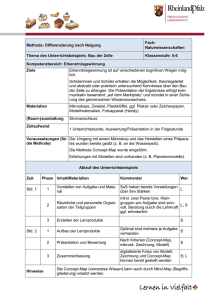
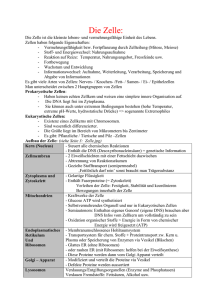
Immunmonitoring und Immuntherapie mit HIV
Werbung

Immunmonitoring und Immuntherapie mit HIV-spezifischen mRNAs Der Naturwissenschaftlichen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades Dr. rer. nat. vorgelegt von Jennifer Etschel aus Ansbach Als Dissertation genehmigt von der Naturwissenschaftlichen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg Tag der mündlichen Prüfung: 08.04.2013 Vorsitzender der Prüfungskommission: Prof. Dr. Johannes Barth Erstberichterstatter: Prof. Dr. Thomas Winkler Zweitberichterstatter: Prof. Dr. Thomas Harrer Inhaltsverzeichnis 1 ZUSAMMENFASSUNG / SUMMARY ................................................................ 1 Zusammenfassung ......................................................................................................................... 1 Summary ........................................................................................................................................ 3 2 EINLEITUNG ...................................................................................................... 5 2.1 Humanes Immundefizienz Virus (HIV) .................................................................................... 5 2.1.1 Epidemiologie und Tropismus ............................................................................................... 5 2.1.2 Pathogenese und Klinik der HIV-1-Infektion ......................................................................... 6 2.1.3 Aufbau und Replikationszyklus ............................................................................................. 7 2.2 Immunologische und genetische Faktoren, die die HIV-1-Infektion und Progression beeinflussen ............................................................................................................................ 10 2.2.1 Zytotoxische T-Lymphozyten (CTL) .................................................................................... 10 2.2.2 HLA-Allele ............................................................................................................................ 12 2.2.3 Toll-like Rezeptoren in der angeborenen Immunantwort .................................................... 14 + 2.2.4 CD4 T-Helferzellen ............................................................................................................. 14 2.2.5 Genetische Unterschiede im Gen des Chemokinrezeptor CCR5 ....................................... 15 2.2.6 Interaktion von microRNAs und Viren ................................................................................. 15 2.3 Therapie und Behandlungsansätze von HIV-1 .................................................................... 17 2.3.1 Hochaktive antiretrovirale Therapie (HAART) ..................................................................... 17 2.3.2 Vakzine ................................................................................................................................ 18 2.3.3 Verwendung von mRNA als Vakzine .................................................................................. 20 3 ZIELSETZUNG ................................................................................................. 22 4 ERGEBNISSE .................................................................................................. 24 4.1 Elektroporation von PBMC mit HIV-1-spezifischen mRNAs .............................................. 24 4.1.1 Erfolgreiche Generierung von HIV-1-spezifischen mRNAs................................................. 24 4.1.2 Test und Titration von HIV-1-spezifischen Antikörpern in transfizierten HEK293T Zellen .. 25 4.1.3 Elektroporation der HIV-1-spezifischen mRNAs zeigt schnelle und effiziente Proteinexpression ................................................................................................................ 26 4.1.4 PBMC exprimieren HIV-1-Proteine bis zu sechs Tagen nach Elektroporation ................... 28 4.1.5 mRNA-kodierte HIV-1-Proteine sind in vitro immunogen und führen zur Freisetzung von γIFN ....................................................................................................................................... 29 I Inhaltsverzeichnis 4.1.6 Das autologe Virus löst in fünf von zehn Fällen eine stärkere Immunantwort aus als Referenzviren....................................................................................................................... 30 4.1.7 Vergleich der ausgelösten T-Zellantworten zwischen mRNA-kodierten HIV-1-Proteinen und Peptiden, Peptid-Pools und Vaccinia-Viren ......................................................................... 41 4.1.8 mRNA-kodierte gp120-Proteine induzieren geringe Immunantworten ................................ 44 4.1.9 Geringe Mengen an HIV-1-spezifischer mRNA reichen aus, um eine Proteinexpression und T-Zellstimulation zu erhalten ................................................................................................ 46 4.1.10 + Durch mRNA-kodierte HIV-1-Proteine aktivieren HIV-1-spezifische CD8 zytotoxische T-Lymphozyten ohne zusätzliche Effektorzellen ................................................................. 48 4.1.11 Neben γ-IFN induzieren die von HIV-1-spezifischen mRNAs-kodierten Proteine die Sekretion von TNF-α............................................................................................................ 50 4.2 Funktionelle Analysen mit HIV-1-spezifischen mRNAs ...................................................... 53 4.2.1 mRNA-kodierte HIV-1-Proteine stimulieren die Generierung von HIV-1-spezifischen CTL Linien ................................................................................................................................... 53 4.2.2 PBMC proliferieren nicht spezifisch nach Elektroporation mit mRNA ................................. 54 4.2.3 Mit HIV-1-spezifischer mRNA-elektroporierte PBMC zeigen nach Kryokonservierung noch schwache Immunantworten ................................................................................................. 55 4.2.4 mRNA-kodierte Nef-Proteine reduzieren die Expression von CD4, CXCR4 und HLA-I ..... 56 4.2.5 Folgen der simultanen Elektroporation von gag und nef mRNA ......................................... 58 4.2.5.1 Die simultane Elektroporation zweier HIV-1-spezifischer mRNAs in B-Zelllinien hat keine Auswirkungen ..................................................................................................... 58 4.2.5.2 Die simultane Elektroporation zweier HIV-1-spezifischer mRNAs in PBMC führt zu einem Verlust der Immunantworten ............................................................................. 61 4.2.6 Mit mRNA-elektroporierten PBMC kann die CTL-Epitop-Hierarchie studiert werden ......... 63 4.3 Kreuzreaktion HIV-1-spezifischer CTLs mit humanen Proteinen ...................................... 66 4.3.1 Das Nef-Epitop TM9 ähnelt einem Epitop im humanen Protein Fat3.................................. 66 4.3.1.1 Breite CTL-Antwort gegen die Epitope Nef TM9 und Fat3 TM9 T/P ........................... 66 4.3.1.2 pfat3 mRNA Elektroporation führte trotz effizienter Proteinexpression zu keiner Erkennung durch Nef-spezifische CTLs ...................................................................... 68 4.3.1.3 In silico Analysen des fat3 mRNA-Konstrukts sagen eine Prozessierung des Epitops TM9 T/P durch das Proteasom mit einer 50%igen Wahrscheinlichkeit voraus ........... 71 4.3.2 Das Nef-Epitop Nef7v1 ist einem Epitop im humanen Enzym D-Aminosäuren-Oxidase (DAO) ähnlich ...................................................................................................................... 72 4.4 Analyse und Screening von neuen und bekannten CTL-Epitopen ................................... 75 4.4.1 Definition von drei neuen Gag und zwei neuen Nef CTL-Epitopen ..................................... 75 4.4.2 Analyse der neu definierten CTL-Epitope ............................................................................ 77 4.4.2.1 Das Peptid Gag07-113 enthält ein HLA-A*02-restringiertes CTL-Epitop .................... 77 4.4.2.2 Das Peptid Gag07-02 enthält ein HLA-B*41-restringiertes CTL-Epitop ...................... 78 II Inhaltsverzeichnis 4.4.2.3 Die restringierenden HLA-Allele für die Peptide Gag07-11 und Gag07-75 konnten nicht bestimmt werden ................................................................................................. 80 4.4.2.4 Die Peptide Nef07-08 und Nef07-10 enthalten HLA-B*41-Epitope ............................. 81 4.4.3 Das bekannte HLA-C-restringierte Nef-Epitop Nef11T3 ist ein häufig erkanntes Epitop .... 83 4.5 Etablierung alternativer Transfektionsmethoden ............................................................... 85 4.5.1 Chitosan transfiziert DNA, aber keine mRNA in HEK293T ................................................. 85 4.5.2 HIV-1 Tat führt zu keinem Transfer von Nukleinsäuren in Zellen ....................................... 86 4.5.3 Atelokollagen transfiziert weder DNA noch mRNA in HEK293 und HEK293T ................... 86 4.5.4 FuGENE kann neben DNA auch mRNA erfolgreich in HEK293T Zellen transfizieren ....... 87 4.6 Analyse des Sequenzverlaufs und der Immunantworten eines HIV-1-infizierten Patientenpaares, das mit demselben Virus infiziert worden war ...................................... 88 4.6.1 Unterschiedliche Erkennung der homologen und heterologen viralen mRNA-Konstrukten 88 4.6.2 Die viralen autologen Sequenzen erklären die detektierten Immunantworten.................... 90 4.6.3 In Patient 0225 mutierte das Virus im selben Zeitraum deutlich mehr als in Patient 0284 . 93 4.7 Interaktion zwischen HIV-1-spezifischen mRNAs und zellulären miRNAs ...................... 97 + 4.7.1 Einzelne HIV-1-Gene beeinflussen das zelluläre miRNA-Profil in CD4 T-Zellen .............. 97 4.7.1.1 Durch Deep Sequencing konnten zelluläre miRNAs identifiziert werden, deren Expressionsstärke von einzelnen HIV-1-Genen verändert wird .................................. 97 4.7.1.2 Durch HIV-1-Proteine modifizierte miRNAs konnten teilweise in einzelnen Proben durch qRT-PCR validiert werden ............................................................................... 101 4.7.2 CTL Fluchtmutationen in miRNA-Binderegionen beeinflussen die Regulation von miRNAs ........................................................................................................................................... 104 5 DISKUSSION ................................................................................................. 108 5.1 Elektroporation von PBMC mit HIV-1-spezifischen mRNAs ............................................ 108 5.1.1 Immunmonitoring gegen autologe Virusproteine mit HIV-1-spezifischen mRNAs ............ 110 5.1.2 HIV-1-spezifische mRNAs als therapeutischer Impfstoff .................................................. 114 5.2 Funktionelle Analysen mit HIV-1-spezifischen mRNAs .................................................... 116 5.2.1 Wirksamkeit eines Kombinationsimpfstoffes aus gag und nef mRNA .............................. 117 5.2.2 Studien zur CTL-Epitop-Hierarchie identifizieren effizient prozessierte Epitope als mögliche Vakzin-Antigene ................................................................................................................ 119 5.3 Ausschluss von Nebenwirkungen bei einer Impfung ....................................................... 120 5.3.1 Wechselwirkung zwischen HIV-1-spezifischen CTLs und dem humanen Protein Fat3 ... 121 5.3.2 Wechselwirkung zwischen HIV-1-spezifischen CTLs und dem humanen Protein DAO ... 122 5.4 Analyse und Definition bekannter und neuer CTL-Epitope ............................................. 123 III Inhaltsverzeichnis 5.5 Etablierung alternativer Transfektionsmethoden von mRNA .......................................... 125 5.5.1 Transfektion mit Chitosan .................................................................................................. 125 5.5.2 Transfektion über das HIV-1 Tat-Protein ........................................................................... 126 5.5.3 Transfektion mit Hilfe von Atelokollagen ........................................................................... 126 5.5.4 Transfektion mit FuGENE .................................................................................................. 127 5.6 Evolution eines HI-Ausgangsviruses in zwei unterschiedlichen Patienten und die daraus resultierenden Immunantworten ............................................................................ 127 5.6.1 Vergleichende Immunantworten auf die mRNA-kodierten HIV-1-Proteine ....................... 128 5.6.2 Evolution eines HI-Virus in zwei unterschiedlichen Patienten ........................................... 129 5.7 Interaktion von HIV-1-spezifischen mRNAs und zellulären miRNAs .............................. 130 + 5.7.1 Einfluss der HIV-1 Gene gag und nef auf miRNAs in CD4 T-Zellen ................................ 131 5.7.2 Einfluss von CTL Fluchtmutanten in Nef auf die miR-29a-Interaktion............................... 132 5.8 Ausblick ................................................................................................................................. 134 6 MATERIAL UND METHODEN ....................................................................... 136 6.1 Material .................................................................................................................................. 136 6.1.1 Patienten ............................................................................................................................ 136 6.1.2 Verbrauchsmateralien ........................................................................................................ 136 6.1.3 Laborgeräte........................................................................................................................ 137 6.1.4 Chemikalien ....................................................................................................................... 139 6.1.5 Puffer und Lösungen ......................................................................................................... 140 6.1.6 Kulturmedien und Zusätze ................................................................................................. 141 6.1.7 Peptide und Proteine ......................................................................................................... 143 6.1.8 Antikörper........................................................................................................................... 143 6.1.9 Kommerziell erhältliche Kits............................................................................................... 144 6.1.10 Enzyme ...................................................................................................................... 144 6.1.11 Längenstandards für Gelelektrophoresen.................................................................. 145 6.1.12 Vektoren ..................................................................................................................... 145 6.1.13 Bakterien, Viren, Zellen und Zelllinien ....................................................................... 145 6.1.14 Oligonukleotidprimer .................................................................................................. 147 6.1.15 TaqMan MicroRNA Assays ...................................................................................... 148 6.1.16 Datenbanken .............................................................................................................. 148 6.1.17 Software ..................................................................................................................... 149 ® 6.2 Methoden ............................................................................................................................... 149 6.2.1 Molekularbiologische Methoden ........................................................................................ 149 6.2.1.1 Isolation von Nukleinsäuren ....................................................................................... 149 6.2.1.2 Konzentrationsbestimmung von Nukleinsäuren ........................................................ 150 IV Inhaltsverzeichnis 6.2.1.3 Polymerasen-Ketten-Reaktion (PCR) ........................................................................ 150 6.2.1.4 Agarosegelelektrophorese ......................................................................................... 153 6.2.1.5 Sequenzierung ........................................................................................................... 153 6.2.1.6 Klonierungsmethoden ................................................................................................ 154 6.2.1.7 Generierung von mRNA ............................................................................................ 158 6.2.1.8 Hochdurchsatzsequenzierung (Deep Sequencing) ................................................... 160 6.2.1.9 Quantitative Echtzeit (Real-time) PCR (qRT-PCR) ................................................... 160 6.2.1.10 ................................................................................................. HLA Typisierung ................................................................................................................................... 162 6.2.1 Zellbiologische Methoden .................................................................................................. 162 6.2.1.1 Allgemeine Hinweise für Zellkulturverfahren ............................................................. 162 6.2.1.2 Zellselektion ............................................................................................................... 165 6.2.1.3 Transfektionsmethoden ............................................................................................. 166 6.2.1.4 mRNA-Elektroporation ............................................................................................... 167 6.2.1.5 Durchflusszytometrie ................................................................................................. 168 6.2.1.6 Untersuchung der Vitalität von Zellen ........................................................................ 170 6.2.1.7 Bestimmung der Zellproliferation ............................................................................... 170 6.2.1.8 Lösen und Verdünnen von Peptide ........................................................................... 171 6.2.1.9 γ-Interferon ELISPOT ................................................................................................ 171 6.2.1.10 Analyse der Freisetzung von weiteren Zytokine ...................................................... 173 6.2.1.11 Dualer Luciferase-Reporter-Assay ........................................................................... 174 6.2.1.12 Infektion mit rekombinanten Vaccinia-Vektoren....................................................... 175 7 LITERATURVERZEICHNIS ........................................................................... 176 8 ABKÜRZUNGSVERZEICHNIS ...................................................................... 194 9 EIGENE PUBLIKATIONEN, KONFERENZBEITRÄGE UND VORTRÄGE ... 199 10 DANKSAGUNG ............................................................................................. 202 V 1 Zusammenfassung / Summary Zusammenfassung Ein wesentlicher Schwerpunkt der Promotionsarbeit waren Experimente zur Entwicklung von viralen mRNA-Konstrukten zum Immunmonitoring von T-Zellen und zur Entwicklung von Vakzinen. In der vorgelegten Dissertation wurden HIV-1-spezifische mRNAs generiert und erfolgreich zur Elektroporation in PBMC eingesetzt. Es konnte dabei gezeigt werden, dass die Elektroporation von PBMC mit autologen HIV-1-spezifischen mRNAs eine schnelle und effiziente Analyse der T-Zellantworten gegen das eigene HI-Virus erlaubt. Es konnte eine schnelle und effiziente Expression der mRNA-kodierten Proteine in allen Subpopulationen der PBMC nachgewiesen werden, wobei die Proteinexpression bis zu sechs Tage nach Elektroporation detektiert werden konnte. Die mRNA-kodierten HIV-1Proteine waren in vitro hoch immunogen und lösten die Sekretion von γ-IFN und TNF-α aus. Dabei zeigte sich, dass die γ-IFN Freisetzung von HIV-1-spezifischen T-Zellen verursacht wird. Neben der Immunogenität wurde auch die Funktionalität der mRNAkodierten HIV-1-Proteine belegt. Denn die Expression der Moleküle CD4, CXCR4 und HLA-I auf der Zelloberfläche wurde durch die mRNA-kodierten Nef-Proteine deutlich reduziert. Außerdem konnten mRNA-kodierte HIV-1-Proteine CD8+ T-Zellen aktivieren. Die mRNA-kodierten HIV-1-Proteine können als alternative Antigene für das Monitoring von T-Zellantworten bei HIV-1-infizierten Patienten eingesetzt werden. Sie induzierten stärkere CTL-Antworten wie rekombinante Vaccinia-Viren und ähnliche Immunreaktionen wie Peptid-Pools. Im Vergleich zu einzelnen HIV-1-Peptiden rufen sie jedoch geringere T-Zellantworten hervor. Durch den Einsatz autologer HIV-1-spezifischer mRNAs konnten die Immunantworten gegen autologe Virusproteine detektiert und mit denen gegen heterologe Virusproteine verglichen werden. In fünf von zehn Fällen lösten die autologen viralen Proteine stärkere Immunantworten aus als Proteine von HIV-1 Referenzviren. Folglich konnte bewiesen werden, dass die im Monitoring ermittelten Immunreaktionen häufig unterschätzt werden, da mit den Standard-Methoden die CTLAntworten gegen das eigene Virus nicht quantifiziert werden können. In zwei Fällen wurde eine deutlich geringere T-Zellantwort gegen das autologe Virusprotein gemessen, was auf CTL Fluchtmutanten in der autologen Virussequenz zurückgeführt werden konnte. Demzufolge kann es auch zur Überschätzung der Immunantworten beim StandardMonitoring kommen. Neben der Etablierung einer neuen, effizienten Methode zum Monitoring von T-Zellantworten gegen autologe Viren diente die mRNA-Elektroporation auch zur Aufklärung wichtiger Fragestellungen, die den Einsatz HIV-1-spezifischer mRNAs als 1 1 Zusammenfassung / Summary therapeutische Vakzine betreffen. Es konnte bestätigt werden, dass die mRNA-kodierten Proteine Gag und Nef in HIV-1-infizierten Patienten besser erkannt wurden als Env. Allerdings konnte gezeigt werden, dass die gleichzeitige Verabreichung von gag und nef mRNA zu einem drastischen Verlust der induzierten Immunantworten in PBMC aufgrund der HLA-I-Reduktion durch das Nef-Protein führt. Zudem wurden klare Hinweise erhalten, aus welchen Epitopen eine optimale mRNA-Vakzine bestehen soll, da mit dem entwickelten Assay die CTL-Epitop-Hierarchie studiert werden konnte. Außerdem wurde von mir untersucht, ob Kreuzreaktionen von HIV-1-spezifischen CTLs mit humanen Proteinen existieren, die zu Nebenwirkungen bei einer mRNA-Vakzinierung führen könnten. Dazu wurden Nef-spezifische CTL auf die Erkennung von Peptiden aus den humanen Proteinen DAO und Fat3 getestet. Dabei zeigte sich eine deutliche Erkennung der Peptide, jedoch keine Erkennung von Fat3-exprimierenden Zelllinien. Im Rahmen der Promotion konnte ich mehrere CTL-Epitope neu definieren, darunter drei neue CTL-Epitope in Gag und zwei in Nef. Da HIV-1 Nef HLA-A und –B, nicht jedoch HLA-C von der Zelloberfläche herabreguliert und damit die HIV-1-spezifische CTL-Antwort inhibiert, gewinnt die HLA-C-restringierte CTL-Antwort an Bedeutung. Daher wurde eine systematische Analyse der Erkennung eines konservierten HLA-Cw-restringierten CTL in Nef durchgeführt, wobei eine Erkennung des Epitops von ca. der Hälfte der untersuchten HLA-Cw*07-positiven HIV-1-Patienten festgestellt werden konnte. Da bei einer in vivo Applikation die mRNA-Elektroporation eher ungeeignet ist, wurde nach alternativen Transfektionsmethoden gesucht. Dabei konnte mRNA mit Hilfe von FuGENE in die Zelle transfiziert werden, wohingegen Chitosan, Atelokollagen und das HIV-1 Tat-Protein nicht in der Lage waren, mRNA in die Zelle einzuschleusen. Um den Einfluss von viralen Proteinen auf die Expression von zellulären miRNAs zu erfassen, wurden die HIV-1 gag und nef mRNAs in CD4+ T-Zellen elektroporiert und das miRNA-Profil analysiert. Die gewonnenen Daten weisen darauf hin, dass virale Gene aus verschiedenen HIV-1-Patienten einen unterschiedlichen Einfluss auf zelluläre miRNAs haben können. CTL Fluchtmutationen spielen ebenso eine Rolle, da die vorgelegten Ergebnisse zeigen, dass sie die Bindungsaffinität von zellulären miRNAs beeinflussen können. Damit konnte in der vorgelegten Arbeit die Elektroporation von HIV-1-spezifischen mRNAs in PBMC als neue effiziente Methode zum Immunmonitoring etabliert werden. Die Verwendung autologer viraler mRNA erlaubt zusätzlich die Bestimmung der T-Zellantwort gegen das eigene Virus. Darüber hinaus eignet sich die etablierte Technik, um virale mRNAs als Vakzine zur Therapie und Prävention der HIV-1-Infektion zu entwickeln. 2 1 Zusammenfassung / Summary Summary A major focus of the thesis was the production and characterization of viral mRNA constructs for immunomonitoring of T-cells and for the development of RNA-based vaccines. I could demonstrate that the electroporation of PBMC with autologous HIV-1specific mRNA allows a rapid and efficient analysis of the T-cell responses against the autologous virus. Several HIV-1-specific mRNAs encoding Gag, Nef and Env were generated and successfully used for electroporation of PBMC in this thesis. Thereby, a rapid and efficient expression of the mRNA-encoded proteins in all subpopulations of PBMC was demonstrated whereat the protein expression was detected up to six days after electroporation. The mRNA-encoded HIV-1 proteins were highly immunogenic in vitro and triggered the secretion of γ-IFN and TNF-α. It was shown that the release of γ-IFN was induced by HIV-1-specific T-cells. In addition to the immunogenicity the functionality of the mRNA-encoded HIV-1 proteins was also demonstrated. The expression of the molecules CD4, CXCR4 and HLA-I on the cell surface was clearly reduced by mRNA-encoded Nef proteins. Moreover, the mRNA-encoded HIV-1 proteins could activate CD8+ T-cells in vitro. The mRNA-encoded HIV-1 proteins can be used as efficient antigens for the monitoring of T-cell responses in HIV-1-infected patients. They induced stronger CTL responses compared to recombinant vaccinia viruses and similar immune reactions as peptide pools. Using autologous HIV-1-specific mRNAs the immune response against autologous viral proteins was compared to the immune response against heterologous viral proteins. In five of ten cases, the autologous viral proteins induced stronger immune responses than proteins of HIV-1 reference viruses, demonstrating that conventional monitoring using heterologous antigens frequently underestimates the magnitude of the CTL responses against the autologous virus. In two cases, the T-cell response against the heterologous viral antigen exceeded the response against the autologous virus which could be explained by CTL escape mutations in the autologous viral sequence. This observation underlines the important role of CTL escape and it also demonstrates that the use of heterologous antigens can also overestimate the HIV-1-specific immune responses. Besides the establishment of a novel efficient tool for monitoring the T-cell responses against autologous viruses, mRNA-electroporation can also address important issues concerning the use of HIV-1-specific mRNAs as therapeutic vaccines. It could be confirmed that the mRNA-encoded proteins Gag and Nef induced better immune responses than Env and that they are suitable antigens for vaccination. However, it was shown that combined application of gag and nef mRNAs causes a dramatic loss of the 3 1 Zusammenfassung / Summary induced immune responses in PBMC due to the HLA-I reduction by the Nef protein. Moreover, the experiments provided interesting insights about the hierarchy of CTL epitopes and the efficiency of epitope presentation which are useful for the design of vaccines. There are several sequence homologies between HIV-1 and the human genome that could potentially lead to autoimmune reactions after vaccination. Therefore, I investigated the cross-recognition of the human proteins DAO and Fat3 by Nef-specific CTL. I observed cross-recognition of DAO and Fat3-derived peptides by Nef-specific CTL, but I could not detect CTL recognition of cell lines expressing the Fat3 protein. This is suggesting that the cross-reactive peptides are not processed from these proteins. During my thesis I could define three new CTL epitopes in Gag and two in Nef. As HIV-1 Nef downregulates HLA-A and –B molecules from the cell surface, there is a growing interest in HLA-C-restricted CTL responses. Therefore, I investigated the recognition of a HLA-Cw*07-restricted CTL epitope in Nef in a cohort of HIV-1-infected patients. I could demonstrate that this epitope is targeted by approximately half of the HLA-Cw*07-positive patients. As the mRNA-electroporation is not suitable for in vivo application, I investigated alternative transfection methods. The mRNA could be transferred into cells by FuGENE, but not by chitosan, atelocollagen or the HIV-1 protein Tat. To assess the influence of viral proteins on the expression of cellular miRNAs, HIV-1 nef and gag mRNAs were transfected into purified CD4+ T-cells and the miRNA profile was analyzed by sequencing. It could be shown that viral genes from various HIV-1-patients can differently influence cellular miRNAs. CTL escape mutations play also a role not only for CTL recognition, but they can also influence the binding affinity of cellular miRNAs. In my thesis, the electroporation of HIV-1-specific mRNAs in PBMC could be established as a novel, efficient tool for immunomonitoring. The use of autologous viral mRNA allows also the determination of the T-cell response against the autologous virus and the analysis of function of the viral proteins. In addition, the results provide useful information for the development of efficient mRNA-based vaccines for therapy and prevention of HIV-1-infection. 4 2 Einleitung Im Jahre 1983 gelang es Mitarbeitern um Luc Montagnier am Pasteur-Institut in Paris erstmals das Virus aus einem Patienten zu isolieren, welches für das erworbene Immunschwächesyndrom (acquired immunodeficiency syndrome, AIDS) verantwortlich ist (1). Robert Gallo und seiner Gruppe am National Cancer Institute in Bethesda war dies kurze Zeit später möglich. Seit 1986 wird dieses Pathogen humanes Immundefizienz Virus (HIV) genannt und ersetzt die zuvor verwendeten Bezeichnungen wie Lymphadenopathie-assoziiertes Retrovirus (LAV) oder humanes T-Zell Leukämie Virus Typ III (HTLV-III) (2). Luc Montagnier und seine Mitarbeiterin Françoise Barré-Sinoussi wurden 2008 für diese Arbeit mit dem Nobelpreis für Medizin oder Physiologie ausgezeichnet. Seitdem bemühen sich Wissenschaftler darum, die Wirkungsweise des Virus aufzuklären und eine erfolgreiche Therapie zur Bekämpfung des Virus zu entwickeln. Dabei wurde ein komplexes Zusammenspiel zwischen Virus und Wirt entschlüsselt. Das menschliche Immunsystem besitzt viele Mechanismen zur Erkennung und Eindämmung von Viren. Neben CD4+ T-Helferzellen, neutralisierenden Antikörper und zytotoxischen T-Lymphozyten, welche HIV-1-infizierte Zellen erkennen und eliminieren, spielen auch genetische Faktoren des Wirts, wie HLA-Allele und antivirale Faktoren eine wichtige Rolle. Letztendlich gelingt es unserem Immunsystem aber nicht, HIV-1 vollständig zu eliminieren, weshalb eine wirkungsvolle Therapie entwickelt wurde. Obwohl HIV-1 eines der beststudierten Viren ist und dadurch auch eine erfolgreiche Behandlung hervorgebracht wurde, ist die Heilung einer HIV-1-Infektion immer noch nicht möglich. 2.1 Humanes Immundefizienz Virus (HIV) 2.1.1 Epidemiologie und Tropismus Weltweit zählt HIV mit ungefähr 34 Millionen infizierten Menschen (Stand 2010) immer noch zu den häufigsten Infektionskrankheiten. In Zentraleuropa lag die Anzahl der Erkrankten bei ca. 840.000 (770.000-930.000), in Deutschland bei ca. 70.000 (60.00083.000) (3). Seit 2006 kann ein Rückgang der Neuinfektionen konstatiert werden (2011: ca. 2.700), während vor 2006 ein Anstieg zu verzeichnen war (3, 4). Die Übertragung des Virus erfolgt hauptsächlich sexuell. Die parenterale Übertragung durch kontaminierte Blutprodukte oder Nadeln bei Drogenmissbrauch sowie die vertikale Übertragung während der Schwangerschaft kommen in Deutschland kaum mehr vor. 5 2 Einleitung Man unterscheidet zwischen HIV-1 und HIV-2, die sich in ihrer Sequenz bis zu 50% unterscheiden. HIV-2 zeigt ein AIDS-ähnliches Krankheitsbild, die Virulenz ist jedoch deutlich geringer. HIV-2 kommt vor allem in Westafrika und in Indien vor, wohingegen HIV-1 weltweit vertreten ist und sich aufgrund von Sequenzunterschieden in die drei Subtypen M, O und N spaltet (5-7). Die meisten vorkommenden Viren gehören dem MSubtyp (major) an, der sich weiter in die Genotypen A-I unterteilt. Der Genotyp B hat dabei die größte Bedeutung in den westlichen Industrienationen (8). Subtyp O bezeichnet Ausreißer (outlier) und der N-Subtyp umfasst neue Viren (new). Des Weiteren wird HIV-1 in R5- und X4-Stämmen differenziert. Erstere verwenden den Chemokinrezeptor CCR5 als Ko-Rezeptor und infizieren darüber primär Makrophagen und Lymphozyten. Sie kommen insbesondere in den frühen Stadien der Erkrankung vor und bilden keine Synzytien. Daher werden sie auch als NSI (non-syncytium-inducing)bzw. M (Makrophagen)-tropische Stämme bezeichnet. Im Gegensatz dazu kommen X4Stämme, welche den CXC-Motiv-Chemokinrezeptor CXCR4 als Ko-Rezeptor binden, vor allem in den späteren Krankheitsstadien vor und infizieren transformierte CD4+ T-Zelllinien und primäre Lymphozyten. Da sie Synzytien bilden können, werden sie auch als SI (syncytium-inducing)- oder T (T-Zellen)-tropische Stämme bezeichnet. Überdies finden sich auch dual-trope Stämme, die die Eigenschaften beider Stämme vereinen. HIV-1 ist zudem in der Lage durch Mutationen den Tropismus während eines Krankheitsverlaufs zu wechseln (6, 7, 9). 2.1.2 Pathogenese und Klinik der HIV-1-Infektion Die akute Infektion mit HIV-1 erfolgt oft unbemerkt, da Symptome bei nur ca. 20-30% der Infizierten auftreten und sehr unspezifisch wie auch variabel sind. Meist ähneln die Symptome einem grippalen Infekt und machen sich in Form von Fieber, Lymphknotenschwellung oder Erkältungsbeschwerden bemerkbar. In dieser Phase weisen die Patienten eine hohe Virämie mit 106 bis 108 Viruspartikel/ml Blut auf. Kurzfristig sinkt in dieser Phase die Zahl der CD4+ T-Zellen unter 500 Zellen/µl. Nach dieser kurzen, initialen Virämie, welche meist nur wenige Wochen andauert, sinkt die Viruslast und die Zahl der CD4+ T-Zellen normalisiert sich. Es folgt eine symptomfreie Latenzzeit, die mehrere Jahre anhält. Die Anzahl der CD4+ T-Zellen reduziert sich währenddessen stetig (Abbildung 2.1). CD4+ T-Lymphozyten, Makrophagen und dendritsche Zellen (dendritic cells; DCs) werden durch das Virus immer mehr zerstört, so dass es nach und nach zu einem Ausfall der immunologischen Funktionen dieser Zellen 6 2 Einleitung kommt. Fällt die Zahl der CD4+ T-Zellen unter den Schwellenwert von 200 Zellen/µl Blut, kann das Immunsystem freigesetzte Viren nicht mehr abfangen und eliminieren. Abbildung 2.1 Typische Verlaufskurve einer HIV-1-Infektion. Die Infektion gliedert sich in die drei Stadien der Primärinfektion, der klinischen Latenz und dem Endstadium AIDS. Die rote Linie beschreibt den Verlauf + der Viruslast im Plasma über die Zeit, die blaue Linie zeigt die Anzahl der CD4 T-Zellen. Detaillierte Erklärung siehe Text. Abbildung von Wikipedia: http://en.wikipedia.org/wiki/File:Hiv-timecourse_copy.svg (10). Es folgt das Endstadium AIDS. Dieses ist charakterisiert durch wiederholte Infekte mit opportunistischen Erregern und das Auftreten von malignen Tumoren, vor allem von Lymphomen oder von Karposi-Sarkomen. Oft werden auch schwere neurologische Symptome wie Demenz diagnostiziert (7, 11). Nach der CDC-Klassifikation (U.S. Centers for Disease Control and Prevention) lässt sich die HIV-1-Infektion in drei klinische Kategorien einteilen. Kategorie A umfasst das asymptomatische Stadium der Infektion mit >500 CD4+ T-Zellen/µl. Kategorie B beschreibt die symptomatischen Erkrankten ohne AIDS und 200-499 CD4+ T-Zellen/µl und zur letzten Kategorie C gehören Patienten mit Vollbild AIDS und weniger als 200 CD4+ T-Zellen/µl (12). 2.1.3 Aufbau und Replikationszyklus HIV-1 ist der Familie der Retroviren zuzuordnen und gehört der Gattung der Lentiviren an. Sein Genom ist etwa 9.800 Basen groß und liegt im Viruskapsid in zwei Kopien als lineares, einzelsträngiges, plus-orientiertes RNA-Molekül vor. Die virale RNA besitzt eine 7 2 Einleitung cap-Struktur am 5`-Ende und eine Poly-Adenylierung am 3`-Ende, so dass sie mit der mRNA (messenger RNA) von Eukaryonten vergleichbar ist. Neben den Genen gag, pol und env, welche in allen replikationskompetenten Retroviren vorkommen, besitzt HIV-1 sechs zusätzliche, sogenannte akzessorische Gene (vif, vpu, vpr, tat, rev und nef). Alle neun Gene sind von langen terminalen Sequenzwiederholungen (long terminal repeats, LTR) umgegeben, welche zur Integration des Virus in das Wirtsgenom benötigt werden (Abbildung 2.2) (13). Abbildung 2.2 Schematischer Aufbau des HIV-1-Genoms. Die Gene gag (orange), pol (rot) und env (blau), kodieren für die Strukturproteine, die Enzyme und die Hüllproteine des Virus. Zusätzlich besitzt HIV-1 noch sechs akzessorische Gene (grün). Erklärung siehe Text. Adaptiert von Costin, 2007 (13). Das Gen gag (group antigen) trägt die Information für die Proteine p17, p24 und p7, welche die Matrix, das Kapsid und das Nukleokapsid bilden, sowie das Linkerprotein p6. Die Glykoproteine gp120 und gp41 für die Virushülle werden von env (envelope) kodiert. Die vom Virus benötigten Enzyme zur Virusduplikation und -integration wie Protease, Reverse Transkriptase und Integrase werden von pol (polymerase) kodiert. Die akzessorischen Gene vif, vpu, vpr, tat, rev und nef enthalten die Information für Proteine, welche die Replikation wie auch die Infektion des Virus entscheidend beeinflussen können. Das Vif-Protein beispielsweise inaktiviert den zellulären, antiviralen Restriktionsfaktor APOBEC3G. Das Nef-Protein hat unter den akzessorischen Proteinen eine zentrale Rolle, da es mit vielen verschiedenen Wirtsproteinen interagieren kann und zudem entscheidend zur Pathogenität des Virus beiträgt. So wurde gezeigt, dass 8 2 Einleitung Rhesusaffen, die mit nef deletierten Viren infiziert sind, einen stark abgeschwächten Krankheitsverlauf aufweisen. Zudem zeigen Sequenzanalysen von Langzeit- Nichtprogressoren (longterm non-progressors, LTNP) höchst mutierte Nef Sequenzen, die unter anderem große Deletionen im nef Gen aufweisen (7, 11, 14-16). Abbildung 2.3 Schematischer Überblick über den Replikationszyklus von HIV-1. Erklärung siehe Text. Abbildung von Wikipdia: http://de.wikipedia.org/w/index.php?title=Datei:Hiv_gross_german_2.jpg&filetimestamp=20110125195209 (17) Der Lebenszyklus von HIV-1 ist durch mehrere Schritte gekennzeichnet (Abbildung 2.3). Der erste Schritt beschreibt den Eintritt des Virus in die Zielzelle, was über Fusion der viralen Membran mit der Membran der Zielzelle erfolgt. Primär findet diese Verschmelzung bei HIV-1 über Rezeptoren statt, wobei die Oberflächendomäne gp120 des Env-Proteins eine zentrale Rolle spielt. Zunächst bindet gp120 an das CD4-Molekül auf der Oberfläche der Zielzellen, zu welchen CD4+ T-Helferzellen, Makrophagen und Monozyten gehören. Diese Bindung verursacht eine Konformationsänderung in gp120, wodurch die Bindestelle für den Ko-Rezeptor frei wird. Je nach Tropismus des Virus erfolgt die Bindung mit den zellulären Ko-Rezeptor CCR5 oder CXCR4. Dies führt zu einer erneuten Konformationsänderung, welche eine „6-Helices-Struktur“ ausbildet, die die Fusion der zwei Membranen ermöglicht. Nach Eintritt des Nukleokapsids in das Zytoplasma wird das Nukleokapsid entpackt und die virale RNA wird mit Hilfe der Reversen Transkriptase in eine provirale DNA umgewandelt. Diese wandert in Form eines Präintegrationskomplexes bestehend aus zellulären sowie viralen Faktoren in den Zellkern und nach Aktivierung der Zelle wird die provirale DNA durch die Integrase in die 9 2 Einleitung Wirts-DNA eingefügt. Die Integration der proviralen DNA ist Voraussetzung für die Transkription der viralen Gene. Zelluläre Transkriptionsfaktoren wie NF-κB (nuclear factor 'kappa-light-chain-enhancer' of activated B-cells) können die Expression der viralen Gene initiieren, indem sie an den LTR-Bereich der integrierten, proviralen DNA binden. In diesem Bereich befinden sich der Promotor sowie Bindestellen für Transkriptionsfaktoren. Da zunächst regulatorische Proteine wie Tat und Rev exprimiert werden, kann die Expression der viralen Gene in eine frühe und späte Phase unterteilt werden. Rev und Tat stimulieren dabei die weitere Transkription als auch die Ausbildung von langen RNATranskripten. Rev fördert zudem die Expression von strukturellen und enzymatischen Genen, welche zur Bildung von viralen Partikeln benötigt werden. Nach Transkription der viralen Gene im Zellkern findet die Translation an den Ribosomen im Zytoplasma, sowie Spaltung von Vorläuferproteinen durch die Protease oder Glykosilierung der Proteine am endoplasmatischen Retikulum statt. Die Produkte von pol und gag sowie die virale RNA lagern sich zusammen und bilden das Nukleokapsid. Die Produkte von env bilden später die „spikes“ auf der Oberfläche der Viren. Im Anschluss werden die Partikel über den Golgi-Apparat zur Zelloberfläche transportiert, können sich dort von der Zelle abschnüren und als neue, infektiöse Viren weitere Zellen infizieren (7, 11, 18). 2.2 Immunologische und genetische Faktoren, die die HIV-1-Infektion und Progression beeinflussen 2.2.1 Zytotoxische T-Lymphozyten (CTL) Neben B-Zellen gehören zu den Hauptzellen des adaptiven Immunsystems die T-Zellen, welche nach Entstehung im Knochenmark im Thymus heranreifen. Sie gelangen von dort aus in die Blutbahn als reife, naive T-Zellen, welche noch nicht aktiviert sind. Je nach Oberflächenrezeptor unterscheidet man zwischen zwei Subpopulationen, den CD4+ T-Zellen, auch als T-Helferzellen bezeichnet, und den CD8+ T-Zellen, die auch T-Killerzellen oder zytotoxische T-Lymphozyten (cytotoxic T lymphocytes; CTL) genannt werden. Die T-Helferzellen spalten sich noch in weitere Populationen und können die Aktivierung und Proliferation von CTLs wesentlich beeinflussen (19). Die zytotoxische T-Zelle differenziert erst nach Kontakt mit ihrem Antigen zu einer aktiven Effektorzelle. Der Kontakt findet dabei über den spezifischen T-Zell-Rezeptor (T cell receptor; TCR) der CTL und das antigenbeladene HLA Klasse I-Molekül statt, welches antigenpräsentierende Zellen (antigen presenting cells; APC) wie dendritsche Zellen, Makrophagen und B-Zellen 10 2 Einleitung den CD8+ T-Zellen präsentieren. Die aktivierte CTL eliminiert nun Virus-infizierte Zellen, wobei mehrere Mechanismen möglich sind. Zum einen können CTLs Zytokine sezernieren, die antivirale Eigenschaften aufweisen, wie γ-Interferon (γ-IFN) oder TumorNekrosis-Faktor-α (TNF-α). Zum anderen können CTLs über ihren Fas-Liganden an den Fas-Rezeptor der Zielzelle binden und so die Apoptose der Zielzelle induzieren (20). Oft nutzen sie auch den TRAIL-Liganden und binden den dazugehörigen Rezeptor auf der Oberfläche der Zielzelle, den death receptor 4 oder 5, was ebenfalls zur Apoptose der Zielzelle führt (21). In den meisten Fällen erfolgt die zytolytische Aktivität von CTLs über die Sekretion von lytischen Granula, die Perforin enthalten und damit die Zielzelle lysieren können (20, 22). HIV-1-spezifische CTLs spielen eine wichtige Rolle in der Immunantwort gegen HIV-1. Bereits im Tiermodell mit Rhesusmakaken, die mit dem simianen Immundefizienz Virus (SIV) infiziert sind, konnte gezeigt werden, dass während der Primärinfektion die virale Replikation ansteigt, wenn CD8+ T-Zellen depletiert werden (23). Gleichermaßen ist die CTL-Aktivität im Menschen invers korreliert mit der Virämie und dem Krankheitsverlauf (24). Die antigenspezifische Zelllyse, welche von den CTLs ausgelöst wird, ist sehr effizient und zudem schnell genug, um HIV-1-infizierte Lymphozyten abzutöten bevor sie eine maximale Virusproduktion erreichen. Eine erfolgreiche Suppression der viralen Replikation findet man insbesondere bei Gag-spezifischen CTLs, CTLs gegen andere HIV-1-Proteine sind oft weniger effektiv (25). HIV-1-spezifische CTLs treten sehr früh im Verlauf der Infektion auf, meist wenn der initiale Peak der Virämie sinkt. Die erste CD8+ T-Zell-Antwort ist jedoch nur gegen wenige Epitope gerichtet (26). In der chronischen Phase der Infektion nehmen die Bandbreite und Stärke dieser Antwort zu, dennoch kann durch CTLs die virale Replikation nur in wenigen Patienten, wie LTNP, kontrolliert werden. Geht es in das Stadium AIDS über, sinken die CTL-Antworten ab (27). Neben der Frequenz der CTL-Antwort ändert sich auch die CTL-Spezifität im Laufe einer Infektion. Einige CTLs finden sich nur in der akuten Phase der Infektion, während andere CTLs einzig in der chronischen Phase auftauchen (28). Durch Mutationen in der viralen Sequenz können CTL-Epitope entweder verloren gehen, nicht mehr prozessiert werden oder nicht mehr an das HLA-Molekül binden. Auch eine fehlende Erkennung durch den TCR kann Folge einer Mutation sein (27). Diese sogenannten CTL Fluchtmutationen treten sehr häufig auf. Da das Immunsystem nachreift und neue TCR mit anderer Spezifität bildet, kommt es anschließend zu einer zweiten und eventuell auch dritten CTLAntwort. Aufgrund der nachgereiften CTLs können nun verschiedene Virusvarianten, auch Fluchtmutanten, kreuzreaktiv erkannt werden. Patienten, die ein größeres Repertoire an kreuzreaktiven CTLs besitzen, kontrollieren das Virus in der akuten Phase besser. Daher könnte die Effektivität einer CTL-Antwort nicht nur durch die antivirale zytotoxische 11 2 Einleitung Aktivität beeinflusst werden, sondern eben auch durch ihre kreuzreaktiven Eigenschaften (28). Des Weiteren wurde festgestellt, dass Patienten, die HIV-1 gut kontrollieren, CD8+ T-Zellen mit einer höheren Polyfunktionalität besitzen. Diese können mehrere Zytokine wie γ-IFN, TNF-α, Interleukin-2 (IL-2) sowie MIP-1β sekretieren (29, 30). Demzufolge spielen HIV-1-spezifische CTLs eine entscheidende Rolle zur Eliminierung des Virus und insbesondere LTNP weisen meist eine hohe CTL-Aktivität auf. 2.2.2 HLA-Allele Eine Ansammlung von Genen, die auf dem humanen Chromosom 6 liegen, werden als Haupthistokompatibilitätskomplex (major histocompatibility complex, MHC) oder als humanes Leukozytenantigen-System (humane leukocyte antigen, HLA) bezeichnet. Sie kodieren für Proteinkomplexe, die eine entscheidende Rolle in der Immunantwort und der individuellen, zellulären Immunität spielen. So sind diese Proteinkomplexe unter anderem an der Differenzierung zwischen fremden und eigenen Proteinkomponenten beteiligt. Es werden zwei Klassen von Molekülen unterschieden. HLA Klasse I-Moleküle (HLA-I) präsentieren endogene, acht bis neun Aminosäuren lange Peptide den CD8+ T-Zellen, welche das peptidbeladene HLA-Molekül über einen spezifischen TCR erkennen. Dabei werden zunächst Proteine im Zytoplasma, die entweder fehlgefaltet sind oder von einem Virus oder einem Tumor stammen, von der Protease in Peptide gespalten. Anschließend werden im endoplasmatischen Retikulum neusynthetisierte HLA-Moleküle mit passenden Peptiden beladen und über den Golgi-Apparat an die Zelloberfläche transportiert (siehe Abbildung 2.4) (27, 31, 32). Dagegen präsentieren HLA Klasse II-Moleküle (HLA-II) exogene, 12-19 Aminosäuren lange Peptide den CD4+ T-Zellen. Die Aufnahme der Peptide erfolgt über Endozytose. Jeder Mensch besitzt jeweils zwei der HLA-I-Moleküle der drei Hauptgruppen A, B und C. Von den HLA-II-Molekülen werden ebenfalls jeweils zwei Moleküle der Gruppe DQ, DR und DP exprimiert. Die verschiedenen Gruppen differenzieren sich in ihren Peptidbindungseigenschaften. Die Nomenklatur für HLA-Allele setzt sich stets aus der Abkürzung HLA, dem Genort und einer Zahl zusammen. So bezeichnet bei HLA-B*1301 der Buchstabe B den Genort, die Zahl 13 ist die Nummer der Gruppe für das spezifische Antigen und 01 die Subgruppe (33). Assoziationsstudien haben gezeigt, dass es einen Zusammenhang zwischen dem individuellen HLA-Typ und der unterschiedlich starken Virämie bei HIV-1-Patienten gibt (34). Insbesondere konnte dieser Zusammenhang für das HLA-B-Allel beobachtet werden. Die HLA-Allele B*57, B*58, B*62, B*27 und B*1302 sind alle mit einer guten Kontrolle von HIV-1 assoziiert (35). Dabei zeigen HLA-B*5701 und –B*5703 den 12 2 Einleitung stärksten, protektiven Effekt unter Kaukasiern und Afrikanern (36). Diese HLA-Allele binden bestimmte virale Epitope, die Fluchtmutationen nicht tolerieren können, da sonst die Fitness und Replikationsfähigkeit des Virus erheblicher eingeschränkt wird (37). Dagegen gibt es auch HLA-I-Allele, die mit einem schlechteren Krankheitsverlauf sowie einer deutlich schnelleren Progression der Erkrankung assoziiert sind, wie z.B. einige Subtypen von HLA-B*35 oder der Haplotyp A*01-B*08-DR*03 (38-41). Des Weiteren weisen Patienten mit homozygoten HLA-Allelen einen deutlich schlechteren Krankheitsverlauf auf als Patienten mit heterozygoten HLA-Allelen. Dies liegt daran, dass Patienten mit heterozygoten HLA-Allelen ein größeres Repertoire an viralen Epitopen präsentieren können (42, 43). Die Immunantwort hängt folglich sehr stark vom individuellen HLA-Typ ab. Man schätzt, dass allein der HLA-I-Typ der Patienten ungefähr für 6-15% der Variationen in den Viruslasten verantwortlich ist (37). Abbildung 2.4 Schematische Darstellung des HLA Klasse I Signalweges und die Erkennung durch CTL. In einer Virus-infizierten Zelle wird die integrierte provirale DNA zu mRNA transkripiert, wandert aus dem Zellkern in das Zytosol und wird dort zu einem viralen Protein translatiert. Dieses wird unter anderem durch das Proteasom zu kleinen Peptidfragmenten gespalten, welche mit Hilfe von TAP (transporter associated with antigen presenting) in das Endoplasmatische Retikulum gelangen. Einige der Peptide können nun an MHC-IMoleküle binden, woraufhin sie an die Zelloberfläche transportiert werden. Der TCR einer CTL kann spezifisch bestimmte MHC-I-Moleküle mit ihrem gebunden Peptid erkennen und dadurch eine Immunreaktion auslösen. Der funktionsfähige TCR ist ein Heterodimer aus einer α- und einer β-Kette, die beide jeweils aus einer konstanten (C) und einer variablen (V) Domäne bestehen. Das MHC-I Molekül besteht aus einer α-Kette aus drei Domänen und verbindet sich mit dem extrazellulären β2-Mikroglobulin. Adaptiert von Sewell, 2000 (27). 13 2 Einleitung 2.2.3 Toll-like Rezeptoren in der angeborenen Immunantwort Die erste Immunantwort gegen eine HIV-1-Infektion ist die angeborene Immunantwort. Dazu gehören unter anderem auch Toll-like Rezeptoren (TLR), die von Viren und Bakterien bestimmte Motive erkennen und eine direkte Immunaktivierung auslösen. Die einzelsträngige RNA von HIV-1 wird von TLR 7 und 8 erkannt, was zur Produktion von antiviralen Zytokinen führt (44). In erster Linie wird Interferon-α (IFN-α) freigesetzt, welches die Expression von intrinsischen Restriktionsfaktoren erhöht und andere Immunzellen wie B- und T-Zellen reguliert (44, 45). Allerdings spielen auch genetische Faktoren des Wirts eine Rolle. So wurde eine erhöhte Produktion von IFN-α bei Frauen gefunden, was wahrscheinlich auf eine erhöhte Expression von TLR 7 zurückzuführen ist, da sich der Genlokus für TLR 7 auf dem X-Chromosom befindet (46). Darüber hinaus kommen in dem Gen für TLR 7 Einzelpolymorphismen (single nucleotide polymorphism, SNP) vor, welche zu einer geringeren Freisetzung von IFN-α führen. Dies resultiert letztendlich auch in einem schnelleren Krankheitsverlauf bei HIV-1 (47). 2.2.4 CD4+ T-Helferzellen Nachdem lange Zeit die Rolle der CD4+ T-Zellen für den Krankheitsverlauf bei HIV-1 umstritten war, weiß man nun, dass nicht nur CD8+ T-Zellen einen Einfluss auf den Verlauf einer HIV-1-Infektion haben, sondern auch die CD4+ T-Helferzellen sehr wichtig sind. Eine starke HIV-1-spezifische CD4+ T-Zellantwort ist mit einer besseren Kontrolle der Virusreplikation assoziiert. HIV-2, welches eine geringere Pathogenität im Gegensatz zu HIV-1 besitzt, geht stets mit robusten und polyfunktionalen CD4+ T-Zellen einher (48, 49). Zum einen sind CD4+ T-Zellen von Bedeutung, da sie IL-2 produzieren, welches das wichtigste Proliferationssignal für CTLs ist. Zum anderen erkennen CD4+ T-Zellen Epitope, welche von HLA Klasse II präsentiert werden. Dass die daraufhin von den CD4+ T-Zellen ausgelöste Immunantwort eine wichtige Rolle in der Kontrolle der Virusreplikation spielt, zeigen Fluchtmutationen in den HLA Klasse II Epitopen. CD4+ T-Zellen verschwinden zwar nach der initialen Virämie, dennoch üben sie genug Druck aus, dass das Virus Mutationen in diese Epitope einbaut, um den Druck des Immunsystems zu entkommen (50). Außerdem konnten nachgewiesen werden, dass HIV-1-Patienten, die das Virus sehr gut kontrollieren, eine Population von Gedächtnis CD4+ T-Zellen besitzen, die eine hohe Avidität gegen das Gag-Protein aufweisen (51). Obwohl CD4+ T-Zellen bevorzugt von HIV-1 infiziert werden, bleiben die meisten CD4+ T-Zellen auch bei einer hohen Virämie virusfrei (52). 14 2 Einleitung 2.2.5 Genetische Unterschiede im Gen des Chemokinrezeptor CCR5 Neben den bereits genannten Aspekten tragen noch weitere genetische Faktoren des Wirts zum Verlauf einer HIV-1-Infektion bei. So wurde für den Chemokinrezeptor CCR5 eine natürlich auftretende Deletion von 32 Basen beschrieben, die bei ca. 1% der Kaukasier zu finden ist. Diese CCR5Δ32 Mutation resultiert in einem verfrühten Stoppcodon und somit in einem nicht-funktionsfähigen Rezeptor. Da CCR5 der KoRezeptor für den Eintritt der Viren in Zellen ist, können R5- bzw. M-trope Viren Zellen nicht mehr infizieren, X4- bzw. T-trope und dual-trope HIV-Stämme indessen schon (53, 54). Homozygote Merkmalsträger für CCR5Δ32 sind damit resistent gegenüber einer HIV-1-Infektion von R5-tropen Viren. Heterozygote Merkmalsträger zeigen einen langsameren Krankheitsverlauf mit einer Verzögerung in das Stadium AIDS von 2-4 Jahren (55, 56). Wie wichtig dieser genetischer Faktor für die Entwicklung einer Gentherapie bei HIV-1 ist, zeigt der Fall des sog. „Berlin-Patienten“. Dieser konnte mit einer Stammzell-Transplantation eines Spenders, der Merkmalsträger für CCR5Δ32 ist, von HIV-1 geheilt werden. Die Stammzell-Transplantation bei diesem HIV-1-infizierten Patienten aus Berlin wurde durchgeführt, da dieser an Leukämie erkrankt war und die Chemotherapie keinen Erfolg zeigte (57). Seit der Operation im Februar 2007 konnten bei ihm keine Viren mehr nachgewiesen werden (58). Der Erfolg dieser Therapie wird wahrscheinlich ein Einzelfall bleiben. Dennoch konnte aus diesem Fall viel gelernt werden und es öffneten sich daraus neue Perspektiven einer Gentherapie für die Behandlung von HIV-1. 2.2.6 Interaktion von microRNAs und Viren MicroRNAs (miRNA) sind einzelsträngige, 18-25 Nukleotid lange nicht-kodierende RNAs, welche die Genexpression post-transkriptionell regulieren. Zur Zeit sind beim Menschen 1.424 verschiedene miRNAs bekannt, die neben der Regulierung von zellulären Prozessen wie der Entwicklung des Immunsystems (59, 60), der Signaltransduktion (61) oder der Zellproliferation auch eine Rolle in der Tumorgenese (62), der Entstehung inflammatorischer Erkrankungen (63) sowie bei der Replikation von Viren spielen (64). Die Regulation von zellulären mRNAs erfolgt dabei über die Bindung von miRNAs an teilweise komplementäre Basensequenzen meist in der 3’-untranslatierten Region (UTR) oder seltener im offenen Leseraster, was schließlich den Abbau der mRNA oder eine Inhibierung der Translation zur Folge hat (65). 15 2 Einleitung Abbildung 2.5 Die Biogenese von miRNAs. MicroRNAs sind entweder in eigenen Genen kodiert oder aber werden aus Introns gebildet. Dabei werden zunächst die miRNA-Gene zu primären miRNAs (pri-miRNAs) transkripiert. Durch Drosha, eine nukleäre RNase III, werden die pri-miRNAs zu precursor miRNAs (premiRNAs) gespalten, welche anschließend über Exportin aus dem Zellkern in das Zytoplasma gelangen. Durch Dicer wird die precursor miRNA zu einer reifen, doppelsträngigen miRNA prozessiert. Dieser Duplex wird in den RISC Komplex, welcher unter anderem aus Argonaut-Proteinen entsteht, aufgenommen. Darin trennt sich der Duplex auf. Bindet die miRNA an eine komplementäre mRNA-Sequenz mit einer vollständigen Übereinstimmung, so kann die mRNA direkt abgebaut werden. Falls die Bindung zwischen miRNA und mRNA einige Fehlpaarungen enthält, wird die Translation der mRNA inhibiert, die mRNA jedoch nicht degradiert. Abbildung adaptiert von Wikipedia: http://en.wikipedia.org/wiki/File:MiRNA-biogenesis.jpg (66). Einen ersten Hinweis auf eine Interaktion zwischen HIV-1 und miRNAs lieferte die Entdeckung, dass HIV-1 mRNA mit Effektoren des miRNA-Stoffwechselweges in zellulären „p-body“-Strukturen ko-lokalisiert (67). Mehrere Mechanismen sind für eine mögliche Modulation der Wirt-Virus-Interaktion durch miRNAs denkbar (67). Durch virale Infektionen kann das zelluläre miRNA-Milieu verändert werden (68, 69). Eine Virusinfektion induziert den antiviralen Verteidigungsmechanismus einer Zelle. Dabei werden spezifische miRNAs exprimiert, um die Replikation des Virus zu inhibieren. So reduziert z.B. miR-122 spezifisch die Replikation des Hepatitis C-Virus in der Leber (70) und miR-29a inhibiert die Nef-Expression und damit die HIV-1-Replikation in Jurkat T-Zellen durch Bindung der Zielsequenz im nef Gen (71). Eine weitere Möglichkeit wäre, dass Virusinfektionen durch die Induktion von miRNAs die Expression von verschiedenen zellulären Genen verringern, wobei das Virus das miRNA-Muster zu seinen Gunsten verändert. Viren können auch selbst miRNAs kodieren, um die Expression von WirtsmRNAs zu inhibieren. Die Hemmung von viralen mRNAs durch Virus-kodierte miRNAs 16 2 Einleitung stellt eine vierte Möglichkeit dar, wodurch verschiedene Schritte des viralen Lebenszyklus reguliert werden können, wie z.B. der Übergang in die Latenzphase (72, 73). Ein Beispiel bildet die von HIV-1 im nef Gen kodierte miR-N367, welche die HIV-1-Transkription vermindert (72). Außerdem wurde bereits gezeigt, dass das regulatorische HIV-1-Protein Tat eine zentrale Rolle bei der HIV-1-Replikation spielt und einige miRNAs ein verändertes Expressionsmuster nach Tat-Behandlung aufweisen (74). Da das HI-Virus aufgrund seiner hohen Sequenzdiversität sehr variabel ist, kann es der Inhibierung mittels miRNAs durch Mutationen in der Zielsequenz der miRNA entkommen (65). 2.3 Therapie und Behandlungsansätze von HIV-1 2.3.1 Hochaktive antiretrovirale Therapie (HAART) Die hochaktive antiretrovirale Therapie (highly active antiretroviral therapy, HAART) wird seit 1996 zur Behandlung einer HIV-1-Infektion eingesetzt. Die verschiedenen Medikamente werden in fünf Gruppen unterteilt: Nukleosidische Reverse-TranskriptaseInhibitoren (NRTI), Nicht-Nukleosidische Reverse-Transkriptase-Inhibitoren (NNRTI), Protease-Inhibitoren (PI), Integrase-Inhibitoren und Eintritts- bzw. Fusionshemmer (75). Darunter waren NRTI die ersten HIV-1-Medikamente, für welche es bereits 1987 eine Zulassung gab. Sie werden von der Zelle direkt aufgenommen und entfalten ihre Wirkung erst nach intrazellulärer Phosphorylierung (11). Sie bestehen aus alternativen Substraten für die Reverse Transkriptase, so dass diese mit den physiologischen Nukleosiden konkurrieren. Werden diese Nukleosidanaloga in den DNA-Strang während der cDNASynthese eingebaut, kommt es zum Abbruch der reversen Transkription (76). Die Gruppe der NNRTI hingegen, welche das erste Mal 1990 beschrieben wurde, bindet direkt und nicht-kompetitiv an die Reverse Transkriptase (77). Bei den Protease-Inhibitoren handelt es sich um Peptidanaloga, die an das aktive Zentrum der Protease binden und so die Prozessierung von viralen Proteinen verhindern (78). Seit 2007 sind auch IntegraseInhibitoren auf dem Markt, welche die virale Integrase hemmen und dadurch den Einbau der viralen cDNA in die Wirts-DNA unterbinden (79). Ebenfalls erst seit kurzem gibt es Eintritts- bzw. Fusionshemmer, welche den Eintritt der Viren in die Zelle blockieren. Diese bestehen aus Ko-Rezeptor-Antagonisten (CCR5 bzw. CXCR4) und Fusionsinhibitoren, welche die Verschmelzung zwischen Virus und Zellen verhindern (80). Bei der antiretroviralen Therapie handelt es sich um eine Kombinationstherapie aus bis zu fünf verschiedenen Medikamenten. Mit den Jahren hat sich die Kombination von drei 17 2 Einleitung Medikamenten etabliert, wobei meist zwei Reverse-Transkriptase-Inhibitoren und ein Protease-Inhibitor verabreicht werden (81). Die Verwendung von mehreren Medikamenten soll sicherstellen, dass die Wahrscheinlichkeit für die Entwicklung von Resistenzmutationen stark verringert wird (7). Ziel der HAART ist die Unterdrückung der viralen Replikation und die Erhöhung der CD4+ T-Zellen, so dass die Viruslasten im Idealfall unter der Nachweisgrenze von <20 Kopien/ml liegen. Damit kann eine Progression in das Stadium AIDS verhindert werden. Dank der HAART kann heute wahrscheinlich eine weitgehend normale Lebenserwartung trotz HIV-1-Infektion möglich werden, eine Heilung jedoch nicht. Die HAART ist somit sehr effektiv, allerdings können dabei auch starke Nebenwirkungen auftreten. Lipodystrophie, mitochondriale Toxizität, Leberschäden und Herzinfarkte können Folgen der HAART sein (11). Des Weiteren können bei Einnahmefehlern sehr schnell resistente Viren auftreten, die einen Therapiewechsel erfordern (82). Ein weiterer Nachteil der HAART sind die immensen Kosten der Therapie, die sich in Deutschland auf ca. 16.000€ pro Jahr und pro Patient belaufen (83). Aufgrund dieser hohen Kosten ist die Therapie gerade in Entwicklungsländern nicht immer, und wenn nur eingeschränkt, verfügbar (84). 2.3.2 Vakzine Aufgrund der unter 2.3.1 beschriebenen Nachteile der HAART ist die Suche nach wirkungsvollen, therapeutischen Impfstoffen als alternative Therapie immer noch im vollen Gange. Dies zeigen auch die geleisteten finanziellen Investitionen. Im Jahr 2008 wurden beispielsweise weltweit 868 Millionen USD für Forschung und Entwicklung von HIV-1Vakzinen ausgegeben (85). Über die Jahre wurden unterschiedliche Ansätze verfolgt, jedoch gibt es bis heute noch keinen zugelassenen Impfstoff. Das Hauptproblem in der Entwicklung einer erfolgreichen Vakzine liegt in der antigenen Plastizität des Virus und der Fähigkeit sich dem Immunsystem immer wieder entziehen zu können (86, 87). Bei den meisten viralen Infektionen handelt es sich bei den Impfstoffen um attenuierte, tote oder fragmentierte Pathogene, welche eine Antikörper-Antwort induzieren. Diese Antikörper können die Infektion weiterer Zellen verhindern und zusätzlich die zelluläre Immunantwort aktivieren, um Virus-infizierte Zellen zu eliminieren. Neutralisierende Antikörper erkennen und binden Proteine auf der Oberfläche von Viren. Auf der Suche nach neutralisierenden Antikörpern gegen HIV-1 sind neben der Hypervariabilität des Virus, die Instabilität der Env-Moleküle, welche Ziel der neutralisierenden Antikörper sind, und die Unzulänglichkeit der konservierten Zielbereiche entscheidende Hindernisse (85). Der erste neutralisierende Antikörper gegen HIV-1 mit dem Namen b12 wurde 1994 18 2 Einleitung isoliert und ist gegen die CD4-Bindestelle von gp120 gerichtet (88). Es folgten weitere Antikörper, die jedoch alle nicht zur therapeutischen Anwendung kamen. Nachdem sich die Suche nach neutralisierenden Antikörpern zur Vakzinierung als schwierig erwies, richtete sich der Fokus verstärkt auf HIV-1-Vakzine, welche eine T-ZellAntwort, insbesondere CTL-Antwort, induzieren können. Jedoch können CTLs nur bereits infizierte Zellen erkennen und eliminieren. Nachdem in HIV-1-exponierten, nicht-infizierten Menschen HIV-1-spezifische CTLs nachgewiesen wurden, keimte Hoffnung auf, dass man durch eine T-Zell-basierte Vakzine erste, kleine Infektionsherde eindämmen könnte (89). Da sich CTLs dem Immunsystem durch CTL Fluchtmutationen (siehe 2.2.1) entziehen können, ist es für eine effiziente Vakzine wichtig, möglichst hoch konservierte CTL-Epitope zu integrieren. Um CTLs zu aktivieren, stehen verschiedene Techniken zur Verfügung. Man verwendet unter anderem DNA, welche HIV-1-Antigene kodiert. Dazu wird Plasmid-DNA intradermal, subkutan oder intramuskulär verabreicht. Nach Transkription und Translation zu Protein werden die entsprechenden Antigene von den APC präsentiert, wodurch T-Zellen aktiviert werden (90). Allerdings scheiterten erste Studien mit DNA-Vakzinen aufgrund ihrer mangelnden Immunogenität. Dieses Problem wurde versucht zu umgehen, indem rekombinante Viren oder Proteine zur Verbesserung der Immunantworten eingesetzt wurden. Eine wichtige Plattform ist dabei das modifizierte Vaccinia-Virus Ankara (MVA), welches mit HIV-1-Antigenen wie z.B. nef verwendet wurde. Dieses war sehr gut verträglich und induzierte zudem eine erhöhte CTL-Antwort (91). Ein weiterer Vektor, der die Immunogenität von DNA-Vakzinen steigert, ist das humane Adenovirus Serotyp 5 (Ad5). Dieser Vektor wurde in einer klinischen Studie Phase II von Merck, der STEP-Studie, verwendet. Dabei wurden drei Dosen des Vektors mit den HIV-1-Genen gag, pol und nef 3.000 Personen verabreicht. Die Studie wurde allerdings abgebrochen, da es zu einer erhöhten Anzahl von HIV-1-Infektionen in der immunisierten Gruppe kam (87, 92). Eine andere klinische Studie namens RV144 mit 16.400 Freiwilligen wurde in Thailand durchgeführt und verwendete ein rekombinantes Kanarienpockenvirus, welches als Vektor für die HIV-1-Gene gag, pol und zwei Molekülen env gp120 eingesetzt wurde. Nach dreieinhalb Jahren konstatierte man 31% weniger HIV-1-Infektionen in der immunisierten Gruppe im Vergleich zur Placebo-Gruppe (93). Die RV144-Studie ist bis jetzt die einzige, großangelegte Impfstudie im Menschen, die eine moderate Effizienz zeigte. Vorteile der DNA-Vakzine sind ihre Flexibilität, Stabilität, einfache Lagerung und Sicherheit, da sie im Gegensatz zu normalen Impfstoffen keine lebenden oder abgetöteten Viren enthalten. Diese Impfstoffe wären aufgrund von Sicherheitsgründen auch nicht akzeptabel. Auf der anderen Seite gehen DNA-Vakzine auch mit einigen Nachteilen einher. So kann es zum Einbau der eingebrachten DNA in die zelluläre DNA 19 2 Einleitung kommen, wodurch Onkogene aktiviert werden oder Chromosomenbrüche auftreten können. Außerdem kann es zur Entwicklung von Autoimmunitäten und antibiotischen Resistenzen beitragen (90). Alle bisherigen Studien mit Ausnahme der RV144-Studie schlugen fehl, was vermutlich an der immer noch zu geringen Immunogenität der verwendeten Impfstoffe liegt. 2.3.3 Verwendung von mRNA als Vakzine Aufgrund der Nachteile der DNA-Vakzinierung wurde mRNA als alternative Nukleinsäure zur Impfung interessant (94). Da es sich um eine Nukleinsäure handelt, die nur transient in den eingebrachten Zellen exprimiert wird, wird die genetische Information nicht dauerhaft in den Organismus integriert. Demzufolge kommt es zu keiner Integration in die Wirts-DNA, wie es beim Einsatz von DNA der Fall ist. Die in vitro generierte mRNA ist hoch immunogen und benötigt keine weiteren Vektoren wie Vaccinia-Viren. Sowohl in vitro als auch in Tiermodellen konnte die Induktion von CTLs und Antikörpern durch mRNA nachgewiesen werden (95-97). Neben der guten Immunogenität von mRNA wurde in vitro eine starke Stimulierung der TLR 3, 7 und 8 durch RNA detektiert. Diese KoStimulierung könnte dabei als eine Art Adjuvans wirken (44, 98-100). Die Wirkungsweise funktioniert wie bei DNA. Die mRNA, die ein bestimmtes Protein als Antigen kodiert, wird von der Zelle aufgenommen, geht allerdings nicht in den Zellkern, sondern es findet sogleich die Proteintranslation im Zytoplasma statt. Die Antigene werden dann über die HLA-Moleküle den T-Zellen präsentiert, was eine Aktivierung von CTLs zur Folge hat. Der Einsatz von mRNA begann 1995 in der Behandlung von Tumorerkrankungen, was viele klinische Studien nach sich zog (101). Heute findet man die Therapie mit mRNA vorwiegend immer noch im Bereich der Tumorforschung, wobei es nun aber auch erste Studien für deren Einsatz bei Infektionskrankheiten gibt (102, 103). Das größte Problem bei der Verwendung von mRNA stellt die geringe Halbwertszeit dar, da mRNA sehr instabil ist und leicht von RNasen degradiert werden kann (104). Mehrere Methoden wurden zur Stabilisierung der mRNA getestet. Eine Verlängerung der Poly-ASequenz am 3`-Ende der mRNA sowie ein antireverses Cap-Analog (ARCA) tragen zur Stabilitätsverbesserung bei (105, 106). Da RNasen ubiquitär vorhanden sind, muss auch bei Verabreichung der Vakzine die mRNA geschützt werden. Dazu gab es Studien mit mRNA, welche mit Substanzen wie Protamin oder Liposomen geschützt wurde, und erfolgreich CTLs in Mäusen induzieren konnte (107, 108). Die Anwendung von mRNA erfolgt in den meisten klinischen Studien intradermal. Die mRNA kann dem Patienten aber auch subkutan, intravenös oder intranodal zugeführt werden. Zusätzlich zu den direkten 20 2 Einleitung Injektionsarten von mRNA besteht auch die Möglichkeit dendritische Zellen des Patienten zur Therapie zu entnehmen, in vitro mRNA in diese Zellen einzubringen und anschließend dem Patienten wieder zu verabreichen (109). Die neuste Entwicklung im Bereich der mRNA-Therapie sind autologe mRNA-Vakzine (110-112). Diese umgehen das Problem der hohen Sequenzvariabilität der Antigene, da das Antigen direkt aus dem Patienten isoliert und zur Generierung von mRNA verwendet wird. Aufgrund der immer noch fehlenden Vakzine zur Therapie einer HIV-1-Infektion, aber den Fortschritten im Bereich der mRNA-Technologie, rückte die Generierung und Analyse einer autologen, therapeutischen mRNA-Vakzine in den Fokus dieser Arbeit. Vor allem deren Anwendung in PBMC (peripheral blood mononuclear cells; mononukleäre Zellen des peripheren Blutes) würde eine schnelle und kostengünstige Methode erlauben. Da es bisher keine effiziente Strategie für das Monitoring von CTL-Antworten gegen autologe Virusproteine gibt, sollten autologe mRNAs zudem für diesen Einsatz getestet werden. 21 3 Zielsetzung Das primäre Ziel der vorgelegten Dissertation war die Generierung von mRNAs der HIV-1-Gene gag, nef und env und deren in vitro Testung auf Funktionalität und Immunogenität. Dabei sollten sowohl mRNAs aus autologen Virussequenzen von HIV-1infizierten Patienten sowie mRNAs aus HIV-1 Referenzstämmen hergestellt werden. Anschließend sollten diese mRNAs im Hinblick auf eine mögliche Anwendung als therapeutischer Impfstoff präklinisch untersucht und zudem als neue Methode zum Monitoring der Immunantworten von HIV-1-Patienten etabliert werden. Zunächst sollte die mRNA-Elektroporation von PBMC etabliert werden, da die HIV-1spezifischen mRNAs in PBMC exprimiert werden sollten. Um die mRNAs auf ihre Funktionalität und Immunogenität zu untersuchen, sollten zum einen die Expression der mRNA-kodierten Proteine in den Zellen detektiert und zum anderen die transfizierten Zellen in immunologische Assays eingesetzt werden. Da die hohe Sequenzvariabilität von HIV-1 eines der großen Probleme in der Vakzinentwicklung ist, sollten die Immunantworten, welche durch autologe mRNA-kodierte HIV-1-Proteine ausgelöst werden, mit denen verglichen werden, welche durch mRNA-kodierte Proteine von Referenzviren induziert werden. Durch diese Analysen sollte geklärt werden, ob eine therapeutische Vakzine optimaler Weise aus autologen oder heterologen Virussequenzen bestehen soll. Des Weiteren sollte das Risiko von Nebenwirkungen bei einer Impfung möglichst gering sein. Daher sollte ausgeschlossen werden, dass HIV-1-spezifische CTLs, die durch mRNA-kodierte HIV-1-Proteine aktiviert werden, mit humanen Proteinen kreuzreagieren und dadurch auch nicht-infizierte Zellen zerstören. Aus diesem Grunde sollten HIV-1-spezifische CTL Linien generiert und auf ihre Kreuzreaktivität zu humanen, homologen Epitopen überprüft werden. Außerdem ist die Auslösung einer breit gefächerten Immunreaktion ein weiterer, wichtiger Aspekt bei einer Impfung, so dass eine Vakzinierung mit HIV-1-spezifischen mRNAs eine Kombination aus verschiedenen mRNAs sein wird. In diesem Zusammenhang sollte ermittelt werden, wie sich die Verabreichung von zwei unterschiedlichen HIV-1-spezifischen mRNAs auf deren Effektivität und Immunogenität auswirkt. Das Immunmonitoring der HIV-1-spezifischen T-Zellantwort von HIV-1-Patienten ist sehr wichtig für das Verständnis der Pathophysiologie der HIV-1-Infektion. Bisherige Methoden, welche Peptide, Peptid-Pools oder rekombinante Vaccinia-Vektoren verwenden, detektieren jedoch nur die T-Zellantwort gegen HIV-1 Referenzstämme und nicht gegen das autologe Virus. Deswegen sollten die generierten, autologen HIV-1spezifischen mRNAs auch für den Einsatz im Immunmonitoring getestet und mit den Standard-Techniken verglichen werden. Damit könnte eine umfassende Analyse der 22 3 Zielsetzung Quantität der T-Zellantwort gegen das autologe HI-Virus ermöglicht werden. Im Zuge des Immunmonitorings sollten auch neue CTL-Epitope definiert werden. Ein weiterer Ansatzpunkt in der vorgelegten Dissertation sollte die Erprobung von alternativen Transfektionsmethoden sein, um mRNA sicher und gut verträglich in Zellen transportieren zu können. Da die Elektroporation zwar eine gute Vorgehensweise für in vitro Studien repräsentiert, jedoch in vivo nicht zugelassen ist, ist die Suche nach alternativen Transfektionsreagenzien für eine in vivo Applikation unumgänglich. Ferner sollte mit Hilfe der generierten HIV-1-spezifischen mRNAs der Einfluss einzelner HIV-1-Gene auf das zelluläre miRNA-Profil von CD4+ T-Zellen studiert werden. Zwar ist bekannt, dass eine HIV-1-Infektion das zelluläre miRNA-Muster verändert, allerdings wurden in den bisherigen Studien nur komplette HI-Viren eingesetzt. Daher ist keine Aussage möglich, welche HIV-1-Gene welche miRNAs speziell beeinflussen. Die Analyse von CD4+ T-Zellen ist dabei besonders interessant, da es sich um die Zielzellen der HIV-1-Infektion handelt. 23 4 Ergebnisse 4.1 Elektroporation von PBMC mit HIV-1-spezifischen mRNAs 4.1.1 Erfolgreiche Generierung von HIV-1-spezifischen mRNAs Für die Herstellung von HIV-1-spezifischer mRNA wurde zunächst die PCR zur Amplifikation der Gene gag, nef und env etabliert. Nach PCR-Aufreinigung und Sequenzierung wurden die Fragmente in den Vektor pGEM4Z64A bzw. pGEMsigsurvivinDClamp kloniert und in vitro zu mRNA transkripiert. Auf diese Weise wurden insgesamt sechs autologe gag mRNAs, sieben autologe nef mRNAs und zwei autologe env gp120 mRNAs generiert. Von Patient 0143 existierte bereits zu Beginn meiner Arbeit autologe nef mRNA. Von fünf Patienten stehen nun autologe gag und nef mRNAs zur Verfügung, von vier Patienten ist nur jeweils eines der beiden Gene als autologe mRNA vorhanden. Der Grund hierfür liegt in der Amplifikation der PCRProdukte. Trotz Verwendung von verschiedenen Primern zur spezifischen cDNASynthese und PCR konnte kein oder kein ausreichendes Produkt zur Klonierung gewonnen werden. Auch Abwandlungen der PCR-Bedingungen wie Zyklenanzahl oder Annealing-Temperatur und die Veränderung der Salzkonzentration in der PCR-Reaktion führten zu keinem Erfolg. Vermutlich können die verschiedenen Primer nicht binden, da sich in deren Binderegion Mutationen in der Virussequenz des Patienten befinden. Eine zu geringe Ausgangskonzentration der isolierten, viralen RNA könnte ebenfalls eine Rolle spielen. Die Generierung von autologer env gp120 mRNA führte aufgrund der hohen Sequenzvariabilität in env nur in zwei Fällen zum Erfolg. Neben autologen HIV-1spezifischen mRNAs wurden zudem von den HIV-1 Referenzstämmen NL4-3 und SF2 mRNAs generiert. Dies erfolgte über PCR der Plasmide pNL4-3 und pDSF2, welche HIV-1-Gene enthalten. Eine Übersicht der generierten Produkte ist in Tabelle 4.1 dargestellt. 24 4 Ergebnisse Tabelle 4.1 Übersicht über die erfolgreich generierten HIV-1-spezifischen mRNAs Virusisolat NL4-3 SF2 0011 0143 0225 0284 0335 0347 0545 0899 0925 gag nef * env (gp120) - *Nicht in dieser Arbeit generiert, sondern von Fr. Dr. K. Maurer 4.1.2 Test und Titration von HIV-1-spezifischen Antikörpern in transfizierten HEK293T Zellen Zur Bestimmung der Proteinexpression der mRNA-elektroporierten Zellen sollten intrazelluläre Färbungen mit anschließender Analyse am Durchflusszytometer durchgeführt werden. Da die vorhandenen HIV-1-spezifischen Antikörper noch nicht für die Durchflusszytometrie getestet wurden, sollte dies vorher anhand transfizierter HEK293T Zellen erfolgen. Dazu wurden die HIV-1 Gene gag, nef und env von den Referenzviren SF2 und NL4-3 in den Vektor pEGFP-C1 kloniert, so dass sie ein Fusionsprotein mit GFP (green fluorescence protein) bilden. Nach erfolgreicher Generierung der Plasmide wurden diese in HEK293T Zellen mit FuGENE transfiziert. Da die HIV-1 Gene als Fusionsprotein mit GFP gebildet werden, kann anhand der Fluoreszenz von GFP zum einen die erfolgreiche Transfektion detektiert werden und zum anderen, ob der zu untersuchende Antikörper bindet. Daher wurden die Zellen 48 Stunden nach Transfektion zunächst unter einem Fluoreszenzmikroskop betrachtet. Konnte man eine GFP-Fluoreszenz feststellen, wurden die Zellen permeabilisiert und intrazellulär mit den entsprechenden HIV-1-spezifischen Antikörpern und einem PEgekoppelten, sekundären Antikörper gefärbt. Bei der Analyse am Durchflusszytometer konnte anhand der transfizierten, doppelt fluoreszierenden Zellen erkannt werden, ob der Antikörper gebunden hat (Abbildung 4.1). 25 4 Ergebnisse Abbildung 4.1 Der Nef-Antikörper EH1 bindet spezifisch an HEK293T Zellen, die mit dem Plasmid pEGFP-C1 SF2 Nef transfiziert wurden. SF2 Nef liegt als Fusionsprotein mit GFP vor. HEK293T wurden mit FuGENE transfiziert und bei positiver Fluoreszenz des GFP 48 Stunden nach Transfektion permeabilisiert und intrazellulär mit dem Nef-spezifischen Antikörper gefärbt. Somit konnte detektiert werden, ob der Antikörper spezifisch bindet und für Analysen am Durchflusszytometer geeignet ist. Der linke Dot Plot zeigt, dass sich in beinah allen transfizierten Zellen aufgrund der Färbung mit dem Nef-Antikörper eine Nef-Expression (y-Achse) nachweisen lässt. Die Kontrollfärbungen auf untransfizierte HEK293T (rechts) und Zellen, die mit dem Vektor pEGFP-C1 transfiziert wurden (Mitte), demonstrieren keine Nef-Expression. Für Gag- und Env-spezifische Antikörper wurde ebenso verfahren. Insgesamt konnte für Nef ein Antikörper (Nef EH1), für Gag zwei Antikörper (p24 AG3.0, p24 183/H12) und für Env gp120 drei Antikörper (gp120 AD3, gp120 697-3D, gp120-160 Chessie 6) ermittelt werden, die spezifisch an die jeweiligen Proteine binden und für Analysen am Durchflusszytometer genutzt werden können. Durch Titration konnte ich zudem die geeignete Antikörperkonzentration bestimmen. Alle Antikörper und deren zu verwendende Konzentration sind in der Tabelle 6.22 zusammengefasst. 4.1.3 Elektroporation der HIV-1-spezifischen mRNAs zeigt schnelle und effiziente Proteinexpression Nach Elektroporation der PBMC mit HIV-1-spezifischen mRNAs und GFP mRNA als Kontrolle wurden die Zellen für eine intrazelluläre Färbung mit HIV-1-spezifischen Antikörpern und anschließender Durchflusszytometrie verwendet. Hiermit sollte überprüft werden, ob die Elektroporation erfolgreich war und die mRNA-kodierten Proteine in der Zelle exprimiert werden. Dabei konnte bereits nach vier Stunden eine deutliche Expression der Proteine GFP, Gag und Nef detektiert werden (Abbildung 4.2 A). Betrachtet man die Mediane sind 90% der PBMC positiv für GFP (range:67% -93%), 65% der Zellen exprimieren das Gag-Protein (range: 4%-88%) und 32% das Nef-Protein (range: 1%-89%). Hierbei fällt auf, dass in den meisten Versuchen die Expression des 26 4 Ergebnisse Nef-Proteins deutlich geringer ist als die Expression des Gag-Proteins. GFP zeigte konstant hohe Expressionswerte. Da PBMC aus vielen Subpopulationen bestehen, ist neben der Elektroporationseffizienz der Gesamt-PBMC auch die Effizienz in den verschiedenen Zelltypen innerhalb der PBMC interessant. Auf Grund dessen analysierte ich die Expression der Proteine GFP und SF2 Gag in CD4+ T-Lymphozyten, CD8+ T-Lymphozyten, CD19+ B-Zellen und CD14+ Monozyten. Alle Subpopulationen waren etwa zu 90% positiv für GFP, wohingegen die Expression des Gag-Proteins in allen Populationen mit Ausnahme der CD14+ Monozyten geringer war (Abbildung 4.2 B). Die Elektroporationseffizienz mit mRNA ist in T-Lymphozyten und Monozyten sehr gut. Für B-Zellen wurde eine gute Transfektionseffizienz von GFP beobachtetet, vergleichbar zu T-Zellen, während GagProtein in B-Zellen deutlich schlechter exprimiert wurde. Abbildung 4.2 Expression der mRNA-kodierten Proteine GFP, HIV-1 Gag und HIV-1 Nef in PBMC von Normalspendern und HIV-1-infizierten Patienten. Die Expression wurde 4 Stunden nach Elektroporation mittels intrazellulärer Färbung und Durchflusszytometrie bestimmt. A: Repräsentative Histogramme zeigen die Fluoreszenzintensität in PBMC, die mit GFP-, Gag- oder Nef-kodierender mRNA elektoporiert wurden (schwarz) im Gegensatz zur Mock-Elektroporation (grau). B: Expression von GFP und SF2 Gag in den unterschiedlichen Subpopulationen der PBMC. PBMC wurden mit der entsprechenden mRNA elektroporiert, und nach 4 Stunden mit einem Gag-spezifischen Antikörper und verschiedenen CD-Markern gefärbt. Dargestellt sind die prozentualen Frequenzen der GFP- bzw. Gag-exprimierenden Zellen innerhalb der verschiedenen Subpopulationen. Die Grafik zeigt den Median aus drei Experimenten mit den Interquartilen. C: Die Verteilung der PBMC-Subpopulationen wird durch die Elektroporation nicht verändert. Dargestellt sind die Median-Werte aus drei Experimenten mit den Interquartilen. E-PBMC= elektroporierte PBMC. 27 4 Ergebnisse Des Weiteren ist wichtig, dass sich innerhalb der PBMC die Verteilung der Subpopulation nicht durch die Elektroporation verändert. Daher wurden unbehandelte, mockelektroporierte und SF2 gag mRNA-elektroporierte PBMC 24 Stunden nach Elektroporation mit verschiedenen CD-Antikörpern gefärbt und am Durchflusszytometer vermessen. Wie in Abbildung 4.2 C dargestellt, wiesen die Frequenzen der PBMCSubpopulationen keine Veränderungen nach Elektroporation auf. Somit kann konstatiert werden, dass keine der Subpopulationen selektiv durch die Elektroporation getötet wird. 4.1.4 PBMC exprimieren HIV-1-Proteine bis zu sechs Tagen nach Elektroporation Nachdem eine deutliche Proteinexpression bereits vier Stunden nach Elektroporation detektiert werden konnte, stellte sich die Frage, wie lange die exogenen Proteine in den elektroporierten PBMC vorhanden sind. Deswegen wurden PBMC mit mRNA elektroporiert und anschließend die Proteinexpression zu bestimmten Zeitpunkten intrazellulär mit spezifischen Antikörpern bestimmt. In Abbildung 4.3 ist ein repräsentatives Beispiel aus drei Kinetikversuchen mit ähnlichen Ergebnissen dargestellt. Die GFP-Expression ist sehr stabil. Auch sechs Tage nach Elektroporation waren noch über 80% der Zellen positiv für GFP. Für das HIV-1 Gag-Protein ließ sich die höchste Proteinexpression nach 24 Stunden messen. Zu diesem Zeitpunkt exprimierten 61% der PBMC Gag. Anschließend sank die Gag-Proteinmenge kontinuierlich, dennoch exprimierten 140 Stunden nach Elektroporation noch 15% der PBMC das Gag-Protein. Die HIV-1 Nef-Proteinmenge erreichte ein Expressionsmaximum nach 48 Stunden. Danach fiel die Expression sehr schnell ab, so dass nach 140 Stunden kein Nef-Protein mehr nachweisbar war. Diese Ergebnisse demonstrieren, dass mRNA-kodierte Proteine sehr schnell in den Zellen synthetisiert werden und für mehrere Tage nach der Elektroporation in PBMC vorhanden sind. 28 4 Ergebnisse Abbildung 4.3 Expressionskinetik der mRNA-kodierten Proteine GFP, HIV-1 Gag und HIV-1 Nef. PBMC wurden mit den entsprechenden mRNAs elektroporiert. Zu den angegebenen Zeitpunkten wurden die Zellen intrazellulär mit Gag- bzw. Nef-spezifischen Antikörpern gefärbt und durchflusszytometrisch bestimmt. Die gezeigte Darstellung repräsentiert eins aus drei Experimenten mit ähnlichen Ergebnissen. 4.1.5 mRNA-kodierte HIV-1-Proteine sind in vitro immunogen und führen zur Freisetzung von γ-IFN PBMC von HIV-1-infizierten Spendern wurden mit mRNA elektroporiert und vier Stunden später in einen γ-IFN ELISPOT Assay eingesetzt, um die Immunogenität der mRNAkodierten Proteine zu testen. Bei den meisten Patienten konnte nach Elektroporation mit HIV-1-spezifischer mRNA eine deutliche Sekretion von γ-IFN beobachtet werden (Abbildung 4.4 - Abbildung 4.9). Durchgeführte Kontrollen wie die Elektroporation ohne mRNA (Mock) und mit GFP mRNA induzierten in allen durchgeführten Experimenten keine Zytokinfreisetzung, so dass die Immunaktivierung durch die mRNA-kodierten HIV-1-Proteine spezifisch ausgelöst wird und nicht durch unspezifische Signale, die bei der mRNA-Elektroporation entstehen. Jedoch führte bei einigen Patienten die Elektroporation mit HIV-1-spezifischer mRNA trotz nachgewiesener Proteinexpression zu keiner oder nur einer geringen γ-IFN Produktion. Dies war bei den Patienten 0011, 0545 und 0899 der Fall, bei welchen jeweils autologe nef mRNA und SF2 nef mRNA getestet wurden. Für die Patienten 0545 und 0899 konnte im γ-IFN ELISPOT Assay auch keine Reaktion gemessen werden, wenn PBMC mit HIV-1-Peptiden inkubiert wurden, wohingegen 0011 eine gute Reaktion auf das Nef-Peptid FL8 zeigte. 29 4 Ergebnisse 4.1.6 Das autologe Virus löst in fünf von zehn Fällen eine stärkere Immunantwort aus als Referenzviren PBMC von HIV-1-Patienten, von welchen eine autologe HIV-1 mRNA vorhanden ist (siehe Tabelle 4.1), wurden sowohl mit autologer gag oder nef mRNA sowie mit mRNA von HIV-1 Referenzviren elektroporiert. Nach vier Stunden wurden 2x105 Zellen pro Well im γ-IFN ELISPOT Assay analysiert, um die T-Zellantwort gegen autologe virale Proteine mit der T-Zellantwort gegen heterologe virale Proteine zu vergleichen. Zudem wurde stets die Expression der mRNA-kodierten Proteine bestimmt. Eine Übersicht zu den Patientendaten ist in Tabelle 4.2 gegeben. Tabelle 4.2 Patientendaten, zu welchen die T-Zellantwort gegen das autologe Virus bestimmt wurde. Die Daten beziehen sich auf den Zeitpunkt des dargestellten γ-IFN ELISPOT Assays. Patienten ID Geschlecht HLA Klasse I ART Dauer der ART [Jahren] Viruslast [Kopien/ml] CD4/µl 0011 M A*01, A*24, B*08, B*61,Cw*07, Cw*02 ATV, 3TC/ AZT 8 <20 683 0143 W A*02, A*03, B*07, B*60, Cw*03, Cw*07 keine / 220 786 0225 M A*02, A*03, B*07, B*18, Cw*05, Cw*07 EFV, TDF, FTC 4 20 653 0284 M A*02, A*11, B*39, B*57,Cw*06,Cw*07 keine / 350 734 0335 W A*02, A*03, B*62, B*60, Cw*03, Cw*03 keine / 7.600 397 0347 M A*02, A*24, B*13, B*18, Cw*06, Cw*07 0545 M A*02, A*02, B*75, B*62, Cw*03, Cw*08 0899 M A*01, A*02, B*51, B*52, Cw*07, Cw*12 0925 M A*01, A*66, B*41, B*44, Cw*16, Cw*17 TDF, FTC, RAL TDF/ FTC, EFV TDF/ FTC, EFV 9 <20 673 8,5 <20 818 1 <20 1075 keine / 9.000 349 ART: Antiretrovirale Therapie, ATV: Atazanavir, EFV: Efavirenz, RAL: Raltegravir, TDF: Tenofovir DF, FTC: Emtricitabin, AZT: Zidovudin, 3TC: Lamivudin Patient 0011 Da für diesen Patienten nur autologe nef mRNA vorhanden war, wurde die Nefspezifische T-Zellantwort nach Elektroporation mit autologer nef und SF2 nef mRNA miteinander verglichen. Allerdings wurde nach Elektroporation der nef mRNAs trotz 30 4 Ergebnisse nachgewiesener Proteinexpression kaum eine messbare T-Zellantwort quantifiziert (SF2 Nef: 20%; autologes Nef: 40%). Da die Immunantworten so gering waren, konnte kein Vergleich der beiden Nef-Proteine durchgeführt werden (Abbildung 4.4 A). Auch in wiederholten Versuchen konnte keine γ-IFN Freisetzung detektiert werden. Die Inkubation der PBMC mit dem Peptid FL8 (FLKEKGGL) resultierte hingegen in einer guten Nefspezifischen T-Zellantwort (30 SFU=spot forming units) (Abbildung 4.4 A). Das Peptid entspricht der Sequenz eines HLA-B*08 Epitops (113). Der Patient wurde bereits seit mehreren Jahren antiretroviral therapiert, was in der Regel mit einer Reduktion der Zahl an zirkulierenden HIV-1-spezifischen CTLs einhergeht (114). Patient 0143 Trotz großer Sequenzunterschiede zwischen dem autologen Nef von Patient 0143 und SF2 Nef (Abbildung 4.10 B) konnte nur eine geringe Divergenz in der T-Zellantwort auf die beiden Nef-Proteine festgestellt werden (Abbildung 4.4 B). SF2 Nef induzierte 14 SFU, autologes Nef 21 SFU (Abbildung 4.4 B). Allerdings wurden die beiden Nef-Proteine unterschiedlich exprimiert. So exprimierten nur 11% SF2 Nef, 28% der Zellen hingegen exprimierten das autologe Nef-Protein. Dadurch kann die etwas bessere Immunantwort gegen das autologe Nef-Protein von der stärkeren Expression stammen. Das Peptid Nef KL9 (KEKGGLEGL), ein HLA-B*60-restringiertes Epitop (115), zeigte mit 68 SFU eine klare Nef-spezifische T-Zellantwort (Abbildung 4.4 B). 31 4 Ergebnisse Abbildung 4.4 Vergleich der T-Zellantwort der Patienten 0011 und 0143 gegen autologe und heterologe HIV-1-Proteine im γ-IFN ELISPOT Assay. PBMC von HIV-1-infizierten Patienten wurden mit autologer und heterologer HIV-1-spezifischer mRNA elektroporiert und nach vier Stunden in einem ELISPOT Assay analysiert. A: Die Nef-spezifische T-Zellantwort von Patient 0011 war sehr gering, so dass eine vergleichende Aussage nicht möglich war. B: Patient 0143 zeigte eine etwas stärkere T-Zellantwort gegen das autologe NefProtein. SFU=spot forming units Patient 0225 Nach Elektroporation der PBMC dieses antiretroviral therapierten Patienten mit gag mRNA konnte eine starke T-Zellantwort mit 57 SFU auf das autologe Gag-Protein im ELISPOT detektiert werden. SF2 Gag löste mit 16 SFU eine deutlich schwächere Immunantwort aus (Abbildung 4.5 A). Beide Proteine wurden von den Zellen gleich stark exprimiert (SF2 Gag: 79%, autologes Gag: 82%). Gegen das p17 KL14 Peptid (KIRLRPGGKKKQKL) konnte eine gute Immunstimulation gezeigt werden (Abbildung 4.5 A). Dieses enthält die beiden HLA-A*03-restringierten Epitope RK9 (RLRPGGKKK) und KK9 (KIRLRPGGK) (116, 117). Da die Reaktion gegen das Peptid p17 KL14 stärker als die Summe der Reaktionen gegen RK9 und KK9 war, kann vermutet werden, dass das p17 KL14 Peptid möglicherweise weitere, nicht definierte CTL-Epitope bzw. auch HLA-IIEpitope enthält. 32 4 Ergebnisse Abbildung 4.5 Vergleich der T-Zellantworten des Patienten 0225 gegen autologe und heterologe HIV-1Proteine im γ-IFN ELISPOT Assay. A: Die Gag-spezifische Immunantwort des Patienten war deutlich stärker gegen das autologe Gag-Protein. B: Die Nef-spezifische Immunantwort des Patienten war geringer auf das autologe Nef-Protein. Im Hinblick auf die Nef-spezifische T-Zellantwort trat genau das Gegenteil auf. Das autologe Nef aktivierte weniger T-Zellen (8 SFU) als SF2 Nef (37 SFU) (Abbildung 4.5 B), obwohl SF2 Nef von weniger Zellen exprimiert wurde (25%) als das autologe Nef (57%). In Abbildung 4.10 ist ein Sequenzalignment der Aminosäuren der verschiedenen NefProteine dargestellt. In der autologen Nef Sequenz von Patient 0225 findet man in nur einem relevanten CTL-Epitop eine Mutation. Das HLA-A*03-restringierte Epitop QK10 (QVPLRPMTYK) (118) hat an Position acht einen Threonin zu Asparagin Austausch, der zum Verlust der Erkennung führt. Dies wurde sowohl durch die Inkubation der PBMC des Patienten mit den verschiedenen Peptiden nachgewiesen (Abbildung 4.5 B) als auch mit QK10-spezifischen CTL Linien des Patienten (Abbildung 4.6 A). In beiden Fällen resultierte die Stimulation mit dem Peptid QK10 T/N, welches die Mutation des autologen Virus enthält, in einer fehlenden oder schwächeren T-Zellantwort als die Stimulation mit dem Wildtyp Peptid. Dies zeigt zum einen, dass es sich hierbei um eine CTL Fluchtmutante handelt und zum anderen, dass diese Mutation Grund der schlechteren Erkennung des autologen Nef-Proteins ist. 33 4 Ergebnisse Das Immunsystem des Patienten ist allerdings nachgereift, um sich an die veränderten Verhältnisse anzupassen. So konnten drei Jahre nach Plasmaentnahme, welches zur Isolation der viralen RNA und Generierung der autologen mRNA diente, CTLs gegen die mutierte Variante Nef QK10 T/N detektiert werden. Sowohl die Inkubation mit dem Wildtyp Peptid QK10 als auch mit dem mutierten Peptid QK10 T/N führten zur Stimulation von spezifischen CTL Linien (Abbildung 4.6 B). Da nur die QK10 T/N-spezifische CTL Linie eine geringe Kreuzreaktivität zu QK10 zeigte, kann man davon ausgehen, dass es sich um zwei verschiedene TCRs handelt und das Immunsystem von 0225 einen neuen TCR gegen das mutierte Epitop gebildet hat (Abbildung 4.6). Abbildung 4.6 Nef-spezifische CTL Linien des Patienten 0225 zeigten zunächst nur die Erkennung des Wildtyp Peptids von QK10, zu einem späteren Zeitpunkt konnte das Immunsystem nachreifen und es ließen sich CTLs gegen das mutierte Epitop QK10 T/N detektieren. QK10-spezifische (schwarze Balken) bzw. QK10 T/N-spezifische (graue Balken) CTL Linien wurden durch Inkubation der PBMC von Patient 0225 mit den jeweiligen Peptiden generiert. Das Peptid Nef QK10 T/N entspricht der autologen Nef Sequenz des Patienten und stellt eine CTL Fluchtmutante dar. Gezeigt ist hier das Ergebnis einer γ-IFN ELISPOT Analyse 5 mit 1x10 Zellen pro Well. A: Ende des Jahres 2010 konnten nur CTLs gegen das Wildtyp Peptid QK10 quantifiziert werden, die keine Kreuzreaktion zu der mutierten Form zeigten. Das autologe Epitop QK10 T/N enthält eine CTL Fluchtmutante und wurde nicht erkannt. B: Anfang des Jahres 2011 wurden CTLs gegen das mutierte Epitop detektiert. Zudem zeigte die QK10 T/N-spezifische CTL Linie eine mäßige Kreuzerkennung des QK10 Peptids. Das Immunsystem ist somit nachgereift und hat einen neuen TCR für das mutierte Epitop gebildet. Patient 0284 Für den noch unbehandelten Patient 0284 konnte autologe gag und nef mRNA verwendet werden. Die T-Zellantwort gegen autologes Gag war mit 275 SFU mehr als doppelt so stark wie gegen SF2 Gag mit 109 SFU (Abbildung 4.7 A). Die Expression der beiden Proteine war gleich. SF2 Gag wurde von 59% der PBMC exprimiert, autologes Gag von 64% der Zellen. Die eingesetzten Gag-Peptide 34 induzierten eine sehr starke 4 Ergebnisse Immunantwort, insbesondere das Peptid Gag07-41, welches das bekannte HLA-B*57restringierte CTL-Epitop KF11 (KAFSPEVIPMF) enthält (119). Die summierten Frequenzen der Gag-spezifischen T-Zellen, welche durch die Inkubation der PBMC mit diversen Gag-Peptiden beobachtet wurden, überstiegen die Anzahl der Gag-spezifischen T-Zellen, die durch das Protein Gag stimuliert wurden. Dies lässt auf eine bessere Präsentation der exogen zugefügten Peptide schließen. Die Nef-spezifische T-Zellantwort von diesem Patient fiel schwächer aus als die Gagspezifische T-Zellantwort, was vermutlich durch eine geringe Nef-Proteinexpression in den nef mRNA-elektroporierten PBMC verursacht wurde. SF2 Nef wurde von 7% der Zellen exprimiert, autologes Nef von 9% der Zellen. Trotz dieser ähnlichen, sehr niedrigen Proteinexpression induzierte autologes Nef eine zweimal stärker T-Zellantwort als SF2 Nef (Abbildung 4.7 B). Die Stimulation der PBMC mit Nef-Peptiden zeigte eine sehr gute Immunantwort (Abbildung 4.7 B). Abbildung 4.7 Vergleich der T-Zellantworten des Patienten 0284 gegen autologe und heterologe HIV-1Proteine im γ-IFN ELISPOT Assay. Sowohl die Gag-spezifische (A) als auch die Nef-spezifische (B) T-Zellantwort war stärker gegen das autologe Virus. 35 4 Ergebnisse Patient 0335 PBMC des Patienten 0335 zeigten mit 77 SFU eine stärkere Antwort gegen das autologe Gag-Protein als gegen das SF2 Gag-Protein mit 54 SFU (Abbildung 4.8 A). Dabei exprimierten 63% der Zellen das autologe Gag-Protein und 55% das SF2 Gag. Durch die Verwendung von Gag-Peptidpools, die aus jeweils fünf Peptiden bestehen und SF2 entsprechen, und weiteren kürzeren Peptiden konnte das HLA-A*02-restringierte Epitop YL9 (YVDRFYKTL) (120) als einziges CTL-Epitop in SF2 Gag identifiziert werden, welches von diesem Patienten erkannt wurde (Abbildung 4.8 A). Dieses Epitop ist zwischen dem autologen und SF2 Gag-Protein konserviert (Abbildung 4.10 A). Folglich könnte die stärkere T-Zellantwort gegen das autologe Gag ein Resultat von weiteren CTLEpitopen sein, die in SF2 Gag nicht enthalten sind, oder aber das Resultat einer besseren Prozessierung und Präsentation des YL9-Epitops im autologen Kontext. Beide Nef-Proteine stimulierten eine sehr geringe spezifische T-Zellantwort in Patient 0335. Die von SF2 Nef induzierte Antwort (9 SFU) war dabei stärker als die von dem autologen Nef (3 SFU) (Abbildung 4.8 B), wobei die Expression des SF2 Nef-Proteins (20%) etwas höher war als die Expression des autologen Nef (12%). Der Grund für diese sehr geringe Immunantwort nach nef mRNA-Elektroporation könnte die schwache Expression der Nef-Proteine sein. Da das Peptid AK9 (AVDLSHFLK) für das HLA-B*62restringierte Epitop (121) eine sehr starke T-Zellantwort mit 220 SFU induzierte (Abbildung 4.8 B), kann ausgeschlossen werden, dass der Patient generell nur eine sehr schwache Immunantwort gegen Nef aufweist. In dem Epitop findet sich sowohl in der autologen als auch in der SF2 Sequenz ein V-zu-L Austausch an Position 2 (Abbildung 4.10 B), was bei früheren Analysen der Arbeitsgruppe nur zu einer leichten Verminderung der Erkennung durch AK9-spezifische, HLA-B*62-restringierte CTL Linien führte, was die schwache Immunantwort auf die beiden Nef-Proteine daher alleine vermutlich nicht erklärt. Leider konnten hierzu keine weiteren Untersuchungen durchgeführt werden, da der Patient im Verlauf des Projektes verstarb. 36 4 Ergebnisse Abbildung 4.8 Vergleich der T-Zellantworten des Patienten 0335 gegen autologe und heterologe HIV-1Proteine im γ-IFN ELISPOT Assay. A: Die Gag-spezifische Immunantwort des Patienten war stärker gegen das autologe Gag-Protein. B: Die Nef-spezifische Immunantwort des Patienten war geringer auf das autologe Nef-Protein, jedoch fiel die Immunantwort auf das SF2 Nef-Protein ebenfalls sehr gering aus. Patient 0347 Die spezifische T-Zellantwort des Patienten 0347 war mit 10 SFU sehr schwach gegen das autologe Gag-Protein. Hingegen führte SF2 Gag zu einer deutlich größeren Freisetzung von γ-IFN mit 34 SFU (Abbildung 4.9). Die Proteinexpression war beinahe gleich, da 81% der PBMC das autologe Gag exprimierten und 76% SF2 Gag. Der Vergleich der Aminosäurensequenzen der beiden Proteine offenbarte neben anderen Mutationen eine CTL Fluchtmutation in einem HLA-B*13-Epitop namens p15 RI9 (RQANFLGKI) (122). In der autologen Gag Sequenz befindet sich eine Lysin (K) zu Arginin (R) Substitution an Position acht (Abbildung 4.10 A). Die Inkubation von PBMC des Patienten mit dem Wildtyp Peptid p15 RI9 und dem Peptid p15 RI9 K/R, welches die Mutation des autologen Virus enthält, zeigte eine starke Abnahme der T-Zellantwort gegen das Peptid p15 RI9 K/R (Abbildung 4.9 A). CTL Linien des Patienten bestätigten diese Beobachtung. Nur die Inkubation mit dem Wildtyp Peptid p15 RI9 führte zur Generierung einer positiven CTL Linie (Daten nicht gezeigt). Die Inkubation mit dem 37 4 Ergebnisse Peptid p15 RI9 K/R zeigte keine Reaktion, so dass es sich hierbei um eine CTL Fluchtmutante handelt. Die Mutation p15 RI9 K/R in der autologen Gag Sequenz von 0347 könnte somit ein Grund für die schlechtere T-Zellantwort gegen das von der mRNAkodierte autologe Gag-Protein sein. Patienten 0545 und 0899 Wie bereits unter 4.1.5 erwähnt, konnte für diese beiden Patienten keine T-Zellantwort im γ-IFN ELISPOT Assay detektiert werden. PBMC wurden in beiden Fällen mit autologer nef und SF2 nef mRNA elektroporiert. Trotz nachgewiesener Proteinexpression führten die mRNA-kodierten HIV-1-Proteine zu keiner Sekretion von γ-IFN. Jedoch induzierten auch die verwendeten Nef-Peptide keine oder nur eine sehr geringe T-Zellantwort mit maximal 13 SFU. Beide Patienten wurden seit längerem bereits mit einer effektiven antiretroviralen Therapie behandelt. Patient 0925 Die beobachtete Gag-spezifische T-Zellantwort in PBMC des Patienten 0925 fiel relativ schwach aus. Zudem induzierten autologes Gag und SF2 Gag eine identische Immunantwort mit 18 SFU (Abbildung 4.9 B). In 60% der Zellen wurde das autologe GagProtein exprimiert, in 50% das SF2 Gag-Protein. Durch den Einsatz von Peptiden konnte die Erkennung von zwei CTL-Epitopen in Gag nachgewiesen werden: das Peptid Gag07-2, welches das HLA-B*40 Epitop GELDRWEKI (120) enthält, und das Peptid p24/3 mit dem HLA-B*44 Epitop EEKAFSPEV (123) (Abbildung 4.9 B). Obwohl autologe nef mRNA des Patienten vorhanden war, konnte die T-Zellantwort gegen das autologe Nef im Rahmen dieser Arbeit nicht mehr analysiert werden, da der Patient mit HAART behandelt wurde und in Folge dessen keine Immunantworten mehr ohne Vorstimulation der Zellen im γ-IFN ELISPOT zeigte. 38 4 Ergebnisse Abbildung 4.9 Vergleich der T-Zellantworten der Patienten 0347 und 0925 gegen autologe und heterologe HIV-1-Proteine im γ-IFN ELISPOT Assay. A: Die Gag-spezifische Immunantwort des Patienten 0347 war geringer gegen das autologe Gag-Protein. B: Die Gag-spezifische Immunantwort des Patienten 0925 war auf autologes und heterologes Gag-Protein gleich. Reproduzierbarkeit und Zusammenfassung Für jeden Patienten wurden die Versuche mindestens zu zwei verschiedenen Zeitpunkten durchgeführt. Das Erkennungsmuster der autologen mRNA gegenüber der heterologen mRNA konnte in allen Fällen reproduziert werden. Die Unterschiede bezüglich der Proteinexpression zwischen den Zeitpunkten lagen in der Mehrzahl der Experimente (>80%) unter 10% Abweichung. Dies weist auf eine gute Reproduzierbarkeit des Assays hin. Bei den Patienten 0545 und 0899 wurde der Versuch aufgrund der komplett fehlenden T-Zellantwort nicht wiederholt. Die Analyse der T-Zellantwort gegen das autologe Nef-Protein des Patienten 0335 konnte nicht wiederholt werden, da der Patient verstarb. Insgesamt konnten mit PBMC, welche mit autologer HIV-1-spezifischer mRNA elektroporiert waren, sehr effizient die CD8+ T-Zellantwort gegen autologe Virusproteine analysiert werden. Für fünf Patienten konnte eine stärkere T-Zellantwort gegen autologe virale Proteine im Vergleich zur T-Zellantwort gegen Proteine des HIV-1 Referenzstamms SF2 beobachtet werden (Nef von Patient 0143, Gag und Nef von Patient 0284, Gag von 39 4 Ergebnisse Patient 0225 und Gag von Patient 0335). Eine geringere T-Zellantwort gegen autologes Virusprotein konnte in zwei Patienten mit Fluchtmutationen in CTL-Epitopen assoziiert werden (Nef 0225 und Gag 0347). Patient 0335 wies ebenfalls eine geringere Immunantwort gegen das autologe Nef-Protein auf, jedoch konnte hierfür keine Erklärung gefunden werden. Die weiteren Patienten zeigten entweder keine T-Zellreaktion auf die durch mRNA-Elektroporation in die Zellen eingebrachten HIV-1-Proteine (0545 und 0899) oder es konnte kein Unterschied in der Erkennung zwischen autologen und heterologen Proteinen festgestellt werden (Nef 0011 und Gag 0925). 40 4 Ergebnisse Abbildung 4.10 Alignment der Aminosäurensequenzen der autologen Gag- (A) und Nef- (B) Proteine. Grau unterlegte Aminosäuren unterscheiden sich von der Consensus Sequenz. Rot eingerahmte Sequenzbereiche sind CTL-Epitope, welche für die Immunantworten der Patienten eine entscheidende Rolle spielen. Die Consensus Sequenz wurde von der Vector NTI Software generiert. 4.1.7 Vergleich der ausgelösten T-Zellantworten zwischen mRNAkodierten HIV-1-Proteinen und Peptiden, Peptid-Pools und Vaccinia-Viren Zum Monitoring der T-Zellantworten von HIV-1-infizierten Patienten werden üblicherweise HIV-1-Peptide, Peptid-Pools oder rekombinante Vaccinia-Viren verwendet. Da mRNAkodierte HIV-1-Proteine als alternative Antigene zum Immunmonitoring eingesetzt werden könnten, sollte die Kapazität der mRNA-kodierten Proteine zur Stimulation von T-Zellen mit der von Standardantigenen verglichen werden. Dazu wurden PBMC von HIV-1infizierten Patienten mit gag und nef mRNA elektroporiert und in γ-IFN ELISPOT Assays ohne weitere Effektorzellen eingesetzt, um die Aktivierung von Gag- oder Nefspezifischen T-Zellen in den elektroporierten PBMC zu analysieren. Gleichzeitig wurden PBMC des Patienten auch mit einzelnen Peptiden, Peptid-Pools oder rekombinanten Vaccinia-Viren im ELISPOT Assay inkubiert. Die Elektroporation der PBMC mit HIV-1spezifischer mRNA führte zu einer deutlichen Stimulation der HIV-1-spezifischen T-Zellen und ist vergleichbar mit der T-Zellstimulation nach Inkubation mit Peptid-Pools (Abbildung 4.4 A). Jedoch zeigten einzelne Peptide eine höhere Kapazität HIV-1-spezifische T-Zellen zu stimulieren. Auch die Summe der T-Zellantworten gegen mehrere Peptide lag über der T-Zellantwort gegen HIV-1 mRNA-kodierte Proteine (Abbildung 4.5 -Abbildung 4.9). Die 41 4 Ergebnisse Stimulation von T-Zellen nach Elektroporation mit HIV-1-spezifischer mRNA ist deutlich stärker als die nach Infektion der PBMC mit rekombinanten Vaccinia-Viren (Abbildung 4.5 A). Weder mock-elektroporierte PBMC noch GFP mRNA-elektroporierte PBMC bewirkten eine Freisetzung von γ-IFN, daher ist die T-Zellantwort auf gag oder nef mRNA eine spezifische Immunantwort. Da die Infektion von PBMC mit Vaccinia-Viren weniger effizient ist als die von EBVtransformierten lymphoiden B-Zelllinien (B-LCL), haben wir die T-Zell-Erkennung von mRNA-elektroporierten B-LCL mit der Erkennung von Vaccinia-infizierten B-LCL verglichen. Dafür wurde eine HLA-A*03-positive B-LCL mit SF2 gag mRNA elektroporiert oder mit einem rekombinanten Vaccinia-Virus infiziert, welches SF2 Gag exprimiert. Daraufhin wurden die Zellen im γ-IFN ELISPOT Assay mit CTL Linien von verschiedenen HIV-1-Patienten, die positiv für das HLA-A*03-restringierte p17 Epitop KK9 (KIRLRPGGK) sind, inkubiert. Somit konnte die T-Zell-Erkennung durch die CTL Linien verglichen werden. Die verwendeten Negativkontrollen wie unbehandelte B-LCL, mock- elektroporierte B-LCL, GFP mRNA-elektroporierte B-LCL und B-LCL, die mit einem Kontroll Vaccinia-Virus (vSC8) infiziert wurden, lösten durch die Ko-Inkubation eine geringe T-Zellaktivierung aus und stellen Hintergrundwerte dar (Abbildung 4.11 A). Als Positivkontrolle dienten die Peptid-inkubierte CTL Linie und die CTL Linie, welche mit Peptid-inkubierten B-LCL ko-inkubiert wurden. Diese zeigten jeweils eine deutliche γ-IFN Freisetzung, die mit einem Anti-CD8-Antikörper geblockt werden konnte (Abbildung 4.11 A). SF2 gag mRNA elektroporierte B-LCL wurden von beiden KK9-spezifischen CTL Linien merklich besser erkannt als SF2 Gag-Vaccinia infizierte B-LCL (Abbildung 4.11 A). Zwar könnte diese Differenz in der Erkennung auch ein Resultat der unterschiedlich starken Proteinexpression von SF2 Gag sein, da 70% der mRNA-elektroporierten B-LCL positiv für das Gag-Protein waren und nur 50% der Vaccinia-infizierten B-LCL (Abbildung 4.11 B). Durch die Erhöhung der Infektionsdosis der Vaccinia-Viren wurde die Proteinexpression jedoch nicht erhöht, die Viabilität der Zellen allerdings deutlich vermindert. Insgesamt demonstrieren diese Daten, dass mRNAs, die HIV-1-Proteine kodieren, als alternatives Antigen zum Immunmonitoring verwendet werden können, da sie gleiche Mengen an γ-IFN wie Peptidpools freisetzten und besser erkannt wurden als rekombinante Vaccinia-Viren. 42 4 Ergebnisse Abbildung 4.11 SF2 gag mRNA-elektroporierte B-LCL wurden im Gegensatz zu SF2 Gag Vacciniainfizierten B-LCL besser von Gag-spezifischen CTL Linien erkannt. PBMC von zwei verschiedenen HIV1-infizierten Patienten wurden mit dem Peptid p17 KK9 stimuliert und für 10 Tage inkubiert. Ausgewachsene CTL Linien wurden mit dem Peptid p17 KK9 im γ-IFN ELISPOT Assay getestet. B-LCL wurden mock elektroporiert oder mit GFP bzw. SF2 gag mRNA elektroporiert. Zudem wurden B-LCL mit einem SF2 Gag exprimierenden Vaccinia-Virus oder einem Kontroll Vaccinia-Virus (vSC8) infiziert (MOI=5). Vier Stunden nach Elektroporation bzw. 24 Stunden nach Infektion wurden die Zellen zur Analyse in einen γ-IFN ELISPOT Assay und zur intrazellulären Färbung verwendet. A: γ-IFN ELISPOT Assay, um die Erkennung der SF2 gag mRNAelektroporierten B-Zellen mit der Erkennung der SF2 Gag-Vaccinia infizierten B-LCL durch Gag-spezifische 4 4 CTL Linien zu vergleichen. Es wurden 5x10 Zellen der CTL Linie mit 5x10 der B-LCL inkubiert. In diesem Beispiel wurden CTL Linien von zwei verschiedenen Patienten verwendet (schwarze bzw. weiße Balken). B: Intrazelluläre Färbung mit einem spezifischen p24 Antikörper und Durchflusszytometrie der elektroporierten und Vaccinia-infizierten B-LCL. Die Gag Expression ist auf der y-Achse angegeben. Sowohl gag mRNAelektroporierte als auch Gag-Vaccinia infizierte B-LCL exprimierten das SF2 Gag-Protein, jedoch war die Expression in den gag mRNA-elektroporierten B-LCL um 20% höher. B-LCL: EBV-transformierte B-lymphoblastoiden Zelllinien 43 4 Ergebnisse 4.1.8 mRNA-kodierte gp120-Proteine induzieren geringe Immunantworten Es sollten ebenfalls autologe HIV-1 env gp120 Gene amplifiziert und für die mRNASynthese kloniert werden. Allerdings gelang dies nur bei zwei HIV-1-infizierten Patienten (0225 und 0347, Tabelle 4.1). Trotz mehrfacher Abänderung des PCR- Amplifikationsprotokolls und der Verwendung von diversen PCR-Primern konnte kein weiteres Fragment erfolgreich amplifiziert werden. Ob und in welchem Ausmaß die autologen Env gp120-Proteine T-Zellantworten induzieren, sollte in γ-IFN ELISPOT Assays gemessen werden. Dazu wurden PBMC von HIV-1-infizierten Patienten mit 0225 gp120 mRNA elektroporiert und anschließend im ELISPOT inkubiert. Es konnte in allen durchgeführten Versuchen kaum eine Expression des gp120-Proteins detektiert werden (n=4; gp120 exprimierende Zellen 0%-7,6%). Jedoch konnte in drei Patienten eine Immunantwort gegen das gp120-Protein von 0225 Env nachgewiesen werden, so dass die fehlende Detektion der Expression eventuell auf die Variabilität des gp120-Proteins (Abbildung 4.13) und eine dadurch fehlende Erkennung durch den Antikörper zurückzuführen ist. Patient 0566 zeigte dabei die geringste Reaktion mit 15 SFU/2x105 PBMC. Patient 0284 wies eine T-Zellantwort von 30 SFU/2x105 PBMC auf, was sogar einer stärkeren Reaktion entspricht als die CTL-Antwort, welche bei Patient 0225 selbst quantifiziert wurde (20 SFU/2x105 PBMC). Somit induzierte das autologe gp120 bei 0225 eine mäßige Immunantwort, wohingegen bei seinem Partner mit demselben Ausgangsvirus eine etwas bessere Reaktion beobachtet wurde. Autologes gp120 von 0284 stand nicht zur Verfügung. Das gp120-Protein von 0566 ist nicht mit dem gp120-Protein von 0225 verwandt. Daher zeigte 0566 nur geringe T-Zellantworten gegen das mRNA-kodierte gp120 von 0225. Dass dennoch Immunreaktionen auftreten, lässt vermuten, dass CTL-Epitope, die über die HLA-Allele von 0566 restringiert sind, in gp120 von 0225 nicht mutiert sind und daher noch durch CTLs von 0566 erkannt werden können. 44 4 Ergebnisse Abbildung 4.12 Env gp120 löste nur geringe Immunantworten aus, sowohl nach Peptidstimulation als auch nach Elektroporation mit gp120-spezifischer mRNA. PBMC von HIV-1-infizierten Patienten wurden zum einen mit Env-Peptiden im γ-IFN ELISPOT Assay inkubiert und zum anderen mit env gp120-spezifischer mRNA elektroporiert und anschließend im γ-IFN ELISPOT Assay inkubiert. Als Kontrollen dienten mock- und 5 GFP mRNA-elektroporierte PBMC. Es wurden stets 2x10 PBMC im ELISPOT eingesetzt und die autologe gp120 mRNA von 0225 zur Elektroporation verwendet. A: Patient 0225 zeigte auf sein autologes gp120 eine geringe T-Zellreaktion mit 20 SFU. Die Inkubation mit zwei Env-Peptiden löste keine Reaktion aus. B: Patient 0566 zeigte ebenfalls keine Immunantwort nach Peptidinkubation, aber eine leichte T-Zellantwort gegen das gp120 des Patienten 0225 mit 15 SFU. C: Patient 0284 zeigte von den drei Patienten die stärkste T-Zellantwort gegen das gp120 von 0225. Es handelt sich dabei um das gp120 seines Partners, was somit vom selben Ausgangsvirus stammte. Die Inkubation der PBMC mit ausgewählten Env-Peptiden induzierte keine bis schwache Reaktionen (max. 8 SFU/2x105 PBMC). Die Env-Peptide entsprechen der Consensus B bzw. HXB2 Sequenz und wurden anhand der HLA-Allele des Patienten ausgewählt, da sie potentielle CTL-Epitope für diese HLA-Allele enthielten. Aufgrund der schlechten Nachweisbarkeit des Env-Proteins mit diversen Antikörpern und der Immunogenität wurden keine weiteren Analysen dazu durchgeführt. 45 geringen 4 Ergebnisse Abbildung 4.13 Alignment der Aminosäurensequenzen des gp120-Proteins von Env zeigen die hohe Variabilität des Gens. Die gp120 Sequenzen der Referenzviren NL4-3 und SF2 wurden mit den autologen gp120 Sequenzen von 0225 und 0347 verglichen. Es wurde die Software Vector NTI verwendet, welche auch die angegebene Consensus-Sequenz generierte. Grau unterlegt sind Aminosäuren oder Abschnitte, in welchen die Sequenzen nicht übereinstimmen. Bereits der Vergleich von NL4-3 und SF2 offenbart die hohe Variabilität. 4.1.9 Geringe Mengen an HIV-1-spezifischer mRNA reichen aus, um eine Proteinexpression und T-Zellstimulation zu erhalten Welche Mindestmenge an mRNA zur Elektroporation benötigt wird, um eine Proteinexpression zu detektieren und eine T-Zellantwort zu erhalten, sollten Titrationsversuche klären. Dazu wurden PBMC von HIV-negativen und HIV-positiven Spendern mit verschiedenen Mengen an mRNA elektroporiert, nach vier Stunden intrazellulär gefärbt und durchflusszytometrisch analysiert. Eine Fluoreszenz für GFP konnte in über 80% der PBMC bis zu einer Menge von 2,5 µg nachgewiesen werden. Bei Verwendung von 0,5 µg GFP mRNA fluoreszierten immerhin noch 51% der Zellen (Abbildung 4.14 A). Die Frequenz der SF2 Gag-exprimierenden PBMC fällt von 62% bei der Verwendung von 10 µg mRNA auf 16% bei der Verwendung von 1 µg mRNA (Abbildung 4.14 A). Da in den meisten Experimenten weniger Nef-Protein als Gag-Protein nachweisbar war und ausgeschlossen werden sollte, dass es sich hierbei eventuell um eine unterschiedliche Sensitivität der Antikörper handelt, kamen in der Titration der nef mRNA zusätzlich noch steigende Mengen zum Einsatz. Dabei zeigte sich, dass der 46 4 Ergebnisse Einsatz von mehr mRNA auch in einer höheren Proteinexpression resultierte. Während 15 µg SF2 nef mRNA zu 27% Nef-exprimierenden Zellen führten, wurde durch die Verwendung von 30 µg mRNA die Frequenz der Nef-exprimierenden Zellen mit 50% verdoppelt (Abbildung 4.14 B). Dies würde eher gegen eine geringere Sensitivität des Nef-Antikörpers sprechen. Abbildung 4.14 Proteinexpression von mRNA-elektroporierten PBMC in Titrationsversuchen. PBMC von Normalspendern wurden mit unterschiedlichen Mengen an GFP, SF2 gag und SF2 nef mRNA elektroporiert. Anschließend wurde die Expression nach Färbung mit spezifischen Antikörpern am Durchflusszytometer analysiert. A: Proteinexpression der Proteine GFP und SF2 Gag in PBMC nach Elektroporation mit verschiedenen Mengen an mRNA. B: Proteinexpression von SF2 Nef nach Elektroporation mit unterschiedlicher Menge an mRNA. Des Weiteren sollte bestimmt werden, wie viel mRNA in die Zellen eingebracht werden muss, um eine ausreichende Proteinexpression für die Aktivierung von T-Zellen zu erhalten. Da die T-Zellantwort gegen das Gag-Protein deutlich stärker ausfällt und dadurch besser messbar ist, wurden von einem Patienten, von welchem eine gute GagAntwort bekannt war, PBMC mit verschiedenen Mengen an SF2 gag mRNA elektroporiert und nach vier Stunden die γ-IFN Freisetzung im ELISPOT überprüft. Wie erwartet, konnte ein kontinuierlicher Abfall der SFU mit sinkender mRNA-Menge im ELISPOT beobachtet werden. Dennoch reichte 1 µg SF2 gag mRNA aus, um nach Proteinexpression in den Zellen eine T-Zellaktivierung von 12 SFU zu erhalten (Abbildung 4.15). Diese Ergebnisse belegen, dass geringe Mengen an mRNA zur Elektroporation ausreichend sind, um eine Proteinexpression detektieren zu können. Dazu reichen 0,5 µg GFP mRNA und 1 µg gag mRNA aus. Außerdem genügen 1 µg gag mRNA, um durch die anschließende Proteinexpression in den Zellen eine messbare T-Zellaktivierung zu erhalten. 47 4 Ergebnisse Abbildung 4.15 Sekretion von γ-IFN nach Elektroporation von PBMC eines HIV-1-infizierten Patienten mit unterschiedlicher Menge an SF2 gag mRNA. PBMC wurden mit 1 µg, 5 µg und 10 µg SF2 gag mRNA 5 elektroporiert. Nach 4 Stunden wurden 2x10 PBMC pro Well auf einen γ-IFN ELISPOT Assay gegeben, um die Induktion der γ-IFN Antwort zu messen. 4.1.10 Durch mRNA-kodierte HIV-1-Proteine aktivieren HIV-1- spezifische CD8+ zytotoxische T-Lymphozyten ohne zusätzliche Effektorzellen Nachdem eine gute Immunogenität der HIV-1-spezifischen mRNAs in vitro festgestellt wurde, wollten wir wissen, ob die Stimulation von HIV-1-spezifischen T-Zellen durch mRNA-elektroporierte PBMC allein ausreicht oder ob noch zusätzliche Effektorzellen benötigt werden. Daher wurden PBMC eines HIV-1-positiven Spenders (0609) mit heterologer nef mRNA elektroporiert. Diese elektroporierten PBMC wurden einmal mit autologen, unbehandelten PBMC und einmal ohne autologe, unbehandelte PBMC in einem γ-IFN ELISPOT Assay inkubiert. Als Kontrolle wurden Zellen mit dem Peptid Nef RI9 (RQEILDLWI) inkubiert, gegen welches der Patient eine bekannte T-Zellantwort besaß. Die Stimulation von 100.000 PBMC mit dem Peptid Nef RI9 löste mit 609 SFU eine starke Immunreaktion aus. Wie erwartet resultierte die Verdoppelung der Zellzahl auf 200.000 mit 1009 SFU etwa in der Verdopplung der Immunantwort (Abbildung 4.16). Eine starke T-Zellantwort mit einer deutlichen γ-IFN Freisetzung wurde auch durch die nef mRNA-elektroporierten PBMC induziert (212 SFU). Während die Zugabe von 100.000 unbehandelten PBMC zu den 100.000 nef mRNA-elektroporierten PBMC die T-Zellantwort verdoppelte, wurde die Immunreaktion mit 634 SFU verdreifacht, wenn man 200.000 nef mRNA-elektroporierte PBMC ohne die Zugabe von Effektorzellen einsetzte 48 4 Ergebnisse (Abbildung 4.16). Folglich werden keine zusätzlichen Effektorzellen benötigt, um eine T-Zellantwort zu induzieren. PBMC allein, welche mit HIV-1-spezifischer mRNA elektroporiert wurden, sind zur Aktivierung der CTL-Antwort ausreichend und eine Steigerung der Sensitivität kann am besten durch die Erhöhung der Zellzahl an elektroporierten Zellen erreicht werden. Abbildung 4.16 γ-IFN-ELISPOT Assay mit variierenden Zellzahlen zeigte, dass keine zusätzlichen Effektorzellen benötigt werden. PBMC eines HIV-1-infizierten Patienten (0609) wurden mock, mit GFP mRNA oder mit heterologer nef mRNA von Patient 0143 elektroporiert. Die Frequenz der Nef-spezifischen T-Zellen wurde vier Stunden nach Elektroporation im ELISPOT detektiert, indem unterschiedliche Zellzahlen 5 eingesetzt wurden. Die Verwendung von 2x10 nef mRNA-elektroporierten PBMC führte zu einer größeren 5 T-Zellantwort als die Inkubation von 1x10 nef mRNA-elektroporierten PBMC mit unbehandelten PBMC als Effektorzellen. Des Weiteren wurden PBMC von vier HIV-negativen Spendern mit HIV-1-spezifischen mRNAs elektroporiert und im Anschluss auf Freisetzung von γ-IFN im ELISPOT untersucht. Im Gegensatz zu den PBMC der HIV-positiven Spender führten HIV-1spezifische mRNAs in PBMC von HIV-negativen Spendern zu keiner Produktion von γ-IFN. Dies beweist, dass die Immunreaktion, die in Zellen von Patienten ausgelöst wird, durch HIV-1-spezifische T-Zellen induziert wird und nicht durch eine unspezifische Aktivierung von T-Zellen. Daraufhin stellte sich die Frage, ob es sich bei den HIV-1-spezifischen T-Zellen, welche die Immunantworten auf die Antigen-kodierende mRNA hervorrufen, um CD8+ zytotoxische T-Lymphozyten oder um CD4+ T-Lymphozyten handelt. Daher wurden PBMC von HIV-1-Patienten mit HIV-1-spezifischer mRNA elektroporiert und anschließend 49 4 Ergebnisse im ELISPOT Assay mit blockierenden Antikörpern inkubiert. Es konnte eine starke Reduktion der ausgelösten Immunantworten festgestellt werden, wenn die Zellen mit einem Anti-CD8 Antikörper geblockt wurden (Abbildung 4.17). Die Inkubation der Zellen mit einem Anti-CD4 Antikörper zeigte einen geringen inhibitorischen Effekt. Dies konnte in zwei Patienten nachgewiesen werden. Somit lässt sich konstatieren, dass die meisten Immunantworten gegen Antigene, die durch HIV-1-spezifische mRNAs kodiert werden, durch HIV-1-spezifische CTLs induziert werden. + Abbildung 4.17 Durch die Elektroporation mit HIV-1-spezifischen mRNAs wurden CD8 HIV-15 spezifische T-Zellen stimuliert. 2x10 unbehandelte PBMC von HIV-1-positiven Spendern (hier: 0284) 5 wurden mit 2x10 PBMC im γ-IFN ELISPOT Assay inkubiert, die entweder mit SF2 gag mRNA oder autologer gag mRNA elektroporiert wurden. Anschließend wurden blockierende Anti-CD4- bzw. Anti-CD8-Antikörper direkt zu den Zellen gegeben. Die Reduktion der Frequenz der SFU durch diese Antikörper zeigte, dass sowohl SF2 gag mRNA elektroporierte PBMC sowie auch autologe gag mRNA elektroporierte PBMC + + hauptsächlich von CD8 T-Zellen erkannt werden und nur ein kleiner Anteil von CD4 T-Zellen. 4.1.11 Neben γ-IFN induzieren die von HIV-1-spezifischen mRNAs-kodierten Proteine die Sekretion von TNF-α Der Cytometric Bead Array (CBA) Human Th1/Th2 Cytokine Kit erlaubt die Messung mehrerer Zytokine, die in den Überstand der Zellen abgegeben werden können. Daher wurden vier bzw. 24 Stunden nach Elektroporation mit HIV-1-spezifischer mRNA 50µl aus dem Überstand abgenommen und anschließend mit dem oben genannten Kit die Freisetzung der Zytokine IL-2, IL-4, IL-6, IL-10, TNF-α sowie γ-IFN analysiert. Allerdings konnte nur eine deutliche Freisetzung von TNF-α nach Elektroporation mit HIV-1spezifischen mRNAs gemessen werden, die anderen Zytokine zeigten keine messbare 50 4 Ergebnisse Produktion. Die Elektroporation mit gag oder nef mRNA induzierte in allen Patienten eine Abgabe von TNF-α, wobei die Stärke der TNF-α Freisetzung zwischen den Patienten variierte. Außerdem wiesen die verschiedenen mRNA-Konstrukte eine unterschiedliche Kapazität zur Stimulation von TNF-α auf. In Abbildung 4.18 sind repräsentative Beispiele dargestellt. So rief SF2 nef mRNA eine fast dreifach höhere Produktion von TNF-α hervor als SF2 gag (Abbildung 4.18 D). Die autologe gag mRNA in Patient 0284 induzierte eine dreimal höhere Freisetzung von TNF-α als SF2 gag mRNA (Abbildung 4.18 A), wohingegen die autologe gag mRNA von Patient 0347 eine geringere Produktion im Vergleich zu SF2 gag mRNA bewirkte (Abbildung 4.18 C). Die autologe nef mRNA von Patient 0143 stimulierte eine ähnliche TNF-α-Sekretion wie SF2 nef mRNA (Abbildung 4.18 B). Überdies wurden höhere Zytokinlevel vier Stunden nach Elektroporation als nach 24 Stunden detektiert. Unbehandelte PBMC, mock-elektroporierte sowie GFP mRNAelektroporierte PBMC stellen mit geringen Werten an TNF-α Hintergrundwerte dar. Eine Sekretion von TNF-α konnte zudem auch in PBMC von HIV-negativen Spendern nach Elektroporation mit HIV-1-spezifischen mRNAs beobachtet werden (Abbildung 4.18 D). Daher kann man annehmen, dass die TNF-α Produktion zumindest in den HIVnegativen Spendern nicht durch Antigen-spezifische T-Zellen angeregt wird, sondern durch die Triggerung der Toll-like Rezeptoren TLR7 und TLR8. Diese beiden TLRs, die in diversen Zelltypen innerhalb der PBMC exprimiert sind, erkennen einzelsträngige RNAMoleküle, welche bestimmte Nukleotidsequenzen aufweisen. 51 4 Ergebnisse Abbildung 4.18 Sekretion von TNF-α nach Elektroporation der PBMC mit HIV-1-spezifischer mRNA. Vier Stunden nach Elektroporation wurde der Überstand der Zellen auf die Freisetzung von Zytokinen analysiert. Die Abbildungen A-D stellen repräsentative Beispiele dar. A: Patient 0284 B: Patient 0143 C: Patient 0347 D: Normalspender 0119 52 4 Ergebnisse 4.2 Funktionelle Analysen mit HIV-1-spezifischen mRNAs 4.2.1 mRNA-kodierte HIV-1-Proteine stimulieren die Generierung von HIV-1-spezifischen CTL Linien Standardmäßig werden HIV-1-spezifische CTL Linien in vitro generiert, indem PBMC von HIV-1-Patienten mit HIV-1-spezifischen Peptiden, die CTL-Epitope enthalten, inkubiert werden. Nun sollte getestet werden, ob auch durch die Elektroporation von PBMC mit HIV-1-spezifischer mRNA CTL zur Proliferation induziert werden können. Daher wurden PBMC von HIV-1-Patienten mit HIV-1-spezifischer mRNA elektroporiert. Nach vier Stunden wurden unbehandelte PBMC des Patienten mit den elektroporierten PBMC stimuliert. Die Zellen wurden zehn Tage kultiviert und anschließend wurde die CTLAktivität im γ-IFN ELISPOT analysiert. Dazu wurden 1x105 Zellen pro Well auf die ELISPOT-Platte gegeben und mit einem HIV-1-Peptid getestet. Wie exemplarisch in Abbildung 4.19 dargestellt, konnte eine Stimulation der Zellen durch die mRNA-kodierten HIV-1-Proteine erreicht werden. SF2 gag mRNA sowie autologe gag mRNA des Patienten führten zu einer klaren Stimulation von HIV-1-spezifischen CTL Linien, wohingegen GFP mRNA-elektroporierte PBMC keine CTL Linien induzieren konnten. Als Kontrolle wurden PBMC des Patienten mit dem Peptid Gag07-41 inkubiert. Abbildung 4.19 PBMC, welche mit gag mRNA elektroporiert wurden, stimulierten HIV-1-spezifische CTL Linien. PBMC eines HIV-1-Patienten wurden mit mRNA elektroporiert. Nach 4 Stunden wurden unbehandelte PBMC mit den mRNA-elektroporierten PBMC stimuliert. Nach 10-tägiger Kultivierung wurden die Zellen im γ-IFN ELISPOT getestet. Als read-out wurde das Peptid Gag07-41 verwendet. GFP mRNAelektroporierte PBMC zeigten keine Stimulation von Gag-spezifischen CTL Linien. 53 4 Ergebnisse 4.2.2 PBMC proliferieren nicht spezifisch nach Elektroporation mit mRNA Nachdem festgestellt wurde, dass mRNA-elektroporierte PBMC HIV-1-spezifische CTL Linien stimulieren können (siehe 4.2.1), sollte geklärt werden, ob die mRNAelektroporierten PBMC proliferieren. Dazu wurden autologe PBMC mit mRNAelektroporierten PBMC vier Stunden nach Transfektion stimuliert und anschließend mit CFSE (Carboxyfluoreszein-Succinimidyl Ester) markiert. Dabei diffundiert CFSE passiv in die Zellen und nach Abspaltung einer Acetatgruppe entsteht ein Fluoreszenzmolekül. Als Kontrolle wurden PBMC mit PHA oder einem HIV-1-Peptid stimuliert. Die Proliferation der Zellen wurde durch die Detektion der zellteilungsbedingten Abnahme der CFSEFluoreszenz über einen Verlauf von mehreren Tagen im Durchflusszytometer bestimmt. Wie in Abbildung 4.20 exemplarisch dargestellt, konnte nach einer Kultivierung von fünf Tagen eine CFSE-Abnahme festgestellt werden. Allerdings wurde diese CFSE-Abnahme nicht nur wie erwartet bei den nef mRNA-elektroporierten PBMC detektiert, sondern auch in den unbehandelten PBMC und den mock-elektroporierten PBMC. Daher konnte man mit dieser Methode keine spezifische Induktion der Proliferation der mRNA- elektroporierten PBMC erreichen. Abbildung 4.20 Analyse der Proliferation nach mRNA-Elektroporation von PBMC. PBMC eines HIV-1infizierten Patienten wurden mock- bzw. mit HIV-1 nef mRNA elektroporiert. Vier Stunden nach Elektroporation wurden die elektroporierten Zellen als Stimulationszellen für unbehandelte PBMC verwendet und mit CFSE markiert. Die Proliferation der Zellen wurde durch die proliferationsbedingte Abnahme der CFSE-Fluoreszenz am Durchflusszytometer gemessen. Dabei wurde die CFSE-Fluoreszenz am Tag 5 (grau) mit der am Tag 1 (schwarze Linie) verglichen. In allen drei Ansätzen konnte eine geringe Proliferation der Zellen detektiert werden, jedoch kein Unterschied zwischen den unbehandelten PBMC (links), den mockelektroporierten PBMC (Mitte) und den nef mRNA-elektroporierten PBMC festgestellt werden. Dargestellt ist ein repräsentatives Experiment aus drei Versuchen. 54 4 Ergebnisse 4.2.3 Mit HIV-1-spezifischer mRNA-elektroporierte PBMC zeigen nach Kryokonservierung noch schwache Immunantworten Als nächstes sollte untersucht werden, ob kryokonservierte und wieder aufgetaute PBMC, die mit HIV-1-spezifischer mRNA elektroporiert wurden, noch T-Zellreaktionen im γ-IFN ELISPOT auslösen können. Dies ist wichtig, wenn mRNA-elektroporierte PBMC für das Immunmonitoring verwendet oder als Impfstoff wiederholt verabreicht werden sollen. Daher wurden PBMC von HIV-1-infizierten Patienten 24 Stunden nach Elektroporation nach Standardprotokoll (siehe Methoden) kryokonserviert. Bei der nächsten Routineuntersuchung des Patienten wurden frische PBMC aus Citratblut isoliert und diese als Effektorzellen zusammen mit den aufgetauten, mRNA-elektroporierten PBMC im ELISPOT Assay inkubiert. Dabei zeigte sich, dass die kryokonservierten und wieder aufgetauten PBMC noch Immunantworten induzieren können, jedoch die Stärke der induzierten Reaktion deutlich schwächer ist als zum Zeitpunkt vor Kryokonservierung (Abbildung 4.21). Es wurden stärkere T-Zellreaktionen erzielt, wenn zu den aufgetauten, mRNA-elektroporierten PBMC frische, autologe PBMC als Effektorzellen zugegeben wurden (Abbildung 4.21). Im Vergleich zu den ursprünglich ausgelösten Immunantworten vor Kryokonservierung lag die Stärke der Immunantworten nach der Kryokonservierung bei durchschnittlich 19% ohne Effektorzellen und bei 30% mit Effektoren. Insgesamt wurde der Versuch dreimal durchgeführt. In einem Fall konnte überhaupt keine Reaktion im ELISPOT quantifiziert werden. 55 4 Ergebnisse Abbildung 4.21 Kryokonservierte, aufgetaute PBMC, die mit HIV-1-spezifischer mRNA elektroporiert wurden, konnten noch T-Zellantworten induzieren, die jedoch deutlich schwächer sind. PBMC von HIV-1-infizierten Patienten (hier exemplarisch 0225) wurden mit HIV-1-spezifischer mRNA elektroporiert und vier Stunden nach Elektroporation im γ-IFN ELISPOT Assay inkubiert (schwarze Balken). Nach 24 Stunden wurden die Zellen kryokonserviert. Nach Auftauen wurden die kryokonservierten Zellen sogleich in einem ELISPOT Assay eingesetzt (graue Balken). Die induzierten T-Zellreaktionen der aufgetauten PBMC sind deutlich besser, wenn man frische, autologe PBMC des Patienten als Effektorzellen hinzufügt. 4.2.4 mRNA-kodierte Nef-Proteine reduzieren die Expression von CD4, CXCR4 und HLA-I Nachdem eine schnelle, effiziente Expression der mRNA-kodierten Proteine und eine gute Immunogenität der HIV-1-Proteine in vitro nachweisbar war, stellte sich die Frage, ob die durch mRNA-Elektroporation in PBMC eingebrachten HIV-1-Proteine funktionell aktiv sind. Für das HIV-1-Protein Nef sind viele Funktionen bekannt. Unter anderem reduziert Nef die Moleküle HLA-I, CD4 sowie CXCR4 auf der Oberfläche von infizierten Zellen (124-126). Daher sollte nun untersucht werden, ob sich nach Elektroporation von PBMC mit nef mRNAs die Expression dieser Moleküle verändert. Dazu wurden PBMC von Normalspendern und HIV-1-infizierten Patienten mit verschiedenen nef mRNAs elektroporiert und nach 24 Stunden mit spezifischen Antikörpern für CD4, CXCR4 und HLA-A*02 gefärbt. Die Analyse der 56 Fluoreszenzintensitäten erfolgte am 4 Ergebnisse Durchflusszytometer. Wie in Abbildung 4.22 zu sehen ist, konnte eine effiziente Reduktion für alle drei Moleküle nach Elektroporation mit nef mRNA beobachtet werden. Während knapp 50% aller mock-elektroporierten PBMC CD4 positiv sind, sank die Anzahl der CD4 positiven Zellen nach nef mRNA-Elektroporation auf 36%. Dabei blieb nur eine kleine Population von Zellen zurück, die das CD4-Molekül in hohem Ausmaß exprimierten, die meisten Zellen zeigten nur geringe Mengen an CD4 (Abbildung 4.22 A). Für das HLAA*02 Molekül konnte ein Rückgang der Expression bis zu 50% verzeichnet werden (Abbildung 4.22 B). Die CXCR4-Menge wurde durch nef mRNA am wenigsten beeinflusst, dennoch wurde ein Unterschied von bis zu 25% CXCR4-positiver Zellen im Vergleich zu mock-elektroporierten Zellen nachgewiesen (Abbildung 4.22). Bei der Analyse von verschiedenen nef mRNAs konnte festgestellt werden, dass sich die unterschiedlichen Nef-Proteine auch in ihrer Fähigkeit differenzieren, wie effektiv sie die genannten Moleküle regulieren (Daten nicht gezeigt). Folglich sind die generierten nef mRNAs in der Lage die Expression von CD4, CXCR4 und HLA-I entscheidend zu modulieren und somit auch funktionell aktiv. Abbildung 4.22 Expression der Oberflächenmoleküle CD4, HLA-A*02 und CXCR4 wurde nach Elektroporation mit nef mRNA reduziert. PBMC von Normalspendern wurden mit nef mRNA bzw. mockelektroporiert, nach 24 Stunden mit spezifischen Antikörpern gefärbt und durchflusszytometrisch analysiert. A: Expression des Oberflächenmarkers CD4 in PBMC. In den mock-elektroporierten PBMC (links) waren knapp + + 50% CD4 . Nach Elektroporation mit nef mRNA sank die Anzahl CD4 -Zellen auf 36% (rechts). B und C: Die Expression der Moleküle HLA-A*02 (B) und CXCR4 (C) auf der Oberfläche von PBMC verringerte sich nach nef mRNA Elektroporation. 57 4 Ergebnisse 4.2.5 Folgen der simultanen Elektroporation von gag und nef mRNA 4.2.5.1 Die simultane Elektroporation zweier HIV-1-spezifischer mRNAs in B-Zelllinien hat keine Auswirkungen Im Hinblick auf eine Anwendung der mRNA als therapeutische Vakzine ist wichtig zu untersuchen, ob die gleichzeitige Verabreichung von mehreren HIV-1-spezifischen mRNAs die Wirkung der verschiedenen, einzelnen mRNAs beeinflusst. Zum einen könnte die gleichzeitige Impfung mit nef und gag mRNA die Expression eines der beiden Konstrukte verändern oder zum anderen könnte eines der Proteine zu einer schlechteren Induktion der γ-IFN Ausschüttung führen. Daher wurden EBV-transformierte B-Zelllinien einzeln mit gag bzw. nef mRNA elektroporiert. In einem weiteren Ansatz wurde die BZelllinie simultan mit beiden mRNA-Konstrukten elektroporiert. Die Expression der beiden Proteine wurde vier Stunden nach Elektroporation in allen Ansätzen mittels Durchflusszytometrie gemessen und verglichen. Dabei ließ sich ermitteln, dass die gleichzeitige Elektroporation von beiden mRNAs in einem gemeinsamen Ansatz zu einer geringen Reduktion der Expression führte, die jedoch maximal bei 25% lag. In den meisten Fällen war keine Veränderung oder nur eine schwache Minderung von 10% nach gemeinsamer Elektroporation zu beobachten (Abbildung 4.23). Somit gibt es im Hinblick auf die Effektivität der Transfektion nur geringe Einbußen, wenn zwei unterschiedliche HIV-1-spezifische mRNAs gleichzeitig in einem Ansatz verabreicht werden. Die elektroporierten B-Zelllinien wurden zudem in einen γ-IFN ELISPOT Assay eingesetzt. Da B-Zellen zu den Antigen-präsentierenden Zellen gehören, wurden diese mit Gag- bzw. Nef-spezifischen CTL Linien inkubiert. Für die Generierung dieser CTL Linien wurden PBMC von HIV-1-infizierten Patienten, welche zudem Träger des HLA-Allels B*13 waren, mit dem Nef-Peptid Nef RI9 und mit dem Gag-Peptid p15 RI9 stimuliert, beides HLAB*13-restringierte Epitope, und für zehn Tage in R10IL2 kultiviert. Für die Elektroporation wurde daher auch eine HLA-B*13 positive B-Zelllinie verwendet, damit die entsprechenden Epitope von der B-Zelle den CTL Linien präsentiert werden können. Die Ausschüttung von γ-IFN bleibt meist unverändert, unabhängig davon, ob die HIV-1spezifische mRNA einzeln oder zusammen mit einer weiteren HIV-1-spezifischen mRNA verwendet wurde (Abbildung 4.24). Wurde eine geringe Reduktion der Gag- oder NefExpression in den Zellen detektiert, die mit beiden mRNAs elektroporiert wurden, so wurde teilweise eine geringfügig schlechtere Erkennung der elektroporierten B-Zellen durch die CTL Linie beobachtet. Allerdings wird dies wahrscheinlich die Folge der 58 4 Ergebnisse geringeren Proteinexpression sein. Der Versuch wurde dreimal durchgeführt und in allen Versuchen konnte das Resultat bestätigt werden. Abbildung 4.23 B-Zelllinien, welche mit gag und nef mRNA simultan elektroporiert wurden, zeigten im Gegensatz zur Einzelelektroporation der jeweiligen mRNA nur eine geringe Reduktion der Proteinexpression. B-Zelllinien wurden 4 Stunden nach Elektroporation mit den HIV-1-spezifischen Antikörpern (p24 183/H12 bzw. Nef EH1) intrazellulär gefärbt und am Durchflusszytometer analysiert. Die Population der Gag- bzw. Nef-positiven Zellen befindet sich auf der y-Achse. 46% der B-Zellen, die nur mit gag mRNA elektroporiert wurden, exprimierten das Gag-Protein (Mitte links). Im Vergleich dazu waren es 34% Gag-positive Zellen in den B-Zellen, die sowohl mit gag als auch mit nef mRNA elektroporiert wurden (unten links). Für Nef waren 53% der nef mRNA-elektroporierten B-Zellen positiv (Mitte rechts), dagegen sind es in den B-Zellen, welche mit gag und nef mRNA elektroporiert wurden, 38% (unten rechts). Als Kontrolle dienten mock-elektroporierte B-Zelllinien, die ebenfalls mit den HIV-1-spezifischen Antikörpern gefärbt wurden (oben). 59 4 Ergebnisse Abbildung 4.24 Die Erkennung von mRNA-elektroporierten B-Zelllinien durch HIV-1-spezifische CTL Linien im γ-IFN ELISPOT Assay blieb konstant, unabhängig davon, ob die B-Zelllinie mit der mRNA 5 einzeln oder zusammen mit einer weiteren mRNA elektroporiert wurde. 1x10 Zellen einer B-Zelllinie 4 wurden vier Stunden nach Elektroporation zusammen mit 5x10 Zellen einer CTL Linie im γ-IFN ELISPOT Assay inkubiert. A: Eine Nef-spezifische CTL Linie erkannte die B-Zelllinie, welche mit nef mRNA elektroporiert wurde, im gleichen Ausmaß wie die B-Zelllinie, die sowohl mit nef als auch mit gag mRNA elektroporiert wurde. Ebenso wurden die Peptid-inkubierte B-Zelllinie sowie das Peptid erkannt. B: Eine Gagspezifische CTL Linie (Epitop p15 RI9) erkannte die B-Zelllinie, welche mit gag mRNA elektroporiert wurde, gleichermaßen wie die B-Zelllinie, die mit gag und nef mRNA zusammen elektroporiert wurde. Das Peptid alleine und die Peptid-inkubierte B-Zelllinie wurden von der CTL Linie auch erkannt. Alle weiteren Ansätze dienten als Negativ-Kontrolle und zeigten keine Erkennung durch die CTL Linien. E-BZ: elektroporierte B-Zelllinie. 60 4 Ergebnisse 4.2.5.2 Die simultane Elektroporation zweier HIV-1-spezifischer mRNAs in PBMC führt zu einem Verlust der Immunantworten Nachdem keine wesentlichen Auswirkungen der gemeinsamen Elektroporation von gag und nef mRNA in B-Zelllinien festgestellt wurde, analysierte ich die Folgen der simultanen Verabreichung zweier mRNAs in PBMC von HIV-1-infizierten Patienten. Dazu wurden PBMC ebenfalls mit gag und nef mRNA einzeln und in einem weiteren Ansatz zusammen elektroporiert. Vier Stunden nach der Elektroporation wurden die Zellen im γ-IFN ELISPOT analysiert. In Abbildung 4.25 ist ein exemplarisches Ergebnis dargestellt. PBMC des Patienten 0284 zeigten hierbei eine deutliche T-Zellantwort auf sein autologes GagProtein mit 434 SFU/2x105 PBMC. Eine mäßige Nef-spezifische T-Zellantwort konnte ebenso detektiert werden (45 SFU/2x105 PBMC). Bei einer gemeinsamen Elektroporation von gag und nef mRNA könnte man aufgrund der Addition der Gag- und Nef-spezifischen T-Zellantwort eine Erhöhung der Immunantwort erwarten. Allerdings sank die HIV-1spezifische Immunantwort auf 29 SFU/2x105 PBMC (Abbildung 4.25) herab. Die Proteinexpression von Gag und Nef im gemeinsamen Elektroporationsansatz (79% Gagexprimierende Zellen; 80% Nef-exprimierende Zellen) war identisch zur Expression im Einzelansatz, so dass die geringere T-Zellantwort nicht die Folge einer schwächeren Proteinexpression ist. Nef-Proteine, welche von in vitro synthetisierten nef mRNAs kodiert werden, sind in der Lage die Konzentration der HLA-Moleküle A und B auf der Oberfläche zu reduzieren (siehe 4.2.4). Daher kann man vermuten, dass hier ebenfalls dieser Effekt auftritt. Durch Reduktion der HLA-A- und HLA-B-Moleküle werden HLA-A- und HLA-Brestringierte Epitope nicht mehr präsentiert, was zur Abschwächung der allgemeinen Immunantwort führt. Dieser Effekt konnte unter Verwendung der autologen nef mRNA des Patienten ebenfalls beobachtet werden. 61 4 Ergebnisse Abbildung 4.25 Die HIV-1-spezifische T-Zellantwort verringerte sich drastisch bei simultaner Elektroporation von gag und nef mRNA in PBMC. PBMC eines HIV-1-infizierten Patienten (0284) wurden zum einen einzeln mit SF2 nef mRNA oder der autologen gag mRNA elektroporiert, zum anderen wurden die beiden mRNAs vereinigt elektroporiert. Vier Stunden nach Elektroporation wurden die Zellen im ELISPOT eingesetzt. Der Patient zeigte eine mäßige T-Zellantwort auf SF2 Nef und eine sehr starke Reaktion auf das autologe Gag. PBMC, welche mit beiden mRNAs elektroporiert wurden, demonstrierten eine drastische Reduktion der Immunantwort. Als Kontrollen dienten mock-elektroporierte und GFP mRNA-elektroporierte PBMC, die keine γ-IFN-Sekretion zeigten. Aufgrund der Beobachtung, dass durch die simultane Elektroporation von gag und nef mRNA die HIV-1-spezifische Immunantwort in PBMC abnimmt und Nef die Expression der HLA-A- und HLA-B-Moleküle reduzieren kann (127), sollte genauer untersucht werden, ob T-Zellantworten, welche durch HLA-A- und HLA-B-Epitope entstehen, durch die Elektroporation mit nef mRNA verringert werden. Dafür wurden PBMC eines HIV-1infizierten Patienten (0566) mit gag bzw. nef mRNA elektroporiert und diese elektroporierten PBMC im Anschluss in einen γ-IFN ELISPOT mit diversen HIV-1Peptiden inkubiert. Dabei wurden Peptide gewählt, die Epitope zu den jeweiligen HLA-Aund HLA-B-Allelen des Patienten enthalten. Als Kontrolle dienten mock-elektroporierte PBMC. Die Inkubation der mock-elektroporierten PBMC mit den Peptiden führte zu einer deutlichen Sekretion von γ-IFN (Abbildung 4.26). PBMC, welche mit gag mRNA elektroporiert wurden, zeigten ähnliche, wenn auch schwächere T-Zellantworten auf die Peptidstimulationen. Dagegen resultierte die Stimulation mit Peptiden in den nef mRNAelektroporierten PBMC in einer deutlich verringerten T-Zellantwort (Abbildung 4.26). Da die verwendeten Peptide allen Epitopen entsprechen, welche von HLA-A- bzw. HLA-B- 62 4 Ergebnisse Molekülen präsentiert werden, kann die verminderte Expression dieser Moleküle durch Nef Ursache der geringeren T-Zellantwort sein. Zusammenfassend offenbaren diese Ergebnisse, dass durch das mRNA-kodierten NefProtein in PBMC die Expression von HLA-A- und HLA-B-Moleküle reduziert wird, so dass sich die Immunantworten verringern. Abbildung 4.26 PBMC eines HIV-1-infizierten Patienten (0566), welche mit nef mRNA elektroporiert wurden, zeigten eine geringere T-Zellantwort nach Peptidstimulation als mock-elektroporierte PBMC oder gag mRNA-elektroporierte PBMC. PBMC wurden mit gag mRNA bzw. mit nef mRNA elektroporiert und 5 vier Stunden nach Elektroporation wurden 2x10 PBMC mit HIV-1-Peptiden inkubiert. Dabei führte die Stimulation mit Peptiden in den mock- und gag mRNA-elektroporierten PBMC zu einer klaren Immunantwort, wohingegen die nef mRNA-elektroporierten PBMC nur eine schwache T-Zellantwort auf die Peptidstimulation aufwiesen. Die Anzahl der SFU, welche durch die jeweilige Elektroporation verursacht wird, ist in dieser Darstellung bereits subtrahiert. 4.2.6 Mit mRNA-elektroporierten PBMC kann die CTL-EpitopHierarchie studiert werden Da trotz einer vorhandenen CTL Antwort gegen eine Vielzahl von Epitopen viele Patienten AIDS entwickeln, gibt es anscheinend wichtige Unterschiede in der Qualität der Epitope. Manche CTL-Epitope werden viel besser von HIV-1-spezifischen CTLs erkannt, da sie anscheinend besser prozessiert und präsentiert werden. Daher stellte sich die Frage, ob es eine gewisse CTL-Epitop-Hierarchie gibt und welche Epitope besonders gut sind. Dies sollte mit der entwickelten Methode der mRNA-Elektroporation getestet werden. Dazu wurden PBMC eines HIV-1-infizierten Patienten mit gag oder nef mRNA elektroporiert und 63 4 Ergebnisse anschließend zusammen mit autologen Gag- bzw. Nef-spezifischen CTL Linien in einen 5 γ-IFN ELISPOT verwendet. Es wurden 1x10 Zellen der CTL Linie zusammen mit 2x10 5 elektroporierten PBMC pro Well inkubiert. Dabei konnte festgestellt werden, dass die Erkennung der Gag-Epitope, welche von der gag mRNA prozessiert und präsentiert werden, durch die verschiedenen Gag-spezifischen CTL Linien sehr variiert. Das HLAA*02-restringierte SL9v8 Epitop (27), welches in der autologen gag mRNA enthalten ist, wurde im Vergleich zur Peptiderkennung zu 47% von der CTL Linie erkannt (Abbildung 4.27 A). Im Gegensatz dazu wurde das HLA-A*03-restringierte p17 KK9 Epitop (128), welches in der autologen gag mRNA enthalten ist, zu 75% von der spezifischen CTL Linie erkannt (Abbildung 4.27 B). Somit kann man konstatieren, dass das p17 KK9 Epitop in Gag besser prozessiert und präsentiert wird als das SL9 Epitop. Bezüglich des NefProteins konnten keine großen Unterschiede in der Erkennung der verschiedenen, untersuchten Epitope festgestellt werden. Wie in Abbildung 4.27 ersichtlich wird, war die Erkennung von Nef-Epitopen, welche durch nef mRNA-elektroporierte PBMC präsentiert werden, sehr gering und lag zwischen 10% und 20%. Dennoch konnte eine leicht bessere Präsentation des B*18-Epitops RY10 (129) mit 17% im Vergleich zu dem B*07-Epitop Nef7v1 (130) mit 10% eruiert werden (Vergleiche Abbildung 4.27 A und B). Es konnten zudem nicht viele Nef CTL-Epitope miteinander verglichen werden, da die Generierung von Nef-spezifischen CTL Linien, die nicht mit anderen Epitopen kreuzreagierten, sehr schwierig war. In einem anderen Versuchsansatz (siehe 4.1.10) wurde deutlich, dass das HLA-B*13restringierte Nef RI9 Epitop ein gut prozessiertes Nef-Epitop ist. PBMC von dem HIV-1infizierten Patienten 0609 wurden mit heterologer nef mRNA elektroporiert und anschließend im γ-IFN ELISPOT Assay analysiert. Im Vergleich zur Peptiderkennung wird das Nef RI9 Epitop, welches in der nef mRNA enthalten ist, zu 63% erkannt, wenn 200.000 PBMC im Assay eingesetzt wurden. Bei 100.000 PBMC ist der Effekt geringer (Abbildung 4.16). Frühere Versuche der Arbeitsgruppe zeigten, dass sich die Nefspezifische CTL Antwort des Patienten dominant gegen das Peptid Nef RI9 richtet und andere Peptide nicht erkannt wurden. Damit kann die Immunantwort der PBMC gegen die heterologe nef mRNA allein dem Epitop RI9 zugeschrieben werden (Abbildung 4.16). Die gute Prozessierung des Nef-Epitops RI9 könnte auch die gute Kontrolle der Virusreplikation von HLA-B*13-positiven Individuen erklären. Die Daten zeigen, dass die entwickelte Methode gut verwendet werden kann, um die Hierarchie der CTL-Epitope zu studieren. Einige CTL-Epitope werden deutlich besser von den mRNA-elektroporierten PBMC präsentiert und wären daher geeignete Kandidaten bei einer Vakzinierung. 64 4 Ergebnisse Abbildung 4.27 Das p17 KK9 Epitop in Gag wird von gag mRNA-elektroporierten Zellen deutlich besser präsentiert als das SL9 Epitop in Gag, das RY10 Epitop in Nef wird von nef mRNAelektroporierten Zellen leicht besser präsentiert als das Nef7v1 Epitop. PBMC eines HIV-1-infizierten Patienten wurden mit dem p17 KK9 Peptid, einem bekanntem A*03-restringierten Peptid, stimuliert oder mit dem Peptid SL9v8, der autologen Variante des A*02-Epitops SL9, um CTL Linien zu generieren. PBMC des Patienten wurden mit gag mRNA elektroporiert und vier Stunden nach Elektroporation mit autologen Gagspezifischen CTL Linien in einem γ-IFN ELISPOT Assay inkubiert. A: Die relative γ-IFN Sekretion der SL9v8spezifischen CTL Linie. Im Vergleich zur Peptiderkennung lag die Erkennung des Epitops, präsentiert von den gag mRNA-elektroporierten PBMC, bei 47%. B: Die relative γ-IFN Sekretion der p17 KK9-spezifischen CTL Linie. Dabei betrug die Epitoperkennung, präsentiert durch die gag mRNA-elektroporierten PBMC, 75% im Vergleich zur Peptiderkennung der CTL Linie. Für Nef wurden ebenfalls PBMC eines HIV-1-infizierten Patienten mit dem HLA-B*18-restringierten Peptid RY10 und dem HLA-B*07-restringierten Peptid Nef7v1 stimuliert. Die daraus resultierenden CTL Linien wurde dann mit autologer nef mRNA-elektroporierten PBMC des Patienten im ELISPOT inkubiert. C: Die relative Sekretion von γ-IFN der Nef7v1-spezifischen CTL Linie. Im Vergleich zur Peptiderkennung war die Erkennung des Epitops, präsentiert von den nef mRNA-elektroporierten PBMC, sehr gering und lag bei 10%. D: Die relative γ-IFN Sekretion der RY10-spezifischen CTL Linie zeigte im Vergleich zur Peptiderkennung eine Erkennung des Epitops von 17%, wenn es von den nef mRNA-elektroporierten PBMC präsentiert wurde. 65 4 Ergebnisse 4.3 Kreuzreaktion HIV-1-spezifischer CTLs mit humanen Proteinen 4.3.1 Das Nef-Epitop TM9 ähnelt einem Epitop im humanen Protein Fat3 4.3.1.1 Breite CTL-Antwort gegen die Epitope Nef TM9 und Fat3 TM9 T/P Das CTL-Epitop Nef TM9 TPQVPLRPM ist über die HLA-I-Allele B*07 und B*35 restringiert (131, 132). Durch BLAST entdeckten wir eine Sequenzähnlichkeit des Epitops zu dem humanen Protein Fat3 (NCBI-Zugangsnummer: NM_001008781). Die betreffende Sequenz des Fat3 unterscheidet sich dabei nur in zwei Aminosäuren und lautet PPQVPVRPM (TM9 T/P). Aufgrund dieser Sequenzähnlichkeit sollte getestet werden, ob HIV-1-infizierte Patienten CTLs gegen das HIV-1-Epitop TM9 sowie gegen das Fat3Epitop TM9 T/P besitzen und ob diese kreuzreaktiv zueinander sind. Daher wurden PBMC von HIV-1-Patienten, die für HLA-B*07 oder HLA-B*35 positiv sind, mit den Peptiden TM9 und TM9 T/P in vitro stimuliert, um spezifische CTL Linien zu generieren. Das Auswachsen von Antigen-spezifischen CTL Linien wurde im γ-IFN ELISPOT analysiert. Insgesamt wurden 100 Patienten in 214 Tests untersucht. Das Ergebnis der ELISPOT-Screening Assays ist in Tabelle 4.3 zusammengefasst. Eine TM9-spezifische CTL Antwort konnte in 53 Patienten (53%) detektiert werden, eine TM9 T/P-spezifische Antwort in 43 Patienten (43%). Eine starke T-Zellantwort mit mehr als 250 SFU/1x105 Zellen konnte in 26 der 53 TM9-spezifischen CTL Linien beobachtet werden (26%) und 39 TM9-spezifische CTL Linien (39%) zeigten eine deutliche Kreuzreaktivität zu dem TM9 T/P Peptid. Von den 43 TM9 T/P-spezifischen CTL Linien wiesen 19 eine sehr starke T-Zellantwort (19%) auf und 31 der 43 TM9 T/P-spezifischen CTL Linien erkannten das Epitop Nef TM9. Für die Mehrzahl der Patienten konnte eine Immunreaktion gegen beide Peptide quantifiziert werden, wohingegen 11 Patienten eine spezifische Immunantwort nur gegen eins der Peptide zeigte. Zusätzlich habe ich die Daten auch hinsichtlich ihres HLAB-Allels untersucht und gegliedert (Tabelle 4.4 A und B). Dabei konnte kein statistisch signifikanter Unterschied zwischen HLA-B*07- oder HLA-B*35-positiven Individuen bezüglich der Erkennung der Peptide TM9 und TM9 T/P festgestellt werden (Chi-QuadratVierfeldertest). In Abbildung 4.28 sind exemplarische TM9- und TM9 T/P-spezifische CTL Linien dargestellt. Neben Kreuzreaktionen wurden einige CTL Linien auch in Titrationsanalysen eingesetzt. Dabei wurden keine kongruenten Ergebnisse erzielt. Einige CTL Linien erkannten das Peptid noch bei einer Konzentration von 10 ng/ml, andere CTL 66 4 Ergebnisse Linien wiederum konnten das Peptid bereits bei einer Konzentration von 10 µg/ml nicht mehr erkennen (Daten nicht gezeigt). Abbildung 4.28 Exemplarische γ-IFN ELISPOT Assays mit TM9- bzw. TM9 T/P-spezifischen CTL Linien. PBMC von HIV-1-infizierten Patienten wurden mit den Peptiden Nef TM9 (TPQVPLRPM) und TM9 T/P (PPQVPVPM), einem sequenzähnlichen Epitop im humanen Protein Fat3, stimuliert und im γ-IFN ELISPOT analysiert. TM9-spezifische CTL Linien sind in schwarz dargestellt, TM9 T/P-spezifische CTL Linien in Weiß. A: TM9-spezifische CTL Linie mit einer starken Kreuzreaktivität zu TM9 T/P. B: TM9-spezifische CTL Linie ohne Kreuzreaktivität zu TM9 T/P. C: TM9 T/P-spezifische CTL Linie, die ebenso das Peptid Nef TM9 erkennt. D: TM9 T/P-spezifische CTL Linie, die das Nef TM9 Peptid nicht erkennt. Zusammenfassend konnte festgestellt werden, dass das HIV-1 Nef-Epitop TM9 und das sequenzähnliche Epitop TM9 T/P im Fat3-Protein relativ häufig von HLA-B*07 und HLAB*35-positiven HIV-1-Patienten erkannt wurden. In einigen Fällen wurde sogar eine stärkere CTL-Antwort gegen das Fat3-Epitop als gegen das HIV-1 Nef Epitop beobachtet. 67 4 Ergebnisse Tabelle 4.3 Übersicht der γ-IFN ELISPOT Screeningergebnisse. Insgesamt wurden 100 HIV-1-infizierte Patienten, die positiv für das HLA-B*07 oder/und HLA-B*35-Allel sind, auf eine Erkennung der beiden Peptide Nef TM9 und Fat3 TM9 T/P getestet. Eine starke Erkennung definierte sich durch eine Anzahl von mehr als 5 250 SFU/1x10 Zellen. Erkennung von TM9 Starke Erkennung von TM9 Kreuzreaktion zu TM9 T/P Erkennung von TM9 T/P Starke Erkennung von TM9 T/P Kreuzreaktion zu TM9 Anzahl Patienten 53 26 39 43 19 31 Häufigkeit [%] 53% 26% 39% 43% 19% 31% Tabelle 4.4 Übersicht der γ-IFN ELISPOT Screeningergebnisse eingeteilt nach dem entsprechenden HLA-B-Allel. Insgesamt waren von 100 Patienten 57 HLA-B*07-positiv und 48 HLA-B*35-positiv. Davon trugen 5 Patienten beide Allele, HLA-B*07 und HLA-B*35. Es wurde kein statistisch signifikanter Unterschied in der Erkennung der beiden Peptide zwischen den zwei HLA-B-Allelen festgestellt. Eine starke Erkennung 5 definierte sich durch eine Anzahl von mehr als 250 SFU/1x10 Zellen. A: Ergebnisse für das Peptid Nef TM9. B: Ergebnisse für das Peptid Fat3 TM9 T/P. A Erkennung von TM9 Starke Erkennung von TM9 Kreuzreaktion zu TM9 T/P Anzahl Patienten HLA-B*07 Häufigkeit [%] HLA-B*07 32 56,14% 23 47,92% 20 35,09% 7 14,58% 25 43,86% 16 33,33% Anzahl Patienten HLA-B*35 Häufigkeit [%] HLA-B*35 B Erkennung von TM9 T/P Starke Erkennung von TM9 T/P Kreuzreaktion zu TM9 Anzahl Patienten HLA-B*07 Häufigkeit [%] HLA-B*07 Anzahl Patienten HLA-B*35 Häufigkeit [%] HLA-B*35 26 45,61% 20 41,67% 10 17,54% 9 18,75% 22 38,60% 15 31,.25% 4.3.1.2 pfat3 mRNA Elektroporation führte trotz effizienter Proteinexpression zu keiner Erkennung durch Nef-spezifische CTLs Ob TM9- bzw. TM9 T/P-spezifische CTL Linien das Epitop TM9 T/P nicht nur als Peptid sondern auch als prozessiertes Peptid aus dem Protein erkennen, sollte in einer weiteren Analyse untersucht werden. Dazu stand ein bereits im Labor kloniertes Plasmid zur Verfügung, welches 609 Basen des Fat3-Gens als offenen Leserahmen mit einem N-terminalen Flag-tag enthält. Aufgrund der Größe des Fat3-Gens wurde nur ein kleiner Bereich des Gens kloniert, welcher auch das homologe Epitop beinhaltet. Das Plasmid beruht auf dem Vektor pGEM4Z64A und konnte somit zur mRNA-Synthese verwendet 68 4 Ergebnisse werden. Die in vitro transkripierte mRNA pfat3 (partielles fat3) wurde anschließend zur Elektroporation von EBV-transformierten lymphoiden B-Zelllinien eingesetzt. Zudem wurden die Zelllinien auch mit GFP mRNA, HIV-1 nef mRNA (autologes Nef von 0545) sowie mock elektroporiert. Daraufhin wurde die Proteinexpression durch intrazelluläre Färbung mit einem Nef- bzw. Flag-tag-spezifischen Antikörper und anschließender Analyse am Durchflusszytometer überprüft. Die Elektroporation der B-Zelllinien war in allen vier durchgeführten Versuchen sehr effizient und zeigte folgende Mediane der Proteinexpression: 91% GFP-positive Zellen (Range 78%-92%), 59% der Zellen exprimierten das partielle Fat3-Protein (Range: 22%-78%) und 75% der Zellen exprimierten das HIV-1 Nef-Protein (Range: 30-88%) (Abbildung 4.29). Abbildung 4.29 Sowohl das Nef-Protein als auch das partielle Fat3-Protein wurden effizient nach mRNA-Elektroporation in B-Zelllinien exprimiert. B-Zelllinien wurden mock, mit HIV-1 nef mRNA oder pfat3 mRNA elektroporiert. Vier Stunden nach Elektroporation wurden die Zellen permeabilisiert, intrazellulär mit einem Nef- bzw. einem Flag-tag-spezifischen Antikörper gefärbt und im Durchflusszytometer analysiert. Dargestellt ist eine exemplarische Färbung aus vier Versuchen. In der oberen Reihe ist die Färbung mit dem Flag-tag-spezifischen Antikörper ersichtlich, in der unteren Reihe die Färbung mit dem Nef-spezifischen Antikörper. Der linke Dot Plot zeigt jeweils die mock-elektroporierten B-Zellen. Es konnte eine Expression des pfat3 mRNA-kodierten Proteins von 78% detektiert werden. Knapp 88% der nef mRNA-elektroporierten B-Zellen exprimierten das HIV-1 Nef-Protein. Die elektroporierten B-Zelllinien wurden mit den TM9- bzw. TM9 T/P-spezifischen CTL Linien im γ-IFN ELISPOT Assay ko-inkubiert, um die spezifische Erkennung der von den 69 4 Ergebnisse mRNA-kodierten Proteinen durch die CTL Linien zu analysieren. Abbildung 4.30 repräsentiert ein exemplarisches Ergebnis eines ELISPOT Assays mit CTL Linien eines HLA-B*07-positiven Patienten (A) und mit CTL Linien eines HLA-B*35-positiven Patienten (B). Das Experiment wurde je dreimal durchgeführt, auch mit CTL Linien von anderen Patienten. In allen Versuchen konnte man eine deutliche Erkennung des Nef TM9Epitops, welches durch die nef mRNA-elektroporierten B-Zellen prozessiert und präsentiert wurde, durch die TM9- und TM9 T/P-spezifische CTL Linie beobachten. Die Ko-inkubation der TM9- und TM9 T/P-spezifischen CTL Linie mit B-Zellen, welche mit pfat3 mRNA elektroporiert wurden, induzierten nur eine geringe Freisetzung von γ-IFN. Diese war vergleichbar mit der Erkennung von mock-elektroporierten und GFP mRNAelektroporierten B-Zellen durch die CTL Linien und stellten somit Hintergrundwerte dar. Eine starke Sekretion von γ-IFN wurde stets durch die Inkubation der CTL Linien mit Peptiden und mit Peptid-inkubierten B-Zelllinien erreicht (Abbildung 4.30). Folglich wird das Epitop Nef TM9 sowohl als exogenes Peptid als auch als Peptid, welches endogen von den nef mRNA-elektroporierten B-Zellen prozessiert und präsentiert wird, von CTL Linien erkannt. Dagegen wird das Fat3 Epitop TM9 T/P gut als exogenes Peptid von TM9 T/P-spezifischen und TM9-kreuzreaktiven CTL Linien erkannt, jedoch wurden B-Zellen, die mit pfat3 mRNA elektroporiert wurden und das Protein exprimierten, von diesen CTL Linien nicht erkannt. 70 4 Ergebnisse Abbildung 4.30 TM9- und TM9 T/P-spezifische CTL Linien erkennen nef mRNA-elektroporierte BZelllinien, aber nicht pfat3 mRNA-elektroporierte B-Zelllinien. B-Zellen (B-LCL) wurden mock, mit GFP mRNA, HIV-1 nef mRNA oder mit pfat3 mRNA elektroporiert. Vier Stunden nach Elektroporation wurden die elektroporierten B-LCL mit TM9- und TM9 T/P-spezifischen CTL Linien in einem γ-IFN ELISPOT Assay ko4 5 inkubiert. Es wurden jeweils 5x10 Zellen der CTL Linie und 1x10 Zellen der elektroporierten B-LCL eingesetzt. Auf der y-Achse sind die SFU dargestellt. A: Exemplarisches Ergebnis von CTL Linien, die HLAB*07-positiv sind. B: Exemplarisches Ergebnis von CTL Linien, die HLA-B*35-positiv sind. Es wurden jeweils drei Versuche durchgeführt und in keinem konnte eine Erkennung der pfat3 mRNA-elektroporierten B-LCL beobachtet werden. 4.3.1.3 In silico Analysen des fat3 mRNA-Konstrukts sagen eine Prozessierung des Epitops TM9 T/P durch das Proteasom mit einer 50%igen Wahrscheinlichkeit voraus Obwohl eine klare Erkennung des Peptides TM9 T/P durch TM9 T/P-spezifische und TM9-kreuzreaktive CTL Linien detektiert werden konnte, beobachtete ich trotz effizienter Proteinexpression keine Erkennung des pFat3-Proteins, welches durch die mRNA kodiert wird. Eine fehlende oder veränderte Prozessierung des Konstrukts zu dem entsprechenden Epitop durch das Proteasom könnte eine mögliche Erklärung sein. Aus diesem Grund untersuchte ich die 609 Basen lange Sequenz des fat3-Gens, welche in unserem Plasmid enthalten ist, in silico auf Proteasom-Spaltung. Dazu wurde das 71 4 Ergebnisse Programm MAPPP verwendet (siehe Tabelle 6.21). Dieses Programm beruht auf FRAGPREDICT (133, 134) und prognostiziert eine Prozessierung des Epitops TM9 T/P durch das Proteasom Wahrscheinlichkeit (Abbildung mittelmäßig. Das 4.31). Allerdings Nef-Epitop TM9 ist wird die mit vorhergesagte einer 99%igen Wahrscheinlichkeit prozessiert, das TM9 T/P Epitop hingegen nur mit einer Erwartung von 50-59%. Es ist daher zu vermuten, dass das Epitop TM9 T/P aus dem pfat3-Konstrukt nicht durch das Proteasom gespalten wird und es daher auch trotz einer nachweisbaren Proteinexpression nach pfat3 mRNA-Elektroporation in B-Zelllinien zu keiner Erkennung durch CTL Linien kommt. Abbildung 4.31 In silico Analyse auf Proteasomschnittstellen der Proteinsequenzen von HIV-1 Nef und Fat3 offenbarte, dass die Wahrscheinlichkeit einer Prozessierung für das Epitop TM9 bei 99% liegt, für das Epitop TM9 T/P dagegen nur bei maximal 59%. Die Sequenzen der Proteine, die von der mRNA kodiert werden, wurden mit Hilfe des Programms MAPPP analysiert. Dargestellt ist nur der betreffende Ausschnitt aus der Proteinsequenz. Orange hervorgehobene Sequenzen markieren die besagten Epitope. Die Zahlen in Klammern geben die Wahrscheinlichkeit an, mit welcher das entsprechende Peptid prozessiert wird. Je höher der Wert bei 1 liegt, desto wahrscheinlicher wird das Epitop prozessiert. 4.3.2 Das Nef-Epitop Nef7v1 ist einem Epitop im humanen Enzym D-Aminosäuren-Oxidase (DAO) ähnlich Ein weiteres Nef CTL-Epitop, welches eine hohe Sequenzähnlichkeit zu einem Epitop eines humanen Proteins besitzt, ist das Epitop Nef7v1 mit der Sequenz FPVRPQVPL. Es ist über die HLA-I-Allele B*07, B*35 und B*51 restringiert (135, 136) und unterscheidet sich in nur drei Aminosäuren von einem Epitop des humanen Flavoproteins DAminosäuren-Oxidase (DAO) (NCBI-Zugangsnummer: NM_001917) mit der Sequenz RPVRPQIRL (Nef7v4). Es sollte nun getestet werden, ob HIV-1-infizierte Patienten 72 4 Ergebnisse spezifische CTLs aufweisen, die das humane Epitop aus DAO erkennen. Daher wurden PBMC von HIV-1-Patienten mit den entsprechenden HLA-B-Allelen mit den Peptiden Nef7v1 und Nef7v4 in vitro stimuliert und die Generierung von Antigen-spezifischen CTL Linien wurde nach zehntägiger Kultivierung im γ-IFN ELISPOT Assay überprüft. In 149 Tests wurden 104 Patienten untersucht. Von diesen 104 Patienten wiesen 37 Patienten Nef7v1-spezifische CTLs auf (36%), wobei 11 CTL Linien eine sehr starke Reaktion mit über 250 SFU/1x105 Zellen zeigten (11%) und 14 CTL Linien das humane Epitop Nef7v4 kreuzreaktiv erkennen konnten (13%). Die Stimulation von PBMC mit dem Peptid Nef7v4 führte in 26 Patienten zu einer Erkennung (25%). Dabei zeigten sieben CTL Linien (7%), die spezifisch sind für das DAO-Epitop, eine starke Reaktion mit über 250 SFU/1x105 Zellen und 19 wiesen eine Kreuzreaktion zu dem HIV-1 Nef-Epitop Nef7v1 auf (18%). Diese Ergebnisse sind in Tabelle 4.5 zusammengefasst. Tabelle 4.5 Übersicht der γ-IFN ELISPOT Screeningergebnisse. Insgesamt wurden 104 HIV-1-infizierte Patienten, die positiv für das HLA-Allel-B*07, -B*35 und/oder –B*51 sind, auf eine Erkennung der beiden Peptide Nef7v1 und DAO Nef7v4 getestet. Eine starke Erkennung definierte sich durch eine Anzahl von mehr 5 als 250 SFU/1x10 Zellen. Erkennung von Nef7v1 Starke Erkennung von Nef7v1 Kreuzreaktion zu Nef7v4 (DAO) Erkennung von Nef7v4 (DAO) Starke Erkennung von Nef7v4 Kreuzreaktion zu Nef7v1 Anzahl Patienten 37 11 14 26 7 19 Häufigkeit [%] 35,58% 10,58% 13,46% 25,00% 6,73% 18,27% Des Weiteren wurden die Daten nach dem jeweiligen HLA-B-Allel gegliedert. Unter den 104 Patienten waren 45 Patienten für HLA-B*07 positiv, 39 für HLA-B*35 und 25 für HLAB*51. Jedoch konnte zwischen den verschiedenen HLA-B-Allelen kein signifikanter Unterschied in der Erkennung der Epitope detektiert werden (Chi-Quadrat-Vierfeldertest). Die Ergebnisse sind in Tabelle 4.6 dargestellt. Insgesamt konnten in HIV-1-infizierten Patienten spezifische CTLs gegen das DAOEpitop, welches dem HIV-1-Epitop Nef7v1 stark ähnelt, nachgewiesen werden. Allerdings war das Ausmaß sehr gering, denn in nur 25% der Fälle ließ sich eine CTL-Antwort gegen das humane Epitop identifizieren und nur 7% zeigten dabei eine deutliche Reaktion. Aufgrund der geringen Frequenz starker CTL-Reaktionen gegen das humane Nef7v4 Epitop wurden keine weiteren Analysen durchgeführt. 73 4 Ergebnisse Tabelle 4.6 Übersicht der γ-IFN ELISPOT Screeningergebnisse für die Peptide Nef7v1 und DAO Nef7v4 eingeteilt nach dem entsprechenden HLA-B-Allel. Insgesamt waren von 104 Patienten 45 HLA-B*07positiv, 39 HLA-B*35-positiv und 25 HLA-B*51-positiv. Es wurde kein statistisch signifikanter Unterschied in der Erkennung der beiden Peptide zwischen den HLA-B-Allelen festgestellt. Eine starke Erkennung definierte 5 sich durch eine Anzahl von mehr als 250 SFU/1x10 Zellen. A: Ergebnisse für das Peptid Nef7v1. B: Ergebnisse für das Peptid Nef7v4. A Erkennung von Nef7v1 Starke Erkennung von Nef7v1 Kreuzreaktion zu Nef7v4 Anzahl Patienten HLA-B*07 16 6 6 Anzahl Patienten HLA-B*35 17 4 6 Anzahl Patienten HLA-B*51 7 3 3 Erkennung von Nef7v4 (DAO) Starke Erkennung von Nef7v4 Kreuzreaktion zu Nef7v1 Anzahl Patienten HLA-B*07 10 2 4 Anzahl Patienten HLA-B*35 17 4 15 Anzahl Patienten HLA-B*51 5 0 32 B 74 4 Ergebnisse 4.4 Analyse und Screening von neuen und bekannten CTL-Epitopen Bei einer therapeutischen Vakzine, die CTLs induzieren soll, spielen die verwendeten CTL-Epitope eine entscheidende Rolle. Eine effiziente Vakzine sollte CTL-Epitope enthalten, die starke Immunantworten bei möglichst vielen Patienten hervorrufen. Daher war es ein weiteres Ziel dieser Arbeit neue CTL-Epitope zu definieren, wobei der Fokus auf der Analyse von CTL-Epitopen in Patienten mit guter Kontrolle der Virusreplikation lag. Zum anderen sollte die Immunantwort, welche durch bekannte CTL-Epitope ausgelöst wird, in einer möglichst großen Kohorte an HIV-1-Infizierten untersucht werden. Hierbei rückten insbesondere CTL-Epitope ins Interesse, welche über das HLA-C-Molekül präsentiert werden, da diese durch das Nef-Protein nicht negativ beeinflusst werden. 4.4.1 Definition von drei neuen Gag und zwei neuen Nef CTLEpitopen Zwei HIV-1-infizierte Patienten, die noch therapie-naiv sind und bisher eine gute Kontrolle der Virusreplikation aufzeigten, wurden in diese Untersuchung eingeschlossen. Nach Isolation aus Citratblut wurden die PBMC direkt in einen γ-IFN ELISPOT Assay mit Peptidsets, die einen Großteil der HIV-1-Proteine abdecken, inkubiert. Bei den Peptiden handelte es sich stets um Peptide, deren Aminosäurensequenz der Consensus B Sequenz (www.hiv.lanl.gov) von HIV-1 entsprechen. Die Peptide wurden vom NIH (National Institute of Health) bezogen. Anhand des HLA-Typs der Patienten und der Epitop-Datenbank (ELF=epitope location finder, HIV sequence database) konnten die Immunantworten, welche durch die verwendeten Peptide ausgelöst wurden, bestimmten CTL-Epitopen zugeordnet werden. Patient 0284 Für Patient 0284 wurden die Immunantworten gegen HIV-1 Gag, Nef, Tat, Rev, Vif, Vpr, Vpu und gegen die Hälfte von Pol mit Hilfe der Peptidsets analysiert. Die Untersuchung der T-Zellantworten gegen die akzessorischen Moleküle Vif, Vpr, Vpu, Tat und Rev ergab keine Reaktion. Somit wies dieser Patient keine CTL-Antwort gegen diese Proteine auf oder die Reaktion war zu gering, um sie ohne Vorstimulation der Zellen im γ-IFN ELISPOT Assay detektieren zu können. Auch bei der Analyse mit Pol-Peptiden konnte nur eine geringe T-Zellantwort gegen ein einziges, bekanntes Peptid ermittelt werden (Pol07-124 TYQIYQEPFKNLKTG, HLA-A*11, 21 SFU/1x105 PBMC). Im Gegensatz dazu konnten gegen Peptide der Proteine Gag und Nef deutliche T-Zellantworten erfasst werden. So besitzt Patient 0284 CTLs gegen sieben verschiedene Gag-Epitope und ein 75 4 Ergebnisse dominantes Nef-Epitop (Tabelle 4.7). Für das Peptid Gag07-113 war zum damaligen Zeitpunkt noch kein CTL-Epitop für den HLA-Typ des Patienten in der Datenbank (ELF) beschrieben. Tabelle 4.7 Übersicht über die vorhandenen CTL-Antworten des Patienten 0284 nach Inkubation der PBMC mit Gag- und Nef-Peptidsets. Die CTL-Antwort wurde im γ-IFN ELISPOT gemessen. Angegeben sind Name und Sequenz der verwendeten Peptide, ihre ausgelöste Immunantwort im ELISPOT (SFU) sowie darin enthaltene Epitope und das dazugehörige HLA-Allel. Die Epitope wurden mit dem Programm ELF (HIV sequence database) vorhergesagt. Peptidname Peptidsequenz Gag07-20 SLYNTVATLYCVHQR SFU/ 5 1x10 PBMC 9 Gag07-36 QMVHQAISPRTLNAW 26 Gag07-41 Gag07-77 EKAFSPEVIPMFSAL RAEQASQEVKNWMTE 75 39 Gag07-87 EMMTACQGVGGPGHK 51 Gag07-109 Gag07-113 Nef07-26 FLGKIWPSHKGRPGN LQSRPEPTAPPEESF IYSQKRQDILDLWVY 23 36 49 Epitop HLA-Allel SLYNTVATL ISPRTLNAW QAISPRTL KAFSPEVIPMF QASQEVKNW EMMTACQGV ACQGVGGPGHK FLGKIWPSHK ? RQDILDLWVY A*02 B*57 Cw*07 B*57 B*57 A*02 A*11 A*02 ? Cw*07 HLA-I 0284: A*02, A*11, B*39, B*57, Cw*06, Cw*07 Patient 0925 Für Patient 0925 wurden die Immunantworten gegen die HIV-1-Proteine Gag und Nef analysiert. Die Inkubation der PBMC mit den Nef-Peptiden offenbarte T-Zellreaktionen gegen insgesamt vier Epitope, wovon drei kein bekanntes CTL-Epitop zum HLA-Typ des Patienten enthielten, jedoch potentielle Epitope von dem Programm ELF vorgeschlagen wurden (Tabelle 4.8). Für Gag wurde eine Immunreaktion gegen drei Epitope beobachtet, wobei wiederum zwei Peptide noch unbekannte CTL-Epitope innehaben. 76 4 Ergebnisse Tabelle 4.8 Übersicht über die vorhandenen CTL-Antworten des Patienten 0925 nach Inkubation der PBMC mit Gag- und Nef-Peptidsets. Die CTL-Antwort wurde im γ-IFN ELISPOT gemessen. Angegeben sind Name und Sequenz der verwendeten Peptide, ihre ausgelöste Immunantwort im ELISPOT (SFU) sowie darin enthaltene Epitope und das dazugehörige HLA-Allel. Potentielle, aber noch nicht beschriebene CTLEpitope sind kursiv dargestellt. Die Epitope wurden mit dem Programm ELF HIV (sequence database) vorhergesagt. Peptidname Peptidsequenz Gag07-02 ASVLSGGELDRWEKI SFU/ 5 2x10 PBMC 11 Gag07-11 Gag07-75 Nef07-08 Nef07-10 Nef07-20 Nef07-23 LERFAVNPGLLETSE VDRFYKTLRAEQASQ GVGAVSRDLEKHGAI LEKHGAITSSNTAAN RPMTYKAAVDLSHFL HFLKEKGGLEGLIYS 73 15 20 26 30 24 Epitop HLA-Allel VLSGGELDR GELDRWEKI ? ? GAVSRDLEK LEKHGAITS AAVDLSHFL KEKGGLEGL A*66 B*40 ? ? A*66 B*40 Cw*16 B*44 HLA-I 0925: A*01, A*66, B*41, B*44, Cw*16, Cw*17 4.4.2 Analyse der neu definierten CTL-Epitope 4.4.2.1 Das Peptid Gag07-113 enthält ein HLA-A*02-restringiertes CTL-Epitop Durch Inkubation der PBMC von Patient 0284 konnte eine T-Zellantwort für das Peptid Gag07-113 beobachtet werden (4.4.1). Da hierfür noch kein CTL-Epitop beschrieben war, welches zu den HLA-Allelen des Patienten passte, sollte geklärt werden, wie die exakte Sequenz des Epitops lautet und über welches HLA-Allel die Präsentation des Epitops erfolgt. Daher wurden in einer gemeinsamen Arbeit mit Frau A. Hückelhoven mehrmals spezifische CTL Linien von 0284 mit diesem Peptid generiert. Diese wurden in Restriktionsanalysen eingesetzt, um das präsentierende HLA-Molekül zu identifizieren. Dabei erwies sich in wiederholenden Versuchen, auch mit CTL Linien von anderen Spendern, das HLA-Allel A*02 als das restringierende Molekül (Abbildung 4.32). Kreuzreaktionen zu den überlappenden Peptiden Gag07-112 und Gag07-114 traten in keinen getesteten Linien auf, so dass das Epitop auf die Sequenz RPEPTAPPE eingrenzt werden konnte. Jedoch passt diese Sequenz nicht zum bekannten Bindungsmuster des HLA-A*02-Moleküls. Da außerdem die Inkubation der CTL Linie sowohl mit einem AntiCD4 als auch mit einem Anti-CD8 Antikörper die Reaktion inhibierte, handelte es sich offensichtlich um eine Linie, die spezifisch für ein CD4-Epitop und ein CD8-Epitop ist. 77 4 Ergebnisse Abbildung 4.32 Restriktionsanalyse mit einer Gag07-113-spezifischen CTL Linie offenbarte, dass sich in dem Peptid ein bisher unbekanntes HLA-A*02-restringiertes CTL-Epitop befindet. Durch Inkubation mit dem Peptid Gag07-113 und Kultivierung für 10 Tage in R10IL2-Medium wurde aus PBMC von 0284 eine CTL Linie generiert. B-LCL, die möglichst nur ein HLA-Allel mit der CTL Linie teilen, wurden sowohl mit als 4 4 auch ohne Peptid für eine Stunde inkubiert, 3x gewaschen und anschließend wurden 5x10 B-LCL mit 5x10 Zellen der CTL Linie im γ-IFN ELISPOT Assay ko-inkubiert. Nur die Peptid-inkubierte, HLA-A*02-positive B-LCL wurde von der CTL Linie erkannt und zeigte eine deutliche γ-IFN Sekretion. Angegeben sind nur die HLA-Allele, die B-LCL und CTL Linie gemeinsam haben. Schwarze Balken stellen die Peptid-inkubierten B-LCL dar, weiße Balken die Kontrolle ohne Peptid. HLA-I CTL Linie: A*02, A*11, B*39, B*57, Cw*06, Cw*07 4.4.2.2 Das Peptid Gag07-02 enthält ein HLA-B*41-restringiertes CTL-Epitop Da das Peptid Gag07-02 eine T-Zellantwort bei Patient 0925 auslöste und darin kein bekanntes CTL-Epitop vorhanden war, sondern nur potentielle Epitope zu den HLAAllelen A*66 bzw. B*40 aus der Datenbank vorgeschlagen wurden (Tabelle 4.8), wurde eine spezifische CTL Linie zu diesem Peptid ausgehend von PBMC des Patienten 0925 generiert. Diese wurde anschließend in eine Restriktionsanalyse und in Kreuzreaktionen eingesetzt, um sowohl die Sequenz des Epitops eingrenzen zu können sowie auch das restringierende HLA-Allel zu bestimmen. Wie in Abbildung 4.33 A dargestellt, wird das Peptid nur von einer HLA-B*41-positiven B-Zelllinie präsentiert und demzufolge von der spezifischen CTL Linie erkannt. Zudem wies die Inhibierung der Reaktion durch den Anti- 78 4 Ergebnisse CD8 Antikörper ebenfalls auf ein HLA Klasse I-Epitop hin. Kreuzreaktionen mit den Peptiden Gag07-01 und Gag07-03 führten zu keiner Erkennung von Gag07-01, jedoch zu einer schwachen Erkennung von Gag07-03 (Abbildung 4.33 B). Damit lässt sich das Epitop auf die Sequenz SGGELDRWEKI einschränken. Da für das Molekül HLA-B*41 neun Aminosäuren lange Epitope optimal sind und sich an den Ankerpositionen ein Glutamat (Position zwei) bzw. ein Leucin (Position neun) befinden sollen (137), lautet die Sequenz des Epitops vermutlich GELDRWEKI. Darüber hinaus würde dies dem potentiellen HLA-B*40 Epitop (siehe Tabelle 4.8) entsprechen. Allerdings müsste man mit kürzeren Peptiden noch beweisen, ob HLA-B*41 wirklich das gleiche Epitop präsentiert wie HLA-B*40. Abbildung 4.33 Restriktionsanalyse und Kreuzreaktionen einer Gag07-02-spezifischen CTL Linie identifizierten ein HLA-B*41-restringiertes CTL-Epitop, welches vermutlich die Sequenz GELDRWEKI besitzt. PBMC von 0925 wurden durch Peptidinkubation mit Gag07-02 zur CTL Linie generiert. A: Nur die BLCL mit dem HLA-Allel B*41 konnte das Epitop präsentieren und wurde daher von der CTL Linie erkannt. 5 4 1x10 Zellen der CTL Linie wurden zusammen mit 5x10 Zellen von gematchten B-Zelllinien, die möglichst nur ein HLA-Allel mit der CTL Linie teilen, in einem γ-IFN ELISPOT Assay inkubiert. Zuvor wurden die B-Zelllinien entweder mit dem Peptid Gag07-02 (schwarze Balken) oder ohne Peptid (weiße Balken) für eine Stunde inkubiert und daraufhin 3x gewaschen. Angegeben sind nur die HLA-Allele, welche der B-LCL und der CTL Linie gemeinsam sind. B: Die Gag07-02-spezifische CTL Linie erkannte sowohl die Peptide Gag07-02 als 4 auch Gag07-03 und wurde durch einen Anti-CD8 Antikörper geblockt. 1x10 Zellen der CTL Linie wurden allein, mit den überlappenden Peptiden Gag07-01, -02 oder -03 oder einen Anti-CD8-Antikörper in einem γ-IFN ELISPOT Assay inkubiert. HLA-I CTL Linie: A*01, A*66, B*41, B*44, Cw*16, Cw*17 79 4 Ergebnisse 4.4.2.3 Die restringierenden HLA-Allele für die Peptide Gag07-11 und Gag07-75 konnten nicht bestimmt werden Die Inkubation der PBMC von 0925 mit den Peptiden Gag07-11 und Gag07-75 löste eine T-Zellreaktion im ELISPOT aus. Jedoch waren für diese Peptide bisher noch keine CTLEpitope beschrieben. Daher sollten sowohl die exakten Sequenzen der beiden Epitope als auch die HLA-Allele, welche das entsprechende Epitop präsentieren, bestimmt werden. Für das Peptid Gag07-11 wurden Kreuzreaktionen und Restriktionsanalysen mit der spezifischen CTL Linie durchgeführt. Eine Inkubation mit den beiden überlappenden Peptiden Gag07-10 sowie Gag07-12 führte zu keiner Freisetzung von γ-IFN (Abbildung 4.34). Folglich sollte unter der Annahme eines maximal zehn Aminosäuren langen Epitops das Epitop im Sequenzbereich RFAVNPGLLET liegen. Die Reaktion der CTL Linie konnte mit einem Anti-CD8 Antikörper gehemmt werden, so dass es sich um ein HLA Klasse IEpitop handeln muss (Abbildung 4.34). In mehreren Restriktionsanalysen konnte kein restringierendes HLA-Allel identifiziert werden, da die HLA-Allele Cw*16 und Cw*17 ein Alanin an Position zwei des Epitops bevorzugen, könnte es sich um ein HLA-Crestringiertes CTL-Epitop handeln. Weitere Analysen könnten zur Bestätigung dieser Vermutung beitragen. Eine T-Zellantwort auf das Peptid Gag07-75 konnte bereits beim nächsten Untersuchungstermin des Patienten nicht mehr festgestellt werden, so dass keine Studien mehr folgten. 5 Abbildung 4.34 Kreuzreaktionen und CD8-Block der Gag07-11-spezifischen CTL Linie. 1x10 Zellen der spezifisch generierten CTL Linie von Patient 0925 wurden ohne Peptid, mit den dargestellten Peptiden und mit einem Anti-CD8-Antikörper im γ-IFN ELISPOT Assay inkubiert. Es wurde keine Kreuzreaktion zu den überlappenden Peptiden beobachtet, was die Sequenz des Epitops auf RFAVNPGLLET eingrenzt. Die Reaktion konnte mit dem Anti-CD8 Antikörper geblockt werden. 80 4 Ergebnisse 4.4.2.4 Die Peptide Nef07-08 und Nef07-10 enthalten HLA-B*41-Epitope Zwar wurden für die beiden Nef-Peptide Nef07-08 und Nef07-10 potentielle Epitope aus der Datenbank vorgeschlagen, dennoch ist unklar, welches CTL-Epitop in dem jeweiligen Peptid liegt und die T-Zellantwort bei Patient 0925 auslöste (Tabelle 4.8). Daher wurden spezifische CTL Linien von 0925 mit diesen Peptiden generiert und diese nach positiver Testung im γ-IFN ELISPOT in Restriktionsanalysen und Kreuzreaktionen eingesetzt. Beide CTL Linien erkannten dabei nur Peptid-inkubierte B-Zelllinien, die das HLA-Allel B*41 tragen (Abbildung 4.35). Da die Immunreaktionen bei beiden CTL Linien zumindest teilweise mit einem Anti-CD8 Antikörper geblockt werden konnte (Abbildung 4.36), kann man jeweils auf ein CTL-Epitop schließen, das über das Molekül HLA-B*41 restringiert ist. Abbildung 4.35 Restriktionsanalysen mit Nef07-08- und Nef07-10-spezifischen CTL Linien wiesen auf HLA-B*41-restringierende CTL-Epitope in den jeweiligen Peptiden hin. Nach Generierung der spezifischen CTL Linien aus PBMC von Patient 0925 wurden B-LCL ausgewählt, welche möglichst nur ein HLA-Allel mit der CTL Linie gemeinsam haben. Diese wurden einmal mit und einmal ohne Peptid inkubiert, 4 5 gewaschen und 5x10 B-LCL mit 1x10 Zellen der CTL Linie in einem γ-IFN ELISPOT Assay inkubiert. Angegeben sind nur die gemeinsamen HLA-Allele der B-Zelllinie und der CTL Linie. A: Die Restriktionsanalyse mit der Nef07-08-spezifischen CTL Linie zeigte eine klare Reaktion bei Ko-Inkubation mit der HLA-B*41-positiven B-LCL, die zuvor mit dem Peptid Nef07-08 inkubiert wurde (blau). B: Die Restriktionsanalyse mit der Nef07-10-spezifischen CTL Linie demonstrierte ebenso eine klare Erkennung der HLA-B*41-positiven B-LCL, die zuvor mit dem Peptid Nef07-10 inkubiert wurde (magenta). HLA-I CTL Linie: A*01, A*66, B*41, B*44, Cw*16, Cw*17 81 4 Ergebnisse Kreuzreaktionen der Nef07-08-spezifischen CTL Linie mit den Peptiden Nef07-07 und Nef07-09 zeigten keine Erkennung der beiden überlappenden Peptide (Abbildung 4.36 A), so dass das Epitop im Bereich der Sequenz AVSRDLEKHG liegen müsste. Die Ankeraminosäuren würden bei einem HLA-B*41-restringierten Epitop Glutamat an Position zwei und Leucin an Position neun sein (137), welche jedoch in der Sequenz nicht vorkommen. Das Programm SYFPEITHI sagt die Sequenz SRDLEKHGA für das HLAB*41-Epitop vorher. In diesem Fall hätte man aber eine Kreuzreaktion der Nef07-08spezifischen CTL Linie mit dem Peptid Nef07-09 erhalten müssen. Daher ist noch unklar, wie die exakte Sequenz des Epitops ist. Unsere Ergebnisse stimmen nicht mit dem vorgeschlagenen, potentiellen Epitop überein (ELF, HIV sequence database), da wir anstelle einer HLA-A*66-Restriktion eine HLA-B*41-Restriktion feststellten. Kreuzreaktionen der Nef07-10-spezifischen CTL Linie demonstrierten eine Erkennung des überlappenden Peptides Nef07-09, aber nicht des Peptides Nef07-11 (Abbildung 4.36 B). Dadurch muss das Epitop in der Sequenz LEKHGAITSSN liegen, welche sowohl in Nef07-09 als auch Nef07-10 vorhanden ist. Da HLA-B*41 an Position zwei ein Glutamat als Ankeraminosäure bevorzugt und meist Liganden mit einer Länge von neun Aminosäuren bindet, könnte das HLA-B*41 CTL-Epitop in Nef07-10 dem potentiellen HLA-B*40 LEKHGAITS entsprechen (siehe Tabelle 4.8). Dies soll durch die Verwendung von kürzeren Peptiden noch bewiesen werden. Abbildung 4.36 Kreuzreaktionen und CD8-Block mit den CTL Linien, die spezifisch für Nef07-08 bzw. 5 Nef07-10 sind. 1x10 Zellen einer spezifisch generierten CTL Linie von Patient 0925 wurden ohne Peptid, mit den dargestellten Peptiden und mit einem Anti-CD8-Antikörper im γ-IFN ELISPOT Assay inkubiert. A: Die Nef07-08-spezifische CTL Linie (blau) zeigte keine Kreuzreaktion zu den überlappenden Peptiden, was die Sequenz des Epitops auf AVSRDLEKHG eingrenzt. Die T-Zellantwort konnte mit dem Anti-CD8 Antikörper geblockt werden. B: Die Nef07-10-spezifische CTL Linie (magenta) erkannte das überlappende Peptid Nef0709, so dass das CTL-Epitop auf die Sequenz LEKHGAITSSN eingegrenzt werden konnte. Die Inkubation mit dem Anti-CD8-Antikörper inhibierte die Sekretion von γ-IFN um die Hälfte. 82 4 Ergebnisse 4.4.3 Das bekannte HLA-C-restringierte Nef-Epitop Nef11T3 ist ein häufig erkanntes Epitop Das CTL-Epitop Nef11T3 RQDILDLWIY wird über HLA-Cw*07 restringiert (138). Da HLAC-Moleküle durch Nef nicht negativ beeinflusst werden können, sind HLA-Crestringierende Epitope interessante Kandidaten bei einer Vakzinierung zur Induktion von CTLs. Aufgrund der Häufigkeit des HLA-Allels Cw*07 sollte untersucht werden, wie oft und in welchem Ausmaß eine CTL-Antwort gegen dieses Epitop auftritt. Dazu wurden CTL Linien von HIV-1-infizierten Patienten generiert, welche HLA-Cw*07-positiv sind. Nach zehntägiger Kultivierung wurden die Zellen im γ-IFN ELISPOT Assay getestet. Insgesamt wurden 85 Patienten in 92 Assays untersucht. Von diesen 85 Patienten wiesen 36 eine CTL-Antwort gegen dieses Epitop auf (42%), 19 davon sogar eine ausgeprägte Immunantwort mit mehr als 250 SFU (22%). Anhand der autologen Nef Sequenzen, die uns zur Verfügung standen, konnte eine Variante des Epitops festgestellt werden. Bei dieser, Nef11T3 I/V RQDILDLWVY, findet man an Position neun ein Valin (V) anstelle eines Isoleucins (I). Da diese in den autologen Virussequenzen der Patienten häufig zu finden war (siehe auch Abbildung 4.10 B), wurden von 35 Patienten auch CTL Linien mit dem varianten Peptid generiert und analysiert. Bei 15 Patienten (43%) konnte hierbei eine CTL-Antwort gegen das Peptid Nef11T3 I/V detektiert werden, neun davon (26%) wiesen eine starke Immunreaktion mit über 250 SFU auf. Des Weiteren waren alle Nef11T3spezifischen CTL Linien kreuzreaktiv zu Nef11T3 I/V und alle Nef11T3 I/V-spezifischen CTL Linien erkannten das Wildtyp Peptid. Bei Titrationsversuchen konnte eine höhere Affinität der Variante detektiert werden, da eine Erkennung des Peptids zum Teil bis zu einer Konzentration von 10 ng/ml stattfand (Abbildung 4.37 B). Im Falle des Wildtyp Peptids führten nur Konzentrationen bis 100 ng/ml zu einer Sekretion von γ-IFN (Abbildung 4.37 A). Zusammenfassend kann festgehalten werden, dass das HLA-Cw*07-restringierende Peptid Nef11T3 sowie die Variante Nef11T3 I/V sehr häufig von HIV-1-infizierten Patienten erkannt wurden. 83 4 Ergebnisse Abbildung 4.37 Titrationsversuche mit Nef11T3- und Nef11T3 I/V-spezifischen CTL Linien zeigten eine 5 höhere Affinität von Nef11T3 I/V. Von den generierten CTL Linien wurden 1x10 Zellen im γ-IFN ELISPOT Assay mit verschiedenen Konzentrationen von Peptiden inkubiert. A: Titration des Nef11T3-Peptids (schwarz) und Kreuztitration des Peptids Nef11T3 I/V (blau) mit einer Nef11T3-spezifischen CTL Linie. Beide Peptide wurden von der CTL Linie nur bis zu einer Peptidkonzentration von 100 ng/ml erkannt. B: Titration des Peptides Nef11T3 I/V (blau) und Kreuztitration des Peptides Nef11T3 (schwarz) mit einer Nef11T3 I/Vspezifischen CTL Linie. Das Peptid Nef11T3 I/V wurde auch bei einer Konzentration von 10 ng/ml noch deutlich erkannt. 84 4 Ergebnisse 4.5 Etablierung alternativer Transfektionsmethoden Zur Transfektion von mRNA in vitro stellt die Elektroporation die vermutlich schnellste, kostengünstigste und effizienteste Methode dar. Für eine in vivo Applikation fehlen aktuell noch die technischen Voraussetzungen. Daher sollten alternative Transfektionsmethoden etabliert werden, die im Hinblick auf eine in vivo Anwendung sicher und gut verträglich sind. Für alle in dieser Arbeit untersuchten Reagenzien konnte bereits ein erfolgreicher Transfer von DNA und zum Teil von siRNA in Zellen gezeigt werden, ein Transfer von mRNA jedoch noch nicht. 4.5.1 Chitosan transfiziert DNA, aber keine mRNA in HEK293T Bei Chitosan handelt es sich um ein natürlich vorkommendes Polysaccharid aus dem Panzer von Krebsen (139), welches DNA und siRNA in verschiedene Zellen transportieren kann (140, 141). Aufgrund seiner nicht-toxischen, biologisch abbaubaren und biokompatiblen Eigenschaften stellt es eine interessante Möglichkeit zur Transfektion in vivo dar. Daher wurden Transfektionen von GFP mRNA, welche mit Chitosan komplexiert ist, in HEK293T Zellen, PBMC, Makrophagen und Dendritische Zellen (DCs) durchgeführt. Zur Kontrolle wurden die Zellen auch mit der Plasmid-DNA pEGFP-C1 transfiziert. Es konnte ein erfolgreicher Transfer der mit Chitosan komplexierten PlasmidDNA in HEK293T Zellen bereits nach 22 Stunden detektiert werden, welche nach 120 Stunden besonders deutlich war (Abbildung 4.38). Zellen, welche mit komplexierter GFP mRNA transfiziert wurden, zeigten auch nach 120 Stunden keine Fluoreszenz. In PBMC, Makrophagen und DCs konnte weder nach DNA-Transfektion noch nach mRNATransfektion eine GFP-Fluoreszenz gemessen werden. Folglich war mit Chitosan nur die Transfektion von Plasmid-DNA in HEK293T Zellen erfolgreich, alle anderen Versuche schlugen fehl. 85 4 Ergebnisse Abbildung 4.38 Chitosan komplexierte Plasmid-DNA, wodurch eine erfolgreiche Transfektion der DNA in HEK293T Zellen stattfinden konnte. Sowohl GFP mRNA als auch Plasmid-DNA von pEGFP-C1 wurde mit einer Chitosanlösung vermischt und anschließend zu HEK293T Zellen gegeben. Durch Detektion der GFP-Fluoreszenz an einem Fluoreszenzmikroskop konnte der Erfolg der Transfektion ermittelt werden. Die grüne Fluoreszenz ist in der Fluoreszenzaufnahme in weiß dargestellt (rechts) und zeigt wenige GFPexprimierende Zellen. Vergleicht man dazu die Durchlichtaufnahme (links), welche die Dichte der Zellen zeigt, wird deutlich, dass nur wenige Zellen GFP exprimieren. Diese Aufnahmen wurden 120 Stunden nach Transfektion dokumentiert. 4.5.2 HIV-1 Tat führt zu keinem Transfer von Nukleinsäuren in Zellen Verschiedene Studien mit dem HIV-1 Tat-Protein oder einem einzelnen Peptid, welches die Protein-Transduktionsdomäne von Tat enthält, zeigten bereits eine erfolgreiche und sichere in vivo-Transfektion von Proteinen, siRNA und DNA (142-144). In dieser Arbeit wurden HEK293T und Makrophagen zur Transfektion mit Tat-Protein und Tat-Peptid verwendet. Sowohl GFP mRNA als auch GFP-DNA in Form des Plasmides pEGFP-C1 kamen zum Einsatz. Allerdings konnte in keinem Versuch ein erfolgreicher Transfer der Nukleinsäure mit Hilfe des Tat-Proteins bzw. des Tat-Peptides beobachtet werden. Auch nach Veränderung der Ratio zwischen Tat und DNA bzw. mRNA konnte keine GFPFluoreszenz detektiert werden. 4.5.3 Atelokollagen transfiziert weder DNA noch mRNA in HEK293 und HEK293T Atelokollagen ist ein Pepsin-verdautes Typ I Kollagen und kann aufgrund seiner positiven Ladung mit negativ geladenen Nukleinsäuren wie DNA oder siRNA Komplexe bilden und diese in Zellen übertragen. Atelokollagen ist als Biomaterial sehr sicher einsetzbar und weder toxisch noch immunogen (145). Eine Transfektion von siRNA und DNA über diese 86 4 Ergebnisse Form der Komplexbildung wurde bereits mehrfach gezeigt (146, 147). Es wurde sowohl GFP mRNA als auch als Kontrolle GFP-DNA (pEGFP-C1) mit Atelokollagen vermengt und anschließend die Transfektion der Komplexe in HEK293 und HEK293T durchgeführt. Allerdings war keine der durchgeführten Transfektionen erfolgreich und auch diverse Abänderungen im Protokoll der Transfektion führten zu keiner Detektion der GFPFluoreszenz. 4.5.4 FuGENE kann neben DNA auch mRNA erfolgreich in HEK293T Zellen transfizieren Bei allen Transfektionsversuchen (4.5.1 bis 4.5.3) wurde als Kontrolle die Plasmid-DNA pEGFP-C1 mit FuGENE in die verwendeten Zellen HEK293T, HEK293, PBMC, DCs und Makrophagen transfiziert. Zudem wurde auch versucht GFP mRNA mit FuGENE zu transfizieren. Dabei konnte neben der erfolgreichen Transfektion von HEK293T und HEK293 Zellen mit Plasmid-DNA auch ein Transfer der GFP mRNA in HEK293T beobachtet werden. Zwar waren nur wenige Zellen schwach positiv für GFP, aber die Fluoreszenz war dennoch klar zu erkennen (Abbildung 4.39). In den weiter genannten Zelltypen wurde keine GFP Fluoreszenz detektiert. Der Transfer von mRNA wurde für die Verwendung von FuGENE zum damaligen Zeitpunkt noch nicht beschrieben. Abbildung 4.39 GFP mRNA konnte mit Hilfe von FuGENE in HEK293T Zellen transfiziert werden. HEK293T Zellen wurden sowohl mit GFP-DNA als auch mit GFP mRNA transfiziert, indem zuvor die Nukleinsäuren mit FuGENE komplexiert wurden. Die erfolgreiche Transfektion wurde anhand der GFPFluoreszenz an einem Fluoreszenzmikroskop ermittelt. Links ist die Durchlichtmikroskop-Aufnahme zu sehen, im rechten Bild dazu ist der gleich Ausschnitt in der Fluoreszenzaufnahme dargestellt, wobei die grüne Fluoreszenz in weiß dargestellt wird. Wenige Zellen zeigten eine schwache Expression von GFP. Die Aufnahmen wurden 48 Stunden nach Transfektion erstellt. 87 4 Ergebnisse 4.6 Analyse des Sequenzverlaufs Immunantworten eines und der HIV-1-infizierten Patientenpaares, das mit demselben Virus infiziert worden war 4.6.1 Unterschiedliche Erkennung der homologen und heterologen viralen mRNA-Konstrukten Bei den HIV-1-infizierten Patienten 0225 und 0284 handelt es sich um ein homosexuelles Paar mit demselben Ausgangsvirus, wobei 0284 seinen Partner 0225 kurz nach seiner eigenen Infektion infizierte. Jedoch zeigen die beiden Patienten einen divergenten Krankheitsverlauf. So konnte das Immunsystem von 0284 das Virus sehr schnell gut kontrollieren, so dass die Viruslasten stetig sanken und inzwischen bei 80 Kopien/ml liegen. Patient 0225 hingegen konnte die Replikation des Virus von selbst nicht eindämmen, wodurch schon frühzeitig wegen einer persistierenden hohen Virämie eine ART eingeleitet wurde. Von beiden Patienten standen uns nach Klonierung die autologen gag und nef Gene in Form von mRNA zur Verfügung. Ob das eng verwandte Virus des Partners vergleichbare CTL-Antworten induziert wie das autologe Virus, sollte mit Hilfe der mRNA-Elektroporation überprüft werden. PBMC des Patienten 0225 wurden zum einen mit den drei verschiedenen gag mRNAs (autolog, 0284 und SF2) sowie zum anderen mit den drei verschiedenen nef mRNAs (autolog, 0284 und SF2) elektroporiert und anschließend in einen γ-IFN ELISPOT Assay eingesetzt. Ebenso wurde bei Patient 0284 verfahren. Unbehandelte PBMC, mock-elektroporierte und GFP mRNA- elektroporierte PBMC dienten stets als Kontrolle und führten zu keiner γ-IFN Sekretion. In Abbildung 4.40 ist das Ergebnis dieser Analyse dargestellt. Beide Patienten wiesen, wie bereits unter 4.1.6 erwähnt, eine stärkere CTL-Antwort gegen das autologe Gag-Protein im Vergleich zu SF2 auf (Abbildung 4.40 obere Reihe). Allerdings differenzierten sich die Immunantworten im Hinblick auf das Virus des Partners. So zeigte Patient 0284 eine sehr starke CTL-Antwort gegen das Gag-Protein von 0225 (181 SFU/2x105 PBMC) ähnlich wie gegen das autologe Virus (252 SFU/2x105 PBMC). Zellen des Patienten 0225 erkannten dagegen das Gag-Protein seines Partners am schlechtesten (7 SFU/2x105 PBMC). Unterschiede in der Proteinexpression sind für die verschieden stark ausgelösten Immunantworten in den beiden Patienten nicht verantwortlich, da die Expression der verschiedenen Gag-Proteine um maximal 8% variierte (Daten nicht gezeigt). 88 4 Ergebnisse Für die CTL-Antworten gegen das Nef-Protein (Abbildung 4.40 untere Reihe) konnten ebenso keine einheitlichen Ergebnisse erzielt werden. In PBMC des Patienten 0225 wurde trotz der höchsten Expression des autologen Nef-Proteins (58%) eine sehr schwache T-Zellreaktion detektiert (8 SFU/2x105 PBMC). Die beste Immunantwort wurde gegen das SF2 Nef erzielt (37 SFU/2x105 PBMC) (Abbildung 4.40). Das SF2 Nef-Protein konnte in 25% der Zellen detektiert werden, während nur 8% der elektroporierten Zellen für 0284 Nef positiv waren. Daher ist unklar, ob eine vergleichbare starke CTL-Antwort durch das 0284 Nef erreicht werden hätte können, wenn die Expression des 0284 NefProteins stärker gewesen wäre. In PBMC des Patienten 0284 hingegen löste das autologe Nef-Protein die stärkste Immunantwort aus (33 SFU/2x105 PBMC), die Immunreaktionen gegen SF2 Nef und 0225 Nef waren trotz höherer oder gleicher Expression deutlich geringer (Abbildung 4.40). Abbildung 4.40 Virale Gene, welche unterschiedlich sind, jedoch dasselbe Ausgangsvirus haben, lösten nicht immer stärkere Immunantworten aus als Referenzviren. Die Patienten 0225 und 0284 haben dasselbe Ausgangsvirus. PBMC von Patient 0225 und 0284 wurden jeweils mit SF2 nef, 0225 nef und 0284 nef mRNA sowie mit SF2 gag, 0225 gag und 0284 gag mRNA elektroporiert. Nach vier Stunden wurden die Zellen in einem γ-IFN ELISPOT Assay inkubiert, um die T-Zellantworten gegen die verschiedenen mRNA5 kodierten Proteine zu messen. Dargestellt sind die SFU/2x10 PBMC. Die obere Reihe zeigt die T-Zellantworten gegen die Gag-Proteine, die untere die T-Zellantworten gegen die Nef-Proteine (linke Spalte: 0225, rechte Spalte: 0284). 89 4 Ergebnisse Somit kann festgehalten werden, dass die T-Zellantworten auf eng verwandte Viren, die dasselbe Ausgangsvirus haben, in Patienten sehr unterschiedlich sein können und nicht unbedingt zu einer besseren Immunreaktion führen als Referenzviren. 4.6.2 Die viralen autologen Sequenzen erklären die detektierten Immunantworten Weshalb die verschiedenen mRNA-kodierten HIV-1-Proteine zu den kontroversen Immunantworten in den beiden Patienten führten, sollte durch eine vergleichende Analyse der entsprechenden viralen Sequenzen geklärt werden. Patient 0284 zeigte die stärkste Immunreaktion gegen das autologe Gag-Protein, gefolgt von der Immunreaktion gegen das Gag-Proteins seines Partner (0225) und die schwächste Reaktion wurde gegen SF2 Gag detektiert (Abbildung 4.40). In Gag befinden sich viele CTL-Epitope, welche durch HLA-Moleküle des Patienten 0284 präsentiert werden können. Die Mehrheit weist keine bis wenige Mutationen auf (Abbildung 4.41 A). Das HLA-A*02-restringierte Epitop FLGKIWPSHK (AS 435-444) (123) zeigt in der Sequenz des SF2-Proteins einen H-zu-Y Austausch an Position neun. Ebenso findet sich in der SF2 Gag Sequenz ein E-zu-D Austausch an Position fünf des HLA-B*57restringierten Epitops QASQEVKNW (AS 310-318) (120). Die Sequenz des HLA-A*02restringierten Epitops SL9 SLYNTVATL (AS 77-85) (27) unterscheidet sich sowohl in der SF2 Gag Sequenz als auch in der 0225 Gag Sequenz. Zusätzlich zu dem SL9-Epitop weisen auch die Epitope EMMTACQGV (HLA-A*02, AS 347-355) (148), ACQGVGGPGHK (HLA-A*11, AS 351-361) (149) und TLYCVHQR (HLA-A*11, AS 84-91) (150) in der Sequenz von 0225 Aminosäuren-Austausche auf. Insbesondere die R-zu-N Substitution an Position acht des Epitops TLYCVHQR in der Sequenz von 0225 führte wahrscheinlich zum Verlust des Epitops und trug damit zur geringeren T-Zellantwort auf 0225 Gag bei. Somit unterscheiden sich die Sequenzen von SF2 und 0225 zur Sequenz von 0284 in sechs CTL-Epitopen, welche vermutlich durch CTLs von 0284 schlechter erkannt werden wie die autologen Varianten. Auch in der autologen Sequenz von 0284 findet sich im Epitop TSTLQEQIGW (HLA-B*57, AS 242-251) (120) eine CTL Fluchtmutante und zudem eine Prozessierungsmutante vor dem CTL-Epitop ISPRTLNAW (HLA-B*57, AS 149-157) (151). Da die Immunantwort gegen das autologe Gag-Protein dennoch am stärksten war, kann man vermuten, dass der Patient weitere, für seine Sequenz spezifische Epitope erkannte. Patient 0225 zeigte die stärkste Immunreaktion gegen sein autologes Gag-Protein. Die Gag-Proteine von SF2 und seinem Partner 0284 induzierten deutlich geringere CTL- 90 4 Ergebnisse Antworten, wobei 0284 Gag die schwächste T-Zellreaktion in PBMC von 0225 auslöste. Im Alignment der Gag Sequenzen ist ersichtlich, dass bei Patient 0225 aufgrund seiner HLA-Allele weniger publizierte CTL-Epitope in Gag vorliegen als bei Patient 0284 (Abbildung 4.41 A). Außerdem weisen die Epitope SL9 SLYNTVATL (HLA-A*02, AS 7785) (27) und KL14 KIRLRPGGKKKYKL (HLA-A*03, AS 18-31) (116) Mutationen in SF2 Gag und 0284 Gag auf und sind so vermutlich für die schlechtere Erkennung der entsprechenden Proteine verantwortlich. Das autologe Nef-Protein bewirkte eine stärkere T-Zellreaktion in Zellen des Patienten 0284 als das SF2 Nef bzw. das Nef-Protein seines Partners 0225 (Abbildung 4.40). Bei einem Vergleich der Aminosäurensequenzen der verschiedenen Nef-Proteine konnten in drei Epitopen Unterschiede ermittelt werden. Die Epitope AITSSNTAA (HLA-A*02, AS 5260) (152), HTQGYFPDWQ (HLA-B*57, AS 126-135) (153) und AFHHMAREL (HLA-A*02, AS 200-208) (113) haben in der autologen Sequenz von 0284 eine veränderte Aminosäuren-Abfolge als in den beiden heterologen Proteinen. Wenn es sich bei diesen Veränderungen um Fluchtmutationen gehandelt haben sollte, beweist die stärkere Erkennung der autologen Variante, dass das Immunsystem in der Lage ist, neue CTLSpezifitäten zu generieren, welche spezifisch die mutierten Varianten erkennen können. Für Patient 0225 konnte eine sehr geringe Immunreaktion gegen sein autologes Nef-Protein detektiert werden, wohingegen SF2 Nef und 0284 Nef stärkere CTLAntworten induzierten (Abbildung 4.40). Eine vergleichende Analyse der Aminosäurensequenzen offenbart im HLA-A*03-restringierten CTL-Epitop QVPLRPMTYK (AS 83-92) (118) einen T-zu-N Austausch an Position acht in der autologen Sequenz (Abbildung 4.42 A). Es handelt sich dabei um eine CTL Fluchtmutante, welche zur verminderten Erkennung der autologen Nef-Variante führte. Auch die beiden Epitope AVRERMRRT (AS 15-23) (154) und AFHHMAREL (AS 200-208) (113), welche über die HLA-Allele B*07 bzw. A*02 präsentiert werden, beinhalten Mutationen in der autologen Sequenz (Abbildung 4.42 A). Da SF2 Nef und 0284 Nef weder in den genannten Epitopen noch in anderen CTL-Epitopen des Patienten 0225 Aminosäuren-Austausche aufweisen (Abbildung 4.42 A), lösten die beiden heterologen Proteine stärkere T-Zellreaktionen aus als das autologe Protein. Folglich deckte der Vergleich der Proteinsequenzen Unterschiede in CTL-Epitopen auf, welche die divergente Erkennung der HIV-1-Proteine der Patienten 0225 und 0284 erklären könnten. 91 4 Ergebnisse Abbildung 4.41 Alignment der Aminosäurensequenzen des Gag- und des Nef-Proteins von SF2, 0225 und 0284 offenbart in den betreffenden CTL-Epitopen der beiden Patienten CTL Fluchtmutationen. Die Alignments und die Consensus-Sequenz wurden mit der Vector NTI Software erstellt. Grau unterlegte Bereiche stellen Aminosäuren dar, die zwischen den drei Sequenzen unterschiedlich sind. CTL-Epitope zum HLA-Typ von Patient 0225 sind grün umrandet, CTL-Epitope zum HLA-Typ von Patient 0284 sind blau umrandet. Rot umrandet wurden CTL-Epitope, welche von beiden Patienten präsentiert werden können und HLA-A*02 oder HLA-Cw*07 restringiert sind. A: Alignment der Aminosäurensequenzen der verwendeten gag mRNAs. B: Alignment der Aminosäurensequenzen der verwendeten nef mRNAs. 0225 HLA-I: A*02, A*03, B*07, B*18, Cw*05, Cw*07; 0284 HLA-I: A*02, A*11, B*39, B*57, Cw*06, Cw*07 92 4 Ergebnisse 4.6.3 In Patient 0225 mutierte das Virus im selben Zeitraum deutlich mehr als in Patient 0284 Da sich trotz desselben Ausgangsvirus erhebliche Unterschiede in den viralen Gag und Nef Sequenzen der beiden Patienten 0225 und 0284 feststellen ließen (Abbildung 4.41 A und B), stellte sich die Frage, welche Mutationen bereits im Ausgangsvirus vorhanden waren und welche sich erst in einen der beiden Probanden entwickelt haben. Daher wurde aus Plasmaproben der Patienten, welche zum Zeitpunkt der Erstdiagnose (ED) im August 2006 aserviert wurden, virale RNA isoliert und diese nach spezifischer cDNASynthese und PCR sequenziert. Es konnte dabei die vollständige Gag Sequenz von 0284 und die vollständige Nef Sequenz von 0225 erhalten werden. Für Gag 0225 fehlen die Aminosäuren 44 bis 148 und für Nef 0284 die ersten 76 Aminosäuren. In diesen Bereichen wurden mehrere Sequenzen gleichzeitig detektiert, auch die Verwendung von verschiedenen Primern bei der Sequenzierreaktion zeigte keinen Erfolg. In den Gag Sequenzen von 0225 werden viele Ungleichheiten zwischen den beiden Zeitpunkten deutlich (Abbildung 4.42 A). So können bereits zu Beginn der Sequenz Mutationen identifiziert werden, welche durch Selektionsdruck ausgelöst durch CTLs entstanden sind. Die Aminosäure 30 ist ein Beispiel. An vielen anderen Positionen in Gag kann gut verfolgt werden, welche Viren sich in diesem Patienten manifestierten. So kamen zum Zeitpunkt der Erstdiagnose Viren vor, welche als Aminosäure 240 entweder ein Glycin (G) oder ein Arginin (R) trugen. Da sich in der späteren Sequenz nur noch Viren mit Glycin finden, haben sich diese Viren durchgesetzt. Viren mit Isoleucin (I) an Position 282 konnten sich gegen Viren mit Threonin (T) behaupten und die Aminosäure Asparagin (N) an Position 305 konnte sich gegen Threonin (T) manifestieren (Abbildung 4.42 A). Vergleicht man nun die Gag Sequenz von 0284, welche vom Zeitpunkt der Erstdiagnose stammt, mit der Sequenz, die der mRNA entspricht, lassen sich wenige Differenzen ermitteln. Nur 14 Aminosäuren haben sich zwischen den beiden Zeitpunkten verändert (Abbildung 4.42 B). Beispielsweise finden sich bei Erstdiagnose sowohl Viren, die ein Threonin (T) an Aminosäureposition 242 tragen und solche mit einem Asparagin (N), wie es in der mRNA-Sequenz enthalten ist. Da die Aminosäure in einem CTL-Epitop liegt, fand vermutlich ein Selektionsdruck durch CTLs statt, der dann in einer Fluchtmutante resultierte. Die Aminosäure 338 lautete zum Zeitpunkt der Erstdiagnose noch Glycin (G) und mutierte zu Arginin (R), ebenso die Aminosäure 377, welche von Valin (V) zu Methionin (M) mutierte. Ursprünglich fanden sich zunächst Viren, die an Aminosäureposition 396 ein Arginin (R) besaßen. Letztendlich hat sich die Variante mit Lysin (K) durchgesetzt (Abbildung 4.42 B). 93 4 Ergebnisse Wie Gag zeigte auch der Sequenzvergleich für Nef von Patient 0225 sehr viele Differenzen zwischen den beiden Zeitpunkten (Abbildung 4.43 A). Neben vielfach vorkommenden Mutationen, die zu veränderten Aminosäuren führten, entwickelte das Virus auch eine Insertion von zehn Aminosäuren (Abbildung 4.43 A). Dass es sich bei dem T-zu-N Austausch im Epitop QK10 (AS 83-93) um eine CTL Fluchtmutante handelt, wird durch das Fehlen dieser Variante zu Beginn der Infektion bestätigt. Die autologe Sequenz des Epitops AFHHMAREL (AS 200-208) war zum Ausgangszeitpunkt bereits vorhanden. Im CTL-Epitop AIRERMRRA (AS 15-23) jedoch zeigen sich deutliche Veränderungen. Das Nef-Protein des Virus hat sich in Patient 0284 zwischen den beiden Zeitpunkten kaum verändert. In dem Sequenz-Abschnitt, der uns zu Verfügung stand, konnten nur sechs mutierte Aminosäuren beobachtet werden (Abbildung 4.43 B). Das HLA-A*02restringierte CTL-Epitop AFHHMAREL (AS 194-202) wurde dabei sehr stark mutiert, insgesamt drei Aminosäuren-Austausche fanden statt, die letztendlich zum Verlust des Epitops führten. Durch diese Daten wird klar demonstriert, dass sich dasselbe Ausgangsvirus in zwei HIV-1-Infizierten sehr unterschiedlich entwickeln kann. Das HI-Virus hat sich in den Genen gag und nef in Patient 0225 stark verändert, in Patient 0284 im selben Zeitraum dagegen kaum. Da die beiden Patienten einen divergenten Krankheitsverlauf aufzeigen, wird erneut deutlich, dass nicht nur das Virus selbst, sondern auch Faktoren wie das eigene Immunsystem entscheidend zur Progression der Erkrankung beitragen. 94 4 Ergebnisse Abbildung 4.42 Alignment der Aminosäurensequenzen des Gag-Proteins von 0225 und 0284 zum Zeitpunkt der Erstdiagnose (ED) und nach ca. 2 Jahren, welche den mRNA-Sequenzen entsprechen. Die Alignments wurden mit der Vector NTI Software gebildet. Grau unterlegte Bereiche stellen Aminosäuren dar, die zwischen den Gag Sequenzen unterschiedlich sind. Aminosäuren, welche mit X in der Erstdiagnose auftauchen, sind in den beiden Möglichkeiten für X aufgeschlüsselt. A: Alignment der Gag Sequenzen des Patienten 0225. Es konnte keine vollständige Sequenz erhalten werden, so dass der Bereich zwischen den Aminosäuren 44-148 fehlt und mit X als Platzhalter dargestellt ist. Hier ist kein Vergleich der Sequenzen zu den verschiedenen Zeitpunkten möglich. B: Alignment der Gag Sequenzen des Patienten 0284. 95 4 Ergebnisse Abbildung 4.43 Alignment der Aminosäurensequenzen des Nef-Proteins von 0225 und 0284 zum Zeitpunkt der Erstdiagnose (ED) und nach ca. 2 Jahren, welche den mRNA-Sequenzen entsprechen. Die Alignments wurden mit der Vector NTI Software gebildet. Grau unterlegte Bereiche stellen Aminosäuren dar, die zwischen den Nef Sequenzen unterschiedlich sind. Aminosäuren, welche mit X in der Erstdiagnose auftauchen, sind in den beiden Möglichkeiten für X aufgeschlüsselt. A: Alignment der Nef Sequenzen des Patienten 0225. B: Alignment der Nef Sequenzen des Patienten 0284. Es konnte keine vollständige Sequenz erhalten werden, so dass die ersten 76 Aminosäuren fehlen und mit X als Platzhalter dargestellt sind. Hier ist kein Vergleich der Sequenzen zu den verschiedenen Zeitpunkten möglich. 96 4 Ergebnisse 4.7 Interaktion zwischen HIV-1-spezifischen mRNAs und zellulären miRNAs 4.7.1 Einzelne HIV-1-Gene beeinflussen das zelluläre miRNAProfil in CD4+ T-Zellen 4.7.1.1 Durch Deep Sequencing konnten zelluläre miRNAs identifiziert werden, deren Expressionsstärke von einzelnen HIV-1-Genen verändert wird Bisherige Studien zur Interaktion von zellulären miRNAs und HIV-1 haben die Veränderung des miRNA-Profils meist in Zelllinien nach Transfektion mit einem HIV-1 NL4-3 Virusisolat oder in PBMC von HIV-1-infizierten Patienten untersucht (69, 155). In dieser Arbeit sollte nun eruiert werden, ob und in welcher Weise einzelne, virale Gene einen Einfluss auf das zelluläre miRNA-Muster haben. Da es sich bei CD4+ T-Zellen um die Zielzellen des HI-Virus handelt, sollten diese zur Analyse verwendet werden. Zu diesem Anlass wurden PBMC eines gesunden Spenders isoliert und daraus die CD4+ TZellen selektioniert. Die gewonnenen CD4+ T-Zellen wurden mit GFP mRNA, HIV-1 nef sowie HIV-1 gag mRNA elektroporiert. Es wurden dabei zwei nef mRNAs aus verschiedenen Patienten mit unterschiedlichem Krankheitsverlauf verwendet. Patient 0225 konnte das Virus nicht kontrollieren und wurde daher mit ART behandelt, wohingegen Patient 0284 eine gute Kontrolle der Infektion zeigte und seit sechs Jahren keine Therapie benötigt. Durch die Verwendung dieser beiden nef mRNAs sollte ermittelt werden, ob HIV-1-Gene von verschiedenen Probanden miRNAs unterschiedlich beeinflussen können und somit eine Rolle in der Krankheitsprogression spielen. 24 Stunden nach Elektroporation wurde ein Aliquot der Zellen zur Detektion der Proteinexpression verwendet, während die restlichen Zellen geerntet wurden und daraus die Isolierung der gesamten, zellulären RNA erfolgte. Es konnte in allen Elektroporationen eine deutliche Proteinexpression detektiert und genügend RNA isoliert werden (Tabelle 4.9). Zudem wurde die Qualität der RNA mit Hilfe des Agilent Bioanalyzer überprüft und jeweils ein RIN-Wert von über 8 erzielt, was auf eine sehr gute RNA-Qualität hinweist. 97 4 Ergebnisse Tabelle 4.9 Übersicht der generierten Bibliotheken für das Deep Sequencing. Insgesamt wurden vier Bibliotheken generiert, welche GFP bzw. HIV-1-Proteine exprimierten. Die isolierte RNA war ausreichend konzentriert und von guter Qualität. Expression des Proteins [%] 82 56 17 68 Probe GFP 0225 Nef 0284 Nef 0225 Gag Konzentration der isolierten RNA [ng/µl] 185,5 207,5 233 159 Gesamtmenge an RNA [µg] 9,6 10,7 8,2 6 RIN 9,6 8,6 9,2 8,9 Das Deep Sequencing konnte für alle Proben erfolgreich durchgeführt werden. In drei von vier Proben wurden mehr als 15 Mio. Sequenzen detektiert und die Mehrzahl war 18-26 Nukleotide lang (Abbildung 4.44), so dass es sich dabei um miRNAs handelt. Diese wurden in jeder Probe nach ihrer Häufigkeit sortiert und anschließend wurden die Daten mit der Datenbank des „miRanalyzers“ abgeglichen, um die erhaltenen Sequenzen bekannten miRNAs zuzuordnen. Die Anzahl der erhaltenen Sequenzen für jede miRNA wurde prozentual auf die insgesamt detektierten miRNA-Sequenzen umgerechnet. Daraufhin wurden in jeder Probe nur miRNAs weiter analysiert, bei welchen die Anzahl der gelesenen Sequenzen über 0,05% der insgesamt erhaltenen miRNA-Sequenzen liegt. Die Elektroporation mit GFP mRNA diente als Kontrolle, so dass die Expression der verschiedenen miRNAs in den HIV-1 mRNA-elektroporierten CD4+ T-Zellen mit der Expression in den GFP mRNA-elektroporierten Zellen verglichen wurde. Somit konnte eine Zu- oder Abnahme der Expression von einigen miRNAs nach Elektroporation mit HIV-1 mRNA im Vergleich zu GFP gemessen werden, wobei die Unterschiede sehr gering waren. Es wurden 14 miRNAs detektiert, welche ihre Expression nach Elektroporation mit HIV-1 mRNA im Vergleich zu GFP mRNA modifizieren. Allerdings induzierten alle drei verschiedenen HIV-1 mRNAs Veränderungen in die gleiche Richtung (Tabelle 4.10 A). Hierbei könnte es sich um miRNAs handeln, die eine allgemeine Rolle in der viralen Immunantwort spielen und das zelluläre miRNA-Profil in ähnlicher Weise manipulieren. Ein verändertes Expressionsprofil der HIV-1 mRNA-elektroporierten CD4+ T-Zellen im Vergleich zu GFP mRNA-elektroporierten T-Zellen wurde bei 20 weiteren miRNAs nachgewiesen, jedoch gingen die Expressionsunterschiede der miRNAs zwischen den diversen HIV-1-Genen in unterschiedliche Richtungen (Tabelle 4.10 B). In den meisten Fällen resultierte die Elektroporation mit 0284 nef mRNA in einer verringerten Expression der jeweiligen miRNA im Vergleich zur Elektroporation mit GFP mRNA, wohingegen 0225 nef oder 0225 gag mRNA zu einer erhöhten Expression führten oder keinen Effekt zeigten. Dies würde darauf hinweisen, dass HIV-1-Gene aus Patienten mit unterschiedlichem Krankheitsverlauf das miRNA-Profil divergent beeinflussen können. Die Ergebnisse sind in Tabelle 4.10 A und B zusammengefasst. 98 4 Ergebnisse Abbildung 4.44 Darstellung der erhaltenen Sequenzgrößen (x-Achse) gegen ihre Häufigkeit in der Gesamtzahl (y-Achse) beim Deep Sequencing der vier Bibliotheken. Die Mehrzahl der sequenzierten Fragmente ist zwischen 18 und 25 Nukleotide lang, was den miRNAs entspricht. Des Weiteren sind 5% der Sequenzen ca. fünf Nukleotide lang, wobei es sich um degradierte Fragmente handelt. Zudem wurden auch Fragmente mit einer Länge von 28 bis 30 Nukleotiden sequenziert, die den piRNAs (piwi interacting RNA) entsprechen. GWO-1: GFP mRNA, GWO-2: 0225 nef mRNA, GWO-3: 0225 gag mRNA, GWO-4: 0284 nef mRNA Damit enthüllte die Analyse von CD4+ T-Zellen, welche mit HIV-1 mRNA elektroporiert wurden, dass einzelne HIV-1-Gene das zelluläre miRNA-Profil modifizieren können. Das miRNA-Muster der Zelle wurde jedoch nur geringfügig verändert. 99 4 Ergebnisse Tabelle 4.10 Veränderungen der Expression von zellulären miRNAs nach Elektroporation mit HIV-1spezifischen mRNAs. Ein Wert größer 1 bedeutet, dass diese miRNA in der jeweiligen Probe im Vergleich zu den GFP mRNA-elektroporierten Zellen überexprimiert ist. Ein Wert kleiner 1 weist auf eine Abnahme der Expression hin. Dargestellt sind nur miRNAs mit Werten größer 1,1 oder kleiner 0,9. Die Bezeichnung -3p und -5p gibt den betreffenden Strang der miRNA an. A: Die Modifikation der Expression dieser 14 miRNAs geht bei allen Proben in die gleiche Richtung. B: Die Modifikation der Expression dieser miRNAs ist zwischen den Proben unterschiedlich. Die Probe, welche im Vergleich zu den beiden anderen Proben eine andere Modifikation aufweist, ist grau hervorgehoben. In den meisten Fällen handelt es sich dabei um die Zellen, welche mit 0284 nef mRNA elektroporiert wurden. A miRNA 0225 Gag 0225 Nef 0284 Nef let-7a let-7b let-7d let-7f let-7i miR-10a-5p miR-30e-3p miR-103a-3p miR-125a-5p miR-146a-5p miR-146b-5p miR-148a-3p miR-425-5p miR-1246 1,27 1,41 1,15 1,38 1,13 1,16 1,24 1,14 0,86 0,79 0,81 1,14 0,76 0,93 1,28 1,39 1,27 1,39 1,19 1,19 1,25 1,15 0,80 0,79 0,77 1,15 0,77 0,79 1,38 1,42 1,27 1,33 1,15 1,07 1,15 1,08 1,06 0,74 0,74 1,05 0,78 0,72 B miRNA miR-19b-3p miR-21-3p miR-21-5p miR-24-3p miR-26a-5p miR-26b-5p miR-27a-3p miR-29a-3p miR-29c-3p miR-30c-5p miR-92a-3p miR-92b-3p miR-99b-5p miR-101-3p miR-141-3p miR-142-3p miR-142-5p miR-150-5p miR-342-3p miR-342-5p miR-361-3p miR-423-3p miR-423-5p miR-1307-5p 0225 Gag 1,13 1,03 1,08 0,97 1,18 1,44 1,23 1,05 1,09 0,89 0,98 0,96 1,07 1,06 1,01 1,08 1,12 0,97 1,21 0,83 0,97 0,79 0,93 1,12 0225 Nef 1,06 1,07 1,09 0,84 1,17 1,22 1,26 1,09 1,09 0,94 1,01 1,07 0,95 1,11 1,01 1,09 1,12 0,99 1,11 0,94 0,86 0,90 1,10 0,92 100 0284 Nef 0,82 0,81 0,85 0,89 1,02 0,98 0,87 0,87 0,80 1,12 1,20 1,41 1,20 0,84 0,76 0,62 0,81 1,36 1,04 1,07 0,77 0,98 1,37 0,59 4 Ergebnisse 4.7.1.2 Durch HIV-1-Proteine modifizierte miRNAs konnten teilweise in einzelnen Proben durch qRT-PCR validiert werden Zunächst wurden zu den 34 miRNAs, die eine veränderte Expressionsstärke durch einzelne HIV-1-Proteine zeigten, bekannte Funktionen und Krankheitsbilder recherchiert. Dabei wurden miRNAs identifiziert, für welche bereits eine Rolle in der HIV-1-Replikation beschrieben wurde (z.B. miR-30, -125, -150) (155, 156). Ebenso wurden miRNAs gefunden, die eine Rolle bei anderen viralen Infektionen haben (miR-27a, -141) (157, 158), die eine wichtige immunologische Funktion zu haben scheinen (z.B. let-7-Familie, miR-26a, -142, -146a, -148a) (159-162) und für die noch keine Funktion bekannt ist (z.B. miR-1307). Von diesen wurden nun interessante Kandidaten ausgewählt, deren Expression in quantitativer Real time-PCR (qRT-PCR) validiert werden sollte. Die Daten sollten sowohl in Aliquots der RNA, die für das Deep Sequencing gewonnen wurde, als auch in RNA-Proben eines zweiten Spenders erhoben werden. Die RNA des zweiten, gesunden Spenders wurde nach demselben Verfahren gewonnen wie die RNA der Deep Sequencing Proben. Die Expression der HIV-1-Proteine 24 Stunden nach Elektroporation war außer für 0225 Nef bei Spender 2 höher als bei Spender 1 (GFP: 92%, 0225 Gag: 82%, 0225 Nef: 35%, 0284 Nef: 21%; Vergleiche dazu Tabelle 4.9). Aus den gewonnenen RNA-Proben wurde zunächst spezifische cDNA zu jeder miRNA synthetisiert, welche anschließend als Matrize in der qRT-PCR diente. Die Analyse fand in Triplikaten statt. Die kleinen, nukleären RNAs RNU44 und RNU6B dienten als endogene Kontrolle für die quantitativen miRNA-Expressions-Levels. Für alle ausgewählten miRNAs konnte die qRT-PCR erfolgreich durchgeführt werden. Die Triplikate zeigten kaum Abweichungen voneinander. Der Vergleich der miRNAExpression bezog sich auf die RNA-Proben der GFP mRNA-elektroporierten CD4+ T-Zellen, so dass diese auf den Wert 1 gesetzt wurden. Die Ergebnisse sind in Abbildung 4.45 dargestellt. Es wurde deutlich, dass sich die Resultate der Deep SequencingAnalyse nur bei einigen, wenigen miRNAs bestätigen ließen. So konnten die Expressionsveränderungen von let-7b, miR-148a, miR-150 und miR-342 nicht verifiziert werden. Die Beobachtungen der Deep Sequencing-Analyse von miR-26b, -142-5p und -1307 konnten nur in einem Spender bestätigt werden (Abbildung 4.45 C, G und L). Die Ergebnisse der restlichen miRNAs konnten zumindest in einzelnen Proben in beiden Spendern validiert werden. So wurde eine Expressionszunahme von let-7a im Deep Sequencing nach Elektroporation der drei HIV-1-spezifischen mRNAs detektiert. In der qRT-PCR konnte dieser Anstieg für die Probe 0225 Gag in beiden Spendern und für die beiden Nef-Proben in jeweils nur einem Spender bestätigt werden (Abbildung 4.45 A). Ähnlich verhielt es sich bei let-7f, miR-27a, miR-30c und miR-146a (Abbildung 4.45 B, D, 101 4 Ergebnisse E und H). In drei von diesen vier miRNAs konnte stets das Resultat in der Probe 0284 Nef validiert werden. Für eine miRNAs konnte das Ergebnis der Deep Sequencing-Analyse vollständig bestätigt werden. Das Nef-Protein von 0284 führte in beiden Spendern zu einer Reduktion der miR-142-3p (Abbildung 4.45 F). Insgesamt waren die Differenzen von geringem Ausmaß, was mit den Beobachtungen der Deep Sequencing-Analyse übereinstimmte. Somit kann festgehalten werden, dass nicht alle gemessenen miRNA- Expressionsunterschiede im Deep Sequencing durch qRT-PCR validiert werden konnten. Dennoch zeigten einige miRNAs durch HIV-1-spezifische mRNAs eine Veränderung ihrer Expression, was zudem in zwei unterschiedlichen Spendern bestätigt werden konnte. Diese stellen interessante Kandidaten für weitere Analysen dar. 102 4 Ergebnisse 103 4 Ergebnisse Abbildung 4.45 Die Expression verschiedener zellulärer miRNAs wird durch HIV-1-Gene beeinflusst, + jedoch wurden nur einzelne Proben durch qRT-PCR validiert. CD4 T-Zellen zweier gesunder Spender wurden mit GFP mRNA, 0225 gag, 0225 nef und 0284 nef mRNA elektroporiert. Nach 24 Stunden wurden die Zellen geerntet und RNA isoliert. Nach spezifischer cDNA-Synthese für die jeweiligen miRNAs wurde eine qRT-PCR durchgeführt. Die Auswertung erfolgte nach der „ΔΔCT-Methode“, RNU6B und RNU44 dienten als endogene Kontrolle. Schwarze Säulen stellen das Ergebnis der Analyse mit RNA-Proben des Spenders 1 dar, welche auch zur Deep Sequencing-Analyse verwendet wurden. In grau sind die Ergebnisse mit RNA-Proben des Spender 2 dargestellt. Resultate des Deep Sequencing, die durch qRT-PCR bestätigt werden konnten, sind orange umrandet, wobei eine durchgezogene Linie anzeigt, dass es in beiden Spendern reproduziert werden konnte. Eine gestrichelte Linie weist darauf hin, dass es nur in einem der beiden Spender validiert wurde. 4.7.2 CTL Fluchtmutationen in miRNA-Binderegionen beeinflussen die Regulation von miRNAs Da das HI-Virus aufgrund seiner hohen Sequenzdiversität sehr variabel ist, kann es der Inhibierung mittels miRNAs durch Mutationen in der Zielsequenz der miRNA entkommen (65). In erster Linie tragen Fluchtmutationen in CTL-Epitopen zur Variabilität bei. So liegt die Bindesequenz der humanen miR-29a, welche bekannter Weise die Nef-Expression reprimiert und die Virusreplikation hemmt, im offenen Leserahmen des nef Gens (71). In dieser Zielsequenz befindet sich auch das bekannte HLA-A*24-restringierte CTL-Epitop RY10 und das HLA-B*18-restringierte CTL-Epitop YY9 (129, 153). Ob Fluchtmutationen in diesem CTL-Epitop einen Einfluss auf die Bindungsaffinität der miRNA haben, sollte nun untersucht werden. Dazu wurde der offene Leserahmen des nef Gens von SF2 und von vier autologen Patientenisolaten als 3`UTR hinter die cDNA-Sequenz des Luziferasegens in den Vektor psiCHECK3 kloniert. Anschließend wurden die verschiedenen Plasmide zusammen mit einem miR-29a-Expressionsplasmid in HEK293 transfiziert. Nach 48 Stunden wurde ein dualer Luziferase-Reporter-Assay mit den Zellen durchgeführt, bei welchem die Aktivität der Firefly-Luziferase gemessen wurde. Abbildung 4.46 zeigt die 104 4 Ergebnisse Zusammenfassung aus fünf Experimenten, die jeweils in Triplikaten durchgeführt wurden. Dargestellt ist der Mittelwert der relativen Aktivität der Firefly-Luziferase. Je niedriger die Aktivität, desto mehr wurde die Translation der Luziferase-cDNA aufgrund einer Bindung der miR-29a in nef inhibiert. Es wird deutlich, dass die verschiedenen nefs unterschiedlich stark von miR-29a reguliert werden. SF2 nef wurde von miR-29a am stärksten gehemmt (67% Luziferaseaktivität), nef von Patient 0011 dagegen zeigte die schwächste Inhibierung durch diese miRNA (84% Luziferaseaktivität). Die Nukleotidsequenz des nef Gens von 0011 offenbart in der Binderegion von miRNA-29a (Abbildung 4.47 A) vier Nukleotide, die nicht mit der Bindesequenz der miRNA übereinstimmen (sog. mismatches) und anhand der Aminosäurensequenz als CTL Fluchtmutationen identifiziert werden konnten (Abbildung 4.47 B). Allerdings wäre ein Austausch von zwei Nukleotiden für die T-zu-C Mutation an Position fünf des Epitops nicht nötig gewesen. Daher kann spekuliert werden, ob die zweite Nukleotidsubstitution in 0011 nef eine Folge der Selektion durch miR-29a ist. Die anderen nef Sequenzen weisen weniger mismatches auf. Dies liefert einen ersten wichtigen Hinweis, dass CTL Fluchtmutationen einen Einfluss auf die Regulation der viralen Genexpression durch zelluläre miRNAs haben. Abbildung 4.46 Die verschiedenen nef-Varianten wurden von der humanen miRNA-29a unterschiedlich stark inhibiert. Dargestellt ist die relative Luziferaseaktivität in % von HEK293 Zellen 48 Stunden nach Transfektion. nef Sequenzen wurden als 3`UTR nach der cDNA Sequenz des Luziferasegens kloniert und anschließend mit einem Expressionsplasmid für die miR-29a (pmiRVec-29a; gestrichelt) in HEK293 Zellen kotransfiziert. Als Kontrolle wurden die nef-Varianten mit dem pmiRVec-Vektor ko-transfiziert, der die miR-29a nicht exprimiert (grau). Als Positivkontrolle diente ein Plasmid, in welchen die ideale Bindesequenz der miR29a 3x hintereinander nach dem Luziferasegen kloniert wurde. Die geringste Inhibierung durch die miR-29a wurde für 0011 Nef detektiert, gefolgt von 0284 nef und 0225 nef. 105 4 Ergebnisse Da im Luziferase-Reporter-Assay die nef Sequenzen als 3`UTR und nicht als kodierende Sequenz vorliegen, stellte sich die Frage, ob die Expression der verschiedenen NefProteine durch die miR-29a ebenfalls unterschiedlich inhibiert wird. Deswegen wurden die fünf verschiedenen nef-Varianten in den Vektor pcDNA3flag kloniert, in welchem die nef Gene zu Protein translatiert werden und zudem mit einem N-terminalen Flag-tag versehen sind. Dies erlaubt eine einheitliche Detektion der Proteinexpression. Die verschiedenen Konstrukte wurden mit dem miR-29a-Expressionsplasmid in HEK293 ko-transfiziert und nach 48 Stunden wurden die Zellen geerntet, fixiert, permeabilisiert sowie mit einem Flagtag-spezifischen Antikörper Durchflusszytometrie. Auch intrazellulär hier gefärbt. konnte eine Die Detektion unterschiedliche erfolgte über Inhibierung der verschiedenen Nef-Proteine durch die humane miR-29a beobachtet werden (Abbildung 4.48). Die Expression des SF2 Nef-Proteins wird am stärksten durch die miR-29a reduziert (73,5 MFI), was mit den Daten der Luziferase-Reporter-Assays übereinstimmt. Allerdings konnte die schwache Inhibierung von 0011 Nef im Luziferase-Reporter-Assay nicht bestätigt werden, denn hier zeigte miR-29a den geringsten Effekt bei dem NefProtein von 0143 (90,6 MFI). Die Sequenz von 0143 nef weist zwei mismatches zur idealen Bindesequenz von miR-29a auf (Abbildung 4.47 A), wovon nur der erste in einer F-zu-L CTL Fluchtmutation im RY10-Epitop resultiert (Abbildung 4.47 B). Der Versuch wurde dreimal durchgeführt. Es konnte jedoch für 0225 Nef kein Ergebnis erzielt werden, da trotz erfolgreicher Klonierung keine Expression des Proteins nachweisbar war. Auch in einer zur Kontrolle durchgeführten Western-Blot Analyse von Herrn Dr. J. Wittmann konnte kein 0225 Nef-Protein detektiert werden (Daten nicht gezeigt). Abbildung 4.47 Alignments der Nukleotid- und Aminosäuren-Sequenzen aus dem Bereich der miR-29a Binderegion offenbart, dass 0011 und 0143 nef mismatches im Vergleich zur idealen Bindesequenz der miR-29a haben, die CTL Fluchtmutationen sind. Die Alignments wurden mit der Software Vector NTI generiert. Grau unterlegte Nukleotide bzw. Aminosäuren unterscheiden sich von den anderen Sequenzen. A: Targetsequenz der miRNA und der jeweilige Abschnitt in den nef Sequenzen auf Nukleotidebene. 0143 nef zeigt zwei mismatches, 0011 sogar vier. Alle anderen haben einen mismatch (letztes Nukleotid). B: Nef Sequenzen im Bereich des CTL-Epitops RY10 auf Aminosäurenbasis. Nef von 0143U weist zwei CTL Fluchtmutanten auf, wobei die Y-zu-C Mutation nicht mehr in der Binderegion der miR-29a liegt. Nef von 0011 zeigt eine CTL Fluchtmutante in diesem Epitop. 106 4 Ergebnisse Die humane miRNA-29a kann somit verschiedene nef-Varianten unterschiedlich regulieren. Die Analysen zeigten, dass CTL Fluchtmutanten in der bekannten Binderegion der miR-29a die Erkennung verringern können. Luziferase-Reporter-Assays mit den verschiedenen nef-Varianten als 3`UTR detektierten, dass 0011 nef am wenigsten durch die miR-29a inhibiert wird. Dagegen wurde auf Proteinebene nach intrazellulärer Färbung 0143 Nef als die Variante identifiziert, die kaum durch miR-29a inhibiert wird. Obwohl die beiden unterschiedlichen Assays keine übereinstimmenden Ergebnisse aufweisen, kann dennoch eine Gemeinsamkeit festgehalten werden. Die beiden nef Gene 0011 und 0143 weisen im Gegensatz zu den anderen verwendeten nef Genen CTL Fluchtmutanten auf, welche zur verminderten Hemmung durch die miR-29a beitragen. Abbildung 4.48 Die verschiedenen nef-Varianten wurden auf Proteinebene durch die humane miR-29a unterschiedlich stark inhibiert. Dargestellt sind die MFI-Werte (mean fluorescence intensity) nach intrazellulärer Färbung 48 Stunden nach Transfektion von HEK293 Zellen. Die verschiedenen nef Gene wurden in den Vektor pcDNA3flag kloniert. Die erhaltenen Plasmide wurden mit dem Expressionsplasmid der humanen miR-29a (pmiRVec-29a; gestrichelt) in HEK293 Zellen ko-transfiziert. Als Kontrolle wurden die nefVarianten mit dem pmiRVec-Vektor ko-transfiziert, der die miR-29a nicht exprimiert (grau). Nach 48 Stunden wurden die Zellen permeabilisiert und mit einem Flag-tag-spezifischen Antikörper intrazellulär gefärbt. Die Analyse erfolgte über Durchflusszytometrie. Die geringste Inhibierung durch miR-29a konnte für 0143 Nef beobachtet werden. 107 5 Diskussion 5.1 Elektroporation von PBMC mit HIV-1-spezifischen mRNAs Mehrere Studien haben bereits die Elektroporation von PBMC mit mRNA beschrieben, um Tumor-spezifische oder CMV-spezifische T-Zellen zu stimulieren oder zu monitoren (97, 163-165). In der vorgelegten Dissertation konnte nun gezeigt werden, dass die Elektroporation von PBMC mit HIV-1-spezifischer mRNA eine effiziente Methode ist, um T-Zellantworten gegen HIV-1 zu bestimmen. Außerdem konnte zum ersten Mal gezeigt werden, dass die entwickelte Technik für die Analyse der T-Zellantworten gegen autologe HIV-1-Proteine geeignet ist. Dies erlaubt die Immunogenität von viralen Genen, die aus unterschiedlichen Patienten und Laborstämmen isoliert wurden, in einem einfachen Testsystem zu vergleichen. Um Immunantworten gegen autologe HIV-1-Proteine zu detektieren, wurde zunächst die virale RNA aus neun verschiedenen HIV-1-infizierten Patienten isoliert, kloniert und über in vitro Transkription HIV-1-spezifische mRNAs gewonnen. Es konnten sechs autologe gag mRNAs, sieben autologe nef mRNAs und zwei autologe env gp120 mRNAs generiert und erfolgreich zur Elektroporation in PBMC eingesetzt werden. Darüber hinaus stand autologe nef mRNA von einem weiteren Patienten zur Verfügung. Die durchgeführten Versuche zeigten, dass mRNA-elektroporierte PBMC die mRNA-kodierten HIV-1-Proteine synthetisieren. Bisherige Studien dokumentierten eine Expression der mRNA-kodierten Proteine 24 Stunden nach Elektroporation (163). In unseren Analysen konnte bereits nach vier Stunden eine deutliche Expression von GFP und der mRNA-kodierten HIV-1-Proteine über HIV-1-spezifische Antikörper und anschließender Durchflusszytometrie nachgewiesen werden. Dennoch war die Fraktion der GFP-exprimierenden Zellen nach GFP mRNA-Elektroporation höher im Vergleich zur Fraktion der mRNA-kodierten HIV-1Proteine Gag und Nef. Dies liegt vermutlich vor allem an der sensitiveren Detektion der GFP-Fluoreszenz im Gegensatz zu den HIV-1-Proteinen, welche über eine indirekte Färbung mit spezifischen, monoklonalen Antikörpern nach Fixierung und Permeabilisierung der Zellen analysiert werden. Des Weiteren wurde eine effizientere Expression der HIV-1 Gag-Proteine im Vergleich zu den HIV-1-Proteinen Nef oder Env gp120 beobachtet. Die fehlende Expression des autologen Env gp120 in allen durchgeführten Versuchen könnte daran liegen, dass der verwendete monoklonale Antikörper das variable env Gen nicht erkannte (166). Der verwendete Antikörper bindet in den ersten 204 Aminosäuren des gp120-Proteins, die exakte Sequenz des Epitops ist 108 5 Diskussion allerdings nicht bekannt, so dass nicht ausgeschlossen werden kann, dass das Epitop in einer hoch variablen Region liegt und nicht in einen der konservierten Bereiche. Die geringere Expression von Nef im Vergleich zu Gag wird vermutlich nicht durch toxische Effekte des Nef-Proteins oder der nef mRNA verursacht, da im Hinblick auf die Zellviabilität nach Elektroporation keine Unterschiede zwischen Gag- oder Nefkodierender mRNA festgestellt wurden. Titrationsanalysen zur mRNA-Elektroporation zeigten, dass die Verwendung von höheren Mengen an nef mRNA zu einer ähnlichen Proteinexpression wie bei Gag führt. Dies bestärkt die Hypothese, dass die weniger sensitive Detektion des Nef-Proteins von einer geringeren Affinität des Nef-spezifischen Antikörpers im Vergleich zum Gag-spezifischen Antikörper herrührt. Aber auch eine höhere Stabilität oder eine potentiell stärkere Translationsrate der gag mRNA im Vergleich zur nef mRNA sowie ein höherer Umsatz des Nef-Proteins sind mögliche Erklärungen für die effizientere Expression des HIV-1 Gag-Proteins nach mRNA Elektroporation. Dies wurde allerdings in dieser Arbeit nicht weiter untersucht. Eine Optimierung der Codon-Verwendung bei der Translation oder die Einführung von alternativen Startcodons könnte eventuell zu einer effizienteren Expression der mRNAkodierten Nef-Proteine führen (167, 168). Wir und andere Arbeitsgruppen haben bereits über die erfolgreiche mRNA-Elektroporation von B-Zellen und Dendritischen Zellen für die Detektion von HIV-1-spezifischen T-Zellen berichtet (169-172). Jedoch hat die Verwendung von B-Zellen bzw. Dendritischen Zellen als antigen-präsentierende Zellen sehr viele Nachteile. Zum einen benötigt die Anzucht und Kultivierung dieser Zellen sehr viel Zeit und Aufwand. Zum anderen führt die Verwendung von EBV-transformierten B-Zelllinien in ELISPOT Assays häufig zu starken Hintergrundaktivitäten entweder durch das Vorkommen von EBV-spezifischen oder alloreaktiven T-Zellen oder durch die unspezifische T-Zell-Stimulation durch kostimulierende Moleküle. Dagegen hat die mRNA-Elektroporation von PBMC mehrere grundlegende Vorteile. Die schnelle Expression der mRNA-kodierten Proteine in PBMC bereits vier Stunden nach Elektroporation erlaubt die Verwendung von frisch-isolierten PBMC ohne weitere Manipulation oder Vorkultivierung der Zellen. Obwohl ungefähr die Hälfte der Zellen die Elektroporation nicht überleben, können die restlichen Zellen dennoch sowohl als antigen-präsentierende Zellen als auch als antigen-spezifische Effektorzellen fungieren. Färbungen der wichtigsten Subpopulationen der PBMC zeigten, dass alle Zellpopulationen eine ähnliche Expression der mRNA-kodierten Proteine aufweisen. Zudem beeinträchtigt die Elektroporation die Verteilung der verschiedenen Zellpopulationen nicht. Die Verwendung der mRNA-elektroporierten PBMC in ELISPOT Assays demonstrierte eine klare Sekretion von γ-IFN. Damit ist die von uns synthetisierte, autologe HIV-1109 5 Diskussion spezifische mRNA in vitro immunogen, was mit den Erwartungen ausgehend von anderen Studien übereinstimmt (96, 102, 109). Mock und GFP mRNA-elektroporierte PBMC führten zu keiner Zytokinproduktion, so dass die Sekretion von γ-IFN antigen-spezifisch ist und nicht durch unspezifische Signale verursacht wird, die bei der Elektroporation entstehen können. Die Reaktion muss zudem von HIV-1-spezifischen T-Zellen induziert worden sein, da keine Ausschüttung von γ-IFN im ELISPOT nachweisbar war, wenn PBMC von HIV-negativen Individuen mit HIV-1-spezifischer mRNA elektroporiert wurden. Da die in vitro Transkription sehr teuer ist, sollten Titrationsexperimente klären, welche Mindestmenge an mRNA noch zu einer nachweisbaren Proteinexpression führt und eine antigen-spezifische Freisetzung von γ-IFN induziert. Eine durchweg exzellente und gesättigte Expression von Gag konnte bei der Verwendung von 15 µg HIV-1 gag mRNA pro Elektroporation erreicht werden. Dies kann somit als die optimale Konzentration betrachtet werden. Aber auch der Einsatz von nur 1 µg HIV-1 gag mRNA resultierte in einer Gag Expression in mehr als 10% der Zellen und konnte noch eine γ-IFN Antwort im ELISPOT Assay auslösen. Auch geringe Mengen an GFP mRNA bis zu 0,5 µg führten zu einer deutlichen Proteinexpression von GPF. Folglich ist es möglich, weniger GFP und HIV-1 gag mRNA pro Elektroporation zu verwenden, um die Kosten zu reduzieren. Da bei der Verwendung von nef mRNA der Anteil an Nef-exprimierenden Zellen meist geringer war im Vergleich zu gag mRNA, ist eine Reduktion der nef mRNAMenge zur Kostenersparnis nicht sinnvoll. Stattdessen zeigten die Daten, dass der Einsatz von mehr nef mRNA zu einer höheren Expression der Nef-Proteine führt. Ebenso ist es nicht empfehlenswert weniger env gp120 mRNA einzusetzen, da die Immunantworten bei 15 µg mRNA sehr gering waren. 5.1.1 Immunmonitoring gegen autologe Virusproteine mit HIV-1spezifischen mRNAs Da in dieser Arbeit gezeigt werden sollte, dass mRNA-elektroporierte PBMC von HIV-1infizierten Patienten zum Immunmonitoring gegen autologe HIV-1-Stämme verwendet werden können und zudem die Immunantworten gegen autologe Viren mit denen gegen Referenzviren verglichen werden sollten, wurden γ-IFN ELISPOT Analysen durchgeführt. Zunächst konnte dabei festgestellt werden, dass die elektroporierten PBMC alleine ausreichend sind, um eine γ-IFN Freisetzung durch antigen-spezifische T-Zellen zu induzieren. Die Zugabe von weiteren antigen-präsentierenden Zellen oder Effektorzellen wurde nicht benötigt, was beweist, dass die mRNA-elektroporierten PBMC simultan als Stimulatoren sowie als Effektoren im selben Assay genutzt werden können. So 110 5 Diskussion induzierten 200.000 nef mRNA-elektroporierte PBMC allein eine höhere Sekretion von γ-IFN als 100.000 nef mRNA-elektroporierte PBMC mit Zugabe von 100.000 unbehandelten PBMC. Dies liegt vermutlich an dem stärkeren, antigenen Stimulus durch die Verwendung höherer Frequenzen elektroporierter PBMC. Diese Daten demonstrieren, dass die mRNA-elektroporierten Effektorzellen funktionell sind und dass keine weiteren Effektoren in dem Assay zugesetzt werden müssen. Die Verwendung von PBMC ohne die Trennung von antigen-präsentierenden Zellen und Effektorzellen spart sowohl Zeit als auch Arbeit. Außerdem reduziert es die Anzahl der benötigten Zellen und somit die Menge an Blut, welche vom Patienten zum Immunmonitoring abgenommen werden muss. Standardmäßig werden zum Monitoring von HIV-1-spezifischen T-Zellen HIV-1Peptide, Peptid-Pools oder rekombinanten Vaccinia-Vektoren, die HIV-1-Proteine exprimieren, eingesetzt (89, 173-175). Es sollte geklärt werden, ob mRNA-elektroporierte PBMC eine ähnliche Kapazität wie diese Standardmethoden besitzen T-Zellantworten zu induzieren. Das Ausmaß der γ-IFN Antwort, welche durch die Elektroporation mit HIV-1spezifischer mRNA hervorgerufen wurde, entsprach der γ-IFN Antwort von HIV-1Peptiden. Wurde jedoch die gesamte T-Zellantwort von mehreren Peptiden eines HIV-1Proteins summiert, war die T-Zellantwort gegen das mRNA-kodierte Protein deutlich geringer. Da die exogenen Peptide im Überschuss zugegeben werden, stimmte dieses Ergebnis mit unseren Erwartungen überein. Um einzelne Peptide entsprechend auszuwählen, benötigt man den HLA-Typ des Patienten. Daher haben HIV-1-spezifische mRNAs den Vorteil, dass keine Kenntnis des HLA-Typs des Patienten nötig ist, um die Immunantwort gegen ein komplettes HIV-1-Protein zu analysieren. Zudem sind HIV-1spezifische mRNAs für diesen Zweck günstiger, denn der Einsatz von Peptidsets aus überlappenden Peptiden, die ein ganzes Protein umfassen, ist teuer und arbeitsaufwendig. Dieser Arbeitsaufwand kann zwar durch die Verwendung von PeptidPools verringert werden, aber hierbei konnten wir die sinkende Sensitivität der Detektion der Immunantworten gegen spezifische Epitope im ELISPOT Assay bestätigen (163). Die Immunantworten, welche durch die mRNA-kodierten HIV-1-Proteine ausgelöst wurden, waren vergleichbar mit den durch Peptid-Pools induzierten Immunantworten. Rekombinante Vaccinia-Viren werden häufig genutzt, um die T-Zellantwort gegen ein komplettes HIV-1-Protein zu detektieren. Sie können allerdings PBMC schlecht infizieren, so dass die geringe T-Zellantwort gegen Vaccinia-Viren in PBMC oft auf eine fehlende oder schlechte Infektion der Zellen zurückgeht. Daher war vermutlich auch die TZellantwort der mRNA-elektroporierten PBMC deutlich stärker im Vergleich zu den Vaccinia-infizierten PBMC. Deswegen wurden für eine vergleichende Analyse zwischen HIV-1-spezifischen mRNAs und rekombinanten Vaccinia-Viren B-LCL verwendet, welche sich gut mit Vaccinia-Viren infizieren lassen. Dabei führte ein SF2 Gag-exprimierendes 111 5 Diskussion Vaccinia-Virus in B-LCL trotz einer hohen Vaccinia-Dosis zu einer geringeren Gag Expression sowie zu einer geringeren Gag-spezifischen T-Zellantwort als gag mRNAElektroporation. Die SF2 Gag Expression konnte in B-LCL nicht durch den Einsatz höherer Dosen an Vaccinia gesteigert werden, da die Vaccinia-Viren auf B-LCL toxisch wirkten und eine Kompetition der Proteinsynthese zwischen Vaccinia-Proteinen und dem Gag-Protein auftrat (176). Demzufolge ist die gag mRNA-Elektroporation in B-LCL eine effizientere Methode, um Gag-spezifische T-Zellen zu detektieren, als die Verwendung von Gag-exprimierenden rekombinanten Vaccinia-Viren. Ein weiterer Vorteil der mRNAElektroporation im Gegensatz zu Vaccinia-Viren ist das geringere Infektionsrisiko für das Laborpersonal. Das vorgeschriebene S2-Labor beim Umgang mit Vaccinia-Viren wird bei der mRNA-Elektroporation nicht benötigt. Unsere Studie mit autologer HIV-1-spezifischer mRNA beweist deutlich, dass die Elektroporation von PBMC mit autologer viraler mRNA eine effiziente Methode ist, um T-Zellantworten gegen ein hoch variables Virus zu monitoren. Es wurde eine stärkere CTL-Antwort gegen die mRNA-kodierten autologen viralen Proteine in fünf von zehn mRNA-Konstrukten aus sieben Patienten nachgewiesen. Dies zeigt, dass die TZellantwort in einem erheblichen Anteil von HIV-1-infizierten Patienten unterschätzt und mit den Standardmethoden des Immunmonitorings nicht richtig quantifiziert wird. Interessanterweise konnte gegen ein autologes Gag-Protein sowie gegen ein autologes Nef-Protein eine schlechtere T-Zellerkennung als gegen die mRNA-kodierten Proteine von HIV-1 Referenzstämmen im ELISPOT gemessen werden. In diesen beiden Fällen würde die T-Zellantwort bei Verwendung der Standardmethoden überschätzt werden. Durch Sequenzvergleich wurden Mutationen in CTL-Epitopen in diesen beiden autologen Sequenzen ermittelt. Die schlechtere Erkennung des autologen Gag von Zellen des Patienten 0347 war assoziiert mit einer K-zu-R CTL Fluchtmutante im HLA-B*13restringierten Epitop p15 RI9 (RQANFLGI) (122, 177). SF2 Gag, welche die WildtypSequenz des Peptids kodiert, induzierte eine stärkere Immunantwort als autologes Gag. Dies zeigte auch die Inkubation der Zellen mit dem Wildtyp Peptid und der varianten, autologen Form des Peptids. Die schlechtere Erkennung des autologen Nef-Proteins von Zellen des Patienten 0225 war assoziiert mit einer T-zu-N CTL Fluchtmutation im HLAA*03-restringierten Epitop QK10 (QVPLRPMTYK) (118). Andere Epitope waren nicht mutiert und waren zwischen dem SF2 und autologen Nef-Protein konserviert. Daher ist zu vermuten, dass die T-Zellantwort gegen das QK10-Epitop einen wesentlichen Anteil der detektierten Nef-spezifischen Immunantwort ausmacht, was auf eine besonders effiziente Präsentation des QK10-Epitops im Vergleich zu den anderen Nef-Epitopen in diesem Patienten hinweist. 112 5 Diskussion Des Weiteren konnten die T-Zellantworten gegen die mRNA-kodierten Proteine Effekte erklären, welche nach Stimulation der PBMC mit Peptiden im ELISPOT beobachtet wurden. So wurde bei Patient 0284 eine stärkere CTL-Antwort gegen das Peptid Nef0726 als gegen das Peptid Nef11T3 detektiert, obwohl beide das HLA-Cw*07-restringierte Nef-Epitop RQDILDLWIY präsentieren (138). Allerdings findet sich in Nef07-26 eine I-zuV Variante an Position neun des Epitops. Diese existiert auch in der autologen viralen Proteinsequenz des Patienten. Da das autologe Nef-Protein eine stärkere Immunreaktion auslöste, wurde auch die Peptid-Variante, welche der autologen Sequenz entspricht, besser von CTLs erkannt als das Peptid mit der heterologen Sequenz. Bei den Patienten 0545 und 0899 induzierten die mRNA-kodierten HIV-1-Proteine keine nachweisbare T-Zellreaktion. Ebenso zeigte auch Patient 0011 trotz Proteinexpression kaum Immunantworten nach Elektroporation mit nef mRNA. Jedoch konnte eine T-Zellantwort durch die Stimulation der PBMC von Patient 0011 mit dem HLA-B*08restringierten, dominanten HIV-1-Peptid FL8 erreicht werden. Eine Inkubation der PBMC von den Patienten 0545 und 0899 mit HIV-1-Peptiden führte hingegen zu keiner T-Zellantwort. Dies kann darauf zurückgeführt werden, dass exogen zugeführte Peptide höhere Immunantworten induzieren als mRNA-kodierte HIV-1-Proteine. Aus den mRNAkodierten Proteinen wird eine Vielzahl von Epitopen intrazellulär prozessiert und über die HLA-Allele präsentiert. Daher binden auch Peptide an HLA-Moleküle, die eventuell keine oder nur sehr geringe Immunantworten induzieren, so dass weniger freie HLA-Moleküle für die dominanten CTL-Epitope zur Verfügung stehen und folglich die T-Zellantwort gegen dieses Epitop geringer ausfällt als wenn es als exogenes Peptid allein zugegeben worden wäre. Deshalb induzierte das Peptid Nef FL8 bei Patient 0011 eine Immunreaktion, wohingegen die beiden mRNA-kodierten Nef-Proteine kaum eine Reaktion auslösten. Dass auch Peptidstimulationen bei den Patienten 0545 und 0899 zu keiner CTL-Antwort führten, liegt vermutlich an der therapeutischen Behandlung dieser Patienten mit HAART. Denn es wurde bereits gezeigt, dass durch die Unterdrückung der Virämie die antigene Stimulation von HIV-1-spezifischen CTLs stark verringert wird, so dass die Frequenz der HIV-1-spezifischen CTL abnimmt (114). Jeder Versuch wurde mindestens zweimal durchgeführt, die Differenz der Transfektionseffizienz zwischen den zwei getesteten Zeitpunkten eines jeden Patienten lag bei maximal 10%. Daher kann von einer geringen Varianz des Assays ausgegangen werden. Die Ergebnisse der ELISPOT Analysen konnten ebenso reproduziert werden. Dennoch traten kleine Unterschiede in der absoluten Anzahl der produzierten Zytokine auf, die mit dem unterschiedlichen Status des Patienten und der PBMC erklärt werden können. Aber auch andere Faktoren, die jeden biologischen Assay beeinflussen, wie die Qualität der mRNA oder die Durchführung, sind denkbar. Im Hinblick auf die 113 5 Diskussion Transfektionseffizienz zwischen den verschiedenen Individuen kann vermutet werden, dass intra- und extrazelluläre Faktoren, wie Fettsäuren, z.B. Cholesterin, oder miRNAs, die Permeabilität der Zellmembran oder die Translation der mRNA in Protein beeinflussen können (178, 179). Solche Unterschiede können beim Immunmonitoring durch die Bestimmung der Proteinexpression kontrolliert und gegebenenfalls durch Modifikation der verwendeten mRNA-Mengen ausgeglichen werden. Wichtig ist, dass die Expression der mRNA-kodierten Proteine ermittelt wird und die heterologen und autologen Proteine im gleichen Umfang vorhanden sind. Somit kann eine gute Stabilität des entwickelten Assays konstatiert werden. Die Elektroporation von PBMC mit HIV-1-spezifischen mRNAs stellt folglich eine alternative Methode zum Immunmonitoring dar. Es werden ähnliche Ergebnisse im Vergleich zu Peptid-Pools und bessere Ergebnisse im Vergleich zu rekombinanten Vaccinia-Viren erzielt. Außerdem kann durch den Einsatz von autologen HIV-1spezifischen mRNAs auch die T-Zellantwort gegen das eigene Virus ermittelt werden, so dass es nicht mehr zu einer Unter- bzw. Überschätzung der Immunantworten kommt. 5.1.2 HIV-1-spezifische mRNAs als therapeutischer Impfstoff Da HIV-1-spezifische mRNAs in vitro immunogen sind und zur Sekretion von γ-IFN führen, stellen sie interessante Vakzin-Kandidaten dar. Durch Inkubation der HIV-1 mRNA-elektroporierten PBMC mit einem Anti-CD8 Antikörper konnte gezeigt werden, dass es sich bei den meisten γ-IFN sekretierenden Zellen um CD8+ T-Zellen handelt, jedoch wurden auch CD4+ T-Zellen stimuliert. Somit werden auch die Zielzellen einer therapeutischen Impfung durch die Elektroporation der PBMC mit HIV-1-spezifischer mRNA aktiviert. Denn bei einer therapeutischen Impfung sollen vorhandene HIV-1spezifische CTLs stimuliert werden, welche zur Erkennung und Eliminierung von infizierten Zellen beitragen. Eine Expression der mRNA-kodierten HIV-1-Proteine konnte bis zu sechs Tagen nach Elektroporation detektiert werden, wobei insbesondere in den ersten drei Tagen eine sehr gute Expression nachgewiesen wurde. Anschließend gehen viele Zellen vermutlich in Apoptose. Bei einer Impfung mit HIV-1-spezifischen mRNAs würde eine gute Expression bis drei Tage nach Verabreichung ausreichen, da in den ersten 48 Stunden die Antigenpräsentation erfolgt, welche letztendlich zur T-Zellantwort führt und somit die Wirksamkeit der Vakzine entscheidend beeinflusst. Eine Sekretion von TNF-α nach Elektroporation mit HIV-1-spezifischen mRNAs konnte nicht nur im Zellüberstand von HIV-1-infizierten Patienten, sondern auch im Zellüberstand 114 5 Diskussion von gesunden Spendern gemessen werden. Dies weist darauf hin, dass die TNF-αSekretion nicht bzw. nicht nur durch HIV-1-spezifische T-Zellen induziert wird, sondern auch durch Zellen des angeborenen Immunsystems. Bestimmte Sequenzmotive in HIV-1 können TLR7 und TLR8 triggern und darüber ein ko-stimulierendes Signal ausüben (44, 180). Es wurde beobachtet, dass die verschiedenen HIV-1-spezifischen mRNAs eine unterschiedlich starke Sekretion von TNF-α bewirken. Allerdings konnte kein Zusammenhang zwischen der Anzahl der bekannten Sequenzmotive, welche in der jeweiligen mRNA vorkommen, und den gemessenen Zytokin-Konzentrationen hergestellt werden (180). Möglicherweise gibt es noch weitere unbekannte Motive, die dieses Signal triggern können. Des Weiteren war die Menge des sezernierten TNF-α als Antwort auf ein bestimmtes HIV-1-Protein bei verschiedenen Spendern sehr unterschiedlich. Aufgrund der teilweise sehr hohen Produktion von TNF-α kann man vermuten, dass der Effekt vor allem von Monozyten ausgelöst wird und es sich hierbei um eine TLR8-Triggerung handelt (181). Bei einer Impfung hätte dies den Vorteil, dass die virale mRNA durch die Triggerung der Freisetzung von TNF-α selbst als potentes Adjuvans dient (182). Die Analyse der Immunantworten gegen autologe HIV-1-Proteine, die von mRNA kodiert werden, geben wichtige Hinweise darauf, aus welchen Proteinen eine optimale Vakzine bestehen soll. So ist bei Patienten, welche eine stärkere CTL-Antwort gegen das autologe virale Protein aufweisen, eine therapeutische Impfung mit der autologen HIV-1spezifischen mRNA sinnvoll. Wird die autologe HIV-1-spezifische mRNA schlechter erkannt, so macht die Vakzinierung mit dieser immer noch Sinn, solange CTL Fluchtmutationen, welche für die geringere Erkennung der autologen Variante verantwortlich sind, keinen Einfluss auf die Bindung des Peptids an das HLA-Molekül haben. Erst wenn Mutationen in der autologen HIV-1-spezifischen mRNA Ankerpositionen des Epitops betreffen und das Peptid nicht mehr an das HLA-Molekül binden kann, dann macht eine Impfung mit dieser Variante keinen Sinn mehr. Des Weiteren konnte bei den Analysen mit HIV-1-spezifischen mRNA bestätigt werden, dass die stärksten Immunreaktionen gegen das Gag-Protein gerichtet sind und vor allem LTNP wie Patient 0284 gute Immunantworten gegen Gag besitzen (25). Auch Nef-Proteine stimulierten CTL-Antworten, wohingegen Env nur geringe T-Zellantworten auslöste. γ-IFN ELISPOT Analysen mit PBMC von drei unterschiedlichen Patienten zeigten keine oder nur geringe T-Zellantworten gegen HIV-1 Env gp120 sowohl nach env gp120 mRNA Elektroporation als auch nach Peptidinkubation. Allerdings konnte auch bei den unterschiedlichen Versuchen mit env gp120 mRNA keine oder eine nur geringe Expression des Proteins detektiert werden. Daher sind weitere Analysen mit mehr unterschiedlichen env gp120 mRNAs nötig, um zu überprüfen, ob die von uns beobachtete geringe Immunogenität von Env Folge der geringen Expression des Env-Proteins oder der starken Variabilität von Env 115 5 Diskussion ist oder aber ob sie Audruck eines T-Zell-hemmenden Effekts oder einer geringeren KoStimulation ist. Somit decken sich die in dieser Arbeit ermittelten Daten mit Ergebnissen aus früheren Studien (183). Außerdem kann man daraus schließen, dass eine therapeutische Vakzine Gag und Nef als Antigene auf jeden Fall enthalten sollte. 5.2 Funktionelle Analysen mit HIV-1-spezifischen mRNAs HIV-1-spezifische mRNAs werden nach Elektroporation in PBMC effizient zu Protein translatiert und führen zur Sekretion von γ-IFN durch HIV-1-spezifische CTLs. Damit sie sich gut als therapeutischer Impfstoff oder zum Monitoring von T-Zellantworten eignen, müssen sie neben der Immunogenität noch weitere Eigenschaften aufweisen. Um die Kosten und den Arbeitsaufwand möglichst gering zu halten, ist es sinnvoll eine größere Menge an PBMC mit HIV-1-spezifischen mRNAs zu elektroporieren und anschließend bei -80° C zu konservieren. Bei Verabreichung der zweiten Impfdosis oder beim nächsten Besuch des Patienten, zu welchem die HIV-1-spezifische T-Zellantwort analysiert werden soll, müssten die konservierten PBMC nur aufgetaut werden und könnten so direkt im Assay eingesetzt werden. Es sollte daher getestet werden, ob dies möglich ist und ob die konservierten PBMC nach dem Auftauen noch in der Lage sind, Immunantworten zu induzieren. Dabei zeigte sich, dass die mRNA-elektroporierten PBMC kryokonserviert werden können. Der direkte Einsatz der wiederaufgetauten PBMC in γ-IFN ELISPOT Assays führte zu einer Sekretion von γ-IFN, allerdings von geringerer Stärke im Vergleich zu nicht konservierten, mRNA-elektroporierten PBMC. Die Freisetzung von γ-IFN konnte erhöht werden, wenn unbehandelte, frische PBMC hinzugegeben wurden. Van Camp et al. konnte zeigen, dass die Elektroporation von kryokonservierten und wieder aufgetauten PBMC nach Über-Nacht-Inkubation möglich ist, auch wenn die Expression der mRNA-kodierten Proteine deutlich geringer ausfällt und die Zellen somit nach dem Auftauen weniger effizient sind (163). Neben einer geringeren Effizienz der Zellen könnte die verminderte Zytokinfreisetzung auch die Folge der unterschiedlich betrachteten Zeitpunkte sein. Bei allen Versuchen wurden die mRNAelektroporierten PBMC 24 Stunden nach Elektroporation kryokonserviert. Das Immunmonitoring von frischen PBMC erfolgte vier Stunden nach Elektroporation und die Zellen inkubierten daraufhin in der Regel 40 Stunden im ELISPOT Assay. Daher könnte die Zeitverschiebung von 20 Stunden dazu beitragen, dass die Zytokinausschüttung zum Zeitpunkt der Kryokonservierung größtenteils schon stattgefunden hat und die Zellen 116 5 Diskussion daher nach dem Auftauen weniger γ-IFN produzieren. Trotzdem konnte demonstriert werden, dass mRNA-elektroporierte PBMC nach Kryokonservierung noch immunogen sind und T-Zellantworten induzieren können. Eine Wiederverwendung kryokonservierter, elektroporierter PBMC zum Immunmonitoring oder zur Vakzinierung ist daher möglich. Des Weiteren stellte sich die Frage, ob mRNA-kodierte HIV-1-Proteine funktionell aktiv sind und bekannte Funktionen der HIV-1-Proteine ausführen können. Das HIV-1Protein Nef kann in einer infizierten Zelle die Expression der Oberflächenmoleküle CD4, CXCR4 und HLA-I reduzieren (125, 127, 184). Durch Färbung mit spezifischen Antikörpern und Durchflusszytometrie nach nef mRNA-Elektroporation von PBMC konnte beobachtet werden, dass auch Nef-Proteine, welche von in vitro transkripierter nef mRNA kodiert werden, diese Funktionen vollziehen können und die Expression der genannten Moleküle verringern. Somit sind die nef mRNA-Konstrukte nicht nur immunogen, sondern auch funktionell aktiv. Eine weitere Fragestellung hinsichtlich der Funktionalität der HIV-1-spezifischen mRNAs war, ob mRNA-elektroporierte PBMC fähig sind, HIV-1-spezifische CTL Linien in vitro zu stimulieren. In der Regel erfolgt dies über die Inkubation mit Peptiden (150, 185). Die vorgestellten Ergebnisse zeigen, dass die Ko-Inkubation von unbehandelten PBMC mit PBMC, die mit HIV-1-spezifischer mRNA elektroporiert wurden, zur Aktivierung von CTL Linien führte. Somit können mRNA-elektroporierte PBMC nicht nur zum Immunmonitoring verwendet werden, sondern auch zur Generierung von CTL Linien in vitro. Allerdings werden durch die Generierung von CTL Linien über mRNA-Elektroporation zwar Proteinspezifische Linien erhalten, aber keine Epitop-spezifischen Linien. Benötigt man monoklonale Epitop-spezifische CTL Linien, ist die Standardmethode mit Peptiden vorteilhafter. Diese ist auch deutlich kostengünstiger. Des Weiteren konnte in dieser Arbeit keine spezifische Proliferation der mRNA-elektroporierten PBMC im Vergleich zu den mock-elektroporierten PBMC festgestellt werden. 5.2.1 Wirksamkeit eines Kombinationsimpfstoffes aus gag und nef mRNA Bei einer effizienten Vakzine sollte eine möglichst breite Immunantwort induziert werden, was durch die Kombination von mehreren Antigenen erreicht wird. So wurde auch in den jüngsten, großen Vakzin-Studien, wie der STEP-Studie und der RV144-Studie, ein Kombinationsimpfstoff bestehend aus meist drei HIV-1-Genen verabreicht (92, 93). Daher sollte untersucht werden, wie sich die gemeinsame Verabreichung von gag und nef mRNA in vitro auswirkt. Bei der simultanen Elektroporation von HIV-1 gag und nef mRNA 117 5 Diskussion in B-Zelllinien verringerte sich die Expressionsstärke der jeweiligen mRNA-kodierten HIV-1-Proteine um bis zu 25% im Vergleich zur Proteinexpression, wenn die betreffende HIV-1 mRNA einzeln elektroporiert wurde. Diese geringe Reduktion kann z.B. die Folge einer ausgelasteten Proteinsynthese der Zellen sein, da alle Ribosomen besetzt sein könnten und somit keine Proteine mehr translatiert werden. Zudem wurden die HIV-1 mRNA-elektroporierten B-Zelllinien mit HIV-1-spezifischen CTL Linien im γ-IFN ELISPOT Assay ko-inkubiert. Dabei erkannten CTL Linien, die spezifisch für das Gag-Epitop p15 RI9 waren, die B-Zelllinie, die nur mit HIV-1 gag mRNA elektroporiert war, im gleichen Ausmaß wie die B-Zelllinie, die mit HIV-1 gag und nef mRNA elektroporiert wurde. Ebenso verhielt es sich bei B-Zelllinien, die entweder mit nef mRNA oder mit gag und nef mRNA zusammen elektroporiert wurden, und CTL Linien, die spezifische für das Epitop Nef RI9 waren. Dagegen wurde eine klare Reduktion der T-Zellantwort beobachtet, wenn PBMC eines HIV-1-infizierten Patienten mit HIV-1 gag und nef mRNA gemeinsam elektroporiert wurden, obwohl PBMC im Gegensatz zu B-Zelllinien keine verminderte Expression der mRNA-kodierten Proteine nach gemeinsamer Elektroporation zeigten. Einzelne mRNA-kodierte T-Zellantwort, HIV-1-Proteine insbesondere das induzierten Gag-Protein. Die in PBMC drastische eine deutliche Verringerung der Immunantwort bei der gemeinsamen Verabreichung von gag und nef mRNA entsteht durch die Nef-mediierte Reduktion der Expression des HLA-I Moleküls auf der Zelloberfläche (124), wobei Nef nur HLA-A- und HLA-B-Moleküle, nicht aber die HLA-CMoleküle herabreguliert. Daher kann man davon ausgehen, dass die noch vorhandene Immunantwort bei der simultanen Elektroporation von gag und nef mRNA vorwiegend von HLA-C-restringierten Epitopen hervorgerufen wird. Dies konnte auch über Peptidinkubation der mock-elektroporierten, gag mRNA-elektroporierten und nef mRNAelektroporierten PBMC nachgewiesen werden. Die mock- und gag mRNA-elektroporierten PBMC präsentierten und erkannten die HLA-A- bzw. HLA-B-restringierten Epitope und führten zur Freisetzung von γ-IFN. Dagegen zeigten die nef mRNA-elektroporierten PBMC eine verringerte T-Zellantwort nach Stimulation mit den jeweiligen Peptiden. Es konnte zudem festgestellt werden, dass die mock-elektroporierten PBMC zu einer höheren γ-IFN Sekretion nach Peptidinkubation führten als die gag mRNA- elektroporierten PBMC. Ursache dafür sind mRNA-kodierte Gag-Epitope, welche über HLA-I-Moleküle präsentiert werden, so dass dadurch weniger Moleküle zur Bindung der exogenen Peptide zur Verfügung stehen. Allerdings konnte dieser Effekt nur in PBMC beobachtet werden. B-Zelllinien, welche als aktivierte Zellen sehr viele HLA-Moleküle auf ihrer Oberfläche tragen (siehe Expressionsprofil UCSC), zeigten diesen Effekt nicht, da vermutlich trotz Reduktion der HLA-Moleküle durch Nef immer noch genügend Moleküle auf der Oberfläche verbleiben, um die entsprechenden Antigene effizient zu präsentieren. 118 5 Diskussion Dies erklärt, warum über APC wie Dendritische Zellen und B-Zellen in vielen Zellen trotz Nef noch eine starke HLA-A- und –B-restringierte CTL-Antwort induziert werden kann, wohingegen die Effektivität der HLA-A- und –B-restringierten CTL hinsichtlich der Eliminierung von infizierten CD4+ T-Zellen durch Nef stark beeinträchtigt wird. Folglich beweisen diese Ergebnisse, dass bei der Verwendung eines Kombinationsimpfstoffes, der nef mRNA enthält, die induzierten T-Zellantworten aufgrund der Reduktion der HLA-I-Moleküle vermutlich stark dezimiert werden. Damit würde die Immunogenität eines Impfstoffes deutlich vermindert werden, da nur noch HLA-Crestringierte Epitope zur Induktion der CTL-Antwort eine Rolle spielen. Andererseits wäre eine Fokussierung der Immunantwort auf die Epitope, die von Nef nicht herabreguliert werden, vermutlich sinnvoll, da nur diese Epitope auf infizierten Zellen effektiv präsentiert werden. Daher wären weitere Studien in Tiermodellen nötig, um zu prüfen, ob eine gleichzeitige Applikation von Nef die Effektivität eines Impfstoffes behindert oder über die Fokussierung auf Nef-resistente CTL-Epitope steigert. Eine weitere Möglichkeit zur Verbesserung einer T-Zell-basierten Vakzine wäre die Entwicklung von Polytop-Impfstoffen, welche HLA-C-restringierte Epitope der am häufigsten vorkommenden HLA-C-Allele enthält, so dass der hemmende Effekt von Nef vermieden werden kann. Da die Expression der HLA-C-Allele durch genetische Polymorphismen stark beeinflusst werden kann (186), sind hier weitere Untersuchungen nötig, um die Effektivität von HLA-C-restringierten CTL-Antworten bei Patienten mit unterschiedlichen HLA-C-Expressionsstärken zu untersuchen. 5.2.2 Studien zur CTL-Epitop-Hierarchie identifizieren effizient prozessierte Epitope als mögliche Vakzin-Antigene Unsere Analysen zum Monitoring der T-Zellantworten gegen autologe HI-Viren sowie Studien von anderen liefern wichtige Hinweise, dass einige CTL-Epitope besser prozessiert und präsentiert werden als andere Epitope im selben Protein (129, 187). Kommt es zur Mutation eines dieser dominanten Epitope, geht meist die Immunantwort gegen das komplette HIV-1-Protein verloren. So führte z.B. eine CTL Fluchtmutation in dem HLA-A*03-restringierten Epitop QK10 bei Patient 0225 zu einem fast vollständigen Verschwinden der Immunantwort gegen das autologe Nef-Protein. Alle anderen CTLEpitope, die aufgrund seines HLA-Typs eine Rolle spielen könnten, wiesen keine Mutationen auf und waren zwischen der autologen und heterologen Nef Sequenz konserviert. Daher stellten wir die Hypothese auf, dass dieses Epitop in der CTL-EpitopHierarchie sehr weit oben steht und besonders gut prozessiert und präsentiert wird. Die in 119 5 Diskussion dieser Arbeit entwickelte Methode der mRNA-Elektroporation von PBMC eignet sich gut, um die CTL-Epitop-Hierarchie zu studieren. Durch Verwendung von HIV-1-spezifischen CTL Linien kann die Erkennung des einzelnen Peptids mit der Erkennung des Epitops, welches von den mRNA-elektroporierten PBMC präsentiert wird, verglichen werden. Dies gibt Aufschluss darüber, wie gut das Epitop aus der Proteinsequenz prozessiert wird. Es wurde zum einen das HLA-A*03-restringierte Epitop p17 KK9 dem HLA-A*02restringierten Epitop SL9 in Gag gegenübergestellt. Zum anderen wurden die Nef-Epitope Nef7v1 und RY10 verglichen, welche über die HLA-Moleküle B*07 bzw. B*18 präsentiert werden. Die Nef-Epitope, die von den nef mRNA-elektroporierten PBMC prozessiert und präsentiert wurden, zeigten eine insgesamt sehr geringe Erkennung im Vergleich zu Peptiden, obwohl das Zentrum des Nef-Proteins (AS 68-146), in welchem auch die beiden Epitope Nef7v1 und RY10 liegen, als endogen gut prozessierte Region beschrieben wurde (187). Da die beiden Epitope über HLA-B-Moleküle präsentiert werden, ist wahrscheinlich die Reduktion der HLA-B-Expression durch das mRNA-kodierte NefProtein für die insgesamt schwächere Erkennung der Nef-Epitope im Vergleich zu Peptiden verantwortlich. Dennoch schnitt das Epitop Nef RY10 in den Analysen leicht besser ab (17% Erkennung durch die CTL Linie; 10% für Nef7v1). Die Gag-Epitope wurden von den gag mRNA-elektroporierten PBMC sehr gut prozessiert und präsentiert. Außerdem konnte beobachtet werden, dass das p17 KK9 Epitop besser prozessiert und präsentiert wird als das SL9 Epitop (75% Erkennung von KK9 gegenüber 47%). Resultate aus Studien, die die CTL-Epitop-Hierarchie untersuchen, können Epitope identifizieren, die gut prozessiert und präsentiert werden und somit zu einer guten T-Zellantwort führen. Diese stellen daher geeignete Antigene für eine Vakzinierung dar. Schlecht prozessierte CTL-Epitope sollten beim Entwurf einer optimalen therapeutischen Vakzine nicht berücksichtigt werden, da sie nur geringe Immunantworten induzieren und zudem die Prozessierung effizienter Epitope behindern könnten. 5.3 Ausschluss von Nebenwirkungen bei einer Impfung Bei der therapeutischen Vakzinierung mit HIV-1-spezifischen mRNAs sollten denkbare Nebenwirkungen möglichst gering gehalten werden. Dabei stehen bei den zu verhindernden Nebenwirkungen nicht sog. Impfreaktionen, die nur vorübergehend und lokal auftreten, wie Schmerzen und Schwellungen an der Injektionsstelle, im Vordergrund, sondern Komplikationen, die schwerwiegende Folgen haben können. Die große Gefahr bei einem Impfstoff, der HIV-1-spezifische CTLs über verabreichte Antigene aktivieren 120 5 Diskussion soll, ist, dass die induzierten CTLs aufgrund von Sequenzhomologien der erkannten Epitope Kreuzreaktion gegen körpereigene Proteine ausbilden, so dass durch die Zerstörung von nicht-infizierten Zellen Autoimmunreaktionen ausgelöst werden. Da viele Abschnitte aus dem HIV-1-Genom eine hohe Sequenzähnlichkeit zu humanen Proteinen besitzen (188), muss sichergestellt werden, dass HIV-1-spezifische CTLs diese Proteine nicht kreuzreaktiv erkennen und eliminieren. 5.3.1 Wechselwirkung zwischen HIV-1-spezifischen CTLs und dem humanen Protein Fat3 Durch BLAST konnte eine hohe Sequenzähnlichkeit zwischen dem HLA-B*07- sowie HLA-B*35-restringierten Nef-Epitop TM9 (131, 132) und dem humanen Protein Fat3 entdeckt werden. Die beiden Epitope unterscheiden sich dabei in nur zwei Aminosäuren. Das Protein Fat3 ist das dritte Mitglied der Fat-Familie der Säuger und setzt sich aus 4.555 Aminosäuren zusammen (189). Die Funktion des Proteins ist noch unklar. Expressionsstudien konnten Fat3 vor allem im fötalen Gehirn und im adulten, neuralen Gewebe nachweisen, so dass eine Rolle in der Interaktion zwischen den Neuriten während der Entwicklung oder eine Funktion in der Zelladhäsion oder der Modulation der extrazellulären Matrix zwischen Axonen vermutet wird (190-192). Stimulation der PBMC von 100 HIV-1-infizierten Patienten mit den Peptiden Nef TM9 und Fat3 TM9 T/P in vitro demonstrierten, dass eine breite Immunantwort gegen das Fat3-Epitop vorhanden ist. Denn 43% der untersuchten Patienten wiesen eine CTL-Antwort gegen TM9 T/P auf und 20% zeigten sogar eine stark ausgeprägte Reaktion. Zudem erkannten die meisten CTL Linien, die spezifisch für das Nef-Epitop TM9 waren, das Fat3-Epitop kreuzreaktiv. In einigen Fällen war die CTL Antwort gegen das Fat3-Epitop sogar stärker als gegen das Nef-Epitop. Bei zwei der untersuchten Patienten wurden psychische Erkrankungen diagnostiziert, so dass im Hinblick der diskutierten, neurologischen Funktion von Fat3 ein Zusammenhang zwischen den vorhandenen CTLs gegen TM9 sowie TM9 T/P und dem Krankheitsbild bestehen könnte. Eine in silico Analyse sagte eine Prozessierung des Epitops in der pfat3 mRNA mit einer Wahrscheinlichkeit von maximal 59% vorher. Um auszuschließen, dass es bei einer Impfung mit nef mRNA zu Wechselwirkungen zwischen Nef-spezifischen CTLs und dem Protein Fat3 kommt, wurde ein 609 Basenausschnitt von Fat3, der das Epitop TM9 T/P enthält, kloniert und zu mRNA in vitro transkribiert. Anschließend wurde untersucht, ob TM9- und TM9 T/P-spezifische CTL Linien die mRNAkodierten Epitope erkennen, welche von nef mRNA- bzw. pfat3 mRNA-elektroporierten B-Zelllinien präsentiert werden. Es wurde nur das von der nef mRNA-kodierte Epitop TM9 121 5 Diskussion von den CTL Linien erkannt, das von der pfat3 mRNA-kodierte Epitop TM9 T/P wurde weder von kreuzreaktiven TM9-spezifischen noch TM9 T/P-spezifischen CTL Linien erkannt. Dies spricht dafür, dass dieses Epitop entweder in der Zelle nicht richtig prozessiert oder aufgrund der Kompetition mit anderen Epitopen nicht ausreichend prozessiert wird. Damit ist es sehr unwahrscheinlich, dass TM9- oder TM9 T/P-spezifische CTLs, die bei vielen HIV-1-infizierten Patienten vorhanden sind, das körpereigene Protein Fat3 erkennen und Fat3-exprimierende Zellen zerstören. 5.3.2 Wechselwirkung zwischen HIV-1-spezifischen CTLs und dem humanen Protein DAO Zwischen dem Nef CTL-Epitop Nef7v1, welches von den HLA-Molekülen B*07, B*35 und B*51 präsentiert wird (135, 136), und dem humanen Protein der D-Aminosäuren-Oxidase (DAO) konnte eine hohe Sequenzähnlichkeit ermittelt werden. Die Epitope Nef7v1 FPVRPQVPL und Nef7v4 RPVRPQIRL (DAO) unterscheiden sich in drei Aminosäuren. Das Protein DAO gehört zu den Flavoproteinen mit Flavin-Adenin-Dinukleotid (FAD) als prosthetische Gruppe und katalysiert die Deaminierung von D-Aminosäuren (193). Es wird vor allem in unterschiedlichen Bereichen des Gehirns, wie im Cerebellum, in der Amygdala und dem spinalen Cordus exprimiert, aber auch in der Leber und der Niere produziert (194, 195). Bei Schizophrenie-Patienten wurde eine erhöhte Aktivität des Enzyms nachgewiesen, was einen niedrigen D-Serin-Gehalt zur Folge hat und somit zur Erkrankung beitragen könnte (196, 197). Würden HIV-1-Patienten CTLs gegen das DAOEpitop Nef7v4 aufweisen oder Nef7v1-spezifische CTLs das Epitop Nef7v4 kreuzreaktiv erkennen, könnten diese CTLs durch eine Impfung mit nef mRNA verstärkt werden. Dadurch würden DAO-exprimierende Zellen zerstört, was einen geringeren Gehalt von DAO und folglich eine Ansammlung von D-Aminosäuren nach sich ziehen könnte. Daher wurden PBMC von 104 Patienten mit den beiden Peptiden Nef7v1 und Nef7v4 in vitro stimuliert und im γ-IFN ELISPOT getestet. Dabei konnten Nef7v4-spezifische CTLs in 25% der Patienten detektiert werden, aber nur 7% zeigten eine starke CTL-Antwort gegen dieses Epitop. Nef7v1-spezifische CTL Linien wurden bei 36% der Patienten nachgewiesen, wovon nur 13% das DAO-Epitop Nef7v4 kreuzreaktiv erkennen konnten. Rolland et al. konnte zeigen, dass je unterschiedlicher die Nef-Peptide zu den humanen Homologen sind, desto größer ist die durch das entsprechende Nef-Peptid ausgelöste Immunantwort (188). Dies konnten unsere Daten allerdings nicht bestätigen, da wir für das Nef-Epitop TM9, welches sich nur in zwei Aminosäuren von seinem Homolog unterscheidet, deutlich stärkere Immunantworten detektierten als gegen das Epitop 122 5 Diskussion Nef7v1, welches in drei Aminosäuren von seinem Homolog abweicht. Somit finden sich in HIV-1-infizierten Patienten zwar CTLs gegen das sequenzähnliche Epitop im humanen Protein DAO, doch das Ausmaß der Immunantworten ist sehr gering. Daher kann man davon ausgehen, dass bei einer Vakzinierung mit nef mRNA nur sehr wenige Patienten DAO-spezifische oder Nef-spezifische und DAO-kreuzreaktive CTLs aufweisen würden. Weitere Untersuchungen mit DAO-exprimierenden Zelllinien sind nun nötig, um zu prüfen, ob diese Nef7v4-spezifischen CTL DAO-exprimierende Zellen erkennen und lysieren können. 5.4 Analyse und Definition bekannter und neuer CTLEpitope Eine effiziente Vakzine sollte gute CTL-Epitope enthalten, die zum einen in vielen HIV-1infizierten Patienten eine Immunreaktion auslösen und zum anderen selten durch CTL Fluchtmutationen verloren gehen. Daher sollten nicht nur bekannte Epitope auf ihre Erkennung in einer großen Kohorte analysiert werden, sondern auch neue CTL-Epitope definiert werden. Zur Definition neuer Epitope wurden zwei therapie-naive Patienten auf ihre CTL-Antworten gegen verschiedene HIV-1-Proteine mit überlappenden Peptidsets im γ-IFN ELISPOT untersucht. Es wurden jedoch nur CTL-Antworten gegen die Proteine Gag und Nef detektiert, was mit dem Ergebnis von Jia et al. übereinstimmt, dass die meisten CTL-Antworten gegen diese beiden Proteine gerichtet sind (183). Dabei wurden drei neue CTL-Epitope in Gag und zwei in Nef definiert. Restriktionsanalysen ergaben, dass es sich bei den beiden Nef-Epitopen um HLA-B*41-restringierte Epitope handelt. Auch eines der neu definierten Gag-Epitope wird über HLA-B*41 präsentiert. Die Präsentation des zweiten Gag-Epitops erfolgt über das HLA-Molekül A*02. Ein drittes Gag-Epitop konnte bisher keinem HLA-Allel zugeordnet werden. Das HLA-A*02restringierte Gag-Epitop liegt in dem Peptid Gag07-113. Kreuzreaktionen Gag07-113spezifischer CTL Linien mit den benachbarten, überlappenden Peptiden führten zu keiner γ-IFN Sekretion, so dass man vermuten könnte, dass es sich in dem Peptid Gag07-113 um das Epitop mit der Sequenz RPEPTAPPE handelt. Jedoch stimmen in dieser Sequenz die Ankerpositionen nicht mit dem bekannten Bindungsmotiv für HLA-A*02 überein. Da die T-Zellreaktion der CTL Linie sowohl mit einem Anti-CD4 als auch mit einem Anti-CD8 Antikörper inhibiert wurde, liegt der Verdacht nahe, dass in Gag07-113 ein HLA-I- und ein HLA-II-Epitop vorliegen. Das neu definierte CTL-Epitop in Gag07-02, welches über HLAB*41 restringiert ist, konnte durch Kreuzreaktionen auf die Sequenz GELDRWEKI 123 5 Diskussion eingeschränkt werden. Dieses Epitop wurde von ELF als HLA-B*40-restringierendes Epitop vorhergesagt. Die beiden neu definierten Nef-Epitope wurden über Restriktionsanalysen ebenso dem HLA-B*41 zugeordnet. Da jedoch sowohl B*40 als auch B*41 zum gleichen Supertyp gehören, sollten sie ähnliche Bindungsmotive haben. Die Nef07-08-spezifische CTL Linie zeigte keine Kreuzreaktion mit Nef07-07 oder Nef07-09, so dass die Epitop-Sequenz AVSRDLEKHG sein müsste. Allerdings sind in dieser Sequenz die Ankeraminosäuren für das HLA-B*41-Molekül nicht vorhanden. Das Programm SYFPEITHI schlägt als HLA-B*41-Epitop SRDLEKHGA vor. Dann hätte jedoch die Nef07-08-spezifische CTL Linie das Peptid Nef07-09 kreuzreaktiv erkennen müssen. Es kann ausgeschlossen werden, dass das Peptid Nef07-09 nicht funktionsfähig ist, da es bei Inkubation mit der Nef07-10-spezifischen CTL Linie zu einer Kreuzreaktion führte. Somit ist die exakte Epitop-Sequenz für Nef07-08 noch unklar. Allerdings konnte das von ELF vorhergesagte potentielle HLA-A*66-restringierende Epitop nicht bestätigt werden. Aufgrund der Kreuzreaktion der Nef07-10-spezifischen CTL Linie mit dem Peptid Nef07-09 konnte die Epitop-Sequenz auf LEKHGAITSSN eingegrenzt werden. Es handelt sich vermutlich um das von ELF vorgeschlagene, potentielle CTL-Epitop LEKHGAITS, welches über HLA-B*40 präsentiert wird. Dennoch sollte die genaue Sequenz der drei HLA-B*41-restringierten CTL-Epitope in Gag und Nef durch den Einsatz von kürzeren Peptiden bestätigt werden. Der Einsatz von HLA-C-restringierten CTL-Epitopen ist besonders sinnvoll, da diese Epitope durch Nef nicht negativ beeinflusst werden, denn nur die Expression der HLA-Aund -B-Moleküle wird von der Zelloberfläche reduziert (124). Die Analyse des HLACw*07-restringierten CTL-Epitops Nef11T3 (138) zeigte, dass knapp die Hälfte der untersuchten Patienten eine CTL-Antwort gegen dieses Epitop besitzt. Zudem wies ein Fünftel der Patienten sehr starke Immunreaktionen gegen dieses Epitop auf. Auch eine Epitop-Variante, die in der Mehrzahl der autologen Virussequenzen zu finden war, führte in 43% der untersuchten Patienten zu einer deutlichen T-Zellreaktion. Allerdings war die Kohorte, welche mit dem varianten Peptid getestet wurde, mit 34 Patienten noch relativ klein, so dass sich die Frequenz der Erkennung in einer größeren Kohorte deutlich unterscheiden könnte. Dennoch demonstrierten die Ergebnisse eine breite Erkennung des Epitops. Daher könnte dieses Epitop bei einer therapeutischen Vakzinierung gute Immunreaktionen induzieren und zu einer guten Wirksamkeit beitragen. Da das HLACw*07-Allel mit einer durchschnittlichen Häufigkeit von 52% in Deutschland vorkommt (198), könnte eine Vakzinierung mit einem Antigen, welches dieses Epitop enthält, bei vielen Patienten zu einer starken Immunreaktion führen. 124 5 Diskussion 5.5 Etablierung alternativer Transfektionsmethoden von mRNA Die Elektroporation von mRNA stellt in vitro eine effiziente Transfektionsmethode dar. Für eine in vivo Applikation der HIV-1-spezifischen mRNAs als therapeutischer Impfstoff könnte man eine mRNA-Elektroporation der PBMC in vitro vornehmen und die elektroporierten PBMC nach Reinigung dem Patienten zurückinjizieren. Dieses Verfahren wird bereits mit Dendritischen Zellen, welche mit tumor-spezifischen mRNAs als Antigene beladen wurden, angewendet (111). Dennoch wäre eine direkte Verabreichung der mRNA eine schnellere, kostengünstigere und einfachere Methode. Daher sollten alternative Reagenzien zur Transfektion der mRNA getestet werden. Diese sind weder immunogen noch toxisch und sehr gut verträglich, was sie für eine in vivo Anwendung prädestiniert. 5.5.1 Transfektion mit Chitosan Bei Chitosan handelt es sich um ein natürlich vorkommendes Polysaccharid aus dem Panzer von Krebsen (139), welches DNA und siRNA in verschiedene Zellen transportieren kann (140). Es wurde zunächst als Nahrungsergänzungsmittel und Lebensmittelzusatz verwendet (199). Wir konnten die Transfektion von Plasmid-DNA über Chitosan-Nanopartikel in HEK293T Zellen reproduzieren, allerdings konnte kein Transfer von mRNA detektiert werden. Eine effiziente Transfektion von HEK293 Zellen wurde mit Chitosan bereits gezeigt (200). Die Transfektion von Plasmid-DNA in HEK293T Zellen mit Chitosan gilt als weniger effektiv, was mit den Ergebnissen der vorgelegten Arbeit übereinstimmt (140). Denn auch in unseren Versuchen sind nur sehr wenige Zellen erfolgreich transfiziert worden. Der Einsatz von Chitosan ist sehr kompliziert, da viele Faktoren die Effizienz der Transfektion beeinflussen. So ist die Größe der gebildeten Komplexe zwischen DNA und Chitosan ein kritischer Faktor. Es konnte gezeigt werden, dass je kleiner die Komplexe sind, desto besser können sie durch die dann mögliche Endozytose von den Zellen aufgenommen werden (201). Die Transfektionseffizienz kann durch die Modifikation von Chitosan z.B. mit Galaktose erhöht werden (202). Daher ist es zum einen möglich, dass HEK293T in unserem Versuch nicht die geeigneten Zelllinien waren oder zum anderen die gebildeten Komplexe zu groß waren. Des Weiteren könnte auch das Problem bestehen, dass die mRNA-Chitosan-Komplexe zwar von den Zellen aufgenommen worden sind, aber die mRNA in den Endosomen verpackt blieb und daher keine Proteinsynthese stattfand. 125 5 Diskussion 5.5.2 Transfektion über das HIV-1 Tat-Protein Das HIV-1 Tat-Protein besitzt in seiner Aminosäurensequenz eine Protein-TransduktionsDomäne, welche für den Transport von Proteinen und Nukleinsäuren in Zellen verantwortlich ist (144). Verschiedene Studien mit dem kompletten Tat-Protein oder nur dem einzelnen Peptid, welches die Protein-Transduktionsdomäne von Tat enthält, zeigten bereits die erfolgreiche und sichere in vivo Transfektion von siRNA und DNA (142). In der vorgelegten Arbeit konnte weder Plasmid-DNA noch mRNA in HEK293T Zellen oder Makrophagen transfiziert werden. Sowohl HIV-1 Tat-Protein als auch Tat-Peptid wurde getestet. Obwohl bereits die erfolgreiche Transduktion von HEK293 Zellen nachgewiesen wurde (203) und zunächst der Tat-vermittelte Transfer von Nukleinsäuren als Rezeptorund Transporterunabhängig beschrieben wurde (204), wurde in primären Gehirnzellen gezeigt, dass die Transfektionseffizienz von der Expression der Proteoglykane auf der Zelloberfläche abhängt (205). Somit könnte eine geringe Expression von Proteoglykanen auf der Oberfläche von HEK293T Zellen und Makrophagen ein Grund für die fehlende Transfektion durch Tat sein. Die Transduktion wird auch davon limitiert, dass viele Peptide in den Vesikeln bleiben und daher nie im Zytoplasma enden (144). Der Einsatz von chemischen crosslinkern zwischen dem Tat-Peptid und der Nukleinsäure könnte die Komplexbildung erleichtern (143). 5.5.3 Transfektion mit Hilfe von Atelokollagen Atelokollagen ist ein Pepsin-verdautes Typ I Kollagen, welches meist aus der Dermis von Kälbern gewonnen wird (146). Da es sich dabei um ein Biomaterial handelt, wird es vom Körper wieder abgebaut (145). Atelokollagen ist positiv geladen und kann über elektrostatische Wechselwirkungen Komplexe mit negativ geladenen Nukleinsäuren bilden. Ein positiver Nebeneffekt dabei ist, dass die Nukleinsäuren durch die Komplexe vor dem Abbau durch Nukleasen geschützt sind, was bei der Verwendung der sehr instabilen mRNA ein wichtiger Faktor ist. Atelokollagen wird bereits in der Wundheilung klinisch eingesetzt (146). Ein erfolgreicher Transfer mit Atelokollagen wurde für DNA, Proteine und siRNA beschrieben (145, 206). Leider konnte ich in allen durchgeführten Versuchen weder eine erfolgreiche Transfektion von DNA noch von mRNA in HEK293 oder HEK293T Zellen erzielen, obwohl die Transfektion von Plasmid-DNA wie beschrieben durchgeführt wurde und zudem HEK293 Zellen zum Einsatz kamen, für welche bereits ein Transfer durch Atelokollagen gezeigt wurde (207). Dass es dennoch zu keinem positiven Ergebnis kam, könnte entweder an dem Zustand oder der Zellzyklusphase der Zellen liegen oder daran, dass ein Atelokollagen verwendet wurde, 126 5 Diskussion welches speziell für in vivo Anwendungen deklariert ist. Inwieweit sich dieses von dem Atelokollagen unterscheidet, welches für in vitro Anwendungen ausgezeichnet ist, lässt sich nicht nachvollziehen. Ein weiterer Versuchsdurchlauf mit dem „Atelocollagen in vitro Kit“ würde darüber Ausschluss geben. 5.5.4 Transfektion mit FuGENE Ein häufig verwendetes in vitro Transfektionsreagenz ist FuGENE, welches sehr effizient besonders adhärente Zelllinien transfiziert. Die Nukleinsäuren werden über den Prozess der Lipofektion in die Zellen transportiert. Die im FuGENE enthaltenen Lipide fusionieren dabei mit der Plasmamembran der Zelle und gelangen über Endozytose ins Zellinnere (208). Bei allen durchgeführten Transfektionsversuchen mit Chitosan, HIV-1 Tat oder Atelokollagen wurden die Zellen stets als Kontrolle mit FuGENE transfiziert. Neben der erfolgreichen und sehr effizienten Transfektion mit Plasmid-DNA konnte auch ein Transfer von GFP mRNA in HEK293T Zellen beobachtet werden. Die GFP Expression war dabei sehr deutlich, wenn auch nur in wenigen Zellen. Bisher wurde noch kein Transfer von mRNA mittels FuGENE gezeigt, in einer kürzlich erschienen Arbeit wurde jedoch der Transfer von mRNA in Fibroblasten erfolgreich durchgeführt (209). 5.6 Evolution eines unterschiedlichen HI-Ausgangsviruses Patienten und die in zwei daraus resultierenden Immunantworten Der individuelle Einfluss des Immunsystems kann gut an Patientenpaaren studiert werden, die sich zwar mit demselben Ausgangsvirus infiziert haben, aber einen divergenten Krankheitsverlauf zeigen. Mit der in dieser Arbeit neu entwickelten Methode des Immunmonitorings über mRNA-Elektroporation von PBMC kann neben der Immunantwort gegen autologe Virusproteine auch die Immunantwort gegen eng verwandte Virusproteine untersucht werden. 127 5 Diskussion 5.6.1 Vergleichende Immunantworten auf die mRNA-kodierten HIV-1-Proteine Bei der Analyse des Patientenpaares 0225 und 0284 wurde entdeckt, dass autologe virale Proteine des Partners nicht unbedingt stärkere Immunantworten induzieren als Proteine des Referenzviruses SF2. Da das eigene Virus und das Virus des Partners eng verwandt sind, würde man eine stärkere T-Zellreaktion gegen das näher verwandte Virus erwarten. Allerdings induzierten die mRNA-kodierten SF2-Proteine in zwei von vier Fällen eine stärkere Immunantwort im Vergleich zu mRNA-kodierten HIV-1-Proteinen des Partners. Zudem wurde die stärkste CTL-Antwort in drei von vier Fällen gegen autologe mRNAkodierte HIV-1-Proteine detektiert. Durch den Vergleich der Gag und Nef Sequenzen von 0225, 0284 und SF2 konnte festgestellt werden, dass das Virus in Patient 0225 sehr stark mutierte, wobei insbesondere CTL Fluchtmutanten eine Rolle spielten. Daher kann man vermuten, dass das Virus in Patient 0225 einem starken CTL-Druck ausgesetzt war und dadurch Mutationen entwickelte, um diesem Druck zu entkommen. In Patient 0284 hingegen ist dies nicht der Fall, was möglicherweise daran liegt, dass dessen CTL-Epitope in Bereichen des Virus liegen, die ohne den Verlust der viralen Fitness nicht mutiert werden können. So findet sich in dem HLA-B*57-restringierten CTL-Epitop TW10 in der Gag Sequenz von 0284 eine T242N-Mutation, welche die Replikationsfähigkeit enorm einschränkt (210). Durch CTL Fluchtmutationen in den jeweiligen Sequenzen konnten auch die Immunantworten gegen die verschiedenen mRNA-kodierten HIV-1-Proteine erklärt werden. Die kaum noch vorhandene CTL-Antwort gegen das autologe Nef-Protein in Patient 0225 konnte auf eine T90N-Mutation in dem HLA-A*03-restringierten Epitop QK10 zurückgeführt werden. Ebenso erklären Mutationen in dominanten CTL-Epitopen die schlechtere Erkennung der eng verwandten Virusproteine im Vergleich zu SF2Proteinen. Die Zellen des Patienten 0225 zeigten eine schwache Immunantwort gegen SF2 Gag und eine kaum noch vorhandene Immunantwort gegen 0284 Gag. Insbesondere die Veränderungen in den Gag-Epitopen SL9 und KL14 sind vermutlich ausschlaggebend für die schwächere Erkennung der heterologen Proteine im Vergleich zum autologen Protein. Da SF2 Gag in diesen beiden Epitopen Aminosäuren aufweist, welche noch eher eine Erkennung zulassen als die Aminosäuren-Varianten in 0284 Gag, ist damit die schlechtere T-Zellantwort gegen 0284 Gag erklärbar. Bei der Detektion der Nefspezifischen T-Zellantworten von Patient 0284 konnte die geringste Immunreaktion für 0225 Nef beobachtet werden. Dies wird vermutlich vor allem durch die abweichende Sequenz im Epitop Nef AL9 verursacht, welches sich in 0225 Nef im Vergleich zu 0284 Nef in drei Aminosäuren unterscheidet. 128 5 Diskussion 5.6.2 Evolution eines HI-Virus in zwei unterschiedlichen Patienten Durch Vergleich der viralen Gag und Nef Sequenzen der Patienten 0225 und 0284 konnten deutliche Unterschiede festgestellt werden. Es wurden nun zusätzlich die Gag und Nef Sequenzen zum Zeitpunkt der Erstdiagnose (August 2006) und ca. zwei Jahren nach Erstdiagnose (Zeitpunkt der mRNA-Synthese) von beiden Patienten betrachtet. Allerdings war es nicht möglich eine vollständige Sequenz des Gag-Proteins von 0225 und des Nef-Proteins von 0284 zum Zeitpunkt der Erstdiagnose zu erhalten. Die Viruslast lag damals für Patient 0225 bei 200.000 Kopien/ml, für Patient 0284 bei 2.000 Kopien/ml. Aufgrund dieser hohen Virämie von 0225 zu Beginn der Infektion sind vermutlich zahlreiche Virusvarianten in diesem Patienten vorhanden, die replizieren (211). Durch die hohe Fehlerrate der Reversen Transkriptase werden vermehrt falsche Nukleotide eingebaut (212), so dass letztendlich viele verschiedene Viren vorliegen und bei der Sequenzierung detektiert werden. Daher ist es meist bei einer hohen Virämie schwierig, eine einzige virale Sequenz zu erhalten, während bei einer geringeren Viruslast die Frequenz neu entstehender Mutationen geringer sein dürfte. Gründe, warum die Nef Sequenz von 0284 nicht vollständig amplifiziert werden konnte, können z.B. Mutationen in der Binderegion der Primer sein. Der Vergleich zwischen den beiden Zeitpunkten soll Aufschluss darüber geben, wie sich das HI-Virus in zwei verschiedenen Wirtsorganismen entwickelt hat und welche Mutationen bereits bei Infektion vorhanden waren. Auf diese Weise können die Varianten ausfindig gemacht werden, welche durch CTL-Druck entstanden sind und somit Fluchtmutanten repräsentieren. Es können so aber auch Mutationen identifiziert werden, welche Reversionen von übertragenen CTL Fluchtmutanten sind. Dadurch konnte die in 0225 Nef beobachtete T90N-Substitution im Epitop QK10, welche für die geringere T-Zellantwort auf das autologe Nef-Protein verantwortlich ist, als eindeutige CTL Fluchtmutante bestimmt werden. Denn zum Zeitpunkt der Erstdiagnose war diese Variante noch nicht vorhanden. Der Verlust der Immunantwort gegen das autologe NefProtein führt dazu, dass das Virus stärker replizieren kann. Dies könnte dazu beitragen, dass Patient 0225 eine schlechtere Kontrolle der Virusreplikation zeigte, so dass er bei anhaltend hohen Virämien frühzeitig mit antiretroviralen Medikamenten therapiert werden musste. Auch die in 0284 Gag auftretende T242N-Mutation im Epitop TW10 stellt eine CTL Fluchtmutation dar. Man findet zu Beginn der Infektion sowohl Viren mit dem WildtypEpitop als auch Viren mit der Mutante. Dies stimmt mit den Beobachtungen überein, dass sich die frühe CTL-Antwort bei HLA-B*57-positiven HIV-1-Patienten gegen das TW10Epitop richtet und es daher zur Selektion dieser CTL Fluchtmutation kommt (213). Diese ist bei den meisten HLA-B*57- sowie HLA-B*58-positiven HIV-1-Patienten zu finden und 129 5 Diskussion resultiert in einer erheblichen Einschränkung der Replikationsfähigkeit des Virus (210, 213). Um diesen Verlust auszugleichen, gibt es eine Reihe von kompensatorischen Mutationen. Dazu gehört auch die in 0284 Gag auftretende Prozessierungsmutante A148P (214) vor dem ISW9-Epitop. Weiter beschriebene Kompensationsmutationen für die TW10 Fluchtmutante sind in 0284 Gag nicht zu finden (H221Q, I225V, M230I, G248A). Diese sind alle mit einer schnelleren Krankheitsprogression assoziiert. Die verminderte Replikationsfähigkeit durch die T242N-Substitution in TW10 und der fehlende Ausgleich durch kompensatorische Mutationen tragen vermutlich zur guten Kontrolle der Virusreplikation in 0284 bei. Die Prozessierungsmutante A148P allein kann die Wirkung von T242N anscheinend nicht ausgleichen. Der gute Krankheitsverlauf von 0284 geht somit vermutlich stark auf das HLA-B*57 zurück. Es wurde ebenso beschrieben, dass die T242N-Mutation übertragen werden kann, jedoch bei HLA-B*57- bzw. HLA-B*58-negativen Individuen in der chronischen Phase revertiert (213). In der Gag Sequenz von 0225 ist diese Variante zu keinem Zeitpunkt auffindbar. Weiterhin sind in 0225 Gag sog. „Fußabdrücke“ in der Sequenz zu finden, welche stark darauf hinweisen, dass das Ausgangsvirus von einem HLA-B*57- oder HLA-B*58positiven HIV-1-Infizierten stammt. Die kompensatorischen Mutationen H221Q und I225L sind in der Sequenz zum Zeitpunkt der Erstdiagnose nachweisbar und manifestieren sich schließlich. Dies unterstützt die anamnestische Vermutung, dass die Infektion von 0284 auf 0225 übertragen wurde. Unsere Beobachtungen stimmen damit überein, dass CTL Fluchtmutationen eine große Rolle in der Evolution von HIV-1 spielen (138). 5.7 Interaktion von HIV-1-spezifischen mRNAs und zellulären miRNAs Viele Daten demonstrierten bereits eine Interaktion von HIV-1 mit zellulären miRNAs (67). Allerdings beschränkten sich diese Studien auf den Vergleich zellulärer miRNAs zwischen sog. Progressoren und HIV-1-Patienten, welche das Virus kontrollieren, oder aber es wurde die Veränderung von zellulären miRNAs nach Infektion mit HIV-1 NL4-3 in vitro untersucht (68, 215). Daher ist unklar, welche HIV-1-Gene das zelluläre miRNA-Profil beeinflussen und ob verschiedene HIV-1-Varianten einen unterschiedlichen Effekt ausüben. 130 5 Diskussion 5.7.1 Einfluss der HIV-1 Gene gag und nef auf miRNAs in CD4+ T-Zellen Ob die HIV-1-Gene gag und nef allein das zelluläre miRNA-Profil in CD4+ T-Zellen beeinflussen können, wurde bis jetzt noch nicht untersucht. Es ist ebenfalls unbekannt, ob unterschiedliche HIV-1-Virusisolate aus Patienten mit divergentem Krankheitsverlauf auf das Profil der miRNAs anders wirken. Daher wurden gag und nef mRNAs in CD4+ T-Zellen elektroporiert und die daraus isolierte, zelluläre RNA im Deep Sequencing analysiert, um alle vorkommenden miRNAs der Zellen zu detektieren und miteinander zu vergleichen. Dabei konnte festgestellt werden, dass die HIV-1-Gene gag und nef allein das miRNA-Profil von CD4+ T-Zellen verändern können. Unter den miRNAs, die nach der Elektroporation mit nef oder gag mRNA eine unterschiedliche Expression im Vergleich zur GFP mRNA-Elektroporation aufweisen, befinden sich sowohl miRNAs, für welche bereits eine Rolle in der viralen Replikation beschrieben wurde, als auch miRNAs, die immunologische Funktionen regulieren. Da jede Methode ihre Vor- und Nachteile hat, sollten die Ergebnisse der Deep Sequencing-Analyse mittels qRT-PCR validiert werden. Des Weiteren wurden Proben aus einem zweiten HIV-Negativen studiert. Dabei zeigte sich, dass nur einige Beobachtungen der Deep Sequencing-Analyse sich in beiden Spendern reproduzieren ließen. Einige Daten konnten nur in jeweils einem Spender validiert werden. Dabei handelt es sich somit um Spender-spezifische Effekte. Diese können unter anderem durch verschiedene Ausgangsmengen der jeweiligen miRNA in den Zellen der unterschiedlichen Individuen entstehen oder werden von anderen Faktoren noch zusätzlich beeinflusst. Sie kommen somit für eine HIV-1-spezifische Interaktion nicht in Frage. Für die zelluläre miR-150 wurde bereits beschrieben, dass sie HIV-1 in vitro inhibieren kann (216). Eine leicht erhöhte Expression der miR-150 konnte in Zellen nach Elektroporation mit 0284 nef mRNA detektiert werden, was eine erhöhte Inhibierung von HIV-1 zur Folge hätte. Allerdings wurde diese Beobachtung durch qRT-PCR nicht bestätigt. Nach Infektion mit HIV-1 wurde eine geringere Expression der let-7 Familie gemessen (159). Die Deep Sequencing-Daten nach gag und nef mRNA Elektroporation in CD4+ T-Zellen stimmen damit nicht überein, denn es wurde stets eine Expressionszunahme der let-7-Familie detektiert. Jedoch wurden die Ergebnisse nur teilweise durch qRT-PCR-Analysen validiert. Die Expressionszunahme konnte in beiden Spendern nur für let-7a durch 0225 Gag und für let-7f durch 0284 Nef bestätigt werden. Auch für die zelluläre miR-30a wurde schon ein inhibierender Einfluss auf die HIV-1Replikation in vitro nachgewiesen (156). Die Deep Sequencing-Daten zeigten eine erhöhte Expression der miR-30c nach Elektroporation mit 0284 nef mRNA, was mittels 131 5 Diskussion qRT-PCR in beiden Spendern validiert werden konnte. Da 0284 ein Patient mit guter Kontrolle der Virusreplikation ist, kann man vermuten, dass miR-30c eine ähnliche Funktion wie miR-30a vollzieht. Diese Nef-Variante führte zu einer erhöhten Expression der miR-30c, was wiederum eine stärkere Inhibierung der HIV-1-Replikation zur Folge hat. Dies könnte zu der guten Kontrolle der Infektion beitragen. Die Expression der zellulären miR-142-3p wurde deutlich durch das 0284 Nef-Protein dezimiert. Dies wurde auch in Proben aus einem zweiten Spender über qRT-PCR nachgewiesen. Zwar ist noch keine Interaktion dieser miRNA mit HIV-1 beschrieben, aber es wurde eine verringerte Expression der miR-142 in SLE-Patienten als Auslöser für eine B- und T-Zell-Aktivierung festgestellt (162). Deshalb könnte die gute Kontrolle der Virusreplikation des Patienten 0284 dadurch entstehen, dass sein autologes Nef-Protein die Expression der zellulären miR-142 verringert, es demnach zur Aktivierung von B- sowie T-Zellen kommt und durch die gesteigerten Immunantworten das Virus folglich besser in Schach gehalten wird. Das Expressionsprofil dieser beiden miRNAs (miR-30c, -142) konnte nach Elektroporation mit 0225 gag bzw. 0225 nef mRNA nicht in beiden Spendern validiert werden. Somit konnte gezeigt werden, dass die einzelnen HIV-1-Gene gag und nef das zelluläre miRNA-Profil CD4+ in unterschiedliche T-Zellen Beeinflussung modifizieren von können. Virusisolaten aus Außerdem konnte verschiedenen eine Patienten nachgewiesen werden, die zudem zum Krankheitsverlauf beitragen können. Insbesondere die zelluläre miR-142-3p könnte für eine höhere Aktivität des Immunsystems in Patienten mit kontrollierter Virusreplikation verantwortlich sein. 5.7.2 Einfluss von CTL Fluchtmutanten in Nef auf die miR-29aInteraktion Die zelluläre miRNA-29a bindet HIV-1 Nef, was zur Inhibierung der Nef-Expression und der HIV-1-Replikation führt (71). Die Zielsequenz der miR-29a liegt in der kodierenden Region von Nef und befindet sich zudem in dem HLA-A*24- bzw. HLA-B*18-restringierten CTL-Epitop RY10 (129). Durch RY10-spezifische CTLs werden häufig CTL Fluchtmutanten im RY10-Epitop selektioniert. Es stellte sich daher die Frage, welchen Einfluss CTL Fluchtmutationen auf die Bindung der miR-29a haben. Außerdem sollte geklärt werden, ob Mutationen in dieser Region eventuell auch selektioniert wurden, um der Inhibierung durch die miR-29a zu entkommen. In Luziferase-Reporter-Assays von HEK293 Zellen, welche mit fünf verschiedenen Nef-Varianten aus Patienten sowie aus SF2 und der humanen miR-29a ko-transfiziert wurden, konnte eine unterschiedliche Hemmung der Varianten durch miR-29a detektiert werden. Die Nef-Variante aus dem 132 5 Diskussion HLA-A*24-positiven Patienten 0011 konnte durch die miR-29a kaum noch inhibiert werden (nur 16% Inhibierung). Ein Sequenzvergleich offenbarte in diesem Nef-Protein eine CTL Fluchtmutante in der Binderegion der miRNA. Da im Luziferase-Reporter-Assay die nef Sequenzen als nicht-kodierende 3`UTR des Luziferase-Gens vorkommen, sollte ein weiterer Versuch die Inhibierung auf Proteinebene analysieren. Dabei konnte ebenso eine unterschiedliche Regulation der verschiedenen nef-Varianten festgestellt werden. Allerdings wurde in diesem Assay die geringste Inhibierung des Nef-Proteins von Patient 0143 beobachtet. In dieser Nef Sequenz finden sich ebenfalls CTL Fluchtmutanten. Somit konnte in beiden Assays eine unterschiedliche Regulierung der nef-Varianten durch die miR-29a detektiert werden. Da die beiden nef-Varianten 0011 und 0143 jeweils in einem Assay die geringste Inhibierung zeigten und zudem diese beiden Varianten im Gegensatz zu den anderen drei Varianten Fluchtmutanten in der Binderegion der miR-29a aufweisen, kann ein Einfluss von CTL Fluchtmutanten auf die Interaktion von zellulären miRNAs konstatiert werden. Dass es in den beiden durchgeführten Assays zu unterschiedlichen Ergebnissen bezüglich der geringsten Inhibierung kam, kann technische Gründe haben. Obwohl das Expressionsplasmid der miR-29a im Überschuß transfiziert wurde, ist eine unterschiedliche Expression der miR-29a in den verschiedenen Transfektionen möglich. Da nicht nachgewiesen wurde, wie viel miR-29a exprimiert wird, kann nicht ausgeschlossen werden, dass die beobachteten Effekte durch eine unterschiedlich starke Expression der miR-29a hervorgerufen werden. Dies würde auch erklären, warum die nefVarianten der Patienten 0284 und 0225 kaum von miR-29a im Luziferase-Reporter-Assay inhibiert werden, obwohl sie die gleiche Nukleotidsequenz im Bereich der miR-29aBinderegion haben wie SF2 nef. Aufgrund der Nukleotidsequenz der nef Gene kann man vermuten, dass 0011 nef die geringste Reprimierung durch miR-29a zeigen müsste, da insgesamt vier Nukleotide im Vergleich zur idealen Bindesequenz von miR-29a verändert sind. Diese Hypothese wird verstärkt, da sich zwei mismatches im Bereich der seedSequenz der miRNA befinden, die für eine korrekte Bindung der Zielsequenz zuständig ist und immer die Nukleotide zwei bis acht der miRNA umfasst (178). Das nef Gen von 0143 zeigt dagegen nur einen mismatch im Bereich, in welchem die seed-Sequenz der miR-29a bindet. Des Weiteren kann man annehmen, dass das Virus von 0011 die beiden Nukleotide in der nef Sequenz nicht nur mutiert hat, um dem Druck der CTLs zu entkommen, sondern auch um sich der Inhibierung durch die miR-29a zu entziehen. Denn für eine CTL Fluchtmutante des RY10-Epitops hätte der Austausch eines der beiden Nukleotide ausgereicht. Daher scheint der Austausch eines weiteren Nukleotids darin begründet zu sein, dass dadurch die miR-29a schlechter an die virale mRNA bindet. 133 5 Diskussion Jedoch könnte die Interaktion der zellulären miR-29a mit der nef Sequenz von HIV-1 auch andere Folgen haben. So ist bekannt, dass miR-29a an die 3`UTR der mRNA für γ-IFN bindet und so die Sekretion des Zytokins hemmt (217). Bei einer HIV-1-Infektion gehört das Tat-Protein zu den ersten gebildeten Transkripten. Tat kann die Expression von T-bet induzieren. Es handelt sich dabei um einen TH1-spezifischen Transkriptionsfaktor, welcher die Expression von TH1-Zytokinen wie γ-IFN kontrolliert (161). Wird T-bet durch das TatProtein von HIV-1 induziert, kommt es zur erhöhten Produktion von γ-IFN und eine Immunantwort wird eingeleitet. Bindet nun aber die zelluläre miR-29a vermehrt an das auftretende HIV-1 Nef-Protein, wird zusätzliches γ-IFN freigesetzt, da weniger γ-IFNTranskripte durch miR-29a inhibiert werden. Daher könnte die miR-29a nicht nur über die Inhibierung der Nef-Expression einen negativen Einfluss auf die HIV-1-Replikation haben, sondern auch über die Regulation der γ-IFN Freisetzung. Entzieht sich das HI-Virus der Bindung der miR-29a indem es die Zielsequenz in Nef mutiert, führt dies einerseits zu einer erhöhten Nef-Expression und andererseits zu einer verringerten γ-IFN Sekretion, was eine geringere TH1-Immunantwort zur Folge hat. Die HIV-1-Replikation wird demnach gefördert und die Krankheit könnte schneller voranschreiten. 5.8 Ausblick Die Elektroporation von PBMC mit HIV-1-spezifischen mRNAs repräsentiert ein interessantes Testsystem für die Impfstoffentwicklung, da CTL-Epitope vor ihrem Einsatz als Vakzine auf grundlegende Eigenschaften geprüft werden können. So kann zum einen die Effektivität von CTL-Epitopen studiert werden, indem die wichtigsten bekannten Epitope hinsichtlich ihrer Effizienz zur Stimulation von CTL untersucht werden. Niedrig affine CTL-Epitope sind bei einer therapeutischen Vakzinierung vermutlich unnütz und eventuell sogar nachteilig. Denn durch eine Kompetition der verschiedenen CTLSpezifitäten um die Ressourcen des Immunsystems wird die Entwicklung von CTL mit Spezifität für optimalere Epitope vermutlich behindert. Daher wäre es für die Impfstoffentwicklung enorm wichtig, die hoch affinen und damit effizienten CTL-Epitope zu identifizieren. Zum anderen kann das entwickelte Testsystem zur Definition von CTLEpitope genutzt werden, die von CTLs auf infizierten Zellen trotz der inhibitorischen Wirkung von Nef erkannt werden. Dazu gehören sowohl HLA-C-restringierte und Nefresistente CTL-Epitope als auch HLA-A- und HLA-B-restringierte Epitope, bei denen die viralen Peptide mit so hoher Affinität an die HLA-Moleküle binden, dass sie trotz einer verringerten Anzahl an HLA-Molekülen noch zu einer effektiven Stimulierung von CTL in 134 5 Diskussion der Lage sind. Die Ergebnisse solcher Studien könnten einen bedeutsamen Beitrag dazu leisten, dass bei künftigen therapeutischen Vakzinierungen nur hoch affine und effiziente CTL-Epitope zum Einsatz kommen. Aufgrund der guten Immunogenität von mRNA-kodierten HIV-1-Proteinen eignen sich diese sowohl als alternative Antigene für das Monitoring von T-Zellantworten bei HIV-1Patienten als auch als therapeutische Vakzine zur Induktion von CTLs. Die in dieser Arbeit dargelegten Daten zeigten, dass die Immunantworten gegen autologe Viren meist unterschätzt werden. Da in dieser Studie nur wenige Patienten eingeschlossen waren, sollten die T-Zellantworten gegen autologe Viren für mehr HIV-1-Patienten analysiert werden. Ebenso wäre es sinnvoll noch weitere Antigene zu betrachten, wie z.B. die HIV-1-Gene pol, vpu, vpr oder tat, denn bisher wurden hauptsächlich autologe gag und nef mRNA verwendet. Dabei könnte man feststellen, ob neben gag und nef noch andere HIV-1-Gene deutliche CTL-Antworten auslösen und sich daher gut als Antigen eignen. Auch diese Analysen tragen dazu bei, dass man einen genaueren Einblick gewinnt, aus welchen Antigenen eine effiziente Vakzine bestehen sollte. Mit den gewonnen Ergebnissen sollte die Generierung einer optimalen mRNA-Vakzine zur Therapie einer HIV-1-Infektion möglich sein, welche dann in klinische Studien eingesetzt werden kann. Diese mRNA-Konstrukte können sowohl autologe als auch heterologe HIV-1-Gene sein, je nachdem welche mRNA-kodierten Proteine eine stärkere Immunantwort in vitro bei dem jeweiligen Patienten induzierte. Um die Generierung von solchen individuellen Impfstoffen zu vermeiden, könnten Polytop-mRNA-Konstrukte entwickelt werden. Diese sollten für die häufigsten HLA-Allele hoch affine und effiziente CTL-Epitope kodieren, die zudem als Nef-resistente Epitope definiert wurden. Die individuellen mRNA-Konstrukte wären dann nur noch für einzelne Patienten nötig, die z.B. seltene HLA-Allele tragen. 135 6 Material und Methoden 6.1 Material 6.1.1 Patienten Zur Vereinfachung wird in der vorgelegten Dissertation nur „Patient" geschrieben und keine Unterscheidung des Geschlechtes gemacht. Viruslasten und CD4+ T-Zellzahlen wurden routinemäßig bei allen Patienten stets erhoben. Die Ermittlung der Viruslast erfolgte im Institut für Klinische und Molekulare Virologie Erlangen unter Verwendung des Versant HIV-1 RNA Assays (Bayer Diagnostik, Leverkusen). Die CD4+ T-Zellzahl wurde im zellulären Routinelabor (von Frau S. Bergmann) bestimmt. Studien zur Erkennung von HIV-1-Epitopen schlossen insgesamt 167 HIV-1-infizierte Patienten aus der Erlanger HIV-Kohorte ein, die in der Ambulanz der Medizinischen Klinik 3 in Behandlung sind. Zur Untersuchung der T-Zellantworten gegen autologe Virussequenzen wurden neun HIV-1-infizierte Patienten und fünf negative Kontrollprobanden analysiert. Daten über die verwendeten HIV-1-negativen Individuen sind in Tabelle 6.1 zu finden. Daten zu den neun HIV-1-infizierten Patienten gegen welche die Immunantwort gegen autologe Virussequenzen erfolgte sind in Tabelle 4.2 (unter 4.1.6) zusammengefasst. Tabelle 6.1 Daten der verwendeten HIV-1-negativen Kontrollprobanden Kontroll ID Geschlecht HLA Klasse I 0039 W A*03, A*23, B*07, B*62, Cw*04, Cw*07 0119 W A*01, A*03, B*07, B*08, Cw*07, Cw*07 0204 M A*01, A*11, B*08, B*35, Cw*04, Cw*07 0238 W A*02, A*02, B*39, B*60, Cw*03, Cw*12 0314 W A*02, A*02, B*18, B*57, Cw*05, Cw*07 6.1.2 Verbrauchsmateralien Tabelle 6.2 Verwendete Verbrauchsmaterialien Material ® ABgene PCR plates; ThermoFast® 96 detection plates ABsolute QPCR Seal (Folien) Autoklavierband Combitips plus Firma, Stadt, Land Thermo Scientific, Surrey, UK Thermo Scientific, Surrey, UK Sericlin®, Feuchtwangen Eppendorf, Hamburg 136 6 Material und Methoden 0,5ml; 2,5ml; 5ml; 12,5ml Cryo Röhrchen 2ml Einmalspritze 1ml Omnifix Duo Elektroporationsküvetten, 4mm e.p. Tips Standard 0,1-1µl; 0,5-20µl Falcons 50ml Falcons 15ml Filter Tip Universal 1000µl; 200µl; 100µl; 20µl; 10µl Gewebekulturschale 20mm MACS Separation columns MS/LS Minisart® Filter 0,45µm MultiscreenHTS Filter Plates, 96well Nunc Immuno™ Modules PCR-Reaktikonsgefäße Petrischale 92x16 mm ® PipetteTip Gilson Style blue/yellow, steril Polystyrol, Rundboden-Röhrchen 5ml Reaktionsgefäße 1,5ml Röhre 13ml 100x16mm Serologische Pipetten 2ml, 5ml, 10ml, 25ml Skalpell No. 10 Spritze 10ml/20ml Stericup Filtrationssystem 0,22µm; 180ml & 500ml Sterile Einweghandschuhe, Nitril UVette® Zahnstocher Zellschaber Zellsieb 30µm; 70µm Zellkulturflaschen 25cm2, 75cm2, 175cm2 Zellkulturplatten 6-, 12-, 24-, 48-, 96-well Greiner bio-one, Frickenhausen B. Braun Melsungen AG, Melsungen BioRad, München Eppendorf, Hamburg BD Falcons, Heidelberg Greiner bio-one, Frickenhausen Greiner bio-one, Frickenhausen Greiner bio-one, Frickenhausen Miltenyi, Bergisch Gladbach Sartorius, Göttingen Merck Millipore, Darmstadt Nuncbrand, Roskilde, Dänemark Kisker Biotech GmbH, Steinfurt Sarstedt, Numbrecht Greiner bio-one, Frickenhausen Sarstedt, Numbrecht Sarstedt, Numbrecht Sarstedt, Numbrecht Sarstedt, Numbrecht Feacher Safety Razor, Osaka, Japan BD Discardit TM II, Heidelberg Millipore, Schwalbach Medline Industries, Mundelein, USA Eppendorf, Hamburg Carl Roth, Karlsruhe Greiner bio-one, Frickenhausen BD, Heidelberg Greiner bio-one, Frickenhausen Greiner bio-one, Frickenhausen 6.1.3 Laborgeräte Tabelle 6.3 Verwendete Laborgeräte Gerät Firma, Stadt, Land 7300 Real Time PCR System AID EliSpot Reader Autoklav Brutschrank (Bakterien) Brutschrank (Zellkultur) Durchflusszytometer: FACS Aria FACS Calibur MoFlow™ Eismaschine Elektroporator Gene Pulser Xcell Gelkammer Klicker Kühleinrichtungen: Gefrierschränke (-20° C) Applied Biosystems, Foster City, USA Autoimmun Diagnostika GmbH, Strassberg Webeco, Selmsdorf Heraeus ,Hanau Heraeus, Hanau BD, Heidelberg BD, Heidelberg ® Beckman Coulter , Krefeld Ziegra, Isernhagen BioRad, München Febikon GmbH, Wermelskirchen Eppendorf, Hamburg Bosch, Liebherr, Siemens 137 6 Material und Methoden Gefrierschränke (-80° C) Kühlschrank (4° C) Kryokonservierungsbehälter MACS-Apparatur Multi-Stand Mikroskope: Auflichtmikroskop Auflichtmikroskop (Labovert) Fluoreszenzmikroskop (IX70) Minizentrifuge NanoDrop® ND 1000 Neubauer-Zählkammer PCR-Maschinen: MyCycler™ thermal cycler UNO-Thermoblock ® Gene Amp PCR System 9700 pH-Meter (Checker) Photometer Pipetboy Pipetten Schüttler: ® CERTOMAT BS-T (Bakterien) VIBRAX-VXR (Platten) Sirius (Luminometer) Spannungsgeräte: GPS 200/400 PowerPAC 300 SPECTRA MAX 190 ® Sterilbank (LaminAir HB2472) TECAN (Elispotwasher) Thermoblock (DB1010) Thermomixer TQ-Prep Workstation UV-Tisch (MultiImage™ Light Cabinet) Vortexer: Labdancer MS1 Minishaker VF2 Waagen: Feinwaage Grobwaage (EMB600-2) Wasseraufbereitungsanlage Wasserbad Zentrifugen: Biofuge 28S Centrifuge 5415D Megafuge 1.0 Rotanta 460R ROTIXA RP Varifuge GL Heraeus Herafreeze, Hanau Liebherr, Ochsenhausen Thermo Fisher Scientific NALGENE, Langenselbach Miltenyi, Bergisch-Gladbach Carl Zeiss, Oberkochen Leica, Wetzlar Olympus, Hamburg Carl Roth, Karlsruhe Peqlab, Erlangen Carl Roth, Karlsruhe BioRad, München Biometra GmbH, Göttingen Applied Biosystems, Foster City, USA ® Hanna Instruments, Woonsocket, USA Eppendorf, Hamburg IBS Integra Bioscience, Fernwald Eppendorf, Hamburg Gilson, Bad Camberg B.Braun Biotech International, Melsungen ® IKA JK, Staufen Berthold Detection Systems, Pforzheim Pharmacia LKB, Freiburg BioRad, München Molecular Devices, Sunnyvale, USA Heraeus, Hanau TECAN, Crailsheim Alpha Laboratories, Hampshire, UK HLC Biotech, Göttingen ® Beckman Coulter , Krefeld Biozym, Oldendorf ® IKA JK, Staufen ® IKA JK, Staufen ® Janke&Kunkel Labortechnik IKA , Staufen Mettler/Spoerhase, Gießen Kern, Balingen Millipore, Schwalbach Memmert, Schwabach Heraeus, Hanau Eppendorf, Hamburg Heraeus, Hanau Hettich Zentrifugen, Tuttlingen Hettich Zentrifugen, Tuttlingen Heraeus Christ, Hanau 138 6 Material und Methoden 6.1.4 Chemikalien Tabelle 6.4 Verwendete Chemikalien Chemikalie Firma, Stadt, Land AEC- Substrat (3-Amino-9-Ethylcarbazol) Agarose Ampicillin Aqua Atelokollagen Sigma-Aldrich, Steinheim Avidin/Peroxidase Lösung Borsäure (H3BO3) BSA (Bovine Serum Albumin) (Albumin Fraktion V, mind. 98%) BSA (Bis-trimethylsilylacetamid) (10x) β -Mercaptoethanol Calciumchlorid (CaCl2) Chitosan Chloroform DAPI (4′,6-Diamidin-2-phenylindol) DEPC (Diethylpyrocarbonat) DMF (N,N-Dimethylformamid) DMSO (Dimethylsulfoxid) mind. 99,5 % GC DMSO (Dimethylsulfoxid) (für PCR) dATP, dCTP, dGTP, dTTP (je 100 mM) dNTP-Mix (10 mM) DTT (Dithiothreitol) EDTA (Ethylendiamintetraacetat) Essigsäure (C2H4O2) Ethanol Ethidiumbromidlösung (1%) FuGene HD Transfection Reagent FuGene HD Transfection Reagent Glucose D+ Glycerol Isoamylalkohol Isopropanol Kaliumacetat Kaliumchlorid (KCl) Kanamycin Gel loading Dye blue (6x) LB-Agar (Lennox) LB-Medium (Lennox) Lymphoflot Magnesiumchlorid (MgCl2) (50 mM) Magnesiumchloridlösung (MgCl2) Magnesiumsulfatlösung (Mg2SO4) Manganchlorid (MnCl2) Methanol MOPS (3-Morpholinopropansulonsäure) Natriumacetat (C2H3NaO2) Natriumacetat-Trihydrat Carl Roth, Karlsruhe Carl Roth, Karlsruhe B.Braun Melsungen AG, Melsungen Koken Co. Ltd, Tokio, Japan (M. Tomcik, AG Distler) Sigma-Aldrich, Steinheim Merck, Darmstadt Carl Roth, Karlsruhe NEB, Frankfurt a.M. Sigma-Aldrich, Steinheim Sigma-Aldrich, Steinheim Sigma-Aldrich, Steinheim (K. Goldmann, AG Spriewald) Merck, Darmstadt Roche, Mannheim Carl Roth, Karlsruhe Merck, Darmstadt Merck, Darmstadt Invitrogen, Karlsruhe PAN Biotech GmbH, Aidenbach BioRad, München Sigma-Aldrich, Steinheim Merck, Darmstadt Merck, Darmstadt Merck, Darmstadt Merck, Darmstadt Roche, Mannheim Promega, Madison, USA Merck, Darmstadt Sigma-Aldrich, Steinheim Merck, Darmstadt Merck, Darmstadt Merck, Darmstadt Sigma-Aldrich, Steinheim ® Gibco life technologies, Karlsruhe NEB, Frankfurt a.M. Carl Roth, Karlsruhe Carl Roth, Karlsruhe BioRad, Dreieich Inno-Train Diagnostik GmbH, Kronberg/Taunus Merck, Darmstadt Merck, Darmstadt Sigma-Aldrich, Steinheim Sigma-Aldrich, Steinheim Merck, Darmstadt Merck, Darmstadt Merck, Darmstadt 139 6 Material und Methoden (Na(CH3OO)*3 H2O) Natriumchlorid (NaCl) Natriumhydrogencarbonat (NaHCO3) Natriumhydroxid (NaOH) Plätzchen NEB Buffer 1-4 (10x) Oligo dT Primer DPBS (Phosphat-buffer saline) Phenol-Chloroform-Isoamylalkohol (25:24:1) PFA (Paraformaldehyd) Pfu-Puffer (10x) pNPP (p-Nitrophenyl Phosphate) Puffer S (10x) Propidiumjodid Random Primers (500µg/ml) RNA Loading Dye Salzsäure (HCl) SAP-Buffer (10x) (shrimp alkaline phosphatase) Saponin SDS-Lösung (10%) (Natriumdodecylsulfat) SOB ssRNA Ladder Loading buffer (2x) T4 Ligase buffer (10x) Tris (Tris-(hydroxymethyl)-aminomethan) Trypanblau Tween-20 Wasserstoffperoxid (H2O2) Sigma-Aldrich, Steinheim Merck, Darmstadt Carl Roth, Karlsruhe NEB, Frankfurt a.M. Biomers.net, Ulm ® Gibco life technologies, Karlsruhe Amresco, Solon, USA Carl Roth, Karlsruhe Genaxxon GmbH, Ulm Sigma-Aldrich, Steinheim Genaxxon GmbH, Ulm Sigma-Aldrich, Steinheim Promega, Madison, USA NEB, Frankfurt a.M. Merck, Darmstadt Roche, Mannheim Sigma-Aldrich, Steinheim AppliChem GmbH, Darmstadt Carl Roth, Karlsruhe NEB, Frankfurt a.M. NEB, Frankfurt a.M. Carl Roth, Karlsruhe Merck, Darmstadt Merck, Darmstadt Merck, Darmstadt 6.1.5 Puffer und Lösungen Tabelle 6.5 Verwendete Puffer und Lösungen Puffer/Lösung Zusammensetzung Coating Solution (ELISA) 0,1 M NaHCO3, pH=8,2 Lösung 1 (Plasmidpräperation) 50 mM Tris pH=7,5 10 mM EDTA 100 µg/ml RNase A Lösung 2 (Plasmidpräperation) 10% SDS 0,2 M NaOH Lösung 3 (Plasmidpräperation) 1,32 M Kaliumacetat pH=4,8 (Eisessig zur pH-Einstellung) Lösung AB (Elispot), Acetatpuffer Lösung A: 100 mM Natriumactetat-Trihydrat Lösung B: 100%Essigsäure 0,5% (v/v) Lösung A und B im Verhältnis1:1 MACS-Puffer PBS 10% BSA MOPS (10x) 200 mM MOPS 1 M Sodiumacetat 0,5 M EDTA (pH=8,0) RNase-freies Wasser 140 1% (v/v) 0,5% (v/v) 6 Material und Methoden Permeabilisierungspuffer Sekretionsassay-Puffer TBE (10x) PBS Saponin 10% BSA PBS 10% BSA 2 mM EDTA 0,3% (v/v) 1% (v/v) 0,5% (v/v) 1 M Tris 0,83 M Borsäure 0,01 M EDTA 6.1.6 Kulturmedien und Zusätze Tabelle 6.6 Verwendete Medien und deren Zusätze Medium/Zusatz Firma, Stadt, Land DMEM (1x) Fötales Kälberserum (FCS) (SeraPlus S), hitzeinaktiviert GM-CSF L-Glutamin Hanks Lösung Hepes Humanes AB Serum Interleukin-1β Interleukin-2 (rhIL2) 5 Interleukin-4 (10 U/ml) Interleukin-6 OptiMEM® I Reduced Serum Medium Penicillin/Streptavidin PGE2 PHA RPMI1640 TNF-α Trypsin-ETDA-Solution (10x) Gibco life technologies, Karlsruhe PAN Biotech GmbH, Aidenbach ® Sigma-Aldrich, Steinheim ® Gibco life technologies, Karlsruhe Apotheke des Universitätsklinikums Erlangen Merck, Darmstadt Sigma-Aldrich, Steinheim Sigma-Aldrich, Steinheim Chiron BV, Amsterdam, Niederlande PeProTech Inc., Rocky Hill, USA Sigma-Aldrich, Steinheim ® Gibco life technologies, Karlsruhe ® Gibco life technologies, Karlsruhe Sigma-Aldrich, Steinheim Sigma-Aldrich, Steinheim ® Gibco life technologies, Karlsruhe Sigma-Aldrich, Steinheim Sigma-Aldrich, Steinheim Tabelle 6.7 Verwendete Zellkulturmedien und deren Zusammensetzung Zellkulturmedium Zusammensetzung D10 DMEM Penicillin/Streptavidin FCS, hitzeinaktiviert FCS, hitzeinaktiviert DMSO 1% (v/v) 10% (v/v) 40ml 10ml RPMI1640 Humanes AB Serum Penicillin/Streptavidin L-Glutamin 1% (v/v) 1% (v/v) 1% (v/v) Einfriermedium Leu-Medium 141 6 Material und Methoden R10 R10IL2 R20 R5AB RPMI1640 FCS, hitzeinaktiviert Penicillin/Streptavidin Hepes 1 M L-Glutamin (2 mmol/l) 10% (v/v) 1% (v/v) 1% (v/v) 1% (v/v) RPMI1640 FCS, hitzeinaktiviert Penicillin/Streptavidin Hepes 1 M L-Glutamin (2 mmol/l) Interleukin-2 10% (v/v) 1% (v/v) 1% (v/v) 1% (v/v) 6 1x10 U/ml RPMI1640 FCS, hitzeinaktiviert Penicillin/Streptavidin Hepes 1 M L-Glutamin (2 mmol/l) 20% (v/v) 1% (v/v) 1% (v/v) 1% (v/v) RPMI1640 Humanes AB Serum Penicillin/Streptavidin Hepes 1 M L-Glutamin (2 mmol/l) 5% (v/v) 1% (v/v) 1% (v/v) 1% (v/v) Tabelle 6.8 Verwendete Medien zur Kultivierung von Bakterien Bakterienmedium Zusammensetzung LB-Amp-Medium LB-Medium (Lennox) H20 autoklavieren 200 mg/ml Ampicillin LB-Kan-Medium LB-Amp-Agar LB-Kan-Agar LB-Medium (Lennox) H20 autoklavieren 50 mg/ml Kanamycin LB-Agar (Lennox) H20 autoklavieren 50 mg/ml Kanamycin LB-Agar (Lennox) H20 autoklavieren 50 mg/ml Kanamycin SOC-Medium 20mM Glucose SOB mit H2O auffüllen auf 10ml TbF I 0,03 M Kaliumacetat 0,05 M MnCl2 0,01 M CaCl2 Glycerol 0,1 M KCl 142 20g 1l 100 µg/ml 20g 1l 50 µg/ml 35g 1l 50 µg/ml 35g 1l 50 µg/ml 36mg 307mg 15% (w/v) 6 Material und Methoden TbF II 0,01 M Na-MOPS pH=7,0 75 mM CaCl2 Glycerol 0,01 M KCl Tym-Broth 15% (w/v) 2% Trypton-Pepton 0,5% Bacto-Yeast-Extrakt 0,1 M NaCl 0,01 M MgCl2 oder MgSO4 6.1.7 Peptide und Proteine Peptide wurden entweder von der Firma EMC microcollections (Tübingen) synthetisch als C-terminale Carboxamide hergestellt oder sie wurden vom NIH AIDS Research and Reference Reagent Program, vom AIDS Reagent Project of the Medical Research Council (MRC, Herts, UK) oder dem European Community EVA Programme zur Verfügung gestellt. Die Peptide p17, p15 und p24 des MRC entsprechen der Sequenz von HIV-1 SF2, Nef Peptide des MRC der Sequenz von HIV-1 LAI. Sie sind entweder 20 (Nef, p24) oder 15 Aminosäuren (p17, p15) lang und überlappen in 10 Aminosäuren miteinander. Peptidsets vom NIH bestehen aus 15 Aminosäuren langen Peptiden, die der Consensus B Sequenz entsprechen und in 11 Aminosäuren miteinander überlappen. Peptid-Pools bestehen aus bis zu fünf verschiedenen Peptiden mit einer Endkonzentration von jeweils 0,5 mg/ml. Die verwendeten Proteine, wie HIV-1 Tat und HIV-1 Nef, stammen ebenfalls vom NIH. 6.1.8 Antikörper Tabelle 6.9 Verwendete Antikörper Antikörper Firma, Stadt, Land Anti-CD4 Mouse IgG2a Anti-Flag clone M2 Anti-human IFN-γ mAb1-D1K Anti-human IFN-γ mAb7-B6-1-Biotin Anti-human TNF-α Biotin Mouse anti-human TNF-α IgG1-APC IgG1-FITC IgG1, Kappa from murine myeloma IgG1-PE IgG2a-FITC IgG2a-PE Anti-human CD3-FITC Anti-human CD3-PE Anti-human CD4-APC Anti-human CD4-FITC Anti-human CD4-PE Acris Antibodies GmbH, Herford Sigma-Aldrich Chemie GmbH, München MABTECH AB, Hamburg MABTECH AB, Hamburg BD Pharmingen, San Diego, USA BD Pharmingen, San Diego, USA Beckman Coulter, Fulleton, USA Beckman Coulter, Marseille, Frankreich Sigma-Aldrich Chemie GmbH, München Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich 143 6 Material und Methoden Anti-human CD8-FITC Anti-human CD8-PE Anti-human CD8 pure Anti-human CD14-FITC Anti-human CD14-PE Anti-human CD16-FITC Anti-human CD16-PE Anti-human CD19-FITC Anti-human CD19-PE Anti-human CD56-FITC Anti-human CD56-PE Anti-human CXCR4-FITC gp120 AD3 gp120 697-3D gp120-160 Chessie 6 HLA A2, 28 IgG2a FITC Nef EH1 p24 AG3.0 p24 183/H12 Goat Anti-Maus IgG FITC Rat Anti-Maus IgG1 PE Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich BD Bioscience, San Jose, USA Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich Beckman Coulter, Marseille, Frankreich R&D Systems, Minneapolis, USA NIH, Bethesda, USA NIH, Bethesda, USA NIH, Bethesda, USA One Lamda Inc., Canoga Park, USA NIH, Bethesda, USA NIH, Bethesda, USA NIH, Bethesda, USA Southern Biotech, Birmingham, USA BD Pharmingen, San Diego, USA 6.1.9 Kommerziell erhältliche Kits Tabelle 6.10 Verwendete kommerziell erhältliche Kits Kit Firma, Stadt, Land ABsolute qPCR Rox Mix Agilent RNA 6000 Nano Kit BigDye Sequenzierkit + CD4 T cell isolation Kit human CellTrace™ CFSE Cell Proliferation Kit Dual-Luciferase Reporter Assay System Fixation and Dead Cell Discrimination Kit Human Th1/Th2 Cytokine Kit II CBA miRNeasy Mini Kit ® mMESSAGE mMACHINE T7 Ultra Kit ® NucleoBond PC500 ® NucleoSpin Blood ® NucleoSpin Extract II PureLink™ RNA Mini Kit QIAamp Viral RNA isolation Mini Kit RNeasy Mini Kit ® TaqMan MicroRNA Reverse Transcription Kit Vectastain ABC Kit Thermo Scientific, Surrey, UK Agilent, Böblingen Applied Biosystems, Weiterstadt Miltenyi Biotec, Bergisch Gladbach Molecular Probes, Eugene, USA Promega, Mannheim Miltenyi Biotec, Bergisch Gladbach BD, Heidelberg Qiagen, Hilden AMBION, Austin Texas, USA Macherey-Nagel, Düren Macherey-Nagel, Düren Macherey-Nagel, Düren AMBION, Austin Texas, USA Qiagen, Hilden Qiagen, Hilden Applied Biosystems, Foster City, USA 6.1.10 Linearis, Wertheim Enzyme Tabelle 6.11 Verwendete Enzyme Enzym Firma, Stadt, Land DNase-freie RNase Pfunds Polymerase (2,5U/µl) Invitrogen, Darmstadt Genaxxon Bioscience, Ulm 144 6 Material und Methoden Restriktionsenzyme BamHI, EcoRI, NotI, SacI, SpeI, XbaI RNasin® (recombinant ribonuclease inhibitor) SAP (shrimp alkaline phophatase) (10U/µl) Superscript II Reverse Transcriptase Superscript III Reverse Transcriptase Taq Polymerase (5U/µl) T4 DNA Ligase (20.000U) 6.1.11 NEB, Frankfurt a.M. Promega, Madison, USA Roche, Mannheim Invitrogen, Darmstadt Invitrogen, Darmstadt Genaxxon Bioscience, Ulm NEB, Frankfurt a.M. Längenstandards für Gelelektrophoresen Tabelle 6.12 Verwendete Größenstandards Längenstandard Firma, Stadt 1kb DNA Ladder 100bp DNA Ladder ssRNA Ladder NEB, Frankfurt a.M. NEB, Frankfurt a.M. NEB, Frankfurt a.M. 6.1.12 Vektoren Tabelle 6.13 Verwendete Vektoren Verwendete Vektoren Herkunft (Name, Institut / Firma, Stadt) DpSF2 pcDNA3 pcDNA3 flag pDrive Cloning Vector pEGFP-C1 pGEM4Z64A pGEMsigsurvivinDClamp pmirVec-29a pNL4-3Δenv pNL4-3ΔPR psiCHECK3 6-pHXB2-18 AG Schubert, Virologie, Erlangen Invitrogen, Darmstadt J.Wittmann, Immunologie, Erlangen Qiagen, Hilden BD, Heidelberg J.Dörrie, N.Schaft, Dermatologie, Erlangen J.Dörrie, N.Schaft, Dermatologie, Erlangen J.Wittmann, Immunologie, Erlangen Virologie, Erlangen Virologie, Erlangen J.Wittmann, Immunologie, Erlangen AG Schubert, Virologie, Erlangen 6.1.13 Bakterien, Viren, Zellen und Zelllinien Tabelle 6.14 Verwendete Bakterienstämme Bakterienstamm Herkunft E. coli SCS110 E. coli XL2 Blue E. coli XL10 Gold Stratagene, La Jolla, USA Stratagene, La Jolla, USA Stratagene, La Jolla, USA 145 6 Material und Methoden Tabelle 6.15 Verwendete Vaccinia-Viren Viren Beschreibung Herkunft Vacc SF2 gag Rekombinates Vaccinia Virus, welches das Gag-Protein von HIV-1 SF2 exprimiert NIH AIDS Reagent Program, Bethesda, USA vP1218 Rekombinates Vaccinia Virus, welches das Nef-Protein von HIV-1 MN exprimiert NIH AIDS Reagent Program, Bethesda, USA Vsc8 Rekombinates Vaccinia Virus, welches das β-Galaktosidase Protein exprimiert NIH AIDS Reagent Program, Bethesda, USA Tabelle 6.16 Verwendete Zellen und Zelllinien Zellen/Zelllinie Beschreibung Herkunft EBV-transformierte lymphoide B-Zelllinien (B-LCL) Humane Blutlymphozyten, welche mit Cyclosporin und Überstand der Zelllinie B95-8, die hohe Titer an EBV freisetzt, transformiert wurden HIV-positive/ -negative Spender der AG Harrer Dendritische Zellen PBMC, welche durch die Kultivierung mit IL-1β, IL-4, IL-6, GM-CSF-4, TNF-α, PGE2 zu humanen dendritische Zellen gereift sind und positiv für die Oberflächenmoleküle CD83 und CD86 sind HIV-negative Spender der AG Harrer HEK293 Humane embryonale Nierenzelllinie, welche mit Adenovirus Typ 5 transformiert wurde NIH AIDS Reagent Program, Bethesda, USA HEK293T Derivat der humanen embryonalen Nierenzelllinie (293), welche zusätzlich ein Plasmid mit dem SV40 large TAntigen enthält NIH AIDS Reagent Program, Bethesda, USA Makrophagen Humane Makrophagen, welche positiv sind für CD16 und CD61, isoliert aus PBMC HIV-negative Spender Primäre CD4 T-Zellen Humane T-Zellen, welche positiv anhand des CD4Oberflächenmoleküls sortiert wurden; isoliert aus PBMC HIV-negative Spender PBMC Periphere mononukleäre Blutzellen, die aus Citratblut isoliert wurden HIV-positive/ -negative Spender der AG Harrer + 146 6 Material und Methoden 6.1.14 Oligonukleotidprimer Tabelle 6.17 Verwendete Oligonukleotidprimer für PCR und Klonierung Name mit Richtung Sequenz mit Schnittstelle Anwendung env6122s env7815a gtt gtg tgg tcc ata gta atc ata g cca tag tgc ttc ctg ctg ctc c env6193 XbaI s env7783 EcoRI a env6214 o. ATG BamHI s env7783 XbaI a gag764s PR2352a cgt cta gat aga cta ata gaa aga gca gaa gac cgg aat tca gga aca aag ctc cta ttc cca ctt a cgg gat cca gag tga agg aga agt atc agc ac PCR 1.Runde RT-PCR, PCR 1.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde gag771 XbaI s gag2311 EcoRI a gag771 BamHI s gag2305 SacI a gag771 EcoRV s gag2311 XbaI a gag793 o. ATG BamHI s GFP BamHI flag s cgt cta gag gag gct aga agg aga gag at cgg aat tcc ctt tag ttg ccc ccc tat ct cgg gat ccg gag gct aga agg aga gag at cat gag ctc tgc ccc cct atc ttt att gtg cgg ata tcg gag gct aga agg aga gag at cgt cta gac ctt tag ttg ccc ccc tat ct cgg gat ccg gtg cga gag cgt cgg tat t cgt cta gaa gga aca aag ctc cta ttc cca ctt a gac tag cgg agg cta gaa g cta ata ctg tat cat ctg ctc ctg nef8675s nef9524a cgg gat ccg cgc cac cat gga tta caa gga tga cga cga taa ggt gag caa ggg cga gga gct gtt cac c gca gta gct gag ggg aca gat agg tgg tct aac cag aga gac cca gta c nef9545a gag acc cag tac agg caa aaa gc nef8636 BamHI s nef8636 XbaI s nef8780 XbaI flag s cgg gat ccg cca cat acc tag aag aat aag aca gc cgt cta gac aca ta cct aga aga ata aga cag c cgt cta gag cgc cac cat gga tta caa gga tga cga cga taa ggg ggg caa gtg gtc aaa aaa tag tct ggc cga att ccc acg cct ccc tgg aaa gtc cc cgg gat ccg gtg gca agt ggt caa aag gta nef9439 EcoRI a nef8780 o. ATG BamHIs LTR9687 EcoRI a tat5819 BamHI flag s tat8474 EcoRI a cgg aat tca aag ggt ctg agg gat ctc tag tt cgg gat ccg cgc cac cat gga tta caa gga tga cga cga taa gga gcc agt aga tcc taa tct aga gcc ctg g cgg aat tcc taa tcg aac gga tct gtc tc Tabelle 6.18 Verwendete Oligonukleotidprimer zur Sequenzierung Name und Richtung Sequenz env6342s env7002s env7337a gag880s gag1001a gag1095a gag1171s gag1196a gag1406s gag1484a gag1819s gag2044a tat ggg gta cct gtg tgg ctg tta aat ggc agt cta gca gaa aat ttc trg atc ccc tcc tg tra aac aty tag tat ggg caa gca g cct gtc tga agg gat ggt tgt agc tg gtc taa rgc ttc ytt ggt gtc gtc agc caa aat tat cct ata gt tgc act ata ggg taa ttt tg atg agg aag ctg cag aat ggg gtt cct gct atg tca ctt cc gaa gaa atg atg aca gca tgt cag gg gaa gga cac caa atg aaa gat g 147 PCR 2.Runde PCR 1.Runde RT-PCR, PCR 1.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde PCR 1.Runde RT-PCR, PCR 1.Runde RT-PCR, PCR 1.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde PCR 2.Runde 6 Material und Methoden nef8950s gtg cct ggc trg aag cac aag a nef9035a gtc att ggt ctc aaa ggt acc tgy gg nef9133s tca ggg aar tag cct tgt gtg t nef9148s caa ggc tac ttc cct gat tgg ca nef9169a gcc aat cag gga agw agc ctt gtg t pEGFP-N1 seq s gcc cca ttg acg caa atg ggc ggt ag pEGFP-N1 seq s tgg cat cgc cct cgc cct cgc cgg aca cgc Sp6a gat tta ggt gac act ata g T7s taa tac gac tca cta tag gg s: sense (forward Primer), a: antisense (reverser Primer), eingebrachte Schnittstellen sind mit einem Unterstrich dargestellt 6.1.15 TaqMan® MicroRNA Assays ® Tabelle 6.19 Verwendete TaqMan MicroRNA Assays für qRT-PCR TaqMan® MicroRNA Assay Firma, Stadt, Land let-7a let-7b let-7f hsa-miR-26b has-miR-27a hsa-miR-30c hsa-miR-142-3p has-miR-142-5p has-miR-146a has-miR-148a has-miR-150 has-miR-342 Has-miR-1307 RNU6B RNU44 Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA Applied Biosystems, Foster City, USA 6.1.16 Datenbanken Tabelle 6.20 Verwendete Datenbanken Name Link EXPASY HIV Database MAPPP MicroInspector miRanalyzer NCBI Pubmed SYFPEITHI UCSC Virtual PRA calculation http://www.expasy.ch/ http://www.hiv.lanl.gov/content/index http://www-alt.mpiib-berlin.mpg.de/MAPPP/cleavage.html http://bioinfo.uni-plovdiv.bg/microinspector/ http://bioinfo2.ugr.es/miRanalyzer_dev/miRanalyzer.php http://www.ncbi.nlm.nih.gov/ http://www.ncbi.nlm.nih.gov/sites/entrez?db=PubMed http://www.syfpeithi.de/ http://genome.ucsc.edu/ http://www.etrl.org/etrlpra/webform1.aspx 148 6 Material und Methoden 6.1.17 Software Tabelle 6.21 Verwendete Software Software Firma, Stadt, Land 2100 Expert Software ABI7300 System Software EliSpot Reader 5.0 FACS Express V3 Vector NTI 9 Agilent Technologies, Böblingen Applied Biosystems, Foster City, USA Autoimmun Diagnostika, Strassberg DeNovo Software, California, USA Informax/Invitrogen, Carlsbad, USA 6.2 Methoden 6.2.1 Molekularbiologische Methoden 6.2.1.1 Isolation von Nukleinsäuren Isolation von viraler RNA aus Plasma Um virale RNA aus Plasma von Patienten zu isolieren, wurde der Qiagen-Kit „QIAamp Viral RNA Mini Kit“ verwendet. Es wurde nach Herstellerprotokoll vorgegangen. Insgesamt wurden vier Ansätze von jeweils 140µl Plasma auf einer Säule vereinigt, um die Ausbeute der RNA zu erhöhen, insbesondere bei Patienten mit geringen Viruslasten. Isolation von DNA bzw. RNA aus Zellen Die Isolation genomischer DNA aus Zellen erfolgte mit Hilfe des Kits „NucleoSpin® Blood“ von Machery-Nagel. Zur Gewinnung von zellulärer RNA diente der „PureLink™ RNA Mini Kit“ (AMBION). Für die Analyse von microRNAs wurde jedoch der „miRNeasy Mini Kit“ von Qiagen zur Isolation von zellulärer RNA verwendet, da dieser auch kleine RNAMoleküle wie miRNAs berücksichtigt und hoch effizient aufreinigt. Es wurde stets nach Herstellerprotokoll vorgegangen. Isolation von Plasmid-DNA Minipräperationen Die Zellen einer Übernachtkultur (5ml LB-Medium + Antibiotika und Bakterienstamm) wurden am nächsten Tag durch Zentrifugation (3.000rpm, 10min, Raumtemperatur; Zentrifuge ROTIXA RP) geerntet. Die folgenden Zentrifugationsschritte wurden alle in der Centrifuge 5415D bei Raumtemperatur durchgeführt. Die Zellpellets wurden in 300µl Lösung 1 resuspendiert und in ein 1,5ml Reaktionsgefäß überführt. Nach Zugabe von 300µl Lösung 2 und kurzem Mischen wurden 300µl Lösung 3 hinzugefügt, erneut 149 6 Material und Methoden gemischt und 5 Minuten auf Eis inkubiert. Die Ansätze wurden zentrifugiert (13.000rpm, 10min) und der Überstand in ein neues Reaktionsgefäß überführt. Nach Zugabe von 450µl Isopropanol und erneuter Zentrifugation (13.000rpm, 15min) wurde das Pellet einmal mit 1ml 70% Ethanol gewaschen (13.000rpm, 5min). Nachdem das Pellet vollständig getrocknet war, wurde es in 35µl RNase-freiem Wasser gelöst. Die Plasmidisolation wurde anhand eines 1%igen Agarosegels überprüft. Maxipräperationen Es wurden 300ml LB-Medium inklusive dem notwendigen Antibiotika mit der Bakterienkolonie inokuliert und über Nacht bei 37° C sowie 200rpm inkubiert. Am nächsten Tag wurden die Zellen der Übernachtkultur geerntet (4.000rpm, 10min, 4° C). Anschließend wurde die Plasmid-DNA nach Herstellerprotokoll des Kits „NucleoBond® PC500“ (Macherey-Nagel) isoliert. Alle Zentrifugationen wurden in der Zentrifuge ROTIXA RP ausgeführt, allerdings mit einer maximalen Umdrehungszahl von 4.000rpm, so dass die Zentrifugationszeiten dementsprechend verlängert wurden. Zum Schluss wurde die DNA in 150-200µl RNase-freiem Wasser gelöst und die Konzentration photometrisch bestimmt. 6.2.1.2 Konzentrationsbestimmung von Nukleinsäuren Da die Basen der Nukleinsäuren ultraviolettes Licht bei einem Absorbtionsmaximum von 260nm absorbieren, kann die Konzentration von Nukleinsäuren mit Hilfe eines Photometers, welches Licht der Wellenlänge von 260nm ausstrahlt, bestimmt werden. Hierfür wurden Lösungen von Nukleinsäuren meist 1:50 mit Wasser verdünnt und die optische Dichte (OD) in einer Küvette (UVette®) am Photometer gemessen, woraus sogleich die Konzentration der DNA bzw. RNA errechnet wird. Die Konzentration der zellulären RNA, welche für Deep Sequencing oder qRT-PCR verwendet wurden, wurde am NanoDrop® bestimmt. 6.2.1.3 Polymerasen-Ketten-Reaktion (PCR) Oligonukleotide Oligonukleotid-Primer der Firma biomers.net GmbH wurden als lyophilisiertes Pellet geliefert und in sterilem, RNase-freiem Wasser gelöst. Die erhaltenen Stocklösungen (c=1 µg/µl) wurden für 1-3 Stunden bei Raumtemperatur und 800rpm geschüttelt. Daraufhin wurden die Lösungen auf eine Konzentration von 100 ng/µl verdünnt und mit 150 6 Material und Methoden einer Endkonzentration von 2 ng/µl in die Reaktion eingesetzt. Die Lagerung der Primerlösungen erfolgte bei -20° C. Reverse Transkription Die reverse Transkription (RT-PCR) beschreibt einen Vorgang, bei welchem aus einer beliebigen RNA-Sequenz eine komplementäre DNA-Sequenz (cDNA) synthetisiert wird, die als Matrize für eine PCR dienen kann. Dieser Schritt wird benötigt, wenn man virale oder zelluläre RNA amplifizieren möchte. Dazu wurden jeweils 11µl isolierte RNA (viral bzw. zellulär) in den Standardansatz des Herstellers der Superscript® II Reverse Transkriptase bzw. der Superscript® III Reverse Transkriptase (Invitrogen™) eingesetzt. Anstelle von 1µl RNaseOUT™ wurde allerdings 0,25µl RNasin® verwendet. Die cDNASynthese der HIV-1 Gene gag, nef oder env erfolgte stets mit Gen-spezifischen Primer (siehe 6.1.14). Die Amplifikationszeiten und -temperaturen liefen ebenfalls nach Protokoll des Herstellers ab. Nested-PCR Für die Amplifikation der autologen HIV-1 Gene zur Sequenzierung und Klonierung wurde eine nested-PCR angewendet, bei welcher in zwei PCR-Schritten die DNA amplifiziert wird. Dieses Verfahren ist besonders hilfreich, wenn wenig Probenmaterial vorhanden ist. Dabei liegen die Primer der zweiten PCR-Runde innerhalb des Abschnittes, welcher in der ersten PCR-Runde amplifiziert wird. Die Primer sind unter 6.1.14 nachzusehen. Es wurde die Pfunds Polymerase verwendet, welche eine 3’-5’ Exonukleaseaktivität besitzt, so dass die natürliche Fehlerrate der Polymerase um das zehnfache gesenkt wird. Ausgangsmaterial war in allen Fällen spezifische cDNA aus viraler RNA. Meist wurde der unten stehende Standardansatz pipettiert. Erhielt man jedoch nach Amplifikation kein PCR-Produkt, wurde in einigen Fällen die Menge der eingesetzten cDNA und des PCRProduktes verändert, um ein Amplifikat zu erhalten, oder alternative Primer wurden verwendet. 151 6 Material und Methoden HIV PCR 1.Runde: cDNA 8,0µl Primer forward 1,0µl Primer reverse 1,0µl Pfunds Puffer (10x) 5,0µl dNTPs (2,5 mM each) 4,0µl Pfunds Polymerase 0,4µl RNase-freies Wasser 30,6µl Gesamtvolumen 50,0µl HIV PCR 2.Runde: PCR-Produkt aus 1.Runde 5,0µl Primer forward 1,0µl Primer reverse 1,0µl Pfunds Puffer (10x) 5,0µl dNTPs (2,5 mM each) 4,0µl Pfunds Polymerase 0,4µl RNase-freies Wasser 33,6µl Gesamtvolumen 50,0µl Die Bedingungen der Amplifikation in beiden Runden waren identisch. Die AnnealingTemperatur betrug je nach Primerpaar zwischen 58° C und 60° C. Zur Amplifikation des env Gens wurde die Denaturierung im primären Schritt auf 10 Minuten verlängert. 94° C 2min 94° C 20sek 58°-60° C 30sek 72° C 2min 94° C 20sek 58°-60° C 30sek + 20sek/Zyklus 72° C 2min 72° C 10min 10 Zyklen 20 Zyklen PCR aus Plasmid-DNA Für die Amplifikation der HIV-1 Gene von Referenzviren standen Plasmide aus dem Institut für Klinische und Molekulare Virologie zur Verfügung. Es wurde nur eine PCR mit Plasmid-DNA aus Minipräperation als Ausgangsmaterial durchgeführt. Die PCR wurde 152 6 Material und Methoden gemäß der oben stehenden „HIV PCR 2.Runde“ durchgeführt, mit 5µl Plasmid-DNA und unter denselben Bedingungen. Zur Amplifikation von NL43 gag wurde das Plasmid pNL4-3Δenv verwendet. Die Amplifikation von NL43 env und nef erfolgte mit DNA des Plasmids pNL4-3ΔPR. Die Amplifikation von SF2 gag erfolgte mit Hilfe des Plasmids DpSF2. Die Klonierung von SF2 nef in pGEM4Z64A wurde bereits von Frau K. Zitzelsberger erfolgreich abgeschlossen. Des Weiteren wurden die HIV-1 Gene der Referenzviren auch in den Vektor pEGFP-C1 kloniert. Dazu wurde die PCR der HIV-1 Gene ebenfalls mit 5µl DNA aus Minipräperationen der genannten Plasmide durchgeführt. Jedoch wurden Primer verwendet, die kein ATG amplifizieren (siehe 6.1.14), da das amplifizierte Gen im Anschluss hinter die GFP-Sequenz im pEGFP-C1 kloniert wurde. 6.2.1.4 Agarosegelelektrophorese Sowohl die Aufreinigung als auch die Größen- und Mengenbestimmung von DNAFragmenten erfolgte über die Agarosegelelektrophorese. Dazu wurden 1-2%ige Agarosegele mit 1xTBE-Puffer und 0,1% Ethidiumbromid angesetzt. Die DNA-Fragmente wurden mit Auftragspuffer (6x) vermischt und in die Taschen des Gels pipettiert. Als Größenstandards wurden 3-8µl der 100bp bzw. 1kb DNA-Leitern mit auf das Gel aufgetragen. Die Elektrophorese fand in einer horizontalen Laufkammer statt, welche mit 1xTBE gefüllt war. Die angelegte Spannung betrug zwischen 80V und 140V. Da sich der rote Farbstoff Ethidiumbromid in die hydrophobe Umgebung der Basen der DNA einlagert, können die unterschiedlich großen DNA-Fragmente durch Bestrahlung mit UV-Licht sichtbar gemacht und mittels einer CCD-Kamera fotografisch dokumentiert werden. Aufreinigung von PCR-Produkten Vor Klonierung oder Sequenzierung der PCR-Produkte wurden die Fragmente zunächst über ein 1-1,5%iges Agarosegel aufgetrennt, die richtige Bande aus dem Gel ausgeschnitten und mit Hilfe des Kits „NucleoSpin® Extract II“ von Macherey-Nagel aufgereinigt, um Rückstände von Primern, dNTPs und Puffer aus der Amplifikationsreaktion zu entfernen. Dabei wurde nach Angaben des Herstellers vorgegangen. Die Elution der gebundenen DNA aus der Säule erfolgte jedoch mit 50µl RNase-freiem Wasser. Die Reinheit der PCR-Fragmente wurde anschließend mittels Agarosegelelektrophorese überprüft. 6.2.1.5 Sequenzierung Es wurde stets die Didesoxysequenzierung nach Sanger durchgeführt (218). Bei der Sequenzierung von PCR-Produkten wurden meist die Primer verwendet, welche auch in 153 6 Material und Methoden der PCR-Amplifikation eingesetzt wurden. Zudem standen weitere Primer zur Verfügung, welche unter 6.1.14 aufgelistet sind. Zur Sequenzierung von Plasmiden wurde entweder der Vektor-spezifische Primer T7sense oder ein Insert-spezifischer Primer benutzt. Die Menge der eingesetzten DNA von PCR-Produkten lag zwischen 1-5µl. Zur Sequenzierung von Plasmid-DNA wurden entweder 2µl (bei Minipräperationen) oder 0,5-1µl (bei Maxipräperationen) eingesetzt, so dass sich der unten stehende Ansatz ergibt: DNA X,Xµl Primer 0,50µl TM-Puffer (5x) 1,75µl BigDye 0,50µl RNase-freies Wasser X,Xµl Gesamtvolumen 10,00µl Die Amplifikation erfolgte unter folgenden Bedingungen: 96° C 1min 96° C 10sek 50° C 10sek 30 Zyklen 60° C 4min Nach der Reaktion wurden die Sequenzen im Humangenetischen Institut Erlangen gereinigt und am ABI3730 analysiert. Die Auswertung der Sequenzen erfolgte stets mit der Software Vector NTI. 6.2.1.6 Klonierungsmethoden Herstellung chemisch kompetenter Bakterien Es wurde zunächst eine Vorkultur angelegt. Dazu wurden 5ml Tym-Broth-Medium mit einer geringen Menge der Bakterien des Stammes E.coli XL 1 blue bzw. E.coli XL10 Gold inokuliert und über Nacht bei 37° C und 200rpm inkubiert. Diese Vorkultur wurde am nächsten Tag in 250ml Tym-Broth-Medium gegeben und für etwa weitere 3 Stunden bei 37° C und 200rpm inkubiert, bis eine OD600 von 0,5-0,6 erreicht war. Die Zellen wurden durch Zentrifugation geerntet (3.000rpm, 10min, 4° C; ROTIXA RP) und pro 100ml zentrifugierter Bakteriensuspension wurde das Zellpellet in 30ml Tbf I vorsichtig resuspendiert. Nach einer Inkubationszeit von 90 Minuten auf Eis wurde die Lösung erneut zentrifugiert (3.000rpm, 10min, 4° C). Nun wurde das Pellet pro 100ml 154 6 Material und Methoden zentrifugierter Bakteriensuspension in 4ml Tbf II vorsichtig resuspendiert und auf Eis in 1,5ml Reaktionsgefäße aliquotiert. Zuletzt wurden die Bakterien in flüssigem Stickstoff schockgefroren und bei -80° C gelagert. Restriktionsverdau des PCR Produktes Der Restriktionsverdau des gereinigten PCR-Produktes erfolgte in einem Gesamtvolumen von 20µl bzw. 50µl abhängig von der vorhandenen Menge des PCR-Fragmentes. Die spezifischen Schnittstellen wurden über die PCR-Primer (siehe 6.1.14) eingebracht und nach der multiple cloning site des Vektors pGEM4Z64A oder des Vektors pGEM4ZsigsurvivinDClamp ausgewählt. Die Auswahl des Puffers und der eventuelle Einsatz von BSA erfolgte nach Angaben des Herstellers NEB zu den entsprechenden Restriktionsenzymen. DNA aus PCR-Aufreinigung 30-35µl 10-14µl Puffer (10x) = NEB Buffer 5,0µl 2µl (BSA (10x) 5,0µl 2µl) Restriktionsenzym 1 2,5µl 1µl Restriktionsenzym 2 2,5µl 1µl 50,0µl 20µl RNase-freies Wasser auffüllen auf Die PCR-Produkte wurden anschließend bei 37° C für 1-3 Stunden inkubiert. Restriktionsverdau und Dephosphorylierung des Klonierungsvektors Der Klonierungsvektor (pGEM4Z64A bzw. pGEM4ZsigsurvivinDClamp oder pEGFP-C1) wurde mit den passenden Restriktionsenzymen entsprechend dem jeweiligen PCRKonstrukt verdaut. Dabei wurde stets folgendes Pipettierschema verwendet: Plasmid-DNA 14-16µl Puffer (10x) = NEB-Buffer 2µl (BSA (10x) 2µl) Restriktionsenzym 1 1µl Restriktionsenzym 2 1µl Gesamtvolumen 20µl Die Inkubation erfolgte bei 37° C für 1-3 Stunden. 155 6 Material und Methoden Im Anschluss wurden 2µl SAP-Puffer (10x) und 2µl SAP (shrimp alkaline phosphatase) zum Ansatz gegeben und für weitere 10 Minuten bei 37° C inkubiert. Daraufhin wurde die Reaktion bei 65° C für 15 Minuten abgestoppt. Aufreinigung von gespaltenen DNA-Fragmenten Die PCR-Produkte und der Klonierungsvektor wurden nach dem Restriktionsverdau über ein Agarosegel analysiert und mit Hilfe des Kits „NucleoSpin® Extract II“ von MachereyNagel nach Protokoll aufgereinigt. Die Elution erfolgte in 30µl RNase-freiem Wasser. Zur Mengenabschätzung für die Ligation wurden 3µl der gespaltenen Fragmente mit 5µl Auftragspuffer vermischt und auf ein Agarosegel aufgetragen. Ligation von DNA-Fragmenten Insert (PCR-Produkt) und Vektor wurden meist in einem molekularen Mengenverhältnis von 5:2 eingesetzt. Die Reaktion wurde in einem Gesamtvolumen von 20µl durchgeführt. Gespaltenes PCR-Produkt XXµl Gespaltener Vektor XXµl T4 Ligase-Puffer (10x) 2µl T4 Ligase 1µl RNase-freies Wasser auffüllen auf 20µl Die Ligation erfolgte bei Raumtemperatur für 20-60 Minuten. Transformation von chemisch kompetenten Bakterien Chemisch kompetente Bakterien vom Stamm E.coli XL1 Blue oder XL10 Gold wurden auf Eis aufgetaut und 50µl in ein 1,5ml Reaktionsgefäß überführt. Zu dieser Bakteriensuspension wurden 1-3µl der zu transformierenden DNA (Ligationsansatz) gegeben und auf Eis für 30 Minuten inkubiert. Im Anschluss folgte ein Hitzeschock bei 42° C für 30 Sekunden und eine weitere Inkubation auf Eis für 2 Minuten. Daraufhin wurden 900µl 42° C warmes SOC-Medium zu den Bakterien gegeben und diese für 1 Stunde bei 37° C und 200rpm inkubiert. Nach Zentrifugation (13.000rpm, 30sek, Raumtemperatur; Centrifuge 5415D) wurde der Überstand verworfen und das Zellpellet im Rücklauf des Überstandes resuspendiert. Schließlich wurden die Bakterien auf LBAmp- bzw. LB-Kan-Platten ausplattiert und über Nacht bei 37° C im Brutschrank inkubiert. 156 6 Material und Methoden Colony-PCR Waren nach Transformation und Über-Nacht-Inkubation Bakterienkolonien auf der LBPlatte gewachsen, wurde zur Überprüfung, ob die Kolonien positiv sind und somit das entsprechende Insert enthalten, eine Colony-PCR durchgeführt. Dazu wurde pro Kolonie folgender Ansatz pipettiert, welcher in der Regel einen Vektor-spezifischen Primer (sense), meist der T7sense Primer, und einen Insert-spezifischen Primer (antisense) enthielt: sense Primer 0,10µl antisense Primer 0,10µl Puffer S (10x) 1,00µl dNTPs (2,5 mM each) 1,00µl MgCl2 (50 mM) 0,25µl Taq Polymerase 0,10µl RNase-freies Wasser 7,25µl Gesamtvolumen 10,00µl Die Kolonie wurde mit Hilfe eines Zahnstochers gepickt, auf einer neuen LB-Amp-Platte ausgestrichen und der Rest der Bakterienkolonie wurde zum oben pipettierten Reagenzansatz gegeben. Die Reaktion fand unter folgenden Bedingungen statt: 94° C 1min 94° C 30sek 58° C 30sek 35 Zyklen 68° C 1min 68° C 10min Die PCR-Produkte wurden im Anschluss mit 5µl Auftragspuffer (5x) vermengt und in einem 1-1,5%iges Agarosegel aufgetrennt. Kolonien, welche das Insert enthielten, wurden in 5ml LB-Medium mit Ampicillin angeimpft und über Nacht bei 37° C und 200rpm inkubiert. Die Zellen der Übernachtkulturen wurden am nächsten Tag durch Zentrifugation (3.000rpm, 10min, Raumtemperatur; Zentrifuge ROTIXA RP) geerntet und im Anschluss folgte die Isolation der Plasmid-DNA (Minipräperation) wie unter 6.2.1.1 beschrieben. 157 6 Material und Methoden Sequenzierung Im letzten Schritt der Klonierung wurde die isolierte Plasmid-DNA der Kolonien in eine Sequenzierreaktion eingesetzt, um zu überprüfen, ob das erhaltene Plasmid die richtige Sequenz enthält (siehe 6.2.1.5). Kryokonservierung von Bakterien Bakterien, welche die klonierten Plasmide enthielten, wurden zur Lagerung bei -80° C eingefroren. Dazu wurden 1ml 99%-iges Glycerol und 1ml der Bakteriensuspension (Übernachtkultur) in ein 2ml Kryoröhrchen überführt, kurz gemischt und anschließend sofort im -80° C Schrank kryokonserviert. 6.2.1.7 Generierung von mRNA Isolation der Plasmid-DNA (Maxipräperation) Für die Gewinnung von mRNA wurden zunächst 300ml LB-Medium plus Ampicillin mit der Bakterienkolonie, welche das gewünschte Plasmid enthält, inokuliert und über Nacht bei 37° C und 200rpm inkubiert. Die Isolation der Plasmid-DNA erfolgte wie unter 6.2.1.1 (Maxipräperation) beschrieben. Linearisierung der Plasmid-DNA Zur Linearisierung der Plasmid-DNA wurden 100 µg DNA eingesetzt. Je nach Klonierungs-Vektor wurde der Restriktionsverdau entweder mit dem Enzym NotI (für pGEM4ZsigsurvivinDClamp) oder SpeI (für pGEM4Z64A) durchgeführt, so dass die Schnittstellen jeweils downstream des zu transkribierenden Inserts liegen. Entsprechend den Vorgaben des Herstellers NEB wurde der passende Puffer ausgesucht. Der Ansatz wurde wie folgt pipettiert: Plasmid-DNA (100 µg) XXµl Puffer (10x) =NEB Buffer 20µl BSA (100x) 2µl SpeI (10U/µl) / NotI (10U/µl) 10µl RNase-freies Wasser XXµl Gesamtvolumen 200µl Der Restriktionsverdau wurde über Nacht bei 37° C inkubiert und am nächsten Tag aufgereinigt, wobei die Aufreinigung auf einer Phenol/Chloroform Extraktion basiert. Dafür wurden zunächst 200µl einer Lösung von Phenol:Chloroform:Isoamylalkohol (25:24:1) 158 6 Material und Methoden zum Restriktionsverdau gegeben, eine Minute lang gemischt und zentrifugiert (22.000rpm, 2min, 4° C). Die obere Phase wurde vorsichtig abgenommen, in ein neues Reaktionsgefäß überführt und mit ihrem Volumen eines Chloroform:IsoamylalkoholGemisches (Verhältnis 24:1) vermengt. Nach 1-minütigem Vortexen und wiederholter Zentrifugation wurde die obere Phase abgenommen und 1/10 des Volumens 3M Kaliumacetat (pH 5,6) sowie 2,5 Volumen 100% Ethanol für die Fällung der DNA zugegeben. Nach Fällung der DNA bei -20° C für mindestens eine Stunde erfolgte eine Zentrifugation bei 22.000rpm, 4° C für 30 Minuten. Anschließend wurde das Pellet zweimal mit 500µl 70% Ethanol gewaschen (22.000rpm, 15min, 4° C) und einmal mit 500µl 100% Ethanol (22.000rpm, 5min, 4° C). Nach vollständigem Trocknen des Pellets wurde die DNA in 33µl RNase-freiem Wasser gelöst. Zum Schluss wurde die Konzentration der aufgereinigten, linearisierten Plasmid-DNA photometrisch bestimmt und die Linearisierung anhand eines 1%-igen Agarosegels mit einer Elektrophoresedauer von 2,5 Stunden bei 120V überprüft. In vitro Transkription Die aufgereinigte, linearisierte Plasmid-DNA diente als Matrize für die in vitro Transkription. Für diesen Schritt wurde das „mMESSAGE mMACHINE® T7 Ultra Kit“ von AMBION verwendet und nach Protokoll vorgegangen. Es basiert auf einer T7-RNAPolymerase, welche den T7-Promotor des Plasmids verwendet. Außerdem enthält das Kit eine Poly-A-Polymerase (E-PAP), so dass am Ende der Reaktion eine mRNA inklusive 5’Cap und 3’Poly-A-Schwanz vorliegt. Anschließend wurde die mRNA mit dem „PureLink™ RNA Mini Kit“ (AMBION) aufgereinigt. Es wurde die Anleitung für flüssige Proben befolgt und am Schluss mit 35µl RNase-freiem Wasser eluiert. Zuletzt wurde die Konzentration photometrisch bestimmt, die mRNA in geeigneten Mengen aliquotiert und bei -80° C gelagert. RNA Gel Um Reinheit und Größen der RNA-Fragmente zu bestimmen, wurde die in vitro transkripierte mRNA auf einem 1%-igen Agarosegel aufgetragen. Das Agarosegel wurde mit 1xMOPS-Puffer gegossen und Ethidiumbromid (1µl/10ml) zugesetzt. Von der mRNA wurden 1-2µl mit 5µl 5x RNA-Auftragspuffer und 3-4µl RNase-freiem Wasser gemischt, so dass sich ein Gesamtvolumen von 10µl ergab. Zur Denaturierung von RNASekundärstrukturen wurden die Proben 2 Minuten auf Eis, 3 Minuten bei 95° C und erneut 2 Minuten auf Eis inkubiert. Als Größenstandard wurden 3µl des ssRNA Ladder (NEB) mit 3µl des ssRNA Ladder Loading Buffer (2x) vermischt und ebenfalls nach dem oben genannten Schema denaturiert. Die Elektrophorese zur Auftrennung der Fragmente 159 6 Material und Methoden erfolgte eine Stunde bei 100V in einer horizontalen Kammer, wobei 1xMOPS-Puffer als Laufpuffer verwendet wurde. 6.2.1.8 Hochdurchsatzsequenzierung (Deep Sequencing) Für die Analyse des zellulären miRNA-Profils wurde die Methode der Hochdurchsatzsequenzierung (Deep Sequencing) gewählt. Dieser Ansatz erlaubt die Erstellung des kompletten Inventars der kleinen RNA-Moleküle einer Zelle. Im Gegensatz zu miRNA-Microarrays oder qRT-PCR können so auch unbekannte miRNAs identifiziert werden. Dabei werden aus der Gesamt-RNA einer Zelle die kleinen RNA-Moleküle angereichert und nacheinander an das 3`- und das 5`- Ende ein RNA-Adapter ligiert. Die entstandene RNA wird im Anschluss in cDNA umgeschrieben und mit wenigen PCRZyklen amplifiziert. Zudem kann jede sog. miRNA-Bibliothek eine individuelle Marke (tag) bekommen, so dass mehrere Bibliotheken zusammen gepoolt und mittels der Illumina (Solexa)-Technik im Hochdurchsatz in einem Kanal sequenziert werden können. Dies hält den Sequenzierbedarf möglichst gering. Für unsere Analysen wurde Gesamt-RNA aus CD4+ T-Zellen 24 Stunden nach mRNA-Elektroporation wie unter 6.2.1.1 beschrieben isoliert. Die Qualität der RNA wurde im Humangenetischen Institut Erlangen (von Frau P. Rothe) mit dem Agilent Bioanalyzer 6000 Nano bestimmt. Im Anschluss wurden 4 µg Gesamt-RNA in einem Volumen von 20µl an die Firma FASTERIS gesendet, welche die Hochdurchsatzsequenzierung unserer Proben durchführte. Die Auswertung der Daten erfolgte unter anderem mit Hilfe des miRanalyzer. 6.2.1.9 Quantitative Echtzeit (Real-time) PCR (qRT-PCR) Zur Validierung der Deep Sequencing Daten wurde die Expression ausgewählter miRNAs in den RNA-Proben mittels quantitativer Echtzeit (Real-time) PCR (qRT-PCR) untersucht. Dies ermöglicht die relative Quantifizierung von verschiedenen Ziel-RNAs unterschiedlicher Proben zueinander. Als Ausgangssubstrat diente cDNA, die zuvor aus Gesamt-RNA gewonnen wurde. Für unsere Analysen wurden TaqMan® MicroRNA Assays verwendet (siehe 6.1.15), die speziell auf die Detektierung und Quantifizierung von miRNAs ausgerichtet sind. Die zu untersuchende miRNA wurde über einen spezifischen Primer, welcher eine Haarnadelstruktur besitzt und so an die Ziel-miRNA hybridisiert, während der RT-PCR von einer Reverse Transkriptase in cDNA umgeschrieben. In der darauf folgenden TaqMan PCR wurden spezifische vorwärts- und rückwärts-gerichtete Primer eingesetzt, um die cDNA zu amplifizieren. Dieser Schritt konnte dann über die Fluoreszenz-Signalbildung einer spezifischen, komplementären, fluoreszenzmarkierten 160 6 Material und Methoden TaqMan Sonde quantifiziert werden. Das Fluoreszenzsignal einer intakten Sonde wurde durch einen nicht-fluoreszierenden Quencher verhindert. Bei der Durchführung der qRTPCR wurden stets die Angaben des Herstellers befolgt. Pro Reaktion wurden 10 ng RNA eingesetzt. Das Gesamtvolumen der Reaktion betrug 15µl und setzte sich wie folgt zusammen: 100mM dNTPs 0,15µl MultiScribe™ Reverse Transkriptase (50U/µl) 1,00µl 10x Reverse Transkriptase Puffer 1,50µl RNase Inhibitor (20U/µl) 0,19µl RNase-freies Wasser 4,16µl RNA (2 ng/µl) 5,00µl miR-spezifischer RT-Primer Mix (5x) 3,00µl Gesamtvolumen 15,00µl Die ersten vier genannten Komponenten wurden dem Reverse Transkription Kit (Applied Biosystems) entnommen. Zuerst wurde ein x-facher Sammelansatz aus dNTPs, Reverser Transkriptase, 10x Puffer, RNase Inhibitor und RNase-freiem Wasser pipettiert, von welchem pro Reaktion jeweils 7µl mit 5µl RNA vermischt wurden. Zuletzt wurden 3µl des miR-spezifische RT-Primer Mix hinzugegeben. Die Proben wurden für 5 Minuten auf Eis inkubiert und anschließend erfolgte die Reaktion im Thermocyler unter folgenden Bedingungen: 16° C 30min 42° C 30min 85° C 5min Die erhaltene cDNA wurde 1:15 mit RNase-freiem Wasser verdünnt und in einen 20µl PCR-Ansatz eingesetzt. Von einer cDNA-Probe wurden in der TaqMan PCR jeweils Triplikate gemessen. Pro Reaktionsansatz galt folgende Zusammensetzung: TaqMan MicroRNA Assay (20x) 1,00µl RT-PCR Produkt (cDNA 1:15) 1,33µl ABsolute qPCR Rox Mix 10,00µl Nuklease-freies Wasser 7,67µl Gesamtvolumen 20,00µl 161 6 Material und Methoden Die Ansätze wurden in eine optische 96-Loch Reaktionsplatte pipettiert und diese mit einer optischen, adhäsiven Folie versiegelt. Daraufhin wurde die Platte bei 4° C und 1.000rpm für eine Minute zentrifugiert. Die Analyse erfolgte in einem 7300 Real Time PCR System mit den unten stehenden Parametern. Die Auswertung der Daten wurde nach der „ΔΔCT Methode“ durchgeführt. 95° C 10min 95° C 15sek 40 Zyklen 60° C 60sek 6.2.1.10 HLA Typisierung Sowohl alle Patienten der Erlanger HIV-Kohorte als auch zahlreiche Normalspender sind HLA Klasse I typisiert. Die Typisierungen wurden vom HLA-Labor der Medizinischen Klinik 5 unter Leitung von PD Dr. Bernd Spriewald durchgeführt und erfolgten mit standardisierten serologischen Techniken (Biotest AG, Dreieich) oder genotypischen Analysen (Abbott GmbH&Co. KG, Wiesbaden). 6.2.1 Zellbiologische Methoden 6.2.1.1 Allgemeine Hinweise für Zellkulturverfahren Kultivierung von eukaryotischen Zellen Alle eukaryotischen Zellen wurden im Brutschrank unter den folgenden Bedingungen kultiviert: 37° C, 7% CO2, 80% relative Luftfeuchtigkeit. Das Medium wurde alle 2-5 Tage ausgetauscht. Zentrifugation von Zellen Alle Zentrifugationsschritte mit Zellen und Zelllinien erfolgten in den Zentrifugen ROTIXA RP oder Rotanta 460R. Die Standardzentrifugation wurde bei 1.600rpm für 10 Minuten unter Raumtemperatur und maximaler Beschleunigung und Abbremsung durchgeführt. Abweichungen dazu wurden gesondert angegeben. Kryokonservierung von Zellen Zur Kryokonservierung wurden die entsprechenden Zellen zentrifugiert und der Überstand verworfen. Das Zellpellet wurde in 500µl R10-Medium resuspendiert und in ein CryoRöhrchen überführt. Nach 10-minütiger Inkubation bei 4° C wurden 500µl Einfriermedium 162 6 Material und Methoden hinzugefügt. Daraufhin wurden die Zellen in eine mit Isopropanol gefüllte Einfrierbox gegeben, welche ein kontinuierliches und dadurch schonendes Einfrieren der Zellen bei -80° C ermöglicht. Auftauen von Zellen Um kryokonservierte Zellen wieder in Kultur zu nehmen, wurden die Zellproben aufgetaut und dreimal mit je 10ml PBS gewaschen, damit das im Einfriermedium enthaltene DMSO entfernt wird. Im Anschluss wurde das Zellpellet in einem dem Zelltyp entsprechenden Medium resuspendiert, wobei das Volumen von der Zellzahl abhängig war. Generierung von PHA-Blasten Zur Herstellung von PHA-Blasten wurden 5x106 PBMC mit einer Konzentration von 1x106 PBMC/ml in R10IL2-Medium für 3 Tage kultiviert. Die Stimulation erfolgte durch die Zugabe von PHA (5 µg/ml). Aufgrund des Zusatzes von IL-2 und PHA kommt es zur Aktivierung und erhöhten Proliferation von T-Zellen. Anlegen von CTL Linien Um HIV-1-spezifische CTL Linien zu generieren wurden in der Regel 3-5x106 PBMC von HIV-1-infizierten Patienten in 1ml R10IL2-Medium aufgenommen und mit 3µl des entsprechenden HIV-1 Peptides (6 µg) stimuliert. Die Zellen wurden für ca. 10 Tage in einer 24well-Platte im Brutschrank kultiviert und anschließend im γ-IFN ELISPOT getestet. Waren nur sehr wenige PBMC zur Verfügung, wurden 1x106 PBMC in 500µl R10IL2 mit 6 µg des Peptides stimuliert und in einer 48well-Platte kultiviert. Nach 7 Tagen waren die Zellen meist sehr dicht gewachsen und konnten in eine 24well-Platte überführt werden. Restimulation von CTL Linien CTL Linien, welche sich bereits 2-3 Wochen in Kulturen befanden und weiter kultiviert werden sollten, wurden restimuliert. Ebenso musste mit kryokonservierten CTL Linien nach dem Auftauen verfahren werden. Eine Restimulation kann entweder unspezifisch mit PHA erfolgen oder spezifisch mit einer Peptid-inkubierten B-Zelllinie, wobei die letzte Variante bevorzugt wurde. Dazu wurden 1x106 Zellen einer B-Zelllinie, welche über das erforderliche HLA-Allel verfügt, zentrifugiert. Zu dem Zellpellet wurden 5µl des jeweiligen Peptides (10 µg) gegeben und gemischt. Nach einer Inkubation bei 37° C für eine Stunde wurden die Zellen mit 5ml R10-Medium gewaschen, in 0,5-1ml R10IL2 aufgenommen und in ein Cryo-Röhrchen überführt. Nach Bestrahlung mit 90 Gray wurden die B-Zellen zur restimulierenden CTL Linie gegeben. Des Weiteren wurden 5x106 PBMC eines gesunden 163 6 Material und Methoden Spenders in 0,5ml R10IL2, welche zuvor bei 30 Gray bestrahlt wurden, als feeder Zellen zur CTL Linie hinzugefügt. Bei einer unspezifischen Restimulation wurden nur bestrahlte feeder Zellen und 0,5 µg/ml PHA zur CTL Linie dazu gegeben. Nach 7 Tagen wurde im γ-IFN ELISPOT getestet, ob die CTL Linien noch aktiv sind. Anlegen von EBV-transformierten lymphoiden B-Zelllinien (B-LCL) Mindestens 10x106 PBMC wurden verwendet, um daraus eine EBV-transformierte lymphoide B-Zelllinie zu generieren. Nach Zentrifugation der PBMC wurde das Zellpellet in 3ml des gefilterten Überstandes der B-lymphoblastoiden Zelllinie 95-8 aufgenommen. Es wurden 20 µg/ml Cyclosporin zugefügt. Die Kultivierung erfolgte zunächst in einer kleinen Zellkulturflasche für ein bis zwei Tage. Wenn nötig wurde neues R20-Medium und Cyclosporin dazugegeben. Falls die Zellen noch nicht sehr dicht gewachsen waren, wurden nur 120µl Cyclosporin beigefügt. Kultivierung von humanen Makrophagen 2,5x107 PBMC eines gesunden Spenders wurden in 10ml Leu-Medium in eine große Zellkulturschale (85mm) gegeben und für 1,5 Stunden bei 37° C inkubiert. Anschließend wurde das Medium inklusive den Suspensionszellen vorsichtig abgenommen, so dass die adhärierenden Zellen in der Zellkulturschale verbleiben. Zu diesen wurden 10ml neues Leu-Medium gegeben und 8.000 Einheiten GM-CSF hinzugefügt. Nach Kultivierung unter Standardbedingungen für 2 Tage wurden die Zellen mit Hilfe eines Zellschabers entfernt. Durch Oberflächenfärbung von CD16 und CD61 und anschließender Durchflusszytometrie konnte die Anzucht der Makrophagen überprüft werden. Anzucht von Dendritischen Zellen Um aus PBMC Dendritische Zellen (DC) anzureichern, wurden zuerst 30-40x106 der isolierten PBMC in 10ml Leu-Medium auf einer großen Zellkulturschale (85mm) ausgesät. Nach einer Stunde Inkubation der Zellen im Brutschrank konnte ein Adhärieren der Zellen beobachtet werden. Die nicht-adhärierenden Zellen wurden zweimal mit je 5ml unversetztem RPMI1640 abgewaschen. Danach wurden zur Zellkulturschale erneut 10ml Leu-Medium gegeben. Am nächsten Tag (Tag 1) wurden 2ml Leu-Medium mit GM-CSF (72 IU) und IL-4 (400 U) hinzugefügt. Sowohl an Tag 3 als auch an Tag 5 erfolgte jeweils eine Zugabe von 4ml Leu-Medium mit GM-CSF (94 IU) und IL-4 (800 U). Am sechsten Tag wurden folgende Zusätze beigemengt: IL-1β (100 IU/µl), TNF-α (10 ng/ml), IL-6 (1.000 U/µl) und PGE2 (0,1 mg/ml). Ob die Anzucht der DCs erfolgreich war, wurde mittels Oberflächenfärbung von CD86 und CD83 überprüft. 164 6 Material und Methoden 6.2.1.2 Zellselektion Isolation von PBMC mittels Dichtegradient-Zentrifugation Die peripheren, mononukleären Blutzellen können aus Vollblut mittels einer Dichtegradient-Zentrifugation gewonnen werden. Dabei wurden 15ml des Reagenz Lymphoflot in ein 50ml Falcon vorgelegt und mit 35ml Vollblut überschichtet. Waren weniger als 35ml Vollblut vorhanden, wurde das Volumen entsprechend mit HanksLösung aufgefüllt. Im Anschluss wurden die Zellen bei Raumtemperatur mit 1.920rpm für 20 Minuten zentrifugiert, wobei die Abbremsung der Zentrifuge ausgeschaltet (decel 0) war. Es kommt hierbei zu einer Auftrennung der Zellpopulationen. Dabei enthält die unterste Schicht die Erythrozyten, darüber befindet sich die sog. Ficoll-Schicht, gefolgt von einem Ring mit den mononukleären Zellen und ganz oben sammelt sich das Plasma an. Die Schicht mit den mononukleären Zellen wurde abgenommen, in ein neues 50ml Falcon überführt, mit Hanks-Lösung auf 50ml aufgefüllt und bei 1.720rpm für 10 Minuten gewaschen. Anschließend wurde das Zellpellet noch einmal mit 25ml Hanks-Lösung gewaschen (1.720rpm, 10min). Isolation von CD4+ T-Zellen Aus PBMC können mittels magnetischer Auftrennung einzelne Zellpopulationen spezifisch isoliert werden. Dabei können die gewünschten Populationen entweder positiv oder negativ sortiert werden. Man spricht von einer positiven Selektion, wenn die gewünschte Zellpopulation mit einem magnetisch gekoppelten, spezifischen Antikörper inkubiert wird. Bei einer negativen Selektion bleibt die gewünschte Zellpopulation „unangetastet“, womit eine mögliche Aktivierung der Zellen durch die Selektion ausgeschlossen wird. Zur Isolation von CD4+ T-Zellen wurde der CD4+ T cell isolation kit von Miltenyi verwendet, bei welchem es sich um eine negative Depletion der CD4+ T-Zellen handelt. Es wurde nach dem Protokoll des Herstellers vorgegangen. Die Reinheit der selektionierten Zellen wurde mittels Durchflusszytometrie bestimmt. Bestimmung der Zellzahl Um die Zellzahl zu bestimmen, wurden 90µl einer 0,1%-igen Trypanblau-Lösung mit 10µl der zuvor gut gemischten Zellsuspension vermengt. Anschließend wurden davon 10µl auf eine Neubauer-Zählkammer aufgetragen und unter einem Lichtmikroskop die lebenden Zellen in mindestens zwei Großquadranten ausgezählt. Die Zellzahl pro ml ergibt sich aus der folgenden Formel: 165 6 Material und Methoden 6.2.1.3 Transfektionsmethoden Transfektion mit FuGENE Zur Transfektion von HEK293- bzw. HEK293T-Zellen mit DNA bzw. RNA wurden die Zellen am Tag zuvor auf einer Zellkulturplatte ausplattiert. Je nach Größe der Zellkulturplatte wurde eine Zelldichte von 2x105 (24well-Platte) bis zu 2x106 Zellen (6wellPlatte) gewählt. War am nächsten Tag eine Konfluenz der Zellen von 40-80% erreicht, erfolgte die Transfektion der Zellen. Dabei wurde nach Herstellerangaben von FuGENE HD vorgegangen. Die Detektion, ob die Zellen erfolgreich mit DNA bzw. RNA transfiziert wurden, erfolgte in der Regel nach 48 Stunden am Fluoreszenzmikroskop, wenn GFP analysiert werden sollten, oder über Durchflusszytometrie nach intrazellulärer Färbung mit spezifischen Antikörpern. Transfektion mit Chitosan Um DNA bzw. RNA mit Hilfe einer Chitosanlösung in Zellen zu transfizieren, wurde zunächst die Lösung frisch angesetzt. Dazu wurden 36 mg Chitosan mit 290µl Eisessig und 1710µl Nuklease-freiem Wasser vermengt und anschließend über Nacht bei 37° C und 220rpm inkubiert. Am nächsten Tag wurde der pH-Wert der Lösung auf 5,5 mit 1M NaOH eingestellt, das Volumen mit Nuklease-freiem Wasser auf 200ml aufgefüllt und der gesamte Ansatz steril filtriert. Für die Transfektion wurden die Zellen am Vortag auf eine 24well-Zellkulturplatte mit einer Konzentration von 1x105 Zellen/ml ausplattiert. Anschließend wurden 50 µg der Nukleinsäurelösung (1 µg/µl) mit 200µl Wasser und 250µl NaSO 4 vermengt und auf 55° C erhitzt. Die Chitosanlösung wurde ebenfalls erhitzt. Daraufhin wurden Chitosanlösung und Nukleinsäurenlösung in einem Verhältnis von 1:1 gemischt und bis zur Verwendung bei Raumtemperatur aufbewahrt. Kritisch bei dieser Art der Transfektion ist vor allem der pHWert des Mediums, welcher bei genau 6,5 liegen sollte. Daher wurde neues Zellkulturmedium auf diesen pH-Wert eingestellt und steril filtriert. Das Medium, welches sich auf den Zellen befindet, wurde abgenommen, die Zellen einmal mit 500µl PBS/Well gewaschen und anschließend pro Well 333µl des sterilen Mediums (pH=6,5) zugegeben. Pro Well wurden nun 166µl der Nanopartikel tropfenweise zu den Zellen beigemengt. Die Zellen wurden mindestens 48 Stunden im Brutschrank inkubiert. Transfektion mit HIV-1 Tat Des Weiteren wurde versucht DNA und mRNA mit Hilfe des HIV-1 Tat-Proteins bzw. dem Tat-Peptid Tat07-12 in HEK293T bzw. in humane Makrophagen zu transfizieren. HEK293T wurden am Tag zuvor in eine 24well-Platte gegeben (0,2-0,5Mio/ml), Makrophagen vorher wie unter 6.2.1.1 beschrieben kultiviert. Zuerst wurde die DNA- bzw. 166 6 Material und Methoden die RNA-Lösung sowie Protein- bzw. Peptidlösung auf eine gleiche Konzentration von 2 µg/µl eingestellt. Nun wurden unterschiedliche Ratio zwischen Tat-Protein- bzw. TatPeptid-Menge und Nukleinsäuren ausgetestet, wie 1:1, 1:2, 1:5, 1:10, 1:20, 1:30 und 2:3, 2:6 und 2:8 (Nukleinsäure:Tat). Nach Mischen der entsprechenden Mengen erfolgte die Komplexbildung bei Raumtemperatur für 30 Minuten. Anschließend wurde von den zu transfizierenden Zellen das Medium abgenommen, die Zellen einmal mit Medium ohne Zusätze vorsichtig gewaschen und Medium ohne Zusätze zu den Zellen gegeben. Zum Schluss wurden die Komplexe vorsichtig auf die Zellen getropft und diese für mindestens 48 Stunden kultiviert. Transfektion mit Atelokollagen Am Tag vor der Transfektion wurde die Stocklösung des Atelokollagen auf eine Konzentration von 0,016% bzw. 0,008% mit PBS verdünnt. Die DNA-Lösung wurde ebenfalls auf 100 µg/ml bzw. 50 µg/ml mit PBS verdünnt. Die Ansätze wurden auf Eis pipettiert und anschließend noch weitere 5 Minuten auf Eis inkubiert. Die Komplexbildung erfolgte durch Mischen der Verdünnungen von DNA und Atelokollagen in einem Verhältnis von 1:1. Daraufhin wurden die Ansätze bei 4° C und 600rpm für 20 Minuten inkubiert. Nach Zentrifugation (10.000rpm, 1min, Centrifuge 5415D) wurden die Komplexe bei 4° C für mindestens 16 Stunden aufbewahrt. Am nächsten Tag wurden pro Well 70µl der Komplexlösung auf eine 24well-Platte vorgegeben und bei Raumtemperatur luftgetrocknet. Anschließend wurde das getrocknete Gel mit 2,5x105 Zellen in 1ml Medium überschichtet und für mindestens 48 Stunden im Brutschrank inkubiert. Bei längerer Inkubation wurde das Medium ausgetauscht und die Zellen gesplittet. 6.2.1.4 mRNA-Elektroporation mRNA-Elektroporation von PBMC und CD4+ T-Zellen Zur mRNA-Elektroporation wurden 30-40x106 PBMC bzw. CD4+ T-Zellen in 10ml LeuMedium in einer großen Gewebekulturschale (85mm) ausgesät und über Nacht im Brutschrank inkubiert. Am nächsten Tag wurden die Zellen durch mehrmaliges Waschen mit unversetztem RPMI1640 geerntet. Zum Ernten der adhärierenden Zellen wurde zusätzlich ein Zellschaber verwendet. Alle nachfolgenden Zentrifugationen erfolgten bei 1.000rpm für 10 Minuten bei Raumtemperatur. Nach Zentrifugation wurden die Zellen in 10ml RPMI1640 aufgenommen und gezählt. Nach erneuter Zentrifugation der benötigten Zellmenge wurden die Zellen noch einmal mit 5ml OptiMEM gewaschen. Nach Abnahme des vollständigen Überstandes wurden die Zellen in OptiMEM resuspendiert und auf eine Konzentration von maximal 10x106 Zellen/100µl OptiMEM eingestellt. Die Elektroporation 167 6 Material und Methoden wurde unter den folgenden Bedingungen durchgeführt: Square wave, 500V, 3ms, Pulse 1, Cuvette 4mm. Dazu wurden zunächst 15 µg HIV-1 mRNA bzw. 10 µg GFP mRNA in eine 4mm Küvette mit gestopften Spitzen vorgelegt und 100µl der Zellsuspension mit 5-10x106 Zellen zugegeben. Nach Elektroporation wurden die Zellen umgehend in kleine Zellkulturflaschen mit vorgewärmtem Leu-Medium gegeben und für mindestens 3,5 Stunden im Brutschrank inkubiert. mRNA-Elektroporation von B-Zellen (B-LCL) Am Tag vor der Elektroporation wurde den EBV-transformierten lymphoiden B-Zelllinien R20-Medium zugefügt. Am nächsten Tag wurden pro Elektroporationsansatz 5-10x106 Zellen abzentrifugiert, zweimal mit mindestens 10ml unversetzten RPMI1640 und einmal mit 5ml OptiMEM gewaschen (900rpm, 10min). Das weitere Vorgehen folgte dem Protokoll der mRNA-Elektroporation für PBMC und CD4+ T-Zellen. Jedoch wurden die B-Zellen nach Elektroporation in R20-Medium anstelle von Leu-Medium kultiviert. 6.2.1.5 Durchflusszytometrie Direkte, extrazelluläre Immunfluoreszenzfärbung Zur Unterscheidung bestimmter Zellpopulationen können Oberflächenmoleküle der Zellen direkt mit spezifischen Antikörpern gefärbt werden. Im Anschluss können die Zellen im Durchflusszytometer bezüglich ihrer Größe, Struktur und ihren Fluoreszenzeigenschaften untersucht werden. Zur Analyse von Lymphozytensubpopulationen wurde die entsprechende Zellzahl (Minimum 200.000) in ein 5ml Polystyrol Rundboden-Röhrchen überführt und zentrifugiert. Die Zellen wurden nun entweder mit dem TQ-Prep fixiert und erneut zentrifugiert (2.000rpm, 2min) oder der Antikörper wurde gleich hinzugefügt. Nach Zugabe von 3-5µl des jeweiligen CD-Antikörpers wurden die Zellen unter Lichtausschluss 15 Minuten bei Raumtemperatur inkubiert. Nach dem Waschen der Zellen mit 2ml PBS (2.000rpm, 2min) erfolgte eine Fixierung der Proben mit 2%-iger PFA-Lösung. Die Analyse der Zellen wurde stets am FACS Calibur durchgeführt und die ermittelten Daten mit der FACS Express V3 Software ausgewertet. Die extrazelluläre Färbung des HLA-A2 Moleküls folgte unter den gleichen Bedingungen, wobei allerdings nur 3µl des HLA-A2 Antikörpers sowie der Isotypkontrolle verwendet wurden. Die CXCR4-Färbung wurde nach Angaben des Herstellers durchgeführt. Indirekte, intrazelluläre Immunfluoreszenzfärbung Um intrazelluläre Proteine detektieren zu können, werden Zellen permeabilisiert. Anschließend können die Proteine mit fluoreszierenden Antikörpern gefärbt und am 168 6 Material und Methoden Durchflusszytometer analysiert werden. Diese Art der Immunfluoreszenzfärbung kam sowohl nach mRNA-Elektroporation der Zellen als auch nach Transfektion zum Einsatz. Dafür wurden mindestens 200.000 Zellen in ein 5ml Polystyrol Rundboden-Röhrchen überführt und zentrifugiert. Die Zellen wurden im TQ-Prep fixiert, zentrifugiert (2.000rpm, 2min) und mit 2ml Permeabilisierungspuffer gewaschen (2.000rpm, 2min). Nach Zugabe des primären Antikörpers wurden die Zellen mindestens 30 Minuten bei Raumtemperatur inkubiert und im Anschluss mit 2ml Permeabilisierungspuffer gewaschen (2.000rpm, 2min). Daraufhin wurde der fluoreszenzmarkierte, sekundäre Antikörper dazugegeben und die Zellen wurden unter Lichtausschluss für weitere 30 Minuten bei Raumtemperatur inkubiert. Zum Schluss erfolgte ein weiterer Waschschritt mit 2ml Permeablisierungspuffer. Zur Fixierung der Zellen wurden 100µl einer 2%-igen PFALösung zugegeben. Bis zur Analyse am FACS Calibur wurden die gefärbten Zellen bei 4° C aufbewahrt. Bei der Analyse von GFP mRNA elektroporierten Zellen wurde nach der Fixierung durch den TQ-Prep, eine Zentrifugation bei 2.000rpm für 2 Minuten durchgeführt, der Überstand verworfen und gleich 100µl PFA (2%) zu den Zellen gegeben. Die nachstehende Tabelle zeigt, in welcher Konzentration die Antikörper zur Färbung verwendet wurden. Anstelle des sekundären Antikörpers Anti-Maus IgG1 PE wurde in einigen Färbungen auch der Anti-Maus IgG FITC in der gleichen Konzentration verwendet. Die optimale Konzentration aller Antikörper wurde jeweils vor Verwendung durch Titration ermittelt. Dazu wurden transfizierte HEK293T-Zellen verwendet (siehe 6.2.1.3). Tabelle 6.22 Antikörper und deren eingesetzte Verdünnung in intrazellulären Färbungen Antikörper Eingesetzte Verdünnung Sekundärer Antikörper Flag M2 Nef EH1 p24 AG3.0 p24 183/H12 gp120 AD3 gp120 697-3D gp120-160 Chessie 6 Anti-Maus IgG1 PE Anti-Maus IgG FITC 1:5.000 1:200 1:40 1:1.000 1:20 1:20 1:20 1:500 1:500 Anti-Maus IgG1 PE Anti-Maus IgG1 PE Anti-Maus IgG1 PE Anti-Maus IgG1 PE Anti-Maus IgG1 PE Anti-Maus IgG1 PE Anti-Maus IgG1 PE - Zellsortierung (FACS) Neben der magnetischen Selektion (siehe 6.2.1.2) können einzelne Zellpopulationen auch mittels einer bestimmten Form der Durchflusszytometrie, der FACS (fluorscence activated cell sorting), sortiert werden. Dabei werden die gewünschten Zellen mit einem 169 6 Material und Methoden fluoreszenzmarkierten Antikörper spezifisch gefärbt und anschließend entsprechend ihrer Fluoreszenz in verschiedene Röhrchen sortiert. In den durchgeführten Versuchen wurde entweder der MoFlow™ oder der Aria verwendet. Zur Selektion von lebenden CD4+ T-Zellen wurden 10-20x106 PBMC sowohl mit einem Lebendmarker (siehe 6.2.1.6) als auch mit einem Anti-CD4-Antikörper gefärbt. 6.2.1.6 Untersuchung der Vitalität von Zellen Fixation and Death Cell Discrimination Kit Um die Vitalität von Zellen zu bestimmen wurde der Fixation and Dead Cell Discrimination Kit verwendet und nach Herstellerprotokoll vorgegangen. Dabei werden Zellen mit einem Fluoreszenzfarbstoff gefärbt, welcher aufgrund der beschädigten Zellmembran nur in tote Zellen eindringen kann und an die DNA bindet. Die gefärbten Zellen wurden anschließend zur Zellsortierung verwendet, so dass dabei ein Ausschluss von toten Zellen möglich war. Färbung mit DAPI Ein weiteres Vorgehen, um tote und lebende Zellen voneinander zu unterscheiden, ist das Anfärben der Zellen mit dem Fluoreszenzfarbstoff 4',6-diamidino-2-phenylindole (DAPI), welcher ebenfalls die DNA im Zellkern färbt. Dabei wurden die Zellen in 1ml einer DAPILösung (Konzentration 3 µg/ml) aufgenommen und für 15 Minuten im Brutschrank inkubiert. Danach wurden die Zellen mit 3ml PBS gewaschen und bei 1.600rpm für 5 Minuten zentrifugiert. Im Anschluss wurden die Zellen in Sekretionsassay-Puffer aufgenommen, mit einem 30µm Zellsieb filtriert und zur Zellsortierung am MoFlow™ verwendet. Färbung mit Propidiumjodid Ebenfalls zum Einsatz kam die Färbung von Zellen mit Propidiumjodid (PI), einem Fluoreszenzfarbstoff, welcher in die DNA interkaliert. Dazu wurden die Zellen mit 10 µg/ml Propidiumjodid gefärbt und anschließend für 30 Minuten bei 37° C inkubiert. Die Zellen wurden daraufhin mit 2ml PBS gewaschen, zentrifugiert und in PBS aufgenommen. Nach Filtrierung der Zellsuspension erfolgte die Sortierung am MoFlow™ oder am Aria, wobei nur lebende Zellen sortiert wurden. 6.2.1.7 Bestimmung der Zellproliferation Mit Hilfe des CellTrace™ CFSE Cell Proliferation Kit konnte überprüft werden, ob die KoKultivierung von PBMC und PBMC, welche mit mRNA elektroporiert wurden, nicht nur zur 170 6 Material und Methoden Stimulation, sondern auch zu einer Proliferation der Zellen führt. Das im Kit enthaltene Carboxyfluoreszein-Succinimidyl Ester (CFSE) kann passiv in Zellen diffundieren und durch Abspaltung der Acetatgruppe durch intrazelluläre Esterasen entsteht ein fluoreszendierendes Molekül. Nach Zellteilung wird der Farbstoff an die Tochterzellen abgegeben, so dass die Fluoreszenzintensität bei Proliferation der Zellen abnimmt. Bei der Durchführung wurden die Herstellerangaben berücksichtigt. Die verschiedenen Zellpopulationen wurden in einem Verhältnis von 1:1 in 1ml Medium (R10 bzw. R10IL2) in einer 48well-Platte kultiviert. Zur Kontrolle wurden PBMC entweder mit 3µl Peptid oder 5µl PHA ebenfalls in 1ml Medium stimuliert, wobei darauf geachtet werden musste, dass die Gesamtmenge von PBMC pro Well gleich ist. Zur Analyse wurden 1x105 Zellen an den Tagen 3, 5, 7 und 10 aus der Kultur entnommen, mit 2ml PBS gewaschen (2.000rpm, 2min) und anschließend mit 100µl 2%-iger PFA-Lösung fixiert. Die Messung erfolgte am FACS Calibur, die Auswertung der Rohdaten mit Hilfe der Software FCS Express V3. 6.2.1.8 Lösen und Verdünnen von Peptide Die in Pulverform gelieferten Peptide wurden zunächst in DMSO gelöst und dadurch auf eine Konzentration von 20 mg/ml eingestellt. Diese Stocklösungen wurden auf eine Konzentration von 2 mg/ml verdünnt. Dazu wurden 100µl Peptid-Stocklösung mit 10µl DTT und 890µl Wasser vermischt. Peptide, welche vom NIH zur Verfügung gestellt wurden, wurden in 50µl DMSO gelöst und anschließend 445µl Wasser und 5µl DTT dazugegeben, um ebenfalls eine Konzentration von 2 mg/ml zu erhalten. 6.2.1.9 γ-Interferon ELISPOT Beschichten und Entwickeln der Mikrotiterplatten 96well-Mikrotiterplatten mit Nitrozellulose-Membranfilter wurden für 30 Sekunden mit 20µl 70% Methanol aktiviert, zweimal mit je 200µl PBS gewaschen und mit 50µl des γ-IFN Antikörpers (Klon 1-D1K, Konzentration 10 µg/ml) beschichtet. Nach einer Inkubation bei 4° C über Nacht wurden die Platten zwei- bis viermal mit je 200µl PBS gewaschen und daraufhin mit 150µl R5AB-Medium für eine Stunde bei 37° C geblockt. Die beschichteten Platten wurden bei -20° C gelagert. Nach Inkubation der Zellen von 16-40 Stunden auf der ELISPOT-Platte wurde die Platte 6x mit je 300µl PBS/Tween 0,05% gewaschen. Ein gegen γ-IFN gerichteter mit Biotin gekoppelter Antikörper (mAb7-B6-1-Biotin) wurde in MACS-Puffer auf eine Konzentration von 2 µg/ml verdünnt, davon 100µl pro Well zugegeben und für zwei Stunden bei 171 6 Material und Methoden Raumtemperatur inkubiert. Die Platte wurde erneut 6x mit je 300µl PBS/Tween 0,05% gewaschen. Anschließend wurden 100µl Enzymgemisch pro Well zugegeben, welches bereits 30 Minuten vorher angesetzt wurde und aus 10ml PBS/Tween 0,1% mit jeweils einem Tropfen der Lösungen A und B aus dem Vectastain ABC Kit besteht. Nach einer Inkubation von 1-3 Stunden bei Raumtemperatur wurde die Platte 3x mit 300µl PBS/Tween 0,05%, 3x mit 300µl PBS gewaschen und pro Well 100µl Substrat zugefügt. Dieses wurde zuvor aus einer Spatelspitze ACE-Substrat angesetzt, welches in 2,5ml DMF gelöst und auf 30ml mit Lösung AB aufgefüllt wurde. Nach Mischen und Filtrierung der Substratlösung durch einen 0,2µm-Filter wurde die Lösung durch die Zugabe von 20µl H2O2 (30%)/10ml aktiviert. Die Substratlösung wurde auf der Platte für mindestens 4 Minuten bei Raumtemperatur inkubiert. Sobald eine rosa Färbung der Filtermembran zu erkennen war, wurde die Platte 3x mit je 300µl Aqua dest. gewaschen, um die Reaktion zu stoppen. Zum Schluss wurden die Platten bei Raumtemperatur luftgetrocknet und die Spots mit dem AID EliSpot Reader quantifiziert. Die Auswertung erfolgte mit dem Programm AID EliSpot Reader (Version 5.0). Analyse der T-Zellantwort von frischen PBMC Für die Bestimmung der γ-IFN Ausschüttung von PBMC sowie von PBMC, welche mit mRNA elektroporiert wurden, wurden 1x105 bzw. 2x105 PBMC pro Well in einem Gesamtvolumen von 100µl R5AB-Medium auf eine ELISPOT-Platte aufgetragen. Peptide wurden in einer Konzentration von 20 µg/ml zugegeben. Peptid-Pools wurden in einer Konzentration von 25 µg/ml aufgetragen. PHA diente als Positivkontrolle, wobei 1,2 µg pro Well eingesetzt wurden. Die verschiedenen Ansätze wurden meist in Duplikaten analysiert. Die Zellen wurden anschließend bei 37° C im Brutschrank für mindestens 16 Stunden, wenn möglich für 40 Stunden, inkubiert. Testen von CTL Linien Nach Anlegen der CTL Linie und einer 10-tägigen Kultivierung wurde getestet, ob HIV-1spezifische CTL Linien in der Kultur enthalten sind. Dazu wurden jeweils 50µl der Zellsuspension auf eine ELISPOT-Platte aufgetragen, welche bereits 50µl R5AB-Medium enthielt. Es wurde zum einen die CTL Linie allein und zum anderen die CTL Linie mit Peptid (20 µg/ml) in Unikaten getestet. Bestimmung von Kreuzreaktionen, CD8+- bzw. CD4+-Spezifität und Titration Um zu testen, ob HIV-1-spezifische CTL Linien zu anderen, ähnlichen Epitopen kreuzreaktiv sind und diese somit erkennen, wurden 0,5-1x105 Zellen in 100µl R5ABMedium pro Well zur ELISPOT-Platte gegeben. Die Stimulation erfolgte durch Zugabe 172 6 Material und Methoden von 20 µg/ml der zu testenden Peptide und durch eine Inkubation im Brutschrank für mindestens 16 Stunden. Durch Zugabe von 50 µg/ml eines Anti-CD4 oder 625 ng/ml eines Anti-CD8 Antikörpers zu der CTL Linie kann detektiert werden, ob es sich bei der CTL Linie um einen CD8+ oder um einen CD4+ T-Zell-Klon handelt. Des Weiteren wurde bei Bedarf die Affinität der Peptide zu den jeweiligen CTL Linien in Titrationsexperimenten getestet. Dabei wurden wie bei Kreuzreaktionen 0,5-1x105 Zellen der Linie in 100µl R5AB-Medium pro Well zur ELISPOT-Platte gegeben. Die entsprechenden Peptide wurden in Konzentrationen zwischen 0,01 und 20 μg/ml zugegeben. Restriktion Löst die Stimulation von Zellen mit Peptiden, welche keine bekannten Epitope enthalten, eine γ-IFN Freisetzung aus, so wird eine Restriktion durchgeführt. Diese soll klären, über welches HLA Klasse I Allel das Epitop restringiert ist. Dazu wurden EBV-transformierte BZelllinien ausgesucht, welche im idealen Fall jeweils nur ein HLA-Allel mit der zu untersuchenden CTL Linie teilten. Von diesen sechs verschiedenen B-Zelllinien wurden zweimal jeweils 1x106 Zellen zentrifugiert und einmal ohne und einmal mit 10 µg des zu untersuchenden Peptides für eine Stunde im Brutschrank inkubiert. Im Anschluss wurden die Zellen dreimal mit je 10ml PBS gewaschen, 800µl R5AB-Medium zugefügt und 50.000 Zellen pro Well auf die ELISPOT-Platte gegeben. Von der CTL Linie wurden je nach Aktivität 0,5-1x105 Zellen pro Well auf der Platte mit den verschiedenen B-Zelllinien inkubiert. Nur die B-Zelllinie mit dem passenden HLA Klasse I Allel kann nach Peptidinkubation das Peptid präsentieren und wird von der CTL Linie erkannt, was zur Ausschüttung von γ-IFN führt. 6.2.1.10 Analyse der Freisetzung von weiteren Zytokine Cytometric Bead Array (CBA) Die Freisetzung von γ-IFN nach Elektroporation der Zellen mit mRNA wurde wie unter 6.2.1.9 beschrieben mittels ELISPOT bestimmt. Für die Messung weiterer sekretierter Zytokine wurde der human TH1/TH2 CBA Kit verwendet. Dazu wurden 4 und 24 Stunden nach Elektroporation 50µl aus dem Überstand der Zellen abgenommen und zunächst bis zur Messung bei -20° C gelagert. Die Analyse der Proben erfolgte in der Dermatologie Erlangen und wurde von Herrn Dr. C. Hofmann durchgeführt. Dabei wurde stets nach Herstellerangaben (BD) vorgegangen. 173 6 Material und Methoden TNF-α ELISA Die Sekretion von TNF-α in den Zellüberstand wurde auch mittels eines indirekten ELISAAssays gemessen. Die nachfolgenden Volumenangaben (in µl) beziehen sich stets auf ein Well, alle Inkubationen wurden bei Raumtemperatur und unter leichtem Schwenken durchgeführt. Abweichungen davon sind angegeben. Zunächst wurden Nunc Immuno Module mit einem Anti-human-TNF-α Antikörper (2 µg/ml in coating Solution) beschichtet, über Nacht bei 4° C inkubiert und am nächsten Tag zweimal mit PBS/0,05% Tween gewaschen. Danach wurde der Antikörper mit 200µl PBS/10%FCS für 2 Stunden geblockt. Nach erneutem zweimaligem Waschen mit PBS/0,05% Tween wurden 100µl des Standards (2 µg/ml) oder 100µl der Proben aufgetragen und über Nacht bei 4° C inkubiert. Am nächsten Tag wurde viermal mit PBS/0,05% Tween gewaschen, 100µl des biotinylierten Anti-TNF-α Antikörpers (1 µg/ml) zugegeben. Nach 45-minütiger Inkubation wurde sechsmal mit PBS/0,05% Tween gewaschen. Anschließend wurden 100µl einer Avidin-Phosphatase (Endverdünnung 1:70.000 in PBS/10% FCS) für 1-2 Stunden inkubiert, dann achtmal mit PBS/0,05% Tween gewaschen und 200µl der Substratlösung pNPP auf die Platte gegeben. Diese wurde nun für 15 bis 30 Minuten bei Raumtemperatur und unter Lichtausschluss inkubiert. Falls kein sichtbarer Farbumschlag auftrat, wurde die Reaktion für weitere 15 Minuten inkubiert. Die Messung der Biolumineszenz erfolgte bei einer OD von 405nm im SPECTRA MAX 190. Wenn keine sofortige Messung der Biolumineszenz möglich war, wurde die Reaktion mit 50µl 3N NaOH abgestoppt. TNF-α Messung mit dem Immunolight® Modul Chemilumineszenz-Substrat Eine weitere Methode für die Messung von TNF-α, das von Zellen sekretiert wird, ist das Immunolight® Modul Chemilumineszenz-Substrat, welches im Routinelabor der Medizinischen Klinik 3 angewandt wird. Dafür wurden 4 und 24 Stunden nach mRNAElektroporation der PBMC jeweils 200µl aus dem Überstand der Zellen abgenommen und zur Analyse in das Routinelabor gegeben. 6.2.1.11 Dualer Luciferase-Reporter-Assay Zur Untersuchung des inhibierenden Effektes der humanen miR-29a auf Nef wurden nefGene aus verschiedenen HIV-1-infizierten Patienten in den Vektor psiCHECK3 kloniert. Dieser Vektor enthält die Gene der Firefly und Renilla Luziferase und dient daher als Reporterkonstrukt. Die unterschiedlichen nef-Plasmide wurden mit dem pmiR-Vec-29a, welcher die humane miR-29a kodiert, in HEK293 mittels FuGENE kotransfiziert. Als Kontrolle diente die Transfektion mit einem Plasmid, in welchem die perfekte 174 6 Material und Methoden Bindesequenz der miR-29a dreimal hintereinander vorliegt. Nach 48 Stunden wurden die Zellen lysiert und geerntet. Es wurde nach Anleitung des Dual-Luciferase® Reporter Assay vorgegangen. Die Luziferase-Aktivität wurde bestimmt durch Zugabe von 100µl Luziferin (LARII) zu 20µl des Zelllysat-Überstandes und anschließender BiolumineszenzMessung am Sirius. Es wurden stets Triplikate angesetzt und gemessen. 6.2.1.12 Infektion mit rekombinanten Vaccinia-Vektoren Um B-Zelllinien mit rekombinanten Vaccinia-Vektoren, welche HIV-1 Proteine exprimieren, zu infizieren, wurden 1x106 B-Zellen zentrifugiert, die Zellen in 100µl R10Medium aufgenommen und die entsprechende Menge des Vaccinia-Vektors mit einer MOI von 5 bzw. 10 dazugegeben. Die MOI wurde wie folgt berechnet: Nach 1,5 Stunden Inkubation der Zellen im Brutschrank wurden 900µl R10-Medium hinzugefügt und der gesamte Ansatz in einer 24well-Platte über Nacht im Brutschrank kultiviert. Die Zellen wurden 24 Stunden nach Infektion in einer indirekten, intrazellulären Immunfluoreszenzfärbung analysiert, um die erfolgreiche Infektion der Zellen nachzuweisen, und in ELISPOT-Analysen eingesetzt. Bei der Infektion von PBMC wurden die Zellen direkt mit der entsprechenden Menge des Vaccinia-Vektors (MOI=12) im γ-IFN ELISPOT Assay eingesetzt. 175 7 Literaturverzeichnis 1. Barre-Sinoussi, F., J. C. Chermann, F. Rey, M. T. Nugeyre, S. Chamaret, J. Gruest, C. Dauguet, C. Axler-Blin, F. Vezinet-Brun, C. Rouzioux, W. Rozenbaum, and L. Montagnier. 1983. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220:868-871. 2. Coffin, J., A. Haase, J. A. Levy, L. Montagnier, S. Oroszlan, N. Teich, H. Temin, K. Toyoshima, H. Varmus, P. Vogt, and et al. 1986. What to call the AIDS virus? Nature 321:10. 3. (RKI), R. K.-I. 2011. 4. (WHO), W. H. O. 2011. 5. Clavel, F., D. Guetard, F. Brun-Vezinet, S. Chamaret, M. A. Rey, M. O. SantosFerreira, A. G. Laurent, C. Dauguet, C. Katlama, C. Rouzioux, and et al. 1986. Isolation of a new human retrovirus from West African patients with AIDS. Science 233:343-346. 6. Flint, S. J. E., L.W.; Racaniello, V.R.; and Skalka, A.M. 2004. Principles of Virology: Molecular Biology, Pathogenesis, and Control of Animal Viruses. 7. Modrow, S. a. F., D. 1998. Molekulare Virologie. 8. Holmes, E. C. 2001. On the origin and evolution of the human immunodeficiency virus (HIV). Biological reviews of the Cambridge Philosophical Society 76:239-254. 9. Moore, J. P., A. Trkola, and T. Dragic. 1997. Co-receptors for HIV-1 entry. Curr Opin Immunol 9:551-562. 10. Bigbug. 2009. Verlauf_einer_HIV-Infektion.png. http://en.wikipedia.org/wiki/File:Hiv-timecourse_copy.svg 2009, April 23. 11. Hoffmann, C. a. R., J.K. 2010. HIV 2010. 12. Castro, D. K. W., J.; Slutsker, L.; et al. 1992. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR. Recommendations and reports : Morbidity and mortality weekly report. Recommendations and reports / Centers for Disease Control 41:1-19. 13. Costin, J. M. 2007. Cytopathic mechanisms of HIV-1. Virology journal 4:100. 14. Kestler, H. W., 3rd, D. J. Ringler, K. Mori, D. L. Panicali, P. K. Sehgal, M. D. Daniel, and R. C. Desrosiers. 1991. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 65:651-662. 15. Kirchhoff, F., T. C. Greenough, D. B. Brettler, J. L. Sullivan, and R. C. Desrosiers. 1995. Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. The New England journal of medicine 332:228-232. 16. Frankel, A. D., and J. A. Young. 1998. HIV-1: fifteen proteins and an RNA. Annual review of biochemistry 67:1-25. 176 7 Literaturverzeichnis 17. Jacobus21. 2011. Hiv_gross_german_2.jpg. http://de.wikipedia.org/w/index.php?title=Datei:Hiv_gross_german_2.jpg&filetimestamp=2 0110125195209 2011, Januar 25. 18. Peterlin, B. M., and D. Trono. 2003. Hide, shield and strike back: how HIV-infected cells avoid immune eradication. Nat Rev Immunol 3:97-107. 19. Castellino, F., and R. N. Germain. 2006. Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annual review of immunology 24:519-540. 20. Andersen, M. H., D. Schrama, P. Thor Straten, and J. C. Becker. 2006. Cytotoxic T cells. The Journal of investigative dermatology 126:32-41. 21. Falschlehner, C., U. Schaefer, and H. Walczak. 2009. Following TRAIL's path in the immune system. Immunology 127:145-154. 22. Janeway, C. A. M., K.M.; Travers, P.; Walport, M. 2009. Immunologie. 23. Schmitz, J. E., M. J. Kuroda, S. Santra, V. G. Sasseville, M. A. Simon, M. A. Lifton, P. Racz, K. Tenner-Racz, M. Dalesandro, B. J. Scallon, J. Ghrayeb, M. A. Forman, D. C. Montefiori, E. P. Rieber, N. L. Letvin, and K. A. Reimann. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283:857-860. 24. Ogg, G. S., X. Jin, S. Bonhoeffer, P. R. Dunbar, M. A. Nowak, S. Monard, J. P. Segal, Y. Cao, S. L. Rowland-Jones, V. Cerundolo, A. Hurley, M. Markowitz, D. D. Ho, D. F. Nixon, and A. J. McMichael. 1998. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science 279:2103-2106. 25. Saez-Cirion, A., M. Sinet, S. Y. Shin, A. Urrutia, P. Versmisse, C. Lacabaratz, F. Boufassa, V. Avettand-Fenoel, C. Rouzioux, J. F. Delfraissy, F. Barre-Sinoussi, O. Lambotte, A. Venet, and G. Pancino. 2009. Heterogeneity in HIV suppression by CD8 T cells from HIV controllers: association with Gag-specific CD8 T cell responses. J Immunol 182:7828-7837. 26. Streeck, H., J. S. Jolin, Y. Qi, B. Yassine-Diab, R. C. Johnson, D. S. Kwon, M. M. Addo, C. Brumme, J. P. Routy, S. Little, H. K. Jessen, A. D. Kelleher, F. M. Hecht, R. P. Sekaly, E. S. Rosenberg, B. D. Walker, M. Carrington, and M. Altfeld. 2009. Human immunodeficiency virus type 1-specific CD8+ T-cell responses during primary infection are major determinants of the viral set point and loss of CD4+ T cells. J Virol 83:7641-7648. 27. Sewell, A. K., D. A. Price, A. Oxenius, A. D. Kelleher, and R. E. Phillips. 2000. Cytotoxic T lymphocyte responses to human immunodeficiency virus: control and escape. Stem Cells 18:230-244. 28. Motozono, C., P. Mwimanzi, and T. Ueno. 2010. Dynamic interplay between viral adaptation and immune recognition during HIV-1 infection. Protein & cell 1:514-519. 29. Betts, M. R., M. C. Nason, S. M. West, S. C. De Rosa, S. A. Migueles, J. Abraham, M. M. Lederman, J. M. Benito, P. A. Goepfert, M. Connors, M. Roederer, and R. A. Koup. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781-4789. 30. Ferre, A. L., P. W. Hunt, J. W. Critchfield, D. H. Young, M. M. Morris, J. C. Garcia, R. B. Pollard, H. F. Yee, Jr., J. N. Martin, S. G. Deeks, and B. L. Shacklett. 2009. Mucosal 177 7 Literaturverzeichnis immune responses to HIV-1 in elite controllers: a potential correlate of immune control. Blood 113:3978-3989. 31. Rock, K. L., D. J. Farfan-Arribas, and L. Shen. 2010. Proteases in MHC class I presentation and cross-presentation. J Immunol 184:9-15. 32. Germain, R. N. 1994. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell 76:287-299. 33. Waßmuth, R. 2005. Einführung in das HLA-System. 34. Kaslow, R. A., M. Carrington, R. Apple, L. Park, A. Munoz, A. J. Saah, J. J. Goedert, C. Winkler, S. J. O'Brien, C. Rinaldo, R. Detels, W. Blattner, J. Phair, H. Erlich, and D. L. Mann. 1996. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat Med 2:405-411. 35. Kaur, G., and N. Mehra. 2009. Genetic determinants of HIV-1 infection and progression to AIDS: immune response genes. Tissue Antigens 74:373-385. 36. Chakrabarti, L. A., and V. Simon. 2010. Immune mechanisms of HIV control. Curr Opin Immunol 22:488-496. 37. Bangham, C. R. 2009. CTL quality and the control of human retroviral infections. Eur J Immunol 39:1700-1712. 38. Kaslow, R. A., R. Duquesnoy, M. VanRaden, L. Kingsley, M. Marrari, H. Friedman, S. Su, A. J. Saah, R. Detels, J. Phair, and et al. 1990. A1, Cw7, B8, DR3 HLA antigen combination associated with rapid decline of T-helper lymphocytes in HIV-1 infection. A report from the Multicenter AIDS Cohort Study. Lancet 335:927-930. 39. Steel, C. M., C. A. Ludlam, D. Beatson, J. F. Peutherer, R. J. Cuthbert, P. Simmonds, H. Morrison, and M. Jones. 1988. HLA haplotype A1 B8 DR3 as a risk factor for HIV-related disease. Lancet 1:1185-1188. 40. Itescu, S., U. Mathur-Wagh, M. L. Skovron, L. J. Brancato, M. Marmor, A. Zeleniuch-Jacquotte, and R. Winchester. 1992. HLA-B35 is associated with accelerated progression to AIDS. J Acquir Immune Defic Syndr 5:37-45. 41. Gao, X., G. W. Nelson, P. Karacki, M. P. Martin, J. Phair, R. Kaslow, J. J. Goedert, S. Buchbinder, K. Hoots, D. Vlahov, S. J. O'Brien, and M. Carrington. 2001. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. The New England journal of medicine 344:1668-1675. 42. Carrington, M., G. W. Nelson, M. P. Martin, T. Kissner, D. Vlahov, J. J. Goedert, R. Kaslow, S. Buchbinder, K. Hoots, and S. J. O'Brien. 1999. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 283:1748-1752. 43. Tang, J., C. Costello, I. P. Keet, C. Rivers, S. Leblanc, E. Karita, S. Allen, and R. A. Kaslow. 1999. HLA class I homozygosity accelerates disease progression in human immunodeficiency virus type 1 infection. AIDS research and human retroviruses 15:317324. 44. Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer. 2004. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303:1526-1529. 178 7 Literaturverzeichnis 45. Chang, J. J., and M. Altfeld. 2010. Innate immune activation in primary HIV-1 infection. J Infect Dis 202 Suppl 2:S297-301. 46. Meier, A., J. J. Chang, E. S. Chan, R. B. Pollard, H. K. Sidhu, S. Kulkarni, T. F. Wen, R. J. Lindsay, L. Orellana, D. Mildvan, S. Bazner, H. Streeck, G. Alter, J. D. Lifson, M. Carrington, R. J. Bosch, G. K. Robbins, and M. Altfeld. 2009. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med 15:955-959. 47. Oh, D. Y., K. Baumann, O. Hamouda, J. K. Eckert, K. Neumann, C. Kucherer, B. Bartmeyer, G. Poggensee, N. Oh, A. Pruss, H. Jessen, and R. R. Schumann. 2009. A frequent functional toll-like receptor 7 polymorphism is associated with accelerated HIV-1 disease progression. AIDS 23:297-307. 48. Duvall, M. G., A. Jaye, T. Dong, J. M. Brenchley, A. S. Alabi, D. J. Jeffries, M. van der Sande, T. O. Togun, S. J. McConkey, D. C. Douek, A. J. McMichael, H. C. Whittle, R. A. Koup, and S. L. Rowland-Jones. 2006. Maintenance of HIV-specific CD4+ T cell help distinguishes HIV-2 from HIV-1 infection. J Immunol 176:6973-6981. 49. Streeck, H., and D. F. Nixon. 2010. T cell immunity in acute HIV-1 infection. J Infect Dis 202 Suppl 2:S302-308. 50. Rychert, J., S. Saindon, S. Placek, D. Daskalakis, and E. Rosenberg. 2007. Sequence variation occurs in CD4 epitopes during early HIV infection. J Acquir Immune Defic Syndr 46:261-267. 51. Vingert, B., S. Perez-Patrigeon, P. Jeannin, O. Lambotte, F. Boufassa, F. Lemaitre, W. W. Kwok, I. Theodorou, J. F. Delfraissy, J. Theze, and L. A. Chakrabarti. 2010. HIV controller CD4+ T cells respond to minimal amounts of Gag antigen due to high TCR avidity. PLoS Pathog 6:e1000780. 52. Douek, D. C., J. M. Brenchley, M. R. Betts, D. R. Ambrozak, B. J. Hill, Y. Okamoto, J. P. Casazza, J. Kuruppu, K. Kunstman, S. Wolinsky, Z. Grossman, M. Dybul, A. Oxenius, D. A. Price, M. Connors, and R. A. Koup. 2002. HIV preferentially infects HIVspecific CD4+ T cells. Nature 417:95-98. 53. Samson, M., F. Libert, B. J. Doranz, J. Rucker, C. Liesnard, C. M. Farber, S. Saragosti, C. Lapoumeroulie, J. Cognaux, C. Forceille, G. Muyldermans, C. Verhofstede, G. Burtonboy, M. Georges, T. Imai, S. Rana, Y. Yi, R. J. Smyth, R. G. Collman, R. W. Doms, G. Vassart, and M. Parmentier. 1996. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382:722-725. 54. Dean, M., M. Carrington, C. Winkler, G. A. Huttley, M. W. Smith, R. Allikmets, J. J. Goedert, S. P. Buchbinder, E. Vittinghoff, E. Gomperts, S. Donfield, D. Vlahov, R. Kaslow, A. Saah, C. Rinaldo, R. Detels, and S. J. O'Brien. 1996. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 273:18561862. 55. Liu, R., W. A. Paxton, S. Choe, D. Ceradini, S. R. Martin, R. Horuk, M. E. MacDonald, H. Stuhlmann, R. A. Koup, and N. R. Landau. 1996. Homozygous defect in 179 7 Literaturverzeichnis HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86:367-377. 56. Kaur, G., and N. Mehra. 2009. Genetic determinants of HIV-1 infection and progression to AIDS: susceptibility to HIV infection. Tissue Antigens 73:289-301. 57. Hutter, G., D. Nowak, M. Mossner, S. Ganepola, A. Mussig, K. Allers, T. Schneider, J. Hofmann, C. Kucherer, O. Blau, I. W. Blau, W. K. Hofmann, and E. Thiel. 2009. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. The New England journal of medicine 360:692-698. 58. Hutter, G., and E. Thiel. 2011. Allogeneic transplantation of CCR5-deficient progenitor cells in a patient with HIV infection: an update after 3 years and the search for patient no. 2. AIDS 25:273-274. 59. Brandl, A., J. Wittmann, and H. M. Jack. 2011. A facile method to increase titers of miRNA-encoding retroviruses by inhibition of the RNaseIII enzyme Drosha. Eur J Immunol 41:549-551. 60. Wittmann, J., H. M. Jack, and M. F. Mashreghi. 2011. [MicroRNAs in B-cells and T-cells as regulators of inflammation]. Zeitschrift fur Rheumatologie 70:507-510. 61. Callis, T. E., J. F. Chen, and D. Z. Wang. 2007. MicroRNAs in skeletal and cardiac muscle development. DNA Cell Biol 26:219-225. 62. Wittmann, J., and H. M. Jack. 2010. Serum microRNAs as powerful cancer biomarkers. Biochim Biophys Acta 1806:200-207. 63. Wittmann, J., and H. M. Jack. 2011. microRNAs in rheumatoid arthritis: midget RNAs with a giant impact. Annals of the rheumatic diseases 70 Suppl 1:i92-96. 64. Chable-Bessia, C., O. Meziane, D. Latreille, R. Triboulet, A. Zamborlini, A. Wagschal, J. M. Jacquet, J. Reynes, Y. Levy, A. Saib, Y. Bennasser, and M. Benkirane. 2009. Suppression of HIV-1 replication by microRNA effectors. Retrovirology 6:26. 65. Liu, Y. P., J. Gruber, J. Haasnoot, P. Konstantinova, and B. Berkhout. 2009. RNAimediated inhibition of HIV-1 by targeting partially complementary viral sequences. Nucleic Acids Res 37:6194-6204. 66. Narayanese. 2012. MiRNA-biogenesis.jpg. http://en.wikipedia.org/wiki/File:MiRNAbiogenesis.jpg 2012, Juni 28. 67. Nathans, R., C. Y. Chu, A. K. Serquina, C. C. Lu, H. Cao, and T. M. Rana. 2009. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol Cell 34:696-709. 68. Houzet, L., M. L. Yeung, V. de Lame, D. Desai, S. M. Smith, and K. T. Jeang. 2008. MicroRNA profile changes in human immunodeficiency virus type 1 (HIV-1) seropositive individuals. Retrovirology 5:118. 69. Yeung, M. L., Y. Bennasser, T. G. Myers, G. Jiang, M. Benkirane, and K. T. Jeang. 2005. Changes in microRNA expression profiles in HIV-1-transfected human cells. Retrovirology 2:81. 180 7 Literaturverzeichnis 70. Jopling, C. L., M. Yi, A. M. Lancaster, S. M. Lemon, and P. Sarnow. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309:1577-1581. 71. Ahluwalia, J. K., S. Z. Khan, K. Soni, P. Rawat, A. Gupta, M. Hariharan, V. Scaria, M. Lalwani, B. Pillai, D. Mitra, and S. K. Brahmachari. 2008. Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology 5:117. 72. Omoto, S., and Y. R. Fujii. 2005. Regulation of human immunodeficiency virus 1 transcription by nef microRNA. J Gen Virol 86:751-755. 73. Umbach, J. L., M. F. Kramer, I. Jurak, H. W. Karnowski, D. M. Coen, and B. R. Cullen. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780-783. 74. Eletto, D., G. Russo, G. Passiatore, L. Del Valle, A. Giordano, K. Khalili, E. Gualco, and F. Peruzzi. 2008. Inhibition of SNAP25 expression by HIV-1 Tat involves the activity of mir-128a. J Cell Physiol 216:764-770. 75. Temesgen, Z., D. Warnke, and M. J. Kasten. 2006. Current status of antiretroviral therapy. Expert opinion on pharmacotherapy 7:1541-1554. 76. Hammer, S. M. 2006. Nucleoside analogue reverse transcriptase inhibitor options: a re-examination of the class. Topics in HIV medicine : a publication of the International AIDS Society, USA 14:140-143. 77. De Clercq, E. 2004. Non-nucleoside reverse transcriptase inhibitors (NNRTIs): past, present, and future. Chemistry & biodiversity 1:44-64. 78. Hughes, A., T. Barber, and M. Nelson. 2008. New treatment options for HIV salvage patients: an overview of second generation PIs, NNRTIs, integrase inhibitors and CCR5 antagonists. The Journal of infection 57:1-10. 79. Ingale, K. B., and M. S. Bhatia. 2011. HIV-1 integrase inhibitors: a review of their chemical development. Antiviral chemistry & chemotherapy 22:95-105. 80. Sterjovski, J., M. J. Churchill, S. L. Wesselingh, and P. R. Gorry. 2006. HIV-1 entry inhibitors: classes, applications and factors affecting potency. Current HIV research 4:387400. 81. Yeni, P. 2006. Update on HAART in HIV. J Hepatol 44:S100-103. 82. Johnson, V. A., V. Calvez, H. F. Gunthard, R. Paredes, D. Pillay, R. Shafer, A. M. Wensing, and D. D. Richman. 2011. 2011 update of the drug resistance mutations in HIV1. Topics in antiviral medicine 19:156-164. 83. Stoll, M., C. Claes, E. Schulte, J. M. Graf von der Schulenburg, and R. E. Schmidt. 2002. Direct costs for the treatment of HIV-infection in a German cohort after the introduction of HAART. Eur J Med Res 7:463-471. 84. Heath, S. L., and J. M. Kilby. 2006. Therapeutic immunization strategies for HIV infection. Curr Opin HIV AIDS 1:74-81. 85. Koff, W. C. Accelerating HIV vaccine development. Nature 464:161-162. 181 7 Literaturverzeichnis 86. Woolard, S. N., and U. Kumaraguru. 2010. Viral vaccines and CTL response. Journal of biomedicine & biotechnology 2010:141657. 87. 764. Walker, B. D., and D. R. Burton. 2008. Toward an AIDS vaccine. Science 320:760- 88. Burton, D. R., J. Pyati, R. Koduri, S. J. Sharp, G. B. Thornton, P. W. Parren, L. S. Sawyer, R. M. Hendry, N. Dunlop, P. L. Nara, and et al. 1994. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266:1024-1027. 89. Herr, W., U. Protzer, A. W. Lohse, G. Gerken, K. H. Meyer zum Buschenfelde, and T. Wolfel. 1998. Quantification of CD8+ T lymphocytes responsive to human immunodeficiency virus (HIV) peptide antigens in HIV-infected patients and seronegative persons at high risk for recent HIV exposure. J Infect Dis 178:260-265. 90. Kutzler, M. A., and D. B. Weiner. 2008. DNA vaccines: ready for prime time? Nat Rev Genet 9:776-788. 91. Harrer, E., M. Bauerle, B. Ferstl, P. Chaplin, B. Petzold, L. Mateo, A. Handley, M. Tzatzaris, J. Vollmar, S. Bergmann, M. Rittmaier, K. Eismann, S. Muller, J. R. Kalden, B. Spriewald, D. Willbold, and T. Harrer. 2005. Therapeutic vaccination of HIV-1-infected patients on HAART with a recombinant HIV-1 nef-expressing MVA: safety, immunogenicity and influence on viral load during treatment interruption. Antivir Ther 10:285-300. 92. Buchbinder, S. P., D. V. Mehrotra, A. Duerr, D. W. Fitzgerald, R. Mogg, D. Li, P. B. Gilbert, J. R. Lama, M. Marmor, C. Del Rio, M. J. McElrath, D. R. Casimiro, K. M. Gottesdiener, J. A. Chodakewitz, L. Corey, and M. N. Robertson. 2008. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 372:1881-1893. 93. Rerks-Ngarm, S., P. Pitisuttithum, S. Nitayaphan, J. Kaewkungwal, J. Chiu, R. Paris, N. Premsri, C. Namwat, M. de Souza, E. Adams, M. Benenson, S. Gurunathan, J. Tartaglia, J. G. McNeil, D. P. Francis, D. Stablein, D. L. Birx, S. Chunsuttiwat, C. Khamboonruang, P. Thongcharoen, M. L. Robb, N. L. Michael, P. Kunasol, and J. H. Kim. 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. The New England journal of medicine 361:2209-2220. 94. Rabinovich, P. M., M. E. Komarovskaya, Z. J. Ye, C. Imai, D. Campana, E. Bahceci, and S. M. Weissman. 2006. Synthetic messenger RNA as a tool for gene therapy. Hum Gene Ther 17:1027-1035. 95. Hoerr, I., R. Obst, H. G. Rammensee, and G. Jung. 2000. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol 30:1-7. 96. Scheel, B., S. Aulwurm, J. Probst, L. Stitz, I. Hoerr, H. G. Rammensee, M. Weller, and S. Pascolo. 2006. Therapeutic anti-tumor immunity triggered by injections of immunostimulating single-stranded RNA. Eur J Immunol 36:2807-2816. 97. Teufel, R., J. P. Carralot, B. Scheel, J. Probst, S. Walter, G. Jung, I. Hoerr, H. G. Rammensee, and S. Pascolo. 2005. Human peripheral blood mononuclear cells 182 7 Literaturverzeichnis transfected with messenger RNA stimulate antigen-specific cytotoxic T-lymphocytes in vitro. Cell Mol Life Sci 62:1755-1762. 98. Chang, J. J., and M. Altfeld. 2009. TLR-mediated immune activation in HIV. Blood 113:269-270. 99. Diebold, S. S., T. Kaisho, H. Hemmi, S. Akira, and C. Reis e Sousa. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529-1531. 100. Wille-Reece, U., B. J. Flynn, K. Lore, R. A. Koup, A. P. Miles, A. Saul, R. M. Kedl, J. J. Mattapallil, W. R. Weiss, M. Roederer, and R. A. Seder. 2006. Toll-like receptor agonists influence the magnitude and quality of memory T cell responses after primeboost immunization in nonhuman primates. J Exp Med 203:1249-1258. 101. Conry, R. M., A. F. LoBuglio, M. Wright, L. Sumerel, M. J. Pike, F. Johanning, R. Benjamin, D. Lu, and D. T. Curiel. 1995. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res 55:1397-1400. 102. Lorenzi, J. C., A. P. Trombone, C. D. Rocha, L. P. Almeida, R. L. Lousada, T. Malardo, I. C. Fontoura, R. A. Rossetti, A. F. Gembre, A. M. Silva, C. L. Silva, and A. A. Coelho-Castelo. 2010. Intranasal vaccination with messenger RNA as a new approach in gene therapy: use against tuberculosis. BMC Biotechnol 10:77. 103. Lu, W., L. C. Arraes, W. T. Ferreira, and J. M. Andrieu. 2004. Therapeutic dendritic-cell vaccine for chronic HIV-1 infection. Nat Med 10:1359-1365. 104. Probst, J., S. Brechtel, B. Scheel, I. Hoerr, G. Jung, H. G. Rammensee, and S. Pascolo. 2006. Characterization of the ribonuclease activity on the skin surface. Genetic vaccines and therapy 4:4. 105. Mockey, M., C. Goncalves, F. P. Dupuy, F. M. Lemoine, C. Pichon, and P. Midoux. 2006. mRNA transfection of dendritic cells: synergistic effect of ARCA mRNA capping with Poly(A) chains in cis and in trans for a high protein expression level. Biochem Biophys Res Commun 340:1062-1068. 106. Holtkamp, S., S. Kreiter, A. Selmi, P. Simon, M. Koslowski, C. Huber, O. Tureci, and U. Sahin. 2006. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 108:40094017. 107. Scheel, B., R. Teufel, J. Probst, J. P. Carralot, J. Geginat, M. Radsak, D. Jarrossay, H. Wagner, G. Jung, H. G. Rammensee, I. Hoerr, and S. Pascolo. 2005. Tolllike receptor-dependent activation of several human blood cell types by protaminecondensed mRNA. Eur J Immunol 35:1557-1566. 108. Scheel, B., S. Braedel, J. Probst, J. P. Carralot, H. Wagner, H. Schild, G. Jung, H. G. Rammensee, and S. Pascolo. 2004. Immunostimulating capacities of stabilized RNA molecules. Eur J Immunol 34:537-547. 109. Kreiter, S., M. Diken, A. Selmi, O. Tureci, and U. Sahin. 2011. Tumor vaccination using messenger RNA: prospects of a future therapy. Curr Opin Immunol 23:399-406. 110. Zhao, Y., E. Moon, C. Carpenito, C. M. Paulos, X. Liu, A. L. Brennan, A. Chew, R. G. Carroll, J. Scholler, B. L. Levine, S. M. Albelda, and C. H. June. 2010. Multiple 183 7 Literaturverzeichnis injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res 70:9053-9061. 111. Um, S. J., Y. J. Choi, H. J. Shin, C. H. Son, Y. S. Park, M. S. Roh, Y. S. Kim, Y. D. Kim, S. K. Lee, M. H. Jung, M. K. Lee, C. Son, P. J. Choi, J. Chung, C. D. Kang, and E. Y. Lee. 2010. Phase I study of autologous dendritic cell tumor vaccine in patients with nonsmall cell lung cancer. Lung Cancer. 112. Routy, J. P., M. R. Boulassel, B. Yassine-Diab, C. Nicolette, D. Healey, R. Jain, C. Landry, O. Yegorov, I. Tcherepanova, T. Monesmith, L. Finke, and R. P. Sekaly. 2010. Immunologic activity and safety of autologous HIV RNA-electroporated dendritic cells in HIV-1 infected patients receiving antiretroviral therapy. Clin Immunol 134:140-147. 113. Maurer, K., E. G. Harrer, A. Goldwich, K. Eismann, S. Bergmann, M. SchmittHaendle, B. Spriewald, S. M. Mueller, and T. Harrer. 2008. Role of cytotoxic Tlymphocyte-mediated immune selection in a dominant human leukocyte antigen-B8restricted cytotoxic T-lymphocyte epitope in Nef. J Acquir Immune Defic Syndr 48:133141. 114. Gray, C. M., J. M. Schapiro, M. A. Winters, and T. C. Merigan. 1998. Changes in CD4+ and CD8+ T cell subsets in response to highly active antiretroviral therapy in HIV type 1-infected patients with prior protease inhibitor experience. AIDS research and human retroviruses 14:561-569. 115. Altfeld, M. A., A. Trocha, R. L. Eldridge, E. S. Rosenberg, M. N. Phillips, M. M. Addo, R. P. Sekaly, S. A. Kalams, S. A. Burchett, K. McIntosh, B. D. Walker, and P. J. Goulder. 2000. Identification of dominant optimal HLA-B60- and HLA-B61-restricted cytotoxic T-lymphocyte (CTL) epitopes: rapid characterization of CTL responses by enzyme-linked immunospot assay. J Virol 74:8541-8549. 116. Lubaki, N. M., S. C. Ray, B. Dhruva, T. C. Quinn, R. F. Siliciano, and R. C. Bollinger. 1997. Characterization of a polyclonal cytolytic T lymphocyte response to human immunodeficiency virus in persons without clinical progression. J Infect Dis 175:1360-1367. 117. Milicic, A., D. A. Price, P. Zimbwa, B. L. Booth, H. L. Brown, P. J. Easterbrook, K. Olsen, N. Robinson, U. Gileadi, A. K. Sewell, V. Cerundolo, and R. E. Phillips. 2005. CD8+ T cell epitope-flanking mutations disrupt proteasomal processing of HIV-1 Nef. J Immunol 175:4618-4626. 118. Goulder, P., D. Price, M. Nowak, S. Rowland-Jones, R. Phillips, and A. McMichael. 1997. Co-evolution of human immunodeficiency virus and cytotoxic T-lymphocyte responses. Immunol Rev 159:17-29. 119. Goulder, P. J., Y. Tang, S. I. Pelton, and B. D. Walker. 2000. HLA-B57-restricted cytotoxic T-lymphocyte activity in a single infected subject toward two optimal epitopes, one of which is entirely contained within the other. J Virol 74:5291-5299. 120. Altfeld, M., E. T. Kalife, Y. Qi, H. Streeck, M. Lichterfeld, M. N. Johnston, N. Burgett, M. E. Swartz, A. Yang, G. Alter, X. G. Yu, A. Meier, J. K. Rockstroh, T. M. Allen, H. Jessen, E. S. Rosenberg, M. Carrington, and B. D. Walker. 2006. HLA Alleles Associated with Delayed Progression to AIDS Contribute Strongly to the Initial CD8(+) T Cell Response against HIV-1. PLoS Med 3:e403. 184 7 Literaturverzeichnis 121. Culmann-Penciolelli, B., S. Lamhamedi-Cherradi, I. Couillin, N. Guegan, J. P. Levy, J. G. Guillet, and E. Gomard. 1994. Identification of multirestricted immunodominant regions recognized by cytolytic T lymphocytes in the human immunodeficiency virus type 1 Nef protein. J Virol 68:7336-7343. 122. Verheyen, J., F. Schweitzer, E. G. Harrer, E. Knops, S. M. Mueller, M. Daumer, K. Eismann, S. Bergmann, B. M. Spriewald, R. Kaiser, and T. Harrer. 2010. Analysis of immune selection as a potential cause for the presence of cleavage site mutation 431V in treatment-naive HIV type-1 isolates. Antivir Ther 15:907-912. 123. Zhai, S., Y. Zhuang, Y. Song, S. Li, D. Huang, W. Kang, X. Li, Q. Liao, Y. Liu, Z. Zhao, Y. Lu, and Y. Sun. 2008. HIV-1-specific cytotoxic T lymphocyte (CTL) responses against immunodominant optimal epitopes slow the progression of AIDS in China. Current HIV research 6:335-350. 124. Fleis, R., T. Filzen, and K. L. Collins. 2002. Species-specific effects of HIV-1 Nefmediated MHC-I downmodulation. Virology 303:120-129. 125. Sol-Foulon, N., C. Esnault, Y. Percherancier, F. Porrot, P. Metais-Cunha, F. Bachelerie, and O. Schwartz. 2004. The effects of HIV-1 Nef on CD4 surface expression and viral infectivity in lymphoid cells are independent of rafts. J Biol Chem 279:3139831408. 126. Sloan, R. D., D. A. Donahue, B. D. Kuhl, T. Bar-Magen, and M. A. Wainberg. Expression of Nef from unintegrated HIV-1 DNA downregulates cell surface CXCR4 and CCR5 on T-lymphocytes. Retrovirology 7:44. 127. Hung, C. H., L. Thomas, C. E. Ruby, K. M. Atkins, N. P. Morris, Z. A. Knight, I. Scholz, E. Barklis, A. D. Weinberg, K. M. Shokat, and G. Thomas. 2007. HIV-1 Nef assembles a Src family kinase-ZAP-70/Syk-PI3K cascade to downregulate cell-surface MHC-I. Cell Host Microbe 1:121-133. 128. Harrer, T., E. Harrer, S. A. Kalams, P. Barbosa, A. Trocha, R. P. Johnson, T. Elbeik, M. B. Feinberg, S. P. Buchbinder, and B. D. Walker. 1996. Cytotoxic T lymphocytes in asymptomatic long-term nonprogressing HIV-1 infection. Breadth and specificity of the response and relation to in vivo viral quasispecies in a person with prolonged infection and low viral load. J Immunol 156:2616-2623. 129. Choppin, J., W. Cohen, A. Bianco, J. P. Briand, F. Connan, M. Dalod, and J. G. Guillet. 2001. Characteristics of HIV-1 Nef regions containing multiple CD8+ T cell epitopes: wealth of HLA-binding motifs and sensitivity to proteasome degradation. J Immunol 166:6164-6169. 130. Wilson, C. C., W. C. Olson, T. Tuting, C. R. Rinaldo, M. T. Lotze, and W. J. Storkus. 1999. HIV-1-specific CTL responses primed in vitro by blood-derived dendritic cells and Th1-biasing cytokines. J Immunol 162:3070-3078. 131. Kiepiela, P., A. J. Leslie, I. Honeyborne, D. Ramduth, C. Thobakgale, S. Chetty, P. Rathnavalu, C. Moore, K. J. Pfafferott, L. Hilton, P. Zimbwa, S. Moore, T. Allen, C. Brander, M. M. Addo, M. Altfeld, I. James, S. Mallal, M. Bunce, L. D. Barber, J. Szinger, C. Day, P. Klenerman, J. Mullins, B. Korber, H. M. Coovadia, B. D. Walker, and P. J. Goulder. 2004. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432:769-775. 185 7 Literaturverzeichnis 132. Matthews, P. C., A. Prendergast, A. Leslie, H. Crawford, R. Payne, C. Rousseau, M. Rolland, I. Honeyborne, J. Carlson, C. Kadie, C. Brander, K. Bishop, N. Mlotshwa, J. I. Mullins, H. Coovadia, T. Ndung'u, B. D. Walker, D. Heckerman, and P. J. Goulder. 2008. Central role of reverting mutations in HLA associations with human immunodeficiency virus set point. J Virol 82:8548-8559. 133. Holzhutter, H. G., and P. M. Kloetzel. 2000. A kinetic model of vertebrate 20S proteasome accounting for the generation of major proteolytic fragments from oligomeric peptide substrates. Biophysical journal 79:1196-1205. 134. Holzhutter, H. G., C. Frommel, and P. M. Kloetzel. 1999. A theoretical approach towards the identification of cleavage-determining amino acid motifs of the 20 S proteasome. J Mol Biol 286:1251-1265. 135. Reche, P. A., D. B. Keskin, R. E. Hussey, P. Ancuta, D. Gabuzda, and E. L. Reinherz. 2006. Elicitation from virus-naive individuals of cytotoxic T lymphocytes directed against conserved HIV-1 epitopes. Med Immunol 5:1. 136. Tomiyama, H., K. Miwa, H. Shiga, Y. I. Moore, S. Oka, A. Iwamoto, Y. Kaneko, and M. Takiguchi. 1997. Evidence of presentation of multiple HIV-1 cytotoxic T lymphocyte epitopes by HLA-B*3501 molecules that are associated with the accelerated progression of AIDS. J Immunol 158:5026-5034. 137. Bade-Doding, C., A. Theodossis, S. Gras, L. Kjer-Nielsen, B. Eiz-Vesper, A. Seltsam, T. Huyton, J. Rossjohn, J. McCluskey, and R. Blasczyk. 2011. The impact of human leukocyte antigen (HLA) micropolymorphism on ligand specificity within the HLAB*41 allotypic family. Haematologica 96:110-118. 138. Allen, T. M., M. Altfeld, S. C. Geer, E. T. Kalife, C. Moore, M. O'Sullivan K, I. Desouza, M. E. Feeney, R. L. Eldridge, E. L. Maier, D. E. Kaufmann, M. P. Lahaie, L. Reyor, G. Tanzi, M. N. Johnston, C. Brander, R. Draenert, J. K. Rockstroh, H. Jessen, E. S. Rosenberg, S. A. Mallal, and B. D. Walker. 2005. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV1) sequence diversity and reveals constraints on HIV-1 evolution. J Virol 79:13239-13249. 139. Hashimoto, M., M. Morimoto, H. Saimoto, Y. Shigemasa, H. Yanagie, M. Eriguchi, and T. Sato. 2006. Gene transfer by DNA/mannosylated chitosan complexes into mouse peritoneal macrophages. Biotechnol Lett 28:815-821. 140. Lai, W. F., and M. C. Lin. 2009. Nucleic acid delivery with chitosan and its derivatives. J Control Release 134:158-168. 141. Li, G. P., Z. G. Liu, B. Liao, and N. S. Zhong. 2009. Induction of Th1-type immune response by chitosan nanoparticles containing plasmid DNA encoding house dust mite allergen Der p 2 for oral vaccination in mice. Cell Mol Immunol 6:45-50. 142. Eguchi, A., T. Akuta, H. Okuyama, T. Senda, H. Yokoi, H. Inokuchi, S. Fujita, T. Hayakawa, K. Takeda, M. Hasegawa, and M. Nakanishi. 2001. Protein transduction domain of HIV-1 Tat protein promotes efficient delivery of DNA into mammalian cells. J Biol Chem 276:26204-26210. 143. Meng, S., B. Wei, R. Xu, K. Zhang, L. Wang, R. Zhang, and J. Li. 2009. TAT peptides mediated small interfering RNA delivery to Huh-7 cells and efficiently inhibited hepatitis C virus RNA replication. Intervirology 52:135-140. 186 7 Literaturverzeichnis 144. Gump, J. M., and S. F. Dowdy. 2007. TAT transduction: the molecular mechanism and therapeutic prospects. Trends Mol Med 13:443-448. 145. Sano, A., M. Maeda, S. Nagahara, T. Ochiya, K. Honma, H. Itoh, T. Miyata, and K. Fujioka. 2003. Atelocollagen for protein and gene delivery. Adv Drug Deliv Rev 55:16511677. 146. Minakuchi, Y., F. Takeshita, N. Kosaka, H. Sasaki, Y. Yamamoto, M. Kouno, K. Honma, S. Nagahara, K. Hanai, A. Sano, T. Kato, M. Terada, and T. Ochiya. 2004. Atelocollagen-mediated synthetic small interfering RNA delivery for effective gene silencing in vitro and in vivo. Nucleic Acids Res 32:e109. 147. Takeshita, F., Y. Minakuchi, S. Nagahara, K. Honma, H. Sasaki, K. Hirai, T. Teratani, N. Namatame, Y. Yamamoto, K. Hanai, T. Kato, A. Sano, and T. Ochiya. 2005. Efficient delivery of small interfering RNA to bone-metastatic tumors by using atelocollagen in vivo. Proc Natl Acad Sci U S A 102:12177-12182. 148. Corbet, S., H. V. Nielsen, L. Vinner, S. Lauemoller, D. Therrien, S. Tang, G. Kronborg, L. Mathiesen, P. Chaplin, S. Brunak, S. Buus, and A. Fomsgaard. 2003. Optimization and immune recognition of multiple novel conserved HLA-A2, human immunodeficiency virus type 1-specific CTL epitopes. J Gen Virol 84:2409-2421. 149. Goulder, P. J., C. Brander, K. Annamalai, N. Mngqundaniso, U. Govender, Y. Tang, S. He, K. E. Hartman, C. A. O'Callaghan, G. S. Ogg, M. A. Altfeld, E. S. Rosenberg, H. Cao, S. A. Kalams, M. Hammond, M. Bunce, S. I. Pelton, S. A. Burchett, K. McIntosh, H. M. Coovadia, and B. D. Walker. 2000. Differential narrow focusing of immunodominant human immunodeficiency virus gag-specific cytotoxic T-lymphocyte responses in infected African and caucasoid adults and children. J Virol 74:5679-5690. 150. Harrer, T., E. Harrer, P. Barbosa, F. Kaufmann, R. Wagner, S. Bruggemann, J. R. Kalden, M. Feinberg, R. P. Johnson, S. Buchbinder, and B. D. Walker. 1998. Recognition of two overlapping CTL epitopes in HIV-1 p17 by CTL from a long-term nonprogressing HIV-1-infected individual. J Immunol 161:4875-4881. 151. Goulder, P. J., M. Bunce, P. Krausa, K. McIntyre, S. Crowley, B. Morgan, A. Edwards, P. Giangrande, R. E. Phillips, and A. J. McMichael. 1996. Novel, crossrestricted, conserved, and immunodominant cytotoxic T lymphocyte epitopes in slow progressors in HIV type 1 infection. AIDS research and human retroviruses 12:1691-1698. 152. Sandberg, J. K., A. C. Leandersson, C. Devito, B. Kohleisen, V. Erfle, A. Achour, M. Levi, S. Schwartz, K. Karre, B. Wahren, and J. Hinkula. 2000. Human immunodeficiency virus type 1 Nef epitopes recognized in HLA-A2 transgenic mice in response to DNA and peptide immunization. Virology 273:112-119. 153. Culmann, B., E. Gomard, M. P. Kieny, B. Guy, F. Dreyfus, A. G. Saimot, D. Sereni, D. Sicard, and J. P. Levy. 1991. Six epitopes reacting with human cytotoxic CD8+ T cells in the central region of the HIV-1 NEF protein. J Immunol 146:1560-1565. 154. Gray, C. M., M. Mlotshwa, C. Riou, T. Mathebula, D. de Assis Rosa, T. Mashishi, C. Seoighe, N. Ngandu, F. van Loggerenberg, L. Morris, K. Mlisana, C. Williamson, and S. A. Karim. 2009. Human immunodeficiency virus-specific gamma interferon enzyme-linked immunospot assay responses targeting specific regions of the proteome during primary subtype C infection are poor predictors of the course of viremia and set point. J Virol 83:470-478. 187 7 Literaturverzeichnis 155. Witwer, K. W., A. K. Watson, J. N. Blankson, and J. E. Clements. 2012. Relationships of PBMC microRNA expression, plasma viral load, and CD4+ T-cell count in HIV-1-infected elite suppressors and viremic patients. Retrovirology 9:5. 156. Lo, H. L., T. Chang, P. Yam, P. M. Marcovecchio, S. Li, J. A. Zaia, and J. K. Yee. 2007. Inhibition of HIV-1 replication with designed miRNAs expressed from RNA polymerase II promoters. Gene therapy 14:1503-1512. 157. Marcinowski, L., M. Tanguy, A. Krmpotic, B. Radle, V. J. Lisnic, L. Tuddenham, B. Chane-Woon-Ming, Z. Ruzsics, F. Erhard, C. Benkartek, M. Babic, R. Zimmer, J. Trgovcich, U. H. Koszinowski, S. Jonjic, S. Pfeffer, and L. Dolken. 2012. Degradation of cellular mir-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog 8:e1002510. 158. Hu, W., X. Wang, X. Ding, Y. Li, X. Zhang, P. Xie, J. Yang, and S. Wang. 2012. MicroRNA-141 represses HBV replication by targeting PPARA. PLoS One 7:e34165. 159. Swaminathan, S., K. Suzuki, N. Seddiki, W. Kaplan, M. J. Cowley, C. L. Hood, J. L. Clancy, D. D. Murray, C. Mendez, L. Gelgor, B. Anderson, N. Roth, D. A. Cooper, and A. D. Kelleher. 2012. Differential Regulation of the Let-7 Family of MicroRNAs in CD4+ T Cells Alters IL-10 Expression. J Immunol 188:6238-6246. 160. Lu, L. F., M. P. Boldin, A. Chaudhry, L. L. Lin, K. D. Taganov, T. Hanada, A. Yoshimura, D. Baltimore, and A. Y. Rudensky. 2010. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 142:914-929. 161. Kulkarni, S., R. Savan, Y. Qi, X. Gao, Y. Yuki, S. E. Bass, M. P. Martin, P. Hunt, S. G. Deeks, A. Telenti, F. Pereyra, D. Goldstein, S. Wolinsky, B. Walker, H. A. Young, and M. Carrington. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature 472:495-498. 162. Ding, S., Y. Liang, M. Zhao, G. Liang, H. Long, S. Zhao, Y. Wang, H. Yin, P. Zhang, Q. Zhang, and Q. Lu. 2012. Decreased miR-142-3p/5p expression causes CD4(+) T cell activation and B cell hyperstimulation in systemic lupus erythematosus. Arthritis and rheumatism. 163. Van Camp, K., N. Cools, B. Stein, A. Van de Velde, H. Goossens, Z. N. Berneman, and V. Van Tendeloo. 2010. Efficient mRNA electroporation of peripheral blood mononuclear cells to detect memory T cell responses for immunomonitoring purposes. J Immunol Methods. 164. Wilgenhof, S., A. M. Van Nuffel, J. Corthals, C. Heirman, S. Tuyaerts, D. Benteyn, A. De Coninck, I. Van Riet, G. Verfaillie, J. Vandeloo, A. Bonehill, K. Thielemans, and B. Neyns. 2011. Therapeutic vaccination with an autologous mRNA electroporated dendritic cell vaccine in patients with advanced melanoma. J Immunother 34:448-456. 165. Johnson, L. A., B. Heemskerk, D. J. Powell, Jr., C. J. Cohen, R. A. Morgan, M. E. Dudley, P. F. Robbins, and S. A. Rosenberg. 2006. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol 177:6548-6559. 166. Bonhoeffer, S., E. C. Holmes, and M. A. Nowak. 1995. Causes of HIV diversity. Nature 376:125. 188 7 Literaturverzeichnis 167. Kozak, M. 1997. Recognition of AUG and alternative initiator codons is augmented by G in position +4 but is not generally affected by the nucleotides in positions +5 and +6. EMBO J 16:2482-2492. 168. Gustafsson, C., S. Govindarajan, and J. Minshull. 2004. Codon bias and heterologous protein expression. Trends in biotechnology 22:346-353. 169. Kavanagh, D. G., D. E. Kaufmann, S. Sunderji, N. Frahm, S. Le Gall, D. Boczkowski, E. S. Rosenberg, D. R. Stone, M. N. Johnston, B. S. Wagner, M. T. Zaman, C. Brander, E. Gilboa, B. D. Walker, and N. Bhardwaj. 2006. Expansion of HIV-specific CD4+ and CD8+ T cells by dendritic cells transfected with mRNA encoding cytoplasm- or lysosome-targeted Nef. Blood 107:1963-1969. 170. Kyte, J. A., and G. Gaudernack. 2006. Immuno-gene therapy of cancer with tumour-mRNA transfected dendritic cells. Cancer Immunol Immunother 55:1432-1442. 171. Van Gulck, E. R., P. Ponsaerts, L. Heyndrickx, K. Vereecken, F. Moerman, A. De Roo, R. Colebunders, G. Van den Bosch, D. R. Van Bockstaele, V. F. Van Tendeloo, S. Allard, B. Verrier, C. Maranon, G. Hoeffel, A. Hosmalin, Z. N. Berneman, and G. Vanham. 2006. Efficient stimulation of HIV-1-specific T cells using dendritic cells electroporated with mRNA encoding autologous HIV-1 Gag and Env proteins. Blood 107:1818-1827. 172. Hofmann, C., T. Harrer, V. Kubesch, K. Maurer, K. J. Metzner, K. Eismann, S. Bergmann, M. Schmitt-Haendle, G. Schuler, J. Dorrie, and N. Schaft. 2008. Generation of HIV-1-specific T cells by electroporation of T-cell receptor RNA. Aids 22:1577-1582. 173. Tobery, T. W., S. Wang, X. M. Wang, M. P. Neeper, K. U. Jansen, W. L. McClements, and M. J. Caulfield. 2001. A simple and efficient method for the monitoring of antigen-specific T cell responses using peptide pool arrays in a modified ELISpot assay. J Immunol Methods 254:59-66. 174. Kern, F., N. Faulhaber, C. Frommel, E. Khatamzas, S. Prosch, C. Schonemann, I. Kretzschmar, R. Volkmer-Engert, H. D. Volk, and P. Reinke. 2000. Analysis of CD8 T cell reactivity to cytomegalovirus using protein-spanning pools of overlapping pentadecapeptides. Eur J Immunol 30:1676-1682. 175. Larsson, M., X. Jin, B. Ramratnam, G. S. Ogg, J. Engelmayer, M. A. Demoitie, A. J. McMichael, W. I. Cox, R. M. Steinman, D. Nixon, and N. Bhardwaj. 1999. A recombinant vaccinia virus based ELISPOT assay detects high frequencies of Pol-specific CD8 T cells in HIV-1-positive individuals. AIDS 13:767-777. 176. Sun, Y., E. Iglesias, A. Samri, G. Kamkamidze, T. Decoville, G. Carcelain, and B. Autran. 2003. A systematic comparison of methods to measure HIV-1 specific CD8 T cells. J Immunol Methods 272:23-34. 177. Honeyborne, I., A. Prendergast, F. Pereyra, A. Leslie, H. Crawford, R. Payne, S. Reddy, K. Bishop, E. Moodley, K. Nair, M. van der Stok, N. McCarthy, C. M. Rousseau, M. Addo, J. I. Mullins, C. Brander, P. Kiepiela, B. D. Walker, and P. J. Goulder. 2007. Control of human immunodeficiency virus type 1 is associated with HLA-B*13 and targeting of multiple gag-specific CD8+ T-cell epitopes. J Virol 81:3667-3672. 178. Treiber, T., N. Treiber, and G. Meister. 2012. Regulation of microRNA biogenesis and function. Thrombosis and haemostasis 107:605-610. 189 7 Literaturverzeichnis 179. Koronkiewicz, S., and S. Kalinowski. 2004. Influence of cholesterol on electroporation of bilayer lipid membranes: chronopotentiometric studies. Biochim Biophys Acta 1661:196-203. 180. Forsbach, A., J. G. Nemorin, C. Montino, C. Muller, U. Samulowitz, A. P. Vicari, M. Jurk, G. K. Mutwiri, A. M. Krieg, G. B. Lipford, and J. Vollmer. 2008. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J Immunol 180:3729-3738. 181. Gorden, K. B., K. S. Gorski, S. J. Gibson, R. M. Kedl, W. C. Kieper, X. Qiu, M. A. Tomai, S. S. Alkan, and J. P. Vasilakos. 2005. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol 174:1259-1268. 182. Kaisho, T., and S. Akira. 2002. Toll-like receptors as adjuvant receptors. Biochim Biophys Acta 1589:1-13. 183. Jia, M., K. Hong, J. Chen, Y. Ruan, Z. Wang, B. Su, G. Ren, X. Zhang, Z. Liu, Q. Zhao, D. Li, H. Peng, M. Altfeld, B. D. Walker, X. G. Yu, and Y. Shao. 2012. Preferential CTL targeting of Gag is associated with relative viral control in long-term surviving HIV-1 infected former plasma donors from China. Cell research 22:903-914. 184. Venzke, S., N. Michel, I. Allespach, O. T. Fackler, and O. T. Keppler. 2006. Expression of Nef downregulates CXCR4, the major coreceptor of human immunodeficiency virus, from the surfaces of target cells and thereby enhances resistance to superinfection. J Virol 80:11141-11152. 185. Schmitt, M., E. Harrer, A. Goldwich, M. Bauerle, I. Graedner, J. R. Kalden, and T. Harrer. 2000. Specific recognition of lamivudine-resistant HIV-1 by cytotoxic T lymphocytes. AIDS 14:653-658. 186. Fellay, J., K. V. Shianna, D. Ge, S. Colombo, B. Ledergerber, M. Weale, K. Zhang, C. Gumbs, A. Castagna, A. Cossarizza, A. Cozzi-Lepri, A. De Luca, P. Easterbrook, P. Francioli, S. Mallal, J. Martinez-Picado, J. M. Miro, N. Obel, J. P. Smith, J. Wyniger, P. Descombes, S. E. Antonarakis, N. L. Letvin, A. J. McMichael, B. F. Haynes, A. Telenti, and D. B. Goldstein. 2007. A whole-genome association study of major determinants for host control of HIV-1. Science 317:944-947. 187. Lucchiari-Hartz, M., V. Lindo, N. Hitziger, S. Gaedicke, L. Saveanu, P. M. van Endert, F. Greer, K. Eichmann, and G. Niedermann. 2003. Differential proteasomal processing of hydrophobic and hydrophilic protein regions: contribution to cytotoxic T lymphocyte epitope clustering in HIV-1-Nef. Proc Natl Acad Sci U S A 100:7755-7760. 188. Rolland, M., D. C. Nickle, W. Deng, N. Frahm, C. Brander, G. H. Learn, D. Heckerman, N. Jojic, V. Jojic, B. D. Walker, and J. I. Mullins. 2007. Recognition of HIV-1 peptides by host CTL is related to HIV-1 similarity to human proteins. PLoS One 2:e823. 189. Mitsui, K., D. Nakajima, O. Ohara, and M. Nakayama. 2002. Mammalian fat3: a large protein that contains multiple cadherin and EGF-like motifs. Biochem Biophys Res Commun 290:1260-1266. 190. Inoue, T., E. Yaoita, H. Kurihara, F. Shimizu, T. Sakai, T. Kobayashi, K. Ohshiro, H. Kawachi, H. Okada, H. Suzuki, I. Kihara, and T. Yamamoto. 2001. FAT is a component of glomerular slit diaphragms. Kidney Int 59:1003-1012. 190 7 Literaturverzeichnis 191. Katoh, Y., and M. Katoh. 2006. Comparative integromics on FAT1, FAT2, FAT3 and FAT4. Int J Mol Med 18:523-528. 192. Nagae, S., T. Tanoue, and M. Takeichi. 2007. Temporal and spatial expression profiles of the Fat3 protein, a giant cadherin molecule, during mouse development. Dev Dyn 236:534-543. 193. Fukui, K., and Y. Miyake. 1992. Molecular cloning and chromosomal localization of a human gene encoding D-amino-acid oxidase. J Biol Chem 267:18631-18638. 194. Sacchi, S., M. Bernasconi, M. Martineau, J. P. Mothet, M. Ruzzene, M. S. Pilone, L. Pollegioni, and G. Molla. 2008. pLG72 modulates intracellular D-serine levels through its interaction with D-amino acid oxidase: effect on schizophrenia susceptibility. J Biol Chem 283:22244-22256. 195. Liu, X., G. He, X. Wang, Q. Chen, X. Qian, W. Lin, D. Li, N. Gu, G. Feng, and L. He. 2004. Association of DAAO with schizophrenia in the Chinese population. Neurosci Lett 369:228-233. 196. Burnet, P. W., S. L. Eastwood, G. C. Bristow, B. R. Godlewska, P. Sikka, M. Walker, and P. J. Harrison. 2008. D-amino acid oxidase activity and expression are increased in schizophrenia. Mol Psychiatry 13:658-660. 197. Wood, L. S., E. H. Pickering, and B. M. Dechairo. 2007. Significant support for DAO as a schizophrenia susceptibility locus: examination of five genes putatively associated with schizophrenia. Biol Psychiatry 61:1195-1199. 198. http://www.etrl.org/etrlpra/webform1.aspx. 2012, Oktober. 199. Baldrick, P. 2010. The safety of chitosan as a pharmaceutical excipient. Regulatory toxicology and pharmacology : RTP 56:290-299. 200. Corsi, K., F. Chellat, L. Yahia, and J. C. Fernandes. 2003. Mesenchymal stem cells, MG63 and HEK293 transfection using chitosan-DNA nanoparticles. Biomaterials 24:1255-1264. 201. Sato, T., T. Ishii, and Y. Okahata. 2001. In vitro gene delivery mediated by chitosan. effect of pH, serum, and molecular mass of chitosan on the transfection efficiency. Biomaterials 22:2075-2080. 202. Mansouri, S., P. Lavigne, K. Corsi, M. Benderdour, E. Beaumont, and J. C. Fernandes. 2004. Chitosan-DNA nanoparticles as non-viral vectors in gene therapy: strategies to improve transfection efficacy. Eur J Pharm Biopharm 57:1-8. 203. Niesner, U., C. Halin, L. Lozzi, M. Gunthert, P. Neri, H. Wunderli-Allenspach, L. Zardi, and D. Neri. 2002. Quantitation of the tumor-targeting properties of antibody fragments conjugated to cell-permeating HIV-1 TAT peptides. Bioconjugate chemistry 13:729-736. 204. Schwarze, S. R., A. Ho, A. Vocero-Akbani, and S. F. Dowdy. 1999. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285:15691572. 191 7 Literaturverzeichnis 205. Simon, M. J., S. Gao, W. H. Kang, S. Banta, and B. Morrison, 3rd. 2009. TATmediated intracellular protein delivery to primary brain cells is dependent on glycosaminoglycan expression. Biotechnol Bioeng 104:10-19. 206. Kawata, E., E. Ashihara, S. Kimura, K. Takenaka, K. Sato, R. Tanaka, A. Yokota, Y. Kamitsuji, M. Takeuchi, J. Kuroda, F. Tanaka, T. Yoshikawa, and T. Maekawa. 2008. Administration of PLK-1 small interfering RNA with atelocollagen prevents the growth of liver metastases of lung cancer. Mol Cancer Ther 7:2904-2912. 207. Honma, K., T. Ochiya, S. Nagahara, A. Sano, H. Yamamoto, K. Hirai, Y. Aso, and M. Terada. 2001. Atelocollagen-based gene transfer in cells allows high-throughput screening of gene functions. Biochem Biophys Res Commun 289:1075-1081. 208. Jacobsen, L. B., S. A. Calvin, K. E. Colvin, and M. Wright. 2004. FuGENE 6 Transfection Reagent: the gentle power. Methods 33:104-112. 209. Arnold, A. N., Y.M.; Fabian, C.; Wirth, H.; Binder, H.; Nikkhah, G.; Armstrong, L.; Stolzing, A. 2012. Reprogramming of Human Huntington Fibroblasts Using mRNA. ISRN Cell Biology 124878:12. 210. Brockman, M. A., A. Schneidewind, M. Lahaie, A. Schmidt, T. Miura, I. Desouza, F. Ryvkin, C. A. Derdeyn, S. Allen, E. Hunter, J. Mulenga, P. A. Goepfert, B. D. Walker, and T. M. Allen. 2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J Virol 81:12608-12618. 211. Saag, M. S., B. H. Hahn, J. Gibbons, Y. Li, E. S. Parks, W. P. Parks, and G. M. Shaw. 1988. Extensive variation of human immunodeficiency virus type-1 in vivo. Nature 334:440-444. 212. Preston, B. D., B. J. Poiesz, and L. A. Loeb. 1988. Fidelity of HIV-1 reverse transcriptase. Science 242:1168-1171. 213. Leslie, A. J., K. J. Pfafferott, P. Chetty, R. Draenert, M. M. Addo, M. Feeney, Y. Tang, E. C. Holmes, T. Allen, J. G. Prado, M. Altfeld, C. Brander, C. Dixon, D. Ramduth, P. Jeena, S. A. Thomas, A. St John, T. A. Roach, B. Kupfer, G. Luzzi, A. Edwards, G. Taylor, H. Lyall, G. Tudor-Williams, V. Novelli, J. Martinez-Picado, P. Kiepiela, B. D. Walker, and P. J. Goulder. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med 10:282-289. 214. Miura, T., M. A. Brockman, A. Schneidewind, M. Lobritz, F. Pereyra, A. Rathod, B. L. Block, Z. L. Brumme, C. J. Brumme, B. Baker, A. C. Rothchild, B. Li, A. Trocha, E. Cutrell, N. Frahm, C. Brander, I. Toth, E. J. Arts, T. M. Allen, and B. D. Walker. 2009. HLA-B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare gag variants associated with reduced viral replication capacity and strong cytotoxic Tlymphocyte [corrected] recognition. J Virol 83:2743-2755. 215. Gupta, A., P. Nagilla, H. S. Le, C. Bunney, C. Zych, A. Thalamuthu, Z. Bar-Joseph, S. Mathavan, and V. Ayyavoo. Comparative Expression Profile of miRNA and mRNA in Primary Peripheral Blood Mononuclear Cells Infected with Human Immunodeficiency Virus (HIV-1). PLoS One 6:e22730. 216. Huang, J., F. Wang, E. Argyris, K. Chen, Z. Liang, H. Tian, W. Huang, K. Squires, G. Verlinghieri, and H. Zhang. 2007. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med 13:1241-1247. 192 7 Literaturverzeichnis 217. Ma, F., S. Xu, X. Liu, Q. Zhang, X. Xu, M. Liu, M. Hua, N. Li, H. Yao, and X. Cao. 2011. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol 12:861-869. 218. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chainterminating inhibitors. Proc Natl Acad Sci U S A 74:5463-5467. 193 8 Abkürzungsverzeichnis AIDS Amp APC ARCA AS BLAST B-LCL BSA bzw. ca. CBA CCR5 CD CDC cDNA CFSE CMV CTL CXCR4 DAPI DAO DC DEPC DMEM DMF DMSO DNA DPBS DTT EBV E.coli EDTA ELF ELISA ELISPOT Env ER et al. FACS FAD FCS FITC FSC Gag GFP gp H2O HAART HIV HLA acquired immunodeficiency syndrome (erworbenes Immunschwächesyndrom) Ampicillin antigen presenting cell (Antigen-präsentierende Zelle) antireverses Cap-Analog Aminosäure basic local alignment search tool EBV-transformierte lymphoide B-Zelllinien bovine serum albumine (Rinderserumalbumin) beziehungsweise circa cytometric bead array CC Chemokin-Rezeptor 5 cluster of differentiation (Differenzierungscluster) U.S. Centers for Disease Control and Prevention complementary DNA (komplementäre DNS) Carboxyfluoreszein-Succinimidyl Ester Cytomegalie-Virus cytotoxic T lymphocyte (Zytotoxische T-Zelle) CXC-Motiv-Chemokinrezeptor 4 4′,6-Diamidin-2-phenylindol D-Aminosäuren-Oxidase dendritic cell (Dendritische Zelle) Diethylpyrocarbonat Dulbecco's Modified Eagle's Medium Dimethylformamid Dimethylsulfoxid Deoxyribonucleic acid (Desoxyribonukleinsäure=DNS) Phosphat-buffer saline Dithiothreitol Epstein-Barr-Virus Escherichia coli Ethylendiamintetraacetat epitope location finder Enzyme-linked immunosorbent assay Enzyme-linked Immuno Spot Assay Envelope Endoplasmatisches Retikulum et alii (und andere) flourescence activated cell sorting Flavin-Adenin-Dinukleotid fetal calf serum (fötales Kälberserum) fluorescein isotheiocyanate forward scatter group antigen green flourescence protein (grün fluoreszierendes Protein) Glykoprotein Wasser Hoch aktive antiretrovirale Therapie Humanes Immundefizienz Virus Humanes Leukozyten Antigen 194 8 Abkürzungsverzeichnis HTLV-III IFN IL IN KA Kan LAV LB LTNP LTR M MA MACS MFI MHC miRNA/miR MOI MOPS MRC mRNA MVA N NCBI Nef NFκB NIH NK NNRTI NRTI NSI O OD PBMC PCR PE PFA PHA PI PI pNPP Pol PR qRT-PCR RNA rpm RPMI RT RT-PCR SAP SFU Humanes T-Zell Leukämie Virus Typ III Interferon Interleukin Integrase Kapsid Kanamycin Lymphadenopathi-assoziiertes Retrovirus Luria-Bertani longterm non-progressors (Langzeit Nicht-Progressoren) long terminal repeats (lange terminale Wiederholungen) major, Hauptgruppe der Virusisolate Matrix magnetic associated cell sorting median fluorescence intensitiy (Durchschnittliche Fluoreszenzintensität) major histocompatibility complex (Haupthistokompatibilitätskomplex) microRNA multiplicity of infection (Multiplizität der Infektion) 3‘-(N-Morpholino)-propansulfonsäure AIDS Reagent Project of the Medical Research Council messenger RNA (Boten-RNA) modifiziertes Vaccinia-Virus Ankara new, neue Virusisolate National Center for Biotechnology Information negative factor nuclear factor 'kappa-light-chain-enhancer' of activated Bcells National Institute of Health Nukleokapsid Nicht-Nukleosidische Reverse-Transkriptase-Inhibitoren Nukleosidische Reverse-Transkriptase-Inhibitoren non-syncytium-inducing (nicht Synzytien induzierend) outlier, Ausreißer der Virusisolate Optische Dichte peripheral blood mononuclear cells (mononukleäre Zellen des peripheren Blutes) polymerase chain reaction (Polymerasen-Ketten-Reaktion) Phycoerythrin Paraformaldehyd Phytohämagglutinin Protease-Inhibitoren Propidium-Jodid p-Nitrophenyl Phosphate Polymerase Protease quantitative Real time-PCR (quantitative Echtzeit-PCR) Ribonucleic acid (Ribonukleinsäure=RNS) revolutions per minute (Umdrehungen/min) Roswell Park Memorial Institute Reverse Transkriptase Reverse Transkriptase Polymerasen-Ketten-Reaktion shrimp alkaline phosphatase (alkalische ShrimpPhosphatase) spot forming units 195 8 Abkürzungsverzeichnis SI SIV SLE SNP ss SSC sog. TAP Tat TCR TBE TH TLR TNF TRAIL TRIM5 UCSC UV UTR z.B. syncytium-inducing (Synzytien induzierend) Simianes Immundefizienz Virus systemischer Lupus erythematodes single nucleotid polymorphism (Einzelnukleotidpolymorphismen) single stranded (einzelsträngig) side scatter sogenannt transporter associated with antigen presenting transactivator (Transaktivator) T cell receptor (T-Zell-Rezeptor) Tris-Borsäure-EDTA T-Helferzelle Toll like Rezeptoren Tumor-Nekrosis-Faktor TNF-related apoptosis-induced ligand Tripartif-Interaktions-Motiv 5 University of California at Santa Cruz Ultra-violett untranslatierte Region zum Beispiel Einheiten: % bp °C g l M min mg ml mM mm ms µg µl µM µm ng nm pg pH sek SFU U U/min V v/v Prozent Basenpaare Grad Celsius Gramm Liter molar (mol/l) Minute Milligramm Milliliter Millimolar Millimeter Millisekunde Mikrogramm Mikroliter Mikromolar Mikrometer Nanogramm Nanometer Picogramm pondus hydrogenii Sekunde Spot forming units Units (Einheit) Umdrehungen/Minute Volt volume/volume 196 8 Abkürzungsverzeichnis Vorsatzzeichen/Vorsätze: kilo (103) milli (10-3) mikro (10-6) nano (10-9) pico (10-12) k m µ n p Aminosäuren Abkürzungen Aminosäure Dreibuchstabencode Einbuchstabencode Alanin Arginin Asparagin Asparaginsäure Cystein Glutamin Glutaminsäure Glycin Histidin Isoleucin Leucin Lysin Methionin Phenylalanin Prolin Serin Threonin Tryptophan Tyrosin Valin Ala Arg Asn Asp Cys Gln Glu Gly His Ile Leu Lys Met Phe Pro Ser Thr Trp Tyr Val A R N D C Q E G H I L K M F P S T W Y V Nukleotide/Nukleinsäuren: A C G N T U K M R S W Y dNTPs Adenin Cytosin Guanin beliebiges Nukleotid Thymin Uracil G oder T A oder C A oder G C oder G A oder T C oder T Desoxyribonukleotid-triphosphat 197 8 Abkürzungsverzeichnis Griechische Buchstaben: α β γ Δ Κ Alpha Beta Gamma Delta Kappa 198 9 Eigene Publikationen, Konferenzbeiträge und Vorträge Eigene Publikationen: Etschel JK, Hückelhoven AG, Hofmann C, Zitzelsberger K, Maurer K, Bergmann S, Mueller-Schmucker SM, Wittmann J, Spriewald BM, Dörrie J, Schaft N, Harrer T. HIV-1 mRNA electroporation of PBMC: A simple and efficient method to monitor T-cell responses against autologous HIV-1 in HIV-1-infected patients. Journal of Immunological methods 380 (2012), 40-55 Veröffentlichte Kongressberichte: Etschel J., Hückelhoven A., Zitzelsberger K., Hofmann C., Bergmann S., Dörrie J., Schaft N., Müller-Schmucker S., Harrer T. Assessment of CTL hierarchy by analysis T cell responses against autologous viral HIV-1 strains using mRNA electroporation of PBMC. Program & Abstracts of 19th Conference on Retroviruses and Opportunistic Infections (CROI) 2012, Seattle, USA, S.216 Ekici A.B., Etschel J., Uebe S., Pasutto F., Kruse F., Mardin C.Y., Reis A. Determining the genetic architecture of central cornea thickness (CCT) with GWAS using DNA pooling. Medizinische Genetik 1, 2009, S.163f. Ekici A.B., Etschel J., Uebe S., Pasutto F., Kruse F., Mardin C.Y., Reis A. Determining the genetic architecture of central cornea thickness (CCT) with GWAS using DNA pooling. Programm guide of the 58th Annual Meeting of the American Society of Human Genetics 2008, Philadelphia, USA Konferenzbeiträge: Posterbeiträge: Etschel J., Hückelhoven A., Zitzelsberger K., Hofmann C., Bergmann S., Dörrie J., Schaft N., Müller-Schmucker S., Harrer T. Assessment of CTL hierarchy by analysis T cell responses against autologous viral HIV-1 strains using mRNA electroporation of PBMC. 19th Conference on Retroviruses and Opportunistic Infections (CROI), 2012, Seattle, USA 199 9 Eigene Publikationen, Konferenzbeiträge und Vorträge Etschel J., Maurer K., Hofmann C., Zitzelsberger K., Hückelhoven A., Bergmann S., Müller-Schmucker S., Dörrie J., Schaft N., Harrer T. Immunmonitoring von T-Zellantworten gegen autologe HIV-1 Stämme mittels mRNA-Elektroporation von PBMC. 5. Deutsch-Österreichische AIDS-Kongress (DÖAK) 2011, Hannover Hückelhoven A., Etschel J., Zitzelsberger K., Müller-Schmucker S.M., Bergmann S., Harrer E.G., Harrer T. Analyse von HIV-1-spezifischen CTL mit Kreuzreaktion zu Influenza A in einer Kohorte von HIV-1-infizierten Patienten. 5. Deutsch-Österreichische AIDS-Kongress (DÖAK) 2011, Hannover Etschel J., Maurer K., Hofmann C., Eismann K., Dörrie J., Schaft N., Müller S.M., Harrer, T. mRNA electroporation of PBMC is a rapid and efficient method for immunomonitoring of T-cell responses against autologous HIV-1 strains. XVIII International Aids conference 2010, Vienna Hückelhoven A., Bergmann S., Eismann K., Müller S.M., Etschel J., Harrer E.G., Harrer T. Analysis of HIV-1-specific CTL cross-reacting with Influenza A virus in a cohort of HIV-1infected patients. XVIII International Aids conference 2010, Vienna Hückelhoven A., Bergmann S., Eismann K., Müller S.M., Etschel J., Harrer E.G., Harrer T. Analyse von HIV-1-spezifischen CTL mit Kreuzreaktionen zu Influenza A in einer Kohorte von HIV-1 infizierten Patienten. 10. Kongress für Infektionskrankheiten und Tropenmedizin (KIT), 2010, Köln Etschel J., Maurer K., Eismann K., Harrer T. Development of RNA-vaccines for therapy of HIV infection. IZKF-Symposium 2009, Kloster Banz Maurer K., Schmökel J., Kirchhoff F., Etschel J., Antoni S., Bergmann S., Eismann K., Rauch P., Metzner K., Mueller S.M., Harrer E.G., Harrer T. Association of effctive control of HIV-1 with strong CTL selection pressure and a highly mutated nef in a HIV-1-infected patient. DGI-Jahrestagung 2009, Freiburg Vortrag: Etschel J., Maurer K., Hofmann C., Eismann K., Dörrie J., Schaft N., Müller S.M., Harrer T. Einfaches und effizientes Immunmonitoring von T-Zellantworten gegen autologe HIV-1Viren durch mRNA Elektroporation von PBMC. 10. Kongress für Infektionskrankheiten und Tropenmedizin (KIT), 2010, Köln 200 9 Eigene Publikationen, Konferenzbeiträge und Vorträge Weitere wissenschaftliche Vorträge: April 2012 Seminar der Medizinischen Klinik 3 „Immunomonitoring and Immunotherapy with HIV-1-specific mRNA” Mai 2011 GRK1071 Retreat in Schlaifhausen „Immunomonitoring and Immunotherapy with HIV specific mRNA” September 2010 GRK1071 Retreat in Boston „Immunomonitoring and Immunotherapy with HIV specific mRNA” April 2010 Seminar der Medizinischen Klinik 3 „Immunomonitoring and Immunotherapy with HIV specific mRNA” April 2010 GRK1071 Retreat in Schlaifhausen „Immunomonitoring and Immunotherapy with HIV specific mRNA” Juli 2009 Retreat der Medizinischen Klinik 3 in Markt Taschendorf „Development of RNA-vaccines for therapy of HIV infection” 201 10 Danksagung Mein ganz besonderer Dank gilt Herrn Prof. Dr. Thomas Harrer, welcher mir die Arbeit an dieser spannenden Dissertation ermöglichte und stets großes Interesse zeigte. Danke für die Förderung, die anregenden Gespräche und Diskussionen sowie hilfreiche Ratschläge zu diesem Projekt. Außerdem danke ich für die Erstellung des Zweitgutachtens. Bei Herrn Prof. Dr. Thomas Winkler möchte ich mich für die Betreuung meiner Arbeit von Seiten der Naturwissenschaftlichen Fakultät II der Universität Erlangen-Nürnberg sowie für die Erstellung des Erstgutachtens bedanken. Ebenso möchte ich meiner ganzen Arbeitsgruppe, der AG Harrer, für das angenehme Arbeitsklima und die gute Zusammenarbeit herzlich danken. Frau Dr. Sandra MüllerSchmucker danke ich für zahlreiche Hilfestellungen bei Problemen, der Beantwortung meiner Fragen und interessante Gespräche. Den medizinisch-technischen Assistentinnen Silke Bergmann, Kathrin Zitzelsberger und Melanie Leykauf danke ich für ihre Hilfe im Laboralltag und Einarbeitung in die unterschiedlichen Labortechniken. Ihre Erfahrungen in den verschiedenen Bereichen konnten mir oft weiterhelfen. Meinen Mit-Doktorandinnen Angela Hückelhoven und Christiane Mummert sowie dem Diplomanden Andrej Stoll möchte ich einen besonderen Dank aussprechen. Wir waren ein gutes Team, welches sich durch Rücksicht und Hilfsbereitschaft gegenseitig unterstützte. Auch meinem Ex-MitDoktoranden Herrn Dr. Christian Hofmann möchte ich für seine Hilfe bezüglich der Elektroporation und den CBA-Messungen danken. Unseren Kooperationspartnern, der AG Jäck der Medizinischen Klinik 3 und der „RNAGroup“ der Dermatologie Erlangen, danke ich für die gute und reibungslose Zusammenarbeit. Bei Herrn Dr. Jürgen Wittmann und Frau Manuela Hauke möchte ich mich besonders für ihre Hilfestellungen und Unterstützung im Bereich der miRNATechnologie bedanken. Herrn Dr. Jan Dörrie sowie Herrn Dr. Niels Schaft aus der Dermatologie danke ich für die Bereitstellung des Klonierungsvektors und ihrer Hilfe bezüglich der mRNA-Transfer-Technologie. Herrn Prof. Dr. Bernhard Fleckenstein und Frau Dr. Brigitte Biesinger-Zwosta danke ich für die Aufnahme in das Graduiertenkolleg 1071 als assoziiertes Mitglied. Dies ermöglichte mir die Teilnahme an vielen interessanten Workshops und Seminaren, die wertvolle Informationen und Tipps für mich bereithielten. Ebenso gewährte mir das 202 10 Danksagung Graduiertenkolleg den Besuch von internationalen Kongressen, welche mir interessante Einblicke lieferten und eine wichtige Erfahrung für mich waren. Ich danke auch den Mitarbeitern der Ambulanz der Medizinischen Klinik 3. Ohne ihre schnelle sowie auch freundliche Durchführung von spontanen Blutabnahmen wäre die Realisierung und Organisation meiner Arbeit schwierig geworden. Meiner „Kollegin“ und guten Freundin Daniela möchte ich danken, dass wir all die Probleme, die während einer Promotion auftreten können, in langen Telefonaten diskutiert haben und diese an gemeinsamen Wochenenden in München oder Nürnberg versucht haben zu vergessen ;o) Von ganzem Herzen danke ich meinem Freund Norman, der mich in der gesamten Zeit unterstützte und mich in meinem Tun stets ermutigte. Ich danke Dir für dein Verständnis und deine Geduld. Zuletzt möchte ich mich bei meiner ganzen Familie, insbesondere bei meinen Eltern bedanken. Sie haben mich während meines Studiums sowie während meiner Dissertation immer gefördert, indem sie mein Handeln stets bestärkten. 203