112 - Cu6Br6S4(4-Pyridiniumthiolat)4 Table 1. Crystal data and structure refinement for Cu6Br6S4(4-pyridiniumthiolato)4. Empirical formula Formula weight Temperature Wavelength Crystal system Space group Unit cell dimensions C20H20Br6Cu6N4S4 1305.34 170(2) K 0.71073 Å tetragonal I41/a a = 15.3161(9) Å a= 90°. b = 15.3161(9) Å b= 90°. c = 13.2602(8) Å g = 90°. 3110.6(3) Å3 4 2.787 Mg/m3 12.027 mm-1 2464 0.11 x 0.09 x 0.06 mm3 2.66 to 28.03°. -20<=h<=20, -20<=k<=20, -16<=l<=16 13166 1853 [R(int) = 0.0484] 97.9 % Full-matrix least-squares on F2 1853 / 0 / 106 1.032 R1 = 0.0324, wR2 = 0.0723 R1 = 0.0431, wR2 = 0.0763 0.00071(9) 0.759 and -1.006 e.Å-3 Volume Z Density (calculated) Absorption coefficient F(000) Crystal size Theta range for data collection Index ranges Reflections collected Independent reflections Completeness to theta = 28.03° Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2sigma(I)] R indices (all data) Extinction coefficient Largest diff. peak and hole Comments: All non-hydrogen atoms were refined anisotropic. The C-H hydrogen atoms were positioned with idealized geometry and were refined using a riding model. A numerical absorption correction was performed min/max transmission: (0.1366/0.2476). Table 2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). Cu(1) Cu(1') Cu(2) Br(1) Br(2) S(1) C(1) C(2) N(1) C(3) C(4) C(5) x 4617(3) 4786(3) 4886(1) 3972(1) 5000 3843(1) 3113(2) 3238(3) 1928(2) 2632(3) 1787(3) 2365(3) y 5805(3) 5808(4) 7765(1) 4412(1) 7500 6961(1) 6747(2) 6077(3) 6479(3) 5951(3) 7131(4) 7272(3) z 5937(3) 5715(4) 4595(1) 5670(1) 2838(1) 5395(1) 4421(3) 3712(3) 2893(3) 2966(3) 3560(4) 4331(4) U(eq) 42(1) 19(1) 49(1) 21(1) 19(1) 14(1) 14(1) 22(1) 31(1) 27(1) 38(1) 29(1) 7. Publikationen und Konferenzbeiträge - 113 - 7 Publikationen und Konferenzbeiträge 7.1 Publikationen [1] Poly[[aquacopper(I)μ-bromo]-μ-5-methylpyrazine-2-carboxylato)-N4:O,N1-[diaqua-copper(II)]-μ-(5-methylpyrazine-2-carboxylato)-O,N1:N4-[aquacopper(I)-μ-bromo]] Christian Näther, Mario Wriedt und Inke Jeß Acta Cryst., 2001, C57, 1395-1397. [2] Synthesis, Crystal Structures and Properties of the Copper(I)halide Coordination Poly2 1 1 mers ∞ [CuX(μ-2-chloropyrazine)] (X = Cl, Br), ∞ [CuI(μ−2-chloropyrazine)] and ∞ [Cu2I2(μ-2-chloropyrazine)] Christian Näther, Mario Wriedt und Inke Jeß Z. Anorg. Allg. Chem., 2002, 628, 394-400. [3] Dichloro-tetrakis(4-methylimidazole)copper(II) Christian Näther, Mario Wriedt und Inke Jeß Acta Cryst., 2002, E58, m39-m40. [4] Dibromo-tetrakis(4-methylimidazole)copper(II) Christian Näther, Mario Wriedt und Inke Jeß Acta Cryst., 2002, E58, m63-m64. [5] Dibromo-tetrakis(4-methylimidazole)copper(II) dihydrate Christian Näther, Mario Wriedt und Inke Jeß Acta Cryst., 2002, E58, m107-m109. [6] Dimorphism of a New CuI Coordination Polymer: Synthesis, Crystal Structures and Properties of Catena[CuI(2-Iodpyrazine-N)] and Poly[CuI(μ2-2-Iodpyrazine-N,N’)] Christian Näther, Mario Wriedt und Inke Jeß Inorg. Chem., 2003, 42, 2391-2397. 7. Publikationen und Konferenzbeiträge [7] - 114 - Poly[(2-iodopyrazine-κN4)copper(I)]-μ3-thiocyanato-κ3N:S:S] Christian Näther, Mario Wriedt und Inke Jeß Acta Cryst., 2005, E61, m329-m331. [8] Hexabromidoterakis(μ3-pyridiniumthiolato-κ3S:S:S)hexacopper(I) Mario Wriedt, Inke Jeß und Christian Näther Acta Cryst., 2007, E63, m3145. [9] Di-chloro-tetrakis(μ2-bromo)-tetrakis(μ3-pyridinium-4-thiolato-hexa-copper(I) Mario Wriedt, Inke Jeß und Christian Näther CSD Private Communication, 2007, CCDC 668016. [10] Dichlorido(di-2-pyridyl sulfide-κ2N,N‘)zinc(II) Mario Wriedt, Inke Jeß und Christian Näther Acta Cryst., 2008, E64, m10. [11] (Di-2-pyridyl sulfide-κ2N,N‘)diiodidozinc(II) Mario Wriedt, Inke Jeß und Christian Näther Acta Cryst., 2008, E64, m11. [12] Dibromido(di-2-pyridyl disulfide-κ2N,N‘)zinc(II) Mario Wriedt, Inke Jeß und Christian Näther Acta Cryst., 2008, E64, m83. [13] Dibromido(di-2-pyridyl sulfide-κ2N,N‘)zinc(II) Mario Wriedt, Inke Jeß und Christian Näther Acta Cryst., 2008, E64, m315. [14] Synthesis, Crystal Structures and Thermal Properties of New ZnX2(2,5-dimethyl-pyrazine) (X = Cl, Br, I) Coordination Compounds Mario Wriedt, Inke Jeß und Christian Näther Inorg. Chem., 2008, eingereicht. 7. Publikationen und Konferenzbeiträge - 115 - 7.2 Konferenzbeiträge [1] Synthesis, Crystal Structures and Thermal Properties of Inorganic-Organic Coordination Polymers based on Copper(I) halides and Aromatic Amines Christian Näther, Inke Jeß, Jan Greve und Mario Wriedt 8th European Conference on Solid State Chemistry, 2001, Oslo. [2] Dimorphie von CuI(2-Iodpyrazin) Christian Näther, Mario Wriedt und Inke Jeß Workshop-Konferenz des Arbeitskreises “Chemische Kristallographie“ der GDCh, 2001, Essen. [3] Darstellung, Struktur und Eigenschaften zweier Modifikationen von CuI(2-Iodpyrazin) Christian Näther, Mario Wriedt und Inke Jeß 10. Jahrestagung der Deutschen Gesellschaft für Kristallographie, 2002, Kiel. Z. Krist., 2002, Suppl. 17. 147. [4] Darstellung, Eigenschaften und Strukturbestimmung aus Röntgen-Pulverdaten von Cu2I2(2-Chlorpyrazin) Christian Näther, Mario Wriedt, Inke Jeß, Günter Reck und Werner Kraus 10. Jahrestagung der Deutschen Gesellschaft für Kristallographie, 2002, Kiel. Z. Krist., 2002, Suppl. 17. 47. [5] Darstellung, Struktur und Eigenschaften koordinationspolymerer Verbindungen auf der Basis von Kupfer(I)halogeniden und aromatischen Stickstoffbasen. Christian Näther, Inke Jeß, Jan Greve und Mario Wriedt Chemiedozententagung ADUC, 2002, Köln. 8. Literatur - 116 - 8 Literatur [1] O. Hassel, H. Mark, Zeitschrift für Physik 1924, 25, 317. [2] M. E. Straumanis, E. Z. Aka, J. Am. Chem. Soc. 1951, 73, 5643. [3] H. J. Bautsch, J. Bohm, I. Kleber, Einführung in die Kristallographie, Vol. 17, Verlag Technik GmbH, Berlin, Deutschland, 1990. [4] H. Eckert, Angew. Chem. 1989, 101, 1763. [5] H. Eckert, Angew. Chem. Int. Ed. Engl. 1989, 28, 1723. [6] G. Sakane, T. Shibahare, H. W. Hou, X. Q. Xin, S. Shi, Inorg. Chem. 1995, 34, 4785. [7] J. P. Glusker, M. Lewis, M. Rossi, Crystal Structure Analysis for Chemists and Biologists, Verlag Chemie, Weinheim, Deutschland, 1994. [8] in CD Römpp Chemielexikon, Vol. 2, Georg Thieme Verlag, Stuttgart, Deutschland, 1995. [9] J. Maddox, Nature 1988, 335, 201. [10] G. R. Desiraju, Organic Solid State Chemistry, Vol. Studies in Organic Chemistry 32, Elsevier, Amsterdam - Oxford - New York - Tokio, 1987. [11] G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier, Amsterdam - Oxford - New York - Tokio, 1989. [12] G. M. J. Schmidt, Pure Appl. Chem. 1971, 27, 647. [13] J. Bernstein, R. J. Davey, J.-O. Henck, Angew. Chem. 1999, 111, 3646. [14] J. Bernstein, R. J. Davey, J.-O. Henck, Angew. Chem. Int. Ed. Engl. 1999, 38, 3440. [15] J. Haleblian, W. McCrone, J. Pharm. Sci. 1969, 58, 8911. [16] T. Miyazaki, T. Watanabe, S. Miyata, Jpn. J. Appl. Phys. 1988, 27, L1724. [17] K. Tanaka, F. Toda, Chem. Rev. 2000, 100, 1025. [18] F. Toda, Acc. Chem. Res. 1995, 28, 480. [19] F. H. Allen, S. Bellard, M. D. Brice, B. A. Cartwright, A. Doubleday, H. Higgs, T. Hummelink, B. G. Hummelink-Peters, O. Kennard, W. D. S. Motherwell, J. R. Rodgers, D. G. Watson, Acta Cryst. 1979, B35, 2331. [20] F. H. Allen, O. Kennard, R. Taylor, Acc. Chem. Res. 1983, 16, 146. [21] G. Bergerhoff, I. D. Brown, in Crystallographic Databases: Information Content, Software Applications, Scientific Applications, International Union of Crystallography, Bonn - Cambridge - Chester, 1987, pp. 77. [22] G. Bergerhoff, R. Hundt, R. Sievers, I. D. Brown, J. Chem. Inf. Comput. Sci. 1983, 23, 66. 8. Literatur [23] - 117 - I. D. Brown, S. M. Bradley, D. Altermatt, in Molecular Structure: Chemical Reactivity and Biological Activity (Eds.: J. J. Stezowski, J.-L. Huang, M.-C. Shao), Oxford University Press, Oxford, England, 1988, pp. 513. [24] R. E. Rosenfield, R. Parthasarathy, J. D. Dunitz, J. Am. Chem. Soc. 1977, 99, 4860. [25] W. B. Schweitzer, in Crystallographic Computing, Vol. 3 (Eds.: G. M. Sheldrick, C. Kruger, R. Goddard), Clarendon Press, Oxford, England,, 1995, pp. 119. [26] J. Bernstein, J. Phys. D., Appl. Phys. 1993, 26, B66. [27] J. L. Manson, A. M. Arif, J. S. Miller, Chem. Commun. 1999, 1479. [28] C. Näther, J. Greve, J. Solid State Chem. 2003, 176, 259. [29] R. L. Carlin, Magnetochemistry, Springer Verlag, Berlin - Heidelberg - New York - Tokyo, 1986. [30] A. Weiss, H. Witte, Magnetochemie - Grundlagen und Anwendungen, Verlag Chemie, Weinheim, Deutschland, 1973. [31] M. J. Zaworotko, Chem. Soc. Rev. 1994, 23, 283. [32] P. J. Hagrman, D. Hagrman, J. Zubieta, Angew. Chem. 1999, 111, 2798. [33] P. J. Hagrman, D. Hagrman, J. Zubieta, Angew. Chem. Int. Ed. Engl. 1999, 38, 2638. [34] C. B. Aakeröy, A. M. Beatty, Aust. J. Chem. 2001, 54, 409. [35] B. F. Abrahams, B. F. Hoskins, J. Liu, R. Robson, J. Am. Chem. Soc. 1991, 113, 3045. [36] B. F. Abrahams, K. D. Lu, B. Moubaraki, K. S. Murray, R. Robson, J. Chem. Soc., Dalton Trans. 2000, 1793. [37] S. R. Batten, Curr. Opin. Solid State Mater. Sci. 2001, 5, 107. [38] S. R. Batten, Cryst. Eng. Comm. 2001, 3, 67. [39] S. R. Batten, B. F. Hoskins, R. Robson, New. J. Chem. 1998, 22, 173. [40] S. R. Batten, R. Robson, Angew. Chem. 1998, 110, 1558. [41] S. R. Batten, R. Robson, Angew. Chem. Int. Ed. Engl. 1998, 37, 1460. [42] A. J. Blake, N. R. Brooks, N. R. Champness, P. A. Cooke, M. Crew, A. M. Deveson, L. R. Hanton, P. Hubberstey, D. Fenske, M. Schröder, Cryst. Eng. 1999, 2, 181. [43] A. J. Blake, N. R. Champness, P. Hubberstey, W.-S. Li, M. A. Withersby, M. Schröder, Coord. Chem. Rev. 1999, 183, 117. [44] A. J. Blake, N. R. Champness, A. Khlobystov, W.-S. Li, M. Schröder, A. Khlobystov, D. A. Lemenovskii, Chem. Commun. 1997, 2027. [45] D. Braga, L. Maini, M. Polito, L. Scaccianoce, G. Cojazzi, F. Grepioni, Coord. Chem. Rev. 2001, 216, 225. [46] L. Carlucci, G. Ciani, D. M. Proserpio, Chem. Commun. 1999, 449. 8. Literatur [47] - 118 - L. Carlucci, G. Ciani, D. M. Proserpio, A. Sironi, Angew. Chem. Int. Ed. Engl. 1995, 34, 1895. [48] Y.-B. Dong, M. D. Smith, H.-C. z. Loye, Inorg. Chem. 2000, 39, 1943. [49] W. W. Ellis, M. Schmitz, A. A. Arif, P. J. Stang, Inorg. Chem. 2000, 39, 2547. [50] A. Fu, X. Huang, J. Li, T. Yuen, C. L. Lin, Chem. Eur. J. 2002, 8, 2239. [51] R. W. Gable, B. F. Hoskins, R. Robson, J. Chem. Soc., Chem. Commun. 1990, 762. [52] D. Hagrman, R. P. Hammond, R. Haushalter, J. Zubieta, Chem. Mater. 1998, 10, 2091. [53] B. F. Hoskins, R. Robson, J. Am. Chem. Soc. 1990, 112, 1546. [54] C. Janiak, Nachr. Chem. 2000, 48, 245. [55] C. Janiak, Nachr. Chem. 2001, 49, 272. [56] X.-J. Yang, C. Janiak, J. Heinze, F. Drepper, P. Mayer, H. Piotrowski, P. Klüfers, Inorg. Chim. Acta 2001, 318, 103. [57] C. Kaes, M. W. Hosseini, C. E. F. Rickard, B. W. Skelton, A. H. White, Angew. Chem. Int. Ed. Engl. 1998, 37, 920. [58] K. Satoshi, K. Susumu, K. Hitoshi, I. Shinichiro, K. Motomi, Inorg. Chim. Acta 1998, 267, 143. [59] J. F. Keggin, F. D. Miles, Nature 1936, 137, 577. [60] T. Kromp, W. S. Sheldrick, Z. Naturforsch. 1999, 54b, 1175. [61] C.-M. Liu, X.-Z. You, W. Chen, Coord. Chem. 1998, 46, 183. [62] P. Losier, M. J. Zaworotko, Angew. Chem. Int. Ed. Engl. 1996, 35, 2779. [63] J. Lu, T. Paliwala, S. C. Lim, C. Yu, T. Niu, A. J. Jacobson, Inorg. Chem. 1997, 36, 923. [64] M. Mimura, T. Matsuo, T. Nakashima, N. Matsumoto, Inorg. Chem. 1998, 37, 3553. [65] J. M. Moreno, J. Suarez-Varela, E. Colacio, J. C. Avila-Rosón, M. A. Hidalgo, D. Martin-Ramos, Can. J. Chem. 1995, 73, 1591. [66] B. Moulton, M. J. Zaworotko, Curr. Opin. Solid State Mater. Sci. 2002, 6, 117. [67] S.-i. Nishikiori, T. Iwamoto, Bull. Chem. Soc. Jpn. 1983, 56, 3246. [68] S. Nishikiori, T. Iwamoto, Inorg. Chem. 1986, 25, 788. [69] S.-i. Noro, S. Kitagawa, M. Kondo, K. Seki, Angew. Chem. Int. Ed. Engl. 2000, 39, 2081. [70] N. S. Persky, J. M. Chow, K. A. Poschmann, N. N. Lacuesta, S. L. Stoll, S. G. Bott, a. S. Obrey, Inorg. Chem. 2001, 40, 29. [71] R. Robson, in Comprehensive Supramolecular Chemistry, Pergamon, New York, 1996, p. 733. [72] R. Robson, B. F. Abrahams, S. R. Batten, R. W. Grable, B. F. Hoskins, J. Liu, in Supramolecular Architecture, ACS publications, Washington DC, 1992. [73] B. Roßenbeck, W. S. Sheldrick, Z. Naturforsch. 1999, 54b, 1510. 8. Literatur - 119 - [74] B. Roßenbeck, W. S. Sheldrick, Z. Naturforsch. 2000, 55b, 467. [75] R. W. Saalfrank, H. Maid, F. Hampel, K. Peters, Eur. J. Inorg. Chem. 1999, 1859. [76] O. M. Yaghi, G. Li, Angew. Chem. Int. Ed. Engl. 1995, 34, 207. [77] O. M. Yaghi, G. Li, Angew. Chem. 1995, 107, 232. [78] C. B. Aakeröy, A. M. Beatty, Cryst. Eng. 1998, 1, 39. [79] B. Moulton, M. J. Zaworotko, Chem. Rev. 2001, 101, 1629. [80] A. J. Blake, N. R. Brooks, N. R. Champness, P. A. Cooke, A. M. Deveson, D. Fenske, P. Hubberstey, W.-S. Li, M. Schröder, J. Chem. Soc., Dalton Trans. 1999, 2103. [81] S. R. Batten, J. C. Jeffery, M. D. Ward, Inorg. Chim. Acta 1999, 292, 231. [82] P. M. Graham, R. D. Pike, M. Sabat, R. D. Bailey, W. T. Pennington, Inorg. Chem. 2000, 39, 5121. [83] A. J. Blake, N. R. Brooks, N. R. Champness, L. R. Hanton, P. Hubberstey, M. Schröder, Pure Appl. Chem. 1998, 70, 2351. [84] J. Y. Lu, B. R. Cabrera, R.-J. Wang, J. Li, Inorg. Chem. 1999, 38, 4608. [85] J. A. Campbell, C. L. Raston, A. H. White, Aust. J. Chem. 1977, 30, 1937. [86] G. Bhosekar, I. Jeß, C. Näther, Inorg. Chem. 2006, 43, 6508. [87] J. Greve, C. Näther, Z. Naturforsch. 2004, 59b, 1325. [88] I. Jeß, C. Näther, Z. Naturforsch. 2007, 62b, 617. [89] C. Näther, J. Greve, I. Jeß, C. Wickleder, Solid State Sci. 2003, 5, 1167. [90] C. Näther, I. Jeß, Z. Naturforsch. 2002, 57b, 1133. [91] C. Näther, I. Jeß, J. Solid State Chem. 2002, 169, 103. [92] C. Näther, I. Jess, Inorg. Chem. 2003, 42, 2968. [93] C. Näther, I. Jeß, Eur. J. Inorg. Chem. 2004, 2868. [94] F. Allen, O. Kennard, Chem. Des. Autom. News 1993, 8, 31. [95] CSD, 1.2 ed., Conquest, 1999. [96] G. Bhosekar, I. Jeß, C. Näther, Z. Naturforsch. 2006, 61b, 721. [97] G. Bhosekar, I. Jess, C. Näther, Acta Cryst. 2006, E62, m1859. [98] G. Bhosekar, I. Jess, C. Näther, Acta Cryst. 2006, E62, m1315. [99] G. Bhosekar, I. Jess, C. Näther, Acta Cryst. 2006, E62, m2073. [100] C. Näther, G. Bhosekar, I. Jess, Inorg. Chem. 2007, 46, 8079. [101] C. Näther, I. Jeß, unpublizierte Arbeiten, Uni Kiel. [102] C. Näther, I. Jeß, Monatsh. Chem. 2001, 132, 897. [103] C. Näther, I. Jeß, J. Greve, Polyhedron 2001, 20, 1017. [104] C. Näther, I. Jeß, H. Studzinski, Z. Naturforsch. 2001, 56b, 997. [105] C. Näther, M. Wriedt, I. Jeß, Z. Anorg. Allg. Chem. 2002, 628, 394. 8. Literatur - 120 - [106] C. Näther, M. Wriedt, I. Jeß, Acta Cryst. 2005, E61, m329. [107] M. Wriedt, I. Jess, C. Näther, Acta Cryst. 2007, E63, m3145. [108] M. Wriedt, I. Jeß, C. Näther, CSD Private Communication 2007, CCDC 668016. [109] M. Wriedt, I. Jess, C. Näther, Acta Cryst. 2008, E64, m10. [110] M. Wriedt, I. Jess, C. Näther, Acta Cryst. 2008, E64, m11. [111] M. Wriedt, I. Jess, C. Näther, Acta Cryst. 2008, E64, m315. [112] M. Wriedt, I. Jess, C. Näther, Acta Cryst. 2008, E64, m83. [113] G. Bhosekar, I. Jeß, Z. Havlas, C. Näther, Cryst. Grow. & Des. 2007, 7, 2627. [114] G. Bhosekar, I. Jeß, N. Lehnert, C. Näther, Eur. J. Inorg. Chem. 2008, 605. [115] G. Bhosekar, I. Jess, C. Näther, Inorg. Chem. 2006, 45, 6508. [116] G. Bhosekar, I. Jess, C. Näther, Acta Cryst. 2006, E62, m1859. [117] C. Näther, G. Bhosekar, I. Jeß, Eur. J. Inorg. Chem. 2007, 5353. [118] A. J. Blake, N. R. Champness, M. Crew, L. R. Hanton, S. Parsons, M. Schröder, J. Chem. Soc., Dalton Trans. 1998, 1533. [119] A. D. Bond, C. J. McKenzie, Acta Cryst. 2005, C61, m519. [120] M. Du, C.-P. Li, J.-H. Guo, Inorg. Chim. Acta 2006, 359, 2575. [121] M. A. S. Goher, F. A. Mautner, Polyhedron 1999, 18, 1805. [122] M. A. S. Goher, F. A. Mautner, J. Chem. Soc., Dalton Trans. 1999, 1923. [123] T. Liu, Z.-P. Xie, Acta Cryst. 2007, E63, m1908. [124] T. Otieno, J. R. Blanton, K. J. Lanham, S. Parkin, J. Chem. Cryst. 2003, 33, 335. [125] O. Teichert, W. S. Sheldrick, Z. Anorg. Allg. Chem. 1999, 625, 1860. [126] O. Teichert, W. S. Sheldrick, Z. Anorg. Allg. Chem. 2000, 626, 2196. [127] E. Sahin, S. Ide, A. Atac, S. Yurdakul, J. Mol. Struct. 2002, 616, 253. [128] E. Dubler, G. Haenggi, H. Schmalle, Inorg. Chem. 1992, 31, 3728. [129] C. A. Grapperhaus, T. Tuntulani, J. H. Reibenspies, M. Y. Darensbourg, Inorg. Chem. 1998, 37, 4052. [130] U. Siemeling, I. Scheppelmann, B. Neumann, A. Stammler, H.-G. Stammler, J. Frelek, Chem. Commun. 2003, 2236. [131] C. Papatriantafyllopoulou, C. P. Rabtopoulou, A. Terzis, E. Manessi-Zoupa, S. P. Perlepes, Z. Naturforsch. 2006, B61, 37. [132] Y. Yamamoto, T. Suzuki, S. Kaizaki, J. Chem. Soc. Dalton Trans. 2001, 1566. [133] A. S. Antsyshkina, M. A. Porai-Koshits, B. I. Saidov, N. N. Maltseva, N. S. Kedrova, V. N. Ostrikova, Russ. J. Inorg. Chem. 1991, 36, 2291. [134] J. Glerup, P. A. Goodson, D. J. Hodgson, K. Michelsen, U. Rychlewska, H. Weihe, Bull. Chem. Soc. Ethiop. 2000, 14, 129. 8. Literatur - 121 - [135] Y. D. M. Champouret, J.-D. Maréchal, I. Dadhiwala, J. Fawcett, D. Palmer, K. Singh, G. A. Solan, Dalton Trans. 2006, 2350. [136] K.-Y. Ho, W.-Y. Yu, K.-K. Cheung, C.-M. Che, Dalton Trans. 1999, 1581. [137] F. Pezet, I. Sasaki, J.-C. Daran, J. Hydrio, H. Aït-Haddou, G. Balavoine, Eur. J. Inorg. Chem. 2001, 2669. [138] M. Vlasse, T. Rojo, D. Beltran-Porter, Acta Cryst. 1983, C39, 560. [139] M. Wriedt, I. Jeß, C. Näther, Inorg. Chem. 2008, eingereicht. [140] S. A. Bourne, M. Kilkenny, L. R. Nassimbeni, J. Chem. Soc., Dalton Trans. 2001, 1176. [141] B. Staub, J. Pickardt, Z. Naturforsch. 1996, 51b, 947. [142] Y. Song, Y. Niu, H. Hou, Y. Zhu, J. Mol. Struct. 2004, 689, 69. [143] F. Lloret, M. Julve, J. Cano, G. D. Munno, Mol. Cryst. Liq. Cryst. 1999, 334, 569. [144] F. Lloret, G. D. Munno, M. Julve, J. Cano, R. Ruiz, A. Caneschi, Angew. Chem. Int. Ed. Engl. 1998, 37, 135. [145] R. Sieber, S. Decurtins, H. Stoeckli-Evans, C. Wilson, D. Yufit, J. A. K. Howard, S. C. Capelli, A. Hauser, Chemistry 2000, 6, 361. [146] S. Decurtins, H. W. Schmalle, P. Schneuwly, J. Ensling, P. Guetlich, J. Am. Chem. Soc. 1994, 116, 9521. [147] I. Jess, C. Näther, Inorg. Chem. 2006, 7446. [148] C. Näther, J. Greve, I. Jeß, Solid State Sci. 2002, 4, 813. [149] C. Näther, J. Greve, I. Jeß, C. Wickleder, Solid State Sci. 2004, 5, 1167. [150] C. Näther, I. Jeß, N. Lehnert, D. Hinz-Hübner, Solid State Sci. 2003, 5, 1343. [151] J. A. Real, G. D. Munno, M. C. Munoz, M. Julve, Inorg. Chem. 1991, 30, 2701. [152] J.-M. Shi, Y.-M. Sun, X. Zhang, L. Yi, P. Cheng, L.-D. Liu, J. Phys. Chem. A 2006, 110, 1677. [153] J.-K. Cheng, Y.-G. Yao, J. Zhang, Z.-J. Li, Z.-W. Cai, X.-Y. Zhang, Z.-N. Chen, Y.-B. Chen, Y. Kang, Y.-Y. Qin, Y.-H. Wen, J. Am. Chem. Soc. 2004, 126, 7796. [154] M. Kondo, S. Kawata, S. Kitagawa, H. Kiso, M. Munakata, Acta Cryst. 1995, C51, 567. [155] F. Nicolò, G. Bruno, G. Tresoldi, Acta Cryst. 1996, C52, 2188. [156] G. Tresoldi, E. Rotondo, P. Piraino, M. Lanfranchi, A. Tiripicchio, Inorg. Chim. Acta 1992, 194, 233. [157] J. Pickardt, J. v. Chrzanowski, R. Steudel, M. Borowski, S. Beck, Z. Naturforsch. 2005, 60, 373. [158] N. V. Raghavan, K. Seff, Acta Cryst. 1977, B33, 386. [159] A. F. Holleman, E. Wiberg, Lehrbuch der Anorganischen Chemie, Vol. 101, de Gruyter, Berlin - New York, 1995. Erklärung Die vorliegende Diplomarbeit wurde im Zeitraum vom 01.11. bis 02.05.2008 am Institut für Anorganische Chemie der Christian-Albrechts-Universität zu Kiel unter der Leitung von PD Dr. Christian Näther durchgeführt. Ich erkläre, dass ich die vorliegende Arbeit selbständig und nur unter Verwendung der angegebenen Hilfsmittel erstellt habe. Kiel, den 02.05.2008 ______________________________ (Mario Wriedt)