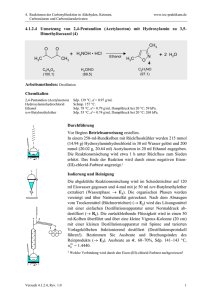
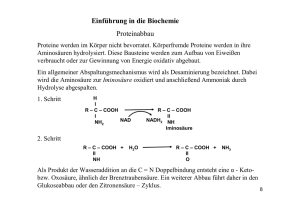
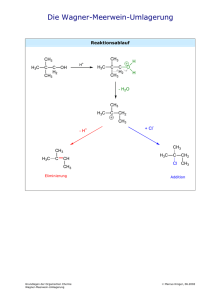
OC Crashkurs Bioinfos
Werbung

Crashkurs Organische Chemie Zweck dieses Crashkurses • Viele Studenten der Bioinformatik, Biophysik und auch der Biologie haben Chemie in der Oberstufe abgewählt. • Große Überraschung zu Beginn des Studiums: ich brauche ja auch Chemie, um Biochemie, Physiologie, Genetik usw. zu verstehen. • Die Studienpläne der o.g. Fächer sehen vor, dass Sie Allgemeine Chemie und Biochemie hören sollen. Biochemie ist aber ohne Organische Chemie nicht machbar. • Fazit: Crashkurs Organische Chemie = 5 Nachmittage mit je 5-6 h OC entspricht einer ca. 1,5-2-stündigen OC-Vorlesung. • In dieser kurzen Zeit kann nur die für die Biochemie relevante OC behandelt werden. Nutzen Sie die anschließende Biochemie-Vorlesung zur Vertiefung der Organischen Chemie mit Hilfe geeigneter Literatur! Literatur • Latscha/Kazmaier – Chemie für Biologen, Springer-Verlag, 3. Aufl. 2008 • Gossauer – Struktur und Reaktivität der Biomoleküle, VCH, 1. Aufl. 2006 Inhaltsverzeichnis 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. Einführung Reaktive Teilchen in der organischen Chemie Alkane Halogenalkane Alkene Alkine Aromaten Alkohole und Ether Aldehyde, Ketone und Derivate Carbonsäuren und Carbonsäurederivate Amine und wichtige Heterocyclen Aminosäuren Biochemie Saccharide 3 1. Einführung 1.1. Was ist Organische Chemie? • Um 1800: Berzelius bezeichnete alle Verbindungen, die von lebenden Organismen produziert werden, als „organisch“. • Aus Elementaranalysen: alle „Organischen Verbindungen“ enthalten als zentrales Element Kohlenstoff C. • 1828: Wöhler wandelt Ammoniumcyanat (anorganisch!) in Harnstoff (organisch!!) um. • Heute: alle organischen Verbindungen enthalten neben C auch immer H. Ausserdem O, N, F, Cl, Br, I, P, S, Se, Li, Mg, B, Al, Ti, Pd, Zn, Zr, Cu, Co u. a. Elemente des PSE (Heteroatome). • Definition: Organische Chemie ist die Chemie der Kohlenstoffverbindungen. Sie beschreibt deren Herstellung, Eigenschaften und Umwandlungen. • Wichtiger Zusammenhang: Biochemie ist Organische Chemie der lebenden Organismen. 4 1.2. Schreibweise in der Organischen Chemie • Wichtig: Strukturformeln sind in der OC viel wichtiger als Summenformeln! • Strukturformeln werden üblicherweise nach bestimmten Regeln gezeichnet, so dass die Formeln die Strukturen realistisch abbilden, schnell gezeichnet werden können und eindeutig sind. • 1) Lange Kohlenstoffketten werden als Zick-Zack-Linien gezeichnet. 2) Jedes Eck entspricht einem C-Atom (C wird nur gezeichnet, wenn man es braucht) 3) Hs werden nur gezeichnet, wenn man sie braucht. 4) Heteroatome werden immer gezeichnet! 5) Freie Elektronenpaare zeichnet man nur, wenn man sie braucht. 6) Positive oder negative Ladungen werden immer angegeben! 7) Ein Strich entspricht einem Elektronenpaar (Lewis-Formel oder Valenz-Strich-Formel). O OH 5 1.3. Eigenschaften von Kohlenstoff in Organischen Verbindungen • Kohlenstoff kann maximal vier Bindungen zu Bindungspartnern ausbilden. Dann ist er neutral (Formalladung 0). • Kohlenstoff kann auch nur drei Bindungen zu Bindungspartnern ausbilden. Dann ist er entweder positiv (Formalladung +1; Carbeniumion) oder negativ (Formalladung -1; Carbanion). Ladungen muss man in Formeln immer angeben. • Kohlenstoff kann auch nur zwei Bindungen zu Bindungspartnern ausbilden. Dann ist er wieder neutral mit einem freien Elektronenpaar (Carben). Zweifach positiv oder zweifach negativ geladene C-Atome kommen in organischen Verbindungen nicht vor. • Kohlenstoff ist in seinen Verbindungen immer hybridisiert. Kohlenstoff kann sp3-hydbridisiert, sp2-hybridisiert oder sp-hybridisiert sein (vgl. Vorlesung „Allgemeine Chemie“). • Je nach Hybridisierung und Ladung gibt es unterschiedliche Strukturen. 6 ” H H C H H H C H H H C H H C N “ N H C H H “ C H O C H ” C H H • sp3-hybridisierter Kohlenstoff ist entweder neutral oder anionisch. sp3hybridisierter Kohlenstoff kann nur Einfachbindungen ausbilden. Das CAtom ist immer tetraedrisch umgeben. • sp2-hybridisierter Kohlenstoff ist entweder neutral oder kationisch. sp2hybridisierter Kohlenstoff kann Einfachund Doppelbindungen ausbilden. Das CAtom ist immer trigonal planar umgeben. H N • sp-hybridisierter Kohlenstoff ist entweder neutral, anionisch oder kationisch. sphybridisierter Kohlenstoff kann Doppelund Dreifachbindungen ausbilden. Das C-Atom ist immer linear umgeben. 7 • Orbitalmodell einer C-CEinfachbindung. Sie ist rotationssymmetrisch bzgl. der Kernverbindungslinie (σBindung). Bindungswinkel: 109°, Länge: 154 pm. Bindungsenergie: 330 kJ/mol. • Orbitalmodell einer C=CDoppelbindung. Bindungswinkel: 120°, Länge: 134 pm. Eine Doppelbindung besteht aus einer rotationssymmetrischen σBindung und einer spiegelsymmetrischen π-Bindung. Bindungsenergie 620 kJ/mol. 8 • Orbitalmodell einer C≡CDreifachbindung. Bindungswinkel: 180°, Länge: 121 pm. Eine Dreifachbindung besteht aus einer rotationssymmetrischen σ-Bindung und zwei spiegelsymmetrischen πBindungen. Bindungsenergie 810 kJ/mol. 9 1.4. Allgemeines zur Struktur Organischer Verbindungen • Die C- und H-Atome bilden das Grundgerüst („Kohlenstoff-Skelett“) organischer Verbindungen. Organische Verbindungen werden primär nach ihrem Grundgerüst eingeteilt. Organische Verbindungen Acyclische Verbindungen Gesättigte Kohlenwasserstoffe Cyclische Verbindungen Ungesättigte Kohlenwasserstoffe Heterocyclen Carbocyclen gesättigt ungesättigt aromatisch Alicyclen gesättigt ungesättigt aromatisch Aromaten 10 • Die Mehrzahl der organischen Verbindungen enthalten neben C und H auch noch Heteroatome (N, P, O, S, Se, F, Cl, Br, I usw.). • Die Heteroatome sind in definierter Weise mit C-Atomen verknüpft und bilden sogenannte Funktionelle Gruppen. Funktionelle Gruppen bestimmen die Reaktionsweisen organischer Moleküle. • Wichtige funktionelle Gruppen in der organischen Chemie: R NH2 R NH–R´ R´ R N R´´ O R OH O R NO R R NO2 R CN R O R F O R Cl R´ R Br O O R NR´R´´ OH OH R H O R O P R OR´ R R OH R SH R I OR´ Cl R SR´ R SO3H 11 • Beispiele: O OH ” O O N “ OH OH OH NH2 COOH HOOC COOH HO COOH HO O HO HO • Heteroatome oder funktionelle Gruppen werden oft auch als Substituenten bezeichnet (weil sie formal ein oder mehrere H-Atome ersetzen). 12 1.5. Ladungsverteilung in Organischen Molekülen − Mesomerie • Kovalente Bindungen zwischen zwei C-Atomen sind unpolar! • Kovalente Bindungen zwischen Kohlenstoff und Heteroatomen sind mehr oder weniger polar! (Elektronegativitätsunterschied!!) • Eine einzige Lewis-Formel (Valenz-Strich-Formel) kann polare Bindungen nicht beschreiben. • Deshalb haben Heisenberg und Ingold das Konzept der Mesomerie entwickelt: man beschreibt die Ladungsverteilung in einem organischen Molekül mit Hilfe von mehreren Mesomeren Grenzstrukturen (die keine reale Existenz haben!!!). Die Überlagerung aller möglichen mesomeren Grenzstrukturen eines Moleküls ergibt die reale Ladungsverteilung des Moleküls. Dabei gewichtet man die einzelnen mesomeren Grenzformeln entsprechend ihrer Stabilität. • Mesomere Grenzformeln kann man durch Verschieben von π-Elektronenpaaren ineinander umwandeln („Elektronenschieben“; die Richtung wird von Elektronenschiebepfeilen angedeutet). 13 –0,5 O +0,1 +0,3 H H O O ” “ Mesomere Grenzformeln H H H H Reale Ladungsverteilung O –0,5 +0,5 Überlagerung 1:1 • Durch Überlagerung zweier mesomerer Grenzformeln von Aceton wird die reale Ladungsverteilung relativ gut beschrieben. • Wichtig: um mesomere Grenzformeln zu erhalten, werden Elektronenpaare immer vom Bindungspartner mit kleiner EN zum Bindungspartner mit großer EN verschoben. • Wichtig: die EN im PSE gelten nur für neutrale Atome!!! Anionen haben kleinere EN, Kationen haben größere EN als Neutralteilchen. 14 • Mesomere Grenzformeln sind in der OC extrem wichtig, um die Ladungsverteilung in den Molekülen und damit deren physikalische und chemische Eigenschaften abzuschätzen • Physikalische Eigenschaften: Siedepunkt, Schmelzpunkt, Löslichkeit, Farbe usw. • Chemische Eigenschaften: Struktur, Reaktivität 1 1 6 2 6 2 5 3 5 3 4 4 Mesomere Grenzformeln • Mesomerie erklärt bei Benzol, warum alle Bindungen gleich lang sind (139 pm). • Mesomerie ist generell wichtig für die Struktur von Molekülen und für Reaktivitäten und Reaktionsmechanismen. Überlagerung 1:1 15 1.6. Ladungsverteilung in Organischen Molekülen Induktive und Mesomere Effekte • Mesomerie beschreibt das Verschieben von π-Elektronenpaaren innerhalb eines Moleküls. Was macht man, wenn man keine πElektronenpaare hat? • Man berücksichtigt Elektronenverschiebungen über σ-Bindungen. Diese finden auf Grund von Elektronegativitätsunterschieden der beteiligten Bindungspartner statt. • Definition: Zieht ein Substituent in einem Molekül σ-Elektronen zu sich und wird dadurch negativ polarisiert, dann übt dieser Substituent einen Negativen Induktiven Effekt (kurz: −I-Effekt; lies: Minus-I-Effekt) auf das Molekül aus. Schiebt ein Substituent in einem Molekül σElektronen von sich und wird dadurch positiv polarisiert, dann übt dieser Substituent einen Positiven Induktiven Effekt (kurz: +I-Effekt; lies: Plus-I-Effekt) auf das Molekül aus. • Die Auswirkung von Induktiven Effekten auf einzelne Atome des Moleküls ist um so größer, je weniger σ-Bindungen zwischen Substituent und betrachtetem Atom liegen. 16 • Beispiel: pKS-Werte von Chlorbuttersäuren COOH COOH Cl COOH Cl pKS 4,8 2,9 4,1 Cl COOH 4,5 • Die Cl-Atome polarisieren die OH-Gruppe der Säuren um so schwächer, je weiter sie entfernt sind. Der −I-Effekt nimmt ab. • Beispiel: pKS-Werte von Chloressigsäuren COOH Cl COOH Cl pKS • 4,7 1,3 Cl COOH Cl Cl 0,8 Je mehr Cl-Atome am gleichen C-Atom sitzen, um so stärker ist der −I-Effekt. 17 • Definition: Zieht ein Substituent in einem Molekül π-Elektronen oder n-Elektronen zu sich und wird dadurch negativ polarisiert, dann übt dieser Substituent einen Negativen Mesomeren Effekt (kurz: −MEffekt; lies: Minus-M-Effekt) auf das Molekül aus. Schiebt ein Substituent in einem Molekül π-Elektronen oder n-Elektronen von sich und wird dadurch positiv polarisiert, dann übt dieser Substituent einen Positiven Mesomeren Effekt (kurz: +M-Effekt; lies: Plus-M-Effekt) auf das Molekül aus. • Die Auswirkung von Mesomeren Effekten auf einzelne Atome des Moleküls ist um so größer, je weniger π-Bindungen zwischen Substituent und betrachtetem Atom liegen. • Mesomere Effekte sind immer stärker als induktive Effekte. Wichtig: Mesomere Effekte hängen nicht von der Elektronegativität ab!!! O H ” H O“ H O“ H O“ δ+ O ” ” δ– H δ– δ– 18 2. Reaktive Teilchen in der Organischen Chemie 2.1. Säuren und Basen • Eine Brönstedt-Säure ist eine Verbindung, die H+ abgeben kann. • Wichtige Brönstedt-Säuren, die in der Organischen Chemie angewendet werden: CH3 COOH CH3 CF3 COOH SO3H CH3 SO3H HOOC COOH CF3 SO3H HO COOH HOOC COOH O HO S OH O H Cl O HO N “ O ” O HO P OH OH 19 • Eine Brönstedt-Base ist eine Verbindung, die H+ aufnehmen kann. • Wichtige Brönstedt-Basen, die in der Organischen Chemie angewendet werden: LiOH NaOMe NH3 NaOH NaH NaOEt NEt3 KOH NaNH2 KOtBu HNiPr2 secBuLi tert.BuLi LDA nBuLi • CaH2 N H N Vorsicht! Die eigentlichen Basen sind OH−, NH2−, H−, nBu− usw. 20 • Eine Lewis-Säure ist ein Elektronenpaar-Acceptor. • Wichtige Lewis-Säuren, die in der Organischen Chemie angewendet werden: LiCl MgCl2 BF3 BCl3 SnCl2 TiCl4 LiBr MgBr2 AlCl3 BBr3 SnCl4 ZrCl4 FeCl3 ZnCl2 Al(OR)3 • Wichtig: die eigentliche Lewis-Säure ist das Metall-Ion! • Eine Lewis-Base ist ein Elektronenpaar-Donor. • Wichtige Lewis-Basen, die in der Organischen Chemie angewendet werden: – H2O HOR R3N Cl OH– OR– R3P Br R2O – I– SR– R2S 21 2.2. Nucleophile und Elektrophile • Ein Nucleophil (Nu−) ist ein „kernsuchendes“ Teilchen. Nucleophile sind entweder Anionen oder Neutralverbindungen mit einem oder mehreren Atomen mit negativer Partialladung. Die meisten BrönstedtBasen und Lewis-Basen sind auch Nucleophile. Eine Ausnahme bilden Basen mit sehr großen Substituenten, wie z.B. LDA. • Generell gilt: Alle Nucleophile sind auch Basen, aber nicht alle Basen sind Nucleophile! C N H N H H H ”N H H O H ” COOR H COOR R1 N H H R1 ”N H R O H ” R δ” LI δ“ δ“ Mg δ” Br 1 N R H H O R O 2 R1 N R2 R3 H S H ” R O H H S R S H O 1 R ” N R2 ” R1 S R S O ” Cl Br O R S H R2 O R ” I ” ” ” O ” R S ” 22 • Ein Elektrophil (E+) ist ein „elektronensuchendes“ Teilchen. Elektrophile sind entweder Kationen oder Neutralverbindungen mit einem oder mehreren Atomen mit positiver Partialladung. Die meisten Brönstedt-Säuren und Lewis-Säuren sind auch Elektrophile. O + H R O H N R´ R H R1 R “ R3 2 • R1 R2 N 1 R O O 1 R 2 OR R Cl R´ 2 R C N R “ N O “ O N O Extrem wichtig: Elektrophile reagieren immer mit Nucleophilen!!! Dadurch lassen sich > 95% aller Reaktionen in der organischen Chemie beschreiben. 23 2.3. Carbanionen, Radikale und Cabokationen • Ein Carbanion ist ein Ion mit einer negativen Ladung an einem CAtom. Carbanionen sind immer basisch und meistens auch nucleophil. • In Carbanionen ist das negativ geladene C sp3-hybridisiert und damit tetraedrisch (wenn man das freie Elektronenpaar mitzählt) oder pyramidal (wenn man nur die drei Substituenten betrachtet). Ausnahme: wenn Mesomerie mit C=C- oder C=X-Doppelbindungen möglich ist). H + 1 R • R 2 R 3 -H R 1 ” 2 R R3 Entfernt man formal ein Elektron aus einem Carbanion (Oxidation!), dann kommt man zu Radikalen. In Radikalen ist das C-Atom mit dem ungepaarten Elektron sp3-hybridisiert und damit tetraedrisch bzw. pyramidal. Ausnahme: wenn Mesomerie mit C=C- oder C=XDoppelbindungen möglich ist). ” R1 2 R3 R - e– • R1 2 R 3 R 24 • Ein Carbokation (= Carbeniumion) ist ein Ion mit einer positiven Ladung an einem C-Atom. Carbeniumionen sind immer elektrophil. • In Carbeniumionen ist das positiv geladene C sp2-hybridisiert und damit trigonal planar. X 1 R • R 2 R3 - X– R1 “ 3 R R2 Carbeniumionen sind um so stabiler, je mehr Alkylsubstituenten sie besitzen. Carbeniumionen sind besonders stabil, wenn die positive Ladung durch Doppelbindungen oder Phenylringe delokalisiert werden kann. “ “ “ “ “ Stabilität von Carbeniumionen nimmt ab • Carbanionen, Radikale und Carbeniumionen sind sehr reaktive Teilchen, die überwiegend nur als reaktive Zwischenstufen bei organischen Reaktionen auftreten. 25 3. Alkane 3.1. Offenkettige Alkane • Offenkettige Alkane sind gesättigte Kohlenwasserstoffe mit der allgemeinen Summenformel CnH2n+2.Die C-Atome sind sp3-hybridisiert und nur über σ-Bindungen miteinander verbunden. H • Das einfachste Alkan ist das Methan CH4. • Das Alkan mit 2 C-Atomen ist das Ethan C2H6. • Vom Ethan gibt es 2 verschiedene Formen, die durch Drehen der beiden CH3-Gruppen um die Einfachbindung ineinander überführbar sind. Beide Forman haben dieselbe Summenformel, aber unterschiedliche räumliche Struktur. • Definition: Moleküle mit der gleichen Summenformel, aber unterschiedlicher Strukturformel, nennt man Isomere. • Definition: Isomere, die durch Drehen eines Teils des Moleküls um eine C-C-Einfachbindung ineinander überführbar sind, nennt man 26 Konformationsisomere. H H H H H H C C H H H • • • Um Konformationsisomere zu zeichnen, gibt es die Newman-Projektion. Man betrachtet dabei das Molekül entlang der C-C-Einfachbindung, um die gedreht wird. Das vordere C-Atom wird als Punkt gezeichnet, das hintere als Kreis. Die Konformation, bei der die H-Atome (oder allgemein irgendwelche Substituenten) „auf Lücke“ stehen, nennt man gestaffelte Konformation (engl.: staggered). Die Konformation, bei der die H-Atome „auf Deckung“ stehen, nennt man ekliptische Konformation (engl.: eclipsed). In der ekliptischen Konformation kommen sich die Wasserstoffe so nah, dass sie sich „gegenseitig spüren“. In der OC nennt man diese Wechselwirkung sterische Hinderung. H H H H H H H H H H H H H H H H H H HH H H HH gestaffelt ekliptisch geringe sterische Hinderung größere sterische Hinderung stabilere Konformation weniger stabile Konformation Über 99% aller Ethanmoleküle liegen bei RT in der gestaffelten Konformation vor. 27 • • Das Alkan mit 3 C-Atomen heißt Propan C3H8. Auch von Propan und allen höheren Alkanen gibt es Konformationsisomere. Die stabilste Form ist die, in der möglichst viele Atome gestaffelt sind. ⇒ zick-zack-Form. Das nächst höhere Alkan ist das Butan C4H10. Vom Butan gibt es neben den Konformationsisomeren noch zwei weitere Isomere, die sich bzgl. der Verknüpfung der C-Atome unterscheiden. • Definition: Isomere, die sich durch die Verknüpfung von Atomen unterscheiden, nennt man Konstitutionsisomere. • Von allen höheren Alkanen gibt es sowohl Konformationsisomere als auch Konstitutionsisomere. • Das nächst höhere Alkan ist das Pentan C5H12. Es gibt folgende Konstitutionsisomere: nPentan, i-Pentan, neo-Pentan. HH H H HH HH n-Butan (normal) i-Butan (iso) n-Pentan i-Pentan neo-Pentan 28 • Das nächst höhere Alkan ist das Hexan C6H14. Es gibt n-Hexan, i-Hexan, 3-Methyl-pentan, 2,3-Diomethylbutan und neo-Hexan (2,2-Dimenthylbutan). • Die nächst höheren Alkane sind Heptan C7H16, Octan C8H18, Nonan C9H20, Decan C10H22 usw. • Entfernt man formal von den Alkanen ein H-Atom, dann kommt man zu den AlkylResten (mit den entAlkan sprechenden stereoMethan CH4 chemischen BezeíchEthan CH3–CH3 nungen n-, iso-, neo-). • Alkane und Alkylgruppen bilden eine homologe Reihe = zwei aufeinanderfolgende Glieder unterscheiden sich um CH2. Alkylrest Methyl CH3– Ethyl CH3–CH2-– n-Propan CH3CH2CH3 n-Propyl CH3CH2CH2– Butan Butyl Pentan Pentyl Hexan Hexyl Heptan Heptyl …. …. 29 3.2. Cycloalkane • Cycloalkane sind gesättigte ringförmige Kohlenwasserstoffe mit der allgemeinen Summenformel CnH2n. .... Cyclopropan • • Cyclobutan Cyclopentan Cyclohexan Cycloheptan Cyclopropan ist eben, der Bindungswinkel beträgt 60° ⇒ hohe Ringspannung (= Baeyer-Spannung). Alle H-Atome stehen ekliptisch ⇒ hohe sterische Hinderung (bei Ringen = Pitzer-Spannung). Cyclobutan weicht der Baeyer-Spannung und der Pitzer-Spannung teilweise aus, indem es eine gewinkelte Konformation einnimmt (Schmetterling-Konformation). ................ H H H H H H H H H H H H H H 30 • Cyclopentan hat die geringste Baeyer-Spannung und weicht der in einem ebenen 5-Ring auftretenden Pitzer-Spannung dadurch aus, dass es eine Briefumschlag-Konformation annimmt. • Cyclohexan ist nicht eben. Es existiert in zwei spiegelbildlichen Sesselkonformationen, die sich leicht ineinander umwandeln können. Dabei wird u.a. die Wannenkonformation durchlaufen. Die Wannenkonformation ist weniger stabil als die Sesselkonformationen. Wichtig: es gibt 2 Arten von H-Atomen in der Sessel-Konformation von Hexan: solche, die axial stehen (ax) und solche, die equatorial stehen (eq). H H H H H H H H H H H H H H H H H H H H H H 120° ax ax eq ax eq f eq eq ax eq ax ax eq eq eq ax ax f eq eq eq ax ax ax ax eq eq eq eq ax ax eq eq eq ax ax 31 • Zwischen den axialen Substituenten treten sogenannte 1,3-diaxiale Wechselwirkungen auf, die diese Positionen destabilisieren. Deshalb bevorzugen substituierte Cyclohexane diejenige Sesselkonformation, bei der der Substituent equatorial steht. ax ax eq ax eq eq eq eq ax eq ax ax CH3 CH3 • Cyclohexanderivate spielen in der Natur eine sehr große Rolle. Dabei kommt ein Cyclohexanring oft nicht einzeln vor, sondern bildet zusammen mit anderen Ringen kondensierte Ringsysteme. C A B D Hβ Hβ Hβ Hα Hα Hα 32 3.2. Radikalische Substitution von Alkanen • Die wichtigste Reaktion der Alkane ist die Radikalische Substitution SR mit Chlor und Brom. • Bei radikalischen Substitutionen wird ein H durch ein reaktionsfähiges Radikal ersetzt. hν Cl Cl H H H H H H H • H H H H H • H H H H H H H H • H H • H •Cl - HCl Cl Cl - Cl• Startreaktion (Kettenstart) 2 Cl• H H • H H H H H H Propagation (Kettenreaktion) Cl H H H H H H •Cl H H H Abbruchreaktion (Radikalkombination) Cl H 33 • Auf diese Weise erhält man aus Alkanen Chloralkane = Alkylchloride und Bromalkane = Alkylbromide, die als Ausgangsmaterialien für viele andere organische Verbindungen wichtig sind. • Mit elementarem Fluor verläuft die Reaktion explosionsartig, elementares Iod reagiert nicht (insgesamt endotherme Reaktion!). • Man erhält generell Produktgemische, die meist destillativ getrennt werden. 34 4. Halogenalkane 4.1. Struktur der Halogenalkane • Halogenalkane erhält man z.B. aus Alkanen durch radikalische Chlorierung oder radikalische Bromierung. CH4 CH3–CH3 Cl2 Cl2 CH3Cl CH2Cl2 + CH3–CH2Cl + + CH3–CHCl2 CHCl3 + + CCl4 ClCH2–CH2Cl + .... • Sie werden bezeichnet durch den Namen des Alkylrestes mit der Endung –chlorid, -bromid usw. oder durch die Vorsilbe Chlor-, Bromusw. und die Endung des entsprechenden Alkans. • CH3Cl = Methylchlorid = Chlormethan; CH3−CHBr2 = 1,1-Dibromethan = Ethylidenbromid; ClCH2−CH2Br = 1-Brom-2-Chlor-ethan • In Halogenalkanen ist die C-Hal-Bindung in Abhängigkeit der Elektronegativität des Halogens (−I-Effekt) mehr oder weniger stark polarisiert. Dadurch bekommt der Kohlenstoff eine positive Partialladung und wird elektrophil (wichtig für Reaktionen der 35 Alkylhalogenide). • Wichtige Halogenalkane sind Methyliodid CH3I, Ethyliodid CH3-CH2I, Dichlormethan CH2Cl2, Chloroform CHCl3, Dichlordifluormethan CCl2F2 (Frigen), 1-Brom-1-Chlor-2,2,2-trifluorethan CHBrCl-CF3 (Halothan) und viele andere. • Komplizierteres Beispiel: Chlorbutan C4H9Cl. Zur Summenformel C4H9Cl gibt es verschiedene mögliche Strukturformeln (= Isomere). Cl 1-Chlorbutan Cl H 2-Chlorbutan H Cl 2-Chlorbutan • Es gibt zwei Isomere von 2-Chlorbutan. Grund: Das C-Atom 2 in 2-Chlorbutan ist ein asymmetrisches C-Atom. • Definition: Ein C-Atom mit 4 verschiedenen Subsstituenten nennt man asymmetrisches C-Atom. • Definition: Isomere mit definierter Konstitution, die sich bzgl. der räumlichen Anordnung der Substituenten an einem asymmetrsichen C-Atom unterscheiden, nennt man Konfigurationsisomere. 36 • Die beiden Konfigurationsisomere von 2-Chlorbutan verhalten sich zueinander wie Bild und Spiegelbild, die man aber nicht zur Deckung bringen kann. • Definition: Moleküle, die sich wie Bild und Spiegelbild verhalten, sich aber nicht zur Deckung bringen lassen, sind chiral. Allgemein: Chiralität ist die Eigenschaft von Gegenständen (auch Molekülen!), nicht mit ihrem Spiegelbild identisch zu sein. „Bild“ Cl H Cl H „Spiegelbild“ Enantiomere • Definition: „Bild“ und „Spiegelbild“ nennt man Enantiomere. Enantiomere sind Konfigurationsisomere, die sich nicht mit ihrem Spiegelbild zur Deckung bringen lassen. • Zur Bestimmung der Konfiguration von asymmertischen C-Atomen haben Cahn, Ingold und Prelog die sogenannte Sequenzregel entwickelt. 37 • Sequenzregel: Man ordnet jedem Substituenten an einem asymmetrischen C-Atom eine Rangordnung zu, die sich aus der Ordnungszahl der Atome der Substituenten ergibt. Nun orientiert man das Molekül so, dass der rangniedrigste Substituent nach hinten zeigt. Man sieht einen „Merzedes-Stern“, bei dem man sich nun vom ranghöchsten zum rangniedrigsten Substituenten bewegen muss. Erfolgt diese Bewegung im Uhrzeigersinn, dann besitzt das asymmetrische C-Atom R-Konfiguration, erfolgt diese Bewegung im Gegenuhrzeigersinn, dann besitzt das asymmetrische C-Atom SKonfiguration. S-2-Chlorbutan • Cl H Cl H S R R-2-Chlorbutan Enantiomere sind nicht identisch und haben unterschiedliche Eigenschaften in chiraler Umgebung! In achiraler Umgebung sind alle Eigenschaften der beiden Enantiomere gleich, wenn sie nicht von der 38 Raumrichtung abhängen. • Chirale Verbindungen sind optisch aktiv, d.h. sie drehen die Ebene des linear polarisierten Lichts um einen bestimmten Winkel α. • Das eine Enantiomer dreht die Ebene des linear polarisierten Lichts nach rechts (+), das andere Enantiomer nach links (−). • Wichtig: (+) und (-) haben nichts mit R und S zu tun!!! • Definition: Eine 1:1-Mischung von zwei Enantiomeren nennt man Racemat. Racemate sind optisch inaktiv. • Wichtig: die meisten Biomoleküle (Aminosäuren, Zucker, DNA usw.) sind chiral und optisch aktiv. Racemate treten in der Natur höchst selten auf!!! 39 • Noch komplizierteres Beispiel: 2-Brom-3-Chlorbutan. Cl H H Cl Enantiomere BrH H Br Diastereomere Cl H H Br H Cl Enantiomere BrH • Alle Konfigurations- und Konformationsisomere fasst man zu den Stereoisomeren zusammen. Stereoisomere kann man auch unterteilen in Enantiomere und Diastereomere. Enantiomere sind Stereoisomere, die sich wie Bild und Spiegelbild verhalten, sich aber nicht zur Deckung bringen lassen. Diastereomere sind alle anderen Stereoisomere. • Enantiomere und Diastereomere treten bei Aminosäuren und Zuckern auf. 40 4.2. Reaktionen der Halogenalkane • Die Halogene ziehen auf Grund von ihrem −IEffekt Ladung von den benachbarten C-Atomen ab. Dadurch erhalten diese C-Atome eine positive Partialladung und werden elektrophil und können mit Nucleophilen reagieren. δδδ+ δ+ δ– δδ+ X X = F, Cl, Br, I 4.2.1. Nucleophile Substitution SN • Wenn man Alkylhalogenide mit Nucleophilen reagieren lässt, dann wird das Halogenid durch das Nucleophil ersetzt. Solche „Ersetzungsreaktionen“ bezeichnet man als Nucleophile Substitution SN. • Beispiel: Williamson-Ether-Synthese O H + Na - H2 Abgangsgruppe ” O “ Na Nucleophil = Edukt 1 I ” -I Elektrophil = Edukt 2 O Produkt 41 • Es gibt 2 Arten von SN-Reaktionen: SN1 und SN2. • SN1-Reaktionen (Nucleophile Substitution 1. Ordnung) verlaufen nach einem Zeitgesetz 1. Ordnung. vR = k [ R − X ] • Die Reaktionsgeschwindigkeit hängt also nur von der Konzentration des Elektrophils (hier: Alkylhalogenid) ab. D.h., der geschwindigkeitsbestimmende Schritt ist der Bruch der C-X-Bindung. - X” Allgemeiner SN1Mechanismus • R X langsam Bei SN1-Reaktionen tritt als Zwischenstufe immer ein Carbeniumion auf! Reaktive Zwischenstufen können prinzipiell isoliert werden. Zwischenstufen sind lokale Minima im Reaktionsprofil! R + Nu” “ R schnell Nu Zwischenstufe R R X + Nu ” “ R ” Nu + X 42 • Eine SN1-Reaktion läuft nur ab, wenn ein stabiles Carbeniumion als Zwischenverbindung auftritt und eine gute Abgangsgruppe vorliegt. “ “ “ “ “ Stabilität von Carbeniumionen nimmt ab • SN1-Reaktion verlaufen um so leichter, je besser die Abgangsgruppe das Molekül verlässt. Die „Güte“ einer Abgangsgruppe nennt man Nucleofugie. RN2“ ROSO2CF3 ROSO2CH3 ROSO2 CH3 RI RBr ROH2“ Nucleofugie nimmt ab RCl “ RORH “ RNR3 RF ROOCR´ ROH ROR´ RNH2 Nucleofugie nimmt ab • Wichtig: man kann schlechte Abgangsgruppen durch Protonieren in 43 gute Abgangsgruppen umwandeln. • SN2-Reaktionen (Nucleophile Substitution 2. Ordnung) verlaufen nach einem Zeitgesetz 2. Ordnung. vR = k[ R − X ][ Nu ] • Die Reaktionsgeschwindigkeit hängt also sowohl von der Konzentration des Elektrophils (hier: Alkylhalogenid) als auch der Konzentration des Nucleophils ab. SN2-Reaktionen bestehen nur aus einem Schritt mit Rückseitenangriff! HH Allgemeiner SN2Mechanismus R X H ” + Nu Nu H HH X R - X” Nu R Übergangszustand • Bei SN2-Reaktionen tritt nur ein Übergangszustand auf, der nicht isoliert werden kann. Übergangszustände sind Maxima im Reaktionsprofil und können niemals isoliert werden! 44 • Eine SN2-Reaktion läuft nur ab, wenn kein stabiles Carbeniumion als Zwischenverbindung auftreten kann und wenn ein gutes Nucleophil an der Reaktion teilnimmt. • Die „Güte“ (Stärke) eines Nucleophils nennt man Nucleophilie. Für häufig verwendete Nucleophile findet man folgende Reihenfolge: RSe” RS” CN” I” PhNH2 OH” N3” Pyridin Br” PhO” AcO” Cl” F” NO3” H2O Nucleophilie nimmt ab • Aus experimentell bestimmten Reaktionsgeschwindigkeiten bei SN2Reaktionen kann man folgende allgemeine Regeln ableiten: 1) Ein Nucleophil mit einer negativen Ladung ist besser als seine konjugierte Säure. 2) Im Periodensystem nimmt die Nucleophilie innerhalb einer Periode von links nach rechts ab (genauso wie Basizität). 3) Im Periodensystem nimmt die Nucleophile innerhalb einer Gruppe von oben nach unten zu (umgekehrt zur Basizität). 4) Je größer die Substituenten in einem Nucleophil sind, um so geringer ist die Nucleophile. 45 • Die häufigsten Nucleophile in der Biochemie sind H2O, R-NH2 und RSH. Die häufigsten Abgangsgruppen in der Biochemie sind H2O, R-NH2, R-SHund vor allem PO43−. • Komplizierteres Beispiel: Cl H • = H Cl O-H HO Cl H – Cl ” HO H Setzt man z.B. enantiomerenreines S-2-Chlorbutan bei einer SN2Reaktion ein, dann erhält man R-2-Butanol. Bei der SN2-Reaktion kehrt sich durch den planaren Übergangszustand die Konfiguration am asymmetrischen C-Atom um! Dies bezeichnet man nach ihrem Entdecker als Walden´sche Umkehr. 46 4.2.2. Eliminierung E • Wenn man Alkylhalogenide mit Basen reagieren lässt, dann wird Halogenwasserstoff aus dem Molekül abgespalten. Solche Abspaltungsreaktionen nennt man Eliminierungen E. • Beispiel: ” HH O K“ Br HH - KBr - HO • Es gibt 3 Arten von Eliminierungen: E1, E2 und E1cB. • Bei E1-Eliminierungen dissoziiert zuerst die Abgangsgruppe (hier: Halogenid-Ion) ab und es bildet sich ein Carbeniumion. Dies ist der geschwindigkeitsbestimmende Schritt, so dass die E1-Eliminierung auch einem Zeitgesetz 1. Ordnung gehorcht. vR = k [ R − X ] 47 • Aus dem Carbeniumion wird dann von einer Base ein H+ entfernt, so dass sich eine Doppelbindung ausbildet. H ” B Allgemeiner E1Mechanismus - BH XH ” H HH HH -X H “ H HH H H HH HH Zwischenstufe H B” - BH H H HH • Man erhält Produktgemische, bei denen i.d.R. das Produkt mit der stabileren Doppelbindung überwiegt (vgl. Kap. 5) • Bei E2-Eliminierungen verläuft die Reaktion nach einem Zeitgesetz 2. Ordnung über einen Übergangszustand, den man nicht isolieren kann! Die Reaktionsgeschwindigkeit hängt von der Konzentration des Alkylhalogenids und von der Konzentration der Base ab. vR = k[ R − X ][ B ] 48 • Die C-H- und die C-X-Bindung werden gleichzeitig gebrochen. Man spricht von einem Synchronmechanismus oder von einem konzertierten Prozess. Wichtig: H und X müssen antiperiplanar angeordnet sein. Allgemeiner E2Mechanismus B H XH H HH HH Rotation um C-CBindung H X H B” H H H H X - BH H Übergangszustand • Auch hier erhält man Produktgemische, bei denen i.d.R. das Produkt mit der stabileren Doppelbindung überwiegt (vgl. Kap. 5) • Bei E2-Eliminierungen verläuft die Reaktion nach einem Zeitgesetz 2. Ordnung über einen Übergangszustand, den man nicht isolieren kann! Die Reaktionsgeschwindigkeit hängt von der Konzentration des Alkylhalogenids und von der Konzentration der Base ab. 49 • Für E1cB-Eliminierungen wird im Molekül noch eine stark elektronenziehende Gruppe (−M-Effekt) benötigt. Dann kann ein Proton durch eine Base abstrahiert werden und das entstehende Carbanion wird mesomeriestabilisiert. Im nächsten Schritt wird ddann die Abgangsgruppe abgespalten. X Allgemeiner E1cBMechanismus • O ” B X O R HH ” R H O - X” R H Biochemische Eliminierungen verlaufen häufig nach einem E1cBMechanismus. 50 5. Alkene 5.1. Struktur der offenkettigen Alkene • Offenkettige Alkene sind ungesättigte Kohlenwasserstoffe mit einer Doppelbindung (vgl. Kap. 1.3., Folie 8). Allgemeine Summenformel: CnH2n. Die C-Atome der Doppelbindung sind sp2-hybridisiert. • Der Name der Alkene leitet sich von der Vorsilbe für das entsprechende Kohlenstoffgerüst ab ( Eth, Prop, But, Pent,...) und der Endung –en für Alken. H H • Das einfachste Alken ist das Ethen C2H4. • Das nächsthöhere Alken ist das Propen C3H6. • Vom Buten C4H8 gibt es mehrere Isomere. Die Doppelbindung kann zwischen den CAtomen 1 und 2 liegen oder zwischen den C-Atomen 2 und 3. 1-Buten und 2-Buten sind Konstitutionsisomere (speziell: Regioisomere). Ein zusätzliches Konstitutionsisomer ist das Isobuten (2-Methyl-propen). H H H2C CH CH2 CH3 1-Buten H3C CH CH CH3 2-Buten Isobuten 51 • Vom 2-Buten gibt es zwei Isomere, die sich bezüglich der Stellung der Methylcis-2-Buten Gruppen an der Doppelbindung untercis-transscheiden. Das Isomer, bei dem die beiden Isomere Methylgruppen auf derselben Seite liegen, heißt cis-2-Buten, das Isomer mit den trans-2-Buten Methylgruppen auf unterschiedlichen Seiten heißt trans-2-Buten. cis- und trans-2-Buten kann man nicht durch Drehen um die Doppelbindung ineinander überführen! Um cis-2Buten in trans-2-Buten umzuwandeln, muss man die π-Bindung brechen. Dies kann man durch Bestrahlen mit UV-Licht erreichen. hν • • • • • Definition: Isomere mit definierter Konstitution, die sich in der räumlichen Anordnung von Substituenten um ein starres Molekülgerüst (Doppelbindung, Ring, C-Atom) unterscheiden, nennt man Konfigurationsisomere. • cis- und trans-Isomere sind Konfigurationsisomere. Oft spricht man 52 auch von geometrischen Isomeren. • Wichtig: Konfigurationsismere lassen sich nur durch Brechen und Neuknüpfen von Bindungen ineinander umwandeln. • Von allen höheren offenkettigen Alkenen gibt es ebenfalls cis-transIsomere (neben den Konstitutionsismeren). • cis- und trans-Isomere haben unterschiedliche Eigenschaften (Siedepunkte, Schmelzpunkte, Löslichkeit, Reaktivität, Stabilität usw.). trans-Isomere sind stabiler als cis-Isomere! H Sterische Hinderung • H H H H H H H H H H H H H H H Keine sterische Hinderung Auch die unterschiedlichen Konstitutionsismere von Alkenen sind unterschiedlich stabil. Endständige Doppelbindungen sind weniger stabil als innere Doppelbindungen. Grund: Hyperkonjugation. Stabilität nimmt ab 53 • Beispiel für ein kompliziertes Alken: H3C Cl HOOC Cl HOOC H H3C H • Sind das cis-trans-Isomere? Es sind sicher Konfigurationsisomere (speziell: geometrische Isomere), aber die Bezeichnungen cis und trans kann man nur für 1,2-disubstituierte Doppelbindungen mit zwei gleichen Substituenten verwenden. • Für höher substituierte Alkene: Z/E-Nomenklatur. • Nach Cahn, Ingold und Prelog wird den beiden Substituenten an jedem C-Atom der Doppelbindung eine Rangfolge zugeordnet. Der Rang eines Substituenten ergibt sich aus der Ordnungszahl der Atome. Liegen die beiden ranghöchsten Substituenten auf derselben Seite der Doppelbindung, dann besitzt die Doppelbindung ZKonfiguration (Z = zusammen), liegen die beiden ranghöchsten Substituenten auf entgegengesetzten Seiten der Doppelbindung, dann besitzt die Doppelbindung E-Konfiguration (E = entgegen). E-3-Chlor-2-methylpropensäure 2 H3C 1 Cl HOOC 1 H 2 Rangordnung 1 HOOC 1 Cl H3C 2 H 2 Z-3-Chlor-2-methylpropensäure 54 5.2. Struktur der cyclischen Alkene • Cyclische Alkene haben die allgemeine Summenformel CnH2n-2. Analog zu den Cycloalkanen gibt es unterschiedliche Ringgrößen. .... Cyclopropen • Cyclobuten Cyclopenten Cyclohexen Cyclohepten Bis zur Ringgröße 7 können Cycloalkene nur cisDoppelbindungen enthalten. Für Cycloocten gibt es beide Möglichkeiten. • Cyclohexenringe treten in der Natur häufiger auf, z.B. Cholesterin oder Provitamin-D2 (Ergosterol). ................ H H H H H H HO H H H H HO H 55 5.3. Herstellung der Alkene • Alkene kann man generell durch Eliminierung aus substituierten Alkanen herstellen. Dabei bildet sich immer das stabilste Alken (am höchsten substituierte Doppelbindung, wenn möglich in der transKonfiguration) bevorzugt, wenn man gute Abgangsgruppen verwendet. (Regel von Saytzeff). Bei schlechten Abgangsgruppen bildet sich bildet sich bevorzugt das Produkt mit der weniger stabilen Doppelbindung (Regel von Hoffmann). KOtBu - HCl Saytzeff-Produkt Cl KOtBu Hoffmann-Produkt NMe3 “ • - NMe3 Als Konkurrenzreaktionen zu Eliminierungen können immer auch Nucleophile Substitutionen ablaufen. Durch Erhitzen und voluminöse 56 Basen begünstigt man die Eliminierung. • Man kann auch sauer eliminieren. Dies wird immer bei der Eliminierung von Alkoholen durchgeführt, weil man OH− nicht basisch eliminieren kann (ganz schlechte Abgangsgruppe). OH H3PO4 • Man kann auch sauer eliminieren. Dies wird immer bei der Eliminierung von Alkoholen durchgeführt, weil man OH− nicht basisch eliminieren kann (ganz schlechte Abgangsgruppe). Durch Protonieren wird die schlechte Abgangsgruppe OH− in die gute Abgangsgruppe H2O überführt, die dann nach einem E1-Mechanismus eliminiert. Derartige Eliminierungen kommen in der Biochemie häufig vor. • Weitere Möglichkeiten zur Erzeugung von Doppelbindungen werden in späteren Kapiteln besprochen. 57 5.4. Reaktionen der Alkene • • Die wichtigsten Reaktionen der Alkene sind Additionsreaktionen. R1 R3 R2 R4 X–Y X R3 4 R 1 R 2 R Y Die Substituenten R1 bis R4 beeinflussen die Ladungsdichte der Doppelbindung. Elektronenreiche Doppelbindungen gehen elektrophile Additionen ein, elektronenarme Doppelbindungen reagieren in nucleophilen Additionen. 5.4.1. Elektrophile Additionen AdE • Man kann sowohl symmetrische Teilchen X-X als auch unsymmetrische Teilchen X-Y an Doppelbindungen addieren. Die wichtigsten symmetrischen Teilchen sind Halogene (außer Fluor) und Wasserstoff. Die wichtigsten unsymmetrischen Teilchen sind H2O, Halogenwasserstoffsäuren, Alkohole R-OH, Thiole R-SH, Aldehyde und Ketone. 58 • Symmetrische Teilchen haben zunächst kein elektrophiles Zentrum. Bei der Annäherung an eine Doppelbindung erfolgt jedoch eine Polarisierung auf Grund der Abstoßung der π-Elektronen und der Elektronen des angreifenden Teilchens. Es bildet sich ein so genannter π-Komplex. Die gezeigte Brom-Addition wurde lange Zeit als Nachweis für Alkene verwendet (Brom wird schnell entfärbt). Br δ” R1 R2 δ” R3 R4 Br–Br R1 R2 Br δ“ δ” “ Br R3 R4 Br 3 1 R R4 R 2 R π-Komplex Br ” R1 R2 R3 4 R Br Bromoniumion − • Dann wird aus dem polarisierten Teilchen ein Br abgespalten und das Br+ im Bromoniumion gebunden. Das abgespaltene Bromidion greift nun von hinten (SN2) an und öffnet das Bromoniumion zum trans-Produkt (trans-Addition). • Beispiel: BrH Br2 H Br Br Br + 59 • Die Addition von H2 benötigt Katalysatoren, da die π-Bindung von Alkenen die σ-Bindung im H2-Molekül nicht ausreichend polarisieren kann (warum?), um einen π-Komplex zu bilden. Als Katalysatoren verwendet man vorwiegend Ni, Pd, oder Pt. • An der Katalysatoroberfläche dissoziiert das H2-Molekül in 2 H-Atome, die dann auf das Alken übertragen werden können. H H H H • Pd • Pd • Pd • Pd • Pd • Pd H Pd H Pd • Pd • Pd H Pd H Pd Katalysecyclus H H H Pd • H Pd Pd Pd H Pd H Pd H Pd H Pd • Pd • Pd Wichtig: die katalytische Hydrierung ist eine cis-Addition! H Pd H Pd 60 • Die Addition unsymmetrischer Teilchen HX erfolgt an unsymmetrisch substituierte Doppelbindungen immer so, dass dasjenige C-Atom, das mehr Hs trägt, den Wasserstoff aus HX bindet, das andere bekommt die Gruppe X. Wer hat, dem wird gegeben! (Regel von Markownikow). H3 C H3 C • CH2 H3C “ H3C + H–X X ” H3C CH3 H3C H3C X Moderne Erklärung dafür: Die Reaktion verläuft so, dass sich alös Zwischenstufe das stabilste Carbeniumion bildet. Das gilt auch für die Addition von H2O, die allerdings durch katalytische Mengen H2SO4 katalysiert werden muss (warum?). H3C H3C H3C OH H2O kat. H2SO4 H3C H3C CH2 HBr H3C H3C H3C Br 61 5.4.2. Nucleophile Additionen AdN • Nucleophile Additionen funktionieren nur gut bei elektronenarmen Doppelbindungen, also bei Alkenen mit elektronenziehenden Substituenten. O R “ R´ R O – OH • ” R O ” R´ O ” O O H H O R O O ” R´ Scheffer-Weitz-Reaktion R´ In der Biochemie finden nucleophile Additionen an elektronenarme Doppelbindungen bevorzugt mit Wasser oder Aminen oder Thiolen als Nucleophilen statt. Additionen von C-Nucleophilen werden in späteren Kapitel besprochen. 62 5.4.3. Radikalische Additionen AdR • Radikalische Additionen verlaufen ebenfalls sehr leicht mit elektronenreichen Doppelbindungen. Halogenwasserstoffsäuren lassen sich auch in Gegenwart von Luftsauerstoff radikalisch an Doppelbindungen addieren. Statt Luftsauerstoff kann man auch einen Radikalkettenstarter verwenden wie z.B. Dibenzoylperoxid. • O2• Br• H Br – HOO• H3C H3C • •Br CH2 H3C H3C • + H Br CH2 Br – Br• H3C H CH2 Br H3C Wichtig: Die radikalische Addition von HX verläuft nach AntiMarkownikow. Grund: Reaktion verläuft über das stabilste Radikal als Zwischenstufe. 63 6. Alkine 6.1. Struktur der offenkettigen Alkine • Offenkettige Alkine sind ungesättigte Kohlenwasserstoffe mit einer Dreifachbindung (vgl. Kap. 1.3., Folie 8). Allgemeine Summenformel: CnH2n-2. Die C-Atome der Dreifachbindung sind sp-hybridisiert. • Der Name der Alkine leitet sich von der Vorsilbe für das entsprechende Kohlenstoffgerüst ab (Eth, Prop, But, Pent,...) und der Endung –in für Alkin. • • • Das einfachste Alkin ist das Ethin C2H2. Das nächsthöhere Alkin ist das Propin C3H4. Vom Propin gibt es noch ein Konstitutionsisomer mit zwei Doppelbindungen: Allen Vom Butin C4H6 gibt es nur zwei Isomere, mit einer Dreifachbindung: 1-Butin und 2-Butin. H3C H2C H H3C H H H3C H H • H H H CH3 64 • Vom Butin gibt es noch zwei weitere Konstitutionsisomere mit je zwei Doppelbindungen: Methylallen und 1,3-Butadien. H3C HC • CH2 • Die Doppelbindungen in Allenen nennt man kumuliert, die in 1,3Dienen nennt man konjugiert. • Zwei konjugierte Doppelbindungen sind stabiler als zwei kumulierte Doppelbindungen, weil erstere mesomeriestabilisiert sind (alle 4 CAtome sind sp2-hybridisiert und können miteinander überlappen). 6.2. Struktur der cyclischen Alkine • Cyclische Alkine gibt es erst ab einer Ringgröße von 9 C-Atomen. Das gilt ebenfalls für cyclische Allene. Cyclische Diene gibt es ab 4 Ringgliedern. 65 6.3. Herstellung der Alkine • Ethin kann man aus Ethen herstellen, indem man zuerst Brom addiert und dann zwei mal HBr eliminiert. Ethin ist in reiner Form explosiv. Br2 • KOtBu Br Br – 2 HBr Man kann Ethin auch aus Calciumcarbid CaC2 durch Hydrolyse mit Wasser erzeugen (früher: Carbidlampen). CaC2 • 2 H2O Ca(OH)2 + C2H2 Alle höheren Alkine werden aus Ethin hergestellt. Dabei nutzt man aus, dass H-Atome von endständigen Dreifachbindungen leicht mit Basen als H+ abgespalten werden können. Man erhält dann AcetylidAnionen oder sogar Acetylid-dianionen (Carbidionen). H H NaNH2 NH3 fl. H ” Na “ NaNH2 NH3 fl. Na “ ” ” Na “ 66 • Die Acetylid-Ionen werden z. B. mit Alkylhalogeniden alkyliert. X H ” Na “ R R´ R H R´ NaNH2 R NH3 fl. R´ H Y R ” Na R´ “ R1 R2 R R 1 R´ R 2 67 6.4. Reaktionen der Alkine • Acetylid-Ionen sind nucleophil. Man kann sie deshalb mit Elektrophilen umsetzen (Reppe-Reaktionen). Vgl. Kap. 9. • Alkine gehen elektrophile Additionen ein. Z. B. kann man HCl addieren und gelangt dann zu Vinylchlorid. HCl H • H H Cl H Vinylchlorid = Ausgangsmaterial für PVC Polyvinylchlorid Nucleophile Additionen an Dreifachbindungen funktionieren sowohl bei elektronenarmen Dreifachbindungen als auch bei alkylsubstituierten Dreifachbindungen. R • H R´´ OH H O R´´ cat. KOH R R´ R´ Als Nucleophile können Alkohole, Thiole, Carbonsäuren u.a. verwendet werden. 68 • Die Addition von Wasser erfordert Hg2+-Ionen als Katalysator. Dabei entsteht zunächst ein Enol, das sofort zum Keton isomerisiert. 2“ Hg R 2“ Hg R´ R R – Hg • 2“ H R Hg“ R´ H “ O H R – H+ R´ O H “ Hg R´ O H H R´ R R´ O Lindlar-Hydrierung: Man kann Alkine mit desaktiviertem Pd und H2 zu cis-Alkenen hydrieren. R • H O Pd / Chinolin R R' BaSO4 / H2 H H R´ Wenn man nicht-desaktiviertes Pd verwendet, dann erhält man Alkane (wird nicht als Lindlar-Hydrierung bezeichnet). 69 7. Aromaten • In den Anfängen der organischen Chemie wurden zahlreiche Verbindungen aus Pflanzen isoliert, die alle einen aromatischen Geruch gemeinsam hatten. Man bezeichnete diese Verbindungen als Aromaten und stellt erst sehr viel später fest, dass sie gemeinsame strukturelle Merkmale besitzen. 7.1. Struktur der wichtigsten Aromaten • Die erste genau charakterisierte aromatische Verbindung war Benzol. Benzol besitzt die Summenformel C6H6 und muss demnach stark ungesättigt sein. Chemisch verhält es sich aber nicht wie erwartet. Z.B. wird Brom nicht entfärbt. • Von 1862 bis 1872 gab es mehrere Strukturvorschläge für Benzol. Kekulé 1862 Dewar 1867 Ladenburg 1869 Kekulé 1872 70 • Heute wissen wir, dass kein Gleichgewicht zwischen den beiden Strukturen vorliegt. Man muss beide Formeln als mesomere Grenzformeln ansehen. Energie 71 • Im Benzol liegen nicht abwechselnd Doppelbindungen und Einfachbindungen vor, sondern alle Bindungen sind gleich lang:139 pm. Das entspricht einer 1,5-fach-Bindung. Alle 6 C-Atome sind sp2hybridisiert. • Benzol ist um ca. 150 kJ/mol stabiler als ein fiktives Cyclohexatrien mit alternierenden Einfach- und Doppelbindungen. • Inzwischen kennt man sehr viele Aromatische Verbindungen, die sich alle vom Benzol ableiten und (4n+2) π-Elektronen besitzen. • Hückel-Regel: Aromatische Verbindungen besitzen (4n+2) πElektronen. Im einfachsten Fall liegt ein so genanntes aromatisches π-Elektronensextett vor. • Diese π-Elektronen sind cyclisch konjugiert und verleihen dem Molekül eine besondere Stabilität. • Beispiele für substituierte monocyclische Aromaten: CH3 CH3 CH3 CH3 CH3 CH3 CH3 Toluol o-Xylol m-Xylol CH3 p-Xylol H3C CH3 Mesitylen 72 OH OH OH OH OH OH OH Phenol Brenzkatechin NO2 NH2 Resorzin OH Hydrochinon COOH COOH HO OH Phloroglucin CN HO Nitrobenzol OH Annilin CHO Benzoesäure CHO Salicylsäure Benzonitril CHO OCH3 OCH3 Benzylalkohol Benzaldehyd Anisaldehyd OH Vanillin Styrol 73 • Bei zweifach substituierten Benzolen bezeichnet man die verschiedenen Substitutionsmuster als ortho- (1,2-disubstituiert), meta- (1,3-disubstituiert) und para- (1,4-disubstituiert). • In der Biochemie findet man aromatische Verbindungen oft als Pflanzeninhaltsstoffe, Farbstoffe, Vitamine und auch als Hormone. Es gibt auch Aminosäuren, die aromatische Reste enthalten. • Polycyclische Aromatische Verbindungen werden gelegentlich in Pilzen gefunden und sind für Menschen oft wichtige Antibiotika oder Cytosatika. Solche aromatischen Naturstoffe leiten sich von industriell wichtigen Grundchemikalien ab. Naphthalin • Azulen Anthracen Phenanthren Aromatische Grundchemikalien werden aus Erdöl, Steinkohlenteer und Braunkohle gewonnen. Daraus werden dann viele substituierte Aromaten hergestellt. 74 7.2. Reaktionen von Aromaten • Die wichtigsten Reaktionen von Aromaten sind a) die Elektrophile Aromatische Substitution SEAr und b) die Nucleophile Aromatische Substitution SNAr. • Wichtig: Aromaten reagieren bevorzugt in Substitutionsreaktionen, weil dabei das aromatische π-Elektronensextett erhalten bleibt. 7.2.1. Elektrophile Aromatische Substitution • Auf Grund des aromatischen π-Systems können Aromaten von Elektrophilen angegriffen werden. Dabei wird immer ein H+ durch ein Elektrophil ersetzt. +E + E – H+ 75 • Diese Reaktion läuft in mehreren Teilschritten ab. 1) Das Elektrophil setzt sich locker auf die π-Elektronenwolke und bildet einen so genannten π-Komplex. 2) Der π-Komplex reagiert weiter zum so genannten σ-Komplex (Wheland-Komplex), indem das Elektrophil an ein konkretes C-Atom bindet. Dabei wird vorübergehend der stabile aromatische Zustand aufgehoben. Der σ-Komplex ist aber mesomeriestabilisiert! 3) H+ wird abgespalten, wobei der stabile aromatische Zustand wieder hergestellt wird. Das H+ bleibt kurzfristig noch in einem zweiten π-Komplex gebunden, bevor es das Molekül vollständig verlässt. HE + E+ E “ “ σ-Komplex π-Komplex I E E H π-Komplex II “ –H + 76 • Wenn man bereits von substituierten Aromaten ausgeht, beeinflusst der bereits vorhandene Substituent die Position der Zweitsubstitution. a) elektronenschiebende Substituenten erleichtern die Reaktion des Elektrophils und dirigieren es bevorzugt in ortho- und para-Position. EDG +E EDG + EDG E + – H+ E Reaktion ist schneller als bei Benzol; i.d.R. bildet sich mehr pals o-Produkt. a) elektronenziehende Substituenten erschweren die Reaktion des Elektrophils und dirigieren es bevorzugt in meta-Position. EWG + +E + –H EWG E Reaktion ist langsamer als bei Benzol 77 • Beispiele für Elektrophile Aromatische Substitutionen. 1) Halogenierung. Halogenierungen erfordern i.d.R. eine LewisSäure als Katalysator (meist FeCl3 oder AlCl3). Cl H Cl Cl Cl AlCl3 δ” δ“ “ AlCl4” Cl3Al Cl Cl – AlCl3 – HCl 2) Friedel-Crafts-Alkylierung. Bei der Friedel-Crafts-Alkylierung setzt man Alkylhalogenide zusammen mit katalytischen Mengen einer Lewis-Säure mit Aromaten um. R HR R Cl AlCl3 δ” δ“ Cl3Al Cl R “ AlCl4” – AlCl3 – HCl Tertiäre Alkylhalogenide reagieren über die freien Carbeniumionen. 78 3) Friedel-Crafts-Acylierung. Bei der Friedel-Crafts-Acylierung setzt man Carbonsäurehalogenide oder Anhydride zusammen mit stöchiometrischen Mengen einer Lewis-Säure mit Aromaten um. Dabei entstehen Aromatische Ketone. ” AlCl3 O Cl H R O Cl AlCl3 R O Cl ” “ Cl3Al O R ” O Cl3Al Cl R “ – H“ R H2O 3“ – Al – Cl” 4) Vilsmeier-Formylierung. Bei der Vilsmeier-Formylierung werden Aromaten mit Dimethylformamid und Phosphoroxychlorid POCl3 in aromatische Aldehyde überführt. 79 Cl H “ Me N Me Cl H H NMe2 “ H O H2O 5) Nitrierung. Aromaten werden durch Nitriersäure (H2SO4 konz. + HNO3 konz.) nitriert. ” O“ ” HN O O N O “ O“ O N “ - H“ 5) Azokupplung. Aromaten werden durch Diazoniumsalze unterhalb von 0 °C zu Azoverbindungen gekuppelt. N N “ HN N “ -H “ N N 80 7.2.2. Nucleophile Aromatische Substitution • Elektronenarme aromatische π-Systeme mit geeigneter Abgangsgruppe X können von Nucleophilen angegriffen werden. Dabei wird die Abgangsgruppe immer durch das Nucleophil ersetzt. X Nu – + Nu EWG EWG – X– • Für Nucleophile Aromatische Substitutionen gibt es prinzipiell zwei verschiedene Mechanismen: a) Additions-EliminierungsMechanismus und b) Eliminierungs-Additions-Mechanismus. • Additions-Eliminierungs-Mechanismus Das Nucleophil greift den elektronenarmen Aromaten unter Ausbildung eines σ-Komplexes (Meisenheimer-Komplex) an der Position an, an der die Abgangsgruppe sitzt. Dabei wird der aromaatische Zustand vorübergehend aufgehoben. Der σ-Komplex ist jedoch auch mesomeriestabilisiert (vor allem über vorhandene −M-Substituenten wie NO2. Im nächsten Schritt wird die Abgangsgruppe abgespalten. 81 H H R Cl N “ ” NO2 Cl NO2 H2N R H Cl N NO2 • NHR NO2 ” -H R “ NO2 - Cl” NO2 NO2 NO2 Eliminierungs-Additions-Mechanismus (Arin-Mechanismus) Aromaten, die nur schwach elektronenarm sind, reagieren oft nach dem Eliminierungs-Additions-Mechanismus. Das angreifende Nucleophil ist i.d.R. auch eine sehr starke Base. Als Zwischenverbindung tritt ein so genanntes Arin auf. Cl H H ” O H O H ” O H OH H KOH/NaOHSchmelze – H2O – Cl ” Arin 82 8. Alkohole, Phenole und Ether • Alkohole besitzen eine oder mehrere OH-Gruppen (Alkohol-Gruppe, Hydroxy-Gruppe), die an einen Alkylrest gebunden sind. Ist eine OHGruppe an einen Arylrest gebunden, dann spricht man von Phenolen. • Wichtig: Nie 2 OH-Gruppen am gleichen C-Atom! 8.1. Struktur der wichtigsten Alkohole • Der einfachste Alkohol ist Methanol CH3OH. Methanol wird industriell aus CO und H2 und einem Cu/Zn/Al-Katalysator bei 250 °C und 50 bar Druck hergestellt. • Der nächst höhere Alkohol ist Ethanol C2H5OH. Ethanol wird aus Ethen durch Addition von Wasser hergestellt. H H + H2SO4 konz. H3C CH2 H • H O O S OH O + H2O H3C CH2 OH – H2SO4 verd. Die Firma Karlsberg stellt Ethanol durch alkoholische Gärung aus Traubenzucker her. 83 • Der nächst höhere Alkohol ist Propanol C3H7OH. Von Propanol gibt es zwei Konstitutionsisomere. Primäres C-Atom ⇒ primärer Alkohol • OH OH n-Propanol i-Propanol Vom Butanol gibt es 4 Konstitutionsisomere: n-Butanol, sec.Butanol, i-Butanol und tert.Butanol (ein tertiärer Alkohol). Vom sec.Butanol existieren 2 Stereoisomere, nämlich die beiden Enantiomere. OH n-Butanol OH OH i-Butanol sec.Butanol HO H • Secundäres C-Atom ⇒ secundärer Alkohol OH tert.Butanol H OH Von allen Alkoholen mit mehr als 4 C-Atomen gibt es ebenfalls Konstitutionsisomere und Stereoisomere (sowohl Enantiomere als auch Diastereomere). 84 • n-Hexanol C6H13OH, n-Octanol C8H17OH und n-Dodecanol C12H25OH (Laurylalkohol) findet man in Pflanzensäften. In natürlichen Wachsen kommen z.B. n-Hexadecanol (Cetylalkohol) C16H33OH und nTriacontanol C30H61OH und n-Dotriacontanol C32H65OH vor. • Wichtige ungesättigte Alkohole sind Allylalkohol C3H5OH und Crotylalkohol C4H7OH. OH OH Allylalkohol • Der Sexuallockstoff des Seidenspinners Bombyx mori ist ein zweifach ungesättigter Alkohol (Bombykol). OH • Crotylalkohol Bombykol Die Cyclischen Alkohole spielen für synthetische Zwecke eine wichtige Rolle. In der Natur treten vor allem Cyclohexanolderivate wie z.B. das Cholesterin auf. Viele Steroidhormone sind Cyclohexanolderivate. H Cholesterin H HO H H 85 • • In Fetten findet man (in Form von Fettsäureestern) das Glycerin (1,2,3-Propantriol). HO HO In vielen Stoffwechselwegen spielen Substanzen mit OH-Gruppen (die noch weitere funktionelle Gruppen besitzen) eine wichtige Rolle. Z. B. kann Traubenzucker auch als 5-fach-Alkohol (Pentaol) aufgefasst werden. HO HO HO OH O OH OH 8.2. Synthese von Alkoholen • Hydrolyse von Alkylhalogeniden KOH / H2O R X – KX • OH R Addition von Wasser an Alkene OH H2O / H+ cat. R R 86 • Reduktion von Aldehyden, Ketonen und Carbonsäurederivaten LiAlH4 O R R H R R R´ LiAlH4 O R R OR´ • LiAlH4 O O H H H O H H R´ O H H H Weitere Methoden werden in den Kap. 9 und 10 besprochen. 8.3. Reaktionen von Alkoholen 8.3.1. Nucleophile Substitution • Die OH-Gruppe lässt sich nach überführen in eine bessere Abgangsgruppe durch Nucleophile ersetzen. R O S Cl O OH N R O S O O ” C N – TsO” R CN 87 • Alkohole bzw. in deprotonierter Form als Alkoholate können auch selbst als Nucleophile fungieren. Cl H N H3C O H –H “ H3C ” O Me – Cl HO Ph ” Me Ph 8.3.2. Eliminierung • Die OH-Gruppe lässt sich nach überführen in eine bessere Abgangsgruppe mit starken, nicht-nucleophilen Basen auch eliminieren. O S Cl O HO TsO H TsO N + H“ – H2O H H ” “ O K H H H “ “ –H H H 88 8.3.3. Oxidation • Primäre Alkohole lassen sich leicht zu Aldehyden oder Carbonsäuren oxidieren. Sekundäre Alkohole können zu Ketonen oxidiert werden. 1) Oxidation Primärer Alkohole zu Carbonsäuren Primäre Alkohole werden sehr leicht von Chromat CrO42−,Dichromat Cr2O72− oder Chromsäureanhydrid CrO3 (Chromtrioxid) in verdünnter Schwefelsäure mit Aceton als Lösungsvermittler zu Carbonsäuren oxidiert (Jones-Oxidation). Zunächst bildet sich aus dem Alkohol und einem Derivat der Chromsäure H2CrO4 ein Chromsäureester (Cr(+VI)). In diesem greift ein doppelt gebundener Sauerstoff in einem 5-gliedrigen Übergangszustand ein H der CH2-Gruppe an und spaltet dieses als H+ ab. Das zugehörige Elektronenpaar bildet eine C=ODoppelbindung und verdrängt dadurch die Bindung zum Chrom. Es bildet sich eine Cr(+IV)-Verbindung, die weiter in Cr(+III) und Cr(+VI) disproportioniert, und ein Aldehyd als Zwischenverbindung. Dieser reagiert in der wässrig-sauren Lösung zu einem Aldehydhydrat (zwei OH-Gruppen am gleichen C-Atom nennt man Hydrat (!!!), nicht Alkohol (!!!)). 89 O O Cr O H O HH R O H + H“ H ” H O O O Cr O R H O H H – H H “ ” H O O O Cr O R H O R O Cr O O H “ +H H R O Aldehyd “ O H H O O O Cr O R H O H H ” H O O O Cr O R H O –H H O H “ – H2O H “O H R H O H H R H O – H“ R O Cr O O O H H O H Aldehydhydrat Das Aldehydhydrat wird erneut von einer Cr(+VI)-Verbindung oxidiert, so dass letztlich eine Carbonsäure entsteht. Wichtig: das Aldehydhydrat wird oxidiert, nicht der Aldehyd! 90 O O Cr O H O H OH R O H + H“ HO H ” O O O Cr O R H O OH H – R O Cr O O H H “ ” HO O O O Cr O R H O HO H – H“ HO H O O O Cr O R H O H ” O O O Cr O R H O – H2 O HO R H O O Cr O O Carbonsäure O Im Labor kann man diese Reaktion mit bloßem Auge verfolgen. Am Anfang ist die Reaktionsmischung orangerot (Cr+VI), am Ende dunkelgrün (Cr+III). 91 2) Oxidation primärer Alkohole zu Aldehyden Nachdem nicht der freie Aldehyd zur Carbonsäure oxidiert wird, sondern dessen Hydrat, muss man nur von wässrigen Reaktionsmischungen zu wasserfreien Reaktionsmischungen wechseln, um den Aldehyd herzustellen. Man verwendet Dichlormethan als Lösungsmittel und Pyridiniumdichromat als Oxidationsmittel (weil Alkalimetallchromate und Dichromate als auch CrO3 in CH2Cl2 nicht löslich sind!). Vom vorigen Mechanismus läuft nur der erste Teil ab. Um entstehendes Reaktionswasser zu entfernen, setzt man pulverisiertes Molekularsieb zu. H OH R H O ” ” O “ “ O Cr O O Cr H N N H O O CH2Cl2 / Molekularsieb / RT O R H Aldehyd 92 O O Cr O H O HH R O H + H“ H ” H O O O Cr O R H O H H – R O Cr O O O H H “ ” H O O O Cr O R H O H H “ –H H O O O Cr O R H O H ” H O O O Cr O R H O – H2O H R' H O O Cr O O Wichtig: wasserfreie Lösungsmittel verwenden und ein Trockenmittel (Molsieb) verwenden! 93 3) Oxidation secundärer Alkohole zu Ketonen Secundäre Alkohole kann man nach Jones (CrO3/H2SO4/H2O/Aceton) oder in wasserfreiem Dichlormethan mit Pyridiniumdichromat zu Ketonen oxidieren. Ketone können zwaar auch Hydrate bilden, diese Ketonhydrate können unter diesen Bedingungen jedoch nicht weiter oxidiert werden. O O Cr O H O R' H R O H +H “ H ” R' O O O Cr O R H O H H “ ” R' O O O Cr O R H O R' H –H H O O O Cr O R H O “ H ” R' O O O Cr O R H O – H2O R' R H O O Cr O O R' H – R O Cr O O O 94 8.4. Struktur der wichtigsten Phenole • Der Grundkörper der Phenole ist das Phenol Ph-OH. Die Eigenschaften der Phenole werden einerseits durch die OH-Gruppe, andrerseits auch durch den aromatischen Ring bestimmt. So beeinflusst der Phenylring z. B. die pKS-Werte der Phenole (pKS ≈ 9), während Alkohole schwächer sauer sind (pKS von 15 bis 17). • Weitere wichtige Phenole: OH OH OH OH OH OH OH OH Phenol α-Naphthol HO β-Naphthol Brenzkatechin Resorzin OH Hydrochinon I H NH2 COOH I O HO I H NH2 COOH I Thyrosin Thyroxin 95 8.5. Synthese von Phenolen • Hock´sche Phenolsynthese. Benzol wird mit Propen in Gegenwart von Phosphorsäure zu Cumol alkyliert. Dieses reagiert mit Sauerstoff in einer Radikalreaktion zu Cumolhydroperoxid, das bei Behandlung mit Schwefelsäure zu Phenol und Aceton reagiert. O O H OH H2SO4 O2 O + H3PO4 Cumol • Cumolhydroperoxid Dow-Phenolsynthese. Chlorbenzol wird mit NaOH in einer nucleophilen aromatischen Substitution nach dem Arin-Mechanismus in Phenol überführt. Cl KOH/NaOHSchmelze OH 96 • Phenolverkochung. Anillin wird mit NaNO2 und HCl in Phenyldiazoniumchlorid umgewandelt. Phenyldiazoniumsalze sind nur bis ca. 0 °C stabil. Erwärmt man sie auf Raumtemperatur oder höher, spalten sie N2 ab und bilden ein Phenylkation, das mit Wasser zu Phenol reagiert. NH2 “ N O N + H“ N “ N H2N O HN N – H“ H O“ H + H“ N “N ∆ – H2O H “ N O HN O – N2 N N O H – H“ “ OH + H2O – H“ Der Mechanismus der Phenolverkochung ist eine Variante des Eliminierung-Additions-Mechanismus von SNAr-Reaktionen, bei dem kein AArin auftritt. 97 8.6. Reaktionen von Phenolen • Nucleophile Substitution. Phenole können nach Deprotonieren mit Alkylhalogeniden zu Phenylalkylethern umgesetzt werden. I + NaOH OH ONa O – H2O Wichtig: die OH-Gruppe von Phenolen kann nur durch spezielle übergangsmetallkatalysierte Reaktionen substituiert werden. Phenolische OH-Gruppen können nicht eliminiert werden. • Elektrophile Aromatische Substitution. Phenole reagieren sehr gut mit Elektrophilen. Dabei wird der neue Substituent bevorzugt in o- und pStellung zur phenolischen OH-Gruppe dirigiert. Eine wichtige SEArReaktion von Phenol ist die Kolbe-Schmitt-Reaktion, bei der aus Natriumphenolat und CO2 Salicylsäure hergestellt wird. O C O O ” “ Na H O O O O ” ” O O +H O H “ OH O H 98 • Oxidation von Diphenolen. ortho-Diphenole und para-Diphenole können leicht zu den so genannten Chinonen oxidiert werden. mDiphenole reagieren nicht oder zersetzen sich beim Veruch, sie zu oxidieren. OH Ag2O / Na2SO4 Diethylether O OH O OH K2CrO4 / H2SO4 O OH O Chinone spiele eine wichtige Rolle bei Oxidationsprozessen in der Biochemie. 8.7. Ether • Ether sind „Wassermoleküle“, bei denen die beiden Hs durch Alkyloder Arylreste ausgetauscht wurden. Ether spielen in der Chemie eine wichtige Rolle als Lösungsmittel. Viele natürliche Duftstoffe sind Ether. 99 O O O O Diethylether O O Tetrahydrofuran Dioxan Anisol Diphenylether • Ether werden überwiegend durch Williamson-Ethersynthese hergestellt. • An der Luft bilden Ether leicht so genannte Etherperoxide. HH O – • H • O O• O – HO2• O O O OH • HO O + HO2• O H O n Mit konzentrierter Iodwasserstoffsäure können Ether wieder gespalten werden. O HI OH + I 100 • Spezielle Ether sind die Epoxide oder Oxirane. Es handelt sich im dreigliedrige Ringe, wobei ein Atom ein Sauerstoff ist. Solche gespannten, reaktionsfähigen Ether können durch intrramolekulare Nucleophile Substitution oder durch Prileshaev-Reaktion hergestellt werden. ” O O H O ” ” O O O Cl – HCO3” H O • Cl – Cl” O O R O Prileshaev-Reaktion Epoxide sind gute Alkylierungsmittel und reagieren sehr leicht mit Nucleopohilen unter Ringöffnung. In der Leber werden Alkene im Rahmen von Entgiftungsreaktionen zunächst epoxidiert, wodurch sie schädlicher werden als die Alkene zuvor. Erst wenn das Epoxid mit Wasser zum Diol geöffnet wurde, werden die Verbindungen ungiftiger. 101 9. Aldehyde, Ketone und Derivate • Aldehyde, Ketone und Chinone haben alle eine Carbonylgruppe, die mit keinem weiteren Heteroatom verbunden ist. Die Carbonylgruppe gehört zu den wichtigsten funktionellen Gruppen in der Organischen Chemie und in der Biochemie. δ” δ” R O δ“ H Aldehyd δ” R O δ“ R' Keton O δ“ δ” δ” O δ“ O δ“ δ“ o-Chinon O δ” p-Chinon Wichtig: die Carbonylgruppe ist so polarisiert, dass das Carbonyl-C elektrophil ist. Carbonyl-C-Atome sind sp2-hybridisiert. Carbonylgruppen sind eben. 9.1. Struktur der wichtigsten Aldehyde und Ketone • Der einfachste Aldehyd ist Formaldehyd CH2O. Formaldehyd ist gasförmig und löst sich gut in Wasser (Formalin-Lösung). Dabei bildet Formaldehyd ein Hydrat. Beim Eindampfen von Formalin-Lösung scheidet sich polymerer Paraformaldehyd als weißes Pulver ab. 102 H O O ” H H H H HO • O O O H O“ H H ” – H“ H O + H“ O H H H – H2O Hydrat Paraformaldehyd O O H O H H Die nächst höheren Aldehyde sind Acetaldehyd, Propionaldehyd, Butyraldehyd und Isobutyraldehyd (ranzige Butter), n-Valeraldehyd (Baldrian), n-Capronaldehyd, n-Caprylaldehyd und n-Caprinaldehyd. Sie sind teilweise wichtige Synthesechemikalien, kommen aber auch in der Natur vor. O H Acetaldehyd CHO CHO Propionaldehyd n-Butyraldehyd CHO Capronaldehyd Benzaldehyd CHO i-Butyraldehyd n-Valerianaldehyd CHO CHO Caprylaldehyd CHO CHO Caprinaldehyd O CHO Anisaldehyd 103 • Es gibt natürlich auch ungesättigte Aldehyde, die auch wichtige Synthesechemikalien darstellen. CHO Acrolein • CHO CHO Crotonaldehyd Propargylaldehyd Neben diesen monofunktionellen Aldehyden gibt es auch Aldehyde, die mehrere Aldehydgruppen enthalten. OHC CHO Glyoxal • OHC CHO CHO OHC Malonaldehyd Succinaldehyd Wichtig: Malonaldehyd ist eine 1,3-Dicarbonylverbindung. Durch die beiden ziehenden Aldehydgruppen werden die H-Atome der CH2Gruppe azide und reagieren unter Keto-Enol-Tautomerie in die EnolForm, die bei Malonaldehyd sehr instabil ist. HH O HH O O H H HH O H H H ” O O H H O O H H H O O H H H 104 • Aldehyde können auch noch Hydroxygruppen als zweite funktionelle Gruppe enthalten. HO H HO CHO HO CHO Glycolaldehyd • HO H Lactaldehyd CHO D(+)-Glycerinaldehyd HO HO H OH H CHO OHH OH Traubenzucker Das einfachste Keton ist das Aceton. Es ist eine wichtige Grundchemikalie und ein wichtiges Lösungsmittel. Das nächst höhere Keton ist Butanon, das ebenfalls für Synthesen verwendet wird. Bei den höheren Ketonen muss man im Namen die Position der Carbonylgruppe durch eine Zahl angeben, z.B. 2-Pentanon oder 3-Pentanon. O O O O Aceton Butanon 2-Pentanon 3-Pentanon O O Acetophenon O Propiophenon Benzophenon 105 • Wichtige ungesättigte Ketone O O O Methylvinylketon • 2-Cyclopentenon 2-Cyclohexenon Wichtige Diketone O O O O O O O Diacetyl Benzil O Acetylaceton Acetonylaceton Acetylaceton ist (wie Malonaldehyd) auch eine 1,3-Dicarbonylverbindung, die im Gleichgewicht mit ihrer Enolform steht. Die Enolform ist sowohl mesomeriestabilisiert als auch über eine intramolekulare Wassserstoffbrückenbindung begünstigt. O O 15% O H O 85% 106 • Wichtiges Triketon: Ninhydrin zum Aminosäurenachweis. Ninhydrin ist ein 1,2,3-Triketon, das nur als Hydrat stabil ist. δ“ δ“ O H δ“ O H δ“ O O O δ“ O H “O O ” H – H“ + H“ δ“ δ“ O OH OH O • Allgemein bilden Caronylgruppen, die zu stark elektronenziehenden Gruppen benachbart sind, sehr leicht Hydrate. • In der Natur und in der Biochemie spielen Ketone mit zusätzlichen funktionellen Gruppen eine große Rolle. O HO H OH H O HO O HO H HO H Acetoin Benzoin HO OH Dihydroxyaceton OH H OH O Fructose 107 9.3. Synthese von Aldehyden und Ketonen • Aldehyde kann man durch Oxidation von primären Alkoholen mit Pyridiniumdichromat in wasserfreiem Dichlormethan und Zustaz eines Trockenmittels herstellen (vgl. Kap. 8, Folien 92, 92). • Industriell kann man Alkohole mit geeigneten Katalysatoren dehydrogenieren. Davon leitet sich der Name ab (Alkohol dehydrogenatus). Cu O H R H H O R H 250 - 300 °C – H2 • Reduktion von Carbonsäurederivaten. Man kann Carbonsäureester mit geeigneten Reduktionsmitteln (z.B. DIBALH) bei tiefer Temperatur (−80 °C) zu Aldehyden reduzieren. O R OR' Al H –80 °C R O Al H OR' + H2O R O H H OR' O R – R'OH H 108 • Ketone kann man durch Oxidation von secundären Alkoholen mittels Jones-Oxidation oder mit Pyridiniumdichromat in wasserfreiem Dichlormethan und Zustaz eines Trockenmittels herstellen (vgl. Kap. 8, Folien 92, 92). • Industriell kann man auch secundäre Alkohole mit geeigneten Katalysatoren dehydrogenieren. O H R R' H Cu O R 250 - 300 °C R' – H2 • Friedel-Crafts-Acylierung liefert Aryl-Alkyl-Ketone oder Diarylketone (vgl. Kap. 7, Folie 79). • Addition von Wasser an innere Alkine ergibt Enole, die zu Ketonen tautomerisieren (vgl. Kap. 6, Folie 69). • Hock´sche Phenolsynthese liefert als Nebenprodukt Aceton (Kap. 8, Folie 96). 109 9.4. Reaktionen von Aldehyden und Ketonen • Auf Grund der Polarität der C=O-Gruppe ist das Carbonyl-C-Atom elektrophil, so dass man mit einem Nucleophil angreifen kann. nucleophil O R O H nucleophil ” O R R “ H elektrophil O O H HH pKS ≈ 16 • R' ” R “ R' elektrophil O H O O O O H HH pKS ≈ 5 HH HH pKS ≈ 20 HH HH symbolisiert den −I-Effekt pKS ≈ 9 Durch den −I-Effekt der Carbonylgruppe werden die α-H-Atome acidifiziert, so dass man sie leicht abspalten kann und mit den erhaltenen Enolen/Enolaten Reaktionen in α-Posotion zur C=O110 Gruppe ausführen kann 9.4.1.Nucleophile Additionen von HeteroatomNucleophilen • Reaktionen an der Aldehydgruppe sind nucleophile Additionen, die über tetraedrische Zwischenstufen verlaufen (AdNCO) und tetraedrische Endprodukte ergeben. • Als Nucleophile können die üblichen Nucleophile der organischen Chemie verwendet werden. Die wichtigsten sind H2O, R−OH, R−SH, R−NH2, R−NH−R‘, R−NH−NH2, NH2OH, CN− und andere wichtige CNucleophile. • Hydratbildung: Die Addition von Wasser führt zu den Hydraten von Aldehyden und Ketonen. Die Hydratbildung ist i.d.R. reversibel (steht in Einklang mit der Erlenmeyer-Regel). Je stärker elektronenziehend die Seitenketten, um so leichter bilden sich Hydrate und um so stabiler sind sie. O R H O ” H R R' O H O“ R' H ” – H“ R O + H“ O R R' H O H O R' H 111 • Beispiele: H H • O OH OH Cl3C HO OH OH H OH F3C OH OH CF3 O Acetalbildung: Aldehyde und Ketone reagieren mit Alkoholen in Gegenwart von katalytischen Mengen Säure unter Wasserabspaltung zu Acetalen. Alle Teilschritte des Reaktionsmechanismus sind reversibel, so dass man das abgespaltene Wasser aus der Reaktionsmischung entfernen muss, um gute Ausbeuten zu erhalten. O R +H R' “ O H H H O R'' HO R R “ R' “ O R'' HO OR'' – H“ R' R + H“ R' Halbacetal H “O H R O–R'' R' R'' O“ – H2O R R'' H H O R'' R'' O R “ O R'' – H“ R' R''O OR'' R R' Acetal 112 • Wichtig: Weil die Acetalbildung reversibel ist, kann man durch Zusatz von Wasser und katalytischen Mengen Säure Acetale auch wieder spalten in Alkohol und Carbonylverbindung. • Wichtig: Offenkettige Halbacetale sind instabil !!! (Ausnahme?) Cyclische Halbacetale sind stabil und können isoliert werden, wenn sie 5 oder 6 Ringglieder besitzen. O H H H OH O O H O O O H OH H • 5-gliedriges cyclisches Halbacetal = 1-Hydroxytetrahydrofuran 6-gliedriges cyclisches Halbacetal = 1-Hydroxytetrahydropyran Wichtig: Zucker liegen als stabile Halbacetale vor!!! HO HO H OH H H O OHH OH HO HO HO O OH OH 113 • Thioacetalbildung: Aldehyde und Ketone reagieren mit Thiolen R−SH in Gegenwart von katalytischen Mengen Säure unter Wasserabspaltung zu Dithioacetalen. Alle Teilschritte des Reaktionsmechanismus sind reversibel, so dass man das abgespaltene Wasser aus der Reaktionsmischung entfernen muss, um gute Ausbeuten zu erhalten. Der Mechanismus entspricht dem der Acetalbildung. • Bildung von Iminen. Setzt man Aldehyde oder Ketone mit primären Aminen in Gegenwart katalytischer Mengen Säure um, dann bilden sich Imine. H O R +H R' “ O N R'' H H H HO R R “ R' “ N R'' H R' – H“ H HO N R'' R “ +H R' Halbaminal H H “O N R'' H R R' – H2O H “ R'' N R R' – H“ N R R'' Imin (R‘‘ ≠ H) R' 114 • Analog zu den primären Aminen reagieren andere Stickstoffderivate wie z.B. Hydroxylamin (als Hydrochlorid), Semicarbazid, 2,4-Dinitrophenylhydrazin und Hydrazin. H2NOH • HCl O R R' – H2O OH N Oxim R R' O H2N NH O R R' N NH2 – H2O R NH NH2 Semicarbazon R' O O2N H2N NH O R R' NO2 NO2 N R – H2 O NH 2,4-Dinitrophenylhydrazon NO2 R' O H2N NH2 O R R' – H2 O N R R NH2 R R' – H2O Hydrazon R' N R R' N R' Azin 115 • Bildung von Enaminen: Aldehyde oder Ketone ergeben Enamine, wenn man sie mit secundären Aminen und katalytischen Mengen H+ umsetzt. 1 R “ O R +H R' H R1 “O N R2 H R R' O R H H HO R “ R' R1 “ R2 N – H2O R HH • N R2 H R' –H R1 “ R “ 2 N R R1 R' N – H“ R1 2 HO N R R “ +H R' R2 R' Enamin H Bildung von Aminalen: Aldehyde oder Ketone ohne α-H-Atome ergeben Aminale, wenn man sie mit secundären Aminen und katalytischen Mengen H+ umsetzt. Die Reaktion verläuft analog zu obigem Mechanismus bis zum Iminiumion. Dieses kann sich aber weder zum Imin noch zum Enamin stabilisieren, so dass nur die Möglichkeit der Addition eines zweiten Amins zum Aminal bleibt. 116 + H“ O R R' H R1 “O N R2 H R R' O R1 2 N R H H H HO R R “ R' – H2O R1 “ R2 N R R' R1 2 N R H 1 “ 2 N R R1 R' 2R – H“ R 2 HO N R R R' 1 R “ 2 R 1 N N R H R R' – H“ + H“ R2 R1 N R1 N R R' Tertiäre Amine reagieren nicht mit Aldehyden oder Ketonen. • Reduktion von Aldehyden und Ketonen: Aldehyde und Ketone kann man mit hydrid-übertragenden Reagenzien wie z.B. NaBH4 oder LiAlH4 zu Alkoholen reduzieren. Aldehyde ergeben primäre Alkohole nach wässriger Aufarbeitung. Ketone ergeben secundäre Alkohole nach wässriger Aufarbeitung. • In der Biochemie erfolgt die Reduktion ebenfalls durch Hydridübertragung oder durch die schrittweise Übertragung von zwei Elektronen und anschließend einem H+. 117 R 2 H” “ H Al H Li H O R ” R' R O R' H” H AL “ Li O H H “ Li + Al H H H R R' H H R' R H O H O Al ” O R' R R' R O H “ Li H2O R R' H“ oder OH R R' H OH” 9.4.2.Nucleophile Additionen von C-H-aziden Verbindungen • C-H-azide Verbindungen sind Verbindungen des Typs, bei denen durch die −I- und −M-Substituenten die HAtome so sauer werden, dass man sie leicht abspalten kann (pKS-Werte zwischen 5 und 25). Dafür stehen Basen mit pKS-Werten von 10-45 zur Verfügung. H R EWG EWG 118 • Cyanhydrinbildung: Blausäure (HCN) kann als Nucleophil an Aldehyde oder Ketone addieren. Dabei bilden sich so gennante Cyanhydrine. H C N O R H O“ R' – C N ” R O C N ” R R' H R' CN Cyanhydrin (z.B. in Leinsamen (!)) • Wichtig: Cyanhydrine spalten beim Erhitzen oder in Gegenwart von Säure wieder HCN ab! • Benzoinkondensation: Setzt man Benzaldehyd mit katalytischen Mengen KCN um, dann bildet sich nur intermediär ein Cyanhydrin, das dann aber mit noch vorhandenem Benzaldehyd weiter reagiert. ” O C N H C N ” O H O H ” O O H O C N O H OH Benzoin CN H – C N ” H 119 Analog reagieren andere Aldehyde ohne α-H-Atome. • Strecker-Synthese: Aldehyde werden mit Ammoniumcarbonat und Kaliumcyanid umgesetzt. Nach saurer Hydrolyse des gebildeten Aminonitrils (vgl. Kap. 10.1., Folie 132) bilden sich Aminosäuren. O R H • N H + HCN + NH3 R R H – H2 O H NH2 + H2O / H+ CN H NH2 R COOH Wittig-Reaktion: Setzt man Alkylhalogenide mit Triphenylphosphin um, so bilden sich in einer SN2-Reaktion Alkyltriphenyl-phosphoniumsalze. Durch den positiv geladenen Phosphor (starker −I und −MEffekt) werden die H-Atome der benachbarten CH2- oder CH-Gruppe azide. Man kann sie mit Basen abspalten und erhält dann so genannte Phosphor-Ylide (lies: Ülid), die man an Carbonylgruppen addieren kann. PPh3 R X R “ ” PPh3 X H H Wittig-Salz LDA “ –H R “ PPh3 ” H Ylid R PPh3 H Ylen 120 O R R' PPh3 Ph3P O H H R H H R' – Ph3P=O R H H R' Die Wittig-Reaktion ist eine der wichtigsten Methoden, um Alkene herzustellen. • Reppe-Reaktion: Endständige Acetylene kann man leicht deprotonieren. Die Acetylid-Ionen sind gute Nucleophile und addieren sich leicht an Aldehyde und Ketone. Man erhält so genannte Propargylalkohole. Reppe-Reaktionen sind industriell sehr wichtig. O R H Na“ H ” – H2 R ” R ” R' O R R R' H2O / H“ OH R R R' 121 • Aldol-Reaktion: Wenn man Acetaldehyd mit KOH deprotoniert, dann erhält man Kalium-Enolat des Acetaldehyds. Dieses reagiert mit noch vorhandenem Acetaldehyd zu Aldol. H H O ” O O H H H Acetaldehyd • H O ” H H H ” CH3 O O H3C Enolat + H2O H – OH” OH O H3C Aldolat H Aldol Diese Reaktion funktioniert auch bei höheren Aldehyden. Von gekreuzter Aldolreaktion spricht man, wenn man zwei verschiedene Carbonylverbindungen miteinander umsetzt. Besonders wichtig sind Reaktionen zwischen Enolaten von Ketonen und Aldehyden. Die umgekehrte Reaktion funktioniert nicht! (warum?). O R N R' HH Keton ” Li “ O R ” O R'' ” H R' H Ketonenolat O O R'' R' RH Aldolat OH O + H2O ” – OH R'' R' RH Aldol 122 • Wichtig: Um das Aldolprodukt zu isolieren, muss man die Reaktionsmischung vorsichtig neutralisieren. Bei zu starkem ansäuern wird die OH-Gruppe im Aldol noch sauer eliminiert. Man spricht dann insgesamt von Aldolkondensation, die eine α,β-ungesättigtes Dicarbonylverbindung ergibt. OH O H “ H O O + H“ R'' R' R'' RH • O “ R' – H2O RH R'' O R' RH –H “ R'' R' R Knoevenagel-Kondensation: Wenn man eine 1,3-Dicarbonylverbindung deprotoniert und als Enolatkomponente in der Reaktion mit einem Aldehyd einsetzt, dann erhält man α,β-ungesättigte 1,3Dicarbonylverbindungen. Als 1,3-Dicarbonylverbindung verwendet man meist Ester der Malonsäure. O H H COOR COOR ” – H–O–R 1 R – H2O O R 2 R COOR COOR H COOR ” COOR 1 R ” 2 R O 1 R 2 R COOR + H–O–R H COOR – O R ” HO R1 2 R COOR H COOR 123 • Mannich-Reaktion: Man setzt ein secundäres Amin mit einem Aldehyd um in Gegenwart katalytischer Mengen H+. Dabei bildet sich ein Iminiumion, das mit einem Keton in der Enol-Form reagiert und ein β-Aminoketon bildet. 1 R N H O R H “ H / – H2O O R' HH R2 + H“ R'' –H “ 2 R1 “ R N H R O R' R1 H – H“ R H R2 N O R'' R' H R'' Durch Zugabe von Säure oder einfaches Erhitzen der Mannich-Base kann man das secundäre Amin eliminieren und erhält ebenfalls α,βungesättigte Ketone. 124 9.5. Chinone • Chinone kann man aus o-Dihydroxybenzolderivaten oder pDihydroxybenzolderivaten durch Oxidation erhalten. Umgekehrt kann man Chinone wieder in die Dihydroxybenzole durch Reduktion umwandeln. Diese Prozesse laufen auch in der Zelle mit speziellen Chinonen an, wie z.B. Ubichinon. O ” “ + H” (2 e + H ) H3CO H3CO H O 10 ” “ – H” (2 e + H ) O ” H3CO H3CO H OH 10 Ubichinon (Vitamin Q10) Analoge Reaktionen laufen mit Vitamin E und Vitamin K ab (vgl. Biochemie). 125 10. Carbonsäuren und Carbonsäurederivate 10.1. Strukturen der wichtigsten Carbonsäuren • Carbonsäuren enthalten die Carboxyl-Gruppe COOH. Die Carbonsäuren kann man unterteilen in Monocarbonsäuren (nur eine COOH-Gruppe), Dicarbonsäuren (zwei COOH-Gruppen im Molekül), Tricarbonsäuren (drei COOH-Gruppen) usw. O Ameisensäure (C1) Essigsäure (C2) H O OH Capronsäure (C6) O O OH Önanthsäure (C7) O Propionsäure (C3) Caprylsäure (C8) O OH O OH Pelargonsäure (C9) O Valeriansäure (C5) OH O OH Buttersäure (C4) OH OH O OH Caprinsäure (C10) 126 OH O Laurinsäure (C12) OH O Myristinsäure (C14) OH O Palmitinsäure (C16) OH O Margarinsäure (C17) OH O Stearinsäure (C18) OH O Arachinsäure (C20) OH 127 • Es gibt auch zahlreiche ungesättigte Carbonsäuren, die aus pflanzlichen oder tierischen Fetten isoliert wurden. Acrylsäure O O OH OH Elaidinsäure O Crotonsäure OH O O Methacrylsäure OH OH Ölsäure (Oleinsäure) O Tiglinsäure • OH O Angelikasäure OH Langkettige ungesättigte Fettsäuren mit cisDoppelbindung senken den Cholesterinspiegel, auch mehrfach ungesättigte Fettsäuren mit cisDoppelbindungen. 128 • Insbesondere mehrfach ungesättigte Fettsäuren sind in der Biochemie von Bedeutung (Membranbestandteile). O Ölsäure (C18) Linolsäure (C18) α-Linolensäure (C18) γ-Linolensäure (C18) Nomenklatur der Fett-säuren: OH entweder verwendet man Trivialnamen oder man verwendet systematische Namen oder man verwendet O Abkürzungen, die die Zahl der C-Atome, die Zahl der OH Doppelbindungen und die Position der Doppelbindungen angeben (18:3 ∆9,12,15) = αO Linolensäure. Bezeichnet man die C-Aome OH nach dem gr. Alphabet, dann ist das letzte C-Atom immer ω. Das letzte C-Atom der letzten OH Doppelbindung ist dann ω−3 oder ω−6 usw. O O OH Arachidonsäure (C20) 129 • Dicarbonsäuren sind ebenfalls sehr wichtige Substanzen, sowohl für die chemische Industrie als auch für den Stoffwechsel. Oxalsäure (C2) HOOC HOOC COOH COOH Pimelinsäure (C7) Malonsäure (C3) HOOC Bernsteinsäure (C4) HOOC Glutarsäure (C5) HOOC COOH HOOC COOH COOH Suberinsäure (C8) HOOC COOH Azelainsäure (C9) COOH HOOC COOH Sebacinsäure (C10) Adipinsäure (C6) • HOOC COOH Ungesättigte Dicarbonsäuren Maleinsäure (cis) HOOC COOH Fumarsäure (trans) HOOC COOH 130 • Wichtige Hydroxy-Carbonsäuren und Keto-Carbonsäuren O O HO H OH OH O H OH (+)-Äpfelsäure (+)-Milchsäure Ox. Ox. Ox. O O O HO OH OH O H Glyoxylsäure • HO OH OH Glycolsäure O O OH O O α-Ketoglutarsäure Brenztraubensäure Wichtige Dihydroxy-Dicarbonsäure: Weinsäure H HO COOH HO H COOH H HO COOH HO H COOH H HO COOH HO H COOH HO H COOH H HO COOH , -Weinsäure , -Weinsäure , -Weinsäure , -Weinsäure meso -Weinsäure 131 10.2. Synthese von Carbonsäuren • Oxidation von primären Alkoholen (vgl. Kap. 8.3.3., Folien 89-91). • Hydrolyse von Nitrilen. Alkylhalogenide kann man mit KCN in Alkylnitrile überführen. Nitrile können sauer hydrolysiert werden, wobei Carbonsäuren entstehen. ” R X O O R • R ” –X N R C N H + H“ C N H O H H – NH3 R N R O H “ H H N“ H H H +H “ H C O R O H “ H H N “ H H H O O – H“ R H “ H O H – H“ R N O H H H “ O H C N H O R –H “ H H + H“ N O H H O R O H Hydrolyse von Cyanhydrinen liefert α-Hydroxycarbonsäuren. 132 • Malonestersynthese. Malonsäurediethylester können mit Natriumethanolat deprotoniert werden. Die gebildeten Carbanionen/Esterenolate können mit Alkylhalogeniden alkyliert werden. Nach Hydrolyse des Alkylmalonsäurediethylesters wird decarboxyliert, so dass man eine verlängerte Carbonsäure erhält. H H COOEt ” O COOEt H – EtOH COOEt ” COOEt R X ” –X O – EtO ” R H R H COOEt HO O R H COOEt O OH R R H O ” C OEt O H O H O – CO2 O R ” O OH OEt COOEt H OH H OH HH Analog kann man die Produkte der Knoevenagel-Kondensation hydrolysieren und decarboxylieren. Man erhält α,β- ungesättigte 133 Carbonsäuren. 10.3. Reaktionen von Carbonsäuren • Die wichtigesten Reaktionen von Carbonsäuren führen zu folgenden Carbonsäurederivaten: O R O Cl R Carbonsäurechlorid O O O R R Anhydrid O O R' R Ester N R'' R' Amid 10.3.1. Bildung von Carbonsäurechloriden • Kocht man Carbonsäuren mit Thionylchlorid, dann entstehen Carbonsäurechloride (neben HCl und SO2). O R Cl O H O S – HCl – SO2 O Cl R Cl 134 10.3.2. Bildung von Carbonsäureanydriden • Carbonsäurechloride und Natriumsalze der Carbonsäuren reagieren miteinander zu Carbonsäureanhydriden. Der Reaktionsmechanismus entspricht einer nucleophilen Substitution an einer Carbonylgruppe und wird als SN2t bezeichnet, weil er nach einem Zeitgesetz 2. Ordnung und über eine tetraedrische Zwischenverbindung verläuft. O O Allg. SN2t-Mechanismus R ” O R Cl O O O R Cl – Cl” Tetraedrische Zwischenstufe sp3 sp2 • R ” O R O O R sp2 Kocht man Carbonsäuren mit P4O10 (Phosphor-V-oxid = Wasser entziehendes Mittel), dann wird aus zwei Carbonsäuren ein H2O abgespalten und man erhält dieselbe Verbindung (anhydros = ohne Wasser). O R O OH HO + P4O10 R – H2O O R O O R 135 10.3.3. Bildung von Carbonsäureestern • Ester sind Reaktionsprodukte von Carbonsäuren und Alkoholen. Die Reaktion erfolgt ausschließlich säurekatalysiert unter Wasserabspaltung. Das Wasser muss aus dem Reaktionsgemisch entfernt werden. O R +H O H “ H O R • O H “H O “ H O R R' O – H2O H O R' H R O “ R O O H –H O “ R H O O H R' + H“ O “ R' H H O“ R' O H –H R O R' Auch Säurechloride und Alkohole ergeben Ester. Dabei muss man noch eine Base zusetzen, um den Alkohol zu deprotonieren und um die freiwerdende HCl zu binden. O R ” O Cl R' ” O R O Cl O R' – Cl” R O R' 136 • Wenn die Alkohol-Gruppe und die Säuregruppe Bestandteil desselben Moleküls sind, dann bilden sich cyclische Ester (= Lactone) besonders leicht, wenn sich 5- oder 6-gliedrige Ringe ausbilden können. +H HO O OH “ – H2O O O + H“ HO HO O – H2O Butyrolacton • O O Valerolacton Ester und Lactone treten in der Natur oft als Riechstoffe oder Aromastoffe auf. O O O O Essigsäure-isobutylester (Bananen-Aroma) Exaltolid (moschusartiger Geruch) 137 • Eine wichtige Klasse von Estern sind Fette, Öle und andere aus Fettsäuren aufgebaute Erster (zusammenfassend als Lipide bezeichnet), die in lebenden Organismen meist als Energiespeicher und in Membranen vorkommen. Lipide Speicherlipide Fette Öle Membranlipide Phospholipide Phosphoglyceride = Glycerophospholipide • Sphingomyeline = Spingophospholipide Glycolipide Cholesterin Ganglioside Sphingoglyco lipide Fette: Ester von Fettsäuren und Glycerin. Es kommen überwiegend gesättigte Fettsäuren vor. Es müssen nicht gleichartige Fettsäuren mit einem Glycerinmolekül verknüpft sein. O O O O O O 138 • Öle: Ester von Fettsäuren und Glycerin. Es kommen überwiegend ungesättigte Fettsäuren vor. Je mehr ungesättigte Fettsäuren enthalten sind, um so dünnflüssiger wird das Öl. Es müssen nicht gleichartige Fettsäuren mit einem Glycerinmolekül verknüpft sein. O O O O O O • Phosphoglyceride: Glycerin ist nur mit zwei Fettsäuren (gesättigt oder ungesättigt) verestert. Die dritte OH-Gruppe ist mit Phosphorsäure verestert, die zusätzlich noch mit anderen geladenen Gruppen OH verestert sein kann. O P OH O O O O O 139 ” O O P O O Lecithin (= Phosphatidylcholin) H3C CH3 N “ CH3 O O O O • Sphingomyeline: Anstatt Glycerin enthalten Sphingomyeline Sphingosin, das nur mit einer Fettsäure (gesättigt oder ungesättigt) verknüpft ist (meist Palmitinsäure). Eine OH-Gruppe ist mit Phosphorsäure verestert, die zusätzlich noch mit anderen geladenen Gruppen verestert sein kann. OH OH Ceramid O N H O N H Sphingosin H2N OH OH O O P O ”O CH3 ein Sphingomyelin N CH3 “ CH 3 140 OH • Sphingoglycolipide: Anstatt Glycerin enthalten Sphingoglycolipide Sphingosin, das nur mit einer Fettsäure (gesättigt oder ungesättigt) verknüpft ist (meist Palmitinsäure). Eine OH-Gruppe ist mit einem Monosaccharid verestert. Liegt statt des Monosaccharids ein Di- oder Oligosaccharid vor, dann nennt man die Verbindung Gangliosid (weil sie in den Membranen von Nervenzellen vorkommen). OH ein Sphingoglycolipid • O N H HO O Glucosylceramid OH OH OH O Gangliosid: Anstatt Glycerin enthalten Ganglioside Sphingosin, das nur mit einer Fettsäure (gesättigt oder ungesättigt) verknüpft ist (meist Palmitinsäure). Eine OH-Gruppe ist mit einem Di- oder Oligosaccharid verknüpft. Sie kommen in den Membranen von Nervenzellen vor). OH O N H O Disaccharid oder Oligosaccharid 141 10.3.4. Bildung von Carbonsäureamiden • Carbonsäurechloride, Carbonsäureanhydride oder Carbonsäureester kann man mit Ammoniak oder mit primären oder secundären Aminen zu den Carbonsäureamiden umsetzen. R1 H N R2 O R X ” H O R 1 ” N R2 – H“ O “R X R O 1 N R2 R X – X” R N R2 R1 Die neu entstandene C-N-Bindung nennt man Amidbindung. Falls die Aminogruppe von einer Aminosäure stammt, spricht man auch von Peptidbindung (vgl. Biochemie). • Wichtig: Amidbindungen haben eingeschränkte Drehbarkeit, weil auf Grund des +M-Effektes der Amingruppierung zusammen mit dem −MEffekt der Carbonylgruppe die C-N-Bindung partiellen Doppelbindungscharakter besitzt. ” O R N R2 R1 R δ” O O “ R1 N R2 R R1 N δ“ R2 142 10.4. Reaktionen von Carbonsäurederivaten • Umwandlung in Carbonsäuren. Alle Carbonsäurederivate können durch Reaktion mit Wasser und H+ oder OH− wieder in Carbonsäuren überführt werden. 1) Carbonsäurehalogenide. Aliphatische Carbonsäurehalogenide reagieren heftig mit Wasser (sowohl sauer als auch alkalisch), aromatische Carbonsäurehalogenide reagieren langsam mit Wasser (sowohl sauer als auch alkalisch). 2) Carbonsäureanhydride. Aliphatische Carbonsäureanhydride reagieren heftig mit Wasser (sowohl sauer als auch alkalisch), aromatische Carbonsäureanhydride reagieren langsam mit Wasser (sowohl sauer als auch alkalisch). 3) Ester. Die säurekatalysierte Esterhydrolyse ist die exakte Umkehrung der Esterbildung und führt deshalb nur zu einer Gleichgewichtsmischung. Die alkalische Esterhydrolyse verläuft vollständig, da die entstehende Carbonsäure im letzten Schritt deprotoniert wird, was die Reaktion irreversibel macht. Wichtig: deshalb kann man Ester nicht basenkatalysiert aus Carbonsäuren und Alkoholen herstellen!!! 143 O R ” ” O H O R' O O O R' R “ ” – O R' H ” O H O R O H O R O ” O H2O / H saure Aufarbeitung R O H 4) Carbonsäureamide. Carbonsäureamide werden leichter im sauren hydrolysiert als im alkalischen (warum?). Verglichen mit den anderen Carbonsäurederivaten erfolgt die Hydrolyse von Amiden deutlich langsamer (warum?) O R N R2 R 1 ” ” O H 1 H – R O N R1 R ” 2 N R O R O H R 1 ” N R2 O R O ” R2 H2O / H“ saure Aufarbeitung O O R O H 144 O R – • N 2 R R H 1 “ H O “ 1 R 1 R H N R2 “ O R N R2 H O R H O O – H“ R R2 – H“ H H H O O “ H R N 1 R H O H H H N R1 H O O H “ R N 1 R 2 H R “ R2 O R O H Claisen-Kondensation. Bei der Claisen-Kondensation werden Ester mit aziden α-H-Atomen mit dem Alkoholat, das die Alkoholkomponente des Esters ist, deprotoniert. Da so erhaltene Esterenolat reagiert mit einem nicht deprotonierten Ester nach einem SN2tMechanismus zu β-Ketoestern. ” O R O O R' O R' R HH – O R' R' O R O R O ” O O R' R O R O R' ” R' O ” O O R R' 145 10.5. Kohlensäure und Derivate • Kohlensäure H2CO3 ist nur unter hohem CO2-Druck stabil. Unter Normalbedingungen zerfällt sie in H2O und CO2. Es gibt jedoch zahlreiche stabile Kohlensäurederivate. Das wichtigste davon ist Phosgen COCl2 (zählt zu den anorganischen Verbindungen). CO + Cl2 Aktivkohle • O 200 °C Cl Cl Aus Phosgen kann man fast alle anderen wichtigen (organischen) Kohlensäurederivate herstellen. 1) Chlorameisensäureester O Cl 1 R–O–H Cl O R – HCl O Cl 2) Kohlensäureester (Dialkylcarbonate, Diarylcarbonate oder Alkylarylcarbonate) O R O O 1 R'–O–H Cl R – HCl O O R' 146 3) Carbamate (Urethane) O R O 1 R'–NH2 O Cl R – HCl O N H R' 4) Harnstoff und Harnstoffderivate O 2 NH3 Cl Cl O Cl O H2N – 2 HCl 2 R–NH2 Cl NH2 O R – 2 HCl N H N H R 5) Isocyanate O Cl 1 R–NH2 Cl O R – 1 HCl N H Cl R N C O – 1 HCl 147 6) Guanidin 2 NH3 O Cl Cl – 2 HCl O H2N 1 NH3 NH2 – H2O formal NH H2N 1 NH3 NH2 Guanidin real H2N C N Cyanamid Wichtige Guanidin-Derivate sind z.B. die Aminosäure Arginin oder die Nucleinbase Guanin. O NH H2N NH COOH NH2 Arginin H H2N N N N N H Guanin 148 11. Amine und wichtige Heterocyclen 11.1. Strukturen der wichtigsten Amine • Amine sind Alkyl- oder Arylderivate von Ammoniak NH3 und sind Basen mit pKS-Werten zwischen 4 und 11. • Ist im NH3 ein H durch einen Rest R ersetzt, dann spricht man von e primären Aminen. Sind zwei Hs durch zwei Reste R und R‘ ersetzt, nennt man die Verbindung secundäres Amin. Sind alle drei Hs durch Reste ersetzt, dann liegt ein tertiäres Amin vor. Trägt der Stickstoff vier Reste, dann handelt es sich um ein quartäres Ammonium-Ion. H R N H primäres Amin • R' R N H R' R N R'' secundäres Amin R1 4 tertiäres Amin Beispiele: R2 “ N R3 R quartäres Ammoniumion N NH2 n-Butylamin N H Diisopropylamin Ethylmethylnaphthylamin 149 NH2 “ N CH3 HO ” Cl Benzyltriethylammoniumchlorid Anilin “N CH3 O ” CH3 OH CH3 O Cholin “N CH3 CH3 ” OH Acetylcholin 11.2. Herstellung der Amine • Alkylierung von Ammoniak. Ammoniak und Amine sind nicht nur basisch, sondern auch nucleophil (Ausnahme?). D.h., man kann sie mit Alkylhalogeniden umsetzen. Nachteil: durch jede eingefüührte Alkylgruppe wird der Stickstoff nucleophiler und reagiert immer schneller mit noch vorhandenem Alkylhalogenid, bis man schließlich zum quartären Ammoniumsalz kommt. H N H H R–Cl SN2 R N H H R–Cl SN2 R R–Cl N H R SN2 Reaktionsgeschwindigkeit nimmt zu R R–Cl N R R SN2 R ” “ N R Cl R R 150 • Gabriel-Synthese. Man geht von Phthalimid aus, deprotoniert mit KOH und setzt das so erhaltene Kaliumphthalimid mit Alkylhalogeniden um. Im Kaliumphthalimid kann der Stickstoff nur einmal mit Alkylhalogeniden reagieren (warum?) und man erhält nach Hydrolyse nur primäre Amine. KOH N H O O O O – H2O N ” R–Cl – Cl O ” N R H2O R NH2 O Alternativ wird im letzten Schritt statt Wasser Hydrazin H2N−NH2 eingesetzt (liefert bessere Ausbeuten). • Sekundäre und tertiäre Amine werden entweder durch Alkylierung von primären oder secundären Aminen hergestellt und das erhaltene Gemisch durch Destillation getrennt oder durch spezielle Verfahren erzeugt. 151 11.3. Strukturen wichtiger N-Heterocyclen • Heterocyclen sind cyclische Verbindungen, bei denen mindestens ein C-Atom durch ein Heteroatom (die wichtigsten sind N, O, S) ersetzt wurde. • Beispiele: Carbocyclus formal Ersetzen eines CHx durch NHx-1 Heterocyclus N N H Pyrrolidin Vorkommen Prolin N H N H N N H S Pyrrol Indol Imidazol Thiazol Porphyrine Trypthophan Histidin Vitamin B1 (Hämin, Chlorophyll) Serotonin 152 Carbocyclus formal Ersetzen eines CHx durch NHx-1 Vorkommen N H N Piperidin Pyridin Pyrimidin Chinolin Vitamin B6 NAD NADP Vitamin B1 Nucleinbasen Chinin Piperin Coniin • N N N N Heterocyclus N N N H Purin Nucleinbasen Coffein Harnsäure Auch Sauerstoff- und Schwefel-haltige Heterocyclen haben biologische Bedeutung. Vorkommen O S Tetrahydropyran Thiolan Vitamin E Vitamin H 153


![6.3.1 1-Oxa-spiro[2.5]octan - Institut für Organische Chemie](http://s1.studylibde.com/store/data/001356875_1-96e669e5c88ad586db9f9f199d424d05-300x300.png)