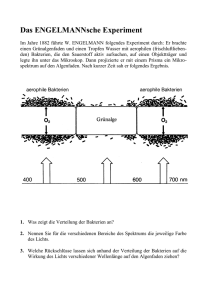
Alanin Razemase Gene von Staphylococcus aureus Inaugural
Werbung

AUS DEM INSTITUT FÜR MEDIZINISCHE MIKROBIOLOGIE, IMMUNOLOGIE UND HYGIENE DER UNIVERSITÄT ZU KÖLN DIREKTOR: UNIVERSITÄTSPROFESSOR DR. MED. M. KRÖNKE Alanin Razemase Gene von Staphylococcus aureus Inaugural-Dissertation zur Erlangung der Doktorwürde der Hohen Medizinischen Fakultät der Universität zu Köln vorgelegt von Quoc-Bao Ha Ngoc aus Bonn Promoviert am 18. August 2010 M & S Copy-Druckhaus, Köln Gedruckt mit Genehmigung der Medizinischen Fakultät der Universität zu Köln, 2010 Dekan: Universitätsprofessor Dr. med J. Klosterkötter 1. Berichterstatter: Universitätsprofessor Dr. med M. Krönke 2. Berichterstatter: Professor Dr. rer. nat. F.-G. Hanisch Erklärung Ich erkläre hiermit, dass ich die vorliegende Arbeit ohne unzulässige Hilfe Dritter und ohne Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe; die aus fremden Quellen direkt oder indirekt übernommenen Gedanken sind als solche kenntlich gemacht. Bei der Auswahl und Auswertung des Materials sowie der Herstellung des Manuskriptes habe ich keine Unterstützungsleistungen erhalten. Weitere Personen waren an der geistigen Herstellung der vorliegenden Arbeit nicht beteilligt. Insbesondere habe ich nicht die Hilfe eines Promotionsberaters in Anspruch genommen. Dritte haben von mir weder unmittelbar noch mittelbar geldwerte Leistungen für Arbeiten erhalten, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen. Die Arbeit wurde von mir bisher weder im Inland noch im Ausland in gleicher Form einer anderen Prüfungsbehörde vorgelegt und ist auch noch nicht veröffentlicht. Quoc-Bao Ha Ngoc Köln, den 2. April 2007 iii Die vorliegende Dissertation wurde angefertigt in der Zeit von August 1997 bis Mai 2003 im Department of Biology and Biochemistry der University of Houston Lehrstuhl: Professor Dr. Michael Benedik und von Februar 2007 bis April 2007 am Institut für medizinische Mikrobiologie, Immunologie und Hygiene der Universität zu Köln Lehrstuhl: Professor Dr. med. Martin Krönke. Die in dieser Arbeit angegebenen Untersuchungen sind nach entsprechender Anleitung durch Herrn Professor Dr. Michael Benedik und Herrn Dr. Ulrich Strych durchgeführt worden. Die dieser Arbeit zugrunde liegenden Experimente sind von mir mit Unterstützung von Herrn Dr. Ulrich Strych durchgeführt worden. iv DANKSAGUNG Herrn Prof. Dr. med. Martin Krönke und Herrn Prof. Dr. rer. nat. Hanisch danke ich herzlich für die freundliche Unterstützung und die Bereitschaft meine Arbeit zu begutachten. Herrn Prof. Dr. Michael Benedik danke ich herzlich für diese interessante Aufgabenstellung, und die Möglichkeit diese Arbeit durchführen zu können. Mein besonderer Dank gilt Herrn Dr. Ulrich Strych, der mir bei dem gesamten Projekt behilflich war, sowohl durch theoretische Erklärungen, als auch durch praktische Hilfen. Seine Hilfe hat mir ein Basiswissen über die Mikrobiologie und Biochemie verschafft. Besonders bedanke ich mich bei meinen Eltern für ihre Geduld, ihre Unterstützung und ihre Aufopferung für mich während meiner gesamten Zeit als Student. Ich bedanke mich auch bei meinem Bruder für seine Hilfe und seinen Revisionen dieser Arbeit. Danke. v INHALTSVERZEICHNIS Titelblatt......................................................................................................................................i Erklärung...................................................................................................................................iii Danksagung................................................................................................................................v Inhaltsverzeichnis......................................................................................................................vi Abbildungen............................................................................................................................viii Tabellen.....................................................................................................................................ix Abkürzungen..............................................................................................................................x 1. Einleitung 1 1.1. Übersicht von Mikroorganismen.................................................................1 1.2. Bedeutung von Mikroorganismen...............................................................2 1.3. Normalflora.................................................................................................3 1.4. Bakterielle Infektion....................................................................................5 1.4.1. Invasive Bakterien........................................................................7 1.4.2. Toxine...........................................................................................8 1.5. Immunabwehr..............................................................................................9 1.6. Staphylococcus aureus...............................................................................10 1.6.1. Koagulase...................................................................................11 1.6.2. Toxisches Schocksyndrom.........................................................11 1.6.3. Lebensmittelvergiftung..............................................................12 1.6.4. Staphylogenes Lyell-Syndrom und andere Infektionen.............12 1.6.5. Akute Endokarditis.....................................................................13 1.6.6. Antibiotische Resistenz von Staphylococcus aureus.................14 1.6.7. Nosokomiale Infektionen und AIDS..........................................14 1.7. Aufbau der Zellwand.................................................................................15 1.8. Antibiotika.................................................................................................21 1.9. Alanin Razemasen.....................................................................................22 vi 1.10. Forschungsziele.......................................................................................23 2. Methodik und Materialien 25 2.1. DNA Extraktion.........................................................................................25 2.2. Speed Preparation......................................................................................26 2.3. Amplifikation der Alanin Razemase Gene durch Polymerase-Kettenreaktion.......................................................................26 2.4. Chroma SpinTM .........................................................................................27 2.5. Klonierung..................................................................................................28 2.5.1. Restriktion der Alanin Razemase Gene......................................28 2.5.2. Ligation.......................................................................................28 2.5.3. Transformation............................................................................29 2.5.4. Blau-Weiss-Screening.................................................................29 2.6. Agarose-Gelelektrophorese von Salr1 und Salr2......................................30 2.7. DNA-Sequenzierung.................................................................................30 2.8. Komplementation vom E. coli alr-Mutant................................................31 2.9. IPTG induzierte Überexprimierung von Alanin Razemase.......................32 2.10. SDS-Polyacrylamid-Gelelektrophorese der Alanin Razemase Proteine.................................................................32 3. Ergebnisse 34 4. Diskussion 44 5. Literaturverzeichnis 47 6. Zusammenfassung 49 7. Curriculum Vitae 50 vii ABBILDUNGEN Abbildung 1-1. Anordnung und Form von Bakterien......................................................2 Abbildung 1-2. Staphylococcus aureus .........................................................................10 Abbildung 1-3. N-Acetylglucosamin verbunden mit N-Acetylmuraminsäure........................................................................16 Abbildung 1-4. L-Alanin und D-Alanin........................................................................17 Abbildung 1-5. Peptidoglycan Querbrücken bei Bakterien...........................................18 Abbildung 1-6. Peptidoglycan bei grampositven Bakterien..........................................19 Abbildung 1-7. Zellwand bei grampositiven Bakterien.................................................20 Abbildung 1-8. Zellwand bei gramnegativen Bakterien................................................20 Abbildung 1-9. Alanin Razemase von Bacillus stearothermophilus.............................24 Abbildung 3-1. Agarose-Gelelektrophorese der Alanin Razemase Gene......................36 Abbildung 3-2a. DNA-Sequenzierung von Alanin Razemase in Staphylococcus aureus......................................................................37 Abbildung 3-2b. DNA-Sequenzierung (Fortsetztung).....................................................38 Abbildung 3-2c. DNA-Sequenzierung (Fortgesetzung)..................................................39 Abbildung 3-3a. Komplementation von E. coli dadX- alrts Mutanten.............................40 Abbildung 3-3b. Komplementation von E. coli dadX- alrts Mutanten.............................40 Abbildung 3-4. Agarose-Gelelektrophorese von Restriktionsprodukten.......................41 Abbildung 3-5. IPTG Induzierte Überexprimierung von Alanin Razemase in E. coli..................................................................43 Abbildung 4-1. Theoretischer Inhibitor von Alanin Razemase.....................................46 viii TABELLEN Tabelle 1-1. Krankheitserreger and Krankheiten...................................................................6 Tabelle 3-1. Bakterienstämme und Vektoren ......................................................................41 ix ABKÜRZUNGEN APS - Ammoniumpersulfat BamHI - Bacillus amyloliquefaciens H Restriktions-Endonuklease dNTP - Mix: dATP (2’-Desoxyadenosin 5’Triphosphat, Natrium Salz) dGTP (2’-Desoxyguanosin 5’triphosphate, Natrium Salz) dTTP (2’-Desoxythymidin 5’triphosphate, Natrium Salz) dCTP (2’-Desoxycytidin 5’triphosphate, Natrium Salz) EDTA - Ethylendiamintetraessigsäure IPTG - Isopropyl-β-D-thiogalactopyranosid LB - Luria-Bertani Medium pBS - Bluescript Klonierungs-Vektor PCI - Phenol: Chloroform: Isoamyl-Alkohol SDS - Natriumlaurylsulfat (C12 H25 Na O4 S) SDS-PAGE - Natriumlaurylsulfat Polyacrylamid-Gelelektrophorese SPM - Speed Preparation Mix TAE Puffer - TBE-Puffer, TRIS-Borat-EDTA-Puffer Taq - Thermus aquaticus DNA-Polymerase TE - TRIS EDTA (Ethylendiamintetraessigsäure) TEMED - N, N, N’, N’ - Tetramethylethylendiamin TMF - Transformation Puffer (40 mM MnCl2, 100 mM CaCl2, 50 mM RbCl) Tris Base - Tris(hydroxymethyl)-aminomethan (C4 H11 NO3) X-gal - 5-Brom-4-chlor-3-indoxyl-β-D-galactopyranosid (C14 H15 Br Cl NO6) XhoI - Xanthomonas holcicola Restriktions-Endonuklease x 1. EINLEITUNG 1.1. Übersicht von Mikroorganismen Mikroorganismen sind kleinste Lebewesen, die in der Regel ohne technischen Aufwand nicht für das menschlich Auge sichtbar sind. Obwohl sie fast überall vorzufinden sind, war die Existenz von Mikroorganismen vor der Erfindung des Mikroskopes unbekannt. Über mehrere Jahrhunderte wurde die grundlegende Ursache von Epidemien, die tausenden von Menschen den Tod brachten, nie verstanden. Verdorbene Nahrungsmittel und der Mangel an Impfungen und Antibiotika förderten die Bildung von Krankheiten und Seuchen durch pathogene Mikroorganismen, und obwohl nur einige wenige Mikroorganismen pathogen sind, sind es gerade diese, welche die häufigsten infektiösen Krankheiten beim Menschen erzeugen. Eine Hauptgruppe von Mikroorganismen wird repräsentiert von der Gruppe der Bakterien (Tortora, Funke, and Case, 2001, S.2-3). Bakterien sind einzelzellige Organismen, deren genetisches Material nicht von einer speziellen Kernmembran umhüllt sind (Tortora et al., 2001, S.3). Sie sind von einer Zellwand umschlossen, die zum gröβten Teil aus Peptidoglykan besteht. Das Erscheinungsbild von Bakterien spielt eine wesentliche Rolle bei ihrer Identifikation. Der Form nach können Bakterien in drei Hauptgruppen unterteilt werden: Kokken (kugelige Bakterien), Stäbchen (längliche, zylindrische Bakterien) und Spirillen (spiralige, wendelförmige Bakterien). Eine Ausnahme bilden pleomorphe Bakterien, die ihre Form je nach Umgebung oder Entwicklungsstadium ändern können. Weiterhin wichtig für die Klassifikation ist die Anordnung und Organisation der Bakterien: Diplokokken bilden Paare, Streptokokken bilden Ketten und Staphylokokken bilden Haufen (Abb. 1-1). Im Allgemeinen haben Bakterien eine Länge zwischen 0,2 bis 5 µm (Levinson & Jawetz, 1998, S.3; Tortora et al., 2001, S.3-4). 1 Abbildung 1-1. Anordnung und Form von Bakterien. 1: Kokken in Haufen, Ketten oder Paaren 2: Stäbchen: gerade, keulenförmig, zugespitzt oder gebogen 3: Spirillen: leicht geschlängelt oder stark gewunden (Levinson & Jawetz, 1998, S.3) 1.2. Bedeutung von Mikroorganismen Die meisten Mikroorganismen sind dem Menschen und dessen Umwelt von Nutzen, da sie zum biologischen und chemischen Gleichgewicht beitragen. Einige Bakterien helfen zum Beispiel bei der Beseitigung von menschlichen Abfällen oder bei der Eingliederung von freiem Stickstoff in organische Substanzen (Tortora et al., 2001, S.3). Andere Mikroorganismen werden von Säugetieren benötigt, da sie die Verdauung unterstützen oder wichtige Vitamine wie Vitamin B und Vitamin K synthetisieren, welche für Stoffwechsel und Blutgerinnung notwendig sind. Trotz ihres 2 allgemeinen Nutzens, sind einge Mikroorganismen pathogen, und diese sind verantwortlich für einen groβen Teil von Krankheiten im Menschen (Tortora et al., 2001, S.2-3). Ein Mikroorganismus wird als pathogen oder als Krankheitserreger bezeichnet, sobald er eine Krankheit verursachen kann. Einige Krankheitserreger ziehen nach Exposition oft eine Krankheit nach sich, während dies bei anderen seltener ist. Auch opportunistische Krankheitserreger können bei gesunden Menschen nachgewiesen werden. Sie verursachen generell aber keine Krankheiten. Bei immunsupprimierten Personen können diese Organismen jedoch schwere Krankheiten mit immensen Komplikationen auslösen. Die meisten opportunistischen Krankheitserreger sind Teil der menschlichen Normalflora, und sie stellen eine groβe Bedrohung für Menschen mit Immunschwächekrankheiten wie AIDS dar (Levinson & Jawetz, 1998, S.25). 1.3. Normalflora Als Normalflora gelten Bakterien und Pilze, die permanent spezifische Körperregionen besiedeln wie zum Beispiel Haut, Oropharynx, Kolon und Vagina (Levinson & Jawetz, 1998, S.21). Für Menschen haben die Bakterien der Normalflora in drei Fällen eine Bedeutung: Erstens können sie zum pathogenen Erreger werden, wenn beim Menschen eine Immunschwäche besteht, oder wenn sie in eine für sie untypische Körperregion vordringen wie zum Beispiel die inneren Organe. Zweitens können sie den Menschen vor anderen Krankheitserregern schützen, indem sie ökologische Nischen besetzen, und somit die Ansiedlung und Vermehrung anderer Keime erschweren. Drittens dienen sie dem Menschen bei der Produktion von Vitaminen wie Vitamin B und Vitamin K (Levinson & Jawetz, 1998, S.21-22). Die Normalflora kann in vier Hauptbesiedlungsregionen unterteilt werden: Haut, Respirationstrakt, Gastrointestinaltrakt und Urogenitaltrakt. Die Normalflora der Haut ist hauptsächlich vertreten durch Staphylococcus epidermidis. Auf der Hautoberfläche ist das Bakterium nicht pathogen. In den tieferen Hautschichten ist es allerdings als opportunistischer Krankheitserreger meist Ursache von Wund- und Katheterinfektionen. Im 3 Blutkreislauf kann der Erreger, nach Ansiedlung an der Herzinnenwand, eine Endokarditis auslösen. Staphylococcus aureus und anaerobische Organismen wie Peptococcus and Propionibacterium machen nur einen kleinen Teil der Hautflora aus (Levinson & Jawetz, 1998, S.22). Zu den überwiegend mit Normalflora besiedelten Regionen des Respirationstraktes gehören Nase, Mund und Rachen. Eines der wichtigsten Bakterien in der Nasalregion ist Staphylococcus aureus, auf welches später noch genauer eingegangen wird. Im Rachen befinden sich vor allem Staphylococcus epidermidis und Gattungen der Neisseria, welche generell das Wachstum von möglichen Krankheitserregern verhindern. Neisseria meningitidis gilt jedoch unter anderem als einer der häufigsten Erreger der eitrigen Meningitis. Etwa zur Hälfte aller Bakterien in der Mundregion werden von Streptococcus viridans repräsentiert. Diese Bakterien sind ein Teil des Zahnbelages, welcher als Präkursor für Karies gilt. Sie gelten auch als Risiko für eine subakute bakterielle Endokarditis meist in Folge von zahnärztlichen Behandlungen (Levinson & Jawetz, 1998, S.22-23). Die bakterielle Besiedlung des Gastrointestinaltraktes ist je nach Region unterschiedlich. Im Magen sind nur relativ wenige Bakterien vorhanden. Ein gröβerer Teil befindet sich im Ileum terminale, und im Kolon sind die meisten Bakterien vorzufinden. Diese bestehen vor allem aus Escherichia coli, Bacteroides fragilis und Streptococcus faecalis und sind die Hauptverursacher von Harnwegsinfektionen und bakterieller Peritonitis. Streptococcus faecalis kann gegebenfalls auch zur Endokarditis führen (Levinson & Jawetz, 1998, S.23). Die Scheidenflora einer erwachsenen Frau wird vorwiegend durch Gattungen des Lactobacillus besiedelt. Diese verhindern das Wachstum von potenziellen Krankheitserregern in der Vagina, indem sie pH-senkende Säuren produzieren und somit ein saures Milieu erzeugen. Zu den potenziellen Krankheitserregern in dieser Region gehören die B-Streptokokken (Beta-hämolysierende Streptokokken der Serogruppe B). In der Gegenwart von B-Streptokokken können bei einer Geburt Komplikationen wie Sepsis und Meningitis auftreten, da diese Keime bei der Passage durch den Geburtskanal auf das Neugeborene übertragen werden können (Levinson & Jawetz, 1998, S.24). 4 1.4. Bakterielle Infektion Bakterien verursachen Krankheiten, indem sie Toxine produzieren oder, durch Eindringen in den Wirt, eine Entzündung hervorrufen. Toxine können in Exotoxine und Endotoxine eingeteilt werden. Exotoxine sind vom Bakterium freigesetzte Polypeptide, und Endotoxine sind Lipopolysaccharide, die einen Teil der Zellwand bilden. Beide Toxine können allgemeine Symptome wie Fieber oder Schock auslösen selbst wenn der Krankheitserreger selbst nicht mehr präsent ist. Invasive Bakterien verursachen Entzündungen, indem sie sich lokal zu einer groβen Vielzahl vermehren. Sie können generelle Symptome wie Erytheme, Ödeme, Wärme und Schmerzen hervorrufen (Levinson & Jawetz, 1998, S.25). Viele Krankheiten können von Wirt zu Wirt übertragen werden. Wenn die Übertragungsrate hoch ist, wird die Krankheit als ansteckend bezeichnet, wie im Fall der Tuberkulose, bei der die Bakterien durch Tröpfcheninfektion übertragen werden. Die meisten bakteriellen Infektionen stammen aus externen Quellen, aber ein gewisser Teil entsteht auch durch invasive Übergriffe der Normalflora (Levinson & Jawetz, 1998, S.25-26). Erfolgt die Infektion durch ein Bakterium der Normalflora, wurde der Erreger meist von seinem ursprünglichen Platz an eine andere Stelle des Körpers fortbewegt. Ausgangs- und Einganspforte sind für die Übertragung von Krankheitserregern von groβer Bedeutung. Krankheitserreger verlassen ihren Wirt oftmals durch Respirations- und Gastrointestinaltrakt, aber sie können auch durch sexuellen Kontakt, Hautkontakt, Urin, Blutinfusionen, kontaminierten Nadeln oder Insektenstiche übertragen werden (Levinson & Jawetz, 1998, S.26). Die Erreger können den Wirt an verschiedenen Stellen des Körpers befallen. Eine Liste von Krankheitserregern mit den korrespondierenden Krankheiten und ihren Eintrittspforten wird in Tabelle 1-1 geführt. 5 Tabelle 1-1. Krankheitserreger and Krankheiten. Portal of Entry Respiratory tract Pathogen Disease Coccidioides immitis Epstein-Barr virus Coccidioidomycosis Infectious mononucleosis Haemophilus influenzae Histoplasma capsulatum Meningitis Histoplasmosis Influenza virus Influenza Mycobacterium tuberculosis Tuberculosis Neisseria meningitidis Meningitis Common cold Rhinovirus Streptococcus pneumoniae Pneumonia Gastrointestinal tract Skin Genital tract Hepatitis A virus Poliovirus Infectious hepatitis Poliomyelitis Salmonella typhi Shigella dysenteriae Typhoid fever Dysentery Trichinella spiralis Trichinosis Vibrio cholerae Clostridium tetani Cholera Tetanus Plasmodium vivax Malaria Rabies virus Rickettsia rickettsii Rabies Rocky Mountain spotted fever Trichophyton rubrum Candida albicans Tinea pedis (athlete's foot) Vaginitis Chlamydia trachomatis Urethritis Neisseria gonorrhoeae Gonorrhea Treponema pallidum Syphilis (Levinson & Jawetz, 1998, S.27) 6 Levinson & Jawetz (1998, S.26) beschreiben sieben Infektionsstadien: 1. Übertragung von einer externen Quelle an die Eintrittspforte 2. Umgehung der ersten Verteidigungslinie des Wirts (z.B. Haut, Magensäure) 3. Haftung an Schleimhäute, meist durch Pili der Bakterien 4. Kolonisation durch Vermehrung der Bakterien an der Haftungsstelle 5. Krankheitssymptome werden verursacht durch Produktion von Toxinen oder Invasion gefolgt von Entzündungsreaktionen 6. Unspezifische und spezifische Immunantwort des Wirts im Stadium 3,4,5 7. Fortschreiten oder Auflösen der Krankheit 1.4.1. Invasive Bakterien Bakterien verursachen Krankheiten durch Invasion des Gewebes mit darauffolgender Entzündungsreaktion. Invasive Bakterien setzen verschiedene Enzyme frei, die den Wirt angreifen und schädigen können (Levinson & Jawetz, 1998, S.27). Kollagenasen und Hyaluronidasen sind wichtige Enzyme, die zum Beispiel von Streptococcus pyogenes, einem Erreger von Scharlach und Mandelentzündungen, freigesetzt werden. Diese Enzyme zersetzen Kollagen bzw. Hyaluronsäure, was dem Bakterium das Durchbrechen der Subkutis ermöglicht. Es folgt oft eine Zellulitis (Levinson & Jawetz, 1998, S.27). Die Freisetzung von Koagulase durch Staphylococcus aureus fördert die Ausfällung von Fibrinogen zu Fibrin. Dieser Fibrinmantel umhüllt dann das Bakterium, wodurch das Bakterium vor Phagozytose geschützt wird. Einen direkten Schutz vor Phagozytose erlangen Staphylokokken durch die Produktion von Leukozidin, das neutrophile Granulozyten und Makrophagen zerstört (Levinson & Jawetz, 1998, S.27; Tortora et al., 2001, S.582). Neisseria gonorrhoeae, Haemophilus influenzae, und Streptococcus pneumoniae produzieren Immunglobulin A (IgA) Proteasen, die IgA zersetzen und es den Bakterien ermöglichen, sich an Schleimhäute anzusiedeln (Levinson & Jawetz, 1998, S.27). 7 1.4.2. Toxine Die von Bakterien produzierten Toxine können in Exotoxine und Endotoxine unterteilt werden. Exotoxine werden von grampositiven und gramnegativen Bakterien produziert. Sie sind von Bakterien abgesonderte Polypeptide, deren zugehörige Gene normalerweise auf Plasmiden oder Bakteriophagen zu finden sind. Exotoxine gehören mit zu den giftigsten bekannten Stoffen. Die geringe Menge von nur 1 µg Tetanustoxin kann für Menschen bereits tödlich sein (Levinson & Jawetz, 1998, S.29-33). Das von Clostridium tetani produzierte Tetanustoxin blockiert die Freisetzung des inhibitorischen Neurotransmitters Glycin, ein Vorgang der zu Muskelkrämpfen und spastischer Paralyse führt. Ein weiteres wichtiges Exotoxin ist das Botulinumtoxin, das von Clostridium botulinum produziert wird. Dieses Nervengift verursacht Paralyse, indem es die Freisetzung von Acetylcholin an der Synapse verhindert. In kleineren Mengen wird dieses Gift auch zu kosmetischen Zwecken benutzt, wobei es meist unter dem Namen Botox bekannt ist. Auf das von Staphylococcus aureus produzierte Toxische Schocksyndrom Toxin wird später noch eingegangen. Endotoxine sind nur in gramnegativen Bakterien vorhanden. Es sind Lipopolysaccharide, die in die Zellwand eingebaut sind. Die Gene für die entsprechenden Lipopolysaccharide sind im bakteriellen Chromosom zu finden. Obwohl Endotoxine, relativ zu den Exotoxinen, weniger giftig auf den menschlichen Körper wirken, können auch sie Symptome wie Schock oder Fieber hervorrufen. Die meisten Fälle septischer Schocks werden wahrscheinlich durch Endotoxine gramnegativer Bakterien verursacht (Levinson & Jawetz, 1998, S.29-36). 8 1.5. Immunabwehr Der menschliche Körper besitzt eine Vielzahl von Abwehrmechanismen, um sich gegen Krankheitserreger zu schützen. Unspezifische und spezifische Immunabwehr schützen den Körper generell vor Mikroorganismen und bestimmten Erregern. Die Haut bildet den wichtigsten Teil der unspezifischen Immunabwehr, da sie eine physikalische Barriere bildet. Sie enthält Fettsäuren, die durch ihren niedrigen pH antimikrobiell wirken. Der Respirationstrakt wird geschützt durch Zilien und Schleim, welche Bakterien abfangen und ausstoβen können. Desweiteren gehören Granulozyten, natürliche Killerzellen, Makrophagen, Lysozyme, Komplemente und Interferone zur unspezifischen Immunabwehr. Zur spezifischen Immunabwehr des Körpers gehören Antikörper und T-Lymphozyten (Levinson & Jawetz, 1998, S.38-39). T-Lymphozyten schützen den Menschen durch direkte Abtötung von Krankheitserregern, durch Mobilisation von Phagozyten oder anderen Lymphozyten und durch Sekretion von Chemikalien. Antikörper schützen den Körper durch Neutralisation von Toxinen, durch Inhibition der Anhaftung von Krankheitserregern an die Wirtszelle und durch Hilfe bei der Lyse von Krankheitserregern (Tortora e tal., 2001, S.421). Durch Evolutionsprozesse haben die meisten Bakterien Mechanismen zur Umgehung der menschlichen Immunabwehr entwickelt. Einige Bakterien produzieren eine Kapsel um ihre Zellwand, um die Anbindung von Phagozyten zu verhindern. Staphylococcus aureus benutzt Leukozidin und einen Fibrinmantel, um sich vor Phagozytose beziehungsweise vor Leukozyten und Makrophagen zu schützen (Levinson & Jawetz, 1998, S.27-28). Ist das Immunsystem des Menschen stark geschwächt, können bakterielle Infektionen meist nur noch mit Antibiotika therapiert werden. Die erfolgreiche Behandlung mit Antibiotika wurde jedoch in den letzten Jahren erheblich beeinträchtigt, durch die immer gröβer werdende Anzahl von bakteriellen Resistenzen. Klinisch ist zum Beispiel Staphylococcus aureus von groβer Bedeutung, da es relativ schnell eine Resistenz gegen Penicilline entwickelt (Tortora et al., 2001, S.324). 9 1.6. Staphylococcus aureus Das grampositive fakultativ anaerobe Bakterium Staphylococcus aureus (Abb. 1-2) wurde nach seinen gelb gefärbten und in Trauben angeordneten Kolonien benannt. Es wächst relativ gut in einer Umgebung mit hohem osmotischen Druck und niedriger Feuchtigkeit. Durch diese Eigenschaften können Staphylokokken in Nasenschleim, auf der Haut und in Essen mit hoch osmotischem Druck wachsen (Tortora et al., 2001, S.323). Staphylokokken sind relativ resistent gegen Umweltbelastungen. Sie können in vegetativer Form eine Temperatur von 60oC bis zu 30 Minuten lang überleben. Die gelbe Farbe schützt vor Trockenheit und Strahlung, und sie erleichtert das Überleben der Bakterien auf der Haut (Tortora et al., 2001, S.690). |-----------------------------------------| ~ 2 µm Abbildung 1-2. © Dr. Tony Brain/ SPL/ Custom Medical Stock Staphylococcus aureus. S. aureus in Trauben angeordneten Kolonien. Jedes einzelne Bakterium besitzt eine runde Form. Die gold-gelbe Farbe kann auf dem schwarz-weiss Foto nicht gesehen werden. (Tortora et al., 2001, S.324). 10 1.6.1. Koagulase Als Teil der Normalflora des Respirationstraktes und der Haut kann Staphylococcus aureus potenziell zum Krankheitserreger werden, wenn die Hautschranke durch physische Belastungen oder medizinische Eingriffe geschädigt wird. Man unterscheidet Koagulase produzierende (Koagulase positive) und nicht Koagulase produzierende (Koagulase negative) Bakterienstämme. Koagulase katalysiert die Bildung von Fibringerinnsel aus Fibrinogen. Das Bakterium wird so vor Phagozytose geschützt, da der entzündete Bereich durch einen Fibrinmantel abgedeckt ist. Unter den Staphylokokkenspezies hat Staphylococcus aureus die höchste pathogene Potenz. Fast alle pathogenen Bakterienstämme produzieren Koagulase (Levinson & Jawetz, 1998, p.27; Tortora et al., 2001, S.582). 1.6.2. Toxisches Schocksyndrom Bestimmte Bakterienstämme von Staphylococcus aureus produzieren das Toxische Schocksyndrom Toxin (TSST), welches eine Infektion mit hohem Fieber, Erbrechen und in einigen Fällen den Tod hervorrufen kann. TSST heftet sich an MHC-Klasse-II (HauptHistokompatibilitätskomplex) Proteine. Die Komplexverbindung kann mit den β-Ketten von T-Zell-Rezeptoren interagieren, wonach hohe Mengen von Interleukinen ausgeschüttet werden. Symptome wie Fieber, Hautausschlag oder Schock können die Folge sein (Levinson & Jawetz, 1998, S.34; Tortora et al., 2001, S.324, 584). 11 1.6.3. Lebensmittelvergiftung Ein Enterotoxin von Staphylococcus aureus ist eines der Hauptursachen von Gastroenteritis. Das Toxin steuert das Brechzentrum des Gehirns an, und verursacht Bauchkrämpfe und Diarrhoe (Tortora et al., 2001, S.691). In den meisten Fällen kann sich die betroffene Person schon nach 24 Stunden erholen (Tortora et al., 2001, S.324, 691). Da Staphylococcus aureus einen Teil der Normalflora von Haut und Nase darstellt, ist dessen Verbreitung auf Nahrungsmittel während ihrer Zubereitung nicht selten. Wird die Nahrung nur unter 60°C erhitzt oder mit Salz zubereitet (hoher osmotischer Druck), so kann Staphylococcus aureus konkurrierende Bakterien überwachsen. Erfolgt die Lagerung des Essens so lange, dass sich die Bakterien auf eine Anzahl von eine Million pro Gramm vermehren können, ist bereits genügend Toxin vorhanden, um eine Krankheit hervorzurufen. Auch wenn die Nahrung wieder erhitzt wird und dadurch alle Bakterien absterben, bleibt das Toxin jedoch erhalten, weil es bis zu einer halben Stunde unter kochenden Temperaturen standhalten kann. Das Wiederaufwärmen von Essen bietet demnach keinen Schutz vor einer möglichen Lebensmittelvergiftung (Tortora et al., 2001, S.691). Obwohl die Gastroenteritis bei gesunden Menschen generell nicht tödlich ist, kann sie zur Austrocknung (Exsikkose) führen und stellt daher für ältere Menschen oder für Bewohner von Pflegeheimen eine mögliche Lebensgefahr dar (Tortora et al., 2001, S.691). 1.6.4. Staphylogenes Lyell-Syndrom und andere Infektionen Das Staphylogene Lyell-Syndrom umfasst schwere Hauterkrankungen, die von Exfoliatin Toxin bildenden Stämmen des Staphylococcus aureus, meist vom Phagentyp 71, hervorgerufen werden. Meistens sind Regionen um Mund und Nase befallen. Die Haut wird faltig und löst sich ab. Das darunter liegende Gewebe ist hellrot und glänzend. Der Prozess weitet sich auf die umliegende Haut aus, und nach zwei Tagen löst sich die Haut schon bei Berührung in Schichten ab. Die schwerste Form des Staphylogenen Lyell-Syndroms wird bei Neugeborenen oder Kleinkindern vorgefunden (Dermatitis exfoliativa neonatorum oder 12 Morbus Ritter). Der Ursprung der Krankheitserreger kann auf das Personal der Säuglingsstation zurückverfolgt werden. Die Übertragung erfolgt meist über Nase, Haut oder perianalen Kontakt (Levinson & Jawetz, 1998, S.22). Auch Impetigo contagiosa ist meist auf das Personal der Säuglingsstation zurückzuführen. Bei der Impetigo bilden sich auf der Haut juckende rote Ausschläge mit Pusteln. Durch Kratzen wird der Erreger auf andere Hautstellen übertragen. Nach Eintrocknung der Pusteln entstehen dann Krusten (Tortora et al., 2001, S.444, 582-583). Auf der Haut kann Staphylococcus aureus in natürliche Hautöffnungen wie Haarfollikel eindringen, wodurch eine Follikulitis entstehen kann. Eine Follikulitis an den Augenwimpern wird auch Gerstenkorn oder Hordeolum genannt. Weitet sich die Follikulitis aus können sich Furunkel oder Abszesse bilden, welche operativ behandelt werden müssen (Tortora et al., 2001, S.583). Operative Wundinfektion, Hämatosepsis (Blutvergiftung), Pneumonie und Harnwegsinfektion sind weitere Beispiele von Infektionen, die durch Staphylococcus aureus verursacht werden (Tortora et al., 2001, S.421). 1.6.5. Akute Endokarditis Eine Endokarditis ist eine Entzündung der inneren Muskelschicht des Herzens. Ist die Entwicklung der Entzündung relativ schnell und durch ein Bakterium verursacht, handelt es sich um eine akute bakterielle Endokarditis (Tortora et al., 2001, S.628). Da sich das Bakterium Staphylococcus aureus massenhaft auf der menschlichen Haut befindet, besteht eine hohe Wahrscheinlichkeit, dass das Bakterium bei Hautdefekten in die Blutbahn gelangen kann. Die Infektion kann sich vom Eintrittspunkt über die Blutbahn bis an die Herzklappen ausbreiten. Setzen sich die Keime dort nieder, dann kann es schnell zur einer Schädigung der Herzklappen führen. Unbehandelt kann dieser Zustand für die betroffene Person tödlich enden. Als Endokarditisprophylaxe verschreibt man oft Penicillin vor medizinischen Eingriffen wie zum Beispiel einer Tonsillektomie. Das Risiko einer Endokarditis besteht jedoch trotzdem, da viele Stämme von Staphylococcus aureus eine Resistenz gegen Penicilline entwickelt haben (Tortora et al., 2001, S.324, 628). 13 1.6.6. Antibiotische Resistenz von Staphylococcus aureus Als Krankheitserreger stellt Staphylococcus aureus im Krankenhaus eine besonders groβe Bedrohung dar. Das Bakterium befindet sich auf Patienten, Krankenhauspersonal und Besuchern. Ständig besteht die Gefahr einer Infektion von operativen Wunden. Eine Behandlung von solchen Infektionen erweist sich als extrem schwierig, da durch die groβe Präsenz an Krankenhausantibiotika sehr viele Bakterienstämme Resistenzen entwickelt haben. Über die letzten Jahrzehnte hat Staphylococcus aureus Resistenzen gegen jegliche Arten von Penicillinen entwickelt. Nur noch ungefähr 10% aller Stämme sind Penicillin empfindlich. Das Antibiotikum Vancomycin wird manchmal als letzte Instanz zur Behandlung von penicillinresistenten Stämmen verwendet. Vancomycin hemmt die Synthese der bakteriellen Zellwand. Das Antibiotikum wirkt relativ toxisch auf den menschlichen Körper und darf nur mit Vorsicht verschrieben werden (Tortora et al., 2001, S.559, 583). Das kürzliche Auftreten von vancomycinresistenten S. aureus Stämmen stellt eine medizinische Notfallsituation dar. In den USA hat die Food and Drug Administration (FDA) daraufhin das Antibiotikum Synercid® zugelassen. Synercid® hemmt die Proteinsynthese durch Angriff auf bakterielle Ribosomen. Das Antibiotikum hat jedoch nur einen eingeschränkten Nutzen, da es sehr teuer und sehr viele Nebenwirkungen hat (Tortora et al., 2001, S.559). Offensichtlich spielt die Forschung nach neuen Antibiotika wegen der ständig wachsenden Anzahl bakterieller Resistenzentwicklungen eine wesentliche Rolle in der Zukunft der Medizin. 1.6.7. Nosokomiale Infektionen und AIDS Eine nosokomiale Infektion ist eine Infektion, die durch einen Krankenhausaufenthalt entsteht und mit diesem zeitlich in Verbindung steht. Solch eine Infektion erfordert gewöhnlich das Zusammenspiel dreier Faktoren: Bakterien im Krankenhaus-Milieu, kompromittierter Patient und Übertragungsweg. Die Prävalenz von nosokomialen Infektionen 14 wird auf 5-10 % der Patienten geschätzt. In den USA sind jährlich etwa zwei Millionen Patienten von nosokomialen Infektionen betroffen, wobei davon fast 90.000 Fälle tödlich enden (Ayliffe et al., 1992, S.1-11; Tortora et al., 2001, S.420). In den vierziger- und fünfziger Jahren war Staphylococcus aureus der Haupterreger für nosokomiale Infektionen. Patienten wurden erfolgreich mit Penicillin behandelt. In den achtziger Jahren jedoch tauchten antibiotikaresistente Stämme auf, die heute bis zu 34% der nosokomialen Infektionen ausmachen (Tortora et al., 2001, S.420). Bei gesunden Menschen kann sich das Immunsystem gegen die meisten Bakterienstämme wehren. T-Lymphozyten können Krankheitserreger auf direktem oder indirektem Wege angreifen, indem sie Chemikalien absondern oder Phagozyten und andere Lymphozyten anregen. HIV ist ein Virus, der bestimmte T-Lymphozyten zerstört, die sogenannten T-Helferzellen. Das Immunsystem des Betroffenen wird erheblich geschwächt und somit hoch anfällig für opportunistische Krankheitserreger. Typische Erreger sind Streptococcus pneumoniae und Haemophilus influenzae, die beide Pneumonien verursachen können. Pseudomonas aeruginosa und Mycobacterium tuberculosis sind noch gefährlicher für HIVinfizierte Patienten, da eine akute Pneumonie mit hoher Sterblichkeitsrate oder eine Tuberkulose folgen kann (Deodhar, 2003, ¶ 5-6; Tortora et al., 2001, S.420-421). Die Zunahme von nosokomialen Infektionen und die Entwicklung antibiotischer Resistenzen stellt ein groβes klinisches Problem dar, besonders für HIV-infizierte Patienten. Die Forschung nach neuen antibiotischen Medikamenten ist daher unumgänglich. 1.7. Aufbau der Zellwand Für die Entwicklung von wirksamen Antibiotika muss der Aufbau von Bakterien genau analysiert werden. Fast alle Prokaryoten besitzen eine Zellwand. Die Zellwand besitzt eine groβe Anzahl von verschiedenen Funktionen: sie bestimmt die Form und das Aussehen von Bakterien, sie schützt die Bakterien vor Belastungen und Veränderungen der Umwelt, sie umhüllt die fragile Plasmamembran, sie schützt vor osmotischem Druck, sie verhindert den 15 Verlust von Zytoplasma und sie erhält das Bakterium als separate Entität (Moat & Foster, 1995, S.2; Tortora et al., 2001, S.85-86). Ein grundlegender Bestandteil der Zellwand ist das Peptidoglycan. Peptidoglycan ist ein lineares Polymer aufgebaut aus wechselnden Einheiten von N-Acetylglucosamin (NAG) und N-Acetylmuraminsäure (NAM) (Moat & Foster, 1995, S.2) (Abb. 1-3). An den NAMs befinden sich Seitenketten, die aus vier sich zwischen D- und L-Form abwechselnden Aminosäuren bestehen (Abb. 1-4). Je nach Organismus sind diese Seitenketten direkt miteinander verbunden oder sie können durch kurze Aminosäureketten quer verbrückt sein. Bei Staphylococcus aureus kann die Anzahl von Querbrücken zwischen den angrenzenden Peptiden nachezu 100% ausmachen (Moat & Foster, 1995, S.3; Tortora et al., 2001, S.86). Abbildung 1-3. N-Acetylglucosamin (NAG) verbunden mit NAcetylmuraminsäure (NAM).(Tortora et al., 2001, S.47) 16 Abbildung 1-4. L-Alanin und D-Alanin. (Tortora et al., 2001, S.86) Man beachte die unterschiedliche dreidimensionale Anordnung. Man kann zwischen Bakterien mit grampositiven und gramnegativen Zellwänden unterscheiden. Die Gram-Färbung basiert auf eine differenzierende Färbung der Bakterienzellwand, wobei bestimmte Bakterien nach einer Spülung mit einer 95% Ethanollösung einen kristallvioletten Farbstoff beibehalten. Grampositive Bakterien behalten den Farbstoff, während bei gramnegativen Bakterien der Farbstoff wieder ausgespült wird. Diese beiden Arten von Bakterien unterscheiden sich grundsätzlich (Moat & Foster, 1995, S.2). Die äuβere Schicht von grampositiven Bakterien besteht aus einer Zellmembran und einer Zellwand, welche die Zellmembran umfasst. Viele Schichten aus Peptidoglycan bilden eine relativ dicke Zellwand. Im Gegensatz zu gramnegativen Bakterien, besitzen grampositive Bakterien eine höhere Anzahl von Peptidoglycan Querbrücken (Abb.1-5 und 1-6). Eine besondere Eigenschaft ist die Einbindung von Teichonsäuren in die Zellwand. Teichonsäuren 17 werden aufgeteilt in Lipo-Teichonsäuren, welche mit der Plasmamembran verbunden sind und sich durch die gesamte Peptidoglycanschicht erstrecken, und Wand-Teichonsäuren, die mit der Peptidoglycanschicht verankert sind (Moat & Foster, 1995, S.2-3; Tortora et al., 2001, S.86) (Abb. 1-7). Abbildung 1-5. Peptidoglycan Querbrücken bei Bakterien. G = N-Acetylglucosamin (NAG) und M = N-Acetylmuraminsäure (NAM) diagonale Linie: β-1,4 Verbindung zwischen NAG und NAM vertikale Linie: Seitenkette horizontale Linie: Querbrücke Querbrücken kommen häufiger vor bei grampositiver Zellwand (Moat & Foster, 1995, S.4) Die Zellwand gramnegativer Bakterien hat einen komplexeren Aufbau im Vergleich zu grampositiven Bakterien. Die Peptidoglycanschicht ist relativ dünn und besteht bei manchen Bakterien nur aus einer einzigen Schicht. Eine Lipiddoppelmembran, die sogenannte äuβere Membran, bildet die äuβerste Schicht der Zellwand. Die äuβere Membran besteht aus Phospholipiden, Lipopolysacchariden, Enzymen und anderen Proteinen wie den Porinen (Abb. 1-8). 18 Abbildung 1-6. Peptidoglycan bei grampositven Bakterien. In jeder Seitenkette befinden sich vier Aminosäuren; das Kohlenhydratgerüst besteht aus einem abwechselnden Muster von β-1,4 verbundenen NAGs and NAMs; die Querbrücken bestehen aus kurzen Aminosäureketten. (Tortora et al., 2001, S.87) Der Lipidabschnitt der Lipopolysaccharide ist für den Organismus giftig und wird als Endotoxin bezeichnet. Hauptsächlich schützt die äuβere Membran das Bakterium vor Phagozytose, Verdauungsenzymen, Detergentien, Schwermetallen und Gallensalzen. Zwischen äuβerer und innerer Membran befindet sich der periplasmatische Raum, in dem auch die dünne Peptidoglycanschicht liegt. Hier können auβerdem degradierende Enzyme und Transportproteine vorgefunden werden (Moat & Foster, 1995, p.2-3; Tortora et al., 2001, p.86-88). Im allgemeinen sind grampositive Bakterien relativ widerstandsfähig gegen physische Beanspruchung, reagieren aber empfindlich gegen schädliche Chemikalien, Enzyme oder Antibiotika. Im Gegensatz dazu sind gramnegative Baketrien mehr vor Chemikalien und Molekülen geschützt, jedoch sind sie anfälliger für physische Belastungen (Tortora et al., 2001, p.89). 19 Abbildung 1-7. Zellwand bei grampositiven Bakterien. (Tortora et al., 2001, S.87) Mehrere Peptidoglycanschichten sind durch Teichonsäuren untereinander und mit der Plasmamembran verbunden. Abbildung 1-8. Zellwand bei gramnegativen Bakterien. (Tortora et al., 2001, p.87) Die Zellwand von gramnegativen ist etwas komplexer als bei grampositiven Bakterien, jedoch ist die Peptidoglycanschicht relativ dünn und simpel aufgebaut. Teichonsäuren sind nicht vorhanden. 20 1.8. Antibiotika Bei der Entwicklung eines Antibiotikums ist selektive Toxizität ein grundlegendes Konzept. Das Ziel ist die Abtötung oder Wachstumshemmung von Bakterien, ohne dabei den Menschen zu schädigen. Dazu müssen Unterschiede zwischen menschlichen und bakteriellen Zellen untersucht werden. Bakterielle Zellwand, Ribosomen, Nucleinsäuren und Zellmembrane haben sich als wirksame Angriffspunkte für Antibiotika erwiesen, da sich diese Einheiten wesentlich von denen der menschlichen Zellen unterscheiden. Die bakterielle Zellwand wird zum Beispiel durch Penicillin angegriffen. Penicillin hemmt die Peptidoglycansynthese durch Inhibition einer Transpeptidase, die für die Vernetzung von Peptidoglycan benötigt wird. Die bakterielle Zellwand ist somit geschwächt und Wasser kann frei in das hoch osmotische Innere der Zelle eindringen. Das Bakterium platzt und stirbt (Levinson & Jawetz, 1998, S.48-49). Die bakterielle Proteinsynthese wird durch Tetrazyklin gehemmt, welche die ribosomale 30S-Untereinheit angreift. Quinolone und Rifampicine inhibieren die Nucleinsäurensynthese von DNA beziehungsweise mRNA. Polymyxin unterbricht das Gefüge der bakteriellen Zellmembran, indem es mit Phospholipiden interagiert. Diese Medikamente töten oder hemmen das Wachtum von Bakterien, indem sie die für Prokaryoten spezifischen Eigenschaften angreifen (Levinson & Jawetz, 1998, S.49). Die Einnahme von Antibiotika führt nicht immer zum gewünschten Therapieerfolg. Die Wirksamkeit von Penicillinen zum Beispiel wird durch verschiedene Faktoren vermindert. Die Wirkung von Penicillin basiert auf einen β-Lactam-Ring, der aber durch β-Lactamasen inaktiviert werden kann. Bestimmte Bakterienstämme werden penicillinresistent, indem sie β-Lactamasen produzieren. Ebenso wirkt Penicillin nicht gegen gramnegative Bakterien, da diese über ihrer Peptidoglycanschicht noch zusätzlich eine äußere Membran besitzen. Ein weiterer Nachteil des Penicillins ist die relativ häufige allergische Reaktion der Patienten gegen dieses Medikament (Levinson & Jawetz, 1998, S.51). Neuere Medikamente greifen verschiedentliche Bereiche der Zellwandsynthese an. Vancomycin wird zur Therapie von resistenten Bakterienstämmen verwendet. Es verhindert den Einbau von biologischen Vorstufen in das Peptidoglycan. Vancomycin ist jedoch relativ 21 toxisch, so dass es nicht regelmässig verordnet oder eingenommen werden darf. Cycloserin hemmt die Synthese vom D-Alanyl-D-Alanin Dipeptid. Es ist ein D-Alanin Analogstoff, der Alanin Razemase kompetitiv hemmt. Auch dieses Medikament hat viele Nebenwirkungen und wird daher in der Klinik nur zur Therapie von resistenten Bakterien benutzt (Heritage, 2003, ¶ 5; Levinson & Jawetz, 1998, S.53). 1.9. Alanin Razemasen Alanin Razemasen (EC 5.1.1.1) [Abb. 1-9] sind Pyridoxal 5’-Phosphat abhängige Enzyme, welche die Razemisierung zwischen L- und D-Alanin katalysieren (Strych et al., 2000, p.290). Sie sind der einzig beschriebene biosynthetische Weg für die Produktion der Aminosäure D-Alanin, die eine notwendige biologische Vorstufe der Synthese von Peptidoglycan in grampositiven und gramnegativen Bakterien ist (Strych et al., 2000, p.290; Wasserman et al., 1984). D-Alanin liegt üblicherweise als D-Alanyl-D-Alanin Dipeptid vor in der C-terminalen Position vom UDP-N-Acetylmuramyl (MurNAc)-Pentapeptid Präkursor von Peptidoglycan (van Heijenoort, 1994). Der vorletzte D-Alanin Rückstand dieses Präkursors ist direkt beteiligt an der Vernetzung von angrenzenden Peptidoglycansträngen während der Zellwandsynthese (Matsuhashi, 1994). Alanin Razemasen sind sowohl ubiquitär als auch unentbehrlich für Bakterien. Dadurch sind diese Enzyme ein logisches Ziel für die Entwicklung von antibiotischen Medikamenten (Lambert & Neuhaus, 1972). In Escherichia coli werden zwei Alanin Razemasen beschrieben. Die alr-kodierte Alanin Razemase (biosynthetische Razemase) wird grundlegend exprimiert, wogegen das dadXkodierte Enzym (katabolische Razemase) für den L-Alanin Katabolismus notwendig ist, um ein Substrat für die D-Alanin spezifische Dehydrogenase, die durch das dadA Gen kodiert wird, bereitzustellen (Lilley et al., 1993; Wijsman, 1972; Wild et al., 1985). Das dadX und das dadA Gen bilden ein Operon, das durch L-Alanin positiv stimuliert und durch Glukose unterdrückt wird (Lobocka et al., 1994; Wild et al., 1985). 22 Das Produkt des dadX Gens ist die Hauptquelle der Alanin Razemase Aktivität (85% der gesamten Aktivität) und es dient wahrscheinlich als eine D-Alanin Nebenquelle für die Biosynthese der Zellwand (Wasserman et al., 1984). Nur ein Alr - DadX – Doppelmutant ist D-Alanin abhängig um zu wachsen (Wild et al., 1985). Die Entwicklung eines Moleküls oder eines Medikamentes, das Alanin Razemasen inhibieren kann, würde sich als wirkungsvolles Antibiotikum erweisen. Ohne eine D-Alanin Synthese kann auch keine Peptidoglycansynthese erfolgen. Dies wäre letal für grampositive und gramnegative Bakterien, so dass ein solches Medikament als Breitband-Antibiotikum eingesetzt werden könnte. 1.10. Forschungsziele Das Hauptziel dieses Projektes ist das Protein Alanin Razemase von verschiedenen Bakterien zu isolieren, klonieren und zu reinigen, um die Proteinstruktur mittels Röntgenkristallographie zu analysieren. Im Durchschnitt, variiert die Primärstruktur der Razemasen bei verschiedenen Bakterien bis zu 60 Prozent. Ein Vergleich der Proteinstruktur der unterschiedlichen Razemasen kann einen Aufschluss über das aktive Zentrum geben. Informationen über die Lokalisation und Eigenschaften des aktiven Zentrums sind notwendig, um einen effektiven Inhibitor des Enzyms zu synthetisieren. Da menschliche Zellen kein D-Alanin benötigen, wäre ein effektiver Inhibitor des Enzyms als neues und potentes Antibiotikum gegen grampositive und gramnegative Bakterien geeignet. Bei dieser Arbeit wurden Alanin Razemase Gene des grampositiven Bakteriums Staphylococcus aureus extrahiert und kloniert. Anschlieβend wurden die Alanin Razemase Gene in ein Überexprimierungssystem transformiert. Die exprimierten Proteine wurden gereinigt und zur weiteren Analyse an die Kristallographie übergeben. 23 Abbildung 1-9. Alanin Razemase von Bacillus stearothermophilus. Ein computergeneriertes 3D-Modell mit Alpha-Helix und BetaFaltblatt des Enzyms (http://www.biochem.ucl.ac.uk/bsm/pdbsum/1bd0/tracel.html, 9. Mai, 2002) 24 2. METHODIK UND MATERIALIEN 2.1. DNA Extraktion Staphylococcus aureus wurde von einer einzelnen Kolonie aufgepickt und über Nacht in mit 2 mL LB-Medium (Luria-Bertani-Medium) gefüllten Reagenzgläsern angewachsen. Am nächsten Tag wurden die Proben in Eppendorf Röhrchen umgefüllt, und 3 Minuten lang bei 14.000 Upm in einer Eppendorf Zentrifuge zentrifugiert. Der Überstand wurde durch Vakuum Absaugung entfernt und das Pellet am Röhrchenboden durch wirbelnde Bewegungen in 200 µL Zellsuspensionslösung I resuspendiert. 20 µL kaltes Lysozym wurde hinzugefügt und 2 Minuten bei Raumtemperatur belassen. Nachdem 10 µL von 10% SDS (Sigma) und 200 µL von Zell-Lyselösung II hinzugefügt wurden, wurde die Mixtur 35 Minuten lang bei 37 0C inkubiert. 400 µL gepuffertes Phenol kam hinzu und die Mixtur wurde wiederholt mit 1-minütigen Zwischenpausen gevortext. Die Lösung wurde dann 2 Minuten lang bei 14.000 Upm zentrifugiert und etwa 350 µL des RNA und DNA beinhaltenden Überstandes wurde in ein frisches Eppendorf Röhrchen transferiert. Ein Volumen einer Phenol-Trichloromethan (50:50) Lösung wurde addiert. Die Lösung wurde gevortext und dann 2 Minuten bei 14.000 Upm zentrifugiert. Etwa 350 µL des Überstandes wurde isoliert und 3 M Natrium Acetat (1/10 Volumen des Überstandes) und 100% Ethanol (2-faches Volumen des Überstandes) wurden addiert. Die Lösung wurde gevortext und anschliessend 10 Minuten bei 14.000 Upm zentrifugiert. 300 µL Ethanol wurden zum Pellet addiert und die Lösung wurde gevortext und für weitere 10 Minuten bei 14.000 Upm zentrifugiert. Der Überstand wurde verworfen und das Pellet durch wiederholte Invertierung des Röhrchens in 500 µL Ethanol resuspendiert. Die Lösung wurde für 10 Minuten bei 14.000 Upm zentrifugiert und der Überstand wurde verworfen. Das Pellet wurde luftgetrocknet und für den weiteren Gebrauch in TE-Puffer resuspendiert. 25 2.2. Speed Preparation Eine Mikro-Pipette wurde verwendet, um 1,5 mL jeder einzelnen Übernacht-Kultur in ein separates frisches Eppendorf Röhrchen zu transferieren. Die Röhrchen wurden für 2 Minuten bei 14.000 Upm zentrifugiert und der Überstand wurde verworfen. 400 µL von Speed Preparation Mix (SPM) und 400 µL PCI wurden unter einer Absaughaube zum Pellet addiert. Das SPM beinhaltet Lithiumchlorid, EDTA, TRIS-Puffer (pH 8,0) und Tween Detergens. Das PCI beinhaltet 25 Volumen Phenol, 24 Volumen Chloroform und 1 Volumen IsoamylAlkohol. Die Mixtur wurde gevortext und anschliessend 5 Minuten bei 14.000 Upm zentrifugiert. 200 µL des Überstandes wurde in frische beschriftete Eppendorf Röhrchen transferiert. 500 µL von 100% Ethanol wurde hinzugefügt und die Mixtur wurde anschliessend gevortext und 5 Minuten bei 14.000 Upm zentrifugiert. Das überschüssige Ethanol wurde verworfen und 200 µL von 70% Ethanol wurde zum Pellet addiert. Die Eppendorf Röhrchen wurden gevortext und anschliessend 2 Minuten bei 14.000 Upm zentrifugiert. Das Ethanol wurde Vakuum-entfernt, die Eppendorf Röhrchen wurden luftgetrocknet und die Pellets wurden auf Eis belassen, bevor sie in 50 µL TE-Puffer resuspendiert wurden. 2.3. Amplifikation der Alanin Razemase Gene durch Polymerase-Kettenreaktion (PCR) Für die Polymerase-Kettenreaktion (PCR) wurde eine Konzentration von 100 nM Oligonucleotid Primer verwendet. DNA Konzentration wurden durch UV- Spektroskopie bei 260 nm Wellenlänge bestimmt. Die PCR Mixtur enthielt 10 µL von 10X Puffer, 5 µL dNTPs, 2 µL von S. aureus DNA, 80 µL destilliertes Wasser und 1 µL Taq-Polymerase. Die Mixtur wurde in zwei PCR Röhrchen mit jeweils 48 µL geteilt. 1 µL von Vorwärts-Primer und 1 µL 26 von Rückwärts-Primer wurden jeweils für jedes der zwei Alanin Razemase Gene (Salr1 and Salr2) der PCR Mixtur beigefügt. Die PCR Maschine wurde eingestellt auf: 5 Minuten bei 96 0C gefolgt von 20 Zyklen mit 30 Sekunden bei 96 0C, 30 Sekunden bei 55 0C und 1 Sekunde bei 720 C. Der letzte Schritt wurde auf 5 Minuten bei 72 0C gestellt, und die Lagerungstemperatur betrug 4 0C. 2.4. Chroma SpinTM Chroma SpinTM wurde zur Entfernung von dNTPs und Oligonucleotid Kontaminanten von kleiner als 100 Basen verwendet. Die Chroma SpinTM Säule wurde zwei Mal invertiert, um die Gel Matrix zu resuspendieren. Das Ende der Säule wurde abgebrochen und die Säule wurde oben auf ein 2 mL Mikro-Zentrifuge Röhrchen gesetzt. Die Säule wurde zusammen mit dem Röhrchen 5 Minuten lang bei 700 x g zentrifugiert, um die Flüssigkeit von der Matrix zu trennen. Das Mikro-Zentrifuge Röhrchen wurden verworfen und ein frisches Sammelröhrchen wurde unter die Säule gelegt. Die DNA Probe wurde langsam in die Mitte der Gel-Fläche getropft. Die Säule wurde zusammen mit dem Röhrchen 5 Minuten lang bei 700 x g zentrifugiert und das gereinigte DNA wurde gesammelt. 27 2.5. Klonierung 2.5.1. Restriktion von Alanin Razemase Gene Eine Restriktionsenzym-Mixtur mit 12 µL von 10X Puffer Nummer 4, 60 µL destilliertes Wasser, 1 µL XhoI, 1 µL BamHI und 1 µL BSA (100X) wurde vorbereitet. Jeweils 30 µL dieser Mixtur wurde zu jeder der 20 µL Proben Salr1 DNA und Salr2 DNA hinzugefügt. Die Proben wurden 3 Stunden bei 37 0C inkubiert. Mit Bezug auf die DNA Menge, wurden 0,1 Volumen 3 M Kalium Acetat (KAc) und 2,5 Volumen 100% Ethanol zu den Proben addiert. Die Mixtur wurde 10 Minuten bei 14.000 Upm zentrifugiert, und das Pellet in 500 µL von 70% Ethanol resuspendiert. Bei gleicher Umdrehungszahl wurde dann 2 Minuten zentrifugiert und das Pellet wurde in 10 µL destilliertem Wasser resuspendiert. Eine Erhitzung auf 72 0C für 10 Minuten deaktivierte die Restriktionsenzyme. 1 µL Vektor-DNA (pBS) wurde mit 12 µL Restriktionsenzym-Mixtur und 8 µL destilliertem Wasser gemischt. Die Mixtur wurde 3 Stunden bei 37 0C inkubiert und anschliessend auf 72 0C für 25 Minuten erhitzt, um die Restriktionsenzyme zu deaktivieren. Die DNA Proben und der Vektor wurden dann auf Eis gelagert. 2.5.2. Ligation 10 µL der Proben, genannt Salr1 und Salr2, wurden jeweils gemischt mit 3 µL pBS Vektor, 1,5 µL von 10X DNA Ligase Puffer und 0,5 µL T4 DNA Ligase Puffer. Eine Kontroll-Mixtur wurde vorbereitet, indem 10 µL destilliertes Wasser anstatt 10 µL der Proben benutzt wurde. Die Mixturen wurden 30 Minuten bei Raumtemperatur inkubiert und bei 4 0C über Nacht gelagert. 28 2.5.3. Transformation Vor der Transformation wurden kompetente Zellen (DH5α) generiert. E. coli Zellen wurden bis zur log-Phase gewachsen, was ungefähr bestimmt wurde durch einen Messwert von 0,3 – 0,5 Absorbierungseinheiten eines Spektrophotometer bei 600 nm. Die Zellen wurden 15 Minuten lang bei 4 0C zentrifugiert. Ein halbes Volumen TMF wurde benutzt, um das Pellet zu resuspendieren. Die Zellen wurden 60 Minuten auf Eis gelegt und dann 15 Minuten bei 4 0C zentrifugiert. Das Pellet wurde in 0,1 Volumen TMF resuspendiert. Zur Verdünnung wurde genügend Glycerol hinzugefügt, bis eine End-Konzentration von 20% erreicht wurde. Die Zellen wurden in Aliquote aufgeteilt und bei -80 0C gelagert. 150 µL kompetenter DH5α Zellen wurden zu den auf Eis gekühlten Röhrchen mit den DNA Proben hinzugefügt. Die Zellproben wurden für 20 Minuten auf Eis gehalten. Dann wurde ein Hitze Schock für 100 Sekunden bei 42 0C durchgeführt. 1 mL LB-Medium wurde dazugemischt und für eine Stunde bei 37 0C inkubiert. 40 µL X-gal (50 µg/ml) wurde gleichmäβig auf Ampicillin-LB-Platten verteilt. Jeweils 0,5 mL der LB-Brühen wurden auf den vorbereiteten Platten verteilt und über Nacht bei 37 0C inkubiert. Für den weiteren Gebrauch wurden diese Platten beschriftet und bei 4 0C im Kühlschrank gehalten. 2.5.4. Blau-Weiss-Screening Weisse Kolonien, die nicht dicht von blauen oder weissen Satellit-Kolonien umgeben waren, wurden mit sterilen Zahnstochern gepickt. Sterile Zangen wurden benutzt, um die Zahnstocher in Reagenzgläser, die mit 3 mL LB-Medium und 1,5 µL Ampicillin (100 µg/ml) gefüllt waren, zu platzieren. Die Reagenzgläser wurden beschriftet und für 2 Tage unter Bewegung bei 30 0C inkubiert. 29 2.6. Agarose-Gelelektrophorese von Salr1 und Salr2 Die Agarose-Gelelektrophorese wurde mit 0,7% Agarose durchgeführt. 50 mL eines 1X TAE Puffer wurde mit 0,35g Agarose gemischt und anschlieβend für zwei Mal 1,5 Minuten in einer Mikrowelle bei höchster Stufe erhitzt. Die Mixtur wurde vorsichtig gerührt und es wurde gewartet bis sich die Mixtur bei Raumtemperatur auf 50 0C abgekühlt hatte. Die Mixtur wurde in einen 6 x 9 cm Plastikguss gegossen und ein Gel-Kamm wurde etwa 1 cm unter dem oberen Ende des Gels platziert. Nach der Polymerisation wurde der Kamm entfernt und die so entstandenen Taschen mit Puffer gespült. Das Gel wurde in eine mit Laufpuffer gefüllte Elektrophoresekammer transferiert. 15 µL der DNA-Proben wurden mit 2 µL von 10X Farbmarkern gemischt. Die DNA-Proben und 10 µL einer 1 Kb DNA-Leiter wurden mit einer Pipette jeweils in eine Tasche gespritzt. Eine konstante elektrische Spannung von 100 Volt wurde für ungefähr 45 Minuten auf das Gel angelegt. Das Gel wurde dann in verdünntem Ethidiumbromid gewaschen. Nachdem das Gel einige Male mit destilliertem Wasser gespült wurde, wurde ein Bild mit einer Eagle Eye Kamera gemacht. 2.7. DNA-Sequenzierung DNA-Sequenzierung wurde zum Vergleich der klonierten Genproben-Sequenz mit der publizierten S. aureus Sequenz durchgeführt. Das Wizard Mini Prep Paket wurde verwendet, um hochgereinigtes Plasmid-DNA für die DNA-Sequenzierung zu gewinnen. Der Reinheitsgrad der DNA-Proben wurde durch UV-Spektroskopie bestimmt. 5 µL DNA wurde mit 995 µL destilliertem Wasser in eine Küvette platziert. Das Spektrophotometer wurde mit einer Leerprobe von 1 mL destilliertem Wasser kalibriert und auf eine Wellenlänge von 260 nm beziehungsweise 280 nm gestellt, um die optische Dichte (OD) der beiden DNAProben zu bestimmen. Die OD bei 260 nm wurde verwendet um die Konzentration von DNA in der Probe zu messen. 30 Die Sequenzierungsreaktionen wurden an einer PCR Maschine durchgeführt. Jeweils ein Volumen mit einer Menge von 500 ng DNA wurde in zwei PCR Röhrchen eingefüllt. Dazu kamen 8 µL Reaktionsmixtur und 1 pmol/µL von jeweils Vorwärts-Primer und RückwärtsPrimer in jedes Röhrchen. Die Röhrchen wurden bis 20 µL mit destilliertem Wasser aufgefüllt. Die PCR Maschine wurde eingestellt auf: 2 Minuten bei 96 0C gefolgt von 30 Zyklen mit 10 Sekunden bei 96 0C, 30 Sekunden bei 50 0C und 4 Sekunden bei 60 0C. Nach der Beendigung wurden die Röhrchen auf 4 0C gehalten. Vor dem Übertragen in frische Eppendorf Röhrchen, wurden die DNA-Proben kurz gedreht. 2 µL einer hohen Salzlösung und 50 µL von 100% Ethanol wurden addiert, und die Mixtur wurde gevortext. Die Proben wurden 10 Minuten auf Eis gelegt und anschliessend 30 Minuten bei 14.000 Upm zentrifugiert. Der Überstand wurde mit einer Nadel abgesaugt und 250 µL von 70% Ethanol wurde addiert. Die DNA-Proben wurden kurz gevortext und 5 Minuten bei 14.000 Upm zentrifugiert. Der Überstand wurde vorsichtig mit einer frischen Nadel abgesaugt. Um die DNA-Proben zu trocknen, wurden sie 30 Minuten in ein erhitztes Speedvacuum gelegt. Die Proben wurden bis zum Lauf am ABI 3700 DNA-Sequenzierer im Tiefkühlschrank gelagert. 2.8. Komplementation vom E. coli alr-Mutant Die Plamide mit den klonierten Alanin Razemase Gene von S. aureus wurden in E. coli alr-Mutant dadX- alrts (MB 1460) transformiert. E. coli (MB 1460) ist ein Alanin Razemase defizienter Stamm, der generell bei 30 0C wächst. Bei 42 0C kann dieser Bakterienstamm jedoch nicht überleben, aufgrund seiner Alanin Auxotrophie. Vier LB-Platten mit Ampicillin und 10 µg/mL Thymin wurden vorbereitet. Auf zwei Platten wurden als Kontrolle E. coli (MB 1460) ohne transformierte Plasmide verteilt, und auf den anderen zwei Platten wurden E. coli (MB 1460) mit transformierte Plasmide verteilt. Jeweils eine transformierte und eine nicht-transformierte Platte wurden bei 30 0C beziehungsweise 31 42 0C übernacht inkubiert. E. coli mit funktionsfähigen Alanin Razemase Gene würden bei 42 0C überleben. 2.9. IPTG induzierte Überexprimierung von Alanin Razemase 5 µL des rekombinanten Expressions-Plasmid pET17b, der das Alanin Razemase PCRFragment trägt, wurde in E. coli BL21(DE3) transformiert. Die Zellen wurden auf einer LBPlatte mit Ampicillin verteilt und übernacht bei 37 0C inkubiert. Die resultierenden Kolonien wurden in 5 mL einer Ampicillin-LB Brühe geimpft. Das Wachstum der Zellen wurde anhand eines Spektrophotometers kontrolliert. Nachdem ein OD600 (optische Dichte bei 600 nm Wellenlänge) Wert von etwa 0,5 erreicht wurde, wurden die Zellen mit IPTG (1 mM End-Konzentration) induziert. Rifampicin (150 µL/mL) wurde 30 Minuten nach der Induktion hinzugefügt und die Zellen wurden danach 3 Stunden inkubiert. 100 µL der Probe wurde in ein frisches Eppendorf Röhrchen übertragen und 2 Minuten bei 14.000 Upm zentrifugiert. Das Pellet wurde in 30 µL SDS-PAGE Puffer resuspendiert, wovon dann 10 µL auf ein SDS-PAGE Gel geladen wurde. 2.10. SDS-Polyacrylamid-Gelelektrophorese der Alanin Razemase Proteine Die Trenngel-Mischung bestand aus: 12 mL 30% Acrylamid/Bisacrylamid (37.5:1), 7,5 mL 1.5 M Tris-Puffer (pH 8,8), 300 µL 10% SDS und 9,975 mL distilliertes Wasser. Die Mischung wurde gefiltert und mit einer Vakuum Flasche entgast. Etwa 225 µL von 10% Ammoniumpersulfat (APS) und 15 µL Tetramethylethylendiamin (TEMED) wurden zur Mischung gefügt. Ohne Bläschen zu erzeugen, wurde das Gel zügig mit Handschuhen 32 zwischen zwei abgedichtete Glasscheiben gegossen. Die Scheiben wurden durch einen Abstandhalter (Spacer) auseinander gehalten, und sie mussten während des gesamten Vorgangs sauber und fettfrei gehalten werden. Das Gel wurde mit etwas Butanol überschichtet. Nach der Polymerisation des Gels wurde das Butanol vorsichtig ausgekippt. Die Sammelgel-Mischung bestand aus: 1,25 mL 30% Acrylamid/Bisacrylamid (37.5:1), 2,5 mL 0.5 M Tris-Puffer (pH 6,8), 100 µL 10% SDS und 6 mL distilliertes Wasser. 75 µL von 10% APS und 7,5 µL TEMED wurden zur Mischung gefügt. Das Sammelgel wurde vorsichtig über das Trenngel gegossen und oben wurde ein spezieller Kamm eingesteckt. Die Polymerisation des Gels dauerte mindestens eine Stunde. 100 µL der Protein-Probe wurde in ein frisches Eppendorf Röhrchen übertragen und 2 Minuten bei 14.000 Upm zentrifugiert. Um die Proteine zu denaturieren, wurde das Pellet in 30 µL von 2X SDS-PAGE Puffer resuspendiert und dann für 10 Minuten auf 30 0C erhitzt. Als Proteinstandard wurde ein Molekulargewichtsmarker (Life Technologies Inc.) verwendet. Mit einer Pipette wurden 10 µL von jeder Probe und 15 µL Molekulargewichtsmarker in die Taschen des Gels gefüllt. Bromphenolblau wurde zugesetzt und das Gel lief übernacht bei 80 Volt bis das Bromphenolblau das Gel durchlaufen hatte. Das Gel wurde aus der Elektrophorese-Apparatur entfernt und anschliessend mit Coomassie-Brillant-Blau gefärbt. Zum Trocknen wurde das Gel mit klarer Folie überdeckt und auf weisses Filterpapier gelegt. Durch einen Scanner konnte dann ein Bild von dem getrockneten Gel gemacht werden. 33 3. ERGEBNISSE Im ersten Schritt dieses Projektes wurden die Alanin Razemase Gene von Staphylococcus aureus isoliert. S. aureus Bakterien wurden über Nacht in LB-Medium gewachsen und die DNA durch Phenolextraktion und Ethanolpräzipitation isoliert. Bei der PCR wurden Vorwärts-Primer und Rückwärts-Primer mit einer Länge von 20 beziehungsweise 22 Nucleotiden benutzt, um die zu amplifizierenden DNA-Abschnitte zu flankieren. Die PCR Produkte wurden durch Agarose-Gelelektrophorese analysiert. Das Agarosegel (Abb. 3-1) zeigt zwei Banden, die der Gröβe der Alanin Razemase Gene entsprechen: S. aureus Alanin Razemase 1 (~1,0 kB) auf der linken Bande und S. aureus Alanin Razemase 2 (~1,1 kB) auf der rechten Bande. Von hier an werden die beiden Gene als Salr1 und Salr2 bezeichnet. Es konnte nicht bestimmt werden, welches der beiden Gene katabolisch oder biosynthetisch war. Mittels Zentrifugation durch eine Chroma SpinTM Säule wurde die DNA gereinigt. Die Säule entfernte dNTPs und Oligonucleotid Kontaminanten kleiner als 100 Basen von den DNA Proben. Im nächsten Schritt wurden die Alanin Razemase Gene in einen pBS Vektor geklont. Eine Restriktion mit XhoI und BamHI wurde bei den DNA-Proben und pBS Vektor durchgeführt. Die Fragmente wurden mit Ethanol präzipitiert und jeweils an einen Vektor gebunden. Die Vektoren mit Fragment wurden in kompetente DH5α Zellen (MB 503) transformiert, welche dann auf X-gal/Ampicillin LB-Platten verteilt und bei 37 0C inkubiert wurden. Der pBS Vektor und das X-gal erlaubten ein Blau-Weiss Screening der transformierten Kolonien. Die weissen Kolonien enthielten den Vektor mit dem Genfragment. Diese wurden dann gepickt und übernacht in LB-Medium mit Ampicillin gewachsen. Die Zellen wurden lysiert, mit Ethanol präzipitiert und mit Restriktionsendonukleasen geschnitten. Die richtige Gröβe des Genfragmentes wurde durch Agarose-Gelelektrophorese gesichert. Die Kolonien mit Vektor und richtigem Genfragment wurden nochmals gewachsen. Durch das Wizard Mini Prep Paket konnte hoch gereinigtes Plasmid-DNA von diesen Zellen gewonnen werden. Eine DNA-Sequenzierung des PCR Produktes folgte, um einen Vergleich mit der S. aureus Genom Sequenz zu erstellen und eventuelle Mutationen oder Deletionen zu 34 finden. Abbildungen 3-2a bis 3-2c zeigen das Ergebnis der DNA-Sequenzierung des Alanin Razemase Gens Salr2. Die DNA-Sequenzierung des Alanin Razemase Gens Salr1 zeigte so viele Mutationen und Deletionen im Vergleich zur publizierten S. aureus Genom Sequenz, dass sie an dieser Stelle nicht mehr aufgeführt wurde. Nachfolgende Experimente zeigten auch, dass Salr1 nicht funktionstüchtig war. Die Salr2 Sequenz war fast identisch (Veränderung von 1 Base) mit der publizierten S. aureus Genom Sequenz. Obwohl der Ausdruck die Deletion einer Base zeigte, schien dies entweder eine stille Mutation oder ein Fehler bei der Sequenzierung zu sein, da die Funktionstüchtigkeit dieses Gens in den nachfolgenden Schritten gezeigt wurde. Die Funktionstüchtigkeit dieses Gens wurde durch eine Komplementation von einem Alanin Razemase defizienten E. coli Mutanten (MB1460) geprüft. Transformierte Zellen mit Salr2 konnten normal bei 30 0C und 42 0C wachsen. Die Kontrollgruppe zeigte kein Wachstum bei 42 0C. Abbildungen 3-3a und 3-3b zeigen die Komplementation auf vier Platten, wobei die beiden Platten in Abbildungen 3-3b die Funktionstüchtigkeit von Salr2 zeigen. Sämtliche Komplementationsversuche mit Salr1 waren erfolglos und das Projekt wurde ausschlieβlich mit Salr2 fortgeführt. Durch eine Restriktion wurde sowohl Salr2 vom pBS Vektor geschnitten als auch ein pET17 Expressionsvektor angeschnitten. Die Fragmente durchliefen ein Agarose Gel, wobei die Banden von Salr2 (~1,1 kB), pBS Vektor (~3,0 kB) und pET17 Vektor (~3,0 kB) die erwartete Gröβe zeigten (Abb. 3-4). Die DNA-Banden von Salr2 und pET17 Vektor wurden aus dem Gel geschnitten, isoliert und für eine Ligation durch das Wizard Mini Prep Paket aufgereinigt. Nach der Ligation wurde das fertige Plasmid zur Expression in einen TMFkompetenten E. coli-Stamm (BL21[DE3]) transformiert. Alle Bakterienstämme und Vektoren sind in Tabelle 3-1 aufgelistet. 35 Agarose-Gelelektrophorese der Alanin Razemase Gene Abbildung 3-1. Eagle Eye Bild einer Agarose-Gelelektrophorese. Gezeigt sind die PCR Produkte von Alanin Razemase in Staphylococcus aureus. Auf der ersten Bahn befindet sich der Molekulargewichtsmarker. Die Banden sind mit den entsprechenden DNA Gröβen links angegeben. Die zweite Bahn zeigt eine Salr1 Bande auf 1,0 kb. Die dritte Bahn zeigt eine Salr2 Bande auf 1,1 kb. Die kräftigen Banden unter 1,0 kb repräsentieren DNA Rückstände der PCR. 36 DNA-Sequenzierung von Alanin Razemase in Staphylococcus aureus Abbildung 3-2a. Staph-2U und Staph-1U zeigen die DNA Sequenz vom VorwärtsPrimer. Staph-alr2 zeigt die publizierte S. aureus Genom Sequenz. STPH-alr2 zeigt die kombinierte Sequenz von Vorwärts-Primer und Rückwärts Primer. 37 Abbildung 3-2b. Die Pfeile zeigen auf Veränderungen in der DNA Sequenz: 1) eine Basendeletion in der Sequenz, 2) ein Fehler beim Ausdruck; Verifizierung einer Adenin (A) Base am Computerbildschirm. 38 Abbildung 3-2c. Staph-2R und Staph-1R zeigen die DNA Sequenz vom RückwärtsPrimer. Staph-alr2 zeigt die publizierte S. aureus Genom Sequenz. STPH-alr2 zeigt die kombinierte Sequenz von Vorwärts-Primer und Rückwärts-Primer. Ein Vergleich mit der publizierten S. aureus Genom Sequenz zeigt eine 99% Übereinstimmung. 39 Komplementation von E. coli dadX- alrts Mutanten Abbildung 3-3a. Agarplatten mit D-Alanin auxotrophen E. coli dadX- alrts Mutanten (MB1460) nach 16 Stunden Inkubation bei 30 0C und 42 0C. Bakterien wuchsen nur bei 30 0C. Abbildung 3-3b. Agarplatten mit D-Alanin auxotrophen E. coli dadX- alrts Mutanten (MB1460), die mit Salr2 transformiert wurden, nach 16 Stunden Inkubation bei 30 0C und 42 0C. Bakterien wuchsen bei beiden Temperaturen, so dass eine Funktionsfähigkeit des Gens gezeigt werden konnte. 40 Agarose-Gelelektrophorese von Restriktionsprodukten Abbildung 3-4. Agarose-Gelelektrophorese von den BamHI und XhoI Restriktionsprodukten. Bahn MW zeigt den Molekulargewichtsmarker mit entsprechenden DNA Gröβen. Bahn 1 und 2 zeigen das Salr2 Fragment bei ~1,1 kB und den pBS Vektor bei ~3 kB. Bahn 3, 4 und 5 zeigen den pET17 Expressions-Vektor bei ~3 kB. Die Salr2 und pET17 Banden wurden aus dem Gel geschnitten, und dann isoliert und für die Ligation aufgereinigt. Tabelle 3-1. Verwendete Bakterienstämme und Vektoren Stämme und Vektoren BL21(DE3) Genotyp Referenz E. coli B F- dcm ompT hsdS(RB- MB-)gal λ (DE3) - ts MB 1460 dadX , alr MB 503 DH5α rec A+ Novagen, Madison, WI, USA Labor Bestand Labor Bestand R pBS T7 Promoter, T3 Promoter, Ap pET17b T7lac Promoter kontrol. Expressions Vektor, ApR 41 Novagen, Madison, WI, USA Novagen, Madison, WI, USA Die Überexprimierung vom Alanin Razemase Gen Salr2 in E. coli Stamm BL21(DE3) war erfolglos. Zahlreiche Überexprimierungen wurden vorgenommen, aber keines der SDS-PAGE Gele zeigte einen bedeutsamen Unterschied der Proteinexpression zwischen induzierten und nicht-induzierten Zellen. Alle SDS-PAGE Gele zeigten eine annähernd gleiche Menge von Proteinen in allen Bahnen, so dass diese Gele verworfen werden mussten. Um die Funktionsfähigkeit des E. coli Stammes BL21(DE3) zu verifizieren, wurde der Versuch mit einem Alanin Razemase Gen von E. coli wiederholt. Dieses Gen wurde in einem anderen Projekt dieses Labors erfolgreich isoliert und kloniert. Abbildung 3-5 zeigt ein SDS-PAGE Gel einer erfolgreichen Überexprimierung des E. coli Alanin Razemase Gens. Die Addition von IPTG zu E. coli BL21(DE3) induzierte die Expression der T7-RNA Polymerase dieses Stammes, so dass durch den T7 Promoter des pET Vektors eine hohe Transkriptionsrate von Salr2 erfolgte. Durch die hohe Anzahl von mRNA erhöhte sich auch die Translationsrate der Alanin Razemase. Rifampicin ist ein Antibiotikum, das die RNA Polymerase, aber nicht die T7RNA Polymerase inhibiert. Die Zugabe von Rifampicin reduzierte somit die Produktion von anderen Proteinen, so dass der Hintergrund des SDS-PAGE Gels frei von anderen Proteinen blieb. Die erfolgreiche Überexprimierung des E. coli Alanin Razemase Gens wurde auf einem SDS-PAGE Gel verifiziert (Abb. 3-5). IPTG induzierte Zellen zeigten eine dichte Proteinbande bei ~39 kDa, während nichtinduzierte Zellen eine relativ dünne Proteinbande auf ~39 kDa zeigten. Die Zellkulturen mit der höchsten Überexprimierung (Kulturen der Bahn 10) wurden in 2 L von LB-Medium gewachsen. Diese Zellen wurden mit IPTG induziert, bei 30 0C inkubiert und dann zur weiteren Proteinanalyse einem anderen Labor übergeben. Bei dieser Arbeit war die Überexprimierung von E. coli BL21(DE3) somit erfolgreich für Alanin Razemase von E. coli, jedoch war sie nicht erfolgreich für Alanin Razemase von S. aureus. Ein Jahr später wurde diese Arbeit im selben Labor mit der selben Methodik und denselben Materialien von Dr. Ulrich Strych wiederholt. Dieses Mal war die Überexprimierung von S. aureus Alanin Razemase erfolgreich. 42 IPTG Induzierte Überexprimierung von Alanin Razemase in E. coli Abbildung 3-5. Überexprimierung von Alanin Razemase in E. coli BL21(DE3). Bahn 1: nicht-induzierte negative Kontrolle [BL21(DE3) ohne Salr2] Bahn 2: induzierte negative Kontrolle [BL21(DE3) ohne Salr2] Bahn 3: induzierte positive Kontrolle [BL21(DE3) mit pET17 + Gen, das ein 20 kDa Protein produziert] Bahn 4: nicht-induzierte positive Kontrolle [BL21(DE3) mit pET17 + Gen, das ein 20 kDa Protein produziert] Bahn 5, 7, 9, 11, 13, 15: BL21(DE3) mit Salr2 ohne IPTG Induktion, nach 3 Stunden Inkubation Bahn 6, 8, 10, 12, 14, 16: BL21(DE3) mit Salr2 nach 1 mM IPTG Induktion, nach 3 Stunden Inkubation Bahn MW: Molekulargewichtsmarker (Life Technologies Inc.) Bahnen 5+6, 7+8, 9+10, 11+12, 13+14 und 15+16 kamen jeweils von einer Kultur, wobei ein Teil induziert und ein Teil nicht induziert wurde. Die Kultur auf Bahn 9+10 zeigt eine Bande mit bezeichnender Überexprimierung auf ~39 kDa. Diese Kultur wurde für weitere Protein Analysen verwendet. 43 4. DISKUSSION Das Ziel des gesamten Projektes war die Isolation, Klonierung und Reinigung von Alanin Razemase von verschieden Bakterien. Die Proteinstrukturen sollten mittels Röntgenkristallographie analysiert werden, um einen Alanin Razemase inhibierenden Stoff zu entwickeln, welcher potenziell als neues Antibiotikum eingesetzt werden könnte. Bei dieser Arbeit wurden Alanin Razemase Gene vom grampositiven Bakterium Staphylococcus aureus extrahiert und kloniert. Anschliessend wurden die Alanin Razemase Gene in ein Überexprimierungs-System transformiert. Die exprimierten Proteine wurden gereinigt und zur weiteren Analyse an die Kristallographie gegeben. Salr1 wurde erfolgreich von S. aureus isoliert durch Extraktion und PCR Amplifikation. Das PCR entsprach der erwarteten Gröβe von etwa 1,0 kB. Das Gen wurde dann in einen pBS Vektor geklont. Die Transformation des Plasmids (Vektor + Salr1) in kompetente DH5α Zellen wurde erreicht. Darauf folgte ein Blau-Weiss-Screening der transformierten Zellen. Anschlieβende Extraktion, Restriktion und Agarose-Gelelektrophorese verifizierten zwar die richtige Gröβe des Gens, jedoch zeigte die DNA-Sequenzierung sehr viele Mutationen und Deletionen. Auch alle Komplementationsversuche mit Salr1 im Alanin Razemase defizienten E. coli Mutanten scheiterten. Kein funktionsfähiges Salr1 konnte produziert werden. Salr2 wurde erfolgreich von S. aureus extrahiert und isoliert. Das Gen wurde mit PCR amplifiziert. Das PCR Produkt entsprach der erwarteten Gröβe von etwa 1,1 kB. Ein pBS Vektor wurde zum Klonen des Gens verwendet. Die Transformation des Plasmids (Vektor + Salr2) in kompetenten DH5α Zellen wurde erreicht. Durch Blau-Weiss-Screening konnten erfolgreich transformierte Zellen ausgewählt werden. Anschlieβende Extraktion, Restriktion und Agarose-Gelelektrophorese verifizierten die richtige Gröβe des Gens. Die DNA-Sequenzierung zeigte zwar eine mögliche Deletion, jedoch wurde die Funktionsfähigkeit von Salr2 beim Komplementationsversuch im Alanin Razemase defizienten E. coli Mutanten gezeigt. Zum Schluss war die Überexprimierung in E. coli des Stammes BL21(DE3) erfolglos. Die Überexprimierung wurde viele Male wiederholt, aber 44 keines der SDS-PAGE Gele zeigte einen bedeutsamen Unterschied der Proteinexpression zwischen induzierten und nicht-induzierten Zellen. Ein möglicher Grund des Misserfolges könnte der ursprüngliche Primer gewesen sein, der für die DNA Amplifikation verwendet wurde. Wie sich später herausstellte, bestand ein Fehler am Primer. Bei der Bestellung des Primers fehlte eine Base an der Schnittstelle des Primers. Da die Restriktions- und Ligationsversuche in den folgenden Schritten erfolgreich waren, schien dies die Primer als Ursache des Misserfolges auszuschlieβen. Eine zweite Möglichkeit des Defektes war die einzelne Basendeletion (Abb. 3-2). Die erfolgreiche Komplementation von Salr2 im Alanin Razemase defizienten E. coli Mutanten zeigte jedoch die Funktionsfähigkeit von Salr2. Es handelte sich möglicherweise um eine stille Mutation oder einen Fehler des Sequenzierungscomputers. Als dritte Möglichkeit wurde postuliert, dass eine zu hohe Konzentration von Alanin Razemase toxisch oder letal auf die Zellen wirken kann. Dies würde erklären warum die Komplementation erfolgreich und die Überexprimierung erfolglos war. Später wurde diese Arbeit jedoch auf exakte Weise erfolgreich wiederholt, was auch diese Möglichkeit als Fehlerquelle ausschlieβt. In nachfolgenden Arbeiten wurden Alanin Razemasen von Mykobakterien und Pseudomonas aeruginosa erfolgreich geklont und exprimiert. Es gelang die Herstellung von Proteinkristallen bei P. aeruginosa Alanin Razemase (Strych et al., 2000; Strych et al., 2001). Nachkommende Arbeiten befassten sich mit der Strukturanalyse des Enzyms. Die Ergebnisse wurden zum besseren Verständnis des Enzyms und Enzymmechnismus verwendet, um einen potenziellen Inhibitor zu entwickeln. Als Inhibitor käme ein Analogon der Aminosäure Alanin in Frage. Zum Beispiel ist Cycloserin ein D-Alanin Analogstoff, der Alanin Razemase kompetitiv hemmt. Cycloserin ist jedoch höchst toxisch und kann nur unter Bedenken verabreicht werden. 45 Da Alanin Razemase die Razemisierung zwischen L- und D-Alanin katalysiert, könnte ein möglicher Inhibitor ein Alanin Analogon sein, das eine Resonanz zwischen L- und D-Form bildet. Die -CH3 Gruppe von Alanin könnte durch eine -NH-R Gruppe ersetzt werden, wobei die -R Gruppe zyklisch an die -NH2 Gruppe gebunden ist (Abb. 4-1). Theoretisch würde dieses Molekül ein Alaninanalogon darstellen, welches eine L- und D-Resonanzform bildet. Bei einer Entwicklung einer bakteriellen Resistenz könnte die -R Gruppe einfach modifiziert werden. Abbildung 4-1. Theoretischer Inhibitor von Alanin Razemase. Eine -NH-R Gruppe ist als Ringstruktur an die -NH2 Gruppe der Aminosäure gebunden. Als L- und D-Resonanzform könnte dieses Molekül sowohl ein L- als auch D-Alanin Analogon sein. 46 5. LITERATURVERZEICHNIS 1) Ayliffe, G. A. J., Lowbury, E. J. L., Geddes, A. M., and J. D. Williams. Third edition. 1992. Control of Hospital Infection. A practical handbook. London: Chapman and Hall. 2) Deodhar, L. (n.d.). Opportunistic infections in AIDS. Abgerufen 10. Februar, 2003, von http://www.bhj.org/journal/1997/3901_jan/special_123.htm 3) Heritage, J. 2003. Antimicrobial Chemotherapy. Abgerufen 13. Februar, 2003, von http://www.bmb.leeds.ac.uk/mbiology/ug/ugteach/icu8/antibiotics/wall.html 4) Lambert, M. P., and F. C. Neuhaus. 1972. Mechanism of D-cycloserine action: alanine racemase from Escherichia coli. J. Bacteriol. 110:978-987. 5) Levinson, W., and E. Jawetz. Fifth Edition. 1998. Examination & Board Review : Medical Microbiology & Immunology. Stamford, Connecticut : Appleton & Lange. 6) Lilley, P. E., N. P. Stamford, S. G. Vasudevan, and N. E. Dixon. 1993. The 92-min region of the Escherichia coli chromosome: location of the ubiA and alr genes. Gene129: 9-16. 7) Lobocka, M., J. Hennig, J. Wild, and T. Klopotowski. 1994. Organization and expression of the Escherichia coli K12 dad operon encoding the smaller subunit of the D-amino acid dehydrogenase and the catabolic alanine racemase. J. Bacteriol. 176:1500-1510. 47 8) Matsuhashi, M. 1994. Utilization of the lipid-linked precursors and the formation of the peptidoglycan in the process of cell growth and division: membrane enzymes involved in the final steps of peptidoglycan synthesis and the mechanism of their regulation, p. 55-71. J.-M. Ghuysen and R. Hakenbeck (ed.), Bacterial cell wall. Elsevier Sciences B. V., Amsterdam, The Netherlands. 9) Moat, A. G., and J. W. Foster. Third Edition. 1995. Microbial Physiology. New York : Wiley-Liss. 10) Strych, U., H. C. Huang, K. L. Krause, and M. J. Benedik. 2000. Characterization of the Alanine Racemases from Pseudomonas aeruginosa PAO1. Current Microbiology. 41:290-294. 11) Tortora, G. J., Funke, B. R., and C. L. Case. Seventh Edition. 2001. Microbiology : an introduction media update. San Francisco : Benjamin Cummings. 12) van Heijenoort, J. 1994. Biosynthesis of the bacterial peptidoglycan unit, p. 39-54. J.-M. Ghuysen and R. Hakenbeck (ed.), Bacterial cell wall. Elsevier Sciences B. V., Amsterdam, The Netherlands. 13) Wasserman, S. A., E. Daub, P. Grisafi, C. T. Walsh, and D. Botstein. 1984. Catabolic alanine racemase from Salmonella typhimurium: DNA sequence, enzyme purification and characterization. Biochemistry 23:5182-5187. 14) Wijsman, H. J. W. 1972. The characterization of an alanine racemase mutant of Escherichia coli. Genet Res. 20:269-277. 15) Wild, J., J. Hennig, M. Lobocka, W. Walcsak, and T. Klopotowski. 1985. Identification of the dadX gene for the predominant isoenzyme of alanine racemase in Escherichia coli K12. Mol. Gen. Genet. 198:315-322. 48 6. ZUSAMMENFASSUNG Die Synthese der bakteriellen Zellwand benötigt D-Alanin, ein Isomer der im Menschen natürlich vorkommenden Aminosäure L-Alanin. Die Razemisierung zwischen L-Alanin und D-Alanin wird durch eine Gruppe von Enzymen, der Alanin Razemasen, katalysiert. Die Entwicklung eines Inhibitors von Alanin Razemasen kann potentiell effektive Antibiotika produzieren, da Alanin Razemasen zwar universell in allen Prokaryoten vorkommen, das Vorkommen jedoch fast nur auf Prokaryoten beschränkt ist. Die Alanin Razemase Gene von Staphylococcus aureus wurden durch PCR vervielfältigt, und anschlieβend in Escherichia coli kloniert. Die DNA-Sequenzierung von einem Alanin Razemase Gen zeigte eine fast identische Sequenz (Veränderung von einer Base) zum bekannten S. aureus Genom. Die Aktivität des klonierten Genes wurde demonstriert anhand einer Komplementation eines D-Alanin auxotrophen Escherichia coli Mutanten. Die Überexprimierung des Proteins wurde im 49 E. coli BL21 System durchgeführt. 7. CURRICULUM VITAE Quoc - Bao Ha Ngoc Merowingerstr. 151 50374 Erftstadt Tel 02235 / 461368 E-mail: [email protected] Karrierevorstellung Niederlassung als Facharzt der Neurologie. Bildungsgang 2010 Universität zu Köln Medizinische Doktorprüfung Promotion am 18. August 2010 Köln 2000 - 2010 Universität zu Köln Studium Humanmedizin Abschluss April 2010 Köln 2002 - 2003 University of Houston Senior Honors Thesis in Biochemistry Houston, USA Prof. Dr. Michael Benedik 1993 - 1999 University of Houston Bachelor of Science in Biochemistry Honors in Major, Cum Laude Houston, USA 1993 - 1999 University of Houston Bachelor of Science in Psychology Cum Laude Houston, USA 1992 - 1993 Sharpstown High School Houston, USA Abitur Abschluss (Highschool Diploma) Notenschnitt: 1,0 50 Berufserfahrung 1996 - 1998 privat Houston, USA Nachhilfelehrer Biologie, Chemie, organische Chemie, Physik, Mathematik (Calculus – Integral und Differential Mathematik) 1997 - 1998 Klavierlehrer privat Houston, USA 1994 - 1996 University of Houston Houston, USA University Tutor (Nachhilfe Lehrer) Biologie, Chemie, organische Chemie, Physik, Mathematik (Calculus – Integral und Differential Mathematik) 1994 - 1995 Cineplex Odeon Houston, USA Movie Theater Kontrolle der Tickets und Sauberhaltung der Kinos Famulatur und Praktikum Juni 2010 Universität zu Köln Neurochirugie, Stereotaxie Hospitation Köln April – July 2008 Universität zu Köln Neurologie Praktisches Jahr, Stationsarbeit, Ambulanz, Stroke Unit Köln Jan. – April 2008 Baylor College of Med. Houston, USA Surgery Praktisches Jahr, Oncological Surgery, Pediatric Surgery, Vascular Surgery Aug. – Dez. 2007 UT Medical Branch Galveston, USA Internal Medicine Praktisches Jahr, Rheumatology, Allergy and Immunology, Infectious Diseases, General Internal Medicine Sep. – Okt. 2006 Clinic for Internal Med. Houston, USA Famulant Untersuchungen, Fallvorstellungen, Notfälle, Aufnahmen 51 Famulatur und Praktikum (Fortsetzung) März – April 2005 Clinic for Pediatrics Houston, USA Famulant Untersuchung von Patienten, Dokumentation, Fallvorstellung, Impfungen July – Dez. 1996 MD Anderson Cancer Houston, USA Clinic Aide - Volunteer Patienten Versorgung, Kommunikation zwischen Patienten und medizinischem Personal, Transport von Pharmaka und Blut Mikrobiologische Labor Erfahrung 1998 - 2000 University of Houston Undergraduate Senior Honors Thesis PCR FPLC Elektrophorese Spektrophotometer Enzymatisches Essay Überexpressions System Transformation Sequenzierung Houston, USA Sprachkenntnisse Deutsch, Englisch, Vietnamesisch, Französisch, Latein Interessen und Aktivitäten Mitglied: Psi Chi - National Honor Society in Psychology (November 1998 - lebenslang) Präsident und Gründer: Kappa Rho Omega - Asian Service Fraternity (University of Houston, Januar 1996 – Dezember 1997) Gründer und Mitglied: VSA - Vietnamese Student Association (University of Houston, Januar 1995 – Dezember 1999) Sport Manager: VSA - Vietnamese Student Association (University of Houston, Winter Semester 1995 und Sommer Semester 1999) 52 Hobbies und Sonstiges Tennis: 1. - 3. Platz in allen Universitätsturnieren (University of Houston, Intramural Tournaments) von 1993 – 1998; Bezirksliga - Verbandsliga für Tennis Medenspiele, Verband Mittelrhein von 2000 - 2010 Tischtennis: 1. - 3. Platz in allen Universitätsturnieren (University of Houston, Intramural Tournaments) von 1993 – 1998; ACUI (Texas, Louisiana, Oklahoma) Universitäts Tischtennismannschaft 1995, 1998 und 1999 Klavier: Spiele regelmässig seit dem 6. Lebensjahr Bowling: Silbermedaille Gewinner des ACUI Bowlingturniers für Bundesstaaten Texas, Louisiana und Oklahoma im Februar 2000 Tae-Kwon-Do: Silbermedaille Gewinner des Kim’s Wanderpokal, Düsseldorf 1992 53