Skript 12
Werbung

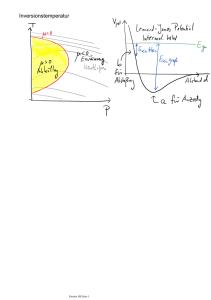
12. Vorlesung, Do, 1.12.2011 12.1 Verschiedene Formulierungen des zweiten Hauptsatzes Rudolf Clausius: Wärme kann nie von selbst von einem kälteren in ein wärmeres Reservoir übergehen. William Thomson Kelvin: Unmöglichkeit eines Perpetuum mobile 2ter Art. Perpetuum mobile 1ter Art: Erzeugung von Arbeit aus dem Nichts (Widerspruch zum 1ten Hauptsatz) Perpetuum mobile 2ter Art: Periodisch arbeitende Maschine, die die zugeführte Wärme zu 100% in Arbeit umwandelt. Für ein abgeschlossenes System bestehend aus Wärmereservoir R, Maschine M und Speicher S der gewonnenen Arbeit gilt für den Maschinenprozess: ΔS= ΔSR+ΔSM+ ΔSS >= 0 Der Speicher habe einen oder wenige Freiheitsgrade (z.B. ein Freiheitsgrad für die Dehnung einer Feder-> ΔSS~kB ). Das Wärmereservoir hat sehr viel mehr Freiheitsgrade (ΔSR >> ΔSS). Die Maschine hat nach einem Zyklus die gleiche Entropie (ΔSM=0). Für das Wärmereservoir gilt im quasistatisch-reversiblen Fall: ΔSR=-q/T. Diese von der Maschine aufgenommene Wärme wird in Arbeit umgewandelt und gespeichert. ΔS= ΔSR+ΔSM+ ΔSS = -q/T < 0 Mittels eines P.M. 2ter Art könnte man aus einem Reservoir bei T1 Wärme entziehen und in einem Carnot-Prozess in Arbeit umwandeln. Dies könnte man nutzen, um ein heißeres Reservoir zu erwärmen (entspricht Fluss von Wärme von einem kälterem zu einem wärmeren Bad). D.h. Clausius-Definition ist äquivalent zu Kelvin-Definition Umgekehrt: Könnte Wärme von einem kälteren in ein heißeres Reservoir fließen, so könnte man diese in einem Carnot-Prozess verwenden, und die Wärme dann wieder an das kalte Reservoir zurückführen. Dabei hätte man effektiv nur dem kälteren Reservoir Wärme entzogen (und damit Arbeit geleistet)-> Perpetuum Mobile 2ter Art (Kelvin äquivalent zu Clausius). Der zweite H.S. legt einen Zeitpfeil fest, d.h. obwohl die mikroskopischen Naturgesetze i.A. invariant gegenüber Zeitumkehr sind, können irreversible makroskopische Prozesse nur in einen Richtung laufen. 12.2. Carnot-Maschine ist optimal Carnot 1 Q T1 1 1 ; T2 Q2 dS Q T ; Q dS 0 T Wir betrachten den allgemeinen Kreisprozess, bei dem Wärmeaustausch mit der Umgebung stattfinden kann (nicht notwendigerweise nur bi Maximal- und Minimaltemperatur). Wir zerlegen den Prozess in Abschnitte mit dQ > 0 und Wärmeabgabe dQ < 0 und erlauben auch irreversible Prozessführung. Für die bestmögliche Umsetzung von Wärme in Arbeit gilt: W Q Q Q Q Q 2 Q 0 Q1 Q0 zweiter Hauptsatz : 0 Q T Q 0 Q T Q0 Q T Q2 Q1 T2 T1 gilt wegen : T1 T T2 0 Q2 Q1 T Q 1 1 T2 T1 T2 Q2 Q2 Q1 Q T 1 1 1 1 Carnot Q2 Q2 T2 Der Wirkungsgrad ist also nur dann gleich dem Carnot-Wirkungsgrad, wenn Wärmeübertragung nur bei der Maximal- und Minimaltemperatur erfolgt und wenn alle Teilprozesse reversible erfolgen. Jede reversibel zwischen zwei Temperaturen arbeitende Maschine erreicht den Carnot-Wirkungsgrad. Dies kann man durch Zusammenschalten der Maschine mit einer als Wärmepumpe arbeitenden Carnot-Maschine beweisen. Das Resultat zeigt, dass dies auch unabhängig von der Arbeitssubstanz gilt. Man kann dies nutzen, um eine Temperaturskala zu definieren (unter Einbeziehung einer Referenztemperatur). Der absolute Nullpunkt entspricht dann der Temperatur für das kältere Reservoir einer Carnot-Maschine mit Wirkungsgrad 1 (Wirkungsgrad Null bei der Referenztemperatur). 11.3. Dritter Hauptsatz der Thermodynamik Wir betrachten ein System mit sehr kleinen Werten der inneren Energie. Die Quantenmechanik sagt uns, es gibt einen Grundzustand mit Energie Eo. Wenn wir die Möglichkeit der Entartung von Zuständen zunächst außer Acht lassen, entspricht dies einem einzigen Mikrozustand. Ω(E->Eo)=1 => S(E->Eo)= kBln Ω(E->Eo)=0 Wenn man ausgehend vom Grundzustand höhere Niveaus anregt: Ω(E) ~(E-Eo)f, f~3N/2 1/kBT~ f/(E-Eo) -> ∞ für E-> Eo => S(T->0)=0 Entartung des Grundzustands kann auftreten, z.B. in einem System von nicht (oder fast nicht) mit einander wechselwirkender Objekte (z.B. Kernspin). Beim Abkühlen ergeben sich (für Spin ½) 2N Einstellmöglichkeiten der N Kernspins (2N-fache Entartung des Grundzustands). Dies führt zu einer „Restentropie“ So= kBln2N=2 kBln2 (Spin-Spin-Wechselwirkung ist allerdings bei sehr tiefen Temperaturen u.U. nicht mehr vernachlässigbar). Fazit im Grenzfall T-> 0 mit Werten von kBT < Anregungsenergien des Systems ändert sich die Entropie nicht mehr, d.h. ΔS(T->0+) =0. Dies ist der dritte Hauptsatz oder auch Nernst’sches Theorem. 11.4. Phasenübergänge Bisher haben wir Zustandsänderungen für einen bestimmten Aggregatzustand eines Stoffes betrachtet (d.h. die Zustandsgrößen ändern sich kontinuierlich). In der Realität haben wir es mit verschiedenen Aggregatzuständen (auch genannt Phasen: gasförmig, flüssig, fest) zu tun. Gleichgewichtszustände von Substanzen können durch Angabe von Volumen, Energie und Anzahl an Teilchen beschrieben werden. Praktisch sinnvoller ist die Angabe des Druckes p und der Temperatur T (und Teilchenzahl) als Zustandsvariablen für eine Substanz. Es besteht die Möglichkeit im Falle verschiedener Aggregatzustände, dass es für bestimmte werte paare der Variablen p, T zwei oder mehr Lösungen der Zustandsgleichungen gibt (z.B. V(p,T) ist nicht unbedingt eindeutig). Für diesen Fall können auch zwei oder mehr Phasen einer Substanz koexistieren. Man erhält dann im einfachsten Fall im p-T-Phasendiagramm eine Koexistenzlinie. Bewegt man sich über die Koexistenzlinie, entspricht dies einem Phasenübergang (z.B. fest-flüssig: Sublimation; fest-flüssig: Schmelzen; flüssig-gasförmig: Verdampfen). Dabei ändern sich einige Zustandsgrößen in nicht-stetiger Weise. Tripelpunkt: Falls sich 3 Koexistenzlinien an einem Punkt treffen, spricht man von Tripelpunkt (dort können drei Phasen koexistieren). Das Ende einer Koexistenzlinie ist ein kritischer Punkt. Wir haben bereits gezeigt, dass für zwei Teilsysteme A, B (gleich groß) im Gleichgewicht ein Zustand mit TA=TB, PA=PB, μA=μB eingenommen wird. D.h. die Teilchen verteilen sich so, das ein geeignetes thermodynamisches Potential (in unserem Fall G, da wir p, T, N vorgeben) ein Minimum einnimmt. Für homogene System (jede Phase betrachten wir als homogen) galt die Gibbs-Duhem-Gleichung: G(N,T,P) = NA μA(p,T)+ NB μB(p,T) Mit N=NA+NB Das vollständige Differential des chemischen Potentials im homogenen System war: dμ=-(S/N) dT+(V/N)dP Wir betrachten zwei mögliche Phasen (z.B. flüssig und gasförmig) A, B und interessieren uns für den Übergang zunächst entlang variablem T. Da die Struktur der Phasen unterschiedlich ist, wird der Verlauf des chemischen Potentials als Funktion von p,T unterschiedlich sein für jede Phase. Bei Koexistenz gilt μA(p,T)=μB(p,T). Mit G = NA μA(p,T)+ NB μB(p,T) minimal im Gleichgewicht, gehen für einen beliebigen Punkt im p-T-Phasendiagramm alle Teilchen in die Phase mit dem niedrigerem chemischen Potential über, also: μ(p,T)= μA(p,T), falls am gewählten p, T-Punkt μA<μB (dann ist Gmin= N μA(p,T)) μ(p,T)= μB(p,T), falls am gewählten p, T-Punkt μA>μB (dann ist Gmin= N μB(p,T)) Wie verläuft μ(p,T) ? Betrachte zunächst bei festem Druck po als Funktion der Temperatur. Informationen dazu können wir aus der ersten und zweiten Ableitung des chemischen Potentials nach T erhalten. S 0; N T p da S 0 Cp 1 2 1 S 2 N T p N T T p Die beiden Ableitungen legen Verlauf der Krümmung und Steigung der chemischen Potentiale für die beiden Aggregatzustände fest. Sei Phase B das Gas (und A der flüssige Zustand): Für T<To (entspricht der Koexistenztemperatur ) ist μA<μB (d.h. Phase A, wird eingenommen). Für T>To entsprechend der umgekehrte Fall. D. h. die chemischen Potentialkurven schneiden sich bei To und es gibt einen Wechsel (Umwandlung der Phasen zur Phase mit der jeweils niedrigeren freien Energie). Dies impliziert einen Knick für das chemische Potential bei To und die (negative) Ableitung nach der Temperatur (entspricht der spezifischen Entropie pro Teilchen) macht einen Sprung (Unstetigkeitstelle). Analoges Verhalten kann man zeigen für die Variation des Druckes bei festgehaltener Temperatur (man beobachtet dann einen Sprung im Volumen). Am kritischen Punkt im Phasendiagramm verschwindet die Unstetigkeit der ersten Ableitung (zweite Ableitungen bleiben unstetig). Klassifizierung von Phasenübergängen nach Ehrenfest: Sind alle mten Ableitungen mit m< n vom chemischen Potential μ stetig, die nten Ableitung aber unstetig, spricht man von einem Phasenübergang nter-Ordnung.