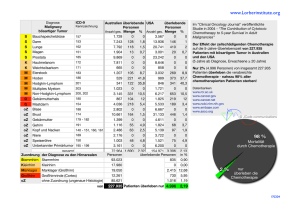
Get cached PDF
Werbung

Chemotherapeutische Beeinflussung des zellulären Immunstatus bei Patienten mit erstmanifestierten soliden Tumoren des Gastrointestinaltraktes Dissertation zur Erlangung des akademischen Grades Dr. med. an der Medizinischen Fakultät der Universität Leipzig Eingereicht von: Karoline Grunemann geb. am 14.10.1976 in Frankfurt (Oder) Angefertigt am: Institut für Klinische Immunologie der Medizinischen Fakultät der Universität Leipzig und dem Zentrum für Innere Medizin des Klinikums Altenburger Land GmbH Betreuer: Prof. Dr. med. Ulrich Sack Beschluss über die Verleihung des Doktorgrades vom: 24.05.2011 1 Für Bart 2 Inhaltsverzeichnis Inhaltsverzeichnis 3 I Bibliografische Beschreibung 6 II Abbildungsverzeichnis 8 III Tabellenverzeichnis 9 IV Abkürzungsverzeichnis 10 1. EINLEITUNG 13 1. 1 Das Immunsystem 13 1.1.1 Angeborenes, unspezifisches Immunsystem 13 1.1.2 Erworbenes, spezifisches Immunsystem 16 1.1.3 Humane Leukozyten-Antigene (HLA-Moleküle) 19 1.1.4 Zytokine und andere Mediatoren 19 1.2 Tumorerkrankungen und das Immunsystem 20 1.2.1 Tumorerkennung und Tumorescape 20 1.2.2 Tumorassoziierte Antigene gastrointestinaler Tumoren 22 1.2.3 Therapiekonzepte 22 1.2.3.1 Immuntherapie maligner Tumoren 22 1.2.3.2 Konventionelle Polychemotherapie 23 1.2.4 Chemotherapeutisch induzierte Effekte auf das Immunsystem 1.3 Gastrointestinale Tumore und deren Therapie 24 25 1.3.1 Ösophaguskarzinom 25 1.3.2 Magenkarzinom 26 1.3.3 Kolonkarzinom 27 1.4 Zielsetzung 28 1.5 Theoretischer Hintergrund der Methoden 29 1.5.1 Dichtegradientenzentrifugation 29 1.5.2 IFN-γ-ELISPOT-Assay (Enzyme linked immunospot assay) 29 2. PROBANDEN, MATERIAL UND METHODEN 33 3 2.1 Probanden 33 2.1.1 Kontrollgruppe 33 2.1.2 Patientengruppe 35 2.2 Material 39 2.2.1 Blutentnahme 39 2.2.2 IFN-γ-ELISPOT–Assay 40 2.3 Methoden 43 2.3.1 Materialgewinnung und Lymphozytenseparation 43 2.3.2 Kryokonservierung der Zellen 44 2.3.4 Bestimmung der vitalen Zellzahl 44 2.3.5 IFN-γ-ELISPOT-Assay 46 2.3.6 Statistische Analyse der Messdaten 49 3. ERGEBNISSE 50 3.1 Patienten 50 3.2 ELISPOT-Testergebnisse 52 3.2.1 Vergleich der Spotanzahl (n) von Kontrollgruppe und Tumorpatienten vor Chemotherapie 52 3.2.2 Vergleich der Stimulationsindizes [n] von Kontrollgruppe und Tumorpatienten vor Chemotherapie 55 3.2.3 Vergleich der Spotintensität (A) von Kontrollgruppe und Tumorpatienten vor Chemotherapie 57 3.2.4 Vergleich der Stimulationsindizes [A] von Kontrollgruppe und Tumorpatienten vor Chemotherapie 59 3.2.5 Vergleich der Spotanzahl (n) von Tumorpatienten vor und nach Chemotherapie 61 3.2.6 Vergleich der Stimulationsindizes [n] von Tumorpatienten vor und nach Chemotherapie 63 3.2.7 Vergleich der Spotintensität (A) von Tumorpatienten vor und nach Chemotherapie 65 3.2.8 Vergleich der Stimulationsindizes [A] von Tumorpatienten vor und nach Chemotherapie 67 4 3.2.9 Zusammenfassung der Ergebnisse 69 4. DISKUSSION 70 4.1 Stellenwert des ELISPOT- Assays bei der Messung zellulärer Immunität _70 4.2 Korrelation zwischen zellulärer und serologischer Immunität 72 4.3 Diskussion der Ergebnisse von Kontrollgruppe und Tumorpatienten vor Chemotherapie 72 4.4 Diskussion der Ergebnisse von Tumorpatienten vor und nach Chemotherapie 73 4.6 Schlussfolgerung 75 5. ZUSAMMENFASSUNG 77 6. SUMMARY 80 7. LITERATURVERZEICHNIS 83 ANLAGEN 107 Erklärung über die eigenständige Abfassung der Arbeit 119 LEBENSLAUF 120 DANKSAGUNG 122 5 I Bibliografische Beschreibung Grunemann, Karoline “Chemotherapeutische Beeinflussung des zellulären Immunstatus bei Patienten mit erstmanifestierten soliden Tumoren des Gastrointestinaltraktes” Universität Leipzig, Dissertation 123 Seiten, 190 Literaturangaben, 9 Grafiken, 8 Tabellen, 6 Anlagen In der vorliegenden Arbeit wurde der zelluläre Immunstatus von 17 Patienten mit Erstdiagnose eines soliden gastrointestinalen Tumors vor und nach intravenöser Applikation von drei Zyklen einer konventionellen Polychemotherapie untersucht. Verglichen wurde zu Beginn der Therapie mit einer Kontrollgruppe, bestehend aus 21 nicht onkologisch vorerkrankten Probanden. Zur Messung der individuellen T-Zellvermittelten Immunantwort auf Einzelzellebene wird auf die Methode des IFN-γELISPOT-Assays zurückgegriffen. Die zentrale Frage war, ob die Applikation einer Polychemotherapie einen messbaren Effekt auf die Immunantwort des einzelnen Individuums hat und ob sich daraus Therapieempfehlungen ableiten lassen. Zudem sollte untersucht werden, ob generelle Unterschiede zwischen Patienten mit einer unbehandelten Tumorerkrankung und gesunden Probanden bzw. allgemein internistisch erkrankten Patienten zu erkennen sind. In Zusammenschau der Ergebnisse sind trotz überwiegend unveränderter T-ZellAntwort auf die meisten der eingesetzten Antigene einige statistisch signifikante Unterschiede festzuhalten. So zeigte die Gruppe der Tumorpatienten vor Applikation der Chemotherapie im Vergleich zur Kontrollgruppe eine signifikant erhöhte Spotintensität und einen höheren Stimulationsindex [A] in Bezug auf das TuberkuloseAntigen CFP-10. Diese Veränderungen waren nach Applikation der Chemotherapie nicht mehr nachzuweisen. Des W eiteren ergaben sich bei den Tumorpatienten vor und nach Chemotherapie signifikante Veränderungen der T-Zell-Antwort bezüglich des 6 Antigens Tetanus-Toxoid. Nach Applikation von 3 Zyklen Chemotherapie kam es zu einer Verminderung des Stimulationsindex [A]. Es wird daher die Vermutung nahe gelegt, dass sich gerade in Bezug auf bakterielle Infektionen die T-Zell-Antwort der Tumorpatienten signifikant ändert. Daraus könnten sich Konsequenzen bezüglich einer Antibiotikaprophylaxe vor Beginn einer Chemotherapie ableiten. Die klinische Relevanz müsste jedoch anhand gezielter Messung auf spezifische bakterielle Antigene in größer angelegten Studien überprüft werden. 7 II Abbildungsverzeichnis Abbildung 1: Schematische Darstellung des ELISPOT Abbildung 2: Boxplots: Spotanzahl (n) von Kontrollgruppe und Tumorpatienten vor Chemotherapie Abbildung 3: 63 Boxplots: Spotintensität (A) von Tumorpatienten vor und nach Chemotherapie Abbildung 9: 61 Boxplots: Stimulationsindizes [n] von Tumorpatienten vor und nach Chemotherapie Abbildung 8: 59 Boxplots: Spotanzahl (n) von Tumorpatienten vor und nach Chemotherapie Abbildung 7: 57 Boxplots: Stimulationsindizes [A] von Kontrollgruppe und Tumorpatienten vor Chemotherapie Abbildung 6: 55 Boxplots: Spotintensität (A) von Kontrollgruppe und Tumorpatienten vor Chemotherapie Abbildung 5: 52 Boxplots: Stimulationsindizes [n] von Kontrollgruppe und Tumorpatienten vor Chemotherapie Abbildung 4: 31 65 Boxplots: Stimulationsindex [A] von Tumorpatienten vor und nach Chemotherapie 67 8 III Tabellenverzeichnis Tabelle 1: Patienten, Alter, Geschlecht, Tumorerkrankungen, Therapieform 36 Tabelle 2: Verwendete Chemotherapien und Inhaltsstoffe 38 Tabelle 3: Verwendete Materialien zur Blutentnahme und Bezugsquellen 39 Tabelle 4: Verwendete Reagenzien für den ELISPOT-Assay und Bezugsquellen 40 Tabelle 5: Verwendete Substrate, Enzyme für den ELISPOT-Assay und Bezugsquellen 40 Tabelle 6: Verwendete Antikörper, Antigene für den ELISPOT-Assay und Bezugsquellen 41 Tabelle 7: Verwendete Laborgeräte, Software für den ELISPOT-Assay und Bezugsquellen Tabelle 8: Verwendete Puffer und Medien für den ELISPOT-Assay 41 43 9 IV Abkürzungsverzeichnis AEG adenocarcinoma of esophagogastric junction (engl., Adenokarzinom des ösophago-gastralen Übergangs) AG Aktiengesellschaft APC antigen presenting cell (engl., antigenpräsentierende Zelle) ATP Adenosintriphosphat Aufl. Auflage BCIP/ NBT 5- Bromo-4-Chloro-3-Indolylphosphat / Nitroblau Tetrazolium BSA bovines Serumalbumin bzw. beziehungsweise ca. circa CA 19-9 carbohydrate antigen (engl., Kohlenhydrat-Antigen) CD cluster of differentiation (engl., Zelldifferenzierungsmarker) CEA karzinoembryonales Antigen CFP-10 culture filtrate antigen - 10 CFU-GEMM colony forming units for granulocytes, eosinophilic granulocytes, monocytes, macrophages (engl., koloniebildende Einheit für Granulozyten, Eosinophile, Monozyten und Makrophagen) CMV Zytomegalievirus CSF colony stimulating factor (engl., koloniestimulierender Faktor) diff. differenziert d. h. das heisst DMSO Dimethylsulfoxid dt. deutsch ECF Epirubicin, Cisplatin, 5-Fluorouracil EGFR epidermal growth factor receptor (engl., epidermaler W achstumsfaktorrezeptor) engl. englisch ELISPOT Enzyme linked immunospot assay ELISA Enzyme linked immunosorbent assay 10 EOX Epirubicin, Oxaliplatin, Xeloda = Capecitabine ESAT-6 early secretory antigenic target - 6 et al. et alii (lat., und andere) FAP familiäre adenomatöse Polyposis FKS fötales Kälberserum FOLFIRI 5-Fluorouracil, Folinsäure, Irinotecan FOLFOX-4 5 - Fluorouracil, Folinsäure, Oxaliplatin FOXP3 forkhead box protein 3 5-FU 5-Fluorouracil gr. griechisch HIV human immunodeficiency virus HLA human leucocyte antigen (engl., humanes Leukozytenantigen) HNPCC hereditäres nicht-polypöses kolorektales Karzinom IFN Interferon Ig Immunglobulin lat. latein Lf limes flocculationis (Flockungseinheit) MAK Membranangriffskomplex MHC Major histocompatibility complex (engl., Haupthistokompatibilitätskomplex) MMS Monozyten-Makrophagen-System NK-Zelle natürliche Killerzelle Nr. Nummer o. g. oben genannt PAMP pathogen associated molecular pattern (engl., Pathogen assoziiertes molekulares Muster) PBMC peripheral blood mononuclear cells (engl., periphere mononuleäre Blutzellen) PRR pattern recognition receptor, (engl. Mustererkennungsrezeptor) PVDF Polyvinylidenfluorid SI Stimulationsindex sog. so genannt TCR T-Zell-Rezeptor 11 TLR toll like receptor TNM Tumor - Lymph node - Metastasis (Tumor-Lymphknoten-Metastase; classification of malignant tumors, engl. Klassifikation malignen Tumoren) TNF- Tumornekrosefaktor alpha Treg regulatorische T-Zellen u. a. unter anderem UICC International Union Against Cancer (engl., Internationale Union gegen Krebs) VE voll entsalzt WHO World Health Organisation (engl., W eltgesundheitsorganisation) z. B. zum Beispiel Z. n. Zustand nach 12 1. EINLEITUNG 1. 1 Das Immunsystem Der menschliche Organismus ist ein offenes biologisches System, da er in ständigem stofflichen Austausch mit seiner Umgebung steht. Dabei kommt er auch mit potenziell pathogenen Erregern und Substanzen in Berührung (Elsaie ML et al., 2008; Caso JR et al., 2008; Adkinson NF jr. et al., 1977). Um sich vor diesen zu schützen, hat sich im Laufe der Evolution ein außerordentlich angepasstes und effektives System entwickelt, das Immunsystem (immunis, lat.: von etwas frei sein). Mit ihm gelingt es, auf zellulärer und molekularer Ebene, wirksame Abwehrmechanismen zur Verfügung zu stellen (Prescott SL, 2009). Man unterscheidet das angeborene und das erworbene Immunsystem. Alle zellulären Komponenten des Immunsystems gehen auf die hämatopoetische, pluripotente Stammzelle des Knochenmarks zurück, aus der sich über verschiedene Signalwege, u. a. von löslichen Mediatoren (Zytokinen), multipotente Progenitorzellen, sog. CFU-GEMM (colony forming units for granulocytes, eosinophilic leucocytes, monocytes, macrophages) der myeloischen und lymphatischen Zellreihe differenzieren (Palacios R et al., 1993; Dresch C et al., 1985). 1.1.1 Angeborenes, unspezifisches Immunsystem Der stammesgeschichtlich ältere Teil des Immunsystems kann einer Vielzahl von Erregern in sehr kurzer Latenzzeit ein gut etabliertes Abwehrsystem gegenüberstellen (Haslett C et al., 1989). Diese Abwehrmechanismen tragen wesentlich zur Unterscheidung von körpereigenen und körperfremden Antigenen bei. Der Nachteil ist, dass hierbei nur ein begrenztes Repertoire nicht wandlungsfähiger Antigenrezeptoren zu Verfügung steht, sog. Mustererkennungsrezeptoren (PRRs, engl: pattern recognition receptors). Sie sind in der Lage, pathogenassoziierte molekulare Muster zu erkennen (PAMPs, engl: pathogen associated molecular patterns) (Seya T et al., 2010; Seya T et 13 al., 2001). Das sind Aminosäuresequenzen, die hochkonserviert in Mikroorganismen, nicht jedoch im Wirtsgewebe zu finden sind (Morgensen TH, 2009). Diese nichtklonalen Abwehrmechanismen sind gegen eine ganze Gruppe von Antigenen gerichtet und somit relativ unspezifisch (Cooper EL et al., 2002). Das angeborene Immunsystem entwickelt kein immunologisches Gedächtnis. Das erste Hindernis für eindringende Pathogene sind physikalische Barrieren wie die intakte Haut, Schleimhäute, Flimmerepithelien und das antimikrobielle Enzymsystem (Crawford RM et al., 1994). Der zelluläre Anteil des angeborenen Immunsystems umfasst Granulozyten, Monozyten, Makrophagen und dendritische Zellen, die sich aus der pluripotenten myeloischen Progenitorzelle über myeloische Vorläuferzellen entwickeln (Stanley ER et al., 1975). Granulozyten werden durch humorale Signale wie Interleukine und Komplementfaktoren aus dem Knochenmark freigesetzt (Colotta F et al., 1992; Cannistra SA et al., 1988). Unterschieden werden nach Färbeverhalten neutrophile (polymorphkernige), basophile und eosinophile Granulozyten. Neutrophile Granulozyten identifizieren, phagozytieren und töten Erreger. Durch Kennzeichnung der zu phagozytierenden Zellen mittels Immunglobulinen und Komplementfaktoren, die Opsonierung, gelingt ihnen die Differenzierung zwischen körperfremden und körpereigenen Zellen (Kemper C et al., 2008; Brouwer N et al., 2006). Eosinophile Granulozyten spielen eine besondere Rolle in der Abwehr von Parasiten (Spry CJ, 1978), bei allergischen Reaktionen und beim Abbau von Fibrin im Rahmen von Entzündungsreaktionen. Sie scheinen ebenfalls eine wichtige immunmodulatorische Rolle als antigenpräsentierende Zellen zu spielen (Akuthota P et al., 2010). Durch Degranulation sind sie befähigt, ihre Enzyme (Zytokine, Chemokine, Peroxidasen, Leukotriene und Plättchen aktivierende Faktoren) nach außen abzugeben und somit größere extrazelluläre Organismen zu töten (Neves JL et al., 2009). Basophile Granulozyten besitzen Oberflächenrezeptoren für das Fc-Fragment des IgE (Marone G et al., 1981) und haben eine unter Steuerung einer Vielzahl von Zytokinen ähnliche Funktion wie Mastzellen bei der IgE-vermittelten allergischen Sofortreaktion, bei der sie durch Degranulation u. a. Histamin ausschütten können (Valent P at al., 2010). 14 Monozyten und Makrophagen bilden das MMS (Monozyten-Makrophagen-System). Ihre Aufgabe ist die Phagozytose und Abtötung von Mikroorganismen und gealterten oder entarteten körpereigenen Zellen. Die Erkennung von Mikroorganismen erfolgt über spezielle Toll-like-Rezeptoren (TLRs), Transmembranrezeptoren aus der Familie der PRRs, die außerordentlich viele Signal gebende Triggerfunktionen erfüllen (Kulkarni R et al., 2010; Kaczorowski DJ et al., 2009). Makrophagen produzieren zudem einige Zytokine wie IL-1, IL-6, IL-8 und TNF- (Crawford RM et al., 1994). Eine wichtige Eigenschaft ist die Fähigkeit zur Antigenpräsentation und somit zur Stimulation von TLymphozyten. (Barker RN et al., 2002; Janeway CA et al., 2002). Ihrerseits werden sie wiederum durch T-Zell-Zytokine (z. B. Interferon-γ) aktiviert. Die wichtigsten antigenpräsentierenden Zellen sind die dendritischen Zellen, die sich aus myeloischen und lymphatischen Vorläuferzellen entwickeln (Chesnut RW et al., 1985). Sie stellen neben den NK-(natürlichen Killer) Zellen ein Bindeglied zwischen angeborenem und erworbenem Immunsystem dar. Man unterscheidet LangerhansZellen in Haut und Schleimhaut, interdigitierende dendritische Zellen als APC (antigenpräsentierende Zellen) für T-Lymphozyten und follikuläre dendritische Zellen als APC für B-Lymphozyten und somit beteiligt an der Ausbildung eines immunologischen Gedächtnisses (Hoefsmit EC et al., 1982). Der zweite, humorale Teil des angeborenen Immunsystems umfasst eine Kaskade von Plasmaproteinen, die man als Komplementsystem bezeichnet, und die letztlich in der Lyse eingedrungener Bakterien oder Zellen gipfelt. Die meisten dieser Plasmaproteine sind Zymogene (Bezeichnung mit dem Buchstaben C), d. h. Pro-Enzyme, die durch proteolytische Spaltung aktiviert werden. Man unterscheidet den klassischen und alternativen Signalweg der Komplement-Aktivierung. Der klassische W eg ist antikörperabhängig: Ein Aggregat von Immunglobulinen bewirkt eine kaskadenartige Aktivierung verschiedener Komplement-Proteine (Czermak BJ et al., 1998; Fällmann M et al., 1993). Der alternative Weg wird direkt durch die Zelloberfläche des Mikroorganismus ausgelöst. Endprodukte sind jeweils zwei C5-Konvertasen, die durch Bindung an weitere Komplementproteine eine proteolytische Terminalsequenz auf der Zellmembran bilden, den sog. Membranangriffskomplex (MAK) (Kolb WP et al., 1975 und 1973), der durch Poren in der Zellmembran zur direkten Lyse führt. W eitere Effekte der 15 Komplementaktivierung sind die Anaphylatoxin-vermittelte Kontraktion der glatten Muskulatur, die Erhöhung der Gefäßpermeabilität, die Degranulation von Basophilen und Mastzellen, die Chemotaxis von Granulozyten und die B-Zell-Stimulation (Pezuotto A et al., 2007; Swierzko AS et al., 2003). 1.1.2 Erworbenes, spezifisches Immunsystem Im Gegensatz zum angeborenen Immunsystem kann das erworbene bzw. adaptive Immunsystem ein immunologisches Gedächtnis entwickeln. Durch hochspezifische Antigenrezeptoren ist es möglich, eine maßgefertigte Immunantwort auf ein determiniertes Antigen zu generieren (Bonilla FA et al., 2010). Beispielsweise interagiert der T-Zell-Rezeptor (TCR) mit den passenden HLA-Molekülen (humane leucocyte antigen) auf Epithelien und ist so in der Lage, spezifische Oberflächenstrukturen von Fremdantigenen wiederzuerkennen (Miller JF, 1976). Die Anpassung des TCR erfolgt durch genetisches Rearrangement der antigenerkennenden Oberflächenmoleküle (Hedrick SM et al., 1985; Sims JE et al., 1984). Die große Vielfalt der ca. 1015 verschiedenen T-Zell-Rezeptoren ermöglicht dem Organismus im Falle eines erneuten Kontaktes mit demselben Antigen, eine schnelle und zielgerichtete Immunantwort zu erzeugen. Wesentlich für die erworbene Immunität ist das Prinzip der klonalen Selektion. Jeder Lymphozyt besitzt einen Rezeptortyp mit eigener Spezifität. Potenziell autoreaktive unreife Leukozyten werden ausgemustert, bevor die Ausreifung abgeschlossen ist (Janeway CA Jr et al., 1989). Trifft nun ein Antigen auf einen naiven Lymphozyten, erfolgt die Zellteilung und dadurch die Entstehung eines Klons identischer Nachkommen. Man kann das adaptive Immunsystem ebenfalls in eine zelluläre und humorale Immunantwort unterteilen, wobei Letztere durch das Serum übertragbar ist. Humorale Immunantwort durch B-Lymphozyten B-Lymphozyten sind in der Lage, Immunglobuline (Antikörper) zu sezernieren. Durch genetische Rekombination der schweren und leichten Immunglobulinketten entsteht eine enorme Vielfalt von insgesamt ca. 106 Immunglobulinen (Johnson K et al., 2009). Diese leiten durch ihre Bindung an das Antigen (Opsonierung) die Aktivierung des 16 klassischen Komplementsystems und anderer Komponenten des angeborenen Immunsystems ein (Boackle SA et al., 1998). Bei Kontakt mit einem Antigen kommt es zu einer Aktivierung und Proliferation der BLymphozyten (Williamson AR et al., 1976). Zur Erkennung eines Antigens benötigt der naive B-Lymphozyt jedoch APCs. Die Aktivierung erfolgt T-Zell-abhängig oder -unabhängig. Nach intrazellulärer Zerlegung des Antigens und Präsentierung mittels HLA-Klasse-II-Moleküle an der B-Zell-Oberfläche können CD4+ (cluster of differentiation) -T-Helferzellen den B-Lymphozyten über Oberflächenrezeptoren und Zytokinausschüttung aktivieren (Janeway CA Jr, 1980). Dabei erfolgt die Produktion von Antikörperklassen zytokinspezifisch. T-Zell-unabhängig ist die Aktivierung der BLymphozyten über direkte Bindung des Antigens an membranständige Rezeptoren möglich. Man unterscheidet zwei verschiedene Typen von B-Effektorzellen: Plasmazellen sind für die spezifische monoklonale Synthese und Sezernierung von Antikörpern zuständig. B-Gedächtniszellen speichern Informationen über bestimmte Antigenstrukturen und können bei erneutem Kontakt mit diesem Antigen mit Hilfe von CD4+ -T-Helferzellen und T-Gedächtniszellen in kürzerer Latenzzeit große Mengen präzise ausgerichteter Antikörper produzieren. Zelluläre Immunantwort durch T-Lymphozyten Die lymphoiden Progenitorzellen der T-Lymphozyten erfahren ihre Ausreifung im Thymus. Charakteristika sind der o. g. antigenspezifische T-Zellrezeptor und der Oberflächenmarker CD3+. Sie benötigen zur Antigenerkennung eine professionelle APC, die das Antigen auf dem entsprechenden HLA-Komplex präsentiert. Auch hier sind zusätzlich costimulierende Zytokine bei der Aktivierung beteiligt. Diesen Vorgang nennt man Priming (Lenschow DJ et al., 1997). Nur bei Vorliegen costimulatorischer Signale kommt es zu einer Proliferation und Differenzierung der T-Zellen zu bewaffneten T-Effektorzellen. Fehlt ein Signal, können diese Zellen anerg werden (Guerder S et al., 1994). Je nach zytokinspezifischer Signalgebung erfolgt nun die Differenzierung in CD4+ Helferzellen und CD8+ - zytotoxische Zellen, wobei diese in einem festen Verhältnis von ca. 2-3 / 1 stehen, dem CD4 / CD8- Quotienten. Die Beschreibung dieser funktionellen Dichotomie der T-Helferzellen, die sich wechselseitig regulieren können, löste das 17 zuvor favorisierte Konzept der T-Supressorzelle vollständig ab (Mosmann TR et al., 1986). Es konnte aber ein Teil der T-Helferzellpopulation identifiziert welche, welche als natürlich vorkommende und adaptive regulatorische T-Zellen (Treg) unterschieden werden und vermutlich die Entstehung autoimmuner (Zhou X et al., 2010; Jain N et al., 2010) und kanzerogener Prozesse verhindern können (Ha TY, 2009). CD4+ -Helferzellen besitzen den CD4-Rezeptor, der als Co-Rezeptor mit dem HLA-IIKomplex der APC in Verbindung tritt (Kalish RS, 1995). Sie verstärken die Immunantwort durch Steigerung der Expression von B-Zellen und CD8+ - zytotoxischen Zellen. Nach ihrem Zytokinmuster unterteilt man die CD4+ -Helferzellen in Th1Helferzellen, welche die Zytokine IL-2, IFN-γ und TNF- produzieren, und Th2- Helferzellen, die IL-4, -10 und -13 produzieren. Ein Teil der CD4+ -Helferzellen wandelt sich zu T-Gedächtniszellen. CD8+ - zytotoxische Zellen besitzen den CD8-Rezeptor als Co-Rezeptor zum HLA-IKomplex (Marusic-Galesic S et al., 1988) auf pathologisch veränderten kernhaltigen Zellen und können diese durch zytolytische Mediatoren direkt durch Einleitung einer Apoptose oder Nekrose abtöten. Natürliche Killerzellen NK-Zellen können weder den B- noch den T-Lymphozyten zugeordnet werden, da sie sich schon früh von der lymphatischen Stammzelle differenzieren. Sie sind CD 3-, haben keinen TCR und können sowohl antikörpervermittelt als auch nicht- antikörpervermittelt zytotoxisch wirksam werden. Zielzellen ohne HLA-Moleküle, wie virusinfizierte oder tumoröse Zellen, werden von ihnen abgetötet. Sie können innerhalb von Minuten aktiviert werden, sind allerdings aufgrund ihres eingeschränkten Antigenrepertoires weniger effektiv als CD8+ - zytotoxische Zellen. Offenbar sind sie in der Lage, eine adaptive Immunantwort zu generieren (Paust S et al., 2010). 18 1.1.3 Humane Leukozyten-Antigene (HLA-Moleküle) Intrazelluläre Antigene sind in Form eines extrem polymorphen Genclusters, den MHCMolekülen (major histocompatibility complex) auf dem Chromosom 6 codiert (Bakker E at al., 1979). Sie werden als HLA (human leukocyte antigen), membranständige Glykoproteine, auf der Epitheloberfläche aller Körperzellen präsentiert. Jeder Mensch besitzt ein individuelles, genetisch determiniertes HLA-Muster. Man unterteilt zwei Klassen von HLA-Molekülen. HLA-Klasse-I-Moleküle (Bjorkman PJ et al., 1987) werden auf allen kernhaltigen Zellen sowie den kernlosen Thrombozyten exprimiert und präsentieren vorwiegend Peptide zytosolischen Ursprungs, z. B. Viruspartikel, zytosolische Parasiten oder + Tumorantigene. Sie werden von zytotoxischen T-CD8 - Zellen erkannt und eliminiert. HLA-Klasse-II-Moleküle werden primär auf professionellen APCs wie Makrophagen, dendritischen Zellen, aktivierten B-Zellen und Thymusepithelzellen exprimiert und präsentieren vor allem Peptide aus vesikulären Zellkompartimenten und somit auch extrazelluläre Antigene, z. B. Bakterien- und Parasitenfragmente (Pieters J, 1997). Sie werden ausschließlich von T-CD4+-Helferzellen erkannt, die wiederum B-Zellen aktivieren und damit weitere Kaskaden des Immunsystems auslösen. 1.1.4 Zytokine und andere Mediatoren Zytokine sind eine heterogene Gruppe von Steuer- und Kommunikationsproteinen. Dazu gehören Interleukine (IL 1-12), Tumornekrosefaktoren (TNFInterferone (IFN- , IFN- , IFN-γ) und hämatopoetische Wachstumsfaktoren (G-CSF, GM-CSF). Sie werden von vielen immunkompetenten Lymphozyten, und TNF- ), Zellen wie B- und T- Zellen des MMS aber auch von Parenchym- und Epithelzellen synthetisiert und entfalten ihre parakrine, autokrine und endokrine Wirkung über Zytokinrezeptoren. Diese werden nach der CD-(cluster of differentiation) Klassifikation eingeteilt. Ihre Funktionen reichen über Stimulation, Aktivierung und Proliferation von Entzündungszellen bis zur endokrinen ZNS-Stimulation. Interferon-γ (IFN-γ) kommt dabei eine besondere Rolle zu. Es wurde erstmals 1965 aufgrund seiner antiviralen Aktivität beschrieben (Wheelock EF, 1965). Es wird von 19 CD4+ Th1-Helferzellen, CD8+-zytotoxischen Zellen und NK-Zellen nach Stimulation durch dendritische Zellen über IL-12 sezerniert (Lederer JA, 1996). Es fördert die proinflammatorische und antimikrobielle Aktivität von Makrophagen, bevor das adaptive Immunsystem entsprechend entwickelt ist und verstärkt zudem durch Expression von MHC-I- und MHC-II-Molekülen (Wedgwood JF et al., 1988) die Wirkung zytotoxischer T-Lymphozyten. Es kann sogar direkt zytotoxisch wirksam werden und spielt offenbar eine entscheidende Rolle in der Aufrechterhaltung der gesunden gastrointestinalen Mukosa (Carol M et al., 1998) und in der Protektion vor zahlreichen Autoimmunerkrankungen, z. B. der autoimmunen Endokarditis (Barin JG et al., 2010). 1.2 Tumorerkrankungen und das Immunsystem Ein maligner Tumor entsteht durch den vielschichtigen Prozess der malignen Transformation durch multiple Mutationen in Tumorsupressor-Genen, Proto-Onkogenen und Transkriptionsregulatoren einer gesunden Zelle und führt zu einer Änderung der intrazellulären Proteinkonstellation. Diese Proteine werden als Tumorantigene bezeichnet und nach intrazellulärem Abbau über MHC-I-Klasse-Moleküle den CD8+zytotoxischen T-Zellen präsentiert. 1.2.1 Tumorerkennung und Tumorescape Das Immunsystem ist prinzipiell in der Lage, diese Tumorantigene zu erkennen (Rosenberg SA et al., 1988). Es konnte gezeigt werden, dass Tumorzellen in vitro von humanen Immunzellen erkannt und zerstört werden können (Itoh K,1987). Allerdings konnte in vivo keine vergleichbar effiziente Eliminierung entarteter Zellen erreicht werden. Tumorzellen verfügen über ein beeindruckendes System, sich dem Angriff des Immunsystems zu entziehen, man bezeichnet dies auch als Tumorescape (Palena C et al., 2010). Bei vielen der über MHC-Moleküle präsentierten Tumorantigene handelt es sich um überexprimierte Produkte des Zellstoffwechsels und nicht um mutierte 20 Proteine. Aufgrund des Phänomens der klonalen Selektion, bei dem eine Prägung des Immunsystems auf "selbst" und "nicht selbst" erfolgte, haben diese Antigene nur eine geringe Immunogenität (Seya T et al., 2010). Außerdem kommt es zu einer stetigen genetischen Veränderung der Tumorzelle und deren Oberflächenstrukturen, sodass sogar entstandene Immunantworten wieder gelöscht werden können. Offenbar erzeugen Tumorzellen durch sog. Immunoediting ein immunsuppressives, inflammatorisches Mikroklima, um sich zu schützen (Arum CJ et al., 2010; Yaqub S et al., 2009; Dunn GP et al., 2002). Dies ist ein sich ständig adaptierender Prozess. Des Weiteren konnte bewiesen werden, dass Tumorzellen durch verschiedene Mechanismen die Expression von HLA herunterregulieren können und somit die Antigenpräsentation und die damit verbundene T-Zell-Aktivierung ausbleibt (Bogerding A et al., 2010; Ye Q et al., 2010; Sers C et al., 2009). Durch den Verlust costimulatorischer Zytokinsignale, wird ebenfalls die T-Zellantwort supprimiert (Fujiwara K et al., 2004). Zudem sind Tumorzellen in der Lage, die Reifung dendritischer Zellen zu unterdrücken (Perrot I et al., 2007). Durch ein verändertes Zytokinmillieu kann beispielsweise die Funktionsfähigkeit von Makrophagen beeinträchtigt werden (Elgert KD, 1998). Ein weiteres bemerkenswertes Phänomen + + ist die tumorbedingte Zunahme + Treg(regulatorischer) Zellen (sog. CD4 CD25 FOXP3 Treg) (Clarke SL et al., 2006), die jedoch teilweise anerg gegenüber einer T-Zell-Rezeptorstimulation sind (W olf AM et al., 2003) und daher keine Zytokine produzieren können (Lee PP et al., 1999). Aber das Immunsystem spielt, wenn auch mit begrenzter Effizienz, eine Rolle in der Kontrolle des Tumorwachstums. Scheinbar wirkt sich beispielsweise eine Infiltration des Tumors durch immunkompetente T-Zellen, vor allem CD8+ -zytotoxische Zellen, prognostisch günstig in Bezug auf die Überlebensrate der Patienten aus (Yamada N et al., 2010; Simpson JA et al., 2010; Lee W S et al., 2010; Mortarini R et al., 2003) und bietet somit einen Ansatz in der Immuntherapie maligner Tumoren (Vanderlocht J et al., 2010). 21 1.2.2 Tumorassoziierte Antigene gastrointestinaler Tumoren Durch genetische Veränderungen exprimieren Tumorzellen qualitativ und quantitativ veränderte Antigene im Vergleich zu gesundem Gewebe. Der Nachweis dieser tumorspezifischen Antigene, sog. Tumormarker, hat große Bedeutung in Diagnostik, Therapie, Verlaufsbeurteilung und Prognose der erkrankten Patienten (Seleznick MJ, 1992; Kuusela P et al., 1984). Das Karzinoembryonale Antigen (CEA) ist ein onkofetales Antigen, welches während der Embryonal- und Fetalzeit gebildet wird und physiologischer Weise im fetalen Gastrointestinaltrakt und Serum vorkommt. Die Bildung wird nach der Geburt supprimiert. 95% der gastrointestinalen Tumoren exprimieren CEA, wobei keine Organspezifität besteht. Die Serumwerte korrelieren mit der Tumorausdehnung (Cooper EH et al., 1975; Herrera MA et al., 1976). Das Carbohydrate Antigen (CA 19-9) ist ein Glykolipid und kommt im fetalen Epithel von Magen, Darm und Pankreas vor. Es ist nachweisbar bei einer Reihe gastrointestinaler Tumoren, v. a. beim Magen- und Kolonkarzinom, aber auch bei Karzinomen der Gallenblase und Gallenwege, des Pankreas (Haglund C et al., 1986) und bei benignen entzündlichen Erkrankungen wie z. B. der Pankreatitis (Del Villan BC et al., 1983). 1.2.3 Therapiekonzepte 1.2.3.1 Immuntherapie maligner Tumoren Zahlreiche Fortschritte konnten in den letzten dreißig Jahren auf dem Gebiet der Immuntherapie von Tumoren verzeichnet werden. Dabei versucht man hauptsächlich, die vorhandenen spontanen Immunantworten der Patienten gegen spezifische Tumorantigene durch Impfungen mit Antigenen und Adjuvantien zu verstärken (van den Broek M et al., 2010). Zwei generelle Prinzipien antigenunabhängige vom der Immuntherapie antigenabhängigen werden (Rosenberg unterschieden: SA, 1999). Bei das der antigenunabhängigen Therapie werden in der klinischen Praxis bereits erfolgreich spezielle Adjuvanzien verwendet, die Immunantworten von B- und T-Zellen bzw. die 22 Reifung und Ausdifferenzierung von APCs bewirken. Ein Beispiel dafür ist der GM-CSF (granulocyte-macrophage colony stimulating factor) (Disis ML et al., 1996) oder der Einsatz von Zytokinen wie IL-2 , Interferon- und IL-12 (Akaza H et al., 2010; Huang X et al., 2010; Hiroishi K et al., 2010). Bei der antigenabhängigen Therapie erfolgt z. B. die Applikation antigenspezifischer Peptide verschiedener Tumorantigene direkt oder in Kombination mit Zytokinen wie IFN-γ (Fallarino F et al., 1999) oder Viren (W ang M et al., 1995). Zahlreiche Arbeiten beschreiben die Beladung APCs wie dendritischer Zellen mit o. g. Peptiden, wobei dies bisher nicht evidenzbasiert ist (Petersen TR et al., 2010). Ein weiteres Prinzip ist die passive Verwendung von Antikörpern wie z. B. Herceptin gegen Her2/ neu-Rezeptoren beim Mammakarzinom (Baselga J et al, 1998), Cetuximab, einem Antikörper gegen EGFR (epithelial growth factor receptor) additional bei der Therapie solider Tumoren (Huang SM et al., 1999) oder Rituximab, einem AntiCD20-Antikörper, der in der Therapie maligner Lymphome breite Anwendung findet (Leopardo D et al., 2010; Dotan E et al., 2010). 1.2.3.2 Konventionelle Polychemotherapie Unter einer Chemotherapie versteht man die Applikation von Substanzen, die in den Zellstoffwechsel maligner Zellen und deren Zellteilungsvorgänge entweder zytostatisch oder zytotoxisch eingreifen. Prinzipiell wirkt sich dies auf alle Körperzellen, hautsächlich jedoch auf schnell proliferierende Zellen wie die des Tumors, aber auch anderer Gewebe wie Schleimhautepithelien, Haarzellen und Zellen des hämatopoetischen Systems aus. Man unterscheidet neo-adjuvante Therapieapplikationen, d. h. einer Operation oder Radiatio vorgeschaltet um die Tumormasse zu verkleinern und Ansprechraten zu verbessern, und adjuvante Applikationen, z. B. um nach potenziell kurativen Operationen das Rezidivrisiko zu minimieren. Kurative Therapien verfolgen das Ziel einer Heilung bzw. langen Remissionsfreiheit. Palliative Ansätze sollen mit einer Verlängerung der Überlebenszeit und Reduktion tumorbedingter Symptome zu Verbesserung der Lebensqualität bei fortgeschrittener Erkrankung beitragen. 23 Heutzutage wird meist eine Kombination mehrerer Zytostatika, eine sog. Polychemotherapie, verabreicht. Vorteile sind die Reduktion der Resistenzentwicklung durch unterschiedliche Wirkmechanismen, synergistische oder additive Effekte durch unterschiedlichen Angriff im Zellzyklus und unterschiedliche Toxizitätsspektren (nonoverlapping toxicity), um volle Dosierungen zu ermöglichen (Hind D et al., 2008). Intermittierende Applikationen sollen bei gleichzeitiger Maximierung der Zellabtötung die Immunsuppression reduzieren. 1.2.4 Chemotherapeutisch induzierte Effekte auf das Immunsystem Es ist bekannt, dass der Tumor selbst, aber auch applizierte konventionelle zytotoxische Chemotherapeutika Immunantworten signifikant beeinflussen (Gardner RV, 1999; Campbell MJ et al., 2005; Caras I et al., 2004). So zeigt sich bei vielen Patienten unter Therapie eine deutliche passagere oder anhaltende Reduktion einiger Lymphozytenpopulationen (Kotsakis A et al., 2000; Kreienberg R et al., 1979), wobei andere Patienten dadurch unbeeinflusst zu bleiben scheinen (Mellios T et al., 2010). Leukopenien sind neben Anämie und Thrombozytopenie bekannte und gefürchtete Nebenwirkungen der hoch dosierten Polychemotherapien (Crawford J et al., 2004). Sie stellen für die Patienten ein erhöhtes Risiko dar, an bakteriellen, viralen oder opportunistischen Infektionen, wie Pilzinfektionen, zu erkranken (O´Brien SN et al., 2003). Fieberhafte Neutropenien sind bei diesen immundefizienten Patienten mit einer deutlich erhöhten Morbidität verbunden. Die Applikation der Chemotherapeutika hat jedoch im Gegensatz dazu offensichtlich auch einen messbar positiven Effekt auf die Immunantwort, wie z. B. die Zunahme von T-Gedächtniszellen und Treg im peripheren Blut. Die chemotherapieinduzierte Infiltration mit sog. TregFOXP3+ T-Lymphozyten scheint mit einer längeren Rezidivfreiheit und Überlebensdauer zu korrelieren (Correale P et al., 2010; Liu W M et al., 2010). Interessant erscheint der Aspekt, dass in Modellversuchen Chemotherapeutika wie Doxorubicin und Paclitaxel in nicht zytotoxischer Dosierung indirekt zur Aktivierung dendritischer Zellen und somit zur Stimulation von T-Effektorzellen beitragen können (Shurin GV et al., 2009). So bewirkt die Chemotherapie über verschiedene 24 Mechanismen, wie z. B. die Freisetzung von ATP (Adenosintriphosphat) oder bestimmter Proteine aus der Tumorzelle einen apoptotischen Zelltod (Martins I et al., 2009; Tesniere A et al., 2010). Der massive Anfall apoptotischen Zellmaterials bewirkt eine Verbesserung der Antigenpräsentation durch APCs, was letztlich auf die Wirksamkeit einer kombinierten Chemo - Immuntherapie hinweist (van der Most RG et al., 2006). 1.3 Gastrointestinale Tumore und deren Therapie Gastrointestinale Tumore sind maligne Erkrankungen des Gastrointestinaltraktes, einschließlich des Ösophagus- und Magenkarzinoms, des Colon- und Rektumkarzinoms bzw. neuroendokriner Tumore. Die Tumore, der in dieser Studie eingeschlossenen Patienten, sollen im Folgenden näher erläutert werden. 1.3.1 Ösophaguskarzinom Ösophaguskarzinome werden in 2 histologische Gruppen eingeteilt: das Plattenepithelkarzinom und das Adenokarzinom des gastroösophagealen Übergangs. Das Plattenepithelkarzinom des Ösophagus ist der häufigste Tumor der Speiseröhre. In Deutschland beträgt die Inzidenz bei Männern bei 4-5 / 100.000 Einwohner, bei Frauen 0,5-1/100.000 Einwohner, mit einem medianen Erkrankungsalter von 65 Jahren. Männer sind etwa 4-mal häufiger betroffen als Frauen. Bei Diagnosestellung sind Ösophaguskarzinome in über 70% der Fälle in fortgeschrittenen UICC-Stadien II-IV, somit haben die Patienten eine 5-Jahres-Überlebensrate von maximal 5%. In der westlichen Gesellschaft ist das Auftreten eines Ösophaguskarzinoms zu über 80% mit chronischem Alkohol- und Nikotinabusus assoziiert (Weikert C et al., 2009; Franke A et al., 2005). Prädisponierend ist außerdem das Plummer-Vinson-Syndrom, die glutensensitive Enteropathie, die Sklerodermie, die Achalasie, Ösophagusdivertikel, vorangegangene Säure- und Laugenverätzungen der Speiseröhre und eine stattgehabte Strahlentherapie in Hals- und Thoraxbereich (Herold G et al., 2009). 25 Therapeutisch kommen in sehr frühen Tumorstadien (uT1b) endoskopische Verfahren (Kurokawa J et al., 2009); bei fortgeschritteneren Tumoren Radio-, kombinierte Radiochemotherapien und operative Verfahren in Betracht (Zimmermann F et al., 2006). In fortgeschrittenen Stadien können dem Patienten systemische Cisplatin- oder Oxaliplatin-basierte Chemotherapien angeboten werden (Ajani JA et al. 2010; Kachele V et al. 2010; Laak E, 2005). Die Applikation erfolgt derzeit meist nach dem TCFSchema (Docetaxel, Cisplatin, 5-FU) (Shim HJ et al., 2010; Ajani JA et al., 2005). Bei Karzinomen des gastrointestinalen Übergangs (AEG) handelt es sich histologisch um Adenokarzinome. Man unterscheidet das AEG I (Barrett-Karzinom), welches auf dem Boden einer Zylinderzellmetaplasie nach chronischer Refluxösophagitis entsteht, das AEG II (Kardiakarzinom) und das subkardiale Karzinom AEG III. Analog zum Plattenepithelkarzinom kommen chirurgische, adjuvante, neoadjuvante und palliative Therapieoptionen infrage. zweiwöchentliche Applikation (=Folinsäure), Oxaliplatin, So zeigt bei nach dem Docetaxel) metastasierten Adenokarzinomen FLOT-Schema eine deutliche (5-FU, die Leucovorin Minderung der Tumorprogressionsrate und eine Verlängerung der mittleren Überlebenszeit (Al Batran SE et al., 2008). 1.3.2 Magenkarzinom Das Magenkarzinom ist die 5.-häufigste Todesursache bei Frauen und die 6.-häufigste bei Männern. Das mittlere Erkrankungsalter liegt bei Männern bei 68, bei Frauen bei 74 Jahren bei einer 5-Jahresüberlebensrate von 34,4 %. Man unterscheidet zwei histologische Subtypen, das Adenokarzinom und das szirrhöse Karzinom. Distal lokalisierte Magenkarzinome sind mit Abstand am häufigsten und zeigen einen signifikanten Zusammenhang mit der Infektion durch Helicobacter pylori, einem gramnegativen Stäbchenbakterium (Uemura N, 2002). W eitere Risikofaktoren sind stattgehabte Magenteilresektionen und eine salzreiche Ernährung, v. a. der Genuss von Nitritpökelsalz (Herold G et al., 2009). Therapeutisch kommen bei Magenkarzinomen im frühen Stadium endoskopisch etablierte Verfahren wie die EMR (endoscopic mucosal resection) in Betracht (Crumley AB et al., 2010). Bei fortgeschrittenen Stadien bestehen chirurgische Therapieoptionen. 26 Die neoadjuvante Chemo- bzw. Radiochemotherapie gilt dabei als prognoseverbessernd (Sastre J et al., 2006). Sie ist Cisplatin- oder Oxaliplatin-basiert (Zhang AM et al., 2007; Sendler A et al., 2006) und wird meist nach dem ECFProtokoll (Epirubicin, Cisplatin, 5-FU) (Leong T et al., 2003; Zaniboni A et al., 1995) oder EOX-Protokoll (Epirubicin, Oxaliplatin, Xeloda = Capecitabine) (Xiang XJ et al., 2010) verabreicht. Auch adjuvante Therapieansätze bewiesen Überlebensvorteile (Paoletti X et al., 2010; W aters JS et al., 1999). Auf dem palliativen Sektor zeigte zumindest die systemische Chemotherapie einen Vorteil gegenüber rein supportiven Methoden (W agner AD, 2010). 1.3.3 Kolonkarzinom Das Adenokarzinom des Kolons ist eines der häufigsten Malignome der westlichen Industriegesellschaften, das zweithäufigste der Frau und das dritthäufigste beim Mann. Die Inzidenz in Deutschland beträgt etwa 70/100.000 Einwohner/Jahr. Der mittlere Erkrankungsgipfel bei Männern liegt um das 67., bei Frauen um das 72. Lebensjahr. Davon abzugrenzen ist das Rektumkarzinom. Man unterscheidet in 10% der Fälle hereditäre Karzinomsyndrome wie die FAP (familiäre adenomatöse Polyposis), das HNPCC (hereditäres nicht-polypöses kolorektales Karzinom) und hamartöse Polyposissyndrome wie das Peutz-Jeghers-Syndrom. Weitaus häufiger sind sporadisch auftretende Kolonkarzinome, die sich insbesondere aus tubulo-villösen oder villösen Adenomen entwickeln. Risikofaktoren sind eine positive Familienanamnese und chronisch-entzündliche Darmerkrankungen, v. a. die Colitis ulcerosa. Ernährungsfaktoren wie eine ballaststoffarme Kost und Bewegungsmangel spielen ebenfalls eine prädisponierende Rolle (Schalhorn A, 2006). Ein kurativer Therapieansatz ist primär die operative en-bloc-Entfernung, auch hierbei gewinnen endoskopische Verfahren wie die EMR zunehmend an Bedeutung. Eine adjuvante Chemotherapie ist nach potenziell kurativer Operation Prognose verbessernd (Blinman P, 2010). Dabei sind hocheffektive adjuvante und palliative Therapieformen das sog. FOLFOX 4-Protokoll (5-Fluorouracil, Folinsäure (=Leucovorin), Oxaliplatin) und das FOLFIRI-Protokoll (5-FU / Folinsäure / Irinotecan) (Goldberg RM, 2005; de Gramont A et al., 2000) . 27 1.4 Zielsetzung Die zelluläre Immunität von Patienten unter Einfluss einer Hochdosischemotherapie hat eine große prognostische Bedeutung. Mit wachsender Immunsuppression steigt die Gefahr opportunistischer und z. T. lebensbedrohlicher Infektionen, vor denen die Patienten möglicherweise bereits im Vorfeld mittels Impfungen oder Antibiotikaprophylaxe geschützt werden können. Zudem ist eine ausreichende Immunität entscheidend für Fortsetzung der Therapie und Applikation der notwendigen Zytostatikadosis, um somit die volle Ausschöpfung konservativer Therapiemethoden zu gewährleisten. Ziel dieser Arbeit ist die Messung der zellulären Immunität unbehandelter Patienten mit erstdiagnostiziertem soliden Tumor des Gastrointestinaltraktes zu den Zeitpunkten vor und nach intravenöser Applikation einer konventionellen Polychemotherapie. Dabei sollen folgende Fragen beantwortet werden: • Ist die zelluläre Immunität bei Tumorpatienten messbar? • Sind Unterschiede in der Immunität tumorerkrankter Patienten im Vergleich zu einer vermeintlich tumorfreien Gruppe zu messen? • Läßt sich ein messbarer Effekt der Chemotherapie auf die zelluläre Immunität bestimmen? • Können immunologische Grenzwerte definiert werden, die Komplikationen prädiktiv anzeigen? 28 1.5 Theoretischer Hintergrund der Methoden 1.5.1 Dichtegradientenzentrifugation Die Dichtegradientenzentrifugation ist ein physikalisches Trennverfahren, welches verschieden gelöste Makromoleküle mithilfe einer Ultrazentrifuge entsprechend ihres Molekulargewichtes unter dem Einfluss starker Zentrifugalkräfte trennt. So können z. B. mononukleäre Zellen von Granulozyten, Erythrozyten und Thrombozyten isoliert werden. W ährend des Trennungsprozesses sedimentieren die Moleküle mit unterschiedlicher Geschwindigkeit in Form von Banden im Lösungsmittel, sodass man sie an definierter Stelle entnehmen kann. Durch Überschichtung mit Dispersionsmitteln unterschiedlicher Dichte und Einstellung eines Dichtegradienten ist die Verkürzung der Untersuchungszeiten möglich (Lange H et al., 1980). Das Lymphozytenseparationsmedium LSM 1077, welches hier verwendet wurde, basiert auf Ficoll™, einem hydrophilen Polymer mit einem Molekulargewicht von 400.000 Dalton. Periphere mononukleäre Zellen lagern sich als "Interphase" auf der "Ficoll-Phase" ab. 1.5.2 IFN-γ-ELISPOT-Assay (Enzyme linked immunospot assay) Der ELISPOT-Assay ermöglicht die Darstellung der Zytokinproduktion (z. B. Interferonγ) einzelner Zellen und erlaubt so einen hochspezifischen quantitativen Nachweis von T-Zellantworten auf ein determiniertes Antigen. Die Methode des ELISPOT-Assay wurde erstmals von zwei unabhängigen Gruppen im Jahr 1983 publiziert (Czerkinsky C et al., 1983; Sedgwick JD, 1983) und basiert auf dem zu dieser Zeit bereits gut etablierten, traditionellen Prinzip des Sandwich-ELISAs (Enzyme linked immunosorbent assay) (Sedgwick JD et al., 1983; Kalyuzhny AE, 2005), bei dem im Gegensatz zum ELISPOT-Assay die Messung der IFN-γKonzentration im Vordergrund steht. 29 Mithilfe des ELISPOT-Assays ist man in der Lage, die Zytokinproduktion einzelner, sowohl adhärenter als auch nicht adhärenter Zellen mit einer bis zu 200-mal höheren Sensitivität nachzuweisen (Tanguay S et al., 1994). Initial diente der Test vorrangig dem Nachweis durch B-Zellen sezernierter Antikörper. Über die folgenden Jahre wurde der ELISPOT-Assay zu einem Verfahren entwickelt, welches verschiedenste Zytokine, wie TNF (Tumornekrosefaktor alpha), IFN-γ, zahlreiche Interleukine und andere lösliche Mediatoren einer Vielzahl von Zellen nachweisen kann. Es erlaubt ex vivo eine quantitative Bestimmung der Zytokin produzierenden Zellen auf Einzelzellebene, ohne dass in vitro eine Expansion oder Manipulation der Zellpopulation erforderlich ist (Cox JH, 2006; Janetzki S et al., 2005). Die niedrige Detektionsgrenze von 1/105 PBMCs (peripheral blood mononuclear cells) ermöglicht somit den Nachweis kleinster Zellmengen (Scheibenbogen C et al., 2000), was in der klinischen Praxis von größter Bedeutung ist. So findet der ELISPOT-Assay unter anderem breite Anwendung in der Impfstoffentwicklung und Kontrolle des Impfstatus (Allmendinger J et al., 2010; Danziger IL et al., 2010), der Diagnose einer latenten bzw. manifesten Tuberkulose (Kim HS et al., 2010; Lienhardt C et al., 2010), der HIV-Diagnostik und -Therapie (Steeck H et al., 2009), der Tumordiagnostik (Lion E et al., 2009) und in Diagnostik und Therapie von Autoimmunerkrankungen. Neuere Studien zeigen eine sehr gute Präzision automatisch durchgeführter ELISPOT-Assays, die es ermöglichen, mit der sonst sehr zeitintensiven Labormethode eine große Anzahl an Patientenproben in kürzerer Zeit zu bearbeiten (Almeida CA et al., 2009). Das Prinzip des IFN-γ-ELISPOT-Assays beruht auf der spezifischen Erkennung des von mononukleären Zellen sezernierten Zytokins durch Antikörper, die auf einer Membran immobilisiert sind. Diese speziellen hydrophoben Membranen befinden sich am Boden einer sterilen 96-Well-Filterplatte und bestehen aus Polyvinylidenfluorid (PVDF) oder Nitrozellulose mit hoher Bindungskapazität für Biomoleküle. PVDFMembranen unterstützen in vitro kultivierte Zellen ohne Auswirkung auf ihre Physiologie zu haben (Kalyuzhny AE, 2009). 30 Die Platten werden zunächst mit Antikörpern gegen IFN-γ, sog. capture-Antikörpern, werden die zu beschichtet. untersuchenden Anschließend isolierten, meist kryokonservierten PBMCs, beispielweise Lymphozyten, zugegeben. Es konnte bewiesen werden, dass kein signifikanter Unterschied in der Zytokinsekretion zwischen frisch isolierten bzw. zuvor kryokonservierten Zellen besteht (Smith JG, et al. 2001). Ebenso konnte die Gruppe um Disis et al. am Beispiel der funktionellen TZell-Antwort auf Tetanus-Toxoid insbesondere unter Verwendung bestätigen, dass es von FKS (fötalem Kälberserum) zu keiner signifikanten Reduktion der vitalen Zellen nach Kryokonservierung kommt (Disis ML et al., 2005). Während der Inkubationszeit von mehreren Stunden bei 37°C werden die T-Zellen aktiviert. Sie sind nun in der Lage, das zugegebene Antigen spezifisch zu erkennen und mit der Sekretion von Zytokinen, u. a. von IFN-γ zu beginnen. Dieses wiederum bindet an die auf der PVDFMembran immobilisierten IFN-γ-Antikörper. Nach Entfernen der ungebundenen Zellen durch W aschen mit einem speziellen W aschpuffer erfolgt die Zugabe eines zweiten, ebenfalls auf IFN-γ gerichteten Antikörpers. Dieser sogenannte Detektions-Antikörper wurde zuvor biotinyliert. Abbildung 1: schematische Darstellung des ELISPOT (Janeway CA et al., 2002) Biotinylierung ist ein chemisches Verfahren, bei dem Biotin, das wasserlösliche Vitamin H, an verschiedene Makromoleküle wie Antikörper, Enzyme und Nukleinsäuren gebunden werden kann. Die Besonderheit ist dabei, dass durch die Bindung die chemischen, immunologischen und physikalischen Eigenschaften dieser Moleküle nicht verändert werden. 31 Nun wird das Enzym alkalische Phosphatase, welches über spezifische Linker an Streptavidin (aus Streptomyces avidinii gewonnen) gebunden wurde, zu den Ansätzen gegeben. Durch die hohe Bindungsaffinität von Streptavidin an Biotin resultiert ein gebundener Streptavidin – alkalische Phosphatase - Komplex an den biotinylierten Detektionsantikörpern. Die alkalische Phosphatase katalysiert nun über das zugegebene Substrat BCIP / NBT plus (5-Bromo-4-Chloro-3-Indolylphosphat / Nitroblau Tetrazolium) eine Farbreaktion an den Stellen des Plattenbodens, wo eine Bindung des sezernierten Zytokins durch die IFN-γ-capture-Antikörper stattgefunden hat. Jeder dieser Farbflecke, im Folgenden auch als Spot bezeichnet, entspricht letztlich einer vitalen, Zytokin produzierenden Zelle. Mithilfe eines hochauflösenden Phasenkontrastmikroskops in Kombination mit entsprechender Software kann sowohl die Dichte als auch die Anzahl der Spots ausgemessen und ausgezählt werden. 32 2. PROBANDEN, MATERIAL UND METHODEN 2.1 Probanden Von Juni 2008 bis April 2010 wurde zur Durchführung der paraklinischen immunologischen Untersuchungen im Rahmen dieser Studie von insgesamt 38 Probanden und Patienten des Klinikums Altenburger Land anonymisiert Blut entnommen. Bei allen Patienten erfolgte die Blutentnahme in Zusammenhang mit einer im Vorfeld routinemäßig geplanten stationären oder ambulanten Blutentnahme. Die Probanden und Patienten wurden vor der Blutentnahme mithilfe eines Aufklärungsbogens im Rahmen eines ausführlichen ärztlichen Aufklärungsgespräches über die Indikationsstellung und die Risiken der peripheren venösen Blutentnahme aufgeklärt und bestätigten ihre freiwillige Teilnahme durch ihre Unterschrift. Für die Blutentnahme liegt das Votum der Ethikkommission (Nr.: 065 – 2008, “Lymphozytenfunktion bei chemotherapierten Patienten”) der Medizinischen Fakultät der Universität Leipzig vor. Die Patienten und Probanden beider im folgenden dargestellten Gruppen waren zu Beginn der Studie zwischen 21 und 84 Jahren alt. Das Durchschnittsalter betrug 51,8 Jahre (+/- 16,3 Jahre, medianes Alter 56,5 Jahre) 2.1.1 Kontrollgruppe In der Zeit von Juni bis September 2008 erfolgte die Blutentnahme bei den 21 Probanden der Kontrollgruppe im Alter zwischen 21 und 67 Jahren. Das Durchschnittsalter betrug 43,2 Jahre (+/- 15,5 Jahre, medianes Alter 39 Jahre). Die Gruppe setzte sich zusammen aus insgesamt 12 gesunden, nicht-stationären freiwilligen Probanden im Alter von 21 bis 48 Jahren, davon zehn weiblichen und zwei männlichen Geschlechts. Das Durchschnittsalter betrug hier 32,2 Jahre (+/- 7,4 Jahre, medianes Alter 31,5 Jahre). Des W eiteren wurden dieser Gruppe 9 allgemein internistisch erkrankte, stationäre Patienten, zum Entnahmezeitpunkt im Alter von 41 bis 67 Jahren, davon 4 weiblichen 33 und 5 männlichen Geschlechts, zugeordnet. Das Durchschnittsalter betrug 58 Jahre (+/- 8,7 Jahre, medianes Alter 60 Jahre). Diese Patienten befanden sich aufgrund akuter Beschwerden in stationärer internistischer Behandlung. Dabei handelte es sich überwiegend um kardiologische, gastroenterologische und endokrinologische Krankheitsbilder. Die häufigsten Diagnosen sind im Folgenden zusammengefasst: • Koronare Herzkrankheit, Z. n. 3-fach-Bypass-Operation, Z. n. Myokardinfarkt, ischämische Kardiomyopathie • arterielle Hypertonie • Diabetes mellitus Typ 2 • Hypothyreose, Hyperthyreose • Refluxösophagitis • Alkohol- und Nikotinabusus Alle Probanden und Patienten befanden sich in einem guten klinischen Zustand ohne aktuelle Anzeichen einer akuten Infektionskrankheit. Einschlusskriterien: • Mindestalter 18 Jahre • vorliegende schriftliche Einverständniserklärung Ausschlusskriterien: • floride oder stattgehabte Tumorerkrankung • floride Infektionskrankheit • Autoimmunerkrankung • Einnahme immunsuppressiver Medikamente (einschließlich systemischer Prednisolongaben) in den letzten 12 Monaten • fehlende Einverständniserklärung 34 2.1.2 Patientengruppe In der Zeit von Juni 2008 bis April 2010 wurde bei insgesamt 17 Patienten mit Erstmanifestation eines soliden Tumors im Bereich des Gastrointestinaltraktes zu zwei verschiedenen Zeitpunkten Blut entnommen. Die Patienten waren zum Zeitpunkt der ersten Blutentnahme zwischen 43 und 84 Jahren alt. Das Durchschnittsalter betrug 62,3 Jahre (+/- 11,0 Jahre, medianes Alter 63 Jahre). Die erste Blutentnahme erfolgte beim unbehandelten Patienten unmittelbar am Tag der Applikation des 1. Chemotherapiezyklus. Die zweite Blutentnahme wurde nach Applikation von jeweils drei kompletten Chemotherapie-Zyklen durchgeführt. Dieser Zeitpunkt erwies sich am praktikabelsten, weil somit neoadjuvante Applikationen, die oft nur 3 Zyklen umfassen, eingeschlossen werden konnten. Die Therapiedauer, über die sich die 3 Zyklen erstreckten, betrug zwischen 29 und 85 Tagen, durchschnittlich 38,9 Tage (+/- 17,6 Tage, Median 29,5 Tage). Die 2. Blutentnahme erfolgte mindestens 6 Tage nach Beendigung des 3. Zyklus, in einem Ausnahmefall maximal 51 Tage, durchschnittlich 17,9 Tage (+/- 14,5 Tage, Median 12,0 Tage). Die Patienten waren an folgenden Tumoren erkrankt: • Ösophaguskarzinom (1 Patient; männlich) • Magenkarzinom (9 Patienten; 7 männlich, 2 weiblich) • Colorektalkarzinom (7 Patienten; 4 männlich, 3 weiblich) Einschlusskriterien: • erstmanifestierter, histologisch gesicherter Tumor des Gastrointestinaltraktes (Ösophagus-, Magen- oder Colorektalkarzinom) • vorliegende schriftliche Einverständniserklärung Ausschlusskriterien: • vorangegangene maligne Tumorerkrankung in der Eigenanamnese • vorangegangene Chemotherapie • Vorliegen einer Autoimmunerkrankung • floride Infektionskrankheit 35 • Einnahme immunsuppressiver Medikamente (einschließlich systemischer Prednisolongaben) in den letzten 12 Monaten • fehlende schriftliche Einverständniserklärung Die Patienten erhielten jeweils konventionelle Polychemotherapien in üblicher neoadjuvanter, adjuvanter oder palliativer Therapieplanung. Alle Therapien basierten auf 5-FU (5-Fluorouracil) und einem Platinderivat. Die verwendeten ChemotherapieProtokolle sind im Anhang aufgelistet. Zusammenfassend erhielten 6 Patienten eine Therapie nach dem FOLFOX-4- Protokoll (5 mit 100%, 1 mit 75%), 4 Patienten eine Therapie nach dem FLOT-Protokoll (3 mit 100%, 1 mit 75%), 3 Patienten eine Therapie nach dem TCF-Protokoll (je 100%), 2 Patienten eine Therapie nach dem ECF-Protokoll (je 100%), 1 Patient eine Therapie nach dem FUFOX-Protokoll (100%) und 1 Patient erhielt neoadjuvant Cisplatin und 5FU (100%). Bei einem Patienten (12) erfolgte aufgrund einer Unverträglichkeit nach einer Applikation des 1. Zyklus der Chemotherapie nach EOX-Protokoll die Umstellung auf die Alternativtherapie nach ECF-Protokoll. Folgende Tabelle zeigt die Verteilung der Tumorerkrankungen in Bezug auf Alter und Geschlecht der Patienten einschließlich der applizierten Chemotherapien. Die Prozentangaben beziehen sich auf die individuell applizierte Dosis in Bezug zur ursprünglichen Standarddosis. Tabelle 1: Patienten, Alter, Geschlecht, Tumorerkrankungen, Therapieform patient age sex tumor 1 63 m Magen therapy schlecht diff. palliativ TCF, Adenokarzinom pTx, 100% pN1, pM1 2 43 m Magen Kardiakarzinom neoadjuvant TCF, 100% 3 50 m Magen metast. palliativ TCF, Adenokarzinom 100% 36 4 52 m Rektum mäßig diff. palliativ FUFOX + Adenokarzinom Cetuximab, 100% Rektum (upT3, pN1, pM1) 5 6 76 66 w w Kolon Kolon mäßig diff. adjuvant FOLFOX- Adenokarzinom Colon 4 (de Gramont), ascendens 75% mäßig diff. adjuvant FOLFOX- Adenokarzinom Colon 4 (de Gramont), ascendens, pT3, pN2, 100% pM0, G2, UICC II 7 68 m Kolon, Zökumkarzinom pT1, adjuvant FOLFOX- Rektum pN0, R0, UICC I; 4 (de Gramont), Rektumkarzinom pT2, 100% pN1, R0, UICC III 8 43 m Ösophagus mäßig-schlecht diff. neoadjuvant tubuläres Cisplatin, 5-FU, Adenokarzinom, 100% pT2b, pN3, cM0, G3 9 71 m Magen schlecht diff. palliativ FLOT, Adenokarzinom mit 100% Siegelringzellbildung pT1, pN1, pMx, R0, G3 10 84 m Magen metastasiertes gering palliativ FLOT, diff. Adenokarzinom, 75% pT2b, pN2, M1, R0 11 59 w Magen mäßig-schlecht-diff. neoadjuvant ECF, Adenkarzinom, pT2b, 100% pN0, pM0, G3 37 12 13 60 66 m m Magen Kolon schlecht diff. neoadjuvant 1. Adenokarzinom mit Zyklus EOX Siegelringzellbildung 100%, pT3, pN2, pM1, UICC Umstellung auf IV ECF, 100% mäßig diff. adjuvant FOLFOX- Adenokarzinom Colon 4 (de Gramont), transversum pT3, 100% pN2, pM0, G2 14 72 w Kolon schlecht diff. adjuvant FOLFOX- Adenokarzinom rekto- 4 (de Gramont), sigmoidaler 100% Übergang, pT3, pN0, cM0, G3 15 59 m Kolon Zökumkarzinom adjuvant FOLFOX4 (de Gramont), 100% 16 17 67 60 m w Magen Magen mäßig-schlecht diff. palliativ FLOT, Adenokarzinom Cardia 100% schlecht-diff. palliativ FLOT, Adenokarzinom, G3 100% m = männlich w = weiblich Tabelle 2: Verwendete Chemotherapien und Inhaltsstoffe Abkürzung Zusammensetzung TCF Docetaxel 75 mg/m², Cisplatin 75 mg/m², 5-Fluorouracil 750 mg/m², Dexamethason ECF Epirubicin 50 mg/m², Cisplatin 60 mg/m², 5-Fluorouracil 200 mg/m² EOX Epirubicin 50 mg/m², Oxaliplatin 130 mg/m², Xeloda = Capecitabine 500 - 625 mg/d p.o. kontinuierlich 38 FOLFOX-4 5-Fluorouracil 400 - 600 mg/m², Calciumfolinat 200 mg/m², Oxaliplatin 85 mg/m² FUFOX 5-Fluorouracil 2000 mg/m², Calciumfolinat 500 mg/m², Oxaliplatin 50 mg/m², Cetuximab (Erbitux) 250 - 400 mg/m² FLOT 5-Fluorouracil 2600 mg/m², Oxaliplatin 85 mg/m², Calciumfolinat 200 mg/m², Docetaxel 50 mg/m² 2.2 Material 2.2.1 Blutentnahme Tabelle 3: Verwendete Materialien zur Blutentnahme und Bezugsquellen Material Bezugsquelle SST™ II Avance Tube with Silica Clot Activator, Polymer Gel, silicone-coated interior, 8,5 ml BD GmbH, Heidelberg, Deutschland Plasma Tube, 90 USP Units of Lithium Heparin (spray-coated), 6,0 ml BD GmbH, Heidelberg, Deutschland BD Vacutainer® Safety-Lok™ Sicherheitsblutentnahmeset, Flügelkanüle mit flexiblem Entnahmeschlauch und Luer-Adapter, steril BD GmbH, Heidelberg, Deutschland 39 2.2.2 IFN-γ-ELISPOT–Assay Tabelle 4: Verwendete Reagenzien für den ELISPOT-Assay und Bezugsquellen Reagenzien Bezugsquelle BSA (bovines Serumalbumin) Serva Electrophoresis GmbH, Heidelberg, Deutschland DMSO (Dimethyl Sulfoxide) Merck AG, Hohenbrunn, Deutschland Ethanol Zentralapotheke des Universitätsklinikums Leipzig, Deutschland FKS (fötales Kälberserum)= FBS (fötales bovines Serum) Biochrom KG seromed®, Berlin, Deutschland Isopropanol J.T. Baker, Deventer, Niederlande LSM 1077 (lymphocyte separation medium) PAA Laboratories GmbH, Österreich PBS, steril (phosphate buffered saline) Biochrom KG seromed®, Berlin, Deutschland RPMI 1640 (Roswell Park Memorial Institute, Zellkulturmedium für humane Lymphozyten) PAA Laboratories GmbH, Österreich Trypanblau Sigma-Aldrich Chemie, München, Deutschland Tween 20 (Polyoxyethylen-20sorbitan- Monolaurat) Sigma-Aldrich Chemie, München, Deutschland Tabelle 5: Verwendete Substrate, Enzyme für den ELISPOT-Assay und Bezugsquellen Substrate und Enzyme Bezugsquelle BCIP® / NBT (Liquid Substrate System) Sigma-Aldrich Chemie GmbH, München, Deutschland Streptavidin-Alkalische Phosphatase MABTECH, Hamburg, Deutschland 40 Tabelle 6: Verwendete Antikörper, Antigene für den ELISPOT-Assay und Bezugsquellen Antikörper und Antigene Bezugsquelle Anti-Human IFN-γ mAb 1-D1K MABTECH, Hamburg, Deutschland Anti-Human IFN-γ mAb 7-B6-1-Biotin, Stammkonzentration 1 mg/ml) MABTECH, Hamburg, Deutschland PHA (Phytohämagglutinin) Universität Leipzig, Deutschland CEF Peptide Pool (CytomegalieEbstein Barr- Flu), MHC class I restricted T-cell epitopes from human CMV, EBV and Influenza Virus) AID Autoimmun Diagnostika GmbH, Straßberg, Deutschland Candida albicans AID Autoimmun Diagnostika GmbH, Straßberg, Deutschland CFP 10 (culture filtrate protein) AID Autoimmun Diagnostika GmbH, Straßberg, Deutschland ESAT-6 (early secretory antigenic target-6) AID Autoimmun Diagnostika GmbH, Straßberg, Deutschland Tetanus Toxoid, highly purified, detoxified with formaldehyde (700-1200 Lf/ml, protein) content: 1,75-3,00 mg/ml Statens Serum Institute, Dänemark Tabelle 7: Verwendete Laborgeräte, Software für den ELISPOT-Assay und Bezugsquellen Laborgeräte und Software Bezugsquelle Adobe Photoshop 7.0 Adobe Systems Inc., San Jose, CA, USA AID EliSpot Reader System AID Autoimmun Diagnostika GmbH, Straßberg, Deutschland AID EliSpot Software 3.5 AID Autoimmun Diagnostika GmbH, Straßberg, Deutschland Canon Power Shot G5 Digitalkamera Canon Inc., Tokio, Japan 41 Einfrierbox Nalgene® Nunc International, Rochester, USA Einfrierröhrchen (1 ml) Nunc TM, Dänemark Inkubationsschrank Forma Scientific, Marietta, Ohio, USA Multikanalpipetten (100-200 µl) Eppendorf AG, Hamburg, Deutschland Multiscreenplatten (Multiscreen HTS -IP) Millipore, Bredford Massachusetts, USA Neubauer-Zählkammer Fein Optik, Bad Blankenburg, Deutschland Phasenkontrastmikroskop Olympus Deutschland GmbH, Hamburg, Deutschland Präzisionspipetten (2 -1000 µl) Eppendorf AG, Hamburg Reaktionsgefäße (0,5 – 2 ml) Sarstedt AG & Co., Nürnbrecht Standardspitzen (10, 100, 1000 µl) Eppendorf AG, Hamburg, Deutschland Stemi 2000 Stereomikroskop Carl Zeiss AG, Oberkochen, Deutschland Sterilbank (laminar flow cabinet) Clean Air Techniek B.V., JA W oerden, Niederlande Sterile Röhrchen (15 ml, 50 ml), Polysterol, konisch Greiner Bio-one GmbH, Frickenhausen, Deutschland Sterilpipetten (1 ml, 2 ml, 5 ml, 10 ml, 25 ml), serologisch Greiner Bio-one GmbH, Frickenhausen, Deutschland Variable Pipetten (100 – 1000 µl) Eppendorf AG, Hamburg, Deutschland Zellkulturflaschen Sarstedt AG & Co., Nümbrecht, Deutschland 42 Tabelle 8: Verwendete Puffer und Medien für den ELISPOT-Assay Puffer und Medien Verdünnungspuffer PBS + 0,5 BSA Waschpuffer PBS + 0,01% Tween20 Blockierlösung RPMI-1640 + 10% FKS Einfriermedium 10 % DMSO (Dimethylsulfoxid) in FKS Zellkulturmedium RPMI-1640, 10% FKS 2.3 Methoden 2.3.1 Materialgewinnung und Lymphozytenseparation Die Entnahme des venösen Vollblutes der Probanden wurde mittels Einmalpunktion unter sterilen Kautelen aus einer peripheren Vene des Armes vorgenommen. Eine Blutentnahme umfasste die Entnahme von 1 mal 8,5 ml Serum unter Gelzusatz und 1 mal 6 ml unter Lithium-Heparin-Zusatz. Anschließend erfolgte der Transport der Blutproben bei Raumtemperatur in lichtgeschützten Transportbehältern schnellstmöglich, spätestens jedoch innerhalb von 6 Stunden, vom Klinikum Altenburger Land zur Weiterverarbeitung in das immunologische Labor des Max-Bürger-Forschungszentrums der Universität Leipzig. Unmittelbar anschließend wurden die Blutproben in Lithium-Heparin-Röhrchen einer sterilen Dichtegradientenzentrifugation zugeführt. Das Blut wurde mit dem doppelten Volumenanteil sterilem PBS (phosphatgepufferte Salzlösung) vermischt und nachfolgend vorsichtig auf 4 ml LSM-1077 (Lymphozytenseparationsmedium) aufgetragen, ohne mit diesem vermischt zu werden. Der Trennungsprozess erfolgte durch eine sich anschließende Zentrifugation mit einer Winkelbeschleunigung von 500 g über 20 Minuten. Die auf 70-100 % angereicherten Lymphozyten bilden sich in der sogenannten Interphase, einer weißen Schicht zwischen Plasma und Separationsmedium ab. Sie wurden nachfolgend mittels steriler 43 Pipetten extrahiert, zweimalig in sterilem PBS-Kulturmedium gewaschen und erneut bei mit einer Winkelbeschleunigung von 320 g über 10 Minuten zentrifugiert. Nach Verwerfung des Überstandes wurden die Zellen resuspendiert und für die Kryokonservierung vorbereitet. 2.3.2 Kryokonservierung der Zellen Als erster Schritt wurden die Zellen in das Zellkulturmedium RPMI 1640 und 10% FKS und anschließend in ein Einfriermedium, bestehend aus 400 µl FKS und 10% Dimethylsulfoxid (DMSO) gegeben. DMSO ist ein organisches Lösungsmittel und wird bei der Kryokonservierung als Gefrierschutzmittel eingesetzt. Es gehört in die Gruppe der klassischen penetrierenden Kryoprotektiva und verhindert eine extreme intrazelluläre Eisbildung und osmotische Dehydrierung der Zellen während des Einfriervorganges (Bakhach J, 2009). Die Einfrierröhrchen wurden anschließend in einer mit Isopropanolol gefüllten Nalgene® Einfrierbox für mindestens 4 Stunden bei einer Temperatur von 2-8 °C gelagert. Dann erfolgte die Umlagerung in einen Gefrierschrank bei einer Temperatur von -80 °C. Die Zellsuspensionen wurden zügig und schonend in einem 37°C warmen W asserbad zur Hälfte aufgetaut. Nach Überführung in ein 15-ml-Röhrchen mit 1 ml vorgewärmtem FKS und Zugabe von 10 ml RPMI 1640 wurden die Zellen bei 320 g und Raumtemperatur für 5 Minuten zentrifugiert. Nach Verwerfung des Zellüberstandes und Resuspension wurde die Zentrifugation wiederholt. Diese Arbeitsschritte wurden, ausgenommen der Zentrifugation, an einer Sterilbank ausgeführt. 2.3.4 Bestimmung der vitalen Zellzahl Zur Bestimmung der Anzahl der vitalen Zellen diente die Trypanblau-Methode. Trypanblau, auch als Benzaminblau bezeichnet, ist ein anionischer, synthetischer Diazofarbstoff, der an Proteine bindet. Vitale Zellen nehmen den Farbstoff nicht auf. In abgestorbene Zellen hingegen dringt der Farbstoff ein und färbt deren Zellkerne und Zytoplasma dunkelblau an. Diese Eigenschaft macht man sich bei der Vitalzählung zunutze. Zur Auszählung wurde ein Standard-Hämozytometer, die Neubauer- 44 Zählkammer, verwendet. Die Zählkammer besitzt eine Tiefe von 0,1 mm und besteht aus 3 x 3 großen Quadraten mit einer Fläche von 1 mm², die wiederum in jeweils 16 Kleinquadrate unterteilt sind. Für die Leukozytenzählung werden üblicherweise die 4 großen Eckquadrate verwendet. 50 µl der Zellsuspension wurden mit 100 µl Trypanblau-Lösung vermischt und anschließend in die Zählkammer pipettiert. Mithilfe eines Phasenkontrastmikroskops wurden bei 10-facher Vergrößerung zweimal 16 Kleinquadrate der 4 großen Eckquadrate in diagonaler Richtung und in üblicher W eise ausgezählt. Nachfolgend konnte die Zellzahl pro Milliliter anhand folgender Formel berechnet werden: Zellen ml = ausgezählte Zellzahl x 1/Tiefe (mm) Fläche (mm²) x Verdünnung - Fläche Großquadrat: 1mm² - Tiefe Zählkammer: 0,1 mm - Vedünnungsfaktor: 2 Die Gesamtzellzahl ergibt sich aus dem errechneten W ert der Zellen/ml multipliziert mit dem verwendeten Suspensionsvolumen. 45 2.3.5 IFN-γ-ELISPOT-Assay Die Durchführung des ELISPOT-Testes erfolgte gemäß des Protkolls für “INFy Elispot Assays on Multiscreen IP Plate” der Firma Millipore Bredford Massachusetts, USA. Verwendet wurden sterile 96-W ell Millipore Corp. MultiScreen-HTS-IP-Filterplatten mit Immobilon®-P-Membran (MSIP S4510). Vorbereitend wurden die Filterplatten mit 30 µl 35%igen Ethanols pro W ell für eine Minute behandelt und dadurch aktiviert. Im folgenden Schritt wurde die Platte dreimalig mit jeweils 150 µl sterilem PBS je W ell gewaschen und danach vorsichtig ausgeklopft. Nun konnte die ELISPOT-Platte mit dem IFN-γ-Capture-Antikörper beschichtet werden. Dazu wurde ein Mischungsverhältnis von 6,0 ml PBS und 60 µl des IFN-γ-Antikörpers, davon wurden 60 µg in jedes W ell pipettiert. Dabei war darauf zu achten, dass der Boden eines jeden W ells vollständig bedeckt war. Die Platte wurde anschließend über Nacht bei 4°C inkubiert. Am darauf folgenden Tag wurden die inkubierte Platte erneut 3-mal mit je 200 µl sterilem PBS gewaschen, um ungebundene Antikörper zu entfernen. Zur Minderung der unspezifischen Bindung erfolgte das Blockieren der Platte mit einer Mischung aus einem Zellkulturmedium für humane Lymphozyten (RPMI) und 10% FKS. Dabei wurden je Well 150 µl der Mischung pipettiert und die Platte nachfolgend für 2,5 Stunden bei 37°C inkubiert. Nach Überführung der beschichteten Platte auf Raumtemperatur wurde die Blockierlösung durch vorsichtiges Ausklopfen aus den W ells entfernt. Nun erfolgte das Einstellen der Zellen auf eine Konzentration von 2 x 106 Zellen pro Milliliter des gemischten Zellkulturmediums, bestehend aus RPMI und 10% FKS. In jedes W ell wurden je 100 µl dieser Zellsuspension pipettiert. Aufgrund der teilweise geringen Zellanzahl konnten nur wenige Proben doppelt angesetzt werden. Aus den zwei Ergebniswerten wurde anschließend der Mittelwert gebildet, der für die nachfolgenden Berechnungen herangezogen wurde. Als Leerwert diente eine Probe des Zellkulturmediums des entsprechenden Patienten, lediglich unter Zugabe von 100 µl RPMI und 10% FKS ohne Antigen. Es folgte eine sogenannte Postivkontrolle. Dabei wurden 100 µl einer Mischung aus 10 µl PHA (Phytohämagglutinin) pro ml RPMI und 10% FKS zugegeben. Phytohämagglutinin ist ein pflanzliches Polysaccharid, welches in der Lage ist, in Lymphozyten eine blastische Transformation und Zellteilung anzuregen (Nowell PC, 46 1960). Es regt die in der Probe befindlichen vitalen Leukozyten zu einer unspezifischen Maximalstimulation an. Anschließend wurden die für diese Studie ausgewählten Antigene zugegeben. Die verwendeten Antigene waren: ESAT-6, CFP-10, CEF, Candida albicans und TetanusToxoid. Bei ESAT-6 und CFP-10 handelt es sich um hochspezifische Tuberkuloseantigene. Sie treten frühzeitig zu Beginn einer Tuberkuloseinfektion auf und stellen einen ausgesprochenen Virulenzfaktor dar. Diese sekretorischen Proteine induzieren sowohl in- als auch ex-vivo eine starke Immunantwort der T-Lymphozyten und finden daher in der ELISPOT-basierten Tuberkulosediagnostik eine breite Verwendung (Hoffmann M et al., 2003). Sie sind auch in kommerziellen Test-Kits erhältlich. Vor allem die kombinierte Anwendung beider Antigene ist mit hoher diagnostischer Sensitivität verbunden (Fox A et al., 2007). CEF ist eine determinierte Mischung aus 23 hoch immunogenen Peptiden, entwickelt aus humanpathogenen Zytomegalie-, Ebstein-Barr- und Influenza-Viren. Dieser Peptidpool dient der Stimulation von CD8+ T-Zellen, um vorzugsweise IFN-γ zu produzieren. Candida albicans Antigen und Tetanus-Toxoid zählen zu den sogenannten RecallAntigenen. Das sind Antigene, mit denen ein Individuum bereits im Vorfeld sensitiviert wurde. Sie sind Teil des immunologischen Gedächtnisses. Candida albicans-Antigen ist ein Antigen, entwickelt aus Candida albicans, einem fakultativ pathogenen Hefepilz. Tetanus-Toxoid ist ein hochpotentes biologisches Toxin, welches von Clostridium tetani, einem grampositiven anaeroben Bakterium produziert wird. Es ist ebenfalls in der Lage, eine starke T-Zell-vermittelte Immunantwort auszulösen (Mayer S et al., 2002). Jeweils 100 µl des entsprechenden Antigens, gelöst in antibiotikafreiem Medium (RPMI und 10% FKS), wurden je Well pipettiert und mit der Zellsuspension vermischt. Anschließend wurde die Platte bei 37°C, 5% CO2 und 95% Luftfeuchte über 24 Stunden inkubiert. Die fertig inkubierte ELISPOT-Platte wurde am folgenden Tag zunächst ausgeschüttet, behutsam ausgeklopft und anschließend 6-mal mit 200 µl / Well W aschpuffer, 47 bestehend aus PBS und 0,01% Tween20, gewaschen. Bei Tween20 handelt es sich um ein nichtionisches Tensid (Polyoxyethylen-20-sorbitan-Monolaurat), welches hierbei als Waschpuffer zur Solubilisierung von Proteinen und zum Vermeiden unspezifischer Bindungen eingesetzt wurde. Nun konnte der biotinylierte IFN-γ-Detection- Antikörper (Mischungsverhältnis: 2 µl Antikörper “7-B6-1biotin” / ml PBS + 0,5% BSA) aufgetragen werden. Für 100 W ells wurde zunächst eine Mischung aus 7,5 ml PBS / 0,5 % BSA und 15 µl des biotinylierten IFN-γ-Antikörpers hergestellt. Davon wurden je 60 µl in jedes W ell pipettiert. Die Platte durchlief anschließend eine Inkubationszeit von mindestens 3 Stunden bei 37°C. Nachfolgend wurde die Platte nach behutsamem Ausklopfen erneut 6-mal mit 200 µl / Well W aschpuffer (PBS mit 0,01 % Tween20) gewaschen. Nach Ausschütten und behutsamem Ausklopfen der Platte wurden im nächsten Schritt 100 µl / W ell der Streptavidin-alkalischen-Phosphatase (1:1000 mit PBS verdünnt) zu den Ansätzen gegeben und für 50 Minuten bei Raumtemperatur im Dunkeln inkubiert. Nach abgeschlossener Inkubation wurde die Platte ausgeschüttet, behutsam auf Papier ausgeklopft und 3-mal mit 200 µl / W ell Waschpuffer (PBS mit 0,01 % Tween20) und 3-mal lediglich mit 200 µl / Well PBS gewaschen, um die bis zu diesem Zeitpunkt ungebundene Streptavidin-alkalische-Phosphatase zu entfernen. Nun konnte das Substrat der alkalischen Phosphatase (BCIP / NBT plus) mit je 75 µl / Well aufgetragen und die Platte zur Spotentwicklung für 20 Minuten bei Raumtemperatur im Dunkeln inkubiert werden. Zur Unterbrechung der Reaktion erfolgte die 10-malige Spülung der Platte unter fließendem VE-Wasser (vollentsalztes, demineralisiertes, durch Ionenaustauscher gereinigtes, destillatgleiches W asser) und anschließendes Trocknen. Die ELISPOT-Platte konnte nun ausgelesen und bewertet werden. Die entstandenen Spots wurden mithilfe eines Stemi 2000 Stereomikroskops mit aufgesetzter Canon Power Shot G5 Digitalkamera in hochauflösender Qualität fotografiert. Unter Verwendung des Bildbearbeitungsprogrammes Adobe Photoshop 7.0 erfolgte die exakte Bestimmung der Grenzen der einzelnen Spots und deren Fläche. Die Analyse und Auszählung der Spotanzahl und -dichte erfolgte mittels AID Elispot Software 3,5 und des AID-Elispot-Reader Systems. 48 2.3.6 Statistische Analyse der Messdaten Die erhobenen Ergebnisse wurden zunächst in die Microsoft Office Excel Version für Windows 1997-2003 übertragen und verwaltet. Die Darstellung der Ergebnisse erfolgte mittels deskriptiver Statistik als Minimalwerte, Maximalwerte und Mediane. Die statistischen Berechnungen wurden mit Hilfe des Programmpaketes SigmaPlot Version 11 (Systat- Software, GmbH, Erkrath, Deutschland) erstellt. Aufgrund der geringen Patientenanzahl und der größtenteils nicht normal verteilten Werte kamen durchgehend nicht-parametrische Tests zum Einsatz. Zur Untersuchung der Gruppenvergleiche diente dabei vorwiegend der Mann - W hitney - U – Test (= Mann-W hitney Rank Sum Test). P-W erte < 0,050 wurden als statistisch signifikant gewertet. Boxplots unter Angaben der 5 % und der 95 % Perzentile sollen die statistischen Ergebnisse verdeutlichen. 49 3. ERGEBNISSE 3.1 Patienten Im Rahmen dieser Studie konnte von Juni 2008 bis April 2010 bei insgesamt 21 Probanden der Kontrollgruppe und 17 Tumorpatienten Blut entnommen werden. In der Kontrollgruppe erfolgte die Blutentnahme einmalig. Bei allen dieser 21 Probanden konnte eine Bestimmung erfolgen. In einer zweiten Gruppe wurden die Tumorpatienten zusammengefasst. Von den 17 Tumorpatienten wurde zu jeweils 2 Zeitpunkten Blut entnommen, d. h. vor und nach intravenöser Applikation von 3 Zyklen einer standardisierten Polychemotherapie. Bei 3 Tumorpatienten konnte leider aufgrund logistischer Probleme bei der Übersendung der Proben keine Bestimmung zum Zeitpunkt nach der Chemotherapie stattfinden. Bei einem Patienten konnte aufgrund dieser Probleme keine Bestimmung Zeitpunkt vor der Chemotherapie stattfinden. Bei 3 weiteren Patienten konnte mittels ELISPOT-Assay kein verwertbares Ergebnis zum Zeitpunkt nach der Chemotherapie erhoben werden. 16 von 17 Patientenproben standen letztlich zum Zeitpunkt vor der Chemotherapie, und 11 von 17 Patientenproben standen zum Zeitpunkt nach der Chemotherapie zur Verfügung. Um die Reaktionsfähigkeit der T-Zellen nach Zellisolierung und Kryokonservierung zu bestätigen, wurde als Positivkontrolle die Proliferation der T-Zellen auf ein unspezifisches Mitogen (Phytohämagglutinin, PHA) getestet. Des W eiteren wurden TZellen in einem Well nur mit Medium inkubiert. Die dabei gemessene individuelle spontane Proliferation lieferte den Leerwert. Neben der Spotanzahl (n) wurde die Spotintensität (A) gemessen. Diese kann bei zweifelhaft positiven Reaktionen hinzugezogen werden. Zeigt die Reaktion auf 50 Phytohämagglutinin (PHA) deutlich erhöhte W erte auch bei der Spotintensität, bestätigt dies die positive Testung grenzwertig erscheinender W erte. Von den erhobenen W erten wurden die jeweiligen individuellen Leerwerte subtrahiert. Diese korrigierten W erte wurden nachfolgend für alle weiteren Berechnungen verwendet. Die Ergebnisse sind als spot forming cells (n) pro 2 x 10 5 PBMC, als Spotintensität (A) und als Stimulationsindizes (SI), dem Verhältnis der Spotanzahl bzw. der Spotintensität des jeweiligen stimulierten Antigens zum Leerwert dargestellt. Als positives Ergebnis wurde gewertet, wenn mehr als 5 sfc/ 2 x 105 PBMC nachweisbar waren. Dies entspricht einem Stimulationsindex (SI) von > 2. 51 3.2 ELISPOT-Testergebnisse 3.2.1 Vergleich der Spotanzahl (n) von Kontrollgruppe und Tumorpatienten vor Chemotherapie Hierbei wurden die Testergebnisse der gesunden Kontrollgruppe (21 Patienten) und der Tumorpatienten vor Applikation der Chemotherapie (16 Patienten) für den Parameter Spotanzahl (n, spot forming cells) in Bezug auf den Leerwert, die unspezifische Maximalstimulation (PHA, Phytohämagglutinin) und die fünf verwendeten Antigene (ESAT-6, CFP-10, CEF, Candida-Antigen, Tetanus-Toxoid) gegenübergestellt. Die folgende Abbildung veranschaulicht die Häufigkeitsverteilung der Testergebnisse beider Gruppen anhand von Boxplots. Abbildung 2: Boxplots: Spotanzahl (n) von Kontrollgruppe und Tumorpatienten vor Chemotherapie 52 Die Spotanzahl der Kontrollgruppe für den Leerwert lag zwischen 0 und 65 (Median 14,50). Die Spotanzahl der Tumorgruppe vor Applikation der Chemotherapie streute zwischen 0 und 81 Spots (Median 10,00). Der Man-Whitney Rank Sum Test ergab keinen statistisch signifikanten Unterschied beider Gruppen (p= 0,373). Die Spotanzahl der Kontrollgruppe für das Antigen PHA lag zwischen -2 und 489 (Median 416,25). Die Spotanzahl der Tumorgruppe lag für dieses Antigen zwischen -4 und 496 (Median 184,00). Im Man-Whitney Rank Sum Test zeigte sich kein statistisch signifikanter Unterschied (p= 0,068). Die Spotanzahl der Kontrollgruppe für das Antigen ESAT-6 lag zwischen -65,00 und 58,00 (Median -3,00). Die Tumorgruppe zeigte zwischen -47,00 und 85,00 (Median 2,50) Spots. Im Man-Whitney Rank Sum Test zeigte konnte auch hier kein statistisch signifikanter Unterschied beider Gruppen verzeichnet werden (p= 0,724). Die Spotanzahl der Kontrollgruppe für das Antigen CFP-10 lag zwischen -65,00 und 34,00 (Median -8,00). Die Spotanzahl der Tumorgruppe ergab zwischen -72,00 und 119,00 (Median -0,50). Der Man-W hitney Rank Sum Test zeigte keinen statistisch signifikanten Unterschied (p= 0,198). Beim Antigen CEF lag die Spotanzahl der Kontrollgruppe zwischen -65,00 und 148,00 (Median 1,00) und die Spotanzahl der Tumorgruppe zwischen -13,00 und 630 (Median 31,00). Im Man-Whitney Rank Sum Test ergab sich ebenfalls kein statistisch signifikanter Unterschied beider Gruppen (p= 0,149). Die Kontrollgruppe zeigte für das Candida-Antigen zwischen -60,00 und 17,00 Spots (Median -2,00). Die Spotanzahl der Tumorgruppe lag hier zwischen -41,00 und 89,00 (Median -3,00). Im Man-W hitney Rank Sum Test konnte bei einem Median von -2,00 in der Kontrollgruppe und einem Median von -3,00 in der Tumorgruppe kein statistisch signifikanter Unterschied (p= 0,914) erhoben werden. Die Spotanzahl der Kontrollgruppe für das Antigen Tetanus-Toxoid lag zwischen 61,00 und 14,00 (Median -4,00). Die Tumorgruppe zeigte zwischen -57,00 und 25,00 53 (Median 0,50) Spots. Auch hier zeigte der Man-W hitney Rank Sum Test keinen statistisch signifikanten Unterschied zwischen beiden Gruppen (p= 0,581) bezüglich der Spotanzahl. Zusammenfassend zeigte sich kein statistisch signifikanter Unterschied Spotanzahl von Kontrollgruppe und Tumorgruppe vor Applikation einer Chemotherapie. 54 3.2.2 Vergleich der Stimulationsindizes [n] von Kontrollgruppe und Tumorpatienten vor Chemotherapie Hierbei wurden die Stimulationsindizes Kontrollgruppe (21 Patienten) denen [n] für die Spotanzahl der Tumorpatienten der gesunden vor Applikation der Chemotherapie (16 Patienten) gegenübergestellt. Gezeigt werden die Reaktion auf PHA (unspezifische Maximalstimulation) und die Reaktion auf die fünf verwendeten Antigene (ESAT-6, CFP-10, CEF, Candida-Antigen, Tetanus-Toxoid). Die folgende Abbildung verdeutlicht das Testergebnis mittels Boxplot. Abbildung 3: Boxplots: Stimulationsindizes [n] von Kontrollgruppe und Tumorpatienten vor Chemotherapie Der SI [n] der Kontrollgruppe für die Maximalstimulation PHA lag zwischen –0,50 und 205,00 (Median 19,29). Die Tumorgruppe zeigte vor Chemotherapie einen SI zwischen 55 -1,00 und 247,00 (Median 17,00). Der Man-W hitney Rank Sum Test ergab keinen statistisch signifikanten Unterschied (p= 0,920). Beim Antigen ESAT-6 rangierte der SI [n] der Kontrollgruppe zwischen –1,00 und 19,33 (Median -0,92). Der SI der Tumorgruppe lag vor Chemotherapie zwischen -1,00 und 3,10 (Median 0,83). Im Man-Whitney Rank Sum Test konnte kein statistisch signifikanter Unterschied beider Gruppen (p= 0,477) verzeichnet werden. Ein statistisch signifikanter Unterschied (p= 0,027) im SI [n] beider Gruppen ergab sich bezüglich des Antigens CFP-10. Der SI [n] der Kontrollgruppe lag hier zwischen -1,00 und 6,80 (Median -0,92). Die Tumorgruppe zeigte im Vergleich dazu bei einer Schwankungsbreite zwischen -1,00 und 18,50 (Median –0,33) einen signifikant höheren SI [n] vor Applikation der Chemotherapie. Der SI [n] der Kontrollgruppe für das Antigen CEF lag zwischen -1,00 und 15,33 (Median 0,006). Die Tumorgruppe zeigte hier Werte zwischen -1,00 und 82,67 (Median 1,72). Mittels des Man-W hitney Rank Sum Tests konnte jedoch kein statistisch signifikanter Unterschied beider Gruppen (p= 0,160) errechnet werden. Beim Candida-Antigen lag der SI [n] der Kontrollgruppe zwischen -1,00 und 4,00 (Median –0,52). Bei den Tumorpatienten streuten die W erte zwischen -1,00 und 3,33 (Median –0,25). Der Man-W hitney Rank Sum Test ergab ebenfalls keinen statistisch signifikanten Unterschied (p= 0,802). Der SI [n] der Kontrollgruppe für das Antigen Tetanus-Toxoid lag zwischen –1,00 und 4,67 (Median –0,70). Der SI [n] der Tumorgruppe lag zwischen -1,00 und 5,50 (Median 0,08). Es bestand kein signifikanter Unterschied (p = 0,285). Zusammenfassend konnte in nahezu allen Parametern kein statistisch signifikanter Unterschied im Stimulationsindex [n] der Kontrollgruppe im Vergleich zur Tumorgruppe vor Chemotherapie verzeichnet werden. Lediglich im Stimulationsindex [n] beim Antigen CFP-10 zeigte sich ein signifikanter Unterschied zugunsten eines höheren SI in der Tumorgruppe. 56 3.2.3 Vergleich der Spotintensität (A) von Kontrollgruppe und Tumorpatienten vor Chemotherapie Die Spotintensitäten (A) der gesunden Kontrollgruppe (21 Patienten) und der Tumorpatienten vor Applikation der Chemotherapie (16 Patienten) wurden nun gegenübergestellt. Gezeigt werden der Leerwert, die Reaktion auf PHA und die die fünf Antigene (ESAT-6, CFP-10, CEF, Candida-Antigen, Tetanus-Toxoid). Die Abbildung veranschaulicht die Häufigkeitsverteilung der Testergebnisse beider Gruppen. Abbildung 4: Boxplots: Spotintensität (A) von Kontrollgruppe und Tumorpatienten vor Chemotherapie Die Kontrollgruppe zeigte bei der Spotintensität Leerwerte zwischen 0,00 und 1350,00 (Median 105,00). Die Spotintensität der Tumorgruppe für diesen Parameter lag zwischen 0,00 und 1307,00 (Median 117,25). Im Man-W hitney Rank Sum Test ergaben sich keine statistisch signifikanten Unterschiede (p= 0,500). 57 Bei unspezifischer Maximalstimulation PHA lag die Spotintensität der Kontrollgruppe zwischen -46,00 und 4948,00 (Median 3531,00). Die Tumorgruppe zeigte hier W erte zwischen -16,00 und 8955,00 (Median 4590,50). Auch hier ergab sich kein signifikanter Unterschied im Man-Whitney Rank Sum Test (p= 0,453). Die Spotintensität der Kontrollgruppe für das Antigen ESAT-6 lag zwischen -1350,00 und 1159,00 (Median -11,00). Die Tumorgruppe bot W erte zwischen -504,00 und 4514,00 (Median 143,50). Im Man-Whitney Rank Sum Test ergaben sich keine statistisch signifikanten Unterschiede beider Gruppen (p= 0,086). Auch im Vergleich der Spotintensitäten von Kontrollgruppe und Tumorpatienten für das Antigen CFP-10 ergab sich erneut ein statistisch signifikanter Unterschied beider Gruppen (p= 0,029). Die Tumorpatienten zeigten mit Werten zwischen 994,00 und 4514,00 (Median 121,50) signifikant höhere Spotintensitäten als die Kontrollgruppe mit Werten zwischen -1350,00 und 345,00 (Median -3,00). Beim Antigen CEF lag die Spotintensität der Kontrollgruppe zwischen 863,00 und 3896,00 (Median 275,00). Die Spotintensität der Tumorgruppe zeigte Werte zwischen 184,00 und 4968,00 (Median 854,50). Der Man-Whitney Rank Sum Test ergab dabei keinen statistisch signifikanten Unterschied (p= 0,334). Die Spotintensität der Kontrollgruppe für das Candida-Antigen lag zwischen -54,00 und 1350,00 (Median 100,00). Die Spotintensität der Tumorgruppe rangierte zwischen 333,00 und 2350,00 (Median 135,50). Auch hier ergab sich wiederum kein statistisch signifikanter Unterschied beider Gruppen (p= 0,304). Auch beim Antigen Tetanus-Toxoid zeigte der Man-W hitney Rank Sum Test keine signifikanten Unterschiede (p= 0,064). Die Spotintensität der Kontrollgruppe lag hier zwischen -1350,00 und 481,00 (Median 68,00), die der Tumorgruppe zwischen -333,00 und 545,00 (Median 216,00). Zusammenfassend besteht analog zur Spotanzahl auch in der Spotintensität in nahezu allen gemessenen Parametern kein statistisch signifikanter Unterschied zwischen 58 Kontrollgruppe und unbehandelten Tumorpatienten. Lediglich beim Antigen CFP-10 zeigte sich eine signifikant erhöhte Spotintensität zugunsten der Tumorgruppe. 3.2.4 Vergleich der Stimulationsindizes [A] von Kontrollgruppe und Tumorpatienten vor Chemotherapie Die Stimulationsindizes [A] für die Spotintensität der gesunden Kontrollgruppe (21 Patienten) wurden nun denen der unbehandelten Tumorpatienten (16 Patienten) gegenübergestellt. Gezeigt werden die Reaktion auf PHA (unspezifische Maximalstimulation) und die Reaktion auf die fünf verwendeten Antigene (ESAT-6, CFP-10, CEF, Candida-Antigen, Tetanus-Toxoid). Die folgende Abbildung verdeutlicht das Testergebnis. Abbildung 5: Boxplots: Stimulationsindizes [A] von Kontrollgruppe und Tumorpatienten vor Chemotherapie 59 Bei Maximalstimulation (PHA) zeigte die Kontrollgruppe einen SI [A] zwischen -0,52 und 95,15 (Median 27,08). Der SI [A] der Tumorgruppe vor lag hier zwischen -1,00 und 453,55 (Median 14,02). Der Man-W hitney Rank Sum Test ergab jedoch keinen statistisch signifikanten Unterschied (p= 0,474). Der SI [A] der Kontrollgruppe für das Antigen ESAT-6 lag zwischen -1,00 und 15,94 (Median -0,43). Die unbehandelten Tumorpatienten zeigten W erte zwischen -1,00 und 16,57 (Median 0,87). Auch hier ergab sich rechnerisch kein statistisch signifikanter Unterschied beider Gruppen (p= 0,245). Beim Antigen CFP-10 konnten bei der Kontrollgruppe W erte zwischen -1,00 und 10,46 (Median -0,52) gemessen werden. Der SI [A] der Tumorgruppe lag im Vergleich dazu zwischen -1,00 und 53,09 (Median 0,816). Dies ergab einen statistisch signifikanten Unterschied beider Gruppen (p= 0,045) im Man-Whitney Rank Sum Test. Der SI [A] der Kontrollgruppe für das Antigen CEF rangierte zwischen -1,00 und 44,49 (Median 2,49). Die Tumorpatienten hatten diesbezüglich Werte zwischen -1,00 und 155,25 (Median 4,03). Das ergab im Man-Whitney Rank Sum Test keinen statistisch signifikanten Unterschied (p= 0,413). Der SI [A] der Kontrollgruppe für das Candida-Antigen lag zwischen -1,00 und 12,73 (Median 0,52). Der SI [A] der Tumorgruppe lag hier zwischen -1,00 und 9,91 (Median 0,75), was ebenfalls keinen statistisch signifikanten Unterschied (p= 0,414) darstellte. Auch für das Antigen Tetanus-Toxoid konnten keine statistisch signifikanten Unterschiede im Vergleich der Stimulationsindizes [A] erhoben werden (p = 0,210). Die Kontrollgruppe zeigte hier Werte zwischen -1,00 und 10,61 (Median 0,57), die Tumorgruppe Werte zwischen -1,00 und 16,46 (Median 1,04). Zusammenfassend besteht in nahezu allen Parametern kein statistisch signifikanter Unterschied im SI [A] der Kontrollgruppe im Vergleich zur Tumorgruppe vor Chemotherapie. Lediglich im SI [A] bei Antigen CFP-10 zeigte sich analog zum SI [n] ein signifikanter erhöhter SI [A] in der Tumorgruppe. 60 3.2.5 Vergleich der Spotanzahl (n) von Tumorpatienten vor und nach Chemotherapie Die Spotanzahl (n) der Tumorpatienten vor (16 Patienten) und nach Applikation der Chemotherapie (11 Patienten) wurden nun gegenübergestellt. Abbildung 6: Boxplots: Spotanzahl (n) von Tumorpatienten vor und nach Chemotherapie Im Leerwert zeigten die unbehandelten Tumorpatienten Werte zwischen 0 und 81 (Median 14,50). Nach 3 Zyklen Chemotherapie lag die Spotanzahl zwischen 0,00 und 100,00 (Median 40,00). Der Man-W hitney Rank Sum Test zeigte keinen statistisch signifikanten Unterschied (p= 0,288). Die Spotanzahl der Tumorgruppe vor Applikation der Chemotherapie bezüglich der Maximalstimulation PHA lag zwischen -4 und 496 (Median 416,25). Nach Applikation der Chemotherapie konnten W erte zwischen 0,00 und 434,00 (Median 255,00) 61 gemessen werden. Auch hier ergab sich kein statistisch signifikanter Unterschied (p= 0,415). Für das Antigen ESAT-6 zeigten unbehandelte Tumorpatienten Spotanzahlen zwischen -47,00 und 85,00 (Median -2,50). Nach 3 Zyklen Chemotherapie rangierte die Spotanzahl zwischen -42,00 und 59,00 (Median 2,00), was rechnerisch ebenfalls keinen signifikanten Unterschied darstellte (p= 0,531). Vor Applikation der Chemotherapie konnten in der Tumorgruppe bezüglich des Antigens CFP-10 Spotanzahlen zwischen -72,00 und 119 (Median -0,50) verzeichnet werden. Nach Therapie lag die Spotanzahl zwischen -64,00 und 98,50 (Median -5,00). Auch hier bedeutete dies entsprechend des Man-W hitney Rank Sum Tests keinen statistisch signifikanten Unterschied beider Gruppen (p= 0,324). Für das Antigen CEF konnten vor Chemotherapie Spotananzahlen zwischen -13,00 und 630 (Median 31,00) gemessen werden. Nach der Chemotherapie lag die Spotanzahl zwischen -17,00 und 264,50 (Median 18,00), was keinen statistisch signifikanten Unterschied darstellte (p= 0,882). Bezüglich des Candida-Antigens ergaben sich bei Spotanzahlen zwischen -41,00 und 89 (Median -3,00) vor und zwischen -59,00 und 38,00 (Median -4,00) nach Chemotherapie ebenfalls keine statistisch signifikanten Unterschiede (p= 0,444). Beim Tetanus-Toxoid zeigte sich bezüglich der Spotanzahl ebenfalls kein signifikanter Unterschied (p=0,271). Vor Chemotherapie lag die Spotanzahl im Median bei 11,50, nach Therapie bei 2,00. Zusammenfassend zeigte sich kein statistisch signifikanter Unterschied in der Anzahl der spot forming cells vor und nach Applikation einer Chemotherapie. 62 3.2.6 Vergleich der Stimulationsindizes [n] von Tumorpatienten vor und nach Chemotherapie Hier wurde der SI [n] für die Spotanzahl der Tumorpatienten vor (16 Patienten) und nach (11 Patienten) Applikation von drei Zyklen Chemotherapie gegenübergestellt. Der folgende Boxplot verdeutlicht die Untersuchungsergebnisse. Abbildung 7: Boxplots: Stimulationsindizes [n] von Tumorpatienten vor und nach Chemotherapie Der SI [n] der Tumorpatienten vor Applikation der Chemotherapie für die Maximalstimulation PHA lag zwischen -1,00 und 247,00 (Median 17,00). Nach Applikation von 3 Zyklen Chemotherapie lag der SI [n] zwischen 0,99 und 58,00 (Median 7,26). Das ergab laut Man-Whitney Rank Sum Test keinen statistisch signifikanten Unterschied (p= 0,170). 63 Auch beim Antigen ESAT-6 konnten keine signifikanten Veränderungen des SI [n] vor und nach Chemotherapie verzeichnet werden (p= 0,365). Vor Therapie lagen die W erte zwischen –1,00 und 3,10 (Median –0,83), nach Therapie zwischen -1,00 und 4,75 (Median 0,09). Der SI [n] der unbehandelten Tumorpatienten bezüglich des Antigens CFP-10 lag zwischen -1,00 und 18,50 (Median –0,33). Nach Applikation von 3 Zyklen Chemotherapie lag der SI [n] zwischen -1,00 und 8,25 (Median –0,64). Der ManWhitney Rank zeigte auch hier keinen statistisch signifikanten Unterschied vor und nach Therapie (p= 0,321). Beim Antigen CEF lag der SI [n] vor Chemotherapie zwischen -1,00 und 82,67 (Median 1,71). Nach der Chemotherapie ergaben sich W erte zwischen –0,17 und 5,23 (Median 1,40), was ebenfalls kein signifikanter Unterschied war (p= 0,858). Analog dazu zeigte sich auch beim Candida-Antigen keine statistisch signifikante Änderung des Stimulationsindex [n] vor und nach Chemotherapie(p= 0,512). Es konnten vor Therapie W erte zwischen -1,00 und 3,33 (Median –0,25) und nach Therapie Werte zwischen -1,00 und 3,00 (Median –0,67) vezeichnet werden. Auch beim Antigen Tetanus-Toxoid zeigte sich keine signifikante Dynamik im Stimulationsindex [n] (p= 0,063). Vor Chemotherapie ergaben sich Werte zwischen -1,00 und 5,50 (Median 0,08), nach Applikation der 3 Zyklen lagen die W erte zwischen -1,00 und 0,20 (Median -0,62). Zusammenfassend bestand auch bezüglich des Stimulationsindex [n] der Tumorpatienten vor und nach Applikation der Chemotherapie bei allen Parametern kein statistisch signifikanter Unterschied vor und nach Applikation der Chemotherapie. 64 3.2.7 Vergleich der Spotintensität (A) von Tumorpatienten vor und nach Chemotherapie Nun wurde die Spotintensität (A) der Tumorpatienten vor (16 Patienten) und nach (11 Patienten) Applikation der Chemotherapie gegenübergestellt. Abbildung 8: Boxplots: Spotintensität (A) vonTumorpatienten vor und nach Chemotherapie Die Spotintensität der Tumorgruppe rangierte im Leerwert zwischen 0,00 und 1307,00 (Median 117,25). Nach Applikation von 3 Zyklen Chemotherapie lag die Spotintensität zwischen 0,00 und 1921,00 (Median 383,00). Kein statistisch signifikanter Unterschied konnte im Man-Whitney Rank Sum Test errechnet werden (p= 0,311). Bei der unspezifischen Maximalstimulation PHA zeigten sich vor der Chemotherapie Werte zwischen -16,00 und 8955,00 (Median 4590,50). Nach Therapie lag die 65 Spotintensität zwischen 0,00 und 8794,50 (Median 4375,00). Auch hier ergab sich kein statistisch signifikanter Unterschied beider Gruppen (p= 0,444). Die Spotintensität der Tumorpatienten vor Chemotherapie für das Antigen ESAT-6 lag zwischen -504,00 und 4514,00 (Median 143,50). Nach der Chemotherapie lag die Spotintensität zwischen -348,00 und 3608,00 (Median 237,00). Auch dies ergibt im Man-W hitney Rank Sum Test keinen statistisch signifikanten Unterschied (p= 0,388). Beim Antigen CFP-10 lag die Spotintensität der Tumorpatienten zwischen -994,00 und 4514,00 (Median 121,50), nach der Chemotherapie rangierte sie zwischen -1426,00 und 4249,00 (Median 4,00). Im Man-W hitney Rank Sum Test zeigte sich auch hier kein statistisch signifikanter Unterschied beider Gruppen (p= 0,675). Auch beim Antigen CEF ergaben sich keine signifikanten Unterschiede in der Spotintensität vor und nach Chemotherapie (p= 0,882). Hier lagen die W erte vor Therapie zwischen -184,00 und 4968,00 (Median 854,50) und nach Therapie zwischen -362,00 und 8803,50 (Median 725,00). Die Spotintensität der Tumorpatienten bezüglich des Candida-Antigens lag zwischen 333,00 und 2350,00 (Median 135,50). Nach Applikation von 3 Zyklen Chemotherapie ergaben sich W erte zwischen -383,00 und 629,00 (Median 11,00). Entsprechend des Man-W hitney Rank Sum Tests stellten sich auch hier keine statistisch signifikanten Unterschiede dar (p= 0,126). Analog dazu ergaben sich auch beim Antigen Tetanus-Toxoid keine signifikanten Veränderungen (p = 0,089). Vor Chemotherapie lag die Spotintensität diesbezüglich im Median bei 334,00. Nach Chemotherapie ergab sich ein Median von 22,00. Zusammenfassend zeigte sich kein statistisch signifikanter Unterschied in der Spotintensität bezüglich aller eingesetzter Antigene. 66 3.2.8 Vergleich der Stimulationsindizes [A] von Tumorpatienten vor und nach Chemotherapie Nun wurde der SI [A] für die Spotintensität der Tumorpatienten vor (16 Patienten) und nach (11 Patienten) Applikation von drei Zyklen Chemotherapie gegenübergestellt. Der folgende Boxplot verdeutlicht die Untersuchungsergebnisse. Abbildung 9: Boxplots: Stimulationsindizes [A] von Tumorpatienten vor und nach Chemotherapie Der SI [A] der Tumorpatienten vor Applikation der Chemotherapie für die Maximalstimulation PHA lag zwischen -1,00 und 453,55 (Median 14,02). Nach Applikation von 3 Zyklen Chemotherapie zeigten sich Werte zwischen 1,52 und 58,33 (Median 8,05). Der Man-Whitney Rank Sum Test ergab dabei keinen statistisch signifikanten Unterschied (p= 0,340). 67 Beim Antigen ESAT-6 lag der SI [A] der Tumorpatienten vor Chemotherapie zwischen -1,00 und 16,57 (Median 0,87). Nach Applikation von 3 Zyklen Chemotherapie konnten bei Werten zwischen -1,00 und 4,98 (Median 0,69) ebenfalls keine statistisch signifikanten Unterschiede errechnet werden (p= 0,952). Auch beim zweiten Tuberkuloseantigen CFP-10 ergaben sich im Verlauf der Chemotherapie keine signifikanten Unterschiede im SI [A] (p= 0,293). Vor Therapie bestanden W erte zwischen -1,00 und 53,09 (Median 0,82), nach Therapie lagen die Werte zwischen -1,00 und 10,83 (Median 0,22). In Reaktion auf das Antigen CEF konnte ebenfalls keine signifikante Veränderung verzeichnet werden (p= 0,952). Der SI [A] rangierte vor Chemotherapie zwischen -1,00 und 155,25 (Median 4,03) und nach Therapie zwischen -0,19 und 11,28 (Median 4,37). Analog dazu ergab sich kein signifikanter Unterschied bezüglich des SI [A] in Reaktion auf das Candida-Antigen (p= 0,095). Vor Therapie lagen die Werte zwischen 1,00 und 9,91 (Median 0,75). Nach Applikation von 3 Zyklen Chemotherapie lag der SI zwischen -1,00 und 2,76 (Median 0,31). Der SI [A] der unbehandelten Tumorpatienten für das Antigen Tetanus-Toxoid lag zwischen -1,00 und 16,46 (Median 1,04). Nach Applikation von 3 Zyklen Chemotherapie lag der SI zwischen -1,00 und 3,32 (Median -0,72). Der Man-W hitney Rank Sum Test zeigte hier einen statistisch signifikanten Unterschied beider Gruppen (p= 0,010). Zusammenfassend besteht bezüglich des SI [A] vor und nach Applikation der Chemotherapie bei den meisten Parametern kein statistisch signifikanter Unterschied. Beim Antigen Tetanus-Toxoid zeigte sich im Gegensatz zum SI [n] eine deutliche Verminderung des SI [A] nach Applikation der Chemotherapie. 68 3.2.9 Zusammenfassung der Ergebnisse In Zusammenschau aller erhobenen Ergebnisse zeigte sich eine überwiegend unveränderte T-Zell-Antwort auf die meisten der eingesetzten Antigene vor und nach Applikation der Chemotherapie bzw. im Vergleich der unbehandelten Tumorpatienten mit der gesunden Kontrollgruppe. Dennoch sind einige statistisch signifikante Unterschiede festzuhalten. So zeigte die Gruppe der Tumorpatienten vor Applikation der Chemotherapie im Vergleich zur Kontrollgruppe eine signifikant erhöhte Spotintensität (A) und erhöhte Stimulationsindizes [A] und [n] in Bezug auf das Tuberkulose-Antigen CFP-10. Die Spotanzahl (n) bei CFP-10 ergab keine Differenz beider Gruppen. Diese Veränderungen waren nach Applikation der Chemotherapie nicht mehr nachzuweisen. Des W eiteren ergaben sich bei den Tumorpatienten vor und nach Chemotherapie signifikante Veränderungen der T-Zell-Antwort bezüglich des Antigens Tetanus-Toxoid. Nach Applikation von 3 Zyklen Chemotherapie kam es zu einer signifikanten Verminderung des Stimulationsindex [A]. 69 4. DISKUSSION 4.1 Stellenwert des ELISPOT- Assays bei der Messung zellulärer Immunität Das diagnostische Verfahren des ELISPOT-Assays liefert, wie im Vorfeld beschrieben, seit Jahren verlässliche Ergebnisse in der immunologischen Forschung, der Stammzellforschung, bei verschiedensten klinischen Fragestellungen, wie z. B. in der Impfstoffentwicklung und Kontrolle des Impfstatus (Allmendinger J et al., 2010; Danziger IL et al., 2010), der Tuberkulosediagnostik (Kim HS et al., 2010; Lienhardt C et al., 2010), der HIV-Diagnostik und -Therapie (Steeck H et al., 2009), der Tumordiagnostik (Lion E et al., 2009), in Diagnostik und Therapie von Autoimmunerkrankungen und der Verlaufsbeobachtung immunsupprimierter Patienten nach Organ- oder Knochenmarktransplantation (Nickel P et al., 2009). Die Messung der zellulären Immunität, das sogenannte Immunmonitoring, gewinnt gerade auch für Tumorpatienten unter konventioneller Chemotherapie oder begleitender Immuntherapie zunehmend an Bedeutung, denn der Immunstatus ist ein wesentlicher prognostischer Faktor bezüglich der Überlebenszeit der Tumorpatienten (Deschoolmeester V et al., 2010; McMullen TP et al., 2010; Wilke CM et al., 2010). Vor allem hat sich in letzter Zeit die Bedeutung immunsuppressiver Treg-CD4+-CD25+Lymphozyten im Verhältnis zu den schützenden CD4+Th-Zellen, die sogenante CD4+/CD4+CD25+-ratio, als wichtiger Parameter zur Darstellung antitumoraler Immunität herauskristallisiert (Lissoni P et al., 2009). Die Entwicklung von Methoden immunologischer antitumoraler für die genaue Bestimmung Marker mittels ex vivo aussagekräftiger T-Zell-Assays ist eine Herausforderung. Der IFN-γ-ELISPOT-Assay, die Zytokin-Durchflusszytometrie und der (51)Chromium Release Assay sind trotz Einschränkungen in Sensitivität und Spezifität die dabei bisher am häufigsten verwendeten Verfahren (Walker EB et al., 2003, Whiteside TL et al., 2003). Sie erlauben jedoch nicht die exakte Differenzierung 70 spezifischer T-Effektorzellen und die simultane Messung verschiedener Parameter, die zu einem umfassenden Monitoring benötigt werden. Neue immunologische Assays wie die Multifaktor-Durchflusszytometrie, die dies ermöglichen, könnten Alternativen darstellen (Zaritskaya L et al., 2010). Für die Messung einer tumorspezifischen Immunität konnten in den letzten Jahren entsprechende Tumorantigene im Rahmen von ex vivo-Analysen eingesetzt werden (Knutson KL et al., 2006). Aufgrund der generell niedrigeren Immunität von Tumorantigenen werden außerdem Verfahren benötigt, die eine große Bandbreite von T-Zell-Antworten nachweisen können. Eine Zellexpansion ex vivo konnte die Sensitivität des ELISPOT steigern (Schultes BC et al., 2003). Eine weitere Möglichkeit zur Steigerung der Sensitivität wäre, weitere Zytokine wie IL-4 oder IL-5, einzusetzen (Goodell V et al., 2007). Bezüglich der Reproduzierbarkeit der ELISPOT-Ergebnisse gab es in den letzten Jahren immer wieder enttäuschende Untersuchungen. Zhang W et al. konnten jedoch in einer großen multizentrisch angelegten Studie in den USA und Europa diese These widerlegen und beweisen, dass unter standardisierter Prozedur und Datenanalyse durchaus reproduzierbare ELISPOT-Ergebnisse erreicht werden können (Zhang W et al., 2009). Zusammenfassend ist der ELISPOT-Assay immer noch eine verlässliche und gut reproduzierbare Methode, die zur Messung der individuellen T-Zell-Antwort auch bei Tumorpatienten herangezogen werden kann. Dies bestätigt sich auch in unserer Untersuchung. Die zelluläre Stimulation gegenüber den ausgewählten Antigenen war reproduzierbar und mit vergleichbaren Ergebnissen spezifisch nachweisbar. Studien ergaben jedoch Sensitivitätsverluste im Bereich stark verminderter T-ZellAntworten ab, wie sie bei immunsupprimierten Patienten auftreten können (Goodell V et al., 2007). Bei gewünschter spezifischer T-Zelldifferenzierung auch in Bezug auf Tumorantigene müsste gegebenenfalls auf andere Methoden zurückgegriffen werden. 71 4.2 Korrelation zwischen zellulärer und serologischer Immunität Die Messung der antigenspezifischen humoralen Immunität ist ein Indikator für die Entwicklung einer Immunität bei Infektionskrankheiten. Es erscheint jedoch fraglich, ob die serologische Immunität direkte Rückschlüsse auf den zellulären Immunstatus erlaubt. Ebenso ist unklar, ob eine signifikante Korrelation bezüglich tumorassoziierter Antigene besteht. Bei der Kontrolle des Impfstatus von Kindern zeigte sich in einer aktuellen Studie zumindest in Bezug auf Tetanus- und Hepatitis-B-Antigene keine Korrelation zellulärer und serologischer Immunität (Fischer A, 2009). Candon et al. konnten in einer 2009 veröffentlichten Studie an nierentransplantierten Patienten im Rahmen einer InfluenzaImmunisierung ebenfalls keinen Zusammenhang zwischen serologischer und zellulärer Immunität herstellen (Candon S et al., 2009). Zu dem gleichen Schluß kamen Tosh et al. beim Vergleich des Röteln-Antikörpertiters mit der entsprechenden T-Zell- vermittelten Zytokinsekretion (Tosh PK et al., 2009). Auch Chinchai et al. fanden keinen Zusammenhang zwischen zellulärer und humoraler Immunität nach Hepatitis-BImmunisierung (Chinchai T et al., 2009). Aus Mangel an signifikanten Beweisen für eine tatsächlich bestehende Korrelation zellulärer und humoraler Immunität wurde in dieser Studie bewusst auf einen zusätzlichen serologischen Vergleich verzichtet. 4.3 Diskussion der Ergebnisse von Kontrollgruppe und Tumorpatienten vor Chemotherapie In dieser Studie wurde eine Gruppe, bestehend aus 21 nicht-onkologisch erkrankten und gesunden Probanden, zunächst 17 Patienten mit unbehandelten, neu manifestierten soliden Tumoren des Gastrointestinaltraktes gegenübergestellt. Dabei standen 16 von ursprünglich 17 Blutproben der Tumorpatienten dem ELISPOT-Test zur Verfügung. 72 Bei der Mehrzahl der eingesetzten Antigene konnte kein signifikanter Unterschied in der T-Zell-Antwort beider Gruppen verzeichnet werden. Jedoch zeichnete sich eine Signifikanz in Bezug auf das Tuberkulose-Antigen CFP-10 ab. Hier zeigten die unbehandelten Tumorpatienten eine gesteigerte Reaktivität. Das ist ungewöhnlich, zumal bei keinem der Patienten ein Hinweis auf Tuberkulose bestand. Eine zuverlässige Aussage bezüglich einer Tuberkuloseinfektion ist prinzipiell auch bei Tumorpatienten mittels ELISPOT-Test möglich (Lai CC et al., 2010). Sie sollte jedoch, entsprechend der üblichen Tuberkulosediagnostik mindestens in Kombination zweier Tuberkulose-Antigene, meist aus dem Fusionsprotein ESAT-6 / CFP-10, getroffen werden (Kalra M et al., 2010; Liu F et al., 2009; Hill PC et al., 2005). Die T-Zellantwort auf ESAT-6 war hier jedoch im Vergleich zur Kontrollgruppe unbeeinträchtigt. Ebenso lässt die unveränderte Anzahl der spot forming cells im Verhältnis zu erhöhter Spotintensität und Stimulationsindex keine verwertbaren Rückschlüsse zu. Kreuzreaktivitäten mit nicht tuberkulösen Mykobakteriosen könnten hier eine Rolle spielen (Vordermeier HM et al., 2007; Geluk A et al., 2004). Im Gegensatz dazu konnte in verschiedenen Studien eine tumorbedingte verminderte T-Zell-Antwort bei Tumorpatienten nachgewiesen werden. Dabei fiel eine hohe Rate spontaner Apoptosen aktivierter antitumoraler T-Effektorzellen bei Tumorpatienten im Vergleich zu gesunden Spendern auf (Hoffmann T et al., 2002). Dies könnte ein genereller Grund für die geringe Anzahl detektierbarer antigenspezifischer T-Zellen sein, wie in Untersuchungen nach Impfungen von Tumorpatienten nachgewiesen wurde (Whiteside TL, 2004; Reichert TE et a., 2003). 4.4 Diskussion der Ergebnisse von Tumorpatienten vor und nach Chemotherapie Von den insgesamt 17 Blutproben der Tumorpatienten vor Chemotherapie konnten nach intravenöser Applikation von 3 Zyklen konventioneller Polychemotherapie nur noch 11 Patientenproben dem ELISPOT-Test zugeführt werden. Bei 2 von diesen 11 73 Patienten (P06 und P19) war selbst bei unspezifischer Maximalstimulation durch PHA keine Zytokinsekretion mehr detektierbar. Diese T-Zell-Depletion könnte eine mögliche Auswirkung der Chemotherapie darstellen. Bei den verbliebenen 9 Patienten zeigte sich auf die Mehrheit der in dieser Studie eingesetzten Antigene keine wesentliche Änderung der T-Zell-Antwort. Hervorzuheben sind jedoch folgende signifikante Veränderungen in Bezug auf das Antigen Tetanus Verminderung Toxoid: Es ergab sich eine statistisch signifikant messbare des Stimulationsindexes [A]. Spotanzahl, Spotintensität und Stimulationsindex [n] zeigten sich hingegen unbeeinflusst. Dies zeigt deutlich, dass bei einer umfassenden Analyse immer auch die jeweiligen Stimulationsindizes berücksichtigt werden sollten, da anhand der alleinigen Bestimmung von Spotanzahl und -intensität richtungsweisende Veränderungen nicht aufgedeckt werden könnten. In Anbetracht der Ergebnisse wird die Vermutung nahegelegt, dass sich die T-ZellAntwort im Verlauf der Chemotherapie zumindest auf ausgewählte Antigene verringert. In Bezug auf das Antigen Tetanus Toxoid könnte dies eine verminderte Resistenzlage gegenüber bakteriellen Infektionen vermuten lassen. Ähnliche Ergebnisse ergaben Untersuchungen der T-Zell-Antwort auf Aspergillus-Antigen bei Patienten mit hämatologischen Malignomen (Hebart H et al., 2002). In dem untersuchten Zeitraum konnte jedoch bei den Patienten dieser Studie keine erhöhte Inzidenz bakterieller Infektionen verzeichnet werden. Sowohl der Stimulationsindex als auch die Messung der IFN-γ-Sekretion sind signifikante Indikatoren der Antigen-spezifischen Immunität. Kritisch geprüft werden sollte gerade in der Messung der antitumoralen Immunität offenbar die W ahl der Antigene. Es zeigte sich in einer Untersuchung bei Mammakarzinom-Patientinnen, denen zuvor Impfstoffe gegen Her-2/neu und Tetanus verabreicht wurden, dass die qualitative und quantitative Messung der IFN-γ-Sekretion ein signifikanter und verlässlicher Marker bei robuster T-Zell-Antwort, wie z. B. auf CMV (Zytomegalievirus), war. Bei geringer oder moderater T-Zell-Antwort, wie z. B. üblicherweise auf Tetanus- 74 Toxoid, bestand jedoch deutlich verminderte Sensitivität des ELISPOT-Testes (Goodell V et al., 2007). Daher sind die in dieser Arbeit erhobenen Ergebnisse bezüglich des Tetanus-Toxoids mit Vorsicht zu interpretieren, zumal gerade Tumorpatienten generell zu einer geringeren T-Zell-Antwort tendieren (Hoffmann T et al., 2002). Einschränkend ist zu bemerken, dass in dieser Studie eine relativ kleine Patientenpopulation untersucht wurde, sodass Rückschlüsse auf größere Kollektive unter Vorbehalt zu ziehen sind. Aufgrund des kleinen Patientenkollektives wurde bewusst auf einen Vergleich der individuellen T-Zell-Antworten in Bezug auf die Zusammensetzung und Dosis der applizierten Polychemotherapien verzichtet. Außer Acht gelassen wurden aus diesem Grund weitere Einflussfaktoren wie Alter und Geschlecht der untersuchten Patienten. Die Interpretation erschwert beispielsweise, dass das mediane Alter der Tumorpatienten mit 63 Jahren deutlich über dem der Kontrollgruppe liegt (medianes Alter 39 Jahre). Zudem besteht eine Heterogenität bezüglich der Therapiedauer und dem Zeitpunkt der 2. Blutentnahme innerhalb der Tumorgruppe. Nicht zuletzt könnten auch Unterschiede der verschiedenen eingeschlossenen Tumorerkrankungen untereinander bestehen. Es darf vermutet werden, dass sich die Immunantwort im weiteren Verlauf der Chemotherapie und bei möglichem Progress der Tumorerkrankung weiter modifiziert, sodass sich nach einem größeren Zeitintervall zusätzliche messbare Veränderungen manifestieren. 4.6 Schlussfolgerung Abschließend lässt sich zusammenfassen, dass die Methode des ELISPOT-Assays, trotz einiger Einschränkungen der Sensitivität im Bereich stark verminderter TZellantworten, durchaus für die Messung des zellulären Immunstatus von Tumorpatienten geeignet ist. Die im Rahmen dieser Arbeit untersuchten Tumorpatienten zeigten im Vergleich zu einer nicht onkologisch vorerkrankten Kontrollgruppe bis auf eine Ausnahme keine signifikanten Unterschiede in der zellulären Immunantwort auf die eingesetzten Antigene. Einzig auffällig war eine gesteigerte Reaktivität der unbehandelten 75 Tumorpatienten Kreuzreaktivitäten gegenüber dem Tuberkuloseantigen zurückzuführen Tuberkuloseinfektion ist. Verwertbare CFP-10, die eventuell auf Aussagen bezüglich einer bzw. vermehrten Infektanfälligkeit lassen sich jedoch nicht ableiten. Auch bezüglich der drei applizierten Chemotherapie-Zyklen konnte zumindest für die Mehrheit der Antigene keine signifikante Änderung des zellulären Immunstatus verzeichnet werden. In Bezug auf das Antigen Tetanus Toxoid zeigte sich jedoch eine signifikant messbare Verminderung des Stimulationsindex [A] nach Chemotherapie. Ob diese singuläre Veränderung als Marker für eine gesteigerte Infektanfälligkeit bezüglich bakterieller Infektionen gilt und sich daraus therapeutische Konsequenzen für den Klinikalltag ergeben, lässt sich aufgrund der geringen Datenanzahl und o. g. Einschränkungen nicht schlussfolgern. Weitere und über einen längeren Zeitraum angelegte Untersuchungen an größeren Patientenkollektiven, möglicherweise auch unter Verwendung weiterer Antigene, ist zu empfehlen. 76 5. ZUSAMMENFASSUNG Dissertation zur Erlangung des akademischen Grades Dr. med. “Chemotherapeutische Beeinflussung des zellulären Immunstatus bei Patienten mit erstmanifestierten soliden Tumoren des Gastrointestinaltraktes” eingereicht von: Karoline Grunemann angefertigt am: Institut für Klinische Immunologie der Medizinischen Fakultät der Universität Leipzig und dem Zentrum für Innere Medizin des Klinikums Altenburger Land GmbH betreut von: Prof. Dr. med. Ulrich Sack August 2010 In der vorliegenden Arbeit wurde der zelluläre Immunstatus von Patienten mit verschiedenen neu manifestierten soliden Tumoren im Bereich des Gastrointestinaltraktes vor und nach intravenöser Applikation von 3 Zyklen einer konventionellen Polychemotherapie untersucht. Bei den Tumoren handelte es sich ausschließlich um Ösophagus-, Magen- und Kolorektalkarzinome. Die Tumorpatienten waren weder chemotherapeutisch noch immunsuppressiv vorbehandelt. Als Vergleichsgruppe dienten nicht-onkologisch vorerkrankte Probanden. Ziel der Arbeit war es, mögliche Unterschiede des zellulären Immunstatus der Tumorpatienten vor Chemotherapie im Vergleich zu einer vermeintlich gesunden 77 Kontrollgruppe aufzudecken, vorhandene Auswirkungen der Chemotherapie auf die Immunantwort nachzuweisen und gegebenenfalls therapeutische Konsequenzen abzuleiten. Die Messung der zellulären Immunität, das sogenannte Immunmonitoring, gewinnt insbesondere für Tumorpatienten unter konventioneller Chemotherapie oder begleitender Immuntherapie zunehmend an Bedeutung. Es konnte nachgewiesen werden, dass der Immunstatus prädiktiv für das Auftreten bakterieller und viraler Infektionen, aber auch für die antitumorale Immunantwort ist und somit einen wesentlichen prognostischen Parameter für die Überlebenszeit dieser Patienten darstellt. Im Zeitraum von Juni 2008 bis April 2010 erfolgte bei 21 Probanden der Kontrollgruppe und 17 Tumorpatienten die Entnahme von 8,5 ml Serumblut und 6,0 ml Heparinblut zur Durchführung der immunologischen Testverfahren dieser Studie. Die 21 Probanden waren zwischen 21 und 67 Jahren alt (Median 39 Jahre, +/- 15,5 Jahre). Die 17 Tumorpatienten waren zwischen 43 und 84 Jahren alt (Median 63 Jahre, +/- 11,0 Jahre). Die Tumorpatienten erhielten eine für den jeweiligen Tumor übliche konventionelle Polychemotherapie, dabei wurden 6 verschiedene Schemata appliziert. Alle basierten auf 5-Fluorouracil (5-FU) und einem Platinderivat. Die Therapiedauer betrug im Durchschnitt 38,9 Tage. Die erste Blutentnahme erfolgte am 1. Tag der Chemotherapie am unbehandelten Patienten, die 2. Entnahme durchschnittlich 17,9 Tage nach Vollendung des 3. Zyklus. Zur Messung der individuellen T-Zell-vermittelten Immunantwort diente die in der klinischen Praxis gut etablierte Methode des IFN-γ-ELISPOT (Enzyme linked Immunospot) -Assays, die es erlaubt, hochspezifisch quantitativ T-Zell-Antworten auf Einzelzellebene nachzuweisen. Es wurden 5 herkömmliche Antigene als Stimulatoren für die T-Zell-vermittelte Zytokinsekretion, in diesem Fall IFN-γ, eingesetzt: ESAT-6, CFP-10, CEF, Candida albicans und Tetanus Toxoid. In Zusammenschau aller erhobenen Ergebnisse zeigte sich eine überwiegend unveränderte T-Zell-Antwort auf die meisten der eingesetzten Antigene vor und nach Applikation der Chemotherapie bzw. im Vergleich der unbehandelten Tumorpatienten mit der gesunden Kontrollgruppe. Dennoch sind einige statistisch signifikante Unterschiede festzuhalten. 78 So zeigte die Gruppe der Tumorpatienten vor Applikation der Chemotherapie im Vergleich zur Kontrollgruppe eine signifikant erhöhte Spotintensität (A) und erhöhte Stimulationsindizes [A] und [n] in Bezug auf das Tuberkuloseantigen CFP-10. Die Spotanzahl (n) bei CFP-10 ergab keine Differenz beider Gruppen. Diese Veränderungen waren nach Applikation der Chemotherapie nicht mehr nachzuweisen. Aufgrund unveränderter Spotanzahl (n) auf CFP-10 und unveränderter T-Zellantwort auf das 2. Tuberkuloseantigen ESAT-6 lassen sich keine verwertbaren Rückschlüsse ableiten. Kreuzreaktivitäten könnten eine ursächliche Rolle spielen. Des W eiteren ergaben sich bei den Tumorpatienten vor und nach Chemotherapie signifikante Veränderungen der T-Zell-Antwort bezüglich des Antigens Tetanus-Toxoid. Nach Applikation der Chemotherapie kam es zu einer Verminderung des Stimulationsindex [A]. Das könnte die Vermutung nahelegen, dass sich im Verlauf der Chemotherapie zumindest auf ausgewählte Antigene die T-Zell-Antwort verringert. In Bezug auf das Antigen Tetanus Toxoid könnte dies eine verminderte Resistenzlage gegenüber bakteriellen Infektionen vermuten lassen. Ob diese singuläre Veränderung jedoch als allgemeingültiger Marker für eine gesteigerte Infektanfälligkeit bezüglich bakterieller Infektionen gilt, darf bezweifelt werden. Auch die Ableitung therapeutischer Konsequenzen für den Klinikalltag ist auf der Basis dieser Ergebnisse nicht möglich. Abschließend läßt sich zusammenfassen, dass die Methode des ELISPOT-Assays, trotz einiger in anderen Studien erwiesener Einschränkungen der Sensitivität im Bereich stark verminderter T-Zellantworten, durchaus für die Messung des zellulären Immunstatus von Tumorpatienten geeignet ist. Weitere und über einen längeren Zeitraum angelegte Untersuchungen an größeren Patientenkollektiven, eventuell auch unter Verwendung weiterer Antigene, sind sinnvoll, um mögliche Unterschiede herauszuarbeiten. 79 6. SUMMARY Dissertation submitted to attain the academic degree Dr. med. “Influence of Chemotherapy on the Cellular Immunity of Patients with first Manifestation of Solid Tumors of the Gastrointestinal Tract” submitted from: Karoline Grunemann prepared at: Institute of Clinical Immunology of the Faculty of Medicine of the University of Leipzig and the Centre for Internal Medicine of the Hospital “Altenburger Land”, Altenburg supervised by: Prof. Dr. med. Ulrich Sack august 2010 The present work examined the cellular immune status of patients with different new manifested tumors of the gastrointestinal tract before and after intravenous application of three cycles of a conventional chemotherapy. The work exclusively involves esophageal, gastric and colorectal cancer. The tumor patients were not pretreated neither chemotherapeutic or immunosuppressive. Healthy volunteers served as a comparative group. Aim of the study was to discover possible differences in the cellular immune status of tumor patients before starting chemotherapy compared with supposedly healthy individuals, to demonstrate existing impacts on the immune response and to derive therapeutic consequences. The measurement of cellular immunity, the immune monitoring, becomes increasingly important, especially for tumor patients treated with conventional chemotherapy or 80 immunotherapy. The immune status provides predictive information about bacterial and viral infections and anti-tumoral immune response. It is an essential prognostic parameter for the survival time of these patients. In the period from june 2008 to april 2010 6,0 ml heparinized and 8,5 ml serum blood samples were taken from 21 probands of the control group and 17 tumor patients. These blood samples were supplied to the immunologic tests of this study. The 21 probands were aged between 21 and 67 years (+/- 15,5 years, median 39 years). The 17 tumor patients differed in ages between 43 and 84 years (+/- 11,0 years, median 63 years). The tumor patients received a conventional chemotherapy treatment for the particular tumor. 6 different chemotherapy regimens, all based on 5-fluorouracil (5-FU) and platinum derivative, have been applied. The average duration of the therapy was 38,9 days. The first blood samples were taken from the untreatet patient on the first day of chemotherapy. The second samples were taken within an average time span of 17,9 days after completion of the 3rd cycle. For the measurement of the individual T-cell mediated response the well established method of IFN-γ-ELISPOT- (enzyme linked immunospot) assay has been used. This method provides a highly specific quantitative detection of T-cell-respones on the single cell level. 5 traditional antigens were used as stimulators for the T-cell mediated cytokine secretion: ESAT-6, CFP-10, CEF, candida albicans and tetanus toxoid. The results show a mainly unaltered T-cell-response to most of the antigens before and after the chemotherapy treatment and by comparison of the untreated tumor patients with a healthy control group. But there have to be shown some statistically significant differences. In comparison with the control group the untreated tumor patients showed a significantly increased spot intensity (A) and stimulation indices [A] and [n] concerning the tuberculosis antigen CFP-10. The spot count (n) has not been influenced. All of those alterations were no longer detectable after chemotherapy. Considering the unchanged spot count (n) on CFP-10 and unaltered T-cell-response to the second tuberculosis antigen ESAT-6, there can not be drawn utilisable conclusions. Cross reactivities could play a possible role. Furthermore the tumor patients had significant changes in their T-cell-response to the antigen tetanus toxoid after receiving a chemotherapy. A significantly decreased 81 stimulation index [A] has been registered. The spot count, spot intensity and the stimulation index [n] have not been altered. These results may presume the increase of T-cell-responses to selected antigens during chemotherapy. Concerning the antigen tetanus toxoid there could exsist a diminished resistance to bacterial infections. It must be questioned if this singular change can be taken as a generally valid marker for a decreased resistance to bacterial infections. Based on these results it is not possible to derive practical clinical conclusions. In summary it can be said, that the method of ELISPOT-assay is suitable for measuring the cellular immune status of tumor patients, despite some limitations in sensitivity in low T-cell-responses, as described in other studies. Further investigations over a longer period and on a lager group of patients, would be useful to work out possible differences. 82 7. LITERATURVERZEICHNIS Adkinson NF Jr, (1979), Environmental influences on the immune system and allergic reactions. Environ Health Perspect.; 20: 97-103. Ajani JA, Correa AM, Walsh GL, Komaki R, Lee JH, Vaporciyan AA, Rice DC, Yao JC, Maru DM, Hofstetter WL, Phan AT, Swisher SG, (2010), Trimodality therapy without a platinum compound for localized carcinoma of the esophagus and gastroesophageal junction.Cancer. 116(7): 1656-1663. Ajani JA, Fodor MB, Tjulandin SA, Moiseyenko VM, Chao Y, Cabral Filho S, Majlis A, Assadourian S, Van Cutsem E, (2005), Phase II multi-institutional randomized trial of docetaxel plus cisplatin with or without fluorouracil in patients with untreated, advanced gastric, or gastroesophageal adenocarcinoma. J Clin Oncol.; 23(24): 5660-5667. Akaza H, Kawai K, Tsukamoto T, Fujioka T, Tomita Y, Kitamura T, Ozono S, Miki T, Naito S, Zembutsu H, Nakamura Y, (2010), Successful outcomes using combination therapy of interleukin-2 and interferon-alpha for renal cell carcinoma patients with lung metastasis. Jpn J Clin Oncol.; 40(7): 684-689. Akuthota P, W ang H, Weller PF, (2010), Eosinophils as antigen-presenting cells in allergic upper airway disease.Curr Opin Allergy Clin Immunol.; 10(1): 14-19. Al-Batran SE, Hartmann JT, Hofheinz R, Homann N, Rethwisch V, Probst S, Stoehlmacher J, Clemens MR, Mahlberg R, Fritz M, Seipelt G, Sievert M, Pauligk C, Atmaca A, Jäger E, (2008), Biweekly fluorouracil, leucovorin, oxaliplatin, and docetaxel (FLOT) for patients with metastatic adenocarcinoma of the stomach or esophagogastric junction: a phase II trial of the Arbeitsgemeinschaft Internistische Onkologie. Ann Oncol. 19(11): 1882-1887. 83 Allmendinger J, Paradies F, Kamprad M, Richter T, Pustowoit B, Liebert UG, (2010), Determination of rubella virus-specific cell-mediated immunity using IFN gammaELISPOT. J Med Virology 82(2): 335-340. Almeida CA, Roberts SG, Laird R, McKinnon E, Ahmed I, Pfafferott K, Turley J, Keane NM, Lucas A, Rushton B, Chopra A, Mallal S, John M, (2009), Automation of the ELISpot assay for high-throughput detection of antigen-specific T-cell responses. J Immunol Methods.; 344(1): 1-5. Arum CJ, Anderssen E, Viset T, Kodama Y, Lundgren S, Chen D, Zhao CM, (2010), Cancer immunoediting from immunosurveillance to tumor escape in microvillus-formed niche: a study of syngeneic orthotopic rat bladder cancer model in comparison with human bladder cancer. Neoplasia.; 12(6): 434-442. Bakhach J, (2009), The cryopreservation of composite tissues: Principles and recent advancement on cryopreservation of different types of tissues, Organogenesis 5(3): 119-126. Bakker E, Pearson PL, Meera Khan P, Schreuder GM, Madan K, (1979), Orientation of major histocompatibility (MHC) genes relative to the centromere of human chromosome 6. Clin Genet.; 15(2): 198-202. Barin JG, Talor MV, Baldeviano GC, Kimura M, Rose MR, Ciháková D, (2010), Mechanisms of IFNgamma regulation of autoimmune myokarditis, Ex Mol Pathol. Abstract. Epub ahead of print. Barker RN, Erwig LP, Hill KS, Devine A, Pearce WP, Rees AJ, (2002), Antigen presentation by macrophages is enhanced by the uptake of necrotic, but not apoptotic, cells. Clin Exp Immunol.; 127(2): 220-225. 84 Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J, (1998), Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res.; 58(13): 2825-2831. Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC, (1987), Structure of the human class I histocompatibility antigen, HLA-A2. Nature 329(6139): 506-512. Blinman P, Duric V, Nowak AK, Beale P, Clarke S, Briscoe K, Boyce A, Goldstein D, Hudson M, Stockler M, (2010), Adjuvant chemotherapy for early colon cancer: what survival benefits make it worthwhile? Eur J Cancer., 46(10): 1800-1807. Boackle SA, Morris MA, Holers VM, Karp DR, (1998), Complement opsonization is required for presentation of immune complexes by resting peripheral blood B cells. J Immunol.; 161(12): 6537-6543. Bonilla FA, Oettgen HC, (2010), Adaptive immunity. J Allergy Clin Immunol.; 125(2 Suppl 2): S33-40. Borgerding A, Hasenkamp J, Engelke M, Burkhart N, Trümper L, Wienands J, Glass B, (2010), B-lymphoma cells escape rituximab-triggered elimination by NK cells through increased HLA class I expression. Exp Hematol.; 38(3): 213-221. Brouwer N, Dolman KM, van Zwieten R, Nieuwenhuys E, Hart M, Aarden LA, Roos D, Kuijpers TW, (2006), Mannan-binding lectin (MBL)-mediated opsonization is enhanced by the alternative pathway amplification loop.Mol Immunol.; 43(13): 2051-2060. Campbell MJ, Scott J, Maecker HT, Park JW, Esserman LJ, (2005), Immune dysfunction and micrometastases in women with breast cancer. Breast Cancer Res Treat.; 91(2): 163-171. 85 Candon S, Thervet E, Lebon P, Suberbielle C, Zuber J, Lima C, Charron D, Legendre C, Chatenoud L, (2009), Humoral and cellular immune responses after influenza vaccination in kidney transplant recipients. Am J Transplant.; 9(10): 2346-2354. Cannistra SA, Griffin JD, (1988), Regulation of the production and function of granulocytes and monocytes. Semin Hematol. ; 25(3):173-188. Caras I, Grigorescu A, Stavaru C, Radu DL, Mogos I, Szegli G, Salageanu A, (2004), Evidence for immune defects in breast and lung cancer patients.Cancer Immunol Immunother.; 53(12): 1146-1452. Carol M, Lambrechts A, Van Gossum A, Libin M, Goldman M, Mascart-Lemone F. (1998), Spontaneous secretion of interferon gamma and interleukin 4 by human intraepithelial and lamina propria gut lymphocytes. Gut. 42(5): 643-649. Caso JR, Leza JC, Menchén L, (2008), The effects of physical and psychological stress on the gastro-intestinal tract: lessons from animal models. Curr Mol Med.; 8(4): 299312. Chesnut RW , Grey HM, (1985), Antigen presenting cells and mechanisms of antigen presentation. Crit Rev Immunol.; 5(3): 263-316. Chinchai T, Chirathaworn C, Praianantathavorn K, Theamboonlers A, Hutagalung Y, Bock PH, Thantiworasit P, Poovorawan Y, (2009), Long-term humoral and cellular immune response to hepatitis B vaccine in high-risk children 18-20 years after neonatal immunization. Viral Immunol.; 22(2): 125-130. Clarke SL, Betts GJ, Plant A, W right KL, El-Shanawany TM, Harrop R, Torkington J, Rees BI, Williams GT, Gallimore AM, Godkin AJ, (2006), CD4+CD25+FOXP3+ Regulatory T Cells Suppress Anti-Tumor Immune Responses in Patients with Colorectal Cancer. PLoS ONE 1(1): e129. 1-6. 86 Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A, (1992), Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood.; 80(8): 2012-2020. Cooper EH, Turner R, Steele L, Neville AM, Mackay AM, (1975), The contribution of serum enzymes and carcinoembryonic antigen to the early diagnosis of metastatic colorectal cancer. Br J Cancer.; 31(1): 111-117. Correale P, Rotundo MS, Del Vecchio MT, Remondo C, Migali C, Ginanneschi C, Tsang KY, Licchetta A, Mannucci S, Loiacono L, Tassone P, Francini G, Tagliaferri P. (2010), Regulatory (FoxP3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. J Immunother.; 33(4): 435-441. Cooper EL, Kauschke E, Cossarizza A, (2002), Digging for innate immunity since Darwin and Metchnikoff. Bioessays.; 24(4): 319-33. Cox JH, Ferrari G, Janetzki S, (2006), Measurement of cytokine release at single cell level using the ELISPOT assay, 38(4): 274-282 Crawford J, Dale DC, Lyman GH, (2004), Chemotherapy-induced neutropenia: risks, consequences, and new directions for its management. Cancer.; 100(2): 228-237. Crawford RM, Leiby DA, Green SJ, Nacy CA, Fortier AH, Meltzer MS, (1994), Macrophage activation: a riddle of immunological resistance.Immunol Ser. ;60:29-46. Crumley AB, Going JJ, McEwan K, McKernan M, Abela JE, Shearer CJ, Stanley AJ, Stuart RC, (2010), Endoscopic mucosal resection for gastroesophageal cancer in a U.K. population. Long-term follow-up of a consecutive series. Surg Endosc. Abstract. Epub ahead of print. Czerkinsky C, Rees AS, Bergmeier LA, Challacombe SJ, (1983), The detection and specificity of class specific antibodies to whole bacterial cells using a solid phase radioimunoassay, Clin. exp. Immunology 53, 192-300. 87 Czermak BJ, Lentsch AB, Bless NM, Schmal H, Friedl HP, Ward PA, (1998), Role of complement in in vitro and in vivo lung inflammatory reactions. J Leukoc Biol.; 64(1): 40-48. Danziger IL, Cherkassky L, Siegel H, McManamon M, Kramer K, Budev M, Sawinsky D, Augustine JJ, Hricik DE, Fairchild R, Heeger PS, Poggio ED, (2010), Effects of influenza immunization on humoral and cellular alloreactivity in humans, Transplantation, 89(7): 838-844. de Gramont A, Figer A, Seymour M, Homerin M, Hmissi A, Cassidy J, Boni C, CortesFunes H, Cervantes A, Freyer G, Papamichael D, Le Bail N, Louvet C, Hendler D, de Braud F, W ilson C, Morvan F, Bonetti A., (2000), Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol.; 18(16): 2938-2947. Del Villano BC, Brennan S, Brock P, Bucher C, Liu V, McClure M, Rake B, Space S, Westrick B, Schoemaker H, Zurawski VR Jr, (1983), Radioimmunometric assay for a monoclonal antibody-defined tumor marker, CA 19-9. Clin Chem.; 29(3): 549-552. Deschoolmeester V, Baay M, Van Marck E, Weyler J, Vermeulen P, Lardon F, Vermorken JB, (2010), Tumor infiltrating lymphocytes: an intriguing player in the survival of colorectal cancer patients. BMC Immunol.; 11:19. Disis ML, Bernhard H, Shiota FM, Hand SL, Gralow JR, Huseby ES, Gillis S, Cheever MA, (1996), Granulocyte-macrophage colony-stimulating factor: an effective adjuvant for protein and peptide-based vaccines. Blood.; 88(1): 202-210. Dotan E, Aggarwal C, Smith MR, (2010), Impact of Rituximab (Rituxan) on the Treatment of B-Cell Non-Hodgkin's Lymphoma. P T.; 35(3): 148-157. Dresch C, Barreau P, (1985), Differentiation of mixed colony-forming cells in normal human bone marrow and blood. Exp Hematol.; 13(11): 1143-1151. 88 Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD, (2002), Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol.; 3(11): 991-998. Elgert KD, Alleva DG, Mullins DW., (1998), Tumor-induced immune dysfunction: the macrophage connection. J Leukoc Biol.; 64(3): 275-290. Elsaie ML, Olasz E, Jacob SE. (2008); Cytokines and Langerhans cells in allergic contact dermatitis. G Ital Dermatol Venereol.; 143(3): 195-205. Fallarino F, Gajewski TF, (1999), Cutting edge: differentiation of antitumor CTL in vivo requires host expression of Stat1. J Immunol.; 163(8): 4109-4113. Fällman M, Andersson R, Andersson T, (1993), Signaling properties of CR3 (CD11b/CD18) and CR1 (CD35) in relation to phagocytosis of complement-opsonized particles. J Immunol.; 151(1): 330-338. Fischer A, (2009), Immunstatus im zellulären und serologischen Vergleich bei Kindern unter chirurgischen Eingriffen, Diss. Univ. Leipzig: 51-56. Fox A, Jeffries DJ, Hill PC, Hammond AS, Lugos MD, Jackson-Sillah D, Donkor SA, Owiafe PK, McAdam KP, Brookes RH, (2007), ESAT-6 and CFP-10 can be combined to reduce the cost of testing for Mycobacterium tuberculosis infection, but CFP-10 responses associate with active disease., Trans R Soc Trop Med Hyg. 101(7): 691-698. Franke A, Teyssen S, Singer MV, (2005), Alcohol-related diseases of the esophagus and stomach.; Dig Dis 23(3-4): 204-213. Fujiwara K, Higashi T, Nouso K, Nakatsukasa H, Kobayashi Y, Uemura M, Nakamura S, Sato S, Hanafusa T, Yumoto Y, Naito I, Shiratori Y, (2004), Decreased expression of B7 costimulatory molecules and major histocompatibility complex class-I in human hepatocellular carcinoma. J Gastroenterol Hepatol.; 19(10): 1121-1127. Ha TY, (2009), The role of regulatory T cells in cancer. Immune Netw.; 9(6): 209-235. 89 Gardner RV, (1999), Long term hematopoietic damage after chemotherapy and cytokine. Front Biosci.,; 4: e47-57. Geluk A, van Meijgaarden KE, Franken KL, W ieles B, Arend SM, Faber WR, Naafs B, Ottenhoff TH, (2004), Immunological crossreactivity of the Mycobacterium leprae CFP10 with its homologue in Mycobacterium tuberculosis. Scand J Immunol.; 59(1): 66-70. Goodell V, dela Rosa C, Slota M, MacLeod B, Disis ML, (2007), Sensitivity and specificity of tritiated thymidine incorporation and ELISPOT assays in identifying antigen specific T cell immune responses. BMC Immunol.; 8: 21. Guerder S, Meyerhoff J, Flavell R, (1994), The role of the T cell costimulator B7-1 in autoimmunity and the induction and maintenance of tolerance to peripheral antigen. Immunity.; 1(2): 155-166. Ha TY, (2009), The role of regulatory T cells in cancer. Immune Netw.; 9(6): 209-235. Haglund C, Roberts PJ, Kuusela P, Scheinin TM, Mäkelä O, Jalanko H, (1986), Evaluation of CA 19-9 as a serum tumour marker in pancreatic cancer. Br J Cancer.; 53(2): 197-202. Haslett C, Savill JS, Meagher L, (1989), The neutrophil. Curr Opin Immunol.; 2(1): 1018. Hebart H, Bollinger C, Fisch P, Sarfati J, Meisner C, Baur M, Loeffler J, Monod M, Latgé JP, Einsele H, (2002), Analysis of T-cell responses to Aspergillus fumigatus antigens in healthy individuals and patients with hematologic malignancies. Blood.; 100(13): 4521-4528. 90 Hedrick SM, Germain RN, Bevan MJ, Dorf M, Engel I, Fink P, Gascoigne N, Heber-Katz E, Kapp J, Kaufmann Y, et al., (1985), Rearrangement and transcription of a T-cell receptor beta-chain gene in different T-cell subsets. Proc Natl Acad Sci U S A. ; 82(2): 531-535. Herold G et al., (2009), Innere Medizin- eine vorlesungsorientierte Darstellung unter Berücksichtigung des Gegenstandskataloges für die Ärztliche Prüfung, Ausgabe 2010. Herrera MA, Chu TM, Holyoke ED, (1976), Carcinoembryonic antigen (CEA) as a prognostic and monitoring test in clinically complete resection of colorectal carcinoma. Ann Surg.; 183(1): 5-9. Hill PC, Jackson-Sillah D, Fox A, Franken KL, Lugos MD, Jeffries DJ, Donkor SA, Hammond AS, Adegbola RA, Ottenhoff TH, Klein MR, Brookes RH, (2005), ESAT6/CFP-10 fusion protein and peptides for optimal diagnosis of mycobacterium tuberculosis infection by ex vivo enzyme-linked immunospot assay in the Gambia. J Clin Microbiol.; 43(5): 2070-2074. Hind D, Tappenden P, Tumur I, Eggington S, Sutcliffe P, Ryan A, (2008), The use of irinotecan, oxaliplatin and raltitrexed for the treatment of advanced colorectal cancer: systematic review and economic evaluation. Health Technol Assess.; 12(15): iii-ix, xi162. Hiroishi K, Eguchi J, Baba T, Shimazaki T, Ishii S, Hiraide A, Sakaki M, Doi H, Uozumi S, Omori R, Matsumura T, Yanagawa T, Ito T, Imawari M, (2010), Strong CD8(+) T-cell responses against tumor-associated antigens prolong the recurrence-free interval after tumor treatment in patients with hepatocellular carcinoma., J Gastroenterol.; 45(4): 451-458. Hoefsmit EC, Duijvestijn AM, Kamperdijk EW, (1982), Relation between langerhans cells, veiled cells, and interdigitating cells. Immunobiology.; 161(3-4): 255-265. 91 Hoffmann M, Vernazza P, Fierz W, (2003), Diagnostik der latenten Tuberkulose im neuen Millenium, Schweiz Med Forum 50: 1244-1247. Hoffmann TK, Dworacki G, Meidenbauer N, Gooding W , Johnson JT, W hiteside TL, (2002), Spontaneous apoptosis of circulating T lymphocytes in patients with head and neck cancer and its clinical importance. Clin. Cancer Res., 8: 2553-2562. Huang SM, Harari PM, (1999), Epidermal growth factor receptor inhibition in cancer therapy: biology, rationale and preliminary clinical results. Invest New Drugs.; 17(3): 259-269. Huang X, Yu HS, Chen Z, Li JL, Hu ZM, Gao JM, (2010), A novel immunotherapy for superficial bladder cancer by the immobilization of streptavidin-tagged bioactive IL-2 on the biotinylated mucosal surface of the bladder wall. Chin J Cancer. 29(6): 611-616 Itoh K, (1987), Tumor-infiltrating lymphocytes from human metastatic melanoma, Gan To Kagaku Ryoho, (6 Pt 2): 2119-2126. Jain N, Nguyen H, Chambers C, Kang J, (2010), Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc Natl Acad Sci U S A.; 107(4): 1524-1528. Janetzki S, Cox JH, Oden N, Ferrari G, (2005), Standartisation and validation issues of the ELISPOT assay, Methods Mol Biol, 302: 51-86. Janeway CA, Travers P, Walport M, Shlomchik M, (2002), Immunologie, Spektrum Akademischer Verlag Heidelberg GmbH, 5. Aufl., 686-687. Janeway CA Jr, Dianzani U, Portoles P, Rath S, Reich EP, Rojo J, Yagi J, Murphy DB, (1989), Cross-linking and conformational change in T-cell receptors: role in activation and in repertoire selection. Cold Spring Harb Symp Quant Biol.; 54 Pt 2: 657-666. 92 Janeway CA Jr, (1980), Idiotypes, T-cell receptors, and T-B cooperation. Contemp Top Immunobiol.; 9:171-203. Johnson K, Reddy KL, Singh H, (2009), Molecular pathways and mechanisms regulating the recombination of immunoglobulin genes during B-lymphocyte development. Adv Exp Med Biol.; 650: 133-147. Kaczorowski DJ, Nakao A, McCurry KR, Billiar TR, (2009), Toll-like receptors and myocardial ischemia/reperfusion, inflammation, and injury. Curr Cardiol Rev.; 5(3): 196202. Kaechele V, Moehler M, Lutz MP, von Wichert G, Eisele M, Klaus J, Galle PR, Adler G, Seufferlein T, (2010), A phase I/II study of oxaliplatin and paclitaxel in patients with nonresectable cancer of the oesophagus and adenocarcinoma of the gastro-oesophageal junction: a study of the Arbeitsgemeinschaft Internistische Onkologie. Cancer Chemother Pharmacol. 66(1): 191-195. Kalish RS, (1995), Antigen processing: the gateway to the immune response. J Am Acad Dermatol.; 32(4): 640-652. Kalyuzhny AE, (2009), ELISPOT assay on membrane microplates, Methods Mol Biol, 536: 355-365. Kalra M, Khuller GK, Grover A, Behera D, Wanchu A, Verma I, (2010), Utility of a combination of RD1 and RD2 antigens as a diagnostic marker for tuberculosis. Diagn Microbiol Infect Dis.; 66(2): 153-1561. Kalyuzhny AE, (2005), Chemistry and biology of the ELISPOT assay, Methods Mol Biol., 302: 15-31. Kemper C, Mitchell LM, Zhang L, Hourcade DE, (2008), The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci U S A.; 105(26): 9023-9028. 93 Kim HS, Kim CH, Hur M, Hyon IG, Park MJ, Song W, Park JY, Hang, HJ, Lee KM, (2010), Clinical usefullness of T-SPOT.TB test for diagnosis of tuberculosis, Korean J Lab Med., 30 (2): 171-177. Knutson KL, dela Rosa C, Disis ML, (2006), Laboratory analysis of T-cell immunity. Front Biosci.; 11: 193219-193244. Kolb W P, Haxby JA, Arroyave CM, Müller-Eberhard HJ, (1973), The membrane attack mechanism of complement. Reversible interactions among the five native components in free solution. J Exp Med.; 138(2): 428-437. Kolb W P, Haxby JA, Arroyave CM, Müller-Eberhard HJ, (1972), Molecular analysis of the membrane attack mechanism of complement., J Exp Med.; 135(3): 549-566. Kotsakis A, Sarra E, Peraki M, Koukourakis M, Apostolaki S, Souglakos J, Mavromanomakis E, Vlachonikolis J, Georgoulias V, (2000), Docetaxel-induced lymphopenia in patients with solid tumors: a prospective phenotypic analysis. Cancer. ; 89(6): 1380-1386. Kreienberg R, Melchert F, Lemmel EM, (1979), Monitoring the immune status of breast cancer patients under adjuvant chemotherapy and polychemotherapy, Onkologie, 2(5): 181-186. Kulkarni R, Behboudi S, Sharif S, (2010), Insights into the role of Toll-like receptors in modulation of T cell responses. Cell Tissue Res. Abstract. Epub ahead of print. Kurokawa Y, Muto M, Minashi K, Boku N, Fukuda H (2009), A phase II trial of combined treatment of endoscopic mucosal resection and chemoradiotherapy for clinical stage I esophageal carcinoma, Jpn J Clin Oncol., 39(10): 686-689. Kuusela P, Jalanko H, Roberts P, Sipponen P, Mecklin JP, Pitkänen R, Mäkelä O., (1984), Comparison of CA 19-9 and carcinoembryonic antigen (CEA) levels in the serum of patients with colorectal diseases. Br J Cancer.; 49(2): 135-139. 94 Laack E, Andritzky B, Dürk H, Burkholder I, Edler L, Schuch G, Boeters I, Görn M, Lipp R, Horst H, Popp J, Hossfeld DK, (2005), Docetaxel and cisplatin as first-line treatment for patients with metastatic esophageal cancer: a pilot study. Onkologie. 28(12): 64750. Lai CC, Tan CK, Lin SH, Liu W L, Liao CH, W ang CY, W ang JY, Huang YT, Lin HI, (2010), Diagnostic usefulness of enzyme-linked immunospot assay for interferongamma for tuberculosis in cancer patients. Hsueh PRScand J Infect Dis. Abstract. Epub ahead of print. Lange H, (1980), Schnelle Dichtegradienten-Zentrifugation dispergierter Teilchen, Colloid & Polymer Science, 1077-1085. Lederer JA, Perez VL, DesRoches L, Kim SM, Abbas AK, Lichtman AH, (1996), Cytokine transcriptional events during helper T cell subset differentiation. J Exp Med.; 184(2): 397-406. Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, W eber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, Roederer M, Davis MM, (1999), Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med.; 5(6): 677-685. Lee W S, Park S, Lee W Y, Yun SH, Chun HK, (2010), Clinical impact of tumor-infiltrating lymphocytes for survival in stage II colon cancer. Cancer. Abstract. Epub ahead of print. Lenschow DJ, Walunas TL, Bluestone JA, (1996), CD28/B7 system of T cell costimulation. Annu Rev Immunol.; 4: 33-258. Leong T, Michael M, Foo K, Thompson A, Lim Joon D, Weih L, Ngan S, Thomas R, Zalcberg J, (2003), Adjuvant and neoadjuvant therapy for gastric cancer using epirubicin/cisplatin/5-fluorouracil (ECF) and alternative regimens before and after chemoradiation. Br J Cancer; 89(8): 1433-1438. 95 Leopardo D, Di Lorenzo G, De Renzo A, Federico P, Luponio S, Buonerba C, Matano E, Merola G, Imbimbo M, Montesarchio E, Rea A, Merola MC, De Placido S, Palmieri G, (2010), Efficacy of rituximab in gastric diffuse large B cell lymphoma patients.W orld J Gastroenterol.; 16(20): 2526-2530. Lienhardt C, Fielding K, Hane A, Niang A, Ndao CT, Karam F, Fletcher H, Mbow F, Gomis JF, Diadhiou R, Toupane M, Dieye T, Mboup S, (2010), Evaluation of the Prognostic Value of IFN-c Release Assay and Tuberculin Skin Test in Household Contacts of Infectious Tuberculosis Cases in Senegal, PLoS One, 5(5): e10508 Lion E, Smits EL, Berneman ZN, Van Tendeloo VF, (2009), Quantification of IFNgamma produced by human purified NK cells following tumor cell stimulation: comparison of three IFN-gamma assays., J Immunol Methods.; 350(1-2): 89-96. Lissoni P, Brivio F, Fumagalli L, Messina G, Meregalli S, Porro G, Rovelli F, Vigorè L, Tisi E, D'Amico G, (2009), Effects of the conventional antitumor therapies surgery, chemotherapy, radiotherapy and immunotherapy on regulatory T lymphocytes in cancer patients. Anticancer Res.; 29(5): 1847-1852. Liu F, Zhang ZD, Gao MQ, Ma LP, Wang QF, Wu XG, Jia HY, Gu SX, Ma Y, (2009), The comparison of different antigens in enzyme-linked immunospot assay for the diagnosis of tuberculosis. Zhonghua Jie He He Hu Xi Za Zhi.; 32(11): 838-841. Liu WM, Fowler DW, Smith P, Dalgleish AG, (2010), Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer.; 102(1): 115-123. Marone G, Kagey-Sobotka A, Lichtenstein LM, (1981), IgE-mediated histamine release from human basophils: differences between antigen E- and anti-IgE-induced secretion. Int Arch Allergy Appl Immunol.; 65(3): 339-348. 96 Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Séror C, Métivier D, Perfettini JL, Zitvogel L, Kroemer G, (2009), Chemotherapy induces ATP release from tumor cells. Cell Cycle.; 8(22): 3723-3728. Marusić-Galesić S, Stephany DA, Longo DL, Kruisbeek AM, (1988), Development of CD4-CD8+ cytotoxic T cells requires interactions with class I MHC determinants. Nature.; 333(6169): 180-183. Mayer S, Laumer M, Mackensen A, Andreesen R, Krause SW , (2002), Analysis of the immune Response against Tetanus toxoid: enumeration of specific T helper cells by the Elispot assay, Immunobiology, 205(3): 282-289. McMullen TP, Lai R, Dabbagh L, W allace TM, de Gara CJ, (2010), Survival in rectal cancer is predicted by T cell infiltration of tumour-associated lymphoid nodules. Clin Exp Immunol.; 161(1): 81-88. Mellios T, Ko HL, Beuth J, (2010), Impact of adjuvant chemo- and radiotherapy on the cellular immune system of breast cancer patients. In Vivo. (2): 227-230. Miller JF, (1976), Antigen perception by T lymphocytes. Influence of the major histocompatibility complex. Ric Clin Lab.; 6(4): 285-299. Mogensen TH, (2009), Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev.; 22(2): 240-273. Mortarini R, Piris A, Maurichi A, Molla A, Bersani I, Bono A, Bartoli C, Santinami M, Lombardo C, Ravagnani F, Cascinelli N, Parmiani G, Anichini A, (2003), Lack of terminally differentiated tumor-specific CD8+ T cells at tumor site in spite of antitumor immunity to self-antigens in human metastatic melanoma. Cancer Res. 15; 63(10): 2535-2545. 97 Mosmann TR, Cherwinski H, Bond MW , Giedlin MA, Coffman RL, (1986), Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol.,175(1): 5-14. Murphy K, Travers P, Walport M, (2009), Janeway Immunologie, 7. Auflage, dt., Spektrum Akademischer Verlag Heidelberg GmbH., 37-96 Neves JS, Weller PF, (2009), Functional extracellular eosinophil granules: novel implications in eosinophil immunobiology. Curr Opin Immunol.; 21(6): 694-699. Nickel P, Bestard O, Volk HD, Reinke P, (2009), Diagnostic value of T-cell monitoring assays in kidney transplantation. Curr Opin Organ Transplant.; 14(4): 426-431. Nowell PC, (1960), Phytohemagglutinin: an initator of mitosis in cultures of normal human leukocytes, Cancer research 20: 462-466. O'Brien SN, Blijlevens NM, Mahfouz TH, Anaissie EJ, (2003), Infections in patients with hematological cancer: recent developments.Hematology Am Soc Hematol Educ Program.: 438-472. Palacios R, Samaridis J, (1993), Bone marrow clones representing an intermediate stage of development between hematopoietic stem cells and pro-T-lymphocyte or proB-lymphocyte progenitors. Blood.; 81(5): 1222-1238. Palena C, Schlom J,(2010), Vaccines against human carcinomas: strategies to improve antitumor immune responses. J Biomed Biotechnol.; 380697. Paoletti X, Oba K, Burzykowski T, Michiels S, Ohashi Y, Pignon JP, Rougier P, Sakamoto J, Sargent D, Sasako M, Van Cutsem E, Buyse M, GASTRIC (Global Advanced/Adjuvant Stomach Tumor Research International Collaboration) Group, (2010), Benefit of adjuvant chemotherapy for resectable gastric cancer: a metaanalysis. JAMA.; 303(17): 1729-1737. 98 Paust S, Senman B, von Andrian UH, (2010), Adaptive immune responses mediated by natural killer cells. Immunol Rev.; 235(1): 286-296. Perrot I, Blanchard D, Freymond N, Isaac S, Guibert B, Pachéco Y, Lebecque S, (2007), Dendritic cells infiltrating human non-small cell lung cancer are blocked at immature stage. J Immunol.; 178(5): 2763-2769. Petersen TR, Dickgreber N, Hermans IF, (2010), Tumor antigen presentation by dendritic cells. Crit Rev Immunol.; 30(4): 345-386. Pezzuoto A, Ulrichs T, Burmester GR, (2007), Taschenatlas der Immunologie, Grundlagen-Labor-Klinik, Georg Thieme Verlag Stuttgart, 2. Aufl.; 1. engl. Aufl. 2003, Color Atlas of Immunology. 164-167. Pieters J, (1997), MHC class II compartments: specialized organelles of the endocytic pathway in antigen presenting cells, Biol Chem. 378(8): 751-758. Prescott SL, (2009), Role of dietary immunomodulatory factors in the development of immune tolerance. Nestle Nutr W orkshop Ser Pediatr Program.; 64:185-94; discussion 194-200, 251-257. Reichert TE, Strauss L, W agner EM, Gooding W, Whiteside TL, (2002), Signaling abnormalities and reduced proliferation of circulating and tumor-infiltrating lymphocytes in patients with oral carcinoma. Clin. Cancer Res., 8: 3137-3145. Reichert TE, Strauss L, W agner EM, Gooding W, Whiteside TL, (2002), Signaling abnormalities and reduced proliferation of circulating and tumor-infiltrating lymphocytes in patients with oral carcinoma. Clin. Cancer Res., 8: 3137-3145. 99 Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA et al., (1988), Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med.; 319(25): 1676-1680. Rosenberg SA, (1999), A New Era of Cancer Immunotherapy: Converting Theory to Performance, CA-A Cancer J for Cl 49(2): 70-73 Sastre J, Garcia-Saenz JA, Diaz-Rubio E, (2006), Chemotherapy for gastric cancer. World J Gastroenterol.; 12(2): 204-213. Schalhorn A, Kolligs F, Tympner C., Gross, M, Wagner A, W ichmann M, Lersch C, Zellmann K, (2006), Kolonkarzinom, Manual Gastrointestinale Tumoren, Tumorzentrum München, W . Zuckschwerdt Verlag GmbH, 7. Aufll., 117-160. Scheibenbogen C, Romero P, Rivoltini L, Herr W, Schmittel A, Cerottini JC, Woelfel T, Eggermont AM, Keilholz U,(2000), Quantitation of antigen-reactive T-cells in peripheral blood by IFNgamma-ELISPOT assay and chromium-release assay, a four-centre comparative trial, J. Immunol. Methods, 244 (1-2), 81-89. Schultes BC, W hiteside TL, (2003), Monitoring of immune responses to CA125 with an IFN-gamma ELISPOT assay. J Immunol Methods.; 279(1-2): 1-15. Sedgwick JD, (2005), ELISPOT assay: a personal retrospektive, Methods Mol Biol. 302, 3-14 Sedgwick JD, Holt PG, (1983), A solid-phase immunoenzymatic technique for the enumeration of specific antibody-secreting cells, J 57 (1-3), 301-309. Seleznick MJ, (1992), Tumor markers. Prim Care.; 9(4): 715-726. Sendler A, Prinz C, Janetschek P, Becker K, Grützner U, Lordick F, Schuhmacher C, Zimmermann F, (2006), Magenkarzinom, Manual Gastrointestinale Tumoren, Tumorzentrum München, W. Zuckschwerdt Verlag GmbH, 7. Aufll., 26-53. 100 Sers C, Kuner R, Falk CS, Lund P, Sueltmann H, Braun M, Buness A, Ruschhaupt M, Conrad J, Mang-Fatehi S, Stelniec I, Krapfenbauer U, Poustka A, Schäfer R, (2009), Down-regulation of HLA Class I and NKG2D ligands through a concerted action of MAPK and DNA methyltransferases in colorectal cancer cells. Int J Cancer.; 125(7): 1626-1639. Seya T, Matsumoto M, Tsuji S, Begum NA, Nomura M, Azuma I, Hayashi A, Toyoshima K, (2001), Two receptor theory in innate immune activation: studies on the receptors for bacillus Culmet Guillen-cell wall skeleton. Arch Immunol Ther Exp (W arsz).; 49 Suppl 1: S13-21. Seya T, Shime H, Ebihara T, Oshiumi H, Matsumoto M, (2010), Pattern recognition receptors of innate immunity and their application to tumor immunotherapy. Cancer Sci.; 101(2): 313-320. Shim HJ, Cho SH, Hwang JE, Bae W K, Song SY, Cho SB, Lee W S, Joo YE, Na KJ, Chung IJ, (2010), Phase II Study of Docetaxel and Cisplatin Chemotherapy in 5Fluorouracil/Cisplatin Pretreated Esophageal Cancer. Am J Clin Oncol. Abstract. Epub ahead of print. Shurin GV, Tourkova IL, Kaneno R, Shurin MR, (2009), Chemotherapeutic agents in noncytotoxic concentrations increase antigen presentation by dendritic cells via an IL12-dependent mechanism. J Immunol.; 183(1): 137-144. Simpson JA, Al-Attar A, Watson NF, Scholefield JH, Ilyas M, Durrant LG, (2010), Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut.; 59(7): 926-33. Sims JE, Tunnacliffe A, Smith WJ, Rabbitts TH, (1984), Complexity of human T-cell antigen receptor beta-chain constant- and variable-region genes. Nature.; 312(5994): 541-545. 101 Smith JG, Liu X, Kaufhold RM, Clair J, Caulfield MJ, (2001), Development and Validation of a Gamma Interferon ELISPOT Assay for Quantitation of Cellular Immune Responses to Varizella-Zoster-Virus, Clinical and Diagnostic Laboratory Immunology, 8 (5), 871-879. Spry CJ, (1978), Eosinophils as effector cells in disease. Schweiz Med W ochenschr.; 108(41): 1572-1576. Stanley ER, Hansen G, Woodcock J, Metcalf D, (1975), Colony stimulating factor and the regulation of granulopoiesis and macrophage production. Fed Proc.; 34(13): 22722278. Stein H, Sarbia M, (2006), Adenokarzinome des distalen Ösophagus und des ösophago-gastralen Übergangs, Manual Gastrointestinale Tumoren, Tumorzentrum München, W . Zuckschwerdt Verlag GmbH, 7. Aufll., 19-25. Streeck H, Frahm N, W alker BD, (2009), The role of IFN-gamma ELISPOT assay in HIV vaccine research, Nat Protoc., 4(4): 461-469. Swierzko AS, Cedzyński M, Kirikae T, Nakano M, Klink M, Kirikae F, Ziółkowski A, Vinogradov EV, Kawakami M, (2003), Role of the complement-lectin pathway in anaphylactoid reaction induced with lipopolysaccharide in mice. Eur J Immunol.; 33(10): 2842-2852. Tanguay S, Killion JJ, (1994), Direct comparison of ELISPOT and ELISA-based assays for detection of individual cytokine-secreting cells, Lymphokine Cytokine Res., 13(4), 259-263. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, Mendiboure J, Pignon JP, Jooste V, van Endert P, Ducreux M, Zitvogel L, Piard F, Kroemer G, (2010), Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene.; 29(4): 482-491. 102 Tosh PK, Kennedy RB, Vierkant RA, Jacobson RM, Poland GA, (2009), Correlation between rubella antibody levels and cytokine measures of cell-mediated immunity. Viral Immunol. 22(6): 451-456. Uemura N, Okamoto S, Yamamoto S, (2002), H. pylori infection and the development of gastric cancer. Keio J Med.; 51 Suppl 2: 63-68. van den Broek M, von Boehmer L, Knuth A, (2010), Developments in cancer immunotherapy. Dig Dis.; 28(1): 51-56. Valent P, Dahinden CA, (2010), Role of interleukins in the regulation of basophil development and secretion. Curr Opin Hematol.; 17(1): 60-66. Vanderlocht J, Van Elssen CH, Senden-Gijsbers BL, Meek B, Cloosen S, Libon C, Bos GM, Germeraad WT, (2010), Increased tumor-specific CD8+ T cell induction by dendritic cells matured with a clinical grade TLR-agonist in combination with IFNgamma. Int J Immunopathol Pharmacol.; 23(1): 35-50. van der Most RG, Currie A, Robinson BW, Lake RA, (2006), Cranking the immunologic engine with chemotherapy: using context to drive tumor antigen cross-presentation towards useful antitumor immunity. Cancer Res.; 66(2): 601-604. Vordermeier HM, Brown J, Cockle PJ, Franken WP, Drijfhout JW , Arend SM, Ottenhoff TH, Jahans K, Hewinson RG, (2007), Assessment of cross-reactivity between Mycobacterium bovis and M. kansasii ESAT-6 and CFP-10 at the T-cell epitope level. Clin Vaccine Immunol.;14(9): 1203-1209. Wagner AD, Unverzagt S, Grothe W , Kleber G, Grothey A, Haerting J, Fleig W E, (2010) Chemotherapy for advanced gastric cancer, Cochrane Database Syst Rev. 17;3: CD004064. Walker EB, Disis ML, (2003), Monitoring immune responses in cancer patients receiving tumor vaccines. Int Rev Immunol.; 22(3-4): 283-319. 103 Wang M, Bronte V, Chen PW, Gritz L, Panicali D, Rosenberg SA, Restifo NP, (1995), Active Immunotherapy of Cancer with a Nonreplicating Recombinant Fowlpox Virus Encoding a Model Tumor-Associated Antigen, J Immunol.; 154(9): 4685–4692. Waters JS, Norman A, Cunningham D, Scarffe JH, W ebb A, Harper P, Joffe JK, Mackean M, Mansi J, Leahy M, Hill A, Oates J, Rao S, Nicolson M, Hickish T, (1999), Long-term survival after epirubicin, cisplatin and fluorouracil for gastric cancer: results of a randomized trial. Br J Cancer.; 80(1-2): 269-672. Wedgwood JF, Hatam L, Bonagura VR (1988), Effect of interferon-gamma and tumor necrosis factor on the expression of class I and class II major histocompatibility molecules by cultured human umbilical vein endothelial cells. Cell Immunol. 111(1): 1-9. Weikert C, Dietrich T, Boeing H, Bergmann MM, Boutron-Ruault MC, Clavel-Chapelon F, Allan N, Key T, Lund E, Olsen A, Tjonneland A, Overvad K, Rohrmann S, Linseisen J, Pischon T, Trichopoulou A, W einehall, Johannson I, Sánchez MJ, Agudo A, Barricarte A, Amiano P, Chirlaque MD, Quirós JR, Wirfalt E, Peeters PH, Bueno-deMesquita HB, Vrieling A, Pala V, Palli D, Veineis P, Tumino R, Panico S, Bingham S, Khaw KT, Norat T, Jenab M, Ferrari P, Slimani M, Riboli E, (2009), Lifetime and baseline alcohol intake and risk of cancer of the upper aero-digestive tract in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Int J Cancer 125 (2): 406-412. Wheelock EF, (1965), Interferon-like virus-inhibitor induced in human leukocytes by phytohemagglutinin. Science. 149: 310-311. Whiteside TL, (2004), Methods to monitor immune response and quality control. Dev Biol (Basel).; 116: 219-28; discussion 229-236. 104 Whiteside TL, Zhao Y, Tsukishiro T, Elder EM, Gooding W, Baar J, (2003), Enzymelinked immunospot, cytokine flow cytometry, and tetramers in the detection of T-cell responses to a dendritic cell-based multipeptide vaccine in patients with melanoma. Clin Cancer Res.; 9(2): 641-649. Wilke CM, W u K, Zhao E, W ang G, Zou W , (2010), Prognostic significance of regulatory T cells in tumor. Int J Cancer.;127(4): 748-758. Williamson AR, Zitron IM, McMichael AJ, (1976), Clones of B lymphocytes: their natural selection and expansion. Fed Proc.; 35(10): 2195-2201. Wolf AM, W olf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B, (2003), Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res.; 9(2): 606-612. Xiang XJ, Qiu F, Xiong JP, Zhang L, Yu F, Feng M, Zhan ZY, (2010), A phase II trial of Epirubicin, Oxaliplatin, and Capecitabine (EOX) as first-line chemotherapy in advanced gastric cancer: a Chinese single-center experience.; 56(3): 171-177. Yamada N, Oizumi S, Kikuchi E, Shinagawa N, Konishi-Sakakibara J, Ishimine A, Aoe K, Gemba K, Kishimoto T, Torigoe T, Nishimura M, (2010), CD8(+) tumor-infiltrating lymphocytes predict favorable prognosis in malignant pleural mesothelioma after resection. Cancer Immunol Immunother., 59(10): 1543-1549. Yaqub S, Aandahl EM, (2009), Inflammation versus adaptive immunity in cancer pathogenesis. Crit Rev Oncog.; 15(1-2): 43-63. Ye Q, Shen Y, W ang X, Yang J, Miao F, Shen C, Zhang J, (2010), Hypermethylation of HLA class I gene is associated with HLA class I down-regulation in human gastric cancer. Tissue Antigens. 75(1): 30-39. 105 Zaniboni A, Barni S, Labianca R, Marini G, Pancera G, Giaccon G, Piazza E, Signaroldi A, Legnani W , Luporini G, (1995), Epirubicin, cisplatin, and continuous infusion 5fluorouracil is an active and safe regimen for patients with advanced gastric cancer. An Italian Group for the Study of Digestive Tract Cancer (GISCAD) report. Cancer.; 76(10): 1694-1699. Zaritskaya L, Shurin MR, Sayers TJ, Malyguine AM, (2010), New flow cytometric assays for monitoring cell-mediated cytotoxicity. Expert Rev Vaccines.; 9(6): 601-616. Zhang AM, Ye S, (2007), W eekly dosage of docetaxel combined with cisplatin and 5fluorouracil in the treatment of advanced gastric cancer, Nan Fang Yi Ke Da Xue Xue Bao.; 27(10): 1555-1557. Zhang W , Caspell R, Karulin AY, Ahmad M, Haicheur N, Abdelsalam A, Johannesen K, Vignard V, Dudzik P, Georgakopoulou K, Mihaylova A, Silina K, Aptsiauri N, Adams V, Lehmann PV, McArdle S, (2009), ELISPOT assays provide reproducible results among different laboratories for T-cell immune monitoring--even in hands of ELISPOTinexperienced investigators. J Immunotoxicol.; 6(4): 227-234. Zhou X, Kong N, Zou H, Brand D, Li X, Liu Z, Zheng SG, (2010), Therapeutic potential of TGF-beta-induced CD4(+) Foxp3(+) regulatory T cells in autoimmune diseases. Autoimmunity. Abstract. Epub ahead of print. Zimmermann F, Meining A, Lordick F, Sarbia M, Brücher B, (2006), ÖsophagusPlattenepithelkarzinom, Manual Gastrointestinale Tumoren, Tumorzentrum München, W. Zuckschwerdt Verlag GmbH, 7. Aufll., 1-18. 106 ANLAGEN Chemotherapieprotokolle: Protokoll ECF 107 Protokoll EOX Datum: Name: Vorname: Geb.·Dat.: Diagnose: Schema: EOX Alter: Gescht.: GrOI!e: Gewicht: KOF: Zyktus Nr.: Dieser Plan gilt fOr Zyklus 1 bis 7. Schema: Standard: Faktor: lndividuell· 2 Oxaliplatin (Eioxalin) 130 mg/m Epirubicin 50 mg/m2 2 Capecitabine (Xeloda) 625 mg/m aufgetei/1 auf 2 Gaben t:Jgl. morgens + abends, jeweits 30 min 100% 130 mg/rrf 100"/o 50 mg/rrr 100% 625 mg/m2 nach der Mahlzeit Tage: 1 1 1·21 Wiederholung ab Tag 22 Kreatinin: Clearance: Ta·Datum Zeit I . 06.08.2009 Stunde 08:00 Uhr Therapie Zofran 8 mg in 100 mlNaCI 0,9% o.v. I. 06.08.2009 Uhr (00:00) Capecltablne (Xeloda) 312,5 mg/m2, entsprlcht 566 mg absolut entsprlcht gerundet 1 Tablette(n) zu je 500 mg p.o. 1. 06.08.2009 Uhr (00:00) 1. 06.08.2009 Uhr (00:00) 50 ml Glukose 5% 1. 1. Uhr (02:00) 1. 06.08.2009 06.08.2009 06.08.2009 1. 06.08.2009 20:00 Uhr 50 mlGlukose 5% 1. 06.08.2009 20:00 Uhr Capecitablne (Xeloda) 312,5 mg/m2, entsprlcht 566 mg absolut entsprlcht gerundet 1 Tablette(n) zu je 500 mg p.o. 2. bis bls bis bis 21' 07.08.2009 08:00 Uhr Paspertin 20 gil 08:00 Uhr Capectlablne (Xeloda) 312,5 mg/m2,entsprlcht 566 mg absolut ::ntsFl !';t f<.:>rundel1 Tablottein) : .u je 500 m;p.o. 20:00 Uhr Capectiabine (Xeloda) 312,5 mg/m2, entsprlcht 566 mg absolut entsprlcht gerundet 1Tablette(n) zu je 500 mg p.o. 26.08.2009 Epirubicin 50 mg/m2,entspricht 91 mg absolut in 100 ml Glukose 5% uber 1 h (100 ml/h) Oxaliplatin (Eioxatin) 130 mg/m2,entspricht 235 mg absolut in 500 ml Glukose 5% uber 2 h (250 ml/h) Alleine infundierenlll 1·2 x wOchll. kleines Blulbild, Krealinin.Harnstoff, E'lyte, Bilirubin,Transaminasen zusalzllch: Zofran per os bei Bedarf Zyklus Nr.2 geplanlab: 108 Protokoll FLOT Datum: Name: Vorname: Geb.-Dat.: Alter: Geschlecht: Grone: Gewicht: KOF: Diagnose: Schema: FLOT (nach Al-8atran 2007) Zyklus Nr.: Schema: Standard: 2600 mg/m2 5-FU Oxaliplatin Calciumfolinat Faktor: lndividuell: 2 100% 2600 mg/m 100% 85 mg/m 100% 200 mg/m 2 Docetaxel 2 85 mg/ m 2 200 mg/m2 50 mg/m Voraussetzungen: Leukozyten 2: 2,5 GPT/1 Thrombozyten 2: 80 GPT/1 G-CSF 48 h vor Fortsetz. abgesetzt 100% lst-Werte an Tag +1 Leukos (GPT/1) Thrombos (GPT/1) Laborkontrollen: 2x!Woche kl. 88, 1x/Woche kl. Labor Tage: 1 1 2 2 50 mg/m Imussen eing [ragen warden Wiederholung Tag+ 15 -1. 08.02.2010 20:00 Uhr 1. 09.02.2010 Uhr (00:00) 250 ml NaCI 0,9 % + 8 mg Zofran + 8 mg Dexamethason i.v. Ober 30 min 1. 09.02.2010 Uhr (00:00) Tavegil 2 mg i.v. 1. 09.02.2010 Uhr (00:00) Ranitidin 1 Amp. i.v. 1. 09.02.2010 Uhr (00:05) Docetaxel 50 mg/m2, entspricht 81,4 mg absolut in in 250 ml NaCI0,9% iiber 60 min streng i.v. (250 ml pro Std.) Streng i.v.! 1. 00.01.1900 Uhr (01:00) 500 ml NaCI 0,9% i.v. Obt:r 1 h 1. 1. Dexamethason 8 mg p.o. 0-0-1 Uhr (02:00) Glukose 5% 50 ml i.v. 09.02.2010 Uhr (02:00) Oxaliplatin 85 mg/m2, entspricht 138,4 mg absolut in in 500 ml Glucose 5% iiber 120 min streng i.v. (250 ml pro Std.) Streng i.v.! 1. Uhr (04:00) Glukose 5% 50 ml i.v. 1. 09.02.2010 Uhr (04:00) Calciumfolinat 200 mg/m2, entspricht 325,5 mg absolut in in 250 ml NaCI 0,9% iiber 60 min i.v. (500 ml pro Std.) 1. 09.02.2010 Uhr (05:00) 5·FU 2600 mg/m2, entspricht 4232,1 mg absolut in in 250 ml NaCI 0,9% "(Baxterpumpe)" iiber 24 h c.l. 20:00 Uhr 1. 09.02. 2010 2. 10.02.2010 Dexamethason 4 mg p.o. 1-0-0 3. 11.02.2010 Dexamethason 4 mg p.o. 1-0-0 3. Zyklus 8eginn Dexamethason 4 mg p.o. 23.02.2010 109 Protokoll FOLFOX 4 (De Gramont- Schema) Datum: Name: Vorname: Geb.-Dat.: Alter: Geschlecht: Gror..e: Gewicht: KOF: Oxaliplatin Calciumfolinat 5-FU: 5-FU: Diagnose: Schema: FOLFOX4 (De Gramont-Schema) Zyklus Nr.: Applikation: Standard: 85 mg/m2 2 200 mg/m 400 mg/m2 2 600 mg/m Faktor: lndividuell: 75% 63,75 mg/m2 75% 150 mg/m2 75% 300 mg/m2 75% 450 mg/m2 Kreatinin: Clearance: ll9 Datum Zeit Tage: 1 1 1 1-2 Wiederholg. ab Tag +15 1. 22.06.2009 Stunde Therapie Uhr (00:00) Zofran 8 mg i.v. 1. 22.06.2009 Uhr (00:00) Glukose 5% 50 ml i.v. 1. 22.06.2009 Uhr (00:00) Oxaliplatin 64mg/m2 in 250 ml G5% i.v. 2h entspricht 99,1 mg Oxaliplatin absolut Alleine infundierenl 1. 22.06.2009 Uhr (02:00) Glukose 5% 50 ml i.v. 1. 22.06.2009 Uhr (02:00) Calciumfolinat 150mg/m2 in 250 ml NaCI i.v. 2h entspricht 233,27 mg Calciumfolinat absolut 1.-2. 22.06.2009 Uhr (04:00) 5-FU 300 mg/m2 i.v.-push (3-5 min) entspricht 466,5 mg 5-FU absolut in 50 ml NaCI 0,9% 1.-2. 22.06.2009 Uhr (04:00) 5-FU 450mg/m2 in 250 ml NaCI c.i. 22 h, entspricht 699,82 mg 5-FU absolut in 250 ml NaCI 0,9% (Baxterpumpe) 1. 22.06.2009 2. 23.06.2009 Uhr (00:00) Zofran 8 mg i.v. 2. 23.06.2009 Uhr (00:10) Calciumfolinat 150mg/m2 in 250 ml NaCI i.v. 2h entspricht 233,27 mg Calciumfolinat absolut 2.-3 23.06.2009 Uhr (02:10) 5-FU 300 mg/m2 i.v.-push (3-5 min) entspricht 466,5 mg 5-FU absolut in 50 ml NaCI 0,9% 2.-3 23.06.2009 Uhr (02:15) 5-FU 450mg/m2 in 250 ml NaCI c.i. 22 h, entspricht 699,82 mg 5-FU absolut in 250 ml NaCI 0,9% (Baxterpumpe) 2. 23.06.2009 20:00 Uhr Paspertin Tropfen 0-0-20 bei Bedarf 3. 24.06.2009 08:00 Uhr Paspertin Tropfen 20-20-20 bei Bedarf 4. 25.06.2009 08:00 Uhr Paspertin Tropfen 20-20-20 bei Bedarf 20:00 Uhr Paspertin Tropfen 0-0-20 bei Bedarf Laborkontrolle: BB, Na, K, Mg, Krea, Harnsaure 1x/Woche Zyklus Nr : 4 geplant ab: 06.07.2009 110 Protokoll FUFOX + Cetuximab Datum: Name: Vorname: Geb.-Dat.: Alter: Geschlecht: Grof1e: Gewicht: KOF: Diagnose: Schema: FUFOX + Cetuximab Zyklus Nr.: Applikation: Standard: Oxaliplatin Calciumfolinat 5-FU: Cetuximab (Erbitux ) Cetuximab (Erbitux) 50 mg/m2 500 mg/m2 2000 mg/m2 400 mg/m3 250 mg/m4 Faktor: lndividuell: 100% 50 mg/m2 100% 500 mg/m 2 100% 2000 mg/m2 0% 0 mg/m2 Tage: 0 mg/m2 0% Kreatinin: Clearance: Ie,g Datum Zeit 1. - 21.04.2009 1. 21.04.2009 21.04.2009 1. 21.04.2009 1. 21.04.2009 1. 21.04.2009 1. 1,8,15,22 1,8,15,22 1,8,15,22 1 8,15,22,29 Wiederholg. ab Tag 36 Stunde Therapie Uhr (00:00) Paracetamol1000 mg p.o. Uhr (00:00) Uhr (00:00) Tavegil 2 mg i.v. Ranitidin 1 Amp. i.v. Uhr (00:00) Cetuximab (Erbitux) 400 mg/m2, entspricht 0 mg absolut in 250 ml NaCI 0,9% iiber 2 h (125 ml/h) Ab Appl. 2: Cetuximab 0 mg/m2 entsprlcht 0 mg absolut Uhr (02:00) NaCI 0,9% 500 ml Ober 60 min Lv. CAVE: 60 min. Therapiepause nach Cetuximab einhalten 21.04.2009 Uhr (00:00) Zofran 8 mg i.v. 1. 21.04.2009 Uhr (01:00) Glukose 5%50 ml i.v. 1. 21.04.2009 Uhr (01:00) Oxaliplatin 50mg/m2 in 500 ml G5% i.v. 2h entspricht 108,4 mg Oxaliplatin absolut Alleine infundierenl 1. 21.04.2009 Uhr (03:00) Glukose 5% 50 ml i.v. 21.04.2009 Uhr (02:00) Calciumfolinat 500mg/m2 in 250 ml NaCI 0,9% entspricht 1084, mg Calciumfolinat absolut Ober 2 h i.v. 1.-2. 21.04.2009 Uhr (02:00) 5-FU: 2000 mg/m2, entspricht 4336 mg absolut in Zytostatikapumpe (250 ml) c.i. 24 h 1. nach ROcksprache evtl. Prednisolut 100 mg i.v. 1. 21.04.2009 20:00 Uhr Paspertin 20 Tropfen bei Bedarf 2. 22.04.2009 08:00 Uhr Paspertin Tropfen 20-20-20 bei Bedarf 3. 22.04.2009 08:00 Uhr Paspertin Tropfen 20-20-20 bei Bedarf Laborkontrolle: Chemo Labor am Tag vor Chemo Achtung: bei Durchfall, Mukositis, Krea-Anstieg keine Chemo Appl. Nr. 2 Cetux. 250 mg/m2 ab: Appl. Nr. 3 Cetux. 250 mg/m2 ab: Appl. Nr. 4 Cetux. 250 mg/m2 ab: Appl. Nr. 5 Cetux. 250 mg/m2 ab: 28.04.2009 05.05.2009 12.05.2009 19.05.2009 Zyklus Nr. 2 geplant ab: 26.05.2009 Appl. Nr. 2 gegeben am: Dosismodifikation: Appl. Nr. 3 gegeben am: Dosismodifikation: Appl. Nr. 4 gegeben am: Dosismodifikation: ACHTUNG: NUR CETUXIMABIII 111 Protokoll TCF Datum: Name: Vorname: Geb.-Dat.: Alter: Geschlecht: GrMe: Gewicht: Diagnose: Schema: TCF Emetogenes Potential: Stufe 5 Zyklus Nr.: 1 Applikation 1 KOF: Standard: 2 75 mg/m 75 mg/m2 2 750 mg/m Schema: Cisplatin Docetaxel 5-FU Faktor: lndividuell: 2 100% 75 mg/m 2 100% 75 mg/m 2 100% 750 mg/m Tage: 1 1 1-5 WH Tag +22 Kreatinin: Clearance: !MJ Datum Zeit Stunde Therapie Uhr (020:00) Dexamethason 4 mg Tbl. 0-0-1 -1. 10.06.2008 1. 11.06.2008 1. 11.06.2008 1. 11.06.2008 Uhr (02:00) Cisplatin 75 mg/m2, entspricht 160,1 mg absolut in NaCI 0,9% 500 ml iiber 1 h (500 ml/h) 1. 11.06.2008 Uhr (01:32) Tavegil 2 mg i.v. 1. 11.06.2008 Uhr (01:34) Ranitidin 1 Amp. i.v. 1. 11.06.2008 Uhr (02:00) Docetaxel 75 mg/m2 in NaCI 0,9% 250 ml in NaCI 0,9% 250 ml iiber 1 h (250 ml/h) 1.-5. 5. 11.06.2008 bis 15.06.2008 Uhr (02:00) 5-FU 750 mg/m2/Tag Ober 5 Tage in 250 ml NaCI 0,9% entspricht 8007 mg 5-FU absolut iiber 120 h i.v. (0,035 ml/min) iiber BaxterPumpe (lt. Auskunft Fa. Baxter mehrere Pumpen zur Probe fiir Apotheke Gera zur VerfOgung gestelltl) 1. 11.06.2008 20:00 Uhr Dexamethason 4 mg p.o. 1. 11.06.2008 20:00 Uhr Zofran 8 mg p.o. bei 8edarf 1. 11.06.2008 Uhr (00:00) 500 ml E 154 + 1 Amp. Zofran 8 mg + 8 mg Dexamethason + 100 ml Mannitol 15% + 1 Amp. Magnerot Ober 2h i.v. Emend 125 mg Tbl. 1-0-0 Ein-/Ausfuhrkontrolle. 8ilanz: 0-0-1 ml (Falls>+ 1000 ml: Lasix 20 mg i.v.) 2. 12.06.2008 08:00 Uhr Dexamethason 4 mg p.o. 1-0-1; 3. 13.06.2008 08:00 Uhr Dexamethason 4 mg p.o. 1-0..Q; Emend 80 mg Tbl. Emend 80 mg Tbl. 1-0..Q 1-0-0 Laborkontrolle: 88, Na, K, Mg, Krea, Harnsaure 1x/Woche zusatzlich: Zofran nabzw. MCP-Tropfen + 8 mg Dexamethason nach 8edarf Zyklus Nr. 2 geplant ab: 02.07.2008 112 ELISPOT- Testergebnisse der Kontrollgruppe Patient K01 K02 K03 K04 K05 K06 K07 K08 K09 K10 K11 K12 K13 K14 K15 K16 K17 K18 K19 K20 K21 LW -n LW -A PHA-n 16,00 199,00 135,00 21,00 274,00 259,00 65,00 863,00 435,00 12,00 148,00 448,00 8,00 124,00 184,00 11,00 105,00 489,00 13,00 191,00 399,00 3,00 31,00 41,00 12,00 122,00 332,00 14,00 179,00 294,00 10,00 98,00 404,00 2,00 21,00 41,00 5,00 52,00 132,00 0,00 0,00 145,00 14,00 128,00 246,00 60,00 1.350,00 440,00 5,00 101,00 36,00 4,00 88,00 -2,00 2,00 11,00 34,00 5,00 33,00 31,00 3,00 47,00 113,00 Patient CFP 10-A CEF-n K01 -199,00 -8,00 K02 -3,00 46,00 K03 -863,00 -65,00 K04 -148,00 1,00 K05 -124,00 -6,00 K06 -105,00 -4,00 K07 -191,00 90,00 K08 70,00 11,00 K09 -122,00 6,00 K10 -179,00 -14,00 K11 141,00 11,00 K12 -1,00 -2,00 K13 6,00 -3,00 K14 106,00 16,00 K15 87,00 -1,00 K16 -1.350,00 148,00 K17 -101,00 2,00 K18 62,00 -1,00 K19 38,00 -2,00 K20 345,00 22,00 K21 3,00 46,00 LW: n: A: Cand: Tet: PHA-A ESAT 6-n ESAT 6-A CFP 10-n 3.158,00 -16,00 -199,00 -16,00 4.726,00 -21,00 -274,00 -17,00 4.137,00 -65,00 -863,00 -65,00 4.852,00 -7,00 56,00 -12,00 3.377,00 33,00 1.159,00 -8,00 4.895,00 -11,00 -105,00 -11,00 4.809,00 -13,00 -191,00 -13,00 464,00 2,00 72,00 2,00 4.878,00 -10,00 17,00 -12,00 4.821,00 -14,00 -179,00 -14,00 4.902,00 4,00 545,00 -6,00 912,00 -2,00 -21,00 -1,00 4.948,00 -3,00 7,00 -4,00 2.064,00 3,00 22,00 11,00 3.531,00 1,00 248,00 -8,00 3.650,00 -60,00 -1.350,00 -60,00 863,00 -5,00 -101,00 -5,00 -46,00 -2,00 -18,00 1,00 562,00 -2,00 -11,00 1,00 385,00 -2,00 52,00 34,00 1.753,00 58,00 749,00 -2,00 CEF-A Cand-n Cand-A Tet-n Tet-A 342,00 -13,00 -54,00 -16,00 -199,00 1.977,00 -8,00 119,00 -17,00 13,00 -863,00 -52,00 108,00 -61,00 20,00 275,00 2,00 198,00 3,00 209,00 18,00 2,00 104,00 10,00 242,00 381,00 -1,00 100,00 -7,00 100,00 3.896,00 -7,00 100,00 -9,00 26,00 255,00 12,00 173,00 9,00 226,00 528,00 -8,00 64,00 -6,00 140,00 -179,00 -8,00 8,00 -13,00 21,00 464,00 -2,00 78,00 -8,00 68,00 -21,00 -1,00 11,00 -2,00 -21,00 10,00 -3,00 0,00 -4,00 -4,00 229,00 17,00 154,00 12,00 232,00 299,00 -8,00 31,00 -10,00 56,00 3.650,00 -60,00 1.350,00 -60,00 -1.350,00 266,00 -4,00 39,00 4,00 204,00 1,00 1,00 75,00 2,00 100,00 -11,00 -2,00 -11,00 -2,00 -11,00 669,00 4,00 420,00 4,00 350,00 2.091,00 12,00 327,00 14,00 481,00 Leerwert Spotanzahl Spotdichte Candida Antigen Tetanus Antigen 113 Stimulationsindizes der Kontrollgruppe K.-gruppe SI PHA-n SI PHA-A SI ESAT 6-n SI ESAT 6-A SI CFP 10-n SI CFP 10-A K01 8,44 15,87 -1,00 -1,00 -1,00 -1,00 K02 12,33 17,25 -1,00 -1,00 -0,81 -0,01 K03 6,69 4,79 -1,00 -1,00 -1,00 -1,00 K04 37,33 32,78 -0,58 0,38 -1,00 -1,00 K05 23,00 27,23 4,13 9,35 -1,00 -1,00 K6 44,45 46,62 -1,00 -1,00 -1,00 -1,00 K07 30,69 25,18 -1,00 -1,00 -1,00 -1,00 K08 13,67 14,97 -0,67 2,32 0,67 2,26 K09 27,67 39,98 -0,83 0,14 -1,00 -1,00 K10 21,00 26,93 -1,00 -1,00 -1,00 -1,00 K11 40,40 50,02 0,40 5,56 -0,60 1,44 K12 205,00 43,43 -1,00 -1,00 -0,50 -0,05 K13 26,40 95,15 -0,60 0,13 -0,80 0,12 K14 K15 17,57 27,56 0,07 1,94 -0,57 0,68 K16 7,33 2,70 -1,00 -1,00 -1,00 -1,00 K17 7,20 8,54 -1,00 -1,00 -1,00 -1,00 K18 -0,50 -0,52 -0,50 0,20 0,25 0,70 K19 17,00 51,09 -1,00 -1,00 0,50 3,45 K20 6,20 11,67 -0,40 1,58 6,80 10,45 K21 37,67 37,30 19,33 15,94 -0,67 0,06 K.-gruppe SI CEF-n SI CEF-A SI CAND-n K01 -0,50 1,72 -0,81 K02 2,19 7,22 -0,38 K03 -1,00 -1,00 -0,80 K04 0,08 1,86 0,17 K05 -0,75 0,15 0,25 K6 -0,36 3,62 -0,09 K07 6,92 20,40 -0,54 K08 3,67 8,23 4,00 K09 0,50 4,33 -0,67 K10 -1,00 -1,00 -0,57 K11 1,10 4,73 -0,20 K12 -1,00 -1,00 -0,50 K13 -0,60 0,19 -0,60 K14 K15 -0,07 2,34 -0,57 K16 2,47 2,70 -1,00 K17 0,40 2,63 -0,80 K18 -0,25 0,01 0,25 K19 -1,00 -1,00 -1,00 K20 4,40 20,27 0,80 K21 15,33 44,49 4,00 LW: SI: n: A: Cand: leeres Feld: SI CAND-A -0,27 0,43 0,13 1,34 0,84 0,95 0,52 5,58 0,52 0,05 0,80 0,52 0,00 SI TET-n -1,00 -0,81 -0,94 0,25 1,25 -0,64 -0,69 3,00 -0,50 -0,93 -0,80 -1,00 -0,80 SI TET-A -1,00 0,05 0,02 1,41 1,95 0,95 0,14 7,29 1,15 0,12 0,70 -1,00 -0,08 0,24 -1,00 0,39 0,85 -1,00 12,73 6,96 -0,71 -1,00 0,80 0,50 -1,00 0,80 4,67 0,44 -1,00 2,02 1,14 -1,00 10,62 10,23 Leerwert Stimulationsindex Spotanzahl Spotdichte Candida Antigen Tet: Tetanus Antigen keine Werte messbar 114 ELISPOT- Testergebnisse der Tumorpatienten vor Chemotherapie Patient P01-1 P02-1 P03-1 P05-1 P06-1 P07-1 P08-1 P09-1 P10-1 P11-1 P12-1 P13-1 P14-1 P15-1 P18-1 P19-1 LW -n LW -A PHA-n 30,00 278,00 470,00 13,00 105,00 296,00 0,00 0,00 1,00 37,00 486,00 463,00 10,00 109,00 490,00 16,50 125,50 410,50 16,00 269,00 484,00 72,00 994,00 381,00 3,00 32,00 51,00 2,00 11,00 494,00 5,00 100,00 -4,00 4,00 35,00 496,00 16,00 333,00 484,00 24,00 504,00 311,00 1,00 16,00 -1,00 81,00 1.307,00 422,00 Patient P01-1 P02-1 P03-1 P05-1 P06-1 P07-1 P08-1 P09-1 P10-1 P11-1 P12-1 P13-1 P14-1 P15-1 P18-1 P19-1 CEF-n 276,00 -13,00 0,00 -13,00 630,00 16,00 -10,00 264,00 248,00 54,00 -5,00 0,00 82,00 46,00 0,00 139,00 LW: n: A: Cand: Tet: Leerwert Spotanzahl Spotdichte Candida Antigen Tetanus Antigen PHA-A ESAT 6-n ESAT 6-A CFP 10-n CFP 10-A 4.722,00 -25,00 188,00 -16,00 184,00 4.507,00 -12,00 43,00 -13,00 -105,00 27,00 17,00 405,00 2,00 35,00 4.514,00 53,00 4.514,00 119,00 4.514,00 4.891,00 31,00 1.806,00 63,00 2.049,00 4.874,50 -16,00 -24,50 4,00 259,50 4.731,00 -3,00 234,00 -8,00 295,00 4.006,00 -47,00 1.246,00 -72,00 -994,00 346,00 9,00 156,00 -1,00 69,00 4.989,00 -2,00 -11,00 37,00 584,00 -16,00 -5,00 -100,00 -3,00 -8,00 4.965,00 0,00 131,00 5,00 174,00 4.667,00 -16,00 -333,00 -16,00 -333,00 4.496,00 -24,00 -504,00 -19,00 -73,00 -16,00 -1,00 -16,00 0,00 11,00 8.955,00 85,00 2.569,00 100,00 2.964,00 CEF-A Cand-n Cand-A Tet-n Tet-A 4.722,00 -21,00 95,00 -12,00 268,00 -105,00 -9,00 46,00 1,00 147,00 0,00 2,00 72,00 0,00 0,00 -184,00 4,00 758,00 3,00 505,00 4.891,00 -9,00 86,00 4,00 545,00 485,00 -1,00 181,50 8,50 290,50 121,00 -12,00 127,00 -14,00 269,00 4.006,00 -41,00 742,00 -57,00 506,00 4.968,00 10,00 202,00 3,00 86,00 1.224,00 0,00 1,00 11,00 181,00 -100,00 -5,00 -100,00 -5,00 -100,00 33,00 4,00 347,00 5,00 205,00 3.340,00 -16,00 -333,00 -16,00 -333,00 3.075,00 -6,00 360,00 -14,00 227,00 -14,00 2,00 144,00 0,00 48,00 3.726,00 89,00 2.350,00 25,00 487,00 115 ELISPOT-Testergebnisse der Tumorpatienten nach Chemotherapie Patient LW -n LW -A PHA-n PHA-A ESAT 6-n ESAT 6-A CFP 10-n CFP 10-A P01-2 P02-2 P03-2 66,00 575,00 434,00 4.425,00 -42,00 174,00 -42,00 251,00 P04-2 20,00 348,00 255,00 4.652,00 -20,00 -348,00 -20,00 -348,00 P05-2 P06-2 0,00 0,00 0,00 0,00 1,00 237,00 1,00 4,00 P07-2 P08-2 5,00 28,00 73,00 657,00 -5,00 -28,00 -5,00 -28,00 P09-2 40,00 383,00 369,00 4.617,00 -13,00 1.907,00 -40,00 -383,00 P10-2 70,00 1.392,00 430,00 3.608,00 6,00 3.608,00 -48,00 312,00 P11-2 94,00 1.429,00 414,00 8.794,50 59,00 1.430,50 98,50 4.249,00 P12-2 P13-2 53,00 823,00 357,50 6.268,50 56,50 547,50 61,00 761,00 P14-2 100,00 1.921,00 99,00 2.912,00 56,00 1.016,00 -64,00 -1.426,00 P15-2 4,00 75,00 232,00 4.375,00 19,00 226,00 33,00 812,00 P18-2 P19-2 0,00 0,00 0,00 0,00 2,00 23,00 0,00 0,00 Patient P01-2 P02-2 P03-2 P04-2 P05-2 P06-2 P07-2 P08-2 P09-2 P10-2 P11-2 P12-2 P13-2 P14-2 P15-2 P18-2 P19-2 CEF-n CEF-A Cand-n Cand-A Tet-n 43,00 4.425,00 -44,00 -2,00 725,00 -19,00 0,00 0,00 1,00 Tet-A 180,00 1,00 -66,00 -20,00 -575,00 -348,00 11,00 2,00 22,00 7,00 129,00 -4,00 24,00 76,00 3.431,00 -40,00 -383,00 253,00 3.608,00 -59,00 629,00 34,00 1.622,00 -23,00 -379,50 1,00 93,00 -40,00 -383,00 -70,00 -1.392,00 2,50 114,00 264,50 8.803,50 -17,00 -362,00 18,00 328,00 -17,00 -276,00 -62,00 -1.385,00 -1,00 -52,00 2,00 LW: n: A: Cand: leeres Feld: 17,00 38,00 343,00 1,00 -209,00 12,00 207,00 1,00 4,00 0,00 0,00 Leerwert Spotanzahl Spotdichte Candida Antigen Tet: Tetanus Antigen keine Werte messbar 116 Stimulationsindizes der Tumorpatienten vor Chemotherapie Patient P01-1 P02-1 P03-1 P04-1 P05-1 P06-1 P07-1 P08-1 P09-1 P10-1 P11-1 P12-1 P13-1 P14-1 P15-1 P18-1 P19-1 Patient P01-1 P02-1 P03-1 P04-1 P05-1 P06-1 P07-1 P08-1 P09-1 P10-1 P11-1 P12-1 P13-1 P14-1 P15-1 P18-1 P19-1 LW: SI: n: A: Cand: leeres Feld: SI PHA-n SI PHA-A SI ESAT 6-n SI ESAT 6-A SI CFP 10-n SI CFP 10-A 15,67 16,99 -0,83 0,68 -0,53 0,66 22,77 42,92 -0,92 0,41 -1,00 -1,00 12,51 49,00 39,31 30,25 5,29 17,00 247,00 -0,80 124,00 30,25 12,96 -1,00 5,21 9,29 44,87 74,90 17,59 4,03 10,81 453,55 -0,16 141,86 14,02 8,92 -1,00 6,85 SI CEF-n SI CEF-A 9,20 16,99 -1,00 -1,00 -0,35 63,00 1,63 -0,63 3,67 82,67 27,00 -1,00 0,00 5,13 1,92 0,00 1,72 -0,38 44,87 5,95 0,45 4,03 155,25 111,27 -1,00 0,94 10,03 6,10 -0,88 2,85 1,43 3,10 -0,92 -0,19 -0,65 3,00 -1,00 -1,00 0,00 -1,00 -1,00 -1,00 1,05 9,29 16,57 1,59 0,87 1,25 4,88 -1,00 -1,00 3,74 -1,00 -1,00 -1,00 1,97 3,22 6,30 -0,24 -0,50 -1,00 -0,33 18,50 -0,60 1,25 -1,00 -0,79 0,00 1,23 9,29 18,80 0,82 1,10 -1,00 2,16 53,09 -0,08 4,97 -1,00 -0,14 0,69 2,27 SI Cand-n -0,70 -0,69 SI Cand-A 0,34 0,44 SI Tet-n -0,40 0,08 SI Tet-A 0,96 1,40 0,11 -0,90 1,06 -0,75 -0,57 3,33 0,00 -1,00 1,00 -1,00 -0,25 2,00 1,10 1,56 0,79 3,45 0,47 0,75 6,31 0,09 -1,00 9,91 -1,00 0,71 9,00 1,80 0,08 0,40 1,16 -0,88 -0,79 1,00 5,50 -1,00 1,25 -1,00 -0,58 0,00 0,31 1,04 5,00 4,08 1,00 0,51 2,69 16,45 -1,00 5,86 -1,00 0,45 3,00 0,37 Leerwert Stimulationsindex Spotanzahl Spotdichte Candida Antigen Tet: Tetanus Antigen keine Werte messber 117 Stimulationsindizes der Tumorpatienten nach Chemotherapie Patient P01-2 P02-2 P03-2 P04-2 P05-2 P06-2 P07-2 P08-2 P09-2 P10-2 P11-2 P12-2 P13-2 P14-2 P15-2 P18-2 P19-2 Patient P01-2 P02-2 P03-2 P04-2 P05-2 P06-2 P07-2 P08-2 P09-2 P10-2 P11-2 P12-2 P13-2 P14-2 P15-2 P18-2 P19-2 LW: SI: n: A: Cand: Tet: SI PHA-n SI PHA-A SI ESAT 6-n SI ESAT 6-A SI CFP 10-n SI CFP 10-A 6,57 12,75 7,70 13,37 -0,64 -1,00 0,30 -1,00 -0,64 -1,00 0,44 -1,00 14,60 9,23 6,14 4,44 23,46 12,05 2,59 6,64 -1,00 -0,33 0,09 0,63 -1,00 4,98 2,59 1,04 -1,00 -1,00 -0,69 1,05 -1,00 -1,00 0,22 3,00 7,26 0,99 58,00 8,05 1,52 58,33 1,07 0,56 4,75 0,68 0,53 3,01 1,21 -0,64 8,25 0,93 -0,74 10,83 SI Cand-n SI Cand-A SI Tet-n SI Tet-A SI CEF-n SI CEF-A 0,65 -0,10 7,70 2,08 -0,67 -0,95 0,31 0,00 -1,00 -1,00 -1,00 -1,00 1,40 1,90 3,61 0,37 4,61 8,96 2,59 1,24 -0,80 -1,00 -0,84 -0,24 0,86 -1,00 0,45 -0,22 0,20 -1,00 -1,00 0,03 3,32 -1,00 -1,00 0,11 5,23 -0,17 4,50 11,28 -0,19 4,37 0,65 0,01 3,00 0,34 -0,11 2,76 -0,35 -0,62 -0,25 -0,38 -0,72 -0,69 Leerwert Stimulationsindex Spotanzahl Spotdichte Candida Antigen Tetanus Antigen 118 Erklärung über die eigenständige Abfassung der Arbeit Hiermit erkläre ich, dass ich die vorliegende Arbeit selbstständig und ohne unzulässige Hilfe oder Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe. Ich versichere, dass Dritte von mir weder unmittelbar noch mittelbar geldwerte Leistungen für Arbeiten erhalten haben, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen, und dass die vorgelegte Arbeit weder im Inland noch im Ausland in gleicher oder ähnlicher Form einer anderen Prüfungsbehörde zum Zweck einer Promotion oder eines anderen Prüfungsverfahrens vorgelegt wurde. Alles aus anderen Quellen und von anderen Personen übernommene Material, das in der Arbeit verwendet wurde oder auf das direkt Bezug genommen wird, wurde als solches kenntlich gemacht. Insbesondere wurden alle Personen genannt, die direkt an der Entstehung der vorliegenden Arbeit beteiligt waren. ................................. .................................... Datum Unterschrift 119 120 121 DANKSAGUNG Hiermit möchte ich mich herzlichst bei allen bedanken, die zum Entstehen dieser Dissertation beigetragen haben. Ich danke Herrn Prof. Dr. med. Frank Emmrich für die Möglichkeit, meine Dissertation am Institut für klinische Immunologie der Universität Leipzig durchführen zu können. Mein herzlicher Dank gilt Herrn Prof. Dr. med. Ulrich Sack für die Betreuung meiner Arbeit, die unkomplizierte Zusammenarbeit sowie die fachliche und persönliche Unterstützung. Besonders bedanken möchte ich allen ProbandInnen und PatientInnen des Klinikums Altenburger Land danken, die durch Ihre freiwillige Teilnahme diese Studie erst ermöglicht haben. Vor allem aber werden mir die onkologischen PatientInnen mit ihrem Leid und ihrer Kraft unvergessen bleiben. Ebenso bedanke ich mich bei Herrn Chefarzt Dr. med. Armin Schulz-Abelius für die Möglichkeit, Patienten seiner Klinik in die Studie einschließen zu können und die Bereitstellung der benötigten Patientendaten. Ich danke vor allem bei meinen Kollegen, Pfleger Jens Zwoch und allen Schwestern der Station 31 und der onkologischen Ambulanz des Klinikums Altenburger Land für ihre ausdauernde Unterstützung bei der Suche nach neuen Patienten, den Blutentnahmen und die Organisation des zeitnahen Transportes der Proben. Auch dem Medizinischen Zentrallabor Altenburg danke ich für die Bereitstellung der Blutröhrchen und die Organisation des Transportes durch dessen Kurierdienst. 122 Ein großes Dankeschön gilt auch den Mitarbeiterinnen und Mitarbeitern des Institutes für Klinische Immunologie der Universität Leipzig für ihre Hilfsbereitschaft und Unterstützung. Nicht zuletzt möchte ich meinen Eltern danken. Ihr habt mir immer den nötigen Rückhalt und Unterstützung geboten und das Gefühl gegeben, dass ich auf Euch zählen kann. Ein riesiges Dankeschön geht an Thomas Kirsche. Du hast meiner Arbeit mit Deinem technischen Know-how den letzten Schliff gegeben und warst mir bis zum letzten Tag eine große Hilfe. Mein besonderer Dank gilt Bart Fosselle. Danke, dass Du an mich geglaubt und mich auch in schwierigen Phasen immer wieder motiviert hast. Dir ist diese Arbeit gewidmet. 123