9 Reaktionen CH acider Verbindungen
Werbung

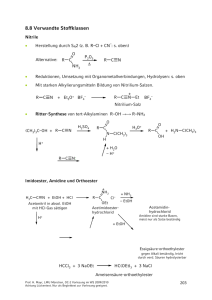
9 Reaktionen CH acider Verbindungen 9.1 Brønsted-Aciditäten Die Stärke von Brønsted-Säuren wird durch ihren pKa-Wert quantifiziert, der stark vom Lösungsmittel abhängt. Beachten Sie, dass die Konzentration des Lösungsmittels nicht in die Gleichgewichtskonstante Ka eingeht. oder KAH = AH + H2O A– + H3O+ BH+ + H2O B + H3O+ [A–]·[H3O+] ; [AH] pKAH = –lg KAH [B]·[H3O+] [BH] KBH + = pKBH + = –lg KBH+ allgemein: pKa = –lg Ka Als Faustregel sollten Sie sich merken: O H 3C R–SO3H R–CO2H Ph–OH R–OH + + Ph NH3 R NH3 pKa ~0 ~5 ~10 ~15 O C R H3C ~20 C OR Ph–NH2 NH3 in DMSO in fl. NH3 ~30 ~35 ~25 Bitte prägen Sie sich keine pKB-Werte ein, weil Sie sonst die beiden Skalen durcheinander mischen! pKa-Werte weiterer CH-acider Verbindungen O O H3C–NO2 H H 9 10.2 H H 16.0 H H C C H N N O O O OEt EtO O H H H H 11.2 11.0 13.3 O S Ph C C H H3C–CN H C C H 21 ~25 ~26 H3C H H OEt CH3 33 H3C–CH3 H ~44 ~50 Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 209 9.2 Halogenierungen 4 3 2 γ β α 1 O R CH2 CH2 CH2 C Beachten Sie die Zählweise: H Die Abspaltung des α-Protons ist möglich, weil dabei ein Resonanz-stabilisiertes Carbanion entsteht. H R C H O + HB+ C ..B R' Enolat-Ion Die Bezeichnung Enolat leitet sich vom Enol ab, das bei gewöhnlichen Aldehyden und Ketonen in kleiner Gleichgewichtskonzentration neben der Carbonylkomponente vorliegt (Keto-Enol-Tautomerie). H R C H O C H Carbonyl-Form Enol-Form Beim Behandeln von Acetaldehyd mit Cl2, Br2 oder I2 im Alkalischen entsteht durch elektrophile Halogenierung des intermediären Enolations zunächst Chloracetaldehyd (bzw. die entsprechende Brom- oder Iodverbindung), der allerdings nicht isoliert werden kann, sondern zu Trichloracetaldehyd (Chloral) weiterreagiert. Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 210 H O C H + H2O C H H + O H Cl H2O Cl O C C Cl Cl 2 Cl2/OH– H Cl Chloral Chloralhydrat OH– OH– Haloform-Reaktion: Wird die Halogenierung in Gegenwart von Kalilauge durchgeführt, kommt es zur Spaltung der CC-Bindung und Bildung von HCHal3. Die Haloform-Reaktion dient zum Nachweis von Methylketonen und sekundären Alkan-2-olen. O OH R CH CH3). (Strukturelemente R C CH3 und O H3C C CH3 3 I2/3 OH– OH– Die Halogenierung von Carbonylverbindungen kann im Sauren auf der Stufe der Monohalogenierungsprodukte angehalten werden. Unter Säurekatalyse erfolgt hierbei die Bildung einer kleinen Gleichgewichtskonzentration des Enols, das mit Halogenen reagiert. Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 211 Halogenierung im Sauren: ·· O: H H C H H+ C H C H H H O H C C H H O C H H – H+ H H C H O H H C C H H ·· :Br ·· – H+ H H C C H ·· Br : ·· O C H Br OH H3O+ O H C C H O H O H Br2 + HBr isolierbar, langsamere Folgereaktion + Br2/H+ O Br Br α,α-Dichloride O R PCl5 R' Cl R Cl R' + POCl3 Die Reaktion mit PBr5 liefert schlechte Ausbeuten der analogen Dibromide. BBr3 reagiert in dieser Weise mit Ar Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. O C . H 212 9.3 Alkylierungen CH-acider Verbindungen H C C H 1) NaNH2 1) EtMgBr 2) R–Hal 2) R–Br Zur Alkylierung von Ketonen kann mit starker Base (z. B. Li N(iPr)2) = LDA) quantitativ das Enolat-Ion erzeugt werden. O H H Cl LDA Et2O Et2O Die Deprotonierung des Ketons durch LDA ist hierbei schneller als die Reaktion des gebildeten Enolats mit noch unumgesetztem Keton. O O Li O langsamer LiO Unsymmetrische Ketone lassen sich regioselektiv deprotonieren. Details dazu in Kap. 9.4. O O ? O – – Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. oder 213 Wegen der höheren Elektrophilie von Aldehyden ist die Herstellung von LithiumEnolaten aus Aldehyden i. A. nicht möglich, da Aldehyde rascher mit Enolat-Ionen reagieren als sie durch LDA deprotoniert werden. O Li AldolSelbstkondensation O H R R Carbonsäuren werden durch LDA zuerst an der Carboxylgruppe deprotoniert, bevor durch ein zweites Equivalent LDA der α-Waserstoff abgespalten wird. H ·· O: R C C :O H H ·· 2 LiN(iPr)2 – ·· 2 Li+ R C ·· O: C : H :O ·· – R'–X R'X = prim. Alkyl-, Allylod. Benzylhalogenid Alternativ zu Lithium-Enolaten können Enamine Equivalente von Enolat-Ionen verwendet werden. – ·· :O ·· ·· R'2N ·· :O und Silylenolether als ·· Me3SiO ·· – R R R ·· R Enolat-Ion R R Enamin R R Silylenolether Die zwitterionische Grenzstruktur weist auf die Nucleophilie des β-Kohlenstoffs hin. Zur Herstellung von Enaminen aus Carbonylverbindungen und sekundären Aminen s. Kap. 7 (S. 161) und Praktikum (Präparat 28). Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 214 O N H + ·· N + kat. H+ N + H2O H2O Br In gleicher Weise wie Allylhalogenide reagieren andere SN2-aktive Verbindungen, wie Benzylhalogenide und α-Halogencarbonylverbindungen. [H+] ·· iBu2N O OEt Br H2O Das nach der Hydrolyse erhaltene Produkt entspricht also dem einer α-Alkylierung der Carbonylverbindung. Einfache Alkylhalogenide, wie Methyliodid, greifen zu einem erheblichen Ausmaß den Stickstoff des Enamins an. O ··N Alkyl X X Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. X X R 215 Die Acylierung von Enaminen erlaubt die Herstellung von 1,3-Diketonen. Die Mechanismen sind sehr komplex (über intermediäre N-Acylierung oder über Ketene und [2+2]-Cycloaddition). Der nachfolgend formulierte „Mechanismus“ ist nur die Rationalisierung des Endergebnisses. O ·· N O + O C R Cl H2O O R Die einfachste Herstellung von Silylenolethern gelingt durch Erwärmen von Carbonylverbindungen mit Me3SiCl und Triethylamin. O NEt3 Cl–SiMe3 Silylenolether sind deutlich weniger nucleophil als Enamine oder gar LithiumEnolate. Daher reagieren sie nur mit stärkeren Elektrophilen, z. B. tert.Alkylkationen, die intermediär aus tertiären Alkylchloriden und Lewis-Säuren erzeugt werden. OSiMe3 Cl [TiCl4] Silylenolether sind die Substrate der Wahl für die tert.-Alkylierung von Carbonylverbindungen, da tert.-Alkylierungen von Lithium-Enolaten oder Enaminen nicht gelingen (Warum ?). Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 216 Alternative: Malonester- und Acetessigester-Synthesen H H C NaOEt/ CO2Et EtOH R–X konz. HCl O R H CO2Et Malonsäurediethylester O H Δ O HO 1) NaOEt/EtOH 2) R'X konz. HCl 115 °C Die Malonester-Synthese ermöglicht die Umwandlung R-X R-CH2-COOH sowie die Herstellung dialkyl-substituierter Essigsäuren R CH CO2H . R' Da der Alkylierungsschritt eine SN2-Reaktion ist, sind primäre Alkylderivate R–X als Reaktionspartner bevorzugt. Wie können Sie durch eine Malonestersynthese Cycloalkancarbonsäuren herstellen? CO2H Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 217 Acetessigester-Synthese H H NaOEt/ EtOH CO2Et C C 1) verd. NaOH 2) H2SO4/25 °C R–X O R CH3 CH3 (Ketonspaltung) O Acetessigester 1) NaOEt/EtOH 2) R'X 1) verd. NaOH 2) H2SO4/25 °C O Die Acetessigester-Synthese ermöglicht die Umwandlung R-X R CH2 C CH3 O sowie die Herstellung von Verbindungen des Typs R CH C CH3 . R' Bisenolate Verwendet man einen Überschuss sehr starker Basen, erfolgt eine zweifache Deprotonierung. Die Alkylierung erfolgt dann zunächst am basischeren C-Atom. O LDA O tBuO Li ·· O: ·· LDA O tBuO tBuO Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. Li ·· O: ·· R–X R'–X ·· O: ·· Li 218 Regioselektivität der Alkylierung von Enolaten Elektrophile können Enolat-Ionen am Kohlenstoff oder Sauerstoff angreifen. E+ : O O O E oder E O-Angriff Enolat-Ion O C-Angriff Die ambidente Reaktivität der Enolat-Ionen wird üblicherweise durch Pearsons HSAB-Konzept erklärt. Harte Elektrophile (H+, Cl-SiMe3) greifen bevorzugt am harten Sauerstoff-Zentrum des Enolats an, während weiche Elektrophile R–I bevorzugt am Kohlenstoff angreifen. O + H3O+ O + Me3SiCl Bei Alkylierungen treten häufig Gemische auf. O – Na+ O Ph Ph Na+ – C4H9-X OC4H9 Ph O Ph C4H9 X I Br Cl OSO3CH3 OTos OPO3Bu2 F Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. O-Angriff HMPT DMSO 15 10 40 23 67 46 83 –– 85 44 93 78 96 –– Et2O 1 5 18 11 20 36 219 Generell gilt: Je freier das Anion, umso mehr O-Angriff. Ph O (MeO)2SO2 DME Li + Ph Ph O OH ohne 12-Krone 4 45% 55% + 12-Krone 4 75% 25% 9.4 Reaktionen CH-acider Verbindungen mit der Carbonylgruppe von Aldehyden und Ketonen Ph C C H 1) BuLi 2) O R R' 3) H3O+ + R + H+ [Base] O Fulven – H2O R' R R' Aldol-Reaktion OH– H 2O O 2 H OH O H 5 °C, 4–5 h (50 %) Aldol Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. [Essigsäure] 80–100 °C O H Crotonaldehyd 220 Aldol-Reaktion bezeichnet allgemein die Addition eines Enols oder Enolat-Ions eines Aldehyds oder Ketons an die Carbonylgruppe eines anderen Aldehyds oder Ketons. Führt man die Aldol-Reaktion bei höheren Temperaturen durch (100 °C), kommt es zur Basen-katalysierten Wasser-Abspaltung und man erhält α,β-ungesättigte Aldehyde (Ketone). Aldol-Addition und Aldol-Kondensation Mechanismus der Aldolreaktion am Beispiel der Basen-katalysierten Aldoladdition des Acetaldehyds. • Bildung des Enolats • Nucleophiler Angriff des Enolats auf die Carbonylgruppe eines zweiten Aldehyds • Protonenübertragung H O C C H H + O H H δ– O δ+ C H O C H H H OH H C C C H H C H +H2O, –OH– H H H Aldol (enthält Aldehyd und Alkohol-Funktion) Die vorstehend beschriebene Aldol-Addition verläuft reversibel; vor allem wenn Ketone als Carbonylkomponenten eingesetzt werden, liegt das Gleichgewicht weitgehend auf der Seite der Reaktanten. Bei Temperaturerhöhung schließt sich an die Aldol-Addition ein α,β-ungesättigte Eliminierungsschritt an (E1cB-Reaktion), wobei eine Carbonylverbindung entsteht. Man spricht dann von der Aldol-Kondensation (Kondensation bezeichnet die Vereinigung zweier Moleküle unter Abspaltung eines kleinen Teilchens, z. B. Wasser). Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 221 Mechanismus der Wasserabspaltung im Basischen: O H H H H O C C H C C H H OH H trans-But-2-enal (Crotonaldehyd) Die Aldolkondensation kann auch durch Säuren katalysiert werden. Unter sauren Bedingungen greift das Enol als Nucleophil an der protonierten Carbonylgruppe des zweiten Aldehyd-Moleküls an. Bei der Säure-katalysierten Aldol-Reaktion kommt es fast immer zur nachfolgenden Wasser-Abspaltung, und die primär gebildeten Aldole lassen sich nicht isolieren. H HO C C H H H H O C CH3 Crotonaldehyd Ungünstige Gleichgewichtslage bei Aldol-Additionen von Ketonen OH O O 2 Gleichgewicht wurde von beiden Seiten aus eingestellt. [BaO] 94 % 6% Additionsprodukt Unter schärferen Bedingungen kommt es zur Kondensationsprodukts, wenn das Wasser entfernt wird. 2 O [NaOH] Bildung des Aldol- + H2O Δ Kondensationsprodukt Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 222 Gekreuzte Aldol-Reaktion: OH R O R H OH– O R' H R' R + + OH H + H O R' OH O O R OH + H O R' H R R' 4 Aldol-Additionsprodukte Falls eine der Carbonylkomponenten keinen α-Wasserstoff besitzt, reduziert sich die Zahl möglicher Produkte auf 2. Eine selektive Reaktion wird möglich, wenn die enolisierbare Komponente langsam zu einer nicht-enolisierbaren Carbonylverbindung gegeben wird. O O + H H NaOH H2O O Δ H O H + H 65 % + H2O H Gekreuzte Aldol-Kondensationen erfolgen dann selektiv, wenn • eine der Carbonylkomponenten keinen α-Wasserstoff besitzt und • die Carbonylkomponente ohne α-Wasserstoff elektrophiler ist. Aromatische Aldehyde und Formaldehyd eignen sich daher besonders gut für gekreuzte Aldol-Reaktionen. - Herstellung von Dibenzalaceton aus Aceton und Benzaldehyd. H C H O H2C H C O CH2 O C H Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 223 Gezielte Aldol-Reaktionen auch dadurch möglich, dass die CH-acide Komponente mit starken Basen quantitativ ins Enolat überführt wird (s. S. 213). O LDA THF –78 °C ·· :O : Li O H H2O R Neben LDA finden dabei Anwendung: Diese Basen werden aus den entsprechenden Aminen und n-BuLi (bzw. NaH) hergestellt. Regioselektivität der Deprotonierung Stehen mehrere Protonen zur Verfügung, stellt sich die Frage, wo eine Base ein Proton abstrahieren wird. Die Struktur des gebildeten Enolats hängt häufig davon ab, ob die Deprotonierung unter reversiblen (thermodynamische Kontrolle) oder irreversiblen Bedingungen (kinetische Kontrolle) erfolgt. Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 224 Ob es sich bei einer Reaktion um eine thermodynamisch oder kinetisch kontrollierte Reaktion handelt, kann beeinflusst werden durch • die Base, • das Lösungsmittel, • die Temperatur • sowie die Struktur des Edukts. LDA liefert in THF immer das kinetische Produkt. Dies liegt daran, dass die Lithiumverbindung an den Sauerstoff koordiniert und so als Base das direkt benachbarte Proton abstrahiert. Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 225 LDA THF O O LDA/THF O Es entsteht ausschließlich das kinetische Produkt, obwohl das thermodynamische deutlich besser resonanzstabilisiert ist. O kinetisch bevorzugt thermodynamisch günstiger Durch Silylierung lassen sich die so erhaltenen Enolate fixieren. O O LDA THF, -78°C, Ar H+ -H2O TMSCl TiCl4 O O Si O HO O Manchmal lässt sich die Regioselektivität auch dadurch steuern, dass alternativ unter Basen- oder Säure-Katalyse gearbeitet wird. O H R OH– Ph bevorzugt unter Basen-Katalyse Ph bevorzugt unter Säure-Katalyse :O : ·· R O O H+ H R OH Intramolekulare Aldolkondensationen können zur Synthese 5-, 6- und 7-gliedriger Ringe genutzt werden. O O NaOH NaOH H2O 100 °C H2O 100 °C Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 226 Gezielte Aldol-Reaktion nach Mukaiyama OSiMe3 O Ph H OH 1) TiCl4 + O Ph 2) H2O TiCl4 H2O OSiMe3 Knoevenagel-Kondensation CO2Et R Herstellung von C H R oder C CO2Et H C C CO2H H durch Umsetzung von Carbonylverbindungen mit Malonestern und anderen Verbindungen hoher CH-Acidität. Beachten Sie die Analogie zur gekreuzten Aldol-Kondensation. R O + H R O H + H H H H CO2Et C CO2Et CO2H C CO2H Piperidin Benzol Δ Piperidin Δ R CO2H H CO2H Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. R CO2Et H CO2Et 227 Analoge Reaktionen: O + H C H CN 80 % CN O H O + H H C H Ph Ph 98 % O Pr 74 % H CO2Et CN O Ph 95 % H CO2Et Umsetzungen mit Nitroalkanen O H C N O H H B – HB+ ·· :O: O N O HO O N O Addition Bei aromatischen Aldehyden folgt rasche Dehydratisierung O H CH3NO2 NaOH, MeOH Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. NO2 85 % 228 Perkin-Reaktion Zimtsäure aus Benzaldehyd und Acetanhydrid O O O + Ar CO2H Ar 180 °C O H Na+ –OAc Komplexer Mechanismus; CC-Verknüpfung durch nucleophilen O O Angriff von an der Carbonylgruppe des Aldehyds. C C CH3 H2C O ·· Darzens-Kondensation R Cl O R' + H H O OEt Base (NaNH2 oder KOtBu) R O CO2Et 2) H+ R' H Glycidester B– 1) OH– R O R' H OH– H+ O R O ·· O: ·· R' H Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 229 Reaktionen CH-acider Verbindungen mit Iminium-Ionen (Mannich-Reaktion) Umsetzung eines Aldehyds (meist Formaldehyd) mit einem primären oder sekundären Amin und CH-acider Komponente. O R CH3 ·· H2C=O + HNR2 Formalin-Lsg. oder Paraformaldehyd R NR2 Mannich-Base H+ – H2O Iminium-Ion + ·· + H2C–NR2 O R + H2C=O + HNR2 O H+ CH3 Salz der Mannich-Base • Problem: Statt des Amins kann auch die CH-acide Verbindung mit dem Formaldehyd reagieren: Aldol-Reaktion statt Aminoalkylierung. • Die relative Reaktivität von Amin und CH-acider Verbindung hängt stark vom pH ab. Meist günstige Bedingungen für Mannich-Reaktion, wenn Amin als Hydrochlorid eingesetzt wird. • In Kap. 7 wurde bereits die Mannich-Reaktion mit elektronenreichen Arenen besprochen. Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2009/2010 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 230