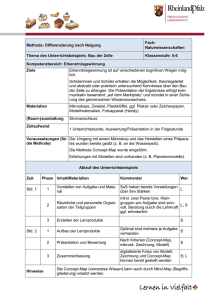
Die Herabregulation des CD4-Rezeptors - OPARU
Werbung

Universität Ulm Institut für Virologie Leiter: Prof. Dr. Thomas Mertens Die Herabregulation des CD4-Rezeptors schützt HIV-1 infizierte T-Zellen vor Superinfektion und frühzeitiger Apoptose Dissertation zur Erlangung des Doktorgrades der Humanbiologie (Dr. biol. hum.) der Medizinischen Fakultät der Universität Ulm vorgelegt von Steffen Wildum aus Ulm 2006 Amtierender Dekan: Prof. Dr. Klaus-Michael Debatin 1. Berichterstatter: Prof. Dr. Frank Kirchhoff 2. Berichterstatter: PD Dr. Wolfgang Weidemann Tag der Promotion: 17.11.2006 „Die Zeit wird kommen, wo unsere Nachkommen sich wundern, dass wir so offenbare Dinge nicht gewusst haben.“ Lucius A. Seneca [4 v.Chr. – 65 n.Chr.] Widmung Meinen Eltern, die ihren Teil zu dieser Arbeit beigetragen haben. 1. INHALTSVERZEICHNIS 1 1. Inhaltsverzeichnis 1 2. Abkürzungsverzeichnis 3 3. Einleitung 6 3.1 Humane Immundefizienzviren: Entdeckung und Epidemiologie 6 3.2 Morphologie und Genomaufbau der Humanen Immundefizienzviren 7 3.3 CD4-Herabregulation durch HIV-1 8 3.6 Fragestellung 9 4. Material 10 4.1 Eukaryotische Zelllinien 10 4.2 Bakterien 10 4.3 Nukleinsäuren 10 4.4 Enzyme 11 4.5 Reagenzien und Laborhilfsmittel 12 4.6 Reagenzsysteme (Kits) 13 4.7 Medien 13 4.8. Lösungen und Puffer 14 4.9 Antikörper für die Durchflusszytometrie 14 5. Methoden 15 5.1 DNA-Methoden 15 5.2 Zellkultur 16 5.3 Bakterienkultur 16 5.4 Herstellung rekombinanter HIV-1 NL4-3 Viren 17 5.5 Isolierung primärer Lymphozyten aus Buffy Coat 18 5.6 Herstellung von Virusstocks und Infektionen 18 5.7 FACS-Analysen 20 5.8 Nachweis viraler Proteine durch Western Blot Analyse 20 5.9 Computerprogramme und Datenquellen 21 1. INHALTSVERZEICHNIS 6. Ergebnisse 2 23 6.1 Rekombinante HIV-1 NL4-3 Viren mit defekten vpu, env und nef Genen 23 6.2 CD4-Modulation in HIV-1 infizierten Zellen 24 6.3 Die Herabregulation von CD4 schützt HIV-1 infizierte T-Zellen vor einer Superinfektion 29 6.4 Nef schützt infizierte Zellen durch die Modulation von CD4 und mittels eines anderen Mechanismus vor einer Superinfektion 32 6.5 Auch die Nef-vermittelte CxCR4-Herabregulation trägt, neben weiteren Faktoren, zur Superinfektionsresistenz bei 34 6.6 Die Resistenz gegen eine Superinfektion schützt HIV-1 infizierte T-Zellen vor dem frühzeitigen Tod durch Apoptose 37 7. Diskussion 39 8. Zusammenfassung 44 9. Literaturverzeichnis 46 2. ABKÜRZUNGSVERZEICHNIS 2. Abkürzungsverzeichnis A As Abb AIDS APC bp BSA °C CII-TA CD CHO Zellen cm CMV CTL DMEM DMSO DTT DNA dNTP E.coli EDTA eGFP ELISA engl. env EtOH FACS FKS g gag Gp h HBS HEPES HIV HLA HRP ID Ig Ii IL IRES Ampere Aminosäure Abbildung engl.: acquired immunodeficiency syndrome engl.: antigen presenting cell Basenpaar Rinderserumalbumin Grad Celsius engl.: major histocompatibility complex class-II transactivator engl.: cluster designation Zellen aus den Ovarien chinesischer Hamster Zentimeter engl.: Cytomegalovirus engl.: cytotoxic T-Lymphocyte engl.: Dulbecco`s modified eagle medium Dimethylsulfoxid Dithiothreitol Desoxyribonukleinsäure Desoxynukleosidtriphosphate Escherichia coli Ethylendiamintetraacetat engl.: enhanced green fluorescent protein engl.: enzyme linked immunosorbent assay englisch engl.: envelope Ethanol engl.: fluorescence activated cell sorter fötales Kälberserum Gramm engl.: group specific antigen Glykoprotein engl.: hour engl.: HEPES buffered saline Hydroxyethylpiperazinethansulfonsäure Humanes Immundefizienzvirus engl.: human leucozyte antigen engl.: horse raddish peroxidase Identifikation Immunglobulin engl.: invariant chain (CD74) Interleukin engl.: internal ribosomal entry site 3 2. ABKÜRZUNGSVERZEICHNIS kB kg kD l LB LTR m µ M MFI MHC min n N NaOH nef ORF PBMC PBS PCR PE Pen/Strep PFA PHA P pol PPT PR R5-tropes Virus Rev RNA rpm RT s SDS SDS-PAGE SIV SOC SOE-PCR Stabw SV40 tat Tab. TEMED Kilobasen Kilogramm Kilo-Dalton Liter Luria Bertani engl.: long terminal repeat milli (10-3) mikro (10-6) Molar (mol/l) mittlere Fluoreszenzintensität engl.: major histocompatibility complex Minute nano (10-9) Normal Natronlauge engl.: negative factor engl.: open reading frame engl.: peripheral blood mononuclear cell engl.: phosphate buffered saline engl.: polymerase chain reaction Phycoerythrin Penicillin/Streptomycin Paraformaldehyd Phytohämagglutinin pico (10-12) Polymerase Polypurintrakt Protease Virus, das den CCR5-Rezeptor für die Infektion einer Zelle benötigt engl.: regulator of expression of virion proteins Ribonukleinsäure engl:: rounds per minute Reverse Transkriptase engl.: second Natriumdodecylsulfat SDS-Polyacrylamid-Gelelektrophorese engl.: simian immunodeficiency virus Salz-optimiertes Medium mit Glukose (salt-optimized + carbon) engl.: splice overlap extension-PCR Standardabweichung engl.: simian virus 40 engl.: transactivator of transcription Tabelle Tetramethylendiamin 4 2. ABKÜRZUNGSVERZEICHNIS Tris TM U vif Vol vpr vpu VSV VSV-G (v/v) wt (w/v) X4-tropes Virus A C D E F G H Ala Cys Asp Glu Phe Gly His 5 Trishydroxymethylaminomethan Transmembranprotein Unit engl.: viral infectivity factor Volumen engl.: viral protein rapid engl.: viral protein out engl.: vesicular stomatitis virus engl.: vesicular stomatitis virus glycoprotein Volumenanteil am Gesamtvolumen (engl.: volume per volume) Wildtyp Masse pro Volumen (engl.: weight per volume) Virus, das den CxCR4-Rezeptor für die Infektion einer Zelle benötigt Alanin Cystein Asparaginsäure Glutaminsäure Phenylalanin Glycin Histidin I K L M N P Q Ile Lys Leu Met Ans Pro Gln Isoleucin Lysin Leucin Methionin Asparagin Prolin Glutamin R S T V W Y Arg Ser Thr Val Trp Tyr Arginin Serin Threonin Valin Tryptophan Tyrosin 3. EINLEITUNG 6 3. Einleitung 3.1 Humane Immundefizienzviren: Entdeckung und Epidemiologie Im Jahr 1981 traten in New York und Los Angeles mysteriöse Todesfälle von jungen homosexuellen Männern auf, die häufig an einem seltenen Hauttumor, dem so genannten Kaposi-Sarkom, erkrankt waren und schließlich an opportunistischen Infektionen verstarben. Da die betroffenen Personen die Symptome eines defekten Immunsystems zeigten, wurde die Erkrankung als AIDS (engl.: acquired immunodeficiency syndrome) bezeichnet (Gottlieb et al., 1981). Zwei Jahre lang wurde nach der Ursache der erworbenen Immunschwäche geforscht, bis 1983 von der Arbeitsgruppe um Luc Montagnier ein Retrovirus als Erreger der Krankheit identifiziert wurde (Barré-Sinoussi et al., 1983). Erst drei Jahre später wurde dieses Virus der Familie der Lentiviren zugeordnet und als „Humanes Immundefizienzvirus Typ 1“ (HIV-1) bezeichnet (Coffin et al., 1986). Im gleichen Jahr wurde aus Blutlymphozyten eines afrikanischen AIDS-Patienten ein mit HIV-1 verwandtes Immundefizienzvirus isoliert und als „Humanes Immundefizienzvirus Typ 2“ (HIV-2) bezeichnet (Clavel et al., 1986). Ursprünglich entstand das HI-Virus durch die Übertragung von natürlich vorkommenden Affen-Immundefizienzviren (engl.: simian immunodeficiency virus, SIV) auf den Menschen. Das älteste bekannte HIV-1 positive menschliche Blutserum stammt aus dem Jahr 1959 (Zhu et al., 1998). Seitdem hat sich AIDS zu einer weltweiten Pandemie entwickelt. Wie aus dem jährlichen UNAIDS-Report ersichtlich wird (www.unaids.org), waren 2005 weltweit etwa 38,6 Millionen Menschen mit HIV infiziert, wovon sich etwa 4,1 Millionen neu infiziert hatten. Die Mehrheit der Infizierten (etwa 26 Millionen) lebt im südlichen Afrika, wo in manchen Regionen bereits jeder fünfte Erwachsene HIV-positiv ist. AIDS gehört mit 2,8 Millionen Toten im Jahr 2005 weltweit zu einer der häufigsten Todesursachen. Dies liegt auch daran, dass trotz intensiver Forschung noch immer kein Impfstoff oder heilendes Medikament gegen das HI-Virus existiert. Seit wenigen Jahren steht zwar eine Kombinationstherapie zur Verfügung (hoch aktive antiretrovirale Therapie, HAART), die an verschiedenen Stellen im Replikationszyklus des Virus ansetzt und zu einer deutlich gesteigerten Lebenserwartung der Patienten führt (Palella et al., 1998), aber die Behandlung ist sehr kostenintensiv und die Nebenwirkungen sind beträchtlich. Eine vollständige Heilung durch die Eliminierung des Virus aus dem Organismus scheint 3. EINLEITUNG 7 bislang nicht möglich, da das Virus sein Genom in das Wirtszellgenom integriert und in langlebigen CD4+ Zellpopulationen persistiert (Ho et al., 1998). Die dringend notwendige Entwicklung eines wirksamen Impfstoffes ist nach heutigem Kenntnisstand selbst auf lange Sicht nicht zu erwarten (McMichael u. Hanke, 2003). 3.2 Morphologie und Genomaufbau der Humanen Immundefizienzviren HIV-Partikel haben einen Durchmesser von ca. 100 nm und sind von einer Phospholipiddoppelmembran umgeben, die der zellulären Zytoplasmamembran entstammt. In diese ist das virale Transmembranprotein gp41 eingelagert, an welches das externe Hüllprotein gp120 als Trimer nicht kovalent gebunden ist. Mit der Membraninnenseite sind die p17 Matrixproteine über Myristinsäurereste verbunden. Das virale Genom in Form zweier einzelsträngiger ca. 9,2 kB großer RNA Moleküle in Plusstrangorientierung befindet sich im aus p24 Kapsidproteinen aufgebauten konischen Kapsid, welches über p6Proteine mit der Membran verbunden ist. Das RNA-Genom ist mit den Nukleokapsidproteinen p7 und p9 komplexiert und bildet zusammen mit den viralen Enzymen Intergrase (IN), Reverse Transkriptase (RT) und Protease (PR) das Nukleokapsid (DeVita et al., 1997). Neben den Strukturproteinen werden auch Nef, Vpr und Vif in das Viruspartikel inkorporiert (Cohen et al., 1990; Liu et al., 1995; Welker et al., 1996). Das provirale Genom von HIV wird von langen Sequenzwiederholungen, den so genannten 5´ und 3´ LTRs (engl.: long terminal repeat), flankiert (Abb. 1). Abb. 1: Genomorganisation von HIV-1. Abkürzungen: LTR: long terminal repeat; gag: group specific antigen; pol: polymerase, vif: viral infectivity factor; tat: transactivator of transcription; r: vpr, viral protein r; u: vpu, viral protein u; env: envelope; rev: regulator of expression of viral proteins ; nef: negative factor. Die 5´ LTR dient als Promoter für die zelluläre RNA-Polymerase II. Neben den für die Retroviren typischen Strukturgenen gag, pol und env kodiert das HIV-1 Genom noch 3. EINLEITUNG 8 sechs weitere Gene. Hierbei handelt es sich um die regulatorischen Gene tat und rev sowie die akzessorischen Gene vif, vpr, vpu und nef. Diese akzessorischen Proteine sind für die Replikation des Virus in vitro nicht essentiell. 3.3 CD4-Herabregulation durch HIV-1 HIV-1 verwendet drei seiner Proteine (Vpu, Env und Nef) zur CD4Herabregulation (Lama, 2003). Nef wird in einem sehr frühen Stadium der Infektion gebildet und entfernt als einziges der drei Proteine CD4-Moleküle direkt von der Zelloberfläche. Dabei wirkt Nef als Verbindung zwischen CD4 und dem Adaptorprotein AP-2 und bewirkt die Endozytose von CD4 und seine Degradation in Lysosomen (Mangasarian et al., 1997; Piguet et al., 1998). Env und Vpu werden dagegen erst später synthetisiert und blockieren den Transport von neu gebildetem CD4 zur Zelloberfläche, indem Env das CD4-Molekül im Endoplasmatischen Retikulum zurückhält und Vpu die Ubiquitinilierung und damit den schnellen Abbau der Env-CD4-Komplexe durch das Proteasom veranlasst (Levesque et al., 2004). Die CD4-Herabregulation bringt für das Virus mehrer Vorteile mit sich. Sie erleichtert die Partikelfreisetzung aus der infizierten Zelle, da knospende Partikel mit geringerer Wahrscheinlichkeit mit ihren eingelagerten viralen Hüllproteinen mit den CD4Molekülen auf der Wirtszellmembran interagieren. Eine Bindung von Env an CD4 würde zu einer Fusion der Wirtszellmembran mit der sich abschnürenden Virusmembran führen und somit das Knospen des Partikels verhindern (Bour et al., 1999; Ross et al., 1999). Des weiteren wird durch die CD4-Herabregulation in der Virus produzierenden Zelle die Infektiosität der Nachkommenviren gesteigert, da diese weniger CD4 in ihrer Membran eingelagert haben, das direkt mit den Env-Proteinen interagieren könnte (Cortes et al., 2002; Levesque et al., 2003). Außerdem soll durch eine hohe CD4-Expression die Einlagerung von Env in die Virusmembran vermindert werden, wodurch die Infektiosität reifer Partikel sinken würde (Lama et al., 1999). Weiterhin verhindert die CD4-Modulation die effektive Stimulation des Immunsystems und schwächt damit die antivirale Immunantwort, da CD4 mit dem MHC-Klasse II Komplex (engl. major histocompatibility complex) einer Antigen-präsentierenden Zelle (APC) interagiert und ein wichtiger kostimulatorischer Faktor der T-Zell-Rezeptor vermittelten T-Zell-Aktivierung ist (Weiss et al., 1994). Schließlich soll durch die CD4-Herabregulation eine Superinfektion der Wirtszelle vermieden (Benson et al. 1993; Michel et al., 2005) und damit möglicherweise 3. EINLEITUNG 9 der vorzeitige Tod durch Apoptose, induziert durch zu große Mengen an viraler DNA oder viralen Proteinen im Zytoplasma, verhindert werden. 3.4 Fragestellung Das Humane Immundefizienzvirus Typ 1 verwendet drei seiner Proteine (Vpu, Env und Nef) zur Herabregulation von CD4 von der Zelloberfläche (Lama, 2003). Da CD4 der primäre Rezeptor von HIV-1 und notwendig für eine Infektion der Zelle ist, wird seit langem postuliert, dass die HIV-1 vermittelte CD4 Modulation eine als Superinfektion bezeichnete zweite Infektion bereits infizierter Zellen verhindert. In bislang erfolgten Studien wurden zur experimentellen Untersuchung dieser Hypothese chronisch infizierte Zellen oder transfizierte Zellen, die Env oder Nef von einem Expressionsplasmid exprimieren, verwendet (LeGuern et al., 1992; Benson et al., 1993; Little et al., 1994; Pham et al., 2004; Michel et al., 2005). Es wurde jedoch noch nie experimentell gezeigt, dass die Herabregulation von CD4 in produktiv infizierten T-Zellen eine anschließende Superinfektion verhindert. In der vorliegenden Promotionsarbeit sollte deshalb unter Verwendung rekombinanter Viren mit einzelnen oder kombinierten Defekten in vpu, env und/oder nef zunächst der Beitrag dieser drei viralen Gene zur CD4 Herabregulation bestimmt werden. Anschließend sollte untersucht werden, wie effektiv Vpu, Env und verschiedene HIV-1, HIV-2 und SIV Nef-Proteine HIV-1 infizierte T-Zellen gegen eine Superinfektion schützen und ob diese Resistenz gegen eine Superinfektion die Zellen möglicherweise vor dem frühzeitigen Tod durch Apoptose bewahrt. 4. MATERIAL 10 4. Material 4.1 Eukaryote Zelllinien Jurkat: humane T Zelllinie 293T: humane Nierenepithelzellen, die mit Adenovirus Typ 5 transformiert wurden und das SV40 (simian virus 40) large T-Antigen exprimieren (DuBridge et al., 1987). HeLa CII-TA: humane Krebszelllinie aus einem Gebärmutterhalskarzinom, die stabil mit dem CII-TA Plasmid transfiziert ist und humanes MHCKlasse II auf der Zelloberfläche exprimiert (Stumptner-Cuvelette et al., 2001). 4.2 Bakterien Escherichia coli XL2-Blue TM: recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac[F’ proAB lacIqZ∆M15 Tn10 (Tetr) Amy Camr] (Bullock et al., 1987) (Stratagene; Heidelberg). 4.3 Nukleinsäuren 4.3.1 Oligonukleotide Folgende Oligonukleotide wurden von Biomers Deutschland (Ulm) bezogen, wichtige Schnittstellen sind kursiv gezeichnet: 1 2 3 4 5 6 7 8 9 10 11 dsred2PspAI dsred2NcoI dsred2NcoIuni dsred2NcoIrev 5pBRnef HpaI 3pBRNL43nef MluI 3pBRNA7nef MluI 5pBRenv-NdeI 3pBRenv-NdeI NL43PflMI NL43StuI GTCCCGGGTTATCTCGATCCGGTGGATCCTGGGC CACCATGGCCTCCTCCGAGAACGTC GAAGAAGACAATGGGCTGGGAGGC GCCTCCCAGCCCATTGTCTTCTTC GCTGTTAACTTGCTCAATGCCACAGCC CGACGCGTTCAGCAGTTCTTG CGACGCGTTCAGCAGTCTTTG GATGCTAAAGCATATATGATACAGAGG CCTCTGTATCATATATGCTTTAGCATC GTCTCCATAGAATGGAGGAAAAAG CCTTTGGACAGGCCTGTGTAATG 4.3.2 Plasmide Alle verwendeten Plasmide enthalten das Ampicillinresistenzgen zur Selektion in Bakterienstämmen. 4. MATERIAL pDSRed2-C1 11 Vektor zur Amplifikation des DsRed2-Gens von Clontech (Mountain View, CA; USA). PCR2.1-TOPO-TA Vektor: Vektor zum Klonieren von PCR-Fragmenten von Invitrogen (Karlsruhe). pBR-NL4-3 vpu-: pBR322 Vektor, der das HIV-1 NL4-3 Provirus enthält. Das vpu-Gen ist durch eine 40 Aminosäuren umfassende Deletion (von As 1 bis 40) zerstört (Rücker et al., 2004). pBR-NL4-3-IRES-eGFP: pBR322 Vektor, der das HIV-1 NL4-3 Provirus enthält. Dieses wurde so verändert, dass Nef und eGFP mittels eines IRES-Elements von einer bicistronischen mRNA exprimiert werden (Schindler et al., 2005). pBR-NL4-3 nef- -IRES-eGFP: pBR-NL43-IRES-eGFP Vektor mit einem defekten nef-Gen (Schindler et al., 2005). pHIT60: Expressionsvektor, der das Hüllprotein des Vesikulären Stomatitis Virus (VSV-G) exprimiert (Hiebenthal-Millow u. Kirchhoff., 2002). 4.3.3 Längenstandard ″1-kb-Leiter″ Invitrogen/Gibco (Karlsruhe) 4.3.4 Nukleotide für die Polymerase-Ketten-Reaktion dNTPs für die Polymerase-Ketten-Reaktion wurden von Invitrogen (Karlsruhe) bezogen. 4.4 Enzyme 4.4.1 Restriktionsendonukleasen Restriktionsendonukleasen wurden von New England Biolabs (Schwalbach, Traunstein) bezogen und in den vom Hersteller empfohlenen Puffersystemen verwendet. 4.4.2 Sonstige Enzyme Taq-DNA-Polymerase Stratagene Europe AG (Heidelberg) T4-DNA-Ligase Invitrogen/Gibco (Karlsruhe) 4. MATERIAL 12 RNAse A aus Rinderpankreas Boehringer (Mannheim) Trypsin aus Rinderpankreas Stratagene Europe AG (Heidelberg) Alkalische Phosphatase aus Kälberdarm Boehringer (Mannheim) 4.5 Reagenzien und Laborhilfsmittel Agarose Invitrogen/Gibco (Karlsruhe) Ampicillin Bayer (Leverkusen) AnnexinV Binding Puffer BD Biosciences AnnexinV-APC Caltag BCIP/NBT Phosphatase Substrat KPL Biocoll Separating Solution Biochrom (Berlin) Dimethylsulfoxid (DMSO) Fluka (Neu-Ulm) DMRIE-C Invitrogen/Gibco (Karlsruhe) Dithiothreitol (DTT) Amersham Pharmacia (Freiburg) Ethidiumbromid (EtBr) Sigma (München) Ethylen-Diamin-Tetraacetat (EDTA) Sigma (München) Fötales Kälberserum (FKS) Invitrogen/Gibco (Karlsruhe) Glycerin Merck (Darmstadt) Interleukin-2 (IL-2) Strathmann (Dengelsberg) Isopropanol Merck (Darmstadt) L-Glutamin Invitrogen/Gibco (Karlsruhe) Natriumazid Merck (Darmstadt) Natriumchlorid Sigma (München) Natriumdodecylsulfat (SDS) Sigma (München) Natriumfluorid Sigma (München) N, N’-Methylen-Bisacrylamid Sigma (München) N, N, N’, N’,-Tetramethylendiamin (TEMED) Sigma (München) Nonidet P 40 (NP40) Sigma (München) NuPAGE Novex Bis-Tris Gele Invitrogen/Gibco (Karlsruhe) NuPAGE LDS Sample Puffer Invitrogen/Gibco (Karlsruhe) NuPAGE Reducing Agent Invitrogen/Gibco (Karlsruhe) Paraformaldehyd (PFA) Sigma (München) Penicillin G Sigma (München) 4. MATERIAL 13 Phosphat-Buffered-Saline (PBS) Invitrogen/Gibco (Karlsruhe) Phytohämaglutinin (PHA) Remel (Lenexa,USA) Reaktionsgefäße Eppendorf (Hamburg) SureBlueTM TMB Microwell Peroxidase Substrat KPL Triton X-100 Sigma (München) Tween 20 Roth (Karlsruhe) Whatman Papier Whatman (Maidstone, Kent; England) 4.6 Reagenzsysteme (Kits) RosetteSep Human CD4+ T Cell Enrichment Cocktail StemCell Technologies TA Cloning Kit Invitrogen/Gibco (Karlsruhe) Takara DNA Ligationskit Böhringer Ingelheim (Heidelberg) Ultra CleanTM 15 Mobio Inc. (Solana, CA; USA) WizardTM Plus Midipreps Promega (Madison, WI; USA) 4.7 Medien 4.7.1 Zellkulturmedien Adhärente Zellen: DMEM (Dulbecco’s modified Eagle Medium; Invitrogen/Gibco) mit 350µg/ml L-Glutamin, 120µg/ml Streptomycinsulfat, 120µg/ml Penicillin und 10% (v/v) hitzeinaktiviertem FKS. Suspensionszellen: RPMI-1640 Medium (Invitrogen/Gibco) mit 350µg/ml LGlutamin, 120µg/ml Streptomycinsulfat, 120µg/ml Penicillin und 10% (v/v) hitzeinaktiviertem FKS. 4.7.2 Bakterienkulturmedien LB-Medium: 10g/l Bacto-Trypton, 5g/l Hefeextrakt, 8g/l NaCl, 1g/l Glukose; Zugabe von 100mg/l Ampicillin vor Gebrauch. Der pH-Wert wurde mit NaOH auf 7,2 eingestellt LBAMPAgar: 15g/l Agar und 100mg/l Ampicillin in LB-Medium; SOC-Medium: 20g/l Bacto-Trypton, 5g/l Hefeextrakt, 2,5mM NaCl, 10mM MgCl2, 10mM, MgSO4, 20mM Glukose. 4. MATERIAL 14 4.8 Lösungen und Puffer 4.8.1 Isolierung von Plasmid-DNA aus Bakterien Resuspensionspuffer (P1), Lysispuffer (P2), Neutralisationspuffer (P3) (Qiagen, Hilden), 70% EtOH, Isopropanol und LiChrosolv-H2O. 4.8.2 Calcium-Phosphat-Transfektion 2x HBS (10x Stock): 8,18% NaCl (w/v); 5,94% HEPES (w/v); 0,2% Na2HPO4 (w/v). Die Lösung wurde sterilfiltriert und der pH-Wert mit 1M NaOH auf 7,12 eingestellt. 2M CaCl2 4.8.3 HIV p24 Kapsid-Antigen-ELISA Lösung zur Lyse der Viruspartikel: 10ml Triton X-100 (Sigma, München), ad 100ml H2O Waschpuffer: 25ml 20x Waschkonzentrat (KPL, Maryland), ad 500ml mQ-H2O; Probenpuffer: RPMI Medium plus 0,2% Tween 20 Substrat: TMB Peroxidase Substrat System (KPL, Maryland); Stoplösung: 4N H2SO4 4.8.4 Herstellung von Zelllysaten Lysepuffer: 1% Triton X-100; 0,15 M NaCl; 50 mM Tris-HCl pH 7.4; 5 mM EDTA; 1 mM NaF; 1 mM Na2VO3 4.8.5 Sonstige Lösungen und Puffer FACS-Puffer: 1*PBS, 1%FKS, 0,1%Na-Azid PBS: 137mM NaCl; 7,3mM Na2HPO4; 1,47mM K2HPO4; 1mM CaCl2; 2mM MgCl2 50x TAE-Puffer: 500mM Tris, 250mM Natriumacetat, 50mM Na2EDTA. 4.9 Antikörper für die Durchflusszytometrie Anti-CD4-PE (Serotec, Düsseldorf) Anti-CxCR4-PE (BD Bioscience) Anti-CCR5-PE (BD Bioscience) 5. METHODEN 15 5. Methoden 5.1 DNA-Methoden 5.1.1 Standardmethoden Folgende Methoden wurden nach Protokollen von Maniatis et al., 1989, durchgeführt: - Plasmid-DNA-Isolierung nach alkalischer Lyse der Bakterien - Ethanol- und Isopropanolfällung der DNA - Konzentrationsbestimmung der Nukleinsäuren - Dephosphorylierung von DNA 5' Enden mit alkalischer Phosphatase - Spaltung von DNA mit Restriktionsendonukleasen - Ligation von DNA-Fragmenten mit T4-DNA-Ligase - Auftrennung von Nukleinsäuren durch Gelelektrophorese 5.1.2 Isolierung von Plasmid-DNA aus Bakterien Plasmid-DNA für Klonierungen wurde durch alkalische Lyse von Bakterien und anschließende Isolierung und Reinigung der DNA gewonnen (Maniatis et al., 1989). Für Sequenzierreaktionen oder die Transfektion eukaryoter Zellen wurden die Präparationen mit dem WizardTM Plus Midipreps Kit (Promega; Madsion, WI, USA) oder dem QIAwell 8 Miniprep Kit (Qiagen; Hilden) nach Angaben der Hersteller durchgeführt. Die DNAKonzentrationen wurden in einem Spektralphotometer (Eppendorf) bestimmt. 5.1.3 Isolierung von DNA aus Agarosegelen Die DNA-Fragmente wurden in einem Agarosegel elektrophoretisch aufgetrennt, unter Bestrahlung mit langwelligem UV-Licht (366nm) sichtbar gemacht und die DNABanden mit einem Skalpell ausgeschnitten. Anschließend wurde die DNA mit dem Geneclean® II Kit (Dianova, Hamburg) entsprechend den Anweisungen des Herstellers isoliert. 5.1.4 Ligation von DNA Ligationen wurden mit dem Takara DNA-Ligationskit (Boehringer Ingelheim, Heidelberg) durchgeführt. Plasmid-DNA und Insert-DNA wurden im Verhältnis von 1:3 nach den Protokollen der Hersteller eingesetzt. 5. METHODEN 16 5.1.5 DNA-Sequenzierung Sequenzierreaktionen wurden kommerziell bei der Firma MWG-Biotech AG in Auftrag gegeben. 2µg DNA wurden bei 60°C eingedampft und eingeschickt. 5.1.6 Polymerase-Ketten-Reaktion (PCR) Alle PCR-Reaktionen wurden nach dem Protokoll von Saiki et al. (1988), in einem Thermocycler (PTC-100TM, MJ Research) durchgeführt. 5.2 Zellkultur 5.2.1 Kultur adhärenter Zellen Die adhärenten Zellen wurden in 25cm2 Zellkulturflaschen in DMEM bei 37°C und 5% CO2 kultiviert. Die Zellen wurden zweimal wöchentlich 1:10 bis 1:20 gesplittet. 5.2.2 Kultur von Suspensionszellen Die Suspensionszellen wurden in 25cm2 Zellkulturflaschen in RPMI-1640 bei 37°C und 5% CO2 kultiviert. Die Zellen wurden zweimal wöchentlich 1:10 bis 1:20 gesplittet. 5.3 Bakterienkultur Alle verwendeten Plasmide enthalten ein Ampicillin-Resistenzgen. Transformierte Bakterien wurden daher durch Zusatz von Ampicillin (100µg/ml) zum Kulturmedium selektioniert. Verwendet wurde der Escherichia coli-Stamm XL2-BlueTM. Die Kulturen wurden für mindestens 12h bei 37 °C in LBamp-Flüssigmedium geschüttelt oder auf LBampAgarplatten inkubiert. Bakterienstocks wurden durch Vermischen von 500µl Bakterienkultur mit 500µl Glycerin erstellt und bei -80 °C eingefroren. 5.3.1 Transformation von Escherichia coli XL2TM-Blue Nach dem Auftauen der kompetenten Zellen auf Eis wurden je 10µl davon zu 5µl Ligationsansatz pipettiert und die Ansätze für 30min auf Eis inkubiert. Anschließend erfolgte ein Hitzeschock der Bakterien für 30s bei 42°C im Wasserbad und eine 2 Minuten dauernde Inkubation auf Eis. Nach Zugabe von 180µl SOC-Medium und einer Inkubation von 30min bei 37°C wurde die Bakteriensuspension auf LBamp-Agarplatten ausplattiert und bei 37°C über Nacht inkubiert. 5. METHODEN 17 5.4 Herstellung rekombinanter HIV-1 NL4-3 Viren 5.4.1 Herstellung rekombinanter HIV-1 NL4-3-IRES-eGFP Viren Zur Herstellung der HIV-1 NL4-3-IRES-eGFP Viren, die in den Genen vpu, env und nef einzeln oder kombiniert defekt sind, wurde zuerst aus dem bestehenden Vektor pBR-NL4-3 vpu- der das vpu-Gen umfassende Bereich über PflMI und StuI in die Vektoren pBR-NL4-3-IRES-eGFP und pBR-NL4-3 nef- -IRES-eGFP umkloniert. Anschließend wurde mittels splice overlap extension (SOE)-PCR das env-Gen durch eine Insertion von zwei Nukleotiden in die NdeI-Schnittstelle, durch die es zu einer Verschiebung des env-Leserahmens und damit zu einem vorzeitigen Stoppcodon kommt, mutiert (env-). Dazu wurden in zwei PCR-Ansätzen mit den Primerpaaren NL43PflMI / 3pBRenv-NdeI bzw. NL43StuI / 5pBRenv-NdeI (siehe 4.3.1) die DNA-Bereiche von der PflMI- bis zur NdeI-Schnittstelle im env und von dort bis zur StuI-Schnittstelle im env aus den entsprechenden rekombinanten pBR-NL4-3-IRES-eGFP Vektoren amplifiziert. Die Produkte wurden über Agarosegelelektrophorese aufgereinigt, die DNA aus dem Gel isoliert und beide Fragmente als „template“ in einer zweiten PCR mit den zugehörigen Primern NL43PflMI und NL43StuI eingesetzt. Dieses Amplikon wurde wiederum aufgereinigt, mit PflMI und StuI verdaut und in die ebenso präparierten rekombinanten pBR-NL4-3-IRES-eGFP Vektoren kloniert. Zur Herstellung von env-defekten HIV-1 NL4-3-IRES-eGFP Viren, die unterschiedliche nef-Allele exprimieren, wurden die entsprechenden nef-Allele aus pBRNL4-3 Vektoren mit dem 5’ Primer 5pBRnef HpaI und dem nef-spezifischem 3’ Primer 3pBRNL43nef MluI bzw. 3pBRNA7nef MluI (siehe 4.3.1) in einer Standard-PCRReaktion amplifiziert und über HpaI und MluI in den pBR-NL4-3 env- -IRES-eGFP Vektor kloniert. 5.4.2 Herstellung rekombinanter HIV-1-NL4-3-IRES-DsRed2 Viren Als Ausgangskonstrukte wurden die unter 5.4.1 beschriebenen IRES-eGFP Viren verwendet, bei denen das eGFP Gen durch das DsRed2 Gen ersetzt wurde. Dazu wurde mittels SOE-PCR das DsRed2 Gen in zwei Schritten amplifiziert und dabei die enthaltene interne NcoI-Schnittstelle still mutiert. Als erstes wurde das DsRed2-Gen aus dem Vektor pDSRed2-C1 (Clontech) mit den Primerpaaren dsred2NcoI / dsred2NcoIrev bzw. dsred2PspAI / dsred2NcoIuni (siehe 4.3.1) in zwei PCR-Ansätzen amplifiziert. Die Produkte aus diesen PCR-Reaktionen wurden über Agarosegelelektrophorese aufgereinigt, 5. METHODEN 18 die DNA aus dem Gel isoliert und beide Fragmente als „template“ in einer zweiten PCR mit den zugehörigen Primern dsred2NcoI und dsred2PspAI eingesetzt. Dieses Amplikon wurde wiederum aufgereinigt, in den pCR2.1-TOPO-TA Vektor (Invitrogen) kloniert und sequenziert. Anschließend erfolgte die Umklonierung des DsRed2-Gens über NcoI und PspAI in die ebenso verdauten proviralen IRES-eGFP Vektoren. 5.5 Isolierung primärer Lymphozyten aus Buffy Coat Zur Isolierung primärer Blutlymphozyten (PBMC „peripheral blood mononuclear cells“) und CD4+-T-Zellen wurde Buffy Coat (Lymphozytenkonzentrat aus 500ml Vollblut) von der Blutspendenzentrale Ulm bezogen und 1:3 in PBS verdünnt. Für die Gewinnung von CD4+-T-Zellen wurde das verdünnte Blut außerdem mit einer Antikörpermischung (RosetteSep von StemCell Technologies) vermengt und 30min bei Raumtemperatur inkubiert, wobei alle CD4 negativen Zellen an die Erythrozyten gebunden werden. Das verdünnte Buffy Coat wurde dann über Biocoll Separating Solution (Biochrom) geschichtet und durch Dichtegradientenzentrifugation für 30min bei 2500rpm aufgetrennt. Die angereicherten Lymphozyten wurden aus der Interphase abgenommen, zweimal in PBS gewaschen und in RPMI1640 mit 10% FKS und 1% Pen/Strep in einer Konzentration von 2*106 Zellen/ml aufgenommen. Zur Stimulation der Lymphozyten erfolgte die Zugabe von 10ng/ml IL-2 und 3µg/ml PHA. 5.6 Herstellung von Virusstocks und Infektionen 5.6.1 Kalzium-Phosphat-Transfektion von 293T-Zellen Die Transfektion der 293T-Zellen wurde nach der Kalzium-Phosphat-PräzipitatMethode durchgeführt. Am Tag vor der Transfektion wurden 0,2 x 106 Zellen in 6-WellPlatten ausgesät und über Nacht bei 37°C und 5%igem CO2-Gehalt inkubiert. Die Zellen waren zum Zeitpunkt der Transfektion zu 50-75% konfluent. Für die DNA-Arbeitslösung wurden 5µg DNA mit 13µl 2M CaCl2 gemischt und mit H2O auf 100µl aufgefüllt. Diese Lösung wurde tropfenweise zu 100µl 2 x HBS zugegeben. Nach 5min Inkubation bei Raumtemperatur bildete sich ein milchiges Präzipitat. Der Transfektionscocktail wurde auf das Medium der Zellkulturen getropft und die Zellen über Nacht bei 37°C inkubiert. Nach etwa 16h wurde das Medium (DMEM: 10% FKS, Glutamin, SP) erneuert. Die Virusstocks wurden am dritten Tag abgenommen und 3min bei 2000rpm abzentrifugiert. 5. METHODEN 19 5.6.2 HIV p24 Kapsid Antigen-ELISA Die Konzentration viralen Antigens in Virusüberständen wurde mittels HIV p24 Kapsid Antigen-ELISA (AIDS Repository; Frederick, MD; USA) bestimmt. Das durch Lyse mit Triton X-100 freigesetzte HIV-Kapsidprotein p24 bindet an einen monoklonalen Maus-Antikörper. Ungebundenes Material wurde durch Waschen entfernt. Die Quantifizierung des Kapsidproteins erfolgt durch die sogenannte „sandwich“-ELISA Methode. Zuerst wurde ein polyklonaler Antikörper aus Kaninchen zugegeben, der an das Kapsid-Protein p24 bindet. Der gebundene Antikörper wurde mit Anti-KaninchenAntikörper, der mit einer Peroxidase gekoppelt ist, inkubiert. Nach Zugabe des Substrates (SureBlueTM TMB Microwell Peroxidase Substrate, KPL) kommt es zu einer Farbänderung der Lösung. Die enzymatische Reaktion wurde durch 1N Salzsäure gestoppt und die photometrische Quantifizierung des gefärbten Produktes erfolgte bei den Wellenlängen 450 und 650nm. Die gemessene Farbkonzentration ist proportional zu der Menge an Kapsidprotein und wird in ng/ml p24 angegeben. 5.6.3 Infektion primärer Lymphozyten und Jurkat-Zellen, Zur Infektion wurden 1 Million prästimulierter PBMCs oder Jurkat Zellen in 50ng p24 Virusstock aufgenommen und für 4-6h bei 37°C inkubiert. Dann erfolgte die Zugabe von 3ml RPMI 1640 mit 10% FKS und 1% Pen/Strep. PBMCs wurden zusätzlich mit 10ng/ml IL-2 stimuliert. Die FACS-Analysen erfolgten zu verschiedenen Zeitpunkten innerhalb von 3 Tagen nach Infektion. 5.6.4 Superinfektion von T-Zellen Jurkat T-Zellen wurden wie unter 5.6.3 beschrieben mit unterschiedlichen VSV-G pseudotypisierten HIV-1 NL4-3-IRES-DsRed2 Viren infiziert. 3 Tage später wurden die Zellen bei 1300rpm für 3 min zentrifugiert, in 300ng p24 HIV-1 NL4-3-IRES-eGFP Virusstock resuspendiert und für 4-6h bei 37°C inkubiert. Dann erfolgte die Zugabe von 3ml RPMI 1640 mit 10% FKS und 1% Pen/Strep. Nach weiteren 2 Tagen wurden die Zellen im FACS analysiert. 5. METHODEN 20 5.7 FACS-Analysen 5.7.1 Modulation von Oberfächenmarkern auf T-Zellen Zur FACS-Färbung wurden Zellen einer Transfektion oder Infektion in die entsprechende Zahl an Färbungen aufgeteilt und einmal mit 800µl FACS Puffer gewaschen. Nach der Zentrifugation für 5min bei 1400rpm wurden die Zellen in 100µl FACS-Puffer + 5-10µl PE konjugiertem Antikörper (nach Herstellerangaben) aufgenommen und für 30min bei 4°C gefärbt. Nach einem Waschschritt mit 1ml FACSPuffer wurden die Zellen in 200µl FACS-Puffer mit 2% PFA für 15min bei 4°C fixiert und im FACS (FACSCalibur, Becton-Dickinson) vermessen. 5.7.2 Apoptose superinfizierter T-Zellen Jurkat oder CD4+-T-Zellen wurden wie in 5.6.4 beschrieben superinfiziert und die Messung der Apoptoserate erfolgte über die Bindung von AnnexinV an apoptotische Zellen. Dazu wurden Aliquots der infizierten Zellen in 800µl PBS gewaschen, in 100µl AnnexinV Bindepuffer (Apoptosis Detection Kit, BD Bioscience) plus 5µl AnnexinVAPC (Caltag) aufgenommen und nach 15min Inkubation bei Raumtemperatur im Dunkeln in 200µl AnnexinV Bindepuffer plus 3% PFA fixiert. Anschließend wurde im FACS der Anteil an apoptotischen Zellen (AnnexinV+) in den DsRed2+, eGFP+, DsRed2+/eGFP+ und nicht infizierten Zellpopulationen bestimmt. 5.8 Nachweis viraler Proteine durch Western Blot Analyse 5.8.1 Herstellung von Zell- und Viruslysaten Zur Herstellung von Zelllysaten wurden in 6-Well-Platten mit 5µg proviraler DNA transfizierte 293T-Zellen oder mit VSV-G-Pseudotypen infizierte HeLa-Zellen verwendet. 1 Tag nach der Transfektion bzw. 3 Tage nach der Infektion wurde das Kulturmedium abgenommen und die Zellen in 200µl Lysepuffer lysiert. Vorhandene Zellreste wurden durch Zentrifugation für 5min bei 2000rpm pelletiert. Zur Herstellung von Viruslysaten wurden Virusstocks, die 150ng p24 enthielten, für 3h bei 14000rpm zentrifugiert und das Viruspellet anschließend in 80µl Lysepuffer lysiert. 5. METHODEN 21 5.8.2 Auftrennung der Proteine durch SDS-PAGE und Nachweis im Western Blot Je 20µl des jeweiligen Lysats wurden mit 5µl NuPAGE LDS Sample Puffer und 2µl NuPAGE Reducing Agent gemischt, für 10min bei 80°C inkubiert und mit Hilfe eines NuPAGE Novex Bis-Tris Polyacrylamidgels in einer XCell SureLock Mini-Cell Elektrophoresekammer aufgetrennt (Invitrogen). Nach dem Prinzip des Semi-Dry-Blots wurden die aufgetrennten Proteine dann mit einem Trans-Blot SD Gerät (BioRad) auf eine Immobilon PVDF Transfer Membran (Millipore) übertragen und diese in 25ml Detector Block Solution (KPL) leicht geschwenkt. Nach 2h wurde der für das zu detektierende Protein spezifische Antikörper entsprechend den Herstellerangaben zugesetzt und die Membran für eine weitere Stunde geschwenkt, bevor die ungebundenen Antikörper in drei Waschschritten a 5min mit Wash Solution (KPL) entfernt wurden. Durch Inkubation der Membran für 1h mit einem spezifisch an den ersten Antikörper bindenden zweiten Antikörper, der mit alkalischer Phosphatase markiert war (KPL), wurden nach weiteren drei Waschschritten die Proteine durch Zugabe von BCIP/NBT Substrat (KPL) in einer enzymatischen Reaktion direkt auf der Membran nachgewiesen. 5.9 Computerprogramme und Datenquellen 5.9.1 Computerprogramme zur Sequenzanalyse Für die Auswertung von Aminosäure und DNA-Sequenzen wurden folgende Programme aus dem Internet benutzt: Sequence Reverse Complementor (http://bioinformatics.org/sms/rev_comp.html/), Sequenz-Alignment Programm MultAlin V5.4.1 (http://prodes.toulouse.inra.fr/multalin/) und das DNA/Aminosäuresequenz Programm Expasy-Tool (http://www.expasy.org/tools/dna.html). Außerdem wurden die Programme Bioedit V5.0.9 von Tom Hall, Department of Microbiology, North Carolina State University, Raleigh, NC, USA, und Gene Construction Kit V2.0 von Bob Gross und Anders Putte zur Sequenzanalyse verwendet. 5.9.2 Computerprogramme zur FACS-Auswertung Für die FACS-Auswertung wurde das Programm CellQuest-Pro von Becton Dickinson benutzt, sowie Microsoft Excel Office XP zur Tabellenkalkulation. 5. METHODEN 22 5.9.3 Statistische Auswertungen und Korrelationsanalysen Für alle statistischen Analysen wurde das GraphPad PRISM Programm Version 4.0 (Abacus Concepts, Berkeley, CA, USA) benutzt. P-Werte wurden mit dem beidseitigen Students-T-Test und Mann-Whitney–Test berechnet. 6. ERGEBNISSE 23 6. Ergebnisse 6.1 Rekombinante HIV-1 NL4-3 Viren mit defekten vpu, env und nef Genen HIV-1 verwendet drei seiner Proteine zur Herabregulation von CD4 von der Zelloberfläche, was unter anderem eine Superinfektion verhindern soll (Benson et al., 1993). Um den Beitrag der drei viralen Proteine Vpu, Env und Nef zur CD4Herabregulation und der damit verbundenen Resistenz gegen eine Superinfektion zu bestimmen, wurden rekombinante Humane Immundefizienzviren mit einzelnen und kombinierten Defekten in den entsprechenden drei Genen hergestellt. Dazu wurden diese Gene in dem proviralen Vektor pBR-NL4-3-IRES-eGFP wie unter 5.4.1 beschrieben zerstört, wobei alle anderen viralen Proteine im natürlichen Kontext unter Kontrolle des LTR-Promoters und der ursprünglichen Spleißstellen exprimiert werden. Außerdem koexprimieren alle proviralen Konstrukte Nef und eGFP von einer bicistronischen mRNA, wodurch die Unterscheidung von infizierten und nicht infizierten Zellen ermöglicht wird (Abb. 2). Abb. 2: Rekombinante provirale HIV-1 NL4-3 Konstrukte mit defekten vpu, env und nef Genen. Es wurden sieben provirale Konstrukte mit einzelnen oder kombinierten Defekten in den Genen vpu, env und nef hergestellt. Der schwarze Balken zeigt die Lage der Deletion im vpu-Gen und die Sterne geben die Position der vorzeitigen Stopp-Codons im env- und nef-Gen wieder. Alle Konstrukte exprimieren eGFP über ein IRES-Element. LTR, long terminal repeat; r, vpr; u, vpu; IRES, interne Ribosomeneintrittstelle; FP, Fluoreszenzprotein. Zur Kontrolle der viralen Genexpression wurden die proviralen Konstrukte in 293T Zellen transfiziert und die Proteine Nef, p24 und eGFP sowie das zelluläre Protein ß-Actin in den Zelllysaten durch Western Blot Analyse nachgewiesen (Abb. 3). Vpu konnte in den 293T-Lysaten aufgrund einer auf gleicher Höhe laufenden unspezifischen Bande nicht 6. ERGEBNISSE 24 detektiert werden, weshalb hierfür Zelllysate von mit VSV-G Pseudotypen infizierten HeLa-Zellen verwendet wurden. Das virale Env Protein gp120 wurde in Viruslysaten detektiert. Wie in Abbildung 3 dargestellt, zeigten die verschiedenen Konstrukte die erwarteten Unterschiede in ihrer Fähigkeit zur Expression von Vpu, Env und Nef. p24 und eGFP wurde von allen proviralen Konstrukten in ähnlichen Mengen exprimiert. eGFP diente als Kontrolle für die Transfektionseffizienz und die ß-Actin Banden zeigen, dass gleiche Zelllysatmengen verwendet wurden. Abb. 3: Proteinexpression der re- kombinanten HIV-1 NL4-3 Konstrukte. Nef, p24, eGFP und ß-Actin wurden in Zelllysaten transfizierter 293T-Zellen und Vpu in Lysaten infizierter HeLa-Zellen durch Western Blot Analyse detektiert. Der Nachweis des Env Proteins gp120 erfolgte in Viruslysat. Mock, ohne provirale DNA. 6.2 CD4-Modulation in HIV-1 infizierten Zellen Um den Beitrag der viralen Proteine Vpu, Env und Nef zur CD4-Modulation zu ermitteln wurden unter Verwendung der Konstrukte mit einzelnen und kombinierten Defekten in den entsprechenden Genen (Abb. 2) VSV-G pseudotypisierte Viren hergestellt. Diese tragen das Hüllprotein des Vesikulären Stomatitis Virus (VSV) auf ihrer Oberfläche, was den CD4-unabhängigen Eintritt des Partikels in die Zielzelle und somit auch die Untersuchung der env-defekten Viren erlaubt. Außerdem können Zielzellen so unter Umgehung der Nef-vermittelten Erhöhung der Partikelinfektiosität mit vergleichbar hoher Effizienz transduziert werden (Lama, 2003). Mit den VSV-G pseudotypisierten Viren wurden Jurkat T-Zellen infiziert, die nach der Markierung des CD4 auf der 6. ERGEBNISSE 25 Zelloberfläche mit Antikörpern im FACS zu unterschiedlichen Zeitpunkten analysiert wurden (Abb. 4). Abb. 4: Kinetik der CD4-Modulation in HIV-1 infizierten Jurkat T-Zellen. (A) FACS-Analyse (24 bzw. 48 Stunden nach der Infektion) von Jurkat T-Zellen, die mit rekombinanten HIV-1 NL4-3 Viren transduziert wurden, die in den Genen vpu, env und nef einzeln und kombiniert defekt sind. Die Zahlen geben die roten MFIs für den entsprechenden eGFP Bereich an. (B) Quantitative Auswertung der CD4Herabregulation. Erläuterung zur Berechnung der X-fachen CD4-Modulation im Text. Es wurde jeweils die CD4-Modulation bei niedriger (links) und mittlerer (rechts) eGFP Expression (siehe A) zu den angegebenen Zeitpunkten bestimmt. E, env; U, vpu; N, nef. 24 Stunden nach der Infektion konnten im FACS erstmals grüne (d.h. infizierte) Zellen nachgewiesen werden. Ab diesem Zeitpunkt enthielten alle Zellen, die mit nefintakten Viren infiziert waren, reduzierte Mengen an CD4 auf der Zelloberfläche (Abb. 4A, Spalte 2 und 4 bis 6). Zellen, die das virale Env Protein exprimierten zeigten unabhängig von Nef bei starker eGFP- und somit auch viraler Genexpression ebenfalls verminderte CD4 Oberflächenlevel (Abb. 4A, Spalte 3 und 7), wogegen die Expression von Vpu nur zu einer minimalen Abnahme von CD4 führte. Da infizierte Zellen Nef und 6. ERGEBNISSE 26 eGFP von einer bicistronischen mRNA in einem konstanten stöchiometrischen Verhältnis koexprimieren, kann die Nef-Expression mit der Rezeptormodulation korreliert werden. Zur Kalkulation der X-fachen CD4-Modulation wurde die rote mittlere Fluoreszenzintensität (MFI) eines definierten Bereichs der eGFP Expression (s. Abb. 4A) des vpu, env und nef defekten Kontrollkonstrukts (E-U-N-) durch die MFI des entsprechenden Bereichs der Zellen geteilt, die unterschiedliche Kombinationen dieser drei Gene exprimieren. Die quantitative Auswertung der CD4-Modulation durch die acht untersuchten Viren ist in Abbildung 4B dargestellt. Es zeigte sich, dass bei niedrigen eGFP Expressionsleveln Nef eine vier- bis fünffache Herabregulation von CD4 bewirkt, wogegen Env und Vpu keinen nennenswerten Einfluss auf CD4 hatten (Abb. 4B, links). Bei mittleren Expressionsleveln bewirkte Nef eine fünfzehnfache Reduktion von CD4 und zusammen mit Env wurde die CD4-Menge sogar um das fünfundzwanzigfache verringert. Die Expression von Env in Abwesenheit von Nef bewirkte eine sechsfache Verminderung der CD4-Expression. Vpu hatte auch bei gesteigerter Proteinexpression keine signifikante Wirkung (Abb. 4B, rechts). Nef regulierte CD4 somit in Jurkat Zellen effizient herab, wogegen Env nur eine schwache Wirkung zeigte und Vpu keinen messbaren Effekt hatte. Die Effizienz der CD4-Herabregulation durch Vpu, Env und Nef unterscheidet sich möglicherweise in immortalisierten Zellen (wie z.B. Jurkat T-Zellen) und Primärzellen. Deshalb wurden auch humane PBMCs (engl. peripheral blood mononuclear cell), die mit HIV1 infiziert wurden, zur Analyse der CD4-Modulation verwendet. Wie in Abbildung 5A dargestellt, konnte nach Markierung der PBMCs mit anti-CD4-Antikörpern im FACS eine CD4+ und eine CD4-/CD4niedrig Zellpopulation unterschieden werden. Beide PBMCPopulationen wurden mit den VSV-G pseudotypisierten HIV-1 Viren transduziert und 32 Stunden später konnten die ersten eGFP-positiven, infizierten Zellen detektiert werden. PBMCs, die mit dem E-U-N--Kontrollkonstrukt infiziert waren, exprimierten auch bei hoher viraler Proteinexpression konstante CD4-Mengen auf ihrer Zelloberfläche (Abb. 5A, Spalte 1). Die Expression von Vpu oder Env in den Primärzellen reduzierte bei hohen Expressionsleveln die CD4-Menge in etwa fünffach (Abb. 5A, Spalten 7 und 8; Abb. 5B, rechts). Allerdings führte nur die Expression von Nef zum vollständigen Verschwinden der CD4+ Zellpopulationen (Abb. 5A, Spalten 2 und 4 bis 6). Nef alleine bewirkte eine 6. ERGEBNISSE 27 vierzehnfache Reduktion der CD4-Menge (Abb. 5B, rechts) und war damit hauptsächlich für die CD4-Modulation verantwortlich. Die Koexpression von Vpu und Nef verdoppelte diesen Effekt schon zu frühen Zeitpunkten. In Gegenwart von Env und Nef zeigte Vpu diese Steigerung der CD4-Herabregulation jedoch erst später – ca. 53 Stunden nach der Infektion (Abb. 5B, rechts). Somit sind Nef und Env in Primärzellen ähnlich effektiv bei der CD4-Herabregulation wie in immortalisierten Jurkat Zellen, wogegen Vpu in PBMCs deutlich mehr zur CD4-Herabregulation beiträgt. Abb. 5: Kinetik der CD4-Modulation in HIV-1 infizierten humanen PBMCs. (A) Prästimulierte PBMCs wurden mit den verschiedenen HIV-1 NL4-3-IRES-eGFP Konstrukten transduziert und zu den angegebenen Zeitpunkten im FACS analysiert. Die Zahlen geben die roten MFIs für den entsprechenden eGFP Bereich an. (B) Quantitative Auswertung der CD4-Herabregulation. Erläuterung zur Berechnung der X-fachen CD4-Modulation im Text. Es wurde jeweils die CD4-Modulation bei niedriger (links) und mittlerer (rechts) eGFP Expression (siehe A) bestimmt. E, env; U, vpu; N, nef. 6. ERGEBNISSE 28 Nef entfernt als einziges der drei untersuchten Proteine CD4 direkt von der Zelloberfläche und leitet es zum Abbau in Lysosomen, wogegen Env und Vpu den Transport von neu synthetisiertem CD4 zur Zelloberfläche stören (Piguet et al., 1998; Doms et al., 2000; Lama, 2003). Um den Effekt der drei Proteine auf den Transport von neu synthetisiertem CD4 zur Zelloberfläche zu untersuchen, wurden 293T Zellen, die kein CD4 exprimieren, mit den verschiedenen proviralen Konstrukten und einem CD4Expressionsplasmid kotransfiziert und im FACS analysiert (Abb. 6). Abb. 6: Blockierung der CD4-Expression in transfizierten 293T Zellen. (A) 293T Zellen wurden mit den angegebenen proviralen HIV-1 NL4-3-IRES-eGFP Konstrukten und 1 µg eines CD4- Expressionsplasmids kotransfiziert und 48 h später im FACS analysiert. Die Zahlen geben die roten MFIs innerhalb der gezeigten eGFP Bereiche an. (B) X-fache Blockierung (Mittelwert +/- Stabw., n=3) der CD4-Expression durch die angegebenen proviralen Konstrukte. Zur Berechnung wurde die MFI des E-U-N--Konstruktes durch die entsprechenden MFIs der anderen Konstrukte geteilt. (C) Korrelation zwischen CD4-Herabregulation in infizierten Jurkat Zellen (gemessen 48 h nach Infektion bei mittlerer eGFP Expression, Abb.3A) und Blockierung der CD4-Expression in transfizierten 293T Zellen. (D) CD4-Modulation durch Vpu, Env und Nef bei unterschiedlichen CD4-Mengen. 293T Zellen wurden mit 4µg proviraler DNA und 0,1, 0,5 bzw. 1 µg CD4-Expressionsplasmid kotransfiziert. Mock, nicht transfiziert; E, env; U, vpu; N, nef. 6. ERGEBNISSE 29 In Abwesenheit von Vpu, Env und Nef exprimierten die transfizierten 293T Zellen große Mengen an CD4 auf der Oberfläche (Abb. 6A, Spalte 1). Zellen, die mit dem Wildtyp-Konstrukt (E+U+N+) transfiziert wurden zeigten dagegen eine fünffache Verminderung der CD4-Menge. Die Expression von Vpu oder Nef reduzierte CD4 um das Zwei- bis Dreifache, wogegen Env keine Wirkung hatte (Abb. 6B). Da Vpu, Env und Nef wie beschrieben unterschiedliche Mechanismen zur CD4-Modulation verwenden, überrascht es nicht, dass die Effizienzen, mit denen die proviralen Konstrukte CD4 in infizierten Jurkat Zellen herabregulieren und in transfizierten 293T Zellen die CD4Expression blockieren, nicht korrelieren (Abb. 6C). Unerwartet war jedoch die Beobachtung, dass Env keinen Effekt auf CD4 zeigte, obwohl es den Transport von CD4 zur Zelloberfläche behindern soll (Lama et al., 1999). Da unter diesen experimentellen Bedingungen die CD4-Expressionslevel möglicherweise höher sind als in Jurkat oder Primärzellen, wurden in einem weiteren Experiment 293T Zellen mit unterschiedlichen Mengen des CD4-Expressionsplasmids kotransfiziert. Wie in Abbildung 6D gezeigt, reduzierten Vpu und Nef die CD4-Expression bei der Kotransfektion von sowohl 0,1 als auch 0,5 µg CD4-Plasmid. Dagegen zeigte Env nur bei der Kotransfektion von 0,1 µg CD4-Plasmid einen deutlichen Effekt. Diese Ergebnisse bestätigen, dass die drei untersuchten viralen Proteine CD4 unabhängig voneinander herabregulieren (Chen et al., 1996). Außerdem scheint der Mechanismus mit dem Env die CD4-Expression blockiert bei niedrigeren CD4-Mengen gesättigt zu sein, als dies bei Nef oder Vpu der Fall ist. 6.3 Die Herabregulation von CD4 schützt HIV-1 infizierte T-Zellen vor einer Superinfektion Da CD4 der primäre Rezeptor von HIV-1 ist, dient die CD4-Herabregulation möglicherweise auch dem Schutz einer HIV-1 infizierten Zelle vor einer erneuten Infektion – der so genannten Superinfektion (Benson et al., 1993; Michel et al., 2005). Da ein experimenteller Nachweis dieser Hypothese in infizierten Zellen bislang fehlte, wurde im folgenden Experiment untersucht, ob die durch Vpu, Env und Nef vermittelte CD4- 6. ERGEBNISSE 30 Herabregulation eine Superinfektion infizierter Zellen verhindert und welchen Anteil an diesem Effekt die einzelnen Proteine dabei besitzen. Dazu wurde in den in Abschnitt 6.1 beschriebenen (Abb. 2) proviralen Konstrukten das eGFP- durch das DsRed2-Reportergen ersetzt (5.4.2). Diese Konstrukte dienten anschließend zur Herstellung von VSV-G pseudotypisierten Viren mit denen Jurkat TZellen transduziert wurden. Drei Tage nach dieser Infektion wurden die Zellen mit Wildtyp HIV-1-IRES-eGFP Viren inkubiert und zwei Tage später erfolgte die Auswertung am FACS. Zellen, die nur initial transduziert wurden, leuchteten rot und Zellen, die erst durch die zweite Infektion nach drei Tagen infiziert wurden, leuchteten grün. Superinfizierte Zellen wurden anhand ihrer roten und grünen Fluoreszenz (DsRed2+/eGFP+) detektiert. Abbildung 7 zeigt den Versuchsaufbau. Abb. 7: Messung der Superinfektion. Jurkat Zellen wurden mit den VSV-G pseudotypisierten HIV-1 NL4-3-IRESDsRed2 vpu, env und nef Mutanten transduziert und 3 Tage später mit eGFP exprimierenden HIV-1 NL4-3 Wildtypviren infiziert. 2 Tage später erfolgte die Analyse der super- infizierten Zellen im FACS. Das DsRed2 Protein wurde für die initiale Infektion verwendet, da es als Tetramer aktiv ist und mehr Zeit für die Reifung benötigt als das eGFP Protein. Andere rote Fluoreszenzproteine die ebenfalls getestet wurden, wie z.B. ein Monomer (Campbell et al., 2002), konnten im FACS nicht detektiert werden oder zeigten eine zu geringe Fluoreszenzintensität. Wie in Abbildung 8A gezeigt, wurden fünf Tage nach der initialen Infektion etwa 3 % rote Zellen detektiert. Die Inkubation der Zellen mit hohen Dosen des X4-tropen HIV-1 NL4-3-IRES-eGFP Viruses führte zwei Tage später ebenfalls zu einer Infektionsrate von 3 %. Zellen, die initial mit dem vpu, env und nef intakten Virus infiziert wurden, waren 6. ERGEBNISSE 31 effizient gegen eine Superinfektion durch das HIV-1 Wildtypvirus, nicht jedoch durch VSV-G pseudotypisierte Viren geschützt (Abb. 8A). Die Expression von Nef in infizierten Zellen verringerte die Zahl an superinfizierten Zellen sechsfach, wogegen Vpu und Env die Superinfektion in Abwesenheit von Nef nur etwa zweifach reduzierten (Abb. 8B). In Gegenwart von Nef zeigten Vpu und Env keine zusätzliche Wirkung. Übereinstimmend mit seiner starken Aktivität bei der Modulation von CD4 (Abschnitt 6.2) spielte Nef somit auch bei der Verhinderung der Superinfektion die wichtigste Rolle. Abb. 8: Beitrag von Vpu, Env und Nef zur Verhinderung der Superinfektion. (A) Jurkat T-Zellen wurden initial mit den verschiedenen DsRed2 exprimierenden vpu, env und/oder nef defekten Viren infiziert, 3 Tage später mit eGFP exprimierendem HIV-1 Wildtypvirus (HIV) oder VSV-G Pseudopartikeln (VSV) inkubiert und 2 Tage danach im FACS analysiert. Zur Berechnung der Anzahl an superinfizierten Zellen (in Prozent) wurde die Anzahl der DsRed2+/eGFP+ Zellen (Bereich R3) durch die Anzahl aller zumindest DsRed2+ Zellen (Bereich R2+R3) geteilt. (B) Quantitative Auswertung der Anzahl an superinfizierten Jurkat Zellen (DsRed2+/eGFP+), die initial mit den angegebenen Vpu, Env und Nef Mutanten transduziert und anschließend mit HIV-1 Wildtypviren infiziert wurden. Die Zahlen (Mittelwerte +/- Stabw.; n=3) geben die X-fache Reduktion der Superinfektion im Vergleich zur Kontrolle (E-U-N-) an. Mock, nicht infiziert; DsRed2 und eGFP, Zellen infiziert mit VSV-G pseudotypisierten HIV-1 NL4-3-IRES-DsRed2 oder Wildtyp eGFP Viren; E, env; U, vpu; N, nef. 6. ERGEBNISSE 32 6.4 Nef schützt infizierte Zellen durch die Modulation von CD4 und mittels eines anderen Mechanismus vor einer Superinfektion Nef entfernt effizient CD4 von der Zelloberfläche und schützt infizierte Zellen somit effektiv vor einer Superinfektion (Abschnitt 6.2 und 6.3). Zur Korrelation der Nefvermittelten CD4-Herabregulation mit der Resistenz gegen eine Superinfektion wurden in einem weiteren Experiment zehn HIV-1 nef-Allele untersucht, die sich in ihrer Aktivität bezüglich der CD4-Modulation unterscheiden. Dies waren das effektiv CD4 herabregulierende NA7 Nef (Schindler et al., 2003) sowie Mutanten, die schwächer (85A, 105A) oder nicht (174AA) aktiv sind. Weiter das primäre nef-Allel LTNP4-1 von einem Langzeitüberlebenden welches CD4 nicht moduliert und die Mutante LTNP4-8 (D56A K174E), die schwach aktiv ist (Mariani et al., 1996). Außerdem nef-Allele, die aus einem HIV-1-Infizierten mit schnell voranschreitender Krankheit vor (P2-87) und nach (P2-93) dem Ausbruch von AIDS isoliert wurden und CD4 effektiv herabregulieren (Schindler et al., 2003). Und schließlich die Allele NPex und Pex, die die Konsensussequenzen einer großen Anzahl von HIV-1 Subtyp B Sequenzen repräsentieren, hoch aktiv CD4 herabregulieren und sich durch die Aminosäurenaustausche T15A, K39R, N51T, H102Y, N157T, S163C, S169N, L170Q und E182M unterscheiden, die mit unterschiedlichen Stadien des Krankheitsverlaufs assoziiert sind (Carl et al., 2001; Kirchhoff et al., 1999). Diese nef-Allele wurden zuerst mittels PCR amplifiziert, sequenziert und in die env-defekten proviralen Vektoren pBR-HIV-1 NL4-3-IRES-eGFP bzw. DsRed2 kloniert. Da zu den verwendeten Allelen bis zu diesem Zeitpunkt nur Daten aus Experimenten mit Nef Expressionsplasmiden vorlagen (Mariani et al., 1996; Greenberg et al., 1998; Carl et al., 2001; Schindler et al., 2003), wurden diese Allele zunächst auf ihre Aktivität im proviralen Kontext untersucht. Zu diesem Zweck wurden Jurkat T-Zellen mit den eGFP exprimierenden VSV-G pseudotypisierten Viren infiziert und die CD4-Modulation durch die verschiedenen nef-Allele im FACS bestimmt (Abb. 9A, obere Reihe). Am effektivsten entfernten die NL4-3, NA7, NA7-82A, P2-93 und Pex nef-Gene CD4 von der 6. ERGEBNISSE 33 Zelloberfläche, die NA7-105, P2-87 und NPex nef-Allele waren ein wenig schwächer aktiv und die NA-174AA und LTNP4-1 Nefs hatten praktisch keine Wirkung auf CD4. Abb. 9: Nef-vermittelte CD4-Herabregulation und die damit verbundene Resistenz gegen eine Superinfektion. (A) CD4-Herabregulation (obere Reihe) und superinfizierte Jurkat Zellen (untere Reihe), die die angegebenen nef-Allele exprimieren. Die Zahlen geben die rote MFI (obere Reihe) und den Prozentsatz an superinfizierten Zellen (untere Reihe) an. Die Kontrollen zeigen nicht (oben) und nur mit dem HIV-1 NL4-3-IRES-DsRed2 Konstrukt infizierte (unten) Zellen. (B) Anteil superinfizierter Zellen (in %), die initial mit den angegebenen HIV-1 nef-Varianten infiziert wurden (Mittelwert +/Stabw.; n=3). Die Zahlen geben die X-fache Reduktion der Superinfektion im Vergleich zur nef-defekten Kontrolle an. LTNP, „long-term nonprogressor“; Pr, HIV-1 infizierter Progressor; Cons., Konsensus nefAllele. (C) Korrelation zwischen CD4-Herabregulation und Superinfektion. Das offene Symbol zeigt die Anzahl an superinfizierten Zellen, die initial mit dem nef-defekten Virus infiziert wurden. Das kleine Diagramm zeigt die Korrelation zwischen Superinfektion und CD4-Herabregulation für die rekombinanten Viren mit intakten nef-Genen. Unter Verwendung der DsRed2 exprimierenden Viren wurden diese nef-Allele anschließend auf ihren Beitrag zur Verhinderung der Superinfektion infizierter Jurkat T- 6. ERGEBNISSE 34 Zellen untersucht (Abb. 9A, untere Reihe). Alle zehn nef-Allele machten die infizierten Zellen, wenn auch unterschiedlich effizient, resistent gegen eine Superinfektion. Die NL43, NA7, NA7-82A, NA7-105A, P2-87, P2-93, NPex und Pex Nef Proteine waren sehr effektiv und reduzierten die Zahl an superinfizierten Zellen um das Zehn- bis Zwanzigfache (Abb. 9B). Die NA7 Mutanten 82A und 105A waren dabei etwas schwächer aktiv als das wildtyp NA7 Nef und auch das P2-87 Nef, das in der chronischen Phase der Infektion isoliert wurde, zeigte im Vergleich zu dem nach Ausbruch von AIDS isolierten P2-93 Nef eine etwas verringerte Aktivität. Die Expression der nef-Allele, die aus dem HIV-1 Infizierten mit rascher Progression zu AIDS stammten (P2), schützte infizierte Zellen dreimal so gut vor anschließender Superinfektion wie die der nef-Gene des HIV-1 infizierten Langzeitüberlebenden (LTNP4). Obwohl die NA7-174AA und LTNP4-1 Nef Proteine praktisch keine Wirkung auf CD4 zeigten (Abb. 9A, obere Reihe), schützten auch sie infizierte Zellen vor einer anschließenden Superinfektion (Abb. 9B). Die Anwesenheit von Nef in infizierten Zellen reduzierte somit unabhängig von der CD4-Modulation die Superinfektionshäufigkeit um etwa den Faktor drei (Abb. 9C). Dies bedeutet, dass an der Nef-vermittelten Resistenz gegen eine Superinfektion neben dem CD4-abhängigen auch ein CD4-unabhängiger Mechanismus beteiligt ist. 6.5 Auch die Nef-vermittelte CxCR4-Herabregulation trägt, neben weiteren Faktoren, zur Superinfektionsresistenz bei HIV-1 benötigt für eine effiziente Infektion von Zellen neben dem primären Rezeptor CD4 zusätzlich einen der beiden Chemokinrezeptoren CCR5 oder CxCR4 als Korezeptor. In zwei kürzlich erschienen Veröffentlichungen wurde beschrieben, dass Nef beide Rezeptoren von der Zelloberfläche entfernt (Michel et al., 2005; Hrecka et al., 2005). Da es sich bei den für die vorhergehenden Experimente verwendeten HIV-1 NL4-3 Konstrukten um X4-trope Viren handelt, könnte auch die Modulation von CxCR4 in infizierten Zellen eine Superinfektion verhindern und den beobachteten CD4unabhängigen Effekt von Nef erklären. Zur Überprüfung dieser Möglichkeit wurde ein 6. ERGEBNISSE 35 weiteres Superinfektionsexperiment mit nef-Allelen durchgeführt, die unterschiedliche Effekte auf CD4 und CxCR4 zeigen (Tab.1). Die entsprechenden nef-Allele wurden zuerst aus schon bestehenden proviralen pBR-NL4-3-IRES-eGFP Vektoren in die env-defekten Vektoren pBR-HIV-1 NL4-3-IRESeGFP bzw. DsRed2 umkloniert. Mit den eGFP exprimierenden Viren wurden anschließend Jurkat Zellen infiziert und im FACS analysiert. Wie schon in Hrecka et al. beschrieben, entfernte das NA7 Nef CD4 effizient von der Zelloberfläche, wogegen CxCR4 nur schwach herunterreguliert wurde (Abb. 10A und B). Die Mutante 174AA hingegen war in beiden Funktionen inaktiv. Eine starke Wirkung auf beide Rezeptoren zeigte das SIVmac239 Nef, wogegen das SIVcol Nef nur CxCR4 sehr effizient herunterregulierte. Das NA7 Nef reduzierte die Superinfektion in Übereinstimmung mit den Ergebnissen aus Abschnitt 6.4 um das Zwölf- und die 174AA Mutante um das Dreifache (Abb. 10C). Da die 174AA Mutante weder den CD4 noch den CxCR4 Rezeptor herunterreguliert, muss noch ein weiterer, bis jetzt noch unbekannter Mechanismus zur maximalen Nefvermittelten Superinfektionsresistenz beitragen. Die Expression des SIVcol Nefs schützte infizierte Zellen unabhängig von einer Wirkung auf CD4 effizient vor einer Superinfektion. Da HIV-1 nef-Allele verglichen mit HIV-2 oder SIV Nefs den CxCR4 Rezeptor jedoch nur schwach herunterregulieren (Hrecka et al., 2005), spielt diese 6. ERGEBNISSE 36 Funktion für die Resistenz von HIV-1 infizierten Zellen gegen Superinfektion wahrscheinlich nur eine untergeordnete Rolle. Abb. 10: Beitrag der nef-vermittelten Modulation von CxCR4 zum Schutz vor Superinfektion. FACS-Analyse (A) und quantitative Wirkung (B) von verschiedenen nef-Allelen auf die CD4 und CxCR4 Zelloberflächenexpression von Jurkat Zellen. Legende: NA7; 174AA; mac239 und col Nef. Die Bereiche für keine (N), niedrige (L), mittlere (M) und hohe (H) eGFP Expression sind aus Abbildung A ersichtlich. (C) Anzahl superinfizierter Zellen (in %), die initial mit den angegebenen HIV1 nef Varianten infiziert wurden. Die Zahlen geben die Reduktion der Superinfektion (X-fach) im Vergleich zur nef-defekten Kontrolle an. Ein unabhängiges Experiment lieferte ähnliche Ergebnisse. In mehreren Veröffentlichungen wurde eine Koinfektion von Zellen mit HIV-1 und HIV-2 beschrieben (Blackard et al., 2002; Allen et al., 2003; Gross et al., 2004; Nethe et al., 2005; Smith et al., 2005), weshalb auch der Effekt eines HIV-2 und eines SIV nef- 6. ERGEBNISSE 37 Allels auf die Superinfektion untersucht wurde. Das HIV-2 Ben und das SIVagm tan1 Nef entfernten sowohl den CD4 als auch den CxCR4 Rezeptor mit hoher Effizienz von der Zelloberfläche und schützten damit auch wie erwartet infizierte Zellen vor einer Superinfektion (Tab.1 und Abb. 10C). Dies zeigt, dass mit HIV-2 oder anderen Primatenlentiviren infizierte T-Zellen möglicherweise gegen eine zweite Infektion durch HIV-1 geschützt sind. 6.6 Die Resistenz gegen eine Superinfektion schützt HIV-1 infizierte T-Zellen vor dem vorzeitigen Tod durch Apoptose Bei der Infektion einer Zelle durch HIV-1 wird zunächst das RNA-Genom des Virus in doppelsträngige DNA umgeschrieben und diese anschließend in das Wirtszellgenom integriert. Nicht integrierte provirale DNA wird möglicherweise durch die Zelle als DNA-Schädigung erkannt, die letztlich zur Induktion der Apoptose führt (Daniel et al., 1999). Durch eine zweite Infektion einer bereits infizierten Zelle akkumuliert sich möglicherweise nicht integrierte DNA in der Zelle und steigert die Apoptosewahrscheinlichkeit. Somit könnte die Verhinderung einer Superinfektion eine bereits infizierte Zelle vor dem vorzeitigen Tod durch Apoptose schützen und damit eine längere Partikelfreisetzung aus der infizierten Zelle und eine effiziente Vermehrung des Virus gewährleisten. Um diese Hypothese erstmalig zu überprüfen, wurden Jurkat und primäre CD4+ TZellen mit nef-defekten VSV-G pseudotypisierten HIV-1 NL4-3-IRES-DsRed2 Viren transduziert, drei Tage später mit dem eGFP exprimierenden NL4-3 Wildtypvirus infiziert und nach weiteren zwei Tagen im FACS analysiert. 35 % der nur DsRed2+ Jurkat Zellen waren apoptotisch und zeigten damit eine etwa zweifach höhere Apoptoserate als nicht infizierte Zellen (15,9 %). In der superinfizierten Zellpopulation war etwa die Hälfte (52 %) der Zellen apoptotisch (Abb. 11A). Diese Unterschiede der Apoptoserate zwischen nicht, einfach und doppelt infizierten Zellen waren in primären CD4+ T-Zellen noch einmal deutlich gesteigert (Abb. 11B). Dort bewirkte eine initiale HIV-1 Infektion der Zellen einen fünffachen Anstieg der Apoptoserate (nicht infiziert: 4,5 %; einfach infiziert: 6. ERGEBNISSE 38 24,8 % (DsRed2) bzw. 29,9 % (eGFP)) und eine anschließende Superinfektion erhöhte diesen Effekt noch einmal um etwa den Faktor drei auf 81,4 %. Diese Ergebnisse zeigen direkt, dass die Verhinderung einer Superinfektion infizierte Zellen vor dem vorzeitigen Tod durch Apoptose schützt. Abb. 11: Superinfizierte T-Zellen induzieren vermehrt Apoptose. (A) Jurkat Zellen und (B) primäre CD4+ TZellen wurden mit nef-defekten VSVG pseudotypisierten HIV-1 NL4-3IRES-DsRed2 Viren transduziert und drei Tage später mit wildtyp HIV-1 NL4-3-IRES-eGFP Viren infiziert. Zwei Tage später wurde die Anzahl an apoptotischen Zellen in der eGFP+, DsRed2+ und doppelt-positiven Zellpopulation im FACS bestimmt. In der quantitativen Auswertung (rechts) sind die Mittelwerte (+/- Stabw.) von je drei Infektionen gezeigt. 7. DISKUSSION 39 7. Diskussion Der CD4-Rezeptor spielt im Replikationszyklus von HIV-1 eine bedeutende Rolle, da er einerseits essentiell für die Infektion des Virus ist, aber andererseits die Virusfreisetzung beinträchtigen kann. Schon bald nachdem HIV-1 als Ursache von AIDS entdeckt worden war, wurde das CD4-Protein als der primäre Rezeptor von HIV-1 identifiziert (Dalgleish et al., 1984; Klatzmann et al., 1984). Zwei Jahre später wurde jedoch gezeigt, dass in HIV-1 infizierten Zellen die CD4-Expression blockiert ist (Hoxie et al., 1986). Inzwischen weiß man, dass HIV-1 drei virale Proteine (Vpu, Env und Nef) zur CD4-Herabregulation verwendet (Chen et al., 1996) und dies für das Virus große Vorteile hat. Die CD4-Modulation erleichtert einerseits die Partikelfreisetzung aus der infizierten Zelle (Bour et al., 1999; Ross et al., 1999). Andererseits können hohe CD4Expressionslevel die Infektiosität der Nachkommenviren beeinträchtigen, da hierdurch viel CD4 in die Virusmembran eingelagert wird und dort mit dem viralen Hüllprotein Env interagiert (Lama et al., 1999; Cortes et al., 2002; Levesque et al., 2003). Da CD4 auch eine wichtige Rolle bei der Aktivierung des Immunsystems durch T-Zellen spielt (Weiss et al., 1994), wird durch die CD4-Modulation möglicherweise auch die antivirale Immunantwort geschwächt. Schließlich soll die verminderte CD4-Expression auch eine Superinfektion blockieren (Benson et al., 1993) und die HIV-1 infizierten Zellen vor einer dadurch induzierten frühzeitigen Apoptose schützen. Da für diese Hypothesen bislang nur Daten von chronisch infizierten Zellen oder transfizierten Zellen, die Env oder Nef von einem Expressionsplasmid exprimieren, existierten (LeGuern et al., 1992; Benson et al., 1993; Little et al., 1994; Pham et al., 2004; Michel et al., 2005), war das Ziel dieser Doktorarbeit, den Beitrag der drei viralen Proteine Vpu, Env und Nef zur CD4Herabregulation und Superinfektionsresistenz in produktiv infizierten T-Zellen zu untersuchen. Zur Analyse der CD4-Herabregulation wurden zunächst rekombinante HIV-1 NL43-IRES-eGFP Viren mit einzelnen und kombinierten Defekten in vpu, env und nef hergestellt. Das Virusisolat NL4-3 ist X4-trop, gut charakterisiert und wird weltweit als Standard zur Infektion von T-Zelllinien und Primärzellen mit HIV-1 genutzt. Über ein einkloniertes IRES-Element wird eGFP zusammen mit Nef von einer bicistronischen mRNA koexprimiert. Dieses System ermöglicht die einfache Identifikation infizierter Zellen anhand ihrer grünen Fluoreszenz und die Korrelation der viralen Protein-Expression mit der CD4-Modulation (Schindler et al., 2005). Zur Standardisierung der Infektion und 7. DISKUSSION 40 zur Analyse der env-defekten Viren wurden Viruspartikel mit dem Hüllprotein des Vesikulären Stomatitisvirus pseudotypisiert. Dies erlaubt die vergleichbare Infektion von Zielzellen unter Umgehung des Nef-vermittelten Einflusses auf die Infektiosität (Chazal et al., 2001). Nef trug in allen untersuchten Zellarten hauptsächlich zur CD4-Herabregulation bei und führte schon in der frühen Phase der Infektion (24 bis 32 Stunden nach der Infektion) zu einer verminderten CD4-Expression. Der Effekt von Env und Vpu auf CD4 war insgesamt deutlich schwächer. Dies bestätigt die Ergebnisse einer früheren Studie, in der alkalische Phosphatase exprimierende Reporterviren zur Analyse der vpu-, env- und nefvermittelten CD4-Herabregulation verwendet wurden (Chen et al., 1996). Allerdings war in dieser Studie die CD4-Modulation in Jurkat Zellen stärker als in PBMCs, was nicht mit den Ergebnissen dieser Doktorarbeit übereinstimmt. Bis zur maximalen CD4Herabregulation dauerte es in PBMCs zwar einen Tag länger als in Jurkat Zellen, aber drei Tage nach der Infektion wurden vergleichbare Level der CD4-Herabregulation erreicht. Passend dazu wurde in einer kürzlich erschienenen Veröffentlichung gezeigt, dass Nef die CD4-Expression in primären CD4+-T-Zellen drastisch reduziert (Keppler et al., 2006). Auch war in Experimenten dieser Doktorarbeit im Gegensatz zu früheren Ergebnissen der Effekt von Vpu auf CD4 in PBMCs stärker als in Jurkat Zellen (Abb. 4 und 5). Diese Unterschiede könnten sich durch die verschiedenen verwendeten experimentellen Systeme einschließlich der proviralen Konstrukte erklären lassen. So wird z.B. bei den von Chen et al. verwendeten Viren Nef über ein IRES-Element exprimiert, wogegen bei den in dieser Arbeit hergestellten Konstrukten (Abb. 2) alle viralen Proteine im physiologischen Kontext unter Kontrolle des LTR-Promoters und der natürlichen Spleißstellen exprimiert werden. In den mit dem CD4-Expressionsplasmid kotransfizierten 293T Zellen, die normalerweise kein CD4 exprimieren, konnte der Effekt der viralen Proteine auf den Transport von neu synthetisiertem CD4 zur Zelloberfläche untersucht werden. In Übereinstimmung mit der Beobachtung, dass Env und Vpu CD4 im Endoplasmatischen Retikulum zurückhalten und dadurch diesen Transport hemmen (Levesque et al., 2004), waren in diesem System bei niedrigen CD4-Mengen Env und Vpu in etwa so aktiv in der Blockierung der CD4-Expression wie Nef. Der Mechanismus, mit dem Env die CD4Expression blockiert, scheint allerdings durch größere CD4-Mengen schnell gesättigt zu werden (Abb. 6). 7. DISKUSSION 41 Durch die Verwendung von Viren, die grüne (eGFP) und rote (DsRed2) Fluoreszenzproteine exprimieren, konnte im Rahmen dieser Doktorarbeit gezeigt werden, dass die CD4-Herabregulation HIV-1 infizierte Zellen vor einer Superinfektion schützt (Abb. 7). Dies bestätigt die Daten aus früheren Studien, die allerdings mit chronisch infizierten Zellen oder transfizierten Zellen durchgeführt wurden (LeGuern et al., 1992; Benson et al., 1993; Little et al., 1994; Pham et al., 2004; Michel et al., 2005). Nach der initialen Infektion dauert es etwa 16 bis 24 Stunden, bis die CD4-Modulation in Jurkat Zellen erstmals nachweisbar ist (Abb.4 und Lama et al., 2003). In PBMCs dauert es noch etwas länger (Abb.5). Die durchschnittliche Lebenserwartung einer produktiv HIV-1 infizierten primären CD4+-T-Zelle wird in vivo auf etwa 2,2 Tage geschätzt, wobei die Zeit, nach der die Hälfte aller infizierten Zellen gestorben sind, etwa 1 bis 2 Tage beträgt (Perelson et al., 1996; Cavert et al., 1997). Da die Zeitspanne, in der die CD4Herabregulation vor einer Superinfektion schützt, somit nur sehr kurz ist bevor die infizierten Zellen sterben, stellt sich die Frage, ob dieser Effekt physiologisch von Bedeutung ist (Lama et al., 2003). Für das Virus kann eine Superinfektion sowohl vor- als auch nachteilig sein. Wird eine bereits infizierte Zelle ein zweites Mal durch ein Virus eines anderen HIV-Stammes infiziert, so ermöglicht dies die genetische Rekombination und erleichtert dem Virus die Anpassung an veränderte Umweltbedingungen. Die Nachkommenviren können so z.B. schnell eine Resistenz gegen antivirale Medikamente entwickeln oder besser der Immunantwort entgehen. HIV-1 zeigt eine hohe Rekombinationsrate (Levy et al., 2004) und es sind viele Rekombinationsereignisse zwischen den verschiedenen HIV-Stämmen beschrieben (Blackard et al., 2002). So entstand wahrscheinlich sogar der Vorläufer von HIV-1, das Immundefizienvirus der Schimpansen (SIVcpz), durch eine Rekombination von zwei SIV-Stämmen von kleineren Affen (Bailes et al., 2003). Allerdings kann eine Superinfektion zu einem späten Zeitpunkt im Replikationszyklus des Virus dazu führen, dass die Zelle abstirbt bevor die Virusproduktion und die Freisetzung der Nachkommenviren beendet sind. Die Blockierung der Superinfektion in einer späten Phase des Replikationszyklus ist somit vorteilhaft für das Virus, da sie die Zellen vor dem vorzeitigen Tod durch Apoptose schützt (Abb. 11) und eine effektive Replikation ermöglicht. In infizierten Zellen mit einer kurzen Lebensdauer beeinflusst die CD4Herabregulation die Rekombinationsrate wahrscheinlich wenig. Diese Situation ändert sich allerdings wahrscheinlich, wenn das Virus in Zellen mit einer langen Lebensdauer (z.B. 7. DISKUSSION 42 Makrophagen oder ruhende T-Zellen) persistiert, wie es in Patienten der Fall ist, die mit der hoch-aktiven antiretroviralen Therapie (HAART) behandelt werden (Stevenson, 2003). Wenn die Virusreplikation durch die HAART in langlebige Makrophagen oder T-Zellen verlagert wird und die CD4-Herabregulation dort effizient eine Superinfektion verhindert, erschwert dies die Entstehung von resistenten Virusvarianten. Dass Nef CD4 auch in ruhenden T-Zellen effektiv von der Zelloberfläche entfernt, wurde erst kürzlich gezeigt (Keppler et al., 2006). Die CD4-Herabregulation und die dadurch erreichte Blockierung einer Superinfektion wird hauptsächlich von Nef bewirkt (Abb. 8). Die Analyse verschiedener nef-Allele mit unterschiedlicher CD4-Modulationsaktivität ergab, dass der Anteil an superinfizierten Zellen umso kleiner war, je mehr CD4 die jeweiligen Nef-Proteine von der Zelloberfläche entfernten normalerweise effizient (Abb. CD4 9). Nef-Allele herab, von wogegen Nefs AIDS-Patienten aus HIV-1 regulieren infizierten Langzeitüberlebenden in dieser Funktion inaktiv sind (Mariani et al., 1996; Carl et al., 2001; Tobiume et al., 2002; Arganaraz et al., 2003). Dementsprechend wurde von den Allelen NA7 und P2 aus AIDS-Patienten auch die Superinfektion um das Drei- bis Siebenfache stärker blockiert als von denen, die aus einem Langzeitüberlebenden (LTNP4) stammten (Abb. 9). In AIDS-Patienten ist die Zahl an nicht infizierten CD4+-T-Zellen gering, weshalb es für das Virus in dieser Umgebung von Vorteil sein kann, eine Superinfektion verstärkt zu verhindern. Dagegen wird in Langzeitüberlebenden die HIVInfektion durch das Immunsystem effizient kontrolliert. Unter diesen Bedingungen scheint für das Virus eine Superinfektion vorteilhaft zu sein, da durch die gesteigerte Rekombinationsrate vermehrt neue HIV-Varianten entstehen, die leichter der Immunantwort entgehen können. Auch nef-Allele die kaum CD4 von der Zelloberfläche entfernen, reduzierten die Superinfektion um etwa den Faktor drei (Abb. 9, NA7-174AA und LTNP4-1). Daraus lässt sich schließen, dass neben der CD4-Herabregulation mindestens ein weiterer Mechanismus zur Nef-vermittelten Superinfektionsresistenz beiträgt. Das in diesem Experiment verwendete X4-trope HI-Virus NL4-3 benötigt neben CD4 noch den CxCR4 Korezeptor zur Infektion einer Zelle, der auch durch Nef moduliert wird (Hrecka et al., 2005; Michel et al., 2005). Wie man anhand des SIVcol Nefs sieht (das nur CxCR4, nicht aber CD4 moduliert), kann die CxCR4-Herabregulation ebenfalls eine Superinfektion verhindern (Abb.10). Dies wird durch eine kürzlich veröffentlichte Studie bestätigt, in der ein FACS- 7. DISKUSSION 43 basierendes Virus-Zell-Fusionsexperiment zeigt, dass SIVCol Nef exprimierende T-Zellen deutlich seltener mit HIV-Reporterviren fusionieren als die entsprechenden Kontrollzellen (Venzke et al., 2006). Die Allele NA7-174AA und LTNP4-1 modulieren allerdings weder CD4 noch CxCR4. Es kann zwar nicht vollständig ausgeschlossen werden, dass die minimalen Effekte von NA7-174AA und LTNP4-1 auf CD4 oder CxCR4 zur Verhinderung einer Superinfektion beitragen, aber es scheint unwahrscheinlich, dass sie maßgeblich für die Superinfektionsresistenz verantwortlich sind. Dazu trägt wohl eher eine der vielen bekannten Rezeptor-unabhängigen Funktionen von Nef bei, wie z.B. die Modifikation des Zytoskeletts (Fackler et al., 1999; Campbell et al., 2004), die Veränderung der Zellmembranbeschaffenheit (Zheng et al., 2003; van’t Wout et al., 2005) oder die Beeinflussung von intrazellulären Signal- und Reaktionswegen (Doms et al., 2000; Lama et al., 2003). Der CxCR4-Rezeptor wird von HIV-1 Nefs nur schwach herabreguliert, wogegen HIV-2 und SIV Nefs in dieser Funktion stark aktiv sind (Hrecka et al., 2005). Die Ergebnisse mit dem SIVcol Nef zeigen, dass die CxCR4-Herabregulation signifikant die Superinfektion reduziert (Abb. 10). Allerdings verwenden die Affen-Immundefizienzviren (SIV), von denen die in dieser Funktion stark aktiven nef-Allele kodiert werden, CxCR4 normalerweise nicht als Korezeptor. Die physiologische Bedeutung der CxCR4Herabregulation scheint somit nicht die Verhinderung der Superinfektion zu sein, sondern z.B. wie erst kürzlich beschrieben die Blockierung der T-Zell-Migration (Hrecka et al., 2005). Da auch CCR5 von Nef moduliert werden soll (Michel et al., 2005), wurde im Rahmen dieser Doktorarbeit auch versucht, die Auswirkung der CCR5-Herabregulation auf die Superinfektion zu bestimmen. Dabei konnte die Nef-vermittelte CCR5-Modulation in stabil transfizierten CHO Zellen, die diesen Rezeptor überexprimieren, bestätigt werden. Da die CCR5-Herabregulation in anderen Zelllinien oder primären Zellen allerdings nur schwach ist (Michel et al., 2005), spielt sie bei der Superinfektionsresistenz, wenn überhaupt, wahrscheinlich nur eine sehr geringe Rolle. Zusammenfassend zeigen die Daten dieser Doktorarbeit, dass Nef HIV-1 infizierte Zellen effektiv vor einer Superinfektion schützt. Dazu trägt sowohl die CD4Herabregulation als auch ein bislang unbekannter CD4-unabhängiger Mechanismus bei. Die Blockierung einer Superinfektion schützt die Zellen vor dem vorzeitigen Tod durch Apoptose, verlängert dadurch die Zeitspanne in der die Nachkommenviren freigesetzt werden können und ermöglicht dem Virus somit eine effektive Replikation. 8.ZUSAMMENFASSUNG 44 8. Zusammenfassung Das Humane Immundefizienzvirus Typ 1 (HIV-1) verwendet drei seiner Proteine (Vpu, Env und Nef) zur Herabregulation von CD4 von der Zelloberfläche. Dies führt zu einer verstärkten Freisetzung von Viren und einer erhöhten Infektiosität der Partikel. Da CD4 der primäre Rezeptor von HIV-1 und neben den Korezeptoren CxCR4 oder CCR5 notwendig für eine Infektion der Zelle ist, wird seit langem postuliert, dass die HIV-1 vermittelte CD4-Modulation eine als Superinfektion bezeichnete zweite Infektion bereits infizierter Zellen verhindert. In der vorliegenden Promotionsarbeit sollte der Beitrag der viralen Proteine Vpu, Env und Nef zur CD4-Herabregulation und die daraus resultierende Resistenz von HIV-1 infizierten T-Zellen gegen eine Superinfektion untersucht werden. Hierzu wurden Viren mit einzelnen und kombinierten Defekten in den vpu, env und nef Genen hergestellt, die das grüne Fluoreszenzprotein (eGFP) über eine interne Ribosomeneintrittsstelle (IRES) exprimieren. FACS Analysen infizierter Jurkat T-Zellen, primärer Blutlymphozyten (PBMC) und mit CD4 kotransfizierter 293T Zellen zeigten, dass alle drei Proteine zur effektiven Entfernung von CD4 von der Zelloberfläche beitragen. Nef entfernte CD4 dabei immer sehr effektiv von der Zelloberfläche, wogegen Env eine schwächere Wirkung hatte. Vpu war in Jurkat Zellen nicht und in PBMCs schwach aktiv. In 293T Zellen blockierte es dagegen effizient den Transport von neu synthetisiertem CD4 zur Zelloberfläche. Zur Untersuchung der Superinfektion wurde eine auf zwei unterschiedlichen Fluoreszenzproteinen basierende FACS-Analyse verwendet. Dazu wurden Jurkat T-Zellen initial mit rekombinanten DsRed2 exprimierenden HIV-1 Viren transduziert, drei Tage später mit eGFP exprimierenden Wildtyp-HIV-1-Viren infiziert und nach weiteren zwei Tagen im FACS analysiert. Nef schützte in Übereinstimmung mit seiner starken CD4Modulationsaktivität HIV-1 infizierte Zellen effektiv vor einer Superinfektion, wogegen Vpu und Env nur eine geringe schützende Wirkung zeigten. Die Analyse von zehn verschiedenen nef-Allelen, die CD4 unterschiedlich stark herabregulieren, ergab, dass die nef-vermittelte CD4-Herabregulation mit der Resistenz gegen eine Superinfektion korreliert. Des Weiteren zeigte die Untersuchung von nef-Allelen mit unterschiedlicher Wirkung auf CD4 und CxCR4, dass auch die Herabregulation von CxCR4 Zellen vor einer Superinfektion mit HI-Viren schützt, die diesen Korezeptor verwenden (X4-trope Viren). Diese Analysen ergaben außerdem, dass zusätzlich zu den rezeptorabhängigen Mechanismen ein bislang unbekannter rezeptorunabhängiger Mechanismus an der Nef- 8.ZUSAMMENFASSUNG 45 vermittelten Resistenz gegen eine Superinfektion beteiligt ist, da auch nef-Allele, die weder CD4 noch CxCR4 von der Zelloberfläche entfernten, den Anteil an superinfizierten Zellen reduzierten. Weitere Untersuchungen zeigten, dass superinfizierte T-Zellen deutlich häufiger apoptotisch sind und durch den programmierten Zelltod sterben als einfach oder nicht infizierte Zellen. Zusammengefasst zeigen die Ergebnisse dieser Promotionsarbeit, dass Nef im Vergleich zu Vpu und Env den primären viralen Rezeptor CD4 am effektivsten von der Zelloberfläche entfernt und damit HIV-infizierte Zellen vor einer Superinfektion und dem vorzeitigen Tod durch Apoptose schützt. Dies verlängert die Zeitspanne der Partikelfreisetzung aus der infizierten Zelle und ermöglicht damit eine effiziente Vermehrung des Virus. 9. LITERATURVERZEICHNIS 46 9. Literaturverzeichnis Allen, T. M., and M. Altfeld. 2003. HIV-1 superinfection. J. Allergy Clin. Immunol. 112:829-835. Argañaraz, E. R., M. Schindler, F. Kirchhoff, and J. Lama. 2003. Enhanced CD4 down-modulation by late-stage HIV-1 nef alleles is associated with increased Env incorporation and viral replication. J. Biol. Chem. 36:33912-33919. Bailes, E., F. Gao, F. Bibollet-Ruche, V. Courgnaud, M. Peeters, P. A. Marx, B. H. Hahn, and P. M. Sharp. 2003. Hybrid origin of SIV in chimpanzees. Science 300:1713. Benson, R. E., A. Sanfridson, J. S. Ottinger, C. Doyle, and B. R. Cullen. 1993. Downregulation of cell surface CD4 expression by simian immunodeficiency virus Nef prevents viral super infection. J. Exp. Med. 177:1561-1566. Barré-Sinoussi, F., J. C. Chermann, F. Rey, M. T. Nugeyre, S. Chamaret, J. Gruest, C. Dauget, C. Axler-Blin, F. Vezinét-Brun, C. Rouzioux, W. Rozenbaum, and L. Montagnier. 1983. Isolation of a T-lymphotropic retrovirus form a patient at risk for AIDS. Science 220: 868-871. Blackard, J. T., D. E. Cohen, and K. H. Mayer. 2002. Human immunodeficiency virus superinfection and recombination: current state of knowledge and potential clinical consequences. Clin. Infect. Dis. 34:1108-1114. Bour, S., C. Perrin, and K. Strebel. 1999. Cell surface CD4 inhibits HIV-1 particle release by interfering with Vpu activity. J. Biol. Chem. 274:33800-33806. Campbell, E. M., R. Nunez, and T. J. Hope. 2004. Disruption of the actin cytoskeleton can complement the ability of Nef to enhance human immunodeficiency virus type 1 infectivity. J. Virol. 78:5745-5755. Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA.99:7877-7882. Carl, S., T. C. Greenough, M. Krumbiegel, M. Greenberg, J. Skowronski, J. L. Sullivan, and F. Kirchhoff. 2001. Modulation of different human immunodeficiency virus type 1 Nef functions during progression to AIDS. J. Virol. 75:3657-3665. 9. LITERATURVERZEICHNIS 47 Cavert, W., D. W. Notermans, K. Staskus, S. W. Wietgrefe, M. Zupancic, K. Gebhard, K. Henry, Z. Q. Zhang, R. Mills, H. McDade, J. Goudsmit, S. A. Danner, and A. T. Haase. 1997. Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV-1 infection. Science 276:960-964. Chazal, N., G. Singer, C. Aiken, M. L. Hammarskjold, and D. Rekosh. 2001. Human immunodeficiency virus type 1 particles pseudotyped with envelope proteins that fuse at low pH no longer require Nef for optimal infectivity. J Virol. 75:4014-8. Chen, B. K., R. T. Gandhi, and D. Baltimore. 1996. CD4 down-modulation during infection of human T cells with human immunodeficiency virus type 1 involves independent activities of vpu, env, and nef. J. Virol. 70:6044-6053. Clavel, F., M. Guyader, D. Guetard, M. Salle, L. Montagnier, and M. Alizon. 1986. Molecular cloning and polymorphism of the human immune deficiency virus type 2. Nature 324(6098):691-5. Coffin, J., A. Haase, J. A. Levy, L. Montagnier, S. Oroszlan, N. Teich, H. Temin, K. Toyoshima, H. Varmus, and P. L. Vogt. 1986. What to call the AIDS virus? Nature 321:10. Cohen, E. A., G. Dehni, J. G. Sodroski, and W. A. Haseltine. 1990. Human immunodeficiency virus vpr product is a virion-associated regulatory protein. J Virol. 64(6):3097-9. Cortes, M. J., F. Wong-Staal, and J. Lama. 2002. Cell surface CD4 interferes with the infectivity of HIV-1 particles released from T cells. J. Biol. Chem. 277:1770-1779. Dalgleish, A. G., P. C. Beverley, P. R. Clapham, D. H. Crawford, M. F. Greaves, and R. A. Weiss. 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312(5996):763-7. Daniel, R., R. A. Katz, and A. M. Skalka. 1999. A role for DNA-PK in retroviral DNA integration. Science 284:644-647. DeVita, V.T., S. Hellmann and S. Rosenberg. 1997. AIDS. Lipincott-Raven Publishers, Philadelphia, New York. Doms, R. D., and D. Trono. 2000. The plasma membrane as a combat zone in the HIV battlefield. Genes Dev. 14:2677-2688. 9. LITERATURVERZEICHNIS 48 DuBridge, R. B., P. Tang, H. C. Hsia, P. M. Leong, J. H. Miller, and M. P. Calos. 1987. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol.Cell Biol., 7, 379-387. Fackler, O. T., W. Luo, M. Geyer, A. S. Alberts, and P. M. Peterlin. 1999. Activation of Vav by Nef induces cytoskeletal rearrangements and downstream effector functions. Mol. Cell. 3:729-739. Gottlieb, M. S., R. Schroff, H. M. Schanler, J. D. Weisman, P. T. Fan, R. A. Wolf, and A. Sanyon. 1981. Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexuall men: evidence of a new acquired cellular immunodeficiency. N. Engl. J. Med. 305: 1426-1431. Greenberg, M. E., A. J. Iafrate, and J. Skowronski. 1998. The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. EMBO J. 17:2777-2789. Greenberg, M. E., L. DeTulleo, I. Rapoport, J. Skowronski, and T. Kirchhausen. 1998. A dileucine motif in HIV-1 Nef is essential for sorting into clathrin-coated pits and for downregulation of CD4. Curr. Biol. 8:1239-1242. Gross, K. L., T. C. Porco, and R. M. Grant. 2004. HIV-1 superinfection and viral diversity. AIDS 18:1513-1520. Hiebenthal-Millow, K., and F. Kirchhoff. 2002. The most frequent naturally occurring length polymorphism in the HIV-1 LTR has little effect on proviral transcription and viral replication. Virol. 292(1):169-75. Ho, D. D. 1998. Toward HIV eradication or remission: the tasks ahead. Science 280(5371):1866-7. Hoxie, J. A., J. D. Alpers, J. L. Rackowski, K. Huebner, B. S. Haggarty, A. J. Cedarbaum, and J. C. Reed. 1986. Alterations in T4 (CD4) protein and mRNA synthesis in cells infected with HIV. Science 234(4780):1123-7. Hrecka, K., T. Swigut, M. Schindler, F. Kirchhoff, and J. Skowronski. 2005. Nef proteins from diverse groups of primate lentiviruses downmodulate CXCR4 to inhibit migration to SDF-1 chemokine. J. Virol. 79:10650-10659. 9. LITERATURVERZEICHNIS 49 Keppler, O. T. , N. Tibroni, S. Venzke, S. Rauch, and O. T. Fackler. 2006. Modulation of specific surface receptors and activation sensitization in primary resting CD4+ T lymphocytes by the Nef protein of HIV-1. J. Leukoc. Biol. 79(3):616-27. Kirchhoff, F., P. J. Easterbrook, N. Douglas, M. Troop, T. C. Greenough, J. Weber, S. Carl, J. L. Sullivan, and R. S. Daniels. 1999. Sequence variations in human immunodeficiency virus type 1 Nef are associated with different stages of disease. J. Virol. 73:5497-5508. Klatzmann, D., E. Champagne, S. Chamaret, J. Gruest, D. Guetard, T. Hercend, J. C. Gluckman, and L. Montagnier. 1984. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 312(5996):767-8. Lama, J. 2003. The physiological relevance of CD4 receptor down-modulation during HIV infection. Curr. HIV Res. 1:167-184. Lama, J., A. Mangasarian, and D. Trono. 1999. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr. Biol. 9:622-631. LeGuern, M., and J. Levy. 1992. Human immunodeficiency virus (HIV) type 1 can superinfect HIV-2-infected cells: pseudotype virions produced with expanded cellular host range. Proc. Natl. Acad. Sci. USA 89:363-367. Levesque, K., Y. S. Zhao, and E. A. Cohen. 2003. Vpu exerts a positive effect on HIV-1 infectivity by down-modulating CD4 receptor molecules at the surface of HIV-1-producing cells. J. Biol. Chem. 278:28346-28353. Levesque, K., A. Finzi, J. Binette, and E. A. Cohen. 2004 Role of CD4 receptor downmodulation during HIV-1 infection. Curr HIV Res. 2(1):51-9. Levy, D. N., G. M. Aldrovandi, O. Kutsch, and G. M. Shaw. 2004. Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. USA 101:4204-4209. Little, S. J., N. L. Riggs, M. Y. Chowers, N. J. Fitch, D. D. Richman, C. A. Spina, and J. C. Guatelli. 1994. Cell surface CD4 downregulation and resistance to superinfection induced by a defective provirus of HIV-1. Virology 205:578-582. 9. LITERATURVERZEICHNIS 50 Liu, H., X. Wu, M. Newman, G. M. Shaw, B. H. Hahn, and J. C. Kappes. 1995. The Vif protein of human and simian immunodeficiency viruses is packaged into virions and associates with viral core structures. J Virol. 69, 7630-7638 Mangasarian, A., M. Foti, C. Aiken, D. Chin, J.-L. Carpentier, and D. Trono. 1997. The HIV-1 Nef protein acts as a connector with sorting pathways in the Golgi and at the plasma membrane. Immunity 6:67-77. Maniatis, T., J. Sambrook and E. F. Fritsch. 1989. Molecular cloning. Cold Spring Harbor laboratory Press, New York. Mariani, R., F. Kirchhoff, T. C. Greenough, J. L. Sullivan, R. C. Desrosiers, and J. Skowronski. 1996. High frequency of defective nef alleles in a long-term survivor with nonprogressive human immunodeficiency virus type 1 infection. J. Virol.70:7752–7764. McMichael, A. J., and T. Hanke. 2003. HIV vaccines 1983-2003. Nat Med.; 9(7):874-80 Michel, N., I. Allespach, S. Venzke, O. T. Fackler, and O. T. Keppler. 2005. The Nef protein of human immunodeficiency virus establishes superinfection immunity by a dual strategy to downregulate cell-surface CCR5 and CD4. Curr Biol. 15:714-723. Nethe, M., B. Berkhout, and A. C. van der Kuyl. 2005. Retroviral superinfection resistance. Retrovirology 2:52-65. Palella, F. J. Jr., K. M. Delaney, A. C. Moorman, M. O. Loveless, J. Fuhrer, G. A. Satten, D. J. Aschman, and S. D. Holmberg. 1998. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N. Engl. J. Med., 338, 853-860. Perelson, A. S., P. Essunger, Y. Cao, M. Vesanen, A. Hurley, K. Saksela, M. Markowitz, and D. D. Ho. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188-191. Perelson, A. S., A. U. Neumann, M. Markowitz, J. M. Leonard, and D. D. Ho. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582-1586. Pham, H. M., E. R. Arganaraz, B. Groschel, D. Trono, and J. Lama. 2004. Lentiviral vectors interfering with virus-induced CD4 down-modulation potently block human 9. LITERATURVERZEICHNIS 51 immunodeficiency virus type 1 replication in primary lymphocytes. J Virol. 78:1307213081. Piguet, V., Y.-L. Chen, A. Mangasarian, M. Foti, J.-L. Carpentier, and D. Trono. 1998. Mechanism of Nef-induced CD4 endocytosis: Nef connects CD4 with the µ chain of adaptor complexes. EMBO J. 17:2472-2481. Ross, T. M., A. E. Oran, and B. R. Cullen. 1999. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr. Biol. 9:613-621. Rücker, E., J. Münch, J. C. Grivel, F. Kirchhoff, and L. Margolis. 2004. Vpr and Vpu are important for efficient HIV-1 replication and CD4+ T cell depletion in human lymphoid tissue ex vivo. J. Virol. 78:12689-12693. Schindler, M., S. Wuerfl, P. Benaroch, T. C. Greenough, R. Daniels, P. Easterbrook, M. Brenner, J. Münch, and F. Kirchhoff. 2003. Down-modulation of mature MHC class II and up-regulation of invariant chain cell surface expression are well conserved functions of human and simian immunodeficiency virus nef alleles. J. Virol. 77:10548-10556. Schindler, M., J. Münch, and F. Kirchhoff. 2005. HIV-1 inhibits DNA damage triggered apoptosis by a Nef-independent mechanism. J. Virol. 79:5489-5498. Smith, D. M., D. D. Richman, and S. J. Little. 2005. HIV superinfection. J. Infect. Dis. 192:438-444. Stevenson, M. 2003. HIV-1 pathogenesis. Nat. Med. 9:853-860. Tobiume, M., M. Takahoko, T. Yamada, M. Tatsumi, A. Iwamoto, and M. Matsuda. 2002. Inefficient enhancement of viral infectivity and CD4 downregulation by human immunodeficiency virus type 1 Nef from Japanese long-term nonprogressors. J. Virol. 76:5959-5965. Stumptner-Cuvelette, P., S. Morchoisne, M. Dugast, S. Le Gall, G. Raposo, O. Schwartz, and P. Benaroch. 2001. HIV-1 Nef impairs MHC class II antigen presentation and surface expression. Proc Natl Acad Sci. 98(21):12144-9. van 't Wout, A.B., J. V. Swain, M. Schindler, U. Rao, M. S. Pathmajeyan, J. I. Mullins, and F. Kirchhoff. 2005. Nef induces multiple genes involved in cholesterol 9. LITERATURVERZEICHNIS 52 synthesis and uptake in human immunodeficiency virus type 1-infected T cells. J. Virol. 79:10053-10058. Ventzke S., N. Michel, I. Allespach, O.T. Fackler, and O. T. Keppler. 2006. Expression of Nef downregulates CXCR4, the major coreceptor of human immunodeficiency virus, from the surface of target cells and therby enhances resistance to superinfection. J. Virol. Aug 23 [Epub ahead of print]. Weiss, A., and D. R. Littman. 1994. Signal transduction by lymphocyte antigen receptors. Cell. 76:263-274. Welker, R., H. Kottler, H. R. Kalbitzer, and H. G. Kräusslich. 1996. Human immunodeficiency virus type 1 Nef is incorporated into virus particles and specifically cleaved by the viral proteinase. Virol. 219, 228-236. Zheng, Y. H., A. Plemenitas, C. J. Fielding, and B. M. Peterlin. 2003. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV-1 progeny virions. Proc. Natl. Acad. Sci. USA 100:8460-8465. Zhu, T., B. T. Korber, A. J. Nahmias, E. Hooper, P. M. Sharp, and D. D. Ho. 1998. An African HIV-1 sequence from 1959 and implications for the origin of the epidemic. Nature, 391: 594-597.