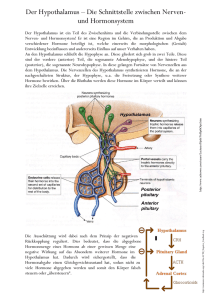
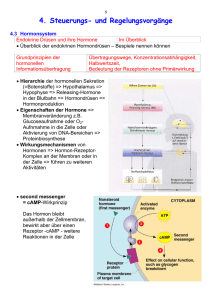
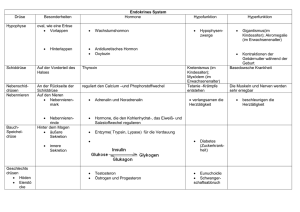
DIE HORMONELLE STEUERUNG DES ORGANISMUS
Werbung

Diplomarbeit DIE HORMONELLE STEUERUNG DES ORGANISMUS Endokrine Regelkreise und ihre pharmakologische Relevanz eingereicht von Katarina Michelitsch zur Erlangung des akademischen Grades Doktorin der gesamten Heilkunde (Dr.in med. univ.) an der Medizinischen Universität Graz ausgeführt am Institut für Experimentelle und Klinische Pharmakologie unter der Anleitung von Univ.-Prof. Mag. Pharm. Dr. phil. Eckhard Beubler Graz, 23.12.2016 Eidesstattliche Erklärung Ich erkläre ehrenwörtlich, dass ich die vorliegende Arbeit selbstständig und ohne fremde Hilfe verfasst habe, andere als die angegebenen Quellen nicht verwendet habe und die den benutzten Quellen wörtlich oder inhaltlich entnommenen Stellen als solche kenntlich gemacht habe. Graz, am 23.12.2016 Katarina Michelitsch eh i Danksagungen Ich möchte mich herzlich bei meinem Diplomarbeitsbetreuer Univ.-Prof. Mag. pharm. Dr. phil. Eckhard Beubler für sein Interesse und die hilfreiche Betreuung bedanken. Des Weiteren möchte ich meiner Familie, vor allem meinen lieben Eltern und meinem lieben Bruder, für ihre Unterstützung, die vielen Gespräche, Ermutigungen und Ratschläge während des gesamten Studiums danken. Ein großer Dank gebührt außerdem meinen lieben FreundInnen für ihre positive Zusprache, die anregenden Diskussionen, die kritischen Auseinandersetzungen und die vielen lachenden Momente. Merci! ii Zusammenfassung Titel Die hormonelle Steuerung des Organismus - Endokrine Regelkreise und ihre pharmakologische Relevanz Einleitung Verschiedenste Prozesse des menschlichen Organismus werden durch eine Vielzahl von Hormonen und Neurotransmittern gesteuert, deren Komplexität, Integrität und Konnex besonders im Prinzip hypothalamo-hypophysärer Achsen deutlich wird. Hormone und Hormon-Analoga werden heutzutage therapeutisch ubiquitär eingesetzt- im besonderen Fokus dieser Arbeit stehen hierbei die physiologischen und pathophysiologischen Mechanismen der Organ- und Hormonsysteme Schilddrüse, Nebennierenrinde und Gonaden, sowie vor allem deren pharmakologische Eigenschaften, aktuelle Therapieschemata und Anwendungsgebiete. Ziel Diese Arbeit soll grundlegendes und aktuelles Wissen zur hormonellen Steuerung des menschlichen Organismus mit besonderem Fokus auf die hypothalamo-hypophysären Regelkreise, sowie deren pharmakologische Relevanz im klinischen Kontext zusammenfassen und veranschaulichen. Im Besonderen soll hierbei auf die hypothalamo-hypophysäre thyreoidale Achse, die hypothalamo-hypophysäre-adrenotrophe Achse und die hypothalamo-hypophysäre-gonadale Achse eingegangen werden. Material und Methodik Diese Diplomarbeit ist eine Literaturrecherche, die basierend auf relevanten und aktuellen Forschungsergebnissen den derzeitigen Wissensstand zusammenfassen und erläutern soll. Für die Recherche wurde vorwiegend auf den Lehr- und Fachbuchbestand der Medizinischen Universität Graz, sowie auf diverse Fachjournale wie das European Journal of Endocrinology, das European Journal of Pharmacology und das Journal of Clinical Endocrinology and Metabolism, sowie die online Datenbanken PubMed und Google Scholar zugegriffen. Ergebnisse Die pharmakologische Relevanz hypothalamo-hypophysärer Regelkreise im klinischen Kontext konnte deutlich analysiert und konkretisiert werden- eine Dysfunktion oder Dysbalance hormoneller Regelkreise führt zu weitgreifenden Auswirkungen auf den Organismus. Die iii pharmakologischen Anwendungsformen, Wirkungen und Nebenwirkungen der einzelnen Hormone, sowie deren Interaktionen untereinander sind weitreichend und wurden explizit erläutert. Schlussfolgerung Immer noch sind viele Mechanismen und Zusammenhänge endokriner Achsen unklar, weitere Forschung auf diesem Gebiet ist somit unerlässlich. In Zukunft werden molekularbiologische und technische Innovationen wie genetische Sequenzierungen und die Einbeziehung hormoneller CoAktivatoren das Wissen über physiologische und pathophysiologische Mechanismen, sowie deren pharmakologischen Nutzen noch erweitern. iv Abstract Title The hormonal regulation of the human organism – the endocrine axis and its pharmacological relevance Introduction Many of the physiologic processes in the human organism are triggered and regulated by multiple hormones and neurotransmitters of which the complexity, integrity and nexus are particulary noticeable in the principle of hypothalamic-pituitary axes. Hormones and hormon-analogues are used ubiquitary in pharmacologic therapy today- the emphasis is particulary on the physiologic and pathophysiologic mechanisms of the organ- and hormone-systems of the thyroid, the adrenal cortex and the gonads as well as especially their pharmacologic characteristics and their current and relevant state in pharmacological and therapeutical use. Aim The aim of this thesis is to give an overview, to discuss and summarize the main actions of the hypothalamic-pituitary axes and their consequences for the human organism as well as to focus on the pharmacological use of the different hormones of these systems in respect of the clinical context. Detailed examination of the hypothalamic-pituitary-thyroid axis, the hypothalamicpituitary-adrenal axis and the hypothalamc-pituitary-gonadal axis shall be made. Methods This thesis is based on a literature research and shall represent the current state of knowledge. The research was conducted via the library of the Medical University of Graz. The databases PubMed and Google Scholar were used for the extended search of recent publications, articles, clinical trials, reviews, etc. Furthermore medical journals such as the European Journal of Endocrinology, the European Journal of Pharmacology and the Journal of Clincal Endocrinology and Metabolism have been accessed and offered insight into current research progress. Results The pharmacological relevance of the hypothalamic-pituitary axes in the clinical context became clear- a dysfunction or dysbalance of hormonal feedback-mechanisms has profound effects on the organism. Pharmacological use, actions and adverse effects of the different hormones as well as their interactions are extensive and have been explicitly elucidated. v Conclusion Many of the described hormonal processes and their correlations are still unknown, additional research is therefore necessary. In the future molecularbiological and technical innovations such as genome sequencing and the incorporation of hormonal co-activators will extend the knowledge about physiological and pathophysiological mechanisms of action as well as their pharmacolgical use and benefit. vi Inhaltsverzeichnis Danksagungen .................................................................................................................................... ii Zusammenfassung ............................................................................................................................. iii Abstract .............................................................................................................................................. v Inhaltsverzeichnis ............................................................................................................................. vii Glossar und Abkürzungen ................................................................................................................. ix Abbildungsverzeichnis ...................................................................................................................... xi 1 Einleitung ................................................................................................................................... 1 1.1 Hormone ............................................................................................................................. 1 1.1.1 Synthese und chemische Struktur............................................................................... 1 1.1.2 Transport, Wirkung und Rezeptorklassen .................................................................. 3 1.1.3 Regulation und hormonelle Regelkreise .................................................................... 5 1.2 Das hypothalamo-hypophysäre System ............................................................................. 8 1.2.1 Aufbau und Zielgewebe ............................................................................................. 8 1.2.2 Steuerung und Rückkopplungsmechanismen ............................................................. 9 1.2.3 Funktionstests ........................................................................................................... 10 1.3 Der Hypothalamus............................................................................................................ 11 1.3.1 GHRH (growth hormone releasing hormone) und GHRIH (Somatostatin). ............ 12 1.3.2 TRH (thyreotropin releasing hormone) .................................................................... 15 1.3.3 CRH (corticotropin releasing hormone, Corticoliberin) .......................................... 15 1.3.4 GnRH (gonadotropin releasing hormone, Gonadoliberin) ....................................... 15 1.4 Die Hypophyse (Glandula pituitaria, Hirnanhangsdrüse) ................................................ 19 1.4.1 ADH (Antidiuretisches Hormon, Vasopressin) ........................................................ 20 1.4.2 Oxytocin ................................................................................................................... 23 1.4.3 GH (growth hormone) und IGF-1 (Insulin-like growth factor-1) ............................ 25 1.4.4 Prolaktin und Dopamin ............................................................................................ 28 1.4.5 TSH (Thyreotropin).................................................................................................. 29 1.4.6 ACTH (Adrenocorticotropes Hormon, Corticotropin) ............................................. 30 1.4.7 Gonadotropine .......................................................................................................... 31 2 Material und Methoden ............................................................................................................ 34 3 Ergebnisse ................................................................................................................................ 35 3.1 Die hypothalamo-hypophysäre-thyreoidale Achse .......................................................... 35 3.1.1 Trijodthyronin und Thyroxin.................................................................................... 36 3.1.2 Hyperthyreose und ihre pharmakologische Therapie ............................................... 37 vii 3.1.3 3.2 Hypothyreose und ihre pharmakologische Therapie ................................................ 39 Die hypothalamo-hypophysäre-adrenotrophe Achse ....................................................... 42 3.2.1 Glucocorticoide ........................................................................................................ 43 3.2.2 Mineralcorticoide ..................................................................................................... 47 3.3 Die hypothalamo-hypophysäre-gonadale Achse .............................................................. 49 3.3.1 Physiologie des ovulatorischen Zyklus .................................................................... 50 3.3.2 Estrogene .................................................................................................................. 51 3.3.3 Antiestrogene ........................................................................................................... 52 3.3.4 Gestagene ................................................................................................................. 53 3.3.5 Antigestagene und SPRM (selektive Progesteronrezeptor-Modulatoren)................ 55 3.3.6 Androgene ................................................................................................................ 55 3.3.7 Antiandrogen wirksame Substanzen ........................................................................ 57 3.3.8 Die hormonelle Kontrazeption ................................................................................. 58 4 Diskussion ................................................................................................................................ 63 5 Literaturverzeichnis .................................................................................................................. 64 viii Glossar und Abkürzungen 5-HT 5-Hydroxytryptamin A. Arteria Aa. Arteriae ACTH Adrenocorticotropes Hormon ADH GHRH hormone GHRIH Growth hormone release-inhibiting hormone GHQ Anti-Diuretisches Hormon Growth hormone releasing General Health Questionnaire GnRH Gonadotropin releasing ATP Adenosintriphosphat bzw. beziehungsweise GTP Guanosintriphosphat C Kohlenstoff H+ Wasserstoff ca. circa HCG human chorionic cAMP Cyclisches Adenosin-Monophosphat CBG hormone gonadotropine HCS Corticosteroid-binding human chorionic somatotropin globulin HDL high density lipoprotein COX-2 Cyclooxygenase Typ 2 HMG human menopausal CRH Corticotropin releasing hormone gonadotropine HPA Hypothalamic-pituitary- CYP Cytochrom-Protein adrenal (axis) D Dopamin d.h. das heißt DDVAP 1-Desamino-8-D-Arginin- HPL human placental lactogen Vasopressin IE Internationale Einheit DHEA Dehydroepiandrosteron IGF Insulin-like growth factor DHT Dehydrotestosteron IL Interleukin DIT Diiodtyrosin i.m. intramuskulär EEG Elektroencephalogramm IU International Unit ER Estrogen-Rezeptor IUP Intrauterinpessar etc. etcetera i.v. intravenös FSH Follikel-stimulierendes Jak 2 Januskinase 2 HPG Hypothalamic-pituitaryGonadal (axis) + Hormon K Kalium GABA γ-Aminobuttersäure kg Kilogramm GDP Guanosindiphosphat L Liter GH Growth hormone LDL low density lipoprotein ix LH Luteinisierendes Hormon LPH Lipotropin Progesteronrezeptor- Mb. Morbus Modulator min. Minute MIT Monoiodtyrosin MSH Melanophoren- sst1-5 Somatostatin-Rezeptoren stimulierendes Hormon T3 Trijodthyronin N. SPRM SRIF Selektiver somatotropin releaseinhibiting factor Nervus T4 Thyroxin + Natrium TBG Thyroxin-binding Ncl. Nucleus Ncll. Nuclei NF Nuclear Factor NSAR Nichtsteroidales Anti- Na globulin TBPA Präalbumin TIA Rheumatikum O2 Sauerstoff OHSS ovarielles PCO Thyroxin-bindendes Transitorische ischämische Attacke TIDA tuberoinfundibuläre dopaminerge Neurone Hyperstimulations-Syndrom TNF Tumor-Nekrose-Faktor Polycystisches TRH Thyreotropin-Releasing Ovarsyndrom Hormon p.o. per os TSH POMC Proopiomelanocortin PRL Prolaktin U Unit PTU Propylthiouracil V1a,1b,2 Vasopressin-Rezeptoren REM Rapid-Eye-Movement V. Vena s.c. subkutan Vv. Venae SERD Selektiver VLDL very low density hormone EstrogenrezeptorSERM Thyroidea stimulating lipoprotein Downregulator vWF von-Willebrand-Faktor Selektiver z.B. zum Beispiel EstrogenrezeptorModulator SIADH Syndrom der inadäquaten ADH-Sekretion SGA small for gestational age SHBG Sexualhormon-bindendes Globulin x Abbildungsverzeichnis Abbildung 1: Schematische Darstellung der vaskulären und neuronalen Verbindungen zwischen Hypothalamus und Hypophyse.(9) ..................................................................................................... 8 Abbildung 2: Prinzip der negativen Rückkopplung.(4) ..................................................................... 9 Abbildung 3: Schematische Darstellung der hypothalamischen Kerngebiete.(9) ............................ 12 Abbildung 4: Hypothalamo-hypophysäre-thyreoidale Achse.(4) .................................................... 35 Abbildung 5. Hypothalamo-hypophysäre-adrenotrophe Achse.(4) ................................................. 43 Abbildung 6: Steroidale Strukturformeln.(28) ................................................................................. 45 Abbildung 7: Hypothalamo-hypophysäre-gonadale Achse.(4) ........................................................ 49 Abbildung 8: Schematische Darstellung des weiblichen Menstruationszyklus.(39) ....................... 51 xi 1 Einleitung Der menschliche Organismus unterliegt humoralen Prozessen und endokrinen Regelkreisen, welche über ein vielfältiges und komplexes physiologisches Wirkungsspektrum verfügen und ebenso hohe Relevanz in Bezug auf die pharmakologische Therapie endokriner Störungen aufweisen. Ziel dieser Arbeit ist es, relevante und neueste wissenschaftliche Erkenntnisse für das pharmakologische Verständnis endokriner Regelkreise zusammenzufassen und zu veranschaulichen. Dabei sollen grundlegende Begriffe und biologische Prozesse geklärt und erläutert werden, sowie in Folge auf die spezifischen endokrinen Achsen mit besonderem Fokus auf die hypothalamo-hypophysäre-thyreoidale Achse, die hypothalamo-hypophysäre-adrenotrophe Achse und die hypothalamo-hypophysäre-gonadale Achse in Hinsicht auf ihren pharmakologischen Nutzen und Gebrauch eingegangen werden. 1.1 Hormone Hormone werden definiert als chemische Signalmoleküle, die in geregelter Weise mit dem Blutstrom an spezifische Zielorgane und –gewebe transportiert werden. Die hormonelle Signalübertragung stellt einen speziellen Prozess der Kommunikation zwischen verschiedenen Zellen des Organismus dar. Molekularbiologisch betrachtet, funktioniert dies mittels Ligand-RezeptorBindungen, wobei generell die Auffassung besteht, dass Hormone von sekretorischen Drüsen produziert werden und infolgedessen Rezeptoren an der Zellmembran oder im Zellkern aktivieren. Jüngste Forschungsarbeiten legen den Verdacht nahe, dass die Wirkung von Hormonen und deren Regulation wesentlich komplexer ist, als bisher angenommen.(1) Prinzipiell unterscheidet man je nach Signalübertragungsweg zwischen endokriner, neuroendokriner, parakriner und autokriner Hormonsekretion. Unter endokriner Signalübertragung versteht man die Freisetzung und den Transport des Hormons mittels Blutstrom, mit Wirkung des Hormons an einem vom Syntheseort entfernten Wirkort. Bei der neuroendokrinen Sekretion erfolgt die Hormonabgabe aus einer Nervenendigung in den Blutstrom. Die parakrine Sekretion erfolgt mittels Diffusion, da der Wirkort des Hormones in Nähe des Syntheseorts liegt. Eine autokrine Signalübertragung liegt vor, wenn die horomonproduzierende Zelle gleichzeitig Zielort des von ihr produzierten Hormons ist. Eine eindeutige Abgrenzung zwischen den einzelnen biologischen Signalübertragungswegen ist jedoch nicht immer möglich, da manche Hormone sowohl endokrin, als auch parakrin wirken. Dies konnte beispielsweise bei Hormonen der Adenohypophyse gezeigt werden. IGF-1 (Insulin-like growth factor-1) besitzt andererseits parakrine und autokrine Eigenschaften.(2,3) 1.1.1 Synthese und chemische Struktur Die Bildung von Hormonen findet entweder in lokalisierten spezifischen Zellen und Zellverbänden von endokrinen Drüsen, oder in Zellen mit zusätzlicher sekretorischer Funktion statt. 1 Hormonbildende Zellen enthalten üblicherweise spezielle sekretorische Granula, um größere Mengen an Hormonen zu speichern und diese dann als Antwort auf ein spezifisches Signal zu sezernieren.(1) Zu den klassischen Hormonbildungsorganen zählen der Hypothalamus, die Hypophyse, die Schilddrüse und die Nebenschilddrüse, die Nebennieren, die Langerhans-Inseln des Pankreas, die Gonaden und die Plazenta. Die Bildung einzelner Hormonmoleküle beginnt in den spezifischen endokrinen Drüsen mittels Vorläufermolekülen. Im Fall der Steroidhormone beispielsweise, ist Cholesterol das Vorläufermolekül, welches durch multiple Hydroxylierungen, Methylierungen und Demethylierungen basierend auf Cytochrom P450-Reaktionen zu Glucocorticoiden, Androgenen, Estrogenen und deren aktive Metaboliten umgewandelt wird.(1,2) Jedoch werden nicht alle Hormone in spezialisierten endokrinen Drüsen gebildet. Beispiel dafür ist das Proteinhormon Leptin, das Appetit und Energiehaushalt steuert. In Adipozyten gebildet, signalisiert es dem zentralnervösen System den Ernährungszustand des Organismus. Ebenso ein Beispiel ist das Cholesterol-Derivat 7-Dehydrocholesterol, Vorstufe des Vitamin-D, welches in den Keratinozyten der Haut durch photochemische Reaktionen gebildet wird.(1) Unter ektoper Hormonproduktion versteht man die meist unkontrollierte Synthese eines Hormons durch maligne entartete Zellen in nicht-endokrinen Geweben. Typisches Beispiel hierfür ist das kleinzellige Bronchialkarzinom, dessen Zellen häufig Corticotropin bilden.(2) 1.1.1.1 Chemischer Aufbau Aus chemischer Sicht unterscheidet man hydrophile und lipophile Hormone nach ihrer Löslichkeit in Wasser, sowie je nach Aufbau: Glykoproteine, Protein- und Peptidhormone, Steroidhormone und Derivate der Aminosäuren Tyrosin und Tryptophan. Vitamin-A-Säure, eine ungesättigte Fettsäure, wird in manchen Einteilungen zu den Hormonen mit intrazellulärem Rezeptor gerechnet.(2-4) Hydrophile Hormone werden in sekretorischen Vesikeln gespeichert, können nach Exozytose in freier Form im Plasma transportiert werden und binden meist an membranständige Rezeptoren. Sie haben eine kurze Halbwertszeit und die Zellantworten sind meist schnell. Protein- und Peptidhormone zählen zu den hydrophilen Hormonen. Lipophile Hormone sind im Plasma an Transportproteine gebunden und binden meist an intrazelluläre Rezeptoren. Sie können intrazellulär nicht gespeichert werden, haben eine lange Halbwertszeit und Zellantworten sind meist langsam. Zu den lipophilen Hormonen zählen die Steroidhormone und die Aminosäurenderivate. Steroidhormone werden aufgrund der nichtvorhandenen Speicherkapazität erst bei Bedarf neu gebildet und direkt in den Blutstrom sezerniert.(2,3) Protein- und Peptidhormone werden durch Proteinbiosynthese mittels Transkription und Translation, meist als biologisch noch inaktive Vorläufermoleküle produziert. Aus einem Vorläufermolekül entsteht durch Abspaltung eines Teilpeptids ein Prohormon, in dem die aktive Form des eigentlichen Hormons als Teilsequenz enthalten ist und welches durch gezielte Hydrolyse 2 freigesetzt wird. Durch weitere Modifikation (sogenannte posttranslationale Prozessierung) können aus einem Vorläufermolekül verschiedene aktive Hormone synthetisiert werden. Glykosylierungsreaktionen nach Verknüpfung der Aminosäuren spielen für die Aktivität von Proteinhormonen eine zentrale Rolle. Beispiele für Protein- und Peptidhormone sind ACTH (Adrenocorticotropes Hormon), Insulin, Prolaktin, Wachstumshormon (GH), Calcitonin, sowie die Releasing-Hormone des Hypothalamus. Zu den Glykoproteinen zählen TSH (Thyreoideastimulierendes Hormon), sowie die Gonadotropine LH (Luteinisierendes Hormon), FSH (Follikelstimulierendes Hormon) und HCG (human chorionic gonadotropine).(2-4) Steroidhormone werden aus Cholesterin synthetisiert. Dazu zählen die Nebennierenrindenhormone, Sexualhormone und Vitamin-D-Derivate. Als Grundstruktur aller Steroidhormone fungiert Cyclopentanoperhydrophenanthren mit vier Ringen (A, B, C, D). Jedes Steroidhormon enthält eine ungesättigte C=C-Doppelbindung, mit Ausnahme der Estrogene, welche einen aromatisierten ARing enthalten. Steroidhormone können auch nach Zahl ihrer C-Atome eingeteilt werden: C-21Steroide (Glucocorticoide, Mineralcorticoide und Progesteron), C-19-Steroide (Androgene) und C18-Steroide (Estrogene).(3) Derivate der Aminosäure Tyrosin sind Schilddrüsenhormone (Thyroxin, Trijodthyronin) und Katecholamine (Adrenalin, Noradrenalin, Dopamin), Abkömmlinge der Aminsäure Tryptophan sind Serotonin und Melatonin.(2,3) 1.1.2 Transport, Wirkung und Rezeptorklassen Steroidhormone und Schilddrüsenhormone sind im Blut an Transportproteine gekoppelt. Sexualhormone Glucocorticoide werden mittels mit Sexualhormon-bindendem CBG (Corticosteroid-binding Globulin (SHBG) globulin, transportiert, Transcortin) und Schilddrüsenhormone beispielsweise mit Thyroxin-bindendem Globulin (TBG) und Thyroxinbindendem Präalbumin (TBPA). Protein- und Peptidhormone werden aufgrund ihrer hydrophilen Eigenschaften frei im Plasma transportiert und benötigen keine Transportproteine.(2) Hormone können bereits voll aktiviert sein, wenn sie in den Blutstrom sezerniert werden (z.B. GH, Insulin) oder erst nach Aktivierung durch bestimmte Zellen ihre biologischen Effekte erlangen. Diese Aktivierungsprozesse sind meist komplex gesteuert z.B. T3 (Trijodthyronin), T4 (Thyroxin) oder Testosteron. Ob eine Zelle auf ein Hormon reagiert, hängt von der Expression des jeweiligen hochspezifischen Rezeptors, sowie von intrazellulären Signalwegen ab, die durch die Bindung des Hormons aktiviert werden, wobei Hormonbindungsmechanismen grundsätzlich aus einer Primärreaktion zwischen Hormon und spezifischem Rezeptor und einer anschließenden Folgereaktion in der Zielzelle bestehen. Rezeptorproteine können an der Zellmembran, im Zytoplasma oder am Zellkern lokalisiert sein. Generell binden Peptid- und Proteinhormone aufgrund ihrer chemischen Struktur eher an Rezeptoren der Zellmembranen und Steroidhormone eher an intrazelluläre Rezeptoren.(1,2) 3 1.1.2.1 Rezeptorklassen Hormone können am jeweiligen Rezeptor agonistisch (d.h. die Bindung des Hormons führt zu einer spezifischen Zellantwort) oder antagonistisch (d.h. die Bindung des Hormons führt zur Unterdrückung einer spezifischen Zellantwort) wirken. Durch die Bindung des Hormons an den jeweiligen Rezeptor wird die sterische Konformation des Rezeptormoleküls verändert und es kommt zur intrazellulären Signaltransduktion: eine Kaskade von intrazellulären Mechanismen wird ausgelöst, die in einem zellulären Effekt resultiert. Bei der intrazellulären Signalweiterleitung spielen Second Messenger (intrazelluläre Botenstoffe) eine wichtige Rolle, indem sie das Signal eines Hormons, nach dessen Bindung an den jeweiligen Rezeptor, in der Zelle weiterleiten, unterdrücken oder verstärken.(3) Grundsätzlich sind membranständige von intrazellulären Rezeptoren zu unterscheiden. Rezeptoren im Zellinneren sind Rezeptoren für lipophile Hormone wie Steroid- und Schilddrüsenhormone. Man unterscheidet Typ-1- von Typ-2-Rezeptoren, sie sind Rezeptoren für Transkriptionsfaktoren. Membranrezeptoren befinden sich an der Oberfläche der Zelle. An ihnen binden hydrophile Hormone (Protein-und Petidhormone), die nicht die Zellmembran durchdringen können. Sie sind aus einer oder mehreren extrazellulären Domänen (Hormonbindungsstellen), transmembranösen Domänen und intrazellulären Domänen aufgebaut; diese führen zur Aktivierung der intrazellulären Signaltransduktion. Man unterscheidet molekularbiologisch vier Klassen der Membranrezeptoren, wobei alle vier als Hormonrezeptor wirksam sein können: Ionenkanal-gekoppelte Rezeptoren, GProtein-gekoppelte Rezeptoren, Rezeptoren mit Enzymaktivität und Rezeptoren für Transportproteine.(3) Bei ligandengesteuerten Ionenkanälen induziert die Bindung des spezifischen Hormons direkt die Öffnung des jeweiligen Rezeptorkanals. Beispiele für diese Rezeptorklasse sind der nikotinische Acetylcholinrezeptor, sowie der 5-HT3-Rezeptor (ein durch Serotonin gesteuerter Ionenkanal für Natriumionen).(3,4) G-Protein-gekoppelte Rezeptoren sind aufgebaut aus einer Polypeptidkette mit sieben transmembranösen α-helikalen Domänen, einem extrazellulären N-Terminus und einem intrazellulären C-Terminus. Am C-terminalen Ende kann der Rezeptor mit GTP-bindenden Proteinen (Guanosintriphosphat, G-Proteine) interagieren, welche nachfolgende Effektormoleküle aktivieren oder suppressiv beeinflussen. Diese Wechselwirkung zwischen G-Protein und nachfolgendem Effektormolekül spielt eine zentrale Rolle für die anschließende intrazelluläre Signaltransduktion. Unter G-Protein versteht man ein aus den drei Proteinuntereinheiten Gα, Gβ, Gγ bestehendes, heterotrimeres Molekül. Gα-Untereinheiten variieren in zwanzig unterschiedlichen Formen, welche wiederum auf unterschiedliche Effektormoleküle einwirken. Ebenso kann die Gβγ-Untereinheit an spezielle Effektoren binden. Es besteht außerdem eine Rezeptorpromiskuität; das heisst, dass ein Rezeptortyp mit einem oder mehreren verschiedenen G-Protein-Typen in Wechselwirkung treten kann. Ist kein Ligand an das G-Protein gebunden, liegt dieses in seiner inaktiven Form vor: die drei 4 Proteinuntereinheiten sind zu einem Komplex verbunden, wobei die Gα-Einheit GDP (Guanosindiphosphat) gebunden hat. Dockt nun ein Ligand an den Rezeptor an, wird GDP zu GTP phosphoryliert und der Proteinkomplex teilt sich in die nun aktivierte Gα-GTP-Einheit und in die Gβγ-Einheit. Die sich im aktivierten Zustand befindende Gα-Einheit hat nun die Fähigkeit an Effektoren zu binden und verfügt über eine intrinsische GTPase-Aktivität, wodurch GTP wieder zu GDP dephosphoryliert wird. Die Gα-Untereinheit inaktiviert sich somit selbst und führt so zur Termination der Hormonwirkung. Gα-GDP und Gβγ verbinden sich wieder zum ursprünglichen heterotrimeren Komplex und das G-Protein steht für den nächsten Reaktionszyklus bereit. Zu den G-Protein-gekoppelten Rezeptoren zählen die Rezeptoren von Peptidhormonen wie Vasopressin, Somatostatin, GnRH (gonadotropin releasing hormone) und Oxytocin, sowie die Rezeptoren von Glykoproteinhormonen wie Adrenalin, Dopamin, LH, FSH und TSH.(3,4) Enzymgekoppelte Rezeptoren werden unterteilt in Rezeptoren mit ligandengesteuerter eigenständiger Enzymaktivität (Rezeptor-Tyrosinkinasen; sie verfügen über eine integrierte Tyrosinkinaseaktivität) und assoziierter Enzymaktivität (Tyrosinkinase-gekoppelte Rezeptoren).(3,4) 1.1.3 Regulation und hormonelle Regelkreise 1.1.3.1 Sekretionsmuster und zirkadiane Rhythmen Anatomisch unterschiedliche endokrine Drüsen bestehen aus hochdifferenzierten Zellen, welche Hormone synthetisieren, speichern und sezernieren. Bestehende zirkulierende Hormonkonzentrationen sind Folge von glandulären Sekretionsmustern und spezifischen HormonClearance-Raten. Die Hormonausschüttung ist streng reguliert, um die jeweils benötigten Hormonspiegel zu erreichen und ideal an Zielgewebe und Zielkonzentration anzupassen. Jeweilige Zielkonzentrationen sind unter anderem abhängig von der Stoffwechselsituation des Organismus, wobei bereits geringe Alterationen zu Hormonüber- oder -unterfunktion mit wesentlichen Konsequenzen für das Individuum führen können. Die zirkulierenden Hormonkonzentrationen sind inhomogen, wobei unterschiedliche und angepasste Sekretionsmuster die einwandfreie physiologische Funktion determinieren. Beispielsweise wird Gonadotropin durch einen hypothalamischen Impulsgeber episodisch sezerniert, die Insulinsekretion erfolgt nach Aufnahme von Glucose und anderen Signalen und die Ausschüttung von Prolaktin ist durch sekretorische Peaks (aufgrund der Stimulation der Brustdrüse durch den Säugling) gekennzeichnet.(1) Die endogene Sekretion von Hormonen unterliegt außerdem zirkadianen Rhythmen. Diese dienen zur angepassten Antwort auf Umwelteinflüsse und werden über zeitliche zirkadiane Stimuli gesteuert. Licht gilt als Hauptfaktor solcher umgebungsbedingter Einflüsse auf das endogene System. Dabei spielt der Tractus retinohypothalamicus eine zentrale Rolle: er stimuliert zirkadiane Impulsgeber in den hypothalamischen suprachiasmatischen Nuclei. Diese Signale beeinflussen maßgeblich den Schlaf-Wach-Rhythmus, sowie Hormonsekretion und –wirkung. Ein gestörter zirkadianer Rhythmus oder eine Veränderung des rhythmischen Musters resultiert in einer 5 Beeinträchtigung des Hormonhaushaltes und kann Anzeichen einer endokrinen Dysfunktion oder einer Läsion der Impulsgeber (sprich Hypothalamus oder Hypophyse) sein. Die meisten hypophysären Hormone unterliegen zirkadianen Rhythmen, häufig Tag-Nacht-Rhythmen. Beispielsweise werden ACTH-Spitzen vor neun Uhr morgens gemessen. Der Verlust der zirkadianen ACTH-Sekretion mit hohen nächtlichen Cortisol-Spiegeln ist typisch bei Mb. Cushing.(1) Ein weiterer wichtiger Faktor, der die Pulsatilität der Hormonsekretion nachweislich beeinflusst, ist Schlaf. So erfolgt beispielsweise etwa 70% der GH-Sekretion während der Tiefschlaf-Phase und die Pulsationsspitzen der pubertären LH-Sekretion korrelieren mit den REM-Schlafphasen.(1,4,5) 1.1.3.2 Die Auswirkungen von Stress auf das Endokrinum Die hypothalamo-hypophysäre-adrenotrophe (HPA) Achse (auch: „Stress-Achse“) wird über unterschiedliche Stressfaktoren stimuliert. Hierzu zählen beispielsweise körperliche Betätigung, Infektionen, Operationen, Hämorrhagien und Hypoglykämien. Die Hypophyse reagiert auf periphere und zentrale Stresssignale mittels eines lebensnotwendigen Mechanismus, indem sie die Glucocorticoid-Synthese in der Nebennierenrinde steigert. Chronischer Stress, wie beispielsweise im Fall einer Depression, aktiviert ebenso die HPA-Achse, jedoch führt eine persistierende Stimulation zu einer Beeinträchtigung des physiologischen Feedback-Mechanismus. Hierbei wirken multiple molekulare Prozesse, wie beispielsweise die Down-Regulation von zentralen Glucocorticoid-Rezeptoren, welche den physiologischen andrenotrophen Regelkreis determinieren. Somit kommt es bei chronischer Aktivierung zu einem Defizit der negativen Rückkopplung und zu einer kontinuerlichen CRH- und ACTH-Ausschüttung.(6) Es konnte in weiterer Folge gezeigt werden, dass sich die hypothalamo-hypophysäre-adrenotrophe Achse und die hypothalamo-hypophysäre-gonadale (HPG) Achse gegenseitig beeinflussen und Zusammenhänge bestehen. Einerseits modulieren Estrogene und Testosteron Signale der HPAAchse, andererseits führt die (vor allem wiederholte bzw. chronische) Aktivierung der StressAchse zu einer Hemmung der Estrogen- und Testosteron-Sekretion. Am Tiermodell konnte darüber hinaus gezeigt werden, dass die Reaktivität der HPA-Achse und damit die Sensitivität auf Stressfaktoren bereits kurz nach der Geburt durch Unterschiede in der mütterlichen Fürsorge beeinflusst werden.(7) 1.1.3.3 Rezeptorregulation Für die Regulation der Hormonsekretion auf molekularer Ebene sind drei Prinzipien von Bedeutung: 1. Die Desensibilisierung („desensitization“): Bei anhaltend hohen Hormonkonzentrationen verringert eine Zelle die von ihr exprimierte Rezeptorzahl entweder durch rezeptorvermittelte Endozytose oder durch Einschränkung der Rezeptorsynthese. Die Zelle wird somit für das entsprechende Hormon unempfindlich.(3) 6 2. Die Inaktivierung von Rezeptoren: Hier wird die Weiterleitung eines Signals blockiert. Beispielsweise erfolgt dies bei G-Protein-gekoppelten Rezeptoren durch Proteinkinase-Avermittelte Phosphorylierung.(3) 3. Das Rebound-Phänomen: Eine lang andauernde Hemmung von Rezeptoren resultiert folglich in einer Überempfindlichkeit der Zelle für das jeweilige Hormon. Wird die Hemmung der Rezeptoren abrupt beendet, können auch Hormonkonzentrationen im Normbereich zu einer übermäßigen Reaktion führen.(3) 1.1.3.4 Hormonelle Regelkreise Als endokrine Achse bezeichnet man einen hormonellen Regelkreis, dessen Funktion der Erhalt einer bestimmten Hormonkonzentration innerhalb bestimmter Grenzen ist. Ein Regelkreis auf endokriner Ebene besteht aus dem hierarchischen Aufbau von Hypothalamus, Hypophyse, peripherer Hormondrüse und Zielzellen. Die jeweils übergeordneten Hormone regulieren Produktion und Sekretion der untergeordneten Hormone.(3) Zu den typisch hierarchisch aufgebauten endokrinen Achsen zählen die Hormonsysteme der Schilddrüse, der Nebennieren und der Gonaden. Die hormonelle Kontrolle obliegt jeweils gewissen Strukturen des Zentralnervensystems: dem Hypothalamus und der Hypophyse, sowie großteils noch unbekannten übergeordneten Zentren. Im Hypothalamus befinden sich neurosekretorische Zellen, die Reize integrieren und verarbeiten. Dieser Prozess führt zur Hormonausschüttung in die Blutkapillaren der Eminentia mediana, wodurch Rezeptoren an der Hypophyse aktiviert werden und es wiederum zur Freisetzung weiterer Hormone kommt, welche in Folge hämatogen zu Schilddrüse, Nebenniere oder Gonaden transportiert werden und dort ebenfalls eine Hormonsynthese und -sekretion bewirken.(8) Diese Prozesse und ihre Auswirkungen sollen in den folgenden Kapiteln genauer erläutert werden. 7 1.2 Das hypothalamo-hypophysäre System 1.2.1 Aufbau und Zielgewebe Die Systemeinheit Hypothalamus und Hypophyse erfasst den peripheren Hormon- und Stoffwechselstatus und kann regulierend in diesen eingreifen. Die hierarchische Strukturierung des endokrinen Systems ermöglicht somit eine ideale Anpassung des Organismus an aktuelle Bedürfnisse und Stoffwechselsituationen. Die zirkuläre und lineare Darstellung der endokrinen Achse ist dennoch eine stark vereinfachte, da das hypothalamo-hypophysäre System mit vielfachen anderen hormonellen Systemen, dem Immunsystem und dem neuronalen System in Verbindung steht und durch diese beeinflusst wird. Die Komplexität dieser Prozesse wird außerdem dadurch gesteigert, dass die Freisetzung eines Hormons durch mehrere Signale reguliert werden kann. Umgekehrt kann ein physiologischer Parameter ebenso der Freisetzung mehrerer Hormone unterliegen. Ein Hormon ist außerdem imstande, an unterschiedlichen Endorganen und unterschiedlichen Zellen desselben Gewebes verschiedene Wirkungen hervorzurufen.(3,9) Abbildung 1: Schematische Darstellung der vaskulären und neuronalen Verbindungen zwischen Hypothalamus und Hypophyse.(10) Durch Neurone im Hypothalamus werden sogenannte Releasing-Hormone (Freisetzungshormone, Liberine) und Inhibiting-Hormone (Hemmhormone, Statine) gebildet, welche die Freisetzung von hypophysären Hormonen (Tropine) aktivieren oder hemmen. Die Zellkörper der Neurone liegen im Hypothalamus, die Nervenendigungen der Neurone liegen in der äußeren Schicht der Eminentia mediana und sezernieren Releasing- oder Inhibiting-Hormone in das erste Kapillargebiet des hypophysären Pfortaderkreislaufs, wodurch diese zum Hypophysenvorderlappen gelangen. Dort induzieren die hyopthalamischen Hormone über spezifische Rezeptoren eine Freisetzung der hypophysären Hormone.(4,9,11) Hypothalamische Releasing-Hormone sind: GHRH (growth hormone releasing hormone), welches die Freisetzung von GH (growth hormone, Wachstumshormon) in der Hypophyse induziert, TRH 8 (thyreotropin releasing hormone), das die Sekretion von TSH (thyreotropes Hormon, Thyreoptropin) reguliert, CRH (corticotropin releasing hormone), welches zur Ausschüttung von ACTH (Adrenocorticotropes Hormon) führt, sowie GnRH (gonadotropin releasing hormone), das die Abgabe von FH (Follikel-stimulierendes Hormon) und LH (luteinisierendes Hormon) aus der Hypophyse steuert. Zu den hypothalamischen Inhibiting-Hormonen zählen Somatostatin und Dopamin.(4) 1.2.2 Steuerung und Rückkopplungsmechanismen Hormonsynthese, -sekretion und -wirkung stehen eng miteinander in Verbindung. Zirkulierende Hormonkonzentrationen werden innerhalb endokriner Achsen von Zellen des Hypothalamus und der Hypophyse reguliert. Diese Zellen sezernieren Signalmoleküle, welche spezifische Prozesse der Synthese und Freisetzung von Hormonen in den Zielorganen aktivieren oder hemmen und können somit Zellteilung und Zellvermehrung induzieren, wobei dies wiederum in einem Wachstum des Zielorgans resultiert. So führt beispielsweise eine übermäßige TSH-Sekretion bei Vorliegen einer Hypothyreose aufgrund von Jodmangel zu einer Hyperplasie der Schilddrüse.(1) Hypothalamus: Synthese und Ausschüttung von Releasing Hormonen Hypothalamus Hypophyse: Synthese und Ausschüttung von Tropinen Adeno- Erfolgsorgan: Schilddrüse, Nebennierenrinde, Gonaden Synthese und Ausschüttung von z.B. T3,T4 (Schilddrüse) Negative Rückkopplung auf hypothalamische Zentren auf Hypophyse hypophyse periphere Hormonkonzentration Erfolgsorgan Abbildung 2: Prinzip der negativen Rückkopplung.(4) Die Zellen des Hypothalamus und der Hypophyse unterliegen verschiedenen Rückkopplungsmechanismen (sogenannten Feedback-Mechanismen). Innerhalb der endokrinen Achse werden auf allen hierarchischen Ebenen metabolische, funktionelle und hormonelle Änderungen rückgemeldet. Die hormonproduzierende Drüse kann, ebenso wie die übergeordneten Hormondrüsen, die frei zirkulierende Hormonkonzentration messen und somit die jeweilige Hormonsynthese an die gegebenen Konzentrationen anpassen. Kortex, Hypothalamus und andere übergeordnete Strukturen empfangen über humorale und neuronale Rückmeldungen außerdem noch Informationen über zu regulierende Stoffwechselfunktionen. Unter negativer Rückkopplung (bzw. negativem Feedback) versteht man den hemmenden Effekt auf die Zellen der Hypophyse und des Hypothalamus bei einem Anstieg der Konzentration eines peripheren Hormons. Die Produktion 9 der zugehörigen Releasing-Hormone und glandotropen Hormone wird eingeschränkt und führt somit wiederum zu einer Suppression des Zielorgans. Die Hormonsynthese wird vermindert und die freie Hormonkonzentration sinkt. Bei der positiven Rückkopplung (bzw. positivem Feedback) verläuft dieser Mechanismus umgekehrt- eine verringerte freie Hormonkonzentration führt zu gesteigerter Hormonproduktion auf den verschiedenen Ebenen. Dies findet sich beispielsweise bei der Regulierung des Menstruationszyklus.(3) 1.2.3 Funktionstests Um die Funktionalität der endokrinen Achse zu überprüfen, eignen sich klinische Funktionstests. Die Adenohypophyse lässt sich mittels Stimulations- beziehungsweise Suppressionstests untersuchen. Stimulationstests werden bei Verdacht auf Hormondrüsenunterfunktion eingesetzt, wobei durch die Gabe von hypothalamischen Releasing-Hormonen eine Stimulation der Adenohypophyse erreicht wird (z.B. Metopirontest zur Untersuchung der Funktionalität der hypothalamo-hypophysären-adrenotrophen Achse). Bei intakter Hypophyse führt dies zu einem Anstieg der korrelierenden hypophysären Hormonkonzentration und bei intakter peripherer Hormondrüse wiederum zu einem Anstieg der jeweiligen peripheren Hormonkonzentration. Beispielsweise wird der GnRH-Test differentialdiagnostisch bei Verdacht auf hypogonadotropen Hypogonadismus bei Frau und Mann eingesetzt. GnRH-Gabe soll die hypophysäre Sekretion von FSH und LH stimulieren. Dabei werden 100 μg GnRH i.v. appliziert und unmittelbar vor, sowie 30 und 60 Minuten nach der Applikation LH-Spiegel bestimmt. Wenn LH um den 3- bis 4-fachen Faktor ansteigt, bezeichnet man das Testergebnis als positiv.(3,4) Ebenso wird ein Entzug Rückkopplungsmechanismus des in peripheren einem Hormons Anstieg der durch den hypophysären intakten positiven Hormonkonzentration resultieren. Die Hormonsekretion aus der Hypophyse kann auch durch Veränderung gewisser Stoffwechselparameter stimuliert werden. So werden bei einer Hypoglykämie vermehrt GH, Prolaktin und ACTH sezerniert (diesen Umstand macht man sich beim Insulinhypoglykämietest zu Nutze).(3,4) Suppressionstests werden bei Verdacht auf eine Drüsenüberfunktion durchgeführt, meist mittels Gabe eines peripheren Hormons oder Analogons (z.B. Dexamethason-Hemmtest) oder eines Inhibiting-Hormons. Die Neurohypophyse bzw. die Funktionalität der ADH-Achse kann durch Urin- und Plasmaosmolaritätsbestimmungen nach Infusion von hypertoner Kochsalzlösung oder nach Dürsten geprüft werden.(3) 10 1.3 Der Hypothalamus Der Hypothalamus ist die zentrale Struktur für die Regulation von Organ- und Stoffwechselfunktionen, sowie das übergeordnete Koordinationszentrum vieler hormoneller und vegetativer Funktionen. Diese Prozesse werden neurohumeral über das neuroendokrine System, sowie nerval über Neurone und Leitungsbahnen des vegetativen Systems gesteuert. Der Hypothalamus ist Teil des Diencephalons und liegt ventral des Thalamus, von dem er durch den Sulcus hypothalamicus abgegrenzt wird. Er besteht aus Kerngruppen und bildet die seitliche und distale Grenze des III. Ventrikels. Man unterscheidet anatomisch drei große, klinisch wichtige Kerngebiete (von oral nach caudal), wobei bestimmten Kerngebieten bestimmte Funktionen zugeordnet werden können.(9,11) 1. Anteriore, rostrale Kerngruppe: Diese Kerngruppe synthetisiert Hormone, die in Folge in der Neurohypophyse freigesetzt werden und besteht aus dem Nucleus preopticus, dem Nucleus paraventricularis und dem Nucleus supraopticus. Die vordere präoptische Region steuert die Konstanthaltung der Körpertemperatur, wobei eine Schädigung dieser Struktur zu zentraler Hyperthermie führt. Bei Stimulation der paraventrikulären Kerne kommt es zu einer Aktivierung des Parasympathikus. Supraoptische und paraventrikuläre Kerne regulieren außerdem den Wasserhaushalt des Organismus. Eine Schädigung dieser Strukturen kann zu zentralem Diabetes insipidus, aber auch zu Hyponatriämie aufgrund eines fehlenden Durstempfindens führen. Die vorderen Kerne wirken außerdem regulativ auf die Nahrungsaufnahme. Eine Läsion des medialen Abschnitts führt zu Übergewicht, eine Läsion des lateralen Abschnitts zu Appetitlosigkeit und Gewichtsverlust.(11) 2. Tuberale, mittlere Kerngruppe: Hier wird die Hormonfreisetzung aus der Adenohypophyse gesteuert. Sie besteht aus dem Nucleus dorsomedialis, dem Nucleus ventromedialis und den Nuclei tuberales.(11) 3. Posteriore, mammilläre Kerngruppe (dynamogene Zone): Bei Stimulierung dieser Gruppe wird der Sympathikus aktiviert. Anteile sind der Nucleus posterior und die in den Corpora mammillaria gelegenen Nuclei mammillares. Die hintere Region steuert die Reaktion des Organismus auf Temperaturänderung, beispielsweise Schwitzen. Bei Schädigung kann es zu Hypothermie kommen.(11) Es erfolgt eine zusätzliche Unterteilung des Hypothalamus in einen medialen und lateralen Abschnitt durch die Fornixbahn. Die drei großen Kerngruppen bilden den medialen Abschnitt, der laterale Abschnitt kann nicht in definierte Kerngruppen eingeteilt werden. Aufgrund seiner Funktion als Koordinationszentrum aller vegetativen Funktionen ist der Hypothalamus mit vielen Hirnregionen afferent und efferent verbunden. Wichtige afferente Verbindungen bilden der Hippocampus, die olfaktorischen Areale, die Amygdala, sowie viszerale Afferenzen und solche aus erogenen Zonen (Genitalien, Brustwarze). Wichtige efferente Verbindungen sind der Hirnstamm und dessen parasympathische Kerngebiete, das Tegmentum des Mittelhirns (Vermittlung 11 vegetativer Informationen zwischen Hypothalamus, Hirnnervenkernen und Spinalmark), der Nucleus anterior thalami (Verbindung zum limbischen System) und die Hypophyse. Der Hypothalamus steht efferent über den Tractus supraopticohypophysialis und den Tractus tuberohypophysialis mit der Hypophyse in Verbindung.(11) Komplexe Vorgänge wie Sättigung und Hunger werden über teilweise noch unbekannte Regelkreise vor allem durch den posterioren Anteil des Hypothalamus gesteuert, unter anderem über das Hormon Leptin. Ebenfalls wurde nachgewiesen, dass dieser Anteil an der hormonellen Steuerung von Affektzuständen wie Aggression und dem Sexualverhalten beteiligt ist. Der Abschnitt ist eng mit der Hippocampusformation und dem Nucleus anterior thalami (limbisches System) verbunden.(9) Abbildung 3: Schematische Darstellung der hypothalamischen Kerngebiete.(10) Der Hypothalamus sezerniert Releasing- und Inhibiting-Hormone (hauptsächlich Peptidhormone), welche über die Portalgefäße direkt zur Hypophyse gelangen und dort die Hormonsekretion regulieren. Diese Steuerhormone werden vor allem im vorderen und medialen Hypothalamus (Ncll. infundibularis et paraventricularis) synthetisiert. Diese werden dann über Axone, die sich zum Tractus tuberoinfundibularis verbinden, über die Eminentia mediana zum Infundibulum und weiter über das hypophysäre Pfortadersystem zur Adenohypophyse geleitet.(4,9) Hypothalamische Hormone werden im klinischen Alltag hauptsächlich zu diagnostischen Zwecken genützt, therapeutische Anwendungen stehen im Hintergrund.(4) 1.3.1 GHRH (growth hormone releasing hormone) und GHRIH (Somatostatin). Die Hauptwirkung von GHRH bezieht sich auf die Induktion der Freisetzung von GH-Pulsen. Die Sekretion von GHRH wird durch Hypoglykämie, Thyroidektomie, Hypophysektomie und Stimulation der α2-Adrenozeptoren gesteigert, sowie durch Somatostatin, IGF-1 und GABA (γAminobuttersäure) gehemmt.(12) 12 Die somatotrope Funktion der Adenohypophyse lässt sich durch einen diagnostischen Test mittels GHRH überprüfen, welcher hypothalamische von hypophysären Störungen abgrenzen soll. GHRH setzt direkt an Rezeptoren der Adenohypophyse an; somit ist es möglich, eine Lokalisationsdiagnostik (Etagendiagnostik) durchzuführen. Maximalkonzentrationen von GH werden bei intakter Hypophyse und Ansprechen auf GHRH nach etwa 30 Minuten erreicht. Bei suprasellären Läsionen (z.B. bei Kraniopharyngeom) kommt es zwar zu einer normalen GHSekretion nach GHRH-Gabe, dies ist jedoch nicht der Fall, wenn hypothalamisch ansetzende Stimuli angewendet werden. Die üblicherweise verwendete Dosis von GHRH ist 50 μg i.v.(4) Bei idiopathischem hypophysärem Minderwuchs wurde häufig ein Ansprechen auf GHRH beobachtet. Diese Beobachtung lässt vermuten, dass es sich bei diesen PatientInnen um einen hypothalamisch-bedingten GH-Mangel, durch das Fehlen von GHRH, handelt. Klinische Therapien mit GHRH haben sich jedoch bis heute nur teilweise bewährt. Es konnte gezeigt werden, dass bei Kindern mit einem hypothalamisch bedingten GH-Defizit eine GHRH-Substitution durchaus sinnvoll sein kann, bei hypophysär bedingten GH-Defiziten ist eine GHRH-Substitution jedoch verständlicherweise vollkommen ineffizient.(4,12) 1.3.1.1 Somatostatin und seine pharmakologische Anwendung Somatostatin, auch GHRIH (growth hormone release-inhibiting hormone) oder SRIF (somatotropin release-inhibiting factor) genannt, ist ein Tetradekapeptid und wirkt multifunktional. Es hemmt die Sekretion von GH (vermutlich durch Inhibierung der GHRH-Wirkung an der Hypophyse), TSH, ACTH, Glucagon, Insulin, Renin, Pepsin, Sekretin, Cholezystokinin, sowie die Wirkung von Histamin und Pentagastrin auf die Synthese von Magensäure. Außerdem verringert es, vermutlich durch lokale Vasokonstriktion, die Durchblutung im Splanchnikusgebiet, was bei PatientInnen mit Cirrhosis hepatis und portalem Hypertonus zu einer Verminderung des portalen Drucks und des Blutflusses durch die V. azygos und die Leber führt.(4) Die Freisetzung von Somatostatin aus dem Hypothalamus wird induziert durch GHRH, GH und IGF-1, sowie durch GABA und Opiate gesenkt.(12) Bisher wurden fünf Subtypen von spezifischen Somatostatin-Rezeptoren (sst1-5) beschrieben. Alle Somatostatin-Rezeptoren sind G-Protein-gekoppelt. Es wurde nachgewiesen, dass viele endokrin aktive Tumoren des Gastrointestinaltrakts und der Hypophyse Somatostatinrezeptoren (besonders sst2) exprimieren. Somit konnten Somatostatin und Somatostatin-Analoga teilweise klinisch erfolgreich zur Einschränkung des Wachstums dieser Tumoren, sowie zur Sekretionshemmung eingesetzt werden. Weiters erlaubt es die Expression von Somatostatin-Rezeptoren, bei Somatostatin-Rezeptor-positiven Tumoren, diese mittels radioaktiv markierten SomatostatinAnaloga zu detektieren und darzustellen. Gezielte Strahlen- und Chemotherapiekonzepte, deren Fokus auf der Implementation von subtypspezifischen Somatostatin-Analoga liegt, welche mit Chemotherapeutika oder Radionukliden verknüpft sind und selektiv an Somatostatin-Rezeptorpositive Tumoren binden, befinden sich derzeit in der Entwicklungsphase.(4) 13 Somatostatin muss per i.v.-Dauerinfusion appliziert werden, die Halbwertszeit beträgt nur etwa drei Minuten, weshalb modifizierte Somatostatin-Analoga klinisch bevorzugt angewendet werden. Somatostatin wird therapeutisch erfolgreich bei schweren Ulkusblutungen, hämorrhagischer Gastritis und Ösophagusvarizenblutungen eingesetzt. Hierfür wird eine initiale Sättigungsdosis von 3,5 μg als einmalige i.v.-Injektion, gefolgt von einer anschließenden Dauerinfusion von 250 μg/h, verabreicht.(4,12) Hauptnebenwirkung von Somatostatin ist die initiale Hypoglykämie aufgrund der Hemmung der Glukagonsekretion. Nach zwei bis drei Stunden kommt es zu einem Anstieg des Blutzuckers bedingt durch die gleichzeitige Hemmung der Insulinsekretion. Regelmäßige Kontrollen des Blutzuckerspiegels sind daher notwendig. Bei zu rascher Infusion kann es außerdem zu Hitzegefühl und Übelkeit kommen.(4) 1.3.1.2 Somatostatin-Analoga Octreotid ist ein Somatostatin-Analogon, ein Octapeptid und besitzt eine verlängerte Halbwertszeit von etwa zwei Stunden. Es bindet selektiv und agonostitisch an sst2-Subtypen des SomatostatinRezeptors. Octreotid wird subkutan oder intramuskulär (länger wirksames mikroverkapseltes Monatsdepot) injiziert. Hauptindikation von Octreotid ist die Akromegalie. Initial werden zwei bis drei Mal täglich 0,05-0,1 mg subkutan verabreicht. Die Dosierung wird dann entsprechend der GHund IGF-1-Spiegel angepasst. Im weiteren Therapieverlauf kann auf die langwirksame mikroverkapselte Form von Octreotid umgestellt werden. Des Weiteren wird Octreotid zur symptomatischen oder neoadjuvanten Behandlung von endokrin aktiven Tumoren des Gastrointestinaltrakts und des Mediastinums angewandt, beispielsweise bei metastasierenden Karzinoiden, VIPomen und Glukagonomen. Hierfür kann Octreotid alleine oder in Kombination mit Interferon-α oder Tyrosinkinaserezeptorinhibitoren eingesetzt werden. Bei endokrin aktiven Tumoren beträgt die initiale Dosis 0,05 mg und wird ein bis zwei Mal täglich subkutan appliziert. Die Dosierung kann auf 0,1-0,2 mg drei Mal pro Tag erhöht werden.(4,13) Nebenwirkungen von Octreotid sind Übelkeit, Diarrhoe und krampfartige Bauchschmerzen. Bei längerfristiger Gabe kann es zu Cholelithiasis kommen; vermutlich aufgrund der verminderten Gallenblasenkontraktion. Sonografische Kontrollen sind indiziert. Octreotid beeinflusst die Insulinsekretion weniger als Somatostatin, somit sind Blutzuckerentgleisungen selten. Interaktionen mit anderen Arzneimitteln sind bekannt: Octreotid erhöht die Bioverfügbarkeit von Bromocriptin, vermindert die intestinale Resorption von Ciclosporin und verzögert die Aufnahme von Cimetidin.(4) Lanreotid ist ein mit Octreotid vergleichbares Somatostatin-Analogon, welches ebenfalls als Depotpräparat im Handel ist und eine ähnliche Rezeptorselektivität besitzt. Parsireotid (SOM-230), ein weiteres Somatostatin-Analogon, besitzt die höchste Affinität zum sst5-Rezeptorsubtyp. Es wird zur Therapie des Mb. Cushing eingesetzt, da ACTH-produzierende Hypophysenadenome häufig sst5-Rezeptoren exprimieren. Nebenwirkung von Parsireotid sind Hyperglykämien.(4,12) 14 Kontraindikation für die Gabe von Somatostatin oder eines Somatostatin-Analogons ist eine bestehende Schwangerschaft.(4) 1.3.2 TRH (thyreotropin releasing hormone) Das TRH-Molekül ist ein Tripeptid und stimuliert in der Adenohypophyse die Synthese und Sekretion von TSH, sowie von Prolaktin. Es fungiert als Neurohormon und Neurotransmitter und ist der zentrale Regulator der hypothalamo-hypophysären-thyreoidalen Achse (siehe Kapitel 3.1).(4,14) TRH-Neurone befinden sich im Nucleus paraventricularis des Hypothalamus. Die Sekretion von TRH wird physiologisch Schilddrüsenhormone durch gesteuert. positives Dies oder erfordert negatives ein Feedback komplexes der peripheren Zusammenspiel der hypophyseotropen TRH-Neuronen, dem vaskulären System, spezifischen Glia-Zellen (Tanyzyten), sowie dem Liquor cerebrospinalis.(14) TRH wird diagnostisch bei Verdacht auf Schilddrüsenerkrankungen genutzt. Es wird üblicherweise i.v. oder nasal verabreicht. Häufige Nebenwirkungen bei i.v. Gabe sind Schwindel, Harndrang und Nausea. Beim TRH-Test kommt es bei Vorliegen einer primären Hypothyreose durch die Verabreichung von TRH zu einem starken Anstieg von TSH. Liegt eine Hypophyseninsuffizienz vor, bleibt der TSH-Anstieg aus. Ist der TSH-Anstieg nach TRH-Gabe stark verlangsamt, spricht dies für ein Problem auf Ebene des Hypothalamus. Bei Hyperthyreose wird der TSH-Anstieg durch den negativen Rückkopplungsmechanismus inhibiert.(4) 1.3.3 CRH (corticotropin releasing hormone, Corticoliberin) CRH (corticotropin releasing hormone) ist ein wichtiger physiologischer Stimulator der ACTHSekretion und ACTH–Synthese in der Adenohypophyse. Seine Freisetzung wird durch multiple afferente Signale reguliert, beispielsweise wirken Katecholamine über β- und α1-Adrenozeptoren, Serotonin, Acetylcholin und die Zytokine Interleukin-1 (IL-1) und IL-6 stimulierend; Glucocorticoide, GABA und Katecholamine wirken über α2-Adrenozeptoren hingegen inhibierend.(6) Für den therapeutischen Einsatz von CRH gibt es derzeit keine Indikationen, jedoch wird CRH in der Diagnostik eingesetzt, beispielsweise beim CRH-Stimulationstest bei Verdacht auf CushingSyndrom. Bei Vorliegen eines hypothalamisch-hypophysär bedingten (zentralen) CushingSyndroms führt eine CRH-Gabe zu einem starken ACTH- und Cortisol-Anstieg. Bei Vorliegen eines Cortisol-produzierenden Nebennnierentumors mit unveränderter Cortisol-Sekretion, kommt es zu keinem Anstieg der ACTH-Spiegel. Bei ektoper ACTH-Produktion bleibt die CortisolSekretion konstant und die ACTH-Konzentration wird nicht bzw. nur gering erhöht.(4,6) 1.3.4 GnRH (gonadotropin releasing hormone, Gonadoliberin) GnRH ist ein Dekapeptid und das hypothalamische Releasing-Hormon der Gonadotropine FSH und LH. Es bindet an der Adenohypophyse an spezifische G-Protein-gekoppelte Rezeptoren, wobei 15 dies als Stimulus für Produktion und Sekretion der Gonadotropine wirkt, welche hauptsächlich über eine Aktivierung der Phospholipase-C und einer Steigerung der intrazellulären Calciumkonzentration induziert werden. Die Abgabe von GnRH unterliegt einer pulsatilen Sekretion, die beim Mann etwa mit einem Puls pro zwei Stunden und bei der Frau während der Follikelphase des Menstruationszyklus etwa alle 90 Minuten, während der Lutealphase allerdings verlangsamt, stattfindet. Generell variiert die Freisetzung in Abhängigkeit von Zyklusphase, Tageszeit und Lebensalter. Die Halbwertszeit von GnRH beträgt lediglich vier Minuten, da es im Plasma sehr schnell hydrolysiert wird. Seine Elimination erfolgt renal.(2,4,15) Ein Mangel an GnRH führt zu hypothalamisch bedingten Fertilisationsstörungen und sekundärer Amenorrhoe. Neuroendokrine Ursachen für Infertilität und Amenorrhoe sind jedoch vielfältig und komplex, insofern empfiehlt sich eine gezielte Therapie je nach Ätiologie: Physiologische Auslöser für Infertilität und Amenorrhoe sind eine Hyperprolaktinämie während der Schwangerschaft und Stillzeit und die funktionelle hypothalamische Amenorrhoe z.B. aufgrund von Stress. Pathophysiologische Ursachen sind eine Hyperprolaktinämie beispielsweise aufgrund eines Prolaktin-sezernierenden Tumors, primäre Hypothyreose, traumatische, neoplastische oder entzündliche Prozesse im Allgemeinen und genetische Gründe (idiopathischer hypogonadotroper Hypogonadismus, kongenitale adrenale Hyperplasie). Iatrogene Ursachen können eine GnRHSuppression durch Opioide oder Glucocorticoide, aber auch Radiatio oder Operationen der Hypophyse sein. Der idiopathische hypogonadotrope Hypogonadismus kann durch die pulsatile Applikation von GnRH alle 90 min bei der Frau und alle 120 min beim Mann mittels eines Infusionsgeräts therapiert werden. Dabei treten bei dieser Therapieform Mehrlingsschwangerschaften und Überstimulierung der Ovarien seltener auf, als bei einer Therapie mit Gonadotropinen.(4,16) Die pulsatile GnRH-Substitution wurde in den letzten Jahren vor allem in gynäkologischen und geburtshilflichen Fachbereichen intensiver erforscht. Dafür wurde ein Konzept für die subkutane pulsatile Applikation von GnRH mittels einer automatisierten Pumpe in Form eines mit GnRHLösung gefüllten Klebe-Pods entwickelt, wobei dieser alle drei Tage gewechselt werden muss. Die anfänglichen Dosierungen betragen meist 10-15 μl, alle 90 min. Korrekt gewählte Zeitintervalle der Applikation sind für den Behandlungserfolg entscheidend, wobei in klinischen Studien gezeigt werden konnte, dass zufriedenstellende Ovulations- und Schwangerschaftsraten bei Pulsintervallen von 90 bzw. 120 min erreicht werden konnten. Die Indikationen für eine pulsatile GnRHSubstitution fokussieren sich derzeit auf die primäre oder sekundäre idiopathische hypogonadotrope Ovarialinsuffizienz und die hypothalamische Amenorrhoe infolge von Essstörungen. Seltenere Indikationen sind das Kallmann-Syndrom, das Syndrom polyzystischer Ovarien (PCO), hypothalamische Funktionsverluste aufgrund intrakranieller Tumoren oder eine Induktion des ovulatorischen Zyklus bei therapieresistenter Hyperprolaktinämie.(17) 16 1.3.4.1 GnRH-Analoga und ihre pharmakologische Anwendung Therapeutisch werden neben GnRH auch GnRH-Analoga mit agonistischer oder antagonistischer Aktivität am GnRH-Rezeptor eingesetzt. GnRH-Agonisten können die Gonadotropin-Sekretion sowohl stimulieren, als auch hemmen. Bei einmaliger Gabe, sowie bei wiederholter längerfristiger und stoßweiser Gabe (Imitation der physiologischen GnRH-Sekretion) wird die Ausschüttung von FSH und LH aus der Hypophyse stimuliert und somit auch die periphere Sexualhormon-Synthese. Der eventuelle initiale Testosteron-Surge kann einerseits das Risiko eines Tumorwachstums und andererseits das Risiko für eine Zunahme der krankheitsbedingten Beschwerden erhöhen. Eine kontinuierliche GnRH-Analoga-Therapie hingegen, führt nach einer kurzzeitigen Phase der Stimulation („flare-up“-Phänomen), durch eine Desensibilisierung und Downregulierung der GnRH-Rezeptoren zu einer Reduktion der Ausschüttung von FSH und LH mit zeitgleicher reversibler Abnahme der Sexualhormon-Spiegel (sogenannte chemische Kastration). Wirkdauer und Wirksamkeit von GnRH konnten durch die Modifikation des Moleküls an den Positionen 6 und 10 erheblich gesteigert werden (z.B. besitzt Goselerin eine Halbwertszeit von 3 Stunden). Diese sogenannten langwirksamen GnRH-Analoga werden daher zur langfristigen Reduktion der Gonadotropin-und Sexualhormonspiegel eingesetzt, beispielsweise zur Androgendeprivation in der palliativen Therapie des Prostatakarzinoms. Damit der initiale Testosteronanstieg verhindert wird, verabreicht man parallel zum GnRH-Analogon ein Antiandrogen für maximal vier Wochen. Als Langzeittherapie werden Goserelin, Buserelin, Triptorelin und Leuprorelin als Depotimplantate alle drei Wochen injiziert. Histrelin wird subkutan ein Mal jährlich appliziert.(4,18,19) Bei Kryptorchismus stellt die nasale GnRH-Behandlung eine Ergänzung zur operativen Therapie dar. In den letzten Jahren hat sich herausgestellt, dass eine alleinige Hormontherapie für die Erreichung des Descensus testis aufgrund der niedrigen Erfolgsrate von nur 20% nicht mehr indiziert ist. Eine Ausnahme stellt die Behandlung des Pendelhodens („retractile testis“) dar. Sie besteht aus einer täglichen intermittierenden Applikation von 1-2 mg GnRH verteilt auf mehrere Einzeldosen für vier Wochen. Eine neoadjuvante Therapie mittels GnRH-Analoga wirkt sich darüber hinaus positiv auf das androgenabhängige Gewebe aus. Die Anwendung von HCG zur Behandlung eines Kryptorchismus ist laut aktueller Leitlinien nicht mehr indiziert.(20) Einige sexualhormonabhängige Tumoren exprimieren GnRH und GnRH-Rezeptoren, darunter Tumore der Mamma, der Prostata, des Ovars und des Endometriums. GnRH-Analoga können entsprechende Rezeptoren antagonisieren und somit die Proliferation der Tumorzellen hemmen. Weitere Indikationen für eine Therapie mit GnRH-Agonisten sind Pubertas praecox, Endometriose, Standardisierung der Ovulation bei In-vitro-Fertilisation und Uterus myomatosus.(4) GnRH-Rezeptor-Antagonisten der zweiten Generation wie Cetrorelix und Ganirelix können subkutan appliziert werden. Die Nebenwirkungsrate wurde gegenüber der ersten Generation deutlich reduziert. GnRH-Rezeptor-Antagonisten erreichen im Vergleich zu GnRH-RezeptorAgonisten eine schnellere Senkung der Gonadotropin- und Sexualhormonspiegel und verursachen 17 kein initiales „flare-up“-Phänomen. Eine Indikation für die Anwendung von GnRH-RezeptorAntagonisten stellt die kontrollierte ovarielle Stimulation bei assistierter Reproduktion bzw. Invitro-Fertilisation dar. Hier verhindern GnRH-Rezeptor-Antagonisten die vorzeitige Ovulation und die Behandlungsdauer ist gegenüber der Anwendung von GnRH-Rezeptor-Agonisten deutlich verkürzt. Beim fortgeschrittenen hormonabhängigen Prostatakarzinom sind Abarelix und Degarelix zugelassen. Diese werden ein Mal im Monat in Depotform verabreicht, wobei aufgrund von häufig auftretenden anaphylaktischen Reaktionen eine Beobachtungszeit des Patienten von 30 Minuten eingehalten werden muss. Als Nebenwirkungen werden alle Konsequenzen des Testosteronabfalls, sowie eventuelle Verlängerungen der QT-Zeit, Diarrhoe und Anämie beschrieben.(4,18) 18 1.4 Die Hypophyse (Glandula pituitaria, Hirnanhangsdrüse) Die Hirnanhangsdrüse liegt in der Fossa hypophysialis cranial des Sinus sphenoidalis in der Sella turcica und ist etwa 0,6-0,8 g schwer. Sie wird unterteilt in einen Lobus anterior (Adenohypophyse) und einen Lobus posterior (Neurohypophyse). Der Vorderlappen ist der hormonproduzierende Anteil der Hypophyse und stammt vom Epithel des Rachendachs ab. Der Hinterlappen ist der hormonsezernierende Anteil und eine Ausstülpung des Diencephalons. Über das Infundibulum (Hypophysenstiel) sind Adeno- und Neurohypophyse mit dem Hypothalamus verbunden.(9) Die Adenohypophyse nimmt etwa drei Viertel des Organs ein und lässt sich in eine Pars tuberalis, Pars distalis und Pars intermedia unterteilen. Histologisch können drei Zelltypen der Adenohypophyse differenziert werden: basophile, azidophile und chromophobe Zellen. Basophile Zellen stimulieren nachgeordnete endokrine Zellen (glandotrope Hormone), azidophile Zellen wirken durch ihre Hormone direkt an Zielzellen (nicht-glandotrope Hormone) und chromophobe, wenig anfärbbare Zellen sind Stammzellen bzw. Zellen, welche ihre Hormone bereits sezerniert haben. Die Pars intermedia der Adenohypophyse macht etwa 2% der Hypophyse aus und enthält kolloidgefüllte Zysten und Follikel, sowie tubulöse Drüsen.(9,11) Die Kommunikation zwischen Hypothalamus und Adenohypophyse erfolgt hämatogen über die Regulationshormone. Die Axone von Neuronen aus hypothalamischen Kerngebieten enden im Infundibulum an einem Gefäßnetz, das durch die Aa. hypophysiales superiores gebildet wird. Aus den Axonen werden Steuerhormone in das Gefäßsystem freigesetzt, in Venolen angereichert und über einen zweiten venösen Kreislauf (hypophysärer Portalkreislauf) zur Adenohypophyse transportiert. Etwa 80% der Hypophyse werden über diesen Kreislauf versorgt. Die restlichen 20% werden von den Ästen der A. hypophysialis inferior übernommen. Die hypothalamischen Releasing- und Inhibiting-Hormone regulieren somit an den nachgeschalteten hypophysären Effektorzellen die Sekretion von Hormonen der Adenohypophyse.(9,11) Die Hormone der Adenohypophyse regulieren die endokrine Funktion der Schilddrüse (TSH), der Nebennierenrinde (ACTH) und der Gonaden (Gonadotropine). Außerdem werden die Hormone Prolaktin und GH synthetisiert, deren Wirkungen sich nicht auf ein einzelnes Organ beschränken. Neben der hypothalamischen Kontrolle ist vor allem die negative oder positive Rückkopplung der freien Hormonkonzentration regulierender Faktor von Synthese und Sekretion der Hypophysenvorderlappenhormone.(4) Die Neurohypophyse bildet die Pars nervosa und somit den dorsalen Teil der Hypophyse und des Infundibulums. Im zur Neurohypophyse gehörenden Teil des Infundibulums finden sich Pituizyten (mit Astrozyten verwandte Gliazellen), sowie die meist marklosen Fasern des Hypothalamus. Die Kommunikation zwischen Hypothalamus und Neurohypophyse erfolgt mittels Neurosekretion (axonale Freisetzung). Hypothalamische Neurone des Nucleus paraventrikularis und des Nucleus supraopticus synthetisieren die zwei Peptidhormone Oxytocin und Antidiuretisches Hormon (ADH, Vasopressin), welche über Axone des Tractus supraopticohypophysialis in die Neurohypophyse 19 gelangen. Der Tractus supraopticohypophysialis verläuft im Hypophysenstiel. Die Neurohypophyse selbst fungiert somit nicht als Syntheseort, die Hormone werden hier lediglich gespeichert und bei Bedarf freigesetzt. Die Sekretion von Oxytocin erfolgt axonal aus den Neuronen des Nucleus paraventricularis und diejenige von ADH aus den Neuronen des Nucleus supraopticus. Die beiden Peptidhormone werden, an sogenannte Neurophysine gebunden, aus dem Perikaryon der neurosekretorischen Neuronen der jeweiligen hypothalamischen Kerngebiete in Vesikeln (Gomori-positive Herring-Körperchen) anterograd axoplasmatisch in die Neurohypophyse transportiert. Die gefäßnahe Freisetzung des Vesikelinhalts findet bei Stimulation durch Exozytose statt.(9,11) Die arterielle Blutversorgung der Hypophyse erfolgt aus den jeweils paarig angelegten A. hypophysialis superior und A. hypophysialis inferior, welche der A. carotis interna entspringen. Die A. hypophysialis superior zieht zum Infundibulum, wobei rechte und linke Arterie einen Gefäßring bilden. Die A. hypophysialis inferior versorgt die Neurohypophyse. Die Adenohypophyse wird nicht direkt arteriell versorgt, sondern indirekt über die Portalgefäße aus Infundibulum und Neurohypophyse. Dabei breiten sich die Kapillarkonvolute nach Vereinigung als zweites Kapillarnetz in Sinusoiden um die Zellen der Adenohypophyse aus. Über die Sinus durae matris erfolgt der venöse Abfluss.(9) 1.4.1 ADH (Antidiuretisches Hormon, Vasopressin) Das Antidiuretische Hormon (ADH, Vasopressin) ist ein Peptidhormon aus neun Aminosäuren, beim Menschen ist es das sogenannte 8-Arginin-Vasopressin. Es zirkuliert vorwiegend frei im Blut, wobei die Plasmaspiegel bei weniger als 1-10 pmol liegen. Die Konzentration von ADH sollte mit der Plasma-Osmolalität korreliert werden. Die Halbwertszeit beträgt etwa eine Minute. Die Inaktivierung erfolgt hauptsächlich renal und hepatisch, wobei nur 5-20% im Urin ausgeschieden werden. ADH wird hauptsächlich von magnozellulären Neuronen des Ncl. paraventricularis und des Ncl. supraopticus synthetisiert, in geringen Mengen allerdings auch von parvozellulären Neuronen.(4,21,22) ADH wirkt antidiuretisch und vasokonstriktorisch. Es reguliert die Rückresorption von Wasser in der Niere, wobei ein Signal für die Sekretion von ADH der Anstieg der Osmolarität der Körperflüssigkeiten ist (sogenannte Osmoregulation). Diese wird über Osmorezeptoren beispielsweise im Hypothalamus (Ncl. supraopticus) und in der V. portae hepatis gemessen. Eine Steigerung des osmotischen Drucks um nur 2% führt bereits zu einer Ausschüttung von ADH. Die Informationen der peripheren Rezeptoren werden vermutlich über den N. vagus zu ADHproduzierenden Neuronen mittels eines derzeit noch unbekannten Mechanismus geleitet. Kommt es durch die Stimulation der Neurone zur Auslösung eines Aktionspotentials, führt dies zur Freisetzung des gespeicherten ADH ins Blut. Eine Läsion dieses Regelkreises kann zu exzessivem Wasserverlust (Diabetes insipidus) und Hyponatriämie führen. Volums- und Barorezeptoren regulieren ebenfalls die Sekretion von ADH. Diese finden sich im Sinus caroticus, im 20 Aortenbogen, im linken Vorhof und in den Pulmonalvenen. Durch sie erfolgt bei einer Abnahme des zirkulierenden Blutvolumens (z.B. durch Reduktion des Herzzeitvolumens, Blutungen oder Hypalbuminämie, etc.) ebenfalls eine Ausschüttung von ADH. Dies geschieht ebenso bei Vorliegen einer hypotonen Dehydratation, wobei hier primär die Volumsregulation noch vor der Osmoregulation ausschlaggebend ist.(4,9,21) Stress, Trauma, Schmerz, Übelkeit, Erbrechen, Hypoglykämie sowie auditive, visuelle, psychische und olfaktorische Reize können die Sekretion von ADH und auch von Oxytocin triggern. Ebenso verschiedene Pharmaka wie Antidepressiva, Antikonvulsiva, Barbiturate und cholinerge Agonisten (Nicotin, Acteylcholin). Glucocorticoide, Chlorpromazin, Phenytoin und Alkohol wirken hingegen hemmend auf die Freisetzung von ADH.(4,21) 1.4.1.1 ADH-Rezeptoren Von den Vasopressin-Rezeptoren sind drei Subtypen bekannt: V1a, V1b und V2. Je nach Literaturquelle werden sie auch als V1-, V2- und V3-Rezeptoren bezeichnet. Alle drei sind GProtein-gekoppelte Rezeptoren und binden ADH mit ähnlicher Affinität. Die Aktivierung von V 1aund V1b-Rezeptoren steigert die Aktivität der Phospholipase-C und erhöht die intrazelluläre Calciumkonzentration. V1a-Rezeptoren findet man unter anderem an der glatten Muskulatur von Gefäßen, Darm und Uterus, an Thrombozyten und Hepatozyten. Über diesen Rezeptortyp ist ADH durch Konstriktion von Arteriolen und Kapillaren im Splanchnikusgebiet, der Skelettmuskulatur, der Haut und der Koronarien vasokonstriktorisch wirksam. V1b-Rezeptoren werden unter anderem von corticotropen Zellen der Adenohypophyse exprimiert, wobei ADH hier die Sekretion von ACTH stimuliert. Der V2-Rezeptor wird in den Sammelrohren der Nieren exprimiert (antidiuretische Wirkung von ADH), ist an eine Adenylatcyclase gekoppelt und wirkt cAMPvermittelt. In höheren Konzentrationen bewirkt ADH über diesen Rezeptor außerdem einen Anstieg der zirkulierenden Konzentrationen der Gerinnungsfaktoren vWF (von-Willebrand-Faktor) und Faktor VII bzw. Faktor VIII. In der Leber kommt es darüber hinaus durch ADH zu einer Aktivierung der Glykogenphosphorylase mit der Folge einer Freisetzung von Glucose in den Blutkreislauf.(4,21) 1.4.1.2 ADH-Analoga und ihre pharmakologische Anwendung Therapeutisch werden vorwiegend Vasopressin-Analoga eingesetzt, da sie selektiver, besser verträglich und länger wirksam als physiologisches ADH sind. Desmopressin (1-Desamino-8-DArginin-Vasopressin, DDVAP) wirkt über V2-Rezeptoren und somit stark antidiuretisch. An V1aRezeptoren agiert es als potenter Antagonist, weshalb die vasokonstriktorische Wirkung ausbleibt und somit keine wesentlichen kardiovaskulären Nebenwirkungen bestehen. Desmopressin hat eine stärkere metabolische Stabilität als Vasopressin. Die Plasmahalbwertszeit liegt bei drei Stunden und die übliche antidiuretische Wirkdauer nach i.v.-Applikation beträgt 5-20 Stunden. Es wird wegen der fast fehlenden vasokonstriktorischen Wirkung für die Therapie des primären (idiopathischen,familiären) und sekundären Diabetes insipidus centralis (als Folge von Tumoren, 21 Infektionen des hypothalamo-hypophysären Systems, Schädel-Hirn-Traumata) eingesetzt, da hier, anders als beim nephrogenen Diabetes insipidus (durch ADH-Resistenz der Tubuli und Sammelrohre), ein Mangel an Vasopressin besteht. Desmopressin kann hierfür i.v., subkutan oder intramuskulär (Dosierung: 0,5-2 μg, 2xtäglich), aber auch intranasal (Dosierung: 5-20 μg) appliziert werden. Bei primärer Enuresis nocturna vermindert Desmopressin die Häufigkeit des Einnässens um mindestens eine Nacht pro Woche während einer laufenden Behandlung (Dosierung: 10-40 μg intranasal oder 0,2-0,4 mg peroral). Für die zahnärztliche Leitungs- bzw. Infiltrationsanästhesie ist Desmopressin in fester Kombination mit Prilocain verfügbar (Xylonest® 3% DENTAL mit Octapressin®). Des Weiteren wird Desmopressin bei Gerinnungsstörungen wie der Von-Willebrand-Jürgens-Krankheit und leichter bis mittlerschwerer Hämophilie A, aufgrund seiner freisetzenden Wirkung auf die Sekretion von vWF und Faktor VIII, eingesetzt. Desmopressin steigert die vorhandene Faktor VIII-Aktivität um das 4- bis 6-fache. Voraussetzung ist jedoch eine gewisse Restaktivität des Faktor VIII von mindestens 5%. Für die Therapie ist eine vergleichsweise hohe Dosis notwendig: 0,4 μg Desmopressin pro kg Körpergewicht langsam i.v. über einen Zeitraum von 30 Minuten, gegebenenfalls alle 12-24 Stunden über maximal 7 Tage, da es bei mehrfacher Gabe in kurzen Zeitabständen zu einem Wirkungsverlust kommen kann. Die Indikation zur Anwendung besteht somit hauptsächlich bei akuten prä- und postoperativen Situationen. Bei Überdosierung und hohen Dosen von Desmopressin kann es zu Flüssigkeitsretention bis hin zur Wasserintoxikation mit Hyponatriämie, Bewusstseinsverlust und Krämpfen kommen. Kontraindikationen sind die psychogene Polydipsie und das Von-WillebrandJürgens-Syndrom vom Subtyp 2b aufgrund der erhöhten Gefahr von Thrombozytopenien.(4,15,21) Terlipressin (N-Triglycil-8-Lysin-Vasopressin) ist ein vorwiegend inaktives Hormonogen, aus welchem protrahiert Lysin-Vasopressin durch enzymatische Spaltung freigesetzt wird. Gefährliche Plasmaspiegelpeaks können im Gegensatz zur Gabe von Vasopressin vermieden werden. Es hat eine Plasmahalbwertszeit von etwa einer Stunde und eine Wirkdauer von 4-6 Stunden. Therapeutisch wird Terlipressin bei Ösophagusvarizenblutungen eingesetzt, da ADH den Druck in der Pfortader durch Vasokonstriktion im Splanchnikusgebiet senkt. Dies kann zusätzlich zur Kontraktion der Ösophagusmuskulatur mit konsekutiver Kompression der Ösophagusvarizen zu einem Blutungsstillstand beitragen. Bei akuten Blutungen steht, neben der vielerorts angestrebten Sklerosierung der Varizen, Terlipressin als medikamentöse Alternative zur Verfügung. Es ist das bisher einzige vasoaktive Pharmakon, das sich nachweislich positiv auf die Blutstillung und Senkung der Mortalität auswirkt. Es wird auch zur medikamentösen Prophylaxe einer Rezidivblutung in Kombination mit einem Nitratester (Isosorbidmononitrat) und einem nichtkardioselektiven β-Adrenozeptor-Antagonisten eingesetzt. Die Dosierung beträgt initial 1-2 mg Terlipressin i.v. und als Erhaltungsdosis 1 mg in 4-bis 6-stündigem Abstand. Unerwünschte Wirkungen sind Blutdruckanstiege und myokardiale Ischämien aufgrund der vasokonstriktorischen Effekte, weshalb eine enge Überwachung von PatientInnen mit Gefäßerkrankungen, Hypertonie, 22 Herzrhythmusstörungen und Asthma bronchiale erforderlich ist. Kontraindikationen für Terlipressin sind das Vorliegen einer Schwangerschaft und der septische Schock.(4) Felypressin (2-Phenylalanin-8-Lysin-Vasopressin) wirkt vorwiegend vasokonstriktorisch und kann daher, als Alternative zu Katecholaminen, Lokalanästhetika zugesetzt werden.(4) Eine erhöhte Ausschüttung von ADH wird als Syndrom der inadäquaten ADH-Sekretion (SIADH) bezeichnet. Diese führt zu einer Hypoosmolarität als Folge einer Hyponatriämie. Ziel der Therapie ist hier die sogenannte „Aquarese“ (gesteigerte Ausscheidung von Wasser) ohne begleitende Natriurese oder Kaliurese. Dies gelingt mit dem Einsatz selektiver V 2- bzw. kombinierter V1a- und V2-Rezeptor-Antagonisten. ADH ist ebenfalls Teil der neurohumoralen Gegenregulation bei Erkrankungen wie dem nephrotischen Syndrom, bei Herzinsuffizienz und Leberzirrhose. Derzeit erforscht wird der Einfluss von Vasopressin auf Zustände wie Hypertonie, Dysmenorrhoe, Raynaud-Syndrom, Angstzustände und Depression. Hierfür wurden Vasopressin-RezeptorAntagonisten für alle drei Rezeptor-Subtypen entwickelt: Relcovaptan (V1a), Tolvaptan (V2), Conivaptan (V1a und V2) und SSR149415 (V1b). Zur oralen Therapie der Hyponatriämie als Folge von SIADH ist Tolvaptan zugelassen, allerdings aufgrund der erforderlichen Kontrollen des Serum-Natriums und des Volumenstatus nur in stationärem Setting sinnvoll. Die Plasmakonzentrationen von Tolvaptan sind CYP3A4-abhängig. Häufige unerwünschte Wirkungen sind Hypernatriämien, Pollakisurie, Mundtrockenheit und Durst.(4,21) 1.4.2 Oxytocin Oxytocin ist ein Neuropeptid aus neun Aminosäuren und wirkt über spezifische G-Proteingekoppelte Rezeptoren, welche vorwiegend in den glatten Muskelzellen des Myometriums des Uterus nachgewiesen wurden. Es verursacht eine Zunahme der Kontraktionskraft des Uterus, sowie eine Steigerung der Kontraktionsfrequenz. Estrogene erhöhen die Sensitivität des Uterus für Oxytocin, Gestagene vermindern diese. Während der Schwangerschaft kommt es zu einer Empfindlichkeitszunahme des Uterus für Oxytocin mit einer gesteigerten Expression von Oxytocin-Rezeptoren. Im Myometrium führt Oxytocin zu einer Sekretion von Prostaglandinen, wobei noch unklar ist, ob dies ein primärer oder sekundärer Effekt (als Folge der gesteigerten Uteruskontraktion) ist. Ein Anstieg von Oxytocin verursacht somit ein Einsetzen der Geburtswehen, weshalb es zur Geburtseinleitung verwendet wird. Es wirkt des Weiteren konstriktorisch auf das Myoepithel der Brustdrüse und steuert somit den Milchfluss, wobei hierfür das Saugen an der Brustdrüse den Sekretionsreiz darstellt. Man spricht auch vom nervalhormonellen Reflexbogen der Milchejektion, da es über mechanosensible Bahnen zur Reizung hypothalamischer Kerngebiete kommt, welche dann wiederum die Sekretion von Oxytocin in der Hypophyse initiieren. Im weiblichen Genitaltrakt wird die Spermatozoenaszension durch Oxytocin begünstigt. Stimuli für die Oxytocinausschüttung sind die Dilatation der Vagina und die Stimulation der Zervix (Geburtsvorgang, Koitus). Beim Mann scheint Oxytocin bei Erektion und Ejakulation eine Rolle zu spielen.(4,9,23) 23 Das Vorkommen von Oxytocin im menschlichen Organismus beschränkt sich nicht nur auf die hypothalamischen Kerne Ncl. paraventricularis und Ncl. supraopticus, sondern konnte auch in anderen Hirnstrukturen wie dem limbischen System und den autonomen Kernen des Hirnstamms nachgewiesen werden. Oxytocin agiert somit als Neurotransmitter und Neuromodulator für psychische Funktionen und soll bei Angst- und Stressreduktion, Emotionsregulation, sozialem Bindungsverhalten und Schmerzempfinden eine Rolle spielen. Besonders für die moderne Schmerztherapie ist interessant, dass Oxytocin-Rezeptoren im Bereich von Hirnstamm und Rückenmark, sowie im Hippocampus und in den Amygdala nachgewiesen werden konnten. Die Mechanismen der positiven Wirkungen von Oxytocin sind vielfältig und derzeit größtenteils noch ungeklärt.(23) 1.4.2.1 Pharmakologische Anwendung von Oxytocin Gesicherte Indikationen für die Gabe von Oxytocin sind die Geburtseinleitung, sowie eine primäre und sekundäre Wehenschwäche, wobei die Verabreichung mittels i.v. Dauerinfusion erfolgt (0,5-20 mIE/min.). Außerdem wird Oxytocin in der Nachgeburtsperiode für die Förderung und Abstoßung der Plazenta eingesetzt (3-10 IE i.m. oder langsam i.v.). Bei postpartaler Uterusatonie werden heutzutage meist Prostaglandine oder Methylergometrin verwendet. Der Großteil der derzeitigen Studien zu Oxytocin und Schmerzempfindung zeigt eine positive schmerzreduzierende Wirkung, weshalb Oxytocin in Zukunft eventuell in die Behandlung von chronischen Schmerzen einbezogen werden wird.(4,23) Nebenwirkungen bei Überdosierung sind Uterus-, Zervix- oder Dammrupturen, sowie die uterine Tetanie, wodurch die utero-plazentare Zirkulation vermindert wird. Durch die verstärkten Uteruskontraktionen kann es zu schnellen Geburten kommen, die das Risiko von intrakraniellen Blutungen und Hypoxien für das Kind bergen. Oxytocin bewirkt in hohen Dosen eine Relaxation der Gefäßmuskulatur mit Abfall vor allem des diastolischen Blutdrucks, reflektorischer Tachykardie und gesteigertem Herzzeitvolumen. Bei längerfristiger hochdosierter Infusionstherapie folgt auf die Hypotonie eine anhaltende Hypertonie, sowie antidiuretische Effekte, die auf die ADH-ähnliche Wirkung zurückzuführen sind. Als Folge können Wasserintoxikationen mit Hyponatriämien, Krämpfen, Koma und cerebralen Ödemen auftreten.(4) Kontraindikationen für die Gabe von Oxytocin sind jegliche Komplikationen des Geburtsvorgangs, Lageanomalien, Präeklampsie, potenzielle Uterusrupturen, vorzeitige Plazentalösung, sowie potenzielle kindliche Asphyxie. Außerdem sollte es nicht zur Beschleunigung des Geburtsvorgangs verwendet werden.(4) 1.4.2.2 Oxytocin-Rezeptor-Antagonist: Atosiban Atosiban ist ein Oxytocin-Rezeptor-Antagonist und hemmt kompetitiv die Wirkung von Oxytocin am Rezeptor. Mit größerer Affinität bindet es allerdings am ADH-V1a-Rezeptor, an welchem es die Wirkung von ADH hemmt. Es wird i.v. appliziert (initiale Bolusgabe von 6,75 mg, danach Sättigungsinfusion mit 5 mg über 3 Stunden, anschließende Dauerinfusion von 100 µg/min bis zu 24 45 Stunden). Die Plasmahalbwertszeit beträgt etwa dreizehn Minuten. Atosiban kann vorzeitige Uteruskontraktionen hemmen (Tokolyse) und wird zur Verzögerung potenzieller Frühgeburten eingesetzt. Zugelassen ist es zur Tokolyse für 48 Stunden. Nebenwirkungen für die Schwangere sind Tachykardie, Hyperglykämie, Übelkeit, Erbrechen, sowie Kopfschmerz und Schwindel. Klinische Untersuchungen ergaben eine vergleichbare Wirksamkeit wie β-Sympathomimetika und Calciumantagonisten bei geringeren kardialen Nebenwirkungen für die Mutter. Es konnten allerdings keine Vorteile in Bezug auf Schwangerschaftsverlängerung und neonatales Outcome im Vergleich zu β-Sympathomimetika, Calciumantagonisten oder Placebo aufgezeigt werden.(4,24) Aufgrund der signifikant höheren Behandlungskosten im Vergleich zu äquieffektiven Pharmaka wie z.B. Calciumantagonisten wird Atosiban vornehmlich bei Hochrisikoschwangerschaften mit z.B. kardiopulmonalen Erkrankungen oder Diabetes mellitus Typ I eingesetzt.(24) 1.4.3 GH (growth hormone) und IGF-1 (Insulin-like growth factor-1) Das Wachstumshormon GH (growth hormone, Somatotropin) ist ein Proteinhormon aus 191 Aminosäuren und wirkt nicht spezifisch auf den Stoffwechsel eines einzigen Organ- bzw. Zellsystems, sondern auf beinahe alle Zellen des Organismus. Vor allem beeinflusst es Protein-, Kohlenhydrat- und Fettstoffwechsel und fördert das Wachstum. Im Körper ist GH teilweise an ein Bindungsprotein gebunden, welches den extrazellulären Teil des GH-Rezeptors enthält. Der GHRezeptor wird ubiquitär exprimiert und gehört in die Klasse der Enzym-gekoppelten Rezeptoren. GH ist bivalent (d.h. es besitzt zwei Bindungsstellen für den Rezeptor und bindet an zwei Rezeptormoleküle). Seine Bindung bewirkt eine ligandeninduzierte Dimerisierung des Rezeptors und führt so durch Transphosphorylierung zur Aktivierung der mit den Rezeptormolekülen assoziierten Tyrosinkinase Jak 2 (Januskinase 2), wodurch verschiedene Signalwege in Gang gebracht werden. Die Plasmahalbwertszeit von GH beträgt etwa 20 Minuten, die Ausscheidung erfolgt zu 25-50% renal. Die Anwendung von Referenzbereichen der basalen GH-Konzentration wird aufgrund der tageszeitlichen Pulsationen nicht zur Diagnostik empfohlen. Der Referenzbereich von IGF-1 ist von Lebensalter und Geschlecht abhängig.(4,22) 1.4.3.1 Steuerung und Wirkung von GH und IGF-1 Die Sekretion von GH regulieren die hypothalamischen Peptidhormone GHRH und Somatostatin, sowie das pleiotrope Peptidhormon Ghrelin. Die Ausschüttung von GH wird außerdem gesteigert durch Stress, Hunger, körperliche Aktivität, Dopamin, Insulin-induzierte Hypoglykämie und Aminosäureinfusionen. Gehemmt wird die GH-Sekretion durch Glucose, Glucocorticoide und freie Fettsäuren. Durch GH wird in peripheren Organen IGF-1 (Insulin-like growth factor) gebildet, welches wiederum durch negative oder positive Rückkopplung auf Adenohypophyse und Hypothalamus wirkt. Die Freisetzung von GH erreicht ihr Maximum während der Tiefschlafphase und wird außerdem vom Alter beeinflusst, wobei in peripubertären Phasen die höchste Freisetzungsrate erreicht wird und diese kontinuierlich mit dem Alter abnimmt. Bei mangelhafter oder fehlender GH-Sekretion im Kindesalter kommt es zu Kleinwuchs. Bei erhöhter GH-Sekretion 25 im Erwachsenenalter (z.B. durch ein GH-produzierendes Hypophysenadenom) kommt es zu Akromegalie.(1,4,12) GH stimuliert direkt die Lipolyse an Adipozyten (Anstieg der freien Fettsäuren im Serum) und die Gluconeogenese an Hepatozyten. Indirekt wirkt es über IGF-1, welches in vielen Geweben produziert wird, wobei der Maximalanteil der freien IGF-1-Menge in der Leber synthetisert wird. Der IGF-1-Rezeptor ist ein Typ-Ι-Rezeptor, hat eine hohe strukturelle Ähnlichkeit mit dem InsulinRezeptor und wird weitverbreitet exprimiert.(1,4) Generell steigert GH den Proteinaufbau, sowie die Lipolyse und hält Kohlenhydratspeicher konstant. Im Hungerzustand stellt GH durch die Mobilisierung der Fettspeicher Energie bereit, andererseits reduziert es den peripheren Glucoseverbrauch. Auf den Kohlenhydratstoffwechsel wirkt GH, indem es eine Abnahme der Glucoseutilisation verursacht und sich somit antagonistisch zum Insulin verhält, wovon seine diabetogene Wirkung abzuleiten ist. Durch die gehemmte Glucoseutilisation bleibt der Blutglucosespiegel erhöht und die β-Zellen des Pankreas werden permanent zur Insulinsekretion angeregt. Insulin und GH wirken synergistisch auf den Glykogenaufbau und antagonistisch auf den Fett- und Kohlenhydratstoffwechsel (Insulin fördert die Umwandlung von Kohlenhydraten zu Fetten, GH hemmt diese).(4) GH besitzt eine anabole Wirkung und fördert die Proteinsynthese, unter anderem durch eine gesteigerte intrazelluläre Aufnahme von Aminosäuren. Durch die erhöhte Retention von Stickstoff kommt es zu einer Zunahme von Kalium, Natrium, Phosphat, Chlorid und Calcium, sowie anderer Elemente. Insulin und GH wirken hier wiederum synergistisch.(4) 1.4.3.2 Pharmakologische Anwendung von GH und IGF-1 Therapeutisch wird rekombinant hergestelltes, GH erfolgreich zur Behandlung des hypophysären Zwerg- und Minderwuchs eingesetzt, wobei es vor allem zur Steigerung des Längenwachstums beiträgt. Die erforderliche therapeutische Dosis liegt bei 0,025-0,05 mg/kg Körpergewicht/Tag (subkutane Verabreichung, 1xtäglich). Ein primärer Mangel von IGF-1 ist nur selten Ursache eines Minderwuchses. Hintergründe hierfür können Mutationen im IGF-1-Gen, im GH-Rezeptor oder der nachgeschalteten Signaltransduktion, sowie GH-Antikörper sein. Für diesen Zweck hat sich das rekombinante IGF-1 Mecasermin (RhIGF-1) bewährt (subkutane Applikation, 2xtäglich). Zu den häufigsten Nebenwirkungen zählen Hypoglykämien, weshalb auf eine adäquate Nahrungsaufnahme vor und nach den Injektionen zu achten ist. Beschriebene seltene Nebenwirkungen von Langzeittherapien mit rekombinantem IGF-1 sind das Auftreten von Neoplasien, Katarakt und renalen Hypertrophien.(4,25) Eine weitere therapeutische Indikation für rekombinant hergestelltes GH ist die Substitutionstherapie von Erwachsenen mit evidentem GH-Mangel durch organisch manifeste und morphologisch nachweisbare Schädigung von Hypothalamus und/oder Hypophyse. Therapeutische Effekte sind eine Zunahme von Muskelmasse bei gleichzeitiger Reduktion des Körperfetts (vor allem des viszeralen Fetts), sowie eine mittelfristige Zunahme der Knochendichte durch 26 Knochenumbau, außerdem eine Verbesserung des HDL-/LDL-Cholesterin-Verhältnisses, der Sauerstoffaufnahme, der körperlichen Leistungsfähigkeit und der Lebensqualität der betroffenen PatientInnen. Die Substitution sollte mit niedrigen Dosen begonnen (0,15-0,3 mg täglich) und individuell durch Bestimmung der IGF-1-Konzentration angepasst werden. Häufige Nebenwirkungen bei Erwachsenen sind periphere Ödeme aufgrund von Wasserretention, Arthralgie, Parästhesie und Myalgie, wobei die Symptome spontan oder bei Reduktion der Dosis abnehmen. Die diabetogenen Effekte von GH haben nur selten relevante Konsequenzen. Kontraindikation für die Therapie mit GH sind jegliche Anzeichen einer Tumoraktivität.(4) Weitere Indikationen für eine Behandlung mit rekombinantem GH sind eine chronische Niereninsuffizienz, das Turner-Syndrom, das Prader-Willi-Syndrom, sowie intrauterine Wachstumsverzögerungen (SGA, small for gestational age).(12) Die Modeerscheinung, GH als Anti-Aging-Produkt gegen den physiologischen Alterungsprozess zur Steigerung der Leistungsfähigkeit einzusetzen, konnte durch klinische Studien nicht belegt werden. Der Einsatz ist jedenfalls aufgrund der IGF-1-vermittelten mitogenen und insulinantagonistischen Wirkung stark abzulehnen, zudem gibt es keine zugelassene medizinische Indikation. Klinische Studien zur anabolen Wirkung von GH bei älteren, unterernährten, gebrechlichen PatientInnen und IntensivpatientInnen wurden aufgrund von erhöhter Mortalität und gefährlicher Nebenwirkungen abgebrochen. Die Anwendung im Leistungssport zur Vermehrung der Muskelmasse ist ebenfalls abzulehnen.(4) Derzeit befinden sich lang-wirksame GH-Präparate, sowie alternative Applikationsformen in Entwicklung, wobei hierbei in Zukunft vor allem transdermale Anwendungsformen vielversprechend scheinen.(12,26) 1.4.3.3 GH-Antagonist: Pegvisomant Der Wachstumsrezeptor-Antagonist Pegvisomant (Somavert®) ist ein gentechnisch hergestelltes und gezielt verändertes GH-Molekül. Aufgrund von acht Mutationen, welche bewirken, dass Pegvisomant zwar an den GH-Rezeptor binden kann, die Dimerisierung und Aktivierung des Rezeptors jedoch verhindert, ist es ein klinisch sehr effektiver Antagonist. IGF-1-Plasmaspiegel sinken aufgrund der Aufhebung der GH-Wirkung um bis zu 97%, GH-Plasmaspiegel steigen jedoch, aufgrund der nicht vorhandenen negativen Rückkopplung durch IGF-1, an. Indikation für die Therapie mit Pegvisomant ist die Akromegalie bei nicht ausreichendem Therapieerfolg von Operation, Radiatio oder Somatostatin-Analoga. Pegvisomant wird subkutan ein Mal täglich appliziert, die Plasmahalbwertszeit beträgt 74-172 Stunden. Die Anfangsdosis beträgt 80 mg s.c., an den nachfolgenden Tagen werden 10 mg/Tag s.c. verabreicht. Pegvisomant kann auch mit Somatostatin-Analoga kombiniert verabreicht werden. Die Dosisanpassung erfolgt unter Kontrolle der IGF-1-Spiegel um je 5 mg nach etwa fünf Wochen, die Tageshöchstdosis beträgt 30 mg.(4,12) Bisherige klinische Studien zeigten bei PatientInnen, die mit Pegvisomant über 30 Monate therapiert wurden, bei 17% das Auftreten von niedrigtitrigen Antikörpern, wobei deren klinische 27 Relevanz noch unbekannt ist. Ein befürchtetes Wachstum des Resttumors eines Hypophysenadenoms durch erhöhte GH-Spiegel, aufgrund der ausbleibenden negativen Rückkopplung, konnte bisher nicht gezeigt werden. Weitere Untersuchungen auf diesem Gebiet sind jedoch notwendig.(4) Eine erst kürzlich veröffentlichte Studie zur Sicherheit der Langzeittherapie mit Pegvisomant von PatientInnen mit Akromegalie und unterschiedlichem Bedarf an Pegvisomant um IGF-1Konzentrationen zu normalisieren, ergab, dass bei PatientInnen mit einem erhöhtem Bedarf an Pegvisomant, ebenso höhere Basis-IGF-1-Level, sowie aggressivere Formen der Erkrankung vorliegen und die Betroffenen häufiger unter Hypertonie, Schlafapnoe, Diabetes mellitus, sowie Übergewicht leiden. Diese Ergebnisse verfügen insofern über klinische und therapeutische Relevanz, als dass sie ein besseres Verständnis der Dosis-Wirkungs-Beziehung von Pegvisomant bewirken und inadäquate Dosierungen und Dosisanpassungen, sowie unkontrollierten IGF-1Konzentrationen vorbeugen können.(27) 1.4.4 Prolaktin und Dopamin Das Peptidhormon Prolaktin (PRL) besteht aus 199 Aminosäuren. Die Synthese von Prolaktin erfolgt in den laktotropen Zellen der Adenohypophyse, seine Sekretion unterliegt der Kontrolle des Hypothalamus. Die Freisetzung wird durch das hypothalamische Inhibiting-Hormon Dopamin reguliert, welches direkt über Dopamin-D2-Rezeptoren an den laktotropen Zellen wirkt und einerseits den Calciumeinstrom in die Zelle hemmt und damit zu einer Hyperpolarisation der Zellmembran führt, andererseits blockiert Dopamin die Adenylatzyklase und bewirkt somit eine verminderte Expression des PRL-Gens. Hypothalamische dopaminerge Neurone sind im Nucleus arcuatus lokalisiert, wobei vorwiegend die tuberoinfundibulären dopaminergen (TIDA) Neurone für das negative Feedback und die Hemmung der Prolaktin-Sekretion verantwortlich sind. Prolaktin selbst fördert wiederum die hypothalamische Synthese von Dopamin (sogenannter Short-LoopMechanismus). Stimuli der Prolaktin-Freisetzung sind Saugreiz, TRH, Estrogene, Stress und Hypoglykämie. Generell sind zirkadiane Rhythmen von Bedeutung: Die Prolaktin-Konzentration unterliegt einem Tag-Nacht-Rhythmus, sie fällt im Verlauf des Tages ab, steigt während dem Schlaf an und erreicht ihr Maximum am frühen Morgen. Die Halbwertszeit im Plasma liegt bei etwa 50 min. Erhöhte Prolaktinspiegel außerhalb von Schwangerschaft und Stillzeit sind meist pathologisch, eine Ausnahme stellt die Hyperprolaktinämie aufgrund von Stress dar. Stress aktiviert nicht nur die corticotrope Achse und die Ausschüttung von Cortisol aus der Nebennierenrinde, sondern auch die Freisetzung von Prolaktin, wobei Prolaktin wiederum inhibierend auf die corticotrope Achse wirkt. Als Grund dafür wird eine zu vermeidende Überexposition des Fetus gegenüber Cortisol vermutet. Prolaktin ist prä- und postpartal gemeinsam mit Estrogenen und Progesteron beteiligt an der Laktopoese und der Mammogenese. Beim Mann konnte bisher keine physiologische Wirkung nachgewiesen werden.(4,22,28) 28 Prolaktinspiegel steigen während der Schwangerschaft stark an, wobei sie zum Zeitpunkt der Geburt ihren Höhepunkt erreichen und in Folge abfallen, jedoch bleiben sie über die gesamte Stillzeit erhöht. Durch die zunehmende Unempfindlichkeit der TIDA-Neurone gegenüber Prolaktin, durch den Aktivitätsverlust des Short-Loop-Mechanismus und damit Ausfall der Hemmung der PRL-Ausschüttung, sowie durch die Hochregulierung des PRL-Gens aufgrund der hohen Estrogen-Spiegel, kommt es während der Schwangerschaft zu einer Zunahme der Anzahl und Aktivität der laktotropen Zellen. Außerdem inhibieren Estrogene durch Bindung spezifischer Rezeptoren zusätzlich die Freisetzung von Dopamin aus den TIDA-Neuronen.(4,28) Bei Therapien mit Dopamin-Rezeptor-Antagonisten (D2-Antagonisten, Neuroleptika) und bei Vorliegen eines Prolaktinoms (prolaktinsezernierender Tumor der Adenohypophyse) kann es zu Störungen der Prolaktinfreisetzung und zu Hyperprolaktinämien kommen. Erhöhte Prolaktinspiegeln führen zu einem Abfall der Gonadotropinsekretion und damit zu Infertilität oder sekundärer Amenorrhoe (physiologischer Mechanismus der sogenannten Laktationsamenorrhoe während der Stillzeit). Mäßige Hyperprolaktinämien (bis etwa 60 ng/ml) sind häufig Ursache von Störungen der Schilddrüsen- oder Nierenfunktion, Stress oder funktionellen Störungen des Hypophysen-Stiels bei Beeinträchtigung der dopaminergen Suppression.(4,15) 1.4.5 TSH (Thyreotropin) TSH (thyroidea stimulating hormone, Thyreotropin) ist ein Glykoprotein aus einer α- und einer βEinheit und reguliert die Schilddrüsenfunktion. Seine Synthese, Sekretion und Aktivität wird durch das hypothalamische Releasing-Hormon TRH gesteuert. Über die Bindung an den Typ-1-TRHRezeptor stimuliert TRH primär die Freisetzung des prä-synthetisierten TSH, sowie in zweiter Linie die Synthese der beiden Glykoprotein-Untereinheiten.(14) TSH bindet an einen G-Protein-gekoppelten Rezeptor und bewirkt so die Iodaufnahme in die Schilddrüse, sowie die Synthese und Freisetzung von Trijodthyronin (T 3) und Thyroxin (T4). Die freien Schilddrüsenhormone hemmen wiederum durch negative Rückkopplung die TRH- und TSHFreisetzung aus der Adenohypophyse (siehe Kapitel 3.1).(4,14) Die Normwerte von TSH bei Erwachsenen liegen je nach Literatur zwischen 0,4 – 4,5 mU/L. TSHKonzentrationen unterliegen zirkadianen Rhythmen und erreichen ihr Maximum am frühen Morgen, die geringsten Werte werden am späten Nachmittag gemessen. Die Plasmahalbwertszeit beträgt etwa eine Stunde. Serum-TSH-Konzentrationen spiegeln indirekt die aktuelle Hormonausschüttung aus der Schilddrüse wider und sind folglich ein wichtiger Parameter zur Einschätzung der Schilddrüsenfunktion. Bei verstärkter und lang andauernder Stimulation der Schilddrüse durch TSH kommt es zu Hypertrophie der Thyreozyten und vermehrter Vaskularisierung durch den TSH-vermittelten wachstumsfördernden Effekt. Der Einfluss von TSH auf die Hyperplasie der Thyreozyten ist allerdings umstritten. Bei einer Schilddrüsenüberfunktion durch Schilddrüsenadenome kommt es durch somatische Mutationen zu einer konstitutiven TSHRezeptor-Aktivierung.(4,29,30) 29 In der Diagnostik von Schilddrüsenkarzinomrezidiven bei PatientInnen, die nach einer Thyreoidektomie eine Substitutionstherapie mit Schilddrüsenhormon erhalten, verwendet man ein gentechnisch hergestelltes TSH (Thyrotropin alpha). In Kombination mit einem SerumThyreoglobulin-Test wird Thyrotropin alpha zur Stimulation der Iodaufnahme bei der RadioiodidGanzkörperszintigraphie angewandt. So können Gewebsreste und Tumore dargestellt werden. TSH kann prätherapeutisch auch in Kombination mit Radioiodid zur Ablation von Restgewebe bei vortherapierten PatientInnen mit Schilddrüsenkarzinom, im gut differenzierten Stadium und ohne Fernmetastasen, eingesetzt werden.(4) 1.4.6 ACTH (Adrenocorticotropes Hormon, Corticotropin) ACTH ensteht aus dem Vorläufermolekül Proopiomelanocortin (POMC), aus welchem unter anderem auch β-Endorphin, α-, β- und γ-MSH (Melanophoren-stimulierendes Hormon), β-LPH (Lipotropin) und Metenkephalin synthetisiert werden. ACTH besteht aus 39 Aminosäuren, wobei die Aminosäuresequenz von α-MSH ident mit einer Teilsequenz des ACTH ist. Liegt eine Nebennierenrindeninsuffizienz (Mb. Addison) vor, werden ACTH und MSH gegenregulatorisch vermehrt freigesetzt, wodurch die braune Hautpigmentierung der betroffenen PatientInnen erklärt werden kann.(4,6) ACTH bindet an einen spezifischen G-Protein-gekoppelten Rezeptor, dessen Aktivierung zur Freisetzung von cAMP führt und stimuliert die Produktion und Freisetzung von Hormonen aus der Nebennierenrinde, vorwiegend von Glucocorticoiden, ebenso von Mineralcorticoiden und Androgenen. Das hypothalamische Releasing-Hormon CRH stimuliert die Freisetzung von ACTH aus der Adenohypophyse. Die Hemmung erfolgt mittels negativer Rückkopplung durch Glucocorticoide. Pro Tag werden etwa 20 mg ACTH freigesetzt. Die ACTH-Konzentration im Plasma beträgt etwa 0,9-11,3 pmol/l, diejenigen seiner direkten Vorläufer (POMC und Pro-ACTH) sind ebenfalls im Plasma nachweisbar und etwa 5-fach höher (POMC: 5-33 pmol/l). Bei langfristig erhöhten ACTH-Plasmaspiegeln kommt es zu einer Hypertrophie und Hyperplasie der inneren Zone der Nebennierenrinde. ACTH-Spiegel werden zirkadian reguliert, Maximalwerte werden am Morgen, Minimalwerte am Abend gemessen. Stress ist ein wichtiger beeinflussender Faktor der ACTH-Sekretion; innerhalb von wenigen Minuten kann es zu einer vermehrten Freisetzung von ACTH und Glucocorticoiden kommen (siehe Kapitel 1.1.3.2). Stimulationsfaktoren sind Infektionen, Operationen, Geburt, Kälte, Traumata, psychischer Stress und körperliche Arbeit.(4,6,22) 1.4.6.1 Pharmakologische Anwendung von ACTH In der Diagnostik wird ACTH zur Überprüfung der Funktion der Nebennierenrinde eingesetzt, wobei hier der Anstieg der Serum-Cortisolspiegel ausschlaggebend ist. Der Kurzzeit-TetracosactidTest (0,25 mg Tetracosactid i.m. oder i.v., Blutprobennahme unmittelbar vor und 30 min nach Injektion) spiegelt die Nebennierenrindenfunktion wieder und wird als normal angesehen, wenn der Plasma-Cortisolspiegel 500 nmol/L nach 30 min übersteigt. Tetracosactid (β1-24-Corticotropin, 30 Synacthen®) ist aus den ersten 24 Aminosäuren von ACTH aufgebaut und stimuliert ebenso die adrenokortikale Synthese von Corticosteroiden. Tetracosactid wird darüber hinaus in der Therapie der generalisierten malignen Epilepsie (West-Syndrom) als Depotform i.m. oder i.v. eingesetzt.(4,22) In der Behandlung der sekundären Nebennierenrindeninsuffizienz hat die Substitution mit ACTH gegenüber jener mit Glucocorticoiden keine Vorteile. Unerwünschte Nebenwirkungen sind bei einer Langzeittherapie mit ACTH dieselben wie bei der Therapie mit Glucocorticoiden. Zusätzlich kommt es jedoch durch die gesteigerte Freisetzung von Mineralcorticoiden zu einer stärkeren hypokaliämischen Alkalose und zu einer vermehrten Wasser- und Natriumretention. Außerdem kann es bei Frauen durch die vermehrte Sekretion von Androgenen zum Auftreten von Hirsutismus und Akne kommen.(4) 1.4.7 Gonadotropine Die Gonadotropine FSH (Follikel-stimulierendes Hormon, Follitropin) und LH (Luteinisierendes Hormon, Lutropin) sind Glykoproteine. Sie werden von der Adenohypophyse synthetisiert und stimulieren primär die Produktion der Sexualhormone in den Gonaden. Die freigesetzten Sexualhormone, sowie die ovariell sezernierten Substanzen Aktivin und Inhibin, beeinflussen wiederum die Sekretion von GnRH aus dem Hypothalamus und der Gonadotropine aus der Hypophyse über negative und positive Rückkopplung.(4,31) Zu den Gonadotropinen werden neben FSH und LH auch HMG (human menopausal gonadotropine), sowie die plazentaren Glykoprotein- und Peptidormone HCG (human chorionic gonadotropine) und HPL (human placental lactogen) oder HCS (human chorionic somatotropin) gezählt. Gonadotropine bestehen aus zwei Untereinheiten (α und β), wobei die α-Einheit (92 Aminosäuren) von FSH, LH, HCG, sowie von TSH ident sind, die β-Untereinheiten sind hormonspezifisch. Sie wirken über G-Protein-gekoppelte Rezeptoren, welche über einen großen extrazellulären N-Terminus verfügen. FSH, LH und HCG werden in aktiver Form renal ausgeschieden, wodurch eine Bestimmung im Harn möglich ist. Die Plasmahalbwertszeit von FSH beträgt etwa 24 Stunden, für LH etwa 12 Stunden, jene von HCG etwa 30 Stunden.(4) FSH reguliert die Gonadenfunktion: bei der Frau die Follikelreifung, beim Mann die Spermatogenese. Klinisch wird FSH bei weiblicher Infertilität zur Stimulation der Follikelbildung bei Follikelreifungsstörungen verwendet, alternativ kann auch HMG eingesetzt werden. HMG ist ein Gemisch aus FSH und LH und wurde vermehrt im Urin von menopausalen Frauen nachgewiesen. Bei weiblicher Sterilität und Anovulation werden FSH und HMG individuell dosiert und die Follikelreifung in den Ovarien engmaschig mittels Laborkontrollen und Sonografie überwacht. Die Ovulation wird mittels HCG ausgelöst. Generell wird mit einer täglichen Dosis von 75-150 IE FSH oder 75-150 IE HMG begonnen. Für die Auslösung der Ovulation werden nach ein bis zwei Tagen nach der letzten Gabe von FSH oder HMG 10.000 IE HCG injiziert. Eine Komplikation der Ovulations-stimulierenden Pharmaka stellt das ovarielle 31 Hyperstimulationssyndrom (OHSS) dar. Hierbei kann es durch die Überstimulierung bis zur Ruptur der Ovarien und somit zu lebensbedrohlichen Zuständen kommen. Die Anwendung von GnRH zur Induktion des Eisprungs soll seltener OHSS auslösen, jedoch erhöhen Gonadotropine die Wahrscheinlichkeit von Mehrlingsschwangerschaften. Kontraindikationen sind sexualhormonabhängige Tumoren, sowie Zysten oder Vergrößerungen der Ovarien, welche nicht im Rahmen eines PCO (polyzystisches Ovarialsyndrom) auftreten.(4,15) Das Sekretionsmuster von LH ändert sich im Verlauf der Pubertät. Im Kindesalter liegen LH-Werte bei Mädchen um etwa 2-4 mIE/ml ohne tageszeitliche Peaks. In der Pubertät erhöht sich vor allem in den Schlafphasen die pulsatile GnRH-Sekretion und somit auch LH-Sekretion mit Werten zwischen 8-12 mIE/ml, wobei die Pulsationsspitzen mit den REM-Phasen korrelieren. Tagsüber liegen die LH-Werte bei etwa 4 mIE/ml. Bei der erwachsenen Frau schwankt die LH-Sekretion je nach Zyklusphase zwischen 5-12 mIE/ml. Beim Mann bewirkt LH eine Stimulation der Testosteronbiosynthese durch eine vermehrte Umwandlung von Cholesterin in Pregnenolon, sowie die Synthese von Testosteron aus DHEA.(5,15) HCG ist das luteotrope Hormon während der Gravidität und besitzt vorwiegend LH-Aktivität. Es wird zur Ovulationsauslösung, sowie zur Differenzialdiagnostik von männlicher und weiblicher Sterilität (HCG-Test) eingesetzt. Seine Anwendung in der Therapie des Kryptorchismus wird nicht mehr empfohlen. Der HCG-Test wird in Fällen von männlichem Hypogonadismus zur Abklärung der Gonadenfunktion, insbesondere zur Differenzialdiagnostik der Anorchie und des Maldescensus testis, verwendet. Bei intakter Funktion kommt es beim Mann nach der Gabe von HCG (5000 IE i.m.) zu einem Anstieg von Testosteron. Bei männlicher Sterilität und Hypophysenunterfunktion (hypogonadotroper Hypogonadismus) können Gonadotropine therapeutisch angewandt werden; es erfolgt die Kombination von FSH (150 IE, drei Mal pro Woche) und HCG (1500 bis 6000 IE pro Woche) für mindestens vier Monate. Die Behandlungsdauer bis zur Induktion der Spermatogonese kann 18 Monate und länger betragen. Kontrolliert werden Ejakulat und Testosteronkonzentrationen.(4,20) HPL (oder HCS) besitzt unter anderem GH-ähnliche Aktivität und spielt vermutlich in der Mammogenese während der Schwangerschaft eine Rolle. Klinisch wird es nur in Ausnahmefällen eingesetzt.(4) 1.4.7.1 Pharmakologische Anwendung der Gonadotropine Für In-vitro-Fertilisationen ist ein Heranreifen mehrerer Follikel für die extrakorporale und assistierte Befruchtung essentiell. Hier werden FSH und HMG in hohen Dosierungen eingesetzt. Zusätzlich erfolgt meist eine Behandlung mit GnRH-Analoga (GnRH-Rezeptor-Antagonisten oder desensitivierende GnRH-Rezeptor-Agonisten), um eine frühzeitige Ovulation durch endogene LHFreisetzung zu verhindern. Nach Auslösung des Eisprungs mittels HCG werden die Oozyten entnommen. Nach erfolgreicher in-vitro Befruchtung erfolgt die Implantation von zwei bis drei Embryonen in den Uterus der Patientin.(4) 32 Die konservative Behandlung eines Kryptorchismus mittels HCG wird laut aktueller Leitlinien nicht mehr empfohlen.(20) FSH, LH und HCG werden gentechnisch hergestellt, um pharmakokinetische und pharmakodynamische Eigenschaften zu verbessern. Ein Beispiel ist Corifollitropin alpha, an welches an die β- Untereinheit von FSH das C-terminale Peptid der β-Untereinheit von HCG angefügt wurde und so eine Verlängerung der Plasmahalbwertszeit auf 69 Stunden erreicht werden konnte. Durch einmalige subkutane Gabe in der ersten Woche einer ovariellen Stimulation konnten vergleichbare Erfolge beobachtet werden wie mit der täglichen Gabe von FSH-Präparaten. Ovarielle Überstimulationen wurden allerdings häufiger berichtet. Kontraindikation für Corifollitropin alpha ist eine eingeschränkte Nierenfunktion.(4,32) 33 2 Material und Methoden Diese Diplomarbeit ist eine Literaturrecherche, die grundlegendes sowie aktuelles Fachwissen zum Thema der hormonellen Steuerung des menschlichen Organismus und hypothalamo-hypophysärer Regelkreise aus pharmakologischer Sicht und Relevanz erläutern, zusammenfassen und veranschaulichen soll. Ein besonderer Fokus liegt auf der derzeitigen pharmakologischen Anwendung der unterschiedlichen Hormone und Hormon-Analoga. Das Material für diese Arbeit wurde im Zeitraum von August bis Dezember 2016 gesammelt. Aktuelle Forschungsergebnisse der letzten 10 Jahre wurden, soweit möglich und verfügbar, ausgewertet, analysiert und in die Arbeit einbezogen. Dabei wurde vor allem auf den Fach- und Lehrbuchbestand der Bibliothek der Medizinischen Universität Graz zurückgegriffen. Die wissenschaftlichen Datenbanken PubMed, Cochrane Collaboration, UpToDate und GoogleScholar, sowie die elektronische Plattform des Springer Verlags (Springer e-med) wurden zur Recherche neuerer Publikationen, wissenschaftlicher Artikel, Reviews und aktueller Studien verwendet. Diverse Fachjournale wie das European Journal of Endocrinology, das European Journal of Pharmacology und das Journal of Clinical Endocrinology and Metabolism fanden ebenfalls Einzug. Limitationen in der Recherche bestehen dahingehend, dass diese Diplomarbeit einen Überblick über aktuelles Fachwissen darlegen soll und detaillierte Ausführungen den Rahmen dieser Arbeit, auch aufgrund der Masse des zu diesem Thema bereits veröffentlichten Materials, überschreiten würden. Dennoch wurde versucht, auf die wesentlichsten Punkte einzugehen und diese fokussiert herauszuarbeiten. 34 3 Ergebnisse 3.1 Die hypothalamo-hypophysäre-thyreoidale Achse Primäre Funktion der hypothalamo-hypophysären-thyreoidalen Achse ist die Aufrechterhaltung zirkulierender, adaptierter Spiegel von Schilddrüsenhormonen, deren Konzentrationen essentiell für die biologische Funktion vieler Gewebe des menschlichen Organismus sind, wie beispielsweise der physiologischen Regulation der kardiovaskulären Funktion, der Leber- und Knochenfunktion, des Energiestoffwechsels, aber ebenso der Entwicklung cerebraler Strukturen.(14) Der zentrale Regulationsmechanismus der hypothalamo-hypophysären-thyreoidalen Achse ist unter physiologischen Bedingungen das im Hypothalamus gebildete Releasing-Hormon TRH, welches im Hypophysenvorderlappen die Synthese, Sekretion und biologische Aktivität von TSH reguliert. TSH bewirkt wiederum innerhalb von Minuten die Freisetzung der Schilddrüsenhormone Trijodthyronin (Liothyronin, T3,) und Thyroxin (Levothyroxin, Tetraiodthyronin, T4), außerdem führt es zu einer gesteigerten Hormonsynthese und Iod-Aufnahme. Durch negative Rückkopplung hemmen die Schilddrüsenhormone wiederum die TRH- und TSH-Freisetzung (siehe Abbildung 5), vor allem durch eine Suppression der TRH-Genexpression in hypophyseotropen Neuronen des Hypothalamus. Diese Regulation der Transkription des TRH-Gens scheint relativ schnell zu erfolgen. Es konnte nachgewiesen werden, dass die exogene Zufuhr von Schilddrüsenhormonen die Gentranskription im Nucleus paraventricularis innerhalb von fünf Stunden senkt.(4,14) Hypothalamus Hypothalamus TRH Adenohypophyse Hypothalamus TRH Adenohypophyse TSH TRH Adenohypophyse TSH TSH Schilddrüse: T3,T4 Schilddrüse: T3,T4 Schilddrüse: T3,T4 T A T B T C Abbildung 4: Hypothalamo-hypophysäre-thyreoidale Achse.(4) A. Unter physiologischen Bedingungen wird durch negative Rückkopplung das Gleichgewicht zwischen TRH, TSH und der peripheren Hormonkonzentration geregelt. TRH- und TSH-Synthese werden durch T3 und T4 gehemmt. B. Hypothyreose (z.B. durch Autoimmunthyreoiditis, Radioiodtherapie, Thyreoidektomie). Bei Abfall der peripheren Konzentration der Schilddrüsenhormone wird die Sekretion von TRH aus dem Hypothalamus gesteigert und folglich ebenso die TSH-Sekretion aus der Adenohypophyse. Eine konstante TSH-Stimulation der Schilddrüse führt zu Hypertrophie. C. Hyperthyreose. Eine vermehrte Synthese von Schilddrüsenhormonen führt zu einer verminderten Produktion von TRH und TSH. 35 Die Funktion der Schilddrüse wird neben der Sekretion von TRH und TSH ebenso vom autonomen Nervensystem beeinflusst, da die Schilddrüse sowohl von adrenergen Nervenfasern des Sympathikus, als auch von cholinergen Axonen des N. vagus innerviert wird. Genauere Vorgänge und Auswirkungen des vegetativen Nervensystems auf die Schilddrüse sind allerdings noch unzureichend erforscht, jedoch soll der Einfluss des Sympathikus einen hemmenden Effekt verursachen, da hierdurch der Blutfluss durch die Schilddrüse verringert wird. Weiters hemmt Noradrenalin in vitro ebenfalls die Wirkung von TSH an den Zellen der Schilddrüse und in vivo konnte eine verminderte Sekretion der Schilddrüsenhormone festgestellt werden. Umgekehrt führt eine Stimulation der parasympathischen Nervenfasern zu einer Zunahme des Blutflusses in der Schilddrüse.(14) 3.1.1 Trijodthyronin und Thyroxin Schilddrüsenhormone sind Derivate der Aminosäure L-Tyrosin. Täglich werden etwa 90 µg T4 und 8 µg T3 von der Schilddrüse sezerniert. Die Serum-Konzentration von T4 liegt bei 6-8 µg/100 ml (75-100 nmol/L) und von T3 bei 100-150 ng/100 ml (1,5-2,3 nmol/L). Nur ein geringer Anteil des Serum-T3 stammt aus der Schilddrüse selbst, etwa 80% des zirkulierenden T 3 entsteht in peripheren Geweben durch Konversion aus T4. Thyreoglobulin (ein Glykoprotein aus 134 Tyrosin-Resten und die Speicherform von T3 und T4) wird ebenfalls von der Schilddrüse freigesetzt und wird unter anderem nach radikaler Thyroidektomie für den eventuellen Nachweis von Metastasen bei Schilddrüsenkarzinomen bestimmt.(4,29) Die Produktion, Speicherung und Sekretion der Schilddrüsenhormone ist komplex gesteuert, da diese Prozesse von der Versorgung des Körpers mit Iodid, sowie der bereits erwähnten TSHFreisetzung abhängen. Aus dem Blut wird Iodid in der Schilddrüse aktiv über den Natrium-IodSymporter in die Follikelepithelzellen transportiert und dort gespeichert. Iodid wird mit Wasserstoffperoxid durch eine membranständige Peroxidase oxidiert und in Tyrosinreste des Thyreoglobulins eingebaut. Hierbei entstehen MIT- (3-Monoiodtyrosin) und DIT- (3,5Diiodtyrosin) -Reste, welche durch die Peroxidase zu T3 und T4 verbunden werden. Thyreoglobulin wird im Kolloid der Schilddrüsenfollikel gespeichert und gelangt durch TSH-Stimulation mittels Pinozytose in die Schilddrüsenzelle. Mittels hydrolytischer Spaltung in Phagolysosomen werden T3 und T4 in die Blutbahn freigesetzt. Durch die Iodtyrosin-Deiodase werden sezernierte MIT und DIT deiodiert. Das frei werdende Iodid gelangt teilweise in die Blutbahn, teilweise wird es wieder in Thyreoglobulin eingebaut. Im Blut werden T3 und T4 hauptsächlich gebunden an TBG (Thyroxinbindendes Globulin), TBPA (Thyroxin-bindendes Präalbumin) und Albumin transportiert, nur etwa 0,5‰ von T4 und 5‰ von T3 liegen ungebunden vor. Einige Substanzen beeinflussen die Bindung der Schilddrüsenhormone an die Transportproteine: in Konkurrenz um die Bindung an TBG stehen Heparin, Salicylate, Sulfonylharnstoffe, Phenytoin, Phenylbutazon und Diazepam. Hingegen 36 erhöhen Estrogene, Heroin, Methadon, 5-Fluorouracil und Clofibrat die Bindungskapazität von TBG.(4) 3.1.1.1 Wirkungsspektrum der Schilddrüsenhormone Schilddrüsenhormone haben eine breite Wirkung auf den Organismus. Physiologisches Wachstum, Reifung, sowie der Metabolismus von Proteinen, Kohlenhydraten, Lipiden, Nukleinsäuren, Vitaminen und Ionen unterliegen ihrem Einfluss. Schilddrüsenhormone führen zur Erhöhung des Grundumsatzes- sie steigern die Wärmeproduktion und den Sauerstoffverbrauch, beispielsweise aktiviert T3 die Produktion der Na+-K+-ATPase unter erhöhtem O2-Verbrauch. In höheren Konzentrationen führen T3 und T4 zu einer Abnahme des Glykogengehalts von Leber und Muskulatur, zu einer verminderten Glucosetoleranz und Insulinresistenz, sowie zu einem Anstieg der freien Fettsäuren und Abfall der Triglyceride im Blut. In niedrigen Konzentrationen stimulieren sie die Proteinsynthese, in hohen Konzentrationen wird diese gehemmt und die Proteolyse gesteigert. Weiters fördern sie den Cholesterinabbau zu Gallensäuren. Der Calcium- und PhosphatStoffwechsel wird aktiviert, Osteoklasten und Osteoblasten angeregt und es kommt zu vermehrter Ausscheidung von Calcium. Außerdem beeinflussen Schilddrüsenhormone die physiologische Entwicklung und Funktion des zentralen Nervensystems. Bei ausgeprägtem Mangel kommt es zum Kretinismus (körperlicher und geistiger Entwicklungsstillstand, irreversible mentale Retardierung). Schilddrüsenhormone steigern die Kontraktilität der Herzmuskelfasern und somit Herzfrequenz und Schlagvolumen, sowie den Sauerstoffverbrauch des Myokards. Durch das erhöhte Herzzeitvolumen und den verminderten totalen peripheren Widerstand kommt es zum Anstieg der Blutdruckamplitude. Aufgrund der erhöhten Erregbarkeit des Reizleitungssystems haben PatientInnen mit Hyperthyreose ein erhöhtes Risiko für Vorhofflimmerarrhythmien und Extrasystolen. Es liegt meist ebenfalls eine Katecholamin-Sensitivität vor, unter anderem als Folge der vermehrten Synthese von β-Adrenorezeptoren.(4) Die meisten Vorgänge werden durch die Bindung (überwiegend von T 3) an intrazelluläre Schilddrüsenhormon-Rezeptoren als ligandengesteuerte Transkriptionsfaktoren vermittelt, der Wirkungseintritt ist folglich verzögert. Für Schilddrüsenhormone wurden bereits aber ebenso schnelle, nicht-transkriptions- bzw. -translations-abhängige Wirkungen beschrieben, wobei diese sich auf die Plasmamembran, das Zytoskelett und die Mitochondrien beziehen.(4) 3.1.2 Hyperthyreose und ihre pharmakologische Therapie Die pathologische Überfunktion der Schilddrüse ist gekennzeichnet durch eine überschießende Sekretion von Schilddrüsenhormonen. Man kann zwischen Bildungshyperthyreosen (funktionelle Autonomie oder Mb. Basedow) und Freisetzungshyperthyreosen (Initialstadium der HashimotoThyreoiditis) unterscheiden. Funktionelle Schilddrüsenautonomien können differenziert werden in unifokale, multifokale oder disseminierte Autonomien. Der Mb. Basedow und die HashimotoThyreoiditis stellen Immunthyreopathien dar. Entzündlichen Ursachen einer Hyperthyreose sind die subakute Thyreoiditis de Quervain und die Strahlenthyreoiditis. Eine Schilddrüsenüberfunktion 37 kann auch im Zusammenhang mit Iodexzess bzw. vermehrter exogoner Hormonzufuhr (Hyperthyreose factitia) entstehen. Auf hypophysärer Ebene führt ein TSH-Überschuss zu einer gesteigerten Freisetzung der Schilddrüsenhormone, ebenso wie TSH-sezernierende Neoplasien.(29) Generell kann eine Hyperthyreose medikamentös, chirurgisch oder durch Radioiodtherapie behandelt werden. Diese Arbeit beschränkt sich auf die pharmakologische Therapie der Schilddrüsenüberfunktion. 3.1.2.1 Thyreostatika: Dosierungen und Nebenwirkungen Die medikamentöse Therapie der Hyperthyreose kann durch drei Medikamentengruppen mit thyreostatischer Wirkung erfolgen. Durch antithyreoidale Pharmaka wie Thioamide (Carbimazol oder Thiamazol) und Propylthiouracil (PTU) wird dosisabhängig die Iodination von Tyrosin (katalysiert durch die Peroxidase) und folglich der Einbau von Iod in Thyreoglobulin gehemmt und so eine Blockade der Hormonsynthese initiiert. Carbimazol ist ein Vorläufermolekül und wird in der Leber zu Thiamazol umgewandelt. Thiamazol wird frei, vollständig resorbiert und in der Schilddrüse, den Nieren und der Leber gespeichert. Die Wirkdauer beträgt etwa 24 Stunden, somit ist eine einmalige Applikation pro Tag ausreichend. PTU verfügt über eine kürzere Halbwertszeit von 12-24 Stunden und wird demnach zwei Mal täglich verabreicht. Die initiale Dosis des Thyreostatikums ist abhängig von der Schwere des Krankheitsbildes und dem geschätzten Grad der Iodversorgung bzw. -belastung. Nebenwirkungen sind dosisabhängig. Eine quasi sichere komplette Blockade der Schilddrüse bei unbekannter Iodversorgung erreicht man mit einer Initialdosis von 20 mg Thiamazol pro Tag. Bei ausgeprägtem Krankheitsbild oder höherer Iodzufuhr kann eine initiale Dosis von 30-40 mg Thiamazol pro Tag verabreicht werden. Im Falle einer Iodkontamination (z.B. durch Kontrastmittel) muss die Dosis auf ≥ 40 mg pro Tag erhöht und eine Kombination von Thiamazol mit Natriumperchlorat (Irenat®-Tropfen) als sinnvoll erachtet werden. Natriumperchlorat blockiert den Natrium-Iod-Symporter, das Iod kann somit nicht mehr in die Schilddrüsenfollikel eindringen. Die Dosis beträgt 900-1200 mg/Tag (drei Mal 15-20 Tropfen).(29) Bei einer Dosis von 10 mg Thiamazol/Tag liegt die Nebenwirkungsrate bei <10%, bei 60 mg/Tag bei >30%. Beschriebene (meist reversible) Nebenwirkungen sind Fieber, Pruritus und Arzneimittelexantheme, Gelenk- Geschmacksstörungen, Cholestase, Thrombopenie, Leukopenie bis oder Muskelschwächen, Erhöhung zur der Agranulozytose allergische Leberenzyme, (die aplastische Wahrscheinlichkeit Vaskulitis, Anämie, für eine Agranulozytose bei Einnahme von Thiamizol liegt bei 0,35%, bei PTU 0,37%). Bis zu 30% der PatientInnen unter PTU-Therapie entwickeln eine transiente Transaminasenerhöhung. Beim Auftreten von schweren Nebenwirkungen sollte eine Umstellung der Thyreostatika erfolgen. Kinder und Jugendliche sollten aufgrund der erhöhten Lebertoxizität nur mit Thioamiden therapiert werden.(29) 38 Zur symptomatischen Therapie einer Hyperthyreose werden β-Blocker eingesetzt, vorwiegend Propranolol (80-160 mg täglich), vor allem bei Hinweis auf gesteigerte Adrenalinsensitivität mit Tachykardie und Arrhythmien.(29) 3.1.3 Hypothyreose und ihre pharmakologische Therapie Bei manifester Unterfunktion der Schilddrüse ist eine (meist lebenslange) Substitutionstherapie erforderlich. Ziel der Therapie ist eine Verbesserung der Symptome und der euthyreote Zustand der PatientInnen (Normalisierung der TSH-, T3- und T4- Konzentrationen). Eine adäquate Behandlung beseitigt im Normalfall alle Symptome einer Hypothyreose, wobei neuromuskuläre und psychische Beschwerden noch über einige Monate nach Therapiebeginn persistieren können. Die Substitutionstherapie erfolgt meist mit synthetischen T 4-Präparaten (Levothyroxin). Die Plasmahalbwertszeit von Levothyroxin beträgt sieben Tage, somit ist eine einmalige Applikation pro Tag ausreichend für konstante Serum-T4- und T3-Spiegel. T4 ist ein Prohormon mit kleiner intrinsischer Aktivität und wird in peripheren Geweben zur aktiven Form T3 deiodiert. Dieser Deiodisierungs-Prozess stellt etwa 80% der täglichen T3-Produktion dar, somit ist eine alleinige Substitutionstherapie mit T4 ausreichend für konstante T4- und T3-Spiegel, dies konnte auch in klinischen Studien gezeigt werden. Die Substitution von T4 birgt außerdem den Vorteil, dass körpereigene physiologische Prozesse die Synthese der aktiven Hormonform regulieren.(33,34) 3.1.3.1 Substitutionstherapie: Dosierungen und Nebenwirkungen T4 ist in variablen Darreichungsformen erhältlich (Tabletten, Kapseln, Liquide). Grundsätzlich ist die Darreichungsform irrelevant, allerdings sollten regelmäßige Spiegel-Kontrollen durchgeführt werden und bei einem Wechsel des Präparats auf etwaige Abänderungen der Bioverfügbarkeit geachtet werden. Nebenwirkungen bei T4-Substitution, sowie Allergien auf Farbstoffe oder Arzneistoffträger, sind selten, solange die zugeführte Dosis im therapeutischen Bereich liegt. Überdosierungen verursachen subklinische bis klinisch manifeste Hyperthyreosen und sollten unbedingt vermieden werden. Das Hauptrisiko der subklinischen Hyperthyreose bei älteren PatientInnen mit Serum-TSH-Werten von < 0,1 mU/L besteht in dem drei Mal häufigeren Vorkommen von Vorhofflimmern. Ein weiterer unerwünschter Effekt der iatrogenen subklinischen Hyperthyreose ist der gesteigerte Knochenabbau (vor allem bei postmenopausalen Frauen).(33) Die durchschnittliche Substitutionsdosis von T4 bei Erwachsenen beträgt etwa 1,6 µg/kg Körpergewicht pro Tag (112 µg/Tag bei einem 70 kg schweren Erwachsenen), doch der Bereich der erforderlichen therapeutischen Dosis variiert individuell stark zwischen 50 bis ≥ 200 µg/Tag und kann auch abhängig von der Ursache der Hypothyreose sein. So konnte beispielsweise in Studien gezeigt werden, dass PatientInnen mit einer chronischen Autoimmunthyreoditis, einer Radioiod-Therapie oder einer zentralen Hypothyreose als Ursache der Schilddrüsenunterfunktion weniger Bedarf an T4 haben, als PatientInnen nach subtotaler Thyreoidektomie bei Schilddrüsentumor. Diese Ergebnisse legen den Verdacht nahe, dass sowohl die physiologischen Konzentrationen von TSH, als auch das Vorliegen von Schilddrüsenrestgewebe, Einfluss auf die 39 jeweilig erforderliche T4-Dosis haben. Im Allgemeinen korreliert der Bedarf an T 4 besser mit dem Lean Body Mass (fettfreie Körpermasse) als mit dem totalen Körpergewicht. Neugeborene und Kinder benötigen weitaus höhere Dosen pro kg Körpergewicht, bei älteren PatientInnen sollte initial restriktiv substituiert werden (25-50 µg/Tag).(33) Erste Verbesserungen bei T4-Substitutionstherapie sind etwa zwei Wochen nach Therapiebeginn zu erwarten, eine vollkommene Regeneration kann je nach Schweregrad der Hypothyreose jedoch einige Monate in Anspruch nehmen. Obwohl eine Symptomverbesserung bereits nach ein bis zwei Wochen eintreten kann, werden konstante TSH-Konzentrationen erst nach mindestens sechs Wochen erreicht. Folglich sollte bei Initiation einer T 4-Substitutionstherapie nach sechs Wochen eine Reevaluierung der Patientin oder des Patienten, sowie eine Messung des TSH-Spiegels durchgeführt werden. Bei PatientInnen deren Symptome nach zwei- bis drei-wöchiger Therapiedauer andauern, können freie T4- und TSH-Spiegel im Serum bereits nach drei Wochen erneut bestimmt werden. Ist die freie T4-Konzentration unter dem Normbereich, kann die Dosis nach drei Wochen ohne zusätzliche Kontrollen gesteigert werden, jedoch im Bewusstsein, dass die gemessenen T4- und TSH-Spiegel zu diesem Zeitpunkt noch variieren und keine konstanten Werte darstellen. Normale TSH-Werte liegen zwischen 0,5 – 5,0 mU/L, wobei die obere Grenze je nach Literatur zwischen 4,5 - 5,0 mU/L variiert. Außerdem gibt es einen altersabhängigen Shift zu höheren TSH-Spiegeln, weshalb die obere TSH-Grenze bei 80-jährigen PatientInnen bei etwa 7,5 mU/L liegt. Wurde eine adäquate Erhaltungsdosis ermittelt, werden jährliche Kontrollen des Serum-TSH empfohlen. In einigen Situationen ist eine Anpassung der Substitutionstherapie erforderlich. So ist beispielsweise während der Schwangerschaft, bei Gewichtszunahme, Estrogentherapie, Absorptionsstörungen (z.B. Zöliakie), erhöhter Exkretion (z.B. Nephrotisches Syndrom) oder gesteigerter Stoffwechselrate (z.B. bei Therapie mit Rifampizin, Carbamazepin, Imatinib, Phenytoin oder Phenobarbital) der Bedarf an Schilddrüsenhormonen erhöht. Umgekehrt erfordern Gewichtsabnahme, zunehmendes Alter und eine Therapie mit Androgenen eine Dosisreduktion. T4 sollte nicht gleichzeitig mit Medikamenten eingenommen werden, die dessen Absorption beeinträchtigen (beispielsweise Eisensulfate, Calciumkarbonat, Cholestyramin, Sertralin, Raloxifen und Omeprazol).(33) 3.1.3.2 T3-T4-Kombinationstherapien und reine T3-Präparate Eine Kombinationstherapie aus T3- und T4-Präparaten ist vorhanden, wird derzeit aber nicht bzw. nur unter besonderen Bedingungen empfohlen. Einige PatinentInnen zeigen trotz T4-Stubstitution und normalen Serum-TSH-Konzentrationen Symptome der Hypothyreose. Mehrere randomisierte klinische Studien haben die Effekte einer kombinierten T 3-T4-Therapie bei Symptompersistenz untersucht; fast alle (acht von neun klinischen Studien) mit dem Resultat, dass eine Kombinationstherapie gegenüber der T4-Monotherapie keinerlei Vorteile in Bezug auf die Lebensqualität, den Gemütszustand und die psychometrische Leistung der Betroffenen erweist. Eine Metaanalyse von elf publizierten randomisierten Studien (insgesamt 1216 PatientInnen) ergab 40 keinen Benefit der Kombinationstherapie bezüglich Müdigkeit, körperliche Schmerzen, Angst, Depression und Lebensqualität.(35) Eine T3-T4-Komibinationstherapie (Liotrix, 4:1 Mischung aus T4 und T3) wird derzeit nur für die seltene Subgruppe von PatientInnen mit einem Polymorphismus der Typ-2-Deiodinase erwogen. Hierbei handelt es sich um einen Defekt der Konvertierung von T 4 zu T3. Eine Analyse des Zusammenhangs des Typ-2-Deiodinase-Polymorphismus und dem psychischem Wohlbefinden (gemessen nach GHQ-Score) ergab bei den betroffenen PatientInnen ein besseres Ansprechen auf die Kombinationstherapie als auf die T4-Monotherapie.(33) Des Weiteren könnten PatientInnen nach einer Thyreoidektomie mit keiner verbleibenden endogenen T3-Produktion von einer zusätzlichen reinen T3-Therapie (Liothyronin) profitieren. Für den Großteil der PatientInnen mit Hypothyreose wird eine reine Therapie mit T 3-Präparaten aufgrund der großen tageszeitlichen Fluktuationsunterschiede von T3, der raschen gastrointestinalen Absorption und der relativ kurzen Plasmahalbwertszeit (etwa ein Tag) nicht empfohlen. Die temporäre Behandlung mit T3 von PatientInnen mit Schilddrüsentumor bei geplanter Radioiodid-Gabe hat sich mit dem Ergebnis der Verringerung der hypothyreotischen Phasen bewährt.(33) 41 3.2 Die hypothalamo-hypophysäre-adrenotrophe Achse Corticosteroide sind Steroidhormone der Nebennierenrinde und zudem ihre synthetischen Derivate, wobei alle Corticosteroide mit glucocorticoider oder mineralcorticoider Wirkung aus 21 C-Atomen aufgebaut sind. Die Steroidhormone der Nebennierenrinde lassen sich nach ihren biologischen Wirkungen in Glucocorticoide (gebildet in der Zona fasciculata, Kohlenhydratstoffwechsel als primärer Wirkort) und Mineralcorticoide (gebildet in der Zona glomerulosa, Elektrolythaushalt als primärer Wirkort) einteilen. Beide Hormongruppen sind lebensnotwendig, bei Mangelerscheinungen ist eine Substitutionstherapie erforderlich. Zu den Steroidhormonen der Nebennierenrinde zählen außerdem Androgene wie Androstendion und DHEA (Dehydroepiandrosteron), ein Prohormon, das in anderen Geweben zu Estradiol, Estron, Dihydrotestosteron (DHT) und Testosteron umgewandelt wird.(4,22) Cholesterin bildet die Ausgangssubstanz für die Biosynthese der Steroidhormone. Der geschwindigkeitsbestimmende Schritt ist hierbei die Umwandlung von Cholesterin in Pregnenolon, der Großteil, der an der Steroidbiosynthese beteiligten Enzyme, sind Cytochrom-P450-Enzyme. Die Nebennierenrinde speichert keine Hormone; die Biosynthese und die Sekretion der Glucocorticoide und adrenalen Androgene erfolgen insofern direkt nach der Stimulation durch ACTH aus der Hypophyse. Die Sekretion von ACTH wird wiederum durch das hypothalamische Releasing-Hormon CRH reguliert, wobei dessen Freisetzung ebenso Einflüssen von Neurotransmittern aus übergeordneten Zentren unterliegt. Sezernierte Glucocorticoide wirken auf allen Ebenen dieses Regelkreises inhibierend. Dieser negative Rückkopplungsmechanismus besteht aus zwei Phasen: die erste, schnelle Phase ist gekennzeichnet durch die sofortige Suppression der endogenen Cortisolsynthese beim Anfluten des Glucocorticoids, vermutlich durch die Inhibierung der CRH- und ACTH-Sekretion. Die verzögerte, zweite Phase erfolgt durch Hemmung der CRHund ACTH-Synthese. Generell unterliegt die Cortisolsekretion, durch die hypothalamo- hypophysäre-adrenotrophe Achse stimuliert, zwei Mechanismen: einerseits der basalen zirkadianen Sekretion und andererseits der verstärkten Freisetzung in Stresssituationen.(4,22) ACTH stimuliert akut darüber hinaus die Synthese und Sekretion von Aldosteron. Bei kontinuierlich erhöhten ACTH-Spiegeln kommt es allerdings zu einem Rückgang der AldosteronKonzentration zum Normwert (sogenannter „ACTH-Escape“). Die Synthese von Aldosteron wird ansonsten vorwiegend über die extrazelluläre Kalium-Konzentration und Angiotensin II gesteuert. Folglich kommt es bei Inhibierung der ACTH-Freisetzung zu keiner Atrophie der Zona glomerulosa.(4,22) 42 Hypothalamus Hypothalamus CRH Hypothalamus CRH Adenohypophyse Adenohypophyse ACTH Hypothalamus CRH Adenohypophyse ACTH CRH Adenohypophyse ACTH Nebenniere: CORTISOL Nebenniere: CORTISOL Nebenniere: CORTISOL T A T B T C ACTH Nebenniere: CORTISOL T D Abbildung 5. Hypothalamo-hypophysäre-adrenotrophe Achse.(4) A. Unter physiologischen Bedingungen regulieren die peripheren Konzentrationen an Glucocorticoiden die Synthese und Sekretion von CRH und ACTH durch negative Rückkopplung. B. Bei Mb. Addison (primäre Nebennierenrindeninsuffizienz) kommt es zu einem Abfall der peripheren GlucocrticoidKonzentration. Es wird vermehrt CRH und ACTH gebildet. C. Primäres Cushing-Syndrom (Nebennierentumor). Die stark vermehrte Glucocorticoid-Sekretion führt bei intaktem Regelkreis zur Verminderung von CRH und ACTH. D. Morbus Cushing. Bei vermehrter Sekretion von ACTH (z.B. durch ein ACTH-sezernierendes Hypophysenadenom) kommt es zu einer bilateralen Nebennierenrinden-Hyperplasie, wobei die vermehrte Freisetzung von Glucocorticoiden zu keiner zentralen Suppression führt. 3.2.1 Glucocorticoide Die chemische Konfiguration Delta-4,3-Keto-11-Beta,17-Alpha,21-Trihydroxyl ist essentiell für die Aktivität der Glucocorticoide und Bestandteil sowohl der endogenen, als auch der synthetischen Glucocorticoide. Endogene Glucocorticoide werden in der Zona fasciculata der Nebennierenrinde gebildet, das wohl wichtigste körpereigene Glucocorticoid ist Cortisol. Das meiste Cortisol im Serum liegt Protein-gebunden, entweder an CBG (corticosteroid-binding globulin) oder Albumin, vor. Synthetische Glucocorticoide wie Prednisolon binden entweder nur schwach an Albumin oder zirkulieren frei. Die tägliche Sekretionsrate von Cortisol unterliegt Schwankungen von 5-20 μg/100 ml und beträgt etwa 10-20 mg/Tag, von Corticosteron etwa 1-4 mg/Tag. Die Plasmahalbwertszeit von Cortisol beträgt etwa 80 Minuten. Unter physiologischen Bedingungen ist die zirkadiane Rhythmik der Cortisolsekretion stabil. Die Cortisolkonzentration erreicht zwischen 3.00 Uhr und 10.00 Uhr ihr Maximum, fällt im Laufe des Tages wieder ab und erreicht ihr Minimum zwischen Mitternacht und 3.00 Uhr. Entsprechend dem zirkadianen Rhythmus ändert sich ebenso die Sensitivität der übergeordneten Zentren des Rückkopplungssystems. In Stresssituationen, wie beispielsweise bei Traumata, Infektionen, Operationen, Geburt, schwerer körperlicher Arbeit, psychischem Stress oder Kälte, kommt es durch die Freisetzung von Entzündungs- und Immunmediatoren wie TNF-α (Tumor-Nekrose- 43 Faktor-α), IL-1 und IL-6 zu einer Aktivierung der Hypothalamus-Hypophysen-NebennierenrindenAchse und einem starken, lebensnotwendigen Anstieg der Freisetzung von CRH und ACTH. Die verzögerte Phase der Rückkopplung der Glucocorticoide wird hierbei übergangen. (4,36) Inaktives Cortison wird durch die 11β-Hydroxysteroid-Dehydrogenase Typ 1 in aktives Cortisol konvertiert. Dieses Isoenzym wird von vielen Zielgeweben der Glucocorticoide exprimiert. Die 11β-Hydroxysteroid-Dehydrogenase Typ 2 konvertiert Cortisol zu Cortison. Man findet sie hauptsächlich in Zielgeweben der Mineralcorticoide wie z.B. Nieren, Colon, Speicheldrüsen, ebenso in der Plazenta, wo sie die Cortisol-Aktivierung der Zellen unterdrückt. Dexamethason, Fludrocortison, Betamethason und Methylprednisolon können aufgrund ihrer chemischen Struktur nicht durch die 11β-Hydroxysteroid-Dehydrogenase Typ 2 inaktiviert werden. Prednisolon wird hingegen im Vergleich zu Cortison effektiver oxidiert.(36) Die Bindung von Glucocorticoiden erfolgt unter anderem an den der Klasse der ligandengesteuerten Transkriptionsfaktoren zugehörigen Glucocorticoid-Rezeptor. In inaktiver Form liegt der Rezeptor intrazellulär und zytosolisch an einen Proteinkomplex gebunden vor. In aktiver Form (bei Bindung von Cortisol) wandert er in den Zellkern und reguliert die Transkription von Genen über DNA-Bindungs-abhängige, sowie DNA-Bindungs-unabhängige Mechanismen. Glucocorticoide verfügen ebenso über nicht-genomische Effekte, welche sich hauptsächlich aus schnellen, unspezifischen Interaktionen mit Zellmembranen, den nicht-genomischen Eigenschaften des zytosolischen Glucocorticoid-Rezeptors und den spezifischen Interaktionen mit Membrangebundenen Glucocorticoid-Rezeptoren ergeben.(4,37) 3.2.1.1 Pharmakologische Anwendung von Glucocorticoiden Endogene und synthetische Glucocorticoide werden pharmakologisch bei endokrinen und nichtendokrinen Erkrankungen eingesetzt. Endokrinologische Indikationen sind beispielsweise die Diagnostik eines Cushing-Syndroms oder Mb. Cushing, die Therapie einer Nebennierenrindeninsuffizienz und die kongenitale adrenale Hypoplasie. Glucocorticoide werden therapeutisch ebenso bei entzündlichen und allergischen Reaktionen, sowie Autoimmunerkrankungen eingesetzt. Generell ist bei chronischer Anwendung mit mäßig ausgeprägten bis zu starken Nebenwirkungen zu rechnen.(36) Zahlreiche synthetische Glucocorticoide wurden ausgehend von der molekularen Struktur von Cortisol hergestellt. Systemisch wirksam sind Prednisolon, Methylprednisolon, Dexamethason und Triamcinolon. Lokal wirksame und besonders zur inhalativen Therapie eingesetzte Agonisten sind Budesonid, Beclometason und Ciclesonid. Die Synthese von Prednisolon (Delta-1-Hydrocortison) erfolgte mittels Einführung einer Doppelbindung zwischen erster und zweiter Position von Hydrocortison (Cortisol). Synthetische Glucorticoide besitzen längere Plasmahalbwertszeiten als Cortisol, reichend von etwa einer Stunde (Prednisolon) bis zu über vier Stunden (Dexamethason), wobei individuelle Variationen berücksichtigt werden müssen (z.B. verlängerte Clearance bei 44 älteren PatientInnen). Dosis und Art der Applikation hängen von der zu behandelnden Erkrankung ab.(4,36) Abbildung 6: Steroidale Strukturformeln.(36) Strukturformeln von Cortisol, Cortison und anderen häufig eingesetzten synthetischen Glucocorticoiden. 3.2.1.2 Wirkungen und Nebenwirkungen Eine Therapie mit Glucocorticoiden führt zur Hemmung der ACTH-Sekretion durch negative Rückkopplung und kann in einer Atrophie der Zona fasciculata und der Zona reticularis der Nebennierenrinde und damit in einem Rückgang der endogenen Biosynthese von Glucocorticoiden und adrenalen Androgenen resultieren. Nach Absetzen des Pharmakons kommt es je nach Dauer und Dosis der Therapie wiederum zur Normalisierung der ACTH-Sekretion und Nebennierenrindenfunktion. Andererseits kommt es bei konstant erhöhten ACTH-Konzentrationen (z.B. bei Mb. Cushing) zu einer Hyperplasie und Hypertrophie der inneren Zonen der Nebennierenrinde und folglich zu einer exzessiven Produktion von Cortisol und adrenalen Androgenen.(4) Entscheidende Wirkungen und Wirkungsmechanismen der Glucocorticoide sind teilweise immer noch unklar. Synthetische Glucocorticoide finden jedoch aufgrund ihrer entzündungshemmenden und immunsuppressiven Wirkung ein breites Anwendungsgebiet. Glucocorticoide inhibieren lokal früh- und spätentzündliche Reaktionen unabhängig von der auslösenden Noxe, sowie Immunreaktionen, unter anderem durch die Hemmung des Transkriptionsfaktors NF-κB (NuclearFactor-κB) und von Genen proinflammatorischer Proteine wie COX-2 (Cyclooxygenase-2) und IL- 45 1. Es kommt außerdem zur Aktivierung von Genen regulativer und antiinflammatorischer Proteine wie IL-10, IκBα und IL-1-Rezeptor-Antagonist. Glucocorticoide wirken regulativ auf den Kohlenhydrat-, Fett- und Proteinstoffwechsel. Cortisol besitzt außerdem eine klinisch bedeutsame mineralcorticoide Wirkung mit renaler Retention von Natrium und daraus resultierenden peripheren Ödemen. Von ihren vielen Wirkorten lassen sich auch die glucocorticoiden Nebenwirkungen ableiten, wobei deren Ausmaß und Reversibilität stark von Dauer und Dosis der Therapie abhängen.(4,37) Glucocorticoide verfügen über permissive Effekte, beispielsweise hat Cortisol selbst kaum eine lipolytische Wirkung im Fettgewebe, verstärkt aber die lipolytischen Eigenschaften von Adrenalin, Noradrenalin und GH. Hohe Konzentrationen führen zu Appetit- und Gewichtszunahme, sowie zu einer Umverteilung des Fettgewebes; es kommt zu einer Zunahme von Fett in Gesicht und Nacken, sowie zu einem Fettverlust an den Extremitäten (typisches Bild der Stammfettsucht bei CushingSyndrom), wobei dies hauptsächlich in den ersten zwei Monaten nach Therapiebeginn auftritt. Glucocorticoide wirken katabol, fördern somit die Proteolyse und Gluconeogenese und führen zu einer negativen Stickstoffbilanz, teilweise bedingt durch die gesteigerte Ausscheidung von Aminosäuren und Harnsäure. Unter hochdosierter Glucococorticoid-Therapie kann es zu Muskelatrophien kommen (sogenannte Steroidmyopathie). An der Haut verursachen Glucocorticoide Atrophien, Purpura, Hypertrichose, Striae, sowie die sogenannte Steroid-Akne. Glucosetoleranz und Insulinsensitivität nehmen ab, der Glucoseumsatz und die Plasma-Glucosekonzentration steigen, somit kann ein Prädiabetes unter Glucocorticoid-Gabe in einen latenten bzw. klinisch manifesten Diabetes mellitus exazerbieren. Ebenso beeinflussen Glucocorticoide den Knochenstoffwechsel, indem sie die Resorption von Calcium im Darm hemmen, die Phosphat-Clearance, sowie die renale Calcium- und Phosphat-Ausscheidung steigern und somit das Risiko für Osteoporose, Frakturen und Osteonekrose erhöhen. Glucocorticoide wirken positiv inotrop und erhöhen die vaskuläre Wirkung von Katecholaminen und β2-Agonisten und verbessern folglich die Mirkozirkulation bei akuten Schockzuständen. Bei systemischer, kontinuierlicher Therapie kommt es jedoch zu einem erhöhten Risiko von kardiovaskulären Nebenwirkungen; vor allem durch gesteigerte Atherosklerose, Hypertonus, ischämische Herzerkrankungen, kardiale Dekompensationen und Vorhofflimmerarrhythmien. Die Atherosklerose-fördernden Effekte von Glucocrticoiden werden teilweise vermutlich durch erhöhte Lipoprotein-Spiegel getriggert, wobei noch unklare und unterschiedliche Studienergebnisse vorliegen. Im Gastrointestinaltrakt erhöhen Glucocorticoide das Risiko für unerwünschte Wirkungen, wie Gastritis, gastrointestinale Blutungen und Ulzera, wobei vor allem die Kombination von Glucocorticoiden mit NSAR (nicht-steroidale Anti-Rheumatika) zu einem signifikanten synergistischen Anstieg dieser Effekte führt und nicht empfohlen wird. Am zentralen Nervensystem bewirken Glucocorticoide neben ihren indirekten Effekten durch Einfluss auf den Metabolismus, das Herz-Kreislauf-System und den Elektrolythaushalt, auch eine Senkung der Reizschwelle für bestimmte Stimuli. EEG-Veränderungen wurden beobachtet, bei einigen 46 PatientInnen kommt es zum Auftreten von Dysphorien und Depressionen, aber auch Euphorien, erhöhte motorische Aktivität und Schlafstörungen können ebenso vorkommen. Das endokrine Psychosyndrom mit akuter Zyklothymie und Antriebsschwankungen ist selten.(4,37,38) Eine weitere, meist nicht reversible Nebenwirkung einer systemischen Glucocorticoid-Gabe ist das Dosis-abhängige Auftreten von Katarakt und Glaukom. Erhöhter intraokulärer Druck und Glaukome wurden bisweilen hauptsächlich bei PatientInnen mit Glucocorticoid-hältigen Augentropfen festgestellt. Das Risiko für die Entwicklung einer Katarakt ist bei prolongierter Therapie (> 1 Jahr) erhöht. Glucocorticoid-induzierte Katarakte finden sich typischerweise posterior subkapsulär, bilateral und entwickeln sich langsam, wobei Kinder eine erhöhte Anfälligkeit zeigen.(37) Aufgrund ihrer vielen potenten Effekte, aber ebenso nicht zu unterschätzenden Nebenwirkungen, muss der Indikation einer Therapie mit Glucocorticoiden immer ein Abwägen des Nutzen-RisikoVerhältnisses vorausgehen. Generelle aktuelle Empfehlungen lassen sich zusammenfassen auf: eine möglichst geringe Dosierung für die kürzest mögliche Zeitdauer, um ein Therapieziel zu erreichen, die zusätzliche Therapie von Komorbiditäten, welche das Risiko von Nebenwirkungen einer Behandlung mit Glucocorticoiden erhöhen, sowie regelmäßige Kontrollen und Monitoring von PatientInnen.(37) 3.2.2 Mineralcorticoide Das wichtigste, in der Zona glomerulosa der Nebennnierenrinde gebildete, endogene Mineralcorticoid ist Aldosteron. Aldosteron bindet in der Niere an den Mineralcorticoid-Rezeptor epithelialer Zellen des distalen Tubulus und der Sammelrohre und reguliert die Elektrolyt- und Flüssigkeitshomöostase. Aldosteron wird in Leber und Niere zu Aldosteron-18-Glucuronid und Tetrahydroaldosteron-3-Glucuronid konjugiert, beide Formen werden renal ausgeschieden. Die Sekretionsrate von Aldosteron beträgt etwa 50-150 μg/Tag, diejenige seiner Vorstufe Desoxycorticosteron beträgt etwa 50-200 μg/Tag, wobei Desoxycorticosteron viel weniger wirksam ist als Aldosteron. Die Plasma-Aldosteron-Konzentration liegt bei etwa 10 ng/100 ml, die Plasmahalbwertszeit beträgt weniger als 30 Minuten.(4,22) 3.2.2.1 Wirkungen und Nebenwirkungen der Mineralcorticoide Der Mineralcorticoid-Rezeptor ist ein Rezeptor der Klasse der ligandengesteuerten Transkriptionsfaktoren. Er wird in einigen Geweben besonders stark exprimiert, beispielsweise in der Niere, in Schweiß- und Speicheldrüsen, im Colon, im Fettgewebe, in Gefäßen, im Herz und im Hippocampus. Der Mineralcorticoid-Rezeptor bindet neben Aldosteron auch Cortisol und Progesteron (Progesteron fungiert als endogener Aldosteron-Antagonist). Aldosteron und in geringerem Maße Cortisol, sowie synthetische Analoga steigern in distalen Tubuli und Sammelrohren der Niere die Natrium-Reabsorption (durch Steigerung der Wasser- und Natriumretention kommt es zur Volumenzunahme des Extrazellulärraums) und fördern gleichzeitig die Ausscheidung von K+ und H+ (dies führt zu Hypokaliämie und metabolischer 47 Alkalose). Ebenso fördern Mineralcorticoide die Magnesiumausscheidung. Die Natrium-Retentioninduzierende Wirkung resultiert in einem Blutdruckanstieg. Kardiovaskuläre Schädigungen entstehen außerdem durch die direkte Induktion von Entzündung, oxidativem Stress und Fibrose.(4,39) Der primäre Hyperaldosteronismus ist meist Folge eines Aldosteron-produzierenden Adenoms der Nebennierenrinde (sogenanntes Conn-Syndrom). Symptome eines Hyperaldosteronismus sind Hypertonie, Zunahme des Extrazellulärvolumens, Hypokaliämie und metabolische Alkalose. Als sekundären Hyperaldosteronismus bezeichnet man die pathologische Stimulation der AldosteronSekretion oder eine Hemmung des Aldosteron-Abbaus außerhalb der Nebennierenrinde. Die Stimulation der Freisetzung von Aldosteron erfolgt großteils über das Renin-AngiotensinAldosteron-System. Eine pathologische Überstimulation ist somit meist Resultat einer gesteigerten Renin-Sekretion, entweder durch Verminderung des zirkulierenden Blutvolumens (z.B. bei Herzinsuffizienz, nephrotischem Syndrom, Cirrhosis hepatis) oder einer reduzierten Nierenperfusion (z.B. bei Nierenarterienstenose, maligner Hypertonie).(4) 3.2.2.2 Mineralcorticoid-Rezeptor-Antagonisten Der Mineralcorticoid-Rezeptor-Antagonist Spironolacton konnte die Mortalität bei schwerer Herzinsuffizienz um bis zu 30% senken. Am Tiermodell konnte dies durch die Hemmung der Mineralcorticoid-vermittelten Wirkung auf Herz (kardiale Hypertrophie) und Koronargefäße (entzündliche Veränderungen und Fibrose) erklärt werden. Ein weiterer Mineralcorticoid-RezeptorAntagonist mit größerer Selektivität für den Mineralcorticoid-Rezeptor ist Eplerenon. Eplerenon konnte die Morbidität und Mortalität von PatientInnen nach akutem Myokardinfarkt, linksventrikulärer Dysfunktion und dekompensierter Herzinsuffizienz signifikant reduzieren.(4) In den letzten Jahren wurden einige nicht-steroidale Mineralcorticoid-Rezeptor-Antagonisten entwickelt. Ein neuer, hochpotenter, langwirksamer, selektiver und nicht-steroidaler Mineralcorticoid-Rezeptor-Antagonist ist CS-3150, der bei Hypertonie, sowie kardiovaskulären und renalen Erkrankungen eingesetzt werden soll.(39) 48 3.3 Die hypothalamo-hypophysäre-gonadale Achse Der hypothalamo-hypophysäre-gonadale Regelkreis besteht aus einem komplexen Zusammenspiel von Sexualhormonen, aber auch externen Einflüssen, wie psychischen und physischen Faktoren oder Umwelteinflüssen. Sexualhormone sind Steroidhormone: zu ihnen zählen Estrogene, Gestagene und Androgene, welche nicht nur in den Gonaden, sondern auch in der Plazenta und den Nebennieren produziert werden. Allen Sexualhormonen gemeinsam ist ihre Wirkung über Steroidhormonrezeptoren, die vorwiegend intrazellulär lokalisiert sind, wobei geringe hormonspezifische Unterschiede vorliegen.(4,15) Die Synthese und Sekretion der Sexualhormone unterliegen dem Einfluss von Hypothalamus und Hypophyse. Das im Hypothalamus gebildete und pulsatil sezernierte GnRH beeinflusst Produktion und Freisetzung der Gonadotropine FSH und LH aus der Adenohypophyse, welche wiederum die Hormonsynthese in Ovarien und Leydig-Zellen der Hoden regulieren. Bei der Frau steuern Gonadotropine: die Follikulogenese, den Erhalt des Corpus luteum, sowie die ovarielle Hormonproduktion. Beim Mann vermitteln sie die Spermatogenese und die Testosteron-Sekretion. Die GnRH-Freisetzung aus dem Hypothalamus unterliegt wiederum der regulativen Wirkung von Neurotransmittern und anderen Hormonen: Noradrenalin stimuliert die GnRH-Sekretion, GABA und Dopamin führen zur Suppression, CRH und vom ACTH-Prohormon Proopiomelanocortin (POMC) abgeleitete Peptide, wie z.B. β-Endorphine, inhibieren die GnRH-Sekretion. Der hypothalamische GnRH-Pulsgeber wird zusätzlich über Kisspeptin-1 gesteuert (ein Neuropeptid, das aus den Nuclei paraventricularis und arcuatus freigesetzt wird).(4,15) Hypothalamus GnRH Adenohypophyse FSH,LH Ovar: ESTRADIOL, PROGESTERON TA Hypothalamus GnRH Adenohypophyse FSH,LH Hoden: TESTOSTERON B Abbildung 7: Hypothalamo-hypophysäre-gonadale Achse.(4) A. Bei der Frau. B. Beim Mann. 49 Während der Kindheit ist die hypothalamo-hypophysäre-gonadale Achse hoch sensibel gegenüber den negativen Rückkopplungsmechanismen, somit reichen geringe Konzentrationen der Sexualhormone aus, um die zentrale GnRH- und Gonadotropinproduktion zu hemmen. Diese Sensibilität nimmt während der Pubertät graduell ab, wobei in Folge für eine Suppression der GnRH- und Gonadotropinsekretion zehnfach höhere Sexualhormonmengen erreicht werden müssen.(5) Die hypothalamo-hypophysäre-gonadale Achse darf, wie kein neuroendokriner Regelkreis, isoliert betrachtet werden. Endokrine und externe Einflüsse sind vielfältig und miteinander vernetzt. So konnte beispielsweise gezeigt werden, dass etwa 50% der sekundären Amenorrhoen durch physiologische, pathologische und iatrogene Störungen der hypothalamo-hypophysären- adrenotrophen Achse verursacht werden.(16) 3.3.1 Physiologie des ovulatorischen Zyklus Der weibliche Menstruationszyklus unterteilt sich in vier verschiedene Zyklusphasen mit unterschiedlichen Hormonkonzentrationen, welche auch das Endometrium des Uterus zyklisch beeinflussen. Diese sind: Menstruation, Follikelphase, Ovulation und Lutealphase.(15) Die genauen Prozesse der verschiedenen Zyklusphasen können in dieser Arbeit nicht beschrieben werden, da dies das Ausmaß weitaus überschreiten würde; grundlegende Vorgänge können somit nur peripher gestreift werden, sollen aber zumindest Erwähnung finden. Der weibliche Zyklus ist gekennzeichnet durch eine relativ konstante, niedrigdosierte Sekretion von FSH und LH, die durch Rückmeldung an Hypothalamus und Hypophyse reguliert wird, wobei Estradiol-Konzentrationen, die vom sich entwickelnden Follikel sezerniert werden, die zentrale Funktion des Rückkopplungsmechanismus übernehmen. Während der Follikelphase besteht eine hohe Frequenz von LH-Pulsen mit niedriger Amplitude, während der Lutealphase kommt es zu einer Frequenzabnahme und gleichzeitiger Amplitudenzunahme. Die Effekte von Estrogen und Progesteron auf Hypophyse und Hypothalamus sind die entscheidenden Faktoren der pulsatilen LH-Freisetzung, wobei die Frequenz der LH-Ausschüttung hauptsächlich über die inhibitorische Wirkung von Progesteron auf den Hypothalamus bestimmt wird. Die Konzentration von Progesteron steigt in der Lutealphase stark an und bewirkt eine Abnahme der Pulsfrequenz von GnRH. Die Ovulation am Ende der follikulären Phase des Menstruationszyklus ist getriggert durch eine massive und prolongierte Freisetzung von Gonadotropinen.(4,15,40) 50 Abbildung 8: Schematische Darstellung des weiblichen Menstruationszyklus.(52) (a) Zeitlicher Verlauf der Sekretion der Gonadotropine FSH und LH und der Sexualhormone Estradiol und Progesteron. (b) Auswirkungen der Hormonsekretion auf das Endometrium des Uterus. 3.3.2 Estrogene Estrogene werden in Ovarien und Plazenta, sowie in geringeren Mengen auch in den Nebennieren und Testes, gebildet. In den Granulosazellen des Ovars erfolgt die Synthese durch die Aromatase P450arom (CYP19). Durch Aromatisierung von Androgenen können sie darüber hinaus in beträchtlichen Mengen im Fettgewebe produziert werden. Estrogene regulieren zusammen mit Gestagenen die neuroendokrine Steuerung des weiblichen Zyklus, die Ausbildung sekundärer Geschlechtsmerkmale, sowie die Fertilisations- und Implantationsvorgänge bei der Frau, die Reproduktion beim Mann, die Embryonalentwicklung, zentrale metabolische und kardiovaskuläre Prozesse, sowie den Stütz- und Bewegungsapparat.(4,15,41) Estrogene sind Steroidhormone mit 18 Kohlenstoffatomen. Das physiologische und biologisch aktivste Estrogen ist 17β-Estradiol. Die Estrogene Estron und Estriol besitzen eine weitaus geringere biologische Aktivität, wobei dies Folge der unterschiedlichen Bindungsaffinitäten zu Estrogen-Rezeptoren (ERα und ERβ) ist. Bei der Frau beträgt die tägliche Sekretionsrate von Estrogenen je nach Zyklusphase etwa 25-100 µg (90-350 nmol). Während der Schwangerschaft werden Werte bis 30 mg/Tag erreicht. Bei postmenopausalen Frauen beträgt die 51 Estrogenkonzentration etwa 5-10 µg/Tag (17-35 nmol). Männer sezernieren täglich etwa 2-25 µg (7-90 nmol) Estrogen.(4,42) Oral wirksame Estrogen-Derivate wie 17α-Ethinylestradiol und sein 3-Methylether (Mestranol) wurden durch Alkylsubstitution hergestellt; sie spielen in der oralen hormonellen Kontrazeption eine zentrale Rolle. Estradiolvalerat (entsteht durch Veresterung der Hydroxylgruppe an C-17) kann als intramuskuläres Depot injiziert werden, woraus Estradiol hydrolytisch freigesetzt wird. Estradiolvalerat und konjugierte Estrogen-Präperate können allerdings auch oral verabreicht werden. Konjugierte Estrogene sind Ester, enthalten hauptsächlich Sulfate von Estron, 7,8Dehydroestron (Equilin), sowie 17α-Dihydroequilin und werden bei oraler Anwendung im Colon gespalten.(4) Estrogene wurden früher aufgrund ihrer osteoprotektiven Effekte häufig in der Therapie der postmenopausalen Osteoporose eingesetzt, heutzutage sind sie aufgrund hoher Nebenwirkungsraten nur noch in zweiter Linie relevant. Dies trifft auf PatientInnen mit hohem Frakturrisiko und Kontraindikationen bzw. Intoleranzen gegenüber primär empfohlenen Pharmaka zu. Potentielle Nebenwirkungen der postmenopausalen Estrogen-Therapie sind Mamma- und Lungenkarzinome, kardiovaskuläre Erkrankungen wie Apoplexien oder venöse Thromboembolien, sowie Erkrankungen der Gallenblase.(43) 3.3.3 Antiestrogene Man unterscheidet im Allgemeinen reine Antiestrogene und selektive EstrogenrezeptorModulatoren (SERM), wobei reine Antiestrogene über die direkte Bindung an den Estrogenrezeptor wirken und SERM zu einer Konformationsänderung des Rezeptors mit entsprechender Veränderung der Transkription von Estrogen-abhängigen Genen führen. SERM sind nicht-steroidale Substanzen, deren Anwendungsgebiet von der Therapie der Osteopenie und Osteoporose (in Europa) bis zur Prävention des Mammakarzinoms bei erhöhtem Risiko (in den USA) reicht.(44) Ein kompetitiver Estrogenrezeptor-Antagonist ist beispielsweise Fulvestrant, der als Therapie bei Estrogenrezeptor-positiven, Tamoxifen-resistenten, metastasierenden Mammakarzinomen bei postmenopausalen Frauen zugelassen ist. Fulvestrant verfügt über eine etwa 30-fach höhere Affinität als Tamoxifen (SERM, Triphenylethylen) und eine mit Estradiol vergleichbare Affinität zu ERα und ERβ, somit kommt es zur kompletten Hemmung der Estrogen-vermittelten Gentranskription (Inhibierung der Dimerisierung und Translokation des Estrogen-Rezeptors), ohne agonistische Wirkung. Fulvestrant ist außerdem ein selektiver Estrogenrezeptor-Down-Regulator (SERD), da es einen Abbau des Antagonist-Rezeptor-Komplexes im Cytoplasma bewirkt, wobei dies in einer Down-Regulation der Estrogen-Rezeptoren resultiert.(4,44) SERM sind verschiedene chemische Substanzklassen, wie z.B. Raloxifen (Benzothiophen), Clomifen (Chlorethylen) oder Bazedoxifen (Indol).(44) 52 3.3.4 Gestagene Als Gestagene bezeichnet man die Stoffgruppe der Sexualhormone, welche Progesteron-ähnliche Wirkungen aufweisen, wobei deren biologische Effekte fast alle aus dem Zusammenspiel mit Estrogenen resultieren. Progesteron-Rezeptoren werden von Estrogen aktiviert, somit sind eine ausreichende Estrogenwirkung, das Estrogen-Gestagen-Verhältnis, sowie die zeitliche Sequenz des Zusammenwirkens ausschlaggebend für die endogene Aktivität.(4,41) 3.3.4.1 Progesteron Das physiologische endogene Gestagen ist Progesteron, welches über 21 Kohlenstoffatome verfügt. Sein Vorstufenmolekül ist Pregnenolon. Tägliche Sekretionsraten von Progesteron reichen von nur einigen Milligramm in der Follikelphase, bis zu 20 mg (60 μmol) in der Lutealphase und bis zu einigen 100 mmol während der Schwangerschaft. Bei der Frau wird es hauptsächlich im Corpus luteum des Ovars, jedoch auch in der Plazenta und den Nebennieren, gebildet. Die ovarielle Synthese ist dabei stark von Gonadotropinen, vor allem von LH, abhängig. Die plazentäre Progesteronsynthese ist derzeit noch nicht hinreichend geklärt. Bei der männlichen Testosteronbiosynthese ist Progesteron ein Zwischenprodukt. Die Plasmahalbwertszeit liegt bei etwa 20 Minuten. 80% des Progesterons sind im Blut an Albumin, 18% an CBG (Transkortin) gebunden, etwa 2% liegen frei vor. Der Progesteronabbau findet durch die Reduktion der Doppelbindungen und Ketogruppen vorwiegend in der Leber statt, wobei Metaboliten wie Pregnandiole (3α,20α-Pregnandiol) und Pregantriol (Hauptmetabolit von 17α-Hydroxyprogesteron) entstehen und als Glucuronide und Sulfate renal ausgeschieden werden.(4,42) Progesteron ist zusammen mit Estradiol das zentrale Hormon der neuroendokrinen Regulation des weiblichen Zyklus. Progesteron wirkt hemmend auf den hypothalamischen GnRH-Pulsgeber, erhöht die Amplitude der LH-Pulse aus der Adenohypophyse und blockiert über Down-Regulation von hypophysären Estrogen-Rezeptoren die estradiolabhängige, massive LH-Freisetzung um den vierzehnten Tag des Zyklus. Letztere Wirkung stellt die Basis der Funktion der hormonellen Kontrazeption dar. Progesteron beeinflusst außerdem den zyklischen Auf- und Abbau des Endometriums, wobei in der ersten Zyklushälfte vorwiegend Estrogene wirksam sind (induzieren die Proliferation des Endometriums). Nach der Ovulation kommt es zur sekretorischen Umwandlung des Endometriums, welche durch die vermehrte Progesteron-Synthese bedingt ist. Des Weiteren ist Progesteron an einer Vielzahl der weiblichen Reproduktionsvorgänge beteiligt, wie beispielsweise der Veränderungen des Vaginalepithels, der Zusammensetzung des Uterus-, Tuben- und Cerivicalsekrets, dem Oozytentransport, dem Nidationsvorgang, sowie der schwangerschaftserhaltenden Wirkung. Ein vorzeitiger Abfall der Progesteron-Konzentration führt zum Abort.(4,41,42) Die thermogenetische Wirkung mit einem Anstieg der Körpertemperatur (0,6-1°C) in der zweiten Hälfte des weiblichen Zyklus ist charakteristisch für Progesteron. In hohen Konzentrationen wirkt 53 es außerdem katabol, stimuliert die Lipoproteinlipase und die Fetteinlagerung und kann zu verminderter Glucosetoleranz führen.(4) 3.3.4.2 Synthetische Gestagene Synthetische Gestagene unterscheiden sich stark in Wirkqualität und –stärke und verfügen über unterschiedliche Partialwirkungen, welche therapeutisch gezielt nutzbar sind. Oral oder parenteral applizierbare Präparate sind entweder Derivate androgener Vorläufer wie z.B. des 19Nortestosterons (Estrane, wie z.B. Norethisteron, Lynestrenol, etc.), des Norgestrels (Gestane, wie z.B. Levonorgestrel, Desogestrel, Dienogest, etc. mit verringerter androgener Partialwirkung) oder Derivate des Progesterons und des 17α-Hydroprogesterons, welche nur eine minimale androgene oder anabole Partialwirkung besitzen. Progesteron selbst kann oral nur in mikronisierter Form verabreicht werden. 17α-Hydroprogesteron hingegen kann in Form von 17α-Hydroprogesteroncaproat parenteral appliziert werden.(4) Das Wirkungsspektrum von synthetischen Gestagenen unterscheidet sich insofern von demjenigen von Progesteron, als dass sie neben den klassischen gestagenen auch estrogene, antiestrogene, androgene, antiandrogene, glucocorticoide und mineralcorticoide Wirkungen besitzen. Beispielsweise verfügt Drospirenon, ein dem Spironolacton verwandtes Gestagen, über sowohl antiandrogene, als auch antimineralcorticoide Effekte und führt zu einer Verminderung prämenstrueller Ödeme. Gestagene Derivate des 19-Nortestosterons sind Bestandteil von vielen oralen Kontrazeptiva, wobei einige anabole Restwirkungen aufweisen und teilweise in geringem Ausmaß zu Estrogenen metabolisiert werden. Nebenwirkungen dieser Präparate sind Androgenisierungserscheinungen und Gewichtszunahme. Dienogest, ein Norgestrel-Derivat, wirkt antiandrogen. Neuere Norgestrel-Derivate (z.B. Desogestrel, Etonogestrel, Gestoden, Norgestimat) sind charakterisiert durch eine vollständig fehlende androgene Wirkung. Sie werden als orale Kontrazeptiva der dritten Generation eingesetzt.(4,31) Das wichtigste Anwendungsgebiet der Gestagene ist die hormonelle Kontrazeption. Weitere Indikationen für Gestagene oder Estrogen-Gestagen-Kombinationen sind dysfunktionelle Blutungen (z.B. langandauernde Blutungen bei anovulatorischem Zyklus), Dys- oder Polymenorrhoe, das prämenstruelle Syndrom, Mastodynie, Mastopathie, Menstruationsverschiebungen, die Therapie einer sekundären Amenorrhoe (nach sicherem Ausschluss einer Schwangerschaft), die palliative Therapie eines metastasierten Mamma- und Endometriumkarzinoms (z.B. mit Medroxyprogesteron-actetat oder Norethisteron-actetat), Endometriose (für die Therapie einer Endometriose sind relativ hohe Gestagendosen erforderlich; z.B. Norethisteron-actetat 10-20 mg/Tag ab 5. Zyklustag über sechs Monate, Dienogest oder GnRH-Analoga), sowie weibliche Androgenisierungserscheinungen (hierfür werden Präparate mit antiandrogener Teilwirkung eingesetzt, wie z.B. Dienogest, Cyproteron-actetat, Chlormadinonactetat).(4,31) 54 Die Kontraindikationen für die Anwendung von Gestagenen entsprechen nahezu gänzlich jenen der Estrogene und gelten somit auch für Estrogen-Gestagen-Kombinationen (siehe Kapitel 3.3.8.3). Gestagene mit antiandrogener Wirkung und 19-Nortestosteron-Derivate sind während einer Schwangerschaft kontraindiziert (Gefahr der Feminisierung bzw. Virilisierung des Fetus).(4) 3.3.5 Antigestagene und SPRM (selektive Progesteronrezeptor-Modulatoren) Man unterscheidet reine Antigestagene und SPRM (selektive Progesteronrezeptor-Modulatoren). SPRM fungieren als Liganden des Progesteron-Rezeptors, welche über Veränderung der Transkription von Progesteron-abhängigen Genen teils Progesteron-antagonistische, teils Progesteron-agonistische Wirkungen vermitteln. Neben dem Progesteron-Rezeptor binden sie auch an andere Steroidrezeptoren, wodurch entsprechende Partialwirkungen abzuleiten sind.(44) Der Gestagen-Antagonist und SPRM Mifepriston (auch RU-486 genannt) wird als Abortivum im Frühstadium der Schwangerschaft eingesetzt (50-200 mg), wobei es bei einem Einsatz vor der 7. Schwangerschaftswoche bei 80% der betroffenen Frauen zu einem kompletten Schwangerschaftsabbruch führt. Nach der 8. Schwangerschaftswoche reduziert sich die Abortrate jedoch auf ein Drittel. Eine Kombination mit Prostaglandinen führt bei fast 100% der Frauen zu einem kompletten Abort.(4,44) Ein weiterer SPRM ist Ulipristal-actetat, welches innerhalb von fünf Tagen postkoital zur Notfallkontrazeption zugelassen ist. Es ist somit länger wirksam, als das bisweilen eingesetzte Levonorgestrel (wirkt bis zu drei Tage). Ulipristal verhindert die Ovulation (Dosierung: 30 mg). In klinischen Studien konnte Ulipristal außerdem das Blutungsrisiko bei Myomen des Uterus signifikant senken und wird deshalb zunehmend bei Vorliegen eines Uterus myomatosus therapeutisch eingesetzt. Als Nebenwirkungen werden abdominale Schmerzen und Menstruationsstörungen beschrieben.(4,44) 3.3.6 Androgene Androgene werden in den Leydig-Zellen des Hodens, in den Nebennieren, sowie in Stroma- und Thekazellen des Ovars gebildet und in der Peripherie in geringen Mengen durch Aromatisierung zu Estrogenen umgewandelt. Androgene bestehen aus 19 Kohlenstoffatomen. Beim Mann zirkuliert vorwiegend Testosteron, in vielen Zielorganen ist jedoch seine reduzierte Form, das 5αDihydrotestosteron (DHT), biologisch wirksam. Die tägliche Testosteronsynthese beim erwachsenen Mann beträgt etwa 3-10 mg, wobei Serum-Testosteronkonzentrationen aufgrund der zirkadianen Rhythmik morgens um ca. 20-40% höher sind als am Abend. Ebenso spielen Umwelteinflüsse, zirkulierende SHBG-Konzentrationen, sowie posturale Effekte auf die hepatische Blutversorgung eine Rolle. Im Ovar werden hauptsächlich Androstendion und Dehydroepiandrosteron (DHEA) produziert. Die Testosteronproduktion der erwachsenen Frau beträgt im Normalfall etwa 5-10% der männlichen Testosteronproduktion (prämenopausal 200-800 ng/l).(4,22,45) 55 Dihydrotestosteron und Testosteron binden an den Androgenrezeptor bzw. bewirken alternativ, wie auch die anderen Sexualhormone, ebenso rasche zelluläre Effekte an der Zellmembran, eventuell über G-Protein-gekoppelte Rezeptoren, wobei diese Mechanismen nach wie vor ungeklärt sind. Die Testosteronwirkung besteht aus einem Zusammenspiel von Testosteron und seinen aktiven Metaboliten Estradiol und DHT. Sie determinieren die sexuelle Differenzierung der Geschlechtsorgane in der Embryonalphase, die Ausprägung sekundärer männlicher Geschlechtsmerkmale und die Funktion von Prostata und Samenblasen bzw. die Spermatogenese. Sexualunspezifische Wirkungen sind anabole Effekte auf Muskulatur und Knochenreifung, sowie die Stimulation der Hämatopoese.(4,45) Testosteron wird hauptsächlich hepatisch durch Oxidation am Kohlenstoffatom 17 zu Androstendion, Androsteron, Eticholanolon und Eticholandiol metabolisiert (sogenannte 17-KetoSteroide). Die meisten Metaboliten liegen als Sulfate oder Glucuronide vor, ihre Ausscheidung erfolgt renal.(4) 3.3.6.1 Pharmakologische Anwendung von Androgenen Zur Herstellung von Androgen-Depotpräparaten erfolgt eine Veresterung der OH-Gruppe in C-17Position mit langkettigen Fettsäuren z.B. Undecanoat oder Enantat. Testosteron-enantat (Testoviron®-Depot-250) wird intramuskulär alle zwei bis drei Wochen appliziert, um den Serumspiegel im Normbereich zu halten, Schwankungen sind jedoch häufig. Bei oraler Gabe wird Testosteron durch den First-Pass-Effekt der Leber vollständig metabolisiert; diesen Prozess umgeht man bei transdermaler Applikation von Testosteronpflastern oder Testosterongelen (Testogel ® 25 mg/50 mg, Androtop® Gel 50 mg). Das oral wirksame Testosteron-undecanoat (Andriol®) gelangt durch seine aliphatische Seitenkette teilweise über die Lymphe in die Zirkulation (Dosierung: zwei bis vier Kapseln à 40 mg/Tag).(4) Indikationen für die Verabreichung von Testosteron sind der primäre und sekundäre männliche Hypogonadismus, wobei sich für die Substitutionstherapie vor allem injizierbare Depotpräparate bewährt haben. Ein weiteres Anwendungsgebiet ist der off-label-Gebrauch bei übermäßigem Längenwachstum, meist im Alter von 12-16 Jahren bei einer zu erwartenden Körpergröße von zwei Metern (Testosteron-enantat 250 mg i.m./Woche oder 500 mg/zwei Wochen, für ein Jahr).(4) Untersuchungen zur Testosteron-Substitution bei älteren Männern mit einem physiologischen altersbedingten Testosteronabfall konnten bisher keine eindeutigen Resultate liefern. Eine 2016 veröffentlichte Analyse von sieben Placebo-kontrollierten Studien untersuchte die Auswirkungen einer Testosteron-Therapie auf die Sexualfunktion bei Männern ≥ 65 Jahren mit reduzierten Testosteron-Spiegeln (durchschnittlich <275 ng/dL) mit dem Ergebnis einer subjektiv signifikanten Verbesserung der Sexualaktivität, der Libido und der erektilen Funktion, jedoch ohne klare Evidenz von Testosteron-Schwellenwerten, welche zu signifikanten Verbesserungen führen.(46) Bekannte Nebenwirkungen bei Androgentherapien sind Störungen der Leberfunktion und Cholestase (vor allem bei Anwendung von C-17-alkylierten, oral wirksamen Androgenen wie z.B. 56 Methyltestosteron), Störungen des Elektrolythaushalts (Retention von Natrium, Kalium, Chlorid, Wasser, Calcium und anorganischem Phosphat) und Ödembildung. Kardiovaskuläre Nebenwirkungen und negative Einflüsse auf das Lipoproteinprofil werden vermutet, ihr Mechanismus ist derzeit jedoch noch unklar. Eine aktuelle Studie ergab ein erhöhtes Risiko von venösen Thromboembolien bei Testosteron-Therapie, vor allem in den ersten sechs Monaten. Bei Frauen führt eine Therapie mit Androgenen vermehrt zu Virilisierung mit verstärktem Haar- und Bartwuchs, sowie Akne, etc.(4,47) Kontraindikationen für die Anwendung von Androgenen sind eine bekannte Hyperplasie oder Tumore der Prostata und das Vorliegen einer Schwangerschaft.(4) 3.3.7 Antiandrogen wirksame Substanzen Indikationen für antiandrogen-wirksame Substanzen sind die Therapien von Erkrankungen, welche durch Androgene verursacht oder ungünstig beeinflusst werden, z.B. Tumoren oder Hyperplasie der Prostata, sowie die Therapie von Androgenisierungserscheinungen, z.B. Akne, androgene Alopezie oder Hirsutismus.(4,44) Antiandrogene Wirkungen beruhen auf einer selektiven Modulation der Androgenrezeptoren, einem kompetitiven Antagonismus oder der Inhibierung der DHT-Synthese. Beispielsweise verfügen Spironolacton (ein Aldosteronrezeptor-Antagonist), sowie einige Gestagene über antiandrogene Eigenschaften. Zur wachstumshemmenden Therapie des Prostatakarzinoms verwendet man z.B. Cyproteron-acetat (200-300 mg/Tag p.o. oder 300 mg/Woche i.m). Cyproteron-actetat ist ein 17-Hydroxyprogesteron-Derivat und führt zu einer kompetitiven Hemmung des Androgenrezeptors, sowie zu einer schwachen Hemmung der 5-α-Reduktase. Zur Therapie der Androgenisierung ist es nur in Kombination mit einem Estrogen zugelassen.(4,44) Androgenrezeptor-Antagonisten mit hochpotenten antiandrogenen Wirkungen sind die nicht- steroidalen Substanzen Flutamid, Bicalutamid und Nilutamid (in Europa nicht zugelassen), welche in Kombination mit GnRH-Analoga zur Therapie des metastasierenden Prostatakarzinoms eingesetzt werden. Die Kombination mit GnRH-Analoga bewirkt, neben der adrenalen Hemmung der Androgene durch das Antiandrogen, eine zusätzliche Hemmung der testikulären Androgene durch die nicht-pulsatile Gabe von GnRH und somit eine maximale Androgenblockade. Flutamid wurde jedoch eine hohe Hepatotoxizität nachgewiesen und Bicalutamid führte in einigen klinischen Studien zur Behandlung des lokalisierten Prostatakarzinoms zu höherer Mortalität und wird nicht mehr empfohlen.(4) Finasterid ist ein 5α-Reduktase-Hemmer und blockiert somit spezifisch die Umwandlung von Testosteron in Dihydrotestosteron durch das Enzym 5α-Reduktase-Typ-II. Finasterid wird zur Therapie der benignen Prostatahyperplasie (Dosierung: 5 mg) und bei männlicher androgenetischer Alopezie (Dosierung: 1 mg/Tag über zwei Jahre) eingesetzt, zur Prävention eines Prostatakarzinoms wird es nicht empfohlen. Ein weiterer 5α-Reduktase-Hemmer zur Behandlung der benignen Prostatahyperplasie ist Dutasterid, wobei dieses sowohl die 5α-Reduktase-Typ-II, als 57 auch –Typ-I hemmt (dualer Inhibitor). Seine Halbwertszeit beträgt drei bis fünf Wochen, es führt zu einer Größenverminderung der Prostata und verbessert die Diurese. Zugelassen ist Dutasterid zur Therapie und Fortschreitungsprophylaxe der benignen Prostatahyperplasie alleine (Dosierung: 0,5 mg) oder in Kombination mit Tamsulosin, einem α1-Adrenozeptor-Antagonist, sein Einsatz bei Androgenisierung ist ein „off-label use“.(4,44) Zu den häufigen Nebenwirkungen bei Antiandrogen-Therapie zählen Libidoverlust, Potenzstörungen, Gynäkomastie und Exantheme.(4) 3.3.8 Die hormonelle Kontrazeption Ziel und Hauptwirkung der hormonellen Kontrazeption ist die reversible funktionelle Sterilität. Generell wird die Wirksamkeit und Zuverlässigkeit von kontrazeptiven Methoden durch den sogenannten Pearl-Index angegeben. Er beschreibt die Wahrscheinlichkeit für eine Schwangerschaft, wenn 100 Frauen über ein Jahr eine bestimmte kontrazeptive Methode anwenden würden (Anzahl ungewollter Schwangerschaften pro 1200 Anwendungsmonate bzw. 100 Frauenjahre).(4,5) 3.3.8.1 Anwendungs- und Darreichungsformen Man unterscheidet im Allgemeinen orale und parenterale hormonelle Kontrazeptiva. Zu den oralen Kontrazeptiva zählen Estrogen-Gestagen-Kombinationspräparate, sowie reine Gestagen-Präparate (sogenannte Minipille), wobei die kombinierten Präparate einen besseren Schutz bieten (PearlIndex 0,1), als die reinen Gestagen-Präparate (Pearl-Index 0,5). Der Estrogen-Anteil der oralen Kontrazeptiva besteht meist aus Ethinylestradiol oder Mestranol (3-Methylether des Ethinylestradiols). Gestagen-Anteile sind entweder eine dem Norgestrel-ähnliche Substanz (z.B. Levonorgestrel, Norgestrel, Desogestrel, Gestoden, etc.) oder ein Norethisteron-Derivat (z.B. Lynestrenol, Dienogest, Etynodiol-diacetat, Noretynodrel, etc.) oder Derivate des Hydroxyprogesterons mit antiandrogenen Effekten (z.B. Cyproteron-acetat, Chlormadinon-actetat). Die drei Generationen der oralen Kontrazeptiva differenzieren sich hauptsächlich durch den unterschiedlichen Estrogengehalt: die erste Generation bestand aus mehr als 50 μg Ethinylestradiol, die zweite Generation aus etwa 30 μg Ethinylestradiol und in der dritten Generation konnte der Ethinylestradiol-Anteil auf 20-30 μg reduziert werden. Heutzutage werden vorwiegend Präparate mit einem Ethynilestradiolgehalt von unter 50 μg verwendet (sogenannte „Mikropillen“) und nur in Einzelfällen auf höhere Dosierungen zurückgegriffen. Parenterale Kontrazeptiva sind Depotpräparate aus reinen, hochdosierten, intramuskulär injizierbaren Gestagenen (z.B. Medroxyprogesteron, Norethisteron-enantat). Die Hauptwirkung bezieht sich auf eine Ovulationshemmung, sowie eine Veränderung des Cervicalsekrets und Endometriums, ihre Wirkdauer beträgt acht bis zwölf Wochen. Häufige Nebenwirkungen sind jedoch ein unregelmäßiger Zyklusablauf, Kopfschmerzen, Gewichtszunahme, Depression, sowie Verluste der Knochenmasse, weshalb Akzeptanz und Einsatz begrenzt sind. Prinzipiell sind sie nur dann 58 indiziert, wenn Methoden der oralen Kontrazeption oder ein Intrauterinpessar keine Alternativen darstellen.(4,31) Levonorgestrel oder Etonogestrel enthaltende und subkutan zu applizierende Implantate verfügen über eine kontrazeptive Wirkung von drei bis fünf Jahren, wobei ihr Nebenwirkungsprofil jenem der intramuskulären Depots entspricht. Sie führen zu keiner vollständigen Suppression der ovariellen Aktivität, wodurch ihr Vorteil in einer bestehenden basalen Estrogensekretion besteht und es somit zu keiner Veränderung der Knochendichte oder Endometriumatrophie kommt.(4,31) Intrauterinpessare (IUP, „Spirale“), die Levonorgestrel enthalten, bewirken eine sichere Kontrazeption (Pearl-Index 0,1), jedoch auch eine Endometriumatrophie mit konsekutiver Amenorrhoe. Serumkonzentrationen von Levonorgestrel betragen hierbei ein Fünftel des am niedrigsten dosierten oralen Kontrazeptivums, allerdings führt ihre Anwendung zu einem erhöhten Risiko für Uterusperforationen, ektope Gravidität und Zyklusunregelmäßigkeiten.(4,31) Transdermale hormonelle Pflaster (EVRA®-Pflaster) enthalten 0,6 mg Ethinylestradiol und 6 mg Norelgestromin (Metabolit von Norgestimat). Die täglichen Abgaberaten betragen etwa 20 μg Ethinylestradiol und 150 μg Norelgestromin, wobei das Pflaster 21 Tage getragen und anschließend 7 Tage pausiert wird. Die kontrazeptive Sicherheit ist vergleichbar mit oralen Präparaten.(4,31) Intravaginale Silastic-Ringe (Nuva Ring®) setzen täglich etwa 15 μg Ethinylestradiol und 120 μg Etonogestrel frei. Der Ring muss alle sieben Tage ausgetauscht werden, nach drei Wochen folgt eine Woche Pause. Der Pearl-Index dieser Methode beträgt 0,65.(4,31) 3.3.8.2 Unerwünschte Wirkungen Nebenwirkung und Komplikation von hormonellen Kontrazeptiva ist unter anderem ein erhöhtes Risiko für thrombembolische und kardiovaskuläre Ereignisse. Durch die Einnahme oraler Estrogene steigen die Konzentrationen bestimmter Gerinnungsfaktoren, wie Faktor VII und Faktor VIII, an und die Konzentrationen gerinnungshemmender Faktoren, wie z.B. Antithrombin, sinken dosisabhängig. Alle oralen Kontrazeptiva führen zu einem erhöhten Risiko für venöse Thromboembolien, unabhängig vom verwendeten Gestagen. Das Risiko für arterielle Thromboembolien, transitorische ischämische Attacken (TIA) und Apoplexien potenziert sich bei der Kombination von Nikotinabusus und der Einnahme von Estrogenen. Durch die niedrigen Estrogenanteile der modernen Kontrazeptiva kommt es zu keinem klinisch signifikanten Blutdruckanstieg; ein präexistenter Hypertonus ist jedoch ein zusätzlicher Risikofaktor für das Auftreten von Apoplexien bei Einnahme oraler Kontrazeptiva. Hoch dosierte Gestagene mit androgener Partialwirkung führen zu Veränderungen des Lipoproteinstoffwechsels mit einem ungünstigen Verhältnis von VLDL- zu HDL-Cholesterin. Bei neueren Gestagenen wie Levonorgestrel, Norgestimat, Desogestrel und Gestoden konnte ein geringer Triglyceridanstieg beobachtet werden. Die Kanzerogenität oraler Kontrazeptiva ist immer noch unzureichend geklärt. Als gesichert gilt, dass das relative Risiko für ein Endometriumkarzinom bei Einnahme 59 hormoneller Kontrazeptiva deutlich gesenkt wird und auch hinsichtlich des Ovarialkarzinoms eine protektive Wirkung besteht. Allerdings scheint das relative Risiko für Mammatumore erhöht zu sein. Hormonelle Kontrazeptiva können zu Leberzelladenomen führen, die sich aber nach Absetzen der Einnahme wieder zurückbilden. Die Inzidenz beträgt etwa 1:200 000. Messbare Veränderungen oder Einschränkungen der Leberfunktion konnten nicht nachgewiesen werden. Weitere unerwünschte Wirkungen hormoneller Kontrazeptiva sind Kopfschmerzen, Müdigkeit, Wasserretention und Ödembildung, Appetitsteigerung, Libidoverlust, Depressionen, Akne, Nausea und Emesis, cervicale Hypersekretion, Soorkolpitis, Dys-, Hyper-, Hypo- und Amenorrhoe.(4,43) Die molekularen Mechanismen der Wasserretention bei kombinierter oraler Kontrazeption konnten erst vor kurzem geklärt werden: über Histon-Code-Modifikationen wird die Transkriptionsaktivität von Mineralcorticoid-Rezeptoren reguliert.(48) 3.3.8.3 Kontraindikationen und Wechselwirkungen Absolute Kontraindikationen für die Anwendung hormoneller Kontrazeptiva sind eine bestehende Gravidität, kardiovaskuläre Erkrankungen und Risikofaktoren wie z.B. präexistente latente oder manifeste thrombembolische Prozesse, arterielle Hypertonie, Nikotinabusus, Migräne mit fokalen neurologischen Symptomen, Diabetes mellitus, eine dekompensierte, akute und schwere chronische Hepatitis, Cirrhosis Lebertumoren), hepatis, onkologische Leberstoffwechselstörungen Erkrankungen (Mammakarzinom, (Dubin-Johnson-Syndrom, maligne Rotor-Syndrom, idiopathischer Schwangerschaftsikterus, Schwangerschaftspruritus) und Herpes gestationis.(4,31) Zu den relativen Kontraindikationen zählen mäßige kardiovaskuläre Erkrankungen und Risikofaktoren (z.B. Hypercholesterinämie, medikamentös eingestellter Hypertonus, Thrombophlebitis, starke Varikosis), Epilepsie, Migräne ohne fokale neurologische Symptome und Stoffwechselerkrankungen (z.B. ausgeprägte Adipositas, gestörte Glucosetoleranz).(31) Bei gleichzeitiger Einnahme von Barbituraten, Phenytoin, Rifampicin oder Phenylbutazon kann es zur Induktion von Cytochrom-P450-Enzymen und somit zu einem beschleunigten Abbau hormoneller Kontrazeptiva (vor allem von Estrogenen) und damit zur Verminderung der kontrazeptiven Wirkung kommen.(4) 3.3.8.4 Estrogen-Gestagen-Kombinationspräparate Estrogen-Gestagen-Kombinationen werden in Einphasen-, abgestufte Einphasen- (Zwei- bzw. Dreistufenpräparate) und Zweiphasen- oder Sequentialpräpatate unterteilt. Sie unterscheiden sich sowohl in der Zusammensetzung, als auch in der Dosierung der hormonellen Teilkomponenten.(4) Einphasenpräparate (klassische Kombinationspräparate) enthalten konstante Konzentrationen von Estrogenen und Gestagenen über den gesamten Einnahmezyklus. Sie bewirken eine Hemmung der Gonadotropinsekretion aus der Adenohypophyse und des Ovulations-auslösenden LH-Peaks, wobei die Estrogenkomponente hauptsächlich die Freisetzung von FSH, sowie die Selektion des dominanten Follikels hemmt und die Gestagenkomponente die LH-Sekretion inhibiert. Eine Ovulationshemmung kann auch mit reinen Gestagenpräparaten erreicht werden (siehe Kapitel 60 3.3.7.4). Vorteile der zusätzlichen Estrogenkomponente sind jedoch die stabilisierende Wirkung auf das Endometrium, die Verhinderung von uterinen Blutungen (sogenannten Druchbruchblutungen) und die Induzierung der Expression von Progesteron-Rezeptoren als Voraussetzung für die Wirksamkeit des Gestagens.(4) Bei abgestuften Einphasenpräparaten (Zwei- und Dreistufenpräparate) variiert die Dosis von Estrogenen und Gestagenen im Verlauf des Einnahmezyklus. Zweistufenpräparate sind durch eine niedrige Gestagendosis in den ersten elf Tagen und konstante Estrogendosen charakterisiert. Dreistufenpräparate sollen sich durch die Variierung von Gestagen- und Estrogendosen noch mehr dem natürlichen Zyklus annähern.(4) Zweiphasenpräparate imitieren ebenso den physiologischen Verlauf des Zyklus, indem während der ersten sieben bis elf Tage nur estrogenhaltige Tabletten, in weiterer Folge Estrogen-GestagenKombinationen eingenommen werden. Bei diesen Präparaten beruht der Effekt der Ovulationshemmung hauptsächlich auf dem Estrogen-Anteil, wodurch die meisten verfügbaren Pharmaka tägliche Ethinylestradioldosen von 50 μg enthalten und auf mögliche Nebenwirkungen besonders zu achten ist.(4) 3.3.8.5 Reine Gestagen-Präparate Die kontrazeptive Wirkung von niedrigen Gestagendosen (sogenannte „Minipille“) resultiert einerseits aus der hemmenden Wirkung auf die gonadotrope Funktion der Hypophyse und andererseits aus der Gestagen-vermittelten Veränderung des Cervicalsekrets, welche die Spermienaszension erschwert, sowie durch Implantations-inhibierende Effekte auf das Endometrium. Bei nur etwa 30% der Frauen wird die Ovulation gehemmt; eine Ausnahme bildet das reine Gestagen-Präparat Desogestrel (Cerazette®), welches kontinuierlich eingenommen wird und antiovulatorisch wirkt.(4,31) Vorteil der Minipille ist die geringe Hormonbelastung, sowie der komplette Verzicht auf die estrogene Wirkkomponente. Nachteile sind eine verminderte kontrazeptive Sicherheit, eine schlechtere Zykluskontrolle und die Notwendigkeit der zeitgenauen Einnahme.(4,31) 3.3.8.6 Die hormonelle Kontrazeption des Mannes Kontrazeptive Methoden für Männer beschränken sich derzeit auf Präservative (Pearl-Index zwischen 2 und 12) und die Sterilisation durch die chirurgische Durchtrennung der Ductus deferentes; die Entwicklung einer sicheren und reversiblen hormonellen Kontrazeption für den Mann ist bis heute nicht gelungen.(31) Bisherige klinische Studien zeigten, dass die Gabe von Testosteron ähnliche und vergleichbare kontrazeptive Effekte aufweist, wie angewandte weibliche Kontrazeptiva, allerdings mussten bisher sehr hohe Testosteron-Konzentrationen zugeführt werden, welche das Risiko für potentielle Nebenwirkungen erheblich steigen ließen.(49) Eine erst kürzlich veröffentlichte, prospektive und multizentrische Phase-II-Studie mit dem Ziel eine neue kontrazeptive Methode für den Mann mittels geringer Testosteron-Konzentration zu 61 entwickeln, ergab, dass die Injektion der Kombination Testosteron-Undecanoat mit dem Gestagen Norethisteronenanthat die Spermiogenese wirksam und reversibel unterdrückt. Der Pearl-Index dieser Methode wurde auf 2,18 berechnet. 320 gesunde Männer im Alter zwischen 18 und 45 Jahren wurden an insgesamt zehn Zentren in Deutschland, Italien, Großbritannien, Australien, Chile, Indien und Indonesien in die Studie einbezogen. Die Studienteilnehmer erhielten intramuskuläre Injektionen aus 200 mg Norethisteronenanthat und 1000 mg TestosteronUndecanoat in 8-wöchigen Abständen über insgesamt 82 Wochen, gefolgt von einer Erholungsphase von bis zu einem Jahr. Die 2008 begonnene Studie wurde allerdings, aufgrund unerwünschter Nebenwirkungen wie das Auftreten von Akne, Schmerzen an der Injektionsstelle, gesteigerte Libido, Dysphorie und Depressionen, teilweise schon 2011 beendet. Es konnte aber gezeigt werden, dass die Anzahl der Spermien signifikant und längerfristig supprimiert werden kann. Weitere Studien in diese Richtung sind somit in Zukunft zu erwarten.(49) 62 4 Diskussion Endokrine und neuroendokrine Prozesse sind komplex und multifaktoriell gesteuert. Im Diskurs um die hormonelle Steuerung des menschlichen Organsimus sind viele der beschriebenen Vorgänge immer noch unzureichend erforscht; trotz unzähliger neuer Studien bestehen wesentliche Wissensdefizite um Zusammenhänge aufzuzeigen und empirisch gesicherte Aussagen treffen zu können. Verschiedenste Komponenten, genetische Grundlagen und Funktionen der endokrinen Regulationsprozesse konnten teilweise bereits konstatiert werden. In Zukunft werden Innovationen wie genetische Sequenzierungen das Wissen über physiologische und pathologische Vorgänge endokriner Achsen noch erweitern. Fakt ist, dass neuroendokrine Achsen und Regelkreise, wie sie bisher gelehrt und in der Literatur beschrieben wurden, nur einzelne Anhaltspunkte symbolisieren und physiologische Mechanismen zudem nur sehr vereinfacht dargestellt werden können. Viele neue klinische Studien beschäftigen sich bereits heute mit den Einflüssen von Hormonen nicht nur in Bezug auf den Hypothalamus, sondern ebenso auf andere Gehirnregionen und deren kognitive und emotionale Funktionen unter Berücksichtigung des Lebensalters, der sozialen Situation und psychischer Faktoren. Dadurch werden neuroendokrine, autonome, metabolische Systeme, sowie Immunsysteme zusehends verknüpft. Epigenetische Mechanismen in Berücksichtigung genomischer, sowie nicht-genomischer Prozesse spielen hierbei eine zentrale Rolle.(50) Die Erkenntnis, dass Hormonrezeptoren meist nicht spezifisch ein einzelnes Hormon binden, führt zunehmend zu der Ansicht, dass auch viele Co-Aktivatoren und Co-Faktoren die Wirkung der einzelnen Hormonrezeptoren beeinflussen, wie es beispielsweise bei der Aktivierung von Estrogenund Progesteron-Rezeptoren der Fall ist. Diese Einsichten sind unter anderem Anhaltspunkte für die Entwicklung neuer Pharmaka.(41) Spannend bleibt, wie zukünftige Verhaltensweisen und Gesellschaftsstrukturen das menschliche Endokrinum beeinflussen und verändern werden, wobei hier der generelle Lebensstil eines Individuums eine zentrale Rolle spielt. Unsere Gesellschaft wird nachweislich älter, jedoch nicht gesünder; Krankheiten wie Adipositas, Diabetes mellitus und Osteoporose sind im Zunehmen und ihre Auswirkungen auf das Hormonsystem derzeit nur teilweise abschätzbar. Beispielsweise führt Adipositas bei Frauen zu Zyklusunregelmäßigkeiten, welche auf endokrine Veränderungen zurückzuführen sind, wobei sich diesbezüglich hohe Konzentrationen von Insulin, Testosteron und anderen Androgenen, sowie erniedrigte Konzentrationen von SHBG negativ auf das weibliche Endokrinum auswirken.(51) Das hormonelle System, die Auswirkungen seiner Dysfunktion, sowie die vielseitigen pharmakologischen Ansatzpunkte sind in jedem Fall nicht zu unterschätzen und werden in Zukunft weiterhin an medizinischer, sowie sozioökonomischer Relevanz gewinnen. 63 5 Literaturverzeichnis (1) Kronenberg HM, Melmed S, Larsen PR, Polonsky KS. Principles of Endocrinology. In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, editors. Williams Textbook of Endocrinology. 13th ed. Philadelphia: Elsevier; 2016. p. 2-11. (2) Vaupel P, Schaible H, Mutschler E. Hormonelles System. In: Vaupel P, Schaible H, Mutschler E, editors. Anatomie, Physiologie, Pathophysiologie des Menschen. 7th ed. Stuttgart: Wissenschaftliche Verlagsgesellschaft Stuttgart; 2015. p. 565-626. (3) Ritter M. Hormone. In: Speckmann E, Hescheler J, Köhling R, editors. Physiologie. 6th ed. München: Elsevier Urban&Fischer; 2013. p. 691-744. (4) Aktories K, Förstermann U, Hofmann FB, Starke K. Allgemeine und spezielle Pharmakologie und Toxikologie. 11th ed. München: Elsevier Urban&Fischer; 2013. (5) Krieg J, et al. Regulation der Fortpflanzungsfunktion. In: Kaufmann M, Costa SD, Scharl A, editors. Die Gynäkologie. 3rd ed. Berlin Heidelberg: Springer Medizin; 2013. p. 73-77. (6) Harno E, White A. Adrenocorticotropic Hormone. In: Jameson JL, De Groot LJ, editors. Endocrinology - Adult and Pediatric, Volume I. 7th ed. Philadelphia: Elsevier Saunders; 2016. p. 129-146. (7) Toufexis D, Rivarola M, Lara H, Viau V. Stress and the reproductive axis. J Neuroendocrinol 2014 Sep;26(9):573-586. (8) Kleine B, Rossmanith W editors. Hormone und Hormonsysteme - Lehrbuch der Endokrinologie. 3rd ed. Berlin, Heidelberg: Springer Verlag; 2014. (9) Anderhuber F, Pera F, et al. editors. Waldeyer - Anatomie des Menschen. Lehrbuch und Atlas in einem Band. 19th ed. Berlin, Boston: De Gruyter; 2012. (10) Welt CK. Hypothalamic-pituitary axis. 2016; Available at: https://www.uptodate.com/contents/hypothalamic-pituitary-axis. Accessed Nov 21st, 2016. (11) Schünke M, Schulte E, Schumacher U. Prometheus - Kopf,Hals und Neuroanatomie. 4th ed. Stuttgart: Thieme; 2015. (12) Murray PG, Higham CE, Clayton PE. 60 YEARS OF NEUROENDOCRINOLOGY: The hypothalamo-GH axis: the past 60 years. J Endocrinol 2015;226(2):T123-T140. (13) Brcic L, Heidinger M, Popper H. Neuroendokrine Tumoren des Mediastinums. Pathologe 2016;37(5):434-440. (14) Fekete C, Lechan RM. Central Regulation of Hypothalamic-Pituitary-Thyroid Axis Under Physiological and Pathophysiological Conditions. Endocr Rev 2014 Apr;35(2):159-194. (15) Dadak C editor. Sexualität, Reproduktion, Schwangerschaft, Geburt. 8th ed. Wien: Facultas; 2015. (16) Fourman LT, Fazeli PK. Neuroendocrine Causes of Amenorrhea—An Update. J Clin Endocrinol Metab 2015 Mar;100(3):812-824. 64 (17) Strowitzki T. Die pulsatile Gonadotropin-Releasing-Hormon-Behandlung. Gynäkologische Endokrinologie 2016 Sep;14:245-248. (18) Wörmann B, Wolff JM. Die systemische Therapie des metastasierten Prostatakarzinoms. Urologe 2010 Feb;49:221-227. (19) Wolff JM. GnRH-Antagonisten- Eine neue Therapieoption beim fortgeschrittenen Prostatakarzinom. Aktuel Urol 2009 Apr;40(3):159-163. (20) Oswald J. Leitlinienempfehlung zur Therapie des Kryptorchismus, der Hydrocele und der Varicocele. Pädiatrie & Pädologie 2008;2:24-28. (21) Hannon MJ, Thompson CJ. Vasopressin, Diabetes insipidus, and the Syndrome of Inappropriate Antidiuresis. In: Jameson JL, De Groot LJ, editors. Endocrinology - Adult and Pediatric, Volume I. 7th ed. Philadelphia: Elsevier Saunders; 2016. p. 298-311. (22) Lothar T. Labor und Diagnose. 8th ed. Frankfurt: TH-Books; 2012. (23) Pfeifer A, Ditzen B, Neubauer E, Schiltenwolf M. Wirkung von Oxytocin auf das menschliche Schmerzerleben. Schmerz 2016 Oct;30(5):457-469. (24) Voigt F, Farrokh A, et al. Tokolyse- Update 2016. Gynäkologe 2016;49(3):201-211. (25) Aguirre G.A., et al. Insulin-like growth factor-1 deficiency and metabolic syndrome. J Transl Med 2016;14:3-25. (26) Christiansen JS, et al. Growth Hormone Research Society perspective on the development of long-acting growth hormone preparations. Eur J Endocrinol 2016 Jun;174(6):C1-C8. (27) Van der Lely AJ, et al. Treatment with high doses of pegvisomant in 56 patients with acromegaly: experience from ACROSTUDY. Eur J Endocrinol 2016 Oct;175(4):239-245. (28) Bervini S, Valente L, Chris E. Neues von Prolaktin. Gynäkologische Endokrinologie 2016;4:239-244. (29) Schott M. Hyperthyreose. Internist 2013 Mar;54(3):315-327. (30) Okosieme O, et al. Management of primary hypothyroidism: statement by the British Thyroid Association Executive Committee. Clin Endocrinol 2016 Jun;84(6):799-808. (31) Kaufmann M, Costa SD, Scharl A. Die Gynäkologie. 3rd ed. Berlin Heidelberg: Springer Medizin; 2013. (32) Fensore S, et al. Corifollitropin alfa compared to daily FSH in controlled ovarian stimulation for in vitro fertilization: a meta-analysis. J Ovarian Res 2015;8:33-41. (33) Ross DS. Treatment of hypothyroidism. 2016; Available at: https://www.uptodate.com/contents/treatment-of-hypothyroidism. Accessed Oct 15th, 2016. (34) Jonklaas J, Davidson B, Bhagat S, Soldin SJ. Triiodothyronine Levels in Athyreotic Individuals During Levothyroxine Therapy. JAMA 2008 Feb;299(7):769-777. 65 (35) Grozinsky-Glasberg S, Fraser A, Nahshoni E, Weizman A, Leibovici L. ThyroxineTriiodothyronine Combination Therapy Versus Thyroxine Monotherapy for Clinical Hypothyroidism: Meta-Analysis of Randomized Controlled Trials. J Clin Endocrinol Metab 2006 Jul;91(7):2592-2599. (36) Nieman LK. Pharmacologic use of glucocorticoids. 2016; Available at: https://www.uptodate.com/contents/pharmacologic-use-of-glucocorticoids. Accessed Nov 21st, 2016. (37) Saag KG, Furst DE. Major side effects of systemic glucocorticoids. 2016; Available at: https://www.uptodate.com/contents/major-side-effects-of-systemic-glucocorticoids. Accessed Nov 21st, 2016. (38) Christiansen C, Christensen S, Mehnert F, Cummings S, Chapurlat R, Sørensen H. Glucocorticoid Use and Risk of Atrial Fibrillation or Flutter: A Population-Based, Case-Control Study. Arch Intern Med 2009;169(18):1677-1683. (39) Arai K, Homma T, Morikawa Y, Ubukata N, Tsuruoka H, Aoki K, et al. Pharmacological profile of CS-3150, a novel, highly potent and selective non-steroidal mineralocorticoid receptor antagonist. Eur J Pharmacol 2015 Aug;761:226-234. (40) Plant TM. 60 YEARS OF NEUROENDOCRINOLOGY: The hypothalamo-pituitary–gonadal axis. J Endocrinol 2015 Aug;226(2):T41-T54. (41) Weigel NL, Smith CL. Estrogen and Progesterone Actions. In: Jameson JL, De Groot LJ, editors. Endocrinology - Adult and Pediatric, Volume II. 7th ed. Philadelphia: Elsevier Saunders; 2016. p. 2207-2215. (42) McGee EA, Strauss III JF. Ovarian Hormone Synthesis. In: Jameson JL, De Groot LJ, editors. Endocrinology - Adult and Pediatric, Volume II. 7th ed. Philadelphia: Elsevier Saunders; 2016. p. 2192-2206. (43) Herrero S, Pico Y. Treatments for post-menopausal osteoporotic women, what`s new? How can we manage long-term treatment? Eur J Pharmacol 2016 May;779:8-21. (44) Von Wolff M, Stute P. Gynäkologische Endokrinologie und Reproduktionsmedizin. 1st ed. Stuttgart: Schattauer; 2013. (45) Handelsman DJ. Androgen Phiosiology, Pharmacology and Abuse. In: Jameson JL, De Groot LJ, editors. Endocrinology - Adult and Pediatric, Volume II. 7th ed. Philadelphia: Elsevier Saunders; 2016. p. 2368-2393. (46) Cunningham GR, Stephens-Shields A, et al. Testosterone Treatment and Sexual Function in Older Men With Low Testosterone Levels. J Clin Endocrinol Metab 2016 Aug;101(8):3096-3104. (47) Martinez C, Suissa S, et al. Testosterone treatment and risk of venous thromboembolism: population based case-control study. BMJ 2016 Nov;355:i5968. (48) Igunnu A, Seok Y, Olatunji LA, Kang S, Kim I. Combined oral contraceptive synergistically activates mineralocorticoid receptor through histone code modifications. Eur J Pharmacol 2015 Dec;769:48-54. (49) Behre HM, et al. Efficacy and Safety of an Injectable Combination Hormonal Contraceptive for Men. J Clin Endocrinol Metab 2016;101(12):4479-4788. 66 (50) McEwen BS, Gray JD, Nasca C. 60 YEARS OF NEUROENDOCRINOLOGY: Redefining neuroendocrinology: stress, sex and cognitive and emotional regulation. J Endocrinol 2015 Aug;226(2):T67-T83. (51) Schäfer-Graf U. Adipositas und Schwangerschaft. Diabetologe 2016;12:6-12. (52) Sonntag B. Zyklusstörungen- Diagnostik und Therapie. Gynäkologe 2016;49:357-372. 67