Qualitative Analyse Theoretische Grundlagen
Werbung

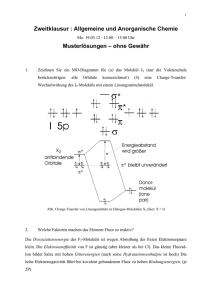
Institut für Anorganische Chemie - Materialchemie der Universität Wien F. Gehringer, H. Mikler, H. Schicketanz Chemisches Grundpraktikum Theoretische Grundlagen / 2005 Qualitative Analyse Theoretische Grundlagen 1. Einleitung Die qualitative Analyse behandelt die Trennung von Ionengemischen, sowie die Nachweise von Ionen und Elementen. Trennoperationen beruhen im allgemeinen auf einer Verteilung der einzelnen Stoffe in verschiedenen Phasen. Bei der qualitativen Analyse handelt es sich meistens darum, dass bestimmte Stoffe als Niederschläge abgeschieden werden, während andere in Lösung bleiben. Durch Nachweis-reaktionen soll das Vorhandensein eines Stoffes (Elements, Ions) angezeigt werden. Es geht dabei insbesondere um das Ausfallen eines Niederschlages, um eine Gasentwicklung oder um eine Farbänderung durch Komplexbildung. Die als Trennoperationen und Nachweise verwendeten chemischen Reaktionen kann man auf vier Grundtypen zurückführen: 1. Niederschlagsbildung 2. Säure-Basenreaktionen 3. Komplexbildung 4. Redoxreaktionen Die meisten in der qualitativen Analyse verwendeten Reaktionen gehören zu diesen Grundtypen oder können als Kombinationen mehrerer Grundtypen beschrieben werden. Für jeden Reaktionstyp werden im Text einige, für die qualitative Analyse wichtige Beispiele behandelt, wobei die Diskussion der zugrunde liegenden Gleichgewichte und deren Beschreibung durch das Massenwirkungsgesetz im Vordergrund steht. 2. Das Massenwirkungsgesetz (MWG) Mit dem Massenwirkungsgesetz (empirisch gefunden von Guldberg und Waage im Jahr 1867) wird die Lage des chemischen Gleichgewichts beschrieben. Für eine einfache Reaktion: A+Bô C+D sei zunächst die Bildung der Endstoffe C und D aus den Ausgangsstoffen A und B betrachtet: v d[A] d[B] d[C] d[D] dt dt dt dt Die Reaktionsgeschwindigkeit v der Hin- oder Bildungsreaktion drückt die Abnahme der Konzentrationen [A] und [B] der Ausgangsstoffe A und B bzw. die Zunahme der Konzentrationen [C] und [D] der Endstoffe C und D je Zeiteinheit aus. Da im homogenen Medium (Gase oder gelöste Stoffe) A mit B reagieren muss, um C und D zu bilden, ist v der Konzentration der Reaktanden (Edukte) proportional: v k .[A] [B] k wird als Geschwindigkeitskonstante der Hinreakton bezeichnet. Sind bei einer reversiblen (umkehrbaren) Reaktion C und D in einer für die Reaktion ausreichenden Menge entstanden, so setzt die Rück- oder Zerfallsreaktion ein: v d[A] d[B] d[C] d[D] dt dt dt dt Die Reaktionsgeschwindigkeit v der Rück- oder Zerfallsreaktion drückt daher die Abnahme der Konzentrationen [C] und [D] bzw. die Zunahme der Konzentrationen [A] und [B] aus. Dementsprechend gilt auch v k [C] [D] Für die Gesamtreaktion gilt daher allgemein: Die Geschwindigkeit r der Gesamtreaktion setzt sich aus der Differenz der Geschwindigkeiten der Teilreaktionen zusammen. r v v Nach Beginn einer chemischen Umsetzung nimmt v dauernd ab, v dagegen zu. Mit v v ist das chemische Gleichgewicht erreicht. v v und r = 0 Dabei handelt es sich um ein dynamisches Gleichgewicht, bei dem Hin- und Rückreaktion gleichzeitig stattfindet, sich aber der gegenseitige Umsatz gerade aufhebt, sodass nach außen hin keine Veränderung des Systems zu beobachten ist; daher gilt: k [A] [B] k [C] [D] K k [C] [D] k [A] [B] Eine chemische Reaktion befindet sich dann im Gleichgewichtszustand, wenn der Quotient aus dem Produkt der Konzentrationen der Endstoffe (Produkte) und dem Produkt der Konzentrationen der Ausgangsstoffe (Edukte) einen charakteristischen Zahlenwert K, die Gleichgewichtskonstante oder Massenwirkungskonstante, erreicht hat. K ist konzentrationsunabhängig, ändert sich jedoch im allgemeinen mit Temperatur und Druck (Gasreaktionen). Umkehrreaktion (C D) ô (A B) Wird eine Reaktionsgleichung mit -1 multipliziert, so muss sie in umgekehrter Richtung gelesen werden: AB ô CD ·(-1) ergibt CD ô AB K Umkehr daraus folgt 1 [A] [B] K [C] [D] Die Gleichgewichtskonstante der Umkehrreaktion entspricht daher dem reziproken Wert der Gleichgewichtskonstante der ursprünglichen Reaktion. Beteiligen sich an einer Einzelreaktion mehrere Moleküle derselben Art, so ist im Massenwirkungsgesetz die Konzentration der Moleküle zur Potenz zu erheben. Für eine allgemeine Reaktion nA mB ô pC qD gilt daher: [C] p [D]q K [A] n [B] m Hinweis: Bei Anwendung des MWG auf homogene Lösungen ist die Aktivität oder „wirksame Konzentration“ a(i) einzusetzen, die aufgrund der interionischen Wechselwirkungen kleiner als die wirkliche Konzentration c(i) ist. Für die Aktivität eines gelösten Stoffes gilt daher: c(i)mol l 1 c(i) a(i) f(i) f(i) c o mol l 1 co f(i) ist der Aktivitätskoeffizient, durch den die Wechselwirkungskräfte berücksichtigt werden. Für „ideale Lösungen“, d.h. bei großer Verdünnung, nähert sich der Aktivitätskoeffizient dem Wert 1; d.h. f(i) 1 . c° bedeutet die Standardkonzentration, man versteht darunter eine Lösung der Konzentration c° = 1 mol·l-1 bei 25°C (1 l = 1 dm3). Zur Vereinfachung wird in vielen Texten auf das Anschreiben von c° verzichtet und f(i) 1 angenommen. Bei Gleichungen, die aus dem MWG abgeleitet sind, tritt dann die Schreibweise [Reaktand] an die Stelle von: a(i) c(i) = [i] = [Reaktand] co mit c o 1mol l 1 [Reaktand] bedeutet aber dann nur den Zahlenwert. In diesem Text wird für alle Betrachtungen die Schreibweise [Reaktand] verwendet. 3. Niederschlagsbildung Die Trennung eines Systems in Niederschlag und Lösung ist die wichtigste Trennmethode der klassischen Analyse. Wir beschränken uns auf ionische Verbindungen: Dabei wird angenommen, dass die Ionen des gelösten Salzes lediglich unter Hydratisierung reagieren (diese tritt prinzipiell bei allen Ionen ein, wird aber in den Formeln nicht mit angegeben), nicht aber unter Hydrolyse (Protolyse). Komplexbildung oder Redoxreaktionen mit etwa vorhandenen Fremdionen sollen auch ausgeschlossen sein. Es wird angenommen, dass der aus der Lösung ausfallende Niederschlag eine reine Phase ist. 3.1 Das Löslichkeitsprodukt (Lp) Das Löslichkeitsprodukt ist der Ausdruck des MWG für heterogene Lösungsgleichgewichte. Die hier betrachteten Systeme bestehen aus Lösungen und festen Stoffen; ist ein Bodenkörper vorhanden, so steht dieser mit der gesättigten Lösung bei gegebener Temperatur im Gleichgewicht. In wässriger Lösung spalten viele Stoffe in Ionen auf. Daher gilt für den Gleichgewichtszustand einer einfachen Reaktion: AB(s) ô A B Aus der Oberfläche des Niederschlages werden in der Zeiteinheit je Fläche jene Zahl von Kationen A+ und Anionen B- in Lösung gehen, die eine zum Verlassen der Oberfläche notwendige Energie besitzen. Die Zunahme der Konzentrationen [A+] und [B-] in der Lösung ist daher der Oberfläche S proportional: d[A ] d[B ] dt dt k1 S k 1 : Proportionalitätskonstante der Auflösungsreaktion Gleichzeitig werden aus der Lösung (aus Elektroneutralitätsgründen paarweise) Ionen an die Oberfläche des Niederschlages angelagert. Die Abnahme der Konzentrationen [A+] und [B-] ist daher dem Produkt der Konzentrationen der beiden Ionen und der Oberfläche S proportional: d[A ] dt d[B ] dt k 2 S [A ] [B ] k 2 : Proportionalitätskonstante der Abscheidungsreaktion Im Gleichgewichtsfall sind die Geschwindigkeiten für Auflösung und Abscheidung gleich groß: k 1 S k 2 S [A ] [B ] k1 Lp [A ] [B ] k2 Daher ist in der gesättigten Lösung eines Salzes AB das Produkt der Ionenkonzentrationen bei gegebener Temperatur konstant. Für den allgemeinen Fall eines Salzes AmBn gilt die Gleichung: A m B n ô mA a nB b Wenn AmBn in m Kationen Aa+ und n Anionen Bb- aufspaltet, so ist m·a = n·b und für das Löslichkeitsprodukt erhält man: Lp = [Aa+]m · [Bb-]n 3.1.1 Ausfällen von Salzen Die Fällungsreaktion: Mischt man zwei leicht lösliche Salze A+B- mit C+D-, so fällt aus dem Lösungsgemisch A+D- bzw. C+B- aus, wenn die Löslichkeit von A+D- bzw. C+B- in Wasser (oder einem Lösungsmittel) kleiner ist, als die von A+B- bzw. C+D-. Beispiel: BaCl2 + Na2SO4 ô BaSO4(s) + 2 NaCl Die Gleichgewichtskonstante für die Fällungsreaktion Ba2+ + SO42- ô BaSO4(s) ist daher gegeben mit: K Fällung 1 Lp 3.2 Zusammenhang zwischen Löslichkeit und Löslichkeitsprodukt 3.2.1 Löslichkeit in reinem Lösungsmittel a) Für ein Salz AB, das in wässriger Lösung in A+ und B- dissoziiert, gilt in der gesättigten Lösung: Lp = [A+]·[B-]. Nun ist in der Lösung des Salzes in Wasser (Lösungsmittel) aus stöchiometrischen Gründen die Konzentration von A+ gleich der von B- und diese gleich der Konzentration des gelösten Salzes, c: [A+] = [B-] = c (c: Stoffmengenkonzentration von AB (mol·l-1)). Lp = [A+] · [B-] = c2; bzw. c = Lp b) Für ein Salz A2B erhalten wir: [A ] [B2 ] c 2 Lp [A ]2 [B 2 ] (2c) 2 c 4c 3 bzw. c 3 Lp 4 c) Allgemein gilt für ein Salz AmBn: [A a + ] [B b ] c m n Lp [A a ] m [ B b ] n ( mc) m ( nc) n m m n n c m n , bzw. c m n Lp mm . n n 3.2.2 Löslichkeit bei gleichionigem Zusatz Setzt man der Lösung von AB, die sich im Gleichgewicht mit dem Niederschlag befindet, ein Salz mit einem gemeinsamen Ion, z.B. XB zu, so wird durch die Zugabe der B--Ionen das Gleichgewicht gestört. Es erfolgt die Reaktion A+ + B- AB(s) und festes AB fällt aus; die Löslichkeit, c = [A+], wird daher durch gleichionigen Zusatz verringert. Für das Gleichgewicht gilt: Lp = [A+] · [B-]; jetzt ist aber [B-] größer als [A+], daher ergibt sich: c [A ] Lp [ B ] Die Löslichkeit eines Salzes in reinem Wasser ist ein Maximum gegenüber der Löslichkeit in Salzlösungen mit einem gemeinsamen Kation oder Anion. In manchen Fällen tritt bei sehr hoher Konzentration eines gleichionigen Zusatzes Komplexbildung ein, sodass eine Löslichkeitserhöhung erfolgt. 3.2.3 Löslichkeitsberechnungen - Zahlenbeispiele Beipiel A: Kaliumperchlorat, KClO4, hat in Wasser bei 0°C eine Löslichkeit von 7,5 g·l-1 (Molmasse 138,56 g·mol-1). Wie groß ist das Löslichkeitsprodukt? c 7,5 0,054 mol l 1 138,56 Da es sich um ein Salz AB handelt, gilt: Lp = c2 = (0,054)2 = 2,9·10-3 Beispiel B: Wie groß ist die Löslichkeit c von KClO4 in 0,20 M ClO4-, d.h. wenn man festes KClO4 in einer Lösung 0,20 M an ClO4- (Anfangskonzentration) suspendiert? c [K ] Lp [ClO 4 ] Als erste Näherung setzen wir [ClO4-] = 0,20 M; d.h. wir vernachlässigen die aus dem schwerlöslichen Salz stammende Konzentration an ClO4-, und erhalten: c1 2 ,9 10 3 2 ,0 10 1 1,45 10 2 mol l 1 Für die zweite Näherung berücksichtigt man die aus dem gelösten KClO4 stammende Konzentration an ClO4-, sie beträgt c1 = 1,45·10-2 mol·l-1 und ist zur Anfangskonzentration [ClO4-] = 0,20 mol·l-1 hinzuzurechnen: c2 2,9 10 3 2 ,145 10 1 1,35 10 2 mol. l 1 In analoger Weise kann in dritter Näherung berechnet werden: c3 2,9 10 3 2,135 10 1 1,36 10 2 mol l 1 Die exakte Berechnung (ohne Näherung) wird folgendermaßen durchgeführt: c [K ] Lp 0,20 c c2 + 0,20c - 0,0029 = 0 c 1,( 2 ) 0,1 1,29 10 2 c1 = -0,1 + 0,11358 = 1,358·10-2 mol·l-1 Die hier angegebene Zahl signifikanter Stellen ist ohne praktische Bedeutung, insbesondere, da Zahlenwerte der Löslichkeitsprodukte nicht so genau bekannt sind. Aus diesem Grunde wird für viele Zwecke bereits mit der ersten Näherung ein ausreichend genaues Ergebnis erhalten. 3.3 Niederschlagsbildung bei Zugabe eines Fällungsmittels 3.3.1 Bildung eines Niederschlages Wir wollen nun den Vorgang der Niederschlagsbildung betrachten, also das Ausfällen eines schwerlöslichen Salzes AB aus einer Lösung eines seiner Ionen. Bei Zugabe von B--Ionen (in Form eines gelösten, in Ionen dissoziierten Salzes XB) zu einer Lösung von A+-Ionen (in Form eines gelösten, in Ionen dissoziierten Salzes AY) läuft folgende Reaktion ab: A+ + B- ô AB(s) Es genügt, die an der Niederschlagsbildung beteiligten Ionen anzuschreiben. Die Gleichgewichtskonstante des in umgekehrter Richtung verlaufenden Vorganges ist das Löslichkeitsprodukt. Die Ausgangskonzentration von A+, [A+]o, ist vorgegeben. Es lassen sich verschiedene Bereiche unterscheiden: a) Bei der Zugabe von B- zur Lösung tritt solange kein Niederschlag auf, „bis das Löslichkeitsprodukt von AB erreicht ist“. Die hierzu nötige Konzentration [B-] ergibt sich zu [ B ] c Lp [A ]0 d.h. sie ist umso kleiner, je kleiner Lp und je größer die Ausgangskonzentration [A+]o ist. Wird der schwerlösliche Niederschlag zum Nachweis des Ions A+ erzeugt, so ist die Fällung also am empfindlichsten in konzentrierteren Lösungen und für möglichst schwerlösliche Salze. b) Bei fortgesetzter Zugabe von B- fällt weiter festes AB aus, wodurch die Konzentration [A+] sinkt und die im Gleichgewicht befindliche Konzentration [B-] ansteigt. c) Am Äquivalenzpunkt sind beide Ionen in chemisch äquivalenten Mengen vorhanden, für ein Salz AB ist [A+] = [B-]. Daraus folgt, dass auch die in Lösung befindlichen Konzentrationen von A+ und Beinander gleich sind: [A ] [ B ] c Lp d) Bei weiterem Zusatz von XB wird die Gleichgewichtskonzentration [B-] erhöht, die Konzentration [A+], deren Zahlenwert gleich ist dem Zahlenwert der Löslichkeit c, nimmt weiter ab: [A ] c Lp [B ] Die im Gleichgewicht vorliegende Konzentration [A+] wird umso kleiner, je größer die über den Äquivalenzpunkt hinaus zugesetzte Menge an B- ist. Dieser Löslichkeitserniedrigung, die ja im allgemeinen gewünscht wird, sind jedoch aus praktischen Gründen Grenzen gesetzt, weil Ionenkonzentrationen wegen eintretender Sättigung grundsätzlich nicht beliebig gesteigert werden können. Bei sehr hoher Konzentration [B-] kann Komplexbildung mit entsprechender Löslichkeitserhöhung eintreten. Da Gleichgewichte von beiden Seiten her erreicht werden können, gilt das oben Gesagte sinngemäß auch für die Auflösung eines Niederschlages. Wird die Konzentration [A+] oder [B-], die mit dem schwerlöslichen Niederschlag AB im Gleichgewicht ist, unterschritten, so tritt Auflösung des Niederschlages ein, bis diese Gleichgewichtskonzentrationen wiederhergestellt sind. Können sie nicht erreicht werden, so löst sich der gesamte Niederschlag auf. Das kann zum Beispiel geschehen, wenn reines Lösungsmittel zugesetzt wird, ist aber bei schwerlöslichen Salzen wenig wirksam (für ein Salz mit Lp = 10-10 erhält man c = 10-5 mol·l-1, d.h. man braucht 105 Liter Wasser, um 1 Mol Niederschlag in Lösung zu bringen). Viel effektiver können die Konzentrationen vermindert werden, wenn Stoffe zugesetzt werden, die mit A+ oder B- unter Bildung wenig dissoziierter Verbindungen zusammentreten können, z.B. H+ oder Komplexbildner, oder die Konzentration wird durch eine Redoxreaktion herabgesetzt. Daraus ergeben sich für die Ausführung einer möglichst quantitativen Fällung folgende Gesichtspunkte: Die zu fällende Lösung soll, wenn möglich, nicht allzu verdünnt, aber auch nicht zu konzentriert sein, um Mitreißen oder Einschließen von Fremdionen (bei Trennungen) zu vermeiden. Die Fällungslösung wird tropfenweise in der Wärme und unter gutem Rühren zugesetzt, bis der Niederschlag auszufallen beginnt. Sodann wird langsam weiter Fällungsmittel zugesetzt bis die Fällung offenbar beendet ist und ein kleiner Überschuss des Fällungsmittels zugegeben, um die Löslichkeit weiter zu erniedrigen. 3.3.2 Niederschlagsbildung bei der Fällung zweier schwerlöslicher Salze nebeneinander Das Ion A+ möge mit dem Ion B- ein Salz mit dem Löslichkeitsprodukt Lp(AB) und mit dem Ion C- eines mit dem Löslichkeitsprodukt Lp(AC) bilden, wobei Mischkristallbildung AB + AC nicht auftreten darf. A+ + B- ô AB(s) A+ + C- ô AC(s) Lp(AB) sei kleiner als Lp(AC). Vorgelegt sei eine gemischte Lösung vergleichbarer Konzentration von B- und C-. Wie verhalten sich deren Konzentrationen bei allmählichem Zusatz von A+ ? Bei der Konzentration [A ]1 Lp(AB) beginnt AB auszufallen. [ B ]0 Diese Konzentration ist aber zu klein, als dass auch AC ausfallen könnte, denn hierzu wäre eine Konzentration von [A ]2 Lp(AC) [C ]0 erforderlich. Da nun nach obiger Annahme Lp(AC) größer ist als Lp(AB), fällt zunächst nur AB aus. Bei fortdauernder Fällung von AB wird die Konzentration von B- kleiner, während die im Gleichgewicht befindliche von A+ langsam anwächst. Wenn diese Konzentration [A+]2 erreicht, beginnt schließlich auch AC, das leichter lösliche der beiden Salze, mitzufallen. Wie groß ist der Anteil von B-, der zu diesem Zeitpunkt noch nicht ausgefallen ist? Da die jeweilige Konzentration von A+ mit beiden Niederschlägen im Gleichgewicht steht, gilt für diesen Fall: [A ]2 Lp(AB) Lp(AC) ; daraus folgt: [B ] [C ] [ B ] Lp(AB) [C ] Lp(AC) Die Fällung von C- beginnt aber erst, daher ist seine Konzentration noch gleich der Anfangskonzentration [C-]o; mit [C-] = [C-]o erhalten wir: [B ] Lp(AB) [C ]0 Lp(AC) Das Verhältnis der noch in Lösung vorhandenen Konzentration [B-] zur ursprünglich vorhandenen [B-]o ist also [ B ] Lp(AB) [C ]0 [ B ]0 Lp(AC) [ B ]0 Für den in Lösung verbliebenen Teil von B- ist also das Verhältnis der beiden Lp bestimmend; für eine praktisch quantitative Fällung des schwerer löslichen der beiden Salze (99,9%, [B-]/[B-]o = 10-3) ist daher bei gleichen Anfangskonzentrationen von B- und C- ein Verhältnis Lp(AC)/Lp(AB) von mindestens 103 erforderlich. Salze, deren Lp sich um mindestens drei Zehnerpotenzen unterscheiden, werden also aus einer gemeinsamen Lösung praktisch nacheinander gefällt. (Zur analytischen Verwertung muss jedoch eine Möglichkeit vorhanden sein, den Endpunkt der ersten bzw. den Beginn der zweiten Fällung genau festzustellen, z.B. durch andere Färbung von AC). Bemerkung: Bei der obigen Diskussion wurden die Mitfällungen außer acht gelassen. Sie bewirken, dass in der Praxis zur quantitativen Trennung meist ein größeres Verhältnis der Löslichkeitsprodukte nötig ist als 103. Zusammenfassung: Ein schwerlöslicher Niederschlag fällt bei allmählichem Zusatz der Fällungslösung aus, sobald das Löslichkeitsprodukt überschritten wird. Ein Salz ist daher umso schwerer löslich, je kleiner sein Lp ist. Sind mehrere Ionenarten vorhanden, die mit dem zugesetzten Ion schwerlösliche Niederschläge bilden können, so fällt zunächst derjenige Niederschlag aus, dessen Lp zuerst überschritten wird, also bei gleichen Ausgangskonzentrationen und gleichen Salztypen der Niederschläge (1:1, 2:1, usw.) der mit dem kleineren Lp. Es gilt auch die Umkehrung, dass sich ein Niederschlag auflöst, sobald in der darüber befindlichen Lösung das Lp unterschritten wird, z.B. durch Verdünnung, Komplexbildung oder Redoxreaktion. 3.4 Niederschlagsbildung, gekoppelt mit anderen Reaktionen Ist ein Reaktand gleichzeitig an verschiedenen Reaktionen beteiligt, so sind diese Reaktionen gekoppelt und ihre durch das MWG beschriebenen Gleichgewichte sind über die Konzentration des gemeinsamen Reaktanden verknüpft. Lässt sich eine chemische Reaktion durch Addition anderer Reaktionsgleichungen erhalten, so berechnet man die Gleichgewichtskonstante der Gesamtreaktion durch Multiplikation der Konstanten der einzelnen Teilgleichungen. Wurden die Teilgleichungen vor der Addition mit einem Faktor multipliziert, so sind ihre Konstanten vor der Multiplikation mit demselben Faktor zu potenzieren. Als Spezialfall ist hier auch die Umkehrreaktion enthalten: Man erhält die Gleichung der Umkehrreaktion durch Multiplikation mit -1; die Gleichgewichtskonstante der Umkehrreaktion ist gleich dem reziproken Wert der Gleichgewichtskonstante der ursprünglichen Reaktion. 4. Niederschlagsbildung, gekoppelt mit Säure-Base-Reaktion 4.1 Fällung von Sulfiden in Lösungen verschiedener H+-Konzentration Die Sulfide der meisten Elemente unterscheiden sich hinsichtlich ihrer Löslichkeitsprodukte in charakteristischer Weise voneinander, sodass durch zweckmäßige Wahl des pH-Wertes (siehe unten) zahlreiche Trennungen ausgeführt werden können. In der qualitativen Analyse benutzt man die Fällung mit H2S zur Trennung der Metalle in Gruppen: 1. In 1 bis 2 M HCl fallen mit H2S die Sulfide von As, Sb, Sn, Hg, Bi, Pb, Cu und Cd aus (kleines Löslichkeitsprodukt). 2. Aus ammoniakalischer Lösung fallen die Sulfide von Fe, Zn, Mn, Co und Ni aus (größeres Löslichkeitsprodukt). Die Sulfide von Cr und Al haben eine größere Löslichkeit als die Hydroxide dieser Elemente, sodass bei der eintretenden Konkurrenz der Ionen OH- und S2- um das Kation zuerst die Hydroxide ausfallen. Eine interessante Besonderheit tritt bei den Sulfiden von Co und Ni auf: Obwohl sie in saurer Lösung wegen ihres verhältnismäßig hohen Löslichkeitsproduktes nicht ausfallen und daher erst in ammoniakalischer Lösung durch H2S gefällt werden, lösen sie sich, im Gegensatz zu den Sulfiden von Fe, Mn und Zn, bei nachfolgender Behandlung mit 2 M HCl nicht mehr auf. Dieses Verhalten wird bei einigen Analysengängen zu ihrer Abtrennung benützt. Der Grund für dieses Verhalten liegt in einer irreversiblen Veränderung dieser Sulfide beim Stehen an Luft. 3. Die Sulfide der Alkali- und Erdalkalimetalle sind wasserlöslich. Die Fällung von Sulfiden erfolgt sowohl aus saurer (Punkt 1) als auch aus alkalischer Lösung (Punkt 2) mit H2S. H2S reagiert als zweiprotonige Säure: H2S ô H+ + HSHS- ô H+ + S2________________ K1 = 10-7 K2 = 10-131 H2S ô 2 H+ + S2- KS = K1·K2 = 10-20 [ H ]2 [S 2 ] KS K 1 K 2 10 20 [ H 2 S] Drückt man aus dieser Beziehung die Sulfidionenkonzentration [S2-] aus, so erhält man [S 2 ] K S [ H 2 S] [ H ]2 Wenn bei Sättigung mit H2S (p(H2S) = 1,013 bar) die H2S-Konzentration c(H2S, 25°C) = 0,1 mol·l-1 beträgt, so erhält man für [S2-]: 10 21 [S ] [ H ]2 2 Diese Beziehung drückt die Abhängigkeit der Sulfidionenkonzentration von der H+-Ionenkonzentration bzw. vom pH-Wert aus. Die hier beschriebene Überlegung beruht ausschließlich auf dieser einfachen Beziehung. Unsicherheiten hinsichtlich des Zahlenwertes von K2 sowie Protolyse-Gleichgewichte des S2--Ions, vor allem in alkalischen Lösungen, werden nicht berücksichtigt. 4.1.1 Fällung von Sulfiden aus saurer Lösung Bei pH = 0 ([H+] = 1 mol·l-1) beträgt 10 21 [S 2 ] 2 10 21 mol l 1 1 Ein Sulfid AS AS(s) ô A2+ + S2Lp = [A2+] · [S2-] kann nur dann ausfallen, wenn bei einer Ausgangskonzentration von [A2+] = 1 mol·l-1 Lp < 10-21 ist. 1 Für K2 werden in der Literatur unterschiedliche Zahlenwerte angegeben. Die am häufigsten verwendeten Werte liegen bei 10-13 bis 10-14. Nur von Rollie J. Myers [J. Chem. Education, 63 (8) 687 (1986)] werden Zahlenwerte von 10-17 bis 10-23 angenommen. 4.1.2 Fällung von Sulfiden aus alkalischer Lösung Wird der pH-Wert der Lösung durch Zugabe von NH3 auf pH = 8 steigt die Sulfidionenkonzentration: ([H+] = 10-8 mol·l-1) gebracht, so 10 21 [S ] 16 10 5 mol l 1 10 2 sodass auch Sulfide mit Lp > 10-21 bei einer Ausgangskonzentration von [A2+] = 1 mol·l-1 ausfallen können. 4.2 Löslichkeit von Sulfiden in Lösungen verschiedener H+-Konzentration Für die Löslichkeit eines Sulfids AS in H2S-gesättigter saurer Lösung werden folgende Gleichgewichte herangezogen: ô A2+ + S2- AS(s) Lp 1 1 20 K S 10 S2- + 2 H+ ô H2S ______________________ AS(s) + 2 H+ ô A2+ + H2S K K Lp Lp 20 K S 10 [A 2 ] [ H 2 S] Lp 10 20 [ H ]2 mit [H2S] = 0,1 mol·l-1 (siehe oben), Lp = [A2+] · [S2-] und c = [A2+] erhält man daher für die Löslichkeit (= Stoffmengenkonzentration des Metallkations): Lp 10 20 c 10 -1 [H + ]2 c = 1021 · Lp · [H+]2 Für saure Lösungen, z.B. pH = 0 ([H+] = 1 mol·l-1), ist c für Lp < 10-21 klein; das Gleichgewicht der oben genannten Reaktion liegt auf der linken Seite, d.h. das Sulfid ist schwer löslich. Sulfide mit Lp > 10-21 sind dagegen bei diesen Bedingungen löslich. In alkalischen Lösungen, z.B. pH = 8 ([H+] = 108 mol·l-1), sind auch Sulfide mit Lp > 10-21 schwer löslich. Ein Beispiel aus dem Analysengang soll aufgrund der unterschiedlichen Löslichkeitsprodukte die Fällung von Sulfiden aus saurer bzw. alkalischer Lösung zeigen: in saurer Lösung fallen aus HgS: Lp = 10-52 CuS: Lp = 10-36 CdS: Lp = 10-26 in alkalischer Lösung fallen aus ¦ ¦ FeS: Lp = 10-17 MnS: Lp = 10-14 ZnS: Lp = 10-24 - 10-22 Die saure Lösung der Cu-Gruppe muss daher nach dem Einleiten von H2S auf ca. das vierfache ihres Volumens mit H2O verdünnt werden (um den pH-Wert zu erhöhen), damit CdS ausfällt. Würde man auf ein größeres Volumen verdünnen, so könnte auch ZnS ausfallen. Für die Löslichkeit eines Sulfids A2S3 in H2S gesättigter saurer Lösung werden folgende Gleichgewichte herangezogen: A2S3(s) ô 2 A3+ + 3 S2- 6 H+ + 3 S2- ô 3 H2S Lp 1 KS 3 10 60 ___________________________ K A2S3(s) + 6 H+ ô 2 A3+ + 3 H2S K mit Lp = [A3+]2 [S2-]3 [A 3 2 und c ] [ H 2 S] 3 6 [H ] [A 3 Lp Lp 10 60 3 KS Lp 10 60 ] 2 erhält man: 2 Lp 10 c 60 Lp 4 ( 2 c ) [ H 2 S] 6 [H ] 3 ; [H2S] = 10-1moll-1 10 63 [ H ] 6 1,58 10 31 Lp [ H ]3 Die Löslichkeit des Sulfids ist klein, wenn das Löslichkeitsprodukt und die H+-Ionenkonzentration klein sind. 4.3 Löslichkeit von Sulfiden in reinem Wasser Hierbei spielt Hydrolyse der Sulfidionen S2- + H2O ô HS- + OHHS- + H2O ô H2S + OHeine wesentliche Rolle. Manche Sulfide mit größerem Lp sind bereits in Wasser etwas löslich. Daher ist es allgemeine Praxis, Sulfidniederschläge nicht mit reinem Wasser, sondern mit H2S- oder (NH4)2Sx-hältigem Wasser zu waschen. 4.4 Fällung zweier schwerlöslicher Sulfide nebeneinander Gegeben seien zwei Kationen A2+ und B2+, wobei Lp(AS) kleiner als Lp(BS) sein soll. Wir betrachten nun eine mit H2S gesättigte saure Lösung und denken uns die H+-Ionen-konzentration allmählich erniedrigt. Bei welcher H+-Konzentration beginnt AS auszufallen? Entsprechend Abschnitt 4.2., gilt folgende Gleichung: [A2+] = 1021 · Lp(AS) · [H+]2 und damit [ H ] 3,16 10 11 [A 2 ] Lp(AS) Setzen wir nun für [A2+] die Ausgangskonzentration ein, so erhalten wir den pH-Wert knapp vor Beginn der Fällung. Der entsprechende pH-Wert für den Fällungsbeginn von B2+ muss größer sein, d.h. die H+-Ionenkonzentration muss kleiner sein, da sich die für die Fällung notwendige H+-Ionenkonzentrationen nach dem Obigen umgekehrt verhalten wie die Wurzeln der Löslichkeitsprodukte. Wie groß ist der Anteil an A2+, der noch nicht ausgefällt wurde, wenn B2+ auszufallen beginnt? Es gilt (siehe 4.2): [A2+] = 1021 · Lp(AS) · [H+]2 [B2+] = 1021 · Lp(BS) · [H+]2 Für [B2+] können wir [B2+]o setzen, da die Fällung von B2+ gerade erst begonnen hat. Division der Gleichungen ergibt [A 2 ] Lp(AS) [ B 2 ]0 Lp( BS) Der gesuchte nicht ausgefällte Anteil von A2+ ist daher: [A 2 ] Lp(AS) [ B 2 ]0 [A 2 ]0 Lp( BS) [A 2 ]0 Diese Formel entspricht also der in Abschnitt 3.3.2. für zwei schwerlösliche, gegen H+-Ionen nicht empfindliche Salze hergeleiteten. Dort ist aber die Durchführung nur in Form einer allmählichen Zugabe des gemeinsamen Ions bis zum Beginn der Ausfällung des leichter löslichen der beiden Salze möglich, also in Form einer Titration. Hier können wir dagegen durch Einstellung eines definierten pH-Wertes erreichen, dass die S2--Ionenkonzentration praktisch nur für die Fällung von A2+ ausreicht, ohne dass B2+ mitfällt (vorausgesetzt wird Sättigung mit H2S). Nach der obigen Formel ist bei gleicher Ausgangskonzentration von A2+ und B2+ für 99,9 % Fällung, also [A2+]/[A2+]o = 1·10-3, ein Unterschied zwischen den beiden Lp-Werten von mindestens drei Zehnerpotenzen nötig (in der Praxis meist wesentlich mehr, da Einschließen von Fremdionen in Niederschläge mit großer Oberfläche oder Alterung der Niederschläge erfolgen kann). Darauf beruht die Trennung der Arsen- (As(III,V), Sb(III,V), Sn (II,IV)) und Kupfergruppe (Hg2+, Cu2+, Bi3+, Cd2+) von der Eisengruppe. 4.5 Hydroxidfällung Die Ausfällung von Hydroxiden durch Zusatz von OH--haltigen oder H+-aufnehmenden Stoffen wird in der qualitativen Analyse häufig durchgeführt; so fallen z.B. in der Eisengruppe Cr(OH)3 und Al(OH)3 durch das zugesetzte NH3 trotz der Gegenwart von (NH4)2S aus, da die Hydroxide dieser Elemente schwerer löslich sind als ihre Sulfide und daher zuerst ausfallen (siehe Seite 4.1). Weiters gehört hierher das Ausbleiben der Fällung von Mg(OH)2 durch NH3 in Gegenwart von NH4+. NH4+ kann hier den pH-Wert der Lösung durch Pufferwirkung so einstellen, dass die OH--Ionenkonzentration für die Ausfällung nicht ausreicht, d.h. das Löslichkeitsprodukt nicht erreicht wird. Die Fällung von Hydroxiden unterscheidet sich nicht grundsätzlich von der anderer schwerlöslicher Salze. Für die Löslichkeitsprodukte gilt z.B.: Lp(A(OH)2) = [A2+] · [OH-]2 Lp(A(OH)3) = [A3+] · [OH-]3 Die Niederschlagsbildung wird aber hier mit dem Gleichgewicht für die Eigendissoziation des Wassers sowie mit anderen in der Lösung ablaufenden Protonierungs- und Deprotonierungsreaktionen gekoppelt sein. Es wird meistens vorteilhaft sein, eine Hydroxidfällung unter Einbeziehung der Protonierungs- bzw. Deprotonierungsreaktion eines eingesetzten Puffergemisches zu formulieren. Als Beispiel sei die Auflösung von Mg(OH)2 in einem Puffergemisch NH4Cl-NH3 diskutiert. Mg(OH)2(s) 2 NH4+ + 2 OH- ô Mg2+ + 2 OH- Lp (Lp = 1 · 10-11) ô 2 NH3 + 2 H2O 1 KB2 (KB = 2 · 10-5) K Lp 2,5 10 2 KB2 ___________________________________________ Mg(OH)2(s) + 2 NH4+ ô Mg2+ + 2 NH3 + 2 H2O [ Mg 2 ] [ NH 3 ]2 Lp 2 K 2 2 2,5 10 KB [ NH 4 ] Die Löslichkeit von Mg(OH)2, d.h. die im Gleichgewicht vorliegende Konzentration an Mg2+, wird hier durch das Verhältnis [NH4+]/[NH3] bestimmt: c [ Mg 2 ] K [ NH 4 ]2 [ NH 3 ]2 4.6 Löslichkeitsgleichgewichte, gekoppelt mit Komplexbildung Komplexbildung besteht in der aufeinanderfolgenden Anlagerung von Liganden an ein Zentralatom (bei Kationen in wässrigen Lösungen handelt es sich meist um Ersatz von H2O-Molekülen der ersten Hydratationssphäre durch Liganden-Ionen oder -Moleküle). Es gilt z.B. für drei Neutralliganden (siehe Beispiel 3: Komplexbildungsreaktionen): ô [AL]n+ An+ + L n+ [AL] + L ô [AL2] n+ [AL2]n+ + L ô [AL3]n+ _____________________ ô [AL3]n+ An+ + 3 L 3 K1 K2 K3 ß3 = K1 · K2 · K3 [[AL 3 ] n ] [ A n ] [ L] 3 (Entsprechendes gilt für mehr Liganden). K1, K2, K3 sind die Bildungskonstanten der einzelnen Komplexe, ß3 wird als Bruttobildungskonstante bezeichnet. Reaktionen, bei denen die Komplexbildung mit Löslichkeitsgleichgewichten gekoppelt ist, sind für die Analyse sehr wichtig; es kann durch Zusatz eines Komplexbildners ein in Wasser sehr schwerlöslicher Niederschlag in Lösung gebracht werden, bzw. es wird die Bildung eines solchen Niederschlages durch den Komplexbildner verhindert. 4.6.1 Auflösung von AgCl in NH3 AgCl(s) ô Ag+ + Cl+ Ag + 2 NH3 ô [Ag(NH3)2]+ ________________________________ AgCl(s) + 2 NH3 ô [Ag(NH3)2]+ + Cl- Lp ß2 (Lp = 1·10-10) (ß2 = 1,6 ·107) K = Lp · ß2 = 1,6 ·10-3 [[Ag( NH 3 ) 2 ] ] [Cl ] K Lp 2 1,6 10 3 2 [ NH 3 ] Man kann nun die Löslichkeit c von AgCl in einer NH3-Lösung der Ausgangskonzentration co berechnen, wobei folgende Gleichgewichtskonzentrationen eingehen: c([Ag(NH3)2]+) = c(Cl-) = c; c(NH3) = (co - 2c) c2 K ; ( c 0 2 c) 2 Nimmt man z.B. an co = 2,0 mol·l-1 , so ergibt sich c = 0,074 mol·l-1 4.6.2 Auflösung von Sulfiden durch Komplexbildung mit Sulfidionen Einige Metalle, die schwerlösliche Sulfide bilden, geben mit einem Überschuss von Sulfidionen lösliche Komplexe (Thiokomplexe, Thiosalze). Im folgenden soll die Auflösung von Sulfiden durch solche Komplexbildungsprozesse in einfacher Weise betrachtet werden, d.h. es wird angenommen, dass nur die in den Reaktionsgleichungen angeführten Spezies eine Rolle spielen. A2S5(s) ô 2 A5+ + 5 S25+ 22A +8S ô 2 [AS4]3 __________________________ Lp (A = As, Sb) ß4 2 A2S5(s) + 3 S2- ô 2 [AS4]3- K = Lp · ß42 [[AS 4 ]3 ]2 K Lp 4 2 [S 2 ]3 Aus dieser Gleichgewichtskonstante folgt daher für die Auflösung eines Sulfids unter Thiosalzbildung, dass die Reaktion bei genügend großer Sulfidionenkonzentration von links nach rechts ablaufen muss. Fügt man H+-Ionen zu (starke Säure) so sinkt [S2-]: [S 2 ] 10 21 [ H ]2 (Siehe 4.1) Dadurch wird die Reaktion rückläufig; die Thiosalze zersetzen sich beim Ansäuern, d.h. Sulfid fällt aus und H2S wird freigesetzt. Die Bruttogleichung lautet: 2 [AS4]3- + 6 [H+] ô 3 H2S + A2S5 Wegen der Löslichkeit ihrer Sulfide in S2--Lösungen hoher Konzentration können die zur Thiosalzbildung befähigten Elemente As, Sb und Sn(IV) von Hg, Pb, Bi, Cu und Cd getrennt werden. Da auch das HgS eine gewisse Tendenz zur Thiosalzbildung zeigt, darf zum Abtrennen nur die weniger basische Ammoniumsulfidlösung verwendet werden; eine Natriumsulfidlösung würde eine zu hohe OH--Konzentration und damit auch eine zu hohe S2--Konzentration besitzen. Um die Oxidation von Sn(II), das keine Thiosalze bildet, zum Sn(IV) zu sichern, wird gelbes Ammoniumpolysulfid, (NH4)2Sx, verwendet. Hierdurch werden gleichzeitig As(III) zu As(V) und Sb(III) zu Sb(V) oxidiert, wobei Schwefel der Oxidationszahl 0 in solchen der Oxidationszahl -II übergeht. 4.6.3 Hydroxokomplexe Eine Anzahl von Hydroxiden bildet mit überschüssigen OH--Ionen wasserlösliche Hydroxokomplexe: A(OH)n + m OH- ô [A(OH)n+m]mZn(OH)2 + 2 OH- ô [Zn(OH)4]2(Es treten auch Zwischenstufen wie [Zn(OH)3]- auf.) Für die Auflösung von Zn(OH)2 in starker Lauge (NaOH) kann man - bei Nichtberücksichtigung von [Zn(OH)3]- - formulieren: Zn(OH)2(s) ô Zn2+ + 2 OH2+ Zn + 4 OH ô [Zn(OH)4]2______________________________ Zn(OH)2(s) + 2 OH- ô [Zn(OH)4]2- K Lp (Lp = 10-17) ß4 (ß4 = 1016) K = Lp·ß4 = 10-1 [[ Zn(OH ) 4 ]2 ] = Lp·ß4 = 10-1 [OH ]2 Derartige Hydroxide vermögen daher sowohl mit Säuren als auch mit Basen zu reagieren, sie sind amphoter. Zu diesen gehören die Hydroxide von Al, Zn, Sn(II), Sn(IV) und Pb(II). Aus den Komplexsalzlösungen kann durch Verschiebung des Komplexgleichgewichtes infolge Verringerung der OH--Konzentration wieder das Hydroxid ausgefällt werden; hierzu sind alle Stoffe imstande, die H+Ionen abgeben können (Säuren nach Brnstedt), z.B. das Ion NH4+. Für die Ausfällung von Al(OH)3 aus einer Lösung von [Al(OH)4]- kann man - wieder unter Nichtberücksichtigung von Zwischenstufen - formulieren: Al3+ + 3 OH- ô Al(OH)3(s) [Al(OH)4]- ô Al3+ + 4 OH- NH4+ + OH- ô NH3 + H2O 1 Lp 1 4 1 KB (Lp = 10-33) (ß4 = 1032) (KB = 2 ·10-5) _________________________________________ [Al(OH)4]- + NH4+ ô K Al(OH)3(s) + NH3 + H2O K 1 5 105 4 Lp K B [ NH 3 ] 1 5 105 [[Al(OH ) 4 ] ] [ NH 4 ] 4 Lp K B 5. Redoxreaktionen In diesem Abschnitt werden die Beziehung zwischen den Gleichgewichtskonstanten von Redoxreaktionen und den elektrochemischen Potentialen sowie einige Anwendungen dieser Beziehung behandelt. 5.1 Oxidation und Reduktion Redoxreaktionen sind Prozesse, bei denen Elektronenübertragung zwischen Ionen bzw. Atomen oder Molekülen stattfindet. In jeder Redoxreaktion sind ein Reduktions- und ein Oxidationsprozess miteinander gekoppelt, d.h. es nehmen zwei „Redoxpaare“ an einem solchen Prozess teil. Als Redoxpaar bezeichnet man die reduzierte und oxidierte Form eines Stoffes, die durch Elektronenabgabe bzw. -aufnahme ineinander überführt werden. (Siehe Beispiel 4a: Redoxreaktionen). Daraus wird ersichtlich, dass bei einem chemischen Vorgang keine Oxidation ohne gleichzeitige Reduktion und umgekehrt stattfinden kann. Es sei daran erinnert, dass sich ein Zinkblech, welches in eine CuSO4- oder Cu(NO3)2-Lösung taucht, mit metallischem Kupfer überzieht. Allgemein ist die Abscheidung von Metallen aus ihren Lösungen durch Zugabe weniger edler Metalle möglich. Einige Nachweise in der qualitativen Analyse können mit solchen Redoxreaktionen im Reagenzglas durchgeführt werden. z.B. wird Antimon auf einem Eisennagel oder Quecksilber und Silber auf einem Kupferblech abgeschieden. 5.2 Elektromotorische Kraft (EMK), Nernstsche Gleichung In einem galvanischen Element laufen Oxidations- und Reduktionsreaktionen räumlich getrennt an geeigneten Elektroden ab.z.B. taucht beim Daniell-Element ein Zinkstab in eine ZnSO4- und ein Kupferstab in eine CuSO4-Lösung ein. Beide Lösungen sind durch ein Diaphragma oder einen Stromschlüssel von einander getrennt. Werden die beiden Stäbe durch einen Schließungsdraht elektrisch leitend verbunden, so fließen die Elektronen vom Zink zum Kupfer; Kupfer scheidet sich am Kupferstab ab, Zink geht vom Zinkstab in Lösung. Der chemische Vorgang ist derselbe wie beim vorhin erwähnten Versuch im Reagenzglas. Wird jedoch ein hochohmiges Messinstrument in den äußeren Leiter gebracht, so lässt sich die Spannung dieser galvanischen Kette nahezu stromlos messen. Diese gemessene Spannung oder Potentialdifferenz zwischen den beiden Halbelementen (Metallstäben) ist die elektromotorische Kraft der galvanischen Kette, für die die Nernstsche Gleichung gilt: E E0 RT ln Q zF (1) Es bedeutet: E die EMK der Kette (V), E0 die Standard EMK oder das Standardpotential (V), R die Gaskonstante (J·mol-1·K-1), T die absolute Temperatur (K), z die Zahl der umgesetzten Elektronen pro Formelumsatz, F die Faradaykonstante (C·mol-1), Q den Reaktionsquotienten (Konzentrationsquotient) der an der Reaktion beteiligten Stoffe. Die Nernstsche Gleichung beschreibt für die elektromotorische Kraft (Redoxpotential) eines Redoxsystems. Die Standardpotentiale E0 haben für verschiedene Redoxsysteme charakteristische Werte. Der Reaktionsquotient Q drückt die Konzentrationsabhängigkeit der EMK (Potential) eines Redoxsystems aus. Aufgrund der auftretenden EMK kann die galvanische Kette elektrische Arbeit leisten. Lässt man daher den Strom fließen, so sinkt die gemessene Spannung (elektromotorische Kraft) E auf Null ab, d.h. die galvanische Kette erreicht den Gleichgewichtszustand. Der Reaktions-quotient (Q) geht in die Gleichgewichtskonstante (K) über. Somit gilt für die Nernstsche Gleichung: E0 RT ln K zF (2) 5.3 Standardpotentiale Die Standardpotentiale von Redoxsystemen erhält man durch Messung der EMK eines galvanischen Elements, bei dem ein Standardhalbelement (c = 1 mol · l-1) gegen eine Standardwasserstoffelektrode geschaltet ist. Für die gemessene elektromotorische Kraft (E12) gilt: E12 = E1 - E2 (3) Aus der Stellung der Redoxsysteme in der Spannungsreihe kann vorausgesagt werden, welche Redoxreaktion möglich ist. Als Beispiel werden die zwei Redoxpaare Cu2+|Cu und H+|H2 verwendet. Die beiden Teilgleichungen werden zunächst als Reduktion angeschrieben: Reduktionsschreibweise (Elektronen links). Cu2+ + 2 e- ô Cu E1 2 H+ + 2 e- ô H2 E2 In diesem Fall ist die Zahl der ausgetauschten Elektronen in beiden Redoxgleichungen gleich. Da Kupfer edler als Wasserstoff ist (siehe Spannungsreihe), werden Cu2+-Ionen zu Cu-Metall reduziert, H2 dagegen wird zu H+-Ionen oxidiert. Um beide Gleichungen zu addieren, multipliziert man die Gleichung des Redoxpaares, an dem Oxidation erfolgt, mit (-1) (Umkehrreaktion: siehe Seite 3). Bei einer Umkehrreaktion ändert sich das Vorzeichen des Potentials (E). Cu2+ + 2 e- ô Cu H2 ô 2 H+ + 2 e_____________________ Cu2+ + H2 ô Cu + 2 H+ E1 -E2 = -E0(H+|H2) = 0 E12 = E1 - E2 = E1 - 0 Mit Gleichung (1) ergibt sich: E 1 E 10 1 RT ln 2 F [Cu 2 ] und mit der Bedingung c(Cu2+) = 1 mol ·l-1 (siehe oben) wird E1 = E 10 = E0 (Cu2+|Cu) Die gemessene EMK E12 ist gleich dem Standardpotential E0 (Cu2+|Cu). Standardpotentiale sind Relativwerte bezogen auf die Standardwasserstoffelektrode, deren Standardpotential willkürlich null gesetzt wurde. 5.4. Potentiale von Halbelementen Wird in einer galvanischen Kette ein Redoxpaar beliebiger Konzentration gegen eine Standardwasserstoffelektrode geschaltet, so gilt für die gemessene elektromotorische Kraft Gleichung (3). Die Reaktionsgleichungen der Redoxpaare Ox|Red und H+|H2 werden zunächst wieder in der Reduktionsschreibweise angeschrieben: Ox + z e- ô Red 2H+ + 2 e ô H2 E1 E2 Ox bedeutet die oxidierte und Red die reduzierte Form des Redoxpaares. Da hier die Zahl der ausgetauschten Elektronen in beiden Gleichungen unterschiedlich ist, wird die Reaktionsgleichung für das Redoxpaar Ox|Red mit 2 und jene für das Redoxpaar H+|H2 mit z multipliziert. Die Potentiale E1 und E2 bleiben bei dieser Multiplikation unverändert. Nimmt man an, dass E1 positiver ist als E2, so wird Ox durch H2 reduziert. Daher wird die Gleichung für das Redoxpaar H+|H2 in umgekehrter Richtung angeschrieben. Die Gleichungen für die beiden Redoxpaare werden nun addiert: Ox + z e- ô Red ·1 ½H2 ô H+ + e·z __________________________ Ox + ½z H2 ô Red + z H+ E 1 E 10 E1 -E2 = -E0 (H+|H2) = 0 E12 = E1 - E2 = E1 - 0 RT [Re d] RT [Ox ] ln ln E 10 zF [Ox ] zF [Re d ] (4) Die gemessene EMK ist gleich dem Potential des Halbelementes. Enthält die Reaktionsgleichung eines Redoxpaares auch andere Ionen, die für die Umwandlung der beiden Formen ineinander notwendig sind, so sind deren Konzentrationen zu berücksichtigen. Z.B. gilt für das Redoxpaar MnO4-|Mn2+ (gemessen gegen die Standardwasserstoffelektrode) folgende Reaktionsgleichung: MnO4- + 8 H+ + 5e- ô Mn2+ + 4 H2O Für das Potential des Halbelementes ergibt sich daher: E 1 E 0 (MnO 4 |Mn 2 ) RT [Mn 2 ] ln 5F [MnO 4 ][H ]8 5.5. Standardpotentiale von Elementen mit mehreren Oxidationsstufen. Gibt es von Elementen Ionen mit mehreren unterschiedlichen Ladungen, so sind auch mehrere Redoxpaare zu berücksichtigen. Allgemein: Für n Oxidationsstufen gibt es ½ · n · (n-1) Redoxpaare. Ein gewünschtes Standardpotential kann aus den n-1 tabellierten Standard-potentialen berechnet werden. Da die Zahlenwerte der Standardpotentiale in der Literatur etwas schwanken, werden in diesem Text nur 2 Nachkommastellen angegeben. Als Beispiel werden die drei Oxidationsstufen des Eisens herangezogen: Fe, Fe2+, Fe3+. E0(Fe3+|Fe) bedeutet das Standardpotential der Redoxgleichung: Fe3+ + 3e- ô Fe (Reduktionsschreibweise!) Will man nun das Standardpotential E0(Fe3+|Fe) aus den tabellierten Standardpotentialen E0(Fe3+|Fe2+) und E0(Fe2+|Fe) berechnen, so werden die Redoxgleichungen wie folgt angeschrieben. Fe3+ + eô Fe2+ 2+ Fe + 2 e- ô Fe __________________ Fe3+ + 3 e- ô Fe E0(Fe3+|Fe2+) = +0,77 V E0(Fe2+|Fe) = -0,44 V E0(Fe3+|Fe) = ? 1 E0(Fe3+|Fe2+) + 2 E0(Fe2+|Fe) = 3 E0(Fe3+|Fe) E 0 ( Fe 3 | Fe) 0,77 2 ( 0,44) 0,037 V 0,04 V 3 Treten nicht nur in den Teilgleichungen sondern auch in der Endgleichung Elektronen auf, so sind bei der Addition der Gleichungen die Standardpotentiale der jeweiligen Gleichung mit der Zahl der Elektronen dieser Gleichung zu multiplizieren. Begründung: Bei der Addition von Gleichungen können die vom Weg unabhängigen freien Enthalpien (Gibbssche Energie) ebenfalls addiert werden und ergeben die freie Enthalpie der Endgleichung. Für die oben angeführten Gleichungen der Oxidationsstufen des Eisens erhält man: G0(Fe3+|Fe2+) + G0(Fe2+|Fe) = G0(Fe3+|Fe) mit G0 = -zFE0 folgt -1FE0(Fe3+|Fe2+) + (-2FE0(Fe2+|Fe)) = -3FE0(Fe3+|Fe) 5.6. Berechnung der Gleichgewichtskonstanten von Redoxreaktionen Mit Gleichung (2) erhält man aus der Standard-EMK einer galvanischen Kette die Gleichgewichtskonstante der entsprechenden Redoxreaktion. Die Standard-EMK von 0 Ketten ( E 12 ) wird aus den Standardpotentialen der beiden Redoxpaare ( E 10 , E 02 ) berechnet. Die meisten Standardpotentiale von Redoxpaaren sind tabelliert. Es gilt: 0 E 12 = E 10 - E 02 (5) Die Gleichung für die Redoxreaktion (Gesamtreaktion) ergibt sich aus den Gleichungen (Teilgleichungen) der beiden Redoxpaare, die in der Reduktionsschreibweise angegeben werden. Ox und Red bezeichnen die oxidierten bzw. reduzierten Formen der Redoxpaare. Ox1 + z1e- ô Red1 - Ox2 + z2e ô Red2 E 10 E 02 (6) (7) Man nimmt an, dass Gleichung (6) eine Reduktion, Gleichung (7) eine Oxidation beschreibt (Umkehr von Gleichung (7) gibt (7')). Um die Gesamtreaktion (8) zu erhalten, werden die Teilgleichungen (6) und (7') addiert und in bezug auf die ausgetauschten Elektronen ausgeglichen. Die Gleichung für das erste Redoxpaar (6) wird mit z2, die für das zweite (7') mit z1 multipliziert. Von dieser Multiplikation werden die Standardpotentiale ( E 10 und E 02 ) nicht berührt. Ox1 + z1 e- ô Red1 ·z2 E 10 - E 02 (6) Red2 ô Ox2 + z2 e·z1 (7') __________________________________ Niederschlages. Dieses Verhalten ist auch die Erklärung für die Kalomelreaktion zum Nachweis von Hg22+: Hg2Cl2(s) + 2 NH3 = Hg + HgNH2Cl + NH4Cl