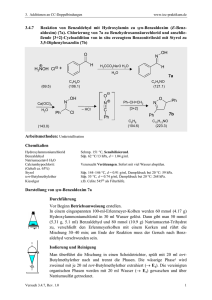
Dokument_18.
Werbung

Entwicklung von neuen metall- und organokatalysierten Methoden zur enantioselektiven Sulfidoxidation mit H2O2 Der Naturwissenschaftlichen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades Dr. rer. nat. vorgelegt von Dipl.-Chem. Kerstin Angela Stingl aus Neustadt a.d. Aisch Als Dissertation genehmigt von der Naturwissenschaftlichen Fakultät der Friedrich-AlexanderUniversität Erlangen-Nürnberg. Tag der mündlichen Prüfung: 22. Februar 2013 Vorsitzender der Prüfungskommission: Prof. Dr. Johannes Barth Erstberichterstatter: Prof. Dr. Svetlana Tsogoeva Zweitberichterstatter: PD. Dr. Norbert Jux II Die vorliegende Arbeit wurde am Institut für Organische Chemie des Departments Chemie und Pharmazie der Friedrich-Alexander-Universität Erlangen-Nürnberg unter Leitung von Prof. Dr. Svetlana Tsogoeva in der Zeit von Juli 2008 bis Juni 2012 erstellt. III Teile dieser Arbeit sind bereits veröffentlicht: Kerstin A. Stingl, Svetlana B. Tsogoeva, Recent Advances in Sulfoxidation Reactions: A Metal-free Approach, Tetrahedron: Asymmetry 2010, 21, 1055-1074. Shengwei Wei, Kerstin A. Stingl, Katharina M. Weiß, Svetlana B. Tsogoeva, Bifunctional Organocatalysis with N-Formyl-L-Proline: A Novel Approach to Epoxide Ring Opening and Sulfide Oxidation, Synlett 2010, 707-711. Kerstin A. Stingl, Katharina M. Weiß, Svetlana B. Tsogoeva, Asymmetric vanadium- and iron-catalyzed oxidations: new mild (R)-modafinil synthesis and formation of epoxides using aqueous H2O2 as a terminal oxidant, Tetrahedron 2012, 68, 8493-8501. Poster: Poster-Präsentation: Kerstin A. Stingl, Shengwei Wei, Katharina M. Weiß, Svetlana B. Tsogoeva, Bifunctional Organocatalysis with N-Formyl-L-Proline: A Novel Approach to Epoxide Ring Opening and Sulfide Oxidation “3rd EuCheMS Chemistry Congress”, Nürnberg, 29/08/2010 - 02/09/2010. Poster-Präsentation: Kerstin A. Stingl, Svetlana B. Tsogoeva, New Iron (III) Complexes for Asymmetric Sulfide Oxidation with Aqueous Hydrogen Peroxide “3rd Erlangen Symposium on Redox-Active Metal Complexes: Control of Reactivity via Molecular Architecture”, Erlangen, 05/10/2011 - 08/09/2011. Vorträge: SFB 583 - Kolloquium, Redoxactive Non-Heme Metal Complexes for Enantioselective Sulfoxidation and Epoxidation Reactions, Erlangen, 2008 und 2009. IV Für meine Eltern V “Auch aus Steinen, die dir in den Weg gelegt werden, kannst du etwas Schönes bauen.” Erich Kästner VI Inhaltsverzeichnis 1 Allgemeine Einleitung ......................................................................................................1 1.1 Chirale Sulfoxide – Eine fundamentale Betrachtung .......................................................... 1 1.2 Asymmetrische Sulfidoxidation ........................................................................................... 3 1.2.1 Metallkatalysierte Oxidation von prochiralen Sulfiden ........................................................ 5 1.2.1.1 Vanadiumhaltige Katalysatoren in der enantioselektiven Sulfidoxidation: H2O2 als Oxidationsmittel und mechanistische Aspekte ................................................................... 6 1.2.1.2 Eisenhaltige Katalysatoren in der enantioselektiven Sulfidoxidation: Stand der Wissenschaft ....................................................................................................................... 9 1.2.2 Organokatalysierte Sulfidoxidation ................................................................................... 15 1.2.2.1 Organokatalysatoren in der enantioselektiven Sulfidoxidation: Stand der Wissenschaft . 16 1.3 Sulfoxide als biologisch aktive Verbindungen - Anwendung in der pharmazeutischen Industrie am Beispiel von Esomeprazol und Armodafinil ................................................. 21 2 Motivation und Zielsetzung ............................................................................................25 3 Ergebnisse und Diskussion ...........................................................................................29 3.1 Design und Synthese chiraler Liganden und deren Anwendung auf die metallkatalysierte Oxidation von Thioanisol ................................................................................................... 29 3.1.1 Liganden basierend auf Formamiden, Harnstoffen und Peptiden .................................... 30 3.1.2 Liganden basierend auf Schiffschen-Basen und deren Derivaten ................................... 44 3.1.3 Liganden basierend auf Hybridverbindungen (peptidhaltige Schiffsche-Basen) .............. 79 3.2 Organokatalysierte Oxidation von Thioanisol ................................................................... 91 3.2.1 N-Formyl-L-Prolin als Organokatalysator ......................................................................... 91 3.2.2 BINOL-Phosphate als Organokatalysatoren ..................................................................... 95 VII 3.3 Anwendung ausgewählter metall- und organokatalytischer Systeme auf die Synthese von biologisch aktiven Verbindungen: (S)-Omeprazol und (R)-Modafinil................................ 98 3.3.1 Substratsynthesen: Omeprazol und Modafinil .................................................................. 98 3.3.2 Katalysereaktionen ......................................................................................................... 100 4 Zusammenfassung ...................................................................................................... 105 4 Summary .................................................................................................................... 111 5 Experimenteller Teil .................................................................................................... 116 5.1 Synthesen der Substrate und Referenzverbindungen .................................................... 119 5.2 Synthesen und katalytische Experimente (Kapitel 3.1) .................................................. 124 5.2.1 Synthese von Formamid- und Harnstoffliganden ........................................................... 124 5.2.2 Katalysereaktionen mit Formamid- und Harnstoffliganden ............................................. 130 5.2.3 Synthese von Schiffsche-Base Liganden, deren Derivaten und Metallkomplexen ........ 132 5.2.4 Katalysereaktionen mit Schiffsche-Base Liganden und deren Derivaten ....................... 149 5.2.5 Synthese von Aminosäure- bzw. Peptidbausteinen und Hybridverbindungen ............... 151 5.2.6 Katalysereaktionen mit Hybridverbindungen .................................................................. 169 5.3 Katalytische Experimente unter Organokatalyse (Kapitel 3.2) ....................................... 170 5.4 Katalytische Experimente zur enantioselektiven Synthese von (S)-Omeprazol und (R)Modafinil (Kapitel 3.3) ..................................................................................................... 171 6 Referenzen ................................................................................................................. 172 7 Anhang ....................................................................................................................... 183 VIII Abkürzungsverzeichnis abs. absolutiert acac Acetylacetonat ATR Attenuated Total Reflection API Active Pharmaceutical Ingredient Äq. Äquivalente Bn Benzyl Boc t-Butyloxycarbonyl Boc2O Di-tert-butyldicarbonat Cbz Carboxybenzyl CDI N,N’-Carbonyldiimidazol CHP Cumolhydroperoxid δ chemische Verschiebung d Tag DC Dünnschichtchromatographie DCM Dichlormethan DET Diethyltartrat DMF N,N‘-Dimethylformamid DMM Dimethoxymethan DMSO Dimethylsulfoxid EA Elementaranalyse EI Elektronenstoßionisation ESI Elektronensprayionisation FAB Fast Atom Bombardment GC Gaschromatographie IX Gly Glycin h Stunde His Histidin HPLC High Performance Liquid Chromatography H2Pydic 2,6-Pyridindicarbonsäure HR High Resolution IR Infrarotspektroskopie J skalare Kopplungskonstante LM Lösungsmittel MALDI-TOF Matrix Assisted Laser Desorption/Ionization - Time of Flight Me Methyl MeCN Acetonitril m-CPBA meta-Chlorperbenzoesäure Min Minute MS Massenspektrometrie m/z Verhältnis von Masse zu Ladung n.b. nicht bestimmt NMR Nuclear Magnetic Resonance p para PE Petrolether Ph Phenyl Phe Phenylalanin ppm Parts Per Million quant. quantitativ RT Raumtemperatur rpm Revolutions Per Minute X Ser Serin Smp Schmelzpunkt TBHP t-Butylhydroperoxid TEA Triethylamin Temp. Temperatur TFA Trifluoressigsäure THF Tetrahydrofuran Thr Threonin Tos Tosyl t bzw. tert tertiär UHP Urea Hydroperoxide Val Valin [Wert] Substratkonzentration XI KAPITEL 1 1 1.1 Allgemeine Einleitung Chirale Sulfoxide – Eine fundamentale Betrachtung Aufgrund ihrer außerordentlich vielseitigen Verwendbarkeit stellen chirale Sulfoxide eine äußerst wichtige und attraktive Verbindungsklasse in der modernen organischen Chemie und Biochemie dar.[1] Zum einen exisitieren zahlreiche schwefelhaltige Arzneimittel, bei deren Synthese Sulfoxide als Intermediate fungieren, zum anderen werden chirale Sulfoxide selbst als pharmazeutisch aktive Zielverbindungen eingesetzt.[2] Weitere Anwendung finden diese in der organischen Synthese in Form von chiralen Auxiliaren[2b, 3], chiralen Schwefelliganden[4] und Organokatalysatoren[5] (Abbildung 1-1). Abbildung 1-1: Ausgewählte chirale Sulfoxide in ihrer Anwendung als pharmazeutischem Wirkstoff ((R)-Modafinil (1)), als chiralem Auxiliar (2) und als Organokatalysator (3). Sulfoxide gehören zur Klasse der Schwefelverbindungen und besitzen die allgemeine Struktur R1-(S=O)-R2, in der die Sulfinylgruppe an zwei organische Reste gebunden ist. Sulfoxide werden oft in Analogie zum Strukturmotiv von Carbonylverbindungen beschrieben, wobei anders als bei Carbonylverbindungen durch konstitutiv verschiedene Substituenten (R1 ≠ R2) am Schwefel ein 1 Kapitel 1 Allgemeine Einleitung chirales Zentrum erzeugt wird. Hierbei dient das freie Elektronenpaar des Schwefels als vierter Substituent, da nicht alle Valenzelektronen des Schwefels an Bindungen beteiligt sind (Abbildung 1-2).[6] Abbildung 1-2: Darstellung der Chiralität von Sulfoxiden anhand beider möglichen enantiomeren Formen. Im Unterschied zur planaren Carbonylfunktion weisen Sulfoxide jedoch eine trigonal pyramidale Geometrie auf. Dieser Unterschied ist zum einen auf eine Überlappung eines p-Orbitals des Sauerstoffatoms mit einem d-Orbital des Schwefelatoms zurückzuführen (d-π-Wechselwirkung), zum anderen fungiert das freie Elektronenpaar am Schwefel als ein vierter Substituent - durch die Abstoßung der Bindungselektronen und dem freien Elektronenpaar wird eine tetrahedrale sp3Hybridisierung hervorgerufen. Die S-O-Bindung wird durch elektrostatische Wechselwirkungen zwischen dem partiell positiven Schwefelatom und dem partiell negativen Sauerstoffatom polarisiert, sodass zwei mögliche Grenzstrukturen die Bindungsverhältnisse im Sulfoxid beschreiben.[6-7] Aufgrund der synthetischen Verfügbarkeit chiraler Sulfoxide in beiden enantiomeren Formen, ihrer hohen optischen Stabilität und der Tatsache, dass sie als effiziente Träger chiraler Information fungieren, haben sich chirale Sulfoxide als leistungsfähige „chiral controller" in verschiedenen asymmetrischen Reaktionen etabliert. Die hohe optische Stabilität begründet sich dadurch, dass diese im Gegensatz zu anderen Heteroatomchiralitätszentren, wie beispielsweise stickstoffhaltigen Verbindungen der allgemeinen Form NR3, eine hohe Inversionsbarriere aufweisen und somit keiner „pyramidalen Inversion“ bei RT oder höheren Temperaturen unterliegen. Somit racemisieren enantiomerenreine Sulfoxide mit nur wenigen Ausnahmen, wie beispielsweise Benzyl- oder 2 Kapitel 1 Allgemeine Einleitung Allylsulfoxiden, erst ab einer Temperatur von über 200 °C. Die Charakterisierung von chiralen Sulfoxiden als effiziente Träger der chiralen Information wird dadurch begründet, dass durch eine große stereoelektronische Divergenz der am Schwefel befindlichen Substituenten (Sauerstoffatom, freies Elektronenpaar und zwei organische Gruppen) eine wohldefinierte chirale Umgebung um das Schwefelatom erzeugt werden kann. Weiterhin kann eine wirkungsvolle Übertragung der chiralen Information durch die Möglichkeit der Koordination des Schwefel- oder Sauerstoffatoms der Sulfoxideinheit aufgrund ihrer hohen Polarisierbarkeit an Lewis-Säuren oder Übergangsmetalle erreicht werden.[2b] Basierend auf diesen Eigenschaften und den daraus resultierenden Anwendungen wurde eine Vielzahl an Methoden entwickelt, um Sulfoxide in ihrer enantiomerenreinen Form zu erhalten. Dieser Sachverhalt wurde bereits ausführlich in verschiedenen Reviews beschrieben. [2b, 8] Unter diesen Methoden befinden sich beispielsweise die Spaltung racemischer Sulfoxide und asymmetrische Syntheseverfahren. Da aufgrund der schlechten natürlichen Verfügbarkeit eine Darstellung aus dem „chiral pool“ eher ungeeignet und die Spaltung racemischer Sulfoxide wegen des limitierenden Faktors bezüglich der chemischen Ausbeute (50%) grundsätzlich weniger attraktiv ist, hat sich die asymmetrische Synthese als eine gute Alternative etabliert.[2a] 1.2 Asymmetrische Sulfidoxidation In der asymmetrischen Synthese werden zur Herstellung von optisch aktiven Sulfoxiden grundsätzlich zwei Strategien verfolgt, welche entweder auf dem Gebrauch von chiralen Auxiliaren/Reagenzien oder auf der Anwendung eines asymmetrischen Katalyseverfahrens beruhen.[2a] Im Rahmen der ersten Strategie wurde eine bis zum heutigen Zeitpunkt bedeutende Methode Anfang der 60er Jahre entwickelt, bei der chirale Sulfoxide durch nukleophile Substitution an optisch aktiven Sulfinaten mittels metallorganischen Verbindungen (Grignard Reagenzien) 3 Kapitel 1 Allgemeine Einleitung synthetisiert werden (Andersen-Methode, 1962).[9] Obwohl diese Methode chirale Sulfoxide mit hohen Enantioselektivitäten liefert, ist sie jedoch überwiegend auf die Synthese von Alkyl-Aryloder Aryl-Aryl-Sulfoxiden beschränkt. Den Schlüsselschritt der Andersen-Methode stellt die Synthese der jeweiligen Menthylsulfinatester dar, wodurch die Entwicklung von neuen Sulfinatestern sowie deren Derivaten in Hinblick auf die Substraterweiterung stetig voran getrieben wird.[8b] Eine weitere Variante beinhaltet den Gebrauch stöchiometrischer Mengen von chiralen Oxidationsmitteln (z.B. Oxaziridinen).[10] Ihre Anwendung ist jedoch mit relativ hohen Kosten verbunden und zudem sind diese in ihrer Verfügbarkeit limitiert.[2a] Eine der attraktivsten Synthesemethoden zur Darstellung von chiralen Sulfoxiden stellt zweifelslos die katalytische enantioselektive Oxidation von prochiralen Sulfiden dar, welche einen einfachen sowie direkten und atomeffizienten Zugang der wertvollen Produkte ermöglicht.[1a, 8b] Oxidation kann auf unterschiedliche Weisen durchgeführt werden: metallkatalysiert,[1a, Diese 2b, 8, 11] metallfrei[8b, 12] oder durch biologische Oxidation.[2b, 13] Da sich die Anwendung von Biokatalysatoren (isolierte Enzyme, Ganzzellkulturen) oft auf ihr natürliches Substrat begrenzt oder diese zu empfindlich gegenüber variablen Reaktionsbedingungen sind, hat sich die Entwicklung von synthetischen Katalysatoren, wie z.B. nicht-porphyrinhaltigen Metallkomplexen oder den in jüngerer Zeit entwickelten Organokatalysatoren, bewährt. Vorteile liegen hier in der hohen Variationsmöglichkeit der Liganden, ihrer Robustheit sowie der Anwendbarkeit auf ein größeres Substratspektrum. Auf diesem Weg wird auch ein einfacherer Zugang zu beiden Enantiomeren ermöglicht. 4 Kapitel 1 1.2.1 Allgemeine Einleitung Metallkatalysierte Oxidation von prochiralen Sulfiden Die metallkatalysierte asymmetrische Sulfidoxidation hat sich seit dem Beginn der 80er Jahre als eine der erfolgreichsten Methoden in der Erzeugung optisch aktiver Sulfoxide etabliert. Die Pionierarbeiten sind dabei Kagan[14] und Modena[15] zuzuordnen, welche unabhängig voneinander modifizierte Sharpless-Reagenzien[16] in der enantioselektiven Sulfidoxidation untersuchten. Beide Systeme beruhen mit wenigen Ausnahmen (u. a. dem Zusatz von H2O im Kagan-System) auf der Verwendung von Ti(Oi-Pr)4 als Metallquelle, Diethyltartrat (DET) als chiralem Auxiliar und t-Butylhydroperoxid (TBHP) als terminalem Oxidationsmittel und führen in der Oxidation von einigen prochiralen Sulfiden zu hohen Enantioselektivitäten.[17] Basierend auf diesen Arbeiten wurde bis dato eine Reihe verschiedener chiraler chelatisierender Liganden, wie beispielsweise bidentate Diole (4)[18], tridentate Schiffsche-Basen (5)[19] und tetradentate salenartige Liganden (6)[20] synthetisiert (Abbildung 1-3), welche in Kombination mit diversen Metallzentren, vorwiegend Titan und Vanadium, effektive katalytische Systeme in der Oxidation von prochiralen Sulfiden bildeten.[8a] Abbildung 1-3: Exemplarische Darstellung von verschiedenen polydentaten Liganden. Eine Problematik in der Oxidation von Sulfiden zu Sulfoxiden stellt die potentielle Überoxidation zu Sulfonen dar. Ist während des Oxidationsprozesses eine Sulfonbildung in Kombination mit einer Enantioselektivität bezüglich des gewünschten Sulfoxids zu beobachten, so kann diese Enantioselektivität auch teilweise durch kinetische Racematspaltung hervorgerufen worden sein (Schema 1.2-1). Das heißt, die gebildeten 5 Enantiomere besitzen unterschiedliche Kapitel 1 Allgemeine Einleitung Geschwindigkeitskonstanten bezüglich der Weiteroxidation zum Sulfon, wodurch das langsamer reagierende Enantiomer im Reaktionsgemisch angereichert wird und einen höheren ee-Wert produziert. Die Ausbeute des Sulfoxids wird jedoch gleichzeitig gesenkt. Außerdem kann die Gegenwart des Sulfons die Isolierung des Sulfoxids in einem hohen Reinheitsgrad erschweren.[8a] Schema 1.2-1: Schematische Darstellung der kinetischen Racematspaltung von Sulfoxiden. Dieses Phänomen wird beispielsweise unter Verwendung von verschiedenen titanhaltigen Katalysatoren,[18b, 21] aber auch in der Vanadiumkatalyse beobachtet.[22] So fordert diese Problematik, sowie die Oxidation von anspruchsvollen Substraten wie z.B. Alkyl-Alkyl- bzw. hochfunktionalisierten Sulfiden und/oder die Verwendung von H2O2 als einfachem Oxidationsmittel, die Entwicklung von neuen effizienteren Systemen weiterhin heraus. 1.2.1.1 Vanadiumhaltige Katalysatoren in der enantioselektiven Sulfidoxidation: H2O2 als Oxidationsmittel und mechanistische Aspekte Die Entwicklung von praktikablen Synthesen, welche sich unter anderem durch ihre einfache Handhabung, eine leichte Verfügbarkeit und geringe Toxizität der Reaktanden auszeichnen, rückte in jüngster Zeit immer mehr in den Fokus der Wissenschaft. Erste Ansätze dazu lieferten beispielsweise Jacobsen im Jahr 1992,[20] gefolgt von Katsuki 1994,[23] durch den Einsatz von H2O2 in Kombination mit Mangan(III)salenkomplexen in der Oxidation von prochiralen Alkyl-Aryl-Sulfiden. Obwohl H2O2 als Oxidationsmittel geeignet war, wurden mit PhIO höhere Enantioselektivitäten 6 Kapitel 1 Allgemeine Einleitung erzielt.[23-24] Ein Meilenstein in der vanadiumkatalysierten Sulfidoxidation gelang Bolm und Bienewald im Jahr 1995 durch den erfolgreichen Einsatz von wässrigem H2O2 und dessen Aktivierung unter der Verwendung von einfach zugänglichen Schiffsche-Base Liganden (abgeleitet von chiralem t-Leucinol und verschieden substituierten Salicylaldehyden) und VO(acac)2.[19a] Der Metallkatalysator wurde in situ generiert und die Oxidation unter einfachen und milden Bedingungen bei RT sowie an Luft durchgeführt, wobei Enantioselektivitäten von bis zu 85% ee erreicht wurden. In Schema 1.2-2 ist exemplarisch für diese Arbeiten die Oxidation von 2-Phenyl1,3-dithian dargestellt. Schema 1.2-2: Vanadiumkatalysierte Sulfidoxidation mit H2O2 entwickelt von Bolm und Bienewald.[19a] Inspiriert von diesen Ergebnissen[19, 25] erfolgten zahlreiche Modifikationen der verwendeten Schiffschen-Basen und Untersuchungen zu deren Einsatz in der vanadiumkatalysierten Sulfidoxidation,[22c, 26] sowie die Betrachtung von mechanistischen Fragestellungen. [27] So postulierten beispielsweise Bolm und Bienewald die Existenz von zwei möglichen Vanadium(V)Spezies, welche Oxoperoxy-Gruppen beinhalten und aufgrund der unterschiedlichen Stellungsmöglichkeit der Oxo-Funktion bezüglich des Restes „R“ zu zwei diastereomeren Anordungen führen können (7 und 8, Abbildung 1-4).[25] 7 Kapitel 1 Allgemeine Einleitung Abbildung 1-4: Vanadium(V)-oxomonoperoxyverbindungen 7 und 8 als katalytisch aktive Spezies in der vanadiumkatalysierten Sulfidoxidation postuliert durch Bolm und Bienewald.[25] Einen möglichen Reaktionsmechanismus für die enantioselektive vanadiumkatalysierte Sulfidoxidation formulierten Zeng et al. ausgehend von einem 5-fach koordinierten OxovanadiumSchiffsche-Base Komplex (Schema 1.2-3).[28] Schema 1.2-3: Postulierter Reaktionsmechanismus für die enantioselektive Sulfidoxidation unter der Verwendung von Vanadium-Schiffsche-Base Komplexen nach Zeng und Zhao.[28] Der Reaktionszyklus beginnt mit der Koordination eines Peroxidmoleküls an das Metallzentrum (Vanadium) mit anschließender Ankoordination des Substrats (II), welche den geschwindigkeitsbestimmenden Schritt darstellt. Als nächstes erfolgen die Sauerstoffübertragung vom Peroxid zum Schwefelatom des Substrats (III) und die Dissoziation des Sulfoxidprodukts (IV), wodurch der Oxovanadiumkomplex (I) zurückgebildet wird und ein neuer Zyklus beginnen kann. 8 Kapitel 1 Allgemeine Einleitung Abweichend von den von Bolm entwickelten Liganden wurden, erstmals von Vetter und Berkessel (Ligand 9)[29], später von Katsuki (Ligand 10)[30] und Ahn (Ligand 11)[31], Schiffsche-Base Liganden mit einem zusätzlichen Chiralitätselement (Binaphthyl- oder davon abgeleitetes Gerüst) in der vanadiumkatalysierten Oxidation von Thioanisol mittels H2O2 erfolgreich eingesetzt. Unter ähnlicher Reaktionsführung (1-2 mol% Ligand, 1 mol% VO(acac)2, DCM, 1.1-1.2 Äq. H2O2) wurde für Ligand 9 Methylphenylsulfoxid in 92% Ausbeute und 78% ee, für Ligand 10 81% Ausbeute und 88% ee und für Ligand 11 90% Ausbeute und 86% ee erhalten. Die verwendeten Liganden sind in Abbildung 1-5 dargestellt. Abbildung 1-5: Repräsentative Schiffsche-Base Liganden mit jeweils zwei Chiralitätselementen. Die hohe Bandbreite und Vielfalt an Katalysatoren, die in der vanadiumkatalysierten Sulfidoxidation entwickelt wurden, lässt auch auf gute bzw. bessere Resultate bei anderen Redoxmetallen hoffen. 1.2.1.2 Eisenhaltige Katalysatoren in der enantioselektiven Sulfidoxidation: Stand der Wissenschaft Im Gegensatz zu den bekannten Beispielen von titan- und vanadiumhaltigen Katalysatoren ist bis dato über die Verwendung von eisenhaltigen Katalysatoren in der enantioselektiven Sulfidoxidation nur wenig bekannt.[8] Dabei stellt besonders die Entwicklung von umweltfreundlichen und günstigen 9 Kapitel 1 Allgemeine Einleitung Katalysatoren in der chemischen Forschung eine große Herausforderung dar. In diesem Kontext liegen die Vorteile der Verwendung von Eisen und seinen Salzen einerseits darin, dass diese sehr leicht verfügbar und dadurch kostengünstig zu erweben sind (zweithäufigstes Metall nach Aluminium und mit 4.7 Gew.-% in der Erdhülle vorhanden),[32] andererseits zeichnet sich Eisen gegenüber anderen Metallen wie z.B. Vanadium durch seine geringe Toxizität aus. Die Leistungsfähigkeit und Bedeutung die Eisen in Redoxprozessen zukommt wird zweifelsfrei durch das Vorkommen von Eisen in Proteinstrukturen deutlich, wo Eisen als Redoxzentrum am Sauerstofftransport, dem Sauerstoffspeicher und an Elektronentransferprozessen beteiligt ist. Eisenproteine werden in Häm- und Nichthäm-Proteine unterteilt, wobei es sich bei ersteren um Eisenporphyrin-Komplexe handelt, wie sie unter anderem im Hämoglobin, Cytochromen bzw. Oxygenasen und Peroxidasen zu finden sind. Nichthäm-Proteine sind aus Eisen-Schwefel-Clustern oder reinen Eisen-Protein-Komplexen aufgebaut, wie z.B in Nitrogenasen oder Ferritinen.[32] Die Erforschung von Oxidationsreaktionen hat sich im Wesentlichen auf Eisenporphyrin-Komplexe konzentriert,[1a, 33] während über die Anwendung von synthetischen Nichthäm-Eisenkomplexen als enantioselektive Katalysatoren weniger bekannt ist. So wurden Anfang der 90er Jahre verschiedene auf natürlichen Vorbildern basierende hämhaltige Eisenkatalysatoren untersucht, welche die Oxidation von Sulfiden im Allgemeinen mit moderaten Enantioselektivitäten katalysieren.[34] In den Jahren 1997[35] und 1999[36] berichteten Fontecave et al. zum ersten Mal von der Verwendung eines Nichthäm-Dieisen(III)komplexes (Komplex 12, Abbildung 1-6), welcher ein chirales Bipyridinderivat enthält. Dabei wurden die katalytischen Eigenschaften in der enantioselektiven Sulfidoxidation von verschiedenen Aryl-Alkyl-Sulfiden in Abhängigkeit von H2O2 untersucht. Eine hohe Ausbeute von 90% und ein moderater ee-Wert von 40% konnten nur im Falle von p-Bromophenylmethylsulfid erreicht werden. 10 Kapitel 1 Allgemeine Einleitung Abbildung 1-6: Nichthäm-Eisenkomplexe 12 und 13 in der enantioselektiven Sulfidoxidation untersucht von Fontecave et al.[35-37] Zwei Jahre später wurde von derselben Arbeitsgruppe ein dem Dieisen(III)komplex analoger einkerniger Eisen(II)komplex (Komplex 13, Abbildung 1-6) entwickelt und in der enantioselektiven Oxidation von Sulfiden eingesetzt. Dieser zeigte jedoch, im Vergleich zur µ-oxo Spezies, eine geringere Reaktivität und Stereospezifität (≤48% Ausbeute und ≤5% ee).[37] Einen großen Erfolg in der enantioselektiven eisenkatalysierten Sulfidoxidation mittels H2O2 erzielten Legros und Bolm im Jahr 2003,[38] indem sie die von ihnen zuvor entwickelten SchiffscheBase Liganden der vanadiumkatalysierten Sulfidoxidation in Kombination mit Fe(acac)3 in der Oxidation von Sulfiden einsetzten. Nach einem Screening der verschiedenen von t-Leucinol abgeleiteten Schiffsche-Base Liganden lieferte Ligand 14 unter ähnlichen Reaktionsbedingungen wie in der Vanadiumkatalyse zunächst nur mäßig gute Ausbeuten von 21-44% für eine Reihe verschiedener Sulfide. Die erreichten Enantioselektivitäten lagen dabei in einem Bereich von 2790% ee.[38] Kurz darauf zeigten Legros und Bolm, dass durch den Zusatz von 1 mol% eines Additivs (4-Methoxybenzoesäure in Form ihres Lithiumsalzes), die Effizienz des katalytischen Systems im Hinblick auf Ausbeute (<78%) und Enantioselektivität (<96% ee) deutlich gesteigert werden konnte (siehe Schema 1.2-4).[39] 11 Kapitel 1 Allgemeine Einleitung Schema 1.2-4: Eisenkatalysierte Sulfidoxidation mit H2O2 entwickelt von Legros und Bolm.[39] Analog zur vanadiumkatalysierten Oxidation zeichnet sich dieses System durch seine leichte Handhabung (in situ Bildung des Katalysators und Reaktionsführung an Luft), den milden Reaktionsbedingungen (RT) und einer niedrigen Katalysatorbeladung von 2 mol% aus. Der genaue Mechanismus bzw. die Rolle des Additivs ist jedoch noch nicht bekannt. Da aber eine wie die in Schema 1.2-4 dargestellte elektronenreiche Carbonsäure die besten Resultate lieferte, nehmen die Autoren an, dass diese als Co-Ligand fungieren und somit ein monocarboxylatverbrückter Dieisen(III)komplex eine Schlüsselrolle im katalytischen Oxidationsprozess einnehmen könnte. Bryliakov und Talsi berichteten in den Jahren 2004 und 2007 von der Anwendung verschiedener chiraler Eisen(III)salenkomplexe auf die Oxidation von Sulfiden mittels PhIO.[40] Dabei produzierten die einkernigen Eisen(III)komplexe 15 und 16 (Abbildung 1-7) Benzylphenylsulfoxid in MeCN nach einer Reaktionszeit von 2 h mit einem maximalen ee-Wert von 62%. Die Reaktion wurde bei 0 °C und einer sehr geringen Katalysatorbeladung von 1 mol% durchgeführt. Die Umsätze betrugen 95% bei Komplex 15 und 91% bei Komplex 16. In Verbindung mit H2O2 wurden allerdings racemische Produkte erhalten. 12 Kapitel 1 Allgemeine Einleitung Abbildung 1-7: Eisen(III)salenkomplexe entwickelt von Bryliakov und Talsi.[40] Unter Verwendung des zweikernigen Dieisen(III)salenkomplexes 17 konnte speziell das Oxidationsprodukt i-Propylphenylsulfoxid mit 84% ee und 69% Umsatz erhalten werden. Die Reaktion wurde in diesem Falle bei -21 °C mit einer Katalysatorbeladung von 2 mol% durchgeführt. MesIO wurde als terminales Oxidationsmittel verwendet. Im Jahr 2007 enwickelten Egami und Katsuki[41] den neuartigen chiralen Eisen(III)salankomplex 18, welcher erfolgreich in der enantioselektiven Oxidation sowohl von verschiedenen Alkyl-ArylSulfiden als auch von Alkyl-Alkyl-Sulfiden eingesetzt wurde (Schema 1.2-5). Die Verwendung von H2O2 als Oxidationsmittel und die Reaktionsführung in H2O machen dieses katalytische System besonders aus ökologischer Sicht wertvoll. Die Katalysatorbeladung ist mit nur 1 mol% sehr gering. 13 Kapitel 1 Allgemeine Einleitung Schema 1.2-5: Neuartiger Eisen(III)salankomplex in der enantioselektiven Sulfidoxidation entwickelt von Katsuki.[41] Die unter diesen Bedingungen signifikante Sulfonbildung (<24%) konnte durch weitere Optimierung des Systems (Durchführung der Reaktion bei 0 °C, Verringerung der Menge an H2O als Lösungsmittel und Verringerung der Katalysatorbeladung auf 0.2 mol%) minimiert (<9%) und somit höhere Ausbeuten für die entsprechenden Sulfoxide (z.B. für Methyloctylsulfoxid 96% statt 82% Ausbeute) erreicht werden. Auch die Substratbreite konnte unter diesen Bedingungen erweitert werden (Toleranz von funktionellen Gruppen wie z.B. Hydroxy-, Alkenyl-, Ester, etc.).[42] Den jüngsten Bericht in der eisenkatalysierten enantioselektiven Sulfidoxidation lieferten Liao und List[43] mit der Entwicklung des neuartigen Ionenpaar-Katalysators 19, welcher aus einem achiralen Eisen(III)salen-Kation und einem chiralen BINOL-Phosphat-Gegenanion aufgebaut ist. Verschiedene Alkyl-Aryl-Sulfide, insbesondere elektronenarme und sterisch anspruchsvolle Substrate, konnten in Gegenwart von PhIO in Benzol und mit einer geringen Katalysatorbeladung von 1 mol% mit hohen Ausbeuten (88-95%) und Enantioselektivitäten (66-96% ee) oxidiert werden (Schema 1.2-6). Der neu synthetisierte Ionenpaar-Katalysator und sein erfolgreicher Einsatz in der enantioselektiven Sulfidoxidation basiert auf dem von den Autoren entwickelten Konzept der 14 Kapitel 1 Allgemeine Einleitung „asymmetrischen Gegenion-vermittelten Katalyse“ (engl. Asymmetric Counteranion-Directed Catalysis - ACDC-Konzept). Schema 1.2-6: Neuer Eisen(III)salen-Phosphat-Komplex in der enantioselektiven Sulfidoxidation entwickelt von Liao und List.[43] 1.2.2 Organokatalysierte Sulfidoxidation Die Entwicklung von metallfreien organokatalytischen Methoden in der asymmetrischen Katalyse stellt seit ihrem enormen Aufschwung Ende der 90er Jahre eine wertvolle Alternative zu konventionellen Prozessen, also der Anwendung von synthetischen Metall- oder Biokatalysatoren, dar.[12a, 44] Diese Entwicklung wird in großen Teilen durch die Notwendigkeit der pharmazeutischen Industrie, durch Katalysatorrückstände verursachte Metallbelastungen aus ihren Produkten zu entfernen, entscheidend vorangetrieben. Bei der Verwendung von Metallkatalysatoren im Herstellungsprozess eines Wirkstoffes sind aufwendige Reinigungsprozesse von Nöten.[45] Dabei spielt gerade heute, zu einem Zeitpunkt in dem das ökologische Bewusstsein aller so ausgeprägt ist wie selten zuvor, eine ökologische Denkweise in der Entwicklung von schadstoffarmen Systemen eine erhebliche Rolle und gibt so Anstoß, die Nutzung toxischer und umweltbelastender 15 Kapitel 1 Allgemeine Einleitung Stoffe weitestgehend zu vermeiden. Darüber hinaus ist aus ökonomischer Sicht der Gebrauch von Organokatalysatoren sehr attraktiv, da diese oft in wenigen Schritten aus leicht zugänglichen Ausgangsmaterialen herstellbar sind. Zudem weisen sie häufig eine hohe Stabilität auf, sind kaum toxisch und in der Regel weniger luft- und feuchtigkeitsempfindlich.[46] Aus diesen Gesichtspunkten heraus ist es daher umso erstaunlicher, dass bis dato organokatalytische Methoden in der Sulfidoxidation im Gegensatz zu metallkatalytischen Varianten weit weniger erforscht wurden. In den vergangenen Jahren wurden metallfreie, nicht jedoch organokatalytische Methoden für die Synthese von Sulfoxiden entwickelt.[12b, 47] Dabei wurden Sulfoxide mit hoher Enantioselektivität vor allem unter Verwendung von chiralen Imin- oder Oxaziridin-Reagenzien und Katalysatoren erhalten.[10, 12b, 48] Da diese Systeme durch beispielsweise eine geringe Substrattoleranz nur eingeschränkt anwendbar sind und oft der Einsatz von stöchiometrischen Mengen der Reagenzien notwendig ist, gibt dies immer wieder Anreiz zur Entwicklung von neuen metallfreien organokatalytischen Systemen. 1.2.2.1 Organokatalysatoren in der enantioselektiven Sulfidoxidation: Stand der Wissenschaft Bei einigen der bisher entwickelten organokatalytischen Synthesen zur Darstellung von Sulfoxiden handelt es sich um nicht-stereoselektive Methoden, welche beispielsweise von Bäckvall (Einsatz von Flaviniumverbindungen)[49] oder von Lattanzi (Einsatz eines Thioharnstoffs)[50] veröffentlicht wurden. Deren Anzahl, dabei insbesondere die von asymmetrischen organokatalytischen Sulfidoxidationen, ist bis heute stark limitiert.[8b] Die Spannweite der entwickelten Katalysatoren reicht unter anderem von hypervalenten Iodverbindungen (in Kombination mit substöchiometrischen Mengen von chiralem Informationsträger),[51] über chirale Dioxirane,[52] chirale Flaviniumverbindungen,[53] chirale Iminiumsalze,[54] bis hin zu Phosphaten.[55] Im Folgenden werden ausgewählte Beispiele näher diskutiert. 16 chiralen BINOL- Kapitel 1 Allgemeine Einleitung Rozwadowska et al.[54] synthetisierten das enantiomerenreine Iminiumsalz 20 (Familie der Isoquinoliniumsalze) und prüften dessen Effizienz als Organokatalysator in der Oxidation von Methyl-p-Tolylsulfid zum korrespondierenden Sulfoxid. Die Verwendung von Oxon als Oxidationsmittel führte unter variabler Reaktionsführung (Temperatur, Menge an Oxidationsmittel, LM und Basenzusatz) zu moderaten bis guten Ausbeuten von 50-84%, die ee-Werte blieben jedoch gering bis moderat (14-42% ee) (siehe Schema 1.2-7). Die Verwendung von m-CPBA als Oxidationsmittel führte zu einem racemischen oder zu einem Produkt mit minimaler asymmetrischer Induktion von 8% ee. Schema 1.2-7: Iminiumsalz 20 als Organokatalysator in der Oxidation von Methyl-p-Tolylsulfid. Im Jahr 2005 berichteten Colonna und Malacria[52a] zum ersten Mal von der Anwendung eines in situ generierten chiralen Dioxirans (hergestellt aus dem fruktosehaltigen Keton 21 und Oxon) auf die enantioselektive Synthese von Thiosulfinaten und azyklischen AlkylidenmethylensulfidSulfoxiden. Die Oxidation von verschiedenen Dithioethern lieferte unter der Verwendung von 30 mol% des chiralen Precursors 21 und 1.4 Äquivalenten Oxon in MeCN/DMM (1:2) bei 0 °C (Reaktionsbedingungen analog der Shi-Epoxidierung)[56] die korrespondierenden Produkte mit Umsätzen von 18-98% und Enantioselektivitäten von 0-75% ee (siehe Abbildung 1-8). Das Sulfoxid eines Schwefel-Schwefel-Ketenacetals konnte 17 in der Monooxidation unter veränderten Kapitel 1 Allgemeine Einleitung Reaktionsbedingungen (2.1 Äq. KHSO5, MeCN/DMM (1:2), Borax, H2O, K2CO3, 0 °C) in 70% Ausbeute und 46% ee mit (R)-Konfiguration erhalten werden. Abbildung 1-8: Anwendung von einem in situ generierten chiralen Dioxiran auf die Synthese verschiedener Disulfide. Die Anwendung von 21 wurde 2007 durch Khiar und Fernández[52b] erweitert und in der Oxidation von funktionalisierten, sterisch anspruchsvollen Disulfiden eingesetzt. In Abhängigkeit der jeweiligen Struktur des Substrates konnten die entsprechenden Sulfoxide in Ausbeuten von 2089% und hohen Enantioselektivitäten (70-96% ee) erhalten werden. Die neuesten Entdeckungen in diesem Bereich aus dem Jahr 2012 beruhen auf den Arbeiten von Tao und Wang[55a] sowie von List,[55b] welche unabhängig voneinander zum ersten Mal chirale BINOL-Phosphate als Brønstedt-Säure-Katalysatoren in der asymmetrischen Sulfidoxidation untersuchten. Tao und Wang führten zunächst ein Screening mit verschieden substituierten chiralen BINOL-Phosphaten in der Oxidation von Thioanisol in CHCl3 unter Verwendung von H2O2 durch. Nach der Ermittlung des besten Katalysators und unter optimierten Reaktionsbedingungen (10 mol% von 22, 1.5 Äq. H2O2, CHCl3, -40 °C) wurde eine Reihe von Aryl-Alkyl-Sulfiden, als auch 1,3-Dithiane, in der Oxidation getestet. Dabei wurden Aryl-Alkyl-Sulfoxide in Ausbeuten von 3592% isoliert, mit gleichzeitig moderaten bis guten Enantioselektivitäten von 66-82% ee (Schema 1.2-8). 18 Kapitel 1 Allgemeine Einleitung Schema 1.2-8: Enantioselektive Oxidation von Aryl-Alkyl-Sulfiden unter Verwendung des BINOLPhosphats 22. Die asymmetrische Oxidation von 1,3-Dithianen lieferte unter denselben Reaktionsbedingungen die jeweiligen Monooxidationsprodukte in guten bis exzellenten Ausbeuten (60-99%) und Diasteriosowie Enantioselektivitäten (88:12-99:1 dr, 56-74% ee). In Abbildung 1-9 sind vier Monooxidationsprodukte exemplarisch dargestellt. Abbildung 1-9: Asymmetrische Oxidation von 1,3-Dithianen mittels BINOL-Phosphat 22. List et al.[55b] untersuchten zunächst verschiedene chirale BINOL-Phosphate in der Oxidation von Thioanisol in Cyclohexan unter Verwendung von 1.1 Äquivalenten H2O2 bei RT. Die besten Ergebnisse lieferte in diesem Fall das Imidodiphosphorsäurederivat 23. Exzellente Ausbeuten (8919 Kapitel 1 Allgemeine Einleitung 99%) und Enantioselektivitäten (85-98% ee) konnten so in der Oxidation verschiedener Sulfide unter optimierten Reaktionsbedingungen erhalten werden (Schema 1.2-9). Schema 1.2-9: Enantioselektive Oxidation verschiedener Sulfide unter Verwendung der Imidodiphosphorsäure 23. Der durch beide Arbeitsgruppen postulierte Mechanismus der beschriebenen Reaktion beruht auf einer dualen Aktivierung von H2O2 über die Ausbildung von Wasserstoffbrückenbindungen durch die Brønstedt-Säure- und Lewis-Base-Funktionen in den bifunktionellen Organokatalysatoren 22 bzw. 23. Während die erreichten hohen Stereoselektivitäten der Sulfidoxidation bei Wang und Tao hauptsächlich durch die chirale Umgebung im BINOL-Phosphat und einer zusätzlichen sterischen Kontrolle der enthaltenen Substituenten (R = 1-Naphthyl) erklärt werden können, werden die exzellenten Selektivitäten bei List et al. durch die von der Imidodiphosphorsäure 23 geschaffene Cavität („chiral pocket“) erklärt, in welcher die Reaktanden (Sulfid und H2O2) zur Reaktion gebracht werden (siehe Abbildung 1-10). Durch dieses Bauprinzip wird die räumliche Orientierung der Reaktanden zueinander auf wenige Möglichkeiten beschränkt, was die Anzahl der produktbildenden Übergangszustände limitiert und final zu einer eindrucksvollen Erhöhung der Enantioselektivität führt. 20 Kapitel 1 Allgemeine Einleitung Abbildung 1-10: Der von List et al. postulierte Übergangszustand.[55b] Das von List et al. entwickelte organokatalytische Verfahren konkurriert zum ersten Mal mit den in der Metallkatalyse etablierten Systemen hinsichtlich katalytischer Aktivität, Effizienz und Substratbreite.[55b] Dennoch ist keines der bekannten Systeme universell einsetzbar und somit bleibt weiterhin Raum für Verbesserungen und die Entwicklung von neuen Systemen. 1.3 Sulfoxide als biologisch aktive Verbindungen - Anwendung in der pharmazeutischen Industrie am Beispiel von Esomeprazol und Armodafinil Die Erkenntnis, dass eine Vielzahl von Sulfoxiden eine hohe biologische Aktivität aufweist, hat ein breites Spektrum von sulfoxidhaltigen Wirkstoffen für die pharmazeutische Industrie erschlossen.[2] Aufgrund der strukturellen Eigenschaften sulfoxidhaltiger Verbindungen (siehe Kapitel 1.1) liegen diese als ein Paar von Enantiomeren vor. Die Enantiomere können sich aber in ihrer biologischen Aktivität (z.B. Metabolisierung oder Enzymhemmung) unterscheiden; das heißt, dass ein Enantiomer eine geringere oder keine gewünschte Effektivität aufzeigt oder sogar negative Effekte zur Folge haben kann. Daher ist es grundsätzlich von großer Bedeutung, beide Enantiomere einer Verbindung zu synthetisieren und auf ihre biologische Aktivität hin zu prüfen. So ist seit der Grundsatzerklärung im Jahr 1992 durch die FDA („Food and Drug Administration“) über die 21 Kapitel 1 Bedeutung Allgemeine Einleitung von Medikamenten in ihrer enantiomerenreinen Form der Verkauf von enantiomerenreinen Verbindungen deutlich gestiegen.[2a, 57] Eines der prominentesten Beispiele für sulfoxidhaltige Pharmazeutika ist Esomeprazol (Abbildung 1-11), erhältlich unter dem Handelsnamen Nexium®, welches sich seit 2006 auf Platz 4 der weltweit meistverkauften Medikamente befindet.[58] Esomeprazol, das (S)-Enantiomer von Omeprazol, gehört zur Gruppe der Protonenpumpeninhibitoren (PPI) und wird unter anderem zur Behandlung von Magen- und Zwölffingerdarmgeschwüren sowie gegen Magensäureüberproduktion eingesetzt. Im Gegensatz zu racemischem Omeprazol wird Esomeprazol im Körper langsamer metabolisiert und zeigt durch die höhere Bioverfügbarkeit eine gesteigerte pharmakologische Wirkung.[59] Zur Herstellung von Esomeprazol wurde bis heute eine Reihe verschiedener Synthesemethoden beschrieben. Die ersten Synthesen von optisch aktivem Omeprazol wurden mittels Racematspaltung durchgeführt.[60] Später wurde die Methodik der asymmetrischen Oxidation von prochiralen Sulfiden auf die Herstellung von Esomeprazol angewendet, wobei ein Großteil auf dem Nutzen von titanhaltigen Katalysatoren beruht.[61] Ein bedeutender und im industriellen Maßstab anwendbarer Prozess wurde von Cotton et al. (AstraZeneca) entwickelt.[61a] Hierbei handelt es sich um ein modifiziertes Kagan-ModenaSystem,[14a, 15] worin die Abwandlung auf der Verwendung von Hünigs-Base beruht (siehe Schema 1.3-1). 22 Kapitel 1 Allgemeine Einleitung Schema 1.3-1: Syntheseverfahren von Esomeprazol nach Cotton et al. (AstraZeneca).[61a] Das ursprüngliche Kagan-System lieferte aufgrund des fehlenden unterschiedlichen sterischen Anspruchs der zwei Substituenten des Sulfid-Precursors nahezu keine Enantioselektivitäten.[61a] Abbildung 1-11: Arzneiwirkstoffe Esomeprazol und Armodafinil.[62] Ein weiteres Beispiel neuerer sulfoxidhaltiger Medikamente ist Armodafinil, bekannt unter dem Handelsnamen Nuvigil®, welches im Jahr 2005 von dem Pharmaunternehmen Cephalon patentiert und 2007 von der FDA in den USA zugelassen wurde (Abbildung 1-11). Armodafinil, das (R)-Enantiomer von Modafinil, wird in der Behandlung von Schlafstörungen verbunden mit Narkolepsie, obstruktivem Schlafapnoe/Hypopnoe-Syndrom oder durch Schichtarbeit hervorgerufene Schlafstörungen eingesetzt.[63] Auch im Falle von Armodafinil wird eine höhere 23 Kapitel 1 Allgemeine Einleitung Aktivität gegenüber dem Racemat (Modafinil) beobachtet, welche sich durch die langsamere Metabolisierung und eine dadurch längere Verfügbarkeit des Wirkstoffs im Organismus begründet.[64] Der Schlüsselschritt der von Rebiere et al. entwickelten Methode zur Synthese von Armodafinil und ihre Anwendung im industriellen Maßstab (Cephalon) basiert, ebenso wie bei Esomeprazol, auf den anfänglichen Arbeiten von Kagan und Mitarbeitern; das heißt auf einer asymmetrischen titankatalysierten Sulfidoxidation. In diesem Prozess wird Armodafinil in einer 4-Stufensynthese mit lediglich zwei Isolationsschritten in 88% Ausbeute und einer Enantioselektivität von >99.5% ee erhalten.[63] Von wenigen weiteren Herstellungsverfahren, wie beispielsweise von Enantiomerentrennung (z.B. durch Chromatographie,[65] fraktioneller Kristallisation[65-66] oder einer Trennung über Diastereomerenbildung[66-67]) und der Oxidation von prochiralen Sulfiden (mittels Metallkatalysatoren,[63a, 65, 68] chiralen Oxaziridinen,[68] mikrobieller[69] oder biokatalytischer Oxidation[70]), wurde in der Literatur berichtet. Einige dieser Methoden weisen allerdings eine Reihe gravierender Nachteile im Hinblick auf die erreichten Ausbeuten bzw. Enantioselektivitäten (<60% ee) sowie die gewählten Reaktionsführungen (unter anderem die Verwendung toxischer Reagenzien wie z.B. Dimethylsulfat[67a] oder Iodomethan[71]) auf. Daher bietet die Entwicklung von neuen umweltfreundlichen und effizienten Systemen in der Synthese sulfoxidhaltiger Wirkstoffe, wie am Beispiel von Esomeprazol und Armodafinil gezeigt, nachwievor ein großes Forschungspotential. 24 KAPITEL 2 2 Motivation und Zielsetzung Die enantioselektive Sulfidoxidation repräsentiert eine bedeutende Methode zur Synthese chiraler Sulfoxide, welche wertvolle Bausteine in der organischen Synthesechemie und biologisch aktiven Verbindungen darstellen und sogar als pharmazeutische Wirkstoffe Einsatz finden.[2] Es wurde bis dato eine Reihe asymmetrischer Methoden zur Synthese chiraler Sulfoxide entwickelt, wobei die meisten Systeme aus den Pionierarbeiten von Kagan[14, 17a] und Modena[15, 17b] und somit aus der Verwendung von chiralen Metallkomplexen hervorgegangen sind. Dabei stellt der Gebrauch von kostengünstigen und gleichzeitig umweltfreundlichen Oxidationsmitteln, wie beispielsweise Wasserstoffperoxid, bei den bekannten enantioselektiven Verfahren immer noch die Minderheit dar. Einen alternativen Weg zur klassischen metallkatalysierten Sulfidoxidation ermöglicht die Entwicklung von organokatalytischen Methoden, welche in jüngster Zeit immer mehr an Bedeutung gewonnen haben. Dabei wurde das Gebiet der asymmetrischen organokatalytischen Sulfidoxidation trotz des starken Aufschwungs der Organokatalyse in den 90er Jahren bis heute wenig erforscht. Somit bleibt die Entwicklung neuer effizienter Katalysatoren, mit Fokus auf einem umweltfreundlichen Oxidationsprozess, nachwievor eine große Herausforderung. In diesem Zusammenhang bestand ein Ziel der Dissertation darin, neue chirale Liganden zu synthetisieren, welche im Anschluss unter der Verwendung des umweltfreundlichen Oxidationsmittels Wasserstoffperoxid auf ihre Effizienz in der eisen- oder vanadiumkatalysierten Sulfidoxidation hin geprüft werden sollten. 25 Außerdem stand die Entwicklung neuer Kapitel 2 Motivation und Zielsetzung organokatalytischer Ansätze als alternative metallfreie Methode in der Synthese chiraler Sulfoxide im Mittelpunkt dieser Arbeit (Abbildung 2-1). Abbildung 2-1: Entwicklung von metall-und organokatalytischen Methoden zur enantioselektiven Sulfidoxidation unter Verwendung von H2O2 als umweltfreundlichem Oxidationsmittel. Für die katalytischen Studien sollte Thioanisol als gängiges Modellsubstrat in der Oxidation von prochiralen Sulfiden zu Sulfoxiden dienen. Als primäres Oxidationsmittel wurde H2O2 gewählt, da dieses während der Reaktion zu Wasser reduziert wird, welches als unproblematischer Abfallstoff einzustufen ist. Weitere Vorteile des Oxidationsmittels begründen sich in der kostengünstigen Verfügbarkeit und leichten Handhabbarkeit. Als Ausgangsbasis für die Entwicklung von neuen hochenantioselektiven Redox- Metallkatalysatoren sollten chirale nicht-porphyrinhaltige Liganden wie beispielsweise (Thio-) Harnstoffe und Bisformamide dienen, welche bereits erfolgreich im Arbeitskreis Tsogoeva als chirale Organokatalysatoren[72] in verschiedenen Reaktionen eingesetzt wurden, um so organound metallkatalytische Ansätze miteinander zu kombinieren. Inspiriert von bereits etablierten Ligandensystemen (welche z.B. Schiffsche-Basen im Ligandgerüst enthalten bzw. aus peptidhaltigen Schiffsche-Base Liganden (sogenannten Hybriden) aufgebaut sind)[19a, 26 73] sollten Kapitel 2 Motivation und Zielsetzung weiterhin neue chirale Liganden entwickelt und auf ihre katalytische Aktivität in der Oxidation von Thioanisol untersucht werden. Abbildung 2-2: Allgemeine Übersicht potentieller chiraler Ligand-Zielstrukturen. Als Metallquellen sollten Metallionen dienen, die aus ökologischer und ökonomischer Sicht leicht verfügbar, kostengünstig und wenig toxisch sind. Im Mittelpunkt sollte Eisen stehen, ein Metall, das seine hohe katalytische Effizienz und Redoxaktivität in natürlichen Systemen (z.B. Hämoglobin oder Cytochrom-C-Oxidase) eindrucksvoll unter Beweis stellt.[74] Darüberhinaus sollten sich die entwickelten metall- und organokatalytischen Verfahren dadurch auszeichnen, dass diese in ihrer Reaktionsdurchführung leicht zu handhaben sind. Somit sollten die potentiellen Metallkomplexe in erster Linie in situ generiert und die Reaktionen wenn möglich unter milden Bedingungen sowie an Luft durchgeführt werden. Die entwickelten metall- und organokatalytischen Systeme sollten anschließend in der Synthese sulfoxidhaltiger Pharmazeutika am Beispiel von Esomeprazol und (R)-Modafinil Anwendung finden (Abbildung 2-3). 27 Kapitel 2 Motivation und Zielsetzung Abbildung 2-3: Anwendung der entwickelten Verfahren auf die Synthese von sulfoxidhaltigen Pharmazeutika. 28 KAPITEL 3 3 3.1 Ergebnisse und Diskussion Design und Synthese chiraler Liganden und deren Anwendung auf die metallkatalysierte Oxidation von Thioanisol Chirale Metallkomplexe, welche als Katalysatoren in der asymmetrischen Synthese verwendet werden, setzen sich allgemein aus einem chiralen Liganden (Träger der stereochemischen Information) und einer Metallquelle zusammen. Die Metall-Ligand-Komplexe können entweder als Präkatalysatoren oder direkt in situ generiert und ohne weitere Isolierung für Katalysereaktionen eingesetzt werden.[75] In der vorliegenden Arbeit wurden die Metallkomplexe hauptsächlich in situ hergestellt und bezüglich ihrer Effizienz in der enantioselektiven Oxidation von Thioanisol (Modellsubstrat) mittels H2O2 untersucht (Schema 3.1-1). Schema 3.1-1: Modellreaktion – Oxidation von Thioanisol (24) mit H2O2 unter der Verwendung von in situ generierten Metallkomplexen. Als Vergleichssubstanzen wurden zunächst das racemische Oxidationsprodukt Methylphenylsulfoxid (25) und das entsprechende Sulfon 26, welches durch eine mögliche Überoxidation gebildet werden kann, nach einem literaturbekannten Syntheseverfahren hergestellt (Abbildung 3-1).[76] 29 Kapitel 3 Ergebnisse und Diskussion Abbildung 3-1: Methylphenylsulfon (26) als mögliches Nebenprodukt infolge einer Überoxidation. Ein entscheidender Faktor für den asymmetrischen Verlauf der Oxidationsreaktion ist die Wechselwirkung zwischen der Struktur des Liganden und des Metallzentrums und somit ist die „richtige“ Wahl des chiralen Liganden von großer Bedeutung.[75] In diesem Abschnitt der Dissertation wird die Synthese verschiedener Ligandensysteme beschrieben, welche als Liganden für die Entwicklung neuer Nichthäm-Redoxsysteme dienen sollten. Die Redox-Metallkatalysatoren, deren Bildung in erster Linie in situ erfolgte, wurden in der Sulfidoxidation untersucht und die erhaltenen Ergebnisse im Anschluss diskutiert. 3.1.1 Liganden basierend auf Formamiden, Harnstoffen und Peptiden Die neu entwickelten chiralen Liganden basierend auf einer formamid- bzw. harnstoffhaltigen Struktur sind in Abbildung 3-2 dargestellt. Abbildung 3-2: Neu entwickelte Liganden 27 und 28 auf Formamid- und Harnstoffbasis. 30 Kapitel 3 Ergebnisse und Diskussion Der polydentate Ligand 28 wurde in Kooperation mit Prof. Dr. Ivana Ivanović-Burmazović entworfen, welcher einerseits in der Sulfidoxidation, als auch in der Dismutierung von Superoxid[77] untersucht werden sollte. Diese Art von Ligand wurde bis zum heutigen Zeitpunkt nicht in der metallkatalysierten Sulfidoxidation eingesetzt. 3.1.1.1 Synthese des Bisformamidliganden 27 Der in Schema 3.1-2 dargestellte chirale Bisformamidligand 27 wurde, ausgehend von den kommerziell erhältlichen Aminosäuren L-Phenylalanin (29) und L-Prolin (33), in einer 5-stufigen Synthese hergestellt („chiral pool“). Schema 3.1-2: Darstellung des Bisformamidliganden 27. Der Aufbau des chiralen Linkers 32, der die beiden N-Formyl-L-Prolin-Einheiten (34) miteinander verknüpft, erfolgte nach literaturbekannten Synthesen.[78] Dabei fand im ersten Schritt eine Veresterung am C-Terminus von L-Phenylalanin (29) mittels Thionylchlorid in MeOH statt.[78a] Das entstandene Hydrochlorid-Salz 30 wurde in einer klassischen Amidierung mit Hilfe einer Ammoniumhydroxidlösung in 31 überführt.[78b] Die Reduktion mittels LiAlH4 in absolutem THF lieferte den chiralen Linker 32.[78a] Auf die Formylierung von L-Prolin (33) wurde eine modifizierte 31 Kapitel 3 Ergebnisse und Diskussion literaturbekannte Synthesemethode[72e] angewandt. Hierfür wurde L-Prolin (33) in Ameisensäure gelöst und dieses anschließend zu einer auf 0 °C gekühlten Lösung aus Essigsäureanhydrid in Ameisensäure zugetropft. Das nach der Aufarbeitung erhaltene leicht gelbliche Öl wurde in Petrolether gerührt, wobei mit der Zeit ein weißer Feststoff ausfiel. Zum Schluss wurden die beiden Peptidbindungen im Zielmolekül 27 über eine CDI-vermittelte Kupplungsreaktion[79] von 32 und 34 geknüpft. Der Ligand 27 wurde in einer Gesamtausbeute von 8% erhalten. Zur Identifikation der hygroskopischen Verbindung wurde eine vollständige Charakterisierung mittels Standardmessmethoden durchgeführt. Besonders interessant waren dabei die 1H- bzw. 13CNMR Messungen bei RT und 100 °C. Die mehrfachen Signalsätze im 1H- bzw. 13C-NMR Spektrum bei RT geben Hinweis darauf, dass Verbindung 27, aufgrund der Möglichkeit von cis/transIsomerie,[80] als Mischung von verschiedenen Isomeren vorliegt. Bei einer Hochtemperaturmessung werden die eingeschränkten Rotationen der Bindungen und eventuell auftretende inter- und intramolekulare Wechselwirkungen im Molekül aufgehoben, was den Übergang der Signale ineinander zur Folge hat (Koaleszenz). Dieser Sachverhalt wird in Abbildung 3-3 anhand der 13C-NMR Spektren bei RT und 100 °C gezeigt. 32 Kapitel 3 Ergebnisse und Diskussion Abbildung 3-3: 13C-NMR Spektren von Ligand 27 gemessen bei RT und 100 °C in DMSO-d6. Bei der Hochtemperaturmessung gehen beispielsweise die Signale im Bereich von 161-162 ppm und 170-172 ppm partiell ineinander über (siehe vergrößerter Bereich). 3.1.1.2 Anwendung formamidhaltiger Liganden in der Katalyse Eine Reihe verschiedener formamidhaltiger Liganden, inklusive des literaturunbekannten chiralen Bisformamids 27, wurden als Liganden in der metallkatalysierten Oxidation von Thioanisol eingesetzt und auf ihre Effizienz hin überprüft. Die Ergebnisse sind in Tabelle 3.1-1 zusammengefasst. 33 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-1: Screening verschiedener formamidhaltiger Liganden unter variablen Reaktionsbedingungen. Eintrag Ligand MXn LM Zeit [h] Ausbeute [%]a ee [%]b 1 35 VO(acac)2 CHCl3 48 4 rac 2 36 VO(acac)2 CHCl3 60 29 rac 3 36 FeCl3 x 6 H2O MeOH 4 78 rac 4 34 FeCl3 x 6 H2O MeOH 48 67 rac 5 27 FeCl3 x 6 H2O MeOH 48 87 rac 6 27 Fe(acac)3 MeOH 48 70 rac 7 - FeCl3 x 6 H2O MeOH 48 82 rac 8 27 - MeOH 48 50 rac 9 - - MeOH 48 61 n.b. a Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD). Erste Experimente wurden im Vorfeld mit den bereits in der Arbeitsgruppe hergestellten Bisformamidliganden 35[81] und 36[72d] in Kombination mit VO(acac)2 als Metallionenquelle in CHCl3 durchgeführt. Das Verhältnis zwischen Ligand und Metall betrug dabei 3:2, da die Vermutung besteht, dass es sich um eine ligandenbeschleunigte Reaktion handelt. [19a] Außerdem ist generell bekannt, dass VO(acac)2 mit H2O2 eine achirale Spezies bilden und diese unter bestimmten Reaktionsbedingungen in Konkurrenz mit der Aktivierung von H2O2 am in situ gebildeten 34 Kapitel 3 Ergebnisse und Diskussion Metallkatalysator treten kann und so final zu einer Verringerung des ee-Werts führt.[27b, 82] Aufgrund der Tatsache, dass die Liganden 35 und 36 unter den in Anlehnung an Jackson et al.[22b] entwickelten Reaktionsbedingungen keine guten Resultate lieferten (Einträge 1 und 2), wurde in den nächsten Experimenten VO(acac)2 durch die Metallionenquelle FeCl3 x 6 H2O ersetzt. Da dieses Salz jedoch nur mäßig in CHCl3 löslich ist, wurde MeOH als polareres Lösungsmittel gewählt, in dem beide Reaktanden in Lösung vorliegen und so eine Koordination der Liganden am Metall ermöglicht wird. Der Ligand 36 liefert Methylphenylsulfoxid bereits nach wenigen Stunden in guten Ausbeuten von 78%, wobei keine chirale Induktion zu beobachten war (Eintrag 3). Während N-Formyl-L-Prolin (34) das Sulfoxid nach einer Reaktionszeit von 48 h in 67% Ausbeute produzierte, wurden höhere Ausbeuten mit Ligand 27, welcher einen flexiblen chiralen Linker enthält, erzielt. Beide Produkte lagen jedoch in Form eines Racemats vor (Einträge 4 und 5). Durch den Ersatz von FeCl3 x 6 H2O gegen Fe(acac)3 in Kombination mit Ligand 27 wurde Methylphenylsulfoxid in 70% Ausbeute als Racemat isoliert (Eintrag 6). Ein Grund für die fehlende chirale Induktion in MeOH könnte die Folge einer zu schnell ablaufenden Hintergrundreaktion sein, da FeCl3 x 6 H2O selbstständig die Reaktion in MeOH katalysiert (82% Ausbeute, Eintrag 7). Prinzipiell ist aber eine enantioselektive Oxidation von Sulfiden in MeOH unter der Verwendung von H2O2 möglich.[41] Der Einsatz von Ligand 27 ohne die Verwendung einer Metallquelle lieferte das Oxidationsprodukt in 50% Ausbeute (Eintrag 8) als Racemat, während die Hintergrundreaktion (ohne Ligand und Metallquelle) bereits nach 48 h Reaktionszeit das Sulfoxid in 61% Ausbeute produzierte (Eintrag 9). Hinweise auf eine erfolgreiche Komplexierung des polydentaten Liganden 27 an das Zentralmetall lieferten massenspektrometrische Untersuchungen. Die zugehörige Testreihe zur Komplexierung unter variablen Reaktionsbedingungen ist in Tabelle 3.1-2 dargestellt. 35 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-2: Methoden zur Komplexierung von Ligand 27 mit einem Eisensalz unter variablen Reaktionsbedingungen. Methode a MXn LM Temp. MALDI-MS [°C] Charakteristische Signale (Int%) 1a FeCl2 THF 75 491 [M1]∙+ (25%) 526 [M2]∙+ (10%) 2b FeCl2 THF RT 491 [M1]∙+ (40%) 526 [M2]∙+ (30%) 3b FeCl2 MeOH RT 491 [M1]∙+ (100%) 526 [M2]∙+ (10%) 4b FeCl3 x 6 H2O MeOH RT 491 [M1]∙+ (100%) 526 [M2]∙+ (19%) (FeCl2/Ligand 27/2,6-Lutidin (5:1:2.2)); 16 h; b (MXn/Ligand 27 (1:1)), 3 h. Nach jedem Experiment wurde das Lösungsmittel entfernt und der Rückstand analysiert. In den MS Spektren sind jeweils zwei charakteristische Signale bei m/z = 491 [M1]∙+ und m/z = 526 [M2]∙+ zu erkennen, die aufgrund ihres typischen Isotopenmusters auf zwei Monoeisen(III)komplexe hinweisen. Es wird postuliert, dass in beiden Fällen ein Ligandmolekül 27 an das Zentralatom koordiniert und die Komplexe sich jeweils in der Anzahl an gebundenen Chlorliganden unterscheiden. Je nach der verwendeten Methode zur Komplexierung liegen die möglichen Komplexe laut MS in unterschiedlichen Verhältnissen vor. In MeOH dominiert das Signal bei m/z = 491 [M1]∙+, während in THF beide Komplexe in einem ähnlichen prozentualen Verhältnis zueinander vorliegen. Die Isolierung bzw. Reinigung der laut Massenspektrometrie entstandenen Komplexverbindungen blieb erfolglos. Auch der direkte Einsatz des isolierten Komplexes (ohne weitere Reinigung) in der Katalyse erwies sich als problematisch, da dieser nur in DMSO vollständig, teilweise in H2O und in keinem anderen der getesteten organischen Lösungsmittel löslich ist. Somit konnte für die Katalysereaktion unter der Verwendung von Ligand 27 und FeCl3 x 6 H2O in MeOH, trotz massenspektrometrischem Hinweis auf eine erfolgte Komplexierung und somit die Entstehung einer potentiell Chiralität übertragenden Spezies, keine asymmetrische Induktion am Produkt beobachtet werden. In Abbildung 3-4 ist ein MALDI-MS Spektrum der Komplexierung des Liganden 27 nach Methode 1 exemplarisch dargestellt. Eine genaue 36 Kapitel 3 Ergebnisse und Diskussion strukturelle Darstellung der Komplexe ist jedoch nicht möglich, da keine geeigneten Einkristalle zur Röntgenstrukturanalyse erhalten wurden. Abbildung 3-4: MALDI-MS Spektrum der Komplexierung von Ligand 27 nach Methode 1 (Tabelle 3.1-2). 37 Kapitel 3 3.1.1.3 Ergebnisse und Diskussion Synthese des Harnstoffliganden 28 Die Synthese des neuen harnstoffhaltigen Liganden 28 wurde in Anlehnung an literaturbekannte Syntheseverfahren über drei Stufen entwickelt. Die Syntheseroute ist in Schema 3.1-3 dargestellt. Schema 3.1-3: Synthesemethode zur Darstellung des Harnstoffliganden 28. Der erste Syntheseschritt, die Bildung des primär-aminhaltigen Harnstoffs 38, beinhaltet das langsame Zutropfen von Ethylisocyanat zu einer Lösung von 37 in absolutem DCM bei einer Temperatur von -30 °C. Aufgrund der Bildung eines weißen Niederschlags während der Reaktion wurde mittels KPG-Rührer gerührt. Die Reaktion wurde über Nacht bei tiefen Temperaturen gerührt und Verbindung 38 nach entsprechender Aufarbeitung und Reinigung erhalten.[83] Der Dialkohol 39 wurde in einer Swern-Oxidation mittels Oxalylchlorid, DMSO und TEA in den Dialdehyden 40 überführt.[84] Abschließend führte die Reaktion der Bausteine 38 und 40 in einer reduktiven 38 Kapitel 3 Ergebnisse und Diskussion Aminierung zur gewünschten Zielverbindung 28. Hier war zu beachten, dass die selektive Reduktion des Imins mit NaCNBH3 in einem pH-Bereich von 5-8 stattfindet.[85] Das bei der Hydrolyse entstandene HCN wurde in eine mit wässriger KOH gefüllte Waschflasche geleitet. Ligand 28 wurde in drei Syntheseschritten mit einer Gesamtausbeute von 13% erhalten. 3.1.1.4 Anwendung des harnstoffhaltigen Liganden 28 in der Katalyse Der neue harnstoffhaltige Ligand 28 wurde in Kombination mit verschiedenen Metallquellen (VO(acac)2, Fe(ClO4)3, FeCl3 x 6 H2O, Fe(acac)3 oder Mn(acac)3) unter der Verwendung unterschiedlicher Lösungsmittel auf seine katalytische Aktivität in der Oxidation von Thioanisol geprüft. Die Reaktionsbedingungen ähnelten den in der Katalyse mittels formamidhaltigen Liganden beschriebenen Bedingungen. Die Ergebnisse sind in Tabelle 3.1-3 zusammengefasst. Wie in Tabelle 3.1-3 gezeigt wird, lässt sich ein analoger Trend in den Ergebnissen der Katalysen, verglichen mit den unter Verwendung formamidhaltiger Liganden aus Tabelle 3.1-1, erkennen. In chlorierten Lösungsmitteln lieferte Ligand 28 in Verbindung mit VO(acac)2 oder Fe(ClO4)3 das entsprechende Sulfoxid, auch nach mehreren Stunden, nur in geringen Ausbeuten (38% bzw. 10%, Einträge 2 und 3), während in MeOH die Ausbeuten in Kombination mit FeCl 3 x 6 H2O deutlich gesteigert werden konnten (85%, Eintrag 5). Mit den Metallquellen Fe(acac)3 und Mn(acac)3 wurde das Produkt stattdessen in 56% bzw. 17% Ausbeute erzeugt (Einträge 6 und 7). Ferner wurde der primär-aminhaltige Baustein 38 in Kombination mit VO(acac)2 in der Oxidation überprüft. Die Ausbeute war mit 25% gering (Eintrag 1). In keiner der Reaktionen konnte ein Enantiomerenüberschuss beobachten werden. Der alleinige Einsatz von Ligand 28, ohne die Verwendung einer Metallquelle, lieferte das Oxidationsprodukt in 61% Ausbeute ebenso als Racemat (Eintrag 8). 39 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-3: Einsatz der Harnstoffverbindungen 28 und 38 unter variablen Reaktionsbedingungen. Eintrag Ligand MXn LM Zeit [h] Ausbeute [%]a ee [%]b 1 28 VO(acac)2 CHCl3 72 38 rac 2 28 Fe(ClO4)3 DCM 72 10 rac 3 28 Fe(ClO4)3 MeOH 72 48 rac 4 28 FeCl3 x 6H2O MeOH 48 85 rac 5 28 Fe(acac)3 MeOH 48 56 rac 6 28 Mn(acac)3 MeOH 48 17 rac 7c 38 VO(acac)2 CHCl3 48 25 rac 8 28 -- MeOH 48 61 rac a Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c c(Substrat) = 0.8 mol/L. Basierend auf diesen Ergebnissen wurde im Anschluss die Effizienz des Liganden 28 in Kombination mit FeCl3 x 6 H2O (Verhältnis Ligand/Metall 3:2) in weiteren Lösungsmitteln überprüft (Tabelle 3.1-4). Nur in MeCN konnte Methylphenylsulfoxid in ähnlich hohen Ausbeuten wie in MeOH isoliert werden (72%, Eintrag 4). Das Produkt war auch in diesem Fall racemisch. In chlorierten Lösungsmitteln (DCM und CHCl3) sowie in Toluol war das Produkt nur in sehr geringen Ausbeuten (4% bzw. 2%) isolierbar (Einträge 2, 3 und 5). Maßgeblich hierfür könnte die schlechte 40 Kapitel 3 Ergebnisse und Diskussion Löslichkeit eines der Reaktanden sein, wodurch eine Komplexierung in situ erschwert oder ganz verhindert wird. Tabelle 3.1-4: Screening von Lösungsmitteln. a Eintrag LM Zeit [h] Ausbeute [%]a ee [%]b 1 MeOH 48 85 rac 2 DCM 60 4 rac 3 CHCl3 60 4 rac 4c MeCN 60 72 rac 5c Toluol 60 2 rac Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c schlechte Löslichkeit der Liganden (Zugabe von DCM im Volumenverhältnis 1:5 (DCM/LM)). 3.1.1.5 Kurzkettige Oligopeptide als Liganden in der Katalyse Neben formamid- und harnstoffhaltigen Verbindungen sollten auch kurzkettige Oligopeptide als Liganden in der eisenkatalysierten Sulfidoxidation eingesetzt und auf ihre katalytische Effizienz geprüft werden. Diese Idee wurde durch den erfolgreichen Einsatz kurzer Peptide im Arbeitskreis Tsogoeva als Organokatalysatoren in der enantioselektiven nitro-Michael-Addition inspiriert.[86] Als Liganden in der eisenkatalysierten Sulfidoxidation fanden diese bisher noch keine Verwendung. 41 Kapitel 3 Ergebnisse und Diskussion Erste Versuche wurden ausgehend von den Tripeptidliganden 41 (H-D-Pro-L-Pro-L-Asp-OH) und 42 (H-D-Pro-L-Pro-L-Asp-NH2), welche aus einer arbeitskreisinternen Kooperation bereitgestellt wurden, durchgeführt und unter verschiedenen Reaktionsbedingungen in der Oxidation von Thioanisol mit H2O2 getestet. In Tabelle 3.1-5 sind die Ergebnisse zusammengefasst. Tabelle 3.1-5: Untersuchung der Tripeptidliganden 41 und 42 in der Sulfidoxidation. Eintrag Ligand Additiv LM Zeit [h] Ausbeute [%]a ee [%]b 1 41 TEA (9 mol%) DCM 48 Spuren - 2 41 TEA (9 mol%) MeOH 48 90 rac 3 41 - MeOH 48 79 rac 4 41 TEA (9 mol%) MeCN 48 7 n.b. 5 41 - MeCN 48 59 rac 6 - TEA (9 mol%) MeCN 48 Spuren - 7 41 TEA (9 mol%) THF 48 3 n.b. 8 41 - THF 48 25 rac 9 41 TEA (9 mol%) H2O 48 19 rac 10 42 TEA (6 mol%) MeOH 2 78 rac 11 42 TEA (3 mol%) MeOH 2 67 rac a Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD). 42 Kapitel 3 Ergebnisse und Diskussion Da die Tripeptide 41 und 42 als Zwitterionen vorliegen, wurde in beiden Fällen ein prozentualer Anteil einer Base (TEA) zur Reaktion zugegeben, um eine Koordination der polydentaten Liganden an das Metall zu erleichtern. Hohe Ausbeuten von bis zu 90% konnten mit Tripeptid 41 in MeOH nach 48 h erhalten werden (Eintrag 2). In anderen Lösungsmitteln (DCM, MeCN, THF oder H2O) wurden mit selbigem Liganden in Kombination mit dem Additiv TEA keine bzw. geringe Ausbeuten (<19%) erhalten (Einträge 1, 4, 6 und 8). Der Zusatz von TEA hat einen positiven Effekt auf die katalytische Aktivität beider Liganden in MeOH (Einträge 2 vs. 3 und 10 vs. 11). In MeCN wird stattdessen für Ligand 41 ein gegenläufiger Effekt beobachtet, das heißt ohne die Zugabe von TEA konnte das Produkt nach 48 h in 59% anstatt 7% Ausbeute isoliert werden (Einträge 5 vs. 4). Da sich der Ligand in MeCN und trotz der Zugabe von TEA nicht löst, wird angenommen, dass die Reaktion allein durch FeCl3 x 6 H2O katalysiert wird. In keiner der Reaktionen konnte eine asymmetrische Induktion beobachtet werden. Interessant ist in diesem Zusammenhang aber die Untersuchung der möglichen Hintergrundreaktion im Falle von Ligand 41 im Reaktionsmedium MeOH. Dieser Sachverhalt ist in Tabelle 3.1-6 zusammengefasst. Die Oxidation von Methylphenylsulfid mit 1.2 Äquivalenten H 2O2 in reinem MeOH bzw. unter varierendem Zusatz der übrigen Reaktanden (Ligand 41, FeCl3 x 6 H2O oder TEA) führte nach 48 h zu moderaten Ausbeuten des Produkts von 58-67% (Einträge 1-4). Die in den Einträgen 5-7 gezeigten Experimente geben Aufschluss über eine mögliche Komplexierung des Tripeptides 41 mit FeCl3 x 6 H2O in MeOH. Während die Oxidation durch die Verwendung von Tripeptid 41 als Ligand mit FeCl3 x 6 H2O bzw. in Kombination mit TEA als Additiv das Produkt in jeweils guten bis sehr guten Ausbeuten lieferte (79%, 90%, Einträge 5 und 7), wurde die Oxidation in einem Kontrollexperiment durch die Verwendung von FeCl3 x 6 H2O und TEA fast vollständig unterdrückt (4%, Eintrag 6). Demzufolge wird angenommen, dass die Aktivierung von H 2O2 und somit die Oxidation von Methylphenylsulfid durch die sichtlich gesteigerten Ausbeuten in den Einträgen 5 43 Kapitel 3 Ergebnisse und Diskussion und 7 (79% und 90%) möglicherweise über einen in situ gebildeten Eisenkomplex verläuft. Eine asymmetrische Induktion am Produkt blieb jedoch aus. Tabelle 3.1-6: Hintergrundreaktionen in MeOH. Eintrag 41 [mol%] FeCl3 x 6 H2O [mol%] TEA [mol%] Ausbeute [%]a ee [%]b 1 - - - 61 n.b. 2 3 - - 64 rac 3 - 2 - 67 n.b. 4 - - 3 58 n.b. 5 3 2 - 79 rac 6 - 2 3 4 n.b. 7 3 2 3 90 rac a Isolierte 3.1.2 Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD). Liganden basierend auf Schiffschen-Basen und deren Derivaten Da der Einsatz von formamid-, harnstoff- und peptidhaltigen Liganden in der eisenkatalysierten Oxidation von Thioanisol wenig zufriedenstellende Ergebnisse lieferte, wurde eine Reihe verschiedener iminhaltiger Liganden und von diesen Grundstrukturen abgeleitete Systeme 44 Kapitel 3 Ergebnisse und Diskussion synthetisiert und anschließend in der vanadium- und eisenkatalysierten Sulfidoxidation auf ihre Effizienz hin untersucht. Auf Basis von literaturbekannten Strukturen[19a, 41, 87] wurden die neuen Liganden 45, 48, 49, 56, 57, 58, 59, 60 und 61 entwickelt. Eine Darstellung der verschiedenen iminhaltigen Verbindungen und ihrer Derivate ist in Übersicht 3.1-1 gezeigt, wobei Liganden mit ähnlichen strukturellen Merkmalen jeweils zusammengefasst und durch einen farblichen Rahmen gekennzeichnet sind. Übersicht 3.1-1: Synthetisierte iminhaltige Liganden und deren Derivate. 45 Kapitel 3 Ergebnisse und Diskussion Im ersten Block befinden sich jene Liganden, welche sich von Aminoalkoholen ableiten lassen (tridentate Imin- bzw. Aminliganden), im zweiten Block sind verschiedene polydentate Liganden zusammengefasst, welche ausgehend von chiralem Diphenylethylendiamin aufgebaut sind. Der dritte Block umfasst verschiedenartige primär-aminhaltige Liganden. Die Synthese der Liganden wird im nächsten Abschnitt beschrieben. 46 Kapitel 3 3.1.2.1 Ergebnisse und Diskussion Synthesen der verschiedenen Ligandensysteme Die in Abbildung 3-5 dargestellten Imin-Liganden wurden über die in Schema 3.1-4 gezeigte allgemeine Methode in Anlehnung an literaturbekannte Verfahren synthetisiert (siehe Experimenteller Teil, Kapitel 5). Schema 3.1-4: Allgemeine Synthesemethode zur Darstellung von Iminen. In einer Kondensationsreaktion wurde der jeweilige aromatische Aldehyd mit einem primären Amin (Aminoalkohol bzw. Diamin) verknüpft und das dabei freiwerdende Wasser mittels geeignetem Trocknungsmittel aus der Reaktionsmischung entfernt, um das Gleichgewicht der Reaktion möglichst auf die Seite der Produkte zu verschieben. In der Synthese der Imine 43-45, welche sich von Aminoalkoholen ableiten, als auch des Liganden 51, dessen Aminofunktion mit einer p-TosylGruppe geschützt ist, wurde MeOH als Lösungsmittel verwendet, sowie Na2SO4 oder MgSO4 als Trocknungsmittel. Nach einer Reaktionszeit von 2 h (51) bzw. 3 d bei RT (43, 44, 45) konnten die jeweiligen Liganden in 42-87% Ausbeute erhalten werden. 47 Kapitel 3 Ergebnisse und Diskussion Abbildung 3-5: Synthetisierte Imin-Liganden. Die primär-aminhaltigen Liganden 53, 56, 57, 60 und 61 wurden hingegen, mit Ausnahme von Ligand 55, in DCM synthetisiert. Dabei diente Molekularsieb (3Å) als Trocknungsmittel. Nach einer Reaktionszeit von 3 h und einer schnellen Reinigung mittels Flash-Säulenchromatographie konnten die gewünschten Liganden in 26-87% Ausbeute erhalten werden. In den Fällen 53, 55, 57, 60 und 61 waren die Ausbeuten lediglich gering bis moderat. In der Literatur wird beschrieben, dass primär-aminhaltige Liganden in Lösung disproportionieren können, wodurch hauptsächlich das stabilere Bis-Imin gebildet und das Diamin zurückgewonnen wird. [88] So waren die iminaminhaltigen Liganden immer mit einem gewissen prozentualen Anteil (3-15%) ihres zugehörigen Bis-Imins, im Falle von Ligand 57 sogar mit bis zu 44%, verunreinigt. Als alternative Route zur Darstellung des Liganden 53 wurde die in Schema 3.1-5 gezeigte dreistufige Synthese in 48 Kapitel 3 Ergebnisse und Diskussion Anlehnung an ein literaturbekanntes Verfahren durchgeführt.[89] Trotz des längeren Syntheseweges wurde eine beachtliche Gesamtausbeute von 79% erhalten. Schema 3.1-5: Alternativer Syntheseweg von Imin-Amin-Ligand 53.[89] Der primär-aminhaltige Ligand 53 stellte die Basis für die neuen polydentaten Liganden 48 und 49 dar (Abbildung 3-6). So wurde Ligand 48 in einer weiteren Kondensationsreaktion des Imin-AminLiganden 53 mit 6-Methylpyridin in DCM erhalten. Die geringe Ausbeute von 25% begründet sich möglicherweise durch die bedingt durch die Einführung der Pyridin-Einheit entstandene Instabilität des Produktes. Ligand 49 hingegen wurde ausgehend von Imin-Amin-Liganden 53 und N-Formyl- L-Prolin (34), dessen Synthese zu einem späteren Zeitpunkt beschrieben wird, in einer CDIvermittelten Kupplungsreaktion hergestellt und in einer moderaten Ausbeute von 62% erhalten. Der Salenligand 47 konnte als Nebenprodukt in der Synthese des Imin-Amin-Liganden 53 in 34% Ausbeute isoliert werden (siehe auch Abbildung 3-6). 49 Kapitel 3 Ergebnisse und Diskussion Abbildung 3-6: Synthetisierte polydentate Liganden 47, 48 und 49. Die in Abbildung 3-7 dargestellten Amin-Liganden 46, 52, 54 und 58 wurden mittels klassischer Reduktionsmethoden in Anlehnung an literaturbekannte Verfahren synthetisiert (siehe Experimenteller Teil). Schema 3.1-6: Allgemeine Synthesemethode zur Reduktion von Iminen. Zur Reduktion der Liganden 52, 54 und 58 wurde NaBH4 in MeOH verwendet - Ligand 46 wurde mittels LiBH4 in THF reduziert. Eine erfolgreiche Reduktion wurde durch den Farbumschlag der gelb-grünlichen Reaktionslösung zu einer farblosen Lösung erkennbar. Die Liganden 46, 52, 54 und 58 konnten in guten Ausbeuten (65-76%) isoliert werden. Abbildung 3-7: Synthetisierte Amin-Liganden 46, 52, 54 und 58. Ferner wurde der in Schema 3.1-7 dargestellte primär-aminhaltige Ligand 59 in einer CDIvermittelten Kupplungsreaktion[79] synthetisiert. Dazu wurde zunächst der C-Terminus von Boc-L50 Kapitel 3 Ergebnisse und Diskussion His-OH mit CDI in THF aktiviert und anschließend eine Lösung von Diphenylethylendiamin (62) in DCM langsam zugetropft, um die Wahrscheinlichkeit einer Zweifachkondensation möglichst gering zu halten. Ligand 59 wurde in einer Ausbeute von 32% erhalten. Schema 3.1-7: Synthese des Liganden 59. 3.1.2.2 Synthese von neuen primär-aminhaltigen Eisen(III)komplexen Im Rahmen der Dissertation wurden insgesamt vier neue primär-aminhaltige Eisen(III)komplexe nach den in Schema 3.1-8 und Schema 3.1-9 gezeigten Synthesemethoden hergestellt. Diese Komplexe sollten zu einem späteren Zeitpunkt im Vergleich zu den in situ generierten Komplexen in der Oxidation von Thioanisol mittels H2O2 eingesetzt werden. Schema 3.1-8: Synthesestrategie zur Darstellung der neuen Eisen(III)komplexe 65, 66 und 67 mit primär-aminhaltigen Schiffsche-Base Liganden. Zur Synthese der in Schema 3.1-8 dargestellten Komplexe 65-67 wurden 2.0 Äquivalente des jeweiligen Imin-Amin-Liganden 53, 60 oder 61 und 1.0 Äquivalente FeCl3 x 6 H2O verwendet (in 51 Kapitel 3 Ergebnisse und Diskussion Anlehnung an die Reaktionsbedingung beschrieben in Abschnitt 3.1.2.3.2.3). Die Komplexierung wurde dabei unter milden Bedingungen (bei RT und an Luft) in THF innerhalb von 2 h durchgeführt. Nach dem Entfernen des Lösungsmittels wurden die erhaltenen tief violetten Komplexe ohne weitere Reinigung mittels IR-Spektroskopie und Massenspektrometrie analysiert und charakterisiert. In Abbildung 3-8 sind die hochauflösenden ESI-MS Spektren der postulierten einkernigen Eisenkomplexe 65-67 (Schema 3.1-8) dargestellt. Der jeweilige Produktpeak (für Komplex 65 bei m/z = 910.48 [M-Cl]+ und für Komplexe 66 und 67 bei m/z = 1282.57) lässt auf eine zweifache Koordination der tridentaten Schiffsche-Base Liganden an das Eisen(III)-Zentralatom schließen. Zusätzlich sind die Signale der freien Liganden (Ligand 53 bei m/z = 429.28 [M+H]+ und Liganden 60 bzw. 61 bei m/z = 429.28 [M+H]+) im Spektrum zu erkennen. Die theoretischen Isotopenmuster der postulierten Komplexe 65-67 stimmen mit den gemessenen sehr gut überein. Die vorgeschlagene Struktur konnte noch nicht per Röntgenstrukturanalyse bestätigt werden, da keine geeigneten Kristalle erhalten wurden. 52 Kapitel 3 Ergebnisse und Diskussion Abbildung 3-8: Exemplarische Darstellung der ESI-MS Spektren der postulierten einkernigen Eisenkomplexe 65-67. Der in Schema 3.1-9 gezeigte mononukleare Eisen(III)komplex 68 wurde in Anlehnung an die Arbeiten von Katsuki synthetisiert.[41] Dazu wurden 2.0 Äquivalente des Amin-Liganden 58 und 1.0 Äquivalente FeCl2 in EtOH für 1.5 h gerührt. Nach dem Entfernen des Lösungsmittels wurde der erhaltene Rückstand via IR-Spektroskopie und Massenspektrometrie analysiert. Das hochauflösende Massenspektrum (Abbildung 3-9) zeigt einen charakteristischen Peak bei m/z = 1194.4540 was erneut auf die Koordination zweier tridentater Liganden an das Eisen(III)Zentralatom hinweist. Das theoretische Isotopenmuster des postulierten Komplexes 68 stimmt mit 53 Kapitel 3 Ergebnisse und Diskussion dem gemessenen sehr gut überein, jedoch ist auch hier die tatsächliche Struktur noch nicht bekannt. Schema 3.1-9: Synthesemethode zu Darstellung des neuen Eisen(III)komplexes 68 nach Katsuki.[41] Abbildung 3-9: ESI-MS Spektrum des postulierten einkernigen Eisenkomplexes 68. 54 Kapitel 3 3.1.2.3 Ergebnisse und Diskussion Anwendung Schiffsche-basehaltiger Liganden in der Katalyse Im folgenden Abschnitt der Dissertation werden die Ergebnisse der Anwendung der im vorigen Abschnitt beschriebenen Ligandensysteme auf ihre Effizienz in der vanadium- und eisenkatalysierten Oxidation von Thioanisol mit H2O2 diskutiert. 3.1.2.3.1 3.1.2.3.1.1 Anwendung von chiralen Liganden auf die vanadiumkatalysierte Sulfidoxidation Screening von chiralen Liganden abgeleitet von β-Aminoalkoholen Basierend auf den wegweisenden Arbeiten von Bolm und Bienewald[19, 25] wurden erste katalytische Experimente ausgehend von den in Tabelle 3.1-7 dargestellten Imin- bzw. AminLiganden, welche aus einem Salicylaldehyd-Derivat und einem chiralen 1,2-Aminoalkohol aufgebaut sind, durchgeführt. Zur Oxidation von Thioanisol wurden 2 mol% VO(acac)2 als Metallquelle, 3 mol% des jeweiligen chiralen Liganden, sowie 1.2 Äquivalente H2O2 in CHCl3 eingesetzt. Wie aus Tabelle 3.1-7 ersichtlich ist, wurden zunächst die Imine 43-46 mit variierendem Substitutionsmuster an der Salicylaldehyd-Einheit auf dessen Einfluss in der Oxidation von Thioanisol untersucht. Ligand 44 lieferte Methylphenylsulfoxid in 61% Ausbeute und 40% ee nach 24 h Reaktionszeit (Eintrag 1). Der Ersatz der Iod-Substituenten durch die sterisch anspruchsvolleren t-Butylgruppen in Positionen 3 und 5 der Salicylaldehyd-Einheit führte zu besseren Ausbeuten und einem leicht erhöhten ee-Wert (79% Ausbeute, 45% ee, Eintrag 2). Ferner ist anzumerken, dass sowohl die Ausbeute als auch die Enantioselektivität des Produkts unter diesen Reaktionsbedingungen mit Ligand 43 im Vergleich zu den von Somanathan et al.[26b] berichteten Ergebnissen gesteigert werden konnten (Lit.:[26b] 64% Ausbeute, 28% ee vs. 79% Ausbeute, 45% ee, Eintrag 2). Eine Änderung der Zugabereihenfolge von H2O2 und Thioanisol bewirkte eine Verringerung des ee-Werts des Produkts auf 34% ee (Eintrag 3 vs. Eintrag 2). Weiterhin führte die Verlängerung der Reaktionszeit von 24 h auf 48 h zu schlechteren Ausbeuten 55 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-7: Screening chiraler tridentater Liganden (mit unterschiedlichen Substituenten). a Eintrag Ligand Zeit [h] Ausbeute [%]a ee [%]b 1 44 24 61 40 2 43 24 79 45 3c 43 24 81 34 4 43 48 67 43 5d 43 48 72 16 6 45 24 32 rac 7 46 24 76 12 Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c Zugabereihenfolge geändert: Ligand 43 und VO(acac)2 wurden für 1 h gerührt, H2O2 wurde vor Thioanisol zugegeben; d Reaktion wurde in MeCN durchgeführt. (67%), wobei der ee-Wert (43%) weitestgehend konstant blieb (Eintrag 4 vs. Eintrag 2). Die Durchführung der Reaktion in einem polareren Lösungsmittel (MeCN) führte zu einer deutlichen Verschlechterung des ee-Wertes (72% Ausbeute, 16% ee, Eintrag 5 vs. Eintrag 4). Der Einfluss der Hydroxygruppe und der Art des Halogensubstituenten in der Salicyleinheit auf das katalytische System wird durch die Oxidation mit Ligand 45 verdeutlicht. Der Ersatz der Hydroxy- und Iodsubstituenten durch Fluoratome, welche andere elektronische und koordinative Eigenschaften besitzen, führte zu einem vollständigen Verlust der Enantioselektivität. Auch die Ausbeute wurde 56 Kapitel 3 Ergebnisse und Diskussion deutlich verschlechtert (Eintrag 6 vs. Eintrag 1). Der zum Amin reduzierte Ligand 46 lieferte Methylphenylsulfoxid in 76% Ausbeute mit nur 12% ee (Eintrag 7). 3.1.2.3.1.2 Screening polydentater Liganden abgeleitet von chiralen 1,2-Diaminen In diesem Abschnitt werden die Ergebnisse des Einsatzes verschiedener tri- bzw. polydentater Liganden (48, 49, 51, 52, 53 und 55), welche sich von einem chiralen 1,2-Diamin (bzw. 1.4-Diamin) ableiten, in der vanadiumkatalysierten enantioselektiven Oxidation von Thioanisol beschrieben. Die erhaltenen Resultate sind in Tabelle 3.1-8 zusammengefasst. Im Gegensatz zu Ligand 43 (Eintrag 4, Tabelle 3.1-7) lieferte Ligand 53 (Ersatz der Hydroxyfunktion durch ein primäres Amin) unter gleichen Reaktionsbedingungen nach 48 h Reaktionszeit zwar bessere Ausbeuten (79%), jedoch betrug die Enantioselektivität des Produkts nur noch 9% ee (Eintrag 1). Ebenso führte die Einführung einer Pyridineinheit in diesen Liganden (48) zu keiner Verbesserung hinsichtlich Ausbeute und Enantioselektivität (78% Ausbeute, 9% ee, Eintrag 2), obwohl eine zusätzliche Koordinationsstelle zum Metall geschaffen wurde. Im Fall von Ligand 49 hatte der Einbau eines N-Formyl-L-Prolin-Substituenten sogar den vollständigen Verlust der Enantioselektivität im Produkt zur Folge (60% Ausbeute, rac, Eintrag 3). Die Liganden 51 und 52, in denen die freie Aminogruppe durch eine p-Tosyl-Einheit geschützt vorliegt, lieferten Methylphenylsulfoxid in nahezu gleichen Ausbeuten, wobei die reduzierte Form (Ligand 52) einen minimalen ee-Wert von 4% erzeugt (Eintrag 5). Auch Ligand 55, der eine chirale BinaphthylGruppe, also eine zusätzliche Quelle chiraler Information enthält, führte zu keinem erfolgreichen Resultat (70% Ausbeute, rac, Eintrag 6 und 66% Ausbeute, rac, Eintrag 7). 57 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-8: Screening polydentater Liganden abgeleitet von chiralen 1,2-Diaminen (bzw. 1,4Diamin). Eintrag Ligand Zeit [h] Ausbeute [%]a ee [%]b 1 53 48 79 9 2 48 48 78 9 3 49 48 60 rac 4 51 60 62 rac 5 52 48 67 4 6 55 24 70 rac 7c 55 24 66 rac a Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c Zugabereihenfolge geändert: Ligand 55 und VO(acac)2 wurden für 1 h gerührt, H2O2 wurde vor Thioanisol zugegeben. 58 Kapitel 3 3.1.2.3.2 Ergebnisse und Diskussion Anwendung von primär-aminhaltigen Liganden auf die eisenkatalysierte Sulfidoxidation Im Gegensatz zum Fortschritt in der metallkatalysierten Sulfidoxidation, welcher mit symmetrischen Schiffsche-Base Liganden erreicht wurde, ist die Verwendung unsymmetrischer Schiffsche-Base Liganden in diesem Gebiet weitaus seltener. Im folgenden Abschnitt wird zum ersten Mal von der Anwendung unsymmetrischer Sulfidoxidation berichtet. Es primär-aminhaltiger ist bekannt, dass Liganden auf Übergangsmetalle die in eisenkatalysierte Oxygenase- und Peroxigenaseenzymen an Donoratome von Peptidketten in einer nicht-symmetrischen Umgebung gebunden sind, wodurch die Entwicklung von neuen unsymmetrischen eisenhaltigen Katalysatoren auch in dieser Arbeit motiviert und inspiriert wird. Primär-aminhaltige nicht-symmetrische Liganden wurden lediglich in der vanadium- nicht aber der eisenkatalysierten Sulfidoxidation von Romanowski et al. im Jahr 2007 eingesetzt,[87a] welche durch den Gebrauch von chiralen Dioxovanadium(V)komplexen sowie CHP[87a, 90] oder H2O2[91] als Oxidationsmittel in DMSO die entsprechenden Sulfoxide mit Enantioselektivitäten von 19-39% ee produzierten. 3.1.2.3.2.1 Screening von chiralen Liganden und verschiedenen Eisenquellen Aufgrund der richtungsweisenden Resultate in der vanadiumkatalysierten Oxidation von Thioanisol mittels H2O2 (Eintrag 1, Tabelle 3.1-8 und Einträge 1 und 2, Tabelle 3.1-7) wurden ausgewählte Liganden einem Screening von verschiedenen eisenhaltigen Metallquellen unterworfen. Die Ergebnisse sind in Tabelle 3.1-9 zusammengefasst. 59 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-9: Screening verschiedener chiraler Liganden mit unterschiedlichen Eisenquellen. Eintrag Ligand MXn LM Ausbeute [%]a ee [%]b 1 43 (3 mol%) FeCl3 x 6 H2O CHCl3 8 28 2 43 (3 mol%) FeCl3 x 6 H2O MeCN 64 13 3 43 (3 mol%) Fe(acac)3 CHCl3 15 13 4c 43 (4 mol%) Fe(acac)3 CHCl3 2 n.b. 5c, d 44 (4 mol%) Fe(acac)3 DCM 28 12 6 53 (3 mol%) FeCl3 x 6 H2O CHCl3 20 33 7 53 (3 mol%) Fe(acac)3 CHCl3 31 14 8 55 (3 mol%) FeCl3 x 6 H2O CHCl3 7 2 9 47 (3 mol%) FeCl3 x 6 H2O CHCl3 25 rac a Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c Benzoesäure als Additiv verwendet (1 mol%); d c(Substrat) = 0.6 mol/L. Erste Experimente mit Ligand 43 in Kombination mit FeCl3 x 6 H2O oder Fe(acac)3 lieferten Methylphenylsulfoxid in CHCl3 in geringen Ausbeuten (2-15%, Einträge 1, 3 und 4) mit einem 60 Kapitel 3 Ergebnisse und Diskussion maximalen ee-Wert von 28% (Eintrag 1). In MeCN konnte die Ausbeute zwar auf 64% gesteigert werden, jedoch blieb der ee-Wert mit 13% verhältnismäßig gering (Eintrag 2). Auch Versuche, die in Anlehnung an die von Legros und Bolm entwickelten Reaktionsbedingungen durchgeführt wurden, welche durch den Zusatz einer Carbonsäure bzw. deren Li-Salz als Additiv die Oxidiation von Thioanisol mit H2O2 im Hinblick auf Ausbeute und Enantioselektivität erheblich steigerten,[39] führten in der Oxidation mit Ligand 43 und 44 zu keiner Verbesserung (Einträge 4 und 5). Das vielversprechendste Ergebnis bezüglich Ausbeute und Enantioselektivität lieferte der primäraminhaltige Ligand 53 in Kombination mit FeCl3 x 6 H2O in CHCl3 (20% Ausbeute, 33% ee, Eintrag 6). Weiterhin ist anzumerken, dass der salenartige tetradentate Ligand 47 (Bis-Addukt von 53) unter den genannten Bedingungen ein racemisches Produkt mit einer Ausbeute von 25% produzierte (Eintrag 9). Demzufolge ist eine negative Beeinflussung des ee-Wertes bei der Verwendung des primär-aminhaltigen Liganden 53, entsprechend dem Grad der Verunreinigung mit seinem Bis-Addukt (siehe Abschnitt 3.1.2.2.1) zu erwarten. 3.1.2.3.2.2 Optimierung der Reaktionsbedingungen mit dem primär-aminhaltigen Liganden 53 Ausgehend von dem erfolgsversprechenden Resultat aus Tabelle 3.1-9 (20% Ausbeute, 33% ee, Eintrag 6), wurde der primär-aminhaltige Ligand 53 in Kombination mit FeCl3 x 6 H2O einem Screening von verschiedenen Lösungsmitteln, Additiven, sowie anderen Oxidationsmitteln unterworfen, um die Reaktionsbedingungen im Hinblick auf Ausbeuten und Enantioselektivitäten weiter zu optimieren. a. Screening von Lösungsmitteln Die Optimierung begann mit einem Screening verschiedener Lösungsmittel, die Ergebnisse sind in Tabelle 3.1-10 dargestellt. 61 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-10: Screening verschiedener Lösungsmittel mit ausgewähltem Imin-Amin-Ligand 53. a Eintrag Ligand 53 [mol%] LM Ausbeute [%]a ee [%]b 1 3 CHCl3 20 33 2 3 Toluol 13 21 3 3 DCM 27 28 4 3 MeCN 62 16 5 3 CHCl3/MeCN (2:1) 26 31 6 3 CHCl3/MeCN (4:1) 29 33 7 4 CHCl3/MeCN (4:1) 40 35 8c 4 CHCl3/MeCN (4:1) 37 35 9d 4 CHCl3/MeCN (4:1) 11 7 10e 4 MeOH 73 6 11e 4 THF 50 34 Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c Ligand 53 wurde in einem Schritt synthetisiert im Gegensatz zur alternativen 3-Stufensyntheseroute (siehe Kapitel 3.1.2.1); d Zugabe von 6 x 8.22 µL H2O2 über einen Zeitraum von 2 d; e Reaktionszeit: 24 h. Die Oxidation von Thioanisol mit Ligand 53 lieferte Methylphenylsulfoxid in chlorierten Lösungsmitteln in geringen Ausbeuten (<27%) und mäßig guten ee-Werten (<33% ee) (Einträge 1 und 3). In dem unpolaren Lösungsmittel Toluol wurde das Produkt nur noch in 13% Ausbeute und 21% ee erhalten (Eintrag 2). Da die Ausbeute des Produkts in MeCN erheblich gesteigert werden konnte (62%, Eintrag 4), wurde eine Mischung von MeCN und CHCl3 verwendet und die Oxidation 62 Kapitel 3 Ergebnisse und Diskussion in verschiedenen Mischungsverhältnissen (CHCl3/MeCN 2:1, 4:1) durchgeführt, mit dem Ziel Ausbeute und ee-Wert des Sulfoxids zu steigern (Einträge 5-9). Eine zusätzliche Änderung des Ligand/Metall-Verhältnisses von 3:2 auf 4:2 bewirkte weiterhin eine Erhöhung der Ausbeute, wobei der ee-Wert relativ konstant blieb (40% Ausbeute, 35% ee, Eintrag 7 vs. Einträge 5 und 6). Der primär-aminhaltige Ligand 53, welcher auf zwei verschiedene Weisen hergestellt werden kann, führte in der eisenkatalysierten Oxidation von Alkenen[92] mit H2O2 je nach Synthesemethode zu unterschiedlichen Resultaten hinsichtlich Ausbeute und Enantioselektivität. Daher wurden die aus verschiedenen Syntheserouten gewonnenen Liganden 53 auch in der Oxidation von Thioanisol unabhängig voneinander überprüft. Beide Liganden lieferten in dieser Reaktion vergleichbare Ausbeuten und Enantioselektivitäten (40% Ausbeute, 35% ee, Eintrag 7 vs. 37% Ausbeute, 35% ee, Eintrag 8). Die portionsweise Zugabe von H2O2 (6 x 8.22 µL) innerhalb eines Zeitraums von 48 h führt zu keiner Verbesserung des Reaktionsergebnisses (11% Ausbeute, 7% ee, Eintrag 9). In MeOH wurde das Produkt nach 24 h mit guten Ausbeuten von 73% und einer Enantioselektivität von 6% ee isoliert (Eintrag 10). Einen zufriedenstellenden Kompromiss in Bezug auf Enantioselektivität und Ausbeute lieferte die Verwendung von THF, in dem das Sulfoxid in moderaten Ausbeuten (50%) und einem akzeptablen ee-Wert von 34% ee isoliert werden konnte (Eintrag 11). b. Screening von Additiven und Oxidationsmitteln Analog zum Screening verschiedener Lösungsmittel erfolgte parallel ein weiteres Screening von verschiedenen Additiven (Tabelle 3.1-11) und Oxidationsmitteln (Tabelle 3.1-12). Wie in Tabelle 3.1-11 zu sehen ist, wurden 1 mol% der jeweiligen Säure bzw. Base als Additiv eingesetzt. Mit L-Ascorbinsäure wurde Ausbeute wie auch Enantioselektivität des Produkts im Lösungsmittelgemisch CHCl3/MeCN (4:1) deutlich verschlechtert (25% Ausbeute, 4% ee, Eintrag 1). Auch das vielversprechende Additiv 4-Methoxybenzoesäure, welches von Bolm erfolgreich in der eisenkatalysierten Oxidation mit aminoalkoholhaltigen Liganden eingesetzt 63 Kapitel 3 Ergebnisse und Diskussion wurde,[39] trug zu keiner Verbesserung bei (39% Ausbeute, 33% ee, Eintrag 2 und 28% Ausbeute, 11% ee, Eintrag 3). Der Einsatz von 2,6-Pyridindicabonsäure (H2Pydic), welches erfolgreich durch die Arbeitsgruppe von Beller in der eisenkatalysierten Epoxidierung verwendet wurde,[87b] lieferte unabhängig vom Lösungsmittel moderate bis gute Ausbeuten von 62-79%, jedoch ohne chirale Induktion am Produkt (Einträge 4-6). Weiterhin wurde auch die Base trans-2,5-Dimethylpiperazin als Additiv in der Oxidation geprüft. Sowohl Ausbeute als auch Enantioselektivität des Produkts in THF waren gering (18% Ausbeute, 8% ee, Eintrag 7). Tabelle 3.1-11: Screening von Additiven in verschiedenen Lösungsmitteln. Eintrag Additiv LM Ausbeute [%]a ee [%]b 1 L-Ascorbinsäure CHCl3/MeCN (4:1) 25 4 2 4-Methoxybenzoesäure CHCl3/MeCN (4:1) 39 33 3c 4-Methoxybenzoesäure DCM 28 11 4 H2Pydic CHCl3/MeCN (4:1) 62 rac 5 H2Pydic DCM 75 rac 6d H2Pydic THF 79 rac THF 18 8 7d a Isolierte trans-2,5Dimethylpiperazin Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c Reaktionsbedingungen von Legros und Bolm: (Fe(acac)3, DCM, 16 h, Rühren mit 150 rpm, c(Substrat) = 0.5 mol/L)[39a]; d Reaktionszeit 24 h. 64 Kapitel 3 Ergebnisse und Diskussion Neben den umweltfreundlichen Oxidationsmitteln H2O2 oder UHP (an Harnstoff gebundenes H2O2) wurden im Vergleich auch andere Oxidationsmittel (CHP, TBHP und m-CPBA) getestet. Die Ergebnisse sind in Tabelle 3.1-12 zusammengefasst. UHP lieferte das Oxidationsprodukt im Vergleich zu H2O2 trotz längerer Reaktionszeit in geringerer Ausbeute und Enantioselektivität (30% Ausbeute, 14% ee, Eintrag 2 vs. 39% Ausbeute, 34% ee, Eintrag 1). Auch der Einsatz von CHP, welches als Oxidationsmittel erfolgreich unter anderem in der titankatalysierten Sulfidoxidation verwendet wird,[8a, 61a] zeigte keine Verbesserung des katalytischen Systems (35% Ausbeute, rac, Eintrag 3). Andere Oxidationsmittel wie beispielsweise TBHP oder m-CPBA lieferten Methylphenylsulfoxid zwar in höheren Ausbeuten von 59% und 79%, jedoch auch in diesen Fällen in seiner racemischen Form (Einträge 4 und 5). Somit bleibt wässriges H2O2 das Oxidationsmittel der Wahl. Tabelle 3.1-12: Screening von Oxidationsmitteln. Eintrag Oxidationsmittel LM Zeit [h] Ausbeute [%]a ee [%]b 1 H2O2 CHCl3/MeCN (4:1) 24 39 34 2 UHP CHCl3/MeCN (4:1) 48 30 14 3 CHP CHCl3/MeCN (4:1) 48 35 rac 4 TBHP CHCl3/MeCN (4:1) 48 59 rac 5 m-CPBA CHCl3/MeCN (4:1) 24 79 rac a Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD). 65 Kapitel 3 3.1.2.3.2.3 Ergebnisse und Diskussion Untersuchung weiterer Liganden in der eisenkatalysierten Oxidation von Thioanisol unter optimierten Reaktionsbedingungen Aus dem vorherigen intensiven Screening (LM, Additiv, Oxidationsmittel) mit dem primäraminhaltigen Liganden 53 in Kombination mit FeCl3 x 6 H2O als Metallquelle ging hervor, dass die Oxidation von Thioanisol in THF einen guten Kompromiss bezüglich Ausbeute und Enantioselektivität des Produkts darstellt (50%, 34% ee, Tabelle 3.1-13, Eintrag 1). Aus diesem Grund wurden erneut ausgewählte chirale Liganden unter den zu diesem Zeitpunkt besten Reaktionsbedingungen untersucht. Die Ergebnisse sind in Tabelle 3.1-13 zusammengefasst. Tabelle 3.1-13: Screening verschiedener chiraler Liganden in THF. 66 Kapitel 3 Ergebnisse und Diskussion Daten zu Tabelle 3.1-13: Eintrag Ligand Ausbeute [%]a ee [%]b 1 53 50 34 (R) 2 62 17 3 (S) 3c 53 51 36 (R) 4 47 36 rac 5 54 48 6 ( R) 6 50 49 21 (R) 7 43 68 6 ( R) 8 44 72 7 ( R) 9 55 74 rac 10d 56 54 8 ( R) 11e 53 9 rac 12 - 78 rac 13e - 13 n.b. a Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c Reaktion wurde unter Inertbedingungen (N2) durchgeführt; d Zugabe von H2O2 erfolgte via Spritzenpumpe (10 µL/h) über einen Zeitraum von 5 h (Ligand 56 war mit 25% Bis-Addukt verunreinigt); e Reaktion wurde ohne Eisensalz durchgeführt. Im Vergleich zum primär-aminhaltigen Liganden 53 lieferte das kommerziell erhältliche chirale Diamin 62, welches den Grundbaustein des Liganden 53 darstellt, das Oxidationsprodukt in 17% Ausbeute als nahezu racemische Verbindung (3% ee (S), Eintrag 2). Auch die Durchführung der Katalyse mit Ligand 53 unter Inertbedingungen brachte keine weiteren Verbesserungen im Hinblick auf Ausbeute und Enantioselektivität (51% Ausbeute, 36% ee, Eintrag 3). Das Einführen einer zweiten Salicyleinheit zum sogenannten Bis-Addukt 47 führte im Produkt zu Einbußen in der Ausbeute (36%) und zum vollständigen Verlust der chiralen Information (Eintrag 4). Der reduzierte Ligand 54 lieferte Methylphenylsulfoxid zwar in vergleichbarer Ausbeute wie Ligand 53 (48%, Eintrag 5 vs. 50%, Eintrag 1), die Enantioselektivität sank jedoch auf 6% ee (Eintrag 5), was die Bedeutung der Iminfunktion im Katalysator verdeutlicht. Um die Bedeutung der freien Aminofunktion weiter herauszustellen, wurde der primär-aminhaltige Ligand als HCl-Salz (50) 67 Kapitel 3 Ergebnisse und Diskussion geschützt. Dies wirkte sich nur auf die chirale Induktion im Produkt aus, die Ausbeute blieb unverändert (49%, 21% ee, Eintrag 6). Es wird angenommen, dass während der Komplexbildung in situ HCl freigesetzt werden kann, wodurch die freie Aminofunktion teilweise zurück gebildet wird, was eine sichtbare chirale Induktion zur Folge hat. Diese ist jedoch im Vergleich zu Ligand 53 weniger ausgeprägt, was darauf schließen lässt, dass die Aminofunktion zumindest teilweise protoniert vorliegt (21% ee vs. 34% ee, Eintrag 6 vs. Eintrag 1). Zusätzlich wurden bekannte Schiffsche-Base Liganden (43 und 44), welche sich von Aminoalkoholen ableiten, untersucht. Die Ausbeuten stiegen auf 68% bzw 72% an, der ee-Wert blieb jedoch verhältnismäßig gering (6% ee, Eintrag 7 und 7% ee, Eintrag 8). Ligand 55, welcher eine sterisch anspruchsvollere BinaphthylEinheit im Austausch gegen (1R,2R)-(+)-1,2-Diphenylethylendiamin als chiralen Baustein enthält, führte zu einer guten Ausbeute des Sulfoxids von 74%. Eine chirale Induktion konnte aber nicht beobachtet werden (Eintrag 9). Auch Ligand 56 enthält eine chirale Binaphthyl-Einheit, die hier aber den Salicylaldehydbaustein des Liganden 53 ersetzt und so zur Existenz einer zusätzlichen chiralen Informationsquelle führt. Methylphenylsulfoxid wurde in 54% Ausbeute und einer geringen Enantioselektivität von 8% ee gebildet (Eintrag 10). Zum Vergleich wurden auch die einzelnen Komponenten des katalytischen Systems, sowie die Hintergrundreaktion in THF untersucht (Einträge 11-13). Der alleinige Einsatz des primär-aminhaltigen Liganden 53 in der Oxidation von Thioanisol führte lediglich zu 9% Ausbeute des Produkts in Form des Racemats (Eintrag 11). Die Verwendung von FeCl3 x 6 H2O in Abwesenheit eines Liganden produziert Methylphenylsulfoxid in hohen Ausbeuten (78%, Eintrag 12), während die Hintergrundreaktion in THF das Produkt in geringen Ausbeuten von 13% liefert (Eintrag 13). Zusammenfassend bleibt zu sagen, dass die zuvor erhaltenen Ergebnisse mit Ligand 53 durch ein weiteres Ligandenscreening nicht übertroffen werden konnten. 68 Kapitel 3 3.1.2.3.2.4 Ergebnisse und Diskussion Weitere Optimierung der Reaktionsbedingungen mit Ligand 53 Nachdem sich durch den Test verschiedener Liganden Ligand 53 als der leistungsfähigste herauskristallisiert hat, wurden weitere Variationen der Reaktionsparameter vorgenommen, um die bisherigen Resultate zu verbessern. Eine zusätzliche Optimierung der Reaktionsbedingungen erfolgte durch eine Verringerung der Reaktionszeit von 24 auf 5 h (siehe Tabelle 3.1-14), wodurch nahezu keine Einbußen in Ausbeute und Enantioselektivität zu verzeichnen waren (50% Ausbeute, 34% ee, Eintrag 1 vs. 52% Ausbeute, 31% ee, Eintrag 2). Darüberhinaus wurde H2O2 nicht mehr in einer Portion zugegeben, sondern mithilfe einer Spritzenpumpe langsam über einen Zeitraum von 5 h zugetropft. Diese Methode bewirkte eine Steigerung des ee-Werts auf insgesamt 43% (Eintrag 3). Der Grund dafür ist, dass durch die langsame Zugabe des Oxidationsmittels der Überschuss an nicht katalytisch wirksamem H2O2 geringer ist und somit ein unkatalysierter Oxidationsprozess verhindert bzw. minimiert werden kann.[27b] Zusätzliche Bemühungen, die Ausbeute und Enantioselektivität des Oxidationsprodukts zu steigern, blieben erfolglos (Einträge 4-9). In diesem Zusammenhang wurde auch der primär-aminhaltige Ligand 53 mit verschiedenen prozentualen Verunreinigungen seines Bis-Addukts 47 (siehe vorheriges Kapitel) in der Oxidation von Thioanisol untersucht, da dieses Methylphenylsulfoxid nach 24 h in 36% Ausbeute als Racemat liefert und somit Auswirkungen auf Enantioselektivität und Ausbeute des Produkts haben kann (siehe Tabelle 3.1-13, Eintrag 4). Die Verwendung des Liganden 53 mit jeweils einer maximalen Verunreinigung von 12% des Bis-Addukts 47 im Vergleich zu 3% Verunreinigung zeigten jedoch kaum einen Einfluss auf Enantioselektivität sowie Ausbeute (52% Ausbeute, 43% ee, Eintrag 3 vs. 48% Ausbeute, 44% ee, Eintrag 4). Auch die weitere Verringerung der Reaktionszeit von 5 auf 3 h bzw. 1 h führte lediglich zu schlechteren Ausbeuten und ee-Werten im Produkt (Einträge 10 und 11). 69 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-14: Weitere Optimierung des Systems mit dem primär-aminhaltigen Liganden 53. a Eintrag H2O2 [Äq.] c [mol/L] Zeit [h] Ausbeute [%]a ee [%]b 1 1.2 0.8 24 50 34 2 1.2 0.8 5 52 31 3c 1.2 0.8 5 52 43 4c,d 1.2 0.8 5 48 44 5c,e 1.2 0.8 5 52 32 6c 1.2 1.2 5 39 49 7c 2.0 0.8 5 56 35 8c,f 1.2 0.8 5 28 24 9c,g 1.2 0.8 5 31 24 10c 1.2 0.8 3 46 39 11c 1.2 0.8 1 41 36 Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c H2O2-Zugabe via Spritzenpumpe (10 µL/h); d Ligand 53 war mit 3% des Bis-Imins 47 verunreinigt; e Reaktion wurde in MeCN durchgeführt; f Reaktion wurde bei 0 °C durchgeführt; g Thioanisol wurde gleichzeitig per Spritzenpumpe zugegeben. Wie aus Tabelle 3.1-15 hervorgeht, hat auch die Art der Eisenquelle einen entscheidenden Einfluss auf das katalytische System. Wurde statt FeCl3 x 6 H2O die Metallquelle Fe(acac)3 verwendet, sind sowohl die Ausbeute als auch der ee-Wert im Sulfoxid stark gesunken (33% Ausbeute, 10% ee, 70 Kapitel 3 Ergebnisse und Diskussion Eintrag 2 vs. Eintrag 1). Die Verwendung von wasserfreiem FeCl2 lieferte vergleichbare Werte wie FeCl3 x 6 H2O (45% Ausbeute, 30% ee, Eintrag 3 vs. Eintrag 2). Tabelle 3.1-15: Screening verschiedener Eisenquellen mit dem ausgewählten Liganden 53. Eintrag FeXn Ausbeute [%]a ee [%]b 1 FeCl3 x 6 H2O 50 34 2 Fe(acac)3 33 10 3 FeCl2 45 30 a Isolierte 3.1.2.3.2.5 Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD). Anwendung neuer primär-aminhaltiger Liganden 60 und 61 auf die Oxidation von Thioanisol Im Anschluss an das intensive Screening mit dem primär-aminhaltigen Liganden 53 wurde seine Struktur weiter modifiziert und sterisch anspruchsvollere Salicylaldehyd-Derivate eingeführt, um Ausbeuten und Enantioselektivitäten der Katalysen weiter zu erhöhen. Dieser Sachverhalt wird in Tabelle 3.1-16 diskutiert. 71 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-16: Anwendung von neuen primär-aminhaltigen Liganden 60 und 61 auf die Oxidation von Thioanisol. a Eintrag Ligand Ausbeute [%]a ee [%]b 1 53 52 43 2 61 65 36 3 60 66 53 4c 60 69 54 Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c Reaktionszeit 7 h: nach 5 bzw. 6 h wurden weitere 10 µL H2O2 per Spritzenpumpe (10 µL/h) zugegeben (insgesamt 1.7 Äq. H2O2). Die Anwendung des neuen Liganden 61 auf die Oxidation von Thioanisol lieferte das Sulfoxid nach 5 h Reaktionszeit in THF unter der langsamen Zugabe von 1.2 Äquivalenten H2O2 in 65% Ausbeute und einem ee-Wert von 36% (Eintrag 2). Das beste Ergebnis wurde mit Ligand 60 erhalten, in dem die Tritylgruppe in para-Position zur Hydroxyfunktion im Salicylaldehyd positioniert ist. Hier konnte das gewünschte Produkt in moderater Ausbeute und Enantioselektivität isoliert werden (66% Ausbeute, 53% ee, Eintrag 3). Die Zugabe von weiteren 0.5 Äquivalenten H2O2 erbrachte keine erheblichen Verbesserungen hinsichtlich Ausbeute und Enantioselektivität (69% Ausbeute, 54% ee, Eintrag 4). 72 Kapitel 3 Ergebnisse und Diskussion 3.1.2.3.2.6 Anwendung des isolierten Eisenkomplexes auf die Oxidation von Thioanisol Weiterhin sollte untersucht werden, welchen Einfluss die isolierten Eisen(III)komplexe auf die Modellreaktion im Vergleich zu den in situ generierten Komplexen besitzen. Dazu wurde eine kleine exemplarische katalytische Studie anhand des isolierten Eisen(III)komplexes 65 durchgeführt (Tabelle 3.1-17). Tabelle 3.1-17: Oxidation von Thioanisol mit dem isolierten Eisenkomplex 65. Eintraga Zugabe von X [mol%] des Komplexes 65 (Reaktionszeit) Ausbeute [%]b ee [%]c 1 2.0 (zu Beginn) 49 42 2 2.0 (zu Beginn) + 2.0 (2 h) 54 48 3 2.0 (zu Beginn) + 2.0 (2 h) + 2.0 (4 h) 54 50 4 6.0 (zu Beginn) 41 48 5 2.0 (zu Beginn) + 1.0 (1 h) + 1.0 (2 h) + 1.0 (3 h) + 1.0 (4 h) 60 53 a Gesamte Reaktionszeit beträgt 5 h; b Isolierte Ausbeute; c ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD). Zunächst wurde die Aktivität des Eisenkomplexes 65 im Vergleich zu dem in situ generierten in der Oxidation von Thioanisol überprüft (Eintrag 1, Tabelle 3.1-17 vs. Eintrag 1, Tabelle 3.1-16). Es wurden vergleichbare Resultate für das Oxidationsprodukt hinsichtlich Ausbeute und Enantioselektivität erhalten (49% Ausbeute, 42% ee vs. 52% Ausbeute, 43% ee). Daher wurde in 73 Kapitel 3 Ergebnisse und Diskussion weiteren Experimenten die kontinuierliche Erhöhung der Katalysatormenge in bestimmten Zeitabständen und deren Auswirkung auf Ausbeute und Enantioselektivität untersucht (Einträge 25). Es wurde deutlich, dass die stete Zugabe einer kleinen Menge an Komplex 65 über einen gewissen Zeitraum sowohl die Ausbeute (60%), als auch den ee-Wert des Sulfoxids (53% ee) steigern kann (Eintrag 5). Hingegen lieferte die Zugabe der gleichen Menge an Komplex 65 (6 mol%) in einer Portion bereits zu Beginn der Katalyse das Produkt in deutlich geringeren Ausbeuten (41%) sowie einem niedrigeren ee-Wert von 48% (Eintrag 4 vs. Eintrag 5). Diese Untersuchung weist auf eine geringe Wechselzahl (engl. Turnover Number, TON) des Katalysators hin, welche ein Maß für die Effizienz eines Katalysators darstellt. [75] Als finales Resultat kann somit auf eine teilweise Zersetzung bzw. Inaktivierung des Komplexes 65 während der Reaktion geschlossen werden. 3.1.2.3.2.7 Einsatz bifunktioneller Organokatalysatoren als Co-Katalysatoren in der eisenkatalysierten Oxidation von Thioanisol: N-Formyl-L-Prolin 34 und BINOL-Phosphat 69 Ein Ansatz, die Ergebnisse der eisenkatalysierten Oxidation von Thioanisol mit dem primäraminhaltigen Liganden 60 zu verbessern, war die Verbindung N-Formyl-L-Prolin 34 sowie das BINOL-Phosphat 69 als potentielle Co-Katalysatoren zu prüfen (Tabelle 3.1-18). Den Grund dieses Vorhabens lieferte der erfolgreiche Einsatz der bifunktionellen Brønsted-Säure/Lewis-BaseKatalysatoren in der organokatalysierten Oxidation von Thioanisol mittels H 2O2, welcher ausführlich in Kapitel 3.2 beschrieben wird. 74 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-18: Untersuchung der Co-Katalysatoren 34 und 69 in der Oxidation von Thioanisol. a Eintrag Co-Kat. LM Temp. [°C] Zeit [h] Ausbeute [%]a ee [%]b 1c,d 34 (1 mol%) THF RT 5 60 52 2c,d 69 (1 mol%) THF RT 5 15 32 3e 34 (1 mol%) DCM RT 24 48 37 4e 69 (1 mol%) DCM -10 24 22 25 Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c c(Substrat) = 0.8 mol/L; Zutropfen von H2O2 per Spritzenpumpe (10 µL/h); c(Substrat) = 0.48 mol/L. d e Der Zusatz von 1 mol% N-Formyl-L-Prolin (34) als Co-Katalysator zeigte keinen positiven Einfluss auf den Reaktionsausgang. Die Ausbeute des Produkts war im Vergleich zum Experiment ohne Verwendung eines Co-Katalysators geringer (66% vs. 60%), während der ee-Wert nahezu konstant blieb (53% vs. 52% ee) (siehe Eintrag 3, Tabelle 3.1-16 vs. Eintrag 1, Tabelle 3.1-18). Als nächstes wurde das zweifach phenylsubstituierte BINOL-Phosphat 69 untersucht. Es konnte eine signifikante Verringerung der Ausbeute des Produkts von 66% auf 15% sowie eine Verringerung der Enantioselektivität von 53% auf 32% ee beobachtet werden (Eintrag 3, Tabelle 3.1-16 vs. 75 Kapitel 3 Ergebnisse und Diskussion Eintrag 2, Tabelle 3.1-18). Basierend auf der Tatsache, dass N-Formyl-L-Prolin (34) die Oxidation von Thioanisol mit H2O2 in DCM begünstigt (siehe Kapitel 3.2.1), wurde dieses als Co-Katalysator auch in diesem Lösungsmittel (anstatt THF) untersucht. Nach 24 h Reaktionszeit wurde Methylphenylsulfoxid in moderater Ausbeute (48%) und geringer Enantioselektivität (37% ee) isoliert (Eintrag 3). Auch die Verwendung des Co-Katalysators 69 in DCM trug zu keiner weiteren Verbesserung des katalytischen Systems bei (Eintrag 4). Während das Produkt in der eisenkatalysierten Oxidation unter der Verwendung des primär-aminhaltigen Liganden 60 in Form des (R)-Enantiomers isoliert wurde (siehe Eintrag 3, Tabelle 3.1-16), wurde Methylphenylsulfoxid in der organokatalysierten Oxidation in Gegenwart des BINOL-Phosphats 69 mit entgegengesetzter (S)-Konfiguration erhalten (siehe Kapitel 3.2.2, Tabelle 3.2-3, Eintrag 5). Aufgrund der unterschiedlichen asymmetrischen Induktion der einzelnen Katalysatoren wirken diese in Kombination einander entgegen. Dieser Sachverhalt könnte den negativen Einfluss des CoKatalysators auf die eisenkatalysierte Sulfidoxidation erklären (Mismatch-Effekt). 3.1.2.3.2.8 Screening zusätzlicher polydentater Liganden auf potentielle Aktivität in der eisenkatalysierten Oxidation von Thioanisol Unter den optimierten Reaktionsbedingungen, wie sie für Ligand 53 in der Oxidation von Thioanisol gefunden wurden, sollte eine Reihe leicht verfügbarer Liganden mit variierendem Bauprinzip auf Effizienz in der Sulfidoxidation getestet werden. Hierdurch wurde versucht, neue Ligandensysteme für diese Reaktion zu erschließen. Die Ergebnisse sind in Tabelle 3.1-19 zusammengefasst. 76 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-19: Screening weiterer polydentater Liganden. Eintrag Ligand Ausbeute [%]a ee [%]b 1 59 7 rac 2c 59 49 rac 3 Boc-L-His-OH 66 rac 4 H-L-His-OH 20 rac 5d 53/70 60 23 (R) 6 71 71 rac 7 57 58 7 (S) 8e 68 51 rac 9f 68 69 35 (R) a Isolierte Ausbeute; durchgeführt in b H2O; ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); d Hybridverbindung 70 wurde als Co-Ligand eingesetzt: c Reaktion Ligand 53/70 (2 mol%:2 mol%); e 5 h Reaktionszeit, Zutropfen von H2O2 per Spritzenpumpe (10 µL/h) über einen Zeitraum von 5 h; f Reaktion in H2O, 3 h (Angaben analog Lit.[41]). 77 Kapitel 3 Ergebnisse und Diskussion Die Testreihe begann mit Versuchen unter Einsatz des neuen primär-aminhaltigen Liganden 59. Dieser Ligand enthält das bereits bekannte phenylsubstituierte Diamin als chiralen Baustein, an welchen eine Histidinfunktionalität gebunden ist. Durch die Schaffung zusätzlicher andersgestaltiger Koordinationsstellen wurde eine erhöhte Effizienz erhofft. Diese Vermutung konnte experimentell nicht bestätigt werden, Methylphenylsulfoxid wurde mit 7% Ausbeute in racemischer Form isoliert (Eintrag 1). Aufgrund der mäßig guten Löslichkeit des Liganden 59 in THF wurde die Oxidation auch in H2O durchgeführt. Die Oxidation führte nach 24 h zum vollständigen Umsatz von Thioanisol (Reaktionsverfolgung per DC), jedoch konnte das Sulfoxid nur in moderater Ausbeute (49%) und als racemische Verbindung isoliert werden, da eine deutlich sichtbare Überoxidation zum Sulfon stattfand (Eintrag 2). Die einzelnen Komponenten des Liganden sollten der Vollständigkeit halber auch auf ihre Effizienz geprüft werden. Unter Einsatz von Boc-L-His-OH konnte Methylphenylsulfoxid in 66% Ausbeute als Racemat isoliert werden (Eintrag 3). H-L-His-OH produzierte ebenfalls ein racemisches Produkt in einer Ausbeute von lediglich 20% (Eintrag 4). Auf weitere Versuche mit Ligand 59 wurde aufgrund der unzufriedenstellenden Ergebnisse verzichtet. Da Ligand 53 bereits erfolgreich in der Oxidation von Thioanisol eingesetzt wurde, sollte mit der Hybridverbindung 70 ein weiterer potentieller CoKatalysator getestet werden. Wie in Kapitel 3.1.3.2 beschrieben, katalysiert 70 die Oxidation von Thioanisol mit einer guten Ausbeute von 76%, allerdings in racemischer Form. Durch die Kombination der beiden Liganden wurde eine synergistische Zunahme von Ausbeute und ee-Wert erwartet. Ligand 53 und Ligand 70 wurden in einem Verhältnis von 1:1 mit jeweils 2 mol% eingesetzt. Die Ausbeute des Sulfoxids konnte auf 60% erhöht werden, jedoch sank dabei gleichzeitig der ee-Wert auf 23% (Eintrag 5 vs. Eintrag 1, Tabelle 3.1-16). Eine weitere Modifikation des Grundgerüsts von Ligand 53 wurde mit dem pentadentaten Liganden 71 verwirklicht, in welchem statt einer freien Amino- eine iminverbrückte hydroxysubstituierte Chinolineinheit vorliegt. Die neue Funktionalität stellt weitere potentielle Koordinationsmöglichkeiten des Liganden ans Metall bereit. In der Katalyse lieferte allerdings auch 71 racemisches Methylphenylsulfoxid in einer 78 Kapitel 3 Ergebnisse und Diskussion Ausbeute von 71% (Eintrag 6). In Anlehnung an Arbeiten von Katsuki[41] wurde Komplex 68 synthetisiert, in dem zwei Moleküle des Liganden 58, der die reduzierte Form des Imin-Liganden 57 darstellt, an das zentrale Eisenatom koordinieren. Während in THF das Oxidationsprodukt trotz langsamer Zugabe von H2O2 per Spritzenpumpe nach 5 h Reaktionszeit nur in moderaten Ausbeuten (51%, Eintrag 8) als Racemat isoliert werden konnte, konnte in H2O nach bereits 3 h durch die Zugabe von H2O2 in einer Portion ein ee-Wert von 35% beobachtet und eine Produktausbeute von 69% erhalten werden (Eintrag 9). Die Methode von Katsuki konnte jedoch mit dem neuen primär-aminhaltigen Eisenkomplex 68 nicht verbessert werden (Lit.: 89% Ausbeute, 88% ee)[41]. Im Zuge der Herstellung des Komplexes 68 wurde auch der genannte Ligand 57 in der Oxidation eingesetzt. Dieser besitzt die entgegengesetzte Konfiguration des bereits diskutierten Liganden 56. Wie zu erwarten ist, lieferte 57 Methylphenylsulfoxid in (S)- statt in (R)-Konfiguration (7% ee) mit vergleichbarer Ausbeute (Eintrag 7, 58% vs. 54% mit 56). Es bleibt festzuhalten, dass keine der beschriebenen Modifikationen die Effizienz des Liganden 53 unter optimierten Bedingungen zu überteffen vermochte. 3.1.3 Liganden basierend auf Hybridverbindungen (peptidhaltige Schiffsche-Basen) Im Rahmen dieser Arbeit sollten ferner neue chirale Hybridverbindungen, welche sich aus einer Aldehyd- und Peptid- (bzw. Aminosäure-) Einheit zusammensetzen, entwickelt und als Liganden in der metallkatalysierten Sulfidoxidation eingesetzt werden. Über die Anwendung solcher peptidhaltiger Schiffscher-Basen in der metallkatalysierten Sulfidoxidation wurde erstmals von Jackson et al. berichtet.[73d] Basierend auf dieser und früheren Arbeiten von Hoveyda und Snapper,[73b, 93] welche diese Liganden erfolgreich in verschiedenen C-C- Bindungsknüpfungsreaktionen eingesetzt haben, wurden nun weitere Modifikationen der Liganden durchgeführt und eine kleine Bibliothek neuer Hybridverbindungen erstellt. Ein Überblick der synthetisierten Liganden ist in Abbildung 3-10 dargestellt. 79 Kapitel 3 Ergebnisse und Diskussion Abbildung 3-10: Hybridverbindungen im Überblick. 3.1.3.1 3.1.3.1.1 Zur Synthese der Liganden Eduktsynthesen Herstellung der Hybridverbindungen Peptidbausteine, dienten, wurden welche gängige Schutzgruppenchemie angewendet.[94] 80 als Ausgangsstoffe Methoden der zur klassischen Synthese der Peptid- und Kapitel 3 Ergebnisse und Diskussion Das Tripeptid H-L-Val-L-Thr(Bn)-Gly-OMe (87) wurde ausgehend von den kommerziell erhältlichen Aminosäurebausteinen (Boc-L-Thr(Bn)-OH (79), H-Gly-OMe Hydrochlorid (80) und H-L-Val-OH (83)) innerhalb einer 6-stufigen Synthese in Anlehnung an bekannte Literaturmethoden aufgebaut (siehe Schema 3.1-10). Schema 3.1-10: Syntheseroute zur Darstellung von Tripeptid 87. Die Synthese begann mit einer einer CDI-vermittelten Kupplungsreaktion[79] von 79 und 80, gefolgt von der Entschützung des N-Terminus von 81 mittels TFA.[95] Das so erhaltene Dipeptid 82 wurde in einer erneuten CDI-Kupplungsreaktion[79] mit Boc-L-Val-OH (84), welches durch die Reaktion von H-L-Val-OH (83) mit Boc2O hergestellt wurde,[96] umgesetzt. In einer darauffolgenden Entschützung des N-Terminus von Boc-L-Val-L-Thr(Bn)-Gly-OMe (85) durch TFA und der 81 Kapitel 3 Ergebnisse und Diskussion Freisetzung der Aminofunktion mittels 20%-iger K2CO3-Lösung konnte die Zielverbindung 87 in einer Gesamtausbeute von 34% isoliert werden.[95, 97] Zur Lagerung wurde das Tripeptid 87 in Form seines TFA-Salzes 86 aufbewahrt. Um die Synthesebausteine 88 und 89 zu erhalten wurde zunächst der C-Terminus der jeweiligen Aminosäuren, L-Threonin bzw. L-Serin, mit Thionylchlorid in MeOH verestert.[98] Die Regeneration der freien Aminofunktion, welche nach der Veresterung als Hydrochloridsalz vorlag, wurde im Falle von H-L-Thr-OMe (88) durch den Umsatz mit einer 20%-igen K2CO3-Lösung (pH 10) und anschließender Extraktion erreicht.[97] Diese Methode war auf H-L-Ser-OMe (89) nicht übertragbar, da sich das Produkt aus der wässrigen Phase nicht mehr extrahieren lies. Deshalb wurde auf einen basischen Ionentauscher (Amberlyst(OH)-26) zurückgegriffen, welcher nach Einstellung der Lösung auf einen pH-Wert von 9-10 abfiltriert werden konnte. H-L-Ser-OMe (89) wurde nach anschließender säulenchromatographischer Reinigung in 58% Ausbeute erhalten (siehe Abbildung 3-11). Abbildung 3-11: Synthetisierte Aminosäurebausteine. Die Synthese der beiden Dipeptide 96 und 97 (Schema 3.1-11), welche sich in einem stereogenen Zentrum unterscheiden, erfolgte über zwei Stufen. Zuerst wurden die Aminogruppen von L-Threonin (90) bzw. L-allo-Threonin (91) mit Benzylchlorformiat geschützt (92 und 93)[99] und in einer anschließenden Kupplungsreaktion mit H-D-Ph-OMe[78a] (95) mittels i-Butylchlorformiat in die Dipeptide 96 und 97 überführt.[100] 82 Kapitel 3 Ergebnisse und Diskussion Schema 3.1-11: Syntheseroute zur Darstellung der Dipeptide 96 und 97. 3.1.3.1.2 Synthese neuer Hybridverbindungen Die Synthesen der verschiedenen Hybridverbindungen 70, 72-78, welche in Abbildung 3-10 dargestellt sind, werden im folgenden Abschnitt beschrieben. Es wurden sowohl tri- als auch dipeptid- und mono-aminosäurehaltige Schiffsche-Basen hergestellt. Die Darstellung der tripeptidhaltigen Schiffschen-Basen 72, 73 und 74 wurde, wie im allgemeinen Schema 3.1-12 beschrieben, erfolgreich durchgeführt. Schema 3.1-12: Allgemeine Syntheseroute zur Darstellung der tripeptidhaltigen Schiffschen-Basen 72, 73 und 74. 83 Kapitel 3 Ergebnisse und Diskussion H-L-Val-L-Thr(Bn)-Gly-OMe (87) wurde durch die Umsetzung des TFA-Salzes 86 mit 20%-iger K2CO3-Lösung und anschließender Extraktion aus der wässrigen Phase für jeden Reaktionsansatz frisch isoliert. Das freie Amin 87 wurde nach Trocknen im Hochvakuum ohne weitere Reinigung in MeOH (abs.) aufgenommen und mit Na2SO4 sowie dem jeweiligen Aldehyd portionsweise versetzt. Nach entsprechender Aufarbeitung wurden die tripeptidhaltigen Schiffschen-Basen 72, 73 und 74 in guten bis exzellenten Ausbeuten erhalten (siehe Abbildung 3-12). Abbildung 3-12: Synthetisierte tripeptidhaltige Schiffsche-Basen 72, 73 und 74. Die Zielverbindungen 75 und 76 wurden in einer Kondensationsreaktion aus den jeweiligen Aminosäurebausteinen (H-L-Thr-OMe (88) oder H-L-Ser-OMe (89)) und (R)-2-Hydroxy-2'-phenyl[1,1'-binaphthalen]-3-carbaldehyd[101] in exzellenten Ausbeuten erhalten (siehe Abbildung 3-13). 84 Kapitel 3 Ergebnisse und Diskussion Abbildung 3-13: Synthetisierte monoaminosäurehaltige Schiffsche-Basen 75 und 76. Die dipeptidhaltigen Schiffschen-Basen 77, 70 und 78 (Abbildung 3-10) wurden in Anlehnung an ein literaturbekanntes Syntheseverfahren[100] hergestellt (siehe Schema 3.1-13). Schema 3.1-13: Allgemeine Syntheseroute zur Darstellung von dipeptidhaltigen Schiffschen-Basen nach dem Verfahren von Inoue.[100] Die Cbz-geschützten Dipeptide 96 und 97 wurden in absolutem MeOH gelöst und anschließend die freie Aminofunktion durch katalytische Hydrierung an Pd/C mittels H2-Gas in situ regeneriert. Anschließend wurden die freien Amine mit einer Lösung von (R)- oder (S)-2-Hydroxy-2'-phenyl[1,1'-binaphthalen]-3-carbaldehyd[101] in absolutem DCM versetzt und die Zielverbindungen 70, 77 und 78 nach der Reinigung mittels Flash-Säulenchromatographie in sehr guten Ausbeuten erhalten (siehe Abbildung 3-14). 85 Kapitel 3 Ergebnisse und Diskussion Abbildung 3-14: Synthetisierte dipeptidhaltige Schiffsche-Basen 70, 77 und 78. 3.1.3.2 Anwendung peptidhaltiger Schiffsche-Base Liganden in der Katalyse In Anlehnung an die Arbeit von Jackson[73d] sollten die modifizierten Liganden bezüglich ihrer Effizienz in der enantioselektiven Oxidation von Thioanisol überprüft werden. Das Screening der verschiedenen Liganden und Metallquellen erfolgte in DCM als Lösungsmittel. Der Verlauf der Reaktion wurde mittels Dünnschichtchromatographie verfolgt. Die erste katalytische Studie wurde ausgehend vom Tripeptid-Liganden 72 in Kombination mit verschiedenen Metallquellen (VO(acac)2, Ti(Oi-Pr)4, Fe(acac)3, FeCl3 x 6 H2O, Mn(acac)3 oder Mn(OAc)2 x 4 H2O) durchgeführt (Einträge 1-5, Tabelle 3.1-20). Dabei war die Prüfung der Eisen- und Mangansalze als potentiell redox-aktive Metallzentren von besonderem Interesse, da diese im Vergleich zu VO(acac)2 oder Ti(Oi-Pr)4 in Verbindung mit peptidhaltigen Iminen für diese Reaktion, soweit bekannt, noch nicht getestet wurden. Weiterhin wurden 1.2 Äquivalente H2O2 zugegeben und mit einer geringen Katalysatorbeladung von <5 mol% gearbeitet. Die Ergebnisse sind in Tabelle 3.1-20 zusammengefasst. 86 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-20: Screening von tripeptidhaltigen Schiffschen-Basen 72, 73, 74 und verschiedenen Metallquellen. Eintrag Ligand MXn Zeit Ausbeute [%]a ee [%]b 1 72 (3 mol%) FeCl3 x 6 H2O (2 mol%) 4d 2 n.b. 2 72 (5 mol%) Mn(OAc)2 x 4 H2O (5 mol%) 4d Spuren - 3 72 (5 mol%) Mn(acac)3 (5 mol%) 3d Spuren - 4 72 (5 mol%) VO(acac)2 (5 mol%) 3d 71 2 (S) 5c 72 (5 mol%) Ti(Oi-Pr)4 (5 mol%) 2d 52 2 (S) 6 73 (5 mol%) Fe(acac)3 (5 mol%) 3d 22 3 ( R) 7 74 (5 mol%) Fe(acac)3 (5 mol%) 24 h 17 rac 8 74 (5 mol%) FeCl3 x 6 H2O (5 mol%) 24 h 2 n.b. 9 74 (5 mol%) Mn(acac)3 (5 mol%) 24 h Spuren - 10 74 (5 mol%) VO(acac)2 (5 mol%) 24 h 57 2 (S) 11c 74 (5 mol%) Ti(Oi-Pr)4 (5 mol%) 24 h 45 32 (R) a Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c Durchführung unter Stickstoffatmosphäre. Die ersten Experimente lieferten unter der Verwendung von FeCl3 x 6 H2O, Mn(OAc)2 x 4 H2O oder Mn(acac)3 in Kombination mit Ligand 72 (Ligand/Metall, 1.5:1 bzw. 1:1) auch nach mehreren Tagen nahezu keinen Umsatz von Thioanisol (Einträge 1-3). Erst durch den Einsatz von VO(acac)2 konnte Methylphenylsulfoxid mit einer Ausbeute von 71% nach 3 Tagen bzw. im Falle von 87 Kapitel 3 Ergebnisse und Diskussion Ti(Oi-Pr)4 mit einer Ausbeute von 52% nach 2 Tagen Reaktionszeit isoliert werden. In beiden Fällen wurde ein nahezu racemisches Produkt erhalten (Einträge 4 und 5). Als nächstes wurde Ligand 73, der sich strukturell nur in den Substituenten an der Salicyleinheit vom Liganden 72 unterscheidet, in Kombination mit Fe(acac)3 getestet. Nach einer Reaktionszeit von 3 Tagen wurde das Sulfoxid mit einer geringen Ausbeute von 22% isoliert, wobei eine minimale chirale Induktion von 3% ee zu beobachten war (Eintrag 6). Die achirale Salicyleinheit wurde durch eine chirale, sterisch-anspruchsvollere Binaphthyl-Einheit ersetzt (Schaffung einer neuen chiralen Umgebung) und der resultierende Ligand 74 unter gleichen Bedingungen auf seine Effizienz hin überprüft (Einträge 7-11). Es musste festgestellt werden, dass ausgehend von Ligand 74 in Kombination mit den Metallsalzen Fe(acac)3, FeCl3 x 6 H2O oder Mn(acac)3 kaum bzw. keine katalytische Aktivität zu beobachten war (Einträge 7-9). Im Fall von Fe(acac)3 konnte das Sulfoxid nach 24 h mit 17% Ausbeute isoliert werden, unter Verwendung von VO(acac)2 mit einer moderaten Ausbeute von 57% (Eintrag 10). Beide Produkte lagen in nahezu racemischer Form vor. Erst Ti(Oi-Pr)4 bewirkte in Verbindung mit dem tripeptidhaltigen Liganden 74 eine asymmetrische Induktion am Substrat. Hier konnte Methylphenylsulfoxid mit einer moderaten Ausbeute von 45% und einem geringen Enantiomerenüberschuss von 32% ee isoliert werden (Eintrag 11). Aufgrund der Tatsache, dass nur in Verbindung mit Ti(Oi-Pr)4 und Ligand 74 eine asymmetrische Induktion am Substrat zu beobachten war, entstand die Überlegung, die sterisch-anspruchsvollere Binaphthyl-Einheit von Ligand 74 (Tabelle 3.1-20) mit den von Jackson[73d] getesteten Aminosäuren bzw. Peptiden zu vereinen, mit der Zuversicht ein neues, hoch-effizientes katalytisches System zu schaffen. Die Ergebnisse der Katalysen mit den neu synthetisierten aminosäure- bzw. peptidhaltigen Liganden 70, 75-78 sind in Tabelle 3.1-21 zusammengefasst. 88 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.1-21: Screening von weiteren aminosäure- und peptidhaltigen Iminen mit einer Binaphthyl-Einheit. Eintrag Ligand Ausbeute [%]a ee [%]b 1c 75 62 rac 2c 76 62 8 3c 78 63 10 4c 77 68 21 5c 70 76 rac a Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c Durchführung unter Stickstoffatmosphäre. 89 Kapitel 3 Ergebnisse und Diskussion Ligand 75 lieferte in Verbindung mit Ti(Oi-Pr)4 bei einer Katalysatorbeladung von 5 mol% (Ligand/Metall (1:1)), in DCM nach 24 h das entsprechende Sulfoxid in 62% Ausbeute in racemischer Form (Eintrag 1). Der Austausch der Aminosäure L-Threonin gegen L-Serin in Ligand 76 führte zu keiner Verbesserung der Ausbeute, jedoch konnte ein geringer Enantiomerenüberschuss von 8% ee beobachtet werden (Eintrag 2). Auch die Erprobung des Liganden 78, bestehend aus einer Binaphthyl-Einheit und dem Dipeptid L-Thr-D-Phe-OMe, führte zu keiner deutlichen Verbesserung des katalytischen Systems (63% Ausbeute, 10% ee, Eintrag 3). Erst durch die Verwendung des gegensätzlichen Enantiomers der Binaphthyl-Einheit (S statt R) in Ligand 77 konnte der Enantiomerenüberschuss des Sulfoxids um 10% erhöht werden (68% Ausbeute, 21% ee, Eintrag 4). Die Verwendung des Liganden 70, in dem das Dipeptid H-L-Thr-DPh-OMe gegen das von Jackson erfolgreich eingesetzte Dipeptid H-allo-Thr-D-Ph-OMe ausgetauscht wurde, führte zu einem unzufriedenstellenden Ergebnis. Die Ausbeute konnte zwar auf 76% gesteigert werden, das Produkt war jedoch racemisch (Eintrag 5). Zum Vergleich ist der in Abbildung 3-15 von Jackson et al. entwickelte peptidhaltige Schiffsche-Base Ligand, welcher in der titankatalysierten Sulfidoxidation mit H2O2 eingesetzt wurde, dargestellt.[73d] Abbildung 3-15: Peptidhaltige Schiffsche-Base Liganden A und B entwickelt von Jackson.[73d] 90 Kapitel 3 3.2 Ergebnisse und Diskussion Organokatalysierte Oxidation von Thioanisol Während die Entwicklungen auf dem Gebiet der metallkatalysierten Sulfidoxidation in Verbindung mit verschiedenen Oxidationsmitteln auch in jüngster Zeit weiter vorangetrieben wurden,[1-2, 8, 73a] ist der Entwicklung von organokatalytischen Methoden für diese Reaktion vergleichsweise wenig Aufmerksamkeit geschenkt worden (siehe Kapitel 1.2.2). Aus diesem Grunde besteht bis dato weiterhin der Bedarf nach neuen Methoden zur enantioselektiven Oxidation von Sulfiden, auch unter milden und umweltfreundlichen Bedingungen, zu forschen. 3.2.1 N-Formyl-L-Prolin als Organokatalysator Das aus dem „chiral pool“ leicht zugängliche N-Formyl-L-Prolin (34) wurde bereits von Tsogoeva et al. in verschiedenen organokatalytischen Reaktionen (Strecker-Reaktion von Aldiminen und Hydrosilylierung von Ketiminen[81], sowie Aminolyse von Epoxiden[92]) eingesetzt. Dementsprechend sollte N-Formyl-L-Prolin (34) auch als potentieller Organokatalysator in der Oxidation von Sulfiden untersucht werden. Als Modellsubstrat diente hierfür Thioanisol, welches in Anwesenheit verschiedener Lewis-basischer Verbindungen (98, 34, 99 und 100) unter milden Bedingungen und unter der Verwendung von H2O2 als umweltfreundlichem Oxidationsmittel oxidiert werden sollte. Das Screening wurde in DCM unter Zusatz von 20 mol% des jeweiligen Organokatalysators durchgeführt. Die Ergebnisse sind in Tabelle 3.2-1 zusammengefasst. 91 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.2-1: Screening verschiedener Organokatalysatoren. Eintrag Katalysator Umsatz [%]a Verhältnis von 25/26a 1 98 15 100:0 2 34 65 100:0 3 99 7 n.b. 4 100 15 100:0 5 - 9 n.b. a Umsatz und Verhältnis (Sulfoxid 25/Sulfon 26) wurden mittels GC bestimmt. Während Methylphenylsulfoxid (25) in der Oxidation von Thioanisol mit den kommerziell erhältlichen Verbindungen Pyridin-N-Oxid (98), L-Prolin (99) und N-Formyl-L-Pyrrolidin (100) nach 24 h nur in geringer Menge gebildet wurde (<15%, Einträge 1, 3 und 4), lieferte N-Formyl-L-Prolin (34) als Organokatalysator das Oxidationsprodukt in moderatem Umsatz von 65% (Eintrag 2). Auch die Oxidation ohne Zusatz eines Katalysators (Hintergrundreaktion) zeigte nur einen geringen Umsatz von 9% (Eintrag 5). Mit diesen Experimenten konnte gezeigt werden, dass allein durch das Zusammenspiel der Formamid- und Carboxyl-Einheit wie sie in N-Formyl-L-Prolin (34) vorliegt, die Oxidation von Thioanisol begünstigt wird (bifunktionelle Aktivierung). Ferner wurde keine Überoxidation zum Sulfon beobachtet (Eintrag 2). Basierend auf den Ergebnissen aus Tabelle 3.2-1 wurde im nächsten Schritt N-Formyl-L-Prolin (34) in der Oxidation von Thioanisol in verschiedenen Lösungsmitteln (CHCl3, Hexan, Toluol, MeOH und H2O) getestet (siehe Tabelle 3.2-2). 92 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.2-2: Screening verschiedener Lösungsmittel mit N-Formyl-L-Prolin (34) als Organokatalysator. Eintrag LM Temp. [°C] Zeit [h] 1 CHCl3 RT 24 64 100:0 2 Hexan RT 24 7 n.b. 3 Toluol RT 24 36 94:6 4 MeOH RT 24 64b n.b. 5 H2O RT 24 45b n.b. 6 H2O RT 120 70b n.b. 7 DCM RT 120 82b n.b. 8 CHCl3 RT 120 88b n.b. 9 DCM 40 24 97 98:2 10 CHCl3 40 24 82 100:0 11 H2O 40 24 72b n.b. a Umsatz [%]a Verhältnis von 25/26a Umsatz und Verhältnis (Sulfoxid 25/Sulfon 26) wurden mittels GC bestimmt; b isolierte Ausbeute. Während in CHCl3 ein ähnlicher Umsatz wie in DCM beobachtet werden konnte (64%, Eintrag 1, vs. 65%, Eintrag 2, Tabelle 3.2-1), zeigte N-Formyl-L-Prolin (34) in den Lösungsmitteln Hexan und Toluol eine weitaus geringere Aktivität (7% und 36%, Einträge 2 und 3). Ferner wurde in Toluol eine leichte Überoxidation zum Sulfon (94:6 Sulfoxid 25/Sulfon 26) im Gegensatz zu den chlorierten Lösungsmitteln beobachtet (Eintrag 3 vs. Einträge 1 und 2, Tabelle 3.2-1). In polareren Lösungsmitteln wie MeOH oder H 2O wurde Methylphenylsulfoxid nach 24 h in 64% bzw. 45% Ausbeute erhalten (Einträge 4 und 5). Durch Verlängerung der Reaktionszeit auf 120 h konnten die Ausbeuten in den Lösungsmitteln H2O, DCM und CHCl3 verbessert werden (70%, 82% und 88%, 93 Kapitel 3 Ergebnisse und Diskussion Einträge 6, 7 und 8). Eine Beschleunigung des Oxidationsprozesses wurde in DCM durch eine Erhöhung der Temperatur auf 40 °C erreicht. So konnte Methylphenylsulfoxid nach 24 h in DCM (bei 40 °C) in einem hohen Umsatz (97%) und einer hohen Chemoselektivität (98:2 Sulfoxid/Sulfon) erhalten werden (Eintrag 9). Eine derartige Beschleunigung unter erhöhter Temperatur konnte in den Lösungsmitteln CHCl3 und H2O nicht gezeigt werden (Einträge 10, 11 vs. 6, 8). Während gute bis exzellente Ausbeuten in der Oxidation von Thioanisol unter Verwendung des bifunktionellen Katalysators N-Formyl-L-Prolin (34) erzielt werden konnten, wurde das Produkt lediglich in racemischer Form erhalten. Ein möglicher Ablauf des Oxidationsprozesses wird in Abbildung 3-16 postuliert, welcher auch die fehlende chirale Induktion erklären könnte. Abbildung 3-16: Postulierter Reaktionsmechanismus für die N-Formyl-L-Prolin katalysierte Oxidation von Thioanisol mittels H2O2. Das Oxidationsmittel H2O2 kann durch die Ausbildung zweier Wasserstoffbrückenbindungen mit dem bifunktionellen Organokatalysator N-Formyl-L-Prolin (34) in Wechselwirkung treten und aktiviert werden. Aufgrund der möglichen fehlenden Katalysator-Substratwechselwirkung und einer zu geringen sterischen Hinderung verursacht durch N-Formyl-L-Prolin (34), zeigt diese Reaktion einen racemischen Verlauf. 94 Kapitel 3 3.2.2 Ergebnisse und Diskussion BINOL-Phosphate als Organokatalysatoren Basierend auf dem in Kapitel 3.2.1 neu entwickelten Konzept der Lewis-Base/Brønsted-Säure Katalyse in der Oxidation von Thioanisol mittels H2O2 sollten neben N-Formyl-L-Prolin (34) zusätzlich verschiedene BINOL-Phosphate als potentielle bifunktionelle Organokatalysatoren untersucht werden. BINOL-Phosphate wurden in der Vergangenheit bereits, auch von Tsogoeva et al.,[102] erfolgreich in verschiedenen Reaktionen als asymmetrische Organokatalysatoren eingesetzt.[103] Aufgrund der stattfindenden Aktivierung von H2O2 mittels N-Formyl-L-Prolin (34) wurde das Konzept auf ein starreres System mit axial chiralem Rückgrat (Binaphthyl-Gerüst) erweitert. Die Übertragung des konzeptionellen Ansatzes der N-Formyl-L-Prolin (34) vermittelten Sulfidoxidation mittels H2O2 auf die Verwendung von BINOL-Phosphaten ist in Abbildung 3-17 gezeigt. Abbildung 3-17: Lewis-Base/Brønsted-Säure Katalyse: BINOL-Phosphate als bifunktionelle Organokatalysatoren in der Oxidation von Thioanisol unter Verwendung von H2O2 als Oxidationsmittel. Die Ergebnisse des Screenings der verschiedenen BINOL-Phosphate 101-105[104] sind in Tabelle 3.2-3 zusammengefasst. Dabei wurden die Reaktionsbedingungen in Anlehnung an die zuvor entwickelte organokatalytische Oxidation von Thioanisol mittels N-Formyl-L-Prolin (34) gewählt. 95 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.2-3: Screening von verschiedenen BINOL-Phosphaten. a Eintrag Organokatalysator Ausbeute [%]a ee [%]b 1 101 (20 mol%) 44 9 (S) 2 101 (10 mol%) 26 10 (S) 3c 101 (20 mol%) Spuren n.b 4 102 (20 mol%) 98 20 (S) 5 103 (20 mol%) 68 36 (S) 6 104 (20 mol%) 73 30 (S) 7 105 (20 mol%) Spuren n.b. 8 - Spuren n.b. Isolierte Ausbeute; b ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (OD); c Reaktion bei -20 °C durchgeführt. Unter Verwendung von BINOL-Phosphat 101 wurde Methylphenylsulfoxid in einer moderaten Ausbeute von 44% und einem geringen Enantiomerenüberschuss von 9% ee isoliert (Eintrag 1). Eine Reduktion der Katalysatorbeladung von 20 mol% auf 10 mol% führte zu einer geringeren Ausbeute von 26%, wobei der ee-Wert konstant blieb (10% ee, Eintrag 2). Bei der Durchführung der Reaktion bei einer niedrigeren Temperatur (-20 °C) wurde nahezu kein Umsatz beobachtet (Eintrag 3). BINOL-Phosphat 102, welches statt einem SiPh3 einen 2,4-(NO2)2-C6H4-Rest enthält, lieferte das Oxidationsprodukt in exzellenten Ausbeuten (98%) mit einem Enantiomerenüberschuss von 20% (Eintrag 4). Darauf aufbauend wurden die BINOL-Phosphate 103, 104, 105 auf ihre Effizienz überprüft. Im Falle von 103 konnte der ee-Wert des Produkts auf 36% gesteigert werden, 96 Kapitel 3 Ergebnisse und Diskussion wobei sich die Ausbeute auf 68% verringerte (Eintrag 5). Während mit dem Einsatz von BINOLPhosphat 104 ein ähnliches Resultat erhalten wurde (73% Ausbeute, 30% ee, Eintrag 6), führte die Einführung des sterisch anspruchsvollen Anthracen-9-yl-Rests in 105 zu einem vollständigen Verlust der Aktivität (Eintrag 7). Anzumerken ist, dass während der Entwicklung dieser Arbeit zwei neue Publikationen von Tao und Wang et al.[55a] und List et al.[55b] auf diesem Gebiet erschienen sind, in welchen zum ersten Mal über den erfolgreichen Einsatz von BINOL-Phosphaten als chirale Organokatalysatoren in der enantioselektiven Sulfidoxidation berichtet wird (siehe Kapitel 1.2.2.1). Somit wurde der Grundstein zur Entwicklung hocheffizienter binaphthylhaltiger bifunktioneller Organokatalysatoren in der metallfreien enantioselektiven Sulfidoxidation gelegt. 97 Kapitel 3 3.3 Ergebnisse und Diskussion Anwendung ausgewählter metall- und organokatalytischer Systeme auf die Synthese von biologisch aktiven Verbindungen: (S)-Omeprazol und (R)-Modafinil Wie bereits in den Kapiteln 1.1 und 1.3 beschrieben, sind chirale Sulfoxide in zahlreichen Arzneimitteln enthalten. Zu den bekanntesten Beispielen sulfoxidhaltiger Pharmazeutika gehören Esomeprazol und Armodafinil. Ausgewählte Liganden-Systeme sollten auf die Synthese der genannten Wirkstoffe angewendet werden. Zuvor mussten die zur Oxidation benötigten prochiralen Sulfide, wie auch die racemischen Referenzverbindungen synthetisiert werden, was im Folgenden beschrieben wird. 3.3.1 Substratsynthesen: Omeprazol und Modafinil Die beiden Wirkstoffe Omeprazol und Modafinil sowie die zugehörigen prochiralen Sulfide, welche die Ausgangsverbindungen in der enantioselektiven Oxidation darstellen, wurden nach literaturbekannten Syntheseverfahren hergestellt. In Schema 3.3-1 wird die zweistufige Synthese von Omeprazol gezeigt.[105] Schema 3.3-1: Darstellung von Omeprazol (rac-109). 98 Kapitel 3 Ergebnisse und Diskussion Dabei wurde zuerst der Omeprazol-Precursor 108 in einer SN2-Reaktion durch die Verknüpfung der Bausteine 106 und 107 gebildet und dieser anschließend mittels m-CPBA zu Omeprazol (rac-109) oxidiert. Omeprazol (rac-109) wurde mit einer Gesamtausbeute von 54% erhalten. Bei der Synthese von Modafinil (rac-115) (Schema 3.3-2) wurde die Verbindung 112 in einer Kondensationsreaktion ausgehend von Benzhydrol (110) und Mercaptoessigsäure (111) in TFA aufgebaut. Im nächsten Schritt wurde die Carboxylgruppe von 112 mittels SOCl2 aktiviert. Dabei wurde die Reaktion abweichend von der Literatur in Toluol statt in Benzol durchgeführt. Die direkte Umsetzung des Säurechlorids 113 mit einer wässrigen Ammoniaklösung (25%) zum Amid 114 lieferte das Produkt in einer Ausbeute von 33%.[106] In der abschließenden Oxidationsreaktion des prochiralen Sulfides mittels H2O2 in THF wurde Modafinil (rac-115) in 80% Ausbeute erhalten.[107] Die Gesamtausbeute der Modafinilsynthese (rac-115) betrug 26%. Schema 3.3-2: Darstellung von Modafinil (rac-115). Zusätzlich wurden zu den jeweiligen Referenzverbindungen rac-109 und rac-115 die entsprechenden Sulfone 116 und 117 synthetisiert (Abbildung 3-18), da diese eventuell in den Katalysereaktionen in Folge einer Überoxidation als Nebenprodukte entstehen können. 99 Kapitel 3 Ergebnisse und Diskussion Abbildung 3-18: Mögliche gebildete Nebenprodukte 116 und 117. Die Oxidation zum Omeprazol-Sulfon 116 wurde ausgehend von Omeprazol (rac-109) mittels eines weiteren Äquivalents m-CPBA durchgeführt. Das Modafinil-Sulfon 117 hingegen wurde ausgehend vom prochiralen Sulfid 114 durch einen Überschuss an H2O2 (4 Äq.) in Essigsäure erhalten.[108] 3.3.2 Katalysereaktionen Im Anschluss an die Substratsynthesen wurden die in den Kapiteln 3.1 und 3.2 entwickelten bzw. optimierten metall- und organokatalytischen Systeme auf ihre Effizienz in der enantioselektiven Synthese von (S)-Omeprazol und (R)-Modafinil getestet. Die Ergebnisse der Untersuchung der verschiedenen Methoden im Vergleich zur enantioselektiven Oxidation von Thioanisol sind in Tabelle 3.3-1 zusammengefasst. Die zugehörigen chiralen Liganden sind in Abbildung 3-19 dargestellt. Der Einsatz des neu entwickelten primär-aminhaltigen Liganden 53 in Kombination mit FeCl3 x 6 H2O lieferte Omeprazol in THF nach 5 h Reaktionszeit trotz der langsamen Zugabe von H 2O2 in einer geringen Ausbeute von 16% mit einem sehr geringen Enantiomerenüberschuss von 3% ee. Durch den Einsatz des Eisen(III)komplexes 67, welcher im Salicylaldehydbaustein statt zwei t-Butylgruppen eine Tritylgruppe in Position 5 enthält, konnte die Ausbeute bei gleichen Reaktionsbedingungen minimal auf 20% und der ee-Wert auf 19% gesteigert werden. 100 Kapitel 3 Ergebnisse und Diskussion Tabelle 3.3-1: Überblick der Anwendung verschiedener katalytischer Systeme. Metallkatalyse V-Katalysea Omeprazol Modafinil Organokatalyse Fe-Katalyse Ti-Katalyseb BINOL-Phosphate - 16% (3% ee (R))c 84% (rac) Spurend - 20% (19% ee (R))e 92% (33% ee (R)) 57% (rac)e - - - - 96% (rac) 87% (rac)f - quant. (10% ee (R))g PhS(O)Me a 66 (53% ee (R))c 68% (21% ee (S)) 68 (36% ee (S))f VO(acac)2 (2 mol%), Ligand 43 (3 mol%), H2O2 (1.2 Äq.), CHCl3, 24 h; c(Substrat) = 0.8 mol/L; b Ti(Oi-Pr)4 c 79% (45% ee (R)) (5 mol%), 77 (5 mol%), H2O2 (1.2 Äq.), DCM, 24 h; c(Substrat) = 0.4 mol/L; FeCl3 x 6 H2O (2 mol%), Ligand 53 (4 mol%), H2O2 (1.2 Äq.): Spritzenpumpe (10 µL/h), THF, 5 h; c(Substrat) = 0.8 mol/L; d BINOL-Phosphat 101 mit R = SiPh3 (20 mol%), DCM, -10 °C, 24 h; c(Substrat) = 0.48 mol/L; e Eisen(III)komplex 67 (2 mol%), H2O2 (1.2 Äq.): Spritzenpumpe (10 µL/h), THF, 5 h; c(Substrat) = 0.8 mol/L; f BINOL-Phosphat 103 mit R = Ph (20 mol%), DCM, -10 °C, 24 h; c(Substrat) = 0.48 mol/L; g BINOL-Phosphat 104 mit R = C6H5-p-Naphthyl (20 mol%), DCM, -10 °C, 24 h; c(Substrat) = 0.48 mol/L. In beiden Fällen wurde jedoch das (R)-Enantiomer erhalten. Modafinil wurde unter gleichen Reaktionsbedingungen in moderaten Ausbeuten von 57% produziert, wobei aber keine chirale Induktion zu beobachten war. Die Anwendung des peptidhaltigen Schiffsche-Base Liganden 77 in Kombination mit Ti(Oi-Pr)4 als Metallquelle führte zwar zu hohen Ausbeuten von Omeprazol (84%) und Modafinil (96%), jedoch lagen beide Produkte in racemischer Form vor. Ein erfreuliches Ergebnis bezüglich der Synthese von (R)-Modafinil wurde in der vanadiumkatalysierten Oxidation mit dem aus einem Aminoalkohol abgeleiteten Schiffsche-Base Liganden 43 erhalten. Dabei wurde (R)-Modafinil in einer exzellenten Ausbeute von 92% und einem Enantiomerenüberschuss von 101 Kapitel 3 Ergebnisse und Diskussion 33% ee isoliert (Vergleich Lit.: 45%, 12% ee)[68]. Die Anwendung dieses Systems auf die Synthese von optisch aktivem Omeprazol wurde nicht untersucht, da das klassische Bolm-System[19a] bereits in der Literatur aufgrund geringer Ausbeuten und beträchtlichen Nebenreaktionen als nicht praktikabel in dieser Synthese beschrieben wurde.[61e] Abbildung 3-19: Verwendete Liganden bzw. Komplexe zur enantioselektiven Synthese von Esomeprazol und Armodafinil. Neben den metallkatalysierten Systemen wurden verschiedene BINOL-Phosphate als potentielle Organokatalysatoren, welche eine bifunktionelle Aktivierung von H2O2 ermöglichen, in der Synthese von (R)-Modafinil und (S)-Omeprazol überprüft. Ein erfreuliches Resultat wurde mit BINOL-Phosphat 104 erhalten, welches (R)-Modafinil in quantitativer Ausbeute mit einem Enantiomerenüberschuss von 10% lieferte. Durch den Ersatz des sterisch anspruchsvollen Restes R = C6H5-p-Naphthyl im BINOL-Phosphat 104 gegen einen kleineren Rest R = C6H5 (103) wurde (R)-Modafinil nur noch mit 87% Ausbeute in racemischer Form isoliert. Bei der Anwendung des BINOL-Phosphats 101 auf die Synthese von Esomeprazol konnte keine Aktivität beobachtet werden. 102 Kapitel 3 Ergebnisse und Diskussion Aufgrund der richtungsweisenden Ergebnisse aus Tabelle 3.3-1 wurde die Oxidationsmethodik in der Synthese von (R)-Modafinil (bezüglich der Eisen- und Vanadiumkatalyse) weiter optimiert. Die Ergebnisse sind in Tabelle 3.3-2 zusammengestellt. Tabelle 3.3-2: Anwendung der Liganden 53 und 43 auf die Synthese von enantiomerenangereichertem (R)-Modafinil. a Eintrag Ligand MXn LM Zeit [h] Ausbeute [%]a ee-Wert [%]b 1c 53 FeCl3 x 6 H2O THF 24 45 rac 2 53 VO(acac)2 CHCl3 24 89 9 ( R) 3 43 VO(acac)2 CHCl3 24 92 33 (R) 4d 43 VO(acac)2 CHCl3 24 >99 22 (R) 5 43 VO(acac)2 CHCl3 3.5 93 35 (R) 6 43 VO(acac)2 CHCl3 0.25 >99 32 (R) 7e 43 VO(acac)2 CHCl3 24 Spuren Isolierte Ausbeute; ee-Wert bestimmt durch Analyse an chiraler HPLC-Säule (AS); b n.b. c Verwendung des Liganden 53 (4 mol%); Reaktion wurde bei 0 °C durchgeführt; PhIO wurde als Oxidationsmittel verwendet. d e Hinsichtlich der eisenkatalysierten Oxidation unter der Verwendung des primär-aminhaltigen Liganden 53 wurde kein zufriedenstellendes Resultat in der Synthese von (R)-Modafinil erhalten (45%, rac, Eintrag 1), obwohl sich dieses katalytische System in der Oxidation von Thioanisol als durchaus aktiv zeigte. Als nächstes wurde FeCl 3 x 6 H2O durch VO(acac)2 ersetzt und der primäraminhaltige Ligand 53 in Chloroform untersucht. Es konnten zwar hohe Ausbeuten von 89% erzielt werden, der ee-Wert blieb jedoch auf verhältnismäßig geringem Niveau (9% ee) (Eintrag 2). Das 103 Kapitel 3 Ergebnisse und Diskussion beste Resultat lieferte der Schiffsche-Base Ligand 43 (welcher sich vom Aminoalkohol (1S,2R)-2Amino-1,2-diphenylethanol ableitet) in Kombination mit VO(acac)2 in Chloroform. Nach 24 h konnte (R)-Modafinil in sehr hohen Ausbeuten (92%) mit einem Enantiomerenüberschuss von 33% isoliert werden (Eintrag 3). Die Durchführung der Katalyse bei einer niedrigeren Temperatur (0 °C) führte zwar nach 24 h Reaktionszeit zu exzellenten Ausbeuten von 99%, jedoch zu einer gleichzeitigen Abnahme des ee-Wertes auf 22% (Eintrag 4). Erfreuliche Resultate im Hinblick auf Ausbeute und Enantioselektivität zeigten sich durch eine weitere Verringerung der Reaktionszeit auf 3.5 h (Eintrag 5) bzw. 15 Minuten (Eintrag 6). So konnte (R)-Modafinil in quantitativen Ausbeuten mit einem Enantiomerenüberschuss von 32% unter milden Bedingungen innerhalb von 15 Minuten isoliert werden (Eintrag 6). Bemerkenswert ist weiterhin, dass beim Ersatz von H2O2 durch das Oxidationsmittel Iodosobenzen (PhIO) nach 24 h Reaktionszeit kein Oxidationsprodukt zu beobachten war (Eintrag 7). Somit wurde eine schonende und sehr attraktive Synthesemethode von (R)-Modafinil, basierend auf einem verbesserten vanadiumkatalysierten System, in dem das Produkt enantiomeren-angereichert (32% ee) und in exzellenten Ausbeuten innerhalb einer kurzen Reaktionszeit (15 Minuten) erhalten wurde, etabliert. 104 KAPITEL 4 4 Zusammenfassung Die Verbindungsklasse der chiralen Sulfoxide weist einen enormen Stellenwert in der modernen organischen Chemie und Biochemie auf,[1] welcher sich in ihrer vielseitigen Anwendung unter anderem als chirale Auxiliare[2b, 3], Organokatalysatoren[5] und in der Verwendung sulfoxidhaltiger Verbindungen als pharmakologische Wirkstoffe begründet.[2] Ein bekanntes und erfolgreiches Active Pharmaceutical Ingredient (API) unter den sulfoxidhaltigen Arzneimitteln ist der Protonenpumpeninhibitor Esomeprazol, welcher sich auf Platz 4 der weltweit meist verkauften Medikamente befindet und unter anderem zur Behandlung von Refluxkrankheiten und Magensowie Zwölfingerdarmgeschwüren eingesetzt wird.[58-59] Unter den zahlreichen entwickelten Methoden zur Erzeugung chiraler Sulfoxide gilt die enantioselektive Oxidation von prochiralen Sulfiden in der Gegenwart eines chiralen Katalysators mittels H2O2 als eine der attraktivsten Synthesemethoden aufgrund ihrer hohen Atomökonomie und der Verwendung eines „grünen Oxidationsmittels“.[109] Das Ziel dieser Dissertationsarbeit war, basierend auf metall- und organokatalytischen Verfahren, neue Katalysatoren für die enantioselektive Sulfidoxidation zu entwickeln und auf ihre Effizienz zu untersuchen. Die Verwendung von H2O2 als umweltfreundlichem Oxidationsmittel, sowie eine einfache Handhabung in der Reaktionsführung standen dabei im Vordergrund. Die verwendete Modellreaktion ist untenstehend gezeigt: 105 Kapitel 4 Zusammenfassung Die erfolgreich entwickelten bzw. verbesserten Systeme und deren Anwendung werden nun im Folgenden zusammengefasst. Eisen- und Vanadiumkatalysatoren in der asymmetrischen Sulfidoxidation Eisen und Vanadium sind als redoxaktive Metallzentren in aktiven Zentren von Enzymen enthalten, wie z.B. in der Dioxygenase und Vanadium-Bromoperoxidase.[110] Die Aktivität und Selektivität dieser Enzyme mit synthetischen Metallkatalysatoren zu imitieren ist daher seit geraumer Zeit als eine große Herausforderung anzusehen. Besonders nicht-porphyrinhaltige Metallkatalysatoren speziell in Kombination mit Eisen als Redoxmetall sind für die enantioselektive Sulfidoxidation nur wenig erforscht. Zu Beginn der Arbeit wurde eine Vielzahl verschiedener Ligandensysteme (Bisformamid-und Harnstoffliganden sowie verschiedene Schiffsche-Base Liganden) neu entwickelt und charakterisiert. Unter letzteren befinden sich teils bereits etablierte Liganden für die asymmetrische Sulfidoxidation, welche in dieser Arbeit die Grundlage für das Design neuer eisenhaltiger Katalysatoren bildeten bzw. die Verbesserung von bestehenden Methoden insbesondere in ihrer Anwendung auf die Synthese von pharmakologischen Wirkstoffen ermöglichten. Nach der Untersuchung der verschiedenen neuen bzw. bekannten Liganden in Verbindung mit Eisen und Vanadium als redoxaktive Metallzentren stellte sich heraus, dass primär-aminhaltige Liganden in Kombination mit FeCl3 x 6 H2O für die enantioselektive Sulfidoxidation geeignet sind. In dieser Arbeit wird zum ersten Mal von primär-aminhaltigen Eisenkatalysatoren berichtet, welche unter milden Bedingungen und unter der Verwendung 106 einer wässrigen H2O2-Lösung als Kapitel 4 Zusammenfassung umweltfreundlichem Oxidationsmittel die Oxidation von Thioanisol zum korrespondierenden Sulfoxid enantioselektiv katalysieren. Unter einfacher Reaktionsführung (Generierung des Katalysators in situ und Durchführung der Reaktion an Luft) konnten in Verbindung mit Ligand 60 moderate Ausbeuten bis zu 69%, sowie Enantioselektivitäten bis zu 54% ee für Methylphenylsulfoxid erzielt werden. Die unsymmetrische Natur der Liganden, die freie Aminofunktion, sowie die langsame Zugabe des Wasserstoffperoxids sind entscheidend für den enantioselektiven Verlauf der Reaktion. Weiterhin konnten drei neue primär-aminhaltige Eisen(III)komplexe erfolgreich isoliert werden, deren Bildung durch massenspektrometrische Untersuchungen (HR-ESI-MS) nachgewiesen wurde. Ferner wurde in der vorliegenden Arbeit ein verbessertes vanadiumbasiertes katalytisches System entwickelt, welches erfolgreich in der Synthese des pharmakologischen Wirkstoffs (R)-Modafinil eingesetzt wurde. Das katalytische System, bestehend aus VO(acac) 2 und Ligand 43, lieferte in der Oxidation von Thioanisol mit H2O2 innerhalb von 24 h das Oxidationsprodukt in guten Ausbeuten von 79-81% und moderaten Enantioselektivitäten von 34-45% ee (vs. Lit.[26b] 64%, 28% ee). Als katalytisch sehr aktiv zeigte sich dieses System in seiner Anwendung auf die Synthese von (R)-Modafinil. Das Produkt wurde innerhalb einer sehr kurzen Reaktionszeit von nur 15 Minuten 107 Kapitel 4 Zusammenfassung und mit einer geringen Katalysatorbeladung in quantitativen Ausbeuten und akzeptablen eeWerten von bis zu 35% ee gebildet (vs. Lit.[68] 45%, 12% ee, 16 h). Organokatalysatoren für die asymmetrische Sulfidoxidation Während die Entwicklung von metallbasierten Methoden auf dem Gebiet der asymmetrischen Sulfidoxidation stetig voran getrieben wurde, wurden vergleichsweise wenige Fortschritte in der Entwicklung von metallfreien Ansätzen verzeichnet. Somit ist bis heute die Erforschung von neuen organokatalytischen Methoden zur Synthese von chiralen Sulfoxiden eine große Herausforderung. In der vorliegenden Arbeit wurde ein neues Konzept für die Sulfidoxidation entwickelt, welches auf der Verwendung von H2O2 mittels bifunktioneller Katalysatoren beruht. Es wurde gezeigt, dass unter der Verwendung des Organokatalysators N-Formyl-L-Prolin (34), Produktumsätze von bis zu 97% in der Oxidation von Thioanisol erzielt werden können. Die durch Überoxidation hervorgerufene Sulfonbildung kann unter bestimmten Bedingungen vollständig unterdrückt werden und somit ist diese Reaktion hoch chemoselektiv in der Bildung von Methylphenylsulfoxid. Mit dem aus dem „chiral pool“ leicht zugänglichen N-Formyl-L-Prolin (34) konnte keine asymmetrische Induktion in der Oxidation von Thioanisol beobachtet werden. Im Gegensatz dazu 108 Kapitel 4 Zusammenfassung konnten chirale BINOL-Phosphate, welche analog zu N-Formyl-L-Prolin (34) eine Lewis-Base und Brønsted-Säure Funktion in einem Molekül aufweisen, erfolgreich als bifunktionelle Organokatalysatoren in der Oxidation von Thioanisol eingesetzt werden. Dabei zeigte das BINOLPhosphat 102 mit R = 2,4-Dinitrophenyl die größte Aktivität, während mit BINOL-Phosphat 103 (R = Ph) eine Enantioselektivität von 36% erreicht wurde. Das postulierte Aktivierungsprinzip von H2O2 durch die bifunktionellen Katalysatoren ist in der folgenden Abbildung dargestellt: In beiden Fällen wird eine duale Aktivierung von H 2O2 durch die Ausbildung von Wasserstoffbrückenbindungen mit der Lewis-Base und Brønsted-Säure Einheit im bifunktionellen Katalysator erwartet. In der Anwendung von chiralen BINOL-Phosphaten auf die Synthese von (R)-Modafinil zeigte sich der Organokatalysator 104 mit R = C6H5-p-Naphthyl am erfolgreichsten. (R)-Modafinil konnte in quantitativen Ausbeuten und mit 10% ee unter den im folgenden Schema gezeigten Reaktionsbedingungen isoliert werden. 109 Kapitel 4 Zusammenfassung 110 CHAPTER 4 4 Summary Chiral sulfoxides can be considered as an important class of compounds with a huge role in modern organic chemistry and biochemistry which is based on their versatility;[1] they can be applied as chiral auxiliaries[2b, 3], organocatalysts[5] and particularly as pharmaceutical agents.[2] A widely applied sulfoxide containing agent is the proton pump inhibitor esomeprazole, located at number 4 of the world's top-selling drugs.[58] It is used for example in the treatment of gastric refluxand peptic ulcer diseases.[59] Among the numerous methods developed for the generation of chiral sulfoxides, the enantioselective oxidation of prochiral sulfides in the presence of a chiral catalyst with H2O2 is one of the most attractive synthetic methods due to their high atom economy and the utilization of a “green oxidant”.[109] The aim of this work was to develop new metal- and organocatalysts for the enantioselective sulfide oxidation and to examine their catalytic efficiency. The design of the new catalytic systems focused on the specific activation of the environmentally friendly oxidant H 2O2, an easy handling of the reaction and the ability to operate under mild conditions. The applied model reaction is shown below. The successfully developed or improved systems and their applications are summarized in the following. 111 Chapter 4 Summary Iron-and Vanadiumcatalysts in the Asymmetric Sulfoxidation Iron and vanadium as redox-active metal centers are present in the active centers of enzymes, such as dioxygenase and vanadium bromoperoxidase. [110] The mimicry of the enzymes’ efficiencies with artificial metal catalysts is therefore considered a major challenge for synthetic chemists. However, in the field of non-porphyrin containing catalysts for enantioselective sulfoxidations, especially in combination with iron as a redox metal, little research has been done yet. At the beginning of this work, a variety of different ligand systems (bisformamide and urea ligands and different Schiff bases) was developed and characterized. Among the Schiff base ligands, there are some compounds that have been used successfully in the asymmetric sulfide oxidation, which formed the basis for the development of new iron-containing catalysts or the improvement of existing methods and were applied to the synthesis of pharmacological agents. After the examination of various new and known ligands in combination with primarily iron and vanadium as redox-active metal centers, it was found that primary-amine containing ligands show great potential in the enantioselective sulfide oxidation in combination with FeCl3 x 6 H2O. In the present study, new primary-amine containing iron catalysts are described for the first time which catalyze the oxidation of thioanisole to the corresponding sulfoxide enantioselectively under mild conditions and with the use of an aqueous solution of H2O2 as an environmentally friendly oxidant: 112 Chapter 4 Summary In a simple reaction procedure (in situ generation of the metal catalysts under air) methyl phenyl sulfoxide was obtained in moderate yields up to 69% and enantioselectivities up to 54% ee using ligand 60. The dissymmetric nature of the ligands, the free amino group and the slow addition of hydrogen peroxide were found to be crucial for the enantioselective outcome of the reaction. Furthermore, three new primary-amine containing iron(III) complexes could be isolated successfully; their formation was proven by mass spectrometry (HR-ESI-MS). Additionally, an improved vanadium-based catalytic system was developed, which was successfully applied to the synthesis of the pharmaceutically active ingredient (R)-modafinil. The catalytic system consisting of VO(acac)2 and ligand 43, employed in the oxidation of thioanisole with H2O2, led to the oxidation product within 24 h in good yields of 79-81% and moderate enantioselectivities of 34-45% ee (vs. ref.[26b] 64%, 28% ee). This system showed a high catalytic activity when applied to the synthesis of (R)-modafinil – the product was formed in quantitative yields and with acceptable enantioselectivities of up to 35% ee within a very short reaction time (15 minutes) and a low catalyst loading (vs. ref.[68] 45%, 12% ee, 16 h). 113 Chapter 4 Summary Organocatalysts for the Asymmetric Sulfoxidation While a lot of progress has been made in the development of metal-based methods in the field of asymmetric sulfide oxidation, comparatively less research has been done regarding metal-free sulfoxidations. Thus the exploration of new organocatalytic approaches towards chiral sulfoxides remains a major challenge for modern chemists. Within this work, a new concept for sulfoxidation reactions has been established which is based on the activation of H2O2 with the use of bifunctional catalysts. It was found that the organocatalyst N-formyl-L-proline (34) is able to oxidize thioanisole in a yield of up to 97%. The overoxidation to the corresponding sulfone can be completely suppressed under certain reaction conditions which allows the highly chemoselective production of methyl phenyl sulfoxide. The organocatalyst N-formyl-L-proline (34), which is easily available from the chiral pool, showed no asymmetric induction in the oxidation of thioanisole. In contrast, BINOL-phosphates, which represent another example for bifunctional Lewis base/Brønsted acid catalysts, showed the desired activity in the enantioselective sulfoxidation. BINOL-phosphates with the substitution pattern R = 2,4-Dinitrophenyl (102) or Ph (103), respectively, revealed the greatest potential in this context. 114 Chapter 4 Summary The hypothetical principle of the activation of H2O2 by the employed bifunctional organocatalysts is shown in the following figure: In both cases, a dual activation of H2O2 through the formation of hydrogen bonds with the bifunctional catalyst’s Lewis base and Brønsted acid unit is proposed. In the application of chiral BINOL-phosphate catalysts to the synthesis of (R)-modafinil, the catalyst 104 with a C6H5-p-naphthyl residue has delivered the most significant results. (R)-modafinil could be isolated in quantitative yields with an enantiomeric excess of 10%. The detailed reaction conditions are given in the scheme below: 115 KAPITEL 5 5 Experimenteller Teil Vorbemerkungen Kommerziell erhältliche Chemikalien wurden ohne weitere Reinigung eingesetzt. Weiterhin wurden, soweit nicht anders vermerkt, die Reaktionen bei RT in nach Standardvorschriften gereinigten Lösungsmitteln durchgeführt. Absolutierte Lösungsmittel wurden über Trocknungsmittel destilliert (DCM: P2O5, THF: Na). Für die analytischen und spektroskopischen Untersuchungen wurden folgende Geräte eingesetzt: NMR-Spektroskopie: NMR-Spektren wurden auf Bruker Avance-Geräten (300 MHz oder 400 MHz bzw. 75 MHz oder 100 MHz) oder einem Jeol-Gerät (400 MHz) aufgenommen. Alle Spektren wurden bei RT in deuterierten Lösungsmitteln (CDCl3: 1H 7.24 ppm, 39.5 ppm; MeOH-d4: 1H 3.30 ppm, 13C 13C 77.0 ppm; DMSO-d6: 1H 2.49 ppm, 13C 49.0 ppm; D2O: 1H 4.80 ppm) als Standard vermessen. Die chemischen Verschiebungen der 1H-NMR und 13C-NMR Spektren wurden in ppm angegeben. Spinmultiplizitäten werden als s (Singulett), bs (breites Singulett), d (Dublett), dd (Dublett vom Dublett), ddd (Dublett vom Dublett vom Dublett), t (Triplett), q (Quartett) oder m (Multiplett) abgekürzt, die Ergänzung p kennzeichnet pseudo-Spinmultiplizitäten. Die Rohdaten wurden mit dem Programm MestRe-C (3.6.9.0) bearbeitet. 116 Kapitel 5 Experimenteller Teil Massenspektrometrie (MS): Für die Massenspektrometrie wurden folgende Geräte verwendet: FAB-Massenspektrometer Micromass ZABSPEC, EI-Massenspektrometer MAT 95 XP Finnigan, MALDI- Massenspektrometer Shimadzu Biotech AXIMA Confidence, ESI-Massenspektrometer Bruker Daltoniks maXis oder BRUKER micrOTOF II. Die High Performance Liquid Chromatography (HPLC): Chromatogramme wurden auf einem Agilent Technologies 1200 Series-Gerät aufgenommen. Chirale Säulen: IA (Daicel Chiralpak), OD (Macherey-Nagel), AS (Daicel Chiralpak). Die Angabe der Retentionszeit erfolgt in Minuten. Gaschromatographie (GC): Gaschromatographie (GC) wurde auf einem Thermo Instrument Trace GC Ultra durchgeführt (achirale Säule: TR 5, 7 m x 0.25 mm ID (Macherey-Nagel)). Die Angabe der Retentionszeit erfolgt in Minuten. Infrarot-Spektroskopie (IR): IR-Spektren wurden als dünne Filme in Substanz auf einem Varian IR-660 Spektrometer aufgenommen. Die Angabe der Absorption erfolgt in Wellenzahlen [cm-1]. Elementaranalyse (EA): Die Elementaranalysen wurden an einem CE Instruments EA 1119 CHNS Gerät durchgeführt. Polarimeter: Die Bestimmung des optischen Drehwertes chiraler Verbindungen erfolgte mit einem Polarimeter Model 341 der Firma Perkin Elmer. Als Lichtquelle diente eine Natrium-Lampe mit einer Wellenlänge von λ = 589 nm. 117 Kapitel 5 Experimenteller Teil Schmelzpunkt: Die Schmelzpunkte wurden auf einem Electrothermal Appendix B IA 9100 Gerät bestimmt. Folgende Hilfsmittel wurden für präparative Zwecke verwendet: Dünnschichtchromatographie (DC): Für die Dünnschichtchromatographie wurden ALUGRAM® SIL G/UV254 (Macherey-Nagel) DC-Platten verwendet. Die Detektion der Verbindungen erfolgte durch UV-Licht der Wellenlängen 254 nm und 366 nm. Bei nicht UV-aktiven Verbindungen wurden die DC-Platten in einer Ninhydrin- (0.3 g Ninhydrin in 100 mL BuOH und 3.0 mL Eisessig) oder einer Molybdatophosphorsäurelösung (10% in EtOH) entwickelt. Flash- und Säulenchromatographie: Für die säulenchromatographische Reinigung wurde Kieselgel 60 M (Macherey-Nagel) oder basisches Aluminiumoxid (Al2O3, aktiviert, Brockmann I) als stationäre Phase verwendet. 118 Kapitel 5 5.1 Experimenteller Teil Synthesen der Substrate und Referenzverbindungen rac-Methylphenylsulfoxid (25):[76] In einem Rundhalskolben wurden Thioanisol (1.18 mL, 10.0 mmol), MnSO4 x H2O (0.02 g, 0.10 mmol) und H2O2 (30%, 5.11 mL, 50.0 mmol) in MeCN (20 mL) gelöst und für 24 h gerührt. Der Überschuss an H 2O2 wurde mit Na2S2O3 zerstört. Das Lösungsmittel wurde am Rotationsverdampfer vollständig entfernt, der Rückstand in H 2O (10 mL) aufgenommen und mit EtOAc (3 x 40 mL) extrahiert. Die vereinigten organischen Phasen wurden über MgSO4 getrocknet und das Rohprodukt säulenchromatographisch (SiO 2, EtOAc/PE 1:1) gereinigt. Das Produkt wurde als weißer Feststoff erhalten (1.31 g, 7.70 mmol, 77%). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[111] 1H-NMR (300 MHz, CDCl3): δ = 2.70 (s, 3H), 7.47-7.54 (m, 3H), 7.60-7.64 (m, 2H). Methylphenylsulfon (26):[76] In einem Rundhalskolben wurden Thioanisol (0.59 mL, 5.00 mmol), MnSO4 x H2O (0.01 g, 0.05 mmol), eine wässrige NaHCO3-Lösung (0.2 M, 50 mL) und H2O2 (30%, 2.55 mL, 25.0 mmol) in MeCN (25 mL) gelöst und für 48 h gerührt. Der Überschuss an H2O2 wurde mit Na2S2O3 zerstört. Das Lösungsmittel wurde am Rotationsverdampfer vollständig entfernt, der Rückstand in H2O (10 mL) aufgenommen und mit EtOAc (3 x 40 mL) extrahiert. Die vereinigten organischen Phasen wurden über MgSO4 getrocknet und das Rohprodukt säulenchromatographisch (SiO2, EtOAc/PE 1:1) gereinigt. Das Produkt wurde als weißer Feststoff erhalten (0.62 g, 3.98 mmol, 80%). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[112] 119 Kapitel 5 Experimenteller Teil 1H-NMR (300 MHz, CDCl3): δ = 3.03 (s, 3H), 7.55-7.60 (m, 2H), 7.63-7.67 (m, 1H), 7.94 (d, J = 7.3 Hz, 2H). 5-Methoxy-2-(((4-methoxy-3,5-dimethylpyridin-2-yl)methyl)thio)-1H-benzimidazol (108):[105a] 2-Mercapto-5-methoxybenzimidazol (106) (1.00 g, 5.50 mmol) und 2-Chloromethyl-4-methoxy-3,5-dimethyl-pyridin Hydrochlorid (107) (1.20 g, 5.50 mmol) wurden in einer Mischung von Aceton/EtOH (7 mL/21 mL) gelöst und mit NaOH (0.44 g, 11.0 mmol) und NaI (27.5 mg, 0.18 mmol) versetzt. Die Reaktionslösung wurde für 1.5 h refluxiert, das Lösungsmittel wurde am Rotationsverdampfer entfernt und der Rückstand in H2O (20 mL) aufgenommen. Die wässrige Phase wurde mit EtOAc (3 x 25 mL) extrahiert und die vereinigten organischen Phasen mit H 2O (3 x 30 mL) gewaschen. Die organische Phase wurde über Na2SO4 getrocknet und das Lösungsmittel vollständig entfernt. Das Rohprodukt wurde säulenchromatographisch (SiO2, EtOAc/PE 10:1, DCM/MeOH 10:1) gereinigt, wonach das Produkt als weißer Feststoff erhalten wurde (1.60 g, 4.97 mmol, 90%). 1H-NMR (300 MHz, CDCl3): δ = 2.25 (s, 3H), 2.29 (s, 3H), 3.76 (s, 3H), 3.82 (s, 3H), 4.33 (s, 2H), 6.79 (dd, J = 8.7 Hz, 2.4 Hz, 1H), 7.01 (d, J = 2.0 Hz, 1H), 7.38 (d, J = 8.7 Hz, 1H), 8.24 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 11.2, 13.3, 34.9, 55.8, 60.0, 110.9, 125.5, 126.6, 148.5, 156.0, 156.1, 165.2; MS (MALDI-TOF): m/z = 330 [M]+. 120 Kapitel 5 Experimenteller Teil rac-5-Methoxy-2-(((4-methoxy-3,5-dimethylpyridin-2-yl)methyl)sulfin-yl)-1H-benzo[d]imidazole (rac109):[113] 108 (1.08 g, 3.29 mmol) wurde in EtOAc (11 mL) suspendiert. Die Suspension wurde auf -9 °C abgekühlt und langsam mit m-CPBA (70%, 0.81 g, 3.29 mmol) versetzt ohne die Temperatur über 5 °C ansteigen zu lassen. Der ausgefallene Feststoff wurde abfiltriert, mit gekühltem EtOAc (3 x 20 mL) gewaschen und getrocknet (0.76 g, 2.20 mmol, 67%). Das Rohprodukt (0.76 g, 2.20 mmol) wurde in H2O (3 mL) aufgenommen und mit einer wässrigen Methylaminlösung (40%, 0.61 mL) versetzt. Anschließend wurde Aceton (5 mL) zugegeben und der pH-Wert der Lösung mittels 2 M HCl auf 7-8 eingestellt. Die Lösung wurde mit H2O (11 mL) verdünnt, filtriert und der Feststoff zügig mit H 2O gewaschen. Das Produkt wurde als weißer Feststoff erhalten (0.68 g, 1.97 mmol, 60%). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[114] 1H-NMR (300 MHz, DMSO-d6): δ = 2.16 (s, 3H), 2.18 (s, 3H), 3.68 (s, 3H), 3.80 (s, 3H), 4.72 (q, J = 13.6 Hz, 2H), 6.92 (dd, J = 8.9 Hz, 2.3 Hz, 1H), 7.09 (s, 1H), 7.54 (d, J = 8.8 Hz, 1H), 8.18 (s, 1H), 13.44 (bs, 1H). 5-Methoxy-2-(((4-methoxy-3,5-dimethylpyridin-2-yl)methyl)sulfonyl)-1H-benzo[d]imidazol (116): In einem Rundhalskolben wurde rac-109 (0.20 g, 0.58 mmol) in DCM (8 mL) gelöst und bei 0 °C mit m-CPBA (77%, 0.13 g, 0.58 mmol) versetzt. Die Reaktionsmischung wurde für 1 h gerührt, wobei der Reaktionsfortschritt mittels DC verfolgt wurde (SiO2, 121 Kapitel 5 Experimenteller Teil CHCl3/MeOH 10:1). Anschließend wurde die Reaktionslösung mit einer gesättigten NaHCO3Lösung (2 x 10 mL) gewaschen. Nach dem Trocknen mit Na2SO4 wurde das Lösungsmittel am Rotationsverdampfer entfernt und das Rohprodukt säulenchromatographisch (Al2O3, CHCl3/MeOH 10:1) gereinigt. Das Produkt wurde als weißer Feststoff erhalten (0.13 g, 0.35 mmol, 60%). MS (MALDI-TOF): m/z = 362 [M+H]+. 2-(Benzhydrylthio)essigsäure (112):[106] Eine Lösung von Benzhydrol (110) (3.00 g, 16.3 mmol) und Mercaptoessigsäure (111) (1.50 g, 16.3 mmol) in TFA (13 mL) wurde für 3 h bei RT gerührt. Das Lösungsmittel wurde am Rotationsverdampfer entfernt und der Rückstand in H2O (18 mL) aufgenommen. Der ausgefallene Niederschlag wurde abfiltriert, mit Hexan (50 mL) gewaschen und im Vakuum getrocknet. Das Produkt wurde als weißer Feststoff erhalten (4.20 g, 16.3 mmol, quant.). Die NMR Spektren stimmen mit den in der Literatur berichteten überein.[68] H-NMR (300 MHz, DMSO-d6): δ = 3.05 (s, 2H), 5.37 (s, 1H), 7.21-7.43 (m, 10H), 12.41 (bs, 1H); 1 C-NMR (75 MHz, DMSO-d6): δ = 33.8, 52.9, 127.3, 128.0, 128.7, 140.9, 170.9. 13 2-(Benzhydrylthio)acetamid (114):[106] 2-(Benzhydrylthio)essigsäure (112) (1.50 g, 5.76 mmol) wurde in Toluol (10 mL) gelöst und langsam mit einer Lösung von Thionylchlorid (1.46 mL, 20.2 mmol) in Toluol (2 mL) versetzt. Die Reaktionsmischung wurde für 2 h auf 80 °C erhitzt. Das Lösungsmittel wurde am Rotationsverdampfer vollständig entfernt und 2-(Benzhydrylthio)acetyl- 122 Kapitel 5 Experimenteller Teil chlorid (113) als oranges Öl erhalten. Dieses wurde in DCM (6 mL) gelöst und zu einer wässrigen Ammoniaklösung (25%, 17.5 mL) zugetropft. Die Reaktionslösung wurde für 2 h bei RT gerührt, die Phasen anschließend getrennt und die wässrige Phase mit DCM (2 x 10 mL) extrahiert. Die vereinigten organischen Phasen wurden mit einer NaHCO 3- (5%, 3 x 10 mL) und einer gesättigten NaCl-Lösung (10 mL) gewaschen, über Na2SO4 getrocknet und das Lösungsmittel am Rotationsverdampfer vollständig entfernt. Nach Umkristallisation in Isopropylether wurde das Produkt als weißer Feststoff erhalten (0.49 g, 1.88 mmol, 33 %). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[115] 1H-NMR (300 MHz, CDCl3): δ = 3.03 (s, 2H), 5.13 (s, 1H), 5.85 (bs, 1H), 6.49 (bs, 1H), 7.17-7.38 (m, 10H). rac-2-(Benzhydrylsulfinyl)acetamid (rac-115):[107] 2-(Benzhydrylthio)acetamid (114) (0.15 g, 0.57 mmol) wurde in THF (2.30 mL) gelöst und mit wässrigem H2O2 (30%, 59.0 µL, 0.58 mmol) versetzt. Die Reaktionslösung wurde für 2 h refluxiert. Nach dem Abkühlen auf RT wurde die Lösung mit DCM (3 x 5 mL) extrahiert, die vereinten organischen Phasen über Na2SO4 getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Das Rohprodukt wurde säulenchromatographisch (SiO2, EtOAc) gereinigt, wonach das Produkt als weißer Feststoff erhalten wurde (0.13 g, 0.46 mmol, 80%). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[68] 1H-NMR (300 MHz, DMSO-d6): δ = 3.21 (d, J = 13.5 Hz, 1H), 3.36 (d, J = 13.5 Hz, 1H), 5.34 (s, 1H), 7.32-7.43 (m, 7H), 7.50-7.52 (m, 4H), 7.68 (s, 1H); MS (MALDI-TOF): m/z = 296 [M+Na]+, 312 [M+K]+. 123 Kapitel 5 Experimenteller Teil 2-(Benzhydrylsulfonyl)acetamid (117):[108] 2-(Benzhydrylthio)acetamid (114) (0.15 g, 0.58 mmol) wurde in Essigsäure (1 mL) gelöst und langsam mit wässrigem H2O2 (30%, 0.18 mL, 1.78 mmol) versetzt. Die Reaktionslösung wurde für 5 h gerührt und anschließend stehen gelassen. Nach 24 h wurde nochmal wässriges H2O2 (30%, 59.0 µL, 0.58 mmol) zugetropft und die Lösung für weitere 24 h stehen gelassen. Der entstandene Niederschlag wurde in EtOH umkristallisiert und das Produkt als weißer Feststoff erhalten (57.5 mg, 0.19 mmol, 34%). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[115] 1H-NMR (400 MHz, DMSO-d6): δ = 3.75 (s, 2H), 6.09 (s, 1H), 7.35-7.43 (m, 6H), 7.50 (s, 1H), 7.63- 7.65 (m, 4H), 7.72 (s, 1H); MS (MALDI-TOF): m/z = 312 [M+Na]+, 328 [M+K]+. 5.2 5.2.1 Synthesen und katalytische Experimente (Kapitel 3.1) Synthese von Formamid- und Harnstoffliganden (S)-1-Methoxy-1-oxo-3-phenylpropan-2-aminium Hydrochlorid (30):[78a] In einem Rundhalskolben wurde zu auf 0 °C gekühltem absolutem Methanol (20 mL) vorsichtig Thionylchlorid (3.70 mL, 51.0 mmol) zugetropft und die Reaktionslösung anschließend portionsweise mit L-Phenylalanin (29) (6.00 g, 36.3 mmol) versetzt. Die Reaktionslösung wurde 4 h unter Rückfluss erhitzt. Das Lösungsmittel wurde am Rotationsverdampfer vollständig entfernt, wonach das Produkt als weißer Feststoff erhalten wurde (7.62 g, 35.4 mmol, 98%). Die NMR Spektren stimmen mit den in der Literatur berichteten überein.[78a] 124 Kapitel 5 Experimenteller Teil 1H-NMR (400 MHz, D2O): δ = 3.25 (dd, J = 14.5 Hz, 7.5 Hz, 1H), 3.36 (dd, J = 14.5 Hz, 5.8 Hz, 1H), 3.85 (s, 3H), 4.45 (dd, J = 7.4 Hz, 5.9 Hz, 1H), 7.40-7.47 (m, 5H); 13C-NMR (100 MHz, D2O): δ = 33.6, 51.6, 52.2, 126.2, 127.3, 127.4, 131.8, 168.1. (S)-2-Amino-3-phenylpropanamid (31):[78b] (S)-Methyl-2-amino-3-phenylpropanoat (30) (4.90 g, 30.0 mmol) wurde in Toluol (48 mL) gelöst und anschließend mit wässriger Ammoniaklösung (25%, 13.4 mL) versetzt. Der Reaktionsfortschritt wurde mittels DC verfolgt (EtOAc/MeOH 4:1). Nach 3 d wurde das Lösungsmittel wurde vollständig am Rotationsverdampfer entfernt und der Rückstand mit Et2O (2 x 20 mL) gewaschen. Das Lösungsmittel wurde entfernt und der Rückstand in H 2O (40 mL) aufgenommen. Der pH-Wert der Lösung wurde mit einer NaOH-Lösung (30%) auf 11 eingestellt. Die wässrige Phase wurde mit DCM (9 x 15 mL) extrahiert. Die vereinigten organischen Phasen wurden über MgSO 4 getrocknet und filtriert. Nach vollständigem Entfernen des Lösungsmittels wurde das Produkt als weißer Feststoff erhalten (2.78 g, 0.02 mmol, 63%). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[78a] 1H-NMR (300 MHz, D2O): δ = 2.89 (dd, J = 13.5 Hz, 7.1 Hz, 1H), 2.97 (dd, J = 13.5 Hz, 6.6 Hz, 1H), 3.65 (t, J = 6.8 Hz, 1H), 7.13-7.67 (m, 5H); MS (MALDI-TOF): m/z = 165 [M+1]+. 125 Kapitel 5 Experimenteller Teil (S)-3-Phenylpropan-1,2-diamin (32):[78a] In einem Rundhalskolben wurde unter Stickstoffatmosphäre LiAlH4 (4.10 g, 0.11 mol) in absolutem THF (60 mL) suspendiert. (S)-2-Amino-3phenylpropanamid (31) (3.70 g, 22.5 mmol) wurde portionsweise zugegeben und die Reaktionslösung für 24 h refluxiert. Bei 0 °C wurde überschüssiges LiAlH4 mit einer NaOH-Lösung (5%, 12 mL) und mit reinem H2O (1 mL) vorsichtig hydrolysiert. Der ausgefallene Niederschlag wurde abfiltriert und mit THF gewaschen. Das Filtrat wurde vollständig eingeengt, der Rückstand in DCM aufgenommen und über Na2SO4 getrocknet. Nach dem Entfernen des Lösungsmittels am Rotationsverdampfer wurde das Rohprodukt durch Vakuumdestillation (12.0 mbar, 155-157 C) gereinigt. Das Produkt wurde als farbloses Öl erhalten (1.05 g, 6.99 mmol, 31%). Die NMR Spektren stimmen mit den in der Literatur berichteten überein.[78a] 1H-NMR (300 MHz, CDCl3): δ = 1.36 (s, 4H), 2.41-2.54 (m, 2H), 2.71-2.79 (m, 2H), 2.91-2.94 (m, 1H), 7.14-7.29 (m, 5H); 13C-NMR (100 MHz, CDCl3): δ = 42.2, 48.0, 55.0, 126.1, 128.4, 129.1, 139.1. (S)-1-Formylpyrrolidin-2-carboxysäure (N-Formyl-L-Prolin) (34):[72e] In einem Rundhalskolben wurde Essigsäureanhydrid (6.00 mL, 63.5 mmol) in Ameisensäure (85%, 5.00 mL, 0.11 mol) gelöst und auf 0 °C abgekühlt. Zu dieser Lösung wurde eine Lösung von L-Prolin (33) (1.00 g, 8.67 mmol) in Ameisensäure (85%, 10.0 mL, 0.23 mol) zugetropft.[80] Die Reaktionsmischung wurde für 3 d bei 0 °C gerührt, die Lösung wurde mit H2O (9 mL, 0 °C) versetzt und das Lösungsmittel am Rotationsverdampfer entfernt. Der Rückstand wurde in MeOH (40 mL) aufgenommen und das Lösungsmittel am Rotationsverdampfer 126 Kapitel 5 Experimenteller Teil anschließend entfernt. Das leicht gelbliche Öl wurde in PE gerührt, wobei mit der Zeit ein weißer Feststoff ausfiel. Das Lösungsmittel wurde entfernt und das Produkt als weißer Feststoff erhalten (1.17 g, 8.20 mmol, 94%). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[72e] 1H-NMR (400 MHz, CDCl3): δ = 1.87-2.30 (m, 4H), 3.49-3.71 (m, 2H), 4.40-4.47 (m, 1H), 8.25 (s, 1H), 8.28 (s, 1H), 11.10 (bs, 1H); 13C-NMR (100 MHz, CDCl3): δ = 22.7, 23.9, 29.0, 29.6, 44.3, 46.9, 57.0, 59.0, 162.2, 162.9, 173.8, 174.3. (2S,2'S)-N,N'-((S)-3-phenylpropan-1,2-diyl)bis(1-formylpyrrolidin-2-carboxamid) (27): Unter Stickstoffatmosphäre wurde N-Formyl-L-Prolin (34) (0.54 g, 3.73 mmol) in absolutem THF (3.80 mL) gelöst und mit CDI [79] (0.59 g, 4.10 mmol) versetzt. Nach vollendeter CO 2-Entwicklung wurde (S)-3-Phenylpropan-1,2-diamin (32) (0.30 g, 1.86 mmol) zugetropft und die Reaktionslösung über Nacht gerührt. Das Lösungsmittel wurde entfernt und das Rohprodukt säulenchromatographisch (SiO2, EtOAc/MeOH 10:1, EtOAc/MeOH 4:1, DCM/MeOH 9:1) gereinigt. Das Produkt wurde als weißer Feststoff erhalten (0.32 g, 0.80 mmol, 43%). [α]D20 = -136° (c = 0.15, CHCl3); 1H-NMR (400 MHz, DMSO, 100 °C): 1.71-2.12 (m, 8H), 2.69-2.82 (m, 2H), 2.99-3.59 (m, 6H), 3.95-4.35 (m, 3H), 7.01 (s, 1H), 7.15-7.27 (m, 5H), 7.34-7.50 (m, 1H), 7.62-7.89 (m, 1H), 8.10-8.21 (m, 1H); 13C-NMR (100 MHz, DMSO, 100 °C): 21.8, 22.0, 22.8, 22.9, 28.5, 28.6, 29.2, 29.3, 36.9, 37.1, 41.8, 42.1, 43.0, 45.6, 49.9, 57.1, 57.2, 58.9, 120.9, 125.4, 127.4, 128.5, 138.0,160.8, 170.2, 170.6, 171.3; MS (EI): m/z = 423 [M+Na]+; HRMS (EI): Gemessen: m/z = 400.2098; Berechnet für [C21H28N4O4]+ m/z = 400.2111; IR (dünner Film): ṽ = 3310, 3059, 2974, 127 Kapitel 5 Experimenteller Teil 2895, 2356, 1653, 1633, 1534, 1377, 1323, 1234, 1186, 1154, 1092, 1063, 1030, 979, 914, 748, 700, 663, 617, 519, 477 cm-1. 1-((1S,2S)-2-Aminocyclohexyl)-3-ethylurea (38): In einem Rundhalskolben wurde (1S,2S)-Cyclohexan-1,2-diamin (37) (0.63 g, 5.53 mmol) in absolutem DCM (140 mL) gelöst. Die Lösung wurde auf -30 °C gekühlt und über 2 h langsam mit Ethylisocyanat (0.44 mL, 5.53 mmol) versetzt, woraufhin ein weißer Niederschlag ausfiel (KPG-Rührer). Die Reaktionslösung wurde für 6 h bei -30 °C und über Nacht bei -15 °C gerührt. Das Lösungsmittel wurde am Rotationsverdampfer entfernt, der Rückstand in DCM aufgenommen und filtriert.[83] Das Rohprodukt wurde säulenchromatographisch (SiO2, CHCl3/MeOH 1:1) gereinigt, wonach das Produkt als weißer Feststoff erhalten wurde (0.51 g, 2.74 mmol, 50%). 1H-NMR (300 MHz, CDCl3): δ = 1.04-1.27 (m, 8H), 1.65 (bs, 4H), 1.89-1.92 (m, 2H), 2.31-2.39 (m, 1H), 3.12-3.24 (m, 3H), 4.65 (d, J = 8.5 Hz, 1H), 5.22 (bs, 1H); 13C-NMR (100 MHz, CDCl3): δ = 15.4, 24.9, 25.2, 33.0, 34.9, 35.2, 56.2, 57.5; MS (FAB): m/z = 186 [M+H]+. Pyridin-2,6-dicarbaldehyd (40):[84] Unter Stickstoffatmosphäre wurde Oxalylchlorid (3.93 g, 2.65 mL, 31.0 mmol) in absolutem DCM (45 mL) gelöst. Nach dem Abkühlen der Lösung auf -78 °C wurde langsam eine Lösung von DMSO (6.60 g, 6.00 mL, 84.5 mmol) in DCM (9 mL) zugetropft. Nach beendeter Zugabe wurde die Lösung für 5 Min gerührt und mit 2,6Pyridindimethanol (39) (1.50 g, 10.0 mmol) in DMSO/DCM (6 mL/9 mL) über einen Zeitraum von 128 Kapitel 5 Experimenteller Teil 15 Min versetzt. Nachdem die Reaktionsmischung für weitere 20 Min gerührt wurde, wurde TEA (15 mL) zugetropft und die Lösung auf RT erwärmt. Zur Aufarbeitung wurde H 2O (75 mL) zugetropft und die wässrige Phase mit DCM (3 x 50 mL) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter NaCl-Lösung (3 x 50 mL) gewaschen, über MgSO4 getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Das Rohprodukt wurde mittels Säulenchromatographie (SiO2, PE/EA 1:1) und Vakuumsublimation gereinigt, wonach das Produkt als weißer Feststoff erhalten wurde (1.12 g, 8.26 mmol, 76%). Die NMR Spektren stimmen mit den in der Literatur berichteten überein.[84] 1H-NMR (300 MHz, CDCl3): δ = 7.86-8.08 (m, 1H), 8.15 (d, J = 6.6 Hz, 2H), 10.14 (s, 2H); 13C-NMR (100 MHz, DMSO-d6): δ = 125.8, 139.6, 152.6, 192.8; MS (FAB): m/z = 136 [M+H]+. 1,1'-(((Pyridin-2,6-diyl-bis(methylen))bis(azandiyl))bis((1 S,2S)-cyclohexan-2,1-diyl))bis(3ethylharnstoff) (28): In einem Rundhalskolben wurde der Harnstoff 38 (0.30 g, 1.62 mmol) in absolutem MeOH (3 mL) gelöst und portionsweise mit Pyridin-2,6-dicarbaldehyd (40) (0.11 g, 0.81 mmol) versetzt, wodurch ein weißer, voluminöser Niederschlag ausfiel. Nach der Zugabe von weiterem absolutem MeOH (8 mL) wurde die Reaktionsmischung für 30 Min gerührt. Anschließend erfolgte die Zugabe von Essigsäure (92.2 µL, 1.62 mmol). Der pH-Wert der Reaktionslösung sollte im Bereich von 5-8 liegen, um eine selektive Reduktion zu ermöglichen. Die Reaktionslösung wurde auf 0 °C gekühlt, mit NaCNBH 4 (0.16 g, 2.43 mmol) versetzt und anschließend auf RT erwärmt, woraufhin sich der Niederschlag auflöste. Nach der Kontrolle des pH-Werts wurde weitere Essigsäure (46.3 µL, 0.81 mmol) zugegeben. Die Reaktionsmischung wurde über Nacht bei RT gerührt und anschließend auf 0 °C 129 Kapitel 5 Experimenteller Teil gekühlt. Die Lösung wurde mit HCl (6 M, 4 mL) auf pH 1 eingestellt, für 4 h bei RT gerührt und für 30 Min mit Stickstoff entgast. Bei 0 °C wurde die Reaktionslösung mit einer NaOH-Lösung (15%, 11 mL) auf pH 12 eingestellt, mit NaCl versetzt und mit DCM (9 x 15 mL) ausgeschüttelt. Die vereinigten organischen Phasen wurden über K2CO3 getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Das Rohprodukt wurde säulenchromatographisch (SiO2, DCM/MeOH 3:1) gereinigt, wonach das Produkt als weißer Feststoff erhalten wurde (0.14 g, 0.28 mmol, 35%).[85] [α]D20 = +28° (c = 0.15, MeOH); 1H-NMR (300 MHz, MeOH-d4): δ = 0.96 (t, J = 7.2 Hz, 6H), 1.071.17 (m, 10H), 1.57-1-61 (m, 6H), 1.73-1.85 (m, 2H), 1.85-2.10 (m, 2H), 2.19-2.40 (m, 2H), 2.993.06 (m, 4H), 3.20-3.41 (m, 2H), 3.70 (dd, J = 53.8 Hz, 14.1 Hz, 4H), 7.28 (d, J = 7.3 Hz, 2H), 7.62 (t, J = 7.7 Hz, 1H); 13C-NMR (100 MHz, MeOH-d4): δ = 15.8, 25.5, 26.1, 31.8, 33.9, 35.8, 52.1, 54.6, 62.1, 122.2, 138.7, 159.7, 161.1; MS (FAB): 474 [M+H]+; HRMS (EI): Gemessen: m/z = 473.3476; Berechnet für [C25H43N7O2]+ m/z = 473.3478; EA: Gemessen: C, 61.63; H, 9.01; N, 19.69; Berechnet für [C25H43N7O2 + MeOH]: C, 61.75; H, 9.37; N, 19.39; Smp: 55 °C; IR (KBr): ṽ = 3309, 2931, 2920, 2856, 2819, 1624, 1560, 1448, 1249, 1229, 1098, 750, 694 cm-1. 5.2.2 Katalysereaktionen mit Formamid- und Harnstoffliganden Allgemeine Arbeitsvorschrift 1 (AAV1): Vanadium– und eisenkatalysierte Oxidation von Thioanisol unter der Verwendung von Formamid- und Harnstoffliganden In einem Rundhalskolben wurden MXn (2 mol%), der jeweilige Ligand (3 mol%) und das LM vorgelegt. Nach 30-minütigem Rühren der Lösung bei RT wurde zuerst Thioanisol (1.0 Äq., 0.40 mmol) und anschließend 30%iges H2O2 (1.2 Äq.) zugegeben und die Reaktionsmischung unter der angegebenen Temperatur, Reaktionszeit, sowie Substratkonzentration gerührt. Das Rohprodukt wurde säulenchromatographisch gereinigt 130 (SiO2, PE/EtOAc 1:4) und der Kapitel 5 Experimenteller Teil Enantiomerenüberschuss mittels HPLC-Analyse (OD-Säule, Hexan/i-PrOH 93:7, Fluss 1.0 mL/Min, 30-32 bar, 25 °C, t(R-Enantiomer) = 18.0 Min, t(S-Enantiomer) = 22.4 Min) bestimmt. Zur Identifikation des Produkts wurde das 1H-NMR Spektrum mit literaturbekannten Daten[111] verglichen. Allgemeine Arbeitsvorschrift 2 (AAV2): Eisenkatalysierte Oxidation von Thioanisol unter der Verwendung von Tripeptidliganden In einem Rundhalskolben wurden FeCl3 x 6 H2O (2 mol%), der jeweilige Ligand (3 mol%), das LM (c(Substrat) = 0.48 mol/L) und TEA (3-9 mol%) vorgelegt. Nach 15-minütigem Rühren der Lösung bei RT wurde zuerst Thioanisol (1.0 Äq., 0.24 mmol) und anschließend 30%iges H2O2 (1.2 Äq.) zugegeben und die Reaktionsmischung unter angegebener Reaktionszeit gerührt. Das Rohprodukt wurde säulenchromatographisch gereinigt (SiO2, PE/EtOAc 1:4) und der Enantiomerenüberschuss mittels HPLC-Analyse (OD-Säule, Hexan/i-PrOH 93:7, Fluss 1.0 mL/Min, 30-32 bar, 25 °C, t(REnantiomer) = 18.0 Min, t(S-Enantiomer) = 22.4 Min) bestimmt. Zur Identifikation des Produkts wurde das 1H- NMR Spektrum mit literaturbekannten Daten[111] verglichen. 131 Kapitel 5 5.2.3 Experimenteller Teil Synthese von Schiffsche-Base Liganden, deren Derivaten und Metallkomplexen N-((1S,2S)-2-((E)-(3,5-Di-tert-butyl-2-hydroxybenzyliden)amino)-1,2-diphenylethyl)-4-methylbenzensulfonamid (51):[116] Unter Stickstoffatmosphäre wurde (1S,2S)-N-(4-Toluensulfonyl)-1,2-diphenylethylendiamin (0.12 g, 0.34 mmol) in absolutem MeOH (0.75 mL) gelöst. Anschließend wurde wasserfreies Na2SO4 (0.19 g, 1.31 mmol) zugegeben und eine Lösung von 3,5-Di-tert-butyl-2-hydroxybenzaldehyd (64) (0.08 g, 0.32 mmol) in absolutem MeOH (1.35 mL) zugetropft. Die Reaktionslösung wurde für 2 h bei RT gerührt. Das Lösungsmittel wurde am Rotationsverdampfer entfernt, der Rückstand in PE aufgenommen und anschließend filtriert. Das Lösungsmittel des Filtrats wurde am Rotationsverdampfer vollständig entfernt woraufhin das Produkt als gelber Feststoff erhalten wurde (0.16 g, 0.28 mmol, 86%). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[116] 1H-NMR (300 MHz, CDCl3): δ = 1.25 (s, 9H), 1.46 (s, 9H), 2.31 (s, 3H), 4.51 (d, J = 5.7 Hz, 1H), 4.69 (pt, J = 6.0 Hz, 1H), 5.18 (d, J = 6.4 Hz, 1H), 6.91-6.93 (m, 3H), 7.01-7.18 (m, 10H), 7.41 (d, J = 8.2 Hz, 3H), 8.17 (s, 1H), 12.9 (s, 1H). 132 Kapitel 5 Experimenteller Teil N-((1S,2S)-2-((3,5-Di-tert-butyl-2-hydroxybenzyl)amino)-1,2-diphenylethyl)-4-methylbenzensulfonamid (52):[117] Das Imin 51 (0.25 g, 0.43 mmol) wurde unter Stickstoffatmosphäre in absolutem MeOH (4.20 mL) gelöst und die Lösung anschließend auf 0 °C gekühlt. NaBH4 (0.05 g, 1.28 mmol) wurde in kleinen Portionen zugegeben und die Reaktionslösung über Nacht gerührt. Zur Aufarbeitung[118] wurde der Reaktionsansatz mit H 2O (4 mL) versetzt und die Lösung anschließend mit 1 M HCl auf pH 7 eingestellt. Nach der Extraktion mit EtOAc (3 x 10 mL) wurden die vereinigten organischen Phasen über MgSO4 getrocknet. Das Rohprodukt wurde mittels Säulenchromatographie (SiO2, PE/EtOAc 3:1) gereinigt, wonach das Produkt als weißer Feststoff erhalten wurde (0.18 mg, 0.31 mmol, 72%). 1H-NMR (300 MHz, CDCl3): δ = 1.21 (s, 9H), 1.44 (s, 9H), 2.26 (s, 3H), 3.57 (d, J = 13.2 Hz, 1H), 3.75 (d, J = 13.2 Hz, 1H), 3.88 (d, J = 7.6 Hz, 1H), 4.57 (d, J = 7.6 Hz, 1H), 6.62-6.68 (m, 3H), 6.89-7.00 (m, 7H), 7.17-7.20 (m, 4H), 7.43 (d, J = 8.7 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.4, 29.6, 31.6, 34.1, 34.9, 51.1, 62.3, 66.5, 121.6, 123.1, 123.5, 127.0, 127.4, 127.6, 128.1, 128.2, 128.5, 129.3, 135.9, 136.8, 137.0, 140.6, 143.1, 154.0. 2-((E)-(((1R,2R)-2-Amino-1,2-diphenylethyl)imino)methyl)-4,6-di-tert-butylphenol (53):[88a] (1R,2R)-(+)-1,2-Diphenylethylendiamin (62) (0.50 g, 2.35 mmol) wurde unter Stickstoffatmosphäre in absolutem DCM (8 mL) gelöst. Anschließend wurde Molekularsieb (3Å, 5.30 g) zugegeben und eine Lösung von 3,5-Di-tert-butylsalicylaldehyd (64) (0.55 mg, 2.33 mmol) in DCM (10 mL) über einen Zeitraum 133 Kapitel 5 Experimenteller Teil von 30 Min zugetropft. Die Reaktionslösung wurde für 3 h gerührt und anschließend am Rotationsverdampfer (Wasserbadtemperatur 30 °C) auf ca. 1 mL eingeengt. Das Rohprodukt wurde zügig aufgrund der Instabilität des Produktes (Disproportionierung) mittels FlashSäulenchromatographie (SiO2, PE/EtOAc 7:3) gereinigt, wonach das Produkt als gelber Feststoff erhalten wurde (0.37 g, 0.86 mmol, 37%). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[88a] 1H-NMR (400 MHz, CDCl3): δ = 1.27 (s, 9H), 1.46 (s, 9H), 1.64 (bs, 2H), 4.29 (d, J = 7.8 Hz, 1H), 4.40 (d, J = 7.8 Hz, 1H), 7.06 (d, J = 2.4 Hz, 1H), 7.11-7.20 (m, 10H), 7.38 (d, J = 2.4 Hz, 1H), 8.45 (s, 1H), 13.59 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 29.4, 31.4, 34.0, 34.9, 62.1, 82.2, 117.8, 126.4, 127.2, 127.3, 127.4, 127.7, 127.8, 128.1, 128.2, 136.6, 140.3, 140.6, 141.8, 158.0, 167.2. 6,6'-((1E,1'E)-(((1R,2R)-1,2-Diphenylethan-1,2-diyl)-bis-(azanylyliden))-bis-(methanylyliden))-bis(2,4-di-tert-butylphenol) (47): Der Salenligand 47 wurde als Nebenprodukt in der Synthese von Ligand 53 erzeugt (siehe Vorschrift Ligand 53).[88a] Ligand 47 wurde als gelber Feststoff erhalten (0.51 g, 0.79 mmol, 34%). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[119] 1H-NMR (300 MHz, CDCl3): δ = 1.21 (s, 18H), 1.41 (s, 18H), 4.72 (s, 2H), 6.98 (d, J = 2.4 Hz, 2H), 7.16-7.19 (m, 10H), 7.30 (d, J = 2.4 Hz, 2H), 8.40 (s, 2H), 13.6 (s, 2H); 13C-NMR (75 MHz, CDCl3): δ = 29.4, 31.4, 34.0, 34.9, 80.0, 117.8, 126.3, 127.1, 127.3, 127.9, 128.2, 136.3, 139.7, 139.9, 157.9, 167.2; MS (MALDI-TOF): m/z = 645 [M+H]+. 134 Kapitel 5 Experimenteller Teil 2-((((1R,2R)-2-Amino-1,2-diphenylethyl)amino)methyl)-4,6-di-tert-butylphenol (54):[92] Der Schiffsche-Base Ligand 53 (0.20 g, 0.46 mmol) wurde unter Stickstoffatmosphäre in absolutem MeOH (5 mL) gelöst und die Lösung anschließend auf 0 °C gekühlt. NaBH4 (71.0 mg, 1.87 mmol) wurde in kleinen Portionen zugegeben und die Reaktionsmischung über Nacht bei RT gerührt. Die Reaktionslösung wurde mit H2O (10 mL) versetzt und das Produkt mit DCM (3 x 20 mL) extrahiert. Die vereinigten organischen Phasen wurden über MgSO4 getrocknet. Anschließend wurde das Lösungsmittel am Rotationsverdampfer vollständig entfernt und das Produkt ohne weitere Reinigung als weißer Feststoff erhalten (0.15 g, 0.38 mmol, 76%). Die NMR Spektren stimmen mit den in der Literatur berichteten überein.[92] 1H-NMR (300 MHz, CDCl3): δ = 1.22 (s, 9H), 1.39 (s, 9H), 3.53-3.62 (m, 1H), 3.74-3.80 (m, 2H), 4.04-4.14 (m, 1H), 6.61-6.68 (m, 1H), 6.91-7.28 (m, 12H); 13C-NMR (100 MHz, CDCl3): δ = 29.6, 31.6, 34.0, 34.8, 51.3, 61.0, 68.9, 122.5, 122.9, 123.4, 127.0, 127.3, 127.5, 127.9, 128.3, 128.4, 135.9, 139.5, 140.5, 142.9, 154.5. 2-((E)-(((1R,2R)-2-Amino-1,2-diphenylethyl)imino)methyl)-4,6-di-tert-butylphenol Hydrochlorid (50): (1R,2R)‐1,2‐Diphenylethylendiamin Hydrochlorid[89] (63) (0.35 g, 1.40 mmol) wurde in einer Mischung von absolutem MeOH/EtOH (9 mL/9 mL) gelöst. Nach der Zugabe von 3,5-Di- tert-butylsalicylaldehyd (64) (0.33 g, 1.40 mmol) wurde die Reaktionslösung für 24 h gerührt. Das Lösungsmittel wurde am Rotationsverdampfer auf ca. 1 mL eingeengt. Der 135 Kapitel 5 Experimenteller Teil erhaltene Feststoff wurde abfiltriert und mit EtOH gewaschen. Anschließend wurde das Filtrat am Rotationsverdampfer vollständig eingeengt und der resultierende Niederschlag mit Et 2O gewaschen.[89] Das Produkt wurde als schwach gelber Feststoff erhalten (0.57 g, 1.22 mmol, 87%). 1H-NMR (300 MHz, MeOH-d4): δ = 1.30 (s, 9H), 1.46 (s, 9H), 4.87 (s, 1H), 4.92 (d, J = 9.9 Hz, 1H), 7.16-7.27 (m, 6H), 7.32 (s, 5H), 7.45 (d, J = 2.4 Hz, 1H), 8.71 (s, 1H); MS (MALDI-TOF): m/z = 429 [M-Cl]+. 2,4-Di-tert-butyl-6-((E)-(((1R,2S)-2-hydroxy-1,2-diphenylethyl)imino)methyl)phenol (43):[120] In einem Rundhalskolben wurde (1S,2R)-(+)-2-Amino-1,2- diphenylethanol (0.40 g, 1.87 mmol) in absolutem MeOH (10 mL) gelöst und mit Na2SO4 (1.00 g, 7.48 mmol) versetzt. Anschließend erfolgte die tropfenweise Zugabe von 3,5-Di-tertbutylsalicylaldehyd (64) (0.44 g, 1.87 mmol) in absolutem MeOH (2 mL). Die Reaktionslösung wurde bei RT für 3 d gerührt. Das Lösungsmittel wurde am Rotationsverdampfer vollständig entfernt und das Rohprodukt säulenchromatographisch (SiO2, PE/EtOAc 3:1) gereinigt, wonach das Produkt als gelber Feststoff erhalten wurde (0.70 g, 1.62 mmol, 87%). Die NMR Spektren stimmen mit den in der Literatur berichteten überein.[121] 1H-NMR (400 MHz, CDCl3): δ = 1.28 (s, 9H), 1.47 (s, 9H), 2.08 (d, J = 3.0 Hz, 1H), 4.54 (d, J = 6.8 Hz, 1H), 5.10 (dd, J = 6.8 Hz, 3.0 Hz, 1H), 6.96 (d, J = 2.4 Hz, 1H), 7.26-7.40 (m, 12H), 8.17 (s, 1H), 13.43 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 29.3, 31.3, 33.9, 34.9, 78.3, 80.0, 117.8, 126.3, 127.2, 127.9, 127.9, 128.1, 128.1, 128.7, 136.6, 139.6, 140.1, 140.2, 157.9, 167.1; MS (MALDITOF): m/z = 431 [M+H]+. 136 Kapitel 5 Experimenteller Teil 2,4-Di-tert-butyl-6-((((1R,2S)-2-hydroxy-1,2-diphenylethyl)amino)methyl)phenol (46):[122] Der Schiffsche-Base Ligand 43 (0.30 g, 0.70 mmol) wurde unter Stickstoffatmosphäre in absolutem THF (6 mL) gelöst und die Lösung anschließend auf 0 °C gekühlt. LiBH 4 (61.0 mg, 2.79 mmol) wurde in kleinen Portionen zugegeben und die Reaktionsmischung über Nacht bei RT gerührt. Die Reaktion wurde durch Zugabe von H2O gequencht. Anschließend wurde die Lösung mit 1 M HCl auf pH 1 eingestellt, mit einer gesättigten NaHCO3-Lösung neutralisiert und mit DCM (4 x 25 mL) extrahiert. Die vereinigten organischen Phasen wurden mit H2O gewaschen und anschließend über MgSO 4 getrocknet. Das Lösungsmittel wurde vollständig am Rotationsverdampfer entfernt, woraufhin das Produkt als weißer Feststoff erhalten wurde (0.21 g, 0.49 mmol, 71%). Die NMR Spektren stimmen mit den in der Literatur berichteten überein.[122] 1H-NMR (300 MHz, CDCl3): δ = 1.23 (s, 9H), 1.42 (s, 9H), 3.56 (d, J = 13.3 Hz, 1H), 3.78 (d, J = 13.3 Hz, 1H), 3.89 (d, J = 6.2 Hz, 1H), 4.91 (d, J = 6.2 Hz, 1H), 6.60 (s, 1H), 7.18-7.35 (m, 11H); 13C-NMR (100 MHz, CDCl3): δ = 29.6, 31.6, 34.0, 34.9, 51.1, 68.2, 121.9, 122.9, 123.3, 126.8, 128.1, 128.2, 128.4, 128.5, 128.6, 135.9, 137.8, 140.5, 154.3. 2-((E)-(((1R,2S)-2-Hydroxy-1,2-diphenylethyl)imino)methyl)-4,6-diiodophenol (44):[26c] In einem Rundhalskolben wurde (1S,2R)-(+)-2-Amino-1,2-diphenylethanol (0.20 g, 0.94 mmol) in absolutem MeOH (3 mL) gelöst und mit MgSO4 (0.56 g, 4.70 mmol) versetzt. Anschließend erfolgte die tropfenweise Zugabe von 3,5-Diiodosalicylaldehyd (0.35 g, 0.94 mmol) in absolutem MeOH (14 mL). Die Reaktionslösung 137 Kapitel 5 Experimenteller Teil wurde für 3 d gerührt. Das MgSO4 wurde abfiltriert, das Lösungsmittel am Rotationsverdampfer vollständig entfernt und das Rohprodukt anschließend säulenchromatographisch (SiO2, PE/EtOAc 3:1) gereinigt. Das Produkt wurde als gelber Feststoff erhalten (0.30 g, 0.53 mmol, 56%). Die NMR Spektren stimmen mit den in der Literatur berichteten überein.[26c] 1H-NMR (300 MHz, CDCl3): δ = 2.04 (bs, 1H), 4.48 (d, J = 7.3 Hz, 1H), 5.00 (d, J = 7.3 Hz, 1H), 7.19-7.39 (m, 11H), 7.77 (s, 1H), 7.98 (d, J = 2.0 Hz, 1H), 14.44 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 78.0, 79.3, 79.7, 87.3, 119.8, 127.0, 127.9, 128.3, 128.4, 128.9, 138.4, 139.7, 139.9, 148.7, 160.4, 163.6; MS (FAB): m/z = 570 [M+H]+. (1S,2R)-1,2-Diphenyl-2-((E)-(2,3,5-trifluorobenzyliden)amino)ethanol (45): In einem Rundkolben wurde 2,3,5-Trifluorobenzaldehyd (0.13 g, 0.81 mmol) in absolutem MeOH (5 mL) gelöst und mit MgSO4 (0.39 g, 3.20 mmol) versetzt. Anschließend erfolgten die portionsweisen Zugaben von (1S,2R)-2-Amino-1,2-diphenylethanol (0.17 g, 0.81 mmol) und absolutem MeOH (5 mL). Die Reaktionsmischung wurde für 3 d bei RT gerührt. Das Lösungsmittel wurde am Rotationsverdampfer entfernt, der Rückstand in DCM aufgenommen und das MgSO 4 abfiltriert. Das Filtrat wurde am Rotationsverdampfer eingeengt und das Rohprodukt säulenchromatographisch gereinigt (SiO2, PE/EtOAc/TEA 8:4:0.1) gereinigt. Das Produkt wurde als weißer Feststoff erhalten (0.12 g, 0.34 mmol, 42%). [α]D20 = -41° (c = 0.15, CHCl3); 1H-NMR (300 MHz, CDCl3): δ = 2.27 (d, J = 2.6 Hz, 1H), 4.49 (d, J = 6.8 Hz, 1H), 5.03 (dd, J = 6.7 Hz, 2.5 Hz, 1H), 6.92-6.95 (m, 1H), 7.22-7.67 (m, 11H), 8.23 (d, J = 2.2 Hz, 1H); 13C-NMR (100 MHz, CDCl3): δ = 61.6, 78.2, 107.3, 107.5, 107.6, 107.8, 108.3, 108.5, 127.1, 127.9, 128.0, 128.1, 128.2, 128.5, 139.7, 140.3, 152.9; 138 19F-NMR (282 MHz, CDCl3): δ = Kapitel 5 Experimenteller Teil -114.21, -133.16, -150.87; MS (FAB): m/z = 355 [M+H]+; EA: Gemessen: C, 70.94; H, 4.78; N: 3.88; Berechnet für C21H16F3NO: C, 70.98; H, 4.54; O, 4.50; N, 3.93; Smp. = 90 °C; IR (dünner Film): ṽ = 3368, 3061, 3058, 2900, 2882, 2360, 1736, 1648, 1600, 1489, 1450, 1377, 1344, 1211, 1122, 1050, 1038, 1025, 993, 851, 800, 756, 778, 696, 605, 598, 532 cm-1. (E)-2-(((2'-Amino-[1,1'-binaphthalen]-2-yl)imino)methyl)-4,6-di-tert-butylphenol (55):[123] In einem Rundkolben wurde R-(+)-2,2`-Diamino-1,1binaphthalen (0.20 g, 0.70 mmol) in einer Mischung aus absolutem MeOH/Toluol (4 mL/4 mL) gelöst und mit MgSO4 (0.42 g, 3.50 mmol) versetzt. Anschließend erfolgte die tropfenweise Zugabe von 3,5-Di-tert- butylsalicylaldehyd (64) (0.16 g, 0.70 mmol) in absolutem DCM (4 mL). Die Reaktionsmischung wurde für 3 d bei RT gerührt. Das MgSO4 wurde abfiltriert und das Lösungsmittel am Rotationsverdampfer vollständig entfernt. Das Rohprodukt wurde säulenchromatographisch (SiO2, PE/EtOAc/TEA 8:2:0.1) gereinigt, woraufhin das Produkt als oranger Feststoff erhalten wurde (92.0 mg, 0.18 mmol, 26%). Das 1HNMR Spektrum stimmt mit dem in der Literatur berichteten überein.[123] 1H-NMR (300 MHz, CDCl3): δ = 1.23 (s, 9H), 1.24 (s, 9H), 3.60 (s, 2H), 6.88-6.91 (m, 1H), 7.02 (d, J = 2.4 Hz, 1H), 7.08-7.19 (m, 3H), 7.27 (d, J = 2.4 Hz, 1H), 7.30-7.35 (m, 1H), 7.43-7.49 (m, 2H), 7.63 (d, J = 8.8 Hz, 1H), 7.73-7.78 (m, 2H), 7.94 (d, J = 8.1 Hz, 1H), 8.03 (d, J = 8.6 Hz, 1H), 8.64 (s, 1H), 12.75 (s, 1H); MS (MALDI-TOF): m/z = 502 [M+H]+. 139 Kapitel 5 Experimenteller Teil 2-((E)-(((1R,2R)-2-Amino-1,2-diphenylethyl)imino)methyl)-4-tert-butyl-6-trityl-phenol (61): (1R,2R)-(+)-1,2-Diphenylethylendiamin (62) (0.15 g, 0.71 mmol) wurde unter Stickstoffatmosphäre in absolutem DCM (8 mL) gelöst. Anschließend wurde Molekularsieb (3Å, 1.70 g) zugegeben und eine Lösung von 5-tert-Butyl-2-hydroxy-3-tritylbenzaldehyd[124] (0.29 g, 0.69 mmol) in absolutem DCM (7 mL) über einen Zeitraum von 10 Min zugetropft. Die Reaktionsmischung wurde für 3h gerührt und anschließend am Rotationsverdampfer (Wasserbadtemperatur 30 °C) auf ca. 1 mL eingeengt. Das Rohprodukt wurde mittels Flash-Säulenchromatographie (SiO2, PE/EtOAc 7:3, 1:1) gereinigt. Das Produkt wurde als gelber Feststoff erhalten (0.19 g, 0.30 mmol, 42%). [α]D20 = +74° (c = 0.10, THF); 1H-NMR (300 MHz, CDCl3): δ = 1.11 (s, 9H), 1.85 (bs, 2H), 4.21 (s, 2H), 6.94-7.21 (m, 26H), 7.29 (d, J = 2.4 Hz, 1H), 8.34 (s, 1H), 13.10 (bs, 1H); 13C-NMR (75 MHz, CDCl3): δ = 31.2, 34.0, 62.0, 63.4, 82.0, 118.1, 125.5, 127.0, 127.1, 127.2, 127.4, 127.5, 127.6, 127.8, 127.9, 128.0, 131.0, 132.0, 134.0, 140.0, 140.3, 141.6, 145.5, 157.5, 166.7; HRMS (ESI): Gemessen: m/z = 615.3381; Berechnet für [C44H42N2O+H]+ m/z = 615.3369; EA: Gemessen: C, 85.22; H, 6.99; N, 4.39; Berechnet für C44H42N2O: C, 85.96; H, 6.89; N, 4.56; IR (dünner Fim): ṽ = 3726, 3707, 3626, 3600, 3055, 3026, 2957, 2864, 2360, 2340, 1624, 1595, 1490, 1444, 1392, 1361, 1265, 1213, 1184, 1155, 1027, 881, 826, 771, 746, 686, 658, 647, 557, 515 cm -1. 140 Kapitel 5 Experimenteller Teil 2-((E)-(((1R,2R)-2-Amino-1,2-diphenylethyl)imino)methyl)-6-tert-butyl-4-trityl-phenol (60): (1R,2R)-(+)-1,2-Diphenylethylendiamin (62) (0.20 g, 0.94 mmol) wurde unter Stickstoffatmosphäre in absolutem DCM (10 mL) gelöst. Anschließend wurde Molekularsieb (3Å, 2.20 g) zugegeben und eine Lösung von 3-tert-Butyl-2-hydroxy-5tritylbenzaldehyd[124] (0.40 g, 0.94 mmol) in absolutem DCM (10 mL) über einen Zeitraum von 30 Min zugetropft. Die Reaktionsmischung wurde für 3 h gerührt und anschließend am Rotationsverdampfer (Wasserbadtemperatur 30 °C) auf ca. 1 mL eingeengt. Das Rohprodukt wurde zügig mittels Flash-Säulenchromatographie (SiO2, PE/EtOAc 7:3) gereinigt. Das Produkt wurde als gelber Feststoff erhalten (0.21 g, 0.35 mmol, 37%). [α]D20 = +36° (c = 0.10, THF); 1H-NMR (300 MHz, CDCl3): δ = 1.26 (s, 9H), 1.95 (bs, 2H), 4.19 (d, J = 8.2 Hz, 1H), 4.37 (d, J = 8.2 Hz, 1H), 6.89 (d, J = 2.4 Hz, 1H), 7.04-7.23 (m, 26H), 8.27 (s, 1H), 13.75 (s, 1H); 13C-NMR (75 MHz, CDCl3): δ = 29.3, 34.9, 62.1, 64.3, 117.3, 125.9, 127.3, 127.4, 127.6, 128.0, 128.1, 131.0, 131.9, 133.8, 136.0, 136.1, 146.8, 158.4; HRMS (ESI): Gemessen: m/z = 615.3368; Berechnet für [C44H42N2O+H]+ m/z = 615.3369; EA: Gemessen: C, 86.40; H, 7.14; N, 4.41; Berechnet für C44H42N2O: C, 85.96; H, 6.89; N, 4.56; IR (dünner Film): ṽ = 3727, 3672, 3626, 3055, 3027, 2954, 2866, 2360, 2340, 1622, 1593, 1490, 1439, 1390, 1359, 1275, 1201, 1182, 1156, 1140, 1084, 1028, 873, 832, 803, 749, 698, 652, 636, 555, 521 cm -1. 141 Kapitel 5 Experimenteller Teil 2,4-Di-tert-butyl-6-((E)-(((1R,2R)-2-((E)-((6-methylpyridin-2-yl)methylen)amino)-1,2-diphenylethyl)imino)methyl)phenol (48): In einem Rundhalskolben wurde Imin 53 (0.10 g, 0.23 mmol) in absolutem DCM (5 mL) gelöst und mit Molekularsieb (3Å, 1.50 g), sowie mit 6-Methyl- pyridinaldehyd (0.03 g, 0.23 mmol) versetzt. Nachdem die Reaktionsmischung für 5 h rührte, wurde das Lösungsmittel am Rotationsverdampfer entfernt und das Produkt anschließend säulenchromatographisch (SiO2, PE/EtOAc 6:4, DCM/MeOH 9:4) gereinigt. Das Produkt wurde als gelber Feststoff erhalten (30.0 mg, 0.06 mmol, 25%). 1H-NMR (300 MHz, CDCl3): δ = 1.20 (s, 9H), 1.39 (s, 9H), 2.48 (s, 3H), 4.77 (q, J = 8.0 Hz, 2H), 6.92 (s, 1H), 7.07-7.22 (m, 10H), 7.27 (s, 2H), 7.55 (t, J = 7.7 Hz, 1H), 7.87 (d, J = 7.9 Hz, 1H), 8.34 (d, J = 6.8 Hz, 2H), 13.75 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 24.2, 29.4, 31.4, 34.0, 34.9, 79.6, 81.2, 117.8, 118.4, 124.2, 126.0, 126.8, 127.2, 127.3, 128.0, 128.2, 128.3, 128.9, 136.5, 139.7, 140.0, 140.5, 153.9, 157.8, 158.1, 163.3, 166.7; MS (FAB): m/z = 533 [M+H]+; HRMS (EI): Gemessen: m/z = 531.3239; Berechnet für [C36H41N3O]+ m/z = 531.3250. (S)-N-((1R,2R)-2-((E)-(3,5-Di-tert-butyl-2-hydroxybenzyliden)amino)-1,2-diphenyl-ethyl)-1-formylpyrrolidin-2-carboxamid (49): N-Formyl-L-Prolin (34) (67.0 mg, 0.47 mmol) wurde in absolutem THF (3 mL) gelöst, portionsweise mit CDI (76.0 mg, 0.47 mmol) versetzt und für 1 h gerührt. Anschließend wurden Imin 53 (0.20 g, 0.47 mmol) und weiteres THF (2 mL) zugegeben. Die Reaktionslösung 142 Kapitel 5 Experimenteller Teil wurde für 20 h bei RT gerührt. Das Lösungsmittel wurde am Rotationsverdampfer vollständig entfernt und das Rohprodukt säulenchromatographisch gereinigt (SiO 2, PE/EtOAc 7:3, EtOAc/PE 9:1). Das Produkt wurde als hellgelber Feststoff erhalten (0.16 g, 0.29 mmol, 62%). [α]D20 = -13° (c = 0.15, CHCl3); 1H-NMR (300 MHz, CDCl3): δ = 1.26 (s, 9H), 1.43 (s, 9H), 1.55-1.89 (m, 4H), 2.45-2.50 (m, 1H), 2.76-2.83 (m, 1H), 3.13-3.20 (m, 1H), 4.39-4.45 (m, 2H), 5.49 (t, J = 8.9 Hz, 1H), 7.02 (s, 1H), 7.06 (d, J = 5.7 Hz, 2H), 7.08-7.22 (m, 8H), 7.35 (s, 1H), 7.95 (s, 1H), 8.04 (d, J = 9.3 Hz, 1H), 8.30 (s, 1H), 13.29 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 23.9, 26.0, 29.4, 31.5, 34.1, 35.1, 46.2, 57.8, 59.1, 79.8, 117.5, 126.2, 127.2, 127.4, 127.5, 128.3, 128.5, 136.7, 138.7, 139.5, 140.3, 157.9, 162.7, 166.9, 169.0; MS (FAB): m/z = 554 [M+H]+; HRMS (EI): Gemessen: m/z = 553.3327; Berechnet für [C35H43N3O3]+ m/z = 553.3304; Smp.= 95 °C; IR (dünner Film): ṽ = 2953, 1700, 1578, 1638, 1626, 1560, 1543, 1525, 1450, 1439, 1421, 1376, 1362, 1272, 1248, 1201, 1172, 1028, 879, 827, 773, 697, 633, 560, 528, 435 cm -1. (S)-3-((E)-(((1R,2R)-2-Amino-1,2-diphenylethyl)imino)methyl)-2'-phenyl-[1,1'-binaphthalen]-2-ol (56): (1R,2R)-(+)-1,2-Diphenylethylendiamin (62) (86.0 mg, 0.40 mmol) wurde unter Stickstoffatmosphäre in absolutem DCM (8 mL) gelöst und mit Molekularsieb (3Å, 1.70 g) versetzt. Eine Lösung von (S)-2-Hydroxy-2'-phenyl-[1,1'-binaphthalen]-3-carbaldehyd[92] (0.15 g, 0.40 mmol) in absolutem DCM (5 mL) wurde über einen Zeitraum von 30 Min zugetropft. Die Reaktionslösung wurde für 2.5 h gerührt und anschließend am Rotationsverdampfer (Wasserbadtemperatur 30 °C) auf ca. 1 mL eingeengt. Das Rohprodukt wurde zügig mittels FlashSäulenchromatographie (SiO2, PE/EtOAc 1:1) gereinigt.[88a] Das Produkt wurde als grüner Feststoff erhalten (0.14 g, 0.24 mmol, 61%). 143 Kapitel 5 Experimenteller Teil [α]D20 = +18° (c = 0.15, CHCl3); 1H-NMR (300 MHz, CDCl3): δ = 1.70 (bs, 2H), 4.34 (dd, J = 9.9 Hz, 6.5 Hz, 2H), 7.05-7.31 (m, 17H), 7.40-7.49 (m, 2H), 7.65-7.77 (m, 4H), 7.94-8.06 (m, 3H), 8.35 (s, 1H), 12.09 (bs, 1H); 13C-NMR (100 MHz, CDCl3): δ = 61.8 (62.3), 81.3 (81.9), 119.8 (120.1), 123.2 (124.7), 125.7-143.4 (29C), 154.4 (154.6); HRMS (ESI): Gemessen: m/z = 569.25650; Berechnet für [C41H32N2O+H]+ m/z = 569.25874; IR (dünner Film): ṽ = 3728, 3651, 3050, 3025, 2981, 2925, 2853, 2360, 2337, 2020, 1953, 1729, 1625, 1492, 1443, 1382, 1342, 1320, 1248, 1180, 1147, 1117, 1025, 939, 890, 859, 820, 748, 696, 523, 435 cm -1. (R)-3-((E)-(((1S,2S)-2-Amino-1,2-diphenylethyl)imino)methyl)-2'-phenyl-[1,1'-binaphthalen]-2-ol (57): Synthesevorschrift analog Ligand 56. Das Produkt wurde als grüner Feststoff erhalten (0.19 g, 0.32 mmol, 48%). 1H-NMR (300 MHz, CDCl3): δ = 1.60 (bs, 2H), 4.34 (dd, J = 10.2 Hz, 6.3 Hz, 2H), 6.97-7.30 (m, 17H), 7.50-7.52 (m, 2H), 7.64-7.73 (m, 4H), 7.94-8.06 (m, 3H), 8.35 (s, 1H), 12.09 (bs, 1H). 144 Kapitel 5 Experimenteller Teil 3-((((1S,2S)-2-Amino-1,2-diphenylethyl)amino)methyl)-2'-phenyl-[1,1'-binaphthalen]-2-ol (58): Der Schiffsche-Base Ligand 57 (0.17 g, 0.29 mmol) wurde unter Stickstoffatmosphäre in einer Mischung von absolutem THF/MeOH (4 mL/2 mL) gelöst. NaBH4 (78.0 mg, 2.07 mmol) wurde in kleinen Portionen zugegeben und die Reaktionsmischung über Nacht bei RT gerührt. Die Reaktion wurde mit H2O (6 mL) gequencht und das Produkt mit DCM (4 x 15 mL) extrahiert.[125] Die vereinigten organischen Phasen wurden über Na2SO4 getrocknet. Anschließend wurde das Lösungsmittel am Rotationsverdampfer vollständig entfernt. Das Rohprodukt wurde säulenchromatographisch (SiO 2, DCM/MeOH 10:1) und durch Umfällen (DCM/Pentan) gereinigt. Das Produkt wurde als weißer Feststoff erhalten (0.11 g, 0.19 mmol, 65%). [α]D20 = +55° (c = 0.15, DCM); 1H-NMR (300 MHz, CDCl3): δ = 3.27 (bs, 2H), 3.61-3.68 (m, 2H), 3.97-4.06 (m, 2H), 6.94-7.46 (m, 22H), 7.59-7.68 (m, 2H), 7.98 (dd, J = 23.3 Hz, 8.3 Hz, 2H); 13C- NMR (75 MHz, CDCl3): δ = 50.3, 60.7, 68.3, 119.4, 122.9, 124.6, 124.9, 125.7, 126.1, 126.2, 126.4, 126.8, 127.1, 127.2, 127.4, 127.5, 127.7, 127.8, 127.9, 128.0, 128.2, 128.3, 128.4, 128.9, 131.5, 132.9, 133.1, 134.3, 137.9, 140.1, 142.4, 153.1; HRMS (ESI): Gemessen: m/z = 571.2740; Berechnet für [C41H34N2O+H]+ m/z = 571.2744; IR (dünner Film): ṽ = 3053, 3026, 2953, 2851, 2363, 2339, 1627, 1599, 1493, 1453, 1432, 1375, 1353, 1309, 1252, 1208, 1149, 1109, 1071, 1027, 939, 864, 821, 760, 749, 698, 603, 583, 525, 437 cm-1. 145 Kapitel 5 Experimenteller Teil tert-Butyl-((S)-1-(((1R,2R)-2-amino-1,2-diphenylethyl)amino)-3-(1H-imidazol-5-yl)-1-oxopropan-2yl)-carbamat (59): In einem Rundhalskolben wurde Boc-L-His-OH (0.24 g, 0.94 mmol) unter Stickstoffatmosphäre in einer Mischung von absolutem DCM/THF (5 mL/2 mL) gelöst und mit CDI[79] (0.15 g, 0.95 mmol) versetzt. Die Reaktionsmischung wurde solange gerührt, bis keine CO2-Entwicklung mehr zu beobachten war. Nach der langsamen Zugabe von (1R,2R)-(+)-1,2-Diphenylethanylendiamin (62) (0.20 g, 0.94 mmol) wurde die Reaktionslösung über Nacht gerührt. Das Lösungsmittel wurde am Rotationsverdampfer entfernt und das Rohprodukt säulenchromatographisch (SiO2, EtOAc/MeOH 7:1, EtOAc/MeOH 2:1) gereinigt. Das Produkt wurde als weißer Feststoff erhalten (0.14 g, 0.30 mmol, 32%). [α]D20 = -10° (c = 0.15, CHCl3); 1H-NMR (300 MHz, CDCl3): δ = 1.40 (s, 9H), 2.84-2.89 (m, 1H), 3.08-3.14 (m, 1H), 4.30-5.56 (m, 5H), 6.66 (s, 1H), 7.00-7.43 (m, 12H), 7.84 (s, 1H), 8.11 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 28.2, 28.6, 54.3, 58.9, 60.4, 80.3, 120.0, 126.8, 126.9, 127.5, 127.7, 128.4, 128.5, 131.3, 135.4, 139.4, 141.2, 155.8, 171.9; MS (MALDI-TOF): m/z = 450 [M]+; HRMS (ESI): Gemessen: m/z = 450.2499 bzw. m/z = 472.2319; Berechnet für [C25H31N5O3+H]+ m/z = 450.2503 bzw für [C25H31N5O3+Na]+ m/z = 472.2328; IR (dünner Film): ṽ = 3255, 3029, 2975, 2929, 2359, 2336, 1675, 1648, 1493, 1452, 1365, 1299, 1247, 1160, 1045, 1018, 943, 916, 823, 758, 697, 662, 640, 619, 575, 510, 460, 426, 403 cm-1. 146 Kapitel 5 Experimenteller Teil Synthese von primär-aminhaltigen Eisen(III)komplexen 65-67 Allgemeine Arbeitsvorschrift: Eine Lösung von Schiffsche-Base Ligand 53, 60 oder 61 (2.0 Äq., 0.60 mmol) in THF (14 mL) wurde mit FeCl3 x 6 H2O (1.0 Äq., 0.30 mmol) versetzt. Die Reaktionsmischung wurde für 2 h an Luft gerührt und nach anschließendem Entfernen des Lösungsmittels am Rotationsverdampfer wurde der entsprechende Komplex 65, 66 oder 67 als violetter Feststoff erhalten (0.30 mmol, quant.). Komplex 65: HRMS (ESI): Gemessen: m/z = 910.4851; Berechnet für [C58H70FeN4O2-Cl]+ m/z = 910.4844; IR (dünner Film): ṽ = 3061, 3031, 2950, 2902, 2864, 2340, 2359, 1759, 1725, 1600, 1250, 1170, 772, 697 cm-1. Komplex 66: HRMS (ESI): Berechnet für Gemessen: m/z = [C88H82FeN4O2-Cl]+ 1282.576; m/z = 1282.5785; IR (dünner Film): ṽ = 3055, 3028, 2953, 2866, 2359, 2339, 1768, 1721, 1598, 1536, 1491, 1441, 1316, 1163, 1032, 990, 872, 750, 698, 653, 568 cm-1. 147 Kapitel 5 Experimenteller Teil Komplex 67: HRMS (ESI): Gemessen: m/z = 1282.5789; Berechnet für [C88H82FeN4O2-Cl]+ m/z = 1282.5785; IR (dünner Film): ṽ = 3054, 3027, 2957, 2864, 2359, 2340, 1768, 1719, 1599, 1536, 1491, 1442, 1257, 1163, 1030, 697, 573 cm-1. Komplex 68: Eine Lösung von Ligand 58 (94.0 mg, 0.16 mmol) in EtOH (25 mL) wurde mit FeCl2 (10.5 mg, 0.08 mmol) versetzt. Die Reaktionsmischung wurde für 1.5 h an Luft gerührt, anschließend Lösungsmittel vollständig am wurde das Rotationsverdampfer entfernt.[41] Der Rückstand wurde in DCM aufgenommen und nach erneutem Entfernen des Lösungsmittels wurde das Produkt als grün-blauer Feststoff erhalten (98.5 mg, 0.08 mmol, quant.). HRMS (ESI): Gemessen: m/z = 1194.4540; Berechnet für [C82H66FeN4O2-Cl]+ m/z = 1194.4532; IR (dünner Film): ṽ = 3236, 3218, 3051, 3029, 2358, 2341, 1590, 1567, 1493, 1446, 1419, 1352, 1261, 1113, 997, 950, 923, 863, 822, 790, 749, 696, 573, 516 cm -1. 148 Kapitel 5 5.2.4 Experimenteller Teil Katalysereaktionen mit Schiffsche-Base Liganden und deren Derivaten Allgemeine Arbeitsvorschrift 3 (AAV3): Vanadiumkatalysierte Oxidation von Thioanisol In einem Rundhalskolben wurden VO(acac)2 (2 mol%), der jeweilige Ligand (3 mol%) und CHCl3 (0.5 mL) vorgelegt. Nach 60-minütigem Rühren der Lösung bei RT wurde diese mit Thioanisol (1.0 Äq., 0.40 mmol) und anschließend 30%igem H2O2 (1.2 Äq.) versetzt. Die Reaktionsmischung wurde bei RT und unter angegebener Reaktionszeit gerührt. Das Rohprodukt wurde säulenchromatographisch gereinigt (SiO2, PE/EtOAc 1:4) und der Enantiomerenüberschuss mittels HPLC-Analyse (OD-Säule, Hexan/i-PrOH 93:7, Fluss 1.0 mL/Min, 30-32 bar, 25 °C, t(R-Enantiomer) = 18.0 Min, t(S-Enantiomer) = 22.4 Min) bestimmt. Zur Identifikation des Produkts wurde das 1H-NMR Spektrum mit literaturbekannten Daten[111] verglichen. Allgemeine Arbeitsvorschrift 4 (AAV4): Eisenkatalysierte Oxidation von Thioanisol unter der Verwendung verschiedener Liganden – Zugabe von H2O2 in einer Portion In einem Rundhalskolben wurden FeXn (2 mol%), der jeweilge Ligand (4 mol%) und THF (c(Substrat) = 0.8 mol/L) vorgelegt. Nach 30-minütigem Rühren der Lösung bei RT wurde sie zuerst mit Thioanisol (1.0 Äq., 0.40 mmol) und anschließend mit 30%igem H2O2 (1.2 Äq.) versetzt und unter der angegebenen Temperatur und Zeit gerührt. Das Rohprodukt wurde säulenchromatographisch gereinigt (SiO2, PE/EtOAc 1:4) und der Enantiomerenüberschuss mittels HPLC-Analyse (OD-Säule, Hexan/i-PrOH 93:7, Fluss 1.0 mL/Min, 30-32 bar, 25 °C, t(R-Enantiomer) = 18.0 Min, t(S-Enantiomer) = 22.4 Min) bestimmt. Zur Identifikation des Produkts wurde das 1H-NMR Spektrum mit literaturbekannten Daten[111] verglichen. 149 Kapitel 5 Experimenteller Teil Allgemeine Arbeitsvorschrift 5 (AAV5): Eisenkatalysierte Oxidation von Thioanisol unter der Verwendung verschiedener Liganden – langsame Zugabe von H2O2 In einem Rundhalskolben wurden FeCl3 x 6 H2O (2 mol%), der jeweilige Ligand (4 mol%) und THF (0.5 mL) vorgelegt. Nach 30-minütigem Rühren der Lösung bei RT wurden zuerst Thioanisol (1.0 Äq., 0.40 mmol) und anschließend 30%iges H2O2 (1.2 Äq.) tropfenweise über einen Zeitraum von 5 h zugegeben (via Spritzenpumpe). Das Rohprodukt wurde säulenchromatographisch gereinigt (SiO2, PE/EtOAc 1:4) und der Enantiomerenüberschuss mittels HPLC-Analyse (ODSäule, Hexan/i-PrOH 93:7, Fluss 1.0 mL/Min, 30-32 bar, 25 °C, t(R-Enantiomer) = 18.0 Min, t(S-Enantiomer) = 22.4 Min) bestimmt. Zur Identifikation des Produkts wurde das 1H-NMR Spektrum mit literaturbekannten Daten[111] verglichen. Allgemeine Arbeitsvorschrift 6 (AAV6): Oxidation von Thioanisol unter der Verwendung von Eisen(III)komplexen In einem Rundhalskolben wurden der jeweilige Metallkomplex (2-6 mol%), das Lösungsmittel (c(Substrat) = 0.8 mol/L) und Thioanisol (1.0 Äq., 0.40 mmol) vorgelegt. Die Art der Zugabe von 30%igem H2O2 (1.2 Äq.) und die Reaktionszeit ist dem jeweiligen Experiment in der zugehörigen Tabelle zu entnehmen. Die Reaktion wurde bei RT durchgeführt. Das Rohprodukt wurde säulenchromatographisch gereinigt (SiO2, PE/EtOAc 1:4) und der Enantiomerenüberschuss mittels HPLC-Analyse (OD-Säule, Hexan/i-PrOH 93:7, Fluss 1.0 mL/Min, 30-32 bar, 25 °C, t(R-Enantiomer) = 18.0 Min, t(S-Enantiomer) = 22.4 Min) bestimmt. Zur Identifikation des Produkts wurde das 1H-NMR Spektrum mit literaturbekannten Daten[111] verglichen. 150 Kapitel 5 Experimenteller Teil Allgemeine Arbeitsvorschrift 7 (AAV7): Oxidation von Thioanisol unter der Verwendung von CoKatalysatoren In einem Rundhalskolben wurden FeCl3 x 6 H2O (2 mol%), der jeweilige Ligand (4 mol%) und das Lösungsmittel vorgelegt. Nach 30-minütigem Rühren der Lösung bei RT wurde diese mit dem jeweiligen Co-Katalysator (1 mol%), dann Thioanisol (1.0 Äq., 0.40 mmol) und anschließend 30%igem H2O2 (1.2 Äq.) versetzt. Die Art der Zugabe von H2O2, die Temperatur, die Reaktionszeit und die Substratkonzentration ist dem jeweiligen Experiment in der zugehörigen Tabelle zu entnehmen. Das Rohprodukt wurde säulenchromatographisch gereinigt (SiO 2, PE/EtOAc 1:4) und der Enantiomerenüberschuss mittels HPLC-Analyse (OD-Säule, Hexan/i-PrOH 93:7, Fluss 1.0 mL/Min, 30-32 bar, 25 °C, t(R-Enantiomer) = 18.0 Min, t(S-Enantiomer) = 22.4 Min) bestimmt. Zur Identifikation des Produkts wurde das 1H-NMR Spektrum mit literaturbekannten Daten[111] verglichen. 5.2.5 Synthese von Aminosäure- bzw. Peptidbausteinen und Hybridverbindungen H-L-Thr-OMe (88): Unter Stickstoffatmosphäre wurde Thionylchlorid (1.90 mL, 25.2 mmol) zu auf 0 °C gekühltem absolutem MeOH (27 mL) zugetropft. Die Reaktionsmischung wurde für 30 Min bei 0 °C gerührt und mit L-Threonin (90) (3.00 g, 25.2 mmol) versetzt. Nachdem die Lösung für 24 h bei RT rührte, wurde das Lösungsmittel am Rotationsverdampfer entfernt, der Rückstand in MeOH gelöst und das Lösungsmittel erneut entfernt. Das Produkt wurde als farbloses hochviskoses Öl erhalten (4.22 g, 24.8 mmol, 99%).[98a] Die NMR Spektren stimmen mit den in der Literatur berichteten Spektren überein.[126] (1H-NMR (300 MHz, MeOH-d4): δ = 1.31 (d, J = 6.6 Hz, 151 Kapitel 5 Experimenteller Teil 3H), 3.84 (s, 3H), 3.92 (d, J = 4.2 Hz, 1H), 4.23-4.31 (m, 1H); 13C-NMR (100 MHz, MeOH-d4 ): δ = 20.5, 53.7, 59.8, 66.3, 169.6.) H-L-Thr-OMe Hydrochlorid (2.26 g, 13.3 mmol) wurde in wenig H2O gelöst und die Lösung anschließend mit K2CO3 (20%) auf pH 10 eingestellt. Die wässrige Phase wurde mit DCM (13 x 10 mL) und EtOAc (2 x 10 mL) extrahiert. Die vereinigten organischen Phasen wurden über Na2SO4 getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Der Rückstand wurde in Et2O aufgenommen und nach erneutem Entfernen des Lösungsmittels wurde das Produkt als farbloses Öl erhalten (0.63 g, 4.76 mmol, 36%).[97] Die NMR Spektren stimmen mit den in der Literatur berichteten überein.[127] 1H-NMR (300 MHz, CDCl3): δ = 1.18 (d, J = 6.3 Hz, 3H), 2.15 (bs, 3H), 3.23 (d, J = 5.3 Hz, 1H), 3.71 (s, 3H), 3.80-3.88 (m, 1H); 13C-NMR (100 MHz, CDCl3 ): δ = 19.7, 52.1, 59.8, 68.2, 174.6. H-L-Ser-OMe (89): Unter Stickstoffatmosphäre wurde Thionylchlorid (0.75 mL, 10.3 mmol) zu auf 0 °C gekühltem absolutem MeOH (10 mL) zugetropft. Die Reaktionsmischung wurde für 30 Min bei 0 °C gerührt und anschließend mit L-Serin (1.00 g, 9.51 mmol) versetzt. Nachdem die Lösung für 24 h bei RT rührte, wurde das Lösungsmittel am Rotationsverdampfer entfernt, der Rückstand in MeOH aufgenommen und das Lösungsmittel anschließend erneut entfernt. Nach Umfällen (MeOH/Ether) wurde das Produkt in Form eines weißen Feststoffs erhalten (1.40 g, 9.38 mmol, 99%).[98b] Die NMR Spektren stimmen mit den in der Literatur berichteten überein.[128] (1H-NMR (300 MHz, MeOH-d4): δ = 3.84 (s, 3H), 3.90 (dd, J = 11.8 Hz, 3.5 Hz, 1H), 4.00 (dd, J = 11.8 Hz, 4.4 Hz, 1H), 4.13-4.15 (m, 1H); 13C-NMR 169.4.) 152 (100 MHz, MeOH-d4 ): δ = 53.7, 56.1, 60.7, Kapitel 5 Experimenteller Teil H-L-Ser-OMe Hydrochlorid (0.61 g, 3.90 mmol) wurde in MeOH (13 mL) gelöst und die Lösung portionsweise mit Amberlyst(OH)-26 versetzt (Ionentauscher wurde vorher mit MeOH (3 x 10 mL) gewaschen). Nachdem die Lösung einen pH von 9-10 erreicht hatte, wurde der Ionentauscher abfiltriert und mit MeOH gewaschen. Das Rohprodukt wurde säulenchromatographisch (SiO 2, CHCl3/MeOH 9:1) gereinigt. Das Produkt wurde als farbloses Öl erhalten (0.27 g, 2.28 mmol, 58%). 1H-NMR (300 MHz, MeOH-d4): δ = 3.49 (t, J = 4.4 Hz, 1H), 3.70 (dd, J = 10.9 Hz, 4.1 Hz, 1H), 3.73 (s, 3H), 3.78 (dd, J = 10.9 Hz, 4.7 Hz, 1H); 13C-NMR (100 MHz, MeOH-d4): δ = 57.2, 64.8, 64.9, 175.3. H-D-Phe-OMe Hydrochlorid (95):[129] In absolutem MeOH (6 mL) wurde H-D-Phe-OH (2.00 g, 12.1 mmol) unter Stickstoffatmosphäre suspendiert. Die Suspension wurde auf 0 °C gekühlt, mit Thionylchlorid (0.88 mL, 12.1 mmol) versetzt, für 15 Min bei 0 °C gerührt und anschließend für 2 h refluxiert. Das Lösungsmittel wurde am Rotationsverdampfer entfernt und das Produkt als weißer Feststoff erhalten (2.60 g, 12.1 mmol, quant.).[78a] Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[129] 1H-NMR (300 MHz, MeOH-d4): δ = 3.16 (dd, J = 14.3 Hz, 7.3 Hz, 1H), 3.25 (dd, J = 14.4 Hz, 6.2 Hz, 1H), 3.79 (s, 3H), 4.32 (dd, J = 7.3 Hz, 6.2 Hz, 1H), 7.24-7.40 (m, 5H). 153 Kapitel 5 Experimenteller Teil Cbz-L-Thr-OH (92):[99] L-Threonin (90) (1.50 g, 12.6 mmol) wurde in H2O (130 mL) gelöst und mit NaHCO3 (2.60 g, 31.5 mmol) versetzt. Benzylchlorformiat (2.00 mL, 13.9 mmol) wurde innerhalb von 15 Min zugetropft und die Reaktionslösung für 4 h gerührt. Die Lösung wurde mit Et2O (3 x 50 mL) gewaschen und anschließend mit konzentrierter HCl auf pH 2 eingestellt. Die wässrige Phase wurde mit Et2O (3 x 100 mL) extrahiert, die vereinigten organischen Phasen mit gesättigter NaCl-Lösung gewaschen und anschließend über Na2SO4 getrocknet. Das Lösungsmittel wurde am Rotationsverdampfer vollständig entfernt und das Produkt als viskoses Öl erhalten (3.00 g, 12.0 mmol, 95%). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[99] 1H-NMR (300 MHz, CDCl3): δ = 1.15 (d, J = 6.5 Hz, 3H), 4.16-4.37 (m, 2H), 5.04 (s, 2H), 6.02 (d, J = 9.0 Hz, 1H), 6.38 (d, J = 9.0 Hz, 1H), 7.05 (bs, 1H), 7.25-7.31 (m, 5H). Cbz-L-allo-Thr-OH (93):[99] Synthese analog Cbz-L-Thr-OH (92). Das Produkt wurde als viskoses Öl erhalten (0.54 g, 2.12 mmol, 84%). 1H-NMR (400 MHz, CDCl3): δ = 1.23 (d, J = 6.5 Hz, 3H), 4.13-4.20 (m, 1H), 4.36-4.39 (m, 1H), 5.07 (s, 2H), 5.84 (d, J = 8.0 Hz, 1H), 7.26-7.34 (m, 5H). 154 Kapitel 5 Experimenteller Teil Cbz-L-Thr-D-Phe-OMe (96): H-D-Phe-OMe Hydrochlorid (95) (0.66 g, 3.07 mmol) wurde unter Stickstoffatmosphäre in absolutem THF (6 mL) suspendiert, mit TEA (0.43 mL, 3.07 mmol) versetzt und für 1 h gerührt. Cbz-L-Thr-OH (92) (0.78 g, 3.07 mmol) wurde in absolutem THF gelöst, die Lösung auf 0 °C abgekühlt und unter starkem Rühren mit TEA (0.43 mL, 3.07 mmol) versetzt. Zu dieser Lösung wurden bei 0 °C Isobutylchlorformiat (0.40 mL, 3.07 mmol) und die Suspension aus H-DPhe-OMe in THF zugetropft. Die Reaktionsmischung wurde für 2 h bei 0 °C und für 16 h bei RT gerührt,[100] anschließend filtriert, mit EtOAc gewaschen und das Lösungsmittel am Rotationsverdampfer entfernt. Das Rohprodukt wurde säulenchromatographisch (SiO 2, PE/EtOAc 2:1, PE/EtOAc 1:9) gereinigt. Das Produkt wurde als weißer Feststoff erhalten (0.52 g, 1.25 mmol, 41%).[130] 1H-NMR (300 MHz, CDCl3): δ = 1.06 (d, J = 6.4 Hz, 3H), 2.92-3.15 (m, 3H), 3.68 (s, 3H), 4.07 (d, J = 7.8 Hz, 1H), 4.23-4.29 (m, 1H), 4.78-4.84 (m, 1H), 5.09 (d, J = 2.4 Hz, 2H), 5.71 (d, J = 7.8 Hz, 1H), 6.87 (d, J = 7.8 Hz, 1H), 7.07-7.09 (m, 2H), 7.20-7.33 (m, 8H); 13C-NMR (100 MHz, CDCl3): δ = 18.3, 37.5, 52.4, 53.1, 58.9, 66.8, 67.2, 127.2, 128.0, 128.2, 128.5, 128.7, 129.0, 135.5, 136.0, 156.7, 170.9, 171.8; MS (MALDI-TOF): m/z = 415 [M+H]+, 437 [M+Na]+, 453 [M+K]+. 155 Kapitel 5 Experimenteller Teil Cbz-L-allo-Thr-D-Phe-OMe (97): Synthesevorschrift analog Cbz-L-Thr-D-Phe-OMe (96). Das Produkt wurde als weißer Feststoff erhalten (0.26 g, 0.63 mmol, 29%). 1H-NMR (300 MHz, CDCl3): δ = 1.11 (d, J = 6.3 Hz, 3H), 2.98-3.18 (m, 2H), 3.29-3.31 (m, 1H), 3.70 (s, 3H), 3.813.86 (m, 1H), 4.04-4.08 (m, 1H), 4.83 (dd, J = 13.2 Hz, 7.5 Hz, 1H), 5.08 (s, 2H), 5.67 (d, J = 7.3 Hz, 1H), 6.76 (d, J = 7.0 Hz, 1H), 7.08 (d, J = 6.2 Hz, 2H), 7.21-7.33 (m, 8H). Boc-L-Val-OH (84):[96] L-Valin (2.00 g, 17.1 mmol) wurde in Dioxan (34 mL) und H2O (17 mL) gelöst und mit einer NaOH-Lösung (1 M, 17.1 mL) versetzt. Die Lösung wurde auf 0 °C gekühlt, mit Di-tert-butyldicarbonat (4.10 g, 18.8 mmol) versetzt und über Nacht bei RT gerührt. Das Dioxan wurde am Rotationsverdampfer entfernt und die wässrige Phase mit PE gewaschen (3 x 20 mL). Anschließend wurde der pH mit KHSO4 (20%) auf 2 eingestellt und die Lösung mit EtOAc (4 x 30 mL) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter NaCl-Lösung (2 x 20 mL) gewaschen und über Na2SO4 getrocknet. Das Lösungsmittel wurde am Rotationsverdampfer vollständig entfernt und das Produkt als hochviskoses farbloses Öl erhalten (3.30 g, 15.2 mmol, 89%). Das 1H-NMR Spektrum stimmt mit dem in der Literatur berichteten überein.[96] 1H-NMR (300 MHz, CDCl3): δ = 0.91 (d, J = 6.9 Hz, 3H), 0.98 (d, J = 6.8 Hz, 3H), 1.42 (s, 9H), 2.15-2.19 (m, 1H), 4.24 (dd, J = 9.0 Hz, 4.5 Hz, 1H), 5.01 (d, J = 8.9 Hz, 1H). 156 Kapitel 5 Experimenteller Teil Boc-L-Thr(OBn)-Gly-OMe (81): Zu einer Lösung von Boc-L-Thr(OBn)-OH (79) (1.00 g, 3.23 mmol) in absolutem DCM (15 mL) wurde unter Stickstoffatmosphäre CDI[79] (0.53 g, 3.26 mmol) zugegeben und die Lösung für 1 h bei RT gerührt. Die Reaktionslösung wurde mit H-Gly-OMe Hydrochlorid (80) (0.41 g, 3.23 mmol) und TEA (0.44 mL, 3.20 mmol) versetzt und über Nacht gerührt. Das Lösungsmittel wurde am Rotationsverdampfer vollständig entfernt und der Rückstand in H2O suspendiert. Die Suspension wurde mit KHSO4 (20%) auf pH 2 eingestellt und mit DCM (5 x 20 mL) extrahiert. Das Lösungsmittel wurde am Rotationsverdampfer entfernt, der Rückstand in EtOAc aufgenommen und anschließend mit K2CO3 (5%, 3 x 20 mL) und H2O (2 x 20 mL) gewaschen. Die vereinigten organischen Phasen wurden über Na2SO4 getrocknet, das Lösungsmittel vollständig entfernt und das Produkt als weißer Feststoff erhalten (1.00 g, 2.63 mmol, 81%). 1H-NMR (300 MHz, MeOH-d4): δ = 1.20 (d, J = 6.3 Hz, 3H), 1.44 (s, 9H), 3.30 (s, 3H), 4.04 (d, J = 6.2 Hz, 2H), 4.06-4.16 (m, 1H), 4.17 (s, 1H), 4.53 (q, J = 11.7 Hz, 2H), 7.23-7.33 (m, 5H); 13C-NMR (100 MHz, MeOH-d4): δ = 16.9, 28.6, 41.9, 52.5, 60.3, 72.4, 76.1, 80.9, 128.6, 129.0, 129.3, 139.7, 157.9, 171.4, 173.6. H-L-Thr(OBn)-Gly-OMe x TFA (82): Eine Lösung 2.47 mmol) von in Boc-L-Thr(OBn)-Gly-OMe absolutem DCM (5 mL) (81) wurde (0.94 bei g, 0 °C tropfenweise mit TFA (3.70 mL, 49.4 mmol) versetzt.[95] Die Reaktionslösung wurde für 6 h bei RT gerührt. Das Lösungsmittel wurde am Rotationsverdampfer vollständig entfernt und das 157 Kapitel 5 Experimenteller Teil Rohprodukt anschließend säulenchromatographisch (SiO2, EtOAc/MeOH 4:1) gereinigt. Das Produkt wurde als hellgelber Feststoff erhalten (0.98 g, 2.48 mmol, quant.). 1H-NMR (300 MHz, D2O): δ = 1.34 (d, J = 5.9 Hz, 3H), 3.70 (s, 3H), 3.96 (s, 2H), 4.00-4.08 (m, 2H), 4.50 (d, J = 11.6 Hz, 1H), 4.68 (d, J = 11.6 Hz, 1H), 7.35-7.40 (m, 5H); 13C-NMR (100 MHz, D2O): δ = 13.4, 39.3, 50.9, 55.5, 69.0, 70.7, 126.5, 126.6, 126.8, 135.1, 166.3, 169.5. Boc-L-Val-L-Thr(OBn)-Gly-OMe (85): Zu einer Lösung von Boc-L-Val-OH (84) (0.60 g, 2.77 mmol) in absolutem DCM (12 mL) wurde unter Stickstoffatmosphäre CDI[79] (0.45 g, 2.79 mmol) zugegeben und die Reaktionslösung für 75 Min gerührt. Die Lösung wurde mit H-L-Thr(OBn)-Gly-OMe x TFA (82) (1.10 g, 2.77 mmol) und TEA (0.38 mL, 2.77 mmol) versetzt und über Nacht gerührt. Das Lösungsmittel wurde vollständig am Rotationsverdampfer entfernt, der Rückstand in H2O suspendiert und die wässrige Phase mit KHSO4 (20%) auf pH 2 eingestellt und mit DCM (4 x 20 mL) extrahiert. Das Lösungsmittel wurde entfernt, der Rückstand in EtOAc aufgenommen und mit K2CO3 (5%, 3 x 20 mL) und H2O (2 x 20 mL) gewaschen. Die vereinigten organischen Phasen wurden über Na2SO4 getrocknet, das Lösungsmittel vollständig am Rotationsverdampfer entfernt und das Produkt als hellgelber Feststoff erhalten (0.91 g, 1.90 mmol, 68%). 1H-NMR (300 MHz, CDCl3): δ = 0.92 (d, J = 6.9 Hz, 3H), 0.99 (d, J = 6.9 Hz, 3H), 1.16 (d, J = 6.4 Hz, 3H), 1.34 (s, 9H), 2.14-2.26 (m, 1H), 3.28 (s, 3H), 3.92-4.02 (m, 3H), 4.26-4.28 (m, 1H), 4.51-4.53 (m, 1H), 4.59 (s, 2H), 4.96 (d, J = 6.0 Hz, 1H), 6.88 (d, J = 7.4 Hz, 1H), 7.26-7.33 (m, 6H); 13C-NMR (100 MHz, CDCl3): δ = 16.2, 17.6, 19.4, 28.2, 30.3, 41.3, 52.2, 56.5, 60.7, 71.7, 73.8, 80.5, 127.4, 127.8, 128.4, 137.9, 156.4, 169.7, 170.1, 171.6. 158 Kapitel 5 Experimenteller Teil H-L-Val-L-Thr(OBn)-Gly-OMe x TFA (86): Eine Lösung von Boc-L-Val-L-Thr(OBn)-Gly-OMe (85) (0.88 g, 1.83 mmol) in absolutem DCM (5 mL) wurde bei 0 °C tropfenweise mit TFA (2.70 mL, 36.6 mmol) versetzt.[95] Die Reaktionslösung wurde für 6 h gerührt, anschließend wurde das Lösungsmittel am Rotationsverdampfer vollständig entfernt und das Rohprodukt säulenchromatographisch (SiO2, DCM/MeOH 9:1) gereinigt. Das Produkt wurde als weißer, kristalliner Feststoff erhalten (1.10 g, 2.20 mmol, quant.). 1H-NMR (300 MHz, MeOH-d4): δ = 1.02 (d, J = 6.9 Hz, 3H), 1.07 (d, J = 6.9 Hz, 3H), 1.26 (d, J = 6.3 Hz, 3H), 2.19-2.29 (m, 1H), 3.69 (s, 3H), 3.83-3.86 (m, 1H), 3.90-3.93 (m, 1H), 3.94-4.04 (m, 2H), 4.52-4.65 (m, 3H), 7.22-7.37 (m, 5H); 13C-NMR (100 MHz, MeOH-d4): δ = 16.6, 17.6, 18.9, 31.6, 41.9, 52.6, 58.6, 59.5, 72.3, 76.0, 128.8, 129.1, 129.3, 139.6, 169.9, 171.4, 171.9. H-L-Val-L-Thr(OBn)-Gly-OMe (87): H-L-Val-L-Thr(OBn)-Gly-OMe x TFA (86) (0.20 g, 0.41 mmol) wurde in H2O (5 mL) gelöst und die Lösung mit K2CO3 (20%) auf pH 8-10 eingestellt. Die Lösung wurde mit DCM (5 x 20 mL) extrahiert, die vereinigten organischen Phasen mit H2O (2 x 30 mL) gewaschen und über Na2SO4 getrocknet.[97] Das Lösungsmittel wurde vollständig am Rotationsverdampfer entfernt und das Produkt als farbloses Öl erhalten (0.10 g, 0.27 mmol, 67%). 1H-NMR (300 MHz, CDCl3): δ = 0.78 (d, J = 6.9 Hz, 3H), 0.94 (d, J = 6.9 Hz, 3H), 1.14 (d, J = 6.4 Hz, 3H), 1.49 (bs, 2H), 2.19-2.25 (m, 1H), 3.24 (bs, 1H), 3.67 (s, 3H), 3.91 (dd, J = 18.1 Hz, 159 Kapitel 5 Experimenteller Teil 5.3 Hz, 1H), 4.02 (dd, J = 18.2 Hz, 5.7 Hz, 1H), 4.10-4.13 (m, 1H), 4.56-4.66 (m, 3H), 7.12 (t, J = 5.1 Hz, 1H), 7.22-7.32 (m, 5H), 8.11 (d, J = 7.0 Hz, 1H); 13C-NMR (100 MHz, CDCl3): δ = 15.3, 16.1, 19.5, 30.9, 41.1, 52.2, 55.8, 60.2, 71.5, 74.1, 127.6, 127.7, 128.3, 137.9, 169.8, 169.9, 174.9. Methyl-2-((2S,3R)-3-(benzyloxy)-2-((S)-2-((E)-(3,5-di-tert-butyl-2-hydroxybenzyliden)amino)-3methylbutanamido)butanamido)acetat (72): Unter Stickstoffatmosphäre wurde H-L-Val-L- Thr(OBn)-Gly-OMe (87) (98.0 mg, 0.26 mmol) in absolutem MeOH (3 mL) gelöst und mit Na2SO4 (0.15 g, 1.04 mmol) versetzt. 3,5-Di-tert- butylsalicylaldehyd (64) (60.0 mg, 0.26 mmol) wurde in MeOH (3 mL) gelöst und langsam zum freien Amin 87 getropft. Die Reaktionsmischung wurde über Nacht bei RT gerührt, das Lösungsmittel am Rotationsverdampfer entfernt und der Rückstand in PE aufgenommen. Die Suspension wurde über eine Glasfritte filtriert und das Lösungsmittel des Filtrats vollständig entfernt.[73c] Das Produkt wurde als gelber Feststoff erhalten (0.15 g, 0.26 mmol, quant.). [α]D20 = +58° (c = 0.15, CHCl3); 1H-NMR (300 MHz, CDCl3): δ = 0.92 (d, J = 6.9 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H), 1.20 (d, J = 6.6 Hz, 3H), 1.30 (s, 9H), 1.42 (s, 9H), 2.42-2.53 (m, 1H), 3.66 (s, 3H), 3.70 (d, J = 4.3 Hz, 1H), 3.86 (dd, J = 18.2 Hz, 5.1 Hz, 1H), 4.02 (dd, J = 18.1 Hz, 5.6 Hz, 1H), 4.15-4.23 (m, 1H), 4.57-4.68 (m, 3H), 6.95 (t, J = 5.6 Hz, 1H), 7.03 (d, J = 6.8 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 7.23-7.32 (m, 5H), 7.42 (d, J = 2.4 Hz, 1H), 8.34 (s, 1H), 12.88 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 15.5, 17.4, 19.6, 29.4, 31.4, 32.0, 34.1, 35.1, 41.2, 52.2, 56.2, 71.5, 73.7, 79.5, 117.6, 126.7, 127.7, 128.0, 128.4, 136.9, 137.8, 140.6, 158.0, 169.2, 169.4, 169.8, 171.8; HRMS (ESI): Gemessen: m/z = 596.3699; Berechnet für [C34H49N3O6+H]+ m/z = 596.3694; IR (dünner Film): ṽ = 3410, 3288, 2956, 2869, 2359, 2341, 1751, 1647, 1623, 1539, 1496, 1455, 160 Kapitel 5 Experimenteller Teil 1437, 1387, 1361, 1339, 1271, 1249, 1202, 1172, 1078, 1026, 985, 879, 826, 801, 771, 732, 696, 668, 644, 614, 418 cm-1. Methyl-2-((2S,3R)-3-(benzyloxy)-2-((S)-2-((E)-(2-hydroxy-3,5-di-iodobenzyliden)amino)-3-methylbutanamido)butanamido)acetat (73): H-L-Val-L-Thr(OBn)-Gly-OMe x TFA (86) (0.20 g, 0.41 mmol) wurde in wenig H2O gelöst und die Lösung mit K2CO3 (20%) auf pH 8-10 eingestellt. Die wässrige Lösung wurde mit DCM (6 x 20 mL) extrahiert, die vereinigten organischen Phasen mit H2O (2 x 30 mL) gewaschen und über Na2SO4 getrocknet.[97] Das Lösungsmittel wurde am Rotationsverdampfer vollständig entfernt. Das freie Amin wurde als Öl erhalten und ohne weitere Reinigung direkt umgesetzt (0.10 g, 0.27 mmol, 64%). Unter Stickstoffatmosphäre wurde H-L-Val-L-Thr(OBn)-Gly-OMe (87) (0.10 g, 0.27 mmol) in absolutem MeOH (5 mL) gelöst und mit Na2SO4 (0.15 g, 1.08 mmol) versetzt. 3,5Diiodosalicylaldehyd (97.0 mg, 0.26 mmol) wurde portionsweise zum freien Amin gegeben und die Reaktionsmischung mit MeOH (5 mL) versetzt. Die Reaktionslösung wurde über Nacht gerührt, wobei ein voluminöser, gelber Niederschlag ausfiel. Anschließend wurde über eine Fritte filtriert, mit DCM gewaschen und das Filtrat vollständig eingeengt.[73c] Das Rohprodukt wurde säulenchromatographisch (SiO2, EtOAc/PE 1:1, EtOAc/PE 4:1) gereinigt, wonach das Produkt als gelber Feststoff erhalten wurde (0.14 g, 0.19 mmol, 71%). [α]D20 = +49° (c = 0.15, CHCl3); 1H-NMR (300 MHz, CDCl3): δ = 0.90 (d, J = 6.9 Hz, 3H), 0.95 (d, J = 6.9 Hz, 3H), 1.18 (d, J = 6.4 Hz, 3H), 2.37-2.51 (m, 1H), 3.69 (s, 3H), 3.75 (d, J = 4.7 Hz, 1H), 3.87 (dd, J = 18.2 Hz, 5.7 Hz, 1H), 4.04 (dd, J = 18.2 Hz, 5.7 Hz, 1H), 4.11-4.18 (m, 1H), 4.59-4.70 (m, 3H), 6.88 (d, J = 6.4 Hz, 1H), 7.02 (t, J = 5.5 Hz, 1H), 7.26-7.34 (m, 5H), 7.57 (d, J = 2.1 Hz, 161 Kapitel 5 Experimenteller Teil 1H), 8.07 (d, J = 2.1 Hz, 1H), 8.16 (s, 1H), 13.61 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 15.1, 17.5, 19.6, 32.1, 41.2, 52.3, 55.8, 71.6, 73.8, 79.1, 79.9, 87.1, 119.9, 127.8, 128.5, 137.6, 140.6, 149.3, 159.9, 165.9, 169.2, 169.7, 170.4; HRMS (ESI): Gemessen: m/z = 758.0199; Berechnet für [C26H31I2N3O6+Na]+ m/z = 758.0194; IR (dünner Film): ṽ = 3274, 3062, 2965, 2875, 2359, 2332, 1743, 1734, 1636, 1549, 1435, 1374, 1282, 1210, 1157, 1120, 1036, 1027, 1009, 900, 861, 730, 694, 657, 607, 549, 446, 418 cm-1. Methyl-2-((2S,3R)-3-(benzyloxy)-2-((2S)-2-((E)-(((R)-2-hydroxy-2'-phenyl-[1,1'-binaphthalen]-3-yl)methylen)amino)-3-methylbutanamido)butanamido)acetat (74): H-L-Val-L-Thr(OBn)-Gly-OMe x TFA (86) (0.23 g, 0.47 mmol) wurde in wenig H2O gelöst und die Lösung mit K2CO3 (20%) auf pH 8-10 eingestellt. Die wässrige Lösung wurde mit DCM (6 x 20 mL) extrahiert, die vereinigten organischen Phasen mit H2O (2 x 30 mL) gewaschen und über Na2SO4 getrocknet.[97] Das Lösungsmittel wurde am Rotationsverdampfer vollständig entfernt, das als freie Amin Öl erhalten und ohne weitere Reinigung direkt umgesetzt (0.12 g, 0.32 mmol, 67%). Unter Stickstoffatmosphäre wurde H-L-Val-L-Thr(OBn)-Gly-OMe (87) (0.12 g, 0.32 mmol) in absolutem MeOH (3 mL) gelöst und mit Na2SO4 (0.18 g, 1.28 mmol) versetzt. (R)-2-Hydroxy-2'-phenyl-[1,1'binaphthalen]-3-carbaldehyd[101] (0.12 g, 0.32 mmol) wurde in einer Mischung von MeOH/DCM (10 mL/3 mL) gelöst. Diese Lösung wurde tropfenweise zum freien Amin zugegeben, die Reaktionsmischung über Nacht gerührt und über eine Fritte filtriert. Das Lösungsmittel des Filtrats wurde vollständig entfernt.[73c] Der Rückstand wurde in Et2O resuspendiert und dieser anschließend am Rotationsverdampfer wieder entfernt. 162 Das Rohprodukt wurde mittels Flash- Kapitel 5 Experimenteller Teil Säulenchromatographie (SiO2, DCM, DCM/EtOAc 1:1) gereinigt, wonach das Produkt als gelber Feststoff erhalten wurde (0.23 g, 0.32 mmol, quant.). [α]D20 = -10° (c = 0.15, CHCl3); 1H-NMR (300 MHz, CDCl3): δ = 0.90-0.96 (m, 6H), 1.11 (d, J = 6.4 Hz, 3H), 2.39-2.48 (m, 1H), 3.62 (s, 3H), 3.75 (d, J = 4.4 Hz, 1H), 3.89 (dd, J = 18.1 Hz, 5.0 Hz, 1H), 4.02-4.13 (m, 2H), 4.61-4.68 (m, 3H), 6.91-7.32 (m, 17H), 7.42-7.48 (m, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.69-7.72 (m, 1H), 7.85 (s, 1H), 7.95 (d, J = 8.2 Hz, 1H), 8.01 (d, J = 8.4 Hz, 1H), 8.52 (s, 1H), 12.14 (s, 1H); 13C-NMR (75 MHz, CDCl3): δ = 14.6, 17.5, 19.7, 32.3, 41.2, 52.2, 55.3, 71.6, 73.9, 79.9, 110.6, 119.6, 120.5, 123.4, 124.9, 125.7, 126.3, 126.4, 127.1, 127.4, 127.8, 127.9, 128.1, 128.3, 128.5, 128.6, 130.9, 132.8, 132.9, 134.6, 135.0, 137.7, 140.8, 141.9, 154.3, 168.3, 168.9, 169.7, 170.9; HRMS (ESI): Gemessen: m/z = 736.3383; Berechnet für [C46H45N3O6+H]+ m/z = 736.3381; IR (dünner Film): ṽ = 3300, 3056, 2961, 2931, 2869, 2356, 2341, 1748, 1734, 1653, 1627, 1507, 1494, 1457, 1436, 1368, 1340, 1319, 1288, 1259, 1205, 1184, 1141, 1119, 1078, 1026, 940, 892, 821, 763, 747, 695, 668, 613, 599, 548, 526, 433 cm -1. (2S)-Methyl-3-hydroxy-2-((E)-(((R)-2-hydroxy-2'-phenyl-[1,1'-bi-naphthalen]-3-yl)methylen)amino)propanoat (76): Unter Stickstoffatmosphäre wurde H-L-Ser-OMe (89) (0.02 g, 0.17 mmol) in absolutem MeOH (3 mL) gelöst und mit Na2SO4 (99.0 mg, 0.68 mmol) versetzt. (R)-2-Hydroxy-2'-phenyl-[1,1'binaphthalen]-3-carbaldehyd[101] (64.0 mg, 0.17 mmol) wurde in absolutem DCM (3 mL) gelöst und langsam zum freien Amin zugetropft. Die Reaktionsmischung wurde über Nacht gerührt, das Lösungsmittel am Rotationsverdampfer entfernt, der Rückstand in DCM aufgenommen und über eine Glasfritte filtriert.[73c] Der Feststoff wurde mit DCM (2 x 5 mL) gewaschen und das Filtrat vollständig 163 Kapitel 5 Experimenteller Teil eingeengt. Der Rückstand wurde in DCM resuspendiert und nach Entfernen des Lösungsmittels wurde das Produkt als gelber Feststoff erhalten (81.0 mg, 0.17 mmol, quant.). [α]D20 = -91° (c = 0.15, CHCl3); 1H-NMR (300 MHz, CDCl3): δ = 2.14 (bs, 1H), 3.74 (s, 3H), 3.864.07 (m, 2H), 4.18 (dd, J = 6.7 Hz, 5.3 Hz, 1H), 6.97-7.12 (m, 5H), 7.17-7.31 (m, 6H), 7.41-7.48 (m, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.72-7.75 (m, 1H), 7.84 (s, 1H), 7.93 (d, J = 8.2 Hz, 1H), 8.01 (d, J = 8.4 Hz, 1H), 8.56 (s, 1H), 12.38 (bs, 1H); 13C-NMR (100 MHz, CDCl3): δ = 52.6, 63.5, 72.5, 119.8, 120.3, 123.3, 125.0, 125.6, 126.2, 126.3, 126.4, 126.9, 127.2, 128.1, 128.3, 128.5, 128.6, 128.7, 130.8, 132.7, 132.9, 134.2, 135.7, 140.4, 142.0, 154.3, 168.8, 170.0; MS (MALDI-TOF): m/z = 476 [M+H]+; HRMS (ESI): Gemessen: m/z = 476.1856; Berechnet für [C31H25NO4+H]+ m/z = 476.1860; IR (dünner Film): ṽ = 3052, 2950, 2850, 2393, 2332, 1734, 1628, 1558, 1539, 1507, 1493, 1457, 1436, 1387, 1340, 1312, 1289, 1260, 1208, 1181, 1148, 1109, 1070, 1045, 1027, 978, 940, 891, 863, 821, 793, 762, 747, 698, 609, 547, 526, 508, 435 cm-1. (2S,3R)-Methyl-3-hydroxy-2-((E)-(((R)-2-hydroxy-2'-phenyl-[1,1'-binaphthalen]-3-yl)methylen)amino)butanoat (75): Unter Stickstoffatmosphäre wurde H-L-Thr-OMe (88) (29.0 mg, 0.22 mmol) in absolutem MeOH (7 mL) gelöst und mit Na2SO4 (0.12 g, 0.88 mmol) versetzt. (R)-2-Hydroxy-2'-phenyl-[1,1'binaphthalen]-3-carbaldehyd[101] (0.08 g, 0.21 mmol) wurde in absolutem DCM (3 mL) gelöst und langsam zum freien Amin zugetropft. Die Reaktionsmischung wurde über Nacht gerührt, das Lösungsmittel am Rotationsverdampfer vollständig entfernt, der Rückstand in DCM aufgenommen und über eine Glasfritte filtriert, mit DCM gewaschen und das Filtrat vollständig eingeengt.[73c] Der Rückstand wurde mittels Flash-Säulenchromatographie 164 Kapitel 5 Experimenteller Teil (SiO2, DCM, DCM/EtOAc 1:1) gereinigt, wonach das Produkt als gelber Feststoff erhalten wurde (0.11 g, 0.22 mmol, quant.). [α]D20 = -105° (c = 0.15, CHCl3); 1H-NMR (300 MHz, CDCl3): δ = 1.23 (d, J = 6.4 Hz, 3H), 2.28 (bs, 1H), 3.74 (s, 1H), 3.94 (d, J = 5.7 Hz, 1H), 4.20 (bs, 1H), 6.96-7.00 (m, 3H), 7.04-7.09 (m, 1H), 7.16-7.32 (m, 6H), 7.42-7.47 (m, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.72-7.75 (m, 1H), 7.84 (s, 1H), 7.93 (d, J = 8.2 Hz, 1H), 8.01 (d, J = 8.4 Hz, 1H), 8.54 (s, 1H), 12.36 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 19.4, 52.4, 63.7, 68.5, 119.8, 120.3, 123.3, 125.0, 125.6, 126.2, 126.4, 126.9, 127.1, 128.1, 128.2, 128.3, 128.5, 128.7, 130.8, 132.7, 132.9, 134.2, 135.7, 139.8, 140.4, 142.0, 154.2, 168.7, 170.2; MS (MALDI-TOF): m/z = 490 [M+H]+; HRMS (ESI): Gemessen: m/z = 512.1842; Berechnet für [C32H27NO4+Na]+ m/z = 512.1832; IR (dünner Film): ṽ = 3030, 2966, 2890, 2359, 2330, 1734, 1718, 1628, 1576, 1558, 1539, 1507, 1495, 1457, 1436, 1374, 1344, 1312, 1288, 1260, 1202, 1185, 1118, 1085, 1046, 1018, 979, 940, 892, 865, 820, 795, 763, 698, 668, 611, 550, 526, 467, 433 cm-1. (2R)-Methyl-2-((2S,3R)-3-hydroxy-2-((E)-(((R)-2-hydroxy-2'-phenyl-[1,1'-binaphthalen]-3-yl)methylen)amino)butanamido)-3-phenylpropanoat (78): Cbz-L-Thr-D-Phe-OMe (96) (0.08 g, 0.19 mmol) wurde unter Stickstoffatmosphäre in absolutem MeOH (5 mL) gelöst und mit 10%-Pd/C (21.0 mg, 10.0 mol%) versetzt. Die erfolgte durch Abspaltung Hydrierung Atmosphärendruck. Der der bei Cbz-Schutzgruppe RT und unter Reaktionsfortschritt wurde mittels DC verfolgt (DCM/MeOH 10:1). Nach 4 h wurde die Reaktionslösung über Celite filtriert und mit MeOH gewaschen. Das Filtrat wurde am Rotationsverdampfer eingeengt. Der Rückstand wurde mit Na2SO4 (0.11 g, 0.76 mmol) und einer 165 Kapitel 5 Experimenteller Teil Lösung von (R)-2-Hydroxy-2'-phenyl-[1,1'-binaphthalen]-3-carbaldehyd[101] (72.0 mg, 0.19 mmol) in absolutem DCM (3 mL) versetzt.[100] Die Reaktionsmischung wurde über Nacht gerührt, über eine Fritte filtriert und mit DCM gewaschen. Das Rohprodukt wurde mittels Flash- Säulenchromatographie (SiO2, DCM, DCM/EtOAc 1:1) gereinigt, wonach das Produkt als gelber Feststoff erhalten wurde (0.10 g, 0.16 mmol, 84%). [α]D20 = +9° (c = 0.15, CHCl3); 1H-NMR (300 MHz, CDCl3): δ = 1.16 (d, J = 6.4 Hz, 3H), 2.84-2.92 (m, 1H), 3.12-3.18 (m, 2H), 3.71 (s, 3H), 3.83 (d, J = 3.8 Hz, 1H), 4.04-4.08 (m, 1H), 4.72-4.79 (m, 1H), 6.49 (d, J = 8.0 Hz, 1H), 6.89-7.16 (m, 9H), 7.23-7.31 (m, 5H), 7.44-7.49 (m, 1H), 7.64-7.69 (m, 1H), 7.76-7.81 (m, 1H), 7.89 (s, 1H), 7.96 (d, J = 8.2 Hz, 1H), 8.04 (d, J = 8.5 Hz, 1H), 8.42 (s, 1H), 11.86 (bs, 1H); 13C-NMR (75 MHz, CDCl3): δ = 19.1, 37.4, 52.6, 53.4, 60.4, 69.7, 78.2, 119.7, 120.6, 123.7, 125.2, 125.8, 126.3, 126.5, 126.6, 127.1, 127.2, 127.3, 128.2, 128.4, 128.7, 128.8, 128.9, 130.6, 132.7, 132.9, 134.7, 135.4, 136.1, 140.3, 142.0, 153.8, 169.1, 170.5, 171.6; MS (MALDI-TOF): m/z = 637 [M+H]+; HRMS (ESI): Gemessen: m/z = 637.2703; Berechnet für [C41H36N2O5+H]+ m/z = 637.2697; IR (dünner Film): ṽ = 3394, 3313, 3051, 2972, 2926, 2864, 2360, 2336, 1735, 1660, 1625, 1497, 1438, 1341, 1284, 1213, 1180, 1115, 1075, 1027, 990, 938, 897, 862, 820, 747, 698, 605, 524, 486, 434 cm-1. 166 Kapitel 5 Experimenteller Teil (R)-Methyl-2-((2S,3R)-3-hydroxy-2-((E)-(((S)-2-hydroxy-2'-phenyl-[1,1'-binaphthalen]-3-yl)methylen)amino)butanamido)-3-phenylpropanoat (77): Cbz-L-Thr-D-Phe-OMe (96) (0.24 g, 0.59 mmol) wurde unter Stickstoffatmosphäre in absolutem MeOH (10 mL) gelöst und mit 10%-Pd/C (63.0 mg, 10 mol%) versetzt. Die Abspaltung der Cbz-Schutzgruppe erfolgte durch Hydrierung bei RT und unter Atmosphärendruck. Der Reaktionsfortschritt wurde mittels DC verfolgt (DCM/MeOH 10:1). Nach 4 h wurde die Reaktionslösung über Celite filtriert und mit MeOH gewaschen. Das Filtrat wurde am Rotationsverdampfer eingeengt. Der Rückstand wurde mit Na2SO4 (0.34 g, 2.36 mmol) und einer Lösung von (S)-2-Hydroxy-2'-phenyl[1,1'-binaphthalen]-3-carbaldehyd[101] (0.22 g, 0.59 mmol) in absolutem DCM (5 mL) versetzt. Die Reaktionsmischung wurde über Nacht gerührt, über eine Fritte filtriert und mit DCM gewaschen.[100] Das Rohprodukt wurde mittels Flash-Säulenchromatographie (SiO2, DCM, DCM/EtOAc 1:1) gereinigt, wonach das Produkt als gelber Feststoff erhalten wurde (0.32 g, 0.50 mmol, 85%). [α]D20 = +163° (c = 0.15, CHCl3); 1H-NMR (300 MHz, CDCl3): δ = 1.11 (d, J = 6.4 Hz, 3H), 2.84-2.92 (m, 1H), 3.08 (dd, J = 13.9 Hz, 4.9 Hz, 1H), 3.18 (d, J = 6.3 Hz, 1H), 3.68 (s, 3H), 3.81 (d, J = 3.4 Hz, 1H), 4.04-4.08 (m, 1H), 4.64-4.71 (m, 1H), 6.46 (d, J = 7.9 Hz, 1H), 6.85-7.30 (m, 14H), 7.39-7.46 (m, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.71-7.80 (m, 1H), 7.83 (s, 1H), 7.97 (d, J = 8.1 Hz, 1H), 8.05 (d, J = 8.5 Hz, 1H), 8.40 (s, 1H), 11.89 (bs, 1H); 13C-NMR (75 MHz, CDCl3): δ = 18.9, 36.9, 52.7, 53.4, 69.6, 78.4, 119.6, 120.6, 123.6, 125.2, 125.7, 126.2, 126.3, 126.4, 126.9, 127.1, 128.2, 128.3, 128.6, 128.7, 128.8, 128.9, 130.7, 132.8, 132.9, 134.6, 135.3, 136.0, 140.7, 141.9, 153.8, 168.8, 170.6, 171.6; HRMS (ESI): Gemessen: m/z = 637.2703; Berechnet für [C41H36N2O5+H]+ m/z = 637.2697; IR (dünner Film): 3403, 3052, 3025, 2969, 2880, 2360, 2335, 167 Kapitel 5 Experimenteller Teil 2228, 2188, 2037, 1957, 1739, 1659, 1626, 1497, 1438, 1340, 1282, 1210, 1115, 1077, 1026, 1002, 939, 892, 862, 819, 747, 698, 601, 485, 431 cm-1. (R)-Methyl-2-((2S,3S)-3-hydroxy-2-((E)-(((S)-2-hydroxy-2'-phenyl-[1,1'-binaphthalen]-3-yl)methylen)amino)butanamido)-3-phenylpropanoat (70): Cbz-L-allo-Thr-D-Phe-OMe (97) (0.26 g, 0.62 mmol) wurde unter Stickstoffatmosphäre in absolutem MeOH (7 mL) gelöst und mit 10%-Pd/C (66.0 mg, 10 mol%) versetzt. Die erfolgte durch Abspaltung der Hydrierung Cbz-Schutzgruppe bei RT und unter Atmosphärendruck. Der Reaktionsfortschritt wurde mittels DC verfolgt (DCM/MeOH 10:1). Nach 4 h wurde die Reaktionslösung über Celite filtriert und mit MeOH gewaschen. Das Filtrat wurde am Rotationsverdampfer eingeengt. Der Rückstand wurde mit Na2SO4 (0.35 g, 2.48 mmol) und einer Lösung von (S)-2-Hydroxy-2'-phenyl-[1,1'-binaphthalen]-3-carbaldehyd[101] (0.23 g, 0.62 mmol) in absolutem DCM (6 mL) versetzt. Die Reaktionsmischung wurde über Nacht gerührt, über eine Fritte filtriert und mit DCM gewaschen.[100] Das Rohprodukt wurde mittels Flash- Säulenchromatographie (SiO2, DCM/MeOH/TEA 30:1:0.1) gereinigt, wonach das Produkt als gelber Feststoff erhalten wurde (0.36 g, 0.57 mmol, 92%). [α]D20 = +140° (c = 0.15, CHCl3); 1H-NMR (300 MHz, CDCl3): δ = 1.12 (d, J = 6.3 Hz, 3H), 2.86-2.92 (m, 2H), 3.08 (dd, J = 13.9 Hz, 5.2 Hz, 1H), 3.64 (s, 3H), 3.75 (d, J = 5.7 Hz, 1H), 4.14-4.21 (m, 1H), 4.66-4.72 (m, 1H), 6.46 (d, J = 7.8 Hz, 1H), 6.92-6.98 (m, 8H), 7.15-7.18 (m, 3H), 7.26-7.32 (m, 3H), 7.39-7.47 (m, 2H), 7.67 (d, J = 8.5 Hz, 1H), 7.78-7.80 (m, 1H), 7.83 (s, 1H), 7.97 (d, J = 8.1 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 8.40 (s, 1H), 11.81 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 19.3, 37.4, 52.7, 53.4, 69.5, 78.8, 119.8, 120.8, 123.9, 125.5, 125.9, 126.5, 126.6, 126.7, 127.3, 168 Kapitel 5 Experimenteller Teil 127.4, 128.5, 128.9, 128.9, 129.0, 129.1, 129.2, 130.9, 133.0, 133.3, 134.9, 135.6, 136.3, 140.9, 142.2, 153.9, 169.0, 170.2, 171.4; HRMS (ESI): Gemessen: m/z = 637.2681; Berechnet für [C41H36N2O5+H]+ m/z = 637.2697; IR (dünner Film): ṽ = 3409, 3351, 3053, 3027, 2968, 2878, 2359, 2334, 1953, 1740, 1661, 1627, 1505, 1439, 1342, 1317, 1285, 1261, 1214, 1182, 1116, 1080, 1026, 941, 894, 862, 820, 748, 699, 599, 526, 486, 432 cm-1. 5.2.6 Katalysereaktionen mit Hybridverbindungen Allgemeine Arbeitsvorschrift 8 (AAV 8): Oxidation von Thioanisol unter der Verwendung von Hybridverbindungen In einem Rundhalskolben wurden MXn (2 oder 5 mol%), der jeweilige Ligand (3 oder 5 mol%) und DCM (1 mL) vorgelegt und die Lösung für 30 Min bei RT gerührt. Anschließend wurde Thioanisol (1.0 Äq., 0.40 mmol), dann 30%iges H2O2 (1.2 Äq.) zugegeben und die Reaktionsmischung bei RT unter der angegebenen Zeit gerührt. Das Rohprodukt wurde säulenchromatographisch gereinigt (SiO2, PE/EtOAc 1:4) und der Enantiomerenüberschuss mittels HPLC-Analyse (OD-Säule, Hexan/i-PrOH 93:7, Fluss 1.0 mL/Min, 30-32 bar, 25 °C, t(R-Enantiomer) = 18.0 Min, t(S-Enantiomer) = 22.4 Min) bestimmt. Zur Identifikation des Produkts wurde das literaturbekannten Daten[111] verglichen. 169 1H-NMR Spektrum mit Kapitel 5 5.3 Experimenteller Teil Katalytische Experimente unter Organokatalyse (Kapitel 3.2) Allgemeine Arbeitsvorschrift 9 (AAV 9): Oxidation von Thioanisol unter der Verwendung von NFormyl-L-Prolin (34) In einem Rundhalskolben wurden der jeweilige Organokatalysator (20 mol%), das Lösungsmittel (c(Substrat) = 0.48 mol/L) und Thioanisol (1.0 Äq., 0.24 mmol) vorgelegt. Zu dieser Mischung wurde 30%iges H2O2 (1.2 Äq.) in einer Portion zugegeben und die Reaktionsmischung unter der angegebenen Temperatur, sowie angegebener Zeit gerührt. Die Reaktion wurde durch die Zugabe von Na2SO3 beendet und der Umsatz mittels GC-Analyse bestimmt (Säule: TR 5, 7 m x 0.32 mm (Thermo), Standard: Hexadekan 99%). Zur Bestimmung der Ausbeute wurde das Rohprodukt säulenchromatographisch gereinigt (SiO2, PE/EtOAc 1:1). Zur Identifikation des Produkts wurde das 1H-NMR Spektrum mit literaturbekannten Daten[111] verglichen. Allgemeine Arbeitsvorschrift 10 (AAV 10): Oxidation von Thioanisol unter der Verwendung von BINOL-Phosphaten In einem Rundhalskolben wurden der jeweilige Organokatalysator (10-20 mol%), DCM (c(Substrat) = 0.48 mol/L) und Thioanisol (1.0 Äq., 0.24 mmol) vorgelegt. Zu dieser Mischung wurde 30%iges H2O2 (1.2 Äq.) in einer Portion zugegeben und die Reaktionsmischung bei der angegebenen Temperatur gerührt. Nach 24 h Reaktionszeit wurde das Rohprodukt säulenchromatographisch gereinigt (SiO2, PE/EtOAc 1:1) und der Enantiomerenüberschuss mittels HPLC-Analyse (OD-Säule, Hexan/i-PrOH 93:7, Fluss 1.0 mL/Min, 30-32 bar, 25 °C, t(R-Enantiomer) = 18.0 Min, t(S-Enantiomer) = 22.4 Min) bestimmt. Zur Identifikation des Produkts wurde das 1H-NMR Spektrum mit literaturbekannten Daten[111] verglichen. 170 Kapitel 5 5.4 Experimenteller Teil Katalytische Experimente zur enantioselektiven Synthese von (S)-Omeprazol und (R)-Modafinil (Kapitel 3.3) Allgemeine Arbeitsvorschrift 11 (AAV 11): Synthese von (S)-Omeprazol Die Oxidation des prochiralen Sulfids 108 mittels H2O2 wurde, wie in den Allgemeinen Arbeitsvorschriften AAV5, AAV6, AAV8 und AAV10 beschrieben, unter der Anwendung des jeweiligen metall- bzw. organokatalytischen Verfahrens durchgeführt. Die genauen Reaktionsbedingungen sind Tabelle 3.3-1 in Kapitel 3.3.2 zu entnehmen. Die Reaktion wurde durch die Zugabe von Na2SO3 beendet, die Lösung mit DCM (3 x 5 mL) extrahiert und das Rohprodukt anschließend säulenchromatographisch gereinigt (Al2O3, EtOAc/MeOH 10:1). Der Enantiomerenüberschuss wurde mittels HPLC-Analyse (AS-Säule, Hexan/i-PrOH 85:15, Fluss 1.1 mL/Min, 25 bar, 25 °C, t(R-Enantiomer) = 26.5 Min, t(S-Enantiomer) = 52.6 Min) bestimmt. Zur Identifikation des Produkts wurde das 1H-NMR Spektrum mit literaturbekannten Daten[114] verglichen. Allgemeine Arbeitsvorschrift 12 (AAV 12): Synthese von (R)-Modafinil Die Oxidation des prochiralen Sulfids 114 mittels H2O2 wurde, wie in den Allgemeinen Arbeistvorschriften AAV3, AAV6, AAV8 und AAV10 beschrieben, unter der Anwendung des jeweiligen metall- bzw. organokatalytischen Verfahrens durchgeführt. Die genauen Reaktionsbedingungen sind Tabelle 3.3-1 in Kapitel 3.3.2 zu entnehmen. Das Rohprodukt wurde säulenchromatographisch gereinigt (SiO2, EtOAc) und der Enantiomerenüberschuss mittels HPLCAnalyse (AS-Säule, Hexan/i-PrOH 60:40, Fluss 0.9 mL/Min, 31 bar, 25 °C, t(S-Enantiomer) = 17.2 Min, t(R-Enantiomer) = 39.0 Min) bestimmt. Zur Identifikation des Produkts wurde das 1H-NMR Spektrum mit literaturbekannten Daten[68] verglichen. 171 KAPITEL 6 6 [1] Referenzen a) H. B. Kagan, in Catalytic Asymmetric Synthesis, 2nd ed. (Ed.: I. Ojima), Wiley-VCH, New York, 2000; b) B. Ferber, H. B. Kagan, Adv. Synth. Catal. 2007, 349, 493-507. [2] a) J. Legros, J. R. Dehli, C. Bolm, Adv. Synth. Catal. 2005, 347, 19-31; b) I. Fernández, N. Khiar, Chem. Rev. 2003, 103, 3651-3706. [3] a) D. A. Evans, M. M. Faul, L. Colombo, J. J. Bisaha, J. Clardy, D. Cherry, J. Am. Chem. Soc. 1992, 114, 5977-5985; b) M. C. Carreno, Chem. Rev. 1995, 95, 1717-1760; c) H. Pellissier, Tetrahedron 2006, 62, 5559-5601; d) M. Carmen Carreno, G. HernandezTorres, M. Ribagorda, A. Urbano, Chem. Commun. 2009, 6129-6144. [4] a) M. Mellah, A. Voituriez, E. Schulz, Chem. Rev. 2007, 107, 5133-5209; b) H. Pellissier, Tetrahedron 2007, 63, 1297-1330. [5] a) S. Kobayashi, C. Ogawa, H. Konishi, M. Sugiura, J. Am. Chem. Soc. 2003, 125, 66106611; b) A. Massa, M. R. Acocella, V. D. Sio, R. Villano, A. Scettri, Tetrahedron: Asymmetry 2009, 20, 202-204. [6] a) A. Gossauer, Struktur und Reaktivität der Biomoleküle, Helvetica Chimica Acta, Zürich, 2006; b) A. Young, http://www.chemistry.illinois.edu/research/organic/seminar_extracts/2007_2008/Abstract_S pring2008_Andrew_Young.pdf (aufgerufen am 25.10.2012). [7] A. Streitwieser, C. H. Heathcock, E. M. Kosower, Organische Chemie, 2. Aufl., Wiley-VCH, Weinheim, 1994. 172 Kapitel 6 [8] Referenzen a) E. b. Wojaczy ska, J. Wojaczy ski, Chem. Rev. 2010, 110, 4303-4356; b) G. E. O`Mahony, P. Kelly, S. E. Lawrence, A. R. Maguire, Arkivoc 2011, i, 1-110. [9] a) K. K. Andersen, Tetrahedron Lett. 1962, 3, 93-95; b) K. K. Andersen, W. Gaffield, N. E. Papanikolaou, J. W. Foley, R. I. Perkins, J. Am. Chem. Soc. 1964, 86, 5637-5646. [10] F. A. Davis, A. C. Sheppard, Tetrahedron 1989, 45, 5703-5742. [11] K. Kaczorowska, Z. Kolarska, K. Mitka, P. Kowalski, Tetrahedron 2005, 61, 8315-8327. [12] a) P. I. Dalko, L. Moisan, Angew. Chem. Int. Ed. 2001, 40, 3726-3748; b) F. G. Gelalcha, Chem. Rev. 2007, 107, 3338-3361. [13] a) H. L. Holland, Chem. Rev. 1988, 88, 473-485; b) H. L. Holland, Nat. Prod. Rep. 2001, 18, 171-181. [14] a) P. Pitchen, H. B. Kagan, Tetrahedron Lett. 1984, 25, 1049-1052; b) P. Pitchen, E. Dunach, M. N. Deshmukh, H. B. Kagan, J. Am. Chem. Soc. 1984, 106, 8188-8193. [15] F. Di Furia, G. Modena, R. Seraglia, Synthesis 1984, 1984, 325-326. [16] T. Katsuki, K. B. Sharpless, J. Am. Chem. Soc. 1980, 102, 5974-5976. [17] a) H. B. Kagan, E. Duñach, C. Nemecek, P. Pitchen, O. Samuel, S.-H. Zhao, Pure Appl. Chem. 1985, 57, 1911-1916; b) O. Bortolini, F. Di Furia, G. Licini, G. Modena, M. Rossi, Tetrahedron Lett. 1986, 27, 6257-6260. [18] a) N. Komatsu, Y. Nishibayashi, T. Sugita, S. Uemura, Tetrahedron Lett. 1992, 33, 53915394; b) N. Komatsu, M. Hashizume, T. Sugita, S. Uemura, J. Org. Chem. 1993, 58, 45294533. [19] a) C. Bolm, F. Bienewald, Angew. Chem. Int. Ed. 1996, 34, 2640-2642; b) C. Bolm, G. Schlingloff, F. Bienewald, J. Mol. Catal. A: Chem. 1997, 117, 347-350. [20] M. Palucki, P. Hanson, E. N. Jacobsen, Tetrahedron Lett. 1992, 33, 7111-7114. [21] a) A. Massa, F. R. Siniscalchi, V. Bugatti, A. Lattanzi, A. Scettri, Tetrahedron: Asymmetry 2002, 13, 1277-1283; b) X. Jia, X. Li, L. Xu, Y. Li, Q. Shi, T. T. L. Au-Yeung, C. W. Yip, X. Yao, A. S. C. Chan, Adv. Synth. Catal. 2004, 346, 723-726. 173 Kapitel 6 [22] Referenzen a) J. Sun, C. Zhu, Z. Dai, M. Yang, Y. Pan, H. Hu, J. Org. Chem. 2004, 69, 8500-8503; b) C. Drago, L. Caggiano, R. F. W. Jackson, Angew. Chem. Int. Ed. 2005, 44, 7221-7223; c) Q. Zeng, H. Wang, T. Wang, Y. Cai, W. Weng, Y. Zhao, Adv. Synth. Catal. 2005, 347, 1933-1936. [23] K. Noda, N. Hosoya, R. Irie, Y. Yamashita, T. Katsuki, Tetrahedron 1994, 50, 9609-9618. [24] C. Kokubo, T. Katsuki, Tetrahedron 1996, 52, 13895-13900. [25] C. Bolm, F. Bienewald, Synlett 1998, 1998, 1327-1328. [26] a) B. Pelotier, M. S. Anson, I. B. Campbell, S. J. F. Macdonald, G. Priem, R. F. W. Jackson, Synlett 2002, 2002, 1055-1060; b) A. Gama, L. Z. Flores-López, G. Aguirre, M. Parra-Hake, L. H. Hellberg, R. Somanathan, Arkivoc 2003, xi, 4-15; c) Y. Wu, J. Liu, X. Li, A. S. C. Chan, Eur. J. Org. Chem. 2009, 2009, 2607-2610. [27] a) K. P. Bryliakov, N. N. Karpyshev, S. A. Fominsky, A. G. Tolstikov, E. P. Talsi, J. Mol. Catal. A: Chem. 2001, 171, 73-80; b) S. A. Blum, R. G. Bergman, J. A. Ellman, J. Org. Chem. 2002, 68, 150-155; c) D. Balcells, F. Maseras, G. Ujaque, J. Am. Chem. Soc. 2005, 127, 3624-3634. [28] Q. Zeng, H. Wang, W. Weng, W. Lin, Y. Gao, X. Huang, Y. Zhao, New J. Chem. 2005, 29, 1125-1127. [29] A. H. Vetter, A. Berkessel, Tetrahedron Lett. 1998, 39, 1741-1744. [30] C. Ohta, H. Shimizu, A. Kondo, T. Katsuki, Synlett 2002, 2002, 161-163. [31] Y.-C. Jeong, S. Choi, Y. D. Hwang, K.-H. Ahn, Tetrahedron Lett. 2004, 45, 9249-9252. [32] A. F. Hollemann, E. Wiberg, Lehrbuch der Anorganischen Chemie, Walter de Gruyter, Berlin, 1995. [33] a) G. Helmchen, R. W. Hoffmann, J. Mulzer, E. Schaumann, Methods of Organic Chemistry (Houben Weyl), Vol. E21 e, Thieme, Stuttgart/New York, 1995; b) T. Katsuki, in Catalytic Asymmetric Synthesis, 2nd ed. (Ed.: I. Ojima), Wiley-VCH, New York, 2000. 174 Kapitel 6 [34] Referenzen a) J. T. Groves, P. Viski, J. Org. Chem. 1990, 55, 3628-3634; b) Y. Naruta, F. Tani, K. Maruyama, J. Chem. Soc., Chem. Commun. 1990, 1378-1380; c) Y. Naruta, F. Tani, K. Maruyama, Tetrahedron: Asymmetry 1991, 2, 533-542; d) L.-c. Chiang, K. Konishi, T. Aida, S. Inoue, J. Chem. Soc., Chem. Commun. 1992, 254-256. [35] C. Duboc-Toia, S. Ménage, C. Lambeaux, M. Fontecave, Tetrahedron Lett. 1997, 38, 3727-3730. [36] C. Duboc-Toia, S. Ménage, R. Y. N. Ho, L. Que, C. Lambeaux, M. Fontecave, Inorg. Chem. 1999, 38, 1261-1268. [37] Y. Mekmouche, H. Hummel, R. Y. N. Ho, J. L. Que, V. Schünemann, F. Thomas, A. X. Trautwein, C. Lebrun, K. Gorgy, J.-C. Leprêtre, M.-N. Collomb, A. Deronzier, M. Fontecave, S. Ménage, Chem. – Eur. J. 2002, 8, 1196-1204. [38] J. Legros, C. Bolm, Angew. Chem. Int. Ed. 2003, 42, 5487-5489. [39] a) J. Legros, C. Bolm, Angew. Chem. Int. Ed. 2004, 43, 4225-4228; b) J. Legros, C. Bolm, Chem. – Eur. J. 2005, 11, 1086-1092. [40] a) K. P. Bryliakov, E. P. Talsi, Angew. Chem. Int. Ed. 2004, 43, 5228-5230; b) K. P. Bryliakov, E. P. Talsi, Chem. – Eur. J. 2007, 13, 8045-8050. [41] H. Egami, T. Katsuki, J. Am. Chem. Soc. 2007, 129, 8940-8941. [42] T. Katsuki, H. Egami, Synlett 2008, 2008, 1543-1546. [43] S. Liao, B. List, Adv. Synth. Catal. 2012, 2363-2367. [44] a) P. I. Dalko, L. Moisan, Angew. Chem. Int. Ed. 2004, 43, 5138-5175; b) K. N. Houk, B. List, Acc. Chem. Res. 2004, 37, 487-487; c) A. Berkessel, H. Gröger, Asymmetric Organocatalysis, Wiley-VCH, Weinheim, 2005; d) B. List, J. W. Yang, Science 2006, 313, 1584-1586; e) P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007; f) H. Pellissier, Tetrahedron 2007, 63, 9267-9331; g) A. Dondoni, A. Massi, Angew. Chem. Int. Ed. 2008, 47, 4638-4660. 175 Kapitel 6 [45] Referenzen idw/Universität Regensburg, http://idw-online.de/pages/de/news376211 (aufgerufen am 29.10.2012). [46] A. Berkessel, http://www.chemanager-online.com/themen/chemikaliendistribution/metallfrei-effektiv-sicher-und-kostenguenstig (aufgerufen am 29.10.2012). [47] a) W. Adam, C. R. Saha-Möller, P. A. Ganeshpure, Chem. Rev. 2001, 101, 3499-3548; b) J.-E. Bäckvall, Modern Oxidation Methods, Wiley-VCH, Weinheim, 2004; c) P. Kowalski, K. Mitka, K. Ossowska, Z. Kolarska, Tetrahedron 2005, 61, 1933-1953. [48] F. A. Davis, R. T. Reddy, W. Han, P. J. Carroll, J. Am. Chem. Soc. 1992, 114, 1428-1437. [49] a) A. B. E. Minidis, J.-E. Bäckvall, Chem. – Eur. J. 2001, 7, 297-302; b) A. A. Lindén, N. Hermanns, S. Ott, L. Krüger, J.-E. Bäckvall, Chem. – Eur. J. 2005, 11, 112-119; c) A. A. Lindén, M. Johansson, N. Hermanns, J.-E. Bäckvall, J. Org. Chem. 2006, 71, 3849-3853. [50] A. Russo, A. Lattanzi, Adv. Synth. Catal. 2009, 351, 521-524. [51] a) H. Tohma, S. Takizawa, H. Watanabe, Y. Fukuoka, T. Maegawa, Y. Kita, J. Org. Chem. 1999, 64, 3519-3523; b) A. Ozanne-Beaudenon, S. Quideau, Tetrahedron Lett. 2006, 47, 5869-5873. [52] a) S. Colonna, V. Pironti, J. Drabowicz, F. Brebion, L. Fensterbank, M. Malacria, Eur. J. Org. Chem. 2005, 2005, 1727-1730; b) N. Khiar, S. Mallouk, V. Valdivia, K. Bougrin, M. Soufiaoui, I. Fernández, Org. Lett. 2007, 9, 1255-1258. [53] a) R. Jurok, R. Cibulka, H. Dvořáková, F. Hampl, J. Hodačová, Eur. J. Org. Chem. 2010, 2010, 5217-5224 und die darin zitierten Referenzen; b) V. Mojr, M. Budesinsky, R. Cibulka, T. Kraus, Org. Biomol. Chem. 2011, 9, 7318-7326 und die darin zitierten Referenzen. [54] A. Głuszy ska, I. Maćkowska, M. D. Rozwadowska, W. Sienniak, Tetrahedron: Asymmetry 2004, 15, 2499-2505. [55] a) Z.-M. Liu, H. Zhao, M.-Q. Li, Y.-B. Lan, Q.-B. Yao, J.-C. Tao, X.-W. Wang, Adv. Synth. Catal. 2012, 354, 1012-1022; b) S. Liao, I. Coric, Q. Wang, B. List, J. Am. Chem. Soc. 2012, 134, 10765-10768. 176 Kapitel 6 Referenzen [56] Y. Shi, Acc. Chem. Res. 2004, 37, 488-496 und die darin zitierten Referenzen. [57] I. Agranat, H. Caner, Drug Discovery Today 1999, 4, 313-321 und die darin zitierten Referenzen. [58] http://en.wikipedia.org/wiki/Top_selling_drugs (aufgerufen am 01.11.2012). [59] L. Olbe, E. Carlsson, P. Lindberg, Nat. Rev. Drug Discovery 2003, 2, 132-139. [60] a) P. Lindberg, S. von Unge, WO 94/27988, 1994; b) S. von Unge, V. Langer, L. Sjölin, Tetrahedron: Asymmetry 1997, 8, 1967-1970; c) J. Deng, Y. Chi, F. Fu, X. Cui, K. Yu, J. Zhu, Y. Jiang, Tetrahedron: Asymmetry 2000, 11, 1729-1732. [61] a) H. Cotton, T. Elebring, M. Larsson, L. Li, H. Sörensen, S. von Unge, Tetrahedron: Asymmetry 2000, 11, 3819-3825 und die darin zitierten Referenzen; b) M. Seenivasaperumal, H.-J. Federsel, A. Ertan, K. J. Szabo, Chem. Commun. 2007, 21872189; c) M. Seenivasaperumal, H.-J. Federsel, K. J. Szabó, Adv. Synth. Catal. 2009, 351, 903-919; d) T. Khomenko, K. Volcho, N. Komarova, N. Salakhutdinov, Russ. J. Org. Chem. 2008, 44, 124-127; e) E. A. Koneva, T. M. Khomenko, S. Y. Kurbakova, N. I. Komarova, D. V. Korchagina, K. P. Volcho, N. F. Salakhutdinov, A. G. Tolstikov, G. A. Tolstikov, Russ. Chem. Bull. 2008, 57, 1680-1685; f) B. Jiang, X.-L. Zhao, J.-J. Dong, W.-J. Wang, Eur. J. Org. Chem. 2009, 2009, 987-991; g) J. Sun, M. Yang, Z. Dai, C. Zhu, H. Hu, Synthesis 2008, 2008, 2513-2518; h) J. Y. Choi, G.-S. Hwang, B. K. Senapati, D. H. Ryu, Bull. Korean Chem. Soc. 2008, 29, 1879-1880; i) R. D. Mahale, M. R. Rajput, G. C. Maikap, M. K. Gurjar, Org. Process Res. Dev. 2010, 14, 1264-1268. [62] a) http://www.flickr.com/photos/erica_marshall/398265962/ (Verwendung mit freundlicher Genehmigung von Erica Marshall); b) http://multivu.prnewswire.com/mnr/cephalon/49928/ (aufgerufen am 01.11.2012). [63] a) F. Rebiere, G. Duret, L. Prat, W.O. 028428, 2005; b) K. K. C. Liu, S. M. Sakya, C. J. O’Donnell, A. C. Flick, J. Li, Bioorg. Med. Chem. 2011, 19, 1136-1154. 177 Kapitel 6 [64] Referenzen Y. Wong, S. King, D. Simcoe, S. Gorman, W. Laughton, G. McCormick, P. Grebow, J. Clin. Pharmacol. 1999, 39, 281-288. [65] W. Hauck, P. Adam, C. Bobier, N. Landmesser, Chirality 2008, 20, 896-899. [66] A. Osorio-Lozada, T. Prisinzano, H. F. Olivo, Tetrahedron: Asymmetry 2004, 15, 38113815. [67] a) L. Lafon, U.S. 4,927,855, 1990; b) R. Moshkovits-Kaptsan, V. Braude, K. Chen, W.O. Patent 103221 A2, 2007. [68] J. Ternois, F. Guillen, J.-C. Plaquevent, G. Coquerel, Tetrahedron: Asymmetry 2007, 18, 2959-2964. [69] H. F. Olivo, A. Osorio-Lozada, T. L. Peeples, Tetrahedron: Asymmetry 2005, 16, 35073511. [70] I. Nigel-Etinger, A. Mahammed, Z. Gross, Catal. Sci. Technol. 2011, 1, 578-581. [71] O. Neckebrock, L. Courvoisier, S. Graf, G. Serrure, G. Coquerel, S. Rose, C. Besselievre , WO 04/060858, 2004. [72] a) S. B. Tsogoeva, D. A. Yalalov, M. J. Hateley, C. Weckbecker, K. Huthmacher, Eur. J. Org. Chem. 2005, 2005, 4995-5000; b) S. B. Tsogoeva, S. Wei, Chem. Commun. 2006, 1451-1453; c) S. Wei, D. A. Yalalov, S. B. Tsogoeva, S. Schmatz, Catal. Today 2007, 121, 151-157; d) S. B. Jagtap, S. B. Tsogoeva, Chem. Commun. 2006, 4747-4749; e) C. Baudequin, D. Chaturvedi, S. B. Tsogoeva, Eur. J. Org. Chem. 2007, 2007, 2623-2629. [73] a) C. Bolm, Coord. Chem. Rev. 2003, 237, 245-256; b) C. A. Krueger, K. W. Kuntz, C. D. Dzierba, W. G. Wirschun, J. D. Gleason, M. L. Snapper, A. H. Hoveyda, J. Am. Chem. Soc. 1999, 121, 4284-4285; c) J. T. Su, P. Vachal, E. N. Jacobsen, Adv. Synth. Catal. 2001, 343, 197-200; d) S. D. Green, C. Monti, R. F. W. Jackson, M. S. Anson, S. J. F. Macdonald, Chem. Commun. 2001, 2594-2595. [74] a) M. F. Perutz, M. G. Rossmann, A. F. Cullis, H. Muirhead, G. Will, A. C. T. North, Nature 1960, 185, 416-422; b) T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. 178 Kapitel 6 Referenzen Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono, S. Yoshikawa, Science 1995, 269, 1069-1074. [75] K. Muñiz, Chem. unserer Zeit 2006, 40, 112-124. [76] I. Mohammadpoor-Baltork, M. Hill, L. Caggiano, R. F. W. Jackson, Synlett 2006, 2006, 3540-3544. [77] K. Dürr, D. A. Yalalov, F. W. Heinemann, S. B. Tsogoeva, I. Ivanovic-Burmazovic, Z. Naturforsch. B 2010, 65b, 258-262 und die darin zitierten Referenzen. [78] a) M. J. Robertson, G. N. De Iuliis, M. Maeder, G. A. Lawrance, Inorg. Chim. Acta 2004, 357, 557-570; b) D. J. Ager, I. Prakash, Synth. Commun. 1996, 26, 3865-3868. [79] R. Paul, G. W. Anderson, J. Am. Chem. Soc. 1960, 82, 4596-4600. [80] M. P. Hinderaker, R. T. Raines, Protein Sci. 2003, 12, 1188-1194. [81] K. Stingl, Diplomarbeit, Universität Erlangen-Nürnberg 2008. [82] a) N. N. Karpyshev, O. D. Yakovleva, E. P. Talsi, K. P. Bryliakov, O. V. Tolstikova, A. G. Tolstikov, J. Mol. Catal. A: Chem. 2000, 157, 91-95; b) V. Conte, F. D. Furia, S. Moro, J. Mol. Catal. A: Chem. 1997, 117, 139-149. [83] C. Bied, J. J. E. Moreau, M. Wong Chi Man, Tetrahedron: Asymmetry 2001, 12, 329-336. [84] R. G. Hicks, B. D. Koivisto, M. T. Lemaire, Org. Lett. 2004, 6, 1887-1890. [85] a) A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff, R. D. Shah, J. Org. Chem. 1996, 61, 3849-3862; b) K. H. Baggaley, R. Fears, H. Ferres, G. R. Geen, I. K. Hatton, L. J. A. Jennings, A. W. R. Tyrrell, Eur. Med. Chem. 1988, 23, 523-531. [86] M. Freund, S. Schenker, S. B. Tsogoeva, Org. Biomol. Chem. 2009, 7, 4279-4284. [87] a) E. Kwiatkowski, G. Romanowski, W. Nowicki, M. Kwiatkowski, K. Suwi ska, Polyhedron 2007, 26, 2559-2568; b) F. G. Gelalcha, B. Bitterlich, G. Anilkumar, M. K. Tse, M. Beller, Angew. Chem. Int. Ed. 2007, 46, 7293-7296. [88] a) M. Nielsen, K. V. Gothelf, J. Chem. Soc, Perkin Trans. 1 2001, 2440-2444; b) M. F. Renehan, H.-J. Schanz, E. M. McGarrigle, C. T. Dalton, A. M. Daly, D. G. Gilheany, J. Mol. 179 Kapitel 6 Referenzen Catal. A: Chem. 2005, 231, 205-220; c) M. Holbach, X. Zheng, C. Burd, C. W. Jones, M. Weck, J. Org. Chem. 2006, 71, 2903-2906. [89] E. J. Campbell, S. T. Nguyen, Tetrahedron Lett. 2001, 42, 1221-1225. [90] G. Romanowski, E. Kwiatkowski, W. Nowicki, M. Kwiatkowski, T. Lis, Polyhedron 2008, 27, 1601-1609. [91] G. Romanowski, M. Wera, Polyhedron 2010, 29, 2747-2754. [92] K. M. Weiß, Dissertation 2011, Universität Erlangen-Nürnberg. [93] a) B. M. Cole, K. D. Shimizu, C. A. Krueger, J. P. A. Harrity, M. L. Snapper, A. H. Hoveyda, Angew. Chem. Int. Ed. 1996, 35, 1668-1671; b) K. D. Shimizu, B. M. Cole, C. A. Krueger, K. W. Kuntz, M. L. Snapper, A. H. Hoveyda, Angew. Chem. Int. Ed. 1997, 36, 1704-1707; c) K. D. Shimizu, M. L. Snapper, A. H. Hoveyda, Chem.– Eur. J. 1998, 4, 1885-1889; d) J. R. Porter, W. G. Wirschun, K. W. Kuntz, M. L. Snapper, A. H. Hoveyda, J. Am. Chem. Soc. 2000, 122, 2657-2658; e) J. R. Porter, J. F. Traverse, A. H. Hoveyda, M. L. Snapper, J. Am. Chem. Soc. 2001, 123, 984-985. [94] a) C. A. G. N. Montalbetti, V. Falque, Tetrahedron 2005, 61, 10827-10852; b) T. W. Greene, P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd ed., John Wiley & Sons, New York, 1999. [95] B. I. Morinaka, M. N. Masuno, J. R. Pawlik, T. F. Molinski, Org. Lett. 2007, 9, 5219-5222. [96] A. Thaqi, A. McCluskey, J. L. Scott, Tetrahedron Lett. 2008, 49, 6962-6964. [97] K. Omata, S. Aoyagi, K. Kabuto, Tetrahedron: Asymmetry 2004, 15, 2351-2356. [98] a) L. Bi, M. Zhao, K. Gu, C. Wang, J. Ju, S. Peng, Bioorg. Med. Chem. 2008, 16, 17641774; b) K. Gu, L. Bi, M. Zhao, C. Wang, J. Ju, S. Peng, Bioorg. Med. Chem. 2007, 15, 6273-6290. [99] M. Akhtar, D. Gani, Tetrahedron 1987, 43, 5341-5349. [100] H. Nitta, D. Yu, M. Kudo, A. Mori, S. Inoue, J. Am. Chem. Soc. 1992, 114, 7969-7975. 180 Kapitel 6 [101] Referenzen Der Aldehyd-Baustein wurde mit freundlicher Genehmigung von Dr. Katharina Weiß zur Verfügung gestellt. [102] a) A. Zamfir, S. B. Tsogoeva, Org. Lett. 2009, 12, 188-191; b) A. Zamfir, S. B. Tsogoeva, Synthesis 2011, 2011, 1988-1992; c) O. V. Serdyuk, A. Zamfir, F. Hampel, S. B. Tsogoeva, Adv. Synth. Catal. 2012, 354, 3115-3121. [103] a) T. Akiyama, J. Itoh, K. Yokota, K. Fuchibe, Angew. Chem. Int. Ed. 2004, 43, 1566-1568; b) D. Uraguchi, M. Terada, J. Am. Chem. Soc. 2004, 126, 5356-5357; c) D. Uraguchi, K. Sorimachi, M. Terada, J. Am. Chem. Soc. 2004, 126, 11804-11805; d) T. Akiyama, Chem. Rev. 2007, 107, 5744-5758; e) M. Terada, Chem. Commun. 2008, 4097-4112; f) A. Zamfir, S. Schenker, M. Freund, S. B. Tsogoeva, Org. Biomol. Chem. 2010, 8, 5262-5276. [104] Die BINOL-Phosphate wurden mit freundlicher Genehmigung von Dr. Alexandru Zamfir, Dr. Katharina Weiß, Dr. Olga Serdyuk und Dipl.-Chem. Felix Held zur Verfügung gestellt. [105] a) J. Deng, CN 101486706, 2009; b) N. Hafner Milac, D. Jereb, WO 2000002876, 2000. [106] T. Prisinzano, J. Podobinski, K. Tidgewell, M. Luo, D. Swenson, Tetrahedron: Asymmetry 2004, 15, 1053-1058. [107] a) F. Shi, M. K. Tse, H. M. Kaiser, M. Beller, Adv. Synth. Catal. 2007, 349, 2425-2430; b) S. Liao, Dissertation 2011, Universität Köln. [108] L. Lafon, CA 1296021, 1988. [109] Y.-C. Jeong, D.-J. Ahn, W.-S. Lee, S.-H. Lee, K.-H. Ahn, Bull. Korean Chem. Soc. 2011, 32, 1063-1066. [110] a) T. D. H. Bugg, Tetrahedron 2003, 59, 7075-7101; b) A. Butler, J. N. Carter-Franklin, Nat. Prod. Rep. 2004, 21, 180-188. [111] A. Lattanzi, F. Bonadies, A. Senatore, A. Soriente, A. Scettri, Tetrahedron: Asymmetry 1997, 8, 2473-2478. [112] F. Schoenebeck, J. A. Murphy, S.-z. Zhou, Y. Uenoyama, Y. Miclo, T. Tuttle, J. Am. Chem. Soc. 2007, 129, 13368-13369. 181 Kapitel 6 Referenzen [113] M. Hafner, WO 00/02876, 2000. [114] C. Yang, Q. Jin, H. Zhang, J. Liao, J. Zhu, B. Yu, J. Deng, Green Chemistry 2009, 11, 1401. [115] N. Chatterjie, J. Stables, H. Wang, G. Alexander, Neurochem. Res. 2004, 29, 1481-1486. [116] B. Liu, J. Liu, X. Jia, L. Huang, X. Li, A. S. C. Chan, Tetrahedron: Asymmetry 2007, 18, 1124-1128. [117] K. Kubota, C. L. Hamblett, X. Wang, J. L. Leighton, Tetrahedron 2006, 62, 11397-11401. [118] J.-L. Panayides, R. Pathak, H. Panagiotopoulos, H. Davids, M. A. Fernandes, C. B. de Koning, W. A. L. van Otterlo, Tetrahedron 2007, 63, 4737-4747. [119] F. P. Ballistreri, L. Brinchi, R. Germani, G. Savelli, G. A. Tomaselli, R. M. Toscano, Tetrahedron 2008, 64, 10239-10243. [120] Y. Jiang, X. Zhou, W. Hu, L. Wu, A. Mi, Tetrahedron: Asymmetry 1995, 6, 405-408. [121] Q. Fan, L. Lin, J. Liu, Y. Huang, X. Feng, Eur. J. Org. Chem. 2005, 2005, 3542-3552. [122] Y. Li, B. He, B. Qin, X. Feng, G. Zhang, J. Org. Chem. 2004, 69, 7910-7913. [123] X.-G. Zhou, J.-S. Huang, P.-H. Ko, K.-K. Cheung, C.-M. Che, J. Chem. Soc., Dalton Trans. 1999, 3303-3309. [124] A. Kochnev, I. Oleynik, S. Ivanchev, G. Tolstikov, Russ. Chem. Bull. 2007, 56, 1125-1129. [125] J. Balsells, P. J. Carroll, P. J. Walsh, Inorg. Chem. 2001, 40, 5568-5574. [126] K. J. Beresford, N. J. Church, D. W. Young, Org. Biomol. Chem. 2006, 4, 2888-2897. [127] I. T. Devedjiev, S. G. Bairyamov, V. S. Videva, Heteroat. Chem. 2008, 19, 252-255. [128] J. L. Sorrells, F. M. Menger, J. Am. Chem. Soc. 2008, 130, 10072-10073. [129] H. S. Okumura, B. Philmus, C. Portmann, T. K. Hemscheidt, J. Nat. Prod. 2009, 72, 172176. [130] Z. Tang, F. Jiang, L.-T. Yu, X. Cui, L.-Z. Gong, A.-Q. Mi, Y.-Z. Jiang, Y.-D. Wu, J. Am. Chem. Soc. 2003, 125, 5262-5263. 182 KAPITEL 7 7 Anhang Danksagung Der Tag ist gekommen…die Arbeit ist fertig!!! Aus diesem Anlass heraus möchte ich mich herzlichst bei den Menschen bedanken, die mich während dieser Zeit begleitet und unterstützt haben und mir mit Rat und Tat zur Seite standen. An dieser Stelle möchte ich mich zuallererst bei meiner Doktormutter Frau Prof. Dr. Svetlana B. Tsogoeva bedanken, für die freundliche Aufnahme in ihren Arbeitskreis, die interessante Themenstellung und ihre Unterstützung, sowie stetes Interesse am Fortgang dieser Arbeit. Alle fachlichen Diskussionen und ihre Ideen haben maßgeblich zum Erfolg dieser Arbeit beigetragen. Сердечно благодарю за все! Einen großen Dank möchte ich auch meinen Arbeitskollegen aussprechen: vor allem Kascha (Dr. Katharina Maria Weiß), die sich mit mir 3 Jahre lang einen Abzug geteilt hat und in dieser Zeit (und immer noch!) ein sehr gute Freundin geworden ist. Aber auch meinen ehemaligen Kollegen Dr. Alexandru Zamfir (für die gemeinsame Zeit mit den Lehrämtlern), Dr. Matthias Freund („Bucovina Club“), Dr. Shengwei Wei („echte“), Dr. Sebastian Schenker (den man immer vergessen hat ), sowie den Neuen: Christoph Reiter (der Schwäble), Felix Held (alias Heldbremse…und der für 183 Kapitel 7 Anhang jeden Unsinn zu haben ist), Anja Fingerhut („Yippie, yippie yeah..Krawall und remmidemmi…“), Christina Heckel („und bitte den Kaffepad aus der Maschine nehmen“: du bist eine würdige Nachfolgerin ), Dr. Olga Serdyuk (…die mir die russische Sprache näher bringen wollte: с ас бо Olya!), Sonia Lopez Molina (such a pity that you're gone already, I wish you all the best!!! Jajajajajajajaja…) und allen weiteren Ehemaligen, die nicht so lange bei uns waren: Dominik Lieb, Bernd Kranzer, Regina Messerer, Nicole Brückner, Kathrin Eder und Michael Grunst (des Baddeln woar schee…) für die Unterstützung und eine schöne und angenehme Zeit im Labor. Des Weiteren bedanke ich mich bei allen Angestellten des Instituts für Organische Chemie für Ihre Hilfe und Unterstützung. Mein Dank gilt Christian Placht, Wilfried Schätzke und Harald Maid (NMRSpektroskopie), Eva Hergenröder (Elementaranalyse), Margarete Dzialach und Wolfgang Donaubauer (Massenspektrometrie), Hannelore Oschmann, Robert Panzer und Detlef Schagen (Chemikalienausgabe), Stefan Fronius und Bahram Saberi (Glasbläserei), Eberhard Rupprecht, Erwin Schreier und Horst Meyer (mechanische Werkstatt..und für den maßgeschneiderten Abzug für die Lehrämtler), Holger Wohlfahrt (elektrische Werkstatt), Dr. Frank Hampel, Pamela Hampel und Christiane Brandl-Ritter (Verwaltung). Zuletzt gilt mein besonderer Dank meiner Familie und meinen Freunden, die immer an mich geglaubt haben und mich in allen Lebenslagen unterstützt und zur Seite gestanden haben. Vielen Dank für alles!!! 184