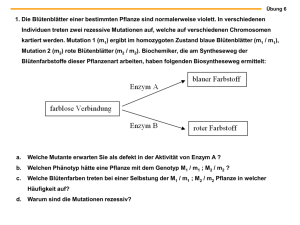
Aufklärung des enzymatischen Mechanismus der L-Fuculose
Werbung

Aufklärung des enzymatischen Mechanismus der L-Fuculose-1-phosphat-Aldolase aus Escherichia coli durch ortsgerichtete Mutagenese und röntgenographische Untersuchungen INAUGURALDISSERTATION zur Erlangung der Doktorwürde der Fakultät für Chemie und Pharmazie der Albert-Ludwigs-Universität Freiburg im Breisgau vorgelegt von Diplom-Chemiker Andreas C. Jörger Freiburg im Breisgau Dezember 1999 Dekan: Prof. Dr. R. Schubert Referent: Prof. Dr. G. E. Schulz Korreferent: Prof. Dr. D. Jahn Tag der Bekanntgabe des mündlichen Prüfungsergebnisses: 17.02.2000 1 Inhaltsverzeichnis 1 Einleitung............................................................................................................................................................. 5 1.1 Aldolasen ..................................................................................................................................................... 5 1.2 Aldolasen und ihre Bedeutung als Biokatalysatoren in der organischen Chemie......................................... 8 1.3 L-Fuculose-1-phosphat-Aldolase aus Escherichia coli ................................................................................ 9 1.4 Die Röntgenstruktur der L-Fuculose-1-phosphat-Aldolase........................................................................ 11 1.5 Ziele der Arbeit .......................................................................................................................................... 13 2 Materialien ......................................................................................................................................................... 14 2.1 Geräte......................................................................................................................................................... 14 2.2 Chemikalien, Enzyme und Kits.................................................................................................................. 15 2.3 Bakterienstämme und Vektoren ................................................................................................................. 17 2.4 Lösungen, Puffer und Nährmedien............................................................................................................. 19 3 Methoden ........................................................................................................................................................... 25 3.1 Gentechnische Methoden ........................................................................................................................... 25 3.1.1 Reinigung von Oligonukleotiden........................................................................................................ 26 3.1.2 Spaltung von DNA mit Restriktionsendonukleasen ........................................................................... 27 3.1.3 Agarose-Gelelektrophorese ................................................................................................................ 28 3.1.4 Ligation von DNA-Fragmenten.......................................................................................................... 29 3.1.5 Transformation ................................................................................................................................... 29 3.1.6 Präparation von Plasmid-DNA und M13-dsDNA .............................................................................. 31 3.1.7 Präparation von M13ss-DNA ............................................................................................................. 31 3.1.8 Präparation von Uracil-haltiger M13-ssDNA..................................................................................... 32 3.1.9 Mutagenese......................................................................................................................................... 33 3.1.10 DNA-Sequenzierung ........................................................................................................................ 36 3.2 Proteinpräparation ...................................................................................................................................... 37 3.2.1 Stammkulturen und Expressionstests ................................................................................................. 37 3.2.2 Bakterienkultivierung ......................................................................................................................... 39 3.2.3 Bakterienaufschluß und Abtrennung von Nukleinsäuren ................................................................... 39 3.2.4 Anionenaustauscher-Chromatographie............................................................................................... 39 3.2.5 Gelpermeations-Chromatographie...................................................................................................... 40 3.2.6 Dialyse................................................................................................................................................ 41 3.2.7 Konzentrierung von Proteinlösungen ................................................................................................. 41 3.3 Proteinanalytik ........................................................................................................................................... 42 3.3.1 Diskontinuierliche SDS-Polyacrylamid-Gelelektrophorese ............................................................... 42 3.3.2 Isoelektrische Fokussierung (IEF)...................................................................................................... 42 3.3.3 Bestimmung der Proteinkonzentration ............................................................................................... 43 3.3.4 Röntgenfluoreszenz-Spektroskopie .................................................................................................... 44 3.3.5 Bestimmung der enzymatischen Aktivität .......................................................................................... 44 3.3.6 Bestimmung der kinetischen Parameter.............................................................................................. 47 3.4 Kristallisation und Röntgenstrukturanalyse................................................................................................ 49 3.4.1 Kristallisation nach der Hängetropfenmethode .................................................................................. 50 3.4.2 Kristallmontage .................................................................................................................................. 51 3.4.3 Kristallsysteme ................................................................................................................................... 51 3.4.4 Theorie der Röntgenstreuung ............................................................................................................. 52 3.4.5 Reziprokes Gitter und Ewald-Konstruktion ....................................................................................... 55 3.4.6 Flächenzählermessung und Auswertung............................................................................................. 56 3.4.7 Skalierung von Datensätzen ............................................................................................................... 59 3.4.8 Strukturfaktor und Fourier-Transformation........................................................................................ 59 3.4.9 Temperaturfaktor................................................................................................................................ 61 3.4.10 Modellbau und Elektronendichtekarten............................................................................................ 62 3.4.11 Verfeinerung..................................................................................................................................... 63 3.4.12 Verfeinerung mit X-PLOR ............................................................................................................... 66 3.4.13 Verfeinerung mit REFMAC ............................................................................................................. 68 3.4.14 Darstellung von Proteinstrukturen .................................................................................................... 68 2 4 Ergebnisse und Diskussion ................................................................................................................................ 70 4.1 Planung der Mutanten ................................................................................................................................ 70 4.1.1 Mutationen der postulierten katalytischen Reste ................................................................................ 70 4.1.2 Mutation am flexiblen Loop(23-27) ................................................................................................... 73 4.1.3 Mutationen am flexiblen C-terminalen Kettenende............................................................................ 74 4.1.4 Mutationen zur Veränderung der Stereospezifität der Reaktion......................................................... 75 4.1.5 Mutationen zur Veränderung der Selektivität auf Seite der DHAP-Komponente .............................. 77 4.2 Mutagenese ................................................................................................................................................ 81 4.2.1 Wahl der Mutagenese-Oligonukleotide .............................................................................................. 81 4.2.2 Mutagenese nach Eckstein bzw. Kunkel ............................................................................................ 81 4.2.3 Nachweis der Mutationen auf DNA-Ebene ........................................................................................ 84 4.2.4 Rückklonierung und dsDNA-Sequenzierung...................................................................................... 85 4.3 Proteinpräparation der Mutanten................................................................................................................ 86 4.3.1 Expressionstest und Bakterienkultivierung......................................................................................... 86 4.3.2 Ionenaustauscher-Chromatographie ................................................................................................... 88 4.3.3 Gelpermeations-Chromatographie...................................................................................................... 91 4.3.4 Isoelektrische Fokussierung ............................................................................................................... 92 4.3.5 Spektroskopische Untersuchung der Mutante Y113F ........................................................................ 93 4.4 Kristallisation ............................................................................................................................................. 96 4.4.1 Kristallform T..................................................................................................................................... 96 4.4.2 Weitere Kristallformen ..................................................................................................................... 101 4.5 Röntgenstrukturanalyse der FucA-Mutanten............................................................................................ 103 4.5.1 Das C-terminale Kettenende............................................................................................................. 106 4.5.2 Struktur der Mutanten Y113F und Y113F/Y209F ........................................................................... 109 4.5.3 Struktur der Mutante E73S ............................................................................................................... 111 4.5.4 Struktur der Mutante E73Q .............................................................................................................. 113 4.5.5 Struktur der Mutante E73Q/Y113F/Y209F ...................................................................................... 116 4.5.6 Strukturen bei Mutationen im flexiblen Loop(23-27) ...................................................................... 118 4.5.7 Struktur der Mutante F131A ............................................................................................................ 121 4.5.8 Die Phosphatbindungstasche der Mutanten N29L/S71A, N29Q und S71Q ..................................... 122 4.6 Akzeptanz nicht-natürlicher Substrate...................................................................................................... 131 4.7 Ergebnisse der Enzymkinetik für L-Fuculose-1-phosphat........................................................................ 133 4.7.1 Kinetische Parameter des Wildtyps .................................................................................................. 134 4.7.2 Mutationen am flexiblen C-terminalen Kettenende und an den postulierten katalytischen Resten .. 138 4.7.3 Mutationen in der hydrophoben Wand ............................................................................................. 141 4.7.4 Mutationen im flexiblen Loop(23-27) und in der Phosphatbindungstasche ..................................... 141 4.7.5 Modell für das Binden von L-Fuculose-1-phosphat und die Rolle des C-terminalen Kettenendes bei der Katalyse................................................................................................................................ 143 4.7.6 Vergleich mit anderen Aldolasen ..................................................................................................... 145 4.7.7 Vergleich mit der L-Ribulose-5-phosphat-4-Epimerase................................................................... 148 4.8 Selektivität von FucA-Mutanten in bioorganischen Synthesen................................................................ 152 5 Zusammenfassung und Ausb lick ........................................................................................................... 157 6 Literatur ........................................................................................................................................................... 159 7 Anhang............................................................................................................................................................. 169 7.1 DNA- und Aminosäuresequenz der FucA wie in M13mp19 kloniert ...................................................... 169 7.2 Vektoren................................................................................................................................................... 170 3 Abkürzungen A260 APS BCIP B-Faktor β-ME BFG BHP BSA dNTP ddNTP DIG DHA DHAP dsDNA EDTA ELISA Fuc1P FucA FruA(I), FruA(II) GDH GPC GPDH GA3P IEC IPTG kb LDH NBT NCS MES OD578 PAGE PEG PGH RhuA rmsd Rcryst Rfree RF Ru5P RNAse RT SDS ssDNA Taq TEMED Tris ü. N. Vt WT X-Phosphat Xu5P Absorption bei einer Wellenlänge von 260 nm Ammoniumperoxodisulfat 5-Brom-4-chlor-3-indolylphosphat kristallographischer Temperaturfaktor β-Mercaptoethanol Bakterienfeuchtgewicht β-Hydroxypyruvat Rinderserumalbumin (Bovine Serum Albumin) 2'-Desoxynukleosid-5'-triphosphat 2',3'-Didesoxynukleosid-5'-triphosphat Digoxigenin Dihydroxyaceton Dihydroxyacetonphosphat doppelsträngige DNA Ethylendiamintetraacetat Enzym-gekoppelter Immunadsorptionsnachweis (Enzyme-Linked ImmunosorbantAssay) L-Fuculose-1-phosphat L-Fuculose-1-phosphat-Aldolase Fructose-1,6-bisphosphat-Aldolase Klasse-I/Klasse-II Glycerin-Dehydrogenase Gelpermeations-Chromatographie Glycerinphosphat-Dehydrogenase Glyceraldehyd-3-phosphat Ionenaustauscher-Chromatographie (Ionic Exchange Chromatography) Isopropyl-β-D-thiogalactopyranosid Kilobasen Lactat-Dehydrogenase Nitroblue-tetrazoliumchlorid Nicht-kristallographische Symmetrie (Non-Crystallographic Symmetry) Morpholinoethansulfonsäure Optische Dichte bei einer Wellenlänge von 578 nm Polyacrylamidgel-Elektrophorese Polyethylenglykol Phosphoglycolohydroxamat L-Rhamnulose-1-phosphat-Aldolase mittlere quadratische Abweichung (root mean square deviation) kristallographischer R-Faktor (konventionell) freier R-Faktor replikative Form Ribulose-5-phosphat Ribonuklease Raumtemperatur Natriumdodecylsulfat (Sodium Dodecyl Sulfate) einzelsträngige DNA (singlestrand DNA) Thermus aquaticus N,N,N',N'-Tetramethylethylendiamin Tris-(hydroxymethyl)-aminomethan über Nacht Säulenvolumen Wildtyp 5-Brom-4-chlor-3-indoxyl-phosphat Xylulose-5-phosphat Abkürzungen, die im European Journal of Biochemistry ohne Definition verwendet werden, sind nicht aufgeführt (Information for Authors (1996). Eur. J. Biochem. 235, 1-7). 4 Erläuterung englischer Fachtermini Glossar alignment Ausrichtung ähnlicher Aminosäuresequenzen mit eingefügten Lücken annealing Zusammenlagerung zweier komplementärer einzelsträngiger Nukleinsäureketten zu einem Doppelstrang backbone Rückgrat; die Hauptkette des Proteins barrel Faß; z. B. β-Faltblatt mit faßartiger Struktur brass plate Messingplatte blotting Transfer von DNA oder Proteinen von einem Gel auf eine Trägermembran bulk solvent correction Näherungsweise Beschreibung der Solvensbereiche im Kristall für das Modell constrained Festgehalten; eine Abweichung ist nicht zugelassen cluster Anhäufung von Punkten flood field Anordnung zur Intensitätseichung des Flächenzählers frame Rotationsaufnahme über einen bestimmten Winkelbereich hanging drop Hängetropfen induced fit Umfassende Veränderung der Proteinstruktur ausgelöst durch Substratbindung interface Kontaktregion label Markierung eines Moleküls zur Detektion loop Schleife innerhalb einer Polynukleotid- oder Polypeptidkette least squares kleinste Quadrate map Elektronendichtekarte maximum likelihood maximierte Wahrscheinlichkeit model bias Einfluß des eingesetzten Modells nicking Einzelstrangbruch durch Spaltung einer Phosphodiesterbindung innerhalb doppelsträngiger DNA omit map Elektronendichtekarte, welche unter Weglassen eines Teils des Modells berechnet wurde. overfitting unkontrollierte Modelländerung bei zu vielen freien Parametern peak relativer oder absoluter Maximalwert primer kurzes DNA- oder RNA-Stück, von dem aus DNA-Polymerasen den zu der einzelsträngigen Matrize komplementären Strang synthetisieren rendering Photorealistische Verbesserung von Graphiken durch Beleuchtung restrained Eingeschränkt; eine Abweichung ist bei Überwindung einer festgesetzten Energiebarriere möglich ribbon Band; Bänderdarstellung der Sekundärstrukturelemente in Proteinen rigid body Starrer Körper bei gleichnamiger Verfeinerungsprozedur screening Reihentests; Überprüfung eines weiten Feldes von Möglichkeiten durch ein sinnvolles Netz von Stichproben. sheet Sekundärstrukturelement von Proteinen: Faltblatt simulated annealing Simuliertes Aufheizen und Abkühlen eines Proteinmodells multiple cloning site Polyklonierungsstelle; kurzer DNA-Abschnitt auf Vektoren, der viele Restriktionsschnittstellen aufweist und sich somit zum Einklonieren von Fremd-DNA eignet torsion angle refinement Verfeinerung durch Änderung von Torsionswinkeln definierter Atomgruppe 5 1 Einleitung 1.1 Aldolasen Aldolasen sind ubiquitäre Enzyme, die zur Enzymklasse der C-C-verknüpfenden Lyasen zählen. Sie ermöglichen aldolartige Additionen (bzw. Spaltungen) von Aldehyden an kleine Kohlenstofffragmente und katalysieren zentrale Schritte im Stoffwechsel (Horecker et al., 1972). Derzeit sind etwa 40 verschiedene Aldolasen bekannt (Enzyme Handbook, 1998), von denen die Mehrheit am Metabolismus von Kohlenhydraten, Aminosäuren oder Hydroxysäuren beteiligt ist. Der bekannteste und am besten untersuchte Vertreter ist die Fructose-1,6-bisphosphatAldolase (FruA), die Fructose-1,6-bisphosphat zu Dihydroxyacetonphosphat (DHAP) und Glyceraldehyd-3-phosphat spaltet (Glykolyse) bzw. im Rahmen der Gluconeogenese auch die umgekehrte Reaktion in Syntheserichtung katalysiert. Sie tritt bei Vertebraten in drei genetisch unabhängigen Isoenzymen auf (Aldolase-A, -B und -C), die sich durch ihre Expression in unterschiedlichen Geweben aber auch durch ihre Substratspezifität unterscheiden. So findet man das A-Isoenzym hauptsächlich in Muskelgewebe und Erythrozyten, das B-Isoenzym in der Leber und das C-Isoenzym im Gehirn (Penhoet et al., 1966; 1969). Mutationen in den entsprechenden Genen können letale Folgen haben (erbliche FructoseIntoleranz), sofern keine spezielle Fructose-freie Diät verabreicht wird. So wurde erst kürzlich ein neues, Fructose-Intoleranz auslösendes Allel der humanen Aldolase-B identifiziert und das entsprechend mutierte Enzym charakterisiert (Rellos et al., 1999). Die Beobachtung, daß die durch Aldolasen katalysierten Reaktionen bzw. die Aktivierung und Stabilisierung der Zwischenstufen auf zwei verschiedenen Wegen erfolgen kann, führte bereits in den 60er Jahren zur Einteilung der Aldolasen in zwei Klassen: Klasse-I und Klasse-II (Rutter, 1964). Klasse-I-Aldolasen bilden ein kovalentes Schiff-BasenIntermediat zwischen der ε-Aminogruppe einer Lysin-Seitenkette und dem Carbonylkohlenstoff des Zuckers. Bei Klasse-II-Aldolasen erfolgt die Aktivierung des Zuckers über ein zweiwertiges Metall-Ion, hauptsächlich Zink, weshalb Vertreter der Klasse-II auch als Metallo-Aldolasen bezeichnet werden. Neben Zink werden aber auch andere zweiwertige Metall-Ionen verwendet, wie z. B. Fe(II) in Halobacterium mediterraneum (Dhar & Altekar, 1986) Die beiden Klassen haben sich nach heutigem Kenntnisstand aus zwei evolutionär nicht verwandten Vorgängern durch konvergente Evolution entwickelt (Perham, 1990; Marsh & Lebherz, 1992; Fothergill-Gilmore & Michels, 1993). So zeigt z. B. der Sequenzvergleich von FruA(I) und FruA(II) aus E. coli keine signifikanten Homologien (Alefounder et al., 1989). Dabei stellen die Skelettmuskel-Aldolasen der Vertebraten die mit am höchsten konservierten Enzyme dar, wobei ein Austausch von nur 2 % der Reste pro 100 Millionen 6 Jahre stattgefunden hat (Fothergill-Gilmore, 1987; Freemont et al., 1988). Während man in den 60er Jahren noch davon ausging, daß die Klasse-II-Aldolasen entwicklungsgeschichtlich älter sind (Rutter, 1964), haben die Untersuchungen von Marsh & Lebherz (1992) gezeigt, daß dieses Urteil revidiert werden muß. Denn in allen drei Reichen des Lebens, in den Archaebakterien, den Eukaryonten und den Eubakterien werden beide Klassen gefunden, was darauf schließen läßt, daß beide Klassen bereits in der ursprünglichen Lebensform vorhanden waren. Aus der Tatsache, daß die Divergenz der drei Reiche bereits in einem sehr frühen Stadium der Evolution begonnen hat, kann deshalb gefolgert werden, daß beide Klassen entwicklungsgeschichtlich etwa gleich alt sind. Die meisten gegenwärtigen Organismen exprimieren allerdings nur jeweils den Vertreter einer Klasse: Klasse-I-Aldolasen bei Tieren, Pflanzen oder Protozoen und Klasse-II-Aldolasen z. B. in Hefen oder Pilzen. Einige Organismen sind aber unter bestimmten Wachstumsbedingungen immer noch fähig, beide Klassen zu exprimieren (wie die einzellige Grünalge Chlamydomonas mundana oder einige Eubakterien wie E. coli oder Lactobacillus casei) und spiegeln somit eine enzymatische Redundanz wider, die von den meisten gegenwärtigen Lebensformen im Laufe der Evolution eliminiert wurde; sei es durch die konstitutive Repression des Gens, das für die Aldolase einer Klasse kodiert, durch Modifikation des Strukturgens einer Klasse, das dadurch den Status eines Pseudogens einnimmt oder durch die völlige Eliminierung des Strukturgens einer Klasse. Aldolasen liegen in der Regel als Homo-Oligomere vor, wobei die Größe der Untereinheiten und der Oligomerisierungsgrad stark variieren können. So liegt z. B. die Klasse-II Fructose-1,6-bisphosphat-Aldolase (FruA(II)) aus E. coli als Dimer vor, während das Klasse-I-Enzym aus dem gleichen Organismus tetramer ist (Stribling & Perham, 1973); genauso wie bei Kaninchen und Mensch. Eine trimere Variante wurde mit der 2-Keto-3deoxy-6-phosphogluconat-Aldolase aus Pseudomonas putida gefunden. Höhere Oligomerisierungszustände wie z. B. Oktamere wurden bei der 7,8-Dihydroneopterin-Aldolase aus Staphylococcus aureus (Hennig et al., 1998) gefunden. Dekamere bildet die FruA(II) aus Haloarcula vallismortis (Krishnan & Altekar, 1991). Eine monomere Variante der FruA(I) wurde im Eubakterium Micrococcus aerogenes beobachtet (Marsh & Lebherz, 1992). Mit dem Fortschritt in der Röntgenstrukturanalyse wurden auch die dreidimensionalen Strukturen der Aldolasen zugänglich, wobei zunächst nur Strukturen von Klasse-I-Aldolasen gelöst wurden. Eine Übersicht mit den entsprechenden Referenzen zeigt Tabelle 1. Den Anfang machte die Struktur der 2-Keto-3-deoxy-6-phosphogluconat-Aldolase aus Pseudomonas putida. Es folgten u. a. die Strukturen der FruA(I) aus Kaninchenmuskel, Humanmuskel, Drosophila melanogaster sowie erst kürzlich aus Plasmodium falciparum, dem Erreger von Malaria. Letztere Struktur könnte von großem Nutzen für den Entwurf von AntiMalaria-Wirkstoffen sein. Der Malaria-Parasit durchläuft sowohl in seinem Überträger, dem Moskito Anopheles, als auch in seinem menschlichen Wirt einen komplexen Lebenszyklus. Nach Infektion des menschlichen Wirts nimmt er in den Erythrozyten ein Stadium seines 7 Lebenszyklus ein, das über keinen funktionsfähigen Zitronensäurezyklus verfügt, so daß seine ATP- und somit Energieproduktion ausschließlich von der Glykolyse abhängt. Aus diesem Grund wird angenommen, daß selektive Inhibitoren gegen glykolytische Enzyme von Plasmodium falciparum den Parasiten besonders wirksam vernichten. Weitere Strukturen von Klasse-I-Aldolasen sind die der 7,8-Dihydroneopterin-Aldolase und der N-AcetylneuraminatLyase. Sie alle besitzen als gemeinsames zentrales Strukturmotiv ein (βα)8-barrel (TIMbarrel). Die Struktur der L-Fuculose-1-phosphat-Aldolase (FucA) aus E. coli war die erste Struktur einer Klasse-II-Aldolase (Dreyer & Schulz, 1993; 1996). Inzwischen sind mit der L-Rhamnulose-1-phosphat-Aldolase (RhuA) aus E. coli (Krömer & Schulz, persönliche Mitteilung) und der FruA(II) aus E. coli (Blom et al., 1996; Hall et al, 1999) zwei weitere Strukturen von Klasse-II-Vertretern bekannt. Im Gegensatz zu Klasse-I-Aldolasen tritt jedoch kein gemeinsames Strukturmotiv auf. Die FruA(II) bildet wie die Klasse-I-Vertreter ein (βα)8-barrel, wobei das aktive Zentrum im Gegensatz zu den Klasse-I-Vertretern nicht in der Mitte sondern auf der C-terminalen Seite des barrels liegt, während RhuA und FucA ein zentrales β-Faltblatt aufweisen, das von Helices eingebettet wird. Tabelle 1 Röntgenstrukturen von Klasse-I- und Klasse-II-Aldolasen Struktur Klasse Auflösung Referenz [Å] 2-Keto-3-deoxy-6-phosphogluconat-Aldolase aus Ps. putida I 2.8 Mavridis et al., 1982 Fructose-1,6-bisphosphat-Aldolase aus Kaninchenmuskel I 2.7 Sygusch et al., 1987 1.9 Blom & Sygusch, 1997 I 2.0 Gamblin et al., 1990;1991 I 2.8 Dalby et al., 1999 Fructose-1,6-bisphosphat-Aldolase aus Drosophila melanogaster I 2.5 Hester et al., 1991 Fructose-1,6-bisphosphat-Aldolase aus Plasmodium falciparum I 3.0 Kim et al., 1998 N-Acetylneuraminat-Lyase aus E. coli I 2.2 Izard et al., 1994 2.5 Lawrence et al., 1995 1.9 Jia et al., 1996 2.2 Jia et al., 1997 im Komplex mit DHAP Fructose-1,6-bisphosphat-Aldolase aus Humanmuskel im Komplex mit Fructose-1,6-bisphosphat im Komplex mit Pyruvat bzw. Hydroxypyruvat Transaldolase-B aus E. coli I reduziertes Schiff-Base-Intermediat katalytischer Antikörper IGG2A Fab-Fragment I 2.2 Barbas et al., 1997 7,8-Dihydroneopterin-Aldolase aus Staphylococcus aureus I 1.7 Hennig et al., 1998 Fuculose-1-phosphat-Aldolase aus E. coli II 1.9 Dreyer & Schulz, 1993; 1996a 2.4 Dreyer & Schulz, 1996b im Komplex mit PGH Rhamnulose-1-phosphat-Aldolase aus E. coli II 1.4 Krömer & Schulz (pers. Mitt.) Fructose-1,6-bisphosphat-Aldolase aus E. coli II 1.7 Blom et al., 1996 2.0 Hall et al., 1999 im Komplex mit PGH 8 1.2 Aldolasen und ihre Bedeutung als Biokatalysatoren in der organischen Chemie Die hohe Regio- und Stereoselektivität, milde Reaktionsbedingungen sowie ökonomische und ökologische Aspekte haben in den letzten Jahren zum verstärkten Einsatz von Enzymen in der organischen Synthese geführt. Die Fortschritte in der Gentechnologie haben darüber hinaus immer mehr Enzyme in großen Mengen zugänglich gemacht, so daß viele industrielle Prozesse inzwischen enzymkatalytisch durchgeführt werden. Es seien an dieser Stelle nur einige Beispiele genannt wie z. B. die Produktion von Mais-Sirup mit hohem Fructosegehalt durch Gluco-Amylase und Gluco-Isomerase (Getränkeindustrie), die Trypsinspaltung zur Umwandlung von Schweine-Insulin in menschliches Insulin, die Synthese des Süßstoffs Aspartam mit Thermolysin oder die Herstellung von Penicillinanaloga durch die PenicillinAcylase. Aldolasen spielen vor allem bei der Synthese komplexer Kohlenhydrate eine Rolle, die von immer größerem Interesse in der medizinischen Forschung bzw. bei der Entwicklung von Pharmazeutika im Rahmen von Wirkstoff-Screenings werden. Denn die Oligosaccharide auf Zelloberflächen sind an einer Vielzahl von zentralen biologischen Erkennungsprozessen beteiligt: angefangen bei Signalübertragung, interzellulärer Kommunikation und Zelladhäsion über virale Infektionen und Immunerkennung bis hin zu Zelldifferenzierung bei der Entwicklung von Organen und bei der Karzinogenese. Konventionelle Verfahren zur Herstellung solcher komplexen Kohlenhydrate beinhalten einen wiederholten Einsatz von Schutzgruppen und weisen in der Regel unzureichende Stereokontrolle auf. Übersichten über die vielfältigen Anwendungsmöglichkeiten von Aldolasen bei asymmetrischen C-C-Verknüpfungen finden sich bei Wong et al. (1995), Fessner & Walter (1996) sowie bei Petersen et al. (1997). Eine besondere Stellung nehmen die vier bekannten bakteriellen DHAP-Lyasen (alle Klasse-II) ein: D-Fructose-1,6-bisphosphat-Aldolase, L-Fuculose-1-phosphat-Aldolase, L-Rhamnulose-1-phosphat-Aldolase und D-Tagatose-1,6-bisphosphat-Aldolase. Bei der katalysierten Aldoladdition werden jeweils zwei neue Stereozentren mit definierter Konfiguration gebildet. Das von Fessner (1992) vorgeschlagene „Baukastensystem“ der DHAPEnzyme (Abbildung 1) macht alle vier möglichen Konfigurationen zugänglich. Durch die vergleichsweise geringe Spezifität der Enzyme auf Seite der Aldehyd-Komponente eignen sie sich besonders gut für synthetische Zwecke, da dadurch auch ein breites Spektrum an nichtnatürlichen Substraten akzeptiert wird. Nur tertiäre, aromatische oder α,β-ungesättigte Verbindungen werden in der Regel nicht umgesetzt (Fessner, 1993). Dagegen ist die SubstratToleranz auf Seite der DHAP-Komponente stark begrenzt (Arth, 1997; Arth & Fessner, 1998). FucA und RhuA besitzen außerdem eine deutliche kinetische Präferenz für L-konfigurierte 2-Hydroxyaldehyde (>95:5), so daß sie zur kinetischen Racematspaltung 9 solcher Verbindungen verwendet werden können; d. h. in einem Schritt können gleich drei neue Stereozentren determiniert werden (Fessner et al., 1992; 1993). OH OH O 2- O 2- 2OPO3 O3PO OH OPO3 FruA OH OH RhuA D-Fructose-1,6-bisphosphat OH L-Rhamnulose-1-phosphat (D-threo) (L-threo) 3S, 4R O O X H HO 3R, 4S 2OPO3 OH OH 2- O 2- O3PO TagA FucA OH O 2- OPO3 OPO3 OH OH D-Tagatose-1,6-bisphosphat OH OH L-Fuculose-1-phosphat (L-erythro) (D-erythro) 3S, 4S 3R, 4R Abbildung 1 Baukastensystem der komplementären DHAP-Aldolasen (Fessner, 1992). Neben den DHAP-Aldolasen sind auch die Pyruvat-Lyasen wie die N-Acetylneuraminsäure-Aldolase (Klasse-I) von großem synthetischen Interesse, da sie zur Synthese von Sialinsäurederivaten eingesetzt werden kann, deren Bedeutung in der Zellbiologie stark gestiegen ist (Kragl et al. 1994; v. Itzstein & Thomson, 1997). Wie die DHAP-Aldolasen, so besitzen auch die Pyruvat-Lyasen eine breite Toleranz gegenüber der Aldehydkomponente und eine hohe Spezifität auf Seite der Donorkomponente Pyruvat. 1.3 L-Fuculose-1-phosphat-Aldolase aus Escherichia coli L-Fuculose-1-phosphat-Aldolase (FucA, E.C. 4.1.2.17) aus E. coli katalysiert die reversible Aldolspaltung von L-Fuculose-1-phosphat zu L-Lactaldehyd und Dihydroxyacetonphosphat (DHAP; Ghalambor & Heath, 1962; 1966). Diese Reaktion nimmt eine zentrale Stellung im L-Fucose-Stoffwechsel einiger Bakterien ein (Abbildung 2) und kann als bakterielles Äquivalent zur glykolytischen FruA bezeichnet werden (Lin, 1987). Dabei wird L-Fucose im ersten Schritt durch die L-Fucose-Isomerase (EC 5.3.1.3) von einer Aldose zur Ketose L-Fuculose isomerisiert, die im Folgeschritt mittels L-Fuculose-Kinase (EC 2.7.1.51) 10 phosphoryliert wird. Das dabei entstandene L-Fuculose-1-phosphat (Fuc1P) wird durch FucA in C3-Bausteine gespalten, wodurch es weiteren Stoffwechselwegen zugeführt wird. OH OH OH O OH OH Isomerase H OH OH L-Fucose FucA O + Kinase OH O 2- 2- HO OPO 3 OPO3 OH L-Lactaldehyd OH L-Fuculose O H O OH DHAP OH Fuc1P Abbildung 2 Der L-Fucose-Stoffwechsel in E. coli nach Ghalambor & Heath (1962) FucA zählt zu den zinkbindenden Klasse-II-Aldolasen und bildet ein Homotetramer mit 215 Aminosäuren und einem Zink-Ion pro Untereinheit. Das Struktur-Gen fucA wurde von Chen et al. (1987; 1989) kloniert (Plasmid pfuc41a) und sequenziert. Es folgte die Sequenzierung des kompletten Fucose-Regulons in der gleichen Arbeitsgruppe (Lu & Lin, 1989). In der Zwischenzeit wurde im vollständig sequenzierten Genom von Haemophilus influenzae (Fleischmann et al., 1995) eine weitere FucA-Sequenz postuliert, wobei das entsprechende Enzym eine Sequenzhomologie von 81% aufweisen soll. Desweiteren zeigt FucA eine hohe Sequenzhomologie zu RhuA sowie zu dem Enzym L-Ribulose-5-phosphat-4Epimerase aus E. coli (Mineno et al., 1990) und Salmonella typhimurium (Lin et al., 1985). Erste FucA-katalysierte Synthesen unter Verwendung ganzer Zellen (E. coli O-111 B4) wurden von Drueckhammer et al. (1989) durchgeführt. Es folgten synthetische Anwendungen unter Verwendung des nicht näher spezifizierten Expressionssystems E. coli JM105/pFUCA-5 durch Ozaki et al. (1990), wobei partiell gereinigtes Enzym eingesetzt wurde. Ausgehend von dem Plasmid pfuc41a entwickelte Dreyer die leistungsfähigen Expressionssysteme E. coli HB101/pKFA (Dreyer, 1991) sowie E. coli JM105/pKKFA2 (Dreyer, 1995), bei denen es sich um pKK223-3 Abkömmling handelt (Brosius & Holy, 1984). Dadurch stand FucA in großen Mengen zur Verfügung; zum einen für die Kristallisation und erfolgreiche Röntgenstrukturanalyse (siehe folgendes Kapitel), zum anderen zur systematischen Untersuchung des Substratspektrums sowie zum Einsatz in organischen Synthesen (Schneider, 1994). Dabei weist das Enzym wie die anderen Aldolasen eine große Toleranz für unterschiedlich konfigurierte und substituierte Aldehyde auf, wobei die Diastereoselektivität der Reaktion bei sterisch anspruchsvollen oder unpolaren 11 Substituenten in α- oder β-Stellung zur Carbonylgruppe beeinträchtigt wird. FucA zeigt darüber hinaus eine hohe Stabilität in Gegenwart organischer Cosolventien, wodurch auch Synthesen mit hydrophoben Verbindungen möglich sind. Inzwischen ist das Enzym kommerziell erhältlich und wird u. a. als FucA Aldol Reaction Kit vertrieben (Fa. Roche, 1999). Dabei wird das Cosubstrat DHAP in situ durch enzymatische Oxidation von L-Glycerol-3-phosphat mit dem System L-Glycerophosphat-Oxidase/Katalase erzeugt. 1.4 Die Röntgenstruktur der L-Fuculose-1-phosphat-Aldolase Die Röntgenstruktur der FucA wurde von Dreyer & Schulz (1993; 1996) in zwei verschiedenen Kristallformen durch multiplen isomorphen Ersatz bzw. mittels molekularem Ersatz gelöst und bis zu einer Auflösung von 1.9 Å (Kristallform T, Raumgruppe P4212) bzw. 2.7 Å (Kristallform K, Raumgruppe F432) verfeinert. Im Gegensatz zur hochauflösenden tetragonalen Kristallform, erwies sich die kubische Kristallform als stabil bei Tränkexperimenten mit dem Übergangszustandsanalogon Phosphoglycolohydroxamat (PGH), wodurch die Analyse der Komplexstruktur ermöglicht wurde, die bei einer Auflösung von 2.4 Å verfeinert wurde. Alle drei Modelle umfassen jeweils die Reste 1-206; d. h. die neun C-terminalen Aminosäuren (207-215) waren in der Elektronendichte aufgrund erhöhter Flexibilität nicht detektierbar. Eine Ausnahme bildet die Struktur des Wildtyps (WT) ohne PGH, bei der das katalytische Zink-Ion gegen Cobalt ausgetauscht wurde. Dort konnte das Modell bis einschließlich Aminosäure 210 erweitert werden. Abbildung 3 Stereobild der ribbon-Darstellung des FucA-Tetramers (Dreyer & Schulz, 1996) mit Blick entlang der vierzähligen Achse. Das Zink-Ion, seine Liganden sowie das Substratanalogon PGH sind als gelbe ball-and-stick-Modelle dargestellt. 12 Die FucA-Untereinheit besteht aus einem zentralen neunsträngigen, nahezu durchgehend antiparallelem β-Faltblatt, das zwischen zwei Schichten von α-Helices eingebettet liegt. Das allgemeine Faltungsmuster der FucA unterscheidet sich somit grundlegend von dem der Klasse-I-Aldolasen bzw. FruA(II), die alle als zentrales Faltungsmuster ein (βα)8-barrel besitzen. Das aus vier identischen Untereinheiten aufgebaute FucATetramer weist die relativ seltene molekulare C4-Symmetrie auf, wobei die aktiven Zentren an der Grenzfläche zwischen zwei Untereinheiten liegen und von Resten benachbarter Untereinheiten gebildet werden (Abbildung 3). (a) (b) Abbildung 4 Stereodarstellung des aktiven Zentrums von FucA: (a) ohne Liganden, (b) im Komplex mit dem Übergangszustandsanalogon PGH (Dreyer & Schulz 1993; 1996). Reste von der benachbarten Untereinheit sind mit ‘ gekennzeichnet . 13 In der Inhibitor-freien Struktur wird das katalytische Zink-Ion annähernd tetraedrisch von drei Histidin-Seitenketten (His92, His94 und His155) und der Carboxylatgruppe von Glu73 koordiniert. Darüber hinaus ragt Tyr113’ von der gegenüberliegenden Untereinheit ins aktive Zentrum und reicht mit seiner phenolischen Hydroxygruppe in die unmittelbare Nähe des Zn2+, ist jedoch kein Zink-Ligand (Zn-O-Abstand = 3.5 Å). Von einer Seite her wird das aktive Zentrum von einer Art hydrophober Wand abgeschirmt, die von den aromatischen Seitenketten der Reste Tyr113’, Phe131 und Phe206’ gebildet wird. Desweiteren konnte ein Sulfat-Ion, das aus dem Kristallisationspuffer stammt, im aktiven Zentrum identifiziert werden (Abbildung 4a). Die Position des Sulfat-Ions in der unligierten Struktur wird in der Inhibitorstruktur von der Phosphatgruppe des PGH eingenommen. Durch die chelatartige Bindung von PGH wird Glu73 aus der Koordinationssphäre des Zink-Ions verdrängt. Zusätzlich kann ein Art induced fit beobachtet werden, der u. a. zur Umlagerung und Fixierung des Loops(23-27) führt, was sich in den erniedrigten B-Faktoren dieses Loops in der PGH-Struktur widerspiegelt (Abbildung 4b). Dabei ist die größte Bewegung für Thr26 zu beobachten (3.5 Å für Thr26-Oγ). Der Vergleich der beiden Strukturen ließ Dreyer & Schulz (1996b) einen Mechanismus für die von FucA katalysierte Reaktion vorschlagen, bei dem Glu73 als allgemeine Base und Tyr113’ als allgemeine Säure fungiert. 1.5 Ziele der Arbeit Ziel der vorliegenden Arbeit war zunächst die Kontrolle des von Dreyer & Schulz (1996b) auf Basis der Röntgenstrukturen des Wildtyps vorgeschlagenen Reaktionsmechanismus von FucA durch ortsgerichtete Mutagenese. In diesem Zusammenhang sollte auch die Rolle der in der Röntgenstruktur nicht detektierten neun C-terminalen Aminosäuren (207-215) aufgeklärt werden. Aufbauend auf den dabei gewonnenen Erkenntnissen, waren im zweiten Teil der Arbeit Mutanten zu planen, um die kinetische Enantioselektivität und Diastereoselektivität der Reaktion für unterschiedliche Aldehyd-Substrate besser zu verstehen bzw. gezielt in eine Richtung zu verschieben und dadurch das Spektrum enzymkatalysierter organischer Synthesen zu erweitern. Desweiteren sollte untersucht werden, ob es möglich ist, die Selektivität auf Seite des Aldol-Donors DHAP hin zu anderen Donoren zu verändern; einerseits um neue Produkte zu synthetisieren, aber auch um das teure DHAP zu ersetzen. Die auf DNA-Ebene generierten Mutanten sollten produziert, isoliert und bezüglich ihrer enzymatischen Aktivitäten für natürliche bzw. nicht-natürliche Substrate getestet werden. Desweiteren sollten Kristallisationsexperimente durchgeführt und bei positivem Resultat die entsprechenden Mutanten röntgenographisch untersucht werden. 14 2 Materialien 2.1 Geräte Gerät Typ Hersteller Bakterienanzucht Dampfsterilisator Brutschrank Bakterienschüttler Fermenter Photometer Varioautoklav 500 EV BK 8 RW 650 Vibrax VXR Biostat B PR 2210 H & P Labortechnik W. Ehret New Brunswick & Scientific IKA Labortechnik B. Braun Eppendorf DNA-Sequenzierung Sequenzierungsgerät Spannungsquelle Nylon-Membran UV-Crosslinker Entwicklungstrommel Hybridisierungsofen GATC 1500-System ECPS 3000/150 Direct-Blotting-Membrane GATC-Link Eigenbau Mini 10 GATC Pharmacia GATC GATC Institutswerkstatt Hybaid Gentechnik Thermocycler Mehrzweck-Saugeinrichtung Agarose-Gelektrophorese-Kammer Spannungsquellen Zentrifugen OmniGene HB-TR3-CM TouchDown Vac-Man Laboratory Vacuum Manifold GNA 100 EPS 500/400 ECPS 3000/150 5415 C Rotanta SpeedVac Concentrator Hybaid Hybaid Promega Pharmacia Pharmacia Pharmacia Eppendorf Hettich Savant Isoelektrische Fokussierung Kryomat IEF-Anlage IEF-Gele Spannungsquelle Julabo FC 200 Multiphor 2117 Servalyte Precotes 2197 Power Supply Julabo Labortechnik Pharmacia Pharmacia Pharmacia LKB Kristallisation Dialyseknopf Kristallisationsbox Deckgläser Mikroskop Eigenbau Linbro Zellkulturbox ∅ 21 mm, Stärke 2 Laborlux 12 Pol S Institutswerkstatt ICN Hecht Assistent Leitz-Leica Proteinpräparation Ultraschallgenerator Ultra-Kryomat Zentrifugen Modell 7100 TK-30D Biofuge 28 RS Labofuge I Measuring & Scientific Equipment Lauda Heraeus Heraeus Christ 15 Ultraschallbad Säulen Pumpen FPLC-Anlage Gradientenmischer Detektoren Fraktionssammler Schreiber Spektrophotometer Quarzküvetten Konzentratoren Dialyseschlauch Ultrafiltrationszelle Ultrafiltrationsmembran Sorvall RC2-B, RC-5B Sonorex RK106 Super Eigenbau GPC-200 2232 Microperpex S P-3 Eigenbau EM-1 Econo UVIS 204 7000 Ultrorac SE 130 Lambda 5 104-QS Centriprep-30, Centricon-30 20/32, Wandstärke 20 µm, ∅ 16 mm Magnetrührzelle 402 YM5/PM10 DuPont Bandelin Institutswerkstatt Pharmacia LKB Pharmacia Institutswerkstatt Bio Rad Linear Instruments Pharmacia LKB BBC Goerz-Metrawatt Perkin Elmer Hellma Amicon Roth Amicon Amicon Röntgenstrukturanalyse Glaskapillaren Mikroskop Hartwachs Röntgengeneratoren Drahtflächenzähler Rechner Graphikbildschirme Wandstärke 1/100 mm Stemi 2000 Deiberit 502 M18X HF RU 200 B X 1000 RISC 6000 Indigo2 W. Müller Zeiss Böhme Siemens Rigaku Nicolet/Siemens IBM Silicon Graphics SDS-PAGE Elektrophorese-Apparatur Spannungsquelle Geltrockner Mini Protean II Power Pac 3000 Slab Drier GSD-4 2.2 Chemikalien, Enzyme und Kits Chemikalien Acrylamid, ultra pure Agar-Agar Agarose, research grade Ampicillin, Natriumsalz APS Bromphenolblau, Natriumsalz ATP, Natriumsalz BSA Caseinpepton Chloramphenicol, research grade Coomassie Brilliant Blue R-250 DC-Platten, Kieselgel 60 F254, 0.2 mm DEAE-Sepharose-CL6B Gerbu Gibco BRL Serva Merck Merck Serva Aldrich Sigma Gibco BRL Serva Serva Merck Pharmacia Bio Rad Bio Rad Pharmacia 16 DNA-Längenstandard (λ-Marker, Bst EII-geschnitten) EDTA, Dinatrium-Salz Ethidiumbromid Glykolaldehyd-Dimer Hefeextrakt Hexamincobalt-(III)-chlorid β-Hydroxypyruvat, Lithiumsalz IEF-Marker (pI 3-10) IPTG, biotechnical grade LMW-Marker (low molecular weight) MES, Kaliumsalz 3-Methacryloxypropyltrimethoxysilan NADH NBT N,N'-Methylenbisacrylamid, research grade Oligonukleotide Oligonukleotide (5’ DIG-markiert) PEG-8000 SDS Superdex S200 prep grade, HiLoad 16/60 TEMED Thiamin, Hydrochlorid Tris X-Phosphat, p-Toluidinsalz New England Biolabs Gerbu Merck Fluka Gibco BRL Sigma Fluka Pharmacia Gerbu Pharmacia Serva GATC Gerbu Gerbu Merck Dr. G. Igloi, Inst. f. Biologie-III, Uni. Freiburg MWG Fluka, Serva, Sigma Merck Pharmacia Merck Serva Roth Gerbu Gängige Laborchemikalien sind nicht aufgeführt. Soweit nicht anders beschrieben, waren alle Chemikalien vom Reinheitsgrad p. a. Enzyme Glycerin-Dehydrogenase (EC 1.1.1.6) Boehringer Mannheim Glycerin-3-phosphat-Dehydrogenase (EC 1.1.1.8) Fluka/Boehringer Mannheim Lactat-Dehydrogenase (EC 1.1.1.27) Boehringer Mannheim Klenow-Polymerase (EC 2.7.7.7) Boehringer Mannheim Streptavidin-Alkalische-Phosphatase-Konjugat GATC T4-DNA-Ligase (EC 6.5.1.1) Ligase-Puffer Boehringer Mannheim T4-Polynukleotid-Kinase (EC 2.7.1.78) Kinase-Puffer New England Biolabs Taq-DNA-Polymerase (EC 2.7.7.7) Taq-Puffer Amersham Thermo-Sequenase Sequenase-Verdünnungspuffer Amersham EcoR I Boehringer Mannheim Pst I Restriktionspuffer-H Boehringer Mannheim Die Zusammensetzung der mitgelieferten Puffer kann der Beschreibung zum jeweiligen Enzym entnommen werden. 17 Kits DIG Nucleic Acid Detection Kit Boehringer Mannheim Anti-DIG-Alkalische-Phosphatase-Konjugat, NBT/X-Phosphat-Lösung, Blockierungspuffer DIG Taq DNA Sequencing Kit for Standard and Cycle Sequencing Boehringer Mannheim Reaktionspuffer, Taq-DNA-Polymerase, Terminations-Mix-ddGTP/dGTP, -ddATP/dGTP, -ddTTP/dGTP, -ddCTP/dGTP, Formamid-Puffer GATC-BioCycle Sequencing Kit ddGTP-Mix, ddATP-, ddTTP-, ddCTP-, dITP-, Reaktionspuffer, Stop-Mix GATC QIAprep Spin M13 Kit M13-Präzipitations-Puffer, M13-Lyse & Bindungs-, PE-Waschpuffer Qiagen QIAprep Spin Plasmid Kit Puffer-P1, -P2, -N3, -PB, -PE, RNase-A Qiagen QIAquick Gel Extraction Kit Puffer-QG, -PE, -EB Qiagen Sculptor in vitro mutagenesis system Amersham Native T7-DNA-Polymerase, Klenow-Polymerase, T4-DNA-Ligase, T5-Exonuklease, Nci I, Exonuklease-III (EC 3.1.11.2), DNA-Polymerase-I, Puffer-A, -B, -C, -D, dNTP-Mix-A, -Mix-B, E. coli TG1 Die Zusammensetzung von Puffern und Lösungen kann der Beschreibung zum jeweiligen Kit entnommen werden (soweit angegeben). Nur verwendete Puffer und Lösungen sind aufgezählt, Verbrauchsgegenstände (z. B. QIAprep-Säulen) sind nicht angegeben. 2.3 Bakterienstämme und Vektoren Bakterienstämme CJ236 Stammsammlung der Abteilung Ein dut ung -Stamm, der zur Erzeugung Uracil-haltiger DNA für Mutagenese-Experimente verwendet wird (Kunkel et al., 1987). Er enthält das F’-Plasmid. Bei Wachstum in Chloramphenicol findet eine Selektion zu zugunsten der Retention des F’-Plasmids statt (Sambrook et al., 1989). - - JM 101 Stammsammlung der Abteilung Unterstützt die Vermehrung von Vektoren mit Amber-Mutationen und enthält das F’-Plasmid (Sambrook et al., 1989) JM105 Stammsammlung der Abteilung Unterstützt die Vermehrung von Vektoren mit Amber-Mutationen und enthält das F’-Plasmid (Sambrook et al., 1989) TG1 Amersham Ein EcoK -Derivat von JM101, das restriktions- und modifikations-defizient ist. Enthält das F’-Plasmid und zeigt hohe Transformationsrate für Phosphorothioat-DNA (Sambrook et al., 1989). - 18 XL1-Blue Stratagene Rekombinations-defizienter Stamm, der die Vermehrung von Vektoren mit Amber-Mutationen unterstützt. Liefert sehr reine Plasmid-DNA und enthält ein modifiziertes (TetracyclinResistenz) F’-Plasmid (Sambrook et al., 1989). Bei allen Stämmen handelt es sich um E. coli K12. Das F’-Plasmid ermöglicht ihnen die Ausbildung von Fertilitätspili, die als Erkennungsstellen für M13-Phagen fungieren und somit die Voraussetzung für die Infektion der Bakterienzellen mit M13-Phagen sind. Das F’-Plasmid trägt darüber hinaus noch weitere Gene wie z. B. das lacIq-Gen für die Überproduktion des lac-Repressors, das für die αKomplementation (Blau/Weiß-Selektion) wichtige Fragment der β-Galaktosidase (lacZ∆M15) sowie für die Prolinbiosynthese wichtige Gene (proAB+) zur Selektion des F’-Plasmids auf prolinfreiem Minimal(M9)-Agar. Vektoren M13mp19-dsDNA M13mp19-fuca pKK223-3 pKKFA2 Pharmacia C. Müller-Dieckmann, Inst. für Org. Chemie und Biochemie, Univ. Freiburg Pharmacia Dr. M. Dreyer, Inst. für Org. Chemie und Biochemie, Univ. Freiburg 19 2.4 Lösungen, Puffer und Nährmedien Agarose-Gelelektrophorese 50×TAE-Puffer Tris Essigsäure 0.5 M EDTA pH 8.0 (RT, 5 M NaOH) deion. H2O 242 g 57.1 ml 100 ml ad 1000 ml Ethidiumbromid-Lösung Ethidiumbromid Agarosegel 1 % (w/v) Agarose 1×TAE-Puffer Ethidiumbromid-Lösung Probenpuffer Tris EDTA Glycerin Bromphenolblau DNA-Längenstandard Bst EII-verdaute λ-Phagen-DNA (40 ng/µl in Proben-Puffer) DNA-Fragmentlängen [bp]: 14120, 8454, 7242, 6369, 5686, 4822, 4342, 3675, 2323, 1929, 1371, 1264, 702, 224, 117 TE-Puffer Tris EDTA 1 mg/ml 0.5 g 50 ml 25 µl (halber Ansatz für kleines Gel) 10 mM 1 mM 50 % (v/v) 0.05 % (w/v) pH 7.5 (RT, 6 M HCl) 10 mM 1 mM pH 8.0 (RT, 6 M HCl) Aktivitätstests Assay-Lösung-Fuc1P Tris KCl NADH L-Fuculose-1-phosphat (Dicyclohexylammoniumsalz) 60 60 1 2.4 mM mM mM mM pH 7.5 (RT, 6 M HCl) Assay-Lösung-DHA Tris KCl Glykolaldehyd Dihydroxyaceton 50 mM 20 mM 50 mM 5 mM pH 8.0 (RT, 6 M HCl) Assay-Lösung-BHP Tris KCl Glykolaldehyd β-Hydroxypyruvat 50 mM 20 mM 50 mM 11.25 mM pH 8.0 (RT, 6 M HCl) 20 Lösung-A KCl Tris Lösung-B Glykolaldehyd 100 mM nach Spaltung des Glykolaldehyd-Dimers durch 10min. Kochen Lösung-C Dihydroxyaceton oder β-Hydroxypyruvat Lösung-D Tris NADH 80 mM 200 mM pH 8.0 (RT, 6 M HCl) 20 mM 45 mM 50 mM 0.27 mM pH 7.5 (RT, 6 M HCl) Die Assay-Lösungen-DHA/-BHP wurden aus den entsprechenden Volumina der Lösungen A bis C zusammengesetzt. DNA-Sequenzierung 50 µl 30 µl 300 µl ad 10 ml Silanlösung 3-Methacryloxypropyltrimethoxysilan Essigsäure bidest. H2O Ethanol Harnstoff-Diluent Harnstoff 10×TBE-Puffer bidest. H2O Acrylamid-Lösung-1 Acrylamid N,N'-Methylenbisacrylamid Sequenzierungsgel 6 % (w/v) Acrylamid-Lösung-1 Harnstoff-Diluent 10×TBE-Puffer Tris Borsäure EDTA, Dinatrium-Salz bidest. H2O Maleinsäure-Puffer Maleinsäure NaCl NaOH bidest. H2O Blockierungspuffer Maleinsäure-Puffer mit Blockierungs-Reagenz 84.0 g 18.7 g 61.6 g 30 % (w/v) 0.8 % (w/v) 10 ml 40 ml 154.5 g 26.2 g 9.0 g ad 1000 ml pH 8.3 (stellt sich ein) 23.21 g 17.53 g 16 g ad 2000 ml pH 7.5 (RT, 5 M NaOH) 10 % (w/v) 21 Reaktionspuffer Tris NaCl MgCl2 . 6 H2O bidest. H2O 12 g 5.84 g 2g ad 1000 ml pH 9.5 (RT, 6 M HCl) Isoelektrische Fokussierung Anodenlösung L-Asparaginsäure L-Glutaminsäure 3.4 g/l 3.6 g/l Kathodenlösung L-Arginin L-Lysin Ethylendiamin Fixierer Trichloressigsäure Sulfosalicylsäure Färber Coomassie-Brilliant-Blue R-250 Ethanol Essigsäure 4 % (w/v) 25 % (w/v) 8 % (w/v) Entfärbelösung Ethanol Essigsäure 40 % (w/v) 10 % (w/v) IEF-Längenstandard Amyloglucosidase (pI = 3.50), Sojabohnen-Trypsin-Inhibitor (pI = 4.55), β-Lactoglobulin (pI = 5.20), Carboanhydrase-B aus Rind (pI = 5.85), humane Carboanhydrase-B (pI = 6.55), Myoglobin, saure Bande (pI = 7.35), Myoglobin, basische Bande (pI = 8.15), Linsen-Lectin, saure Bande (pI = 8.15), Linsen-Lecin, mittlere Bande (pI = 8.45), Linsen-Lectin, basische Bande (pI = 8.65), Trypsinogen (pI = 9.30) 4.4 g/l 3.6 g/l 12 % (v/v) 12.5 % (w/v) 3.5 % (w/v) Kristallisation/Röntgenstrukturanalyse Puffer-A Ammoniumsulfat KH2PO4 β-ME ZnCl2 25 % (w/v) 20 mM 4 mM 1 mM pH 7.8 (RT, 6 M KOH) Puffer-B KH2PO4 β-ME ZnCl2 20 mM 4 mM 1 mM pH 7.8 (RT, 6 M KOH) Silikonisierungslösung Dichlordimethylsilan in Toluol 2 % (v/v) 22 Mutagenese nach Kunkel 5xPEG/NaCl-Lösung PEG-8000 NaCl 15 % (w/v) 2.5 M 10xASB-Puffer Tris MgCl2 DTE NaCl 200 mM 100 mM 10 mM 500 mM pH 7.5 (RT, 6 M HCl) 2xEB-Puffer Tris MgCl2 DTE dGTP dATP dTTP dCTP ATP 40 mM 20 mM 4 mM 1 mM 1 mM 1 mM 1 mM 2 mM pH 8.0 (RT, 6 M HCl) Nährmedien LB-Medium Caseinpepton Hefeextrakt NaCl 10 g/l 5 g/l 10 g/l pH 7.5 (RT, 5 M NaOH) LB-Agar LB-Medium mit Agar-Agar 15 g/l LB-Top-Agar LB-Medium mit Agar-Agar 7.5 g/l 2xYT-Medium Caseinpepton Hefeextrakt NaCl 10xM9-Salzlösung Na2HPO4 · 12 H2O KH2PO4 NH4Cl NaCl 161 g/l 30 g/l 10 g/l 5 g/l M9-Agar LB-Agar-Agar 10xM9-Salzlösung 20 % (w/v) Glucose 1 M MgSO4 1 M Thiamin 0.1 M CaCl2 225 ml 25 ml 10 ml 1 ml 1 ml 1 ml Ampicillin-Lösung Ampicillin (Endkonzentration 100 µg/ml) 16 g/l 10 g/l 5 g/l pH 7.0 (RT, 5 M NaOH) 25 mg/ml Proteinpräparation TMZ-Puffer-I Tris β-ME ZnCl2 20 mM 10 mM 1 mM pH 7.6 (RT, 6 M HCl) 23 TMZ-Puffer-II TMZ-Puffer-III TMZ-Puffer-IV TMZ-Puffer-V TMZ-Puffer-VI Polymin-P-Lösung TMZ-Puffer-I NaCl TMZ-Puffer-I NaCl TMZ-Puffer-I NaCl TMZ-Puffer-I NaCl TMZ-Puffer-I NaCl Polymin-P 50 mM pH 7.6 (RT, 6 M HCl) 150 mM pH 7.6 (RT, 6 M HCl) 400 mM pH 7.6 (RT, 6 M HCl) 500 mM pH 7.6 (RT, 6 M HCl) 1.5 M pH 7.6 (RT, 6 M HCl) 18 % (w/v) pH 7.0 (RT, 6 M HCl) SDS-PAGE Acrylamid-Lösung-2 Acrylamid N,N’-Methylenbisacrylamid Trenngel-Puffer Tris SDS Trenngel 12 % (w/v) Acrylamid-Lösung-2 Trenngel-Puffer deion. H2O 10 % (w/v) APS TEMED Sammelgel-Puffer Tris SDS Sammelgel 6 % (w/v) Acrylamidlösung-2 Sammelgel-Puffer deion. H2O 10 % (w/v) APS TEMED SDS-Probenpuffer Glycerin Sammelgel-Puffer 16 % (w/v) SDS 0.2 % (w/v) Bromphenolblau β-ME 39 % (w/v) 1 % (w/v) 1.5 M 0.4 % (w/v) pH 8.8 (RT, 6 M HCl) 3 ml 2.5 ml 4.5 ml 100 µl 10 µl 0.5 M 0.4 % (w/v) pH 6.8 (RT, 6 M HCl) 0.75 ml 1.25 ml 3 ml 50 µl 5 µl 4 ml 2 ml 2 ml 1 ml 1 ml 24 LMW-Marker Mr [kDa]: 94, 67, 43, 30, 20.1, 14.4 Elektrophorese-Puffer Tris Glycin SDS Coomassie-Lösung Coomassie Brilliant Blue R-250 Ethanol Essigsäure Entfärbelösung-2 Ethanol Essigsäure 50 mM 380 mM 0.1 % (w/v) pH 8.3 (RT, 6 M HCl) 0.25 % (w/v) 25 % (v/v) 10 % (v/v) 30 % (v/v) 10 % (v/v) Transformation TFB-Puffer MES KCl MnCl2 CaCl2 Hexamincobalt-(III)-chlorid 10 mM 100 mM 45 mM 10 mM 3 mM pH 6.2 (RT, 6 M KOH) 25 3 Methoden 3.1 Gentechnische Methoden Mutagenese-Oligonukleotide Reinigung der Oligonukleotide Phosphorylierung der Oligonukleotide Präparation der M13mp19-fuca-ssDNA Mutagenese Transformation in E. coli TG1 pKK223-3 Präparation der mutierten M13mp19-fuca-dsDNA Präparation der mutierten M13mp19-fuca-ssDNA Restriktionsspaltung Restriktionsspaltung DNA-Sequenzierung Isolierung des Vektorfragments Isolierung des mutierten Genfragments Mutagenese Mehrfachmutanten Ligation Transformation in E. coli TG1 Präparation des mutierten Plasmids pKKFA2 Abbildung 5 Schematische Darstellung der gentechnischen Arbeiten zur Generierung der FucAMutanten (exemplarisch für die Mutagenese nach Eckstein) sowie zur Rückklonierung in den Expressionsvektor pKK223-3. Modifizierungen, die bei der Mutagenese nach Kunkel nötig waren, werden an entsprechender Stelle gesondert erwähnt. 26 Alle zur Erzeugung der Mutanten erforderlichen Schritte werden in Abbildung 5 gezeigt. Bei den beiden in dieser Arbeit angewendeten Mutagenese-Verfahren (Eckstein/Kunkel) handelte es sich um Oligonukleotid-dirigierte Verfahren. Ausgangspunkt war dabei das in den Bakteriophagen M13mp19 klonierte Gen des WT als Templat (M13mp19-fuca). Mit dieser Templat-DNA und den entsprechend konstruierten Mutagenese-Oligonukleotiden wurde dann die Mutagenese nach Eckstein bzw. Kunkel durchgeführt. Nach Transformation des Mutagenese-Ansatzes in E. coli TG1 (Mutagenese nach Eckstein) wurden die positiven Klone durch Sequenzierung der viralen DNA im Bereich des FucA-Gens identifiziert und die entsprechende doppelsträngige DNA isoliert. Mittels der Restriktionsenzyme EcoR I und Pst I wurde das mutierte Gen daraufhin in den Expressionsvektor pKK223-3 kloniert und das neue Konstrukt in E. coli TG1 transformiert. Aus E. coli TG1 konnte der Expressionsvektor dann für weitere Analysen bzw. für die Transformation in den Expressionsstamm E. coli JM105 in ausreichend großen Mengen isoliert werden. 3.1.1 Reinigung von Oligonukleotiden Die in dieser Arbeit für die Mutagenese verwendeten Oligonukleotide wurden im Institut für Biologie III der Universität Freiburg nach dem Phosphoramidit-Verfahren hergestellt. Die Dimethoxytrityl-Schutzgruppe war bei Erhalt bereits abgespalten. Zur Reinigung wurde die in lyophilisierter Form erhaltene DNA (Synthesemaßstab 40 nmol) in 100 µl TE-Puffer aufgenommen und unlösliche Bestandteile durch Zentrifugation abgetrennt (5 min, 15800∗g). Zur Fällung der DNA wurden 40 µl der Oligonukleotidlösung mit 100 µl Ethanol und 0.5 µl 5 M NaCl versetzt und mindestens 1 h bei -20 °C inkubiert. Nach Zentrifugation (20 min, 15800∗g, 4 °C) wurde das Pellet mit 500 µl zuvor bei -20 °C gelagertem 70 %igem (v/v) Ethanol salzfrei gewaschen und erneut zentrifugiert (10 min, 15800∗g, 4 °C). Das Pellet wurde im Vakuum getrocknet und in 40 µl TE-Puffer aufgenommen. Die für die Sequenzierung verwendeten DIG-markierten Oligonukleotide wurden bereits HPLC-gereinigt geliefert, so daß keine weitere Aufreinigung erforderlich war. Die lyophilisierten Oligonukleotide wurden nach Erhalt entsprechend den Herstellerangaben mit bidest. H2O auf 100 pmol/µl verdünnt. Die Konzentrationsbestimmung erfolgte jeweils photometrisch bei 260 nm. Konzentrationsbestimmung von Oligonukleotidlösungen Voraussetzung für die photometrische Konzentrationsbestimmung eines Oligonukleotids ist die Kenntnis des molaren Extinktionskoeffizienten. Dieser läßt sich über die Extinktionskoeffizienten der einzelnen Nukleotide additiv berechnen (εdATP = 15400 M-1cm-1, εdGTP = 11700 M-1cm-1, εdTTP = 8800 M-1cm-1, εdCTP = 7300 M-1cm-1). Allerdings kann die 27 Konzentration einer Oligonukleotidlösung gemäß der Gleichungen (1) und (2) mit Hilfe der beiden folgenden Näherungen auch vereinfacht und ohne Berücksichtigung der Nukleotidzusammensetzung ermittelt werden (Sambrook et al., 1989): (i) Nukleotide besitzen eine mittlere Molmasse von 340 g/mol und (ii) Nukleotidlösungen mit einer Konzentration von 35 µg/ml besitzen im Durchschnitt eine Absorption A260 von 1. c1 [µg/ml] = A260 . 35 . V (1) c1 . 1000 N . 340 (2) c2 [pmol/µl] = V: N: Verdünnung der Oligonukleotidlösung Anzahl der Nukleotide im Oligonukleotid 3.1.2 Spaltung von DNA mit Restriktionsendonukleasen Restriktionsendonukleasen erkennen die DNA an spezifischen Stellen und schneiden beide Stränge der Helix durch Spaltung der Phosphodiesterbindung. Sie stellen somit ein wichtiges Werkzeug in der Gentechnik dar. Ihre ursprüngliche Aufgabe ist es, ihren Wirt vor FremdDNA (Infektion mit Phagen) zu schützen, wobei die wirtseigene DNA durch Methylierung an der Erkennungsstelle vor der Spaltung geschützt wird (Adams et al., 1986). In dieser Arbeit wurden ausschließlich Enzyme des Typs-II verwendet. Diese erkennen eine 4 - 8 Basenpaare lange in der Regel palindrome (zweizählige Rotationssymmetrie) DNA-Sequenz und spalten die DNA innerhalb dieser Sequenz. Im Falle der verwendeten Restriktionsenzyme EcoR I (Schnittstelle: G↓AATTC) und Pst I (Schnittstelle: CTGCA↓G) entstehen dabei überstehende kohäsive Enden, die eine gezielte Rekombination unterschiedlicher DNA-Fragmente ermöglichen (Bukhari et al., 1977). Die Spaltung mit Restriktionsenzymen wurde sowohl für analytische als auch für präparative Zwecke verwendet. Analytische Restriktionsverdaue hatten dabei die folgende Zusammensetzung: DNA (ca. 100 - 400 ng/µl; 0.5 - 1 µg Gesamtmenge) Restriktionspuffer-H BSA (1 mg/ml) bidest. H2O EcoR I Pst I x µl 1 µl 1 µl (8-x) µl 5U 5U 28 Analytische Ansätze wurden ca. 60 min bei 37 °C inkubiert. Bei präparativen Ansätzen wurde mit einem Gesamtvolumen von 20 µl bei DNA-Mengen von ca. 1 - 5 µg gearbeitet und die Inkubationszeit wurde auf 3 h ausgedehnt. Die Lagerung der Ansätze erfolgte schließlich bei -20 °C. 3.1.3 Agarose-Gelelektrophorese Agarose ist ein lineares Polymer mit alternierenden β-1,4-verknüpften D-Galaktose- und 3,6Anhydro-L-Galaktoseeinheiten als Grundgerüst. Wird Agarose in einem geeigneten Puffer unter Erhitzen gelöst, so erstarrt es beim Abkühlen in Form einer Matrix, deren Porengröße eine Funktion der Agarosekonzentration ist. Die Agarose-Gelelektrophorese eignet sich zur Trennung von DNA-Fragmenten mit einer Größe von 500 - 20000 bp, wobei die Wanderungsgeschwindigkeit der DNA-Fragmente von deren Größe und Konformation abhängt (Sambrook et al., 1989). Die Detektion der DNA kann mit Ethidiumbromid erfolgen. Dabei handelt es sich um einen Fluoreszenzfarbstoff, der zwischen die Basen der DNA interkaliert und bei Bestrahlung mit UV-Licht im Gegensatz zum freien Farbstoff orange-rote Fluoreszenz zeigt (Sharp et al., 1973). Zur analytischen und präparativen Auftrennung von DNA-Fragmenten wurden 0.9 %ige Agarosegele verwendet. Dazu wurde die entsprechende Menge an Agarose suspendiert und bis zur Ausbildung einer klaren Lösung in 1xTAE-Puffer erhitzt. Nach Ersetzen des Wasserverlustes und Abkühlen auf ca. 60 °C wurde Ethidiumbromid-Lösung bis zu einer Endkonzentration von 0.5 µg/ml zugegeben. Daraufhin wurde die Lösung in einen mit Klebeband abgedichteten Gelträger gegossen und der Kamm zur Ausbildung der Probentaschen eingehängt. Nach Erstarren des Gels wurde das Klebeband entfernt, der Gelträger in die Elektrophorese-Apparatur eingesetzt, mit 1xTAE-Puffer überschichtet und der Kamm vorsichtig entfernt. Die zu untersuchenden DNA-Lösungen wurden mit Probenpuffer versetzt (30 Vol.%), durchmischt und in die Probentaschen pipettiert. Die Elektrophorese wurde dann 50 - 60 min bei ca. 80 V und 110 mA durchgeführt. Zur Auswertung wurden analytische Gele bei 254 nm photographiert. Isolierung von DNA aus Agarosegelen Die Isolierung von DNA-Fragmenten aus Agarosegelen erfolgte mit dem QIAquick Gel Extraction Kit. Die Methode basiert auf der Auflösung des Agarosegels mit Hilfe eines chaotropen Puffers, der das Binden der DNA an Silicagel im Folgeschritt ermöglicht (Vogelstein & Gillespie, 1979). Das gewünschte DNA-Fragment wurde mit einem Skalpell so knapp wie möglich aus dem präparativen Gel ausgeschnitten und in ein 2-ml-Reaktionsgefäß überführt. Um Strahlenschäden zu vermeiden, geschah dies unter längerwelligem UV-Licht (366 nm). Alle 29 weiteren Schritte wurden wie in der vom Hersteller mitgelieferten Anleitung durchgeführt (Fa. Qiagen, 1995; 1997). Die DNA-Fragmente wurden mit ca. 25 - 30 µl TE-Puffer bzw. mit dem mitgelieferten Elutionspuffer eluiert und bis zur weiteren Verwendung bei -20 °C gelagert. Konzentration und Reinheit der DNA-Lösungen wurden mittels Agarose-Gelelektrophorese bestimmt. 3.1.4 Ligation von DNA-Fragmenten Ligasen katalysieren die Bildung von Phosphodiesterbindungen zwischen 3’-Hydroxy- und 5’-Phosphatgruppen von dsDNA-Fragmenten. Die T4-DNA-Ligase benötigt für diese Reaktion ATP. Bei der Ligation von pKK-Vektorfragment und FucA-Genfragment war durch die Verwendung verschiedener nicht kompatibler Enden vor und hinter dem Gen, als Folge eines Restriktionsverdaus mit zwei unterschiedlichen Restriktionsenzymen (EcoR I und Pst I), die Orientierung des Inserts vorgegeben. Da somit auch eine Selbstligation ausgeschlossen war, konnte auf eine Dephosphorylierung der DNA-Fragmente mit alkalischer Phosphatase verzichtet werden. Zur Verbesserung der Ligationsausbeute wurde das Genfragment in fünf- bis siebenfachem molarem Überschuß eingesetzt. Ein Ligationsansatz hatte typischerweise die folgende Zusammensetzung: Genfragment Vektorfragment BSA (1 mg/ml) Ligase-Puffer bidest. H2O T4-DNA-Ligase 10 - 30 ng 10 - 30 ng 1 µl 1 µl ad 10 µl 1U Der Ansatz wurde über Nacht bei 16 °C inkubiert (Ferretti & Sgaramella, 1981). 3.1.5 Transformation Das Einschleusen von Plasmiden oder Phagen-DNA in Bakterien wird als Transformation bezeichnet. Dabei ist die Fähigkeit DNA aufzunehmen (Kompetenz) von mehreren Faktoren wie Bakterienstamm, Wachstumsbedingungen bzw. Wachstumsphase abhängig. In dieser Arbeit wurde die Methode nach Hanahan (1983) angewendet. Die Bakterienzellen werden dabei durch Behandlung mit eiskalter Salzlösung (TFB-Puffer) während der frühen log-Phase und anschließendem kurzen Hitzeschock (2 min, 42 °C) durch Veränderungen in der 30 Zellmembran aufnahmebereit (kompetent) gemacht. Dabei wurde folgendermaßen vorgegangen: In einem 50-ml-Kulturröhrchen wurden 10 ml 2xYT-Medium mit 100 µl einer Stammkultur bzw. einer Kolonie von einer Agarplatte angeimpft und bis zu einer OD578 = 0.3 bis 0.4 kultiviert. Daraufhin wurden 5 ml in ein neues Kulturröhrchen überführt und die restlichen Zellen weiter kultiviert. Nach Zentrifugation (5 min, 3000∗g) wurde das Nährmedium abgegossen, das verbleibende Pellet in 1.7 ml eiskaltem TFB-Puffer resuspendiert und 5 min auf Eis gestellt. Die Suspension wurde dann auf zwei 1.5-mlReaktionsgefäße verteilt und erneut zentrifugiert (5 min, 2000∗g, 4 °C). Der Überstand wurde wieder abgenommen, das Pellet in jeweils 212 µl TFB-Puffer und 2 µl 750 mM β-ME suspendiert und für mindestens 10 min auf Eis gestellt. Zur Transformation wurden dann 100 µl der kompetenten Zellen mit 1 - 5 µl DNA-Lösung versetzt und 60 - 90 min auf Eis gestellt. Der Hitzeschock wurde über einen Zeitraum von 2 min bei 42 °C ausgeführt. Im Anschluß daran wurden die Zellen sofort für 60 sec auf Eis gestellt. Plattierung von Plasmid-DNA Nach beendeter Transformation wurden die Transformationsansätze mit 300 µl 2xYTMedium versetzt und 30 min bei 37 °C inkubiert. Dadurch sollte die phänotypische Ausbildung der plasmidkodierten Ampicillin-Resistenz ermöglicht werden. Schließlich wurden die Ansätze mit einem Drigalski-Spatel gleichmäßig auf LBA-Agar-Platten ausgestrichen und ü. N. bei 37 °C inkubiert. Plattierung von RF-DNA (Top-Agar-Plattierung) Bakterienzellen, die von M13-Phagen infiziert worden sind, werden nicht lysiert. Allerdings wird der Stoffwechsel und somit das Wachstum der Wirtszelle bei der Vermehrung der Phagen verlangsamt. Deshalb erscheinen diese in einem homogenen Bakterienrasen als transparente Flecken (Plaques). Dies kann man sich zu Nutze machen, um Phagen-haltige Zellen mittels Top-Agar-Plattierung zu selektieren: Ca. 4 ml flüssiges Top-Agar wurden in einem sterilen Reagenzglas auf 42 °C temperiert. Nach Zugabe des Transformationsansatzes sowie 200 µl einer hochgewachsenen Bakterienkultur (weiterkultivierte Zellen s. o.) wurde der Top-Agar gleichmäßig auf einer LB-Agar-Platte ausgegossen und nach dem Erstarren ü. N. bei 37 °C inkubiert. Am folgenden Tag wurden gut isolierte Plaques gepickt, die entsprechenden Phagen vermehrt und anschließend die Phagen-DNA isoliert (cf. Kapitel 3.1.6 und 3.1.7). 31 3.1.6 Präparation von Plasmid-DNA und M13-dsDNA Plasmide sind zirkuläre extrachromosomale DNA-Moleküle, die natürlicherweise in Bakterien und einigen anderen Organismen vorkommen. Sie werden in veränderter Form als Klonierungsvektoren für DNA-Fragmente benutzt. Der Bakteriophage M13 ist ein filamentöser, nicht-lytischer Virus. Seine zirkuläre DNA liegt in den Wirtszellen in ihrer replikativen, doppelsträngigen Form vor (RF-DNA), während der verpackte und ins extrazelluläre Medium sekretierte Virus nur einzelsträngige DNA enthält. Diese Tatsache ermöglicht es, selektiv nur ssDNA bzw. dsDNA zu präparieren. Zur Isolierung von Plasmiden bzw. RF-DNA wurde in dieser Arbeit ein Kit der Firma Qiagen verwendet (QIAprep Spin Plasmid Kit). Dabei handelt es sich um eine Modifizierung der Methode von Birnboim & Doly (Birnboim & Doly, 1979; Vogelstein & Gillespie, 1979). Die Methode beruht auf einer selektiven Denaturierung höhermolekularer DNA im Alkalischen bei pH 12.0 - 12.5, während Plasmid-DNA unter diesen Bedingungen intakt bleibt. Bei der Neutralisation renaturiert die chromosomale DNA unter Ausbildung eines unlöslichen Netzwerks. Sie wird abgetrennt und die verbleibende DNA bei hoher Salzkonzentration in einer Minisäule an Silicagel gebunden (Chen & Thomas, 1980), gewaschen und eluiert. Überschüssiges SDS wird als Kaliumsalz gefällt und RNA gleich nach der Zell-Lyse durch RNAse abgebaut. In der Regel wurden ca. 5 ml einer ü. N. kultivierten Bakteriensupension 5 min bei 3000∗g zentrifugiert und das Pellet (im Falle von RF-DNA das entsprechende Bakterienpellet aus der Präparation der ssDNA; siehe Kapitel 3.1.7) in 250 µl Puffer-P1 resuspendiert. Alle folgenden Schritte wurden wie in der vom Hersteller mitgelieferten Beschreibung durchgeführt (Fa. Qiagen, 1995). Die Elution der DNA erfolgte mit 50 µl TE-Puffer. Die Konzentration und Reinheit der erhaltenen Plasmidlösungen wurden mittels AgaroseGelelektrophorese bestimmt. Die Lagerung erfolgte bei -20 °C. 3.1.7 Präparation von M13ss-DNA Die Präparation von M13ss-DNA erfolgte mit dem QIAprep Spin M13 Kit (Fa. Qiagen, 1994). Dazu wurde ein Phagenplaque zusammen mit 200 µl einer dicht gewachsenen Bakteriensuspension von E. coli TG1 bzw. XL1 in 5 ml 2xYT-Medium kultiviert (50-mlKulturröhrchen) und 6 h bei 37 °C waagerecht geschüttelt. Nach Zentrifugation (10 min, 3000∗g) wurde der Überstand auf 2-ml-Reaktionsgefäße verteilt und erneut zentrifugiert (10 min, 15800∗g). Das Zellpellet wurde für eine eventuelle spätere Isolierung der M13dsDNA bei -20 °C gelagert. Jeweils 1.6 ml des Überstandes wurden abgenommen, mit 16 µl M13-Präzipitationspuffer versetzt und 2 min bei RT inkubiert. Alle folgenden Schritte wurden 32 wie in der Anleitung zum Kit beschrieben durchgeführt. Die Elution der DNA erfolgte nach 5 min Inkubation auf der Säule mit 30 - 50 µl TE-Puffer. Die Konzentration der erhaltenen ssDNA wurde dann über Agarose-Gelelektrophorese durch den Vergleich mit den Bandenintensitäten verschieden konzentrierter M13-ssDNA-Lösungen bestimmt. Die Konzentration des Standards wurde zuvor photometrisch über die Absorption bei 260 nm bestimmt. Bis zur weiteren Verwendung wurde die DNA bei -20 °C gelagert. 3.1.8 Präparation von Uracil-haltiger M13-ssDNA E. coli synthetisiert normalerweise ein Enzym (Uracil-N-glycosylase), das in DNA eingebautes Uracil entfernt, wodurch diese Stellen zum Angriffspunkt für spezifische Nukleasen werden, welche die DNA dann weiter abbauen können (Lindahl, 1974; Duncan et al., 1978). In Uracil-N-glycoslase defizienten Stämmen (ung--Stämme) allerdings wird Uracil nicht entfernt, und die bakterielle DNA enthält somit einen geringen Anteil an Uracil. In dutung- F’-Stämmen ist dieser Anteil noch erhöht, da der dUTP-Spiegel in diesen Stämmen aufgrund fehlender dUTPase erhöht ist. M13-Bakteriophagen, die durch Infektion solcher Stämme gewonnen werden, enthalten in ihrer DNA in der Regel 20 - 30 Uracile. Zur Präparation Uracil-haltiger M13-ssDNA als Templat für die Mutagenese nach Kunkel wurden 20 ml 2xYT-Medium (0.25 µg/ml Uridin, 35 µg/ml Chloramphenicol) in einem 100-ml-Kolben mit 100 µl einer Übernachtkultur (2xYT-Medium, 34 µg/ml Chloramphenicol) von E. coli CJ236 (dut- ung- F’) angeimpft (Kunkel et al., 1987). Nach Zugabe von 40 µl M13mp19-fuca-Phagenstammlösung, die zuvor 10 min bei 65 °C inkubiert worden war, wurde 7.5 Stunden unter heftigem Schütteln kultiviert. Daraufhin wurden die Zellen abzentrifugiert (Sorvall-SS34, 10 min, 12000∗g, 4 °C) und aus dem Überstand die Phagen durch Zugabe eines Viertels des Volumens an 5xPEG/NaCl-Lösung gefällt. Zur vollständigen Fällung wurde der Ansatz über Nacht bei 4°C stehengelassen. Nach Zentrifugation (Sorvall-SS34, 10 min, 48000∗g, 4 °C) wurde der Überstand abgesaugt und das Phagenpellet in 200 µl TE-Puffer aufgenommen. Unlösliche Bestandteile wurden durch kurze Zentrifugation abgetrennt. Die Phagenlösung wurde mit jeweils 200 µl ss-Phenol, ss-Phenol/Chloroform (1:1) und Chloroform-Isoamylalkohol (24:1) extrahiert. Daraufhin wurde die Phagen-DNA durch Zugabe von 540 µl Ethanol und 18 µl 7.5 M Ammoniumacetat über Nacht bei -20 °C gefällt. Nach Zentrifugation (20 min, 15800∗g, 4 °C) wurde das DNAPellet in 20 µl TE-Puffer aufgenommen und die Reinheit bzw. die Ausbeute der ssDNA mittels Agarosegel bestimmt. 33 3.1.9 Mutagenese 5’-Phosphorylierung von Oligonukleotiden Die Ligation des bei der Mutagenese in vitro synthetisierten DNA-Stranges zu einem zirkulären Produkt erfordert 5’-phosphorylierte Enden. Deshalb wurden die zur Mutagenese eingesetzten Oligonukleotide mit einer Kinase am 5’-Ende phosphoryliert. Dazu wurde der folgende Ansatz 30 min bei 37 °C inkubiert und dann zur Inaktivierung der Kinase 10 min auf 70 °C erhitzt, worauf er bis zur weiteren Verwendung bei -20 °C gelagert wurde: Oligonkleotid (10 pmol/µl) Kinase-Puffer 10 mM ATP 100 mM DTE H2O T4-Polynukleotid-Kinase 5 µl 3 µl 3 µl 1.5 µl 17 µl 5U Mutagenese nach Kunkel Bei dieser Methode (Kunkel, 1985; Kunkel et al., 1987) verwendet man einzelsträngige Templat-DNA, bei der ein geringer Prozentsatz an Thymin durch Uracil ersetzt ist. Die Kunkel-Methode beruht dabei auf der starken Selektion gegen Uracil-haltige DNA in E. coli ung+-Stämmen. Die Templat DNA wird in einem dut- ung- F’-Stamm (z. B. E. coli CJ236) erzeugt. Die resultierende Uracil-haltige M13-ssDNA dient dann als Templat für die Mutagenese, bei der in vitro ein Heteroduplex erzeugt wird; mit Uracil im Templat-Strang und Thymin im neu synthetisierten Strang. Dabei wird die gewünschte Mutation durch ein entsprechend konstruiertes Mutagenese-Oligonukleotid eingeführt. Die Transformation der DNA in einen ung+-Stamm hat dann eine Zerstörung des Templat-Strangs und somit Unterdrückung der Produktion von WT-Phagen zur Folge. Annealing: 5’-phoshoryliertes Oligonukleotid (0.33 pmol/µl) Uracil-haltige Templat-DNA (0.03 pmol/µl) 10xASB-Puffer H2O → 3 min, 70°C; Abkühlen bei RT 2 µl 2 µl 1 µl 5 µl 34 Elongation + Ligation: 2xEB-Puffer Klenow-Fragment (1 U/µl) T4-DNA-Ligase (1 U/µl) → über Nacht bei 15°C 12 µl 1 µl 1 µl Die Mutagenese-Ansätze werden mit 0.5 µl 0.5 M EDTA versetzt, 10 min auf 65 °C erhitzt und in E. coli JM101 transformiert. Mutagenese nach Eckstein Die Mutagenese nach Eckstein (Taylor et al., 1985; Sayers & Eckstein, 1989; 1991) wurde mit einem Kit der Firma Amersham durchgeführt. Die dazu benötigten Mutagenese-Primer wurden entsprechend der mitgelieferten Anleitung geplant (Fa. Amersham, 1995). Der schematische Verlauf der Mutagenese ist in Abbildung 6 gezeigt. Im ersten Schritt hybridisiert das mutagene Oligonukleotid an die einzelsträngige M13-Templat-DNA und wird im Folgeschritt durch T7-DNA-Polymerase und T4-DNA-Ligase unter Verwendung von dCTPαS anstelle von dCTP zum Heteroduplex verlängert. Danach wird überschüssige Templat-DNA durch T5-Exonuklease abgebaut. Im nächsten Schritt, dem sogenannten nicking, wird die Heteroduplex-DNA mit Nci I geschnitten. Dieses Restriktionsenzym (Erkennungssequenz und Schnittstelle: CC↓(C/G)GG) kann keine Phosphorothioatbindungen spalten, so daß nur der nicht-mutierte WT-Strang geschnitten wird. Diese Schnittstelle im WT-Strang ist nun Angriffspunkt für die im nächsten Schritt verwendete Exonuklease-III, die ausgehend vom freien 3’-Ende den nicht mutierten Strang des Heteroduplex in 3’→5’ Richtung abbaut. Die Dauer des Exonuklease-III-Abbaus des parentalen Strangs ist dabei so gewählt, daß noch ein kurzes doppelsträngiges DNA-Stück erhalten bleibt, das dann im letzten Schritt als Primer für die DNA-Polymerase-I fungiert, wodurch eine Repolymerisierung zum Homoduplex mit mutierter Sequenz erfolgt. Die einzelnen Teilschritte der Mutagenese wurden, wie in der mitgelieferten Anleitung beschrieben, durchgeführt. Dabei wurden geringfügige Modifikationen vorgenommen. Der annealing-Ansatz wurde nach 3 min Inkubation bei 75 - 80 °C nicht sofort, sondern im Wasserbad über einen Zeitraum von ca. 40 min langsam abgekühlt. Dadurch wird vor allem bei Oligonukleotiden, die zur Ausbildung stabiler Sekundärstrukturen neigen, die kinetisch bevorzugte Ausbildung interner Loop-Strukturen zugunsten einer Hybridisierung mit der Templat-DNA verschoben. Die Extension zum Heteroduplex im Folgeschritt kann wahlweise mit T7-DNA-Ligase oder Klenow-Fragment erfolgen. Bei den in dieser Arbeit erzeugten 35 Mutanten geschah dies ausnahmslos mit T7-DNA-Ligase. Die Transformation des Mutagenese-Ansatzes erfolgte in E. coli TG1. Um eine hohe Transformationsrate zu erreichen, wurde das Medium mit einer auf M9-Minimalmedium gewachsenen Kolonie, die nicht älter als einen Monat war, angeimpft. Pro Transformationsansatz wurden 100 µl kompetente E. coli TG1-Zellen zu 2 - 5 µl mutierter M13-dsDNA gegeben. In einigen Fällen mußte der Mutagenese-Ansatz in Verdünnung zugegeben werden, um bei der anschließenden Plattierung ausreichend separierte Phagenplaques zu erhalten. mutagenes Oligonukleotid x Annealing 70 °C, 3 min 37 °C, 30 min x rekombinantes ssDNA-Templat 0.8 U T4-DNA-Polymerase 2.5 U Ligase RT, 10 min 37 °C, 30 min 70 °C, 15 min Extension und Ligation mit dCTPαS x Entfernung von ssDNA-Templat 2000 U T5-Exonuklease 37 °C, 30 min 70 °C, 15 min Nicking des WT-Strangs 5 U Nci I 37 °C, 90 min x Nci I 160 U Exonuklease-III 37 °C, 30 min 70 °C, 15 min Abbau des WT-Strangs x x x Ligation 3.5 U DNA-Polymerase-I 2.5 U Ligase 37 °C, 60 min Transformation Abbildung 6 Schematische Darstellung der Mutagenese nach Eckstein (nach Fa. Amersham, 1995). 36 3.1.10 DNA-Sequenzierung Die DNA-Sequenzierung erfolgt heute fast ausschließlich nach der Didesoxymethode (Sanger et al., 1977). Sie beruht auf der in-vitro-Synthese komplementärer DNA-Stränge. Durch den Einbau von 2’,3’-Didesoxynukleotiden wird das weitere Kettenwachstum blockiert. Da der Einbau des Didesoxyanalogons statistisch erfolgt, entstehen unterschiedlich lange Fragmente, die jedoch entsprechend dem verwendeten Didesoxynukleotid immer die gleiche terminale Base besitzen. In vier parallelen Ansätzen, wobei jeweils eines der vier ddNTPs zugesetzt wird, können so die Abbruchfragmente nach allen vier Basen erhalten werden. Die DNA-Fragmente aus den vier Ansätzen werden über eine denaturierende Polyacrylamid-Gelektrophorese der Größe nach aufgetrennt. Die Detektion der Fragmente kann durch Einbau von radioaktiv markiertem [α-35S]-dATP während der Elongation des neu synthetisierten Strangs (Biggin et al., 1983) oder durch Verwendung eines kovalent an die Sequenzierungsprimer bzw. die ddNTPS gebundenen, nicht-radioaktiven labels erfolgen. Als label können dabei z. B. Biotin, Fluorescein (Cohen et al., 1993) oder Digoxigenin (Sagner, 1993) verwendet werden. Die Sequenz kann dann auf dem Sequenzierungsgel bzw. auf der blotting-Membran in 5’→3’-Richtung abgelesen werden. Um eine radioaktive Sequenzierung zu umgehen, wurden in dieser Arbeit zunächst 5’-DIG-markierte Primer benutzt, später dann nur noch Biotin-markierte ddNTPs. Für die DIG-Variante wurde das DIG Taq DNA Sequencing Kit for Standard and Cycle Sequencing (Fa. Boehringer Mannheim) verwendet, für die Biotin-Variante das GATC-BioCycle Sequencing Kit (Fa. GATC) in Kombination mit der Thermo-Sequenase. Die Sequenzierungsreaktionen wurden im Thermocycler nach dem Cycle-SequencingVerfahren durchgeführt. Dabei wird durch zyklische Wiederholung von annealing, Elongation und Denaturierung weniger DNA benötigt als bei Verwendung der T7-DNA-Polymerase, da eine lineare Amplifizierung stattfindet. Die Verwendung der Thermo-Sequenase, einer thermostabilen DNA-Polymerase, ermöglicht dabei eine Elongation bei höheren Temperaturen. Dadurch wird auch das Auftreten sogenannter Stop-Artefakte reduziert, die durch die Termination an stabilen Sekundärstrukturen entstehen. In dieser Arbeit erfolgte das Cycle-Sequencing mit dem folgenden Temperaturprogramm: 1) 95 °C, 3 min 2) 58 °C, 30 sec; 95 °C, 30 sec (30 Zyklen) 3) 60 °C, 4 min Die Elektrophorese wurde mit Hilfe des GATC 1500-Systems durchgeführt. Bei diesem Gerät werden die aus dem Gel austretenden DNA-Fragmente direkt auf eine an der Gelunterkante vorbeilaufende Nylonmembran geblottet. Nach der Vernetzung (durch UV) der 37 DNA mit der Membran erfolgt die Detektion der DNA mit Anti-DIG-Alkalische-PhosphataseKonjugat im Falle von Digoxigenin bzw. mit Streptavidin-Alkalische-Phosphatase-Konjugat im Falle von Biotin. Diese Konjugate binden an das label und die darin enthaltene Alkalische Phosphatase katalysiert die Umsetzung des chromogenen Substrats X-Phosphat zu 5-Brom-4chlor-indigo. Durch gleichzeitige Zugabe von NBT entsteht dabei ein unlöslicher violettbrauner Formazan-Niederschlag. Da alle Reaktionsschritte beginnend mit dem CycleSequencing (Fa. Boehringer Mannheim, 1994; Fa. GATC, 1996) über die Elektrophorese (Fa. GATC, 1995) bis hin zur Entwicklung der blotting-Membran (Fa. Boehringer Mannheim, 1993; Fa. GATC, 1996) entsprechend den Herstellerangaben durch-geführt wurden, kann an dieser Stelle auf eine explizite Beschreibung verzichtet werden. 3.2 Proteinpräparation Die Vorgehensweise bei der Produktion und Reinigung der FucA und ihrer Mutanten ist in Abbildung 7 schematisch dargestellt. Als Expressionsstamm wurde E. coli JM105 verwendet, wobei jeweils in 1-l-Schüttelkolben kultiviert wurde. Nach Zellaufschluß mit Ultraschall und Fällung der DNA mit Polymin-P wurden die Mutanten in zwei Schritten wie von Dreyer (1995) für den WT beschrieben gereinigt: zunächst mit einer AnionenaustauscherChromatographie (IEC) an DEAE-Sepharose-CL6B und daran anschließend mittels Gelpermeations-Chromatographie (GPC) an Superdex S200. 3.2.1 Stammkulturen und Expressionstests Die pKKFA2-Plasmide mit den entsprechend mutierten fuca-Genen wurden in E. coli JM105 transformiert und auf LBA-Agar-Platten nach positiven Klonen selektioniert. Eine einzelne Kolonie wurde von der entsprechenden Agar-Platte gepickt und bei 37 °C in 6 ml LBAMedium (50-ml-Kulturröhrchen) bis zu einer OD578 = 0.6 kultiviert. 1.5 ml wurden entnommen, mit IPTG (Endkonzentration 1 mM) induziert und über Nacht bei 37 °C kultiviert. Der Rest wurde in 1-ml-Portionen aufgeteilt, mit 500 µl 90 %igem Glycerin (v/v) versetzt, durchmischt und nach 30 min Inkubation bei RT schließlich bis zur weiteren Verwendung als Startkulturen bei -20 °C gelagert. Am folgenden Tag wurden 100 µl des induzierten Ansatzes zentrifugiert (5 min, 8000∗g), das Bakterienpellet in 40 µl SDSProbenpuffer aufgenommen und mittels SDS-PAGE auf Überexpression von FucA getestet. Der Rest wurde mit Toluol/Desoxycholat aufgeschlossen und auf FucA-Aktivität getestet. 38 pKKFA2 Transformation in E. coli JM 105 Startkulturen/Expressionstests Kultivierung in E. coli JM105 in 1-l-Schüttelkolben (4 x 300 ml) Zentrifugation 30 min, 10800∗g Resuspension der Bakterienzellen in 25 ml TMZ-Puffer, Ultraschallaufschluß Zentrifugation 30 min, 27000∗g Polymin-P-Fällung 45 min, 4 °C Zentrifugation 30 min, 19000∗g Anionenaustauscher-Chromatographie DEAE-Sepharose-CL6B Elution mit NaCl-Gradient Aufkonzentrierung auf 30 - 40 mg/ml Gelpermeations-Chromatographie Superdex S200 Dialyse gegen TMZ-Puffer Analytik Aufkonzentrierung auf 10 mg/ml Kristallisation Abbildung 7 Schematische Darstellung der Schritte zur Präparation der FucAMutanten und anschließender Kristallisation bzw. Analytik der Mutanten 39 3.2.2 Bakterienkultivierung Die präparative Kultivierung der FucA-Mutanten erfolgte in 1-l-Schüttelkolben. Dazu wurden zunächst 4 x 10 ml LBA-Medium (50-ml-Kulturröhrchen) mit 100 - 300 µl einer bei -20 °C gelagerten Startkultur angeimpft und bei -37 °C so lange kultiviert, bis die OD578 = 0.6 betrug. Mit diesen Vorkulturen wurden daraufhin vier 1-l-Schüttelkolben mit 300 ml LBA-Medium angeimpft und bei 37 °C weiterkultiviert bis wiederum ein OD578 = 0.6 - 0.8 erreicht war. Daraufhin wurde durch Zugabe von 150 µl 1 M IPTG pro Kolben (Endkonzentration 0.5 mM) induziert. Es wurde weitere 8 - 11 h bei 37 °C unter Schütteln kultiviert und die Zellen schließlich abzentrifugiert (30 min, 10800∗g, 8 °C). Das erhalten Bakterienpellet wurde entweder gleich aufgeschlossen oder bei -20 °C gelagert. 3.2.3 Bakterienaufschluß und Abtrennung von Nukleinsäuren Das Bakterienpellet (vgl. Kapitel 3.2.2) wurde mit 4 - 5 ml TMZ-Puffer-I pro Gramm Feuchtzellen bis zur Homogenität resuspendiert. Der Zellaufschluß erfolgte dann durch 2 x 7 min Beschallen unter Kühlung (4 °C) bei maximaler Geräteleistung. Zelltrümmer und unlösliche Bestandteile wurden durch anschließende Zentrifugation abgetrennt (30 min, 27000∗g, 8 °C). Um die Kapazität der Anionenaustauscher-Säule im ersten Reinigungsschritt nicht unnötig zu erniedrigen, wurde direkt nach dem Zellaufschluß eine Fällung der Nukleinsäuren mit Polymin-P durchgeführt. Dazu wurde der Überstand abgenommen und mit 5 µl/ml Polymin-P-Lösung versetzt. Nach 45 min Inkubation bei 4 °C wurde erneut zentrifugiert (30 min, 19000∗g, 8 °C) und der Überstand nach der Fällung der Nukleinsäuren unverzüglich auf die Ionenaustauscher-Säule aufgetragen. Die rasche Aufarbeitung war nötig, da auf Zusatz des Proteaseinhibitors PMSF verzichtet wurde, um eine Reaktion mit im aktiven Zentrum vorhandenen Serinen auszuschließen. 3.2.4 Anionenaustauscher-Chromatographie Ionenaustauscher bestehen aus chemisch modifizierten Polymeren, die kovalent gebundene, positiv oder negativ geladene funktionelle Gruppen enthalten. Diese werden dann in Lösung durch die entsprechenden Gegenionen elektrostatisch abgesättigt (Sternbach, 1991). Der erste Reinigungsschritt der FucA und ihrer Mutanten bestand in einer Anionenaustauscher-Chromatographie (IEC) an DEAE-Sepharose-CL6B. Dabei besteht das Säulenmaterial aus positiv geladenen Diethylaminoethylgruppen, die kovalent mit einer Agarose-Matrix verknüpft sind. Aufgrund des pI-Werts von 6.55 der FucA und des in der 40 gleichen Größenordnung erwarteten pI-Werts für die Mutanten, war ein Arbeiten bei physiologischen Bedingungen möglich (pH = 7.6). Die Parameter der IEC sind in Tabelle 2 aufgeführt. Der Rohextrakt wurde nach Polymin-P-Fällung und Zentrifugation direkt auf die zuvor mit ca. 9 Vt TMZ-Puffer-I äquilibrierte Ionenaustauscher-Säule aufgetragen, die Säule anschließend mit 3 - 4 Vt TMZPuffer-I gewaschen und die Elution von Proteinen durch Messung der Absorption bei 280 nm verfolgt. Die Elution der FucA erfolgte dann durch Anlegen eines linearen Salzgradienten von 0 - 500 mM NaCl (Gesamtvolumen des Gradienten ca. 9 Vt) oder im späteren Verlauf der Arbeit durch Anlegen eines Stufengradienten von 50 mM NaCl und anschließendem linearen Gradienten von 50 - 400 mM NaCl. Die Flußrate betrug jeweils 120 ml/h, wobei nach Anlegen des NaCl-Gradienten fraktioniert wurde (8-ml-Fraktionen). Die Fraktionen wurden mittels SDS-PAGE auf FucA untersucht, die entsprechenden Fraktionen vereinigt und zur weiteren Aufreinigung mittels GPC auf eine Konzentration von ca. 30 - 50 mg/ml eingeengt. Nach Beendigung der Chromatographie wurde die Säule mit 5 Vt TMZ-Puffer-VI regeneriert. Nach 4 Läufen wurde zusätzlich eine Batch-Regenerierung mit 0.1 M NaOH vorgenommen. Tabelle 2 Übersicht über die Parameter bei der Anionenaustauscher-Chromatographie Säulenmaterial Säulenhöhe Säulendurchmesser Gelvolumen Vt Temperatur Flußrate Äquilibrierung Auftrag Waschen Salzgradient [mM] Regenerierung DEAE-Sepharose-CL6B 26 cm 3 cm 184 ml 4 °C 120 ml/h (17 cm/h) 9 Vt TMZ-Puffer-I 25 - 30 ml Rohextrakt nach Polymin-P-Fällung 3 - 4 Vt TMZ-Puffer-I 0 - 500 mM NaCl 4.5 Vt TMZ-Puffer-I 4.5 Vt TMZ-Puffer-V oder 50 mM NaCl 1.5 - 2 Vt TMZ-Puffer-II 50 - 400 mM NaCl 4.5 Vt TMZ-Puffer-II 4.5 Vt TMZ-Puffer-IV 5 Vt TMZ-Puffer-VI 3.2.5 Gelpermeations-Chromatographie Bei der Gelpermeations-Chromatographie werden die Proteine aufgrund ihrer unterschiedlichen Größen getrennt. Die Elution erfolgt gemäß der Molekularmasse, wobei Proteine mit höherer Molekularmasse zuerst eluiert werden. Das Säulenmaterial besteht aus porösen 41 Kügelchen aus einem unlöslichen, aber stark hydratisierten Polymer. Bei der in dieser Arbeit verwendeten Säule (Superdex S200) handelt es sich um ein Dextran, das kovalent an stark vernetzte Agarose gebunden ist, und eine Trennung von Proteinen im Molekularmassenbereich von 10 bis 600 kDa erlaubt. Die vereinigten Fraktionen der IEC wurden nach Aufkonzentrierung und Zentrifugation (10 min, 15800∗g, 4 °C) mit einer Spritze über eine Injektionsschleife auf die GPC-Säule aufgetragen. Einen Überblick über die Säulenparameter gibt Tabelle 3. Pro Lauf wurden 20 - 40 mg Protein aufgetragen. Die Elution erfolgte isokratisch mit TMZ-Puffer-III bei einem Fluß von 36 ml/h, wobei jeweils 1.8 bzw. 2.4 ml fraktioniert wurden. Die Detektion erfolgte bei 280 nm und die weitere Analyse der proteinhaltigen Fraktionen via SDS-PAGE. Tabelle 3 Übersicht über die Parameter bei der Gelpermeations-Chromatographie Säulenmaterial Säulenhöhe Säulendurchmesser Gelvolumen Temperatur Flußrate Äquilibrierung Auftrag Elution Superdex S200 prep grade 60 cm 1.6 cm 121 ml RT 36 ml/min (18 cm/h) 4 Vt TMZ-Puffer-III 700 - 900 µl Proteinlösung nach IEC (max. 40 mg) 1 Vt TMZ-Puffer-III 3.2.6 Dialyse Das Entsalzen von Proteinlösungen erfolgte mittels Dialyse. Dabei wurde die Proteinlösung in einem Dialyseknopf (Volumen ≤ 500 µl) oder in einem Dialyseschlauch mindestens zweimal 8 h gegen das 100 - 200fache Volumen des entsprechenden salzfreien Puffers dialysiert. Um einen eventuellen Proteinverlust aufgrund eines undichten Dialyseschlauches zu erkennen und entsprechend reagieren zu können, wurde unmittelbar vor und nach der Dialyse die Proteinkonzentration bestimmt. 3.2.7 Konzentrierung von Proteinlösungen Für die Konzentrierung von Proteinlösungen wurden Centriprep-30- bzw. Centricon-30Konzentratoren mit einem Fassungsvermögen von 15 bzw. 2.5 ml verwendet. Größere Volumina wurden dabei in der Regel durch Mehrfachbeladungen eingeengt. In Extremfällen wurde jedoch auf Amicon-Ultrafiltrationszellen mit einer YM5/PM10-Membran zurück- 42 gegriffen, bei denen unter leichtem Rühren durch Anlegen eines Stickstoff-Überdrucks (2 bar) aufkonzentriert wurde. 3.3 Proteinanalytik 3.3.1 Diskontinuierliche SDS-Polyacrylamid-Gelelektrophorese Die SDS-PAGE ermöglicht es, Proteine in ihrer denaturierten Form entsprechend ihren Molmassen zu trennen (Laemmli, 1970). Dabei werden die Proteine vor dem Auftragen auf das Gel mit SDS denaturiert. Es bindet 1.4 g SDS pro g Protein, was zur Folge hat, daß die elektrophoretische Mobilität im wesentlichen durch Molekularsiebeffekte bestimmt wird und in einem linearen Zusammenhang zum Logarithmus der Molmasse steht. Die verwendeten Polyacrylamidgele entstehen durch Copolymerisation von Acrylamid und einem geeigneten Vernetzer wie N,N’-Methylenbisacrylamid zu einem dreidimensionalen Gitter. Durch Variation der Totalacrylamidkonzentration T und des Vernetzungsgrades C kann die Größe der entstehenden Poren gezielt verändert werden (Westermeyer, 1990). Durch Fokussierung der Proteine in einem großporigen Sammelgel und Trennen in einem kleinporigen Trenngel (diskontinuierliche SDS-PAGE) lassen sich zusätzlich Bandenschärfe und Trenneffekte verbessern (Righetti et al., 1990). Bei der diskontinuierlichen SDS-PAGE im Rahmen dieser Arbeit kamen Gele mit den Dimensionen 8 cm x 7 cm x 0.75 mm zur Verwendung. Die Totalacrylamidkonzentration des Sammelgels betrug T = 6 %, die des Trenngels T = 12 % und der Vernetzungsgrad jeweils C = 2.5 %. Pro Probentasche wurden 10 - 20 µl Proteinlösung (5 - 30 µg Protein) aufgetragen. Die Lösungen enthielten mindestens 25 % (v/v) SDS-Probenpuffer und wurden vor dem Auftragen gekocht (5 min, 100 °C). Die Elektrophorese erfolgte bei einer konstanten Stromstärke von 27 mA pro Gel sowie einer Spannung von ca. 60 V (Beginn) bis 220 V (Ende). Nach beendeter Elektrophorese wurden die Gele 20 min in Coomassie-Lösung (zuvor in der Mikrowelle erwärmt) gefärbt, 20 min in ebenfalls warmer Entfärbelösung entfärbt und zur endgültigen Entfärbung ü. N. in 10 %ige (v/v) Essigsäure gelegt. Zur Konservierung wurden die Gele ca. 5 min in dest. Wasser gelegt und dann in einem Geltrockner zwischen zwei Cellophanfolien getrocknet. 3.3.2 Isoelektrische Fokussierung (IEF) Die IEF ist eine hochauflösende Methode, bei der in Gegenwart eines konstanten pHGradienten die Proteine im elektrischen Feld nach ihren isoelektrischen Punkten getrennt werden. Sie wandern unter diesen Bedingungen so lange, bis sie den pH-Wert erreicht haben, 43 an dem ihre Nettoladung null ist (pH = pI). An diesem Punkt findet dann gleichzeitig eine Fokussierung zu schärferen Banden statt. Um Molekularsiebeffekte zu vermeiden, müssen die verwendeten Gele eine niedrige Acrylamidkonzentration besitzen (T = 3 - 5 %). Die Ausbildung des pH-Gradienten erfolgt mittels sogenannter Trägerampholyte (Dunn, 1989; Righetti et al., 1990). Die IEF wurde bei 7 °C mit “Servalyte Precotes“-Gelen (pH-Bereich 3 - 10) nach der vom Hersteller mitgelieferten Vorschrift durchgeführt. Der Kontakt zwischen Elektrode und Gel wurde durch passend zurechtgeschnittene Filterpapierstreifen hergestellt, wobei diese Streifen mit der entsprechenden Anoden- bzw. Kathodenlösung getränkt wurden. Das Gel wurde vor dem Probenauftrag 30 min bei 200 V vorfokussiert. Die Proteinlösungen (15 µl; 0.5 - 1.5 mg/ml), die zuvor wegen der erforderlichen geringen Ionenstärke gegen TMZPuffer-I dialysiert worden waren, wurden über spezielle Applikatorstreifen aufgetragen. Bei einer Leistungsbegrenzung von 8 W wurde so lange fokussiert, bis die maximale Spannung von etwa 2000 V erreicht war (2 - 2.5 h). Anschließend wurde das Gel 30 min in Fixierlösung, 2 min in Entfärbelösung, 30 min in Färbelösung und dann bis zur vollständigen Entfärbung des Hintergrundes (zweimal 15 min) in Entfärbelösung gebadet. Zur Konservierung wurde das Gel an der Luft getrocknet. 3.3.3 Bestimmung der Proteinkonzentration Die Konzentration einer gereinigten Proteinlösung läßt sich bei bekannter Aminosäurezusammensetzung über die Absorption bei 280 nm bestimmen. Aus der Anzahl der bei 280 nm absorbierenden Tryptophan-, Tyrosin- und Cystin-Reste berechnet sich der molare Extinktionskoeffizient in guter Näherung nach Gleichung 3 (Gill & von Hippel, 1989). ε280nm = nTrp · εTrp + nTyr · εTyr + nCys · εCys nx: εx: (3) Anzahl der jeweiligen Aminosäure im Protein Extinktionskoeffizient der jeweiligen Aminosäure bei 280 nm (εTrp = 5690 M-1cm-1, εTyr = 1280 M-1cm-1, εCys = 60 M-1cm-1) Der molare Extinktionskoeffizient ε280 für die FucA aus E. coli mit zwei Tryptophan-, sieben Tyrosin- und einem Cystin-Rest (Cys14 bildet, wie aus der Röntgenstruktur ersichtlich ist, eine Disulfidbrücke mit β-ME aus dem Präparationspuffer) errechnet sich somit zu 20380 M-1cm-1. Für Mutanten, bei denen die Anzahl dieser Aminosäurenreste verändert wurde, 44 müssen die entsprechenden Korrekturen mitberücksichtigt werden. Die Proteinkonzentration kann dann über das Lambert-Beer’sche-Gesetz nach Gleichung (4) berechnet werden. c[mg/ml] = A 280 ⋅ Mr 280 ⋅ d (4) d: Schichtdicke der Küvette [cm] 3.3.4 Röntgenfluoreszenz-Spektroskopie Die Röntgenfluoreszenz-Spektroskopie ist eine Möglichkeit zur qualitativen und semiquantitativen Analyse von Pulverpresslingen. Wird ein Elektron durch energiereiche Röntgenstrahlung aus einem Festkörper herausgeschlagen, so fällt ein Elektron aus einem höheren Orbital in das leere Orbital und emittiert dabei ein Photon (Röntgenfluoreszenz). Die Wellenlänge des emittierten Photons hängt von der Energiedifferenz der beiden Orbitale ab, die wiederum bei den K-Schalen leicht elementspezifisch analysiert werden kann. Zur Probenvorbereitung wurden die Proteinlösungen (insgesamt 50 - 200 mg) dreimal gegen 5 mM Tris-Puffer (pH 7.6) dialysiert und anschließend lyophilisiert. Eine Dialyse gegen dest. H2O war nicht möglich, da die FucA-Proben unter diesen Bedingungen im Dialyseschlauch ausfielen. Die Messungen selbst wurden freundlicherweise von der Firma Siemens durchgeführt. Verwendet wurde dabei das Röntgenfluoreszenzphotometer SRS3000 in seiner Standardkonfiguration unter Verwendung der Basissoftware SPECTRA3000 und des universell einsetzbaren Analysenprogramms SSQ3000 (Siemens SemiQuant). Zunächst wurde das gesamte Elementspektrum von Natrium bis Uran kontinuierlich durchlaufen. Bei der semiquantitativen Auswertung wurden dann aus den Scans automatisch die Nettointensitäten der vorhandenen Elementlinien bestimmt und daraus unter Anwendung einer probenspezifischen Matrixkorrektur (als Matrix wurde H7C3NO2 für Protein verwendet) und universeller Geräteempfindlichkeit die Konzentrationen iterativ berechnet. 3.3.5 Bestimmung der enzymatischen Aktivität Die Bestimmung der enzymatischen Aktivität der FucA und ihrer Mutanten mit dem natürlichen Substrat L-Fuculose-1-phosphat (Fuc1P) erfolgte über einen kontinuierlichen, gekoppelten, zweistufigen Test. Im ersten Schritt des Tests wird Fuc1P durch FucA in einer Aldolspaltung zu Dihydroxyacetonphosphat (DHAP) und L-Lactaldehyd umgesetzt. In der Folgereaktion wird das gebildete DHAP durch Glycerol-3-phosphat-Dehydrogenase (GPDH) zu Glycerol-3-phosphat reduziert. GPDH benötigt für diese Reaktion NADH als Cofaktor, das 45 zu NAD+ oxidiert wird (Abbildung 8). Die Abnahme von NADH läßt sich photometrisch bei 366 nm verfolgen (Ghalambor & Heath, 1966). Da das Gleichgewicht bei der Folgereaktion nahezu vollständig auf der Seite des L-Glycerol-3-phosphats liegt (K = 5.8·1012), wird DHAP dem Aldolgleichgewicht entzogen (Michal & Lang, 1974), und der Verbrauch an Fuc1P ist demnach direkt proportional zum NADH-Verbrauch. L-Fuculose-1-phosphat FucA L-Lactaldehyd + DHAP L-Glycerolphosphat GPDH NADH + H + NAD + Abbildung 8 Schematische Darstellung des Aktivitätstests für FucA mit dem natürlichen Substrat L-Fuculose-1-phosphat Zu 500 µl der zuvor 10 min bei 37 °C inkubierten Assay-Lösung wurden 5 µl GPDHSuspension und 5 - 20 µl der zu untersuchenden Proteinlösung hinzupipettiert. Nach kurzem Schütteln wurde die Lösung sofort photometrisch vermessen und der Absorptionsverlauf bei 366 nm mit einem Schreiber aufgezeichnet. Um eine Reproduzierbarkeit der Tests zu gewährleisten, wurde die Enzymkonzentration jeweils so gewählt, daß die beobachtete Extinktionsänderung pro Minute im Bereich zwischen 0.05 und 0.3 lag. Die Bestimmung der enzymatischen Aktivität erfolgte dann gemäß Gleichung (5). A[ U / ml] = U: ∆E/∆T: εNADH: Vt: Vp: d: ∆E / ∆T ⋅ Vt NADH ⋅ d ⋅ Vp Enzymmenge, die 1 µmol Substrat pro min umsetzt Extinktionsabnahme pro min molarer Extinktionskoeffizient von NADH bei 366 nm = 3300 M-1cm-1 Gesamtvolumen Volumen der zugegebenen Enzymlösung Küvettendurchmesser [cm] (5) 46 Die enzymatische Aktivität für die nicht-natürlichen Substrate Dihydroxyaceton (DHA) und β-Hydroxypyruvat wurde in Syntheserichtung in einem diskontinuierlichen Test über den Nachweis der Edukte getestet (Abbildung 9). Statt L-Lactaldehyd wurde bei diesen Tests der leichter zugängliche Glykolaldehyd verwendet, der mit 38 % relativ zu L-Lactaldehyd eine noch ausreichende Umsatzgeschwindigkeit besitzt (Schneider, 1994). Die Bestimmung der Aktivität erfolgte dabei über den Nachweis der verbliebenen Menge des Edukts DHA bzw. β-Hydroxypyruvat durch Reduktion mit Glycerolphosphat-Dehydrogenase (GDH) bzw. Lactat-Dehydrogenase (LDH). Beide Hilfsenzyme benötigen als Cofaktor NADH, wodurch sich die Reaktionen photometrisch bei 366 nm verfolgen lassen. 400 µl der jeweiligen Assay-Lösung wurden mit 50 µl FucA-Lösung (Proteinkonzentration 3 - 4 mg/ml) versetzt und der Reaktionsansatz bei 37 °C inkubiert, wobei zu verschiedenen Zeitpunkten t nach dem Starten der Reaktion 50-µl-Proben entnommen wurden. Im Falle von DHA als Substrat wurden diese Proben zur Säuredenaturierung von FucA mit 30 µl 0.7 M HClO4 vermischt und danach mit 20 µl 1 M NaOH neutralisiert. Diese Mischung wurde mit 1000 µl Lösung-D versetzt und der noch vorhandene Anteil an DHA nach Zugabe von 2 µl GDH-Suspension photometrisch über die Extinktionsabnahme bei 366 nm bestimmt. Da β-Hydroxypyruvat empfindlich gegen starke Schwankungen des pHWerts ist, wurde in diesem Fall statt auf die Säuredenaturierung der FucA auf eine Hitzedenaturierung der Proben zurückgegriffen. Die zu den verschiedenen Zeiten entnommenen Proben (50 µl) wurden 1 min im kochenden Wasserbad erhitzt, kurz geschüttelt und 1 min zentrifugiert. 25 µl des Überstands wurden mit 1000 µl Lösung-D versetzt, und 5 min nach Zugabe von 2 µl LDH-Suspension wurde die verbliebene Menge an β-Hydroxypyruvat photometrisch bestimmt. Glykolaldehyd + Glycerol Dihydroxyaceton oder Glycerat β-Hydroxypyruvat GDH LDH FucA NADH + H + NADH + H + NAD + NAD + X Abbildung 9 Schematische Darstellung des Aktivitätstests für FucA mit den nicht-natürlichen Substraten Dihydroxyaceton bzw. β-Hydroxypyruvat. Es handelt sich hierbei um einen diskontinuierlichen Test, bei dem zu verschiedenen Zeitpunkten die noch nicht umgesetzte Menge des Eduktes DHA bzw. β-Hydroxypyruvat bestimmt wird. 47 3.3.6 Bestimmung der kinetischen Parameter Das kinetische Verhalten vieler Enzyme läßt sich über das von Michaelis und Menten (1913) erarbeitete Modell erklären. Es beschreibt die Reaktionsgeschwindigkeit einer enzymatischen Katalyse in Abhängigkeit von der Substratkonzentration. Die fundamentalen Reaktionen sind dabei Bildung und Zerfall des zunächst gebildeten Enzym-Substrat-Komplexes (ES): k3 1 E+S ES E+P (6) k E: S: ES: P: k1, k2, k3: Enzym Substrat Enzym-Substrat-Komplex Produkt Geschwindigkeitskonstanten Der Enzym-Substrat-Komplex bildet sich mit der Geschwindigkeitskonstante k1 aus Substrat (S) und Enzym (E). Der Zerfall des Komplexes kann dann entweder mit Geschwindigkeitskonstante k3 in Enzym (E) und Produkt (P) oder mit k2 zurück in die Edukte erfolgen. Unter der Annahme irreversibler Produktbildung, was für die Anfangsphase einer Reaktion zutrifft, und eines Fließgleichgewichtes (steady state) läßt sich gemäß dem Modell von Michaelis und Menten folgende Beziehung zwischen Reaktionsgeschwindigkeit und Substratkonzentration herleiten: ⋅ [S] KM + [S] (7) = k 3 ⋅ [Etotal] (8) = max ν: νmax: K M: [S]: k3: [Etotal]: max Reaktionsgeschwindigkeit maximale Reaktionsgeschwindigkeit Michaelis-Menten-Konstante Substratkonzentration Geschwindigkeitskonstante für den Verfall des Enzym-Substrat-Komplexes in Enzym und Produkt. Gesamt-Enzymkonzentration 48 Für hohe Substratkonzentrationen strebt die Michealis-Menten-Gleichung einen Maximalwert (νmax) an, für den Gleichung (8) gilt. Dabei entspricht die Geschwindigkeitskonstante k3 der Wechselzahl kcat, die für ein gegebenes Enzym die Anzahl der Reaktionszyklen angibt, die ein Molekül bei Substratsättigung pro Sekunde katalysieren kann. Die Michaelis-Menten-Konstante KM entspricht der Substratkonzentration, bei der die Reaktionsgeschwindigkeit ν die Hälfte der Maximalgeschwindigkeit νmax erreicht. Für den Fall, daß k3<<k2 kann KM ein Maß für die Stabilität des Enzym-Substrat-Komplexes sein. KM und νmax dienen somit zur kinetischen Charakterisierung von Enzymen. Sie lassen sich graphisch aus dem (ν versus [S])-Plot ermitteln. Da νmax experimentell nicht erreicht werden kann, wurden durch Umformung der Michaelis-Menten-Gleichung Linearisierungsverfahren entwickelt. Dabei können die kinetischen Parameter direkt aus der Geradensteigung bzw. aus den Achsenschnittpunkten ermittelt werden. Die kinetischen Parameter für die FucAMutanten wurden in dieser Arbeit über drei verschiedene Linearisierungsverfahren ermittelt: über den Lineweaver-Burk-Plot (1/ν versus 1/[S]) gemäß Gleichung (9), den Hanes-WoolfPlot ([S]/ν versus [S]) gemäß Gleichung (10) und den Eadie-Scatchard-Plot (ν/[S] versus ν) gemäß Gleichung (11): 1 1 = KM + max [S] = KM max + max [S] = ⋅ max KM 1 1 [S] (9) ⋅ [S] (10) max − 1 ⋅ KM (11) Die Bestimmung der Reaktionsgeschwindigkeit erfolgte über den im vorherigen Abschnitt beschriebenen gekoppelten Test und läßt sich aus der umgewandelten LambertBeer’schen-Beziehung berechnen: = F 7 = ( 7 NADH ⋅ d (12) Bei der Bestimmung der Anfangsgeschwindigkeit der katalysierten Reaktion wird die Enzymkonzentration konstant gehalten, während die Substratkonzentration variiert wird. Die Substratkonzentrationen sollten dabei nach Möglichkeit einen Bereich von 0.1 bis 10 KM abdecken. 49 3.4 Kristallisation und Röntgenstrukturanalyse Voraussetzung für die Röntgenstrukturanalyse von Biomakromolekülen sind Einkristalle. Die Kristallisation von Proteinen erfolgt durch langsame Übersättigung einer Proteinlösung mit Hilfe geeigneter Fällungsmittel wie anorganische Salze (beispielsweise (NH4)2SO4), organische Lösungsmittel oder Polyethylenglykole unterschiedlicher Molekularmasse. Dabei konkurrieren die Fällungsmittel mit den Proteinen um Wassermoleküle. Bestimmte Fällungsmittel- und Proteinkonzentrationen haben Protein-Protein-Wechselwirkungen zur Folge, die im Idealfall die Anordnung zum Proteinkristall bewirken. Ideale Kristallisationsbedingungen liegen dann vor, wenn nur wenige Kristallkeime in der labilen Phase des übersättigten Systems gebildet werden und das System anschließend durch fortschreitendes Kristallwachstum und der damit verbundenen Abnahme der Proteinkonzentration in die metastabile Phase übergeht. In dieser Phase des Systems findet Kristallwachstum, aber keine Bildung weiterer Kristallkeime statt. Neben Größe und Streuvermögen sind auch eine geringe Mosaizität und die Stabilität gegen Strahlenschäden entscheidende Parameter für die Kristallqualität. Bisher ist es noch nicht möglich, die Kristallisationsbedingungen eines gegebenen Proteins vorherzusagen, da die Bildung von Proteinkristallen durch eine Vielzahl von Parametern wie z. B. Protein-, Fällungsmittel- und Salzkonzentrationen, Puffersubstanz, pHWert, Temperatur oder auch durch Zusatz von organischen Lösungsmitteln beeinflußt wird (Gilliland & Ladner, 1996). Aus diesem Grund ist die Kristallisation nach wie vor eine zeitintensive empirische Methode. Durch molekularbiologische Methoden wie Klonierung und Überexpression sowie immer effizientere Reinigungsmethoden sind jedoch von den meisten Proteinen größere Mengen zugänglich, so daß ein systematisches Vorgehen möglich ist. Die Suche nach initialen Kristallisationsbedingungen erfolgt in der Regel durch sogenannte Screenings, bei denen die Vielfalt der Parameter punktuell abgetastet wird (Carter & Carter, 1979). So wurde von Jankaric & Kim (1991) aus den Bedingungen von erfolgreichen Kristallisationsexperimenten ein solches Screening entwickelt, das von Cudney et al. (1994) erweitert wurde. Die entsprechenden Screenings sind inzwischen bei Hampton Research kommerziell erhältlich. Sind einmal initiale Bedingungen mit erfolgversprechenden Kristallen gefunden worden, so können die Bedingungen in feinen Abstufungen optimiert werden. Bei der Kristallisation von Mutanten können in der Regel die WT-Bedingungen als Startpunkt dienen, es sei denn Mutationen an der Proteinoberfläche (z. B. an Kristallkontakten) bzw. drastische strukturelle Änderungen in der Mutantenstruktur machen die Suche nach neuen Bedingungen erforderlich. 50 3.4.1 Kristallisation nach der Hängetropfenmethode Die Kristallisation der FucA und ihrer Mutanten erfolgte nach der Hängetropfenmethode (McPherson, 1990). Diese Methode beruht auf dem Prinzip der Gasphasendiffusion. Dabei hängt ein Tropfen der Proteinlösung mit einer niedrigen Fällungsmittelkonzentration an einem Deckglas über einem Reservoir mit hoher Fällungsmittelkonzentration (Abbildung 10). Zum Konzentrationsausgleich diffundiert Wasser aus dem Tropfen in den Reservoirpuffer, so daß die Fällungsmittelkonzentration im Tropfen langsam die Sättigungskonzentration erreicht. Da das Volumen des Reservoirs um ein Vielfaches größer ist als das des Tropfens, entspricht die Endkonzentration des Fällungsmittels im Tropfen in etwa der Anfangskonzentration im Reservoir. Abbildung 10 Schematische Darstellung der Hängetropfenmethode. Bei den Kristallisationsexperimenten in dieser Arbeit wurden Kristallisationsboxen der Firma ICN Biomedicals verwendet, wobei der überstehende Rand vor der Kristallisation mit Schmirgelpapier geglättet und anschließend mit Silikonöl bestrichen wurde. Die Vertiefungen wurden mit 1000 µl Reservoirpuffer gefüllt und ein Deckplättchen mit anhängendem Tropfen aufgesetzt. Die Tropfen wurden dabei durch Vermischen von 5 µl Fällungsmittellösung und 5 µl Proteinlösung hergestellt. Die Kristallisation erfolgte dann bei 20 °C. Silikonisieren von Deckplättchen und Glaskapillaren Durch das Silikonisieren der Glasoberflächen kann das Verlaufen der Tropfen auf den Deckplättchen bzw. das Rutschen der Kristalle in Kapillaren verhindert werden. Dazu wurden die Deckplättchen bzw. die Kapillaren kurz in Silikonisierungslösung getaucht, an der Luft getrocknet und schließlich 1 h auf 160 °C erhitzt. 51 3.4.2 Kristallmontage Proteinkristalle sind gegenüber mechanischen Belastungen und Veränderungen der sie umgebenden Mutterlauge empfindlich. Aufgrund ihres hohen Solvensgehalts von bis zu 80 % müssen sie auch gegen Austrocknung geschützt werden. Für die bei RT durchgeführten Streuexperimente werden Proteinkristalle deswegen in Glaskapillaren überführt, in denen sie über die Gasphase im Diffusionsgleichgewicht mit der Mutterlauge stehen (Abbildung 11). Zur röntgenographischen Untersuchung wurden die FucA-Kristalle direkt aus dem Tropfen montiert. Dazu wurden zu dem Tropfen circa 30 µl Reservoirlösung pipettiert und der Kristall unter dem Mikroskop in eine silikonisierte Glaskapillare gesaugt (∅ 0.7 - 1.0 mm). Nach dem Ansaugen des Kristalls wurde noch etwas Luft und nochmals Reservoirlösung angesaugt. Auf der dünnen Seite wurde die Kapillare daraufhin mit Hartwachs verschlossen und von der anderen Seite her die Mutterlauge um den Kristall mit dünnen Filterpapierstreifen nahezu vollständig entfernt. Um das Austrocknen des Kristalls während der Messung zu verhindern, wurde in einigem Abstand vom Kristall mittels einer Glaskapillare (∅ 0.3 - 0.5 mm) etwas Reservoirlösung zugegeben und diese Seite der Kapillare ebenfalls mit Hartwachs verschlossen. Flüssigkeitssäule Hartwachssiegel Kristall mit Resten von Mutterlauge Abbildung 11 Kapillare mit montiertem Kristall 3.4.3 Kristallsysteme Kristalle stellen eine regelmäßige dreidimensionale Anordnung gleicher Bausteine dar, die v v v sich durch Translation um die Einheitsvektoren a , b und c ineinander überführen lassen. Diese Einheitsvektoren spannen die Elementarzelle als kleinste Untereinheit des Kristallgitters auf, wobei die Achsen der Elementarzelle mit a, b und c, die Winkel mit α, β und γ bezeichnet werden. Zusätzlich zur Translationssymmetrie können Kristalle noch weitere Symmetrieelemente wie Inversionszentren, Spiegelebenen, Drehachsen, Gleitspiegelebenen 52 und Schraubenachsen aufweisen. Die Elementarzelle läßt sich in asymmetrische Einheiten unterteilen, aus denen nach Anwendung der entsprechenden Symmetrieoperationen des Kristalls wieder die Elementarzelle entsteht. Ziel der Röntgenstrukturanalyse ist es, die atomare Anordnung innerhalb der asymmetrischen Einheit zu bestimmen. Aus der Kombination von Punktsymmetrien und Translation ergeben sich die 230 kristallographischen Raumgruppen (International Tables for Crystallography, 1996). Bei Biomakromolekülen sind aufgrund der Chiralität der Bausteine (L-Aminosäuren, D-Zucker) Spiegelebenen und Inversionszentren nicht möglich. Die Zahl der 230 kristallographischen Raumgruppen reduziert sich somit auf 65 in Proteinkristallen auftretende "biologische" Raumgruppen. 3.4.4 Theorie der Röntgenstreuung Röntgenstrahlen treten beim Durchtritt durch Materie aufgrund ihres energiereichen oszillierenden elektromagnetischen Feldes mit Elektronen in Wechselwirkung. Die Elektronen werden in Schwingungen versetzt und emittieren als oszillierende Dipole phasengekoppelte Röntgenstrahlung gleicher Wellenlänge (kohärente Strahlung oder Thomson-Streuung). Die Emission der Sekundärstrahlung erfolgt als konzentrische sphärische Wellen in alle Raumrichtungen, wobei es zu Interferenzen der Streustrahlungen der einzelnen Atome kommt. In Abhängigkeit von der Elektronendichteverteilung treten in bestimmten Winkeln bezüglich des Primärstrahls Intensitätsmaxima und -minima auf. Abbildung 12 Schematische Darstellung der Röntgenstreuung gemäß dem Bragg’schen Gesetz. Röntgenstrahlen, die von benachbarten Netzebenen mit dem Abstand d gestreut werden, haben einen Gangunterschied von 2d·sinΘ. Positive Interferenz tritt auf, wenn dieser einem ganzzahligen Vielfachen n der Wellenlänge des einfallenden Strahles λ entspricht. 53 Wie Bragg (1913) zeigte, kann die Streuung von Röntgenstrahlen in Kristallen als Reflektion an Netzebenen beschrieben werden. Die entsprechenden geometrischen Verhältnisse sind in Abbildung 12 veranschaulicht. Der einfallende monochromatische Röntgenstrahl trifft unter dem Glanzwinkel Θ auf eine Netzebenenschar und wird an den einzelnen Netzebenen reflektiert. Erfüllt der Glanzwinkel Θ die Bragg'sche Gleichung (13), so ist der Gangunterschied ein ganzzahliges Vielfaches der Wellenlänge. Es kommt zu konstruktiver Interferenz, die als Reflex detektiert werden kann. n . λ = 2 . d . sin Θ (13) n: Reflektionsordnung (n = 1, 2, 3, ...) λ: Wellenlänge [Å] d: Netzebenenabstand [Å] Θ: Glanzwinkel [°] Eine weitere Möglichkeit der Veranschaulichung ist in Abbildung 13 durch Betrachtung von Röntgenstrahlung als eine ebene Welle dargestellt. Dabei wird dem einfallenden v v Primärstrahl der Richtungsvektor k 0 , der gestreuten Welle der Richtungsvektor k ’ zugev ordnet. Die Streuamplitude E in einem Volumenelement d r ist abhängig von der Anzahl der v v streuenden Elektronen ρel ( r )dr . Integriert man über alle Volumenelemente, ergibt sich die v Streuamplitude E am Ort R nach Gleichung (14). Abbildung 13 Schematische Darstellung der Röntgenstreuung an einem Materiebrocken. Die Röntgenstrahlung wird als ebene Welle behandelt 54 vv E = const ⋅ ei( ωt − kR) ∫ v i(kv − kv 0 )vr v ( dr el r ) ⋅ e (14) Vol ω = 2πc/λ: t: r k0 : r k: ρ el ( vr ): Kreisfrequenz ω und Wellenlänge λ der Röntgenstrahlung Zeit v v Wellenvektor des einfallenden Strahls k 0 = k = 2π/λ Wellenvektor des gestreuten Strahls Elektronendichte am Ort vr Die Terme vor dem Integral enthalten physikalische Informationen über die Ausbreitung der Welle. Im Integral sind die Informationen über die Struktur der Elektronendichte im betrachteten Volumenelement des Streuers enthalten, weshalb es auch als Strukturfaktor v v v bezeichnet wird. Zur Vereinfachung wird der Streuvektor 2π s = k − k 0 eingeführt, wodurch der Strukturfaktor dann folgende Form erhält: v F (s) = ∫ v 2 πisvvr v ( dr el r ) ⋅ e (15) Vol Dieses Integral ist mathematisch gesehen eine Fourier-Analyse. Durch diese wird die v v ortsabhängige Elektronendichte ρel ( r ) des realen Raumes in die komplexen, von s v abhängigen Strukturfaktoren F ( s ) des reziproken Raumes überführt. Bei der Röntgenstreuung an Kristallen kommt es nur dann zu positiven Interferenzen, wenn die Phasenverschiebung ein Vielfaches von 2π ist, woraus die sogenannten Laue-Bedingungen resultieren: v v a⋅s = h v v b⋅s = k (16) v v c⋅s = l h,k,l = ...,-2,-1,0,1,2... Die Zahlen h, k und l werden als Miller’sche Indizes bezeichnet. Sie beschreiben v Netzebenen im realen Raum (siehe unten). Durch die Darstellung des Ortsvektors r in fraktionellen Koordinaten des realen Raums ergibt sich für die Strukturfaktor-Gleichung folgende Form: 55 1 F ( h, k, l) = VEZ ∫ 1 ∫ 1 ∫ ρel ( x , y, z ) ⋅ e 2 πi( hx + ky + lz ) dxdydz (17) x =0 y= 0 z =0 3.4.5 Reziprokes Gitter und Ewald-Konstruktion Jedem Kristallgitter kann man ein anderes fiktives Gitter zuordnen: sein reziprokes Gitter. Durch dieses neue Gitter lassen sich einige Eigenschaften des Originalgitters bei der Beschreibung der Röntgenstreuung vereinfachen. Durch einen Kristall läßt sich eine Vielzahl von Netzebenenscharen legen. Jede Netzebenenschar besitzt einen bestimmten Abstand dhkl und kann durch einen Vektor beschrieben werden, der senkrecht auf den Netzebenen steht und den Abstand 1/dhkl besitzt. Die Reflexe eines dreidimensionalen Kristallgitters bilden wiederum ein dreidimensionales Gitter. Dieses fiktive Gitter steht zu dem realen Kristallgitter in fester Beziehung und wird reziprokes Gitter genannt. Jedem periodischen Ebenenschar-Abstand im realen Gitter entspricht eine senkrecht zur Ebenenschar gerichtete Translation im reziproken Gitter. Die Beträge des Abstands und der Translation verhalten sich reziprok zueinander. Die Netzebenenschar mit den Miller'schen Indizes hkl und dem Netzebenenabstand dhkl wird im reziproken Raum durch den Gitterpunkt Phkl dargestellt. Jedem Gitterpunkt Phkl ist v v der Streuvektor s zugeordnet. Die Richtung des Streuvektors s im reziproken Raum ist parallel zur Flächennormalen der Netzebenen hkl im realen Raum. Die Länge des Vektors v entspricht dem Reziproken des Netzebenabstands dhkl. Der Streuvektor s kann auch als v v v ganzzahliges Vielfaches der reziproken Basisvektoren a * , b* und c* beschrieben werden, wobei die reziproken Basisvektoren in einem festen Zusammenhang mit den Basisvektoren v v v a , b und c des realen Gitters stehen. Es gilt: v v v v s = ha * + kb * + lc * a* = v v v a * , b* , c * : v s : h,k,l: V: b×c , V b* = c×a , V c* = (18) a×b V Einheitsvektoren im reziproken Raum Streuvektor Miller’sche Indizes v v v* ( a × b )· c 56 Die Ewald-Konstruktion (Ewald, 1921) überträgt die Bragg'schen Reflektionsbedingungen in den reziproken Raum (Abbildung 14). Um den Kristall wird eine Kugel mit dem Radius 1/λ gelegt. Der Röntgenstrahl tritt am Punkt I in die Kugel ein und trifft unter dem Glanzwinkel Θ auf den Kristall. In den Austrittspunkt des primären Röntgenstrahls wird der Ursprung O des reziproken Gitters gelegt. Aufgrund dieser Konstruktion erfüllen bei vorgegebenem Winkel gerade die Punkte des reziproken Gitters die Bragg'sche Gleichung, die mit der Kugeloberfläche zusammenfallen (Punkt P). I Abbildung 14 Darstellung der Verbindung von realem mit reziprokem Raum über die EwaldKonstruktion. Der vollständige Röntgendatensatz kann durch Drehen des Kristalls erhalten werden. Dabei ist zu beachten, daß das Reflexmuster eines Kristalls zusätzlich zur Symmetrie der realen Elementarzelle noch ein Inversionszentrum besitzt. 3.4.6 Flächenzählermessung und Auswertung Flächenzählermessungen wurden mit einem Drei- und einem Vierkreisgoniometersystem durchgeführt. Dabei läßt sich der Detektor in der 2θ-Ebene drehen, die Winkel ω und ϕ sind variabel. Der Winkel χ ist bei Vierkreissystemen ebenfalls variabel, bei Dreikreissystemen auf χ = 45° fixiert (Abbildung 15). Als Röntgenquelle diente Cu-Kα-Strahlung (λ = 1.542 Å) an einer rotierenden, wassergekühlten Kupferanode (RU200B, Fa. Rigaku und M18XHF, Fa. Siemens). Die Cu-Kα-Linie wurde mit einem Graphit-Monochromator selektiert und auf einen Strahldurchmesser von 0.5 mm kollimiert. Der Generator wurde dabei maximal mit einer Spannung von 40 kV und einem Röhrenstrom von 120 mA betrieben. 57 Zur Detektion der Reflexe wird ein zweidimensionaler Proportionalzähler verwendet. Dieser enthält in einer Xenon-gefüllten Kammer drei Elektroden, die jeweils aus einer Vielzahl dünner Drähte im Abstand von ca. 1 mm bestehen. Tritt Röntgenstrahlung durch das Berylliumfenster dieser Kammer, so entstehen durch Stoßionisation freie Elektronen und Xenon-Kationen. Die frei gewordenen Elektronen ionisieren weitere Teilchen, so daß pro Photon mehrere Hundert Elektronen und Kationen gebildet werden. Durch Beschleunigung der Elektronen im elektrischen Feld zwischen der ersten Kathode und der Anode kommt es zur Signalverstärkung. Dadurch kommt es zu einer sekundären Ionisationskaskade mit einer ausreichend großen Anzahl an Ladungsträgern, die an der Anode bzw. zweiten Kathode entladen werden und dabei ein elektrisches Signal erzeugen. Da die Drahtgitter von Anode und zweiter Kathode in einem Winkel von 90° zueinander stehen, kann der Eintrittspunkt der Röntgenphotonen bestimmt werden. Die Detektoroberfläche ist so in ein Raster von 512 x 512 Bildpunkte (pixel) unterteilt, wobei ein pixel, die kleinste mit diesem Detektor auflösbare Flächeneinheit, eine Fläche von ca. 190 x 190 µm2 hat. Da diese Detektortypen nur über einen beschränkten dynamischen Bereich verfügen, d. h. ab einer gewissen Anzahl von Photonen pro Fläche eine Sättigung erreicht wird, muß die Leistung der Röntgenröhre dementsprechend an die Streukraft der Kristalle angepaßt werden. Abbildung 15 Schematische Darstellung der Meßanordnung eines Dreikreissystems (χ = 45°) mit einem Flächenzähler. Vor der Datensammlung muß die Ortsempfindlichkeit des Detektors durch gleichmäßige Bestrahlung mit einer amorphen 55Fe-Quelle kalibriert werden (flood field correction). Dazu wird der Primärstrahl auf eine dünnes Plättchen amorphen 55Fe gerichtet und über einen Zeitraum von 5 min die homogene Röntgenfluoreszenz detektiert. Die daraus resultierende Korrekturtabelle wird während der Messung automatisch auf die Streubilder angewandt. Um räumliche Verzerrungen des Detektors zu korrigieren, wird zusätzlich vor jeder Messung die Ortsgenauigkeit geeicht (brass plate correction). Dazu wird eine 58 Lochplatte aus Messing mit genau definierten Bohrungen in Gitteranordnung auf den Detektor geschraubt und 10 min die Röntgenfluoreszenstrahlung des amorphen 55Fe detektiert. Das gemessene Muster wird mit dem Lochmuster der Messingplatte verglichen und anhand der ermittelten Daten eine Korrektur für die räumliche Verzerrung (Parallaxe) berechnet (Derewenda & Helliwell, 1989). Der Abstand von Kristall und Detektor wurde so gewählt, daß die Reflexe durch den Detektor noch klar voneinander getrennt abgebildet werden konnten. Er sollte nach Howard et al. (1987) etwa ein Achtel (in [cm]) der längsten Elementarzellachse (in [Å]) betragen. Für die Messung der FucA-Mutanten wurde deshalb ein Detektorabstand von 12 cm gewählt. Durch Schwenken des Kristalls um 2θ wird die maximal zu detektierende Auflösung festgelegt. Durch die Winkel χ, ϕ und ω erfolgt dann die Orientierung des Kristalls in Bezug auf den einfallenden Strahl. Mit den Programmen SADIE/ASTRO läßt sich die optimale Orientierung des Kristalls zur Minimierung des Meßaufwands bei maximaler Vollständigkeit der Messung bestimmen. Dies ist vor allem bei hochsymmetrischen Raumgruppen wie im Falle tetragonaler FucAKristalle von Vorteil. Die Messung erfolgte nach der Rotationsmethode (Arndt et al., 1973). Der Kristall wird dabei mit einer Schrittweite von 0.1° bis 0.3° um die ω- oder ϕ-Achse gedreht. Im Falle des Vierkreissystems ist auch eine Drehung um χ möglich. Ein Teil des reziproken Gitters durchstößt während der Drehung innerhalb einer Aufnahme (frame) die Ewaldkugel. Die entsprechenden Reflexe werden vom Detektor erfaßt und ihre Intensität wird ermittelt. Abhängig von der Streukraft der Kristalle, ihrer Größe, dem gewählten KristallDetektor-Abstand und des eingestellten 2θ-Winkels beträgt die Expositionszeit für ein frame 1 bis 4 min. Die Auswertung der Meßdaten erfolgte mit dem Programm XDS (Kabsch, 1988). Dabei werden zuerst die geometrischen Korrekturterme aus der brass-plate-Aufnahme berechnet (CORRECT), eine initiale Intensitätsverteilung des Hintergrundes bestimmt (INIT) und Reflexpositionen, d. h. die Position auf dem Detektor und die Stellung der Rotationsachse für die nachfolgende Indizierung gesammelt (COLSPOT). Dann wird die Orientierung des reziproken Gitters relativ zum Laborsystem berechnet. Aus einer Liste von geclusterten Differenzvektoren zwischen Reflexen werden die drei besten Basisvektoren ermittelt. Nach Bestimmung und Verfeinerung der primitiven Elementarzelle wird zusammen mit einem Qualitätsindex eine Auflistung aller möglichen Bravaisgitter ausgegeben (INDEX). Die auf mehreren frames verteilten partiellen Reflexen werden dann zu dreidimensionalen Reflexprofilen zusammengebaut und die gemittelten Profile von starken Reflexen angezeigt. Gleichzeitig wird alle 15 frames die Orientierungsmatrix neu berechnet, um mechanisch bedingte Abweichungen oder ein Rutschen des Kristalls zu korrigieren (COLPROF). Anschließend werden die individuellen Profile der einzelnen Reflexe mit den zugehörigen gemittelten Profilen verglichen und daraus ihre Intensitäten bestimmt (PROFIT). Die 59 Intensitäten werden nun entsprechend der Meßanordnung korrigiert, die Reflexe in Gruppen von mehreren frames skaliert und die Intensität symmetrieäquivalenter Reflexe verglichen (CORRECT). Reflexe mit schlechter Korrelation werden dabei aussortiert. Zuletzt werden anhand der stärksten Reflexe der gesamten Messung Zellparameter, Orientierungsmatrix und Kristall-Detektor-Abstand verfeinert (GLOREF). 3.4.7 Skalierung von Datensätzen Die Skalierung der Daten von einem oder mehreren Kristallen wurde mit den Programmen MERGE (B. Dijkstra, Groningen), XSCALE (Kabsch, 1988) oder SCALA/AGROVATA (Evans, 1993; CCP4, 1994) durchgeführt. Daran anschließend wurden die Reflexintensitäten mit dem Programm TRUNCATE (CCP4, 1994) in Strukturfaktoramplituden Fobs umgewandelt. Daten aus einer oder mehreren Messungen müssen wegen der auftretenden Strahlenschäden, der ungleichen Strahlungsdosis und der orientierungsabhängigen Kristallgröße aufeinander skaliert werden. Dazu werden die Reflexe einer Anzahl von aufeinanderfolgenden frames mit Skalierungsfaktoren versehen und aufeinander skaliert. Als Maß für die Güte eines Datensatzes wird neben der Vollständigkeit, der Redundanz und den relativen Intensitäten der Reflexe üblicherweise auch der Rsym für symmetrieverwandte Reflexe angegeben (Gleichung 19): ∑ ∑ I( hkl) − I( hkl) = ∑ ∑ I( hkl) n R sym hkl i i n hkl i (19) i Der Wert des R-Faktors Rsym zeigt jedoch eine deutliche Abhängigkeit von der jeweiligen Raumgruppe und der erreichten Redundanz des Datensatzes. Aus diesem Grund wurden von Diederichs & Karplus (1997) Vorschläge für einen modifizierten R-Faktor Rint gemacht, die in den Programmen bisher allerdings noch nicht berücksichtigt wurden. 3.4.8 Strukturfaktor und Fourier-Transformation Jedem Reflex kann ein Strukturfaktor zugeordnet werden. Dieser enthält die Koordinaten des Reflexes hkl im reziproken Raum sowie die Intensität und Phase des Reflexes. Mathematisch läßt sich der Strukturfaktor nach Gleichung (20) als Fourier-Transformation der Elektronendichte beschreiben. 60 _F hkl = ⌠ ρel (x,y,z) . e2πi(hx+ky+lz) dV ⌡ V (20) F hkl: Strukturfaktor des Reflexes hkl ρel(x,y,z) : Elektronendichte am Ort x,y,z Umgekehrt läßt sich die Elektronendichte über eine Fourier-Synthese aus den Strukturfaktoren und dem Volumen der Elementarzelle V nach Gleichung (21) berechnen. Der Strukturfaktor ist eine komplexe Größe, die sich aus der Strukturfaktoramplitude und der Phase des Reflexes zusammensetzt (Gleichung (22)). el (x, y, z) = 1 ⋅ ∑ F(h, k, l) ⋅ e −2πi(hx + ky +lz) V hkl F( h, k , l) = F( h, k , l) ⋅ e iϕ hkl (21) (22) F(h,k,l): Strukturfaktoramplitude Strukturfaktorphase ϕhkl: Experimentell können nur die Intensitäten Iobs(h,k,l) der einzelnen Reflexe hkl gemessen werden. Diese sind proportional zum Absolut-Quadrat des Strukturfaktors (Gleichung 23). Die Phaseninformation geht daher bei der Messung verloren. F(h, k, l) 2 = F(h, k, l) ⋅ F* (h, k, l) = const ⋅ I obs (h, k, l) (23) Zur Berechnung der Elektronendichte muß das sogenannte "Phasenproblem", ein intrinsisches Problem der Röntgenstrukturanalyse, überwunden werden. Dabei eignen sich im wesentlichen zwei Methoden für die Phasenbestimmung in Proteinkristallen. Bei der Methode des Multiplen Isomorphen Ersatzes (MIR) wird aus den Differenzen der Reflexintensitäten zwischen Datensätzen von nativen Kristallen und isomorphen Schweratomderivaten die Position der Schweratome bestimmt. Anhand dieser Positionen lassen sich die Phasen der Strukturfaktoren näherungsweise bestimmen. 61 Voraussetzung für eine Strukturlösung nach der Methode des Molekularen Ersatzes (MR) ist ein strukturell hinreichend ähnliches Proteinmodell (Suchmodell). Das Suchmodell muß in die Elementarzelle des unbekannten Kristalls richtig orientiert (Rotationsfunktionssuche) und plaziert (Translationssuche) werden. Durch die stetig zunehmende Anzahl bekannter Proteinstrukturen gewinnt die Methode des Molekularen Ersatzes für die Strukturaufklärung immer mehr an Bedeutung. 3.4.9 Temperaturfaktor Der Strukturfaktor F( h , k , l ) läßt sich als Summe der Beiträge aller Atome in der Elementarzelle ausdrücken: F(hkl) = ∑ f j ⋅ e − 1 2 Bs 4 ⋅e vv 2πi s rj (24) j j: fj: B: v rj : Index über alle Atome Atomformfaktor Temperaturfaktor Ortsvektor des Atoms j Der atomare Streufaktor f beschreibt ein Atom im Ruhezustand. Im realen Kristall schwingen jedoch alle Atome infolge ihrer thermischen Energie; und je höher die Temperatur ist, desto größer ist diese Bewegung. Neben dieser dynamischen Fehlordnung trägt auch eine statische Fehlordnung durch Kristallfehler zu einer Abweichung der Atome von ihrer Gleichgewichtslage bei. Diese Änderungen gegenüber dem idealen Atom im Ruhezustand werden − 1 Bs 2 beschrieben. Der Term B wird als Temperaturfaktor durch den Korrekturterm e 4 bezeichnet und hängt direkt mit der mittleren quadratischen Verschiebung u 2 des Atoms um seine Gleichgewichtslage zusammen: B = 8π 2 u 2 (25) Bei sehr hoch aufgelösten Strukturen können anisotrope B-Faktoren bestimmt werden, welche die unterschiedlichen Schwingungen des Atoms in den drei Raumrichtungen beschreiben. Ein mittlerer B-Faktor von 30 Ų entspricht somit einem mittleren Verschiebungsquadrat von 0.62 Å. Nach Wilson (1949) stehen der mittlere B-Faktor und die absolute Skalierung der Intensitäten in folgender Beziehung: 62 r I( s ) sin 2 Θ ln = ln − 2 C B λ2 ∑ fi2 (s) (26) i Der Skalierungsfaktor C und der mittlere B-Faktor B können durch Auftragung des v natürlichen Logarithmus des Quotienten aus der gemessenen mittleren Intensität I( s ) und der unter Vernachlässigung der thermischen Bewegung gegebenen mittleren Intensität ∑fi2(s) (Wilson, 1949) gegen (sin Θ / λ ) 2 erhalten werden. 3.4.10 Modellbau und Elektronendichtekarten Zu Beginn des Modellbaus wurde die von Dreyer in Kristallform T gelöste Struktur des WT verwendet. Da dieses Modell für die einzelnen Mutanten naturgemäß noch mit Fehlern behaftet ist, wurde es in einer zyklischen Prozedur abwechselnd manuell verändert bzw. verfeinert. Das Einfügen der Aminosäurenaustausche bzw. struktureller Veränderungen in den Mutanten erfolgte mit Hilfe des interaktiven Graphikprogramms O (Jones et al., 1991). Dabei wurden die folgenden Elektronedichtekarten zur Korrektur bzw. Ergänzung des Modells verwendet: (Fo-Fc)-Elektronendichtekarte Die (Fo-Fc)-Elektronendichtekarte wird mit dem Koeffizient (Fobs-Fcal)·exp(iϕcalc) berechnet. Diese Karten zeigen den Unterschied zwischen Modell und der tatsächlich experimentell ermittelten Elektronendichte. Fehlende Atome erscheinen dabei als positive, falsch plazierte entsprechend mit negativer Differenzdichte. Die (Fo-Fc)-Karten werden daher für den Einbau fehlender Aminosäuren, Wassermoleküle und Liganden verwendet bzw. wie in dieser Arbeit auch zur Orientierung der Seitenketten neu eingeführter Aminosäuren in Mutantenstrukturen. 2(Fo-Fc)-Elektronendichtekarte Die (2Fo-Fc)-Elektronendichtekarte wird mit dem Koeffizient (2Fobs-Fcal)·exp(iϕcalc) berechnet und entspricht somit der Addition einer (Fobs-Fcal)·exp(iϕcalc)-Differenz-Elektronendichtekarte zu einer Fobs·exp(iϕcalc)-Karte. Neben der modellierten Elektronendichte erscheinen Differenzen zur Modellstruktur auf halbem Dichteniveau. Allerdings werden (2Fo-Fc)-Karten stark vom Modell beeinflußt (model bias). 63 Simulated-annealing-omit-Elektronendichtekarte Zur Klärung von unsicheren Bereichen innerhalb des Modells sind omit-Karten geeignet. Hierzu werden die unsicheren Bereiche aus dem Proteinmodell entfernt (omit). Nach einer Molekulardynamik-Simulation (Brünger et al., 1987) lassen sich (Fo-Fc)- und (2Fo-Fc)Elektronendichtkarten berechnen, deren model bias reduziert ist. Diese Karten sind vor allem für den automatischen Einbau von Atomen geeignet (Lamzin & Wilson, 1993), aber auch zum Bauen flexibler Loops. σA-gewichtete Elektronendichtekarten Der model bias der Elektronendichtekarte bei der Kombination von Phaseninformation aus verschiedenen Quellen läßt sich reduzieren, indem die Fehler der partiellen Struktur bei der Gewichtung miteinbezogen werden. Die zur Gewichtung verwendeten Terme m und D lassen sich nach Read (1986; 1990) durch einen σA-Term ausdrücken. In dieser Arbeit wurden vorwiegend σA-gewichtete (2mFo-DFc)- bzw. (mFo-DFc)-Elektronendichtekarten verwendet, die mit den CCP4 Programmen SIGMAA, REFMAC und FFT berechnet wurden (CCP4, 1994). 3.4.11 Verfeinerung Fehler in der Elektronendichte, die durch Multiplen Isomorphen Ersatz bzw. Molekularen Ersatz erhalten wurden, führen zwangsläufig zu einem fehlerhaften Modell des entsprechenden Proteins. Ziel der Verfeinerung ist es daher, die Modellparameter an die Meßwerte anzupassen. Dabei werden die Ortskoordinaten und B-Faktoren der Proteinatome so verändert, daß die aus dem Modell berechneten Strukturfaktoramplituden optimal mit den gemessenen übereinstimmen. Die Zahl der Observablen (Röntgenreflexe) sollte bei der Verfeinerung die Zahl der zu verfeinernden Parameter übersteigen. Da die Zahl der Röntgenreflexe jedoch durch die Auflösungsgrenze der Kristalle, die Vollständigkeit der Daten sowie durch den Aufbau des Kristalls begrenzt ist, müssen zusätzliche Nebenbedingungen in die Verfeinerung eingeführt werden. Aus der Röntgenstrukturanalyse kleiner Moleküle hat man sehr genaue Kenntnisse über die stereochemischen Parameter von Peptidstrukturen (Bindungslängen und -winkel, Chiralität, Torsionswinkel). Die so erhaltenen Idealwerte können daher zur Einschränkung der Verfeinerungsparameter verwendet werden. Diese sogenannten restraints wirken während der Verfeinerung durch die Einschränkung der freien Parameter wie künstliche zusätzliche Observablen. Analog dazu kann auch die Ähnlichkeit der Konformationen NCS-verwandter Peptidketten zur Begrenzung der freien Verfeinerungsparameter verwendet werden, was allerdings bei der im Rahmen dieser Arbeit aufgetretenen Kristallform nicht ausgenutzt werden konnte. Das Kriterium für die Qualität der Verfeinerung ist der R-Faktor (Gleichung 27). 64 R= k= ∑ ∑ hkl hkl Fobs ( hkl ) − k ⋅ Fcalc ( hkl ) ∑ F ( hkl ) hkl obs (27) w ( hkl) ⋅ Fobs ( hkl) ⋅ Fcalc ( hkl) ∑ hkl w ( hkl) ⋅ Fcalc ( hkl) 2 w(hkl): auflösungsabhängiger Gewichtungsfaktor Dabei ist k ein Skalierungsfaktor zwischen den observierten Strukturfaktoramplituden Fobs und den berechneten Amplituden Fcalc. Bei hochverfeinerten Strukturen liegt der R-Faktor zwischen 10 und 20 %. Neben dem kristallographischen R-Faktor, bei dem alle Reflexe bei der Verfeinerung berücksichtigt werden, wird auch der freie R-Faktor (Brünger, 1992a; 1993) zur Beurteilung der Qualität einer Struktur herangezogen. Dabei wird ein statistisch gewählter Satz von 5 - 10 % der Reflexe nicht in die Verfeinerung einbezogen (test-set). Mit diesen Reflexen wird dann der freie R-Faktor Rfree in Analogie zum kristallographischen R-Faktor berechnet. Dadurch kann ein overfitting des Modells durch eine Freigabe von zu vielen Parametern während der Verfeinerung verhindert werden. Liegt overfitting vor, so sinkt zwar der kristallographische R-Faktor, der freie bleibt dagegen konstant oder steigt gar an. Da der Rfree mit dem mittleren Fehler der Modellphasen korreliert, kann er direkt als Kriterium für Fortschritt und Qualität der Verfeinerung angesehen werden (Kleywegt & Brünger, 1996). Die Methode des kleinsten quadratischen Fehlers (least squares) Der zur Zeit bei der Verfeinerung noch am häufigsten verwendete Algorithmus ist die Minimierung des kleinsten quadratischen Fehlers (least squares minimizing). Dabei werden die Differenzen zwischen den Strukturfaktoren Fobs und Fcalc gemäß Gleichung (28) minimiert. Dies geschieht unter der Annahme, daß die Abweichungen zwischen Fobs und Fcalc einer GaußVerteilung entsprechen. Q = ∑ w ( hkl) ⋅ ( Fobs ( hkl) − Fcalc ( hkl) )2 hkl w(hkl): auflösungsabhängiger Gewichtungsfaktor (28) 65 Der Vorteil dieser Methode liegt in der relativ geringen Rechenzeit. Es ist allerdings häufigeres manuelles Eingreifen in die Strukturverfeinerung nötig, da diese Methode nur über einen geringen Konvergenzradius verfügt. Bei dem neueren Programm REFMAC (Murshudov et al., 1997) kommt statt der least-squares-Verfeinerung ein maximumlikelihood-Algorithmus zum Einsatz. Dabei handelt es sich im Prinzip um eine Verallgemeinerung der least-squares-Methode, die eine bessere Annäherung an die Realität darstellt. Molekulardynamik (slowcool) Molekulardynamik-Methoden für die Verfeinerung von Proteinstrukturen wurden von Brünger et al. (1987) im Programm X-PLOR implementiert. Im Gegensatz zur least-squaresMethode bieten sie den Vorteil großer Konvergenzradien, so daß im Laufe der Strukturverfeinerung, z. B. bei Mutantenstrukturen mit größeren strukturellen Veränderungen, lokale Minima ohne manuelle Veränderungen überwunden werden können. Dabei werden den Atomen zunächst initiale Geschwindigkeiten zugeordnet, die einer Maxwell-Verteilung z. B. bei 2000 K entsprechen. Anschließend werden die Newton’schen Bewegungsgleichungen für alle Atome i des Moleküls numerisch für bestimmte Zeitintervalle (etwa 1 fs) gelöst. Dabei wird die Energiefunktion Epot des durch die umgebenden Atome verursachten Kraftfeldes näherungsweise berechnet. mi ⋅ d 2 ri δE pot = δri dt 2 (29) mi: Masse des Atoms i ri: Ortskoordinaten des Atoms i Nachdem in ca. 0.5 - 1.0 ps ein Gleichgewichtszustand erreicht wurde, wird die Temperatur des Systems erniedrigt. Sobald eine festgelegte Änderung der Koordinaten, z. B. um 1/10 der höchsten Auflösung, erreicht wurde, werden neue Werte für Fcalc berechnet. Ein typisches slowcool-Protokoll umfaßt ein Temperaturintervall von 2000 K bis 300 K. Das Verhältnis von Verfeinerungsparametern zu Observablen bei der Molekulardynamik läßt sich dadurch verbessern, daß lediglich die Änderungen in den Torsionswinkeln einer Proteinstruktur berücksichtigt werden (torsion angle refinement). Mit diesem Verfahren können darüber hinaus wesentlich höhere Simulationstemperaturen verwendet werden, was wiederum erhöhte Konvergenzradien zur Folge hat (Rice & Brünger, 1994). 66 Verfeinerung von Temperaturfaktoren Statistische Fehlordnungen und thermische Bewegungen der Atome im Protein haben eine auflösungsabhängige Änderung der Reflexintensitäten zur Folge (Kapitel 3.4.9). Diese läßt sich jedoch durch eine least-squares-Minimierung der Temperaturfaktoren modellieren. Um die Zahl der Verfeinerungsparameter niedrig zu halten, wird bei Auflösungen oberhalb 3 Å nur ein mittlerer B-Faktor für das gesamte Protein bestimmt. Bei Auflösungen zwischen 3 und 2.5 Å werden meist zwei B-Faktoren pro Aminosäure verfeinert (je ein Wert für alle Hauptund alle Seitenkettenatome). Individuelle isotrope B-Faktoren lassen sich ab einer Auflösung von ca. 2.5 Å sinnvoll verfeinern. Dabei werden üblicherweise Einschränkungen für die Abweichungen benachbarter Atom angenommen, da sich deren B-Faktoren nicht beliebig stark voneinander unterscheiden können. Bei atomarer Auflösung kleiner als 1.5 Å können darüber hinaus auch anisotrope thermische Bewegungen einzelner Atome verfeinert werden. Die Auflösungen der Mutanten-Strukturen in dieser Arbeit lagen zwischen 1.66 und 2.55 Å, so daß jeweils eine individuelle isotrope Verfeinerung der B-Faktoren vorgenommen wurde. 3.4.12 Verfeinerung mit X-PLOR Das Programm X-PLOR (Brünger, 1992b) ist ein sehr vielseitiges Programm zur Verfeinerung und Analyse makromolekularer Strukturen. Dabei können experimentelle Daten sowohl aus NMR- als auch aus Röntgendiffraktionsmessungen verwendet werden. Es beinhaltet die oben aufgeführten Verfeinerungsmethoden. Dabei kann über einen eigenen Befehlssyntax die Reihenfolge sowie Details der einzelnen Verfeinerungsschritte individuell festgelegt werden. Unabhängig von der Verfeinerungsmethode minimiert X-PLOR eine Energiefunktion Etotal. Die Gewichtungsfaktoren für die experimentellen Strukturfaktoramplituden wA und Phasen wP werden durch die Routine CHECK abgeschätzt und eingesetzt. In der Regel sind jedoch die Modellphasen so gut, daß der Term EPXray nicht mehr berücksichtigt wird. Bei den geometrischen Energietermen in Eempirical wird der von Engh & Huber (1991) zusammengestellte Parametersatz zugrunde gelegt. Als Ausgangsmodell für die Verfeinerung der Mutanten-Strukturen diente die von Dreyer & Schulz (1996a) bis 1.92 Å aufgelöste Struktur des Wildtyps. Zur Verfeinerung wurde sowohl das Programmpaket X-PLOR als auch das Programm REFMAC (Murshudov et al., 1997) benutzt. Das Programm X-PLOR wurde jeweils zu Beginn der Verfeinerung verwendet. Tabelle 4 zeigt exemplarisch das Verfeinerungsprotokoll für eine Verfeinerung mit eingefügtem Molekulardynamik-Schritt. Dies war jedoch nur bei Mutanten-Strukturen mit größeren strukturellen Veränderungen der Fall. Ansonsten wurde mit X-PLOR nur eine rigid- 67 body-Verfeinerung durchgeführt und für alle weiteren Verfeinerungsschritte wurde das Programm REFMAC verwendet. E total = Eeffective + E empirical (30) E effective = w A ⋅ E AXray + w P ⋅ E PXray + w N ⋅ E NCS E empirical = w bonds E bonds + w angle E angles + w dihe E dihe + w impr E impr + w vdw E vdw + w elec E elec + w pvdw E pvdw + w pelec E pelec EXray: ENCS: wA,wP: wN: wbond, etc: Ebond: Eangle: Eimpr: Edihe: Evdw: Eelec: Epvdw, Epelec: Pseudo-Energie, wodurch die Amplituden und Phasen der Röntgendaten beschrieben werden Pseudo-Energieterm, der NCS-Abweichungen bestraft Gewichtungsfaktoren für die Strukturfaktoramplituden und Phasen Gewichtungsfaktor für Abweichungen von der NCS Gewichtungsfaktoren der geometrischen Energieterme Bindungsenergie Bindungswinkelenergie Chiralitätsenergie Rotamerenenergie van-der-Waals-Energie elektrostatische Energie Wechselwirkungen mit symmetrieäquivalenten Molekülen Tabelle 4 Beispielhafte Verkettung der Unterprogramme bei der Verfeinerung mit X-PLOR GENERATE CHECK RIGID BULKSOL POSITIONAL SLOWCOOL POSITIONAL B-FAKTOR Erstellung einer Protein-Struktur-Datei, die von X-PLOR benötigt wird Bestimmung des Gewichtungsfaktors wA Least-squares-Verfeinerung der Position von starren Atomgruppen (rigid body) in der Elementarzelle zur Berechnung der Zellparameterdifferenzen zwischen WTund Mutanten-Struktur Berechnung partieller Strukturfaktoren für den kontinuierlichen Solvensanteil (bulk solvent) Least-squares-Verfeinerung der Atompositionen Simulated annealing (Molekulardynamik) Erneute least-squares-Verfeinerung der Atompositionen Verfeinerung individueller B-Faktoren nach dem least-squares-Verfahren 68 3.4.13 Verfeinerung mit REFMAC Nach Abschluß der simulated-annealing-Verfeinerung wurde mit dem CCP4-Programm REFMAC (Murshudov et al., 1997) weiter verfeinert, das auf einem maximum-likelihoodAlgorithmus beruht (Kapitel 3.4.11). Die geometrischen restraints, die zur Verfeinerung benötigt werden, müssen zunächst mit dem Programm PROTIN (CCP4, 1994) festgelegt werden. Dabei werden die dazu idealisierten Molekülparameter für die Standardaminosäuren, die auf dem Parametersatz von Engh & Huber (1991) basieren, und die Cofaktoren einem sogenannten dictionary entnommen. Um neue Strukturbausteine in die Verfeinerung miteinzubeziehen, wurde zunächst ein Modell der Verbindung mit Hilfe des Programms SYBYL (Tripos Associates Inc., St. Louis, MO, USA, 1992) energieminimiert. Daraufhin wurde das Standard-dictionary um diese idealisierten Molekülparameter erweitert. Strukturbausteine, die in dieser Arbeit in das Standard-dictionary aufgenommen werden mußten, waren Dihydroxyacetonphosphat als Substrat sowie ein Cystein, das mit einem Molekül β-Mercaptoethanol kovalent verknüpft war. Automatischer Einbau von Wassermolekülen Zu Beginn der Verfeinerung wurden die Positionen der Wassermoleküle aus der WT-Struktur übernommen. Im weiteren Verlauf der Verfeinerung wurden in der Umgebung der Mutationsstelle zunächst manuell Wassermoleküle entfernt oder hinzugefügt. Nach einigen Verfeinerungsrunden wurde das Entfernen oder Einfügen der Solvensmoleküle automatisch mit dem Programm ARPP (Lamzin & Wilson, 1993) durchgeführt. Die so erhaltenen Positionen für Wassermoleküle wurden nach der Verfeinerung zusätzlich auf ihre physikalische Plausibilität überprüft. So durften geeignete Wassermoleküle nicht näher als 2.2 Å zu anderen Atomen liegen (Ausnahme in der Zink-Koordinationssphäre) und mußten in 2.2 bis 3.5 Å Entfernung zu einem potentiellen H-Brücken-Partner liegen (Stickstoff oder Sauerstoff). 3.4.14 Darstellung von Proteinstrukturen Eine atomare Darstellung von Proteinen ist aufgrund der Molekülgröße nur in kleinen Ausschnitten möglich. Sollen größere Bereiche dargestellt werden, so ist es aus Gründen der Anschaulichkeit nötig, daß Abstraktionen eingeführt werden. Dabei werden Sekundärstrukturelemente zumeist als Bändermodelle (ribbon-Diagramme) dargestellt. Die Zuordnung der einzelnen Aminosäurereste zu den jeweiligen Sekundärstrukturelementen kann mit den Programmen DSSP (Kabsch & Sander, 1983) oder STRIDE (Frishman & Argos, 1995) 69 automatisch erfolgen. Zur Darstellung von atomaren Modellen, Cα-backbone-Verläufen und ribbon-Darstellungen wurde im Rahmen dieser Arbeit das Programm MOLSCRIPT (Kraulis, 1991) benutzt. Zum photorealistischen rendering der Abbildungen aus MOLSCRIPT wurde das Programm RENDER aus der Programmsuite Raster3D (Merritt & Bacon, 1997) verwendet. 70 4 Ergebnisse und Diskussion 4.1 Planung der Mutanten 4.1.1 Mutationen der postulierten katalytischen Reste Basierend auf dem Vergleich der Röntgenstrukturen der FucA in unligierter Form und mit dem Übergangszustandsanalogon PGH (Abbildung 4, Seite 12) wurde von Dreyer & Schulz (1996b) ein Reaktionsmechanismus postuliert, der eine Beteiligung von Glu73 und Tyr113’ am Katalysezyklus beinhaltet (Abbildung 16). Unter Berücksichtigung weiterer enzymologischer Studien mit Anhydroalditolphosphaten als Fuc1P-Analoga schlossen sich auch Fessner et al. (1996) diesem Mechanismus an. Aus der Lage des PGH in einer Bindungstasche am interface kann gefolgert werden, daß DHAP vor der Aldehydkomponente bindet, da ein gebundener Aldehyd den Zugang für DHAP stark erschweren würde. DHAP muß nun durch Deprotonierung für den nukleophilen Angriff am Carbonylkohlenstoff des Aldehyds aktiviert werden. Die Deprotonierung erfolgt am C3-Atom, das aufgrund der benachbarten Ketogruppe eine erhöhte C-H-Azidität besitzt. Wie die Untersuchungen von Kimura et al. (1999) zeigen, wird der pKa-Wert durch die Komplexierung an das Zink um etwa 10 Größenordnungen erniedrigt. Während die entsprechenden Aldehyde in Lösung einen pKa-Wert von ca. 19 besitzen, konnte für den Modellkomplex 1-(4-Bromophenacyl)-1,4,7,10-tetraazacyclododecan ein pKa-Wert von 8.4 ermittelt werden, was sehr gut mit dem von Schneider (1994) bestimmten pH-Optimum für die FucA-katalysierte Reaktion korrespondiert. Als Base im aktiven Zentrum kommt nur Glu73 in Frage, das bei Substratbindung aus der Koordinationssphäre des Zink-Ions verdrängt wird. Das Oε1-Atom ist nach der Übertragung des Protons nicht mehr solvenszugänglich und der ungeladene Zustand der Seitenkette kann durch die relativ hydrophobe Umgebung zwischen Phe76, Phe131, Ile131-Cβ und His155-Cε stabilisiert werden. Die Carboxylatgruppe wäre somit in H-Brücken-Distanz zur 3-Hydroxygruppe von DHAP sowie zum Hauptkettenstickstoff von Gly132. Die negative Ladung an C3 des DHAP wird mesomeriestabiliert, wobei es zur Bildung eines Chelatkomplexes mit dem Zink-Ion kommt. DHAP ist somit für den nukleophilen Angriff am positivierten Carbonylkohlenstoff aktiviert. Über die Lage der Aldehydkomponente sind keine strukturellen Daten zugänglich. Durch Modellieren eines offenkettigen Fuculosemoleküls bei optimaler Überlagerung mit PGH in die Komplexstruktur sowie unter Berücksichtigung der Tatsache, daß der nukleophile Angriff auf der Si-Seite der Carbonylgruppe erfolgen muß (ergibt sich aus der Stereochemie des Produktes), kann auf die ungefähre Lage der Aldehydkomponente vor der Bindungs- 71 bildung geschlossen werden. Demnach kommt die Carbonylgruppe der Aldehydkomponente in unmittelbarer Nähe zur phenolischen OH-Gruppe von Tyr113’ zu liegen. Hieraus folgerten Dreyer & Schulz, daß während bzw. unmittelbar nach dem Knüpfen der C-C-Bindung Tyr113’ sein phenolisches Proton auf den Aldehyd übertragen könnte. Nach Freisetzen des Produktes Fuc1P und Wiederherstellung der Protonierungszustände im aktiven Zentrum wäre das Enzym dann für einen neuen Katalysezyklus bereit. Die Wiederherstellung der Protonierungszustände könnte entweder durch einen direkten Protonentransfer zwischen der Carbonsäuregruppe von Glu73 und dem Phenolat-Ion von Tyr113’ bei der Rückbewegung von Glu73 in die Koordinationssphäre des Zink-Ions oder, was wahrscheinlicher wäre, indirekt über Solvensmoleküle erfolgen. Abbildung 16 Schematische Darstellung des Katalysezyklus der FucA basierend auf der Struktur des WT mit und ohne Inhibitor Phosphoglycolohydroxamat (PGH) aus Dreyer & Schulz (1996b). Dabei wird die Reaktion als Aldoladdition (im Uhrzeigersinn) beschrieben. Zum Vergleich ist die Struktur des Inhibitors in einem Kasten neben DHAP in der äquivalenten Konformation dargestellt. Ausgangspunkt ist die Substrat-freie Struktur (oben) mit Glu73 in der Koordinationssphäre des Zink-Ions. Die Hauptschritte sind dann: Deprotonierung am C3-Atom von DHAP durch Glu73 und Stabilisierung des Endiolat-Intermediats durch das Zink-Ion (rechts), Binden des Aldehyds mit seiner Carbonylgruppe nahe Tyr113’, so daß er mit seiner Si-Seite zum DHAP zeigt (unten), Knüpfen der C-C-Bindung und Protonierung des Sauerstoffs durch Tyr113’ (links), Freisetzung des Produktes und Wiederherstellung der Protonierungszustände im aktiven Zentrum. Die Ringbildung erfolgt gemäß diesem Mechanismus nach Freisetzung des Fuc1P ins Medium. 72 Diesen Mechanismus galt es nun durch entsprechende Mutationen zu kommentieren bzw. zu modifizieren. Das Ergebnis einer ersten Mutante lag zu Beginn der Arbeit bereits vor. Der Austausch von Glu73 zu Gln hatte einen drastischen Reaktivitätseinbruch auf ca. 0.1 % der WT-Aktivität zur Folge (Müller-Dieckmann, 1994), was die Rolle von Glu73 in obigem Mechanismus zu bestätigen scheint. Die Restaktivität dieser Mutante könnte z. B. auf partielle Desamidierung zurückzuführen sein. Ausgehend davon wurde die weniger konservative Mutante E73S geplant, wodurch gemäß dem vorgeschlagenen Mechanismus eine völlig inaktive Mutante erwartet wurde. Diese Mutante war auch im Hinblick auf eine mögliche Cokristallisation mit dem natürlichen Substrat L-Fuculose-1-phosphat interessant. Die Rolle von Tyr113’ als Protonendonor sollte durch die Mutanten Y113F und Y113T untersucht werden. Der Austausch zu Thr war von Interesse, weil ein Sequenzvergleich mit der homologen RhuA zeigte, daß RhuA (Raumstruktur zu Beginn dieser Arbeit noch nicht bekannt) an der entsprechenden Stelle ein Threonin besitzt (Abbildung 17). FucA RhuA FucA RhuA 10 20 30 40 50 60 | | | | | | HHHHHHHHHHHHHHHHHH SSSSSS SSSSSS 333 333 SSSS αA β1 β2 αa αb β3 ---MERNKLARQIIDTCLEMTRLGLNQGTAGNVSVRYQD-----------------------------GMLITPTGIPYEK--LTESH---IVFID--GN * * ** * * * * * ** * * MQNITQSWFVQGMIKATTDAWLKGWDERNGGNLTLRLDDADIAPYHDNFHQQPRYIPLSQPMPLLANTPFIVTGSGKFFRNVQLDPAANLGIVKVDSDGA αA β1 αB αa β2 αb β3 αc αC β4 333 HHHHHHHHHHHHHHH SSSSS HHHH333333 SSSS 333 SSSSS 333 HHHHSSSSSS | | | | | | | | | | 10 20 30 40 50 60 70 80 90 100 70 80 90 100 110 120 130 140 | | | | | | | | SSS HHHHHHHHH SSSSS HHHHHHHHH S 333333 SSSS HHHHHHHHHH β4 αB β5 αC β6 αc β7 αD GKHEEG-----KLPSSEWRFHMAAYQSRP-----DANAVVHNHAVHCTAVSIL--------NRSIPAIHYMIAAAGGNSIPCAPYATFGTRELSEHVALA * * * ** * * * ** * * * ** * * GYHILWGLTNEAVPTSELPAHFLSHCERIKATNGKDRVIMHCHATNLIALTYVLENDTAVFTRQLWEGSTECLVVFPDGVGILPWMVPGTDEIGQATAQE β5 αD β6 αE αF αG β7 αH SSSSSSS HHHHHHHHHHHHHHH SSSSS HHHHHH HHHHHHHHHH HHHH SSS HHHHHHHHHH | | | | | | | | | | 110 120 130 140 150 160 170 180 190 200 150 160 170 180 190 200 210 | | | | | | | SSSSS SSSSSSS HHHHHHHHHHHHHHHHHHHHHH HHHHHHHHHHHHHHH β8 β9 αE αF LKNRKATLLQHHGLIACEVNLEKALWLAHEVEVLAQLYLTTLAITDPVPVLSDEEIAVVLEKFKTYGLRIEE-* ** * * * ** * ** * * MQKHSLVLWPFHGVFGSGPTLDETFGLIDTAEKSAQVLVKVYSMGGMKQTISREELIALGKRFGVTPLASALAL β8 β9 αI αJ αK H SSSS SSSSSS HHHHHHHHHHHHHHHHHHHHHHH HHHHHHHHHHH HHHH | | | | | | | 210 220 230 240 250 260 270 H FucA RhuA Abbildung 17 Sequenzvergleich zwischen FucA und RhuA; bereits auf der Basis der Raumstrukturen (Krömer & Schulz, pers. Mitteilung). Identische Aminosäuren sind mit einem Stern markiert. Die Zuordnung einzelner Reste zu Sekundärstrukturelementen ist mit Großbuchstaben gekennzeichnet: H für α-Helices, 3 für 310-Helices und S für β-Faltblattstränge. Das alignment wurde freundlicherweise von Dipl. Chem. Markus Krömer zur Verfügung gestellt. Zu Beginn dieser Arbeit war die Raumstruktur der RhuA allerdings noch unbekannt. 73 Wie bereits oben erwähnt, wurden die Mutationen an den katalytischen Resten nicht nur zur Bestätigung des Reaktionsmechanismus geplant. Inaktive Mutanten, bei denen der Aktivitätsverlust ausschließlich auf dem Verlust einer für die Katalyse essentiellen Seitenkette beruht, die ansonsten aber ein intaktes aktives Zentrum aufweisen, bieten eine Möglichkeit, die Struktur der Mutanten im Komplex mit dem natürlichen Substrat aufzuklären. Dies wäre gerade im Falle der FucA von großem Interesse, da nur die Struktur mit PGH, einem Analogon der DHAP-Komponente, bekannt war. Eine Übersicht über alle geplanten Mutanten, die Veränderungen an den Positionen 73 und 113’ enthalten, gibt Tabelle 5. Ein Großteil der aufgeführten Mutanten wurde dabei zu einem späteren Zeitpunkt der Arbeit geplant und basiert bereits auf Ergebnissen, die andere Mutanten ergaben, so daß sie an dieser Stelle nur der Vollständigkeit halber aufgeführt sind und nicht explizit diskutiert werden. Tabelle 5 Übersicht über die Mutationen der postulierten katalytischen Reste an den Positionen 73 und 113’ Mutante a) b) c) ohne Kombination mit anderen Resten in Kombination mit Aminosäure 209’ in Kombination mit Aminosäuren 206’ und 209’c) E73Q a) E73S b) Y113F Y113T E73Q/Y113F b) Y113F/Y209F E73Q/Y113F/Y209F b) F206Y Y113F/F206Y F206Y/Y209F Y113F/F206Y/Y209F Mutante von Müller-Dieckmann (1994). Planung für Tränkversuche bzw. zur Cokristallisation mit dem natürlichen Substrat Fuc1P. Mutanten mit Permutation der Tyrosine an den Positionen 113’, 206’ und 209’. 4.1.2 Mutation am flexiblen Loop(23-27) Neben der Bewegung der Seitenkette von Glu73 wird bei der Inhibitorbindung auch eine Veränderung der Konformation und Flexibilität der an der PGH-Bindung beteiligten Loops beobachtet. Die größten Änderungen weist dabei der flexible Loop(23-27) auf. In der unligierten Struktur besitzt dieser Loop eine große Flexibilität, was sich in den relativ hohen B-Faktoren und der relativ schlecht definierten Elektronendichte in diesem Bereich zeigt. Ein Vergleich der hochaufgelösten Struktur in Kristallform T und der Struktur in Kristallform K mit gebundenem PGH (siehe Kapitel 1.4) zeigt, daß sich Loop(23-27) bei PGH-Bindung in Richtung PGH verschiebt und fixiert wird (Abbildung 18). Die größte Bewegung wird dabei von Thr26 vollzogen (3.5 Å für Thr26-Oγ), das in der Komplexstruktur über eine H-Brücke mit dem Nε-Atom von Trp169’ aus der benachbarten Untereinheit fixiert wird (2.9 Å Abstand). Da eine Konformation, die näher am Inhibitor liegt, stabilisiert wird, wäre es 74 denkbar, daß diese Bewegung durch Binden der Aldehydkomponente oder Fuc1P noch verstärkt wird und Thr26 somit als potentieller H-Brücken-Donor für das Substrat in Frage kommen könnte. Um den Einfluß von Thr26 im flexiblen Loop bei Substratbindung und Katalyse zu untersuchen, wurde deshalb ein Austausch gegen Ala vorgenommen. Abbildung 18 Die Fixierung des flexiblen Loops(23-27) bei Bindung des Inhibitors PGH. Schwarz: Lage des Loops, wie sie in der Struktur ohne Inhibitor in der Kristallform T verfeinert wurde. Rot: Lage des Loops nach Bindung des Inhibitors in Kristallform K. Die Peptidbindung zwischen Ala27 und Gly28 hat sich um 180° gedreht. Die Konformation des Loops in der Komplexstruktur wird zusätzlich durch H-Brücken über die Seitenketten von Asn24 und Thr26 mit Tyr47 bzw. Trp169’ stabilisiert. Die Überlagerung der beiden Strukturen erfolgte anhand des zentralen β-Faltblattes. 4.1.3 Mutationen am flexiblen C-terminalen Kettenende Die Modelle des WT umfassen nur die Aminosäuren 1 bis 206. Die neun C-terminalen Aminosäuren konnten bei der Strukturlösung aufgrund ihrer erhöhten Flexibilität nicht detektiert werden. Die relative Nähe der letzten in der Röntgenstruktur detektierten Aminosäure Phe206’ zum aktiven Zentrum ließ eine mögliche Beteiligung des C-terminalen Kettenendes bei der Katalyse vermuten. Eine erste Bestätigung dieser Hypothese lieferte eine C-terminale Deletionsmutante, bei der alle Aminosäuren nach Phe206’ abgespalten wurden, was einen deutlichen Abfall der enzymatischen Aktivität zur Folge hatte. So wurde für die Spaltung von Fuc1P nur ca. 0.5 % der WT-Aktivität nachgewiesen (Müller-Dieckmann, 1994). Das Phänomen eines flexiblen C-terminalen Kettenendes, das unterschiedliche Konformationen einnehmen kann, wird auch für die mechanistisch verschiedenen Klasse-I-Aldolasen beschrieben (Gamblin et al., 1991). Dabei legt eine Vielzahl von Daten eine Bewegung bei Substratbindung bzw. Freisetzung der Reaktionsprodukte und eine mögliche Involvierung bei der Katalyse nahe (Littlechild & Watson, 1993; Dalby et al., 1998). Die Aufgabe war es nun, systematisch Mutationen am C-terminalen Kettenende einzuführen, um den Aktivitätseinbruch bei der Deletionsmutante entsprechenden Resten 75 zuordnen zu können und die mögliche Rolle des C-terminalen Kettenendes durch die kinetischen Parameter der entsprechenden Mutanten genauer aufzuklären. Die Sequenz des C-terminalen Kettenendes sowie eine Übersicht über alle C-terminalen Mutanten ist in Abbildung 19 schematisch dargestellt. FucA Reste 1-206 207 215 -Lys-Thr-Tyr-Gly-Leu-Arg-Ile-Glu-Glu-OH Ala Phe Ala Ala Ala Asp Gln Leu Abbildung 19 Aminosäuresequenz des flexiblen C-terminalen Kettenendes (Reste 207-215). Die Positionen der Aminosäureaustausche sind mit einem Pfeil markiert, ebenso die Schnittstellen bei den beiden C-terminalen Deletionsmutanten. 4.1.4 Mutationen zur Veränderung der Stereospezifität der Reaktion Die Untersuchungen von Schneider (1994) zeigen, daß FucA auf Seite der Aldehydkomponente eine Vielzahl nicht-natürlicher Aldehydsubstrate akzeptiert. Dabei weist FucA für Aldehydkomponenten, die an α- oder β-Position eine OH-Gruppe bzw. einen anderen H-Brücken-Akzeptor besitzen, eine hohe Stereoselektivität auf: Es werden ausschließlich D-erythro-konfigurierte Produkte gebildet (Tabelle 6). Für Aldehydsubstrate mit aliphatischen Resten werden dagegen unterschiedliche Verhältnisse an D-erythro- und L-threo-konfigurierten Produkten gefunden. Um zu den entsprechenden L-threo-Produkten zu gelangen, muß jedoch der nukleophile Angriff an der Re-Seite der Carbonylgruppe erfolgen. Angewendet auf das in Abbildung 18 dargestellte Modell für die Bindung der Aldehydkomponente, würde dies einer Drehung des Aldehyds von 180° um die CO-Doppelbindung entsprechen. Die räumlichen Verhältnisse im aktiven Zentrum der FucA sind nun so beschaffen, daß die entsprechende Seite des aktiven Zentrums durch eine hydrophobe Wand abgeschirmt wird, die von den aromatischen Seitenketten der Reste Tyr113’, Phe131 und Phe206’ gebildet wird (Abbildung 20). Aldehyde mit aliphatischen Substituenten könnten entsprechend ihrem Platzbedarf unterschiedlich gut mit dieser hydrophoben Wand wechselwirken, was die Verschiebung des Produktspektrums zugunsten der L-threo-Produkte zur Folge haben könnte. 76 Tabelle 6 Produktverhältnisse bei FucA-katalysierten Synthesen für unterschiedlich substituierte Aldehyde gemäß Schneider (1994) Aldehydkomponente Produktverhältnis Aldehydkomponente D-erythro/L-threo a) Produktverhältnis D-erythro/L-threo a) O O 97:3 95:5 H H OH O O 97:3 HO 50:50 H HO H O O 97:3 30:70 H H O O 97:3 68:32 H H N a) Vgl. Abbildung 1: D-erythro = 3R,4R, L-threo = 3R,4S; die Nachweisgrenze liegt jeweils bei 3 %. Y113’ F206’ F131 HO O R2 R1 O 2- O O 3PO Abbildung 20 Die Orientierung der Aldehydkomponente beim nukleophilen Angriff von DHAP und die Position der postulierten hydrophoben Wand, gebildet aus den aromatischen Ringen von Tyr113’, Phe131 und Phe206’. Reste, die von der benachbarten Untereinheit kommen, sind mit einem Strich gekennzeichnet. Nimmt der Wasserstoff am Carbonyl-C-Atom die Position R2 ein (wie für das natürliche Substrat L-Lactaldehyd), so erfolgt der nukleophile Angriff an der Si-Seite und es entstehen Produkte mit D-erythro-Konfiguration (3R,4R). Für den Fall, daß R1 gleich H ist, erfolgt der nukleophile Angriff an der Re-Seite und es werden L-threo-Produkte (Tabelle 6) gebildet. Bei einer derartigen Orientierung treten die Aldehydsubstituenten in Wechselwirkung mit der hydrophoben Wand, die das aktive Zentrum abschirmt. 77 Mutationen im Bereich der hydrophoben Wand sollten demnach das Diastereomerenverhältnis für aliphatische Aldehyde nachhaltig beeinflussen. So könnte z. B. das schrittweise Verkürzen der Seitenkette von Phe131 für einige aliphatische Aldehyde ein Verschieben des Diastereomerenverhältnis zu Gunsten des L-threo-konfigurierten Produktes bewirken. Auch eine Ausdehnung der hydrophoben Wand, beispielsweise durch Einfügen eines Tryptophans an Position 206’, sollte das Diastereomerenverhältnis erheblich verändern. Um diese Hypothese zu überprüfen und um dadurch auch ein detaillierteres Verständnis der Stereoselektivität des Enzyms zu erhalten, wurden die folgenden fünf Mutanten geplant: F131A, F131V, F131L, F206W und die Doppelmutante F131A/F206W. 4.1.5 Mutationen zur Veränderung der Selektivität auf Seite der DHAP-Komponente Während die FucA auf Seite der Aldehydkomponente eine Vielzahl an Substraten akzeptiert, zeigt sie eine hohe Selektivität auf Seite der DHAP-Komponente. Bei der Verwendung des Enzyms in der organischen Synthese ist jedoch DHAP ein entscheidender Kostenfaktor. Aus diesem Grunde wurden Mutanten im Hinblick auf eine Substitution der DHAP-Komponente geplant. Dabei wurden als potentielle Alternativsubstrate Dihydroxyaceton (DHA) und β-Hydroxypyruvat ausgewählt (Abbildung 21), die synthetisch leichter zugänglich und somit kostengünstiger sind. O O H + OH HO OPO3 OPO3 OH OH O 2- 2- OH DHAP L-Lactaldehyd L-Fuculose-1-phosphat O HO OH DHA O O - HO O β-Hydroxypyruvat Abbildung 21 Potentielle nicht-natürliche Substrate auf Seite der DHAP-Komponente. Die strukturellen Unterschiede im Vergleich zu DHAP sind durch einen Kasten hervorgehoben. 78 Die Planung der entsprechenden Mutanten erfolgte auf Basis der Struktur des WT im Komplex mit PGH. Zunächst wurden mit dem Programm O die jeweiligen Substrate mit den äquivalenten PGH-Atomen überlagert. Anschließend wurden systematisch Aminosäurenaustausche im Bereich der Phosphatbindungstasche simuliert, die zum einen den Boden der Bindungstasche räumlich ausfüllen, ohne jedoch zu Kollisionen mit benachbarten Resten zu führen, zum anderen aber gleichzeitig zusätzliche stabilisierende Wechselwirkungen mit dem neuen Substrat ermöglichen sollten. Ausgangspunkte für die mögliche Lage der Seitenketten waren die vom Programm O vorgeschlagenen häufigsten Rotamere der entsprechenden Reste (z. B. drei Rotamere im Falle von Glu und Gln), basierend auf einer Untersuchung von Ponder & Richards (1987) an 19 hochaufgelösten Proteinstrukturen. Die so generierten Modelle wurden dann zusätzlich mit dem Programm X-PLOR bezüglich ihrer Energie minimiert. Tabelle 7 gibt einen Überblick über alle in dieser Hinsicht geplanten Mutanten. Tabelle 7 Übersicht über die geplanten Mutanten zur Spezifitätsänderung bei der DHAP-Komponente erwünschte Spezifität DHA β-Hydroxypyruvat geplante Mutante: N29Q N29E S71Q, T43V/S71Q a) S71E T41R S71H S71K S71R a) Del(27) Del(27)/S71H Del(27)/S71K Del(27)/S71R Die Mutante wurde erst auf Basis der Röntgenstruktur der Mutante S71Q geplant (vgl. Kapitel 4.5.8). Abbildung 22 Energie-minimiertes Modell der Mutante S71Q im Komplex mit Dihydroxyaceton (DHA). Die Wechselwirkungen zwischen dem Zink-Ion und seinen Liganden sind durch gepunktete Linien hervorgehoben, ebenso alle H-Brücken zwischen der modellierten Seitenkette und DHA bzw. benachbarten Aminosäuren (Abstand ≤ 3.5 Å). 79 Abbildung 23 Energie-minimiertes Modell der Mutante N29Q im Komplex mit Dihydroxyaceton (DHA). Die Wechselwirkungen zwischen dem Zink-Ion und seinen Liganden sind durch gepunktete Linien hervorgehoben, ebenso alle H-Brücken zwischen der modellierten Seitenkette und DHA bzw. benachbarten Aminosäuren (Abstand ≤ 3.5 Å). Im Hinblick auf eine Spezifitätsänderung zu DHA erwiesen sich zwei Positionen als besonders günstig und erfolgversprechend: Aminosäure 29 und Aminosäure 71. Der Ersatz von Asn29 durch Glu oder Gln (Abbildung 23) bzw. Ser71 durch Glu oder Gln (Abbildung 22) sollte zum Verschließen der Phosphatbindungstasche führen. Außerdem wären die neuen Seitenketten so positioniert, daß eine Stabilisierung der Bindung von DHA an das Enzym über eine zusätzliche H-Brücke zwischen der freien OH-Gruppe des DHA und den Oε-Atomen der mutierten Aminosäuren erfolgen könnte. Im Falle der beiden Glu-Mutanten würde durch die neu eingeführte Carboxylatgruppe auch gleichzeitig der Wegfall des negativen Potentials der Phosphatgruppe kompensiert werden, das eventuell einen gewissen Einfluß auf die Katalyse haben könnte. Bei den Mutanten zur Verwendung von β-Hydroxypyruvat war das Hauptziel das Einführen einer positiv geladenen Seitenkette an die entsprechende Stelle in der Phosphatbindungstasche, um eine Stabilisierung des Substrates über eine Salzbrücke mit der Carboxylatgruppe des β-Hydroxypyruvats zu ermöglichen. Dies könnte beispielsweise durch Ersetzen von Ser71 durch His, Lys oder Arg geschehen (Abbildung 24). Das Modell für Mutante S71R weist zwar einen zu geringen Abstand zwischen der Guanidingruppe des Arginins und der Carboxylatgruppe auf, berücksichtigt allerdings nicht die mögliche Flexibilität der Hauptkettenatome der Phosphatbindungstasche. Unter Berücksichtigung der dynamischen Prozesse in Proteinstrukturen wurde diese Mutante deshalb in die Planung miteinbezogen. 80 Abbildung 24 Energie-minimiertes Modell der Mutante S71K im Komplex mit β-Hydroxypyruvat (BHP). Die Wechselwirkungen zwischen dem Zink-Ion und seinen Liganden sind durch gepunktete Linien hervorgehoben, ebenso alle H-Brücken und Salzbrücken zwischen der modellierten Seitenkette und β-Hydroxypyruvat bzw. benachbarten Aminosäuren (Abstand ≤ 3.5 Å). Abbildung 25 Energie-minimiertes Modell der Mutante T41R im Komplex mit β-Hydroxypyruvat (BHP). Die Wechselwirkungen zwischen dem Zink-Ion und seinen Liganden sind durch gepunktete Linien hervorgehoben, ebenso alle H-Brücken und Salzbrücken zwischen der modellierten Seitenkette und β-Hydroxypyruvat bzw. benachbarten Aminosäuren (Abstand ≤ 3.5 Å). Die H-Brücke zwischen dem Nη1-Atom von Arg41 und dem Carbonylsauerstoff von Ile45 ist aus Gründen der Übersichtlichkeit nicht dargestellt. Eine weitere Möglichkeit, eine Guanidingruppe in eine Distanz von etwa 2.5 - 3 Å zur Carboxylatgruppe zu bringen, könnte der Austausch von Thr41 zu Arg bieten. Das Einführen eines Arginins an dieser Position scheint hinsichtlich der sterischen Verhältnisse in der Phosphatbindungstasche auf den ersten Blick am gewagtesten. Es konnte allerdings eine Konformation für die Seitenkette gefunden werden, in der es zu keinen Kollisionen mit 81 benachbarten Resten kommt und gleichzeitig die Guanidingruppe so liegt, daß eine Salzbrücke mit der Carboxylatgruppe des Hydroxypyruvats gebildet werden kann (Abbildung 25). Diese Konformation kann darüber hinaus durch ein Netzwerk von H-Brücken mit benachbarten Resten stabilisiert werden. Um möglicherweise etwas mehr Platz für die Carboxylatgruppe des β-Hydroxypyruvats zu schaffen, wurde als zusätzliche Variante auch eine Verengung des Loops(23-27) durch Deletion einer Aminosäure (Ala27) in Kombination mit den oben beschriebenen Mutationen in die Planung miteinbezogen. 4.2 Mutagenese 4.2.1 Wahl der Mutagenese-Oligonukleotide Die Sequenzen der für die Eckstein- bzw. Kunkel-Mutagenese verwendeten Oligonukleotide sind in Tabelle 8 aufgeführt. Da in der als Templat dienenden M13mp19-fuca-ssDNA der (-)-Strang des fuca-Gens vorhanden ist, müssen die Oligonukleotide zum (-)-Strang komplementär, also Teil des (+)-Strangs sein. Teilweise waren stille Mutationen erforderlich, um die Ausbildung von Sekundärstrukturen zu verhindern (Oligo-F131A) bzw. um die Ausbildung einer EcoR I-Schnittstelle zu unterbinden (Oligo-E214Q), die eine erfolgreiche Rückklonierung verhindern würde. 4.2.2 Mutagenese nach Eckstein bzw. Kunkel Eine Übersicht über die verschiedenen Mutagenese-Ansätze gibt Tabelle 9. Zu Beginn der Arbeit wurde die Methode nach Kunkel angewendet (Mutanten R212A, E214A und E215A). Aufgrund der eher geringen Mutationsrate bzw. dem Scheitern der Mutagenese bei der Mutante Y113F kam im folgenden dann nur noch die Methode nach Eckstein zur Anwendung, die sich durch eine sehr hohe Erfolgsrate auszeichnete. So wurde dann auch (gemittelt über alle mit dieser Methode erzeugten Mutanten) eine Mutationsrate von 90 % erreicht. Die Mutagenese nach Eckstein hatte darüber hinaus den Vorteil, daß das so generierte mutierte Templat direkt als Templat zur Erzeugung von Mehrfachmutanten verwendet werden konnte. 82 Tabelle 8 Sequenzen der verwendeten Mutagenese-Oligonukleotide Bezeichnung Oligo-T26A Oligo-Del(27) Oligo-N29E Oligo-N29Q Oligo-T41R Oligo-T43V Oligo-S71E Oligo-S71H Oligo-S71K Oligo-S71Q Oligo-S71R Oligo-E73Q Oligo-E73S Oligo-Y113F Oligo-Y113T Oligo-F131A Oligo-F131L Oligo-F131V Oligo-F206W Oligo-F206Y Oligo-F206Y/Y209F Oligo-K207A Oligo-Y209F Oligo-Del(211-215) Oligo-R212A Oligo-E214A Oligo-E214D Oligo-E214L Oligo-E214Q Oligo-E215A a) Sequenz a) Länge ** AACCAGGGGGCAGCGGGGAAC 21 GGGACTGAACCAGGGGACA_GGGAACGTCAGTGTAC * * GGACAGCGGGGGAGGTCAGTGTACG * * GGACAGCGGGGCAGGTCAGTGTACG ** GGATGCTGATTAGACCTACAGGC * ** GATGCTGATTACACCTGTAGGCATTCC ** GAAAGCTCCCCGAAAGCGAATGGCG *** GGAAAGCTCCCCCACAGCGAATGGCG ** GGAAAGCTCCCCAAAAGCGAATG ** GAAAGCTCCCCCAAAGCGAATG ** GGAAAGCTCCCCAGAAGCGAATG * GCTCCCCTCAAGCCAATGGCGTTTCC ** CTCCCCTCAAGCTCATGGCGTTTTC * GCTATTCACTTCATGATTGCG ** GCTATTCACACCATGATTGCG *** GCCTTATGCGACTGCTGGAACACG * GCCTTATGCGACCTTAGGAACACG * GCCTTATGCGACCGTTGGAACACG ** CTGGAGAAATGGAAAACCTATGG * CTGGAGAAATACAAAACCTATGGG * * CTGGAGAAATACAAAACCTTTGGGTTACG ** GAGAAATTCGCAACCTATGGG * AGAAATTCAAAACCTTTGGGT * CAAAACCTATGGGTAACGAATTGAAG ** TATGGGTTAGCAATTGAAGAG * GGGTTACGAATTGCAGAGTTA * GGGTTACGAATTGATGAGTAATTTC ** GGGTTACGAATTTTAGAGTAATTTC ** GGGTTACGAATCCAAGAGTAATTTC * ATTGAAGCGTAATTTCGTAC 35 25 25 23 27 25 26 23 22 23 26 25 21 21 24 24 24 23 24 29 21 21 26 21 21 25 25 25 20 Die Basen der zu mutierenden Aminosäuren sind fett gedruckt, die Sternchen markieren die Basenfehlpaarungen. Sternchen über nicht fett gedruckten Basen markieren stille Mutationen. 83 Tabelle 9 Übersicht über die durchgeführten Mutagenese-Reaktionen Mutante a) Templat-DNA Oligonukleotid b) T26A Del(27) Del(27)/S71H Del(27)/S71K Del(27)/S71R N29E N29Q T41R T43V/S71Q S71E S71H S71K S71Q S71R E73Q/Y113F E73Q/Y113F/Y209F E73S Y113F c) Y113F Y113F/F206Y Y113F/F206Y/Y209F Y113F/Y209F Y113T F131A F131A/F206W F131L F131V F206W F206Y F206Y/Y209F K207A Y209F Del(211-215) R212A c) E214A c) E214D E214L E214Q E215A c) M13mp19-fuca M13mp19-fuca Del(27) Del(27) Del(27) M13mp19-fuca M13mp19-fuca M13mp19-fuca S71Q M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca Y113F Y113F/Y209F M13mp19-fuca M13mp19-fuca M13mp19-fuca Y113F Y113F/F206Y Y209F M13mp19-fuca M13mp19-fuca F206W M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca M13mp19-fuca Oligo-T26A Oligo-Del(27) Oligo-S71H Oligo-S71K Oligo-S71R Oligo-N29E Oligo-N29Q Oligo-T41R Oligo-T43V Oligo-S71E Oligo-S71H Oligo-S71K Oligo-S71Q Oligo-S71R Oligo-E73Q Oligo-E73Q Oligo-E73S Oligo-Y113F Oligo-Y113F Oligo-F206Y Oligo-F206Y/Y209F Oligo-Y113F Oligo-Y113T Oligo-F131A Oligo-F131A Oligo-F131L Oligo-F131V Oligo-F206W Oligo-F206Y Oligo-F206Y/Y209F Oligo-K207A Oligo-Y209F Oligo-Del(211-215) Oligo-R212A Oligo-E214A Oligo-E214D Oligo-E214L Oligo-E214Q Oligo-E215A a) b) c) d) Sequenzierte Klone 2 3 1 1 1 3 4 2 3 3 1 1 2 2 2 2 2 14 5 4 2 5 2 3 1 2 1 3 2 11 2 7 3 3 3 3 3 3 2 Positive Klone 2 3 1 1 1 3 3 2 3 2 1 1 2 1 2 2 2 5 2 3 2 2 1 2 1 2 1 d) 3 d) 2 5 3 1 3 3 3 3 1 Die Zahlen stehen für die Positionen der ausgetauschten Aminosäuren. Der Buchstabe vor einer Zahl bezeichnet die WT-Aminosäure, der dahinter die neu eingeführte Aminosäure. Mit Del sind DeletionsMutanten bezeichnet. Mehrfach-Mutanten sind durch einen Schrägstrich unterteilt. Vgl. Tabelle 8. Keine Eckstein-Mutagenese, sondern Mutagenese nach Kunkel. Aufgrund von zwei zunächst nicht homogen gelieferten Mutagenese-Oligonukleotiden wurden zusätzlich die Mutanten E204A/F206Y/Y209F, F206Y/Y209C/L211I, K205N/F206Y/Y209F, K205Q/F206Y/Y209L, F206Y/K207N/Y209F sowie Del(206-215) erhalten, welche aber nicht weiter untersucht wurden. 84 4.2.3 Nachweis der Mutationen auf DNA-Ebene Im Anschluß an die Mutagenese wurde die erhaltene M13mp19-fuca-ssDNA im Bereich der jeweiligen Mutationen sequenziert. Ausschnitte aus der blotting-Membran der ssDNASequenzierung ausgewählter Mutanten sind in Abbildung 26 dargestellt. G A T C G A T C G A T C G A T C G A T C → → → → → → → (a) (b) (c) (d) (e) Abbildung 26 Ausschnitte aus der blotting-Membran der ssDNA-Sequenzierung der Mutanten T43V/S71Q (a, b), F206Y/Y209F (c), Y209F (d) und E214Q (e). Die Sequenzen sind jeweils von unten nach oben zu lesen (5’→3’). Die Mutationsstellen sind mit Pfeilen markiert. Zur Sequenzierung wurden der M13-Universal-Primer für Mutationen an den Positionen 26 bis 73, der Primer FucA-Prim1 für die Positionen 113 und 131 sowie der Primer FucAPrim2 für die Positionen 206 bis 215 verwendet (Tabelle 10). Beim M13-Universal-Primer standen sowohl 5’-DIG-markierte als auch unmarkierte Primer zur Verfügung. Bei den beiden anderen Primern handelte es sich um 5’-DIG-markierte Primer. Diese wurden auch bei Sequenzierungen mit Biotin-Markierung verwendet. Das Ergebnis der Sequenzierungen ist in Tabelle 9 gezeigt. Während mit der EcksteinMethode eine Mutationsquote von 90 % erreicht wurde, lag diese bei der Kunkel-Methode bei nur 22 %. Die niedrige Quote ist vor allem auf das Scheitern der Mutation im Falle von Mutante Y113F zurückzuführen. Stichprobenartig wurden auch immer wieder Bereiche außerhalb der Mutationsstelle sequenziert. Bei den Mutanten N29E, S71Q und S71E wurde dabei eine zusätzliche Mutation im Codon für Pro42 von CCT nach CCG gefunden, was jedoch keine veränderte Aminosäure an dieser Position zur Folge hat. Da die Mutagenese für diese drei Mutanten ausgehend von M13mp19-fuca-ssDNA aus der gleichen Präparation 85 durchgeführt wurde und andere Mutanten diese stille Mutation nicht enthielten, ist davon auszugehen, daß die Mutation bereits im Templat vorhanden war und bei der Kultivierung der zugrunde liegenden M13-Phagen entstanden ist. Tabelle 10 Sequenzen der verwendeten Sequenzierungsprimer a) Bezeichnung a) Sequenz Länge M13-Universal-Primer GTAAAACGACGGCCAGT 17 FucA-Prim1 CTCCCCTCAAGCGAATGG 18 FucA-Prim2 CGCTGGCTCTCAAAAATC 18 FucA-Prim1 und FucA-Prim2 sind 5’-DIG-markierte Primer. Die Bindungsstellen können dem Anhang entnommen werden. Der M13-Universal-Primer wurde sowohl in DIG-markierter als auch in unmarkierter Form verwendet. 4.2.4 Rückklonierung und dsDNA-Sequenzierung Die aus den E. coli TG1- bzw. XL1-Zellen isolierte M13mp19-fuca-dsDNA wurde mit EcoR I und Pst I gespalten und in das mit den gleichen Restriktionsenzymen gespaltene Expressionsplasmid pKK223-3 kloniert. Die so erhaltenen Plasmide (pKKFA2-Homologe) wurden aus E. coli TG1-Zellen präpariert, und das Vorhandensein des fuca-Gens wurde durch Restriktionsanalyse mit EcoR I und Pst I überprüft. Wenn mehrere Mutanten parallel erzeugt bzw. kloniert worden waren, wurden die Mutationen zusätzlich auch direkt im Expressionsvektor nachgewiesen, um Verwechslungen auszuschließen. Dazu wurden die Expressionsvektoren im Bereich der Mutationsstelle sequenziert. Bei Mutationen im ersten Drittel des Gens wurde als Sequenzierungsprimer das Mutagenese-Oligonukleotid Oligo-T26A verwendet. Die Sequenzierung wurde wie bei der M13-ssDNA durchgeführt. Der einzige Unterschied bestand darin, daß die Entwicklung der blotting-Membran im Falle der dsDNA-Sequenzierungen in der Regel etwas länger dauerte (2 - 5 h). 86 4.3 Proteinpräparation der Mutanten 4.3.1 Expressionstest und Bakterienkultivierung Die Präparation der Mutanten erfolgte nach den von Dreyer ausgearbeiteten Bedingungen (Dreyer, 1995). Dazu wurden alle auf DNA-Ebene generierten Mutanten in E. coli JM105 transformiert und auf Expression von FucA-Mutanten getestet. Die Expressionsraten waren für alle untersuchten Mutanten etwa in der gleichen Größenordnung. Abbildung 27 zeigt exemplarisch das SDS-Gel der Expressionstests für die Mutanten R212A, E214A und E215A. 1 2 3 4 5 6 7 8 9 10 11 94 67 43 30 20.1 14.4 Bahn 1, 11: LMW-Marker (kDa) Bahn 2: Mutante R212A; Klon 1 Bahn 3: Mutante R212A; Klon 2 Bahn 4: Mutante R212A; Klon 3 Bahn 5: Mutante E214A; Klon 1 Bahn 6: Bahn 7: Bahn 8: Bahn 9: Bahn 10: Mutante E214A; Klon 2 Mutante E214A; Klon 3 Mutante E215A; Klon 1 Mutante E215A; Klon 2 Mutante E215A; Klon 3 Abbildung 27 SDS-Gel der Expressionstests der Mutanten R212A, E214A und E215A. Ausgehend von den bei -80 °C eingefrorenen Startkulturen der positiv getesteten Klone wurden die Mutanten in Schüttelkolben kultiviert. Aufgrund der sehr hohen Expressionsrate wurden bis zu drei Mutanten parallel in jeweils vier 1-l-Schüttelkolben à 300 ml LBAMedium kultiviert. In Einzelfällen wurden auch nur zwei Kolben pro Mutante verwendet. Eine Übersicht über die durchgeführten Präparationen zeigt Tabelle 11. Die Induktion der Zellen erfolgte bei einer OD578 zwischen 0.6 und 0.8 zu einer Endkonzentration von 0.5 mM IPTG. Nach der Induktion wurde für weitere 8 - 11 h kultiviert und dann geerntet. Die Ausbeuten lagen zwischen 3.3 und 6.2 g Bakterien (BFG) pro Liter Kulturmedium. Die Zellen wurden resuspendiert und sofort danach mittels Ultraschall aufgeschlossen. Dabei wurden relativ lange Beschallungszeiten gewählt (jeweils insgesamt 14 min), um einen möglichst vollständigen Zellaufschluß zu gewährleisten. 87 Tabelle 11 Ergebnisse der durchgeführten Präparationen Mutations- Anzahl der stelle a) Präparationen WT T26A Del(27) Del(27)/S71H Del(27)/S71K Del(27)/S71R N29E N29Q T41R T43V/S71Q S71E S71H S71K S71Q S71R E73Q/Y113F E73Q/Y113F/Y209F E73S Y113F Y113F/Y209F Y113T F131A F131A/F206W F206W Del(207-215) d) K207A Y209F Del(211-215) R212A E214A E214D E214L E214Q E215A e) a) b) c) d) e) L L L+P L+P L+P P P P P P P P P P A A+C A A A+C A H H H C C C C C C C C C C 3 1 1 1 1 1 1 1 2 1 1 2 2 1 1 1 2 2 2 1 1 2 1 2 2 2 2 1 1 2 1 1 1 1 Volumen Medium (Liter) BFG 1.2, 1.2, 0.6 4.8, 5.6, 2.8 7.8 1.2 5.1 1.2 3.5 0.9 5.5 1.2 4.0 1.2 5.2 1.2 6.5 1.2 4.2, 3.8 1.2, 0.9 5.2 1.2 5.8 1.2 5.5, 4.5 1.2, 1.2 5.2, 2.3 1.2, 0.6 5.7 1.2 5.7 1.2 5.0 1.2 4.8, 6.8 1.2, 1.2 6.5, 5.8 1.2, 1.2 7.5, 5.9 1.2, 1.2 5.5 1.2 4.8 1.2 5.5, 7.2 1.2, 1.2 4.5 1.2 5.8, 5,4 1.2, 1.2 5.5, 2.6 1.2, 0.6 8.0, 2.6 1.2, 0.6 4.0, 3.1 1.2, 0.6 4.5 1.2 6.8 1.2 7.0, 5.2 1.2, 1.2 3.2 0.6 5.5 1.2 2.8 0.6 6.5 1.2 gereinigtes Protein b) (mg) 175, 49, 28 340 42 66 97 30 40 235 - c) 31 55 38, 33 28, 25 53 37 87 28, 33 28, 104 20, 208 34 110 85, 180 32 143, 65 144, 13 35, 15 134, 14 191 155 320, 146 8 128 13 120 A = postulierter katalytischer Rest (Dreyer & Schulz, 1996b), C = flexibles C-terminales Kettenende, H = hydrophobe Wand, L = flexibler Loop(23-27), P = Phosphatbindungstasche. Bei den angegebenen Mengen handelt es sich zumeist nicht um die maximal mögliche Ausbeute, sondern um die Menge, die letztendlich gereinigt wurde. Die großen Unterschiede erklären sich dadurch, daß nach dem ersten Reinigungsschritt mittels IEC unterschiedlich eng geschnitten wurde und nur die jeweils benötigte Menge mittels GPC nachgereinigt wurde. Bildung von Einschlußkörpern aufgrund inkorrekter Faltung. Stammkultur von Müller-Dieckmann (1994). Reinigung nur über IEC. 88 4.3.2 Ionenaustauscher-Chromatographie Die Reinigung der Mutanten erfolgte in der Regel über zwei Säulenschritte (cf. Abbildung 7). Nach Zellaufschluß und Fällung der Nukleinsäuren mit Polymin-P wurde der Rohextrakt nach Filtration direkt auf die Anionenaustauscher-Säule (DEAE-Sepharose-CL6B) aufgetragen. Die Elution der Mutanten erfolgte dann durch Anlegen eines NaCl-Gradienten. Zu Beginn der Arbeit wurde mit einem linearen Gradienten von 0 - 500 mM NaCl eluiert, später wurde dann auch teilweise zunächst ein Stufengradient (50 mM NaCl) vorgeschaltet und schließlich mittels linearem Gradienten von 50 - 400 mM NaCl eluiert. Die Elution des WT erfolgte bei ca. 150 - 160 mM NaCl, während alle Mutanten (abhängig von etwaigen Ladungsveränderungen durch die Mutation) bei einer NaClKonzentration zwischen 130 und 190 mM eluiert wurden. So wurde beispielsweise die Mutante E214A, deren Elutionsdiagramm in Abbildung 28 exemplarisch aufgeführt ist, bei 140 mM NaCl eluiert. Das SDS-Gel der peak-Fraktionen (Abbildung 29) zeigt den sehr guten Reinigungseffekt dieses Säulenschrittes. Dabei erwiesen sich im Hinblick auf die Reinheit der FucA-haltigen Fraktionen vor allem solche Mutationen als besonders günstig, die eine Verschiebung der Elution zu niedrigeren NaCl-Konzentrationen zur Folge hatten, wie z. B. bei der Mutante E214A. Besonderheiten bei der Präparation einzelner Mutanten werden im folgenden gesondert aufgeführt. 1.6 250 Absorption (280 nm) 1.2 200 1.0 150 0.8 100 0.6 0.4 50 NaCl-Konzentration [mM] 280 nm NaCl-Gradient vereinigte Fraktionen 1.4 0.2 0 0 0 200 400 600 800 1000 1200 1400 Elutionsvolumen [ml] Abbildung 28 Elutionsdiagramm der IEC (DEAE-Sepharose-CL6B) der Mutante E214A. 89 1 2 3 4 5 6 7 8 9 10 11 12 94 67 43 30 20.1 14.4 Bahn 1: Bahn 2: Bahn 3: Bahn 4: Bahn 5: Bahn 6: Auftrag (Rohextrakt) Elutionsvolumen 1208-1216 ml Elutionsvolumen 1224-1232 ml Elutionsvolumen 1240-1248 ml Elutionsvolumen 1256-1264 ml Elutionsvolumen 1272-1280 ml Bahn 7: Bahn 8: Bahn 9: Bahn 10: Bahn 11: Bahn 12: Elutionsvolumen 1288-1288 ml Elutionsvolumen 1296-1304 ml Elutionsvolumen 1312-1220 ml Elutionsvolumen 1336-1244 ml Elutionsvolumen 1360-1368 ml LMW-Marker [kDa] Abbildung 29 SDS-PAGE ausgesuchter Fraktionen nach der IEC der Mutante E214A. Mutante T41R: Vortests hatten ergeben, daß die Mutante T41R ähnlich gut wie die übrigen Mutanten exprimiert wird. Der erwartete FucA-peak bei einer NaCl-Konzentration von ca. 150 mM war nur sehr schwach ausgeprägt (Abbildung 30), und das entsprechende SDS-Gel der Fraktionen zeigte nur geringe Mengen an Protein mit einer Molekularmasse, die der Mutante zugeordnet werden konnte. 0.5 250 280 nm Absorption (280 nm) 200 NaCl-Gradient 0.3 150 0.2 100 0.1 50 0 NaCl-Konzentration [mM] 0.4 0 0 200 400 600 800 1000 1200 1400 Elutionsvolumen [ml] Abbildung 30 Elutionsdiagramm der IEC (DEAE-Sepharose-CL6B) der Mutante T41R. 90 1 2 3 4 5 6 7 8 9 10 11 12 13 94 67 43 30 20.1 Bahn 1: Bahn 2: Bahn 3: Bahn 4: Bahn 6: Bahn 5: Bahn 7: LMW-Marker [kDa] feste Zellen vor dem Abernten Auftrag (Rohextrakt) Elutionsvolumen 1140-1148 Elutionsvolumen 1172-1180 Elutionsvolumen 1156-1164 Elutionsvolumen 1188-1196 Bahn 8: Bahn 9: Bahn 10: Bahn 12: Bahn 11: Bahn 13: Elutionsvolumen 1204-1212 Elutionsvolumen 1220-1228 Elutionsvolumen 1236-1244 Elutionsvolumen 1268-1276 Elutionsvolumen 1252-1260 Elutionsvolumen 1284-1292 Abbildung 31 SDS-PAGE ausgesuchter Fraktionen der IEC der Mutante T41R. Eine Erklärung zeigt der Vergleich der Bahnen 2 und 3 des SDS-Gels in Abbildung 31. Eine Zellprobe, die unmittelbar vor dem Ernten entnommen und direkt in SDS-Probenpuffer aufgeschlossen wurde, zeigt eine deutliche Überexpression der Mutante T41R. Im Rohextrakt nach Zellaufschluß und Polymin-P-Fällung ist diese Bande jedoch nahezu vollständig verschwunden. Dies legt den Schluß nahe, daß die Mutante durch die Einführung des Arginins in der Phosphatbindungstasche größtenteils nicht mehr korrekt faltet und es zur Bildung von Einschlußkörpern kommt. Dieser Befund wurde bei einer Wiederholung der Präparation bestätigt. Mutante Y113F: Das Elutionsverhalten der Mutante bei der IonenaustauscherChromatographie entsprach dem der übrigen Mutanten; so erfolgte die Elution bei einer NaClKonzentration von 150 bzw. 160 mM (2 Präparationen). Die Mutante fiel jedoch durch eine deutliche Blaufärbung auf. Nach Auftrag des grünlich schimmernden Rohextraktes auf die Säule bildete sich beim Äquilibrieren ein blau gefärbter Ring am oberen Rand der Säule, der sich im Laufe des NaCl-Gradienten die Säule entlang bewegte und schließlich der Mutante zugeordnet werden konnte. Die Farbe blieb im Laufe der Präparation und auch in den Wochen danach stabil. Erst nach einigen Monaten Lagerung verschwand die Blaufärbung. 91 Mutante E215A: Die Mutante zeigte eine deutlich verringerte Löslichkeit gegenüber dem WT und den anderen Mutanten. Die Elution von der Ionenaustauscher-Säule dieser Mutante erfolgte über Nacht. Am nächsten Morgen hatte sich in den FucA-haltigen Fraktionen (bei ca. 7 °C) ein Niederschlag gebildet, da die Mutante eine geringere Löslichkeit als der WT aufweist und demzufolge in höher konzentrierten Lösungen teilweise ausfällt. Die Konzentration in den Fraktionen der Ionenaustauscher-Chromatographie lag in der Regel zwischen 4 und 8 mg/ml. Aufgrund der geringen Löslichkeit wurde auf eine weitere Reinigung mittels GPC verzichtet, um ein Ausfallen des Proteins auf der Säule (bei RT) zu verhindern. Alle Untersuchungen der Mutante wurden mit den saubersten Fraktionen der IECSäule nach vorherigem Abzentrifugieren des ausgefallenen Proteins durchgeführt. 4.3.3 Gelpermeations-Chromatographie Die saubersten Fraktionen der IEC wurden im zweiten Reinigungsschritt über eine Gelpermeations-Chromatographie an Sephadex S200 nachgereinigt. Der Verlauf der Elution und die Reinheit der nach der GPC erhaltenen Proteinproben ist am Beispiel der Mutante K207A (Abbildungen 32 & 33) exemplarisch dargestellt. Das Elutionsvolumen aller untersuchter Mutanten stand dabei in Übereinstimmung mit einem tetrameren Oligomerisierungszustand. Die peak-Fraktionen wurden nach Reinheitskontrolle über SDS-PAGE vereinigt und zur weiteren Verwendung gegen TMZ-Puffer-I dialysiert. 2.5 280 nm vereinigte Fraktionen Absorption (280 nm) 2.0 1.5 1.0 0.5 0 0 20 40 60 80 100 120 Elutionsvolumen [ml] Abbildung 32 Elutionsdiagramm der GPC der Mutante K207A. Das Elutionsvolumen der Mutante von 67 ml entspricht dabei (gemäß Eichung der Säule) einer apparenten Molekularmasse von 91 ± 8 kDa. Der rechnerisch ermittelte Wert für das tetramere Enzym beträgt 95 kDa. 92 1 2 3 4 5 6 94 67 Bahn 1: LMW-Marker [kDa] Bahn 2: Elutionsvolumen 62.0-63.8 ml Bahn 3: Elutionsvolumen 63.8-65.6 ml Bahn 4: Elutionsvolumen 65.6-67.4 ml Bahn 5: Elutionsvolumen 67.4-69.2 ml Bahn 6: Elutionsvolumen 69.2-71.0 ml 43 30 20.1 14.4 Abbildung 33 SDS-PAGE ausgesuchter Fraktionen der GPC der Mutante K207A 4.3.4 Isoelektrische Fokussierung 1 2 3 4 5 6 7 8 9 10 11 12 9.30 Bahn 1: Bahn 2: Bahn 3: Bahn 4: Bahn 5: Bahn 6: Bahn 7: Bahn 8: Bahn 9: Bahn 10: Bahn 11: Bahn 12: Mutante Y209F Mutante E214Q Mutante E214D Mutante K207A IEF-Marker Mutante S71H IEF-Marker Mutante N29Q Mutante N29E Mutante S71Q Mutante S71E IEF-Marker 8.65 8.45 8.15 7.35 6.85 6.55 5.85 → ← 4.55 3.50 Abbildung 34 markiert. IEF ausgewählter FucA-Mutanten. Die Probenauftragspunkte sind mit Pfeilen Als weiteres Reinheitskriterium neben der SDS-PAGE bzw. auch zur weiteren Charakterisierung der Mutanten wurde eine IEF durchgeführt: Wie der WT, so spalten auch die Mutanten bei der IEF in mehrere diskrete, nahe beieinanderliegende Banden auf (3 Hauptbanden). In Übereinstimmung mit der Theorie weisen Mutanten, bei denen keine Veränderung des Verhältnisses von positiv zu negativ geladenen Aminosäuren vorliegt, den 93 gleichen pI wie der WT auf, der etwa bei pH 6.6 liegt. Wie aus Abbildung 34 hervorgeht, spiegeln sich die Austausche von bzw. hin zu geladenen Aminosäuren erwartungsgemäß auch in einem veränderten pI wider. So hat der Austausch von Ser71 gegen His oder Glu ein Sinken des pI von 6.6 über 6.55 auf einen Wert von etwa 6.3 zur Folge. Eine Verschiebung des pI in die gleiche Richtung ist auch bei Wegnahme einer positiven Ladung wie in Mutante K207A zu beobachten, während dementsprechend z. B. bei Mutante E214Q (pI = 6.85) eine Verschiebung zu höherem pI auftritt. Eine Ausnahme bildet Mutante S71Q. Da durch die Mutation die Ladungsbilanz der Mutante nicht verändert wurde, sollte sie den gleichen pI wie der WT annehmen. Tatsächlich jedoch ist der pI der Mutante (pI = 6.35) um etwa 0.2 pHEinheiten ins Saure verschoben. Das wird möglicherweise durch die erheblichen Strukturänderungen bei Mutante S71Q verursacht (vgl. Kapitel 4.5.8). 4.3.5 Spektroskopische Untersuchung der Mutante Y113F Die Mutante Y113F fiel bei der Präparation durch eine blaue Färbung auf. Das Absorptionsspektrum der Mutante im Spektralbereich von 300 - 700 nm zeigt eine breite Bande mit einem Absorptionsmaximum bei 618 nm (Abbildung 35) und einem molaren Extinktionskoeffizienten von ε618 = 220 M-1cm-1 (bezogen auf die eingesetzte Menge an Mutante Y113F). Dieser Wert wurde bei zwei unabhängigen Präparationen der Mutante ermittelt. Darüber hinaus weist das Spektrum eine Schulter bei ca. 360 nm auf. Es handelt sich somit um ein Spektrum, wie es typischerweise bei Charge-Transfer-Komplexen gefunden wird. Wird neben Tyr113 auch noch Tyr209 zu Phe mutiert, so verschwindet die Blaufärbung überraschenderweise wieder. 0.3 0.3 0.3 Mutante Y113F 0.1 0 300 400 500 600 Wellenlänge [nm] 700 Mutante Y113F/Y209F Absorption 0.2 Absorption Absorption Mutante Y209F 0.2 0.1 0 300 400 500 600 Wellenlänge [nm] 700 0.2 0.1 0 300 400 500 600 700 Wellenlänge [nm] Abbildung 35 Absorptionsspektren der Mutanten Y209F, Y113F und Y113F/Y209F im Bereich von 300 - 700 nm. Die Messung erfolgte mit 1-cm-Küvetten und bei Proteinkonzentrationen von 7.2 mg/ml (Mutante Y209F), 8.9 mg/ml (Mutante Y113F) und 8.6 mg/ml (Mutante Y113F/Y209F). 94 Um weitere Hinweise auf die Ursache der Blaufärbung der Mutante zu bekommen, wurde ihre Zusammensetzung mittels Röntgenfluoreszenz-Spektroskopie bestimmt. Dazu wurden die Mutante Y113F sowie Mutante T26A und der WT als Referenzproben in lyophilisierter Form an die Firma Siemens AG gesandt, wo dann unter Verwendung des Röntgenfluoreszenzspektrometers SRS3000 eine semiquantitative Analyse durchgeführt wurde. Bei der Messung wurde das gesamte Elementspektrum von Na bis Uran kontinuierlich durchlaufen und dann anhand der Nettointensitäten der gefundenen Elementlinien die Konzentrationen iterativ berechnet (Tabelle 12). Neben den erwarteten Elementen Schwefel und Zink wurden in allen drei Proben auch geringfügige Mengen mitgeschleppter Ionen wie z. B. Kalium und Chlorid gefunden. Abweichend von den anderen beiden Proben wurden jedoch bei Mutante Y113F auch Spuren von Eisen gefunden. Tabelle 12 Ergebnisse der Röntgenfluoreszenzspektroskopie a) „Protein“ b) Si P S Cl K Ca Fe Zn a) b) WT Gew. % T26A Gew. % Y113F Gew. % 93.8 0.020 0.048 1.93 3.01 0.014 0.050 1.16 95.6 0.027 0.010 2.78 0.75 0.010 0.019 0.778 96.5 0.017 0.009 1.65 1.32 0.030 0.039 0.401 Elemente, die in der Tabelle nicht aufgeführt sind, lagen unterhalb der Nachweisgrenze. Bei der semiquantitativen Auswertung wurden aus den Scans automatisch die Nettointensitäten der vorhandenen Elementlinien bestimmt und unter Anwendung einer probenspezifischen Matrixkorrektur und universeller Geräteempfindlichkeiten die Konzentrationen iterativ berechnet. Als Matrix wurde H7C3NO2 unter „Protein“ angenommen. Dies ist nötig, da die für die einzelnen Elemente gemessenen Intensitäten nicht nur von der relativen Konzentration des Elements sondern auch von der Umgebung der Probe abhängig sind. Die entsprechenden Konzentrationen der gefundenen Elemente in Tabelle 12 sind in Gewichtsprozent bezogen auf die Matrix H7C3NO2 angegeben. Um die Menge an gefundenem Eisen in Relation zur Menge an FucA zu setzen, wurde als Bezugspunkt nicht die Matrix benutzt, die nur eine relativ ungenaue Angabe der tatsächlich vorhandenen Menge an FucA ist, sondern die Menge an gefundenem Schwefel unter Berücksichtigung der Amino- 95 säuresequenz der FucA: Bei 4 Cysteinen, 5 Methioninen sowie einem gebundenen β-ME ergeben sich somit 10 Schwefelatome pro Monomer. Unter dieser Berechnungsgrundlage ergeben die Messungen für Mutante T26A 1.37 Zinkatome pro Untereinheit und für Mutante Y113F 1.18 Zinkatome und 0.13 Eisenatome pro Untereinheit. Bezogen auf die Matrix H7C3NO2 berechnet sich für Mutante Y113F ein Wert von 1.5 Zinkatomen und 0.17 Eisenatomen pro Untereinheit. Skaliert auf den Faktor 1 kann dies bedeuten, daß in ca. 10 % der aktiven Zentren von Mutante Y113F Zink durch Eisen ersetzt sein könnte. Fe(II) als Ursache für die blaue Farbe würde auch eine plausible Erklärung für das Verschwinden der Farbe nach einigen Monaten Lagerung durch Oxidation von Fe(II) zu Fe(III) liefern. 96 4.4 Kristallisation 4.4.1 Kristallform T Die Kristallisation der Mutanten folgte im wesentlichen den von Dreyer (1995) ausgearbeiteten Bedingungen für die hochauflösende Kristallform T des WT. Dabei wurden sowohl der pH-Wert als auch die Konzentration des Fällungsmittels Ammoniumsulfat variiert. Als optimal erwies sich dabei ein pH-Wert zwischen 7.8 und 8.3 sowie eine Fällungsmittelkonzentration zwischen 1.6 und 1.75 M Ammoniumsulfat bei 50 %iger Vorsättigung (Tabelle 13). Eine Übersicht über die Ergebnisse der Kristallisation der einzelnen Mutanten ist in Tabelle 14 gegeben. So bildeten sich bei Mutante R212A schon nach drei Tagen erste Kristalle, die nach ca. 2 Wochen ihre maximale Größe erreicht hatten. In anderen Fällen (z. B. Mutante F131A) dauerte es mehrere Monate bis erste Kristalle auftraten. Bei Mutante E73Q bildeten sich erst nach ca. 2 Jahren Kristalle mit tetragonalem Habitus. Dabei handelte es sich um Kristalle aus Kristallisationsexperimenten von Müller-Dieckmann (1994; siehe dazu auch Kapitel 4.1.1). Daß es sich bei den unterschiedlichen Kristallisationsdauern nicht zwangsläufig um Charakteristika der jeweiligen Mutante handelt, wird am Beispiel der Mutante T26A ersichtlich. So entstanden bei Kristallisationsansätzen mit identischen Bedingungen in einigen Fällen schon nach einigen Tagen Kristalle, in anderen aber erst nach weit über einem Jahr, wobei die erst später gewachsenen Kristalle zumeist deutlich größere Dimensionen aufwiesen. Teilweise bildete sich zunächst ein Präzipitat, aus dem später Kristalle wuchsen. Oft wuchsen die Kristalle jedoch nicht mit vollständig ausgebildeter quadratischer Grundfläche. Dafür war aber in solchen Kristallen die dritte Dimension deutlich stärker ausgebildet, was sich in der Regel in besserem Streuvermögen der Kristalle widerspiegelte (Abbildung 38). Tabelle 13 Standard-Kristallisationsbedingungen für die FucA-Mutanten in der Kristallform T a) Reservoirlösung: Proteinlösung: 20 mM KH2PO4/KOH pH 7.8 - 8.3 1.60 - 1.75 M (NH4)2SO4 4 mM β-ME 1 mM ZnCl2 8.5 - 10 mg/ml Protein 20 mM Tris/HCl pH 7.6 10 mM β-ME 1 mM ZnCl2 a) Die Hängetropfen setzten sich aus gleichen Volumina an Reservoir- und Proteinlösung zusammen. 97 Tabelle 14 Übersicht über das Ergebnis der Kristallisationsexperimente für die FucA-Mutanten Größe b), Habitus, Besonderheiten... Mutationsstelle a) Auftreten von Kristallform T röntgentauglich röntgenogr. untersucht/ Datensatzaufnahme T26A L + + +/+ große Kristalle, regelmäßiger Habitus Del(27) L + + +/+ große Kristalle, regelmäßiger Habitus Del(27)/S71H L+P - - - nur Niederschlag Del(27)/S71R L+P + - - N29Q P + + +/+ nur kleine flache Kristalle, stark schichtenförmig verwachsen große Kristalle, regelmäßiger Habitus N29E P + - - nur kleine Kristalle, regelmäßiger Habitus T43V/S71Q P + + - S71E P + c) - - mittelgroße Kristalle, die allerdings nur sehr selten auftraten, regelmäßiger Habitus nur kleine Kristalle, regelmäßiger Habitus S71H P - - - nur Niederschlag S71K P + - +/- S71Q P + d) + +/+ S71R P + - - E73Q/Y113F A + + +/- A+C + + +/+ E73S A + + +/+ Y113F A + + +/+ mittelgroße bis große Kristalle, aber oft mit Verwachsungen und Rissen große Kristalle, regelmäßiger Habitus A+C + + +/+ große Kristalle, regelmäßiger Habitus Y113T A + - - F131A H + + +/+ F131A/F206W H + e) + - Y209F C + + - R212A C + + +/+ große Kristalle, regelmäßiger Habitus E214A C + + +/+ große Kristalle, regelmäßiger Habitus E214L C + + +/- Kristalle mittlere Größe, Tendenz zu Verwachsungen, Auflösungsgrenze bei ca. 3 Å E73Q/Y113F/Y209F Y113F/Y209F a) b) c) d) e) Kristalle mittlerer Größe mit Rissen, Auflösungsgrenze bei ca. 3 Å große Kristalle, regelmäßiger Habitus nur kleine Kristalle, schichtenförmig verwachsen mittlere Größe, regelmäßiger Habitus, Auflösungsgrenze bei ca. 3 Å große Kristalle, regelmäßiger Habitus Größe maximal 70 x 70 x 20 µm3, schichtenförmig verwachsen mittelgroße Kristalle, die nur sehr selten auftraten, regelmäßiger Habitus mittelgroße Kristalle mit ausgeprägter dritten Dimension: (250 x 250 x 200 µm3) große Kristalle, regelmäßiger Habitus A = postulierter katalytischer Rest (Dreyer & Schulz, 1996b), C = flexibles C-terminales Kettenende, H = hydrophobe Wand, L = flexibler Loop(23-27), P = Phosphatbindungstasche. Als „klein“ werden an dieser Stelle Kristalle bezeichnet, deren längste Ausdehnung 150 µm nicht überstieg; als „groß“ solche, deren längste Ausdehnung mindestens 500 µm betrug und die über eine deutliche dritte Dimension (≥ 200 µm) verfügten. Zunächst hatte sich eine neue sehr instabile Kristallform gebildet, die im Laufe einiger Wochen in den Tropfen zerfiel, wobei ein dunkler Niederschlag zurückblieb. Erst nach über einem Jahr hatten sich in einigen dieser Tropfen kleine Kristalle der Kristallform T gebildet. Kristalle der Mutante S71Q traten erst bei deutlich höherer Fällungsmittelkonzentration auf (vgl. Tabelle 15). Tetragonale Kristalle konnten erst mittels Jancarik-Screening (Jancarik & Kim, 1991) gezüchtet werden (Bedingung Nr. 4, siehe Tabelle 16), wobei sie auch dort erst nach mehreren Monaten auftraten. Eine Verfeinerung der Bedingungen konnte dann aus Zeitgründen nicht mehr durchgeführt werden. 98 Abbildung 36 Kristall der Mutante E73Q/Y113F/Y209F (Kristallform T); Abbildung 37 Kristall der Mutante S71Q (Kristallform T); || || 100 µm 100 µm 99 Abbildung 38 Kristall der Mutante T26A (Kristallform T); Abbildung 39 Kristalle der Mutante R212A (Kristallform T); || || 100 µm 100 µm 100 Alle Mutanten, die in der Kristallform T kristallisiert werden konnten, weisen unter dem Polarisationsmikroskop ein typisches, schon beim WT beobachtetes Phänomen auf: Bei Aufsicht auf die quadratische Grundfläche, also entlang der vierzähligen Achse, tritt Doppelbrechung auf (Abbildung 36), obwohl die c-Achse in tetragonalen Kristallsystemen definitionsgemäß optisch isotrop ist. Darüber hinaus zeigen die Kristalle ein an einen Briefumschlag erinnerndes Muster, d. h. man erkennt zwei Paare von sich gegenüberliegenden Dreiecken, wobei sich die Paare jeweils optisch gleich verhalten. Kristalle, die über eine ausreichende dritte Dimension verfügen, bieten von der Seite betrachtet ein ähnliches Bild (Abbildung 37 & 39). Für eine ausführlichere Diskussion dieses Phänomens sei auf Dreyer (1995) verwiesen. Tabelle 15 modifizierte Kristallisationsbedingungen für Mutante S71Q in Kristallform T a) b) Reservoirlösung: Proteinlösung: 20 mM KH2PO4/KOH pH 8.1 1.9 M (NH4)2SO4 4 mM β-ME 1 mM ZnCl2 10 mg/ml Protein 20 mM Tris/HCl pH 7.6 20 mM L-Fucitol b) 10 mM β-ME 1 mM ZnCl2 a) Die Hängetropfen setzten sich aus gleichen Volumina an Reservoir- und Proteinlösung zusammen. L-Fucitol wurde freundlicherweise von Schneider (1994) zur Verfügung gestellt. Schwierigkeiten bei der Kristallisation traten vor allem bei Mutationen in der Phosphatbindungstasche auf. Von den zahlreichen Mutanten mit veränderter Phosphatbindungstasche, mit denen Kristallisationsexperimente durchgeführt wurden, konnten nur für Mutante N29Q unter den in Tabelle 13 angegebenen Bedingungen röntgentaugliche Kristalle erhalten werden. Diese erreichten eine maximale Größe von bis zu 1000 x 500 x 500 µm3. Mutante S71Q zeigte ein von den anderen Mutanten abweichendes Kristallisationsverhalten. Kristalle traten erst bei einer Ammoniumsulfatkonzentration von mindestens 1.75 M auf, was diese Mutante deutlich von allen anderen in der Kristallform T kristallisierenden Mutanten abhebt. Die besten Kristalle mit einer Dimension von ca. 600 x 400 x 400 µm3 wurden schließlich durch Zusatz von 20 mM L-Fucitol bei einer Fällungsmittelkonzentration von 1.9 M Ammoniumsulfat erhalten (Tabelle 15; Abbildung 37). Bei allen anderen Mutanten mit Mutationen in der Phosphatbindungstasche scheiterten die Versuche, röntgentaugliche Kristalle zu erhalten. Es bildeten sich entweder nur Niederschläge (z. B. Mutante S71H oder N29E), nur kleine parallel zur quadratischen Grundfläche stark schichtenförmig verwachsene Kristalle (z. B. Mutante S71R) oder Kristalle mittlerer Größe, die jedoch starke Rißbildungen zeigten (Mutante S71K) und demzufolge maximal bis 3 Å streuten. 101 Bei Mutanten, die unter den Standardbedingungen keine röntgentauglichen Kristalle ergaben, wurde zusätzlich ein Jancarik-Screening durchgeführt, um nach neuen Bedingungen zu suchen. Für Mutante F131A/F206W konnte so eine Bedingung (Tabelle 16) gefunden werden, die zu röntgentauglichen Kristallen führte. Die dabei erhaltenen Kristalle erreichten eine Größe von 250 x 250 x 200 µm3. Eine Verfeinerung der Bedingungen und eine Aufklärung der Elementarzelle konnte jedoch aus Zeitgründen nicht mehr durchgeführt werden. Der Habitus der Kristalle sowie das Doppelbrechungsmuster lassen jedoch den Schluß zu, daß es sich hierbei auch um Kristallform T handelt. Tabelle 16 modifizierte Kristallisationsbedingungen für Mutante F131A/F206W in Kristallform T a) Reservoirlösung: Proteinlösung: 100 mM Tris/HCl pH 8.5 2 M (NH4)2SO4 (Jancarik-Screening Bedingung Nr. 4) 10 mg/ml Protein 20 mM Tris/HCl pH 7.6 10 mM β-ME 1 mM ZnCl2 a) Die Hängetropfen setzten sich aus gleichen Volumina an Reservoir- und Proteinlösung zusammen. 4.4.2 Weitere Kristallformen Neben der Kristallform T war auch die kubische Kristallform K, die einen linsenförmigen bis nahezu perfekt kugelförmigen Habitus aufweist, für die Mutanten von Interesse. Diese streut zwar deutlich schlechter als Kristallform T, hat sich aber für den WT als stabil bei Tränkexperimenten mit dem Inhibitor PGH erwiesen, während in der Kristallform T sofort Risse auftraten und die Kristalle schließlich zerstört wurden. Die Kristalle erscheinen beim WT unter den gleichen Bedingungen wie Kristallform T, jedoch deutlich seltener und nicht gezielt reproduzierbar. Dies war auch bei den Mutanten der Fall. So trat die Kristallform K nur in Ausnahmefällen auf. Darüber hinaus bildeten sich dann in den Tropfen nie vereinzelte große Kristalle, wie für den WT beschrieben, sondern immer nur eine sehr große Zahl kleiner Kristalle mit einem Durchmesser von ca. 50 µm. Die Kristalle waren somit nicht röntgentauglich. In einem einzelnen Tropfen wurden bei Mutante T26A auch einige größere Kristalle gefunden (Abbildung 40a). Sie erwiesen sich allerdings bei Tränkversuchen mit PGH als nicht stabil und zeigten sofort Rißbildung. Bei weiteren Kristallisationsexperimenten mit dem WT traten unter den Bedingungen aus Tabelle 13, bei denen der pH-Wert der Reservoirlösung auf einen Wert zwischen pH 6 und 6.5 erniedrigt wurde, die in Abbildung 40b gezeigten Kristalle auf. Vom Habitus scheinen sie eine Art Hybridform zwischen Kristallform T und K darzustellen. So sind sie leicht 102 rundlich, erinnern aber immer noch an die quadratische Grundfläche von Kristallform T. Unter dem Polarisationsmikroskop zeigen sie auch das für Kristallform T typische Doppelbrechungsmuster, wobei die einzelnen Bereiche allerdings nicht mehr so scharf gegeneinander abgegrenzt sind. Wie die röntgenographische Untersuchung eines derartigen Kristalls zeigte, handelt es sich wie bei Kristallform K um die Raumgruppe F432 mit a = b = c = 183.3 Å. Es konnte ein Datensatz bis zu 2.83 Å Auflösung (Rsym = 7.3 %) mit einer Vollständigkeit von 97 % aufgenommen werden. Im Gegensatz zu den bereits bekannten Kristallen dieser Raumgruppe waren die reproduzierbaren Kristalle leider wie Kristallform T nicht stabil gegen Tränkexperimente mit PGH, so daß dieser Weg dann für die Mutanten nicht weiter verfolgt wurde. Auch die von Dreyer beschriebenen Kristallformen W und H wurden bei einzelnen Mutanten beobachtet. So konnte beispielsweise die Kristallform W für die Mutante E214A reproduziert werden. Nach Zusatz von 4 % 2-Methylpentandiol zu den unter Tabelle 13 aufgeführten Bedingungen wuchsen würfelförmige Kristalle bis zu einer Größe von 600 x 600 x 600 µm3. Wie aufgrund des hohen Solvensgehalts von 76 % in dieser Kristallform zu erwarten, lag die Auflösungsgrenze wie für die WT-Kristalle nur bei ca. 3 Å. Von Kristallform H traten nur sehr vereinzelt Kristalle auf, die dann auch nicht sehr groß wurden und demgemäß nicht röntgenographisch untersucht werden konnten. (a) (c) (b) (d) Abbildung 40 Kristalle der Mutante T26A in Kristallform K (a), des WT in einer Hybridform zwischen Kristallform T und K (b), der Mutante T26A in Kristallform D (c) sowie der Mutante F131A/F206W in Kristallform D2 (d); || 100 µm 103 Neben den bereits für den WT beschriebenen Kristallformen traten bei den Mutanten auch noch weitere Kristallformen auf. Beispielsweise wuchsen unter den Bedingungen für die Kristallform T bei einigen Mutanten auch Kristalle, deren Habitus zwar innerhalb desselben Tropfens etwas variierte, aber dabei immer regelmäßige Dreiecksflächen aufwies: Kristallform D (Abbildung 40c). Teilweise wuchsen solche Kristalle auch als flache Plättchen (Kristallform D2) wie in Abbildung 40d für Mutante F131A/F206W gezeigt. Die Kristalle erreichten in Einzelfällen eine maximale Größe von 400 x 400 x 300 µm3, waren jedoch mechanisch instabil und zerfielen größtenteils nach einigen Wochen wie im Falle von Mutante S71E. Für Mutante E73Q wurde ein Kristall vom Typ-D, der noch aus Kristallisationsexperimenten von Müller-Dieckmann (1994) stammte, röntgenographisch untersucht. Er zeigte jedoch schon beim Montieren aus der Reservoirlösung starke Rißbildung und streute nur bis maximal 4 Å, wobei das Streuvermögen im Strahl dann sehr rasch abnahm, so daß nur knapp 50 frames aufgenommen und ausgewertet werden konnten. Die Auswertung dieser frames mit dem Programm XDS legt als mögliche Raumgruppe ein tetragonal innenzentriertes Gitter mit Achsenlängen von a = b = 134 Å und c = 191 Å nahe. 4.5 Röntgenstrukturanalyse der FucA-Mutanten Von insgesamt 13 verschiedenen Mutanten konnten Datensätze aufgenommen werden. Röntgentaugliche Kristalle von drei weiteren Mutanten, die erst nach sehr langer Zeit gewachsen waren, konnten dann aus Zeitgründen nicht mehr vermessen werden. Die Tabellen 17 & 18 geben einen Überblick über die Statistik der Datensammlung. Alle untersuchten Mutanten gehören wie der WT zur Raumgruppe P4212 mit teilweise gegenüber dem WT leicht abweichenden Zellparametern: Die Abweichungen betrugen für die a-Achse maximal 0.5 % und für die c-Achse bis zu 1.6 %. Die aufgenommenen Datensätze reichten bis zu Auflösungen zwischen 2.55 Å für Mutante E73S und 1.66 Å für Mutante S71Q. Die Rsym-Werte für die einzelnen Datensätze betrugen zwischen 3.4 und 8.7 %. Die Mutante S71Q hat somit eine deutlich bessere Auflösung als der WT und dabei einen sehr niedrigen Rsym-Wert. Mittels dieser Mutante könnte daher unter Kryobedingungen mit energiereicher Synchrotronstrahlung die Auflösungsgrenze beträchtlich verbessert werden. 104 Tabelle 17 Statistik der Datensammlung T26A Del(27) N29Q N29L/S71A a) S71Q E73S E73Q a) a=b 93.8 93.3 93.7 93.8 93.5 94.1 93.9 c 43.0 43.4 43.0 43.5 42.4 43.5 43.0 10 - 2.09 10 - 2.15 10 - 1.84 10 - 2.53 10 - 1.66 10 - 2.55 10 - 2.15 letzte Schale (Å) 2.16 - 2.09 2.22 - 2.15 1.90 - 1.84 2.61 - 2.53 1.72 - 1.66 2.63 - 2.55 2.22 - 2.15 unabhängige Reflexe c) 10121 (822) 9818 (795) 16459 (1428) 6246 (487) 21716 (1782) 6347 (521) 10320 (861) Redundanz 3.1 (2.3) 4.9 (2.9) 3.9 (2.2) 4.5 (2.6) 3.7 (2.0) 5.3 (3.4) 3.4 (2.1) Rsym (%) 6.6 (15) 6.8 (19) 5.4 (17) 7.7 (20) 3.4 (16) 6.4 (23) 4.4 (13) mittleres I/σ 8.6 (4.8) 9.7 (4.0) 10.4 (4.4) 8.8 (3.7) 17.2 (4.7) 10.8 (3.3) 15.5 (5.7) Vollständigkeit (%) 87 (78) 92 (82) 97 (91) 92 (80) 96 (84) 95 (89) 95 (87) Zellparameter (Å) b) Auflösung (Å) a) b) c) Kristalle von Müller-Dieckmann (1994). Alle Mutanten gehören zur Raumgruppe P4212 (wie der WT). Alle Werte in Klammern beziehen sich auf die letzte Auflösungsschale. Tabelle 18 Statistik der Datensammlung E73Q/ Y113F Y113F/Y209F F131A R212A E214A WT-FucA a) Y113F/Y209F Zellparameter [Å] b) a=b 93.7 93.7 93.7 93.9 93.7 93.8 93.7 c 43.2 42.6 42.8 43.3 42.6 42.9 42.8 10 - 1.97 10 - 1.92 10 - 1.86 10 - 2.17 10 - 2.18 10 - 2.44 10 - 1.92 2.04 - 1.97 1.96 - 1.92 1.92 - 1.86 2.24 - 2.17 2.24 - 2.18 2.52 - 2.44 1.96 - 1.92 13420 (1143) 13096 (503) 15037 (1037) 10014 (840) 9269 (463) 6829 (568) 14541 Redundanz 4.2 (2.3) 4.7 (1.7) 4.9 (2.0) 3.2 (1.8) 3.9 (1.9) 4.1 (2.3) 9 Rsym [%] 5.4 (19) 4.3 (18) 6.4 (17) 6.3 (13) 3.4 (17) 8.7 (21) 6.5 (28) 12.4 (4.0) - 8.5 (4.4) 10.1 (5.4) - 8.2 (3.3) - 96 (89) 87 (63) 92 (69) 95 (87) 89 (65) 92 (85) 97 (93) Auflösung [Å] letzte Schale [Å] unabhängige Reflexe c) mittleres I/σ d) Vollständigkeit (%) a) b) c) d) Werte von Dreyer (1995). Alle Mutanten gehören zur Raumgruppe P4212 (wie der WT). Alle Werte in Klammern beziehen sich auf die letzte Auflösungsschale. Bei Datensätzen, die mit dem Programm MERGE ausgewertet wurden, wird in der entsprechenden Ausgabedatei kein mittleres I/σ angegeben. 105 Die charakteristischen Daten der Mutantenstrukturen im Vergleich zu denen des WT sind in den Tabellen 19 & 20 aufgelistet. Die r.m.s.-Abweichungen für die Cα-Atome gegenüber der WT-Struktur liegen zwischen 0.14 Å2 für Mutante T26A und 0.61 Å2 für Mutante S71Q, also teilweise innerhalb des Koordinatenfehlers. Die Unterschiede der r.m.s.Abweichungen bei den einzelnen Mutanten sind dabei vor allem auf veränderte Konformationen des flexiblen Loops(23-27) zurückzuführen. Bei Mutante S71Q treten darüber hinaus größere Veränderungen im gesamten Bereich der Phosphatbindungstasche auf. Wie in der WT-Struktur so war auch bei allen untersuchten Mutanten die Seitenkette von Cys14 modifiziert, wobei ein Molekül β-Mercaptoethanol kovalent an Sγ gebunden war. Die in der WT-Struktur beobachteten alternativen Konformationen bei fünf Aminosäureresten wurden auch für alle Mutantenstrukturen bei der Verfeinerung berücksichtigt, wobei die relativen Besetzungszahlen der WT-Struktur beibehalten wurden. Bei Mutante S71Q erlaubte die hohe Auflösung der Struktur das Einführen alternativer Seitenketten für vier weitere Reste (Asn146, Leu152, Leu157 und His172). In der Struktur von Mutante E73Q/Y113F/Y209F wurden schließlich auch alternative Konformationen für das neu eingeführte Glutamin beobachtet (s. u.). Tabelle 19 Charakteristische Daten der verfeinerten Strukturen T26A Del(27) N29Q N29L/S71A S71Q E73S E73Q 10 - 2.09 10 - 2.15 10 - 1.84 10 - 2.53 10 - 1.66 10 - 2.55 10 - 2.15 R-Faktor [%] Freier R-Faktor [%] a) 15.0 20.7 15.6 22.3 16.5 20.4 14.2 - 16.4 20.0 14.9 22.9 15.0 20.5 Nichtwasserstoffatome: b) Proteinatome Zink SO42Wassermoleküle 1623 1 10 109 1592 1 10 100 1598 1 10 113 1596 1 5 93 1600 1 5 136 1594 1 5 101 1597 1 10 118 rmsd Bindungslängen [Å] rmsd Bindungswinkel [°] 0.009 2.0 0.010 2.3 0.008 1.6 0.013 2.9 0.007 1.4 0.011 2.5 0.009 1.9 Mittlere B-Faktoren: Protein [Å2] Zink [Å2] SO42- [Å2] Wassermoleküle [Å2] 18 11 26 32 24 13 41 35 22 14 38 36 25 18 50 34 19 12 74 36 29 23 78 40 27 15 53 40 rmsd Cα-Atome [Å] c) letzter Rest d) abweichende Reste f) 0.14 209 - 0.47 206 e) 24-26 0.34 206 24-26 0.24 206 25 0.61 206 24-26, 42-47, 53, 67, 69-72 0.40 206 25-26 0.16 206 - Auflösung a) b) c) d) e) f) Das Test-Set umfaßte 6% der Reflexe. Bei Mutante E73Q wurde im aktiven Zentrum zusätzlich ein Molekül DHAP mit einer relativen Besetzungszahl von 0.25 eingebaut. Abweichungen gegenüber der Struktur des WT. C-terminaler Rest mit definierter Konformation. Die Numerierung der Reste wurde trotz der Deletion von Ala27 gemäß den Positionen im WT vorgenommen. Reste mit einer Abweichung ≥ 1 Å für das Cα-Atom relativ zur entsprechenden Position im WT. 106 Tabelle 20 Charakteristische Daten der verfeinerten Strukturen E73Q/ Y113F/Y209F Y113F Y113F/Y209F F131A R212A E214A WT-FucA a) 10 - 1.97 10 - 1.92 10 - 1.86 10 - 2.17 10 - 2.18 10 - 2.44 10 - 1.92 R-Faktor [%] Freier R-Faktor [%] b) 17.1 22.1 15.9 20.7 16.8 21.6 15.2 21.8 14.4 20.0 14.9 22.6 18.6 - Nichtwasserstoffatome: c) Proteinatome Zink SO42Wassermoleküle 1596 1 110 1612 1 10 108 1596 1 5 110 1591 1 10 101 1613 1 10 99 1597 1 10 101 1597 1 10 119 rmsd Bindungslängen [Å] rmsd Bindungswinkel [°] 0.11 2.1 0.008 1.9 0.008 1.8 0.010 2.0 0.009 2.1 0.010 2.4 0.013 1.7 Mittlere B-Faktoren: Protein [Å2] Zink [Å2] SO42- [Å2] Wassermoleküle [Å2] 25 17 39 27 22 50 42 26 21 40 40 25 17 42 37 25 17 48 36 17 13 24 28 19 12 24 36 rmsd Cα-Atome [Å] d) letzter Rest e) abweichende Reste f) 0.21 206 - 0.36 208 24-26 0.37 206 24-26 0.16 206 - 0.33 208 25-26 0.17 206 - 206 - Auflösung a) b) c) d) e) f) Struktur von Dreyer (1995) gelöst. Das Test-Set umfaßte 6% der Reflexe. Bei Mutante E73Q/Y113F/209F wurde im aktiven Zentrum zusätzlich ein Molekül DHAP mit einer relativen Besetzungszahl von 0.7 eingebaut. Abweichungen gegenüber der Struktur des WT. C-terminaler Rest mit definierter Konformation. Reste mit einer Abweichung ≥1 Å für das Cα-Atom relativ zur entsprechenden Position im WT. 4.5.1 Das C-terminale Kettenende Da die neun C-terminalen Aminosäuren in der Struktur des WT nicht detektiert werden konnten, war es von besonderem Interesse, wie der C-terminale Bereich in den Mutantenstrukturen aussehen würde. Eine Fixierung des vollständigen C-terminalen Kettenendes ist in keiner der Mutantenstrukturen zu beobachten. Dies ist vor allem im Falle der vier Mutanten interessant, die eine C-terminale Mutation enthalten: nämlich bei den Mutanten R212A, E214A, Y113F/Y209F sowie E73Q/Y113F/Y209F. Dennoch war bei einigen Mutanten am C-terminalen Kettenende zusätzliche Elektronendichte vorhanden, die es schließlich erlaubte, die Modelle über Phe206’ hinaus auszudehnen. So konnte bei Mutante T26A die C-terminale Helix bis Tyr209’ erweitert werden und in den Mutanten Y113F bzw. R212A bis Thr208’. Exemplarisch seien an dieser Stelle die (2Fo-Fc)-Elektronendichten bei einem Konturniveau von 1 σ im C-terminalen Bereich der Mutanten E214A (Abbildung 41), R212A (Abbildung 42) und T26A (Abbildung 43) gezeigt. Die Orientierung der drei zusätzlichen 107 Reste bei Mutante T26A ist die gleiche wie in der Struktur des WT, nachdem das katalytische Zink-Ion gegen Cobalt ausgetauscht wurde (Abbildung 44). In beiden Strukturen zeigt die Seitenkette von Tyr209’ vom aktiven Zentrum weg, was auf den ersten Blick einer Beteiligung des Restes an der Katalyse entgegenzustehen scheint. Eine genauere Betrachtung der Strukturen enthüllt jedoch, daß die phenolische OH-Gruppe des Tyrosins jeweils eine H-Brücke zu einem Sulfat, das an einem Kristallkontakt sitzt, ausbildet. Tyr209’ und somit auch das C-terminale Kettenende sind in diesen Kristallen in einer Konformation stabilisiert, die keineswegs repräsentativ für die Verhältnisse während der Katalyse sein muß. Die Überlagerung der verschiedenen C-terminalen Fragmente zeigt auch, daß sich bei Abwesenheit der Wechselwirkung zwischen Tyr209’ und dem Sulfat am Kristallkontakt die C-terminale Helix etwas zum aktiven Zentrum hinbewegt. Auf der Grundlage dieser Strukturen fällt es leicht, sich vorzustellen, daß sich Tyr209’ bei Substratbindung in Richtung Substrat dreht und sich die C-terminale Helix dabei als Ganzes leicht mitbewegt. Das legen auch die kinetischen Daten der C-terminalen Mutanten nahe (siehe unten). In allen vier bekannten Strukturen mit partiell fixiertem C-terminalem Kettenende besitzt die Seitenkette von Lys207’ nach wie vor eine sehr hohe Flexibilität. Bei einem σ-cut off von 1.0 σ ist bei allen vier Mutanten über das Cβ-Atom hinaus keine Elektronendichte vorhanden. Die Beobachtung, daß in keiner Struktur eine definierte Elektronendichte über Gly210’ hinaus vorhanden ist, erlaubt die Unterteilung der C-terminalen Kette in zwei Segmente: Segment-I mit den Resten 207-209 und Segment-II mit den Resten 211-215. Die Rolle von Gly210’ könnte dabei die eines Scharniers bzw. flexiblen Linkers zwischen diesen beiden Segmenten sein, wodurch Segment-II besonders beweglich wird. Wie sich an späterer Stelle noch zeigen wird, ist diese Unterteilung auch unter mechanistischen Gesichtspunkten gerechtfertigt. Abbildung 41 Die C-terminalen Reste (Asp204 bis Phe206) bei Mutante E214A mit σA-gewichteter (2Fo-Fc)-Elektronendichte bei einem Konturniveau von 1.0 σ. 108 Abbildung 42 Die C-terminalen Reste (Lys205 bis Thr208) bei Mutante R212A mit σA-gewichteter (2Fo-Fc)-Elektronendichte bei einem Konturniveau von 1.0 σ. Abbildung 43 Die C-terminalen Reste (Lys205 bis Tyr209) bei Mutante T26A mit σA-gewichteter (2Fo-Fc)-Elektronendichte bei einem Konturniveau von 1.0 σ. Ebenfalls dargestellt ist das in einen Kristallkontakt involvierte Sulfat-Ion, das über den Kristallisationspuffer mitgeschleppt wurde. Abbildung 44 Überlagerung der C-terminalen Kettenenden aus verschiedenen FucA-Strukturen: Wildtyp mit gebundenem Zink-Ion (schwarz; Dreyer, 1995), Wildtyp nach Metallaustausch von Zink nach Cobalt (grün; Dreyer, 1995), Mutante Y113F (orange), Mutante R212A (gelb) und Mutante T26A (rot). 109 4.5.2 Struktur der Mutanten Y113F und Y113F/Y209F Die Abbildung 45 zeigt für Mutante Y113F die sehr gut definierte (2Fo-Fc)-Elektronendichte im Bereich der Mutationsstelle nach der Verfeinerung. Wie die Überlagerung (überlagert wurde anhand des zentralen ß-Faltblattes) der Mutantenstruktur mit der des WT zeigt, treten durch die Mutation, abgesehen vom Aminosäureaustausch, keine signifikanten strukturellen Veränderungen in der Umgebung der Mutationsstelle auf (Abbildung 46). Die Positionen der Hauptkettenatome bzw. die Position der Atome des aromatischen Rings im neu eingeführten Phenylalanin haben sich verglichen zu denen des Tyrosins im WT nur geringfügig verschoben. Dabei tritt im wesentlichen eine Drehung von 11° um den Winkel χ1 auf. Abbildung 45 Verfeinertes Modell der Mutante Y113F mit σA-gewichteter (2Fo-Fc)-Elektronendichte bei einem Konturniveau von 2.0 σ. Gezeigt sind die Mutationsstelle sowie die Zink-Koordinationssphäre. Abbildung 46 Das aktive Zentrum im verfeinerten Modell der Mutante Y113F (durchgezogene Linie) überlagert mit der WT-Struktur (gepunktete Linie). Überlagert wurde anhand des zentralen βFaltblattes. Das katalytische Zink-Ion ist als graue Kugel dargestellt. 110 Daneben zeigt die Mutantenstruktur auch geringfügige Veränderungen im Bereich der Zink-Koordinationssphäre. Im Gegensatz zum WT, wo die beiden Zn-Oε(Glu73)-Distanzen um 0.5 Å differieren, ist Glu73 in der Mutantenstruktur ein nahezu perfekter zweizähniger Ligand. Das Ausbleiben größerer struktureller Veränderungen erlaubt somit eine eindeutigere Interpretation der kinetischen Eigenschaften der Mutante im folgenden. Denn diese können nun direkt über den Aminosäureaustausch, also das Fehlen der phenolischen OH-Gruppe, erklärt werden. Sekundäreffekte wie strukturelle Umordnungen in der Nachbarschaft der mutierten Aminosäure spielen keine Rolle. Abbildung 47 Konformation des flexiblen Loops(23-27) im WT (schwarz), im WT mit gebundenem PGH (rot) sowie in Mutante Y113F (grün). Überlagert wurde anhand des zentralen β-Faltblattes. Deutliche Unterschiede zur WT-Struktur weist das Modell der Mutante im Bereich des flexiblen Loops(23-27) auf. So besitzt dieser Loop in der Mutantenstruktur eine Konformation ähnlich der des WT mit gebundenem PGH, ohne jedoch die in der PGH-Struktur beobachtete 180°-Drehung der Peptidbindung zwischen Ala27 und Gly28 zu zeigen (Abbildung 47). Der Peptidflip zwischen Gly25 und Thr26 hingegen ist sowohl in der PGH-Struktur des WT als auch in der Struktur von Mutante Y113F zu beobachten. Es muß jedoch in diesem Zusammenhang erwähnt werden, daß der Loop bei Mutante Y113F im Gegensatz zur PGH-Struktur nicht fixiert wird. So ist die Elektronendichte in diesem Bereich deutlich schlechter definiert als in der PGH-Struktur des WT und die B-Faktoren sind entsprechend deutlich höher. Dies zeigt aber auch, daß es sich bei den unterschiedlichen Konformationen des Loops im WT mit und ohne PGH nicht um eine Bewegung zwischen zwei klar definierten Konformationen handelt, sondern vielmehr um eine Fixierung eines vorher flexiblen und schlecht definierten Segments durch die Inhibitorbindung. Die Struktur der Doppelmutante Y113F/Y209F ist nahezu identisch zu der, wie sie für Mutante Y113F beschrieben wurde; auch im Bereich des flexiblen Loops(23-27). Die r.m.s.Abweichung zwischen den Cα-Atomen der beiden Mutanten beträgt dabei 0.09 Å2. Auf eine detaillierte Darstellung der Struktur wird deshalb an dieser Stelle verzichtet. Eine 111 Verlängerung des Modells am C-terminalen Kettenende über Phe206’ hinaus wie in Mutante Y113F war für Mutante Y113F/Y209F allerdings nicht möglich, wenngleich positive Differenzdichte jenseits von Phe206’ vorhanden war. Diese konnte jedoch nicht eindeutig interpretiert werden. Ein weiterer Unterschied in der verfeinerten Struktur der Doppelmutante ist die Tatsache, daß das Sulfat aus dem Kristallkontakt nicht mehr enthalten ist. Die Abwesenheit des Sulfats hat allerdings keine signifikanten strukturellen Folgen. Das Binden des Sulfat-Ions ist also nicht essentiell für die Ausbildung des entsprechenden Kristallkontaktes, was sich ja bereits aus der nur partiellen Besetzung der Bindungsstelle (Besetzungszahl 0.71) im WT ableiten ließ. Die genaue Analyse der Struktur der Mutante Y113F ist auch unter dem Gesichtspunkt der blauen Farbe der Mutante von Interesse (vgl. Kapitel 4.3.5). Schließlich zeigte auch der Kristall, der zur Strukturlösung verwendet wurde, genauso wie das Protein in Lösung eine blaue Färbung, jedoch sehr viel intensiver, was aufgrund der dichten Packung der Moleküle im Kristall nicht verwunderlich ist. Nach Beendigung der Messung war die blaue Farbe allerdings verschwunden. Die Röntgenstruktur alleine gibt keinen eindeutigen Aufschluß darüber, worauf die Blaufärbung beruhen könnte, schließt allerdings auch Möglichkeiten aus wie beispielsweise ein durch die Mutante gebundenes externes Chromophor oder die Ausbildung eines internen Chromophors. Die Hypothese, daß die Farbe auf Fe(II) an der Zink(II)-Position beruhen könnte, steht aber durchaus im Einklang mit der Struktur der Mutante. Eine partielle Substitution des katalytischen Zink-Ions durch Eisen läßt sich jedoch durch die röntgenographischen Daten nicht beweisen, da die elektronischen Effekte zu gering sind. Das Verschwinden der Farbe im Laufe der Messung würde gemäß obiger Hypothese einer Oxidation von Fe(II) zu Fe(III) entsprechen. 4.5.3 Struktur der Mutante E73S Bei Mutante E73S konnte man darauf gespannt sein, wie sich der Verlust des Zink-Liganden Glu73 auf die strukturelle Integrität im Bereich der Koordinationssphäre des Zink-Ions auswirken würde. Der fehlende Zink-Ligand wird in der Mutanten-Struktur durch ein Wassermolekül ersetzt (Abbildungen 48 & 49), wobei der Abstand zwischen dem Zink-Ion und dem Wasser-Liganden 1.9 Å beträgt. Darüber hinaus hat der Imidazol-Ring von His94 eine Drehung von etwa 20° um den Winkel χ2 erfahren. Daraus resultiert nun eine verzerrt tetraedrische Koordination des Zink-Ions und die Ligandensphäre entspricht der, wie sie in Carboanhydrase-II gefunden wird (Liljas et al., 1972; Håkansson et al., 1992). Das an das Zink-Ion koordinierte Wassermolekül, das wohl als Hydroxid-Ion vorliegen wird, bildet eine H-Brücke zu einem benachbarten Wassermolekül aus. Dieses wiederum steht in H-Brücken-Wechselwirkung zu dem Oγ-Atom von Ser73. Da der Rest 73 über seine Seiten- 112 kette nicht mehr direkt sondern nur noch indirekt über eine H-Brücken-Kette mit dem ZinkIon verankert ist, haben sich auch die Positionen der Hauptkettenatome leicht verändert. So hat sich beispielsweise bei Überlagerung des zentralen β-Faltblattes die Position des Cα-Atoms von Ser73 relativ zum WT um 0.7 Å verschoben. Zusätzlich scheinen sich die Seitenketten von Ser73 und Phe131, verglichen mit der Situation im WT, gegenseitig abzustoßen. Der Abstand zwischen dem Cδ1-Atom von Phe131 auf der einen Seite und dem Cγ-Atom von Glu73 bzw. dem Oγ-Atom von Ser73 auf der anderen Seite beträgt 5.0 bzw. 6.7 Å. Abbildung 48 Die Koordinationssphäre des Zink-Ions im verfeinerten Modell der Mutante E73S mit σA-gewichteter (2Fo-Fc)-Elektronendichte bei einem Konturniveau von 1.2 σ. Abbildung 49 Das aktive Zentrum im verfeinerten Modell der Mutante E73S (durchgezogene Linie) überlagert mit der WT-Struktur (gepunktete Linie). Überlagert wurde anhand des zentralen β-Faltblattes. Das katalytische Zink-Ion ist als graue Kugel dargestellt. Ebenfalls gezeigt sind zwei Wassermoleküle (kleine Kugeln) im Bereich der Mutationsstelle, die im WT nicht vorhanden sind. Die B-Faktoren im Bereich der Mutationsstelle und der Zink-Koordinationssphäre sind drastisch erhöht. Sie müssen jedoch im Zusammenhang mit dem mittleren B-Faktor der Struktur insgesamt relativ zum WT bzw. den anderen Mutanten gesehen werden. So liegt der mittlere B-Faktor für die Struktur von Mutante E73S mit einem Wert von 29 Å2 deutlich über dem Wert von 19 Å2 des WT bzw. anderer Mutanten (Tabellen 19 & 20). Signifikant erhöht 113 sind die B-Faktoren um Phe131. Wie der Vergleich mit den B-Faktoren für Mutante E73Q zeigt, kann dies direkt mit dem Verlust der Wechselwirkungen des aromatischen Rings von Phe131 mit Glu73 im WT korreliert werden (Abbildung 52). Ein weiterer Unterschied zur WT-Struktur ist die veränderte Konformation des flexiblen Loops(23-27), die sich auch deutlich von der in der Struktur von Mutante Y113F beobachteten unterscheidet und einen Übergang zwischen den beiden zuvor beobachteten Konformationen darstellt. Dies soll jedoch nicht ausgeführt werden. 4.5.4 Struktur der Mutante E73Q In der Struktur von Mutante E73Q, bei der die postulierte katalytische Base Glutamat gegen Glutamin ausgetauscht worden war (siehe Kapitel 4.1.1 und 4.4.1), ist das Glutamin im Gegensatz zum Glutamat im WT kein Zink-Ligand mehr, sondern ist vom Zink-Ion weggedreht (Abbildungen 50 & 51). Dies beruht hauptsächlich auf einer Drehung der Seitenkette um die Winkel χ2 (115°) und χ3 (140°), jeweils relativ zur Position des Glutamats im WT. Diese Orientierung der Seitenkette wird durch H-Brücken-Wechselwirkungen der Amidgruppe stabilisiert. So nimmt das Oε-Atom näherungsweise die Position eines Wassermoleküls in der WT-Struktur ein und bildet eine H-Brücke zum Hauptkettenstickstoff von Gly132 mit einer Distanz von 3.3 Å. Das Nε-Atom ist in eine etwas stärkere H-Brücke mit einer Distanz von 3.0 Å zum Oγ-Atom von Ser72 involviert (Abbildung 51a). Abbildung 50 Die Umgebung der Mutationsstelle im verfeinerten Modell der Mutante E73Q mit σA-gewichteter (2Fo-Fc)-Elektronendichte bei einem Konturniveau von 1.2 σ (blau); ebenso dargestellt ist in grün die σA-gewichtete (Fo-Fc)-Elektronendichte bei 3.0 σ. Das katalytische Zink-Ion ist als graue Kugel dargestellt, das Wassermolekül in der Zink-Koordinationssphäre als kleine gelbe Kugel. 114 Diese Orientierung des Glutamins steht im Einklang mit der Vermutung, daß Glu73 im WT nach Protonierung die Zink-Koordinationssphäre verläßt und mit der Hauptkette von Gly132 interagiert. Der Unterschied der Konformation des Glutamins in der Mutantenstruktur zu der des Glutamats in der PGH-Struktur des WT (Abbildung 51b) ist auf die unterschiedlichen H-Brücken-Ketten der Amidgruppe bzw. der Carboxylatgruppe zurückzuführen. Letztere bildet eine H-Brücken-Kette zwischen dem Hauptkettenstickstoff von Gly132 und dem O3-Atom des PGH aus. Der freie Platz in der Zink-Koordinationssphäre wird bei Mutante E73Q von einem Wassermolekül eingenommen, so daß eine verzerrt tetraedrische Koordination des Zink-Ions mit drei Histidin- und einem Wasser- bzw. Hydroxid-Liganden resultiert. Der Abstand zwischen dem Zink-Ion und dem Wassermolekül beträgt 2.1 Å. Damit entspricht die Zink-Koordinationssphäre derjenigen der Mutante E73S. (a) (b) Abbildung 51 Überlagerungen des aktiven Zentrums im verfeinerten Modell der Mutante E73Q (überlagert wurde jeweils anhand des zentralen β-Faltblattes): (a) Überlagerung von E73Q (durchgezogene Linie) mit angepaßtem Wasser (kleine graue Kugel; 75 % Besetzung) mit dem WT (gepunktete Linie); (b) Überlagerung von E73Q (durchgezogene Linie) mit angepaßtem DHAP (durchgezogene Linie; 25 % Besetzung) mit der PGH-Struktur des WT (gepunktete Linie). Das katalytische Zink-Ion ist jeweils als graue Kugel dargestellt. Wie Abbildung 50 zeigt, kann die für Mutante E73Q beobachtete Elektronendichte durch dieses Modell jedoch noch nicht völlig befriedigend erklärt werden. So war auch nach der Verfeinerung zwischen dem Sulfat- und dem Zink-Ion noch deutliche positive Differenzdichte bei einem Konturniveau von 3 σ zu beobachten, die sich auch nicht mit dem Einbau 115 ° B-Faktor [A2 ] weiterer Wassermoleküle erklären ließ. Es hat vielmehr den Anschein, als ob man die Differenzdichte über einen zweizähnigen Liganden, der mit einer niedrigen relativen Besetzung gebunden hat, erklären könnte. Eine Möglichkeit, die verbliebene Differenzdichte zu deuten, wäre somit über die partielle Besetzung der aktiven Zentren mit dem Substrat DHAP gegeben. Es wurde jedoch zu keinem Zeitpunkt der Präparation DHAP oder Fuc1P zugesetzt, so daß das DHAP aus den E. coli-Zellen mitgeschleppt worden sein müßte. Da DHAP einen zentralen Baustein im Stoffwechsel darstellt, ist es in den Zellen in größeren Mengen vorhanden. Diese Hypothese erscheint somit durchaus plausibel. In einer weiteren Verfeinerungsrunde wurde deshalb DHAP (in seiner Endiolat-Form) in das Modell eingebaut und mit einer relativen Besetzungszahl von 0.25 verfeinert. Die Orientierung des DHAP nach der Verfeinerung entsprach etwa der des PGH in der WT-Struktur (Abbildung 51b). Aminosäurereste Abbildung 52 Mittlere B-Faktoren für die Hauptkettenatome von Mutante E73Q (dicke durchgezogene Linie), Mutante E73S (dünne gepunktete Linie) und dem WT in der gleichen Kristallform (dünne durchgezogene Linie). Wie der Verlauf der B-Faktorverteilung für die Hauptkettenatome in Abbildung 52 zeigt, weist Mutante E73Q vor allem in den an der Phosphatbindung beteiligten Loops deutlich höhere B-Faktoren als der WT auf. Beim flexiblen Loop(23-27) tritt darüber hinaus eine Unterbrechung der (2Fo-Fc)-Elektronendichte (bei einem Konturniveau von 1.0 σ) zwischen den Resten 23 und 26 auf. Dies ist ein Indiz dafür, daß der Loop in dieser Struktur unterschiedliche Konformationen einnimmt, was möglicherweise die Folge der partiellen Besetzung der aktiven Zentren mit DHAP ist. 116 4.5.5 Struktur der Mutante E73Q/Y113F/Y209F Bei der Verfeinerung von Mutante E73Q/Y113F/Y209F war auch nach der ersten Verfeinerungsrunde signifikante positive Differenzdiche im aktiven Zentrum insbesondere um die Zink-Koordinationssphäre vorhanden. Diese ließ sich auch nicht über den Einbau weiterer Wassermoleküle erklären. Abbildung 53 zeigt die positive Differenzdichte bei einem Konturniveau von 3.0 σ, nachdem sowohl das Sulfat-Ion als auch die Wassermoleküle zwischen der Position des Sulfat- und des Zink-Ions aus dem Modell entfernt worden waren. Wie man der Abbildung entnehmen kann, läßt sich diese Dichte sehr gut mit einem an das Zink-Ion gebundenem DHAP-Molekül vereinbaren. Dieses wurde dann in den folgenden Verfeinerungsrunden in seiner Endiolat-Form in das Modell eingebaut und schließlich mit einer relativen Besetzungszahl von 0.7 verfeinert. Ebenso wie bei Mutante E73Q wurde zu keiner Zeit DHAP oder Fuc1P zugesetzt, so daß es auch in diesem Fall aus den Zellen mitgeschleppt worden sein muß. Abbildung 53 Das aktive Zentrum der Mutante E73Q/Y113F/Y209F mit (Fo-Fc)-Elektronendichte bei einem Konturniveau von 3.0 σ nachdem das Sulfat-Ion sowie die Wassermoleküle zwischen Sulfat- und Zink-Ion aus dem Modell entfernt wurden. Ebenfalls dargestellt ist die an die positive Differenzdichte angepaßte Position des mutmaßlich gebundenen DHAP (Besetzung 70 %). Gln73 wurde in zwei Konformationen mit relativen Besetzungszahlen von 0.7 bzw. 0.3 verfeinert. Die dargestellte Konformation der Seitenkette von Gln73 entspricht der, wie sie mit einer Besetzungszahl von 0.7 verfeinert wurde. Sie sollte bei DHAP-Bindung vorliegen. Wie Abbildung 54 zeigt, unterscheidet sich jedoch die Bindung des vermutlich gebundenen DHAP bei Mutante E73Q/Y113F/Y209F deutlich von der des PGH im WT bzw. des DHAP in Mutante E73Q (siehe oben). Der Unterschied besteht im wesentlichen darin, daß die Hydroxamat- bzw. Endiolat-Ebenen um ca. 130° gegeneinander verdreht sind. Ursache hierfür ist die fehlende phenolische Hydroxygruppe von Tyr113’ als Folge des Austauschs gegen Phenylalanin. Bei einer entsprechenden Orientierung des DHAP im WT analog zu der 117 in Mutante E73Q/Y113F/Y209F würde die phenolische Hydroxygruppe von Tyr113’ mit dem O2-Atom des DHAP kollidieren. Die Seitenkette von Gln73 ist wie in Mutante E73Q vom Zink weg orientiert. In diesem Fall aber konnte die Seitenkette entsprechend der partiellen Besetzung der aktiven Zentren mit DHAP in zwei alternativen Konformationen verfeinert werden: zum einen in einer Konformation, die der in Mutante E73Q entspricht (siehe oben), also mit H-BrückenWechselwirkungen des Oε-Atoms mit dem Hauptkettenstickstoff von Gly132 (Distanz 3.2 Å) und des Nε-Atoms mit dem Oγ-Atom von Ser72 (Distanz 2.8 Å). Diese wurde folglich der Orientierung in den aktiven Zentren ohne gebundenem DHAP zugeschrieben (relative Besetzungszahl 0.3). Zum anderen in einer Konformation, die mit einer relativen Besetzungszahl von 0.7 verfeinert wurde, in der das Nε-Atom in Richtung Zink zeigt (Zn-Nε-Distanz = 3.0 Å). Dabei interagiert das Nε-Atom von Gln73 ebenfalls mit den O2- und O3-Atomen des eingebauten DHAP (Distanz 2.8 Å bzw. 3.0 Å). Abbildung 54 Überlagerung der Struktur von Mutante E73Q/Y113F/Y209F mit angepaßtem DHAP mit 70 % Besetzung (durchgezogene Linie) mit der des WT (gepunktete Linie). Überlagert wurde anhand des zentralen β-Faltblattes. Gln73 ist in zwei alternativen Konformationen dargestellt. Dabei liegt die mit einer relativen Besetzungszahl von 0.7 verfeinerte Konformation näher am Zink. Sie sollte bei DHAP-Bindung vorliegen. Die mit einer relativen Besetzungszahl von 0.3 verfeinerte Konformation liegt näher an Gly132 und entspricht der Konformation, die auch bei Mutante E73Q beobachtet wurde (siehe Abbildung 51). Ebenfalls gezeigt ist das PGH in der entsprechenden Komplex-Struktur des WT (gestrichelte Linie). Die Elektronendichte im Bereich des flexiblen Loops(23-27) läßt auf unterschiedliche Konformationen des Loops als Folge der partiellen Besetzung des aktiven Zentrums mit DHAP schließen. Da die Dichte jedoch teilweise sehr schlecht definiert war und eine einwandfreie Zuordnung zu unterschiedlichen Konformationen nicht zweifelsfrei möglich war, wurde auf eine Verfeinerung alternativer Loop-Konformationen verzichtet und der Loop(23-27) nur in einer Konformation analog zum WT ohne gebundenem PGH verfeinert. 118 4.5.6 Strukturen bei Mutationen im flexiblen Loop(23-27) Der Loop(23-27) besitzt beim WT eine sehr hohe Flexibilität, was in der Röntgenstruktur an den hohen B-Faktoren einerseits und der relativ schlecht definierten Elektronendichte andererseits abgelesen werden kann. Die bereits beschriebenen Mutanten-Strukturen zeigen außerdem, daß Loop(23-27) auch in Abwesenheit von PGH unterschiedliche Konformationen einnehmen kann. Mit den Mutanten T26A und Del(27) gelang es, die Röntgenstrukturen bei zwei Mutationen im flexiblen Loop aufzuklären, wobei die Auflösungen für beide Strukturen mit 2.09 Å bzw. 2.15 Å in der gleichen Größenordnung liegen. Mutante T26A Wie wirkt sich der Austausch von Thr26 gegen Ala auf die Konformation bzw. Flexibilität des Loops(23-27) aus? Hat der Verlust der Hydroxy- und Methylgruppen eine noch höhere Flexibilität als im WT und eine folglich noch schlechter definierte Elektronendichte zur Folge? Die Antwort gibt Abbildung 55. Die Elektronendichte im Bereich der Mutationsstelle ist relativ gut definiert. Die Überlagerung mit der WT-Struktur (Abbildung 56) zeigt darüber hinaus, daß keine signifikanten Veränderungen in der Konformation des flexiblen Loops aufgetreten sind. Es scheint vielmehr, daß in der Mutanten-Struktur eine Versteifung des Loops(23-27) stattgefunden hat, was sich auch im Verlauf der mittleren B-Faktoren für die Hauptkettenatome zeigt (Abbildung 57). Abbildung 55 Die Umgebung der Mutationsstelle im verfeinerten Modell der Mutante T26A mit σA-gewichteter (2Fo-Fc)-Elektronendichte bei einem Konturniveau von 1.0 σ. An dieser Stelle muß angemerkt werden, daß die Versteifung des flexiblen Loops nicht notwendigerweise eine Folge des Aminosäureaustausches sein muß. Es könnte primär auch eine Folge des veränderten interface sein. Obwohl Thr26 in einen Kontakt zwischen zwei Untereinheiten des Enzyms involviert ist, zieht die Mutation keine strukturellen Störungen in der benachbarten Untereinheit nach sich. Eine Besonderheit bei der Struktur von Mutante 119 T26A, die bereits in Kapitel 4.5.1 ausgeführt wurde, ist die Tatsache, daß die C-terminale Helix über Phe206’ hinaus um drei Reste erweitert werden konnte. ° B-Faktor [A2 ] Abbildung 56 Der Loop(23-27) und die Zink-Koordinationssphäre im verfeinerten Modell der Mutante T26A (durchgezogene Linie) überlagert mit der WT-Struktur (gepunktete Linie). Überlagert wurde anhand des zentralen β-Faltblattes. Das katalytische Zink-Ion ist als graue Kugel dargestellt. Aminosäurereste Abbildung 57 Mittlere B-Faktoren für die Hauptkettenatome von Mutante T26A (dicke durchgezogene Linie) und dem WT in der gleichen Kristallform (dünne durchgezogene Linie). Mutante Del(27) Mutante Del(27) bzw. die entsprechenden Mehrfachmutanten (Tabelle 7, Seite 78) waren im Hinblick auf eine Selektivitätsänderung auf Seite der DHAP-Komponente geplant worden (Kapitel 4.4.4), um mehr Platz für die Carboxylatgruppe von β-Hydroxypyruvat zu schaffen. Die Röntgenstruktur der Mutante sollte nun Aufschluß darüber geben, wie sich die Verkürzung des Loops auf die räumlichen Verhältnisse im aktiven Zentrum auswirkt; d. h. ob 120 die Deletion von Ala27 nur eine veränderte Konformation des flexiblen Loops zur Folge hat, oder ob sie strukturelle Veränderungen über den Loop hinaus induziert. Bereits bei der WT-Struktur ist die Elektronendichte im Bereich des flexiblen Loops teilweise sehr schlecht definiert. Daran ändert sich auch nach Verkürzung des Loops nichts (Abbildung 58). Wie die schlecht definierte Elektronendichte vor allem für die Reste Gln24 und Gly25 zeigt, scheinen die Verhältnisse in der Deletionsmutante eher noch ungünstiger zu sein. Folglich sind auch die B-Faktoren deutlich erhöht und nehmen für die Reste 23 bis 25 Werte zwischen 60 und 70 Å2 an. Abbildung 58 Die Umgebung der Deletionsstelle im verfeinerten Modell der Mutante Del(27) mit σA-gewichteter (2Fo-Fc)-Elektronendichte bei einem Konturniveau von 1.0 σ. Die Numerierung der Aminosäuren erfolgt gemäß der Position des jeweiligen Restes im WT. Abbildung 59 Der flexible Loop und die Zink-Koordinationssphäre im verfeinerten Modell der Mutante Del(27) (durchgezogene Linie) überlagert mit der WT-Struktur (gepunktete Linie). Überlagert wurde anhand des zentralen β-Faltblattes. Das katalytische Zink-Ion ist als graue Kugel dargestellt. Die Numerierung der Aminosäuren erfolgt gemäß der Position des jeweiligen Restes im WT. 121 Wie die Überlagerung der Struktur von Mutante Del(27) mit der des WT zeigt, bewirkt die Deletion von Ala27 keine strukturellen Veränderungen in der Umgebung der ZinkKoordinationssphäre (Abbildung 59). Im Bereich des flexiblen Loops nimmt Thr26 in der Deletionsmutante in etwa die Position von Ala27 im WT ein. Dabei hat sich die Position des Cα-Atoms von Thr26 relativ zum WT um 3.2 Å und die Position des Oγ-Atoms gar um knapp 6 Å verschoben. Die größte Abweichung für die Cα-Atome wird mit 4.9 Å für Gly25 beobachtet. Die Position des Glycins auf der C-terminalen Seite der Deletion (Gly28) entspricht mit geringfügigen Unterschieden der des Glycins in der WT-Struktur. Beim folgenden Asparagin (Asn29), der ersten Aminosäure von β-Strang β1 (Abbildung 17, Seite 72), sind die Positionen in WT und Mutante wieder identisch. Der Verlust von Ala27 wird also, wie auch erwartet wurde, durch eine Veränderung der Konformation des flexiblen Loops kompensiert. Bei den Resten der benachbarten Untereinheit, die in unmittelbarer Nähe zum flexiblen Loop(23-27) liegen, treten nur geringfügige strukturelle Veränderungen auf, was sich insbesondere in den veränderten Positionen der Seitenketten von Trp169’, His172’ und Glu173’ zeigt. Über den flexiblen Loop hinaus hat die Mutation keine nachhaltigen strukturellen Veränderungen zur Folge. Die kinetischen Eigenschaften der Mutante, d. h. das Sinken der Aktivität für Fuc1P auf 1.7 % des WT-Wertes (siehe Tabelle 21, Seite 137), sollten also über die Verkürzung und die daraus resultierenden konformationellen Änderungen des Loops erklärt werden. 4.5.7 Struktur der Mutante F131A Bei Mutante F131A war es von besonderem Interesse wie sich der Austausch von Phe zu Ala auf die geometrischen Verhältnisse in der postulierten hydrophoben Wand (Kapitel 4.1.4) auswirken würde. Die Röntgenstruktur der Mutante zeigt, daß der Verlust der Phenylgruppe keinen entscheidenden Einfluß auf die Konformation der beiden benachbarten aromatischen Seitenketten von Tyr113’ und Phe206’ hat, die beim WT Van-der-Waals-Wechselwirkungen mit dem Phenylring von Phe131 ausbilden (Abbildungen 60 & 61). Das bedeutet, durch den Austausch von Phe zu Ala an Position 131 wird keine weitreichende strukturelle Störung induziert, sondern die hydrophobe Wand wird in eine Mulde umgeformt. Außerdem zeigt die Mutantenstruktur ein Wassermolekül in einem Abstand von 4.2 Å vom Cβ-Atom von Ala131. Dieses Wassermolekül ist in der WT-Struktur nicht vorhanden, da es dort eine destabilisierende Wechselwirkung mit dem aromatischen Ring von Phe131 haben würde. Stabilisiert wird dieses Wasser über eine H-Brücke zum Oγ-Atom von Ser72 (Distanz 3.1 Å) und eine H-Brücke zu einem weiteren Solvensmolekül (Distanz 2.4 Å), das wiederum eine H-Brücke (Distanz 2.6 Å) zur phenolischen Hydroxy-Gruppe von Tyr113’ ausbildet. Eine B-FaktorVerteilung der Mutante F131A ist weiter unten in Abbildung 64 dargestellt. 122 Abbildung 60 Die Umgebung der Mutationsstelle im verfeinerten Modell der Mutante F131A mit σA-gewichteter (2Fo-Fc)-Elektronendichte bei einem Konturniveau von 1.5 σ. Das mit W bezeichnete Wassermolekül ist in der WT-Struktur nicht vorhanden. Abbildung 61 Das aktive Zentrum im verfeinerten Modell der Mutante F131A (durchgezogene Linie) überlagert mit der WT-Struktur (gepunktete Linie). Überlagert wurde anhand des zentralen β-Faltblattes. Das katalytische Zink-Ion ist als graue Kugel dargestellt. Ein Wassermolekül nahe der Mutationsstelle, das in der WT-Struktur nicht vorhanden ist, ist als kleine dunkle Kugel dargestellt. Das andere Wassermolekül (kleine helle Kugel) ist auch in der Struktur des WT (mit und ohne PGH) enthalten. Es befindet sich nahe der Position des Carbonylsauerstoffs in den im folgenden noch entwickelten Modellen für das Binden der Aldehydkomponente (Abbildung 84, Seite 155). Dieses zusätzliche Wassermolekül sowie die veränderten räumlichen und geometrischen Verhältnisse in der hydrophoben Wand in Mutante F131A sind von großer Bedeutung für die kinetischen Eigenschaften der Mutante sowie für die Diastereoselektivität der Reaktion mit nicht-natürlichen aliphatischen Aldehydsubstraten (s. u. in den Kapiteln 4.7.3 und 4.8). 4.5.8 Die Phosphatbindungstasche der Mutanten N29L/S71A, N29Q und S71Q Unter den zahlreichen Mutanten innerhalb der Phosphatbindungstasche konnte von insgesamt drei Mutanten die Röntgenstruktur gelöst werden. Dies sind die Mutanten N29L/S71A, N29Q und S71Q. Dabei stammten die Kristalle der Mutante N29L/S71A aus Kristallisations- 123 experimenten von Müller-Dieckmann (1994). Während bei Mutante N29L/S71A mit 2.55 Å nur eine mittlere Auflösung erreicht wurde, handelt es sich bei den Mutanten N29Q und S71Q um hochaufgelöste Strukturen, deren Auflösungen mit 1.84 Å bzw. 1.66 Å noch über der WTStruktur liegen. Mutante N29L/S71A Die Mutante N29L/S71A war eine der ersten FucA-Mutanten und wurde von MüllerDieckmann (1994) zu einem Zeitpunkt geplant, als die Struktur des WT im Komplex mit PGH noch nicht bekannt war. Sie sollte den Beweis erbringen, daß die Position des Sulfats nahe dem katalytischen Zink-Ion in der unligierten Struktur der FucA während der Katalyse von der Phosphatgruppe des Substrates eingenommen wird. Durch die beiden Aminosäurenaustausche an den Positionen 29 und 71 wurden in dieser Mutante zwei Liganden entfernt, die H-Brücken zu dem Sulfat-Ion ausbilden. Darüber hinaus erhält die Bindungstasche einen hydrophoberen Charakter. Sollte die Phosphatgruppe während des Katalysezyklus die entsprechende Bindungsstelle besetzen, so wäre ein drastischer Aktivitätsverlust bei der Mutante die Folge. Wie die Untersuchungen zur Enzymkinetik von Müller-Dieckmann zeigten, ist dies für die Mutante N29L/S71A auch der Fall. Die spezifische Aktivität der Mutante ist verglichen mit dem WT um mehr als das 5000fache erniedrigt. Daß es sich definitiv um die Phosphatbindungstasche handelt, wurde schließlich durch die Struktur des WT mit dem Übergangszustandsanalogon PGH verifiziert. Abbildung 62 Die Umgebung der Mutationsstellen im verfeinerten Modell der Mutante N29L/S71A mit σA-gewichteter (2Fo-Fc)-Elektronendichte bei einem Konturniveau von 1.2 σ. 124 Abbildung 63 Die Umgebung der Phosphatbindungstasche im verfeinerten Modell der Mutante N29L/S71A (durchgezogene Linie) überlagert mit der WT-Struktur (gepunktete Linie). Überlagert wurde anhand des zentralen β-Faltblattes. Das katalytische Zink-Ion ist als graue Kugel dargestellt. Ebenfalls gezeigt ist ein Wassermolekül (kleine Kugel) im Bereich der Mutationsstelle, das in der Struktur des WT nicht vorhanden ist. Die Röntgenstruktur der Mutante N29L/S71A zeigt, daß als Folge der erhöhten Hydrophobie innerhalb der Phosphatbindungstasche durch die beiden neu eingeführten Aminosäuren kein Sulfat aus dem Kristallisationspuffer mehr gebunden wird (Abbildungen 62 & 63). Statt dessen wird ein zusätzliches Solvensmolekül über den Hauptkettenstickstoff von Ser72 gebunden, das in der Nähe der Position des O3-Atoms des Sulfat-Ions in der WTStruktur zu liegen kommt. Das neu eingeführte Leucin, in etwa isosterisch zum Asparagin im WT, nimmt eine Konformation ein, in der die Seitenkette in Van-der-Waals-Wechselwirkungen mit den Cα-, Cβ- und Cγ-Atomen von Glu73 involviert ist. Der Verlust der Wechselwirkungen mit dem Sulfat-Ion hat eine strukturelle Destabilisierung der gesamten Bindungstasche zur Folge. Zwar ändern sich die geometrischen Verhältnisse in der Bindungstasche nur geringfügig, dafür zeigen die Bereiche, die im WT an der Sulfatbindung beteiligt sind, in der Mutante eine deutlich erhöhte Flexibilität. Dies schlägt sich in einer weniger gut definierten Elektronendichte nieder und spiegelt sich folglich auch in den teilweise drastisch erhöhten B-Faktoren für den Loop um Thr43 sowie den Loop um die Mutationsstelle Ala71 und den angrenzenden Bereichen wider. Abbildung 64 zeigt den Verlauf der mittleren B-Faktoren für die Hauptkettenatome der Mutante N29L/S71A im Vergleich zum WT und zur Mutante F131A, die den gleichen mittleren B-Faktor über alle Proteinatome wie Mutante N29L/S71A zeigt (Tabelle 19, Seite 105). Der flexible Loop(23-27) weist dagegen eine ähnliche thermische Beweglichkeit wie im WT auf und nimmt im Gegensatz zu einigen anderen Mutanten auch die gleiche Konformation wie in der WT-Struktur an. ° B-Faktor [A2 ] 125 Aminosäurereste Abbildung 64 Mittlere B-Faktoren für die Hauptkettenatome von Mutante N29L/S71A (dicke durchgezogene Linie), Mutante F131A (dünne gepunktete Linie) und dem WT in der gleichen Kristallform (dünne durchgezogene Linie). Mutante N29Q Die Mutante N29Q ist eine der Mutanten, die im Hinblick auf eine Spezifitätsänderung von DHAP zu DHA geplant wurden (Kapitel 4.1.5). Gemäß dem in Abbildung 23 (Seite 79) dargestellten Modell für die Mutante wurde erwartet, daß die neu eingeführte GlutaminSeitenkette aufgrund ihres erhöhten Platzbedarfs in die Phosphatbindungstasche hineinragt und in dieser Konformation möglicherweise über H-Brücken-Wechselwirkungen mit Thr41 stabilisiert wird. Abbildung 65 Die Umgebung der Mutationsstelle im verfeinerten Modell der Mutante N29Q mit σA-gewichteter (2Fo-Fc)-Elektronendichte bei einem Konturniveau von 1.5 σ. 126 Abbildung 66 Die Umgebung der Phosphatbindungstasche im verfeinerten Modell der Mutante N29Q (durchgezogene Linie) überlagert mit der WT-Struktur (gepunktete Linie). Überlagert wurde anhand des zentralen β-Faltblattes. Das katalytische Zink-Ion ist als graue Kugel dargestellt. Die hochaufgelöste Röntgenstruktur der Mutante (1.84 Å Auflösung) enthüllt nun, daß die Seitenkette des neu eingeführten Glutamins trotz der zusätzlichen Methylengruppe die gleichen Wechselwirkungen eingeht wie das Asparagin im WT (Abbildungen 65 & 66). Auch in Mutante N29Q ist die Phosphatbindungstasche mit einem Sulfat-Ion aus dem Kristallisationspuffer besetzt, was aufgrund des strukturellen Modells der Mutante nicht unbedingt zu erwarten war. Die Amidgruppe der Glutamin-Seitenkette zeigt das gleiche H-Brücken-Muster wie das Asparagin im WT; und zwar H-Brücken mit dem O2-Atom des Sulfat-Ions, dem Imidazol-Stickstoff von His77 sowie mit dem Hauptkettensauerstoff von Ala27 (letztere Aminosäure ist aus Gründen der Übersichtlichkeit in Abbildung 66 nicht dargestellt). Um den erhöhten Platzbedarf durch die zusätzliche Methylengruppe auszugleichen, kommt es auch zu kleineren Verschiebungen der Positionen der Hauptkettenatome. So hat sich die Position des Cα-Atoms von Gln29 verglichen mit der WT-Struktur um 0.4 Å verschoben. Verschiebungen in der gleichen Größenordnung werden auch für das Sulfat-Ion sowie für den gegenüberliegenden Loop zwischen den Aminosäuren 71 und 73 beobachtet. Die durch das Einfügen einer Methylengruppe induzierte vergleichsweise große Veränderung wird also durch eine Vielzahl von kleineren Bewegungen innerhalb der gesamten Phosphatbindungstasche kompensiert, um die im WT vorhandenen Wechselwirkungen aufrecht erhalten zu können. Eine besondere Bedeutung scheint in diesem Zusammenhang auch der H-Brücke zwischen dem Oε-Atom von Gln29 und dem Nε-Atom von His77 zuzukommen. Diese H-Brücke ist Teil einer Einheit von vier Resten, nämlich Gln29 (bzw. Asn29 im WT), His77, 127 Ser31 und Tyr81, die über eine parallel zum β1-Strang verlaufende H-Brücken-Kette miteinander verbunden sind. Die Seitenketten von His77 und Ser31 sind nicht solvenszugänglich und benötigen somit in ihrer Umgebung polare Gruppen zur Stabilisierung. Mutante S71Q ° ∆Cα [A] Die Mutante S71Q ist die zweite Mutante, die im Hinblick auf eine Spezifitätsänderung von DHAP zu DHA geplant wurde und von der die Struktur bestimmt werden konnte. Gemäß dem für die Mutante entwickelten Modell wurde keine größere strukturelle Veränderung durch die Mutation erwartet, da die Seitenkette des neu eingeführten Glutamins optimal den Raum in der Phosphatbindungstasche füllen könnte. Darüber hinaus sollte sie in einer günstigen Konformation über H-Brücken der Amidgruppe mit dem Hauptkettenstickstoff von Ser72 bzw. dem Carbonylsauerstoff von Gly28 stabilisiert werden (Abbildung 22, Seite 78). Aminosäurereste Abbildung 67 ∆Cα-Plot der Überlagerung der Struktur von Mutante S71Q mit dem WT Wie jedoch bereits aus Tabelle 19 (Seite 105) entnommen werden konnte, zeigt die Struktur von Mutante S71Q mit einem Wert von 0.61 Å2 von allen Mutantenstrukturen die größte r.m.s.-Abweichung für die Cα-Atome (verglichen mit dem WT). Es müssen also doch größere Konformationsänderungen eingetreten sein. Der ∆Cα-Plot für die Mutante (Abbildung 67) zeigt, daß es in der N-terminalen Hälfte des Proteins drei hervorstechende Bereiche mit signifikanten Abweichungen gibt: den flexiblen Loop(23-27) mit einer maximalen Abweichung von 3.7 Å für Gly25, den Loop(43-46) am Boden der Phosphatbindungstasche (maximale Abweichung für Gly44) sowie den Loop um die Mutationsstelle mit einer maximalen Abweichung von 3.4 Å für Pro70. Die Veränderungen und geometrischen Verhältnisse in der Phosphatbindungstasche sind detailliert in Abbildung 69 & 70 dargestellt. ° B-Faktor [A2 ] 128 Aminosäurereste Abbildung 68 Mittlere B-Faktoren für die Hauptkettenatome von Mutante S71Q (dicke durchgezogene Linie), Mutante N29L/S71A (dünne gepunktete Linie) und dem WT in der gleichen Kristallform (dünne durchgezogene Linie). Abbildung 69 Die Umgebung der Mutationsstelle und die Zink-Koordinationssphäre im verfeinerten Modell der Mutante S71Q mit σA-gewichteter (2Fo-Fc)-Elektronendichte bei einem Konturnivau von 2.0 σ. Wie aufgrund des Modells der Mutante erwartet, wird durch das neu eingeführte Glutamin das Binden von Sulfat aus dem Kristallisationspuffer verhindert. Statt dessen werden in der Bindungstasche der Mutante zwei sehr gut definierte Wassermoleküle detektiert, die in H-Brücken-Distanz zur Amidgruppe von Gln71 liegen. Die Seitenkette des Glutamins nimmt außerdem eine ähnliche Konformation wie im Modell ein, die über H-Brücken des Oε-Atoms mit den Hauptkettenstickstoffen von Ser72 und Glu73 stabilisiert wird. Der entscheidende Unterschied zum Modell, der wohl auch der Auslöser für die größeren strukturellen Veränderungen sein könnte, ist die Tatsache, daß Gln71 über sein 129 Nε-Atom Wechselwirkungen mit dem Carbonylsauerstoff von Thr41 sowie der Seitenkette von Thr43 eingeht. Dadurch könnte ein Art Dominoeffekt ausgelöst werden: Der Boden der Bindungstasche um Thr43 bewegt sich nach oben in Richtung Zink und induziert so Bewegungen in seiner Umgebung, die dann in abgeschwächter Form auch an entferntere Bereiche des Proteins weitergegeben werden; z. B. die Bewegung der Seitenkette von His64 bzw. der 310-Helix um Ser53. Die größte Bewegung insgesamt ist mit knapp 7 Å für das Cδ1-Atom von Leu69 zu konstatieren. Die Reste Leu69 und Pro70 lagern sich aufgrund des durch die Bewegung des Loops um Thr43 freigewordenen Raumes um, was sich dahingehend auswirkt, daß auch die Reste in C-terminaler Richtung vom Zink wegbewegt werden. Dadurch verschiebt sich die relative Lage des neu eingeführten Glutamins verglichen mit dem WT bzw. dem für die Mutante entwickelten Strukturmodell. Darüber hinaus hat dies auch eine Veränderung in der Zink-Koordinationssphäre zur Folge, was weiter unten noch einmal aufgegriffen wird. Abbildung 70 Die Umgebung der Phosphatbindungstasche im verfeinerten Modell der Mutante S71Q (durchgezogene Linie) überlagert mit der WT-Struktur (gepunktete Linie). Überlagert wurde anhand des zentralen β-Faltblattes. Das katalytische Zink-Ion ist als graue Kugel dargestellt. Ebenfalls gezeigt sind Wassermoleküle (kleine Kugeln) im Bereich der Mutationsstelle bzw. der ZinkKoordinationssphäre, die in der Struktur des WT nicht vorhanden sind. Um die oben veranschaulichte Bewegung modulieren und eventuell umkehren zu können, so daß die Seitenkette von Gln71 näherungsweise wie im entwickelten Modell beschrieben positioniert werden kann, wurde auf Basis der oben ausgeführten Analyse der Bewegungen eine weitere Mutation eingeführt. Dies führte zur Generierung der Mutante T43V/S71Q (vgl. Tabelle 7, Seite 78). Die Struktur dieser Mutante ist leider noch nicht vorhanden. 130 Die Konformation des flexiblen Loops(23-27) in der Mutante S71Q entspricht in etwa der von Mutante Y113F (s. o.). Sie ist also ähnlich zur Struktur des WT mit gebundenem PGH, allerdings ohne den Peptidflip zwischen Ala27 und Gly28. Wie der B-Faktorplot für die Mutante zeigt, erfährt diese Konformation in Mutante S71Q eine deutliche Stabilisierung, was sich in den relativ niedrigen B-Faktoren widerspiegelt (Abbildung 68). Bei Mutante Y113F war dies nicht der Fall. Abbildung 71 Vergleich der Koordinationssphäre des katalytischen Zink-Ions in Mutante S71Q (schwarze durchgezogene Linie) und in der Struktur des WT mit gebundenem PGH (schwarze gestrichelte Linie). Überlagert wurde anhand des zentralen β-Faltblattes. Das Zink-Ion ist als große graue Kugel dargestellt, das Wassermolekül in der Zink-Koordinationssphäre als kleine graue Kugel. An dieser Stelle sei noch einmal besonders die veränderte Koordinationssphäre des Zink-Ions in Mutante S71Q hervorgehoben. Das katalytische Zink-Ion ist nicht mehr von nur vier sondern von fünf Liganden koordiniert, nämlich von His92, His94, His155, Glu73 und einem Wassermolekül, wobei eine quadratisch pyramidale Koordination vorliegt. Berücksichtigt man zusätzlich Tyr113’, dessen phenolische OH-Gruppe mit einer Distanz von 3.3 Å allerdings zu weit vom Zink-Ion entfernt ist, um als Ligand im eigentlichen Sinne bezeichnet werden zu können, ergibt sich eine verzerrt oktaedrische Koordination. Bei Überlagerung der Struktur von Mutante S71Q mit der PGH-Struktur des WT (Abbildung 71) wird deutlich, daß der koordinierende Oε-Sauerstoff von Glu73 und das Wassermolekül in etwa die Positionen der beiden Ligandenatome des PGH einnehmen. Dies wirft die Frage auf, ob es sich bei der Koordinationssphäre in Mutante S71Q nicht um eine Momentaufnahme während der Katalyse handeln könnte. Möglicherweise nimmt der Carbonylsauerstoff des DHAP während der Katalyse zunächst die Position des Wassermoleküls in der Zink-Koordinationssphäre ein, wodurch das azide Proton an C3 in die Nähe zur Carboxylatgruppe von Glu73 gebracht wird, so daß in einem Schritt Protonentransfer und Verdrängung des Glu73 aus der Zink-Koordinationssphäre durch das O3-Atom von DHAP erfolgen könnte. 131 4.6 Akzeptanz nicht-natürlicher Substrate Wie bereits bei der Mutantenplanung (Kapitel 4.1.5) ausgeführt wurde, wäre eine Selektivitätsänderung bei der DHAP-Komponente wünschenswert. Als potentielle Alternativsubstrate wurden in dieser Arbeit Dihydroxyaceton und β-Hydroxypyruvat mit den für diesen Zweck entworfenen Mutanten (Tabelle 7, Seite 78) getestet. Die Aktivitäten mit DHA und β-Hydroxypyruvat wurden dabei in Syntheserichtung mit einem diskontinuierlichen Test über den Nachweis des Eduktes untersucht (Kapitel 3.3.5). Um das Gleichgewicht in Richtung des Kondensationsproduktes zu verschieben, wurde das zweite Edukt Glykolaldehyd in 10fachem (für DHA) bzw. 4.5fachem (für β-Hydroxypyruvat) molarem Überschuß eingesetzt. N29Q N29E S71Q S71E ∆E (366 nm) 0.08 0.06 10 0.04 5 0.02 Umsatz DHA [%] 15 0.10 0 0 0 50 100 150 200 t [min] Abbildung 72 Zeitlicher Verlauf der Aktivitätstests für die Mutanten N29Q, N29E, S71Q sowie S71E mit dem nicht-natürlichen Substrat Dihydroxyaceton (DHA) bei 37 °C und pH 8.0. Gemessen wurde die noch vorhandene Menge des Eduktes DHA zu verschiedenen Zeitpunkten durch Reduktion mit GDH, die dabei NADH verbraucht. Aufgetragen sind die ∆E-Werte bei 366 nm zwischen den entsprechenden Proben der Assays nach unterschiedlichen Reaktionszeiten und dem entsprechenden Wert zu Beginn der Reaktion ohne Enzymzugabe (Mittelwert aus jeweils drei Messungen). Ein positiver Wert für ∆E entspricht dabei einer geringeren Restmenge an DHA. Der prozentuale Umsatz an DHA läßt sich dann über die ursprünglich eingesetzte Substratmenge direkt aus den ∆E-Werten ermitteln. Wie aus den Abbildungen 72 & 73 entnommen werden kann, konnte unter den gegebenen Bedingungen für keine der untersuchten Mutanten eine signifikante Aktivität für DHA bzw. β-Hydroxypyruvat nachgewiesen werden. Allerdings können mit diesen Tests sehr geringe Aktivitäten bzw. sehr ungünstige Gleichgewichtslagen der Reaktion - also Gleichgewichte, die nahezu vollständig auf Seite der Edukte liegen - nicht erfaßt werden. Als Maß für den Fehler und somit die Nachweisgrenze des Tests können die ∆E-Differenzen zwischen den einzelnen Zeitproben herangezogen werden. Hochgerechnet auf die eingesetzte Menge des jeweiligen Eduktes sowie die Zeitspanne der Messungen (210 min für DHA und 120 min 132 6 wt S71K Del(27)/S71K Del(27)/S71H Del(27)/S71R ∆E (366 nm) 0.04 0.02 4 2 0 0 -0.02 0 20 40 60 80 100 Umsatz β-Hydroxypyruvat [%] für β-Hydroxypyruvat) entspricht dies einer Nachweisgrenze von 0.002 U/mg für DHA und 0.005 U/mg für β-Hydroxypyruvat unter den gegebenen Reaktionsbedingungen. Zum Vergleich dazu sei die Mutante N29L/S71A angeführt, bei der zwei Reste, welche H-Brücken zur Phosphatgruppe von Fuc1P ausbilden, gegen hydrophobe Reste ausgetauscht wurden. Diese zeigt in dem kontinuierlichen Test für Fuc1P (Kapitel 3.3.5) noch eine Restaktivität von 0.004 U/mg (Müller-Dieckmann, 1994). 120 t [min] Abbildung 73 Zeitlicher Verlauf der Aktivitätstests für den WT sowie die Mutanten S71K, Del(27)/S71K, Del(27)/S71H und Del(27)/S71R mit dem nicht-natürlichen Substrat β-Hydroxypyruvat (bei 37 °C und pH 8.0). Gemessen wurde die noch vorhandene Menge an Edukt zu verschiedenen Zeitpunkten durch Reduktion mit LDH, die dabei NADH verbraucht. Aufgetragen sind die ∆E-Werte bei 366 nm zwischen den entsprechenden Proben der Assays mit und ohne FucA nach unterschiedlichen Reaktionszeiten (Mittelwert aus jeweils drei Messungen). Ein positiver Wert für ∆E entspricht dabei einer geringeren Restmenge des Edukts β-Hydroxypyruvat. Der prozentuale Umsatz an β-Hydroxypyruvat läßt sich dann über die ursprünglich eingesetzte Substratmenge direkt aus den ∆E-Werten ermitteln. Von zwei der in Hinsicht auf eine Spezifitätsänderung zu DHA geplanten Mutanten, nämlich Mutante N29Q und S71Q, wurden bereits im vorherigen Kapitel die Röntgenstrukturen vorgestellt. Der Vergleich zwischen dem auf Basis der WT-Struktur erarbeiteten Strukturmodell für die Mutanten und der tatsächlichen experimentell ermittelten Struktur zeigt doch drastische Unterschiede bzw. im Falle von Mutante S71Q ein völlige Veränderung der strukturellen Verhältnisse im Bereich der Phosphatbindungstasche, die sich auch auf die Koordinationssphäre des Zink-Ions auswirkt. Damit fallen zumindest für diese beiden Mutanten auch die erwarteten stabilisierenden Wechselwirkungen für DHA als potentielles Substrat weg, die basierend auf dem entwickelten Strukturmodell erwartet wurden. Die ausbleibenden Reaktivitäten dieser beiden Mutanten sind somit folgerichtig. Der Befund für diese beiden Mutanten läßt auch für die anderen unter diesem Gesichtspunkt geplanten Mutanten größere Abweichungen von den konstruierten Strukturmodellen erwarten. 133 Trotz des negativen Befunds für die untersuchten Mutanten erscheint eine Spezifitätsänderung jedoch nach wie vor möglich. Die im Falle von Mutante S71Q eingetretenen Veränderungen zeigen, daß man bei der Planung derartiger Mutanten aber mit größeren strukturellen Fluktuationen rechnen muß. Wie im folgenden noch gezeigt werden wird, könnte sich eine eventuelle Strukturlösung der L-Ribulose-5-phosphat-4-Epimerase, die eine hohe Sequenzhomologie zu Bereichen des aktiven Zentrums von FucA zeigt, als sehr hilfreich bei der Planung weiterer Mutanten erweisen. 4.7 Ergebnisse der Enzymkinetik für L-Fuculose-1-phosphat Die kinetischen Parameter für den WT und die Mutanten wurden wie unter Kaptitel 3.3.6 beschrieben bestimmt, wobei je nach Mutante die Aktivitäten bei 7 bis 12 verschiedenen Substratkonzentrationen gemessen wurde (Abbildung 74). Die Abbildungen 75 & 76 zeigen exemplarisch für den WT bzw. die Mutante E214Q die Diagramme der drei für die Auswertung herangezogenen Linearisierungsverfahren zur Bestimmung von KM und kcat für das natürliche Substrat L-Fuculose-1-phosphat. Die relativen Aktivitäten der einzelnen Mutanten sowie die Ergebnisse aus den Bestimmungen der KM- und kcat-Werte sind schließlich in Tabelle 21 zusammengefaßt. Angegeben sind jeweils die Mittelwerte aus den drei verschiedenen Auftragungen einschließlich der Fehler in Klammern. Je nach Mutante lagen die Fehler bei der Bestimmung der kinetischen Parameter zwischen 5 % und 18 %; in zwei Ausnahmefällen allerdings deutlich höher: ein Fehler von 40 % bei Mutante E214L und ein Fehler von 70 % bei Mutante N29Q. Die unterschiedlichen Fehler resultieren zum einen aus der unterschiedlichen Anzahl an Meßpunkten für die einzelnen Mutanten. Entscheidender ist aber, daß sie in etwa mit dem Wert von KM korrelieren; d. h. je höher der KM-Wert der Mutante, desto höher die Fehler bei der Messung. Denn mit zunehmendem KM wurde auch eine Größenordnung erreicht, bei der es nicht mehr möglich war, bei optimalen Substratkonzentrationen zu arbeiten, die ja bei Bestimmung der kinetischen Parameter mittels Linearisierungsverfahren eine Substratkonzentration zwischen 0.1 und 10 KM überstreichen sollten. Ein Problem ist z. B. die zunehmende Viskosität der Assay-Lösung mit zunehmender Konzentration an Fuc1P und daraus resultierende Pipettierfehler. Bei Mutationen an den Resten Tyr113’ und Tyr209’ bestand außerdem die Schwierigkeit, daß der Test im Gegensatz zum WT und anderen Mutanten nicht linear verlief (Abbildung 74). Mit zunehmender Reaktionsdauer wurde eine geringere Aktivität gemessen, so daß bei diesen Mutanten auf eine Inaktivierung bzw. Inhibierung des Enzyms im Laufe des Tests geschlossen werden muß. Zur Auswertung wurden daher immer nur die während der ersten Minute des Tests ermittelten Aktivitäten herangezogen. 134 (a) (b) E (366 nm) 0.6 0.5 0.4 1 2 t [min] 3 1 2 t [min] Abbildung 74 Zeitlicher Verlauf der Extinktionsabnahme bei den Aktivitätstests für Mutante Y113F (a) und E214L (b) bei einer Substratkonzentration von 2.4 mM Fuc1P und pH 7.5 (cf. Kapitel 3.3.5). Eingesetzt wurden 5 µl Proteinlösung (jeweils 2.8 mg/ml). 4.7.1 Kinetische Parameter des Wildtyps Um einen verläßlichen Bezugspunkt für die bei den Mutanten ermittelten Werte zu haben, wurden zunächst die kinetischen Parameter für den WT bestimmt. Dabei wurde für den WT ein KM von 2.2 mM und ein Wert für kcat von 19.3 s-1 pro Untereinheit bestimmt. Ein ähnlicher Wert für KM unter den gleichen Assay-Bedingungen wurde auch von Dreyer bestimmt (Dreyer, pers. Mitteilung). In der Literatur wird dagegen für den WT ein KM-Wert für Fuc1P von 0.7 mM beschrieben (Ghalambor & Heath, 1966; Schneider, 1994). Dabei erfolgten die Messungen von Ghalambor & Heath bei einem pH-Wert von 7.2 bei 37 °C unter Zusatz von 12.5 mM NaF (Endkonzentration). Der Wert von Schneider bezieht sich auf einen pH-Wert von 7.5 bei 25 °C. Ein Wert für νmax wird in beiden Fällen nicht angegeben. Der Unterschied zum Wert von Schneider könnte durch die unterschiedliche Temperatur verursacht sein. Bei den von Ghalambor & Heath ermittelten Werten muß berücksichtigt werden, daß es sich nicht um rekombinantes Protein, sondern um die aus E. coli Stamm-O-111-B4 isolierte Aldolase handelt. Auffallend sind auch die deutlich unterschiedlichen pH-Optima, die von Ghalambor & Heath bzw. Schneider angegeben werden. Nach Ghalambor & Heath liegt dies für die FucA aus E. coli Stamm-O-111-B4 etwa bei pH 7, für die rekombinante FucA nach Schneider bei pH 8.5 in Spaltungsrichtung und bei pH 9 in Syntheserichtung. 135 10 -3 1/v in [min . 10 ] 8 (a) 6 4 2 0 0 4 3 2 1 1/[S] in [1/mM] (b) [S]/v in [mM . min . 10-2] 1.6 1.2 0.8 0.4 0 0 15 10 5 [S] in [mM] (c) v/[S] in [min-1. mM -1.10 2 ] 5 4 3 2 1 0 0 2 4 6 8 10 12 v in [min-1.10 2 ] Abbildung 75 Bestimmung der KM- und kcat-Werte für L-Fuculose-1-phosphat des WT. Gezeigt sind der Lineweaver-Burk-Plot (a), der Hanes-Woolf-Plot (b) und der Eadie-Scatchard-Plot (c). Die entsprechenden Ausgleichsgeraden wurden jeweils mittels linearer Regression ermittelt. 136 10 (a) 1/v in [min . 10-3] 8 6 4 2 0 0 0.5 1.5 1.0 1/[S] in [1/mM] (b) [S]/v in [mM . min . 10-2] 3.0 2.5 2.0 1.5 1.0 0.5 0 0 10 20 [S] in [mM] (c) v/[S] in [min-1. mM -1.10 2 ] 2.5 2.0 1.5 1.0 0.5 0 0 2 4 6 8 10 12 v in [min-1.10 2 ] Abbildung 76 Bestimmung der KM- und kcat-Werte für L-Fuculose-1-phosphat der Mutante E214Q. Gezeigt sind der Lineweaver-Burk-Plot (a), der Hanes-Woolf-Plot (b) und der Eadie-ScatchardPlot (c). Die entsprechenden Ausgleichsgeraden wurden jeweils mittels linearer Regression ermittelt. 137 Tabelle 21 Kinetik für die Spaltung von L-Fuculose-1-phosphat Mutationsa) Del(27) -1 KM (mM) 100 19.3 (2.3) 2.2 (0.2) L 14 4.0 (0.3) 5.8 (0.4) Wildtyp d) e) b) kcat c) (s ) stelle T26A Relative spezifische Aktivität (%) L 1.7 - - e) P 0.95 5.5 (3) 130 (90) E73Q/Y113F A 0 - - E73Q/Y113F/Y209F A+C 0 - - E73S N29Q A 0 - - Y113F f) A 4.3 0.4 (0.15) 0.5 (0.3) Y113T f) A 0.7 - - Y113F/Y209F f) A+C 0.2 - - F131A H 0.6 0.6 (0.1) 22 (4) F131A/F206W H 0.1 - - F206W H 16 8.3 (0.8) 11 (1.0) Del(207-215) C 0.5 1.5 (0.3) 87 (14) Del(211-215) C 7 7.0 (1.0) 21 (3) K207A C 62 19.5 (1.5) 6.8 (0.5) Y209F f) C 5 1.3 (0.1) 4.4 (0.6) R212A C 86 12.0 (1.3) 1.8 (0.2) E214D C 95 19.5 (1.0) 3.0 (0.2) E214Q C 71 20.5 (1.0) 6 (0.3) E214A C 48 18.5 (3) 8 (1.3) E214L C 6 12.8 (5) 74 (30) E215A C 90 21.5 (3) 3.1 (0.4) a) b) c) d) e) f) A = postulierter katalytischer Rest (Dreyer & Schulz, 1996b), C = flexibles C-terminales Kettenende, H = hydrophobe Wand, L = flexibler Loop(23-27), P = Phosphatbindungstasche. Gemessen mit 2.4 mM Fuc1P bei pH 7.5 und 37 °C. Die spezifische Aktivität des WT beträgt dabei 21 U/mg. Der Wert bezieht sich auf eine Untereinheit. Die kinetischen Parameter für den WT setzen sich aus den Messungen mit zwei unabhängig voneinander durchgeführten Präparationen des WT zusammen, wobei das Substrat Fuc1P ebenfalls aus zwei unabhängigen Präparationen stammte. Die Mutanten Del(27) und N29Q sind die einzigen Mutanten, die zur Spezifitätsänderung auf Seite der DHAP-Komponente geplant wurden, die noch signifikante Aktivität für das natürliche Substrat Fuc1P zeigen. Alle anderen Mutanten aus Tabelle 7 besitzen - wenn überhaupt - nur noch minimale Hintergrundaktivität und sind deshalb an dieser Stelle nicht mehr explizit aufgeführt. Initiale Aktivität, die innerhalb der ersten Minute nach Enzymzugabe gemessen wurde. 138 4.7.2 Mutationen am flexiblen C-terminalen Kettenende und an den postulierten katalytischen Resten Die Mutante, bei welcher das C-terminale Kettenende komplett deletiert wurde, zeigt nur 0.5 % WT-Aktivität. Nahezu der gleiche Wert wurde auch von Müller-Dieckmann (1994) bestimmt. Die kinetischen Parameter zeigen, daß das C-terminale Kettenende sowohl an der Katalyse als auch an der Substratbindung beteiligt ist, da sowohl KM als auch kcat stark verändert sind: KM ist um den Faktor 38 erhöht und kcat um den Faktor 13 erniedrigt; d. h. die Mutante besitzt eine geringere Katalyserate als der Wildtyp und eine deutlich verringerte Affinität zum Substrat Fuc1P. Das C-terminale Kettenende kann weiterhin in zwei Segmente unterteilt werden, nämlich Segment-I von Rest 207 bis 209 und Segment-II von Rest 211 bis 215, wobei Gly210’ als flexibler Linker fungieren könnte. Ein Vergleich mit den kinetischen Daten der Mutante Del(211-215), bei der nur Segment-II entfernt wurde, legt den Schluß nahe, daß Segment-II hauptsächlich an der Substratbindung und Segment-I dagegen direkt an der Katalyse beteiligt ist. Das Bild vervollständigt sich bei Betrachtung der kinetischen Parameter der entsprechenden Punktmutanten. Beim Ersetzen von Arg212’ und Glu215’ gegen Ala wird keine signifikante Auswirkung auf die enzymatische Aktivität beobachtet. Wird jedoch Glu214’ sukzessive gegen andere Reste ausgetauscht, so ist ein deutlicher Aktivitätsverlust zu registrieren; und zwar in der Reihe Glu→Asp→Gln→Ala→Leu, was hauptsächlich auf einen Anstieg von KM zurückzuführen ist (2.2→3.0→5.7→8.4→74 mM). Dies ist ein deutlicher Hinweis darauf, daß Glu214’ auf irgendeine Weise an der Substratbindung beteiligt ist. Ein ähnlich erhöhter KM-Wert bei unverändertem kcat tritt auch beim Austausch von Lys207’ gegen Ala auf (KM = 6.8 mM). Verlängert man die C-terminale Helix über Phe206’ hinaus, so wie es in der Cobalt-Struktur sowie in den Strukturen der Mutanten T26A, Y113F und R212A (Abbildung 44) beobachtet wurde, so zeigt Lys207’ vom aktiven Zentrum weg, und es erscheint unwahrscheinlich, daß Lys207’ während der Katalyse mit dem Substrat interagieren kann. Es ist vielmehr naheliegender, daß der Effekt bei Mutante K207A auf einen sekundären Effekt von Lys207’ bei der Substratbindung zurückzuführen ist; sei es, daß Lys207’ zum richtigen Falten des vollständigen C-terminalen Kettenendes bei der Substratbindung beiträgt oder am Transport des Substrats ins aktive Zentrum bzw. an der Freisetzung der Edukte beteiligt ist. Der stärkste Effekt bei den Punktmutanten tritt bei Mutante Y209F auf, die nur 5 % der WT-Aktivität zeigt. Dies ist in erster Linie auf ein 15fach erniedrigtes kcat zurückzuführen, während sich KM in etwa verdoppelt hat. Zusätzlich kann eine leichte Inaktivierung des Enzyms im Verlauf des Aktivitätstests beobachtet werden. Das Argument, daß die verminderte Aktivität auf eine strukturelle Störung der Bindungsstelle zurückzuführen sein 139 könnte, z. B. durch eine veränderte Wechselwirkung der hydrophoben Seitenkette mit den Resten Phe131 oder Phe206’ aus der hydrophoben Wand, läßt sich durch den Vergleich mit den kinetischen Daten der beiden C-terminalen Deletionsmutanten entkräften. Aus den kinetischen Parametern kann somit direkt abgeleitet werden, daß Tyr209’ an der Katalyse beteiligt ist. Die Orientierung der Seitenkette von Tyr209’ in der Cobalt-Struktur des WT bzw. der Struktur von Mutante T26A (Abbildung 44) scheint auf den ersten Blick einer Beteiligung von Tyr209’ an der Katalyse entgegenzustehen. Wie jedoch in Kapitel 4.5.1. bereits ausführlicher diskutiert wurde, handelt es sich bei dieser Orientierung von Tyr209’ vielmehr um einen Effekt der Kristallpackung, und die Konformation der C-terminalen Reste in den beiden Strukturen ist keineswegs ein repräsentatives Abbild der Verhältnisse während der Katalyse. Zusammenfassend kann festgestellt werden, daß die kinetischen Daten der Mutanten am flexiblen C-terminalen Kettenende ein umfassendes Bild von der Funktion und Bedeutung desselbigen bei der Katalyse geben, über das die Röntgenstrukturen nur wenig Auskunft gaben. Sie legen nahe, daß das C-terminale Kettenende bei der Katalyse als eine Art Deckel fungiert, der sich nach Substratbindung schließt (induced fit) und das aktive Zentrum während der Katalyse gegen das umgebende Medium abschirmt. Via Tyr209’ ist es darüber hinaus auch noch direkt an der Katalyse beteiligt. Nach erfolgter Katalyse könnte das Kettenende in Umkehrung der Effekte bei Substratbindung auch die Freisetzung der Produkte unterstützen. Diese Funktion erfordert jedoch eine großes Maß an Flexibilität, d. h. die Fähigkeit, alternierend unterschiedliche Konformationen einnehmen zu können. Und gerade dies spiegelt sich ja in der Tatsache wider, daß die neun C-terminalen Aminosäuren in der Röntgenstruktur des WT nicht oder nur partiell detektiert werden konnten. Wie auf Basis des von Dreyer & Schulz (1996b) vorgeschlagenen Mechanismus erwartet werden konnte, führt der Ersatz von Glu73 durch Ser zu einer gänzlich inaktiven Mutante, was die Rolle von Glu73 als katalytische Base bestätigt. Wie bereits in Kapitel 4.1.1 erwähnt, besitzt die entsprechende Gln-Mutante noch 0.1 % WT-Aktivität. Bei der von Dreyer & Schulz vorgeschlagenen katalytischen Säure Tyr113’ ist die Situation komplizierter: Die Mutante Y113F zeigt eine initiale Aktivität von 4 % des WT-Wertes, aber wie für Mutante Y209F ist auch hier eine Inaktivierung des Enzyms im Laufe des Enzymtests zu beobachten. Diese ist deutlich stärker ausgeprägt als im Falle von Mutante Y209F. Das erschwert die genaue Bestimmung der kinetischen Parameter, die demzufolge mit größeren Fehlern behaftet sind (Tabelle 21). Erstaunlicherweise zeigt Mutante Y113F als einzige Mutante einen erniedrigten Wert für KM, also eine höhere Affinität zu Fuc1P als der WT; kcat dagegen ist um etwa einen Faktor 50 erniedrigt. Die Röntgenstruktur dieser Mutante (Kapitel 4.5.2) zeigt, daß die beobachteten Effekte nur auf dem Verlust der phenolischen Hydroxygruppe und nicht auf sekundären 140 Effekten (d. h. strukturellen Störungen in der Umgebung der Mutationsstelle) beruhen. Die erhöhte Affinität der Mutante zu Fuc1P könnte auf eine energetisch günstigere Koordination von Fuc1P an das Zink-Ion als Folge der durch den Wegfall der phenolischen Hydroxygruppe erhöhten Zugänglichkeit der Zink-Koordinationssphäre zurückzuführen sein. Bei Mutante Y113T, die auf Basis des Sequenzvergleichs mit der homologen RhuA geplant wurde, ist der Aktivitätsverlust noch deutlicher als bei Mutante Y113F. Y113T zeigt nur noch 0.7 % WT-Aktivität. Auch für Tyr113’ bestätigen die kinetischen Daten der entsprechenden Mutanten also eine Beteiligung an der Katalyse. Wenn beide katalytisch wichtigen Tyrosine gegen Phenylalanin ausgetauscht werden (Mutante Y113F/Y209F), sinkt die Aktivität um den Faktor 500. Verglichen mit den beiden Einzelmutanten tritt also ein in etwa multiplikativer Effekt auf. Die Mutantendaten legen somit den Schluß nahe, daß FucA nicht nur wie von der Röntgenstruktur abgeleitet zwei sondern drei katalytische Reste besitzt: Glu73, Tyr113’ und Tyr209’. Die Rolle eines Tyrosins als katalytische Säure in Syntheserichtung steht durchaus im Einklang mit dem pH-Optimum der durch FucA katalysierten Reaktion, das in Spaltungsrichtung bei pH 8.5 und in Synthesesrichtung bei pH 9 liegt (Schneider, 1994). Tyrosin besitzt einen pKa-Wert von ungefähr 10. Für Tyr113’ ist jedoch eine Absenkung des pKa-Werts zu erwarten, da die phenolische OH-Gruppe nur 3.5 Å vom positiv geladenen Zink-Ion entfernt ist. Darüber hinaus ist die Basizität des durch die C-C-Bindungsbildung entstehenden Alkoholat-Ions (pKa ca. 15) um einige Größenordnungen stärker als die des Phenolat-Ions (pKa < 10). Zu Beginn der Spaltungsreaktion müßte Tyr113’ gemäß obigem Mechanismus bereits deprotoniert vorliegen, um das Proton an der 4-Hydroxygruppe abstrahieren zu können. Ob die Stärke des Zink-Ions als Lewissäure ausreicht, trotz der Entfernung von ca. 3.5 Å das Phenolat genügend zu stabilisieren, bleibt fraglich. Aufschluß über die genauen Protonierungszustände im aktiven Zentrum unter physiologischen Bedingungen könnte unter Umständen eine sehr hoch aufgelöste Röntgenstruktur des WT liefern, die es ermöglicht, auch Wasserstoffatome zu detektieren. Ein Argument gegen die Funktion von Tyr113’ als katalytische Base in Spaltungsrichtung ist die Größenordnung des Aktivitätsverlustes bei Mutante Y113F. Wäre Tyr113’ die katalytische Base, so würde man einen viel drastischeren Aktivitätsverlust erwarten. Die Röntgenstruktur der Mutante gibt keine Hinweise darauf, daß man die Restaktivität damit erklären könnte, daß ein Wassermolekül die Rolle der phenolischen OH-Gruppe übernimmt. Bei Substratbindung könnte die Bewegung des C-terminalen Kettenendes die phenolischen OH-Gruppen von Tyr113’ und Tyr209’ näher zusammenführen, so daß sie im Katalysezyklus die Koordination und Orientierung der Carbonylgruppe des Aldehyds bzw. Stabilisierung des Alkoholat-Ions an C4 von Fuc1P übernehmen könnten. Demnach müßte Glu73 innerhalb eines Katalysezyklus sowohl die Rolle des Protonendonors als auch 141 -akzeptors übernehmen. Um diese Aufgabe übernehmen zu können, muß sich die Seitenkette von Glu73 so bewegen können bzw. orientiert sein, daß die Carboxylatgruppe sowohl das C3-Atom als auch die 4-Hydroxygruppe des Substrats während der Katalyse erreichen kann. 4.7.3 Mutationen in der hydrophoben Wand Die katalytische Aktivität wird erheblich von Mutationen in der hydrophoben Wand (Kapitel 4.1.4) beeinflußt. Der Austausch von Phe131 gegen Ala bewirkt einen drastischen Aktivitätseinbruch auf 0.6 % der WT-Aktivität. Dabei ist KM um den Faktor 6 erhöht und kcat um mehr als das 30fache erniedrigt. Bei der Mutante F206W, bei der diese hydrophobe Wand vergrößert wurde, sind die Effekte weniger stark, da immerhin noch 16 % der WT-Aktivität erreicht wird. Während eine 5fache Erhöhung von KM auftritt, ist kcat nur geringfügig verändert. Die entsprechende Doppelmutante F131A/F206W liegt mit ihrer Aktivität unter der der beiden Einzelmutanten. Die Aktivitätseinbrüche können durch veränderte geometrische Verhältnisse im aktiven Zentrum wie z. B. im Falle der Mutante F206W durch eine Störung der Bewegung des C-terminalen Kettenendes bei der Katalyse erklärt werden. Da von Mutante F206W keine röntgentauglichen Kristalle erhalten wurden, können die beobachteten kinetischen Effekte nicht strukturell diskutiert werden. Bei der Mutante F131A erlaubt die bekannte Röntgenstruktur (Kapitel 4.5.7) eine etwas detailliertere Betrachtung der veränderten kinetischen Parameter und des drastischen Aktivitätsverlustes. Ein Einfluß der Mutation auf die Konformation des katalytisch aktiven Tyr113’ kann auf Grundlage der Röntgenstruktur zweifelsfrei ausgeschlossen werden. Dafür bieten sich zwei andere Erklärungsmöglichkeiten an: Erstens könnte die korrekte Bewegung des C-terminalen Kettenendes bei Substratbindung, die im WT vermutlich die Seitenkette von Tyr209’ in die Nähe des aromatischen Rings von Phe131 bringt, durch die Mutation gestört sein. Entscheidender dürfte jedoch zweitens das in der Mutantenstruktur nahe dem Cβ-Atom von Ala131 am Eingang zur hydrophoben Tasche sitzende Wassermolekül sein. Dieses kann bei Substratbindung in das aktive Zentrum eingeschlossen bleiben und dort zu ungünstigen Wechselwirkungen mit dem Substrat bzw. zu einer Störung der Protonierungszustände während der Katalyse führen. 4.7.4 Mutationen im flexiblen Loop(23-27) und in der Phosphatbindungstasche Wird der bei PGH-Bindung am weitesten bewegte Rest Thr26 des flexiblen Loops(23-27) gegen Ala ausgetauscht, so sinkt die spezifische Aktivität auf 17 % des WT-Wertes. Dabei sind sowohl Substratbindung (KM) als auch Katalyse (kcat) betroffen. Allerdings sind die 142 Effekte zu gering, um Thr26 eine direkte Beteiligung als H-Brücken-Donor bei der Katalyse zuzuschreiben. Die verminderte Aktivität ist wohl eher auf eine strukturelle Störung zurückzuführen, da ja Thr26 nach Substratbindung fixiert wird (Abbildung 18, Seite 74). In dieser Konformation könnte dann Thr26 nach dem induced fit des C-terminalen Kettenendes mit Glu214’ interagieren. Die Struktur der Mutante zeigt, daß der Loop(23-27) die gleiche Konformation wie in der Inhibitor-freien Struktur des WT einnimmt, wobei mit den niedrigeren B-Faktoren diese Konformation offenbar stabilisiert wird. Daraus Rückschlüsse auf die verminderte Aktivität der Mutante zu ziehen, wäre jedoch zu weit gegriffen. Eine weitere Ursache für den Aktivitätsverlust bei Mutante T26A könnte sein, daß Thr26 im WT eine für die Katalyse notwendige relative Bewegung der Untereinheiten zueinander erleichtert. Für die Mutante Del(27), bei der Ala27 entfernt wurde, wird nur noch 1.7 % der Aktivität des WT erreicht. Daß der Wert deutlich unter der Aktivität für Mutante T26A liegt, ist nicht verwunderlich, da durch das Entfernen einer Aminosäure eine Verkürzung des flexiblen Loops und somit größere Veränderungen der Positionen sämtlicher Reste des flexiblen Loops zu erwarten sind. Dies wird auch durch die Röntgenstruktur der Mutante bestätigt: So zeigt das Modell der Mutante beispielsweise eine deutliche Verschiebung der Position von Thr26 (vgl. Kapitel 4.5.6). Es sei an dieser Stelle noch einmal erwähnt, daß diese Mutante nicht zur Aufklärung des Mechanismus, sondern zur Akzeptanz des nicht-natürlichen Substrats β-Hydroxypyruvat geplant wurde. Bei allen Mutanten in der Phosphatbindungstasche, die im Hinblick auf eine Spezifitätsänderung auf Seite der DHAP-Komponente geplant wurden, wurde mit drastischen Aktivitätsverlusten gerechnet, da die Bindungstasche für den Phosphat-Anker blockiert sein sollte. Mit einer Ausnahme, nämlich Mutante N29Q, ist dies auch der Fall. Mutante N29Q dagegen besitzt immerhin noch knapp 1 % WT-Aktivität, wobei der Aktivitätsverlust hauptsächlich auf eine Erhöhung von KM (d. h. eine deutlich niedrigere Affinität zu Fuc1P) zurückzuführen ist. Die kinetischen Daten zeigen also, daß die Mutante immer noch fähig ist, Fuc1P zu binden, wenn auch mit deutlich niedrigerer Affinität. Die Restaktivität für Mutante N29Q steht im Einklang mit der Röntgenstruktur der Mutante (Kapitel 4.5.8), die zeigte, daß die Seitenkette des neu eingeführten Glutamins trotz der zusätzlichen Methylengruppe die gleichen H-Brücken-Wechselwirkungen wie das Asparagin im WT eingeht. So konnte hier auch ein Sulfat-Ion aus dem Kristallisationspuffer in der Phosphatbindungstasche detektiert werden. Die geringfügigen Veränderungen in der Bindungstasche scheinen jedoch auszureichen, um die Affinität zu Fuc1P deutlich zu erniedrigen. Sie nehmen überdies Einfluß auf die Diastereoselektivität der katalysierten Aldolkondensation (Kapitel 4.8). 143 4.7.5 Modell für das Binden von L-Fuculose-1-phosphat und die Rolle des C-terminalen Kettenendes bei der Katalyse Auf Basis der Struktur des WT mit PGH und der kinetischen Daten der einzelnen Mutanten wurde ein Modell für die Bindung des Substrats Fuc1P im aktiven Zentrum und für die Rolle des C-terminalen Kettenendes entwickelt. Fuc1P in seiner offenkettigen Form wurde dabei so in die PGH-Struktur modelliert, daß es in etwa mit der Position des PGHs überlagert und gleichzeitig die Ergebnisse der Mutagenese-Studien befriedigend erklärt. Beim Modellieren des C-terminalen Kettenendes wurde zunächst die C-terminale Helix über Phe206’ hinaus bis zu Tyr209’ verlängert, also analog zur Struktur der C-terminalen Extension in den Mutanten T26A, Y113F und R212A. Da die Orientierung der Seitenkette von Tyr209’ in Mutante T26A einem Kristallartefakt zugeschrieben wurde (Kapitel 4.5.1), erschien es zulässig, sie im Modell so weit wie möglich in Richtung aktives Zentrum zu drehen. Alle weiteren Reste wurden zunächst manuell mit dem Programm O gebaut, wobei die entsprechenden Bindungsgeometrien interaktiv verfeinert wurden. % aE E E aE aO O / D ψ $ aD $5* aE aS E S aE φ Abbildung 77 Ramachandran-Diagramm des modellierten C-terminalen Kettenendes ab Rest 207. Das Glycin ist dabei als Dreieck dargestellt, alle nicht-Glycin-Reste als Quadrate. Die (φ,ψ)-Konformationen sind in vier Bereiche unterteilt (Laskowski et al., 1993) und zwar mit zunehmender Helligkeit in “energetisch günstige“, “erlaubte“, “zusätzlich erlaubte“ und “verbotene“ Bereiche. Bei der Suche nach möglichen Konformationen des C-terminalen Kettenendes wurden folgende Kriterien berücksichtigt: (i) Konformationsanalyse der dihedralen Winkel φ und ψ mittels Ramachandran-Diagramm, (ii) sterische Kriterien und Berücksichtigung der Wechselwirkungen mit dem Rest des Proteins sowie (iii) eine Orientierung der einzelnen Seitenketten im Einklang mit den kinetischen Daten der Mutanten. Das Ramachandran-Diagramm 144 (Abbildung 77) für das finale Modell zeigt für die Hauptkette der modellierten C-terminalen Reste eine unter stereochemischen Gesichtspunkten zufriedenstellende Konformation. Nur ein Rest, nämlich Arg212’, liegt in einem weniger günstigen (generously allowed) Bereich, während alle anderen Reste in den am meisten bevorzugten Bereichen liegen (most favoured nach Laskowski et al., 1993). Gemäß dem entwickelten Modell für den induced fit des C-terminalen Kettenendes bei Substratbindung kommen nach erfolgter Bindung von Fuc1P zwei Reste im aktiven Zentrum zum liegen: Tyr209’ und Glu214’. Beide Reste könnten demnach über ein H-BrückenNetzwerk miteinander verbunden sein, in das auch Thr26 involviert ist. Die Seitenketten von Lys207’, Thr208’ und Arg212’ zeigen in das umgebende Solvens. Die beiden hydrophoben Reste Leu211’ und Ile213’ wechselwirken mit hydrophoben Bereichen aus der anderen Untereinheit (Ile45 und Leu69 bzw. Pro46), die ansonsten teilweise zum Solvens exponiert wären, und vergrößern damit die Kontaktfläche zwischen den Untereinheiten. Abbildung 78 Modell für das Binden von Fuc1P und die Konformation des flexiblen C-terminalen Kettenendes (Reste 207-215) während der Katalyse. Überlagert sind die C-terminalen Kettenenden aus verschiedenen FucA-Strukturen: Wildtyp mit gebundenem Zink (schwarz; Dreyer, 1995), Wildtyp nach Metallaustausch von Zink nach Cobalt (grün; Dreyer, 1995), Mutante Y113F (orange), Mutante R212A (gelb) und Mutante T26A (rot) gemäß Abbildung 44. Zusätzlich dargestellt in rosa ist das modellierte C-terminale Kettenende mit den Seitenketten von Tyr209’ und Glu214’ sowie die möglichen Wechselwirkungen des gebundenen Substrats Fuc1P mit dem Zink-Ion, Glu73, Tyr113’ sowie dem C-terminalen Kettenende während der Katalyse. Die Orientierung der Seitenkette von Glu73 entspricht der Konformation in der PGH-Struktur des WT (Dreyer, 1995). Das C-terminale Glu215’ ist wieder vom aktiven Zentrum weg orientiert. Das Modell steht also im Einklang mit den kinetischen Daten der entsprechenden Mutanten. Die räumlichen und geometrischen Verhältnisse im aktiven Zentrum schließen aus, daß die beiden katalytisch kompetenten Tyrosine direkt miteinander in Wechselwirkung treten können. Eine 145 unmittelbare wechselseitige Aktivierung kann somit ausgeschlossen werden. Vielmehr spannen sie ein H-Brücken-Netzwerk auf, wodurch die 4-Hydroxygruppe von Fuc1P koordiniert werden könnte, und es erscheint wahrscheinlicher, daß sie als unabhängige Dipole fungieren, die zur Stabilisierung des Übergangszustands dienen. Tyr113’ kann darüber hinaus als potentieller H-Brücken-Partner für die 5-Hydroxygruppe fungieren. Glu73 wäre demnach sowohl für die Übertragung des C3-Protons als auch des Protons der 4-Hydroxygruppe verantwortlich. Um aber das Proton bei der Spaltungsreaktion an der 4-Hydroxygruppe abstrahieren zu können, muß sich die Seitenkette von Glu73 (Glu73Konformation der PGH-Struktur des WT) in Richtung 4-Hydroxygruppe bewegen können (Abbildung 78), da der Abstand mit ca. 4.5 Å zu groß ist. Dies könnte auch eine Bewegung der Rückgrat-Atome beinhalten. Daß derartige Bewegungen möglich sind, hat insbesondere die Struktur von Mutante S71Q (Kapitel 4.5.8) ergeben, die deutlich aufzeigt, daß das Enzym am aktiven Zentrum keinesfalls als starres Polypeptid angesehen werden darf. Vielmehr ist davon auszugehen, daß das Enzym während der Katalyse mehrere Kettenkonformationen in komplizierter Weise durchläuft. 4.7.6 Vergleich mit anderen Aldolasen Die Existenz eines flexiblen C-terminalen Kettenendes mit einem Tyrosin, das eine katalytische Funktion übernimmt, wird auch für die mechanistisch und strukturell verschiedenen Klasse-I-Aldolasen beschrieben, bei denen das Substrat durch Ausbildung einer Schiff-Base mit einem Lysin aktiviert wird (Abbildung 79). Dort nimmt man an, daß das flexible C-terminale Kettenende über ein hochkonserviertes Tyrosin möglicherweise direkt am Protonentransfer an der 4-Hydroxygruppe bzw. am C3-Atom bei der Spaltung bzw. Knüpfung der C-C-Bindung zwischen C3 und C4 beteiligt ist (Gamblin et al., 1991; Littlechild & Watson, 1993). Ersetzt man das entsprechende Tyrosin in der humanen FruA(I) durch Serin (Mutante Y363S), so sinkt νmax für Fructose-1,6-bisphosphat im A-Isoenzym um den Faktor 16, während νmax im B-Isoenzym unverändert bleibt (Takahashi et al., 1989). Die Restaktivität in der Mutante des A-Isoenzyms wird damit erklärt, daß in dieser Mutante ein Wasser die Rolle des Protonendonors übernimmt. Aus der Tatsache, daß das B-Isoenzym keine veränderte Aktivität bei Austausch des C-terminalen Tyrosins aufweist, folgern die Autoren, daß die Protonenübertragung in diesem Isoenzym von vornherein über ein Wassermolekül erfolgt. Andere Autoren schlagen auf Basis von Mutagenese-Studien bzw. eines strukturellen alignments mit der Transaldolase-B (Jia et al., 1996; 1997) vor, daß die Protonierung bzw. Deprotonierung der 4-Hydroxygruppe über einen Aspartatrest (Asp33) erfolgen könnte (Morris & Tolan, 1993; Dalby et al., 1999). Desweiteren wurde auch eine intramolekulare Protonenübertragung durch die 1-Phosphatgruppe des Substrates selbst diskutiert (Hupe, 146 1984). Das C-terminale Kettenende würde demnach vielmehr einen Beitrag zur Stabilisierung des Übergangszustands leisten (Heyduk et al., 1991), so wie es auch hier für FucA vorgeschlagen wird. Das C-terminale Kettenende ist bei FruA(I) außerdem auch zu einem gewissen Teil für die Spezifitäten der verschiedenen Isoenzyme verantwortlich (Berthiaume et al., 1991). OPO3 2- OPO3 Aktivierung von DHAP über Imin-Bildung (Schiff-Base-Intermediat) 2- OPO3 2- + OH NH NH OH O K229 K229 OH OH Binden von GA3P 2- 2- OH O 3PO H3N OPO 3 2- 1 5 O3PO 6 4 K229 HO O 2 Si 3 O OH B-H 2- O3PO OH OPO3 2- OPO3 OH Si N H K229 OH 2- C-C-Verknüpfung Protonentransfer + NH K229 HO OH B Abbildung 79 Schematische Darstellung des enzymatischen Mechanismus von Klasse-I-Aldolasen am Beispiel des A-Isoenzyms der humanen FruA(I); modifizierte Darstellung nach Gefflaut et al. (1995). Der Mechanismus ist in Syntheserichtung (Uhrzeigersinn) ausgehend von Lys229 im freien Zustand (links) dargestellt und beinhaltet: (i) Binden von DHAP und Ausbildung eines Carbinolamins durch die Ketogruppe des Substrats mit der Seitenkette von Lys229, (ii) Ausbildung einer Imin/Immonium-Zwischenstufe unter Wasserabspaltung (Schiff-Base-Intermediat), (iii) Binden der Aldehydkomponente und Isomerisierung zum Enolamin, (iv) nukleophiler Angriff des C3-Atoms des Enolamins an der Si-Seite der Carbonylgruppe von GA3P unter Knüpfen der C-C-Bindung und (v) Hydrolyse der Imin-Zwischenstufe in Umkehrung der Ausbildung der Schiff-Base in den ersten beiden Reaktionsschritten gefolgt von der Freisetzung des Produktes Fructose-1,6-bisphosphat. Dagegen scheint das C-terminale Kettenende bei der mechanistisch und strukturell homologen RhuA keine katalytische Funktion zu besitzen, da es im Kristall eine definierte Konformation einnimmt, die vom aktiven Zentrum weg zeigt und über zahlreiche Wechselwirkungen mit anderen Resten der gleichen Untereinheit stabilisiert wird (Krömer & Schulz, persönliche Mitteilung). Wie die Untersuchungen von Merkel (1999) zeigen, besitzt die Mutante Del(264-274), bei der die 11 C-terminalen Aminosäuren entfernt wurden, nahezu die gleiche enzymatische Aktivität wie der WT. Das C-terminale Kettenende zeigt auch deutliche Sequenzunterschiede verglichen mit FucA: Im Gegensatz zu FucA besitzt es keine 147 geladenen sondern hauptsächlich hydrophobe Reste, und an Stelle von Tyr209’ besitzt RhuA ein Threonin (vgl. Abbildung 17, Seite 72). Die katalytische Base Glu73 ist in RhuA konserviert (Glu117). An Stelle von Tyr113’ besitzt RhuA ein Threonin (Thr170’; Kapitel 4.1.1). Der Abstand zwischen Thr170’-Oγ und dem katalytischen Zink-Ion beträgt jedoch ca. 7.8 Å. Es ist in dieser Position darüber hinaus über ein H-Brücken-Netzwerk stabilisiert. Als möglicher katalytischer Rest kommt dagegen Glu171’ in Frage. In der Röntgenstruktur zeigt der Rest vom Zink weg und bildet eine H-Brücke zu einer Asparaginsäure aus der anderen Untereinheit (Asp26). Dabei muß jedoch berücksichtigt werden, daß die zur Strukturlösung verwendeten Kristalle bei einem pH-Wert von 4.6 gezüchtet wurden und sich die beiden Carboxylatgruppen unter physiologischen Bedingungen vermutlich abstoßen. Dadurch könnte die Carboxylatgruppe von Glu171’ in etwa die Position der phenolischen OH-Gruppe von Tyr113’ in FucA einnehmen und somit an der Katalyse partizipieren, sei es durch Stabilisierung des Übergangszustands (wie Tyr113’ in FucA) oder als zweiter Protonendonor/-akzeptor neben Glu117. Diese Hypothese wird durch die Mutante E171Q unterstützt, die einen um den Faktor 500 erniedrigten kcat-Wert aufweist (Merkel, 1999). Der Aktivitätsverlust der Mutante E171Q von RhuA liegt somit in der Größenordnung der Mutante Y113F/Y209F bei FucA, bei der die beiden katalytisch kompetenten Tyrosine ausgetauscht wurden. 2- OPO3 OPO 3 Glu182 -COO Deprotonierung von DHAP O H H H O H Zn O His H His O H H His O Asp109 Glu182 -COOH O Asp109 O 2- Zn His His His O Binden von GA3P 2- 2- O 3PO 2- 1 5 6 2 4 HO H O Asp109 OPO 3 OH O 3 O H O3PO O Zn His Si His His 2- OPO 3 OH O C-C-Verknüpfung Protonentransfer O H H O Asp109 O Si Zn His His His O Abbildung 80 Postulierter katalytischer Mechanismus der FruA(II) aus E. coli; dargestellt in Syntheserichtung nach Hall et al. (1999). Nach erfolgter Bindung von DHAP und Komplexierung an das Zink-Ion erfolgt demnach im ersten Schritt eine Deprotonierung an C3 von DHAP durch die Seitenkette von Glu182 und Stabilisierung der Endiolat-Zwischenstufe durch das Zink-Ion. Nach Binden des Aldol-Akzeptors GA3P im nächsten Schritt erfolgt die C-C-Verknüpfung dann durch nukleophilen Angriff des C3-Atoms von DHAP an der Si-Seite der Carbonylgruppe von GA3P. Die Reprotonierung des dabei intermediär entstehenden Alkoholat-Ions an C4 soll durch die Seitenkette von Asp109 erfolgen. 148 Bei der mechanistisch homologen, strukturell aber verschiedenen FruA(II) aus E. coli werden als katalytische Reste wie bei RhuA zwei Seitenketten mit Carboxylgruppen postuliert (Asp109 und Glu182; Hall et al., 1999). Dabei soll Glu182 für die Deprotonierung des DHAP verantwortlich sein, Asp109 für die Polarisierung der Carbonylgruppe des Glycerol-3phosphats bzw. Reprotonierung des Alkoholat-Ions (Abbildung 80). Die Rolle von Asp109 wird durch Mutagenese-Studien bestätigt (Plater et al., 1999), während die Rolle von Glu182 ausschließlich auf Basis der Röntgenstruktur diskutiert wird. Wie bei RhuA so findet auch bei FruA(II) keine Beteiligung des C-terminalen Kettenendes an der Katalyse bzw. Substratbindung statt. Durch die Beteiligung des flexiblen C-terminalen Kettenendes an der Katalyse und aufgrund des Auftretens zweier Tyrosine als katalytisch kompetente Reste nimmt FucA somit nach heutigem Wissen eine Sonderstellung unter den Klasse-II-Aldolasen ein. Ungeklärt bleibt, ob bei der Spaltungsreaktion die zyklische oder die offenkettige Form von Fuc1P gebunden wird. Wenn die Ringöffnung überhaupt vom Enzym katalysiert wird, so lassen die sterischen Verhältnisse im aktiven Zentrum vermuten, daß sie stattfindet bevor das Substrat an das Zink-Ion bindet. Wie bereits von Dreyer (1995) gezeigt wurde, kommen sich das O4-Atom der Aldehydkomponente und die phenolische OH-Gruppe von Tyr113’ zu nahe, wenn man ein Fuc1P-Molekül in seiner furanoiden Form so in das aktive Zentrum modelliert, daß eine möglichst gute Überlagerung des DHAP-Teils mit PGH stattfindet. Im Gegensatz dazu hat die offenkettige Form mehr konformationelle Freiheit, um sich den geometrischen Verhältnissen im aktiven Zentrum anzupassen und besitzt außerdem bereits eine Ketofunktion an C2. Würde diese erst nach erfolgter Bindung aus der furanoiden Form erzeugt, so wäre ein zusätzlicher Protonenakzeptor in der Bindungstasche notwendig. Potentielle Protonenakzeptoren wären mit His92 oder His94 zwar in unmittelbarer Nähe vorhanden, allerdings müßte dafür die entsprechende Imidazol-Bindung zum Zink-Ion gelöst werden, was unwahrscheinlich erscheint. 4.7.7 Vergleich mit der L-Ribulose-5-phosphat-4-Epimerase Wie die Datenbank-Recherchen von Dreyer (1995) ergaben, zeigt vor allem die N-terminale Hälfte von FucA eine hohe Homologie zur L-Ribulose-5-phosphat(L-Ru5P)-4-Epimerase aus E. coli und Salmonella typhimurium. Unter Verwendung des Programms SEQSEE (Wishart et al., 1994) konnten von Johnson & Tanner (1998) auch Übereinstimmungen der Sequenz des Enzyms in der C-terminalen Hälfte aufgezeigt werden (Abbildung 81). Dabei fällt auf, daß vor allem die an der PGH-Bindung und alle an der Zink-Koordination beteiligten Reste konserviert sind. An Stelle des katalytischen Glu73 besitzt die L-Ru5P-4-Epimerase ein Aspartat (Asp76). Nicht konserviert sind die beiden bei FucA katalytisch kompetenten Tyrosine (Tyr113’ und Tyr209’). An Stelle von Tyr113’ besitzt die L-Ru5P-4-Epimerase wie 149 auch die RhuA ein Threonin. An Stelle von Tyr209’ steht ein Histidin. Im weiteren Verlauf des C-terminalen Kettenendes treten zwei benachbarte Tyrosine auf. Für die Reste His95 und His97 sowie Asp76 konnte die Rolle als Zink-Liganden bzw. als katalytische Reste in L-Ru5P-4-Epimerase inzwischen durch Mutagenese-Studien verifiziert werden (Johnson & Tanner, 1998). Die L-Ru5P-4-Epimerase katalysiert im Rahmen des bakteriellen ArabinoseMetabolismus die Umwandlung von L-Ribulose-5-phosphat zu D-Xylulose-5-phosphat (Xu5P), also die Inversion der Stereochemie am C4-Atom des Zuckers. Somit wird das Inversionszentrum in diesem Fall nicht durch eine benachbarte Carbonyl- oder Carboxylatgruppe aktiviert. Da die L-Ru5P-4-Epimerase NAD+-unabhängig ist (Deupree & Wood, 1970) und auch keinen primären kinetischen Isotopeneffekt sowohl bei der Epimerisierung von L[4-3H]Ru5P als auch D[4-3H]Xu5P zeigt (Salo et al., 1972), wurden von Deupree & Wood (1972) zwei mögliche Reaktionsmechanismen vorgeschlagen: (i) eine Dehydratisierung unter Ausbildung einer Doppelbindung zwischen C3 und C4 mit anschließender Rehydratisierung und (ii) eine Epimerisierung unter intermediärer Spaltung der C3-C4-Kohlenstoffbindung, also ein Retroaldol-/Aldol-Mechanismus. FucA AraD 10 20 30 40 50 60 | | | | | | MERNKLARQIIDTCLEMTRLGLNQGTAGNVSV--RYQDGMLITPTGIPYEKLTESHIVFIDGN-GKHEEG * **· · * · · * * **** * ·* * *· * ·* * · * ** -MLEDLKRQVLEANLALPKHNLVTLTWGNVSAVDRERGVFVIKPSGVDYSVMTADDMVVVSIETGEVVEG | | | | | | 10 20 30 40 50 60 70 80 90 100 110 120 130 | | | | | | | FucA -KLPSSEWRFHMAAYQSRPDANAVVHNHAVHCTAVSILNRSIPAIHYMIAAAGGNSIPCAPYAT-----* ***· * **· * ·** *· * * · **** * *** * AraD TKKPSSDTPTHRLLYQAFPSIGGIVHTHSRHATIWAQAGQSIPATGTTHADYFYGTIPCTRKMTDAEING | | | | | | | 70 80 90 100 110 120 130 140 150 160 170 180 190 | | | | | | FucA ---FGTR----ELSEHVALALKNRKATLLQHHGLIACEVNLEKALWLAHEVEVLAQLYLTTLAITDPVPVL * * * · *· ** ·* * * *· * ·* * *· · * * AraD EYEWETGNVIVETFEKQGIDAAQMPGVLVHSHGPFAWGKNAEDAVHNAIVLEEVA--YMGIFC-RQLAPQL | | | | | | | 140 150 160 170 180 190 200 200 210 | | FucA SDEEIAVVLEKF-KTYGLRIEE * · ·· · · * · AraD PDMQQTLLDKHYLRKHGAKAYYGQ | | | 210 220 230 Abbildung 81 Sequenzvergleich zwischen FucA und der L-Ru5P-4-Epimerase (AraD) aus E. coli nach Johnson & Tanner (1998). Das alignment erfolgte dabei mit dem Programm SEQSEE (Wishart et al., 1994). Identische Aminosäuren sind mit einem Stern, ähnliche Aminosäuren mit einem Punkt markiert. Reste von FucA, die an der Zink-Koordination, an der Bindung des Inhibitors PGH oder am katalytischen Zyklus beteiligt sind, sind fett gedruckt. 150 Aufgrund der Sequenzhomologie zu FucA wurde von Dreyer (1995) sowie Johnson & Tanner (1998) der Retroaldol-/Aldol-Mechanismus favorisiert. Der kcat-Wert von 20.4 s-1 für die Epimerisierung von L-Ru5P zu D-Xu5P ist dabei nahezu identisch zu dem für die Spaltung von Fuc1P durch FucA unter vergleichbaren Assay-Bedingungen gemessenen Wert (vgl. Kapitel 4.7.1). Beide Enzyme zeigen also unter gleichen Bedingungen die gleiche Katalyserate, was auf Basis der Homologie beider Enzyme als zusätzliches Indiz für einen analogen Mechanismus angesehen werden kann. Gemäß dem Katalysemodell von Johnson & Tanner (Abbildung 82) bindet L-Ru5P zunächst mit seinen O2- und O3-Atomen an das katalytische Zink-Ion. Im ersten Reaktionsschritt wird daraufhin das Proton der Hydroxylgruppe an C4 durch die Carboxylatgruppe von Asp76 unter Spaltung der C3-C4-Bindung abstrahiert. Die Stabilisierung des dabei entstehenden Endiolats erfolgt wie bei FucA und anderen Klasse-IIAldolasen durch Komplexierung an das Zink-Ion. Im Gegensatz zu Klasse-II-Aldolasen werden dann jedoch die Spaltprodukte nicht freigesetzt, sondern bleiben durch das Enzym vom Solvens abgeschirmt. Statt dessen wird ein Rotation der Carbonylgruppe der Aldehydkomponente vorgeschlagen. Daraufhin erfolgt die Reprotonierung der Carbonylgruppe durch Asp76 unter neuerlicher Ausbildung der C3-C4-Bindung, also eine Aldolkondensation. OH 1 OH O 2 Zn 5 2- O3PO 3 4 H His O H O Zn His His His O O H 2- O3PO His His O L-Ru5P O O Asp76 Rotation OH OH His O Zn 2- O3PO OH O H Zn His His His O 2- O H O O3PO His His H O D-Xu5P O Asp76 Abbildung 82 Postulierter Retroaldol-/Aldol-Mechanismus für die durch die L-Ribulose-5-phosphat4-Epimerase katalysierte Umwandlung von L-Ribulose-5-phosphat (L-Ru5P) in D-Xylulose-5phosphat (D-Xu5P) unter Beteiligung von Asp76 als katalytische Base bzw. Säure (Johnson & Tanner, 1998). 151 Die Verifizierung des Retroaldol-/Aldol-Mechanismus gelang Johnson & Tanner durch experimentellen Nachweis der beiden Teilreaktionen. So konnten sie zeigen, daß L-Ru5P-4Epimerase ausgehend von den beiden postulierten Zwischenprodukten DHA und Glykolaldehydphosphat die Kondensation sowohl zu L-Ru5P als auch D-Xu5P katalysiert. Während die Aktivitäten dabei für den WT allerdings nur geringfügig über der Nachweisgrenze lagen, konnte mit Mutante H97N eine höhere Aktivität von etwa 0.004 U/mg bei pH 7.6 und 37 °C erreicht werden. Die Aldol-Donorkomponente DHA in diesem Mechanismus entspricht DHAP in FucA. Aus diesem Grunde könnte die L-Ru5P-4-Epimerase auch ein vielversprechender Ansatzpunkt bei weiteren Planungen zur Selektivitätsänderung von DHAP zu DHA bei FucA sein (vgl. Kapitel 4.1.5 & 4.6). Dabei müßte jedoch zunächst einmal geklärt werden, ob die Donorkomponente DHA in L-Ru5P-4-Epimerase überhaupt analog zu DHAP in FucA bindet. Denn das würde ja heißen, daß gemäß dem Sequenzvergleich die Reste der Phosphatbindungstasche zwar größtenteils konserviert sind, die Bindungstasche aber nicht besetzt wird. Alternativ wurde von Dreyer vorgeschlagen, daß die 5-Phosphatgruppe von L-Ru5P die Phosphatbindungstasche besetzt. In diesem Fall könnte jedoch die Carbonylgruppe am C2-Atom bei ähnlichen räumlichen Verhältnissen nicht an das Zink-Ion koordinieren, und eine Stabilisierung des intermediär gebildeten Endiolats über das Zink-Ion wäre nicht möglich. Eine abschließende Diskussion dieses Punktes sowie der erhöhten Aldolaseaktivität für die Mutante H97N - einschließlich der Übertragung der Verhältnisse auf FucA im Hinblick auf weiteres Protein-Engineering - sind jedoch nur mit Informationen über die Röntgenstruktur der L-Ru5P-4-Epimerase möglich. Erste kristallographische Studien wurden zwar bereits von Andersson et al. (1995) veröffentlicht, über eine Aufklärung der Struktur ist bisher aber nichts bekannt. 152 4.8 Selektivität von FucA-Mutanten in bioorganischen Synthesen Wie bereits mehrfach erwähnt, zeigt FucA in Syntheserichtung eine breite Akzeptanz für nicht-natürliche Aldehydsubstrate. Dabei zeigt der WT eine starke kinetische Präferenz für L-konfigurierte 2-Hydroxyaldehyde (Fessner et al., 1992; 1993). Unter Reaktionsbedingungen mit kinetischer Kontrolle wird ausgehend von rac-Lactaldehyd nur das L-Enantiomer prozessiert. Bei Vorhandensein eines H-Brücken-Akzeptors in α- oder β-Position zur Carbonylgruppe verläuft die Reaktion stereoselektiv, und es entstehen ausschließlich D-erythro-konfigurierte Produkte. Ist ein derartiger Substituent nicht vorhanden, so entstehen Gemische aus D-erythro- und L-threo-Produkten, wobei das Produktverhältnis vom jeweiligen Aldehydrest abhängt (Tabelle 6, Seite 76). Um Informationen über die Koordination der Aldehydkomponente zu erhalten, wurden die Selektivitäten einiger Mutanten untersucht. Die Messungen wurden freundlicherweise von Goße (1998) in der Arbeitsgruppe Fessner an der RWTH-Aachen durchgeführt. Abbildung 83 zeigt schematisch die Auswahl der untersuchten Aldehydsubstrate einschließlich der Stereochemie der beobachteten Produkte. Die für die einzelnen Mutanten bestimmten Selektivitäten sind dann in Tabelle 22 aufgeführt. (a) DHAP O OPO3 O OH DHAP O 2- OPO3 O + OH H (b) 2- OH HO OH HO OH L D OH O OH OPO3 H 2- O 2- + OH OPO3 OH L-threo D-erythro (c) DHAP O OH O OH OPO3 H O 2- 2- OPO3 + OH OH L-threo D-erythro (d) DHAP O OH O OH OPO3 H OH D-erythro 2- O 2- OPO 3 + OH L-threo Abbildung 83 Schematische Darstellung der möglichen Produkte in FucA-katalysierten organischen Synthesen mit DHAP und rac-Lactaldehyd (a), Acetaldehyd (b), Propionaldehyd (c) und isoButyraldehyd (d). 153 Tabelle 22 Selektivität von FucA-Mutanten in Syntheserichtung für verschiedene Aldehydkomponenten a) Aldehyd b) Lactaldehyd Acetaldehyd Propionaldehyd iso-Butyraldehyd Produktverhältnis c) L:D Wildtyp 97:3 95:5 50:50 68:32 T26A 85:15 75:25 - - N29Q 90:10 42:58 - - Y113F 10:90 d) 35:65 - - F131A 32:68 e) 30:70 3:97 3:97 F206W 70:30 50:50 36:64 60:40 65:35 - - D-erythro/L-threo D-erythro/L-threo D-erythro/L-threo d) Del(207-215) 40:60 Del(211-215) 80:20 54:56 - - Y209F 50:50 35:65 - - R212A 95:5 95:5 - - E214A 71:29 86:14 60:40 75:25 E214L 89:11 70:30 58:42 48:52 E215A 90:10 85:15 - - a) b) c) d) e) Die Werte wurden freundlicherweise von Goße (1998) zur Verfügung gestellt. Aldehydkomponente in der FucA-katalysierten Aldolkondensation mit DHAP als Donorkomponente. Die ebenfalls untersuchten Aldehydsubstrate Pivaldehyd und Benzaldehyd wurden weder vom WT noch von den Mutanten umgesetzt und werden in der Tabelle nicht mehr explizit aufgeführt. Verhältnis der beobachteten Produkte (cf. Abbildung 83). Die 1H-NMR-Daten ließen keine Unterscheidung zwischen dem erwarteten D-erythro- und einem möglicherweise auch auftretenden L-threo-Produkt zu. Die 1H-NMR-Daten zeigen eindeutig, daß es sich um das D-erythro-Produkt handelt. Unter kinetisch kontrollierter Reaktionsführung mit rac-Lactaldehyd zeigt der WT eine starke kinetische Präferenz für L-Lactaldehyd und es wird dementsprechend nur das L-konfigurierte Produkt beobachtet. Im Gegensatz dazu diskriminieren die Mutanten weniger scharf zwischen den D- und L-Enantiomeren, so daß auch unterschiedliche Anteile des D-konfigurierten Produkts (Psicose-1-phosphat) gefunden werden. Bei der Mutante Y113F hat sich die kinetische Enantioselektivität nahezu vollständig umgekehrt, da zu 90 % D-konfiguriertes Produkt gebildet wird (L:D = 10:90). Der Verlust des kompletten C-terminalen Kettenendes (Mutante Del(207-215)) hat ebenfalls eine Umkehr der kinetischen Enantioselektivität zur Folge (L:D = 40:60), wenn auch nicht ganz so stark wie bei Mutante Y113F. Das gleiche gilt für Mutante F131A (L:D = 32:68). Mutante Y209F zeigt überhaupt keine kinetische Diskriminierung mehr zwischen den beiden racemischen Antipoden (L:D = 50:50). Bei allen anderen untersuchten Mutanten bleibt die kinetische Präferenz für L-Lactaldehyd bestehen, wenngleich sie zumeist schwächer ausgeprägt ist als beim WT. 154 Ähnliche Effekte treten bei der Diastereoselektivität (definiert durch das D-erythro/Lthreo-Verhältnis) für die drei aliphatischen Aldehyde Acet-, Propion- und iso-Butyraldehyd auf. Der WT liefert für Acetaldehyd hauptsächlich D-erythro-konfiguriertes Produkt (95:5). Für den sterisch anspruchsvollen iso-Butyraldehyd gilt, wenn auch schon deutlich abgeschwächt, das gleiche, während für Propionaldehyd beide Diastereomere zu gleichen Anteilen gebildet werden. Bei den Mutanten sind die Verhältnisse jedoch teilweise komplett umgedreht. So zeigen im Falle von Acetaldehyd die Mutanten N29Q, Y113F, Y209F und F131A eine Inversion der Diastereoselektivität, wobei der Anteil des L-threo-Produktes zwischen 58 % und 70 % liegt. Mutante F206W bildet beide Diastereomere zu gleichen Teilen. Alle anderen Mutanten zeigen unterschiedliche Verhältnisse zugunsten des D-erythro-Produktes, begünstigen also eine Orientierung der Aldehydkomponente analog zu der im WT. Bei Propion- und isoButyraldehyd wird im Falle von Mutante F131A der drastischste Effekt erreicht. Mutante F131A zeigt für diese beiden Aldehyde eine völlige Inversion der Diastereoselektivität und bildet ausschließlich jeweils das L-threo-konfigurierte Produkt. Die Ergebnisse der Selektivitätsmessungen zeigen drei herausragende Reste: Tyr113’, Phe131 und Tyr209’. Die kinetische Enantioselektivität für rac-Lactaldehyd und das invertierte Produktspektrum für diese Mutanten legen nahe, daß diese Reste unmittelbar an der Positionierung der Aldehydkomponente beteiligt sind. Die Umkehr der Diastereoselektivität für Mutante N29Q muß auf Basis der Röntgenstruktur der Mutante, die nur geringfügige Veränderungen innerhalb der Phosphatbindungstasche zeigt, primär wohl eher auf eine etwas veränderte Position der DHAP-Komponente zurückgeführt werden. Auf der Grundlage obiger Daten konnte in Kombination mit der PGH-Struktur des WT ein detaillierteres Modell für das Binden der Aldehydkomponente erarbeitet werden, das in Abbildung 84a dargestellt ist. Dieses Modell beinhaltet: (i) die näherungsweise coplanare Anordnung des DHAPEndiolats und der Carbonylgruppe von L-Lactaldehyd mit H-Brücken-Wechselwirkungen zwischen der phenolischen OH-Gruppe von Tyr113’ und sowohl der Carbonyl- als auch Hydroxygruppe des Aldehyds, (ii) die räumliche Nähe des C3-Atoms von DHAP und des Carbonylkohlenstoffs des Aldehyds, so daß die Ausbildung einer C-C-Bindung durch nukleophilen Angriff ermöglicht wird und (iii) eine Orientierung des Aldehyds, so daß er mit seiner Si-Seite zum C3-Atom von DHAP zeigt, was in einer Bildung des natürlich beobachteten D-erythro-Diastereomers resultiert. Dieses Modell erklärt sehr gut, warum FucA-WT Aldehyde mit einer α-Hydroxygruppe selektiv zum jeweiligen D-erythro-konfigurierten Produkt umsetzt, da die Hydroxygruppe in diesem Fall mit Tyr113’ und etwas abgeschwächt auch mit dem Zink-Ion interagieren kann. Bei den Mutanten kann durch die unterschiedlich starke Störung der geometrischen Verhältnisse eventuell auch die α-Hydroxygruppe des D-Enantiomers an Tyr113’ binden, was 155 den partiellen Verlust der Enantioselektivität erklären würde. Diese Störung tritt, den veränderten Enantioselektivitäten zufolge, auch bei Mutationen im flexiblen C-terminalen Kettenende auf. (a) (b) Abbildung 84 Modell für das Binden der Aldehydkomponente und die mögliche Beteiligung des flexiblen C-terminalen Kettenendes über die Seitenkette von Tyr209’ (orange): (a) WT mit modelliertem L-Lactaldehyd (orange); (b) iso-Butyraldehyd wie er in Mutante F131A gebunden wird, so daß ausschließlich das L-threo-Diastereomer gebildet wird. Das Modellieren der Aldehyde erfolgte manuell auf Basis der Strukturen des WT (Dreyer & Schulz, 1996) bzw. der Mutante F131A. Die Orientierung von Tyr209’ ist analog zu dem unter Kapitel 4.7.5 bereits beschriebenen Modell des gesamten C-terminalen Kettenendes. Mutante F131A sticht auf besondere Weise hervor. So zeigt sie nicht nur einen drastischen Aktivitätsverlust für das natürliche Substrat Fuc1P (Kapitel 4.7.3), sondern kehrt auch die Enantioselektivität für Lactaldehyd um und weist gar eine völlige Inversion der Diastereoselektivität für große aliphatische Aldehyde auf. Letzteres kann sehr gut mit der Struktur der Mutante unter Anwendung des gerade entwickelten Modells erklärt werden. Wie bereits erwähnt (Kapitel 4.5.7), formt diese Mutation die hydrophobe Wand in eine hydrophobe Tasche um. Dadurch wird, wie man Abbildung 84 entnehmen kann, genug Platz für 156 den aliphatischen Rest des Aldehyds in einer Orientierung geschaffen, die einen Angriff des Carbanions an der Re-Seite der Carbonylgruppe erlaubt. 157 5 Zusammenfassung und Ausblick Im Rahmen dieser Arbeit wurde der enzymatische Mechanismus sowie die Kontrolle der Diastereoselektivität der L-Fuculose-1-phosphat-Aldolase aus E. coli durch MutageneseStudien untersucht. Grundlage bei der Planung der entsprechenden Mutanten waren die bis zu atomarer Auflösung bestimmten Röntgenstrukturen des Wildtyps in unligierter Form sowie im Komplex mit dem Übergangszustandsanalogon Phosphoglycolohydroxamat. Diese hatten gezeigt, daß die vier aktiven Zentren in dem tetrameren Enzym an den Grenzflächen zwischen je zwei Untereinheiten liegen und jeweils Reste beider Untereinheiten an ihrem Aufbau beteiligt sind. Die entsprechend geplanten Mutanten wurden auf DNA-Ebene erzeugt und die mutierten Gene in den Expressionsvektor pKK223-3 kloniert. Im folgenden wurden sie im Überexpressionsstamm E. coli JM105 hergestellt, daraus isoliert und auf ihre enzymatische Aktivität getestet. Ergänzend dazu wurden für einige Mutanten von der Arbeitsgruppe Fessner an der RWTH-Aachen die Enantio- sowie Diastereoselektivitäten in organischen Synthesen mit unterschiedlichen Aldehydkomponenten bestimmt. Parallel zur kinetischen Charakterisierung der Mutanten wurden Kristallisationsexperimente durchgeführt. Die dabei erhaltenen Kristalle erlaubten es, die Röntgenstrukturen von insgesamt 13 Mutanten zu lösen, wobei die Auflösungen zwischen 1.66 und 2.55 Å lagen. Die Auflösungen der Mutantenstrukturen waren somit teilweise deutlich besser als die der WT-Struktur, die bei 1.92 Å aufgeklärt wurde. Mittels der Strukturen und der kinetischen Daten der Mutanten konnte ein so detailliertes Verständnis der FucA-katalysierten Reaktion erlangt werden, daß gute Modelle für das Binden von L-Lactaldehyd bzw. L-Fuculose-1-phosphat im aktiven Zentrum aufgestellt werden konnten. Desweiteren konnte gezeigt werden, daß das C-terminale Kettenende aus den Resten 207 bis 215 an der Katalyse partizipiert und nach erfolgter Substratbindung das aktive Zentrum durch einen induced fit verschließt. In der Struktur des WT hatten diese Reste keine definierte Konformation. Durch die kinetischen Daten der C-terminalen Mutanten konnte jedoch nachgewiesen werden, daß die Reste Tyr209’ und Glu214’ höchstwahrscheinlich mit dem Substrat interagieren. Der zuvor veröffentlichte Mechanismus, dem nur die Strukturen des WT zugrunde lagen, wurde aufgrund dieser neuen Daten modifiziert. Während Dreyer & Schulz für den katalytischen Mechanismus in Syntheserichtung zwei katalytische Reste postuliert hatten (Glu73 als katalytische Base und Tyr113’ als katalytische Säure), konnte nun gezeigt werden, daß FucA über drei katalytisch kompetente Reste verfügt. Dies sind Glu73, Tyr113’ und Tyr209’. Die beiden Tyrosin-Reste fungieren dabei als unabhängige Dipole und sind gemäß der beobachteten kinetischen Effekte bzw. Diastereoselektivitäten an der Koordination der Carbonylgruppe der Aldol-Akzeptorkomponente beteiligt bzw. stabilisieren in Spaltungs- 158 richtung die negative Ladung an der 4-Hydroxygruppe von L-Fuculose-1-phosphat. Tyr113’ konnte außerdem eine Rolle bei der kinetischen Enantioselektivität und Diastereoselektivität der Reaktion zugeschrieben werden. Glu73 muß demzufolge den Protonentransfer sowohl am C3-Atom als auch an der 4-Hydroxygruppe des Substrates bewerkstelligen. Der Vergleich mit anderen Klasse-II-Aldolasen wie der Rhamnulose-1-phosphat-Aldolase aus E. coli bzw. der Fructose-1,6-bisphosphat-Aldolase aus E. coli zeigt, daß FucA mit seinen beiden katalytisch kompetenten Tyrosin-Resten nach heutigem Kenntnisstand eine Sonderstellung in dieser Enzymklasse einnimmt. Neben der Rolle des C-terminalen Kettenendes sowie einzelner Reste, konnte auch die Bedeutung eines hydrophoben Bereichs im aktiven Zentrum aufgeklärt werden. So konnte durch Mutationen in diesem Bereich die Diastereoselektivität des Enzyms für ausgewählte Aldehyde gezielt manipuliert werden. Beispielsweise verschob sich bei der Mutante F131A die Diastereoselektivität der Reaktion für große aliphatische Aldehyde vollständig auf die Seite L-threo-konfigurierter Produkte. Weiterhin wurde versucht, durch Mutationen in der Phosphatbindungstasche die Selektivität in Bezug auf den Aldol-Donor von DHAP zu den unter ökonomischen Gesichtspunkten günstigeren Substraten Dihydroxyaceton und β-Hydroxypyruvat zu verändern. Mit den auf der Basis der PGH-Struktur des Wildtyps geplanten Mutanten gelang dies jedoch nicht. Wie die Kristallstruktur der Mutante S71Q enthüllte, ist dies primär eine Folge größerer struktureller Veränderungen in der Phosphatbindungstasche und den angrenzenden Bereichen des Proteins. Die Analyse der strukturellen Umordnungen zeigte auf deutliche Weise, daß das aktive Zentrum von FucA keinesfalls als starres Polypeptid-Gerüst angesehen werden darf, wie es die Kristallstrukturen des Wildtyps suggerieren könnten. Darüber hinaus gab die Struktur der Mutante S71Q Einblicke in die möglichen konformationellen Veränderungen während eines Katalysezyklus. Trotz oder gerade wegen der strukturellen Fluktuation im aktiven Zentrum während der Katalyse erscheinen Spezifitätsänderungen mittels weiterer Mutanten durchaus möglich. In diesem Zusammenhang werden sicher auch die Strukturen anderer Klasse-II-Aldolasen sowie der homologen L-Ribulose-5phosphat-4-Epimerase von großem Interesse sein. Neben dem Einsatz von FucA in der organischen Synthese, der in Zukunft sicher noch verstärkt stattfinden wird, gibt es als ganz anderes Fachgebiet noch die Nanotechnologie, für die FucA aufgrund ihrer strukturellen Eigenheiten geradezu prädestiniert scheint. Denn durch ihre molekulare C4-Symmetrie, die ansonsten in der Natur relativ selten anzutreffen ist, stellt FucA ein mögliches Zielobjekt beim Design strukturierter makromolekularer Netzwerke dar. 159 6 Literatur Adams, R. L. P., Knowler, J. T. & Leader, D. P. (1986). The biochemistry of the nucleic acids, 10th ed., Chapman and Hall, London, New York. Agarwal, C. R. (1978). A new least-squares refinement technique based on the fast Fourier transform algorithm. Acta Crystallogr. Sect. A 34, 791-809. Alefounder, P. R., Baldwin, S. A., Perham, R. & Short, N. J. (1989). Cloning, sequence analysis and over-expression of the gene for the class II fructose-1,6-bisphosphate aldolase of Escherichia coli. Biochem. J. 257, 529-534. Andersson, A., Schneider, G. & Lindqvist, Y. (1995). Purification and preliminary X-ray crystallographic studies of recombinant L-ribulose-5-phosphate 4-epimerase from Escherichia coli. Protein Sci. 4, 1648-1650. Arndt, U. W., Champness, J. N., Phizackerley, R. P. & Wonacott, A. J. (1973). A single-crystal oscillation camera for large unit cells. J. Appl. Cryst. 6, 457-463. Arth, H.-L. (1997). Synthesestudien zu Dihydroxyacetonphosphat-Analoga. Dissertation. RWTH Aachen. Arth, H.-L. & Fessner, W.-D. (1998). Practical synthesis of 4-hydroxy-3-oxobutylphosphonic acid and its evaluation as a bio-isosteric substrate of DHAP aldolase. Carbohydr. Res. 305, 313-321. Bachmann, B. J. (1983). Linkage map of Escherichia coli K-12, ed. 7. Microbiol. Rev. 47, 180-230. Barbas III, C. F., Heine, A., Zhong, G., Hoffmann, T., Gramatikova, S., Björnestedt, R., List, B., Anderson, J., Stura, E. A., Wilson, I. A. & Lerner, R. A. (1997). Immune versus natural selection: antibody aldolases with enzymatic rates but broader scope. Science 278, 2085-2092. Beck, S. & Pohl, F. M. (1984). DNA sequencing with direct blotting electrophoresis. EMBO J. 3, 2905-2909. Bednarski, M. D., Simon, E. S., Bischofberger, N., Fessner, W.-D., Kim, M.-J., Lees, W., Saito, T., Waldmann, H. & Whitesides, G. M. (1989). Rabbit muscle aldolase as a catalyst in organic syntheses. J. Am. Chem. Soc. 111, 627-635. Berthiaume, L., Loisel, T. P. & Sygusch, J. (1991). Carboxyl terminus region modulates catalytic activity of recombinant maize aldolase. J. Biol. Chem. 266, 17099-17105. Biggin, M. D., Gibson, T. J. & Hong, G. F. (1983). Buffer gradient gels and 35S label as an aid to rapid DNA sequence determination. Proc. Natl. Acad. Sci. USA 80, 3963-3965. Birnboim, H. & Doly, S. (1979). A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7, 1513-1523. Blom, N. S., Tétreault, S., Coulombe, R. & Sygusch, J. (1996). Novel active site in Escherichia coli fructose 1,6-bisphosphate aldolase. Nat. Struct. Biol. 3, 856-862. Blom, N. & Sygusch, J. (1997). Product binding and role of the C-terminal region in class I D-fructose 1,6-bisphosphate aldolase. Nat. Struct. Biol. 4, 36-39. Blundell, T. L. & Johnson, L. N. (1976). Protein crystallography, Academic Press, New York. 160 Bragg, W. H. & Bragg, W. L. (1913). Proc. Roy. Soc. (London) A 88, 428-438. Brosius, J. & Holy, A. (1984). Regulation of the ribosomal RNA promoters with a synthetic lac operator. Proc. Natl. Acad. Sci. USA 81, 6929-6933. Brünger, A. T., Kuriyan, J. & Karplus, M. (1987). Crystallographic R-factor refinement by molecular dynamics. Science 235, 458-460. Brünger, A. T. (1992a). The free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472-474. Brünger, A. T. (1992b). X-PLOR Version 3.1. A system for X-ray crystallography and NMR. Yale University Press, New Haven and London. Brünger, A. T. (1993). Assessment of phase accuracy by cross validation: the free R value. Methods and applications. Acta Crystallogr. Sect. D 49, 24-36. Bukhari, A. I., Shapiro, J. A. & Adhya, A. L. (1977). DNA insertion elements, plasmids and episomes. pp. 757-768, Cold Spring Harbor Laboratory Press. Busch & Lukas (1991). Wissenschaftliche Software. Freiburg im Breisgau. CCP4 (1994). The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D. 50, 760-763. Carter, C. W. Jr. & Carter, C. W. (1979). Protein crystallization using incomplete factorial experiments. J. Biol. Chem. 254, 12219-12223. Chen, B. & Thomas, A. (1980). Recovery of DNA segments from agarose gels. Anal. Biochem. 191, 339-341. Chen, Y.-M., Zhu, Y. & Lin, E. C. C. (1987). The organization of the fuc regulon specifying L-fucose dissimilation in Escherichia coli K12 as determined by gene cloning. Mol. Gen. Genet. 210, 331337. Chen, Y.-M., Lu, Z. & Lin, E. C. C. (1989). Constitutive activation of the fucAO operon and silencing of the divergently transcribed fucPIK operon by an IS5 element in Escherichia coli mutants selected for growth on L-1,2-propanediol. J. Bacteriol. 171, 6097-6105. Cohen, D., Chumakov, I. & Weissenbach, J. (1993). A first-generation physical map of the human genome. Nature 366, 698-701. Cooper, S. J., Leonard, G. A., McSweeney, S. M., Thompson, A. W., Naismith, J. H., Qamar, A., Plater, A., Berry, A. & Hunter, W. H. (1996). The crystal structure of class II fructose-1,6bisphosphate aldolase shows a novel binuclear metal-binding active site embedded in a familiar fold. Structure 14, 1303-1315. Cudney, R., Patel, S., Weisgraber, K. & Newhouse, Y. (1994). Screening and optimization strategies for macromolecular crystal growth. Acta Crystallogr. Sect. D 50, 414-423. Dalby, A., Dauter, Z. & Littlechild, J. A. (1999). Crystal structure of human muscle aldolase complexed with fructose 1,6-bisphosphate: Mechanistic implications. Protein Sci. 8, 291-297. Derewenda, Z. & Helliwell, J. R. (1989). Calibration tests and use of a Nicolet/Xentronics imaging proportional chamber mounted on a conventional source for protein crystallography. J. Appl. Cryst. 22, 123-137. 161 Deupree, J. D. & Wood, W. A. (1970). L-Ribulose 5-phosphate 4-epimerase from Aerobacter aerogenes. Evidence for nicotinamide adenine dinucleotide-independent 4-epimerization by the crystalline enzyme. J. Biol. Chem. 245, 3988-3995. Deupree, J. D. & Wood, W. A. (1972). L-Ribulose 5-phosphate 4-epimerase from Aerobacter aerogenes. Evidence for a role of divalent metal ions in the epimerization reaction. J. Biol. Chem. 247, 3093-3097. Dhar, N. M. & Altekar, W. (1986). Distribution of class II fructose-1,6-bisphosphate aldolases in halophilic archaebacteria. FEMS Microbiol. Lett. 35, 177-181. Diederichs, K. & Karplus, P. A. (1997). Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4, 269-275. Dreyer, M. (1991). Überexpression, Reinigung, Kristallisation und röntgenographische Untersuchung der L-Fuculose-1-phosphat-Aldolase aus Escherichia coli sowie Überexpression und Reinigung der L-Fucose-Isomerase aus Escherichia coli. Diplomarbeit. Universität Freiburg im Breisgau. Dreyer, M. K. & Schulz, G. E. (1993). The spatial structure of the class II L-fuculose-1-phosphate aldolase from Escherichia coli. J. Mol. Biol. 231, 549-553. Dreyer, M. K. (1995). Kristallstrukturen und enzymatischer Mechanismus der Fuculose-1-phosphatAldolase aus Escherichia coli. Dissertation. Universität Freiburg im Breisgau. Dreyer, M. K. & Schulz, G. E. (1996a). Refined high-resolution structure of the metal-ion dependent L-fuculose-1-phosphate aldolase (class II) from Escherichia coli. Acta Crystallogr. Sect D 52, 1082-1091. Dreyer, M. K. & Schulz, G. E. (1996b). Catalytic mechanism of the metal-dependent fuculose aldolase from Escherichia coli as derived from the structure. J. Mol. Biol. 259, 458-466. Drueckhammer, D. G., Durrwachter, J. R., Pederson, R. L., Crans D. C., Daniels, L. & Wong, C.-H. (1989). Reversible and in situ formation of organic arsenates and vanadates as organic phosphate mimics in enzymatic reactions: mechanistic investigation of aldol reactions and synthetic applications. J. Org. Chem. 54, 70-77. Ducruix, A. & Giegé, R. (1992). Crystallization of nucleic acids and proteins: a practical approach, Oxford University Press, Oxford. Duncan, B. K., Rockstroh, P. A. & Warner, H. R. (1978). Escherichia coli K-12 mutants deficient in uracil-DNA glycosylase. J. Bacteriol. 134, 1039-1045. Dunn, M. J. (1989). Protein Purification Methods – A Practical Approach, (Harris, E. L. V. & Augal, S., eds.), pp. 18-37, IRS Press at Oxford University Press, Oxford. Engh, R. A. & Huber, R. (1991). Accurate bond and angle parameters for X-ray protein-structure refinement. Acta Crystallogr. Sect. A 47, 392-400. Enzyme Handbook (1998). (Schomburg, D. & Stephan, D., eds.), Springer-Verlag, Berlin, Heidelberg. Evans, P. R. (1993). Data reduction. In: Proceedings of the CCP4 study weekend: data collection and processing. (Sawyer, L., Isaacs, N. & Burley, S., eds.), pp. 114-122, Daresbury Laboratory, Warrington, UK. Ewald, P. P. (1921). Das „reziproke“ Gitter in der Strukturtheorie. Z. Kristallogr. 56, 129-156. 162 Fa. Amersham (1995). Protokoll zum SculptorTM in vitro mutagenesis system RPN 1526. Amersham International, Buckinghamshire, England. Fa. Boehringer Mannheim (1993). DIG Nucleic Acid Detection Kit. Detection of digoxigenin-labeled nucleic acids by enzyme immunoassay and enzyme-catalysed color detection. Boehringer Mannheim Biochemica, Mannheim. Fa. Boehringer Mannheim (1994). DIG Taq DNA Sequencing Kit for Standard Cylce Sequencing. For the non-radioactive sequencing of single- and double-stranded DNA with digoxigenin-labeled primer. Boehringer Mannheim Biochemica, Mannheim. Fa. GATC (1995). Gebrauchsanleitung GATC® 1500-System Direct Blotting Electrophoresis (DNASequencer). GATC – Gesellschaft für Analyse-Technik und Consulting, Konstanz. Fa. GATC (1996). Gebrauchsanleitung GATC®-BioCycle Sequencing Kit. Cycle Seqeuncing using biotinylated terminators and Thermo SequenaseTM optimated for the GATC 1500 System. GATC – Gesellschaft für Analyse-Technik und Consulting, Konstanz. Fa. Qiagen (1994). QIAprep Spin M13 Single-Stranded DNA Purification Protocol (For QIAprep Spin M13 Kits). Qiagen, Hilden. Fa. Qiagen (1995). QIAprep Plasmid Handbook (For Standard Plasmid Minipreparation). Qiagen, Hilden. Fa. Qiagen (1997). QIAquick Spin Handbook (For QIAquick PCR Purification Kit QIAquick Nucleotide Removal Kit QIAquick Gel Extraction Kit). Qiagen, Hilden. Fa. Roche (1999). Chirazyme BioCatalysts for Industry 1999/2000. Boehringer Mannheim GmbH, Roche Molecular Biochemicals, Mannheim. Ferretti, L. & Sgaramella, V. (1981). Temperature dependence of the joining by T4 DNA ligase of termini produced by type II restriction endonucleases. Nucleic Acids Res. 9, 85-93. Fessner, W.-D., Sinerius, G., Schneider, A., Dreyer, M., Schulz, G. E., Badia, J. & Aguilar, J. (1991). Diastereoselective enzymatic aldol additions: L-rhamnulose and L-fuculose-1-phosphate aldolases from E. coli. Angew. Chem. Int. Ed. Engl. 30, 555-558. Fessner, W.-D. (1992). A building-block strategy for asymmetric synthesis: The DHAP aldolases. In: Microbial Reagents in Organic Synthesis (Servi, S., ed.), pp. 43-55, Kluwer Academic Publishers, Dordrecht. Fessner, W.-D., Badia, J., Eyrisch, O., Schneider, A. & Sinerius, G. (1992). Enzymatic syntheses of rare ketose 1-phosphates. Tetrahedron Lett. 33, 5231-5234. Fessner, W.-D. (1993). Biokatalytische de novo-Synthese von Kohlenhydraten. GIT Fachz. Lab. 11, 951-956. Fessner, W.-D., Schneider, A., Eyrisch, O., Sinerius, G. & Badia, J. (1993). 6-Deoxy-L-lyxo- and 6Deoxy-L-arabino-hexulose 1-phosphates. Enzyme syntheses by antagonistic metabolic pathways. Tetrahedron Asymm. 4, 1183-1192. Fessner, W.-D. (1995). Houben-Weyl Methods of Organic Chemistry (Helmchen, G., Hoffmann, R. W., Mulzer, J. & Schaumann, E. eds.) vol E21b, pp 1736-1747, Thieme Verlag, Stuttgart. Fessner, W.-D. & Walter, C. (1996). Enzymatic C-C bond formation in asymmetric synthesis. Top. Curr. Chem. 184, 97-194. 163 Fessner, W.-D., Schneider, A., Held, H., Sinerius, G., Walter, C., Hixon, M. & Schloss, J. V. (1996). The mechanism of class II, metal-dependent aldolases. Angew. Chem. Int. Ed. Engl. 35, 22192221. Fleischmann et al., R. D. & Venter, C. (1995). Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269, 496-512. Fothergill-Gilmore, L. A. (1987). Evolution in glycolysis. Biochem. Soc. Trans. 15, 993-995. Fothergill-Gilmore, L. A. & Michels, P. A. M. (1993). Evolution of glycolysis. Prog. Biophys. Mol. Biol. 59, 105-235. Freemont, P. S., Dunbar, B. & Fothergill-Gilmore, L. A. (1988). The complete amino acid sequence of human skeletal-muscle fructose-bisphosphate aldolase. Biochem. J. 249, 779-788. Frishman, D. & Argos, P. (1995). Knowledge-based protein secondary structure assignment. Proteins: Struct. Funct. Genet. 23, 566-579. Gamblin, S. J., Cooper, B., Millar, J. R., Davies, G. J., Littlechild, J. A. & Watson, H. C. (1990). The crystal structure of human muscle aldolase at 3.0 Å resolution. FEBS Lett. 262, 282-286. Gamblin, S. J., Davies, G. J., Grimes, J. M., Jackson, R. M., Littlechild, J. A. & Watson, H. C. (1991). Activity and specificity of human aldolases. J. Mol. Biol. 219, 573-576. Gefflaut, T., Blonski, C., Perie, J. & Willson, M. (1995). Class I aldolases: Substrate specificity, mechanism, inhibitors and structural aspects. Prog. Biophys. Mol. Biol. 63, 301-340. Ghalambor, M. A. & Heath, E. C. (1962). The metabolism of L-fucose. J. Biol. Chem. 237, 24272433. Ghalambor, M. A. & Heath, E. C. (1966). L-Fuculose-1-phosphate aldolase. Methods Enzymol. 9, 538-542. Gill, S. C. & von Hippel, P. H. (1989). Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319-326. Gilliland, G. L. & Ladner, J. E. (1996). Crystallization of biological macromolecules for X-ray diffraction studies. Curr. Opin. Struct. Biol. 6, 595-603. Goße, C. (1997). Mechanistische Studien zu metallabhängigen Aldolasen und Isomerasen. Dissertation, RWTH Aachen. Håkansson, K., Carlsson, M., Svensson, L. A. & Liljas, A. (1992). Structure of native and apo carbonic anhydrase II and structure of some of its anion-ligand complexes. J. Mol. Biol. 227, 11921204. Hall, R. D., Leonard, G. A., Reed, C. D., Watt, C. I., Berry, A. & Hunter, W. N. (1999). The crystal structure of Escherichia coli class II fructose-1,6-bisphosphate aldolase in complex with phosphoglycolohydroxamate reveals details of mechanism and specificity. J. Mol. Biol. 287, 383394. Hanahan, D. (1983). Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557-580. 164 Hennig, M., D’Arcy, A., Hampele, I. C., Page, M. G. P., Oefner, C. & Dale, G. E. (1998). Crystal structure and reaction mechanism of 7,8-dihydroneopterin aldolase from Staphylococcus aureus. Nat. Struct. Biol. 5, 357-362. Hester, G., Brenner-Holzach, O., Rossi, F. A., Struck-Donatz, M., Winterhalter, K. H., Smit, J. D. G. & Piontek, K. (1991). The crystal structure of fructose-1,6-bisphosphate aldolase from Drosophila melanogaster at 2.5 Å resolution. FEBS Lett. 292, 237-242. Heyduk, T., Michalczyk, R. & Kochman, M. (1991). Long-range effects and conformational flexibility of aldolase. J. Biol. Chem. 266, 15650-15655. Howard, A. J., Gilliland, G. L., Finzel, B. C., Poulos, T. L., Ohlendorf, D. H. & Salemme, F. R. (1987). The use of an imaging proportional counter in macromolecular crystallography. J. Appl. Cryst. 20, 383-387. Hupe, D. J. (1984). Enzyme reactions involving imine formation. New Comprehensive Biochem. 6, 271-301. International Tables for Crystallography (1996). Vol. A, Space-Group Symmetry, 4th corrected edition, (Hahn, T., ed.), Kluwer Academic Publishers, Dordrecht, Boston, London. von Itzstein, M. & Thomson, R. J. (1997). The synthesis of novel sialic acids as biological probes. Top. Curr. Chem. 186, 119-170. Izard, T., Lawrence, M. C., Malby, R., Lilley, G. G. & Colman, P. M. (1994). The three-dimensional structure of N-acetylneuraminate lyase from Escherichia coli. Structure 2, 361-369. Jancarik, J. & Kim, S.-H. (1991). Sparse matrix sampling: a screening method for crystallization of proteins. J. Appl. Cryst. 24, 409-411. Jia, J., Huang, W., Schörken, U., Sahm, H., Sprenger, G. A., Lindqvist, Y. & Schneider, G. (1996). Crystal structure of transaldolase B from Escherichia coli suggests a circular permutation of the α/β barrel within the class I aldolase family. Structure 4, 715-724. Jia, J., Schörken, U., Lindqvist, Y., Sprenger, G. A. & Schneider, G. (1997). Crystal structure of the reduced Schiff-base intermediate complex of transaldolase B from Escherichia coli: Mechanistic implications for class I aldolases. Protein Sci. 6, 119-124. Johnson, A. E. & Tanner, M. E. (1998). Epimerization via carbon-carbon bond cleavage. L-Ribulose5-phosphate 4-epimerase as a masked class II aldolase. Biochemistry 37, 5746-5754. Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. Sect. A 47, 110-119. Kabsch, W. (1988a). Automatic indexing of rotation diffraction patterns. J. Appl. Cryst. 21, 67-71. Kabsch, W. (1988b). Evaluation of single crystal X-ray diffraction data from a position-sensitive detector. J. Appl. Cryst. 21, 916-924. Kabsch, W. & Sander, C. (1983). Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577-2637. Kim, H., Certa, U., Döbeli, H., Jakob, P. & Hol, W. G. J. (1998). Crystal structure of fructose-1,6bisphosphate aldolase from the human malaria parasite Plasmodium falciparum. Biochemistry 37, 4388-4396. 165 Kimura, E., Gotoh, T., Koike, T. & Shiro, M. (1999). Dynamic enolate recognition in aqueous solution by zinc(II) in a phenacyl-pendant cyclen complex: Implications for the role of zinc(II) in class II aldolases. J. Am. Chem. Soc. 121, 1267-1274. Kirkpatrick, S., Gelatt, C. D. & Vecchi, M. P. (1983). Optimization by simulated annealing. Science 220, 671-680. Kleywegt, G. J. & Brünger, A. T. (1996). Checking your imagination: applications of the free R value. Structure 4, 897-904. Kragl, U., Gödde, A., Wandrey, C., Lubin, N. & Augé, C. (1994). New synthetic applications of sialic acid aldolase, a useful catalyst for KDO synthesis. Relation between substrate conformation and enzyme stereoselectivity. J. Chem. Soc. Perkin Trans. 1, 119-124. Kraulis, P. J. (1991). MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24, 946-950. Krishnan, B. R., Blakesley, R. & Berg, D. E. (1991). Linear amplification DNA sequencing from single phage plaques and bacterial colonies. Nucleic Acids Res. 19, 1153. Krishnan, G. & Altekar, W. (1991). Unusual class I fructose bisphosphate aldolase from the halophilic archaebacterium Haloarcula vallismortis. Eur. J. Biochem. 195, 343-350. Kunkel, T. A. (1985). Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. 82, 488-492. Kunkel, T. A., Roberts, J. D. & Zakour, R. A. (1987). Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 154, 367-382. Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680-685. Lamzin, V. S. & Wilson, K. S. (1993). Automated refinement of protein models. Acta Crystallogr. Sect. D 49, 129-147. Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283-291. Liljas, A., Kannan, K. K., Bergstén, P.-C., Waara, I., Fridborg, K., Strandberg, B., Carlbom, U., Järup, L., Lövgren, S. & Petef, M. (1972). Crystal structure of human carbonic anhydrase C. Nature New Biol. 235, 131-137. Lin, H. C., Lei, S. P., Studnicka, G. & Wilcox, G. (1985). The araBAD operon of Salmonella typhimurium LT2. III. Nucleotide sequence of araD and its flanking regions, and primary structure of its product L-ribulose-5-phosphate 4-epimerase. Gene 34, 129-134. Lindahl, T. (1974). An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc. Natl. Acad. Sci. USA 71, 3649-3653. Littlechild, J. A. & Watson H. C. (1993). A data-based reaction mechanism for type I fructose bisphosphate aldolase. Trends Biochem. Sci. 18, 36-39. Lu, Z. & Lin E. C. C. (1989). The nucleotide sequence of Escherichia coli genes for L-fucose dissimilation. Nucleic. Acids Res. 17, 4883-4884. 166 Marsh, J. J. & Lebherz, H. G. (1992). Fructose-bisphosphate aldolases: an evolutionary history. Trends Biochem. Sci. 17, 110-113. Mavridis, I. M., Hatada, M. H., Tulinski, A. & Lebioda, L. (1982). Structure of 2-keto-3-deoxy-6phosphogluconat aldolase at 2.8 Å resolution. J. Mol. Biol. 162, 419-444. McPherson, A. (1990). Current approaches to macromolecular crystallization. Eur. J. Biochem. 189, 1-23. Merkel, I. (1999). Mutagenese, röntgenographische und kinetische Untersuchungen an Rhamnulose-1phosphat-Aldolase aus Escherichia coli. Diplomarbeit, Universität Freiburg im Breisgau. Merritt, E. A. & Bacon, D. J. (1997). RASTER 3D: Photorealistic molecular graphics. Methods Enzymol. 277, 505-524. Messing, J. (1983). New M13 vectors for cloning. Methods Enzymol. 101, 20-78. Michal, G. & Lang, G. (1974). L-(-)-Glycerin-3-phosphat. In: Methoden der enzymatischen Analyse. (Bergmeyer, H. U., Hrsg.), Vol. 2, pp. 1460-1463, VCH, Weinheim. Michaelis, L. & Menten, M. L. (1913). Kinetik der Invertinwirkung. Biochem. Z. 49, 333-369. Mineno, J., Fukui, H., Ishino, Y. & Shinagawa, H. (1990). Nucleotide sequence of the araD gene of Escherichia coli K12 encoding the L-ribulose 5-phosphate 4-epimerase. Nucleic Acids Res. 18, 6722-6722. Morris, A. J. & Tolan, D. R. (1993). Site-directed mutagenesis identifies Aspartate 33 as a previously unidentified critical residue in the catalytic mechanism of rabbit aldolase A. J. Biol. Chem. 268, 1095-1100. Müller-Dieckmann, C. (1994). Gezielte Mutagenese, Präparation, kinetische Untersuchungen sowie Kristallisation von Fuculose-1-phosphat Aldolase aus Escherichia coli. Diplomarbeit. Universität Freiburg im Breisgau. Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D 53, 240-255. Nakamaye, K. L. & Eckstein, F. (1986). Inhibition of restriction endonuclease Nci I cleavage by phosphorothioate groups and its application to oligonucleotide-directed mutagenesis. Nucleic Acids Res. 14, 9679-9698. Ozaki, A., Toone, E. J., von der Osten, C. H., Sinskey, A. J. & Whitesides, G. M. (1990). Overproduction and substrate specificity of a bacterial fuculose-1-phosphate aldolase: a new enzymatic catalyst for stereocontrolled aldol condensation. J. Am. Chem. Soc. 112, 4970-4971. Petersen, M., Zannetti, M. T. & Fessner, W.-D. (1997). Tandem asymmetric C-C bond formations by enzyme catalysis. Top. Curr. Chem. 186, 87-117. Penhoet, E. E., Kochman, M. & Rutter, W. J. (1966). Molecular and catalytic properties of aldolase C. Biochemistry 8, 4391-4402. Penhoet, E. E., Rajkumar, T. & Rutter, W. J. (1969). Multiple forms of aldolase in mammalian tissues. Proc. Natl. Acad. Sci. USA 56, 1275-1282. Perham, R. N. (1990). The fructose-1,6-bisphosphate aldolases: same reaction, different enzymes. Biochem. Soc. Trans. 18, 185-187. 167 Ponder, J. W. & Richards, F. M. (1987). Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J. Mol. Biol. 195, 775-791. Powell, M. J. D. (1977). Restart procedures for the conjugate gradient method. Mathematical Programming 12, 241-254. Plater, A., Zgiby, S. M., Thomson, G. J., Qamar, S., Wharton, C. W. & Berry, A. (1999). Conserved residues in the mechanism of E. coli class II FBP-aldolase. J. Mol. Biol. 285, 843-855. Read, R. J. (1986). Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. Sect. A 42, 878-884. Read, R. J. (1990). Structure factor probabilities for related structures. Acta Crystallogr. Sect. A 46, 900-912. Rellos, P., Ali, M., Vidailhet, M., Sygusch, J. & Cox, T. M. (1999). Alteration of substrate specificity by a naturally-occuring aldolase B mutation (Ala337→Val) in fructose intolerance. Biochem. J. 340, 321-327. Rice, L. M. & Brünger, A. T. (1994). Torsion angle dynamics: reduced variable conformational sampling enhances crystallographic refinement. Proteins: Struct. Funct. Genet. 19, 277-290. Righetti, P. G., Gianazza, E., Gelfi, C. & Chiari, M. (1990). In Gel electrophoresis of proteins: a practical approach, (Hames, B. D. & Rickwood, D., eds.) 2nd ed., pp. 149-214, Oxford University Press, Oxford. Rutter, W. J. (1964). Evolution of aldolases. Federation Proc. 23, 1248-1257. Rychlik, W. (1992). Primer Analysis Software: Oligo 4.1. Plymouth, England. Sagner, G. (1993). Nichtradioaktive Cycle-Sequenzierung mit dem DIG Taq DNA sequencing kit. Boehringer Mannheim Biochemica Information 91, 19-21. Saiki, R. K., Gelfand, D. H., Stoffel, S., Scharf, S. J., Higuchi, R., Horn, G. T., Mullis, K. B. & Ehrlich, H. A. (1988). Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239, 487-491. Salo, W. L., Fossitt, D. D., Bevill, R. D., Kirkwood, S. & Wood, W. A. (1972). L-Ribulose 5phosphate 4-epimerase from Aerobacter aerogenes. Kinetic isotope effect with tritiated substrate. J. Biol. Chem. 247, 3098-3100. Sambrook, J., Fritsch E. F. & Maniatis, T. (1989). Molecular cloning: a laboratory manual, 2nd ed., Cold Spring Harbor Laboratory Press, New York. Sanger, F., Nicklen, F. & Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74, 5463-5467. Sanger, F. (1981). Determination of nucleotide sequences in DNA. Science 214, 1205-1210. Sayers, J. R. & Eckstein, F. (1989). Site-directed mutagenesis, based on the phosphorothioate approach. In Protein function: a practical approach (Creighton, T. E., ed.), pp. 279-295, Oxford University Press, Oxford. Sayers, J. R. & Eckstein, F. (1991). A single-strand specific endonuclease activity copurifies with overexpressed T5 D15 exonuclease. Nucleic Acids Res. 19, 4127-4132. 168 Schneider, A. (1994). Isolierung und Charakterisierung der L-Fuculose-1-phosphat-Aldolase und der Rhamnulose-Kinase. Enzymatische Synthese seltener Ketose-phosphate. Dissertation. Universität Freiburg im Breisgau. Sharp, P. A., Sudgen, B. & Sambrook, J. (1973). Detection of two restriction endonuclease activities in Haemophilus parainfluenzae using analytical agarose ethidium bromide electrophoresis. Biochemistry 12, 3055-3063. Sternbach, H. (1991). Chromographische Methoden in der Biochemie, Thieme Verlag, Stuttgart. Stribling, D. & Perham, R. N. (1973). Purification and characterization of two fructose diphosphate aldolases from Escherichia coli (Crookes' strain). Biochem. J. 131, 833-841. Sygusch, J., Beaudry, D. & Allaire, M. (1987). Molecular architecture of rabbit muscle aldolase at 2.7 Å resolution. Proc. Natl. Acad. Sci. USA 84, 7846-7850. Takahashi, I., Takasaki, Y. & Hori, K. (1989). Site-directed mutagenesis of human aldolase isoenzymes: the role of Cys-72 and Cys-338 residues in aldolase A and the carboxy-terminal Tyr residues in aldolases A and B. J. Biochem. 105, 281-286. Taylor, J., Ott, J. & Eckstein, F. (1985). The rapid generation of oligonucleotide-directed mutations at high frequency using phosphorothioate-modified DNA. Nucleic Acids Res. 13, 8765-8785. Vogelstein, B. & Gillespie, D. (1979). Preparative and analytical purification of DNA from agarose. Proc. Natl. Acad. Sci. USA 76, 615-619. Westermeier, R. (1990). Elektrophorese-Praktikum, VCH, Weinheim. Whitesides, G. M. & Wong, C.-H. (1985). Enzyme in der organischen Synthese. Angew. Chem. 97, 617-638. Wilson, A. C. C. (1949). The probability distributions of X-ray intensities. Acta Crystallogr. 2, 318321. Wishart, D. S., Boyko R. F. & Sykes, B. D. (1994). Constrained multiple sequence alignment using XALIGN. Computer Appl. Biosci. 10, 121-132. Wong, C.-H., Halcomb, R. L., Ichikawa, Y. & Kajimoto, T. (1995a). Enzyme in der organischen Synthese: das Problem der molekularen Erkennung von Kohlenhydraten (Teil I). Angew. Chem. 107, 453-474. Wong, C.-H., Halcomb, R. L., Ichikawa, Y. & Kajimoto, T. (1995b). Enzyme in der organischen Synthese: das Problem der molekularen Erkennung von Kohlenhydraten (Teil II). Angew. Chem. 107, 569-593. 169 7 Anhang 7.1 DNA- und Aminosäuresequenz der FucA wie in M13mp19 kloniert 5’-GG TGT AAA ACG ACG GCC AGT GAA TTC ATG GAA CGA AAT AAA CTT GCT CGT CAG ATT ATT GAC ACT TGC CTG GAA Met Glu Arg Asn Lys Leu Ala Arg Gln Ile Ile Asp Thr Cys Leu Glu 1 10 ATG ACC CGC CTG GGA CTG AAC CAG GGG ACA GCG GGG AAC GTC AGT GTA Met Thr Arg Leu Gly Leu Asn Gln Gly Thr Ala Gly Asn Val Ser Val 20 30 CGT TAT CAG GAT GGG ATG CTG ATT ACG CCT ACA GGC ATT CCA TAT GAA Arg Tyr Gln Asp Gly Met Leu Ile Thr Pro Thr Gly Ile Pro Tyr Glu 40 AAA CTG ACG GAG TCG CAT ATT GTC TTT ATT GAT Lys Leu Thr Glu Ser His Ile Val Phe Ile Asp 50 -------FucA-Prim1------> GAG GAA GGA AAG CTC CCC TCA AGC GAA TGG CGT Glu Glu Gly Lys Leu Pro Ser Ser Glu Trp Arg 70 GGC AAC GGT AAA CAT Gly Asn Gly Lys His 60 TTC CAT ATG GCA GCC Phe His Met Ala Ala 80 TAT CAA AGC AGA CCG GAT GCC AAC GCG GTT GTT CAC AAT CAT GCC GTT Tyr Gln Ser Arg Pro Asp Ala Asn Ala Val Val His Asn His Ala Val 90 CAT TGC ACG GCA GTT TCC ATT CTT AAC CGA TCG ATC CCC GCT ATT CAC His Cys Thr Ala Val Ser Ile Leu Asn Arg Ser Ile Pro Ala Ile His 100 110 TAC ATG ATT GCG GCG GCT GGC GGT AAT TCT ATT CCT TGC GCG CCT TAT Tyr Met Ile Ala Ala Ala Gly Gly Asn Ser Ile Pro Cys Ala Pro Tyr 120 ----FucA-Prim2 GCG ACC TTT GGA ACA CGC GAA CTT TCT GAA CAT GTT G CG CTG GCT CTC Ala Thr Phe Gly Thr Arg Glu Leu Ser Glu His Val Ala Leu Ala Leu 130 140 --------> AAA AAT CGT AAG GCA ACT TTG TTA CAA CAT CAT GGG CTT ATC GCT TGT Lys Asn Arg Lys Ala Thr Leu Leu Gln His His Gly Leu Ile Ala Cys 150 160 GAG GTG AAT CTG GAA AAA GCG TTA TGG CTG GCG CAT GAA GTT GAA GTG Glu Val Asn Leu Glu Lys Ala Leu Trp Leu Ala His Glu Val Glu Val 170 CTG GCG CAA CTT TAC CTG ACG ACC CTG GCG ATT ACG GAC CCG GTG CCA Leu Ala Gln Leu Tyr Leu Thr Thr Leu Ala Ile Thr Asp Pro Val Pro 180 190 GTG CTG AGC GAT GAA GAG ATT GCC GTA GTG CTG GAG AAA TTC AAA ACC Val Leu Ser Asp Glu Glu Ile Ala Val Val Leu Glu Lys Phe Lys Thr 200 TAT GGG TTA CGA ATT GAA GAG TAA-3´ Tyr Gly Leu Arg Ile Glu Glu 210 215 170 7.2 Vektoren Der Phage M13mp- (aus Fa. Amersham Pharmacia Biotech, 1998) mit der Sequenz der multiple cloning site in M13mp18 (In M13mp19 liegt die multiple cloning site invertiert vor, d. h. 5’- und 3’-Enden sind vertauscht) Der Vektor pKK223-3 (aus Fa. Amersham Pharmacia Biotech, 1998) Danksagung Die vorliegende Arbeit wurde im Arbeitskreis von Prof. Dr. Georg E. Schulz am Institut für Organische Chemie und Biochemie der Albert-Ludwigs-Universität Freiburg im Breisgau angefertigt. Bei Prof. Schulz möchte ich mich für die Vergabe des interessanten und vielseitigen Themas sowie seine immerwährende Unterstützung und Diskussionsbereitschaft bedanken. Herrn Dr. Matthias Dreyer danke ich zum einen dafür, daß er mit der Aufklärung der Kristallstruktur der L-Fuculose-1-phosphat-Aldolase die Grundlage für diese Arbeit geschaffen hat, und zum anderen auch für die zahlreichen Tips und hilfreichen Diskussionen zu Beginn der Arbeit. Herrn Dr. Claudius Goße im Arbeitskreis von Prof. Dr. Wolf-Dieter Fessner sei für die Selektivitätsmessungen zahlreicher Mutanten gedankt. Prof. Fessner möchte ich für die gute Zusammenarbeit im Rahmen dieses Projektes danken sowie dafür, daß mir die Möglichkeit gegeben wurde, in seinem damaligen Arbeitskreis an der RWTH-Aachen die Synthese von Fuculose-1-phosphat durchzuführen. Dr. Clemens Vonrhein und Dr. Laurent Maveyraud danke ich für die Bereitstellung hervorragend ausgearbeiteter Skripte, die das Arbeiten an den Computern erheblich vereinfacht haben. An dieser Stelle sei auch Markus Krömer gedankt, der sehr viel Zeit investierte, um das Computersystem der Abteilung immer auf dem neuesten Stand zu halten. Darüber hinaus war er auch bei wissenschaftlichen Fragen immer der richtige Ansprechpartner und das nicht nur wenn es um das Thema Aldolasen ging. Dr. Benedikt Schmid danke ich für die geduldige Einweisung in die molekularbiologischen Arbeitsmethoden. An dieser Stelle nicht unerwähnt bleiben darf Gerd Laule, der immer sehr große Sorgfalt walten ließ, wenn es darum ging, optimale Bedingungen für die Proteinpräparation zu schaffen. Besonders bedanken möchte ich mich vor allem bei Stjepan Jelakovic und Christoph Müller-Dieckmann: dafür, daß sie mir bei Fragen aller Art immer mit Rat und Tat zur Seite standen und für die Freundschaft, die sie mir während meiner Studien- und Promotionszeit entgegenbrachten. Vielen lieben Dank an Stephanie Jürgens für die Hilfe beim Korrekturlesen dieser Arbeit! Allen Mitgliedern des Arbeitskreises sei für das angenehme Arbeitsklima gedankt.