Sachverständigen-Ausschuss für Verschreibungspflicht
Werbung

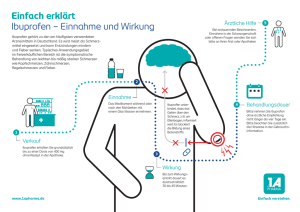
Sachverständigen-Ausschuss für Verschreibungspflicht nach § 53 Absatz 2 des Arzneimittelgesetzes Empfehlung zur Änderung der Arzneimittelverschreibungsverordnung 77. Sitzung 17. Januar 2017 Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM), Bonn 4. Zubereitung aus Aciclovir und Hydrocortison Antrag auf Entlassung aus der Verschreibungspflicht und Änderung der Positionen zu Aciclovir sowie Hydrocortison in Anlage 1 der Arzneimittelverschreibungsverordnung (AMVV) wie folgt: „Aciclovir - ausgenommen in Zubereitungen als Creme in der Kombination mit Hydrocortison in der Konzentration von 1 % Hydrocortison zur Behandlung von Herpes labialis zur Verringerung des Risikos von ulzerativen Läsionen bei Erwachsenen und Jugendlichen ab 12 Jahren, in Packungsgrößen bis zu 2 g und einem Wirkstoffgehalt bis zu 100 mg Aciclovir je abgeteilter Arzneiform -“ „Hydrocortison und seine Ester - ausgenommen in der Kombination mit Aciclovir - siehe Position Aciclovir“ Empfehlung des Sachverständigen-Ausschusses Der Sachverständigen-Ausschuss für Verschreibungspflicht empfiehlt einstimmig, den Antrag auf Entlassung aus der Verschreibungspflicht nach § 48 des Arzneimittelgesetzes anzunehmen und die bestehenden Positionen in Anlage 1 der AMVV wie folgt zu ergänzen: „Aciclovir […] - ausgenommen in Zubereitungen als Creme in Kombination mit Hydrocortison in einer Konzentration von 1 % Hydrocortison zur Behandlung von Herpes labialis zur Verringerung des Risikos von ulzerativen Läsionen bei Erwachsenen und Jugendlichen ab 12 Jahren, in Packungsgrößen bis zu 2 g und mit einem Wirkstoffgehalt bis zu 100 mg Aciclovir je abgeteilter Arzneiform -“ Seite 1 von 5 „Hydrocortison und seine Ester […] - ausgenommen in Zubereitungen als Creme in Kombination mit Aciclovir in einer Konzentration von 1 % Hydrocortison zur Behandlung von Herpes labialis zur Verringerung des Risikos von ulzerativen Läsionen bei Erwachsenen und Jugendlichen ab 12 Jahren, in Packungsgrößen bis zu 2 g und mit einem Wirkstoffgehalt bis zu 100 mg Aciclovir je abgeteilter Arzneiform -“ Begründung Regulatorischer Hintergrund Der Wirkstoff Aciclovir wird seit Langem in Zubereitungen als Creme bei Herpes labialis angewendet. In Deutschland befinden sich mehrere Cremes mit 5 % Aciclovir in Packungsgrößen von 2 g auf dem Markt. Entsprechende Monopräparate in Packungsgrößen bis zu 2 g und mit einem Wirkstoffgehalt bis zu 100 mg je abgeteilter Arzneiform sind von der Verschreibungspflicht ausgenommen und apothekenpflichtig. Seit 2009 wurde in 14 europäischen Ländern im Rahmen eines Dezentralen Verfahrens (Decentralised Procedure – DCP) eine Zubereitung aus Aciclovir (5 %) und Hydrocortison (1 %) in einer Packungsgröße von 2 g zugelassen. In Deutschland wurde die Zulassung im Jahr 2010 erteilt, seit Mai 2013 wird das Kombinationspräparat hierzulande vermarktet. 1 g Creme enthält 50 mg Aciclovir und 10 mg Hydrocortison. Die Anwendungsgebiete umfassen laut Fachinformation die „Behandlung früher Anzeichen und Symptome von rezidivierendem Herpes labialis (Lippenherpes) zur Senkung der Progression von Lippenherpesepisoden zu ulzerativen Läsionen bei immunkompetenten Erwachsenen und Jugendlichen (12 Jahre und älter)“. Die Behandlungsdauer ist auf fünf Tage limitiert. Während die Zulassung in Deutschland als verschreibungspflichtiges Arzneimittel erfolgte, sind entsprechende Arzneimittel in den folgenden acht der 14 am DCP-Verfahren beteiligten Mitgliedstatten der Europäischen Union bereits von der Verschreibungspflicht ausgenommen: Tschechische Republik, Dänemark, Finland, Polen, Portugal, Slowakische Republik, Schweden, Spanien. Die aktuellen Formulierungen der Positionen für Aciclovir und Hydrocortison in Anlage 1 der AMVV lauten wie folgt: „Aciclovir - ausgenommen in Zubereitungen als Creme zur Anwendung bei Herpes labialis in Packungsgrößen bis zu 2 g und einem Wirkstoffgehalt bis zu 100 mg je abgeteilter Arzneiform -“ „Hydrocortison und seine Ester - ausgenommen in Zubereitungen zum äußeren Gebrauch a) in einer Konzentration bis zu 0,25 % Hydrocortison oder Hydrocortisonacetat, berechnet als Base und in Packungsgrößen bis zu 50 g, sowie b) in einer Konzentration von über 0,25 bis zu 0,5 % Hydrocortison oder Hydrocortisonacetat, berechnet als Base und in Packungsgrößen bis zu 30 g zur kurzzeitigen (maximal zwei Wochen andauernden) äußerlichen Anwendung zur Behandlung von mäßig ausgeprägten entzündlichen, allergischen oder juckenden Hauterkrankungen, und sofern auf Behältnissen und äußeren Umhüllungen eine Beschränkung der Anwendung auf Erwachsene und Kinder ab dem vollendeten 6. Lebensjahr angegeben ist -“ Die o. g. aciclovirhaltigen Monopräparate zur Behandlung des Herpes labialis sind damit in Packungsgrößen bis zu 2 g nicht verschreibungspflichtig, nicht jedoch die Zubereitung aus Aciclovir und Hydrocortison. Seite 2 von 5 Klinische Aspekte Ein Großteil der erwachsenen Bevölkerung ist latent Träger einer Infektion mit dem Herpessimplex-Virus. Zwischen 15 % und 40 % der Infizierten erkranken mehr oder weniger häufig an Rezidiven; etwa ein Drittel der Patienten mit Herpes labialis recidivans leidet unter mehr als fünf Rezidiven/Jahr. Der Herpes labialis verläuft unbehandelt in fünf Phasen, wobei sich virale und inflammatorische Prozesse überlagern [nach Hull, 2014]: 1. Prodromalphase (Kribbeln) bis Rötung/Papel (Tag 0 – 1) 2. Bläschenphase (Tag 1 – 2) 3. Wunde und erste Verkrustung (Tag 2 – 3) 4. Harte Kruste (Tag 4 – 8) 5. Abschwellung Tag (9 – 10) Für die Selbstmedikation eines Herpes labialis stehen in Deutschland Monopräparate zur topischen Anwendung mit den Wirkstoffen Aciclovir sowie Penciclovir – beide wirken ausschließlich antiviral – zur Verfügung. Eine möglichst frühzeitige Anwendung, idealerweise bereits in der Prodromalphase, ist hinsichtlich eines optimalen Behandlungserfolgs sinnvoll. Die verschreibungspflichtige Zubereitung aus Aciclovir und Hydrocortison stellt eine Alternative dar und kombiniert die antivirale bzw. antientzündliche Wirkung der beiden Einzelwirkstoffe. Hierdurch soll die Bläschenbildung verhindert und der Krankheitsverlauf insgesamt günstig beeinflusst bzw. die Episodendauer verkürzt werden. Wie auch bei den Monopräparaten ist eine frühzeitige Anwendung angezeigt, um die Bläschenphase möglichst zu vermeiden. Hydrocortison gehört zur Gruppe der schwachen Glucocorticoide (Klasse I). Für die topische Anwendung liegen umfangreiche klinische Erfahrungen vor. Monopräparate mit dem Wirkstoff Hydrocortison in einer Konzentration zwischen 0,25 % und 0,5 % sind im Indikationsbereich „äußerliche Anwendung zur Behandlung von mäßig ausgeprägten entzündlichen, allergischen oder juckenden Hauterkrankungen" bereits seit 1996 (0,25 %) bzw. seit 2006 (0,5 %) apothekenpflichtig. Monopräparate, die Hydrocortison in einer Konzentration von 1 % im selben Indikationsbereich enthalten, sind in Deutschland hingegen verschreibungspflichtig, nicht jedoch in den folgenden zehn Mitgliedstaaten der Europäischen Union: Belgien, Tschechische Republik, Dänemark, Irland, Italien, Portugal (nur in Kombination mit Aciclovir), Slowakische Republik, Spanien, Schweden und Großbritannien. Die Indikation der Zubereitung aus Aciclovir und Hydrocortison 1 % unterscheidet sich jedoch von Monopräparaten mit Hydrocortison, da erstere ausschließlich für die Behandlung von klar abgegrenztem Herpes labialis auf Lippen und im Gesicht indiziert ist, nicht jedoch für eine großflächigere Anwendung. Erfahrungen aus der Selbstmedikation mit Arzneimitteln, die ausschließlich Hydrocortison enthalten, sind daher nur von begrenzter Aussagekraft im Hinblick auf die Bewertung für das Kombinationspräparat. Bei Entzündungsreaktionen, die mit einer bakteriellen, viralen oder mykotischen Infektion assoziiert sind, ist eine topische Monotherapie mit Glucocorticoiden aufgrund des Risikos einer Exazerbation der Infektion durch die immunsuppressive Wirkung der Glucocorticoide kontraindiziert. Eine kurzzeitige Kombinationstherapie mit antibakteriellen bzw. antimykotischen Wirkstoffen und Glucocorticoiden wird hingegen aufgrund der dualen (antiinfektiösen und antientzündlichen) Wirkung in der klinischen Praxis angewendet. Entsprechende Fertigarzneimittel sind verfügbar. Eine Anwendung von Hydrocortison auf ausgedehnten Arealen im Gesicht über einen längeren Zeitraum wird durch die Indikation des Kombinationspräparates – Herpes labialis – und die damit einhergehende Begrenzung auf einen eindeutig erkennbaren, kleinen Hautbereich verhindert. Weiterhin ist die Anwendungsdauer gemäß Produktinformation auf maximal fünf Tage begrenzt. Zusätzlich wird eine Packungsgrößenbegrenzung auf 2 g (maximal 20 mg HydrocortiSeite 3 von 5 son pro Packung) vorgeschlagen. Bei einer vorgesehenen Anwendung des Arzneimittels fünfmal täglich für fünf Tage ist eine Packung von 2 g für die Behandlung einer Herpesepisode aufgebraucht. Für eine Entlassung aus der Verschreibungspflicht ist eine sichere und eindeutige Selbstdiagnose des Herpes labialis erforderlich. Die meisten Patienten zeigen charakteristische Symptome, eine Selbstdiagnose und anschließende Selbstbehandlung erscheint insbesondere bei rezidivierendem Herpes labialis somit unproblematisch. Die Erkrankung ist zudem in der Regel selbstlimitierend und erfordert auch in dieser Hinsicht keinen Arztbesuch. Bezüglich Selbstdiagnose durch den Patienten liegen langjährige Erfahrungen mit rezeptfreien, antiviralen Monopräparaten zur Behandlung des Herpes labialis vor, die auch auf die Zubereitung aus Aciclovir und Hydrocortison übertragen werden können, da es sich um dieselbe Erkrankung handelt. Weiterhin bestehen bereits Erfahrungen aus der verschreibungsfreien Vermarktung des Kombinationsarzneimittels in anderen europäischen Ländern, die bisher bezüglich der Arzneimittelanwendung ohne direkte Überwachung durch den Arzt keine Hinweise auf negative Aspekte gezeigt haben. In Deutschland besteht für die verschreibungsfreie Anwendung von Aciclovir als Monotherapie des Herpes Labialis keine Altersbeschränkung. Hydrocortison in Konzentrationen bis zu 0,5 % ist ab dem vollendeten 6. Lebensjahr von der Verschreibungspflicht ausgenommen. Das Kombinationspräparat aus Aciclovir und Hydrocortison ist ausschließlich für Erwachsene und Kinder ab 12 Jahren zugelassen, da für Kinder unter 12 Jahren keine kontrollierten Wirksamkeitsdaten generiert wurden. Eine Sicherheitsstudie an Kindern von 6 bis 11 Jahren ergab, dass die Creme insgesamt gut vertragen und nur wenige Nebenwirkungen, darunter keine schwerwiegenden Fälle, beobachtet wurden. Vor diesem Hintergrund erscheint ein zusätzlicher Hinweis auf die Altersbegrenzung auf Behältnissen und äußeren Umhüllungen, wie es die AMVV-Position zu Hydrocortison als Monosubstanz vorsieht, als nicht notwendig. Nebenwirkungen und Risiken Es liegen langjährige, umfangreiche Erfahrungen zu den beiden Wirkstoffen Aciclovir und Hydrocortison in der verschreibungsfreien Anwendung vor. Da wesentliche Unterschiede zwischen hydrocortisonhaltigen Monoarzneimitteln und dem hier betrachteten Kombinationspräparat hinsichtlich Indikation, Packungsgröße sowie Anwendungsdauer bestehen, sind für die Bewertung der Sicherheit im Wesentlichen die Erfahrungen zu Aciclovir als Monotherapie des Herpes labialis von Bedeutung. Die Nebenwirkungsprofile des Mono- und des Kombinationspräparates sind nahezu deckungsgleich und umfassen hauptsächlich Lokalreaktionen. Vereinzelt wurden Überempfindlichkeitsreaktionen vom Soforttyp einschließlich Angioödem beobachtet. Einzige Unterschiede sind eine andere Häufigkeit für Austrocknung und Abschuppung der Haut und das zusätzliche Auftreten von Pigmentveränderungen (bekannte Nebenwirkung von Hydrocortison) bei der Kombination. Die Erfahrungen nach Zulassung im Rahmen der Pharmakovigilanz zeigen, dass die Anzahl der gemeldeten Nebenwirkungen gering ist. Von den seit 2012 berichteten 27 Nebenwirkungsfällen wurden drei als schwerwiegend eingestuft, bei denen es sich jedoch durchweg um lokale Reaktionen handelte. Weiterhin gibt es seit Markteinführung des Kombinationspräparates keine Hinweise auf beabsichtigten oder unbeabsichtigten Fehlgebrauch in der Selbstmedikation: Eine Verwechslung mit einer anderen entzündlichen Hauterkrankung im Gesicht wie beispielsweise Impetigo contagiosa wäre denkbar. Hierfür bestünde ein theoretisches Risiko, da es sich hierbei um eine bakterielle Infektion handelt, die auch perioral auftreten kann. Dies erscheint jedoch unwahrscheinlich, da die Impetigo contagiosa vorwiegend bei Kindern mit einem Erkrankungsgipfel von 3 bis 8 Jahren auftritt, keine Beteiligung der sensorischen Nerven stattfindet und sich das klinische Bild bezüglich Ausbreitungsfläche (mehrere Herde), Lokalisation (zentroSeite 4 von 5 fazial) und Effloreszenzen (honiggelbe Krusten) vom Herpes labialis unterscheidet. Weiterhin wurden dem Antragsteller diesbezüglich keine entsprechenden Fälle berichtet. Die Anwendung des Kombinationspräparates bei Herpesinfektionen in anderen Körperregionen (z. B. Genitalbereich) ist gemäß Produktinformation explizit kontraindiziert. Zudem erlaubt die Packungsgrößenbegrenzung auf 2 g letztlich keine großflächige Anwendung. Zusammen mit dem Antrag wurden drei Periodic Safety Update Reports (PSURs) für das Kombinationspräparat vorgelegt, welche den Zeitraum von Dezember 2011 bis Juli 2014 abdecken. In der Berichtsperiode ergaben sich keine sicherheitsrelevanten Aspekte hinsichtlich der Verkaufsabgrenzung. Ein möglicher Off-Label-Use ist Gegenstand des Routinemonitorings im Rahmen der PSURs. Insgesamt liegen keine Hinweise auf direkte oder indirekte Gefahren bei Anwendung des Kombinationspräparates in der Selbstmedikation vor. Diskussion und Zusammenfassung Episoden eines Herpes labialis sind für den Patienten aufgrund der charakteristischen Symptome leicht diagnostizierbar und damit für eine Selbstmedikation geeignet. Eine möglichst frühzeitige Anwendung, idealerweise bereits in der Prodromalphase, ist hinsichtlich eines optimalen Behandlungserfolgs sinnvoll. Aciclovirhaltige Arzneimittel zur Behandlung des Herpes labialis sind seit Langem verschreibungsfrei erhältlich. Für die Kombination aus Aciclovir und Hydrocortison sind nach derzeitigem Kenntnisstand keine weitergehenden Risiken im Vergleich zu den von der Verschreibungspflicht bereits ausgenommenen Monopräparaten zu erkennen. Aus den Anwendungserfahrungen in anderen europäischen Ländern ergeben sich keine Hinweise darauf, dass von einer Selbstmedikation direkte oder indirekte Gefahren für den Patienten ausgehen. Der Antrag auf Entlassung aus der Verschreibungspflicht für die Zubereitung aus Aciclovir und Hydrocortison wird daher befürwortet. Für Anlage 1 der AMVV wird der folgende Wortlaut empfohlen: „Aciclovir - ausgenommen in Zubereitungen als Creme in Kombination mit Hydrocortison in einer Konzentration von 1 % Hydrocortison zur Behandlung von Herpes labialis zur Verringerung des Risikos von ulzerativen Läsionen bei Erwachsenen und Jugendlichen ab 12 Jahren, in Packungsgrößen bis zu 2 g und mit einem Wirkstoffgehalt bis zu 100 mg Aciclovir je abgeteilter Arzneiform -“ „Hydrocortison und seine Ester - ausgenommen in Zubereitungen als Creme in Kombination mit Aciclovir in einer Konzentration von 1 % Hydrocortison zur Behandlung von Herpes labialis zur Verringerung des Risikos von ulzerativen Läsionen bei Erwachsenen und Jugendlichen ab 12 Jahren, in Packungsgrößen bis zu 2 g und mit einem Wirkstoffgehalt bis zu 100 mg Aciclovir je abgeteilter Arzneiform -“ Literatur Hull CM, Levin MJ, Tyring SK, Spruance SL. Novel composite efficacy measure to demonstrate the rationale and efficacy of combination antiviral-anti-inflammatory treatment for recurrent herpes simplex labialis. Antimicrob Agents Chemother 2014; 58(3): 1273-8. Seite 5 von 5 Sachverständigen-Ausschuss für Verschreibungspflicht nach § 53 Absatz 2 des Arzneimittelgesetzes Empfehlung zur Änderung der Arzneimittelverschreibungsverordnung 77. Sitzung 17. Januar 2017 Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM), Bonn 6. Ibuprofen Antrag auf Entlassung aus der Verschreibungspflicht „- zur trans-dermalen Anwendung als Pflaster ohne Zusatz weiterer arzneilich wirksamer Bestandteile in einer Konzentration bis zu 200 mg Ibuprofen je abgeteilter Arzneiform -“ Empfehlung des Sachverständigen-Ausschusses Der Sachverständigen-Ausschuss für Verschreibungspflicht empfiehlt einstimmig, den Antrag auf Entlassung aus der Verschreibungspflicht nach § 48 des Arzneimittelgesetzes anzunehmen und die bestehende Position in Anlage 1 der Arzneimittelverschreibungsverordnung (AMVV) wie folgt zu ändern: „Ibuprofen - ausgenommen zum äußeren Gebrauch (einschließlich Pflaster) in einer Konzentration bis zu 6 Gewichtsprozenten […]“ Begründung Wirkstoff und pharmakologische Wirkung Ibuprofen ist ein Propionsäurederivat und gehört zur Gruppe der nicht-steroidalen Antirheumatika/Antiphlogistika (NSAR, auch NSAIDs – Non Steroidal Antiinflammatory Drugs). Die Substanz wirkt als nicht-selektiver Cyclooxygenasehemmer und führt durch einen Eingriff in die Prostaglandinsynthese zu einer Entzündungshemmung. Indikation, Zulassungsstatus und Verkaufsabgrenzung in Deutschland Ibuprofen wird zur Therapie von Schmerzen, Entzündungen und Fieber eingesetzt. Der empfohlene Dosisbereich für Erwachsene und Jugendliche ab 15 Jahren liegt bei systemischer Applikation zwischen 1200 und 2400 mg Ibuprofen pro Tag. Ibuprofenhaltige Präparate waren 2014 die am häufigsten verordneten nicht-steroidalen Antiphlogistika [Schwabe & Paffrath, 2015]. Seite 1 von 4 Ibuprofen wird seit mehreren Jahrzehnten in Deutschland vermarktet. Die Substanz wurde in oraler Darreichungsform für die Indikationen Fieber und Schmerzen in Dosierung von 200 mg je abgeteilter Form sowie zur äußeren Anwendung in einer Konzentration bis zu 5 % bereits 1989 aus der Verschreibungspflicht entlassen. Entsprechend Anlage 1 der AMVV ist Ibuprofen „- zur oralen Anwendung ohne Zusatz weiterer arzneilich wirksamer Bestandteile in einer Konzentration bis zu 400 mg je abgeteilter Form und in einer Tagesdosis bis zu 1200 mg bei leichten bis mäßig starken Schmerzen und Fieber -“ von der Verschreibungspflicht ausgenommen. Weitere Freistellungen bestehen für orale flüssige Zubereitungen, rektale Zubereitungen, Kombinationen mit Pseudoephedrin sowie Zubereitungen in der Indikation Migräne (Wortlaut der AMVV zu Ibuprofen siehe Anhang). In Deutschland wird eine Vielzahl ibuprofenhaltiger Monopräparate (200 – 400 mg) im OTC-Bereich angeboten. Für Säuglinge und Kinder stehen apothekenpflichtige ibuprofenhaltige Suppositorien und Suspensionen zur Verfügung. Präparate mit höherer Wirkstoffkonzentration je abgeteilter Form und/oder solche, die zur Therapie u. a. von (rheumatoider) Arthritis eingesetzt werden, unterliegen der Verschreibungspflicht. Neben den genannten systemischen Anwendungen ist Ibuprofen „- zum äußeren Gebrauch in einer Konzentration bis zu 5 Gewichtsprozenten -“ von der Verschreibungspflicht ausgenommen. Der vorliegende Antrag betrifft ebenfalls eine äußere Applikationsform – die transdermale Anwendung als Pflaster. Als topische Zubereitung fällt das Ibuprofenpflaster nicht unter die o. g. Ausnahme, da die Konzentration in der Matrix 6 % beträgt. Derzeit ist nur das Arzneimittel des Antragstellers als ibuprofenhaltiges Pflaster in Deutschland zugelassen. Die Zulassung erfolgte am 15. August 2016. Der Wirkstoffgehalt je Pflaster beträgt 200 mg Ibuprofen, innerhalb von 24 Stunden soll ein Pflaster angewendet werden. Die Behandlungsdauer sollte 5 Tage nicht überschreiten. Die 10 x 14 cm großen Pflaster sind in Packungsgrößen mit 2, 4, 6, 8 und 10 Pflastern zugelassen. Die Indikation lautet wie folgt: „…wird bei Erwachsenen und Jugendlichen ab 16 Jahren angewendet zur kurzzeitigen, symptomatischen Behandlung - von lokalen Schmerzen bei akuten Zerrungen, - in der Nähe der Gelenke verstauchter Gliedmaßen nach einem stumpfen Trauma.“ Nebenwirkungen und Risiken Zu den häufigsten Nebenwirkungen von Ibuprofen bei systemischer Anwendung gehören gastrointestinale Störungen (z. B. Sodbrennen, Bauchschmerzen, Übelkeit, Erbrechen, Ulzera), zentralnervöse Störungen wie Kopfschmerzen, Schwindel und Schlaflosigkeit sowie Überempfindlichkeitsreaktionen. Eine Reihe weiterer Nebenwirkungen treten selten oder sehr selten auf z. B. Hypertonie, Tinnitus und Störungen der Blutbildung. Bei der topischen Anwendung von Ibuprofen sind die am häufigsten auftretenden Nebenwirkungen Reaktionen an der Applikationsstelle und Hautreaktionen. Bei der Anwendung des Pflasters wird nach 24 Stunden eine Ibuprofen-Plasmakonzentration von 0,49 µg/ml erreicht. Nach 5 Behandlungstagen beträgt die mittlere Cmax 0,51 µg/ml. Verglichen mit oral angewendetem Ibuprofen sind die mittleren Cmax und die Bioverfügbarkeit gering. Die Cmax nach oraler Einnahme von 200 – 400 mg Ibuprofen liegt in der Regel bei 20 – 50 µg/ml. Seite 2 von 4 Aufgrund der geringen systemischen Verfügbarkeit ist die Wahrscheinlichkeit für das Auftreten schwerer Nebenwirkungen bei äußerer Anwendung von Ibuprofen gering. Diskussion Ibuprofenhaltige Präparate zur oralen, rektalen und topischen Anwendung sind mit bestimmten Einschränkungen seit Langem für Patienten als OTC-Präparate zugänglich. Patienten sind somit prinzipiell mit der Anwendung von Ibuprofen in der Selbstmedikation vertraut. Der überwiegende Teil der unerwünschten Wirkungen von Ibuprofen ist dosisabhängig und beruht auf der systemischen Wirkung von Ibuprofen. Die systemische Verfügbarkeit von Ibuprofen nach Anwendung des Pflasters ist gering und bewegt sich in gleicher Größenordnung wie nach Anwendung anderer topischer (und bereits von der Verschreibungspflicht freigestellter) Applikationsformen (Creme/Gel). Aufgrund der geringen systemischen Verfügbarkeit des Wirkstoffes nach Applikation als Pflaster sind schwerwiegende Nebenwirkungen kaum zu erwarten. Die kutanen Applikationsformen in Form von Gel oder Creme werden im Regelfall dreimal täglich aufgetragen, wobei je nach Größe des zu behandelnden Areals jeweils 100 – 250 mg Ibuprofen appliziert werden. Die täglich angewendete Gesamtmenge soll 750 – 1000 mg Ibuprofen (15 – 20 g Gel/Creme bei einer Wirkstoffkonzentration von 5 %) nicht übersteigen. Die täglich lokal applizierte Menge Ibuprofen ist bei diesen, von der Verschreibungspflicht bereits freigestellten Arzneimitteln, somit höher als bei Anwendung des Pflasters. Die Indikationen für die Anwendung des Pflasters sind ähnlich denen der anderen, seit Langem in der Selbstmedikation angewendeten topischen Darreichungsformen mit Ibuprofen. Für Diclofenac, einen anderen zur Gruppe der NSAR zählenden Wirkstoff, ist bereits seit mehreren Jahren ein Pflaster im OTC-Bereich verfügbar. Die Indikation „symptomatische Behandlung von Schmerzen bei akuten Prellungen, Zerrungen oder Verstauchungen in Folge eines stumpfen Traumas“ entspricht weitgehend der des Ibuprofenpflasters. Dem Antrag auf Entlassung aus der Verschreibungspflicht für ibuprofenhaltige Pflaster wird generell zugestimmt. Im Gegensatz zum Antragsteller, der einen eigenen Wortlaut explizit für das Pflaster beantragt hat, wird allerdings eine Erweiterung der bisherigen Position zu Ibuprofen – äußerer Gebrauch – mit folgendem Wortlaut vorgeschlagen (neu aufzunehmende Passagen sind unterstrichen, zu streichende Passagen durchgestrichen): „Ibuprofen ausgenommen zum äußeren Gebrauch (einschließlich Pflaster) in einer Konzentration bis zu 5 6 Gewichtsprozenten -“ Diese Erweiterung der bisherigen Position erfolgt in Anlehnung an die Position zu Diclofenac in Anlage 1 der AMVV, bei der für die verschiedenen Formulierungen zur äußeren Anwendung ebenfalls nur ein Positionseintrag vorhanden ist, der Pflaster einschließt. Zudem dient die Vermeidung eines separaten Eintrages der besseren Lesbarkeit der Gesamtposition zu Ibuprofen. Der Klammerzusatz „einschließlich Pflaster“ ist beim Ibuprofen (im Gegensatz zum Diclofenac) erforderlich, da hier (anders als beim Diclofenac) die Wirkstoffkonzentration nicht in den Produktinformationen und/oder auf dem äußeren Behältnis angegeben ist. Zusammenfassung Ibuprofenhaltige Arzneimittel sind seit Langem in verschiedenen Darreichungsformen (inklusive kutaner Applikationsformen) mit bestimmten Einschränkungen als OTC-Präparate in Deutschland verfügbar. Das zur Freistellung von der Verschreibungspflicht beantragte Pflaster enthält 200 mg Ibuprofen und wird über einen Zeitraum von 24 Stunden angewendet. Die applizierte Wirkstoffmenge ist Seite 3 von 4 somit deutlich geringer als bei oralen Ibuprofenprodukten in der Selbstmedikation, die in Einzeldosen von 200 – 400 mg und einer maximalen Tagesdosis von 1200 mg verabreicht werden. Das Pflaster weist ein günstiges Risikoprofil auf, die systemische Verfügbarkeit des Wirkstoffes ist gering. Der Antrag auf Freistellung von der Verschreibungspflicht wird unterstützt. Abweichend vom Vorschlag des pharmazeutischen Unternehmers wird eine Erweiterung der bisherigen Position zu Ibuprofen – äußerer Gebrauch – mit folgendem Wortlaut vorgeschlagen: „Ibuprofen - ausgenommen zum äußeren Gebrauch (einschließlich Pflaster) in einer Konzentration bis zu 6 Gewichtsprozenten -“ Literatur Schwabe U, Paffrath D (Hrsg.): Arzneiverordnungs-Report 2015. Springer-Verlag Berlin Heidelberg 2015. Seite 4 von 4 Sachverständigen-Ausschuss für Verschreibungspflicht nach § 53 Absatz 2 des Arzneimittelgesetzes Empfehlung zur Änderung der Arzneimittelverschreibungsverordnung 77. Sitzung 17. Januar 2017 Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM), Bonn 7. Selen Überprüfung der freigestellten Tagesdosis zum inneren Gebrauch Empfehlung des Sachverständigen-Ausschusses Der Sachverständigen-Ausschuss für Verschreibungspflicht empfiehlt mehrheitlich, die freigestellte Tagesdosis für Selenverbindungen in Zubereitungen zum inneren Gebrauch auf 70 Mikrogramm Selen anzuheben. Begründung Hintergrund Selen ist ein essentielles Spurenelement für die Ernährung des Menschen, da es als Teil selenhaltiger Aminosäuren wichtige katalytische Vorgänge vermittelt. Selen kann über die Nahrung in Form organischer Verbindungen wie vorwiegend Selenomethionin aus pflanzlichen Lebensmitteln und wie vorwiegend Selenocystein aus tierischen Nahrungsmitteln aufgenommen werden. Anorganische Selenverbindungen wie Natriumselenit, Natriumhydrogenselenit und Natriumselenat finden Verwendung in Nahrungsergänzungsmitteln oder Arzneimitteln zur Supplementierung bzw. zur Therapie [Ekmekcioglu, 2000]. Aufgrund des geringen natürlichen Selengehaltes von in Deutschland erzeugten pflanzlichen Lebensmitteln könnte es bei vegetarisch oder vegan lebenden Menschen, die sich von regionalen Lebensmitteln ernähren, möglicherweise zu einem Selenmangel kommen. Da in der Europäischen Union die Zufütterung von selenreichen Mineralstoffmischungen weit verbreitet ist, weisen Lebensmittel tierischen Ursprungs kaum Schwankungen auf. Im Trinkwasser ist Selen kaum enthalten, liegt dann aber vorrangig als Selenat vor [EFSA, 2014]. Die Referenzwerte für die Nährstoffzufuhr wurden von den Gesellschaften für Ernährung in Deutschland (DGE), Österreich (ÖGE) und der Schweiz (SGE) – abgekürzt D-A-CH – 2015 überarbeitet und gemeinsam herausgegeben. Für männliche Jugendliche ab 15 Jahren und für Erwachsene beträgt der Referenzwert für eine angemessene Zufuhr von Selen danach 70 µg pro Tag, bei weiblichen Jugendlichen und Erwachsenen 60 µg pro Tag [D-A-CH, 2015]. Seite 1 von 5 In Deutschland besteht Verschreibungspflicht für Arzneimittel, die eine höhere Tagesdosis als 50 µg enthalten und die damit unter der angemessenen täglichen Zufuhrmenge liegen. Wirkstoff Menschen, Tiere und viele Bakterien sind auf die Zufuhr von Selen als essentielles Spurenelement angewiesen. Es findet sich in der Natur in anorganischen und organischen Verbindungen in den Oxidationsstufen - 2, + 4 und + 6 [EFSA, 2014]. Zu den anorganischen Selenverbindungen gehören Selenid, Selenit und Selenat, wobei am häufigsten Selenit und Selenat vorkommen. Zu den organischen Selenverbindungen gehören Selenide, selenhaltige Aminosäuren (L-Selenomethionin, L-Selenocystein) sowie Selenoproteine und selenhaltige Proteine [EFSA, 2014]. In selenhaltigen Proteinen erfolgt ein unspezifischer Einbau der Aminosäure Selenomethionin anstelle von Methionin. Strukturell unterscheidet sich Selenomethionin nur durch den Ersatz des Schwefels durch Selen von Methionin. Selenocystein ist fester Bestandteil in sogenannten Selenoproteinen, die antioxidativ wirken und im Körper wichtige Funktionen ausüben. Pharmakologische Wirkung Zu den Selenoproteinen mit wichtigen katalytischen Funktionen gehören die Familien der Glutathionperoxidasen, der Thioredoxinreduktasen und der Dejodasen. Dabei führen die fünf humanen, selenhaltigen Glutathionperoxidasen die Reduktion von organischen Hydroperoxiden aus [Brigelius-Flohe, 2001]. Da die Glutathionperoxidase des Subtyps 4 auch als Strukturbestandteil von Spermien fungiert, ist Selen auch für die männliche Fertilität von Bedeutung [Ursini, 1999]. Selen ist in hohen Konzentrationen in der Schilddrüse vorhanden und trägt als Bestandteil der Glutathionperoxidase zum Schutz dieses Organs vor Hydroperoxidexposition während der Schilddrüsenhormonsynthese bei. Eine ausreichende Selenzufuhr ist somit Voraussetzung für eine normale Schilddrüsenfunktion. Drei Dejodasen katalysieren die Dejodierung von Schilddrüsenhormonen, was zu deren Aktivierung (5‘-Dejodierung) bzw. Inaktivierung (5-Dejodierung) führt. Durch Abspaltung von Jodid aus dem inaktiven Prohormon Thyroxin (T4) entsteht so entweder das aktive Hormon Trijodthyronin (T3) oder inaktives reverses T3(rT3). Die Dejodasen regulieren also maßgeblich die Schilddrüsenhormonhomöostase [Köhrle, 2000; D-A-CH, 2015]. Bei unzureichender Selenversorgung erhöht sich das Verhältnis von T4 zu T3 im Serum, was zu Störungen in der Schilddrüsenfunktion führen und als funktioneller Marker des Selenstatus genutzt werden kann [Brown, 2001]. Früher wurde als Hauptkriterium für die Selenversorgung des Körpers die Aktivität der Glutathionperoxidase GPx angelegt, deren maximale Aktivität bei einer Serum- oder Plasmakonzentration von 90 – 100 µg Selen/l festgestellt wurde [Ashton, 2009]. Aktuellere Ableitungen erfolgen auf der Basis der Konzentration von Selenoprotein P (SEPP1) [Persson-Moschos, 1995]. Bei Selenoprotein P handelt es sich um ein selentransportierendes Protein, das die Versorgung von Organen mit Selen vermittelt [Hill, 2003]. Aus den zahlreichen katalytischen und antioxidativen Funktionen der Selenoproteine resultieren die postulierten förderlichen Effekte für die Gesundheit einer genügenden Selenzufuhr. Indikation und Anwendung Als Indikation selenhaltiger Arzneimittel ist der nachgewiesene Selenmangel zu nennen, der ernährungsbedingt nicht behoben werden kann und entweder durch Verdauungs- und Verwertungsstörungen oder durch Fehl- und Mangelernährung (z. B. durch eine totale parenterale Ernährung) verursacht ist. In den in dieser Indikation angewendeten Fertigarzneimitteln ist als Seite 2 von 5 Wirkstoff ausschließlich Natriumselenit-Pentahydrat in Einzeldosierungen von bis zu 999 µg (entsprechend 300 µg Selen) enthalten. Die üblichen Tagesdosierungen liegen zwischen 50 µg und 300 µg Selen. Die Schätzwerte für eine angemessene Zufuhr („Adequate Intake“) von Selen wurden von verschiedenen ernährungswissenschaftlichen Institutionen nach unterschiedlichen Methoden abgeleitet. Die European Food Safety Authority (EFSA) verwendete für die Ableitung der aktuellen Schätzwerte die SEPP1-Konzentration im Plasma [EFSA, 2014], und auch die D-A-CHEmpfehlung 2015 basiert auf dieser Methode. Für Kinder wurden die Schätzwerte für die angemessene Selenzufuhr von den Werten für Erwachsene unter Berücksichtigung der Unterschiede im Körpergewicht sowie unterschiedlicher Wachstumsfaktoren extrapoliert [D-A-CH, 2015]. Schätzwerte der D-A-CH in µg [D-A-CH, 2015] Alter [in Jahren] Männlich Weiblich Säuglinge 0– 4– < 4* < 12* 10 15 10 15 1– <4 15 15 Kinder 7– 10 – < 10 < 13 30 45 30 45 4– <7 20 20 13 – < 15 60 60 15 – < 19 70 60 > 19 70 60 Erwachsene SchwanStilgere lende 60 75 *Alter in Monaten Schätzwerte der EFSA in µg [EFSA, 2014] Alter [in Jahren] Säuglinge 0– 7– 6* 11* 15 1– 3 15 4– 6 20 Kinder 7– 10 30 11 – 14 55 15 – 17 70 ≥ 18 70 Erwachsene SchwanStillende gere 70 85 *Alter in Monaten Bezüglich des Versorgungsstatus der Bevölkerung in Deutschland wurde anhand von Duplikatstudien eine mittlere tägliche Selenzufuhr von 30 µg für Frauen und 41 µg für Männer ermittelt [Drobner, 1996]. Zulassungsstatus und Verkaufsabgrenzung in Deutschland Arzneimittel mit Selen zum inneren Gebrauch unterliegen ab einer Tagesdosierung von über 50 µg Selen der Verschreibungspflicht Die derzeitige Positionsformulierung in Anlage 1 der Arzneimittelverschreibungsverordnung (AMVV) lautet: „Selenverbindungen - ausgenommen in Zubereitungen zum inneren Gebrauch mit einer Tagesdosis bis zu 50 µg Selen -“ In Deutschland gibt es mit Stand von 23. November 2016 zehn verschreibungspflichtige selenhaltige Arzneimittel, von denen sieben eine Tagesdosis von bis zu 100 µg aufweisen. Nebenwirkungen und Risiken Auch bei langfristiger Einnahme von bis zu 850 µg Selen/Tag zeigten sich in Studien keine unerwünschten Wirkungen; diese wurden erst ab einer dauerhaften Tageszufuhrmenge von circa 1200 µg Selen und höher beobachtet [EFSA, 2006]. Bei dauerhafter Zufuhr hoher Selenmengen kann es zu einer sogenannten Selenose kommen, die sich in neurologischen Störungen, Müdigkeit, Gelenkschmerzen, Übelkeit und Durchfall äußert. Im späteren Verlauf geht eine Selenose mit Symptomen wie dem Verlust von Haaren, gestörter Nagelbildung und einem charakteristiSeite 3 von 5 schen knoblauchartigen Geruch der Atemluft einher. Eine akute Selenvergiftung durch Zufuhr von mehreren Gramm Selen kann zu Herzversagen sowie Kammerflimmern und damit zum Tod führen [Nuttall, 2006]. Das US-amerikanische Food and Nutrition Board hat für Erwachsene eine tolerierbare Gesamtzufuhrmenge von 400 µg/Tag bzw. 7 µg/kg Körpergewicht/Tag festgelegt („Tolerable Upper Intake Level“) [Institute of Medicine, Food and Nutrition Board, 2000], während der von der EFSA vorgeschlagene Wert bei 300 µg/Tag beträgt [EFSA, 2006]. Diskussion und Zusammenfassung Der für Deutschland ermittelte Versorgungsstatus an Selen durch die Nahrungsmittelzufuhr liegt mit 30 µg für Frauen und 41 µg für Männer unter den von der D-A-CH und EFSA angegebenen Schätzwerten für eine angemessene Selenzufuhr. Diese werden von der D-A-CH mit 70 µg für Männer und 60 µg für Frauen und mit 70 µg unabhängig vom Geschlecht von der EFSA angegeben. Vor dem Hintergrund der aktuellen Schätzwerte für eine angemessene Selenzufuhr kann eine Erhöhung der verschreibungspflichtigen Tagesdosis in der AMVV empfohlen werden. Eine Anhebung der freigestellten Tagesdosis für Selenverbindungen in Zubereitungen zum inneren Gebrauch auf 70 Mikrogramm Selen wird daher befürwortet. Literatur Ashton K, Hooper L, Harvey LJ, Hurst R., Casgrain A., Fairweather-Tait SJ. Methods of assessment of selenium status in humans: a systematic review. Am J Clin Nutr 2009; 89: 2025S-2039S. Brigelius-Flohe R, Maiorino M, Ursino F, Flohe L. Selenium: an antioxidant? In: Cadenas E, Packer L (Hrsg.). Handbook of antioxidants. Marcel Dekker, New York, 2., revidierte und erweiterte Auflage (2001) 633-664. Brown KM, Arthur JR. Selenium, selenoproteins and human health: a review. Public Health Nutr 2001; 4: 593-599. D-A-CH – Deutsche Gesellschaft für Ernährung, Österreichische Gesellschaft für Ernährung, Schweizerische Gesellschaft für Ernährung (Hrsg.). Referenzwerte für die Nährstoffzufuhr. Bonn, 2. Auflage, 1. Ausgabe (2015). Drobner C, Anke M, Thomas G. Selenversorgung und Selenbilanz Erwachsener in Deutschland. In: Anke M, Arnold W, Bergmann H (Hrsg.). Mengen- und Spurenelemente. Schubert, Leipzig (1996) 627-634. Ekmekcioglu C. Spurenelemente auf dem Weg ins 21. Jahrhundert – zunehmende Bedeutung von Eisen, Kupfer, Selen und Zink. J. Ernährungsmed. 2000; 2: 18-23. European Food Safety Authority (EFSA). Tolerable upper intake levels for vitamins and minerals. 2006. www.efsa.europa.eu/de/ndatopics/docs/ndatolerableuil.pdf [aufgerufen am: 09. Dezember 2016]. European Food Safety Authority (EFSA). Scientific opinion on dietary reference values for selenium. EFSA Journal 2014; 12: 3846. Hill KE, Zhou J, McMahan VVJ et al. Deletion of selenoprotein P alters distribution of selenium in the mouse. J Biol Chem 2003; 278: 13640-13646. Institute of Medicine, Food and Nutrition Board. Dietary Reference Intakes: Vitamin C, Vitamin E, Selenium, and Carotenoids. National Academy Press, Washington, DC, 2000 Seite 4 von 5 Köhrle J. The deioclinase family: selenoenzymes regulating thyroid hormone availability and action. Cell Mol Life Sci 2000; 57: 1853-1863. Nuttall KL. Evaluating selenium poisoning. Ann Clin Lab Sci 2006; 36: 409-420. Persson-Moschos M, Huang W, Srikumar TS, Åkesson B, Lindeberg S. Selenoprotein P in serum as a biochemical marker of selenium status. Analyst 1995; 120: 833-836. Ursini F, Heim S, Kiess M at al. Dual function of the seleno-protein PHGPx during sperm maturation. Science 1999; 285: 1393-1396. Seite 5 von 5 Sachverständigen-Ausschuss für Verschreibungspflicht nach § 53 Absatz 2 des Arzneimittelgesetzes Empfehlung zur Änderung der Arzneimittelverschreibungsverordnung 77. Sitzung 17. Januar 2017 Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM), Bonn 9. Zubereitung aus Methopren und Fipronil - zur Anwendung bei Hunden und Katzen Anträge auf Entlassung aus der Verschreibungspflicht Empfehlung des Sachverständigen-Ausschusses Der Sachverständigen-Ausschuss für Verschreibungspflicht empfiehlt mehrheitlich, Zubereitungen aus Methopren und Fipronil - zur Anwendung bei Hunden und Katzen - aus der Verschreibungspflicht nach § 48 des Arzneimittelgesetzes (AMG) zu entlassen. Begründung Wirkstoffe Fipronil Fipronil ist ein Ektoparasitikum und gehört zur Gruppe der Phenylpyrazole und ist ein schnell wirkendes Kontaktgift mit insektiziden und akariziden Eigenschaften. Seine Wirkung beruht auf einer Interaktion mit Rezeptoren der Chloridionenkanäle, insbesondere mit solchen, die auf den Neurotransmitter Gamma-Aminobuttersäure (GABA) ansprechen. Hierbei kommt es zu einer Hemmung des prä- und postsynaptischen Chloridionenaustausches durch die Zellmembranen. In Folge der gestörten ZNS-Aktivität tritt der Tod der Insekten und Acari (Milben und Zecken) ein. Fipronil hat eine höhere, selektive Spezifität für GABA-Rezeptoren der Wirbellosen, als für GABA-Rezeptoren von Säugern. Somit verfügen fipronilhaltige Veterinärprodukte über eine sehr gute Verträglichkeit und geringe Toxizität bei Säugetieren (Moffat, 1993). Ausnahme: Wesentlich empfindlicher sind Kaninchen. Bereits nach oraler Gabe von 8 mg/kg der Reinsubstanz kann es zu Todesfällen kommen. Unverträglichkeiten sind auch bei Hühnervögeln und geschwächten Igeln beschrieben (Löscher et al., 2014). Fipronil ist weder teratogen noch mutagen und kann somit bei trächtigen und laktierenden Hunden und Katzen angewendet werden (Curtis, 1996; Tanner, 1997). Fipronil erfasst auch frühe Entwicklungsstadien und verhindert die Fortpflanzung des erwachsenen Flohs, der noch vor der Eiablage getötet wird. Durch die Unterbrechung des FlohSeite 1 von 7 Lebenszyklus eignet sich Fipronil auch zur Sanierung flohverseuchter Haushalte über das Tier. Nach drei bis fünf Behandlungen in monatlichem Abstand wird dies ohne zusätzliche Umgebungsbehandlung erreicht (Wiedemann, 2000). Bei Hunden und Katzen ist innerhalb von 24 Stunden nach Applikation eine 100%ige Residualwirkung aufgebaut (Wiedemann, 2000). Grundlage für die lange Wirkungsdauer ist die Speicherung des Wirkstoffes. Fipronil reichert sich in den Talgdrüsen an, löst sich im Talg und wird kontinuierlich an das Haar und die oberen Hautschichten abgegeben (Birckel, 1997). Fipronil wird aus den Formulierungen (Spot on oder Spray) nur in geringen Mengen nach dermaler und oraler Verabreichung resorbiert. Daher sind nach Ablecken nur geringe Mengen systemisch bioverfügbar, die schnell umfangreich metabolisiert und vorwiegend biliär ausgeschieden werden (Löscher et al., 2014). Fipronil tötet Flöhe (Ctenocephalides canis, Ctenocephalides felis) innerhalb 24 Stunden und Zecken (Dermacentor reticulatus, Dermacentor variabilis, Rhipicephalus sanguineus, Ixodes scapularis, Ixodes ricinus, Haemaphysalis longicornis, Haemaphysalis flava, Haemaphysalis campanulata) und Haarlinge innerhalb 48 Stunden nach Kontakt ab. Methopren (S)-Methopren ist ein Wachstumsregulator für Insekten aus der Wirkstoffgruppe der Juvenilhormon-Analoga, der die Entwicklung der unreifen Stadien der Insekten hemmt. Diese Substanz imitiert die Wirkung des Juvenilhormons und führt zu einer gestörten Entwicklung und damit zum Tod der unreifen Flohstadien. Methopren weist eine sehr geringe Toxizität für Säugetiere auf. Eine orale Gabe von 34.000 mg/kg Körpergewicht bei der Ratte und 5000 mg/kg Körpergewicht beim Hund hat zu keinen toxischen Effekten geführt (Adams, 1995). Es kommt zu keiner insektiziden Wirkung auf adulte Insekten und bestimmte Larvenstadien, so dass kein sofortiger Effekt auftritt. Es besteht jedoch aufgrund der Schädigung der Tochtergeneration eine gute Residualwirkung. Methopren besitzt bei Schweinen, Schafen, Hamstern, Ratten und Kaninchen keine teratogene Wirkung. Es ist lichtempfindlich und bis zu 75 Tage stabil in der Umwelt (Barragry, 1994). Methopren wird in der Veterinärmedizin hauptsächlich zur Flohbekämpfung bei Hund und Katze eingesetzt (Löscher et al., 2014). Kombination Fipronil/Methopren Durch die kombinierte Verabreichung des Insektenwachstumhemmers Methopren mit larvizider und ovizider Wirkung werden auch die Entwicklungsstadien von Flöhen abgetötet, die von dem nur adultizid wirkenden Fipronil nicht erreicht werden und es wird die Flohentwicklung bis sechs Wochen gehemmt (Löscher et al., 2014). Verkaufsabgrenzung Fipronil Fipronil wurde erstmalig zugelassen am 13.05.1996 und damit automatisch verschreibungspflichtig. Mit Wirkung vom 01.07.2001 wurde dieser Stoff aus der Verschreibungspflicht entlassen (apothekenpflichtig). Methopren Methopren wurde erstmalig zugelassen in einem Tierarzneimittel am 16.12.2003 in Kombination mit Fipronil. Methopren wurde nicht als Einzeleintrag in die Arzneimittelverschreibungsverordnung aufgenommen. Es gibt keine methoprenhaltigen Monopräparate. Seite 2 von 7 Kombination Fipronil/Methopren Bei Erstzulassung der Kombination von Fipronil und Methopren wurde diese aufgrund § 49 Absatz 1 Nummer 3 AMG („…Stoffe mit in der medizinischen Wissenschaft nicht allgemein bekannten Wirkungen …“) der Verschreibungspflicht unterstellt (64. Verordnung zur Änderung der Verordnung über die automatische Verschreibungspflicht vom 21.06.2004, Nr. 1754 - Zubereitungen aus Methopren und Fipronil - zur Anwendung bei Hunden und Katzen -). Zulassungsstatus Es gibt derzeit (Stand 18.10.2016) in Deutschland 27 zugelassene Tierarzneimittel mit der Kombination aus Fipronil und Methopren. Weiterhin gibt es eine Vielzahl zugelassener fipronilhaltiger Tierarzneimittel (Monopräparate). Fipronilhaltige Monopräparate werden seit 2001 in der Apotheke vertrieben; daher werden die vorliegenden Erfahrungen und Daten zur Bewertung mit herangezogen. Anwendungsgebiete Floh- und Zeckenbefall bei Hund und Katze Haarlingsbefall bei der Katze Diagnose durch den Tierhalter Für die Anwendung der Präparate bei Befall mit Ektoparasiten ist keine vorherige tierärztliche Diagnose notwendig. Einen Befall mit Ektoparasiten (Floh-, Haarlings- und/oder Zeckenbefall) kann der Tierhalter ohne besonderes Fachwissen feststellen. Fipronilhaltige Monopräparate mit den gleichen Indikationen bezüglich der Abtötung von adulten Flöhen, Zecken und Haarlingen sind seit 2001 apothekenpflichtig. Bezüglich der Flohstichallergie siehe Punkt „Erschwerung späterer diagnostischer oder therapeutischer Maßnahmen“. Anwendersicherheit bei bestimmungsgemäßem Gebrauch Die Darreichungsform für das Kombinationsprodukt (alle Stärken) ist eine Einmalpipette mit den entsprechenden Wirkstoffmengen für die jeweilige Stärke. Der Inhalt einer Pipette ist für die einmalige Anwendung für die jeweilige Tierart und Gewichtsklasse konzipiert. Die Anwendung ist einfach und erlaubt dem Laien eine korrekte Anwendung. Das Risiko für eine inkorrekte Anwendung wie Überdosierung ist gering. Die Anwendung des Produktes erfordert keine Sachkenntnis. Es sind die folgenden Vorsichtsmaßnahmen für den Anwender formuliert: „Dieses Tierarzneimittel kann Schleimhaut-, Haut- und Augenreizungen verursachen. Deshalb jeden Kontakt mit Mund, Haut und Augen vermeiden. Tiere und Anwender, von denen eine Überempfindlichkeit gegen Insektizide oder Alkohol bekannt ist, sollten nicht in Kontakt mit [Produktname] kommen. Kontakt mit den Fingern vermeiden. Falls dies doch geschieht, Hände mit Wasser und Seife waschen. Nach versehentlichem Kontakt mit dem Auge dieses sofort sorgfältig mit klarem Wasser ausspülen. Hände nach der Anwendung waschen. Behandelte Tiere sollten nicht berührt werden und Kinder sollten nicht mit behandelten Tieren spielen, bevor die Applikationsstelle trocken ist. Deshalb empfiehlt es sich, Tiere nicht während Seite 3 von 7 des Tages, sondern in den frühen Abendstunden zu behandeln. Frisch behandelte Tiere sollten nicht in engem Kontakt mit den Besitzern, insbesondere nicht mit Kindern, schlafen. Während der Anwendung nicht rauchen, trinken oder essen.“ Die Vorsichtsmaßnahmen entsprechen denen der fipronilhaltigen Monopräparate. Während der Zeiträume der Periodic Safety Update Reports (PSURs) von 2010 bis 2016 waren die beim Menschen am häufigsten aufgetretenen Symptome (VeDDRA) Augenirritationen, Geschmacksveränderungen, Parästhesie, Dermatitis und Juckreiz. Die meisten Symptome verschwanden ohne zurückbleibende Schäden. In den meisten Fällen traten die Nebenwirkungen nach akzidenteller topikaler, dermaler oder okularer Exposition auf. Die Gesamtinzidenz (Anzahl betroffener Menschen im Verhältnis zur Anzahl behandelter Tiere) für den gesamten Zeitraum 2010 bis 2016 beträgt 0,00026 %. Zusammenfassend werden die genannten Vorsichtsmaßnahmen für den Anwender als ausreichend erachtet. Sicherheitsprofil Die neu zugelassenen Produkte werden erst im Juli 2019 den ersten PSUR einreichen (Data Lock Point Mai 2019). Parallelimporte sind von der gesetzlichen Verpflichtung, PSURs einzureichen, befreit. Im nationalen spontanen Meldesystem sind keine Meldungen für diese Produkte eingegangen (Stand 18.10.2016). Daher liegen Verkaufszahlen und langjährige Erfahrungen durch PSURs ausschließlich für die Produkte mit Erstzulassung in 2003 vor. Während der PSUR-Zeiträume (2010 bis 2016) waren die bei Hund und Katze am häufigsten aufgetretenen Symptome (VeDDRA-Terms) Veränderungen an der Applikationsstelle (Haarveränderungen, Juckreiz, Läsionen, Erytheme), Reaktionen an Haut- und Gliedmaßen (meist Juckreiz), Verhaltensänderungen (meist Hyperaktivität), systemische Veränderungen (Lethargie, Anorexie), Störungen im Verdauungstrakt und Störungen des Immunsystems (Urtikaria). Selten wurden neurologische Symptome beobachtet wie Krämpfe und Ataxie. Die beobachtete Hyperaktivität und die Verhaltensänderungen treten meist aufgrund des Juckreizes und der lokalen Hautirritationen auf. Lethargie und Anorexie sind bekannte Symptome, die in das Profil der Wirkstoffe passen. Symptome im Bereich des Verdauungstrakts (Erbrechen, Diarrhoe, Speicheln) treten üblicherweise nach Ablecken und Abschlucken der Präparate auf. Die aufgetretenen Symptome entsprechen denen der Monopräparate. Die Schlussfolgerung nach Auswertung aller Daten zur Kombination von Fipronil und Methopren zur Anwendung bei Hund und Katze im EU-weiten PSUR-Worksharing 2013 ist, dass das Nutzen-Risiko-Verhältnis positiv ist und keine Maßnahmen erforderlich sind. Die Kombination kann während der Trächtigkeit und Laktation angewendet werden. Es sind keine Wechselwirkungen bekannt. Resistenzentwicklung Derzeit gibt es keine Anhaltspunkte für die Existenz von Resistenzen gegen Fipronil (Pfister, 2009). Zitat Pfister (2009): „Die immer wieder von Tierhaltern beobachtete, als „nachlassend“ bezeichnete Wirksamkeit ist auf andere Ursachen zurückzuführen: Im Vordergrund stehen mangelhafte oder inadäquate Applikation des Produkts, falsche Gewichtsschätzung des zu behandelnden Patienten, Unterdosierung oder Auswaschen der Substanz bei häufiger Durchnässung. Es ist deshalb unerlässlich, dass bei Feststellen einer als „nachlassend“ bezeichneten Wirkung das Behandlungsintervall bzw. die Anwendungsweise entsprechend angepasst bzw. konkret verkürzt wird.“ Sowohl in adulten als auch larvalen Bioassays gab es keine Hinweise auf eine verringerte Empfindlichkeit von Flöhen gegenüber Fipronil (Rust, 2016).Daher ist derzeit durch die Entlas- Seite 4 von 7 sung der Kombination von Fipronil und Methopren aus der Verschreibungspflicht nicht von einer beschleunigten Resistenzentwicklung auszugehen. Ökotoxikologische Auswirkungen Fipronil wird in vielen Ländern als Wirkstoff in Bioziden und begrenzt in Pflanzenschutzmitteln eingesetzt ((EU) No 781/2013). Es ist allerdings aufgrund seiner toxischen Wirkung auf Bienen umstritten. Derzeit wird Fipronil von der Europäischen Behörde für Lebensmittelsicherheit (EFSA) erneut auf seine Bienentoxizität bewertet, ein „Call for Data“ der EFSA endete im Januar 2016. Durch die Darreichungsform und die Art der Anwendung von Fipronil als Spot on beim Tier wird kein Risiko für Bienen gesehen. Fipronilhaltige Spot-on-Monopräparate sind seit 2001 apothekenpflichtig. Es liegen keine Meldungen aus dem spontanen Meldesystem zu Bienen vor. Methopren ist lichtempfindlich und daher nicht stabil in der Umwelt. Es ist nicht toxisch für Bienen und Fische (Löscher et al, 2014). In der Fachinformation der Kombinationspräparate ist unter Punkt 4.4 „Besondere Warnhinweise für jede Zieltierart“ u. a. aufgeführt: „Hunde sollten für zwei Tage nach der Behandlung nicht in Gewässern schwimmen (siehe Punkt 6.6).“ Und unter Punkt 6.6 „Besondere Vorsichtsmaßnahmen für die Entsorgung nicht verwendeter Tierarzneimittel oder bei der Anwendung entstehender Abfälle“ heißt es: „Fipronil und (S)-Methopren können im Wasser lebende Organismen schädigen. Deshalb dürfen Teiche, Gewässer oder Bäche nicht mit dem Tierarzneimittel oder leeren Behältnissen verunreinigt werden. Nicht aufgebrauchte Tierarzneimittel sind vorzugsweise bei Schadstoffsammelstellen abzugeben. Bei gemeinsamer Entsorgung mit dem Hausmüll ist sicherzustellen, dass kein missbräuchlicher Zugriff auf diese Abfälle erfolgen kann. Tierarzneimittel dürfen nicht mit dem Abwasser bzw. über die Kanalisation entsorgt werden.“ In den PSUR-Zeiträumen zwischen 2010 und 2016 sind im spontanen Meldesystem keine Meldung zur Ökotoxizität zu den Kombinations- oder Monopräparaten eingegangen. Das Risiko für die Umwelt durch Anwendung von fipronil- und methoprenhaltigen Tierarzneimitteln wird daher als gering angesehen. Gefahr durch häufigen nicht bestimmungsgemäßen Gebrauch Die Ursachen von nicht bestimmungsgemäßem Gebrauch waren in den meisten Fällen ein Nichtbeachten des vom Zulassungsinhaber empfohlenen Behandlungsschemas, Unter- und Überdosierung sowie die Verabreichung auf eine von der Fachinformation abweichende Art (ungeeignete topische Anwendung, Verabreichung oral oder ins Auge). In der Fachinformation der Kombinationspräparate sind umfangreiche Vorsichtsmaßnahmen sowie Hinweise zum Schutz der behandelten Tiere formuliert. Diese entsprechen denen der Monopräparate und werden als ausreichend erachtet. In seltenen Fällen wurde die Kombination an Nicht-Zieltierarten verabreicht. Anwendung bei Nicht-Zieltierarten Fipronil hat eine geringe therapeutische Breite bei Kaninchen, Igeln und Hühnervögeln und ist bei Kaninchen und Hühnervögeln kontraindiziert (Löscher et al., 2014). Das Kaninchen als Hobbytier wird häufig gehalten (geschätzte 5,1 Millionen Kleintiere [Meerschweinchen, Kaninchen, Seite 5 von 7 Goldhamster] 2015 in Deutschland)1 und auch ggf. mit Hunden und Katzen im selben Haushalt. Daher wird in allen fipronilhaltigen Präparaten die Gegenanzeige „Nicht bei Kaninchen anwenden, da es zu Unverträglichkeiten, u. U. auch mit Todesfolge, kommen kann.“ aufgeführt. Im spontanen Meldesystem gibt es eine geringe Anzahl Meldungen zur Anwendung bei NichtZieltierarten. Die bei den betroffenen Kaninchen am häufigsten aufgetretenen Symptome waren Anorexia, Lethargie, Konvulsionen und Tod. Diese Symptome entsprechen dem pharmakologisch-toxikologischen Profil von Fipronil bei dieser Tierart. Die Inzidenz für Kaninchen beträgt 0,00006 % für beide PSUR-Intervalle. Im direkten Vergleich mit apothekenpflichtigen fipronilhaltigen Monopräparaten zeigt sich, dass die Inzidenz für Kaninchen bei einer Apothekenpflicht von Fipronil nicht signifikant höher ist. Anwendung bei Lebensmittel liefernden Tieren Fipronil und auch Methopren ist nicht für die Anwendung bei Lebensmittel liefernden Tieren zugelassen. Daher ist die Gegenanzeige „Nicht bei Tieren anwenden, die der Gewinnung von Lebensmitteln dienen.“ aufgeführt. Eine häufige Anwendung bei Lebensmittel liefernden Tieren ist unwahrscheinlich (Indikationen, Körpergewicht, Kontraindikationen). Es liegen keine Daten aus dem spontanen Meldesystem oder der Literatur über eine gehäufte missbräuchliche Anwendung der Kombination vor. Zusammenfassend kann man davon ausgehen, dass die Gefahr durch häufigen nicht bestimmungsgemäßen Gebrauch gering ist. Erschwerung späterer diagnostischer oder therapeutischer Maßnahmen Es besteht die Möglichkeit der Verwechslung einer häufig vorkommenden lokalen Reaktion auf Inhaltsstoffe des Tierarzneimittels mit lokalen Hautirritationen an der Applikationsstelle bis zu einer lokalen Dermatitis mit Juckreiz und einer durch Flohbisse ausgelösten Flohspeichelallergie. Diese tritt durch die allergenen Bestandteile des Flohspeichels auf. Symptome sind generalisierter Juckreiz, papulokrustöse Dermatitis, Lymphknotenschwellungen (bei der Katze) und Sekundärinfektionen, Schuppen und Haarausfall durch Kratzen. Charakteristisch ist die Lokalisation auf dem Rücken in der Lumbosakralregion, am Unterbauch, den Flanken und der Innenseite der Oberschenkel und am Hals. Die Therapie besteht unter anderem in der Flohbekämpfung (Reedy et al., 2002). Die meisten Tierhalter wissen, dass ihr Tier mit Flöhen befallen werden kann und werden bei sich entwickelndem Juckreiz das Tier auf Flöhe untersuchen. Die meisten Tierhalter behandeln die Flöhe, ohne den Tierarzt aufzusuchen (Reedy et al., 2002). Eine Indikation für Fipronil und Methopren ist die Anwendung des Tierarzneimittels als Teil einer Behandlungsstrategie zur Behandlung und Kontrolle der Flohstichallergie. In Fällen, in denen der Tierhalter die Symptome nicht in den Griff bekommt, ist davon auszugehen, dass er einen Tierarzt konsultieren wird. Die Diagnose wird durch eine Vorbehandlung nicht erschwert oder überlagert. Zudem sind fipronilhaltige Monopräparate mit der gleichen Indikation seit 2001 apothekenpflichtig. Schlussfolgerung 2001 wurde Fipronil aus der Verschreibungspflicht entlassen. Es liegen somit langjährige Erfahrungen zu einem Vertrieb über die Apotheke vor. Die Präparate, die die Kombination von Fipronil und Methopren enthalten, wurden 2003 erstmals zugelassen. Anwendungsgebiete, Darreichungsform und Art der Anwendung sind nahezu identisch mit denen der Monopräparate. Durch die Kombination mit Methopren ist von keiner Änderung des Sicherheitsprofils auszugehen. Die PSURs der Kombinationspräparate zeigen, dass keine unerwarteten Wechselwirkungen 1 http://www.ivh-online.de/de/der-verband/daten-fakten.html Seite 6 von 7 aufgetreten sind oder sich ein anderes toxikologisches Profil gegenüber den Monopräparaten entwickelt hat. Fazit Das Risiko für die Gesundheit des behandelnden Menschen, des Tieres oder der Umwelt durch die Entlassung der Kombination von Methopren und Fipronil aus der Verschreibungspflicht wird als gering angesehen. Es ist nicht von einem nicht bestimmungsgemäßen Gebrauch in erheblichem Umfang auszugehen und die Anwendung erfordert keine vorherige tierärztliche Diagnose. Daher werden die Voraussetzungen nach § 48 Absatz 2 Nummer 3 des Arzneimittelgesetzes als erfüllt angesehen und die Entlassung der Kombination von Fipronil und Methopren aus der Verschreibungspflicht empfohlen. Literatur Adams, H. R. (Ed.) (1995): Veterinary pharmacology and therapeutics. 7th Ed., Ames, Iowa State Univ. Pr. Barragry TB (1994): Veterinary drug therapy. Philadelphia u. a., Lea & Febiger. Birckel P, Cochet P & Weil A: Skin and hair distribution of 14C-Fipronil by microautoradiography following topical administration to the Beagle dog. In: Proceedings of the European Association of Veterinary Pharmacology and Toxicology International Congress 7th (Madrid): pp 7980, 1997. Blagburn BL & Lindsay DS: Ectoparasiticides. In: Veterinary Pharmacology and Therapeutics (HR Adams, ed) Iowa State University Press, Ames (USA), 8. Edition: pp 1017-1039, 2001. Curtis CF: Use of 0,25 per cent fipronil spray to treat sarcoptic manage in a litter of five-week-old puppies. Vet Rec 139: 43-44, 1996 (Journal: The Veterinary Record). Colliot F, Kukorowski KA et al: Fipronil. A new soil and foliar broad spectrum insecticide. Proc Brighton Crop Prot Conf; 29-34, Journal: Proceedings of the Brighton Crop Protection Conference. Garg RC & Donahue WA: Pharmacologic profile of methoprene, an insect growth regulator, in cattle, dogs, and cats. J Am Vet Med Assoc 194: 410-414, 1989. Löscher W, Ungemach FR, Kroker R (2014): Pharmakotherapie bei Haus- und Nutztieren. 9., aktualisiert und erweiterte Aufl., Enke Verlag, Stuttgart. Moffat AS: New chemicals seek to outwit insect pest. Science 261: 550-551, 1993. Pfister K: Veterinär spiegel 2009; 19(03): 166-168. DOI: 10.1055/s-0029-1186049. Reedy LM, Miller WH, Willemse T (2002): Allergische Hauterkrankungen bei Hund und Katze. Hannover, Schlütersche. Tanner PA, Meo NJ, Sparer D, Romano MN & Keister DM: Advances in the treatment of heartworm, fleas and ticks. Canine Pract 22(2-3): 40-47, 1997. Wiedemann C: Zur Wirksamkeit von Fipronil (Frontline) gegen Ektoparasiten: Teil I – Flohbefall. Tierarztl Umsch 55: 118-123, 2000. Seite 7 von 7