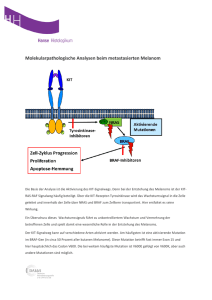
ZIP
Werbung

Aus der Medizinischen Universitätsklinik - Abteilung IVder Albert-Ludwigs-Universität Freiburg i. Brsg. Molekulargenetische Untersuchung des c-MET Proto-Onkogens bei papillären Nierenkarzinomen INAUGURAL – DISSERTATION zur Erlangung des Medizinischen Doktorgrades der Medizinischen Fakultät der Albert-Ludwigs-Universität Freiburg i. Brsg. Vorgelegt 2005 von Susanne Munk-Schulenburg geboren in Nordenham Dekan Prof. Dr. med. C. Peters 1. Gutachter Prof. Dr. med. H.P.H. Neumann 2. Gutachter Prof. Dr. med. U. Wetterauer Jahr der Promotion 2006 INHALT 1 Einleitung 1 1.1 Klinische Aspekte der Nierenkarzinome 1 1.1.1 Ätiologische Faktoren 1 1.1.2 Histologische Einteilung 4 1.2 Papilläre Nierenzellkarzinome 6 1.2.1 Morphologie der papillären Nierenzellkarzinome 6 1.2.2 Hereditäre papilläre Nierenkarzinome 7 1.2.3 Klinische Präsentation 7 1.2.4 Diagnostik 11 1.2.5 Therapie und Prognose 11 1.3 Pathogenese der papillären Nierenkarzinome 14 1.3.1 Das c-MET-Proto-Onkogen und sein Produkt der MET-Rezeptor 14 1.3.2 Bekannte Mutationen des c-MET-Proto-Onkogens in Bezug auf papilläre Nierenkarzinome 1.3.3 Zytogenetische Befunde bei papillären Nierenkarzinomen 1.4. Fragestellung 2 Material und Methoden 2.1 Material 18 21 22 23 23 2.1.1 Patientengut 23 2.1.2 Geräte 25 2.1.3 Enzyme 26 2.1.4 Nukleotide und Nukleinsäuren 26 2.1.5 Chemikalien 26 2.1.6 Lösungen 28 2.2 Methoden 30 2.2.1 Register 30 2.2.2 DNA-Extraktion aus Vollblut 31 2.2.3 Polymerasekettenreaktion 32 2.2.4 Agarosegel-Elektrophorese 35 II 2.2.5 Single-Stranded Conformational Polymorphism (SSCP) Analyse 36 2.2.6 Acrylamidgel-Elektrophorese 36 2.2.7 Silberfärbung der Acrylamidgele 38 2.2.8 Sequenzierung 38 2.2.9 Kontrolle anhand Normalproben 39 3 Ergebnisse 40 3.1 Register 40 3.2 Ergebnisse der SSCP- Analysen der Positivproben 43 3.3 Fallbeschreibungen der Patienten mit der Mutation A3529G in Exon 16 49 3.4 Ergebnisse der SSCP- Analysen der DNA-Proben 52 3.5 Fallbeschreibung des Patienten mit der Mutation G698T in Exon 2 56 3.6 Weitere Polymorphismen 57 3.7 SSCP- Analysen der Patienten mit bekannten somatischen Mutationen 58 3.7.1 Fallbeschreibung des Patienten mit der somatischen Mutation G3522A in Exon 16 60 3.7.2 Fallbeschreibung des Patienten mit der somatischen Mutation T3936G in Exon 19 3.8 Sensitivität der SSCP- Analyse 4 Diskussion 60 61 62 4.1 Konstitutionelle- Mutationen des MET- Gens bei sporadischen papillären Nierenkarzinomen 62 4.2 Konstitutionelle- Mutationen des Met- Gens bei papillären Nierenkarzinomen mit unklarer und positiver familiärer Anamnese 4.3 SSCP- Analyse der Positivkontrollen 65 67 4.4 Konstitutionelle- Mutationen des MET- Gens bei Patienten mit somatischen- Mutationen im Tumorgewebe 68 4.5 Histologische- Klinische Korrelation bei sporadischen und familiären papillären Nierenkarzinomen 4.6 MET- Polymorphismus 69 70 III 5 Zusammenfassung 71 6 Anhang 72 7 Abkürzungen 73 8 Literaturverzeichnis 74 9 Lebenslauf 84 10 Danksagung 85 1 1 Einleitung EINLEITUNG Der Entstehungsmechanismus menschlicher Tumoren ist sehr vielfältig und bis heute noch nicht bei allen Tumorarten vollständig aufgeklärt. Genetische Veränderungen werden allgemein als Grundlage einer Tumorentstehung angesehen, wobei vor allem den Genen eine bedeutende Rolle zugetragen wird, die die Proliferation und Differenzierung einer Zelle beeinflussen. Insbesondere die Inaktivierung von Tumorsuppressorgenen (Marshall 1991) und die Aktivierung zellulärer Proto-Onkogenen (Bishop 1991) werden als einen wichtigen Bestandteil der Pathogenese von Neoplasien angesehen. Durch eine solche Mutation können Zellen einen Wachstumsvorteil gegenüber gesunden Zellen erhalten und somit gesundes Gewebe überwuchern und verdrängen. Solche Genveränderungen können sporadisch auftreten oder im Rahmen eines familiären, erblichen Tumorleidens. Die vorliegende Arbeit beschäftigt sich mit den sporadischen und familiären papillären Nierenkarzinomen, welche mit Veränderungen im so genannten c-MET Proto-Onkogen einhergehen können. 1.1 Klinische Aspekte der Nierenkarzinome 1.1.1 Ätiologische Faktoren Neben den erblichen Faktoren im Rahmen eines familiären Tumorsyndroms wie z.B. bei dem Von-Hippel-Lindau-Syndrom oder den familiären papillären Nierenkarzinomen spielen eine Reihe von anderen Ursachen bei der Krankheitsentstehung eine bedeutende Rolle, da die Mehrzahl der Nierenkarzinome und auch der papillären Nierenkarzinome auf dem Boden einer sporadischen Erkrankung entstehen. Dabei scheinen Alter, Geschlecht, Umwelteinflüsse und geographische Faktoren eine Rolle zu spielen. Das Auftreten von Nierenkarzinomen steht in einem engen Verhältnis zum Alter der Betroffenen. Daten von einer standardisierten Studie zeigen einen Anstieg der Frequenz nach dem dreißigsten Lebensjahr, mit einer minimalen Inzidenz davor (De Reijke et al. 1987). Ein gehäuftes Auftreten erscheint im sechzigsten und siebzigsten Lebensjahr (Abb. 1.1). Einleitung 2 45 40 35 30 25 Frequenz 20 15 10 5 0 30 40 50 60 70 Alter in Jahren 80 90 Abbildung 1.1: Altersverteilung von Nierenzellkarzinomen in 118 Fällen (aus: De Reijke et al. 1987) Ein weiterer wichtiger Faktor der sporadischen Nierenzellkarzinome ist die Geschlechtszugehörigkeit. Die Häufigkeit von sporadischen Nierenkarzinomen ist bei Männern dreimal höher als bei Frauen. Nach der Menopause zeigt sich eine leichte Erhöhung des Männer-Frauen Verhältnis zu Ungunsten der Frauen (Manuel Urrutia Avisrror, Oxford Textbook of Clinical Nephrology, 2nd ed. 1998). Eine vergleichende Studie über die Häufigkeit von Nierenkarzinomen in verschiedenen Ländern zeigt eine erhöhte Inzidenz in den hoch entwickelten Industrieländern. Dies scheint mit vielen Faktoren vergesellschaftet zu sein, z.B. mit der Verstädterung und der ständigen Belastung des Organismus durch verschiedene karzinogene Substanzen aus Umwelt und Industrie (Paganini-Hill et al. 1983). Eine weitere Rolle spielen zudem unterschiedliche Lebensgewohnheiten und das Eßverhalten. So konnte vor einigen Jahren in einer epidemiologischen Studie gezeigt werden, daß adipöse Frauen ein erhöhtes Risiko besitzen, an einem Nierenkarzinom zu erkranken (Mellemgard et al. 1994). Ein häufig diskutierter Risikofaktor ist das Tabakrauchen (Bennington und Laubscher 1968; Kreiger et al. 1993), wobei der pathogenetische Mechanismus hierbei noch nicht vollständig aufgeklärt ist. Ein erhöhtes Risiko wurde zudem bei einer langjährigen Einnahme verschiedener Medikamente berichtet, wie Phenacetin enthaltende Analgetika (Armstrong et al. 1976) oder bei ständigem 3 Einleitung Gebrauch von Diuretika (Weinmann 1993). Auch bei langjährigen Hämodialyse-Patienten konnte ein erhöhtes Risiko dokumentiert werden (Ishikawa 1993). Als weiteren Risikofaktor werden physikalische und chemische Einflüsse diskutiert, wie z.B. die chronische Belastung durch Cadmium, Blei oder Asbest, welche in Tierexperimenten eine karzinogene Wirkung zeigten und somit auch beim Menschen als Karzinogen in Betracht kommen. Auch Aflatoxin, von Aspergillus-Pilzgattungen gebildet und in verschiedenen Nahrungsmitteln enthalten, erzeugte in Experimenten an Ratten verschiedene Arten von Nierenkarzinomen (Epstein et al. 1969). Für das Lösungsmittel Trichloroethylen konnte in einer Studie ein mutagener Effekt auf das VHL-Tumorsuppressorgen gezeigt werden. Bei 44 Patienten mit einem klarzelligen Nierenkarzinom und nachgewiesener chronischer Trichloroethylen – Exposition konnten in 33 Fällen (75%) somatische Mutationen im VHLGen nachgewiesen werden. Bei den Patienten konnten jeweils mehrere unterschiedliche Mutationen im VHL-Gen festgestellt werden und die jeweilige Anzahl zeigte eine Korrelation mit der Schwere der Trichloroethylen – Exposition der Betroffenen (Brauch et al. 1999). Eine physikalische Substanz, der eine krankheitsauslösende Wirkung zugeschrieben wird, ist Thorotrast, welches früher unter anderem als Kontrastmittel z.B. bei retrograden Pyelographien benutzt wurde (Alken et al. 1960; Ruiz-Marcellan et al. 1979). Weiterhin wird eine hormonell bedingte Ätiologie diskutiert, wobei dem Östrogen eine protektive Rolle zugeschrieben wird. Diese Theorie basiert auf dem gehäuften Auftreten von Nierenkarzinomen bei Männern und bei Frauen erst nach der Menopause und wird durch das Vorhandensein von Hormonrezeptoren auf normalem Nierengewebe und Karzinomgewebe unterstützt. Eine Hormonbehandlung bei betroffenen Patienten war bisher jedoch nur wenig erfolgreich (Manuel Urrutia Avisrror, Oxford Textbook of Clinical Nephrology, 2nd ed., 1998). Anhand von Tierexperimenten wird weiterhin eine mögliche Virus-induzierte Genese von Adenokarzinomen der Niere diskutiert (Granhoff et al. 1973). Ein gehäuftes familiäres Auftreten von Nierenkarzinomen ist ein weiterer wichtiger Anhaltspunkt in der Klärung ihrer Ätiologie. In den letzten Jahren konnten anhand verschiedener Studien mit betroffenen Familien genetische Veränderungen nachgewiesen werden, die für bestimmte Nierenkarzinome prädisponierend zu sein scheinen. Bei den molekulargenetischen Entdeckungen, die folgten, hatten die Patienten häufig Tumore im Zusammenhang mit bestimmten erblichen Krankheitssyndromen, wie der Von-HippelLindau-Krankheit (Neumann et al. 1998), den familiären papillären Nierenkarzinomen 4 Einleitung (Schmidt et al. 1997, 1999) oder im Rahmen der familiären Farbenblindheit (Griffin et al. 1967). Abschließend ist zu der Ätiologie der Nierenkarzinome zu sagen, daß wahrscheinlich häufig erst durch das Zusammenspiel mehrerer Risikofaktoren eine Tumorentstehung folgt (multifaktorielle Genese), wobei mit Sicherheit noch weitere als die o.g. Faktoren existieren, bei denen bisher noch kein Zusammenhang nachgewiesen werden konnte. 1.1.2 Histologische Einteilung Eine histologische Einteilung der Nierenkarzinome erweist sich als schwierig, da ständig neue Erkenntnisse über Genetik und Pathogenese veröffentlicht werden und auch in die zytomorphologische Untergliederung einfließen. Somit finden sich bei verschiedenen Autoren abweichende Einteilungen. Eine allgemein verbreitete histologische Klassifizierung soll im Folgenden dargestellt werden. Die Heidelberger Klassifikation (The Heidelberg Classification of Renal Cell Tumors, Kovacs et al. 1997) der Nierenkarzinome unterteilt benigne und maligne Tumore in verschiedene Untergruppen. Es sollen hierbei die malignen Tumore etwas genauer ausgeführt werden. Zu den benignen Tumoren zählen: 1. Metanephritisches Adenom/ Adenofibrom 2. Papilläres Nierenzelladenom 3. Renales Onkozytom Zu den malignen Tumoren zählen: 1. Allgemeines oder gewöhnliches Nierenzellkarzinom Dieser Typ besteht vor allem aus Zellen mit klarem Zytoplasma, wobei das Wachstumsmuster solide, trabekulär, tubulär oder zystisch sein kann. Auch kleine papilläre Areale können vorkommen. Das gewöhnliche Nierenkarzinom umfasst 75% aller malignen Nierenneoplasien und ist somit die häufigste Art. Es wird aufgrund des klaren Zytoplasmas der Zellen in der Färbung auch Klarzellkarzinom genannt. Einleitung 5 2. Papilläres Nierenzellkarzinom Die Zellen dieses Tumors können klein sein mit spärlichem Zytoplasma, jedoch kommen auch häufig Zellen vor, die ein mäßig ausgeprägtes bis reichliches Zytoplasma besitzen mit basophilen, eosinophilen oder blassen Färbeeigenschaften. Das vorherrschende Wachstumsmuster ist papillär, es kann daneben aber auch eine tubulopapilläre und solide Struktur gesehen werden. Mit 10% aller Nierenzellkarzinome stellt dieser Typ die zweithäufigste Art dar. 3. Chromophobes Nierenzellkarzinom Dieser Tumor wächst gewöhnlich in großen, soliden Flächen. Die Zellen besitzen ein charakteristisches blasses oder eosinophiles, granuläres Zytoplasma, entsprechend den zahlreichen zytoplasmatischen Mikrovesikeln. Dieser Typ umfasst ca. 5% der Nierenzellkarzinome. 4. Sammelrohr-Karzinom (Ductus Bellini-Typ) Das Erscheinungsbild dieses Tumors ist charakterisiert durch ungleichmäßige Furchen, umrahmt von hoch atypischen Epithelzellen, in einem dysplastischen Stroma liegend. Es kann eine fokale Schleimproduktion beobachtet werden. Das kürzlich beschriebene medulläre Nierenkarzinom wird als eine Variante des SammelrohrKarzinoms angesehen. Diese Tumorart macht ca. 1% aller Nierenzellkarzinome aus. 5. Unklassifizierte Nierenzellkarzinome Hierzu werden alle Nierenkarzinome gezählt, die sich auch nach einer genetischen Analyse nicht einer der Kategorien zuordnen lassen. Das mögliche Auftreten einer sarkomatoiden Veränderung wurde bei allen Nierenkarzinomtypen dieser Untergruppen gefunden und ein sarkomatoides Erscheinungsbild kann somit nicht eine Kategorie für sich bilden. Jedoch scheint ein Vorkommen ein Anzeichen für eine Progression des Tumors zu sein. Bei einem überwiegenden Anteil der sarkomatoiden Struktur, werden solche Tumore von einigen Autoren als nur sarkomatoid eingestuft. 6 1.2. Einleitung Papilläre Nierenzellkarzinome (papillary renal cell carcinoma, PRCC) 1.2.1 Morphologie der papillären Nierenzellkarzinome Papilläre Nierenkarzinome definieren sich über die Anwesenheit eines vorherrschenden charakteristischen Musters des Zellwachstums (Amin et al. 1997; Delahunt und Eble 1997; Kovacs 1989; Kovacs et al. 1997; Mancilla-Jimenez et al. 1976). Die Tumorzellen sind angeordnet wie Fingerfortsätze, angeheftet an eine Basismembran mit einem zentralen, fibrovaskulären Kern und bedeckt von einer oberflächlichen Schicht aus neoplastischen, epithelialen Zellen. Die Tumorzellen besitzen ein basophiles oder eosinophiles Zytoplasma. Ein papilläres Wachstumsmuster ist dominierend, wobei der Anteil der papillären Strukturen sehr variabel sein kann. Verschiedene Autoren haben deswegen vorgeschlagen, daß ein Tumor mehr als 50% aus papillären Strukturen bestehen muss, um als ein papilläres Nierenkarzinom klassifiziert werden zu können. Abb. 1.2 zeigt jeweils einen Ausschnitt eines histologischen Bildes eines papillären Nierenkarzinoms. Eine weitere Einteilung der PRCC in zwei Subtypen wurde von Delahunt und Eble (The Heidelberg Classification of Renal Cell Tumors, Kovacs et al. 1997) eingeführt, nach ihren morphologischen und immunhistochemischen Charakteristika differenziert. Typ 1 ist gekennzeichnet durch das Auftreten von Papillen, die mit einer einfachen oder doppelten Schicht bekleidet sind, bestehend aus kleinen, epithelialen Zellen mit einem spärlichen klaren oder blassen Zytoplasma. Typ 2 zeichnet sich aus durch Papillen, bedeckt von einer Schicht aus epithelialen Zellen mit reichlichem eosinophilen Zytoplasma, die in Pseudoschichten oder unregelmäßigen Schichten angeordnet sind. Abbildung 1.2: Histologische Bilder eines PRCC ( A: Typ 1, B: Typ 2) (aus: Gunawan et al. 2003) 7 Einleitung 1.2.2 Hereditäre papilläre Nierenkarzinome Hereditäre papilläre Nierenkarzinome sind durch das Auftreten von multiplen, bilateralen Nierentumoren gekennzeichnet (Zbar et al. 1994; Schmidt et al. 1997), im Gegensatz zu den sporadischen papillären Nierenkarzinomen, die meistens solitär vorkommen. Die Läsionen variieren dabei in der Größe von dysplastischen Veränderungen, die nur mikroskopisch gesehen werden können, bis zu Läsionen mit einer Größe von bis zu 18 cm. Die Anzahl der Tumoren kann dabei zwischen 1 bis zu 50 Tumoren pro Niere variieren (s. Abbildung 1.3) Die Morphologie erscheint gleich wie bei den sporadischen PRCC. Der Erbgang ist autosomal dominant mit variierend starker Penetranz, abhängig von dem krankheitsauslösenden Gen und der spezifischen Mutation des entsprechenden Gens. A: sporadische PRCC B: hereditäre PRCC Abbildung 1.3: Schematische Zeichnung des Erscheinungsbildes von sporadischen und hereditären Nierenkarzinomen (aus: Zbar et al. 1998) und makroskopisches Erscheinungsbild eines hereditären papillären RCC (aus: Lubensky et al. 1999) 1.2.3 Klinische Präsentation Die klassischen klinischen Anzeichen eines Nierenkarzinoms werden als eine Trias beschrieben, bestehend aus den Symptomen Schmerzen im Nierenlager, Makrohämaturie und einem palpablen Tumor. Diese Trias wird heute jedoch nur in 5% bis 10% aller Fälle beobachtet und deutet leider auf ein schon fortgeschrittenes Stadium der Krankheit hin. Jeweils eine einzelne dieser Komponenten wird dennoch in ca. 50% der Fälle als ein Einleitung 8 einzelnes Symptom Routinekontrollen beschrieben. durch Nierenkarzinome Sonographie oder im werden weitaus häufiger bei Rahmen anderer diagnostischer Untersuchungen entdeckt, als durch die klinische Verdachtsdiagnose aufgrund der Symptomkonstellation. Nach einer Studie von Skinner und Gibbsons ist das häufigste Symptom die Hämaturie, gefolgt von einer spürbaren abdominellen Tumormasse, Schmerzen und Gewichtsverlust. Symptome Patienten in % Hämaturie 59 Palpierbarer Tumor 45 Schmerzen 41 Gewichtsverlust 28 Symptome durch Metastasen 10 Klassische Trias 9 Akute Varikozele 2 Tabelle 1.1: Symptome bei 309 Patienten mit Nierenzellkarzinomen (Daten nach Skinner, Calvin, Vermillion et al. 1971) Die Hämaturie ist ein wichtiges diagnostisches Zeichen, wobei andere Blutungsquellen aus dem unteren urethralen Trakt auszuschließen sind. Hierbei ist das begleitende Auftreten von wurmartigen Klümpchen im Urin, bestehend aus kleinen Zellhaufen, ein zusätzliches Anzeichen. Der Schmerz verbunden mit Nierenkarzinomen wird als dumpf und kontinuierlich beschrieben, wobei er häufig einer Hämaturie folgt. Während einer Makrohämaturie können Blutklümpchen eine akute Ureterobstruktion hervorrufen, welche zu den typischen Symptomen einer Nierenkolik führen kann. Das plötzliche Auftreten einer skrotalen Varikozele wurde in einer Studie bei 11% der Fälle dokumentiert (Pinals und Krane 1962), wobei sich die Varikozele bei der Mehrheit der Patienten auf der linken Seite befand. Dies ist auf eine Obstruktion der gonadalen Vene an ihrem Eintrittspunkt in die Nierenvene zurückzuführen. Nierenkarzinome können sich häufig auch durch unspezifische aber tumorbedingte Symptome äußern, die auch auf andere Erkrankungen hinweisend sein können. Diese Manifestationen werden als paraneoplastische Syndrome bezeichnet. Dazu zählen unter Einleitung 9 anderem Gewichtsverlust, Fieber, Erythrozytose, Anämie oder Symptome einer Hyperkalzämie. Diese paraneoplastischen Syndrome werden in drei Gruppen eingeteilt: nicht spezifische Symptome, spezifische Symptome und gemischte Symptome. Nicht spezifische Symptome Fieber gehört, wie auch der Gewichtsverlust, zu Symptomen, die viele bösartige Tumore gemeinsam haben. Es wird als intermittierend und mit einer wechselnden Höhe der Temperatur beschrieben. Das Fieber wird durch die Freisetzung von einem endogenen Pyrogen aus den Tumorzellen oder aus Leukozyten innerhalb des Tumors erklärt (Rawlins et al.1970). Das Fieber verschwindet nach einer erfolgreichen Tumorentfernung und kann somit als Verlaufsparameter genutzt werden. Auch der Gewichtsverlust scheint durch eine systemisch wirkende, toxische Substanz aus den Tumorzellen zu resultieren. Eine Anämie wird bei ca. einem Drittel der Patienten festgestellt und ist vom normochromennormozytären Charakter. Chisholm und Roy (1971) stellten die These auf, daß ein Mangel an Erythropoetin, durch einen Nierenzell-Untergang bedingt, eine mögliche Ursache ist oder ein toxischer Einfluss des Karzinoms auf das Knochenmark besteht. Andere hämatologische Symptome wie eine erhöhte leukämoide Reaktionen und Erythrozyten-Sedimentationsrate, Thrombozytopenie, Gerinnungsstörungen sind häufig und kommen auch bei verschiedenen anderen Karzinomen und chronischen entzündlichen Krankheiten vor. Bei ca. 5% der Patienten mit Nierenkarzinomen wird eine Amyloidose der Niere, Leber, Milz und der Nebenniere beobachtet, welche sich auch nach Heilung nicht zurückbildet. Die Ätiologie ist unklar und der Befund deutet auf eine schlechte Prognose hin (Chisholm, Roy 1971). Eine reversible Leberfunktionsstörung, das so genannte Stauffer- Syndrom, wurde bei ca. 15% aller Patienten dokumentiert und äußert sich durch veränderte Werte bei der Leberfunktionsprüfung. Die Ursache ist nicht bekannt, jedoch kann eine Normalisierung der Werte nach einer Tumorentfernung beobachtet werden, sofern keine Lebermetastasen vorhanden sind (Ritchie, Chisholm 1983). Spezifische Symptome Hypertonie ist ein charakteristisches Symptom und tritt bei bis zu 40% der Patienten auf. Dieser Bluthochdruck kommt durch eine spezielle humorale Aktivität des Tumors zustande, eine autonome Reninproduktion (Surfin et al. 1977). Bei einem Drittel (ca. 33%) der Einleitung 10 Patienten mit hochgradigem Nierenkarzinom wurde ein erhöhter Reninspiegel unabhängig vom Blutdruck gefunden, was mit einer schlechten Prognose einherzugehen scheint (Surfin et al.1977). Weitere spezifische Symptome sind Polyzythämie und Hyperkalzämie, welche durch verschiedene aktivierende Substanzen ausgelöst werden, die aus Tumorzellen freigesetzt werden oder aus einer fortschreitenden Niereninsuffizienz resultieren. Eine Polyzythämie wird jedoch nur bei ca. 4% der Patienten beobachtet und wird einer erhöhten Freisetzung von Erythropoetin oder einer Erythropoetin ähnlichen Substanz zugeschrieben. Gegensätzlich wird bei ungefähr 63% aller Patienten eine Erythropoetinerniedrigung beschrieben (Surfin et al. 1977). Hyperkalzämie kann durch eine dem Parathormon ähnliche Substanz aus den Tumorzellen verursacht werden oder durch einen sekundären Hyperparathyreoidismus in Folge einer Niereninsuffizienz. Erhöhte Kalzium-Werte normalisieren sich in der Regel nach einer Therapie und können als Verlaufsparameter genutzt werden. Gemischte Symptome Viele Symptome weisen auf das Krankheitsbild eines Nierenkarzinoms hin, doch nur wenige können direkt mit dieser speziellen Tumorentwicklung in Verbindung gebracht werden. Dazu gehören die Befunde einer leukämoiden Reaktion, einer Neuropathie, einer Nephritis mit Salzverlustsyndrom und gastrointestinalen Beschwerden. Eine Übersicht der Häufigkeit systemischer Auswirkungen gibt Tabelle 1.2 Symptom Häufigkeit in % Erhöhte Erythrozyten-Sedimentationsrate 55,6 Anämie 41,3 Hypertonie 37,6 Kachexie 34,5 Fieber 17,2 Abnormaler Leberfunktionstest 15,0 Erhöhte alkalische Phosphatase 14,7 Hyperkalzämie 5,7 Polyzythämie 3,7 Neuromyopathie 3,3 Amyloidose 2,1 Tabelle 1.2: Frequenz systemischer Auswirkungen (Daten modifiziert nach Chisholm und Roy 1971) Einleitung 11 1.2.4 Diagnostik Heutzutage steht eine Anzahl von nicht-invasiven Methoden zu Verfügung, die eine sehr genaue Diagnostik der Nierenkarzinome zulassen. Ultrasonographie, Computertomographie, Magnetresonanztomographie und Angiographie gehören zu den sensitivsten Methoden der heutigen Diagnostik. An der Universitätsklinik Freiburg ist die Magnetresonanztomographie Methode der Wahl. Die endgültige Diagnose wird mittels histo-pathologischer Untersuchung von Tumorgewebe gestellt. Auch für das Staging des Karzinoms und dem Erstellen eines Therapieplanes spielen diese Verfahren eine wichtige Rolle, da Nierenzellkarzinome häufig hämatogen in verschiedene Organe (besonders in Lunge, Hirn und Knochen) metastasieren und so eine genaue Ausbreitung und Größe des Tumors festgestellt werden kann. An der Universitätsklinik Freiburg werden hierbei die Computertomographie des Thorax, die Magnetresonanztomographie des Kopfes und die Knochenszintigraphie angewendet. So kann ein optimales Therapieschema für den Patienten erstellt werden. 1.2.5 Therapie und Prognose Die Therapie der Wahl ist das chirurgische Entfernen des Tumors. Während bis vor wenigen Jahren die Nephrektomie die Methode der Wahl war, wird zunehmend zur organerhaltenden Tumorresektion übergegangen. Eine solche Enukleation des Tumors kann bei unilateralen, kleinen, nicht in die umgebenden Gefäße und Lymphknoten eingewachsenen Tumoren erfolgen. Hat der Tumor umgebende Strukturen infiltriert, ist weiterhin die radikale Nephrektomie die Methode der Wahl. Gleichzeitig wird eine regionale Lymphadenektomie durchgeführt, da bei vielen Patienten die regionalen und periaortalen Lymphknoten befallen sind. Sind beide Nieren betroffen, so wird die Entfernung der Tumore bei Erhaltung eines Nierenteils angestrebt. Lokale Rezidive sowie Metastasen werden soweit möglich operativ entfernt. Zu der chirurgischen Behandlung können bei Metastasen adjuvante Therapieformen in Erwägung gezogen werden, wie Radiotherapie, Chemotherapie, Immuntherapie oder eine Hormontherapie. Metastasierende Nierenzellkarzinome sind jedoch leider häufig gegen die gängigen adjuvanten Therapien resistent. Einleitung 12 Der wichtigste Prognosefaktor eines Patienten mit Nierenzellkarzinom ist das Stadium des Tumors bei Diagnosestellung und Therapiebeginn. Je höher das Stadium, umso niedriger ist die 5-Jahres-Überlebensrate (Tabelle 1.3). Weiterhin ist die Prognose abhängig vom histologischen Typ des Karzinoms. Chromophobe Karzinomtypen haben eine bessere 5-Jahres-Überlebensrate als papilläre und klarzellige Typen. Die schlechteste Prognose haben die Karzinome mit sarkomatoiden Anteilen (Moch et al. 2000). Speziell für papilläre Nierenkarzinome scheinen Tumore vom Subtyp II eine schlechtere Prognose zu haben als Tumore vom Subtyp I. Zudem konnte eine kürzere Überlebensrate bei Patienten mit zusätzlichem Verlust des Chromosom Xp nachgewiesen werden (Moch et al. 2000; Jiang et al. 1998). Stadium Patientenzahl Überlebensrate (%) I 36 51,0 II 54 58,5 Nierenvene infiltriert 21 64,0 Vena Cava infiltriert 7 25,4 III 8 12,3 IV 32 0 Tabelle 1.3: Korrelation des pathologischen Stadium mit der 5-Jahres-Überlebensrate 13 Einleitung Das Staging der Nierenkarzinome wird hauptsächlich anhand zweier Klassifikationen durchgeführt. Die Erste wurde 1969 von Robson eingeführt und orientiert sich an den makroskopisch sichtbaren pathologischen Befunden, die während der Operation eines Tumors erhoben werden können. Tabelle 1.4 Grad I : Der Tumor liegt innerhalb der Nierenkapsel Grad II : Infiltration des perinephritischen Fettgewebes, der Tumor liegt noch innerhalb der GerotaFaszie Grad III : Beteiligung der regionalen Lymphknoten und/oder der Nierenvene oder Vena Cava Grad IV : Beteiligung der angrenzenden Organe und Fernmetastasen Tabelle 1.4: Stadiumeinteilung des Nierenzellkarzinoms nach Robson et al. 1969 Die internationale TNM Klassifikation von 1997 unterteilt in mehrere Stadien, jeweils für den Tumor, Lymphknoten und Metastasen. Tabelle 1.5 Tumor T1 T2 T3a T3b T3c T4 Tumor 7cm oder kleiner, auf die Niere beschränkt Tumor größer als 7cm, auf die Niere beschränkt Tumorausdehnung ins perirenale Fettgewebe oder Nebenniere, aber nicht über Gerota-Faszie hinaus Infiltration der Nierenvene oder Vena Cava unterhalb des Diaphragma Infiltration der Vena Cava oberhalb des Diaphragma Tumorausdehnung außerhalb der Gerota-Faszie Lymphknoten NX N0 N1 N2 Regionale Lymphknoten können nicht eingeschätzt werden Kein Lymphknotenbefall Metastasierung in einen regionalen Lymphknoten Metastasierung in mehrere regionale Lymphknoten Metastasen MX M0 M1 Keine Einschätzbarkeit von Metastasierung Keine Fernmetastasen Fernmetastasen Tabelle 1.5: TNM Klassifikation der Nierenkarzinome (TNM-Classification of malignant tumors, 5th Ed. 1997) 14 1.3 Einleitung Pathogenese der papillären Nierenkarzinome Das papilläre Nierenkarzinom tritt fast ausschließlich sporadisch, d.h. als Einzelfall in einer Familie, auf. In den letzten Jahren konnten einige somatische, sowie konstitutionelle Mutationen lokalisiert werden, die im Zusammenhang mit der Pathogenese dieser Tumoren stehen. Dies hat zu einem neuen Verständnis der Nierenkarzinome geführt, wodurch nun neben der morphologischen Einteilung auch in zunehmendem Maße eine molekulargenetische Charakterisierung möglich wird. Als eine neue Entität wurde 1994 das familiäre papilläre Nierenkarzinom (Hereditary Papillary Renal Cell Carcinoma, HPRCC) veröffentlicht (Zbar et al. 1994). Ein zugrunde liegender Gendefekt konnte lokalisiert werden (Schmidt et al. 1997). Es wurden Mutationen im so genannten c-MET Proto-Onkogen gefunden. Hierbei konnte bei hereditären Tumoren eine konstitutionelle Mutation des MET-Gens festgestellt werden (Schmidt et al. 1997). Auch bei papillären Nierenkarzinomen ohne eine positive familiäre Anamnese konnten in 13% der Fälle Mutationen im MET-Gen nachgewiesen werden (Lubensky et al. 1999). MET Proto-Onkogen Mutationen prädisponieren somit sowohl für das hereditäre papilläre Nierenkarzinom als auch für das sporadische papilläre Nierenkarzinom. 1.3.1 Das c-MET Proto-Onkogen und sein Produkt der MET-Rezeptor Proto-Onkogene kodieren für spezielle Proteine, deren Aufgabe in der zellulären Rezeption, Transduktion und Umsetzung von Wachstumssignalen besteht. Onkogene entstehen infolge von Mutation aus Proto-Onkogenen und können autonomes Zellwachstum induzieren (Bishop 1991). Die molekulargenetischen Veränderungen, die zu einer Aktivierung der ProtoOnkogene führen, können Struktur wie auch Expression der betroffenen Gene verändern. Die entsprechenden Genprodukte sind insbesondere Wachstumsfaktoren und ihre entsprechenden Rezeptoren. Das MET-Gen kodiert für einen transmembranären Tyrosinkinase-Rezeptor, MET. Dieser Rezeptor, wird in vielen Geweben epithelialer Herkunft exprimiert, unter anderem in der Niere, Darm, Brustgewebe, Leber, Muskeln und Monozyten (Sonnenberg et al. 1993). Zudem wird er in vielen epithelialen Tumoren überexprimiert gefunden, so auch in unterschiedlichen Typen von Nierenkarzinomen, weswegen das MET-Gen als Proto-Onkogen klassifiziert Einleitung 15 wurde (Park et al. 1986; Di Renzo et al. 1992; 1994; Prat et al. 1991). Der MET-Rezeptor besteht aus einer Polypeptidkette aus 1390 Aminosäuren, die sich in ein zweikettiges Heterodimer gliedert. Die Ketten des Dimers sind über Disulfidbrücken (S-S) verbunden (Birchmeier, Gherardi 1998). Insgesamt besteht der MET-Rezeptor aus drei Untereinheiten, der extrazellulären Domäne, der transmembranären Domäne und der zytoplasmatischen (Tyrosinkinase-) Domäne. Die extrazelluläre Domäne enthält die α-Kette mit einem Nterminalen Ende und die transmembranäre β-Kette mit einem C-terminalen Ende an der extramembranären Sequenz. Die β-Kette enthält neben der transmembranären Domäne eine zytoplasmatische Protein-Kinase, die für die intrazelluläre Signal-Transduktion verantwortlich ist. Für die Rezeptoraktivierung finden sich hier zwei spezifische TyrosinPhosphorylierungsstellen bei Y1234 und Y1235. Der entsprechende Rezeptor-Ligand ist der Hepatocyte growth factor/ Scatter factor (HGF/SF) (Bottaro et al. 1991; Naldini et al. 1992). Nach einer proteolytischen Spaltung kann HGF an den Rezeptor binden. HGF ist ein multifunktionaler Faktor, der epitheliale Zell-Mitogenese, Zellbeweglichkeit, Zellwachstum und Morphogenese stimuliert (Bladt et al. 1995). Die extrazelluläre Domäne ist für die Ligandenbindung und die Weiterleitung des Signals zur zytoplasmatischen Domäne verantwortlich, was über eine Rezeptor-Oligomerisation geschieht. In der zytoplasmatischen Domäne findet über eine Autophophorylierung der katalytischen Kinase eine Übertragung des Signals an intrazelluläre Zielproteine statt. Hierbei spielen die downstream gelegenen Tyrosine eine wesentliche Rolle, die je nach Phosphorylierung die Interaktion mit verschiedenen Signaltransduktoren vermitteln (Takada et al. 1998). Die inaktive Konformation des Rezeptors wird durch ein Zusammenspiel verschiedener Faktoren bestimmt. In der katalytisch inaktiven, unphosphorylierten Form ist der Zugang zu dem für die Reaktion benötigtem ATP und zur Protein-bindenden Seite durch eine so genannte „activation loop“ blockiert, die erst durch Ligandenbindung mit folgender Phosphorylierung aktiviert wird. Die Substrat- bindende Tasche, wird dabei von den Resten der Pi-„loop“ (Phosphorylierungsstellen) gebildet. In der „activation loop“ in der inaktiven Form ist die Substrat- bindende Region durch ein Zugangshindernis zu dem für eine Reaktion benötigten ATP blockiert. Die ATP- bindende Tasche wird durch eine Kinase besetzt und bildet ein sterisches Hindernis, wodurch die Substrat- bindende Tasche und die ATPbindende Tasche auseinander gehalten werden. Diese gespaltene Konformation wird zusätzlich durch chemische Wechselwirkungen zwischen den entsprechenden Taschen stabilisiert (Schmidt et al. 1999)(siehe Figur 1). Einleitung 16 Durch Ligandenbindung kommt es zu einer Rezeptor-Oligomerisation mit Konformationsänderung. Hierdurch erfolgt eine Phosphorylisierung und als Folge die Aufhebung der Blockade zwischen der Substrat- bindenden und ATP- bindenden Tasche. Durch den Zugang zum ATP kann nun eine katalytische Reaktion und Bindung eines zytoplasmatischen Moleküls erfolgen. In der aktiven Form kann die Tyrosin-Kinase an zahlreiche Ziel-Moleküle (Signaltransduktoren) binden. Es wird vermutet, daß dies die Grundlage für die vielfältigen Zell-Antworten auf HGF bildet. Figur 1: Schematische Darstellung des MET- Rezeptors und HGF (aus: Birchmeier, Gheradi 1998) HGF/SF: Hepatocyte Growth Factor/Scatter Factor Pi: Phosphorylierungsstellen, intrazellulär (Y1234/Y1235) Gab 1 / Grb 2: Beispiele für Zielmoleküle PI 3-K / MAPK: Phosphoinositide 3 Kinase / Mitogen- aktivierende Protein- Kinase S-S: Disulfidbrücken Bei MET-Mutanten konnten eine erhöhte Tyrosin-Autophosphorylisierung und eine verstärkte Aktivität der Kinase nachgewiesen werden, wodurch vermutet wird, daß die Tumorgenese quantitativ mit der Größe der Aktivierung des MET durch Mutation zusammenhängt (Jeffers et al. 1997). Die Aktivierung oder Amplifikation des MET-Rezeptors Einleitung 17 infolge Mutation erfolgt wahrscheinlich durch eine Destabilisierung der hemmenden „activation loop“ und somit Überführung in eine dauerhaft aktive Form mit Aktivierung der Tyrosin-Kinase in einer alternativen Konformation (Hubbard et al. 1994). Belegend hierfür konnte bei den bisher gefundenen Mutationen des MET-Gens keine Rezeptor-Inaktivierung gefunden werden. Weiterhin konnte ein Zusammenhang gefunden werden, daß HGF über den MET- Rezeptor die Differenzierung und Organisation von Nierenzellen zu sich verzweigenden Tubuli stimuliert und dabei eine frühembryonale Entwicklung von normalen Nephronen simuliert (Montesano et al. 1991). Es wird daher vermutet, daß eine vererbte Mutation des MET- Gens, zu einer verzögerten Differenzierung von vielen Blastenzellen während der embryonalen Entwicklung führt. Diese undifferenzierten Zellen behalten somit ihre Proliferationskapazität und bilden embryonale Reste i. Nierengewebe, die während des Lebens langsam weiter wachsen können (Fischer et al. 1998). Das MET-Proto-Onkogen selbst besteht aus 21 Exons mit einer Minimumgröße von 110kb (Schmidt et al.1997), wobei das erste Exon die 5`UTR-Sequenz enthält und nicht kodierend ist (Gambarotta et al. 1994). Exon 2 bis 21 enthalten die kodierenden Exons, Exon 21 enthält die 3`UTR-Sequenz. Die Größe der kodierenden Exons bewegt sich in einem Bereich von 81 bis 231 Basenpaaren(bp), mit der Ausnahme von Exon 2, welches 1214 bp besitzt. Die Lokalisation des MET-Gens ist auf dem langen Arm des Chromosom 7, im Bereich von 7q31.1 in einem 27-centimorgen Intervall zwischen D7S496 und D7S183 (Schmidt et al. 1997) (siehe Figur 2). Lokalisation des c-MET Gens Figur 2: Lokalisation des MET- Gens auf Chromosom 7 (aus OMIM Database) 18 Einleitung Die extrazelluläre Domäne des MET-Rezeptors ist in Exon 2 bis 13 lokalisiert (Codon 25923). Die transmembranäre Domäne befindet sich zusammen mit dem Ende der extrazellulären und dem Anfang der intrazellulären Domäne in Exon 13 (Codon 933-955). Die Exons 15 bis 21 enthalten die intrazelluläre Domäne (Codon 956-1390) mit der TyrosinKinase Domäne (Codon 1078-1345) (Schmidt et al., 1997) (siehe Figur 3). Figur 3: Exon und Intron Grenzen des MET- Gens mit Darstellung der einzelnen Domänen und Nukleotide (aus: Duh et al. 1997) Die menschliche MET cDNA kodiert insgesamt für ein Protein von 1408 Aminosäuren (aa). Die extrazelluläre Domäne enthält 950 Aminosäuren, die transmembranäre Domäne enthält die Aminosäuren 951 bis 973 und die Tyrosin- Kinase Domäne enthält die Aminosäuren 1102 bis 1351 (Cooper et al. 1984; Park et al. 1987). 1.3.2 Bekannte Mutationen des MET Proto-Onkogens in Bezug auf papilläre RCC Es wurden bisher 15 verschiedene konstitutionelle und somatische Mutationen des MET-Gens identifiziert. Alle Mutationen beschränken sich auf die Exons 16, 17, 18 und 19 des MET- 19 Einleitung Gens. Die Mutationen sind ausschließlich vom Missense-Typ und in der Tyrosin-Kinase Domäne des Gens lokalisiert. Bei keiner dieser Mutationen konnte eine Inaktivierung des MET-Rezeptors nachgewiesen werden. Die ersten Mutationen des MET-Gens in Bezug auf papilläre RCC wurden 1997 von Schmidt et al. beschrieben. Es wurden Mutationen bei Patienten mit familiärem papillären NCC und sporadischem papillären RCC gefunden. Untersucht wurden Exon 2 und Exon 9 bis 21 (Schmidt et al. 1997). In der gleichen Studie konnte zudem eine sehr häufig zusätzlich auftretende Trisomie des Chromosom 7 bestätigt werden, die charakteristisch für sporadische papilläre RCC ist (Kovacs 1993). Es wurden 9 Mutationen gefunden, lokalisiert in den Exons 17, 18 und 19 in der Tyrosin-Kinase Domäne. Davon waren 5 konstitutionelle Mutationen und 4 somatische Mutationen. Insgesamt wurden konstitutionelle Mutationen der Keimbahn bei 4 von 7 betroffenen Familien gefunden, bei einem Patienten ohne positive familiäre Anamnese und einem Patienten, dessen Mutter einen nicht genau klassifizierten Nierentumor hatte. Somatische Mutationen wurden bei 3 von 60 (5%) Patienten mit sporadischen papillären NCC identifiziert und bei 2 von 3 Tumoren eines Patienten mit bilateralem papillären RCC. Eine weitere Studie der gleichen Gruppe konnte bei einem Patientengut von 128 Patienten weitere 5 neue Missense-Mutationen bei 8 Patienten identifizieren, 5 konstitutionelle und 3 somatische Mutationen, wobei nur sporadische papilläre RCC untersucht wurden. Bei dieser Studie wurde zudem ein Polymorphismus in Exon 15 entdeckt, der mit einem Nukleotidaustausch von Cytosin zu Thyrosin bei nt 3223 einhergeht. Die Mutationsanalyse konzentrierte sich bei dieser Studie auf die Exons 16 bis 19, die Exons 5, 7 und 13 wurden zusätzlich sequenziert (Schmidt et al. 1999). Eine weitere neue Mutation vom Missense-Typ wurde bei einer Untersuchung von zwei großen nordamerikanischen Familien gefunden, wobei Exon 2, 4, 7 und 9 bis 21 untersucht wurden (Schmidt et al. 1998). In der ersten Familie waren 9 Personen betroffen, in der zweiten Familie 13 Personen. Die Mutation ist bei beiden Familien identisch, was zu der Annahme eines gemeinsamen Vorfahren führte. Nach Sequenzierung wurde ein Nukleotidaustausch von Histidin zu Argenin identifiziert, was jedoch nicht zu einer Aminosäurenvertauschung führt. Bei der klinischen Untersuchung hatten 22 von 32 Familienmitgliedern mit der Mutation einen Nierentumor, keine Person ohne die Mutation hatte einen Nierentumor und 12 von 13 Personen über 50 Jahre die Mutationsträger waren, hatten einen Nierentumor. Einleitung 20 Insgesamt zeigten 75% der betroffenen Patienten dieser Studien multiple, bilaterale Tumore, Metastasenbildung war selten. Das durchschnittliche Alter bei Entdeckung der Tumore war 40 Jahre. Die Tumore ließen sich alle dem Subtyp 1 der papilläre Nierenkarzinome zuordnen. Die Studien zeigten weiterhin, daß eine Mutation des MET- Gen scheinbar mit einer verminderten Penetranz der Erkrankung und einem spätem Manifestationsalter der klinischen Symptome vergesellschaftet ist. Eine Zusammenfassung aller bisher identifizierten Mutationen des c-MET Proto-Onkogens zeigt Tabelle 1.6 Exon NukleotidVeränderung Codon Effekt auf Aminosäure 16 G3522A V1110I Val Æ Ile 16 C3528T H1112Y His Æ Tyr 16 A3529T H1112L His Æ Leu 16 C3564G H1124D His Æ Asp 18 C3831G L1213V Leu Æ Val 19 G3930C D1246H Asp Æ His 19 T3936C Y1248H Tyr Æ His 19 T3936G Y1248D Tyr Æ Asp 19 A3937G Y1248C Tyr Æ Cys 19 T3997C M1268T Met Æ Thr 16 G3522A V1110I Val Æ Ile 16 C3528T H1112Y His Æ Tyr 16 A3529G H1112R His Æ Arg 17 T3640C M1149T Met Æ Thr 18 G3810T V1206L Val Æ Leu 19 G3906A V1238I Val Æ Ile 19 G3930A D1246N Asp Æ Asn 19 T3936G Y1248D Tyr Æ Asp 19 A3937G Y1248C Tyr Æ Cys 19 T3997C M1268T Met Æ Thr C3223T T1010I ThrÆ Ile somatisch konstitutionell MET-Polymorphismus 15 Tabelle 1.6: Bekannte Mutationen und Polymorphismen des c-MET Proto-Onkogens (aus: Schmidt et al. 1997, Schmidt et al. 1999) Einleitung 21 1.3.3 Zytogenetische Befunde bei papillären Nierenkarzinomen Verschiedene Studien konnten bei papillären Nierenkarzinomen eine Polysomie der Chromosomen 3q, 7, 8, 12, 16, 17 und 20 und bei Männern einen Verlust des Y-Chromosoms nachweisen (Kovacs 1993). Eine Kombination von Trisomie 7 und 17 scheint dabei besonders charakteristisch für papilläre RCC zu sein (Jiang et al. 1998, Corless et al. 1996) und eine Progression des Tumors scheint mit einer Trisomie 20 einherzugehen (Palmedo et al. 1999). Insgesamt konnte bei dem Großteil der Patienten mit papillären Nierenkarzinomen und MET-Mutationen zusätzlich eine Trisomie 7 beobachtet werden (Zhuang et al. 1998; Kovacs et al.1993). Diese Polysomie scheint für einen Wachstumsvorteil für normale Zellen verantwortlich zu sein (Ermis et al. 1995). Das MET- Allel auf Chromosom 7 wird dabei vervielfacht und kann für eine Erhöhung der Zellproliferation verantwortlich sein (Ermis et al. 1995). Dijkhuizen (1997) wies in zwei Fällen von sporadischen papillären RCC eine Trisomie des Chromosoms 17 nach und nahm somit eine Beteiligung eines weiteren Gens lokalisiert auf Chromosom 17 in der Pathogenese dieser Tumore an. Bei einer Untergruppe von sporadischen papillären Nierenkarzinomen wurde eine chromosomale Translokation (X;1)(p11; q21) nachgewiesen (Meloni et al. 1993). Van Kessel`s und Cooper`s Arbeitsgruppe zeigten, daß diese Translokation zu einer Fusion des Transkriptionsfaktor TFE3-Gens bei p11.2 auf dem X-Chromosom mit einem neuen Gen führt, genannt PRCC, auf Chromosom 1q21.2 . Weitere Untersuchungen des TFE3-Gen in diesen Tumoren führten zu der Entdeckung, daß TFE3 mit PRCC verbunden ist und daß das PRCC-TFE3 Fusionsprotein offensichtlich eine N-terminale Transkriptionsfaktor- aktivierende Domäne darstellt, die mit der TFE3 DNA-bindenden Domäne verbunden ist (Beckmann und Kadesch 1990; Sidhar et al. 1996). Ein Charakteristikum dieser Translokation ist der Verlust des normalen TFE3-Transkripts und Übrigbleiben des hauptsächlich aktivierenden Fusionsproteins. Diese Translokation wurde bei papillären RCC des Subtyp 1 gefunden, die mit einem unterschiedlichen klarzelligen Erscheinungsbild einhergehen. MET, PRCC und TFE3 sind somit die bisher bekannten Gene, deren Mutationen für ein papilläres Nierenkarzinom prädisponieren. PRCC- und TFE3-Mutationen scheinen eine Rolle bei der Pathogenese der sporadischen papillären Nierenkarzinome zu spielen, METMutationen stehen im Zusammenhang mit sporadischen sowie hereditären papillären Nierenkarzinomen. Einleitung 22 1.4 Fragestellung Die vorliegende Arbeit beschäftigt sich mit der molekulargenetischen Diagnostik der hereditären und sporadischen papillären Nierenkarzinome in Bezug auf das c-MET-ProtoOnkogen. Eine Identifizierung von Patienten mit familiären Nierenkarzinomen ist besonders für den Patienten und seine Angehörigen wichtig, da sich aus einer solchen Diagnose wichtige Folgen hinsichtlich der Früherkennung für noch nicht erkrankte, aber mutationstragende Angehörige ergeben. Auch der wissenschaftliche Aspekt ist bedeutsam, da bisher noch nicht viele hereditäre papilläre Nierenkarzinome identifiziert wurden, auch aufgrund schwieriger anamnestischer Evaluation. Da das papilläre Nierenkarzinom erst seit einiger Zeit als eine eigene Entität angesehen wird und das familiäre papilläre Nierenkarzinom sehr selten auftritt, gibt es noch nicht allzu viele molekularpathologische Studien zum MET-Gen. Bisher wurden 15 unterschiedliche Mutationen des MET-Gens vom Missense-Typ gefunden, alle in einer kleinen Region des Gens lokalisiert, wobei jedoch noch nicht alle Exons des MET-Gens analysiert wurden. Vergleiche der klinischen Aspekte mit den histologischen Befunden und den molekulargenetischen Ergebnissen könnte hilfreich für eine Prognosestellung sein und weitere Hinweise auf eine molekulargenetische Pathogenese liefern. Es ergaben sich folgende Fragen: 1. In welchem Protzentsatz finden sich bei Patienten mit histologisch gesichertem papillären Nierenkarzinom Mutationen im MET-Gen? 2. Existieren weitere Mutationen in anderen Regionen des MET-Gens bei Patienten mit papillären Nierenkarzinomen? 3. Wie korrelieren das pathologische Erscheinungsbild des Tumors und die histologische Klassifizierung miteinander, insbesondere in Bezug auf die familiären papillären Nierenkarzinome, verglichen mit den bisherigen Studien? 23 2 MATERIAL UND METHODEN 2.1 Material Material und Methoden 2.1.1 Patientengut Es wurden die schon im Vorfeld gesammelten Blutproben des Registers der Jahre 1984 bis 1994 (Prof. Dr. H.P.H. Neumann 1984-1994) und die über die Hausärzte der Patienten angeforderten Blutproben des erweiterten Registers verwendet. Die Proben wurden bei – 80° C tiefgefroren gelagert. Nach Aufarbeitung der DNA wurde das Material bei 4° C im Kühlschrank aufbewahrt. Zusätzlich wurden 10 DNA-Positivproben mit gesicherten Mutationen der Arbeitsgruppe von Prof. Dr. L. Schmidt an den National Instituts of Health (NIH) als Vergleichsproben genutzt. Insgesamt wurden somit DNA-Proben von 70 Patienten molekulargenetisch untersucht. Aufgrund der histologischen und anamnestischen Befunde wurden die Patienten in vier Gruppen unterteilt: 10 Patienten mit anamnestisch, histologisch und molekulargenetisch gesicherten papillären – Nierenkarzinomen. Diese Patienten wurden schon im Vorfeld von einer anderen Arbeitsgruppe analysiert und es wurden Mutationen im MET-Gen identifiziert (Schmidt et al. 1997, Schmidt et al. 1999). Diese Gruppe diente somit als Positivprobe für die entsprechenden Exons. Es wurden hierbei bei 8 Patienten nur die mutationstragenden Exons als positive Vergleichsprobe untersucht. Diese Fälle stammen aus dem Register der Arbeitsgruppe von Prof. Dr. L. Schmidt. Zwei Patienten der zehn Fälle stammen aus dem eigenen Register der Jahre 1984 bis 1994 und waren zur Mutationsanalyse an die Arbeitsgruppe von Prof. Dr. L. Schmidt an den NIH abgegeben worden. Bei diesen Patienten wurden nochmals die Exons 2 und 4 bis 21 analysiert. Bei vier dieser Fälle waren Mutationen in Exon 16 bekannt, darunter die beiden Fälle des eigenen Registers, die die gleiche Mutation tragen. Eine weitere Positivprobe des Exons 16 trägt ebenfalls die gleiche Mutation. Bei einem Fall liegt eine Mutation im Exon 17 vor, bei einem weiteren Fall eine Mutation in Exon 18 und bei vier weiteren Fällen eine Mutation in Exon 19, wobei zwei Patienten die gleiche Mutation tragen. 2 Patienten mit einer bekannten somatischen Mutation, welche im Tumorgewebe dieser Patienten von der Arbeitsgruppe von Prof. Dr. L. Schmidt identifiziert wurde, ohne eine positive familiäre Anamnese. Beide Patienten stammen aus dem eigenen Register 24 Material und Methoden der Jahre 1984 bis 1994. Eine molekulargenetische Analyse der Blut-DNA war durch diese Arbeitsgruppe nicht erfolgt, da nur Tumorgewebe zur Verfügung stand. Entsprechende Blutproben wurden daher nachträglich über die Hausärzte der Patienten angefordert. Einer der beiden Patienten wurde im Loretto-Krankenhaus Freiburg operiert, das histologische Gutachten wurde vom Pathologischen Institut der Universität Freiburg erstellt. 9 Patienten mit histologisch rein papillären Nierenkarzinomen, ohne positive familiäre Anamnese. Alle Patienten stammen aus dem eigenen Register. Die Operation wurde immer in der Urologischen Abteilung der Universitätsklinik Freiburg durchgeführt, die histologischen Gutachten wurden vom Pathologischen Institut der Universität Freiburg erstellt. 49 Patienten mit histologisch größtenteils papillär strukturierten Nierenkarzinomen ohne positive familiäre Anamnese. Alle Patienten aus dem eigenen Register. Die Operation wurde immer in der Urologischen Abteilung der Universitätsklinik Freiburg durchgeführt, die histologischen Gutachten wurden vom Pathologischen Institut der Universität Freiburg erstellt. Zusammenfassende Darstellung der verwendeten DNA-Proben Proben- Bekannte Mutation anzahl 10 A3529G 2 Exon Register 16 Eigenes Register Untersuchte Arbeit 2, 4-21 A3529G 16 Eigenes Register 2, 4-21 G3522A 16 NIH 16 A3529G 16 NIH 16 T3640C 17 NIH 17 G3810T 18 NIH 18 G3930C 19 NIH 19 G3930A 19 NIH 19 G3930A 19 NIH 19 T3936C 19 NIH 19 G3522A (somatisch) 16 Eigenes Register 2, 4-21 T3936G (somatisch) 19 Eigenes Register 2, 4-21 50 - - Eigenes Register 2, 4-21 8 - - Eigenes Register 16, 17, 18, 19 Exons in dieser 25 Material und Methoden Für die molekulargenetischen Untersuchungen wurden die im Folgenden aufgeführten Geräte und Reagenzien benutzt, welche für diese Arbeit im Labor N5 der Abteilung IV der Medizinischen Universitätsklinik zur Verfügung standen. 2.1.2 Geräte Agarose – Elektrophoresekammer E 92, Biometra, Göttingen Geltrockner SE 1160, Hoefer Scientific Instruments Gene Phor – Gelkammer Pharmacia Biotech Heizblock DB 3A, Techne, Cambridge, UK Membran – Vakuumpumpe MZ2C/ 2,4m3/ h, Vacuubrand, Wertheim PCR – Maschine 60/ 2, Biomed, Theres PCR – Maschine Touchdown, Hybaid Polaroid – Kamera PHC 34, Hoefer Polyacrylamidgel – Elektrophoresekammer SE 1600, Hoefer Spannungsgeräte Power – All 3000V/ 200mA, Serva, Heidelberg UV – Lampe UVTM – 25, Hoefer Zentrifugen Biofuge 13, Heraeus Rotixa/ RP 4200 Hettich, Tuttlingen 26 Material und Methoden 2.1.3 Enzyme Proteinase K Boehringer Mannheim Taq – DNA – Polymerase Gibco 2.1.4 Nukleotide und Nukleinsäuren dATP, dGTP, TTP, dCTP Sigma, Deisenhofen Längenmarker: DNA Längenstandard V Boehringer Mannheim Primer – Oligonukleotide MWG, Biotech, München 2.1.5 Chemikalien Acrylamid ( 2x kristall. ) Serva, Heidelberg Acrylease Stratagene Agarose ( SeaKem LE ) Biozym Diagnostik, Hameln Ammoniumpersulfat Sigma Borsäure Merck Bromphenolblau Merck, Darmstadt Chloroform J.T. Baker EDTA ( Ethylendiamintetraacetat ) Sigma 27 Material und Methoden Essigsäure Roth Ethanol J.T. Baker Ethidiumbromid Sigma Ficoll ( Typ 400 ) Glycerol Sigma Harnstoff Roth Igepal Sigma Magnesiumchlorid Sigma MDE – Gel Solution FMC Bioproducts Europe Natriumcarbonat ( wasserfrei, reinst ) Merck Paraffin ( dickflüssig ) Merck Phenol ( kristall., reinst ) Merck SDS ( Dodecylsulfat ) Merck Salpetersäure Merck TEMED ( N, N, N`, N`,- Tetramethylmethylendiamin ) Sigma Tris ( hydroxymethyl ) aminomethan Sigma Xylencyanol Merck 28 Material und Methoden 2.1.6 Lösungen 2x Acrylamidgel – Ladepuffer 49ml Formamid 12,5mg Bromphenolblau 12,5mg Xylencyanol 1,0ml 0,5 M EDTA 2% Agarosegel 7g Agarose 1,0 x TBE ad 350ml, kurz aufkochen 6 x Agarosegel – Ladepuffer 12,5mg Bromphenolblau 12,5mg Xylencyanol 7,5g Ficoll (Typ 400) ad 50ml ddH2O 20 % Ammoniumpersulfat 0,2g APS 1,0ml ddH2O Mutation Detection Enhancement 62,4ml H2O ( MDE ) Gel – Lösung für SSCP 22,5ml MDE 5,6ml 10 x TBE 60 μl TEMED 600 μl APS 10 x dNTP – Lösung 2mM je dNTP in ddH2O 10 x PCR – Puffer 100mM Tris – HCl (pH 8,3) 500mM KCl 10 – 25mM MgCl2 0,1% Gelatine Reduktionslösung für Silberfärbung 60g Na2CO3 1,0ml Formaldehyd (37%) ad 2l ddH2O 29 2x Saccharose – Lösung Material und Methoden 218g Saccharose 2,4g Tris 2,03g MgCl2 20ml Triton X – 100 ddH2O ad 1l, mit HCl auf pH 7,6 Salz – EDTA 2,19g NaCl 4,46g EDTA mit 1M NaOH auf pH 8 einstellen, ad 500ml ddH2O Silbernitrat – Lösung 12mM 2g ad 1l ddH2O 1 x TBE 86,4g Tris 44g Borsäure 32ml 0,5mM EDTA (pH 8,0) ad 8l ddH2O 0,6 x TBE 51,8g Tris 26,4g Borsäure 19,2ml EDTA ad 8l ddH2O TE – Puffer pH 8,0 1,0ml 0,5 M Tris –HCL (pH 8,0) 1,0ml 50mM EDTA (pH 8,0) 48ml ddH2O 10 x Warner – Lösung 81,1g NaCl 3,7g KCl 0,3g MgCl2 ddH2O ad 1l 30 2.2 Material und Methoden Methoden 2.2.1 Register Ziel war es, alle Patienten mit papillären Nierenkarzinomen der Universitätsklinik Freiburg von 1994 bis einschließlich 1998 zu erfassen und dem Register der Jahre 1984 bis 1994 anzufügen. Zudem sollten verschiedene Ergebnisse der diagnostischen Untersuchungen und die genauen histologischen Befunde dokumentiert werden. Anschließend sollten die bisher gesammelten Blutproben der Patienten bis 1998 ergänzt werden. Dies geschah in mehreren Schritten : 1. Da kein Register für Patienten mit papillären Nierenkarzinomen für diesen Zeitraum existierte, wurden zunächst alle Patienten, die sich einer Operation aufgrund der Diagnose Nierenkarzinom unterzogen hatten, registriert. Dazu wurden alle Operationsberichte der Jahre 1994 bis 1998 der Urologischen Universitätsklinik durchgesehen. 2. Nachdem alle Patienten erfasst waren, wurden im Archiv der Urologischen Klinik alle Krankenakten dieser Patienten herausgesucht und nach dem histologischen Befund geordnet. Es wurde ein Register der Patienten mit papillären, sowie mit klarzelligen, onkozytären, überwiegend sarkomatoiden und chromophoben Nierenkarzinomen sowie Angiomyolipomen, erstellt. Aus den histologischen Gutachten und Untersuchungsbögen der Patienten mit papillären Nierenkarzinomen wurden verschiedene Befunde entnommen und in einer gesonderten Liste zusammengestellt. 3. Da der überwiegende Teil der Akten keine sicheren Aussagen zur Familienanamnese und zum Verlauf zuließen, wurden die jeweils zuständigen Hausärzte ermittelt und angeschrieben. Eine erstellte Einverständniserklärung für die Patienten, ein Informationsschreiben zu der geplanten Untersuchung und ein Fragebogen mit den wichtigsten Punkten zur Familienanamnese, Verlauf und Begleiterkrankungen wurden beigefügt. In einem zweiten Schreiben wurden die Hausärzte um die Zusendung von Patienten – EDTA - Blut gebeten. Die aus den Antworten erhaltenen Informationen wurden erfasst und auf einer entsprechenden Liste ergänzt. Zu den schon vorhandenen Blutproben kamen somit noch 28 Proben von neu erfassten Patienten für eine molekulargenetische Untersuchung hinzu, zusätzlich 8 angeforderte Positivproben aus der Arbeitsgruppe von Prof. Dr. L. Schmidt an den NIH, Bethesda/Frederick, Maryland. 31 Material und Methoden 2.2.2 DNA – Extraktion aus Vollblut ( EDTA ) Gewinnung von DNA aus peripheren Blutleukozyten durch eine Phenol- Chloroformextraktion nach Sambrook (1989). 1. Es wird ein 50 ml – Falcon mit 20 ml einer 2x Saccharose- Lösung, 10 ml einer 1 x Warner`s – Lösung und 0,5 ml Igepal gefüllt. Das Patienten EDTA – Vollblut wird gut durchmischt und anschließend 10 ml davon in die Mischung dazugegeben. Der Falcon wird gut gemischt und dann bei Raumtemperatur ca. 15 – 20 min rotiert, so daß die Plasmamembran durch Igepal aufgelöst wird und eine Lyse sichtbar wird. 2. Das Gemisch wird nun bei 4° C 20 min mit 3500 rpm zentrifugiert. Danach wird der Überstand vorsichtig dekantiert, so dass das Pellet im Falcon zurückbleibt. 3. Das Pellet wird einmal mit 15 ml einer 1 x Warner`s – Lösung gewaschen und dann 10 min mit 3500 rpm zentrifugiert. Die überstehende Waschlösung wird dekantiert. 4. Es wird ein 3,5 ml Mix, bestehend aus 2,7 ml TNE, 0,2 ml Proteinase K, 0,6 ml SDS (10% Stock) und 2,5 ml Natrium – EDTA zu dem im Falcon verbliebenem Pellet hinzugegeben. Das Pellet sollte vom Boden gelöst werden und evtl. mit Hilfe einer Pasteurpipette zerkleinert werden. Um einen optimalen Verdau der Zellkernmembran und ihrer Proteine durch SDS und der Proteinase K zu erreichen, wird das Röhrchen über Nacht in einem Wasserbad bei 55° C inkubiert. 5. Ist ein ausreichender Verdau erfolgt, so ist eine Lyse deutlich sichtbar. Dem Lysat werden 6 ml Phenol zugegeben und das Gemisch wird 10 – 15 min langsam rotiert. Durch diesen Vorgang werden Proteine in die organische Phenolphase extrahiert, welche sich im unteren Teil des Röhrchens bildet. Die DNA verbleibt dabei in der oberen, wässrigen Phase und kann nach anschließendem 10- minütigem Zentrifugieren mit 2500rpm bei ca. 20° C als Überstand abgenommen werden. Der Überstand wird in ein neues Röhrchen pipettiert und diese Prozedur wird nochmals mit Phenol wiederholt und danach zweimal mit reinem Chloroform. 6. Nach der letzten Chloroform – Extraktion wird der nun gereinigte, wässrige Überstand in 50 ml Falcons pipettiert. 7. Zum Ausfällen der DNA werden 1/10 Vol. Natriumacetat und ca. 2 Vol. Ethanol zugefügt. 8. Nun kann die DNA mit einer ausgezogenen Pasteur – Pipette herausgeangelt und in ein Eppendorfcup mit 0,2 – 1,5 ml TE – Puffer, je nach Menge der DNA, überführt werden, worin sie bei 4° C gelagert wird. 32 Material und Methoden 2.2.3 Polymerasekettenreaktion (PCR) (Saiki et al., 1985) Die PCR ist ein gängiges Verfahren der Gentechnologie, bei dem selektiv bestimmte DNA – Abschnitte vervielfältigt werden. Nach mehrmaliger Wiederholung des Verfahrens können selbst kleinste DNA – Mengen sichtbar gemacht werden, oder für weitere Diagnostik verwendet werden. So kommt es z.B. bei 30 Zyklen zu einer exponentiellen Zunahme der DNA auf das 2 30 – fache der ursprünglichen Menge. Um die DNA oder beziehungsweise gewünschte Exons mittels PCR zu amplifizieren, benötigt man ein entsprechendes Reaktionsgemisch, wofür hier ein 0,2 ml Eppendorf – Röhrchen benutzt wird mit einem Ansatzvolumen von 10,1 μl, welches sich wie folgend zusammensetzt : • 5 μl ddH2O • 1 μl DNA – Verdünnung (die DNA wurde hierfür in einem TE – Puffer 1 : 10 verdünnt) • 1 μl 10 x PCR – Puffer (verschiedene Konzentrationen, entsprechend den optimalen Bedingungen jedes einzelnen Primerpaares, welche experimentell bestimmt wurden und 1,0 mM und 1,5 mM MgCl2 betrugen) • 1 μl 10 x dNTP – Gemisch • 1,0 μl forward Primer • 1,0 μl reverse Primer • 0,1 μl Taq – Polymerase (thermostabil) Um eine Polymerasekettenreaktion zu erreichen, muss das Reaktionsgemisch in der PCR – Maschine immer wieder erhitzt und abgekühlt werden, was hier innerhalb 30 Zyklen geschah, die jeweils aus drei Teilschritten bestehen : 1. Zuerst wird das Gemisch 30 sec. auf 95° C erhitzt, um eine Auftrennung des DNA – Doppelstranges in zwei Einzelstränge zu erreichen (Denaturierung). Für eine verlängerte Denaturierung wird vor dem ersten Zyklus 5 min auf 95° C erhitzt. 2. Dann wird die Temperatur für 40 sec. auf z.B. 62° C heruntergesetzt. Dies ist die Annealing – Temperatur der beiden Primer – Oligonukleotide, die für die Hybridisierung der Primer an ihren komplementären DNA – Abschnitt nötig ist. Bei der Hybridisierung setzen die Primer an ihrem komplementären DNA – Abschnitt an Strang und Gegenstrang an und markieren so die zu amplifizierende DNA – Sequenz. Zudem wird so gleichzeitig wieder ein Stück Doppelstrang an der Ansatzstelle der Primer gebildet, welches als 33 Material und Methoden Ausgangspunkt für die Polymerase nötig ist. Jeder Primer hat eine eigene optimale Annealing – Temperatur und es wurden jeweils die experimentell ermittelten Temperaturen verwendet, die sich zwischen 58° C und 62° C bewegen. 3. Im dritten Schritt wird die Temperatur für 30 sec auf 72° C erhöht, um den höchsten Aktivitätsbereich der Taq – Polymerase zu erreichen. Die Polymerase synthetisiert, von den Primern ausgehend, in beide Richtungen neue DNA – Stränge, unter Verwendung der Desoxynucleosid – triphosphate (dNTP`s). Im letzten Zyklus wird eine verlängerte Polymerase – Synthese Phase eingesetzt, durch Erhöhung der Zeit auf 10 min. In der vorliegenden Arbeit wurden 19 der 21 Exons des MET-Gens untersucht. Für Exon 3 konnten bisher keine entsprechenden Primer hergestellt werden und Exon 1 ist nicht kodierend und somit nicht relevant. Diese beiden Exons wurden daher nicht untersucht. Aufgrund der Größe von Exon 2 ist es in sieben Fragmente aufgeteilt, für die jeweils entsprechende Primer existieren. Exon 9, Exon 13, Exon 14, Exon 15 und Exon 21 sind gleichfalls jeweils in zwei Fragmente unterteilt, da ihre Produkte ebenfalls sehr groß sind. Die optimalen PCR-Bedingungen der einzelnen Primer, ihre genauen Sequenzen und die Größe des Produktes sind der folgenden Tabelle zu entnehmen. F: forward primer bp: Basenpaare R: reverse primer – jeweils in 5`Æ 3`Richtung mM: Millimolar Exon Primersequenz 2–1 F: GTC CAG TTG GGA AGC TTT ATT TC R: CAT AAA TGT AGT TAG TGG CAC CAA G F: TTC ACC GCG GAA ACA CCC ATC R: AGT AGG TGT CGA CAA CTA GAG CC F: GCC AAT TTA TCA GGA GGT GTT TGG R: CAC AGT CAG GAC ACT GGC TG F: CTC CCC ACA GAT AGA AGA GCC R: GGG TAA GAA TCT CTG AAC TCA GG F: CCA GTC CTA CAT TGA TGT TTT ACC R: GCT GAC ATA CGC AGC CTG AAG TAT F: ACA TGG AAA TGC CTC TGG AGT G R: TTT GAT AGG GAA TGC ACA CAT GGC F: CCG AAC CAA TGG ATC GAT CTG C R: TAT ACC TCA CAG TTT ATA AGT GGG F: AAA CTG AGC TTG TTG GAA TAA GG R: GTG ACA CTG GTT GTA AAT ATG CA MgCl2Konz. 2–2 2–3 2–4 2–5 2–6 2–7 4 Annealing- Größe des Temperatur PCR-Produkts 1,0 mM 62 °C 335 bp 1,5 mM 60 °C 241 bp 1,5 mM 60 °C 214 bp 1,5 mM 60 °C 252 bp 1,0 mM 62 °C 277 bp 1,0 mM 62 °C 233 bp 1,0 mM 62 °C 181 bp 1,0 mM 62 °C 254 bp 34 Exon Primersequenz 5 F: CAA ACT AGA TAC CCC TCC TGG R: ACA AAA ACA TGT ATG CCA GCT GT F: TTT GTT CGT TTT CCA TAA ATG TGA R: AAG CAC ACC CCA GCA AAG CAT F: TCC CTT GGA TTT GTC ATG TAT TAA R: CAA AAC AAA GTA ACT TGT TCA TCT F: GAA GAT TCT ATC CTA TCA TGT TTG R: TTT TTC TTC TCC ACA CAC ACA CAC F: AAG GTA AGT TTG AGA TCC AGT CTG R: CGG TAA CTG AAG ATG CTT GTC TC F: ATT GAC TTA GCC AAC CGA GAG AC R: CCT CTT GAC TAT TCT ACA GC F: CTC TGA CCT GTA ATC AGT GCA R: GTT AGA GGC AAA GAT GCA GAG F: GGA TGT TGC CAA GCT GTA TTC R: GGC CAA GTA CAA TTG TAT TC F: TGT AAG CAT TCC TGC AGA ACT G R: ACT GGC TTC CTT ATT TAC ATC AT F: ATC TAT CAT GGG TAA ATG CTG AC R: TCC TGT GAA ATT CTG ATC TGG TTG F: AGC AAT TTC TTC AAC CGT CCT TGG R: TGA CTG TGT TCT TAA GGT AAC TTC F: GGG CCC ATG ATA GCC GTC TTT AA R: GTT GGG CTT ACA CTT CGG GCA C F: ACT CCT CAT TTG GAT AGG CTT G R: TAC ACA ACA ATG TCA CAA CCC A F: CCA TTA AAT GAG GTT TTA CTG TTG T R: AGG TCA ATG TGG ACA GTA TTT TGC F: CAT CCT AAC TAG TGG GGA CTC TG R: CAA AGG CCA AAG ATA AAA TGC TTA F: ATT AAA TGT TAC GCA GTG CTA AC R: GGT TGC AAA CCA CAA AAG TAT F: GTA TTC ACT GTT CCA TAA TGA AGT R: GAT GGC TGG CTT ACA GCT AGT T F: AAC AGT AGA TGC TTA GTT TAT GCT R: AAC AGA TTC CTC CTT GTC ACT T F: TTC TAT TTC AGC CAC GGG TAA T R: ATG AAA GTA AAA GAG GAG AAA CTC F: TAT GTA TGG TCA CAT CTC TCA C R: CCT TTG AAG GCA GGC ATT TCT GT F: ACA GAA ATG CCT GCC TTC AAA GG R: CGG AGC GAC ACA TTT TAC GTT CAC F: CAC CCT AAA GCC GAA ATG CG R: CAA GGA GCA AAG AAT ATC GAT GGC Material und Methoden MgCl2Konz. 6 7 8 9–1 9–2 10 11 12 13 – 1 13 – 2 14 – 1 14 – 2 15 – 1 15 – 2 16 17 18 19 20 21 – 1 21 – 2 Annealing- Größe des Temperatur PCR-Produkts 1,0 mM 57 °C 259 bp 1,0 mM 60 °C 268 bp 1,5 mM 55 °C 225 bp 1,5 mM 62 °C 273 bp 1,5 mM 60 °C 286 bp 1,5 mM 60 °C 163 bp 1,0 mM 62 °C 259 bp 1,0 mM 62 °C 360 bp 1,5 mM 57 °C 252 bp 1,5 mM 60 °C 115 bp 1,0 mM 62 °C 200 bp 1,5 mM 50 °C 139 bp 1,5 mM 55 °C 170 bp 1,5 mM 55 °Co 189 bp 1,5 mM 55 °C 229 bp 1,5 mM 50 °C 176 bp 1,5 mM 50 °C 286 bp 1,5 mM 55 °C 214 bp 1,5 mM 57 °C 252 bp 1,5 mM 50 °C 250 bp 1,5 mM 57 °C 213 bp 1,5 mM 57 °C 307 bp Tabelle 2.1: Benutzte Primer zur Amplifikation des MET-Gens (Referenz : Duh et al., 1997) 35 Material und Methoden 2.2.4 Agarosegel – Elektrophorese Anhand der Agarosegel – Elektrophorese kann der Erfolg einer PCR überprüft werden. Es werden jeweils 4 μl der verschiedenen Amplifikationsprodukte mit je 3 μl eines 6 x Agarosegel – Ladepuffers vermischt. Jedes einzelne Gemisch wird dann in eine Geltasche auf ein 2%- iges Agarosegel aufgetragen, welches in einer Gelkammer liegt und mit 1 x TBE – Puffer bedeckt wird. Gleichzeitig wird am Rand ein Längenmarker (Boehringer Mannheim, Längenstandard V) gleicher Konzentration aufgetragen. Bei 250 Volt und einer Laufzeit von ca. 4 min werden die DNA – Fragmente aufgetrennt. Anschließend wird das Gel auf eine UV – Lampe bei 366 nm gelegt und durch das im Gel vorhandene Ethidiumbromid können die aufgetrennten Fragmente sichtbar gemacht werden. Ethidiumbromid ist ein fluoreszierender Stoff, der mit der DNA interkaliert und unter UV – Licht leuchtet. Die DNA – Fragmente können nun durch Vergleich mit dem Längenstandard beurteilt werden. Zur Dokumentation werden die Gele mit einer Polaroid – Kamera fotografiert. Bild 2.1 Bild 2.1: Agarosegelelektrophorese des Exons 12 mit Proben, Längenmarker links im Bild Für die Herstellung des 2%- igen Agarosegels werden 350 ml 1 x TBE mit 7 g Agarose und 15 μl Ethidiumbromid vermischt und in der Mikrowelle bei 500 Watt erhitzt, bis sich die Agarose vollständig gelöst hat. Nach kurzer Abkühlung wird die Mischung zum Erhärten in eine Gießvorrichtung gegossen und mit einem Taschenkamm versehen. Ca. 1 Stunde später kann das Gel verwendet werden. 36 Material und Methoden Wie sich bei dieser Erfolgskontrolle zeigte, traten bei einigen Primerpaaren nur unspezifische Bindungen auf, welche durch das Auftreten von mehreren Banden (Produkten) in verschiedenen Größenbereichen (Vergleich mit Längenmarker) sichtbar wurden. Um ein zufrieden stellendes Ergebnis zu erreichen, wurden für jedes Primerpaar die optimalen Bedingungen bezüglich Pufferkonzentration und Annealing – Temperatur experimentell bestimmt. Ein gutes Bindungsverhalten wird durch eine scharfe Bande im Agarosegel angezeigt, die sich in Höhe der berechneten Größe der Reaktionsprodukte befindet. 2.2.5 SSCP – Analyse (Ainsworth et al., 1991) Die SSCP – Methode ermöglicht es, kleinste Abweichungen in der Primärsequenz von DNAEinzelsträngen (aus der PCR hervorgegangen) zu erkennen. Dabei wird sich ein ganz spezielles Faltungsverhalten von DNA – Einzelsträngen zu nutze gemacht. Unter nicht – denaturierenden Bedingungen faltet sich ein DNA – Einzelstrang aufgrund intramolekularer Wechselwirkungen zu einer ganz bestimmten Konformation. Ist seine Primärsequenz jedoch verändert, z.B. durch eine Punktmutation, faltet sich der Einzelstrang anders. Im Gegensatz zu den nicht veränderten Wildtyp - Einzelsträngen zeigt diese neue Konformation ein anderes Laufverhalten in der Gelelektrophorese, da es unterschiedlich schnell durch das Gel wandert. Im Gel zeigt sich somit bei mutierten Einzelsträngen ein verändertes Bandenmuster, es können eine oder mehrere zusätzliche Banden auftreten (SSCP – shift). Für die Elektrophorese wurden Acrylamidgele verwendet. Zum Teil wurden die Gele zusätzlich mit Glycerol versetzt. Glycerol verändert die Gelmatrix und somit auch die Laufeigenschaften. Es wirkt stabilisierend auf Sekundärstrukturen, die eine höhere Variabilität haben. 2.2.6 Acrylamidgel – Elektrophorese Für die Elektrophorese benötigt man Flachbettgele (370 x 290 x 0,25 mm), die vertikal in die Gelkammern eingespannt werden. Zur Herstellung eines Gels bedarf es zweier Glasplatten, von denen beide vorher 1 x mit 70%-igen und 1 x mit 100%- igen Alkohol gereinigt werden. Eine Glasplatte wird zusätzlich noch mit Acrylease abgerieben, um eine spätere Abhebung der Platte vom Gel zu erleichtern. Beide Glasplatten werden, getrennt durch zwei schmale 37 Material und Methoden Abstandshalter (0,25 mm dick), übereinander gelegt und an den langen Seiten und einer kurzen mit Klebeband gründlich abgedichtet. Die vorbereitete Gelmischung wird luftblasenfrei zwischen die Glasplatten gegossen und anschließend ein Taschenkamm eingesetzt. Für die Gelmischung werden verwendet: • 22,5 ml einer MDE – Gellösung (Mutation Detection Enhancement) • 62,4 ml ddH2O • 5,6 ml 10 x TBE • 60 μl TEMED (Tetramethylendiamid) • 600 μl APS (Ammoniumpersulfat, 20%- ig) Für mit Glycerol versetzte Gele wurde die Menge an ddH2O auf 44,4 ml reduziert und zusätzlich 18 ml 50%- iges Glycerol zugegeben. Nach ca. einer halben Stunde ist ein Gel dieser Zusammensetzung auspolymerisiert, wobei diese Zeitspanne von der Konzentration des APS in der Lösung abhängt. Zur Elektrophorese werden die Gele in die vertikalen Gelkammern eingespannt und der Kamm mit einem Scalpell herausgezogen. Es wird ein 0,6 x TBE Laufpuffer verwendet. Um Verschmutzungen der Gele möglichst gering zu halten, wird ein ca. halbstündiger Vorlauf mit 400 Volt durchgeführt. Vor dem Auftragen der DNA – Proben werden die Geltaschen mit Laufpuffer mittels einer Mikropipette gespült. Die 10 μl PCR – Produkte werden jeweils mit 15 μl SSCP – Ladepuffer vermischt und 5 min bei 96° C denaturiert. Anschließend muss das Gemisch sofort in einem Eisbad abgekühlt werden, um eine Bildung von Doppelsträngen zu verhindern. Für die Beladung des Gels werden pro Tasche 8 μl des Gemisches verwendet. Bei einer Spannung von 250 Volt beträgt der Gellauf unter nicht denaturierenden Bedingungen ca. 16 Stunden. 38 Material und Methoden 2.2.7 Silberfärbung der Acrylamidgele (Budowle et al., 1991) Die Silberfärbung dient der Anfärbung der PCR – Produkte. Nach abgelaufener Elektrophorese werden die Glasplatten der Gelkammer entnommen und waagrecht auf die Platte der Färbevorrichtung (Bender et al., 1994) gelegt. Dabei wird die mit Acrealyse behandelte Glasplatte nach oben gelegt, so daß diese gelöst werden kann und das Gel auf der unteren Platte verbleibt. Anschließend wird der Färberahmen in die Vorrichtung eingespannt, so daß das Gel auf allen Seiten abgedichtet wird. Im ersten Schritt wird das Gel mit 1% - iger Salpetersäure behandelt, welche nach 3 min aus der Färbevorrichtung abgegossen wird. Danach wird das Gel 3 x mit Aqua destillatum gewaschen und eine 12 mMolare Silbernitratlösung wird zugegeben. Das Gel wird lichtgeschützt bedeckt und verbleibt so für 20 min. Nach Abgießen der Silbernitratlösung wieder 3- maliges Waschen mit Aqua dest. Dann wird eine Reduktionslösung zugegeben, bestehend aus Natriumcarbonat und Formaldehyd. Diese Lösung bewirkt eine Reduktion der Silberionen welche sich an die DNA gebunden haben und es tritt eine sichtbare Färbung ein. Die PCR- Produkte stellen sich so als dunkle Banden dar. Die Reduktionslösung wird danach sofort abgegossen und es erfolgt eine Spülung mit Aqua dest, welches 5 min auf dem Gel belassen wird. Zum Abbrechen und Fixieren der Färbereaktion wird das Gel kurz mit 10%- iger Essigsäure bedeckt und anschließend mit Hilfe eines auf das Gel aufgelegten Filterpapiers (Schleicher und Schuell) von der Glasplatte abgezogen. Das Gel mit Filterpapier wird für 1,5 Stunden auf einem Vakuum – Geltrockner bei 80° C getrocknet. 2.2.8 Sequenzierung Diejenigen Proben, die in der SSCP- Analyse aberrierende Banden zeigten, sowie die Positivproben, wurden zur DNA- Sequenzierung in ein darauf spezialisiertes Labor geschickt (Labor für DNA-Analytik, PD Dr. Juliane Alt-Mörbe, Freiburg). Hierfür wurden die auffälligen Banden aus den entsprechenden SSCP- Gelen ausgeschnitten und über 12 Stunden in TE- Puffer gelagert. Die DNA diffundiert dabei in den Puffer und kann anschließend mittels PCR und entsprechenden Primern wie unter 2.2.4 beschrieben reamplifiziert werden. Nach anschließender Erfolgskontrolle mittels Agarosegel- Elektrophorese wurden die Proben verschickt. Alternativ kann das PCR-Produkt auch direkt zur Sequenzierung gegeben werden. Die Sequenzierung erfolgte in einem halbautomatischen Sequenzierer mit Fluoreszenz- 39 Material und Methoden markierten Primern nach der Dideoxymethode. Außer den Ergebnissen stellt das Labor auch die entsprechenden Sequenz-Diagramme zur Verfügung. 2.2.9 Kontrolle anhand Normalproben Grundsätzlich wird im Labor von Prof. Dr. Neumann jede festgestellte Sequenz-Variante unklarer Bedeutung durch 100 Normalkontrollen (DNA gewonnen von gesunden Blutspendern, nach Einverständnis) hinsichtlich des Vorliegens einer möglicherweise pathogenen Mutation bzw. eines Polymorphismus überprüft. Dies ist auch für diese Arbeit mittels SSCP-Analyse und nachfolgender Sequenzierung nach o.g. Methoden erfolgt. 40 3 ERGEBNISSE 3.1 Register Ergebnisse Es wurden alle Patienten einbezogen, die in dem Zeitraum von 1994 bis 1998 wegen einem Nierenkarzinom in der Urologischen Abteilung der Chirurgischen Universitätsklinik Freiburg operiert wurden. Zusätzlich wurde ein Fall mit bekanntem papillären Nierenkarzinom aus dem Loretto-Krankenhaus Freiburg dem Register hinzugefügt. Insgesamt wurden 234 Fälle erfasst. Für den Zeitraum von 1984 bis 1994 konnte auf eine vorhandene Registerauswertung zurückgegriffen werden (Neumann et al. 1998). Als nächster Schritt erfolgte eine Einteilung nach dem histologischen Befund. Von den 234 Fällen fanden sich 46 Patienten mit papillären Nierenkarzinomen, 128 Patienten mit einem Nierenkarzinom vom klarzelligen Typ, 7 Patienten mit einem Karzinom vom chromophoben Typ und 6 Patienten mit einem vorwiegend sarkomatoiden Typus. Des Weiteren fanden sich 8 Fälle mit einem Onkozytom, 8 Patienten mit einem Angiomyolipom und weitere 31 Fälle mit nicht exakt klassifizierbaren Nierenkarzinomen. Tabelle 3.1 zeigt eine prozentuale Verteilung der Einteilung. Histologie Häufigkeit klarzellig 128 (55%) papillär 46 (20%) chromophob 7 (3%) sarkomatoid 6 (3%) Onkozytom 8 (3%) Angiomyolipom 8 (3%) nicht klassifizierbar 31 (13%) Tabelle 3.1: Histologischer Befund bei 234 Patienten mit Nierenzellkarzinomen (Abteilung Urologie der Universitätsklinik Freiburg 1994-1998) Durch Hinzufügen der 46 neu erfassten Fälle von Patienten mit papillären Nierenkarzinomen zu den schon vorhandenen Fällen der Jahre 1984 bis 1993, ergab sich eine Anzahl von 103 Patienten mit papillären Nierenkarzinomen in den Jahren 1984 bis 1998, die in der 41 Ergebnisse Universitätsklinik Freiburg in diesem Zeitraum operiert worden sind, einschließlich des auswärtigen Falles aus dem Loretto-Krankenhaus Freiburg. Alle Tumore wurden histopathologisch als papilläre oder überwiegend papilläre Nierenkarzinome klassifiziert. Anhand des histologischen Befundes, der Patientenakte und eines speziell erstellten Fragebogens an die Hausärzte der Patienten, wurden von allen Patienten mit papillären Nierenkarzinomen die klinischen Daten erfasst. Es konnten jedoch nicht sämtliche Daten erhoben werden, da manche Befunde nicht zugänglich waren, keine Rückantwort durch die Hausärzte erfolgte oder manche Patienten verstorben waren. Somit waren die erhobenen Daten in vielen Fällen inkomplett. In einem gesonderten Schreiben an die Hausärzte wurde jeweils eine EDTA- Blutprobe der entsprechenden Patienten angefordert. Die histo-pathologische Einteilung nach Delahunt und Eble wurde nicht überprüft, da eine solche Klassifikation in den histologischen Befunden nicht vorgenommen wurde. Tabelle 3.2 zeigt, welche Angaben in welcher Häufigkeit von den 103 Patienten evaluiert werden konnten. Patientendaten Häufigkeit n= 103 Geburtsdatum (103) 100% Anschrift (100) 97% Hausarzt (94) 91% Alter bei erster OP (101) 98% verstorben laut Hausarzt (19) 18% Antwort vom Hausarzt (64) 62% Blutprobe erhalten vom Hausarzt (28) 27% Tabelle 3.2: Angaben von 103 Patienten Das Alter der Patienten bei der ersten Operation lag zwischen 23 und 88 Jahren mit einem Mittelwert von 61,4 Jahren. Die Geschlechtsverteilung war 77/ 26 (männlich/ weiblich). 42 Ergebnisse Bezüglich des Tumors konnten folgende Daten erhoben werden, zusammengestellt in Tabelle 3.3 Tumor Häufigkeit n= 103 unilateral 97 (94%) bilateral 5 (5%) multifokal 7 (7%) sporadisch 80 (78%) familiär 1 (1%) unklar 22 (21%) TNM 99 (96%) OP-Art: N / E 99/4 (96%/4%) Tabelle 3.3: Daten zu 103 Tumoren N : Nephrektomie E : Enukleation Die Größe der Tumoren variierte zwischen 2,1 und 18 cm. Nach der TNM-Klassifikation variierte das Tumorstadium zwischen T1 und T4. Eine Übersicht der Häufigkeit der einzelnen Tumorstadien nach der TNM-Klassifikation bei 99 Patienten gibt Tabelle 3.4 Häufigkeit (n=99) Tumorstadium (7) (39) (32) (18) (0) (3) 6% 36% 29% 7% 0% 3% Lymphknotenbefall (11) 11% Fernmetastasen (20) 20% T1 T2 T3a T3b T3c T4 Tabelle 3.4: Häufigkeit der einzelnen Tumorstadien nach TNM bei OP-Datum bei 99 Patienten (TNMReferenz nach der TNM- Classification of malignant tumors, 1997, erläutert in Kapitel 1.2.5, Tab. 1.5) 43 Ergebnisse Von den 19 erfassten verstorbenen Patienten, erlagen 12 den nachfolgenden Komplikationen der Krankheit, 7 verstarben an anderer Ursache. Bei den an den Folgen des Nierenkarzinoms gestorbenen Patienten fanden sich ausschließlich die Tumorstadien T3a und T3b nach der TNM-Klassifikation bei OP-Datum. 3.2 Ergebnisse der SSCP- Analyse der Positivproben Die SSCP – Analyse wurde bei allen unter 2.1.1 beschriebenen Positivproben (Schmidt et al. 1997, Schmidt et al. 1999) als Screeningverfahren eingesetzt. Die Positivproben wurden als Vergleichskontrollen in den entsprechenden Exons genutzt und die Reproduzierbarkeit der erwarteten Ergebnisse wurde überprüft. Untersucht wurden alle 10 Positivproben der Exons 16, 17, 18 und 19, wobei für Exon 16 vier Positivproben vorhanden sind, für Exon 17 und 18 jeweils eine und für Exon 19 vier Positivproben (Schmidt et al. 1997, Schmidt et al. 1999). Bei drei Positivproben des Exons 16 handelt es sich um die gleiche Mutation. Zwei der entsprechenden Patienten gehören einer Familie an und stammen aus dem eigenen Register für papilläre RCC der Jahre 1984 bis 1998. Zwei Positivproben des Exons 19 tragen ebenfalls die gleiche Mutation. Die Mutationsanalyse erfolgte jeweils in der Arbeitsgruppe von Prof. Dr. L. Schmidt an den National Institutes of Health. Die weiteren Positivproben stammen aus derselben Arbeitsgruppe. Die Positivproben für die Exons 16, 18 und 19 fielen jeweils durch eine aberrierende Bande auf und konnten somit als positiv angenommen werden. Anschließende entsprechende Sequenzierungen konnten die bekannten Ergebnisse der Mutationsanalysen der Arbeitsgruppe von Prof. Dr. L. Schmidt bestätigen. In Exon 17 konnte die Positivprobe mittels SSCP – Analyse nicht bestätigt werden. Eine aberrierende Bande konnte nicht gesehen werden. Eine Sequenzierung konnte nicht durchgeführt werden, da keine DNA dieser Probe mehr vorhanden war. Im Folgenden sollen die Ergebnisse der Positivproben für die einzelnen Exons dargestellt werden. 44 Ergebnisse Positivkontrollen des Exon 16 Abbildung 3.1 zeigt einen Ausschnitt der SSCP- Analyse des Exon 16 mit den zwei Positivproben der gleichen Mutation und einer Normalprobe. In der Mitte des Bildes der Normalbefund, rechts und links davon fallen die zwei Positivproben mit einer zusätzlichen Bande auf (Pfeile). Die anschließende Sequenzierung einer der Positivproben ergab nebenstehenden Ausschnitt der DNA- Sequenz. Durch einen Vergleich mit der Wildtypsequenz erkennt man die Nukleotidveränderung von A zu G bei dem Nukleotid 3529. Die bekannte Mutation (Mutation A3529G) der Patienten konnte bestätigt werden. Bei diesen beiden Fällen wurden auch Exon 2 und Exon 4 bis 21 untersucht. Hierbei ergab sich jeweils ein Normalbefund. Referenz der Wildtypsequenz nach Park et al. 1987 (siehe Abbildung 3.1). Mutierte Sequenz: T A T A T C G T G G Wildtyp- Sequenz: T A T A T C A T G G Nukleotid: 3523 3524 3525 3526 3527 3528 3529 3530 3531 3532 Abbildung 3.1: Ausschnitt der SSCP- Analyse des Exons 16 mit zwei Positivproben (Pfeile) und Ergebnis der Sequenzierung einer der Positivproben Bei den hier abgebildeten Positivproben handelt es sich um die zwei Patientenproben aus dem erstellten Register der Jahre 1984 bis 1998, welche zur weiteren Diagnostik weitergeleitet worden waren. Die entsprechenden Mutationen dieser Patienten wurden in der Arbeitsgruppe von Prof. Dr. L. Schmidt an den NIH identifiziert und wurden daher als Positivproben genutzt. Bei den beiden weiteren Positivproben des Exons 16 zeigte sich in der SSCP- Analyse ebenfalls ein jeweils verändertes Laufverhalten. Die anschließenden Sequenzierungen 45 Ergebnisse konnten die bekannten Mutationen (Mutation A3529G und Mutation G3522A) der entsprechenden Proben bestätigen. Positivkontrolle des Exons 17 Die Positivprobe des Exons 17 zeigte in der SSCP – Analyse kein verändertes Laufverhalten gegenüber den anderen Proben (s. Abbildung 3.2). Auch zwei weitere SSCP–Analysen des Exons 17 mit veränderten Versuchsbedingungen hinsichtlich der Gelzusammensetzung und der Lauftemperatur, konnten kein abweichendes Laufverhalten der Positivprobe darstellen. Bezüglich der Gelzusammensetzung wurden reine Acrylamid- Gele, sowie Acrylamid- Gele mit 10% Glycerol versetzt, verwendet. Eine PCR zur Reamplifizierung dieser Probe anhand des ausgeschnittenen SSCP-Produkts misslang. Eine weitere Analyse konnte nicht durchgeführt werden, da keine DNA mehr vorhanden war. Eine Sequenzierung dieser Probe konnte somit nicht durchgeführt werden. Der Versuch an eine weitere DNA-Probe dieses Patienten zu gelangen verlief leider ebenfalls erfolglos. Abbildung 3.2: Ausschnitt der SSCP– Analyse des Exons 17 mit einer Positivprobe (Pfeil) und zwei Normalproben. Die Positivprobe stellt sich in dem mit Glycerol versetzten Gel genauso wie die Normalproben dar. Es ist keine zusätzliche Bande sichtbar. 46 Ergebnisse Positivkontrolle des Exons 18 Abbildung 3.3 zeigt das Ergebnis der SSCP– Analyse des Exons 18 mit einer Positivprobe, in einem mit Glycerol versetzten Gel. Das Ergebnis der Sequenzierung der Positivprobe zeigte einen Nukleotidaustausch an der Position 3810 von G nach T. Die bekannte Mutation (Mutation G3810T) des Patienten wurde somit bestätigt (siehe Abbildung 3.3). Die Positivprobe kann deutlich an der zusätzlichen Bande erkannt werden. Das wellige Erscheinungsbild der beiden Normalproben links im Bild ist bedingt durch ein erschwertes Abziehen des SSCP- Gels von der Glasplatte. Abbildung 3.3: Ausschnitt der SSCP– Analyse des Exons 18 mit einer Positivprobe (Pfeil) und drei Normalproben 47 Ergebnisse Positivkontrollen des Exons 19 Alle vier vorhandenen Positivproben des Exons 19 zeigten in der entsprechenden SSCP– Analyse aberrierende Banden. Zwei Positivproben tragen die gleiche Mutation (Mutation G3930A). Abbildung 3.4 zeigt jeweils einen Ausschnitt der SSCP– Analyse des Exons 19 aus unterschiedlichen Gelelektrophoresen und den Ergebnissen der Sequenzierungen der Positivproben mit den drei unterschiedlichen Mutationen. Alle bekannten Mutationen der Patienten konnten bestätigt werden. Die Positivproben können deutlich an der zusätzlichen Bande (Pfeile) erkannt werden. Die Normalproben zeigen ein anderes Bandenmuster. Mutierte Sequenz: G A C Wildtyp-Sequenz: G A G Nukleotid: 3928 3929 3930 A C A C 3931 3932 Mutierte Sequenz: G A A A C Wildtyp-Sequenz: G A G A C Nukleotid: 3928 3929 3930 3931 3932 Mutierte Sequenz: T G C Wildtyp-Sequenz: T G T Nukleotid: 3934 3935 3936 A T A T 3937 3938 Abbildung 3.4: Ausschnitt der SSCP – Analyse des Exon 19 mit vier Positivproben (Pfeile) und sieben Normalproben und den Ergebnissen der Sequenzierungen der drei unterschiedlichen Positivproben als Peak- Diagramme. 48 Ergebnisse Tabelle 3.5 zeigt eine Zusammenfassung der Ergebnisse der SSCP- Analyse der Positivproben der Exons 16, 17, 18 und 19 und die entsprechenden Mutationen. Positivproben/ DNA-Nr. Exon Nukleotid Mutation SSCP-Analyse Sequenzierung 2437 16 3529 A3529G + + 2557 16 3529 A3529G + + 5080 16 3522 G3522A + + 5081 16 3529 A3529G + + 5082 17 3640 T3640C - Nicht möglich 5083 18 3810 G3810T + + 5084 19 3930 G3930C + + 5085 19 3930 G3930A + + 5086 19 3930 G3930A + + 5087 19 3936 T3936C + + Tabelle 3.5: Zusammenfassung der Ergebnisse der SSCP – Analyse der Positivproben 49 Ergebnisse 3.3 Fallbeschreibungen der Patienten mit der Mutation A3529G in Exon 16 Beide Patienten gehören derselben Familie an. Es handelt sich hierbei um den zum Zeitpunkt der Operation (1997) 46- jährigen Vater als Indexpatienten und seinen 24 Jahre alten Sohn, der auf seinen Wunsch hin prädiktiv molekulargenetisch untersucht wurde. Der Indexpatient stammt aus dem erstellten Register für papilläre Nierenkarzinome. Die molekulargenetischen Analysen wurden zunächst durch die Arbeitsgruppe von Prof. Dr. L. Schmidt an den NIH durchgeführt. In dieser Arbeit konnten die gefundenen Mutationen durch eine SSCP – Analyse mit anschließender Sequenzierung bestätigt werden. In dieser Familie wurde auch die 26 jährige Tochter des Indexpatienten prädiktiv molekulargenetisch untersucht, es ließ sich bei ihr keine Mutation feststellen. In der SSCP – Analyse zeigte sich keine aberrierende Bande und es wurde deshalb keine Sequenzierung durchgeführt. Dieses Ergebnis bestätigt den Befund der Arbeitsgruppe von Prof. Dr. L. Schmidt. Vater und Sohn sind somit Mutationsträger der gleichen Mutation (A3529G). Diese Familie stellt somit den einzigen familiären Fall in diesem Register dar. Klinisch zeigte der Indexpatient im MRT multiple, bilaterale Tumore (s. Abbildung 3.6). Der Sohn war asymptomatisch und wurde als Träger derselben Mutation erst durch die prädiktive Diagnostik entdeckt. Die Familienanamnese des Indexpatienten ist unklar. Mit der einzigen Schwester und ihrer Familie soll kein Kontakt bestehen. Der Vater soll an nicht mehr evaluierbarer Ursache verstorben sein, die Mutter soll an den Folgen eines Mamma- Karzinoms verstorben sein. Eine Großmutter des Vaters soll an unbekannter Ursache mit 75 Jahren verstorben sein, ein Großvater soll ebenfalls an unbekannter Ursache, angeblich in jüngeren Jahren, verstorben sein. Weitere Informationen waren nicht evaluierbar. Die Familie lehnte weitere ihnen angebotene diagnostische Untersuchungen bei dem Sohn ab. Ein möglicherweise schon bestehender Tumor bei dem Sohn konnte somit nicht erfasst werden. Abbildung 3.5 zeigt den Stammbaum der Familie mit entsprechendem Ergebnis der SSCP– Analyse und der Sequenzierung der DNA- Proben der betroffenen Personen. 50 Ergebnisse ? ? I:1 I:2 ? II:2 III:2 II:1 II:3 III:1 Mutierte Sequenz: T A T A T C G T G G Wildtyp- Sequenz: T A T A T C A T G G Nukleotid: 3529 Abbildung 3.5: Stammbaum der Familie mit der Mutation A3529G in Exon 16 und Ergebnisse der SSCPAnalysen sowie das Ergebnis der Sequenzierung des Exon 16 des Indexpatienten Der Indexpatient ist mit einem Pfeil gekennzeichnet. Beide Mutationsträger sind dunkel markiert. Die Tochter trägt keine entsprechende Mutation. Die SSCP – Analysen zeigen bei 51 Ergebnisse dem Indexpatienten und seinem Sohn eine eindeutige zusätzliche Bande (siehe Pfeile). Bei der Tochter zeigt sich ein Normalbefund. In der MRT- Aufnahme des Indexpatienten sind nach Kontrastmittelgabe multiple, bilaterale Tumore der Nieren zu erkennen (weiße Pfeile). Die Bilder wurden freundlicherweise von Herrn Prof. Dr. Riedasch, Abteilung Urologie der Universitätsklinik Heidelberg, zur Verfügung gestellt. Abbildung 3.6: MRT- Darstellung des Abdomens des Indexpatienten mit der Mutation A3529G Die histologische Untersuchung der Tumore erbrachte den Befund von bilateralen, multiplen Nierentumoren mit etwas unterschiedlichem Wachstumsmuster. Die rechte Niere wurde durch Nephrektomie entfernt. Es fanden sich hier multiple tubulo-papilläre Adenome und Herde eines tubulo-papillären Adeno-Karzinoms. In der linken Niere wurden zwanzig Tumore entfernt. Acht Tumore wurden histologisch separat aufgearbeitet. Es handelte sich in allen Fällen um papillär klarzellige oder papillär chromophile Tumoren. Der maximale Durchmesser der Tumore war 1,8 cm. 52 3.4 Ergebnisse Ergebnisse der SSCP- Analyse der DNA- Proben Die SSCP– Analyse wurde auch bei allen weiteren 60 Blutproben der Patienten als Screeningverfahren eingesetzt. Insgesamt wurde bei 52 Patienten mit papillären Nierenkarzinomen das Exon 2 und Exon 4 bis 21 des MET– Gens untersucht, bei 8 weiteren Patienten wurden nur die Exons 16, 17, 18 und 19 untersucht, die sich in anderen Studien als „hot spot“ Region zeigten. Die Blutproben dieser 8 Patienten sind zum Abschluss der Arbeit eingegangen und die Diagnostik der meisten Exons war schon abgeschlossen. Im Folgenden sollen die Ergebnisse der SSCP– Analysen der einzelnen Exons zusammengefasst werden. Exon 2 Es wurden insgesamt 52 DNA – Proben untersucht. In der SSCP- Analyse konnte bei der DNA-Nr. 3837, im telomeren Drittel des Exons 2, deutlich in dem nicht mit Glycerol versetzten Gel eine zusätzliche Bande gesehen werden. Eine weitere SSCP- Analyse dieses Exons mit veränderten Versuchsbedingungen (mit Glycerol versetztes Gel) konnte die aberrierende Bande bei derselben Probe bestätigen (siehe Pfeile, Abbildung 3.7). Bei allen anderen DNA – Proben ergab sich ein Normalbefund. SSCP- Gelanalyse ohne Glycerol SSCP- Gelanalyse mit Glycerol Abbildung 3.7: Ausschnitt der SSCP- Analysen des Exons 2 telomeres Drittel mit unterschiedlichen Versuchsbedingungen 53 Ergebnisse Bei der anschließenden Sequenzierung der aberrierenden Bande der DNA-Nr. 3837 in Exon 2 zeigte sich ein Nukleotidaustausch bei der Position 698 des MET- Gens von G zu T, mit einem daraus resultierenden Aminosäurenaustausch von Serin zu Isoleucin (siehe Abbildung 3.8). Eine solche Veränderung im MET – Gen wurde bisher nicht beschrieben. Referenz des Wildtyps nach Park et al. (1987). Mutierte Sequenz: G A A G A T Wildtyp- Sequenz: G A A G A G C C C A Nukleotid: 694 695 697 698 699 700 701 702 693 696 C C C A Abbildung 3.8: Peak- Diagramm der veränderten Sequenz in Exon 2 der Probe 3837, verglichen mit der Wildtyp- Sequenz. Man erkennt einen Nukleotidaustausch von G zu T bei dem Nukleotid 698. Zur weiteren Abklärung der nachgewiesenen Veränderung G698T in Exon 2 des MET- Gens wurden 100 Kontrollproben (DNA von gesunden Blutspendern, nach deren Einverständnis) hinsichtlich derselben Veränderung mit den gleichen Methoden untersucht. Hierbei fand sich bei 8 Patienten der gleiche Nukleotidaustausch. Bei der Sequenzveränderung G698T in Exon 2 handelt es sich somit um einen Polymorphismus. 54 Ergebnisse Exon 3 Exon 3 wurde nicht untersucht, da bisher keine geeigneten Primer für dieses Exon hergestellt werden konnten. Exon 4 Es wurden 52 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 5 Es wurden 52 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 6 Es wurden 52 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 7 Es wurden 52 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 8 Es wurden 52 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 9 Es wurden 52 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 10 Es wurden 52 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 11 Es wurden 52 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 12 Es wurden 52 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 13 Es wurden 52 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 14 Es wurden 52 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 15 Es wurden 52 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. 55 Ergebnisse Exon 16 Es wurden insgesamt 60 DNA – Proben untersucht. In der SSCP- Analyse konnte bei der Probe mit der DNA-Nr. 1291 eine zusätzliche Bande gesehen werden. Zur Kontrolle wurde eine weitere SSCP- Analyse durchgeführt. Es konnte mit veränderten Versuchsbedingungen mit einem mit Glycerol versetzten Gel, sowie nochmals einer SSCP- Analyse ohne Glycerol, keine zusätzliche Bande mehr gesehen werden (siehe Pfeile Abbildung 3.9). SSCP- Gelanalyse ohne Glycerol SSCP- Gelanalyse mit Glycerol Abbildung 3.9: Ausschnitt der SSCP- Analysen des Exons 16 mit unterschiedlichen Versuchsbedingungen Die Sequenzierung der ausgeschnittenen zusätzlichen Bande der Probennummer 1291 in Exon 16 ergab kein Ergebnis, da sich keine DNA in der Probe nachweisen ließ. Bei der aberrierenden Bande handelt es sich hierbei somit wahrscheinlich um ein PCR Artefakt, wofür auch die Tatsache spricht, daß die zusätzliche Bande nur in einer Gelanalyse auftritt. Auch in einer weiteren SSCP – Analyse ohne Glycerol, welche hier nicht dargestellt ist, ließ sich die zusätzliche Bande nicht mehr darstellen. Bei allen anderen Proben ergab sich ein Normalbefund. 56 Ergebnisse Exon 17 Es wurden 60 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 18 Es wurden 60 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 19 Es wurden 60 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 20 Es wurden 54 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. Exon 21 Es wurden 54 DNA – Proben untersucht. Bei allen Proben ergab sich ein Normalbefund. 3.5 Fallbeschreibung des Patienten mit der DNA-Nr. 3837 und dem Polymorphismus G698T in Exon 2: Es handelt sich um einen bei Diagnosestellung 55–jährigen männlichen Patienten. Die Operation aufgrund der Diagnose unilaterales Nierenkarzinom erfolgte 1992 durch eine Nephrektomie. Intraoperativ fand sich ein Tumorstadium 2 nach Robson und ein TNMStadium T3a N0 M0. Die Tumorgröße betrug 7cm. Eine positive Familienanamnese bezüglich Nierenkarzinome ließ sich nicht evaluieren. Der Patient ist weiterhin in Nachsorgebehandlung bei seinem Hausarzt. Es trat bisher kein Tumorrezidiv auf. Weitere Informationen waren nicht erhältlich. 57 3.6 Ergebnisse Weitere Polymorphismen In den Exons 2 (telomeres Viertel), 7 und 20 konnten bei mehreren Proben verschiedene gleiche Zusatzbanden gefunden werden, die durch die Häufigkeit bei den Proben und aufgrund des typischen Bandenmusters von Muster A, B oder AB, auf einen Polymorphismus schließen lassen. Ein Beispiel dafür zeigt Abbildung 3.10 anhand Exon 20 . A B AB Abbildung 3.10: Ausschnitt der SSCP- Analyse des Exons 20 (SSCP- Gelanalyse mit Glycerol) Es können verschiedenartige Banden gesehen werden, die bei verschiedenen Proben mehrmals auftreten. Es tritt ein Polymorphismus- typisches Muster auf, mit der Abwechslung von Muster A, B oder AB. Anhand der Sequenzierungen des Exons 20 verschiedener Patienten mit einem AB- Bandenmuster konnte bei diesen Patienten der Polymorphismus C4106T heterozygot nachgewiesen werden. Im Folgenden wird das Ergebnis der Sequenzierung des Exons 20 des Patienten 6012 mit einem AB- Bandenmuster dargestellt. Es kann eindeutig die Nukleotidveränderung C4106T in heterozygotem Zustand gesehen werden (siehe Abbildung 3.11). Die Sequenzierung der Patientin 5301 mit einem A- Bandenmuster ergab an dieser Nukleotidstelle Homozygotie T4106T (siehe Abbildung 3.11). Die Sequenzierung eines Patienten mit einem B- Bandenmuster ergab an dieser Stelle 58 Ergebnisse Homozygotie C4106C (Sequenzierung nicht dargestellt). Damit konnte ein häufiger Polymorphismus in Exon 20 gezeigt werden. Seq.: C T G A Nukleotid: T/C G T 4106 A A C T G A T G T A A 4106 Abbildung 3.11: Ergebnisse der Sequenzierungen des Exon 20 des Patienten 6012 mit dem heterozygoten Polymorphismus C4106T und der Patientin 5301 mit Homozygotie T4106T Im telomeren Viertel von Exon 2 konnte mit den gleichen Methoden der Polymorphismus C728T in heterozygotem Zustand bei mehreren Proben nachgewiesen werden. In Exon 7 konnte bei mehreren Proben der Polymorphismus A2138G in heterozygotem Zustand nachgewiesen werden. Die einzelnen Ergebnisse der jeweiligen SSCP- Analysen und Sequenzierungen werden hier nicht weiter dargestellt. Die Diagnostik erfolgte jeweils mit den beschriebenen Methoden. Der von Schmidt et al. gefundene Polymorphismus in Exon 15 konnte bei keinem der Patienten gefunden werden. 59 Ergebnisse Tabelle 3.6 zeigt eine Zusammenfassung der Ergebnisse der SSCP- Gelanalysen der einzelnen Exons des MET- Gens bezüglich Mutationen bzw. Polymorphismen, ohne die Positivproben. Exon 2 Sequenzveränderung Fall/ DNA-Nr. positiv 3837 Nukleotid Pathogene Polymorphismus Mutation 698 keine G698T 728 C728T Exon 4 negativ alle Proben keine Exon 5 negativ alle Proben keine Exon 6 negativ alle Proben keine Exon 7 positiv mehrere Proben Exon 8 negativ alle Proben keine Exon 9 negativ alle Proben keine Exon 10 negativ alle Proben keine Exon 11 negativ alle Proben keine Exon 12 negativ alle Proben keine Exon 13 negativ alle Proben keine Exon 14 negativ alle Proben keine Exon 15 negativ alle Proben keine Exon 16 negativ alle Proben keine Exon 17 negativ alle Proben keine Exon 18 negativ alle Proben keine Exon 19 negativ alle Proben keine Exon 20 positiv mehrere Proben Exon 21 negativ alle Proben 2138 4106 keine keine A2138G C4106T keine Tabelle 3.6: Zusammenfassung der Mutationsanalysen der einzelnen Exons des MET-Gens 60 3.7 Ergebnisse SSCP-Analyse der Patienten mit bekannten somatischen Mutationen im Tumorgewebe Bei den beiden Patienten, bei denen eine somatische Mutation in den Exons 16 (DNA-Nr. 3117; Mutation G3522A) und 19 (DNA-Nr. 2871; Mutation T3936G) im Tumorgewebe identifiziert wurde (Schmidt et al., 1997), fanden sich bei der SSCP- Analyse der Blut- DNA der entsprechenden Exons und der restlichen Exons des MET-Gens keine aberrierenden Banden. Ein Patient stammt aus dem eigenen Register für papilläre Nierenkarzinome, der zweite Patient wurde im Loretto-Krankenhaus Freiburg in der Urologischen Abteilung operiert, das histologische Gutachten wurde am Pathologischen Institut der Universitätsklinik Freiburg erstellt. Die entsprechenden Mutationen in den Tumoren wurden in der Arbeitsgruppe von Prof. Dr. L. Schmidt an den NIH identifiziert. Blutproben der Patienten zur SSCP – Analyse wurden nachträglich angefordert. 3.7.1 Fallbeschreibung des Patienten mit der somatischen Mutation G3522A Es handelt sich um einen zum Zeitpunkt der Operation 55-jährigen männlichen Patienten. Die Operation durch eine Nephrektomie erfolgte 1994 am Loretto-Krankenhaus Freiburg. Die Histologie ergab einen unilateralen, 5cm im Durchmesser großen, vorwiegend papilläre strukturierten, abgekapselten Tumor der rechten Niere. Das Tumorstadium nach TNM betrug T2N0MX. Eine Einteilung nach Robson wurde nicht durchgeführt. Weitere Diagnostik ergab keinen Hinweis auf Metastasen. Die Familienanamnese hinsichtlich Nierenkarzinome ist negativ. 3.7.2 Fallbeschreibung des Patienten mit der somatischen Mutation T3936G Bei dem männlichen Patienten erfolgten zwei Operationen aufgrund der Diagnose Nierentumor in der Universitätsklinik Freiburg im Abstand von sieben Jahren. Zum Zeitpunkt der ersten Operation war der Patient 45 Jahre alt. Es fand sich in der linken Niere ein 6cm großer Tumor, der durch eine Nierenteilresektion entfernt wurde. Eine Nephrektomie wurde aufgrund einer reduzierten Funktion der kontralateralen Niere, durch eine hydronephrotisch geschrumpfte Niere mit mehreren Zysten, nicht durchgeführt. Die Histologie ergab einen 61 Ergebnisse tubulo-papillär wachsenden Tumor mit dem Tumorstadium T2N0M0 nach der TNM Klassifikation. Die zweite Operation erfolgte 1994. Es fand sich ein 4cm großer Tumor in der rechten Niere. Aufgrund eines zusätzlich multifokalen Vorkommens von papillären Adenomen und einem Tumoreinbruch in das Nierenbecken, erfolgte eine Nephrektomie der rechten Niere. Histologisch fand sich ein chromophiles, adenopapilläres Nierenzellkarzinom. Das Tumorstadium nach TNM betrug T2N0MX. Eine Stadiumeinteilung der Tumore nach Robson wurde jeweils nicht durchgeführt. Die Familienanamnese des Patienten hinsichtlich Nierentumore ist negativ. Eine Probe dieses zweiten Tumors wurde zur Analyse an die Arbeitsgruppe von Prof. Dr. L. Schmidt an den NIH geschickt. Der Befund stand zum Zeitpunkt dieser Arbeit noch aus. 3.8 Sensitivität der SSCP- Analyse Da sich 9 von den 10 Positivproben in der SSCP- Analyse mit eindeutig aberrierenden Banden darstellten (90%) und die bekannten Mutationen durch Sequenzierungen bestätigt werden konnten, kann von einer relativ hohen Sensitivität dieses Analyseverfahrens für diese Exons ausgegangen werden. Da keine weitere DNA der Positivkontrolle des Exons 17 zu erhalten war und die Reamplifizierung der DNA aus der SSCP- Analyse erfolglos verlief, kann keine genaue Aussage zu der Sensitivität dieses Verfahrens hinsichtlich dieser Positivkontrolle für Exon 17 gemacht werden. Eine Ursache, warum die Positivkontrolle für Exon 17 keine aberrierende Bande gezeigt hat muss somit offen bleiben. 62 4 Diskussion DISKUSSION Um die Bedeutung des c-MET Proto-Onkogens für die Entstehung von sporadischen und familiären papillären Nierenkarzinomen zu eruieren, wurden in der vorliegenden Arbeit erstmals Exon 2 und Exon 4 bis 21 des c-MET Proto-Onkogen eines Patientenkollektives mit papillären Nierenkarzinomen analysiert. Um bisherige Ergebnisse aus anderen Studien zu reevaluieren und die Sensitivität der Untersuchungen anhand der SSCP-Analyse zu überprüfen, wurden 10 positive Proben aus einer anderen Arbeitsgruppe (Schmidt et al. 1997, 1999) als Positivproben in die Untersuchungen eingeschlossen, wobei zwei Patienten aus dem eigenen Register der Jahre 1984 bis 1994 stammen. In allen Fällen wurde die SSCP-Analyse mit Blutproben als Verfahren zur Identifizierung von konstitutionellen- Mutationen in peripheren Lymphozyten eingesetzt, da bisherige Studien eine gute Sensitivität dieses Analyseverfahrens in Bezug auf das c-MET Proto-Onkogen zeigten und die entsprechenden Primerpaare gute Resultate lieferten ( Duh et al. 1997; Schmidt et al. 1997). In dieser Arbeit wurden keine Tumore untersucht und somit wurden keine somatischenMutationen bestimmt. Bei zwei Patienten aus dem eigenen Register der Jahre 1984 bis 1994 der Uniklinik Freiburg sind von der Arbeitsgruppe von Prof. Dr. L .Schmidt an den NIH (1997) somatische Mutationen im Tumorgewebe identifiziert worden und es wurde in dieser Arbeit zusätzlich bei diesen Patienten auf konstitutionelle Mutationen hin untersucht. 4.1 Konstitutionelle Mutationen des MET- Gens bei sporadischen papillären Nierenkarzinomen Bei allen Proben dieses Patientenkollektivs, wiesen weder die sporadischen, noch die familiär unklaren Fälle, eine konstitutionelle pathogene Mutation des c-MET Proto-Onkogens auf. Die Ergebnisse dieser Arbeit zeigen bei den sporadischen papillären Nierenkarzinomen eine Vergleichbarkeit mit weiteren Studien, bei denen eine geringe Häufigkeit von konstitutionellen, sowie auch somatischen Mutationen im MET-Gen bei Patienten mit sporadischen papillären Nierenkarzinomen nachgewiesen werden konnte. In der Arbeitsgruppe von Prof. Dr. L. Schmidt an den NIH konnte in einer Studie mit 128 ausschließlich sporadischen Fällen 16 MET- Mutationen (13%) mittels SSCP- Analyse nachgewiesen werden. Bei 8 von den 16 Patienten fanden sich konstitutionelle Mutationen, d.h. bei ca. 6% des Patientenkollektivs (Schmidt et al. 1999). In einer weiteren Studie wurden 63 Diskussion bei 3 von 60 (5%) Patienten mit sporadischen papillären RCC somatische Mutationen identifiziert, bei 6 von 7 (86%) Patienten mit familiären papillären RCC wurden konstitutionelle Mutationen gefunden (Schmidt et al. 1997). Die Mutationen beschränken sich alle auf die Tyrosin- Kinase Domäne des MET- Gens. Es können verschiedene Ursachen für die insgesamt geringe Häufigkeit von c-MET ProtoOnkogen Mutationen bei sporadischen papillären RCC angenommen werden. So könnten somatische Mutationen zum Beispiel nur in einem der morphologischen Subtypen der papillären RCC vorkommen oder es existieren weitere bisher unbekannte Kandidatengene. Weiterhin werden von unabhängigen Studien Mutationen in den TFE3- und PRCC- Genen für sporadische papilläre RCC verantwortlich gemacht (Sidhar et al.1996; Wetermann et al. 1996). Eine Trisomie des Chromosoms 7 ist eine charakteristische Eigenschaft von papillären RCC. Eine Studie von Kovacs et al. (1993) zeigte eine somatische Trisomie 7 in 95% der Fälle bei papillären Nierenkarzinomen. Verglichen mit der Studie von Schmidt et al. (1999), bei der 6% der Fälle eine somatische- und 6% der Fälle eine konstitutionelle MET- Mutation aufwiesen sowie dieser Studie, bei der 1,7% (1/58) eine MET- Mutation aufwiesen, scheinen die meisten papillären RCC mit einer somatischen Trisomie 7 einherzugehen, ohne eine METMutation zu tragen (siehe Abbildung 4.1). Interphase-FISH einer Trisomie 7 bei einem Patienten mit PRCC Chromosom 7 markiert mit roten Sonden A: MET- Gen ohne Mutation (met) + : leichter Proliferationsreiz B: MET- Gen mit Mutation (MET) +++ : starker Proliferationsreiz Abbildung 4.1: Polysomie des Chromosom 7 mit Duplikation des MET- Allels und zytogenetische Darstellung einer Trisomie 7 mittels Interphase-FISH (nach Lubensky et al. 1999) 64 Diskussion Von besonderem Interesse ist hierbei das Ergebnis einer Studie von Bernues et al. (1995), bei der eine partielle Duplikation auf Chromosom 7 in der Region 7q21-q35, bei einer Familie mit familiärem papillären RCC und MET- Mutation, identifiziert werden konnte. Dies hatte somit eine Vervielfältigung des mutierten MET- Allels zu Folge. In einer Studie mit familiären, sowie auch sporadischen papillären RCC, wurde eine Polysomie des Chromosom 7 und eine Duplikation des mutierten MET- Allels bei allen identifizierten Mutationen gefunden (Fischer et al. 1998). Diese Befunde lassen die Vermutung zu, daß eine Duplikation des mutierten MET- Gens nötig ist, um eine Tumorentstehung zu verursachen. Eine Mutation von einem Allel des MET- Gens scheint keinen Einfluss auf die Funktion des Wildtyp- Allels während der embryonalen Genese zu haben und eine konstitutionelle Mutation allein scheint für eine Entwicklung eines papillären RCC nicht ausreichend zu sein. Ein weiterer Hinweis hierfür sind Familienmitglieder von betroffenen Personen, mit hereditären papillären RCC, die zwar eine Mutation tragen, aber keine Tumoren entwickeln (Fischer et al. 1998). Die niedrige Penetranz der Erkrankung bei konstitutionellen Mutationen des MET- Gens, könnte somit durch diese Hypothese erklärt werden. Wie diese und vorhergegangene Studien zeigten, scheinen Mutationen des c-MET ProtoOnkogen nur für eine kleine Anzahl von sporadischen papillären RCC eine Rolle zu spielen. Eine zusätzliche MET- Mutation scheint jedoch einen weiteren Wachstumsvorteil für papilläre RCC zu bringen, da das mutierte Gen- Allel dupliziert wird. Der Befund in der vorliegenden Arbeit schließt jedoch in dieser relativ kleinen Studie unerkannte konstitutionelle Mutationen bei den untersuchten Patienten nicht aus, da die Sensitivität der SSCP- Analyse allgemein zwischen 35% und bis zu 100% beschrieben wird (Claustres et al.1993; Michaud et al. 1992; Orita et al. 1989; Sheffield 1993; Soto et al.1992). Die Sensitivität dieses Verfahrens hängt in hohem Maße von der Optimierung der experimentellen Bedingungen und der Sequenz des entsprechenden DNA- Abschnitts ab. Zudem kann das SSCP- Verfahren in dieser Arbeit nicht als 100% sensitiv für alle Exons beschrieben werden, da nur für die Exons 16, 17, 18 und 19 Positivkontrollen zur Verfügung standen und hier auch nicht für alle in diesen Exons unterschiedlichen beschriebenen Mutationen (Schmidt et al.1999). 65 4.2 Konstitutionelle Mutationen des Diskussion MET- Gens bei papillären Nierenkarzinomen mit unklarer und positiver familiärer Anamnese Die Diagnose Familiäres Papilläres Nierenkarzinom bereitet oft aufgrund einer schwer zu erhebenden Familienanamnese Schwierigkeiten. Ist die Diagnose einmal gestellt und gibt es in den betroffenen Familien mehrere symptomatische Mitglieder, so kommt der molekulargenetischen Untersuchung dieser Patienten und auch den nicht symptomatischen Angehörigen eine Schlüsselstellung zu. Bei den Patienten mit unklarer familiärer Anamnese fanden sich, wie schon erwähnt, keine konstitutionellen Mutationen, wobei hier nicht erkannte Mutationen mittels der SSCPAnalyse nicht vollständig ausgeschlossen werden können. Somatische Mutationen wurden nicht untersucht. Neben der schon bekannten Familie mit familiären papillären RCC dieses Registers, fanden sich keine weiteren Patienten mit einer eindeutig gesicherten Diagnose des familiären papillären RCC in dem Patientenkollektiv dieser Studie. Auch in den vorangegangenen Studien zeigten sich wenige größere Familien mit hereditärem papillären RCC, die beiden beschriebenen großen nordamerikanischen Familien scheinen dabei auf einen gemeinsamen Vorfahren zurückzuführen zu sein (Schmidt et al.1997; Schmidt et al. 1998). Konstitutionelle Mutationen des MET- Gens wurden jedoch wie schon erwähnt auch bei 3 von 60 Patienten mit unklarer Familienanamnese identifiziert (Schmidt et al.1997) und bei 8 von 128 mit sporadischen papillären RCC (Schmidt et al. 1999). Die bisher identifizierten Mutationen bei familiärem papillären RCC wurden in den Exons 16, 17, 18 und 19 lokalisiert. Diese Mutationen liegen alle im Gebiet der Tyrosin- Kinase Domäne des MET-Gens, lokalisiert in den Exons 15 bis 21 (Duh et al.1997), die eine wichtige Rolle für die intrazelluläre Signaltransduktion spielt (Takada et al. 1998). Da bei den MET- Mutanten eine erhöhte Tyrosin- Autophosphorylisierung und eine verstärkte Aktivität der Kinase gefunden wurde, scheint eine Tumorexpression hier auf eine Veränderung der Tyrosinkinase- Domäne zurückzuführen zu sein (Jeffers et al.1997). Auch weitere positive Befunde einer Mutation in der Tyrosin- Kinase Domäne des MET- Gens bei anderen Tumorarten, untermauern den Zusammenhang der Lokalisation einer Mutation in diesem Bereich des Gens und einer Tumorexpression (Di Renzo et al. 1992, 1994, 2000, Olivero et al. 1996). Eine Polysomie des Chromosom 7 scheint dabei eine weitere entscheidende Rolle zu spielen. Wie schon erwähnt hängt die Stabilität der Konformation der „activation loop“ des MET- Gens von vielen verschiedenen Faktoren ab und bietet somit viele Angriffspunkte für Mutationen in der Tyrosin- Kinase Domäne. Es liegt somit nahe, daß noch 66 Diskussion weitere Mutationen in der Tyrosin- Kinase Domäne existieren können, die bislang unentdeckt sind. Abbildung 4.2 zeigt eine Darstellung der Tyrosin- Kinase Domäne des MET- Gens mit bekannten Lokalisationen von Mutationen und für die Stabilisierung dieser Konformation wichtigen Aminosäuren-Positionen, die als Angriffspunkte für Mutationen dienen können. dunkelgrauer oberer Teil des Bildes: N- terminales Ende hellgrauer unterer Teil des Bildes: C- terminales Ende F1241, D1222, Y1252: wichtige Aminosäuren- Positionen die für die Stabilisierung der Konformation wichtig sind Fette schwarze Punkte: Lokalisationen von MET- Mutationen (Codon 1112, 1124, 1149, 1110, 1238, 1213, 1206, 1268, 1248 und 1246) Abbildung 4.2: Die Tyrosin- Kinase Domäne des MET- Gens in der inaktiven Form mit Lokalisationen von bekannten Mutationen (aus: Schmidt et al. 1999) Da eine Mutation in den weiteren Regionen des MET- Gens noch nicht im Zusammenhang mit papillären Nierenkarzinomen identifiziert werden konnte, scheint der Tyrosin- Kinase Bereich die signifikante Rolle für die Tumorentstehung zu spielen. Erwähnenswert erscheint hier auch die Tatsache, daß vier der bisher gefundenen Mutationen des MET- Gens (M1268T, 67 Diskussion D1246H, D1246N, V1110I) in Codons lokalisiert sind, die homolog zu Codons im c- KIT, im RET- Proto-Onkogen und im c-erbB (Schmidt et al.1999; Shu et al. 1990) sind, welche als Ziele von natürlich vorkommenden Mutationen nachgewiesen werden konnten (Hofstra et al. 1994; Nagata et al. 1995). MET, c-KIT, RET und c-erbB sind alle transmembranäre Tyrosinkinaserezeptoren, welche Mutationen in der Tyrosinkinase- Domäne aufweisen können und entsprechend zu einer karzinomatösen Entartung in verschiedenen Geweben führen können. Diese Beobachtungen legen nahe, daß die Tyrosinkinase- Domäne als „hot spot“ dieser Gene angesehen werden kann und eine Mutation in diesem Bereich zu karzinomatöser Zellproliferation führt. Nicht zu vernachlässigen sind hierbei jedoch auch die transmembranäre und extrazelluläre Domäne des MET-Gens, die in der Kette der Signaltransduktion weitere wichtige Funktionen innehaben. Wie diese Arbeit zeigt, könnte auch eine Mutation in diesen Bereichen zu einer Tumorentstehung führen, welche aufgrund der bisherigen Ergebnisse der Studien jedoch nicht in größerer Häufigkeit zu erwarten wäre. Um mit einer größeren Sicherheit Mutationen in diesen Bereichen für eine Tumorgenese einzuschließen, sind weitere Studien mit großem Patientenkollektiv nötig. Anhand der bisherigen Studien erscheint es eindeutig, daß konstitutionelle Mutationen des MET- Gens auch bei Patienten ohne eine ausgedehnte positive Familienanamnese gefunden werden können, größere Familien mit mehreren betroffenen und symptomatischen Familienmitgliedern scheinen eher ungewöhnlich zu sein. Familiäre papilläre Nierenkarzinome scheinen eher in kleinen Familien vorzukommen, in denen es nur wenige betroffene Personen gibt. Es ist daher davon auszugehen, daß MET Proto-Onkogen Mutationen mit einer niedrigen Penetranz von Tumoren einhergehen, mit einem späten Manifestationsalter. 4.3 SSCP-Analyse der Positivkontrollen Die Positivkontrollen der Exons 16, 18 und 19 zeigten aberrierende Banden in der SSCPAnalyse und die entsprechenden Sequenzierungen konnten die bekannten Mutationen der Arbeitsgruppe von Prof. Dr. L. Schmidt bestätigen. Die Positivprobe des Exons 17 konnte mittels der SSCP- Analyse nicht bestätigt werden, es konnte keine Alteration erkannt werden. Für die SSCP- Analyse wurden hierbei verschiedene Versuchsbedingungen hinsichtlich der Gelzusammensetzung und der Lauftemperatur ausprobiert. Für dieses Ergebnis der 68 Diskussion Positivprobe kommen mehrere Erklärungsmöglichkeiten in Frage. Eine schwierige Differenzierung zum Wildtyp könnte durch das Auftreten einer sehr stabilen, langstieligen Sekundärstruktur der amplifizierten Sequenz dieser Mutation entstehen, die die Einzelstrangmobilität stark behindert. Weiterhin könnten nicht optimale experimentelle Bedingungen für dieses Exon eine weitere Fehlerquelle darstellen. Weiterhin besteht die Möglichkeit, dass die entsprechende Probe nicht die tatsächliche Positivkontrolle enthielt. Da keine Sequenzierung dieser Probe erfolgen konnte, muss die Ursache hierfür offen bleiben. Da 9 von den 10 (90%) Positivproben sich bei der SSCP- Analyse mit eindeutig aberrierenden Banden darstellten und sich die Ergebnisse durch Sequenzierung bestätigten, kann die Sensitivität der angewandten SSCP- Analyse bezüglich dieser Exons als relativ hoch angenommen werden. Da die Positivproben in Acrylamidgelen ohne und mit Zusatz von Glycerol aberrierende Banden zeigten, scheinen beide Gelarten für eine SSCP- Analyse empfehlenswert zu sein. Eine weitere kürzlich entwickelte Methode zur Entdeckung von Heteroduplex in DNA mit Unterschieden zwischen der Wildtyp- DNA und mutierten DNA- Strängen, die so genannte DHPLC- Methode (Denaturing High- Performance Liquid Chromatography), scheint nach einer neueren Studie genauso empfehlenswert zu sein. Es wurde in dieser Studie ein Patientenkollektiv mit den 15 bekannten MET- Mutationen bei papillären Nierenkarzinomen nochmals mit der DHPLC- Methode analysiert und die Methode stellte sich als 100% genau in der Erfassung dieser Mutationen dar (Nickerson et al. 2000). Es ist somit möglich, daß DHPLC noch sensitiver bezüglich MET- Mutationen ist, als die SSCP- Analyse. In einer weiteren Studie wurde ein Vergleich der DHPLC- Analyse mit der SSCP- Analyse zur Entdeckung von Mutationen im TSC2 Gen erstellt, wobei sich DHPLC als sensitiver darstellte (Choy et al. 1999). Auch eine Anwendung dieser Methode bei der Identifizierung von Polymorphismen wird diskutiert, da in Studien gute Ergebnisse geliefert wurden (Zhonghua et al. 2001). Da DHPLC eine schnelle, billige und sensitive Methode darstellt, ist zu erwägen, ob dieser neuen Methode nicht der Vorzug zu geben ist. 4.4 Konstitutionelle Mutationen des MET- Gens bei Patienten mit somatischen Mutationen im Tumorgewebe Bei den beiden Patienten mit somatischen Mutationen im Tumorgewebe zeigten sich keine zusätzlich aberrierenden Banden in der SSCP- Analyse der Blut- DNA. Da bei beiden 69 Diskussion Patienten auch eine negative familiäre Anamnese vorliegt, können somit sporadische papilläre Nierenkarzinome angenommen werden. Wie schon erwähnt, finden sich bei den sporadischen papillären RCC häufiger somatische Mutationen als konstitutionelle Mutationen. Eine unterschiedliche konstitutionelle Mutation zusätzlich zu einer somatischen Mutation scheint unwahrscheinlich zu sein und so eine Konstellation wurde in bisherigen Studien nicht gefunden. Das negative Ergebnis dieser Arbeit bezüglich einer konstitutionellen Mutation bei diesen Patienten, bestätigt die schon identifizierten Mutationen (Schmidt et al. 1997) der Patienten als rein somatische Mutationen. 4.5 Histologische- Klinische Korrelation bei sporadischen und familiären papillären RCC Wie schon eingangs erwähnt sind familiäre papilläre Nierenkarzinome durch ein bestimmtes klinisches Erscheinungsbild gekennzeichnet. Sie treten häufig als multiple, bilaterale Nierentumore auf (Zbar et al. 1994; Schmidt et al.1997). In einer weiteren Studie fand sich jedoch auch dieses typische Erscheinungsbild bei 3 Personen mit papillären RCC und METMutationen mit unklarer familiärer Anamnese (Schmidt et al. 1997). Da jedoch die Familienanamnesen häufig schwer zu erheben sind, können hierbei weitere unerkannte Familienmitglieder mit papillären RCC nicht gänzlich ausgeschlossen werden. Zudem können weitere Familienmitglieder existieren, die als Carrier eine MET- Mutation tragen, ohne ein Karzinom entwickelt zu haben oder noch asymptomatisch sind und so einer Erfassung entgehen. Bei den Befunden dieser Arbeit traten lediglich 4,8 % der Nierentumore bilateral und 7% multifokal auf, die restlichen Nierenkarzinome waren solitär. Dieses Erscheinungsbild entspricht weitgehend der Häufigkeit der sporadischen Tumore dieser Arbeit. Da bei bis auf einen Fall keine konstitutionellen Mutationen im MET-Gen gefunden wurden, müssen weitere Ursachen für multiple, bilaterale papilläre Nierenkarzinome in Betracht gezogen werden, wie z.B. somatische Mutationen im Tumor, Virusinfektionen, Mutationen des TFE3 und PRCCGens oder Mutationen in bisher unbekannten Genen. Kovacs und Kovacs (1993) fanden in einer Studie, daß praktisch alle sporadischen papillären RCC multipel auftreten, auch bei Patienten ohne eine positive familiäre Anamnese. So kann ein einzelner Tumor mit multiplen mikroskopischen papillären Läsionen einhergehen oder es treten größere bilaterale multiple Läsionen auf. Fraglich ist jedoch, ob die Patienten beider 70 Diskussion Gruppen derselben oder verschiedenen Entitäten zuzuordnen sind. Delahunt und Eble (1997) hingegen fanden einen Zusammenhang zwischen dem Auftreten von mikroskopischen papillären Läsionen und der Morphologie des Primärtumors. Mikroskopische Läsionen traten deutlich häufiger bei dem Typ I der papillären Nierenkarzinome auf. Diese Unterschiede machen das Problem der exakten Einteilung der papillären Nierenkarzinome deutlich, bezüglich der histologischen, wie auch der Phänotyp-Genotyp Zuordnung und einem möglichen klinischen Hinweiszeichen für sporadische bzw. hereditäre papillären Nierenkarzinomen. Weitere statistische Daten dieser Studie wie die Geschlechtsverteilung, das Patientenalter bei Erstdiagnose und die Häufigkeit von papillären Nierenkarzinomen an der Gesamtzahl der Nierentumore entsprechen den Daten in der Literatur. 4.6 MET- Polymorphismus Der von Prof. Dr. L. Schmidt an den NIH beschriebene Polymorphismus C3223T (T1010I) in Exon 15 wurde in dieser Studie bei keiner Patientenprobe gefunden und scheint einen eher seltenen Polymorphismus darzustellen, oder kommt in der geographischen Region dieser Arbeit nicht vor. In dieser Arbeit konnten in mehreren Exons verschiedene Muster von aberrierenden Banden gesehen werden. Das hier gefundene Bandenmuster spricht aufgrund des typischen Auftretens der drei Muster A, B oder AB für einen häufigen Polymorphismus. Diese Befunde wurden durch mehrere Sequenzierungen bestätigt, es konnten in den jeweiligen Exons entsprechende Polymorphismen nachgewiesen werden. In der vorliegenden Arbeit wies ein Patient mit einem sporadischen papillären Nierenkarzinom eine Veränderung im c-MET Proto-Onkogen auf, die bei den anderen Patienten nicht gesehen werden konnte. Es handelt sich hierbei um einen seltenen Polymorphismus im telomeren Drittel des Exons 2 an Position 698 mit einem Nukleotidaustausch von G nach T und einem daraus resultierenden Aminosäurenaustausch von Serin zu Isoleucin. Da dieser Nukleotidaustausch bei der Untersuchung von 100 KontrollPatientenproben ebenfalls bei 8 Proben auftrat, muss ebenfalls von einem Polymorphismus und nicht von einer pathogenen Mutation ausgegangen werden. 71 5 Zusammenfassung Zusammenfassung Das papilläre Nierenkarzinom ist nach dem klarzelligen Nierenkarzinom der zweithäufigste maligne Nierentumor. Das papilläre Nierenkarzinom kommt fast ausschließlich sporadisch vor. 1994 wurde als neue Entität das Familiäre Papilläre Nierenkarzinom publiziert und der zugrunde liegende Gendefekt wurde auf dem langen Arm des Chromosom 7 im Bereich von 7q31.1-34 lokalisiert, im c-MET Proto-Onkogen. Die 15 bisher identifizierten Mutationen sind alle vom Missense-Typ und beschränken sich auf die Tyrosin-Kinase Domäne des METGens in den Exons 16, 17, 18 und 19, wobei bei den vorangegangenen Studien nicht alle Exons untersucht worden sind. Die beschriebenen Mutationen fanden sich sowohl in peripheren Lymphozyten als konstitutionelle Mutationen, sowie im Tumorgewebe als somatische Mutationen. Bei den familiären Fällen traten konstitutionelle Mutationen häufiger auf (86%) als bei den sporadischen Fällen (13%), wobei auch der Anteil an somatischen Mutationen im MET- Gen insgesamt bei den sporadischen Fällen als gering eingeschätzt wird. Um die Bedeutung von Mutationen im MET- Gen für die Genese der papillären Nierenkarzinome zu evaluieren und um die Sensitivität der verwendeten molekulargenetischen Diagnostik zu überprüfen, wurde in dieser Arbeit ein Kollektiv von 60 Patienten hinsichtlich Mutationen in den Exons 2 und 4 bis 21 des MET- Gens untersucht. Die Analyse der Positivkontrollen ergab in 9 von 10 Fällen (90%) den Mutationsnachweis. Bei der molekulargenetischen Analyse der 58 als sporadisch klassifizierten Fälle und der beiden Patienten mit schon bekannten somatischen Mutationen im MET- Gen, konnte in keinem Fall eine weiteren konstitutionelle Mutationen identifiziert werden. In Exon 2 konnte der bisher nicht beschriebene Polymorphismus G698T nachgewiesen werden. Anhand der gewonnenen Daten lässt sich das seltene Auftreten von konstitutionellen Mutationen im c-MET ProtoOnkogen bei sporadischen papillären Nierenkarzinomen bestätigen. Die Evaluierung der Patientendaten und der Familienanamnese zeigte jedoch die Schwierigkeit der eindeutigen klinischen Zuordnung von sporadischen und familiären Fällen. Die SSCP- Analyse als angewendetes Diagnostikverfahren lässt sich aufgrund der guten Sensitivität empfehlen. Bei eindeutig familiären papillären Nierenkarzinomen ist aufgrund der Ergebnisse früherer Studien mit Sicherheit eine genetische Diagnostik mittels der SSCP- Analyse zu befürworten, da hieraus frühzeitige Möglichkeiten therapeutischer Intervention resultieren. Bei Patienten mit nur vager positiver Familienanamnese ist eine molekulargenetische Untersuchung abzuwägen. 72 6 Anhang Anhang Im Labor N5 von Prof. Dr. H.P.H. Neumann werden weiterhin Patienten mit papillären Nierenkarzinomen hinsichtlich Mutationen im c- MET Proto–Onkogen untersucht. Die nachfolgende Tabelle (Tabelle 6.1) gibt eine Übersicht über alle bisher gefundenen Mutationen in diesem Patientengut. DNA-Nr.: Exon Nukleotid Mutation Codon 1291 11 2739 T2739C F849L 2557 16 3529 A3529G H1112R 2437 16 3529 A3529G H1112R 6012 17 3603 G3603A G1137R 6069 18 3879 A3879G M1229R 2871 19 3936 T3936G Y1248D Tabelle 6.1: Übersicht aller bisher gefundenen Mutationen aus der Arbeitsgruppe von Prof. Dr. H.P.H. Neumann 73 7 Abkürzungen Arg Arginin Asn Asparagin Asp Asparaginsäure ATP Adenosintriphosphat bp Basenpaare C Kohlenstoff Cys Cystein ddH2O Aqua bidest DNA Desoxyribonukleinsäure dNTP Desoxyribonukleosid – 5`- Triphosphat EDTA Ethylen- Diamine Tetra Acetate His Histidin HGF Hepatocyte growth factor Ile Isoleucin Leu Leucin m Milli (10-3) M Molar Met Methionin MRT Magnetresonanztomogramm N Stickstoff nt Nukleotid μ Mikro (10-6) PCR Polymerasekettenreaktion RCC Renal cell carcinoma (Nierenzellkarzinom) PRCC Papillary renal cell carcinoma (Papilläres Nierenzellkarzinom) SSCP Single Strand conformation Polymorphism TNM Tumor Nodes Metastasis Taq Thermus aquaticus Thr Thryptophan Tyr UTR Tyrosin Untranslated Region Val Valin Abkürzungen 74 8 Literaturverzeichnis Literaturverzeichnis Ainsworth P.J., Surh L.C., Coulter-Mackie M.B. (1991). Diagnostic single strand conformation polymorphism,(SSCP): a simplified non- radioisotopic method as applied to a Tay- Sachs B1 variant. Nucleic Acids Res 19: 405 – 406. Amin M..B., Corless C.L., Renshaw A.A., Tickoo S.K., Kubus J., and Schultz D.S. (1997). Papillary (chromophil) renal cell carcinoma: histomorphologic characteristics and evaluation of conventional pathologic prognostic parameters in 62 cases. Am J Surg Pathol. 21: 621 - 635 Alken C.E., Roucayrol J.C., Oberhausen E., Tauptiz A., and Ueberberg H. (1960). On the problem of carcinogenesis following Thorotrast pyelography. Urol Int 10: 137 – 156. Armstrong B., Garrod A., and Doll R. (1976). A retrospective study of renal cancer with special reference to coffee and animal protein consumption. Br J Cancer 33: 127 – 136. Avisrror Manuel Urrutia. Renal carcinoma and other tumours. The Oxford Textbook of Clinical Nephrology: 2574 – 2594. Bender B., Wiestler O.D., von Deimling A. (1994). A device for processing large acrylamide gels. Biotechniques 16: 204 – 206. Bennington J.L. and Laubscher F.A. (1968). Epidemiologic studies on carcinoma of the kidney. Cancer 21: 1069 – 1071. Bernues M., Casadevall C., Miro R., Caballin M.R., Villavicencio H., Salvador J., Zamarron A., Egozcue J. (1995). Cytogenetic characterization of a familial renal cell carcinoma. Cancer Genet Cytogenet 84: 123 – 127 Birchmeier C., Gherardi E. (1998). Developmental roles of HGF/SF and its receptor, the cMet tyrosine kinase. Trends Cell Biol 8: 404 – 410. 75 Literaturverzeichnis Bishop JM (1991). Molecular themes in oncogenesis. Cell 64: 235 – 248. Bladt F., Riethmacher D., Isenmann S., Sguzzi A. and Birchmeier C. (1995). Essential role for the c-met receptor in the migration of myogenic prcursor cells into the limb bud. Nature 376: 768 – 771. Bottaro D.P., Rubin J.S., Faletto D.L., Chan A.M., Kniecik T.E., Vande Woude G.F., Aaronson S.A. (1991). Identification of the hepatocyte growth factor as the c-met protooncogene product. Science 251: 802 – 804. Brauch H., Weirich G., Hornauer M.A., Storkel S., Woh, T., Bruning T. (1999). Trichloroethylene exposure and specific somatic mutations in patients with renal cell carcinoma. J Natl Cancer Inst. 91: 854 – 861. Budowle B., Chakraborty R., Giusti A.M., Eisenberg A.J., Allen R.C. (1991). Analysis of the VNTR locus D1S80 by the PCR followed by high- resolution PAGE. Am J Hum Genet 48: 137 – 144. Chisholm G.D., Roy R.R. (1971). The systemic effects of malignant renal tumors. Br. J. Urol 43: 687. Choy Y.S., Dabora S.L., Hall F., Ramesh V., Niida Y., Franz D., Kasprzyk-Obara J., Reeve M.P., Kwiatkowski D.J. (1999). Superiority of Denaturing High Performance Liquid Chromatography over single- stranded conformation and conformation-sensitive gel electrophoreseis for mutation detection in TSC2. Ann Hum Genet 63(5): 383 – 391. Comoglio P.M., Di Renzo M.F., Gaudino G. (1987). Protein tyrosin kinases associated with human malignancys. Am NY Acad Sci 511: 256 – 261. Cooper C.S. (1992). The met oncogene: from detection by transfection to transmembrane receptor for hepatocyte growth factor. Oncogene 7: 3 – 7. 76 Literaturverzeichnis Cooper C.S., Park M., Blair D.G., Tainsky M.A., Juebner K., Croce C.M. and Vande Woude G.F. (1984). Molecular cloning of a new transforming gene from chemically transformed human cell line. Nature 311: 29 – 33. Corless C.L., Aburatani H., Fletcher J.A., Housman D.E., Amin M.B., Weinberg D.S. (1996). Papillary renal carcinoma. Quantitation of chromosomes 7 and 17 by FISH analysis of chromosome 3p for LOH and DNA ploidy. Diagn Mol Pathol. 5: 53 – 64. Delahunt B., Eble J.N. (1997). Papillary renal cell carcinoma: a clinicopathologic and immunhistochemical study of 105 tumors. Mod. Pathol 10: 537 – 544. De Reijke T.M., Schlatmann T.J., and Dabhoiwala N.F. (1987). Adenocarcinoma of the kidney in childhood. Urol Int 42: 220 – 223. Dijkhuizen T., van den Berg E., Storkel S., de Vries B., van der Veen AY., Wilbrink M., Geurts van Kessel A., de Jong B. (1997). Renal oncocytoma with t(5;12;11), der(1)1;8) and add(19): „true“ oncocytoma or chromophobe adenoma? Int J Cancer 73: 521 – 524. Di Renzo M.F., Olivero M., Prat M., Bongarzone I., Pilott S., Belfiore A., Costantino A., Vigneri R., Pierotti M.A. and Comoglio P.M. (1992). Overexpression of the c-met/HGF receptor in human thyroid carcinomas. Oncogene 7: 2549 – 2553. Di Renzo M.F., Olivero M., Katsaros D., Crepaldi T., Gaglia P., Zola P., Sismondi P. and Comoglio P.M. (1994). Overexpression of the c-met/HGF receptor in ovarian cancer. Int J Cancer 58: 658 – 662. Di Renzo M.F., Olivero M., Martone T., Maffe A., Maggiora P., Stefani A.D., Valente G., Giordano S., Cortegina G., Comoglio P.M. (2000). Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene 19: 1547 – 1555. Duh F.M., Scherer S.W., Tsu, L-C., Lerman M., Zbar B., and Schmidt L. ( 1997). Gene structure of the human MET proto-oncogene. Oncogene 15: 1583 – 1584. 77 Literaturverzeichnis Epstein S.M., Bartus B., and Farber E. (1969). Renal epithelial neoplasma induced in male Wistar rats by oral aflotoxin – B1. Cancer Res 29: 1045 – 1050. Ermis A., Henn W., Remberger K., Hopf C., Hopf T., Zang KD.(1995). Proliferation enhancement by spontaneous multiplication of chromosome 7 in rheumatic synovial cells in vitro. Hum Genet 96: 651 – 654. Fischer J., Palmedo G., Knobloch von R., Bugert P., Prayer-Galetti T., Pagano F., Kovacs G. (1998). Duplication and overexpression of the mutant allele of the MET proto-oncogene in multiple hereditary papillary renal cell tumours. Oncogene 17: 733 – 739. Gambarotta G., Pistoi S., Giordano S., Comoglio P.M., and Santoro C. (1994). Structure and inducible regulation of the human MET promoter. J Biol Chem 269: 12852 – 12857. Griffin J.P., Hughes G.V., and Peeling W.B. (1967). A survey of the familial incidence of adenocarcinoma of the kidney. Br J Urol 39: 63 – 66. Gunawan B., von Heydebreck A., Fritsch T., Huber W., Ringert RH., Jakse G., Fuzesi L. (2003). Cytogenetic and Morphologic Typing of 58 Papillary Renal Cell Carcinomas. Cancer Res 63: 6200 – 6205. Hofstra R.M., Landsvater R.M., Ceccherini J., Stulp R.P., Stelwagen T., Luo Y., Pasini B., Hoppner J.W., van Amstel H.K., Romeo G. (1994). A mutation in the RET protooncogene asociated with multiple endocrine neoplasia type 2B and sporadic medullary thyreoid cancer. Nature 367: 375 – 376. Hubbard S.R., Wie L., Ellis L and Hendrikson W.A. (1994). Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 372: 746 – 754. Ishikawa I. (1993). Renal cell carcinoma in chronic haemodialysis patients – a 1990 questionnaire in Japan. Kidney Int Suppl 41: 167 – 169. 78 Literaturverzeichnis Jeffers M., Schmidt L., Nakaigawa N., Webb C.P., Weirich G., Kishida T., Zbar B., vande Woude G.F. (1997). Activating mutations for the met tyrosine kinase receptor in human cancer. Proc. Natl. Acad. Sci. USA 94: 11445 – 11450. Jiang F., Richter J., Schraml P., Bubendorf L., Gasser T., Sauter G., Mihatsch M.J., Moch H. (1998). Chromosomal imbalances in papillary renal cell carcinoma: genetic differences between histological subtypes. Am J Pathol 93: 9154 – 9159. Kovacs G. (1993). Molecular cytogenetics of renal tumors. Adv Cancer Res 62: 89 – 124. Kovacs G. (1989). Papillary renal cell carcinoma. A morphologic and cytogenetic study of 11 cases. Am J Pathol134: 27 – 34. Kovacs G., Akhtar M., Beckwith B.J., Bugert P., Cooper C.S., Delahunt B., Eble J.N., Fleming S., Ljungberg B., Medeiros L.J., Moch H., Reuter V.E., Ritz E., Roos G., Schmidt D., Srigley J.R., Storkel S., van den Berg E., Zbar B. (1997). The Heidelberg classification of renal cell tumours. J Pathol 183(2): 131 – 133. Kreiger N., Marret L.D., Dodds L., Hilditch S., and Darlington G.A. (1993). Risk factor for renal cell carcinoma: results of apopulation – based case – control study. Cancer Causes Control 4 : 101 – 110. Lubensky I.A., Schmidt L., Zhuang Z., Weirich G., Pack S., Zambrano N., Walther MM., Choyke P., Linehan W.M., Zbar B. (1999). Hereditary and Sporadic Papillary Renal Carcinomas with c-Met Mutations Share a Distinct Morphological Phenotype. Am J Pathol 155(2): 517 - 526 Mancilla – Jimenez R., Stanley R.J., Blath R.A. (1976). Papillary renal cell carcinoma: a clinical, radiologic, and pathologic study of 34 cases. Cancer 38: 2469 Marshall CJ (1991). Tumor suppressor genes. Cell 64: 313 – 326. 79 Literaturverzeichnis Mellemgaard A., Engholm G., McLaughlin J.K., and Olsen J.H. (1994). Occupational risk factors for renal cell carcinoma in Denmark. Scand J Work Environ Health. 20(3):160-5. Rawlins M.D., Luff R.H., and Cranston W.I. (1970). Pyrexia in renal carcinoma. Lancet 1 (7661) : 1371 – 1373. Meloni A.M., Dobbs R.M., Pontes J.E., Sandberg A.A. (1993). Translocation (S;1) in papillary renal adenocarcinoma. A new cytogenetic subtype. Cancer Genet Cytogenet 65: 1 – 6. Michaud J., Brody L.C., Steel G., Fontaine G., Martin L.S., Valle D., Mitchell G. (1992). Strand- seperating confirmation polymorphism analysis: efficancy of detection of point mutations in the human ornithine delta- aminotransferase gene. Genomic 13: 389 - 394 Moch H., Gasser T., Amin M.B., Torhorst T., Sauter G., Mihatsch M.J. (2000). Prognostic utility of the recently recommended histologic classification and revised TNM staging system of renal cell carcinoma: a Swiss experience with 588 tumors. Cancer 89(3): 604 – 614. Montero J., Urrutia Avisrror M., and Corrales J. (1987). Renal malignancy in the elderly. Renal function and diseases in the elderly: 400 – 431. Montesano R., Matsumoto K., Nakamura T., Orci L. (1991). Identification of a fibroblastderived epithelial morphogen as hepatocyte growth factor. Cell 67: 901 – 908. Nagata H., Worobec A.S., Oh C.K., Chowdhury B.A., Tannenbaum S., Suzuki Y., Metcalfe D.D. (1995). Identification of a point mutation in the catalytic domain of the protooncogene c- kit in peripheral blood mononuclear cells of patients who have mastocytosis with an hemolytic disorder. Proc Natl Acad Sci USA 92: 10560 – 10564. Naldini L., Tamagnone L., Vigna E., Sachs M., Hartmann G., Birchmeier W., Daikuhara Y., Tsubouchi H., Blasi F., Comoglio P.M. (1992). Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/ scatter factor. EMBO Journal 11: 4825 – 4833. 80 Literaturverzeichnis Neumann H.P.H., Bender B.U., Berger D.P., Laubenberger J., Schultze-Seemann W., Wetterauer U., Ferstl F.J., Herbst E.W., Schwarzkopf G., Hes F.J., Lips C.J., Lamiell J.M., Masek O., Riegler P., Mueller B., Glavac D., Brauch H. (1998). Prevalence, morphology and biology of renal cell carcinoma in Von-Hippel-Lindau disease compared to sporadic renal cell carcinoma. J Urol 160: 1248 – 1254. Nickerson M.L., Weirich G., Zbar B., Schmidt L.S. (2000). Signature- based analysis of MET proto- oncogene mutations using DHPLC. Hum Mutat 16(1): 68 – 76. Olivero M., Rizzo M., Madeddu R., Casadio C., Pennacchietti S., Nicotra M.R., Prat M., Maggi G., Arena N., Natali P.G., Comoglio P.M., Di Renzo M.F. (1996). Overexpression and activation of hepatocyte growth factor/ scatter factor in human non- small- cell lung cancers. Br J Cancer 74: 1862 – 1868. Orita M., Iwahana H., Kanazawa H., Hayashi K., Sekiya T. (1989). Detection of human DNA by gel electrophoresis as single- strand confirmation polymorphism. Proc Natl Acad Sci USA 86: 2766 – 2770. Paganini-Hill A., Ross R.K., and Henderson B.E. (1983). Epidemiology of kidney cancer. In: Urologic cancer. Blackwell Science, Boston, 383 – 407. Palmedo G., Fischer J., Kovacs G. (1999). Duplications of DNA sequences between loci D20S478 and D20S206 at 20q11.2 and between loci D20S902 and D20S480 at 20q13.2 mark new tumor genes in papillary renal cell carcinoma. Lab Invest 79: 311 – 316. Park M., Dean M., Cooper C.S., Schmid, M., O`Brian S.J., Blair D.G., Vande Woude G.F. (1986). Mechanism of met oncogene activation. Cell 45: 895 – 904. Park M., Dean M., Kaul K., Braun M.J., Gonda M.A. and Vande Woude G.F. (1987). Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth factor receptors. Proc. Nat. Acad. Sci. USA 84: 6379 – 6383. 81 Literaturverzeichnis Prat M., Narsinham R.P., Crepaldi T., Nicotra M.R., Natali P.G., Comoglio P.M. (1991). The receptor encoded by the human c- MET oncogene is expressed in hepatocytes, epithelial cells and solid tumors. Int J Cancer 49: 323 – 328. Ruiz – Marcellan F.J., Quintanilla B., Ruiz – Marcellan M.C., Cosme M.A., and Berstham J. (1979). Efectos tardios de la pielografia ascendente con Thorotrast. Actas Urologicas Espanolos 5: 283 – 286. Saiki R.K., Scharf S., Faloona F., Mullis K.B., Horn G.T., Erlich H.A., Arnheim N. (1985). Enzymatic amplification of betaglobin genomic sequences and restriction site analysis for diagnosis of sickle anemia. Science 230: 1350 – 1354. Sambrook J., Fritsch E.F., Maniatis T. (1989). Molecular Cloning. In: Cold Spring Harbor: Cold Spring Harbor Laboratory Press. Schmidt L., Junker K., Nakaigawa N., Kinjerski T., Weirich G., Miller M., Lubensky I., Neumann H.P.H., Brauch H., Decker J., Vocke C., Brown J.A., Jenkins R., Richard S., BergerheimU., Gerrard B., Dean M., Linehan W.M., and Zbar B. (1999). Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene 18: 2343 – 2350. Schmidt L., Duh F-M., Chen F., Kishida T., Glenn G., Choyke P., Scherer S.W., Zhuang Z., Lubensky I., Dean M., Allikmets R., Chidambaram A., Bergerheim U.R., Feltis J.T., Casadevall C., Zamarron A., Bernues M., Richard S., Lips C.J.M., Walther McClellan M., Tsu, L-C., Geil L., Orcutt M.L., Stackhouse T., Lipan J., Slife L., Brauch H., Decker J., Niehans G., Hughson M.D., Moch H., Storkel S., Lerman M.I., Linehan W.M., and Zbar B. (1997). Germline and somatic mutations in the tyrosine kinase domain of the MET protooncogene in papillary renal carcinomas. Nature Genet 16: 68 – 73. Schmidt L., Junker K., Weirich G., Glenn G., Choyke P., Lubensky I., Zhuang Z., Jeffers M., Vande Woude G., Neumann H.P:H., Walther, McClellan, Linehan W.M., and Zbar B. (1998). Two North American Families with Hereditary Papillary Renal Carcinoma and identical Novel Mutations in the MET Proto-Oncogene. Cancer Res 58: 1719 – 1722. 82 Literaturverzeichnis Shu H.K., Pelley R.J., Kung H.J. (1990). Tissue specific transformation by epidermal growth factor receptor: a single point mutation within the ATP- binding pocket of the erbB product increases its intrinsic kinase activity and activates its sarcomagenic potential. Proc Natl Acad Sci USA 87: 9103 – 9107. Sheffield V.C., Beck J.S., Kwitek A.E., Sandstrom D.W., and Stone E.M. (1993). The sensitivity of single – strand conformation polymorphism analysis for detection of single base substitutions. Genomics 16: 325 – 332. Shi J., Yang S., Jiang Z., Jiang H., Chen T., Chen Z., Huang W. (2001). Comparison of denaturing high performance liquid chromatography with direct sequencing in the detection of single nucleotide polymorphism. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 18(3): 198 – 201. Sidhar S.K., Clark J., Gill S., Hamoudi R., Crew A.J., Gwillian R., Ross M., Linehan W.M., Birdsall S., Shipley J., and Cooper C.S. (1996). The t(X;1) (p11.2; q21.2) translocation in papillary renal carcinoma fuses a novel gene PRCC to the TFE3 transcription factor gene. Hum Mol Genet 5: 1333 – 1338. Skinner D.G., Calvin R.B., Vermillion C.D., Pfister R.C., Leadbetter W.F. (1971). Diagnosis and management of renal cell carcinoma. A clinical and pathologic study of 309 cases. Cancer 28: 1165. Skinner D.G., Dekernion J.B. (1978). Clinical manifestations and treatment of renal parenchymal tumors. Genitourinary Cancer : 107. Sonnenberg E., Meyer D., Weidner H.M., Birchmeier C. (1993). Scatter factor/ hepatocyte growth factor and its receptor, the c- met tyrosine kinase, can mediate a single exchange between mesenchym and epithelia during mouse development. J cell Biol 123: 223 – 235. Soto D., and Skumar S. (1992). Improved detection of mutations in the p53- gene in human tumours as single- stranded conformation polymorphs and double stranded heteroduplex DNA. PCR Methods and Applications 2: 96 – 98. 83 Literaturverzeichnis Surfin G., Mirand E.A., Moore R.H., Chu T.M., Murphy G.P. ( 1977). Hormones in renal cancer. J Urol 117: 433 – 438 Tuffery S., Moine P., Demaille J., Claustres M. (1993). Base substitutions in the human dystrophin gene: detection by using the single strand conformational polymorphism (SSCP) technique. Hum Mutat 2: 368 – 374. Weinmann S.A. (1993). Diuretic use and related risk factors for renal cell cancer. Disease Abstract International B 54: 200 Zbar B., Tory K., Merino M. (1994). Hereditary papillary renal cell carcinoma. J Urol 151: 561 – 566. Zhuang Z., Park W.S., Pack S., Schmidt L., Vortmeye, A.O., Pak E., Pham T., Weil R.J., Candidus S., Lubensky I.A., Lineham W.M., Zbar B., Weirich G. (1998). Trisomy 7harbouring non – random duplication of the mutant MET allele in hereditary papillary renal carcinomas. Nature Genet 20: 66 – 6. 84 9 Lebenslauf Lebenslauf Persönliche Daten Name: Susanne Munk-Schulenburg Geburtsname: Schulenburg Geburtsdatum: 24.11.1972 Geburtsort: Nordenham Schulausbildung Pestalozzi Grundschule, Freiburg 1979 – 1982 Johannes – Schwartz Grundschule, Freiburg 1982 – 1983 Wentzinger Gymnasium, Freiburg 1983 – 1993 Abitur 1993 Freiwilliges Soziales Jahr Johanniter – Unfall – Hilfe, Freiburg 1993 – 1994 Studium der Humanmedizin 1994, Universität Freiburg Ärztliche Vorprüfung 1997 1. Abschnitt der Ärztlichen Prüfung 1998 2. Abschnitt der Ärztlichen Prüfung 2000 Praktisches Jahr Städtisches Klinikum Karlsruhe 2000/ 2001 3. Abschnitt der Ärztlichen Prüfung 2001 ÄiP in der Orthopädischen Chirurgie Januar 2002 – Juni 2002 Loretto-Krankenhaus Freiburg ÄiP in der Inneren Medizin des August 2002 – Januar 2003 Diakoniekrankenhaus Freiburg ÄiP in der Humangenetik Februar 2003 – Juli 2003 Assistenzärztin in der Humangenetik August 2003 – April 2005 Institut für Humangenetik und Anthropologie der Universität Freiburg, PD Dr. Kohlhase 85 10 Danksagung Danksagung Ich bedanke mich sehr herzlich bei Herrn Prof. Dr. med. Hartmut P. H. Neumann für die Überlassung des Themas und die großzügige Unterstützung. Das von Prof. Neumann in den Jahren 1984- 1994 erstellte Register und die gesammelten Blutproben der Patienten stellte eine wichtige Grundlage dieser Arbeit dar, ohne die dieses Patientenkollektiv nicht in diesem Rahmen möglich gewesen wäre. Ganz besonders möchte ich mich bei Herrn Dr. Apel für die ausgezeichnete Einarbeitung und die große Unterstützung im Labor bedanken, sowie für die vielen Anregungen bei der Verfassung dieser Arbeit. Ich fand immer kompetente Hilfe und viel Geduld bei ihm. Außerdem bedanke ich mich bei Herrn Dr. Bender für die Beantwortung meiner zahlreicher Fragen. Mein ganz besonderer Dank ist an meine Großmutter Frau Anna Nowara und meine Pateneltern Christine und Andreas Miedniak gerichtet, die mich während meines Studiums finanziell und moralisch unterstützt haben.