Dokument_26.
Werbung

Untersuchung der Interaktion
humaner IgG Antikörper mit humanen FcγR
in vitro und in vivo
Der naturwissenschaftlichen Fakultät
der Friedrich-Alexander-Universität Erlangen-Nürnberg
zur
Erlangung des Doktorgrades Dr. rer. nat.
vorgelegt von
Anja Lux
aus Altenburg
Als Dissertation genehmigt von der
Naturwissenschaftlichen Fakultät der Universität Erlangen-Nürnberg
Tag der mündlichen Prüfung:
22. Dezember 2010
Vorsitzender der Promotionskomission:
Prof. Dr. Rainer Fink
Erstberichterstatter:
Prof. Dr. Falk Nimmerjahn
Zweitberichterstatter:
Prof. Dr. Hans-Martin Jäck
Zwei Dinge sind zu unserer Arbeit nötig:
Unermüdliche Ausdauer und die Bereitschaft, etwas,
in das man viel Zeit und Arbeit gesteckt hat, wieder wegzuwerfen.
Albert Einstein
Inhalt
Inhalt
A. Zusammenfassung ............................................................................1
I.
II.
Zusammenfassung .......................................................................................................1
Summary ......................................................................................................................2
B. Einleitung ...........................................................................................3
I.
Fcγ Rezeptoren.............................................................................................................3
Murine und humane Fcγ Rezeptoren........................................................................3
Induktion von Antikörpereffektorfunktionen ..............................................................5
Einfluss der IgG Glykosylierung auf die FcγR Interaktion.........................................6
Polymorphismen von humanen FcγR .......................................................................7
II. Untersuchungen in humanisierten Mausmodellen .....................................................10
1. Generierung humanisierter Mausmodelle...............................................................10
2. Humanisierte Mausmodelle in der biomedizinischen Forschung............................11
1.
2.
3.
4.
C. Fragestellung ...................................................................................13
D. Ergebnisse .......................................................................................15
I.
Bindung von Immunkomplexen an FcγR in vitro.........................................................15
Herstellung von anti-TNP IgG Subtypen (Klon 7B4)...............................................15
Stabile Expression von humanen FcγR in CHO Zellen ..........................................16
Bindung von Immunkomplexen an FcγR ................................................................17
Einfluss der N297 Glykosylierung auf die FcγR-Bindung .......................................20
II. Untersuchung von Antikörpereffektorfunktionen im humanisierten Mausmodell........24
1. Herstellung von anti-CD20 IgG Subtypen (Klon 1F5).............................................24
2. Eigenschaften von Rag2/γc/Fcγ-/- Mäusen ..............................................................25
3. Aufreinigung und Analyse von humanen hämatopoetischen Stammzellen............27
4. Genotypisierung der FcγR Polymorphismen ..........................................................28
5. Rekonstitution von Rag2/γc/Fcγ-/- Mäusen mit humanen HSC ................................30
6. Charakterisierung der humanisierten Rag2/γc/Fcγ-/- Mäuse.....................................32
6.1) Leukozytenpopulationen im humanen Immunsystem .........................................33
6.2) Leukozytenpopulationen in der humanisierten Maus ..........................................34
6.3) Expression humaner FcγR in humanisierten Rag2/γc/Fcγ-/- Mäusen ...................36
6.4) CD20 Expression in humanisierten Rag2/γc/Fcγ-/- Mäusen .................................38
7. IgG-vermittelte B-Zell-Depletion in humanisierten Mäusen ....................................40
7.1) Fc-Abhängigkeit der 1F5 IgG induzierten B-Zell-Depletion..................................40
7.2) B-Zell-Depletion in Blut, Milz und Knochenmark .................................................41
7.3) Depletion reifer B-Zell-Populationen Milz und Knochenmark ..............................43
7.4) 1F5 IgG vermittelte B-Zell-Depletion im Blut .......................................................45
7.5) Einfluss der FcγRIIA131H/R und FcγRIIIA158F/V Polymorphismen............................47
8. anti-CD20 IgG vermittelte Regulation von FcγR .....................................................48
1.
2.
3.
4.
E. Diskussion........................................................................................51
I.
II.
III.
IV.
In vitro Bindung von Immunkomlexen an humane FcγR ............................................51
Etablierung eines humanisierten Mausmodells ..........................................................53
Anti-CD20 IgG vermittelte B-Zell-Depletion im humanisierten Mausmodell ...............56
Einfluss der FcγR Polymorphismen in vivo.................................................................59
Inhalt
F. Material und Methoden ....................................................................63
I.
Material.......................................................................................................................63
1. Chemikalien, Plastik- und Verbrauchsmaterialen ...................................................63
2. Puffer und Lösungen ..............................................................................................63
3. Kommerzielle Kits ...................................................................................................63
4. Antikörper und Konjugate .......................................................................................64
5. Oligonukleotide.......................................................................................................65
6. Software .................................................................................................................65
II. Methoden....................................................................................................................66
1. Klonierung der 7B4 und 1F5 IgG Antikörper...........................................................66
1.1) Klonierung der variablen Regionen .....................................................................66
1.2) Klonierung der schweren und leichten Ketten.....................................................66
1.3) Herstellung von chemisch kompetenten E.coli TOP10 Bakterien .......................67
1.4) Transformation von E.coli TOP10 Bakterien .......................................................68
1.5) Isolation von Plasmid DNA aus Bakterien...........................................................68
1.6) Sequenzierung ....................................................................................................68
2. Produktion der 7B4 und 1F5 IgG Antikörper...........................................................69
2.1) Transiente Transfektion von HEK293T Zellen ....................................................69
2.2) Aufreinigung der Antikörper.................................................................................69
2.3) Entfernung von LPS-Kontaminationen ................................................................70
3. Proteinmodifikationen .............................................................................................70
3.1) Biotinylierung.......................................................................................................70
3.2) Konjugation von FITC-Fluorochromen ................................................................70
3.3) Konjugation von Alexa647-Fluorochromen .........................................................70
3.4) Herstellung von F(ab)2 Fragmenten ....................................................................70
4. In vitro Analyse der 7B4 IgG Bindung an FcγR ......................................................71
4.1) Affinität der 7B4 IgG für TNP...............................................................................71
4.2) FcγR Expression in stabil transfizierten CHO Zellen ...........................................71
4.3) Anreicherung FcγR exprimierender CHO Zellen .................................................72
4.4) Immunkomplexbindung an FcγR exprimierende CHO Zellen..............................72
4.5) Einfluss der Glykosyslierung auf die Immunkomplexbindung .............................72
5. In vitro Analyse der Bindung von 1F5 IgG an LCL1.11 Zellen................................73
6. In vitro Analyse der C1q Bindung durch 1F5 IgG ...................................................73
7. Humane hämatopoetische Stammzellen ................................................................74
7.1) Aufreinigung humaner hämatopoetischer Stammzellen......................................74
7.2) Durchflusszytometrische Analyse von Nabelschnurblut und HSC ......................74
8. Genotypisierung der FcγR Allele ............................................................................75
8.1) Isolation von genomischer DNA aus humanem Nabelschnurblut .......................75
8.2) Genotypisierung mittels allelspezifischer PCR....................................................75
9. Rekonstitution von humanisierten Rag2/γc/Fcγ-/- Mäusen.......................................76
10. Probengewinnung und durchflusszytometrische Analysen ....................................76
10.1) Präparation von Maus-Leukozyten aus dem Blut..............................................76
10.2) Präparation von humanen Leukozyten aus dem Blut........................................77
10.3) Präparation von Leukozyten aus Milz und Knochenmark .................................77
10.4) Durchflusszytometrische Färbung und Analyse ................................................77
11. Immunfluoreszenz-Analyse der humanisierten Mäuse...........................................78
12. 1F5 IgG vermittelte B-Zell-Depletion ......................................................................78
Inhalt
13. Zellkultur .................................................................................................................79
14. Versuchstierhaltung................................................................................................79
15. Statistik ...................................................................................................................79
G. Literaturverzeichnis ........................................................................81
H. Anhang .............................................................................................89
I.
Sequenzen und Vektorkarten .....................................................................................89
1. Sequenzen der klonierten Antikörper: Schwere Ketten ..........................................89
2. Sequenzen der klonierten Antikörper: Leichte Ketten ............................................91
3. Vektorkarten ...........................................................................................................92
II. Abkürzungsverzeichnis...............................................................................................93
III. Eigene Publikationen..................................................................................................96
1. Zeitschriftenartikel ..................................................................................................96
2. Posterpräsentation und Vorträge............................................................................96
IV. Lebenslauf ..................................................................................................................97
I. Danksagung.......................................................................................98
Zusammenfassung
A. Zusammenfassung
I. Zusammenfassung
Immunglobulin G (IgG) Antikörper sind ein essentieller Bestandteil des adaptiven
Immunsystems.
vollständigen
Genetische Defekte, die zu einer verminderten Bildung bzw. dem
Fehlen dieses
Antikörper-Isotyps
führen,
gehen
mit
einer
erhöhten
Infektionsanfälligkeit einher. Eine Vielzahl von Studien in der Maus deuten darauf hin, dass
insbesondere Fcγ Rezeptoren (FcγR), die auf fast allen Zellen des angeborenen
Immunsystems exprimiert werden, eine wichtige Rolle für die Aktivität von IgG Antikörpern
spielen. In der vorliegenden Arbeit sollte daher untersucht werden, ob die Aktivität von
humanen IgG Antikörpern von ähnlichen Faktoren in vitro und in vivo beeinflusst wird.
Durch in vitro Bindungsstudien von humanen Immunkomplexen an humane FcγR konnte im
ersten Teil dieser Arbeit ein Zusammenhang zwischen der Größe von Immunkomplexen und
der Bindung an FcγRIIA, FcγRIIB und FcγRIIIA, nicht aber an FcγRIA, gezeigt werden.
Weiterhin konnte nachgewiesen werden, dass Immunkomplexe ohne die Glykosylierung am
Asparagin 297 eine verminderte Bindung an die niedrigaffinen FcγR aufweisen. Große
Immunkomplexe konnten die durch Deglykosylierung bedingte reduzierte Affinität teilweise
kompensieren. Einen weiteren möglichen Einflussfaktor auf die IgG-FcγR Interaktion stellen
Polymorphismen in den humanen FcγR dar. In dieser Arbeit wurden genetische Varianten
der aktivierenden FcγR, FcγRIIA131H/R und FcγRIIIA158F/V, untersucht. In in vitro Analysen wies
FcγRIIA131H für alle humanen IgG Isotypen eine erhöhte Affinität auf.
Für die in vivo Analyse IgG-vermittelter Effektorfunktionen wurde ein humanisiertes
Mausmodell etabliert, in dem die genetische Komplexität des humanen Immunsystems
abgebildet wird. Hierfür wurden immundefiziente Rag2/γc/Fcγ-/- Mäuse verwendet, die nach
Transplantation mit humanen hämatopoetischen Stammzellen ein humanes Immunsystem
mit B- und T-Lymphozyten sowie NK-Zellen, Monozyten und dendritischen Zellen
entwickelten. Weiterhin zeigten die humanen Effektorzellen, wie etwa Monozyten und NKZellen, ein dem Menschen vergleichbares Expressionsmuster der humanen FcγR. In diesem
neuen humanisierten Mausmodell wurde der Zusammenhang zwischen den FcγRIIA131H/R
und FcγRIIIA158F/V Polymorphismen und der biologischen Wirksamkeit von IgG beispielhaft
durch die Induktion der Antikörper-induzierten Zell-vermittelten Zytotoxizität (ADCC)
analysiert. Es konnte gezeigt werden, dass die durch Injektion von anti-CD20 IgG
verursachte Depletion von humanen B-Zellen vom konstanten Teil des IgG Antikörpers
abhängt. Zudem zeigten die verschiedenen IgG Subklassen eine unterschiedliche Aktivität
und es konnten erste Hinweise gewonnen werden, die vermuten lassen, dass die IgG2vermittelte Depletion in Gegenwart der hochaffinen Varianten der aktivierenden Fc
Rezeptoren FcγRIIA131H bzw. FcγRIIIA158V verstärkt war.
1
Zusammenfassung
II. Summary
Immunoglobulin G (IgG) antibodies are an essential part of the adaptive immune system.
Genetic defects leading to a reduced production or a complete lack of this antibody isotype
are associated with an increased susceptibility to infectious diseases. Studies of the murine
immune system suggest that the biological activity of IgG is mainly mediated by their binding
to Fcγ receptors (FcγR) expressed on almost all cells of the innate immune system. The aim
of the thesis was to analyse which factors influence the biological activity of IgG in vitro and
in vivo.
We performed in vitro interaction studies of human immune complexes and human FcγR
which suggested a correlation of immune complex size and binding to FcγRIIA, FcγRIIB and
FcγRIIIA, but not FcγRIA. In addition, we could show that a lack of glycosylation on
asparagine 297 impairs binding to low affinity FcγR. However, this reduced binding could be
overcome by increasing the size of the immune complexes. Polymorphisms of the human
FcγR pose another factor influencing the interaction with IgG antibodies. To study this, we
analysed genetic variants of the activating FcγR, FcγRIIA131H/R and FcγRIIIA158F/V. In vitro
binding studies showed an enhanced affinity of FcγRIIA131H for all human IgG subclasses.
To study IgG induced effector functions in vivo we developed a humanized mouse model
which mirrors the genetic complexity of the human immune system. Immunodeficient
Rag2/γc/Fcγ-/- mice transplanted with human hematopoetic stem cells developed a human
immune system as shown by the appearance of B and T lymphocytes, NK cells, myeloid
cells and dendritic cells. Effector cells such as monocytes and NK cells displayed expression
levels of human FcγRs comparable to their human counterparts. This new humanized mouse
model was used to study the influence of FcγRIIA131H/R and FcγRIIIA158F/V polymorphisms on
the biological activity of IgG antibodies such as the induction of antibody induced cell
mediated cytotoxicity (ADCC). B cell depletion induced by the injection of anti-CD20 IgG
antibodies greatly depended on the constant region of the antibody. Furthermore, IgG
subclasses showed a distinct in vivo activity. Preliminary data also suggest an enhanced
IgG2 mediated B cell depletion in the presence of FcγRIIA131H and FcγRIIIA158
2
Einleitung
B. Einleitung
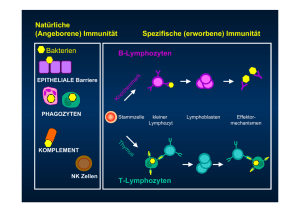
I. Fcγ Rezeptoren
Antikörper, oder Immunglobuline (Ig), sind ein essentieller Bestandteil des adaptiven
Immunsystems zur Abwehr von Viren und Bakterien. Zunehmend werden sie auch zu
therapeutischen Zwecken bei der Behandlung von Entzündungserkrankungen und bei der
Tumortherapie eingesetzt. Die biologische Wirksamkeit von Antikörpern beruht, neben durch
die Antigenbindung ausgelösten Effekten, vor allem auf dem Fc Fragment. Aufgrund dieser
konstanten Domänen können die Immunglobuline in fünf verschiedene Klassen unterteilt
werden: IgM, IgG, IgD, IgE und IgA. Für IgA und IgG existieren jeweils noch weitere
Subtypen. In Abb. 1 sind die vier Varianten von IgG (IgG1-4) dargestellt, die sich durch eine
starke Konservierung ihrer Sequenzen und eine vergleichbare Proteinstruktur auszeichnen.
Dennoch unterscheiden sie sich in Bezug auf die Induktion von Fc-abhängigen
Antikörpereffektorfunktionen. So können IgG1, IgG3 und in vermindertem Maße auch IgG2
beispielsweise an das Protein C1q binden und damit den klassischen Weg der
Komplementkaskade aktivieren. Der Großteil der Fc-abhängigen Effekte wird jedoch über die
Interaktion mit Fc Rezeptoren induziert. Besonders IgG1, IgG2 und IgG3, weniger aber IgG4,
sind in der Lage mit den Fc Rezeptoren für IgG zu interagieren – den FcγR [2].
Abb. 1: Im Menschen existieren
4 Subtypen von Immunglobulin
G (IgG) Antikörpern. Diese
unterscheiden sich in der
Induktion
von
Effektorfunktionen. IgG1, IgG2 und
IgG3 binden an C1q und
aktivieren so den klassischen
Weg der Komplementkaskade.
Vor allem IgG1, IgG2 und IgG3
binden auch an FcγR.
1.
Murine und humane Fcγ Rezeptoren
Als Mitglieder der Immunglobulin-Superfamilie sind FcγR durch extrazelluläre IgG-bindende
Domänen charakterisiert [3]. Die murinen FcγR werden auf vielen Zellen des adaptiven und
angeborenen Immunsystems wie z.B. Monozyten/Makrophagen [4-6], NK-Zellen [7], BLymphozyten [8] und Granulozyten exprimiert [9]. Wie in Abb. 2A dargestellt, werden in der
Maus 4 Klassen von FcγR unterschieden, deren Gene auf Chromosom 1 kodiert sind [10]:
die aktivierenden FcγRI (CD64) [4], FcγRIII (CD16) [3] und FcγRIV [11] und der inhibitorische
FcγRIIB (CD32) [3]. Der auf Makrophagen exprimierte FcγRI bindet aufgrund von drei
3
Einleitung
extrazellulären Domänen mit hoher Affinität an monomere IgG. Dagegen besitzen die
niedrigaffinen FcγRIIB, FcγRIII und FcγRIV zwei extrazelluläre Domänen und interagieren mit
IgG nur in aggregierter Form oder in Form von Immunkomplexen bestehend aus Antigen und
Antikörpern [12, 13]. Ein weiteres wichtiges Merkmal der murinen FcγR ist ihre
unterschiedliche Affinität für die verschiedenen IgG Subklassen in vitro [11] und in vivo [1416].
Abb. 2: Murine und humane FcγR. A) Darstellung der 4 Klassen muriner FcγR. Die aktivierenden
FcγR sind mit der akzessorischen Fcγ Kette assoziiert, die intrazellulär über eine ITAM Domäne
verfügt. Der inhibitorische FcγRIIB ist durch eine intrazelluläre ITIM Sequenz charakterisiert.
B) Darstellung der humanen FcγR. Analog zu den murinen FcγR, sind die aktivierenden FcγRIA und
FcγRIIIA mit der ITAM-tragenden Fcγ Kette assoziiert. Beim aktivierenden FcγRIIA wird die ITAM- und
beim inhibitorischen FcγRIIB die ITIM-Sequenz direkt durch die α Kette kodiert. Die in dieser Arbeit
näher untersuchten Polymorphismen der aktivierenden FcγRIIA und FcγRIIIA sind markiert.
Während die Interaktion von FcγR und IgG durch die extrazellulären IgG-bindenden
Domänen der α Kette vermittelt wird, erfolgt die Signaltransduktion der Rezeptoren durch
intrazelluläre Signalmotive. Das immunoreceptor tyrosine based inhibitory motif (ITIM) des
FcγRIIB befindet sich in der intrazellulären Domäne der α Kette [17]. Anstelle eigener
Signalmotive sind die aktivierenden FcγR der Maus durch ihre Transmembranregionen mit
4
Einleitung
der Fcγ Kette assoziiert [11, 18, 19]. Das intrazelluläre immunoreceptor tyrosine based
activation motif (ITAM) der Fcγ Kette ist für die Signaltransduktion der aktivierenden FcγR
nach IgG-Bindung verantwortlich [20]. Die Assoziation mit der Fcγ Kette erhöht nicht nur die
Affinität der Rezeptoren für IgG [21] sondern ist notwendig für die Expression der
Rezeptoren auf der Zelloberfläche. Fehlt die Fcγ Kette in Immunzellen, so kommt es zum
proteolytischen Abbau der α Ketten [22]. In Fcγ-/- Mäusen kommt es daher zu einer
Reduktion FcγR-induzierter Effektorfunktionen wie der Phagozytose von Immunkomplexen
durch Makrophagen und der Antikörper-induzierten Zell-vermittelten Zytotoxizität (ADCC).
Die Expression von FcγRIIB auf B-Zellen ist dagegen nicht beeinträchtigt [18].
Rezeptoren
für
IgG
finden
sich
auch
auf
den
Zelloberflächen
von
humanen
Monozyten/Makrophagen [23], B-Zellen [24], Granulozyten [25], NK-Zellen [26] und
Thrombozyten [27]. Im Vergleich zur Maus, bei der 4 FcγR bekannt sind, existieren im
humanen Immunsystem 3 Klassen von FcγR, die sich weiter in verschiedene Subtypen
aufteilen lassen. Diese Vielfalt kommt durch Duplikation und Rekombination der Gene [28]
auf Chromosom 1 zustande [29-31]. Die FcγRI-Familie (CD64) besteht aus 3 Genen [32]. Es
wird jedoch nur der hochaffine FcγRIA als funktionelles Produkt exprimiert [33]. Mehrere
Isoformen existieren auch für den niedrigaffinen FcγRII (CD32) [34, 35]: Bei FcγRIIA handelt
es sich um einen aktivierenden FcγR, der in der intrazellulären Domäne über ein ITAM für die
Signaltransduktion verfügt. Analog zur Maus ist der inhibitorische FcγRIIB durch ein
intrazelluläres ITIM charakterisiert [36]. Die beiden Isoformen des niedrigaffinen FcγRIII
(CD16) unterscheiden sich vor allem in ihrer Verankerung in der Zellmembran. Beim
aktivierenden FcγRIIIA handelt es sich um ein Transmembranprotein [37], wogegen FcγRIIIB
über Glykosylphosphatidylinositol (GPI) an die Zellmembran assoziiert ist [37]. Analog zu
den murinen FcγR, sind die humanen aktivierenden FcγRIA und FcγRIIIA mit der Fcγ
Signalkette assoziiert. Diese Interaktion ist sowohl für die Oberflächenexpression als auch
für die Funktionalität der Rezeptoren essentiell [38-40] (Abb. 2B).
2.
Induktion von Antikörpereffektorfunktionen
Antikörpereffektorfunktionen werden durch die Bindung von IgG-Immunkomplexen an FcγR
induziert. Diese Interaktion führt zu einer Kreuzvernetzung der FcγR, gefolgt von der
Rekrutierung in lipid rafts. Bei der Bindung an aktivierende FcγR folgt die Phosphorylierung
des intrazellulären ITAM [41]. Die dadurch ausgelöste Signaltransduktionskaskade führt
letztlich zur Freisetzung von Kalzium [42-44]. Je nach betroffener Effektorzelle kommt es zur
Phagozytose des Immunkomplexes, der Präsentation des aufgenommenen Antigens durch
MHCII-Komplexe, zur Freisetzung von Zytokinen, zur Proliferation oder Reifung der
betroffenen Zelle oder zur Antikörper-induzierten Zell-vermittelten Zytotoxizität (ADCC) [6,
5
Einleitung
45, 46]. Bei Kreuzvernetzung des inhibitorischen FcγRIIB kommt es dagegen zur
Phosphorylierung des ITIM. Dies induziert eine Signalkaskade, die die Signale der
aktivierenden FcγR hemmt [47] (Abb. 3). Die meisten Immunzellen, wie z.B. Makrophagen,
exprimieren sowohl aktivierende wie auch inhibitorische FcγR, wohingegen B-Zellen nur den
inhibitorischen FcγRIIB und NK-Zellen nur den aktivierenden FcγRIII besitzen. Die
Koexpression
aktivierender
und
inhibitorischer
Rezeptoren
beeinflusst
maßgeblich
Antikörper-induzierte Effekte wie z.B. die Zerstörung von Tumorzellen durch ADCC in vivo
[48] oder die Hemmung der Antikörperproduktion durch negative Rückkoppelung [49]. Ein
Fehlen
von
inhibitorischen
Signalen
führt
weiterhin
zur
Entstehung
von
Autoimmunerkrankungen [50]. Daher ist eine Balance zwischen aktivierenden und
hemmenden FcγR-Signalen essentiell für die Regulation der Immunantwort und die
Erhaltung immunologischer Toleranz.
Abb. 3: Die Kreuzvernetzung von
FcγR durch IgG-Immunkomplexe
induziert Antikörpereffektorfunktionen in Abhängigkeit von der
Effektorzelle. Die Koexpression
und
Kreuzvernetzung
des
inhibitorischen FcγRIIB dämpft
die
aktivierende
ITAMSignaltransduktion und bestimmt
damit die Schwelle für die
Immunzellaktivierung.
3.
Einfluss der IgG Glykosylierung auf die FcγR Interaktion
Einen wesentlichen Einfluss auf die Interaktion von IgG mit FcγR hat die N-Glykosylierung
am Asparagin 297 (N297) in der CH2 Domäne der schweren Ketten. Änderungen an ihrer
Zusammensetzung wirken sich auf die Konformation des Fc Fragments und damit auf seine
Funktionalität aus [51]. Dies bestätigen Analysen bei denen durch Behandlung von IgG
Molekülen
mit
verschiedenen
Glykosidasen
die
Zuckerketten
entfernt
werden.
Beispielsweise hydrolysiert die Peptid:N Glykosidase F (PNGaseF) aus Flavobacterium
meningosepticum die Verknüpfung der Zuckerkette mit Asparagin-Resten wie dem N297
[52]. Alternativ schneidet die Endoglykosidase S (EndoS) aus Streptococcus pyogenes nach
N-Acetyl-Glucosamin-Resten innerhalb der Zuckerketten [53] (Abb. 4).
In mehreren Studien wurde gezeigt, dass die N297-Glykosylierung die Affinität für die FcγR
in vitro und die Wirkung von IgG Molekülen in vivo bestimmt. So führt die Deglykosyslierung
6
Einleitung
von IgG Antikörpern mit EndoS zu einer reduzierten Bindung an murine und humane FcγR in
vitro [54, 55]. Die EndoS-Behandlung verursacht außerdem eine Dissoziation der IgG-FcγR
Interaktion
[1,
55].
In
verschiedenen
experimentellen
Modellen
IgG-vermittelter
Autoimmunität wurde durch die Deglykosylierung der IgG mit EndoS eine Besserung der
klinischen Parameter in vivo erreicht [1, 54]. Eine verminderte Interaktion des humanen IgG1
mit humanen FcγR wurde auch erzielt, indem das N297 durch ein Alanin ersetzt wurde [56].
Abb. 4: Variante der IgG Glykosylierung an Aminosäure Asparagin 297 der schweren Ketten.
(adaptiert nach [1]). Eine Behandlung der IgG mit den Enzymen EndoS bzw. PNGaseF trunkiert die
Glykosylierung an den markierten Positionen. (NANA, N-Acetyl-Neuraminsäure; Gal, Galaktose;
GlcNAc , N-Acetyl-Glucosamin; Man, Mannose; Fuc, Fucose)
4.
Polymorphismen von humanen FcγR
Die in vivo Wirkung von Antikörpern wird nicht nur durch unterschiedliche Affinitäten der
FcγR für die IgG Subtypen oder durch das Expressionsniveau der aktivierenden und
inhibitorischen FcγR bestimmt. Polymorphismen in den kodierenden Genen beeinflussen
ebenfalls die Funktionalität der FcγR. So sind für das FcγRIIB-Gen zwei Polymorphismen
bekannt. Ein Polymorphismus im Promotor verursacht die verminderte Transkription des
Rezeptors [57]. Die Rekrutierung des kreuzvernetzten FcγRIIB in lipid rafts wird dagegen
durch eine Mutation in der Transmembranregion verhindert [58]. Beide Polymorphismen
führen zu einer verminderten FcγRIIB-Signaltransduktion.
In den Genen der aktivierenden FcγRIIA und FcγRIIIA sind ebenfalls Polymorphismen
publiziert. Für den humanen aktivierenden FcγRIIA ist eine Punktmutation von Guanin zu
Alanin in der zweiten IgG-bindenden extrazellulären Domäne beschrieben. Diese Mutation
führt zu einem Aminosäureaustausch von Arginin zu Histidin an Position 131 [59]. Aufgrund
ihrer unterschiedlichen Affinität für IgG1 Antikörper der Maus werden die beiden Varianten
als low responder (LR, FcγRIIA131H), die nur schwach mit den Maus-IgG interagieren, bzw.
high responder (HR, FcγRIIA131R), die dagegen eine hohe Affinität für die Maus-IgG
aufweisen, bezeichnet [60]. Die so beschriebenen polymorphen FcγRIIA unterscheiden sich
aber zusätzlich in einem weiteren Aminosäureaustausch von Glutamin zu Tryptophan an
Position 27 [61]. Um die Bindung der humanen IgG in Abhängigkeit des FcγRIIA131H/R
7
Einleitung
Polymorphismus zu untersuchen wurden surface plasmon resonance (SPR) Affinitätsstudien
durchgeführt. Der FcγRIIA131H/R Polymorphismus scheint keinen Einfluss auf die Affinität für
humanes IgG3 und IgG4 zu haben. Allerdings binden humane IgG1 und IgG2 Antikörper
stärker an die FcγRIIA131H-Variante. Zudem interagiert dieser hochaffine FcγRIIA131H verstärkt
mit therapeutischen anti-CD20 Antikörpern [62].
In der Sequenz des humanen aktivierenden FcγRIIIA verursacht die Punktmutation von
Thymin zu Guanin in der zweiten extrazellulären Domäne einen Aminosäureaustausch von
Phenylalanin zu Valin an Position 158. In vitro Experimente zeigen eine verbesserte
Interaktion von FcγRIIIA158V mit IgG1 und IgG3. Dies führt zur Freisetzung von intrazellulärem
Kalzium, einer verstärkten Aktivierung von NK-Zellen und einer Induktion der ADCC. Im
Gegensatz zu FcγRIIIA158F, ist FcγRIIIA158V in der Lage mit IgG4 zu interagieren [63-65]. Die
Bestimmung der Bindungskonstanten zeigt eine erhöhte Affinität von FcγRIIIA158V für alle
humanen IgG Isotypen [62]. Im Gegensatz dazu ist die Beseitigung IgG3-gebundener
Erythrozyten durch FcγRIIIA158V exprimierende Monozyten in vivo vermindert [66].
Die beschriebenen funktionellen Polymorphismen der aktivierenden FcγRIIA und FcγRIIIA
wirken sich auf die in vitro Bindung von IgG aus. Um ihren Einfluss auf Antikörper-vermittelte
Effekte wie z.B. die IgG-induzierte Immunabwehr von Infektionen, der therapeutischen
Anwendung von IgG oder der Entstehung IgG-induzierter Autoimmunerkrankungen in vivo zu
ermitteln, wurden epidemiologische Studien durchgeführt. In einer Studie ist die niedrigaffine
Variante
des
FcγRIIA131R
mit
einer
größeren
Empfindlichkeit
gegen
bakterielle
Atemwegsinfektionen assoziiert, da IgG2-opsonisierte Bakterien schlechter phagozytiert
werden können [67]. Die Wirkung monoklonaler Antikörper z.B. des anti-CD20 IgG1
Rituximab zur Therapie des follikulären Non-Hodgkin Lymphoms wird positiv durch
FcγRIIIA158V beeinflusst [68]. Eine mögliche Erklärung hierfür liefern in vitro Analysen, bei
denen Rituximab mit höherer Affinität an FcγRIIIA158V bindet. Die Zahl FcγRIIIA
exprimierender Effektorzellen in vivo wird nicht durch den Polymorphismus beeinflusst [69].
Weiterhin scheint ein erhöhtes FcγRIII-Expressionsniveau bei FcγRIIIA158V in vivo die
verbesserte Bindung von Rituximab und als Folge dessen eine verstärkte ADCC zu
verursachen [70]. Auch die biologische Wirksamkeit anderer therapeutischer Antikörper wie
z.B. des anti-epidermal growth factor receptor IgG1 Cetuximab wird von Polymorphismen der
aktivierenden FcγR beeinflusst. Während sich die hochaffine Variante von FcγRIIA131H positiv
auf den Therapieerfolg auswirkt, hat der hochaffine FcγRIIIA158V einen negativen Einfluss
[71]. Auch die Anwendung des anti-TNFα IgG1 Infliximab führt bei Expression von
FcγRIIIA158F zu einem besseren therapeutischen Verlauf [72]. Bei anderen Erkrankungen
oder therapeutischen Antikörpern scheinen die FcγR Polymorphismen dagegen keinen
Einfluss zu haben [73, 74].
8
Einleitung
Neben ihren Effekten auf die Immunabwehr und auf monoklonale Antikörpertherapien sind
die beiden Polymorphismen in unterschiedlichen epidemiologischen Studien außerdem mit
der
Entstehung
und
dem
Schweregrad
von
Autoimmun-
und
anderen
Entzündungserkrankungen assoziiert. Ein Beispiel hierfür ist die Rheumatoide Arthritis (RA),
eine systemische Autoimmunerkrankung, die durch die entzündliche Schwellung von
Gelenken
gekennzeichnet
ist.
In
starker
Abhängigkeit
von
der
untersuchten
Bevölkerungsgruppe ist entweder das niedrigaffine [75] oder das hochaffine Allel [76, 77] des
FcγIIIA mit dem Erkrankungsrisiko für RA assoziiert. Der FcγRIIA131H/R Polymorphismus
scheint dagegen nicht für das Auftreten [78] sondern eher für den Krankheitsverlauf der RA
bestimmend zu sein [79] auch wenn das gemeinsame Auftreten von FcγRIIA131H und
FcγRIIIA158V das Risiko an RA zu erkranken erhöht [80]. Sowohl FcγRIIA131R [79] als auch
FcγRIIIA158V [81] exprimierende Patienten zeigen einen deutlich schwereren Verlauf der
Erkrankung.
Eine
weitere
IgG-vermittelte
Autoimmunerkrankung
ist
der
Systemische
Lupus
Erythematodes (SLE). Eine mögliche Ursache von SLE scheinen Defekte bei der
Makrophagen-vermittelten Phagozytose zu sein. Dadurch kommt es zur Ablagerung von
Immunkomplexen im Gewebe, die die Einwanderung von Immunzellen und die Freisetzung
von Entzündungsmediatoren verursachen [82]. In der europäischen Bevölkerung besteht ein
Zusammenhang zwischen dem Auftreten von SLE und der FcγRIIIA158F-Variante [83].
FcγRIIA131R scheint dagegen ein Risikofaktor für die Entstehung einer Lupus-Nephritis oder
der Produktion von Autoantikörpern zu sein [84, 85]. Auch der niedrigaffine FcγRIIIA158F wird
mit einem schlechteren klinischen Phenotyp der SLE in Verbindung gebracht [86].
Bei der Untersuchung anderer IgG-vermittelter Erkrankungen zeigt die niedrigaffine FcγRIIA
Variante einen negativen Einfluss bei der Heparin-induzierten Thrombozytopenie [87], bei
der Entwicklung von Myasthenia gravis [88] und Riesenzell-Arterititis [78]. Außerdem
verstärken FcγRIIA131R und FcγRIIIA158V den Schweregrad der IgA Nephropathie [89].
Auch
wenn
die
Daten
auf
eine
verstärkte
Bindung
von
monomeren
IgG
und
Immunkomplexen an die hochaffinen Rezeptorvarianten hindeuten, lassen die zum Teil
widersprüchlichen Ergebnisse epidemiologischer Studien keinen Schluss auf die tatsächliche
Bedeutung der FcγR Polymorphismen für Antikörper-vermittelte Effekte in vivo zu.
9
Einleitung
II. Untersuchungen in humanisierten Mausmodellen
Humanisierte Mausmodelle dienen der Untersuchung von komplexen Fragestellungen, die in
vitro oder in klassischen Mausmodellen nur unzureichend beantwortet werden können.
Beispiele hierfür sind die Untersuchungen zur Entwicklung und Funktionsweise des
menschlichen Immunsystems oder die Pathogenese von Infektions- und anderen humanen
Erkrankungen. Eine Humanisierung wird durch die Ausstattung der Mäuse mit humanen
Zellen oder Genen erreicht, wobei in dieser Arbeit der Fokus auf zellulär humanisierten
Mausmodellen liegen soll. In den letzten Jahren konnten verschiedene für entsprechende
Fragestellungen optimierte Modelle entwickelt werden.
1.
Generierung humanisierter Mausmodelle
Die Grundlage für die Etablierung humanisierter Mausmodelle war die Entdeckung der
severe combined immunodeficiency (scid) Mutation in Mäusen [90]. Diese Mutation der
DNA-abhängigen
Proteinkinase
p350
führt
zu
einer
Blockade
der
Lymphozyten-
Differenzierung und somit zu einer ausgeprägten Immundefizienz. Die Ursache hierfür liegt
in der essentiellen Rolle von p350 bei der Reparatur von DNA-Doppelstrangbrüchen wie sie
z.B. bei der Rekombination der B- und T-Zell-Rezeptoren vorkommen [90-92]. Nach
Übertragung von isolierten hämatopoetischen Zellen in scid Mäuse kommt es zunächst zur
Proliferation und Differenzierung humaner Immunzellen in vivo [93, 94]. Eine länger
anhaltende Repopulierung des murinen Knochenmarks wird dagegen durch den Transfer
von Zellen aus Nabelschnurblut erreicht [95]. Die Weiterentwicklung des scid Mausmodells
erfolgte durch die Übertragung der Mutation auf diabetis-susceptible nonobese diabetic
(NOD) Mäuse. Der resultierende Stamm zeichnet sich durch eine reduzierte Aktivität von
NK-Zellen, Makrophagen und des Komplementsystems aus [96]. Rekonstituierte NOD/scid
Mäuse zeigen einen 5-10fachen Anstieg des Anteils humaner Zellen in der Milz [97].
Allerdings weisen diese Mäuse auch eine erhöhte Inzidenz von Lymphomen auf [98].
Zusätzlich ist ihre Verwendung in der biomedizinischen Forschung vor allem durch die
sporadische und verzögerte Entwicklung von funktionellen Lymphozyten und der
vorhandenen Restaktivität des angeborenen Immunsystems limitiert [96]. Der negative
Einfluss von NK-Zellen auf den adoptiven Transfer humaner Zellen führte zur zusätzlichen
Deletion des β2 Mikroglobulin Gens [99] bzw. der gemeinsamen Signalkette der Rezeptoren
für Interleukin (IL)-2, IL-4, IL-7, IL-9 und IL-15. Diese sog. γc Kette ist für die NK-ZellEntwicklung essentiell [70, 100]. Die so erzeugten Mausstämme zeigen nicht nur eine
verbesserte Lebenserwartung. Die transferierten humanen Zellen differenzieren auch in alle
hämatopoetischen Linien [101]. Als Alternative zu NOD/scid/γc-/- Mäusen wurde ein weiterer
immundefizienter Mausstamm generiert, der sich durch das vollständige Fehlen aller
10
Einleitung
Lymphozyten - B-, T- und NK-Zellen - auszeichnet. Verursacht wird dieser Phänotyp durch
das Fehlen des recombinase activating gene 2 (RAG2) und die zusätzliche Deletion der IL2R
γc Kette. Durch Verwendung von Rag2/γc-/- Mäusen konnte die Humanisierungseffizienz im
Vergleich zu NOD/scid Mäusen ebenfalls gesteigert werden [102].
Neben dem verwendeten Mausstamm gibt es weitere Faktoren, die das Anwachsen der
humanen Immunzellen beeinflussen. Die Effizienz wird z.B. durch die Art der transplantierten
Zellen bestimmt. Humanisierte Mäuse lassen sich mit hämatopoetischen Zellen aus dem
Knochenmark [103], aus der fötalen Leber, Thymus bzw. Lymphknoten [93] oder aus dem
peripheren Blut [94] rekonstituieren. Die besten Ergebnisse werden jedoch durch Injektion
von CD34+ hämatopoetischen Stammzellen (HSC) aus Nabelschnurblut erzielt [104]. Die
CD34+ Zellen nisten sich dauerhaft im murinen Knochenmark ein [95], das daraufhin zum
primären Ort der humanen Hämatopoese wird. Reife Zellen wandern schließlich in die
Peripherie aus [105]. Der Transfer von CD34+ Zellen führt zu einer dem menschlichen
Immunsystem entsprechenden Entwicklung funktioneller Lymphozyten, dendritischer Zellen,
Monozyten, Erythrozyten und Thrombozyten [101, 104, 106]. Weiterhin kann die
Humanisierungseffizienz durch Transplantation neugeborener, anstelle adulter Mäuse [104,
107] und die Bestrahlung der Mäuse [95, 105] vor der Zellinjektion gesteigert werden. Auch
die Art der Injektion beeinflusst die Humanisierung. Die Zellen können intravenös [96, 102],
intrafemural [108] oder intraperitoneal [109] in adulte Mäuse transferiert werden. Zur
Differenzierung humaner Immunzellen kommt es auch nach der in uteri Applikation der
humanen Zellen [110]. Die Rekonstitution neugeborener Mäuse kann durch intrahepatische
[107] oder intravenöse Injektion in die Gesichtsvene [104] erreicht werden. Letzteres führt zu
einem besonders effizienten Anwachsen des humanen Immunsystems.
2.
Humanisierte
Forschung
Mausmodelle
in
der
biomedizinischen
Humanisierte Mausmodelle in ihren verschiedenen Formen werden in vielen Gebieten der
biomedizinischen Forschung angewendet. Mit ihrer Hilfe können die Entwicklung des
Immunsystems, die Pathogenese von Infektions- und anderen Erkrankungen oder auch die
Wirkung von potentiellen Therapeutika in vivo untersucht werden.
Besonders bei der Untersuchung der Pathogenese humanspezifischer Krankheitserreger wie
beispielsweise dem Dengue-Virus [111] oder Humanen Immundefizienz Virus (HIV) [112]
haben Mausmodelle, die mit einem humanen Immunsystem ausgestattet sind, eine große
Bedeutung. Auch die Entwicklung von Therapeutika z.B. gegen Mycobacterium tuberculosis
[113] und HIV [114] wird durch humanisierte Mausmodelle vereinfacht. Humanisierte Mäuse
eignen sich auch für die Analyse der Wirkmechanismen und potentieller Nebenwirkungen
neuer Medikamente wie z.B. therapeutischer Antikörper vor der Anwendung im Menschen
11
Einleitung
[115, 116]. Ein weiteres Forschungsfeld ist die Entwicklung von zellbasierten Therapien für
Typ-1-Diabetis [117] oder Lebererkrankungen [118] und die Weiterentwicklung der
Gentherapie z.B. bei der Fanconi-Anämie [119]. Natürlich können humanisierte Mausmodelle
auch genutzt werden um die Pathogenese von humanen Erkrankungen wie der Spinalen
Muskelatrophie [120], der Rheumatoiden Arthritis [121] oder Typ-1-Diabetis [122] zu
untersuchen. Weiterhin können Abstoßungsreaktionen nach Organtransplantationen, die
sog. graft versus host disease (GVHD), in ihnen gut abgebildet werden [123]. Auch für die
Tumorforschung haben humanisierte Mausmodelle eine große Bedeutung erlangt. So
werden in xenotransplantierten Mäusen der Einfluss von Onkogenen [124] oder anderen
kanzerogenen Faktoren wie ultraviolettem Licht [125] untersucht oder Medikamente auf ihre
Wirksamkeit überprüft [126].
Zusammenfassend werden humanisierte Mausmodelle heute vielseitig angewendet, nicht
nur
um
die
humane
Hämatopoese,
sondern
v.a.
auch
um
human-spezifische
Krankheitsprozesse in vivo zu untersuchen. Hierbei sind sie in vitro Studien und
herkömmlichen Mausmodellen überlegen. Das variable Anwachsen der humanen Zellen
schränkt ihre Anwendung allerdings weiterhin ein. Auch das Fehlen der humanen Umgebung
in Form von Zytokinprofilen und Stromazellen für die Differenzierung in ein vollständiges,
funktionelles Immunsystem und die verbleibende Aktivität der Maus-Immunzellen ist
problematisch. Die Zielstellung dieser Arbeit besteht in der Analyse von Einflussfaktoren auf
die Interaktion von humanen IgG und FcγR in vivo. Diese Untersuchungen werden zusätzlich
durch die Anwesenheit der murinen FcγR-exprimierenden Immunzellen, die ebenfalls mit
humanem IgG interagieren, kompromittiert. Daher ist es essentiell neue humanisierbare
Mausstämme herzustellen, die eine Untersuchung der Mechanismen der Aktivität humaner
IgG Antikörper in vivo erlauben.
12
Fragestellung
C. Fragestellung
In Form von Immunkomplexen binden Antikörper vom Immunglobulin G Isotyp an FcγRezeptoren (FcγR) auf den Zellen des angeborenen und adaptiven Immunsystems. Diese
Interaktion löst Antikörpereffektorfunktionen wie z.B. die Antikörper-induzierte Zell-vermittelte
Zytotoxizität (ADCC), die Phagozytose von Immunkomplexen oder die Freisetzung von
Sauerstoffradikalen
und
Zytokinen
aus.
Aufgrund
von
Versuchsdaten
in
Mausmodellsystemen wird angenommen, dass die Intensität dieser Effektorantworten von
der Stärke der Bindung der IgG Subklassen an verschiedene FcγR abhängig sein könnte. Im
Rahmen dieser Dissertation sollte untersucht werden, ob ähnliche Einflussfaktoren die
Interaktion von humanen IgG Subklassen mit humanen FcγR in vitro und in vivo
beeinflussen. Einen indirekten Hinweis für einen solchen Zusammenhang geben
epidemiologische Studien, die vermuten lassen, dass Polymorphismen der humanen FcγR,
die zu einer veränderten Affinität zu bestimmten IgG Subklassen führen, mit der Aktivität
bestimmter therapeutischer Antikörper korrelieren.
Um diese Fragestellung zu beantworten, sollte im ersten Teil der Arbeit untersucht werden,
ob die Größe und die Glykosylierung von humanen Immunkomplexen einen Einfluss auf die
Interaktion mit humanen FcγR in vitro haben. Darauf aufbauend sollte im zweiten Teil der
Arbeit ein neues Maus-Modellsystem etabliert werden, in dem der Einfluss der
Polymorphismen in den aktivierenden FcγRIIA131H/R und FcγRIIIA158F/V auf Antikörpervermittelte Effektorfunktionen in vivo untersucht werden kann.
13
Ergebnisse
D. Ergebnisse
I. Bindung von Immunkomplexen an FcγR in vitro
Im ersten Teil der Arbeit wurde die Bindung von humanem Immunglobin G (IgG) an die
humanen Fcγ-Rezeptoren (FcγR) untersucht. Insbesondere wurde analysiert, inwiefern die
Größe und die Glykosylierung von Immunkomplexen die FcγR Bindung beeinflussen.
1.
Herstellung von anti-TNP IgG Subtypen (Klon 7B4)
Um die Bindung von humanen IgG an FcγR zu untersuchen, erfolgte zunächst die Klonierung
des gegen das Hapten 2,4,6-Trinitrophenyl (TNP) gerichteten Antikörpers 7B4. Hierzu wurde
aus der Hybridomzelllinie 7B4 die Gesamt-RNA isoliert, in cDNA umgeschrieben und einer
5´-RACE unterzogen. Mit dieser Methode konnten die variablen Regionen der schweren und
leichten Kette, trotz ihrer unbekannten Sequenz, bestimmt werden (Sequenz siehe Anhang).
Die so erhaltenen VH und VL Domänen wurden dann mittels Polymerasekettenreaktion (PCR)
an die konstanten Domänen (CH bzw. CL) der humanen IgG Subklassen IgG1, IgG2, IgG3
und IgG4 fusioniert und in Expressionsvektoren kloniert. Durch transiente Transfektion der
erhaltenen Konstrukte in HEK293T Zellen und die anschließende Aufreinigung aus dem
Zellkulturüberstand mithilfe von Protein G erhielt man die entsprechenden Antikörper.
Die Integrität und Reinheit der produzierten Antikörper wurde mittels reduzierender
Polyacrylamid (PAA)-gelelektrophorese nachgewiesen. Durch Reduktion der Disulfidbrücken
wurden die zwei schweren Ketten mit einer Größe von ca. 55kDa von den zwei leichten
Ketten mit einer Größe von etwa 30kDa getrennt. Eine Ausnahme bildete hier IgG3 dessen
schwere Kette aufgrund einer verlängerten hinge Region eine Größe von ca. 60kDa
aufweist. Wie in Abb. 5A zu sehen ist, zeigten sich nach Auftrennung von je 2µg Protein und
Färbung mit Coomassie Blau die Fragmente der schweren bzw. leichten Ketten in den
erwarteten Größen. Bei IgG2 war die Bandenintensität merklich vermindert, obwohl die
gleiche Menge Protein aufgetrennt wurde. Bei allen vier Proben war keine Degradation der
Antikörper oder eine Verunreinigung durch andere Proteine sichtbar.
Die Erkennung der hergestellten Antikörper durch einen Sekundärantikörper, der gegen
humanes IgG gerichtet ist, wurde in enzyme-linked immunosorbant assays (ELISA)
überprüft. Die Vertiefungen einer ELISA-Platte wurden mit je 50ng IgG beschichtet und
anschließend mit einem Merretichperoxidase (HRP)-gekoppelten, Fc-spezifischen Antikörper
detektiert. Die gemessene optische Dichte (OD) bei einer Wellenlänge von 450nm betrug,
nach Abzug des Hintergrundes bei einer Wellenlänge von 650nm, im Mittel von 3
unabhängigen Experimenten 0,75±0,08 für IgG1, 0,69±0,08 für IgG2, für IgG3 0,68±0,12 und
15
Ergebnisse
für IgG4 0,58±0,9. Im ELISA konnte demnach keine unterschiedliche Erkennung der
verschiedenen IgG Subklassen durch den Sekundärantikörper festgestellt werden (Abb. 5B).
Abb. 5: Humane anti-TNP IgG Varianten (Klon 7B4). Die variablen Regionen des HybridomAntikörpers 7B4 wurden kloniert und an die humanen konstanten IgG Regionen fusioniert.
A) Gelektrophoretische Auftrennung der produzierten 7B4 IgG1-4 unter reduzierenden Bedingungen.
Die Proteine wurden durch Anfärbung mit Coomassie Blau sichtbar gemacht. B) ELISA zur Kontrolle
vergleichbarer Erkennung der IgG Subklassen durch den Sekundärantikörper (anti-human IgG-HRP).
C) Analyse der Bindung von 7B4 IgG1-4 an TNP-4-BSA bzw. TNP-26-BSA mittels ELISA.
Nicht nur die Integrität der produzierten Antikörper ist für die Herstellung von
Immunkomplexen relevant sondern auch eine vergleichbare Bindung des entsprechenden
Antigens - in diesem Fall TNP. Diese Bindung wurde für die produzierten 7B4 IgG in einem
ELISA bestimmt. Hierfür wurden jeweils 100ng TNP-4-BSA bzw. TNP-26-BSA pro Vertiefung
einer ELISA-Platte beschichtet und anschließend mit 1ng IgG1-4 inkubiert. Der Nachweis
des gebundenen IgG erfolgte mittels einem HRP-konjugiertem, Fc-spezifischen Antikörpers
und Bestimmung der OD bei 450nm. Im ELISA konnte eine signifikant verstärkte TNP-4BSA-Bindung von IgG1 (0,65±0,07) im Vergleich zu IgG2 (0,18±0,09) und IgG4 (0,29±0,04)
gemessen werden. Auch IgG3 (0,55±0,07) interagierte signifikant besser mit TNP-4-BSA als
IgG2 und IgG4. Die Bestimmung der Bindung an TNP-26-BSA zeigte ebenfalls eine
signifikant verbesserte Interaktion von IgG1 (0,64±0,15) im Vergleich zu IgG2 (0,14±0,01)
und IgG4 (0,25±0,05). Die Bindung von IgG3 war an TNP-26-BSA (0,37±0,07) in Bezug auf
TNP-4-BSA signifikant vermindert. Die anderen IgG Subtypen interagierten vergleichbar mit
TNP-4-BSA und TNP-26-BSA (Abb. 5C).
2.
Stabile Expression von humanen FcγR in CHO Zellen
Die humanen Fc Rezeptoren, FcγRIA, die beiden Varianten des FcγRIIA (131H bzw. 131R)
und der inhibitorische FcγRIIB wurden von Frau Anne Bärenwaldt kloniert, stabil in CHO
Zellen exprimiert und für diese Experimente zur Verfügung gestellt. Ein Fusionsprotein aus
den extrazellulären Domänen von FcγRIIIA und den Membran-eingelagerten und
16
Ergebnisse
intrazellulären Bereichen von FcγRIIB (in dieser Arbeit kurz eFcγRIIIA), welches ebenfalls
stabil auf der Oberfläche von CHO Zellen exprimiert wird, wurde von Herrn Jeffrey Ravetch
(New York, USA) bereit gestellt.
Abb. 6: Stabile Expression der humanen FcγR in CHO Zellen. A) Histogrammdarstellung der
durchflusszytometrischen Analyse der FcγR-Expression. B) Quantifizierung des Anteils FcγRexprimierender Zellen aus 3 unabhängigen Experimenten.
Die Kontrolle der Oberflächenexpression der einzelnen stabilen Klone erfolgte per
Durchflusszytometrie mithilfe von FcγR-spezifischen Antikörpern. Abb. 6A zeigt exemplarisch
die Fluoreszenzintensität der Expression, während in Abb. 6B der Anteil FcγR positiver
Zellen an der Gesamtpopulation in 3 unabhängigen Experimenten dargestellt ist. In den
entsprechenden Zelllinien exprimierten 85,6%±6,5 (FcγRIA), 75,8%±1,2 (FcγRIIA131H),
94,3%±1,6 (FcγRIIA131R), 96,1%±1,6 (FcγRIIB) und 96,1%±2,0 (eFcγRIIIA) der Zellen den
jeweiligen FcγR. Eine Verunreinigung der Zelllinien mit den anderen stabilen Klonen war
nicht zu beobachten.
3.
Bindung von Immunkomplexen an FcγR
Mithilfe der FcγR exprimierenden CHO Zellen wurde die Bindung der verschiedenen
humanen IgG Subtypen an FcγR in vitro untersucht. Durch Herstellung der Immunkomplexe
mit TNP-4-BSA bzw. TNP-26-BSA konnte außerdem der Einfluss der Größe dieser
Immunkomplexe
auf
die
Bindung
analysiert
werden.
Nach
der
Inkubation
der
Immunkomplexe mit den FcγR exprimierenden CHO-Zellen erfolgte die Detektion der
gebundenen Immunkomplexe per Durchflusszytometrie mittels gegen den Fc Teil der 7B4
IgG Varianten gerichteter Phycoerythrin (PE)-konjugierter Antikörper. Abb. 7 zeigt die
arithmetischen Mittelwerte der mittleren Fluoreszenzintensität (MFI) aus 3 unabhängig
durchgeführten Experimenten.
17
Ergebnisse
Abb. 7: In vitro Bindungsanalyse humaner IgG Immunkomplexe an stabil exprimierte FcγR.
Immunkomplexe wurden aus 7B4 IgG und TNP-4-BSA bzw. TNP-26-BSA generiert und ihre Bindung
an untransfizierte (A) und mit FcγRIA (B), FcγRIIA131H (C), FcγRIIA131R (D), FcγRIIB (E) und eFcγRIIIA
(F) stabil transfizierte CHO Zellen durchflusszytometrisch bestimmt. Gezeigt sind die arithmetischen
Mittelwerte aus 3 unabhängigen Experimenten.
Der Hintergrund der Bindung von TNP-4-BSA Immunkomplexen an untransfizierte CHOZellen lag bei Fluoreszenzintensitäten von 4,2±0,6 (IgG1), 7,8±1,9 (IgG2), 4,4±0,9 und
14,1±18,2. Bei Untersuchung von TNP-26-BSA Immunkomplexen erhöhte sich der
Hintergrund auf 16,6±6,1 (IgG1), 21,3±14,6 (IgG2), 16,4±7,7 (IgG3) bzw. 19,0±7,1 (IgG4).
Für IgG1 war dieser Anstieg signifikant. Allerdings blieben die gemessenen Werte deutlich
unter den Werten einer spezifischen Interaktion (Vgl. Abb. 7A mit 7B-F).
An FcγRIA exprimierende Zellen banden die getesteten IgG Subtypen vergleichbar stark. Es
gab keine signifikante Veränderung der Fluoreszenzintensitäten bei Verwendung von TNP-4BSA oder TNP-26-BSA. Mit MFI-Werten für IgG1 von 87,9±27,4 im Komplex mit TNP-4-BSA
bzw. 139,3±54,7 im Komplex mit TNP-26-BSA, für IgG2 von 103,4±58,5 (TNP-4-BSA) bzw.
100,4±50,8 (TNP-26-BSA), für IgG3 von 109,0±54,0 (TNP-4-BSA) bzw. 144,7±60,0 (TNP-26BSA) und IgG4 97,2±57,2 (TNP-4-BSA) bzw. 142,6±54,7 (TNP-26-BSA) war eine deutlich
verstärkte Bindung im Vergleich zu untransfizierten Zellen zu erkennen (Abb. 7B).
18
Ergebnisse
Bei der Bindung an die hochaffine Variante des FcγRIIA (FcγRIIA131H) für IgG2 wurden
signifikante Unterschiede sowohl für die unterschiedlichen IgG Subtypen als auch für die
Verstärkung der Interaktion in Abhängigkeit von der Immunkomplexgröße deutlich. Sowohl
IgG1-TNP-4-BSA (147,3±19,9) als auch IgG1-TNP-26-BSA (325,3±62,0) Immunkomplexe
banden signifikant besser an FcγRIIA131H als IgG2-TNP-4-BSA (41,3±1,7) bzw. IgG2-TNP26-BSA (78,3±8,1) Immunkomplexe. Ebenfalls signifikant im Vergleich zu IgG2 war die
stärkere Bindung von IgG3. Komplexe von IgG3 mit TNP-4-BSA banden mit einer
Fluoreszenzintensität von 122,7±29,9, die durch Verwendung von TNP-26-BSA noch auf
479,0±35,4 gesteigert wurde. Im Vergleich zu IgG1 und IgG3 TNP-4-BSA Immunkomplexen
zeigten IgG4 Immunkomplexe eine signifikant schwächere FcγR-Bindung. Mit TNP-4-BSA
wurden MFI-Werte von 43,5±6,4, mit TNP-26-BSA von 252,7±68,4, erzielt. Ein ähnliches
Ergebnis wurde für TNP-26-BSA Immunkomplexe erhalten. Alle mit TNP-26-BSA
komplexierten IgG interagierten signifikant besser mit FcγRIIA131H als die jeweiligen TNP-4BSA Immunkomplexe (Abb. 7C).
Die Fluoreszenzintensitäten für die Immunkomplexbindung an die niedrigaffine Variante des
FcγRIIA (FcγRIIA131R) waren allgemein niedriger als bei den anderen untersuchten FcγR.
Besonders schwach war die Bindung von TNP-4-BSA Immunkomplexen. Sie bewegte sich
für IgG2 (13,3±4,9) und IgG4 (9,3±2,8) im Bereich der Hintergrundbindung an untransfizierte
CHO Zellen. Die mittlere Bindung von IgG1 betrug 37,1±12,9 und war damit signifikant höher
als die Bindung von IgG2 und IgG4. Zu IgG3 (61,6±33,8) gab es keine signifikanten
Differenzen. Mit TNP-26-BSA erzeugte Komplexe banden bei allen Isotypen besser an
FcγRIIA131R aber nur für IgG1 war der Unterschied signifikant. IgG1- (153,3±66,5) und IgG3TNP-26-BSA (203,0±107,1) Immunkomplexe banden signifikant besser als IgG2-TNP-26BSA (20,2±7,9) Immunkomplexe. IgG4 (43,8±23,8) band im Komplex mit TNP-26-BSA zwar
besser als IgG2 aber immer noch sehr schwach.
An den niedrigaffinen inhibitorischen FcγRIIB, der an Aminosäureposition 131 ebenfalls ein
Arginin trägt, banden die Immunkomplexe in einem ähnlichen Muster wie an FcγRIIA131R.
TNP-4-BSA Immunkomplexe mit IgG1 (14,2±5,5), IgG2 (4,8±1,3), IgG3 (31,6±19,4) oder
IgG4 (9,6±4,3) interagierten kaum stärker mit der stabil exprimierenden Zelllinie als mit den
untransfizierten Kontrollzellen. Im Unterschied dazu kam es zu einer signifikant verbesserten
FcγR-Interaktion bei Verwendung von TNP-26-BSA auf 86,3±38,7 (IgG1), 10,4±0,9 (IgG2)
und 44,2±16,6 (IgG4). Die Fluoreszenzintensität von IgG3 (125,5±89,8) Immunkomplexen
war dagegen nur leicht erhöht. Betrachtet man die einzelnen Isotypen, so zeigte sich eine
signifikante Steigerung der Immunkomplexbindung für IgG1 und IgG4 im Vergleich zu IgG2.
Das aus den extrazellulären Domänen von FcγRIIIA und den Transmembran- und
intrazellulären Sequenzen von FcγRIIB bestehende Fusionsprotein (eFcγRIIIA) band nach
19
Ergebnisse
Expression in CHO Zellen, im Vergleich zu den anderen getesteten FcγR, am stärksten an
TNP-4-BSA Immunkomplexe. Nur bei IgG3 und IgG4 war eine weitere signifikante
Verbesserung der Bindung durch Verwendung von TNP-26-BSA zu erreichen. TNP-4-BSA
(IgG1: 262,7±21,1, IgG3: 237,7±22,6) wie auch TNP-26-BSA (IgG1: 287,3±30,9, IgG3:
364,7±53,5) enthaltende Immunkomplexe zeigten erneut eine höhere Fluoreszenzintensität
als die jeweiligen IgG2 (87,4±18,4 für TNP-4-BSA, 91,0±17,4 bei TNP-26-BSA) Komplexe.
IgG4-TNP-4-BSA Immunkomplexe (67,4±8,7) banden signifikant schwächer als die
entsprechenden Immunkomplexe für IgG1 und IgG3. Mit TNP-26-BSA komplexiertes IgG4
(255,0±35,8) zeigte zwar eine signifikant verminderte Bindung bezogen auf IgG3, interagierte
aber signifikant besser mit eFcγRIIIA als IgG2.
Diese
Experimente
weisen
auf
eine
bessere
Interaktion
von
IgG1
und
IgG3
Immunkomplexen mit den niedrigaffinen humanen FcγR hin. Die Verwendung von TNP-26BSA im Gegensatz zu TNP-4-BSA führt zu Immunkomplexen die i.A. verstärkt an FcγR
binden können. Der hochaffine FcγRIA scheint dagegen keine veränderte Affinität für die
einzelnen IgG Subtypen oder Immunkomplexvarianten zu besitzen.
4.
Einfluss der N297 Glykosylierung auf die FcγR-Bindung
Um den Einfluss der IgG Glykosylierung der schweren Ketten auf die Interaktion mit FcγR zu
untersuchen, wurden die Antikörper zunächst mit dem Enzym PNGaseF behandelt, dass die
Zuckerketten direkt am Asparagin 297 (N297) hydrolysiert. Die Vollständigkeit dieser
Deglykosylierung wurde mittels PAA-Gelelektrophorese unter reduzierenden Bedingungen
und anschließendem Lektin Blot unter Verwendung von lens culinaris agglutinin (LCA)
kontrolliert. Nach Färbung des PAA-Gels mit Coomassie Blau zeigten sich die schweren
Ketten der unbehandelten IgG1, IgG2 und IgG4 bei etwa 55kDa und die schwere Kette von
IgG3 bei etwa 60kDa. Die Proteinbanden der PNGaseF behandelten IgG erschienen
dagegen bei einem leicht erhöhten Molekulargewicht. Es waren keine Banden auf Höhe der
unverdauten schweren Ketten zu sehen. LCA bindet an die Mannose-haltige Zuckerketten,
wie sie bei der N297 Glykosylierung der schweren und leichten Ketten auftreten können. Da
der 7B4 Antikörper jedoch keine Asparagin gekoppelten Zuckerketten in der leichten Kette
enthält, wurden im Lektin Blot nur die schweren Ketten der unbehandelten IgG, nicht aber
die leichten Ketten, angefärbt. Nach PNGaseF-Verdau konnten durch LCA keine Zuckerreste
detektiert werden. Dies weist auf eine vollständige Hydrolyse hin. Wie am unveränderten
Laufverhalten bei der Gelelektrophorese zu sehen war, wurden die leichten Ketten mit einer
Größe von etwa 30kDa nicht durch PNGaseF beeinflusst (Abb. 8A).
20
Ergebnisse
Abb. 8: Einfluss der N297 Glykosylierung auf die FcγR Bindung der Immunkomplexe in vitro.
A) Kontrolle der Deglykosylierung der IgG Subklassen durch PNGaseF mittels reduzierender PAAGelektrophorese gefolgt von Coomassie Blau Färbung bzw. Lektin Blot. B-F) Durchflusszytometrische
Analyse der Bindung von TNP-4-BSA bzw. TNP-26-BSA Immunkomplexen in Abhängigkeit vom
Glykosylierungsstatus. Die gemessenen Fluoreszenzintensitäten der PNGaseF-behandelten IgG
wurden auf die Werte der unbehandelten IgG normalisiert. Die relative FcγR Bindung [AU] beträgt 1,0
bei einer gleichen Interaktion von verdauten und unverdauten Immunkomplexen. Dargestellt sind die
arithmetischen Mittelwerte aus je 3 unabhängig durchgeführten Experimenten.
Die durch PNGaseF-Behandlung deglykosylierten Antikörper wurden für die Herstellung von
Immunkomplexen mit TNP-4-BSA bzw. TNP-26-BSA verwendet und die in vitro Bindung an
stabil
exprimierte
FcγR
im
Vergleich
zu
unbehandelten
IgG
Immunkomplexen
durchflusszytometrisch bestimmt. Abb. 8B-F zeigen die relativen Mittelwerte der FcγR
Bindung aus drei unabhängigen Experimenten, wobei eine relative Bindung von 1,0AU
bedeutet, dass es keinen Einfluss der Glykosylierung auf die jeweilige IgG-FcγR Interaktion
gab.
Wie in Abb. 8B zu sehen ist, wurde die Bindung der humanen IgG1 und IgG3 an den
hochaffinen FcγRIA nicht wesentlich von der Deglykosylierung beeinflusst. Nach PNGaseFBehandlung erreichten TNP-4-BSA Immunkomplexe eine Bindung von 0,7AU±0,06 (IgG1)
bzw. 0,72AU±0,08 (IgG3). Mit einer reduzierten relativen Signalintensität von 0,25AU±0,02
21
Ergebnisse
bzw. 0,45AU±0,09 zeigten IgG2 und IgG4 dagegen eine größere Abhängigkeit von den
N297-Zuckerketten. Immunkomplexe, die stattdessen mit TNP-26-BSA generiert wurden,
schienen tendenziell besser mit FcγRIA interagieren zu können. Die relativen Werte stiegen
auf 0,89AU±0,05 für IgG1, 0,44AU±0,03 für IgG2, 0,97AU±0,08 für IgG3 und 0,65AU±0,04
für IgG4. Jedoch ist dieser Unterschied nur für IgG1 und IgG2 signifikant.
Im Gegensatz zu FcγRIA wird die Interaktion der TNP-4-BSA Immunkomplexe mit der
hochaffinen Variante des FcγRIIA131H fast vollständig durch Behandlung der IgG mit
PNGaseF verhindert. IgG1 Immunkomplexe zeigten nach PNGaseF nur noch eine relative
Bindung von 0,01AU±0,0 und IgG2 von 0,05AU±0,01. Auch IgG3 (0,02AU±0,0) und IgG4
(0,06AU±0,04) interagierten praktisch nicht mehr mit FcγRIIA131H. Die Verwendung von TNP26-BSA führte nicht zu einer verstärkten Interaktion. Die relativen Werte stiegen nicht
signifikant auf 0,11AU±0,03 (IgG1), 0,19AU±0,08 (IgG2), 0,14AU±0,05 (IgG3) und
0,08AU±0,06 (IgG4) (Abb. 8C).
Auch die Interaktion von TNP-4-BSA Immunkomplexen mit dem niedrigaffinen FcγRIIA131R
wurde durch Vorbehandlung von IgG1 und IgG3 mit PNGaseF blockiert. So banden verdaute
IgG1 mit einer Stärke von 0,08AU±0,01 und IgG3 mit 0,1AU±0,0. IgG2 (0,45AU±0,13) und
IgG4 (0,34AU±0,13) zeigten dagegen eine bessere Interaktion. Außer für IgG3, bei dem ein
signifikanter Anstieg der relativen Signalintensität auf 0,24AU±0,04 zu beobachten war,
verbesserte sich die Interaktion nicht unter Verwendung von TNP-26-BSA. Sie stagnierte bei
0,18AU±0,06 (IgG1), 0,42AU±0,12 (IgG2) bzw. 0,37AU±0,37 (IgG4) (Abb. 8D).
Wie in Abb. 8E dargestellt, ist die Affinität von FcγRIIB ebenfalls stark vom
Glykosylierungsstatus
der
IgG
abhängig.
Unabhängig
von
der
Generierung
der
Immunkomplexe mit TNP-4-BSA oder TNP-26-BSA zeigten IgG1 (0,11AU±0,02 für TNP-4BSA bzw. 0,22AU±0,06 für TNP-26-BSA), IgG3 (0,11AU±0,02 für TNP-4-BSA bzw.
0,20AU±0,02 für TNP-26-BSA) und IgG4 (0,20AU±0,06 für TNP-4-BSA bzw. 0,28AU±0,12
für TNP-26-BSA) nur eine niedrige relative Signalintensität. Dagegen konnte PNGaseFbehandelter IgG2 (0,73AU±0,09 für TNP-4-BSA bzw. 0,57AU±0,09 für TNP-26-BSA)
vergleichsweise gut mit FcγRIIB interagieren.
Für eFcγRIIIA zeigte sich ein verminderter Einfluss der IgG Glykosylierung. So
verschlechterte sich die relative Bindung von TNP-4-BSA Immunkomplexen für IgG1 nur auf
0,5AU±0,15 und für IgG3 auf 0,29AU±0,31. Deglykosylierte IgG2 (0,08AU±0,03) und IgG4
(0,03AU±0,01) Immunkomplexe banden dagegen nicht an eFcγRIIIA. Die Verwendung von
TNP-26-BSA schien die Defekte bei der FcγR-Interaktion nach PNGaseF-Behandlung für
IgG1 (1,16AU±0,12) und IgG3 (0,96AU±0,32) aufzuheben, während die Bindung von IgG2
22
Ergebnisse
(0,15AU±0,04) und IgG4 (0,07AU±0,04) dadurch nur leicht verbessert wurde. Allerdings war
die Verbesserung für IgG2, nicht aber für IgG1 und IgG3 signifikant (Abb. 8F).
Zusammenfassend wurde im ersten Teil dieser Dissertation eine systematische Analyse der
Bindung von humanen IgG Antikörpern in Form definierter Immunkomplexe an die humanen
FcγR in vitro durchgeführt. Hierbei zeigten die humanen IgG Isotypen IgG1 und IgG3 eine
stärkere Bindung an FcγR als IgG2 und IgG4. Immunkomplexe, die mit TNP-26-BSA
generiert werden, banden im Allgemeinen besser als mit TNP-4-BSA hergestellte
Immunkomplexe (Tab. 1). Die Glykosylierung der IgG schweren Ketten bestimmt dagegen
wesentlich die Interaktion mit den niedrigaffinen FcγR. In vitro schien der Polymorphismus
von FcγRIIA (und FcγRIIB) an Aminosäureposition 131 die Affinität nicht nur für IgG2
sondern auch für die anderen IgG-Subtypen zu beeinflussen.
Tab 1: Übersicht der Interaktion humaner IgG Subklassen mit den humanen FcγR. Die Bindung wurde
wie folgt bewertet: - (MFI <30), + (31 < MFI < 100), ++ (101 < MFI < 200), +++ (301 < MFI).
23
Ergebnisse
II. Untersuchung
von
Antikörpereffektorfunktionen
im
humanisierten Mausmodell
Im zweiten Teil der Arbeit erfolgte die Untersuchung von Einflussfaktoren auf IgG
Effektorfunktionen in vivo. Exemplarisch sollte der Einfluss der FcγR Polymorphismen
FcγRIIA131H/R und FcγRIIIA158F/V auf die Antikörper-induzierte Zell-vermittelte Zytotoxizität
(ADCC) analysiert werden. Hierfür wurde zunächst ein humanisiertes Mausmodell etabliert
und charakterisiert. In diesem experimentellen System erfolgte schließlich die Depletion von
B-Zellen durch Injektion CD20 spezifischer IgG-Fc Varianten.
1.
Herstellung von anti-CD20 IgG Subtypen (Klon 1F5)
In den humanisierten Mäusen sollte die Interaktion von humanem IgG und FcγR in vivo
untersucht werden. Eine durch diese Interaktion induzierte Effektorfunktion ist die ADCC.
Daher wurde beispielhaft die durch IgG induzierte und über ADCC vermittelte B-ZellDepletion ausgewählt. Hierfür wurde zunächst der gegen das humane Oberflächenprotein
CD20 gerichtete Hybridom-Antikörper (Klon 1F5) kloniert, auf die humanen IgG Subklassen
humanisiert und durch transiente Transfektion produziert.
Abb. 9: Anti-CD20 IgG Fc Varianten (Klon 1F5). A) Elektrophoretische Auftrennung unter
reduzierenden Bedingungen gefolgt von der Färbung der Proteine mit Coomassie Blau. B)
Kompetitionsassay zum Vergleich der Antigenbindung. Die hergestellten Antikörper wurden mit dem
parentalen, fluoreszenzmarkierten Hybridomantikörper auf LCL1.11 Zellen koinkubiert. Anschließend
erfolgte die durchflusszytometrische Bestimmung der Reduktion der Fluoreszenzintensität (n=3).
C) Bestimmung der C1q Bindung durch die humanen IgG Subklassen mittels ELISA (n>4).
Die Integrität und Reinheit der produzierten Antikörper wurde mittels PAA-Gelelektrophorese
und nachfolgender Coomassie-Blau-Färbung überprüft. Die schweren Ketten von IgG1, IgG2
und IgG4 erschienen jeweils bei einer Größe von etwa 55kDa. Dagegen war die schwere
24
Ergebnisse
Kette von IgG3 mit 60kDa deutlich größer. Die leichten Ketten zeigten sich etwa bei 30kDa.
Es konnten keine weiteren Proteine oder Spaltprodukte beobachtet werden (Abb. 9A).
Im weiteren Verlauf wurden die produzierten Antikörper auf ihre Antigenbindung getestet, da
Varianzen in der Affinität für CD20 IgG-abhängige Unterschiede in der B-Zell-Depletion
verursachen könnten. Zunächst erfolgte hierfür die Fluoreszenzmarkierung des parentalen
Hybridom-Antikörpers. Dieser wurde mit steigenden Konzentrationen der produzierten
humanen 1F5 IgG gemischt und mit der humanen CD20 exprimierenden B-Zell-Linie
LCL1.11
inkubiert.
Unter
diesen
experimentellen
Bedingungen
konkurrierten
die
verschiedenen Antikörper um die vorhandenen CD20-Moleküle und sowohl die Assoziationsals
auch
die
Dissoziationskonstanten
konnten
verglichen
werden.
Bei
der
durchflusszytometrischen Analyse wurde die maximale Fluoreszenzintensität bei alleiniger
Inkubation mit dem fluoreszenzmarkierten Antikörper erreicht. Die Kompetition mit den
unmarkierten 1F5 IgG führte zu einer Reduktion der Fluoreszenzintensität. In Abb. 9B ist die
sog. CH50 dargestellt, d.h. die Konzentration der 1F5 IgG bei der die mittlere
Fluoreszenzintensität (MFI) 50% betrug. 1F5 IgG1 erreichte die 50%ige Reduktion der MFI
bei einer Konzentration von 47,2ng/µl±13. Der hergestellte 1F5 IgG2 band mit einer CH50
von 107,7ng/µl±43 nur etwa halb so gut. Die CH50 für IgG3 betrug 53,9ng/µl±7,7. Sie war
damit mit der Bindung von IgG1 vergleichbar. Dagegen war die Bindung von IgG4 signifikant
schwächer als bei IgG1 und IgG3. Erst bei einer Konzentration von 99,2ng/µl±13,8 konnte
IgG4 die MFI um 50% vermindern.
Mittels ELISA wurden die produzierten 1F5 IgG weiterhin auf die Bindung des humanen
Komplementfaktors C1q untersucht. Hierfür wurden zunächst je 100ng der 1F5 IgG mit
2ng/µl C1q Protein inkubiert. Das gebundene C1q wurde anschließend mit einem HRPkonjugiertem C1q-spezifischen Antikörper bei einer Wellenlänge von 450nm detektiert. Nach
Abzug des Hintergrundsignals bei einer Wellenlänge von 650nm banden IgG1 (0,12±0,07)
und IgG3 (0,11±0,05) signifikant besser an C1q als IgG2 (0,03±0,02) und IgG4 (0,01±0,0)
(Abb. 9C).
Diese Versuche zeigen, dass die hergestellten 1F5 IgG in ihrer Struktur intakt sind und ihr
Antigen CD20 binden. Bei 1F5 IgG2 und 1F5 IgG4 scheint die Affinität für CD20 allerdings
vermindert.
2.
Eigenschaften von Rag2/γc/Fcγ-/- Mäusen
Für die Etablierung des humanisierten Mausmodells wurde ein bereits im Vorfeld dieser
Dissertation im Labor generierter immundefizienter Mausstamm verwendet. Hierzu wurden
Rag2/γc-/- Mäuse mit Fcγ-/- Mäusen verpaart (Abb. 10A). Eine Deletion des Rag2 Gens führt
zu einer frühen Blockade in der Entwicklung von B- und T-Zellen. Eine Nullmutation im Gen
25
Ergebnisse
der gemeinsamen Signalkette (γc) der Interleukin-Rezeptoren führt zusätzlich zu einer
Inhibierung der NK-Zell-Entwicklung. Nach der Zerstörung der Knochenmarkszellen im
Empfängertier durch Bestrahlung, können die Mäuse daher mit Spender-Stammzellen
rekonstituiert werden ohne dass es zu Abstoßungsreaktionen kommt. Die erzeugten
Rag2/γc/Fcγ-/- Mäuse verfügen zusätzlich über eine Deletion der gemeinsamen Signalkette
der murinen aktivierenden FcγR (Fcγ), die essentiell für deren Oberflächenexpression ist.
Dies ist notwendig, da humane IgG auch an die FcγR der Maus binden und dadurch FcγRvermittelte Effekte auslösen können.
Abb. 10: Charakterisierung von
Rag2/γc/Fcγ-/Mäusen.
A)
Durch
Verpaarung
von
-/-/Rag2/γc und Fcγ Mäusen
wurden Rag2/γc/Fcγ-/- Mäuse
erzeugt. B) Durchflusszytometrische Analyse der murinen
Leukozytenpopulationen
in
Rag2/γc/Fcγ-/- im Vergleich zu
C57BL/6
und
Rag2/γc-/Mäusen.
C)
Histogrammdarstellung der durchflusszytometrischen
Bestimmung
der Expression muriner FcγR
auf PBMCs in Rag2/γc/Fcγ-/- im
Vergleich
zu
Rag2/γc-/Mäusen.
Eine durchflusszytometrische Analyse der mononukleären Zellen des peripheren Blutes
(peripheral blood mononuclear cells, PBMCs) von Rag2/γc/Fcγ-/- Mäusen im Vergleich zu
Rag2/γc-/- und C57BL/6 Mäusen ist in Abb. 10B dargestellt. Die Frequenzen der
Zellpopulationen
im
Blut
wurden
mit
fluoreszenzmarkierten
Antikörpern
gegen
charakteristische Oberflächenantigene in je 3 Mäusen bestimmt. In C57BL/6 Mäusen waren
30,1%±10,5 der lebenden Zellen im Blut TCRß exprimierende T-Zellen und 46,1%±21,7
B220+ B-Zellen. 2,4%±1,0 der Zellen konnten aufgrund des Oberflächenmarkers NK1.1+ als
NK Zellen identifiziert werden. Myeloide Zellen, d.h. Monozyten und Granulozyten, die auf
ihrer Zelloberfläche CD11b nicht aber NK1.1 tragen, machten 21,1%±10,9 der analysierten
Zellen aus. In Rag2/γc-/- und Rag2/γc/Fcγ-/- Mäusen konnten dagegen keine NK1.1+
(0,0%±0,0) Zellen und nur sehr geringe Anteile von TCRβ+ (0,9%±0,9 bzw. 0%±0,0) und
B220 exprimierenden Zellen (0,6±0,2 bzw. 0,4%±0,0) nachgewiesen werden. 98,9%±0,7
26
Ergebnisse
(Rag2/γc-/-) bzw. 98,7%±0,2 (Rag2/γc/Fcγ-/-) der untersuchten Zellen waren CD11b+
Monozyten und Granulozyten.
Zusätzlich zu den Zellpopulationen wurde auch der Expressionsstatus der FcγR mittels
Durchflusszytometrie untersucht. Hierfür wurden je 2 Rag2/γc-/- Mäuse, die über die Fcγ Kette
verfügen und daher alle FcγR exprimieren, mit den erzeugten Rag2/γc/Fcγ-/- Mäusen
verglichen. Während der hochaffine FcγRI in Rag2/γc-/- Mäusen auf dem Großteil der
analysierten PBMCs exprimiert wurde, konnte er auf den PBMCs von Rag2/γc/Fcγ-/- Mäusen
nicht detektiert werden. Der monoklonale Antikörper 2.4G2, der sowohl den inhibitorischen
FcγRIIB als auch den aktivierenden FcγRIII erkennt, band in Rag2/γc-/- Mäusen an alle
analysierten PBMCs. Auch in Rag2/γc/Fcγ-/- Mäusen kam es zu einer Erkennung der Zellen
durch 2.4G2. Das positive Signal bei dieser Färbung wird durch die Expression des
inhibitorischen FcγRIIB verursacht, der nicht Fcγ-abhängig ist. Ein FcγRIII-spezifischer
Antikörper zeigte keine Bindung an Zellen aus Rag2/γc/Fcγ-/- Mäusen im Gegensatz zu
Rag2/γc-/- Mäusen. Der aktivierende FcγRIV wurde ebenfalls nur in Rag2/γc-/- Mäusen nicht
aber in Rag2/γc/Fcγ-/- Mäusen detektiert (Abb. 10C). Die ausgeprägte FcγR-Expression in
Rag2/γc-/- Mäusen hatte ihre Ursache im Fehlen von FcγR negativen Zellen z.B. TLymphozyten. Wie auch bei Rag2/γc/Fcγ-/- Mäusen fanden sich im Blut vor allem CD11b+,
FcγR exprimierende Monozyten und Granulozyten (Abb. 10B).
3.
Aufreinigung und Analyse von humanen hämatopoetischen
Stammzellen
Für die Rekonstitution von humanisierten Mäusen wurden humane CD34+ hämatopoetische
Stammzellen
(HSC)
benötigt,
die
aus
Nabelschnurblut
aufgereinigt
wurden.
Das
Nabelschnurblut wurde für diese Zwecke von Mitarbeitern des Klinikums Fürth unter
Zustimmung der Spender zur Verfügung gestellt. Die Aufreinigung erfolgte über eine
Dichtegradientenzentrifugation zur Aufkonzentrierung von Leukozyten, gefolgt von der
positiven Selektion mittels an magnetische Partikel gekoppelter anti-CD34 Antikörper.
Die Ergebnisse der durchflusszytometrischen Analyse der aufkonzentrierten Leukozyten vor
der
weiteren
Selektion
sowie
der
aufgereinigten Stammzellen
sind
in Abb.
11
zusammengefasst. In den 3 analysierten Nabelschnurblutproben bestand mit 1,8%±1,0 nur
einer kleiner Teil der aufkonzentrierten Leukozyten aus CD34+ HSC (Abb. 11A). Der Anteil
dieser Population wurde durch die spezifische Aufreinigung über CD34-spezifische
Antikörper im Mittel von 8 untersuchten Proben auf 80%±11,6 gesteigert. Der geringe Anteil
CD38+ Zellen von 10,8%±7,3 deutete auf den weitgehend undifferenzierten Zustand dieser
aufgereinigten HSC hin (Abb. 11B). Bei den CD34- Zellen handelte es sich zum größten Teil
um CD19+ B-Zellen (57,2%±16,1). Die aufgereinigte Fraktion enthielt aber auch kleinere
27
Ergebnisse
Populationen von CD3+ T-Zellen (4,5%±2,9), CD56+ NK-Zellen (3,3%±0,9) und durch die
Expression von CD33+ gekennzeichnete myeloide Zellen (16,9%±9,0) (Abb. 11C). Die
übrigen Zellen wurden nicht näher bestimmt. Im arithmetischen Mittel konnten durch die
Aufreinigung ca. 14.000 Zellen/ml Nabelschnurblut gewonnen werden.
Abb. 11: Isolation hämatopoetischer Stammzellen. Durchflusszytometrische Analyse CD34+ Zellen in
der Gesamtzellpopulation im Nabelschnurblut (A) bzw. nach der Aufreinigung der HSC (B).
C) Durchflusszytometrische Identifizierung der CD34 negativen Zellen in der angereicherten
Stammzellpopulation.
4.
Genotypisierung der FcγR Polymorphismen
Die Polymorphismen in den Genen der aktivierenden FcγRIIA und FcγRIIIA wurden nach
Isolation der genomischen DNA aus dem Nabelschnurblut durch Amplifikation mit Allelspezifischen Primern bestimmt. Für die Effizienz der PCR ist dabei die Bindung der letzten
Base des Primers an die Ausgangs-DNA entscheidend. Daher wurden für die Allelspezifische PCR Primer verwendet, die sich ausschließlich in dieser Base unterschieden. Die
Auswertung erfolgte durch elektrophoretische Auftrennung der Amplifikationsprodukte in
Ethidiumbromid-haltigen Agarosegelen und Analyse unter UV-Licht. Da die allelspezischen
Primer bis auf 1 Nukleotid identisch waren, konnten in jedem Fall PCR-Produkte amplifiziert
werden. Allerdings war die Effizienz wesentlich schlechter und die Intensität der DNABanden demnach geringer.
Zur Alleltypisierung von FcγRIIA131H/R wurde in einer ersten PCR zunächst ein 517bp großes,
FcγRIIA spezifisches Fragment amplifiziert, das das Exon 4 und den zu typisierenden
Polymorphismus enthielt. Dieses PCR-Produkt wurde direkt in die zweite, Allel-spezische
PCR eingesetzt. Für die Amplifikation des Arginin-kodierenden Allels wurde ein sense Primer
mit einem endständigen Guanin verwendet, für das Histidin-kodierende Allel verfügte der
28
Ergebnisse
Primer am 3´-Ende stattdessen über ein Adenin. Beispielhaft ist das Ergebnis der drei
möglichen Allelkombinationen in Abb. 12A dargestellt. Die Wasserkontrolle zeigte keine
unspezifschen Banden außer Signalen mit einer Größe von 30-60bp, die durch
überschüssigen Primer in der Reaktion verursacht wurden. Ein spezifisches Produkt mit
einer Größe von 271bp war dagegen bei Zugabe von DNA zu sehen. Im Fall einer
homozygot hochaffinen Probe (FcγRIIA131H) war eine intensive Bande bei Verwendung des
Alanin-Primers zu sehen, aber nur eine sehr schwache Bande bei Verwendung des GuaninPrimers. Bei der homozygot niedrigaffinen Probe (FcγRIIA131R) entstand dagegen mehr PCRProdukt nach Verwendung des Guaninprimers. Vergleichbar intensive Banden bei beiden
PCR-Ansätzen kennzeichneten eine heterozygote Probe (FcγRIIA131H/R).
Abb. 12: FcγRIIA131H/R und FcγRIIIA158F/V Genotypisierung. Gelelektophoretische Auftrennung der
Produkte der Genotypisierungs-PCRs zur Bestimmung von FcγRIIA131H/R (A) bzw. FcγRIIIA158F/V (B).
C) Statistische Verteilung der einzelnen Kombinationen unter 439 typisierten Stammzellproben.
Für die Genotypisierung von FcγRIIIA158F/V wurde ebenfalls zunächst ein FcγRIIIA spezisches
Fragment von 394bp amplifiziert, das die polymorphe Base in Exon 4 enthielt. Die zweite,
Allel-spezische PCR wurde mit Allel-spezifischen antisense Primern durchgeführt. Während
der Primer mit einem Thymin am 3´-Ende seiner Sequenz der Amplifikation des
Phenylalanin-kodierenden Allels diente, führte ein an dieser Stelle befindliches Guanin zur
Amplifikation des Valin-kodierenden Allels. Demzufolge zeigte die Genotypisierung einer
homozygot hochaffinen Probe (FcγRIIIA158V) ein stärkeres Signal mit dem Guaninendständigen Primer. Bei einer homozygot niedrigaffinen Probe (FcγRIIIA158F) entstand mehr
PCR-Produkt bei Verwendung des Thymin-Primers und bei einer heterozygoten Probe
entstanden zwei in ihrer Bandenintensität vergleichbare PCR-Produkte. Weitere Banden z.B.
durch überschüssige Primer oder unspezifische Amplifikation waren nicht zu detektieren
(Abb. 12B). Sowohl für die FcγRIIA als auch für die FcγRIIIA Allel-spezifischen PCR
Reaktionen wurde durch Klonierung und Sequenzierung der erhaltenen PCR-Produkte die
Anwesenheit der jeweiligen Nukleotidaustausche verifiziert.
29
Ergebnisse
Die Frequenzen der Allelkombinationen, die durch die Genotypisierung von 439
Stammzellproben bestimmt wurden, sind in Abb. 12C zusammengefasst. Unter den
gespendeten Nabelschnurblutproben fanden sich alle möglichen Kombinationen der
untersuchten FcγR Polymorphismen. Am häufigsten traten Proben mit der Kombination
FcγRIIA131H/R – FcγRIIIA158F/V auf. Am seltensten war die Kombination FcγRIIA131R/R –
FcγRIIIA158V/V.
Im Durchschnitt lag die Wahrscheinlichkeit des niedrigaffinen, Arginin-
kodierenden Allels für FcγRIIA bei 47,7%±5,7 während das hochaffine, Histidin-kodierende
Allel mit einer Wahrscheinlichkeit von 52,3%±5,7 auftrat. Das niedrigaffine, Phenylalaninkodierende Allel war dagegen für FcγRIIIA wahrscheinlicher (57,2%±5,6) als das hochaffine,
Valin-kodierende
Allel
(42,8%±5,6).
Nach
dem
Hardy-Weinberg-Gesetz
der
Populationsgenetik ergibt sich für eine ideale, ausreichend große Bevölkerungsgruppe eine
gleichmäßige Verteilung des Auftretens der einzelnen Allele. Die Wahrscheinlichkeiten
können anhand einer mathematischen Formel bestimmt werden:
p2 + 2pq + q2 = 1
Hierbei stehen p2 und q2 für die Wahrscheinlichkeiten des homozygoten Auftretens der
beiden Allele und 2pq für die Wahrscheinlichkeit des Heterozygotie. Für die bestimmten
Wahrscheinlichkeiten der Polymorphismen ergab sich demnach eine ideale Verteilung:
5.
p2
2pq
q2
p2 + 2pq + q2 =
FcγRIIA:
0,23
0,5
0,28
1,01
FcγRIIIA:
0,30
0,55
0,15
1,0
Rekonstitution von Rag2/γc/Fcγ-/- Mäusen mit humanen HSC
Für die Rekonstitution von immundefizienten Rag2/γc/Fcγ-/- Mäusen wurden diese zunächst
sublethal in einer Cäsium137 Gammabestrahlungsanlage bestrahlt. Danach erfolgte die
intravenöse Injektion von mindestens 50.000 humanen HSC. Im Alter von ca. 12 Wochen
wurde peripheres Blut entnommen, die PBMCs mittels Dichtegradientenzentrifugation isoliert
und mit spezies-spezifischen Antikörpern, die gegen den Oberflächenmarker CD45 auf
Leukozyten gerichtet sind, angefärbt. In der Maus liegt CD45 in den Allotypen CD45.1 oder
CD45.2 vor, wobei rekonstituierte Rag2/γc/Fcγ-/- Mäuse durch den Marker CD45.2
gekennzeichnet sind. Anschließend wurden die Zellen mittels Durchflusszytometrie
analysiert. Der Grad der Humanisierung wurde über das Verhältnis von hCD45+ humanen
Zellen zu mCD45.2+ hämatopoetischen Zellen der Maus ermittelt. Abb. 13A zeigt beispielhaft
die Verteilung der Zellen in einer peripheren Blutprobe mit 22,4% humanen Zellen und
76,9% hämatopoetischen Zellen der Maus.
30
Ergebnisse
Abb. 13: Rekonstitution von Rag2/γc/Fcγ-/- Mäusen mit humanen HSC. A) Beispielhafte
durchflusszytometrische Bestimmung der Humanisierungseffizienz im peripheren Blut über das
relative Verhältnis von murinen mCD45.2+ und humanen hCD45+ Zellen. B) Rekonstitution
humanisierter Mäuse in Abhängigkeit von der Bestrahlungsdosis. Angegeben ist der Anteil der bis
zum Altern von 12 Wochen verstorbenen und der analysierten Mäuse, die im peripheren Blut
entweder
hCD45+
Zellen
enthielten
(humanisiert)
oder
nicht
humanisiert
waren.
+
C) Durchflusszytometrische Bestimmung des Anteils hCD45 Zellen im peripheren Blut 12 Wochen
alter humanisierter Mäuse in Abhängigkeit von der Bestrahlungsdosis. D) Gesamtüberblick hCD45+
Zellen im peripheren Blut, in der Milz und im Knochenmark der unbehandelten humanisierten Mäuse.
Die arithmetischen Mittelwerte sind angezeigt.
Die Transplantationseffizienz wurde für steigende Bestrahlungsdosen zwischen 3,5Gy und
6Gy ermittelt (Abb 13B). Bei der niedrigsten getesteten Dosis wurden insgesamt 36 Mäuse
rekonstituiert. 36,1% dieser Mäuse verstarben innerhalb von 12 Wochen. Ein Alter von 12
Wochen erreichten demnach 63,9% der Mäuse und konnten somit untersucht werden. Von
allen transplantierten Tieren waren 4 Mäuse humanisiert. Dies entsprach einem Anteil von
31
Ergebnisse
11,1% an der Gesamtpopulation. Der Anteil der humanen Zellen lag bei 10,8%±15,8
(Abb. 13C).
Nach
Bestrahlung
mit
4Gy
und
Transplantation
der
humanen
hämatopoetischen
Stammzellen verstarben von 80 Tieren innerhalb von 12 Wochen 19 Mäuse (23,8%).
Analysiert werden konnten nach 12 Wochen 76,2% der Gesamtpopulation. Im peripheren
Blut von insgesamt 13 Mäusen oder 16,3% fanden sich hCD45+ Zellen. Im arithmetischen
Mittel lag der Anteil der humanen Zellen in diesen Mäusen bei 14,6%±13,1.
6 Mäuse (42,9%) von den 14 transplantierten Tieren verstarben nach der Bestrahlung mit
4,25Gy. In der untersuchten Population befanden sich 4 Mäuse mit durchschnittlich
9,4±8,9% hCD45+ Zellen. Der Anteil der humanisierten Mäuse betrug also 28,6%.
Insgesamt 29 Mäuse wurden vor der Transplantation mit 4,5Gy bestrahlt. Bis zum Alter von
12 Wochen verstarben hiervon 13,8% der Tiere. Von den untersuchten Mäusen waren 5
Tiere, d.h. 17,2% der Population, im arithmetischen Mittel zu 23,6%±13,7 humanisiert.
Die höchste Humanisierungseffizienz in Bezug auf den Anteil humanisierter Mäuse wurde
bei einer Bestrahlungsdosis von 4,75Gy erzielt. Von 29 transplantierten Mäusen verstarben
zwar 13,8% vor dem Alter von 12 Wochen, aber von den verbliebenen Mäusen waren 14
Tiere, also 48,3% der injizierten Mäuse, humanisiert. Auch der Anteil der humanen Zellen
war mit 22,4%±20,7 im Mittel relativ hoch.
Bei der höchsten Bestrahlungsdosis (6Gy) kam es wieder zu einer Abnahme des Anteils
humanisierter Mäuse. Von allen getesteten Bestrahlungsdosen wurden bei 6Gy aber im
Mittel der höchste Humanisierungsgrad erzielt. Nach Rekonstitution von 65 Tieren
verstarben innerhalb von 12 Wochen 25 Mäuse (38,5%). Insgesamt waren 21,5% der Mäuse
(7 Tiere) humanisiert, die mit 32,5%±28,1 humaner Zellen im peripheren Blut aber die beste
Rekonstitution zeigten.
Zusammenfassend zeigt Abb. 13D den Anteil der humanen Zellen in allen für Experimente
verwendeten Mäusen unabhängig von der Bestrahlungsdosis. Der Anteil humaner Zellen
betrug im peripheren Blut durchschnittlich 21,4%±20,8, in der Milz 44,7%±40,8 und im
Knochenmark 55,6%±10,9.
6.
Charakterisierung der humanisierten Rag2/γc/Fcγ-/- Mäuse
Nach der Transplantation der Rag2/γc/Fcγ-/- Mäuse mit humanen hämatopoetischen
Stammzellen entwickelten sich innerhalb von 12 Wochen humane hämatopoetische Zellen.
Im Folgenden sollten diese Zellen näher charakterisiert werden. Hierfür wurden humanisierte
Mäuse hinsichtlich der einzelnen Leukozytenpopulationen im Blut und in den lymphatischen
32
Ergebnisse
Organen Milz und Knochenmark mittels Durchflusszytometrie untersucht. Zum Vergleich
wurden adulte humane Proben analysiert.
6.1) Leukozytenpopulationen im humanen Immunsystem
Die einzelnen Leukozytenpopulationen im humanen Blut sind exemplarisch in Abb. 14
dargestellt. Insgesamt konnten die Proben von 6 Spendern analysiert werden. Die größte
Population im humanen Blut stellten mit 64,9%±12,2 CD3+ T-Zellen dar. Der Anteil CD19+ BZellen betrug 10,0%±2,9. Im Mittel konnten im peripheren Blut zusätzlich CD56+ NK-Zellen
(13,4%±3,5), CD33+ myeloide Zellen (Monozyten bzw. Granulozyten) (5,9%±5,3), und
CD11c exprimierende dendritische Zellen (DC) (5,5%±2,7) identifiziert werden. Der
Oberflächenmarker CD11c war dabei nicht nur auf DCs sondern auch auf CD33+ Zellen
vorhanden. Als DCs wurden daher nur CD11c einzelpositive Zellen gewertet. Im Gegensatz
zum peripheren Blut konnten in den 4 analysierten Milz-Proben 27,5%±8,5 der Leukozyten
als CD3+ T-Zellen identifiziert werden. CD19+ B-Zellen waren dagegen die größte
Leukozytenpopulation (55,9%±9,4). Auch in der Milz konnten im Durchschnitt 19,7%±9,3
CD56+ Zellen detektiert werden. Weiterhin betrug der Anteil CD11c+ DCs 7,3%±2,0 und der
Anteil CD33+ Zellen 8,8%±8,5. Bei der Analyse der Leukozytenpopulationen in 2 humanen
adulten Knochenmarksproben konnten CD19+ B-Zellen im Mittel zu 23,6%±11,4 detektiert
werden. CD3 exprimierende T-Zellen waren zu 34,3%±5,4 vorhanden. 7,9%±3,0 der
Leukozyten konnten als CD56+ NK-Zellen, 5,2%±3,8 als CD11c+ DCs und 15,6%±0,3 als
CD33 exprimierende myeloide Zellen identifiziert werden. Eine weitere Zellpopulation im
Knochenmark waren CD34+ Vorläuferzellen. Ihr Anteil betrug 2,2%±0,4 der analysierten
Zellen.
33
Ergebnisse
Abb. 14: Durchflusszytometrische Identifikation humaner
Leukozytenpopulationen
im
humanen peripheren Blut, in
der Milz und im Knochenmark.
Die entsprechenden einfachpositiven Zellpopulationen B(CD19+), T- (CD3+), NK-Zellen
(CD56+),
myeloide
Zellen
+
(CD33 ), dendritische Zellen
(CD11c+)
und
CD34+
Vorläuferzellen sind umrandet.
6.2) Leukozytenpopulationen in der humanisierten Maus
Das Auftreten der unterschiedlichen Leukozytenpopulationen wurde auch in humanisierten
Rag2/γc/Fcγ-/- Mäusen untersucht. Wegen geringer Zellzahlen wurde das periphere Blut von
15 humanisierten Mäusen zu einer Probe vereint. Für Milz und Knochenmark konnten die
Zellpopulationen in 4 PBS-behandelten, humanisierten Rag2/γc/Fcγ-/- Mäusen im Alter von 12
Wochen bestimmt werden. Wie in Abb. 15 zu sehen, traten im peripheren Blut der
humanisierten Mäuse zu diesen frühen Zeitpunkt der Rekonstitution mit 88,5% fast
ausschließlich CD19+ B-Zellen auf. Nur 5,5% der Leukozyten waren CD3+ T-Zellen.
Weiterhin konnten eine CD33+ Monozytenpopulation (3,3%) und eine CD11c+ DC-Population
(5,3%) identifiziert werden. CD56 exprimierende NK-Zellen waren mit 1,0% der Leukozyten
kaum im Blut detektierbar. Auch in der Milz humanisierter Mäuse traten bevorzugt CD19+ BZellen auf (92,3%±1,6). Eine deutliche T-Zell-Population von 1,7%±0,6 ließ sich aber
abgrenzen. Außerdem konnten wieder myeloide Zellen (1,1%±0,6), NK-Zellen (0,8%±0,5)
und dendritische Zellen (9,4%±11,7) nachgewiesen werden. Im Knochenmark war der Anteil
der CD19+ B-Zellen von den 3 analysierten Organen mit 80,7%±4,4 am geringsten. CD3+ TZellen waren zu 0,4%±0,3 in den analysierten Leukozyten vorhanden. Stattdessen konnte
34
Ergebnisse
eine große Population CD33 exprimierender Zellen (8,8%±1,1) identifiziert werden. CD11c+
DCs waren zu 6,3%±10,4 vorhanden, wogegen erneut kaum CD56+ NK-Zellen (1,6%±1,5)
detektiert werden konnten. Wie auch im humanen Knochenmark fanden sich in den
humanisierten Mäusen auch CD34+ Vorläuferzellen (2,9%±1,8).
Abb. 15: Durchflusszytometrische Identifikation humaner
Leukozytenpopulationen
im
peripheren Blut, in der Milz und
im Knochenmark humanisierter
Rag2/γc/Fcγ-/Mäuse.
Die
entsprechenden
einfachpositiven Zellpopulationen B(CD19+), T- (CD3+), NK-Zellen
(CD56+),
myeloide
Zellen
+
(CD33 ), dendritische Zellen
(CD11c+)
und
CD34+
Vorläuferzellen sind umrandet.
Das Vorhandensein der humanen Leukozyten konnte neben durchflusszytometrischen
Analysen auch durch Immunfluoreszenzfärbungen von Milzschnitten nachgewiesen werden.
Mittels fluoreszenzmarkierter spezifischer Antikörper erfolgte der Nachweis humaner CD19+
bzw. CD20+ B-Zellen in der Milz. Obwohl diese B-Zellen nicht zufällig verteilt schienen, war
eine follikuläre Organisation zu diesem frühen Zeitpunkt noch nicht zu erkennen. Weiterhin
konnten in der Milz humane CD3+ T-Zellen detektiert werden. Durch Verwendung von
Antikörpern gegen CD33 wurden myeloide Zellen, d.h. Monozyten oder Granulozyten,
identifiziert. Diese Zellen schienen zufällig über die Milz verteilt zu sein (Abb. 16).
Zusammenfassend bestätigen durchflusszytometrische Analysen von Blut, Milz und
Knochenmark das Vorhandensein humaner Leukozyten, d.h. B- und T-Lymphozyten, NKZellen, Monozyten und DCs, in den humanisierten Rag2/γc/Fcγ-/- Mäusen. B-Zellen sind dabei
die größte Zellpopulation.
35
Ergebnisse
Abb. 16:
Immunfluoreszenzfärbung der Milz humanisierter
Rag2/γc/Fcγ-/-Mäuse. Die einzelnen Leukozyten wurden mithilfe
fluoreszenzmarkierter
spezifischer Antikörper gegen CD19,
CD20,
CD3
bzw.
CD33
angefärbt.
6.3) Expression humaner FcγR in humanisierten Rag2/γc/Fcγ-/- Mäusen
Da das humanisierte Mausmodell zur Untersuchung von Einflussfaktoren auf die humane
IgG-FcγR Interaktion in vivo verwendet werden soll, wurden die humanisierten Mäuse
hinsichtlich der Oberflächenexpression der humanen FcγRIA, FcγRIIA, FcγRIIB und FcγRIIIA
untersucht.
Hierfür
wurden
die
möglichen
Effektorzellen
der
ADCC,
CD33+
Monozyten/Granulozyten und CD56+ NK-Zellen, in Blut, Milz und Knochenmark 12 Wochen
alter humanisierter Mäuse durchflusszytometrisch analysiert. Für eine bessere Einordnung
der Ergebnisse wurden erneut auch humane Vergleichsproben untersucht (Abb. 17).
CD33+ myeloide Zellen im humanen peripheren Blut zeigten eine deutliche Expression der
aktivierenden FcγRIA und FcγRIIA und des inhibitorischen FcγRIIB. In Bezug auf den
aktivierenden FcγRIIIA konnten zwei Populationen unterschieden werden: Alle Zellen waren
FcγRIIIA positiv, aber ein kleiner Teil der Zellen zeigte ein sehr hohes Expressionsniveau.
Aus humanisierten Mäusen gewonnene CD33 exprimierende Zellen zeichneten sich durch
eine vergleichbare Expression des hochaffinen FcγRIA und des niedrigaffinen FcγRIIIA aus.
Bei letztem fehlte aber die stark positive FcγRIIIA Population. Die Signalintensität bei der
Färbung von FcγRIIA und besonders FcγRIIB war im Vergleich zu humanen Zellen allerdings
vermindert (Abb. 17A). In der humanen Milz wie auch der Milz aus den humanisierten
Mäusen waren CD33+ Zellen durch die Expression von FcγRIA und FcγRIIA charakterisiert.
FcγRIIB konnte, wie schon im Blut, stärker auf humanen Zellen detektiert werden. Auf Zellen
aus den humanisierten Mäusen schien er sehr schwach vorhanden zu sein. FcγRIIIA konnte
nur auf einem kleinen Teil der humanen CD33+ Zellen detektiert werden. Auf der
Zelloberfläche von myeloiden Zellen der humanisierten Mäuse war FcγRIIIA vorhanden (Abb.
17B). Auch im Knochenmark waren humane CD33+ Zellen durch die Expression von FcγRIA
36
Ergebnisse
und FcγRIIA gekennzeichnet. Dagegen schienen FcγRIIB und FcγRIIIA auf einem Großteil
der Zellen nicht exprimiert zu sein. Es konnte jeweils nur eine kleine positive Population
identifiziert werden. Vor allem im Vergleich zu CD33+ Zellen der Milz fand sich in den
humanisierten Mäusen eine stärkere Expression von FcγRIA und FcγRIIIA aber eine
verminderte Signalintensität von FcγRIIA. FcγRIIB konnte nicht detektiert werden (Abb. 17C).
Abb. 17: Expression humaner FcγR auf CD33+ Zellen. Durchflusszytometrische Analyse in
peripherem Blut (A), in der Milz (B) und im Knochenmark (C) von humanen Proben (oben) bzw. in der
humanisierten Maus (unten).
Neben CD33+ myeloiden Zellen wurden als weitere mögliche ADCC-Effektorzellpopulation
auch CD56+ NK-Zellen in Bezug auf die Expression von FcγRIIIA, dem einzig bekannten
FcγR auf NK-Zellen, untersucht (Abb 18). In humanem Blut, Milz und Knochenmark ließen
sich NK-Zellen in 2 Populationen aufteilen. Während jeweils ein Teil der Zellen keine
Expression zeigte, konnte FcγRIII in der zweiten Population sehr stark nachgewiesen
37
Ergebnisse
werden. NK-Zellen aus humanisierten Mäusen wiesen im Blut eine schwache FcγRIIIAExpression auf. In der Milz schienen die CD56+ Zellen dagegen FcγRIIIA negativ zu sein. Im
Knochenmark war die FcγRIIIA-Expression den humanen NK-Zellen vergleichbar.
Abb. 18: Expression humaner FcγR auf NK-Zellen. Durchflusszytometrische Analyse der CD56+
Zellen in peripherem Blut (A), in der Milz (B) und im Knochenmark (C) von humanen Proben (oben)
bzw. in der humanisierten Maus (unten).
Mittels Durchflusszytometrie konnte die grundsätzliche Expression von humanen FcγR auf
den Effektorzellen der ADCC, myeloiden und NK-Zellen, nachgewiesen werden. Das
Expressionsmuster weicht dabei aber teilweise von dem der menschlichen Zellen ab.
6.4) CD20 Expression in humanisierten Rag2/γc/Fcγ-/- Mäusen
Im humanisierten Mausmodell sollte beispielhaft die ADCC-vermittelte B-Zell-Depletion nach
Gabe von anti-CD20 Antikörpern untersucht werden. Daher wurden die IgM- und IgM+ BZellen der humanisierten Mäuse hinsichtlich der Expression des Oberflächenmarkers CD20+
durchflusszytometrisch untersucht. Sowohl im humanen Blut (94,5%) als auch im Blut
humanisierter Mäuse (96,9%) war der Großteil der CD19+ Zellen durch die Expression von
IgM, nicht aber CD10 gekennzeichnet. Innerhalb der CD19+ B-Zell-Population in der Milz
exprimierten dagegen im Mittel 64,1%±21,7 der Zellen sowohl IgM als auch CD10. Im
Knochenmark waren es dagegen nur 36,4%±31,2. Dies weist auf eine Zunahme des Anteils
reiferer B-Zell-Populationen von Knochenmark und Milz in die Peripherie hin (Abb. 19A). Bei
der Analyse der CD20-Expression in den identifizierten B-Zell-Populationen zeigte sich eine
vergleichbar geringe Expression in den IgM-/CD10+ B-Zell-Vorläuferzellen in Milz und
Knochenmark (Abb. 19B). Im Blut konnten IgM-/CD10+ Zellen nicht detektiert werden. Im
Gegensatz dazu war die CD20-Expression in IgM+ B-Zellen im Blut und in der Milz deutlich
stärker ausgeprägt als im Knochenmark (Abb. 19C).
38
Ergebnisse
Abb.
19:
CD20
Expression
in
BZellen. Durchflusszytometrische Analyse von
CD19+ B-Zellen in
humanem Blut und in
Blut,
Milz
und
Knochenmark
humanisierter Mäuse.
A) Dotplot der ausgewählten CD19+ Populationen. B) Histogrammdarstellung der
CD20
Oberflächenexpression IgM- BZellen.
C)
Histogrammdarstellung der
CD20
Oberflächenexpression in IgM+ BZellen.
Im Weiteren erfolgte die durchflusszytometrische Analyse der verschiedenen B-ZellEntwicklungsstadien in den humanisierten Rag2/γc/Fcγ-/- Mäusen. Insbesondere lag der
Fokus der Untersuchungen auf der CD20 Expression in den einzelnen B-Zell-Populationen in
Milz und Knochenmark. Die CD34+/CD10+ B-Zell-Vorläufer, die sog. Pro-B-Zellen, konnten
v.a. im Knochenmark detektiert werden (Abb. 20A). In der Milz fanden sich dagegen die
reiferen, CD19+/IgM+ B-Zell-Stadien. Ein Großteil dieser Zellen exprimierte zusätzlich IgD,
den Marker naiver B-Zellen. In der Milz konnten IgM+/CD27+ Gedächtnis-B-Zellen in geringer
Zahl identifiziert werden (Abb. 20B). CD138+ Plasmazellen waren in der Milz der
untersuchten Mäuse nicht vorhanden (Daten nicht gezeigt). Wie in Abb. 20A zu sehen,
zeigten die frühen B-Zell-Vorläufer im Knochenmark noch keine Expression von CD20.
Dagegen konnte CD20 auf auf den IgM+ B-Zellen der Milz in hoher Intensität nachgewiesen
werden (Abb. 20B).
39
Ergebnisse
Abb. 20: Stadien der B-Zell-Entwicklung in Milz und Knochenmark humanisíerter Mäuse.
A) Durchflusszytometrische Bestimmung früher B-Zell-Vorläuferzellen (CD19+/CD34+/CD10+) im
Knochenmark und CD20-Expression in diesen Zellen. B) Analyse reiferer B-Zell-Populationen in der
Milz und die jeweilige CD20-Expression der markierten Population.
7.
IgG-vermittelte B-Zell-Depletion in humanisierten Mäusen
Nach der Herstellung der anti-CD20 Antikörper und der Charakterisierung der humanisierten
Rag2/γc/Fcγ-/- Mäuse wurde nun die IgG-vermittelte B-Zell-Depletion in vivo untersucht.
Hierfür erfolgte die intravenöse Injektion der produzierten 1F5 IgG Subklassen in
unterschiedlichen Dosen in humanisierte Mäuse. Die Abnahme der B-Zellen im peripheren
Blut und den lymphatischen Organen Milz und Knochenmark konnte daraufhin mittels
Durchflusszytometrie bestimmt werden. Für die Experimente wurden humanisierte Mäuse im
Alter von 11-13 Wochen verwendet, die im peripheren Blut mind. 3% humane Zellen
aufwiesen.
7.1) Fc-Abhängigkeit der 1F5 IgG induzierten B-Zell-Depletion
Anti-CD20 Antikörper depletieren B-Zellen über verschiedene direkte als auch Fc-vermittelte
Mechanismen, wie etwa die Aktivierung der Komplementkaskade oder die Bindung an
zelluläre FcγR. Deswegen sollte zunächst untersucht werden, in welchem Ausmaß die 1F5
IgG induzierte B-Zell-Depletion vom IgG Fc Fragment abhängt. Hierfür wurde das Fc
Fragment des 1F5 Antikörpers, durch Verdau mit Pepsin, entfernt. Das dabei entstehende
F(ab)2-Fragment bindet weiterhin an CD20. Sowohl der unverdaute 1F5 IgG1 als auch das
F(ab)2 Fragment wurden im äquimolaren Verhältnis in humanisierte Mäuse injiziert und die
B-Zell-Depletion im peripheren Blut analysiert. Die nach Injektion verbliebenen CD19+ Zellen
wurden auf den Ausgangswert bezogen. Die errechneten Mittelwerte sind in Abb. 21
dargestellt. Die Injektion von 1F5 IgG1 reduzierte innerhalb von 24 Stunden den Anteil der
CD19+ B-Zellen auf 6,7%±5,6. Diese Depletion war signifikant besser als nach Applikation
40
Ergebnisse
des F(ab)2-Fragments, bei dem der Anteil der B-Zellen im Mittel mit 91,2%±55,5 nahezu
unverändert blieb.
Abb. 21: Fc-Abhängigkeit der B-Zell-Depletion.
Unbehandelter (n=19) und Pepsin-verdauter
anti-CD20 IgG1 (n=4) wurden intravenös in
humanisierte Mäuse injiziert und der Anteil der
CD19+ Zellen vor und 24h nach Injektion
durchflusszytometrisch bestimmt.
Als Alternative zum F(ab)2 Fragment wurde eine mutierte Variante des 1F5 IgG1 injiziert, die
an Aminosäureposition 297 ein Alanin anstelle des Asparagin trägt (N297A). Diese Mutation
verhindert die für eine effiziente Interaktion mit FcγR bzw. C1q notwendige Glykosylierung
der IgG schweren Kette. Während die Injektion des 1F5 IgG1 zu einer Reduktion der BZellen im peripheren Blut auf 6,7%±5,6 führte, zeigte sich bei Gabe von 1F5 IgG1 N297A
eine verschlechterte B-Zell-Depletion. Innerhalb von 24h nach Injektion nahm der Anteil der
CD19+ Zellen auf 15,2%±16,3 ab (Abb. 22).
Abb. 22: Fc-Abhängigkeit der B-Zell-Depletion.
Je 10µg 1F5 IgG1 (n=19) oder 1F5 IgG1
N297A
(n=5)
wurden
intravenös
in
humanisierte Mäuse injiziert und der Anteil der
CD19+ Zellen vor und 24h nach Injektion
durchflusszytometrisch bestimmt.
7.2) B-Zell-Depletion in Blut, Milz und Knochenmark
Die Effizienz der 1F5 IgG vermittelten B-Zell-Depletion wurde durch Injektion von 100µg
Antikörper in humanisierte Mäuse im Blut, in der Milz und im Knochenmark bestimmt. Die
durchflusszytometrische Analyse erfolgte 3 Tage nach der Injektion von PBS bzw. anti-CD20
IgG1 in je 2 Mäuse. Zusätzlich wurde je eine Maus mit den anti-CD20 IgG Subtypen IgG2
und IgG3 injiziert. Außerdem wurde der Anteil der B-Zellen im peripheren Blut von 8
Patienten, die sich einer Behandlung mit einem therapeutischen anti-CD20 IgG1 Antikörper
(Rituximab) unterziehen, vor und 24 Stunden nach Rituximab-Gabe bestimmt.
41
Ergebnisse
Abb. 23: Anti-CD20 IgG induzierte B-Zell-Depletion. A) Exemplarische Darstellung der
durchflusszytometrischen Analyse der CD19+/IgM+ B-Zell-Population im Blut, in der Milz und im
Knochenmark humisierter Mäuse (3 Tage nach Gabe von PBS bzw. 1F5 IgG1) und im Blut von
Rituximab-Patienten (vor bzw. 24h nach Antikörpergabe). B) Quantifizierung des Anteils CD19+/IgM+
Zellen nach Gabe von Rituximab (n=8) bzw. Injektion von PBS (n=4) oder 100µg 1F5 IgG1 (n=2).
C) Immunfluoreszenz CD19+ Zellen in der Milz einer PBS und einer 1F5 IgG1 behandelten
humanisierten Maus. D) Quantifizierung des Anteils CD19+/IgM+ Zellen nach Injektion von 100µg 1F5
IgG2 bzw IgG3 (n=1).
In Abb. 23A ist je ein typisches Beispiel einer PBS- bzw. 1F5 IgG1 behandelten
humanisierten Maus und eines humanen Patienten unter Rituximab-Therapie dargestellt. Die
statistische Auswertung ist in Abb. 23B gezeigt. Die Leukozyten im peripheren Blut der PBSbehandelten humanisierten Mäuse bestanden im Mittel zu 77%±15,9 aus reifen B-Zellen, die
durch die Koexpression von CD19 und IgM auf der Zelloberfläche gekennzeichnet sind. Im
weiteren konnte im Blut mancher humanisierter Mäuse eine CD19+/IgM- Population
identifiziert werden. Im Gegensatz dazu exprimierten im humanen peripheren Blut alle CD19+
42
Ergebnisse
B-Zellen auch IgM. Der Anteil der reifen B-Zellen betrug hier aber nur 16,1%±12,9 der
PBMCs. In der Milz PBS-behandelter humanisierter Mäuse waren 74,5%±9,2 der
Leukozyten
reife
B-Zellen.
Im
Knochenmark
betrug
deren
Anteil
an
der
Leukozytenpopulation 36,5%±21,4. In beiden Organen waren prominente Populationen von
CD19+/IgM- B-Zell-Vorläufern zu beobachten. Die Injektion von 100µg 1F5 IgG1 führte im
peripheren Blut zu einer effizienten Depletion der reifen B-Zellen. Im Vergleich zu den PBSbehandelten Mäusen waren nach 3 Tagen im Mittel nur noch 0,6%±0,3 CD19+/IgM+ Zellen
vorhanden. Im Blut der Rituximab-behandelten Patienten sank der Anteil CD19+/IgM+ Zellen
signifikant auf 9,6%±5,8. In der Milz waren die CD19+/IgM+ Zellen nach Injektion von 1F5
IgG1 auf 8,2%±8,7 reduziert. Im Knochenmark kam es zu einer verminderten Depletion auf
57,1%±15,0 in 1F5 IgG injizierten Tieren (Abb. 23B). Je eine weitere humanisierte
Rag2/γc/Fcγ-/- Maus wurde mit 100µg 1F5 IgG2 bzw. IgG3 injiziert. Die Gabe von 1F5 IgG2
führte zu einer Reduktion der IgM+/CD19+ B-Zellen auf 16,3% in der Milz und 21,8% im
Knochenmark. Nach Injektion von 1F5 IgG3 konnten in der Milz noch 6,6% und im
Knochenmark 21,1% IgM+/CD19+ B-Zellen detektiert werden (Abb. 23D).
Die Depletion von B-Zellen nach anti-CD20 IgG1 Gabe konnte in der Milz auch durch
Immunfluoreszenzfärbung mit einem CD19-spezifischen Antikörper gezeigt werden.
Während in Schnitten PBS-behandelter Mäuse die CD19+ Zellen lose verteilt vorlagen,
konnten diese in IgG-behandelten Milzschnitten nicht mehr beobachtet werden (Abb. 23C).
Zusammenfassend führt die Injektion von anti-CD20 IgG zu einer effizienten Depletion von
CD19+/IgM+ B-Zellen im peripheren Blut, in der Milz und im Knochenmark humanisierter
Rag2/γc/Fcγ-/- Mäuse.
7.3) Depletion reifer B-Zell-Populationen Milz und Knochenmark
In der Milz und im Knochenmark der humanisierten Rag2/γc/Fcγ-/- erfolgte die genauere
durchflusszytometrische Analyse der depletierten B-Zell-Populationen. In Abb. 23 sind
exemplarisch die Ergebnisse je einer PBS- und einer mit 100µg 1F5 IgG1 behandelten Maus
an Tag 3 nach der Injektion dargestellt.
In der Milz humanisierter Mäuse konnten sehr frühe B-Zell-Vorläufer, die CD10+/CD34+ ProB-Zellen, nur in geringer Anzahl in den Kontrollmäusen und in den depletierten Mäusen
identifiziert werden. Die Koexpression von CD10 und IgM kennzeichnet reifere B-ZellEntwicklungsstufen. In der Milz humanisierter Mäuse machen diese Zellen den Großteil der
Leukozyten aus. Die Injektion von 1F5 IgG führt zu einer Verminderung CD10+/IgM+ Zellen
aber nicht zu einer vollständigen Depletion. Dahingegen konnten die CD10+/IgM- B-ZellVorläufer weiter detektiert werden. Naive B-Zellen aus, die bisher keinen Antigenkontakt
hatten, sind durch die Koexpression von IgM und IgD gekennzeichnet. Diese Zellen werden
durch die Injektion von 1F5 IgG fast vollständig depletiert. IgM+/CD27+ Gedächtnis-B-Zellen
43
Ergebnisse
waren ebenfalls nur in Kontrollmäusen nicht aber in IgG behandelten Mäusen vorhanden
(Abb. 24A).
Abb. 24: 1F5 IgG induzierte Depletion von B-Zell-Subpopulationen in humanisierten Rag2/γc/Fcγ-/Mäusen. Dargestellt ist exemplarisch die durchflusszytometrische Analyse früher und reiferer CD19+
B-Zellen in je einer PBS-behandelten Kontrollmaus und einer mit 100µg 1F5 IgG1 injizierten Maus.
A) Milz. B) Knochenmark.
Im Knochenmark wurden CD10+/CD34+ Pro-B-Zellen sowohl nach Gabe von PBS als auch
1F5 IgG identifiziert. Im Vergleich zur Kontrollmaus waren CD10+/IgM+ B-Zellen in der IgGbehandelten Maus kaum vermindert. Die reiferen IgM+/IgD+ Stadien der B-Zell-Entwicklung,
waren dagegen nur in PBS-injizierten Mäusen vorhanden. Gedächtnis-B-Zellen konnten im
Knochenmark nicht detektiert werden (Abb. 24B).
Die Gabe von hohen Dosen (100µg) 1F5 IgG verursacht in der Milz und im Knochenmark
eine Depletion von reifen B-Zellen, die durch die Koexpression von IgM und IgD
gekennzeichnet sind. CD10-exprimierende B-Zellen werden dagegen nur eingeschränkt
depletiert. CD20+ Zellen konnten nach 1F5 IgG Injektion nicht nachgewiesen werden.
44
Ergebnisse
7.4) 1F5 IgG vermittelte B-Zell-Depletion im Blut
Letztendlich wurde das humanisierte Mausmodell verwendet, um den Einfluss der FcγR
Polymorphismen auf die Interaktion von IgG und FcγR und damit die biologische Wirksamkeit
der IgG Subklassen in vivo zu untersuchen. Die Dosis der intravenös injizierten IgG wurde
hierfür von 100µg auf 10µg pro Maus reduziert um mögliche Unterschiede in der
Depletionseffizienz besser verfolgen zu können. Die durchflusszytometrische Bestimmung
der B-Zellen wurde im peripheren Blut vor und 1, 3 und 7 Tage nach Antikörpergabe in je 3
Mäusen durchgeführt. Dabei wurde die Zellzahl aller CD19+ Zellen bestimmt, da B-Zellen im
Blut CD19 und IgM koexprimieren. Zunächst wurde die B-Zell-Depletion der verschiedenen
1F5 IgG Subtypen im genetischen Hintergrund FcγRIIA131H/R und FcγR158F/V verglichen.
Beispielhaft ist die B-Zell-Depletion im peripheren Blut durch IgG1 bzw. IgG4 in Abb. 25A
gezeigt. Vor Injektion (Tag 0) bestanden die PBMCs beider Proben fast ausschließlich aus
CD19+ Zellen. Schon 24h nach Injektion von IgG1 kam es zu einer drastischen Reduktion
der B-Zellen von 2896 auf 226. Bis zu Tag 3 konnte dann ein leichter Anstieg auf 278 Zellen
beobachtet werden. Eine weitere Erholung der CD19+ Zellen konnte im Blut nicht beobachtet
werden. An Tag 7 waren nur 227 der analysierten PBMCs B-Zellen. Die Injektion von 10µg
IgG4 führte in der abgebildeten humanisierten Maus zunächst nur zu einer verminderten
Abnahme der B-Zellen von 1557 auf 814 nach 24h. Bis zu Tag 3 nach Antikörpergabe kam
es nochmals zu einer Reduktion auf 67 CD19+ Zellen. Zur Analyse an Tag 7 kam es zu einer
leichten Erholung. Es konnten 199 B-Zellen detektiert werden.
Abb. 25B zeigt die Quantifizierung und den Zeitverlauf der B-Zell-Depletion für alle IgG
Subklassen. Als Ausgleich für variable Nullwerte der B-Zell-Population, wurden für jede
einzelne Maus die bestimmten B-Zellen vor Injektion auf 100% festgelegt und die Werte
nach Antikörpergabe entsprechend umgerechnet. Im Mittel führte die Gabe von 1F5 IgG1 an
Tag 1 zu einer Depletion auf 7,8%±2,2 der PBMCs. An Tag 3 war die B-Zell-Population auf
8,8%±5,5 reduziert, bevor sie an Tag 7 nochmals leicht auf 6,2%±2,4 sank. IgG2 depletierte
innerhalb von 24h schlechter als IgG1. An Tag 1 betrug der Anteil der B-Zellen an den
PBMCs noch 23,0%±24,8, an Tag 3 fiel er auf 8,9%±9,9 und am Tag 7 nach Antikörpergabe
lag er bei 7,7%±6,2. Auch die Injektion von 1F5 IgG3 verursachte eine Abnahme der B-ZellPopulation auf 22,9%±7,7 nach 24h. Im weiteren Verlauf erreichten die B-Zellen einen Anteil
von 5,2%±1,8 nach 3 Tagen bzw. 10,1%±2,3 an Tag 7 nach Antikörperinjektion. Im Vergleich
zu IgG1, IgG2 und IgG3 war die B-Zell-Depletion innerhalb von 24h nach Gabe von 1F5
IgG4 schwächer ausgeprägt. Während es zunächst zu einer Abnahme der B-Zellen auf
66,7%±42,1 an Tag 1 kam, erreichten die B-Zellen mit 11%±9,0 an Tag 3 und 15%±12,8 an
Tag 7 ähnliche Werte wie bei Injektion von 1F5 IgG1, IgG2 und IgG3.
45
Ergebnisse
Abb. 25: Anti-CD20 IgG induzierte Depletion im peripheren Blut humanisierter Rag2/γc/Fcγ-/(FcγRIIA131H/R, FcγIIIA158F/V). A) Beispiel einer durchflusszytometrischen Analyse des Zeitverlaufs der
B-Zell-Depletion nach Injektion von 1F5 IgG1 bzw. IgG4. B) Quantifizierung CD19+ Zellen während der
IgG induzierten B-Zell-Depletion an Tag 1, Tag 3 und Tag 7 nach Antikörpergabe. Der jeweilige Anteil
der B-Zellen wurde auf den Ausgangswert der individuellen Mäuse umgerechnet (Tag 0: 100%).
Linien markieren den arithmetischen Mittelwert der untersuchten Mäuse.
Diese Experimente zeigen die durch 1F5 IgG induzierte Abnahme der B-Zellen im
peripheren Blut humanisierter Mäuse. Die IgG Subklassen 1, 2 und 3 weisen in Gegenwart
der FcγRIIA131H/R und FcγRIIIA158F/V Varianten eine vergleichbare biologische Wirksamkeit auf.
Auch 1F5 Antikörper der Subklasse IgG4 waren unter diesen experimentellen Bedingungen,
wenn auch verzögert, in der Lage B-Zellen effizient zu depletieren.
46
Ergebnisse
7.5) Einfluss der FcγRIIA131H/R und FcγRIIIA158F/V Polymorphismen
Die IgG induzierte B-Zell-Depletion im peripheren Blut humanisierter Mäuse wurde genutzt
um die biologische Wirksamkeit der IgG Antikörper in Abhängigkeit von FcγRIIA131H/R bzw.
FcγRIIIA158F/V zu analysieren. Humanisierten Mäusen, die entweder homozygot die hochaffine
oder die niedrigaffine Variante des jeweiligen FcγR exprimierten, wurden je 10µg IgG1 oder
IgG2 intravenös injiziert. Mittels Durchflusszytometrie konnte der Anteil der CD19+ Zellen an
den PBMCs vor und 24h nach Antikörpergabe bestimmt werden.
Abb. 26: Anti-CD20 IgG induzierte B-Zell-Depletion im peripheren Blut humanisierter Rag2/γc/Fcγ-/-in
Abhängigkeit vom FcγRIIA131H/R Polymorphismus. Der Anteil der CD19+ Zellen an Tag 1 nach Injektion
von 1F5 IgG1 (A) bzw. IgG2 (B) wurde auf den Ausgangswert der individuellen Mäuse umgerechnet
(Tag 0: 100%). Linien markieren den arithmetischen Mittelwert der untersuchten Mäuse.
Nach Injektion von 1F5 IgG1 kam es in Mäusen, die den niedrigaffinen FcγRIIA131R/R
exprimierten, zu einer Reduktion der B-Zellen auf 7,7%±7,9. In Gegenwart von FcγRIIA131H/H
konnte nur eine minimal verstärkte B-Zell-Depletion beobachtet werden. Der Anteil der
CD19+ Zellen betrug im Durchschnitt 4,0%±3,4 (Abb. 26A). Deutlicher aber dennoch nicht
signifikant war die Allelabhängigkeit der Depletion für 1F5 IgG2. 24h nach Antikörperinjektion
konnten im peripheren Blut von FcγRIIA131R/R Mäusen noch 21,1%±32,1 B-Zellen identifiziert
werden. Bei FcγRIIA131H/H sank der Anteil der CD19+ Zellen dagegen auf 5,3%±3,3 ab (Abb.
26B).
Im peripheren Blut von FcγRIIIA158F/F exprimierenden Mäusen führte die Injektion von IgG1 zu
einer Abnahme der CD19+ auf 5,5%±4,6. Bei Vorhandensein von FcγRIIIA158V/V wurden die
B-Zellen dagegen nur auf 7,4%±9,0 reduziert (Abb. 27A). Auch IgG2 verursachte in
FcγRIIIA158F/F Mäusen eine Depletion der B-Zellen auf 23,0%±31,1. Die biologische
Wirksamkeit von IgG2 erhöhte sich in Anwesenheit des hochaffinen FcγRIIIA158V/V, da der
Anteil der CD19+ Zellen nur noch 9,0%±6,3 betrug (Abb. 27B). Dieser Unterschied war nicht
signifikant.
47
Ergebnisse
Abb. 27: 1F5 IgG induzierte B-Zell-Depletion im peripheren Blut humanisierter Rag2/γc/Fcγ-/- in
Abhängigkeit vom FcγRIIIA158F/V Polymorphismus. Der Anteil der CD19+ Zellen an Tag 1 nach Injektion
von 1F5 IgG1 (A) bzw. IgG2 (B) wurde auf den Ausgangswert der individuellen Mäuse umgerechnet
(Tag 0: 100%). Linien markieren den arithmetischen Mittelwert der untersuchten Mäuse.
Zusammengefasst deuten die Ergebnisse auf einen Einfluss der FcγRIIA131H/R und
FcγRIIIA158F/V Polymorphismen auf die B-Zell-Depletion durch IgG2 hin.
8.
anti-CD20 IgG vermittelte Regulation von FcγR
In den humanisierten Mäusen kommt es nach Injektion von 1F5 IgG zur Depletion von BZellen. Die bisherigen Ergebnisse lassen vermuten, dass dies zumindest teilweise von der
Interaktion der IgG mit FcγR abhängig ist. Im Folgenden sollte untersucht werden, ob sich die
Induktion der ADCC auf die Oberflächenexpression der FcγR auswirkt. In mit PBS bzw.
100µg IgG injizierten humanisierten Mäusen wurde daher die FcγR-Expression von CD33+
myeloiden und CD56+ NK-Zellen in Milz und Knochenmark mittels Durchflusszytometrie
analysiert. Zum Vergleich erfolgten diese Untersuchungen auch im peripheren Blut der
Rituximab-behandelten Patienten.
Abb. 28 zeigt die Veränderungen in der FcγR Expression in 1F5 IgG behandelten
humanisierten Mäusen. Die durchflusszytometrisch bestimmten MFIs der einzelnen FcγR
wurden auf die in PBS-behandelten Kontrollmäusen erhaltenen Werte bezogen. In der Milz
humanisierter Mäuse kam es, nach Injektion der 1F5 IgG, auf der Oberfläche von CD33+
Zellen zu einer Reduktion des FcγRIIA-Signals auf 0,65AU±0,11. Noch ausgeprägter war die
Abnahme des inhibitorischen FcγRIIB (0,43AU±0,2). Dagegen waren im Mittel weder FcγRIA
(0,94AU±0,17) oder FcγRIIIA (1,17AU±1,18) durch die IgG Injektion verändert (Abb. 28A). Im
Knochenmark kam es ebenfalls zu Veränderungen der FcγR Expression auf CD33+ Zellen.
Die Signalintensität des hochaffinen FcγRIA stieg auf 1,54AU±0,76 an. Dagegen konnte
FcγRIIA (0,77AU±0,4) nur vermindert detektiert werden. FcγRIIB wurde
auf der
Zelloberfläche verstärkt identifiziert (1,16AU±0,55) und FcγRIIIA blieb mit 1,0AU±0,41
48
Ergebnisse
unverändert (Abb. 28B). Auf CD56+ NK-Zellen kam es zu einer verstärkten Detektion von
FcγRIIIA nach IgG Injektion. In der Milz konnte im Mittel eine Steigerung auf 2,88AU±1,14
und im Knochenmark auf 1,53AU±2,1 nachgewiesen werden (Abb. 28C).
Abb. 28: Veränderte FcγR Expression auf
CD33+ Zellen in Milz (A), Knochenmark (B)
und auf CD56+ Zellen (C) humanisierter
Rag2/γc/Fcγ-/Mäuse.
Durchflusszytometrische Bestimmung der Oberflächenexpression der FcγR. Die MFI der mit 100µg
1F5 IgG-behandelten Mäuse wurde auf die
MFI der PBS-Kontrollmäuse normalisiert.
Auch im peripheren Blut der Rituximab-Patienten wurde der FcγR Expressionsstatus auf
myeloiden Zellen und NK-Zellen bestimmt. In Abb. 29 sind die MFI vor und 24h nach
Rituximab-Gabe dargestellt. Während es in CD33+ Zellen zu einer signifikant verstärkten
Expression von FcγRIA nach IgG Applikation kam (Abb. 29A), schien die Signalintensität von
FcγRIIA abzunehmen (Abb. 29B). Für FcγRIIB kam es dagegen zu einer signifikanten
Herunterregulation auf der Oberfläche von CD33+ Zellen (Abb. 29C). Auch bei der
Expression von FcγRIIIA konnte eine Abnahme nach Rituximab-Gabe beobachtet werden
(Abb. 29D). In CD56+ NK-Zellen war die MFI von FcγRIIIA signifikant vermindert (Abb. 29E).
Die Ergebnisse zeigen eine veränderte FcγR-Expression auf myeloiden Zellen und NK-Zellen
nach Applikation von anti-CD20 IgG Antikörpern sowohl im humanisierten Mausmodell als
auch in humanen Patienten.
49
Ergebnisse
Abb. 29: Veränderte FcγR Expression in Rituximab-behandelten Patienten vor und 24h nach IgG
Gabe im peripheren Blut. Durchflusszytometrische Bestimmung der Oberflächenexpression (MFI) von
A) FcγRIA, B) FcγRIIA, C) FcγRIIB und D) FcγRIIIA auf CD33+ Zellen. E) Analyse der FcγRIIIAExpression auf CD56+ Zellen.
Zusammenfassend kann festgehalten werden, dass im zweiten Teil der Arbeit ein
humanisiertes Mausmodell etabliert wurde, das durch ein humanes Immunsystem mit B- und
T-Lymphozyten, myeloiden Zellen, NK-Zellen und DCs gekennzeichnet ist. Die Injektion von
1F5 IgG führte zu einer Fc-abhängigen Depletion reifer B-Zellen im Blut, in der Milz und im
Knochenmark. Für den Isotyp IgG2 war diese Depletion durch die FcγRIIA131H/R und
FcγRIIIA158F/V Polymorphismen beeinflusst. Während der B-Zell-Depletion kam es zu
veränderten FcγR-Expressionsmustern auf den CD33+ und CD56+ Effektorzellen der ADCC.
50
Diskussion
E. Diskussion
I. In vitro Bindung von Immunkomlexen an humane FcγR
Im ersten Teil dieser Dissertation wurde die Bindung von humanen IgG Immunkomplexen an
humane FcγR untersucht. IgG Antikörper treten im Serum in monomerer Form oder, in
Verbindung
mit
Bindungsaffinitäten
ihrem
und
Antigen,
als
-spezifitäten
Immunkomplexe
humaner
FcγR
auf.
Publizierte
basieren
Daten
vorwiegend
zu
auf
Untersuchungen zur Interaktion von monomerem IgG und löslichen FcγR mittels surface
plasmon resonance (SPR) und ELISA. So zeigen SPR Analysen eine starke Bindung von
IgG1, 3 und 4, nicht aber von IgG2, an den hochaffinen FcγRIA. IgG1 und IgG3 interagieren
zudem mit FcγRIIA, FcγRIIB und FcγRIIIA, während IgG2 und IgG4 für diese Rezeptoren nur
eine geringe Affinität aufweisen [62, 127]. In ELISA Experimenten kann dagegen keine
Bindung von IgG2 an FcγRIIA und FcγRIIB detektiert werden [55]. In weiterführenden
Studien wurde der Einfluss von Polymorphismen in den Genen der aktivierenden FcγRIIA,
bei dem es zu einem Aminosäureaustausch an Position 131 von Arginin zu Histidin kommt,
und FcγRIIIA, bei dem an Aminosäureposition 158 Phenylalanin oder Valin auftritt,
untersucht. Während FcγRIIA131H die Interaktion mit humanem IgG2 ermöglicht [128], scheint
sich der Polymorphismus mit Bindungskonstanten von 1-4x106M-1 dagegen kaum auf die
Interaktion mit IgG1, IgG3 und IgG4 auszuwirken [62]. In vitro Versuche mittels ELISA zeigen
außerdem eine verbesserte Interaktion von FcγRIIIA158V mit IgG1 und IgG3. Im Gegensatz zu
FcγRIIIA158F, ist FcγRIIIA158V auch in der Lage mit IgG4 zu interagieren [63, 65]. Dies konnte
durch SPR Analysen bestätigt werden, bei denen alle IgG, vor allem aber IgG2, verstärkt mit
FcγRIIIA158V interagieren [62].
Mithilfe dieser Analysen konnte eine gewisse Subklassenspezifität der humanen FcγR für
monomere IgG nachgewiesen werden. Unter in vivo Bedingungen ist vorwiegend die FcγRBindung von Immunkomplexen relevant, da die Aktivierung der Immunzellen und damit die
Induktion von Antikörpereffektorfunktionen durch Kreuzvernetzung mehrerer FcγR initiiert
wird [41]. In der Literatur ist die Herstellung von Immunkomplexen sowohl durch F(ab)2vermittelte Kreuzvernetzung der IgG, durch Hitzeaggregation [62] als auch durch chemische
Kopplung [13] beschrieben. In dieser Arbeit wurden Immunkomplexe gezielt und Antigenspezifisch generiert, indem gegen das Hapten TNP-gerichtete IgG Antikörper (Klon 7B4) und
TNP-gekoppeltes
BSA
koinkubiert
wurden.
Diese
standardisierten
Immunkomplexe
ermöglichen unter Verwendung von TNP-BSA in verschiedenen Kopplungsstufen zusätzlich
eine Steuerung der Immunkomplexgröße. Zur Analyse der Immunkomplexbindung wurden
der hochaffine FcγRIA und die niedrigaffinen FcγRIIA, FcγRIIB und eFcγRIIIA, d.h. ein
Fusionsprotein aus den extrazellulären Domänen von FcγRIIIA und den intrazellulären
51
Diskussion
Domänen von FcγRIIB, stabil in CHO Zellen exprimiert (vgl. Abb. 6). Für FcγRIIA, nicht aber
FcγRIIIA, konnten auch die polymorphen Varianten 131H und 131R untersucht werden. Auf
diese Weise konnten wir eine vergleichbare Interaktion der humanen IgG Subklassen mit
dem hochaffinen FcγRIA zeigen. Dagegen interagierten IgG1 und IgG3 Antikörper besser mit
den niedrigaffinen FcγR als IgG2 und IgG4. Eine Bindung dieser beiden Subklassen konnte
nur für FcγRIIIA und den polymorphen FcγRIIA131H festgestellt werden. Überhaupt wurde für
FcγRIIA131H eine verstärkte Interaktion mit allen IgG beobachtet. Der inhibitorische FcγRIIB,
der an Aminosäureposition 131 ebenfalls ein Arginin trägt, zeigte mit allen humanen IgG
Subklassen die schwächste Interaktion (vgl. Abb. 7). Damit stimmen unsere Ergebnisse mit
bisherigen Studien F(ab)2-vermittelter Immunkomplexe an stabil exprimierte FcγR überein
[62]. Dennoch könnte die in dieser Arbeit bestimmte allgemein verminderte Interaktion von
IgG2 und IgG4 auch auf eine verringerte Bindung an TNP-BSA zurückzuführen sein (vgl.
Abb. 5C). Da für die Klonierung der einzelnen Subklassen die gleichen variablen Domänen
verwendet wurden, kann dies nur durch einen Einfluss der konstanten Regionen auf die
Antigenbindung erklärt werden. In unserem experimentellen System stellten wir zudem eine
verstärkte Bindung von TNP-26-BSA im Vergleich zu TNP-4-BSA Immunkomplexen an die
niedrigaffinen FcγR fest. Dies weist darauf hin, dass nicht nur die Affinität sondern auch die
Avidität einen Einfluss auf die IgG-FcγR Interaktion hat, wodurch Differenzen zwischen in
vitro und in vivo Beobachtungen erklärt werden könnten. Auch wenn in dieser Arbeit nicht
gezeigt werden konnte, dass mit TNP-26-BSA generierte Immunkomplexe tatsächlich größer
sind als mit TNP-4-BSA hergestellte Komplexe, so ist die Bindung einer höheren Anzahl von
anti-TNP IgG an BSA mit 26 TNP-Molekülen und eine stärkere Vernetzung verschiedener
TNP-BSA-Komplexe dennoch wahrscheinlich. Mittels Größenausschluss-Chromatographie
könnte diese Hypothese bewiesen werden. Eine verstärkte Bindung von TNP-26-BSA
Immunkomplexen an die niedrigaffinen FcγR kann dagegen nicht auf eine verbesserte
Interaktion mit 7B4 IgG zurückgeführt werden (vgl. Abb. 5).
Sowohl für murine als auch humane IgG Antikörper ist die Glykosylierung am Asparagin 297
der schweren Ketten als essentiell für eine Interaktion mit FcγR beschrieben. Eine Mutation
des N297 zu Alanin verhindert in SPR Analysen die Bindung an FcγRIA, FcγRIIA, FcγRIIB
und FcγRIIIA [56]. Auch nach der enzymatischen Deglykosylierung mit Endoglykosidase S
(EndoS) kann keine Interaktion der monomeren IgG1, 3 und 4 mit FcγRIIA und FcγRIIB
detektiert werden [55]. In dieser Arbeit wurde der Einfluss der N297 Glykosylierung auf die
Bindung von TNP-BSA Immunkomplexen an stabil exprimierte FcγR untersucht. Hierfür
wurden die 7B4 IgG mit dem Enzym PNGaseF deglykosyliert, das alle mannoseenthaltenden Zuckerketten direkt am Asparagin schneidet. Somit wird, im Gegensatz zu
EndoS, eine vollständige Deglykosylierung ähnlich der N297A Mutation erreicht. Durch die
52
Diskussion
PNGaseF Behandlung konnte die Immunkomplexbindung an die niedrigaffinen FcγRIIA131H,
FcγRIIA131R und FcγRIIB reduziert aber nicht vollständig unterbunden werden. IgG1 und IgG3
Immunkomplexe waren dagegen in ihrer Interaktion mit FcγRIIIA weniger von der
Deglykosylierung beeinflusst. Der hochaffine FcγRIA interagierte mit den deglykosylierten
IgG1 und IgG3 Immunkomplexen nur unwesentlich schlechter als mit den unbehandelten
Antikörpern.
Bei
diesen
Untersuchungen
wurde
weiterhin
deutlich,
dass
größere
Immunkomplexe in vitro eine durch Deglykosylierung verursachte reduzierte Affinität durch
eine höhere Avidität teilweise kompensieren können (vgl. Abb. 8). Für murine IgG ist die
N297 Glykosylierung in vivo essentiell um FcγR-vermittelte Effekte induzieren zu können [1,
54].
Inwiefern
sich
die
Deglykosylierung
von
IgG
auf
die
Induktion
von
Antikörpereffektorfunktionen durch humane FcγR in vivo auswirkt, konnte dagegen bisher
nicht untersucht werden. Auch im Hinblick auf derartige Fragestellungen wurde im
Folgenden ein humanisiertes Mausmodell etabliert, dass der Untersuchung der humanen
IgG-FcγR Interaktion dient.
II. Etablierung eines humanisierten Mausmodells
Im zweiten Teil dieser Arbeit wurde ein humanisiertes Mausmodell etabliert, welches sich
durch ein humanes Immunsystem auszeichnet. Mithilfe dieses Modells war es möglich,
humane Antikörpereffektorfunktionen, die durch die Interaktion von IgG und FcγR induziert
werden, zu analysieren. Inbesondere sollten Einflussfaktoren auf diese Interaktion wie z.B.
Polymorphismen der humanen aktivierenden FcγR untersucht werden. Aufgrund von
fehlenden Modellsystemen konnten derartige Untersuchungen bisher nicht in vivo
durchgeführt werden.
Wie bereits in der Literatur beschrieben, kann durch Transplantation von humanen
hämatopoetischen Stammzellen in Rag2/γc-/- Mäuse eine Rekonstitution des humanen
Immunsystems erreicht werden. Die Immundefizienz dieser Tiere, die durch das Fehlen von
funktionellen B- und T-Lymphozyten und NK-Zellen verursacht wird, verhindert die
Abstoßung der transplantierten Zellen [102, 107]. Für die Etablierung unseres humanisierten
Mausmodells wurden dagegen Rag2/γc/Fcγ-/- Mäuse verwendet. Die zusätzliche Deletion der
Fcγ Kette in diesen Mäusen ist notwendig, da humane IgG an die murinen FcγR binden und
darüber Antikörpereffektorfunktionen auslösen können. Da Rag2/γc/Fcγ-/- keine aktivierenden
murinen FcγR exprimieren (vgl. Abb. 10), werden die zu analysierenden IgG-induzierten
Effekte nur von humanen Immunzellen vermittelt. Vor dem Zelltransfer wurden die
neugeborenen Rag2/γc/Fcγ-/- Mäuse zusätzlich sublethal bestrahlt, um die murinen
Immunzellen zu zerstören und damit eine Einnistung der humanen Zellen zu erleichtern. Die
Rekonstitution der Mäuse erfolgte mit aus Nabelschnurblut aufgereinigten, CD34+ humanen
53
Diskussion
hämatopoetischen
Stammzellen
(HSC).
Die
in
dieser
Arbeit
verwendeten
Stammzellpräparationen enthielten im Mittel 80% weitgehend undifferenzierte CD34+ HSC
(vgl. Abb. 11). Für diese Zellen ist beschrieben, dass sie nach dem Xenotransfer zu einer
langanhaltenden Humanisierung und der Entwicklung aller Leukozytenarten in den
Empfängermäusen führen [107, 129]. Problematisch ist dagegen der Transfer von ebenfalls
in unseren aufgereinigten Stammzellproben enthaltenen T- und NK-Zellen. Diese Zellen
können
direkt
die
sog.
graft
versus
host
disease
auslösen,
d.h.
das
murine
Empfängergewebe schädigen. Dies ist in unserem humanisierten Mausmodell nicht
wahrscheinlich, da die von uns transplantierten Stammzellproben nur sehr geringe Mengen
von T- und NK-Zellen enthielten.
Die Transplantation von HSC in bestrahlte Rag2/γc/Fcγ-/- Mäuse führte zur Entwicklung
humaner Immunzellen, die innerhalb von 3 Monaten im peripheren Blut nachgewiesen
werden konnten. Insgesamt wiesen 24% der xenotransplantierten Mäuse humane Zellen im
Blut auf. Die Effizienz der Humanisierung und der Anteil der humanen Zellen hing dabei stark
von der verwendeten Bestrahlungsdosis ab. Durch Erhöhung der Dosis von 3,5Gy auf 6Gy
konnte der Anteil hCD45 exprimierender Zellen im Mittel auf 32% gesteigert werden.
Allerdings traten große individuelle Schwankungen zwischen den Mäusen auf. Die
Sterblichkeit der Mäuse lag innerhalb von 12 Wochen zwischen 14% (bei 4,5Gy und 4,75Gy)
und 43% (bei 4,25Gy), wobei Bestrahlungsdosen über 6Gy in allen Fällen tödlich waren.
Daraus ergeben sich als optimale Dosen 4,75Gy, bei der die meisten humanisierten Mäuse
generiert werden konnten, und 6Gy, bei der der Anteil humaner Zellen maximal war (vgl.
Abb. 13). Eine mögliche Steigerung der Humanisierungseffizienz durch eine Erhöhung der
Anzahl transplantierter HSC wurde nicht konsequent untersucht. In der Regel wurden 50.000
HSC pro Maus transplantiert. Eine Injektion von bis zu 76.000 Zellen schien die Effizienz
nicht zu verbessern. Bisherige Studien mit humanisierten Mäusen zeigen, dass die
Rekonstitution in Abhängigkeit vom Alter der Mäuse, des verwendeten immundefizienten
Stammes und der injizierten Zellzahl, zu sehr unterschiedlichen Ergebnissen führt: Nach
dem Xenotransfer von HSC in adulte NOD/scid Mäuse konnten 1-30% humane Zellen im
Blut und in der Milz nachgewiesen werden [105]. Eine deutliche Steigerung wurde durch
Verwendung von NOD/scid/γc-/- Mäusen erzielt. Adulte Mäuse enthielten nach der
Transplantation im Blut 25% [106] und neugeborene Mäuse in Blut, Milz und Knochenmark
sogar 50-70% humane Zellen [104]. Der intrahepatische Transfer in neugeborene Rag2/γc-/Mäuse führte mit 60-65% humanen Zellen in Milz und Knochenmark [107] ebenfalls zu sehr
guten Ergebnissen. Trotzdem in dieser Arbeit die scheinbar optimalen Bedingungen auf den
neu
generierten
Rag2/γc/Fcγ-/-
Mausstamm
angewendet
wurden,
ist
die
Humanisierungseffizienz und vor allem der Anteil der humanisierten Mäuse geringer als in
der Literatur beschrieben und bedarf somit weiterer Optimierung. Da als mögliche Ursache
54
Diskussion
eine reduzierte Strahlensensitivität der murinen Zellen in Frage kommt, generieren wir
momentan neue auf Rag2/γc/Fcγ-/- basierende Mausstämme durch Einkreuzung anderer
genetischer Hintergründe mit einer erhöhten Strahlensensitivität. Der wichtige Einfluß des
genetischen Hintergrundes wird durch den Rag2/γc-/- Mausstamm belegt, der auf einen
reinen C57BL/6 Hintergrund gekreuzt wurde und kaum noch humanisierbar ist [130].
Die humanisierten Mäuse wurden mittels Durchflusszytometrie und Immunfluoreszenz auf
das Vorhandensein der humanen Leukozytenpopulationen untersucht. Diese Analysen
wurden hauptsächlich in Blut, Milz und Knochenmark durchgeführt, da es zu diesem frühen
Zeitpunkt noch nicht zur Ausbildung von Lymphknoten kam. Hierbei konnten humane Proben
zum Vergleich herangezogen werden, die von freiwilligen Spendern im Labor oder über Frau
Prof. Dr. Diana Dudziak zur Verfügung gestellt wurden. Im Alter von etwa 12 Wochen
konnten in Blut, Milz und Knochenmark der humanisierten Mäuse humane B- und TLymphozyten, NK-Zellen, dendritische Zellen und CD33 exprimierende myeloide Zellen
identifiziert werden. Anhand von Größe und Granularität der Zellen handelte es sich dabei
vermutlich großteils um Monozyten und nicht um Granulozyten. FcγRIIIB exprimierende
Granulozyten konnten nur in sehr geringem Maße nachgewiesen werden (Daten nicht
gezeigt). Im Knochenmark wurde außerdem eine CD34+ Vorläufer-Zell-Population
identifiziert. Dies deutet auf die erfolgreiche Einnistung der transplantierten Zellen hin und
gewährleistet somit eine andauernde Hämatopoese. In allen drei untersuchten Organen
machten B-Zellen den Großteil der Leukozyten aus (vgl. Abb. 15). Dieser Überschuss an BZellen zu diesem frühen Zeitpunkt nach der Transplantation ist auch für andere humanisierte
Mausmodelle [107] und Menschen nach Knochenmarkstransplantation beschrieben
(persönliche Kommunikation). Diese Zellen schienen den normalen Reifungsprozess vom
Knochenmark ausgehend in die Milz und die Peripherie zu durchlaufen, der durch eine
Zunahme IgM exprimierender B-Zellen gekennzeichnet ist. Bei CD20 handelt es sich um
einen weiteren B-Zell-spezifischen Oberflächenmarker, dessen Expression während der BZell-Differenzierung zunimmt [131], was in dieser Arbeit bestätigt werden konnte (vgl. Abb.
19 und 20). Eine Antigen-vermittelte Aktivierung der B-Zellen und die daraufhin induzierte
Bildung von Gedächtnis-B-Zellen und Plasmazellen konnte in unseren naiven humanisierten
Mäusen kaum beobachtet werden. Zusammenfassend scheint es, wie in der Literatur
beschrieben, nach dem Transfer humaner HSC zur andauernden Differenzierung der
lymphoiden und myeloiden hämatopoetischen Linien zu kommen [104, 105]. Eine
Langzeitbeobachtung unserer humanisierten Rag2/γc/Fcγ-/- Mäuse und eine Überprüfung der
Funktionalität des Immunsystems wurde dagegen bisher nicht durchgeführt. Grundsätzlich
können in humanisierten Mäusen aber B- und T-Zell-vermittelte Immunantworten
nachgewiesen werden [104].
55
Diskussion
Nach Identifikation der verschiedenen Immunzellen wurden die Mäuse auch hinsichtlich der
Expression der humanen FcγR auf myeloiden Zellen und NK-Zellen untersucht. Während
CD33+ myeloide Zellen in Blut, Milz und Knochenmark die humanen aktivierenden FcγRIA
und FcγRIIA in ähnlichem Ausmaß exprimierten wie die humanen Zellen, war das
Expressionsniveau des inhibitorischen FcγRIIB stark reduziert. Auch bei FcγRIIIA gab es
größere Abweichungen, da die stark exprimierende humane Population in den humanisierten
Mäusen nicht identifiziert werden konnte. Stattdessen schienen die Zellen alle intermediär
FcγRIIIA+ zu sein (vgl. Abb. 17). Humane NK-Zellen sind durch eine starke Expression des
aktivierenden FcγRIIIA als einzigem FcγR gekennzeichnet. In der humanisierten Maus
konnte diese Expression vor allem im Knochenmark bestätigt werden. In der Milz
humanisierter Mäuse konnte keine eindeutige NK-Zell-Population identifiziert werden (vgl.
Abb. 18). Die durchflusszytometrischen Analysen deuten sowohl in der humanen Milz als
auch im humanisierten Mausmodell auf eine geringe Anzahl von NK-Zellen hin (vgl. Abb. 14
und 15).
Es bleibt festzuhalten, dass in dieser Arbeit ein neues humanisiertes Mausmodell etabliert
werden konnte, dass sich durch die Anwesenheit von humanen CD20 tragenden BLymphozyten und humanen FcγR exprimierenden Effektorzellen auszeichnet. Die fehlende
Expression der aktivierenden murinen FcγR ermöglicht erstmals die Analyse der humanen
IgG-FcγR Interaktion und von humanen Antikörpereffektorfunktionen wie der IgG-vermittelten
B-Zell-Depletion in vivo.
III.
Anti-CD20 IgG vermittelte B-Zell-Depletion im humanisierten
Mausmodell
Die Depletion von B-Zellen ist ein vielfach angewendetes Verfahren in der Therapie von
Leukämie- und Entzündungskrankheiten, bei dem entartete bzw. pathogene Antikörperproduzierende B-Zellen zerstört werden. Ein klassisches Beispiel hierfür ist die Behandlung
von Non-Hodgkin Lymphomen und der Rheumatoiden Arthritis mit dem therapeutischen
Antikörper Rituximab. Dieser monoklonale IgG der Subklasse 1, der gegen das B-Zellspezifische Oberflächenantigen CD20 gerichtet ist, führt nach Injektion über verschiedene Fc
abhängige und unabhängige Mechanismen zu einer Reduktion der B-Zellen. Hierbei scheint
Rituximab seine Wirkung vorwiegend über die Antikörper-induzierte Zell-vermittelte
Zytotoxizität (ADCC) mithilfe von FcγR auszuüben [131, 132]. Die IgG-induzierte Depletion
von B-Zellen wurde daher als experimentelles System ausgewählt, um im humanisierten
Mausmodell Einflussfaktoren auf die Interaktion von humanem IgG und FcγR in vivo zu
untersuchen.
56
Diskussion
Hierfür wurde zunächst der gegen humanes CD20 gerichtete 1F5 Antikörper, der ebenfalls in
der Therapie von B-Zell-Lymphomen eingesetzt wurde [133], in den humanen IgG
Subklassen kloniert und produziert. Die Injektion dieser 1F5 IgG führt in humanisierten
Mäusen zur Depletion von B-Zellen (vgl. Abb. 23). Zunächst sollte der Wirkmechanismus,
d.h. die Abhängigkeit der Depletion von der Interaktion der Antikörper mit FcγR, ermittelt
werden. Die Applikation des aus 1F5 IgG1 generierten F(ab)2 Fragments führte kaum zu
einer B-Zell-Depletion, wodurch die essentielle Rolle des IgG Fc Fragments gezeigt werden
konnte (vgl. Abb. 21). Die Applikation eines mutierten 1F5 IgG1, dessen schwere Kette keine
Asparagin-verknüpften Glykosylierungen aufweist (N297A), führte im Gegensatz zum F(ab)2
Fragment zu einer effizienten Reduktion der B-Zellen (vgl. Abb. 22). Frühere Publikationen
zeigen, das aufgrund der N297A Mutation keine Interaktion mit FcγR [56] und dem
Komplementfaktor C1q [134] stattfindet. Während die fehlende C1q Bindung in dieser Arbeit
bestätigt wurde (Daten nicht gezeigt), weisen die von uns durchgeführten in vitro
Experimente auf eine Interaktion von FcγRIA und FcγRIIIA mit deglykosyliertem IgG1 hin.
Demzufolge könnte die B-Zell-Depletion nach Gabe von N297A in vivo durch FcγRIA und
FcγRIIIA, nicht aber C1q, vermittelt sein. Ein direkter Einfluss der IgG auf B-Zellen wie
beispielsweise eine Hemmung der Proliferation oder eine Induktion der Apoptose, wie sie für
Rituximab beschrieben wurden [132], könnte zusätzlich zur Fc vermittelten Depletion
stattfinden.
Auch die teilweise verminderte Oberflächenexpression der niedrigaffinen FcγR auf den
myeloiden Effektorzellen deutet auf eine FcγR-Abhängigkeit der B-Zell-Depletion hin. Nach
Gabe von anti-CD20 IgG konnten wir sowohl in den humanisierten Mäusen als auch im
untersuchten Patientenblut eine Abnahme von FcγRIIA und FcγRIIB detektieren (vgl. Abb. 28
und 29). Die Reduktion der FcγR war im Blut stärker ausgeprägt als in der Milz und schien
dadurch mit der Effizienz der B-Zell-Depletion zu korrelieren. Im Knochenmark kam es,
passend zu einer verminderten B-Zell-Depletion, dagegen kaum zu Veränderungen. Zum
einen deutet die reduzierte Oberflächenexpression auf eine Phagozytose der B-ZellImmunkomplexe hin. Andererseits wäre auch eine mangelnde Erkennung der FcγR bei der
durchflusszytometrischen Analyse aufgrund gebundener Immunkomplexe denkbar.
Die Injektion von 100µg 1F5 IgG führte zu einer effizienten Depletion reifer (CD19+/IgM+) BZellen im Blut, in der Milz und im Knochenmark (vgl. Abb. 23). Hierbei konnten bei der
niedrigen Anzahl analysierter Mäuse geringe Unterschiede zwischen den untersuchten IgG
Subtypen festgestellt werden. Während IgM+/CD19+ B-Zellen in der Milz durch IgG1, 2 und 3
vergleichbar depletiert wurden, deuten die Ergebnisse auf eine gute IgG2- und IgG3- nicht
aber IgG1-induzierte Reduktion der Zellen im Knochenmark hin. Die möglicherweise
verminderte Depletion IgM+ B-Zellen im Knochenmark, im Vergleich zu Blut und Milz, ist
57
Diskussion
vermutlich auf eine veränderte Expression des CD20-Moleküls zurückzuführen, da das CD20
Expressionsniveau innerhalb der CD19+/IgM+ Zellen im Vergleich zu Blut und Milz stark
reduziert war (vgl. Abb. 19). Das Expressionsniveau von CD20 wird auch im Menschen als
ein bestimmender Faktor für die Rituximab-Sensitivität angenommen [132]. Erstmals war es
im humanisierten Mausmodell auch möglich, eine genaue Analyse der depletierten humanen
B-Zell-Populationen in Milz und Knochenmark durchzuführen. IgM negative B-Zellen wie z.B.
die CD10 und CD34 exprimierenden Pro-B-Zellen wurden durch 1F5 IgG nicht depletiert, da
diese Zellen kein CD20 auf ihrer Oberfläche tragen. Dagegen wurde eine Reduktion von
CD20 exprimierenden unreifen (CD10+/IgM+) bzw. naiven (IgM+/IgD+) B-Zellen beobachtet.
Die Depletion von Gedächtnis-B-Zellen (CD27+) und Plasmazellen konnte dagegen im
humanisierten Mausmodell nur eingeschränkt analysiert werden, da diese Zellen in den
naiven Mäusen nur selten auftraten (vgl. Abb. 24).
Ein möglicher Einfluss der verwendeten IgG Subklasse auf die Effizienz der B-Zell-Depletion
wurde im Blut durch Injektion von je 10µg 1F5 IgG bestimmt. Diese Dosisreduktion wurde
durchgeführt, da die bisherigen Ergebnisse bei Injektion von 100µg 1F5 IgG darauf
hindeuteten, dass bei dieser Dosis kaum Subklassen-spezifische Unterschiede existieren.
Alle injizierten IgG waren, in unterschiedlichem Ausmaß, in der Lage B-Zellen zu depletieren.
Dabei kam es schon innerhalb von 24h zur Reduktion der peripheren B-Zellen, deren Anteil
bis zum Ende des Beobachtungszeitraums an Tag 7 nach der Injektion stark erniedrigt blieb.
Da die Halbwertszeit von IgG Antikörpern im Serum mehr als 1 Woche beträgt [135], könnte
dies auf eine Depletion von frisch ins Blut eingewanderten B-Zellen zurückzuführen sein.
Vermutlich kommt es dadurch nicht zur Rekonstitution der peripheren B-Zell-Population
durch Zellen aus Milz und Knochenmark. Die ausgeprägteste B-Zell-Depletion an Tag 1 nach
Antikörpergabe trat bei 1F5 IgG1 und IgG3 auf (vgl. Abb. 25). Dies bestätigt die in vitro
Ergebnisse dieser Arbeit, nach denen IgG1 und IgG3 Antikörper effizient mit allen FcγR
interagieren
können.
möglicherweise
über
Außerdem
eine
können
Interaktion
diese
mit
Subklassen
dem
nach
CD20-Bindung
Komplementfaktor
C1q
die
Komplementkaskade aktivieren, was zur Komplement-vermittelten Lyse der B-Zellen führen
würde. Die humanisierten Rag2/γc/Fcγ-/- Mäuse exprimieren die Proteine des murinen
Komplementsystems (Daten nicht gezeigt), die aber eine große Ähnlichkeit mit den humanen
Proteinen aufweisen [136]. Eine Beteiligung des Komplementsystems an der 1F5 IgG
induzierten B-Zell-Depletion ist also wahrscheinlich, wurde aber in dieser Arbeit nicht weiter
untersucht. Auch die Injektion von 1F5 IgG2 führte zu einer effizienten Reduktion der
peripheren B-Zellen. IgG2 Antikörper binden vor allem an den hochaffinen FcγRIA und an die
polymorphen FcγRIIA131H und FcγRIIIA158V. Da die verwendeten Mäuse den genetischen
Hintergrund FcγRIIA131H/R und FcγRIIIA158F/V aufwiesen, ist eine IgG2 vermittelte B-ZellDepletion über diese FcγR wahrscheinlich. Ein Beteiligung des Komplementsystems ist auch
58
Diskussion
für IgG2 möglich, da dieser Isotyp in vitro schwach mit C1q interagierte (vgl. Abb. 9). IgG4
Immunkomplexe zeigten in vitro ebenfalls eine starke Bindung an FcγRIA, FcγRIIA131H und
FcγRIIIA158V. In vivo war bei Injektion von 1F5 IgG4 die schwächste B-Zell-Depletion
innerhalb von 24 Stunden zu beobachten. Erst nach 3 Tagen erreichten die B-Zellen das
Level der IgG1-vermittelten Depletion. Dies deutet darauf hin, dass IgG4, der in vitro nicht
mit C1q interagierte, im humanisierten Mausmodell FcγR-vermittelte Effektorfunktionen
ausübt und widerspricht damit der Anwendung von therapeutischen Antikörpern der
Subklasse IgG4, wenn Fc vermittelte Effekte unerwünscht sind [135]. Die in vitro ermittelte
reduzierte Affinität von 1F5 IgG2 und IgG4 Antikörpern für CD20 ist möglicherweise ein
weiterer Grund für eine verminderte B-Zell-Depletion (vgl. Abb. 9).
Das humanisierte Mausmodell sollte der Untersuchung von Einflussfaktoren auf die IgGFcγR Interaktion in vivo dienen. Unterschiede bei der im Blut bestimmten B-Zell-Depletion
zwischen
einzelnen
Mäusen
könnten
einerseits
durch
eine
hohe
Varianz
der
Humanisierungseffizienz erklärt werden. Im Blut der verwendeten humanisierten Mäuse
traten zwischen 3% und 80% humane Zellen auf. Andererseits bestanden die PBMCs,
bedingt durch ein Alter der Mäuse von nur 12 Wochen, fast ausschließlich aus B-Zellen. Eine
verminderte B-Zell-Depletion könnte somit z.B. durch einen Mangel an FcγR exprimierenden
Effektorzellen bedingt sein. Aufgrund der verschiedenen Stammzellproben, die für die
Rekonstitution benutzt wurden, können außerdem individuelle genetische Einflussfaktoren in
den humanisierten Mäuse nicht ausgeschlossen werden. So besteht die Möglichkeit, dass
humanisierte
Mäuse
z.B.
mit
dem
polymorphen
und
damit
in
seiner
Funktion
232T
eingeschränkten FcγRIIB (FcγRIIB
) generiert worden sind. Eine reduzierte Funktionalität
des inhibitorischen FcγRIIB würde sich direkt positiv auf die B-Zell-Depletion auswirken.
IV.
Einfluss der FcγR Polymorphismen in vivo
Mögliche Einflussfaktoren auf die biologische Wirksamkeit von IgG Antikörpern in vivo sind
Polymorphismen in den Genen der aktivierenden humanen FcγRIIA (131H/R) und FcγRIIIA
(158F/V). Diese Polymorphismen führen zur Expression von FcγR Varianten, die in vitro eine
veränderte Affinität für IgG Antikörper aufweisen. In vivo konnte ein Einfluss der FcγR
Polymorphismen dagegen bisher nicht eindeutig nachgewiesen werden. Einen ersten
Hinweis geben epidemiologische Studien, in denen die polymorphen FcγR Varianten mit dem
therapeutischen Erfolg von monoklonalen Antikörpern, wie beispielsweise Rituximab,
assoziiert sind. Daher wurden die für die Rekonstitution verwendeten hämatopoetischen
Stammzellen
hinsichtlich
der
FcγR
Polymorphismen
typisiert
und
entsprechende
humanisierte Mäuse generiert. Anhand der 1F5 IgG induzierten B-Zell-Depletion konnte
somit die in vivo Relevanz der FcγR Polymorphismen analysiert werden.
59
Diskussion
Obwohl in vitro Experimente eine verstärkte Interaktion von FcγRIIA131H und FcγRIIIA158V mit
den humanen IgG Antikörpern zeigen [62], was in dieser Arbeit für FcγRIIA bestätigt werden
konnte, scheint es im humanisierten Mausmodell keinen signifikanten Einfluss der FcγR
Polymorphismen auf die IgG-induzierte B-Zell-Depletion zu geben. Die intravenöse Gabe
von 10µg 1F5 IgG1 führte in FcγRIIA131H und FcγRIIA131R Mäusen zu einer vergleichbaren
Reduktion der peripheren B-Zellen (vgl. Abb. 26). In humanisierten Mäusen, die den
niedrigaffinen FcγRIIIA158F exprimieren, trat eine leicht verbesserte B-Zell-Depletion im
Vergleich zum hochaffinen FcγRIIIA158V auf (vgl. Abb. 27). Auch bei den bereits in der
Therapie befindlichen monoklonalen Antikörpern handelt es sich zumeist um Antikörper der
Subklasse IgG1 [135]. Epidemiologische Studien von Patienten, die mit Rituximab behandelt
werden, zeigen eine bessere Wirksamkeit des therapeutischen Antikörpers bei der
Behandlung des follikulären Non-Hodgkin Lymphoms in Gegenwart von FcγRIIA131H [137,
138] und FcγRIIIA158V [68, 138]. Unter unseren bisherigen experimentellen Bedingungen
können wir diese Ergebnisse in vivo nicht bestätigen, wofür verschiedene Gründe
ausschlaggebend sein könnten. Ein sehr wichtiger Aspekt ist die Tatsache, dass die
humanisierten Tiere im Alter von 11-13 Wochen selbst kein humanes Serum IgG aufweisen.
Dies spielt insbesondere für die Regulation der Funktion der Aktivität des hochaffinen FcγRIA
eine wichtige Rolle. FcγRIA wird auf Monozyten und Makrophagen exprimiert, denen eine
Beteiligung an der IgG vermittelten B-Zell-Depletion zugeschrieben wird [139]. Durch die
hohen Mengen an Serum IgG (etwa 9mg/ml) ist dieser Rezeptor im Blut normalerweise mit
IgG gesättigt und kann nicht mehr an Immunkomplexe binden. Im humanisierten Mausmodell
fehlt diese Sättigung, wodurch der hochaffine FcγRIA in der Lage ist, mit den 1F5 IgG zu
interagieren. Somit könnte FcγRIA im humanisierten Mausmodell, im Gegensatz zu
therapeutischen Antikörpern im Menschen, an der B-Zell-Depletion beteiligt sein.
Anhaltspunkte hierfür geben auch die in vitro Befunde im ersten Teil der Arbeit, in denen
gezeigt werden konnte, dass FcγRIA mit Immunkomplexen aller IgG Subklassen interagieren
kann. Durch Blockierung des FcγRIA mittels spezischer Antikörper oder Sättigung durch
Gabe von monomerem IgG vor Injektion der 1F5 IgG soll diese Frage geklärt werden.
Eine weitere Erklärung für diese Befunde könnte die von uns verwendete Menge an B-Zell
depletierenden IgG Subklassenvarianten sein. Um den Einfluss der FcγR Polymorphismen
auf die B-Zell-Depletion zu untersuchen, wurden je 10µg IgG pro Maus injiziert. Diese Dosis
wurde gewählt, da bei 10µg eine effiziente aber noch nicht vollständige Depletion der BZellen im peripheren Blut stattfindet und bei höheren Dosen wie z.B. 100µg weder ein
Einfluss der verwendeten IgG Subklasse noch der FcγR Polymorphismen zu erkennen war.
Möglicherweise ist eine Dosis von 10µg, besonders in Anwesenheit von FcγRIA, dennoch zu
hoch um im humanisierten Mausmodell einen Einfluss der FcγR Polymorphismen im Blut zu
60
Diskussion
detektieren. Daher sollen in Zukunft niedrigere Dosen und vor allem auch andere Gewebe in
die Analyse einbezogen werden. Bei der Anwendung von Rituximab im Menschen werden
mit 375mg/m2 wesentlich höhere Dosen des IgG verwendet. Als Vergleich zum
humanisierten Mausmodell bestand im Rahmen einer Kooperation mit Dr. Meidenbauer
(Medizinische Klinik V, Universitätsklinikum Erlangen) die Möglichkeit humanes Patientenblut
vor und 24 Stunden nach der Gabe von Rituximab zu untersuchen. In allen analysierten
Patienten kam es nach Antikörpergabe zu einer effizienten Reduktion der peripheren BZellen. Hierbei konnten ebenso keine Unterschiede zwischen Patienten mit den jeweiligen
FcγR Polymorphismen festgestellt werden (Daten nicht gezeigt). Eine Analyse der B-ZellDepletion in Milz oder Knochenmark konnte nicht durchgeführt werden.
Weiterhin kann nicht ausgeschlossen werden, dass auch eine Komplement-vermittelte BZell-Depletion eine Rolle spielen könnte. Wie bereits erwähnt, sind besonders Antikörper der
Subklassen IgG1 und IgG3 in der Lage Komplement-abhängige Effektorfunktionen zu
induzieren. Zudem ist die Komplement-vermittelte Lyse von B-Zellen für den anti-CD20 IgG1
Rituximab in in vitro Experimenten beschrieben worden [132]. Wegen ihrer eingeschränkten
Bindung an humane FcγR werden IgG2 Antikörper im Gegensatz zur Subklasse IgG1 bisher
nur selten als Therapeutika eingesetzt [135]. Dennoch führte die Injektion von 1F5 IgG2 im
humanisierten Mausmodell zu einer effizienten Reduktion der peripheren B-Zellen. In
Abhängigkeit der FcγR Polymorphismen zeigte sich eine leicht verbesserte Depletion in
Gegenwart von sowohl FcγRIIA131H als auch FcγRIIIA158V (vgl. Abb. 26 und 27). Als mögliche
Erklärungen für die verstärkte B-Zell-Depletion durch diese Varianten in vivo, kommen neben
der höheren Affinität, die eine verstärkte Effektorzellaktivierung bedingt [64], auch ein
erhöhtes Expressionsniveau des FcγR auf den Effektorzellen der B-Zell-Depletion [70] in
Frage. Dies konnte im humanisierten Mausmodell bisher nicht untersucht werden. Da IgG2
Antikörper in vitro schwach mit C1q interagieren können, wäre auch hier ein geringer
Einfluss des Komplementsystems auf die IgG2 induzierte B-Zell-Depletion möglich.
Zusammenfassend wurden im Rahmen dieser Dissertation Einflussfaktoren auf die
Interaktion von humanen IgG Antikörpern und FcγR in vitro und in vivo analysiert. In vitro
wurde die FcγR Interaktion wesentlich durch die verwendete IgG Subklasse, die Größe der
IgG-Immunkomplexe und deren Glykosylierung und durch Polymorphismen in den
aktivierenden FcγRIIA und FcγRIIIA beeinflusst. In einem humanisierten Mausmodell konnte
die biologische Wirksamkeit der IgG Antikörper anhand der Antikörper-induzierten B-ZellDepletion in vivo untersucht werden. Unter diesen experimentellen Bedingungen wurden die
IgG Subklassen hinsichtlich ihrer in vivo Aktivität verglichen. Außerdem wurden erste
Hinweise gewonnen, die auf einen Einfluss der FcγR Polymorphismen bei IgG-vermittelten
Effektorfunktionen hindeuten. Durch eine Optimierung des humanisierten Mausmodells in
61
Diskussion
Bezug auf die Humanisierungseffizienz und eine Anpassung an humane IgG-Serumspiegel,
bietet das in dieser Arbeit etablierte Modell die Möglichkeit weitergehende Untersuchungen
humaner Antikörpereffektorfunktionen in vivo durchzuführen.
62
Material und Methoden
F. Material und Methoden
I. Material
1.
Chemikalien, Plastik- und Verbrauchsmaterialen
Soweit nicht anders angegeben, stammen alle in dieser Arbeit verwendeten Chemikalien,
Reagenzien,
Plastik-
und
Verbrauchsmaterialen
von
den
Firmen
Greiner-Bio-One
(Frickenhausen), Sarstedt (Numbrecht), Nunc (Wiesbaden), Millipore (Schwalbach), VWR
(Ismaning), J. Peske (Aidling-Arnhofen), Roth (Karlsruhe), Eppendorf (Hamburg), Corning
(Wiesbaden), Miltinyi Biotec (Bergisch-Gladbach), Roche (Mannheim), Sigma (Steinheim),
Thermo Fisher Scientific (Rockford, USA), Invitrogen (Darmstadt), New England Biolabs
(Frankfurt).
2.
Puffer und Lösungen
Alle Puffer und Lösungen wurden wie im Methodenteil beschrieben und mit Reinstwasser
(bidest) hergestellt. Fötales Kälberserum (FCS) wurde vor der Verwendung bei 56°C für
30min hitzeinaktiviert.
3.
Kommerzielle Kits
Kit
Firma, Herkunft
5´-RACE System for Rapid Amplification of cDNA ends
Invitrogen, Darmstadt
Alexa FluorR 647 Monoclonal Antibody Labeling Kit
Molecular Probes, Leiden, NL
Direct CD34 Progenitor Cell Isolation Kit, human
Miltenyi Biotec, BergischGladbach
DSB-XTM Biotin Protein Labeling Kit
Invitrogen, Darmstadt
FITC Labeling Kit
KPL, Gaitherburg, USA
PureLink HiPure Maxiprep Kit
Invitrogen, Darmstadt
QiaAmp DSP Blood Mini Kit
Qiagen, Hilden
Qiaquick Gel extraction Kit
Qiagen, Hilden
RNeasy Mini Kit
Qiagen, Hilden
Zero Blunt TOPO PCR Cloning Kit
Invitrogen, Darmstadt
63
Material und Methoden
4.
Antikörper und Konjugate
Im Folgenden sind die in dieser Arbeit verwendeten Antikörper zusammengefasst. Mit *
gekennzeichnete Konjugationen wurden mit den obengenannten Kits vorgenommen.
Gegen humane Antigene gerichtete Antikörper:
64
Antigen
Konjugat
Klon
Anwendung
Verdünnung
Hersteller
CD3
CD3
CD4
CD8
CD10
CD11c
CD11c
CD14
CD16
CD16
CD16B
CD19
CD19
CD19
CD19
CD20
CD20
CD20
CD21
CD23
CD27
CD32
CD32
CD32B
CD33
CD33
CD34
CD34
CD38
CD45
CD45
CD45
CD56
CD56
CD64
CD64
CD89
CD138
IgD
IgM
IgM
IgM
PE
PerCP
APC
PE
PerCP
FITC
biotin
PE
FITC
FITC
PE
FITC
PE
PE
PE-Cy7
FITC*
Alexa647*
Alexa647*
APC
PE
PE
FITC
Alexa647
biotin*
APC
PE-Cy7
FITC
PE
APC
PerCP
APC
APC-H7
FITC
PE
FITC
FITC
PE
PE
biotin
FITC
PE
biotin
UCHT1
UCHT1
RPA-T4
Hit8α
MEM78
3.9
3.9
M5E2
3G8
3G8
CLB-gran11.5
HIB19
HIB19
HIB19
SJ25C1
1F5
1F5
1F5
B-ly4
M-L233
L128
3D3
FUN-2
2B6
P67-6
P67-6
8.G12
8.G12
HB-7
2D1
HI30
2D1
NCAM16.2
NCAM16.2
10.1
10.1
A59
MI-15
IA6-2
G20-127
G20-127
G20-127
IF
FACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
MACS
FACS
FACS
FACS
IF
FACS
FACS
FACS
IF
FACS
FACS
FACS
MACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
MACS
FACS
FACS
FACS
FACS
IF
FACS
1:10
1:200
1:50
1:50
1:50
3µl
1:50
1:50
1:50
1:10
1:25
1:25
1:50
1:10
1:50
1:500
1:600
1:10
1:25
1:25
1:50
1:10
1:400
1:200
1:25
1:50
3µl
1:25
1:300
1:25
1:25
1:300
1:30
1:20
1:50
1:10
1;25
1:25
1:200
1:25
1:10
1:200
IgG Fc
PE
FACS
1:1000
IgG Fc
HRP
ELISA
1:10000
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
ebioscience
ebioscience
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
Eigenproduktion
Eigenproduktion
Eigenproduktion
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
Eigenproduktion
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
BD Pharmingen
Jackson
Immunoresearch
Bethyl
Material und Methoden
Gegen Antigene der Maus gerichtete Antikörper:
Antigen
Konjugat
Klon
Anwendung
Verdünnung
Hersteller
FcγRI
FcγRIIB/III
FcγRIIB/III
FcγRIII
FcγRIV
B220
TCRb
CD11b
NK1.1
CD45.1
CD45.2
CD45.2
Alexa647*
unkonjugiert
PE
PE
Alexa647
APC
FITC
PerCP-Cy5.5
PE
Biotin
FITC
biotin
X54-517.1
2.4G2
2.4G2
275003
9E9
RA3-6B6
H57-597
M1/70
PK136
A20
Ly5.2
104
FACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
FACS
1:400
1:100
1:100
1:50
1:400
1:400
1:200
1:400
1:200
1:100
1:300
1:100
BD Pharmingen
Eigenproduktion
BD Pharmingen
R&D
Eigenproduktion
BD Pharmingen
BD Pharmingen
BD Pharmingen
Biolegend
Biolegend
BD Pharmingen
BD Pharmingen
Verwendete Sekundärantikörper:
Antigen
Konjugat
Klon
Anwendung
Verdünnung
Hersteller
Biotin
Biotin
APC-Cy7
HRP
Streptavidin
Streptavidin
FACS
ELISA
1:1000
1:10000
BD Pharmingen
BD Pharmingen
5.
Oligonukleotide
Die in dieser Arbeit verwendeten Oligonukleotide stammen von der Firma Biomers.net, Ulm.
6.
Software
Software
Firma, Herkunft
Verwendung
Adobe Reader 8.0
Adobe Systems, USA
Textverarbeitung
AxioVision LE 4.6
Zeiss, Jena
Mikroskopie
FACSDiva
Becton Dickinson, USA
Durchflusszytometrie
EasyWin32
Herolab, Wiesloch
Geldokumentaion
Flowjo
Tree Star, USA
Durchflusszytometrie
GraphPad Prism 3.03
GraphPad Software Inc., USA
Graphische Darstellung
MS Excel 2003
Microsoft, USA
Statistik
MS PowerPoint 2003
Microsoft, USA
Präsentation
MS Word 2003
Microsoft, USA
Textverarbeitung
Softmax Pro
Molecular Devices, USA
ELISA
VectorNTI 10.3
Invitrogen, Darmstadt
Sequenzbearbeitung
65
Material und Methoden
II. Methoden
1.
Klonierung der 7B4 und 1F5 IgG Antikörper
1.1) Klonierung der variablen Regionen
Die variablen, für die Antigenerkennung verantwortlichen, Domänen der monoklonalen
Antikörper 1F5 und 7B4 (beide vom IgG2a Isotyp der Maus) wurden aus den
entsprechenden Hybridomzelllinien kloniert. Da die kodierenden Sequenzen der variablen
Bereiche von schwerer (VH) und leichter (VL) Kette unbekannt waren, wurde von der
bekannten Sequenz der konstanten Regionen (CH bzw. CL) ausgehend eine 5´-RACE (Rapid
Amplification of cDNA ends) durchgeführt um das Startcodon zu ermitteln. Hierzu wurde
zunächst die Gesamt-RNA aus 6x106 (1F5) bzw. 1x107 (7B4) Zellen mit dem „RNeasy Mini
Kit“ (Quiagen, Hilden) isoliert. Diese wurde anschließend entsprechend der Anleitung des
Herstellers
mit
dem
„5´-RACE
System
for
Rapid
Amplification
of
cDNA
ends“
weiterverarbeitet (Invitrogen, Darmstadt). Die 5´RACE beinhaltet die Reverse Transkription
von 3µg RNA in komplementäre Desoxyribonukleinsäure (cDNA) unter Verwendung eines
Gen-spezifischen Primers (Primer #1 bzw. #3, sh. Tabelle 2), die Aufreinigung der cDNA und
das Anhängen eines homopolymeren Schwanzes an das 3´-Ende durch das Enzym
Terminale Desoxynukleotid Transferase (TdT). Die so aufbereitete cDNA konnte für die
Amplifikation der variablen Regionen mithilfe eines weiteren Gen-spezischen Primers
(Primer #2 bzw #4) und dem im Kit enthaltenen „Abridged Anchoring Primer“ verwendet
werden. Die Produkte der Polymerasekettenreaktion (PCR) wurden elektrophoretisch in
einem 1%igem Agarosegel mit 40ng/ml Ethidiumbromid aufgetrennt, die entsprechenden
Fragmente einer Größe von je ca. 750bp aus dem Gel ausgeschnitten und mittels dem
„Qiaquick Gel extraction Kit“ (Qiagen, Hilden) entsprechend der Herstellerhinweise
aufgereinigt. Anschließend erfolgte die Klonierung mithilfe des „Zero Blunt TOPO PCR
Cloning Kit“ (Invitrogen, Darmstadt) in den Vektor pCRII-TOPO-blunt. Nach der
Transformation in chemisch kompetente Bakterien („One-Shot chemically competent E.Coli“,
Invitrogen, Darmstadt) wurde die Plasmid-DNA mithilfe der Minilysat-Methode präpariert und
die erhaltene DNA sequenziert. Durchgehende offene Leserahmen wurden anhand einer
BLAST-Analyse hinsichtlich Homologien zu anderen schweren und leichten Ketten getestet.
Anhand der erhaltenen Sequenzen erfolgte das Primerdesign für die Klonierung der
schweren und leichten Ketten.
1.2) Klonierung der schweren und leichten Ketten
Korrekte variable Regionen und die bereits vorhandenen konstanten Domänen
der
humanen IgG Subtypen (bereitgestellt von Prof. Dr. Nimmerjahn) wurden mittels PCR durch
98°C 2min, 30 Zyklen von 98°C 15sec, 56°C 15sec, 72°C 45sec und eine finale Elongation
von 5min bei 72°C amplifiziert. Die Fragmente mit einer Größe von ca. 500bp (variable
66
Material und Methoden
Domänen), ca. 1000bp bzw. etwa 250bp (konstante Domäne der leichten Kette) wurden
anschließend gelelektrophoretisch aufgereinigt. Mithilfe einer zweiten PCR konnten die VH
und CH (schwere Kette) bzw. VL und CL (leichte Kette) Regionen fusioniert werden. Die
Amplifikation erfolgte bei 98°C 2min, 30 Zyklen von 98°C 15sec, 56°C 15sec, 72°C 1min und
eine finale Elongation von 5min bei 72°C. Die Reaktionsbedingungen lauteten für diese
Klonierungsreaktionen je 50µl Ansatz wie folgt: je 1µM Primer, 0,2mM dNTPs (NEB,
Frankfurt), 0,5µl Phusion High Fidelity DNA Polymerase (NEB, Frankfurt), 1x Phusion High
Fidelity Reaktionspuffer.
Sowohl die erhaltenen schweren Ketten als auch der eukaryonte Expressionsvektor pBos
wurden mit den Restriktionsenzymen (alle NEB, Frankfurt) EcoRI und KpnI geschnitten.
Anschließend wurden 200ng der schweren Ketten in 60ng pBos mithilfe von 1µl des Enzyms
T4 DNA Ligase (NEB, Frankfurt) für 1h bei RT ligiert. Die leichten Ketten wurden mit NcoI
und
XhoI
verdaut
und
in
den
mit
NcoI
und
XhoI
geschnittenen
eukaryonten
Expressionsvektor pCMV ligiert.
Primer #
Klonierung
VH und VL
(5´-RACE)
Klonierung
der
schweren
Ketten
Klonierung
der leichten
Ketten
Orientierung
Sequenz (5´Æ3´)
antisense
gacagggatccagagtt
1
Gen-spezifischer Primer 1 (VH)
2
Gen-spezifischer Primer 2 (VH)
antisense
gtactctagaggtcaaggtcactggctca
3
Gen-spezifischer Primer 1 (VL)
antisense
cctgttgaagctcttgaca
4
Gen-spezifischer Primer 2 (VL)
antisense
gtactctagagggtgaagttgatgtcttgtc
5
Amplifikation 7B4 VH
sense
cgtagaattcaccaccatgggttggctgtggaact
6
Amplifikation 1F5 VH
sense
cgtagaattcaccaccatgggatggagttgtatcatc
7
Amplifikation IgG1/2/4 CH
antisense
cggggtacccgtcatttacccggagacagg
8
Amplifikation IgG3 CH
antisense
cggggtacccgctatttacccggagacagg
9
Amplifikation VH
antisense
tgagctcacggtgagagtggtgccttggccccag
10
Amplifikation CH
sense
tcaccgtgagctcagcctccaccaaggggc
11
Amplifikation 7B4 VL
sense
cgtagaattcaccaccatggattttctggtgcagatt
12
Amplifikation 1F5 VL
sense
cgtagaattcaccaccatggattttcaagtgcagattt
13
Amplifikation CL
antisense
cggctcgagcgctaacactctcccctgttgc
14
Amplifikation CL
sense
gggaccaagcttgagatcaaac
15
Amplifikation VL
antisense
tccagcttggtcccccctc
Tabelle 2: Für die Klonierung der 7B4 und 1F5 IgG verwendete Primer.
1.3) Herstellung von chemisch kompetenten E.coli TOP10 Bakterien
Chemisch kompetente E.coli TOP10 Bakterien wurden im Labor erzeugt. Hierfür wurde eine
Übernachtkultur der Bakterien in LB-Medium (Luria/Miller) ohne Antibiotikum bei 37°C und
230rpm durchgeführt. 2ml dieser Kultur wurden in 400ml frisches LB-Medium überführt und
bis zu einer optischen Dichte (OD) von 0,3 bei einer Wellenlänge von 600nm weiter kultiviert.
Anschließend wurden die Bakterien für 15min bei 4000rpm zentrifugiert, in 100ml 100mM
MgCl2 resuspendiert und 5min auf Eis inkubiert. Anschließend wurden die Bakterien für 5min
bei 4000rpm und 4°C zentrifugiert und das Pellet in 100ml 100mM CaCl2 aufgenommen,
gefolgt von einer 20-minütigen Inkubation auf Eis. Nach einer weiteren Zentrifugation wurden
67
Material und Methoden
die Bakterien in 10ml 100mM CaCl2 mit 10% Glycerol resuspendiert, in vorgekühlte 1,5ml
Reaktionsgefäße aliquotiert und sofort in flüssigem Stickstoff eingefroren. Die Lagerung der
chemisch kompetenten Bakterien erfolgte bei -80°C.
1.4) Transformation von E.coli TOP10 Bakterien
Der Ligationsansatz wurde für 30min mit 100µl der chemisch kompetenten TOP10 E.coli auf
Eis inkubiert und anschließend für 45sec einem Hitzeschock bei 42°C unterzogen. Nach
Zugabe von 500µl vorgewärmtem LB-Medium ohne Zusatz von Antibiotika wurden die
Bakterien für mind. 30min bei 37°C und 300rpm kultiviert. Die transformierten Bakterien
wurden schließlich auf LB-Agar-Platten (50ng/ml Carbenicillin) ausplattiert und über Nacht
(ÜN) bei 37°C kultiviert.
1.5) Isolation von Plasmid DNA aus Bakterien
E.coli TOP10 Bakterien wurden ÜN in LB-Medium (5ml für die Plasmidpräpariation im
kleinen Maßstab, 500ml für die Plasmidpräparation im großen Maßstab) mit 50ng/ml
Carbenicillin bei 37°C und 230rpm ÜN kultiviert.
Im kleinen Maßstab wurde Plasmid-DNA mithilfe der Minilysatmethode durch alkalische Lyse
isoliert. Hierfür wurde 1ml der Übernachtkultur für 10min bei 5000rpm zentrifugiert. Das
Bakterienpellet wurde in 100µl Puffer 1 (50mM Tris, 10mM EDTA, 100µg/ml RNAse A, pH
7,5) resuspendiert. Anschließend erfolgte die alkalische Lyse durch Zugabe von Puffer 2
(0,2M NaOH, 1% SDS) für 5min bei Raumtemperatur (RT). Nach der Neutralisierung mit
150µl 3M Kaliumacetat pH 5,5 wurden die lysierten Bakterien für 20min bei 13000rpm
zentrifugiert. Der Überstand mit der Plasmid-DNA wurde dann in 1ml 96% Ethanol überführt
und für 30min bei 13000rpm und 4°C gefällt. Das DNA-Pellet wurde mit 500µl 70% Ethanol
für 5min bei 13000rpm gewaschen und in 50µl bidest aufgenommen.
Im großen Maßstab wurde die Plasmid-DNA mithilfe des „PureLink HiPure Maxiprep Kit“
(Invitrogen, Darmstadt) entsprechend der Anleitung des Herstellers aufgereinigt. Die
Konzentration der isolierten Plasmid-DNA wurde spektrophotometrisch bei einer Wellenlänge
von 260 und 280nm bestimmt.
Zur Kontrolle der aufgereinigten Plasmide wurden sie mit Restriktionsendonukleasen
geschnitten und das Ergebnis mittels Agarose-Gelelektrophorese ausgewertet.
1.6) Sequenzierung
Die klonierten Konstrukte wurden mithilfe der in Tabelle 3 angegebenen Primer sequenziert.
Die Analyse der Sequenzen wurde mit der ContigExpressTM (Invitrogen, Darmstadt) Software
und BLASTTM durchgeführt. Für multiple Sequenzvergleiche wurde ClustalWTM verwendet.
68
Material und Methoden
Primer #
16
17
18
19
20
21
22
23
Orientierung
Sequenz (5´-3´)
Ta
antisense
sense
sense
antisense
sense
antisense
sense
sense
atttaggtgacactatag
aatacgactcactataggg
cggtgggaggtctatataag
caactagaaggcacagtcg
ggagactgaagttagg
ccattataagctgcaataaac
ggatgggctggataaacac
cactgggtaaagcagacacc
46°C
50°C
58°C
58°C
56°C
56°C
55°C
55°C
pCRII-TOPO-Blunt
pCRII-TOPO-Blunt
pCMV
pCMV
PBos
PBos
7B4 IgG, schwere Kette
1F5 IgG, schwere Kette
Tabelle 3: Für die Plasmidsequenzierung verwendete Primer.
2.
Produktion der 7B4 und 1F5 IgG Antikörper
2.1) Transiente Transfektion von HEK293T Zellen
HEK293T
Zellen
wurden
Kalziumphosphatmethode
für
die
transfiziert.
Antikörperproduktion
Hierzu
wurden
transient
HEK293T
mithilfe
Zellen
in
der
14cm
Zellkulturschalen bis zu einer Konfluenz von 70% kultiviert. Vor der Transfektion wurden die
Zellen mit 24ml frischem DMEM-Medium (ergänzt mit 5% FCS, 1% Glutamin und 1%
Penicillin/Streptomycin) versorgt. Pro Schale, wurde für die Transfektion ein DNA Mix aus je
30µg Plasmid (schwere Kette enthaltender pBos-Vektor und leichte Kette enthaltender
pCMV-Vektor), 6µl Helferplasmid, 2,65ml bidest, 300µl 2,5M CaCl2 und 10µl 100µM
Chloroquin hergestellt. Dieser Mix wurde tropfenweise mit 3ml vorgewärmtem 2xHBS (50mM
Hepes, 280mM NaCl2, 1,2mM Na2HPO4, pH 7,09) gemischt und auf die Zellen getropft. Nach
6-8h wurde die Transfektion durch Austauschen des Medium auf 30ml Produktionsmedium
(DMEM mit 10% Nutridoma SP, 1% Penicillin/Streptomycin, 1% Glutamin) abgestoppt. Die
Zellen wurden für 6-7 Tage kultiviert.
2.2) Aufreinigung der Antikörper
Die produzierten Antikörper wurden nach 6-7 Tagen aus dem Zellkulturüberstand der
transfizierten HEK293T-Zellen aufgereinigt. Die Zellkulturüberstände wurden hierfür
zunächst durch Zentrifugation und Sterilfiltration durch einen 22µm Filter von Zelltrümmern
befreit. Die Präzipitation wurde durch schrittweise Zugabe von 60% (w/w) Ammoniumsulfat
unter Rühren bei 4°C erreicht. Am nächsten Tag wurden die Zellkulturüberstände bei
6000rpm und 4°C für 1h zentrifugiert und das Pellet in Phosphate-Buffered Saline (PBS,
137mM NaCl, 3mM KCl, 6,5mM Na2HPO4, 1,5mM KH2PO4) aufgenommen. Die gefällten
Antikörper wurden dann ÜN bei 4°C gegen PBS dialysiert, bevor sie mithilfe von Protein-GSepharose (Roche, Mannheim) ÜN bei 4°C aufgereinigt werden konnten. Nach gründlichem
Waschen der Protein-G-Sepharose mit PBS wurden die Antikörper mit 0,1M Glycin/HCl pH
2,5 eluiert und erneut ÜN bei 4°C gegen PBS dialysiert. Anschließend wurde möglicherweise
vorhandenes LPS entfernt, die Lösung durch Zentrifugation mit VivaSpin 4 (Millipore,
69
Material und Methoden
Schwalbach) einkonzentriert, die Konzentration der Antikörper spektrophotometrisch mithilfe
von „BioRad Protein Assay“ (BioRad, München) bei einer Wellenlänge von 600nm bestimmt
und die Reinheit und Integrität der Präparation mittels Polyacylamidgelelektrophorese
überprüft. Aliquots der aufgereinigten Antikörper wurden bei -80°C aufbewahrt.
2.3) Entfernung von LPS-Kontaminationen
Für die Entfernung einer möglichen Kontamination der produzierten Antikörper durch
bakterielles Lipopolysaccharid (LPS) wurden die kalte Antikörperpräparationen mit 10%
Triton-X114 durch vortexen gemischt und 15min auf Eis inkubiert. Nach erneutem vortexen
erfolgte eine 10min Inkubation bei 42°C bis zur Trübung der Lösung. Anschließend wurde
die Mischung für 5min bei 2000xg zentrifugiert und die obere Phase mitsamt den
produzierten Antikörpern abgenommen.
3.
Proteinmodifikationen
3.1) Biotinylierung
Je 200µg monoklonale Antikörper wurden mit dem „DSB-XTM Biotin Protein Labeling Kit“
(Invitrogen,
Darmstadt)
in
einem
Reaktionsvolumen
von
200µl
entsprechend
der
Herstelleranweisung biotinyliert.
3.2) Konjugation von FITC-Fluorochromen
Je 250µg monoklonale Antikörper wurden mithilfe des „SureLinkTM FITC Labeling Kit“ (KPL,
Gaitherburg, MD, USA) in einem Reaktionsvolumen von 500µl entsprechend der
Anweisungen des Herstellers mit Fluorescein-Isothiocyanat (FITC) Gruppen konjugiert.
3.3) Konjugation von Alexa647-Fluorochromen
Je 100µg monoklonale Antikörper wurden in einem Reaktionsvolumen von 100µl mit
Alexa647-Fluorochromen konjugiert. Hierfür wurde das “Alexa FluorR 647 Monoclonal
Antibody Labeling Kit“ (Molecular Probes, Leiden, NL) laut der Herstelleranweisung
verwendet.
3.4) Herstellung von F(ab)2 Fragmenten
500µg 1F5 IgG1 Antikörper wurden gegen 0,1M Natriumcitrat-Puffer pH 3,5 für 2h bei RT
dialysiert. Anschließend wurde 1µg/µl Pepsin (1mg/ml aufgelöst in 0,1M Natriumcitrat-Puffer
pH 3,5) (Roche, Mannheim) zugegeben, der Ansatz für 2h bei 37°C verdaut und für 2h bei
RT gegen PBS dialysiert.
70
Material und Methoden
4.
In vitro Analyse der 7B4 IgG Bindung an FcγR
4.1) Affinität der 7B4 IgG für TNP
Die Affinität der klonierten und produzierten 7B4 IgG für ihr Antigen, das Hapten 2-,4-,6Trinitrophenyl (TNP), wurde in enzyme-linked immunoabsorbant assays (ELISA) überprüft.
Da TNP ein sehr kleines Molekül ist, wird es für Experimente an größere Proteine wie z.B.
BSA konjugiert. TNP-BSA gibt es in verschiedenen Kopplungsraten. In dieser Arbeit wurde
BSA mit 4 bzw. 26 TNP-Molekülen verwendet.
Für die Quantifizierung der Affinität wurden je 100ng TNP-4-BSA bzw. TNP-26-BSA in 100µl
0,05M Carbonate/Bicarbonate Puffer (pH 9,6) pro Vertiefung einer 96-well high binding
microlon elisa Platte (Greiner-Bio-One, Frickenhausen) gekoppelt. Zwischen den einzelnen
Inkubationsschritten wurden die Vertiefungen je 3x mit PBS / 0,05% Tween-20 gewaschen.
Nach der Kopplung für 1h bei RT, folgte die Blockierung unspezifischer Interaktionen durch
Inkubation mit 200µl PBS / 1% BSA für 1h bei RT. Anschließend wurden je 1ng der 7B4 IgG
in 100µl PBS / 1% BSA zugegeben. Die gebundenen Antikörper wurden mit 100µl 1:10000 in
PBS / 1% BSA verdünntem Meerettich-Peroxidase (HRP) konjugierten F(ab)´2 Fragment,
das gegen die IgG Fc Region gerichtet ist, nachgewiesen. Die Meerettich-Peroxidase
verursachte nach Zugabe von 50µl TMB-Substrat eine Farbreaktion, die durch 50µl 6%
Orthophosphorsäure abgestoppt wurde. Die Intensität des Farbumschlages konnte bei einer
Wellenlänge von 450nm in einem „VersaMax tunable microplate reader“ (Molecular Devices,
Ismaning) detektiert werden. Der Hintergrund wurde bei einer Wellenlänge von 650nm
vermessen.
Um die Konzentration der produzierten 7B4 IgG zu kontrollieren, wurden 100ng der 7B4 IgG
pro Vertiefung aufgebracht, gefolgt vom Blockierungsschritt und der direkten Detektion mit
dem HRP-konjugiertem anti-IgG Fc F(ab)´2. Nach Zugabe von 50µl Substrat wurde die
chemische Reaktion mit 50µl 6% Orthophosphorsäure abgestoppt und die Intensität des
Farbumschlages bei den Wellenlängen 450nm und 650nm bestimmt.
4.2) FcγR Expression in stabil transfizierten CHO Zellen
Die humanen aktivierenden FcγRIA, die beiden polymorphen Varianten des FcγRIIA (131H
und 131R) und der inhibitorische FcγRIIB wurden durch Frau Anne Bärenwaldt kloniert und
stabil in CHO-Zellen exprimiert. Ein Fusionsprotein aus den extrazellulären Domänen von
FcγRIIIA und den Transmembran- und intrazellulären Anteilen von FcγRIIB (in dieser Arbeit
eFcγRIIIA genannt), das ebenfalls stabil in CHO Zellen exprimiert wird, wurde von Jeffrey
Ravetch (New York, USA) zur Verfügung gestellt.
Die Expression der FcγR wurde durchflusszytometrisch bestimmt. Dafür wurden die Zellen
mit 0,05% Trypsin/EDTA von der Zellkulturschale abgelöst. Je 1x105 Zellen wurden in die
71
Material und Methoden
Vertiefungen einer 96-well-Platte gegeben und für 5min bei 4°C und 1400rpm zentrifugiert.
Die Färbung mit den FcγR-spezifischen, Fluoreszenz-markierten Antikörpern (siehe F.I.4)
erfolgte pro Ansatz in 50µl FACS-Puffer (PBS / 2% FCS / 0,05% Natriumazid) für 15min auf
Eis. Es wurden 10.000 lebende Zellen am FACS Calibur (BD, Heidelberg) eingelesen. Für
die Auswertung wurde die „Flow Cytometry Analysis Software“ (Flowjo, Version 7.6.1)
verwendet.
4.3) Anreicherung FcγR exprimierender CHO Zellen
Eine Anreicherung FcγR exprimierender Zellen durch magnetische Zellsortierung wurde
durchgeführt, wenn der Anteil FcγR+ Zellen an der Gesamtpopulation unter 80% sank.
Hierfür wurden die Zellen mit 0,05% Trypsin/EDTA von der Zellkulturschale abgelöst und in
MACS-Puffer (PBS / 2% FCS) aufgenommen. Alle Inkubations- und Waschschritte wurden
mit MACS-Puffer durchgeführt. Nach einer Inkubation mit den jeweiligen FITC-markierten
FcγR-spezifischen Antikörpern für 30min auf Eis wurden anti-FITC MACS (Miltenyi Biotec,
Bergisch-Gladbach) Partikel für weitere 30min auf Eis zugegeben. Die so markierten Zellen
konnten dann über eine MACSR Separation Column (Größe LS, Miltenyi Biotec, BergischGladbach) aufgereinigt und wieder in Kultur genommen werden.
4.4) Immunkomplexbindung an FcγR exprimierende CHO Zellen
Um die Bindung von IgG Immunkomplexen an FcγR zu untersuchen, wurden zunächst
Immunkomplexe aus je 1µg IgG und 0,5µg TNP-4-BSA bzw. TNP-26-BSA in 100µl PBS pro
Ansatz durch Inkubation für 3h bei 60rpm und RT generiert. Die FcγR exprimierenden CHO
Zellen wurden mit 0,05% Trypsin-EDTA von den Zellkulturschalen abgelöst und je 1x105
Zellen vorgelegt. Je 100µl der Immunkomplexe wurden mit den auf Eis vorgekühlten Zellen
vermischt und für 1h bei 4°C unter leichtem Schütteln inkubiert. Die gebundenen
Immunkomplexe wurden durch Inkubation mit Phycoerithrin-konjugierem IgG-Fc spezischem
F(ab)´2 Fragment für 15min auf Eis angefärbt. Für die lebend-tot-Diskriminierung wurden die
Zellen anschließend mit Propidiumjodid (1:5000 in FACS-Puffer) gefärbt. Es wurden 10.000
lebende Zellen am FACS Calibur (Becton Dickonson, Heidelberg) eingelesen. Für die
Auswertung wurde die „Flow Cytometry Analysis Software“ (Flowjo, Version 7.6.1)
verwendet.
4.5) Einfluss der Glykosyslierung auf die Immunkomplexbindung
Um den Einfluss der Glykosyslierung auf die Bindung von TNP-BSA Immunkomplexen an
die humanen FcγR zu analysieren, wurden zunächst die 7B4 IgG mit 50U Peptide:NGlykosidase F (PNGaseF, New England Biolabs, Frankfurt) pro µg IgG ÜN bei 37°C verdaut.
Anschließend wurden die Immunkomplexe wie in Punkt 4.4. beschrieben generiert und ihre
Bindung an FcγR exprimierende CHO Zellen untersucht. Die Kontrolle des PNGaseF72
Material und Methoden
Verdaus erfolgte über die gelelektrophoretische Auftrennung der Antikörper unter
reduzierenden Bedingungen. Anschließend wurden die Proteine auf eine PVDF-Membran
transferiert und ein Lektin-Blot durchgeführt. Hierfür erfolgte zunächst die Blockierung der
Membran mit in Tris-buffered saline (TBS, 0,5M Tris, 0,15M NaCl) verdünntem Western
Blocking Reagent (1:10, Roche, Mannheim). Danach wurde die Membran mit biotinyliertem
lens culinaris agglutinin (LCA, 1:1000 in TBS / 1mM MgCl2 / 1mM CaCl2 / 1mM MnCl2, Vector
Laboratories, USA). Anschließend wurden die Zuckerreste mit Alkalischer Phosphatasekonjugiertem anti-biotin Antikörper (1:1000 in TBS, Sigma, Steinheim) detektiert. Durch die
Zugabe des Substrats NBT/BCIP (1:50, Roche, Mannheim) verdünnt in NBT/X-Puffer (0,1M
Tris-HCl pH 9,5 / 0,1M NaCl / 0,05M MgCl2) kommt es zur Anfärbung glykosylierter Proteine.
5.
In vitro Analyse der Bindung von 1F5 IgG an LCL1.11
Zellen
Die CD20 exprimierende, humane B-Zell-Linie LCL1.11 wurde verwendet, um die
Antigenbindung der klonierten und produzierten anti-CD20 IgG (Klon 1F5) zu testen. Hierfür
wurden 1x105 LCL1.11 entweder mit 10ng/µl Alexa647 konjugierten 1F5 mIgG2a oder mit
einem Mix aus diesem Antikörper und steigenden Mengen (40ng/µl bis 160ng/µl) der zu
testenden 1F5 IgG in 50µl FACS-Puffer für 15min auf Eis inkubiert. Die beiden Antikörper
konkurrieren somit um die Antigenbindung. Dadurch können sowohl die Assoziation als auch
die
Dissoziation
der
Fluoreszenzintensität
produzierten
(MFI)
der
Antikörper
Bindung
verglichen
des
1F5
werden.
Die
mittlere
mIgG2a-Alexa647
wurde
durchflusszytometrisch durch Analyse von 5000 Zellen am FACSCanto II bestimmt.
Rechnerisch wurde schließlich die CH50 der 1F5 IgG ermittelt, d.h. die notwendige
Konzentration um das 1F5 mIgG2a-Alexa647 Signal um 50% zu reduzieren.
6.
In vitro Analyse der C1q Bindung durch 1F5 IgG
Die Bindung der humanen IgG Subklassen an den humanen Komplementfaktor C1q wurde
mittels
ELISA
bestimmt.
Hierfür
wurden
je
100ng
1F5
IgG
in
100µl
0,05M
Carbonate/Bicarbonate Puffer (pH 9,6) pro Vertiefung einer 96-well high binding microlon
elisa
Platte
(Greiner-Bio-One,
Frickenhausen)
gekoppelt.
Zwischen
den
einzelnen
Inkubationsschritten wurden die Vertiefungen je 3x mit PBS / 0,05% Tween-20 gewaschen.
Nach der Kopplung für 1h bei RT, folgte die Blockierung unspezifischer Interaktionen durch
Inkubation mit 200µl PBS / 3% BSA / 0,1% Gelatine / 0,05% Tween-20 für 1h bei RT.
Anschließend wurden je 20ng/µl C1q (AbD Serotec, UK) in 100µl PBS / 3% BSA / 0,1%
Gelatine / 0,05% Tween-20 zugegeben. Das gebundene C1q Protein wurde mit 100µl 1:500
in PBS / 3% BSA / 0,1% Gelatine / 0,05% Tween-20 verdünntem HRP-konjugierten und
gegen humanes C1q gerichteten Antikörper (AbD Serotec, UK) nachgewiesen. Die
73
Material und Methoden
Meerettich-Peroxidase verursachte nach Zugabe von 50µl TMB-Substrat eine Farbreaktion,
die
durch
50µl
6%
Orthophosphorsäure
abgestoppt
wurde.
Die
Intensität
des
Farbumschlages konnte bei einer Wellenlänge von 450nm in einem „VersaMax tunable
microplate reader“ (Molecular Devices, Ismaning) detektiert werden. Der Hintergrund wurde
bei einer Wellenlänge von 650nm vermessen.
7.
Humane hämatopoetische Stammzellen
7.1) Aufreinigung humaner hämatopoetischer Stammzellen
Humane hämatopoetische Stammzellen (HSC) wurden aus Nabelschnurblut, das unter
Einwilligung der Spender für diesen Zweck vom Klinikum Fürth zur Verfügung gestellt wurde,
aufgereinigt. Das Nabelschnurblut wurde bis zur Aufarbeitung bei 4°C max. ÜN gelagert. Für
die Aufreinigung wurden Proben mit >25ml Blut verwendet, die zunächst im Verhältnis 1:1
mit PBS vermischt und dann für 25min bei 1800rpm und 12°C über einen Pancoll
Dichtegradienten durch Zentrifugation aufgetrennt wurden. Anschließend konnte der
Leukozytenring abgenommen und nach einem Waschschritt in PBS für 5min bei 1400rpm
und 4°C die CD34 exprimierenden Zellen mit dem „Direct CD34 Progenitor Cell Isolation Kit,
human“ (Miltenyi Biotec, Bergisch-Gladbach) isoliert werden. Das Protokoll beinhaltet die
Blockierung der humanen Fc Rezeptoren und die Inkubation mit magnetischen anti-CD34
Partikel (je 30min auf Eis). Die an die Partikel gebundenen Stammzellen wurden
anschließend mit MACSR Separation Column (Größe LS, Miltenyi Biotec, BergischGladbach) separiert und in Einfriermedium (FCS / 10% DMSO) aufgenommen. Die
Stammzellproben wurden bis zu ihrer Verwendung in flüssigem Stickstoff aufbewahrt.
7.2) Durchflusszytometrische Analyse von Nabelschnurblut und HSC
Aufgereinigte
hämatopoetische
Stammzellen
wurden
hinsichtlich
der
Qualität
der
Aufreinigung durchflusszytometrisch analysiert. Hierfür wurden die Stammzellproben
aufgetaut, in PBS gewaschen, für 5min bei 1400rpm und 4°C zentrifugiert und in FACSPuffer aufgenommen. Die Zellen wurden jeweils in einem Volumen von 50µl für 15min auf
Eis gefärbt. Die verwendeten Antikörper und die entsprechenden Verdünnungen im Kapitel
E.I. Material aufgeführt. Für die lebend-tot-Diskriminierung wurden die Zellen anschließend
mit dem Kernfarbstoff DAPI inkubiert.
Parallel zu den isolierten Stammzellproben wurden auch Leukozyten aus Nabelschnurblut
vor der CD34-MACS-Aufreinigung durchflusszytometrisch analysiert. Hierfür wurden Proben
nach der Pancoll-Dichtegradientenzentrifugation entnommern, in PBS gewaschen und in
FACS-Puffer aufgenommen. Das Färbeprotokoll war identisch mit der Stammzellanalyse.
Die durchflusszytometrische Analyse von 10000 lebenden Zellen erfolgte am FACS Canto II
(Becton Dickinson).
74
Material und Methoden
8.
Genotypisierung der FcγR Allele
8.1) Isolation von genomischer DNA aus humanem Nabelschnurblut
Vor Beginn der Aufreinigung der humanen hämatopoetischen Stammzellen wurden 200µl
Vollblut entnommen und bis zur Weiterverarbeitung bei -20 °C aufbewahrt. Die genomische
DNA wurde mithilfe des „QiaAmp DSP Blood Mini Kit“ (Qiagen, Hilden) entsprechend der
Herstelleranweisungen isoliert und bei -20 °C gelagert.
8.2) Genotypisierung mittels allelspezifischer PCR
Für die aufgereinigten Nabelschnurblutproben wurden die Polymorphismen in den
kodierenden Genen von FcγRIIA (131H/R) und FcγRIIIA (158F/V) mittels allelspezifischer
PCR bestimmt (Abb. 30).
Primer #
FcγRIIA
FcγRIIIA
24
25
26
27
28
29
30
31
32
33
†
1. PCR
1. PCR*
2. PCR, allelspezifisch*
2. PCR, allelspezifisch*
2. PCR†
1. PCR
1. PCR
2. PCR#
2. PCR, allelspezifisch#
2. PCR, allelspezifisch#
Orientierung
Sequenz (5´Æ3´)
Ta
sense
antisense
sense
sense
antisense
sense
antisense
sense
antisense
antisense
ggagaaaccatcatgctgag
cagcatgggcagctcttc
gaaaatcccagaaatttttcca
gaaaatcccagaaatttttccg
caattttgctgctatgggc
gtgtctttcaggctggctg
cgagattaaactattctggtc
tcacatatttacagaatggcaaagg
tttgggagtaaaaatgtgtcttcagaga
gttgggagtaaaaatgtgtcttcagaga
56°C
56°C
60°C
60°C
60°C
58°C
58°C
64°C
64°C
64°C
Tabelle 4: Verwendete Primer für die Genotypisierung von FcγRIIA131H/R und FcγRIIIA158F/V. Die
Sequenzen der Primer wurden zum Teil verschiedenen Publikationen entnommen: † Norris et al [140],
*
Carlsson et al [87], # Leppers van der Straat et al [141]
Für die Genotypisierung von FcγRIIA131H/R wurde in einer ersten PCR ein 995bp großes,
FcγRIIA spezifisches Fragment amplifiziert. Die Reaktionsbedingungen waren je 25µl Ansatz
wie folgt: 50ng genomische DNA, je 0,4µM Primer, 0,2mM dNTPs, 1µl DMSO, 0,2µl Taq
DNA Polymerase und 2,5µl 10x Taq Standard Reaktionspuffer. Die Amplifikation erfolgte bei
94°C 5min, 56°C 5min, 72°C 5min gefolgt von 30 Zyklen aus 94°C 1min, 56°C 1min und
72°C 2min. Die abschließende Elongation fand 10min bei 72°C statt. 1µl dieser ersten PCR
wurde in die zweite, Allel-spezifische PCR, eingesetzt. Die weiteren Komponenten je 20µl
Ansatz waren: je 0,5µM Primer, 125µM dNTPs, 1µl DMSO, 7,5mM MgCl2, 0,2µl Taq DNA
Polymerase und 2µl 10x Taq Standard Reaktionspuffer. Die Amplifikation des 249bp großen
Fragments erfolgte durch 30 Zyklen von 94°C 15sec, 60°C 15sec, 72°C 30sec und einer
abschließenden Elongation von 72°C für 5min. Die Analyse erfolgte durch elektrophoretische
Auftrennung in einem 1,5%igem Agarosegel.
Analog wurde für die Genotypisierung von FcγRIIIA158F/V in einer ersten PCR ein 1366bp
großes, FcγRIIIA spezifisches Fragment amplifiziert. Hierfür wurden 50ng genomische DNA,
75
Material und Methoden
je 0,5µM Primer, 0,2mM dNTPs, 0,2µl Taq DNA Polymerase und 2,5µl 10x Taq Standard
Reaktionspuffer in einem 25µl Ansatz gemischt. Die Amplifikation erfolgte bei 94°C 5min,
gefolgt von 30 Zyklen bestehend aus 94°C 1min, 58°C 30sec und 72°C 2min. Die finale
Elongation fand für 10min bei 72°C statt. In die Allel-spezifische zweite PCR wurde 1µl der
ersten PCR zusammen mit je 0,5µM Primer, 125µM dNTPs, 0,2µl Taq DNA Polymerase und
2µl 10x Taq Standard Reaktionspuffer in einem Reaktionsvolumen von 20µl gemischt. Die
Amplifikation des 131bp Produkts erfolgte bei einer initialen Denaturierung von 2min bei
94°C, 30 Zyklen von 94°C 15sec, 64°C 15sec und 72°C 30sec und einer letzten Elongation
von 5min bei 72°C. Die Analyse erfolgte durch elektrophoretische Auftrennung in einem
2%igem Agarosegel.
Abb. 30: Graphische Darstellung
der Sequenzen von FcγRIIA (A)
und FcγRIIIA (B) und der für die
Genotypisierung
verwendeten
Primer. Intronsequenzen sind
verkürzt dargestellt. Die Position
der
zu
bestimmenden
Polymorphismen
FcγRIIA131H/R
bzw. FcγRIIIA158F/V sind mit
einem * gekennzeichnet.
9.
Rekonstitution von humanisierten Rag2/γc/Fcγ-/- Mäusen
Neugeborene Rag2/γc/Fcγ-/- Mäuse (nicht älter als 3 Tage) wurden in einer Cäsium137
Gammabestrahlungsanlage mit Dosen zwischen 3,5 und 6Gy bestrahlt. Nach 4-6 Stunden
erfolgte die intravenöse Injektion von 45.000 bis 76.000 HSC in 30µl PBS. Die Mäuse
wurden
im
Alter
von
11
bis
13
Wochen
auf
den
Erfolg
der
Rekonstitution
durchflusszytometrisch untersucht und in Experimenten verwendet.
10. Probengewinnung und durchflusszytometrische Analysen
10.1) Präparation von Maus-Leukozyten aus dem Blut
Peripheres Blut wurde aus dem retro-orbitalen Venenplexus der Maus mithilfe von
Hämatokritkapillaren (Brand, Wertheim) abgenommen und in EDTA-enthaltenden Röhrchen
gesammelt. Für die durchflusszytometrische Analyse wurden die Erythrozyten 2x für 2min
mit 500µl Erythrozyten-Lyse-Puffer (0,15M NH4Cl, 10mM KHCO3, 0,1mM Na2EDTA, pH 7,3)
76
Material und Methoden
lysiert. Die Lyse wurde durch Zugabe von PBS abgestoppt, die Zellen bei 600xg für 5min
zentrifugiert, mit 1ml PBS gewaschen und schließlich in FACS-Puffer aufgenommen.
10.2) Präparation von humanen Leukozyten aus dem Blut
Peripheres Blut wurde aus dem retro-orbitalen Venenplexus der Maus mithilfe von
Hämatokritkapillaren abgenommen und in EDTA-enthaltenden Röhrchen gesammelt.
Humanes Blut wurde aus dem Ohrläppchen von freiwilligen Spendern (Mitarbeiter der
Arbeitsgruppe) gewonnen. Für die durchflusszytometrische Analyse von humanen
Leukozyten (human oder humanisierte Maus) wurde das Blut 1:1 mit PBS verdünnt, auf das
gleiche Volumen Pancoll überschichtet und für 25min bei 1800rpm und 12°C mittels
Dichtegradientenzentrifugation
aufgetrennt.
Danach
konnte
die
Leukozytenschicht
abgenommen und die Zellen in FACS-Puffer aufgenommen werden.
10.3) Präparation von Leukozyten aus Milz und Knochenmark
Leukozyten aus der Milz und dem Knochenmark wurden wie folgt aus humanisierten Mäusen
präpariert: Nach dem Töten der Mäuse durch kraniale Dislokation wurde die Milz entfernt.
Eine Hälfte der Milz wurde für spätere Immunfluoreszenzfärbungen in TissueTekR O.C.T.
Compound (Sakura Finetek, NL) eingebettet, während die andere Hälfte der Milz durch einen
70µm Sieb gestrichen wurde um eine Einzelzellsuspension herzustellen. Die Milzzellen
wurden anschließend durch ein 40µm Sieb filtriert, bevor sie mit PBS für 5min bei 1400rpm
und 4°C gewaschen und in FACS-Puffer aufgenommen wurden. Je nach Anzahl der FACSFärbungen wurde für die Isolation von Leukozyten aus dem Knochenmark zunächst ein oder
beide Hinterbeine abgeschnitten und von Haut und Muskeln befreit. Ober- und
Unterschenkel wurden getrennt, jeweils die Knochenenden abgeschnitten und das
Knochenmark mit einer 20G Kanüle und PBS ausgespült. Eine Einzelzellsuspension wurde
durch Filtration durch ein 40µm Sieb erzielt. Die Zellen wurden mit PBS gewaschen und in
FACS-Puffer aufgenommen.
Humane
Milz-
und
Knochenmarksproben
wurden
für
Forschungszwecke
vom
Universitätsklinikum Erlangen über Prof. Dr. Diana Dudziak (Labor für Biologie Dendritischer
Zellen,
Erlangen)
zur
Verfügung
gestellt.
Diese
Proben
wurden
einer
Pancoll-
Dichtegradienten-Aufreinigung unterzogen, bevor sie für die durchflusszytometrische
Färbung verwendet wurden.
10.4) Durchflusszytometrische Färbung und Analyse
Die FACS-Färbung der isolierten Leukozyten wurde in 96-well-Platten durchgeführt. Gefärbt
wurde jeweils in einem Volumen von 50µl für 15min auf Eis. Mit der Ausnahme der
durchflusszytometrischen Untersuchung der FcγR Expression auf Leukozyten der Maus,
wurden die Zellen vor der FACS-Färbung mit dem monoklonalen Antikörper 2.4G2 inkubiert.
77
Material und Methoden
Diese auch Fc Block genannte Behandlung blockiert die Interaktion der mFcγRIIB und
mFcγRIII mit den Fc Regionen der FACS-Antikörper und verringert damit das Risiko falschpositiver Signale. Die verwendeten Antikörper und die entsprechenden Verdünnungen sind
in Abschnitt E.I. Material aufgeführt. Für die lebend-tot-Diskriminierung wurden die Zellen
anschließend mit dem Kernfarbstoff DAPI inkubiert.
Die durchflusszytometrische Analyse der gefärbten Proben erfolgte am FACS Canto II
(Becton Dickinson). Für die Auswertung der Daten wurde die FACSDiva Software
verwendet. Es wurden ausschließlich lebende Zellen nach dem Duplettenaussschluss
analysiert.
11. Immunfluoreszenz-Analyse der humanisierten Mäuse
Nach dem Töten der Versuchstiere durch kraniale Dislokation wurde die Milz entnommen,
halbiert und eine Hälfte in TissueTekR O.C.T. compound gefüllte TissueTek 4566 Cryomold
Gefäße (beides Sakura Finetek, NL) eingebettet und auf Trockeneis durchgefroren. Die
Organe wurden bis zur Anfertigung von Gewebeschnitten bei -80°C gelagert. 6µm
Kryoschnitte wurden auf SuperFrost plus-Objektträger (Menzel-Gläser, Braunschweig)
aufgezogen, ÜN bei RT getrocknet, 150sec in -20°C kaltem Aceton fixiert und erneut ÜN
getrocknet. Die Schnitte wurden bis zur Färbung bei -20°C gelagert.
Für die Immunfluoreszenzfärbung wurden die Schnitte aufgetaut, mit einem ImmedgePen
(Vector Laboratories, USA) umrandet und nach kurzer Trocknung und Waschen in PBS in
eine feuchte Kammer überführt. Eine unspezifische Antikörperbindung wurde zunächst durch
Inkubation mit PBS / 5% Ziegenserum blockiert, bevor je 20µl der entsprechenden
Antikörperverdünnungen für 1h bei RT aufgebracht wurden. Nach einem letzten Waschen
der Schnitte in PBS wurden sie mit Fluoromount (Sigma, Steinheim) eingedeckelt und nach
kurzer Trocknung am Axiovert 200M (Zeiss, Jena) ausgewertet.
12. 1F5 IgG vermittelte B-Zell-Depletion
Die anti-CD20 1F5 IgG wurden wie beschrieben (siehe Absatz E.II.1.) kloniert und
produziert. Um die IgG-induzierte B-Zell-Depletion im Blut zu untersuchen, wurden 10µg IgG
pro Maus intravenös in die Schwanzvene injiziert. Die Zellzahl der CD19+ B-Zellen wurde
innerhalb der humanen, hCD45+ Leukozytenpopulation durchflusszytometrisch vor und nach
Injektion (Tag 1, Tag 3, Tag 7) bestimmt.
Für eine Analyse der B-Zell-Depletion in Milz und Knochenmark wurden 100µg 1F5 IgG pro
Maus intravenös in die Schwanzvene injiziert und die Mäuse 3 Tage nach Injektion
durchflusszytometrisch untersucht.
78
Material und Methoden
13. Zellkultur
Im Rahmen dieser Arbeit wurden HEK293T (human embryonal kidney), CHO (chinese
hamster ovary) Zellen (adhärente Zelllinien) und die humane B-Zell-Linie LCL1.11
(Suspensionszelllinie) verwendet. FcγR stabil exprimierende CHO Zellen wurden von Frau
Anne Bärenwaldt zur Verfügung gestellt. Die eFcγRIIIA-exprimierenden CHO-Zellen
stammen von Jeffrey Ravetch (New York, USA). Die in Suspension wachsenden 1F5 und
7B4 Hybridomzelllinien wurden bei ATCC erworben bzw. von Birgitta Heyman (Lund,
Schweden) zur Verfügung gestellt.
HEK293T Zellen wurden in DMEM / 5% FCS / 1% Penicillin/Streptomycin / 1% Glutamin
kultiviert. Für die Antikörperproduktion wurden sie in protein-freies Medium (DMEM / 10%
Nutridoma SP / 1% Penicillin/Streptomycin / 1% Glutamin) umgesetzt. Für alle anderen
Zelllinien wurde RPMI1640 Medium verwendet bei dem 10% FCS / 1% Penicillin/
Streptomycin / 1% Glutamin / 1% Natriumpyruvat und 1% non-essential amino acids
zugesetzt wurden.
Die verwendeten Zelllinien wurden unter Standardbedingungen kultiviert (37°C, 5% CO2,
feuchte Atmosphäre). Die Zellen wurden alle 2-3 Tage passagiert, bei adhärenten Zellen
unter Verwendung von 0,25% Trypsin/EDTA. Exakte Zellzahlen wurden mithilfe einer
Neubauer-Zählkammer bestimmt.
14. Versuchstierhaltung
Alle verwendeten Versuchstiere wurden in den zentralen Tierversuchseinrichtungen der
Medizinischen und Naturwissenschaftlichen Fakultäten der Friedrich-Alexander-Universität
Erlangen-Nürnberg, d.h. zunächst im Franz-Penzold-Zentrum (FPZ), später im Biologisch
Technischen Entwicklungsgebäude (BTE), in isolated ventilated cages (IVC) unter specific
pathogen free (SPF)-Bedingungen gehalten. Wie von The Jackson Laboraties empfohlen,
wurde das Trinkwasser der immundefizienten Rag2/γc-/- und Rag2/γc/Fcγ-/- Mäuse durch
Zugabe von HCl auf einen pH Wert von ca. 3,0 eingestellt. Für die Versuche wurden
männliche und weibliche Tiere im Alter von 11-13 Wochen eingesetzt. C57BL/6 Mäuse
wurden von Janvier (Frankreich) bezogen. Die Rag2/γc-/- Mäuse stammen von Hergen Spits
(Niederlande) und die FcγR-/- wurden von Jeffrey Ravetch (USA) zur Verfügung gestellt.
15. Statistik
Für die statistische Auswertung wurden je 2 Datengruppen mit dem Student´schen T-Test
verglichen. P-Werte kleiner 0,05 wurden als signifikant (*), p-Werte zwischen 0,01 und 0,001
79
Material und Methoden
als verstärkt signifikant (**) und p-Werte kleiner 0,001 als hochsignifikant (***) bewertet. Die
statistische Auswertung erfolgte mit Microsoft Office Excel 2003.
80
Literaturverzeichnis
G. Literaturverzeichnis
1.
Albert, H., et al., In vivo enzymatic modulation of IgG glycosylation inhibits
autoimmune disease in an IgG subclass-dependent manner. Proc Natl Acad Sci U S
A, 2008. 105(39): p. 15005-9.
2.
Schroeder, H.W., Jr. and L. Cavacini, Structure and function of immunoglobulins. J
Allergy Clin Immunol. 125(2 Suppl 2): p. S41-52.
3.
Ravetch, J.V., et al., Structural heterogeneity and functional domains of murine
immunoglobulin G Fc receptors. Science, 1986. 234(4777): p. 718-25.
4.
Unkeless, J.C. and H.N. Eisen, Binding of monomeric immunoglobulins to Fc
receptors of mouse macrophages. J Exp Med, 1975. 142(6): p. 1520-33.
5.
Weinshank, R.L., A.D. Luster, and J.V. Ravetch, Function and regulation of a murine
macrophage-specific IgG Fc receptor, Fc gamma R-alpha. J Exp Med, 1988. 167(6):
p. 1909-25.
6.
Diamond, B., B.R. Bloom, and M.D. Scharff, The Fc receptors of primary and cultured
phagocytic cells studied with homogeneous antibodies. J Immunol, 1978. 121(4): p.
1329-33.
7.
Perussia, B., et al., Murine natural killer cells express functional Fc gamma receptor II
encoded by the Fc gamma R alpha gene. J Exp Med, 1989. 170(1): p. 73-86.
8.
Uher, F., M.C. Lamers, and H.B. Dickler, Antigen-antibody complexes bound to Blymphocyte Fc gamma receptors regulate B-lymphocyte differentiation. Cell Immunol,
1985. 95(2): p. 368-79.
9.
Rabellino, E.M., et al., Membrane receptors of mouse leukocytes. II. Sequential
expression of membrane receptors and phagocytic capacity during leukocyte
differentiation. J Exp Med, 1978. 147(2): p. 434-45.
10.
Waterston, R.H., et al., Initial sequencing and comparative analysis of the mouse
genome. Nature, 2002. 420(6915): p. 520-62.
11.
Nimmerjahn, F., et al., FcgammaRIV: a novel FcR with distinct IgG subclass
specificity. Immunity, 2005. 23(1): p. 41-51.
12.
Hulett, M.D., et al., Chimeric Fc receptors identify functional domains of the murine
high affinity receptor for IgG. J Immunol, 1991. 147(6): p. 1863-8.
13.
Heusser, C.H., C.L. Anderson, and H.M. Grey, Receptors for IgG: subclass specificity
of receptors on different mouse cell types and the definition of two distinct receptors
on a macrophage cell line. J Exp Med, 1977. 145(5): p. 1316-27.
14.
Nimmerjahn, F. and J.V. Ravetch, Divergent immunoglobulin g subclass activity
through selective Fc receptor binding. Science, 2005. 310(5753): p. 1510-2.
15.
Fossati-Jimack, L., et al., Markedly different pathogenicity of four immunoglobulin G
isotype-switch variants of an antierythrocyte autoantibody is based on their capacity
to interact in vivo with the low-affinity Fcgamma receptor III. J Exp Med, 2000. 191(8):
p. 1293-302.
16.
Hamaguchi, Y., et al., Antibody isotype-specific engagement of Fcgamma receptors
regulates B lymphocyte depletion during CD20 immunotherapy. J Exp Med, 2006.
203(3): p. 743-53.
17.
Daeron, M., Intracytoplasmic sequences involved in the biological properties of lowaffinity receptors for IgG expressed by murine macrophages. Braz J Med Biol Res,
1995. 28(3): p. 263-74.
81
Literaturverzeichnis
18.
Takai, T., et al., FcR gamma chain deletion results in pleiotrophic effector cell defects.
Cell, 1994. 76(3): p. 519-29.
19.
Ra, C., et al., A macrophage Fc gamma receptor and the mast cell receptor for IgE
share an identical subunit. Nature, 1989. 341(6244): p. 752-4.
20.
Wirthmueller, U., et al., Signal transduction by Fc gamma RIII (CD16) is mediated
through the gamma chain. J Exp Med, 1992. 175(5): p. 1381-90.
21.
Miller, K.L., A.M. Duchemin, and C.L. Anderson, A novel role for the Fc receptor
gamma subunit: enhancement of Fc gamma R ligand affinity. J Exp Med, 1996.
183(5): p. 2227-33.
22.
Kurosaki, T., I. Gander, and J.V. Ravetch, A subunit common to an IgG Fc receptor
and the T-cell receptor mediates assembly through different interactions. Proc Natl
Acad Sci U S A, 1991. 88(9): p. 3837-41.
23.
Anderson, C.L., Isolation of the receptor for IgG from a human monocyte cell line
(U937) and from human peripheral blood monocytes. J Exp Med, 1982. 156(6): p.
1794-806.
24.
Pichler, W.J. and S. Broder, Fc-IgM and Fc-IgG receptors on human circulating B
lymphocytes. J Immunol, 1978. 121(3): p. 887-90.
25.
Looney, R.J., et al., Identification of a second class of IgG Fc receptors on human
neutrophils. A 40 kilodalton molecule also found on eosinophils. J Exp Med, 1986.
163(4): p. 826-36.
26.
Cohen, L., S. Sharp, and A. Kulczycki, Jr., Human monocytes, B lymphocytes, and
non-B lymphocytes each have structurally unique Fc gamma receptors. J Immunol,
1983. 131(1): p. 378-83.
27.
Karas, S.P., W.F. Rosse, and R.J. Kurlander, Characterization of the IgG-Fc receptor
on human platelets. Blood, 1982. 60(6): p. 1277-82.
28.
Qiu, W.Q., et al., Organization of the human and mouse low-affinity Fc gamma R
genes: duplication and recombination. Science, 1990. 248(4956): p. 732-5.
29.
Takai, S., et al., Human high-affinity Fc gamma RI (CD64) gene mapped to
chromosome 1q21.2-q21.3 by fluorescence in situ hybridization. Hum Genet, 1994.
93(1): p. 13-5.
30.
Sammartino, L., et al., Assignment of the gene coding for human FcRII (CD32) to
bands q23q24 on chromosome 1. Immunogenetics, 1988. 28(5): p. 380-1.
31.
Peltz, G.A., et al., Human Fc gamma RIII: cloning, expression, and identification of
the chromosomal locus of two Fc receptors for IgG. Proc Natl Acad Sci U S A, 1989.
86(3): p. 1013-7.
32.
Ernst, L.K., et al., Three genes for the human high affinity Fc receptor for IgG (Fc
gamma RI) encode four distinct transcription products. J Biol Chem, 1992. 267(22): p.
15692-700.
33.
Peltz, G., et al., Characterization of the human monocyte high affinity Fc receptor (hu
FcRI). Mol Immunol, 1988. 25(3): p. 243-50.
34.
Stuart, S.G., et al., Human IgG Fc receptor (hFcRII; CD32) exists as multiple isoforms
in macrophages, lymphocytes and IgG-transporting placental epithelium. Embo J,
1989. 8(12): p. 3657-66.
35.
Brooks, D.G., et al., Structure and expression of human IgG FcRII(CD32). Functional
heterogeneity is encoded by the alternatively spliced products of multiple genes. J
Exp Med, 1989. 170(4): p. 1369-85.
82
Literaturverzeichnis
36.
Van den Herik-Oudijk, I.E., et al., Identification of signaling motifs within human Fc
gamma RIIa and Fc gamma RIIb isoforms. Blood, 1995. 85(8): p. 2202-11.
37.
Ravetch, J.V. and B. Perussia, Alternative membrane forms of Fc gamma RIII(CD16)
on human natural killer cells and neutrophils. Cell type-specific expression of two
genes that differ in single nucleotide substitutions. J Exp Med, 1989. 170(2): p. 48197.
38.
Ernst, L.K., A.M. Duchemin, and C.L. Anderson, Association of the high-affinity
receptor for IgG (Fc gamma RI) with the gamma subunit of the IgE receptor. Proc
Natl Acad Sci U S A, 1993. 90(13): p. 6023-7.
39.
van Vugt, M.J., et al., FcR gamma-chain is essential for both surface expression and
function of human Fc gamma RI (CD64) in vivo. Blood, 1996. 87(9): p. 3593-9.
40.
Kurosaki, T. and J.V. Ravetch, A single amino acid in the glycosyl
phosphatidylinositol attachment domain determines the membrane topology of Fc
gamma RIII. Nature, 1989. 342(6251): p. 805-7.
41.
Duchemin, A.M., L.K. Ernst, and C.L. Anderson, Clustering of the high affinity Fc
receptor for immunoglobulin G (Fc gamma RI) results in phosphorylation of its
associated gamma-chain. J Biol Chem, 1994. 269(16): p. 12111-7.
42.
Rankin, B.M., et al., Stimulation of tyrosine phosphorylation and calcium mobilization
by Fc gamma receptor cross-linking. Regulation by the phosphotyrosine phosphatase
CD45. J Immunol, 1993. 150(2): p. 605-16.
43.
Young, J.D., S.S. Ko, and Z.A. Cohn, The increase in intracellular free calcium
associated with IgG gamma 2b/gamma 1 Fc receptor-ligand interactions: role in
phagocytosis. Proc Natl Acad Sci U S A, 1984. 81(17): p. 5430-4.
44.
Cassatella, M.A., et al., Fc gamma R(CD16) interaction with ligand induces Ca2+
mobilization and phosphoinositide turnover in human natural killer cells. Role of Ca2+
in Fc gamma R(CD16)-induced transcription and expression of lymphokine genes. J
Exp Med, 1989. 169(2): p. 549-67.
45.
Boruchov, A.M., et al., Activating and inhibitory IgG Fc receptors on human DCs
mediate opposing functions. J Clin Invest, 2005. 115(10): p. 2914-23.
46.
Koren, H.S. and M.S. Williams, Natural killing and antibody-dependent cellular
cytotoxicity are mediated by different mechanisms and by different cells. J Immunol,
1978. 121(5): p. 1956-60.
47.
Ono, M., et al., Role of the inositol phosphatase SHIP in negative regulation of the
immune system by the receptor Fc(gamma)RIIB. Nature, 1996. 383(6597): p. 263-6.
48.
Clynes, R.A., et al., Inhibitory Fc receptors modulate in vivo cytoxicity against tumor
targets. Nat Med, 2000. 6(4): p. 443-6.
49.
Takai, T., et al., Augmented humoral and anaphylactic responses in Fc gamma RIIdeficient mice. Nature, 1996. 379(6563): p. 346-9.
50.
Bolland, S. and J.V. Ravetch, Spontaneous autoimmune disease in Fc(gamma)RIIBdeficient mice results from strain-specific epistasis. Immunity, 2000. 13(2): p. 277-85.
51.
Raju, T.S., Terminal sugars of Fc glycans influence antibody effector functions of
IgGs. Curr Opin Immunol, 2008. 20(4): p. 471-8.
52.
Maley, F., et al., Characterization of glycoproteins and their associated
oligosaccharides through the use of endoglycosidases. Anal Biochem, 1989. 180(2):
p. 195-204.
53.
Collin, M. and A. Olsen, EndoS, a novel secreted protein from Streptococcus
pyogenes with endoglycosidase activity on human IgG. Embo J, 2001. 20(12): p.
3046-55.
83
Literaturverzeichnis
54.
Nandakumar, K.S., et al., Endoglycosidase treatment abrogates IgG arthritogenicity:
importance of IgG glycosylation in arthritis. Eur J Immunol, 2007. 37(10): p. 2973-82.
55.
Allhorn, M., et al., Human IgG/Fc gamma R interactions are modulated by
streptococcal IgG glycan hydrolysis. PLoS One, 2008. 3(1): p. e1413.
56.
Shields, R.L., et al., High resolution mapping of the binding site on human IgG1 for Fc
gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants
with improved binding to the Fc gamma R. J Biol Chem, 2001. 276(9): p. 6591-604.
57.
Su, K., et al., A promoter haplotype of the immunoreceptor tyrosine-based inhibitory
motif-bearing FcgammaRIIb alters receptor expression and associates with
autoimmunity. I. Regulatory FCGR2B polymorphisms and their association with
systemic lupus erythematosus. J Immunol, 2004. 172(11): p. 7186-91.
58.
Kono, H., et al., FcgammaRIIB Ile232Thr transmembrane polymorphism associated
with human systemic lupus erythematosus decreases affinity to lipid rafts and
attenuates inhibitory effects on B cell receptor signaling. Hum Mol Genet, 2005.
14(19): p. 2881-92.
59.
Warmerdam, P.A., et al., Molecular basis for a polymorphism of human Fc gamma
receptor II (CD32). J Exp Med, 1990. 172(1): p. 19-25.
60.
Leeuwenberg, J.F., et al., Functional polymorphism of IgG FcRII (CD32) on human
neutrophils. Immunology, 1990. 71(2): p. 301-4.
61.
Parren, P.W., et al., On the interaction of IgG subclasses with the low affinity Fc
gamma RIIa (CD32) on human monocytes, neutrophils, and platelets. Analysis of a
functional polymorphism to human IgG2. J Clin Invest, 1992. 90(4): p. 1537-46.
62.
Bruhns, P., et al., Specificity and affinity of human Fcgamma receptors and their
polymorphic variants for human IgG subclasses. Blood, 2009. 113(16): p. 3716-25.
63.
Vance, B.A., et al., Binding of monomeric human IgG defines an expression
polymorphism of Fc gamma RIII on large granular lymphocyte/natural killer cells. J
Immunol, 1993. 151(11): p. 6429-39.
64.
Wu, J., et al., A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function
and predisposes to autoimmune disease. J Clin Invest, 1997. 100(5): p. 1059-70.
65.
Koene, H.R., et al., Fc gammaRIIIa-158V/F polymorphism influences the binding of
IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa48L/R/H phenotype. Blood, 1997. 90(3): p. 1109-14.
66.
Kumpel, B.M., et al., Clearance of red cells by monoclonal IgG3 anti-D in vivo is
affected by the VF polymorphism of Fcgamma RIIIa (CD16). Clin Exp Immunol, 2003.
132(1): p. 81-6.
67.
Sanders, L.A., et al., Fc gamma receptor IIa (CD32) heterogeneity in patients with
recurrent bacterial respiratory tract infections. J Infect Dis, 1994. 170(4): p. 854-61.
68.
Cartron, G., et al., Therapeutic activity of humanized anti-CD20 monoclonal antibody
and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood, 2002. 99(3): p.
754-8.
69.
Dall'Ozzo, S., et al., Rituximab-dependent cytotoxicity by natural killer cells: influence
of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res,
2004. 64(13): p. 4664-9.
70.
Hatjiharissi, E., et al., Increased natural killer cell expression of CD16, augmented
binding and ADCC activity to rituximab among individuals expressing the
Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood, 2007. 110(7): p. 2561-4.
84
Literaturverzeichnis
71.
Zhang, W., et al., FCGR2A and FCGR3A polymorphisms associated with clinical
outcome of epidermal growth factor receptor expressing metastatic colorectal cancer
patients treated with single-agent cetuximab. J Clin Oncol, 2007. 25(24): p. 3712-8.
72.
Canete, J.D., et al., Influence of variants of Fc gamma receptors IIA and IIIA on the
American College of Rheumatology and European League Against Rheumatism
responses to anti-tumour necrosis factor alpha therapy in rheumatoid arthritis. Ann
Rheum Dis, 2009. 68(10): p. 1547-52.
73.
Lin, T.S., et al., FCGR3A and FCGR2A polymorphisms may not correlate with
response to alemtuzumab in chronic lymphocytic leukemia. Blood, 2005. 105(1): p.
289-91.
74.
Mitrovic, Z., et al., FCgammaRIIIA and FCgammaRIIA polymorphisms are not
associated with response to rituximab and CHOP in patients with diffuse large B-cell
lymphoma. Haematologica, 2007. 92(7): p. 998-9.
75.
Nieto, A., et al., Involvement of Fcgamma receptor IIIA genotypes in susceptibility to
rheumatoid arthritis. Arthritis Rheum, 2000. 43(4): p. 735-9.
76.
Morgan, A.W., et al., Fcgamma receptor type IIIA is associated with rheumatoid
arthritis in two distinct ethnic groups. Arthritis Rheum, 2000. 43(10): p. 2328-34.
77.
Lee, Y.H., J.D. Ji, and G.G. Song, Associations between FCGR3A polymorphisms
and susceptibility to rheumatoid arthritis: a metaanalysis. J Rheumatol, 2008. 35(11):
p. 2129-35.
78.
Morgan, A.W., et al., Association of FCGR2A and FCGR2A-FCGR3A haplotypes with
susceptibility to giant cell arteritis. Arthritis Res Ther, 2006. 8(4): p. R109.
79.
Brun, J.G., T.M. Madland, and C.A. Vedeler, Immunoglobulin G fc-receptor
(FcgammaR) IIA, IIIA, and IIIB polymorphisms related to disease severity in
rheumatoid arthritis. J Rheumatol, 2002. 29(6): p. 1135-40.
80.
Radstake, T.R., et al., Role of Fcgamma receptors IIA, IIIA, and IIIB in susceptibility
to rheumatoid arthritis. J Rheumatol, 2003. 30(5): p. 926-33.
81.
Morgan, A.W., et al., FcgammaRIIIA-158V and rheumatoid arthritis: a confirmation
study. Rheumatology (Oxford), 2003. 42(4): p. 528-33.
82.
Karassa, F.B., T.A. Trikalinos, and J.P. Ioannidis, The role of FcgammaRIIA and IIIA
polymorphisms in autoimmune diseases. Biomed Pharmacother, 2004. 58(5): p. 28691.
83.
Koene, H.R., et al., The Fc gammaRIIIA-158F allele is a risk factor for systemic lupus
erythematosus. Arthritis Rheum, 1998. 41(10): p. 1813-8.
84.
Manger, K., et al., Fcgamma receptor IIa, IIIa, and IIIb polymorphisms in German
patients with systemic lupus erythematosus: association with clinical symptoms. Ann
Rheum Dis, 2002. 61(9): p. 786-92.
85.
Gelmetti, A.P., et al., Polymorphism of the FcgammaRIIalpha IgG receptor in patients
with lupus nephritis and glomerulopathy. J Rheumatol, 2006. 33(3): p. 523-30.
86.
Jonsen, A., et al., Association between SLE nephritis and polymorphic variants of the
CRP and FcgammaRIIIa genes. Rheumatology (Oxford), 2007. 46(9): p. 1417-21.
87.
Carlsson, L.E., et al., Heparin-induced thrombocytopenia: new insights into the
impact of the FcgammaRIIa-R-H131 polymorphism. Blood, 1998. 92(5): p. 1526-31.
88.
van der Pol, W.L., et al., Association of the Fc gamma receptor IIA-R/R131 genotype
with myasthenia gravis in Dutch patients. J Neuroimmunol, 2003. 144(1-2): p. 143-7.
85
Literaturverzeichnis
89.
Tanaka, Y., et al., FcgammaRIIa-131R allele and FcgammaRIIIa-176V/V genotype
are risk factors for progression of IgA nephropathy. Nephrol Dial Transplant, 2005.
20(11): p. 2439-45.
90.
Bosma, G.C., R.P. Custer, and M.J. Bosma, A severe combined immunodeficiency
mutation in the mouse. Nature, 1983. 301(5900): p. 527-30.
91.
Fulop, G.M. and R.A. Phillips, The scid mutation in mice causes a general defect in
DNA repair. Nature, 1990. 347(6292): p. 479-82.
92.
Kirchgessner, C.U., et al., DNA-dependent kinase (p350) as a candidate gene for the
murine SCID defect. Science, 1995. 267(5201): p. 1178-83.
93.
McCune, J.M., et al., The SCID-hu mouse: murine model for the analysis of human
hematolymphoid differentiation and function. Science, 1988. 241(4873): p. 1632-9.
94.
Mosier, D.E., et al., Transfer of a functional human immune system to mice with
severe combined immunodeficiency. Nature, 1988. 335(6187): p. 256-9.
95.
Vormoor, J., et al., Immature human cord blood progenitors engraft and proliferate to
high levels in severe combined immunodeficient mice. Blood, 1994. 83(9): p. 248997.
96.
Shultz, L.D., et al., Multiple defects in innate and adaptive immunologic function in
NOD/LtSz-scid mice. J Immunol, 1995. 154(1): p. 180-91.
97.
Hesselton, R.M., et al., High levels of human peripheral blood mononuclear cell
engraftment and enhanced susceptibility to human immunodeficiency virus type 1
infection in NOD/LtSz-scid/scid mice. J Infect Dis, 1995. 172(4): p. 974-82.
98.
Prochazka, M., et al., The nonobese diabetic scid mouse: model for spontaneous
thymomagenesis associated with immunodeficiency. Proc Natl Acad Sci U S A, 1992.
89(8): p. 3290-4.
99.
Kollet, O., et al., beta2 microglobulin-deficient (B2m(null)) NOD/SCID mice are
excellent recipients for studying human stem cell function. Blood, 2000. 95(10): p.
3102-5.
100.
Ito, M., et al., NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model
for engraftment of human cells. Blood, 2002. 100(9): p. 3175-82.
101.
Shultz, L.D., et al., Human lymphoid and myeloid cell development in NOD/LtSz-scid
IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J
Immunol, 2005. 174(10): p. 6477-89.
102.
Goldman, J.P., et al., Enhanced human cell engraftment in mice deficient in RAG2
and the common cytokine receptor gamma chain. Br J Haematol, 1998. 103(2): p.
335-42.
103.
Kamel-Reid, S. and J.E. Dick, Engraftment of immune-deficient mice with human
hematopoietic stem cells. Science, 1988. 242(4886): p. 1706-9.
104.
Ishikawa, F., et al., Development of functional human blood and immune systems in
NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood, 2005. 106(5): p. 1565-73.
105.
Hogan, C.J., et al., Engraftment and development of human CD34(+)-enriched cells
from umbilical cord blood in NOD/LtSz-scid/scid mice. Blood, 1997. 90(1): p. 85-96.
106.
Hiramatsu, H., et al., Complete reconstitution of human lymphocytes from cord blood
CD34+ cells using the NOD/SCID/gammacnull mice model. Blood, 2003. 102(3): p.
873-80.
107.
Traggiai, E., et al., Development of a human adaptive immune system in cord blood
cell-transplanted mice. Science, 2004. 304(5667): p. 104-7.
86
Literaturverzeichnis
108.
Mazurier, F., et al., Rapid myeloerythroid repopulation after intrafemoral
transplantation of NOD-SCID mice reveals a new class of human stem cells. Nat
Med, 2003. 9(7): p. 959-63.
109.
Gimeno, R., et al., Monitoring the effect of gene silencing by RNA interference in
human CD34+ cells injected into newborn RAG2-/- gammac-/- mice: functional
inactivation of p53 in developing T cells. Blood, 2004. 104(13): p. 3886-93.
110.
Schoeberlein, A., et al., Engraftment kinetics of human cord blood and murine fetal
liver stem cells following in utero transplantation into immunodeficient mice. Stem
Cells Dev, 2004. 13(6): p. 677-84.
111.
Bente, D.A., et al., Dengue fever in humanized NOD/SCID mice. J Virol, 2005.
79(21): p. 13797-9.
112.
Watanabe, S., et al., Hematopoietic stem cell-engrafted NOD/SCID/IL2Rgamma null
mice develop human lymphoid systems and induce long-lasting HIV-1 infection with
specific humoral immune responses. Blood, 2007. 109(1): p. 212-8.
113.
Guirado, E., et al., Passive serum therapy with polyclonal antibodies against
Mycobacterium tuberculosis protects against post-chemotherapy relapse of
tuberculosis infection in SCID mice. Microbes Infect, 2006. 8(5): p. 1252-9.
114.
Ichiyama, K., et al., A duodenally absorbable CXC chemokine receptor 4 antagonist,
KRH-1636, exhibits a potent and selective anti-HIV-1 activity. Proc Natl Acad Sci U S
A, 2003. 100(7): p. 4185-90.
115.
Legrand, N., et al., Transient accumulation of human mature thymocytes and
regulatory T cells with CD28 superagonist in "human immune system" Rag2(-/)gammac(-/-) mice. Blood, 2006. 108(1): p. 238-45.
116.
Bleeker, W.K., et al., Dual mode of action of a human anti-epidermal growth factor
receptor monoclonal antibody for cancer therapy. J Immunol, 2004. 173(7): p. 4699707.
117.
Brolen, G.K., et al., Signals from the embryonic mouse pancreas induce
differentiation of human embryonic stem cells into insulin-producing beta-cell-like
cells. Diabetes, 2005. 54(10): p. 2867-74.
118.
Vassilopoulos, G., P.R. Wang, and D.W. Russell, Transplanted bone marrow
regenerates liver by cell fusion. Nature, 2003. 422(6934): p. 901-4.
119.
Cohen-Haguenauer, O., et al., In vivo repopulation ability of genetically corrected
bone marrow cells from Fanconi anemia patients. Proc Natl Acad Sci U S A, 2006.
103(7): p. 2340-5.
120.
Gladman, J.T., et al., A humanized Smn gene containing the SMN2 nucleotide
alteration in exon 7 mimics SMN2 splicing and the SMA disease phenotype. Hum Mol
Genet.
121.
Maloney, D.G., et al., IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody
therapy in patients with relapsed low-grade non-Hodgkin's lymphoma. Blood, 1997.
90(6): p. 2188-95.
122.
Takaki, T., et al., HLA-A*0201-restricted T cells from humanized NOD mice recognize
autoantigens of potential clinical relevance to type 1 diabetes. J Immunol, 2006.
176(5): p. 3257-65.
123.
van Rijn, R.S., et al., A new xenograft model for graft-versus-host disease by
intravenous transfer of human peripheral blood mononuclear cells in RAG2-/gammac-/- double-mutant mice. Blood, 2003. 102(7): p. 2522-31.
124.
Chalandon, Y., et al., BCR-ABL-transduced human cord blood cells produce
abnormal populations in immunodeficient mice. Leukemia, 2005. 19(3): p. 442-8.
87
Literaturverzeichnis
125.
Nomura, T., et al., Induction of cancer, actinic keratosis, and specific p53 mutations
by UVB light in human skin maintained in severe combined immunodeficient mice.
Cancer Res, 1997. 57(11): p. 2081-4.
126.
O'Reilly, M.S., et al., Angiostatin induces and sustains dormancy of human primary
tumors in mice. Nat Med, 1996. 2(6): p. 689-92.
127.
Maenaka, K., et al., The human low affinity Fcgamma receptors IIa, IIb, and III bind
IgG with fast kinetics and distinct thermodynamic properties. J Biol Chem, 2001.
276(48): p. 44898-904.
128.
Warmerdam, P.A., et al., A single amino acid in the second Ig-like domain of the
human Fc gamma receptor II is critical for human IgG2 binding. J Immunol, 1991.
147(4): p. 1338-43.
129.
Bhatia, M., et al., Purification of primitive human hematopoietic cells capable of
repopulating immune-deficient mice. Proc Natl Acad Sci U S A, 1997. 94(10): p.
5320-5.
130.
Mazurier, F., et al., A novel immunodeficient mouse model--RAG2 x common
cytokine receptor gamma chain double mutants--requiring exogenous cytokine
administration for human hematopoietic stem cell engraftment. J Interferon Cytokine
Res, 1999. 19(5): p. 533-41.
131.
Cragg, M.S., et al., The biology of CD20 and its potential as a target for mAb therapy.
Curr Dir Autoimmun, 2005. 8: p. 140-74.
132.
Renaudineau, Y., et al., Monoclonal anti-CD20 antibodies: mechanisms of action and
monitoring of biological effects. Joint Bone Spine, 2009. 76(5): p. 458-63.
133.
Press, O.W., et al., Monoclonal antibody 1F5 (anti-CD20) serotherapy of human B
cell lymphomas. Blood, 1987. 69(2): p. 584-91.
134.
Duncan, A.R. and G. Winter, The binding site for C1q on IgG. Nature, 1988.
332(6166): p. 738-40.
135.
Salfeld, J.G., Isotype selection in antibody engineering. Nat Biotechnol, 2007. 25(12):
p. 1369-72.
136.
McManus, L.M. and P.K. Nakane, Isolation and characterization of mouse C1q. J
Immunol Methods, 1980. 36(2): p. 159-71.
137.
Paiva, M., et al., FcgammaRIIa polymorphism and clinical response to rituximab in
non-Hodgkin lymphoma patients. Cancer Genet Cytogenet, 2008. 183(1): p. 35-40.
138.
Weng, W.K. and R. Levy, Two immunoglobulin G fragment C receptor polymorphisms
independently predict response to rituximab in patients with follicular lymphoma. J
Clin Oncol, 2003. 21(21): p. 3940-7.
139.
Uchida, J., et al., The innate mononuclear phagocyte network depletes B
lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody
immunotherapy. J Exp Med, 2004. 199(12): p. 1659-69.
140.
Norris, C.F., et al., A naturally occurring mutation in Fc gamma RIIA: a Q to K127
change confers unique IgG binding properties to the R131 allelic form of the receptor.
Blood, 1998. 91(2): p. 656-62.
141.
Leppers-van de Straat, F.G., et al., A novel PCR-based method for direct Fc gamma
receptor IIIa (CD16) allotyping. J Immunol Methods, 2000. 242(1-2): p. 127-32.
88
Anhang
H. Anhang
I. Sequenzen und Vektorkarten
1.
Sequenzen der klonierten Antikörper: Schwere Ketten
Nach Durchführung der 5’-RACE und Klonierung der erhaltenen PCR-Produkte ergab die
Sequenzierung der variablen Regionen der schweren Ketten von 7B4 und 1F5 Antikörpern
folgende DNA-Sequenz:
7B4 VH
atgggttggctgtggaacttgctattcctgatggcagctgcccaaagtgcccaaccaca
60
gatccagttggtacagtctggacctgaggtgaaggagcctggagagacagtcaggatct
120
cctgcaaggcttctggatataccttcacagcccatggaatgggctgggtgaaacaggct
180
ccaggaaagggtttaaggtggatgggctggataaacacctactctggagtgccagcata
240
tgttgatgacttcaagggacggtttgccttttatctggaaacctctgccagcactgtct
300
atttgcagatcaataacgtcaaagatgaagacacggctacatatttctgtggaagatgg
360
gctatggttacgacgtgctttgacacctggggccaaggcaccactctcaccgtgagctc
420
a
421
1F5 VH
atgggatggagttgtatcatcctcttcttggtagcaacagctgcaggtgtccactcccag
60
gtgcaactgcggcagcctggggctgagctggtgaagcctggggcctcagtgaagatgacc
120
tgcaaggcttctggctacacatttaccagttacaatatgcactgggtaaagcagacacct
180
ggacagggcctggaatggattggagctatttatccaggaaatggtgatacttcctacaat
240
cagaagttcaaaggcaaggccacattgactgcagacaaatcctccagcacagcctacatg
300
cagctcagcagtctgacatctgaggactctgcggtctattactgtgcaagatcgcactac
360
ggtagtaactacgtagactactttgactactggggccaaggcaccactctcaccgtgagc
420
tca
423
Diese variablen Regionen wurden an die folgenden konstanten Regionen der humanen
schweren Ketten fusioniert:
IgG1 CH
gcctccaccaagggcccatcggtcttccccctggcaccctcctccaagagcacctctggg
60
ggcacagcggccctgggctgcctggtcaaggactacttccccgaaccggtgacggtgtcg
120
tggaactcaggcgccctgaccagcggcgtgcacaccttcccggctgtcctacagtcctca
180
ggactctactccctcagcagcgtggtgaccgtgccctccagcagcttgggcacccagacc
240
tacatctgcaacgtgaatcacaagcccagcaacaccaaggtggacaagagagttgagccc
300
aaatcttgtgacaaaactcacacatgcccaccgtgcccagcacctgaactcctgggggga
360
ccgtcagtcttcctcttccccccaaaacccaaggacaccctcatgatctcccggacccct
420
gaggtcacatgcgtggtggtggacgtgagccacgaagaccctgaggtcaagttcaactgg
480
tacgtggacggcgtggaggtgcataatgccaagacaaagccgcgggaggagcagtacaac
540
89
Anhang
agcacgtaccgtgtggtcagcgtcctcaccgtcctgcaccaggactggctgaatggcaag
600
gagtacaagtgcaaggtctccaacaaagccctcccagcccccatcgagaaaaccatctcc
660
aaagccaaagggcagccccgagaaccacaggtgtacaccctgcccccatcccgggatgag
720
ctgaccaagaaccaggtcagcctgacctgcctggtcaaaggcttctatcccagcgacatc
780
gccgtggagtgggagagcaatgggcagccggagaacaactacaagaccacgcctcccgtg
840
ctggactccgacggctccttcttcctctacagcaagctcaccgtggacaagagcaggtgg
900
cagcaggggaacgtcttctcatgctccgtgatgcatgaggctctgcacaaccactacacg
960
cagaagagcctctccctgtctccgggtaaatga
999
IgG2 CH
gcctccaccaagggcccatcggtcttccccctggcgccctgctccaggagcacctccgag
60
agcacagcggccctgggctgcctggtcaaggactacttccccgaaccggtgacggtgtcg
120
tggaactcaggcgctctgaccagcggcgtgcacaccttcccggctgtcctacagtcctca
180
ggactctactccctcagcagcgtggtgaccgtgacctccagcaacttcggcacccagacc
240
tacacctgcaacgtagatcacaagcccagcaacaccaaggtggacaagacagttgagcgc
300
aaatgttgtgtcgagtgcccaccgtgcccagcaccacctgtggcaggaccgtcagtcttc
360
ctcttccccccaaaacccaaggacaccctcatgatctcccggacccctgaggtcacgtgc
420
gtggtggtggacgtgagccacgaagaccccgaggtccagttcaactggtacgtggacggc
480
atggaggtgcataatgccaagacaaagccacgggaggagcagttcaacagcacgttccgt
540
gtggtcagcgtcctcaccgtcgtgcaccaggactggctgaacggcaaggagtacaagtgc
600
aaggtctccaacaaaggcctcccagcccccatcgagaaaaccatctccaaaaccaaaggg
660
cagccccgagaaccacaggtgtacaccctgcccccatcccgggaggagatgaccaagaac
720
caggtcagcctgacctgcctggtcaaaggcttctaccccagcgacatcgccgtggagtgg
780
gagagcaatgggcagccggagaacaactacaagaccacacctcccatgctggactccgac
840
ggctccttcttcctctacagcaagctcaccgtggacaagagcaggtggcagcaggggaac
900
gtcttctcatgctccgtgatgcatgaggctctgcacaaccactacacacagaagagcctc
960
tccctgtctccgggtaaatga
981
IgG3 CH
gcctccaccaagggcccatcggtcttccccctggcgccctgctccaggagcacctctggg
60
ggcacagcggccctgggctgcctggtcaaggactacttccccgaaccggtgacggtgtcg
120
tggaactcaggcgccctgaccagcggcgtgcacaccttcccggctgtcctacagtcctca
180
ggactctactccctcagcagcgtggtgaccgtgccctccagcagcttgggcacccagacc
240
tacacctgcaacgtgaatcacaagcccagcaacaccaaggtggacaagagagttgagctc
300
aaaaccccacttggtgacacaactcacacatgcccacggtgcccagagcccaaatcttgt
360
gacacacctcccccgtgcccacggtgcccagagcccaaatcttgtgacacacctccccca
420
tgcccacggtgcccagagcccaaatcttgtgacacacctcccccatgcccacggtgccca
480
gcacctgaactcctgggaggaccgtcagtcttcctcttccccccaaaacccaaggatacc
540
cttatgatttcccggacccctgaggtcacgtgcgtggtggtggacgtgagccacgaagac
600
cccgaggtccagttcaagtggtacgtggacggcgtggaggtgcataatgccaagacaaag
660
ccgcgggaggagcagttcaacagcacgttccgtgtggtcagcgtcctcaccgtcctgcac
720
caggactggctgaacggcaaggagtacaagtgcaaggtctccaacaaagccctcccagcc
780
90
Anhang
cccatcgagaaaaccatctccaaaaccaaaggacagccccgagaaccacaggtgtacacc
840
ctgcccccatcccgggaggagatgaccaagaaccaggtcagcctgacctgcctggtcaaa
900
ggcttctaccccagcgacatcgccgtggagtgggagagcagcgggcagccggagaacaac
960
tacaacaccacgcctcccatgctggactccgacggctccttcttcctctacagcaagctc
1020
accgtggacaagagcaggtggcagcaggggaacatcttctcatgctccgtgatgcatgag
1080
gctctgcacaaccgcttcacgcagaagagcctctccctgtctccgggtaaatag
1134
IgG4 CH
gcctccaccaagggcccatccgtcttccccctggcgccctgctccaggagcacctccgag
60
agcacagccgccctgggctgcctggtcaaggactacttccccgaaccggtgacggtgtcg
120
tggaactcaggcgccctgaccagcggcgtgcacaccttcccggctgtcctacagtcctca
180
ggactctactccctcagcagcgtggtgaccgtgccctccagcagcttgggcacgaagacc
240
tacacctgcaacgtagatcacaagcccagcaacaccaaggtggacaagagagttgagtcc
300
aaatatggtcccccatgcccatcatgcccagcacctgagttcctggggggaccatcagtc
360
ttcctgttccccccaaaacccaaggacactctcatgatctcccggacccctgaggtcacg
420
tgcgtggtggtggacgtgagccaggaagaccccgaggtccagttcaactggtacgtggat
480
ggcgtggaggtgcataatgccaagacaaagccgcgggaggagcagttcaacagcacgtac
540
cgtgtggtcagcgtcctcaccgtcctgcaccaggactggctgaacggcaaggagtacaag
600
tgcaaggtctccaacaaaggcctcccgtcctccatcgagaaaaccatctccaaagccaaa
660
gggcagccccgagagccacaggtgtacaccctgcccccatcccaggaggagatgaccaag
720
aaccaggtcagcctgacctgcctggtcaaaggcttctaccccagcgacatcgccgtggag
780
tgggagagcaatgggcagccggagaacaactacaagaccacgcctcccgtgctggactcc
840
gacggctccttcttcctctacagcaggctaaccgtggacaagagcaggtggcaggagggg
900
aatgtcttctcatgctccgtgatgcatgaggctctgcacaaccactacacacagaagagc
960
ctctccctgtctccgggtaaatga
984
2.
Sequenzen der klonierten Antikörper: Leichte Ketten
Nach Durchführung der 5’-RACE und Klonierung der erhaltenen PCR-Produkte ergab die
Sequenzierung der variablen Regionen der leichten Ketten von 7B4 und 1F5 Antikörpern
folgende DNA-Sequenz:
7B4 VL
atggattttctggtgcagattttcagcttcttgctaatcagtgcctcagtgtcaatgtcc
60
agaggagaaaatgtggtcacccagtctccaggaatcatgtctgcatctccaggggacaag
120
gtcaccatgacctgcagggccagcccaagtgtaacttccagtcacttgcactggtatcag
180
cagaagtcaggtggctcccccaaactctggatttatagcacatacaacttggcttctggg
240
gtccctggtcgcttcagtggcagtgggtctggggcctcttactctctcacaatcagcagt
300
gtggaggctgaagatgctgccacttattactgccagcagtacagaagttattcagtcacg
360
ttcggaggggggaccaagctgga
383
91
Anhang
1F5 VL
atggattttcaagtgcagattttcagcttcctgctaatcagtgctccagtcataatgtcc
60
agaggacaaattgttctctcccagtctccagcaatcctttctgcatctccaggggagaag
120
gtcacaatgacttgcagggccagttcaagtttaagtttcatgcactggtaccagcanaag
180
ccaggatcctcccccaaaccctggatttatgccacatccaacctggcttctggagtccct
240
gctcgcttcagtggcagcgggtctgggacctcttactctctcacaatcagcagagtggag
300
gctgaagatgctgccacttatttctgccatcagtggagtagtaacccgctcacgttcggc
360
gctgggaccaagctgga
377
Diese variablen Regionen wurde mittels PCR an die folgende konstante Region der
κ leichten Kette fusioniert:
CL (κ Leichte Kette)
gatcaaacgaactgtggctgcaccatcggtcttcatcttcccgccatctgatgagcagtt
60
gaaatctggaactgcctctgttgtgtgcctgctgaataacttctatcccagagaggccaa
120
agtacagtggaaggtggataacgccctccaatcgggtaactcccaggagagtgtcacaga
180
gcaggacagcaaggacagcacctacagcctcagcagcaccctgacgctgagcaaagcaga
240
ctacgagaaacacaaagtctacgcctgcgaagtcacccatcagggcctgagctcgcccgt
300
cacaaagagcttcaacaggggagagtgttag
331
3.
Vektorkarten
Abb. 31: In dieser Arbeit wurden für die
Klonierung und Expression der schweren Ketten
der Vektor pBos, für die leichten Ketten der
Vektor pCMV verwendet. Zwischenschritte der
Klonierung erfolgten in pCR-BluntII-TOPO
(Invitrogen, Darmstadt).
92
Anhang
II. Abkürzungsverzeichnis
°C
Grad Celsius
µg
Mikrogramm
µl
Mikroliter
µm
Mikrometer
A
Alanin
A647
Alexa647
Abb.
Abbildung
ADCC
Antikörper-induzierte Zell-vermittelte Zytotoxizität
APC
Allophycocyanin
AU
engl. arbitrary units, willkürliche Einheiten
BLAST
engl. basic local alignment search tool
bp
Basenpaare
BSA
engl. bovine serum albumin, Rinderserumalbumin
bzw.
beziehungsweise
ca.
circa
CD
engl. cluster of differentiation
cDNA
engl. complementary DNA, komplementäre DNA
CH
konstante Domänen der schweren Ketten
CHO
engl. chinese hamster ovary
CL
konstante Domäne der leichten Kette
DAPI
4′,6-Diamidin-2-phenylindol
DC
engl. dendritic cell, dendritische Zelle
DNA
engl. Desoxyribonucleic acid, Desoxyribonukleinsäure
dNTP
Desoxynukleosidtriphosphate
ELISA
engl. enzyme linked immunosorbant assay
EndoS
Endoglykosidase S
engl.
englisch
F
Phenylalanin
FACS
engl. Fluorescence activated cell sorting, Fluoreszenzaktivierte Zellsortierung
Fc
engl. fragment christallyzable
FcR
Fc Rezeptor
FCS
engl. Fetal calf serum, Fötales Kälberserum
FcγR
Fcγ Rezeptor
FITC
Fluoresceinisothiocyanat
g
engl. gravitational force
Gy
Gray
93
Anhang
H
Histidin
h
lat. hora, Stunde
ddH2O
doppelt destilliertes Wasser
HEK
engl. human embryonal kidney
HRP
engl. horseraddish peroxidase
HSC
hämatopoetische Stammzellen
Ig
Immunglobulin
IL
Interleukin
ITAM
engl. immunoreceptor tyrosine based activation motif
ITIM
engl. immunoreceptor tyrosyine based inhibitory motif
kDa
Kilodalton
KM
Knochenmark
lat.
lateinisch
LPS
Lipopolysaccharid
M
Molar
MACS
engl. magnetic assisted cell sorting, Magnetische Zellsortierung
MFI
Mittlere Fluoreszenzintensität
min
Minuten
ml
Milliliter
mM
Nanomolar
N
Asparagin
ng
Nanogramm
NK-Zellen
Natürliche Killer Zellen
nm
Nanometer
NOD
non-obese diabetic
OD
Optische Dichte
PBMC
engl. peripheral blood mononuclear cells
PBS
engl. phosphate-buffered saline, Phosphate-gepufferte Saline
PCR
engl. polymerase chain reaction, Polymerasekettenreaktion
PE
Phycoerythrin
PerCP
Peridininchlorophyll
pH
lat. Pondus Hydrogenii, “Stärke des Wasserstoffs”
PNGaseF
Peptid N:Glykosidase F
R
Arginin
RA
Rheumatoide Arthritis
RACE
engl. rapid amplification of cDNA ends
Rag
engl. recombinase activating gene
RNA
engl. Ribonucleic acid, Ribonukleinsäure
94
Anhang
rpm
engl. rotations per minute
RT
Raumtemperatur
scid
engl. severe combined immunodeficiency
sec
Sekunde
SLE
Systemischer Lupus Erythematodes
sog.
sogenannte
SPR
engl. surface plasmon resonance
TCR
engl. T cell receptor, T-Zell-Rezeptor
TdT
Terminale Desoxynukleotid Transferase
TNP
2,4,6-Trinitrophenyl
U
engl. unit, Einheit
ÜN
über Nacht
UV
ultraviolett
V
Valin
v.a.
vor allem
vgl.
vergleiche
VH
variable Region der schweren Ketten
VL
variable Region der leichten Ketten
z.B.
zum Beispiel
γc
IL 2 Rezeptor Signalkette
95
Anhang
III.
Eigene Publikationen
1.
Zeitschriftenartikel
Lux A, Baerenwaldt A, Albert H, Woigk M, Dudziak D, Nimmerjahn F. A novel humanized
mouse model to study human Immunoglobulin G activity in vivo. (Manuskript eingereicht)
Nimmerjahn F, Lux A, Albert H, Woigk M, Lehmann C, Dudziak D, Smith P, Ravetch JV.
FcγRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. PNAS. 2010.
Lux A, Aschermann S, Biburger M, Nimmerjahn F. The pro and anti-inflammatory activities of
immunoglobulin G. Ann Rheum Dis. 2010 Jan;69 Suppl 1:i92-96.
Aschermann S, Lux A, Baerenwaldt A, Biburger M, Nimmerjahn F. The other side of
immunoglobulin G: suppressor of inflammation. Clin Exp Immunol. 2010 May;160(2):161-7.
2.
04/2009
Posterpräsentation und Vorträge
Vortrag,
7.
Mitarbeiterkolloquium,
Bayrisches
Genomforschungs-
netzwerk (BayGene), München
10/2008
Posterpräsentation, 3rd European Workshop on Immune-Mediated
Inflammatory Diseases, London, UK
06/2008
Vortrag
6.
Mitarbeiterkolloquium,
Bayrisches
Genomforschungs-
Bayrisches
Genomforschungs-
netzwerk (BayGene), München
07/2007
Vortrag,
5.
Mitarbeiterkolloquium,
netzwerk (BayGene), München
96
Anhang
IV.
Lebenslauf
Angaben zur Person
Name
Anja Lux, geb. Richter
Geburtstag
21.09.1982
Geburtsort
Altenburg
Familienstand
verheiratet
Nationalität
deutsch
Schul- und Berufsausbildung
seit 05/2010
Promotion an der Friedrich-Alexander-Universität Erlangen-Nürnberg,
Lehrstuhl für Genetik, Arbeitsgruppe: Prof. Dr. Falk Nimmerjahn
08/2007 – 04/2010
Promotion am Universitätsklinikum Erlangen, Medizinische Klinik III –
Rheumatologie und Immunologie, Arbeitsgruppe Prof. Dr. Falk Nimmerjahn
10/2006 - 04/2007
Diplomarbeit an der Friedrich-Alexander-Universität Erlangen-Nürnberg,
Lehrstuhl für Experimentelle Medizin II, Arbeitsgruppe: Prof. Dr. Jürgen
Behrens
Titel: “Search for Conductin interacting proteins on the mitotic spindle“
08/2006
Ablegen der Diplomprüfungen (Note: 1,0)
08/2004
Ablegen des Vordiploms (Note: 1,5)
10/2002 –04/2007
Studium der Molekularen Medizin an der Friedrich-Alexander-Universität
Erlangen-Nürnberg
08/1993 – 05/2001
Platanengymnasium Altenburg
Abschluss: Allgemeine Hochschulreife (Note: 1,3)
08/1989 – 08/1993
Grundschule Siegfried-Flack-Straße, Altenburg
Auslandsaufenthalte
08/2008 – 12/2008
Laborpraktikum, Nederlands Kanker Instituut, Amsterdam, Niederlande,
Arbeitsgruppe: Dr. Arnoud Sonnenberg
08/2001 – 08/2002
AuPair Aufenthalt in den USA
Zusätzliche Qualifikationen
09/2010
Fortbildungsveranstaltung für Projektleiter und Beauftragte für Biologische
Sicherheit (BBS) (Universität Freiburg)
07/2010
Aufbaukurs für den Umgang mit umschlossenen radioaktiven Stoffen erhöhter
Aktivitäten (Karlsruher Institut für Technologie, Karlsruhe)
03/2006
Seminar „Einführung in die Versuchstierkunde und tierexperimentelle
Techniken“ – Kategorie: B-FELASA (Universität Erlangen-Nürnberg)
02/2006
Grundkurs in Strahlenschutz (Universität Erlangen-Nürnberg)
97
Danksagung
I. Danksagung
Bei meinem Doktorvater Prof. Dr. Falk Nimmerjahn möchte ich mich für die Möglichkeit
bedanken, dass ich dieses spannende aber auch herausfordernde Thema bearbeiten durfte.
Vielen Dank sage ich auch für die ausgezeichnete Betreuung während der letzten Jahre.
Auch heute noch sind Sie immer für Fragen offen und stehen mir stets mit Rat und Tat zur
Seite. Ich bin dankbar, dass ich von Ihnen lernen darf.
Bei Prof. Dr. Hans-Martin Jäck bedanke ich mich für die zeitnahe Erstellung des
Zweitgutachtens dieser Dissertation.
Prof. Dr. Georg Schett und Prof. Dr. Lars Nitschke danke ich für ihre Bereitschaft in diesem
Promotionsverfahren als Prüfer fungiert zu haben.
Bei meiner lieben Kollegin Anne Bärenwaldt, die mir in den letzten Jahren auch zu einer
guten Freundin geworden ist, bedanke ich mich für viele anregende Diskussionen und den
regen Austausch von Ideen, die praktische Unterstützung und nicht zuletzt für das
ausdauernde Korrekturlesen dieser Arbeit. Du weißt, wie schwer ich mich teilweise getan
habe. Noch dazu hattest du immer ein offenes Ohr auch außerhalb des Laboralltags. Danke!
Bei den anderen - den alteingesessenen sowie den neuen und auch vergangenen Mitgliedern der Arbeitsgruppe Heike Albert, Susanne Aschermann, Markus Biburger, Sybille
Böhm, Heike Danzer, Angela Dick, Birgit Lehmann, Johanna Rothamer, Inessa Schwab,
Michaela Seeling, Melissa Woigk und Jessica Zöller bedanke ich mich für die nette
Zusammenarbeit. Jeden Tag gern ins Labor zu gehen, egal wie die Experimente gerade
laufen, hat auch viel mit euch zu tun.
Meiner Familie, allen voran meinen Eltern, danke ich für ihre andauernde Unterstützung bei
allen meinen Projekten. Auch wenn ich mittlerweile mein eigenes Leben führe, seid ihr doch
immer nahe bei mir und steht mir immer mit guten Ratschlägen und konstruktiver Kritik zur
Seite.
Meinem Mann Stefan danke ich, dass er ist, wie er ist. Mit einer unendlichen Geduld und
Verständnis, hast du mich stets wieder aufgerichtet, die Dinge ins rechte Licht gerückt und
mir auch mal den notwendigen „Tritt in den Hintern“ verpasst. Ich liebe dich!
98