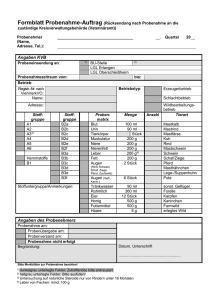
Einblick - Christiani
Werbung

EUROPA-FACHBUCHREIHE für Chemieberufe Fachwissen Chemie 2 Erweiterte Qualifikationen für Laborberufe VERLAG EUROPA-LEHRMITTEL . Nourney, Vollmer GmbH & Co. KG Düsselberger Straße 23 . 42781 Haan-Gruiten Europa-Nr.: 69956 CH2-Titelei.indd 1 07.08.14 12:36 Autoren: Dr. Henrik Althaus Peter Brackmann Dr. Astrid Grote-Wolff Dr. Heinz Hug Helmut Keim Dr. Heribert Keweloh Prof. Dr. Peter Kurzweil Dr. Thomas Meyer StR, Dipl. Chem. Betr. Ausbilder Chemielaboranten Dipl.-Chem. OStR, Dipl.-Chem. OStR, Dipl.-Ing. Priv.-Doz. Dipl.-Chem. OStR, Dipl.-Chem. Stade Bremen Nottuln Wiesbaden Mülheim a. d. R. Oberhausen Amberg Holzminden Unter Mitwirkung von Herrn Dr.-Ing. Eckhard Ignatowitz, Waldbronn Leitung des Arbeitskreises: Dr. Thomas Meyer Verlagslektorat: Dr. Astrid Grote-Wolff Bildbearbeitung: Grafische Produktionen Jürgen Neumann, 97222 Rimpar Zeichenbüro des Verlags Europa-Lehrmittel, 73760 Ostfildern Umschlaggestaltung: Grafische Produktionen Jürgen Neumann, 97222 Rimpar, nach dem Entwurf von Dr. Thomas Meyer 1. Auflage 2014 Druck 5 4 3 2 1 Alle Drucke derselben Auflage sind parallel einsetzbar, da sie bis auf die Behebung von Druckfehlern untereinander unverändert sind. ISBN 978-3-8085-6995-5 Alle Rechte vorbehalten. Das Werk ist urheberrechtlich geschützt. Jede Verwendung außerhalb der gesetzlich geregelten Fälle muss vom Verlag schriftlich genehmigt werden. © 2014 by Verlag Europa-Lehrmittel, Nourney, Vollmer GmbH & Co. KG, 42781 Haan-Gruiten http://www.europa-lehrmittel.de Satz: Grafische Produktionen Jürgen Neumann, 97222 Rimpar Druck: M. P. Media-Print Informationstechnologie, 33100 Paderborn CH2-Titelei.indd 2 07.08.14 12:36 Vorwort Mit der rasanten Entwicklung der Wissenschaft Chemie, der chemischen Analytik und der chemischen Verfahrenstechnik geht die zunehmende fachliche Spezialisierung der Fachkräfte in Chemielaboratorien einher. Bereits während der Ausbildung findet eine Vertiefung der Kenntnisse auf den im fachpraktischen Teil der Ausbildung relevanten Wissensgebieten statt. Das Lehrbuch „Fachwissen Chemie 2: Erweiterte Qualifikationen für Laborberufe“ baut auf den chemischen Grundlagen auf, die im ersten Band dieser Fachbuchreihe vermittelt wurden. Abgestimmt auf den Rahmenlehrplan für die Ausbildung zum Chemielaboranten/zur Chemielaborantin sowie angelehnt an die Lehrpläne der höheren Berufsfachschule Chemie bzw. Chemietechnik für die Ausbildung zum Chemisch-technischen Assistent/zur Chemisch-technischen Assistentin werden die Qualifikationen der Fachstufe bis zum Teil 2 der gestreckten Abschlussprüfung auf einem an der bundeseinheitlichen Prüfung der Chemielaboranten orientierten Niveau vermittelt. Auch Schüler und Auszubildende anderer naturwissenschaftlicher Bildungsgänge sowie Studierende an Hochschulen und Universitäten können mit diesem Buch ihre im Studium zu erwerbenden Fachkenntnisse über ein fundiertes Grundwissen der Chemie hinaus vertiefen. Die fachlichen Schwerpunkte des Lehrwerks: • Qualitätsmanagementsysteme, Qualitätswerkzeuge und mathematisch-statistische Verfahren werden anhand anschaulicher Beispiele und unterstützt durch Grafiken erläutert. • Auf der Basis der vermittelten grundlegenden Kenntnisse zur Probenahme, Probenbehandlung und Probenvorbereitung wird der Leser dazu befähigt, sich anhand aktueller Publikationen das fallspezifische Spezialwissen anzueignen. • Volumetrische und gravimetrische Analyse sowie chromatografische Trenntechniken, wie GC und HPLC, werden genauso behandelt wie die Grundlagen spektrometrischer Verfahren, wie die Atomabsorptionsspektrometrie AAS und die Röntgenfluoreszenzanalyse RFA. • Auf die systematische Darstellung von Reaktionsmechanismen der organischen Chemie folgen die Strukturaufklärung organischer Verbindungen mithilfe der UV/Vis-, IR-, 1H-NMRund 13C-NMR-Spektroskopie sowie der Massenspektrometrie. • Für die Probenahme in großtechnischen Prozessen unerlässlich sind Kenntnisse von Produktionsprozessen, der Darstellung von Produktionsanlagen in RI-Fließbildern sowie der Funktionsweise und Eigenschaften der verschiedenen Anlagenbestandteile. • Im Kapitel Werkstofftechnik erhält der Leser einen Überblick über Werkstoffarten, moderne Werkstoffprüfverfahren sowie über rheologische Bestimmungen in der Praxis. • Grundlegende Kenntnisse zur Elektrochemie und Elektrotechnik werden schrittweise dargestellt. Hierauf aufbauend werden elektrochemische Vorgänge im großtechnischen Prozess, wie Galvanik und Elektrolyse, sowie die elektrochemischen Analyseverfahren verständlich gemacht. • Das Kapitel zur Biotechnologie ermöglicht auf der Basis der biologischen Grundlagen einen Einblick in die modernen Methoden der Mikrobiologie und Biotechnik. Das Buch ist sowohl als den Unterricht begleitendes Werk als auch zum Selbststudium geeignet. Das Verständnis der dargestellten Inhalte wird durch die reichhaltige Bebilderung gefördert. Während der Ausbildung, zur Prüfungsvorbereitung und nach der Ausbildung kann das Buch als wertvoller, umfangreicher Wissensspeicher genutzt werden. Eine Auswahl der genannten Stoffe ist mit ihrer Gefahrstoffkennzeichnung nach GHS im Anhang aufgeführt. Unseren Leserinnen und Lesern wünschen wir viel Freude und Erfolg beim Erlernen und Erforschen der erweiterten Qualifikationen für Laborberufe. Hinweise und Ergänzungen, die zur Verbesserung und Weiterentwicklung des Buches beitragen, werden unter der Verlagsadresse oder per E-Mail ([email protected]) dankbar entgegengenommen. Sommer 2014 Autoren und Verlag 3 CH2-Titelei.indd 3 07.08.14 12:36 Inhaltsverzeichnis 1 Qualität und Qualitätssysteme (Lernfelder 6a, 7, 8, 11) ..................................................11 1.1 1.2 1.3 1.4 1.4.1 1.4.2 1.4.3 1.4.4 1.4.5 1.4.6 1.5 1.5.1 1.5.2 1.5.3 1.5.4 1.5.5 1.6 1.6.1 1.6.2 1.6.3 1.6.4 1.6.5 1.6.6 1.6.7 1.7 1.8 Qualität ................................................................................................................................. 11 Qualitätsmerkmale .............................................................................................................. 15 Fehler .................................................................................................................................... 16 Qualitätssysteme ................................................................................................................. 19 Qualitätskontrolle QK und Qualitätssicherung QS............................................................ 19 Qualitätsmanagement QM .................................................................................................. 20 Totales Qualitätsmanagement TQM................................................................................... 26 Gute Laborpraxis GLP ......................................................................................................... 28 Good Manufacturing Practice GMP.................................................................................... 30 CE-Kennzeichnung ............................................................................................................... 30 Mathematisch-statistische Methoden zur Kontrolle und Überwachung von Qualität. 32 Median m.............................................................................................................................. 32 Arithmetisches Mittel x– ....................................................................................................... 33 Varianz s2 und Standardabweichung s .............................................................................. 34 Variationskoeffizient v ......................................................................................................... 38 Spannweite R ....................................................................................................................... 39 Q7 – Werkzeuge der Qualität.............................................................................................. 39 Fehlersammelliste................................................................................................................ 40 Qualitätsregelkarte............................................................................................................... 41 Histogramm.......................................................................................................................... 44 Korrelationsdiagramm ........................................................................................................ 45 Pareto-Diagramm................................................................................................................. 46 Brainstorming ...................................................................................................................... 47 Ursache-Wirkungs-Diagramm ............................................................................................ 49 Fehlermöglichkeits- und Fehlereinfluss-Analyse .............................................................. 50 Validierung ........................................................................................................................... 52 Aufgaben zu Kapitel 1 ......................................................................................................... 56 2 Probenahme, Probenbehandlung und Probenvorbereitung (Lernfelder 7, 9, 10) .........57 2.1 2.2 2.2.1 2.2.2 2.2.3 2.2.4 2.2.5 2.2.6 2.2.7 2.2.8 2.3 2.4 2.5 2.6 2.6.1 2.6.2 2.6.3 Analyse von Proben............................................................................................................. 58 Probenahme ......................................................................................................................... 59 Ort und Zeit der Probenahme, Festlegung der Grundgesamtheit ................................... 60 Probenahmeverfahren bei Ortsabhängigkeit der Parameter ........................................... 61 Probenahmeverfahren bei Zeitabhängigkeit der Parameter ............................................ 62 Probenahmegeräte für Feststoffe ....................................................................................... 63 Probenahmegeräte für Flüssigkeiten ................................................................................. 64 Probenahmegeräte für Gase ............................................................................................... 65 Probenmenge ....................................................................................................................... 67 Probengefäße ....................................................................................................................... 69 Messungen vor Ort .............................................................................................................. 70 Probenkonservierung und -transport ................................................................................ 71 Probenahmeprotokoll ......................................................................................................... 72 Probenvorbereitung ............................................................................................................ 73 Homogenisierung, Probenverjüngung und Probenteilung .............................................. 74 Lösen der festen Analysenprobe ........................................................................................ 77 Abtrennen von Analyt und Störsubstanzen ...................................................................... 79 4 CH2-Titelei.indd 4 07.08.14 12:36 Inhaltsverzeichnis 2.6.4 2.7 2.8 Einstellen einer geeigneten Analytkonzentration ............................................................. 80 Externe Kalibrierung und Messung ................................................................................... 81 Auswertung, Dokumentation und Qualitätssicherung .................................................... 82 Aufgaben zu Kapitel 2 ......................................................................................................... 82 3 Reaktionen organischer Präparate (Lernfelder 6a, 6b, 11) ..............................................83 3.1 3.1.1 3.1.2 3.1.3 3.2 3.2.1 3.2.2 3.2.3 3.2.4 3.2.5 3.3 3.3.1 3.3.2 3.3.3 3.3.4 3.3.5 3.3.6 3.3.7 3.4. 3.4.1 3.4.2 3.5 3.5.1 3.5.2 3.5.3 3.6 3.6.1 3.6.2 3.6.3 Additionsreaktionen an C-C-Mehrfachbindungen ............................................................ 83 Struktur der Alkene .............................................................................................................. 83 Reaktionen der Alkene......................................................................................................... 84 Reaktionen der Alkine.......................................................................................................... 92 Reaktionen aromatischer Verbindungen ........................................................................... 94 Struktur des Benzols und Aromatizität .............................................................................. 94 Elektrophile aromatische Substitution und Folgereaktionen ........................................... 96 Zweit- und Mehrfachsubstitution ..................................................................................... 103 Reaktionen von Diazoniumverbindungen ....................................................................... 109 Nucleophile aromatische Substitution............................................................................. 112 Substitution und Eliminierung ......................................................................................... 113 Substitutionsreaktionen der Halogenalkane ................................................................... 114 Eliminierungsreaktionen der Halogenalkane .................................................................. 121 Substitutionsreaktionen der Alkohole.............................................................................. 123 Eliminierungsreaktionen von Alkoholen.......................................................................... 124 Oxidation von Alkoholen................................................................................................... 125 Reaktionen der Amine ....................................................................................................... 126 Reaktionen der Ether und Oxirane (Epoxide) .................................................................. 127 Carbonylverbindungen...................................................................................................... 129 Reaktionen der Carbonsäuren und deren Derivate......................................................... 129 Reaktionen der Aldehyde und Ketone ............................................................................. 137 Stereochemie organischer Stoffe .................................................................................... 144 Asymmetrisch substituierte Kohlenstoffatome............................................................... 145 Optische Aktivität ............................................................................................................... 146 Verbindungen mit mehreren chiralen Zentren ................................................................ 147 Makromoleküle .................................................................................................................. 149 Natürliche Makromoleküle ................................................................................................ 150 Synthetische Makromoleküle ........................................................................................... 151 Synthetisch veränderte Naturstoffe ................................................................................. 156 Aufgaben zu Kapitel 3 ....................................................................................................... 158 4 Volumetrische und gravimetrische Analyse (Lernfeld 7) ..............................................161 4.1 4.1.1 4.1.2 4.1.3 4.1.4 4.1.5 4.1.6 4.1.7 4.1.8 Volumetrische Analyse ..................................................................................................... 161 Äquivalenzpunkterkennung .............................................................................................. 162 Maßlösungen ..................................................................................................................... 165 Titrationstechniken ............................................................................................................ 172 Säure-Base-Titration .......................................................................................................... 173 Redoxtitration..................................................................................................................... 180 Komplexometrische Titration ........................................................................................... 187 Fällungstitration ................................................................................................................. 191 Spezielle Titrationen .......................................................................................................... 195 5 CH2-Titelei.indd 5 07.08.14 12:36 Inhaltsverzeichnis 4.2 4.2.1 4.2.2 4.2.3 4.2.4 Gravimetrische Analyse .................................................................................................... 203 Gravimetrische Fällungsanalyse ...................................................................................... 203 Feuchtigkeits- und Trockengehalt, Glührückstand .......................................................... 206 Thermogravimetrie ............................................................................................................ 207 Elektrogravimetrie ............................................................................................................. 208 Aufgaben zu Kapitel 4 ....................................................................................................... 209 5 Chromatografische Trenntechniken (Lernfeld 8) ...........................................................211 5.1 5.1.1 5.1.2 5.1.3 5.1.4 4.1.5 5.1.6 5.2 5.2.1 5.2.2 5.2.3 5.2.4 5.2.5 5.3 5.3.1 5.3.2 Gaschromatografie GC...................................................................................................... 211 Trägergase .......................................................................................................................... 211 Probenaufgabe ................................................................................................................... 214 Injektionssysteme .............................................................................................................. 216 Säulenofen und Säulen ..................................................................................................... 218 Detektoren .......................................................................................................................... 224 Fehlersuche und Optimierung .......................................................................................... 229 Hochleistungs-Flüssigkeits-Chromatografie HPLC......................................................... 231 Eluentenförderung ............................................................................................................. 231 Injektionssystem ................................................................................................................ 233 Säulen und Trennung ........................................................................................................ 234 Detektion............................................................................................................................. 238 Fehlersuche ........................................................................................................................ 240 Spezielle chromatografische Methoden.......................................................................... 241 Ionenchromatografie ......................................................................................................... 241 Elektrophorese ................................................................................................................... 244 Aufgaben zu Kapitel 5 ....................................................................................................... 246 6 Spektroskopie (Lernfeld 9) ...............................................................................................249 6.1 6.2 6.3 6.4 6.4.1 6.4.2 6.5 6.6 6.6.1 6.6.2 6.6.3 6.6.4 6.6.5 6.7 6.7.1 6.7.2 6.7.3 6.8 Grundgrößen der Wellenlehre.......................................................................................... 249 Quantenprinzip und Energie............................................................................................. 251 Spektrenarten .................................................................................................................... 253 Aufbau von Spektralapparaten ........................................................................................ 254 Signalauftrennung ............................................................................................................. 254 Detektion und Auswertung ............................................................................................... 256 Bouguer-Lambert-Beer-Gesetz ......................................................................................... 257 Atomabsorptionsspektrometrie (AAS)............................................................................ 260 Aufbau eines Atomabsorptionsspektrometers ............................................................... 262 Strahlungsquellen.............................................................................................................. 263 Atomisierung...................................................................................................................... 264 Störungen der Atomabsorptionsmessung ...................................................................... 266 AAS-Quantifizierung mittels Standardadditionsverfahren ............................................ 267 Plasma-Emissionsspektrometrie...................................................................................... 268 Plasmafackel....................................................................................................................... 268 Polychromatoren in der Plasma-Emissionsspektroskopie ............................................. 269 Vergleich von AAS und ICP-OES ...................................................................................... 270 Röntgenfluoreszenzanalyse (RFA) ................................................................................... 270 Aufgaben zu Kapitel 6 ....................................................................................................... 273 6 CH2-Titelei.indd 6 07.08.14 12:36 Inhaltsverzeichnis 7 Strukturaufklärung organischer Verbindungen (Lernfeld 10) ......................................275 7.1 7.2 7.3 7.3.1 7.3.2 7.3.3 7.3.4 7.4 7.4.1 7.4.2 7.4.3 7.4.4 7.4.5 7.5 7.5.1 7.5.2 7.6 7.6.1 7.6.2 7.6.3 7.6.4 7.7 7.7.1 7.7.2 7.7.3 Organisch-analytische Vorproben .................................................................................... 275 Elementaranalyse und Molmassenbestimmung ............................................................ 278 UV/Vis-Spektroskopie ...................................................................................................... 280 Anregung von Elektronen in Molekülen .......................................................................... 280 UV/Vis-Spektrometer ......................................................................................................... 282 Absorptionsspektroskopie an Molekülen ........................................................................ 284 Anwendungen der UV/Vis-Spektroskopie ....................................................................... 286 Infrarot-Spektroskopie (IR) ............................................................................................... 290 Molekülschwingungen und Rotationen ........................................................................... 290 Angewandte IR-Spektroskopie ......................................................................................... 291 Auswertung von IR-Spektren ............................................................................................ 293 Nahinfrarot-Spektroskopie (NIR) in der Anwendungspraxis ......................................... 299 Raman-Spektroskopie ....................................................................................................... 300 Massenspektrometrie (MS) .............................................................................................. 301 Molekülpeaks und Fragmente .......................................................................................... 301 Auswertung von Massenspektren .................................................................................... 304 Kernspinresonanz-Spektroskopie (NMR) ........................................................................ 309 Kernspin und NMR-Signale .............................................................................................. 309 Auswertung von 1H-NMR-Spektren ................................................................................. 310 Auswertung von 13C-NMR-Spektren ................................................................................ 314 Spezielle NMR-Techniken in der Anwendungspraxis ..................................................... 316 Strukturaufklärung mit kombinierten Methoden........................................................... 318 Aromastoff.......................................................................................................................... 318 Weckamin ........................................................................................................................... 320 Explosivstoff ....................................................................................................................... 321 Aufgaben zu Kapitel 7 ....................................................................................................... 322 8 Produktionsprozesse überwachen (Lernfeld 12) ...........................................................323 8.1 8.2 8.2.1 8.2.2 8.2.3 8.3 8.4 8.4.1 8.4.2 8.4.3 8.5 8.5.1 8.5.2 8.5.3 8.6 8.6.1 8.6.2 Vom Labormaßstab zum chemischen Produktionsprozess .......................................... 323 Darstellung eines chemischen Produktionsprozesses ................................................... 323 Grundfließbild .................................................................................................................... 324 RI-Fließbild ......................................................................................................................... 324 RI-Fließbild einer Umkristallisation .................................................................................. 326 Komponenten einer chemischen Produktionsanlage .................................................... 328 Rohrleitungen .................................................................................................................... 329 Nennweite DN .................................................................................................................... 329 Nenndruck PN .................................................................................................................... 329 Kennzeichnung von Rohrleitungen .................................................................................. 330 Armaturen .......................................................................................................................... 330 Absperrarmaturen ............................................................................................................. 331 Sicherheitsarmaturen ........................................................................................................ 332 Armaturen mit anderen Aufgaben ................................................................................... 333 Fördereinrichtungen .......................................................................................................... 334 Fördern von Flüssigkeiten ................................................................................................. 334 Zentrifugalpumpen ............................................................................................................ 334 7 CH2-Titelei.indd 7 07.08.14 12:36 Inhaltsverzeichnis 8.6.3 8.6.4 8.6.5 8.6.6 8.7 8.7.1 8.7.2 8.8 8.8.1 8.8.2 8.9 8.9.1 8.9.2 8.9.3 8.10 8.10.1 8.10.2 8.10.3 8.11 8.11.1 8.11.2 8.11.3 8.12 8.12.1 8.12.2 8.12.3 8.12.4 Verdrängerpumpen............................................................................................................ 336 Strahlpumpen .................................................................................................................... 338 Fördern und Verdichten von Gasen, Erzeugung von Unterdruck .................................. 338 Fördern von Feststoffen .................................................................................................... 340 Zerkleinern von Feststoffen .............................................................................................. 341 Brecher................................................................................................................................ 341 Mühlen ................................................................................................................................ 342 Behälter und Reaktoren .................................................................................................... 344 Rührbehälter....................................................................................................................... 344 Reaktoren............................................................................................................................ 345 Heiz- und Kühltechnik ....................................................................................................... 347 Energieträger...................................................................................................................... 347 Heizen und Kühlen von Rührbehältern ............................................................................ 349 Wärmeaustauscher und Kondensatoren ......................................................................... 350 Thermisches Trennen ........................................................................................................ 352 Trocknen ............................................................................................................................. 352 Verdampfen ........................................................................................................................ 354 Kristallisieren...................................................................................................................... 354 Mechanisches Trennen ...................................................................................................... 355 Trennen von Feststoffgemischen ..................................................................................... 355 Trennen von Suspensionen .............................................................................................. 357 Trennen von Emulsionen .................................................................................................. 361 Prozessleittechnik.............................................................................................................. 362 Aufbau eines Prozessleitsystems (PLS) ........................................................................... 362 Messtechnik........................................................................................................................ 364 Steuerungstechnik ............................................................................................................. 374 Regelungstechnik............................................................................................................... 378 Aufgaben zu Kapitel 8 ....................................................................................................... 381 9 Werkstofftechnik (Lernfeld 13) ........................................................................................383 9.1 9.1.1 9.1.2 9.1.3 9.1.4 9.1.5 9.1.6 9.1.7 Einteilung, Aufbau und Eigenschaften der Werkstoffe .................................................. 383 Allgemeine Werkstoffeigenschaften ................................................................................ 383 Polymerwerkstoffe ............................................................................................................. 384 Gläser .................................................................................................................................. 385 Keramiken........................................................................................................................... 385 Verformung der Metalle .................................................................................................... 386 Gitterdefekte und Gefüge .................................................................................................. 386 Legierungen ....................................................................................................................... 387 9.2 9.2.1 Zustandsschaubilder ......................................................................................................... 387 Gibbssche Phasenregel ..................................................................................................... 387 9.2.2 9.2.3 Binäre Systeme .................................................................................................................. 388 Eisen-Kohlenstoff-Legierungen ........................................................................................ 391 9.2.4 Legierte Stähle ................................................................................................................... 394 9.3 9.3.1 Werkstoffprüfung .............................................................................................................. 395 Mechanische Werkstoffprüfung........................................................................................ 396 8 CH2-Titelei.indd 8 07.08.14 12:36 Inhaltsverzeichnis 9.3.2 9.3.3 9.3.4 9.4 9.4.1 9.4.2 9.4.3 9.4.4 9.5 Zerstörungsfreie Werkstoffprüfung .................................................................................. 400 Metallografie und Ätzverfahren ........................................................................................ 403 Chemisch-physikalische Analyseverfahren ..................................................................... 406 Korrosion und Korrosionsschutz...................................................................................... 407 Ursachen der Korrosion .................................................................................................... 407 Erscheinungsformen der Korrosion ................................................................................. 407 Korrosionsschutz ............................................................................................................... 408 Korrosionsbeständige Werkstoffe .................................................................................... 409 Rheologische Bestimmungen in der Praxis .................................................................... 411 Aufgaben zu Kapitel 9 ....................................................................................................... 412 10 Elektrochemie und Elektrotechnik (Lernfelder 15, 20) ...................................................413 10.1 10.1.1 10.1.2 10.1.3 Grundbegriffe ..................................................................................................................... 413 Elektrische Ladung Q ......................................................................................................... 413 Elektrische Spannung U .................................................................................................... 415 Elektrischer Strom Ü........................................................................................................... 416 10.1.4 Elektrischer Widerstand R ................................................................................................. 420 10.2 Stromkreis .......................................................................................................................... 423 10.2.1 10.2.2 10.2.3 10.2.4 10.2.5 10.2.6 10.2.7 Schaltzeichen ..................................................................................................................... 423 Messung von Spannung U und Strom Ü ......................................................................... 423 Reihenschaltung ................................................................................................................ 425 Parallelschaltung................................................................................................................ 427 Brückenschaltung .............................................................................................................. 429 Gleichrichtung .................................................................................................................... 431 Bauelemente ...................................................................................................................... 432 10.3 Elektrochemische Vorgänge ............................................................................................. 437 10.3.1 10.3.2 10.3.3 10.3.4 10.3.5 Daniell-Element .................................................................................................................. 439 Elektrodenvorgänge .......................................................................................................... 441 Standardpotentiale und Elektrochemische Spannungsreihe......................................... 442 Bezugselektroden............................................................................................................... 446 Nernst-Gleichung ............................................................................................................... 448 10.4 Galvanische Elemente ....................................................................................................... 451 10.4.1 Primärelemente.................................................................................................................. 452 10.4.2 Sekundärelemente ............................................................................................................. 453 10.4.3 Brennstoffzellen ................................................................................................................. 454 10.5 Elektrolyse .......................................................................................................................... 455 10.5.1 Prinzipieller Ablauf............................................................................................................. 455 10.5.2 Abscheidungspotentiale und Zersetzungsspannung ..................................................... 456 10.5.3 Faraday-Gesetze................................................................................................................. 458 10.6 Großtechnische Anwendungen ....................................................................................... 461 10.6.1 10.6.2 10.6.3 10.7 Chlor-Alkali-Elektrolyse ..................................................................................................... 461 Kupfer-Raffination .............................................................................................................. 463 Aluminium-Darstellung ..................................................................................................... 464 Korrosion ............................................................................................................................ 464 9 CH2-Titelei.indd 9 07.08.14 12:36 Inhaltsverzeichnis 10.8 10.8.1 10.8.2 10.8.3 10.8.4 10.8.5 10.8.6 10.8.7 Elektrochemische Analyseverfahren ............................................................................... 466 Konduktometrie ................................................................................................................. 466 Potentiometrie.................................................................................................................... 467 Amperometrie .................................................................................................................... 470 Voltammetrie ...................................................................................................................... 472 Polarografie ........................................................................................................................ 474 Coulometrie ........................................................................................................................ 474 Elektrogravimetrie ............................................................................................................. 475 Aufgaben zu Kapitel 10 ..................................................................................................... 476 11 Biotechnologie (Lernfelder 14, 17 ,18) ............................................................................477 11.1 Biologische Grundlagen .................................................................................................... 479 11.1.1 Lebewesen und Eigenschaften lebender Systeme ......................................................... 480 11.1.2 Biologische Stoffklassen ................................................................................................... 483 11.1.3 Biologische Grundstrukturen und -funktionen ................................................................ 490 11.1.4 Zellen der Prokaryonten .................................................................................................... 491 11.1.5 Zellen der Eukaryonten ..................................................................................................... 492 11.1.6 Biologische Membranen ................................................................................................... 495 11.1.7 Viren .................................................................................................................................... 496 11.1.8 Gene als Träger der Erbinformationen............................................................................. 497 11.1.9 Proteinbiosynthese ............................................................................................................ 501 11.1.10 Stoffwechsel und Energieumwandlung........................................................................... 504 11.2 Mikrobiologie ..................................................................................................................... 508 11.2.1 Einordnung und Eigenschaften von Mikroorganismen.................................................. 509 11.2.2 Bedeutung der Mikroorganismen für den Menschen..................................................... 512 11.2.3 Wachstum und Vermehrung von Mikroorganismen ...................................................... 514 11.2.4 Wachstumsvoraussetzungen für Mikroorganismen ....................................................... 516 11.3 Mikrobiologische und biotechnische Methoden ............................................................ 520 11.3.1 Steriles Arbeiten und Sicherheitsvorkehrungen ............................................................. 520 11.3.2 Sterilisationstechniken ...................................................................................................... 522 11.3.3 Kultivierung von Mikroorganismen ................................................................................. 525 11.3.4 Keimzahlbestimmungen und Wachstumsmessungen ................................................... 530 11.3.5 Mikroskopische Methoden ................................................................................................ 532 Aufgaben zu Kapitel 11 ..................................................................................................... 537 Anhang ...............................................................................................................................539 A Liste ausgewählter Gefahrstoffe ................................................................................... 539 B RI-Fließbildsymbole nach DIN EN ISO 10628-2:2013-5 und Kennbuchstaben nach DIN 28000-3:2009-12 ........................................................ 542 Sachwortverzeichnis.......................................................................................................... 544 Bildquellenverzeichnis....................................................................................................... 552 Das Periodensystem der Elemente ................................................... Umschlaginnenseiten 10 CH2-Titelei.indd 10 07.08.14 12:36 1.1 Qualität 1 Qualität und Qualitätssysteme Betriebliche Abläufe werden von einzuhaltenden Vorgaben, Qualitätsdenken oder umfassendem Qualitätsmanagement begleitet. Qualität ist heute überall ein wichtiger Punkt, über den immer wieder gesprochen wird. Grundsätzlich wird davon ausgegangen, dass gute Qualität den Erfolg eines Unternehmens bestimmt. Hierbei steht nicht nur die Qualität der Produkte des Unternehmens im Fokus der Qualitätsgedanken, auch betriebsinterne Abläufe können qualitativ gut sein und auf diese Weise einen betriebswirtschaftlichen Vorteil erbringen. In allen diesen Überlegungen ist der Begriff Qualität enthalten. Er besitzt jedoch in jedem Fall eine andere Bedeutung bzw. unterschiedliche Ausprägung. 1 Qualitätsmanagement Totales Qualitätsmanagement Qualitätssicherung Qualitätskontrolle QK QS QM 1900 1930 Qualität durch Prüfen Prozessverbesserung TQM 2000 Zeitachse Prozess- Umfassendes stabilität Qualitätsund Kundenprinzip orientierung Null-FehlerPhilosophie Im Laufe der historischen Entwicklung hat sich das Qualitätsdenken verändert und weiter entwickelt. Angefangen bei der Qualitätskontrolle (QK) bzw. Qualitätsprüfung und schließlich der Qualitätssicherung (QS), existiert heute in Unternehmen meist ein Qualitätsmanagement (QM) oder sogar ein Total Quality Management (TQM), ein umfassendes Qualitätsmanagement (Bild 1). Bild 1: Entwicklungsstufen von der Qualitätskontolle zum Total Quality Management Der Bereich Qualität und Qualitätssysteme ist sehr komplex. Daher erfolgt die Einführung in diese Thematik schrittweise mit internen Verweisen zu weiterführenden Kapiteln, um das Vertiefen der Kapitelinhalte zu erleichtern. Qualität is t, was der Kunde will ! 1.1 Qualität Q u a li t ät beg i n n t i m K op f. Qualität ist, wenn der Kunde zurückkommt und nicht das Produkt. ss for use. Quality is fitne Quality is a never ending story ... f e n h e it Beschaf ie d t ig n u n g , is t h ih r e r E t e Q u a li t ä c li g ü z e h e it b gesetz e in e r E in g t e u n d v o r a u s e n . le e r g e f ü ll fest n is s e z u Erforder Qualität wird in den meisten Fällen unterschiedlich verstanden. Im Alltag sprechen wir von einer guten Qualität, wenn etwas genau die Eigenschaften besitzt, die wir schätzen. Jeder Hersteller wird beispielsweise von der Bild 2: Beschreibungen für Qualität Qualität seines Produkts überzeugt sein. Seine Kunden werden die Qualität gegebenenfalls vollkommen unterschiedlich beurteilen. Deshalb hängt Qualität mit der Sichtweise oder dem Standpunkt des Beurteilenden zusammen (Bild 2). Der Begriff Qualität stammt vom lateinischen Wort qualitas ab und bedeutet Merkmal, Eigenschaft oder Beschaffenheit. Der eigentliche Begriff Qualität gibt die Eigenschaften eines Stoffs, eines Stoffsystems (Stoffgemisch), eines Prozesses oder beispielsweise auch einer Dienstleistung wieder. Qualität ist damit das Gegenstück zur Quantität (lat. quantitas: Größe, Anzahl, Menge). Qualität gibt die Eigenschaften eines Stoffes, Systems, Prozesses oder einer Dienstleistung an. Quantität bezeichnet die Menge oder Anzahl von Stoffen, Objekten oder Vorgängen. 11 CH2-Kap1.indd 11 01.08.14 08:14 1 Qualität und Qualitätssysteme Fehlererfassung und grafische Darstellung OEG OWG UWG UEG Merkmal 1: Merkmal 2: 2 Qualitätsregelkarte Merkmal 3: H (xi) 1 Masse 3 Histogramm 1 Fehlersammelliste Q7 4 Korrelationsdiagramm Qualität Ursache Wirkung 6 Brainstorming 5 Pareto-Diagramm Merkmal 1 7 Ursache-Wirkungs-Diagramm % Merkmal 2 Fehleranalyse und Lösungsfindung Fehler Bild 1: Die sieben Qualitätswerkzeuge als elementare Werkzeuge der Qualität Mit der Fehlersammelliste, der Qualitätsregelkarte und dem Histogramm werden in der Phase der Fehlererfassung die Informationen zu den Fehlerarten, -häufigkeiten sowie -orten gezählt und grafisch dargestellt. Die elementaren Werkzeuge Korrelationsdiagramm, Pareto-Diagramm, Brainstorming sowie Ursache-Wirkungs-Diagramm werden zur Analyse von Fehlern eingesetzt. Außerdem können mit diesen Werkzeugen die Bedeutung einzelner Fehler sowie die Wechselwirkungen von Fehlern untereinander ausgemacht werden. Diese Werkzeuge dienen somit der Lösungsfindung, um zum qualitativ hochwertigen Prozess zurückzukehren. Auch wenn alle Werkzeuge der Q7 aufeinander aufbauen, können sie trotzdem unabhängig voneinander in der Qualitätsarbeit eingesetzt werden. Die Sieben Qualitätswerkzeuge oder Q7 sind elementare Werkzeuge für die Datenerfassung und -analyse sowie für das Auffinden von Fehlern in Prozessabläufen und Lösungsstrategien. Durch die Q7 können Reaktionen auf Abweichungen optimiert und Reaktionszeiten minimiert werden. Die Q7 dienen der Sicherstellung von Qualitätsstandards. 1.6.1 Fehlersammelliste Fehlersammellisten, auch Fehlersammelkarten oder Strichsammellisten genannt, sind ein Qualitätswerkzeug, das über die Art und die Anzahl von Fehlern Auskunft gibt. Mit Fehlersammellisten können Fehler auf einfache Weise erfasst werden. Die Darstellung ist leicht verständlich. Jedes eintretende Ereignis wird durch einen Strich festgehalten Während sich Untersuchungen mit einem Stichprobenumfang von zehn Messwerten wie im Beispiel der Dickenmessung (Tabelle 1, S. 32) leicht überblicken lassen, sind Stichprobenum40 CH2-Kap1.indd 40 01.08.14 08:14 1.6 Q7 – Werkzeuge und Qualität fänge von hundert oder mehr Messwerten zu umfangreich. Hierzu eignet sich eine Fehlersammelliste. Mit ihr können sowohl unterschiedliche Merkmale (Bild 1) als auch Größenmesswerte erfasst werden (Bild 2). Die Fehlersammelliste zur Erfassung von Größenwerten kann direkt angefertigt werden, wenn bekannt ist, welche Werte die kleinsten und die größten Messwerte annehmen. Ohne diese Extremwerte sind die Messwerte in einer Urliste zusammen zu tragen und anschließend der Größe nach zu ordnen sowie zu klassieren. Dies kann dann in eine Strichsammelliste umgesetzt werden (Bild 3). In der Strichsammelliste werden alle Werte vom Minimal- bis zum Maximalwert untereinander gesetzt und die Häufigkeit des Vorkommens der beobachteten Messwerte durch Striche markiert. Diese geordnete Liste enthält alle beobachteten Werte xi mit den zugehörigen absoluten Häufigkeiten H(xi) bzw. den Besetzungszahlen. Eine Fehlersammelliste, Strichsammelliste oder Fehlersammelkarte ist ein Qualitätswerkzeug zur Erfassung und Darstellung von Ereignissen nach Art und Anzahl. Jedes Ereignis wird durch einen Strich festgehalten. Die geordnete Liste gibt ein anschaulicheres Bild der gemessenen Werte als die Urliste und zeigt sogar eine Verteilung der Werte zwischen dem Minimal- und Maximalwert (Bild 3). Die Fehlersammelliste liefert damit bereits Hinweise zu Prozessabläufen und bildet die Basis für weitere Berechnungen und statistische Analysen. Aus den Fehlersammellisten können oft auch schon Rückschlüsse auf mögliche Fehlerursachen gezogen werden, so dass eine Verbesserung des Prozesses erfolgen kann. Fehler Frühschicht S Spätschicht 1 Temperaturfehler 11 Wägefehler 19 Abweichung Wellenlänge 15 Reißfestigkeit 7 Sonstiges 3 Gesamt: 28 27 55 Bild 1: Fehlersammelliste für unterschiedliche Merkmale Absolute Häufigkeit H (x i ) Wellenlänge x i in nm S 253 7 254 12 255 17 256 14 257 12 258 9 259 6 Bild 2: Fehlersammelliste für Größenwerte Klasse von bis Absolute Häufigkeit H (x i ) 0 5 10 15 S 3 1 51,915 51,944 2 51,945 51,974 6 3 51,975 50,004 5 4 52,005 52,034 11 5 52,035 52,064 12 6 52,065 52,094 6 7 52,095 52,124 6 8 52,125 52,155 1 Bild 3: Fehlersammelliste mit klassierten und geordneten Werten aus der Urliste Voraussetzung für die Aufnahme einer Fehlersammelliste im Rahmen der Qualitätsarbeit ist, dass die äußeren Einflussfaktoren konstant sind und damit alle erfassten Fehler den gleichen grundlegenden Bedingungen unterliegen. 1.6.2 Qualitätsregelkarte Die Qualitätsregelkarte bzw. Regelkarte wird im Qualitätsmanagement zur bildlichen Veranschaulichung und Auswertung von Prüfdaten verwendet, die über einen längeren Zeitraum hinweg fortlaufend aufgenommen werden. Sie ist damit eher ein Datenblatt oder ein Schaubild als eine Karte (Bild 1, S. 42). 41 CH2-Kap1.indd 41 01.08.14 08:14 x– + 3j x– + 2j x– + 1j x– obere Eingriffsgrenze obere Warngrenze Warnbereich Kontroll± 2j bereich ± 3j OEG OWG Toleranzbereich 1 Qualitätsregelkarten werden auch häufig bei der Prozessbeobachtung eingesetzt, um einen laufenden Prozess zu überwachen. Unregelmäßigkeiten im Prozessablauf werden hierin schon früh deutlich, so dass zur Vermeidung von Fehlern und Ausschuss entsprechend eingegriffen werden kann. Konzentration 1 Qualität und Qualitätssysteme Mittelwert In der Qualitätsregelkarte ist für jede Stichumfasst umfasst x– – 1j D 95,4 % probeneinheit der gemessene Wert eines D 99,7 % Merkmals eingetragen, so dass sich ein fortUWG x– – 2j untere Warngrenze laufendes x-y-Achsendiagramm ergibt. Zur – UEG x – 3j Orientierung sind statistische Werte wie der untere Eingriffsgrenze Sollwert oder der arithmetische Mittelwert x– als Mittellinie und auch die StandardabweiNummer der Messung oder Zeitpunkt der Messung chungen mit in die Regelkarte eingezeichnet. Die Lage der Messwerte kann somit direkt in Relation zu diesen statistischen Werten und Bild 1: Qualitätsregelkarte den anderen eingetragenen Messwerten beurteilt werden. Im Einsatz sind unterschiedliche Regelkarten, die von den verfügbaren statistischen Größen und die Stichprobengrößen abhängig sind. Allgemein dienen Regelkarten der Analyse von Messwertlagen und dem Erkennen von Prozessabläufen. Eine Qualitätsregelkarte ist eine grafische Darstellung zur Auswertung von Prozessdaten und zur Prozessbeobachtung. Die Regelkarte wird ausgehend von der Mittellinie in Zonen eingeteilt. Der Abstand der Zonen entspricht beispielsweise jeweils dem Abstand einer Standardabweichung j. Die äußerste Grenze sowohl in positiver als auch in negativer Richtung von der Mittellinie stellt die Toleranzgrenze dar, die die noch tolerierte Abweichung darstellt. Der Bereich innerhalb der 3j-Grenzen wird als der natürliche Toleranzbereich bezeichnet. Innerhalb der Toleranzgrenzen sind im Schaubild Warngrenzen (OWG, UWG) und Eingriffsgrenzen (OEG, UEG) eingezeichnet. Die jeweilige positive und negative Grenze liegt im betragsmäßig –. gleichen Abstand oberhalb und unterhalb des Sollwerts oder des arithmetischen Mittelwerts x Jedes Überschreiten dieser Grenzen weist auf Unregelmäßigkeiten oder Fehler hin. Folgt die Verteilung der Messwerte der Gaußschen Normalverteilung (s. Kap. 1.5.3., S. 37), kennzeichnen die Warngrenzen den Bereich von ± 2j um den Mittelwert. In diesem Bereich sollten rund 95,4 % aller gemessenen Werte liegen. Ein Wert jenseits der Warngrenze kann auf Veränderungen des Prozessablaufs hinweisen, die durch geeignete Maßnahmen wieder korrigiert werden können. Ein Eingriff in den Prozess kann notwendig sein. Die beiden Eingriffsgrenzen stellen dementsprechend den ± 3j-Bereich dar, in dem etwa 99,7 % aller Messwerte enthalten sein sollten. In der Regelkarte sind neben dem Sollwert auch die Warngrenzen (OWG, UWG) und die Eingriffsgrenzen (OEG, UEG) eingezeichnet, so dass die Lage der Messwerte zu den Grenzen deutlich wird. Liegt ein Messwert jenseits der Eingriffsgrenze, so kann der Prozess außer Kontrolle sein. Es ist notwendig, in den Prozessablauf einzugreifen, bevor die Toleranzgrenzen überschritten werden. Die in der Regelkarte gesetzten Grenzen beziehen sich auf statistisch berechnete Werte, so dass von der Gesamtheit aller hergestellten Produkte (100 %) außerhalb des 3j-Bereichs (99,7 %) rein rechnerisch insgesamt 100 % – 99,7 % = 0,3 % aller hergestellten Produkte liegen. 42 CH2-Kap1.indd 42 01.08.14 08:14 2 Probenahme, Probenbehandlung und Probenvorbereitung 2 Probenahme, Probenbehandlung und Probenvorbereitung Mit den stetig steigenden Anforderungen an chemische Produkte, gesunde Nahrungsmittel und eine saubere Umwelt geht eine rasante Weiterentwicklung der Instrumentellen Analytik einher. Durch moderne Gerätetechnik werden innerhalb kurzer Zeit genaue Informationen über die Zusammensetzung einer Probe erzielt. Die Herkunft der zu analysierenden Probe und der Zweck der Untersuchung können sehr unterschiedlich sein, wie folgende Beispiele zeigen: 2 • Die Probe kann einem chemischen Produktionsprozess entnommen werden. In diesem Fall dient das vom Labor des Unternehmens erzielte Messergebnis der Steuerung des Prozesses bzw. der Kontrolle des Produkts. • Umweltproben von Oberflächengewässern, Grundwasser, Boden und Luft werden ebenso wie Proben industrieller und kommunaler Abwassereinleitungen, Abfall- und Abluftproben von Anlagen in den Laboratorien der staatlichen Umweltämter untersucht. Die Analysen dienen der Umwelt- und der Anlagenüberwachung. • Proben z. B. von Lebensmitteln, Futtermitteln, kosmetischen Produkten oder auch Lebensmittelverpackungen liegen bei Verdacht auf eine chemische Belastung oder bei routinemäßigen Untersuchungen den Chemischen und Veterinäruntersuchungsämtern zur Analyse vor. Das Ziel besteht darin, die Bürger vor gesundheitlichen Schäden oder Gefahren sowie vor Irreführung und Täuschung zu schützen. Auch Proben der Körper von Nutz- oder Heimtieren werden hier untersucht, um der Ausbreitung von Tierkrankheiten und -seuchen vorzubeugen. Auftraggeber der Untersuchungen sind die Städte und Gemeinden sowie das Land. • Proben der gesamten landwirtschaftlichen Produktionskette werden in den Laboratorien der Landwirtschaftlichen Untersuchungs- und Forschungsanstalten (LUFA) untersucht. Landwirte und produzierende Gärtner, aber auch Privatpersonen reichen Proben von Boden, Saatgut, Pflanzen, Futtermitteln, Wirtschaftsdüngern (z. B. Gülle, Stallmist), Biogas und Wasser (z. B. Trinkwasser, Tränkwasser, Gießwasser) zur Analyse ein. Der Zweck der Untersuchungen besteht in der Qualitätssicherung der landwirtschaftlichen Produktion. Darüber hinaus kann durch eine qualifizierte Düngeempfehlung der Ertrag maximiert werden. Die Aussagekraft einer Analyse wird maßgeblich durch die Qualität der Probenahme bestimmt. Das Ziel der Probenahme besteht darin, einem zu untersuchenden Stoff eine Stichprobe zu entnehmen, die für die Gesamtmenge des Stoffs repräsentativ ist. Die Gesamtmenge, auf die das Messergebnis übertragen wird, wird Grundgesamtheit genannt. Labor Stichprobe Grundgesamtheit Die Probe muss in Bezug auf den zu messenden Parameter repräsentativ sein, nur dann kann das Messergebnis auf die Grundgesamtheit übertragen werden. Bild 1: Grundgesamtheit und Stichprobe Probenehmer müssen über die erforderliche Sachkunde verfügen. Sie werden in regelmäßigen Schulungen über aktuelle Anforderungen sowie neue Vorschriften und Verfahren informiert. Privatpersonen, z. B. Landwirte, müssen bei der Probenahme die detaillierten Anweisungen des beauftragten Labors beachten. Die meisten Laboratorien bieten auch die Probenahme als zusätzliche Dienstleistung an. 57 CH2-Kap2.indd 57 01.08.14 08:35 2 Probenahme, Probenbehandlung und Probenvorbereitung 2.1 Analyse von Proben Unter dem Begriff Analyse wird eine systematische Untersuchung verstanden, bei der die Eigenschaften eines Objekts sowie seine Beziehungen und Wechselwirkungen mit anderen Objekten bestimmt werden. 2 Das Ziel einer chemischen Analyse besteht in der Bestimmung der qualitativen und quantitativen Zusammensetzung eines Stoffs, also seiner chemischen Parameter. Ein Stoff, z. B. eine Bodenprobe, hat jedoch zahlreiche weitere Eigenschaften, die Auskunft über seine Beschaffenheit und seine Wechselwirkungen mit seinem Umfeld geben. Auch seine sensorischen Parameter, wie z. B. der Geruch und die Farbe, seine physikalischen Parameter, wie z. B. seine Temperatur am Ort der Probenahme, und seine biologischen Parameter, wie z. B. seine Besiedelung mit Lebewesen, lassen wertvolle Rückschlüsse auf die Grundgesamtheit, der die Probe entnommen wurde, zu. Alle erzielten Messwerte und Angaben zu den Charakteristika der Probe sollten sich auf die Probe in ihrem ursprünglichen Zustand, also am Ort und zur Zeit der Probenahme beziehen. Die vollständige Analyse einer Probe betrifft nicht nur die Bestimmung der charakteristischen Parameter. Sie umfasst alle Arbeitsschritte von der Probenahme bis hin zur Auswertung, Dokumentation der erzielten Ergebnisse und Qualitätssicherung der Analyse (Bild 1). Zeitnah zur Probenahme werden die Parameter, die sich in Abhängigkeit von der Zeit, der Temperatur oder/und dem Luftdruck stark verändern können, am Ort der Probenahme bestimmt. Beispiele für Messungen vor Ort sind die Bestimmungen von Farbe und Geruch, der Temperatur sowie des pH-Werts der Probe, aber auch die Bestimmung der Umgebungstemperatur, der Windstärke und der Wetterverhältnisse am Ort und zum Zeitpunkt der Probenahme. Probenahme Messung vor Ort Probenahmeprotokoll Probenkonservierung Probentransport Probenlagerung Probenvorbereitung Auswertung, Dokumentation und Qualitätssicherung Kalibrierung und Messung Bild 1: Einzelschritte der Analyse einer Probe Auch zahlreiche, nur mit der modernen Gerätetechnik des Labors bestimmbare Parameter der Probe unterliegen nach der Probennahme Veränderungen. Mithilfe einer auf die wichtigen Parameter abgestimmten Probenkonservierung können mögliche Veränderungen minimiert werden. Anschließend erfolgt der Probentransport ins Labor. Alle Aspekte der Probenahme, der Probenkonservierung und des Probentransports sowie die Ergebnisse der Messungen vor Ort müssen vom Probenehmer im Probenahmeprotokoll sorgfältig dokumentiert werden, damit ihre Einflüsse auf die im Labor gewonnenen Messergebnisse nachvollziehbar sind. Im Anschluss an eine möglichst kurze Probenlagerung im Labor erfolgt die Probenvorbereitung. Nur eine Teilmenge der Probe wird der Analyse unterzogen, der Rest wird im Hinblick auf weitere Untersuchungen gelagert (Rückstellprobe). Damit die Teilmenge in Bezug auf die zu bestimmenden Parameter repräsentativ ist, erfolgt vor der Teilung der Probe ihre Homogenisierung. Feststoffproben müssen vor der Messung zumeist gelöst werden. Ist der zu bestimmende Stoff, der Analyt, schwer löslich, wird er durch einen Aufschluss in eine leicht lösliche Form überführt. Nach der Probenvorbereitung erfolgen die Kalibrierung des Messgeräts, die Messung, die Auswertung der erzielten Messergebnisse, die Dokumentation sowie die Qualitätssicherung der gesamten Analyse. Das Probenahmeprotokoll ist Bestandteil der vollständigen Unterlagen zur Dokumentation der Analyse. 58 CH2-Kap2.indd 58 01.08.14 08:35 2.2 Probenahme 2.2 Probenahme Das Ziel der Probenahme besteht darin, einer großen Menge an Merkmalsträgern einen kleinen Teil zu entnehmen, dessen Eigenschaften repräsentativ für die Gesamtmenge sind. Der kleine, repräsentative Teil der Merkmalsträger, der untersucht wird, wird Stichprobe genannt. Die große Menge an Merkmalsträgern, auf die das Ergebnis der Untersuchung übertragen wird, wird als Grundgesamtheit bezeichnet. 2 Die Grundgesamtheit ist die Menge aller Merkmalsträger, die sich zu einem bestimmten Zeitpunkt oder innerhalb eines Zeitraums an einem bestimmten Ort befinden. Alle Merkmalsträger weisen einen übereinstimmenden Parameter auf. Nicht alle Merkmalsträger werden untersucht, sondern nur wenige, repräsentative Merkmalsträger werden zur Analyse ausgewählt. Diese werden als Stichproben bezeichnet. Bei der Planung der Probenahme müssen zahlreiche Aspekte berücksichtigt werden (Bild 1). Der Zweck bzw. Anlass der Analyse entscheidet über den Ort sowie den Zeitpunkt bzw. Zeitraum der Probenahme. Auf der Basis von Ort und Zeit wird die Grundgesamtheit festgelegt. Die Grundgesamtheit ist ein genau definierter Teil des Stoffes am Probenahmeort für den Zeitpunkt bzw. Zeitraum der Probenahme. Zweck der Analyse Ort der Probenahme Zeitpunkt bzw. Zeitraum der Probenahme Festlegung der Grundgesamtheit Im nächsten Schritt werden die allen Merkmalsträgern der Grundgesamtheit gemeinsamen Eigenschaften festgelegt: die zu bestimmenden Parameter. Aufgrund der Besonderheiten der festgelegten Parameter kann die Größe der Grundgesamtheit präzisiert werden. Die Auswahl des geeigneten Probenahmeverfahrens richtet sich nach der Orts- und Zeitabhängigkeit der zu bestimmenden Parameter. Die erforderlichen Probenahmegeräte werden durch das gewählte Probenahmeverfahren bestimmt. Zu bestimmende Parameter Orts- bzw. Zeitabhängigkeit Probenahmeverfahren Probenmenge Probenahmegeräte für Feststoffe, Flüssigkeiten und Gase Probengefäße für Feststoffe, Flüssigkeiten und Gase Bild 1: Planung einer Probenahme Über die erforderliche Probenmenge entscheiden die geplanten Messungen. Die Größe und das Material der Probengefäße hängen von der Probenmenge und der notwendigen Konservierung der Probe ab. Die Planung einer Probenahme sollte stets sorgfältig durchgeführt werden. Durch Fehler bei der Probenahme oder auch unvollständige Informationen im Probenahmeprotokoll kann das Messergebnis unbrauchbar werden. Eine lückenhafte Planung kann auch die Probenahme selbst behindern, denn der Ort der Probenahme liegt unter Umständen weit vom Labor entfernt. Alle benötigten Probenahmegeräte und Probengefäße müssen zum Ort der Probenahme mitgebracht werden, damit eine sachgerechte Durchführung der Probenahme erfolgen kann. Für die Bestimmung der Parameter im Labor sind die Proben zu konservieren. Chemische Konservierungsmittel müssen bereitgehalten werden, Möglichkeiten zur physikalischen Konservierung durch Kühlung oder Einfrieren sollten im Fahrzeug des Probenehmers mitgeführt werden. Darüber hinaus müssen alle Geräte für die Messungen vor Ort zum Zeitpunkt der Probenahme vorhanden sein. 59 CH2-Kap2.indd 59 01.08.14 08:35 2 Probenahme, Probenbehandlung und Probenvorbereitung 2.2.1 Ort und Zeit der Probenahme, Festlegung der Grundgesamtheit Bei der Festlegung des Probenahmeorts, des Probenahmezeitpunkts bzw. -zeitraums sowie der Grundgesamtheit muss gut überlegt vorgegangen werden. Obwohl sich die beiden folgenden Beispiele wesentlich voneinander unterscheiden, wird in einer ähnlichen Weise verfahren. 2 Beispiel 1: Bodenuntersuchung Der Probenahmeort ist eine landwirtschaftlich genutzte Fläche. Ihr Ertrag soll mithilfe einer Bodenuntersuchung und einer anschließenden Düngeempfehlung optimiert werden. Labor Stichprobe Mischprobe Das beauftragte Labor empfiehlt die Probenahme zum Zeitpunkt von etwa 3 Wochen vor der geplanten Düngung durchzuführen. m 100 Einzelproben m 0,3 m Die Grundgesamtheit sollte auf 1 Hektar 2 (10 000 m ) Ackerboden begrenzt sein. Die Bodentiefe, bis zu der Proben entnommen werden, muss dem Hauptwurzelbereich der angebauten Pflanzen entsprechen. Beim Anbau von Gemüsepflanzen und Getreide wird eine Bodentiefe von 0 cm bis 30 cm empfohlen. 100 Bild 1: Probenahme zur Bodenuntersuchung Eine für die Grundgesamtheit repräsentative Stichprobe wird gewonnen, indem mindestens 20 Probenahmestellen über die Fläche gleichmäßig verteilt werden (Normalverteilung). Alternativ können die Stellen diagonal oder in Zickzack-Form platziert werden. Die 20 an diesen Stellen gewonnenen Einzelproben werden gesammelt und gründlich gemischt. Der Mischprobe wird eine Stichprobe von etwa 500 g entnommen. Diese Laborprobe wird an das Labor geschickt. Die erzielten Messergebnisse können auf die Grundgesamtheit übertragen werden, d. h. auf den 2 Ackerboden der Fläche 10 000 m bis zu einer Tiefe von 30 cm zum Zeitpunkt der Probenahme. Soll eine größere Fläche untersucht werden, ist eine größere Anzahl Analysen erforderlich. Beispiel 2: Überwachung eines chemischen Produktionsprozesses Der Probenahmeort ist eine chemische Anlage, in der ein kontinuierlich geführter Produktionsprozess erfolgt. Das Ziel der Untersuchung besteht darin, Aufschluss über die mittlere Zusammensetzung des Reaktionsgemisches an zwei charakteristischen Stellen der Anlage innerhalb von 1 Stunde zu bekommen. Der Zeitpunkt der Probenahme ist frei wählbar, der Zeitraum ist aufgrund der Aufgabenstellung auf 1 Stunde festgelegt. Behälter: Behälter: Edukt A Edukt B Ventil Probenahmestelle 1 Mischprobe Labor Analyse 1 Einzelproben Mischprobe Reaktor Probenahmestelle 2 Stichprobe Stichprobe Labor Analyse 2 Grundgesamtheit ist das Reaktionsgemisch, Einzelproben Produkt das über den Zeitraum von 1 Stunde hin eine der beiden charakteristischen Stellen der Anla- Bild 2: Probenahme zur Prozessüberwachung ge passiert. In diesem Beispiel werden somit zwei Grundgesamtheiten festgelegt, denen jeweils eine repräsentative Stichprobe entnommen wird. Das Ziel dieser Untersuchung kann nur mit zwei Analysen erreicht werden. Die repräsentativen Stichproben werden gewonnen, indem an den beiden Probenahmestellen innerhalb von 1 Stunde im Abstand von 10 Minuten Einzelproben entnommen werden. Die sechs Einzelproben werden in einem Behälter gesammelt und gemischt. Der Mischprobe wird eine repräsentative Stichprobe für die Untersuchung im Labor entnommen. 60 CH2-Kap2.indd 60 01.08.14 08:35 3 Reaktionen organischer Präparate Aus Calciumacetylid wird durch Reaktion mit Wasser Ethin gewonnen. Bei dieser stark exothermen Reaktion entsteht zusätzlich Calciumhydroxid: CaC2 + 2 H2O r H– C{C– H + Ca(OH)2 Bei dieser Säure-Base-Reaktion überträgt das in diesem Fall als schwache Säure wirksame 2– Wasser zwei Protonen auf die starke Base Acetylid C2 . Es bilden sich die extrem schwache – Säure Ethin C2H2 und die extrem starke Base Hydroxid OH . Weitere Herstellungsmethoden für Ethin sind das Flammen-Verfahren, bei dem man Methangas CH4 mit einer für eine vollständige Verbrennung nicht ausreichenden Menge Luftsauerstoff bei ca. 1 500 °C reagieren lässt: 3 4 CH4 + O2 r C2H2 + 2 CO + 7 H2 Beim Lichtbogen-Verfahren wird Methan mit einer sehr kurzen Verweilzeit in einen 2000 °C heißen Lichtbogen gebracht. Dabei entstehen Ethin und Wasserstoff: 2 CH4 r C2H2 + 3 H2 Ethin (Acetylen) wird durch die Umsetzung von Calciumcarbid mit Wasser, durch das Flammenverfahren und nach dem Lichtbogen-Verfahren gewonnen. 3.2 Reaktionen aromatischer Verbindungen Der Begriff der aromatischen Verbindungen wurde von Chemikern früherer Zeiten geprägt und für Stoffe verwendet, die man in Ölen von Bäumen und Pflanzen fand und die Benzol, heute auch Benzen genannt, als Molekülbaustein enthielten. 3.2.1 Struktur des Benzols und Aromatizität Benzol wurde im Jahr 1825 von Michael Faraday (engl. Chemiker, 1791 bis 1867) entdeckt. Einige Jahre später fand Mitscherlich (deutscher Chemiker, 1794 bis 1863) aufgrund der qualitativen und quantitativen Analyse sowie einer Molmassenbestimmung, dass die Summenformel von Benzol C6H6 lautet. Verschiedene Wissenschaftler entwarfen mögliche Strukturen des Benzols, bis moderne Strukturaufklärungsmethoden zur Kenntnis des tatsächlichen Molekülaufbaus führten (Bild 1). H 120° H 120° 120° H 109 pm H 139 pm H H Bild 1: Geometrischer Molekülbau des Benzols Das Benzolmolekül ist planar, das heißt alle Kohlenstoff- und Wasserstoffatome liegen in einer Ebene und sind durch j-Bindungen miteinander verbunden. Die sechs Kohlenstoffatome bilden ein regelmäßiges Sechseck. Die Bindungswinkel zwischen allen C-Atomen betragen 120°, die zwischen den C– C- und den C – H-Bindungen ebenfalls. Die Bindungslängen der C– C-Bindungen sind alle gleich und messen 139 pm. Aus diesen Messwerten ergibt sich, dass alle sechs Kohlen2 2 stoffatome sp -hybridisiert sind. Jedes Kohlenstoffatom besitzt drei sp -Hybridorbitale, welche 2 mit den beiden sp -Hybridorbitalen der benachbarten C-Atome und mit je einem s-Orbital eines Wasserstoffatoms überlappen. Jedes Kohlenstoffatom besitzt noch ein nicht-hybridisiertes p-Orbital, wobei jedes dieser p-Orbitale mit den beiden p-Orbitalen der benachbarten C-Atome zur p-Bindung überlappt. 94 CH2-Kap3.indd 94 01.08.14 09:04 3.2 Reaktionen aromatischer Verbindungen So entsteht eine geschlossene, ringförmige p-Elektronenwolke, die sich ober- und unterhalb der Molekülebene um das Gerüst aus Kohlenstoffatomen legt (Bild 1). Diese p-Elektronen sind nicht zwischen zwei bestimmten CAtomen zu lokaliseren, das heißt im BenzolMolekül liegt Mesomerie vor (s. S. 90). H H C C H H C C C C H H Deshalb sind die delokalisierten C–C-Doppel- Bild 1: Delokalisierte p-Elektronen beim Benzol bindungen des Benzols kürzer als die lokalisierten Doppelbindungen in einem Alken. Die Mesomerie im Benzol bewirkt sein chemisches Verhalten, das sich signifikant von dem der Alkene unterscheidet. Während Alkene überwiegend Additionsreaktionen zeigen, finden am Benzol vor allem Substitutionsreaktionen statt. 3 (Cyclohexa-1,3,5-trien) H2 H2 H2 H2 H2 H2 H2 H2 Benzol + 3H2 – 209 kJ – 240 kJ + H2 – 360 kJ (berech.) + 2H2 Cyclohexen 151 kJ + 3H2 Cyclohexa-1,4-dien – 120 kJ Enthalpie Die im Sechsring des Benzols ideal vorliegende Delokalisierung der p-Elektronen stabilisiert das Molekül in hohem Maße. Dies zeigen die Hydrierungswärmen von Cyclohexen, Cyclohexa-1,4-dien und Benzol (Bild 2). Im Cyclohexen und im Cyclohexa-1,4-dien liegen lokalisierte Doppelbindungen vor. Die Hydrierungswärme des Cyclohexa-1,4-diens ist doppelt so groß wie die Hydrierungswärme des Cyclohexens. Erwartungsgemäß müsste die frei werdende Enthalpie bei der Hydrierung von Benzol mit drei „Doppelbindungen“ dann dreimal so groß sein wie bei der Hydrierung von Cyclohexen. –1 Tatsächlich werden aber 151 kJ · mol weniger frei als bei dem für das von Kekule (deutscher Chemiker, 1829 bis 1896) postulierte Cyclohexa-1,3,5-trien. H2 H2 H2 H2 H2 H2 H2 H2 H2 H2 Cyclohexan Cyclohexan Cyclohexan Bild 2: Stabilität des Benzols –1 Die Delokalisierung der p-Elektronen bewirkt, dass Benzol 151 kJ · mol weniger Energie enthält und damit um diesen Energiebetrag stabiler ist als das hypothetische Cyclohexa-1,3,5-trien. Benzol ist eine durch Mesomerie stabilisierte Verbindung, bei der alle sechs Kohlenstoff2 atome sp -hybrisiert sind. Mit Benzol lassen sich vor allem Halogenierungen, Nitrierungen, Sulfonierungen, Acylierungen und Alkylierungen als Substitutionsreaktionen durchführen. Benzol ist der bekannteste Vertreter der aromatischen Verbindungen. Anfänglich wurde Aromatizität nach dem Geruch beurteilt. Nach der Hückel-Regel (deutscher Physiker, 1896 bis 1980) müssen für eine aromatische Verbindung folgende eindeutige Kriterien erfüllt sein: • Es muss ein planares, cyclisches System (Ringsystem) vorliegen. 2 • Alle Kohlenstoffatome des Moleküls müssen sp -hybridisiert sein. • Das Ringsystem muss (4n + 2) p-Elektronen enthalten. Dabei steht n für eine natürliche Zahl (0, 1, 2, 3, 4 …). Anders ausgedrückt: Das Ringsystem muss eine ungerade Anzahl von p-Elektronenpaaren besitzen. + Beispiele für aromatische Stoffe sind außer Benzol das Cyclopropenkation C3H3 (2 p-Elektronen), das Pyridin C5H5N (6 p-Elektronen) oder das Naphthalin C10H8 (10 p-Elektronen). Eine Verbindung ist dann aromatisch, wenn sie cyclisch-planar gebaut ist und aus einem konjugierten p-Elektronensystem besteht, welches der Hückel-Regel folgt. 95 CH2-Kap3.indd 95 01.08.14 09:04 3 Reaktionen organischer Präparate 3.2.2 Elektrophile aromatische Substitution und Folgereaktionen 3 Die p-Elektronenwolke ober- und unterhalb der Ringebene führt zu einem Angriff bzw. zu einer Reaktion mit einem Elektrophil. Dabei bildet sich eine carbokationische Zwischenstufe, die nicht isoliert werden kann. Ihre positive Ladung wird durch die Mesomerie des Ringes, auch Kern genannt, relativ stabilisiert. Das Carbokation hat zwei Möglichkeiten, sich zu einem Produkt zu stabilisieren: Entweder das zum Elektrophil zugehörige Nucleophil – N addiert sich an das zum Elektrophil benachbarte C-Atom (elektrophile Addition) oder das Proton vom Kohlenstoffatom des elektrophilen Angriffs wird abgespalten und verbindet sich mit dem Nucleophil (Bild 1). nicht aromatisches N Additionsprodukt b) + + E+ H a) E carbokationische Zwischenstufe E b) – N a) E + HN aromatisches Substitutionsprodukt Bild 1: Schema der elektrophilen aromatischen Substitution (N –: Nucleophil, E +: Electrophil) Im zweiten Fall entsteht wieder ein stabileres, aromatisches System, deshalb läuft diese Reaktion als (elektrophile) Substitution ab (Bild 2). Der erste Reaktionsschritt verläuft relativ langsam. Er ist endergonisch, da das stabile Benzol in ein instabileres Zwischenprodukt umgewandelt wird. Während der zweite Schritt rasch und exergonisch verläuft, da am Ende der stabile aromatische Zustand zurückgebildet wird. freie Enthalpie Additions- N produkt Folgende fünf Reaktionen gehören zu den häufigsten elektrophilen aromatischen Substitutionen (abgekürzt S E): • Halogenierung • Nitrierung • Sulfonierung • Acylierung • Alkylierung. + H E + – N E Substitutionsprodukt E + HN +E N Reaktionsverlauf Bild 2: Energiediagramm der elektrophilen aromatischen Substitution Allen Reaktionen ist der zweischrittige Mechanismus gemeinsam (Bild 3). + + + E langsam E E H E + schnell H H E + HB+ + B Bild 3: Reaktionsmechanismus der elektrophilen aromatischen Substitution + Das Benzol reagiert mit dem Elektrophil E unter Bildung einer carbokationischen Zwischenstufe, deren positive Ladung delokalisiert ist. Diese Delokalisierung wird durch die drei mesomeren Grenzstrukturen veranschaulicht. + Das Proton H des Kohlenstoffatoms, an dem das Elektrophil E gebunden ist, verbindet sich mit + einer Base B in der Reaktionsmischung zu HB . Das Elektronenpaar, welches zur Bindung des Protons diente, wandert in den Ring und stellt den aromatischen Zustand mit 6 p-Elektronen wieder her. Für die direkte Halogenierung des Benzols ist wegen seiner geringeren Reaktivität im Vergleich zu einem Alken ein Katalysator erforderlich. Dieser Katalysator muss eine Lewis-Säure sein, also ein Stoff, dessen Moleküle mindestens eine Elektronenpaarlücke besitzen. Überträgt ein Chloroder Brom-Molekül eines der freien Elektronenpaare auf diese Elektronenpaarlücke, so entsteht + + ein elektrophiles, positiv geladenes, instabiles Halogenkation (Cl bzw. Br ). Den Ablauf zeigt Bild 1, S. 97, am Beispiel der Bromierung mit Eisen(III)-bromid als Katalysator. 96 CH2-Kap3.indd 96 01.08.14 09:04 3.2 Reaktionen aromatischer Verbindungen Zuerst muss sich der p-Komplex bilden, bei dem eine lockere Wechselwirkung zwischen dem Benzol und dem positivierten Brom-Atom besteht. Der j-Komplex als Zwischenstufe bildet sich durch Bindungstrennung und -bildung am p-Komplex. Durch Abspaltung des Protons am Substitutions-Kohlenstoffatom stabilisiert sich das Ringsystem durch Rückkehr in den aromatischen Zustand. Die Chlorierung des Benzols verläuft analog zur Bromierung, nur dass an Stelle des Eisen(III)-bromids Eisen(III)chlorid als Katalysator benutzt wird. Da beide Katalysatoren hygroskopisch sind, was leicht zu ihrer Inaktivierung führt, gibt man Eisenspäne zur Reaktionsmischung. Diese reagieren mit dem jeweiligen Edukt Brom oder Chlor zu dem benötigten Eisen(III)-halogenid. d+ d– Br [Br ü-Komplex FeBr3] H H H + H Br H H H j-Komplex + Br H + Br + HBr + FeBr3 – Br + [FeBr4] j-Komplex 3 Rückführung in den aromatischen Zustand Bild 1: Bromierung von Benzol Die Halogene Chlor und Brom bilden mit Benzol in Anwesenheit einer Lewis-Säure (meistens AlX3 mit X = Cl, Br) als Katalysator Halogenbenzole (Halogenaryle). Die Nitrierung des Benzols erfolgt mit Salpetersäure wenn überhaupt, dann nur unter Bildung + unerwünschter Nebenprodukte. Salpetersäure enthält nicht das benötigte Elektrophil NO2 . Durch Zugabe von konzentrierter Schwefelsäure entsteht H die sogenannte Nitriersäure, – – + NO2 + H2O + HSO4 HO NO2 + H OSO3H HO+ NO2 + HSO4 ein Gemisch aus Salpeter- und SalpeterSchwefelNitroSchwefelsäure im Stoffmensäure säure niumion – + H HSO4 genverhältnis von etwa 1:1. In NO2 diesem Gemisch bilden sich NO2 + – + + + H2SO4 NO HSO + 2 4 die Nitronium-Ionen NO2 nach folgendem Reaktionsschema (Bild 2): Die Schwefelsäure Bild 2: Nitrierung von Benzol protoniert die Salpetersäure, + wobei Wasser abgespalten wird. Das elektrophile Nitronium-Ion NO2 greift dann das Benzol an. Nach dem im Bild 3, S. 96 dargestellten Reaktionsmechanismus wird Nitrobenzol gebildet. Die Aminogruppe – NH2 lässt sich nur durch Reduktion der Nitrogruppe erhalten, da Ammoniak NH3 als nucleophile Verbindung nicht direkt elektrophil am Benzol substituiert werden kann. Die Reduktion wird mit einem unedlen Metall wie Zink oder Eisen mit einer starken Säure (z. B. Salzsäure) durchgeführt. Der bei diesen Reaktionen kurzfristig entstehende atomare Wasserstoff [H] reduziert die Nitro-Gruppe. Die Reduktion mit Wasserstoff und Platin-Katalysator ist eine andere Möglichkeit, um Aminobenzol herzustellen (Bild 3). NO2 + 6 [H] Nitrobenzol NH2 Zn, HCl + 2 H2O NO2 + 3 H2 NH2 Pt + 2 H2O Anilin Bild 3: Reduktion der Nitro-Gruppe Die Nitrierung ist eine wichtige elektrophile Substitutionsreaktion am Aromaten. Für präparative Zwecke wird die Nitriersäure verwendet, ein Gemisch von konzentrierter Salpetersäure und konzentrierter Schwefelsäure. Eine Aminogruppe am Benzol wird durch die Reduktion der eingeführten Nitrogruppe erreicht. 97 CH2-Kap3.indd 97 01.08.14 09:04 5.1 Gaschromatografie GC 5 Chromatografische Trenntechniken Chromatografische Trennmethoden beruhen auf dem Stoffaustausch zwischen einer stationären und einer mobilen Phase. Die Geschwindigkeit des Transports der an der stationären Phase anhaftenden Probenbestandteile erfolgt unter dem Zwang der mobilen Phase. Man kann deshalb die chromatografischen Methoden nach dem Aggregatzustand der mobilen Phase einteilen in Gas- und Flüssigchromatografie. 5.1 Gaschromatografie GC Das Einsatzgebiet der Gaschromatografie, wie sie in diesem Kapitel dargestellt wird, umfasst die Trennung permanent gasförmiger oder unzersetzt verdampfbarer Stoffgemische im Temperaturbereich von 20 °C bis 300 °C. Daneben gibt es noch die Hochtemperatur-Gaschromatografie HTGC mit einem Temperaturbereich von 300 °C bis ca. 500 °C. Der Konzentrationsbereich ist vom eingesetzten Detektor abhängig und umfasst Hauptbestandteile bis hin zum Ultraspurenbereich (< 1 ppm). Feste Proben können mit der Pyrolyse-Gaschromatografie durch gezielte thermische Zersetzung und Messung der Zersetzungsprodukte bei noch höheren Temperaturen untersucht werden. Gasregler 5 Gas filter Datensystem A/DWandler Säulenofen Bild 1: Prinzipskizze eines Gaschromatografen 5.1.1 Trägergase Als Trägergase finden vor allem Stickstoff, Helium und Wasserstoff Verwendung. Bild 2 zeigt drei Chromatogramme eines Stoffgemisches mit neun Komponenten, die auf der gleichen Säule getrennt wurden. Der einzige Unterschied besteht darin, dass jeweils ein anderes Trägergas mit seiner optimalen mittleren Gasgeschwindigkeit verwendet wurde. Dabei wird deutlich, dass bei Verwendung von Wasserstoff als Trägergas die Analysenzeit erheblich verkürzt ist und die Peakbreiten schmaler sind als beim Stickstoff. Das Helium nimmt hierbei eine Zwischenstellung ein. Allerdings erfordert das Trägergas Wasserstoff erhebliche Sicherheitsvorkehrungen am Gerät und am Arbeitsplatz. Ein weiterer Nachteil: Wasserstoff darf nicht bei Trennungen verwendet werden, bei denen die Möglichkeit von chemischen Reaktionen des Wasserstoffs mit Probenbestandteilen, zum Beispiel einer Addition, besteht. Detektor Injektor Trägergas Bild 1 zeigt den prinzipiellen Aufbau eines Gaschromatografen. Das Trägergas strömt aus der Druckgasflasche durch Reinigungskartuschen (Gasfilter) zur Entfernung von Sauerstoff- und Wasserdampfspuren über einen Gasregler in das Einlass- oder Injektionssystem. Im Injektor findet die Verdampfung der Probe statt. Daran schließt sich unmittelbar die Trennsäule an, die sich in einem thermostatisierbaren Ofen befindet. Am Ende der Trennsäule befindet sich außerhalb des Ofenraums der Detektor. Die analogen Signale des Detektors wandelt ein A/D-Wandler in digitale Signale um, die von einem Datensystem weiterverarbeitet werden. 2 1 6 3 4 Wasserstoff bei 38 cm · s –1 5 7 89 2 1 3 Benzol Toluol Ethenylcyclohexan Ethylbenzol m-Xylol Ethenylbenzol Isopropylbenzol Benzaldehyd n-Propylbenzol 6 4 5 1 Stickstoff bei 12 cm · s –1 0 1. 2. 3. 4. 5. 6. 7. 8. 9. 7 Helium bei 25 cm · s –1 8 9 2 3 6 4 7 5 5 10 8 9 15 Zeit in min Bild 2: Analysezeiten und Trägergase 211 CH2-Kap5.indd 211 01.08.14 08:18 5 Chromatografische Trenntechniken Ein wichtiger Parameter für die gaschromatografische Trennung ist der Volumenstrom (Flussrate) F des Trägergases. F V F = __ t mL · s –1 V t mL s Gl. 5.1 –1 Diese physikalische Größe ist wegen der üblichen Flussraten von weniger als 1 mL · min nicht so leicht messbar. Leichter lässt sich die mittlere lineare Trägergasgeschwindigkeit u– ermitteln. Dazu wird eine Substanz injiziert, von der man weiß, dass sie von der stationären Phase nicht zurückgehalten, retardiert, wird. Aus der Durchflusszeit tM, die diese Substanz braucht, um vom Injektor zum Detektor zu gelangen, und der Säulenlänge ö kann die mittlere lineare Trägergasgeschwindigkeit errechnet werden. u– ö u– = ___ tM –1 cm · s ö tM cm s Gl. 5.2 Mit Wasserstoff erhält man die schnellsten Trennungen, weil seine optimale mittlere Trägergas–1 geschwindigkeit mit 38 cm · s am größten ist. Bei der Ermittlung der optimalen mittleren Trägergasgeschwindigkeit wird bei verschiedenen mittleren Trägergasgeschwindigkeiten jeweils eine Testsubstanz (im Bild 1 Hexadecan) in die gleiche Säule bei gleicher Temperatur injiziert. Testsubstanz: Hexadecan 1,0 H in mm 5 Die Trägergasgeschwindigkeit beeinflusst eine chromatografische Trennung. Die Trennfähigkeit einer Säule ist durch ihre theoretische Bodenzahl N bzw. ihre äquivalente Bodenhöhe H charakterisiert. In der gleichen Säule ist bei gleicher Temperatur die äquivalente Bodenhöhe H von der Trägergasgeschwindigkeit u– abhängig (Bild 1). N2 He 0,5 H2 Für jedes Trägergas entsteht nach dem Auftragen der Trägergasgeschwindigkeit gegen die aus dem Chromatogramm errechnete Bodenhöhe eine H-u–-Kurve mit einem mehr (Stickstoff) oder weniger (Wasserstoff) ausgeprägten Minimum. 12 cm · s–1 25 cm · s–1 0 0 38 cm · s–1 60 20 40 – Mittlere lineare Geschwindigkeit u in cm · s–1 80 Bild 1: Einfluss der Trägergasart auf die H-u–-Kurve Van Deemter hat die dazu gehörende mathematische Funktion entwickelt, die die Kurvenverläufe in Bild 1 beschreibt. An diesem jeweiligen Minimum der Bodenhöhe kann die optimale mittlere Trägergasgeschwin–1 digkeit für das jeweilige Trägergas abgelesen werden. Sie beträgt für Stickstoff 12 cm · s , –1 –1 für Helium 25 cm · s und für Wasserstoff 38 cm · s . Wobei Wasserstoff ein langgezogenes Minimum besitzt, was einem großen Geschwindigkeitsbereich mit optimalen Trenneigenschaften entspricht. Das bedeutet, dass mit Wasserstoff nicht nur die kürzesten Analysezeiten erreicht werden, sondern auch gute Trennungen, was die schmalen Peaks im Bild 1 auf S. 211 belegen. 212 CH2-Kap5.indd 212 01.08.14 08:18 6.3 Spektrenarten 6.3 Spektrenarten Bei dem Emissionsspektrum einer Glühlampe (Bild 1, S. 251) handelt es sich um ein kontinuierliches Spektrum. Kontinuierliche Spektren zeichnen sich durch eine ununterbrochene Folge sämtlicher Wellenlängen aus. Sie werden von festen und flüssigen Körpern oberhalb von 0 K emittiert. Auch stark verdichtete Gase emittieren bei entsprechender Anregung kontinuierliche Spektren. In der Instrumentellen Analytik dienen Strahler, die ein kontinuierliches Spektrum aussenden, vor allem als Strahlungsquellen. Dies kann ein glühender Wolframdraht sowie hochverdichtetes, angeregtes Deuterium in einem UV/Vis-Spektrometer oder ein Globar, ein Stab aus Siliciumcarbid, in einem IR-Spektrometer sein. Ein kontinuierliches Spektrum umfasst in ununterbrochener Folge von Wellenlängen. Jede Absorption und Emission ist bei einem Molekül mit einer Teilchenbewegung (Schwingung, Rotation) verbunden. Eine Molekülschwingung um einen bestimmten Auslenkungsbetrag kann von vielen Molekülrotationen überlagert sein. elektromagnetische Strahlung Anregung Rotation Schwingung Lösemittelmolekül Lösemittelmolekül mit übertragener Energie angeregtes Molekül deaktiviertes Molekül Bild 1: Strahlungslose Deaktivierung Für jede Bewegungsart existieren eigene Energieübergänge. Die Überlagerung der Schwingungsvorgänge und Rotationen hat jedoch zur Folge, dass keine einzelnen Spektrallinien beobachtet werden. Ein Kollektiv von Molekülen emittiert in Form von Bandenspektren. Typische Bandenspektren findet man in der UV/Vis-Spektroskopie und der IR-Spektroskopie. Bild 1 zeigt die strahlungslose Deaktivierung eines Moleküls. Es absorbiert elektromagnetische Strahlung und fängt an zu schwingen und zu rotieren. Die absorbierte Energie wird durch Stöße an Teilchen in der Umgebung (z. B. Lösemittelmoleküle) wieder abgeben. Eine Lösung erwärmt sich und emittiert Wärmestrahlung, die zu einer Verbreiterung der Banden im Bandenspektrum von Molekülen führt. 6 Elektron Moleküle und mehratomige Gase emittieren elektromagnetische Strahlung in Form von Bandenspektren. Ein Atom emittiert elektromagnetische Strahlung einer bestimmten Wellenlänge, wenn ein Elektron von einer Schale mit einer höheren Energie auf eine Schale mit einer geringeren Energie springt (Bild 2). Zur Absorption kommt es, wenn ein Elektron von einer niedrigeren auf eine höhere Bahn gelangt. Angeregte Atome können nicht strahlungslos deaktiviert werden. Sie absorbieren und emittieren üblicherweise in Form von Linienspektren (Bild 1, S. 260). Diese entstehen im einfachsten Fall durch Elektronensprünge zwischen den einzelnen Schalen mit der Hauptquantenzahl n. Daneben gibt es Feinstrukturspektren, die die anderen Quantenzahlen (Nebenquantenzahl ö, Magnetquantenzahl m und Spinquantenzahl s) berücksichtigen. Nur Atome, Atomionen der Edelgase und Metalldämpfe absorbieren und emittieren elektromagnetische Strahlung in Form von Linienspektren. Atomkern – e p+ a) K L M e – Schale Energie + p b) K L M Licht e– p+ c) K L M Bild 2: a) Grundzustand des Wasserstoffs, b) Anregung durch Absorption, c) Emission von Licht 253 CH2-Kap6.indd 253 01.08.14 09:36 6 Spektroskopie 6.4 Aufbau von Spektralapparaten Die in der Instrumentellen Analytik eingesetzten Spektralapparate bzw. Spektrometer arbeiten nach den unterschiedlichsten Prinzipien. Dennoch haben sie einiges gemein. So befindet sich in allen Fällen die Probe im Strahlengang. Auch verfügt jedes Spektrometer über eine Quelle, die elektromagnetische Strahlung oder Elektronen emittiert. Bild 1 zeigt den allgemeinen Aufbau von Spektrometern mit den sechs wichtigsten Funktionsteilen. Quelle Probe im Strahlengang Wolframdraht, Xe-Lampe, D2-Lampe (UV/Vis) Globar (IR) Glühemissionsdraht (Elektronen für MS) Radiosender (NMR) Probenzuführung Signalauftrennung Detektion Auswertung Monochromator (UV/Vis) Fotodioden (UV/Vis) X,Y- Schreiber, Plotter magnetisches/ elektrisches Feld (MS) Empfängerspule (NMR) Computer Spule (NMR) Sekundärelektronenvervielfacher (MS) Küvette (UV/Vis) Zerstäuber (AAS, ICP-OES) Monochromator, Interferometer (IR) Schubstange (MS) Bild 1: Aufbau von Spektrometern 6 Die unterhalb der Kästen aufgeführten Beispiele stellen nur einen kleinen Ausschnitt dar. Bei der Probenzuführung gibt es auch die Möglichkeit, den Ausgang einer Chromatografiesäule mit einem Spektrometer zu koppeln. Die Signalauftrennung kann auch im Durchlicht oder seitlich erfolgen wie z. B. bei der Fluoreszenz-Spektrometrie. 6.4.1 Signalauftrennung Bei den verschiedenen in der Instrumentellen Analytik verwendeten Spektrometern erfolgt die Signalauftrennung, die auch Signalsortierung oder Signalanalyse genannt wird, nach unterschiedlichen Methoden. Bei Fourier-Transform-Spektrometern geschieht dies erst in einem Computer, nachdem das sehr komplexe Signal vom Detektor empfangen wurde. Ein UV/Vis-Spektrometer hingegen verfügt über einen Monochromator, mit dem polychromatisches Licht zerlegt bzw. analysiert wird. Bild 2 zeigt das Grundprinzip eines aus mehreren Bauteilen bestehenden Monochromators zur UV/Vis-Spektroskopie. polychromatisches Licht Eingangsspalt Kollimator Dispersionselement („Zerlegung“) Fokussierelement Ausgangsspalt monochromatisches Licht Bild 2: Grundaufbau eines Monochromators zur UV/Vis-Spektroskopie Heute werden hauptsächlich Gittermonochromatoren verwendet. Zunächst passiert das ankommende Licht einen Eingangsspalt, wodurch ein strichförmiges Lichtband entsteht. Der Kollimator hat die Aufgabe, das Licht parallel auszurichten. Dies geschieht mithilfe einer Linse oder eines gewölbten Spiegels. An dieser Stelle ist das Licht immer noch polychromatisch. Seine Aufspaltung in alle im Spektrum enthaltenen Wellenlängen („Regenbogenfarben“) erfolgt mithilfe einer Dispergiereinrichtung. Hierfür wird ein Prisma oder ein Gitter verwendet. 254 CH2-Kap6.indd 254 01.08.14 09:36 6.4 Aufbau von Spektralapparaten Damit monochromatisches LichtLicht resultiert, ist die Disperquelle giereinrichtung drehbar gelagert. Wird das Prisma oder Gitter so ausgerichtet, dass nur eine bestimmte Wellenlänge, z. B. l = 550,2 nm, den b Kollimator Ausgangsspalt trifft, kann nur EingangsPrisma diese Wellenlänge den Mospalt (drehbar) nochromator verlassen (l1 in Bild 1 und Bild 2). Alle anderen Wellenlängenbereiche werden Bild 1: Prismenmonochromator nach Bunsen ausgeblendet. Damit der Ausgangsspalt punktgenau getroffen wird, enthält Kollimatorspiegel Gehäuse der Monochromator eine Fokussiereinrichtung. Dies kann eine Linse oder ein Hohlspiegel sein. Die Funktion des Prismas beruht auf der Wellenlängenabhängigkeit der Lichtbrechung. So besitzt dunkelrotes Licht (l = 760,8 nm) bei Flintglas den Brechungsindex n = 1,603 und violettes Licht (l = 396,8 nm) den Brechungsindex n = 1,645. Im Ergebnis wird kurzwelliges Licht, z. B. Blau, stärker gebrochen als langwelliges Licht, z. B. Rot. Weil die Beugung (Dispersion) in Kombination mit einer Überlagerung (Interferenz) von der Wellenlänge l abhängt, kommt es bei einem Gitter zur Zerlegung von polychromatischem Licht. Bild 3 zeigt, dass rotes Licht (Bild 3a) stärker gebeugt wird als blaues Licht (Bild 3b). Wird anstelle von monochromatischem Licht polychromatisches Licht eingestrahlt, entsteht eine Reihe von Spektren (Bild 3c). Diese werden ausgehend vom Weißlichtbereich (0. Ordnung) nach links und rechts hin durchnummeriert (1. Ordnung, 2. Ordnung usw.). Je höher die Ordnung (m) ist, desto stärker aufgelöst sind die Farbbänder. Die Spektren höherer Ordnung sind breiter als die Spektren niedriger Ordnung. Ausgangsspalt l2 l1 Fokussierlinse Eingangsspalt Lichtquelle weißes Licht Konkavspiegel Gitter l2 l1 Fokussierspiegel Ausgangsspalt Bild 2: Gittermonochromator nach Czerny-Turner 6 Ordnung a) Linien 4. 3. 2. 1. 0. 1. 2. 3. 4. Ordnung b) Linien 4. 3. 2. 1. 0. 1. 2. 3. 4. Ordnung c) Spektren 4. 3. 2. 1. 0. 1. 2. 3. 4. Bild 3: Beugung des Lichts bei Einsatz eines Gittermonochromators Aus praktischen Gründen werden in der Instrumentellen Analytik meist Spektren 2. Ordnung verwendet. Beim Einsatz eines Prismas wird blaues Licht stärker abgelenkt als rotes Licht. Beim Einsatz eines Gitters wird rotes Licht stärker gebeugt als blaues Licht. Ein wesentliches Kriterium zur Beurteilung eines Monochromators ist die Auflösung R. In der Spektroskopie ist die Auflösung R definiert als das Verhältnis der Wellenlänge l zum zugehörigen kleinsten auflösbaren Wellenlängenabstand dl. l R = ___ dl R l dl 1 m m Gl. 6.7 255 CH2-Kap6.indd 255 01.08.14 09:36 7 Strukturaufklärung organischer Verbindungen 7.3.3 Absorptionsspektroskopie an Molekülen Die UV/Vis-Spektroskopie begleitet die Entwicklung von Farbstoffen und optischen Filtern. Farbstoffe Die zur Absorption im UV- und Vis-Bereich fähigen Atomgruppierungen im Molekül werden als Chromophore bezeichnet. Die wichtigsten farbgebenden Gruppen enthalten Doppelbindungen, z. B. –C=C– in ungesättigten Kohlenwasserstoffen, –N=N– in Azofarbstoffen und –N=O in Nitroverbindungen. antibindend p* H C H H C j Bild 1: C= C-Doppelbindung in Ethen Die Elektronen in Einfach- oder j-Bindungen werden durch gewöhnliches UV-Licht nicht angeregt. Im Vakuum-UV-Bereich (VUV: 40 bis 190 nm) hingegen stört die Absorption der Umgebungsluft, so dass dieser Spektralbereich in der Praxis nicht genutzt wird. bindend prp* bindend j r j* Bild 2: Elektronenübergänge Tabelle 1: Elektronenübergänge Übergang l in nm r r p* r j* r j* > 250 n p n j p* Chromophore > 200 C=O, C=N, C=S, N=O C=C, C=O, C=N f 200 – O – , – S – , >N –, –Halogen < 200 C – C, C – H Tabelle 2: Verschiebung der Absorption in Richtung kurzer Wellen zu langen Wellen hin Blauverschiebung Rotverschiebung Hypsochromie Bathochromie Tabelle 3: Verstärkende Gruppen Auxochrome – O – , NH2, OR, OH, Br, Cl, CH3, F Antiauxochrome NO2, CHO, COCH3, COOH, CN, COO–, SO2NH2 , NH3+ schwach R r stark Tabelle 4: Aufbau von Farbstoffen Chromophor Licht regt Elektronen in Mehrfachbindungen (p r p*) und nichtbindende Elektronen (n r p*) an. n r j* n r p* p j-Bindung Die intensivsten Spektralbanden beruhen auf den wahrscheinlichsten Elektronenübergängen zwischen Orbitalen, die eine möglichst geringe Anregungsenergie benötigen. Das sind z. B.: • p-Elektronenwolken in C=C-Bindungen und • nichtbindende Elektronen in den freien Elektronenpaaren des Sauerstoffs in C = O-Bindungen (Tabelle 1). nicht bindend n H Die Molekülorbitaltheorie (MO-Theorie, s. Band 1, S. 467) beschreibt die C=C-Bindung in Bild 1 als eine Wolke von p-Elektronen, die sich oberhalb und unterhalb der C–C-Einfachoder j-Bindung ausbreitet. Diese p-Elektronen werden durch die Energie des UV-Lichtes in unbesetzte p*-Orbitale höherer Energie angeregt (Bild 2). 7 antibindend j* p-Bindung Auxochrom – CH3 Antiauxochrom – SO2NH2 – OCH3 – COO – > C=S – NHCH3 > C=N– – N(CH3)2 – CoN – CO – OCH3 Benzol > C=O In Farbstoffen schieben ausgewählte Substitu– N=N– – Br – NO2 enten die Absorption des chromophoren Systems in den langwelligen Bereich, d. h. bathochrom in Richtung Rot (Tabelle 2). Verstärkende Gruppen erhöhen die Intensität der Absorption. Technische Farbstoffe kombinieren drei Effekte: • Chromophore, z. B. konjugierte Doppelbindungen und kondensierte Aromaten, verschieben die Absorption bathochrom und erhöhen die Intensität (Hyperchromie). • Elektronenschiebende Gruppen mit freien Elektronenpaaren (Substituenten 1. Ordnung) heißen Auxochrome (Tabellen 3 und 4). • Elektronenziehende Gruppen mit einem Elektronenmangel (Substituenten 2. Ordnung) heißen Antiauxochrome. 284 CH2-Kap7.indd 284 01.08.14 10:19 7.3 UV/Vis-Spektroskopie Spektreninterpretation UV/Vis-Spektren zeigen nicht schmale Linien, wie die Emissionsspektren der Atome, sondern breite Absorptionsbanden (Bild 1), weil elektronische Zustände in sehr viele Schwingungszustände untergliedert sind – das sind die waagrechten Linien mit den strahlungslosen Übergängen im Jablonski -Diagramm (Bild 3, S. 281). p r p* O Extinktionskoeffizient e 10000 n r p* und p r p*-Übergänge sind im Spektrum unterscheidbar, wenn die Prüfsubstanz in Lösemitteln unterschiedlicher Polarität gemessen wird, zum Beispiel in Cyclohexan und in Ethanol. Die Solvatochromie, d. h. die Verschiebung der UV-Banden in polaren Lösemitteln, wirkt sich aus bei: • p r p*-Übergängen zu niedrigeren Wellenlängen (Rotverschiebung), • n r p*-Übergängen zu höheren Wellenlängen (Blauverschiebung). 5000 in Cyclohexan (unpolar) 1000 in Ethanol (polar) 500 n r p* 100 hypsochrom bathochrom 220 240 260 280 300 320 340 360 380 Wellenlänge l in nm Bild 1: UV/Vis-Spektrum von Benzophenon Die Wellenlängen der Bandenmaxima sind wenig stoffspezifisch (Tabelle 1). Die Struktur einer Verbindung lässt sich anhand des UV-Spektrums daher nicht eindeutig bestimmen. Tabelle 1: Absorptionsmaxima chromophorer Gruppen: Wellenlänge l in nm ^ 200 ; 210 ; 220 ; 230 ; 240 ; 250 ; 300 6 200 H2O 167 CH3OH 183 Et-OEt 189 Furan 200 p Ph – OH 211pW Ph – OCH3 217pW – – Ph – O– Ph 255W 252E – – – – – – – – Ph – Hal f 210pW – – – CH3I 258 – – – CH3– S – CH3 228 CH3SH 235 – Et – S – S– Et 250 – – Benzol 180, 204W, 255 Ph– CH3 207E Naphthalin 220E – Ph – Ph 246E Anthracen 256cH – – Ethen 165 RC=CR185 – – – PhCH=CH2 244E – PhCH=CHPh 297E – HC o CR f 173 – – – Ph – CoCH 236W – – – C=O p r p* n r p* CH3COCH3 187p, (273 n) CH3COCH3 200 Gas CH3CONH2 220W Ph – COOH 230W Ph – CHO 242H Ph – COCH3 243E CH3CHO (293 n) – C =S n r p* – Ph – SO3H f 213pE – – – – – CH3CS– CH3 (460) C=N p r p* CH3 C o N < 190 Pyrrol 211 Ph – C o N 224W Ph – NH2 230pW – Pyridin 257pE – N=O p r p* RCH=NOH 190 CH3NO2 210 (271) – – – Ph – NO2 252 R3C– NO3 f 300n Nitrophenol f 318E N =N n r p* – – – – – – Ph – NO2 269W R – N=N – R f 350 Gruppe –O– n r j* =N – n r j* NH3 Et2NH 194 EtNH2 193 Et3N – Hal n r j* CH3Cl CH3Br 173 204 –S– n r j* C=C p r p* 210 213 CHI3 7 349 Et = C2H5, Ph = C6H5, W in Wasser, E in Ethanol, H in Hexan, cH in Cyclohexan, p r p* bzw. n r p*-Übergänge 285 CH2-Kap7.indd 285 01.08.14 10:19 7 Strukturaufklärung organischer Verbindungen 7.3.4 Anwendungen der UV/Vis-Spektroskopie Die quantitative Analyse farbiger oder im Ultraviolett absorbierender Substanzen mithilfe der UV/Vis-Spektroskopie wird als fotometrische Analyse oder Spektralfotometrie bezeichnet. Farbmessungen an organischen Molekülen Die in einem Lösemittel gelöste Probe befindet sich in einer Glasküvette und wird von einer Lichtquelle bestrahlt (Bild 1a). Das prozentuale Verhältnis der durchgelassenen Intensität F zur eingestrahlten Lichtintensität F0 heißt Transmission t oder Transmissionsgrad. Der von der Probe absorbierte Anteil ist der Absorptionsgrad 1 – t. Streuung und Reflexion des auftreffenden Lichts durch Küvette und Probe werden vernachlässigt. Küvette mit absorbierender Probe Ä0(l) Intensität Ä Als Maß für die Lichtschwächung dient die Extinktion A, der negative Logarithmus der Transmission. Das alte Formelzeichen E für die Extinktion darf nicht mehr verwendet werden. Der Vorteil der Extinktion gegenüber der Transmission ist, dass die Auftragung gegen die Konzentration eine Gerade liefert. Die Lichtschwächung durch die Probe bei einer bestimmten Wellenlänge l ist nach dem Bouguer-Lambert-Beer-Gesetz der Konzentration des absorbierenden Stoffes und der Länge des Lichtwegs durch die Küvette proportional. Ä(l) d a) Der molare Extinktionskoeffizient e der Prüfsubstanz ergibt sich aus der Höhe des Absorptionspeaks abzüglich der Basislinie. Für Konzentrationsbestimmungen anhand einer Kalibrierkurve muss e nicht bekannt sein. Statt der Stoffmengenkonzentration dürfen auch Massenkonzentrationen, Massenanteile oder Gasdrücke auf der x-Achse aufgetragen werden (Bild 1c). Amax – Abasis e(l) = ____________ c·d b f·p m = r_____ · w = _____ c = ___ = ______ M R·T M V·M c1 A1 – a __ = ______ c2 A2 – a Gl. 7.8 a b M m V r w f p R Achsenabschnitt der Kalibriergeraden –1 Massenkonzentration der Lösung (g · L ) molare Masse des absorbierenden Stoffes (g · mol –1) Einwaage des absorbierenden Stoffes (g) Volumen der Lösung (L) Dichte der Lösung, # ; 1 000 g · L–1 Massenanteil der Substanz in der Lösung (%) Volumenanteil des Gases in der Mischung (L · L–1 = 1) Druck in der Gasküvette in Pa bei der Temperatur (K) molare Gaskonstante: R = 8,3144 J · mol–1 · K–1 w= 1% 80 % Transmission T Extinktion, dekadisches Absorptionsmaß Wellenlänge des einstrahlenden Lichtes (nm) 2 –1 molarer Extinktionskoeffizient (dm · mol ) Stoffmengenkonzentration des absorbierenden Stoffes (mol · L–1) Schichtdicke der Küvette, meist d = 1 cm = 0,1 dm Lichtintensität (Strahlungsleistung) vor der Küvette (W) Lichtintensität (Strahlungsleistung) hinter der Küvette (W) Transmission, Transmissionsgrad Länge ö 5% 60 % 20 % 40 % 20 % 0% 400 nm 600 nm Wellenlänge l in nm b) 0,6 A = 0,0175 w + 0,0825 Steigung 0,5 Achsenabschnitt 0,4 0,3 unbekannt 7 A l e c d F0 F t Gl. 7.7 100 % Extinktion A A(l) = e · c · d = – lg t F t = ___ F0 0,2 0,1 0 c) 5 10 15 20 35 Massenanteil w in % Bild 1: a) Messaufbau, b) gemessene Transmission, c) Kalibrierkurve 286 CH2-Kap7.indd 286 01.08.14 10:19 10.1 Grundbegriffe 10 Elektrochemie und Elektrotechnik Die beiden Themengebiete Elektrochemie und Elektrotechnik beschäftigen sich mit der Anwendung elektrischer Energie. Elektrische Energie ist die Energie, die über elektrische Ladung übertragen oder in elektrischen Feldern gespeichert wird. In ihren Grundlagen gehen Elektrochemie und Elektrotechnik auf eine gleiche Basis zurück und werden daher in diesem Kapitel gemeinsam behandelt. Die Elektrotechnik ist ein Bereich der Technik, der sich mit technischen Entwicklungen und Geräten befasst, die elektrische Energie nutzen. Hierzu gehört neben elektrischen Maschinen sowie Schaltungen für die Mess-, Steuer- und Regelungstechnik auch die Computertechnik. Physikalische Chemie Elektroorganische Synthesen Elektroanorganische Verfahren Elektroanalytik Organische Chemie Anorganische Chemie Elektrotechnische Prozesse Technische Chemie Die Elektrochemie hingegen umfasst mit den elektrischen Grundlagen und Verfahren mehre- Bild 1: Anwendungsbereiche der Elektrochemie re Teilgebiete der Chemie (Bild 1). Elektrolyse und Elektrosynthese gehören in den Bereich der Physikalischen, Organischen bzw. Anorganischen Chemie. Andere elektrochemische Methoden finden sich in der Laboranalytik und somit im Teilgebiet Analytische Chemie. Großtechnische Prozesse, Batterietechnik oder Brennstoffzellentechnik hingegen werden der Technischen Chemie zugerechnet. Unabhängig von der Zuordnung zu den verschiedenen Bereichen der Elektrochemie wird von einem elektrochemischen Vorgang gesprochen, wenn an den auftretenden Stoffänderungen elektrischer Strom beteiligt ist. Hierbei ist es möglich, dass eine chemische Reaktion durch eine von außen angelegte Spannung erzwungen oder eine elektrische Spannung durch eine chemische Reaktion erzeugt wird. Eine Spannung kann leicht aufgezeichnet und analysiert werden, so dass elektrochemische Verfahren sowohl in der Analytik als auch in der In-Prozess-Kontrolle (s. Kap. 8, S. 366) eingesetzt werden. Der Vorteil dieser Verfahren besteht häufig in einer zerstörungsfreien und kontinuierlichen Messung. Die Elektrotechnik umfasst technische Schaltungen und Geräte, die elektrische Energie nutzen. Die Elektrochemie umfasst chemische Umsetzungen unter Verwendung von elektrischer Energie. 10.1 Grundbegriffe Die elektrischen Größen Strom Ü, Spannung U und Widerstand R sind Basisgrößen, durch die sich viele Grundlagen klären lassen, die für analytische Messungen genutzt werden. Die Voraussetzung für einen Strom ist allerdings das Vorhandensein von elektrischer Ladung. 10.1.1 Elektrische Ladung Q Die elektrische Ladung Q (engl. charge) ist eine grundlegende Größe. Das Formelzeichen Q leitet sich ab vom lateinischen Wort quantum (lat. quantum: Menge). Die Einheit der elektrischen Ladung ist das Coulomb mit dem Einheitenzeichen C. Diese Einheit wurde nach dem französischen Physiker Charles Augustin de Coulomb (1736 bis 1806) benannt. Sie leitet sich direkt von den SI-Basiseinheiten (franz. système international d‘unités, SI, internationales Einheitensystem) Ampere A und Sekunde s ab: 1C=1A·s Q Ü t C A s 10 Gl. 10.1 413 CH2-Kap10.indd 413 01.08.14 11:21 10 Elektrochemie und Elektrotechnik Die elektrische Ladung Q für einen zeitlich konstanten Strom ergibt sich aus der Stromstärke Ü, also der Stärke eines elektrischen Stroms, multipliziert mit der Zeit: Q=Ü·t Q Ü t C A s Gl. 10.2 Als Einheit wird häufig die Einheit Amperestunde Ah verwendet: 3 600 C = 3 600 As = 1 Ah Gl. 10.3 Die elektrische Ladung Q kann vom Menschen nicht über ein Sinnesorgan wahrgenommen werden. Lediglich die Phänomene, die durch elektrische Ladung hervorgerufen werden, können beobachtet oder erfahren werden. Wird beispielsweise ein Kunststofflineal an Kleidungsstücken (elektrischer Isolator) gerieben, können anschließend mit dem Lineal kleine Papierstücke angezogen werden (Bild 1). Fließt elektrische Ladung durch den menschlichen Körper und kommt es durch den Ladungsfluss zum Ladungsausgleich, wird dies durch einen elektrischen Schlag spürbar. Auch Gewitterblitze und Donner sind ein Phänomen, die auf einen schlagartigen Ladungsausgleich elektrischer Ladungen zurückzuführen sind. Der Amerikaner Benjamin Franklin (1706 bis 1790) führte elektrische Phänomene auf das Verschieben von Ladung zurück. Nach Franklin existiert nur eine Ladungsart, die ihren Aufenthaltsort ändern kann. Bei viel vorhandener Ladung kommt es zu einer negativen Aufladung, wofür er den Begriff minus (–) verwendete. Für den anderen Fall benutzte Franklin den Begriff plus (+). Bild 1: Beispiel für die Wirkung elektrischer Ladung Bürette mit Wasser gefüllt Kunststofflineal abgelenkter Wasserstrahl Bild 2: Ablenkung eines feinen Wasserstrahls durch ein polarisiertes Kunststofflineal a) + – b) – – Bild 3: Verhalten von Ladungen mit a) ungleicher Polarität Bei einer Aufladung werden Elektronen verschoben. Es kommt und b) mit gleicher Polazu einer Polarisierung (Ausbildung von verschieden geladenen rität Polen). Eine Anhäufung von Elektronen führt zu einem Elektronenüberschuss und somit zu einer negativen Ladung. Ein Elektronenmangel stellt eine positive Ladung dar. Zwischen unterschiedlich geladenen Bereichen treten Kräfte auf, die durch gegenseitige Anziehung beobachtet werden können (Bild 3a). Bei gleicher Aufladung stoßen sich die Teilchen gegenseitig ab (Bild 3b). Anwendungsbeispiel: Wird mit einem trockenen Baumwolltuch in eine Richtung über ein Kunststofflineal gerieben, kommt es zur Verschiebung von Ladungen. Mit diesem elektrostatisch aufgeladenen Lineal kann ein feiner Wasserstrahl abgelenkt werden, da die Dipolmoleküle des Wassers angezogen werden (Bild 2). 10 Die Verschiebung von elektrischer Ladung führt zu einer Polarisierung. Bei Elektronenüberschuss liegt eine negative elektrostatische Ladung vor: Minuspol. Bei Elektronenmangel liegt eine positive elektrostatische Ladung vor: Pluspol. Zwischen ungleich elektrisch geladenen Bereichen kommt es zum Ladungsfluss bis zum vollständigen Ladungsausgleich. Gleichnamig geladene Teilchen stoßen sich gegenseitig ab. Ungleichnamig geladene Teilchen ziehen sich gegenseitig an. 414 CH2-Kap10.indd 414 01.08.14 11:21 10.1 Grundbegriffe – Das Elektron e ist ein Elementarteilchen mit einem negativen elektrischen Ladungswert Qe– von –19 1,6 · 10 C. Da sich die elektrischen Ladungen der beiden Elementarteilchen Elektron und Proton gegeneinander ausgleichen, muss der Betrag der elektrischen Ladung eines Protons Qp+ gleich – dem Betrag der elektrischen Ladung des Elektrons e sein: |Qe–| = |Qp+| Qe– Qp+ C C Gl. 10.4 + Die beiden Ladungen Qe– und Qp+ unterscheiden sich nur in ihrem Vorzeichen. Das Proton p –19 besitzt daher ebenfalls eine elektrische Ladung Qp+ mit dem Wert von 1,6 · 10 C. Der Wert der elektrischen Ladung wird als Elementarladung e bezeichnet. Elektronen und Protonen besitzen als Elementarteilchen vom Betrag her eine gleich große elektrische Ladung. Diese Ladung wird als Elementarladung e bezeichnet und besitzt einen –19 Wert von e ; 1,602 · 10 C. Eine elektrische Ladung Q ist ein Vielfaches der Elementarladung e. Es gilt: e 1 C = 1 A · s = 6,242 · 1018 · e C Gl. 10.5 Beispiel 10.1: Berechnung einer elektrischen Ladung Q Mit einem Wolltuch wird ein Glasstab abgerieben. Der Glasstab weist danach einen Mangel 5 an 57,6 · 10 Elektronen auf. Wie groß ist die elektrische Ladung? Lösung: Nach Gleichung 10.5 folgt: 18 6,242 · 10 · e v 1 C 5 5 –13 1C · 57,6 · 10 ; 9 · 10 C 57,6 · 10 · e v ___________ 18 6,242 · 10 –13 Die Ladung Q beträgt 9 · 10 C. Da ein Elektronenmangel vorliegt, ist der Stab positiv geladen. Liegt zwischen zwei Bereichen ein Unterschied in der elektrischen Ladung vor, besteht zwischen diesen beiden Bereichen eine elektrische Spannung. Der Ausgleich dieses Ladungsunterschieds ist mit dem Fluss von elektrischer Ladung von einem zum anderen Bereich verbunden. Dieser Ladungsfluss in eine Richtung wird als elektrischer Strom bezeichnet. 10.1.2 Elektrische Spannung U Besteht zwischen zwei Bereichen ein Unterschied in ihrer elektrischen Ladung, liegt zwischen den beiden Bereichen eine elektrische Spannung U (engl. voltage) vor. Sie wird durch eine Anziehungskraft deutlich (Bild 1). Anziehungskräfte + F1 F2 – F1 = F2 Zwischen zwei elektrisch unterschiedlich geladenen Polen besteht eine elektrische Spannung U. Sie entsteht durch die Trennung dieser Ladungen. Bild 1: Kraft zwischen unterschiedlichen elektrischen Ladungen 10 Bildlich lässt sich eine Spannung durch einen Wasserstrom verdeutlichen (Bild 1, S. 416). Fließt Wasser aus einem Reservoir durch einen angeschlossenen Schlauch nach draußen, wird der Wasserstrom durch die Höhe des Wasserstands erzeugt. Das Anheben des Wasserreservoirs erzeugt einen höheren Wasserdruck und somit einen höheren Wasserstrom. Der höhere Wasserdruck entspricht einer höheren Spannung. 415 CH2-Kap10.indd 415 01.08.14 11:21 10.3 Elektrochemische Vorgänge 10.3.1 Daniell-Element Das Daniell-Element, auch als Daniellsches Element bezeichnet, wurde nach dem britischen Chemiker John Frederic Daniell (1790 bis 1845) entwickelt. Eine Zink-Elektrode taucht in einem Behälter aus porösem Material in eine Zinksulfat-Lösung ein. Dieser Behälter steht in einer Kupfersulfat-Lösung mit einer zylinderförmigen Kupfer-Elektrode (Bild 1). Beide Lösungen im Daniell-Element besitzen –1 eine Konzentration von jeweils 1 mol · L . Durch die poröse Trennwand, das Diaphragma, wird eine direkte Vermischung der beiden Lösungen vermieden. Der Austausch von Ladungsträgern ist möglich. Daniell stellte bei seinem Aufbau fest, dass zwischen den beiden leitenden Drähten dieses Elements eine Spannung von rund 1,1 V messbar ist. Aufgrund des hohen Innenwiderstands durch die poröse Trennwand von etwa 10 Q konnte jedoch nur ein Strom von ungefähr 100 mA erreicht werden. Verschiedene Diaphragma-Materialien und alternative Aufbauten für das galvanische Element wurden untersucht. Das Daniell-Element lässt sich auch wie in Bild 2 darstellen. Die beiden Salzlösungen befinden sich in getrennten Behältern, die über ein U-Rohr als Salzleiter oder Salzbrücke verbunden sind. Diese galvanische Zelle besteht aus zwei Halbzellen, einer Zink-Halbzelle und einer Kupfer-Halbzelle. Zink-Elektrode in Zinksulfat-Lösung Kupfer-Elektrode in KupfersulfatLösung Ton-Behälter Bild 1: Aufbau eines Daniell-Elements elektrischer Leiter V e– e– Salzbrücke Zn Cu e– e– Zn2+ SO42– Cl– Cl– e– e– K+ Cu2+ Zn2+ 1 M ZnSO4-Lösung Zn K+ Zn2+ + 2 e– Cu2+ SO42– 1 M CuSO4-Lösung Cu2+ + 2 e– Cu Bild 2: Aufbau eines Daniell-Elements aus zwei räumlich getrennten Halbzellen In einem galvanischen Element müssen die räumlich getrennten Halbzellen über eine Salzbrücke oder ein Diaphragma miteinander verbunden sein, um eine Ionenleitung zu gewährleisten. Zink ist unedler als Kupfer. Daher gehen von der Zink-Elektrode mehr Zink-Ionen in Lösung, als von der Kupfer-Elektrode Kupfer-Ionen in Lösung gehen. Beim Lösungsvorgang gehen Metall-Ionen in die Lösung über und Elektronen bleiben im Metall zurück. Die Metall-Atome werden oxidiert. Wie in Bild 2 dargestellt, wird die Zink-Elektrode durch die zurückbleibenden Elektronen negativ geladen. Die Kupfer-Elektrode trägt nicht so viele Elektronen und ist somit der positiv geladene –1 Pol. Zwischen den beiden Polen existiert bei Lösungen der Konzentration 1 mol · L eine elektrische Spannung von 1,10 V (unter Standardbedingungen). Die überschüssigen Elektronen an der Zink-Elektrode wandern über den elektrischen Leiter zur Kupfer-Elektrode. Hier können die Kupfer-Ionen der Lösung diese Elektronen aufnehmen. Die Kupfer-Ionen werden zu Kupfer-Atomen reduziert, so dass sich an der Elektrode elementares Kupfer anlagert. Da die Kupfersulfat-Lösung an positiv geladenen Kupfer-Ionen verarmt und im Gegenzug die Zinksulfat-Lösung an positiv geladenen Zink-Ionen reicher wird, muss ein Ladungsausgleich zwischen den beiden Halbzellen stattfinden. Dieser Ausgleich geschieht über das Diaphragma bzw. über die Salzbrücke (Bild 1 und 2). Negativ geladene Ionen wandern durch Diffusion von der Kupfer-Halbzelle zur Zink-Halbzelle. Der Stromkreis ist damit geschlossen. Beim Daniell-Element geht das unedlere Zink in Lösung. Das Zink wird oxidiert. Die Zink-Elektrode trägt daher die Bezeichnung Anode. Durch die zurückbleibenden Elektronen ist die Zink-Elektrode negativ geladen. Da diese Halbzelle Elektronen zur Verfügung stellt, ist sie die Donatorhalbzelle. 10 439 CH2-Kap10.indd 439 01.08.14 11:21 10 Elektrochemie und Elektrotechnik In der Kupfer-Halbzelle werden von den KupferIonen Elektronen aufgenommen. Hier findet die Reduktion statt. Die Elektrode wird als Kathode bezeichnet. Die Halbzelle ist die Akzeptorhalbzelle. V e– Salzbrücke Zn Die Elektrode, an der die Oxidation erfolgt, ist die Anode. Die Halbzelle heißt Donatorhalbzelle. Die Elektrode, an der die Reduktion erfolgt, ist die Kathode. Die Halbzelle heißt Akzeptorhalbzelle. e– Cl– Cl– Cl– Cl– Cl– Cl– Zn2+ Cl– Zn2+ Zn2+ K+ Zn2+ Cl– Cl– Cl– Zn2+ K+ SO42– K+ K+ SO42– K+ SO42– Cu K+ K+ K+ SO42– K+ K+ SO42– Bild 1: Daniell-Element nach längerer Standzeit Im Verlauf der Reaktion nimmt die Kupfer-Elektrode aufgrund der sich anlagernden KupferAtome an Größe und Masse zu, während sich die Zink-Elektrode mit der Zeit immer weiter auflöst (Bild 1). Bei den chemischen Vorgängen handelt es sich um eine Redoxreaktion, wobei die Reduktions- und die Oxidationsreaktion räumlich getrennt voneinander ablaufen. An der Zink-Elektrode findet mit der Oxidation die Umsetzung von Zink2+ 2+ Atomen Zn zu Zink-Kationen Zn statt. In der Zink-Halbzelle liegt das Redox-Paar Zn/Zn vor. Die Elektronen wandern von einer Donatorhalbzelle über den elektrischen Leiter zur Akzeptorhalbzelle, so dass an der Kupfer-Elektrode mit der Aufnahme von Elektronen die Reduktion 2+ der Kupfer-Anionen Cu zu elementarem Kupfer Cu erfolgt. In der Kupfer-Halbzelle liegt das 2+ Redox-Paar Cu /Cu vor. Reduktionsreaktion und Oxidationsreaktion der elektrochemischen Redoxreaktion laufen voneinander räumlich getrennt jeweils in einer der beiden Halbzellen ab. z+ vor. In jeder Halbzelle liegt ein Redox-Paar M/M Die beiden im Daniell-Element ablaufenden Reaktionen können getrennt voneinander aufgestellt werden. Die Summe der beiden miteinander verbundenen Teilvorgänge ergibt die Gesamtreaktion: Anode Kathode Zn Cu2+ + 2 e– S Zn + Cu2+ r r r Zn2+ + 2 e– Cu Oxidationsreaktion Reduktionsreaktion Zn2+ + Cu Reduktions-Oxidations-Reaktion (Redoxreaktion) Das Daniell-Element lässt sich kurzgefasst in einem Zellendiagramm darstellen: Zn Zn2+ (1 mol · L–1) Cu2+ (1 mol · L–1) Cu oder Zn Zn2+ (1 mol · L–1) Cu2+ (1 mol · L–1) Cu Das Zellendiagramm gibt der Reihe nach den Aufbau des galvanischen Elements an. Eine Phasengrenzfläche Elektrode – Lösung wird im Zellendiagramm durch einen senkrechten Strich dargestellt ( ). Zwei Doppelstriche ( ) oder eine gestrichelte Linie ( ) kennzeichnen die HalbzellenGrenze. 10 Für den Fall des Daniell-Elements liegt eine Elektrode aus Zink Zn in einer Elektrolytlösung mit 2+ –1 2+ Zn -Kationen der Konzentration 1 mol · L vor. Die Zn -Ionen-haltige Lösung ist durch eine 2+ Trennwand von der Cu -Ionen-haltigen Elektrolytlösung separiert. In die Elektrolytlösung mit 2+ –1 Cu -Ionen der Konzentration 1 mol · L taucht eine Elektrode aus Kupfer Cu. Ein Zellendiagramm gibt unter Berücksichtigung von Phasengrenzflächen und HalbzellenGrenzen den Aufbau eines galvanischen Elements wieder. Das Zellendiagramm selbst verdeutlicht nur den Aufbau eines Elements. Es hält nicht fest, welche Reaktionen in dem Element ablaufen oder welche Vorgänge sich genau an den Phasengrenzflächen bzw. an den Elektroden abspielen. 440 CH2-Kap10.indd 440 01.08.14 11:21 10.3 Elektrochemische Vorgänge 10.3.2 Elektrodenvorgänge Die Doppelschicht ist auf der Seite der Elektrode deutlich dünner als auf der Seite des Elektrolyten (Bild 1). Im Elektronenleiter ist die Schichtdicke nur von der Dichte der Ladungen abhängig, die im Fall von Metallen zwischen den praktisch nicht beweglichen Atomrümpfen vorhanden sind. Auf der Seite des Elektrolyts wird die elektrisch geladene Schicht u. a. von der Ionen-Konzentration im Elektrolyt, der Ionen-Wertigkeit, ihrer Solvatation und der Beweglichkeit der Ionen bestimmt. Betrachtet man die Doppelschicht im Detail, wird auf Seiten des Elektrolyten deutlich, dass diese Schicht komplex aufgebaut ist. Die ablaufenden Vorgänge werden bei der detaillierten Betrachtung deutlich. + + – Metallelektrode negativ geladen Die Phasengrenze Elektrode-Elektrolyt ist die Grenze zwischen einem Elektronenleiter (Elektrode) und einem Ionenleiter (Elektrolyt). An dieser Phasengrenze stehen sich zwei elektrisch geladene Schichten mit entgegengesetztem Vorzeichen gegenüber. Daher wird die Phasengrenze auch als elektrochemische oder elektrolytische Doppelschicht, kurz als Doppelschicht, oder auch als Dipolschicht bezeichnet. Elektrolyt Elektrode – + – – + Wassermoleküle + – + – – – + – + – – hydratisierte Ionen + – – – – + – – + + + – – Anion + Kation – Anion mit Hydrathülle + Kation mit Hydrathülle Bild 1: Ausbildung einer Doppelschicht zwischen Elektrode und Elektrolyt Metallelektrode negativ geladen Stern-Doppelschicht Helmholtz-Schicht freie Elektrolytlösung – + – – Konzentration c des Reaktanten Die Kenntnis über die genauen Vorgänge an den Elektroden und damit an den Phasengrenzflächen gibt die Möglichkeit, elektrochemische Abläufe genauer zu steuern und zu beeinflussen. + Nernstsche Diffusionsschicht – – + – – + – – starre Schicht Elektrode + diffuse Schicht Abstand r von der Elektrode Elektrolyt Bild 2: Aufbau einer Doppelschicht zwischen Elektrode und Elektrolyt Der wichtigste Vorgang zwischen Elektrode und Elektrolyt ist die Durchtrittsreaktion, d. h. der Übergang von Ladungsträgern durch die Phasengrenze. Hierbei kommt es entweder zur Anlagerung eines Teilchens (Adsorption) an der Elektrode oder zur Abgabe eines Teilchens in die Elektrolytlösung verbunden mit einer Solvatation. Dieser Stoffumsatz führt zu Konzentrationsänderungen, denen das System durch Teilchenbewegungen entgegenwirkt. Hierzu müssen Teilchen durch die Lösung in Richtung der Phasengrenzfläche gelangen oder entsprechend entgegengesetzt wandern. Nach dem deutschen Physiker Hermann Ludwig Ferdinand von Helmholtz (1821 bis 1894) besteht die Schicht auf der Elektrolytseite aus einer inneren Helmholtz-Schicht und einer äußeren Helmholtz-Schicht (Bild 2). Die innere Helmholtz-Schicht besteht aus ausgerichteten Ionen direkt an der Elektrodenoberfläche. Diese sind teilweise desolvatisiert und schon an die Oberfläche der Elektrode adsorbiert. Die äußere Helmholtz-Schicht wird aus solvatisierten Ionen der Lösung gebildet. 10 Der deutsche Physiker Otto Stern (1888 bis 1969) brachte verschiedene Modellvorstellungen zusammen. Er konstatierte, dass sich die Doppelschicht aus einer starren Schicht und weiter in Richtung Elektrolytlösung aus der diffusen Schicht zusammensetzt. Diese Kombination wird auch als Stern-Doppelschicht bezeichnet. Die diffuse Schicht trägt nach dem deutschen Physikochemiker Walther Hermann Nernst (1864 bis 1941) auch den Namen Nernstsche Diffusionsschicht. Durch diese Diffusionsschicht können sich Teilchen nur über Diffusion bewegen. Die Schichtdicke der Diffusionsschicht hängt von der Elektrolytkonzentration und der angelegten Spannung ab. 441 CH2-Kap10.indd 441 01.08.14 11:21 11.1 Biologische Grundlagen 11.1.2 Biologische Stoffklassen Die gesamte organische Substanz der Lebewesen wird als Biomasse bezeichnet. Biomasse besteht aus wenigen typischen Substanzklassen. Die in Lebewesen vorkommenden organischen Verbindungen können in hochmolekulare und niedermolekulare Stoffe eingeteilt werden. Die hochmolekularen organischen Substanzen oder Makromoleküle sind zumeist Polymere, die aus gleichen oder gleichartigen Bausteinen, den so genannten Monomeren, bestehen. Die biologischen Polymere sind Proteine, Nukleinsäuren, Polysaccharide und Lipide. Sie kommen zusammen mit ihren Monomeren und weiteren niedermolekularen Stoffwechselprodukten in fast jeder Zelle vor. Proteine und Aminosäuren Proteine oder Eiweiße, die es in großer Vielfalt gibt, sind in lebenden Systemen unentbehrlich. Sie sind alle aus gleichartigen Monomeren, den Aminosäuren, aufgebaut. Sämtliche Aminosäuren bestehen aus Kohlenstoff-, Wasserstoff-, Sauerstoff- und Stickstoffatomen; zwei Aminosäuren enthalten außerdem Schwefel. Bild 1 zeigt den allgemeinen Aufbau einer Aminosäure, die am a-C-Atom eine Aminogruppe und einen Carboxylgruppe trägt. Das aC-Atom ist das C-Atom, das direkt benachbart zum C-Atom der Carboxylgruppe steht. Das aC-Atom besitzt neben einem H-Atom eine Restgruppe R, die bei verschiedenen Aminosäuren unterschiedlich ist. Da alle Aminosäuren bis auf Glycin, die als Rest ein weiteres Wasserstoffatom trägt, vier verschiedene Substituenten am a-C-Atom besitzen, gibt es zwei spiegelbildliche Isomere, die L- und die D-Aminosäuren (Bild 1). Proteine enthalten ausschließlich L-Aminosäuren. Aminogruppe H3+ N Carboxylgruppe CH COO R a – C – Atom Aminosäurerest COO – COO Von Proteinen spricht man, wenn insgesamt mehr als 50 Aminosäuren hintereinander verknüpft sind. Die in Proteinen zu findenden Aminosäuren können gewöhnlich 20 verschiedene Reste enthalten. Somit existieren 20 biologische Aminosäuren Tabelle 1, S. 484), die normalerweise in allen Proteinen in einer charakteristischen Reihenfolge vorhanden sind. – C C H3N + R R NH3 + H H L-Aminosäure Die Aminosäuren sind in den Proteinen über Peptidbindungen verknüpft. Zwischen der Aminogruppe einer Aminosäure und der Carboxylgruppe einer anderen Aminosäure entsteht formal durch Wasserabspaltung und Bildung einer Peptidbindung ein Dipeptid (Bild 2). Peptide und Proteine besitzen immer unterschiedliche Enden, nämlich eine Aminogruppe an einem Ende (N-Ende) und eine Carboxylgruppe am anderen (C-Ende). – D-Aminosäure Bild 1: Aufbau von Aminosäuren H H R N C C H O H OH H R N C C H O OH H2O N-Ende H H R N C H C O H R N C H C O OH C-Ende Bild 2: Peptidbindung zwischen zwei Aminosäuren Proteine sind lange Ketten aus L-Aminosäuren, die über Peptidbindungen miteinander verknüpft sind. 11 483 CH2-Kap11.indd 483 01.08.14 11:22 11 Biotechnologie Die Reste der Aminosäuren können hydrophobe Eigenschaften besitzen, wie z. B. die Aminosäuren mit aromatischen Seitenketten. Andere Aminosäuren haben hydrophile Reste, die Wasserstoffbrücken ausbilden können. Die hydrophilen Aminosäurereste können weiter in ungeladene Reste mit Hydroxylgruppen und in geladene Reste unterteilt werden. Letztere haben entweder eine weitere freie Aminogruppe oder eine Carboxylgruppe. Die Aminosäuren Cystein und Methionin enthalten als einzige Schwefel. Prolin besitzt im Gegensatz zu allen anderen Aminosäuren infolge einer Ringbildung unter Beteiligung der a-Aminogruppe keine freie Aminogruppe. Die Eigenschaften der Proteine werden durch die Eigenschaften der vorhandenen Aminosäurereste bestimmt. Beispielsweise verleiht eine Überzahl an Carboxylgruppen in den Resten dem Protein eine negative Nettoladung im neutralen pHWertbereich einer Zelle. Tabelle 1: In Proteinen vorkommende essentielle Aminosäuren Aminosäure Abkürzung Glycin Alanin Valin Leucin Isoleucin Gly, G Ala, A Val, V Leu, L Ile, I Restgruppe H CH3 CH (CH3)2 CH2 CH (CH3)2 CH CH2 CH3 Hydrophobe Reste CH3 Prolin Methionin Pro, P CH2 Met, M CH2 CH2 Phenylalanin Phe, F CH2 Tyrosin Tyr, Y CH2 Tryptophan Trp, W CH2 CH2 CH2 S CH3 OH Aromatische Reste C HC N H Serin Ser, S CH2 OH OH Threonin Thr, T CH Asparagin Asn, N Glutamin Gln, Q Cystein Cys, C CH2 Lysin Arginin Lys, K Arg, R (CH2)4 (CH2)3 Histidin Asparaginsäure His, H Asp, D CH3 O CH2 C NH2 O (CH2)2 C NH2 SH NH3+ NH C CH2 C +HN CH Hydrophile ungeladene Reste NH2 C H CH NH NH2+ Basische Reste COOH 2 Alle Proteine haben aufgrund Saure Reste (CH2)2 COOH Glutaminsäure Glu, E der Wechselwirkungen ihrer Aminosäuren eine charakteristische räumliche Struktur, die Konformation genannt wird. Der räumliche Aufbau eines Proteins ist für seine Funktion in der Zelle von entscheidender Bedeutung. Strukturell lassen sich vier Betrachtungsebenen unterscheiden (Bild 1, S. 485): • Die Primärstruktur eines Proteins ist die Abfolge oder Sequenz der einzelnen Aminosäuren in der Polypeptidkette (z. B. Ala – Gly – Gln – Trp – Phe –). Die Aminosäuresequenz bestimmt, welche Konformation ein Protein letztendlich einnimmt. • Die Sekundärstruktur ist die räumliche Anordnung von benachbarten Aminosäuren, die durch die Bildung von Wasserstoffbrücken entsteht. Zwei regelmäßige Strukturen, die a-Helix- und die b-Faltblatt-Struktur, sind in vielen Proteinen zu finden. 11 • Unter Tertiärstruktur der Proteine versteht man die übergeordnete räumliche Struktur der gesamten Polypeptidkette. Sie wird auch durch Bindungen von Aminosäureresten bestimmt, die in der Aminosäurekette weit voneinander entfernt liegen können. Beispielsweise können kovalente Bindungen zwischen den Schwefelatomen zweier Cysteinreste, so genannte Disulfidbrücken, die Proteinstruktur wesentlich beeinflussen. Die Aminosäurekette kann sich um eine Nichtproteinverbindung anordnen, die kovalent (prosthetische Gruppe) oder nicht-kovalent (Coenzym) mit dem Protein verbunden sein kann. • Die Quartärstruktur bezeichnet die Zusammenlagerung mehrerer Polypeptidketten (Proteine), die identisch oder unterschiedlich sein können, zu einer gemeinsamen Funktionseinheit. 484 CH2-Kap11.indd 484 01.08.14 11:22 11.1 Biologische Grundlagen Primärstruktur Tertiärstruktur Quartärstruktur alpha-Helix Leu Thr Pro Val Glu Sekundärstruktur Häm (prosthetische Gruppe) beta-Faltblatt His Val Ein Beispiel für ein komplexes Protein mit Quartärstruktur ist der rote Blutfarbstoff Hämoglobin (Bild 1, Quartärstruktur). Er enthält vier Untereinheiten, von denen je zwei identisch sind. Die Untereinheiten selbst bestehen aus einem Proteinanteil und aus einer prosthetische Gruppe, dem Häm, das Eisen als Zentralatom enthält. Die Untereinheiten, die Globine, sind in gleicher und charakteristischer Weise gefaltet. Aminosäure Bild 1: Die vier Strukturebenen eines Proteins Die für die Funktion wichtige räumliche Struktur oder Konformation von Proteinen wird durch die Eigenschaften der Aminosäurereste und deren Beziehungen zueinander bestimmt. Welche Konformation ein Protein einnimmt, ist im Wesentlichen von der Primärstruktur, der Aminosäuresequenz, abhängig. Aufgrund der Vielzahl möglicher Aminosäuresequenzen und infolgedessen von Proteinstrukturen können Proteine in einem Organismus viele unterschiedliche Aufgaben erledigen. • Enzyme dienen als Katalysator, die für sehr viele chemische Reaktionen in der Zelle benötigt werden und den Stoffwechsel gewährleisten. Ein Cofaktor ist ein organisches Molekül oder Metall-Ion, das für die Funktion vieler Enzyme zusätzlich notwendig ist. Es kann fest (prosthetische Gruppe) oder reversibel (Coenzym) an das Enzym gebunden sein. • Strukturproteine, wie z. B. Kollagen und Keratin, bestimmen die Gestalt biologischer Strukturen wie der Haut und Knochen und bieten mechanische Stütz- und Schutzfunktionen. • Transportproteine transportieren spezifisch kleine Moleküle und Ionen beispielsweise in eine Zelle hinein oder wie Hämoglobin, das Sauerstoff binden kann, innerhalb eines Organismus. • Antikörper sind hochspezifische Proteine des Immunsystems zur Abwehr von körperfremden Stoffen wie Krankheitserregern. • Regulator- und Signalproteine sind wichtige Steuerelemente innerhalb und außerhalb der Zelle. Einige Hormone sind Peptide bzw. Proteine und dienen bei Mensch und Tieren der Weitergabe von Signalen über weite Distanzen im Körper. Kohlenhydrate Offenkettige a-Glucose 6 CH2OH Form 5C O Die als Kohlenhydrate zusamH H Ringform O H mengefassten organischen H 1C C 4 Verbindungen sind als Zucker H OH 1C 6 CH2OH HO C OH bekannt oder aus ZuckerbauO 5 3 H C OH C OH 2 2 H H steinen aufgebaut. KohlenH OH H 4C HO C H 1C hydrate kommen überall in 3 H OH 6 CH2OH O der belebten Natur vor und HO C H C OH O 4 5C 3 O 2 machen den weitaus größten H CH H OH H C OH H 5 Teil der Biomasse aus. Sie werC 1C 4 H OH CH2OH den durch Pflanzen, Algen und 6 HO C O H 3 Mikroorganismen bei der Foto2 OH b-Glucose H synthese aus dem Kohlenstoffdioxid der Luft und Bild 2: Strukturformeln von Glucose Wasser aufgebaut. Kohlenhydrate bestehen daher aus Kohlenstoff-, Wasserstoff- und Sauerstoffatomen. Durch unterschiedliche Verknüpfung dieser Atome entstehen viele verschiedene Kohlenhydrate. 11 485 CH2-Kap11.indd 485 01.08.14 11:22 11 Biotechnologie Der Zucker Glucose (Traubenzucker) ist als direktes Produkt der Fotosynthese besonders weit verbreitet und ein wichtiger Energielieferant für Zellen (s. Kap. 11.1.10, S. 505). Glucose gehört mit sechs C-Atomen zu den Hexosen und hat die Summenformel C6H12O6 . CH2OH Hexosen enthalten entweder eine Ketogruppe oder wie im Falle der Glucose eine Aldehydgruppe. Die übrigen Kohlenstoffatome tragen Hydroxygruppen und Wasserstoffatome. Weit verbreitet sind auch die fünf C-Atome enthaltenden Pentosen und die Triosen mit drei C-Atomen. C O HO C H H C OH H C OH CH2OH Bild 1: Fructose Bild 2, S. 485, zeigt die Strukturformel der Glucose, die wie andere Zucker sowohl in Form einer offenen Kette als auch in geschlossener Ringform vorkommen kann. Die Aldehydgruppe geht hierzu mit der Hydroxygruppe am C5-Atom eine Reaktion ein. Dabei können zwei Ringstrukturen entstehen, die a- und die b-Form, je nachdem, ob die Hydroxygruppe am C1-Atom unter oder über der Ringebene angeordnet ist. Die Glucose, die auch Blutzucker genannt wird, da sie im menschlichen Blut enthalten ist, ist ein Einfachzucker oder Monosaccharid. In Früchten kommt die Fructose (Fruchtzucker) vor. Sie ist ebenfalls ein Monosaccharid, besitzt jedoch statt einer Aldehydgruppe eine Ketogruppe (Bild 1). CH2OH 6 CH2OH 6 CH2OH O O O 5 H H H 5 H H H – H 2O H H H 4 1 1 4 OH H OH H OH H O HO HO OH OH H 3 OH a-Glucose H OH OH a-Glucose + H 2 OH Maltose CH2OH O CH2OH O HO O OH OH CH2OH OH OH Lactose OH O O O O O O O O O O O O O O O OH O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O In den beiden Polysacchariden Stärke und Glycogen sind zahlreiche a-Glucosemoleküle zu sehr langen Molekülen miteinander verknüpft. Stärke besteht aus zwei verschiedenen Polymeren, aus der linear verketteten Amylose (a-1,4-glycosidische Bindungen) mit schraubenförmiger oder helikaler Struktur (Bild 3), und aus dem verzweigten Amylopektin mit zusätzlichen a-1,6-glycosidischen Verknüpfungen. Im Verdauungssystem von Mensch und Tieren werden diese Polymere durch Enzyme wieder in Glucosemoleküle gespalten. OH OH CH2OH O O OH O OH CH2OH O Kohlenhydrate können auch Saccharose als Polymere vorliegen. Diese Zuckerpolymere werden als Bild 2: Disaccharide Polysaccharide bezeichnet. Bekannte Vertreter sind Stärke, Glycogen und Cellulose. Stärke dient in vielen Pflanzen als Energiespeicher; Glycogen hat in menschlichen und tierischen Geweben dieselbe Speicherfunktion. 11 3 2 H O CH2OH O H H H OH H HO OH O Reagieren zwei Glucose-Moleküle unter Wasserabspaltung miteinander, entstehen Zweifachzucker oder Disaccharide wie die Maltose (Malzzucker) unter Bildung einer a-1,4-glycosidischen Bindung (Bild 2). Disaccharide sind beispielsweise die als Haushaltszucker bekannte Saccharose (Rohrzucker) und die in der Milch zu findende Lactose (Milchzucker). a) b) Bild 3: Struktur von a) Amylose und b) Cellulose 486 CH2-Kap11.indd 486 01.08.14 11:22 11 Biotechnologie Aufgrund der Eigenarten des genetischen Codes kann trotz Basenaustausch die gleiche Aminosäure im Genprodukt eingefügt werden, da eine Reihe von Aminosäuren identisch kodiert sind (s. Kap. 11.1.9, S. 501). Diese Mutationen werden stille oder stumme Mutationen genannt. Sie führen dazu, dass bestimmte Gene für das gleiche Merkmal bzw. Protein innerhalb einer Population unterschiedliche Nukleotid-Sequenz besitzen. Dieses Phänomen heißt Polymorphismus (griech.: Vielgestaltigkeit). Der durch eine Punktmutation verursachte Austausch einer DNA-Base in einem Gen kann zum Einbau einer abweichenden Aminosäure führen. Dadurch kann das veränderte Protein seine Funktion ganz oder teilweise verlieren. In allen Lebewesen treten Mutationen spontan mit einer bestimmten Häufigkeit auf, die als natürliche Mutationsrate bezeichnet wird. Insbesondere entstehen regelmäßig Ablesefehler durch die DNA-Polymerasen bei der Replikation, dem Ablauf der DNA-Verdoppelung im Zellkern. Diese Fehler können zu Sequenzänderungen in der DNA führen, wenn sie nicht repariert werden. DNA CAA GTA AAC ATA GGA CTT CTT Protein Val His Leu Thr Pro Glu Glu a) Nicht-mutierte Zellen Punktmutation DNA CAA GTA AAC ATA GGA CAT CTT Protein Val His Leu Thr Pro Val Glu b) Mutierte Zellen Hämoglobin Rote Blutzellen Sichelzellen bei Mutationen können auch inSichelzellenanämie duziert, d. h. künstlich erzeugt werden. Dazu können soge- Bild 1: Punktmutation im Hämoglobin bei der Sichelzellenanämie nannte Mutagene wie Strahlung (z. B. radioaktive Strahlung und UV-Strahlen), hohe Temperaturen sowie bestimmte Chemikalien (z. B. a) Bromuracil, Salpetersäure und Akridinfarbstoffe) eingesetzt werden. Endonuklase Durch kurzwellige UV-Strahlung werden zwei auf dem DNA-Strang benachbarte Thyminbasen zu Thymin-Dimeren kovalent verbunden b) DNA(Bild 2a). Die Thymin-Dimere können sich nicht Synthese Polymerase mehr mit den komplementären Adenin-Basen paaren, so dass es zum Abbruch der Replikation kommen würde. Durch enzymatisch gesteuc) erte Prozesse der DNA-Reparatur können allerdings Schäden der DNA erkannt und beseitigt werden. Die Thymin-Dimere werden zusamDNA-Ligase men mit den benachbarten Nukleotiden durch eine Endonuklease herausgeschnitten (Bild 2b). Eine Endonuklease ist ein Enzym, das die DNA d) im Inneren (griech. endo, innen) aufspalten und abbauen kann. Die entstandene Lücke wird Bild 2: Bildung und Reparatur von Thymin-Dimeren durch die DNA-Polymerase, die DNA-Polymere bildet, mit den komplementären Nukleotiden ersetzt (Bild 2c). Das zuletzt eingefügte Nukleotid muss mit dem benachbarten Nukleotid von einem weiteren Enzym, der DNA-Ligase, kovalent verbunden werden, so dass wieder ein geschlossener DNA-Strang vorhanden ist (Bild 2d). 11 Mutagene sind Chemikalien und äußere Einwirkungen wie radioaktive oder UV-Strahlung, die im Erbgut von Organismen Mutationen auslösen können. 500 CH2-Kap11.indd 500 01.08.14 11:22 11.1 Biologische Grundlagen 11.1.9 Proteinbiosynthese Die Biosynthese von Proteinen verläuft in allen Lebewesen in gleichartiger Weise. Dieser Prozess wird auch Genexpression genannt und meint die Exprimierung oder Ausprägung der genetischen Informationen in Proteine. Die Synthese von Proteinen erfolgt immer über die beiden Schritte der Transkription und Translation sowie über das Zwischenprodukt RNA, was zur Aufstellung des zentralen Dogmas der Molekularbiologie führte (Bild 1). Das Dogma ist für alle Lebewesen gültig und fand nur eine Ausnahme bei der Entdeckung der Retroviren (s. Kap. 11.1.7, S. 496). Diese Viren, zu denen auch die AIDS-Erreger gehören, sind in der Lage, ihr RNA-Erbgut in DNA-Moleküle umzuschreiben. Die Proteinbiosynthese erfolgt in prokaryontischen und eukaryontischen Zellen in zwei Stufen (s. Bild 2): Transkription DNA Translation RNA Protein Retroviren Bild 1: Zentrales Dogma der Molekularbiologie • Transkription (Umschreibung): Zunächst werden die DNA-Gene in einen RNA-Strang umgeschrieben. Hierzu werden RNA-Nukleotide, die sich über die komplementäre Basenpaarung (s. Kap. 11.1.2, S. 489) an den geöffneten DNA-Strang anlagern, mithilfe des Enzyms RNA-Polymerase verknüpft. Es entsteht die sogenannte Boten-RNA oder messenger-RNA (kurz: mRNA). • Translation (Übersetzung): Anschließend wird an den Ribosomen, den Eiweißfabriken der Zelle, die Nukleotidsequenz der mRNA in eine spezifische Kette von Aminosäuren übersetzt. Der Informationsfluss von DNA über RNA zum Protein ist das zentrale Dogma der Molekularbiologie. Die Baupläne der Proteine, die als genetische Information in der DNA gespeichert sind, werden in der Transkription auf mRNA umgeschrieben. An den Ribosomen erfolgt in der Translation die eigentliche Synthese des Proteins. Die in allen Lebewesen gültitRNA mit Aminosäure DNA ge Regel, mit der eine mRNA in ein Protein übersetzt wird, wird genetischer Code genannt. Dabei ist jede in ProPeptidkette teinen vorkommende Aminosäure einer Gruppe von drei aufeinanderfolgender RNARibosom Basen, auch Tripletts oder RNACodons genannt, zugeordnet. Polymerase Die RNA besitzt wie die DNA vier verschiedene Basen und mRNA damit vier Buchstaben in der mRNA genetischen Sprache. ProteTranskription Translation ine enthalten gewöhnlich 20 unterschiedliche Aminosäuren Bild 2: Ablauf der Proteinbiosynthese (s. Kap. 11.1.2, S. 483). Die Ko3 dierung der RNA-Sequenz in Dreiergruppen ermöglicht, dass insgesamt 4 = 64 mögliche Codons zur Verfügung stehen. Beispielsweise wird das Triplett CCU auf der RNA in die Aminosäure Prolin übersetzt (Bild 1, S. 502). Bei einer Kodierung der Basen in Zweiergruppen würden nur 2 4 = 16 Codons und damit für alle 20 Aminosäuren zu wenige zur Verfügung stehen. Bei 64 möglichen Codons gibt es allerdings für viele Aminosäuren mehrere Kodierungen. Der genetische Code ist ein Schlüssel, mit dem während der Translation Dreiergruppen aufeinanderfolgender Nukleotide, die Codons genannt werden, in Aminosäuren übersetzt werden. 11 501 CH2-Kap11.indd 501 01.08.14 11:22 11 Biotechnologie Als Startsignal der Translation dient das Triplett AUG der mRNA, das gleichzeitig auch den Einbau der Aminosäure Methionin veranlasst. Drei Codons sind als Stopcodons für die Beendigung (Termination) der Translation verantwortlich. Sie führen zum Abbruch der Synthese, da es keine Aminosäuren zu diesen Codons gibt. Beim Ablauf der Translation (Bild 2) spielen neben der mRNA weitere Zellkomponenten eine wichtige Rolle, vor allem die Ribosomen und eine Gruppe kleiner RNA-Moleküle, die als tRNA zusammengefasst werden. Ribosomen bestehen aus zwei Untereinheiten, die getrennt voneinander im Cytoplasma vorliegen und sich nur bei der Proteinsynthese zusammenlagern. Die Ribosomen bestehen aus verschiedenen Proteinen, den ribosomalen Proteinen, und unterschiedlichen rRNA-Elementen, von denen das längste bei Eukaryonten 4718 Nukleotide enthält. Die tRNA-Moleküle bestehen aus nur 70 bis 80 Nukleotiden. Eine tRNA besitzt eine kleeblattähnliche Form, da einige Nukleotide innerhalb des Moleküls gepaart vorliegen. Die tRNA transportiert die Aminosäuren an den Ort der Proteinsynthese (Bild 2). Die zur Synthese benötigten 20 verschiedenen Aminosäuren sind mit einer jeweils für sie spezifischen tRNA verestert. Durch die Bildung dieser Aminoacyl-tRNA werden die Aminosäuren aktiviert und fähig zur Bildung einer Peptidbindung. Die Aktivierung der Aminosäuren geschieht durch die Aminoacyl-tRNA-Synthetasen, einer Gruppe von Enzymen, die jeweils nur für eine bestimmte Aminosäure spezifisch sind. Erste Base Zweite Base 5´ U C A G 3´-Ende U Phe Phe Leu Leu Leu Leu Leu Leu Ser Ser Ser Ser Pro Pro Pro Pro Tyr Tyr „Stop“ „Stop“ His His Gln Gln Cys Cys „Stop“ Trp Arg Arg Arg Arg U C A G U C A G A Ile Ile Ile Met (Start) Thr Thr Thr Thr Asn Asn Lys Lys Ser Ser Arg Arg U C A G G Val Val Val Val Ala Ala Ala Ala Asp Asp Glu Glu Gly Gly Gly Gly U C A G C Dritte Base Pro = Prolin CCU Bild 1: Der genetische Code 1. Aktivierung der Aminosäure tRNA Aminosäure tRNA mit Peptidkette AminoacyltRNASynthetase 3. Bildung der Peptidbindung tRNA mit Aminosäure Anticodon 2. CodonAnticodonErkennung tRNABindungsstelle 4. Wechsel der Bindungsstelle Ribosom Zur Ausbildung der Peptidbindung am Ribodon Codon Codon Codon Co som müssen die richtigen Aminosäuren in 6 7 5 mRNA 4 räumliche Nähe zueinander gebracht werden. 5. Bewegung der mRNA entlang des Ribosoms Zusätzlich muss das der Aminosäure zugehörige Codon auf der mRNA von der tRNA erkannt Bild 2: Prozesse der Translation werden. Hierzu besitzt jede tRNA ein spezielles Codon, das Anticodon genannt wird und das über Wasserstoffbrückenbindung eine Basenpaarung mit dem komplementären Codon auf der mRNA eingeht. Die Ribosomen besitzen t-RNABindungsstellen, an denen zwei tRNA-Moleküle angelagert werden, eine tRNA mit einer einzelnen Aminosäure und eine, die die entstehende Kette von Aminosäuren gebunden hat. Ein rRNA-Molekül des Ribosoms katalysiert die Bildung einer neuen Peptidbindung zwischen der entstehenden Kette und der einzelnen Aminosäure. Die tRNA mit der verlängerten Peptidkette wechselt dann in die andere Bindungsstelle des Ribosoms über, die von der entladenen tRNA freigegeben wird. Anschließend geht der Zyklus mit der Bindung einer weiteren beladenen tRNA an die nun freie Bindungsstelle weiter. Die mRNA wandert dabei, Codon um Codon, am Ribosom entlang weiter. 11 An den Ribosomen werden tRNA-Moleküle, die mit Aminosäuren beladen sind, mit einer mRNA räumlich zusammengebracht. Dadurch kann sich das Anticodon der tRNA an das komplementäre Codon der mRNA anlagern und eine Peptidbindung kann geschlossen werden. 502 CH2-Kap11.indd 502 01.08.14 11:22