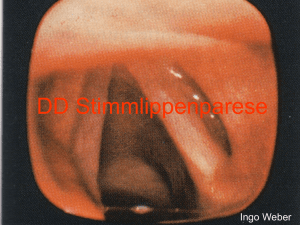
Tumorähnliche Läsionen 7
Werbung

Tumorähnliche Läsionen 7 R. Erlemann 7.1 Nicht-ossifizierendes Knochenfibrom 339 7.2 Solitäre Knochenzyste 345 7.3 Aneurysmatische Knochenzyste 348 7.4 Ganglion 354 7.5 Epidermoidzyste 357 7.6 Fibröse Dysplasie 358 7.7 Osteofibröse Dysplasie 365 7.8 7.8.1 7.8.2 7.8.3 Langerhans-Zell-Histiozytose 368 Eosinophiles Granulom 368 Hand-Schüller-Christian-Erkrankung 373 Abt-Letterer-Siwe-Erkrankung 373 7.9 Diagnose-Schemata 374 Literatur 376 7.1 Nicht-ossifizierendes Knochenfibrom Der fibröse Kortikalisdefekt und das nicht-ossifizierende Knochenfibrom (NOF) sind unterschiedliche Stadien des gleichen nichttumorösen Prozesses. Als Ursache wird eine Entwicklungsstörung vermutet. Definition 왔 Die Läsion ist durch das Vorhan- densein von fibrösem Gewebe in wirbelförmiger Anordnung mit eingelagerten mehrkernigen Riesenzellen, Schaumzellen, Cholesterolkristallen und Hämosiderinablagerungen gekennzeichnet. Man kann diese Läsion als fibrösen (metaphysären) Kortikalisdefekt bezeichnen, wenn nur die Kompakta betroffen ist und die Größe unter 1 cm liegt. Diese Veränderung wird bei vielen Kindern während des normalen Skelettwachstums beobachtet. Ist die Läsion größer und ist auch der Markraum betroffen, wird sie als nicht-ossifizierendes Knochenfibrom bezeichnet. In weniger als 10% der Fälle liegen multiple nicht-ossifizierende Knochenfibrome vor. Eine Koinzidenz von multiplen nicht-ossifizierenden Knochenfibromen und einer Neurofibromatose ist bekannt. Viele Patienten mit einer Neurofibromatose zeigen mehrere nicht-ossifizierende Knochenfibrome, vorzugsweise am Kniegelenk. Daher sollte beim Nachweis multipler nicht-ossifizierender Knochenfibrome eine subtile Neurofibromatosediagnostik durchgeführt werden. Große nichtossifizierende Knochenfibrome werden gelegentlich auch als Xanthofibrome bezeichnet. In diesen ist der Anteil an Schaumzellen höher als in einem nichtossifizierenden Knochenfibrom. Inzidenz Das nicht-ossifizierende Knochenfibrom ist die häufigste tumorähnliche Läsion. In einer normalen Population kann eine Inzidenz von etwa 2% aller Personen angenommen werden. Dabei überwiegen die kleinen intrakortikalen Veränderungen. Viele werden radiologisch richtig diagnostiziert und nicht weiter abgeklärt. Meist größere Läsionen und solche, die radiologisch nicht eindeutig als nicht-ossifizierendes Knochenfibrom zu klassifizieren sind, werden biopsiert und an ein Knochengeschwulstregister gesandt. Sie machen dort bis zu 10% der benignen Raumforderungen aus. Alter und Geschlecht Das nicht-ossifizierende Knochenfibrom tritt typischerweise bei Kindern und Jugendlichen auf. Es liegt ein klarer Altersgipfel in der 2. Lebensdekade vor. Drei Viertel der nicht-ossifizierenden Knochenfibrome werden in der 2. Lebensdekade und mehr als 95% der Läsionen zwischen fünf und 20 Jahren beobachtet. Männer sind etwas häufiger als Frauen betroffen. Klinik Die meisten nicht-ossifizierenden Knochenfibrome verursachen keine Symptome und werden als Zufallsbefund entdeckt. Größere Läsionen können Schmerzen verursachen und gelegentlich zu einer pathologischen Fraktur führen. Lokalisation Zwei Drittel der nicht-ossifizierenden Knochenfibrome finden sich in der Umgebung des Kniegelenks. Dabei sind das distale Femur mit einem Drittel häu- 340 Kapitel 7 Tumorähnliche Läsionen Abb. 7.1. Statistische Daten zum nicht-ossifizierenden Knochenfibrom Abb. 7.2 a, b. Fibröser Kortikalisdefekt (nicht-ossifizierendes Knochenfibrom). a In der Tibiametaphyse stellt sich eine kleine intrakortikale Osteolyse dar, die durch einen dünnen Sklerosesaum vom Markraum abgegrenzt ist. b Das T1-gewichtete SE-Bild bildet die Läsion als muskelisointense Raumforderung in der Kompakta ab b a figer als die proximale Tibia mit einem Viertel und die proximale Fibula mit etwa einem Zehntel der Fälle betroffen. Häufig wird das nicht-ossifizierende Knochenfibrom noch in der distalen Tibia (22%), selten im proximalen Femur (3%), im proximalen Humerus (3%) und in der distalen Fibula (2%) angetroffen. Ein Befall anderer Extremitätenknochen ist eine Rarität. Im Stammskelett tritt das nicht-ossifizierende Knochenfibrom mit extrem wenigen Ausnahmen nicht auf. In den Röhrenknochen wird das nicht-ossifizierende Knochenfibrom in der Hälfte der Fälle diaphysär und in der anderen Hälfte metaphysär oder metadiaphysär angetroffen. Eine Ausdehnung in die Epiphyse ist eine große Rarität. Mehr als vier Fünftel der Läsionen liegen exzentrisch im Knochen. Große nicht-ossifizierende Knochenfibrome können den gesamten Markraum einnehmen, was häufig in der Fibula beobachtet wird (Abb. 7.1). Röntgenmorphologie Der fibröse Kortikalisdefekt stellt sich als exzentrisch in der Kompakta liegende meist ovaläre Osteolyse dar, die glatt oder lobuliert begrenzt ist. Sie kann sich etwas Richtung Markraum vorwölben und ist durch einen feinen Sklerosesaum von diesem abgegrenzt. Auf der Weichteilseite kann eine feine mineralisierte Periostlamelle vorliegen, die allerdings auch fehlen kann (Abb. 7.2 a, b). Das nicht-ossifizierende Knochenfibrom stellt sich als einige Zentimeter große Osteolyse dar, die trabekuliert ist und meist einen lobulierten und nahezu immer einen komplett ausgebildeten feinen sklerotischen Randsaum aufweist.Viele der Läsionen sind ovalär (Abb. 7.3, Abb. 7.4, Abb. 7.5). Die Kompakta ist häufig ausgedünnt und durch Ausbildung einer Periostschale etwas vorgewölbt (Abb. 7.6). Seltener werden glatt begrenzte, nichtlobulierte Osteolysen angetroffen. 7.1 Nicht-ossifizierendes Knochenfibrom Abb. 7.3. Nicht-ossifizierendes Knochenfibrom. Klassische Morphologie einer exzentrisch metadiaphysär gelegenen osteolytischen Läsion, die von einem sklerotischen Randsaum auf der Markraumseite umgeben ist (Lodwick IA). Die Ränder sind lobuliert, und die Kompakta ist ausgedünnt. Einzelne Areale sind verknöchert Abb. 7.4. Nicht-ossifizierendes Knochenfibrom. Typische Morphologie einer exzentrisch metadiaphysär gelegenen Raumforderung, die durch einen kompletten sklerotischen Randsaum vom Markraum abgegrenzt wird und trabekuliert ist (Lodwick IA). Die Kompakta ist aufgelöst, und eine Periostschale ist ausgebildet. Oberhalb kommt ein kleiner satellitenartiger Herd zur Darstellung Abb. 7.5. Nicht-ossifizierendes Knochenfibrom. Größere mehr im Markraum gelegene Läsion, die trabekuliert ist, einen lobulierten sklerotischen Randsaum aufweist und die Kompakta nicht ausgedünnt hat (Lodwick IA) Abb. 7.6. Nicht-ossifizierendes Knochenfibrom. Die Läsion nimmt große Teile des Markraums in Anspruch und hat die Kompakta kaudal nahezu komplett resorbiert 341 342 Kapitel 7 Tumorähnliche Läsionen Abb. 7.7. Nicht-ossifizierendes Knochenfibrom. Die Läsion zeigt ausgedehnte Verknöcherungen, die auf eine Ausheilung hinweisen Abb. 7.8. Multiple nicht-ossifizierende Knochenfibrome. Die Patientin mit einer Neurofibromatose zeigt multiple zentrale und exzentrische Herde,die alle nicht-ossifizierenden Knochenfibromen entsprechen b a Abb. 7.9 a, b. Nicht-ossifizierendes Knochenfibrom. a Im Röntgenbild bietet die Raumforderung eine charakteristische Morphologie. b In der CT kann eine partielle Destruktion der Kompakta nachgewiesen werden, ohne dass eine extraossäre Komponente vorhanden ist. Die Läsion ist durch einen Sklerosesaum zum Markraum abgegrenzt 7.1 Nicht-ossifizierendes Knochenfibrom Die Wachstumsgeschwindigkeit ist niedrig und entspricht nahezu immer einem Grad Lodwick IA. Das nicht-ossifizierende Knochenfibrom bildet sich spontan zurück, indem es ossifiziert. Diese Veränderungen beginnen in der Peripherie und schreiten in Richtung Zentrum fort. In späten Stadien ist die gesamte Läsion mehr oder minder ossifiziert und bietet eine mattglasartige oder deutlich sklerotische Matrix (Abb. 7.7, Abb. 7.8). Das typische nicht-ossifizierende Knochenfibrom ist eine deutlich lobulierte, meist ovaläre Osteolyse mit sklerotischem Randsaum, die exzentrisch metaphysär, metadiaphysär oder metaphysennah diaphysär gelegen ist und bei Kindern und Jugendlichen angetroffen wird. Merke ! Schnittbilddiagnostik Bei einer charakteristischen Röntgenmorphologie ist für die diagnostische Aufarbeitung eines nicht-ossifizierenden Knochenfibroms keine Schnittbilddiagnostik erforderlich. In der CT ist der die Läsion allseits zum Markraum abgrenzende Sklerosesaum sichtbar. Bei größeren Läsionen ist meist auch eine mineralisierte Periostreaktion als Abgrenzung zu den Weichteilen sichtbar, die bei kleinen Läsionen fehlen kann. Im Ausheilungsprozess wird die Matrix zunehmend osteoblastisch (Abb. 7.9 a, b). In der MRT stellt sich das nicht-ossifizierende Knochenfibrom im T1-gewichteten Bild isointentens oder hypointens zur Muskulatur dar. Im T2-gewichteten Bild kann die Läsion isointens zur Muskulatur oder fokal oder weitgehend homogen mäßig hyperintens sein. Diese Variation wird durch unterschied- a b Abb. 7.10 a–c. Nicht-ossifizierendes Knochenfibrom. a Im T1gewichteten SE-Bild weist die Läsion eine vergleichbare Signalintensität wie die benachbarte Muskulatur auf. b Im STIRBild ist sie nur im Randbereich und in einer septalen Struk- liche prozentuale Zusammensetzungen aus fibrotischem Stroma, Riesenzellen, Schaumzellen, Cholesterolkristallen und Hämorrhagie bewirkt. Der sklerotische Randsaum ist bei auf dem T2-gewichteten Bild hyperintensen Läsionen als signalarme Begrenzung sichtbar. Im Heilungsprozess kann gelegentlich zentral Fett eingelagert werden. Das Kontrastmittelenhancement ist eher mäßig, im Randbereich unter dem Sklerosesaum häufig jedoch ausgeprägter (Abb. 7.10 a–c,Abb. 7.11 a, b,Abb. 7.12 a, b). In der Skelettszintigraphie zeigen die Läsionen eine geringe bis mäßige Traceraufnahme. Differenzialdiagnose Das nicht-ossifizierende Knochenfibrom kann immer sicher als benigne Läsion und in den meisten Fällen korrekt diagnostiziert werden. Die Morphologie ist in den meisten Fällen so charakteristisch, dass keine histologische Sicherung notwendig ist. Bei großen Läsionen besteht eine Verwechslungsgefahr mit einer aneurysmatischen Knochenzyste. Ein ausgeprägt lobulierter Rand und ein komplett ausgebildeter feiner Skleroserand sprechen für ein nicht-ossifizierendes Knochenfibrom. Ist das nicht-ossifizierende Knochenfibrom im Stadium der Ausheilung, kann es mit einer fibrösen Dysplasie verwechselt werden. Solange das nicht-ossifizierende Knochenfibrom jedoch noch den stark lobulierten Rand aufweist, wird man es als solches erkennen. Therapie und Prognose Die meisten nicht-ossifizierenden Knochenfibrome bilden sich spontan zurück und bedürfen keiner Therapie. Sie gehören zu den „Leave-me-alone-Läsionen“, die in den meisten Fällen keiner histologischen Sicherung bedürfen. c turierung deutlich signalintensiver als die Muskulatur. c Im kontrastmittelverstärkten T1-gewichteten SE-Bild zeigt sie ein geringes Enhancement 343 344 Kapitel 7 Tumorähnliche Läsionen Abb. 7.11 a, b. Nicht-ossifizierendes Knochenfibrom. Sowohl im a T1-gewichteten SE-Bild als auch im b T2-gewichteten SE-Bild zeigt die Raumforderung eine ähnliche Signalintensität wie die Muskulatur. Die signalarme Darstellung im T2-gewichteten SE-Bild wird bei einigen nicht-ossifizierenden Knochenfibromen angetroffen a b b a Abb. 7.12 a, b. Nicht-ossifizierendes Knochenfibrom mit pathologischer Fraktur. a Im Röntgenbild recht charakteristische Morphologie der Läsion mit einer kortikalen Fraktur. b Im T2-gewichteten SE-Bild liegt frakturbedingt ein ausgedehntes peritumorales Muskelödem vor, das ohne Kenntnis des Röntgenbildes einen aggressiv wachsenden Tumor vortäuschen kann Ein therapeutischer Eingriff ist nach Eintritt oder bei Gefahr einer pathologischen Fraktur erforderlich. Die Gefahr einer pathologischen Fraktur ist gegeben, wenn das nicht-ossifizierende Knochenfibrom mehr als 50% des Knochendurchmessers einnimmt. Eine Kürettage mit einer anschließenden Auffüllung mit Knochenspänen ist dann ausreichend. Da nach eingetretener pathologischer Fraktur allein mit einer Gipsbehandlung eine Frakturheilung erzielt werden kann, ist auch bei einem großen nichtossifizierenden Knochenfibrom die prophylaktische Stabilisierung umstritten. 7.2 Solitäre Knochenzyste Abb. 7.13. Statistische Daten zur solitären Knochenzyste 7.2 Solitäre Knochenzyste Definition 왔 Die solitäre oder juvenile Knochen- zyste (SKZ) ist eine einkammerige Knochenzyste, die mit einer klaren gelblichen Flüssigkeit gefüllt ist. Die Zystenwände sind mit einer dünnen Membran ausgekleidet, die lockeres Bindegewebe und eingestreute osteoklastäre Riesenzellen enthält. Nach einer pathologischen Fraktur ist die Flüssigkeit blutig. Die Pathogenese der solitären Knochenzyste ist unbekannt. Eine Theorie besagt, dass sie als Folge einer vaskulären Störung auftritt, die zu einer venösen Okklusion und zu einem erhöhten intraossären Druck führt. Im weiteren Verlauf kommt es zu einer Knochenresorption und Auffüllung der Höhle mit extrazellulärer Flüssigkeit. In einigen Fällen kann eine solitäre Knochenzyste weder radiologisch noch histologisch von einer aneurysmatischen Knochenzyste sicher abgegrenzt werden. Inzidenz Die solitäre Knochenzyste gehört zu den recht häufig auftretenden benignen ossären Raumforderungen und macht etwa 6% aller benignen Knochentumoren und tumorähnlichen Läsionen aus. Alter und Geschlecht Die solitäre Knochenzyste tritt in der großen Mehrzahl der Fälle bei Kindern und Jugendlichen auf. Der Altersgipfel liegt zwischen dem 5. und 15. Lebensjahr. Etwa 85% der Patienten sind jünger als 20 Jahre. Männer sind etwa doppelt so häufig wie Frauen betroffen. Klinik Die solitäre Knochenzyste verursacht nur geringe oder keine Beschwerden. In vielen Fällen wird sie erst durch eine nach einem banalen Trauma eingetretene pathologische Fraktur auffällig. In anderen Fällen wird sie als Zufallsbefund bei einer Röntgenuntersuchung entdeckt. Schwellungen werden nur selten beobachtet, da die solitäre Knochenzyste keine relevante Knochenauftreibung bewirkt. Eine Einschränkung der Gelenkbeweglichkeit tritt nicht auf. Lokalisation Etwa die Hälfte aller solitären Knochenzysten werden im Humerus angetroffen, davon sind 90% im proximalen Humerus und 10% im Schaft gelegen. Die zweithäufigste Lokalisation ist das Femur, in dem ein Viertel der solitären Knochenzysten, meistens im proximalen Abschnitt, angetroffen werden. Daneben werden die Läsionen mit einer Häufigkeit von 5–10% in den tarsalen Knochen, meist dem Kalkaneus, dem Becken, der Tibia und der Fibula gefunden. Es wird angenommen, dass die solitäre Knochenzyste in der Metaphyse entsteht. Durch das Knochenwachstum entfernt sich die Epiphysenfuge von der Läsion, sodass die solitäre Knochenzyste nach diaphysär wandert. Diese Theorie konnte jedoch bisher noch nicht sicher bewiesen werden. Bei Diagnosestellung sind knapp die Hälfte der solitären Knochenzysten metadiaphysär gelegen, ein Drittel findet sich diaphysär und ein Fünftel metaphysär. Eine Ausdehnung in die Epiphyse wird nur bei etwa 3% der Läsionen beobachtet. Etwa 85% der solitären Knochenzysten sind zentral im Knochen lokalisiert (Abb. 7.13). 345 346 Kapitel 7 Tumorähnliche Läsionen Abb. 7.14. Solitäre Knochenzyste. Es liegt eine zentral diaphysär lokalisierte osteolytische Läsion vor, die allseits von einem sklerotischen Randsaum umgeben ist und Preudotrabekel aufweist (Lodwick IA). Die Kompakta ist fokal gering ausgedünnt Abb. 7.15. Solitäre Knochenzyste. Es findet sich eine metadiaphysär zentral gelegene Osteolyse, die in Kontakt mit der Epiphysenfuge steht und einige Preudotrabekel aufweist (Lodwick IB) Röntgenmorphologie Die solitäre Knochenzyste stellt sich fast ausschließlich als osteolytische Läsion dar, die in knapp der Hälfte der Fälle mehr oder minder ausgeprägte Trabekulierungen oder Pseudotrabekulierungen aufweist. In weniger als 10% der Fälle werden einzelne Verkalkungen oder osteoblastische Bezirke angetroffen. In den langen Röhrenknochen ist die lange Achse der solitären Knochenzyste immer in Richtung der Knochenachse ausgerichtet. Die Osteolyse ist somit fast immer oval (Abb. 7.14, Abb. 7.15). Die solitären Knochenzysten wachsen langsam und weisen häufig einen sklerotischen Randsaum auf. Sie können etwas lobuliert sein. Sie zeigen eine Wachstumsgeschwindigkeit vom Grad Lodwick IA oder IB. In den Röhrenknochen treiben sie in zwei Dritteln der Fälle durch die Bildung einer Periostschale den Knochen leicht bis mäßig auf, wobei die Breite der solitären Knochenzyste meist nicht die Breite der benachbarten Epiphysenfuge übersteigt (Abb. 7.16). Andere Periostreaktionen stellen eine Rarität dar und sind nur bei einer Infraktion vorhanden. Nach eingetretener Fraktur kann ein „Fallen-fragment-Zeichen“ sichtbar sein. Dabei ist ein Knochenfragment in die solitäre Knochenzyste verlagert und setzt sich im abhängigen Abschnitt ab. Dieses ist jedoch nur bei 20% der solitären Knochenzysten nachweisbar. Ein vergleichbares Zeichen ist die „trap door“, bei der das Fragment am Periost angeheftet bleibt, aber sich nach innen in die solitäre Knochenzyste vorwölbt (Abb. 7.17). Im Kalkaneus ist die solitäre Knochenzyste in der Region des Ward-Dreiecks gelegen und nahezu rund, während das Ward-Dreieck (eine physiologische Zone mit deutlich verminderter Trabekeldichte) dreieckförmig und meist nicht komplett trabekelfrei ist (Abb. 7.18 a, b). Das typische Bild einer solitären Knochenzyste ist eine rein osteolytische ovaläre Läsion mit sklerotischem Randsaum oder scharfem Rand, die zentral metadiaphysär oder diaphysär, meist im Humerus bei Kindern angetroffen wird. Merke ! Schnittbilddiagnostik In den Röhrenknochen stellt die konventionelle Röntgendiagnostik die solitäre Knochenzyste ausreichend dar. Im Beckenskelett muss eine CT oder auch MRT durchgeführt werden, um die ventrale und dorsale Kompakta beurteilen zu können. In der CT lassen sich bei nichtfrakturierten Läsionen gelegentlich flüssigkeitsäquivalente Dichtewerte bestimmen. 7.2 Solitäre Knochenzyste Abb. 7.16. Solitäre Knochenzyste. Osteolytische Läsion, die multiple Pseudotrabekel aufweist und den Knochen durch Induktion einer Periostschalenbildung aufgetrieben hat (Lodwick IB). Die differenzialdiagnostische Abgrenzung von einer aneurysmatischen Knochenzyste ist nicht möglich Abb. 7.17. Solitäre Knochenzyste mit pathologischer Fraktur. Recht typische Morphologie einer solitären Knochenzyste, die frakturiert ist. Ein kortikales Fragment ist in die Läsion eingedrungen a Abb. 7.18 a, b. Solitäre Knochenzyste. a Rein osteolytische Läsion im Kalkaneus, die im Gegensatz zu einem intraossären Lipom keine zentrale dystrophe Verkalkung aufweist. b Im STIR-Bild ist nahezu die gesamte Läsion sehr signalintensiv, was auf einen hohen Wassergehalt hinweist b 347 348 Kapitel 7 Tumorähnliche Läsionen In der MRT bestimmt bei der unkomplizierten solitären Knochenzyste der Zysteninhalt die Morphologie. Die solitäre Knochenzyste weist eine recht niedrige Signalintensität im T1-gewichteten und eine extrem hohe Signalintensität im T2-gewichteten Bild auf. Ein in beiden Sequenzen abgrenzbarer dunkler Randsaum entspricht einem Sklerosesaum.Septale Strukturen und eine Weichteilkomponente werden nicht angetroffen.Nach Kontrastmittelgabe zeigte die peripher gelegene Zystenwand ein diskretes Enhancement. Eine Einblutung in die Zyste verändert die Morphologie deutlich. Es können Flüssigkeits-Flüssigkeits-Spiegel und/oder im T1-gewichteten Bild signalintensive Areale auftreten. Bei einer pathologischen Faktur kann eine ödematöse und/oder hämorrhagische Reaktion in den Weichteilen sichtbar sein. Bezirke durch Septen abgegrenzt, müssen diese zusätzlich anpunkiert werden. Normalerweise dauert es 6–12 Monate, bis eine deutliche Regredienz auf dem Röntgenbild sichtbar ist. Gegebenenfalls ist die Steroidinjektion zu wiederholen. Differenzialdiagnose Die solitäre Knochenzyste wird nahezu immer richtig als benigne Läsion eingestuft. Bei einer Lage in der proximalen Humerusmetaphyse oder -diaphyse kann in vielen Fällen die korrekte Diagnose gestellt werden, besonders wenn eine Lodwick-IA-Läsion und/oder eine nur geringe Knochenauftreibung vorliegen. Bei einer deutlicheren Knochenauftreibung ist radiologisch kaum eine Differenzierung von einer aneurysmatischen Knochenzyste möglich. Auch in den anderen großen Röhrenknochen wird man bei einer Lodwick-IA-Läsion und einer nur geringen Knochenauftreibung eher die Diagnose einer solitären Knochenzyste als die einer aneurysmatischen Knochenzyste stellen. Mittels MRT ist häufig eine Differenzierung zwischen solitärer und aneurysmatischer Knochenzyste, die in der Zyste gut vaskularisierte Septen aufweist, möglich. Nach mehreren Frakturen kann die solitäre Knochenzyste durch die reaktive Kallusbildung das Bild einer fibrösen Dysplasie vortäuschen. Im Kalkaneus kann eine solitäre Knochenzyste mit einem Lipom verwechselt werden, das jedoch typischerweise eine zentrale dystrophe Verkalkung aufweist. Mittels CT oder MRT ist durch den Fettnachweis eine sichere Differenzierung möglich. Die Septen werden durch Endothelzellen ausgekleidet und enthalten mehrkernige Riesenzellen und unregelmäßige Knochentrabekel sowie gelegentlich Osteoid. Die exakte Pathogenese der aneurysmatischen Knochenzyste ist unbekannt. Es wird einerseits diskutiert, dass diese Läsion Folge eines veränderten Blutflusses mit einer daraus resultierenden abnormalen lokalen Durchblutung sei.Andererseits wird vermutet, dass sie als Folge einer massiven Einblutung in einen vorbestehenden Tumor entsteht. Die letztere Theorie wird durch die Tatsache unterstützt, dass die aneurysmatische Knochenzyste nicht selten als sekundäre Veränderung bei anderen Knochentumoren und tumorähnlichen Läsionen auftritt. Hierzu zählen das Chondroblastom, der Riesenzelltumor, das nicht-ossifizierende Knochenfibrom, die fibröse Dysplasie, die solitäre Knochenzyste, das Chondromyxoidfibrom, das Osteoblastom und das reparative Riesenzellgranulom. Einige sekundäre aneurysmatische Knochenzysten sind auch in Osteosarkomen, malignen fibrösen Histiozytomen und Fibrosarkomen beobachtet worden. Therapie und Prognose Eine Therapiemöglichkeit besteht in einer sorgfältigen Kürettage und Auffüllung der Höhle mit Knochenspänen. Nach eingetretener pathologischer Fraktur wartet man, bis sich ausreichend Kallus gebildet hat, bevor die Kürettage durchgeführt wird. Eine andere Therapiemöglichkeit besteht in der Punktion der solitären Knochenzyste und der Instillation von Steroiden nach Entleerung der Flüssigkeit. Zur Kontrolle, dass sämtliche Zystenanteile von der Therapie erreicht werden, kann eine Kontrastmittelfüllung der Zyste durchgeführt werden. Sind einzelne 7.3 Aneurysmatische Knochenzyste Definition 왔 Die aneurysmatische Knochenzyste (AKZ) ist eine tumorähnliche Knochenläsion, die aus multiplen mit ungeronnenem Blut gefüllten Hohlräumen besteht, die von Septen umgrenzt werden. Inzidenz Die aneurysmatische Knochenzyste gehört zu den häufigeren Knochenläsionen und macht nahezu 5% aller gutartigen Knochentumoren und tumorähnlichen Läsionen aus. Etwas mehr als 10% der aneurysmatischen Knochenzysten werden als sekundäre Läsionen, besonders bei Chondroblastomen beobachtet. Alter und Geschlecht Die aneurysmatische Knochenzyste wird besonders bei jungen Patienten angetroffen. Die meisten Läsionen werden in der 1. und 2. Lebensdekade diagnostiziert. Drei Viertel der aneurysmatischen Knochenzysten werden bis zum 20. Lebensjahr und 90% bis zum 30. Lebensjahr entdeckt. Ein Auftreten vor dem 5. Lebensjahr ist jedoch eine Rarität. Männer und Frauen sind annähernd gleich häufig betroffen. 7.3 Aneurysmatische Knochenzyste Abb. 7.19. Statistische Daten zur aneurysmatischen Knochenzyste Klinik Die Klinik besteht meist aus nur gering ausgeprägten Schmerzen und häufig in einer tastbaren Schwellung. In einigen Fällen kann die Läsion sehr schnell wachsen, sodass klinisch ein maligner Tumor vermutet wird. Beim Befall eines Wirbelkörpers kann dieser zusammenbrechen, und es können neurologische Symptome auftreten. Die Dauer der Symptome vor Diagnosestellung beträgt meist einige Monate. In einem Viertel der Fälle ist eine pathologische Fraktur das erste bewusst wahrgenommene klinische Symptom. Lokalisation Mehr als die Hälfte der aneurysmatischen Knochenzysten finden sich in den langen Röhrenknochen. Hier sind besonders die Tibia (15%) und das Femur (13%), gefolgt von Fibula (12%) und Humerus (8%) betroffen. Daneben sind die Wirbelsäule (13%), die kleinen Röhrenknochen (10%) und das Becken (7%) nicht selten befallen. In den langen Röhrenknochen findet sich die aneurysmatische Knochenzyste in nahezu der Hälfte der Fälle metadiaphysär, in nahezu einem Drittel rein diaphysär, in knapp 10% metaphysär. In knapp 20% der Fälle liegt eine Ausdehnung in die Epiphyse vor, die jedoch meist erst nach Schluss der Epiphysenfuge beobachtet wird. Eine rein epiphysäre Lage wird nicht angetroffen. In den langen Röhrenknochen sind die aneurysmatischen Knochenzysten in etwa der Hälfte der Fälle zentral gelegen, wobei im Humerus die große Mehrzahl der aneurysmatischen Knochenzysten zentral lokalisiert ist. In den kleinen Röhrenknochen sind die aneurysmatischen Knochenzysten fast ausschließlich zentral gelegen. In der Wirbelsäule sind meist Wirbelbogen und Wirbelkörper betroffen, seltener der Wirbelkörper oder der Wirbelbogen allein. Da sich jedoch häufig die größte Tumormasse im Wirbelbogen befindet, geht man davon aus, dass die aneurysmatische Knochenzyste primär im Wirbelbogen entsteht (Abb. 7.19). Die aneurysmatische Knochenzyste ist die einzige benigne Läsion, die auch in die benachbarten Wirbelkörper einwachsen kann. Merke ! Röntgenmorphologie In der großen Mehrzahl der Fälle stellt sich die aneurysmatische Knochenzyste als reine Osteolyse dar, die nicht selten Trabekulierungen aufweist. Nur in etwa 5% sind neben einer osteolytischen Komponente auch osteoblastische Anteile oder Matrixverkalkungen vorhanden. Die Wachstumsgeschwindigkeit ist eher gering und entspricht meist einem Grad Lodwick IB, seltener einem Grad IA oder IC. Jedoch können bis zu 5% der aneurysmatischen Knochenzysten ein recht aggressives Wachstum zeigen und sogar einen Grad Lodwick II bieten. In bis zur Hälfte der Fälle liegt ein, jedoch meist nur partiell ausgebildeter, sklerotischer Randsaum vor (Abb. 7.20). Einige Läsionen besitzen einen lobulierten Rand. Vier Fünftel der aneurysmatischen Knochenzysten treiben den Knochen durch die Ausbildung einer Periostschale auf. Andere Typen von Periostreaktionen werden kaum beobachtet (Abb. 7.21, Abb. 7.22, Abb. 7.23 a, b). In seltenen Fällen kann die aneurysmatische Knochenzyste die Kompakta ohne Induzierung einer nativradiologisch sichtbar verkalkten periostalen Reaktion auflösen (Abb. 7.24, Abb. 7.25). In der CT ist dann jedoch fast immer eine begrenzende Periostlamelle sichtbar. 349 350 Kapitel 7 Tumorähnliche Läsionen Abb. 7.20. Aneurysmatische Knochenzyste. Es liegt eine exzentrisch metadiaphysär gelegene Läsion vor, die die mediale Kompakta fokal komplett penetriert hat und eine fokale Trabekulierung bietet (Lodwick IC) Abb. 7.22. Aneurysmatische Knochenzyste. Die zentral metadiaphysär gelegene Osteolyse weist eine ausgedehnte Trabekulierung auf und hat fokal die Kompakta komplett penetriert. Die Trabekulierung spricht eher für eine aneurysmatische als für eine solitäre Knochenzyste. Jedoch überschneidet sich die Morphologie beider Entitäten gerade am Humerus sehr stark, sodass häufig keine zuverlässige Differenzialdiagnose zu stellen ist In der Wirbelsäule wächst die aneurysmatische Knochenzyste als osteolytische Läsion, die den befallenen Wirbelabschnitt auftreibt. Sie kann eine Weichteilkomponente ausbilden. Im Beckenskelett ist das typische Bild eine Osteolyse, die meist recht scharfe Grenzen aufweist und den betroffenen Knochen mehr oder minder deutlich auftreibt. In den Fällen, in denen die aneurysmatische Knochenzyste als sekundäre Veränderung in einem anderen Knochentumor auftritt, steht fast immer die Röntgenmorphologie der ursprünglichen Läsion so deutlich im Vordergrund, dass die Koexistenz einer aneurysmatischen Knochenzyste nicht vermutet wird. Das typische Bild einer aneurysmatischen Knochenzyste in den langen Röhrenknochen ist eine rein osteolytische Läsion, die exzentrisch metadiaphysär oder zentral diaphysär gelegen ist, den Knochen auftreibt und vor Schluss der benachbarten Epiphysenfuge auftritt. Merke Abb. 7.21. Aneurysmatische Knochenzyste. Nahezu zentral überwiegend diaphysär gelegene Osteolyse, die eine deutliche Trabekulierung aufweist und den Knochen durch die Induktion einer Periostschale aufgetrieben hat (Lodwick IB) ! 7.3 Aneurysmatische Knochenzyste Abb. 7.23 a, b. Aneurysmatische Knochenzyste. a Die exzentrisch diaphysär gelegene osteolytische Läsion hat die Kompakta ausgedünnt. Gegen ein nicht-ossifizierendes Knochenfibrom spricht der fehlende lobulierte Rand und gegen eine solitäre Knochenzyste die exzentrische Lage. b Zwei Jahre später ist die Läsion deutlich gewachsen und hat die Ausbildung einer Periostschale induziert a Abb. 7.24. Aneurysmatische Knochenzyste. Exzentrisch epimetadiaphysär gelegene Läsion, die zum Markraum die Ausbildung einer Neokompakta induziert hat. Die Raumforderung ist von einer partiell mineralisierten Periostlamelle umgeben b Abb. 7.25. Aneurysmatische Knochenzyste. Die Raumforderung ist kortikal entstanden und hat die Kompakta ausgedünnt. Sie ist von einer partiell mineralisierten Periostlamelle umgeben 351 352 Kapitel 7 Tumorähnliche Läsionen b a Abb. 7.26 a, b. Aneurysmatische Knochenzyste. a Der 5. LWK und der linke Wirbelbogen des 4. LWK sind weitgehend destruiert. Daneben findet sich auch eine Tumormanifestation im rechten kleinen Wirbelgelenk LWK 4/5. b Die CT zeigt die Des- truktionen deutlich. Die Haupttumormasse ist in den Wirbelanhangsgebilden gelegen. Die Läsion bewirkt auch eine Kompression des Duralsacks und breitet sich überwiegend intraspinal im 1. Sakralsegment aus Abb. 7.27 a, b. Aneurysmatische Knochenzyste. a Im T1-gewichteten SE-Bild stellt sich eine ungewöhnlich epimetadiaphysär gelegene Raumforderung dar, die einen signalintensiven Herd bietet, der einer fokalen Einblutung entspricht. b Im T2-gewichteten SE-Bild stellen sich multiple durch Septen getrennte flüssigkeitsgefüllte Hohlräume dar, die teilweise Flüssigkeits-Flüssigkeits-Spiegel aufweisen a b Schnittbilddiagnostik Im peripheren Skelett ist die konventionelle Röntgendiagnostik für die diagnostische Aufarbeitung einer aneurysmatischen Knochenzyste meist ausreichend. Auch wird in den meisten Fällen die Ausdehnung der Läsion hinreichend genau wiedergegeben. In der Wirbelsäule ist jedoch ein Einsatz von CT oder MRT erforderlich. Die CT zeigt an der Wirbelsäule die Destruktion der tumortragenden Knochenabschnitte, die durch ein weichteildichtes Gewebe ersetzt werden. Die betroffenen Wirbelregionen sind meist aufgetrieben, und das Tumorgewebe wird von einer mehr oder minder dicken verkalkten Periostlamelle umgeben (Abb. 7.26 a, b). Die Ausdehnung einer extraossären Komponente ist meist nicht zuverlässig zu bestimmen. Mittels CT 7.3 Aneurysmatische Knochenzyste a b Abb. 7.29. Solide Variante der aneurysmatischen Knochenzyste. Die kontrastmittelverstärkte T1-gewichtete SE-Sequenz stellt eine stark kontrastmittelaufnehmende Läsion im Wirbelbogen von BWK 8 dar, wobei eine kleine Komponente in der Rückseite des Wirbelkörpers abgrenzbar ist. Der Spinalkanal ist deutlich eingeengt. Das weitgehend homogene Enhancement spricht für die sogenannte solide Variante einer aneurysmatischen Knochenzyste c Abb. 7.28 a–c. Aneurysmatische Knochenzyste. a Im T1-gewichteten SE-Bild zeigt die ansonsten zur Muskulatur isointense Läsion einige signalintensive Areale, die fokalen Blutungsherden entsprechen. b Im FS T2*-gewichteten GRE-Bild kommen multiple durch Septen getrennte flüssigkeitsgefüllte Hohlräume zur Darstellung. c Im kontrastmittelverstärkten FS T1-gewichteten SE-Bild lässt sich ein überwiegend septales Enhancement nachweisen, während die Hohlräume kein Enhancement zeigen können oft keine Blutbestandteile im Tumor entdeckt werden. Bei einem engen Fenster sind dagegen in bis zu einem Drittel der Fälle Flüssigkeits-FlüssigkeitsSpiegel vage erkennbar. In einer peripheren Lokalisation kann die CT in den Fällen, in denen auf dem konventionellen Röntgenbild keine abgrenzende Periostlamelle mehr sichtbar ist, diese häufig noch nachweisen, was für die Abgrenzung gegenüber einem malignen Tumor hilfreich sein kann. In der MRT zeigt die aneurysmatische Knochenzyste im T1-gewichteten Bild eine der Skelettmuskulatur vergleichbare Signalintensität. Nicht selten lassen sich einige signalintensive Areale – Methämoglobin – und Flüssigkeits-Flüssigkeits-Spiegel abgrenzen, die aber nicht obligatorisch vorhanden sein müssen. Im T2-gewichteten Bild zeigt die aneurysmatische Knochenzyste eine dem Liquor vergleichbare hohe Signalintensität. Bei entsprechender Dicke sind einzelne Septen in der Läsion nachweisbar. Eisenablagerungen werden nur selten beobachtet. Gerade in dieser Sequenz sind häufig Flüssigkeits-Flüssigkeits-Spiegel sichtbar, die durch die Sedimentation nichtkoagulierter Blutbestandteile entstehen (Abb. 7.27 a, b). Diese sind nicht beweisend für eine aneurysmatische Knochenzyste, da sie auch bei einer Reihe von anderen Läsionen, wie einem Chondroblastom, einem Riesenzelltumor, einer fibrösen Dysplasie, einer solitären Knochenzyste, einem teleangiektatischen Osteosarkom und einem malignen fibrösen Histiozytom gelegentlich beobachtet werden. Die Septen weisen nicht selten kleine, divertikelartige Aussackungen auf. Nach Kontrastmittelgabe zeigen die Septen ein deutliches Kontrastmittelenhancement, während der Zysteninhalt signalarm bleibt (Abb. 7.28 a–c). 353 354 Kapitel 7 Tumorähnliche Läsionen Bei einer Auftreibung des befallenen Knochens ist nicht selten zusätzlich ein Ödem in den benachbarten Weichteilen abgrenzbar. In der Wirbelsäule tritt gelegentlich die sogenannte solide Variante der aneurysmatischen Knochenzyste auf, die nahezu keine Hohlräume besitzt. Diese Läsion stellt sich in der MRT als rein solide Raumforderung dar, die eine weitgehend homogene Kontrastmittelaufnahme zeigt (Abb. 7.29). Solche soliden Bezirke können auch in einer peripheren aneurysmatischen Knochenzyste auftreten, sind hier jedoch wesentlich seltener. In diesem Falle müssen eine sekundäre aneurysmatische Knochenzyste und besonders ein teleangiektatisches Osteosarkom in Erwägung gezogen werden. Differenzialdiagnose In den meisten Fällen wird eine aneurysmatische Knochenzyste korrekt als benigne Läsion eingestuft. Bei einer Lage in den langen Röhrenknochen und in der Wirbelsäule gelingt es häufig, eine korrekte Artdiagnose zu stellen. Dies gilt in den langen Röhrenknochen besonders dann, wenn die aneurysmatische Knochenzyste metadiaphysär exzentrisch oder mitten im Schaft gelegen ist. Dehnt sich die aneurysmatische Knochenzyste in die Epiphyse aus, was fast immer erst nach Schluss der Epiphysenfuge stattfindet, muss ein Riesenzelltumor in Betracht gezogen werden. Hier gilt, je älter der Patient ist, umso wahrscheinlicher ist ein Riesenzelltumor. Dagegen spricht eine Lodwick-IA-Läsion mit einer nur geringen epiphysären Tumormasse eher für eine aneurysmatische Knochenzyste. Bei einer zentralen metadiaphysären Lage im Knochen, die gerade am Humerus besonders häufig auftritt, muss die solitäre Knochenzyste abgegrenzt werden. Ist eine deutliche Knochenauftreibung vorhanden, spricht dies eher für eine aneurysmatische Knochenzyste. Eine Lodwick-IA-Läsion spricht eher für eine solitäre Knochenzyste. In Zweifelsfällen kann mittels MRT durch den Nachweis von kontrastmittelaufnehmenden Septen die aneurysmatische Knochenzyste diagnostiziert werden. Es bleiben jedoch einige Fälle übrig, bei denen keine zuverlässige Differenzierung möglich ist. Bei der aneurysmatischen Knochenzyste der Wirbelsäule müssen der Riesenzelltumor, der häufiger im Wirbelkörper wächst, das Osteoblastom, das nicht selten eine sklerotische Komponente zeigt, und das symptomatische Hämangiom, das eine nicht mehr vom Periost begrenzte Weichteilkomponte aufweisen kann und den Knochen nicht auftreibt sondern eher destruiert, abgegrenzt werden. Therapie und Prognose Da eine aneurysmatische Knochenzyste nicht in allen Fällen sicher von anderen Läsionen abgegrenzt werden kann, sollte vor einem therapeutischen Eingriff eine Biopsie durchgeführt werden. Eine Abgrenzung zu einer solitären Knochenzyste kann mit einer Nadelbiopsie durchgeführt werden. Wird nur seröse Flüssigkeit aspiriert, handelt es sich um eine solitäre Knochenzyste, werden blutige Flüssigkeit und solide Anteile gewonnen, handelt es sich um eine aneurysmatische Knochenzyste oder einen anderen Tumor. Eine sorgfältige Kürettage ist die Therapie der Wahl. Man muss jedoch in bis zu 20% der Fälle mit Rezidiven rechnen. Während der Operation kann es sehr stark bluten, sodass eine präoperative Katheterembolisation der zuführenden Gefäße erwogen werden kann. 7.4 Ganglion Definition 왔 Ein intraossäres Ganglion ist eine benigne nichttumoröse Zyste, die aus fibrösem Gewebe mit massiver mukoider Degeneration besteht. Intraossäre Ganglien können durch Druckarrosionen eines extraossären Ganglions entstehen, die meisten entstehen jedoch im Knochen selbst. Eine Entstehungstheorie geht davon aus, dass das Ganglion aus versprengten synovialen Resten oder durch eine Ausstülpung der Synovialis in den Knochen entsteht. Jedoch ist die genaue Ätiologie unklar. Häufig kann ein intraossäres Ganglion histologisch nicht sicher von einer Arthrosezyste unterschieden werden. Bei einer Arthrosezyste sind jedoch radiologisch die typischen Arthrosezeichen sichtbar, während diese beim Ganglion meist fehlen oder nur gering ausgeprägt sind. Inzidenz Es handelt sich um eine eher seltene benigne tumorähnliche Läsion, deren Inzidenz nicht exakt bekannt ist. Sie macht im Einsendematerial eines Knochengeschwulstregisters weniger als 1% der benignen Raumforderungen aus. Da sicher eine ganze Reihe der Ganglien als Arthrosezyste fehlgedeutet und nicht biopsiert wird, was auch nicht erforderlich ist, muss von einer deutlich höheren Inzidenz ausgegangen werden. 7.4 Ganglion Klinik Leichte bis mäßige Schmerzen sind das führende klinische Symptom. Eine Schwellung oder eine Einschränkung der Gelenkbeweglichkeit gehören nicht zu den typischen Beschwerden. Lokalisation Etwa die Hälfte der Ganglien sind in der Tibia lokalisiert, wobei der distale Abschnitt etwa doppelt so häufig wie der proximale befallen ist. Eine typische Lage ist der Malleolus medialis. Daneben werden Ganglien im Femur, meist proximal, den karpalen Knochen, den tarsalen Knochen und dem Acetabulum angetroffen. In den langen Röhrenknochen sind die Läsionen epiphysär, epimetaphysär oder seltener metaphysär gelegen. Sie können zentral oder exzentrisch im Knochen liegen. Abb. 7.30. Intraossäres Ganglion. Die epimetaphysär gelegene Osteolyse eines 55-jährigen Patienten mit Ausdehnung in den Malleolus medialis weist einen feinen sklerotischen Randsaum auf (Lodwick IA). Die Lage und das Alter sind typisch für ein Ganglion. Bei einem wesentlich jüngeren Patienten käme ein Chondroblastom in Frage Röntgenmorphologie Die Ganglien sind in den meisten Fällen osteolytisch und können Trabekulierungen aufweisen. Sie können rund, oval oder lobuliert sein. Sie weisen nahezu immer scharfe oder sklerotische Grenzen auf und zeigen eine Wachstumsgeschwindigkeit vom Grad Lodwick IA oder seltener IB. Sie können eine lokale Knochenauftreibung bewirken, sind üblicherweise jedoch nicht besonders groß (Abb. 7.30, Abb. 7.31). Gelegentlich kann Gas innerhalb der Läsion beobachtet werden. Es wird angenommen, dass das Gas im Gelenk entsteht und durch Fissuren im Gelenkknorpel in das Ganglion gelangt. Das typische Bild ist eine epiphysär oder epimetaphysär gelegene Osteolyse ohne begleitende Arthrosezeichen bei einem Patienten jenseits des 40. Lebensjahres. Merke Abb. 7.31. Intraossäres Ganglion. Die im Malleolus medialis gelegene Läsion zeigt eine scharfe Begrenzung und angedeutete randständige Trabekel Alter und Geschlecht Ganglien werden zwischen dem 2. und 8. Lebensjahrzehnt angetroffen, zwei Drittel werden in der 4. bis 6. Lebensdekade diagnostiziert werden. Männer scheinen häufiger als Frauen betroffen zu sein. ! Schnittbildmorphologie Eine CT- oder MRT-Untersuchung sind für die Diagnose eines Ganglions nicht notwendig. Durch den mukoiden Inhalt sind in der CT die Dichtewerte höher als die von Wasser, jedoch niedriger als die eines nichtmineralisierten Tumorgewebes. In der CT sind scharfe Grenzen zum benachbarten Knochengewebe nachweisbar (Abb. 7.32 a, b). In der MRT zeigt das Ganglion eine uncharakteristische Morphologie mit in etwa muskelisointenser Signalintensität im T1-gewichteten Bild und sehr hoher Signalintensität im T2-gewichteten Bild. Gelegentlich sind im T2-gewichteten Bild Septen nachweisbar. Nach Kontrastmittelgabe wird nicht selten ein peripheres wandständiges Enhancement beobachtet, das auf eine reaktive Proliferation der Kapillaren zurückgeführt werden kann. Ein periläsionales Ödem wird nicht angetroffen (Abb. 7.33 a–c). 355 356 Kapitel 7 Tumorähnliche Läsionen Abb. 7.32 a, b. Intraossäres Ganglion. a Im Fibulaköpfchen stellt sich eine scharf abgrenzbare Osteolyse dar, die einzelne Trabekel aufweist. b In der CT lässt sich die Trabekulierung deutlich nachweisen. Die Läsion weist nahezu wasseräquivalente Dichtewerte auf b a a b c Abb. 7.33 a–c. Intraossäres Ganglion. a Das Röntgenbild zeigt eine kleine Osteolyse im Malleolus lateralis. b Im T1-gewichteten SE-Bild bietet die Läsion eine nahezu muskelisointense Signalintensität. c Im FS T2-gewichteten SE-Bild liegen flüssigkeitsäquivalente Dichtewerte vor. Daneben demarkiert sich intraläsional ein durch einen signalarmen Saum abgegrenztes Areal Differenzialdiagnose Bei jungen Patienten kommt differenzialdiagnostisch das Chondroblastom in Frage, Ganglien bei Patienten <25 Jahren stellen aber eher eine Rarität dar. Chondroblastome zeigen häufig feine Matrixkalzifikationen. Mittels MRT kann eine weitere Differenzierung erfolgen, da das Chondroblastom im Gegensatz zum Ganglion häufig ein peritumorales Knochenmarködem zeigt und im T2-gewichteten Bild häufig auch größere signalarme Bezirke aufweist. Bei älteren Patienten kommen die Arthrosezyste und die pigmentierte villonoduläre Synovialitis als mögliche Differenzialdiagnosen in Frage. Bei einer Arthrosezyste lässt sich radiologisch immer eine recht deutliche Arthrose nachweisen. Die pigmentierte villonoduläre Synovialitis zeigt bei einem typischen Krankheitsbild multiple Zysten beidseits des Gelenkspalts. In der MRT kann sie auch bei atypischem Röntgenbild anhand ihres Eisengehaltes sicher diagnostiziert werden. 7.5 Epidermoidzyste Eine weitere Differenzialdiagnose ist die Signalzyste bei der rheumatischen Arthritis, die jedoch meist kleiner ist und bevorzugt in den karpalen Knochen auftritt. Intraossäre Gichttophi können das Bild eines Ganglions imitieren, weisen jedoch nicht selten flaue, kaum sichtbare Mineralablagerungen auf. Therapie und Prognose Eine Kürettage ist in den allermeisten Fällen eine ausreichende Therapie. Die Rezidivrate ist gering und wird mit deutlich weniger als 10% angegeben. 7.5 Epidermoidzyste Definition tumoröse ossäre Raumforderung, die aus einer mit verhornendem Plattenepithel ausgekleideten Einzelhöhle besteht, die mit Hornschuppen ausgefüllt sein kann. Die Läsion ist recht selten und wird an der Schädelkalotte und den Phalangen, meist den Nagelkranzphalangen, angetroffen. Bei der Epidermoidzyste der Kalotte geht man davon aus, dass während der embryonalen Entwicklung a b c d e 왔 Die Epidermoidzyste ist eine nicht- Abb. 7.34 a–e. Epidermoidzyste. a Das Röntgenbild zeigt eine weitgehend scharf abgrenzbare Osteolyse in der Kalotte. b In der CT stellt sich die Läsion als Defekt der Tabula externa und der Diploe dar, wobei die Tabula interna nur teilweise ausgedünnt ist. c Im T1-gewichteten SE-Bild ist die Läsion signalärmer als das Hirnparenchym. d Im T2-gewichteten SE-Bild weist die Raumforderung nahezu wasseräquivalente Signalintensitäten auf. e Im kontrastmittelverstärkten FS-Bild ist ein deutliches, etwas inhomogenes Enhancement nachweisbar 357 358 Kapitel 7 Tumorähnliche Läsionen Epithelzellen in den Knochen versprengt worden sind. Die Läsionen wachsen häufig über einen langen Zeitraum und haben bereits eine beträchtliche Größe erreicht, wenn sie klinisch auffällig werden. Die Läsionen in den Phalangen werden auf einen traumatischen Ursprung zurückgeführt und können mit Schmerzen einhergehen. Die Epidermoidzysten zeigen keine charakteristische Altersverteilung, werden aber meist erst jenseits des 20. Lebensjahres diagnostiziert. Röntgenmorphologie In der Kalotte stellt sich eine Epidermoidzyste als Osteolyse dar, die scharfe und meist sklerotische Ränder aufweist. Die Größe kann zwischen wenigen Millimetern und etwa 5 cm variieren. Zentrale Verkalkungen werden in Verseifungsherden beobachtet. In den Phalangen stellt sich die Läsion ebenfalls als Osteolyse dar, die weniger scharf begrenzt ist und häufig die Kompakta destruiert oder aufgetrieben hat. Gelegentlich liegt eine zusätzliche Weichteilschwellung vor. In der MRT zeigen die Läsionen im T1-gewichteten Bild eine niedrigere Signalintensität als die Muskulatur und eine hohe im T2-gewichteten Bild, die typisch für Flüssigkeit ist. Jedoch variiert die Signalintensität, wenn Fett oder hämorrhagische Produkte vorhanden sind (Abb. 7.34 a–e). Differenzialdiagnose An der Kalotte kann eine Epidermoidzyste bei einer engen Lagebeziehung zur Stirnhöhle als Mukozele missgedeutet werden. Weitere Fehldiagnosen können das Hämangiom, das typischerweise ein Radspeichenmuster der Trabekel zeigt, und das eosinophile Granulom, das bei jüngeren Patienten auftritt und meist keinen sklerotischen Randsaum besitzt, sein. An den Phalangen lauten die Differenzialdiagnosen Enchondrom, das meist weniger aggressiv imponiert, Riesenzelltumor und Glomustumor, der meist schärfer definierte Ränder aufweist. Therapie und Prognose Die adäquate Therapie besteht in einer Kürettage. 7.6 Fibröse Dysplasie Definition 왔 Die fibröse Dysplasie ist eine benigne nichttumoröse Erkrankung des Skeletts, bei der ein oder mehrere Herde vorhanden sind, in denen das Knochenmark durch ein fibröses Gewebe ersetzt ist, das bizarr konfigurierte Faserknochenbälkchen enthält. Der Umbau zu Lamellenknochen findet nicht statt. Innerhalb der Matrix können auch Knorpelinseln, Riesenzellen und myxomatöse Degenerationsherde vorhanden sein. Die Herde der fibrösen Dysplasie werden über mehrere Jahre langsam größer. Das Wachstum kann jedoch auch spontan stoppen, häufig dann, wenn die Skelettreife erreicht ist. Meistens tritt die fibröse Dysplasie monostisch auf. Nur etwa 10% der Patienten haben multiple Herde, die mehrere Knochen befallen. Bei der polyostischen Form kann der Patient auch an pigmentierten Hautveränderungen (sehr unregelmäßig begrenzten Café-au-lait-Flecken) und an einer Störung des endokrinen Systems leiden, wobei bei jungen Mädchen eine Pubertas praecox auftritt. Dieser Komplex ist als McCune-Albright-Syndrom bekannt. Selten bei der polyostischen Form und sehr selten bei der monostischen Variante werden zusätzliche intramuskuläre Myxome angetroffen, diese Kombination wird Mazabraud-Syndrom genannt wird. Die fibröse Dysplasie wird als eine angeborene Störung der Knochenentwicklung angesehen. In der Tibia existiert eine Variante, die als osteofibröse Dysplasie bezeichnet wird. Daneben wird diskutiert, ob ein Zusammenhang mit dem ossifizierenden Fibrom des Kiefers besteht. Zwar unterscheiden sich beide Läsionen histologisch, jedoch werden gelegentlich beide Läsionen bei einem Patienten simultan angetroffen. Auch wird der Cherubismus des Kiefers mit der fibrösen Dysplasie in Zusammenhang gebracht. In einem fibrösen Dysplasieherd kann in seltenen Fällen eine aneurysmatische Knochenzyste auftreten. In sehr seltenen Fällen kann die fibröse Dysplasie maligne entarten. Inzidenz Die exakte Inzidenz der fibrösen Dysplasie ist unbekannt, da nicht alle Herde biopsiert werden. In dem Einsendematerial eines Knochengeschwulstregisters hat die fibröse Dysplasie einen Anteil von gut 10% an allen benignen Knochenläsionen. Alter und Geschlecht Die fibröse Dysplasie ist überwiegend eine Erkrankung des jungen Menschen, wird aber in allen Altersgruppen angetroffen. Etwas mehr als die Hälfte der Patienten sind jünger als 20 Jahre,75% der Läsionen werden bis zum 30. Lebensjahr diagnostiziert. Daneben werden aber eine ganze Reihe von Herden bei älteren Patienten diagnostiziert, die als Zufallsbefunde bei einer Röntgenuntersuchung entdeckt wurden. Hierzu zählen besonders Herde in den Rippen und im Schenkelhals. Auch die polyostische fibröse Dysplasie wird überwiegend bei jungen Menschen entdeckt, hier wird 7.6 Fibröse Dysplasie Abb. 7.35. Statistische Daten zur fibrösen Dysplasie die weit überwiegende Anzahl der Erkrankungen bis zum 40. Lebensjahr diagnostiziert. Männer und Frauen sind in etwa gleicher Häufigkeit betroffen. Klinik Die monostische fibröse Dysplasie verursacht keine oder nur wenig Beschwerden, in diesem Fall wird über leichte Schmerzen geklagt. Gelegentlich ist bei einer oberflächennahen Lage eine harte Schwellung tastbar. Nicht selten wird die monostische Form erst durch eine pathologische Fraktur klinisch auffällig. Bei Diagnosestellung kann der betroffene Knochen bereits eine deutliche Deformierung aufweisen,was insbesondere bei der polyostischen Form häufig der Fall ist. Treten pathologische Frakturen auf, sind diese meist nicht disloziert und heilen ohne Verzögerung. An der Kalotte kann durch eine Volumenzunahme der Knochen eine Kompression der Hirnnerven eintreten. Lokalisation Bei der monostischen Form kann die fibröse Dysplasie jeden Knochen befallen. Besonders betroffen sind das Femur (26%), die Tibia (15%), die Rippen (14%), die Kalotte einschließlich Schädelbasis (11%) und der Gesichtsschädel (11%). Selten werden die kleinen Röhrenknochen befallen. In den langen Röhrenknochen liegen die Herde in nahezu der Hälfte der Fälle metadiaphysär und in weiteren 40% diaphysär. Eine rein metaphysäre Lokalisation wird selten und eine Ausdehnung in die Epiphyse in weniger als 10% der Fälle beobachtet. Mehr als 90% der Herde sind zentral im Röhrenknochen lokalisiert. Im Femur liegen wesentlich mehr Herde im proximalen als im distalen Abschnitt. Die polyostische Form befällt primär die untere Extremität. Dabei sind Femur und Tibia oder Becken in 75–90% gleichzeitig betroffen. Die Metatarsalia, die Fibula oder die Fußphalangen sind in 60–70% befallen. Mehr als die Hälfte der Patienten hat Rippen- oder Kalottenherde. Bei einem Befall der oberen Extremität sind der Humerus und die Metacarpalia in mindestens der Hälfte der Fälle betroffen. Der Befall kann sich auf eine Extremität beschränken (monomelische Form), oder es sind eine Vielzahl von Knochen betroffen, wobei häufig eine Seite dominiert (Abb. 7.35). Röntgenmorphologie Die Röntgenmorphologie ist weitgehend von der quantitativen Zusammensetzung aus fibrösem Stroma und der Menge an fehlgebildeten Trabekeln und ihrem Mineralisationsgrad abhängig. Wenn ein Ersatz des normalen Knochens durch ein rein fibröses Stroma vorliegt, stellt sich der Herd als reine Osteolyse dar. Werden jedoch größere Mengen an mineralisierten Trabekeln gebildet, stellt sich die Läsion mattglasartig dar. Bei noch stärkerer Mineralisation imponieren die Herde mäßig bis deutlich osteoblastisch. In vielen Fällen ist die Knochenneubildung jedoch nicht einheitlich in der Läsion verteilt. In diesen Fällen werden zentrale oder randständige osteolytische Bezirke in einer überwiegend mattglasartig imponierenden Läsion angetroffen (Abb. 7.36, Abb. 7.37, Abb. 7.38). Die dichten Anteile der Läsion können sich über einen längeren Abschnitt zungenartig im normalen Knochen ausbreiten, was einer flackernden Kerzenflamme ähnlich sieht (Abb. 7.39). Der Mattglasaspekt ist charakteristisch für eine fibröse Dysplasie.Die Herde sind in einer Reihe von Fällen von einem reaktiven, nicht selten breiten sklerotischen Randsaum umgeben. In den anderen Fällen sind die Herde fast immer scharf zum normalen Knochen abgrenzbar. Seltener findet man einen mattglasartigen 359 360 Kapitel 7 Tumorähnliche Läsionen Abb. 7.36. Fibröse Dysplasie. Die Läsion bietet eine mattglasartige Matrix und ist von einem breiten sklerotischen Randsaum umgeben (Lodwick IA) Abb. 7.37. Fibröse Dysplasie. Die metadiaphysär gelegene Läsion weist eine deutliche Mattglasmatrix auf und ist von einem sklerotischen Randsaum begrenzt (Lodwick IA). Daneben zeigt sie einige ossifizierte Areale Abb. 7.38. Fibröse Dysplasie. Die Läsion weist eine ausgeprägte reaktive Ossifikation auf Abb. 7.39. Fibröse Dysplasie. Die überwiegend diaphysär gelegene Läsion zeigt an ihrem kranialen Rand eine breite reaktive Ossifikationszone, die das Bild einer Kerzenflamme bietet, was recht charakteristisch für eine fibröse Dysplasie ist. Es liegen einzelne osteolytische Areale vor, die durch eine zu geringe Mineralisation des Geflechtknochens zu erklären sind 7.6 Fibröse Dysplasie Abb. 7.40. Fibröse Dysplasie. Die Läsion, die eine typische Mattglasmatrix aufweist, wird durch einen osteolytischen Randsaum vom normalen Markraum abgegrenzt (sequesterartiges Bild). Am Unterrand stellen sich typische Knorpelverkalkungen dar, die einem mineralisierten Knorpelareal entsprechen Abb. 7.41. Fibröse Dysplasie. Die Schichtaufnahme stellt deutlich die Mattglasmatrix dar. Im Rand liegen deutliche Ossifikationen vor. Am Unterrand stellen sich 2 kleine rundliche Verkalkungen dar, die Mineralisationen fokaler Knorpelherde entsprechen oder weitgehend osteoblastischen Herd, der durch einen schmalen osteolytischen Randsaum vom normalen Knochen abgegrenzt wird, wodurch das zentrale Areal wie ein Knochensequester imponiert (Abb. 7.40). Größere Knorpelinseln können sich als osteolytische Bezirke darstellen, die punktförmige oder ringartige Verkalkungen aufweisen können (Abb. 7.41). Bei älteren Patienten werden häufiger fibröse Dysplasieherde im Schenkelhals gefunden, die relativ stark ossifiziert sind. Bei jungen Patienten finden sich dagegen häufiger rein osteolytische Herde, die deutliche Trabekulierungen und keinen Mattglasaspekt aufweisen. Die meisten Läsionen wachsen unter dem Grad Lodwick IA und IB. Eine Auftreibung des Knochens durch die Ausbildung einer Periostschale wird bei größeren Läsionen häufig beobachtet. Aber die Kompakta kann auch breiter werden oder in den Prozess einbezogen werden, wodurch sie nicht mehr als solche abgrenzbar ist. Fokale Knorpelinseln können Matrixverkalkungen aufweisen. Da der neugebildete Knochen funktionell minderwertig ist, treten häufig deutliche Deformierungen des Knochens auf, die sich besonders in den Gewichtsbelastungszonen manifestieren. So ist am Femur die Hirtenstabdeformität bekannt, die durch eine extreme varische Deformierung des Schenkelhalses entsteht. Hier treten häufig Infraktionen und Frakturen auf (Abb. 7.42). Die fibröse Dysplasie kann sich entlang des gesamten Knochens ausdehnen, wächst jedoch erst nach Epiphysenfugenschluss in die Epiphysen ein (Abb. 7.43). In den Rippen stellt sich die fibröse Dysplasie meist als Osteolyse dar, die die Rippe etwas auftreibt. Seltener werden spindelförmige, teilweise mineralisierte Herde beobachtet, die die Kompakta auflösen können. Rein ostoblastische Herde sind in den Rippen selten. Im Beckenskelett bewirkt die fibröse Dysplasie häufig eine Auftreibung des Schambeins und des Sitzbeins (Abb. 7.44). In dieser Region stellen sich die Läsionen meist osteolytisch mit Trabekulierungen und einem sklerotischen Randsaum dar, können jedoch gelegentlich etwas aggressiver imponieren. Am Schädel werden rein osteolytische Herde selten, und wenn dann bevorzugt in der Kalotte und in der Mandibula, angetroffen. Sie weisen meist einen sklerotischen Randsaum auf. An der Kalotte sind die meisten Herde jedoch mineralisiert, gehen von der Tabula externa aus und führen zu einer deutlichen Knochenverbreiterung (Abb. 7.45). Überwiegend sklerotische Herde werden an der Schädelbasis und im Keilbein angetroffen, wobei der befallene Knochen typischerweise deutlich verdickt ist. Meist ist nur eine Seite betroffen. 361 362 Kapitel 7 Tumorähnliche Läsionen Abb. 7.42. Polyostische fibröse Dysplasie. Die fibröse Dysplasie hat langstreckig das Femur befallen. Durch die Schwächung des Knochen hat sich eine Coxa vara ausgebildet (Hirtenstabdeformität). Daneben liegen weitere Herde im Beckenskelett vor Abb. 7.43. Polyostische fibröse Dysplasie. Bei diesem Patienten liegt ein langstreckiger Befall beider Femora und der linken Tibia vor Abb. 7.44. Fibröse Dysplasie. Die Läsion hat das Schambein aufgetrieben und bietet eine typische Mattglasmatrix Das typische Bild einer fibrösen Dysplasie in einem Röhrenknochen ist eine Osteolyse mit einer Mattglasmatrix, die von einem sklerotischen Randsaum umgeben sein kann, meist zentral metadiaphysär oder zentral diaphysär gelegen ist und in der 1. bis 3. Lebensdekade angetroffen wird. Merke ! Abb. 7.45. Fibröse Dysplasie. Die Kalotte ist beidseits parietal deutlich verdickt und weist osteolytische und mattglasartige Areale auf 7.6 Fibröse Dysplasie Abb. 7.46. Fibröse Dysplasie. Es liegt ein Befall der Schädelbasis vor, wobei die betroffenen Knochen aufgetrieben sind und eine Mattglasmatrix bieten Schnittbildmorphologie In den meisten Fällen ist die konventionelle Röntgendiagnostik ausreichend, die fibröse Dysplasie für diagnostische und therapeutische Zwecke darzustellen. Die Skelettszintigraphie kann zum Nachweis von multiplen Herden eingesetzt werden. CT und MRT werden jedoch in vielen Fällen benötigt, die wahre Ausdehnung einer fibrösen Dysplasie im Gesichtsschädel oder in der Schädelbasis zu demonstrieren. Die CT stellt die Auftreibung des befallenen Knochens deutlich dar. Die Matrix der Läsion zeigt mittelhohe Dichtewerte, die zwischen normaler Diploe und Kompakta liegen. Die Struktur ist kleinfleckig inhomogen, wobei häufig kleinste osteolytische Areale und kleinste dichte Areale eingestreut sind (Abb. 7.46). In den langen Röhrenknochen und im Beckenskelett kann die CT eingesetzt werden, wenn die wahre Natur einer osteolytischen Läsion aus dem konventionellen Röntgenbild nicht zuverlässig bestimmt werden kann. Hier findet man eine Tumormatrix, in der mittelhohe Dichtewerte, die zwischen denen von Skelettmuskulatur und Kompakta liegen, dominieren. Die Dichtewerte sind höher als sie ansonsten bei osteolytischen Läsionen gefunden werden. Die Lä- sion zeigt scharfe Grenzen zu dem nicht betroffenen Markraum und kann fokale Verkalkungen aufweisen (Abb. 7.47). In der MRT zeigt die fibröse Dysplasie im T1-gewichteten Bild eine muskelisointense oder etwas niedrigere Signalintensität. Im T2-gewichteten Bild kann die Signalintensität deutlich, auch innerhalb einer Läsion, zwischen nahezu muskelisointenser bis zu deutlich höherer Intensität variieren. Dies ist auf die unterschiedliche Zusammensetzung der Herde aus nichtmineralisiertem Stroma, mit hoher Signalintensität, und mineralisiertem Stroma, mit niedriger Signalintensität, zurückzuführen. Die Läsionen können von einem breiten signalarmen sklerotischen Randsaum umgeben sein. Größere fokale Knorpelinseln weisen eine deutlich höhere Signalintensität im T2-gewichteten Bild auf. Die Läsionen zeigen nahezu immer ein mäßiges bis deutliches Kontrastmittelenhancement (Abb. 7.48 a, b). In der Skelettszintigraphie ist bei bis zu 15% der Herde keine Tracerakkumulation nachweisbar. Differenzialdiagnose Die solitäre fibröse Dysplasie wird in den allermeisten Fällen sicher als benigne Läsion eingestuft. Wenn eine Mattglasmatrix vorliegt, kann die Artdiagnose 363 364 Kapitel 7 Tumorähnliche Läsionen Abb. 7.47. Fibröse Dysplasie. Die CT zeigt die typische Mattglasmatrix. Auf dem oberen linken Bild ist der normale Markraum abgebildet ziemlich sicher gestellt werden. In frühen Phasen, wenn die fibröse Dysplasie rein osteolytisch ist, kann es schwierig bis unmöglich sein, diese von einer solitären Knochenzyste zu differenzieren. Hier kann die CT eingesetzt werden, die höhere als flüssigkeitsäquivalente Dichtewerte nachweist. Gehen die rein osteolytischen Herde mit einer deutlichen Knochenauftreibung einher, kann die Abgrenzung zu einer aneurysmatischen Knochenzyste schwierig sein. In diesen Fällen sollte die MRT hilfreich sein, da keine Blutbestandteile und keine kontrastmittelaufnehmenden Septen vorhanden sind. Sind in einem rein osteolytischen Herd Kalzifikationen vorhanden, kann dieser mit einem Enchondrom verwechselt werden. An der Schädelkalotte besteht bei fokalen osteoblastischen Herden Verwechslungsgefahr mit einem Meningeom und einem Osteom, das jedoch meist in den Nasennebenhöhlen liegt. Ein Meningeom lässt sich mit der CT oder MRT sicher ausschließen. An der Kalotte ist die Differenzialdiagnose zum Morbus Paget, der bei eher älteren Patienten auftritt, schwierig. Das radiologische Bild beider Erkrankungen unterscheidet sich nicht wesentlich. Jedoch befällt die fibröse Dysplasie im Gegensatz zum Morbus Paget nur eine Seite, und ein Befall der Schädelbasis ist beim Morbus Paget selten. Der Morbus Paget zeigt eine extreme Tracerakkumulation in der Skelettszintigraphie. Eine polyostische fibröse Dysplasie muss von einer Enchondromatose, die jedoch in zumindest einzelnen Herden Verkalkungen zeigt und viele Herde in den kleinen Knochen ausbildet, abgegrenzt werden. Die bei älteren Patienten im proximalen Femur nachweisbaren osteoblastischen fibröse Dysplasieherde müssen von einem hier eher selten auftretenden alten Knocheninfarkt abgegrenzt werden. Eine Reihe dieser Läsionen wurde kürzlich als liposklerosierender myxofibröser Tumor reklassifiziert. Inwieweit dies gerechtfertigt ist, kann noch nicht abschließend beurteilt werden. Therapie und Prognose In vielen Fällen ist keine Behandlung der fibrösen Dysplasie notwendig. Liegt ein kleiner typischer Herd vor, von dem keine pathologische Frakturgefahr ausgeht, kann auf eine histologische Sicherung verzichtet werden. Es sollte jedoch gehandelt werden, wenn die Gefahr einer pathologischen Fraktur oder einer Deformierung besteht. In diesen Fällen wird empfohlen, den pathologischen Bezirk durch kortikale Späne zu verstärken. Der Vorteil ist, dass der 7.7 Osteofibröse Dysplasie In sehr seltenen Fällen kann in einer fibrösen Dysplasie eine maligne Transformation, meist in ein Osteosarkom, seltener in ein Fibrosarkom, Chondrosarkom oder malignes fibröses Histiozytom erfolgen. Bei etwa der Hälfte der Fälle war eine Strahlentherapie der fibrösen Dysplasie vorausgegangen. Die Prognose der sekundären Sarkome ist schlecht. 7.7 Osteofibröse Dysplasie a b Abb. 7.48 a, b. Fibröse Dysplasie. a Im T1-gewichteten SE-Bild stellen sich die Läsionen nahezu isointens zur Muskulatur dar. Der proximale Herd hat den Schaft etwas aufgetrieben. b Im kontrastmittelverstärkten T1-gewichteten Bild zeigt der proximale Herd ein deutliches nahezu homogenes Enhancement, der distale dagegen nur ein überwiegend geringes Enhancement kortikale Knochen eingebaut wird, was jedoch lange Zeit in Anspruch nehmen kann. Nach Kürettage eines Herdes verbleibt häufig noch dysplastischer Knochen im Randbereich, der eingebrachte spongiöse Späne resorbiert. Abb. 7.49. Statistische Daten zur osteofibrösen Dysplasie Die osteofibröse Dysplasie wird in der Literatur auch als ossifizierendes Fibrom geführt. Das typische ossifizierende Fibrom ist jedoch eine Läsion, die fast ausschließlich in der Mandibula angetroffen wird und in die Gruppe der Kiefertumoren gehört, auf die an dieser Stelle nicht eingegangen werden soll. In den letzten Jahren hat sich für eine nahezu identisch aufgebaute Läsion außerhalb des Kiefers der Begriff osteofibröse Dysplasie durchgesetzt. Man geht davon aus, dass es sich um eine aktive Variante der fibrösen Dysplasie handelt, da sie dieser histologisch ähnelt. Im Gegensatz zur fibrösen Dysplasie werden die Knochenbälkchen von Osteoblastensäumen umgeben, die osteoid- und geflechtartigen Knochen bilden. Die Läsion ist zwar benigne, wächst aber lokal aggressiv. Es handelt sich um eine seltene Erkrankung des Kindes- und Jugendalters. Klinisch liegt eine schmerzlose, größenprogrediente Raumforderung vor. Etwa drei Viertel der Läsionen werden bis zum 15. Lebensjahr diagnostiziert. Fast alle sind in der Tibia lokalisiert. Gelegentlich finden sich weitere Herde in der Fibula. Fast alle Herde sind in der Diaphyse und meist ventral exzentrisch gelegen (Abb. 7.49). 365 366 Kapitel 7 Tumorähnliche Läsionen Das typische Bild ist eine ventral exzentrisch im Tibiaschaft gelegene lobulierte Osteolyse mit Mattglasmatrix und deutlichen zentralen und peripheren Ossifikationen, die bis zum 15. Lebensjahr angetroffen wird. Merke Abb. 7.50. Osteofibröse Dysplasie. Langstreckige Läsion in der ventralen Kompakta des Tibiaschafts, die aus osteolytischen Bezirken und einer reaktiven Sklerose zusammengesetzt ist, was sie multifokal erscheinen lässt Röntgenmorphologie Die osteofibröse Dysplasie ist eine relativ langsam wachsende Läsion, die eine Wachstumsrate vom Grad Lodwick IA oder IB aufweist. Die Läsion ist meist ovalär oder spindelförmig, wobei die Längsachse in Richtung Knochenachse ausgerichtet ist. In etwa drei Viertel der Fälle bietet sie eine Mattglasmatrix, dabei sind meist zusätzliche breite Verknöcherungsareale in der Peripherie und auch zentral vorhanden. In den restlichen Fällen stellen sich die nicht-ossifizierten Areale rein osteolytisch dar. Die Ränder sind häufig lobuliert, sodass insgesamt das Bild einer multifokalen konfluierenden Tumormanifestation vorgetäuscht wird (Abb. 7.50). In etwa 70% der Fälle ist die Tibia aufgetrieben. Nicht selten erinnert die Morphologie an ein größeres nicht-ossifizierendes Knochenfibrom, das sich in einem mehr oder minder fortgeschrittenen Stadium der Ausheilung befindet, jedoch in einer falschen Lokalisation vorhanden ist. Nicht selten tritt eine ventrale Verbiegung der Tibia auf. Gelegentlich wird eine solitäre lamelläre Periostreaktion angetroffen. ! Schnittbildmorphologie Für die diagnostische Aufarbeitung ist nur selten eine zusätzliche Schnittbilddiagnostik erforderlich. In der CT lässt sich sicher demonstrieren, dass die Läsion eine scharfe Grenze zum normalen Markraum aufweist. Die Matrix imponiert mattglasartig oder weitgehend osteoblastisch. Osteolytische Areale, die keine sklerotische Abgrenzung zu den Weichteilen aufweisen, sollten an ein Adamantinom denken lassen. In der MRT kommt eine sehr lobulierte oder eine spindelförmige Läsion zur Darstellung, die in etwa muskelisointense Signalintensitäten im T1-gewichteten Bild und hohe Signalintensitäten im T2-gewichteten Bild zeigt. Die Grenzen zu dem normalen Fettmark sind scharf definiert. Im T2-gewichteten Bild kann sie mehr oder minder deutliche Inhomogenitäten in Abhängigkeit vom Ausmaß der Matrixmineralisation aufweisen. Der Grad des Kontrastmittelenhancements ist eher gering bis mäßig und in den weniger mineralisierten Anteilen weitgehend homogen (Abb. 7.51 a–c). In der Skelettszintigraphie zeigt die Läsion eine Traceraufnahme. Differenzialdiagnose Bei einem relativ charakteristischen Bild besteht die einzige Differenzialdiagnose in einem Adamantinom, das auch fast ausschließlich in der Tibia auftritt. Sind entweder eine lokale aggressiv wachsende Komponente oder multiple lamelläre Periostreaktionen vorhanden, spricht dies für ein Adamantinom. Patienten mit einem Adamantinom sind tendenziell älter. Eine deutliche Mattglasmatrix und eine ventrale Verbiegung der Tibia sprechen dagegen für eine osteofibröse Dysplasie. Eine zuverlässige Differenzialdiagnose zwischen beiden Entitäten ist radiologisch nicht immer möglich, zumal in den letzten Jahren auch eine neue Variante des Adamantinoms, ein osteofibröse-Dysplasie-ähnliches Adamantinom, mehrfach beschrieben worden ist. Einige Autoren vermuten, dass die osteofibröse Dysplasie und das Adamantinom unterschiedliche Entwicklungsstadien der gleichen Läsion repräsentieren, wobei das osteofibröse-Dysplasie-ähnliche Adamantinom ein Zwischenschritt zwischen beiden Läsionen darstellt (Abb. 7.52 a, b). Diese Theorie und auch die Existenz eines osteofibröse-Dysplasie-ähnlichen Adamantinoms werden jedoch nicht allgemein akzeptiert. 7.7 Osteofibröse Dysplasie b a c rung überwiegend muskelisointense Signalintensitäten. Die Grenze zum Markraum ist scharf. c Im kontrastmittelverstärkten T1-gewichteten SE-Bild zeigt die Läsion ein deutliches Enhancement Abb. 7.51 a–c. Osteofibröse Dysplasie. a Im Röntgenbild stellt sich eine ventral exzentrisch lokalisierte Läsion im Tibiaschaft dar, die osteolytische, mattglasartige und sklerotische Areale aufweist. b Im T1-gewichteten SE-Bild bietet die Raumforde- Abb. 7.52 a, b. OsteofibröseDysplasie-ähnliches Adamantinom. a Im Röntgenbild stellt sich die Raumforderung wie eine osteofibröse Dysplasie dar. b Im T1-gewichteten SE-Bild weist die Läsion eine scharfe Grenze zum Markraum auf. Histologisch wurden jedoch einzelne typische Adamantiomherde in der osteofibrösen Dysplasie nachgewiesen b a 367 368 Kapitel 7 Tumorähnliche Läsionen Therapie und Prognose Die Therapie der osteofibrösen Dysplasie besteht bei älteren Patienten in einer sorgfältigen Kürettage. Anschließend ist eine sorgfältige histologische Aufarbeitung zum Ausschluss eines Adamantinoms erforderlich. Gelegentlich treten Rezidive auf. Bei Patienten mit offenen Wachstumsfugen wird lediglich eine Verlaufsbeobachtung angeraten. Falls eine Verbiegung der Tibia klinisch in den Vordergrund rückt, wird eine Orthesenbehandlung empfohlen. Die osteofibröse Dysplasie metastasiert nicht. 7.8 Langerhans-Zell-Histiozytose 7.8.1 Eosinophiles Granulom Das eosinophile Graulom gehört zu einer Gruppe von Erkrankungen, die auch als Histiocytosis X oder korrekter als Langerhans-Zell-Histiozytose bezeichnet wird. Die Läsionen bestehen aus einem fibrohistiozytären Granulationsgewebe, das einen unterschiedlichen Gehalt an Langerhans-Zellen und eosinophilen Granulozyten aufweist. Daneben finden sich neutrophile Granulozyten, Lymphozyten, Plasmazellen und Riesenzellen. Das eigentliche proliferierende Element ist die Langerhans-Zelle, eine mononukleäre Zelle der dendritischen Linie, die in der Epidermis gefunden wird, sich aber von Prokursorzellen im Knochenmark ableitet. Frische Herde enthalten einen hohen Anteil an Langerhans-Zellen und eosinophilen Granulozyten. Dagegen dominiert in alten Herden ein fibrotisches Gewebe, in dem die Menge an Langerhans-Zellen und eosinophilen Granulozyten gering ist, sodass diese Läsionen mit einer chronischen Osteomyelitis verwechselt werden können. Das eosinophile Granulom ist die benigne und heilbare Variante dieser Erkrankungsgruppe. Es ist entweder auf den Knochen beschränkt oder geht mit weiteren Herden in anderen Organen einher. Die disseminierte akute oder subakute Variante, die häufig tödlich endet, wird als Abt-Letterer-Siwe-Erkrankung bezeichnet. Die disseminierte chronische Variante ist als Hand-Schüller-Christian-Erkrankung bekannt. Die Ätiologie ist unbekannt. Es wird vermutet, dass die Erkrankung durch eine noch nicht bekannte Störung des Immunsystems initiiert wird. Inzidenz Das eosinophile Granulom ist eine seltene Erkrankung mit einer Inzidenz von 1–2 Erkrankungen pro 1 Mio. Einwohner pro Jahr. Es ist die häufigste Varian- te der Langerhans-Zell-Histiozytose und macht etwa 60–80% aller Fälle aus. In den Archiven der Knochengeschwulstregister hat es einen Anteil von knapp 5% unter den benigen Läsionen. Alter und Geschlecht Es handelt sich um eine typische Erkrankung der Kinder und Jugendlichen. Der Altersgipfel mit nahezu der Hälfte der Fälle liegt bereits in der 1. Lebensdekade und etwa zwei Drittel der Erkrankungen treten bis zum 15. Lebensjahr auf. Nahezu 90% der Läsionen werden bis zum 30. Lebensjahr diagnostiziert. Erkrankungen jenseits des 40. Lebensjahres sind Raritäten. Klinik Das führende klinische Symptom sind lokale Schmerzen. Selten wird die Erkrankung erst durch eine pathologische Fraktur klinisch auffällig. Gelegentlich werden pathologische Frakturen relativ kurz nach Schmerzbeginn beobachtet. Der Befall des Felsenbeines kann eine Mittelohrentzündung vortäuschen. Ein Befall des Keilbeines unter Einbeziehung des Hypophysenstiels kann zu einem Diabetes insipidus führen. Ein Befall der Orbita kann einen Exophthalmus hervorrufen. Eine lokale Schwellung gehört nicht zu den führenden klinischen Symptomen. Die Beschwerden bestehen meist erst seit wenigen Wochen, wenn der Patient ärztliche Hilfe in Anspruch nimmt. Lokalisation Ein eosinophiles Granulom kann in jedem Knochen auftreten. Die am häufigsten betroffenen Knochen sind das Femur (22%), die Kalotte (16%) und das Becken (16%). Es folgen mit jeweils etwa 10% der Fälle die Rippen, die Wirbelsäule und der Humerus. In den langen Röhrenknochen liegen zwei Drittel der Läsionen diaphysär und etwa ein Drittel metadiaphysär oder metaphysär. Eine epimetaphysäre Lokalisation stellt eine Rarität dar. Zwei Drittel der Läsionen sind zentral im Röhrenknochen gelegen. An der Wirbelsäule wird bevorzugt der Wirbelkörper allein befallen. Ein alleiniger Befall des Wirbelbogens erfolgt selten. Bei größeren Herden sind sowohl Wirbelkörper als auch -bogen betroffen (Abb. 7.53). Röntgenmorphologie Die allermeisten eosinophilen Granulome wachsen als reine Osteolysen. Nur selten zeigen sie Trabekulierungen oder sklerotische Areale. Etwa zwei Drittel wachsen unter dem Bild einer geographischen Osteolyse meist vom Grad Lodwick IB oder IC. Etwa ein Drittel zeigt ein aggressiveres Wachstum vom Grad Lodwick II und sogar vom Grad III. Üblicherweise 7.8 Langerhans-Zell-Histiozytose Abb. 7.53. Statistische Daten zu eosinophilem Granulom Abb. 7.54. Eosinophiles Granulom. Mottenfraßartige Osteolyse im Femurschaft (Lodwick II), die im Gegensatz zu einem malignen Tumor eine Verdickung der Kompakta induziert hat. Lateral liegt eine nichtunterbrochene lamelläre Periostreaktion vor, die charakteristisch für ein eosinophiles Granulom ist. Differenzialdiagnostisch kommt eine Osteomyelitis in Frage zeigen ältere Herde eine geringere Wachstumsgeschwindigkeit als frische Herde. Typisch sind begleitende lamelläre Periostreaktionen, die nicht unterbrochen sind. Sie werden bei etwa zwei Drittel der Läsionen beobachtet. Selten werden deutliche Knochenauftreibungen gesehen (Abb. 7.54, Abb. 7.55, Abb. 7.56, Abb. 7.57, Abb. 7.58). In der Wirbelsäule führt ein eosinophiles Granulom häufig zu einer Wirbelkörpersinterung, die meist ventral beginnt. In späteren Stadien ist der gesamte Wirbelkörper höhengemindert (Abb. 7.59). Gelegent- lich kann die Höhenminderung so extrem sein, dass nur ein schmales kondensiertes Knochenband übrigbleibt, das auch als Vertebra plana bezeichnet wird. Der Begriff Vertebra plana ist eigentlich mit der Tuberkulose der Wirbelsäule verknüpft. Heute entspricht unter der einheimischen Bevölkerung in Deutschland diese Veränderung jedoch fast immer einem eosinophilen Granulom. Die Höhe der benachbarten Intervertebralräume bleibt erhalten. Gelegentlich ist eine paravertebrale Weichteilkomponente vorhanden. An der Kalotte zerstört das eosinophile Granulom meist sowohl die Tabula externa wie interna. Die Herde haben scharfe Grenzen und wirken wie ausgestanzt (Abb. 7.60). Gelegentlich verbleibt etwas Knochen im Zentrum der Läsion, was auch als Knopfsequester bezeichnet wird. Größere Herde können konfluieren, wodurch die Kalotte landkartenartig imponiert. In den Kiefern kann die Knochendestruktion zum Bild von „schwimmenden Zähnen“ führen. Das typische Bild eines eosinophilen Granuloms in den langen Röhrenknochen ist eine rein osteolytische Läsion, die zentral diaphysär gelegen ist, eine sehr variable Wachstumsgeschwindigkeit aufweisen kann, die von einer nichtunterbrochenen lamellären Periostreaktion begleitet wird und bei Kindern und Jugendlichen angetroffen wird. Merke ! Schnittbilddiagnostik Die im peripheren Skelett gelegenen Läsionen lassen sich mittels konventioneller Röntgendiagnostik meist ausreichend diagnostizieren. Bei aggressiv wachsenden Läsionen ist eine CT oder MRT indiziert. 369 370 Kapitel 7 Tumorähnliche Läsionen Abb. 7.55. Eosinophiles Granulom. Die recht scharf abgrenzbare Osteolyse (Lodwick IB) hat eine reaktive Verdickung der Kompakta induziert Abb. 7.56. Eosinophiles Granulom. Ungewöhnliche, weitgehend in der Querachse des Knochens wachsende Läsion Abb. 7.57. Eosinophiles Granulom. Osteolyse mit Mottenfraßrändern (Lodwick II), die von nichtunterbrochenen lamellären Periostreaktionen begleitet wird In der CT wird man in einer Reihe von Fällen eine nichtdurchbrochene Periostreaktion demonstrieren können. Für die Darstellung einer Weichteilkomponente ist die MRT besser geeignet. Eine Weichteilkomponente ist bei den eosinophilen Granulomen nur klein oder fehlt, bei den malignen Knochentumoren meist jedoch ausgedehnt. An der Wirbelsäule ist zwar die Höhenminderung auf den Röntgenaufnahmen sichtbar, der Tumor im Wir- Abb. 7.58. Eosinophiles Granulom. Osteolyse mit Mottenfraßrändern (Lodwick II). Auch hier kommen am Unterrand der Läsion nichtunterbrochene lamelläre Periostreaktionen zur Darstellung 7.8 Langerhans-Zell-Histiozytose a c Abb. 7.59. Eosinophiles Granulom. Es findet sich eine isolierte ventrale Höhenminderung des 12. BWK, die charakteristisch für ein eosinophiles Granulom ist b Abb. 7.60. Eosinophiles Granulom. Nachweis von mehreren scharf abgrenzbaren Osteolysen in der Kalotte, die bei einem Kind charakteristisch für ein eosinophiles Granulom sind Abb. 7.61 a–c. Eosinophiles Granulom. a Im T1-gewichteten SE-Bild weist der Herd eine vergleichbare Signalintensität wie die Muskulatur auf. Die Kompakta ist penetriert, und lateral ist eine Periostlamelle abgrenzbar. b Im T2-gewichteten SE-Bild lässt sich ein deutliches peritumorales Ödem in den Weichteilen abgrenzen. Neben fokalen Tumorinfiltrationen ist auch eine ödematöse Durchtränkung der Kompakta nachweisbar. c Im kontrastmittelverstärkten T1-gewichteten SE-Bild zeigt die Läsion ein deutliches Enhancement. Daneben ist ein Enhancement der perikortikalen Weichteile sichtbar bel ist jedoch häufig nur in der CT oder MRT abgrenzbar. Die Skelettszintigraphie kann eingesetzt werden, um einen multizentrischen Befall nachzuweisen, kann jedoch nahezu ein Drittel der Herde nicht detektieren. In der MRT stellt sich das eosinophile Granulom im T1-gewichteten Bild weitgehend muskelisointens und im T2-gewichteten Bild hyperintens dar. Die Lä- sion ist häufig ovalär und wird nicht selten von einem ausgedehnten Knochenmarködem umgeben. Sind in den langen Röhrenknochen Periostreaktionen vorhanden, so ist oft auch ein ausgedehntes zirkuläres Weichteilödem vorhanden. Gelegentlich ist eine kleine Weichteilkomponente nachweisbar. Sowohl das eosinophile Granulom als auch die reaktiven Veränderungen zeigen ein deutliches Kontrastmittelenhancement (Abb. 7.61 a–c, Abb. 7.62 a, b). 371 372 Kapitel 7 Tumorähnliche Läsionen Abb. 7.62 a, b. Eosinophiles Granulom. a Ausgeprägte Höhenminderung des 5. HWK, die typisch für ein eosinophiles Granulom ist. b Im kontrastmittelverstärkten T1-gewichteten SE-Bild zeigt der 5. HWK ein deutliches Enhancement. Es lassen sich eine intraspinale und eine prävertebrale gut vaskularisierte Weichteilkomponente abgrenzen a Die MRT-Morphologie ist weitgehend uncharakteristisch und schließt ein Ewing-Sarkom oder eine Osteomyelitis nicht aus. Bei einem Ewing-Sarkom ist jedoch nahezu immer eine solide Weichteilkomponente vorhanden. Zur Detektion weiterer Herde scheint eine MRT-Untersuchung des gesamten zentralen Skeletts mittels STIR-(„short time inversion recovery“-) Sequenz der Skelettszintigraphie überlegen zu sein. Differenzialdiagnose In den meisten Fällen wird man diese Läsion korrekt als benigne einstufen, wobei bei aggressiv wachsenden Läsionen die nichtunterbrochene Periostreaktion das entscheidende Kriterium ist. Eine spezifische Diagnose kann dagegen wesentlich seltener gestellt werden. Wenn eine Osteolyse an der Schädelkalotte oder eine Wirbelkörperhöhenminderung bei Kindern sichtbar ist, spricht dies für ein eosinophiles Granulom. An der Kalotte müssen differenzialdiagnostisch die Epidermoidzyste, wobei die Patienten meist älter sind, das Hämangiom, das typischerweise eine radiäre Trabekelstruktur aufweist, und kongenitale Foramina, die parietookzipital vorkommen können, in Erwägung gezogen werden. Bei einem Wirbelsäulenherd muss vom konventionellen Bild eine Spondylodiszitis abgegrenzt werden, die jedoch mit einer Höhenminderung zumindest eines benachbarten Intervertebralraums einhergeht. Die MRT bringt weitere diagnostische Sicherheit, da die typischen Bandscheibenveränderungen einer Spondylodiszitis fehlen. Das eosinophile Granulom in den anderen Skelettregionen gehört auch bei den Fachleuten zu den Läsionen, die zu selten auf der Liste der möglichen b Diagnosen angegeben werden. Ein im Beckenskelett oder in den Rippen aggressiv wachsendes eosinophiles Granulom wird häufig als Ewing-Sarkom fehlgedeutet. In der MRT ist jedoch beim Ewing-Sarkom regelmäßig eine ausgedehnte Weichteilkomponte nachweisbar. An den langen Röhrenknochen wird man ein eosinophiles Granulom seltener mit einem Ewing-Sarkom verwechseln, da ersteres häufig eine nichtunterbrochene lamelläre Periostreaktion zeigt, die fast immer gegen einen malignen Prozess spricht. Allerdings kann eine floride Osteomyelitis ein nahezu identisches Bild bieten, besonders wenn das eosinophile Granulom epiphysenfugennah gelegen ist. Wenn keine Periostreaktionen vorliegen, sind bei einem langsam wachsenden eosinophilen Granulom bei Kindern mögliche Differenzialdiagnosen die solitäre und aneurysmatische Knochenzyste und auch die fibröse Dysplasie. Je jünger der Patient, umso eher wird man an ein eosinophiles Granulom denken. Tritt das eosinophile Graulom bei älteren Patienten auf, wird es kaum als mögliche Diagnose in Betracht gezogen werden. Hier wird eher an Metastasen, Myelome, maligne fibröse Histiozytome und an Fibrosarkome gedacht. Therapie und Prognose Zunächst sollte ein Versuch mit einer lokalen Steroidinjektion unternommen werden. In vielen Fällen kann dadurch das Wachstum gestoppt werden, und die Läsion heilt aus. Versagt diese Therapie, bieten sich eine sorgfältige Kürettage und anschließende Auffüllung mit Knochenspänen an. Sind einige Läsionen operativ nur sehr schlecht zugänglich, kann eine reine Beobachtung in Erwägung gezogen wer- 7.8 Langerhans-Zell-Histiozytose a b Abb. 7.63 a, b. Hand-Schüller-Christian-Erkrankung. a Im distalen Femur kommen multiple kleine Osteolysen zur Darstellung, die recht scharf begrenzt sind. Es handelt sich eher um multifokale Herde als um Mottenfraßosteolysen. b Daneben können mehrere, teilweise konfluierende Herde in der Kalotte abgegrenzt werden den, da auch spontane Regression und Heilung vorkommen. Bei einem mutizentrischen Befall wird auch eine Chemotherapie durchgeführt. Das eosinophile Granulom ist strahlensensibel und kann mit einer Dosis von 10 Gy therapiert werden. in unterschiedlichen Knochen angetroffen (Abb. 7.63 a, b). Daneben sind auch innere Organe wie Leber, Milz, Lunge und Lymphknoten befallen. Die Krankheit zeigt einen periodischen Verlauf mit aktiven und inaktiven Phasen und verläuft nur bei einer Minderheit der Patienten tödlich. Die ossären Läsionen sehen wie die Herde des eosinophilen Granuloms aus. Häufig sind multiple Herde in einem Knochen vorhanden, die konfluieren können. Die Behandlung besteht in einer Chemotherapie. Hier muss jedoch das Risiko eines strahleninduzierten Sarkoms berücksichtigt werden. CAVE ! Eine durch Steroide induzierte Heilung eines eosinophilen Granuloms erfolgt über mehrere Monate hinweg. Nach Abschluss des Heilungsprozesses bleiben keine oder nur minimale Restveränderungen übrig. Sogar ein deutlich kollabierter Wirbelkörper kann wieder aufgebaut werden. Bei etwa 20% der Patienten, die zunächst nur eine solitäre Läsion hatten, treten weitere Herde auf. Das eosinophile Granulom kann auch in eine generalisierte Form transformieren.Allgemein gilt, dass junge Patienten ein höheres Risiko für das Auftreten multipler Herde haben. Wenn eine solitäre Läsion für mehr als ein Jahr solitär bleibt, kann davon ausgegangen werden, dass keine weiteren Herde auftreten werden. 7.8.2 Hand-Schüller-Christian-Erkrankung Bei dieser sehr seltenen Erkrankung handelt es sich um die disseminierte chronische Form der Langerhans-Zell-Histiozytose. Es werden multiple Herde 7.8.3 Abt-Letterer-Siwe-Erkrankung Bei dieser sehr seltenen Erkrankung handelt es sich um die disseminierte akute oder subakute schwere Variante der Langerhans-Zell-Histiozytose. Um diese Diagnose stellen zu können, müssen folgende Kriterien erfüllt sein: Krankheitsbeginn vor dem 3. Lebensjahr, Fieber, wiederholte bakterielle Infektionen, Anämie, Blutungen, Hepatosplenomegalie, Lymphadenopathie und kutane Veränderungen. Die Erkrankung verläuft meist tödlich. Nicht alle Patienten haben Skelettläsionen, da möglicherweise die Erkrankungszeit zu kurz ist, um diese auszubilden. Sind Skelettveränderungen vorhanden, sind diese multipel und unterscheiden sich nicht von denen des eosinophilen Granuloms. Die Behandlung besteht in einer Chemotherapie. 373 374 Kapitel 7 Tumorähnliche Läsionen 7.9 Diagnose-Schemata epiphysär epimetaphysär epimetadiaphysär metaphysär Osteolyse ohne Verkalkung/Verknöcherung Chondroblastom1 IA IB IC II 䊴 Chondroblastom1 III IA IB IC II 䊴 䊳 10 20 30 40 50 >50 J 䊴 䊳 III IA IB IC II 䊴 䊳 10 20 30 40 50 >50 J 䊴 IA IB IC II III 䊴 10 20 30 40 50 >50 J 䊴 IA IB IC II 䊴 IA IB IC II 䊳 <Chondromyxoidfibrom> III IA IB IC II 䊴 䊳 10 20 30 40 50 >50 J 䊴 䊳 <Osteosarkom> 䊴 䊴 III IA IB IC II III IA IB IC II 䊴 䊳 III 10 20 30 40 50 >50 J 䊳 䊴 Chondromyxoidfibrom IA IB IC II III III 䊴 䊳 䊴 10 20 30 40 50 >50 J 䊳 10 20 30 40 50 >50 J 䊳 䊴 <Chondromyxoidfibrom> IA IB IC II III 䊳 <MFH> IA IB IC II III 䊴 䊳 䊳 10 20 30 40 50 >50 J 䊴 䊳 䊴䊳 䊳 10 20 30 40 50 >50 J 䊴 10 20 30 40 50 >50 J NOF 10 20 30 40 50 >50 J 䊴 III 䊳 䊴 䊴 <MFH> IA IB IC II Ganglion 䊳 䊴 䊳 䊳 䊴 IA IB IC II 䊳 䊴 III <AKZ> IA IB IC II 10 20 30 40 50 >50 J 䊳 10 20 30 40 50 >50 J III 䊳 䊴 䊴 IA IB IC II 䊳 Osteoblastom III 10 20 30 40 50 >50 J 䊳 III 䊳 10 20 30 40 50 >50 J 䊴䊳 Ganglion 䊴 䊳 䊳 䊴 IA IB IC II 䊴 <Fibrosarkom> III 10 20 30 40 50 >50 J 䊳 III 䊳 䊴 IA IB IC II 䊳 SKZ 10 20 30 40 50 >50 J 䊳 Riesenzelltumor Ganglion 䊴 Riesenzelltumor 10 20 30 40 50 >50 J 䊴 䊳 䊳 Osteolyse mit Matrixverkalkung <Chondrosarkom> IA IB IC II III Chondrosarkom IA IB IC II 䊴䊳 䊴 10 20 30 40 50 >50 J 䊴 <Enchondrom> IA IB IC II 䊴 1 gelegentlich kleine Matrixverkalkungen 2 häufig Mattglasmatrix J Lebensjahre Kursiv: häufig exzentrisch 䊳 䊴 <Enchondrom> IA IB IC II III 10 20 30 40 50 >50 J 䊳 䊳 䊴 䊳 III 10 20 30 40 50 >50 J 䊴 häufig seltener <Tumor> eher seltenere Lage oder Morphologe 䊴 䊳 䊳 䊴 䊴 䊳 10 20 30 40 50 >50 J 䊳 䊳 䊴 10 20 30 40 50 >50 J 䊴 䊴 <Enchondrom> IA IB IC II 䊳 䊴 䊳 10 20 30 40 50 >50 J 䊳 III <Chondrosarkom> IA IB IC II III III 䊳 7.9 Diagnose-Schemata metadiaphysär 375 diaphysär Osteolyse ohne Verkalkung/Verknöcherung NOF Ewing Sarkom IA IB IC II III IA IB IC II 䊳 䊴 III 䊴 10 20 30 40 50 >50 J 䊴 䊴 䊳 䊳 III IA IB IC II 䊴 䊳 䊴 10 20 30 40 50 >50 J 䊳 IA IB IC II 䊴 III 䊴 䊳 Osteolytisches Osteosarkom IA IB IC II 䊴 III 䊴 䊴 䊳 10 20 30 40 50 >50 J 䊳 䊴 AKZ IA IB IC II 䊴 IA IB IC II 䊴 III 10 20 30 40 50 >50 J 䊳 䊴 䊳 10 20 30 40 50 >50 J 䊴 Enchondrom IA IB IC II III IA IB IC II 䊴 10 20 30 40 50 >50 J 䊴 䊴 䊳 Enchondrom 䊳 䊴 III 10 20 30 40 50 >50 J 䊳 䊳 III 䊳 10 20 30 40 50 >50 J 䊴 10 20 30 40 50 >50 J 䊴 䊴 䊳 䊳 III 䊳 䊴 10 20 30 40 50 >50 J Chondrosarkom IA IB IC II III 䊳 Plasmozytom IA IB IC II Osteolyse mit Matrixverkalkung Chondrosarkom IA IB IC II 10 20 30 40 50 >50 J 䊴 III III 䊳 䊴 䊳 䊴 䊳 Eosinophiles Granulom IA IB IC II 䊳 䊴 䊳 䊴 III 10 20 30 40 50 >50 J III 䊳 10 20 30 40 50 >50 J 䊴 䊳 AKZ IA IB IC II 䊳 䊴 䊳 䊳 䊳 䊳 䊴 Osteolytisches Osteosarkom 10 20 30 40 50 >50 J 䊴 䊴 IA IB IC II 䊳 10 20 30 40 50 >50 J III 10 20 30 40 50 >50 J III 䊴 IA IB IC II III 䊳 Fibröse Dysplasie2 IA IB IC II 䊳 Fibröse Dysplasie2 䊴 䊳 䊴 10 20 30 40 50 >50 J III IA IB IC II 10 20 30 40 50 >50 J III 䊳 䊴 NHL 䊴 IA IB IC II III 䊴 10 20 30 40 50 >50 J NHL 䊴 Osteoblastom IA IB IC II 䊳 III 䊳 10 20 30 40 50 >50 J 䊴 䊳 IA IB IC II 䊳 䊴 MFH 䊳 䊴 IA IB IC II 䊳 䊳 䊳 Osteoblastom 䊳 III 10 20 30 40 50 >50 J III 䊳 SKZ IA IB IC II 䊴 10 20 30 40 50 >50 J 䊴 䊴 䊴 IA IB IC II 10 20 30 40 50 >50 J 10 20 30 40 50 >50 J 䊳 䊳 䊴 䊳 III 䊴䊳 䊴 䊳 Chondromyxoidfibrom Fibrosarkom IA IB IC II 䊳 10 20 30 40 50 >50 J III 10 20 30 40 50 >50 J 䊴 III Fibrosarkom SKZ MFH 䊴 IA IB IC II 䊴䊳 10 20 30 40 50 >50 J IA IB IC II NOF Ewing Sarkom 䊳 376 Kapitel 7 Tumorähnliche Läsionen epimetadiaphysär metaphysär metadiaphysär diapysär Osteolyse mit ausgeprägter Matrixverknöcherung <Osteosarkom> IA IB IC II Osteosarkom III IA IB IC II 䊴 䊳 䊳 䊳 IA IB IC II 10 20 30 40 50 >50 J III 10 20 30 40 50 >50 J 䊳 IA IB IC II 10 20 30 40 50 >50 J 䊴 <Osteoblastom> IA IB IC II 䊴 III 䊳 III 䊴 䊳 䊴 III 3 extraossär auf der Kompakta 4 Morphologie weitgehend durch reaktive Sklerose bestimmt J Lebensjahre Literatur Abe O, Yoshikawa K, Eguchi M et al. (1997) Intraosseous ganglion communicating with soft tissue counterpart. Radiat Med: 15:125–127 Adler CP (1990) Case report 587: adamantinoma of the tibia mimicking osteofibrous dysplasia. Skeletal Radiol 19:55–58 Ameli NO,Abbassioun K, Saleh H, Eslamdoost (1985) Aneurysmal bone cyst of the spine. Report of 17 cases. J Neurosurg 63:685–690 Arana E, Latorre FF, Revert M et al. (1996) Intradiploic epidermoid cysts. Neuroradiology 38:306–311 Arana E, Marti-Bonmati L (1999) CT and MR imaging of focal calvarial lesions. AJR Am J Roentgenol 172:1683–1688 Arata MA, Peterson HA, Dahlin DC (1981) Pathological fractures through non-ossifying fibromas. Review of the Mayo Clinic experience. J Bone Joint Surg Am 63:980–988 Beltran J, Simon DC, Levy M et al. (1986) Aneurysmal bone cysts: MR imaging at 1.5T. Radiology 158:689–690 Beltran J, Aparisi F, Bonmati LM et al. (1993) Eosinophilic granuloma: MRI manifestation. Skeletal Radiol 22:157–161 Bertoni F, Calderoni P, Bacchini P et al. (1986) Benign fibrous histiocytome of bone. J Bone Joint Surg Am 68:1225–1230 10 20 30 40 50 >50 J 䊳 䊳 <Ewing Sarkom> IA IB IC II 䊳 10 20 30 40 50 >50 J III 䊳 䊴 <Ewing Sarkom> IA IB IC II 䊴 䊳 IA IB IC II 䊴 10 20 30 40 50 >50 J 䊳 10 20 30 40 50 >50 J <Osteoblastom> 䊳 䊴 III 䊳 䊴 IA IB IC II 䊳 IA IB IC II 䊴 <Osteoblastom> 10 20 30 40 50 >50 J 䊴 III 䊳 䊴 䊳 Osteoid-Osteom4 10 20 30 40 50 >50 J 䊳 III 10 20 30 40 50 >50 J 䊴 IA IB IC II 䊴 IA IB IC II 䊳 Osteoid-Osteom4 III 䊳 POS III 10 20 30 40 50 >50 J 䊳 䊳 3 䊴 Osteoid-Osteom4 III 10 20 30 40 50 >50 J 䊴 IA IB IC II 䊳 䊴 䊴 䊳 POS IA IB IC II IA IB IC II 䊴 3 POS III III 10 20 30 40 50 >50 J 䊴 3 IA IB IC II Osteosarkom 䊴䊳 10 20 30 40 50 >50 J 䊴 POS3 䊴 III 䊴 䊳 10 20 30 40 50 >50 J 䊴 Osteosarkom 䊴 III 䊳 10 20 30 40 50 >50 J 䊴 䊳 häufig seltener <Tumor> eher seltenere Lage oder Morphologe 䊴 䊳 䊴 䊳 Bonakdarpour A, Levy WM, Aegerter E (1978) Primary and secondary bone cysts. A radiological study of 75 cases. Radiology 126:75–82 Bosse A, Niesert W, Wuisman P, Roessner A(1993) Fibröse Dysplasie versus osteofibröse Dysplasie. Z Orthop 131:42–50 Bosse A, Russe O,Weber A, Müller KM (1994) Zur Kenntnis der osteofibrösen Dysplasie Campanacci. Radiologe 34:59–62 Burnstein MJ, DeSmet AA, Hafez GR, Heiner JP (1990) Subperiosteal aneurysmal bone cyst of tibia. Case report 611. Skeletal Radiol 19:294–297 Campana RDS, Springfield R, Biagini R, Ruggieri P, Giunti W (1985) Juxtaepiphyseal aneurysmal bone cyst. Skeletal Radiol 13:21–25 Campanacci M, Laus M (1981) Osteofibrous dysplasia of the tibia and fibula. J Bone Joint Surg Am 63:367–375 Campanacci M, Capanna R, Picci P (1986) Unicameral and aneurysmal bone cysts. Clin Orthop 204:25–36 Capanna R, Van Horn J, Ruggieri R, Biagini R (1986) Epiphyseal involvement in unicameral bone cysts. Skeletal Radiol 15:428–432 Ciappetta P, Artico M, Salvati M, Raco A, Gagliardi FM (1990) Intradiploic epidermoid cysts of the skull: report of 10 cases and review of the literature. Acta Neurochir 102:33–37 Literatur Clarke BE, Xipell JUM, Thomas DP (1985) Benign fibrous histiocytoma of bone. Am J Surg Pathol 9:806–815 Cockshott WP, Martin R, Friedman L, Yuen M (1994) Focal fibrocartilaginous dysplasia and tibia vara: a case report. Skeletal Radiol 23:333–335 Constans JP, Meder JF, De Divitiis E, Donzelli R, Maiuri F (1985) Giant intradiploic epidermoid cysts of the skull. Report of two cases. J Neurosurg 2:445–448 Cory DA, Fritsch SA, Cohen MD et al. (1989) Aneurysmal bone cysts: imaging findings and embolotherapy. AJR Am J Roentgenol 153:369–373 Crone-Münzebrock W, Brassow F (1983) A comparison of radiographic and bone-scan findings in histiocytosis X. Skeletal Radiol 9:170–173 Daffner RH, Kirks DB, Gehweiler JA, Heaton DK (1982) Computed tomography of fibrous dysplasia. AJR Am J Roentgenol 139:943–948 De Schepper AMA, Ramon F, Van Marck E (1993) MR imaging of eosinophilic granuloma: report of 11 cases. Skeletal Radiol 22:163–166 Dominguez R, Saucedo J, Fenstermacher M (1989) MRI findings in osteofibrous dysplasia. Magn Reson Imaging 7:567–570 Dungan DH, Seeger LL, Mirra JM (1989) Intraosseous ganglion cyst of the distal end of tibia. Case report 555. Skeletal Radiol 18:385–388 Easley ME, Kneisl JS (1997) Pathologic fractures through nonossifying fibromas: is prophylactic treatment warranted? J Pediatr Orthop 17:808–813 Erlemann R, Fischedick A, Edel G, Peters PE, Galanski M (1987) Neurofibromatose und multiple nicht-ossifizierende Knochenfibrome. RöFo 147:20–24 Erlemann R, Picker S, Müller-Miny H, Wuisman P, Edel G (1993) Aneurysmatische Knochenzyste oder Riesenzelltumor. Wertigkeit der Röntgendiagnostik zur Differentialdiagnose. RöFo 158:343–347 Exner GU, von Hochstetter AR, Uehlinger K (1990) Benign fibrous histiocytoma of the distal femoral metaphysis. Differential diagnosis between neoplasm and growth disorder with identical morphology. Z Orthop 128:308–312 Feldman F, Johnston A (1973) Intraosseous ganglion. AJR Am J Roentgenol 118:328–343 Ferkel RD, Field J, Scherer WP, Bernstein ML, Kasimian D (1999) Intraosseous ganglion cysts of the ankle: a report of three cases with long-term follow-up. Foot Ankle Int 20:384–388 Freeby JA, Reinus WR, Wilson AJ (1995) Quantitative analysis of the plain radiographic appearance of aneurysmal bone cysts. Invest Radiol 30:433–439 Gordon SL, Denton JR, McCann PD, Parisien MV (1987) Unicameral bone cyst of the talus. Clin Orthop 215:201–205 Griffet J, Roche JL, Grellier P, Duplay J (1985) Epidermoid cysts of the cranial vault in children. Apropos of 6 cases. Pediatrics 40:561–564 Guiard JM, Kien P, Colombani S, Caille JM (1986) Intradiploic epidermoid cysts in adults. CT contribution to diagnosis in 6 new cases. J Neuroradiol 13:22–31 Haims AH, Desai P, Present D, Beltran J (1997) Epiphyseal extension of a unicameral bone cyst. Skeletal Radiol 26: 51–54 Halawa M, Aziz AA (1984) Chondrosarcoma in fibrous dysplasia of the pelvis. A case report and review of the literature. J Bone Joint Surg Br 66:760–764 Hefti F, Jundt G (1995) Langerhans-Zell-Histiozytose. Orthopäde 24:73–81 Helwig U, Lang S, Baczynski M, Windhager R (1994) The intraosseous ganglion. A clinical-pathological report on 42 cases. Arch Orthop Trauma Surg 114:14–17 Hoeffel C, Panuel M, Plenat F, Mainard L, Hoeffel JC (1999) Pathological fracture in non-ossifying fibroma with histological features simulating aneurysmal bone cyst. Eur Radiol 9:669–671 Howlett DC, Farrugia MM, Ferner RE, Rankin SC (1998) Multiple lower limb non-ossifying fibromas in siblings with neurofibromatosis. Eur J Radiol 26:280–283 Ishida T, Dorfman HD (1993) Massive chondroid differentiation in fibrous dysplasia of bone. Am J Surg Pathol 17:924– 930 Ishida T, Iijima T, Kikuchi F, Kitagawa T, Tanida T, Imamura T, Machinami R (1992) A clinicopathological and immunohistochemical study of osteofibrous dysplasia, differentiated adamantinoma, and adamantinoma of long bones. Skeletal Radiol 21:493–502 Jee WH, Choi KH, Choe BY, Park JM, Shinn KS (1996) Fibrous dysplasia: MR imaging characteristics with radiopathologic correlation. AJR Am J Roentgenol 167:1523–1527 Komiya S, Inoue A (1993) Aggressive bone tumorous lesion in infancy: osteofibrous dysplasia of the tibia and fibula. J Pediatr Orthop 13:577–581 Kransdorf MJ, Moser RP Jr, Gilkey FW (1990) Fibrous dysplasia. Radiographics 10:519–537 Kransdorf MJ, Sweet DE (1995) Aneurysmal bone cyst: concept, controversy, clinical presentation, and imaging. AJR Am J Roentgenol 164:573–580 Kransdorf MJ, Murphey MD, Sweet DE (1999) Liposclerosing myxofibrous tumor: a radiologic-pathologic-distinct fibroosseous lesion of bone with a marked predilection for the intertrochanteric region of the femur. Radiology 212:693– 698 Kuruvilla G, Steiner GC (1998) Osteofibrous dysplasia-like adamantinoma of bone: a report of five cases with immunohistochemical and ultrastructural studies. Hum Pathol 29:809–814 Lassance Cabral CE, Guedes P, Fonseca T et al. (1998) Polyostotic fibrous dysplasia associated with intramuscular myxomas: Mazabraud´s syndrome. Skeletal Radiol 27:278–282 Lee JH, Reinus WR, Wilson AJ (1999) Quantitative analysis of the plain radiographic appearance of unicameral bone cysts. Invest Radiol 34:28–37 Lieberman PH, Jones CR, Steinman RM et al. (1996) Langerhans cell (eosinophilic) granulomatosis.A clinicopathologic study encompassing 50 years. Am J Surg Pathol 20:519–552 Lopez-Ben R, Pitt MJ, Jaffe KA, Siegal GP (1999) Osteosarcoma in a patient with McCune-Albright syndrome and Mazabraud’s syndrome. Skeletal Radiol 28:522–526 Machida K, Makita K, Nishikawa J, Ohtake T, Iio M (1986) Scintigraphic manifestation of fibrous dysplasia. Clin Nucl Med 11:426–429 Majewski A, Freyschmidt J, Steinmeier R, Ostertag H (1984) Das ossifizierende Knochenfibrom. RöFo 140:179–187 Makley JT, Carter JR (1986) Eosinophilic granuloma of bone. Clin Orthop 189:37–44 Makley JT, Joyce MJ (1989) Unicameral bone cyst (simple bone cyst). Orthop Clin North Am 20:407–415 Martinez V, Sissons HA (1988) Aneurysmal bone cyst.A review of 123 cases including primary lesions and those secondary to other bone pathology. Cancer 61:2291–2304 Matsuo M, Ehara S, Tamakawa Y et al. (1997) Aggressive appearance of non-ossifying fibroma with pathologic fracture: a case report. Radiat Med 15:113–115 Moser Jr RP, Sweet DE, Haseman DB, Madewell JE (1987) Multiple skeletal fibroxanthomas: radiologic-pathologic correlation of 72 cases. Skeletal Radiol 16:353–359 Müller-Miny H, Verstring T, Edel G, Erlemann R, Peters PE (1993) Röntgenmorphologie des eosinophilen Granuloms außerhalb von Schädel und Wirbelsäule. RöFo 158:583–588 Munk PL, Helms CA, Holt RG et al. (1989) MR imaging of aneurysmal bone cysts. AJR Am J Roentgenol 153:99–101 377 378 Kapitel 7 Tumorähnliche Läsionen Nauert C, Zornoza J, Ayala A, Härle TS (1983) Eosinophilic granuloma of bone: diagnosis and management. Skeletal Radiol 10:227–235 Norris MA, Kaplan PA, Pathria M, Greenway G (1990) Fibrous dysplasia: magnetic resonance imaging appearance at 1.5 tesla. Clin Imaging 14:211–215 Oda Y, Hashimoto H, Tsuneyoshi M, Ono N (1992) Case report 742: Intraosseous epidermoid cyst arising in the fifth metacarpal bone. Skeletal Radiol 21:343–345 Ozaki T, Hamada M, Sugihara S et al. (1998) Treatment outcome of osteofibrous dysplasia. J Pediatr Orthop 7:199–202 Park YK, Unni KK, McLeod RA, Pritchard DJ (1993) Osteofibrous dysplasia: clinicopathologic study of 80 cases. Hum Pathol 24:1339–1347 Pope TL, Fechner RE, Keats TE (1989) Intraosseous ganglion. Report of four cases and review of literature. Skeletal Radiol 18:185–187 Remagen W, Niedecker A, Prein J (1986) Gigantic benign fibrous histiocytoma (non-ossifying fibroma). Skeletal Radiol 15:251–253 Ritschl P, Hajek PC, Pechmann U (1989) Fibrous metaphyseal defects. Magnetic resonance imaging appearances. Skeletal Radiol 18:253–259 Ritschl P, Karnel F (1986) Zur Pathogenese des fibrösen Kortikalisdefektes und ossifizierenden Knochenfibroms. Z Orthop 124:682–687 Ritschl P, Karnel F, Hajek P (1988) Fibrous metaphyseal defects – determination of their origin and natural history using a radiomorphological study. Skeletal Radiol 17:8–15 Ruggieri P, Sim FH, Bond JR, Unni KK (1994) Malignancies in fibrous dysplasia. Cancer 73:1411–1424 Sathekge MM, Clauss RP (1995) Criteria and quantification of fibrous dysplasia on MDP scanning. Nuklearmedizin 34: 229–331 Schajowicz F, Clavel Saint M, Slullitel JA (1979) Juxta-articular bone cyst (intraosseous ganglia). A clinico-pathological study of 88 cases. J Bone Joint Surg Br 61:107–116 Shih HN, Chen YJ, Huang TJ, Hsu KY, Hsu RW (1998) Treatment of fibrous dysplasia involving the proximal femur. Orthopedics 21:1263–1266 Springfield DS, Rosenberg AE, Mankin HJ, Mindell ER (1994) Relationship between osteofibrous dysplasia and adamantinoma. Clin Orthop 309:234–244 Stiglbauer R, Ritschl P, Kramer J, Imhof H (1989) Fibröse Dysplasie: Erscheinungsbild im Magnetresonanztomogramm. Röfo 151:338–341 Struhl S, Edelson CD, Pritzker H et al. (1989) Solitary (unicameral) bone cyst. The fallen fragment sign revisited. Skeletal Radiol 18:261–265 Stull MA, Kransdorf MJ, Devaney KO (1992) Langerhans cell histiocytosis of bone. Radiographics 12:801–823 Sundaram M, MCDonald DJ, Marenda G (1989) Intramuscular myxoma: a rare but important association with fibrous dysplasia of bone. AJR Am J Roentgenol 155:107–108 Sweet DE, Vinh TN, Devaney K (1992) Cortical osteofibrous dysplasia of long bone and its relationship to adamantinoma. A clinicopathologic study of 30 cases. Am J Surg Pathol 16:282–290 Taconis WK (1988) Osteosarcoma in fibrous dyplasia of the spine. Skeletal Radiol 17:163–170 Tam W, Resnick D, Haghighi P, Vaughan L (1996) Intraosseous ganglion of the patella. Skeletal Radiol 25:588–591 Tehranzadeh J, Fung Y, Donohue M, Anavim A, Pribram HW (1998) Computed tomography of Paget disease of the skull versus fibrous dysplasia. Skeletal Radiol 27:664–672 Tsai JC, Dalinka MK, Fallon MD, Zlatkin MB, Kressel HY (1990) Fluid-fluid level: a nonspecific finding in tumors of bone and soft tissue. Radiology 175:779–782 Ueda Y, Roessner A, Bosse A et al. (1991) Juvenile intracortical adamantinoma of the tibia with predominant osteofibrous dysplasia-like features. Pathol Res Pract 187:1039–1043 Wakabayashi I, Okada K, Hashimoto M, Sageshima M (1999) Intraosseous ganglion of the metatarsal bone. J Comput Assist Tomogr 23:727–729 Wormer RB, Raney JrRB, D´Angio GJ (1985) Healing rates of treated and untreated bone lesions in histiocytosis X. Pediatrics 76:286–288 Wörtler K, Blasius S, Hillmann A et al. (2000) MR-Morphologie der primären aneurysmatischen Knochenzyste: Retrospektive Analyse von 38 Fällen. RöFo 172:591–596 Wu KK (1992) Epidermoid cysts of the foot: with or without bone involvement. J Foot Surg 31:203–206 Yabut SM, Kenan S, Sisson HA, Lewis MN (1988) Malignant transformation of fibrous dysplasia. A case report and review of the literature. Clin Orthop 228:281–290 Yamaguchi T, Dorfman HD, Eisig S (1999) Cherubism: clinicopathologic features. Skeletal Radiol 28:350–353 Yao L, Eckardt JJ, Seeger LL (1994) Fibrous dysplasia associated with cortical bony destruction: CT and MR findings. J Comput Assist Tomogr 18:91–94