Hauttumoren im Kindesalter
Werbung

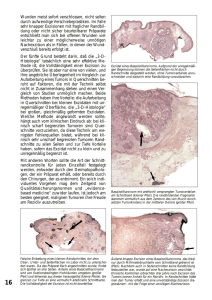
MEDIZIN ÜBERSICHTSARBEIT Hauttumoren im Kindesalter Henning Hamm, Peter H. Höger ZUSAMMENFASSUNG Hintergrund: Dermatologen, Pädiater und Allgemeinmediziner werden häufig von besorgten Eltern wegen eines an der Haut ihres Kindes entstandenen Tumors konsultiert. Methoden: Selektive Literaturrecherche Ergebnisse: Lediglich 1 bis 2 Prozent der bei Kindern exzidierten Hauttumoren stellen sich histologisch als maligne heraus. Anamnestisch-klinische Warnzeichen bestehen in raschem Wachstum, derber Konsistenz, Größe über 3 cm, Ulzeration, fehlender Verschiebbarkeit und Manifestation im Neugeborenenalter. Die häufigsten malignen Hauttumoren des Erwachsenen – Basalzellkarzinom, kutanes Stachelzellkarzinom und Melanom – kommen im Kindesalter sehr selten vor. Kongenitale melanozytäre Nävi und Naevi sebacei bergen ein geringeres malignes Potenzial als früher angenommen, ihre Exzision ist dennoch häufig sinnvoll. Der Spitz-Nävus kann ein Melanom klinisch und histologisch imitieren. Manche benigne Hauttumoren bilden sich über mehrere Jahre spontan zurück, können aber in besonderen Lokalisationen und bei Multiplizität mit Komplikationen einhergehen. Für Hämangiome, bei denen aufgrund drohender Obstruktion und Ulzeration eine Systemtherapie indiziert ist, scheint Propranolol ein wesentlich günstigeres Nutzen-Risiko-Verhältnis aufzuweisen als Kortikosteroide. Schlussfolgerung: Die Entscheidung, ob ein kindlicher Hauttumor einer Exzision, einer anderen Therapie oder einer weitergehenden Diagnostik bedarf, oder ob es sich um einen harmlosen Befund mit Aussicht auf Spontanremission handelt, setzt ein hohes Maß an Sachkenntnis voraus. ►Zitierweise Hamm H, Höger PH: Skin tumors in childhood. Dtsch Arztebl Int 2011; 108(20): 347–53. DOI: 10.3238/arztebl.2011.0347 in Schwerpunkt der heutigen Dermatologie besteht in der Früherkennung und – zumeist operativen – Behandlung maligner Hauttumoren und deren Vorstufen. Bei Erwachsenen sind die mit Abstand häufigsten Hautkrebserkrankungen mit weiterhin steigender Inzidenz das Melanom, Basalzellkarzinom und Stachelzellkarzinom (1). Dagegen stellt sich die Situation im Kindesalter gänzlich anders dar: Der pädiatrische Dermatologe wird mit einer Vielzahl überwiegend benigner Hauttumoren konfrontiert, die möglicherweise dennoch einer Therapie bedürfen, allerdings seltener einer Exzision. In dieser Übersicht wird kurz auf die Rolle eingegangen, die die wichtigsten malignen kutanen Neoplasien des Erwachsenenalters bei Kindern spielen. Nachfolgend werden drei Nävusarten besprochen, die für die Entwicklung maligner Hauttumoren bedeutsam sind beziehungsweise die wichtigste Differenzialdiagnose des kindlichen Melanoms darstellen. Im dritten Teil werden vier typische Hauttumoren des ersten Lebensjahrzehnts thematisiert. Hierzu wurde Literatur herangezogen, die mit Hilfe von PubMed, Medline und PubMed Central ermittelt wurde. Die besprochenen Tumoren wurden aufgrund ihrer hohen Bedeutung in der täglichen klinischen Arbeit der Autoren ausgewählt. E Maligne Hauttumoren im Kindesalter Bei lediglich 1 bis 2 Prozent aller bei Säuglingen und Kindern exzidierten Hauttumoren handelt es sich um maligne Befunde. Zu ihnen zählen: ● Fibrosarkom ● Rhabdomyosarkom ● Angiosarkom ● Neuroblastom ● maligner peripherer Nervenscheidentumor ● kutanes T-Zell- und andere Lymphome sowie die als semimaligne einzustufenden ● infantilen Fibromatosen ● Hämangioendotheliom ● büschelförmiges Angiom ● Dermatofibrosarcoma protuberans. Klinik und Poliklinik für Dermatologie, Venerologie und Allergologie, Universitätsklinikum Würzburg: Prof. Dr. med. Hamm Abteilung Pädiatrische Dermatologie, Katholisches Kinderkrankenhaus Wilhelmstift Hamburg: Prof. Dr. med. Höger Deutsches Ärzteblatt | Jg. 108 | Heft 20 | 20. Mai 2011 Als Warnzeichen gelten rasches Wachstum, Ulzeration, Fixierung oder tiefe Lokalisation auf der Faszie, derbe Konsistenz, Größe über 3 cm und Manifestation im Neugeborenenalter (2). 347 MEDIZIN KASTEN 1 Risikofaktoren für die Entwicklung von Melanomen (3) ● Hellhäutigkeit, rote und blonde Haare, helle Augenfarbe ● Sommersprossen, aktinische Lentigines ● Neigung zu Sonnenbränden bei Exposition gegenüber UV-Licht ● intermittierende intensive Exposition gegenüber UV-Licht ● hohe Zahl gewöhnlicher melanozytärer Nävi ● mehrere atypische melanozytäre Nävi ● kongenitaler melanozytärer Nävus, insbesondere ein riesiger ● Melanome in der Familie ● Störungen der DNA-Reparatur, insbesondere Xeroderma pigmentosum ● Immunsuppression ● frühere maligne Erkrankung KASTEN 2 Risikofaktoren für die Entwicklung von malignen epithelialen Hauttumoren (6) ● Hellhäutigkeit ● UV-Licht ● ionisierende Strahlen ● prädisponierende Genodermatosen: – zum Beispiel Basalzellnävus-Syndrom beim Basalzellkarzinom, – Epidermodysplasia verruciformis beim Stachelzellkarzinom, – Xeroderma pigmentosum bei beiden Tumoren ● Naevus sebaceus ● langfristige Immunsuppression, zum Beispiel nach Organtransplantation ● humane High-risk-Papillomviren ● chemische Karzinogene, zum Beispiel anorganisches Arsen ● chronische Entzündungen, Ulzerationen, Narben und Sklerosen 348 Melanom Die zunehmende Melanominzidenz betrifft das Kindesalter erst ab der Pubertät. 1 bis 4 Prozent aller Melanome entstehen bei unter 20-Jährigen, nur 0,3 bis 0,4 Prozent aller Melanome vor der Pubertät. Abgesehen von den großen kongenitalen melanozytären Nävi führen die meisten der in Kasten 1 zusammengestellten prädisponierenden Faktoren erst im Erwachsenenalter zum Melanom, sind aber schon im Kindesalter vorhanden, so dass Präventionsmaßnahmen unbedingt schon sehr früh beginnen sollten. Auch bei Kindern entstehen die meisten Melanome de novo, im Vergleich zum Erwachsenenalter werden aber mehr untypische, amelanotische und noduläre Melanome beobachtet (4). In der erschwerten Diagnosestellung liegt ein möglicher Grund dafür, dass Melanome bei Kindern zum Zeitpunkt der Exzision eine höhere durchschnittliche Tumordicke aufweisen, ein anderer in der größeren Zurückhaltung, diese seltene Verdachtsdiagnose bei einem Kind zu stellen und eine diagnostische Exzision vorzunehmen. Melanomsimulatoren sind insbesondere der Spitz-Nävus und atypische melanozytäre Nävi. Das therapeutische Vorgehen beim kindlichen Melanom entspricht dem im Erwachsenenalter, einschließlich der heute routinemäßig durchgeführten Sentinel-Lymphknotenbiopsie ab 1 mm Tumordicke. Obwohl positive Sentinel-Lymphknoten-Befunde bei Kindern häufiger sind, unterscheidet sich die Prognose quoad vitam kaum von derjenigen erwachsener Patienten mit gleicher Tumordicke. Die 5-JahresÜberlebensrate bei pädiatrischen Melanomen allgemein wurde mit 74 bis 80 Prozent angegeben (5). Maligne epitheliale Hauttumoren Auch für das semimaligne Basalzellkarzinom (BCC) und das kutane Stachelzellkarzinom (SCC) sind genetische und andere frühzeitig relevante Risikofaktoren bekannt (Kasten 2). Die bedeutendsten Risikofaktoren für beide Krebsarten sind Hellhäutigkeit und UV-Licht-Exposition, auch durch künstliche UV-Quellen, wobei eine intermittierend starke Besonnung mit Auftreten von Sonnenbränden für das BCC und die kumulative Lebenszeit-Exposition für das SCC maßgeblich sind (1). In Abwesenheit zusätzlicher fördernder Umstände wirken sich diese allerdings erst nach jahrzehntelanger Latenz aus. Bereits im Kindesalter auftretende, maligne epitheliale Hauttumoren werden am ehesten auf dem Boden prädisponierender Genodermatosen beobachtet (7). Nävi mit Beziehung zu malignen Hauttumoren Kongenitale melanozytäre Nävi Seit über einem Jahrhundert ist das maligne Potenzial kongenitaler melanozytärer Nävi (KMN) bekannt. Immer klarer wird jedoch, dass die Dimension des Entartungsrisikos fallbezogen äußerst unterschiedlich ist und unter anderem von der Nävusgröße abDeutsches Ärzteblatt | Jg. 108 | Heft 20 | 20. Mai 2011 MEDIZIN hängt. KMN werden daher nach dem größten Durchmesser, den sie voraussichtlich im Erwachsenenalter erreichen, klassifiziert (Tabelle 1). Das Entartungsrisiko kleiner und mittelgroßer KMN ist gering. Melanome entwickeln sich auf ihnen praktisch nie vor der Pubertät und entstehen in der Epidermis, sind also einer Früherkennung zugänglich. In die Indikation zur operativen Entfernung sind aber auch psychosoziale und kosmetische Aspekte einzubeziehen. Deswegen, aber auch aufgrund erleichterter Exzidierbarkeit, ist die – gegebenenfalls serielle – Exzision größerer Nävi oft bereits im Kleinkindesalter angezeigt. Die Problematik bei großen KMN (Abbildung 1) und vor allem Riesennävi ist vielschichtiger (8): ● Vom Nävus ausgehende, kutane Melanome, selten auch andere maligne Tumoren bilden sich in 70 Prozent schon im ersten Lebensjahrzehnt und entstehen oft in tieferen Schichten des Nävus, so dass sie erst später bemerkt werden können. ● Melanome können nicht nur auf dem Nävus entstehen, sondern auch an anderen Stellen, zum Beispiel im Zentralnervensystem. ● Vor allem riesige und große KMN können mit einer neurokutanen Melanose assoziiert sein, einer leptomeningealen Aussaat von Nävuszellen (9). Wichtigster Risikofaktor hierfür ist eine hohe Zahl sogenannter Satellitennävi. Außerdem kommen gehäuft Fehlbildungen des Zentralnervensystems vor. Bei Riesennävi, die die dorsale mediane Körperachse überschreiten und/oder multiple Satelliten aufweisen, wird daher eine kraniale und spinale Kernspintomographie in den ersten vier bis sechs Lebensmonaten empfohlen (10). Eine symptomatische neurokutane Melanose manifestiert sich typischerweise in den ersten zwei bis drei Lebensjahren mit Zeichen des Hirndrucks oder der spinalen Kompression. Die Prognose ist schlecht. Nicht alle Patienten mit einem positiven Befund in der Bildgebung entwickeln aber neurologische Symptome. Die Gefahr, dass eine dieser Komplikationen auftritt, lässt sich für große und riesige KMN auf insgesamt etwa 5 bis 15 Prozent schätzen und ist in den ersten fünf bis zehn Lebensjahren am höchsten. Der therapeutische Umgang mit KMN ist Gegenstand anhaltender Diskussionen, zumal nur das Risiko der Tumorentstehung auf dem kutanen Nävus verringert werden kann. Wenn keine neurokutane Melanose vorliegt, wird von den meisten Experten zu einer frühzeitigen und möglichst vollständigen Exzision großer und riesiger KMN oder zumindest zur Entfernung besonders auffälliger oder schwer kontrollierbarer Areale gegen Ende des ersten Lebensjahres geraten. Unabhängig von der Therapie sind in diesen Fällen zunächst vierteljährliche Kontrolluntersuchungen zu empfehlen. Oberflächlich-ablative Verfahren wie die Dermabrasion dienen primär dem Ziel einer kosmetischen Deutsches Ärzteblatt | Jg. 108 | Heft 20 | 20. Mai 2011 TABELLE 1 Gängige Klassifikation kongenitaler melanozytärer Nävi (KMN) (8) Bezeichnung Größe*1 Inzidenz klein < 1,5 cm etwa 1 : 100 mittelgroß 1,5–19,9 cm etwa 1 : 1 000 groß ≥ 20 cm etwa 1 : 20 000 riesig > 40–50 cm*2 etwa 1 : 500 000 *1 Gemeint ist der größte Durchmesser, der im Erwachsenenalter erreicht wird. Der größte Durchmesser von KMN nimmt vom Neugeborenen- zum Erwachsenenalter am Kopf um den Faktor 1,7 zu, an den Beinen um den Faktor 3,3, und an Rumpf, Armen und Füßen um den Faktor 2,8 (8). *2 kein allgemeiner Konsens Abbildung 1: Großer kongenitaler melanozytärer Nävus bei einem Neugeborenen Verbesserung und sind allenfalls in den ersten Lebensmonaten bei nicht exzidierbaren Befunden sinnvoll. Naturgemäß werden hiermit die tieferen Nävuszellschichten nicht erreicht; auch die nävustypische Hypertrichose wird dadurch nicht verhindert. Eine Lasertherapie sollte nach gegenwärtigem Empfehlungsstand nur in besonders gelagerten Fällen und speziellen Lokalisationen (zum Beispiel Gesicht) in Betracht gezogen werden. Spitz-Nävus Der Spitz-Nävus wird histologisch bei 1 bis 2 Prozent aller exzidierten melanozytären Läsionen diagnostiziert. Seine klinische Variationsbreite reicht von einer kuppelförmigen, hautfarbenen oder rötlichen Papel im Gesicht eines Kleinkindes bis zu einer braun-schwarzen Plaque an der proximalen Extremität eines Jugendlichen. Typisch für Spitz-Nävi und differenzialdiagnostisch bedeutsam ist ihr initial oft rasches Wachstum mit anschließender Persistenz. Der Spitz-Nävus ist weder ein Melanomvorläufer noch ein Melanom, aber der wichtigste, sowohl klinische als auch histologische Melanomsimulator, wie seine ursprüngliche Beschreibung als „juveniles Melanom“ verrät. Die Dignität mancher Hauttumoren mit Spitz-Nävus-ähnlicher Histologie lässt sich selbst von erfahrenen Dermatohistologen trotz Zuhilfenahme immunhistologischer Techniken nicht 349 MEDIZIN apokrine und ekkrine Schweißdrüsentumore, vornehmlich das (pigmentierte) Trichoblastom (Abbildung 2) und das Syringocystadenoma papilliferum. Deutlich seltener als früher angenommen – in retrospektiven Studien in 0 bis 3,5 Prozent der Fälle – entstehen BCCs auf dem Nävus, ausnahmsweise auch Stachelzell- und Adnexkarzinome. Bei Kindern sind nur rund 15 Fälle von BCC auf Naevus sebaceus dokumentiert (13), so dass heute nach Meinung der meisten Autoren keine Indikation zur Entfernung eines Naevus sebaceus im Kindesalter aus prophylaktischen Gründen besteht. Eine frühzeitige Entfernung ist jedoch wegen der erleichterten Exzidierbarkeit (Kapillitium) und aus kosmetischen Gründen (Gesicht) vorteilhaft (14). Methode der Wahl ist die Exzision. Abbildung 2: Pigmentiertes Trichoblastom auf einem Naevus sebaceus Abbildung 3: Hämangiom Auswahl häufiger benigner Hauttumoren immer sicher einordnen („atypischer Spitz-Tumor“). In diesen Fällen haben sich komparative genomische Hybridisierung beziehungsweise Fluoreszenz-in-situ-Hybridisierung als hilfreiche Differenzierungsmethoden erwiesen, sind allerdings bislang nicht routinemäßig verfügbar (11). Im Zweifelsfall sollte therapeutisch wie bei einem gesicherten Melanom verfahren werden. Abhängig von der Tumordicke beinhaltet dies Stagingmaßnahmen und die Sentinel-Lymphknoten-Exstirpation, die bei spitzoiden melanozytären Tumoren in bis zu 50 Prozent der Fälle positiv ausfällt. Der Nachweis von Melanozyten im Lymphknoten ist jedoch nicht gleichbedeutend mit einer Metastase (12). Naevus sebaceus Mit einer Prävalenz von 0,3 Prozent ist der Naevus sebaceus der häufigste organoide Epidermalnävus. „Organoid“ bedeutet, dass mehr als eine Gewebsstruktur, in diesem Fall vor allem Talgdrüsen und Schweißdrüsen, an der Fehlbildung beteiligt ist. Mehr als 90 Prozent der Naevi sebacei sind am Kopf lokalisiert, überwiegend am Kapillitium. An dieser Stelle ist die mit dem Nävus verbundene Alopezie besonders störend. In 10 bis 30 Prozent der Fälle entwickeln sich – zumeist erst im Erwachsenenalter – auf dem Nävus Hauttumore. Größtenteils handelt es sich dabei um benigne Haarfollikel-, Talgdrüsen-, 350 Hämangiom Mit einer Prävalenz von 3 bis 5 Prozent bei Säuglingen stellen Hämangiome die häufigsten Tumoren im Kindesalter dar. Mädchen sind zwei- bis dreimal häufiger betroffen als Jungen, Frühgeborene weisen ein bis zu zehnfach erhöhtes Risiko für Hämangiome auf. Den meisten infantilen Hämangiomen liegt eine Überaktivität des vaskulären endothelialen Wachstumsfaktors zugrunde (15), die durch peripartale Hypoxie getriggert wird. Bei diffusen Hämangiomatosen spielen von plazentaren Hämangiomen (Chorangiomen) ausgehende Endothelzellemboli eine ursächliche Rolle (16). Im differenzialdiagnostisch bedeutsamen Unterschied zu anderen Gefäßtumoren exprimieren „echte“ Hämangiome das immunhistologisch nachweisbare Glukose-Transport-1-Protein (GLUT-1). Infantile Hämangiome weisen eine charakteristische Wachstumsdynamik auf, die sie leicht von vaskulären Fehlbildungen unterscheiden lässt: Kurz nach Geburt entwickelt sich aus einer anfangs weißlichen oder hellroten Macula (Differenzialdiagnose: Gefäßnävus) ein proliferierender, praller, je nach Lage kräftig roter (intrakutan) oder blass-livider (subkutan) Knoten (Abbildung 3). Die Proliferationsphase dauert gewöhnlich sechs bis neun Monate; sie wird gefolgt von einem Wachstumsstopp, bevor im Alter von etwa 12 bis 14 Monaten bei 80 bis 90 Prozent der Hämangiome die spontane Regression einsetzt und sich über mehrere Jahre erstreckt. Die Mehrzahl der Hämangiome bedarf daher keiner Behandlung. Bei drohender Obstruktion (insbesondere periokulär, para- oder intratracheal), Ulzeration (vor allem intertriginös) oder sehr großen Hämangiomen (Gefahr der Herzinsuffizienz, selten auch der Hypothyreose) besteht jedoch eine dringende, absolute Behandlungsindikation, bei Lokalisation im Gesicht ohne Obstruktion aufgrund „kosmetischer“ Beeinträchtigung und zu befürchtender, späterer psychosozialer Stigmatisierung eine relative (Tabelle 2). Als einfach durchführbare und wiederholbare Methode zur Frühtherapie proliferierender Hämangiome Deutsches Ärzteblatt | Jg. 108 | Heft 20 | 20. Mai 2011 MEDIZIN bis zu 1 cm Durchmesser und 3 bis 4 mm Dicke hat sich die Kontaktkryotherapie bewährt. Bei Verwendung von Geräten, bei denen der metallene Applikator elektrisch auf lediglich –32° C gekühlt wird, kommen unerwünschte Wirkungen wie Hypopigmentierung, Blasenbildung und Ulzeration mit nachfolgender Narbenbildung wesentlich seltener vor als bei mit flüssigem Stickstoff gekühlten Metallstäben. Seit der überraschenden Entdeckung des frappierenden Effekts des nichtselektiven Betablockers Propranolol auf proliferierende Hämangiome (17) setzt sich diese Behandlung weltweit bei oben genannten absoluten Indikationen mehr und mehr durch und ersetzt die bislang übliche, hochdosierte Kortikosteroidtherapie. Randomisierte, kontrollierte Studien – und daher auch die arzneirechtliche Zulassung – liegen für dieses Therapieverfahren jedoch noch nicht vor. Schon deswegen sollte die Therapie stationär eingeleitet werden. Aufgrund des offenkundig günstigen Nutzen-Risiko-Profils von Propranolol wurde die entsprechende deutsche Leitlinie der AWMF (18) kürzlich überarbeitet. Auf mögliche Nebenwirkungen wie Hypoglykämie, seltener Hypotension und Bradyarrhythmien, ist unbedingt zu achten. Juveniles Xanthogranulom Das juvenile Xanthogranulom (JXG) stellt die häufigste Form der Nicht-Langerhanszell-Histiozytosen dar. Es handelt sich um gutartige, meist solitäre, derbe, anfangs rötliche, dann gelblich-orangefarbene Papeln und Knoten von 0,5–2 cm Durchmesser, die vorwiegend an Kopf, Hals und Oberkörper lokalisiert sind. Sie werden am häufigsten bei Neugeborenen (5–17 Prozent), Säuglingen und Kleinkindern, nur ausnahmsweise bei älteren Kindern oder Erwachsenen beobachtet. Selten können sie sich auch extrakutan manifestieren: Bei augennahen oder multiplen JXG besteht das Risiko einer Beteiligung der vorderen Augenabschnitte, daher ist in diesen Fällen eine ophthalmologische Untersuchung indiziert. Die Liste weiterer, möglicherweise befallener Organe ist lang: ZNS, Muskulatur, Knochen, Lungen, Leber, Milz, Nieren, Nebennieren, Perikard, Testes und Larynx können betroffen sein (19). Überdurchschnittlich häufig ist die Assoziation mit der Langerhanszell-Histiozytose (20) beziehungsweise mit Neurofibromatose Typ 1 und juveniler MyelomonozytenLeukämie (21). Die Mehrzahl der JXG regrediert spontan im Verlauf der ersten vier bis sechs Lebensjahre und bedarf daher keiner Behandlung. Mastozytom Mastozytome treten entweder solitär oder im Rahmen von Mastozytosen auf, einer heterogenen Gruppe von Erkrankungen, die durch eine Mastzellvermehrung in der Haut, bei einigen – vorwiegend im Erwachsenenalter vorkommenden – Formen auch in Knochenmark, lymphatischen Organen und anderen Organsystemen gekennzeichnet ist. Im Kindesalter werden Deutsches Ärzteblatt | Jg. 108 | Heft 20 | 20. Mai 2011 TABELLE 2 Behandlungsindikation und -optionen für die Therapie infantiler Hämangiome (18) Lokalisation bzw. Typ des Hämangioms Methode der Wahl Alternativen 1. Absolute Behandlungsindikation Drohende Obstruktion (z. B. Auge, Nase, Trachea) Propranolol*1 Nd:YAG-Laser*2 Ulzeration Propranolol*1 Diffuse neonatale Hämangiomatose Propranolol*1 Systemische Kortikosteroide, Exzision umschriebener Hämangiome Systemische Kortikosteroide 2. Relative Behandlungsindikation Lokalisation im Gesicht Kontaktkryotherapie*3 Nd:YAG-Laser*2 3. Keine Behandlungsindikation Unproblematische Hämangiome (z. B. am Stamm und an den proximalen Extremitäten) *1 bisher nicht zugelassen (s. Text) *2 bei flachen Hämangiomen (max. Tiefenausdehnung 1,1 mm) auch gepulster Farbstofflaser *3 max. Tiefenausdehnung 3–4 mm, max. Flächendurchmesser 1 cm Abbildung 4: Solitäres Mastozytom ● solitäre Mastozytome (20 Prozent aller kindlichen Mastozytoseformen), ● die makulopapulöse Mastozytose mit mehr als 5 Mastozytomen (Urticaria pigmentosa, 70 Prozent der Fälle) und ● sehr selten die diffuse kutane Mastozytose (5 Prozent) beobachtet (22). Den verschiedenen Formen liegen aktivierende Mutationen des Proto-Onkogens c-KIT auf Chromosom 4q12 zugrunde, das für einen Mastzell-Wachstumsfaktor-Rezeptor (Stammzellfaktor, SCF) kodiert (23). Solitäre Mastozytome sind in einem Viertel der Fälle bereits bei Geburt vorhanden, die übrigen manifestieren sich innerhalb der ersten beiden Lebensjahre. Sie sind erkennbar als roséfarbene oder bräunlich-orangefarbene Papeln, Knoten oder Plaques von wenigen mm bis einigen cm Durchmesser (Abbildung 4). Ihre Oberfläche ist meist glatt, gelegentlich erinnert sie an Orangenhaut. 351 MEDIZIN Die Diagnose wird durch den Darierschen Reibetest gestellt: Nach Reibung entsteht im und um das Mastozytom innerhalb von drei bis fünf Minuten eine deutliche Schwellung und Rötung, gelegentlich auch eine Blase. Dies kann auch spontan geschehen und insbesondere bei multiplen Mastozytomen zu Flushsymptomen und Blutdruckabfall führen. Eine Hautbiopsie ist nur in Zweifelsfällen erforderlich. Zur Einschätzung der – selten auch systemischen – Mastzellvermehrung dient die Bestimmung der Tryptase, eines typischen Mastzellprodukts, im Serum. Bei gering ausgeprägter Klinik, fehlenden extrakutanen Symptomen und normaler Serum-Tryptase kann bei Kindern auf eine weiterführende Diagnostik, wie sie bei Erwachsenen empfohlen wird, verzichtet werden. In über 70 Prozent bilden sich Mastozytome bis zur Pubertät spontan zurück. Eine Exzision solitärer Tumoren kann bei rezidivierenden Episoden von Blasenbildung und Superinfektion erwogen werden. Wichtig ist die Meidung mastzelldegranulierender Faktoren: ● nicht-immunologische Reize: Reibung, Druck, Sonnenlichtexposition, abrupte Exposition gegenüber kaltem oder warmem Wasser ● immunologische Reize, zum Beispiel Insektenstiche, Impfungen, Fieber ● Histamin-liberierende Substanzen: Für das Kindesalter sind in erster Linie Codein (in Hustensäften), nichtsteroidale Antirheumatika vom Typ Ibuprofen, seltener Narkotika und jodhaltige Kontrastmittel relevant. Pilomatrikom Obwohl das Pilomatrikom mit circa 10 Prozent zu den häufigsten, bei Kindern exzidierten Hauttumoren zählt, wird die Diagnose oft erst histologisch gestellt (14). Es handelt sich um einen langsam wachsenden, meist asymptomatischen, benignen Hauttumor, der von den Matrixzellen des Haarfollikels ausgeht und vornehmlich an Kopf, Hals oder Armen lokalisiert ist (24). Klinisch imponiert ein derber bis steinharter, überwiegend subkutaner Knoten, der mit der Haut adhärent, auf der Unterlage jedoch gut verschiebar ist. Ausdehnung, gelappte Oberfläche und oft bläulicher Farbton lassen sich durch Spannung der Haut über dem Tumor besser erkennen („Zelt-Zeichen“). Die klinische Diagnose lässt sich sonographisch durch den Nachweis echogener Binnenechos erhärten, die Verkalkungszonen entsprechen (daher die veraltete Bezeichnung Epithelioma calcificans Malherbe). Da keine Spontanremission eintritt, besteht die Therapie der Wahl in der vollständigen Exzision. Die Rezidivrate ist aufgrund der guten Abgrenzbarkeit des Knotens mit 0 bis 6 Prozent gering. In 2 bis 3,5 Prozent der Fälle kommt der Tumor in Mehrzahl vor; dann sind verschiedene assoziierte Erkrankungen zu beachten, vor allem myotonische Dystrophie Curschmann-Steinert, Gardner-Syndrom, Rubinstein-Taybi-Syndrom und chromosomale Aberrationen (25). 352 KERNAUSSAGEN ● Maligne Hauttumoren sind im Kindesalter sehr selten. Die Mehrzahl der kindlichen Hauttumoren bedarf daher keiner weiterführenden Diagnostik oder Therapie, sondern nur der klinischen Beobachtung. ● Große und riesige kongenitale melanozytäre Nävi haben ein erhöhtes Entartungs- und Komplikationsrisiko, das sich bereits in den ersten Lebensjahren auswirken kann. Auf Naevi sebacei bilden sich in 10 bis 30 Prozent der Fälle bestimmte, überwiegend benigne Hauttumoren zumeist erst nach dem 20. Lebensjahr. ● Der Spitz-Nävus ist der bedeutendste Melanomsimulator im Kindesalter. ● Im Gegensatz zu den meisten infantilen Hämangiomen bedürfen solche mit drohenden Komplikationen einer raschen, gegebenenfalls systemischen Therapie. ● Mastozytome und juvenile Xanthogranulome regredieren in der Mehrzahl der Fälle spontan. Im Regelfall ist weder eine Biopsie noch eine Exzision erforderlich. ● Syndromale Assoziationen sind bei multiplen juvenilen Xanthogranulomen und Pilomatrikomen möglich. Danksagung Die Autoren danken Prof. Dr. Eva-B. Bröcker, Direktorin der Klinik und Poliklinik für Dermatologie, Venerologie und Allergologie des Universitätsklinikums Würzburg, für die kritische Durchsicht des Manuskripts. Interessenkonflikt Prof. Höger nimmt an einer multizentrischen Studie zur Untersuchung der Wirksamkeit von Propranolol bei Hämangiomen teil, die von der Firma Pierre Fabre, Toulouse/Frankreich, unterstützt wird. Prof. Hamm erklärt, dass kein Interessenkonflikt im Sinne der Richtlinien des International Committee of Medical Journal Editors besteht. Manuskriptdaten eingereicht: 20. 8. 2010, revidierte Fassung angenommen: 24. 8. 2010 LITERATUR 1. Diepgen TL: Epidemiologie von Hauttumoren. Epitheliale Hauttumoren. In: Szeimies RM, Hauschild A, Garbe C, Kaufmann R, Landthaler M (eds.): Tumoren der Haut. Stuttgart: Thieme 2010; 87–92. 2. Knight PJ, Reiner CB: Superficial lumps in children: what, when, and why? Pediatrics 1983; 72: 147–53. 3. Bauer J, Garbe C: Melanozytäre Nävi als Präkursoren und Risikomarker für das maligne Melanom. In: Szeimies RM, Hauschild A, Garbe C, Kaufmann R, Landthaler M (eds.): Tumoren der Haut. Stuttgart: Thieme 2010; 293–9. 4. Mills O, Messina JL: Pediatric melanoma: a review. Cancer Control 2009; 16: 225–33. 5. Strouse JJ, Fears TR, Tucker MA, Wayne AS: Pediatric melanoma: risk factor and survival analysis of the surveillance, epidemiology and end results database. J Clin Oncol 2005; 23: 4735–41. 6. Griffin JR, Cohen PR, Tschen JA, et al.: Basal cell carcinoma in childhood: case report and literature review. J Am Acad Dermatol 2007; 57(Suppl): 97–102. 7. Holman JD, Dyer JA: Genodermatoses with malignant potential. Curr Opin Pediatr 2007; 19: 446–54. Deutsches Ärzteblatt | Jg. 108 | Heft 20 | 20. Mai 2011 MEDIZIN 8. Kovalyshyn I, Braun R, Marghoob A: Congenital melanocytic naevi. Australas J Dermatol 2009; 50: 231–40. 9. Pavlidou E, Hagel C, Papavasilliou A, Giouroukos S, Panteliadis C: Neurocutaneous melanosis: report of three cases and up-todate review. J Child Neurol 2008; 23: 1382–91. 10. Kinsler VA, Chong WK, Aylett SE, Atherton DJ: Complications of congenital melanocytic naevi in children: analysis of 16 years’ experience and clinical practice. Br J Dermatol 2008; 159: 907–14. 11. Gerami P, Jewell SS, Morrison LE, et al.: Fluorescence in situ hybridization (FISH) as an ancillary diagnostic tool in the diagnosis of melanoma. Am J Surg Pathol 2009; 33: 1146–56. 12. Busam KJ, Pulitzer M: Sentinel lymph node biopsy for patients with diagnostically controversial Spitzoid melanocytic tumors? Adv Anat Pathol 2008; 15: 253–62. 13. Altaykan A, Ersoy-Evans S, Erkin G, Ozkaya O: Basal cell carcinoma arising in nevus sebaceous during childhood. Pediatr Dermatol 2008; 25: 616–9. 14. Price HN, Zaenglein AL: Diagnosis and management of benign lumps and bumps in childhood. Curr Opin Pediatr 2007; 19: 420–4. 15. Boye E, Olsen BR: Signaling mechanisms in infantile hemangioma. Curr Opin Hematol 2009; 16: 202–8. 16. Hoeger PH, Maerker JM, Kienast AK Syed SB, Harper JI: Neonatal haemangiomatosis associated with placental chorioangiomas: report of three cases and review of the literature. Clin Exp Dermatol 2009; 34: e78–80. 17. Léauté-Labrèze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taïeb A: Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008; 358: 2649–51. 18. Deutsche Gesellschaft für Kinderchirurgie, Deutsche Gesellschaft für Kinder- und Jugendmedizin, Deutsche Dermatologische Gesellschaft und Arbeitsgemeinschaft Pädiatrische Dermatologie: Leitlinie Hämangiome im Säuglings- und Kleinkindesalter. www.uni-duesseldorf.de/AWMF/ll/006–100.htm 19. Dehner LP: Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol 2003; 27: 579–93. 20. Hoeger PH, Diaz C, Malone M Malone M, Pritchard J, Harper JI: Juvenile xanthogranuloma as a sequel to Langerhans cell histiocytosis: a report of three cases. Clin Exp Dermatol 2001; 26: 391–4. 21. Raygada M, Arthur DC, Wayne AS, Rennert OM, Toretsky JA, Stratakis CA: Juvenile xanthogranuloma in a child with previously unsuspected neurofibromatosis type 1 and juvenile myelomonocytic leukemia. Pediatr Blood Cancer 2010; 54: 173–5. 22. Briley LD, Phillips CM: Cutaneous mastocytosis: a review focusing on the pediatric population. Clin Pediatr (Phila) 2008; 47: 757–61. Deutsches Ärzteblatt | Jg. 108 | Heft 20 | 20. Mai 2011 23. Bodemer C, Hermine O, Palmérini F, et al.: Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol 2010; 130: 804–15. 24. Pirouzmanesh A, Reinisch JF, Gonzalez-Gomez I, Smith EM, Meara JG: Pilomatrixoma: a review of 346 cases. Plast Reconstr Surg 2003; 112: 1784–9. 25. Wachter-Giner T, Bieber I, Warmuth-Metz M, Bröcker EB, Hamm H: Multiple pilomatricomas and gliomatosis cerebri—a new association? Pediatr Dermatol 2009; 26: 75–8. Anschrift für die Verfasser Prof. Dr. med. Henning Hamm Klinik und Poliklinik für Dermatologie, Venerologie und Allergologie Universitätsklinikum Würzburg Josef-Schneider-Straße 2 97080 Würzburg [email protected] SUMMARY Skin Tumors in Childhood Background: Dermatologists, pediatricians, and general practitioners are often consulted by worried parents for the evaluation of a cutaneous tumor. Methods: Selective literature review. Results: Only 1–2% of skin tumors excised in children turn out to be malignant when examined histologically. Warning signs of malignancy include rapid growth, firm consistency, diameter exceeding 3 cm, ulceration, a non-movable mass, and presence in the neonatal period. The more common malignant skin tumors in adults — basal cell carcinoma, cutaneous squamous cell carcinoma, and melanoma — are very rare in childhood. Congenital melanocytic nevi and sebaceous nevi bear a lower malignant potential than previously believed; nevertheless, their excision is often indicated. A Spitz nevus can mimic a melanoma both clinically and histologically. Some benign skin tumors of childhood tend to regress spontaneously within a few years but may cause complications at particular locations and when multiple. For infantile hemangiomas requiring systemic treatment because of imminent obstruction or ulceration, propranolol seems to have a far more favorable risk-benefit ratio than corticosteroids. Conclusion: Physicians need specialized knowledge in order to decide whether a skin tumor in a child should be excised, non-surgically treated, or further evaluated, or whether it can be safely left untreated because of the likelihood of spontaneous remission. Zitierweise Hamm H, Höger PH: Skin tumors in childhood. Dtsch Arztebl Int 2011; 108(20): 347–53. DOI: 10.3238/arztebl.2011.0347 @ The English version of this article is available online: www.aerzteblatt-international.de 353