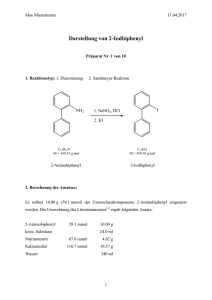
Lineare und cyclische Oligopeptide aus 3α
Werbung

Lineare und Cyclische Oligopeptide aus 3α α-Aminocholansäure-Bausteinen - Synthesen, Konformationen und Komplexierungsverhalten - von Rüdiger Ladberg aus Dortmund Lineare und Cyclische Oligopeptide aus 3α α-Aminocholansäure-Bausteinen - Synthesen, Konformationen und Komplexierungsverhalten - Der Fakultät für Chemie der Ruhr-Universität Bochum zur Erlangung des Doktorgrades Dr. rer. nat. vorgelegt von Rüdiger Ladberg aus Dortmund Als Dissertation genehmigt von der Fakultät für Chemie der Ruhr-Universität Bochum. Tag der Disputation: 14. Dezember 2001 Vorsitzender des Prüfungsausschusses: Referent: Koreferent: Drittprüfer: Prof. Dr. C. Wöll Prof. Dr. M. Feigel Prof. Dr. D. Hasselmann* Prof. Dr. B. Hovemann * in der mündlichen Prüfung vertreten durch Prof. Dr. W. Sander Die vorliegende Arbeit entstand in der Zeit von August 1996 bis Juli 2001 (mit einer zehnmonatigen Unterbrechung von Dezember 1996 bis September 1997) im AK Naturstoffchemie an der Fakultät für Chemie der Ruhr-Universität Bochum. Mein besonderer Dank gilt an dieser Stelle: Herrn Prof. Dr. M. Feigel für die uneingeschränkte wissenschaftliche Betreuung, seine stetige Bereitschaft zu fachlichen Diskussionen und sein großes Interesse am Fortgang dieser Arbeit, das deren Entstehen entscheidend gefördert hat. Außerdem danke ich ihm für die Ermöglichung der Aufnahme von verschiedenen Tätigkeiten außerhalb dieser Arbeit und die Ermutigung hierzu. Diese Aktivitäten haben meine persönliche Weiterentwicklung positiv beeinflußt. Der Deutschen Forschungsgemeinschaft für die finanzielle Unterstützung. Meinen Kollegen und Kolleginnen im Arbeitskreis: Carsten Berghaus, Dr. Bernhard Engels Sabine Felsch, Kerstin Floeder, Dr. Christian Gailer, André Iffland, Andreas Kyas, Heinz Mehlmann, Andrey und Daniel Olschewski, Magnus Ott, Dr. Andreas Rybka, Dr. Claus Weigand und Dr. Maria Wortmann sowie unserem technischen Angestelten Dipl.-Ing. (FH) Kurt Hobert für anregende Diskussionen, stetige Hilfsbereitschaft und das angenehme und freundschaftliche Arbeitsklima. Herrn Prof. Dr. W.S. Sheldrick und seinen Mitarbeitern, insbesondere Frau M. Winter, für die Anfertigung von Röntgenstrukturanalysen. Herrn Dr. W. Dietrich, Herrn M. Gartmann und Herrn Dr. G. Barchan für die Bereitstellung von Meßzeit an den NMR-Geräten und die fachliche Beratung bei auftretenden NMRProblemen. Herrn Dr. D. Müller und seinen Mitarbeitern für die Aufnahme der Massenspektren. Allen hier nicht namentlich genannten Angestellten der Ruhr-Universität, die durch ihre Unterstützung zum Entstehen dieser Arbeit beigetragen haben. Dr. Sabine Bögge, Christine Dümler, Christine Gack, Bernd Graz, Uta Häsel, Markus und Kerstin Münzner, Stefan Parsch, Helge Seehaus, Nicolé Zeggel und allen anderen Freunden für fächerübergreifende Diskussionen sowie für aufbauende Worte während augetretener „Durststrecken“. Der größte Dank jedoch geht an meine Familie, die durch ihre vorbehaltlose Unterstützung mein Promotionsvorhaben erst möglich werden ließ. Inhaltsverzeichnis I Inhaltsverzeichnis 1. Einleitung 1 1.1. Vorbemerkungen 1 1.2. Zielsetzung 8 2. Synthetischer Teil 12 2.1. Synthese der 3α-Aminocholansäuren 5 12 2.1.1. Auswahl der Synthesestrategie 12 2.1.2. Syntheseweg für die 3α-Aminocholansäuren 5 14 2.2. Synthese linearer Oligoamide mit Aminocholansäuren 18 2.2.1. Lineare Oligomere aus Typ-1-Bausteinen 21 2.2.1.1. Lineare Oligomere von 3α-Aminolithocholsäure 5a 22 2.2.1.2. Lineare Oligomere von 3α-Aminodesoxycholsäure 5b 22 2.2.1.3. Lineare Oligomere von 3α-Aminocholsäure 5c 23 2.2.2. Lineare Oligomere aus Typ-2-Bausteinen 24 2.2.2.1. Synthese der Typ-2-Bausteine 24 2.2.2.2. Synthese der linearen Typ-2-Oligomere 26 2.2.2.2.1. Oligomere aus Val-LCS-Bausteinen 26 2.2.2.2.2. Oligomere aus Lys[Z]-LCS-Bausteinen 26 2.2.2.2.3. Oligomere aus Val-DCS-Bausteinen 27 2.2.2.2.4. Oligomere aus Val-CHS-Bausteinen 28 2.2.3. Tabellarische Übersicht über alle synthetisierten linearen Oligoamide 29 2.3. Cyclische Oligoamide mit 3α-Aminocholansäuren 5 30 2.3.1. Unsystematische Oligocyclisierung von Typ-1-Monomeren 31 2.3.1.1. Oligocyclisierung von 3α-Aminolithocholsäure 5a 32 2.3.1.2. Oligocyclisierung von 3α-Aminodesoxycholsäure 5b 33 2.3.1.3. Oligocyclisierung von 3α-Aminocholsäure 5c 34 2.3.2. Cyclisierung linearer Oligoamide 35 2.3.2.1. Cyclisierung von Boc-DCS-DCS-DCS-OMe 16a 35 2.3.2.2. Cyclisierung von Boc-DCS-DCS-DCS-DCS-OMe 17a 36 Inhaltsverzeichnis II 2.3.2.3. Cyclisierung von Boc-CHS-CHS-CHS-OMe 19a 37 2.3.2.4. Cyclisierung von Boc-CHS-CHS-CHS-CHS-OMe 20a 37 2.3.2.5. Cyclisierung von Boc-Val-LCS-Val-LCS-Val-LCS-OMe 26a 38 2.3.2.6. Cyclisierung von Boc-Lys[Z]-LCS- Lys[Z]-LCS- Lys[Z]-LCS-OMe 28a 39 2.3.2.7. Cyclisierung von Boc-Val-DCS-Val-DCS-Val-DCS-OMe 30a 40 2.3.2.8. Cyclisierung von Boc-Val-CHS-Val-CHS-Val-CHS-OMe 32a 41 2.3.3. Übersicht über die synthetisierten Cyclooligoamide 3. Konformationsanalyse und Komplexierungsverhalten 43 44 3.1. Allgemeine Methoden 44 3.1.1. Experimentelle Methoden zur Strukturaufklärung 44 3.1.2. Theoretische Konformationsanalyse 45 3.2. Untersuchung der synthetisierten Verbindungen 47 3.2.1. Lineare Oligomere 47 3.2.1.1. Lineare Oligomere vom Typ 1 49 3.2.1.2. Lineare Oligomere vom Typ 2 54 3.2.2. Cyclische Oligomere 64 3.2.2.1. Macrolactame aus Typ-1-Bausteinen 64 3.2.2.2. Macrolactame aus Typ-2-Bausteinen 75 4. Zusammenfassung und Ausblick 4.1. Zusammenfassung 4.2. Ausblick 94 94 103 5. Experimenteller Teil 105 5.1. Allgemeines 105 5.1.1. Bemerkungen 105 5.1.2. Abkürzungen 107 5.2. Präparative Vorschriften 108 5.2.1. Darstellung der 3α-Aminocholansäuren 5a-c 108 5.2.1.1. 3-Oxo-Cholansäuren 6a-c 108 5.2.1.2. Cholansäure-3-Oxime 10a-c 113 Inhaltsverzeichnis 5.2.1.3. 3α-Aminocholansäuren 5a-c III 114 5.2.2. Peptidsynthesen mit Aminocholansäure-Bausteinen 116 5.2.2.1. Allgemeine Arbeitsvorschriften (AAV) 116 5.2.2.2. Darstellung der Boc-geschützten 3α-Aminocholansäuren 11a-c 119 5.2.2.3. Darstellung der 3α-Aminocholansäuremethylester-Hydrochloride 12a-c 120 5.2.2.4. Lineare Oligoamide aus Aminocholansäure-Bausteinen 121 5.2.2.4.1. Oligopeptide mit 3α-Aminolithocholsäure 122 5.2.2.4.2. Oligopeptide mit 3α-Aminodesoxycholsäure 125 5.2.2.4.3. Oligopeptide mit 3α-Aminocholsäure 128 5.2.2.4.4. Oligopeptide mit N-Valinyl-3α-Aminolithocholsäure 130 5.2.2.4.5. Oligopeptide mit N-[ε-Benzyloxycarbonylamino-Lysinyl]3α-Aminolithocholsäure 133 5.2.2.4.6. Oligopeptide mit N-Valinyl-3α-Aminodesoxycholsäure 137 5.2.2.4.7. Oligopeptide mit N-Valinyl-3α-Aminocholsäure 140 5.2.2.5. Cyclische Oligoamide aus Aminocholansäure-Bausteinen 144 5.2.2.5.1. Unsystematische Oligocyclisierung monomerer 3α-Aminocholansäuren 144 5.2.2.5.2. Systematische Cyclisierung linearer Oligoamide 147 5.2.2.5.2.1. Cyclisierung reiner 3α-Aminocholansäure-Oligoamide (Typ 1) 147 5.2.2.5.2.2. Cyclisierung gemischter L-Aminosäure-/3α-AminocholansäureOligoamide (Typ 2) 151 6. Literaturverzeichnis 156 Anhang A (Röntgenstrukturdaten zu 6b) 160 Anhang B (Röntgenstrukturdaten zu 21a) 166 Anhang C (Röntgenstrukturdaten zu 34a) 173 Anhang D (Ergänzende Überlegungen zur Struktur und Topologie von 38b) 180 Lebenslauf 184 1. Einleitung 1 1. Einleitung 1.1. Vorbemerkungen In der Supramolekularen Chemie liegen neben der Synthese neuer, komplexer Verbindungen insbesondere Phänomene wie Selbstorganisation und molekulare Erkennung im Focus des Interesses, deren Auftreten auf schwachen intra- bzw. intermolekularen Wechselwirkungen beruht1. Für die Gestalt und Funktion von Proteinen sind diese Erscheinungen von zentraler Bedeutung. Wie auch andere Makromoleküle können Proteine ihre eigene Faltung steuern, sich also selbst organisieren, und in Abhängigkeit von der Aminosäure-Sequenz bestimmte lokale Sekundärstrukturen, etwa Helices oder Faltblätter, ausbilden, welche sich dann wiederum zur Tertiärstruktur, der dreidimensionalen Konformation des gesamten Polypeptids, zusammenlagern. Besonders die konformativen Gegebenheiten in den aktiven Zentren von Enzymen, in welchen eine bestimmte Verbindung selektiv gebunden (also „erkannt“), chemisch umgewandelt und anschließend wieder freigesetzt wird, werden derzeit intensiv erforscht2. So können Anhaltspunkte für die Synthese nichtnatürlicher Makomoleküle erhalten werden, welche die Konformation und Funktionalität solcher Zentren in kleinem Maßstab, d.h. ohne das große Proteingerüst, in das die aktiven Zentren eingebettet sind, nachbilden. Solche künstlich hergestellten Makromoleküle, welche ähnlich einem Rezeptorprotein selektiv eine bestimmte Verbindung zu komplexieren vermögen oder sogar – vergleichbar einem Enzym – diese in eine andere Verbindung umwandeln können, stellen eine besondere Herausforderung für die moderne Organische und Metallorganische Chemie dar. Zahlreiche aktuelle Veröffentlichungen auf dem Gebiet der sogenannten Wirt-Gast-Chemie3 sind Beleg für den besonderen Reiz, den dieses Forschungsgebiet auf den Chemiker ausübt. Das Forschungsgebiet der Wirt-Gast-Chemie geht zurück auf die Synthese des ersten Kronenethers durch C.J. Pedersen4 im Jahr 1967 – die erste ungeladene synthetische Verbindung, welche in der Lage war, Alkalimetallionen zu komplexieren. Nachdem J.M. Lehn et al. in der Folgezeit durch die Synthese sowie die Erforschung der Bindungseigenschaften von Cryptanden5 auf das Potential des neuen Forschungsgebietes aufmerksam gemacht hatten, war es schließlich D.J. Cram, der 1977 erstmals eine 1. Einleitung 2 systematische Definition zur Wirt-Gast-Chemie und zur Komplexbildung lieferte, welche noch heute breite Anerkennung findet6: Demnach bestehen Komplexe aus zwei oder mehr Molekülen, die in „einzigartigen strukturellen Beziehungen durch andere elektrostatische Kräfte als die rein kovalenten Bindungen zusammengehalten werden“. Als solche Kräfte gab er für Molekülkomplexe z.B. Wasserstoffbrückenbindungen, Ionenbindungen, π-Säure-π-Base-Wechselwirkungen, MetallLiganden-Bindungen, van-der-Waals-Wechselwirkungen oder partielle kovalente Bindungen (Übergangszustände) an. Zur Wirt-Gast-Beziehung definiert Cram: „Ein hochstrukturierter Molekülkomplex besteht aus mindestens einem Wirt und einem Gast“, wobei „eine hohe strukturelle Organisation [...] gewöhnlich nur durch das Vorhandensein mehrerer Bindungsstellen erreicht“ wird. Zu einer Wirt-Gast-Beziehung gehört nach Cram eine „komplementäre stereoelektronische Anordnung der Bindungsstellen von Wirt und Gast“. Dabei ist eine Wirtverbindung „ein organisches Molekül oder Ion [...], dessen Bindungsstellen im Komplex konvergieren“, eine Gastverbindung ist entsprechend „ein beliebiges Molekül oder Ion, dessen Bindungsstellen im Komplex divergieren“. Diese Verhältnisse werden in Abbildung 1.1. veranschaulicht. Abbildung 1.1. Wirt-Gast-Komplexbildung Das besondere Interesse an der Wirt-Gast-Chemie beruht auf den vielfältigen potentiellen Anwendungsmöglichkeiten dieses Gebiets, beispielsweise in der Biomimetischen Chemie (künstliche Rezeptoren, Enzyme oder Signalmoleküle, die biologischen Systemen nachempfunden sind), der Analytik (etwa der Stofftrennung durch den Einsatz immobilisierter Wirtverbindungen, welche selektiv eine Komponente eines Substanzgemisches komplexieren), der Pharmazie (Verkapseln bzw. Maskieren von Wirkstoffen durch geeignete 1. Einleitung 3 Wirtverbindungen, welche als Carrier das Medikament an den beabsichtigten Wirkort bringen und dort freisetzen) oder der Katalyse (das Gastmolekül wird nicht nur selektiv komplexiert, sondern auch chemisch umgewandelt). Inzwischen wurden zahlreiche verschiedene cyclische und acyclische supramolekulare Systeme synthetisiert und hinsichtlich ihrer Selbstorganisation und ihrer Komplexierungseigenschaften untersucht. Vorteile für die Konstruktion synthetischer molekularer Wirtverbindungen bieten dabei z.B. Steroide als Bausteine. Einige biochemisch wichtige Gruppen von Steroiden sind in der folgenden Liste aufgeführt: - die Sterole (auch Sterine, bekanntestes Beispiel ist das Cholesterin), HO Cholesterin (Cholesterol) - die Steroid-Hormone (Sexualhormone wie beispielsweise Androsteron, sowie Hormone der Nebennierenrinde, auch Corticoide genannt – ein bedeutendes Beispiel ist hier das Cortisol, welches unter dem Namen Hydrocortison als Medikament Verwendung findet), OH O O OH OH OH O Andosteron - Cortisol die herzaktiven Steroide (Cardenolide wie das Digitoxigenin, welches als sein bekannteres Glykosid Digitoxin im roten Fingerhut vorkommt und als solches als Herzmedikament Verwendung findet, sowie Bufadienolide, die wie beispielsweise das Bufotalin als Giftstoffe in Krötensekreten vorkommen [lat. bufo = Kröte]), 1. Einleitung 4 O O O O OH OH O HO HO O Digitoxigenin - Bufotalin die Steroid-Vitamine (Vitamine der D-Reihe bzw. deren Provitamine, wie beispielsweise das Ergosterin (Provitamin D2)), HO Ergosterin - die Cholansäuren (auch Gallensäuren; sie kommen, gebunden an Aminosäuren wie Taurin oder Glycin, in der Galle vor und ermöglichen durch Emulgation von Fetten deren Verdauung; bekanntester Vertreter ist die Cholsäure). O OH OH OH OH Cholsäure Die aufgeführten Beispiele zeigen, dass das tetracyclische Gerüst der Steroide unterschiedliche Konfigurationen aufweisen kann. So können in den vollständig abgesättigten Ringsystemen die Ringe A und B sowie die Ringe C und D jeweils cis- oder trans-verknüpft sein, während die Ringe B und C stets trans-Verknüpfung aufweisen (siehe Abb. 1.2.). 1. Einleitung 5 R R β α 21 A/B-cis C/D-trans H3C CH3 19 CH3 20 23 12 11 A/B-cis C/D-cis 22 18 13 17 C 14 D 16 24 15 1 2 3 A 10 5 4 A/B-trans C/D-trans B 6 H 26 9 8 7 25 H CH3 H3C 27 A/B-trans C/D-cis R R β α Abbildung 1.2. Systematische Numerierung des Steroid-Gerüstes am Beispiel des Cholestans mit Angabe der Konfiguration aller vier möglichen Ringverknüpfungskombinationen (Die beiden rechts abgebildeten Formen kommen in natürlichen Sterinen, deren Stammverbindung das Cholestan ist, nicht vor - eine C/D-cis-Verknüpfung findet sich lediglich bei den herzaktiven Steroiden). Substituenten am Steroidgerüst, die auf der gleichen Seite des Ringsystems wie die beiden Methylgruppen an C-18 und C-19 liegen, werden als β-ständig, Substituenten auf der gegenüberliegenden Seite als α-ständig bezeichnet. Entsprechend wird bei der Nomenklatur der Ringverknüpfungsstellen an C-5 und C-14 verfahren. Aus der unterschiedlichen Verknüpfung ergeben sich bestimmte starre Formen des SteroidGerüstes. Besonders geeignet als Baustein für die Konstruktion supramolekularer Wirtverbindungen ist dabei das A/B-cis und C/D-trans verknüpfte Gerüst der Gallensäuren 1.7 Diese besitzen eine gekrümmte Oberfläche, welche auf der äußeren, konvexen Seite unpolar ist, auf der inneren, konkaven Seite jedoch bis zu drei räumlich fixierte polare Hydroxylgruppen aufweist (vgl. Abb. 1.3.). Letztere können nach Bedarf auch jeweils in andere funktionelle Gruppen umgewandelt werden bzw. als Ankergruppen für komplexere Substituenten dienen. 1. Einleitung 6 Abbildung 1.3. Das starre Gerüst und die zwei unterschiedlichen Seiten der optisch aktiven Cholansäuren machen sie zu hervorragenden Bausteinen für Wirtverbindungen Zudem bietet sich mit der Carbonsäure-Funktion an C-24 eine Möglichkeit zur Anknüpfung weiterer Gallensäure-Einheiten. Ein weiterer Vorteil ist die leichte Verfügbarkeit der drei häufigsten Gallensäuren: Lithocholsäure (1a), Desoxycholsäure (1b) und Cholsäure (1c) sind relativ günstig im Handel erhältlich. Da die systematischen Namen der genannten Cholansäuren 1a-c relativ unübersichtlich sind (der systematische Name der Cholsäure 1c ist beispielsweise 3α, 7α ,12α-Trihydroxy-5β-cholan-24-säure) wird auf deren Nennung im Folgenden verzichtet und auf die kürzeren und in der Naturstoffchemie gebräuchlicheren Trivialnamen Lithocholsäure, Desoxycholsäure und Cholsäure zurückgegriffen. Es sind bereits verschiedene cyclische und acyclische Verbindungen bekannt, die sich der besonderen Eigenschaften des Cholansäuregerüstes bedienen und zum Teil auch als Wirtverbindungen für Ionen oder ungeladene, polare Moleküle fungieren. Dabei wird das Cholansäuremolekül teilweise direkt verwendet, häufiger aber mit Modifikationen am Gerüst, insbesondere an den Positionen C3, C7 und C12 sowie an der Seitenkette an C17. Besonders Verbindungen auf Esterbasis, bei denen die Hydroxylgruppe an C3 mit der SeitenkettenSäuregruppe des nächsten Bausteins verknüpft wurde, und Verbindungen auf Amid-Basis, bei deren Cholansäure-Bausteinen die C3-Hydroxylgruppe direkt oder über eine Brücke zu einer Aminogruppe umfunktionalisiert wurde, wurden hier bislang synthetisiert und untersucht. Unter den Cholansäurehaltigen Macrocyclen auf Esterbasis verdienen besonders die Macrolactone aus in der Regel zwei, drei, oder vier meist an C7 bzw. C12 weiter funktionalisierten Cholansäuren besondere Beachtung. Die meisten dieser Verbindungen 1. Einleitung 7 wurden aus den monomeren Bausteinen dargestellt durch die sogenannte YamaguchiMacrolactonisierung8, einer kinetisch kontrollierten, unter den Reaktionsbedingungen nicht reversiblen Oligocyclisierungsreaktion, welche in unterschiedlichen Anteilen ein Gemisch aus Cholansäure-Macrocyclen vom Dimer bis (nachgewiesen) zum Hexamer liefert. Diese Macrolactone sind beispielsweise in der Lage, Kationen9 oder auch Ferrocen10 zu komplexieren, bzw. als tetramere Schüssel, die auf der einen Seite durch einen Porphyrinring verschlossen ist, sogar Morphin enantioselektiv zu erkennen11. Ein anderer synthetischer Ansatz zur Konstruktion solcher Cholansäure-Macrolactone wird von J.K.M. Sanders et al.12 beschrieben: Er hat einen Weg zur thermodynamisch kontrollierten Macrolactonisierung entwickelt, der auf einer Umesterungs-Reaktion beruht. Da die Cyclisisierungsreaktion bis zu einem Quenching-Schritt reversibel ist, kann durch den Einsatz von Templaten während der Reaktion die bevorzugte Bildung bestimmter Macrolactone induziert werden (beschrieben wurde eine Verschiebung des Produktverhältnisses zugunsten der höheren Cyclen bei Verwendung von Natrium-Ionen als Templat). Bei der Verwendung verschiedener monomerer Cholansäure-Bausteine, welche gemischte Macrolactone liefern13, sieht Sanders darin die Grundlage für den Aufbau dynamischer kombinatorischer Bibliotheken, durch die selektiv und gezielt geeignete Wirtverbindungen für bestimmte Moleküle erhalten werden können. Cyclen auf Amid-Basis aus modifizierten Cholansäuren sind ebenfalls beschrieben und sind beispielsweise in der Lage, Anionen14 oder Kohlenhydrate15 zu komplexieren. Ein Beispiel für eine Kohlenhydrat-komplexierende Wirtverbindung stellt das Cholaphan 2 dar. H OR O N OH OH N H 2 O OR R = H oder R = OBn 1. Einleitung 8 Auch gemischte Macrocyclen wie 3 und 4 aus natürlichen Aminosäuren und jeweils zwei Aminocholansäuren sind bekannt16. Hier dienen die Aminocholansäuren zur Fixierung bestimmter Konformationen der Aminosäure-Komponenten im Cyclus. So wurden zum Beispiel γ-Loops, β-Loops und helicale Konformationen in den dipeptidischen AminosäureKetten der Verbindung 4 postuliert. Bei diesen Systemen scheint jedoch der Raum im Inneren der Cyclen aufgrund der zu geringen Größe nicht für Komplexierungen geeignet zu sein, wie Komplexierungsversuche und Modellingstudien zeigen17. R2 O H R1 R2 H N O H N R1 O H H N N O O H H N N H H O N O N H H H R2 O O N N H R1 H R2 R1 O 3 a) R1 = R2 = H b) R1 = H; R2 = OAc c) R1 = R2 = OAc 4 In acyclischen Systemen dienen Gallensäure-Derivate unter anderem als Gerüst, um biochemische Funktionen räumlich anzuordnen und zu permutieren18, oder um enantio- und regioselektive Reaktionen (kovalent) gebundener Substrate zu induzieren19 oder zu katalysieren20. Außerdem dienen an fester Phase fixierte aminofunktionalisierte Cholansäuren als Linker bzw. Spacer für die kombinatorische Synthese von Peptidsträngen21 mit dem Ziel, ein geeignetes Mimetikum für die katalytische Triade von Serinproteasen zu erhalten. 1.2. Zielsetzung Aufgrund ihrer besonderen Form stellen Cholansäuren exzellente Bausteine für supramolekulare Verbindungen, insbesondere für Wirtverbindungen, dar. Ein entscheidender Nachteil bei der direkten Verwendung ist die relative Empfindlichkeit der aus ihnen 1. Einleitung 9 gebildeten Ester-Oligomere gegen Säuren und Basen, was sie für eine Verwendung als Gerüstbausteine in katalytisch aktiven Systemen oder auch in Carrier-Systemen für Medikamente als eher ungeeignet erscheinen läßt. Daher erschien es interessant, den in der Arbeitsgruppe bereits aufgegriffenen Ansatz weiterzuverfolgen und an C3 α-aminofunktionalisierte Cholansäuren 5 als Bausteine zu verwenden, deren Oligomere durch die amidische Verknüpfung zwei entscheidende Vorteile gegenüber Estern aufweisen sollten: zum einen sind diese Bindungen gegen Säuren und Basen weitgehend unempfindlich, zum anderen bringt der partielle Doppelbindungscharakter der Amidbindungen zusätzliche Rigidität gegenüber der frei rotierbaren Esterbindung. Erste Arbeiten hierzu wurden bereits von D. Albert unternommen, der durch Cyclodimerisierung von linearen Amiden, bestehend aus jeweils einer Aminocholansäure und ein bzw. zwei natürlichen Aminosäuren, die Cyclen 3 und 4 erhalten und untersucht hatte (vgl. Kapitel 1.1.). O R2 OH R1 NH2 5a R1 = R2 = H 5b R1 = H; R2 = OH 5c R1 = R2 = OH In der vorliegenden Arbeit galt das Interesse der Synthese von linearen und cyclischen Oligoamiden unter Verwendung von 3α-Aminolithocholsäure 5a, 3α-Aminodesoxycholsäure 5b und 3α-Aminocholsäure 5c. Hierzu war eine Optimierung des synthetischen Zuganges zu diesen Aminocholansäure-Bausteinen wichtig, um sie auch in großem Maßstab leicht und epimerenrein verfügbar zu machen. Direkte lineare Oligomere der Aminocholansäuren 5 waren noch unbekannt und es erschien interessant, zu untersuchen, ob bereits kleine Systeme mit bis zu vier Bausteinen bereits konformative Besonderheiten aufweisen (z.B. die Ausbildung einer Helix, welche in ihrem Inneren ähnlich wie bei der Jod-StärkeKomplexierung ein Gastmolekül aufnehmen könnte). Ebenso interessant erschien es, zu überprüfen, ob die Aminocholansäuren 5 bei der Cyclisierung analog zur kinetisch kontrollierten Macrolactonisierung ihrer natürlichen Stammverbindungen 1 (vgl. Kap. 1.1.) ebenfalls höhere Amid-Oligocyclen liefern, oder ob hier nur eine reine Cyclodimerisierung der Monomere beobachtet werden kann, wie sie unter den für eine Amid-Bindungsbildung notwendigen Reaktionsbedingungen bislang an vergleichbaren Systemen gefunden worden 1. Einleitung war15, 16 10 . Aufgrund der potentiellen Eignung als Wirtverbindungen sollten die bisher nicht bekannten Cyclooligomere der Aminocholansäuren auch durch systematische Synthese, also durch Cyclisierung der linearen Oligomere, zugänglich gemacht werden und hinsichtlich ihrer Konformation und – exemplarisch – ihres Komplexierungsverhaltens untersucht werden. Ein besonders vielversprechender Ansatz auf dem Weg zu neuen Wirtverbindungen ist die bereits von D. Albert aufgegriffene Kombination von Aminocholansäuren mit natürlichen Aminosäuren. Hier können die Vorteile des starren Steroid-Gerüsts mit seinen Bindungsstellen bzw. Funktionalisierungsmöglichkeiten an C7 und C12 um die durch die verschiedenen Seitenketten natürlicher Aminosäuren gegebene Vielfalt an Funktionalität ergänzt werden und gleichzeitig konformative Besonderheiten in den Oligomeren induziert werden. Im Rahmen der vorliegenden Arbeit sollten zur näheren Untersuchung dieses Potentials lineare und cyclische Oligoamide synthetisiert werden, welche jeweils alternierend aus einer natürlichen Aminosäure und einer Aminocholansäure bestehen. Dazu sollten vorab synthetisierte Bausteine vom Typ Aminosäure-Aminocholansäure zunächst zu linearen Oligomeren bis hin zum Hexaamid (auf die Bausteine bezogen also Trimere) gekoppelt werden, welche dann anschließend systematisch zu den entsprechenden Cyclohexaamiden cyclisiert werden sollten. Die schematischen Darstellungen auf der folgenden Seite (Schema 1.1.) geben einen Überblick über die Ziele der vorliegenden Arbeit. 1. Einleitung Schema 1.1. 11 2. Synthetischer Teil 12 2. Synthetischer Teil 2.1. Synthese der 3α α-Aminocholansäuren O R2 OH R1 NH2 5a R1 = R2 = H 5b R1 = H; R2 = OH 5c R1 = R2 = OH In der Literatur sind verschiedene Wege zur Darstellung von 3α-Aminocholansäuren 5 bekannt. Als Ausgangssubstanz der Synthesewege dienen jeweils die entsprechenden Cholansäuren 1, die im Handel erhältlich sind. Man kann zwei grundsätzliche Synthesestrategien unterscheiden: Erstens die Substitution der OH-Gruppe an C3 durch eine Azidgruppe unter doppelter Inversion mit anschließender Reduktion des Azids, zweitens die Oxidation der Alkohlofunktion an C3 zum entsprechenden Keton, welches weiter zum Oxim umgesetzt wird. Die anschließende Reduktion des Oxims unter geeigneten Bedingungen ergibt dann die entsprechenden 3α-Aminocholansäuren, wobei prinzipiell natürlich auch das 3β-Analogon entstehen kann. Das Syntheseziel für diese Arbeit war eine Ausbeute von 10-20g epimerenreiner 3α-Aminocholansäure pro Ansatz über möglichst wenige Stufen, um damit eine Grundlage für die optionale Übertragung der Synthese in Technikumsmaßstäbe zu schaffen. 2.1.1. Auswahl der Synthesestrategie für die vorliegende Arbeit 2.1.1.1. Synthesen nach der Azid-Route Zu dieser Strategie, welche zunächst die Veresterung der Cholansäuren erfordert, sind bereits mehrere Vorschriften veröffentlicht22. Auch in der hiesigen Arbeitsgruppe bestehen Erfahrungen mit diesem Syntheseweg16, 17. Einen Überblick über das prinzipielle Vorgehen bei dieser Strategie zeigt Schema 2.1. Die doppelte Inversion beruht auf einer zweifachen Mitsunobu-Reaktion. Zum Teil sind die Synthesen auch mit größeren Mengen (Ansätze bis 5g) beschrieben, jedoch bergen sie alle den Nachteil, daß einige Reaktionsschritte sehr 2. Synthetischer Teil 13 empfindlich sind und Schutzgas und absolutierte Lösungsmittel erfordern und daß auf dem Syntheseweg mindestens eine säulenchromatographische Reinigung notwendig ist. Dadurch erschien die Anpassung der Synthese auf große Ansätze, wie es für diese Arbeit gewünscht war, sehr schwierig. Schema 2.1. O R1 OMe Red. HO RO R2 N3 H2N 2.1.1.2. Synthesen nach der Oxim-Route Synthesen nach dieser Strategie (siehe Schema 2.2.) sind schon in den späten 40er Jahren von Jones et al. sowie von Redel et al. beschrieben worden23. Nach der Oxidation an C3 wurde hier jeweils das Keton in das entsprechende Oxim überführt und dieses dann mit Natrium in siedendem Amylalkohol zum Amin reduziert. Zu dieser Zeit konnten jedoch noch keine Angaben über die Stereochemie an C3 der gebildeten Aminocholansäuren gemacht werden. Schema 2.2. O R OH HO R2 Natrium/ Amylalkohol NH2OH/ NaOAc Ox. O HON H2N ? Ein Hinweis darauf, daß dieser Weg die korrekte Stereochemie liefert, fand sich in einer aktuelleren Arbeit von Bellini et al.24: Bellini reduzierte das Oxim von 3-Oxo-Lithocholsäure ebenfalls mit Natrium in Amylalkohol. Zwar entstand hierbei ein Gemisch von 3α- und 3β-Aminolithocholsäure im Verhältnis 90:10, doch gelang es Bellini durch geschickte Aufarbeitung, den größten Teil des 3α-Epimers selektiv und ohne die Notwendigkeit einer 2. Synthetischer Teil chromatographischen 14 Trennung zu isolieren. Zum Vergleich stellte er reine 3β-Aminolithocholsäure durch direkte reduktive Aminierung des Ketons her. Da beide Verbindungen unterschiedliche Kernresonanzspektren liefern müssen (in 3α-Aminolithocholsäure steht das Proton an C3 axial, was eine große Kopplung mit den benachbarten Ringprotonen und damit ein breites Signal erwarten läßt, während das äquatoriale C3-Proton in 3β-Aminolithocholsäure nur kleine Kopplungen zeigen sollte), kann die Zuordnung als zuverlässig angesehen werden. Da Redel und Jones auch die Darstellung von 3-Aminodesoxycholsäure und 3-Aminocholsäure auf diesem Weg beschrieben hatten23, erschien es plausibel, daß auch hier bevorzugt die 3α-Epimeren entstehen sollten. Weil die 3-Oxo-Verbindungen aus den entsprechenden Cholansäuren relativ leicht in wenigen Schritten zugänglich sind und bis zum Oxim keinerlei chromatographische Trennungen erforderlich sind, und da Redel und Jones die Aminocholansäuren bereits in großen Ansätzen (bis ca. 20 g) synthetisiert hatten, erschien es sinnvoll, die Synthese der 3α-Aminocholansäuren 5 für die vorliegende Arbeit nach dieser Strategie zu versuchen, zumal auch in keinem Syntheseschritt die Verwendung von Schutzgas oder größeren Mengen absolutierter Lösungsmittel erforderlich sein sollte. 2.1.2. Syntheseweg für die 3α α-Aminocholansäuren 5 Ausgangspunkt der Synthese waren die im Handel erhältlichen Cholansäuren 1a-c. Zur Synthese der 3α-Aminocholansäuren 5a-c über die Oxim-Route mußten zunächst die entsprechenden 3-Oxo-Cholansäuren 6a-c hergestellt werden, aus denen das Oxim durch Umsetzung mit Hydroxylamin zugänglich ist. 2.1.2.1. 3-Oxo-Cholansäuren 6 Während 3-Oxo-Lithocholsäure 6a durch einfache Jones-Oxidation aus Lithocholsäure selektiv hergestellt werden kann, ist bei Chol- und Desoxycholsäure zu beachten, daß die OH-Gruppen an C7 bzw. C12 nicht ebenfalls oxidiert werden. Dies kann entweder durch selektive Oxidation an C3 erfolgen, z.B. mit Aluminium-tert-butylat23, oder durch Schützen der OH-Gruppen an C7 und C12. In der vorliegenden Arbeit wurde letztere Alternative gewählt. 2. Synthetischer Teil 15 2.1.2.1.1. 3-Oxo-Lithocholsäure 6a Die 3-Oxo-Lithocholsäure 6a wurde nach einer Standardvorschrift25 durch Oxidation von Lithocholsäure mit Jones-Reagenz dargestellt. In Abweichung der Vorschrift wurde ein 1:1-Gemisch von Aceton und Essigsäure als Lösungsmittel verwendet, da Lithocholsäure in reinem Aceton unlöslich ist. Das Keton konnte nach Umkristallisation aus Methanol/Wasser in sehr guter Ausbeute (93.4 %) rein erhalten werden. Die Herstellung auch in großen Ansätzen (getestet bis 20g Lithocholsäure als Edukt) war problemlos möglich. Eine alternative Oxidation mit NaOCl-Lösung wurde versucht, um die Verwendung des krebserregenden CrVI und die Entstehung schwermetallsalzhaltiger Abwässer zu umgehen, lieferte jedoch keine reproduzierbar guten Ergebnisse. 2.1.2.1.2. 3-Oxo-Desoxycholsäure 6b und 3-Oxo-Cholsäure 6c Für die Darstellung der Ketone von Desoxycholsäure und Cholsäure wurde die Synthesestrategie von Leppik et al.25 gewählt. Hierbei wurden zunächst alle OH-Gruppen mit Ameisensäure unter Bildung der Formyloxy-Verbindungen 7b und 7c verestert26. Dieser Schritt verläuft quantitativ und die performyloxylierten Cholansäuren können ohne Reinigungsschritt weiterverwendet werden. Anschließend wurde der Ameisensäureester an C3 selektiv verseift. Dies wurde durch langsames Zutropfen stöchiometrischer Mengen einer 0.2 M NaOH-Lösung zu einer Lösung von 7b bzw. 7c in Aceton erreicht. Die selektive Verseifung auf diese Weise ist deshalb möglich, weil der OH--Angriff an die FormyloxyGruppierungen an C7 und C12 durch das Steroid-Gerüst sterisch gehindert ist, so daß die Formyloxy-Gruppe an C3 am schnellsten angegriffen wird. Im nachfolgenden Schritt wurden die gebildeten Verbindungen 8b und 8c mit Jones-Reagenz oxidiert, woraufhin die verbliebene(n) Formyloxy-Schutzgruppe(n) durch Auflösen in 0.5 M NaOH unter kurzem Erwärmen entfernt werden konnten. Die einzelnen Schritte der Synthese sind im nachfolgenden Schema 2.3. exemplarisch am Beispiel der Cholsäure 1c dargestellt: 2. Synthetischer Teil 16 Schema 2.3. O O OH OH OH OH OH O HCOOH HClO4 O H O O 1c O H H O 2 eq. NaOH 7c O OH O O H O OH O OH OH OH O H NaOH O OH 6c 8c O JonesReagenz O H (Überschuß) O O H O 9c O Die Darstellung der 3-Oxo-Cholansäuren 6b und 6c erfolgte – bezogen auf die eingesetzte Desoxycholsäure 1b bzw. Cholsäure 1c – auf diesem Wege praktisch quantitativ. Hierbei waren selbst Ansätze, die von 50 g Desoxycholsäure bzw. Cholsäure ausgingen, erfolgreich. Bei sorgfältiger Durchführung war in keiner der Synthesestufen ein Reinigungsschritt wie z.B. eine Umkristallisation erforderlich, da die Zwischenprodukte nach der in der Literatur beschriebenen Aufarbeitung jeweils sowohl DC- als auch NMR-rein waren. Die Ketone 6b und 6c kristallisierten bereits beim Einengen der Chloroformlösung aus der Aufarbeitung. Auch hier konnte aufgrund der DC-und NMR-Reinheit auf ein Umkristallisieren verzichtet werden. Umkristallisieren einer kleinen Menge 3-Oxo-Desoxycholsäure lieferte Kristalle, die röntgenstrukturanalytisch untersucht werden konnten. Die ermittelte Röntgenstruktur (Abb. 2.1.) belegt, daß die Oxidation tatsächlich selektiv an C3 erfolge und das übrige Steroidgerüst unverändert erhalten geblieben ist. 2. Synthetischer Teil 17 Abbildung 2.1. Kristallstruktur von 3-Oxo-Desoxycholsäure 6b. Details siehe Anhang A. 2.1.2.2. Cholansäure-3-Oxime 10a-c Die Oxime wurden nach einer Standardvorschrift durch Umsetzung der Ketone mit Hydroxylammoniumchlorid in refluxierendem Methanol hergestellt, welche bereits von Redel erfolgreich angewendet worden war23. Dabei diente Natriumacetat als Hilfsbase. Nach Umkristallisation (Methanol/TBME oder reines Methanol) konnten alle drei Oxime in recht guten Ausbeuten rein (DC, NMR) erhalten werden. Hierbei wurden bis zu 42 g Desoxycholsäure-3-oxim 10b (82.7 % Ausbeute) bzw. 38 g Cholsäure-3-Oxim 10c (74.4 % Ausbeute) bzw. 18 g Lithocholsäure-3-oxim 10a (92.4 % Ausbeute) in einem Ansatz hergestellt. 2.1.2.3. 3α α-Aminocholansäuren 5a-c Die 3α-Aminocholansäuren 5a, 5b und 5c wurden durch Reduktion der entsprechenden Oxime mit Natrium in siedendem Amylalkohol hergestellt. Die Isolierung der reinen 3α-Aminocholansäuren erfolgte in leichter Abweichung der Vorschrift von Bellini et al. durch Waschen der mit Ether verdünnten Reaktionsmischung mit kleinen Portionen Wasser, bis das Waschwasser annähernd neutral reagierte. Anschließend konnten die 3α-Aminocholansäuren durch Zugabe von verdünnter Schwefelsäure aus den vereinigten Waschwässern ausgefällt werden: 3α-Aminolithocholsäure 5a präzipitierte ab pH 7, 3α-Aminodesoxy- cholsäure 5b bei pH 9-11 (bei weiterer Säurezugabe erfolgt jedoch eine erneute Auflösung 2. Synthetischer Teil 18 des Niederschlages) und 3α-Aminocholsäure 5c etwa bei pH 5. Die tatsächliche α-Stellung der Aminogruppe an C3 des Steroidgerüstes konnte für 5a und 5b durch Kristallstrukturen von Folgeverbindungen belegt werden (siehe Kapitel 3), für 5c ergibt sie sich durch Analogieschluß aus dem Vergleich der NMR-Signale der C3-Protonen der drei Aminocholansäuren. Die Ansatzgröße wurde hier lediglich limitiert durch die Handhabbarkeit der nötigen Lösungsmittelmengen bei der Aufarbeitung, da der größte zur Verfügung stehende Scheidetrichter ein Fassungsvermögen von lediglich 5 Litern hatte. Somit war die Ansatzgröße auf maximal ca. 20-22g der Oxime 10a-c begrenzt. Ausbeuten über alle Stufen (ausgehend von den entsprechenden Cholansäuren 1a-c): 3α-Aminolithocholsäure 5a: 59.9 % 3α-Aminodesoxycholsäure 5b: 59.9 % 3α-Aminocholsäure 5c: 44.3 % Damit konnten auf diese Weise sogar bessere Ausbeuten erzielt werden als mit der in der Arbeitsgruppe bisher genutzten Azid-Route (max. 43 % über alle Stufen). 2.2. Synthese linearer Oligoamide mit Aminocholansäuren Die Darstellung der Oligoamide erfolgte nach Standard-Kopplungsmethoden aus der Peptidchemie mit Boc-Schutzgruppenstrategie. Als Bausteine dienten entweder die 3α-Aminocholansäuren selbst (Typ 1), die für die Oligoamid-Synthese selektiv aminoterminal (zu 3α-t-Butyloxycarbonylaminocholansäuren 11a-c) oder carboxyterminal (zu 3α-Aminocholansäuremethylester-Hydrochloriden 12a-c) geschützt wurden, oder die aminoterminal um eine natürliche L-Aminosäure verlängerten Aminocholansäuren (Typ 2) (vgl. hierzu Schema 2.4.). 2. Synthetischer Teil 19 Schema 2.4. In der vorliegenden Arbeit galt es zunächst, Oligomere der Typ-1- und einiger Typ-2Bausteine zu synthetisieren und zu untersuchen. Grundsätzlich sind hier verschiedene Synthesestrategien möglich: ein blockweiser Aufbau (konvergente Synthese, vgl. Schema 2.5.a.) oder ein sequentieller Aufbau, wobei die Oligoamid-Ketten entweder aminoterminal oder carboxyterminal Baustein für Baustein verlängert werden können (siehe Schema 2.5.b.). Der blockweise Aufbau hat dabei – neben den prinzipiellen Vorteilen einer konvergenten Synthese – allerdings den Nachteil, dass grundsätzlich nur geradzahlige Oligomere synthetisiert werden können, weshalb er nur mit Aminolithocholsäure exemplarisch durchgeführt wurde. Bei der sequentiellen Kettenverlängerung ist prinzipiell die aminoterminale Verlängerung, wie sie auch standardmäßig in der Peptidchemie angewandt wird, vorzuziehen, da die hierzu nötige Abspaltung der Boc-Schutzgruppe sehr leicht und im Regelfall quantitativ mit Trifluoressigsäure (TFA) erreicht werden kann, wogegen die zur carboxyterminalen Kettenverlängerung notwendige quantitative Verseifung des Methylesters ungleich schwieriger ist27. 2. Synthetischer Teil Schema 2.5. 20 2. Synthetischer Teil 21 Zur besseren Übersichtlichkeit bei der Beschreibung der Oligoamide in den folgenden Abschnitten wurde, analog der Kurzschreibweise für natürliche Aminosäuren in Peptidketten und Proteinen, in der vorliegenden Arbeit für die verwendeten Aminocholansäuren ebenfalls ein Drei-Buchstaben-Code eingeführt, der in der nachfolgenden Tabelle (Tab. 2.1.) dargestellt ist: Aminocholansäure Abkürzung 3α-Aminolithocholsäure 5a LCS 3α-Aminodesoxycholsäure 5b DCS 3α-Aminocholsäure 5c CHS Tabelle 2.1.: Dreibuchstabencode für 3α-Aminocholansäuren Bei der Angabe der Sequenzabfolge der Oligoamide wird entsprechend der in der Peptidchemie üblichen Konventionen mit dem aminoterminalen Baustein begonnen. 2.2.1. Lineare Oligomere aus Typ-1-Bausteinen Hier wurden Oligomere bis hin zum Tetramer synthetisiert, jeweils auf unterschiedlichen Wegen (vgl. Schema 2.5.). Aufgrund der schlechter werdenden Löslichkeit wurde auf die Synthese höherer Oligomere verzichtet. Zur Knüpfung der Amid-Bindungen wurden verschiedene Kopplungsreagenzien getestet. Mit einigen konnte keine bzw. keine nennenswerte Kopplung erreicht werden, wie z.B. mit CDMT (2-Chlor-4,6-dimethoxy-1,3,5triazin) oder T3P (PPA, Propanphosphonsäureanhydrid). Die besten Ergebnisse konnten durch Einsatz von DEPC (Diethylcyanophosphonat oder Diethylphosphorylcyanid) und EDC (N-Ethyl-N‘-(3-dimethylaminopropyl)-carbodiimid-hydrochlorid) erzielt werden, wobei EDC in den meisten Fällen den Vorzug erhielt, da dessen Reaktionsprodukte bei der Aufreinigung leichter aus dem Reaktionsgemisch zu entfernen sind als die von DEPC und im allgemeinen die Handhabung von DEPC aufgrund seiner hohen Giftigkeit mit größeren Risiken verbunden ist. Die Boc-Abspaltung zur aminoterminalen Kettenverlängerung gelang jeweils quantitativ durch den Einsatz von TFA, die Verseifung der Methylesterschutzgruppe war erheblich aufwendiger, erforderte hohe Überschüsse an KOH und, zur quantitativen Abspaltung, lange 2. Synthetischer Teil 22 Reaktionszeiten (1-3 Tage). Zwar konnten in der vorliegenden Arbeit, von wenigen Ausnahmen abgesehen, bei der Esterverseifung sehr gute Ausbeuten (meist ca. 90-99 %) erzielt werden, jedoch bergen die angegebenen Reaktionsbedingungen bei bestimmten Systemen die Gefahr von Nebenreaktionen (z.B. Eliminierung oder gar die teilweise Spaltung der Amid-Bindungen). 2.2.1.1. Lineare Oligomere von 3α α-Aminolithocholsäure 5a Hier wurde zur Synthese der blockweise Aufbau gewählt und DEPC als Kopplungsreagenz verwendet. Somit wurden hier nur die geradzahligen Oligomere Boc-LCS-LCS-OMe 13a und Boc-LCS-LCS-LCS-LCS-OMe 14a, also das Dimer und das Tetramer, erhalten, welche durch Umfällen bzw. Umkristallisieren rein isoliert werden konnten. Die isolierten Ausbeuten waren zufriedenstellend (Dimer: 71.2 %; Tetramer: 65.3 %), jedoch deutlich kleiner als die tatsächlichen Ausbeuten, da bei der gewählten Reinigungsmethode noch beträchtliche Produktmengen in Lösung verblieben, welche jedoch auf diesem Weg nicht weiter von den Verunreinigungen getrennt werden konnten. Eine säulenchromatographische Reinigung war aufgrund der Empfindlichkeit der Boc-Schutzgruppe nicht möglich. O OCH3 O N H 13a O O N H 2.2.1.2. Lineare Oligomere von 3α α-Aminodesoxycholsäure 5b Hier wurde zur Synthese der sequentielle Aufbau gewählt und EDC als Kopplungsreagenz verwendet. Das in guter Ausbeute (93.8 %) isolierte Dimer Boc-DCS-DCS-OMe 15a wurde zunächst aminoterminal um einen Baustein verlängert, wobei sich jedoch ein Problem ergab: durch den zur aminoterminalen Verlängerung zuerst notwendigen Schritt der 2. Synthetischer Teil 23 Boc-Schutzgruppenabspaltung mit Trifluoressigsäure (TFA) trat eine Nebenreaktion ein, nämlich die teilweise Veresterung der freien OH-Gruppen des Dimers mit TFA, was dazu führte, dass das Trimer 16a zu einem kleinen Teil als einfacher oder zweifacher TFA-Ester vorlag (siehe hierzu das NMR-Spektrum, Abb. 3.3.a, im Kapitel 3). Immerhin war die teilweise Veresterung bei der Zuordnung der NMR-Signale zu den einzelnen Bausteinen hilfreich. Die weitere Kettenverlängerung zum Tetramer wurde dann carboxyterminal durchgeführt, da bei der hier ohnehin notwendigen Verseifung des Methylesters auch die TFA-Ester der OH-Gruppen entfernt werden konnten. Eine Reinigung des Trimers als Methylester gelang nicht, da bei dem Versuch, die sehr labilen TFA-Ester mit feuchtem Natriumcarbonat in Methanol selektiv zu verseifen, auch der Methylester zum Teil mitverseift wurde. Das Trimer, Boc-DCS-DCS-DCS-OMe 16a, und das Tetramer Boc-DCS-DCS-DCSDCS-OMe 17a, konnten jeweils in guten bis sehr guten Ausbeuten isoliert werden (Trimer: 97.8 % (leicht verunreinigt durch TFA-Ester, s.o.); Tetramer: 91.7 %). Vom Trimer wurde zusätzlich das carboxyterminal entschützte Derivat Boc-DCS-DCS-DCS-OH 16b genauer untersucht, da dies im Gegensatz zum Methylester 16a rein isoliert werden konnte. O OCH3 OH O N H OH O N H OH 16a O O N H 2.2.1.3. Lineare Oligomere von 3α α-Aminocholsäure 5c Aufgrund der bei den Aminodesoxycholsäure aufgetretenen Probleme wurde hier von vorneherein die carboxyterminale Kettenverlängerung gewählt, um die eventuelle Bildung von TFA-Estern zu vermeiden. Es wurden in jeweils guten bis sehr guten Ausbeuten das Dimer Boc-CHS-CHS-OMe 18a (Ausbeute: 79.0 %), das Trimer Boc-CHS-CHS-CHS-OMe 2. Synthetischer Teil 24 19a (98.0 %), sowie das Tetramer Boc-CHS-CHS-CHS-CHS-OMe 20a (84.1 %) synthetisiert und charakterisiert. O OCH3 OH O OH N H OH O OH N H OH O OH N H OH O O 20a OH N H 2.2.2. Lineare Oligomere aus Typ-2-Bausteinen Hier wurden Systeme bis zum linearen Trimer (bezogen auf die Typ-2-Bausteine, also insgesamt Hexapeptide) synthetisiert. Basis war wie bei den Typ-1-Oligomeren die sequentielle Kettenverlängerung (siehe Schema 5b), welche je nach System aminoterminal oder carboxyterminal durchgeführt wurde. Als Kopplungsreagenz wurde durchgehend EDC verwendet. Im Übrigen gelten die gleichen allgemeinen Angaben wie in 2.2.1. 2.2.2.1. Synthese der Typ-2-Bausteine Die Synthese der Typ-2-Bausteine erfolgte jeweils aus einer Boc-geschützten L-Aminosäure und einer der Methylester-geschützten 3α-Aminocholansäuren 12a-c mit EDC als Kopplungsreagenz und lieferte jeweils gute bis sehr gute Ausbeuten. Als natürliche Aminosäuren wurden Boc-L-Valin und ε-Benzyloxycarbonylgeschütztes Boc-L-Lysin (Boc-Lys[Z]-OH) verwendet. 2. Synthetischer Teil 25 Übersicht über die synthetisierten Typ-2-Monomere: Monomer Ausbeute O OCH3 Boc-Val-LCS-OMe 21a O 87.9 % N H H N O O O OCH3 Boc-Lys[Z]-LCS-OMe 22a O 84.0 % N H H O N N O O H O O OCH3 OH Boc-Val-DCS-OMe 23a O 94.2 % N H H N O O O OCH3 OH OH Boc-Val-CHS-OMe 24a O 92.2 % N H H N O Das Boc-Val-LCS-OMe-Monomere O 21a kristallisierte aus Methanol und konnte röntgenstrukturanalytisch untersucht werden. Die Kristallstruktur wird im Konformativen Teil diskutiert. 2. Synthetischer Teil 26 Die Abspaltung der Boc-Schutzgruppe zur weiteren Verwendung der Monomere wurde jeweils unter besonders schonenden Bedingungen (0°C, max. 30-45 min Reaktionszeit) durchgeführt, um Nebenreaktionen wie die teilweise TFA-Veresterung an den DCS- und CHS-Monomeren 23a und 24a oder die unwerwünschte Abspaltung der Z-Schutzgruppe an Boc-Lys[Z]-LCS-OMe 22a zu unterdrücken bzw. im Idealfall völlig zu unterbinden. Dies gelang in allen Fällen unter Erhalt der quantitativen Boc-Abspaltung. 2.2.2.2. Synthese der linearen Typ-2-Oligomere 2.2.2.2.1. Oligomere aus Val-LCS-Bausteinen Es wurden auf dem Weg der sequentiellen aminoterminalen Kettenverlängerung das Dimer 25a und das Trimer 26a synthetisiert. Sowohl Boc-Val-LCS-Val-LCS-OMe 25a als auch Boc-Val-LCS-Val-LCS-Val-LCS-OMe 26a konnten in sehr guten Ausbeuten isoliert werden (97.3 % bzw. 90.6 %). O OCH3 O N H H N O O N H 25a H N O O 2.2.2.2.2. Oligomere aus Lys[Z]-LCS-Bausteinen Hier konnten bei der (ebenfalls aminoterminal durchgeführten) Kettenverlängerung keine so guten Ergebnisse erzielt werden. Während das Dimer Boc-Lys[Z]-LCS-Lys[Z]-LCS-OMe 27a noch in sehr guter Ausbeute (92.4 %) isoliert werden konnte, lieferte die weitere 2. Synthetischer Teil 27 Kettenverlängerung zum Trimer Boc-Lys[Z]-LCS-Lys[Z]-LCS-Lys[Z]-LCS-OMe 28a lediglich 72.6 % Ausbeute. Zudem konnte es massenspektrometrisch nicht zweifelsfrei als Trimer identifiziert werden, da der Massenpeak (1998.4) um 7 Einheiten zu hoch lag. Dies könnte aber auch darauf zurückzuführen sein, dass das FAB-Massenspektrometer lediglich einen Messbereich bis etwa 2000 m/z aufweist und es für den Standard-Servicebetrieb auf Massen unterhalb von 1000 kalibriert ist. Diese Vermutung konnte durch das NMR untermauert werden, welches deutliche Hinweise dafür liefert, dass es sich bei der isolierten Verbindung tatsächlich um das gewünschte Trimer handelt. Außerdem lieferte die Cyclisierung der Verbindung zweifelsfrei das cyclische Hexaamid Cyclo-(Lys[Z]-LCS)3 37 (siehe 2.3.2.6. bzw. im Konformativen Teil). O OCH3 O N H H O N N O O O H N H H O N N O O N O H 28a H H O N N O O O H 2.2.2.2.3. Oligomere aus Val-DCS-Bausteinen Aufgrund des Vorhandenseins freier OH-Gruppen wurde hier der Weg der carboxyterminalen Kettenverlängerung gewählt. Das Dimer Boc-Val-DCS-Val-DCS-OMe 29a konnte in sehr guter Ausbeute (92.4 %) isoliert werden, die Verlängerung um einen weiteren Baustein lieferte das Trimer Boc-Val-DCS-Val-DCS-Val-DCS-OMe 30a in immerhin noch guten 85.5 % Ausbeute. 2. Synthetischer Teil 28 O OCH3 OH O N H H N O OH O 29a N H H N O O 2.2.2.2.4. Oligomere aus Val-CHS-Bausteinen Auch hier wurde aufgrund des Vorhandenseins freier OH-Gruppen der Weg der carboxyterminalen Kettenverlängerung für die Darstellung des Dimers und des Trimers gewählt. Das Dimer Boc-Val-CHS-Val-CHS-OMe 31a konnte in sehr guten 95.8 % Ausbeute isoliert werden, die Ausbeute beim Trimer Boc-Val-CHS-Val-CHS-Val-CHS-OMe 32a lag bei recht guten 79.9 %. O OCH3 OH OH O N H H N O OH OH O N H H N O OH OH O N H H N O O 32a 2. Synthetischer Teil 29 2.2.3. Tabellarische Übersicht über alle synthetisierten linearen Oligoamide dargestelltes Oligoamid Ausbeute* lineare Oligoamide vom Typ 1 Boc-LCS-LCS-OMe 13a 71.2 % Boc-DCS-DCS-OMe 15a 93.8 % Boc-CHS-CHS-OMe 18a 79.0 % Boc-DCS-DCS-DCS-OMe 16a 97.8 % Boc-CHS-CHS-CHS-OMe 19a 98.0 % Boc-LCS-LCS-LCS-LCS-OMe 14a 65.3 % Boc-DCS-DCS-DCS-DCS-OMe 17a 91.7 % Boc-CHS-CHS-CHS-CHS-OMe 20a 84.1 % lineare Oligoamide vom Typ 2 Boc-Val-LCS-OMe 21a 87.9 % Boc-Lys[Z]-LCS-OMe 22a 84.0 % Boc-Val-DCS-OMe 23a 94.2 % Boc-Val-CHS-OMe 24a 92.2 % Boc-Val-LCS-Val-LCS-OMe 25a 97.3 % Boc-Lys[Z]-LCS-Lys[Z]-LCS-OMe 27a 92.4 % Boc-Val-DCS-Val-DCS-OMe 29a 92.4 % Boc-Val-CHS-Val-CHS-OMe 31a 95.8 % Boc-Val-LCS-Val-LCS-Val-LCS-OMe 26a 90.6 % Boc-Lys[Z]-LCS-Lys[Z]-LCS-Lys[Z]-LCS-OMe 28a 72.6 % Boc-Val-DCS-Val-DCS-Val-DCS-OMe 30a 85.5 % Boc-Val-CHS-Val-CHS-Val-CHS-OMe 32a 79.9 % *) Die Ausbeuten beziehen sich jeweils auf den letzten Kopplungsschritt. 2. Synthetischer Teil 30 2.3. Cyclische Oligoamide mit 3α α-Aminocholansäuren Zur Synthese cyclischer Oligoamide mit den zur Verfügung stehenden Aminocholansäuren erschienen zwei Zugangswege interessant. Zum einen ist dies die Cyclisierung der entsprechenden linearen Oligomere. Zum anderen erschien es vielversprechend, die monomeren Bausteine vom Typ 1 und 2 den Cyclisierungsbedingungen zu unterwerfen und zu prüfen, inwieweit hier neben der Cyclodimerisierung, wie sie bereits von D. Albert16, 17 und von P. Davis15 an steroidalen Aminosäure-Systemen beobachtet worden war (vgl. Einleitung), auch die Entstehung höherer Oligocyclen zu beobachten ist. Dies war insbesondere interessant, da die Bildung cyclischer Ester-Oligomere mit gewöhnlichen Gallensäuren bereits in der Literatur beschrieben ist9-12 (siehe hierzu die Ausführungen in der Einleitung). Die kinetisch kontrollierte Variante ist als Yamaguchi-Macrolactonisierung8 bekannt und liefert bei Cholansäuren Oligocyclen bis hin zum Hexamer. Eine mögliche Analogie für die Bildung entsprechender Macrolactame aus Aminocholansäuren unter kinetischer Reaktionskontrolle erschien vor diesem Hintergrund plausibel. Deshalb wurden in der vorliegenden Arbeit sowohl lineare Oligomere als auch – zumindest im Fall der Typ-1-Bausteine – die Monomere den Cyclisierungsbedingungen unterworfen. Die beiden Zugangswege zur Cyclooligoamid-Darstellung sind in Schema 2.6. veranschaulicht. Schema 2.6. 2. Synthetischer Teil 31 Als Basisvorschrift für die Cyclisierung wurde die von Grießer, Kroner und Schmidt in Synthesis für die Cyclisierung von Peptiden beschriebene One-Pot-Reaktionsfolge28 mit einer leichten Modifikation im Cyclisierungsschritt gewählt. Dabei wird zunächst das aminoterminal Boc-geschützte Oligoamid (bzw. der monomere Baustein) am Carboxyterminus mit Pentafluorphenol verestert (mittels Dicyclohexylcarbodiimid als Auxiliar), anschließend mit Trifluoressigsäure die Boc-Schutzgruppe abgespalten und gleichzeitig die freiwerdende Aminogruppe als Ammoniumtrifluoracetat erneut geschützt. Die Cyclisierung wird dann in einem intensiv gerührten Zweiphasensystem CHCl3/NaHCO3(aq) durch endgültiges Entschützen der Aminogruppe mittels der wäßrigen HydrogencarbonatLösung initiiert. Dabei ist aufgrund der starken Pentafluorphenolester-Aktivierung der Carboxyfunktion kein weiteres Kopplungsreagenz erforderlich – der Angriff der Aminofunktion und damit die Cyclisierung tritt spontan ein und in der Literaturvorschrift werden lediglich 5 Minuten als Reaktionszeit angegeben. Um ein vollständiges Abreagieren der Edukte sicherzustellen wurde das Reaktionsgemisch bei den Cyclisierungen dieser Arbeit jedoch über zwei Tage gerührt. Dies hat zudem den Vorteil, dass bei Systemen mit freien OH-Gruppen während des Entfernens der Boc-Schutzgruppe möglicherweise entstandene Trifluoressigsäureester (siehe hierzu Abschnitt 2.2.1.2) unter den basischen Reaktionsbedingungen weitgehend wieder gespalten werden können. Die Reaktionsfolge der Cyclisierung ist in Schema 2.7. wiedergegeben. Schema 2.7. 2.3.1. Unsystematische Oligocyclisierung von Typ-1-Monomeren Es wurden alle drei zur Verfügung stehenden Aminocholansäuren hinsichtlich ihres Cyclisierungsverhaltens untersucht. Bei diesen Cyclisierungen wurde der eigentliche Cyclisierungsschritt mit Monomer-Konzentrationen von 3.333 mmol/l (bezogen auf die organische Lösungsmittelkomponente) durchgeführt und das Rohprodukt vor der weiteren 2. Synthetischer Teil 32 Reinigung jeweils massenspektrometrisch auf die enthaltenen cyclischen Oligomere untersucht. 2.3.1.1. Oligocyclisierung von 3α α-Aminolithocholsäure 5a 1 mmol der Boc-geschützten Aminolithocholsäure 11a wurde gemäß Schema 2.6. cyclisiert. Die massenspektrometrische Untersuchung des Rohproduktes zeigte, dass hierbei neben dem Cyclodimer 33a wie erwartet auch noch zwei höhere Oligomere, nämlich das Cyclotrimer 33b und das Cyclotetramer 33d, entstanden sind (vgl. Abb. 2.2.). Abbildung 2.2. FAB-Massenspektrum des Produktgemisches aus der Oligocyclisierung von 3α-Aminolithocholsäure5a. Desweiteren wurden bei der dünnschichtchromatographischen Untersuchung des Rohproduktes noch diverse Nebenprodukte gefunden. Eine Reinigung bzw. Auftrennung des Produktgemisches gestaltete sich jedoch äußerst schwierig. Die beiden Hauptprodukte, das 3α-Aminolithocholsäure-Cyclodiamid 33a und das 3α-Aminolithocholsäure-Cyclotriamid 33b, waren unter allen getesteten flash-säulenchromatographischen Bedingungen praktisch nicht voneinander zu trennen. Das Cyclotetraamid 33c ließ sich auf diesem Wege ebenfalls nicht isolieren. Auf weitere Trennungsversuche des Gemisches (HPLC, Reversed-Phase- 2. Synthetischer Teil 33 Methoden, Kapillarelektrophorese etc.) wurde nach anfänglichen Vorversuchen verzichtet, da der hierfür zu erwartende Aufwand in keinem vertretbaren Verhältnis zum wissenschaftlichen Nutzen stand: Diese Systeme eignen sich mangels einer weiteren Funktionalisierung, wie sie beispielsweise in Cyclooligoamiden von Aminodesoxycholsäure und Aminocholsäure durch die freien OH-Gruppen am Steroid-Gerüst gegeben ist, nicht für eine weitere Verwendung, etwa in Komplexierungsversuchen, weshalb diese Oligocyclisierung ohnehin nur als Modellversuch für die entsprechend weiter funktionalisierten Aminocholansäuren durchgeführt wurde. Ein darüber hinausgehendes Interesse an den AminolithocholsäureCyclooligoamiden 33 bestand nicht. 2.3.1.2. Oligocyclisierung von 3α α-Aminodesoxycholsäure 5b Auch hier wurde 1 mmol der Boc-geschützten 3α-Aminodesoxycholsäure 11b den Cyclisierungsbedingungen unterworfen. Die massenspektrometrische Analyse des Roproduktes zeigte hier, analog zur Aminolithocholsäure-Cyclisierung, ebenfalls das Vorhandensein des Cyclodimers, des Cyclotrimers sowie des Cyclotetramers. Bei der säulenchromatographischen Auftrennung des Gemisches konnten das 3α-Aminodesoxycholsäure-Cyclodiamid 34a und das 3α-Aminodesoxycholsäure-Cyclotriamid 34b isoliert werden. Die Ausbeuten waren für eine unsystematische Cyclisierung sehr zufriedenstellend: Vom Cyclodimer 34a wurden 120 mg erhalten, was bezogen auf das eingesetzte Monomer einer relativen Ausbeute von 32.1 % der Theorie entspricht, vom Cyclotrimer 34b wurden immerhin 34 mg (9.1 %) isoliert. Eine Isolierung des massenspektrometrisch nachgewiesenen Cyclotetramers 34c gelang leider nicht, was wahrscheinlich darin begründet liegt, dass es bei der Cyclisierung wohl nur in sehr geringer Menge entstanden ist. Vom Cyclodimer 34a wurden Kristalle sehr guter Qualität erhalten, die eine röntgenstrukturanalytische Bestimmung der Kristallstruktur ermöglichten (siehe Diskussion im Konformativen Teil). 2. Synthetischer Teil 34 O OH OH O N H N H N H O O N H 34b OH HO OH H 34a N O 2.3.1.3. Oligocyclisierung von 3α α-Aminocholsäure 5c Die Oligocyclisierung von 1 mmol Boc-geschützter 3α-Aminocholsäure 11c führte hier – im Unterschied zu den beiden anderen Aminocholansäuren – fast ausschließlich zur Bildung des 3α-Aminocholsäure-Cyclodiamids 35a. Von diesem konnten 220 mg isoliert werden, was einer sehr guten Ausbeute von 48.5 % entspricht. Hierfür könnten Löslichkeitseffekte verantwortlich sein, die im Cyclisierungsschritt eine Zusammenlagerung zweier Monomerer durch polare Wechselwirkungen (OH---OH) begünstigen. Diese Vermutung wird unterstützt durch die Tatsache, dass das Cyclodimer in Chloroform unlöslich war und aus der Cyclisierungslösung ausfiel. In diesem Präzipitat konnten massenspektrometrisch auch Spuren des Cyclotrimers 35b und des Cyclotetramers 35c nachgewiesen werden, eine Isolierung gelang jedoch nicht, da sie nur in geringer Menge vorhanden waren und eine säulenchromatographische Trennung aufgrund der schlechten Löslichkeit des Präzipitats nicht in Frage kam. Das Cyclodimer 35a konnte durch Kristallisation (es löste sich in der Siedehitze langsam in einem THF/EtOH-Gemisch) aus dem Präzipitat isoliert und gereinigt werden. 2. Synthetischer Teil 35 OH O OH H N N H OH O OH 35a 2.3.2. Cyclisierung linearer Oligoamide Hier wurden gegenüber der unsystematischen Oligocyclisierung die Konzentrationen so angepasst, dass die Konzentration an linearem Oligomer in der Cyclisierungslösung etwa einer 3.3 millimolaren Konzentration an entsprechendem Monomer entsprach. Die carboxyterminale Methylesterschutzgruppe der linearen Oligomere wurde jeweils zunächst verseift, bevor die Oligomere den Cyclisierungsbedingungen gemäß Schema 2.7. unterworfen wurden. 2.3.2.1. Cyclisierung von Boc-DCS-DCS-DCS-OMe 16a Die Cyclisierung des linearen Triamids der Aminodesoxycholsäure lieferte neben dem erwarteten 3α-Aminodesoxycholsäure-Cyclotriamid 34b überraschenderweise auch das Cyclodimerisierungsprodukt, 3α-Aminodesoxycholsäure-Cyclohexaamid 34d, welches aufgrund seiner hohen Molmasse massenspektrometrisch nur durch MALDI nachgewiesen werden konnte. Beide wurden in sehr zufriedenstellenden Ausbeuten (Cyclotrimer: 41.4 %, Cyclohexamer: 9.4 %; jeweils bezogen auf die theoretische Gesamtausbeute) säulenchromatographisch aus dem Reaktionsgemisch isoliert. Weitere Oligocyclen konnten in dieser Reaktion nicht gefunden werden. 2. Synthetischer Teil 36 O N OH H O HO N H O N H OH 34d HO H N O H OH H N O OH N O 2.3.2.2. Cyclisierung von Boc-DCS-DCS-DCS-DCS-OMe 17a Das lineare Tetraamid der Aminodesoxycholsäure zeigte im Gegensatz zum Triamid keine Cyclodimerisierung. Im Reaktionsgemisch wurde neben diversen Nebenprodukten, die nicht näher identifiziert werden konnten, lediglich das 3α-Aminodesoxycholsäure-Cyclotetraamid 34c gefunden. Die säulenchromatographische Auftrennung des Reaktionsgemisches lieferte das Cyclotetramer in recht guter Ausbeute von 31.6 %. 2. Synthetischer Teil 37 O OH N H O N H HO 34c OH H N O H N OH O 2.3.2.3. Cyclisierung von Boc-CHS-CHS-CHS-OMe 19a Erstaunlicherweise entstand hier das Cyclotrimer 35b lediglich in sehr geringer Menge. Es war Bestandteil des Präzipitats von ca. 16 mg, welches sich während der Reaktion gebildet hatte, konnte aus diesem jedoch aufgrund der schlechten Löslichkeit des Präzipiztats nicht isoliert werden. Hauptprodukt dieses Cyclisierungsversuches war das Cyclodimerisierungsprodukt, 3α-Aminocholsäure-Cyclohexaamid 35d (vgl. das DCS-Analogon 34d), welches aus dem in wenig Chloroform gelösten Reaktionsgemisch in kleinen Würfeln auskristallisierte und so rein isoliert werden konnte (Ausbeute: 32.6 %). Eine röntgenstrukturanalytische Untersuchung gelang jedoch nicht, da diese Kristalle keinerlei Beugungsreflexe zeigten. 2.3.2.4. Cyclisierung von Boc-CHS-CHS-CHS-CHS-OMe 20a Bei dieser Cyclisierung trat im Cyclisierungsschritt, ähnlich wie bei der unsystematischen Oligocyclisierung des Aminocholsäure-Monomers, bereits während der Reaktion eine größere Menge Präzipitat auf, die als fast reines 3α-Aminocholsäure-Cyclotetraamid 35c (Ausbeute: 50.7 %) identifiziert werden konnte. Aufgrund der sehr schlechten Löslichkeit der Verbindung (sie löste sich lediglich in DMSO) gelang eine weitere Reinigung jedoch bislang nicht. 2. Synthetischer Teil 38 O OH N OH O H N H HO HO 35c OH OH H N H N O OH OH O 2.3.2.5. Cyclisierung von Boc-Val-LCS-Val-LCS-Val-LCS-OMe 26a Bei der Cyclisierung dieses Typ-2-Oligomers konnte lediglich das erwartete Cyclohexapeptid 3α-Valinylaminolithocholsäure-cyclotriamid 36 gefunden werden. Es wurde säulenchromatographisch in guter Ausbeute (48.3 %) aus dem Reaktionsgemisch isoliert. Die Verbindung kristallisierte zwar als sehr feiner Kristallstaub aus Methylenchlorid/Methanol, eine Bestimmung der Kristallstruktur mittels Röntgenstrukturanalyse gelang jedoch aufgrund der viel zu geringen Größe der Kristalle nicht. 2. Synthetischer Teil 39 H N O O H O N N H N O 36 H O H N N H O 2.3.2.6. Cyclisierung von Boc-Lys[Z]-LCS-Lys[Z]-LCS-Lys[Z]-LCS-OMe 28a Die Cyclisierung dieses Lys[Z]-LCS-Trimers gelang nur sehr schlecht. Die dünnschichtchromatographische Analyse des Reaktionsgemisches zeigte eine Reihe von Nebenprodukten, die nach der Intensität der Spots beurteilt in größeren Mengen bei der Reaktion angefallen waren. Neben dem einfachen Cyclisierungsprodukt konnten darunter jedoch massenspektrometrisch keine höheren Cyclooligomerisierungsaddukte gefunden werden. Eine säulenchromatographische Trennung des Gemisches lieferte das reine Cyclohexapeptid 3α-[ε-Benzyloxycarbonylamino-lysinyl]-aminolithocholsäure-cyclotriamid 37 in 18.6 %iger Ausbeute. 2. Synthetischer Teil 40 O H N N O O O H O O N H H O N N H N O 37 H O H N N H O H N O O 2.3.2.7. Cyclisierung von Boc-Val-DCS-Val-DCS-Val-DCS-OMe 30a Diese Cyclisierung lieferte, wie auch schon die Cyclisierung des reinen Aminodesoxycholsäure-Trimers 16a, neben dem einfachen Cyclisierungsprodukt, 3α-Valinylaminodesoxycholsäure-cyclotriamid 38a, auch wieder das Produkt der Cyclodimerisierung, 3α-Valinylaminodesoxycholsäure-cyclohexaamid 38b, wobei bei letzterem die genaue Struktur bislang nicht völlig sicher geklärt ist (vgl. hierzu die Diskussion im Konformativen Teil). Die Ausbeuten an diesen beiden Produkten waren sehr gut: das Cyclohexapeptid 38a konnte säulenchromatographisch in 32.8 %iger Ausbeute isoliert werden, das Cyclododecapeptid 38b wurde in einer Ausbeute von 21.8 % erhalten. 2. Synthetischer Teil 41 O H N N H O HO OH H O N N H O O N N H O H HO 38b OH H O H N N O O H N N O H OH O OH H N N H O 2.3.2.8. Cyclisierung von Boc-Val-CHS-Val-CHS-Val-CHS-OMe 32a Diese Cyclisierung lieferte ein Produkt, das bislang nicht eindeutig identifiziert werden konnte. Ähnlich wie bei einigen der schon beschriebenen Cyclisierungsversuche trat auch hier bereits während der Cyclisierungsreaktion eine recht große Menge Niederschlag auf, welcher sich lediglich in DMSO löste. Das Kernresonanzspektrum deutet darauf hin, dass es sich dabei um das Cyclisierungsaddukt, 3α-Valinylaminocholsäure-cyclotriamid 39, handeln könnte, jedoch konnte diese Vermutung massenspektrometrisch (FAB, MALDI) nicht abgesichert werden: der vermutliche Massenpeak der Verbindung bei m/z = 1506.1 (FAB) entspricht nicht der Molmasse des Cyclus (Mr=1466.13), sondern liegt um ca. 40 Einheiten zu hoch, und im Bereich der Molmasse konnte weder im FAB noch in der MALDI ein Peak 2. Synthetischer Teil 42 gefunden werden. In beiden fand sich jedoch oberhalb des genannten Massenpeaks jeweils noch der M++Na-Peak, was die Vermutung untermauert, dass es sich bei dem Signal bei 1506.1 tatsächlich um den Massenpeak der unbekannten Verbindung handelt. Bei der Differenz von ca. 40 Einheiten zur Molmasse des erwarteten Cyclus 39 liegt zunächst der Schluß nahe, dass es sich möglicherweise um das erwartete Val-CHS-Cyclotrimer handelt, welches ein Calciumion (Mr=40.08) oder ein Kaliumion (Mr=39.10) komplexiert hat, doch da bei der Cyclisierungseaktion weder mit Calcium- noch mit Kaliumhaltigen Verbindungen gearbeitet wurde, ist dies als eher unwahrscheinlich anzusehen. Zumal in einem solchen Fall auch ein Massenpeak der reinen, unkomplexierten Verbindung auftauchen sollte, welcher jedoch weder im FAB-Spektrum noch im MALDI-Spektrum gefunden wurde. Eine zur sicheren Identifikation notwendige weitere Reinigung und Untersuchung der Verbindung gestaltet sich aufgrund ihrer schlechten Löslichkeit äußerst schwierig und konnte bislang leider nicht erfolgreich durchgeführt werden. H N O O OH O N H OH N H HO N 39 O HO H OH OH O H N N O H 2. Synthetischer Teil 43 2.3.3. Übersicht über die synthetisierten Cyclooligoamide dargestelltes Cyclooligoamid Ausbeute aus Monomer aus lin. Oligomer cyclische Oligoamide vom Typ 1 Cyclo-(LCS)2 33a m.n.a) / n.i.b) - Cyclo-(LCS)3 33b m.n. / n.i. - Cyclo-(LCS)4 33c m.n. / n.i. - Cyclo-(DCS)2 34a 32.1 % - Cyclo-(DCS)3 34b 9.1 % 41.4 % Cyclo-(DCS)4 34c m.n. / n.i. 31.6 % Cyclo-(DCS)6 34d - 9.4 %c) Cyclo-(CHS)2 35a 48.5 % - Cyclo-(CHS)3 35b m.n. / n.i. m.n. / n.i. Cyclo-(CHS)4 35c m.n. / n.i. 50.7 % Cyclo-(CHS)6 35d - 32.6 %c) cyclische Oligoamide vom Typ 2 Cyclo-(Val-LCS)3 36 - 48.3 % Cyclo-(Lys[Z]-LCS)3 37 - 18.6 % Cyclo-(Val-DCS)3 38a - 32.8 % Cyclo-(Val-DCS)6 38b - 21.8 %c) Cyclo-(Val-CHS)3 39 (???) - 54.0 %d) a) m.n. = massenspektrometrisch nachgewiesen; b) n.i. = nicht isolierbar bzw. nicht isoliert; c) als Cyclodimerisierungsprodukt aus Cyclisierung des linearen Trimers; d) unter der Annahme, dass es sich bei dem iolierten Produkt tatsächlich um diese Verbindung handelt. 3. Konformationsanalyse und Komplexierungsverhalten 44 3. Konformationsanalyse und Komplexierungsverhalten 3.1. Allgemeine Methoden 3.1.1. Experimentelle Methoden zur Strukturaufklärung Als sicherste Methode zur experimentellen Strukturaufklärung wird oft die Röntgenstrukturanalyse angesehen. Sie liefert als direkte Methode neben den genauen Strukturdaten (Bindungslängen, Bindungswinkel, Torsionswinkel etc.) auch noch Informationen über die Wechselwirkung mit benachbarten Molekülen, insbesondere von Lösungsmittelmolekülen, sofern diese im Kristallverband eingeschlossen sind. Der grundsätzliche Nachteil dieser Methode ist jedoch, dass sie prinzipiell nur Informationen über die Konformation der untersuchten Verbindung im festen Zustand liefert, welche von den konformativen Gegebenheiten in Lösung deutlich abweichen kann (die im gelösten Molekül vorhandenen intramolekularen Bewegungen werden im Kristallgitter teilweise eingefroren), und dass sie nur für Verbindungen anwendbar ist, die kristallisierbar sind und Einkristalle von hinreichender Größe und Qualität liefern. Darüberhinaus liefert die Röntgenstrukturanalyse aufgrund der langen Meßzeiten pro Winkeleinstellung nur gemittelte Atompositionen, wenn im Kristall schnelle dynamische Prozesse stattfinden. Als wichtigste experimentelle Methode zur Konformationsanalyse in Lösung dient in der heutigen Zeit die Kernresonanzspektroskopie29. Neben den eindimensionalen NMR-Experimenten (1H, 13C, 15N, 31P) liefern insbesondere auch 2D-Experimente wichtige Informationen zur Strukturaufklärung. Unter den homonuklearen 2D-Experimenten sind hier besonders H,HCOSY30, TOCSY31, sowie NOESY32 und ROESY33 zu nennen, weitere strukturelle Informationen können C,H-korrelierten Spektren wie C,H-COSY30, HMQC34 und HMBC35 entnommen werden. Auch N,H-korrelierte Experimente sind möglich, wurden für die vorliegende Arbeit jedoch nicht zur Konformationsanalyse herangezogen. Die NMRSpektroskopie ist vor allem deshalb besonders interessant, da sich mit ihrer Hilfe nicht nur Aussagen über Abstände, Winkel oder die Nachbarschaft von Kernen ableiten lassen, sondern sie auch den Nachweis von Wasserstoffbrücken, dynamischen Konformationsgleichgewichten und Komplexierungen ermöglicht. Durch Kombination verschiedener NMR-Experimente läßt 3. Konformationsanalyse und Komplexierungsverhalten 45 sich beispielsweise zuverlässig die Sequenz von Peptiden sowie die Anwesenheit und Lage sekundärer Strukturmerkmale innerhalb der Peptidkette bestimmen. Neben der NMR-Spektroskopie und der Röntgenstrukturanalyse können noch weitere experimentelle Methoden zur Ermittlung struktur- bzw. vor allem konformationsrelevanter Daten herangezogen werden, die jedoch im Rahmen der vorliegenden Arbeit nicht genutzt wurden. Erwähnenswert sind hier besonders IR- und CD-Spektren. Die IR-Daten von NH-Streckschwingungen beispielsweise geben Informationen über die Beteiligung von Amidprotonen an einer Wasserstoffbrückenbindung36, CD-Spektren können Hinweise auf das Vorhandensein helicaler Strukturen liefern37. 3.1.2. Theoretische Konformationsanalyse Theoretische Berechnungen von Molekülgeometrien und –eigenschaften können in drei große Kategorien eingeteilt werden, welche sich im wesentlichen durch den verwendeten Ansatz und somit den Anteil empirisch bezogener Daten unterscheiden. Man unterscheidet zwischen Ab-initio-Methoden, semiempirischen Methoden und empirischen Methoden. Bei geeigneter Auswahl der Methode sind solche Berechnungen ein wichtiges Hilfsmittel, um die experimentellen Befunde synthetisierter Verbindungen, beispielsweise aus der NMR-Spektroskopie, zu ergänzen und zu untermauern, bzw. um im Vorfeld geplanter Oligomer-Synthesen Informationen über mögliche Konformationen, insbesondere über sekundäre Strukturmerkmale oder die Präorganisation eventueller Hohlräume von Wirtverbindungen, der Kandidaten zu erhalten und somit die Auswahl der zu synthetisierenden Moleküle zu erleichtern. Auch Komplexierungsvorgänge können durch theoretische Berechnungen simuliert werden und die Energie der gebildeten Komplexe kann abgeschätzt werden. Die folgenden Ausführungen geben einen kurzen Überblick über die einzelnen Methoden und ihre Anwendbarkeit auf die in der vorliegenden Arbeit synthetisierten Systeme. 3. Konformationsanalyse und Komplexierungsverhalten 46 3.1.2.1. Ab-initio-Methoden38 Quantenmechanische Ab-initio-Rechnungen kommen ohne empirische Parameter aus und sind bei ausreichendem Basissatz und dunter Berücksichtigung der Elektronenkorrelation die genauesten theoretischen Methoden zur Struktur- und Konformationsbestimmung. Nachteil dieser sehr exakten Rechenmethoden: sie erfordern einen ausgesprochen hohen Rechen- und damit auch Zeitaufwand (die Rechenzeit steigt mit der 5. Potenz der Orbitalzahl!). Daher sind sie nur für sehr kleine Moleküle sinnvoll einsetzbar. Für die großen Systeme in der vorliegenden Arbeit sind Ab-initio-Rechnungen, auch in der Variante der DFT-Methoden, nicht geeignet. 3.1.2.2. Semiempirische Methoden Die verbreitetsten Programme für semiempirische Molekülberechnungen sind MNDO39 (Modified Neglect of Diatomic Differential Overlap), AM140 (Austin Model 1) und PM341 (Parametrisation Model 3). AM1 ist grundsätzlich für Berechnungen peptidischer und peptidähnlicher Systeme eine geeignete Methode, MNDO dagegen liefert keine sinnvolle Beschreibung von Wasserstoffbrücken und ist daher prinzipiell für die Konformationsanalyse der in dieser Arbeit zu untersuchenden Systeme völlig unbrauchbar. Auch semiempirische Methoden erfordern jedoch zur Beschreibung der hier synthetisierten Verbindungen viel zu lange Rechenzeiten, weshalb eine Anwendung hier nicht in Frage kam. Sie finden hauptsächlich Verwendung für Moleküle mit bis zu 100 Atomen. 3.1.2.3. Empirische Methoden Zu den empirischen Methoden zählen in erster Linie Kraftfeldrechnungen. Diese Methoden behandeln Moleküle als starre Kugel-Feder-Modelle. Der besondere Vorteil ist, dass ihre Rechenzeit nur mit der 2. Potenz der Atomzahl ansteigt und somit auch große Moleküle wie die hier synthetisierten Steroid-Oligoamide und -Macrolactame mit vertretbarem Rechenzeitaufwand analysiert werden können. Die Ergebnisse solcher Berechnungen hängen jedoch stark von der Wahl der empirischen Parameter ab, weshalb im Hinblick auf die Authentizität und damit die Qualität der Berechnungen der Wahl des zugrundeliegenden Parametersatzes 3. Konformationsanalyse und Komplexierungsverhalten 47 eine entscheidende Bedeutung zukommt. Einige Kraftfeld-Parametersätze wurden speziell für die Berechnung von Peptiden optimiert. Hierzu gehören beispielsweise MM242, MM343, CHARMM44 und AMBER45. Sie alle berechnen die sterische Energie, welche sich jeweils summarisch aus mehreren Energiebeiträgen (z.B. Beiträge aus Bindungs- oder Winkeldeformationen, aus van-der-Waals-Wechselwirkungen oder elektrostatischen Wechselwirkungen) zusammensetzt46. Wichtig für die möglichst korrekte Analyse der in dieser Arbeit zu untersuchenden Verbindungen ist, dass das verwendete Kraftfeld sowohl den peptidischen als auch den steroidalen Teil der Moleküle richtig beschreibt. Diese Voraussetzung wird am besten durch den Parametersatz des AMBER-Kraftfeldes (AMBER = Assisted Model Building and Energy Refinement) erfüllt, aber auch MM3-Rechnungen sind prinzipiell für solche Verbindungen geeignet. Da die empirischen Parameter des AMBER-Kraftfeldprogramms auf Lösungs-Daten beruhen, während MM3 Gasphasenparameter verwendet, wurde für die synthetisierten Systeme bevorzugt AMBER zur Berechnung genutzt. 3.1.2.4. Molekül-Dynamik-Rechnungen Auch Molekül-Dynamik-Rechnungen (MD) können mit Hilfe eines empirischen Kraftfeldes durchgeführt werden. Die Technik beruht auf der Berechnung der Kraftkonstanten für innere molekulare Bewegungskoordinaten durch das Kraftfeld. Durch Lösen der klassischen Newtonschen Bewegungsgleichung kann dann die Lage jedes Atoms in einem Molekül als Funktion der Zeit beschrieben werden, wodurch die Simulation von inneren Bewegungen des Moleküls, auch bei verschiedenen Temperaturen, möglich wird. Mit MD-Rechnungen können Informationen über verschiedene Regionen einer komplexen Potentialhyperfläche erhalten werden. 3.2. Untersuchung der synthetisierten Verbindungen 3.2.1. Lineare Oligomere Um bei Oligomeren aus linear verketteten Bausteinen eine profunde experimentelle Konformationsanalyse mittels NMR durchführen zu können ist zunächst die Ermittlung der Sequenz notwendig, damit z.B. Aussagen darüber getroffen werden können, welche Protonen 3. Konformationsanalyse und Komplexierungsverhalten 48 von in der Sequenz nicht benachbarten Bausteinen in räumlicher Nachbarschaft zueinander stehen (und somit in Sättigungsexperimenten einen NOE zeigen). Es muß also die Frage beantwortet werden, welche NMR-Signale zum ersten, zweiten, ..., n-ten Baustein gehören. Bei klassischen Peptiden, welche aus amidisch verknüpften natürlichen Aminosäuren bestehen, ist eine solche Sequenzanalyse und -zuordnung in der Regel relativ leicht möglich. Hierzu kann eine Kombination von Ergebnissen aus TOCSY- und ROESY-Experimenten genutzt werden: Das TOCSY-Spektrum gibt Informationen darüber, welche Signale zu jeweils einer Aminosäure gehören und um welche Aminosäure es sich handelt (jede Aminosäure zeigt im TOCSY ein charakteristisches Signalpattern); über den NOE zwischen αH eines Aminosäure-Bausteins und Amid-NH des nächsten Bausteins (Signale aus dem ROESY-Spektrum) können benachbarte Aminosäuren ermittelt werden. Da der aminoterminale Baustein in der Regel leicht identifizierbar ist, kann so die Sequenz unter bestimmten Voraussetzungen (z.B. hinreichend gut unterscheidbare Signale der einzelnen Bausteine in den 2D-Spektren) lückenlos bestimmt und zugeordnet werden. Im Falle der hier synthetisierten Aminocholansäure-Oligomere vom Typ 1 (direkte amidische Steroid-Steroid-Verknüpfung, vgl. Kap. 2.2.1.) ist die vollständige Zuordnung der Signale zu den einzelnen Bausteinen und somit eine exakte Sequenzaufklärung mittels 1H-NMR aus zwei Gründen nicht möglich: • Strukturell bedingt sind, auch bei hohen Meßfrequenzen, (von wenigen Ausnahmen wie Methylprotonen, C3-H und C23-H2 abgesehen) die chemischen Verschiebungen der CH-Protonen des Steroid-Gerüstes sehr ähnlich und daher bereits bei einem einzelnen Baustein stark überlagert (im Bereich von ca. 0.8-2.0 ppm), was schon eine exakte Zuordnung dieser Signale innerhalb eines Bausteins praktisch unmöglich macht. Bei Oligomeren überlagern sich zusätzlich die Signale der verschiedenen Bausteine in diesem Bereich, wodurch die Spektren insgesamt sehr unübersichtlich werden. NOEs zwischen Protonen unterschiedlicher Steroid-Bausteine, welche Aufschlüsse über die vorliegende Konformation geben, können deshalb nicht identifiziert werden. • Meßtechnisch bedingt nimmt im TOCSY-Spektrum die Signalintensität der Crosspeaks mit der Zahl der im Kopplungspfad involvierten Protonen ab. Obwohl zwischen C3 (an dem die Aminofunktion bzw. der Amid-Stickstoff lokalisiert ist) und C23 (an dem die Carboxygruppe hängt) eine lückenlose Kopplungskette über die Protonen des Steroid- 3. Konformationsanalyse und Komplexierungsverhalten 49 Gerüstes besteht, kann daher im TOCSY-Spektrum keine direkte Korrelation zwischen den Protonen an C3 und C23 eines Bausteins beobachtet werden. Durch die Unübersichtlichkeit der 1D- und 2D-Spektren im oben beschriebenen Bereich ist auch ein schrittweises Vorgehen oder die Nutzung anderer NMR-Experimente hier nicht erfolgreich möglich, weshalb die Sequenzaufklärung mittels NMR praktisch unmöglich ist. Entsprechendes gilt für lineare Oligomere vom Typ 2 (alternierende amidische AminosäureSteroid-Verknüpfung, vgl. Kap. 2.2.2.), wobei hier durch das Vorhandensein der natürlichen Aminosäuren in Einzelfällen zumindest teilweise eine Sequenzzuordnung gelang (siehe jeweilige NMR-Daten im experimentellen Teil und in Abschnitt 3.2.1.2.). Aufgrund der genannten Schwierigkeiten konnten bei den synthetisierten linearen Oligoamiden mittels NMR nur einige wenige konformative Aspekte analysiert werden. Da die linearen Oligoamide (mit Ausnahme des Typ-2-Monomers Boc-Val-LCS-OMe 21) auch keine analysierbaren Kristalle lieferten, konnten weitere Informationen zur Konformation nur aus Kraftfeld-Rechnungen abgeleitet werden. In den folgenden beiden Abschnitten sind einige Konformationsdaten zu den linearen Oligomeren anhand ausgewählter Beispiele exemplarisch aufgeführt. Die hier nicht näher aufgeführten Verbindungen zeigten ähnliche Ergebnisse. 3.2.1.1. Lineare Oligomere vom Typ 1 Überraschenderweise zeigte die NMR-Analyse der linearen Oligomere von 3α-Aminolithocholsäure 5a, 3α-Aminodesoxycholsäure 5b und 3α-Aminocholsäure 5c, dass die NMRSpektren des Dimers, des Trimers und des Tetramers sich jeweils stark ähneln, und zwar nicht nur im Bereich von 0.6 - 2.0 ppm, sondern auch bei den charakteristischen Protonen mit Verschiebungen > 2.0 ppm. Besonders die Amid-NH-Protonen und die zugehörigen CHβ-3 Protonen sowie bei den Oligomeren von Aminodesoxycholsäure und Aminocholsäure auch die Protonen C12-H bzw. C7-H und C12-H zeigten im Trimer und im Tetramer jeweils identische Verschiebungen. Für die zwei bzw. drei Amid-NHs im Trimer bzw. im Tetramer konnte im Spektrum jeweils nur ein scharfes Dublett beobachtet werden. Diese Verhältnisse werden durch die Abbildungen 3.1., 3.2. und 3.3. verdeutlicht. 3. Konformationsanalyse und Komplexierungsverhalten 50 Abbildung 3.1. 400MHz-1H-NMR-Spektren von Boc-(LCS)2-OMe 13a und Boc-(LCS)4-OMe 14a (jeweils in CDCl3). Der direkte Vergleich zeigt die Ähnlichkeit der beiden Spektren. Soweit möglich, ist die Signalzuordnung angegeben. Die Bausteine in 14a wurden wie folgt differenziert: Boc-LCS-LCS‘-LCS“-LCS“‘-OMe. Abbildung 3.2. Boc-NH- und Amid-NHSignale der Oligomere 15a, 16b und 17a von Aminodesoxycholsäure im Vergleich (400MHz1 H-NMR, DMSO-d6). Auffällig bei den Amid- NH-Signalen ist die Ähnlichkeit der jeweils aus der Signalaufspaltung abgeleiteten Kopplungskonstanten JNH-CH. Aufgrund der Flexibilität der Ketten (siehe weiter unten) kann daraus jedoch keine strukturelle Information abgeleitet werden, vielmehr sind diese Kopplungskonstanten als zeitliche Mittelwerte über verschiedene Konformationen sowie beim Trimer 16b und beim Tetramer 17a auch als Mittelwerte über alle vorhandenen Amid-NHs aufzufassen. 3. Konformationsanalyse und Komplexierungsverhalten 51 Abbildung 3.3.a) 400MHz-1H-NMR-Spektrum des teilweise TFA-veresterten Aminodesoxycholsäure-Trimers Boc-(DCS)3-OMe 16a sowie der OH-Protonen des Tetramers 17a (jeweils in DMSO-d6). Die durch die teilweise Veresterung bedingte Intensitätsverminderung von zwei OH-Peaks ermöglichte die Zuordnung der OH-Signale zu den einzelnen Bausteinen. Die weiteren Informationen zur sicheren Zuordnung lieferte der Vergleich mit dem Spektrum des Tetramers 17a (die Verschiebung dieser drei OH-Signale ist nahezu identisch mit der der jeweiligen Signale von 16a). Unterscheidung der Bausteine: Boc-DCS-DCS‘-DCS“-OMe bzw. Boc-DCS-DCS‘-DCS“-DCS“‘-OMe Die jeweils abgebildeten Spektren lassen kaum Rückschlüsse auf die Sequenzzuordnung und damit auch nicht auf die konformativen Verhältnisse zu. Lediglich die OH-Protonen der Aminodesoxycholsäure-Oligomere konnten direkt bzw. durch den Vergleich von Spektren sicher den einzelnen Bausteinen zugeordnet werden. Hierbei war auch das NMR-Spektrum des teilweise TFA-veresterten Aminodesoxycholsäure-Trimers 16a (Abb. 3.3.a) hilfreich, da hier zwei OH-Signale, und zwar die des mittleren und des carboxyterminalen Bausteins, durch die teilweise Veresterung des Trimers intensitätsreduziert waren. 3. Konformationsanalyse und Komplexierungsverhalten 52 Abbildung 3.3.b) 400MHz-TOCSY-Spektrum von Boc-(DCS)3-OH 16b (in DMSO-d6). Anhand des Aminodesoxycholsäure-Tetramers 17a wurde exemplarisch die energetisch günstigste Konformation durch eine Kraftfeldrechnung ermittelt. Das globale Minimum der Rechnung ist in Abbildung 3.4.a) gezeigt. Die Ausbildung einer typischen Sekundärstruktur, etwa einer Helix, konnte dabei nicht beobachtet werden, was bei Betrachtung der übrigen lokalen Minima im Bereich bis 30 kJ/mol über dem globalen Minimum plausibel wird. Die Überlagerung der 35 energetisch günstigsten Konformationen innerhalb dieses Bereichs liefert nur noch ein undifferenziertes Knäuel und verdeutlicht die relativ hohe Flexibilität dieses Oligomers (Abb. 3.4.b). Durch diese Flexibilität wird die chemische Umgebung beispielsweise der drei Amid-NH-Protonen im zeitlichen Mittel so ähnlich, dass daraus ein einziges scharfes Dublett im NMR resultiert. 3. Konformationsanalyse und Komplexierungsverhalten 53 Abbildung 3.4.a) Gefundenes Globales Minimum (240.09 kJ/mol) des Tetramers Boc-(DCS)4-OMe 17a einer Monte-Carlo-Suche im AMBER-Kraftfeld mit 7500 analysierten Konformationen. Systematische sekundäre Strukturmerkmale zeigt die Verbindung nicht. Wasserstoffbrückenbindungen sind erkennbar zwischen NH DCS - CO DCS“‘ / OH DCS‘ CO DCS‘ / OH DCS“ - OH DCS“‘ (beteiligte Atome jeweils fett hervorgehoben). Die Bausteine wurden wie folgt unterschieden: Boc-DCS-DCS‘-DCS“-DCS“‘-OMe Abbildung 3.4.b) Überlagerung der 35 energiegünstigsten gefundenen Minima (im Intervall bis 30 kJ/mol über dem globalen Minimum) von 17a aus der Monte-Carlo-Suche. Dieser Vergleich zeigt die Unterschiedlichkeit der verschiedenen Konformationen und damit die hohe Flexibilität der Kette. 3. Konformationsanalyse und Komplexierungsverhalten 54 Abbildung 3.5. 400MHz-TOCSY-Spektrum von Boc-(CHS)4-OMe 20a (in DMSO-d6). 3.2.1.2. Lineare Oligomere vom Typ 2 Wahrscheinlich bedingt durch die in diesen Oligomeren vorhandenen natürlichen Aminosäuren zeigen die 1 H-NMR-Spektren der jeweiligen Verbindungen eine höhere Signaldifferenzierung als die der Typ-1-Oligomere, insbesondere im NH-Bereich. Dadurch konnten die charakteristischen Signale (im Bereich > 2.5 ppm) bis zum Dimer, welches insgesamt ein Tetraamid darstellt, jeweils sicher zugeordnet werden. Bei den Trimeren konnten zumindest die charakteristischen Signale des aminoterminalen Bausteins sicher zugeordnet werden. Die Signale des Aminosäureanteils und des Steroidanteils der beiden 3. Konformationsanalyse und Komplexierungsverhalten 55 anderen Bausteine konnten hier zwar jeweils zusammen je einem Baustein zugeordnet werden, jedoch gelang es aufgrund der in 3.2.1. genannten Schwierigkeiten nicht, zuzuordnen, welche Signale zum mittleren und weoche zum carboxyterminalen Baustein gehören. Auch bei den linearen Typ-2-Oligomeren war eine experimentelle Konformationsanalyse mittels NMR deshalb nur sehr eingeschränkt möglich. Lediglich beim Typ-2-Monomer, dem Diamid Boc-Val-LCS-OMe 21a, gelang eine vollständige Konformationsaufklärung, da sie sich als gut kristallisierbar herausstellte und die Kristalle erfolgreich röntgenstrukturanalytisch untersucht werden konnten. Die ermittelte Konformation im Kristall ist in Abbildung 3.6. wiedergegeben. Die Kristallstruktur liefert auch gleichzeitig den Konfigurationsbeweis an C3 des Steroidgerüstes: die Aminogruppe ist tatsächlich α-ständig. Abbildung 3.6. Kristallstruktur von Boc-Val-LCS-OMe 21a. Die Verbindung kristallisiert in einem monoklinen Gitter in der Raumgruppe P2(1). Der Kristall enthält kein Lösungsmittel. Detaillierte Informationen zur Kristallstruktur sind in Anhang B aufgeführt. 3. Konformationsanalyse und Komplexierungsverhalten 56 Tabelle 3.1. gibt einen Überblick über Winkelbeziehungen charakteristischer Atomgruppen, wie sie in der Kristallstruktur vorgefunden wurden. Im einzelnen sind dies die Torsionswinkel NHLCS-CHβ3LCS und NHVal-CHαVal sowie die zur Charakterisierung von Peptidsträngen üblicherweise angegebenen Torsionswinkel Ψ (hier: NVal-Cα αVal-C(O)Val-NLCS) und Φ (hier: αVal-NVal-C(O)Boc). Ein Vergleich der beiden NH-CH-Torsionswinkel mit NMRC(O)Val-Cα Daten (und damit mit den konformativen Verhältnissen in Lösung) gelang leider nicht, da die jeweiligen NH-Signale im NMR-Spektrum breit waren und keine saubere Dublettaufspaltung zeigten. Dies ist ein Indiz für ein dynamisches Gleichgewicht verschiedener Konformationen in der CDCl3-Lösung. Genausogut könnte die NH-Signalverbreiterung aber auch durch einen schnellen Wasserstoffaustausch bedingt sein. Torsionswinkel bzw. Atomgruppe Winkelmaß NHLCS-CHβ3LCS -168.2° NHVal-CHαVal -173.3° Ψ NVal-Cα αVal-C(O)Val-NLCS 23.3° Φ -132.1° C(O)Val-Cα αVal-NVal-C(O)Boc Tabelle 3.1. Charakteristische Winkelbeziehungen in der Kristallstruktur von Boc-Val-LCS-OMe 21a. Rechtsgängige Winkel sind jeweils positiv dargestellt, bei Ψ und Φ entspricht die ekliptische Anordnung der jeweils endständigen Atome einer Gruppe einem Winkel von 0°. 3. Konformationsanalyse und Komplexierungsverhalten 57 Nachstehend sind exemplarisch einige NMR-Spektren von Typ-2-Oligomeren aufgeführt, wobei der Schwerpunkt auf der Signalzuordnung liegt (Abbildungen 3.7. bis 3.10.). Abbildung 3.7. 400MHz-1H-NMR von Boc-Val-CHS-OMe 24a (in CDCl3). 3. Konformationsanalyse und Komplexierungsverhalten Abbildung 3.8. 400MHz-TOCSY-Spektrum von Boc-Lys[Z]-LCS-OMe 22a (in CDCl3). 58 3. Konformationsanalyse und Komplexierungsverhalten Abbildung 3.9. 400MHz-TOCSY-Spektrum von Boc-(Val-DCS)2-OMe 29a (in CDCl3). Die einzelnen Bausteine wurden wie folgt unterschieden: Boc-Val-DCS-Val‘-DCS‘-OMe 59 3. Konformationsanalyse und Komplexierungsverhalten Abbildung 3.10.a) 400MHz-TOCSY-Spektrum von Boc-(Val-LCS)3-OMe 26a (in CDCl3). Die Bausteine wurden wie folgt unterschieden: Boc-Val-LCS-Val‘-LCS‘-Val“-LCS“-OMe. 60 3. Konformationsanalyse und Komplexierungsverhalten 61 Abbildung 3.10.b) 400 MHz ROESY-Spektrum von Boc-(Val-LCS)3-OMe 26a (in CDCl3). Die Ausschnittvergrößerung zeigt die NOEs zwischen den Amidprotonen der einzelnen Aminolithocholsäure-Bausteine und den αHs der jeweils zugehörigen Valin-Bausteine. Zudem wurde das Hexaamid Boc-(Val-DCS)3-OMe 30a exemplarisch einer theoretischen Konformationsanalyse unterzogen, um eventuelle konformative Besonderheiten aufzufinden, da dies mittels NMR nicht gelang. Das gefundene globale Minimum (90.32 kcal/mol) der Kraftfeldrechnung ist in Abbildung 3.11. aufgeführt. Charakteristische Winkelbeziehungen aus dieser Konformation sind in Tabelle 3.2. a) und b) wiedergegeben. Soweit möglich erfolgte für die NH-CH-Torsionswinkel (Tabelle 3.2.b) jeweils ein Vergleich mit experimentell ermittelten Daten. Dazu wurde aus den NH-Kopplungskonstanten des NMRSpektrums (vgl. Abbildung 3.12.) anhand der Karplus-Kurve ein mögliches Winkelintervall ermittelt. Die relativ gute Entsprechung der NH-CH-Torsionswinkel mit den experimentellen Daten (siehe Tabelle 3.2.b) gibt Anlaß zu der Vermutung, dass das globale Minimum der Kraftfeldrechnung eine gute Näherung der tatsächlichen konformativen Verhältnisse in der CDCl3-Lösung ist. Die unscharfe Signalaufspaltung einiger NH-Protonen im NMR deutet jedoch auch hier auf ein durch die vergleichsweise hohe Flexibilität des Oligoamids bedingtes Konformationsgleichgewicht in Lösung bzw. einen schnellen Wasserstoffaustausch hin. Hinweise auf die Ausbildung einer systematischen repetitiven Sekundärstruktur oder von besonderen lokalen sekundären Strukturelementen (z.B. loop-Geometrien) konnten nicht gefunden werden. 3. Konformationsanalyse und Komplexierungsverhalten 62 Abbildung 3.11. Globales Minimum (90.32 kcal/mol) von Boc-(Val-DCS)3-OMe 30a einer MonteCarlo-Suche im AMBER-Kraftfeld. Die Verbindung zeigt keine systematischen sekundären Strukturmerkmale. Wasserstoffbrücken sind erkennbar zwischen NHDCS-COVal“, CODCS‘-OHDCS‘ und NHVal‘-OHDCS“. Die einzelnen Bausteine wurden wie folgt unterschieden: Boc-Val-DCS-Val‘-DCS‘-Val“-DCS“-OMe. Baustein Val Val‘ Val“ Torsionswinkel und entspr. Atomgruppe Winkelmaß Φ αVal-NVal-C(O)Boc C(O)Val-Cα - 150.9° Ψ αVal-C(O)Val-NDCS NVal-Cα 137.1° Φ‘ C(O)Val‘-Cα αVal‘-NVal‘-C(O)DCS - 160.1° Ψ‘ NVal‘-Cα αVal‘-C(O)Val‘-NDCS‘ 115.7° Φ“ C(O)Val“-Cα αVal“-NVal“-C(O)DCS‘ -143.2° Ψ“ 131.4° NVal“-Cα αVal“-C(O)Val“-NDCS“ Tabelle 3.2.a) Torsionswinkel Φ und Ψ aus dem globalen Minimum der Kraftfeldrechnung von Boc-(Val-DCS)3-OMe 30a. Rechtsgängige Winkel sind jeweils positiv dargestellt, die ekliptische Anordnung der jeweils endständigen Atome einer Gruppe entspricht einem Winkel von 0°. Die Bausteine wurden wie folgt unterschieden: Boc-Val-DCS-Val‘-DCS‘-Val“-DCS“-OMe. 3. Konformationsanalyse und Komplexierungsverhalten 63 Abbildung 3.12. NH-Signale und JNH-CH-Kopplungskonstanten von Boc-(Val-DCS)3-OMe 30a (400MHz-Spektrum, CDCl3). Unsichere Angaben aufgrund der unscharfen Signalaufspaltung sind in Klammern angegeben. Die Bausteine wurden wie folgt unterschieden: Boc-Val-DCS-Val‘-DCS‘-Val“-DCS“-OMe. aus Modelling Baustein aus NMR ϕ(NH-CαH) bzw. ϕ(NH-C3H) JNH-CH entspr. Winkelintervall aus Karplus-Kurve Val 148.5° (8.03) (128.3°-157.1°) DCS 136.0° 7.53 126.9°-153.9° Val‘ 129.3° 8.53 a) 130.4°-160.9° a) DCS‘ 120.4° (6.53) b) (123.1°-148.6°) b) Val“ 152.8° 8.04 a) 128.7°-157.2° a) DCS“ 171.9° (7.03) b) (125.1°-151.2°) b) Tabelle 3.2.b) NH-CH-Torsionswinkel aus dem globalen Minimum der Kraftfeldrechnung von Boc-(Val-DCS)3-OMe 30a und entsprechende mögliche Winkelintervalle aus NMR-Daten, berechnet aus den Kopplungskonstanten JNH-CH mit Hilfe der Karplus-Kurve. Unsichere Werte aufgrund von ungenauen experimentellen Daten sind in Klammern gesetzt. Rechtsgängige Winkel aus dem globalen Minimum sind jeweils positiv dargestellt, bei den aus NMR-Daten berechneten Winkelintervallen handelt es sich um Beträge, daher sind die Winkel stets positiv. Es wurde nur jeweils das Winkelintervall angegeben, das mit dem Winkel des globalen Minimums korrespondiert. Die Bausteine werden wie folgt unterschieden: Boc-Val-DCS-Val‘-DCS‘-Val“-DCS“-OMe. a) und b): Zuordnung im NMR unsicher, möglicherweise sind DCS‘ und DCS“ bzw. Val‘ und Val“ jeweils paarweise vertauscht. 3. Konformationsanalyse und Komplexierungsverhalten 64 3.2.2. Cyclische Oligomere Für die in dieser Arbeit synthetisierten Aminocholansäure-Macrocyclen (Davis schlägt für solche Cholansäurehaltigen Macrolactame den allgemeinen Sammelbegriff „Cyclocholamide“ vor47) wurden aufgrund ihrer Symmetrie durchweg stark vereinfachte NMR-Spektren mit einem einfachen Signalsatz beobachtet (eine Ausnahme). Neben der Konformationsanalyse war bei den cyclischen Komplexierungsversuche Aminocholansäure-Oligoamiden von Bedeutung. Hierzu auch wurden ihre Eignung exemplarisch für einige Untersuchungen durchgeführt. Die Ergebnisse sind in den folgenden beiden Abschnitten dargestellt. 3.2.2.1. Macrolactame aus Typ-1-Bausteinen Die Cyclooligomere der 3α-Aminodesoxycholsäure 5b und der 3α-Aminocholsäure 5c bieten aufgrund ihrer OH-Gruppen am Steroid-Gerüst, welche als (nichtkovalente) Bindungsstellen für kleine organische Gastmoleküle dienen können, prinzipiell gute strukturelle Voraussetzungen für den Einsatz als Wirtverbindungen für Komplexierungen. Weitere wichtige Voraussetzungen für die Eignung als Komplexbildner sind eine gute Löslichkeit und vor allem das Vorhandensein eines Hohlraumes, der hinreichend groß ist für die Aufnahme von Gastmolekülen. Daher war zunächst eine Untersuchung der konformativen Verhältnisse in geeignet erscheinenden Kandidaten interessant. Das 3α-Aminodesoxycholsäure-cyclodiamid 34a war gut kristallisationsfähig und konnte röntgenstrukturanalytisch untersucht werden. Die Kristallstruktur (Abbildung 3.13.a) erbrachte den Beweis dafür, dass auch in der Aminodesoxycholsäure die Aminogruppe α-ständig ist. Zudem lieferte die Kristallstruktur sehr genaue Aufschlüsse über die Größe des im Inneren des Cyclodimers vorhandenen Hohlraumes. Die Projektion der van-der-WaalsOberfläche zeigt, dass der Hohlraum zu klein ist, um ein Gastmolekül aufzunehmen (Abbildung 3.13.b). 3. Konformationsanalyse und Komplexierungsverhalten 65 Abbildung 3.13.a) Schwingungsellipsoid-Darstellung der Kristallstruktur von cyclo-(DCS)2 34a. Der NH-C3H-Torsionswinkel beträgt in der im Kristallverband fixierten Konformation 136.1°. Detaillierte Informationen zur Kristallstruktur sind in Anhang C aufgeführt. Abbildung 3.13.b) Darstellung der van-der-Waals-Oberfläche der im Kristall fixierten Konformation von 34a. Die Abbildung zeigt, dass die Verbindung keinen hinreichend großen Hohlraum zur Aufnahme von Gastmolekülen bietet. Der Kern-Kern-Abstand zwischen den Sauerstoffen der beiden OH-Gruppen beträgt zwar 4.93203 Å, unter Berücksichtigung der van-der-Waals-Radien verbleiben für den Hohlraum jedoch lediglich ca. 1.5 Å. 3. Konformationsanalyse und Komplexierungsverhalten 66 Allerdings ergab die Untersuchung der Kristallstruktur auch, dass pro Cyclodiamid jeweils zwei Moleküle Methanol und zwei Moleküle Wasser in das Kristallgitter eingebaut sind. Diese sind über einen Ring aus Wasserstoffbrücken seitlich an den Cyclus gebunden (Abbildung 3.13.c), wobei die Verbindung zum jeweils nächsten Cyclus im Kristallverband über eine Wasserstoffbrücke vom Wassermolekül zum Carbonylsauerstoff des benachbarten Cyclodiamids hergestellt wird (Abbildung 3.13.d). Bei der Anordnung der Lösungsmittelmoleküle im Kristallgitter handelt es sich gewissermaßen um eine side-onKomplexierung. Abbildung 3.13.c) Je zwei Wassermoleküle und zwei Methanol-Moleküle sind im Kristall über ein Band von Wasserstoffbrückenbindungen mit einem Molekül des Cyclodiamids 34a assoziiert. Die Abbildung zeigt zur besseren Veranschaulichung der Verhältnisse zwei Perspektiven. Die Anordnung der im Kristallverband integrierten Lösungsmittelmoleküle kann als side-on-Komplexierung interpretiert werden. Abbildung 3.13.d) Relative Anordnung der Cyclodiamide 34a zueinander im Kristallgitter. Die Abbildung zeigt, dass zwei benachbarte Cyclen jeweils indirekt über ein Wassermolekül durch Wasserstoffbrückenbindungen miteinander verbunden sind. 3. Konformationsanalyse und Komplexierungsverhalten 67 Das Cyclodiamid 34a wurde zum Vergleich mit der Kristallstruktur auch einer theoretischen Konformationsanalyse mittels einer Kraftfeldrechnung unterzogen. Abbildung 3.14.a zeigt das gefundene globale Minimum der Verbindung. Die abgebildete Konformation zeigt, dass das Cyclodimer gegenüber der Konformation in der Kristallstruktur unsymmetrisch und zur anderen Seite aufgeklappt ist. Die Amidprotonen der Peptidbindung sind in Richtung der offenen Seite gedreht und weisen damit gegenüber der Kristallstruktur in die gegenüberliegende Richtung. Ein lokales Minimum, das nur um 8.2 kJ/mol über dem globalen Minimum liegt, zeigt eine der Kristallstruktur sehr ähnliche Konformation (Abbildung 3.14.b). Abbildung 3.14.a) Gefundenes globales Minimum der Verbindung 34a aus einer MonteCarlo-Suche im Amber-Kraftfeld (172.7 kJ/mol). Das Minimum zeigt eine unsymmetrische Konformation. NH-C3H-Torsionswinkel: - 150.9° und - 142.4°. Abbildung 3.14.b) Lokales Minimum von 34a aus der Monte-Carlo-Suche (179.9 kcal/mol). Diese annähernd C2-symmetrische Konformation wurde 91 mal gefunden und liegt nur um 8.2 kJ/mol über dem gefundenen globalen Minimum. Sie entspricht in etwa der Konformation im Kristall. Es wurden nur 5 Konformationen mit niedrigerer Energie gefunden (incl. globales Minimum). NH-C3H-Torsionswinkel: jeweils 142.8°. 3. Konformationsanalyse und Komplexierungsverhalten 68 Die side-on-Komplexierung der Lösungsmittelmoleküle im Kristallverband machte prinzipiell eine weitere Untersuchung des Komplexierungsverhaltens in Lösung interessant. Bedauerlicherweise erwies sich die durch Kristallisation gereinigte Verbindung als unlöslich in Chloroform (im Gegensatz zur noch verunreinigten Verbindung) und in anderen nichtkompetitiven NMR-Lösungsmitteln, so dass weitere Komplexierungsexperimente nicht durchgeführt werden konnten. Selbst in DMSO war das Cyclodiamid 34a nach der Kristallisation nur noch sehr schlecht löslich. Immerhin war die Löslichkeit ausreichend, um ein NMR-Spektrum von der Verbindung zu erhalten (Abbildung 3.15.). Abbildung 3.15. 400MHz-1H-NMR von cyclo-(DCS)2 34a (in DMSO-d6). Für den Hohlraum im 3α-Aminocholsäure-cyclodiamid 35a sind im Grunde ähnliche Verhältnisse zu erwarten wie in 34a, jedoch mit dem wesentlichen Unterschied, dass ein schüsselartiges Aufklappen des Cyclus zu einer Seite, wie es in der Kristallstruktur von 34a zu beobachten ist, hier aufgrund des sterischen Anspruches der zusätzlichen OH-Gruppen an C7 der beiden Steroid-Gerüste nicht möglich sein sollte. Zudem war auch hier eine Untersuchung des Komplexierungsverhaltens in Lösung durch die Unlöslichkeit der Verbindung in Chloroform oder anderen nichtkompetitiven NMR-Lösungsmitteln erschwert. Die Kristalle, die aus Ethanol/Tetrahydrofuran erhalten wurden, hatten leider keine 3. Konformationsanalyse und Komplexierungsverhalten 69 hinreichend gute Qualität, um röntgenstrukturanalytisch untersucht zu werden. Auch durch wiederholtes Umkristallisieren und die Wahl anderer Lösungsmittelsysteme konnten hierfür keine geeigneten Kristalle erhalten werden. Selbst nach intensiver Trocknung der Kristalle, die aus EtOH/THF erhalten wurden, enthielten sie noch erhebliche Mengen Lösungsmittel, vor allem Ethanol, wie das NMR-Spektrum (Abbildung 3.16.) zeigt Dies ist möglicherweise darauf zurückzuführen, dass die Lösungsmittelmoleküle ähnlich wie in cyclo-(DCS)2 im Kristallgitter integriert sind, dem Intensitätsverhältnis zufolge 1-2 Moleküle EtOH pro Cyclus. Besonders auffällig in diesem NMR-Spektrum ist das im Vergleich zu anderen Aminocholsäure- und Aminodesoxycholsäure-Verbindungen ungewöhnlich weit hochfeldverschobene Proton der C12-OH-Gruppe. Eine abschließende Erklärung für diesen HochfeldShift konnte bislang nicht gefunden werden. Abbildung 3.16. 400MHz-1H-NMR von cyclo-(CHS)2 35a (in DMSO-d6). Der Bereich von 3.0 bis 4.4 ppm ist vergrößert dargestellt. Das Spektrum zeigt, dass die erhaltenen Kristalle noch große Mengen Ethanol enthielten. Die Integration der Signale ergibt ein Verhältnis von etwa 1-2 Molekülen Ethanol pro Molekül 35a. Die C12-OH-Gruppe des Cyclodiamids ist auffällig weit hochfeld-verschoben. Signalzuordnungen erfolgten zum Teil mit Hilfe des TOCSYSpektrums der Verbindung. 3. Konformationsanalyse und Komplexierungsverhalten 70 Im Gegensatz zu den Cyclodiamiden 34a und 35a, die keine hinreichend großen Hohlräume aufweisen, um darin Gastmoleküle aufzunehmen, zeigt eine Modellingstudie des 3a-Aminodesoxycholsäure-Cyclotriamids 34b, dass das Innere dieser Verbindung groß genug ist, um ein Glucosemolekül aufzunehmen (Abbildung 3.17.). Abbildung 3.17. Modellingstudie zur Ermittlung der Hohlraumgröße in Cyclo-(DCS)3 34b. Das Bild zeigt, dass der Tricyclus in seinem Inneren ein Glucosemolekül aufnehmen kann. Dies korrespondiert mit den Erkenntnissen von Davis, der über Kraftfeldrechnungen bei einem geringfügig kleineren Cyclotriamid aus drei bis-nor-Aminocholsäurebausteinen ebenfalls einen hinreichend großen Hohlraum für die Komplexierung von Kohlenhydraten festgestellt hatte47. Aufgrund der guten Löslichkeit des Cyclotriamids 34b in Chloroform ist es zudem gut geeignet für Komplexierungsexperimente. Die Glucose hingegen ist in Chloroform nicht löslich, weshalb eine Komplexierung stattdessen mit dem in Chloroform besser löslichen n-Dodecyl-β-Glucosid 40 versucht wurde. Hierzu wurde ein 1:1 Gemisch von 34b und 40 (Konzentration jeweils 10 mM in CDCl3) NMR-spektroskopisch untersucht und mit den Spektren der reinen Verbindungen verglichen. Der Vergleich der 1H-NMR-Spektren zeigt, dass im Gemisch einige Signale der beiden Verbindungen deutlich gegenüber der jeweis reinen Verbindung verschoben sind (Abbildung 3.18.), was deutlich auf die Ausbildung eines Wirt-Gast-Komplexes hindeutet. Besonders das Signal des NH-Protons erfährt einen erheblichen Tieffeld-Shift um fast 0.4 ppm, außerdem 3. Konformationsanalyse und Komplexierungsverhalten 71 wird das Signal breit und unscharf, was insgesamt auf eine Beteiligung an einer Wasserstoffbrückenbindung hinweisen könnte. Die Komplexbildungskonstante wurde auf ca. 200-9000 l·mol-1 geschätzt. Abbildung 3.18. 600MHz-1H-NMR-Spektren von cyclo-(DCS)3 34b, n-Dodecyl-β-Glucosid 40 und dem 1:1-Gemisch beider Verbindungen (Konzentration jeweils 10 mM in CDCl3). Abbildung 3.19. zeigt eine durch Kraftfeldrechnung ermittelte mögliche Konformation eines Komplexes aus 34b und dem zu 40 analogen Octylglucosid. In der Abbildung sind einige H-H-Entfernungen zwischen C23-H2-Protonen des Cyclotriamids und Ringprotonen des Glucosids gekennzeichnet, die hinreichend klein sind, um in Sättigungsexperimenten einen NOE zu zeigen. Im ROESY-Spektrum können auch tatsächlich NOE-Crosspeaks zwischen dem AB-System an C23 und den Ringprotonen des Glucosids beobachtet werden (Abbildung 3.20.). 3. Konformationsanalyse und Komplexierungsverhalten 72 Abbildung 3.19. Gefundenes Globales Minimum aus einer Docking-Prozedur im AMBERKraftfeld für den Komplex aus 34b und n-Octyl-β-Glucosid. In der Kalottendarstellung sind einige mögliche NOEs zwischen den Protonen an C23 des Cyclotriamids und den Ringprotonen des Glucosids markiert. Abbildung 3.20. Ausschnitt aus dem 600MHz-ROESY-Spektrum des Komplexes aus 34b und 40. Das Spektrum zeigt tatsächlich NOEs zwischen den C23-Protonen von 34b und GlucosidRingprotonen. Zur Signalzuordnung vgl. Abb. 3.18. 3. Konformationsanalyse und Komplexierungsverhalten 73 Das zu 34b analoge Cyclotriamid cyclo-(CHS)3 35b der Aminocholsäure konnte leider nicht rein erhalten werden, weshalb Komplexierungsversuche hiermit nicht in Frage kamen. Bei dem Versuch der Synthese dieser Verbindung war als Hauptprodukt der Hexacyclus cyclo-(CHS)6 35d entstanden. Die übrigen in dieser Arbeit synthetisierten Typ-1-Oligomere der Aminocholansäuren wurden nicht mehr auf ihr Komplexierungsverhalten untersucht, da einerseits mit jedem zusätzlichen Baustein der Hohlraum größer wird und damit nur noch für wenige Gastmoleküle geeignet ist, und da andererseits mit jedem Baustein die Flexibilität des Moleküls größer wird und damit die Präorganisation des Hohlraumes abnimmt, was die Bindungsstärke auch zu gut passenden Gästen prinzipiell verringern sollte. Ein deutlicher Hinweis auf die zunehmende Flexibilität der Cyclen findet sich durch Vergleich der NMR-Signale der Protonen an C23 von Cyclo-(DCS)3 34b, Cyclo-(DCS)4 34c und Cyclo-(DCS)6 34d. Der Vergleich zeigt, dass die beiden Signale des AB-Systems mit steigender Ringgröße unschärfer werden und zusammenrücken, wobei im Cyclohexamer nur noch ein Signal für beide Protonen zu beobachten ist (Abbildung 3.21.). Dieses Phänomen ist mit großer Wahrscheinlichkeit darauf zurückzuführen, dass aufgrund der mit der Ringgröße steigenden Flexibilität der Cyclen das NMR-Spektrum lediglich einen Mittelwert aus zahlreichen (meist unsymmetrischen) sich dynamisch ineinander umwandelnden Konformationen abbildet, in denen die Protonen an C23 der einzelnen Bausteine jeweils unterschiedliche Umgebungen haben. Abschließend ist aus der Reihe der Typ-1-Oligocyclen noch das NMR-Spektrum von Cyclo-(CHS)6 35d abgebildet (Abbildung 3.22.). 3. Konformationsanalyse und Komplexierungsverhalten 74 Abbildung 3.21. Vergleich der 1H-NMR-Spektren von Cyclo-(DCS)3 34b, Cyclo-(DCS)4 34c und Cyclo-(DCS)6 34d (400 MHz, CDCl3). Mit steigender Ringgröße ist eine zunehmende Unschärfe und ein Zusammenrücken der Signale der beiden Protonen an C23 zu erkennen. Abbildung 3.22. 400MHz-1H-NMR-Spektrum von Cyclo-(CHS)6 35d. 3. Konformationsanalyse und Komplexierungsverhalten 75 3.2.2.2. Macrolactame aus Typ-2-Bausteinen Bei der kernresonanzspektroskopischen Untersuchung der drei Typ-2-Cyclotrimere Cyclo-(Val-LCS)3 36, Cyclo-(Lys[Z]-LCS)3 37 und Cyclo-(Val-DCS)3 38a zeigte sich in Chloroform eine ungewöhnlich starke Tieffeld-Verschiebung des jeweiligen Steroid-NHProtons (Abbildungen 3.23. a und b). Zudem war dieses Signal jeweils unscharf und deutlich verbreitert (siehe zum Vergleich auch die NH-Signale in den Spektren der linearen Analoga, Abb. 3.10.a und 3.12.). Abbildung 3.23.a) 400MHz-TOCSY-Spektrum von Cyclo-(Val-LCS)3 36 (in CDCl3). Das Spektrum weist einen einfachen Signalsatz auf, der (im zeitlichen Mittel) auf eine Symmetrie des Cyclus hindeutet. Die Crosspeaks belegen, dass es sich bei dem tieffeldverschobenen NH-Signal um das Amid-Proton der LCS-Bausteine handelt. 3. Konformationsanalyse und Komplexierungsverhalten 76 Abbildung 3.23.b) Charakteristische Signcle aus den 400MHz-1H-NMR-Spektren (jeweils in CDCl3) der Verbindungen Cyclo-(Lys[Z]-LCS)3 37 (unten) und Cyclo-(Val-DCS)3 38a (oben). Die beiden Spektren zeigen analoge Verhältnisse wie das Spektrum von 36. Diese Beobachtungen deuten auf eine Beteiligung dieses Protons an einer Wasserstoffbrücke hin. Eine plausible Möglichkeit hierfür liegt in der Ausbildung einer intramolekularen Wasserstoffbrückenbindung zum Carbonylsauerstoff des nächsten Steroid-Bausteins unter Ausbildung eines γ-loops. Diese lokale Konformation ist in Abbildung 3.24. am Beispiel der Verbindung 36 aufgezeigt. 3. Konformationsanalyse und Komplexierungsverhalten 77 Abbildung 3.24. γ-loop-Konformation (grau unterlegt) in Cyclo-(Val-LCS)3 36. Rechts sind zwei verschiedene mögliche Geometrien, eine mit äquatorial (A) und eine mit axial (B) ständiger Seitenkette, aufgeführt. Zur Überprüfung dieser Vermutung wurde die Verbindung 36 einer theoretischen Konformationsanalyse unterzogen (Amber-Kraftfeld, Monte-Carlo-Suche mit 9845 Schritten). Das globale Minimum und zwei weitere lokale Minima sind in Abbildung 3.25. abgebildet. Während im globalen Minimum (206.89 kJ/mol, 4x gefunden) zwar eine Wasserstoffbrücke, aber keine γ-loop-Konformation zu beobachten ist, zeigt bereits das über zwei Wasserstoffbrücken stabilisierte lokale Minimum mit der zweitniedrigsten Energie (nur um 0.18 kJ/mol höher als das globale Minimum) zumindest an einer Stelle diese Geometrie. In fast allen folgenden lokalen Minima war ebenfalls mindestens ein γ-loop enthalten. Auch lokale Minima mit drei γ-Loops wurden gefunden, eines davon (239.63 kJ/mol) mit zwei äquatorialen und einem axialen γ-turn ist exemplarisch abgebildet. Allen gefundenen Minima gemeinsam ist, dass ausschließlich NH-Protonen des Steroids an Wasserstoffbrücken beteiligt sind, was sich mit den experimentellen Befunden deckt. Die Vielzahl unterschiedlicher Konformationen mit ähnlicher Energie erklärt auch die Unschärfe der Signale im NMR, die sich wahrscheinlich aus einem dynamischen Gleichgewicht dieser Konformationen in Lösung ergibt (ein Wasserstoffaustausch als Ursache für die NH-Signalverbreiterung wird hier aufgrund der Tatsache, dass auch alle anderen Signale keine scharfe Aufspaltung zeigen, als insgesamt eher unwahrscheinlich angesehen, kann aber nicht sicher ausgeschlossen werden). 3. Konformationsanalyse und Komplexierungsverhalten 78 γ γ γ γ Abbildung 3.25. Gefundenes Globales Minimum (A, 206.89 kJ/mol) und zwei weitere lokale Minima (B, 207.07 kJ/mol und C, 239.63 kJ/mol) von Cyclo-(Val-LCS)3 36 aus einer MonteCarlo-Suche im AMBER-Kraftfeld. Die abgebildeten Konformationen zeigen, dass ausschließlich NH-Protonen von LCS-Bausteinen an Wasserstoffbrückenbindungen beteiligt sind. Erst in der Geometrie mit der zweitniedrigsten Energie zeigt sich die vermutete γ-loopKonformation. 3. Konformationsanalyse und Komplexierungsverhalten 79 Winkelmaß Baustein Val 1 LCS 1 Val 2 LCS 2 Val 3 LCS 3 Torsionswinkel A B C ϕ(NH-CαH) 154.5° 9.9° - 145.2° Φ C(O)Val-Cα αVal-NVal-C(O)LCS - 142.1° 69.9° - 78.9° Ψ NVal-Cα αVal-C(O)Val-NLCS 159.8° - 61.3° 67.4° ϕ(NH-C3H) 172.5° - 156.9° 139.6° ϕ(NH-CαH) 152.2° 154.2° - 141.2° Φ‘ αVal-NVal-C(O)LCS C(O)Val-Cα - 138.0° - 147.8° - 78.5° Ψ‘ NVal-Cα αVal-C(O)Val-NLCS 83.5° 136.2° 73.7° ϕ(NH-C3H) 137.4° - 160.8° - 178.8° ϕ(NH-CαH) 153.6° 154.7° 4.3° Φ“ C(O)Val-Cα αVal-NVal-C(O)LCS - 146.0° - 141.7° 70.7° Ψ“ αVal-C(O)Val-NLCS NVal-Cα 134.9° 160.8° - 60.1° ϕ(NH-C3H) -162.7° 178.3° 146.6° Tabelle 3.3. Charakteristische Winkelbeziehungen aus den in Abb. 3.25. gezeigten Minima der Kraftfeldrechnung zu Cyclo-(Val-LCS)3 36. Rechtsgängige Winkel sind jeweils positiv dargestellt, die ekliptische Anordnung der jeweils endständigen Atome einer Gruppe entspricht einem Winkel von 0°. Die Bausteine wurden entsprechend der Abbildung in CO!NH-Richtung von 1 bis 3 durchnumeriert. Bei der Synthese von cyclo-(Val-DCS)3 38a war eine weitere Verbindung (38b) in größerer Menge angefallen, die säulenchromatographisch rein isoliert werden konnte. Die massenspektrometrische Analyse mittels MALDI zeigt, dass diese Verbindung die doppelte Masse besitzt und somit ein Cyclodimerisierungsaddukt darstellt. Besonders ungewöhnlich an dieser Verbindung ist das 1H-NMR-Spektrum, welches gegenüber allen anderen in dieser Arbeit synthetisierten Macrolactamen als einziges keinen einfachen Signalsatz zeigt, sondern einen 3. Konformationsanalyse und Komplexierungsverhalten 80 doppelten, der ein Intensitätsverhältnis von 1:1 aufweist (Abbildung 3.26.a). Es gibt also im Molekül jeweils zwei unterscheidbare Valin- und DCS-Einheiten, woraus insgesamt eine C3-Symmetrie abgelektet werden kann. Bei der Signalzuordnung innerhalb eines Signalsatzes waren vor allem das TOCSY/Spektrum (Abbildung 3.26.b) und das ROESY-Spektrum hilfreich, aber auch Informationen aus dem HMQC (Abbildung 3.26.c) waren zur Signalzuordnung notwendig. Die Frage, ob und ggf. wo die beiden Signalsätze ineinander übergehen, wird später geklärt. Abbildung 3.26.a) 600MHz-1H-NMR-Spektrum der Verbindung 38b (in CDCl3). Die beiden Signalsätze sind in A und B unterschieden, die Zuordnung war durch zweidimensionale NMR-Experimente (TOCSY, ROESY und DQF-COSY sowie HMQC) möglich (vgl. Abb. 3.27.). 3. Konformationsanalyse und Komplexierungsverhalten Abbildung 3.26.b) 600MHz-TOCSY-Spektrum der Verbindung 38b. 81 3. Konformationsanalyse und Komplexierungsverhalten 82 Abbildung 3.26.c) HMQC-Spektrum von 38b. Einige besonders charakteristische Zuordnungen sind angegeben. Auffällig ist die große Verschiebungsdifferenz der beiden Protonen von C22 (evtl. C23, Zuordnung nicht völlig sicher) im Signalsatz A. 3. Konformationsanalyse und Komplexierungsverhalten 83 Abbildung 3.27. Korrelationsbeziehungen, auf denen im NMR von 38b die Signalzuordnung innerhalb eines Signalsatzes (in NH!CO-Richtung von C3-H eines Steroid-Bausteins zu C19-H3 des nächsten Steroid-Bausteins) beruht. Gestrichelte Linien: direkte/indirekte H,H-Korrelation aus TOCSY und DQF-COSY; Durchgezogene Linien: NOEs aus ROESY. Die NMR-Befunde deuten neben der für eine so komplexe Verbindung – immerhin handelt es sich um ein Cyclododecaamid – ungewöhnlich guten Kristallisationsfähigkeit auf eine besondere Struktur mit höherer Ordnung hin. Grundsätzlich kommen hier drei verschiedene Verbindungstypen in Frage (siehe Abbildung 3.28.): ein einfaches Cyclododecaamid (A), ein Catenan aus zwei kettenförmig verknüpften cyclo-(DCS)3-Ringen (B), oder ein verknotetes Cyclododecaamid, ein sogenanntes Knotan (C). Catenane48 und Knotane49 aus AmidMacrocyclen wurden in jüngerer Zeit von Vögtle dargestellt und untersucht. Die Ergebnisse dieser Arbeiten wurden zur Bewertung der verschiedenen Varianten mit herangezogen, wobei allerdings berücksichtigt werden mußte, dass bei der hier untersuchten Verbindung 38b durch die Amid-Bindungen eine Bindungsrichtung vorgegeben war, die als zusätzliche Komponente die Symmetrie und die Zahl und Art der möglichen Stereoisomere beeinflußt. Bei den von Vögtle vorgestellten Catenanen und Knotanen lag eine solche Gesamt-Bindungsrichtung nicht vor, da die Amid-Bindungsrichtungen alternierend waren und sich so insgesamt gegeneinander aufhoben. 3. Konformationsanalyse und Komplexierungsverhalten 84 Abbildung 3.28. Allgemeine Topologie der verschiedenen Varianten von 38b. A: einfacher Cyclus, B: Catenan, C: Knotan. Für die Catenan-Variante (B) und das Knotan (C) sind unter Berücksichtigung der gegebenen Stereochemie der Bausteine jeweils zwei diastereomere Formen denkbar (Abbildung 3.29. a und b). Abbildung 3.29.a) Mögliche Diastereomere von Catenanen mit sich wiederholenden Bausteinen definierter Stereochemie (L-Aminosäuren, Steroid-Gerüst) und vorgegebener Bindungsrichtung (durch Pfeile gekennzeichnet). Zur Herleitung vgl. Anhang D. 3. Konformationsanalyse und Komplexierungsverhalten 85 Abbildung 3.29.b) Mögliche Diastereomere von Knotanen mit sich wiederholenden Bausteinen definierter Stereochemie (L-Aminosäuren, Steroid-Gerüst) und vorgegebener Bindungsrichtung (durch Pfeile gekennzeichnet). Zur Herleitung vgl. Anhang D. Dies ergibt sich aus dem Umstand, dass den einzelnen Cyclen durch die Amid-Bindungen jeweils eine Richtung zugeordnet werden kann. Da eine Lösung der Kristallstruktur aufgrund der Empfindlichkeit der Kristalle und ihres relativ geringen Streuvermögens bisher nicht gelang, war es zur Strukturaufklärung nun zunächst nötig, zu klären, welche der angegebenen Varianten am besten mit den Ergebnissen der NMR-Experimente korrespondiert. Dass der doppelte Signalsatz aus einem Gemisch von Diastereomeren resultiert, konnte mit einiger Sicherheit ausgeschlossen werden, da auch nach mehreren Kristallisationen das Intensitätsverhältnis zwischen beiden Signalsätzen absolut identisch blieb. Es handelt sich also mit großer Wahrscheinlichkeit um eine einzige Verbindung, entweder um den einfachen Cyclus (Variante A aus Abb. 3.28.), oder um eines der beiden Diastereomere des Catenans oder des Knotans. Nach aller Erfahrung sollte ein einfacher Macrocyclus (Variante A), auch ein größerer, lediglich einen einfachen Signalsatz im NMR zeigen, ähnlich wie die anderen in dieser Arbeit synthetisierten höheren Cyclen (z.B. cyclo-(DCS)6 34d und cyclo-(CHS)6 35d). Eine starke Fixierung einer einzelnen Konformation durch Wasserstoffbrückenbindungen, die eine Unterscheidbarkeit von jeweils zwei Valin- und DCS-Bausteinen mit verschiedenen Umgebungen (und damit unterschiedlichen Signalsätzen) zur Folge hätte, kann hier weitgehend ausgeschlossen werden, da aufgrund der insgesamt hohen Flexibilität des großen Ringes in Lösung von einem schnellen dynamischen Gleichgewicht mehrerer Konformationen 3. Konformationsanalyse und Komplexierungsverhalten 86 ausgegangen werden muß – dies würde auf der NMR-Zeitskala wiederum einen einfachen Signalsatz aus einer gemittelten Konformation zur Folge haben. Variante A kann also aufgrund der NMR-Befunde als insgesamt recht unwahrscheinlich angesehen werden. Bei Catenanen sind die Überlegungen etwas komplexer. Hier muß berücksichtigt werden, ob die beiden Ringe schnell gegeneinander rotieren oder ob sie nicht bzw. langsam rotieren (jeweils bezogen auf die NMR-Zeitskala). Im Fall einer schnellen Rotation sollte sich für ein Diastereomer lediglich ein einfacher Signalsatz ergeben, da die beiden Diastereomere jeweils eine zweizählige Drehachse besitzen. Aus dem gleichen Grund sollten bei langsamer bzw. nicht vorhandener Rotation, (diese hatte Vögtle bei den von ihm synthetisierten Catenanen beobachtet48), unterscheidbare Signalsätze auftreten, da an der Kontaktstelle der beiden Ringe die Ausbildung von Wasserstoffbrücken erwartet wird. Somit sollte ein Signalsatz aus den an der Kontaktstelle lokalisierten Bausteinen (pro Ring je eine Valin und eine DCS-Einheit) und (mindestens) ein Signalsatz aus den nicht an der Kontaktstelle lokalisierten Bausteinen resultieren. Drei Signalsätze wären hier ebenfalls möglich, da aufgrund der Richtungskomponente der Amidkette die nicht an der Kontaktstelle lokalisierten Bausteine (pro Ring je zwei Valin- und zwei DCS-Einheiten) jeweils unterschiedliche Umgebungen aufweisen. Wenn sich nur zwei Signalsätze ergeben, sollten sie jedoch im Intensitätsverhältnis 1:2 auftreten, was im Widerspruch zu den NMR-Befunden steht. Da das Vorliegen eines Diastereomerengemisches bereits ausgeschlossen wurde (dies könnte – schnelle Rotation vorausgesetzt – zwei verschiedene Signalsätze, jeder von einem der Diastereomere, erklären) scheidet ein Catenan (B) als mögliche Strukturvariante ebenfalls aus und es bleibt lediglich eines der Knoten-Diastereomere (Variante C) als Möglichkeit übrig. Die Knotan-Variante wird im Folgenden anhand von Modellingstudien und der detaillierten Betrachtung von zweidimensionalen NMR-Experimenten, insbesondere dem ROESY, näher untersucht. Dabei ist zunächst die Frage zu klären, ob in einem Knoten tatsächlich zwei unterscheidbare Baustein-Sätze auftreten und ob diese hinreichend fixiert sind und nicht ihre Position tauschen, denn sonst würde auch die Knotan-Variante als mögliche Struktur den doppelten NMR-Signalsatz im Verhältnis 1:1 nicht erklären können. Ein mögliches Knoten-Diastereomer wurde einer theoretischen Konformationsanalyse unterzogen. Da bei Knoten eine Ringöffnung während der Rechnung vermieden werden mußte, konnten die sonst üblichen Verfahren zur Konformationssuche mit Hilfe von Monte- 3. Konformationsanalyse und Komplexierungsverhalten 87 Carlo-Rechnungen nicht angewendet werden. Stattdessen wurde eine unsymmetrische Startgeometrie des Knotens mittels stochastischer Dynamik (SD) untersucht. Über einen Zeitraum von 6000 ps bei 1200 K wurden insgesamt die Koordinaten von 2000 Geometrien gespeichert. Diese Geometrien wurden anschließend im Kraftfeld optimiert. Das globale Minimum dieser Suche ist in Abbildung 3.30.a) gezeigt. Abbildung 3.30.a) Globales Minimum (218.75 kJ/mol) einer SD-Simulation (AMBERKraftfeld) zur Energieoptimierung einer der beiden diastereomeren Knotengeometrien von 38b (SD = stochastische Dynamik). Die abgebildete Konformation ist über 9 Wasserstoffbrückenbindungen stabilisiert und weist C3-Symmetrie auf. (Das andere Diastereomer wurde auf die gleiche Weise untersucht. Es liegt energetisch um 13,82 kJ/mol höher und zeigt keine C3-Symmetrie; vgl. Abbildung in Anhang D) Man erkennt eine C3-symmetrische Konformation des Knotens, die an den drei Kontaktstellen des zusammenhängenden Stranges jeweils durch drei Wasserstoffbrücken fixiert ist. Dadurch entstehen jeweils zwei unterscheidbare Valin- und DCS-Bausteine. Die lokale Geometrie 3. Konformationsanalyse und Komplexierungsverhalten einer Kontaktstelle ist in Abbildung 3.30.b) 88 wiedergegeben. Dort sind einige H,H-Entfernungen markiert, die einen NOE ergeben sollten, und außerdem die Wasserstoffbrückenbindungen hervorgehoben. Interessant ist in diesem Zusammenhang, dass in einem Strang ein Valin-Amidproton an einer Wasserstoffbrücke beteiligt ist, im anderen ein DCS-Amidproton. Dies korrespondiert mit dem NMR-Spektrum, in dem das ValinAmidproton des Signalsatzes A und das DCS-Amidproton des Signalsatzes B jeweils stark tieffeldverschoben sind. Ein solcher Tieffeld-Shift wird üblicherweise dann beobachtet, wenn die Amidprotonen in Wasserstoffbrücken fixiert sind. Bei der weiteren Diskussion der kraftfeldoptimierten Knoten-Konformation aus Abbildung 3.30.a) wurden deshalb die Valinund DCS-Bausteine jeweils analog der NMR-Zuordnung in A und B unterschieden (siehe Bezeichnung der beiden Stränge in Abbildung 3.30.b). Abbildung 3.30.b) Lokale Geometrie einer der drei identischen Kontaktstellen im Globalen Minimum des Knotans. Wasserstoffbrückenbindungen (gestrichelte Linien) sowie einige charakteristische H,H-Entfernungen, die einen NOE ergeben sollten (Doppelpfeile), sind hervorgehoben. Die beiden Stränge sind entsprechend der Signalzuordnung im NMR in A und B unterschieden. Tabelle 3.4. listet einige charakteristische im Knoten-Modelling (Abb. 3.30.a) gefundene H,H-Entfernungen auf, die einen NOE ergeben sollten (< 3 Å) und gibt an, ob diese theoretisch zu erwartenden NOEs experimentell bestätigt werden konnten. Besonders NOEs, die zwischen jeweils einem Proton von Strang A und Strang B auftreten sollten, werden als 3. Konformationsanalyse und Komplexierungsverhalten 89 charakteristisch angesehen, aber auch NOEs, die aufgrund der besonderen Konformation nur innerhalb eines Stranges auftreten sollten, im anderen aber nicht. Es zeigt sich, dass jeder dieser erwarteten besonders charakteristischen NOEs tatsächlich eine experimentelle Entsprechung im ROESY hat. Exemplarisch sind die NOE-Crosspeaks zwischen den Methylprotonen an C21 beider Signalsätze mit je einem der beiden Protonen von C23-H2 des B-Signalsatzes in einem Ausschnitt des ROESY-Spektrums markiert (Abbildung 3.31.). Im Knoten-Modelling zeigte tatsächlich nur eines der C23B-Protonen (das pro-S-ständige) zu den C21A-Methylprotonen einen für einen NOE hinreichend kleinen Abstand, und zwar das gleiche Proton, das auch zu einem der beiden C23A-Protonen einen Abstand hat, der einen NOE erwarten läßt – und diesen auch im NMR zeigt. Zur C21B-Methylgruppe hingegen zeigt nur das pro-R-ständige Proton einen starken NOE (vgl. Markierungen in Abbildung 3.30.b). betrachtete Wasserstoffatomea) Kernabstände aus Modellingb) NOE-Crosspeak im ROESY αH(ValB) - βH(ValA) 3.00 Å ja C23-HR(DCSB) - C23-HR(DCSA) C23-HR(DCSB) - C23-HS(DCSA) C23-HS(DCSB) - C23-HR(DCSA) C23-HS(DCSB) - C23-HS(DCSA) 5.16 Å 3.74 Å 3.79 Å 2.64 Å nein nein nein ja C23-HR(DCSB) - C21-H3(DCSA) C23-HS(DCSB) - C21-H3(DCSA) 3.98 Å 2.28 Å (nein)c) ja C23-HR(DCSB) - C21-H3(DCSB) C23-HS(DCSB) - C21-H3(DCSB) 2.28 Å 3.27 Å ja (ja)d) NH(ValB) - C22-H2(DCSB)e) 2.19 Å 2.82 Å ja ja a) Bei prochiralen (prostereogenen) Gruppen sind die diastereotopen Protonen jeweils durch die Indices R (für pro-R-chiral) und S (für pro-S-chiral) differenziert. b) Bei mehreren möglichen Abständen ist der, der einen NOE zeigen sollte, fett gedruckt. c) es ist ein sehr schwacher NOE-Crosspeak erkennbar, der jedoch gegenüber dem anderen vernachlässigbar klein ist. (vgl Abb. 3.31.) d) dieser NOE-Crosspeak ist deutlich kleiner als der andere. (vgl. Abb. 3.31.) e) im A-Strang hat die Steroid-Seitenkette eine andere Konformation (vgl. Abb. 3.30.b), in der aufgrund des großen H,H-Abstandes (jeweils > 4 Å) entsprechende NOEs nicht zu erwarten sind und auch nicht beobachtet werden. Auf eine Differenzierung der diastereotopen Protonen von C22 wurde verzichtet, da beide einen NOE zeigen. Tabelle 3.4. Einige charakteristische H,H-Kernabstände aus dem Modelling der Knotenvariante von 38b, die aufgrund ihrer Distanz (bis 3 Å) einen NOE zeigen sollten. 3. Konformationsanalyse und Komplexierungsverhalten 90 pro-R pro-S Abbildung 3.31. Ausschnitt aus dem ROESY-Spektrum von 38b. Die NOE-Crosspeaks der Protonen von C23-H2 des Signalsatzes B zu den Protonen von C21-H3 beider Signalsätze sind markiert. Es zeigt sich – auch hinsichtlich der Intensität – eine deutliche Übereinstimmung mit den aufgrund der Kernabstände aus dem Modelling erwarteten NOEs (siehe auch Tabelle 3.5.b). Eine gute Entsprechung (bei DCSA zumindest tendenziell) zeigt sich auch beim Vergleich der aus der optimierten Knotenkonformation abgelesenen NH-CH-Torsionswinkel mit den Winkelintervallen, die sich jeweils aus den Kopplungskonstanten JNH-CH im NMR-Spektrum ergeben (Tabelle 3.5.a). 3. Konformationsanalyse und Komplexierungsverhalten aus Modelling Baustein 91 aus NMR ϕ(NH-CαH) bzw. ϕ(NH-C3H) JNH-CH entspr. Winkelintervall aus Karplus-Kurve ValA 141.6° 7.38 126.4°-153.2° DCSA 174.9° 9.06 132.2°-166.1° ValB 170.9° 10.20 135.9°-180.0° DCSB 129.7° 5.10 117.1°-141.8° Tabelle 3.5.a) NH-CH-Torsionswinkel aus dem globalen Minimum der Knotanvariante (Abb. 3.30.a) von 38b und entsprechende mögliche Winkelintervalle aus NMR-Daten, berechnet aus den Kopplungskonstanten JNH-CH mit Hilfe der Karplus-Kurve. Rechtsgängige Winkel aus dem globalen Minimum sind jeweils positiv dargestellt, bei den aus NMR-Daten berechneten Winkelintervallen handelt es sich um Beträge, daher sind die Winkel stets positiv. Es wurde nur jeweils das Winkelintervall angegeben, das mit dem Winkel des globalen Minimums korrespondiert. Die Bausteine werden entsprechend Abb. 3.30.b) (Signalzuordnung im NMR) in A und B unterschieden. Baustein ValA ValB Torsionswinkel und entspr. Atomgruppe Winkelmaß Φ C(O)Val-Cα αVal-NVal-C(O)DCS - 92.6° Ψ NVal-Cα αVal-C(O)Val-NDCS 135.7° Φ‘ αVal-NVal-C(O)DCS C(O)Val-Cα - 129.3° Ψ‘ 127.8° NVal-Cα αVal-C(O)Val-NDCS Tabelle 3.5.b) Torsionswinkel Φ und Ψ aus dem globalen Minimum der Knotanvariante von 38b. Rechtsgängige Winkel sind jeweils positiv dargestellt, die ekliptische Anordnung der jeweils endständigen Atome einer Gruppe entspricht einem Winkel von 0°. Die Bausteine wurden analog Tab. 3.5.a) in A und B unterschieden. Die angegebenen Vergleichsdaten zeigen, dass die aus dem Modelling der Knotenvariante von 38b erhaltenen Daten recht gut mit den experimentellen Befunden übereinstimmen. Auch die hohe Rigidität der Knotengeometrie (siehe Abbildung 3.30.c) spricht für diese Variante. 3. Konformationsanalyse und Komplexierungsverhalten 92 Abbildung 3.30.c) Überlagerung von 20 lokalen Minima aus der SD-Rechnung des Knotens innerhalb eines Intervalls von 30 kJ/mol. In diesem Intervall wurden 280 Geometrien gefunden. Dargestellt sind die erste, die 280ste und dazwischen 18 weitere in regelmäßigen Energieabständen. Die Überlagerung zeigt die hohe Rigidität des Knotens: Alle überlagerten Minima haben sehr ähnliche Konformationen. Abschließend war nun noch zu überprüfen, ob im NMR ein Übergang zwischen beiden Signalsätzen identifiziert werden kann, d.h. ob die jeweils zugehörigen Bausteine in einem Molekül aneinanderhängen, und wo dieser Übergang im Molekül jeweils lokalisiert ist. Dieser Übergang konnte im ROESY-Spektrum anhand von sehr schwachen NOE-Crosspeaks zwischen C3-H des einen und den Methylprotonen an C19 des jeweils anderen Signalsatzes gefunden werden (siehe Abbildung 3.32.). Diese Crosspeaks waren zwar ausgesprochen schwach (was in Anbetracht des großen Abstandes der entsprechenden Protonen von über 4 Å 3. Konformationsanalyse und Komplexierungsverhalten 93 zu erwarten war), hoben sich aber deutlich und eindeutig vom Rauschen ab, so dass hier von echten NOE-Crosspeaks ausgegangen werden kann. Abbildung 3.32. Ausschnitt aus dem ROESY-Spektrum von 38b. Die sehr schwachen NOECrosspeaks der C19-Methylgruppen eines Signalsatzes zu den C3-Protonen des jeweils anderen Signalsatzes belegen den Übergang zwischen den Signalsätzen. Die guten Übereinstimmungen aus dem Vergleich der mittels Kraftfeldrechnung ermittelten Konformation des diskutierten Knoten-Diastereomers mit den Befunden aus verschiedenen NMR-Experimenten sind deutliche Indizien dafür, dass es sich bei der Verbindung 38b tatsächlich um den postulierten molekularen Knoten handelt (Das andere Diastereomer zeigt durch die fehlende C3-Symmetrie eine deutlich schlechtere Übereinstimmung; vgl. auch Abbildung in Anhang D). Einen endgültigen Beweis hierfür kann jedoch nur die Lösung der Kristallswruktur erbringen. Diese gelang bisher leider nicht, da die erhaltenen Kristalle trotz sehr guter Qualität ein zu geringes Streuvermögen aufwiesen und während der Messung zerfielen. Weitere Versuche zur Lösung der Kristallstruktur sind im Gange, konnten aber im Rahmen dieser Arbeit nicht mehr erfolgreich abgeschlossen werden. 4. Zusammenfassung und Ausblick 94 4. Zusammenfassung und Ausblick 4.1. Zusammenfassung Die vorliegende Arbeit ist Teil eines Forschungsprojektes mit dem Ziel, lineare und cyclische Oligopeptide zu untersuchen, die 3α-Aminocholansäuren 5 als nichtnatürliche Aminosäurebausteine enthalten. Von konkretem Interesse sind hierbei vor allem die Aspekte Selbstorganisation und Molekulare Erkennung. Im Rahmen dieser Arbeit sollten dazu in einem ersten Ansatz zunächst homologe lineare und cyclische Amid-Oligomere aus zwei verschiedenen Typen von Aminocholansäure-Bausteinen synthetisiert und hinsichtlich ihrer Konformation und exemplarisch auch auf ihr Komplexierungsverhalten hin untersucht werden. Zuerst wurde hierzu eine Synthese für die Aminocholansäuren so optimiert, dass ausgehend von den im Handel erhältlichen Cholansäuren 1 das jeweilige 3α-Epimer 5a-c in wenigen Synthesestufen selektiv, in großer Menge und ohne die Notwendigkeit chromatographischer Reinigungsschritte zugänglich wurde. Als Baustein-Typen für die Oligoamide dienten die reinen Aminocholansäuren (Typ 1) und die aminoterminal um eine natürliche Aminosäure verlängerten Aminocholansäuren (Typ 2). 4. Zusammenfassung und Ausblick 95 Die Synthese der linearen Oligoamide gelang durch Standard-Peptidkopplung mit Boc- und Methylester-Schutzgruppenchemie. Als geeignete Kopplungsreagenzien erwiesen sich DEPC und EDC, wobei u.a. aufgrund der hohen Giftigkeit von DEPC bevorzugt EDC eingesetzt wurde. Bei der Kettenverlängerung war die Wahl einer geeigneten Strategie von Bedeutung. Da der blockweise Aufbau (vgl. Kapitel 2, Schema 2.5.a) nur geradzahlige Oligomere liefert, wurde dieser Weg nur exemplarisch für die Synthese der Aminolithocholsäure-Oligoamide Boc-LCS2-OMe 13a und Boc-LCS4-OMe 14a eingeschlagen. Alle anderen linearen Oligoamide wurden durch sequentielle Kettenverlängerung (Kapitel 2, Schema 2.5.b) hergestellt. In der Peptidchemie werden Oligopeptide dabei üblicherweise aminoterminal verlängert, weil die hierzu notwendige Abspaltung der Boc-Schutzgruppe in der Regel schonend und quantitativ durch den Einsatz von Trifluoressigsäure möglich ist, während die zur carboxyterminalen Verlängerung notwendige quantitative Methylester-Spaltung ungleich schwieriger ist und nur unter erheblich drastischeren Bedingungen erreicht werden kann. Zumindest für die Aminodesoxycholsäure(DCS)- und Aminocholsäure(CHS)-haltigen Oligomere wurde in dieser Arbeit trotzdem fast ausschließlich die carboxyterminale Kettenverlängerung gewählt, da sich herausstellte, dass bei der Abspaltung der Boc-Schutzgruppe in diesen Systemen eine unerwünschte Nebenreaktion eintritt: die freien OH-Gruppen der DCS- bzw. CHS-Bausteine reagieren teilweise mit Trifluoressigsäure zu den entsprechenden Estern, wie bei der Synthese von Boc-DCS3-OMe 16a durch aminoterminale Verlängerung des entsprechenden Dimers 15a deutlich wurde. Die carboxyterminale Kettenverlängerung sollte daher auch in zukünftigen Versuchen die Methode der Wahl sein, wenn Aminocholansäuren mit freien OH-Gruppen zu Oligoamiden gekoppelt werden. Insgesamt konnten bei den einzelnen Kopplungsschritten der Oligoamid-Synthese gute bis sehr gute Ausbeuten erzielt werden. Die folgende Aufstellung zeigt, welche linearen Oligomere im einzelnen synthetisiert wurden: 4. Zusammenfassung und Ausblick Typ-1-Oligomere 96 Typ-2-Oligomere Boc-LCSn-OMe mit n = 2, 4 Boc-(Val-LCS)n-OMe mit n = 1, 2, 3 Boc-DCSn-OMe mit n = 2, 3, 4 Boc-(Lys[Z]-LCS)n-OMe mit n = 1, 2, 3 Boc-CHSn-OMe mit n = 2, 3, 4 Boc-(Val-DCS)n-OMe mit n = 1, 2, 3 Boc-(Val-CHS)n-OMe mit n = 1, 2, 3 Vom gut kristallisationsfähigen Typ-2-Monomer Boc-Val-LCS-OMe 21a konnte die Kristallstruktur gelöst werden (Abbildung 4.1.). Abbildung 4.1. Kristallstruktur des Typ-2-Monomers Boc-Val-LCS-OMe 21a. Eine systematische repetitive Sekundärstruktur bzw. besondere sekundäre Strukturelemente waren weder in den linearen Typ-1-Oligomeren noch in den Typ-2-Oligomeren identifizierbar. Der Grund hierfür liegt wahrscheinlich in der hohen Flexibilität der linearen Oligomere – sie weisen in der theoretischen Konformationsanalyse recht unterschiedliche energieähnliche Konformationen auf, wie das Beispiel des Aminodesoxycholsäure-Tetramers Boc-DCS4-OMe 17a belegt: Die Überlagerung der in einer Monte-Carlo-Rechnung gefundenen 35 Geometrien niedriger Energie zeigt aufgrund der Verschiedenartigkeit der Konformationen nur ein undifferenziertes Knäuel (Abbildung 4.2.). 4. Zusammenfassung und Ausblick 97 Abbildung 4.2. Überlagerung der 35 energiegünstigsten Konformationen (bis 30 kJ/mol über dem globalen Minimum) aus der theoretischen Konformationsanalyse (AMBER-Kraftfeld) von Boc-DCS4-OMe 17a. Das resultierende undifferenzierte Knäuel verdeutlicht die Unterschiedlichkeit der Konformationen und damit die hohe Flexibilität der Kette. Links ist das globale Minimum abgebildet. Bei der Synthese der cyclischen Oligoamide wurden zwei Synthesewege gewählt – zum einen die Oligocyclisierung von Monomeren (nur Typ 1), zum anderen die Cyclisierung linearer Oligomere (Typ 1 und Typ 2) (siehe Kapitel 2, Schema 2.6.). Die Oligocyclisierung der Typ-1-Monomere lieferte wie erwartet bei allen drei Aminocholansäuren neben dem Cyclodimerisierungsprodukt auch noch höhere Oligomere. Massenspektrometrisch konnte jeweils das Cyclotrimer und das Cyclotetramer nachgewiesen werden (siehe z.B. das FAB-Massenspektrum des Produktgemisches aus der Oligocyclisierung von 3α-Aminolithocholsäure 5a in Abbildung 4.3.). 4. Zusammenfassung und Ausblick 98 Abbildung 4.3. FAB-Massenspektrum des Produktgemisches aus der Oligocyclisierung von 3α-Aminolithocholsäure 5a. Isoliert werden konnten bei diesen Oligocyclisierungen jedoch lediglich die Cyclocholamide Cyclo-DCS2 34a, Cyclo-DCS3 34b und Cyclo-CHS2 35a. Bei der Oligocyclisierung von 3α-Aminolithocholsäure wurden keine geeigneten Chromatographiebedingungen für eine Trennung des Gemisches gefunden und die Macrocyclen Cyclo-DCS4 34c, Cyclo-CHS3 35b und Cyclo-CHS4 35c waren bei den jeweiligen Oligocyclisierungs-Reaktionen in zu geringer Menge entstanden, weshalb eine Isolierung aus dem Oligomeren-Gemisch nicht möglich war. Die Cyclooligomere 34c und 35c konnten allerdings durch Cyclisierung der entsprechenden linearen Oligomere in größerer Menge hergestellt und isoliert werden. Cyclo-CHS3 35b war jedoch auch durch Cyclisierung des entsprechenden linearen Oligoamids nicht zugänglich, da es bei dieser Reaktion nur in sehr geriger Menge entstand – Hauptprodukt dieser Cyclisierungsreaktion war das Cyclodimerisierungs-Addukt Cyclo-CHS6 35d. Auch bei der Cyclisierung des linearen DCS-Trimers konnte ein solches Cyclohexamer isoliert werden (Cyclo-DCS6, 34d), hier allerdings nur als Nebenprodukt. Zur Herstellung von Typ-2-Oligomeren wurden lediglich die linearen Trimere Boc-(Val-LCS)3-OMe 26a, Boc-(Lys[Z]-LCS)3-OMe 28a, Boc-(Val-DCS)3-OMe 30a und Boc-(Val-CHS)3-OMe 32a cyclisiert. Als direkte Cyclisierungsprodukte konnten 4. Zusammenfassung und Ausblick 99 Cyclo-(Val-LCS)3 36, Cyclo-(Lsy[Z]-LCS)3 37 und Cyclo-(Val-DCS)3 38a sowie bei der Cyclisierung von 30a auch das Cyclodimerisierungsprodukt Cyclo-(Val-DCS)6 38b isoliert und sicher identifiziert werden. Das Produkt der Cyclisierung des ValinylaminocholsäureTrimers 32a konnte nicht eindeutig als Cyclo-(Val-CHS)3 39 identifiziert werden, da bei der massenspektrometrischen Analyse kein entsprechender Massenpeak gefunden wurde. Eine weitere Untersuchung und Identifizierung der isolierten Verbindung war aufgrund ihrer schlechten Löslichkeit bislang nicht möglich. Die folgende Aufstellung gibt einen Überblick über die synthetisierten Cyclocholamide: Typ-1-Macrolactame Typ-2-Macrolactame Cyclo-LCSn mit n = 2a), 3a), 4a) Cyclo-(Val-LCS)3 Cyclo-DCSn mit n = 2, 3, 4, 6 Cyclo-(Lys[Z]-LCS)3 Cyclo-CHSn mit n = 2, 3a), 4, 6 Cyclo-(Val-DCS)n mit n = 3, 6 Cyclo-(Val-CHS)3b) a) nicht isoliert, aber massenspektrometrisch nachgewiesen. b) fraglich, ob es sich bei dem isolierten Produkt um diese Verbindung handelt. Von dem gut kristallisationsfähigen Aminodesoxycholsäure-cyclodiamid (Cyclo-DCS2) 34a konnte die Kristallstruktur gelöst werden. Es zeigte sich, dass im Kristallgitter mit jedem Molekül 34a jeweils zwei Moleküle Methanol und zwei Wassermoleküle seitlich über ein zusammenhängendes Band von Wasserstoffbrücken assoziiert sind (Abbildung 4.4.). Dabei handelt es sich gewissermaßen um eine side-on-Komplexierung. Die Konformation des Aminodesoxycholsäure-cyclodiamids 34a im Kristall entspricht dabei in etwa der eines lokalen Minimums aus einer Monte-Carlo-Suche im AMBER-Kraftfeld, das um 8.2 kJ/mol über dem gefundenen globalen Minimum liegt (Abbildung 4.4.). 4. Zusammenfassung und Ausblick 100 Abbildung 4.4. Kristallstruktur von Cyclo-DCS2 34a mit seitlich über Wasserstoffbrückenbindungen assoziierten Lösungsmittelmolekülen (links) und lokales Minimum (8.2 kJ/mol über dem globalen Minimum) mit vergleichbarer Konformation aus einer Monte-Carlo-Suche im AMBER-Kraftfeld (rechts). Während der Hohlraum des Cyclodiamids 34a noch zu klein war, um ein Gastmolekül aufzunehmen, zeigte eine Modellingstudie, dass der Hohlraum des Aminodesoxycholsäurecyclotriamids 34b hinreichend groß ist, um ein Glucosemolekül zu komplexieren. Tatsächlich konnte durch NMR-Experimente gezeigt werden, dass Cyclo-DCS3 34b das DodecylGlucosid 40 komplexiert (Abbildung 4.5.). 4. Zusammenfassung und Ausblick 101 Abbildung 4.5. 1H-NMR-Spektrum (600 MHz) eines 1:1-Gemisches aus Cyclo-DCS3 34b und n-Dodecyl-β-Glucosid 40 (jeweils 10 mM in CDCl3). Zum Vergleich sind auch die Spektren der reinen Verbindungen aufgeführt. Eine Besonderheit konnte in den NMR-Spektren der Typ-2-Cyclotrimere Cyclo-(Val-LCS)3 36, Cyclo-(Lys[Z]-LCS)3 37 und Cyclo-(Val-DCS)3 38a gefunden werden: Das NH-Proton des Steroid-Bausteins ist jeweils breit und deutlich tieffeldverschoben (Abbildung 4.6.), was auf eine Beteiligung dieses Protons an Wasserstoffbrückenbindungen hindeutet. Bevorzugt sollten diese in γ-loops lokalisiert sein, wie eine Modellingstudie zeigt. Die Breite des Signals deutet in Verbindung mit der unscharfen Aufspaltung der anderen Signale darauf hin, dass das NMR ein gemitteltes Spektrum verschiedener sich dynamisch ineinander umwandelnder Konformationen abbildet. Eine NH-Signalverbreiterung könnte prinzipiell aber auch aus einem schnellen Wasserstoffaustausch resultieren. Für eine genaue Klärung wären hier weitere Experimente notwendig. 4. Zusammenfassung und Ausblick 102 Abbildung 4.6. 1H-NMR-Spektrum (400 MHz, CDCl3) von Cyclo-(Val-DCS)3 38a. Das NH-Signal des Aminodesoxycholsäure-Bausteins ist tieffeldverschoben und breit. Die beiden Cyclen Cyclo-(Val-LCS)3 36 und Cyclo-(Lys[Z]-LCS)3 37 zeigen analoge Spektren. Bei der Cyclisierung des linearen Typ-2-Trimers Boc-(Val-DCS)3-OMe 30a wurde neben dem direkten Cyclisierungsprodukt 38a noch eine Verbindung 38b mit doppelter Masse isoliert, welche durch NMR-Experimente in Kombination mit Molecular-ModellingMethoden relativ sicher als eines von zwei möglichen Knotan-Diastereomeren identifiziert werden konnte (Abbildung 4.7.a und b). Alternative Varianten mit anderer Topologie wie ein einfacher Cyclus und ein Catenan konnten dabei weitgehend ausgeschlossen werden. Auch das andere mögliche Knotan-Diastereomer zeigte keine so gute Übereinstimmung mit den experimentellen Ergebnissen. Ein endgültiger Beweis dafür, dass Verbindung 38b tatsächlich ein Knotan ist, konnte allerdings bislang nicht erbracht werden. Hierfür wäre die Lösung der Kristallstruktur erforderlich, welche jedoch bisher noch nicht gelang. Die Verbindung bleibt daher bis zur endgültigen Aufklärung der Topologie Gegenstand weiterer Untersuchungen. Abbildung 4.7.a) Mögliche Diastereomere von Knotanen der Verbindung 38b mit sich wiederholenden Bausteinen definierter Stereochemie (L-Aminosäuren, Steroid-Gerüst) und vorgegebener Bindungsrichtung (durch Pfeile gekennzeichnet) (siehe hierzu auch Anhang D). 4. Zusammenfassung und Ausblick 103 Abbildung 4.7.b) Globales Minimum des wahrscheinlich bei Verbindung 38b vorliegenden Knoten-Diastereomers aus einer Modelling-Studie. Das andere Diastereomer zeigt keine C3-Symmetrie und liegt einergetisch um 13,82 kJ/mol höher (siehe Anhang D). 4.2. Ausblick In der vorliegenden Arbeit wurde eine Auswahl homologer linearer und cyclischer Oligoamide mit Aminocholansäure-Bausteinen vorgestellt. Vor allem die hier synthetisierten cyclischen Systeme zeigen vielversprechende Ansätze hinsichtlich der im Forschungsprojekt interessierenden Aspekte Selbstorganisation und Molekulare Erkennung. Deshalb sollten weitere Arbeiten auf dem Gebiet der Aminocholansäurehaltigen Oligoamide folgen. Zunächst wäre es hier interessant, nichthomologe Oligoamide, also solche mit nichtrepetitiven Sequenzen, zu synthetisieren und zu untersuchen. Dabei ist auch die Einführung von zusätzlicher Funktionalität, sowohl über Seitenketten natürlicher L-Aminosäuren als auch 4. Zusammenfassung und Ausblick 104 über an C7 und/oder C12 modifizierte Aminocholansäure-Bausteine von Bedeutung. Beispielhaft war eine solche Weiterfunktionalisierung mit der Synthese homologer Lysinhaltiger Oligoamide in der vorliegenden Arbeit bereits realisiert worden. Solche gezielt funktionalisierten Systeme sollten gerade im Bereich der molekularen Erkennung, möglicherweise auch als künstliche Enzyme, ein hohes Potential haben, besonders hinsichtlich ihrer Selektivität bezüglich ausgewählter Substrate. Insbesondere im Hinblick auf die effiziente und systematische Permutation von Sequenzen mit kombinatorischen Methoden und auf ein systematisches Screening auf Sequenzen mit besonders guten Komplexierungseigenschaften für bestimmte Gastmoleküle sollte dann in einem nächsten Schritt eine Festphasensynthese von (linearen und cyclischen) Oligoamiden mit Aminocholansäuren etabliert werden. Insgesamt bietet der Einsatz von Aminocholansäure-Bausteinen eine Vielfalt an Perspektiven. Die folgende Übersicht zeigt beispielhaft die genannten Möglichkeiten auf: 5. Experimenteller Teil 105 5. Experimenteller Teil 5.1. Allgemeines 5.1.1. Bemerkungen Die für die Synthesen verwendeten α-Aminosäuren wurden in der natürlichen L-Form eingesetzt. Alle verwendeten Lösungsmittel wurden nach Standardmethoden getrocknet und gereinigt bzw. als p.a.-Ware direkt eingesetzt. Soweit nicht ausdrücklich erwähnt, wurden alle Reaktionen ohne Schutzgasatmosphäre durchgeführt. Bei den dargestellten Substanzen handelt es sich, falls nichts anderes angegeben wurde, um farblose bis schwach gelbe Feststoffe. Die EI- und FAB-Massenspektren wurden auf einem Match 5 Auto-Spec Massenspektrometer der Firma Varian aufgenommen. M.A.L.D.I.-Spektren wurden mit dem Gerät VOYAGER DE der Firma RP Biospectrometry gemessen. Kernresonanzspektren wurden (soweit nicht anders erwähnt bei Raumtemperatur) mit den Geräten DPX200 (200 MHz), DRX400 (400 MHz) bzw. DRX600 (600 MHz) der Firoa Bruker aufgenommen. Die chemischen Verschiebungen beziehen sich dabei auf DMSO-d6 (2.49 ppm) bzw. CDCl3 (7.24 ppm) als interner Standard. und sind als δ-Werte in ppm relativ zu TMS (0.0 ppm) angegeben. Zur Charakterisierung der Aufspaltungsmuster wird der übliche Abkürzungscode verwendet: s = Singulett, d = Dublett, t = Triplett, q = Quartett; m = Multiplett, dd = Dublett von Dubletts, dt = Dublett von Tripletts, (br) = breit, (Fk) = Feinkopplung, etc. Bei 1H-NMR-Spektren von Substanzen mit Cholansäuregerüst wurden nur charakteristische Signale zugeordnet (i. d. R. Peaks > 2 ppm sowie die Methylgruppen des Steroidgerüstes). Analytische Dünnschichtchromatographie (DC) wurde auf kieselgelbeschichteten Aluminiumfolien der Firma Merck (Kieselgel 60 F254, d = 0.25 mm) durchgeführt. Zur Detektion wurde Molybdatophosphorsäure/Cer(IV)-Sulfat-Lösung [lit] verwendet. 5. Experimenteller Teil 106 Präparative säulenchromatographische Trennungen wurden, soweit nicht anders erwähnt, unter folgenden Standardbedingungen durchgeführt: Als Trennsäule diente eine 50 cm lange Glassäule (Ø 1 cm), für die jeweils ca. 40-41 g FlashKieselgel (Silica 32-63 mesh) der Firma ICN verwendet wurden; die Trennung erfolgte unter geringem Druck, der durch eine herkömmliche Aquariumpumpe der Firma Schego (Typ M2K 3) erzeugt wurde, mit den jeweils angegebenen Laufmittelgemischen. Die Röntgenstrukturanalysen erfolgten an einem automatischen Vierkreisdiffraktometer vom Typ P4 der Firma Siemens (Verbindungen 6b und 21a) bzw. an einem mit Flächendetektor ausgerüsteten Dreikreisdiffraktometer vom Typ axs CCD 1000 der Firma Bruker (Verbindung 34a) jeweils mit monochromatisierter Mo-Kα-Strahlung. 5. Experimenteller Teil 5.1.2. Abkürzungen AAV abs. Ac aCN Ac2O AcOH AS ber. Boc Boc2O Bz CDMT CH2Cl2 conc. DCC DCHA DCM DEPC DHB DIEA DMF DMSO EDC EE Et2O EtOH eq. Fmoc ges. h HOBt min NMM MeOH PE RT T3P TBME TEA TFA THF TMS verd. Z allgemeine Arbeitsvorschrift absolut Acetyl α-Cyano-Zimtsäure (Matrix für MALDI-Experimente) Essigsäureanhydrid Essigsäure Aminosäure berechnet tert-ButyloxycarbonylDi-tert-Butyldicarbonat Benzyl 2-Chlor-4,6-dimethoxy-1,3,5-triazin Dichlormethan konzentriert N,N‘-Dicyclohexylcarbodiimid Dicyclohexylamin Dichlormethan Diethylcyanophosphonat Dihydroxybenzoesäure (Matrix für MALDI-Experimente) Diiopropylethylamin N,N‘-Dimethylformamid Dimethylsulfoxid N-(3-Dimethylaminopropyl)-N‘-ethyl-carbodiimid-hydrochlorid Essigsäureethylester Diethylether Ethanol Äquivalent(e) 9-Fluorenylmethoxycarbonyl gesättigt Stunde(n) 1-Hydroxybenzotriazol Minute(n) N-Methylmorpholin Methanol Petrolether Raumtemperatur Propanphosphonsäureanhydrid tert-Butylmethylether Triethylamin Trifluoressigsäure Tetrahydrofuran Tetramethylsilan verdünnt Benzyloxycarbonyl- 107 5. Experimenteller Teil 108 5.2. Präparative Vorschriften 5.2.1. Darstellung der 3α α-Aminocholansäuren 5.2.1.1. 3-Oxo-Cholansäuren 6a-c 5.2.1.1.1. 3-Oxo-Lithocholsäure 6a25 20.5 g (54.4 mmol) Lithocholsäure werden in einem Gemisch aus 400 ml Essigsäure und 400 ml Aceton gelöst und die Lösung mit einem Eisbad gekühlt. Unter Rühren wird anschließend Jones-Reagenz (hergestellt aus 7.00 g (70.0 mmol) CrO3, 7 ml conc. H2SO4 und 20 ml H2O) so zugetropft, daß die Temperatur des Reaktionsgemisches bei 0-5 °C bleibt. Danach wird noch weitere 30 min bei 0 °C weitergerührt. Überschüssiges Cr(VI) wird durch Zugabe von 30 ml MeOH und anschließendes 15 min Rühren im Eisbad reduziert. Die Reaktionsmischung wird dann in einen Scheidetrichter überführt, mit 700 ml Wasser verdünnt und mit Chloroform (3x 250 ml) extrahiert. Die vereinigten organischen Phasen werden mit ges. NaCl-Lösung (250 ml) und Wasser (250 ml) gewaschen und anschließend am Rotationsverdampfer eingeengt. Das ölige, noch stark nach Essigsäure riechende Rohprodukt wird aus MeOH/Wasser auskristallisiert. Ausbeute: 19.04 g (50.8 mmol; 93.4 % d. Theorie) HR-MS (FAB): 1 für C24H38O3Na+ 397.270500 ber. 397.271865 H-NMR (CDCl3, 200 MHz): 2.67 (dd, 1H, C4-Hax); 2.46-2.11 (2m, 2H, CH2-23); 2.02, 1.96 (2m, 4H, CH2-2 u. CH2-4); 0.99 (s, 3H, CH3-19); 0.91 (d, 3H, CH3-21); 0.66 (s, 3H, CH3-18) 5. Experimenteller Teil 109 5.2.1.1.2. 3-Oxo-Cholsäure 6b und 3-Oxo-Desoxycholsäure 6c 5.2.1.1.2.1. Performyloxylierung26 50 g der Cholansäure (Desoxycholsäure: 127.4 mmol; Cholsäure: 122.4 mmol) werden in 200 ml Ameisensäure gelöst und mit 40 Tropfen HClO4 (70-72 %ig) versetzt. Die Lösung wird anschließend für 1.5 h bei 55-60 °C gerührt. Danach läßt man auf ca. 40 °C abkühlen und tropft langsam Ac2O zu, bis eine starke Blasenbildung erkennbar wird; während des Zutropfens sollte die Temperatur 55-60 °C nicht übersteigen. Das Reaktionsgemisch wird anschließend auf RT abgekühlt und unter Rühren in 1.5 l Wasser gegossen. Das Präzipitat wird abgesaugt, in Chloroform aufgenommen (500 ml) und die Chloroformphase einige Male mit Wasser gewaschen. Anschließend wird die organische Phase am Rotationsverdampfer eingeengt, wobei das Produkt jeweils in quantitativer Ausbeute (3,12-DiformyloxyDesoxycholsäure 7b: 57.2 g; 3,7,12-Triformyloxy-Cholsäure 7c: 60.3 g) auskristallisiert. Die performyloxylierten Cholansäuren werden ohne weitere Aufreinigung eingesetzt. 3,12-Diformyloxy-Desoxycholsäure 7b: HR-MS (FAB): für C26H40O6Na+ 471.271200 ber. 471.272259 1 H-NMR (CDCl3, 400 MHz): 8.11 (s, 1H, CH-Formyloxy); 8.01 (d(Fk), 1H, CH-Formyloxy); 5.23 (m, 1H, CH-12); 4.82 (m(br), 1H, CH-3); 2.40-2.33 u. 2.26-2.18 (2m, 2H, CH2-23); 0.91 (s, 3H, CH3-19); 0.82 (d, 3H, CH3-21); 0.73 (s, 3H, CH3-18) 3,7,12-Triformyloxy-Cholsäure 7c: HR-MS (FAB): 1 für C27H40O8Na+ 515.262000 ber. 515.262088 H-NMR (CDCl3, 400 MHz): 8.14 (s, 1H, CH-Formyloxy); 8.08 (s, 1H, CH-Formyloxy); 8.00 (d(Fk), 1H, CH-Formyloxy); 5.24 (m, 1H, CH-12); 5.05 (m, 1H, CH-7); 4.69 (m(br), 1H, CH-3); 2.40-2.32 u. 2.26-2.18 (2m, 2H, CH2-23); 0.92 (s, 3H, CH3-19); 0.83 (d, 3H, CH3-21); 0.74 (s, 3H, CH3-18) 5. Experimenteller Teil 110 5.2.1.1.2.2. Selektives Entschützen der Hydroxylgruppe an C325 Die komplette Ausbeute an performyloxylierter Cholansäure aus Versuch 5.2.1.1.2.1. (3,12Diformyloxy-Desoxycholsäure 7b: 57.2 g, 127.4 mmol; 3,7,12-Triformyloxy-Cholsäure 7c: 60.3 g, 122.4 mmol) wird in Aceton (625 ml) gelöst. Unter Rühren wird nun bei RT innerhalb von 2-2.5 h 0.2M NaOH zugetropft (Herstellung für 3,12-Diformyloxy-Desoxycholsäure: 10.25 g NaOH in 1.30 l Wasser; für 3,7,12-Triformyloxy-Cholsäure: 9,95 g NaOH in 1.25 l Wasser). Nach weiteren 0.5 h bei RT wird das Reaktionsgemisch in einen Scheidetrichter überführt, mit 0.5M HCl angesäuert (ca. 500-520 ml) und anschließend mit Chloroform extrahiert (500 ml, dann 2x 250 ml). Die vereinigten Chloroformextrakte werden 3x mit Wasser gewaschen (je 250 ml) und anschließend am Rotationsverdampfer eingeengt. Da die Auswaage für 12-Formyloxy-Desoxycholsäure 8b und 7,12-Diformyloxycholsäure 8c auch nach sehr intensiver Trocknung noch über 100 % liegt (wahrscheinlich aufgrund eingeschlossener Chloroformreste), wird hier jeweils von einer quantitativen Ausbeute ausgegangen. Die Produkte wurden ohne weitere Aufreinigung weiterverwendet. 12-Formyloxy-Desoxycholsäure 8b: HR-MS (FAB): für C25H40O5Na+ 443.276600 ber. 443.277344 1 H-NMR (CDCl3, 400 MHz): 8.09 (s, 1H, CH-Formyloxy); 5.22 (m, 1H, CH-12); 3.60 (m(br), 1H, CH-3); 2.40-2.32 u. 2.25-2.17 (2m, 2H, CH2-23); 0.88 (s, 3H, CH3-19); 0.81 (d, 3H, CH3-21); 0.72 (s, 3H, CH3-18) 7,12-Diformyloxy-Cholsäure 8c: HR-MS (FAB): 1 für C26H40O7Na+ 487.268800 ber. 487.267174 H-NMR (CDCl3, 400 MHz): 8.12 (s, 1H, CH-Formyloxy); 8.07 (s, 1H, CH-Formyloxy); 5.24 (m, 1H, CH-12); 5.04 (m, 1H, CH-7); 3.48 (m(br), 1H, CH-3); 2.39-2.32 u. 2.25-2.17 (2m, 2H, CH2-23); 0.90 (s, 3H, CH3-19); 0.83 (d, 3H, CH3-21); 0.73 (s, 3H, CH3-18) 5. Experimenteller Teil 111 5.2.1.1.2.3. Oxidation an C325 Die komplette Ausbeute des Produktes aus Versuch 5.2.1.1.2.2. (12-FormyloxyDesoxycholsäure 8b: 127.4 mmol; 7,12-Diformyloxy-Cholsäure 8c: 122.4 mmol) wird in 500 ml Aceton gelöst und mit einem Eisbad gekühlt. Unter Rühren wird nun Jones-Reagenz (Herstellung für 12-Formyloxy-Desoxycholsäure: 16.25g CrO3 + 45 ml Wasser + 15.0 ml conc. H2SO4 ; für 7,12-Diformyloxy-Cholsäure: 15.75g CrO3 + 45 ml Wasser + 14.6 ml conc. H2SO4) so zugetropft, daß die Temperatur des Reaktionsgemisches nicht über 10 °C ansteigt. Danach wird noch weitere 30 min unter Eiskühlung weitergerührt, anschließend 150 ml MeOH zugegeben und nochmals für 15 min gerührt. Das Reaktionsgemisch wird dann in einen Scheidetrichter überführt, mit 1 l Wasser verdünnt und mit Chloroform extrahiert (4x 250 ml). Die vereinigten Chloroformphasen werden mit verd. NaCl-Lösung (2x 250 ml) und Wasser (1x 250 ml) gewaschen und anschließend am Rotationsverdampfer eingeengt. Da die Auswaage für 12-Formyloxy-3-oxo-Desoxycholsäure 9b und 7,12-Diformyloxy-3oxo-cholsäure 9c auch nach sehr intensiver Trocknung noch über 100 % liegt (wahrscheinlich aufgrund eingeschlossener Chloroformreste), wird hier jeweils von einer quantitativen Ausbeute ausgegangen. Die Produkte werden ohne weitere Aufreinigung weiterverwendet. 12-Formyloxy-3-oxo-Desoxycholsäure 9b: HR-MS (FAB): für C25H38O5Na+ 441.262100 ber. 441.261694 1 H-NMR (CDCl3, 400 MHz): 8.10 (s, 1H, CH-Formyloxy); 5.27 (m, 1H, CH-12); 2.65 (dd, 1H, C4-Hax); 2.40-2.32 u. 2.26-2.18 (2m, 2H, CH2-23); 0.99 (s, 3H, CH3-19); 0.83 (d, 3H, CH3-21); 0.76 (s, 3H, CH3-18) 7,12-Diformyloxy-3-oxo-Cholsäure 9c: HR-MS (FAB): 1 für C26H38O8Na+ 485.252500 ber. 485.251524 H-NMR (CDCl3, 400 MHz): 8.13 (s, 1H, CH-Formyloxy); 8.07 (s, 1H, CH-Formyloxy); 5.28 (m, 1H, CH-12); 5.13 (m, 1H, CH-7); 2.98 (dd, 1H, C4-Hax); 2.40-2.32 (m, 1H, 1CH2-23); 1.02 (s, 3H, CH3-19); 0.84 (d, 3H, CH3-21); 0.78 (s, 3H, CH3-18) 5. Experimenteller Teil 112 5.2.1.1.2.3. Entschützen der übrigen Hydroxylgruppen25 Die Formyloxygeschützte 3-Oxo-Cholansäure (komplette Ausbeute aus Versuch 5.2.1.1.2.2.; 12-Formyloxy-3-oxo-Desoxycholsäure 9b: 127.4 mmol; 7,12-Diformyloxy-3-oxo-cholsäure 9c: 122.4 mmol) wird in 1.25 l 0.5M NaOH gelöst und für 15-20 min unter Rühren im heißen Wasserbad erhitzt. Danach rührt man noch 30-45 min bei RT. Nach Überführen des Reaktionsgemisches in einen Scheidetrichter wird mit 1M HCl angesäuert (ca. 625 ml) und mit Chloroform extrahiert (500 ml, 2x 250 ml). Die vereinigten organischen Phasen werden mit NaHCO3 verd. (300 ml) und Wasser (250 ml) gewaschen. Anschließend wird das Lösungsmittel am Rotationsverdampfer entfernt, wobei die jeweilige 3-Oxo-Cholansäure vollständig auskristallisiert. Da die Auswaage für 3-Oxo-Desoxycholsäure 6b (51.02 g, 102.5 %) und 3-Oxo-Cholsäure 6c (49.96 g, 100.4 %) auch nach sehr intensiver Trocknung noch über 100 % liegt (wahrscheinlich aufgrund eingeschlossener Chloroformreste), wird hier jeweils von einer quantitativen Ausbeute ausgegangen. Die Produkte werden ohne weitere Aufreinigung weiterverwendet. 3-Oxo-Desoxycholsäure 6b: Die Kristallstrukturdaten sind in Anhang A aufgeführt. HR-MS (EI): für C24H38O4 390.276400 ber. 390.277010 1 H-NMR (CDCl3, 600 MHz): 4.03 (m, 1H, CH-12); 2.70 (dd, 1H, C4-Hax); 2.39 u. 2.27 (2m, 2H, CH2-23); 0.99 (s, 3H, CH3-19); 0.98 (d, 3H, CH3-21); 0.71 (s, 3H, CH3-18) 3-Oxo-Cholsäure 6c: HR-MS (FAB): 1 für C24H38O5Na+ 429.261600 ber. 429.261694 H-NMR (CDCl3, 400 MHz): 4.02 (m, 1H, CH-12); 3.90 (m, 1H, CH-7); 3.36 (dd, 1H, C4-Hax); 0.97 (s, 3H, CH3-19); 0.97 (d, 3H, CH3-21); 0.71 (s, 3H, CH3-18) 5. Experimenteller Teil 113 5.2.1.2. Cholansäure-3-oxime 10a-c (Allgemeine Vorschrift)23 50.0 mmol der 3-Oxo-Cholansäure werden in 175 ml MeOH gelöst. Nach Zugabe von 8.46 g NH2OH·HCl (120 mmol) und 16.53 g NaOAc wasserfrei (200 mmol) wird das Reaktionsgemisch unter Rühren für 2.5 h refluxiert, danach mit 200 ml MeOH verdünnt und in einen Scheidetrichter überführt. Man versetzt nun mit 400 ml Wasser und extrahiert mit Chloroform (500 ml, dann 2x 250 ml). Die vereinigten organischen Phasen werden mit Wasser (2x 200 ml) gewaschen und anschließend am Rotationsverdampfer eingeengt. Das Rohprodukt kann durch Umkristallisation gereinigt werden. Lithocholsäure-3-oxim 10a: Ansatzgröße: 19.04 g (50.8 mmol) 3-Oxo-Lithocholsäure 6a Das Rohprodukt wird aus MeOH/TBME umkristallisiert. Ausbeute: 18.29 g (46.9 mmol, 92.4 % der Theorie) HR-MS (EI): für C24H40NO3+ 390.300500 ber. 390.300820 1 H-NMR (DMSO-d6, 400 MHz): 11.89 (s(br), 1H, COOH); 10.02 (s, 1H, N=OH); 2.76 (dd, 1H, CHeq-4); 2.26-2.18 u. 2.13-2.07 (2m, 2H, CH2-23); 0.91 (s, 3H, CH3-19); 0.87 (d, 3H, CH3-21); 0.62 (s, 3H, CH3-18) Desoxycholsäure-3-oxim 10b: Ansatzgröße: 127.4 mmol 3-Oxo-Desoxycholsäure 6b (Ausbeute aus 5.2.1.1.2.3.) Das Rohprodukt wird aus MeOH/TBME umkristallisiert. Ausbeute: 42.75 g (105.4 mmol, 82.7 % der Theorie) HR-MS (FAB): 1 für C24H39NO4+ 405.288600 ber. 405.287909 H-NMR (DMSO-d6, 400 MHz): 11.88 (s(br), 1H, COOH); 10.01 (s, 1H, N=OH); 4.14 (d, 1H, C12-OH); 3.79 (m, 1H, CH-12); 2.76 (dd, 1H, CHeq-4); 2.25-2.18 u. 2.13-2.04 (2m, 2H, CH2-23); 0.91 (d, 3H, CH3-21); 0.89 (s, 3H, CH3-19); 0.61 (s, 3H, CH3-18) 5. Experimenteller Teil 114 Cholsäure-3-oxim 10c: Ansatzgröße: 122.4 mmol 3-Oxo-Cholsäure 6c Das Rohprodukt wird aus MeOH/TBME umkristallisiert. Ausbeute: 38.42 g (91.1 mmol, 74.4 % der Theorie) HR-MS (FAB): für C25H40NO5+ 422.291900 ber. 422.290649 1 H-NMR (DMSO-d6, 400 MHz): 11.88 (s(br), 1H, COOH); 9.85 (s, 1H, N=OH); 4.18 (d, 1H, C7-OH); 4.10 (d, 1H, C12-OH); 3.79 (m, 1H, CH-12); 3.65 (m, 1H, CH-7); 2.84 (dd, 1H, CHeq-4); 2.73 (dd, 1H, CHax-4); 0.92 (d, 3H, CH3-21); 0.86 (s, 3H, CH3-19); 0.60 (s, 3H, CH3-18) 5.2.1.3. 3α α-Aminocholansäuren (Allgemeine Vorschrift)23, 24 50.0 mmol Cholansäure-3-oxim 10a-c werden unter Erhitzen in 1.2 l Isoamylalkohol gelöst. Zu dem refluxierenden Reaktionsgemisch werden unter starkem Rühren nach und nach 61 g Natrium in kleinen Stücken gegeben. Es werden noch 250 ml Amylalkohol zugegeben und man läßt dann auf RT abkühlen. Das Reaktionsgemisch wird in einen Scheidetrichter überführt und mit 3.0 l Et2O verdünnt. Man extrahiert die Organische Phase mit kleinen Portionen Wasser (jeweils 50-150 ml), bis das Waschwasser neutral reagiert (Die Gesamtmenge an Wasser sollte so gering wie möglich gehalten werden und 1-2 l nicht überschreiten). Die vereinigten wäßrigen Phasen werden dann mit 2M H2SO4 neutralisiert, wobei die Aminocholansäure präzipitiert (Zur Beachtung: 3α-Aminodesoxycholsäure 5b präzipitiert bereits bei einem pH-Wert von etwa 9-11 und geht bei weiterer Säurezugabe wieder in Lösung). Das Präzipitat wird abgesaugt und mehrmals mit Wasser, dann mit Aceton gewaschen. Zur Reinigung wird das Präzipitat mit einer großen Menge Methanol nach und nach aus der Fritte gelöst. Beim Einengen auf ca. 200 ml fällt die jeweilige Aminocholansäure als sehr feiner weißer Niederschlag aus, der abgesaugt, mit wenig Aceton gewaschen und getrocknet wird. 5. Experimenteller Teil 115 3α-Aminolithocholsäure 5a: Ansatzgröße: 18.20 g (46.7 mmol) Lithocholsäure-3-oxim 10a Ausbeute: 12.18 g (32.4 mmol, 69.4 % der Theorie) HR-MS (FAB): für C24H41NO2+ 375.313600 ber. 375.313730 H-NMR (als TFA-Salz, DMSO-d6, 400 MHz): 7.76 (m(br), 3H, NH3+); 2.99 (m(br), 1H, CHβ-3); 2.25-2.18 (m, 1H, 1CH2-23); 2.13-2.05 (m, 1H, 1CH2-23); 0.89 (s, 3H, CH3-19); 0.86 (d, 3H, CH3-21); 0.61 (s, 3H, CH3-18) 1 3α-Aminodesoxycholsäure 5b: Ansatzgröße: 21.00 g (51.8 mmol) Desoxycholsäure-3-oxim 10b Ausbeute: 14.66 g (37.5 mmol, 72.4 % der Theorie) HR-MS (FAB): für C24H42NO3+ 392.314600 ber. 392.316470 H-NMR (als TFA-Salz, DMSO-d6, 400 MHz): 7.76 (m(br), 3H, NH3+); 3.81 (m, 1H, CH-12); 2.97 (m(br), 1H, CHβ-3); 2.25-2.17 (m, 1H, 1CH2-23); 2.13-2.05 (m, 1H, 1CH2-23); 0.91 (d, 3H, CH3-21); 0.87 (s, 3H, CH3-19); 0.60 (s, 3H, CH3-18) 1 3α-Aminocholsäure 5c: Ansatzgröße: 19.00 g (45.1 mmol) 3-Oxo-Cholsäure 10c Ausbeute: 10.96 g (26.9 mmol, 59.6 % der Theorie) HR-MS (FAB): für C24H42NO4+ 408.310000 ber. 408.311384 H-NMR (als TFA-Salz, DMSO-d6, 400 MHz): 7.75 (m(br), 3H, NH3+); 3.80 (m, 1H, CH-12); 3.63 (m, 1H, CH-7); 2.78 (m(br), 1H, CHβ-3); 0.92 (d, 3H, CH3-21); 0.85 (s, 3H, CH3-19); 0.59 (s, 3H, CH3-18) 1 5. Experimenteller Teil 116 5.2.2. Peptidsynthesen mit Aminocholansäure-Bausteinen 5.2.2.1. Allgemeine Arbeitsvorschriften (AAV) 5.2.2.1.1. AAV I: Darstellung von Methylesterhydrochloriden50 20 mmol der Säurekomponente werden in 200 ml MeOH suspendiert und auf 0 °C abgekühlt. Unter Rühren werden nun 4 ml Thionylchlorid zugetropft und das Reaktionsgemisch anschließend für 2-3 h unter Reflux zum Sieden erhitzt. Das Reaktionsgemisch wird dann über Nacht bei RT weitergerührt und anschließend im Vakuum eingeengt. Überschüssiges Thionylchlorid wird entfernt, indem der Rückstand einige Male in Methanol aufgenommen und wieder einrotiert wird. 5.2.2.1.2. AAV II: Verseifung von Methylestern 1 mmol des Methylesters wird in 60 ml MeOH gelöst und unter Rühren mit 5 ml 1M KOH versetzt (Falls der Methylester sich nicht vollständig löst oder nach Zugabe der 1M KOH ausfällt, kann er durch Zugabe geringer Mengen THF in Lösung gebracht werden). Es wird 2 Tage bei RT gerührt und das Lösungsmittel anschließend im Vakuum entfernt. Der Rückstand wird in Wasser aufgenommen und bis zur vollständigen Auflösung mit THF versetzt. Anschließend säuert man mit 1M HCl an und extrahiert (je nach Löslichkeit der freien Säure) mit EE (150 ml, dann 50 ml) oder CHCl3 (150 ml, dann 50 ml). Die vereinigten organischen Phasen werden mit Wasser und ges. NaCl gewaschen, über Natriumsulfat getrocknet und einrotiert. Anmerkung: Diese Vorschrift ist optimiert für Pseudopeptide, deren Carboxyterminus eine Methylestergeschützte Aminocholansäure ist. Eine Zugabe geringerer Mengen KOH führt bei diesen Systemen trotz langer Reaktionszeiten oft nicht zu einer vollständigen Verseifung. Für andere Systeme sollte bei dieser Vorschrift zunächst mit 1.1 Moläquivalenten KOH und kürzeren Reaktionszeiten (ca. 5 h) gearbeitet werden. 5. Experimenteller Teil 117 5.2.2.1.3. AAV III: Einführung der N-terminalen Boc-Schutzgruppe51 10 mmol der Aminokomponente und 0.84 g (10 mmol) NaHCO3 werden in 10 ml Wasser suspendiert. Unter Rühren gibt man eine Lösung von 2.40 g (11 mmol) Boc2O in 20 ml Dioxan zu. Das Reaktionsgemisch wird über Nacht bei RT gerührt, anschließend unter Eiskühlung mit 1M HCl angesäuert und mit EE (3x 100 ml) extrahiert. Die vereinigten organischen Phasen werden je einmal mit verd. NaHCO3 und Wasser gewaschen, über Natriumsulfat getrocknet und anschließend eingeengt. 5.2.2.1.4. AAV IV: Abspaltung der Boc-Schutzgruppe mit TFA52 Das Boc-geschützte (Pseudo-)Peptid (0.25 mmol) wird in 15 ml CH2Cl2 gelöst und unter Rühren mit 7.5 ml TFA versetzt. Es wird für 1-24 h bei RT gerührt (Bei Vorhandensein anderer säurelabiler Schutzgruppen wie z.B. der Z-Schutzgruppe wird für 30 min bei 0°C gerührt) und das Lösungsmittel anschließend im Vakuum entfernt. TFA-Reste werden durch mehrmaliges Aufnehmen des Rückstandes in CH2Cl2 und anschließendes Einrotieren entfernt. Das Produkt kann dann ohne weitere Aufarbeitung direkt weiterverwendet werden. 5.2.2.1.5. AAV V: DEPC-Peptidkopplung53 Zu einer Lösung von 1 mmol Aminokomponente und 1 mmol Carbonsäure-Komponente in 12 ml DMF werden unter Rühren bei -15 °C 1 ml TEA gegeben und anschließend 0.5 ml DEPC, gelöst in weiteren 0.5 ml TEA, zugetropft. Das Reaktionsgemisch wird 2 Tage bei RT gerührt. Die Lösung wird mit Wasser versetzt bis zur vollständigen Präzipitation des Kopplungsproduktes, welches anschließend abfiltriert und mit einigen Portionen Wasser gewaschen wird. Das Produkt wird in EE (30-50 ml) aufgenommen und die organische Lösung nacheinander mit 5%iger KHSO4-Lösung, ges. NaHCO3-Lösung und ges. NaClLösung gewaschen. Nach Trocknung der organischen Phase mit Na2SO4 wird das Lösungsmittel im Vakuum verdampft. 5. Experimenteller Teil 118 5.2.2.1.6. AAV VI: EDC-Peptidkopplung54 Zu einer Lösung von 1 mmol Aminokomponente und 1 mmol Carbonsäure-Komponente in 30 ml CH2Cl2 werden unter Rühren zunächst 1 ml TEA und anschließend 287 mg EDC und 203 mg HOBt (je 1.5 mmol) zugegeben. Das Reaktionsgemisch wird 2 Tage bei RT gerührt und dann in EE (100 ml) aufgenommen. Die organische Lösung wird nacheinander mit 5%iger KHSO4-Lösung, ges. NaHCO3-Lösung und ges. NaCl-Lösung gewaschen. Nach Trocknung der organischen Phase mit Na2SO4 wird das Lösungsmittel im Vakuum verdampft. 5.2.2.1.7. AAV VII: (Oligo-)Cyclisierungsreaktion28 1 mmol der zu cyclisierenden, Boc-geschützten Peptidkomponente wird in 10 ml CH2Cl2 gelöst und mit 1.5 mmol Pentafluorphenol versetzt. Nach Abkühlen auf -20 °C wird unter Rühren eine Lösung von 1.25 mmol DCC in 5 ml CH2Cl2 zugetropft. Das Reaktionsgemisch wird über Nacht bei RT gerührt, wonach der ausgefallene Dicyclohexylharnstoff abfiltriert wird. Das Filtrat wird bei 0 °C mit 10 ml TFA versetzt und 1h gerührt, wonach die Lösung einrotiert wird. Zur Entfernung von TFA-Resten wird der Rückstand einige Male in CH2Cl2 aufgenommen und jeweils wieder einrotiert. Anschließend löst man den Rückstand in 10 ml CH2Cl2 und gibt die Lösung in 240 ml CHCl3. Unter heftigem Rühren wird die organische Lösung nun mit 50 ml 1M NaHCO3-Lösung versetzt und für 3-5 Tage bei RT weitergerührt. Anschließend werden die Phasen getrennt und die organische Phase nacheinander mit 0.1M NaOH, 5%iger KHSO4-Lösung, ges. NaHCO3-Lösung und ges. NaCl-Lösung gewaschen. Nach Trocknung (Na2SO4) wird die organische Phase einrotiert. Der Rückstand wird anschließend säulenchromatographisch gereinigt. 5. Experimenteller Teil 119 5.2.2.2. Darstellung der Boc-geschützten 3α α-Aminocholansäuren nach AAV III 5.2.2.2.1. 3α α-(t-Butyloxycarbonylamino)-Lithocholsäure 11a Ansatzgröße: 2.00 g (5.32 mmol) 3α-Aminolithocholsäure 5a Ausbeute: 1.52 g (3.20 mmol, 60.2 % der Theorie) HR-MS (FAB): für C29H49NO4Na+ 498.356000 ber. 498.355929 1 H-NMR (CDCl3, 400 MHz): 4.42 ((br), 1H, Boc-NH); 3.39 (m(br), 1H, CHβ-3); 2.41-2.34 u. 2.28-2.20 (2m, 2H, CH2-23); 1.42 (s, 9H, Boc-(CH3)3); 0.91 (d, 3H, CH3-21); 0.90 (s, 3H, CH3-19); 0.63 (s, 3H, CH3-18) 1 H-NMR (DMSO-d6, 400 MHz): 12.09 ((br), 1H, COOH); 6.56 (d, 1H, Boc-NH); 3.19 (m(br), 1H, CHβ-3); 2.24-2.17 u. 2.11-2.03 (2m, 2H, CH2-23); 1.35 (s, 9H, Boc-(CH3)3); 0.88 (s, 3H, CH3-19); 0.86 (d, 3H, CH3-21); 0.60 (s, 3H, CH3-18) 5.2.2.2.2. 3α α-(t-Butyloxycarbonylamino)-Desoxycholsäure 11b Ansatzgröße: 2.50 g (6.38 mmol) 3α-Aminodesoxycholsäure 5b Ausbeute: 2.15 g (4.37 mmol, 68.4 % der Theorie) HR-MS (FAB): 1 für C29H49NO5Na+ 514.350500 ber. 514.350844 H-NMR (CDCl3, 400 MHz): 4.46 ((br), 1H, Boc-NH); 3.97 (m, 1H, CH-12); 3.39 (m(br), 1H, CHβ-3); 2.43-2.35 u. 2.29-2.21 (2m, 2H, CH2-23); 1.42 (s, 9H, Boc-(CH3)3); 0.96 (d, 3H, CH3-21); 0.89 (s, 3H, CH3-19); 0.66 (s, 3H, CH3-18) 1 H-NMR (DMSO-d6, 400 MHz): 11.86 ((br), 1H, COOH); 6.65 (d, 1H, Boc-NH); 4.11 (d, 1H, OH-12); 3.78 (m, 1H, CH-12); 3.17 (m(br), 1H, CHβ-3); 2.25-2.17 u. 2.11-2.05 (2m, 2H, CH2-23); 1.35 (s, 9H, Boc-(CH3)3); 0.90 (s, 3H, CH3-19); 0.85 (d, 3H, CH3-21); 0.59 (s, 3H, CH3-18) 5. Experimenteller Teil 120 5.2.2.2.3. 3α α-(t-Butyloxycarbonylamino)-cholsäure 11c Ansatzgröße: 3.00 g (7.36 mmol) 3α-Aminocholsäure 5c Ausbeute: 2.44 g (4.81 mmol, 65.3 % der Theorie) HR-MS (FAB): für C29H49NO6Na+ 530.347200 ber. 530.345759 1 H-NMR (CDCl3, 400 MHz): 4.50 ((br), 1H, Boc-NH); 3.97 (m, 1H, CH-12); 3.83 (m, 1H, CH-7); 3.26 (m(br), 1H, CHβ-3); 2.43-2.35 u. 2.29-2.22 (2m, 2H, CH2-23); 1.41 (s, 9H, Boc(CH3)3); 0.98 (d, 3H, CH3-21); 0.88 (s, 3H, CH3-19); 0.68 (s, 3H, CH3-18) 1 H-NMR (DMSO-d6, 400 MHz): 11.86 ((br), 1H, COOH); 6.62 (d, 1H, Boc-NH); 4.04 (d, 1H, OH-12); 3.97 (d, 1H, OH-7); 3.77 (m, 1H, CH-12); 3.60 (m, 1H, CH-7); 3.00 (m(br), 1H, CHβ-3); 2.25-2.05 (2m, 2H, CH2-23); 1.35 (s, 9H, Boc-(CH3)3); 0.91 (s, 3H, CH3-19); 0.82 (d, 3H, CH3-21); 0.58 (s, 3H, CH3-18) 5.2.2.3. Darstellung der 3α α-Aminocholansäuremethylester-Hydrochloride nach AAV I 5.2.2.3.1. 3α α-Aminolithocholsäuremethylester-hydrochlorid 12a Ansatzgröße: 5.00 g (13.31 mmol) 3α-Aminolithocholsäure 5a Ausbeute: 4.65 g (10.91 mmol, 82.0 % der Theorie) HR-MS (FAB): für C25H44NO2+ 390.334700 ber. 390.337205 H-NMR (CDCl3, 400 MHz): 8.21 (m(br), 3H, NH3+); 3.64 (s, 3H, COOCH3); 3.17 (m(br), 1H, CHβ-3); 2.37-2.29 u. 2.23-2.15 (2m, 2H, CH2-23); 0.92 (s, 3H, CH3-19); 0.89 (d, 3H, CH3-21); 0.62 (s, 3H, CH3-18) 1 5. Experimenteller Teil 121 5.2.2.3.2. 3α α-Aminodesoxycholsäuremethylester-hydrochlorid 12b Ansatzgröße: 12.00 g (30.65 mmol) 3α-Aminodesoxycholsäure 5b Ausbeute: 11.04 g (24.97 mmol, 81.5 % der Theorie) MS (EI, m/e): 405 (M+, 16 %); 390 (21 %); 370 (15 %); 355 (8 %); 331 (16 %); 316 (10 %); 255 (26 %); 82 (51 %); 69 (34 %); 56 (100 %); 36 (51 %) H-NMR (CDCl3, 400 MHz): 8.06 (m(br), 3H, NH3+); 3.92 (m, 1H, CH-12); 3.63 (s, 3H, COOCH3); 3.24 (m(br), 1H, CHβ-3); 2.39-2.31 u. 2.27-2.16 (2m, 2H, CH2-23); 0.94 (d, 3H, CH3-21); 0.90 (s, 3H, CH3-19); 0.64 (s, 3H, CH3-18) 1 5.2.2.3.3. 3α α-Aminocholsäuremethylester-hydrochlorid 12c Ansatzgröße: 9.00 g (22.08 mmol) 3α-Aminocholsäure 5c Ausbeute: 8.69 g (18.97 mmol, 85.9 % der Theorie) MS (EI, m/e): 421 (M+, 28 %); 406 (64 %); 353 (9 %); 314 (7 %); 253 (25 %); 140 (18 %); 98 (52 %); 82 (51 %); 70 (24 %); 56 (61 %); 36 (100 %) H-NMR (CDCl3, 400 MHz): 7.85 (m(br), 3H, NH3+); 3.97 (m, 1H, CH-12); 3.84 (m, 1H, CH-7); 3.63 (s, 3H, COOCH3); 3.13 (m(br), 1H, CHβ-3); 0.95 (d, 3H, CH3-21); 0.89 (s, 3H, CH3-19); 0.64 (s, 3H, CH3-18) 1 5.2.2.4. Lineare Pseudopeptide (bzw. Oligoamide) aus Aminocholansäurebausteinen Bei Pseudopeptiden mit mehr als zwei Kettengliedern wird aus Übersichtlichkeitsgründen auf die Nennung eines systematischen Namens nach der IUPAC-Nomenklatur verzichtet und stattdessen wie bei echten Peptiden üblich die Sequenz im Dreibuchstabencode mit aminoterminaler und carboxyterminaler Schutzgruppe (sofern vorhanden, sonst -NH2 bzw. -OH) dargestellt. Die Abkürzungen, soweit es sich um Aminocholansäuren handelt, bedeuten im Einzelnen: LCS = 3α-Aminolithocholsäure-Baustein, DCS = 3α-AminodesoxycholsäureBaustein, CHS = 3α-Aminocholsäure-Baustein. Für natürliche Aminosäuren wurde der übliche Dreibuchstabencode verwendet. 5. Experimenteller Teil 122 5.2.2.4.1. Oligopeptide mit 3α α-Aminolithocholsäure 5.2.2.4.1.1. N‘-(N-[t-Butyloxycarbonyl-3α α-amino]-Lithocholyl-3α α‘-amino)-Lithocholsäuremethylester (Boc-LCS-LCS-OMe) 13a; DEPC-Kopplung nach AAV V Ansatzgröße: 476 mg (1 mmol) Boc-LCS-OH 11a; 426 mg (1 mmol) HCl·H2N-LCS-OMe 12a Das bräunliche Rohprodukt (ca. 850 mg) wurde aus TBME/Hexan umkristallisiert, woraufhin das reine Produkt als farbloser Feststoff abgesaugt werden konnte. Die Mutterlauge enthielt noch größere Mengen Produkt, jedoch konnte dieses durch die gewählte Aufarbeitung nicht weiter isoliert werden. Auf eine säulenchromatographische Trennung wurde aufgrund der Empfindlichkeit der Boc-Schutzgruppe verzichtet. Ausbeute nach Umkristallisation: 605 mg (0.712 mmol, 71.2 % der Theorie) HR-MS (FAB): 1 für C54H90N2O5Na+ 869.675500 ber. 869.674745 H-NMR (CDCl3, 400 MHz): 5.51 (m(br), 1H, Amid-NH); 4.37 (m(br), 1H, Boc-NH); 3.76 (m(br), 1H, CHβ-3 Amid); 3.64 (s, 3H, OCH3); 3.39 (m(br), 1H, CHβ-3 Boc); 2.36-2.07 (3m, 4H, CH2-23 u. CH2-23‘); 1.42 (s, 9H, (CH3)3 Boc); 0.90 (2d, 6H, CH3-21 u. CH3-21‘); 0.90 (2s, 6H, CH3-19 u. CH3-19‘); 0.63 u. 0.62 (2s, 6H, CH3-18 u. CH3-18‘) 1 H-NMR (DMSO-d6, 400 MHz): 7.54 (d, 1H, Amid-NH); 6.56 (d, 1H, Boc-NH); 3.56 (s, 3H, OCH3); 3.51 (m(br), 1H, CHβ-3‘ (Amid)); 3.20 (m(br), 1H, CHβ-3 (Boc)); 2.36-1.89 (4m, 4H, CH2-23 u. CH2-23‘); 1.36 (s, 9H, (CH3)3 Boc); 0.88 (2d, 6H, CH3-21 u. CH3-21‘); 0.88 (2s, 6H, CH3-19 u. CH3-19‘); 0.61 u. 0.60 (2s, 6H, CH3-18 u. CH3-18‘) 5. Experimenteller Teil 123 5.2.2.4.1.2. N‘-(N-[t-Butyloxycarbonyl-3α α-amino]-Lithocholyl-3α α‘-amino)-Lithocholsäure (Boc-LCS-LCS-OH) 13b; Esterverseifung nach AAV II Ansatzgröße: 300 mg (0,354 mmol) Boc-LCS-LCS-OMe 13a Ausbeute: 293 mg (0.351 mmol, 99.2 % der Theorie) 1 H-NMR (DMSO-d6, 400 MHz): 12.04 (s(br), 1H, COOH); 7.55 (d, 1H, Amid-NH); 6.56 (d, 1H, Boc-NH); 3.51 (m(br), 1H, CHβ-3‘ (Amid)); 3.21 (m(br), 1H, CHβ-3 (Boc)); 2.24-1.89 (3m, 4H, CH2-23 u. CH2-23‘); 1.36 (s, 9H, (CH3)3 Boc); 0.88 (2d, 6H, CH3-21 u. CH3-21‘); 0.88 (2s, 6H, CH3-19 u. CH3-19‘); 0.61 u. 0.60 (2s, 6H, CH3-18 u. CH3-18‘) Da das NMR-Spektrum eindeutig war und kein Methylester-Signal mehr zeigte, wurde in diesem Fall auf eine massenspektrometrische Analyse verzichtet. 5.2.2.4.1.3. N‘-(3α α-amino-Lithocholyl-3α α‘-amino)-Lithocholsäure-Methylester, TFA-Salz (TFA·H2N-LCS-LCS-OMe) 13c; Boc-Abspaltung nach AAV IV Ansatzgröße: 300 mg (0,354 mmol) Boc-LCS-LCS-OMe 13a Ausbeute: nicht exakt bestimmt (als quantitativ angenommen); das NMR-reine Produkt wurde nach der Darstellung direkt weiterverwendet. H-NMR (DMSO-d6, 400 MHz): 7.67 (m, 3H, NH3+); 7.54 (d, 1H, Amid-NH); 3.57 (s, 3H, OCH3); 3.49 (m(br), 1H, CHβ-3‘ (Amid)); 2.99 (m(br), 1H, CHβ-3); 2.24-1.85 (4m, 4H, CH2-23 u. CH2-23‘); 1.36 (s, 9H, (CH3)3 Boc); 0.90 u. 0.89 (2s, 6H, CH3-19 u. CH3-19‘); 0.86 (2d, 6H, CH3-21 u. CH3-21‘); 0.61 (2s, 6H, CH3-18 u. CH3-18‘) Da das NMR-Spektrum eindeutig war und keine Boc-typischen-Signale mehr zeigte, wurde in diesem Fall auf eine massenspektrometrische Analyse verzichtet. 1 5. Experimenteller Teil 124 5.2.2.4.1.4. (Boc-LCS-LCS-LCS-LCS-OMe) 14a; DEPC-Kopplung nach AAV V Ansatzgröße: 290 mg (0.348 mmol) Boc-LCS-LCS-OH 13b; ca. 0.35 mmol (komplette Ausbeute aus 5.2.2.4.1.4.) TFA·H2N-LCS-LCS-OMe 13c Das noch stark verunreinigte rote Rohprodukt wurde in DCM aufgenommen, bis zum Beginn einer leichten Ausfällung mit Hexan versetzt und anschließend einrotiert. Der Rückstand wurde dann in MeOH aufgenommen, wobei sich die Verunreinigungen vollständig lösten, das Produkt jedoch nur zu einem geringen Anteil. Der farblose Feststoff wurde abgesaugt und mit wenig kaltem MeOH gewaschen. Ausbeute (isoliert): 355 mg (0.227 mmol, 65.3 % der Theorie) Die tatsächliche Ausbeute liegt höher, jedoch konnte durch die gewählte Aufarbeitung nicht mehr Produkt isoliert werden - die in der Mutterlauge verbliebenen Produktreste waren noch stark mit DEPC-Resten bzw. deren Abbauprodukten verunreinigt. Aufgrund der Empfindlichkeit der Boc-Schutzgruppe wurde auf eine säulenchromatographische Trennung des Mutterlaugenrückstandes verzichtet. MS (FAB, m/e): 1585.8 (M+ + Na, 8 %); 1562.8 (M+, 3 %); 1462.2 (M+ - Boc 67 %); 1102.8 (5 %); 745.4 (5 %); 358.2 (10 %); 215.1 (12 %); 154.0 (55 %); 107.0 (77 %); 36.0 (100 %) Das MALDI-Spektrum (aufgenommen in DHB-Matrix) zeigt analog zum FAB-Spektrum einen Peak bei M+ + Na, M+ (sehr klein) und bei M+ - Boc. Weitere signifikante Peaks konnten hier nicht gefunden werden. 1 H-NMR (silberstab. CDCl3, 400 MHz): 5.23 (d, 3H, 3 x Amid-NH); 4.38 (m(br), 1H, BocNH); 3.75 (m(br), 3H, 3 x CHβ-3 Amid); 3.64 (s, 3H, OCH3); 3.39 (m(br), 1H, CHβ-3 Boc); 2.37-2.29 u. 2.23-2.15 (2m, 2H, CH2-23 an COOMe); 2.22-2.14 u. 2.03-1.95 (2m, 3 x 2H, 3 x CH2-23 an CONH); 1.41 (s, 9H, (CH3)3 Boc); 0.89 (4d, 4 x 3H, 4 x CH3-21); 0.89 (4s, 4 x 3H, 4 x CH3-19); 0.62 u. 0.61 (3s + 1s, 4 x 3H, 4 x CH3-18) 1 H-NMR (DMSO-d6/CDCl3 ca. 2:1, schlecht gelöst, 400 MHz): 7.43 (d, 3 x 1H, Amid-NH); 6.34 (d(br), 1H, Boc-NH); 3.56 (s, 3H, OCH3); 3.53 (m(br), 3 x 1H, 3 x CHβ-3 (Amid)); 3.19 (m(br), 1H, CHβ-3 (Boc)); 1.35 (s, 9H, (CH3)3 Boc); 0.90-0.85 (8 x 3H, 4 x CH3-21 u. 4 x CH3-19); 0.60 u. 0.59 (4s, 4 x 3H, CH3-18) 5. Experimenteller Teil 125 5.2.2.4.2. Oligopeptide mit 3α α-Aminodesoxycholsäure 5.2.2.4.2.1. N‘-(N-[t-Butyloxycarbonyl-3α α-amino]-Desoxycholyl-3α α‘-amino)-Desoxycholsäure-methylester (Boc-DCS-DCS-OMe) 15a; EDC-Kopplung nach AAV VI Ansatzgröße: 985 mg (2.0 mmol) Boc-DCS-OH 11b; 885 mg (2.0 mmol) HCl·H2N-DCS-OMe 12b Das Rohprodukt wurde aus TBME/Hexan umkristallisiert. Ausbeute: 1.649 g (1.875 mmol, 93.8 % der Theorie), farbloses Pulver MS (FAB, m/e): 901.6 (M+ + Na, 19 %); 878.6 (M+, 3 %); 823.5 (5 %); 779.5 (M+ - Boc, 8 %); 761.5 (11 %); 743.5 (25 %); 255.2 (11 %); 175.5 (14 %); 119.1 (28 %); 55.0 (100 %) 1 H-NMR (DMSO-d6, 400 MHz): 7.62 (d, 1H, Amid-NH); 6.64 (d, 1H, Boc-NH); 4.17 (d, 1H, C12-OH (Aminoterminaler Baustein)); 4.09 (d, 1H, C12‘-OH (Carboxyterminaler Baustein)); 3.78 (2m, 2 x 1H, CH-12); 3.56 (s, 3H, OCH3); 3.48 (m(br), 1H, CHβ-3‘ (Amid)); 3.17 (m(br), 1H, CHβ-3 (Boc)); 2.35-2.27 u. 2.23-2.15 (2m, 2H, CH2-23‘ an COOMe); 2.07-1.99 u. 1.93-1.87 (2m, 2H, CH2-23 an CONH); 1.35 (s, 9H, (CH3)3 Boc); 0.90 (2d, 2 x 3H, CH3-21 u. CH3-21‘); 0.86 u. 0.85 (2s, 2 x 3H, CH3-19 u. CH3-19‘); 0.59 u. 0.58 (2s, 2 x 3H, CH3-18 u. CH3-18‘) 5.2.2.4.2.2. N‘-(3α α-amino-Desoxycholyl-3α α‘-amino)-Desoxycholsäure-Methylester, TFA-Salz (TFA·H2N-DCS-DCS-OMe) 15c; Boc-Abspaltung nach AAV IV Ansatzgröße: 1.60 g (1.82 mmol) Boc-DCS-DCS-OMe 15a Ausbeute: nicht exakt bestimmt (als quantitativ angenommen); das Produkt wurde nach der Darstellung ohne weitere Analysen direkt weiterverwendet. 5. Experimenteller Teil 126 5.2.2.4.2.3. (Boc-DCS-DCS-DCS-OMe) 16a; EDC-Kopplung nach AAV VI Ansatzgröße: 895 mg (1.820 mmol) Boc-DCS-OH 11b; ca. 1.82 mmol (komplette Ausbeute aus 5.2.2.4.2.2.) TFA·H2N-DCS-DCS-OMe 15c Das Produkt wurde mit Hexan aus TBME ausgefällt. Ausbeute (isoliert): 2.233 g (1.78 mmol, 97.8 % der Theorie) Die Auswertung von NMR- und Massensspektren ergab, dass das Produkt zum Teil einfach oder zweifach mit Trifluoressigsäure verestert war. Der Versuch einer Reinigung einer kleinen Probe des Produkts durch kurzes Rühren mit feuchtem K2CO3 in MeOH misslang jedoch, da hierbei zum Teil auch der Methylester verseift wurde. Da als nächster Schritt ohnehin eine Verseifung folgte, wurde zur Vermeidung von Ausbeuteverlusten auf weitere Reinigungsversuche verzichtet und das z.T. TFA-veresterte Produkt direkt weiterverwendet. MS (FAB, m/e): 1466.9 (M(OTf)2+ + Na, 1 %); 1371.6 (M(OTf)+ + Na, 5 %); 1344.6 (M(OTf)2+ - Boc, 4 %); 1274.7 (M+ + Na, 7 %); 1248.7 (M(OTf)+ - Boc, 15 %); 1152.8 (M+ - Boc, 20 %); 743.4 (3 %); 657.3 (3 %); 470.2 (3 %); 371.2 (5 %); 107.0 (70 %); 57.0 (100 %) Das MALDI-Spektrum (aufgenommen in DHB-Matrix) zeigt analog zum FAB-Spektrum einen Peak bei M(OTf)2+ (sehr klein), M(OTf)+ + Na, M+ + Na, M(OTf)+ (sehr klein), M+ + Na, M+ und M+ - Boc. 1 H-NMR (DMSO-d6, 400 MHz): 7.62 (2d, 2 x 1H, Amid-NH); 7.55 (d, Amid-NH der TFAEster); 6.64 (d, 1H, Boc-NH); 5.31 (m, CH-12 der TFA-Ester); 4.17 (d, C12-OH des carboxyterminalen Bausteins)*; 4.15 (d, C12-OH des mittleren Bausteins)*; 4.09 (d, C12-OH des aminoterminalen Bausteins)*; 3.79 (m, CH-12); 3.56 (s, 3H, OCH3); 3.48 (m(br), 2 x 1H, 2 x CHβ-3 (Amid)); 3.17 (m(br), 1H, CHβ-3 (Boc)); 2.35-2.26 u. 2.23-2.12 (2m, CH2-23 an COOMe); 2.07-2.00 u. 1.94-1.86 (2 x 2m, 2 x CH2-23 an CONH); 1.35 (s, 9H, (CH3)3 Boc); 0.91-0.89 (3d, 3 x 3H, 3 x CH3-21); 0.86 u. 0.85 (3s, 3 x 3H, 3 x CH3-19); 0.75 u. 0.73 (2s, CH3-18 der TFA-Ester); 0.59-0.58 (3s, 3 x 3H, CH3-18) * ) die Zuordnung der OH-Protonen erfolgte durch Analogieschluß zum Tetramer (fast identische Verschiebungen der OH-Signale, siehe dort) sowie durch die reduzierte Intensität der OH-Signale der beiden carboxyterminalen Bausteine durch partielle Veresterung mit TFA. 5. Experimenteller Teil 127 5.2.2.4.2.4. (Boc-DCS-DCS-DCS-OH) 16b; Verseifung des Methylesters nach AAV II Ansatzgröße: 1.390 g (1.109 mmol) Boc-DCS-DCS-DCS-OMe 16a (z.T. TFA-verestertes Produkt aus 5.2.2.4.2.3.) Das Produkt wurde mit Hexan aus DCM ausgefällt. Ausbeute (isoliert): 1.285 g (1.037 mmol, ca. 93.5 % der Theorie) MS (FAB, m/e): 1261.1 (M+ + Na, 9 %); 1239.1 (M+, 6 %); 1139.1 (M+ - Boc, 15 %); 307.1 (17 %); 259.1 (20 %); 154.1 (100 %); 57.1 (47 %) 1 H-NMR (DMSO-f6, 400 MHz): 11.87 (s(br), 1H, COOH); 7.65 (2d, 2 x 1H, Amid-NH); 6.68 (d, 1H, Boc-NH); 4.18, 4.17 u. 4.12 (3d, 3 x C12-OH); 3.79 (3m, 3 x 1H, 3 x CH-12); 3.48 (m(br), 2 x 1H, 2 x CHβ-3 (Amid)); 3.17 (m(br), 1H, CHβ-3 (Boc)); 2.24-2.17 u. 2.11-2.04 (2m, CH2-23 an COOH); 2.06-1.99 u. 1.91-1.84 (2 x 2m, 2 x CH2-23 an CONH); 1.35 (s, 9H, (CH3)3 Boc); 0.91 u. 0.89 (3d, 3 x 3H, 3 x CH3-21); 0.86 (2s) u. 0.85 (1s) (3 x 3H, 3 x CH3-19); 0.59, 0.58 u. 0.57 (3s, 3 x 3H, CH3-18) 5.2.2.4.2.5. (Boc-DCS-DCS-DCS-DCS-OMe) 17a; EDC-Kopplung nach AAV VI Ansatzgröße: 1.239 g (1.000 mmol) Boc-DCS-DCS-DCS-OH 16b; 442 mg (1.000 mmol) HCl·H2N-DCS-OMe 12b Das Produkt wurde mit Hexan aus DCM ausgefällt. Ausbeute (isoliert): 1.492 g (0.917 mmol, 91.7 % der Theorie) MS (FAB, m/e): 1648.6 (M+ + Na, 7 %); 1627.1 (M+, 15 %); 1527.6 (M+ - Boc, 20 %); 561.4 (16 %); 255.1 (13 %); 159.1 (29 %); 107.0 (80 %); 57.0 (100 %) 1 H-NMR (DMSO-d6, 400 MHz): 7.62 (3d, 3 x 1H, Amid-NH); 6.64 (d, 1H, Boc-NH); 4.17 (d, C12-OH des carboxyterminalen Bausteins)*; 4.14 (2d, 2 x C12-OH der beiden mittleren Bausteine)*; 4.09 (d, C12-OH des aminoterminalen Bausteins)*; 3.79 (4m, 4 x 1H, 4 x CH-12); 3.56 (s, 3H, OCH3); 3.48 (m(br), 3#x 1H, 3 x CHβ-3 (Amid)); 3.17 (m(br), 1H, CHβ-3 (Boc)); 2.34-2.27 u. 2.23-2.15 (2m, CH2-23 an COOMe); 2.07-2.00 u. 1.93-1.84 (3 x 2m, 3 x CH2-23 an CONH); 1.35 (s, 9H, (CH3)3 Boc); 0.91-0.90 (4d, 4 x 3H, 4 x CH3-21); 0.86-0.85 (4s, 4 x 3H, 4 x CH3-19); 0.59-0.58 (4s, 4 x 3H, 4 x CH3-18) * ) zur Zuordnung der OH-Protonen vgl. auch Boc-DCS-DCS-DCS-OMe 5. Experimenteller Teil 128 5.2.2.4.3. Oligopeptide mit 3α α-Aminocholsäure 5.2.2.4.3.1. N‘-(N-[t-Butyloxycarbonyl-3α α-amino]-Cholyl-3α α‘-amino)-Cholsäuremethylester (Boc-CHS-CHS-OMe) 18a; EDC-Kopplung nach AAV VI Ansatzgröße: 1.015 g (2 mmol) Boc-CHS-OH 11a; 916 mg (2 mmol) HCl·H2N-CHS-OMe 12a Das Rohprodukt (ca. 1.75 g) wurde aus DCM/Hexan umkristallisiert. Ausbeute: 1.439 g (1.579 mmol, 79.0 % der Theorie), farbloses Pulver MS (FAB, m/e): 933.6 (M+ + Na, 11 %); 911.6 (M+, 1 %); 811.5 (M+ - Boc 18 %); 793.5 (10 %); 775.5 (8 %); 757.5 (6 %); 739.5 (11 %); 369.2 (8 %); 314.2 (4 %); 199.1 (14 %); 145.1 (29 %); 107.1 (42 %); 57.1 (100 %) 1 H-NMR (DMSO-d6, 400 MHz): 7.68 (d, 1H, Amid-NH); 6.69 (d, 1H, Boc-NH); 4.18-4.00 (4d, 4 x 1H, 2x C7-OH und 2 x C12-OH); 3.77 (2m, 2 x 1H, CH-12); 3.59 (2m, 2 x 1H, CH-7); 3.56 (s, 3H, OCH3); 3.30 (m(br), 1H, CHβ-3‘ (Amid)); 2.99 (m(br), 1H, CHβ-3 (Boc)); 2.36-1.84 (2 x 2m, 2 x 2H, CH2-23 und CH2-23‘); 1.34 (s, 9H, (CH3)3 Boc); 0.910.89 (2d, 2 x 3H, CH3-21 u. CH3-21‘); 0.82 u. 0.81 (2s, 2 x 3H, CH3-19 u. CH3-19‘); 0.58 u. 0.56 (2s, 2 x 3H, CH3-18 u. CH3-18‘) 1 H-NMR (CDCl3, 400 MHz): 5.56 (d, 1H, Amid-NH); 4.48 (d, 1H, Boc-NH); 3.96 u. 3.94 (2m, 2 x 1H, CH-12); 3.83 (2m, 2 x 1H, CH-7); 3.64 (s, 3H, OCH3); 3.61 (m(br), 1H, CHβ-3‘ (Amid)); 3.27 (m(br), 1H, CHβ-3 (Boc)); 2.39-1.99 (2 x 2m, 2 x 2H, CH2-23 und CH2-23‘); 1.40 (s, 9H, (CH3)3 Boc); 0.97-0.95 (2d, 2 x 3H, CH3-21 u. CH3-21‘); 0.88 (2s, 2 x 3H, CH3-19 u. CH3-19‘); 0.67 u. 0.66 (2s, 2 x 3H, CH3-18 u. CH3-18‘) 5.2.2.4.3.2. N‘-(N-[t-Butyloxycarbonyl-3α α-amino]-Cholyl-3α α‘-amino)-cholsäure (Boc-CHS-CHS-OH) 18b; Esterverseifung nach AAV II Ansatzgröße: 1.367 g (1.50 mmol) Boc-CHS-CHS-OMe 18a Ausbeute: nicht exakt bestimmt (als quantitativ angenommen); das Produkt wurde nach der Darstellung direkt weiterverwendet. 5. Experimenteller Teil 129 MS (FAB, m/e): 919.7 (M+ + Na, 8 %); 797.6 (M+ - Boc 9 %); 779.6 (5 %); 761.5 (5 %); 743.6 (3 %); 725.5 (5 %); 355.2 (6 %); 253.2 (7 %); 213.1 (6 %); 154.0 (28 %); 95.1 (51 %); 57.1 (100 %) 1 H-NMR (CDCl3, 400 MHz): 5.68 (d, 1H, Amid-NH); 4.43 (d, 1H, Boc-NH); 3.95 u. 3.93 (2m, 2 x 1H, 2 x CH-12); 3.83 (2m, 2 x 1H, 2 x CH-7); 3.67 (m(br), 1H, CHβ-3‘ (Amid)); 3.26 (m(br), 1H, CHβ-3 (Boc)); 2.43-2.36 u. 2.29-2.22 (2m, 2H, CH2-23 (an COOH)); 1.41 (s, 9H, (CH3)3 Boc); 1.01-0.96 (2d, 2 x 3H, CH3-21 u. CH3-21‘); 0.88 (2s, 2 x 3H, CH3-19 u. CH3-19‘); 0.68 u. 0.65 (2s, 2 x 3H, CH3-18 u. CH3-18‘) 5.2.2.4.3.3. (Boc-CHS-CHS-CHS-OMe) 19a; EDC-Kopplung nach AAV VI Ansatzgröße: ca. 1.50 mmol (komplette Ausbeute aus 5.2.2.4.2.2.) Boc-CHS-CHS-OH 18b; 687 mg (1.500 mmol) HCl·H2N-CHS-OMe 12c Das Rohprodukt (ca. 2.0 g) wurde durch Lösen in DCM und anschließendes Ausfällen mit Hexan gereinigt. Ausbeute (isoliert): 1.913 g (1.47 mmol, 98.0 % der Theorie) MS (FAB, m/e): 1322.7 (M+ + Na, 7 %); 1300.7 (M+, 2 %); 1270.8 (3 %); 1200.7 (M+ Boc, 29 %); 1182.8 (10 %); 1164.7 (3 %); 369.2 (7 %); 199.1 (15 %); 154.0 (37 %); 107.0 (51 %); 57.1 (100 %) 1 H-NMR (CDCl3, 400 MHz): 5.66 (2d, 2 x 1H, Amid-NH); 4.51 (d, 1H, Boc-NH); 3.95 (3m, 3 x 1H, CH-12); 3.83 (3m, 3 x 1H, CH-7); 3.64 (s, 3H, OCH3); 3.60 (2m(br), 2 x 1H, 2 x CHβ-3 (Amid)); 3.27 (m(br), 1H, CHβ-3 (Boc)); 2.39-1.99 (3 x 2m, 3 x 2H, 3 x CH2-23); 1.40 (s, 9H, (CH3)3 Boc); 0.97-0.94 (3d, 3 x 3H, 3 x CH3-21); 0.88 (3s, 3 x 3H, 3 x CH3-19); 0.67-0.66 (3s, 3 x 3H, 3 x CH3-18) 1 H-NMR (DMSO-d6, 400 MHz): 7.62 (d, 2 x 1H, Amid-NH); 6.58 (d, 1H, Boc-NH); 4.033.95 (6d, 3 x C12-OH u. 3 x C7-OH); 3.78 (3m, 3 x 1H, 3 x CH-12); 3.61 (3m, 3 x 1H, 3 x CH-7); 3.56 (s, 3H, OCH3); 3.31 (m(br), 2 x 1H, 2 x CHβ-3 (Amid)); 3.00 (m(br), 1H, CHβ-3 (Boc)); 2.34-1.86 (3 x 2m, 3 x 2H, 3 x CH2-23); 1.35 (s, 9H, (CH3)3 Boc); 0.91 (3d, 3 x 3H, 3 x CH3-21); 0.82 (3s, 3 x 3H, 3 x CH3-19); 0.58 (3s, 3 x 3H, 3 x CH3-18) 5. Experimenteller Teil 130 5.2.2.4.2.4. (Boc-CHS-CHS-CHS-CHS-OMe) 20a; EDC-Kopplung nach AAV VI Ansatzgröße: 750 mg (0.583 mmol) Boc-CHS-CHS-CHS-OH 19b (aus der Verseifung des Esters Boc-CHS-CHS-CHS-OMe nach AAV II; Ansatzgröße: 1.200 g (0.922 mmol); Ausbeute: 1.034 g (0.795 mmol, 86.2 % der Theorie), lt. NMR vollständig verseift); 267 mg (0.583 mmol) HCl·H2N-CHS-OMe 12c Das Produkt wurde mit Hexan aus DCM ausgefällt. Ausbeute (isoliert): 829 mg (0.490 mmol, 84.1 % der Theorie) MS (FAB, m/e): 1711.8 (M+ + Na, 6 %); 1691.2 (M+, 3 %); 1590.9 (M+ - Boc, 13 %); 1572.9 (6 %); 1556.8 (2 %); 663.4 (4 %); 442.3 (3 %); 391.2 *5 %);"307.1 (11 %); 219.1 (21 %); 154.0 (92 %); 57.0 (100 %) 1 H-NMR (DMSO-d6, 400 MHz): 7.59 (d, 2 x 1H, Amid-NH); 6.49 (d, 1H, Boc-NH); 3.93 (8d, 4 x C12-OH u. 4 x C7-OH); 3.78 (4m, 4 x 1H, 4 x CH-12); 3.61 (4m, 4 x 1H. 4 x CH-7); 3.56 (s, 3H, OCH3); 3.33 (m(br), 3 x 1H, 3 x CHβ-3 (Amid)); 3.01 (m(br), 1H, CHβ-3 (Boc)); 2.32-1.91 (4 x 2m, 4 x 2H, 4 x CH2-23); 1.35 (s, 9H, (CH3)3 Boc); 0.91 (4d, 4 x 3H, 4 x CH3-21); 0.82 (4s, 4 x 3H, 4 x CH3-19); 0.58 (4s, 4 x 3H, 4 x CH3-18) 5.2.2.4.4. Oligopeptide mit N-Valinyl-3α α-Aminolithocholsäure 5.2.2.4.4.1. N-([t-Butyloxycarbonylamino]-valinyl-3α α-amino)-Lithocholsäuremethylester (Boc-Val-LCS-OMe) 21a; EDC-Kopplung nach AAV VI Ansatzgröße: 1.396 g (3.5 mmol) Boc-Val-OH·DCHA 41; 1.491 g (3.5 mmol) HCl·H2N-LCS-OMe 12a Das Produkt kristallisierte in kleinen Prismen aus einer methanolischen Lösung des Rohproduktes (ca. 2.0 g). Hiervon konnte die Kristallstruktur ermittelt werden. Die Röntgenstrukturdaten sind in Anhang B aufgeführt. Isolierte Ausbeute nach Kristallisation: 1.812 g (3.077 mmol, 87.9 % der Theorie) Die tatsächliche Ausbeute liegt höher, da die Mutterlauge noch erhebliche Mengen Produkt enthielt. Diese Produktreste kristallisierten jedoch nicht mehr aus der Mutterlauge aus. Auf eine chromatographische Reinigung des Restes wurde aufgrund der Empfindlichkeit der BocSchutzgruppe verzichtet. 5. Experimenteller Teil HR-MS (FAB): 131 für C35H61N2O5+ 589.457900 ber. 589.458049 1 H-NMR (CDCl3, 400 MHz): 5.65 (d(br),#1H, Amid-NH); 5.01 (d(br), 1H, Boc-NH); 3.76 (m(br), 1H, CHβ-3 (LCS) Amid); 3.76 (dd, 1H, CHα (Val) Boc); 3.64 (s, 3H, OCH3 (LCS)); 2.37-2.29 u. 2.24-2.16 (2m, 2H, CH2-23 (LCS)); 2.09 (m, 1H, CHβ (Val)); 1.43 (s, 9H, (CH3)3 Boc); 0.93 u. 0.89 (2d, 6H, 2 x CH3 (Val)); 0.91 (s, 3H, CH3-19 (LCS)); 0.89 (d, 3H, CH3-21 (LCS)); 0.63 (s, 3H, CH3-18 (LCS)) 5.2.2.4.4.2. N-([t-Butyloxycarbonylamino]-valinyl-3α α-amino)-Lithocholsäure (Boc-Val-LCS-OH) 21b; Esterverseifung nach AAV II Ansatzgröße: 1.200 g (2.038 mmol) Boc-Val-LCS-OMe 21a Das Rohprodukt wurde mit Hexan aus TBME ausgefällt. Ausbeute: 1.170 g (2.035 mmol, 99.9 % der Theorie) MS (FAB, m/e): 619.2 (M+ + 2Na, 3 %); 597.3 (M+ + Na, 44 %); 575.3 (M+, 7 %); 519.2 (M+ + 2Na - Boc, 37 %); 497.3 (M+ + Na - Boc, 13 %); 475.2 (M+ - Boc, 57 %); 374.2 (M+ - Boc - Val, 13 %); 116.0 (35 %); 95.0 (32 %); 72.0 (100 %); 57.0 (89 %) 1 H-NMR (CDCl3, 400 MHz): 5.92 (d(br), 1H, Amid-NH); 5.14 (d(br), 1H, Boc-NH); 3.79 (m(br), 1H, CHα (Val) Boc); 3.74 (m(br), 1H, CHβ-3 (LCS) Amid); 2.41-2.34 u. 2.29-2.21 (2m, 2H, CH2-23 (LCS)); 2.04 (m, 1H, CHβ (Val)); 1.43 (s, 9H, (CH3)3 Boc); 0.93 u. 0.89 (2d, 6H, 2 x CH3 (Val)); 0.92 (s, 3H, CH3-19 (LCS)); 0.92 (d, 3H, CH3-21 (LCS)); 0.63 (s, 3H, CH3-18 (LCS)) 5. Experimenteller Teil 132 5.2.2.4.4.3. N-Valinyl-3α α-Aminolithocholsäure-Methylester (TFA-Salz) (TFA·H2N-Val-LCS-OMe) 21c; Boc-Abspaltung nach AAV IV Ansatzgröße: 400 mg (0.679 mmol) Boc-Val-LCS-OMe 21a Ausbeute: nicht exakt bestimmt (als quantitativ angenommen) MS (FAB, m/e): 511.3 (M+ + Na, 17 %); 489.3 (M+, 100 %); 373.2 (M+ - Val, 3 %); 116.0 (35 %); 95.0 (32 %); 72.1 (26 %); 55.0 (6 %) H-NMR (CDCl3, 400 MHz): 8.15 (m(br), 3H, -NH3+ Val); 7.06 (d, 1H, Amid-NH); 3.76 (d, 1H, CHα (Val)); 3.69 (m(br), 1H, CHβ-3 (LCS) Amid); 3.64 (s, 3H, OCH3 (LCS)); 2.37-2.29 u. 2.24-2.16 (2m, 2H, CH2-23 (LCS)); 2.16 (m, 1H, CHβ (Val)); 1.04 u. 0.98 (2d, 6H, 2 x CH3 (Val)); 0.91 (s, 3H, CH3-19 (LCS)); 0.89 (d, 3H, CH3-21 (LCS)); 0.62 (s, 3H, CH3-18 (LCS)) 1 5.2.2.4.4.4. (Boc-Val-LCS-Val-LCS-OMe) 25a; EDC-Kopplung nach AAV VI Ansatzgröße: 391 mg (0.679 mmol) Boc-Val-LCS-OH 21a; ca. 0.679 mmol (komplette Ausbeute aus 5.2.2.4.4.3.) TFA·H2N-Val-LCS-OMe 21b Das reine Produkt konnte mit Hexan aus einer DCM-Lösung des Rohproduktes (710 mg) ausgefällt werden. Ausbeute: 691 mg (0.661 mmol, 97.3 % der Theorie) MS (FAB, m/e): 1067.7 (M+ + Na, 4 %); 1045.7 (M+, 2 %); 945.6 (M+ - Boc 18 %); 556.4 (6 %); 489.3 (2 %); 457.3 (3 %); 390.2 (6 %); 374.2 (4 %); 107.0 (10 %); 72.0 (100 %) 1 H-NMR (CDCl3, 400 MHz): 6.03 (d, 1H, Amid-NH (Val‘)); 5.71 (d(br), 1H, Amid-NH (LCS)); 5.67 (d, 1H, Amid-NH (LCS‘)); 5.01 (d(br), 1H, Boc-NH (Val)); 4.09 (dd, 1H, CHα (Val‘) Amid); 3.77 (dd, 1H, CHα (Val) Boc); 3.76 (2m(br), 2 x 1H, 2 x CHβ-3 (LCS u. LCS‘) Amid); 3.64 (s, 3H, OCH3 (LCS‘)); 2.37-2.07 (2 x 2m, 2 x 2H, 2 x CH2-23 (LCS u. LCS‘)); 2.09 (m, 1H, CHβ (Val)); 2.02 (m, 1H, CHβ (Val‘)); 1.43 (s, 9H, (CH3)3 Boc); 0.94-0.89 (insges. 24H: 2d, 6H, 2 x CH3 (Val); 2d, 6H, 2 x CH3 (Val‘); 2s, 2 x 3H, 2 x CH3-19 (LCS u. LCS‘); 2d, 2 x 3H, 2 x CH3-21 (LCS u. LCS‘)); 0.62 (2s, 2 x 3H, 2 x CH3-18 (LCS u. LCS‘)) (Zur Differenzierung der NMR-Signale der einzelnen Bausteine (soweit möglich) wurde folgende Bezeichnung gewählt: Boc-Val-LCS-Val‘-LCS‘-OMe) 5. Experimenteller Teil 133 5.2.2.4.4.5. (Boc-Val-LCS-Val-LCS-Val-LCS-OMe) 26a; EDC-Kopplung nach AAV VI Ansatzgröße: 288 mg (0.500 mmol) Boc-Val-LCS-OH 21b; ca. 0.5 mmol TFA·H2N-Val-LCS-Val-LCS-OMe 25c (komplette Ausbeute aus der BocAbspaltung von 523 mg (0.500 mmol) Boc-Val-LCS-Val-LCS-OMe 25a nach AAV IV) Ausbeute: 681 mg (0.453 mmol, 90.6 % der Theorie) MS (FAB, m/e): 1525.6 (M+ + Na, 1 %); 1502.6 (M+, 0.3 %); 1402.3 (M+ - Boc, 6 %); 528.4 (1 %); 457.3 (2 %); 390.3 (2 %); 390.2 (6 %); 154.0 (8 %); 72.0 (100 %) 1 H-NMR (CDCl3, 400 MHz): 6.20 (d, 1H, Amid-NH (LCS‘ o. LCS‘‘)); 6.10 (d, 1H, AmidNH (LCS‘‘ o. LCS‘)); 6.10 (d, 1H, Amid-NH (Val‘‘ o. Val‘)); 6.07 (d, 1H, Amid-NH (Val‘ o. Val‘‘)); 6.01 (d, 1H, Amid-NH (LCS)); 5.06 (d, 1H, Boc-NH (Val)); 4.22 (dd, 1H, CHα (Val‘ o. Val‘‘)); 4.19 (dd, 1H, CHα (Val‘‘ o. Val‘)); 3.83 (dd, 1H, CHα (Val)); 3.73 (3m(br), 3 x 1H, 3 x CHβ-3 (LCS, LCS‘ u. LCS‘‘)); 3.64 (s, 3H, OCH3 (LCS‘‘)); 2.38-1.97 (insges. 9H: 3 x 2m, 3 x 2H, 3 x CH2-23 (LCS, LCS‘ u. LCS‘‘); 3m, 3 x 1H, 3 x CHβ (Val, Val‘ u. Val‘‘)); 1.43 (s, 9H, (CH3)3 Boc); 0.93-0.89 (insges. 36H: 2d, 6H, 2 x CH3 (Val); 2d, 6H, 2 x CH3 (Val‘); 2d, 6H, 2 x CH3 (Val‘‘); 3s, 3 x 3H, 3 x CH3-19 (LCS, LCS‘ u. LCS‘‘); 3d, 3 x 3H, 3 x CH3-21 (LCS, LCS‘ u. LCS‘‘)); 0.62 (3s, 3 x 3H, 3 x CH3-18 (LCS, LCS‘ u. LCS‘‘)) (Zur Differenzierung der NMR-Signale der einzelnen Bausteine (soweit möglich) wurde folgende Bezeichnung gewählt: Boc-Val-LCS-Val‘-LCS‘-Val‘‘-LCS‘‘-OMe) 5.2.2.4.5. Oligopeptide mit N-[εε-Benzyloxycarbonylamino-Lysinyl]-3α α-Aminolithocholsäure 5.2.2.4.5.1. N-(α α-t-Butoxycatbonylamino-[εε-Benzyloxycarbonylamino]-Lysinyl)-3α αAminolithocholsäure-Methylester (Boc-Lys[Z]-LCS-OMe) 22a; EDC-Kopplung nach AAV VI Ansatzgröße: 1.141 g (3.0 mmol) Boc-Lys[Z]-OH 42; 1.278 g (3.0 mmol) HCl·H2N-LCS-OMe 12a Isolierte Ausbeute: 1.895 g (2.520 mmol, 84.0 % der Theorie) 5. Experimenteller Teil 134 MS (FAB, m/e): 774.4 (M+ + Na, 17 %); 752.4 (M+, 12 %); 674.4 (M+ + Na - Boc, 3 %); 652.4 (M+ - Boc, 29 %); 545.4 (5 %); 499.3 (5 %); 388.3 (5 %); 218.1 (10 %); 174.1 (9 %); 91.0 (Bn, 100 %); 57.0 (27 %) 1 H-NMR (CDCl3, 400 MHz): 7.34-7.27 (m, 5H, Phenyl-H der Z-Schutzgruppe) 5.88 (d, 1H, Amid-NH (LCS)); 5.08 (d, 1H, Boc-NH (Lys)); 5.08 (s, 2H, CH2(Z)); 4.80 (t, 1H, Z-NH (Lys)); 3.93 (m, 1H, CHα (Lys)); 3.72 (m(br), 1H, CHβ-3 (LCS)); 3.64 (s, 3H, OCH3 (LCS)); 3.17 (m, 2H, CH2ε (Lys)); 2.37-2.29 u. 2.24-2.16 (2m, 2H, CH2-23 (LCS)); 1.42 (s, 9H, (CH3)3 Boc); 0.90 (s, 3H, CH3-19 (LCS)); 0.89 (d, 3H, CH3-21 (LCS)); 0.62 (s, 3H, CH3-18 (LCS)) 5.2.2.4.5.2. N-(α α-t-Butoxycatbonylamino-[εε-Benzyloxycarbonylamino]-Lysinyl)-3α αAmino-lithocholsäure (Boc-Lys[Z]-LCS-OH) 22b; Esterverseifung AAV II Ansatzgröße: 400 mg (0.532 mmol) Boc-Lys[Z]-LCS-OMe 22a Ausbeute: 375 mg (0.508 mmol, 95.5 % der Theorie) MS (FAB, m/e): 760.5 (M+ + Na, 28 %); 738.5 (M+, 21 %); 660.5 (M+ + Na - Boc, 4 %); 638.5 (M+ - Boc, 20 %); 530.4 (3 %); 485.4 (3 %); 374.3 (3 %); 329.1 (5 %); 307.1 (8 %); 259.1 (7 %); 218.1 (19 %); 175.9 (20 %); 154.0 (45 %); 136.0 (39 %); 91.0 (Bn, 100 %); 57.1 (46 %) 1 H-NMR (CDCl3, 400 MHz): 7.34-7.27 (m, 5H, Phenyl-H der Z-Schutzgruppe) 6.07 (d, 1H, Amid-NH (LCS)); 5.17 (d, 1H, Boc-NH (Lys)); 5.08 (s, 2H, CH2(Z)); 4.84 (t, 1H, Z-NH); 3.97 (m, 1H, CHα (Lys)); 3.72 (m(br), 1H, CHβ-3 (LCS)); 3.17 (m, 2H, CH2ε (Lys)); 2.412.33 u. 2.27-2.19 (2m, 2H, CH2-23 (LCS)); 1.42 (s, 9H, (CH3)3 Boc); 0.91 (s, 3H, CH3-19 (LCS)); 0.91 (d, 3H, CH3-21 (LCS)); 0.63 (s, 3H, CH3-18 (LCS)) 5. Experimenteller Teil 135 5.2.2.4.5.3. N-([εε-Benzyloxycarbonylamino]-Lysinyl)-3α α-Aminolithocholsäure-Methylester, TFA-Salz (TFA·H2N-Lys[Z]-LCS-OMe) 22c; Boc-Abspaltung nach AAV IV Ansatzgröße: 400 mg (0.532 mmol) Boc-Lys[Z]-LCS-OMe 22a Da die Z-Gruppe ebenfalls säurelabil ist, eine Abspaltung dieser Schutzgruppe aber unerwünscht war, wurde die Reaktionszeit auf 45 min (bei 0°C) verkürzt. Ausbeute: nicht exakt bestimmt (als quantitativ angenommen) MS (FAB, m/e): 674.5 (M+ + Na, 7 %); 652.5 (M+, 30 %); 545.6 (5 %); 327.0 (7 %); 281.1 (9 %); 235.1 (14 %); 207.0 (14 %); 175.8 (11 %); 154.1 (20 %); 136.0 (42 %); 91.0 (Bn, 100 %); 73.0 (92 %); 55.0 (51 %) 1 H-NMR (CDCl3, 400 MHz): 7.34-7.27 (m, 5H, Phenyl-H der Z-Schutzgruppe) 7.06 (d, 1H, Amid-NH (LCS)); 5.07 (s, 2H, CH2(Z)); 4.85 (t, 1H, Z-NH); 3.71 (m(br), 1H, CHβ-3 (LCS)); 3.64 (s, 3H, OCH3 (LCS)); 3.40 (m, 1H, CHα (Lys)); 3.17 (m, 2H, CH2ε (Lys)); 2.37-2.29 u. 2.23-2.16 (2m, 2H, CH2-23 (LCS)); 0.91 (s, 3H, CH3-19 (LCS)); 0.89 (d, 3H, CH3-21 (LCS)); 0.62 (s, 3H, CH3-18 (LCS)) 5.2.2.4.5.4. (Boc-Lys[Z]-LCS-Lys[Z]-LCS-OMe) 27a; EDC-Kopplung nach AAV VI Ansatzgröße: 375 mg (0.508 mmol) Boc-Lys[Z]-LCS-OH 21b; ca. 0.53 mmol (komplette Ausbeute aus 5.2.2.4.5.3.) TFA·H2N-Lys[Z]-LCS-OMe 21c Das reine Produkt wurde mit Hexan aus einer DCM-Lösung des Rohproduktes ausgefällt. Ausbeute: 644 mg (0.469 mmol, 92.4 % der Theorie) MS (FAB, m/e): 1393.6 (M+ + Na, 3 %); 1371.6 (M+, 2 %); 1271.6 (M+ - Boc, 7 %); 722.6 (8 %); 492.4 (3 %); 388.2 (3 %); 218.0 (6 %); 136.0 (27 %); 91.0 (100 %) 1 H-NMR (CDCl3. 400 MHz): 7.34-7.27 (2m, 2 x 5J, 2 x Phenyl-H der Z-Schutzgruppen (Z u. Z‘)); 6.12 (d, 1H, Amid-NH (Lys‘)); 5.96 (2d, 2 x 1H, 2 x Amid-NH (LCS u. LCS‘)); 5.08 (d, 1H, Boc-NH (Lys)); 5.08 (2s, 2 x 2H, CH2(Z) u. CH2(Z‘)); 4.83 (2t, 2 x 1H, 2 x Z-NH (Lys u. Lys‘)); 4.30 (q, 1H, CHα (Lys‘)); 3.94 (m, 1H, CHα (Lys)); 3.72 (2m(br), 2 x 1H, 2 x CHβ-3 (LCS u. LCS‘)); 3.64 (s, 3H, OCH3 (LCS‘)); 3.17 (2m, 2 x 2H, 2 x CH2ε (Lys u. Lys‘)); 2.352.04 (4m, 2 x 2H, 2 x CH2-23 (LCS u. LCS‘)); 1.42 (s, 9H, (CH3)3 Boc); 0.90 (2s, 2 x 3H, 2 x CH3-19 (LCS u. LCS‘)); 0.89 (2d, 2 x 3H, 2 x CH3-21 (LCS u. LCS‘)); 0.62 u. 0.61 (2s, 2 x 3H, 2 x CH3-18 (LCS u. LCS‘)) (Zur Differenzierung der NMR-Signale der einzelnen Bausteine (soweit möglich) wurde folgende Bezeichnung gewählt: Boc-Lys[Z]-LCS-Lys[Z]‘-LCS‘-OMe) 5. Experimenteller Teil 136 5.2.2.4.5.5. Boc-Lys[Z]-LCS-Lys[Z]-LCS-Lys[Z]-LCS-OMe 28a; EDC-Kopplung nach AAV VI Ansatzgröße: 323 mg (0.437 mmol) Boc-Lys[Z]-LCS-OH 22b; ca. 0.437 mmol TFA·H2N-Lys[Z]-LCS-Lys[Z]-LCS-OMe (komplette Ausbeute aus der Boc-Abspaltung von 600 mg (0.437 mmol) Boc-Lys[Z]-LCS-Lys[Z]-LCS-OMe nach AAV IV) Das Produkt konnte durch Ausfällen mit Hexan aus einer DCM-Lösung gereinigt werden. Ausbeute: 632 mg (0.317 mmol, 72.6 % der Theorie) MS (FAB, m/e): 1998.4 (M+, 0.1 %); 1888.9 (M+ - Boc, 1 %); 545.4 (1 %); 388.2 (1 %); 174.1 (13 %); 91.0 (100 %) Der Massenpeak (M+) und der (M+ - Boc)-Peak wichen jeweils um +7 Masseneinheiten von der tatsächlichen Masse ab. Dies ist möglicherweise darauf zurückzuführen, dass die molare Masse der Verbindung an der Messrenze des FAB-Geräts liegt und es auf Massen unterhalb von 1000 kalibriert wurde, zumal das NMR deutlich zeigt, dass es sich tatsächlich um die gewünschte Verbindung handelt. 1 H-NMR (DMSO-d6, 400 MHz): 7.75 (2d, 2 x 1H, 2 x Amid-NH (Lys‘ u. Lys‘‘)); 7.55 (3d, 3 x 1H, 3 x Amid-NH (LCS, LCS‘ u. LCS‘‘)); 7.36-7.26 (3m, 3 x 5H, 3 x Phenyl-H der ZSchutzgruppen (Z, Z‘ u. Z‘‘)); 7.16 (3t, 3 x 1H, 3 x Z-NH (Lys, Lys‘ u. Lys‘‘)); 6.59 (d, 1H, Boc-NH (Lys)); 4.99 (3s, 3 x 2H, CH2(Z), CH2(Z‘) u. CH2(Z‘‘)); 4.11 (2q, 2 x 1H, CHα (Lys‘ u. Lys‘‘)); 3.78 (m, 1H, CHα (Lys)); 3.55 (s, 3H, OCH3 (LCS‘)); 3.50 (3m(br), 3 x 1H, 3 x CHβ-3 (LCS, LCS‘ u. LCS‘‘)); 2.95 (3m, 3 x 2H, 3 x CH2ε (Lys, Lys‘ u. Lys‘‘)); 2.36-2.00 (6m, 3 x 2H, 3 x CH2-23 (LCS, LCS‘ u. LCS‘‘)); 1.36 (s, 9H, (CH3)3 Boc); 0.95-0.80 (3s, 3 x 3H, 3 x CH3-19; 3d, 3 x 3H, 3 x CH3-21 (jew. LCS, LCS‘ u. LCS‘‘)); 0.60 u. 0.59 (3s, 3 x 3H, 3 x CH3-18 (LCS, LCS‘ u. LCS‘‘)) (Zur Differenzierung der NMR-Signale der einzelnen Bausteine (soweit möglich) wurde folgende Bezeichnung gewählt: Boc-Lys[Z]-LCS-Lys[Z]‘-LCS‘-Lys[Z]‘‘-LCS‘‘-OMe) 5. Experimenteller Teil 137 5.2.2.4.6. Oligopeptide mit N-Valinyl-3α α-Aminodesoxycholsäure 5.2.2.4.6.1. N-([t-Butyloxycarbonylamino]-valinyl-3α α-amino)-Desoxycholsäuremethylester (Boc-Val-DCS-OMe) 23a; EDC-Kopplung nach AAV VI Ansatzgröße: 798 mg (2 mmol) Boc-Val-OH·DCHA 42; 885 mg (2 mmol) HCl·H2N-DCS-OMe 12b Das Produkt kristallisierte aus einer Ethylacetat-Lösung des Rohproduktes (ca. 1.2 g). Isolierte Ausbeute: 1.139 g (1.883 mmol, 94.2 % der Theorie) MS (FAB, m/e): 627.5 (M+ + Na, 39 %); 605.5 (M+, 17 %); 505.4 (M+ - Boc, 39 %); 487.4 (100 %); 388.3 (13 %); 154.0 (14 %); 116.1 (24 %); 72.1 (86 %); 57.0 (63 %) 1 H-NMR (CDCl3, 400 MHz): 5.70 (d(br), 1H, Amid-NH); 4.98 (d(br), 1H, Boc-NH); 3.97 (m, 1H, CH-12 (DCS)); 3.78 (dd, 1H, CHα (Val) Boc); 3.77 (m(br), 1H, CHβ-3 (DCS) Amid); 3.64 (s, 3H, OCH3 (DCS)); 2.39-2.31 u. 2.25-2.17 (2m, 2H, CH2-23 (DCS)); 2.10 (m, 1H, CHβ (Val)); 1.43 (s, 9H, (CH3)3 Boc); 0.95 (d, 3H, CH3-21 (DCS)); 0.92 u. 0.88 (2d, 6H, 2 x CH3 (Val)); 0.90 (s, 3H, CH3-19 (DCS)); 0.66 (s, 3H, CH3-18 (DCS)) 5.2.2.4.6.2. N-([t-Butyloxycarbonylamino]-valinyl-3α α-amino)-Desoxycholsäure (Boc-Val-DCS-OH) 23b; Esterverseifung nach AAV II Ansatzgröße: 302 mg (0.500 mmol) Boc-Val-DCS-OMe 23a Ausbeute: 288 mg (0.487 mmol, 97.5 % der Theorie) MS (FAB, m/e): 613.4 (M+ + Na, 55 %); 591.4 (M+, 14 %); 491.3 (M+ + Na - Boc, 27 %); 473.3 (72 %); 390.3 (8 %); 374.2 (9 %); 357.2 (8 %); 175.7 (9 %); 154.0 (26 %); 136.0 (22 %); 116.1 (39 %); 72.1 (100 %); 57.0 (83 %) 1 H-NMR (CDCl3, 400 MHz): 5.99 (d(br), 1H, Amid-NH); 5.16 (d(br), 1H, Boc-NH); 3.98 (m, 1H, CH-12 (DCS)); 3.82 (dd, 1H, CHα (Val) Boc); 3.75 (m(br), 1H, CHβ-3 (DCS) Amid); 2.43-2.36 u. 2.29-2.22 (2m, 2H, CH2-23 (DCS)); 2.04 (m, 1H, CHβ (Val)); 1.42 (s, 9H, (CH3)3 Boc); 0.97 (d, 3H, CH3-21 (DCS)); 0.91 u. 0.88 (2d, 6H, 2 x CH3 (Val)); 0.90 (s, 3H, CH3-19 (DCS)); 0.66 (s, 3H, CH3-18 (DCS)) 5. Experimenteller Teil 138 5.2.2.4.6.3. N-Valinyl-3α α-Aminodesoxycholsäure-Methylester (TFA-Salz) (TFA·H2N-Val-DCS-OMe) 23c; Boc-Abspaltung nach AAV IV Ansatzgröße: 302 mg (0.500 mmol) Boc-Val-DCS-OMe 23a Um die mögliche Nebenreaktion (Veresterung der freien OH-Gruppe von DCS mit TFA) zu unterdrücken wurde die Reaktionszeit auf 40 min bei 0°C verkürzt und das Produkt direkt weiterverwendet. Ausbeute: nicht exakt bestimmt (als quantitativ angenommen) MS (FAB, m/e): 527.4 (M+ + Na, 14 %); 505.4 (M+, 33 %); 487.4 (51 %); 388.3 (5 %); 175.9 (6 %); 154.0 (24 %); 136.0 (19 %); 107.1 (12 %); 72.1 (100 %); 55.0 (16 %) 5.2.2.4.6.4. (Boc-Val-DCS-Val-DCS-OMe) 29a; EDC-Kopplung nach AAV VI Ansatzgröße: 288 mg (0.487 mmol) Boc-Val-DCS-OH 23b; ca. 0.5 mmol (komplette Ausbeute aus 5.2.2.4.4.3.) TFA·H2N-Val-DCS-OMe 23c Das reine Produkt konnte mit Hexan aus einer DCM-Lösung des Rohproduktes (516 mg) ausgefällt werden. Ausbeute: 485 mg (0.450 mmol, 92.4 % der Theorie) MS (FAB, m/e): 1099.8 (M+ + Na, 4 %); 1077.8 (M+, 7 %); 977.8 (M+ - Boc 6 %); 554.4 (5 %); 487.4 (3 %); 388.3 (7 %+; 374.2 (5 %); 154.0 (11 %); 107.1 (14 %); 72.0 (100 %) 1 H-NMR (CDCl3, 400 MHz): 6.09 (d, 1H, Amid-NH (Val‘)); 5.79 (2d, 2 x 1H, 2 x Amid-NH (DCS u. DCS‘)); 5.01 (d(br), 1H, Boc-NH (Val)); 4.08 (dd, 1H, CHα (Val‘) Amid); 3.96 (2m, 2 x 1H, 2 x CH-12 (DCS u. DCS‘)); 3.80 (dd, 1H, CHα (Val) Boc); 3.76 (2m(br), 2 x 1H, 2 x CHβ-3 (DCS u. DCS‘) Amid) ; 3.64 (s, 3H, OCH3 (DCS‘)); 2.39-2.07 (2 x 2m, 2 x 2H, 2 x CH2-23 (DCS u. DCS‘)); 2.07 (m, 1H, CHβ (Val)); 2.00 (m, 1H, CHβ (Val‘)); 1.42 (s, 9H, (CH3)3 Boc); 0.96-0.87 (insges. 24H: 2d, 6H, 2 x CH3 (Val); 2d, 6H, 2 x CH3 (Val‘); 2s, 2 x 3H, 2 x CH3-19 (DCS u. DCS‘); 2d, 2 x 3H, 2 x CH3-21 (DCS u. DCS‘)); 0.66 u. 0.65 (2s, 2 x 3H, 2 x CH3-18 (DCS u. DCS‘)) (Zur Differenzierung der NMR-Signale der einzelnen Bausteine (soweit möglich) wurde folgende Bezeichnung gewählt: Boc-Val-DCS-Val‘-DCS‘-OMe) 5. Experimenteller Teil 139 5.2.2.4.6.5. (Boc-Val-DCS-Val-DCS-OH) 29b; Esterverseifung nach AAV II Ansatzgröße: 431 mg (0.400 mmol) Boc-Val-DCS-Val-DCS-OMe 29a Ausbeute: 369 mg (0.347 mmol, 86.7 % der Theorie) MS (FAB, m/e): 1085.8 (M+ + Na, 12 %); 1063.8 (M+, 13 %); 963.8 (M+ - Boc 7 %); 554.4 (5 %); 473.4 (5 %); 374.3 (5 %); 154.0 (32 %); 72.0 (100 %) 1 H-NMR (CDCl3, 400 MHz): 6.66 (d, 1H, Amid-NH (Val‘)); 6.34 u. 6.25 (2d, 2 x 1H, 2 x Amid-NH (DCS u. DCS‘)); 5.16 (d(br), 1H, Boc-NH (Val)); 4.27 (dd, 1H, CHα (Val‘) Amid); 3.96 (2m, 2 x 1H, 2 x CH-12 (DCS u. DCS‘)); 3.87 (dd, 1H, CHα (Val) Boc); 3.71 (2m(br), 2 x 1H, 2 x CHβ-3 (DCS u. DCS‘) Amid) ; 2.41-1.98 (insges. 6H: 2 x 2m, 2 x 2H, 2 x CH2-23 (DCS u. DCS‘); 2m, 2 x 1H, 2 x CHβ (Val u. Val‘)); 1.42 (s, 9H, (CH3)3 Boc); 0.99 (2d, 2 x 3H, 2 x CH3-21 (DCS u. DCS‘)); 0.92-0.88 (insges. 18H: 2d, 6H, 2 x CH3 (Val); 2d, 6H, 2 x CH3 (Val‘); 2s, 2 x 3H, 2 x CH3-19 (DCS u. DCS‘)); 0.66 (2s, 2 x 3H, 2 x CH3-18 (DCS u. DCS‘)) (Zur Differenzierung der NMR-Signale der einzelnen Bausteine (soweit möglich) wurde folgende Bezeichnung gewählt: Boc-Val-DCS-Val‘-DCS‘-OH) 5.2.2.4.6.6. (Boc-Val-DCS-Val-DCS-Val-DCS-OMe) 30a; EDC-Kopplung nach AAV VI Ansatzgröße: 288 mg (0.338 mmol) Boc-Val-DCS-Val-DCS-OH 29b; ca. 0.338 mmol TFA·H2N-Val-DCS-OMe 23c (komplette Ausbeute aus der Boc-Abspaltung von 205 mg (0.338 mmol) Boc-Val-DCS-OMe [] nach AAV IV; vgl. 5.2.2.4.6.3.) Das reine Produkt konnte durch Ausfällen mit Hexan aus einer DCM-Lösung des Rohproduktes (ca. 500 mg) erhalten werden. Ausbeute: 448 mg (0.453 mmol, 85.5 % der Theorie) 5. Experimenteller Teil 140 MS (FAB, m/e): 1572.8 (M+ + Na, 2 %); 1550.8 (M+,"1 %); 1450.7 (M+ - Boc, 4 %); 526.4 (2 %); 455.3 (2 %); 388.3 (4 %); 255.2 (2 %); 154.0 (23 %); 72.0 (100 %) 1 H-NMR (CDCl3, 400 MHz): 6.40 (d, 1H, Amid-NH (DCS‘ o. DCS‘‘)); 6.26 (d, 1H, AmidNH (DCS‘‘ o. DCS‘)); 6.18 (d, 1H, Amid-NH (Val‘‘ o. Val‘)); 6.14 (d, 1H, Amid-NH (Val‘ o. Val‘‘)); 6.04 (d, 1H, Amid-NH (DCS)); 5.05 (d, 1H, Boc-NH (Val)); 4.25 (dd, 1H, CHα (Val‘ o. Val‘‘)); 4.21 (dd, 1H, CHα (Val‘‘ o. Val‘)); 3.95 (3m, 3 x 1H, 3 x CH-12 (DCS, DCS‘ u. DCS‘‘)); 3.86 (dd, 1H, CHα (Val)); 3.72 (3m(br), 3 x 1H, 3 x CHβ-3 (DCS, DCS‘ u. DCS‘‘)); 3.65 (s, 3H, OCH3 (DCS‘‘)); 2.38-2.07 (3 x 2m, 3 x 2H, 3 x CH2-23 (DCS, DCS‘ u. DCS‘‘)); 2.07 (m, 1H, CHβ (Val)); 2.00 (m, 1H, CHβ (Val‘ o. Val‘‘)); 1.97 (m, 1H, CHβ (Val‘‘ o. Val‘)); 1.43 (s, 9H, (CH3)3 Boc); 0.96-0.87 (insges. 36H: 2d, 6H, 2 x CH3 (Val); 2d, 6H, 2 x CH3 (Val‘); 2d, 6H, 2 x CH3 (Val‘‘); 3s, 3 x 3H, 3 x CH3-19 (DCS, DCS‘ u. DCS‘‘); 3d, 3 x 3H, 3 x CH3-21 (DCS, DCS‘ u. DCS‘‘)); 0.66 (3s, 3 x 3H, 3 x CH3-18 (DCS, DCS‘ u. DCS‘‘)) (Zur Differenzierung der NMR-Signale der einzelnen Bausteine (soweit möglich) wurde folgende Bezeichnung gewählt: Boc-Val-DCS-Val‘-DCS‘-Val‘‘-DCS‘‘-OMe) 5.2.2.4.7. Oligopeptide mit N-Valinyl-3α α-Aminocholsäure 5.2.2.4.7.1. N-([t-Butyloxycarbonylamino]-valinyl-3α α-amino)-Cholsäuremethylester (Boc-Val-CHS-OMe) 24a; EDC-Kopplung nach AAV VI Ansatzgröße: 798 mg (2 mmol) Boc-Val-OH·DCHA 41; 916 mg (2 mmol) HCl·H2N-CHS-OMe 12c Das Produkt kristallisierte aus einer Ethylacetat-Lösung des Rohproduktes (ca. 1.35 g). Isolierte Ausbeute: 1.145 g (1.844 mmol, 92.2 % der Theorie) MS (FAB, m/e): 643.3 (M+ + Na, 20 %); 621.4 (M+, 13 %); 521.3 (M+ - Boc, 20 %); 503.3 (6 %); 485.3 (34 %); 386.2 (8 %); 369.2 (10 %); 182.2 (52 %); 154.0 (17 %); 116.0 (30 %); 72.1 (100 %); 57.0 (89 %) 1 H-NMR (CDCl3, 400 MHz): 5.97 (d(br), 1H, Amid-NH); 5.00 (d(br), 1H, Boc-NH); 3.97 (m, 1H, CH-12 (CHS)); 3.84 (m, 1H, CH-7 (CHS)); 3.77 (dd, 1H, CHα (Val) Boc); 3.64 (s, 3H, OCH3 (CHS)); 3.62 (m(br), 1H, CHβ-3 (CHS) Amid); 2.38-2.31 u. 2.25-2.18 (2m, 2H, CH2-23 (CHS)); 2.08 (m, 1H, CHβ (Val)); 1.42 (s, 9H, (CH3)3 Boc); 0.96 (d, 3H, CH3-21 (CHS)); 0.91 u. 0.87 (2d, 6H, 2 x CH3 (Val)); 0.90 (s, 3H, CH3-19 (CHS)); 0.68 (s, 3H, CH3-18 (CHS)) 5. Experimenteller Teil 141 5.2.2.4.7.2. N-([t-Butyloxycarbonylamino]-valinyl-3α α-amino)-Cholsäure (Boc-Val-CHS-OH) 24b; Esterverseifung nach AAV II Ansatzgröße: 311 mg (0.500 mmol) Boc-Val-CHS-OMe 24a Ausbeute: 301 mg (0.496 mmol, 99.2 % der Theorie) MS (FAB, m/e): 629.3 (M+ + Na, 8 %); 607.3 (M+, 5 %); 507.3 (M+ + Na - Boc, 4 %); 489.3 (2 %); 471.3 (7 %); 355.2 (3 %); 289.0 (3 %); 175.7 (11 %); 154.0 (31 %); 136.0 (28 %); 81.0 (55 %); 72.0 (73 %); 57.0 (84 %); 55.0 (100 %); 1 H-NMR (CDCl3, 400 MHz): 6.10 (d(br), 1H, Amid-NH); 5.16 (d(br), 1H, Boc-NH); 3.98 (m, 1H, CH-12 (CHS)); 3.84 (m, 1H, CH-7 (CHS)); 3.82 (dd, 1H, CHα (Val) Boc); 3.61 (m(br), 1H, CHβ-3 (CHS) Amid); 2.42-2.34 u. 2.31-2.22 (2m, 2H, CH2-23 (CHS)); ca. 2.0 (m, 1H, CHβ (Val)); 1.42 (s, 9H, (CH3)3 Boc); 0.99 (d, 3H, CH3-21 (CHS)); 0.91 u. 0.89 (2d, 6H, 2 x CH3 (Val)); 0.90 (s, 3H, CH3-19 (CHS)); 0.68 (s, 3H, CH3-18 (CHS)) 5.2.2.4.7.3. (Boc-Val-CHS-Val-CHS-OMe) 31a; EDC-Kopplung nach AAV VI Ansatzgröße: 301 mg (0.496 mmol) Boc-Val-CHS-OH 24b; ca. 0.5 mmol (komplette Ausbeute aus der Boc-Abspaltung von 205 mg (0.338 mmol) Boc-Val-CHS-OMe 24a nach AAV IV; vgl. 5.2.2.4.6.3.) TFA·H2N-Val-CHS-OMe 24c Das reine Produkt konnte mit Hexan aus einer DCM-Lösung des Rohproduktes ausgefällt werden. Ausbeute: 527 mg (0.475 mmol, 95.8 % der Theorie) 5. Experimenteller Teil 142 MS (FAB, m/e): 1131.7 (M+ + Na, 6 %); 1109.7 (M+, 5 %); 1009.7 (M+ - Boc 7 %); 485.3 (2 %); 422.2 (4 %); 386.2 (4 %); 369.2 (7 %); 154.0 (28 %); 95.1 (47 %); 72.0 (90 %); 55.0 (100 %) 1 H-NMR (CDCl3, 400 MHz): 6.48 (d, 1H, Amid-NH (CHS‘)); 6.37 (d, 1H, Amid-NH (Val‘)); 6.19 (d, 1H, Amid-NH (CHS)); 5.14 (d(br), 1H, Boc-NH (Van)); 4.11 (dd, 1H, CHα (Val‘) Amid); 3.97 (2m, 2 x 1H, 2 x CH-12 (CHS u. CHS‘)); 3.88 (dd, 1H, CHα (Val) Boc); 3.83 (2m, 2 x 1H, 2 x CH-7 (CHS u. CHS‘)); 3.64 (s, 3H, OCH3 (CHS‘)); 3.60 (2m(br), 2 x 1H, 2 x CHβ-3 (CHS u. CHS‘)); 1.42 (s, 9H, (CH3)3 Boc); 1.00 u. 0.96 (2d, 2 x 3H, 2 x CH3-21 (CHS u. CHS‘)); 0.93-0.87 (insges. 18H: 2d, 6H, 2 x CH3 (Val); 2d, 6H, 2 x CH3 (Val‘); 2s, 2 x 3H, 2 x CH3-19 (CHS u. CHS‘)); 0.67 (2s, 2 x 3H, 2 x CH3-18 (CHS u. CHS‘)) 1 H-NMR (DMSO-d6, 400 MHz): 7.74 (d, 1H, Amid-NH (CHS‘)); 7.66 (d, 1H, Amid-NH (CHS)); 7.56 (d, 1H, Amid-NH (Val‘)); 6.36 (d, 1H, Boc-NH (Val)); 4.08 (dd, 1H, CHα (Val‘)); 4.03-3.99 (4d, 4 x 1H, 2 x C7-OH u. 2 x C12-OH (CHS u. CHS‘)); 3.78 (2m, 2 x 1H, 2 x CH-12 (CHS u. CHS‘)); 3.73 (dd, 1H, CHα (Val)); 3.62 (3m, 3 x 1H, 3 x CH-(CHS u. CHS‘)); 3.56 (s, 3H, OCH3 (CHS‘‘)); 3.36 (2m(br), 2 x 1H, 2 x CHβ-3 (CHS u. CHS‘)); 2.342.13 (2 x 2m, 2 x 2H, 2 x CH2-23 (CHS u. CHS‘)); 1.98 (2m, 2 x 1H, CHβ (Val u. Val‘)); 1.37 (s, 9H, (CH3)3 Boc); 0.93-0.90 (2d, 2 x 3H, 2 x CH3-21 (CHS u. CHS‘)); 0.83 (2s, 2 x 2H, 2 x CH3-19 (CHS u. CHS‘)); 0.82-0.77 (2 x 2d, 12H, 2 x CH3 (Val), 2 x CH3 (Val‘)); 0.58 (2s, 2 x 3H, 2 x CH3-18 (CHS u. CHS‘)) (Zur Differenzierung der NMR-Signale der einzelnen Bausteine (soweit möglich) wurde folgende Bezeichnung gewählt: Boc-Val-CHS-Val‘-CHS‘-OMe) 5.2.2.4.7.4. (Boc-Val-CHS-Val-CHS-Val-CHS-OMe) 32a; EDC-Kopplung nach AAV VI Ansatzgröße: 375 mg (0.342 mmol) Boc-Val-CHS-Val-CHS-OH 31b (aus Verseifung von 400 mg (0.360 mmol) des Esters Boc-Val-CHS-Val-CHS-OMe 31a nach AAV II); ca. 0.338 mmol TFA·H2N-Val-CHS-OMe 24c (komplette Ausbeute aus der Boc-Abspaltung von 205 mg (0.338 mmol) Boc-Val-CHS-OMe 24a nach AAV IV; vgl. 5.2.2.4.6.3.) Ausbeute: 437 mg (0.273 mmol, 79.9 % der Theorie) 5. Experimenteller Teil 143 MS (FAB, m/e): 1621.0 (M+ + Na, 8 %); 1599.0 (M+, 4 %); 1499.5 (M+ - Boc, 11 %); 674.3 (2 %); 453.3 (4 %); 386.2 (8 %); 154.0 (16 %); 72.1"(100 %) 1 H-NMR (DMSO-d6, 400 MHz): 7.72 (2d, 2 x 1H, Amid-NH (CHS‘ u. CHS‘‘)); 7.66 (d, 1H, Amid-NH (CHS)); 7.55 (2d, 2 x 1H, Amid-NH (Val‘ u. Val‘‘)); 6.33 (d, 1H, Boc-NH (Val)); 4.08 (2dd, 2 x 1H, CHα (Val‘ u. Val‘‘)); 4.00-3.97 (6d, 6 x 1H, 3 x C7-OH u. 3 x C12-OH (CHS, CHS‘ u. CHS‘‘)); 3.79 (3m, 3 x 1H, 3 x CH-12 (CHS, CHS‘ u. CHS‘‘)); 3.74 (dd, 1H, CHα (Val)); 3.62 (3m, 3 x 1H, 3 x CH-7 (CHS, CHS‘ u. CHS‘‘)); 3.56 (s, 3H, OCH3 (CHS‘‘)); 3.37 (3m(br), 3 x 1H, 3 x CHβ-3 (CHS, CHS‘ u. CHS‘‘)); 2.34-2.14 (3 x 2m, 3 x 2H, 3 x CH2-23 (CHS, CHS‘ u. CHS‘‘)); 1.98 (3m, 3 x 1H, CHβ (Val, Val‘ u. Val‘‘)); 1.37 (s, 9H, (CH3)3 Boc); 0.93-0.91 (3d, 3 x 3H, 3 x CH3-21 (CHS, CHS‘ u. CHS‘‘)); 0.83 (3s, 3 x 3H, 3 x CH3-19 (CHS, CHS‘ u. CHS‘‘)); 0.81-0.78 (3 x 2d, 18H, 2 x CH3 (Val), 2 x CH3 (Val‘), 2 x CH3 (Val‘‘)); 0.58 (3s, 3 x 3H, 3 x CH3-18 (CHS, CHS‘ u. CHS‘‘)) (Zur Differenzierung der NMR-Signale der einzelnen Bausteine (soweit möglich) wurde folgende Bezeichnung gewählt: Boc-Val-CHS-Val‘-CHS‘-Val‘‘-CHS‘‘-OMe) 5. Experimenteller Teil 144 5.2.2.5. Cyclische Pseudopeptide (bzw. Oligoamide) aus Aminocholansäurebausteinen 5.2.2.5.1. Unsystematische Oligocyclisierung monomerer 3α α-Aminocholansäuren 5.2.2.5.1.1. Oligocyclisierung von 3α α-Aminolithocholsäure nach AAV VII Ansatzgröße: 476 mg (1 mmol) 3α-(t-Butyloxycarbonylamino)-Lithocholsäure 11a Rohprodukt: 453 mg Die säulenchromatographische Auftrennung des Produktgemisches, das laut FAB-Massenspektrum Aminolithocholsäure-cyclodiamid 33a, -cyclotriamid 33b und geringe Mengen des Cyclotetramers 33c enthielt, war bislang leider nicht erfolgreich, da die beiden Hauptprodukte, wahrscheinlich Cyclodimer 33a und Cyclotrimer 33b, selbst in verschiedenen getesteten Laufmittelgemischen (DCM:Hexan, DCM:Hexan:MeOH, EE:Hexan, THF:Hexan, THF:Hexan:MeOH, TBME:Hexan, Et2O:Hexan, jeweils in unterschiedlichen Mischungsverhältnissen) sehr ähnliche Laufeigenschaften zeigten (∆Rf sehr gering) und in der FlashSäulenchromatographie nur leichte Antrennungen beobachtet werden konnten. Da die unfunktionalisierten Aminolithocholsäurecyclen nur als Modellsysteme für die auf der Innenseite noch OH-funktionalisierten Aminodesoxycholsäure- und AminocholsäureCyclooligoamide dienten und keine weitere Verwendung geplant war, wurde hier auf weitere Trennversuche (HPLC etc.) verzichtet. Im 1H-NMR-Spektrum des Rohproduktes zeigten sich im Bereich der NH-Protonen drei Signale im Intensitätsverhältnis 1 : 0.85 : 0.17, woraus man auf ein Massenverhältnis der Produkte von 49 % : 42 % : 9 % schließen kann (Annahme: Die Cyclen zeigen aufgrund ihrer Symmetrie jeweils einen einfachen Signalsatz; da die Grundeinheit, der monomere Baustein, jeweils gleich ist, geben die molaren Intensitäten dann direkt das Massenverhältnis der Produkte wieder). Analytischg Daten des Gemisches: MS (FAB, m/e): 1430.3 (M+ Cyclotetramer, 1 %); 1073.0 (M+ Cyclotrimer, 3 %); 715.6 (M+ Cyclodimer, 6 %) 1 H-NMR (DMSO-d6, 400 MHz): 7.80 (d, Amid-NH, Intensität: 0.3870); 7.56 (d, Amid-NH, Intensität: 0.4555); 6.82 (d, Amid-NH, Intensität: 0.0796); 3.37 (m(br), jeweils CHβ-3, Gesamtintensität: 1.000); 0.93-0.86 (jeweils CH3-19 und CH3-21); 0.61 u. 0.57 (jeweils CH3-18) 5. Experimenteller Teil 145 5.2.2.5.1.2. Oligocyclisierung von 3α α-Aminodesoxycholsäure nach AAV VII Ansatzgröße: 492 mg (1 mmol) 3α-(t-Butyloxycarbonylamino)-Desoxycholsäure 11b Rohprodukt: 462 mg Wie bei der Oligocyclisierung von Aminolithocholsäure zeigte auch hier das Massenspektrum des Produktgemisches Massenpeaks des Cyclodimers 34a, des Cyclotrimers 34b und des Cyclotetramers 34c. Das Produktgemisch wurde säulenchromatographisch getrennt. Als Laufmittel diente zunächst THF:Hexan:MeOH (Gradient: 20:90:5 –> 40:80:5.5). Die noch verunreinigten Fraktionen, die Cyclodimer und Cyclotrimer enthielten, wurden danach nochmals mit DCM:Hexan:MeOH (Gradient: 30:90:5 –> 40:40:20) säulenchromatographisch gereinigt. Isolierte Ausbeuten: 3α-Aminodesoxycholsäure-Cyclodiamid 34a: 120 mg (0.161 mmol; 32.1 % der Theorie) 3α-Aminodesoxycholsäure-Cyclotriamid 34b: 34 mg (0.030 mmol; 9.1 % der Theorie) Das Cyclotetramer 34c konnte leider nicht isoliert werden, da es wahrscheinlich in zu geringen Mengen im Rohprodukt enthalten war. Das Cyclodimer 34a und das Cyclotrimer 34b kristallisierten jeweils aus DCM/MeOH. Während jedoch die Kristallstruktur des Cyclodimers gelöst werden konnte, gelang dies beim Cyclotrimer bislang nicht, da die (makroskopisch einkristallin erscheinenden) Kristalle aus zahlreichen übereinandergelagerten einkristallinen(?) Schichten aufgebaut sind, wie eine genauere mikroskopische Analyse ergab. Auch andere Lösemittelgemische lieferten hier bislang keine besseren Ergebnisse. Analytische Daten der beiden Cyclen: 5. Experimenteller Teil 146 3α-Aminodesoxycholsäure-Cyclodiamid 34a: Kristallstrukturdaten des Cyclodimers siehe Anhang C. MS (FAB, m/e): 769.6 (M+ + Na, 14 %); 747.6 (M+, 7 %); 711.6 (36 %); 410.3 (7 %); 281.1 (9 %); 147.1 (34 %); 107.1 (34 %); 81.1 (58 %); 55.0 (100 %) 1 H-NMR (DMSO-d6, 400 MHz): 7.71 (d, 2H, 2 x Amid-NH); 3.83 (d, 2H, 2 x C12-OH); 3.60 (m, 2H, 2 x CH-12); 3.48 (m(br), 2H, 2 x CHβ-3); 0.96 (d, 6H, 2 x CH3-21); 0.88 (s, 6H, 2 x CH3-19); 0.57 (s, 6H, 2 x CH3-18) Anmerkungen zum NMR: Das Spektrum zeigt einen einfachen Signalsatz aufgrund der C2-Symmetrie des Cyclus. Das kristalline Cyclodimer ist in DMSO-d6 nur sehr schlecht löslich, in CDCl3 gar nicht. 3α-Aminodesoxycholsäure-Cyclotriamid 34b: MS (FAB, m/e): 1142.9 (M+ + Na, 15 %); 1120.9 (M+, 25 %); 1102.9 (3 %); 1084.9 (3 %); 1066.9 (17 %); 413.2 (2 %); 329.1 (8 %); 259.1 (13 %); 175.8 (37 %); 152.0 (96 %); 136.0 (97 %); 55.0 (100 %) 1 H-NMR (CDCl3, 400 MHz): 5.22 (d, 3H, 3 x Amid-NH); 3.98 (m, 3H, 3 x CH-12); 3.77 (m(br), 3H, 3 x CHβ-3); 2.21-2.13 u. 2.10-2.02 (2m, 2 x 3H, 3 x CH2-23); 0.98 (d, 9H, 3 x CH3-21); 0.91 (s, 9H, 3 x CH3-19); 0.67 (s, 9H, 3 x CH3-18) Das NMR-Spektrum zeigt einen einfachen Signalsatz aufgrund der C3-Symmetrie des Cyclus. 5.2.2.5.1.3. Oligocyclisierung von 3α α-Aminocholsäure nach AAV VII Ansatzgröße: 508 mg (1 mmol) 3α-(t-Butyloxycarbonylamino)-Cholsäure 11c In Abweichung von der AAV wurde der Ansatz folgendermaßen aufgearbeitet: Zunächst wurde die wäßrige Phase mit NaCl gesättigt und (weitgehend) von der organischen Phase getrennt, anschließend dann das Präzipitat, das sich an der Phasengrenze bzw. in der wäßrigen Phase gebildet hatte, abgesaugt. Die organische Phase wurde gemäß der AAV weiter aufgearbeitet, enthielt aber laut Massenspektrum keine Cyclooligomere der Aminocholsäure. Das Präzipitat hingegen (228 mg) bestand fast ausschließlich aus dem 3α-Aminocholsäurecyclodiamid 35a, vermischt mit geringen Mengen Cyclotrimer, Cyclotetramer und weiteren, nicht näher charakterisierten Verunreinigungen (insgesamt ca. 5-15 %). Die Reinigung erfolgte durch Kristallisation aus THF/EtOH und Dioxan/DCM, hierbei wurde reines Cyclodimer erhalten. Die massenspektrometrisch nachgewiesenen höheren Cyclooligomere (Cyclotrimer und Cyclotetramer) konnten leider nicht isoliert werden, da sie wahrscheinlich in zu geringer Menge im Präzipitat enthalten waren. 5. Experimenteller Teil 147 Isolierte Ausbeute an 3α-Aminocholsäure-Cyclodiamid 35a: 189 mg (0.243 mmol; 48.5 % der Theorie) Analytische Daten von 3α-Aminocholsäure-Cyclodiamid 35a: MS (FAB, m/e): 801.5 (M+ + Na, 25 %); 779.5 (M+, 16 %); 761.5 (2 %); 743.5 (2 %); 725.5 (2 %); 707.5 (19 %); 391.3 (4 %); 329.1 (11 %); 259.1 (17 %); 154.0 (100 %) 1 H-NMR (DMSO-d6, 400 MHz): 7.34 (d, 2H, 2 x Amid-NH); 3.97 (d, 2H, 2 x C7-OH); 3.74 (m, 2H, 2 x CH-12); 3.64 (m, 2H, 2 x CH-7); 3.31 (m(br), 2H, 2 x CHβ-3); 3.15 (d, 2H, 2 x C12-OH); 0.93 (d, 6H, 2 x CH3-21); 0.81 (s, 6H, 2 x CH3-19); 0.57 (s, 6H, 2 x CH3-18) Das Spektrum zeigt einen einfachen Signalsatz aufgrund der C2-Symmetrie des Cyclus. 5.2.2.5.2. Systematische Cyclisierung linearer Oligoamide 5.2.2.5.2.1. Cyclisierung reiner 3α α-Aminocholansäure-Oligoamide (Typ 1) 5.2.2.5.2.1.1. Cyclisierung von Boc-DCS-DCS-DCS-OMe 16a nach AAV VII Ansatzgröße: 500 mg (0.399 mmol) Boc-DCS-DCS-DCS-OMe 16a Der Methylester wurde zunächst nach AAV II verseift und anschleißend ohne weitere Reinigung den Cyclisierungsbedingungen (AAV VII) unterworfen. Rohprodukt: 578 mg Die dünnschichtchromatographische Analyse zeigte eine Reihe von Produkten, jedoch waren klar zwei Hauptprodukte erkennbar. Die säulenchromatographische Trennung des Produktgemisches (zunächst Abtrennung des Dicyclohexylharnstoffes mit 100 % Chloroform, dann Trennung der übrigen Produkte mit DCM:Hexan:MeOH (Gradient: 20:120:5 –> 50:50:10)) lieferte reines Cyclotrimer 34b und eine Mischfraktion (95 mg), die zu einem Massenanteil von ca. 40-50 % (abgeschätzt mittels NMR-Intensitäten) das Cyclohexamer 34d (Produkt der Cyclodimerisierung) enthielt. Letztere wurde nochmals säunenchromatographisch aufgetrennt (Gradient: 10:120:2.5 –> 40:120:5) und lieferte reines Cyclohexamer. 5. Experimenteller Teil 148 Isolierte Ausbeuten: 3α-Aminodesoxycholsäure-Cyclotriamid 34b: 185 mg (0.165 mmol; 41.4 % der Theorie) 3α-Aminodesoxycholsäure-Cyclohexaamid 34d: 42 mg (0.019 mmol; 9.4 % der Theorie) Analytische Daten: 3α-Aminodesoxycholsäure-Cyclotriamid 34b: Die analytischen Daten stimmen mit denen des Cyclotrimers aus 5.2.2.5.1.2. überein. Zusätzlich wurde ein 600MHz-NMR-Spektrum aufgenommen und eine Probe mittels MALDI massenspektrometrisch untersucht: 1 H-NMR (CDCl3, 600 MHz): 5.21 (d, 3H, 3 x Amid-NH); 3.97 (m, 3H, 3 x CH-12); 3.76 (m(br), 3H, 3 x CHβ-3); 2.19-2.13 u. 2.09-2.03 (2m, 2 x 3H, 3 x CH2-23); 0.98 (d, 9H, 3 x CH3-21); 0.90 (s, 9H, 3 x CH3-19); 0.67 (s, 9H, 3 x CH3-18) Das NMR-Spektrum zeigt einen einfachen Signalsatz aufgrund der C3-Symmetrie des Cyclus. Das MALDI-Spektrum bestätigt die Ergebnisse des FAB-Massenspektrums (vgl. 5.2.2.5.1.2.) und zeigt einen Massenpeak sowie einen Peak bei M+ + Na. 3α-Aminodesoxycholsäure-Cyclohexaamid 34d: Da das FAB-Massenspektrometer lediglich bis in einen m/e-Bereich von 2000-2100 mißt, mußte für die massenspektrometrische Analyse des Cyclohexamers auf die MALDI zurückgegriffen werden. MS (MALDI): 1 ein einziger Peak bei 2268.63 (M+ + Na; theoret.: 2264.46), sowie in sehr geringen Intensitäten der Massenpeak und darauffolgend eine Reihe von sechs weiteren Peaks, jeweils im Abstand von ca. 18 m/z-Einheiten (sechsmalige Abspaltung von H2O (aus 6 OH-Gruppen) aus dem Molekül). H-NMR (CDCl3, 400 MHz): 5.88 (d, 3H, 3 x Amid-NH); 3.96 (m, 3H, 3 x CH-12); 3.80 (m(br), 3H, 3 x CHβ-3); 2.11 (2m, 2 x 3H, 3 x CH2-23); 0.98 (d, 9H, 3 x CH3-21); 0.91 (s, 9H, 3 x CH3-19); 0.67 (s, 9H, 3 x CH3-18) Das Spektrum zeigt einen einfachen Signalsatz aufgrund der C6-Symmetrie des Cyclus. 5. Experimenteller Teil 149 5.2.2.5.2.1.2. Cyclisierung von Boc-DCS-DCS-DCS-DCS-OMe 17a nach AAV VII Ansatzgröße: 500 mg (0.307 mmol) Boc-DCS-DCS-DCS-DCS-OMe 17a Der Methylester wurde zunächst nach AAV II verseift und anschleißend ohne weitere Reinigung den Cyclisierungsbedingungen (AAV VII) unterworfen. Rohprodukt: 487 mg Die säulenchromatographische Trennung des Produktgemisches (zunächst Abtrennung des Dicyclohexylharnstoffes mit 100 % Chloroform, dann Trennung der übrigen Produkte mit DCM:Hexan:MeOH (Gradient: 20:120:5 –> 50:50:10)) lieferte das reine Cyclotetramer 34c. Isolierte Ausbeute: 3α-Aminodesoxycholsäure-Cyclotetraamid 34c: 145 mg (0.097 mmol; 31.6 % der Theorie) MS (FAB, m/e): 1516.4 (M+ + Na, 4 %); 1494.4 (M+, 13 %); 460.2 (2 %); 307.0 (20 %); 259.0 (17 %); 154.0 (100 %); 72.1 (86 %) 1 H-NMR (CDCl3, 400 MHz): 5.27 (d, 4H, 4 x Amid-NH); 3.98 (m, 4H, 4 x CH-12); 3.75 (m(br), 4H, 4 x CHβ-3); 2.18-2.10 u. 2.08-2.00 (2m, 2 x 4H, 4 x CH2-23); 0.97 (d, 12H, 4 x CH3-21); 0.90 (s, 12H, 4 x CH3-19); 0.67 (s, 12H, 4 x CH3-18) Das NMR-Spektrum zeigt einen einfachen Signalsatz aufgrund der C4-Symmetrie des Cyclus. 5.2.2.5.2.1.3. Cyclisierung von Boc-CHS-CHS-CHS-OMe 19a nach AAV VII Ansatzgröße: 500 mg (0.389 mmol) Boc-CHS-CHS-CHS-OMe 19a Der Methylester wurde zunächst nach AAV II verseift und anschleißend ohne weitere Reinigung den Cyclisierungsbedingungen (AAV VII) unterworfen. Ähnlich wie bei der unsystematischen Oligocyclisierung trat auch hier bei der Aufarbeitung eine kleine Menge Präzipitat auf (16 mg), welche zunächst isoliert wurde. Die kernresonanzspektroskopische und massenspektrometrische Analyse zeigte, dass es etwa 30-50 % Cyclotrimer 35b enthielt. Eine Reinisolierung gelang jedoch aufgrund der schlechten Löslichkeit nicht. Die organische Phase wurde nach der Aufarbeitung eingeengt und anschließend nochmals in wenig Chloroform aufgenommen. Daraus kristallisierten langsam würfelförmige Kristalle, die mittels MALFI und NMR als Cyclohexamer 35d identifiziert werden konnten. Eine Bestimmung der Kristallstruktur gelang leider bislang nicht. 5. Experimenteller Teil 150 Isolierte Ausbeute: 3α-Aminocholsäure-Cyclohexaamid 35d: 148 mg (0.063 mmol; 32.6 % der Theorie) Analytische Daten: Da das FAB-Massenspektrometer lediglich bis in einen m/e-Bereich von 2000-2100 mißt, mußte für die massenspektrometrische Analyse des Cyclohexamers auf die MALDI zurückgegriffen werden. MS (MALDI): ein einziger Peak bei 2363.41 (M+ + Na; theoret.: 2360.45), sowie in sehr geringer Intensität der Massenpeak. Weitere Peaks traten oberhalb von 500 nicht auf bzw. waren in unterschiedlichen Matrizes nicht reproduzierbar, woraus sich schließen läßt, dass es sich hierbei um Matrixeffekte handelt. 1 H-NMR (CDCl3, 400 MHz): 5.78 (d, 6H, 6 x Amid-NH); 3.95 (m, 6H, 6 x CH-12); 3.95 (m, 6H, 6 x CH-7); 3.64 (m(br), 6H, 6 x CHβ-3); 0.98 (d, 18H, 6 x CH3-21); 0.89 (s, 18H, 6 x CH3-19); 0.67 (s, 18H, 6 x CH3-18) Das Spektrum zeigt einen einfachen Signalsatz aufgrund der C6-Symmetrie des Cyclus. 5.2.2.5.2.1.4. Cyclisierung von Boc-CHS-CHS-CHS-CHS-OMe 20a Ansatzgröße: 500 mg (0.296 mmol) Boc-CHS-CHS-CHS-CHS-OMe 20a Der Methylester wurde zunächst nach AAV II verseift und anschleißend ohne weitere Reinigung den Cyclisierungsbedingungen (AAV VII) unterworfen. Bereits während der Reaktion trat eine größere Menge Präzipitat auf, die zunächst abgetrennt wurde. Auch bei der weiteren Aufarbeitung traten kleinere Mengen Präzipitat auf, die jeweils gesondert untersucht wurden. Das bereits während der Reaktion ausgefallene Präzipitat konnte als Cyclotetramer identifiziert werden. Aufgrund der sehr schlechten Löslichkeit des Präzipitats (es löste sich lediglich in DMSO) gelang eine weitere Reinigung jedoch bislang nicht. Isolierte Ausbeute: 3α-Aminocholsäure-Cyclotetraamid 35c: 234 mg (0.150 mmol; 50.7 % der Theorie) 5. Experimenteller Teil MS (MALDI): 151 Peaks bei 1580.12 (M+ + Na; theoret.: 1580.30), 1602.11 (M+ + 2Na), 1624.13 (M+ + 3Na), sowie in sehr geringer Intensität der Massenpeak. In DHB-Matrix traten oberhalb von 500 weitere Peaks auf, jedoch waren sie in aCN-Matrix nicht reproduzierbar, woraus sich schließen läßt, dass es sich hierbei möglicherweise um Matrixeffekte handelt. 1 H-NMR (DMSO-d6, 400 MHz): 7.65 (d, 4H, 4 x Amid-NH); 4.05 (br, 2 x 4H, C7-OH u. C12-OH); 3.78 (m, 4H, 4 x CH-12); 3.61 (m, 4H, 4 x CH-7); 3.29 (m(br), 4H, 4 x CHβ-3); 0.90 (d, 12H, 4 x CH3-21); 0.83 (s, 12H, 4 x CH3-19); 0.58 (s, 12H, 4 x CH3-18) Das NMR-Spektrum zeigt einen einfachen Signalsatz aufgrund der C4-Symmetrie des Cyclus. 5.2.2.5.2.2. Cyclisierung gemischter L-Aminosäure-/3α α-Aminocholansäure-Oligoamide (Typ 2) 5.2.2.5.2.2.1. Cyclisierung von Boc-Val-LCS-Val-LCS-Val-LCS-OMe 26a Ansatzgröße: 500 mg (0.333 mmol) Boc-Val-LCS-Val-LCS-Val-LCS-OMe 26a Der Methylester wurde zunächst nach AAV II verseift und anschleißend ohne weitere Reinigung den Cyclisierungsbedingungen (AAV VII) unterworfen. Das Rohprodukt DCM:Hexan:MeOH (480 mg) 20:120:2.5; wurde die säulenchromatographisch noch mit gereinigt Dicyclohexylharnstoff (Laufmittel verunreinigte Produktfraktion wurde nochmals chromatographiert. Laufmittel: zunächst Chloroform 100 %, danach DCM:Hexan:MeOH 50:50:10). Das Produkt (Cyclotrimer 36) kristallisiert aus DCM/MeOH. Höhere Cyclooligomere konnten nicht isoliert bzw. gefunden werden. Isolierte Ausbeute: 3α-Valinylaminolithocholsäure-Cyclotriamid 36: 220 mg (0.161 mmol; 48.3 % der Theorie) MS (FAB, m/e): 1369.7 (M+, 6 %); 457.2 (2 %); 412.2 (1 %); 358.2 (2 %); 121.1 (5 %); 72.1 (100 %) 1 H-NMR (CDCl3, 400 MHz): 8.28 (d, 3H, 3 x Amid-NH (LCS)); 6.10 (d, 3H, 3 x Amid-NH (Val)); 4.56 (m, 3H, 3 x CHα (Val)); 3.64 (m(br), 3H, 3 x CHβ-3 (LCS)); 2.21-2.16 u. 2.07-2.01 (2m, 2 x 3H, 3 x CH2-23); 0.95 (d, 9H, 3 x CH3-21 (LCS)); 0.92 (s, 9H, 3 x CH3-19 (LCS)); 0.90 u. 0.89 (2d, 2 x 9H, 6 x CH3 (Val)); 0.67 (s, 9H, 3 x CH3-18 (LCS)) Das NMR-Spektrum zeigt einen einfachen Signalsatz aufgrund der C3-Symmetrie des Cyclus. 5. Experimenteller Teil 152 5.2.2.5.2.2.2. Cyclisierung von Boc-Lys[Z]-LCS-Lys[Z]-LCS-Lys[Z]-LCS-OMe 28a Ansatzgröße: 500 mg (0.251 mmol) Boc-Lys[Z]-LCS-Lys[Z]-LCS-Lys[Z]-LCS-OMe 28a Der Methylester wurde zunächst nach AAV II verseift und anschleißend ohne weitere Reinigung den Cyclisierungsbedingungen (AAV VII) unterworfen. Das Rohprodukt (358 mg) wurde säulenchromatographisch gereinigt (Laufmittel: zunächst Chloroform 100%, dann DCM:Hexan:MeOH, Gradient: 20:240:5 –> 20:120:5 –> 20:60:5). Das Produkt (Cyclotrimer) kristallisiert aus DCM/MeOH. Höhere Cyclooligomere konnten nicht isoliert bzw. gefunden werden. Isolierte Ausbeute an 3α-([ε-benzyloxycarbonylamino]lysinylamino)-Lithocholsäure-Cyclotriamid 37: 87 mg (0.047 mmol; 18.6 % der Theorie) MS (FAB, m/e): 1878.8 (M+ + Na, 0.2 %); 1856.8 (M+ (theor. 1859.7), 0.6 %); 1423.6 (0.6 %); 804.3 (1.5 %); 663.3 (0.7 %); 540.1 (0.8 %); 402.1 (1 %); 225.1 (21.2 %); 154.0 (26 %); 91.0 (100 %) 1 H-NMR (CDCl3, 400 MHz): 8.08 (d, 3 x 1H, 3 x Amid-NH (LCS)); 7.33-7.26 (m, 3 x 5H, 3 x Phenyl-H der Z-Schutzgruppe); 6.17 (d, 3 x 1H, 3 x Amid-NH (Lys)); 5.03 (s, 3 x 2H, 3 x CH2(Z)); 5.03 (t, 3 x 1H, 3 x Z-NH); 4.72 (m, 3 x 1H, 3 x CHα (Lys)); 3.59 (m(br), 3 x 1H, 3 x CHβ-3 (LCS)); 3.09 (m, 3 x 2H, 3 x CH2ε (Lys)); 2.15 u. 1.96 (2m, 3 x 2H, 3 x CH2-23 (LCS)); 0.92 (s, 3 x 3H, 3 x CH3-19 (LCS)); 0.91 (d, 3 x 3H, 3 x CH3-21 (LCS)); 0.65 (s, 3 x 3H, 3 x CH3-18 (LCS)) Das NMR-Spektrum zeigt einen einfachen Signalsatz aufgrund der C3-Symmetrie des Cyclus. 5. Experimenteller Teil 153 5.2.2.5.2.2.3. Cyclisierung von Boc-Val-DCS-Val-DCS-Val-DCS-OMe 30a Ansatzgröße: 400 mg (0.258 mmol) Boc-Val-DCS-Val-DCS-Val-DCS-OMe 30a Der Methylester wurde zunächst nach AAV II verseift (Ausbeute: 373 mg; 0.243 mmol; 94.2 % d. Th.) und anschleißend ohne weitere Reinigung den Cyclisierungsbedingungen (AAV VII) unterworfen. Das Rohprodukt (ca. 380 mg) wurde säulenchromatographisch gereinigt (Laufmittel: zunächst Chloroform 100%, dann DCM:Hexan:MeOH 20:120:5). Dabei konnten zwei Produkte isoliert werden, von denen eines als Cyclotrimer (38a) identifiziert werden konnte und das andere die doppelte Masse und einen doppelten Signalsatz im NMR aufwies. Isolierte Ausbeute: 3α-Valinylaminodesoxycholsäure-Cyclotriamid 38a: 113 mg (0.080 mmol; 32.8 % d. Th.) 3α-Valinylaminodesoxycholsäure-Cyclohexaamid 38b: 75 mg (0.026 mmol; 21.8 % d. Th.) Analytische Daten: 3α-Valinylaminodesoxycholsäure-Cyclotriamid 38a: MS (FAB, m/e): 1418.2 (M+, 5 %); 541.1 (4 %); 397.1 (9 %); 307.1 (22 %); 235.1 (56 %); 163.0 (100 %); 91.0 (76 %) 1 H-NMR (CDCl3, 400 MHz): 8.09 (d, 3H, 3 x Amid-NH (DCS)); 6.07 (d, 3H, 3 x Amid-NH (Val)); 4.58 (m, 3H, 3 x CHα (Val)); 3.93 (m, 3H, 3 x CH-12 (DCS)); 3.50 (m(br), 3H, 3 x CHβ-3 (DCS)); 2.25 u. ~2.03 (2m, 2 x 3H, 3 x CH2-23); 0.98 (d, 9H, 3 x CH3-21 (DCS)); 0.90 (s, 9H, 3 x CH3-19 (DCS)); 0.89 u. 0.87 (2d, 2 x 9H, 6 x CH3 (Val)); 0.66 (s, 9H, 3 x CH3-18 (DCS)) Das NMR-Spektrum zeigt einen einfachen Signalsatz aufgrund der C3-Symmetrie des Cyclus. 5. Experimenteller Teil 154 3α-Valinylaminodesoxycholsäure-Cyclohexaamid 38b: MS (MALDI): in DHB-Matrix ein einziger Peak bei 2864.62 (M+ + Na; theoret.: 2858.26), sowie in sehr geringer Intensität der Massenpeak (bei 2842.44). Weitere Peaks traten oberhalb von 500 nicht auf bzw. waren in aCN-Matrix nicht reproduzierbar, woraus sich schließen läßt, dass es sich hierbei um Matrixeffekte handelt. 1 H-NMR (CDCl3, 600 MHz): zwei Signalsätze (A und B): 7.80 (d, 3H, 3 x Amid-NH (ValA)); 7.76 (d, 3H, 3 x Amid-NH (DCSB)); 6.85 (d, 3H, 3 x Amid-NH (DCSA)); 6.08 (d, 3H, 3 x Amid-NH (ValB)); 4.63 (dd, 3H, 3 x CHα (ValB)); 3.90 (m(br), 3H, 3 x CH-12 (DCSB)); 3.85 (m(br), 3H, 3 x CH-12 (DCSA)); 3.77 (dd, 3H, 3 x CHα (ValA)); 3.77 (m(br), 3H, 3 x CHβ-3 (DCSA)); 3.40 (m(br), 3H, 3 x CHβ-3 (DCSB)); 2.43-2.37 u. 2.32-2.26 (2m, 2 x 3H, 3 x CH2-23 (DCSB)); 1.07 u. 1.00 (2d, 2 x 9H, 6 x CH3 (ValA)); 1.01 (d, 9H, 3 x CH3-21 (DCSB)); 0.97 u. 0.91 (2d, 2 x 9H, 6 x CH3 (ValB)); 0.94 (d, 9H, 3 x CH3-21 (DCSA)); 0.86 (s, 9H, 3 x CH3-19 (DCSB)); 0.84 (s, 9H, 3 x CH3-19 (DCSA)); 0.69 (s, 9H, 3 x CH3-18 (DCSB)); 0.60 (s, 9H, 3 x CH3-18 (DCSA)) 13 C-NMR (CDCl3, 100 MHz): (Es sind nur Verschiebungen angegeben, die sicher zugeordnet werden konnten) 74.39 (C12 DCSA); 73.14 (C12 DCSB); 60.65 (Cα ValA); 58.03 (Cα ValB); 49.61 (C3 DCSB); 46.71 (C3 DCSA); 38.13 (C21 DCSA); 34.52 (C21 DCSB); 34.35 (C23 (o. C22) DCSA); 31.88 (C22 (o. C23) DCSA); 31.78 (Cβ ValA); 31.64 (C23 DCSB); 31.03 (Cβ DCSB); 30.44 (C22 DCSB); 22.57 (C19 DCSB); 22.33 (C19 DCSA); 13.07 (C18 DCSA); 12.43 (C18 DCSB) 5.2.2.5.2.2.4. Cyclisierung von Boc-Val-CHS-Val-CHS-Val-CHS-OMe 32a Ansatzgröße: 400 mg (0.250 mmol) Boc-Val-CHS-Val-CHS-Val-CHS-OMe 32a Der Methylester wurde zunächst nach AAV II verseift und anschleißend ohne weitere Reinigung den Cyclisierungsbedingungen (AAV VII) unterworfen. Bereits während der Reaktion trat eine größere Menge Präzipitat auf, die zunächst abgetrennt wurde. Auch bei der weiteren Aufarbeitung traten kleinere Mengen Präzipitat auf, die jeweils gesondert untersucht wurden. Das bereits während der Reaktion ausgefallene Präzipitat konnte bislang nicht sicher identifiziert werden, es gibt jedoch Hinweise darauf, dass es hauptsächlich aus cyclo-(Val-CHS)3 39 besteht. Aufgrund der sehr schlechten Löslichkeit des Präzipitats (es löste sich lediglich in DMSO) gelang eine Reinisolierung bisher nicht. Isolierte Ausbeute (Präzipitat): 198 mg (0.135 mmol; 54.0 % der Theorie; jeweils unter der Voraussetzung, dass es sich tatsachlich um 3α-Valinylaminocholsäure-Cyclotriamid 39 handelt) 5. Experimenteller Teil 155 analytische Daten (Präzipitat): MS (FAB, m/e): Ein Massenpeak, der der Masse von Cyclo-(CHS)3 entspricht (1466.13), konnte nicht gefunden werden. 1528.5 (5 %); 1506.5 (4 %); 1484.5 (9 %); 329.1 (22 %); 176.0 (56 %); 72.1 (100 %) MALDI: Peaks bei 1531.5 und 1508.5 (in DHB-Matrix). 1 H-NMR (DMSO-d6, 400 MHz): Da das Präzipitat auch in DMSO nur schlecht löslich war, waren alle NMR-Signale stark verbreitert. Eine sichere Zuordnung war deshalb nicht möglich. 6. Literaturverzeichnis 156 6. Literaturverzeichnis [1] Für Übersichten zur Supramolekularen Chemie siehe z.B. J.-M. Lehn, Supramolecular Chemistry, VCH, 1995; F. Vögtle, Supramolekulare Chemie, Teubner, 1989. [2] J. Gante, Angew. Chem. 1994, 106, 1780-1802; D.S. Kemp, Trends Biotechnol. 1990, 8, 249; R. Hirschmann, Angew. Chem. 1991, 103, 1305-1330; A. Giannis, T. Kolter, Angew. Chem. 1993, 105, 1303-1326; M. Kahn, Synlett. 1993, 821; K. Müller, D. Obrecht, A. Knierzinger, C. Stankovic, C. Spiegler, W. Bannwarth, A. Trzeciak, G. Englert, A.M. Labhardt, P. Schönholzer, Perspect. Med. Chem. 1993, 513-531. [3] H.-J. Schneider, A. Yatsimirsky, Principles and Methods in Supramolecular Chemistry, Wiley, 2000; siehe außerdem Ref. [1]. [4] C.J. Pedersen, J. Am. Chem. Soc. 1967, 89, 2495-2496; C.J. Pedersen, J. Am. Chem. Soc. 1967, 89, 7017-7036; C.J. Pedersen, Angew. Chem. 1988, 100, 1053-1059. [5] B. Dietrich, J.-M. Lehn, J.-P. Sauvage, Tetrahedron Lett. 1969, 2885-2888; B. Dietrich, J.-M. Lehn, J.-P. Sauvage, Tetrahedron Lett. 1969, 2889-2892; J.-M. Lehn, Angew. Chem. 1988, 100, 91-116. [6] E.P. Kyba, R.C. Helgeson, K. Modan, G.W. Gokel, T.L. Tarnowski, S.S. Moore, D.J. Cram, J. Am. Chem. Soc. 1977, 99, 2564-2571; D.J. Cram, Angew. Chem. 1988, 100, 1041-1052. [7] Eine Übersicht hierzu gibt beispielsweise: A.P. Davis, Chem. Soc. Rev. 1993, 243-253. [8] J. Inanaga, K. Hirata, H. Saeki, T. Katusi, M. Yamaguchi, Bull Chem. Soc. Jpn. 1979, 52, 1989. [9] R.P. Bonar-Law, J.K.M. Sanders, Tetrahedron Lett. 1992, 33, 2071-2074. [10] K. Lappalainen, E. Kolehmainen, Liebigs Ann. / Recueil 1997, 1965-1968. [11] R.P. Bonar-Law, L.G. Mackay, C.J. Walter, V. Marvaud, J.K.M. Sanders, Pure Appl. Chem. 1994, 66, 803-810. [12] P.A. Brady, J.K.M. Sanders, J. Chem. Soc. Perkin Trans. 1 1997, 3237-3253. [13] S.J. Rowan, J.K.M. Sanders, J. Org. Chem. 1998, 63, 1536-1546. [14] A.P. Davis, J.F. Gilmer, J.J. Perry, Angew. Chem. 1996, 108, 1410-1413. [15] R.P. Bonar-Law, A.P. Davis, B.A. Murray, Angew. Chem. 1990, 102, 1497-1499. R.P. Bonar-Law, A.P. Davis, J. Chem. Soc. Chem. Commun. 1989, 102, 1050-1052. [16] D. Albert, M. Feigel, Helv. Chim. Acta 1997, 80, 2168-2181. D. Albert, M. Feigel, Tetrahedron Lett. 1994, 35, 565-568. [17] D. Albert, Dissertation, Universität Erlangen-Nürnberg, 1997. 6. Literaturverzeichnis 157 [18] G. Wess, K. Bock, H. Kleine, M. Kurz, W. Guba, H. Hemmerle, E. Lopez-Calle, K.H. Baringhaus, H. Glombik, A. Ehnsen, W. Kramer, Angew. Chem. 1996, 108, 2363-2366; R. Hirschmann, P.A. Sprengeler, T. Kawasaki, J.W. Leahy, W.C. Shakespeare, A.B. Smith, Tetrahedron 1993, 9, 3665. [19] U. Maitra, B.G. Bag, P. Rao, D. Powell, J. Chem. Soc. Perkin Trans. 1 1995, 3237-3253; R. Breslow, Acc. Chem. Res. 1980, 13, 170-177. [20] J.P. Guthrie, P.A. Cullimore, R.S. McDonald, S. O’Leary, Can J. Chem. 1982, 60, 747-764; T. Oost, A. Filippazzi, M. Kalesse, Liebigs Ann. / Recueil 1997, 1005-1011. [21] H. De Muynck, A. Madder, N. Farcy, P.J. De Clercq, M. Nieves Pérez-Payán, L.M. Öhberg, A.P. Davis, Angew. Chem. 2000, 112, 149-152. [22] A.P. Davis, S. Dresen, L. J. Lawless, Tetrahedron Lett. 1997, 4305-4308, S. Broderick, A.P. Davis, R.P. Williams, Tetrahedron Lett. 1998, 6083-6086, P.L. Anelli, L. Lattuada, F. Uggeri, Synth. Commun. 1998, 28, 109-117. [23] A.S. Jones, M. Webb, F. Smith, J. Chem. Soc. 1949, 2164-2168. M.J. Redel, A. Bouteville, B. Gauthier, N.H. Quy, Bull. Soc. Chim. Fr. 1949, 877-881. [24] A.M. Bellini, M.P. Quaglio, M. Guarneri, Farmaco Ed. Sci. 1986, 41, 401-407, A.M. Bellini, E. Mencini, M.P. Quaglio, M. Guarneri, A. Fini, Steroids 1991, 56, 395-398. [25] R.A. Leppik, Steroids 1983, 41, 475-484. [26] K.Y. Tserng, P.D. Klein, Steroids 1977, 29, 635-648. [27] J. Jones, Synthese von Aminosäuren und Peptiden, Basistext Cemie 6, VCH 1995, 22f. [28] H. Grießer, M. Kroner, U. Schmidt, Synthesis 1991, 294-300. [29] R. Benn, H. Günther, Angew. Chem. 1983, 95, 381-411, H. Kessler, Angew. Chem. 1982, 94, 509-520. [30] U. Platini, O.W. Sörensen, R.R. Ernst, J. Am. Chem. Soc. 1982, 104, 6800-6801, M.R. Rance, O.W. Sörensen, G. Bodenhauser, G. Wagner, R.R. Ernst, K. Wüthrich, Biochem. Biophys. Rev. Commun. 1983, 117, 458, A.J. Shaka, R. Freeman, J. Magn. Res. 1983, 51, 169-173, N. Müller, R.R. Ernst, K. Wüthrich, J. Am. Chem. Soc. 1986, 108, 6482-6492. [31] L. Braunschweiler, R.R. Ernst, J. Magn. Res. 1983, 53, 521-528, A. Bax, D.G. Davis, J. Magn. Res. 1985, 65, 355-360. [32] D. Neuhaus, M. Williamson, The Nuclear Overhauser Effect in Structural and Conformational Analysis, VCH New York 1995. [33] A.A. Bothner-By, R. Richard, L. Stephens, J. Lee, C.D. Warren, R.W. Jeanloz, J. Am. Chem. Soc. 1984, 106, 811-813, A. Bax, D.G. Davis, J. Magn. Res. 1985, 63, 207-213, D. Neuhaus, J. Keeler, J. Magn. Res. 1986, 68, 568-574, H. Kessler, C. Griesinger, R. Keerseebaum, K. Wagner, R.R. Ernst, J. Am. Chem. Soc. 1987, 109, 607-609, R. Brüschweiler, B. Roux, M. Blackledge, C. Griesinger, M. Karplus, R.R. Ernst, J. Am. Chem. Soc. 1992, 114, 2289-2302. 6. Literaturverzeichnis 158 [34] A. Bax, R.H. Griffey, B.L. Hawkins, J. Magn. Res. 1983, 55, 301-315, A. Bax, S. Subramanian, J. Magn. Res. 1986, 67, 565-569. [35] A. Bax, M.F. Summers, J. Am Chem. Soc. 1986, 108, 2093-2094. [36] T.S. Haque, J.C. Little, S.H. Gellman, J. Am. Chem. Soc. 1996, 118, 6975-6985. [37] M. Madison, M. Atreyi, C.M. Deber, E.R. Blout, J. Am. Chem. Soc. 1974, 96, 6725-6734, L.G. Pease, C. Watson, J. Am. Chem. Soc. 1978, 100, 1279-1286, W. Hehlein, H. Kessler, R. Schuck, J. Am. Chem. Soc. 1982, 104, 4534-4540. [38] W.J. Hehre, L. Radom, P.v.R. Schleyer, J.A. Pople, Ab initio Molecular Orbital Theory, Wiley New York 1986. [39] M.J.S. Dewar, W.J.Thiel, J. Am Chem. Soc. 1977, 99, 4899-4907; M.J.S. Dewar, W.J.Thiel, J. Am Chem. Soc. 1977, 99, 4907-4917; M.J.S. Dewar, M.L. McKee, J. Am Chem. Soc. 1977, 99, 5231-5241. [40] M.J.S. Dewar, E.G. Zoebisch, E.F. Healy, J.J.P. Stewart, J. Am Chem. Soc. 1985, 107, 3902. [41] J.J.P. Stewart, J. Comp. Chem. 1989, 10, 209-220 und 221-264. [42] K.B. Lipkowitz, N.L. Allinger, QCPE Bull. 1987, 7. [43] N.L. Allinger, Y.H. Yuh, H. Lii, J. Am Chem. Soc. 1989, 111, 8551-8566; N.L. Allinger, H. Lii, J. Am Chem. Soc. 1989, 111, 8566-8575; N.L. Allinger, H. Lii, J. Am Chem. Soc. 1989, 111, 8576-8582. [44] B.R. Brooks, R.E. Bruccoleri, B.D. Olafson, D.J. States, S. Swaminathan, M. Karplus, J. Comp. Chem. 1983, 4, 187-217. [45] S.J. Weiner, P.A. Kollman, D.A. Case, U.C. Singh, C. Ghio, G. Alagona, S. Profeta Jr., P. Weiner, J. Am Chem. Soc. 1984, 106, 765-784; S.J. Weiner, P.A. Kollman, D.T. Nguyen, D.A.Case, J. Comp. Chem. 1986, 7, 230-252. [46] E. Osaura, H. Musso, Angew. Chem. 1983, 95, 1-12. [47] A.P. Davis, J.J. Walsh, J. Chem. Soc. Chem. Commun. 1996, 449-451; A.P. Davis, S. Menzer, J.J. Walsh, D.J. Williams, J. Chem. Soc. Chem. Commun. 1996, 453-455. [48] siehe beispielsweise: F. Vögtle, S. Meier, R. Hoss, Angew. Chem. 1992, 104, 1628; S. Ottens-Hildebrandt, S. Meier, W. Schmidt, F. Vögtle, Angew. Chem. 1994, 106, 1818; S. Ottens-Hildebrandt, M. Nieger, K. Rissanen, J. Rouvinen, S. Meier, G. Harder, F. Vögtle, J. Chem. Soc. Chem. Commun. 1995, 777; A. Andrievsky, F. Ahuis, J.L. Sessler, F. Vögtle, D. Gudat, M. Moini, J. Am. Chem. Soc. 1998, 120, 9712-9713; F. Vögtle, O. Safarowsky, C. Heim, A. Affeld, O. Braun, A. Mohry, Pure & Appl. Chem. 1999, 71, 247-251; eine vollständige Literaturübersicht findet sich im Internet unter der Adresse http://www.chemie.uni-bonn.de/oc/ak_vo/catenan/def.htm 6. Literaturverzeichnis 159 [49] O. Safarowsky, M. Nieger, R. Fröhlich, F. Vögtle, Angew. Chem. 2000, 112, 1699-1701; F. Vögtle, A. Hünten, E. Vogel, S. Buschbeck, O. Safarowsky, J. Recker, A.-H. Parham, M. Knott, W.M. Müller, Y. Okamoto, T. Kubota, W. Lindner, E. Francotte, S. Grimme, Angew. Chem. 2001, 113, 2534-2537; siehe außerdem im Internet unter der Adresse: http://www.chemie.uni-bonn.de/oc/ak_vo/knots/def.htm [50] R. Schwyzer, B. Iselin, H. Kappeler, B. Riniker, W. Rittel, H. Zuber, Helv. Chim. Acta 1958, 41, 1273-1286, M. Brenner, W. Huber, Helv. Chim. Acta 1953, 36, 1109-1115. [51] E. Wünsch, Houben-Weyl, Methoden der Organischen Chemie, Bd. XV/1, Synthesen von Peptiden I, 4. Auflage, Thieme Verlag, Stuttgart 1974, O. Keller, W. Keller, G. van Lock, G. Wersin, Org. Synth. 1980, 63, 129. [52] U. Ragnarsson, S. Karlsson, G. Lindeberg, Acta Chem. Scand. 1970, 24, 2821-2825. [53] S. Yamada, Y. Kasai, T. Shioiri, Tetrahedron Lett. 1973, 1595-1598, T. Shioiri, Y. Yokohama, Y. Kasai, S. Yamada, Tetrahedron 1976, 32, 2211-2217, Y. Hamada, S. Rishi, T. Shioiri, S. Yamada, Chem. Pharm. Bull. 1977, 25, 224-230. [54] J.C. Sheehan, S.L. Ledis, J. Am. Chem. Soc. 1973, 95, 875-879. Anhang A 160 Anhang A Röntgenstrukturdaten zu 3-Oxo-Desoxycholsäure 6b. Table 1. Crystal data and structure refinement for C24H30O4.CH3OH 6b. Empirical formula C24 H30 O4 CH3OH Formula weight 450.55 Temperature 293(2) K Wavelength 0.71073 Å Crystal system Orthorhombic Space group P2(1)2(1)2(1) Unit cell dimensions a = 7.0270(14) Å b = 13.274(3) Å c = 26.441(5) Å Volume, Z 2466.3(9) Å3, Density (calculated) 1.213 Mg/m3 Absorption coefficient 0.082 mm-1 F(000) 968 Crystal size 0.30 x 0.25 x 0.20 mm Theta range for data collection 1.54 to 24.98° Limiting indices 0<=h<=8, 0<=k<=15, 0<=l<=31 Reflections collected 2502 Independent reflections 2502 [R(int) = 0.0000] Refinement method Full-matrix least-squares on F2 Data / restraints / parameters 2491 / 0 / 270 Goodness-of-fit on F2 1.020 alpha = 90° beta = 90° gamma = 90° 4 Final R indices [I>2σ(I)] R1 = 0.0795, wR2 = 0.2074 R indices (all data) R1 = 0.1638, wR2 = 0.2884 Absolute structure parameter 4(5) Largest diff. peak and hole 0.620 and -0.203 e.Å-3 Anhang A 161 Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2 x 103) for C24H30O4.CH3OH 6b. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________ C(1) 5974(12) 4874(6) 1473(3) 52(2) C(2) 5228(13) 4780(6) 2018(3) 53(2) C(3) 3418(15) 4182(6) 2030(3) 54(2) O(3) 3158(11) 3512(5) 2343(2) 77(2) C(4) 1950(13) 4446(6) 1642(3) 50(2) C(5) 2731(12) 4607(5) 1108(3) 42(2) C(6) 1174(13) 4982(6) 753(3) 50(2) C(7) 634(13) 6075(5) 852(3) 46(2) C(8) 2330(12) 6796(5) 869(3) 40(2) C(9) 3863(11) 6393(5) 1238(3) 35(2) C(10) 4524(12) 5293(5) 1095(3) 41(2) C(11) 5490(12) 7154(5) 1301(3) 42(2) C(12) 4791(12) 8198(6) 1475(3) 41(2) O(12) 3989(9) 8107(4) 1974(2) 53(2) C(13) 3291(12) 8631(5) 1108(3) 37(2) C(14) 2098(11) 9547(5) 1294(3) 40(2) C(15) 244(13) 9456(6) 962(3) 54(2) C(16) 152(14) 8390(5) 756(3) 54(2) C(17) 1693(11) 7836(5) 1051(3) 37(2) C(18) 4271(13) 8872(6) 601(3) 47(2) C(19) 5456(15) 5261(7) 571(3) 62(3) C(20) 2921(13) 10612(5) 1297(3) 45(2) C(21) 4778(15) 10710(7) 1582(4) 72(3) C(22) 1455(15) 11397(6) 1475(3) 53(3) C(23) 783(16) 11265(7) 2020(3) 64(3) C(24) -704(13) 11999(6) 2170(3) 49(2) O(241) -1492(10) 12602(4) 1902(2) 68(2) O(242) -1172(12) 11890(5) 2659(2) 88(3) C(3') -75(29) 2666(16) -129(7) 95(6) C(2') 3231(31) 2650(16) -134(7) 99(6) C(1') 1591(34) 2375(17) 110(8) 110(7) ________________________________________________________________ Anhang A 162 Table 3a. Bond lengths [Å] for C24H30O4.CH3OH 6b. _____________________________________________________________ C(1)-C(10) C(1)-C(2) C(1)-H(11) C(1)-H(12) C(2)-C(3) C(2)-H(21) C(2)-H(22) C(3)-O(3) C(3)-C(4) C(4)-C(5) C(4)-H(41) C(4)-H(42) C(5)-C(6) C(5)-C(10) C(5)-H(5) C(6)-C(7) C(6)-H(61) C(6)-H(62) C(7)-C(8) C(7)-H(71) C(7)-H(72) C(8)-C(17) C(8)-C(9) C(8)-H(8) C(9)-C(11) C(9)-C(10) C(9)-H(9) C(10)-C(19) C(11)-C(12) C(11)-H(111) C(11)-H(112) C(12)-O(12) C(12)-C(13) C(12)-H(12) O(12)-H(121) C(13)-C(18) C(13)-C(17) 1.531(11) 1.539(11) 0.97 0.97 1.500(13) 0.97 0.97 1.228(9) 1.495(12) 1.531(11) 0.97 0.97 1.524(11) 1.555(11) 0.98 1.523(10) 0.97 0.97 1.529(11) 0.97 0.97 1.529(10) 1.548(10) 0.98 1.534(10) 1.578(9) 0.98 1.533(11) 1.541(10) 0.97 0.97 1.441(9) 1.544(11) 0.98 0.82 1.541(10) 1.548(10) C(13)-C(14) C(14)-C(20) C(14)-C(15) C(14)-H(14) C(15)-C(16) C(15)-H(151) C(15)-H(152) C(16)-C(17) C(16)-H(161) C(16)-H(162) C(17)-H(17) C(18)-H(181) C(18)-H(182) C(18)-H(183) C(19)-H(191) C(19)-H(192) C(19)-H(193) C(20)-C(21) C(20)-C(22) C(20)-H(20) C(21)-H(211) C(21)-H(212) C(21)-H(213) C(22)-C(23) C(22)-H(221) C(22)-H(222) C(23)-C(24) C(23)-H(231) C(23)-H(232) C(24)-O(241) C(24)-O(242) O(242)-H(242) C(3')-C(1') C(3')-C(2')#1 C(2')-C(1') C(2')-C(3')#2 1.556(10) 1.528(10) 1.575(12) 0.98 1.518(11) 0.97 0.97 1.524(11) 0.97 0.97 0.98 0.96 0.96 0.96 0.96 0.96 0.96 1.511(13) 1.539(11) 0.98 0.96 0.96 0.96 1.526(11) 0.97 0.97 1.482(12) 0.97 0.97 1.204(9) 1.343(10) 0.82 1.39(2) 1.44(2) 1.37(3) 1.44(2) _____________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 x-1/2,-y+1/2,-z #2 x+1/2,-y+1/2,-z ı Anhang A 163 Table 3b. Binding angles [°] for C24H30O4.CH3OH 6b. (angles with C-H-bonds not listed) _____________________________________________________________ ı C(10)-C(1)-C(2) 114.4(7) C(3)-C(2)-C(1) 110.6(7) O(3)-C(3)-C(4) 121.9(9) O(3)-C(3)-C(2) 121.6(8) C(4)-C(3)-C(2) 116.5(7) C(3)-C(4)-C(5) 114.7(7) C(6)-C(5)-C(4) 110.8(7) C(6)-C(5)-C(10) 112.2(6) C(4)-C(5)-C(10) 113.1(6) C(7)-C(6)-C(5) 112.6(7) C(6)-C(7)-C(8) 114.0(7) C(7)-C(8)-C(17) 110.2(7) C(7)-C(8)-C(9) 110.2(6) C(17)-C(8)-C(9) 108.5(6) C(11)-C(9)-C(8) 111.1(6) C(11)-C(9)-C(10) 114.6(6) C(8)-C(9)-C(10) 112.0(6) C(1)-C(10)-C(19) 107.1(7) C(1)-C(10)-C(5) 108.2(6) C(19)-C(10)-C(5) 110.4(6) C(1)-C(10)-C(9) 112.1(6) C(19)-C(10)-C(9) 111.6(6) C(5)-C(10)-C(9) 107.4(6) C(9)-C(11)-C(12) 112.8(6) O(12)-C(12)-C(11) 108.9(6) O(12)-C(12)-C(13) 109.9(6) C(11)-C(12)-C(13) 111.4(6) C(12)-O(12)-H(121) 109.5(4) C(18)-C(13)-C(12) 108.6(7) C(18)-C(13)-C(17) 112.5(6) C(12)-C(13)-C(17) 107.6(6) C(18)-C(13)-C(14) 110.7(6) C(12)-C(13)-C(14) 117.4(6) C(17)-C(13)-C(14) 99.9(6) C(20)-C(14)-C(13) 121.4(6) C(20)-C(14)-C(15) 112.8(6) C(13)-C(14)-C(15) 102.2(6) C(16)-C(15)-C(14) 107.8(7) C(15)-C(16)-C(17) 103.7(7) C(16)-C(17)-C(8) 118.8(6) C(16)-C(17)-C(13) 103.7(6) C(8)-C(17)-C(13) 115.7(6) C(21)-C(20)-C(14) 114.2(7) C(21)-C(20)-C(22) 111.6(7) C(14)-C(20)-C(22) 112.0(6) C(23)-C(22)-C(20) 114.7(7) C(24)-C(23)-C(22) 113.3(7) O(241)-C(24)-O(242) 121.7(8) O(241)-C(24)-C(23) 127.2(8) O(242)-C(24)-C(23) 111.1(7) C(24)-O(242)-H(242) 109.5(5) C(1')-C(3')-C(2')#1 113.4(14) C(1')-C(2')-C(3')#2 113.0(14) C(2')-C(1')-C(3') 115(2) _____________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 x-1/2,-y+1/2,-z #2 x+1/2,-y+1/2,-z Anhang A Table 4. Anisotropic displacement parameters (Å2 x 103) for C24H30O4.CH3OH 6b. The anisotropic displacement factor exponent takes the form: -2 π2 [ h2 a*2 U11 + ... + 2 h k a* b* U12 ] _______________________________________________________________________ U11 U22 U33 U23 U13 U12 _______________________________________________________________________ C(1) 50(5) 42(5) 64(6) 2(4) -3(5) 8(4) C(2) 63(6) 44(5) 53(5) 7(4) -10(5) -4(5) C(3) 70(6) 50(5) 41(4) 1(4) -7(5) 3(5) O(3) 88(5) 79(5) 66(4) 40(4) -23(4) -20(4) C(4) 62(6) 40(4) 48(5) 5(4) -7(5) -12(5) C(5) 60(5) 28(4) 39(4) -3(3) 1(4) -4(4) C(6) 70(6) 39(4) 42(5) -4(3) -9(5) -7(5) C(7) 51(5) 43(5) 44(4) 3(4) -13(4) 4(5) C(8) 57(5) 32(4) 31(4) -1(3) 2(4) -9(4) C(9) 46(4) 31(4) 28(3) -4(3) 3(4) -1(4) C(10) 50(5) 34(4) 38(4) 3(3) 2(4) -1(4) C(11) 51(5) 34(4) 41(4) 2(3) -6(4) -4(4) C(12) 39(5) 39(4) 45(4) 2(3) 2(4) -11(4) O(12) 72(4) 55(3) 31(3) -7(2) -2(3) -13(3) C(13) 46(5) 32(4) 33(4) 2(3) 10(4) 0(4) C(14) 53(5) 36(4) 32(4) 1(3) 11(4) 6(4) C(15) 52(5) 39(4) 71(6) 1(4) 4(5) 4(5) C(16) 64(6) 38(4) 60(5) 5(4) -8(5) 2(5) C(17) 46(5) 31(4) 33(4) 1(3) 0(4) -2(4) C(18) 63(5) 39(4) 39(4) 1(3) 7(4) -8(5) C(19) 77(6) 57(5) 51(5) -2(4) 13(5) 9(6) C(20) 62(5) 30(4) 42(4) 0(3) 10(4) -12(4) C(21) 83(8) 50(6) 81(6) -14(5) 6(6) -13(6) C(22) 84(7) 30(4) 44(5) 1(3) 18(5) 5(5) C(23) 79(7) 60(5) 53(5) 2(4) 9(5) 16(6) C(24) 58(6) 34(4) 55(5) 3(4) 5(5) 3(5) O(241) 89(5) 57(4) 59(4) 6(3) 11(4) 12(4) O(242) 132(7) 80(5) 50(4) 14(3) 31(4) 48(5) _______________________________________________________________________ 164 Anhang A Table 5. Hydrogen coordinates ( x 104) and isotropic displacement parameters (Å2 x 103) for C24H30O4.CH3OH 6b. ________________________________________________________________ x y z U(eq) ________________________________________________________________ H(11) 7083(12) 5309(6) 1473(3) 62 H(12) 6380(12) 4214(6) 1358(3) 62 H(21) 6179(13) 4452(6) 2227(3) 64 H(22) 4998(13) 5447(6) 2155(3) 64 H(41) 1306(13) 5056(6) 1749(3) 60 H(42) 1010(13) 3911(6) 1631(3) 60 H(5) 3127(12) 3944(5) 982(3) 51 H(61) 1606(13) 4916(6) 407(3) 60 H(62) 55(13) 4562(6) 794(3) 60 H(71) -39(13) 6114(5) 1172(3) 55 H(72) -233(13) 6296(5) 589(3) 55 H(8) 2879(12) 6856(5) 530(3) 48 H(9) 3246(11) 6342(5) 1569(3) 42 H(111) 6150(12) 7225(5) 980(3) 51 H(112) 6391(12) 6895(5) 1546(3) 51 H(12) 5879(12) 8659(6) 1489(3) 49 H(121) 4716(56) 8360(57) 2181(4) 63 H(14) 1720(11) 9397(5) 1642(3) 48 H(151) -872(13) 9597(6) 1166(3) 65 H(152) 285(13) 9937(6) 686(3) 65 H(161) 415(14) 8378(5) 396(3) 65 H(162) -1089(14) 8093(5) 816(3) 65 H(17) 1177(11) 7734(5) 1392(3) 44 H(181) 5372(41) 9283(31) 662(3) 61 H(182) 3400(26) 9228(32) 386(7) 61 H(183) 4652(62) 8256(6) 440(9) 61 H(191) 5877(74) 4587(12) 501(10) 80 H(192) 6527(52) 5710(34) 565(7) 80 H(193) 4549(29) 5465(41) 320(4) 80 H(20) 3208(13) 10776(5) 944(3) 54 H(211) 5102(45) 11409(7) 1615(20) 93 H(212) 5765(24) 10366(39) 1400(12) 93 H(213) 4645(31) 10416(41) 1912(9) 93 H(221) 2008(15) 12062(6) 1440(3) 64 H(222) 356(15) 11364(6) 1254(3) 64 H(231) 285(16) 10589(7) 2062(3) 77 H(232) 1865(16) 11338(7) 2245(3) 77 H(242) -2106(99) 12238(67) 2723(11) 105 ________________________________________________________________ 165 Anhang B 166 Anhang B Röntgenstrukturdaten zu Boc-Val-LCS-OMe 21a. Table 1. Crystal data and structure refinement for C35H60N2O5 21a Empirical formula C35 H60 N2 O5 Formula weight 588.85 Temperature 293(2) K Wavelength 0.71073 Å Crystal system, space group Monoclinic, Unit cell dimensions a = 12.119(2) Å b = 9.2831(19) Å c = 15.603(3) Å Volume 1749.8(6) Å3 Z, Calculated density 2, Absorption coefficient 0.073 mm-1 F(000) 648 Crystal size 0.42 x 0.36 x 0.33 mm θ range for data collection 2.56 to 25.00° Limiting indices 0<=h<=14, 0<=k<=11, -18<=l<=18 Reflections collected / unique 3427 / 3283 [R(int) = 0.0281] Completeness to θ = 25.00 99.6 % Refinement method Full-matrix least-squares on F2 Data / restraints / parameters 3283 / 3 / 399 Goodness-of-fit on F2 1.040 Final R indices [I>2σ(I)] R1 = 0.0530, wR2 = 0.0982 R indices (all data) R1 = 0.1035, wR2 = 0.1174 Largest diff. peak and hole 0.135 and -0.163 e.Å-3 P2(1) alpha = 90° beta = 94.55(3)° gamma = 90° 1.118 Mg/m3 Anhang B 167 Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2 x 103) for C35H60N2O5 21a. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________ C(1) -2364(4) 2212(5) 2591(3) 56(1) C(2) -1230(4) 1761(5) 2315(3) 53(1) C(3) -1332(3) 1107(5) 1419(3) 47(1) C(4) -2178(3) -80(5) 1362(3) 50(1) C(5) -3306(3) 349(5) 1649(3) 50(1) C(6) -4113(4) -933(6) 1559(3) 65(2) C(7) -3782(4) -2101(6) 2205(3) 59(1) C(8) -3658(4) -1562(5) 3125(3) 50(1) C(9) -2889(4) -224(5) 3227(3) 46(1) C(10) -3226(4) 992(5) 2571(3) 52(1) C(19) -4341(4) 1675(7) 2760(4) 80(2) C(11) -2757(5) 251(5) 4184(3) 69(2) C(12) -2338(5) -969(5) 4783(3) 62(1) C(13) -3068(4) -2332(5) 4696(3) 49(1) C(18) -4190(4) -2034(7) 5057(4) 90(2) C(14) -3177(3) -2723(5) 3731(3) 47(1) C(15) -3692(4) -4227(6) 3713(3) 71(2) C(16) -3169(4) -4936(6) 4540(3) 68(2) C(17) -2553(4) -3751(5) 5079(3) 50(1) C(20) -2546(5) -4051(6) 6048(3) 73(2) C(21) -1930(7) -2907(7) 6591(3) 125(3) C(22) -2072(5) -5544(6) 6288(3) 73(2) C(23) -901(4) -5809(6) 6050(3) 76(2) C(24) -379(5) -7037(6) 6558(4) 69(2) O(24) 16(4) -6961(5) 7288(3) 104(2) O(25) -397(3) -8237(5) 6113(3) 88(1) C(25) 73(5) -9504(7) 6559(4) 93(2) N(3) -273(3) 556(4) 1184(2) 54(1) C(31) 425(3) 1249(5) 705(3) 41(1) O(31) 214(2) 2432(3) 389(2) 52(1) C(32) 1527(3) 494(5) 586(3) 42(1) C(33) 1987(3) 907(5) -271(3) 51(1) C(34) 1219(4) 475(6) -1050(3) 68(2) C(35) 3146(4) 302(7) -323(4) 87(2) N(32) 1453(3) -1052(4) 714(2) 39(1) C(36) 2232(4) -1760(5) 1222(3) 42(1) O(36) 2995(2) -1208(3) 1643(2) 54(1) O(37) 2003(2) -3193(3) 1189(2) 51(1) C(37) 2826(4) -4242(5) 1540(3) 48(1) C(38) 3829(4) -4180(6) 1013(3) 68(2) C(39) 2214(4) -5651(5) 1380(3) 66(2) C(40) 3112(4) -4019(6) 2489(3) 65(1) ________________________________________________________________ Anhang B 168 Table 3a. Bond lengths [Å] for C35H60N2O5 21a _____________________________________________________________ C(1)-C(2) C(1)-C(10) C(1)-H(11) C(1)-H(12) C(2)-C(3) C(2)-H(21) C(2)-H(22) C(3)-N(3) C(3)-C(4) C(3)-H(31) C(4)-C(5) C(4)-H(41) C(4)-H(42) C(5)-C(6) C(5)-C(10) C(5)-H(5) C(6)-C(7) C(6)-H(61) C(6)-H(62) C(7)-C(8) C(7)-H(71) C(7)-H(72) C(8)-C(14) C(8)-C(9) C(8)-H(8) C(9)-C(11) C(9)-C(10) C(9)-H(9) C(10)-C(19) C(19)-H(191) C(19)-H(192) C(19)-H(193) C(11)-C(12) C(11)-H(111) C(11)-H(112) C(12)-C(13) C(12)-H(121) C(12)-H(122) C(13)-C(18) C(13)-C(14) C(13)-C(17) C(18)-H(181) C(18)-H(182) C(18)-H(183) C(14)-C(15) C(14)-H(14) C(15)-C(16) C(15)-H(151) C(15)-H(152) C(16)-C(17) C(16)-H(161) C(16)-H(162) C(17)-C(20) 1.530(6) 1.539(6) 0.9700 0.9700 1.520(6) 0.9700 0.9700 1.456(5) 1.502(6) 0.99(4) 1.525(6) 0.9700 0.9700 1.539(6) 1.553(6) 0.9800 1.512(6) 0.9700 0.9700 1.516(6) 0.9700 0.9700 1.519(6) 1.553(6) 0.9800 1.553(6) 1.557(6) 0.9800 1.542(6) 0.9600 0.9600 0.9600 1.530(6) 0.9700 0.9700 1.543(7) 0.9700 0.9700 1.539(6) 1.543(6) 1.557(6) 0.9600 0.9600 0.9600 1.528(7) 0.9800 1.539(7) 0.9700 0.9700 1.542(6) 0.9700 0.9700 1.535(6) C(17)-H(17) C(20)-C(21) C(20)-C(22) C(20)-H(20) C(21)-H(211) C(21)-H(212) C(21)-H(213) C(22)-C(23) C(22)-H(221) C(22)-H(222) C(23)-C(24) C(23)-H(231) C(23)-H(232) C(24)-O(24) C(24)-O(25) O(25)-C(25) C(25)-H(251) C(25)-H(252) C(25)-H(253) N(3)-C(31) N(3)-H(3) C(31)-O(31) C(31)-C(32) C(32)-N(32) C(32)-C(33) C(32)-H(321) C(33)-C(35) C(33)-C(34) C(33)-H(33) C(34)-H(341) C(34)-H(342) C(34)-H(343) C(35)-H(351) C(35)-H(352) C(35)-H(353) N(32)-C(36) N(32)-H(32) C(36)-O(36) C(36)-O(37) O(37)-C(37) C(37)-C(40) C(37)-C(39) C(37)-C(38) C(38)-H(381) C(38)-H(382) C(38)-H(383) C(39)-H(391) C(39)-H(392) C(39)-H(393) C(40)-H(401) C(40)-H(402) C(40)-H(403) 0.9800 1.518(8) 1.535(7) 0.9800 0.9600 0.9600 0.9600 1.515(7) 0.9700 0.9700 1.500(7) 0.9700 0.9700 1.202(6) 1.312(6) 1.459(7) 0.9600 0.9600 0.9600 1.337(5) 0.859(10) 1.223(5) 1.532(6) 1.453(5) 1.539(6) 0.9800 1.521(6) 1.524(6) 0.9800 0.9600 0.9600 0.9600 0.9600 0.9600 0.9600 1.354(5) 0.855(10) 1.205(5) 1.359(5) 1.469(5) 1.507(6) 1.514(6) 1.522(6) 0.9600 0.9600 0.9600 0.9600 0.9600 0.9600 0.9600 0.9600 0.9600 _____________________________________________________________ Anhang B 169 Table 3b. binding angles [°] for C35H60N2O5 21a (angles with C-H-bonds are not listed) _____________________________________________________________ C(2)-C(1)-C(10) C(3)-C(2)-C(1) N(3)-C(3)-C(4) N(3)-C(3)-C(2) C(4)-C(3)-C(2) C(3)-C(4)-C(5) C(4)-C(5)-C(6) C(4)-C(5)-C(10) C(6)-C(5)-C(10) C(7)-C(6)-C(5) C(6)-C(7)-C(8) C(7)-C(8)-C(14) C(7)-C(8)-C(9) C(14)-C(8)-C(9) C(11)-C(9)-C(8) C(11)-C(9)-C(10) C(8)-C(9)-C(10) C(1)-C(10)-C(19) C(1)-C(10)-C(5) C(19)-C(10)-C(5) C(1)-C(10)-C(9) C(19)-C(10)-C(9) C(5)-C(10)-C(9) C(12)-C(11)-C(9) C(11)-C(12)-C(13) C(18)-C(13)-C(12) C(18)-C(13)-C(14) C(12)-C(13)-C(14) C(18)-C(13)-C(17) C(12)-C(13)-C(17) C(14)-C(13)-C(17) C(8)-C(14)-C(15) C(8)-C(14)-C(13) C(15)-C(14)-C(13) C(14)-C(15)-C(16) C(15)-C(16)-C(17) C(20)-C(17)-C(16) 114.6(4) 111.0(4) 109.8(4) 111.3(4) 110.7(4) 114.5(4) 110.5(4) 112.3(3) 112.0(4) 111.2(3) 113.3(4) 111.1(4) 112.0(4) 107.8(3) 109.9(4) 115.4(4) 112.8(3) 107.3(4) 107.1(3) 110.1(4) 112.1(4) 111.1(4) 109.0(4) 112.4(4) 113.1(4) 109.7(4) 113.0(4) 106.5(3) 110.5(4) 116.7(4) 100.2(4) 120.0(4) 116.0(4) 103.7(4) 103.6(4) 107.6(4) 111.9(4) C(20)-C(17)-C(13) C(16)-C(17)-C(13) C(21)-C(20)-C(22) C(21)-C(20)-C(17) C(22)-C(20)-C(17) C(23)-C(22)-C(20) C(24)-C(23)-C(22) O(24)-C(24)-O(25) O(24)-C(24)-C(23) O(25)-C(24)-C(23) C(24)-O(25)-C(25) C(31)-N(3)-C(3) C(31)-N(3)-H(3) C(3)-N(3)-H(3) O(31)-C(31)-N(3) O(31)-C(31)-C(32) N(3)-C(31)-C(32) N(32)-C(32)-C(31) N(32)-C(32)-C(33) C(31)-C(32)-C(33) C(35)-C(33)-C(34) C(35)-C(33)-C(32) C(34)-C(33)-C(32) C(36)-N(32)-C(32) C(36)-N(32)-H(32) C(32)-N(32)-H(32) O(36)-C(36)-N(32) O(36)-C(36)-O(37) N(32)-C(36)-O(37) C(36)-O(37)-C(37) O(37)-C(37)-C(40) O(37)-C(37)-C(39) C(40)-C(37)-C(39) O(37)-C(37)-C(38) C(40)-C(37)-C(38) C(39)-C(37)-C(38) 120.2(4) 103.4(3) 109.7(5) 112.7(5) 112.2(4) 115.2(5) 111.0(4) 122.7(5) 125.2(6) 112.1(5) 116.2(4) 125.5(4) 114(3) 120(3) 122.4(4) 121.2(4) 116.3(4) 111.8(3) 113.3(4) 111.8(3) 111.6(4) 110.8(4) 112.8(3) 120.8(4) 118(3) 116(3) 125.6(4) 125.7(4) 108.8(4) 120.3(3) 111.9(4) 101.7(3) 110.7(4) 108.7(4) 113.2(4) 110.2(4) _____________________________________________________________ Anhang B Table 4. Anisotropic displacement parameters (Å2 x 103) for C35H60N2O5 21a.The anisotropic displacement factor exponent takes the form: -2 π2 [ h2 a*2 U11 + ... + 2 h k a* b* U12 ] _______________________________________________________________________ U11 U22 U33 U23 U13 U12 _______________________________________________________________________ C(1) 72(3) 46(3) 52(3) -2(2) 15(2) 7(3) C(2) 56(3) 41(3) 61(3) 1(2) 5(2) -6(2) C(3) 43(3) 48(3) 50(3) 7(2) 10(2) 9(2) C(4) 54(3) 52(3) 43(3) -5(2) 9(2) 0(2) C(5) 42(2) 57(3) 51(3) 11(2) 6(2) 6(2) C(6) 49(3) 86(4) 57(3) 15(3) -5(2) -5(3) C(7) 57(3) 64(3) 55(3) 2(3) -9(2) -18(3) C(8) 39(3) 57(3) 54(3) 9(2) 9(2) -2(2) C(9) 45(2) 44(3) 48(3) 1(2) 13(2) 6(2) C(10) 48(3) 49(3) 61(3) 8(2) 18(2) 13(2) C(19) 72(3) 79(4) 92(4) 19(4) 32(3) 25(3) C(11) 108(4) 47(3) 53(3) -2(3) 22(3) 4(3) C(12) 100(4) 43(3) 45(3) -3(2) 11(3) 4(3) C(13) 53(3) 50(3) 47(3) 9(2) 20(2) 9(2) C(18) 82(4) 102(5) 93(4) 33(4) 50(3) 37(4) C(14) 42(2) 48(3) 52(3) 3(2) 7(2) -1(2) C(15) 59(3) 66(4) 85(4) 20(3) -4(3) -17(3) C(16) 69(4) 59(4) 75(4) 20(3) -3(3) -15(3) C(17) 51(3) 54(3) 44(3) 2(2) 11(2) 5(2) C(20) 91(4) 71(4) 58(3) 15(3) 26(3) 22(3) C(21) 251(9) 79(5) 43(4) -4(3) -3(5) 42(6) C(22) 88(4) 77(4) 58(3) 20(3) 19(3) 8(3) C(23) 82(4) 68(4) 76(4) 15(3) 0(3) 0(3) C(24) 67(4) 64(4) 75(4) 4(3) -1(3) -7(3) O(24) 134(4) 90(3) 81(3) 5(3) -33(3) 6(3) O(25) 104(3) 67(3) 90(3) -2(3) -4(2) -5(2) C(25) 99(5) 65(4) 116(5) 13(4) 7(4) 11(4) N(3) 50(2) 42(2) 71(3) 17(2) 19(2) 15(2) C(31) 41(2) 40(3) 41(2) 0(2) 1(2) -4(2) O(31) 47(2) 37(2) 71(2) 14(2) 4(2) 5(2) C(32) 32(2) 37(2) 56(3) 4(2) 3(2) 1(2) C(33) 48(3) 45(3) 60(3) 9(2) 12(2) -2(2) C(34) 91(4) 63(3) 52(3) 5(3) 12(3) 0(3) C(35) 62(3) 107(5) 96(4) 30(4) 33(3) 11(4) N(32) 41(2) 31(2) 43(2) 1(2) -6(2) -1(2) C(36) 40(3) 40(3) 47(3) 1(2) 4(2) -1(2) O(36) 46(2) 50(2) 64(2) 1(2) -13(2) -7(2) O(37) 43(2) 35(2) 71(2) 8(2) -16(2) -2(2) C(37) 51(3) 40(3) 53(3) 4(2) -5(2) 1(2) C(38) 62(3) 70(4) 72(3) -15(3) 9(3) 0(3) C(39) 79(4) 45(3) 70(4) 6(3) -7(3) 0(3) C(40) 76(3) 60(4) 57(3) 7(3) -6(2) 2(3) _______________________________________________________________________ 170 Anhang B Table 5. Hydrogen coordinates ( x 104) and isotropic displacement parameters (Å2 x 103) for C35H60N2O5 21a. ________________________________________________________________ x y z U(eq) ________________________________________________________________ H(11) -2651 2982 2217 67 H(12) -2269 2594 3171 67 H(21) -748 2596 2321 63 H(22) -899 1064 2721 63 H(31) -1570(30) 1880(50) 1010(30) 56 H(41) -1895 -881 1713 59 H(42) -2273 -413 771 59 H(5) -3603 1103 1256 60 H(61) -4855 -603 1646 78 H(62) -4119 -1325 982 78 H(71) -3086 -2518 2064 71 H(72) -4337 -2856 2160 71 H(8) -4390 -1295 3298 59 H(9) -2156 -553 3088 55 H(191) -4882 930 2810 120 H(192) -4585 2314 2300 120 H(193) -4250 2206 3289 120 H(111) -2241 1051 4245 82 H(112) -3466 585 4354 82 H(121) -2311 -632 5373 75 H(122) -1590 -1219 4659 75 H(181) -4630 -2895 5023 135 H(182) -4569 -1285 4728 135 H(183) -4071 -1736 5646 135 H(14) -2417 -2865 3576 57 H(151) -3509 -4757 3207 85 H(152) -4491 -4173 3719 85 H(161) -2657 -5686 4399 82 H(162) -3739 -5365 4860 82 H(17) -1781 -3773 4936 59 H(20) -3317 -4040 6195 87 H(211) -1797 -3249 7171 138(16) H(212) -2367 -2044 6587 138(16) H(213) -1236 -2704 6359 138(16) H(221) -2089 -5673 6904 88 H(222) -2552 -6269 6009 88 H(231) -913 -6026 5441 91 H(232) -463 -4945 6160 91 H(251) 52 -10306 6170 140 H(252) -350 -9729 7037 140 H(253) 826 -9310 6765 140 H(3) -10(30) -230(30) 1410(30) 64 H(321) 2057 857 1042 50 H(33) 2050 1959 -280 61 H(341) 1553 730 -1567 102 H(342) 525 969 -1034 102 H(343) 1096 -546 -1041 102 H(351) 3440 631 -842 131 H(352) 3116 -731 -324 131 H(353) 3614 626 164 131 H(32) 1090(30) -1520(40) 316(19) 48(14) H(381) 3598 -4325 417 101 H(382) 4345 -4919 1204 101 H(383) 4178 -3254 1086 101 H(391) 1597 -5693 1729 98 H(392) 2706 -6440 1524 98 H(393) 1951 -5714 784 98 H(401) 2444 -3943 2777 97 H(402) 3536 -3151 2576 97 H(403) 3539 -4823 2717 97 ________________________________________________________________ 171 Anhang B 172 Table 6. Torsion angles [°] for C35H60N2O5 21a. ________________________________________________________________ C(10)-C(1)-C(2)-C(3) C(1)-C(2)-C(3)-N(3) C(1)-C(2)-C(3)-C(4) N(3)-C(3)-C(4)-C(5) C(2)-C(3)-C(4)-C(5) C(3)-C(4)-C(5)-C(6) C(3)-C(4)-C(5)-C(10) C(4)-C(5)-C(6)-C(7) C(10)-C(5)-C(6)-C(7) C(5)-C(6)-C(7)-C(8) C(6)-C(7)-C(8)-C(14) C(6)-C(7)-C(8)-C(9) C(7)-C(8)-C(9)-C(11) C(14)-C(8)-C(9)-C(11) C(7)-C(8)-C(9)-C(10) C(14)-C(8)-C(9)-C(10) C(2)-C(1)-C(10)-C(19) C(2)-C(1)-C(10)-C(5) C(2)-C(1)-C(10)-C(9) C(4)-C(5)-C(10)-C(1) C(6)-C(5)-C(10)-C(1) C(4)-C(5)-C(10)-C(19) C(6)-C(5)-C(10)-C(19) C(4)-C(5)-C(10)-C(9) C(6)-C(5)-C(10)-C(9) C(11)-C(9)-C(10)-C(1) C(8)-C(9)-C(10)-C(1) C(11)-C(9)-C(10)-C(19) C(8)-C(9)-C(10)-C(19) C(11)-C(9)-C(10)-C(5) C(8)-C(9)-C(10)-C(5) C(8)-C(9)-C(11)-C(12) C(10)-C(9)-C(11)-C(12) C(9)-C(11)-C(12)-C(13) C(11)-C(12)-C(13)-C(18) C(11)-C(12)-C(13)-C(14) C(11)-C(12)-C(13)-C(17) C(7)-C(8)-C(14)-C(15) C(9)-C(8)-C(14)-C(15) C(7)-C(8)-C(14)-C(13) C(9)-C(8)-C(14)-C(13) C(18)-C(13)-C(14)-C(8) C(12)-C(13)-C(14)-C(8) C(17)-C(13)-C(14)-C(8) C(18)-C(13)-C(14)-C(15) 56.5(5) -174.8(4) -52.4(5) 176.4(4) 53.1(5) 179.6(4) -54.7(5) 69.0(5) -57.0(5) 54.4(5) -172.3(4) -51.7(5) -178.0(4) -55.5(5) 51.6(5) 174.1(3) -173.4(4) -55.2(5) 64.3(5) 52.6(5) 177.6(3) 168.9(4) -66.1(5) -68.9(5) 56.0(4) 60.8(5) 171.7(3) -59.3(5) 68.3(5) 179.2(4) -53.3(4) 55.5(5) -175.5(4) -55.4(6) -69.6(5) 52.9(5) 163.8(4) -51.0(6) -174.1(4) -176.8(4) 60.1(5) 62.9(5) -57.6(5) -179.6(4) -70.9(5) C(12)-C(13)-C(14)-C(15) C(17)-C(13)-C(14)-C(15) C(8)-C(14)-C(15)-C(16) C(13)-C(14)-C(15)-C(16) C(14)-C(15)-C(16)-C(17) C(15)-C(16)-C(17)-C(20) C(15)-C(16)-C(17)-C(13) C(18)-C(13)-C(17)-C(20) C(12)-C(13)-C(17)-C(20) C(14)-C(13)-C(17)-C(20) C(18)-C(13)-C(17)-C(16) C(12)-C(13)-C(17)-C(16) C(14)-C(13)-C(17)-C(16) C(16)-C(17)-C(20)-C(21) C(13)-C(17)-C(20)-C(21) C(16)-C(17)-C(20)-C(22) C(13)-C(17)-C(20)-C(22) C(21)-C(20)-C(22)-C(23) C(17)-C(20)-C(22)-C(23) C(20)-C(22)-C(23)-C(24) C(22)-C(23)-C(24)-O(24) C(22)-C(23)-C(24)-O(25) O(24)-C(24)-O(25)-C(25) C(23)-C(24)-O(25)-C(25) C(4)-C(3)-N(3)-C(31) C(2)-C(3)-N(3)-C(31) C(3)-N(3)-C(31)-O(31) C(3)-N(3)-C(31)-C(32) O(31)-C(31)-C(32)-N(32) N(3)-C(31)-C(32)-N(32) O(31)-C(31)-C(32)-C(33) N(3)-C(31)-C(32)-C(33) N(32)-C(32)-C(33)-C(35) C(31)-C(32)-C(33)-C(35) N(32)-C(32)-C(33)-C(34) C(31)-C(32)-C(33)-C(34) C(31)-C(32)-N(32)-C(36) C(33)-C(32)-N(32)-C(36) C(32)-N(32)-C(36)-O(36) C(32)-N(32)-C(36)-O(37) O(36)-C(36)-O(37)-C(37) N(32)-C(36)-O(37)-C(37) C(36)-O(37)-C(37)-C(40) C(36)-O(37)-C(37)-C(39) C(36)-O(37)-C(37)-C(38) 168.6(4) 46.6(4) -166.9(4) -35.4(5) 10.2(5) 149.2(4) 18.4(5) -45.4(6) 80.8(5) -164.8(4) 80.1(5) -153.6(4) -39.2(4) 179.2(5) -59.3(6) 54.8(6) 176.3(4) -67.8(6) 58.3(6) 160.6(5) -79.6(7) 100.4(6) 1.2(8) -178.8(5) 139.4(4) -97.7(5) -2.4(7) 176.3(4) -158.2(4) 23.1(5) -30.0(5) 151.3(4) -59.9(5) 172.7(4) 66.0(5) -61.4(5) -132.1(4) 100.5(5) 4.5(7) -176.4(3) -13.0(7) 167.9(4) 60.9(5) 179.0(4) -64.8(5) ________________________________________________________________ Anhang C 173 Anhang C Röntgenstrukturdaten zu Cyclo-DCS2 34a. Table 1. Crystal data and structure refinement for C50H90N2O8 34a. Empirical formula C50 H90 N2 O8 Formula weight 847.24 Temperature 203(2) K Wavelength 0.71073 Å Crystal system, space group Monoclinic, Unit cell dimensions a = 19.927(5) Å b = 11.156(3) Å c = 13.998(4) Å Volume 2403.3(11) Å3 Z, Calculated density 2, Absorption coefficient 0.077 mm-1 F(000) 936 Crystal size 0.25 x 0.20 x 0.15 mm Theta range for data collection 1.88 to 25.13° Limiting indices -23<=h<=19, -13<=k<=13, -8<=l<=16 Reflections collected / unique 6375 / 4177 [R(int) = 0.0305] Completeness to theta = 25.13 99.7 % Refinement method Full-matrix least-squares on F2 Data / restraints / parameters 4177 / 3 / 284 Goodness-of-fit on F2 0.996 Final R indices [I>2σ (I)] R1 = 0.0410, wR2 = 0.0994 R indices (all data) R1 = 0.0523, wR2 = 0.1063 Absolute structure parameter 0.5(11) Extinction coefficient 0.0019(6) Largest diff. peak and hole 0.179 and -0.215 e.Å-3 C2 alpha = 90° beta = 129.443(7)° gamma = 90° 1.171 Mg/m3 Anhang C Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2 x 103) for C50H90N2O8 34a. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________ O(12) 8418(1) 3206(1) 8853(1) 41(1) O(24) 12401(1) 5338(2) 15916(2) 48(1) N(13) 11617(1) 3915(2) 14472(2) 35(1) C(1) 9206(1) 3958(2) 12816(2) 35(1) C(2) 10075(1) 3456(2) 13289(2) 36(1) C(3) 10781(1) 4401(2) 14029(2) 35(1) C(4) 10537(1) 5508(2) 13235(2) 33(1) C(5) 9659(1) 6035(2) 12756(2) 32(1) C(6) 9461(1) 7180(2) 12017(2) 36(1) C(7) 9227(2) 6931(2) 10769(2) 37(1) C(8) 8464(1) 6062(2) 10015(2) 30(1) C(9) 8680(1) 4880(2) 10743(2) 28(1) C(10) 8907(1) 5109(2) 12020(2) 30(1) C(11) 7964(1) 3934(2) 9962(2) 34(1) C(12) 7694(1) 3743(2) 8679(2) 32(1) C(13) 7424(1) 4934(2) 7963(2) 28(1) C(14) 8206(1) 5786(2) 8754(2) 29(1) C(15) 7968(2) 6843(2) 7888(2) 37(1) C(16) 7418(2) 6268(2) 6585(2) 36(1) C(17) 7254(1) 4936(2) 6717(2) 29(1) C(18) 6616(1) 5421(2) 7755(2) 37(1) C(19) 8109(2) 5565(2) 11861(2) 39(1) C(20) 6373(1) 4482(2) 5555(2) 32(1) C(21) 6180(2) 3183(2) 5679(2) 41(1) C(22) 6245(1) 4632(2) 4361(2) 36(1) C(23) 13190(1) 3836(2) 15768(2) 38(1) C(24) 12374(1) 4431(2) 15389(2) 36(1) O(1) 8566(1) 2111(1) 7234(2) 47(1) O(90) 5350(2) 6701(2) 9156(2) 81(1) C(90) 5583(2) 5496(3) 9394(3) 77(1) ________________________________________________________________ 174 Anhang C 175 Table 3a. Bond lengths [Å] for C50H90N2O8 34a. ______________________________________________________________ O(12)-C(12) O(12)-H(12') O(24)-C(24) N(13)-C(24) N(13)-C(3) N(13)-H(13) C(1)-C(2) C(1)-C(10) C(1)-H(1A) C(1)-H(1B) C(2)-C(3) C(2)-H(2A) C(2)-H(2B) C(3)-C(4) C(3)-H(3) C(4)-C(5) C(4)-H(4A) C(4)-H(4B) C(5)-C(6) C(5)-C(10) C(5)-H(5) C(6)-C(7) C(6)-H(6A) C(6)-H(6B) C(7)-C(8) C(7)-H(7A) C(7)-H(7B) C(8)-C(14) C(8)-C(9) C(8)-H(8) C(9)-C(11) C(9)-C(10) C(9)-H(9) C(10)-C(19) C(11)-C(12) C(11)-H(11A) C(11)-H(11B) C(12)-C(13) C(12)-H(12) 1.434(3) 0.8300 1.233(3) 1.339(3) 1.463(3) 0.8700 1.516(3) 1.550(3) 0.9800 0.9800 1.520(3) 0.9800 0.9800 1.520(3) 0.9900 1.536(3) 0.9800 0.9800 1.529(3) 1.554(3) 0.9900 1.520(3) 0.9800 0.9800 1.526(3) 0.9800 0.9800 1.525(3) 1.550(3) 0.9900 1.538(3) 1.562(3) 0.9900 1.543(3) 1.527(3) 0.9800 0.9800 1.541(3) 0.9900 C(13)-C(14) C(13)-C(18) C(13)-C(17) C(14)-C(15) C(14)-H(14) C(15)-C(16) C(15)-H(15A) C(15)-H(15B) C(16)-C(17) C(16)-H(16A) C(16)-H(16B) C(17)-C(20) C(17)-H(17) C(18)-H(18A) C(18)-H(18B) C(18)-H(18C) C(19)-H(19A) C(19)-H(19B) C(19)-H(19C) C(20)-C(22) C(20)-C(21) C(20)-H(20) C(21)-H(21A) C(21)-H(21B) C(21)-H(21C) C(22)-C(23)#1 C(22)-H(22A) C(22)-H(22B) C(23)-C(24) C(23)-C(22)#1 C(23)-H(23A) C(23)-H(23B) O(1)-H(1W) O(1)-H(2W) O(90)-C(90) C(90)-H(90A) C(90)-H(90B) C(90)-H(90C) 1.538(3) 1.541(3) 1.553(3) 1.534(3) 0.9900 1.549(3) 0.9800 0.9800 1.557(3) 0.9800 0.9800 1.535(3) 0.9900 0.9700 0.9700 0.9700 0.9700 0.9700 0.9700 1.531(3) 1.537(3) 0.9900 0.9700 0.9700 0.9700 1.532(3) 0.9800 0.9800 1.507(3) 1.532(3) 0.9800 0.9800 0.8999(11) 0.9001(11) 1.392(4) 0.9700 0.9700 0.9700 ______________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+2,y,-z+2 Anhang C 176 Table 3b. Binding angles [°] for C50H90N2O8 34a. (angles with C-H-bonds are not listed) _____________________________________________________________ C(12)-O(12)-H(12') C(24)-N(13)-C(3) C(24)-N(13)-H(13) C(3)-N(13)-H(13) C(2)-C(1)-C(10) C(1)-C(2)-C(3) N(13)-C(3)-C(2) N(13)-C(3)-C(4) C(2)-C(3)-C(4) C(3)-C(4)-C(5) C(6)-C(5)-C(4) C(6)-C(5)-C(10) C(4)-C(5)-C(10) C(7)-C(6)-C(5) C(6)-C(7)-C(8) C(14)-C(8)-C(7) C(14)-C(8)-C(9) C(7)-C(8)-C(9) C(11)-C(9)-C(8) C(11)-C(9)-C(10) C(8)-C(9)-C(10) C(19)-C(10)-C(1) C(19)-C(10)-C(5) C(1)-C(10)-C(5) C(19)-C(10)-C(9) C(1)-C(10)-C(9) C(5)-C(10)-C(9) C(12)-C(11)-C(9) O(12)-C(12)-C(11) O(12)-C(12)-C(13) C(11)-C(12)-C(13) C(14)-C(13)-C(12) C(14)-C(13)-C(18) C(12)-C(13)-C(18) C(14)-C(13)-C(17) C(12)-C(13)-C(17) C(18)-C(13)-C(17) C(8)-C(14)-C(15) C(8)-C(14)-C(13) C(15)-C(14)-C(13) C(14)-C(15)-C(16) C(15)-C(16)-C(17) C(20)-C(17)-C(13) C(20)-C(17)-C(16) C(13)-C(17)-C(16) C(22)-C(20)-C(17) C(22)-C(20)-C(21) C(17)-C(20)-C(21) C(20)-C(22)-C(23)#1 C(24)-C(23)-C(22)#1 O(24)-C(24)-N(13) O(24)-C(24)-C(23) N(13)-C(24)-C(23) H(1W)-O(1)-H(2W) 109.5 122.22(19) 118.9 118.9 115.53(17) 110.32(18) 110.07(18) 111.15(18) 109.74(16) 112.14(18) 110.37(18) 112.37(16) 112.74(17) 112.57(18) 110.72(18) 112.60(17) 109.72(16) 110.32(16) 111.83(15) 112.85(17) 111.74(16) 105.48(17) 110.38(17) 107.81(16) 111.49(16) 112.76(17) 108.84(16) 113.96(17) 107.12(17) 111.63(17) 111.15(17) 106.82(15) 112.20(17) 108.59(17) 101.80(15) 117.97(16) 109.35(16) 117.83(17) 113.57(16) 104.09(15) 104.10(16) 107.51(17) 118.40(17) 111.99(17) 102.85(16) 113.90(17) 110.31(18) 113.07(17) 116.07(18) 114.3(2) 121.4(2) 121.5(2) 117.1(2) 103(3) _____________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+2,y,-z+2 Anhang C Table 4. Anisotropic displacement parameters (Å2 x 103) for C50H90N2O8 34a. The anisotropic displacement factor exponent takes the form: -2 π2 [ h2 a*2 U11 + ... + 2 h k a* b* U12 ] _______________________________________________________________________ U11 U22 U33 U23 U13 U12 _______________________________________________________________________ O(12) 50(1) 32(1) 38(1) 0(1) 27(1) 8(1) O(24) 39(1) 51(1) 48(1) -16(1) 25(1) -3(1) N(13) 32(1) 32(1) 36(1) -1(1) 19(1) 1(1) C(1) 39(1) 35(1) 33(1) -4(1) 24(1) -7(1) C(2) 38(1) 30(1) 36(1) 2(1) 21(1) -2(1) C(3) 35(1) 34(1) 33(1) -1(1) 21(1) 2(1) C(4) 32(1) 28(1) 33(1) -5(1) 18(1) -6(1) C(5) 34(1) 29(1) 31(1) -7(1) 19(1) -2(1) C(6) 37(1) 26(1) 36(1) -9(1) 19(1) -4(1) C(7) 41(1) 23(1) 40(1) -5(1) 23(1) -7(1) C(8) 31(1) 22(1) 33(1) -3(1) 19(1) -4(1) C(9) 28(1) 24(1) 31(1) -4(1) 17(1) -3(1) C(10) 31(1) 28(1) 33(1) -4(1) 21(1) -1(1) C(11) 38(1) 27(1) 33(1) -2(1) 21(1) -7(1) C(12) 34(1) 23(1) 33(1) -4(1) 19(1) -5(1) C(13) 29(1) 21(1) 31(1) -2(1) 18(1) -3(1) C(14) 29(1) 22(1) 34(1) -3(1) 19(1) -3(1) C(15) 42(1) 26(1) 37(1) -1(1) 23(1) -6(1) C(16) 41(1) 28(1) 34(1) 2(1) 22(1) -4(1) C(17) 28(1) 24(1) 32(1) -2(1) 18(1) -3(1) C(18) 37(1) 34(1) 39(1) -4(1) 24(1) -2(1) C(19) 38(1) 39(1) 42(1) -8(1) 28(1) -2(1) C(20) 28(1) 32(1) 31(1) -1(1) 16(1) -1(1) C(21) 41(1) 38(1) 37(1) -8(1) 22(1) -16(1) C(22) 30(1) 37(1) 29(1) -2(1) 13(1) -3(1) C(23) 37(1) 39(1) 34(1) 9(1) 21(1) 6(1) C(24) 38(1) 37(1) 33(1) 4(1) 22(1) 0(1) O(1) 55(1) 36(1) 51(1) -4(1) 34(1) -3(1) O(90) 82(2) 56(1) 67(1) -2(1) 29(1) -13(1) C(90) 64(2) 76(2) 92(2) -2(2) 50(2) 7(2) _______________________________________________________________________ 177 Anhang C Table 5. Hydrogen coordinates ( x 104) and isotropic displacement parameters (Å2 x 103) for C50H90N2O8 34a. ________________________________________________________________ x y z U(eq) ________________________________________________________________ H(12') 8292 3092 8169 61 H(13) 11623 3269 14129 42 H(1A) 9235 4130 13528 42 H(1B) 8762 3338 12322 42 H(2A) 10039 3195 12589 44 H(2B) 10222 2756 13814 44 H(3) 10828 4627 14753 42 H(4A) 10507 5298 12529 39 H(4B) 10990 6117 13723 39 H(5) 9720 6267 13490 38 H(6A) 8977 7600 11887 43 H(6B) 9970 7707 12498 43 H(7A) 9731 6589 10895 45 H(7B) 9074 7685 10313 45 H(8) 7959 6436 9880 36 H(9) 9211 4552 10921 34 H(11A) 8168 3169 10408 40 H(11B) 7452 4178 9868 40 H(12) 7196 3181 8209 38 H(14) 8707 5374 8907 35 H(15A) 8491 7223 8101 44 H(15B) 7630 7445 7932 44 H(16A) 7730 6311 6258 43 H(16B) 6864 6693 6016 43 H(17) 7713 4446 6827 35 H(18A) 6405 6130 7237 55 H(18B) 6769 5625 8545 55 H(18C) 6165 4814 7350 55 H(19A) 7840 6220 11271 58 H(19B) 8293 5842 12651 58 H(19C) 7694 4917 11559 58 H(20) 5925 4986 5460 39 H(21A) 6070 3154 6262 61 H(21B) 6675 2680 5974 61 H(21C) 5672 2896 4879 61 H(22A) 6361 5471 4301 43 H(22B) 5634 4472 3659 43 H(23A) 13028 3115 15263 45 H(23B) 13537 3579 16632 45 H(1W) 8205(14) 1476(16) 6930(20) 62(8) H(2W) 9082(13) 1790(40) 7860(20) 123(16) H(90A) 5898 5290 9102 116 H(90B) 5063 5007 8969 116 H(90C) 5949 5350 10277 116 ________________________________________________________________ 178 Anhang C Table 6. 179 Torsion angles [°] for C50H90N2O8 34a. ___________________________________________________________________ C(10)-C(1)-C(2)-C(3) C(24)-N(13)-C(3)-C(2) C(24)-N(13)-C(3)-C(4) C(1)-C(2)-C(3)-N(13) C(1)-C(2)-C(3)-C(4) N(13)-C(3)-C(4)-C(5) C(2)-C(3)-C(4)-C(5) C(3)-C(4)-C(5)-C(6) C(3)-C(4)-C(5)-C(10) C(4)-C(5)-C(6)-C(7) C(10)-C(5)-C(6)-C(7) C(5)-C(6)-C(7)-C(8) C(6)-C(7)-C(8)-C(14) C(6)-C(7)-C(8)-C(9) C(14)-C(8)-C(9)-C(11) C(7)-C(8)-C(9)-C(11) C(14)-C(8)-C(9)-C(10) C(7)-C(8)-C(9)-C(10) C(2)-C(1)-C(10)-C(19) C(2)-C(1)-C(10)-C(5) C(2)-C(1)-C(10)-C(9) C(6)-C(5)-C(10)-C(19) C(4)-C(5)-C(10)-C(19) C(6)-C(5)-C(10)-C(1) C(4)-C(5)-C(10)-C(1) C(6)-C(5)-C(10)-C(9) C(4)-C(5)-C(10)-C(9) C(11)-C(9)-C(10)-C(19) C(8)-C(9)-C(10)-C(19) C(11)-C(9)-C(10)-C(1) C(8)-C(9)-C(10)-C(1) C(11)-C(9)-C(10)-C(5) C(8)-C(9)-C(10)-C(5) C(8)-C(9)-C(11)-C(12) C(10)-C(9)-C(11)-C(12) C(9)-C(11)-C(12)-O(12) C(9)-C(11)-C(12)-C(13) 56.4(2) -162.78(19) 75.4(2) -179.42(17) -56.8(2) 179.92(17) 57.9(2) 177.21(16) -56.2(2) 71.9(2) -54.9(2) 56.0(2) 179.98(17) -57.1(2) -49.7(2) -174.31(18) -177.29(17) 58.1(2) -169.85(17) -51.9(2) 68.3(2) -69.4(2) 165.14(18) 175.91(17) 50.4(2) 53.3(2) -72.2(2) -60.3(2) 66.7(2) 58.1(2) -174.84(16) 177.68(17) -55.3(2) 49.8(2) 176.76(17) 67.5(2) -54.6(2) O(12)-C(12)-C(13)-C(14) -61.7(2) C(11)-C(12)-C(13)-C(14) 57.8(2) O(12)-C(12)-C(13)-C(18) 177.09(16) C(11)-C(12)-C(13)-C(18) -63.4(2) O(12)-C(12)-C(13)-C(17) 52.0(2) C(11)-C(12)-C(13)-C(17) 171.56(17) C(7)-C(8)-C(14)-C(15) -56.6(3) C(9)-C(8)-C(14)-C(15) -179.87(18) C(7)-C(8)-C(14)-C(13) -178.64(17) C(9)-C(8)-C(14)-C(13) 58.1(2) C(12)-C(13)-C(14)-C(8) -61.7(2) C(18)-C(13)-C(14)-C(8) 57.2(2) C(17)-C(13)-C(14)-C(8) 173.99(16) C(12)-C(13)-C(14)-C(15) 168.91(17) C(18)-C(13)-C(14)-C(15) -72.2(2) C(17)-C(13)-C(14)-C(15) 44.6(2) C(8)-C(14)-C(15)-C(16) -158.76(18) C(13)-C(14)-C(15)-C(16) -32.0(2) C(14)-C(15)-C(16)-C(17) 7.3(2) C(14)-C(13)-C(17)-C(20) -162.95(18) C(12)-C(13)-C(17)-C(20) 80.6(2) C(18)-C(13)-C(17)-C(20) -44.1(2) C(14)-C(13)-C(17)-C(16) -38.90(19) C(12)-C(13)-C(17)-C(16) -155.37(17) C(18)-C(13)-C(17)-C(16) 79.95(19) C(15)-C(16)-C(17)-C(20) 147.90(18) C(15)-C(16)-C(17)-C(13) 19.7(2) C(13)-C(17)-C(20)-C(22) 174.63(18) C(16)-C(17)-C(20)-C(22) 55.2(2) C(13)-C(17)-C(20)-C(21) -58.4(3) C(16)-C(17)-C(20)-C(21) -177.82(19) C(17)-C(20)-C(22)-C(23)#1 69.1(2) C(21)-C(20)-C(22)-C(23)#1 -59.3(2) C(3)-N(13)-C(24)-O(24) 1.0(3) C(3)-N(13)-C(24)-C(23) 179.47(18) C(22)#1-C(23)-C(24)-O(24) -60.9(3) C(22)#1-C(23)-C(24)-N(13) 120.6(2) ___________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+2,y,-z+2 Anhang D 180 Anhang D Im Folgenden wird näher auf das Zustandekommen der in Kapitel 3 angegebenen jeweils zwei möglichen Diastereomere für die Catenan-Variante (B) und die Knotan-Variante (C) der Verbindung 38b (Cyclo-(Val-DCS)6) eingegangen (Teile D1 und D2). Für die KnotanVariante wurden zudem die globalen Minima der beiden möglichen Diastereomere aus SD-Simulationen (Prozedur siehe Haupttext in Kapitel 3) angegeben und kurz verglichen (Teil D3). O H N N H O HO OH H O N N H O O N N H O H 38b HO OH H O H N N O O H N N O H OH O OH H N N H O Sowohl bei der Catenan-Variante als auch bei der Knotan-Variante ist durch die gleichgerichteten Amid-Bindungen eine durchgehende Bindungsrichtung in den zusammenhängenden Ketten der einzelnen Cyclen vorgegeben. Da durch die in der Synthese eingesetzten Steroid-Bausteine (DCS) und natürlichen L-Aminosäuren (Val; mit S-Konfiguration an Cα) die Stereochemie bereits vorab determiniert ist, können einige Stereoisomere prinzipiell nicht vorliegen. Zur Vereinfachung ist bei den nachstehenden Abbildungen für die Bindungsrichtung jeweils ein Pfeil angegeben (Richtung: CO –> NH) und zur Kennzeichnung der Stereocheimie exemplarisch die Seitenkette des Valins einmal eingezeichnet. Anhang D 181 Teil D1: Graphische Darstellung von Catenananordnungen zur Ableitung der möglichen Stereoisomere von Catenanen der Verbindung 38b aus zwei zusammenhängenden Cyclen. Doppelt vorhandene Stereoisomere sind einfach durchgestrichen, Isomere mit falscher Stereochemie (Valin mit R-Konfiguration) sind kreuzweise durchgestrichen. Es verbleiben die isomeren Catenane Nr. 1 und 4, die zueinander diastereomer sind. Anhang D 182 Teil D2: Graphische Darstellung von Knotananordnungen zur Ableitung der möglichen Stereoisomere von Knotanen der Verbindung 38b. Doppelt vorhandene Stereoisomere sind einfach durchgestrichen, Isomere mit falscher Stereochemie (Valin mit R-Konfiguration) sind kreuzweise durchgestrichen. Es verbleiben die isomeren Knotane Nr. 1 und 4, die zueinander diastereomer sind. Anhang D 183 Teil D3: Globale Minima (aus SD-Simulationen) der verbleibenden beiden möglichen Knotan-Diastereomere von Verbindung 38b. Das Diastereomer mit der Nummer 1 ist dasjenige, das am besten mit den experimentellen Ergebnissen korrespondiert. Das andere Diastereomer (Nr. 4) weist im Gegensatz zu Diastereomer Nr. 1 keine C3-Symmetrie auf und bildet an den Knoten-Kontaktstellen jeweils unterschiedliche Wasserstoffbrückenbindungen aus, daher war hier eine Identifizierung von nur zwei unterscheidbaren Val- und DCS-Bausteinen nicht möglich (wäre für einen doppelten NMR-Signalsatz zwingend erforderlich). Da für Nr. 4 keine Übereinstimmung mit den experimentellen Ergebnissen gefunden werden konnte, wurde auf eine ausführliche Diskussion in Kapitel 3 verzichtet. Lebenslauf 184 Lebenslauf Rüdiger Ladberg Stockumer Str. 217 44225 Dortmund geboren 18. 11. 1970 in Castrop-Rauxel Staatsangehörigkeit deutsch Familienstand ledig Schulbildung 08/77 - 07/81 Bodelschwingh-Grundschule in Dortmund 08/81 - 05/90 Heinrich-Heine-Gymnasium in Dortmund 05/90 Allgemeine Hochschulreife Grundwehrdienst 07/90 - 07/91 Amphibisches Pionierbataillon 130 in Minden Hochschulbildung 10/91 - 07/96 Studium der Chemie an der Ruhr-Universität Bochum 11/95 - 07/96 Diplomarbeit am Lehrstuhl für Organische Chemie II unter der Leitung von Prof. Dr. D. Hasselmann, Thema: Synthesewege zu einem Methylenoxabicycloheptan-System 07/96 Diplomhauptprüfung 08/96 - 07/01 Promotionsarbeit am Lehrstuhl für Organische Chemie I, AG Naturstoffchemie, unter der Leitung von Prof. Dr. Martin Feigel (unterbrochen von 12/96 bis 09/97) 14. 12. 2001 Promotion zum Dr. rer. nat. seit 03/98 Zusatzstudium der Arbeitswissenschaft an der RuhrUniversität Bochum mit den Schwerpunkten Total Quality Management, Change Management, Information und Kommunikation, Innovationsmanagement, Wirtschaftlichkeitsrechnung sowie Moderne Managementkonzepte und Arbeitsorganisation zur Zeit Diplomarbeit Arbeitswissenschaft im Bereich Total Quality Management 02/93 - 04/96 Dozent für Chemie und Physik für die Firma PhysiKurs GbRmbH - Medizinische Repetitorien, Bochum 12/96 - 01/98 Redakteur und PR-Referent für die Management-Lektüre „8-Seiten-Skripts“ bei der Firma ISI Marketing GmbH, Bochum (ab Oktober 1997 in Teilzeit) 01/98 - 04/98 Assistenztätigkeit in der Marketingabteilung der Euro OTC Pharma GmbH, Kamen (Teilzeit) seit 04/98 Tätigkeit als Freier Journalist für die Zeitung CHEManager des GIT Verlages, Darmstadt; seit 05/01 auch für die Abteilung Unternehmenskommunikation, Referat Fachpresse, der Bayer AG, Leverkusen 07/98 - 02/99 Projektarbeit am Institut für angewandte Innovationsforschung e.V., Bochum (Teilzeit) 04/99 - 04/00 Lehrer für Chemie (2 Kurse) an der Krankenpflegeschule des Elisabeth-Krankenhauses, Recklinghausen Praktische Erfahrung