Chelat-Liganden und ihre Rhodium
Werbung

Asymmetrische Katalysen, 13 [1]
Chelat-Liganden und ihre Rhodium-Komplexe mit seitlichen Asymmetriezentren
und ihre Anwendung in der enantioselektiven Katalyse
Asymmetric Catalyses. 13 [1]
Chelate Ligands and their Rhodium Complexes with Lateral Asymmetrie Centers
and their Use in Enantioselective Catalysis
Henri Brunner* und A. F. M. Mokhlesur Rahman
Institut für Anorganische Chemie der Universität Regensburg,
Universitätsstraße 31, D-8400 Regensburg
Herrn Prof. Dr. Dr. h. c. mult. E. 0. Fischer zum 65. Geburtstag gewidmet
Z. Naturforsch. 38b, 1332-1338 (1983); eingegangen am 16. Mai 1983
Optical Activity, Asymmetrie Induction, Enantioselective Catalysis
The chelate ligands 1 - 3 with lateral asymmetric centers at the nitrogen atoms were
prepared and characterized. In the Rh complexes 4 and 5 the asymmetric centers directly
interact with those coordination positions where during enantioselective catalysis prochiral substrates are converted into optically active products.
The compounds 4 and 5, as well as the in situ catalysts [Rh(COD)Cl]2/2 were used in the
asymmetric hydrogenation of a-N[acetamino]cinnamic acid and in the asymmetric
hydrosilylation of acetophenone with diphenylsilane. The low optical inductions obtained
result from the catalyst loosing its chiral information during catalysis due to racemization
or fragmentation.
Einleitung
Die meisten der bisher in der asymmetrischen
Katalyse mit Übergangsmetall-Komplexen verwendeten Liganden sind Chelatphosphine mit zwei Phenylgruppen am Phosphor und der induzierenden
Chiralität im Chelatgerüst zwischen den beiden
Phosphoratomen [2, 3], in Formel A von Schema 1
durch Punkte gekennzeichnet. Die originäre Chiralität bewirkt meist eine Wellung des Chelatrings
und damit verbunden eine Differenzierung der beiden Phenylgruppen am Phosphor in axiale und
äquatoriale sowie in kanten- bzw. flächenorientierte
Phenylgruppen, wie in A angedeutet [3-8]. Die chirale Einstellung der beiden Phenyl,,ohren" am
Phosphor wird als der eigentliche Überträger der
Chiralität vom Chelatgerüst auf die Koordinationsstellen am Metallatom angesehen, an denen das
prochirale Substrat in das optisch aktive Produkt
umgewandelt wird.
Es wurde bereits auf den Aspekt hingewiesen,
daß im Chelatgerüst der verschiedenen Liganden
zwar verschiedene spezifische Chiralitätselemente
vorhanden sind, aber mit den P(C6Hö)2-Oruppen
* Sonderdruckanforderungen an Prof. Dr. H.J. Keller.
0340-5087/83/1100-1332/$ 01.00/0
immer der gleiche Übertragungsmechanismus benlitzt wird [9-11]. Dies muß auf einen Informationsverlust hinauslaufen. Es wurden auch Modelle
vorgeschlagen, das induzierende Asymmetriezentrum an die Stelle zu rücken, wo in den Chelatphosphinen die P(C6H5)2-Überträger sitzen. Damit wäre
eine direkte Wechselwirkung der induzierenden
Chiralität mit dem prochiralen Substrat ohne Zwischenschaltung eines Überträgers möglich. Dies gilt
zum Beispiel für Chelatliganden, die über einen
Imin-Stickstoff wie in B ans Metallatom gebunden
sind. Geht man bei der Schiffbasenkondensation zu
diesen Liganden von primären optisch aktiven
Aminen aus, in B zum Beispiel von (B)-l-Phenylethylamin, so befindet sich das stickstoffgebundene
Asymmetriezentrum genau dort, wo in den Phosphinen die die Chiralität übertragenden Phenylgruppen lokalisiert sind. Über neue Derivate dieser
Art und ihren Einsatz in der asymmetrischen Katalyse wird im folgenden berichtet.
Diskussion
Darstellung der Liganden 1, 2 und 3
Kondensation von (i?)-( + )-l-Phenvlethylamin
mit Salicvlaldehyd und Acetylaceton ergibt die
Unauthenticated
Download Date | 8/19/17 8:44 AM
H. Brunner et al. • Asymmetrische Katalysen
1333
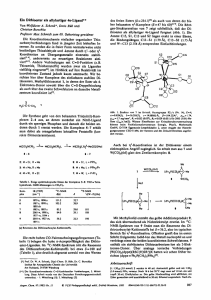
Schema 1.
Verbindungen 1 und 2, die ein seitliches Asymmetriezentrum im Sinne von Formel B, Einleitung,
enthalten, in Ausbeuten von 90% (Schema 2). Das
Salicylaldimin (1) läßt sich mit Tl(OH) oder
Tl(OEt) in das Tl-Salz überführen. Von Verbindung
2 dagegen konnte kein Tl-Salz isoliert werden. Läßt
man 70-proz. HC104 in Ethanol auf 1.1.3.3-Tetramethoxypropan und (i?)-(+)-l-Phenylethylamin
einwirken, so erhält man das Vinamidiniumsalz (3)
(Vinamidin = vinyloges Amidin) als schwachgelbes
Pulver (Schema 2) [12, 13].
Die IR-Spektren der Imine 1-3 enthalten die
C=N-Valenzschwingung bei 1618-1630 cm - 1 , gegenüber den Carbonylverbindungen, von denen sie
sich ableiten, nach niedrigeren Wellenzahlen verschoben. Im Massenspektrum (Feiddesorption,
DMF-Lösung) des Salzes 3 erscheint das dem Vinamidinium-Kation entsprechende Molekülion bei
m/e = 279 [13].
In den iH-NMR-Spektren (Tab. I) zeigen die
Verbindungen 1 und 2 nur je einen Satz von Signa-
(0
len, wie für die H-verbrückten Strukturen in Schema 2 zu erwarten. Das iH-NMR-Spektrum der
Verbindung 3 dagegen enthält alle Signale dreifach,
besonders gut an den drei Dubletts der Methylgruppe zu sehen (Tab. I). Das mittlere Proton des
C-Gerüsts H 2 bildet durch Kopplung mit H 1 Tripletts, von denen zwei gut, das dritte nur andeutungsweise zu erkennen ist, weil es mit anderen
Signalen zusammenfällt. Das Proton H 1 koppelt
mit H 2 und mit den NH-Protonen und ergibt ebenso ein Multiplett wie die Methinprotonen des 1Phenylethylrestes, die zusätzlich zu den Methylprotonen mit den NH-Protonen koppeln.
Aus dem Auftreten von drei Dubletts im Methylbereich folgt, daß zumindest in Lösung mehrere
der isomeren Formen all-cis (U-Form), all-trans (WForm) und cis-trans (Sichel-Form) bezüglich des
Vinamidin-Gerüsts [12, 13] bzw. der cis-trans-Isomeren bezüglich der CN-Doppelbindung vorliegen.
Dabei sind zwei Isomere in fast gleicher Menge
vorhanden, während das dritte deutlich überwiegt.
CH3
11^
/
H
CcHc
V"CH3
C6hS
Schema 2.
Unauthenticated
Download Date | 8/19/17 8:44 AM
1334
H. Brunner et al. • Asymmetrische Katalysen
Tab. I. iH-NMR-Parameter der Verbindungen 1-5. (5-Werte in ppm, Kopplungskonstanten in Hz, i-TMS (Numerierung de
Protonen siehe Schema 2).
Verb. H-aromat. CHI
J H^-RH
CH 2
JH'-H 2
1
6,62-7,53
2
7,05(m)
3
7,25-7,47
7,72-8,12(m)
5,24-5,75(m)
3C
7,25-7,38
7,63-7,72(br)
4
6,31-7,53
5
7,22-7,34
7,91(d)
(2,1)
7,17d
(2,1)
5,46(t)
(11,5)
a
CH3a
JCH3-CH
CH 3 [3, 4] CHa
8,36(s)
4,56(q)
4,83(s)
4,68(t)
(7,1)
Signale des 1-Phenylethylrestes;
b
l,93(s)
l,73(s)
4,60(q)
4,65-4,85(m)
4,64(q)
4,48(q)
4,41(q)
1,65(d)
(9,6)
1,43(d)
(9,6)
1,43(d)
1,47(d)
1,51(d)
(6,8)
1,44(d)
(6,7)
1,67(d)
(7,2)
1,61(d)
(6,8)
Signale der 1.5-Cyclooctadienprotonen;
Auch bei 100 °C ist die Geschwindigkeit der Umwandlung zwischen den verschiedenen Formen in
DMSO-dö immer noch so klein, daß keine NMRKoaleszenz festzustellen ist. Bei 160 °C dagegen ist
die Umwandlung so rasch, daß ein gemitteltes
Spektrum zu beobachten ist, das nur noch ein einziges Methyl-Dublett und ein H 2 -Triplett enthält
(Tab. I).
Darstellung der Komplexe 4 und 5
Komplex 4 (Schema 2) läßt sich durch Einwirken des Tl-Salzes von 1 auf eine Suspension von
[Rh(COD)Cl]2 in Ether erhalten [13]. Nach 24stündigem Rühren wird die gelbe Lösung filtriert
und an Si0 2 chromatographiert. 4 schließt sich in
seinen Eigenschaften an ähnliche Verbindungen an
[14, 15]. Ein entsprechender Komplex mit dem Liganden 2 ließ sich auf diese Weise nicht isolieren.
Auch Versuche, Komplex 5 aus [Rh(COD)Cl]2 und
3 mit einem Überschuß von LiO(^-Bu) bzw. NaH
in THF darzustellen, schlugen fehl. Dagegen gelang
die Synthese von 5 unter den Bedingungen der
Phasentransfer-Katalyse. Eine Suspension von
[Rh(COD)Cl]2, BU 4 N+HS0 4 - und 3 in Methylenchlorid/wäßrige KOH ergibt 5 in 70% Ausbeute
[13]. Im Gegensatz zu Cu", Co11 und Ni 11 [16, 17]
ist bisher nur ein Rhi-Komplex mit einem Vinamidinato-Liganden bekannt [18], der allerdings an
den N-Atomen unsubstituiert ist.
In den iH-NMR-Spektren der Komplexe 4 und 5
(Tab. I) koppelt das Azomethinproton H 1 mit dem
e
= CHb
CH 2 b
Lösungsmittel
CCU
CDC13
DMSO-d
DMSO-d
4,58-4,81(br)
3,62-4,03(br)
4,22-4,63(br)
bei 160 °C;
d
l,81-2,72(br) CDCI3
l,82-2,02(br) CDCI3
2,31-2,56(br)
Dublett eines Dubletts.
Zentralmetall Rhodium und erscheint als Dublett.
Bei Verbindung 5 ergibt das Azomethin-Signal im
250-MHz-iH-NMR-Spektrum vier eng nebeneinanderliegende Linien auf der Hochfeldseite des
Phenylbereichs. Entkoppelt man H 2 bei ö1 =
4,68 ppm, so vereinfacht sich das Azomethinsignal
zu einem Dublett, das durch die Kopplung mit
Rhodium zustandekommt. Dieser Befund erlaubt
sowohl die Zuordnung der <5-Werte als auch der
Kopplungskonstanten Jith-H1 der Azomethinprotonen in 5. Komplex 4 zeigt im olefinischen Bereich
für das 1.5-Cyclooctadien zwei Multipletts, Komplex 5 dagegen nur eines. Dies ist darauf zurückzuführen, daß der Vinamidato-Ligand in 5 im Gegensatz zum Salicylaldiminato-Liganden in 4 eine C2Achse enthält.
Enantioselektive Hxjdrierungen mit den Komplexen
4 und 5
Die isolierten Komplexe 4 und 5 zeigen bei Raumtemperatur und 1,1 bar H 2 -Druck in THF/Methanol-Gemischen keine katalytischen Eigenschaften
bei der homogenen Hydrierung von (Z)-a[N-Acetamino]zimtsäure (AAZ) zu N-Acetylphenvlalanin
[13]. Bei Zusatz von Triphenylphosphin jedoch
katalysieren beide Komplexe 4 und 5 die Hydrierung unter normalen Bedingungen, nicht dagegen
bei Zusatz von i?-(-j-)-iminphos (o-[N-(i2)-l-Phenylethyl]aldiminophenyl-diphenylphosphin
[13,19]).
Mit dem System Komplex5/Triphenylphosphin
(Rh:Ligand-Verhältnis 1:2) zum Beispiel läßt sich
Unauthenticated
Download Date | 8/19/17 8:44 AM
1335 H. Brunner et al. • Asymmetrische Katalysen
AAZ (Katalysator: Substrat-Verhältnis = 1:100)
innerhalb von 20 h vollständig hydrieren [20]. Das
Hydrierprodukt fällt dabei allerdings racemisch an.
Das seitliche Asymmetriezentrum am N-Atom in
5 bewirkt also bei der Produktbildung keine Stereoselektivität. Dies deutet darauf hin, daß entweder
das Asymmetriezentrum während der Hydrierung
racemisiert bzw. abgebaut wird oder das zugesetzte
Phosphin den optisch aktiven Liganden verdrängt,
obwohl dieser als anionischer Chelatligand fest gebunden sein sollte. Zusatz von (—)-Norphos zu
Komplex 5 (Rh: Ligand-Verhältnis 1:1) ergibt mit
95% ee bei relativ langsamem Umsatz eine Enantioselektivität wie die des Systems [Rh(COD)Cl]2/(—)Norphos [21]. x\uch in diesem Fall sind also die
seitlichen Asymmetriezentren an den N-Atomen
ohne Einfluß auf die asymmetrische Induktion.
Enantioselektive Hydrosilylierungen mit Komplex 4
und den Katalysatoren [Rh(COD)Cly2/2
Es waren nur phosphorhaltige optisch aktive
Liganden in die asymmetrische Hydrosilylierung
eingesetzt worden, bis in einer Arbeit dieser Publikationsreihe [11] gezeigt werden konnte, daß Rhodium- und Platin-Komplexe von optisch aktiven
Stickstoff-Liganden mit seitlichem Asymmetriezentrum des Typs B, Einleitung, die asymmetrische
Hydrosilylierung von Acetophenon mit Diphenylsilan katalysieren. Das Hydrolyseprodukt 1-Phenyl ethanol konnte in optischen Ausbeuten bis zu
87,6% ee erhalten werden [22], Demgegenüber ist
der isolierte Komplex 4 bei der Hydrosilylierung
von Acetophenon mit Diphenylsilan [13] zwar katalytisch aktiv, er führt aber zu keiner optischen
Induktion (Tab. II. Nr. 3). Der in sifw-Katalysator
aus [Rh(COD)Cl]2 und einer stöchiometrischen
Menge des Liganden 2 zeigt ebenfalls katalytische
Aktivität, aber auch keine optische Induktion. Dagegen ergibt [Rh(COD)Cl]2 mit einem zehnfachen
Überschuß an 2 eine geringe optische Induktion
von 0,3% ee (Tab. II, Nr. 1, 2).
Vergleich der Chiralitätsübertragung in den
Komplexen 4 und 5
Bei Betrachtung der Struktur von Komplex 4
lassen sich Gründe dafür finden, warum die Hydrosilylierung trotz guter Aktivität des Katalysators
nicht enantioselektiv verläuft. Geht man davon aus,
daß der 0,N-Chelatring bei der Katalyse erhalten
bleibt, der COD-1.5-Ligand dagegen abgespalten
wird, so sind bei oktaedrischer Koordination am
Rh-Atom vier weitere Koordinationsstellen in die
Überlegung einzubeziehen. Drei davon sind cisständig zum N-Atom, eine in der Ebene des Chelatrings, eine oberhalb und eine unterhalb dieser Ebene ; eine Position ist £raws-ständig zu N in der Ebene
des Chelatrings (Schema 2). Die genannten Positionen sind alle voneinander verschieden. Drei, in
eis- Stellung zu N, befinden sich im direkten Einflußbereich des chiralen Substituenten. Eine, in
trans-Stellung zu N, ist dagegen von der induzierenden Chiralität weit entfernt. Komplexe der Art 4
haben damit in der fraws-Position zu N (cis zu O)
eine „achirale" Stelle. Der Neuaufbau des Asymmetriezentrums, bei der asymmetrischen Hydrosilylierung vorwiegend an dieser Position, wäre eine
mögliche Erklärung dafür, daß keine meßbare
Enantioselektivität zustandekommt. Dieser Gedankengang führte uns zur Darstellung eines Liganden, der an beiden N-Atomen des Chelatringes
Tab. II. Asymmetrische Hydrosilylierung von Acetophenon mit Diphenylsilan. Reaktionsbeginn 0 °C, Aufwärmen auf
Raumtemperatur [11].
Rh: Ligand- Rh: Substrat- LösungsVerhältnis
Verhältnis
mittel
Konfiguration
Reakt.zeit % Hydrosi- Opt.
Induktion 1-Phenyllylierung
[h]
ethanol
[%]
[ % ee]
[Rh(COD)Cl]2/2
[Rh(COD)Cl]2/2
4
5
5
5
1
1
1
1
1
1
48
40
41
72
64
64
81
75
82
72
65
53
0
0,3
0
0,1
0,2
0
5/Ä-( + )-iminphos [19]
5/( +)-Norphos [20, 21]
5/(—)-Norphos [20, 21]
1 1
1 1
1 1
68
65
65
92
86
86
6,3
1,9
1,9
NTr. Katalysator
1
2
3
4
5
(5
7
8
9
1
10
1
1
1
1
1
1
1
1
1
1
400
400
400
835
835
835
1 800
1 800
1 800
2
2
2
2
ml
ml
ml
ml
THF
THF
THF
THF
-
5 ml
Petrolether
-
_
R
—
S
S
—
s
R
S
Unauthenticated
Download Date | 8/19/17 8:44 AM
1336
den gleichen optisch aktiven 1-Pheny lethylsubstituenten trägt. In einem Chelatkomplex eines derartigen Liganden gibt es keine „achiralen" Stellen
mehr, denn alle Positionen des Rh-Oktaeders werden von den chiralen Resten direkt beeinflußt.
Dieses Konzept ist im Yinamidin-System der Verbindung 3 und in Komplex 5 (Schema 2) verwirklicht.
Komplexe des Typs 5 bieten gegenüber Verbindungen des Typs 4 einen weiteren Vorteil. Wie erwähnt, sind die bei Chelatkomplexbildung mit einem unsymmetrischen Liganden wie 1 verbleibenden vier Positionen an einem Metalloktaeder alle
vier voneinander verschieden. In einem Chelatkomplex mit einem Liganden wie 3, der eine C2Achse enthält, dagegen, werden die verbleibenden
vier Koordinationsstellen paarweise gleich. In der
Formel 5 von Schema 2 entsprechen die beiden
Positionen oberhalb der Zeichenebene und in der
Zeichenebene rechts in bezug auf den optisch aktiven Rest rechts exakt den beiden Positionen unterhalb der Zeichenebene und in der Zeichenebene
links in bezug auf den optisch aktiven Rest links.
Da sich die Verminderung der konfigurativen und
konform ativen Vielfalt immer wieder als Leitlinie
bei der Verbesserung der Stereoselektivität in
asymmetrischen Reaktionen und Katalysen erwiesen hat, sollte auch unter diesem Aspekt der Katalysator 5 der Verbindung 4 bei der enantioselektiven Hydrosilylierung überlegen sein.
Wie die Versuche zeigten, ist Komplex 5 bei der
Hydrosilylierung von Acetophenon mit Diphenylsilan katalytisch aktiv, aber mit einer enttäuschenden optischen Induktion von 0,1-0,2% ee (Tab. II,
Nr. 4-6). Dieses reproduzierende Ergebnis wTar im
Hinblick auf die oben angestellte Überlegung überraschend und machte folgende weitere Untersuchungen erforderlich.
Drehwertverlust von Komplex 5 während der Katalyse
Ein Zusatz von stöchiometrischen Mengen an
( + )-Norphos, (—)-Norphos bzw. i?-( + )-iminpbos
zu Komplex 5 ergibt zwar denselben Drehsinn,
aber geringere optische Induktionen als in den Systemen aus (-|-)-Norphos, (—)-Norphos bzw. i?-( + )iminphos und [Rh(COD)Cl] 2 (Tab. II, Nr. 7-9). Mit
den gleichen Liganden führen damit die Prokatalysatoren [Rh(COD)Cl]2 und 5 zu verschiedenen Ergebnissen. Daraus muß geschlossen werden, daß
H. Brunner et al. • Asymmetrische Katalysen
im einen Fall der Cl-Ligand und im anderen Fall
der Vinamidinato-Ligand während der Hydrosilylierung im Katalysator verbleiben. Wenn der Vinamidinato-Ligand in 5 während der Katalyse nicht
abgespalten wird, sollte 5 mit (-f-)-Norphos und
(—)-Norphos zwei diastereomere Katalysatoren
ergeben, die unterschiedliche Enantioselektivitäten
bei der Produktbildung zur Folge haben sollten.
Tabelle II (Nr. 8,9) zeigt jedoch, daß beide Systeme
exakt gleiche Enantioselektivität bei unterschiedlicher Produktkonfiguration für ( + )-Norphos und
(—)-Norphos ergeben. Diese Ergebnisse führten
uns zu der Vermutung, daß Komplex 5 unter den
Hydrosilylierungsbedingungen seine optische Aktivität verlieren könnte (durch Racemisierung oder
Fragmentierung [23]).
Zur Überprüfung dieser Hypothese wurde die
Katalysatormenge im Standardansatz der Hydrosilylierung von 2 ml Acetophenon mit 3,5 ml Diphenylsilan [11] soweit vergrößert, daß der Drehwertabfall polarimetrisch verfolgt werden konnte.
Nach Durchmischen aller Komponenten bei Raumtemperatur fiel der Dreh wert ausgehend von 142
Skalenteilen für 8 mg der Verbindung 5 (2-cm-Küvette) in etwa 30 min auf den Wert Null, der sich
innerhalb von 3 h nicht änderte. Bei 0 °C erfolgt
der Drehwertabfall zwar langsamer, der Drehwert
nähert sich aber auch unter diesen Bedingungen im
Verlauf von Stunden dem Wert Null.
Diesen Ergebnissen muß gegenübergestellt werden, daß der Katalysatorkomplex 5 auch nach
24 h bei Raumtemperatur weder in Benzol noch
in THF einen nennenswerten Abfall des Drehwerts
aufwies. Die polarimetrischen Untersuchungen beweisen daher, daß der Katalysatorkomplex unter
den Hydrosilylierungsbedingungen seinen Drehwert verliert. Es ist daher ratsam, alle optisch aktiven Katalysatoren, die bei der asymmetrischen
Katalyse keine hohen optischen Induktionen ergeben [11], auf Drehwertabfall zu testen, bevor man
das Fehlen hoher optischer Induktion ungünstiger
Chiralitätsübertragung vom optisch aktiven Liganden auf das prochirale Substrat zuschreibt .
Experimenteller Teil
Alle Operationen wurden unter N2-Schutz mit
getrockneten Lösungsmitteln durchgeführt. Die
Ausbeuten. Schmelzpunkte und analytischen Daten
der neuen Verbindungen sind in Tab. III zusammengefaßt.
Unauthenticated
Download Date | 8/19/17 8:44 AM
1337 H. Brunner et al. • Asymmetrische Katalysen
Tab. III. Ausbeuten, Schmelzpunkte und analytische Daten der Verbindungen 1-5.
Verbindung
Ausbeute
[%]
Summenformel
Mol.-Gew.
1
89
C15H15NO
225,3
73
2
91
C13H17NO
203,1
-
3
12
Ci9H23C104
4
50
C23H26NORh
5
69
QJ7H33N2RI1
Ber.
Gef.
Ber.
Gef.
Ber.
Gef.
Schmp.
[°C]
378,9
279a
435,4
435b
488,5
488b
230-232
163
178-180
Feiddesorption aus DMF-Lösung, entspricht dem Kation CI 9 H 2 3 + ;
N-[(R)-l-Phenylethyl]-salicylaldimin
(1)
12,2 g (0,1 mol) frisch destillierter Salicylaldehyd
in 30 ml CH 3 OH werden mit 14,1 g (0,12 mol)
(i?)-( + )-l-Phenylethylamin gerührt. Aus der klaren
Lösung fällt innerhalb kurzer Zeit ein gelber Niederschlag aus, der abgefrittet, mit Methanol nachgewaschen und im Hochvakuum getrocknet wird. Ausbeute 20,5 g (89% d.Th.). Schmp. 73 °C [24, 25],
1 ist in allen organischen Solventien außer Methanol
gut löslich. I R : J>c=N 1630 cm" 1 (KBr). Optische
Drehung: [a]§?8 - 1 7 0 ° , [a]||6 - 2 0 5 ° , [a]§ 6 —555°
(1,6 • lO- 2 mol/1, Benzol).
Thallium-Salz von
aldimin
N-[(R)-l-Phenylethyl]-salicyl-
442 mg (2 mmol) Tl(OH) und 450 mg (2 mmol) 1
werden in 50 ml Methanol 20 h am Rückfluß gekocht, bis alles Tl(OH) gelöst ist. Dabei ändert sich
die Farbe der Lösung von gelb nach rot. Die Methanollösung wird abfiltriert und eingeengt. Nach Aufnehmen mit wenig Benzol wird mit Petrolether
ausgefällt. Das Thalliumsalz der Schiffschen Base
stellt ein rotes Pulver dar. Ausbeute 0,8 g (93%
d.Th.).
4-N[( R )-l-Phenylethyl]-amino-3-penten-2-on
(2)
5 g (50 mmol) frisch destilliertes Acetylaceton
und 6,05 g (50 mmol) (i?)-(+)-l-Phenylethylamin
werden in 10 ml Benzol mit 20 mg ^-Toluolsulfonsäure 2 h am Rückfluß gekocht. Nach dem Abziehen
des Lösungsmittels wird das leicht gelbe Öl mit
einer Kugelrohrdestille destilliert (Übergangstemperatur im Hochvakuum 135 °C). Nach der Destillation erstarrt das farblose bis hellgelbe Öl zu einer
kristallinen Masse, die in allen gebräuchlichen Lösungsmitteln gut löslich ist. Ausbeute 9,2 g (91%
d.Th.) [26]. I R : Vc=N 1620 cm- 1 (Film). Optische
Drehung: [a]|f 8 -860°, [a]§l6—1035°, [a]||6—2440°,
[a]|g5 —6990° (6,0 • 10~2 mol/1, Benzol).
b
Analysenwerte [ % ]
N
C
H
Ber.
Gef.
Ber.
Gef.
Ber.
Gef.
Ber.
Gef.
Ber.
Gef.
79,97
80,12
76,89
76,93
60,23
60,06
63,47
63,22
66,38
66,35
6,71
6,72
8,43
8,45
6,12
5,82
6,02
5,96
6,81
6,82
6,21
6,12
6,89
6,90
7,39
7,39
3,22
3,13
5,73
5,79
massenspektroskopisch (70 eV).
( Cyclooctadien-1.5 ) ( N-[ ( R )-l-Phenylethyl ]salicylaldiminato)rhodium (I) (4)
Zur Suspension von 197,2 mg (0,4 mmol)
[Rh(COD)Cl]2 in 20 ml Ether werden 344 mg
(0,8 mmol) des Tl-Salzes von 2 zugegeben. Nach 24 h
Rühren bei Raumtemperatur wird die gelbe Lösung
filtriert, eingeengt und durch Säulenchromatographie an Si0 2 (Akt. I ; 40 • 1,5 cm) aufgetrennt. Dabei
wird mit Benzol zuerst eine gelbe Zone eluiert, die
das Produkt und geringe Mengen unumgesetztes
[Rh(COD)Cl]2 enthält. Nach zweimaliger Kristallisation aus THF/Petrolether 1:5 bei —35 °C ist das
Produkt analysenrein. Ausbeute 175mg (50%d.Th.).
Schmp. 163 °C. Gelbe, im festen Zustand luftstabile
Kristalle; sehr gut löslich in Benzol, THF, CHCI3,
Aceton; gut löslich in Ether; wenig löslich in Petrolether und Methanol. I R : vc=s 1618cm" 1 (KBr).
Optische Drehung: [a]lf8 +215°, [a]||6 + 2 7 5 °
(7,0 • lO" 3 mol/1, Benzol).
N,N'[( R)-1-Phenylethyl J-3-amino-2-propen1-imonium-perchlorat (3)
In eine Lösung aus 5,5 g (33 mmol) 1.1.3.3-Tetramethoxypropan in 5 ml Ethanol läßt man bei
Raumtemperatur 5 ml 70-proz. HCIO4 einfließen.
Nach 15 min Rühren wird die Lösung gelb. Dann
wird die Lösung abgekühlt und mit 6,05 g (50 mmol)
(R)-(Jr) - 1-Phenylethylamin versetzt. Nach 30 min
Rühren bei Raumtemperatur wird noch 30 min bei
40 °C gerührt, wobei sich ein gelber Niederschlag
abscheidet. Man filtriert den Niederschlag ab,
wäscht mit Ether und trocknet im Hochvakuum.
Ein weiterer Teil der Verbindung kann aus der
Mutterlauge gewonnen werden, indem man die
Mutterlauge einengt, mit wenig Ethanol aufnimmt
und soviel Ether zusetzt, bis ein gelber Niederschlag
ausfällt. Ausbeute 1,5g (12% d.Th.). Schmp. 230
bis 232 °C. Das Produkt ist unlöslich in CH2C12 und
Methanol, sehr gut löslich in DMF und DMSO. I R :
vc=n 1620. vuh 3280, vCio4 1100 cm- 1 (KBr). Op-
Unauthenticated
Download Date | 8/19/17 8:44 AM
1338
H. Brunner et al. • Asymmetrische Katalysen
tische Drehung: [a]§f 8 + 4 1 0 ° , [a]fd6 + 5 0 0 ° , [a]|i 6
+ 1125°, [a]§g5 + 2 9 7 0 ° (1,3 • 10~ 2 mol/1, D M F ) .
( Cyclooctadien -1.5 )-{N ,N' [ (R)-l-phe
l-imino-2-propen-3-aminato}-rhodium(I)
nylethyl ] (5)
Zur Suspension v o n 750 m g (2 m m o l ) N , N ' - [ ( i ? ) l-Phenylethyl]-2-amino-3-propen-l-imonium-perchlorat wird in Gegenwart v o n 375 m g (0,75 m m o l )
[Rh(COD)Cl] 2 und 52,5 mg B u 4 N + H S 0 4 - in 15 ml
CH2CI2 unter kräftigem R ü h r e n eine Lösung v o n
500 mg K O H in 5 ml Wasser zugetropft. Es wird 1 h
bei Raumtemperatur gerührt bis die organische
Phase klar ist. Die organische Phase wird abgetrennt
und mit 5 ml Wasser ausgewaschen. E i n d a m p f e n
der organischen Phase liefert einen gelben R ü c k stand, der mehrmals mit Ethanol gewaschen wird.
Nach zweimaliger Kristallisation aus CH2Cl2/EtOH
[1] 12. Mitteilung: siehe ref. [19].
[2] L. Marko und J. Bakos, in R. Ugo (Herausg.):
Aspects of Homogeneous Catalysis, Bd. 4, S. 145,
D. Reidel, Dordrecht, Boston 1981.
[3] H. B. Kagan, in G. Wilkinson, F. G. A. Stone und
E. W . Abel (Herausg.): Comprehensive Organometallic Chemistry, Bd. 8, Kap. 53, S. 463, Pergamon Press, Oxford 1982.
[4] O. Samuel, R. Couffignal, M. Lauer, S. Y . Zhang
und H. B. Kagan, Nouv. J. Chim. 5, 15 (1981).
[5] M. D. Fryzuk und B. Bosnich, J. Am. Chem. Soc.
99, 6262 (1977).
[6] W. S. Knowles, B. D. Vineyard, M. J. Sabacky
und B. R. Stults, in Y . Ishii und M. Tsutsui
(Herausg.): Fundamental Research in Homogeneous Catalysis, Bd. 3, S. 537, Plenum, New
York 1979.
[7] K. E. Koenig, M. J. Sabacky, G. L. Bachman,
W . C. Christopfel, H. D. Barnstoff, R. B. Friedman, W. S. Knowles, B. R. Stults, B. D. Vineyard
und D. J. Weinkauff, in D. W . Slocum und O. R.
Hughes (Herausg.): Transition Metal Mediated
Organic Synthesis, Ann. N. Y . Acad. Sei. 333, 16
(1980).
[8] D. A. Slack, I. Greveling und M. C. Baird, Inorg.
Chem. 18, 3125 (1979).
[9] H. Brunner, G. Agrifoglio, I. Bernal und M.
Creswick, Angew. Chem. 92, 645 (1980); Angew.
Chem. Int. Ed. Engl. 19, 641 (1980).
[10] H. Brunner, in A. Müller und E. Diemann
(Herausg.): Transition Metal Chemistry, S. 265,
Verlag Chemie, Weinheim, Deerfield Beach, Basel
1981.
[11] H. Brunner und G. Riepl, Angew. Chem. Suppl.
1982, 769; Angew. Chem. 94, 369 (1982); Angew.
Chem. Int. Ed. Engl. 21, 377 (1982).
bei — 2 5 °C ist die Verbindung analysenrein. Ausbeute 503 m g ( 6 9 % d . T h . ) . Der im festen Zustand
luftstabile K o m p l e x ist schwerlöslich in E t h a n o l ,
wenig löslich in Petrolether, löslich in Ether, sehr
gut löslich in Benzol, T H F und C H 2 C k Beim Erwärmen in der abgeschmolzenen Kapillare tritt a b
ca. 169 °C langsame Zersetzung ein. Schmp. 178 bis
180 °C (unter Dunkelrotfärbung). I R : vc=N 1605 c m - 1
( K B r ) . Optische Drehung: [ a ] p 8 + 7 2 0 ° , [a]l| 6
+ 930°, [a]436 + 3 0 4 0 ° (6,1 • 10" 3 mol/1, Benzol).
W i r danken der Deutschen Forschungsgemeinschaft, d e m F o n d s der Chemischen Industrie u n d
der B A S F A G für Unterstützung dieser Arbeit.
A . F . M. M. R a h m a n dankt dem Bangladesh Council
of Scientific and Industrial Research Laboratories
f ü r die Freistellung zum Studium und dem D A A D ,
B o n n , für ein Stipendium.
[12] D. Lloyd und H. McNab, Angew. Chem. 88, 496
(1977); Angew. Chem. Int. Ed. Engl. 16, 459
(1977).
[13] A. F. M. M. Rahman, Dissertation, Universität
Regensburg 1983.
[14] R. J. Cozens, K. S. Murray und B. O. West,
J. Organomet. Chem. 27, 399 (1970).
[15] J. T. Mague und M. O. Nutt, J. Organomet.
Chem. 166, 63 (1979).
[16] S. G. McGreachin, Can. J. Chem. 46, 1903 (1968).
[17] R. Knorr und A. Weiß, Chem. Ber. 114, 2104
(1981) und dort zit. Lit.
[18] M. E. Howden, R. D. W. Kemmit und M. D.
Schilling, J. Chem. Soc. Dalton Trans. 1980, 1716.
[19] H. Brunner und A. F. M. M. Rahman, Chem. Ber.,
im Druck.
[20] H. Brunner und W. Pieronczyk, J. Chem. Res. (S)
1980, 76; (M) 1980, 1275.
[21] H. Brunner, W . Pieronczyk, B. Schönhammer,
K. Streng, I. Bernal und J. Korp, Chem. Ber. 114,
1137 (1981).
[22] H. Brunner, G. Riepl und H. Weitzer, Angew.
Chem. Suppl. 1983, 445; Angew. Chem. 95, 326
(1983); Angew. Chem. Int. Ed. Engl. 22, 331
(1983).
[23] G. Riepl, Dissertation, Universität Regensburg
1983.
[24] H. E. Smith, S. L. Cook und M. E. Warren, J.
Org. Chem. 29, 2265 (1964).
[25] H. E. Smith und R. Records, Tetrahedron 22, 813
(1966).
[26] C. Kashima, Y . Tsuda, S. Imada und T. Nishio,
J. Chem. Soc. Perkin Trans. 1, 1866 (1980).
Unauthenticated
Download Date | 8/19/17 8:44 AM