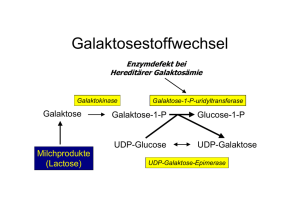
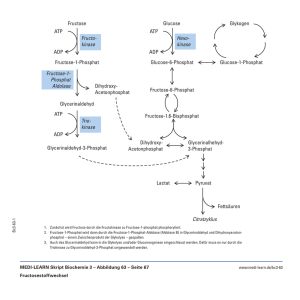
Transiente Mikrokompartimentierung des
Werbung

Transiente Mikrokompartimentierung des pflanzlichen Primärstoffwechsels am Zytoskelett Inaugural-Dissertation zur Erlangung des Doktorgrades des Fachbereiches Biologie/Chemie an der Universität Osnabrück vorgelegt von Anke Scholz, geb. Thilo, aus Berlin Berlin, 2004 Inhaltsverzeichnis I Inhaltsverzeichnis Inhaltsverzeichnis............................................................................................. I 1 Einleitung ................................................................................................... 1 1.1 Mikrokompartimentierung und Channeling ....................................................... 1 1.2 Die Glykolyse ............................................................................................................ 3 1.2.1 1.2.2 1.3 Interaktionen glykolytischer Enzyme......................................................................... 6 Das Enzym Aldolase.................................................................................................... 8 Das pflanzliche Cytoskelett .................................................................................... 11 1.3.1 1.3.2 1.4 2 Mikrotubuli ................................................................................................................12 Mikrofilamente...........................................................................................................12 Fragestellung .......................................................................................................... 15 Material und Methoden........................................................................... 16 2.1 Chemikalien und Materialien ................................................................................ 16 2.2 Pflanzenmaterial: Anzucht und Ernte................................................................... 19 2.3 Molekularbiologische Methoden............................................................................ 19 2.3.1 2.3.2 2.3.3 Bakterien- und Hefestämme ......................................................................................19 Plasmide .....................................................................................................................20 Arbeiten mit Nukleinsäuren ......................................................................................21 2.3.3.1 RNA-Isolierung aus Pflanzenmaterial.................................................................................21 2.3.3.2 Denaturierendes Formaldehyd/Agarose-Gel (RNA-Gel) .....................................................22 2.3.3.3 RNA-Quantifizierung..........................................................................................................23 2.3.3.4 DNA-Quantifizierung..........................................................................................................23 2.3.3.5 Oligonukleotide ...................................................................................................................23 2.3.3.6 Synthese von cDNA.............................................................................................................25 2.3.3.7 DNA-Amplifikation .............................................................................................................25 2.3.3.7.1 Polymerase-Kettenreaktion ..............................................................................................25 2.3.3.7.2 Reverse Transkriptase-PCR .............................................................................................26 2.3.3.8 Reinigung von DNA-Fragmenten .......................................................................................27 2.3.3.9 DNA-Fällung ......................................................................................................................27 2.3.3.10 DNA-Restriktion..................................................................................................................27 2.3.3.11 DNA-Ligation......................................................................................................................28 2.3.3.12 Vektordephosphorylierung ..................................................................................................29 2.3.3.13 Anhängen eines Adenin-Überhangs an ein PCR-Fragment................................................29 2.3.3.14 DNA-Sequenzanalyse ..........................................................................................................29 2.3.3.15 Agarose-Gelelektrophorese und Gelextraktion ....................................................................30 2.3.3.16 Klonierungen ......................................................................................................................31 2.3.3.16.1 Klonierung von ALD_pET-16b...................................................................................31 2.3.3.16.2 Klonierung von ALD_pGBKT7 ..................................................................................32 2.3.3.16.3 Klonierung von ALD_pASK-IBA3 .............................................................................32 2.3.3.16.4 Klonierung von VDAC_pRSET A...............................................................................33 2.3.3.16.5 Klonierung von VDAC_pGBKT7 ...............................................................................34 2.3.3.16.6 Klonierung von Aktin_pGBKT7..................................................................................34 2.3.4 2.3.4.1 Arbeiten mit Bakterien ..............................................................................................35 Bakterienkulturen ...............................................................................................................35 Inhaltsverzeichnis 2.3.4.2 2.3.4.3 2.3.4.4 2.3.4.5 2.3.5 II Herstellung kompetenter E. coli-Zellen ...............................................................................36 Transformation von E. coli-Zellen ......................................................................................38 Blau-Weiß-Selektion ...........................................................................................................40 Plasmidisolierung aus E. coli ..............................................................................................40 Arbeiten mit Hefen.....................................................................................................40 2.3.5.1 Anzucht von S. cerevisiae ....................................................................................................41 2.3.5.2 Herstellung kompetenter Hefezellen und Transformation von Hefen..................................43 2.3.5.3 Plasmidisolierung aus S. cerevisiae .....................................................................................45 2.3.5.4 Herstellung einer cDNA-Bank für das Hefe-2-Hybrid-System ............................................46 2.3.5.4.1 Amplifizierung doppelsträngiger cDNA durch „long distance-PCR“ ................................49 2.3.5.4.2 Reinigung der doppelsträngigen cDNA ............................................................................50 2.3.5.4.3 In vivo-Rekombination.....................................................................................................50 2.3.5.4.4 Ernte der Transformanden................................................................................................51 2.3.5.4.5 Überprüfung der cDNA-Bank-Größe ...............................................................................51 2.3.5.4.6 Test des Fusionsproteinkonstrukts ALD_pGBKT7 auf transkriptionelle Aktivität in S. cerevisiae ........................................................................................................................52 2.3.5.4.7 Toxizitätstest für das Fusionsproteinkonstrukt ALD_pGBKT7 .........................................52 2.3.5.5 Paarung der Hefestämme und Selektion diploider Zellen....................................................53 2.3.5.6 Kontrollen für das Hefe-2-Hybrid-System ...........................................................................55 2.3.6 Biochemische Methoden ............................................................................................57 2.3.6.1 Proteinbestimmung nach BRADFORD...................................................................................57 2.3.6.2 SDS-Gelelektrophorese .......................................................................................................58 2.3.6.3 Western-Blot-Analyse und Immundekoration .....................................................................60 2.3.6.3.1 Färbung geblotteter Proteine mit Ponceau S .....................................................................60 2.3.6.3.2 Immunologischer Nachweis durch Peroxidasereaktion .....................................................61 2.3.6.3.3 Immunologischer Nachweis durch Alkalische-Phosphatase-Reaktion ...............................62 2.3.6.3.4 Immunologischer Nachweis durch Chemolumineszenz.....................................................62 2.3.6.4 Dot-Blot eine Variante des Western-Blots.....................................................................63 2.3.6.5 Far-Western-Blot .............................................................................................................63 2.3.6.6 Heterologe Expression von Proteinen in E. coli BL21(DE3)pLysS......................................63 2.3.6.7 Reinigung der rekombinanten Proteine...............................................................................65 2.3.6.7.1 His-tag-Affinitätschromatographie...................................................................................65 2.3.6.7.2 Reinigung von rekombinantem Protein aus „inclusion bodies“..........................................67 2.3.6.7.3 Reinigung von rekombinantem Protein mit Strep-tag .......................................................68 2.3.6.8 Aldolase-VDAC-Bindeversuch (Affinitätschromatographie) ...............................................69 2.3.7 2.3.8 2.3.9 2.3.10 2.3.11 2.3.12 2.3.13 3 Entsalzen über Dialyse und NAP-5 Säulen................................................................70 Herstellung eines polyklonalen Antiserums ..............................................................70 Copolymerisationsversuche .......................................................................................71 Stöchiometrie der Aldolase-Aktin-Bindung ..............................................................73 Messung der Aldolaseaktivität...................................................................................74 Faktor-Xa -Verdau..................................................................................................76 Bioinformatische Analysen ........................................................................................76 Ergebnisse ................................................................................................ 77 3.1 3.1.1 3.1.2 3.1.3 3.1.4 3.1.5 3.1.6 3.1.7 3.2 3.2.1 3.2.2 3.2.3 Vorarbeiten............................................................................................................. 77 Klonierung des Expressionskonstrukts ALD_pET-16b............................................77 Heterologe Expression und Reinigung von Aldolase (Z. mays) mit His-tag..............78 Faktor-Xa-Verdau .....................................................................................................80 Herstellung eines polyklonalen Antiserums zur Immundetektion von Aldolase......81 Klonierung des Expressionskonstruktes ALD_pASK-IBA3.....................................82 Heterologe Expression und Reinigung von Aldolase (Z. mays) mit Strep-tag ..........83 Katalytische Eigenschaften rekombinanter Aldolase ...............................................85 Copolymerisationsversuche ................................................................................... 87 Einfluss von FBP, F-6P und F-2,6BP auf die Aldolase-Aktin-Bindung....................88 Einfluss des Redox-Milieus auf die Aldolase-Aktin-Bindung ...................................91 Stöchiometrie der Aldolase-Aktin-Bindung ..............................................................94 Inhaltsverzeichnis 3.3 III Ergebnisse der Hefe-2-Hybrid-Analysen............................................................... 96 3.3.1 3.3.2 3.3.3 3.3.4 3.3.5 3.3.6 3.3.7 3.3.8 3.3.9 Klonierung von Aldolase in den Vektor pGBKT7 ....................................................96 Herstellung einer cDNA-Bank ...................................................................................96 Test auf transkriptionale Aktivität des Fusionsproteinkonstruktes und Toxizitätstest ..............................................................................................................97 Analyse der Kontrollen zu den Hefe-2-Hybrid-Versuchen.......................................99 Hefe-2-Hybrid-Analyse der cDNA-Bank mit dem Fusionskonstrukt ALD_pGBKT7 .........................................................................................................101 Analyse der pGADT7-cDNA-Bank-Konstrukte aus den blauen Hefekolonien......102 Klonierung des Expressionskonstruktes VDAC_pRSET A....................................104 Klonierung von VDAC in den Vektor pGBKT7 .....................................................105 Klonierung von Aktin in den Vektor pGBKT7.......................................................106 3.4 Heterologe Expression und Reinigung von VDAC (Z. mays) mit His-tag .......... 107 3.5 Far-Western-Blot.................................................................................................. 109 3.6 Aldolase-VDAC-Bindeversuch (Affinitätschromatographie) ............................. 110 4 Diskussion .............................................................................................. 114 4.1 Sequenzen der Expressionskonstrukte von Aldolase und VDAC....................... 114 4.2 Katalytische Eigenschaften rekombinanter Aldolasen ....................................... 116 4.3 Immunologischer Nachweis von Aldolase ........................................................... 116 4.4 Copolymerisationsversuche ................................................................................. 117 4.4.1 4.4.2 4.5 Einfluss von FBP, F-6P und F-2,6BP auf die Aldolase-Aktin-Bindung..................117 Einfluss des Redox-Milieus auf die Aldolase-Aktin-Bindung .................................119 Protein-Protein-Interaktionen im Hefe-2-Hybrid-System.................................. 121 4.5.1 4.5.2 4.5.3 4.5.4 4.6 Aldolase-Aldolase-Interaktion .................................................................................121 VDAC-Aldolase-Interaktion ....................................................................................121 Interaktionen mit mRNA-Sequenzen unbekannter Funktion und einem Translationsinitiationsfaktor ...................................................................................123 Vermutlich falsch positive Interaktionen ................................................................123 Überprüfung der Protein-Protein-Interaktionen ................................................ 125 5 Zusammenfassung ................................................................................. 127 6 Literatur................................................................................................. 129 7 Danksagung............................................................................................ 154 8 Anhang ................................................................................................... 155 8.1 Abkürzungsverzeichnis ........................................................................................ 155 8.2 Ermittelte Sequenzdaten der Expressionskonstrukte ......................................... 158 8.2.1 Nukleinsäuresequenz des Expressionskonstruktes ALD_pET-16b........................158 Inhaltsverzeichnis 8.2.2 8.2.3 8.2.4 8.2.5 8.2.6 8.2.7 8.2.8 IV Abgeleitete Aminosäuresequenz des Expressionskonstruktes ALD_pET-16b......158 Nukleinsäuresequenz des Expressionskonstruktes ALD_pASK-IBA3 ..................159 Abgeleitete Aminosäuresequenz des Konstruktes ALD_pASK-IBA3....................159 Schematische Dartsellung der Aldolase-Expressionskonstrukte ............................160 Nukleinsäuresequenz des Expressionskonstruktes VDAC_pRSET A....................160 Abgeleitete Aminosäuresequenz des Konstruktes VDAC_pRSET A .....................160 Sequenzvergleiche der Expressionskonstrukte .......................................................161 8.3 Sequenzen der pGADT7-cDNA-Bank-Konstrukte ............................................. 166 8.4 Tabelle zur statistischen Auswertung .................................................................. 173 Lebenslauf..................................................................................................... 174 Eidesstattliche Versicherung ....................................................................... 175 Einleitung 1 1 Einleitung Unter dem Begriff Primärstoffwechsel werden die Biosynthesewege zusammengefasst, deren Produkte für das Überleben der Zellen notwendig sind. Charakteristisch für den pflanzlichen Primärstoffwechsel ist die Umwandlung von Sonnenenergie in chemische Energie durch die Photosynthese und die Synthese hochmolekularer Kohlenhydrate. Auch der Abbau biologischer Moleküle und die Einspeisung von Produkten in den Energiestoffwechsel, hauptsächlich in die Glykolyse und den Citratzyklus, gehören zum Primärstoffwechsel. Dagegen versteht man unter Sekundärstoffwechsel Prozesse, die nur in ganz bestimmten Geweben oder Organen und in ganz bestimmten Entwicklungsstadien erfolgen und nicht der einzelnen Zelle, sondern dem Organismus als Ganzem dienen. Die Grenze zwischen Primärund Sekundärstoffwechsel ist jedoch nicht immer eindeutig zu ziehen. 1.1 Mikrokompartimentierung und Channeling Wie jede eukaryotische Zelle weist auch die Pflanzenzelle eine interne räumliche Struktur auf, die der Abgrenzung einzelner Reaktionsräume oder Kompartimente dient. Durch die Kompartimente einer Zelle, z. B. Organellen wie Mitochondrien und Chloroplasten, ist eine Organisation für bestimmte Stoffwechselwege gegeben. Sie ist jedoch auch für die im Cytosol ablaufenden Prozesse von großer Wichtigkeit. Freie Diffusion, ohne eine spezifische Anordnung von Enzymen, würde zu einem ineffizienten Stoffwechsel führen (SRERE, 1967). Die stattfindende Diffusion wird durch die Mikroumwelt innerhalb des Cytosols beeinflusst. So erfolgt bei der Diffusion von Makromolekülen auf Grund der Partikelgröße und der sterischen Hinderung durch das cytoskelettale Netzwerk innerhalb der Zellen ein Größenausschluss, ähnlich wie durch ein Sieb (LUBY-PHELBS, 1994). Viele Enzyme sind durch die Bildung von Komplexen oder die Assoziation an strukturelle Bestandteile der Zelle sowie die Multifunktionalität dieser Enzyme selbst organisiert. Solche Interaktionen führen zu einer Mikrokompartimentierung von Enzymen. Einige dieser Komplexe sind sehr stabil und gut zu isolieren, wogegen jedoch die Interaktionen in dynamischen Enzymkomplexen wie der Glykolyse oder dem Citratzyklus recht schwach und transient sind (WELCH, 1977). Die Komplexbildung kann mit dem Stoffwechselstatus der Zelle variieren, und eine reversible und dynamische Bindung von Enzymen an Membranen kann zu kinetisch unabhängigen Teilmengen des Enzymassoziationen Enzyms führen (OVÁDI & SRERE, 1996). können konkurrierende Stoffwechselwege, Durch die spezifische im gleichen Kompartiment der Zelle stattfinden, reguliert werden (PLAXTON, 1996), wie z. B. die Einleitung 2 Glykolyse und Gluconeogenese. URETA stellte 1978 die Hypothese auf, dass gemeinsame Zwischenprodukte konkurrierender Stoffwechselwege durch die Beteiligung von Isoenzymen voneinander getrennt werden. 1988 ging OVÁDI von einer dynamischen Interaktion zwischen zwei Enzymen durch die Affinität beider Enzyme zu einem gemeinsamen Zwischenprodukt aus. Der Begriff Metabolon wurde ein Jahr zuvor von SRERE (1987) für einen Komplex aufeinanderfolgender Enzyme geprägt, die leicht oder vorübergehend assoziiert sind. Ein Metabolon ist außerdem eine dicht strukturierte Ansammlung von Aktivitäten, wodurch sich die Möglichkeit ergibt, komplexe Stoffwechselwege zu kanalisieren („channeling“). Darunter wird ein bevorzugter Transfer eines Zwischenprodukts von einem Enzym an ein physikalisch angrenzendes Enzym mit eingeschränkter Diffusion in das umgebende Milieu verstanden (SRIVASTAVA & BERNHARD, 1986; SRERE, 1987). So können z. B. instabile Zwischenprodukte durch die Bindung an ein Protein geschützt werden. „Channeling“ ermöglicht außerdem die Aufrechterhaltung eines Konzentrationsgradienten und verschafft einem Stoffwechselweg Vorteile durch die daraus folgende hohe lokale Konzentration eines Substrates (MATHEWS & SINHA, 1982; OVÁDI & SRERE, 1996). Durch die Konzentrierung von Substraten in definierten Zellbereichen behalten andere Bereiche des Cytosols, in denen die Konzentration gelöster Stoffe geringer ist, die Fähigkeit weitere Stoffe oder Substrate aufzunehmen (FULTON, 1982; SRERE, 1987; MATHEWS, 1993). Durch „Channeling“ wird auch eine schnelle Anpassung der Zelle an geänderte Umweltbedingungen ermöglicht. Die Regulation eines Stoffwechselwegs kann dabei durch die Regulation eines einzigen Enzyms innerhalb des Stoffwechselwegs erfolgen. Von einer Steigerung der Durchflussrate ist aufgrund der schnellen individuellen Reaktionen der Enzyme jedoch nicht auszugehen (MATHEWS, 1993). Die Assoziation von Enzymen kann jedoch auch ausschließlich der Regulation dienen, ohne dass es zum „channeling“ zwischen den aktiven Zentren der beteiligten Enzyme kommt, z. B. indem die katalytische Aktivität eines beteiligten Enzyms reduziert wird, wie es in der Zusammenfassung von WINKEL (2004) für den CysteinSynthase-Komplex beschrieben wurde. Ebenso beteiligen sich z. B. im Calvin-Zyklus kleine, nicht katalytische Proteine (CP12) an der Assoziation eines Multienzymkomplexes bestehend aus Phosphoribulose-Kinase und Glycerinaldehyd-3-phosphat-Dehydrogenase (GAPDH) (SCHEIBE et al., 2002). Einleitung 3 1.2 Die Glykolyse Bei dem seit 1940 vollständig aufgeklärten Stoffwechselweg der Glykolyse handelt es sich um eine Reaktionsfolge zum Abbau von Glukose. Glukose wird dabei in Pyruvat umgewandelt und es entstehen eine geringe Menge Energie in Form von ATP und Reduktionsäquivalente (NADH). Neben der Energiebereitstellung ist es eine wichtige Funktion der Glykolyse, Kohlenstoffgerüste für Biosynthesen zur Verfügung zu stellen. Eine Übersicht über die Reaktionen der Glykolyse sowie einige Verknüpfungen mit anderen Zellkompartimenten und Stoffwechselwegen ist in Abbildung 1.1 dargestellt. Die Reaktionsfolge der Glykolyse findet im Cytosol der Zellen statt und wird von zehn Enzymen katalysiert. In drei Reaktionen wird dabei aus Glucose zunächst Fructose-1,6bisphosphat (FBP) hergestellt. Der erste und der dritte dieser drei Schritte sind irreversibel. Glucose wird als erstes in Glucose-6-phosphat umgewandelt. Diese Reaktion wird durch die Hexokinase katalysiert. Glucose-6-phosphat wird anschließend in Fructose-6-phosphat umgewandelt. Im dritten Schritt dieser Reaktionsfolge entsteht, katalysiert von der Phosphofructokinase, FBP. Dabei handelt es sich um eine Reaktion, die entscheidend die Geschwindigkeit der Glykolyse beeinflusst. Die Aktivität der Phosphofructokinase wird durch die ATP-Konzentration, den pH-Wert und die Menge an Citrat in der Zelle reguliert. Der nächste Schritt der Glykolyse führt zur Spaltung des FBP in zwei Produkte mit je drei Kohlenstoffatomen, Dihydroxyacetonphosphat und Glycerinaldehyd-3-phosphat. Diese Spaltung wird durch das Enzym Aldolase katalysiert. Die energiegewinnenden Schritte, die sich anschließen, beginnen mit der von der GAPDH katalysierten Umwandlung des Glycerinaldehyd-3-phosphats in 1,3-Bisphosphoglycerat. Nach drei weiteren Reaktionen, in denen zwei Moleküle ATP gewonnen werden, erfolgt die abschließende Bildung von Pyruvat. Diese Reaktion, katalysiert von der Pyruvat-Kinase, führt ebenfalls zum Energiegewinn in Form von zwei Molekülen ATP und ist irreversibel. Neben der Reaktion der Phosphofructokinase haben auch die beiden anderen irreversiblen Reaktionen eine regulierende Rolle innerhalb der Glykolyse. Die Hexokinase, die den ersten Schritt der Glykolyse katalysiert, wird von ihrem Produkt, Glucose-6-phosphat, gehemmt. Wird die Phosphofructokinase gehemmt, steigt der Fructose-6-phosphat-Spiegel an, und damit auch der Glucose-6-phosphat-Spiegel. Eine Hemmung der Hexokinase erfolgt also auch, wenn die Phosphofructokinase gehemmt ist. Die Menge an Endprodukt der Glykolyse wird kontrolliert durch die Pyruvat-Kinase. Sie wird u. a. reguliert durch die Konzentration an FBP und ATP. Einleitung 4 Unter aeroben Bedingungen folgen der Glykolyse der Citratzyklus und die Atmungskette, durch die das Pyruvat vollständig zu CO2 und H2O oxidiert wird. Bei dieser vollständigen Oxidation der Glucose entstehen insgesamt 30 Moleküle ATP. Unter anaeroben Bedingungen wird das Pyruvat durch Gärung in Lactat oder Ethanol umgewandelt, wobei die Energieausbeute jedoch nur 2 ATP beträgt (STRYER, 1999). Die Regulation der Glykolyse spielt in der Anpassung eines Organismus eine entscheidende Rolle. Der Kohlenstofffluss muß den Bedarf der Pflanze decken, damit diese alle benötigten Komponenten für ihr Wachstum und Überleben synthetisieren kann (DENNIS et al., 1997). Neben der Funktion der Bereitstellung von Zwischenprodukten für die Biosynthese ist die Energiebereitstellung vor allem im Dunkeln und unter anaeroben Bedingungen wichtig. Gerade bei Sauerstoffmangel, der z. B. durch Überflutung im Wurzelraum auftreten kann, muss eine schnelle Anpassung an die veränderte Situation erfolgen. Die Weiterverarbeitung des Pyruvats mit Hilfe der Gärung ermöglicht unter diesen Bedingungen die Regenerierung des NAD+ zur Aufrechterhaltung der Glykolyse. Würde das NAD+ nicht regeneriert, könnte die Umwandlung des Glycerinaldehyd-3-phosphats durch die GAPDH nicht erfolgen und damit die Glykolyse nicht weiter fortschreiten. Durch die Aufrechterhaltung der Glykolyse kann jedoch zumindest eine geringe Energiemenge bereitgestellt werden, die dem Organismus das Überleben sichern kann. In Maiskeimlingen konnte nachgewiesen werden, dass kurze Zeit nach Anaerobiose die Synthese von ~20 spezifischen Polypeptiden, die am Kohlenstoffstoffwechsel beteiligt sind, erfolgt (SACHS et al., 1980). Dazu gehören unter anderem zwei Isoenzyme der AlkoholDehydrogenase (SACHS & FREELING, 1978), Glucosephosphat-Isomerase (KELLEY & FREELING, 1984a), Aldolase (KELLEY & FREELING, 1984b), Pyruvat-Decarboxylase (LASZLO & ST. LAWRENCE, 1983) und Saccharose-Synthase (SPRINGER et al., 1986; MCCARTY et al., 1986). Einleitung 5 Stärke Glucose ATP Hexokinase ADP Glucose-6-phosphat Cellulose GlucosephosphatIsomerase Plastid • • Fructose-6-phosphat ATP Phosphofructokinase Cytosol Glykolyse FettsäureSynthese u.a. Biosynthesewege • ADP Fructose-1,6-bisphosphat Aldolase Glycerinaldehyd3-phosphat Nucleinsäuren Aminosäuren Lipide Glycerinaldedhyd3-phosphat Dehydrogenase TPI NAD+ + Pi Dihydroxyacetonphosphat NADH + H+ 1,3-Bisphosphoglycerat ADP PhosphoglyceratKinase Plastid ATP s.o. 3-Phosphoglycerat Vakuole PhosphoglyceratMutase 2-Phosphoglycerat Enolase PEP Phosphoenolpyruvat PyruvatKinase Pyruvat Oxalacetat ADP Malat ATP Pyruvat Respiration Pyruvat Aminosäuren Malat Biosynthese Mitochondrion Abb. 1.1: Schema des Stoffwechselwegs der Glykolyse mit Verknüpfungen zu weiteren Biosynthesewegen in der Pflanze (in Anlehnung an PLAXTON, 1996). Die Abbildung erhebt keinen Anspruch auf Vollständigkeit. Abkürzungen sind wie im Text oder wie folgt: TPI: Triosephosphat-Isomerase, PEP: Phosphoenolpyruvat. Einleitung 6 1.2.1 Interaktionen glykolytischer Enzyme Interaktionen von Enzymen der Glykolyse untereinander oder mit anderen Komponenten der Zelle, wie z. B. Organellen oder dem Cytoskelett, wurden vor allem in tierischen Systemen entdeckt und charakterisiert. In letzter Zeit gibt es jedoch auch immer mehr Untersuchungen in pflanzlichen Systemen. Die meisten Enzyme kommen in einer löslichen und einer gebundenen Form in der Zelle vor (PAGLIARO & TAYLOR, 1988; KELETI & OVÁDI, 1988). Durch diese spezifischen Assoziationen werden die Enzymstruktur, die Kinetik und die thermodynamischen Eigenschaften der beteiligten Enzyme beeinflusst (ARNOLD & PETTE, 1970; WALSH et al. 1977, OVÁDI et al., 1986, OVÁDI & SRERE, 1996). MOWBRAY und MOSES zeigten 1976, dass glykolytische Enzyme aus Escherichia coli (E. coli) aggregieren können. In zahlreichen anderen Organismen und Zelltypen - wie z. B. der Bäckerhefe (Saccharomyces cerevisiae), Muskelzellen und Fibroblasten - wurden Interaktionen glykolytischer Enzyme nachgewiesen (TOMPA et al., 1986; OVÁDI & KELETI, 1978; OVÁDI, 1988; MINASCHEK et al., 1992). Die Assoziation glykolytischer Enzyme an strukturelle Proteine wurde als erstes in Muskelzellen beobachtet (ARNOLD & PETTE, 1969), wobei zunächst eine Interaktion der Phosphofructokinase (PFK) mit F-Aktin entdeckt wurde. Diese Interaktion kann durch die Phosphorylierung der PFK verstärkt werden (LUTHER & LEE, 1986). 1970 konnte die Bindung von Aldolase (ALD) an F-Aktin nachgewiesen werden (ARNOLD & PETTE, 1970). Es folgten viele Arbeiten zur näheren Charakterisierung dieser Assoziation, auf die im Einzelnen im nächsten Kapitel eingegangen wird. Der Einfluss von Proteinkonzentration und Ionendichte auf die Assoziation aller zehn an der Glykolyse beteiligten Enzyme auf ihre Bindung an FAktin wurde für Muskelgewebe von CLARKE und MASTERS 1975 untersucht. Neben vielen anderen Interaktionen wurde die der GAPDH mit dem Cytoskelett in Neuronen beschrieben (CLARKE & MORTON, 1982; KNULL et al., 1980) sowie ihre Bindung an F- und G-Aktin, was die katalytische Aktivität beeinflusste (OUPOROV et al., 2001). Ein Modell für die Interaktionen glykolytischer Enzyme und deren Assoziation an F-Aktin in Säugetierzellen wurde von MASTERS und Kollegen 1987 entwickelt. Dabei wird von einer Segmentierung der Glykolyse in Abschnitte mit bis zu vier Enzymen ausgegangen, die auch unabhängig voneinander unter verschiedenen zellulären Bedingungen agieren können. In Abb. 1.2 ist das zweite Segment mit den interagierenden Proteinen ALD, GAPDH Triosephosphat-Isomerase (TPI) und Phosphoglycerat-Kinase (PGK) dargestellt. Neben der Bindung der Enzyme untereinander erfolgt eine Bindung von ALD, GAPDH und PFK (dem letzten Enzym des ersten glykolytischen Segments) an Aktinfilamente. Einleitung 7 Abb. 1.2: Schematische Darstellung interagierender Proteine der Glykolyse an einem Aktin enthaltenden Filament mit der Möglichkeit des „Channelings“ von Metaboliten (aus MASTERS et al., 1987). Abkürzungen wie im Text oder wie folgt: F6P Fructose-6-phosphat; FDP Fructose-1,6bisphosphat; G3P Glycerinaldehyd-3-phosphat, DHAP Dihydroxyacetonphosphat; DPGA 1,3Bisphosphoglycerat, 3PGA 3-Phosphoglycerat. Neben der Assoziation der Enzyme sowohl untereinander als auch an Aktin-enthaltende Filamente wurden weitere Membranbindungen und Bindungen an Mikrotubuli (z. B. DURRIEU et al., 1987; WALSH et al., 1989; HARRIS &WINZOR, 1990; UYEDA, 1992; LOW et al., 1993; GITLITS et al., 2000), Bindungen an Ribosomen (RYAZANOV et al., 1988) und Mitochondrien (z. B. WILSON, 1980; XIE & WILSON, 1990; TAYLOR et al., 2003; GIEGÉ et al., 2003) nachgewiesen. Die korrekte Lokalisation der Enzyme innerhalb der Zelle spielt für die Funktion komplexer Prozesse eine entscheidende Rolle (SRERE & KNULL, 1998). MOORHEAD und PLAXTON konnten 1988 für pflanzliches Gewebe eine Assoziation glykolytischer Enzyme an die subzelluläre, Partikel enthaltende Fraktion nachweisen. Bei der Suche nach Aldolase-Bindeproteinen fanden sie 1992 Interaktionen zwischen Aldolase und PFK. 1995 wurden Enzym-Enzym-Interaktionen von GAPDH, TPI und Aldolase in Chloroplasten nachgewiesen (ANDERSON et al., 1995). In der Cytoskelettfraktion von Maisendosperm konnten Enzyme des Kohlenhydratstoffwechsels wie Aldolase, GAPDH und Saccharose-Synthase nachgewiesen werden (AZAMA et al., 2003). Sieben der zehn glykolytischen Enzyme wurden in Arabidopsis-Zellen an der äußeren Mitochondrienmembran identifiziert (GIEGÉ et al., 2003). An den isolierten Mitochondrien konnte die Glykolyse vollständig ablaufen, was darauf hinweist, dass auch die drei nicht identifizierten Enzyme an den Mitochondrien lokalisiert sind. Von pflanzlicher Hexokinase ist die Bindung an Einleitung 8 Membranen z. B. von Chloroplasten und Mitochondrien bekannt (WIESE et al., 1999, GALINA et al., 1999; DA-SILVA et al., 2001). Durch solche Komplexe glykolytischer Enzyme wäre es möglich, das gebildete Pyruvat direkt in die Mitochondrien zu leiten, wo es als Substrat für die Respiration dient. Nicht nur durch Mikrokompartimentierung, sondern auch durch das Vorkommen mehrerer Enzyme, die eine Reaktion katalysieren können, sowie durch das Vorhandensein der glykolytischen Enzyme in zwei Kompartimenten der Zelle, dem Cytosol und den Plastiden (DENNIS et al., 1997) erreicht der pflanzliche Stoffwechsel eine hohe Flexibilität. 1.2.2 Das Enzym Aldolase Bei der Aldolase handelt es sich um ein glykolytisches Enzym, das an vielen der nachgewiesenen Interaktionen beteiligt ist. Es gibt Klasse-I- und Klasse-II-Aldolasen (Fructose-1,6-bisphosphat-Aldolasen, E.C. 4.1.2.13), die wegen der fehlenden Sequenzhomologie konvergent entstanden sein müssen (MARSH & LEBHERZ, 1992). Klasse-IAldolasen sind typisch für alle Tiere und höheren Pflanzen, Bakterien und Pilze haben dagegen meist Klasse-II-Aldolasen. Es gibt jedoch auch einige Eukaryoten, die beide Aldolase-Typen enthalten (PELZER-REITH et al., 1994). Bei den cytosolischen und chloroplastidären Isoformen höherer Pflanzen handelt es sich um Klasse-I-Aldolasen, deren Homologie auf dem Niveau der Aminosäuresequenz bei 54% liegt (PELZER-REITH et al., 1993). Sie unterscheiden sich durch den Mechanismus der katalysierten Synthese oder Spaltung von Fructose-1,6-bisphosphat von den Klasse-II-Aldolasen. Während Klasse-IIAldolasen Zn2+ oder Fe2+ als elektrophile Gruppe nutzen, zeichnen sich Klasse-I-Aldolasen durch die Bildung einer Schiff´schen Base zwischen einer -Amino-Gruppe von Lysin im katalytischen Zentrum und der Dihydroxyaceton-Einheit des Substrates aus. Das Enzym ist ein Homo-Oligomer, das aus vier Untereinheiten besteht. Im Chloroplasten ist Aldolase aktiv am Calvin-Zyklus beteiligt und katalysiert die Aldolkondensation von Triosephosphaten zu Fructose-1,6-bisphosphat. Dagegen katalysiert sie im Cytosol die Umkehrreaktion, durch die Fructose-1,6-bisphosphat in zwei Triosephosphate (Dihydroxyacetonphosphat und Glycerinaldehyd-3-phosphat) gespalten wird (s. Abb. 1.1). Wird die katalytische Aktivität einer Untereinheit des Aldolase-Tetramers inhibiert, geht die gesamte Aktivität des Tetramers verloren. Dies weist auf die Existenz einer Kommunikation zwischen den Untereinheiten hin (SYGUCH & BEAUDRY, 1997). Die Aminosäuresequenz der anaerob induzierten Aldolase aus Mais wurde 1986 von KELLEY und TOLAN veröffentlicht. Das aktive Zentrum von Kaninchen-Aldolase A liegt in einem Einleitung 9 hochkonservierten Bereich des Proteins, das die Aminosäure Lys-229 einschließt, welche die Schiff´sche Base mit dem Substrat bildet. In cytosolischer Mais-Aldolase befindet sich diese Lysingruppe an Position 225. Auch die Aminosäuren, die mit den Phosphatgruppen des Substrates an Position C-1 und C-6 interagieren, sind identifiziert worden und befinden sich in einem hochkonservierten Bereich des Proteins. Im Aldolase-Gen zeigt eine Sequenz aus 6 Basenpaaren, die sich an Position –70 in Bezug zum Transkriptionsstart befindet, Homologie zu einer Unterregion des „anaerob induzierten Steuerelements“ („anaerobic regulatory element“, ARE) des Alkohol-Dehydrogenase-Gens 1 (Adh1) aus Mais (DENNIS et al., 1988). Dieses ARE ist wichtig für die Induktion der Adh1mRNA (WALKER et al., 1987) und ist in einem weiteren anaerob exprimierten Gen, der Saccharose-Synthase, zu finden (SPRINGER et al., 1986). Die Expression der Aldolase erfolgt unter aeroben Bedingungen in geringem Maße und nimmt unter anaeroben Bedingungen zu (KELLEY & FREELING, 1984a). An vielen der im letzten Kapitel (s. Kap. 1.2.1) beschriebenen Protein-Protein-Interaktionen, von denen die meisten für tierische Systeme beschrieben wurden, ist auch die Aldolase beteiligt. Eine Interaktion zwischen den beiden glykolytischen Enzymen Aldolase und GAPDH verhindert die Aldehyd-Diol-Umwandlung des Glycerinaldehyd-3-phosphats und durch „channeling“ wird das Substrat von der Aldolase an GAPDH direkt weitergeleitet (OVÁDI & KELETI, 1978). Durch die Interaktion zwischen Aldolase und Phosphofructokinase und die Bildung eines Heterokomplexes wird die Kinase vor Inaktivierung geschützt (R S et al., 2000). Aldolase kommt, wie die meisten Enzyme, in löslicher und gebundener Form in Zellen vor (PAGLIARO & TAYLOR, 1988). Von ARNOLD und PETTE wurde 1970 die Aktinbindung der Aldolase beschrieben, die z. B. durch 1 mM FBP vollständig unterdrückt wird. Neben FBP inhibieren u. a. auch anorganisches Phosphat, ATP, ADP, Glucose-1,6-bisphosphat, Fructose1-phosphat, Dihydroxyacetonphosphat sowie erhöhte Ionenstärke die Bindung. Keinen Einfluss auf die Bindung tierischer Aldolase an Aktin zeigten Fructose-6-phosphat, Fructose2,6-bisphosphat, Ribose-5-phosphat und Glycerinaldehyd-3-phosphat (ARNOLD & PETTE, 1970; WANG et al., 1996). Im Jahr 1976 zeigten CLARKE und MORTON, dass Aldolase mit F-Aktin und F-AktinTropomyosin identische Aggregate bildet, die eine Quervernetzung aufweisen. Die Strukturen scheinen durch die Vernetzung nebeneinander liegender Filamente aufgetreten zu sein, was auf mehrere Bindestellen der Aldolase für strukturelle Proteine hinweist. Von WANG et al. (1996) wurde eine Stöchiometrie der Bindung von einem Aldolasemonomer an 25 Einleitung 10 Aktinmonomere beschrieben. Die Aktinbindung wird jedoch durch die vorhandene Konzentration der Aldolase beeinflusst. So kommt es bei geringen Konzentrationen (1 µM) zur Quervernetzung der Aktinfilamente, was in einem hoch vernetzten F-Aktin-AldolaseNetzwerk resultiert. Dagegen entstehen bei höheren Konzentrationen (3 µM) F-Aktinbündel mit gebundener Aldolase höherer Dichte, die jedoch keine Quervernetzung zu einem dichten Netzwerk aufweisen. Durch Inkubation der Aldolase mit Diethylpyrocarbonat, welches mit den Histidinen des Enzyms reagiert, geht dem Enzym die Fähigkeit verloren, FBP zu binden. Mit dem dadurch hervorgerufenen Verlust der Enzymaktivität geht auch die Bindekapazität der Aldolase an Aktin verloren (DON & MASTERS, 1988). In Anwesenheit des Substrates FBP kommt es zu einer Kompetition zwischen der Substrat- und Aktinbindung. Deshalb wird von einer Bündelung der aktinbindenden Reste um das katalytische Zentrum des Aldolasemoleküls oder einer partiellen Überlappung der Bindestellen ausgegangen (WANG et al., 1996; PAGLIARO & TAYLOR, 1988; KNULL & WALSH, 1992; O`REILLY & CLARKE, 1993). Die Binderegion der Aldolase konnte mit Hilfe eines synthetischen Peptids identifiziert werden. Sie weist eine Sequenzhomologie mit dem C-Terminus von Aktin sowie mit identifizierten Aktinbinderegionen anderer Aktinbindeproteine, wie Gelsolin und Severin, auf (O´REILLY & CLARKE, 1993). Auch pflanzliche Aldolasen weisen diese Sequenz auf (WINTER & HUBER, 2000), die pro Aldolasemonomer einmal in der Sequenz auftritt. In Abb. 1.3 ist ein Sequenzvergleich von 10 Aminosäuren des C-Terminus von cytoplasmatischem Aktin ( Aktin), der Aktinbinderegion von Kaninchen-Aldolase A (Muskel-Typ) und zwei Sequenzen pflanzlicher cytosolischer Aldolasen (Arabidopsis thaliana und Zea mays) dargestellt. -Aktin Aldolase Kaninchen Aldolase A. thaliana Aldolase Z. mays AS-Rest 363 34 30 30 D D D D E E E E S S S S Sequenz G P S T G S T E T T G T I I I I V A G G H K K K R R R R Abb. 1.3: Sequenzvergleich der Aktin-Bindestelle tierischer und pflanzlicher Aldolase mit -Aktin. Dick gedruckte Buchstaben zeigen identische Aminosäuren im Vergleich zur Aktin-Sequenz. Bei der Aldolase aus Kaninchen handelt es sich um den Muskel-Typ. Abkürzung: AS = Aminosäure. Bei der Untersuchung menschlicher Aldolase-Isoenzyme zeigte sich eine Gewebespezifität der Aktinbindung (KUSAKABE et al., 1997). Diese deutet auf die Signifikanz der Bindung und die große Bedeutung der Glykolyse hin. Die spezifische Assoziation der Aldolase am Cytoskelett kann eine strukturelle Rolle im Cytoplasma spielen, den Stoffwechsel Einleitung 11 kompartimentieren und/oder die Zellbewegung beeinflussen. Dies zeigt den wahrscheinlich weit verbreiteten funktionalen Dualismus cytosolischer Enzyme (PAGLIARO & TAYLOR, 1992; WANG et al., 1997; JEFFERY, 1999). Neben der Bindung an Aktin wurde auch eine Bindung an ein weiteres Protein des Cytoskeletts, Tubulin, nachgewiesen (KARKHOFF-SCHWEIZER & KNULL, 1987). Tubulin inhibiert die Aktivität der Aldolase (VOLKER & KNULL, 1993). Der mit Aldolase interagierende Bereich befindet sich am C-Terminus von -Untereinheiten von Tubulin und besteht aus 43 Aminosäuren (VOLKER & KNULL, 1997). Die Bindung an Mikrotubuli scheint einen Kontrollmechanismus für die Enzymaktivität darzustellen (SRERE & KNULL, 1998; VÉRTESSY et al., 1997). 1.3 Das pflanzliche Cytoskelett Das Cytosol pflanzlicher und tierischer Zellen wird durch ein dreidimensionales Netzwerk von filamentösen Proteinen, dem Cytoskelett, durchzogen. Es bietet eine räumliche Organisation für Organellen, ein Gerüst für die Wanderung von Organellen und spielt eine wichtige Rolle bei der Aufrechterhaltung der Zellform und der Zelldifferenzierung (TAIZ & ZEIGER, 1999). Mit seiner großen Oberfläche und der Verknüpfung fast aller zellulären Strukturen können Proteine oder andere cytoplasmatische Bestandteile daran binden, und dadurch wird die Immobilisierung von cytoplasmatischen Komponenten möglich (JANMEY, 1998). Das Cytoskelett von Pflanzenzellen setzt sich aus verschiedenen Komponenten zusammen: Mikrotubuli, Mikrofilamenten und Intermediärfilamenten. Mikrotubuli bestehen aus Polymeren des Proteins Tubulin, das sich aus Heterodimeren, dem -Tubulin und dem Tubulin zusammensetzt. Mikrofilamente bestehen aus globulärem Aktin (G-Aktin), einem Protein, das man auch aus Muskelzellen kennt. Ein Mikrofilament besteht aus zwei Ketten von polymerisierten Aktinmolekülen, die sich helikal umeinander winden. Intermediärfilamente setzen sich aus verschiedenen Proteinen zusammen. Zu den beteiligten Proteinen gehört z. B. das Keratin, das in Blättern des Chinakohls gefunden wurde (YANG et al., 1995). Intermediärfilamente sind weniger dynamisch als Mikrotubuli und Mikrofilamente und stellen einen Puffer gegen eine mögliche Deformierung der Zelle dar (CAROTHERS CARRAWAY, 2000). Auf Intermediärfilamente wird im Folgenden nicht weiter eingegangen. Für die Polymerisation von Mikrofilamenten wird ATP, für die von Mikrotubuli GTP benötigt. Interaktionen zwischen Aktin und Mikrotubuli wurden in der Nähe der Plasmamembran oder direkt daran beschrieben (LUNA & HITT, 1992; COLLINGS et al., 1998). Einleitung 12 Die intrazelluläre Bewegung von Mitochondrien beruht in Pflanzen auf der Kontrolle durch F-Aktin, ihre Positionierung im Cytoplasma auf F-Aktin und Mikrotubuli (VAN GESTEL et al., 2002). Das Cytoskelett spielt auch eine Rolle in der Translation, durch die Positionierung von Polysomen in der Zelle (DAVIES et al., 1991; ITO et al., 1994). Neben diesen Funktionen erfüllt das Cytoskelett in Pflanzenzellen noch eine Vielzahl von Aufgaben in der Wahrnehmung und Reaktion auf Signale wie Licht, Schwerkraft, Berührung, Pathogene und Wundheilung (TAKAGI, 2000; BALU KA & HASENSTEIN, 1997; SEDBROOK et al., 1999; FOISSNER et al., 1996). Bei der Cytokinese pflanzlicher und tierischer Zellen spielt das Cytoskelett eine entscheidende Rolle. Das Präprophaseband sowie der Spindelapparat werden aufgebaut. Ein Phragmoplast aus Mikrotubuli und Aktinfilamenten bildet sich in der Äquatorialebene postmitotischer Pflanzenzellen und dehnt sich zentrifugal bis zur Zellwand aus (BOWERMAN & SEVERSON, 1999). 1.3.1 Mikrotubuli Die Organisation der pflanzlichen Mikrotubuli erfolgt nicht wie in tierischen Zellen mit Hilfe von Centriolen, sondern über funktionell analoge „Mikrotubuli-organisierende Zentren“, die viele Mikrotubuli-assoziierte Proteine (MAPs) enthalten, welche die kritische Konzentration für die Polymerisation von Mikrotubuli herabsetzen. Mikrotubuli bestehen aus Tubulin, dessen Molekülmasse 55 kDa beträgt. Kinesin und Dynein können sich, in Abhängigkeit von der Polarität der Mikrotubuli, an diesem entlang bewegen und sind am Gleiten der Mikrotubuli und dem Transport von Proteinen entlang den Mikrotubuli beteiligt (NICK, 1999). Durch die Zellwand werden Mikrotubuli stabilisiert, was auf die Existenz von Transmembranproteinen, die eine Verknüpfung zwischen Zellwand und Mikrotubuli herstellen, hinweist. Die Verbindung zwischen Aktinfilamenten und der Plasmamembran beruht auf der Anwesenheit und Organisation von corticalen Mikrotubuli (COLLINGS et al., 1998). 1.3.2 Mikrofilamente Bei Mikrofilamenten handelt es sich um Polymere des Proteins Aktin. Dieses kommt in einer monomeren Form, dem G-Aktin, und als filamentöses Aktin, F-Aktin, vor. G-Aktin ist benannt nach seiner globulären Form und hat ein Molekulargewicht von 42 kDa und einen Durchmesser von 4 nm. Jede Aktin-Untereinheit (G-Aktin) ist in der Lage, ATP oder ADP zu binden. Die Bindestelle dafür befindet sich im Verbindungsstück zwischen den zwei Domänen des G-Aktins, der größeren C-terminalen und der kleineren N-terminalen Domäne. Einleitung 13 Der hochkonservierte C-terminale Bereich von Aktin spielt bei der Schaffung des engen Kontakts zwischen benachbarten Untereinheiten eine wichtige Rolle (DREWES & FAULSTICH, 1993). Aktin-Monomere verknüpfen sich unter physiologischen Bedingungen zu Filamenten, die einen Durchmesser von 7 nm und eine helikale Struktur von 13-14 Untereinheiten pro halber Drehung aufweisen. Bei der Aggregation von G- zu F-Aktin wird ATP gespalten; das entstehende ADP bleibt am F-Aktin gebunden. Die Enden der Aktinfilamente unterscheiden sich. Es gibt ein schnell wachsendes Plusende oder auch „barbed end“, an dem die Polymerisation überwiegend erfolgt, und ein langsam wachsendes Minusende bzw. „pointed end“. Die kritische Konzentration (CC) beschreibt die minimale Konzentration an G-Aktin, die für die Zusammenlagerung zu F-Aktin notwendig ist. Für die beiden Enden des Filaments bestehen unterschiedliche kritische Konzentrationen. Bei Maispollen beträgt die CC für die Aktinpolymerisation in vitro 0,6 µM (REN et al., 1997), während nicht-pflanzliches Aktin eine CC von 0,1 µM für das „barbed end“ und 0,5 µM für das „pointed end“ aufweist (HATANO, 1994). In Zellen erfolgt die Polymerisation des Aktin an besonderen Nukleationspunkten, die mit Aktin-Bindeproteinen (ABPs) besetzt sind. Verändert sich die Filamentlänge durch gleichzeitiges Binden von Aktinmonomeren am „barbed end“ und Dissoziieren am „pointed end“ nicht, spricht man von „treadmilling“ (WEGNER, 1982). Anders als in gestreiften Muskelzellen, in denen Aktin, Myosin und die assoziierten Proteine in einer festen, parakristallinen Form arrangiert sind, ist die räumliche Organisation in den meisten Nicht-Muskelzellen hoch variabel und flexibel (SCHLIWA, 1981). Die Änderungen der auf Aktin basierenden Netzwerke erfolgen in Bruchteilen einer Sekunde und erlauben die schnelle Anpassung an intrazelluläre und interzelluläre Reize. Eine wichtige Rolle in diesem dynamischen Verhalten des Aktin-Cytoskeletts spielen ABPs, die neben der Fähigkeit, an Aktin zu binden, auch Bindekapazität für verschiedene andere Moleküle besitzen. So kontrollieren ABPs die Größe und Aktivität des G-Aktin-Pools, die subzellulären Orte der Nukleation, die Rate des Filamentumsatzes und die Organisation der Mikrofilamente in höher geordnete Strukturen (DE RUIJTER & EMONS, 1999; STAIGER, 2000; COOPER & SCHAFER, 2000). Eine Gruppierung der ABPs ist aufgrund ihrer Aktivitäten möglich. Die Polymerisation und Depolymerisation der Aktinfilamente regulieren monomerbindende Proteine (z. B. Profilin und Cofilin), indem sie G-Aktin binden und es so dem Aktinpool für die Polymerisation entziehen. Einfluss auf den Filamentumsatz zeigt auch der „AktinDepolymerisations-Faktor“ (ADF), der bevorzugt an die ADP-Form von Aktin bindet und damit die Dissoziation am „pointed end“ und Assoziation am „barbed end“ fördert (CARLIER & PANTALONI, 1997; CHEN et al., 2000). Die Stabilisierung von Filamenten, die Bildung von Einleitung 14 Netzwerken („crosslinking“) und die Bündelung von Aktinfilamenten zu parallelen Strukturen erfolgen mit Hilfe von Proteinen, die an den Seiten der Aktinfilamente binden („side-binding proteins“). In Pflanzen sind die „side-binding“-Proteine Fimbrin, Villin und EF-1 bekannt. Die Möglichkeit, Untereinheiten am Filamentende zu binden oder zu lösen, sowie die Spaltung von Aktinfilamenten kontrollieren „capping“- und „severing“-Proteine. Durch die schon erwähnten Nukleations-Faktoren wird ein Kern von Aktin-Untereinheiten gebildet, der die Polymerisation katalysiert. Motorproteine, wie das Myosin, ermöglichen Bewegungen entlang der Filamente und nehmen Einfluss auf die Bewegung des Aktins. Monomer-Bindeprotein „capping“-Protein „severing“-Protein „side-binding“-Protein Motor-Protein „side-binding“-Protein („crosslinking“) Abb. 1.4: Schematische Darstellung einiger Aktin-Bindeproteine. Aktin-Bindeproteine können als Sensor dienen und stellen eine Verknüpfung zwischen Signalen und der Umorganisation der cytoplasmatischen Architektur dar. An die Plasmamembran assoziierte Aktin-Isoformen sind an der Signalübertragung beteiligt, die auf dem Phosphoinositolweg basiert (TAN & BOSS, 1992). Mehrere Arbeiten zeigen einen Einfluss des Aktin-Cytoskeletts auf die Proteinbiosynthese und die Verteilung von Speicherproteinen (CLORE et al., 1996; WU et al., 1998; MUENCH et al., 1998; KLYACHKO et al., 2000; AZAMA et al., 2003). Für Spitzenwachstum (PIERSON & CRESTI, 1992; BALU KA et al., 2000), Wundheilung (FOISSNER et al., 1996) und intrazelluläre Bewegungen (LICHTSCHEIDL & URL, 1990) ist das Aktin-Cytoskelett essentiell. Grundlage für das cytoplasmatische Strömen, den Transport von Organellen wie Dictyosomen, Mitochondrien und Vesikel, ist ein ausgedehntes F-Aktin-Netzwerk (WILLIAMSON, 1993; VOLKMANN & BALU KA, 1999). Einleitung 15 1.4 Fragestellung Für die Regulation des Stoffwechsels eines Organismus bietet sich die Mikrokompartimentierung an, durch die konkurrierende Stoffwechselwege in der Zelle getrennt und Substrate durch „channeling“ effektiv weitergeleitet werden können. In tierischen Muskelzellen ist dies ein bekanntes Phänomen. Unter anaeroben Bedingungen erfolgt dort die Assoziation glykolytischer Enzyme an das Aktin-Cytoskelett zur gezielten Energiebereitstellung für die Muskelkontraktion. Pflanzen benötigen einen flexiblen Stoffwechsel zur Anpassung an Änderungen von Umweltbedingungen (z. B. Lichtintensität, Sauerstoffmangel, Nährstoffangebot) und zur optimalen Nutzung von Nährstoffen, um überleben zu können. Durch Mikrokompartimentierung und Organisation von Enzymen an zellulären Strukturen könnte eine gezielte Versorgung von zellulären Subkompartimenten mit der benötigten Energie erfolgen. Da die Aminosäure-Sequenz pflanzlicher Aldolase Homologie mit dem C-Terminus von Aktin und bekannten Binderegionen anderer Aktin-Bindeproteine aufweist, sollte eine mögliche Bindung der Aldolase an Aktin in vitro analysiert werden. Außerdem sollten mit Hilfe eines Hefe-2-Hybrid-Systems bislang in Pflanzen unbekannte Protein-ProteinInteraktionen von Enzymen der Glykolyse untereinander sowie mit Bestandteilen des Cytoskeletts gefunden werden. Als Vorausetzung dafür sollte im Rahmen dieser Arbeit ein Hefe-2-Hybrid-System etabliert werden. Da eine Organisation der Glykolyse besonders unter anaeroben Bedingungen an Bedeutung gewinnt, um die benötigte Energie bereitzustellen, wurde die Suche nach interagierenden Enzymen auf Bedingungen unter Sauerstoffmangel fokussiert. Material und Methoden 16 2 Material und Methoden 2.1 Chemikalien und Materialien Vertreiber, Firmensitz Produkt(e) Amersham Bioscience, Freiburg ECL™ Western Blotting Detection Reagent AppliChem, Darmstadt Acrylamid/Bis-Acrylamid, ATP, Coomassie-Brilliantblau G-250, Coomassie-Brilliantblau R-250, DTTred, EDTA, Essigsäure, Formamid, D(+)Glucose, Glycin, GSH, GSSG, Harnstoff, H2O2, Imidazol, KCl, MgCl2, MOPS, NAD+, NADPH, NaCl, NaOH, Paraformaldehyd, D(+)-Saccharose, TEMED, Trichloressigsäure, Tris, Tween-20 Biometra, Göttingen Whatman Chromatographie-Papier Biomol, Hamburg Homidiniumbromid BioRad, München SDS-PAGE-Standards, Ziege-AntiKaninchen IgG (H + L)-MeerrettichPeroxidase-Konjugat, Ziege-AntiKaninchen IgG (H + L)-AlkalischePhosphatase-Konjugat Boehringer, Mannheim Enzyme (GDH, TPI, Aldolase) Clontech, Heidelberg CHROMA SPIN+TE-400 Säulen, MATCHMAKER Library Construction & Screening Kit, Plasmid pGADT7-Rec, Plasmid pGBKT7, Plasmid pGBKT7-Lam, Plasmid pGBKT7-53, X- GAL Cytoskeleton, Denver, CO, USA Aktin Diagonal, Münster Ethanol, Petrischalen Duchefa, Harlem, NL Chloramphenicol Fluka, Buchs, CH Bromphenolblau, Essigsäure, HefeStickstoffbasis ohne Aminosäuren, Methanol Material und Methoden 17 Vertreiber, Firmensitz Produkt(e) GibcoBRL Life Technologies, Karlsruhe Agar, Oligo-dT-Primer, Select Peptone, SuperScript II Reverse Transkriptase, PfxPolymerase IBA, Göttingen Plasmid pASK-IBA3, Strep-TactinSepharose, Anhydrotetracyclin, Hydrochlorid, Desthiobiotin ICN Biomedical Inc., Aurora, OH, USA Ammoniumpersulfat, SDS, Triton X-100 Invitrogen, Karlsruhe Elektroporationsküvetten, Phalloidin, Plasmid pRSET A MBI Fermentas, St. Leon-Rot 5-Bromo-4-Chloro-3-Indolylphosphat (BCIP), dNTPs, λ DNA/PstI, IPTG, 6 × Loading Dye Solution, DNA-Marker, Nitro blau tetrazolium (NBT), Prestained Protein Ladder ~10-180 kDa, Restriktionsendonukleasen, Taq-DNAPolymerase, T4-DNA-Ligase, X-Gal Merck-Schuchardt, Hohenbrunn Merck-Wasser Millipore/Waters, Erkrath Centricon Konzentrierungsröhrchen, MilliQ plus New England Biolabs, Beverly, MA, Calf Intestinal Alkaline Phosphatase USA (CIAP), Protein Marker Novagen, Madison, WI, USA E. coli BL21(DE3)pLysS, Factor-Xa Kit, Plasmid pET-16b PE Biosystems, Foster City, CA, USA ABI Prism®BigDyeTMTerminator Cycle Sequencing Ready Reaction Kit Pharmacia, Uppsala, S Chelating Sepharose , NAP-Säulen Qiagen, Hilden MinElute Gel Extraktion Kit, Plasmid pDrive, QIAprep-spin-Kit, QIAquick PCRPurification Kit, QIAquick Gel Extraktion Kit, RNeasy Plant Mini Kit, Strep-tag Antikörper, Strep-Tactin Superflow Resin Suspension Material und Methoden 18 Vertreiber, Firmensitz Produkt(e) Riedel-de Haën, Seelze CaCl2, Ethanol, HCl, H2O2, Isopropanol, Kaliumacetat, Kaliumhexacyanoferrat (III), KH2PO4, MgSO4, Natriumacetat, Natriumcarbonat, NaH2PO4, NaOH, Natriumthiosulfat, o-Phosphorsäure Roth, Karlsruhe Agar, Agarose NEEO für die Elektrophorese, -Mercaptoethanol, BSA, Dimethylformamid, Glycerin, Hefeextrakt, Lithiumacetat, Pefabloc, Phenol/Chlorophorm/Isoamylalkohol (25:24:1), Roti-Mark 10-150, Trypton Schleicher & Schuell, Dassel Nitrocellulosemembranen, Sterilfilter Serva, Frankfurt Kanamycin, Tetrazyklin Sigma-Aldrich Chemie GmbH, Adeninhemisulfat, Aminosäuren, 4-Chloro- Taufkirchen 1-Naphtol, D-Desthiobiotin, DEPC, DMSO, DTTox, FBP, Fixierer, Glasperlen, 4-Hydroxy-Azo-Benzyl-2-Carboxylsäure, Imidazol, Kodak BioMax MS-1 Film, Kodak professionel HC 110 Entwickler, Hühner-Eiweiß-Lysozym, ß-Mercaptoethanol, MnCl2, NiSO4, p-Nitrophenylphosphat, Oligonukleotide, PMSF, Ponceau-S, Protease Inhibitor Cocktail for Plants, PVPP, Rubidiumchlorid, Silbernitrat Stratagene, Heidelberg E. coli XL1blue, Plasmid pBSK USB, Cleveland, OH, USA Ampicillin Material und Methoden 19 2.2 Pflanzenmaterial: Anzucht und Ernte Das in dieser Arbeit verwendete Pflanzenmaterial (Z. mays L., var. Caramba, Bundesanstalt und Forschungszentrum für Landwirtschaft, Wien) lag allen Arbeiten zugrunde. Zur Isolierung von RNA dienten in Klimakammern angezogene etiolierte, 8-10 Tage alte Keimlinge, von denen die Wurzeln und der Spross geerntet und sofort in flüssigem Stickstoff schockgefroren wurden. Vor der Aussaat wurden die Samen eine Nacht bei 4°C in Wasser gequollen. Die Anzucht erfolgte in Schalen, die mit wasserdurchtränktem Zellstoff ausgelegt waren, im Dunkeln bei 20°C. Um hypoxisches Gewebe herzustellen, wurden die Keimlinge 24 h vor der Ernte mit Flutungspuffer vollständig bedeckt. Flutungspuffer: 5 mM Tris pH 7,5 (HCl) 2.3 Molekularbiologische Methoden 2.3.1 Bakterien- und Hefestämme Der folgenden Tabelle sind die in dieser Arbeit verwendeten Bakterien- und Hefestämme zu entnehmen. Tab. 2.1: Liste der verwendeten Stämme von Escherichia coli (E. coli) und Saccharomyces cerevisiae (S. cerevisiae). Stamm Verwendung und genetische Vertreiber Referenz Klonierungen Stratagene, BULLOCK RecA1, endA1, gyrA96, thi-1, Heidelberg et al., 1987 Charakteristika E. coli XL1blue hsrdR17, supE44, relA1, lac, [F’proAB, laclqZ∆M15Tn10 (tetr)] E. coli Heterologe Expression Novagen, STUDIER BL21(DE3)pLysS F-, ompT, hsdSB(rB-mB-) gal dcm Madison, WI, et al., 1990 (DE3) pLysS, Clmr USA Material und Methoden Stamm 20 Verwendung und genetische Vertreiber Referenz Clontech, JAMES Heidelberg et al., 1996 “Bait“-Plasmid Clontech, HARPER Hefe-2-Hybrid-System Heidelberg et al., 1993 Charakteristika S. cerevisiae AH109 Prey -Plasmid Hefe-2-Hybrid-System MATa, trp1-901, leu2-3, 112, ura3-52, his3-200, gal4∆, gal80∆, LYS2::GAL1UASGAL1TATA-HIS3, GAL2UASGAL2TATA-ADE2, URA3::MEL1UAS-MEL1TATAlacZ, MEL1 S. cerevisiae Y187 MATα, ura3-52, his3-200, ade2-101, trp1-901, leu2-3, 112, gal4∆, met-, gal80∆, URA3::GAL1UAS-GAL1TATAlacZ, MEL1 2.3.2 Plasmide Als Vektoren wurden verschiedene Plasmide eingesetzt, die für die jeweilige Anwendung unterschiedliche Eigenschaften besitzen. Alle verwendeten Vektoren und ihre charakteristischen Eigenschaften sind in Tab. 2.2 aufgelistet. Tab. 2.2: Liste der verwendeten Plasmide. Plasmid Charakteristika Firma pBSK Klonierungsvektor Stratagene, Heidelberg r Amp , lacZ pDrive Klonierungsvektor Qiagen, Hilden Ampr, Kanr, lacZ, T7-Promotor pET-16b Proteinexpressionsvektor r Amp , T7-Promotor, lacI, N-terminaler His-tag, Factor-Xa, IPTG-Induktion, ~5,7 kb Novagen, Madison, WI, USA Material und Methoden 21 Plasmid Charakteristika Firma pRSET A Proteinexpressionsvektor Invitrogen, Karlsruhe r N-terminaler His-tag, Amp , Enterokinase-Schnittstelle, T7-Promotor, IPTG-Induktion, ~ 2,9 kb pASK-IBA3 IBA, Göttingen Proteinexpressionsvektor r C-terminaler Strep-tag II, Amp , Tet-Promotor, ~ 3,3 kb pGADT7-Rec Hefe-Proteinexpressionsvektor Clontech, Heidelberg HA, LEU2, Ampr, GAL4AD, ~ 8,0 kb pGBKT7 Hefe-Proteinexpressionsvektor Clontech, Heidelberg r c-Myc, TRP1, Kan , Gal4(1147)DNA-BD, pGADT7-RecT ~ 7,3 kb Hefe-Proteinexpressionsvektor Clontech, Heidelberg SV40 Large T-antigen, HA, LEU2, Ampr, ~ 8,0 kb pGBKT7-53 Hefe-Proteinexpressionsvektor Clontech, Heidelberg Murein p53, c-Myc, TRP1, Kanr, ~ 8,3 kb pGBKT7-Lam Hefe-Proteinexpressionsvektor Clontech, Heidelberg menschliches Lamin C, c-Myc, TRP1, Kanr, ~ 7,9 kb 2.3.3 Arbeiten mit Nukleinsäuren 2.3.3.1 RNA-Isolierung aus Pflanzenmaterial Die Isolierung von Gesamt-RNA aus Z. mays (L. var. Caramba) erfolgte mit dem „RNeasy Plant Mini Kit“ der Firma Qiagen (Hilden) nach Herstellerangaben. Die Lagerung der RNA erfolgte bei –20°C. Material und Methoden 22 2.3.3.2 Denaturierendes Formaldehyd/Agarose-Gel (RNA-Gel) Um isolierte RNA auf Verunreinigungen mit DNA zu untersuchen, wurde ein Aliquot der RNA-Probe auf ein denaturierendes Formaldehyd/Agarose-Gel aufgetragen. Alle benötigten Geräte wurden vor dem Gebrauch mit 3% H2O2 und anschließend mit Diethyl-PyrocarbonatWasser (DEPC-Wasser) gespült. Für das Gel wurden 1,5 g Agarose mit 72 ml DEPC-Wasser und 10 ml 10 × MOPS/EDTA-Puffer aufgekocht. Nach dem Abkühlen wurden 18 ml 37%iges Formaldehyd zugegeben. Das Gel wurde nach dem Aushärten 5-10 min bei 4°C inkubiert. 10 × MOPS/EDTA-Puffer: 200 mM MOPS pH 7,0 (NaOH) 50 mM Natriumacetat 10 mM EDTA mit DEPC-Wasser angesetzt und autoklaviert. DEPC-Wasser: 0,1% Diethyl-Pyrocarbonat (DEPC) in H2Obidest gelöst und nach 1416 h Inkubation autoklaviert. Die Denaturierung der RNA-Proben erfolgte nach Zugabe von 4 µl 10 × MOPS/EDTA-Puffer 7 µl 37% (v/v) Formaldehyd 20 µl Formamid 1 µl Homidiniumbromid (1 mg/ml) zu 9 µl RNA-Probe (3-8 µg) durch Inkubation der Proben für 10 min bei 56°C. Vor und nach der Inkubation wurden die Proben auf Eis gekühlt. Jede Probe wurde vor der elektrophoretischen Auftrennung mit 4 µl RNA-Gelladepuffer gemischt. Nachdem die Proben auf das Gel auftragen worden waren, wurden die Elektrophoresen bei 70 V in 1 × MOPS/EDTA-Puffer durchgeführt. Alle 30 min wurde der Puffer umgerührt, nach der halben Laufstrecke wurde er erneuert. RNA-Gelladepuffer: 50% (w/v) Glycerin 0,25% Bromphenolblau 1 mM EDTA, pH 8,0 (NaOH) Material und Methoden 23 2.3.3.3 RNA-Quantifizierung Die Bestimmung der RNA-Konzentration und ihrer Reinheit erfolgte photometrisch am Uvikon 810 (Kontron Instruments, Mailand, Italien) bei zwei Wellenlängen (260 nm und 280 nm). Von der RNA-Lösung wurden hierfür 2 µl mit 998 µl DEPC-Wasser gemischt und bei beiden Wellenlängen gemessen. Als Nullwert diente DEPC-Wasser. Mit folgenden Formeln wurden Konzentration und Reinheit der Probe ermittelt: • RNA-Konzentration [CRNA(µg/µl)]: CRNA = E260 nm (Probe) × Verdünnungsfaktor × 40 1000 40 ist der RNA-Faktor (SAMBROOK et al., 1989). • Reinheit der Probe: Reinheit = E260 nm (Probe) E280 nm (Probe) - E280 nm (H2Obidest ) Die Probe wird als rein bezeichnet, wenn der errechnete Wert zwischen 1,7 und 2,0 liegt. Niedrigere Werte weisen auf eine Verunreinigung der RNA mit Proteinen und aromatischen Substanzen (z. B. Phenol) hin. 2.3.3.4 DNA-Quantifizierung Konzentrationen von DNA wurden ebenfalls photometrisch bei 260 nm gemessen. Es wurden hierfür 3 µl DNA-Lösung zu 597 µl H2Obidest gegeben und gut gemischt. Der Nullabgleich erfolgte mit 600 µl H2Obidest. Die Messungen wurden in Quarzküvetten durchgeführt, und die DNA-Konzentration konnte mit folgender Formel berechnet werden: • DNA-Konzentration [CDNA(µg/µl)] E260 nm (Probe) × Verdünnungsfaktor × 50 1000 50 ist der DNA-Faktor. CDNA = 2.3.3.5 Oligonukleotide Zur Amplifikation von DNA-Fragmenten durch Polymerase-Kettenreaktion werden Oligonukleotide als Primer eingesetzt. Es handelt sich dabei entweder um kommerziell erhältliche Standardprimer (z. B. T7-Promotor-Primer, Sp6-Promotor-Primer), die nahe der multiplen Klonierungsstelle (MCS: multiple cloning site ) auf vielen Vektoren binden und Material und Methoden 24 für Sequenzierungsreaktionen eingesetzt werden, oder um genspezifische Primer, die für diese Arbeit konstruiert wurden und an einer definierten Stelle der zu amplifizierenden Sequenz binden. Um gute Ergebnisse erzielen zu können, sollten Primer 15-30 Nukleotide lang sein. Besitzt der Primer am 3’-Ende ein Guanin (G) oder Cytosin (C), bindet er durch die zwischen diesen Nukleotiden sich ausbildenden drei Wasserstoffbrückenbindungen fester an die DNAVorlage („template ), als es bei einem Adenin (A) oder Thymin (T) der Fall ist. Der GCGehalt sollte bei 40-60% und die Schmelztemperatur (Tm) nach Möglichkeit zwischen 55 und 60°C liegen. Die Berechnung der Schmelztemperatur erfolgt nach der Wallace-Regel: Tm = (A + T) × 2°C + (G + C) × 4°C Eine Erhöhung der Spezifität der Primer kann erreicht werden, wenn Tripletts verwendet werden, die für seltene Aminosäuren codieren und nur durch ein oder zwei Codons kodiert werden können. Für eine Klonierung können den Primern Erkennungssequenzen für Restriktionsendonukleasen angehängt werden. Weiterhin muss darauf geachtet werden, dass sich keine Primer-Dimere zwischen den in der PCR eingesetzten Primern und keine Schleifen („loops , hairpins ) innerhalb der Primer ausbilden können. Die in dieser Arbeit verwendeten Primer sind in der folgenden Tabelle zusammengestellt. Tab. 2.3: Liste der verwendeten Primer mit den zugehörigen Schmelztemperaturen. Schnittstellen für Restriktionsendonukleasen sind durch Unterstreichung markiert. Die Abkürzung ALF steht hier für Aldolase. Name Sequenz (5 3) Schmelztemperatur ALF8_Sense GGG AAT TCC ATA TGT CGG CCT ACT GCG GAA AG 66°C ALF_Antisense NNN GGA TCC TCA GTA CTT GTA GTC CTT GAC GTG 70°C AD_Insert-fife GAT GAA GAT ACC CCA CCA AAC CC 70°C AD-Insert3-antisense AGA TGG TGC ACG ATG CAC AG 62°C vdac sense2_Hyb GGG AAT TCC ATA TGG TCG GGC CAG GCC TC 62°C vdacpRSET-For CGC GGA TCC ATG GTC GGG CCA GGC CTC 62°C vdacpREST-Rev CCG GAA TTC TCA GGG CTT GAG GGC CAC 60°C Fw-seq-pASK-IBA AGA GTT ATT TTA CCA CTC CCT 58°C Rev-seq-pASK-IBA GAC GCA GTA GCG GTA AAC G 60°C T7-Promotor Primer TAA TAC GAC TCA CTA TAG GG 56°C T7-Terminator GCT AGT TCA TTG CTC AGC GG 62°C Aktin_sense CGC GGA TCC ATG GCT GAC GAG GAT ATC CAG C 68°C Aktin_antisense AAC TGC AGT TAG AAG CAC TTC ATG TGG ACA TTG 70°C Material und Methoden Name 25 Sequenz (5 3) Schmelztemperatur ALFStrepsense ATG GTA CGT CTC AAA TGT CGG CCT ACT GCG GAA 76°C AGT ACA ALFStrepantisense ATG GTA CGT CTC AGC GCT GTA CTT GTA GTC CTT 68°C GAC GTG GA Oligo-dT-Primer Oligo T(35)-A (PFL 007-009) Oligo T(35)-C - OligoT(35)-G SP6 Promotor Primer CAT ACG ATT TAG GTG ACA CTA TAG 66°C 2.3.3.6 Synthese von cDNA Bei dieser Methode wird mRNA in komplementäre DNA (cDNA) umgeschrieben. Das dafür notwendige Enzym, die Reverse Transkriptase, wurde von der Firma GibcoBRL („Superscript II Rnase H- Reverse Transcriptase“, Karlsruhe) bezogen. Oligo-dT-Primer (GibcoBRL, Karlsruhe) können an den Poly-A-Schwanz der mRNA binden. Mit Hilfe des Enzyms entsteht ein erster Strang cDNA („first-strand cDNA“). Die Durchführung erfolgte nach Herstellerangaben. 2.3.3.7 DNA-Amplifikation 2.3.3.7.1 Polymerase-Kettenreaktion Ein bestimmter DNA-Bereich, der durch zwei begrenzende Oligonukleotide (Primer) definiert ist, kann mit Hilfe der Polymerase-Kettenreaktion (PCR, „polymerase chain reaction ) vervielfältigt (amplifiziert) werden. Eine PCR-Reaktion läuft ab wie folgt: die Denaturierung der DNA in ihre Einzelstränge, die anschließende Anlagerung der spezifischen Primer („Annealing ) und eine Verlängerungsphase („Extension ), bei der mit Hilfe einer hitzestabilen DNA-Polymerase (z. B. Taq-DNA-Polymerase [Thermus aquaticus]), vom Primer ausgehend, ein dem Matrizenstrang komplementärer DNA-Strang synthetisiert wird. Durch Wiederholung der drei Schritte wird eine exponentielle Zunahme der gewünschten DNA-Sequenz erreicht. Material und Methoden 26 Ein Standard-PCR-Ansatz enthielt in einem Gesamtvolumen von 50 µl : 5 µl 10 × PCR Puffer 3 µl MgCl2 (25 mM) 1 µl dNTP (10 mM) 0,25 µl Taq-DNA-Polymerase (5 U/µl) 10-50 pmol Sense-Primer 10-50 pmol Antisense-Primer X µl DNA-„template (ca.10-400 ng) Die PCR-Reaktionen wurden entweder in einem TRIO-Thermoblock TB 1 (Biometra, Göttingen) oder im Mastercycler für PCR (Eppendorf, Hamburg) durchgeführt. Für einige Amplifikationen von DNA-Fragmenten wurde aufgrund ihrer „proof-reading“Aktivität Pfx-DNA-Polymerase (GibcoBRL, Karlsruhe) nach Herstellerangaben genutzt, da diese eine geringere Fehlerquote für den Einbau falscher Nukleinsäuren besitzt. Sie stammt aus Pyrococcus sp. und ist vor allem bei DNA-Fragmenten von einer Länge über 1500 bp der Taq-DNA-Polymerase vorzuziehen. 2.3.3.7.2 Reverse Transkriptase-PCR Es besteht auch die Möglichkeit, die Synthese der „first-strand cDNA“ und die PCR in einem Reaktionsansatz durchzuführen. Diese Reaktion nennt sich Reverse Transkriptase PCR (RTPCR) und ermöglicht Zeit- und Materialersparnis. Dabei wird eine PCR mit mRNA als Matrize durchgeführt, jedoch nicht mit Primern, die an den Poly-A-Schwanz der mRNA binden, sondern mit genspezifischen Primern. Die Primer binden sowohl bei der reversen Transkription als auch in der nachfolgenden Amplifikation spezifisch an derselben Stelle der RNA bzw. cDNA. Die RT-PCR-Reaktion muss in diesem Fall dem eingesetzten Enzymgemisch aus Reverser Transkriptase und Taq-Polymerase angepasst werden und setzt sich aus einem Zyklus zur cDNA-Synthese und der anschließenden PCR-Amplifikation zusammen. Alle RT-PCR Reaktionen wurden mit dem „SuperScript One-Step RT-PCR System“ von GibcoBRL (Karlsruhe) durchgeführt. Material und Methoden 27 2.3.3.8 Reinigung von DNA-Fragmenten Die Reinigung einer Probe von kleineren DNA-Bruchstücken (z. B. Primern) und Enzymen (Polymerasen, Restriktionsendonukleasen) nach einer PCR oder einem Restriktionsverdau erfolgte mit Hilfe des „QIAquick PCR-Purification Kit“ (Qiagen, Hilden). Die Reinigung wurde analog zum Herstellerprotokoll durchgeführt. Eine Lagerung der gereinigten DNA ist bei –20°C möglich. 2.3.3.9 DNA-Fällung Um DNA-Lösungen umzupuffern, zu reinigen oder die Konzentration der Lösung zu erhöhen, wurde DNA gefällt. Dabei wird durch Zugabe von 1/10 Volumen 3 M Natriumacetat (pH 4,8) und 3 Volumina 100%igem Ethanol die DNA gefällt und durch Zentrifugation (15 min, 15800 × g, 4°C) pelletiert. Das Pellet wurde mit 70%igem Ethanol gewaschen und nach erneuter Zentrifugation an der Luft getrocknet. Anschließend konnte es im gewünschten Volumen H2Obidest oder Puffer (TE-Puffer, Qiagen, Hilden) gelöst werden. 2.3.3.10 DNA-Restriktion Doppelsträngige DNA kann mit Hilfe von spezifischen Restriktionsendonukleasen (Restriktionsenzymen) geschnitten werden. Gemeinsame Eigenschaft der Restriktionsenzyme ist die Erkennung einer spezifischen DNA-Sequenz und das Schneiden der DNA. Hierbei können sogenannte „klebrige“ Enden („sticky ends“) oder „glatte“ Enden („blunt ends“) durch das Enzym erzeugt werden. Durch die DNA-Restriktion wurden DNA-Fragmente und Vektoren für eine Ligation vorbereitet und Klonierungen analysiert. Das Schneiden linearer oder ringförmiger DNA wurde nach dem Protokoll des Herstellers des Restriktionsenzyms (MBI Fermentas, St. Leon-Rot) durchgeführt. Standardansatz: 1 µg DNA 2/5 µl 10 × Restriktionspuffer 1-5 U Restriktionsendonuklease auf 20/50 µl mit H2Obidest auffüllen und mindestens 1 h bei 37°C inkubieren. Die Inaktivierung der Restriktionsenzyme erfolgte durch Hitzebehandlung, bei der sich Temperatur und Zeit nach den Herstellerangaben für das eingesetzte Restriktionsenzym richteten. Material und Methoden 28 Tab. 2.4: Liste der verwendeten Restriktionsendonukleasen. Restriktionsendonuklease Erkennungssequenz Bam HI G’GA TCC BsmB I C’GT CTC N Eco RI G’AA TTC Eco 31I G’GT CTC N Hind III A’AG CTT Nde I CA’T ATG Pst I CTG CA’G Xba I T’CT AGA Alle Restriktionsendonukleasen stammten von der Firma MBI Fermentas, St. Leon-Rot. Der Strich in der Erkennungssequenz kennzeichnet die Stelle, an der die DNA durch das Enzym durchtrennt wird. 2.3.3.11 DNA-Ligation Durch die Ligation mit Hilfe des Enzyms T4-DNA-Ligase werden doppelsträngige DNAFragmente miteinander verknüpft. Dabei werden „glatte“, oder „klebrige“ Enden von DNAFragmenten, die mit demselben Restriktionsenzym geschnitten wurden, durch die Bildung einer Phosphodiesterbindung zwischen einem 3`-OH-Ende und dem passenden 5`Phosphatende ligiert. So ist es möglich, in ein Plasmid ein DNA-„Insert (z. B. eine für ein Gen kodierende Nukleotidsequenz) zu integrieren. Durch ein molares Verhältnis zwischen Vektor- und DNA-„Insert -Molekülen von eins zu drei wird eine optimale Insertion erreicht, wenn beide Moleküle etwa gleich groß sind. Die Ligation wurde mit T4-DNA-Ligase (MBI Fermentas, St. Leon-Rot) nach Herstellerangaben durchgeführt. 20 µl-Ansatz: X µl geschnittener Vektor X µl „Insert 1,5 µl T4-DNA-Ligase ( 5 U/µl, MBI Fermentas, St. Leon-Rot) 2 µl 10 × Puffer (MBI Fermentas, St. Leon-Rot) X µl H2Obidest Durch die Inaktivierung der Ligase bei 65°C für 10 min wurde die Ligasereaktion beendet. • Formel zur Berechnung der einzusetzenden Vektor- und „Insert -Mengen für eine Ligation: ng Vektor × kb " Insert" 3 × = g " Insert" kb Vektor 1 Material und Methoden 29 2.3.3.12 Vektordephosphorylierung Um bei einer Ligation die Religation eines geschnittenen Vektors zu verhindern, wurde die geschnittene, gereinigte Vektor-DNA durch Alkalische-Phosphatase (CIAP, New England Biolabs (NEB), Beverly, MA, USA) dephosphoryliert. 50 µl Ansatz: 30-50 ng Vektor-DNA 5 µl 1 × NEB-Puffer (New England Biolabs, Beverly, MA, USA) 0,015-0,025 U Alkalische-Phosphatase X µl H2Obidest Der Ansatz wurde 1 h bei 37°C inkubiert. Die Reaktion wurde anschließend durch 20 minütige Erhitzung auf 65°C gestoppt. 2.3.3.13 Anhängen eines Adenin-Überhangs an ein PCR-Fragment Soll ein PCR-Fragment in den Vektor pDrive (Qiagen, Hilden) kloniert werden, wird am amplifizierten Fragment ein Adenin-Überhang (A-Überhang) benötigt. Dieser kann dann an den Uracil-Überhang (U-Überhang) des linearisierten Vektors binden. Taq-DNA- Polymerasen hängen durch ihre terminale Transferase-Aktivität automatisch ein Adenin an das 3´-Ende von glatten, doppelsträngigen PCR-Fragmenten. Mit Pfx-DNA-Polymerase amplifizierten Fragmenten fehlt jedoch dieser A-Überhang und muss in einer zusätzlichen Reaktion angehängt werden. 100 µl Ansatz: 1 µg DNA-Fragment 1 µl dATP (20 mM) 2,5 U Taq-DNA-Polymerase 10 µl 10 × PCR-Puffer auf 100 µl mit H2Obidest auffüllen Der gut gemischte Reaktionsansatz wurde 30 min bei 72°C inkubiert. 2.3.3.14 DNA-Sequenzanalyse Sequenzanalysen zur Identifizierung von cDNA-Klonen und zur Kontrolle von Klonierungen wurden mit der Didesoxy-Kettenabbruch-Methode (SANGER et al., 1977) durchgeführt. Während einer speziellen PCR mit standardisierten Oligonukleotid-Primern (z. B. T7- oder Sp6-Promotor-Primer, T7-Terminator) oder individuell hergestellten Primern wurden fluoreszierende Didesoxynukleotide, die einen Kettenabbruch hervorrufen, eingesetzt. Die Sequenzierungs-PCR erfolgte mit dem „ABI Prism®BigDyeTMTerminator Cycle Sequencing Ready Reaction Kit“ (PE Biosystems, Foster City, CA, USA). Material und Methoden Sequenzierungs-PCR-Ansatz: 30 4 µl „Ready Reaction Mix 10 pmol Primer 0,4-0,8 µg DNA-„template der PCR-Ansatz wurde mit Merck-Wasser auf 15 oder 20 µl Endvolumen aufgefüllt und dunkel gehalten. Sequenzierungs-PCR-Programm: 96°C 1 min 96°C 30 s 50°C 15 s 30 Zyklen 60°C 4 min 60°C 5 min 10°C Deckelheizung 105°C Durch eine Fällung wurde die amplifizierte DNA von nicht eingebauten Nukleotiden gereinigt. Die Auswertung der Sequenzen erfolgte mit dem ABI Prism® Sequenzierungsautomaten (PE Biosystems, Foster City, USA) durch Ulrike Coja in der Abteilung Spezielle Botanik der Universität Osnabrück. Dabei werden die Produkte der Sequenzierungs-PCR über ein Gel elektrophoretisch aufgetrennt und mit einem Laser abgetastet. Die daraus ermittelte Nukleotidsequenz wurde mit dem Computerprogramm Chromas (Version 1.43) aufgearbeitet. 2.3.3.15 Agarose-Gelelektrophorese und Gelextraktion Zur Auftrennung, Identifizierung und Reinigung von DNA-Molekülen dient die AgaroseGelelektrophorese, die in horizontalen Gelen (10 × 10 × 1 cm) mit 0,5 × TBE als Laufpuffer durchgeführt wurde (SAMBROOK et al., 1989). Die gewählte Agarose-Konzentration richtete sich hierbei nach der Größe der aufzutrennenden Nukleotidfragmente. Als Größenstandard wurde z. B. PstI-geschnittene Lambda-DNA (MBI Fermentas, St. Leon-Rot) verwendet. Zur Extraktion von DNA-Fragmenten aus dem Agarosegel wurde das „QIAquick Gel Extraction Kit“ oder „Min Elute Gel Extraction Kit“ (Qiagen, Hilden) genutzt. Material und Methoden 31 2.3.3.16 Klonierungen 2.3.3.16.1 Klonierung von ALD_pET-16b Für die heterologe Expression rekombinanter Mais-Aldolase (GenBank Acc. Nr.: M16220.1) wurde mit den Primern ALF8_sense und ALF_Antisense eine RT-PCR angesetzt. Reaktionsansatz: 25 µl 2 × „Reaction Mix“ (GibcoBRL, Karlsruhe) 1 µg RNA 10 µM ALF8_Sense 10 µM ALF_Antisense 1 µl RT-Taq Polymerase X µl H2Obidest Das Reaktionsvolumen betrug 50 µl. PCR-Programm: 50°C 30 min 94°C 2 min 94°C 15 s 61°C 30 s 40 Zyklen 72°C 2 min 72°C 10 min 4°C Das PCR-Produkt wurde über ein Agarosegel aufgereinigt, mit den Restriktionsenzymen Nde I und Bam HI geschnitten und mit dem „PCR Purification Kit“ (Qiagen, Hilden) von Nukleotiden gereinigt. Der Vektor pET-16b wurde ebenfalls mit den Restriktionsenzymen Nde I und Bam HI geschnitten und über ein Agarosegel gereinigt. Nach der Ligation des geöffneten Vektors mit dem geschnittenen PCR-Produkt („Insert ) wurden chemisch kompetente Zellen (E. coli XL1blue) mit der Hälfte des 1:5 mit H2Obidest verdünnten Ligationsansatzes transformiert. Von einigen der gewachsenen Kolonien wurden Übernachtkulturen angesetzt, um Plasmidisolierungen durchzuführen. Mit Hilfe von Restriktionsverdau der Plasmide wurden Plasmide mit einem „Insert in der passenden Größe identifiziert. Von einem positiven Klon wurde eine Glycerinkultur angelegt und zur genaueren Identifizierung eine Sequenzierung durchgeführt. Dieser Klon wurde zur heterologen Expression in E. coli BL21(DE3)pLysS transformiert. Material und Methoden 2.3.3.16.2 32 Klonierung von ALD_pGBKT7 Der ALD_pET-16b Klon diente als Ausgangsmaterial für die Klonierung der Aldolase in den Vektor pGBKT7. Da die Schnittstellen so gewählt waren, dass mit ihnen in beide Vektoren kloniert werden konnte, wurde das „Insert aus dem pET-16b Plasmid mit den Restriktionsenzymen Nde I und Bam HI herausgeschnitten, über ein Agarosegel gereinigt und in den ebenfalls mit Nde I und Bam HI geöffneten und gereinigten Vektor pGBKT7 ligiert. Nach der Transformation in E. coli XL1blue wurden auch hier die Plasmide isoliert, geschnitten und mit Hilfe eines Agarosegels analysiert. Positive Klone wurden anschließend ansequenziert. 2.3.3.16.3 Klonierung von ALD_pASK-IBA3 Für die Klonierung der Mais-Aldolase in den Vektor pASK-IBA3 wurde mit Pfx-Polymerase eine PCR mit dem Plasmid ALD_pET-16b angesetzt. Reaktionsansatz (50 µl): 5 µl 10 × Pfx-PCR Puffer 1,5 µl dNTP-Mix (10 mM) 1 µl MgSO4 (50 mM) 50 pmol Primer ALFStrepsense 50 pmol Primer ALFStrepantisense 1 µl „template“ (Plasmid) 1,5 µl Pfx-Polymerase (2,5 U/µl) 0 µl/ 5 µl/ 10 µl Enhancer Solution X µl H2Obidest PCR-Programm: 94°C 2 min 94°C 15 s 68°C 30 s 35 Zyklen 72°C 1 min 72°C 8 min 10°C Die PCR-Produkte wurden gereinigt und für die Ligation in pDrive mit einem A-Überhang versehen (s. Kap. 2.3.3.13). Nach einer weiteren Reinigung wurde das PCR-Fragment in pDrive ligiert und in E. coli XL1blue transformiert. Mit Hilfe der Blau-Weiß-Selektion wurden von vermutlich positiven Kolonien Übernachtkulturen (s. Kap. 2.3.4.1) angesetzt und nach Plasmidisolation durch einen Restriktionsverdau mit der Restriktionsendonuklease Eco RI analysiert. Ein positiver Klon wurde sequenziert. Das nach einem Restriktionsverdau Material und Methoden 33 mit dem Enzym Bsm BI ausgeschnittene „Insert wurde in den mit Eco 31I und Pst I geschnittenen Vektor pASK-IBA3 ligiert und in E. coli XL1blue transformiert. Positive Klone wurden durch Restriktionsanalyse identifiziert und mit den Primern Fw-seq-pASKIBA und Rev-seq-pASK-IBA sequenziert. 2.3.3.16.4 Klonierung von VDAC_pRSET A Für die Klonierung von VDAC aus Mais (Z. mays L., Acc. Nr.: AF178950.1) wurde mit den Oligonukleotiden vdac_pRSET-For und vdac_pRSET-Rev eine PCR mit cDNA als „template angesetzt. Das 830 bp lange Produkt dieser PCR wurde in pDrive kloniert, durch Restriktionsverdau mit Bam HI und Eco RI analysiert und für die Klonierung in den Vektor pRSET A vorbereitet. Der Vektor wurde ebenfalls mit diesen beiden Restriktionsendonukleasen geschnitten und dephosphoryliert, um eine Religation zu verhindern. Die gereinigten DNA-Fragmente wurden in den Vektor ligiert. Nach der Transformation in E. coli XL1blue konnten wieder durch Restriktionsanalysen positive Klone identifiziert werden. Davon wurden Glycerinkulturen angelegt. Durch Sequenzanalysen wurden die Konstrukte überprüft. PCR-Programm: 94°C 2 min 94°C 15 s 75°C 30 s 35 Zyklen 72°C 1 min 72°C 8 min 10°C Für Überexpressionsversuche wurde E. coli BL21(DE3)pLysS transformiert. das Konstrukt VDAC_pRSET A in Material und Methoden 2.3.3.16.5 34 Klonierung von VDAC_pGBKT7 Mit den Primern vdac sense2_Hyb und vdacpRSET-Rev wurde eine PCR mit cDNA als „template“ angesetzt. Die PCR entsprach einer Standard-PCR, wobei jedoch die Primer in einer Konzentration von 100 pmol eingesetzt wurden. Die Temperatur der PCR wurde auf 55°C optimiert. PCR-Programm: 94°C 2 min 94°C 15 s 55°C 30 min 35 Zyklen 72°C 5 min 10°C Das über ein Agarosegel gereinigte PCR-Produkt wurde zunächst in pDrive kloniert und in E. coli XL1blue transformiert. Ein Klon mit einem Plasmid mit „Insert in der entsprechenden Größe wurde sequenziert. Das „Insert wurde mit den Restriktionsenzymen Nde I und Eco RI herausgeschnitten und in den ebenfalls mit diesen Enzymen geöffneten, dephosphorylierten Vektor pGBKT7 ligiert. Nach erfolgter Transformation in E. coli XL1blue und Plasmidisolierung erfolgte die Identifizierung positiver Klone über Restriktionsanalysen und eine Ansequenzierung. 2.3.3.16.6 Klonierung von Aktin_pGBKT7 Bei den PCRs zur Amplifizierung von Aktin aus Mais (Z. mays L., Acc. Nr.: J01238) wurde versucht, durch Optimierung der MgCl2-Konzentration ausreichende Mengen an PCRProdukt zu erhalten. Da trotz der Optimierungsversuche die amplifizierten DNA-Mengen sehr gering blieben, wurden 15 Standard-PCR-Reaktionen mit den Primern Aktin_sense und Aktin_antisense (je 10 pmol) angesetzt. Die Produkte dieser PCR-Reaktionen wurden vereinigt und mit Hilfe des „MinElute Gel Extraktion Kit“ (Qiagen, Hilden) gereinigt. Von dieser Probe wurde die maximal mögliche Menge (13,5 µl) in einen 20 µl Ligationsansatz mit pDrive eingesetzt. Mit Hilfe der Blau-Weiss-Selektion wurde nach der Transformation in E. coli XL1blue nach positiven Transformanden gesucht. Die nach Restriktionsanalyse identifizierten drei positiven Klone wurden als Glycerinkulturen angelegt und ansequenziert. Material und Methoden 35 Mit den Enzymen Pst I und Bam HI wurde dann ein Restriktionsverdau mit dem Vektor pGBKT7 und den pDrive-Plasmiden mit „Insert angesetzt. Die Reinigung der DNA nach dem Restriktionsverdau wurde mit dem „MinElute Gel Extraction Kit“ (Qiagen, Hilden) durchgeführt. Nach Ligation und Transformation in E. coli XL1blue wurden durch Restriktionsanalysen und Sequenzierungen positive Klone gesucht. PCR-Programm: 94°C 2 min 94°C 15 s 61°C 30 min 35 Zyklen 72°C 5 min 10°C 2.3.4 Arbeiten mit Bakterien 2.3.4.1 Bakterienkulturen Die für molekularbiologische und biochemische Untersuchungen verwendeten Bakterienstämme wurden auf verschiedene Art und Weise kultiviert. Übernachtkulturen dienten zur Plasmidisolierung aus Bakterien sowie als Vorkultur für die Herstellung größerer Mengen von Flüssigkulturen. Zum Vereinzeln von Bakterienkulturen sowie zur Vermehrung aus Glycerinkulturen wurden Bakterien auf Agarplatten kultiviert. Glycerinkulturen dienten der Lagerung. Übernachtkultur: In einem Reagenzglas wurden 3-5 ml YT-Flüssigmedium mit dem gewünschten Bakterienstamm inokuliert. Die Bakterien konnten von einer Einzelkolonie auf einer Plattenkultur oder aber aus einer Glycerinkultur stammen und wurden mit Hilfe eines sterilen Zahnstochers übertragen. Bei 37°C erfolgte die Anzucht unter aeroben Bedingungen (Schüttler, ca. 200 Upm) für 16-18 h. Plattenkultur: Die Zellen wurden hierfür mit einer Impföse oder einem Drygalski-Spatel auf einer Platte mit YT-Festmedium ausgestrichen. Dabei variierte die Menge der ausplattierten Bakteriensuspension zwischen 10 und 100 µl. Wenn die vorhandene Flüssigkeit vollständig in die Platte eingezogen war, wurden die Agarplatten mit der Unterseite nach oben, um Kondenswasserablagerung auf dem Medium zu vermeiden, bei 37°C im Brutschrank 16-18 h inkubiert. Material und Methoden 36 Glycerinkultur: Bakterien in mit Glycerin versetztem Medium können bei –70°C eingefroren und über mehrere Monate gelagert werden. Für die Herstellung einer Glycerinkultur wurden 50% (v/v) Bakterien-Flüssigkultur mit 50% (v/v) Glycerinmedium gemischt und in flüssigem Stickstoff eingefroren. Die Lagerung erfolgte bei –70°C. Alle Medien wurden vor der Verwendung sterilautoklaviert (20 min, 121°C, 1 bar) und, wenn möglich, mit einem Selektionsmarker in Form eines Antibiotikums versetzt. Mit Hilfe der Selektionsmarker ist es möglich, Kontaminationen zu verhindern und auf die Anwesenheit eines Plasmids in den E. coli-Stämmen zu selektionieren. Die in dieser Arbeit verwendeten Antibiotika sind in Tabelle 2.5 aufgeführt. YT-Medium: 8 g/l Bacto-Tryptone 5 g/l Hefeextrakt pH 7,0 (NaOH) 2,5 g/l NaCl 15 Glycerinmedium: g/l Agar (für Festmedium) 20% YT-Medium 80% Glycerin Tab. 2.5: Verwendete Antibiotika und ihre Konzentrationen. Antibiotikum Stammlösung Endkonzentration Ampicillin (Amp) 100 mg/ml in 50% Ethanol* 100-200 µg/ml Tetrazyclin (Tet) 5 mg/ml in 50% Ethanol* 10 µg/ml Kanamycin (Kan) 50 mg/ml in sterilem H2O* 25 µg/ml Chloramphenicol (Clm) 12,5 mg/ml in 50% Ethanol** 25 µg/ml *sterilfiltriert, Aufbewahrung bei - 20°C **Aufbewahrung bei + 4°C 2.3.4.2 Herstellung kompetenter E. coli-Zellen Zellen, die in der Lage sind, DNA aufzunehmen, bezeichnet man als kompetent. Die beiden verwendeten Methoden zur Herstellung chemisch und elektrisch kompetenter Zellen sind im Folgenden beschrieben. Material und Methoden 37 a) Chemisch kompetente E. coli-Zellen Die Herstellung chemisch kompetenter E. coli-Zellen durch Behandlung mit Rubidiumchlorid erfolgte nach der Methode von HANAHAN (1983). Mit einer Übernachtkultur mit Tetrazyclin für E. coli XL1blue und Chloramphenicol für E. coli BL21(DE3)pLysS wurden 100 ml YTMedium ohne Antibiotikum im Verhältnis 1:100 angeimpft und bis zu einer optischen Dichte von A600 = 0,5, gegen H2Obidest gemessen, angezogen und anschließend durch Zentrifugation (10 min, 5000 × g, 4°C) geerntet. Die Zellen wurden zunächst in 30 ml TFB1-Lösung vorsichtig resuspendiert und anschließend 5-10 min auf Eis inkubiert. Nach erneuter Zentrifugation wurden die Zellen in 4 ml gekühlter TFB2-Lösung resuspendiert und in 200 µl-Aliquots in flüssigem Stickstoff schockgefroren und bis zur weiteren Verwendung bei –70°C gelagert. TFB1-Lösung: 100 mM RbCl2 pH 5,8 (Essigsäure) 50 mM MnCl2 30 mM Kaliumacetat 10 mM CaCl2 15% (v/v) Glycerin TFB2-Lösung: 75 mM CaCl2 pH 7,0 (NaOH) 10 mM MOPS 10 mM RbCl2 15% (v/v) Glycerin Die Kompetenz der Zellen zur DNA-Aufnahme wurde mit Hilfe eines Kompetenztests quantifiziert. Dafür wurden 200 µl kompetenter Zellen mit 40 ng Plasmid-DNA (pBSK) transformiert (s. Kap. 2.3.4.3). Von diesem Transformationsansatz wurden 10 µl, 50 µl und 100 µl auf selektiven Agarplatten ausplattiert und 16-18 h bei 37°C inkubiert. Bei einer gut bewachsenen Platte wurden die Kolonien ausgezählt. Die Transformationseffizienz (Te) wurde mit folgender Formel berechnet. Te = gezählte Kolonien × 1000 ng × eingesetzte DNA-Menge Die Zellen zeigen eine gute Kompetenz zur Vol gesamt Vol ausplattiert Aufnahme von DNA, Transformationseffizienz zwischen 1×106 und 1×107 liegt. wenn die Material und Methoden 38 b) Elektrisch kompetente E. coli-Zellen Die Herstellung elektrisch kompetenter E. coli-Zellen erfolgte in Anlehnung an das „Electroporator II Manual“ (Invitrogen, Karlsruhe). Um E. coli-Zellen elektrisch kompetent zu machen, wurden 50 ml selektives LB-Medium mit E. coli XL1blue angeimpft und über Nacht bei 37°C im Schüttler (ca. 200 Upm) angezogen. Mit dieser Übernachtkultur wurde 1 Liter selektives LB-Medium angeimpft und bis zu einer optischen Dichte von A550 = 0,5-0,6 angezogen. Die Kultur wurde auf vorgekühlte Zentrifugenbecher verteilt und 30 min auf Eis inkubiert. Anschließend wurden die Zellen durch Zentrifugation (15 min, 4000 × g, 0°C) geerntet. Das Zellpellet wurde in vier Schritten (25 ml + 25 ml + 50 ml + 150 ml) in insgesamt 250 ml sterilem, kaltem H2Obidest resuspendiert. Zwischen den Resuspensionsschritten wurden die Zellen auf Eis aufbewahrt. Nach erneuter Zentrifugation wurden die Zellen in insgesamt 125 ml kaltem H2Obidest resuspendiert (25 ml + 50 ml + 50 ml) und anschließend noch einmal durch Zentrifugation pelletiert. Mit einer gekühlten Glaspipette wurde das Pellet in 20 ml steriler 10%iger Glycerinlösung resuspendiert. In sterilen Zentrifugenbechern aus dem Gefrierschrank (- 20°C) wurden die Zellen noch einmal zentrifugiert. Anschließend wurde das Pellet in 4 ml 10 %iger Glycerinlösung aufgenommen. Diese Zellsuspension wurde in 80 µl Aliquots in flüssigem Stickstoff eingefroren und bis zur Verwendung bei –70°C gelagert. LB-Medium: 10 g/l Select Pepton pH 7,5 (NaOH) 5 g/l Hefeextrakt 5 g/l NaCl 2.3.4.3 Transformation von E. coli-Zellen Bei der Transformation wird freie, ringförmige DNA von einer Zelle aufgenommen. Die beiden verwendeten Methoden der Transformation chemisch und elektrisch kompetenter Zellen sind im Folgenden beschrieben. a) Transformation chemisch kompetenter E. coli-Zellen Chemisch kompetente E. coli-Zellen (100 µl) wurden 20 min auf Eis aufgetaut, mit bis zu 1 µg Plasmid-DNA oder einem fünffach verdünnten Ligationsansatz gemischt und 30 min auf Eis inkubiert. Es folgte ein Hitzeschock bei 42°C für 90 s (E. coli XL1blue) bzw. 20 s (E. coli BL21(DE3)pLysS), an den sich eine Inkubation von 1-2 min auf Eis anschloss. Zu den Zellen wurden dann 1000 µl nicht selektives Flüssigmedium gegeben; die Zellen wurden Material und Methoden 39 zur Regeneration für 1-2 h bei 37°C aerob angezogen. Anschließend wurden 10 µl, 50 µl und 100 µl auf selektiven Agarplatten ausplattiert und über Nacht bei 37°C kultiviert. b) Transformation elektrisch kompetenter E. coli-Zellen Diese Methode diente der Transformation von E. coli-Zellen mit aus Hefen isolierten Plasmiden. Hierbei wird die Plasmamembran der Zellen durch Anlegen einer Spannung kurzzeitig depolarisiert und so für DNA durchlässig. Für die Elektroporation wurden Elektroporationsküvetten (0,1 cm, Invitrogen, Karlsruhe) vorgekühlt. Auf Eis aufgetaute elektrisch kompetente E. coli-Zellen wurden mit 20 µl aus Hefen isolierter Plasmid-DNA (s. Kap. 2.3.5.3) gemischt und in die Küvetten gegeben. Im Elektroporater (Gene PulserTM, Bio-Rad, München) wurden die Zellen bei folgenden Einstellungen der elektrischen Spannung ausgesetzt: Kapazität: 25 F Widerstand: 200 Ω Volt: 1,25 V Zeit: konstant In die Küvetten wurde dann 1 ml SOC-Medium gegeben. Die Probe wurde aus der Küvette in ein steriles 1,5 ml-Reaktionsgefäß pipettiert und ca. 60 min bei 37°C im Schüttler (200 Upm) inkubiert, damit sich die Zellen regenerieren. Von dieser Zellsuspension wurden 100 µl und 200 µl auf selektiven Agarplatten ausplattiert, der Rest ca. 1 min bei 4000 × g abzentrifugiert, das Pellet in ca. 20 µl Medium resuspendiert und ebenfalls ausplattiert. Die Platten wurden 16-18 h im Brutschrank bei 37°C inkubiert. SOC-Medium: 20 g/l Bacto-Tryptone 5 g/l Hefeextrakt 0,584 g/l NaCl 0,186 g/l KCl pH 7,0 (NaOH) auf 980 ml mit H2Obidest auffüllen Vor Gebrauch 10 ml sterile „2 Mg-Lösung“ (aus 1 M MgCl2 und 1 M MgSO4) und 10 ml sterile 2 M Glucose-Lösung zugeben. Material und Methoden 40 2.3.4.4 Blau-Weiß-Selektion Die Blau-Weiß-Selektion hilft bei Klonierungen in den Vektor pDrive, um positive Transformanden zu identifizieren. Dieses Plasmid trägt das Gen für die lacZ-Untereinheit, die auf dem E. coli Chromosom im Stamm XL1blue deletiert ist. Innerhalb der lacZ Untereinheit befindet sich die Klonierungsstelle des Plasmids. Erfolgt die Insertion eines DNA-Fragments in die Klonierungsstelle, kann keine Komplementation stattfinden und die E. coli-Zellen sind dadurch nicht in der Lage, das Substrat 5-Bromo-4-chloro-3-indolyl-β-DGalaktopyranosid (X-Gal) in Galaktose und einen blauen Farbstoff zu spalten. Isopropyl-β-DThiogalaktopyranosid (IPTG) wird als molekularer Induktor des lac-Operons eingesetzt. Mit Hilfe dieses Systems können positive Transformanden anhand ihrer weißen Koloniefarbe identifiziert werden. Um diese Selektion durchführen zu können, wurden die Agarplatten ca. 1 h vor dem Ausplattieren mit 80 µl X-Gal (200 mg/ml) und 8 µl 20%igem IPTG vorbehandelt. 2.3.4.5 Plasmidisolierung aus E. coli Die Isolierung von Plasmid-DNA aus Bakterienzellen für Restriktionsanalysen, Ligationen, Transformationen und Sequenzanalysen wurde nach der Methode von BIRNBOIM und DOLY (1979) mit Hilfe von „QIAprep Spin Plasmid Kits“ (Qiagen, Hilden) nach Herstellerangaben durchgeführt. 2.3.5 Arbeiten mit Hefen Zur Analyse von Protein-Protein-Interaktionen wurde ein 2-Hybrid-System in Hefe (S. cerevisiae) gewählt. Das verwendete „MATCHMAKER Library Construction & Screening Kit” (Clontech, Heidelberg) beruht auf der Grundlage eukaryotischer Transkription, die unter Kontrolle von Transkriptionsfaktoren steht. Transkriptionsfaktoren bestehen in der Regel aus zwei Domänen, der DNA-Bindedomäne (DNA-BD), die für die spezifische Erkennung einer DNA-Sequenz (UAS = „upstream activation sequence“) verantwortlich ist, und einer Aktivierungsdomäne (AD), die die Transkription initiiert. Die künstlich voneinander getrennten Domänen eines Transkriptionsfaktors können die Transkription des spezifischen Gens nur hervorrufen, wenn beide Domänen miteinander wechselwirken, d. h. sich räumlich sehr nahe kommen. Bei dem verwendeten System werden vier Reportergene (MEL1, lacZ, ADE2 und HIS3) durch die Wiederherstellung des Material und Methoden Transkriptionsfaktors transkribiert. Die daraus resultierenden Auxotrophien und die 41 - Galaktosidase-Aktivität ermöglichen eine schnelle Identifizierung positiver Kolonien. Die Fusionsproteine mit den Domänen des Transkriptionsfaktors werden durch Klonierung der entsprechenden DNA-Sequenzen in die Vektoren pGBKT7 und pGADT7-Rec (Clontech, Heidelberg) hergestellt. Bei dem sogenannten „Bait -Protein handelt es sich um das Protein, für das Interaktionspartner gesucht werden und das als Fusionsprotein mit der DNA-BD des Transkriptionsfaktors hergestellt wird. Bei dem verwendeten System wird die AD mit einer cDNA-Bank fusioniert, wodurch ermöglicht wird, in einem Versuch viele verschiedene Interaktionspartner zu finden. Bei diesen Fusionsproteinen aus der cDNA-Bank spricht man von „Prey -Proteinen. Die Plasmide werden dann in Hefezellen verschiedenen Paarungstyps (a und ) transformiert, so dass die Suche nach interagierenden Proteinen über die Paarung der Hefezellen erfolgen kann. 2.3.5.1 Anzucht von S. cerevisiae Die Kultivierung der Hefen erfolgte in Flüssigkultur, auf Agarplatten oder zur Aufbewahrung über einen längeren Zeitraum bei –70°C in Glycerinkulturen. Als Anzuchtmedium wurde ein Vollmedium (YPD) bzw. das Vollmedium mit Zusatz von Adenin (YPDA) oder ein den jeweiligen Auxotrophien entsprechendes Minimalmedium (SD-Medium) gewählt. Der Zusatz von Adenin soll Spontanmutationen des Hefestammes AH109 verhindern. Übernachtkultur: Die angesetzte Menge der Übernachtkultur variierte je nach geplanter Verwendung. Für eine Plasmidisolierung wurden 2 ml des entsprechenden Flüssigmediums mit einer Hefekolonie inokuliert. Für die Herstellung kompetenter Hefezellen wurden 50 ml Kultur benötigt. Es wurde jeweils eine frische, d. h. bis zu zwei Monate alte, Hefekolonie mit einem sterilen Zahnstocher zu 1 oder 2 ml Medium gegeben und durch kräftiges Schütteln aufgelöst. Mit dieser Lösung wurde das restliche Medium angeimpft und 16-18 h bei 30°C im Schüttler bei ca. 200 Upm aerob bis zu einer optischen Dichte von A600 > 1,5 angezogen. Plattenkultur: Auf Agarplatten mit dem entsprechenden Medium wurden die Hefezellen mit einer Impföse ausgestrichen oder, wie in Kapitel 2.3.5.5 beschrieben, nach einer Paarung mit Hilfe eines Drygalski-Spatels ausplattiert. Material und Methoden 42 Glycerinkultur: Zur längeren Aufbewahrung der Hefestämme wurden Glycerinkulturen angelegt. Dafür wurde eine Hefekolonie mit der Impföse in 200 µl des entsprechenden selektiven Mediums durch kräftiges Schütteln gelöst. Zu dieser Hefesuspension wurden 200 µl Glycerin-YPD-Medium gegeben und die Zellen in flüssigem Stickstoff schockgefroren. Die Lagerung bis zur weiteren Verwendung erfolgte bei –70°C. YPD-Medium: 20 g/l Pepton pH 5,8 (HCl) 10 g/l Hefeextrakt 20 g/l Agar (für Festmedium) auf 950 ml mit H2Obidest auffüllen Nach dem Autoklavieren wird auf ca. 55°C abgekühlt und 50 ml 40%ige sterilfiltrierte Glucose-Lösung zugegeben. YPDA-Medium: YPD-Medium, wie oben beschrieben, zubereiten und zusätzlich zur Glucose nach dem Autoklavieren 15 ml einer sterilfiltrierten 0,2%igen Adeninhemisulfat-Lösung zugeben. Glycerin-YPD-Medium: SD-Medium: 50% Glycerin in YPD-Medium 6,7 g/l Hefe-Stickstoff-Basis ohne Aminosäuren 20 pH 5,8 g/l Agar (bei Festmedium) auffüllen auf 850 ml mit H2Obidest Nach dem Autoklavieren und Abkühlen auf ca. 55°C 50 ml sterile 40%ige Glucose-Lösung und 100 ml sterile 10 × „Dropout-Solution“ zugeben. Die Zusammensetzung der 10 × „Dropout-Solution“ ist Tabelle 2.6 zu entnehmen. Je nach „Dropout-Solution“ wurde die entsprechende Aminosäure nicht zugegeben. SD/-Leu Medium wird mit einer „Dropout Solution“ ohne Leucin hergestellt. TDO „Dropout Solution“ („Triple Dropout ) fehlen drei Substanzen (His/Leu/Trp) und QDO („Quadruple Dropout ) enthält kein Adeninhemisulfat, Histidin, Leucin und Tryptophan. Material und Methoden 43 Tab. 2.6: Zusammensetzung der „Dropout-Solution“ zur Selektion auf Hefeauxotrophien. Alle Substanzen stammten von der Firma Sigma-Aldrich Chemie GmbH, Taufkirchen. Substanz 10 x Konzentration L-Adeninhemisulfat 200 mg/l L-Arginin-HCl 200 mg/l L-Histidin-HCl-Monohydrat 200 mg/l L-Isoleucin 300 mg/l L-Leucin 1000 mg/l L-Lysin-HCl 300 mg/l L-Methionin 200 mg/l L-Phenylalanin 500 mg/l L-Threonin 2000 mg/l L-Tryptophan 200 mg/l L-Tyrosin 300 mg/l L-Uracil 200 mg/l L-Valin 1500 mg/l 2.3.5.2 Herstellung kompetenter Hefezellen und Transformation von Hefen Für die Herstellung kompetenter Hefezellen wurde eine 50-ml-Übernachtkultur mit einer optischen Dichte von A600 1,5 benötigt. Diese wurde in dem entsprechendem Medium (YPD oder YPDA) angesetzt. Mit der Übernachtkultur wurden dann 300 ml YPD/YPDA-Medium angeimpft und auf eine optische Dichte von A600 = 0,2-0,3 eingestellt. Diese Kultur wurde nun bei 30°C unter Schütteln mit ca. 230 Upm etwa 3 h bis zu einer optischen Dichte von A600 = 0,4-0,6 angezogen. Die Ernte erfolgte durch Zentrifugation (5 min, 1000 × g, RT). Das Zellpellet wurde gründlich mit ca. 25 ml 1 × TE-Puffer gewaschen und erneut zentrifugiert. Der Überstand musste nun sehr vorsichtig dekantiert werden, da das Pellet sehr weich war. Es wurde anschließend in 1,5 ml frisch hergestellter 1 × TE/LiAc-Lösung aufgenommen. Die Zellen können danach bei Raumtemperatur einige Stunden aufgehoben werden. Für die sich anschließende Transformation der kompetenten Zellen wurden 0,1 µg PlasmidDNA und 0,1 mg „Herring Testes Carrier DNA“ (Clontech, Heidelberg) gemischt. Dazu wurden 100 µl der kompetenten Hefezellen gegeben und wiederum gut gemischt. Nach Zugabe von 600 µl PEG/LiAc-Lösung wurde 10 s kräftig geschüttelt. Die so behandelten Zellen wurden nun für 30 min bei 30°C und 200 Upm geschüttelt. Zu dem Zellgemisch wurden dann 70 µl Dimethyl-Sulfoxid (DMSO) gegeben und durch vorsichtiges Invertieren Material und Methoden 44 gemischt. Anschließend erfolgte ein Hitzeschock für 15 min bei 42°C im Wasserbad und ein Abkühlen auf Eis für 1-2 min. Nach dem Abzentrifugieren der Zellen (5 sec, 15800 × g, RT) wurde das Zellpellet in 0,5 ml TE-Puffer aufgenommen. Von dieser Zellsuspension wurden 100 µl auf den entsprechenden SD-Platten ausplattiert. Um sicherzugehen, dass gut getrennte Einzelkolonien entstehen, und um die Transformationseffizienz zu testen, wurden zusätzlich je 100 µl einer 1:10, 1:100 und 1:1000 Verdünnung ausplattiert. Die Platten wurden bis zum Erscheinen von Kolonien nach ca. 4 Tagen bei 30°C im Brutschrank kultiviert. Die Transformationseffizienz konnte durch Auszählen der Kolonienanzahl von einer Platte mit 30-300 Kolonien ermittelt werden. Mit Hilfe der unten aufgeführten Formel ist die Anzahl der gewachsenen Hefekolonien pro µg DNA zu errechnen. gezählte Kolonien × Vol (Suspension) Vol (ausplattiert) × Verdünnungsfaktor × Plasmid-DNA (µg) 10 × TE-Puffer: 100 mM Tris = Kolonien / µg DNA pH 7,5 (HCl) 10 mM EDTA sterilfiltriert 50% PEG 3350: mit sterilem H2Obidest herstellen (eventuell auf 50°C erwärmen) 10 × LiAc: 1 M Lithiumacetat PEG/LiAc-Lösung: 8 ml 50 % PEG 3350 pH 7,5 (Essigsäure), autoklavieren 1 ml 10 × TE 1 ml 10 × LiAc vor Gebrauch frisch ansetzen Material und Methoden 45 2.3.5.3 Plasmidisolierung aus S. cerevisiae Das Protokoll für die Isolierung von Plasmiden aus S. cerevisiae wurde von Frau Prof. Dr. Doris Rentsch (Molekulare Pflanzenphysiologie, Universität Bern, Schweiz) zur Verfügung gestellt. Eine in selektivem Medium hergestellte 2 ml Übernachtkultur wurde in einem 2 mlReaktionsgefäß abzentrifugiert (3 min, 2000 × g, RT) und das Pellet wurde in 200 ml Aufschlusspuffer resuspendiert. Zu dieser Suspension wurden in Säure gewaschene Glasperlen (0,25-0,7 mm Durchmesser) gegeben, bis noch ca. 1-2 mm Flüssigkeit über den Glasperlen stand. Dazu wurden 200 µl Phenol/Chloroform/Isoamylalkohol (25:24:1) gegeben und mindestens 1,5 min kräftig geschüttelt. Nach einer Zentrifugation (5 min, 14000 × g, RT) wurde die entstandene wässrige Phase abgenommen. Die Phenol/Chloroform-Fällung wurde noch zwei Mal wiederholt, jedoch ohne erneute Zugabe von Glasperlen. Nach der letzten Phenol/Chloroform-Fällung wurden zur wässrigen Phase 0,1 Vol Na-Acetat (pH 5,2) und 2,5 Vol 100%iges Ethanol gegeben. Nach dem Mischen auf Eis wurde 20-30 min inkubiert. Die gefällte DNA wurde durch Zentrifugation (20 min, 14000 × g, 4°C) pelletiert. Nach dem Waschen des Pellets mit 200 µl 70%igem Ethanol und einer erneuten Zentrifugation wurde das Pellet getrocknet. Das trockene Pellet wurde in 20 µl H2Obidest resuspendiert und bei einer Transformation elektrisch kompetenter E. coli-Zellen vollständig verwendet. Aufschlusspuffer: 10 mM Tris pH 8,0 (HCl) 100 mM NaCl 1 mM EDTA 1% SDS 2% Triton X-100 In Säure gewaschene Glasperlen: Glasperlen in einem Erlenmeyerkolben 15 min mit 1 M HCl schütteln und dann so lange mit H2Obidest waschen, bis der pH-Wert nicht mehr im sauren Bereich liegt (pHPapier). Die Perlen zweimal mit 100%igem Ethanol spülen und im Trockenschrank ca. 2 Tage trocknen lassen. Material und Methoden 46 2.3.5.4 Herstellung einer cDNA-Bank für das Hefe-2-Hybrid-System Bei dem in dieser Arbeit genutzten Hefe-2-Hybrid-System wird für ein bekanntes Protein ein Interaktionspartner aus einer cDNA-Bank gesucht. Die Herstellung der cDNA-Bank beruht auf dem Prinzip der homologen Rekombination. Hierfür wird bei der cDNA-Synthese ein Oligo(dT) Primer (CDS III, Clontech, Heidelberg) eingesetzt, der am 3´-Ende der RNA (PolyA-Schwanz) binden kann. Durch RNase-Verdau des 5`-Endes der RNA wird der untranslatierte Bereich der mRNA abgespalten und durch die PowerScript Reverse Transkriptase (Clontech, Heidelberg) werden an das 3´-Ende zusätzliche Nukleotide (Deoxycytidine) angehängt. Daran kann dann der SMART III Primer (Clontech, Heidelberg) binden. Die entstehenden cDNA-Fragmente besitzen am 3´-Ende die Sequenz des SMART III Primers, am 5´-Ende die Sequenz des CDS III Primers. Bei dem linearisierten Vektor pGADT7-Rec enthalten die Enden den Primern komplementäre Sequenzen, so dass durch homologe Rekombination und Reparaturenzyme der Hefe der linearisierte Vektor mit den DNA-Fragmenten in seine zirkuläre Form gebracht wird. Abb. 2.1 gibt einen Überblick über das verwendete System. Material und Methoden 47 Abb. 2.1: Schematische Darstellung des Hefe-2-Hybrid-Systems mit Herstellung der cDNA-Bank und in vivo-Rekombination. Dargestellt ist die Durchführung des Versuchs durch Cotransformation, nicht durch Paarung der Hefen (aus: „MATCHMAKER Library Construction & Screening Kit“ von Clontech, Heidelberg). Material und Methoden 48 Die cDNA-Bank wurde aus hypoxisch angezogenem Sproß- und Wurzelgewebe von Mais (Z. mays L., var. Caramba) hergestellt. Dafür wurde zunächst aus dem fein gemörserten Maisgewebe Gesamt-RNA isoliert (siehe Kap. 2.3.3.1). Die Qualität der isolierten RNA wurde mit Hilfe eines denaturierenden Formaldehyd/Agarose-Gels (RNA-Gel) überprüft. Für die anschließende Synthese der „first-strand cDNA“ wurden in einem 0,25 ml Reaktionsgefäß folgende Substanzen (Clontech, Heidelberg) gemischt: 0,1-2,0 µg RNA-Probe bzw. 1 µl „Human Placenta Poly A+ RNA“ (als Kontroll-RNA) 1 µl CDS III Primer X µl H2Obidest (autoklaviert), so dass das Endvolumen 4 µl betrug. Nach einer Inkubation für 2 min bei 72°C und anschließendem 2-minütigem Abkühlen auf Eis wurde die Probe kurz abzentrifugiert. Folgende Substanzen wurden zugeben: 2,2 µl 5 × „first-strand“ Puffer 1 µl DTT (20 mM) 1 µl dNTP Mix (10 mM) 1 µl RNase I (5 U/ml) 1 µl Powerscript Reverse Transkriptase und nach vorsichtigem Mischen und schwachem Abzentrifugieren 10 min lang bei 42°C inkubiert. Nach Zugabe von 1 µl SMART III Oligonukleotiden folgte eine einstündige Inkubation bei 42°C. Durch Hitzebehandlung (10 min, 75°C) wurde die Synthese beendet. Die Probe wurde auf Raumtemperatur abgekühlt und für einen RNase-Verdau wurden 2 U Rnase H zugegeben und 20 min bei 37°C inkubiert. Die so hergestellte „first-strand cDNA kann zur Lagerung bis zu drei Monate bei –20°C eingefroren werden. Material und Methoden 2.3.5.4.1 49 Amplifizierung doppelsträngiger cDNA durch „long distance-PCR“ Die cDNA (2 µl) wurde auf Eis gekühlt, bevor sich die „long distance-PCR“ (LD-PCR) anschloss. Für die LD-PCR wurden gemischt: 2 µl cDNA 70 µl H2Obidest 10 µl 10 × Advantage 2 PCR Puffer* 2 µl 50 × dNTP Mix* 2 µl 5’ PCR-Primer* 2 µl 3’ PCR-Primer* 10 µl 10 × GC-Melt Lösung* 2 µl 50 × Advantage 2 Polymerase Mix* 100 µl Endvolumen (*Produkte von Clontech, Heidelberg) PCR-Programm: PCR-Block auf 95°C vorheizen 95°C 30 s 95°C 10 s 68°C 6 mina 17 Zyklen 68°C 5 min 4°C a : Bei jedem Zyklus wurde die Verlängerungsphase („Extension ) um 5 sec verlängert, so dass dadurch im 17. Zyklus die „Extension -Zeit 7 min und 20 s betrug. Die Zyklenanzahl dieser LD-PCR richtete sich nach der RNA-Menge (µg), die zur Synthese der “first-strand cDNA“ eingesetzt worden war. Für 1-2 µg Gesamt-RNA sollten 15-20 Zyklen optimal sein. Das Ergebnis der LD-PCR wurde durch Auftragen einer 5 µl Probe auf ein 1,2%iges Agarosegel mit Hilfe von 0,25 µg eines 1-kb Größenstandards (GeneRulerTM 1kb DNA Ladder, MBI Fermentas, St. Leon-Rot) analysiert. Bis zur Weiterverwendung erfolgte die Lagerung der doppelsträngigen (ds) cDNA bei –20°C. Material und Methoden 2.3.5.4.2 50 Reinigung der doppelsträngigen cDNA Die Reinigung der ds cDNA erfolgte mit CHROMA SPIN+TE-400 Säulen (Clontech, Heidelberg) nach Herstellerangaben. Mit dieser Methode werden DNA-Moleküle mit einer Größe von über 200 Basenpaaren (bp) von kleineren DNA-Molekülen gereinigt. Das Prinzip beruht auf der unterschiedlichen Wanderungsgeschwindigkeit verschieden großer Moleküle innerhalb einer Säule auf Grund der Porengröße der Säulenmatrix. Große Moleküle eluieren schneller als kleine Moleküle. Nach dieser Reinigung ist die ds cDNA für die in vivo-Rekombination in den Vektor pGADT7-Rec fertig und kann bei –20°C gelagert werden. 2.3.5.4.3 In vivo-Rekombination Für die Transformation des Hefestammes AH109 mit der ds cDNA und dem linearisierten Vektor pGADT7-Rec mussten zunächst kompetente Hefezellen hergestellt werden (siehe Kap. 2.3.5.2). In einem vorgekühlten 15 ml Reaktionsgefäß wurden gemischt: 20 µl ds cDNA 6 µl pGADT7-Rec* (0,5 µg/µl) 20 µl denaturierte „Herring Testes Carrier DNA“* (*Produkte der Firma Clontech, Heidelberg) und nach Zugabe von 600 µl kompetenter Hefezellen wurde noch einmal vorsichtig gemischt. Nachdem 2,5 ml PEG/LiAc-Lösung zugegeben worden waren, wurde wieder vorsichtig gemischt, und die Zellen wurden anschließend für 45 min bei 30°C inkubiert. Während der Inkubation wurden die Zellen alle 15 min vorsichtig durchmischt. Vor einer Inkubation für 20 min im Wasserbad bei 42°C wurden 160 µl DMSO zugegeben. Bei dieser Inkubation mussten die Zellen alle 10 min gemischt werden. Nach der anschließenden Zentrifugation (5 min, 700 × g, RT) wurde das Zellpellet in 3 ml YPD+Medium (Clontech, Heidelberg) resuspendiert. Es folgte eine Regeneration der Zellen bei 30°C für 90 min mit Schütteln. Nach erneuter Zentrifugation wurde das Zellpellet in 15 ml 0,9%iger NaCl-Lösung resuspendiert. Davon wurden jeweils 150 µl auf eine SD/-Leu Agarplatte (Ø 150 mm) ausplattiert. Um die Transformationseffizienz ermitteln zu können, wurde außerdem eine Platte mit einer 1:1000 Verdünnung der Zelllösung ausplattiert. Alle Platten wurden 3-6 Tage bis zum Erscheinen von Kolonien bei 30°C mit der Unterseite nach oben inkubiert. Material und Methoden 51 Die Ermittlung der Transformationseffizienz erfolgte mit Hilfe folgender Formel und wird pro eingesetzter Vektor-DNA-Menge angegeben: Gezählte Kolonien × 1000 = Anzahl Transformanden / 3 µg pGADT7-Rec Das Ergebnis sollte bei 2.3.5.4.4 1 × 106 Transformanden / 3 µg pGADT7-Rec liegen. Ernte der Transformanden Um die Zellen mit der cDNA-Bank zu ernten, wurden die Platten für 3-4 h bei 4°C gekühlt. Auf jede Platte wurden dann 5 ml Einfriermedium gegeben, und die Kolonien wurden vorsichtig mit einer zum Spatel geformten Pasteurpipette in die Flüssigkeit geschabt. Die Lösungen aller Platten wurden in einer sterilen Flasche vereinigt und gut gemischt. Mit Hilfe eines Hemocytometers (Neubauer-Zählkammer) wurde die Zelldichte überprüft. Liegt die Zelldichte bei 2 × 107 Zellen/ml, muß das Volumen durch Zentrifugation reduziert werden. Die Transformanden werden anschließend in 1 ml Aliquots bei –80°C aufbewahrt. Einfriermedium: 25% Glycerin in YPD Medium 2.3.5.4.5 Überprüfung der cDNA-Bank-Größe Auf SD/-Leu Agarplatten (Ø 100 mm) werden 100 µl Verdünnungen von 1:100, 1:1000 und 1:10 000 der Transformanden ausgestrichen und bei 30°C bis zum Erscheinen von Kolonien inkubiert. Die Zellen müssen anschließend ausgezählt werden; mit Hilfe der folgenden Formel kann die Größe der cDNA-Bank abgeschätzt werden. Kolonien × 1000 µl/ml Vol (ausplattiert) × Verdünnungsfaktor Die Zelldichte der cDNA-Bank sollte = Klone/ml 2 × 107 Zellen/ml entsprechen. Material und Methoden 2.3.5.4.6 52 Test des Fusionsproteinkonstrukts ALD_pGBKT7 auf transkriptionelle Aktivität in S. cerevisiae Da die DNA-BD des Transkriptionsfaktors unter Kontrolle eines konstitutiven Promotors steht, musste getestet werden, ob das Aldolase-Bindedomäne-Fusionsprotein die Reportergene ohne Interaktion mit der Aktivierungsdomäne aktiviert. Für diesen Test wurden die Hefestämme AH109 und Y187 mit dem DNA-BD-Konstrukt (ALD_pGBKT7) transformiert (siehe Kap. 2.3.4.3) und die Transformanden, einschließlich einer negativen Kontrolle (Zellen mit Vektor pGBKT7 ohne „Insert ), auf drei Medien ausplattiert: o SD/-Trp/X-α-Gal o SD/-Trp/-His/X-α-Gal o SD/-Ade/-Trp/X-α-Gal Zeigen die transformierten Kolonien eine weiße Färbung und wachsen nicht auf SD/-Trp/-His und SD/-Ade/-Trp Medium, ist das Fusionsprotein inaktiv. Das Fusionsprotein ist aktiv, wenn die transformierten Zellen eine blaue Färbung aufweisen und auf den Medien SD/-Trp/-His und SD/-Ade/-Trp wachsen. Dann ist eine Suche nach Interaktionspartnern mit dem Hefe-2-Hybrid-System nicht möglich, da die Blaufärbung der Kolonien nicht durch eine Interaktion zweier Proteine hervorgerufen worden ist, sondern das DNA-BD-Konstrukt bereits selbst die Transkription der Reportergene auslöst. 2.3.5.4.7 Toxizitätstest für das Fusionsproteinkonstrukt ALD_pGBKT7 Fusionsproteine können auf die Hefezellen toxisch wirken und damit die Wachstumsrate herabsetzen oder zu einem Absterben der Kultur führen. Um dies zu testen, wurden 50 ml SD/-Trp/Kan Medium mit einer 2-3 mm großen Y187 Kolonie mit inseriertem ALD_pGBKT7 Plasmid angeimpft und für 16-24 h bei 30°C und 250-270 Upm angezogen. Nach dieser Zeit sollte die optische Dichte der Kultur bei A600 0,8, gegen H2Obidest gemessen, liegen. Bei einem viel geringeren Wert kann das Fusionsprotein toxisch sein und für einen Hefe-2-Hybrid-Versuch können die Zellen dann nicht in Flüssigkultur angezogen werden. Es besteht aber die Möglichkeit, die Kolonien in diesem Fall auf Agarplatten anzuziehen. Material und Methoden 53 Dafür wurde eine Kolonie des entsprechenden Stammes (s. o.) in 1 ml SD/-Trp Medium resuspendiert; diese Zellsuspension wurde auf fünf Platten mit dem gleichen Medium ausplattiert. Die Platten wurden bei 30°C inkubiert, bis sie vollständig von einem Zellrasen bewachsen waren. Auf jede Platte wurde 1 ml 0,5 × YPDA gegeben und die Zellen von der Platte in das Medium geschabt. Die Zelldichte wurde mit Hilfe eines Hemocytometers ausgezählt und sollte bei 1 × 109 Zellen/ml liegen. 0,5 × YPDA: 10 g/l Peptone pH 5,8 5 g/l Hefeextrakt mit H2Obidest auf 968 ml auffüllen und nach dem Autoklavieren 25 ml 40%ige Glucose-Lösung und 7,6 ml 0,2%ige Adeninhemisulfat-Lösung zugeben. 2.3.5.5 Paarung der Hefestämme und Selektion diploider Zellen Da die beiden verwendeten Hefestämme AH109 und Y187 zu den zwei verschiedenen HefePaarungstypen a und gehören, kann die Hefe-2-Hybrid-Analyse über die Paarung der Hefen erfolgen. Bei den daraus entstehenden Hefezellen handelt es sich um diploide Zellen, die beide Plasmide enthalten. Anhand der Reportergene kann getestet werden, ob in den Zellen eine Interaktion zwischen dem Protein aus der cDNA-Bank („Prey -Protein) und dem DNABD-Fusionsprotein (DNA-BD-Aldolase, „Bait -Protein) erfolgt ist. Für die Paarung wurde ein 1 ml Aliquot der AH109[cDNA-Bank] ( 2 × 107 Zellen/ml) bei Raumtemperatur aufgetaut und in einem 2 l Erlenmeyerkolben vorsichtig mit den angezogenen Y187 Zellen ( 1 × 109 Zellen/ml) gemischt. Dazu wurden 45 ml 2 × YPDA/Kan gegeben und die Zellen 20-24 h bei 30°C und leichtem Schwenken (3050 Upm) inkubiert. Nach 20 h wurde ein Tropfen der Zellsuspension unter dem Mikroskop auf das Vorhandensein von Zygoten untersucht. Der Kultur wurden weitere 4 h zum wachsen gelassen, wenn noch Zygoten zu sehen waren. Ansonsten wurde die Zellkultur geerntet. Die Zellernte erfolgte durch Zentrifugation (10 min, 1000 × g, RT). Der Überstand wurde verworfen. Der Erlenmeyerkolben, in dem die Zellen kultiviert worden waren, wurde zweimal mit 50 ml 0,5 × YPDA/Kan gespült. Dieses Medium wurde zum Resuspendieren der Zellpellets verwendet; anschließend wurde noch einmal zentrifugiert. Das Pellet wurde in 10 ml 0,5 × YPDA/Kan resuspendiert; das resultierende Volumen wurde bestimmt. Die Selektion der diploiden Hefen erfolgte durch Ausplattieren auf den entsprechenden Minimalmedien. Material und Methoden 54 Dafür wurden je 200 µl des resuspendierten Pellets auf SD/TDO Platten (Ø 150 mm) ausplattiert. Zum Abschätzen der Paarungseffizienz wurden außerdem vier Verdünnungen (1:10, 1:100, 1:1000 und 1:10 000) hergestellt und je 100 µl auf SD/-Trp, SD/-Leu und SD/Leu/-Trp-Medium (Platten Ø 100 mm) ausplattiert. Bei 30°C wurden die Platten mindestens 5 Tage bis zum Erscheinen von Kolonien mit einem Durchmesser von 2 mm inkubiert. Die großen Kolonien (Ø 2 mm) wurden auf SD/QDO/X- -Gal Platten umplattiert und erneut bei 30°C inkubiert (3-8 Tage). Blaue Kolonien wurden von den vierfach selektiven Platten (SD/QDO/X- -Gal) noch 2-3 mal auf SD/-Leu/-Trp/X- -Gal umplattiert und anschließend nochmals auf SD/QDO/X- -Gal. Wenn sie weiterhin blau blieben und gut wuchsen, wurden von ihnen Glycerinkulturen angelegt und Plasmidisolationen durchgeführt. 2 × YPDA: 40 g/l Peptone pH 5,8 (HCl) 20 g/l Hefeextrakt mit H2Obidest auf 870 ml auffüllen nach dem Autoklavieren 100 ml 40%ige Glucoselösung und 30 ml Adeninhemisulfat-Lösung zugeben. Kanamycin-haltiges YPD-/ YPDA-Medium (z. B. YPDA/Kan): Zum Medium wird Kanamycin (50 mg/ml in H2Obidest) in einer Endkonzentration von 10-15 mg/l zugegeben. Um die Paarungseffizienz ermitteln zu können, muss zunächst die Lebensfähigkeit der Kolonien auf dem jeweiligen SD-Medium bestimmt werden. Mit folgender Formel wird die Lebensfähigkeit der Kolonien/ml berechnet: Kolonien × 1000 µl/ml Vol (ausplattiert) × Verdünnungsfaktor = #* lebensfähige Kolonien/ml Der Hefestamm mit der geringeren Lebensfähigkeit ist in einem Versuch der limitierende Partner. Das sollte hier AH109[cDNA-Bank] sein, so dass jeder Klon aus der cDNA-Bank einen Partner des anderen Hefestammes für die Paarung finden konnte. Damit ist sichergestellt, dass innerhalb eines Versuchs möglichst viele unbekannte Protein-ProteinInteraktionen gefunden werden können. (* # steht für Anzahl) Material und Methoden 55 Die Kolonien auf den Platten (SD/-Trp, SD/-Leu und SD/-Trp/-Leu), auf denen die Verdünnungen ausgestrichen worden waren, müssen zur Bestimmung der Paarungseffizienz ausgezählt werden. Für die Auszählung sollten sich zwischen 30 und 300 Kolonien auf den Platten befinden. Die Berechnung der Paarungseffizienz ergibt den prozentualen Anteil diploider Zellen und erfolgt nach folgender Formel: # Kolonien / ml Diploide × 100 = % Diploide # Kolonien / ml limit. Partner Ergibt die Paarungseffizienz einen Wert unter 2%, sollte der Versuch wiederholt werden, da dann die Paarung der Hefezellen nicht erfolgreich war. Die Bestimmung der Anzahl (#) der untersuchten Klone ist durch Auszählen der Platten nach folgender Formel möglich: # Kolonien / ml Diploide × resusp. Vol. = # untersuchter Klone 2.3.5.6 Kontrollen für das Hefe-2-Hybrid-System Als Kontrollen für die Versuche mit dem Hefe-2-Hybrid-System dienen zwei Vektoren (pGBKT7-53, pGBKT7-Lam) und ein DNA-Fragment („SV40 Large T PCR-Fragment ) (Clontech, Heidelberg). Das DNA-Fragment wird, wie die cDNA-Bank mit dem linearisierten Vektor pGADT7-Rec (Clontech, Heidelberg), in den Hefestamm AH109 durch CoTransformation mit in vivo-Rekombination eingebracht. Y187-Zellen werden mit den beiden anderen Plasmiden transformiert. Dafür wurden kompetente Hefezellen hergestellt und mit den in Tabelle 2.7 aufgeführten Komponenten (Clontech, Heidelberg) gemischt. Material und Methoden 56 Tab. 2.7: Zusammensetzung der Proben für die Transformation von Hefezellen. Komponenten SV40 Large T PCR-Fragment AH109 Y187 Y187 [pGADT7-Rec] [pGBKT7-53] [pGBKT7-Lam] 5,0 µl - - 0,5 µl - - pGBKT7-53 (500 ng/µl) - 0,5 µl - pGBKT7-Lam (500 ng/µl) - - 0,5 µl 5,0 µl 5,0 µl 5,0 µl 50 µl - - - 50 µl 50 µl 500 µl 500 µl 500 µl (25 ng/µl) pGADT7-Rec (Sma I-linearisiert, 500 ng/µl) Herring Testes carrier DNA (10 mg/ml) AH109 kompetente Zellen Y187 kompetente Zellen PEG/LiAc-Lösung Die verschiedenen Ansätze wurden gut gemischt und 30 min auf Eis inkubiert, wobei sie alle 10 min vorsichtig geschüttelt wurden. Anschließend wurden 20 µl DMSO in die Ansätze gegeben, gemischt und bei 42°C 15 min inkubiert. Während dieser Inkubation musste alle 5 min vorsichtig geschüttelt werden. Nach einer Zentrifugation bei höchster Geschwindigkeit (15 s, RT) wurde der Überstand verworfen; das Pellet wurde in 1 ml 0,9 %iger NaCl-Lösung aufgenommen. Je 100 µl einer 1:10, 1:100 und 1:1000 Verdünnung wurden auf SD-Medium ausplattiert und bei 30°C 4 Tage inkubiert. Die Selektion der Transformanden erfolgte bei den beiden Y187-Kontroll-Hefestämmen (Y187[pGBKT7-53] und Y187[pGBKT7-Lam]) auf SD/-Trp Medium, die AH109-Kontrolle dagegen wuchs auf SD/-Leu Medium. Die größten Kolonien wurden noch einmal auf dem gleichen Selektionsmedium ausgestrichen. Für den Kontrollversuch zur Paarung der Hefen wurden nur große, frische (< 2 Wochen alt) Kolonien verwendet. Als positive Kontrolle wurden die Hefestämme AH109[pGADT7-RecT] und Y187[pGBKT7-53] gepaart; als Negativkontrolle diente AH109[pGADT7-RecT] mit Y187[pGBKT7-Lam]. Die Kolonien wurden in 500 µl 2 × YPDA-Medium gegeben und vollständig resuspendiert. Unter leichtem Schwenken erfolgte die Inkubation für 20-24 h bei 30°C. 100 µl einer 1:10 und 1:100 Verdünnung wurden dann auf SD/-Leu, SD/-Trp, SD/Leu/-Trp und QDO/X- -Gal Platten ausplattiert. Die Zellen der Positivkontrolle sollten auf dem vierfach selektiven Medium (QDO/X- -Gal) blau werden, die negative Kontrolle sollte Material und Methoden 57 weiß bleiben. Die anderen Platten dienten zum Auszählen für die Berechnung der Paarungseffizienz. 2.3.6 Biochemische Methoden 2.3.6.1 Proteinbestimmung nach BRADFORD Die Proteinkonzentration einer Lösung wurde mit Hilfe der Methode von BRADFORD (1976) photometrisch am Uvicon 810-Photometer (Kontron, Zürich, CH) bestimmt. Durch Bindung des Farbstoffs Coomassie Brilliantblau G-250 an basische Aminosäurereste der Proteine kommt es zu einer Verschiebung des Absorptionsmaximums von 465 nm zu 595 nm. Der aus der Steigung einer Eichgeraden errechnete Faktor (Bradford-Faktor) wird genutzt, um die Proteinkonzentration einer Probe auf Grund der Absorption bei 595 nm zu berechnen. Zur Erstellung einer Eichgerade wurde entfettetes Rinderserumalbumin (BSA, 0-20 µg) eingesetzt. Bradford-Farbreagenz: 50 mg Coomassie-Brilliantblau G-250 10 ml 95%iges Ethanol 175 ml o-Phosphorsäure (85%) mit H2Obidest auf 825 ml auffüllen Proteinbestimmungs-Ansatz: X µl Probe mit H2Obidest auf 500 µl Endvolumen auffüllen 500 µl Bradford-Farbreagenz Der Ansatz muss gut gemischt werden und wird nach ca. 10 min gemessen. Als Referenzwert dient eine Probe ohne Protein. Berechnung der Proteinkonzentration [µg/µl]: C Proteinlösung = OD 595nm (Probe) × Bradford-Faktor [µg] Volumen (Probe )[ l] Der Bradford-Faktor ergibt sich aus 1/Steigung der Eichgerade. Material und Methoden 58 2.3.6.2 SDS-Gelelektrophorese Um Proteingemische aufzutrennen, das Molekulargewicht von Proteinen zu bestimmen oder die Reinheit von Proteinen zu überprüfen, wurde die denaturierende, diskontinuierliche Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese (SDS-PAGE) nach LAEMMLI (1970) durchgeführt. Die vollständige Denaturierung der Probe erfolgte durch 5-minütiges Erhitzen der mit 6 × SDS-Ladepuffer versetzten Probe auf 99°C. Dabei lagern sich die negativ geladenen SDS-Moleküle an die Proteine an und verleihen den Proteinen eine der Molekülmasse entsprechende Ladung. Für die SDS-PAGE wurden vertikale Gele (10,5 × 11,5 × 0,75 cm bzw. 10,5 × 11,5 × 1 cm, Mini Protean II, BioRad, München) verwendet, die sich aus einem 5 %igen Sammelgel und 10 oder 12%igem Trenngel zusammensetzten. Die Elektrophorese wurde bei 120 V mit SDS-Laufpuffer in der Laufkammer durchgeführt. Die Molekülmasse der aufgetrennten Proteine wurde durch Proteinstandardgemische (z. B. Prestained Protein Ladder, ~10-180 kDa, MBI Fermentas, St. Leon-Rot) bestimmt. 6 × SDS-Gelladepuffer: 1,5 mM Tris pH 6,8 (HCl) 30% (w/v) SDS 0,15% (w/v) Bromphenolblau 10% Glycerin Vor Gebrauch werden dem 6 × SDS-Ladepuffer noch 50 µl/ml -Mercaptoethanol (99%) zugegeben. SDS-Laufpuffer: 25 mM Tris 192 mM Glycin 4 mM SDS pH 8,3 (HCl) Material und Methoden 59 Tab. 2.8: Zusammensetzung der SDS-Gele. Trenngel Sammelgel 10% 12% 5% H2Obidest 9,6 ml 8,6 ml 3,6 ml 40% Acrylamid/Bis-Acrylamid (30/0,8) 5,0 ml 6,0 ml 630 µl 1,5 M Tris-HCl pH 8,8 + 0,4% SDS 5,2 ml 5,2 ml - 0,5 M Tris-HCl pH 6,8 + 0,4% SDS - - 680 µl 200 µl 200 µl 50 µl 8 µl 8 µl 5 µl 10% Ammoniumpersulfat 100% TEMED Zum Anfärben der Proteine wurden die Gele für 1-2 h in Coomassiefärbelösung gelegt und anschließend mit Entfärbelösung so lange gewaschen, bis das Gel entfärbt, die Proteinbanden aber gut sichtbar waren. Coomassiefärbelösung: 0,4% (w/v) Coomassie-Brillantblue R-250 9% (v/v) Essigsäure 45,5% (v/v) Methanol Entfärbelösung: 10% (v/v) Essigsäure 45% (v/v) Methanol Für Gele mit sehr geringen Proteinmengen wurde als sensitivere Färbemethode die Silberfärbung nach DAMERVAL et al. (1987) gewählt. Dafür wurden die Gele 15-30 min in Entfärbelösung fixiert und anschließend 2 min in „Farmer s Reducer“ inkubiert. Die Gele wurden bis zum vollständigen Auswaschen der gelben Farbe des „Farmer s Reducer“ dreimal 10 min in H2Obidest gewaschen. Es schloß sich eine Inkubation von 15-30 min in 0,1%iger Silbernitratlösung an. Die Gele wurden dann zweimal 30 sec mit H2Obidest gewaschen und ebenfalls zweimal 30 sec in 2,5% Natriumcarbonat equilibriert. Für die Entwicklung der Gele wurden diese in Entwickler gelegt, bis die Proteinbanden gut zu sehen waren. Der Entwicklungsprozess musste durch Inkubation der Gele in 10%iger Essigsäure gestoppt werden. Material und Methoden 60 „Farmer s Reducer“: 0,03 M Kaliumhexacyanoferrat (III) 0,032 M Natriumthiosulfat-(5-Hydrat) Entwickler: 2,5% Natriumcarbonat 35% Formaldehyd 2.3.6.3 Western-Blot-Analyse und Immundekoration Zum qualitativen Nachweis von Proteinen dient die Western-Blot-Analyse. Hierbei werden durch das Anlegen einer Spannung die Proteine aus einer SDS-PAGE auf eine Nitrocellulosemembran (Protean, Schleicher & Schuell, Dassel) übertragen. Gel, Membran und Filterpapiere wurden für den Blot 15 min in Transferpuffer equilibriert. Anschließend wurden Gel und Membran aufeinandergelegt und von beiden Seiten mit Filterpapier (3 mm, Whatman) bedeckt. In der richtigen Orientierung zu den Polen wurde das „Blot-Sandwich“ in eine mit Transferpuffer gefüllte vertikale „blotting -Nasskammer überführt und bei 4°C und 100 mA 16-18 h geblottet. Transferpuffer: 25 mM Tris 192 mM Glycin 20% (v/v) Methanol 2.3.6.3.1 Färbung geblotteter Proteine mit Ponceau S Mit Hilfe der Ponceau S-Färbung kann nach einem Western-Blot überprüft werden, ob der Proteintransfer auf die Nitrozellulosemembran erfolgreich war. Die Membran wurde dafür in Ponceau-Rot-Lösung (0,2% (w/v) Ponceau S in 5%iger Trichloressigsäure) geschwenkt, bis Proteinbanden zu erkennen waren. Mit H2Obidest wurde die Farbreaktion gestoppt und überschüssige Farblösung abgewaschen. Um nach der Immundekoration eine Molekulargewichtsbestimmung zu ermöglichen, wurden die Banden des Markers mit Bleistiftstrichen auf der Membran gekennzeichnet. Vor dem immunologischen Nachweis wurde die Membran bis zur vollständigen Entfärbung mit 1 × TBS-T-Lösung gewaschen. TBS-Stammlösung (10 ×): 0,5 M Tris pH 7,5 (HCl) 1,5 M NaCl TBS-T-Lösung: 1 × TBS 0,2% (v/v) Tween-20 Material und Methoden 61 Für den anschließenden immunologischen Nachweis der geblotteten Proteine wurde die Membran für mindestens 60 min bei Raumtemperatur unter leichtem Schütteln in BlockierPuffer inkubiert. Dieser Schritt dient dem Absättigen unspezifischer Bindungsstellen auf der Membran. Die anschließende Inkubation mit primärem Antikörper, 1:1000 mit Blokierpuffer verdünnt, wurde bei Raumtemperatur für eine Stunde oder über Nacht bei 4°C durchgeführt. Es folgten drei fünfminütige Waschschritte mit Blockier-Puffer, durch die ungebundene Antikörper ausgewaschen wurden. Der Nachweis des Antikörper-Antigen-Komplexes erfolgte nach einstündiger Inkubation mit sekundärem Antikörper. Die eingesetzte Konzentration des sekundären Antikörpers richtete sich nach der Methode des immunologischen Nachweises (Peroxidasereaktion oder Chemolumineszenz). Durch eine an den zweiten Antikörper gebundene Alkalische-Phosphatase oder Meerrettich-Peroxidase konnte die Antikörper-Antigen-Bindung visualisiert werden. Blockier-Puffer: 1 × TBS 0,2% (v/v) Tween-20 3% 2.3.6.3.2 (w/v) BSAentfettet Immunologischer Nachweis durch Peroxidasereaktion Für die Peroxidasereaktion wurde der mit sekundärem Antikörper, in einer 1:3000 Verdünnung, inkubierte Blot zweimal in TBS gewaschen. Anschließend wurde die Membran bis zum Erscheinen deutlicher Banden in Färbelösung inkubiert. Durch gründliches Waschen der Membran mit H2Obidest wurde die Reaktion gestoppt. Färbelösung: 25 ml TBS 20 mg 4-Chloro-1-Naphthol in 5 ml Methanol 15 µl Wasserstoffperoxid (37%ig) Material und Methoden 2.3.6.3.3 62 Immunologischer Nachweis durch Alkalische-Phosphatase-Reaktion Wie auch bei der Meerrettich-Peroxidase-Reaktion wird bei der Alkalischen-PhosphataseReaktion die Antikörperbindung durch eine Farbreaktion nachgewiesen. Der Blot wurde auch hier nach der Inkubation mit sekundärem Antikörper (in 1:3000 Verdünnung) zweimal mit TBS gewaschen. Die Entwicklung erfolgte in AP-Färbelösung bis zum deutlichen Sichtbarwerden von Banden. AP-Färbelösung: 100 mM NaCl 5 mM MgCl2 0,375 mg/ml NBT in 70%igem Dimethylformamid (DMF) 0,1 mg/ml BCIP in 100%igem DMF mit 100 mM Tris-HCl (pH 8,8) auf das gewünschte Volumen auffüllen. Die Farbreaktion wurde durch Waschen der Membran in H2Obidest gestoppt. 2.3.6.3.4 Immunologischer Nachweis durch Chemolumineszenz Bei dieser Methode wird ein Substrat durch die Meerrettich-Peroxidase umgewandelt und zerfällt, was die Emmission von Licht zur Folge hat (ECL: Enhanced Chemoluminescence ). Das emittierte Licht kann durch Belichtung eines Films sichtbar gemacht werden. Für diese Methode ist eine geringere Konzentration (1:25 000) des sekundären Antikörpers notwendig. Das Waschen der Membran erfolgte zweimal für fünf min in TBS-T-Lösung und anschließend für 5 min in TBS. Für die ECL-Färbung wurde die Membran eine Minute in „ECL Western Blotting Detection Reagent“ (Amersham Bioscience, Freiburg) inkubiert und danach luftblasenfrei in Frischhalte-Folie eingeschlagen. In einer Filmkassette wurde ein Kodak Bio Max Negativfilm (Sigma-Aldrich Chemie GmbH, Taufkirchen) durch die Lichtemission belichtet. Die Belichtungszeit richtete sich nach der Intensität der emittierten Strahlung. Die Entwicklung des Films erfolgte durch Schwenken in Entwicklerlösung, bis deutliche Banden auf dem Film sichtbar wurden. Nach Waschen und Fixieren des Films konnte er an der Luft getrocknet werden. Material und Methoden 2.3.6.4 Dot-Blot 63 eine Variante des Western-Blots Der Unterschied zum Western-Blot liegt darin, dass bei einem Dot-Blot die nachzuweisenden Proteine nicht über eine SDS-PAGE aufgetrennt und anschließend geblottet werden, sondern dass eine Proteinlösung direkt auf die Membran getropft wird. Nach dem Trocknen der Proteinlösung auf der Membran wird diese wie ein normaler Western-Blot behandelt. 2.3.6.5 Far-Western-Blot Mit Hilfe eines „Far-Western-Blots“ oder auch „Overlay-Blots“ können Protein-ProteinInteraktionen nachgewiesen werden. Dabei wird ein Protein oder Proteingemisch auf einer Membran fixiert, was durch SDS-PAGE und anschließendes „blotting oder im Dot-Blot erfolgen kann. Der Blot wird mit einem weiteren Protein überschichtet („overlay ). Bei einem Western-Blot ist das „Overlay -Protein der primäre Antikörper. Im Falle des „Far-Western-“ oder „Overlay-Blots“ handelt es sich um ein Protein, das evtl. mit dem immobilisierten Protein interagiert. Bei den durchgeführten „Far-Western-Blots“ wurden jeweils 2 µg Protein auf eine Nitrozellulosemembran getropft und getrocknet. Anschließend wurde die Membran mindestens 1 h in Blockier-Puffer inkubiert. Danach erfolgte der „Overlay mit 25 nM rekombinanter, gereinigter Strep-tag-Aldolase in Blockier-Puffer bei leichtem Schütteln bei 4°C über Nacht. Nach dreimaligem Waschen für 5 min mit Blockier-Puffer wurde der primäre Antikörper entsprechend einem Western-Blot eingesetzt (siehe Kap. 2.3.6.3). Die weitere Behandlung entsprach dem Western-Blot. Als Kontrolle wurde ein weiterer Blot über Nacht in Blockier-Puffer ohne Aldolase inkubiert. Alle „Far-Western-Blots“ wurden durch Chemolumineszenz entwickelt. 2.3.6.6 Heterologe Expression von Proteinen in E. coli BL21(DE3)pLysS Mit Hilfe von Bakterien können Proteine rekombinant hergestellt werden. Dafür wird die kodierende DNA-Sequenz des gewünschten Proteins in einen Expressionsvektor kloniert und in den Proteinexpressionsstamm E. coli BL21(DE3)pLysS transformiert. Die heterologe Expression der Proteine unterscheidet sich auf Grund der gewählten Vektoren, da diese unterschiedliche Promotoren enthalten. Mit Hilfe von induzierbaren Promotoren ist es möglich, die Synthese des Fremdproteins gezielt auszulösen. In dieser Arbeit kamen zwei verschiedene Expressionssysteme zum Einsatz. Material und Methoden 64 a) Heterologe Expression mit T7-Promoter Bei den Vektoren pET-16b (Novagen, Schwalbach/Ts) und pRSET A (Invitrogen, Karlsruhe) handelt es sich um Vektoren, bei denen die inserierte cDNA unter der Kontrolle des T7-Promotors steht. Die Synthese der T7-Polymerase, die spezifisch diesen Promotor erkennt, kann durch Isopropyl- -D-thiogalactopyranosid (IPTG) induziert werden, so dass die Zellen erst nach Zugabe von IPTG mit der Synthese des Fremdproteins beginnen. Mit Übernachtkulturen wurden 100 ml bis zu 1 l selektives Medium 1:50 angeimpft und bei 37°C aerob kultiviert. Das Wachstum der Zellen wurde stündlich durch Messung der optischen Dichte bei einer Wellenlänge von 600 nm gegen H2O verfolgt. Bei einer optischen Dichte von 0,5-0,8 wurde der Kultur IPTG in einer Endkonzentration von 300 µM zugegeben. Die weitere Kultivierung der Zellen erfolgte nach der Induktion der Proteinexpression für 3-4 h bei 30°C im Schüttler (ca. 200 Upm). Vor der Induktion sowie jede Stunde während der Anzucht nach Induktion wurde ein 1 ml Aliquot der Kultur zur Messung der optischen Dichte und für die Analyse der Überexpression mittels SDSPAGE entnommen. Die Zellernte erfolgte durch Zentrifugation (20 min, 5000 × g, 4°C). Für einen Zellaufschluss mit anschließender Proteinreinigung aus Einschlusskörpern („inclusion bodies ) wurde das Zellpellet bei –20°C eingefroren. Sollten jedoch nach dem Zellaufschluss die löslichen Proteine gewonnen werden, wurde das Zellpellet in 1/5 des Kulturvolumens mit Waschpuffer I resuspendiert. Nach erneuter Zentrifugation wurde das Pellet in 1/30 des Kulturvolumens an Waschpuffer I mit Proteaseinhibitor (0,02 µg/µl Pefabloc) aufgenommen und bei – 20°C eingefroren. Waschpuffer I : 50 mM MOPS 5 mM MgCl2 100 mM Saccharose pH 7,5 (KOH) Material und Methoden 65 b) Heterologe Expression mit tet-Promotor Bei den Strep-tag II-Expressionsvektoren (z. B. IBA-3) befindet sich die Expressionskassette unter Kontrolle des tetA Promotors/Operators. Durch Zugabe von Anhydrotetrazyklin, in Konzentrationen unterhalb antibiotischer Effekte, kann der Promotor vollständig induziert werden. In Abwesenheit des Induktors garantieren die konstitutiv exprimierten tet Repressor-Gene, die sich ebenfalls auf dem Vektor befinden, die vollständige Repression der Expression inserierter Fremdproteine. Mit diesem Expressionssystem kann eine Proteinexpression auch im E. coli Stamm XL1blue erfolgen. Dadurch ist eine Transformation des Stammes BL21(DE3)pLysS mit dem entsprechenden Plasmid nicht unbedingt erforderlich. Flüssigkulturen zwischen 50 und 200 ml wurden mit Übernachtkulturen 1: 50 angeimpft und bis zu einer optischen Dichte von ca. 0,5, gemessen bei 550 nm gegen H2Obidest, bei 37°C aerob kultiviert. Die Induktion der Proteinexpression erfolgte mit 200 µg/l Anhydrotetracyclin-Hydrochlorid (AHT). Nach der Induktion wurden die Zellen für weitere 4 h bei 30°C aerob kultiviert. Geerntet wurden die Zellen durch Zentrifugation (15 min, 4500 × g, 4°C) und anschließend wurde das Zellpellet bei –20°C eingefroren. AHT-Stammlösung: 2 mg/ml AHT in DMF 2.3.6.7 Reinigung der rekombinanten Proteine 2.3.6.7.1 His-tag-Affinitätschromatographie Mit den Expressionsvektoren pET-16b und pRSET A hergestellte rekombinante Proteine lassen sich über Metall-Chelat-Chromatographie reinigen. Diese Vektoren enthalten im Leseraster mit der „Multiple Cloning site“ (MCS) eine codierende His-tag-Sequenz. An die Proteinsequenz wird dadurch bei der Expression N-terminal ein His-tag angehängt. Dieser ermöglicht die Reinigung des Proteins aus einer Proteinlösung durch Nickel-ChelatChromatographie, da die negativ geladenen Histidine des His-tags an Kationen (z. B. NickelIonen) binden. Für den Zellaufschluss wurde dem in 1/30 des Ausgangsvolumens der Bakterienkultur 1 × Waschpuffer gelösten Zellpellet Lysozym (200 µg/ml) zugegeben und bei 35°C aufgetaut. Nach 1,5 h Inkubation mit leichtem Schütteln bei Raumtemperatur und anschließendem Sonifizieren auf Eis (3 × 300 s, 50% Microtip) wurden die löslichen Bestandteile des Rohextrakts von den unlöslichen Zellfragmenten durch Zentrifugation (30 min, 20000 × g, 4°C) getrennt. Dieser geklärte Rohextrakt wurde anschließend auf die vorbereitete NickelChelat-Säule gegeben. Material und Methoden 66 Bei der Vorbereitung der Säule wurden zunächst Nickel-Ionen an chelatisierender Sepharose (Chelating Sepharose®, Pharmacia, Uppsala, Schweden) immobilisiert. Überschüssiges Nickel wurde mit H2Obidest ausgewaschen, und die Säule wurde mit 1 × Bindepuffer equilibriert. Die Bindung der Proteine mit His-tag erfolgte anschließend durch Beladen der Säule mit dem geklärten Rohextrakt. Dieser und alle folgenden Schritte wurden bei 4°C durchgeführt, um ein Denaturieren des Proteins zu verhindern und die Aktivität abbauender Enzyme (wie Proteasen) herabzusetzen. Ungebundene Proteine wurden durch anschließendes Waschen mit 10 Säulenvolumina 1 × Bindepuffer und 6 Säulenvolumina 1 × Waschpuffer entfernt. Die His-tag-Proteine wurden mit Hilfe eines Imidazol-Gradienten eluiert. Imidazol konkurriert mit dem His-tag der Proteine aufgrund der ähnlichen Konformation um die Nickel-Bindestellen des Säulenmaterials und löst so die His-tag-Proteine von der Säule. Dafür wurde die Säule nacheinander mit je 1,5 Säulenvolumina 1× Elutionspuffer mit 150 mM Imidazol, 300 mM Imidazol, 450 mM Imidazol und 1 M Imidazol gespült. Alle Fraktionen wurden für die spätere Analyse aufgefangen. Für die Regeneration der Säulenmatrix erfolgte das Lösen der Nickel-Ionen von der Säulenmatrix durch Durchfluss von 3 Säulenvolumina 1 × „strip off“-Puffer und Waschen mit 10 Säulenvolumina H2Obidest. Nach Behandlung mit 5 Säulenvolumina 1 × Ladepuffer und anschließendem Waschen mit 3 Säulenvolumina H2Obidest war die Säule wieder mit Ni2+-Ionen beladen und konnte für eine erneute Proteinreinigung verwendet werden. 8 × Waschpuffer: 160 mM Tris-HCl pH 7,9 4 M NaCl 480 mM Imidazol 8 × Bindepuffer: 160 mM Tris-HCl pH 7,9 4 M NaCl 40 mM Imidazol 8 × Elutionspuffer: 80 mM Tris-HCl pH 7,9 2 M NaCl Imidazol in den Endkonzentrationen 150, 300, 450 und 1000 mM Material und Methoden 67 4 × „strip off“-Puffer: 80 mM Tris-HCl pH 7,9 100 mM NaCl 400 mM EDTA 8 × Ladepuffer: 2.3.6.7.2 400 mM NiSO4 Reinigung von rekombinantem Protein aus „inclusion bodies“ Nicht alle heterolog überexprimierten Proteine befinden sich nach dem Zellaufschluss und der Zentrifugation als gelöste Proteine im Überstand (geklärtem Rohextrakt). Werden während der Expression die Proteine in den E. coli-Zellen in Einschlusskörper („inclusion bodies“) eingelagert, erfordert das den Aufschluss der unlöslichen Bestandteile des Rohextraktes für die Reinigung der Proteine. Dieser wurde in Anlehnung an das Protokoll „Purification of Polyhistidine-Tagged Proteins“ (Macherey-Nagel, Düren) durchgeführt. Das Pellet der Bakterienkultur wurde dafür in 1/125 des Kulturvolumens 1 × LEW-Puffer mit 1 mg/ml Lysozym versetzt und 30 min auf Eis inkubiert. Nach Aufschluss der Zellen durch Sonifizieren (2 × 60 s, 50% Microtip) wurden die löslichen Bestandteile von den unlöslichen getrennt (Zentrifugation: 30 min, 10000 × g, 4°C). Das Pellet mit den inclusion bodies“ wurde mit 1/50 des Kulturvolumens 1 × LEW-Puffer gewaschen und noch einmal zentrifugiert. Anschließend wurde das Pellet in 1/125 des Kulturvolumens 1 × Denaturierungs-Solubilisierungs-Puffer resuspendiert. Zum vollständigen Resuspendieren des Pellets war ein weiterer Aufschluss durch Sonifizieren (2 × 60 s, 50% Microtip) notwendig. Nach einer Inkubation von 60 min auf Eis folgte eine Zentrifugation (30 min, 10000 × g, RT). Der Überstand dieser Zentrifugation wurde nun auf eine vorbereitete NickelChelat-Säule (vergl. Kap. 2.3.6.7.1) gegeben, die jedoch mit 4 Säulenvolumina Denaturierungs-Solubilisierungs-Puffer equilibriert worden war, und 1 h bei RT geschüttelt. 8 × LEW-Puffer: 400 mM NaH2PO4 pH 8,0 (NaOH) 2,4 M NaCl Denaturierungs-Solubilisierungs-Puffer: 1 × LEW 8 M Harnstoff pH 8,0 Material und Methoden 68 Ungebundene Proteine wurden mit 10 Waschschritten von je 2 Säulenvolumina mit Denaturierungs-Solubilisierungs-Puffer ausgewaschen. Die Elution der gebundenen Proteine erfolgte in 8 Schritten mit je 3 Säulenvolumina Denaturierungs-Solubilisierungs-Puffer mit Imidazol. Der Imidazol-Gradient wurde dabei mit 10 mM, 50 mM, 100 mM und 250 mM Imidazol der Bindung des rekombinanten Proteins an die Nickel-Ionen angepasst. 2.3.6.7.3 Reinigung von rekombinantem Protein mit Strep-tag Das „Two-Step Affinity Purification System“ (Qiagen, Hilden) diente zur Proteinreinigung über Strep-tag-Affinitätschromatographie. Der Zellaufschluss vor der Proteinreinigung erfolgte in Anlehnung an das Protokoll „Preparation of Cleared Lysates from E. coli Cell Cultures“ (Qiagen, Hilden). Dafür musste zunächst das Zellpellet in 15-30 min auf Eis auftauen und anschließend in 1/50 des ursprünglichen Zellkulturvolumens 1 × LEW-Puffer mit 1 mg/ml Lysozym resuspendiert werden. Diese Lösung wurde nach einer Inkubation auf Eis für 30 min sonifiziert (3 × 60 s; 50% Microtip, 4°C). Die aufgeschlossene Probe wurde anschließend durch Zentrifugation (30 min, 10000 × g, 4°C) von Zelltrümmern und unlöslichen Zellbestandteilen getrennt. Der Überstand der Zentrifugation enthielt die löslichen Proteine und wurde als geklärter Rohextrakt der Säulenchromatographie unterworfen. Bei der Säulenchromatographie bindet der Strep-tag des Proteins an Strep-Tactin, und nach Auswaschen ungebundener Proteine wird durch die spezifische Kompetition durch Desthiobiotin das Protein eluiert. Dafür wurde der geklärte Rohextrakt mit 2 ml Säulenmatrix („Strep-Tactin Superflow Resin Suspension , Qiagen, Hilden) gemischt und 1 h bei 4°C geschüttelt. Die Suspension wurde in eine leere Säule gegeben und durch vier Waschschritte mit jeweils 4 ml 1 × LEW-Puffer von ungebundenen Proteinen gereinigt. Die gebundenen Proteine wurden durch Zugabe von 6-8 × 0,5 ml Elutions-Puffer eluiert. Die einzelnen Fraktionen der Reinigung wurden aufgefangen und analysiert. Elutions-Puffer: 1 × LEW-Puffer (s. Kap. 2.3.6.7.2) 2,5 mM Desthiobiotin Material und Methoden 69 Zur Regeneration der Säulenmatrix wurde das Desthiobiotin mit 4-Hydroxy-Azo-Benzyl-2Carboxylsäure (HABA) von der Matrix gelöst und ausgewaschen. Eine erfolgreiche Regeneration ist durch einen Farbumschlag des Regenerationspuffers von gelb zu rot sichtbar. Die Regeneration erfolgte mit 30 ml Regenerations-Puffer und anschließendem Waschen mit 16 ml 1 × LEW-Puffer. Regenerations-Puffer: 50 mM NaH2PO4 pH 8,0 (NaOH) 300 mM NaCl 1 mM 4-Hydroxy-Azo-Benzyl-2-Carboxylsäure (HABA) 2.3.6.8 Aldolase-VDAC-Bindeversuch (Affinitätschromatographie) Die Affinitätschromatographie stellt eine Möglichkeit zur Verifizierung von Protein-ProteinInteraktionen dar. Das Prinzip beruht auf der spezifischen Bindung eines His-tagFusionsproteins an die Säulenmatrix. Wird über diese Säule ein Protein ohne His-tag oder ein Proteingemisch gegeben, sollten diese Proteine nur dann mit dem an die Matrix gebundenen Protein eluieren, wenn eine Bindung zwischen dem an die Matrix gekoppelten Protein und einem anderen Protein vorliegt. Um mit dieser Methode die potentielle Interaktion zwischen dem spannungsabhängigen Anionenkanal VDAC 1 und Aldolase nachzuweisen, wurde rekombinantes, gereinigtes His-tag VDAC-Fusionsprotein an eine Ni-NTA Säulenmatrix gekoppelt und durch 10 Waschschritte (je 2 Säulenvolumina) mit DenaturierungsSolubilisierungs-Puffer von Verunreinigungen befreit. Über eine Strep-Tactin-Säule gereinigte und gegen 1 × LEW-Puffer dialysierte Aldolase-Proteinlösung (ca. 2,5 ml, 0,42 µg/µl) wurde zum an die Ni-NTA-Säule gekoppelten VDAC-Protein gegeben und mindestens 60 min bei RT geschüttelt. Nach erneuten 10 Waschschritten mit DenaturierungsSolubilisierungs-Puffer wurden noch je zwei Waschschritte mit Imidazol (5 mM und 7,5 mM) in Denaturierungs-Solubilisierungs-Puffer angeschlossen, bevor das an die Säule gebundene Protein mit einem Imidazol-Gradienten (10 mM, 50 mM, 100 mM und 250 mM in Denaturierungs-Solubilisierungs-Puffer) eluiert wurde. Alle Fraktionen dieser Säulenchromatographie wurden aufgefangen und mittels SDS-PAGE analysiert. Als Kontrolle wurde parallel dazu eine Säulenchromatographie mit den gleichen Schritten durchgeführt, wobei hier jedoch kein His-tag VDAC-Fusionsprotein an die Ni-NTA-Säule gebunden war. Material und Methoden 70 2.3.7 Entsalzen über Dialyse und NAP-5 Säulen Der Austausch von Puffern, um z. B. hochkonzentrierte Salze aus Proteinlösungen zu entfernen, wurde durch Dialyse oder NAP-5 Säulen (Pharmacia, Uppsala, S) durchgeführt. Bei der Dialyse wird die Proteinlösung in einen Dialyseschlauch, bestehend aus einer Membran mit definierter Porengröße, gefüllt. Auf Grund von Diffusion treten Salze und Flüssigkeiten durch die Membran, größere Moleküle wie Proteine werden jedoch im Inneren des Schlauches gehalten. Die Flüssigkeit, gegen die dialysiert wird, sollte das 1000-fache Probenvolumen haben, um eine ausreichende Verdünnung zu ermöglichen. Die Dialyse wurde unter leichtem Rühren bei 4°C für mindestens 2 h, meist über Nacht, durchgeführt. Dialysiert wurde z. B. nach der Proteinreinigung aus „inclusion bodies“, um aus der Probe Imidazol zu entfernen und die Harnstoffkonzentration zu senken. Das Prinzip der NAP-5 Säulen beruht auf der unterschiedlichen Wanderungsgeschwindigkeit von Molekülen innerhalb einer Säulenmatrix. Bei den NAP-5 Säulen kann ein Probenvolumen von 500 µl in 1000 µl eines anderen Puffers überführt werden. 2.3.8 Herstellung eines polyklonalen Antiserums Die Gewinnung von Auftragsimmunisierung rekombinantem Protein. spezifischen, eines polyklonalen Kaninchens Dafür Antikörpern (BioScience, wurden 500 µg erfolgte Göttingen) Mais-Aldolase mit durch die gereinigtem, (ALD_pET-16b) in E. coli Bl(21)DE3pLysS überexprimiert, gereinigt und auf präparative Gele (SDS-PAGE) aufgetragen. Bei diesen präparativen Gelen gibt es nur zwei Probentaschen, eine kleine für den Marker und eine große für die Probe. Nach der Elektrophorese wurde die Markertasche und ein kleines Stück der Probentasche vom Rest des Gels abgeschnitten und 10-30 min in angewärmter Coomassie-Färbelösung angefärbt. Das restliche Gel wurde in Western-BlotTransferpuffer mit 5% Methanol inkubiert. Nach dem Entfärben und Quellen in H2Obidest wurde mit Hilfe des zuvor abgeschnittenen Gelstückes der Bereich des Gels, in dem sich das gereinigte Protein befand, definiert. Dieser Bereich wurde aus dem ungefärbten Gel ausgeschnitten und für die Immunisierung des Kaninchens an die oben genannte Firma geschickt. Die erhaltenen Kaninchen-Anti-Aldolase-Antikörper wurden an verschiedenen Geweben auf ihre Spezifität getestet. Eine Konzentrationsreihe mit rekombinantem Protein gab Aufschluss über die mit dem Antiserum zu detektierende Menge des nativen Proteins (im „Enzyme-linked Material und Methoden 71 immunosorbent assay“, ELISA) und der denaturierten rekombinanten Aldolase auf Westernblots. 2.3.9 Copolymerisationsversuche Um eine Bindung der Aldolase an Aktin zu untersuchen, wurden Copolymerisationsversuche mit rekombinanter Strep-tag-Aldolase und Kaninchenmuskel-Aktin (Cytoskeleton, Denver, CO, USA) durchgeführt. Für die Copolymerisationsversuche wurde die gereinigte Strep-tagAldolase in 1 × LSB-Puffer („Low-Salt-Buffer“) umgepuffert (NAP-5 Säulen) und anschließend ankonzentriert (Centricon, Millipore Amicon, Eschborn) bis etwa eine Konzentration von 1 mg/ml erreicht war. Das Aktin (1 mg) wurde in 500 µl eiskaltem 1 × LSB gelöst und zur Depolymerisation 1 h auf Eis inkubiert. Beide Proben (Aktin und Aldolase) wurden anschließend 1 h bei 200 000 × g und 4°C zentrifugiert (Beckman Optima Ultrazentrifuge, Beckman Instruments, Palo Alto, CA, USA). Vom Überstand dieser Zentrifugation wurde der Proteingehalt gemessen und, wenn notwendig, noch einmal ankonzentriert. Das Pellet, das unlösliche, denaturierte Proteine enthält, wurde verworfen. Jeweils 0,5 nmol der Proteine wurde, in einer Konzentration von 5 µM, zur Polymerisation mit 300 mM Saccharose, 4 µl Polymerisationsinduzierer und evtl. Zusätzen von z. B. 1 und 10 mM FBP, je 25 mM DTTred und DTTox sowie GSSG und 50 mM GSH auf Eis gemischt und mit 1 × LSB auf ein Endvolumen von 100 µl aufgefüllt. Die Proben wurden dann mindestens 1 h bei 30°C inkubiert. Es folgte eine Zentrifugation für 30 min bei 10 000 × g und 30°C, um Aktin-Bündelungsproteine, unspezifische Bindungen und denaturierte Proteine zu pelletieren. Der Überstand der Proben wurde abgenommen, und in der anschließenden Zentrifugation (1 h, 200 000 × g, 30°C) pelletierten Aktin-Filamente und daran gebundene Proteine. Überstand und Pellet wurden getrennt und für die Analyse der Proben eingefroren. Zu den Pellets wurden vor dem Einfrieren 100 µl H2Obidest gegeben. Durch das Einfrieren bei –20°C depolymerisieren die Proben wieder. Die Auswertung der Proben erfolgte mittels SDSPAGE, wobei die Proteinmengen der Pellets densitometrisch (SpectraMaxPlus; SOFTmax Por Version 3,0, BioRad, München) analysiert wurden. LSB-Puffer (10 ×): 50 mM Tris 2 mM CaCl2 2 mM ATP 5 mM DTTred pH 8,0 (HCl) Material und Methoden Polymerisationsinduzierer: 72 10 mM Tris pH 7,5 (HCl) 1 M KCl 50 mM MgCl2 25 mM ATP Die Auswertung der densitometrisch gemessenen Proben erfolgte durch den Vergleich der verschiedenen Proben eines Proteins untereinander, wobei die in der Kontrolle nachgewiesene Proteinbande gleich 100% gesetzt wurde. Um den Anteil der an das polymerisierte Aktin gebundenen Aldolase ermitteln zu können, wurde außerdem für jede Probe das pelletierte Aktin gleich 100% und das Aldolasepellet in Bezug dazu gesetzt. Nach Berechnung von Mittelwert (MW), Standardabweichung (Sd) und Varianz (sx2) konnten die Proben mit Hilfe des t-Tests mit der Kontrolle verglichen werden. Das daraus resultierende Signifikanzniveau gibt Aufschluss über die Signifikanz der gemessenen Unterschiede zwischen Probe und Kontrolle. Alle Formeln und benutzten Werte sind dem Buch „Biostatistik“ von KÖHLER, SCHACHTEL und VOLESKE (2002) entnommen. Die Varianz sx2 gibt die durchschnittliche quadratische Abweichung der Einzelwerte vom Mittelwert an und ist mit folgender Formel zu berechnen: 2 m ∑ fi x i 1 m i 1 2 2 sx = * ∑ fi x i − = n - 1 i =1 n wobei m die Anzahl verschiedener Messwerte, xi der i-te verschiedene Messwert, fi die Häufigkeit des Messwertes xi, n fi der Stichprobenumfang ist und i der Laufindex von 1 bis m läuft. Material und Methoden 73 Für den Vergleich, ob die Mittelwerte zweier Stichproben signifikant verschieden sind, muß berechnet werden: t versuch = wobei x−y SD × n1 − n 2 n1 + n 2 n1 der Umfang der Stichprobe x, n2 der Umfang der Stichprobe y, x und y die jeweiligen Mittelwerte und 2 ( x) 1 ∑ 2 SD = × ∑x − + n1 + n 2 − 2 n1 ( ) (∑ y ) − (∑n y) 2 2 2 ist. Der Wert tVersuch wird dann mit dem Wert tTab verglichen, wobei die Freiheitsgrade (FG = n1 + n2-2) und das Signifikanzniveau berücksichtigt werden. Ist tVersuch tTab, unterscheiden sich die beiden Stichproben nicht signifikant voneinander. Ist tVersuch > tTab, sind die Stichproben signifikant verschieden. Die Tabelle für die tTab-Werte befindet sich im Anhang. 2.3.10 Stöchiometrie der Aldolase-Aktin-Bindung Um die Stöchiometrie der Bindung von Aldolase an F-Aktin zu bestimmen, wurden Versuche in Anlehnung an VITAVSKA et al. (2003) durchgeführt. Dafür wurde 1 mg Aktin in 500 µl eiskaltem H2Obidest gelöst und bei 4°C eine Stunde depolymerisiert. Strep-tag-Aldolase wurde in Puffer P umgepuffert und, wie das Aktin, 1 h zentrifugiert (200 000 × g, 4°C). Der Proteingehalt des Überstandes der Zentrifugation wurde bestimmt, und das Aktin für die Polymerisation wurde mit Puffer P auf eine Konzentration von 10 µM eingestellt. Die Aktinpolymerisation erfolgte bei 30°C für eine Stunde. Anschließend wurden die Aktinfilamente durch Zugabe von Phalloidin (200 nM) stabilisiert und weitere 15 min bei 30°C inkubiert. Das polymerisierte, stabilisierte Aktin wurde auf eine Konzentration von 200 nM mit Puffer P mit Phalloidin (200 nM) und DTTred (2 mM) eingestellt. Für die Ermittlung der Bindestöchiometrie wurden je 200 nM Aktin mit Aldolase in Konzentrationen von 25 nM bis 800 nM gemischt und bei 20°C 120 min inkubiert. Bei der anschließenden Zentrifugation (200 000 × g, 1 h, 20°C) wurden Aktinfilamente und die daran gebundene Material und Methoden 74 Aldolase pelletiert. Die Proben wurden in Überstand und Pellet getrennt und vor der Analyse mittels SDS-PAGE bei –20°C eingefroren. Da die Proteinkonzentrationen der Proben sehr gering waren, wurden die Gele durch Silberfärbung angefärbt. Puffer P: 5 mM Tris pH 8,0 (HCl) 40 mM KCl 2 mM MgCl2 0,2 mM CaCl2 1,2 mM ATP 2.3.11 Messung der Aldolaseaktivität Die Messungen der Aktivität gereinigter, rekombinanter His-tag- und Strep-tag-Aldolase erfolgten photometrisch an einem Spektralphotometer (Eppendorf 1101 M, Hamburg) bei 25°C mit einer Wellenlänge von 340 nm. Die Aktivität wurde in Anlehnung an die Methode von YELTMAN und HARRIS (1980) bestimmt. Folgende Reaktion in der Glykolyse wird durch die Aldolase katalysiert: Aldolase D-Fructose-1,6-bisphosphat Dihydroxyacetonphosphat + D-Glycerinaldehyd-3-phosphat Da diese Reaktion nicht direkt messbar ist, wird eine nachgeschaltete Indikatorreaktion, katalysiert von der Glycerin-3-phosphat-Dehydrogenase (GDH), gemessen. Die messbare Absorptionsänderung bei der Umwandlung von NADH2 in NAD+ erfolgt durch die Bildung von Glycerin-3-phosphat aus D-Glycerinaldehyd-3-phosphat. Das durch die von der Aldolase katalysierte Reaktion ebenfalls entstehende Dihydroxyacetonphosphat wird durch die Triosephosphat-Isomerase (TPI) in D-Glycerinaldehyd-3-phosphat umgewandelt und steht so der Umwandlung durch GDH ebenfalls zur Verfügung. Alle drei Reaktionen sind in dem unten gezeigten Reaktionsschema dargestellt: Dihydroxyacetonphosphat + D-Glycerinaldehyd-3-phosphat Aldolase D-Fructose-1,6-bisphosphat Triosephosphat-Isomerase Dihydroxyacetonphosphat D-Glycerinaldehyd3-phosphat D-Glycerinaldehyd-3-phosphat Glycerin-3-phosphat-Dehydrogenase NADH2 NAD+ Glycerin-3-phosphat Material und Methoden 75 Für die Aktivitätsmessungen bestand der Reaktionsansatz aus: 100 mM MOPS pH 8,0 (NaOH) 0,25 mM NADH2 je 10 µg GDH/TPI 1 mM Fructose-1,6-bisphosphat 2 µg Aldolase Für die Umsetzung von einem Mol Fructose-1,6-bisphosphat werden jeweils zwei Mol NADH2 verbraucht. Zur Untersuchung einer möglichen Redox-Modulation der Aldolase wurde der Messansatz verändert, so dass oxidierende oder reduzierende Bedingungen erzeugt wurden. Dafür wurde die Aldolase mit DTT oder Glutathion in oxidierter und reduzierter Form in einem Aktivierungsansatz vorinkubiert. Durch die Zugabe der reduzierenden oder oxidierenden Substanzen wurde der Aktivierungsansatz gestartet und nach 5, 10, 20, 40 und 60 min wurden aliquote Anteile (je 100 µl) entnommen und im Aktivitätstest gemessen. Um eine mögliche Absorption oder Bindung von Proteinen an der Oberfläche des Reaktionsgefäßes während der Inkubationszeit des Aktivierungsansatzes zu verhindern, wurde den Proben standardmäßig BSA zugegeben, das keinen Einfluss auf die Reaktion hat. Neben Messungen bei einem pHWert von 8,0 wurden unter reduzierenden Bedingungen zusätzlich Messungen bei pH 9,0 durchgeführt. Aktivierungsansatz (je Probe): Endkonzentration: X µl Probe 2 µg 10 µl 200 mM MOPS mit unterschiedlichem pH-Wert 20 mM 10 µl BSA (10 µg/µl) 10 µg X µl Reduktions- oder Oxidationsmittel (DTT, Glutathion) 10/50 mM mit H2Obidest auf ein Endvolumen von 100 µl aufgefüllt Material und Methoden 76 2.3.12 Faktor-Xa -Verdau Bei dem Vektor pET-16b besteht die Möglichkeit, den His-tag nach der Expression vom rekombinanten Protein durch einen Verdau mit dem Enzym „Faktor-Xa“ (Novagen, Madison, USA) abzuspalten. Die Nukleotidsequenz für die Erkennungssequenz dieser Protease ist zwischen der multiplen Klonierungsstelle und der Sequenz des His-tags auf dem Plasmid enthalten. Um optimale Bedingungen für den Verdau zu finden, wurden Optimierungsversuche mit verschiedenen Enzymkonzentrationen, Inkubationszeiten und -temperaturen durchgeführt. Jeweils 10 µg rekombinantes, gereinigtes Protein wurde mit 0; 0,2; 0,5; 2 und 5 U/µl Enzym 16, 23 und 46 h bei 4°C und RT geschnitten. 50 µl Reaktionsansatz: 10 µg Protein 5 µl 10 × “Cleavage Capture Buffer” 1 µl Enzym (verschiedener Konzentrationen) mit H2Obidest auf 50 µl auffüllen. Die Analyse des Proteaseverdaus erfolgte durch Auftragen der Proben auf SDS-PAGE. 2.3.13 Bioinformatische Analysen Die bekannte Sequenz der cytosolischen Aldolase aus Mais (Z. mays L.) sowie des spannungsabhängigen Mitochondrien-Kanal-Proteins 1 (VDAC) aus Mais (Z. mays L.) der Datenbank GenBank (National Center of Biotechnology Information, NCBI, http://www.ncbi.nlm.nih.gov/) diente der Herstellung von Primern und zur Charakterisierung sequenzierter Proben. Das Programm „Basic Local Alignment Search Tool“ (BLAST 2, http://www.ncbi.nlm.nih.gov/BLAST/gorf/bl2.html, National Center of Biotechnology Information, NCBI) wurde für die Identifizierung homologer Sequenzabschnitte verwendet (ALTSCHUL et al., 1990; ALTSCHUL et al., 1997). Schnittstellen von Restriktionsenzymen innerhalb von Gensequenzen findet (http://www.firstmarket.com/cutter/cut2). das Programm Nukleotidsequenzen in „Webcutter 2.0“ Aminosäuresequenzen umzuwandeln ist mit dem ExPASy Molecular Biology Server (ExPASy: „Expert Protein Analysis System“) auf der Seite http://www.expasy.ch/tools/dna.html möglich, wo auch Molekulargewichte von Proteinen errechnet werden können. Für Klonierungen in den Vektor pASK-IBA3 stellt die Firma IBA (Göttingen) auf ihrer Internetseite (http://www.iba-go.de) ein Programm für die Primerherstellung (“PrimerD´Signer“) zur Verfügung. Ergebnisse 77 3 Ergebnisse Zur Untersuchung der Mikrokompartimentierung des pflanzlichen Primärstoffwechsels am Cytoskelett wurden zwei verschiedene Ansätze gewählt. Zunächst sollte pflanzliche cytosolische Aldolase (E.C. 4.1.2.13) aus Mais (Z. mays L.) auf eine mögliche Interaktion mit Aktinfilamenten untersucht werden. Eine putative Bindedomäne der pflanzlichen Aldolase ergab sich aufgrund der Proteinsequenzhomologie mit der bekannten Aktin-Bindedomäne der tierischen Aldolase (WINTER & HUBER, 2000, s. Kap. 1.2.2). Um Interaktionspartner der Aldolase denkbar wären z. B. andere glykolytische Enzyme zu finden, wurde das Hefe-2- Hybrid-System als Verfahren gewählt. Auf Grund dieser beiden Ansätze gliedert sich der Ergebnisteil in mehrere Bereiche. Im ersten Teil werden notwendige Vorarbeiten für die folgenden Untersuchungen beschrieben. Diese beinhalten die Ergebnisse von Klonierung und Überexpression der Aldolase, Aktivitätsmessungen zur Charakterisierung der rekombinanten Aldolasen und die Herstellung eines Antikörpers zur Immundetektion. Im zweiten Teil folgen die Ergebnisse der Untersuchung zur Bindung der Aldolase an Aktin aus Kaninchenmuskel. Der letzte Teil befasst sich mit den Ergebnissen des Hefe-2-Hybrid-Systems und der Überprüfung einer mit diesem System gefundenen Interaktion mit Hilfe anderer Methoden wie Affinitätschromatographie und Far-Western-Blot. 3.1 Vorarbeiten 3.1.1 Klonierung des Expressionskonstrukts ALD_pET-16b Durch eine RT-PCR wurde ein 1068 bp großes DNA-Fragment amplifiziert (Abb. 3.1 A) und mit Hilfe einer Agarose-Gelelektrophorese analysiert. Nach der Klonierung dieses Fragments in den Vektor pET-16b wurde ein Restriktionsverdau mit den Enzymen Eco RI und Xba I durchgeführt. Dieser führte zu den in Abb. 3.1 B gezeigten zwei Banden, einer Vektorbande bei über 5000 bp und dem Aldolase-Fragment bei ca. 1500 bp. Durch die gewählten Schnittstellen enthält das Aldolase-Fragment zusätzlich ca. 420 bp des Vektors und hat daher nicht die erwartete Größe von ca. 1100 bp. Dieses Plasmidkonstrukt wird im Folgenden ALD_pET-16b genannt. Um die Nukleotidsequenz zu überprüfen und eine mögliche Leserasterverschiebung durch Fehler während der PCR auszuschließen, wurde eine Sequenzierung des Konstrukts durchgeführt. Das Ergebnis dieser Sequenzierung zeigt an neun Stellen einen Nukleotidaustausch im Vergleich zur in der Datenbank GenBank veröffentlichten Sequenz. Dabei handelt es sich jedoch in acht Fällen um sogenannte „stille Ergebnisse 78 Mutationen“, d. h. dass die geänderte Nukleotidsequenz nicht zu einer veränderten Aminosäure führt. Ein Aminosäureaustausch kommt durch einen Nukleotidaustausch des ersten Nukleotids des Tripletts an Position 787 des Klons zustande, wodurch das Codon für ein Leucin anstelle eines Valins entsteht. Bei beiden Aminosäuren handelt es sich um neutrale Aminosäuren. Das Ergebnis der Sequenzanalyse ist im Anhang wiedergegeben. A B bp Ma bp 11500 2838 11500 1700 1700 1093 1093 Ma Vektor pET-16b 2838 Aldolase PCRProdukt 805 Aldolase„Insert 805 Abb. 3.1: Agarosegel (A) zur Überprüfung der Größe des amplifizierten PCR-Produktes und (B) Restriktionsanalyse des klonierten Endkonstruktes ALD_pET-16b. Ma: Lambda DNA/Pst I Marker 24, MBI Fermentas, St. Leon-Rot. 3.1.2 Heterologe Expression und Reinigung von Aldolase (Z. mays) mit His-tag Das ALD_pET-16b Plasmidkonstrukt kodiert das Fusionsprotein Aldolase mit N-terminalem His-tag. Für das Fusionsprotein mit His-tag wurde ein Molekulargewicht von 40 kDa berechnet, wobei der aus zehn Histidinen bestehende His-tag mit etwa 1,6 kDa anzusetzen ist. Zur heterologen Expression wurden E. coli BL21(DE3)pLysS-Zellen mit dem Plasmidkonstrukt ALD_pET-16b transformiert und wie in Kap. 2.3.6.6 beschrieben kultiviert. Nach Anzucht der Zellen bei 37°C, Induktion der Proteinsynthese mit IPTG und darauffolgender Absenkung der Anzuchtstemperatur auf 30°C zeigte sich eine sehr gute Überexpression des Fremdproteins im Vergleich zu den zelleigenen Proteinen (s. Abb. 3.2). Die Induktion der Fremdproteinsynthese erfolgte während der exponentiellen Wachstumsphase der Bakterien ca. 120 min nach Inokulation der Kultur. Durch die Induktion der Proteinsynthese und die Temperatursenkung sinkt die Teilungsrate der Zellen und es erfolgt eine vermehrte Proteinexpression. Das Zellwachstum, gemessen anhand der optischen Dichte der Zellsuspension bei 600 nm gegen H2Obidest, ist der Abb. 3.3 zu entnehmen. Ergebnisse 79 kDa M Ma vor Induktion nach Induktion ÜS ÜS P P 83 62 48 33 25 Abb. 3.2: Heterologe Expression der cytosolischen Aldolase von Z. mays in E. coli mit dem Plasmidkonstrukt ALD_pET-16b. Dargestellt sind verschiedene Fraktionen vor und nach der Induktion der Proteinsynthese, die aus jeweils einem aliquoten Anteil der Bakterienkultur von 1 ml gewonnen wurden. ÜS: Überstand mit löslichen Proteinen, P: Pellet mit unlöslichen Proteinen, Ma: Molekularmassenstandard (Prestained Protein Marker, Broad Range, New England BioLabs, Beverly, MA, USA). Der Pfeil markiert die Höhe der Bande der exprimierten rekombinanten Aldolase. Bei Ernte der Zellen nach einer vierstündigen Synthese des Fremdproteins erreichte die optische Dichte einen Wert über zwei. Mit Hilfe von 1 ml-Anteilen, die während der Proteinsynthese der Zellkultur entnommen worden waren, konnte nach Analyse auf SDSPAGE auf Höhe von etwa 40 kDa eine deutliche Proteinanreicherung nachgewiesen werden. Nach dem Zellaufschluss befand sich das Protein im geklärten Rohextrakt. Mit Hilfe der Metall-Chelat-Affinitätschromatographie konnte es aufgrund seines His-tags gereinigt und angereichert werden. Bei der in Kapitel 2.3.6.7.1 beschriebenen Reinigungsmethode wurde das Optische Dichte bei 600 nm 2,500 Protein gut Verunreinigungen von mit 2,000 anderen Proteinen getrennt, 1,500 und der größte Teil des 1,000 Proteins eluierte bei einer 0,500 Imidazolkonzentration von 0,000 450 mM. Die Ausbeute an 0 30 60 90 120 150 180 210 240 270 300 330 360 Zeit [min] Abb. 3.3: Wachstumskurve von E. coli BL21(DE3)pLysS mit dem Plasmidkonstrukt ALD_pET-16b. Der Pfeil zeigt den Induktionszeitpunkt der Proteinsynthese mit IPTG an. gereinigter Aldolase aus einem Liter Kultur betrug mindestens Abb. 3.4). 5 mg (s. Ergebnisse 80 150 300 450 1000 RE W E mM Imidazol Ma kDa 100 60 40 30 Abb. 3.4: Reinigung der rekombinanten Aldolase des Expressionskonstruktes ALD_pET-16b über MetallChelat-Affinitätschromatographie. SDS-Page-Analyse von geklärtem Rohextrakt vor dem Auftrag auf die Affinitätssäule (RE), Waschschritte 1 und 10 mit Bindepuffer und Waschschritt 16 mit Waschpuffer (W) und nach Elution mit Imidazol in unterschiedlichen Konzentrationen (E). Ma: Molekularmassenstandard (RotiMark 10-150, Roth, Karlsruhe). Aufgetragen wurden 25 µl der jeweiligen Fraktion. 3.1.3 Faktor-Xa-Verdau Wegen der Größe des His-tags und seiner Ladung sollte von dem rekombinanten Protein der Tag proteolytisch entfernt werden. Die Proteaseschnittstelle der Faktor-Xa-Protease zwischen der codierenden Sequenz der Aldolase und dem His-tag sollte die Möglichkeit bieten, den His-tag abzuspalten. Dafür wurden verschiedene Ansätze mit unterschiedlich viel Enzym (s. Kap. 2.3.12) und 10 µg rekombinanter, gereinigter Aldolase angesetzt. Es konnten jedoch keine Bedingungen gefunden werden, die zum Abspalten des His-tags ohne gleichzeitige Degradation der Aldolase führten. Da das rekombinante Protein mit His-tag weder Aktivität noch Aktinbindung zeigte, eignete sich diese Aldolase nicht für die weiteren Versuche, konnte jedoch als Ausgangsmaterial zur Herstellung eines Aldolase-Antikörpers verwendet werden. Eine mögliche Beeinflussung der biochemischen Eigenschaften eines Proteins durch den His-tag wurde bereits von WU & FILUTOWICZ (1999) beschrieben. Ergebnisse 81 3.1.4 Herstellung eines polyklonalen Antiserums zur Immundetektion von Aldolase Die gereinigte, rekombinante Aldolase mit His-tag wurde für die Erzeugung eines polyklonalen Antiserums in Kaninchen genutzt. Das erhaltene Antiserum wurde auf seine Spezifität auf Western-Blots mit Sproß- und Wurzelgewebe von Mais (Z. mays L., var. Caramba), Arabidopsis (A. thaliana L., var. Columbia) und Kartoffel (Solanum tuberosum L., var. Serena) sowie tierischer Aldolase (Boehringer, Mannheim) getestet. Bei allen pflanzlichen Geweben konnte eine deutliche Bande detektiert werden. Gezeigt ist in Abb. 3.5 der positive Nachweis von Aldolase aus Arabidopsis-Blatt und Spross- und Wurzelgewebe von Mais, wogegen tierische Aldolase durch das Serum nicht erkannt wurde. Die detektierte rekombinante Aldolase zeigt einen Größenunterschied zu den anderen Proben, der auf den mit dem Protein fusionierten His-tag zurückzuführen ist. Auch geringe Proteinmengen (50 ng) rekombinanter Strep-tag-Aldolase konnten auf Western-Blots mit Hilfe des Antiserums nachgewiesen werden. Im ELISA, bei dem die Proben nicht denaturiert werden, wurde native rekombinante Aldolase bis zu einer Menge von 3 ng erkannt (Daten nicht gezeigt). 1 2 3 4 5 kDa Ma kDa 120 100 84 75 52 50 -His-tag 36 37 Abb. 3.5: Untersuchung der Spezifität des Mais-Aldolase-Antiserums aus Kaninchen durch Western-BlotAnalyse mit verschiedenen pflanzlichen Geweben (je 20 µg Protein) und tierischer Aldolase. 1: tierische Aldolase (4 µg, Boehringer, Mannheim), 2: A. thaliana, 3: Wurzel Z. mays, 4: Sproß Z. mays, 5: rekombinante Aldolase mit His-tag, Ma: Molekularmassenstandard (Prestained Protein Standard, BioRad, München). Der Größenunterschied der rekombinanten Aldolase beruht auf der Größe des His-tags dieses Proteins ( -His-tag). Ergebnisse 82 3.1.5 Klonierung des Expressionskonstruktes ALD_pASK-IBA3 Für alle weiteren Versuche war es notwendig, Aldolase mit katalytischer Aktivität herzustellen, da für Aldolase mit His-tag, die keine Aktivität zeigte, auch keine Bindung an Aktin nachgewiesen werden konnte. Um den Einfluss des His-tags zu umgehen, wurde ein Strep-tag Vektor gewählt. Bei dem eingesetzten Vektor (pASK-IBA3) ist eine proteolytische Abspaltung des tags nicht möglich. Im Gegensatz zum His-tag wird der Strep-tag bei dem eingesetzten Vektor C-terminal am Protein exprimiert. Bei der Reinigung eines Proteins mit Strep-tag kann unter physiologischen Pufferbedingungen gearbeitet werden. Ebenfalls von Vorteil ist, dass der Strep-tag ungeladen ist (TERPE, 2003). Die Aminosäuresequenz des Strep-tags ist der schematischen Darstellung der Aldolase-Expressionskonstrukte im Anhang zu entnehmen (s. Kap. 7.2.5). Die Klonierung erfolgte wie in Kapitel 2.3.3.16.3 beschrieben. Mit Hilfe eines Restriktionsverdaus mit den Restriktionsendonukleasen Xba I und Hind III konnten positive Klone identifiziert werden (s. Abb. 3.6). Die Nukleotidsequenz eines positiven Klons wurde durch Sequenzierungsreaktionen überprüft. Durch einen Vergleich der Sequenz des Klons mit der Nukleotidsequenz der Aldolase in der Datenbank GenBank ergaben sich sieben unterschiedliche Nukleotide, von denen eins zu einer veränderten Aminosäure in der Proteinsequenz führt (s. Anhang Kap. 7.2.8). Dabei handelt es sich, wie schon bei der Klonierung der Aldolase in den pET-16b Vektor, um Nukleotid 787. Das resultierende Codon ergibt die Aminosäure Valin an Stelle eines Leucins. bp Ma 6 7 8 9 10 11 11500 2838 1700 1093 805 Vektor pASK-IBA3 Aldolase “Insert Abb. 3.6: Identifizierung positiver Klone nach Klonierung von Aldolase in den Vektor pASK-IBA3 durch einen Restriktionsverdau. 6: Klon 6 (enthält kein „Insert ), 7: Klon 7 (wurde für alle weiteren Versuche verwendet), 8: Klon 8, 9: Klon 9, 10: Klon 10, 11: Klon 11, Ma: Lambda DNA/Pst I Marker 24, MBI Fermentas, St. Leon-Rot. Ergebnisse 83 3.1.6 Heterologe Expression und Reinigung von Aldolase (Z. mays) mit Strep-tag Das Fusionsprotein Aldolase mit Strep-tag wurde durch Überexpression des Konstruktes ALD_pASK-IBA3 in E. coli XL1blue-Zellen hergestellt. Der C-terminale Strep-tag ermöglicht die Reinigung des Proteins mit Hilfe der Strep-Tactin-Affinitätsmatrix. StrepTactin ist ein technisches Streptavidin, an das der aus acht Aminosäuren bestehende Strep-tag Optische Dichte bei 550 nm mit hoher Affinität bindet. Die 2,5 Elution des Proteins erfolgt durch 2,0 geringe 1,5 (2,5 mM) Biotin oder Desthio- Konzentrationen biotin. 1,0 Die Wachstumskurve der Zellen 0,5 zeigte auch nach Induktion der 0,0 0 30 60 90 120 150 180 210 240 270 300 330 360 Zeit [m in] Proteinsynthese keine deutliche Abflachung. Die optische Dichte stieg weiter an, die Zellen haben Abb. 3.7: Wachstumskurve von E. coli XL1blue mit dem Plasmidkonstrukt ALD_pASK-IBA3. Der Pfeil zeigt den Induktionszeitpunkt der Proteinsynthese mit AHT. sich weiterhin mit gleicher Teilungsrate vermehrt (Abb. 3.7). Die während des Wachstums vor und nach der Induktion abgenommenen aliquoten Anteile (1 ml) der Zellsuspension zeigen bei einer 12 % SDS-PAGE die Anreicherung einer Proteinbande bei knapp 40 kDa, was der erwarteten Größe der Aldolase mit dem ca. 1,06 kDa großen Strep-tag entspricht (s. Abb 3.8). Nach dem Aufschluss der Zellen befand sich das gewünschte Protein im geklärten Rohextrakt und konnte durch Affinitätschromatographie mit nur sehr geringen Verunreinigungen gewonnen werden. Hundert Milliliter Zellkultur lieferten auf diese Weise ca. 2 mg Protein. Anhand der Abbildung 3.9, bei der einige Fraktionen des Aufschlusses auf eine 12 % SDSPAGE aufgetragen wurden, ist deutlich zu erkennen, dass das gereinigte Protein in den Fraktionen E2 bis E6 eluiert. Ergebnisse 84 kDa 180 Ma 0 4 73 54 35 Aldolase 24 Abb. 3.8: Heterologe Expression rekombinanter Strep-tag Aldolase vor und nach der Induktion der Proteinexpression. SDS-PAGE mit je 10 µl Bakteriensuspension vor der Induktion (0) und 4 h nach Induktion (4). Ma: Molekularmassenstandard (Prestained Protein Ladder, ~10-160kDa, MBI Fermentas, St. Leon-Rot). kDa Ma RE DF W2 E1 E2 E3 E4 E5 E6 118 48 32 19 Abb. 3.9: Reinigung rekombinanter Strep-tag Aldolase über Strep-tag-Affinitätschromatographie. SDS-PAGE-Analyse von geklärtem Rohextrakt (RE; 5 µl) vor Auftrag auf die Säule, Durchfluss ungebundener Proteine nach Auftragen der Probe auf die Säule (DF), Waschschritt 2 (W2) und den Elutionschritten 1-6 (E1-E6) (je 20 µl). Ma: Molekularmassenstandard (Prestained Protein Molecular Weight Marker, MBI Fermentas, St. Leon-Rot). Ergebnisse 85 3.1.7 Katalytische Eigenschaften rekombinanter Aldolase Um die rekombinanten Aldolasen zu charakterisieren, wurden Aktivitätsmessungen mit dem Substrat FBP durchgeführt. Die Aktivitätsmessungen erfolgten nach der in Kapitel 2.3.11 beschriebenen Methode mit den heterolog hergestellten, gereinigten rekombinanten MaisAldolasen. Rekombinant überexprimierte Aldolase mit His-tag des Konstruktes ALD_pET-16b zeigte keine Aktivität. Die gereinigte Aldolase mit Strep-tag (ALD_pASK-IBA3, Reinheit siehe Abb. 3.9) wies Aktivität auf. Diese betrug 1,23 U/mg Protein (+/-0,25 U/mg Protein). Im Gegensatz zu dieser geringen Aktivität wurde eine rekombinante anaerobe cytosolische Aldolase aus Mais mit einer Aktivität von 20,8 U/mg durch BERITAUME et al. (1989) beschrieben. Um eine mögliche Redox-Modulation der Aldolase zu ermitteln, wurden Aktivierungsansätze mit dem synthetischen Redoxreagenz DTT, in reduzierter und oxidierter Form, und dem natürlichen Redoxreagenz Glutathion (GSH und GSSG) bei verschiedenen pH-Werten (pH 8,0 und 9,0) nach jeweils 5, 10, 20, 40 und 60 min Inkubation gemessen. Dabei konnten bei oxidiertem Glutathion und DTT (je 10 mM) keine Änderungen der Aktivität gemessen werden. Auch reduzierende Bedingungen führten zu keiner signifikanten Steigerung der Aktivität (Daten nicht gezeigt). Um den KM-Wert der Aldolase mit Strep-tag zu ermitteln, wurde ihr Substrat, Fructose-1,6bisphosphat, in verschiedenen Konzentrationen von 1M bis 0,00012 mM in Aktivitätsmessungen eingesetzt. Bei Auftragung der Reaktionsgeschwindigkeit [V] (U/mg) gegen die Substratkonzentration [S] (mM) zeigte sich eine hyperbole Kurve (s. Abb. 3.10 A). Die Substratkonzentration bei halbmaximaler Aktivität (KM) konnte anhand eines Lineweaver-Burk-Diagramms ermittelt werden (s. Abb. 3.10 B). Sie entspricht einer Substratkonzentration von 0,5 µM. Die maximale Geschwindigkeit der Reaktion betrug bei sättigender Substratkonzentration unter den gewählten Bedingungen ca. 1,2 U/mg (Vmax) und konnte ab einer Substratkonzentration von 7,4 µM FBP nachgewiesen werden. Ergebnisse 86 A Aldolaseaktivität [U/mg] 1,6 1,4 1,2 1,0 0,8 0,6 0,4 0,2 0 0 0,002 0,004 0,006 0,2 0,4 0,6 0,8 1,0 Substrat [mM] B 1/ Aktivität mg × min 4 µMol 2 0 -2 -4000 -2000 0 2000 4000 6000 8000 1 / [Substrat] (1/mM) Abb. 3.10: Aktivität der rekombinanten Strep-tag Aldolase von Z. mays in Abhängigkeit von der FBP-Konzentration. Die Messungen erfolgten bei pH 8,0. Mit dem Programm Grafit 5.0 erfolgte die Auswertung als Michaelis-Menten-Modell (A) und in einem Lineweaver-Burk-Diagramm (B). Ergebnisse 87 3.2 Copolymerisationsversuche Für die Untersuchung der direkten Wechselwirkung zwischen pflanzlicher Aldolase und Aktinfilamenten wurden Copolymerisationsversuche durchgeführt, wie in Kapitel 2.3.9 beschrieben. Die rekombinante Mais-Aldolase mit Strep-tag bindet an Aktinfilamente (s. Abb. 3.11). Eine Bindung tierischer Aldolase an F-Aktin wurde schon beschrieben und charakterisiert (z. B. ARNOLD & PETTE, 1970; DON & MASTERS, 1988, O´REILLY & CLARKE, kDa Ma 1 1993; WANG et al., 1996). Erst kürzlich konnte auch 2 pflanzliche Aldolase in der Cytoskelettfraktion von 73 Maisendosperm nachgewiesen werden (AZAMA et al., 54 2003). Bei den durchgeführten Versuchen wurde der Einfluss 24 von Fructose-1,6-bisphosphat (FBP, s. Abb. 3.11), welches im tierischen System die Bindung der Aldolase an Aktin bei einer Konzentration von 1 mM Abb. 3.11: SDS-PAGE des Niederschlags bei 200 000 × g nach Copolymerisation rekombinanter Strep-tag-Aldolase von Mais und Aktin aus Kaninchenmuskel. 1: 0,5 nmol Aktin und Aldolase, 2: wie Probe 1 mit Zusatz von 10 mM FBP. Ma: Molekularmassenstandard (Prestained Protein Ladder, ~10160 kDa, MBI Fermentas, St. LeonRot). vollständig unterdrückt (ARNOLD & PETTE, 1970), Fructose-6-phosphat (F-6P) und Fructose-2,6-bisphosphat (F-2,6BP) untersucht. F-6P und F-2,6BP zeigten im tierischen System bei Konzentrationen von 40 µM keinen nachweisbaren Effekt (WANG et al., 1996). Da es Hinweise auf den Einfluss des Redox-Milieus auf Aldolase (OFFERMANN et al., 1984; FRATELLI et al., 2002; ITO et al., 2003) sowie Aktin (HINSHAW et al., 1991; WANG et al., 2001) gibt, wurde zusätzlich das Redox-Milieu während der Versuche durch Glutathion und DTT beeinflusst. Dafür wurde die in die Versuche eingesetzte Aldolase vor Zugabe des Aktins mit den entsprechenden Substanzen vorinkubiert. Für die Auswertung der Ergebnisse gliedert sich dieser Abschnitt in zwei Bereiche. Diese beinhalten den Einfluss von FBP, F-6P und F-2,6BP (s. Kap. 3.2.1) und den Einfluss des Redox-Milieus (s. Kap. 3.2.2) auf die Aldolase-Aktin-Bindung. Die Auswertung der Proben erfolgte wie in Kapitel 2.3.9 beschrieben. Neben dem Vergleich der pelletierten Proteinmengen von Aldolase oder Aktin mit der in der Kontrolle pelletierten Proteinmenge wurde das Aldolase-Pellet in Bezug zum Aktin-Pellet gesetzt. Die Signifikanz der gemessenen Unterschiede wurde mit Hilfe des tTestes der einzelnen Substanzen in Bezug auf die Kontrolle ermittelt. Bei einem Signifikanzniveau von = 0,5 ist ein signifikanter Unterschied mit mind. 50 % Sicherheit aufgetreten und stellt die geringste Signifikanz der durchgeführten Versuche dar. Bei = 0,01 ist in 99 % der Versuche von einem signifikanten Unterschied auszugehen, und bei einem Ergebnisse 88 Signifikanzniveau von = 0,002 erreichen 99,8 % der Versuche einen signifikanten Unterschied zur Kontrolle. Allen hier aufgeführten Ergebnissen liegen mindestens drei unabhängig voneinander durchgeführte Versuche zugrunde. 3.2.1 Einfluss von FBP, F-6P und F-2,6BP auf die Aldolase-Aktin-Bindung Unter Einfluss von 1 mM und 10 mM FBP polymerisierten 82% bzw. 84% des Aktins im Vergleich zur unbehandelten Kontrolle (s. Tab. 3.2). Die mit dem F-Aktin pelletierte Aldolase entsprach jedoch bei 1 mM FBP nur 55% der Menge in der Kontrolle, bei 10 mM FBP pelletierten noch 43%. Dies bedeutet eine Hemmung der Bindung der Aldolase an Aktin unter Einfluss von 1 mM bzw. 10 mM FBP um etwa die Hälfte im Vergleich zur Kontrolle (s. Tab. 3.3). Diese Ergebnisse stehen im Gegensatz zum für tierische Systeme bekannten Einfluss der Metabolitkonzentrationen auf die Bindung (ARNOLD & PETTE, 1970). Tab. 3.2: Pelletierte Aktinmenge in Abhängigkeit von FBP, F-6P und F-2,6BP in Copolymerisationsversuchen von Strep-tag-Aldolase und Aktin im Vergleich zur unbehandelten Kontrolle in %. Exp.: Kontrolle 1 mM FBP 10 mM FBP 10 mM F-6P 10 mM F-2,6BP 1 100 80 97 85 96 2 100 79 71 3 100 4 100 86 83 300 MW 100 82 84 164 106 4 13 118 19 Sd 95 107 128 Tab. 3.3: Pelletierte Aldolasemenge in Abhängigkeit von FBP, F-6P und F-2,6BP in Copolymerisationsversuchen von Strep-tag-Aldolase und Aktin im Vergleich zur unbehandelten Kontrolle in %. Exp.: Kontrolle 1 mM FBP 10 mM FBP 10 mM F-6P 10 mM F-2,6BP 1 100 50 50 46 46 2 100 57 50 3 100 4 100 57 29 11 MW 100 55 43 31 42 4 12 18 11 Sd 50 37 30 Ergebnisse 89 Bezieht man die im Pellet nachgewiesene Aldolase auf das polymerisierte, pelletierte Aktin, so ergeben sich die in Abb. 3.12 dargestellten Anteile. Die in der jeweiligen Probe pelletierte Aktinmenge wurde gleich 100% gesetzt. Unter Kontrollbedingungen bindet mit 34%, in Bezug auf die pelletierte Aktinmenge, der größte Anteil Aldolase an F-Aktin. Unter Einfluss von FBP verringert sich der Anteil der mit dem F-Aktin pelletierten Aldolase auf 21% (1 mM FBP) bzw. 17% (10 mM FBP). Das bedeutet, dass unter Einfluss von 10 mM FBP die halbe Menge der in der Kontrolle nachgewiesenen Aldolase an das Aktin gebunden hat; der Einfluss von 1 mM FBP führte zu einer Bindung von 60% der Aldolasemenge der Kontrolle an das polymerisierte Aktin. Diesen Ergebnissen liegt ein Signifikanzniveau von = 0,01 (1 mM FBP) bzw. 0,002 (10 mM FBP) zugrunde. Neben FBP wurde der Einfluss von F-6P und F-2,6BP auf die Bindung der Aldolase an Aktin untersucht. Bei Einsatz beider Substanzen in einer Endkonzentration von 10 mM kam es zu einer deutlichen Reduktion der Bindung von Aldolase an Aktin. Die Reduktion der Bindung im Vergleich zur pelletierten Aldolasemenge der Kontrolle war durch F-6P am stärksten. Nur ein Drittel der in der Kontrolle nachgewiesenen Aldolasemenge pelletierten unter diesen Bedingungen (s. Abb. 3.12). Trotz der Schwankungen zwischen den Versuchen liegt diesem Ergebnis ein Signifikanzniveau von = 0,01 zugrunde. Der Einfluss von F-6P führte neben der Reduktion der an das F-Aktin bindenden Aldolasemenge zu einer verstärkten Aktinpolymerisation im Vergleich zur Kontrolle (s. Tab. 3.2), die jedoch starken Schwankungen unterlag. Durch F-2,6BP blieb die Polymerisation des Aktin unbeeinflusst (s. Tab. 3.2). Die Aldolasemenge dagegen, die im Pellet nachgewiesen werden konnte, war deutlich reduziert (s. Tab. 3.3). Der an das F-Aktin gebundene Anteil der Aldolase betrug 14% im Vergleich zu 34% in der Kontrolle (s. Abb. 3.12). Damit war der Einfluss von 10 mM F-2,6BP auf die Bindung der Aldolase an F-Aktin stärker als derjenige von 10 mM FBP (s. Abb. 3.12). Ergebnisse 90 Anteil gebundener Aldolase [%] 40 35 30 25 20 15 10 5 0 Kontrolle 1 mM FBP 10 mM FBP 10 mM F-6P 10 mM F-2,6BP Abb. 3.12: An F-Aktin bindender Anteil an Aldolase unter Einfluss von FBP, F-6P und F-2,6BP (in %). Das in den Proben polymerisierte Aktin entspricht 100%. Mit Abb. 3.13 wird die hemmende Wirkung der Substanzen auf die Bindung zwischen Aldolase und Aktin deutlich gemacht. Ausgangsbedingung dafür ist, dass unter Kontrollbedingungen die maximal mit Aktin pelletierte Aldolase nachgewiesen wurde. Der Anteil von 34% gebundener Aldolase an Aktin in der Kontrolle (Abb. 3.12) wird gleich 100% gesetzt. Die hemmende Wirkung beträgt damit in der Kontrolle 0%. Darauf bezogen zeigt sich die stärkste hemmende Wirkung unter Einfluss von F-6P. Sie beträgt knapp 70%. Die hemmende Wirkung von 10 mM FBP ist mit 50% geringer als die von 10 mM F-2,6BP. eingesetzte Substanzen und Konzentrationen Durch 1 mM FBP erfolgte eine Hemmung der Bindung um 38% (s. Abb. 3.13). 10 mM F-2,6BP 10 mM F-6P 10 mM FBP 1 mM FBP Kontrolle 0% 20% 40% 60% 80% hemmende Wirkung [%] Abb. 3.13: Hemmende Wirkung auf die Aldolase-Aktin-Bindung. Dargestellt ist der hemmende Einfluss verschiedener Substanzen auf den Anteil an FAktin gebundener cytosolischer Aldolase in %. Da in der Kontrolle von keiner Hemmung der Bindung auszugehen ist, wurde der Anteil der an Aktin gebundenen Aldolase mit 0% als ungehemmt angesehen. Ergebnisse 91 3.2.2 Einfluss des Redox-Milieus auf die Aldolase-Aktin-Bindung Der Einfluss des Redox-Milieus auf die Bindung der Aldolase an Aktin wurde durch Copolymerisation von Aktin und Aldolase nach Vorinkubation der Aldolase mit Reduktionsbzw. Oxidationsmitteln untersucht. Die Aktinpolymerisation wurde vom oxidierten Glutathion um 10% verringert, wogegen sie unter Einfluss von GSH geringfügig gesteigert wurde (7%, s. Tab. 3.4). Die pelletierte Aktinmenge wies unter Einfluss von GSH die stärksten Schwankungen zwischen den Versuchen auf. Eine Verringerung der Polymerisation von Aktin durch Glutathionylierung wurde bereits von WANG et al. (2001) beschrieben. Inkubation mit reduziertem und oxidiertem DTT führte jedoch zu einem anderen Ergebnis. Die pelletierte Aktinmenge entsprach unter reduzierenden Bedingungen etwa der Kontrolle, jedoch pelletierten unter Einfluss von oxidiertem DTT 24% mehr Aktin als in der Kontrolle. Die im Pellet nachgewiesenen Aldolasemengen waren im Vergleich zur Kontrolle unter Einfluss aller eingesetzten Reduktions- bzw. Oxidationsmittel geringfügig gesteigert. Nur unter Einfluss von GSH konnte eine deutliche Steigerung der pelletierten Aldolase (um 27%) nachgewiesen werden (s. Tab. 3.5). Tab. 3.4: Einfluss des Redox-Milieus auf die pelletierte Aktinmenge im Vergleich zur Kontrolle in %. Untersucht wurde der Einfluss des synthetischen Redoxreagenzes DTT und des natürlichen Redoxreagenzes Glutathion nach Vorinkubation der Aldolase mit diesen auf die Copolymerisationseigenschaften. Exp.: 1 2 3 4 5 MW Sd Kontrolle 100 100 100 100 100 100 GSH 92 125 106 87 127 107 0,81 GSSG 153 46 106 113 33 90 0,5 DTTred 93 125 85 88 DTTox 117 108 67 205 98 0,18 124 0,58 Tab. 3.5: Einfluss des Redox-Milieus auf die pelletierte Aldolasemenge im Vergleich zur Kontrolle in %. Untersucht wurde der Einfluss des synthetischen Redoxreagenzes DTT und des natürlichen Redoxreagenz Glutathion nach Vorinkubation der Aldolase mit diesen auf die Copolymerisationseigenschaften. Exp.: 1 2 3 4 5 6 MW Sd Kontrolle 100 100 100 100 100 GSH 104 120 95 152 162 GSSG 130 60 82 178 71 DTTred 106 120 64 147 DTTox 106 116 81 127 100 127 0,29 104 0,49 109 0,35 108 0,20 Ergebnisse 92 Betrachtet man den Anteil der an das polymerisierte Aktin gebundenen Aldolase, wobei die in der Probe polymerisierte Aktinmenge gleich 100% gesetzt wurde, so zeigt sich eine verstärkte Bindung von Aldolase an Aktin unter Einfluss von Glutathion (red. und ox.) und reduziertem DTT (s. Abb. 3.14). Der Einfluss von oxidiertem DTT liegt in einer leichten Verringerung der Menge gebundener Aldolase. Eine Signifikanz der Ergebnisse im Vergleich zur Kontrolle konnte bei Glutathion, in reduzierter und oxidierter Form, und oxidiertem DTT trotz der starken Schwankungen zwischen den Versuchen mit einem Signifikanzniveau von = 0,5 ermittelt werden. Bei einem Signifikanzniveau von = 0,5 ist von einem signifikanten Unterschied zur Kontrolle in der Hälfte der Versuche auszugehen. Keine Signifikanz zeigt der Unterschied der Bindung von Aldolase an Aktin Anteil der an F-Aktin gebundenen Aldolase [%] unter Einfluss von reduziertem DTT im Vergleich zur Kontrolle. 90,00 80,00 70,00 60,00 50,00 40,00 30,00 20,00 10,00 0,00 Kontrolle GSH GSSG DTT red DTT ox Abb. 3.14: Anteil der an F-Aktin gebundenen Aldolase unter Einfluss von Reduktionsund Oxidationsmitteln im Vergleich zur Kontrolle (in %). Das in den Proben polymerisierte Aktin entspricht 100%. Ergebnisse 93 Um die fördernde oder hemmende Wirkung der reduzierenden und oxidierenden Bedingungen auf die Bindung der Aldolase an Aktin zu verdeutlichen, wurde in Abbildung 3.15 der an das polymerisierte Aktin gebundene Anteil der Aldolase in der Kontrolle gleich 100% gesetzt. Dadurch wird deutlich, dass reduzierende Bedingungen, hervorgerufen durch DTT und Glutathion, zu einer um 15% (DTTred) bzw. 20% (GSH) verstärkten Aktinbindung der Aldolase führten. Zu einer verstärkten Aktinbindung der Aldolase um 40% führte die Inkubation mit oxidiertem Glutathion, wobei hier jedoch zu beachten ist, dass die einzelnen Versuche die größten Schwankungen im Anteil der an Aktin gebundenen Aldolase aufwiesen (s. Abb. 3.14). Eine hemmende Wirkung auf die an eingesetzte Substanzen Aktin gebundene Aldolase wurde durch oxidiertes DTT hervorgerufen. DTT ox DTT red GSSG GSH Kontrolle 0% 50% 100% 150% relativer Anteil der an F-Aktin gebundenen Aldolase [%] Abb. 3.15: Einfluss reduzierender und oxidierender Bedingungen auf den relativen Anteil an FAktin gebundener cytosolischer Aldolase (in %). Der in der Kontrolle an F-Aktin gebundene Anteil der Aldolase wurde gleich 100% gesetzt. Ergebnisse 94 3.2.3 Stöchiometrie der Aldolase-Aktin-Bindung Um die Stöchiometrie der Bindung von Aldolase an F-Aktin zu ermitteln, wurden Versuche wie in Kapitel 2.3.10 beschrieben durchgeführt. Dafür wurden mit Phalloidin stabilisierte Aktinfilamente unter die kritische Konzentration der Aktinpolymerisation verdünnt. Zu diesen Aktinfilamenten wurde Aldolase in verschiedenen Konzentrationen gegeben, und nach Zentrifugation bei 200 000 × g wurden die pelletierten Proteine mittels SDS-PAGE analysiert. Da es unter den gewählten Versuchsbedingungen zur Degradation der Aldolase kam, was auch nicht durch den Einsatz eines Serin-Protease-Inhibitors (Pefabloc 0,6 µg/µl) verhindert wurde, und Aldolase auch ohne eine Bindung an Aktinfilamente pelletierte, konnte mit Hilfe dieser Methode keine Bindestöchiometrie ermittelt werden. Der Einsatz von Detergenz und eines anderen Puffers („Low-Salt-Buffer“), der durch die Copolymerisationsversuche erprobt war, führte zu keiner Verbesserung der Ergebnisse. Der möglicherweise entscheidende Unterschied zwischen den Copolymerisationsversuchen und den Stöchiometrieversuchen besteht im Einsatz des Pilzgiftes Phalloidin. Dieses dient der Stabilisierung von Aktinfilamenten. Da bei den Copolymerisationsversuchen nicht mit Aktinkonzentrationen unterhalb der kritischen Konzentration der Aktinpolymerisation gearbeitet wurde, konnte auf eine Stabilisierung verzichtet werden. Ergebnisse 95 In einem weiteren Copolymerisationsversuch (s. Kap. 2.3.9) wurden 5 µM Aktin mit 0,110 µM Aldolase polymerisiert (s. Abb. 3.15). Die densitometrische Auswertung der Aldolasebanden im Pellet und im Überstand der Proben weist auf eine konzentrationsabhängige Bindung der Aldolase an Aktin hin. Von der eingesetzten Aldolase waren bei Aldolasekonzentrationen von 10 und 5 µM 30-40% an Aktin gebunden. Wurden geringere Aldolasekonzentrationen eingesetzt, erhöhte sich der gebundene Anteil auf 50-60% der eingesetzten Aldolasemenge. Bei allen Proben blieb ein Teil der Aldolase ungebunden im Überstand der Proben nachweisbar. A Aldolase [µM] Ma 10 5 2,5 1,25 0,625 0,313 0,16 0,078 55 kDa Aktin Aldolase 40 kDa B Aldolase [µM] Ma 10 5 2,5 1,25 0,625 0,313 0,16 0,078 55 kDa 40 kDa Aktin Aldolase Abb. 3.16: Überstand (A) und Pellet (B) des Copolymerisationsversuches mit 5 µM Aktin und 0,078-10 µM rekombinanter Aldolase (Z. mays) mit Strep-tag. Alle Proben enthielten 5 µM Aktin. Ma: Molekularmassenstandard (Prestained Protein Ladder, ~10-160kDa, MBI Fermentas, St. Leon-Rot). Ergebnisse 96 3.3 Ergebnisse der Hefe-2-Hybrid-Analysen Um mögliche Protein-Protein-Interaktionen zu finden, wurde das Hefe-2-Hybrid-System gewählt. Dafür wurde das glykolytische Enzym Aldolase in den Vektor pGBKT7 kloniert. Die Ergebnisse dieser Klonierung sowie der Herstellung einer cDNA-Bank und der durchgeführten Hefe-2-Hybrid-Analysen werden im Folgenden beschrieben. 3.3.1 Klonierung von Aldolase in den Vektor pGBKT7 Die Klonierung der Aldolase in den Vektor des Hefe-2-Hybrid-Systems, der die DNABindedomäne des Transkriptionsfaktors enthält (DNA-BD), erfolgte wie in Kapitel 2.3.3.16.2 beschrieben. Als Ausgangsmaterial diente das Plasmid ALD_pET-16b. Die positiven Klone wurden ansequenziert. Bei dem für die weiteren Arbeiten ausgewählten Klon ergab ein Vergleich der sequenzierten 562 Nukleotide des Klons eine Übereinstimmung von 99% mit der Sequenz der Datenbank GenBank. Alle vier Unterschiede zwischen den Nukleotidsequenzen sind stille Mutationen. 3.3.2 Herstellung einer cDNA-Bank Zur Herstellung der cDNA-Bank des Hefe-2-Hybrid-Systems wurde zunächst Gesamt-RNA aus Mais isoliert. Konzentration und Reinheit der Proben wurde photometrisch ermittelt (s. Kap. 2.3.3.3). Um Verunreinigungen mit DNA auszuschließen, wurden die Proben zusätzlich auf einem RNA-Gel analysiert (Abb. 3.17). Da keine Verunreinigungen mit DNA in den Proben nachzuweisen waren, wurde eine Probe, bei der auch keine Verunreinigung mit Proteinen und Probe 1 2 3 4 5 6 7 8 Abb. 3.17: Analyse der isolierten Gesamt-RNA aus verschiedenen Proben von Z.mays auf einem RNA-Gel. Für die Herstellung der cDNA-Bank des Hefe-2-Hybrid-Systems wurde die rot markierte Probe (8) verwendet, die aus einem Gemisch von hypoxisch angezogenem Sproß- und Wurzelgewebe bestand. aromatischen Phenol) Substanzen aufgrund der (z. B. photo- metrischen Messungen nachweisbar war, für die weiteren Arbeiten ausgewählt. Es handelte sich um Probe 8, deren 0,62 µg/µl betrug. Konzentration Ergebnisse 97 Die anschließende Synthese der “first-strand cDNA“ und der Amplifizierung der ds-cDNA durch „long-distance PCR“ erfolgte wie in Kapitel 2.3.5.4.1 beschrieben. Die Proben wurden anhand eines 1,2%igen Agarosegels überprüft und stimmten mit den Erwartungen (Handbuch „Matchmaker Library Construction & Screening Kit“, Clontech, Heidelberg) Abb. 3.18 hergestellt aus zeigt links die Kontrolle (ds-cDNA, überein. „Human Placenta + Poly A RNA , Clontech, Heidelberg), 10000 rechts sind drei Proben der aus Spross- 2500 2000 und 1000 750 angezogenem Mais amplifizierten ds- Wurzel-RNA von hypoxisch cDNA aufgetragen. Nach Reinigung der 500 ds-cDNA, wie in Kapitel 2.3.5.4.2 250 beschrieben, Ma 1 2 3 4 5 6 Ma Abb. 3.18: Analyse der amplifizierten ds-cDNA auf einem 1,2%igen Agarosegel. Links befindet sich in drei Spuren die Kontrolle aus „Human Placenta Poly A+ RNA (1-3), rechts sind drei Proben ds-cDNA aus hypoxischem Sproß- und Wurzelgewebegemisch von Z. mays dargestellt (4-6). Ma: Gene Ruler TM 1 kb DNA Ladder (MBI Fermentas, St. Leon-Rot). wurde die cDNA-Bank durch in-vivo-Rekombination in Hefen fertiggestellt. Die Transformations- effizienz wurde durch Auszählen einer Platte mit einer 1:1000-Verdünnung des Transformationsansatzes ermittelt und lag mit 1 x 104 unter der erwünschten Transformationseffizienz. Die Anzahl der Klone der cDNA-Bank betrug 1 x 105 Klone/ml und wurde nach der in Kapitel 2.3.5.4.5 beschriebenen Formel ermittelt. Nach Ernte der Zellen (Kap. 2.3.5.4.4) wurde die Zelldichte mit Hilfe eines Hemocytometers ausgezählt; dabei wurde ein Wert von 1 x 1010 Zellen/ml ermittelt, der damit über der benötigten MindestZelldichte von 1 x 107 Zellen/ml lag. 3.3.3 Test auf transkriptionale Aktivität des Fusionsproteinkonstruktes und Toxizitätstest Für die Durchführung der Hefe-2-Hybrid-Analysen war es notwendig, das Fusionsproteinkonstrukt aus Aldolase und der DNA-Bindedomäne des Transkriptionsfaktors (ALD_pGBKT7) auf eine eventuelle Toxizität und auf transkriptionelle Aktivierung in Hefen zu überprüfen. Um auszuschließen, dass durch Transkription und Translation des Konstruktes die Reportergene ohne eine Fusion mit der Aktivierungsdomäne angeschaltet werden, wurden beide Hefestämme mit dem Konstrukt transformiert. Die Transformanden wurden auf die Ergebnisse 98 Aktivität der Reportergene (s. Kap. 2.3.5) untersucht, indem sie auf verschiedene selektive Medien ausplattiert wurden. Ohne eine Aktivierung der Reportergene sollten die Hefen nicht auf Medien ohne Histidin und Adenin wachsen können. Durch das in den Medien enthaltene X- -Gal würden Kolonien, bei denen die Reportergene transkribiert und translatiert würden, eine blaue Farbe zeigen. Alle Erscheinungsformen der Kolonien sind in Tabelle 3.6 aufgeführt. Tab. 3.6: Test der transkriptionalen Aktivität der Hefestämme mit dem Plasmidkonstrukt ALD_pGBKT7 und dem Plasmid pGBKT7 ohne „Insert auf selektivem SD-Medium sowie untransformierter Hefen beider Stämme. Hefestamm [mit Plasmidkonstrukt] SD/-Trp/ SD/-Trp/-His/ SD/-Trp/-Ade/ X- -Gal X- -Gal X- -Gal AH109[ALD_pGBKT7] + / weiß AH109[pGBKT7] + / weiß Y187[ALD_pGBKT7] + / weiß Y187[pGBKT7] + / weiß AH109 Y187 : keine Kolonien gewachsen + / Farbe: Kolonien mit dieser Farbe gewachsen Die Hefezellen, die ein Plasmid enthielten, wuchsen nur auf dem Medium SD/-Trp/X- -Gal, und die Kolonien hatten eine weiße Farbe. Dadurch war bestätigt, dass es nicht zu transkriptionaler Aktivierung durch das Bait-Konstrukt (DNA-BD fusioniert mit Aldolase, ALD_pGBKT7) kommt. Hefezellen ohne Plasmid konnten auf keinem der selektiven Medien wachsen. Ergebnisse 99 Für den Toxizitätstest wurden Hefezellen wie in Kap.2.3.5.4.7 beschrieben kultiviert. Die optische Dichte wurde bei 600 nm (OD600) nach 16-24 h gemessen. Der Hefestamm Y187 mit dem Konstrukt ALD_pGBKT7 erreichte nach 24 h eine optische Dichte von nur 0,1 (s.Tab 3.7). Die gewünschte OD600 liegt bei 0,8. Tab. 3.7: Optische Dichte der Hefekultur Y187[ALD_pGBKT7] gemessen bei 600 nm nach 16-24 h Anzucht. Zeit nach Animpfung OD600 nm 17 h 0,038 18 h 0,052 20 h 0,064 24 h 0,114 3.3.4 Analyse der Kontrollen zu den Hefe-2-Hybrid-Versuchen Parallel zu den Hefe-2-Hybrid-Analysen wurden Kontrollen wie in Kapitel 2.3.5.6 beschrieben durchgeführt. Das Fusionsproteinkonstrukt aus DNA-Bindedomäne und Murein p53 (pGBKT7-53) interagiert in einem Hefe-2-Hybrid-System mit dem Fusionsprotein aus Aktivierungsdomäne und der großen Untereinheit des „SV40 large T PCR fragment“ (pGADT7-RecT) (LI & FIELDS, 1993) und diente als Positivkontrolle. Bei der Negativkontrolle wurde ebenfalls das Konstrukt aus der mit dem „SV40 large T PCR fragment“ fusionierten Aktivierungsdomäne eingesetzt. Dieses interagiert nicht mit dem Fusionsproteinkonstrukt aus der mit menschlichem Lamin C fusionierten DNA-Bindedomäne (pGBKT7-Lam). Da Lamin C weder Komplexe bildet noch mit den meisten anderen Proteinen interagiert (BARTEL et al., 1993), können bei Hefekolonien mit diesen Konstrukten die Reportergene nicht aktiviert werden. Durch Auszählen der gewachsenen Kolonien wurden mit Hilfe der in Kap. 2.3.5.5 beschriebenen Formeln die lebensfähigen Kolonien/ml und die Paarungseffizienz ermittelt. Die Paarungseffizienz sollte nach Herstellerangaben über 2% betragen. Anhand der Daten eines Versuchs sind die Ergebnisse beispielhaft für alle durchgeführten Kontrollversuche in den Tabellen 3.8 und 3.9 aufgeführt. Ergebnisse 100 Tab. 3.8: Lebensfähigkeit und Anzahl der diploiden Hefezellen des positiven Kontrollversuchs mit den Hefestämmen AH109[pGADT7-RecT] und Y187[pGBKT7-53]. Medium Gezählte Verdünnung Kolonien Anzahl Lebensfähigkeit lebensfähiger der Hefezellen Kolonien/ml des Stammes Y187 SD/-Leu 4.704 1 :100 4 704.000 SD/-Trp 932 1 :100 932.000 AH109 SD/-Leu/-Trp 326 1 :10 32.600 Diploide v Die Paarungeseffizienz lag bei 3,5%, auf dem Medium SD/QDO/X- -Gal sind blaue Kolonien gewachsen. Tab. 3.9: Lebensfähigkeit und Anzahl der diploiden Hefezellen des negativen Kontrollversuchs mit den Hefestämmen AH109[pGADT7-RecT] und Y187[pGBKT7-Lam]. Medium Gezählte Verdünnung Kolonien Anzahl Lebensfähigkeit lebensfähiger der Hefezellen Kolonien/ml des Stammes Y187 SD/-Leu 7.864 1 :100 7 864.000 SD/-Trp 862 1 :100 862.000 AH109 SD/-Leu/-Trp 239 1 :10 239.000 Diploide v Die Paarungeseffizienz lag bei 28%, auf dem Medium SD/QDO/X- -Gal sind keine Kolonien gewachsen. Mit einer Paarungseffizienz von 3,5% bei der positiven Kontrolle und 28% für die negative Kontrolle war die Paarung erfolgreich. Bei einer erfolgreichen Paarung muss die Paarungseffizienz einen Wert von über 2% erreichen. Die Anzahl der diploiden lebensfähigen Kolonien unterschied sich zwischen den beiden Kontrollversuchen stark. Für die positive Kontrolle wurden 32.600 lebensfähige Kolonien/ml ermittelt, bei dem negativen Kontrollversuch waren es 239.000. In beiden Kontrollversuchen wurden weniger lebensfähige Kolonien/ml für den Stamm AH109 als für den Stamm Y187 nachgewiesen. AH109 war in beiden Fällen der limitierende Partner für die Paarung. Da dieser Stamm als Kontrolle für die cDNA-Bank dient, ist es wichtig, dass er den limitierenden Partner darstellt. Dadurch wird die Möglichkeit gegeben, dass jede Hefezelle des Stammes AH109 eine Hefezelle des anderen Stammes (Y187) für die Paarung finden kann. Ergebnisse 101 3.3.5 Hefe-2-Hybrid-Analyse der cDNA-Bank mit dem Fusionskonstrukt ALD_pGBKT7 Bei den mehrfach AH109[cDNA-Bank] durchgeführten und Hefe-2-Hybrid-Analysen Y187[ALD_pGBKT7] wurden mit den Hefestämmen unterschiedliche Paarungs- effizienzen ermittelt. Die geringste Effizienz lag mit 17,4% noch deutlich über den notwendigen 2%. Mit der Paarungseffizienz schwankte auch die Anzahl der untersuchten Klone, die bei den in Tabelle 3.10 beispielhaft aufgeführten Werten eines Versuches 130.340 Klone umfasste. Der Stamm AH109, der die cDNA-Bank trug und mit der AD des Transkriptionsfaktors fusioniert war, stellte den limitierenden Faktor im Versuch dar, wodurch sichergestellt wurde, dass jeder Klon der cDNA-Bank einen Interaktionspartner mit dem DNA-BD-Fusionsprotein (DNA-BD-Aldolase) zum Paaren finden konnte. Die Anzahl der lebensfähigen Kolonien/ml betrug in diesem Versuch für den Stamm AH109 23.700, bei den diploiden Hefezellen waren es 9.800. Die Paarung erfolgte bei allen durchgeführten Hefe2-Hybrid-Analysen 24 h lang, weil bei mikroskopischen Untersuchungen der Paarungssuspension nach 20 h noch Hefe-Zygoten (s. Abb. 3.19) in der Suspension nachgewiesen werden konnten. Tab. 3.10: Lebensfähigkeit und Anzahl der diploiden Hefezellen eines Hefe-2-Hybrid-Versuchs mit den Hefestämmen AH109[pGADT7-cDNA-Bank] und Y187[ALD_pGBKT7]. Medium Gezählte Verdünnung Kolonien Anzahl Lebensfähigkeit lebensfähiger der Hefezellen Kolonien/ml des Stammes 74 000.000 Y187 SD/-Leu 740 1 :10 000 SD/-Trp 862 1 :100 23.700 AH109 SD/-Leu/-Trp 239 1 :10 9.800 Diploide v Die Paarungseffizienz lag bei 41,4%. Abb. 3.19: Diploide Hefezygote (400fache Vergrößerung) mit der charakteristischen dreilappigen Gestalt. Links und unterhalb der Zygote befinden sich haploide Hefezellen mit kugeliger Gestalt. Ergebnisse 102 Aus der Paarung hervorgegangene Kolonien, die nach mehrfacher Selektion auf verschiedenen Selektionsmedien einen Durchmesser von X- -GAL durch die Aktivität der 2 mm aufwiesen und das Substrat -Galactosidase in einen blauen Farbstoff umwandelten, wurden weiter analysiert. 3.3.6 Analyse der pGADT7-cDNA-Bank-Konstrukte aus den blauen Hefekolonien Die aus den Hefen isolierten Plasmide wurden, da sie Verunreinigungen mit genomischer Hefe-DNA aufwiesen, zunächst in E. coli transformiert. Die transformierten Zellen wurden dann durch Ampicillin auf die Anwesenheit des „Prey -Plasmides (pGADT7-cDNA-Bank) selektioniert. Mit Hilfe der aus diesen E. coli-Zellen isolierten Plasmid-DNA konnte anschließend eine PCR zur Identifizierung der Größe des cDNA-„Inserts und für Sequenzierungen durchgeführt werden. Die Anzahl der Basen, die mit Hilfe der Sequenzierung identifiziert werden konnten, sowie die Ergebnisse der Sequenzvergleiche von den Sequenzen aus den Hefe-2-Hybrid-Versuchen mit den Sequenzen in der Datenbank GenBank sind in Tabelle 3.11 zusammengefasst. Nicht alle Sequenzen wiesen ein StartCodon auf und nur ein „Prey -Plasmid enthielt die vollständige kodierende Sequenz eines Proteins (GOS2, Hefekolonie 2.11). Bei den gefundenen positiven Interaktionspartnern der Aldolase ergaben einige Sequenzierungen rRNA, die als falsch positive Bindepartner interpretiert werden müssen (HONG, 2000). Des Weiteren wurde mehrfach Aldolase als Interaktionspartner identifiziert, was auf Grund der Tetramerbildung der Aldolase zu erwarten war. Als neue Interaktionspartner der Aldolase konnten zwei Proteine identifiziert werden: der Translations-Initiationsfaktor GOS2 aus Mais und das mitochondriale spannungsabhängige Anionenkanalprotein VDAC 1a. Bei diesen beiden Sequenzen lag die Übereinstimmung zur Sequenz in der Datenbank bei 96%. Ein weiteres Ergebniss der Sequenzvergleiche ergab eine Übereinstimmung mit einer Sequenz in der Datenbank, die nur einen kleinen Bereich der für das Protein kodierenden Sequenz umfasste. Dabei wurde eine Methionin-Adenosyltransferase identifiziert. Ergebnisse 103 Tab. 3.11: Ergebnisse der Sequenzanalysen der „Inserts aus dem Vektor pGADT7-cDNA-Bank nach positiver Interaktion in einem Hefe-2-Hybrid-Versuch mit dem Plasmidkonstrukt ALD_pGBKT7. Angegeben sind die mit Hilfe eines Agarosegels bestimmten „Insert -größen, die Anzahl sequenzierter Nukleotide und das Ergebnis der BLAST-Analyse der jeweiligen Hefekolonie. Hefe- Insert- Sequenzierte kolonie größe Größe (bp) Ergebnis des Sequenz-Alignments gi-Nummer kodierende Sequenz für Aldolase aus Mais gi:168419 kodierende Sequenz des spannungs- gi:5929927 (bp) 2.17 1400 421 7.243 1600 700 2.48 1200 493 abhängigen Anionenkanalproteins 1a (vdac1a) aus Mais 2.11a 1000 748 kodierende Sequenz für GOS2 (Translations- gi:21206618 Initiationsfaktor) aus Mais 7.173 900 670 mRNA-Sequenz PC0102485 aus Mais gi:21206618 7.27 850 795 mRNA-Sequenz PC0138904 aus Mais gi:21209073 254 900 330 mRNA-Sequenz CL3683_1 aus Mais gi:21213628 2.43 1000 480 Methionin-Adenosyltransferase aus Mais gi:17017258 2.5 150 150 mitochondriales Gen für 18S rRNA aus Mais gi:13923 2.4 400 120 4D 500 259 7.178 2.7 25S rRNA-Gen und Transposon-ähnliche Sequenz 2.60b 900/ 1800 180 Doppelsignal, kein Ergebnis gi:13397940 Ergebnisse 104 3.3.7 Klonierung des Expressionskonstruktes VDAC_pRSET A Das im Hefe-2-Hybrid-System als putativer Partner für eine Protein-Protein-Interaktion mit der Aldolase gefundene spannungsabhängige Kanalprotein VDAC 1a der äußeren Mitochondrienmembran lag im „Prey“-Vektor des Hefe-2-Hybrid-Systems nicht als vollständige cDNA vor, sondern mit einem Sequenzstück bp Ma aus 5’-untranslatiertem Bereich von 92 Basen und kodierender DNA von 322 Basen. Dadurch konnte diese 3000 cDNA nicht für weitere Klonierungen verwendet werden. 1500 Mit den Primern vdac_pRSET-For und vdac_pRSET-Rev 900 wurde auf Mais-cDNA eine PCR durchgeführt, bei der eine 500 geringe Menge DNA in der gewünschten Größe zwischen 800 und 900 bp amplifiziert wurde (s. Abb. 3.20). Mit dieser erfolgte die Klonierung zunächst in den Vektor pDrive. Nach Analyse positiver Klone wurde das DNA-Fragment in den Vektor pRSET A kloniert und in E. coli XL1blue Abb. 3.20: Agarosegel zur Überprüfung der Größe des amplifizierten für VDAC kodierenden PCR-Produkts. Ma: Gene Ruler 100bp Ladder Plus (MBI Fermentas, St. Leon-Rot). transformiert. Restriktionsanalysen (s. Abb. 3.21) und ein Überexpressionsversuch erfolgten, bevor einer dieser Klone sequenziert wurde. Durch den Vergleich der Sequenz dieses Klons mit der in der Datenbank GenBank veröffentlichten Sequenz des spannungsabhängigen Anionenkanalproteins ergaben sich sechs Nukleotide, die sich unterschieden. Drei davon führen zu keinem Aminosäureaustausch in der Proteinsequenz. Durch einen weiteren Nukleotidunterschied an Position 424 des Klons kodiert das entstehende Triplett die Aminosäure Valin, wogegen durch die DatenbankSequenz ein Leucin kodiert wird. Schließlich führt an Position 812 des Klons ein Nukleotidunterschied zu einem Glycin anstelle eines Alanins. Das Nukleotid 699 des Klons ist ein Adenin, wogegen in der Datenbank-Sequenz sich an dieser Position ein Cytosin befindet. Die daraus resultierende Aminosäure, ein Asparagin, wird im Klon durch ein Lysin ersetzt (s. Anhang Kap. 7.2.8). Ergebnisse 105 bp Ma 11500 1700 805 pRSET A VDAC Abb. 3.21: Identifizierung positiver Klone nach Klonierung des für VDAC kodierenden PCR-Fragments in den Vektor pRSET A durch Restriktionsanalyse. Alle Klone enthalten ein „Insert in der entsprechenden Größe von 831 bp. Ma: Lambda DNA/Pst I Marker 24, MBI Fermentas, St. Leon-Rot. 3.3.8 Klonierung von VDAC in den Vektor pGBKT7 Um das Protein VDAC zukünftig auch in Hefe-2-Hybrid-Analysen einsetzen zu können, musste die kodierende Nukleotidsequenz in den „Bait -Vektor des Hefe-2-Hybrid-Systems kloniert werden. Eine PCR mit Mais-cDNA als „template“ führte zur Amplifikation eines 831 bp großen PCR-Fragments. Dieses DNA-Fragment wurde in den Vektor pDrive ligiert, wobei es nur zu wenigen positiven Insertionen des Fragments in den Vektor kam. In Abb. 3.22 A ist die Bande des „Inserts bei drei Proben schwach zu sehen, nachdem diese mit den Restriktionsendonukleasen Nde I und Bam HI aus dem Vektor geschnitten worden war. Dieses Fragment wurde anschließend in den geöffneten Vektor pGBKT7 ligiert und in E. coli XL1blue kloniert. Die Restriktionsanalyse dieser Klonierung ist in Abb. 3.22 B dargestellt. Von den insgesamt sieben positiven Klonen mit einem Insert in der entsprechenden Größe sind zwei (Probe 10 und 12) auf dem abgebildeten Agarosegel zu sehen. A B bp bp 11500 11500 5077 2838 1700 2838 1700 1093 805 805 Ma 1 2 3 4 5 6 Ma 10 11 12 13 Abb. 3.22: Restriktionsanalysen zur Identifizierung positiver Klone der Klonierung des für VDAC kodierenden PCR-Fragments in pDrive (A) und des Endkonstruktes VDAC in pGBKT7 (B). Der Pfeil markiert das herausgeschnittene „Insert . Ma: Lambda DNA/Pst I Marker 24, MBI Fermentas, St. Leon-Rot. A: Klon 2, 4 und 6 enthalten ein Insert, B: Klon 10 und 12 enthalten ein „Insert von ca. 830 bp. Ergebnisse 106 3.3.9 Klonierung von Aktin in den Vektor pGBKT7 Um Aktin im Hefe-2-Hybrid-System als Bait“ einsetzen zu können, und Proteine, die mit Aktin interagieren, identifizieren zu können, wurde versucht, Aktin in den Vektor pGBKT7 zu klonieren. Nachdem trotz Optimierungsversuchen die mit den Primern Aktin-sense und Aktin-antisense amplifizierten DNA-Mengen sehr gering blieben, wurden die erhaltenen schwachen Banden aus 15 PCR-Ansätzen vereinigt und gemeinsam gereinigt (PCR-Produkte nicht gezeigt). Das PCR-Produkt wurde dann in pDrive ligiert. Nach Transformation des Aktin-pDrive Plasmidkonstruktes in E. coli XL1blue wurden von den mit Blau-WeißSelektion identifizierten positiven Kolonien Übernachtkulturen angesetzt und die Plasmide isoliert. Nach einem Restriktionsverdau mit Bam HI und Pst I konnten von den 17 Kolonien drei als positiv identifiziert werden. Bei diesen Plasmiden war nach dem Restriktionsverdau neben der Vektorbande bei ca. 4000 bp eine Bande bei ca. 1000 bp auf dem Agarosegel zu sehen (Abb. 3.23). Alle drei Proben wurden zur Klonierung in den Vektor pGBKT7 eingesetzt. Dieser wurde vor der Ligation mit den aus pDrive geschnittenen DNA-Fragmenten ebenfalls mit Bam HI und Pst I geschnitten und gereinigt. Obwohl Restriktionsanalysen einige positive Klone zeigten, ergaben Sequenzierungen, dass es sich bei diesen Klonen nicht um Aktin im Vektor pGBKT7 handelt, sondern weiterhin um den Vektor pDrive mit „Insert . Eine erfolgreiche Klonierung dieses Plasmidkonstruktes wurde innerhalb der vorliegenden Arbeit nicht mehr erreicht. bp 5077 2838 1700 805 Ma 1 2 3 4 5 6 7 8 9 10 11 Abb. 3.23: Analyse positiver Klone mit Hilfe von Restriktionsverdau des Konstruktes Aktin in pDrive. Gezeigt sind zwei der drei positiven Klone (4 und 9), die ein „Insert von ca. 1000 bp enthielten. Ma: Lambda DNA/Pst I Marker 24, MBI Fermentas, St. Leon-Rot. Ergebnisse 107 3.4 Heterologe Expression und Reinigung von VDAC (Z. mays) mit His-tag Für die heterologe Expression des VDAC wurde das Plasmidkonstrukt VDAC_pRSET A in E. coli BL21(DE3)pLysS Zellen transformiert und überexprimiert. Die Induktion der Proteinsynthese erfolgte während der exponentiellen Wachstumsphase der Bakterien, deren Zellteilungsrate sich daraufhin verlangsamte. Eine Abflachung der Wachstumskurve ist nach der Induktion zu erkennen (Abb. 3.24). Während der Proteinexpression wurde der Zellsuspension jede Stunde ein aliquoter Anteil von 1 ml Optische Dichte bei 600 nm 2,0 1,8 entnommen. Die Expression 1,6 1,4 wurde 1,2 vor dem Zellauf- schluss und der Protein- 1,0 0,8 reinigung 0,6 0,4 mittels 12%igen 0,2 SDS-PAGE analysiert. 0,0 0 60 120 180 240 300 360 einer Abb. 3.25 (A) 420 Zeit [min] zeigt eine deutliche Bande, die Abb. 3.24: Wachstumskurve von E. coli BL21(DE3)pLysS mit dem Plasmidkonstrukt VDAC_pRSET A gemessen anhand der optischen Dichte bei 600 nm gegen H2O. Der Pfeil markiert den Zeitpunkt der Induktion der Fremdproteinsynthese mit IPTG. nach der zunimmt und Stunden ihre Stärke erreicht befindet sich Induktion nach drei maximale hat. bei Sie etwa 37 kDa. Die errechnete Größe des Proteins mit His-tag betrug 34 kDa. Ein Aufschluss der aliquoten Anteile in geklärtem Rohextrakt und Pellet zeigte, dass sich das Protein vollständig im Pellet befand, was auf einen Einschluss in „inclusion bodies“ schließen lässt (Abb. 3.25 B). Ergebnisse 108 B A kDa Ma1 0h 1h 118 84 51 3h 4h kDa Ma2 RE P 180 73 54 35 27 48 35 Abb. 3.25: Heterologe Expression von VDAC in E. coli BL21(DE3)pLysS in Abhängigkeit von der Dauer der Expression (A) und Aufschluss in geklärtem Rohextrakt und Pellet (B). Ma1: Molekularmassenstandard (Molecular Weight Marker, MBI Fermentas, St. Leon-Rot), 0 h: vor Induktion der Fremdproteinsynthese, 1 h, 2 h, 3 h und 4 h nach der Induktion der Fremdproteinsynthese, jeweils 1 ml der Bakteriensuspension. Ma2: Molekularmassenstandard (Prestained Protein Ladder, ~10160 kDa, MBI Fermentas, St. Leon-Rot), RE: geklärter Rohextrakt, P: Pellet. Dadurch wurde ein Aufschluss der „inclusion bodies“ für die Reinigung des Proteins notwendig. Mit der in Kap. 2.3.6.7.2 beschriebenen Methode wurde dieser Aufschluss mit Harnstoff durchgeführt, und das Protein konnte mit Hilfe seines His-tags über Metall-ChelatAffinitätschromatographie gereinigt werden. Die Fraktionen dieser Reinigung sind in Abb. 3.26 zu sehen. Dabei fällt auf, dass während der Reinigung und der Elution kontinuierlich Protein von der Säule eluiert und nicht eine bestimmte Imidazolkonzentration die Elution hervorruft. Mehrere Elutionsfraktionen, die keine größeren Verunreinigungen aufwiesen, wurden vereinigt, wodurch eine Proteinmenge von ca. 5 mg rekombinantem VDAC gewonnen werden konnte. Ergebnisse kDa 109 Ma 1 2 3 4 5 6 7 8 mM Imidazol: 10 50 100 250 kDa Ma 10 11 12 13 14 15 16 17 18 9 180 73 54 180 73 54 35 35 24 24 Abb. 3.26: Reinigung des rekombinanten VDAC von Mais über Metall-Chelat-Affinitätschromatographie. SDSPAGE-Analyse von je 10 µl der folgenden Fraktionen: 1: geklärter Rohextrakt vor Aufschluss der „inclusion bodies 2: Überstand vom Waschschritt des Pellets vor Aufschluss der inclusion bodies 3: Pellet nach Aufschluss der „inclusion bodies 4: geklärter Rohextrakt (RE2) nach Aufschluss der inclusion bodies 5: Durchfluss des RE2 nach Auftragen auf die Metall-Chelat-Affinitätssäule 6-10: Waschschritte 1 bis 5 (Denaturierungs-Solubilisierungs-Puffer) 11-18: Elutionsschritte 1 bis 8 mit Imidazol in unterschiedlichen Konzentrationen Ma: Molekularmassenstandard (Prestained Protein Ladder, ~10-160 kDa, MBI Fermentas, St. Leon-Rot) 3.5 Far-Western-Blot Um die Protein-Protein-Interaktionen, die durch Hefe-2-Hybrid-Analysen und Copolymerisationsversuche identifiziert wurden, mit einer anderen Methode nachzuweisen, wurden „Far-Western-Blots“ durchgeführt. Bei den auf der Membran durch „Dot-Blot“ immobilisierten Proteinen handelte es sich um jeweils 2 µg Aldolase, BSA, Aktin, Tubulin und VDAC. Diese wurden, wie in Kapitel 2.3.6.5 beschrieben, mit rekombinanter Strep-tagAldolase inkubiert und anschließend entsprechend einem Western-Blot entwickelt. Eine deutliche Immundetektion durch die Aldolase-Antikörper ist bei Aldolase, Aktin und VDAC zu sehen. Die Negativkontrolle BSA zeigt keine Interaktion mit Aldolase. Auf dem Kontrollblot ist deutlich eine Immundekoration der Aldolase, jedoch keines weiteren Proteins zu sehen. Die Detektion des Aktin und VDAC beruht also auf einer Interaktion der immobilisierten Proteine auf der Membran mit der Aldolase, die als Overlay zu den Proteinen gegeben wurde und an diese gebunden hat (s. Abb. 3.27). Ergebnisse A 110 ALD BSA Aktin Tubulin VDAC B ALD BSA Aktin Tubulin VDAC Abb. 3.27: Nachweis von Protein-Protein-Interaktionen mit Hilfe von Far-Western-Blots. Auf dem Kontrollblot (A) und dem Test-Blot (B) wurden als Proben je 2 µg rekombinante Strep-tag-Aldolase (Z. mays; ALD), BSA, Aktin, Tubulin und rekombinanter VDAC (Z. mays; VDAC) im Dot-Blot-Verfahren aufgetragen. Der Blot (B) wurde mit 25 nM rekombinanter Aldolase inkubiert. Die Entwicklung erfolgte durch Chemolumineszenz. 3.6 Aldolase-VDAC-Bindeversuch (Affinitätschromatographie) Für den Nachweis einer direkten Bindung zwischen Aldolase und dem spannungsabhängigen Anionenkanalprotein VDAC 1a der äußeren Mitochondrienmembran, die als putative Interaktionspartner in einem Hefe-2-Hybrid-Versuch identifiziert worden waren, wurden Affinitätschromatographien durchgeführt. Diese Methode diente schon mehrfach der Untersuchung von Protein-Protein-Interaktionen (z. B. KÁLMÁN & BOROSS, 1982; LU et al., 1993). Dafür wurde die überexprimierte Aldolase mit Strep-tag über Affinitätschromatographie gereinigt und auf eine mit VDAC beladene Ni-NTA-Säulenmatrix gegeben. Nach 10 Waschschritten wurden die Proteine eluiert. Die verschiedenen Fraktionen wurden mittels 12 %iger SDS-PAGE analysiert. Das Ergebnis ist in Abb. 3.28 dargestellt. Ergebnisse 111 A B kDa Ma 1 2 3 4 5 6 7 8 9 180 73 A 54 35 24 5 7,5 mM Imidazol kDa Ma 10 11 12 13 14 15 16 17 18 180 73 B 54 35 Aldolase 24 C kDa 180 C 73 54 35 7,5 10 50 100 250 mM Imidazol Ma 19 20 21 22 23 24 25 26 27 Aldolase VDAC 24 Abb. 3.28: Affinitätschromatographie zur Untersuchung der Protein-Protein-Interaktion zwischen VDAC und Aldolase aus Mais. SDS-PAGE Analysen von jeweils 20 µl der jeweiligen Fraktion. A: Bindung des VDAC an die Affinitätsmatrix und verschiedene Waschschritte; B: Zugabe von Aldolase zur Affinitätsmatrix mit daran gekoppeltem VDAC und Waschschritte; C: weitere Waschschritte und Elution der gebundenen Proteine. Ma: Molekularmassenstandard (Prestained Protein Ladder, ~10-160kDa, MBI Fermentas, St. Leon-Rot) A: 1: geklärter Rohextrakt (RE2) von VDAC nach Aufschluss der „inclusion bodies 2: geklärter Rohextrakt vor Aufschluss der „inclusion bodies 3: Durchfluss des RE2 nach Auftrag auf die Metall-Chelat-Affinitätssäule 4 - 9: Waschschritte 1, 3, 5, 7, 9 und 10 (Denaturierungs-Solubilisierungs-Puffer) B: 10: Durchfluss der gereinigten rekombinanten Aldolase 11-15: Waschschritte 1, 4, 6, 8 und 10 (Denaturierungs-Solubilisierungs-Puffer) 16-17: Waschschritte 11und 12 (5 mM Imidazol in Denaturierungs-Solubilisierungs-Puffer) 18: Waschschritt 13 (7,5 mM Imidazol in Denaturierungs-Solubilisierungs-Puffer) C: 19: Waschschritt 14 (wie Waschschritt 13) 20-27: Elutionsschritte 1 bis 8 mit verschiedenen Konzentrationen Imidazol in DenaturierungsSolubilisierungs-Puffer Die Pfeile markieren die Banden der Proteine VDAC (blaue Pfeile) und Aldolase (schwarze Pfeile). Die Ni-NTA-Säule wurde mit einem Überschuss an VDAC-Protein beladen. Überschüssiges Protein und Verunreinigungen wurden vollständig durch die Waschschritte von der Säule entfernt (s. Abb. 3.28 A Spur 9), bevor Aldolase zugegeben wurde. Nach Zugabe der Aldolase wurde in den folgenden Waschschritten immer etwas Aldolase ausgewaschen (s. Abb. 3.28 B Spur 11-15). Bei den Waschschritten mit Imidazol (5 und 7,5 mM, s. Abb. 3.28 B/C Spur 1619) ist deutlich mehr Aldolase und auch ein geringer Teil VDAC in den Fraktionen zu sehen. Während der Elution mit verschiedenen Imidazolkonzentrationen (s. Abb. 3.28 Spur 20-27) Ergebnisse 112 erfolgte die Elution von VDAC in großen Mengen ab Elutionsschritt 4. Die Elution der Aldolase ist im Gegensatz dazu in geringer Menge in allen Fraktionen zu erkennen. Mit Hilfe eines Western-Blots konnte die Bande bei ca. 40 kDa eindeutig als Aldolase identifiziert werden. Sie wurde in allen Elutionsfraktionen detektiert. Das bedeutet, dass bei der Elution der Aldolase kein eindeutiger Zusammenhang mit der Elution des VDAC zu erkennen ist. An VDAC gebundene Aldolase sollte zusammen mit VDAC ab Elutionsschritt 3 von der Säule eluieren, nicht jedoch schon vorher in den Waschschritten mit Imidazol. In einem weiteren Versuch wurde untersucht, ob Aldolase mit Strep-tag auch unspezifisch an die Ni-NTA-Säulenmatrix bindet. Die Ni-NTA-Säulenmatrix wurde entsprechend dem Versuch mit Puffern behandelt, jedoch wurde keine VDAC-Proteinlösung an die Matrix gekoppelt. Nach Zugabe der rekombinanten Aldolase zur Säuelnmatrix und 10 Waschschritten wurde auch hier mit Imidazol eluiert. Die Analyse der Fraktionen erfolgte mittels 12 %iger SDS-PAGE (s. Abb. 3.29). Anhand der SDS-PAGE wird deutlich, dass die Aldolase auch ohne einen His-tag und den putativen Interaktionspartner an die Ni-NTA-Säulenmatrix bindet. Durch die Wasch- und Elutionsschritte mit Imidazol wurde sie vollständig von der Säule eluiert. Ab dem Elutionsschritt E5 (100 mM Imidazol) kann keine Aldolase mehr in den Fraktionen nachgewiesen werden. Dadurch ergibt sich ein deutlicher Unterschied zur Affinitätschromatographie mit VDAC, bei dem die Aldolase nicht in entsprechender Menge in den Waschschritten mit 5 und 7,5 mM Imidazol von der Säule eluiert, dafür aber in geringer Konzentration über die gesamten Elutionsschritte nachzuweisen ist. In den letzten Elutionsschritten mit 100 und 250 mM Imidazol Affinitätschromatographie mit VDAC nachgewiesen werden. kann Aldolase nur bei der Ergebnisse 113 A B 5 kDa 180 73 54 35 Ma 1 2 7,5 3 4 5 6 7 8 9 7,5 kDa 180 10 50 100 250 mM Imidazol Ma 10 11 12 13 14 15 16 17 18 73 54 35 24 24 Abb. 3.29: Bindung der rekombinanten, gereinigten Strep-tag Aldolase an eine Metall-ChelatAffinitätsmatrix als Kontrolle zum Aldolase-VDAC-Bindeversuch mit Affinitätschromatographie. SDS-PAGE Analyse von jeweils 20 µl der entsprechenden Fraktion. Ma: Molekularmassenstandard (Prestained Protein Ladder, ~10-160kDa, MBI Fermentas, St. Leon-Rot) A: 1: geklärter Rohextrakt der Aldolase von Z. mays nach Strep-tag Affinitätschromatographie 2: Durchfluss nach Auftragung auf eine Ni-NTA Säule 3 - 6: Waschschritte 1, 4, 7 und 10 (Denaturierungs-Solubilisierungs-Puffer) 7 - 8: Waschschritte 11und 12 (5 mM Imidazol in Denaturierungs-SolubilisierungsPuffer) 9: Waschschritt 13 (7,5 mM Imidazol in Denaturierungs-Solubilisierungs-Puffer) B: 10: Waschschritt 14 (wie Waschschritt 13) 11-18: Elutionsschritte 1 bis 8 mit unterschiedlichen Imidazolkonzentrationen in Denaturierungs-Solubilisierungs-Puffer Die Pfeile markieren die Bande der Aldolase. Aldolase Diskussion 114 4 Diskussion Um die Regulation eines Stoffwechsels erklären zu können, sind besonders Hinweise auf die Organisation der einzelnen Enzyme, ihre Lokalisation in der Zelle und Interaktionen zwischen Enzymen, die evtl. „channeling“ ermöglichen, von Bedeutung. Mais und Reis sind wichtige Modellpflanzen geworden, um die Rolle des Cytoskeletts z. B. in der Proteinsynthese und Proteinkörperbildung zu verstehen (ABE et al., 1991; STANKOVI et al., 1993, CLORE et al., 1996). Es gibt seit einiger Zeit immer mehr Hinweise auf eine Assoziation von löslichen Enzymen mit den Strukturen des Cytoplasmas. Wegen der komplexen Natur des Cytoplasmas sind Modelle der Organisation des Stoffwechsels jedoch schwer zu erstellen und noch schwerer zu überprüfen. Die Glykolyse wurde in vitro intensiv untersucht, aber es gibt zunehmend Hinweise, dass man die Glykolyse in vivo nicht durch die einfache Übertragung dieser Daten erklären kann. In dieser Arbeit sollte eine Interaktion der pflanzlichen Aldolase mit dem Cytoskelett nachgewiesen werden. Dafür wurden in vitro Bindeversuche mit Kaninchen-Muskel-Aktin durchgeführt (Copolymerisationsversuche, s. Kap. 3.2). Weitere Hinweise auf eine mögliche Metabolonbildung innerhalb der pflanzlichen Glykolyse, vor allem unter Stressbedingungen, sollten Versuche mit einem Hefe-2-Hybrid-System bringen (s. Kap. 3.3). Dieses System eignet sich besonders zur Aufdeckung unbekannter Protein-Protein-Interaktionen. 4.1 Sequenzen der Expressionskonstrukte von Aldolase und VDAC Durch die BLAST-Analyse der Expressionskonstrukte ALD_pET-16b, ALD_pASK-IBA3 und VDAC_pRSET A konnten Unterschiede zwischen den Sequenzen der Konstrukte und den in der Datenbank GenBank veröffentlichten Sequenzen für diese Gene festgestellt werden. Unterschiede zwischen den Nukleotiden der Sequenzen treten hauptsächlich an der dritten Stelle des codierenden Tripletts auf. Die meisten Aminosäuren werden von mehreren Tripletts kodiert (degenerierter Code), bei denen häufig Variationen am dritten Nukleotid auftreten. Codons, die die gleiche Aminosäure codieren, heißen Synonyme. Die Nukleotidaustausche am dritten Nukleotid des Tripletts bezeichnet man als „single nucleotide polymorphism“ (SNP). Ein Nukleotidaustausch, der keine Veränderung in der Aminosäuresequenz hervorruft, wird dementsprechend als „synonymer SNP“ oder stille Mutation bezeichnet. Sie können durch die Verwendung unterschiedlicher Variationen (Ökotypen) des den Sequenzen zugrundeliegenden Materials, in diesem Falle von Maispflanzen, auftreten. Es können aber auch bei der Amplifikation der DNA während der Diskussion 115 PCR oder Sequenzierungs-PCR Fehler aufgetreten sein, die zu diesen verschiedenen Nukleotiden geführt haben. Der Klon ALD_pET-16b unterscheidet sich in neun Nukleotiden von der GenBank-Sequenz. Jedoch nur durch eines dieser unterschiedlichen Nukleotide wird eine veränderte Aminosäure im Protein kodiert. Bei dieser veränderten Aminosäure ist das Nukleotid an erster Position des Tripletts (Position 787) anders als in der GenBank-Sequenz, so dass das Codon für ein Leucin anstelle eines Valins kodiert wird. Bei beiden Aminosäuren handelt es sich um neutrale Aminosäuren mit einer aliphatischen Seitenkette. Ausgangsmaterial für den Klon ALD_pASK-IBA3 war der Klon ALD_pET-16b. Dadurch war zu erwarten, dass der Klon ALD_pASK-IBA3 dieselben Unterschiede in der Nukleotidsequenz aufweist. Es treten innerhalb dieser Sequenz jedoch nur sieben unterschiedliche Nukleotide im Vergleich zur GenBank Sequenz auf. Das deutet darauf hin, dass es sich bei einigen Unterschieden um Fehler handelt, die während der SequenzierungsPCR zustande gekommen sind. Jedoch zeigt sich auch in diesem Klon der Nukleotidaustausch an Position 787, der zu der veränderten Aminosäure führt. Deshalb ist davon auszugehen, dass durch einen Fehler bei der PCR hier das Codon für ein Leucin entstanden ist. Insgesamt sechs Nukleotide unterscheiden sich innerhalb der Nukleotidsequenzen von VDAC1a aus der Datenbank GenBank und dem Klon VDAC_pRSET A. Auch hier besteht die Möglichkeit, dass es sich um Fehler während der Sequenzierungs-PCR handelt. Drei der Unterschiede sind synonyme SNPs, wogegen jedoch die anderen drei unterschiedlichen Nukleotide zu einer veränderten Aminosäuresequenz führen. Dabei führt das Nukleotid an Position 424 zum Codon für ein Valin anstelle eines Leucins, wie es an dieser Position in der Datenbanksequenz veröffentlicht ist. An Position 812 führt ein unterschiedliches Nukleotid zu einem Alanin anstelle eines Glycins in der Aminosäuresequenz. Bei diesen beiden Unterschieden sind jeweils neutrale Aminosäuren durch andere neutrale Aminosäuren ersetzt. Durch den Nukleotidunterschied an Position 699 des Klons entsteht die Aminosäure Asparagin, an deren Stelle sich in der Datenbanksequenz das Codon für ein Lysin befindet. Die basische Seitenkette des Lysins fehlt dadurch in der Aminosäuresequenz des Klons. Inwieweit diese Unterschiede zwischen den Sequenzen Einfluss auf die Faltung oder Eigenschaften der Proteine haben, kann nicht beurteilt werden. Aufschluss darüber könnten Vergleiche zwischen den Eigenschaften der nativen und rekombinanten Proteine bringen. Diskussion 116 4.2 Katalytische Eigenschaften rekombinanter Aldolasen Mit den beiden rekombinant hergestellten Aldolasen der Expressionskonstrukte ALD_pET16b und ALD_pASK-IBA3 wurden Aktivitätsmessungen mit dem Substrat FBP durchgeführt. Wie in Kapitel 3.1.7 beschrieben, zeigte Aldolase mit His-tag (ALD_pET-16b) keine Spaltungsaktivität für FBP. Ursache für die fehlende Aktivität könnte der mit dem Protein exprimierte His-tag am N-Terminus der rekombinanten Aldolase sein. Durch den His-tag befinden sich am N-Terminus des Proteins zehn basische Histidine, die möglicherweise einen Einfluss auf die Proteinstruktur oder direkt auf die katalytische Aktivität haben. Eine schematische Darstellung der Expressionskonstrukte ALD_pET-16b und ALD_pASK-IBA3 befindete sich im Anhang (s. Kap. 7.2.5). Dagegen konnte eine geringe katalytische Aktivität der Aldolase mit Strep-tag des Konstruktes ALD_pASK-IBA3 nachgewiesen werden. Jedoch war diese deutlich geringer als die von BERTHIAUME et al., 1989 bei rekombinanter cytosolischer Mais-Aldolase gemessene (vgl. Kap. 3.1.7). Bei Untersuchungen des Einflusses des C-Terminus auf die Aktivität von Aldolase wurde für wild-typ Mais-Aldolase eine Aktivität von 11,32 U/mg und ein KM-Wert von 7,7 µM für FBP beschrieben. Fehlen der Aldolase die letzten acht Aminosäuren am CTerminus verringert sich die Aktivität auf 1,24 U/mg und der KM-Wert beträgt 0,8 µM (BERTHIAUME et al., 1991). Dies entspricht annähernd den für Strep-tag-Aldolase ermittelten Eigenschaften, für die eine Aktivität von 1,23 U/mg und ein KM-Wert von 0,5 µM ermittelt wurde. Der mit der Enzym-Sequenz exprimierte Strep-tag befindet sich am C-Terminus der Aldolase. Im Gegensatz zur Sequenz für das aktive Zentrum des Enzyms ist die Sequenz des C-Terminus in Vertebraten, Invertebraten und Pflanzen nicht konserviert und könnte die unterschiedlichen Aktivitäten der verschiedenen Aldolasen hervorrufen (SYGUSCH et al., 1987). Aufgrund der Bedeutung des C-Terminus für die Aktivität der Aldolase liegt es nahe, dass die geringe gemessene Aktivität auf dem Einfluss des Strep-tags beruht, der z. B. die notwendige Mobilität des C-Terminus oder die Faltung des Proteins beeinflussen könnte. 4.3 Immunologischer Nachweis von Aldolase Um ein wichtiges Werkzeug für die Durchführung verschiedener Versuche wie Immunpräzipitation, Western-Blotting und ELISA zur Verfügung zu haben, wurde im Rahmen dieser Arbeit ein Antiserum gegen gereinigte, rekombinante Aldolase aus Mais hergestellt. Dieses diente innerhalb der vorliegenden Arbeit zur Detektion von rekombinanter Aldolase im Far-Western-Blot (s. Kap. 3.5). Die Spezifität des Antiserums wurde mittels Western-Blot-Analyse untersucht. In allen getesteten pflanzlichen Geweben aus Mais, Diskussion 117 Arabidopsis und Kartoffel wurde eine einzelne Bande durch das Antiserum spezifisch erkannt. Dagegen wurde durch tierische Aldolase keine Antigen-Antikörper-Reaktion hervorgerufen. Rekombinante Aldolase konnte mit Hilfe des Antiserums in einer Menge von 50 ng auf Western-Blots gut detektiert werden, im ELISA wurden 3 ng nativer rekombinanter Aldolase detektiert. Damit steht ein spezifisches Antiserum zur Immundekoration von cytosolischer Aldolase aus Mais sowie aus den dikotylen Pflanzen Arabidopsis und Kartoffel für verschiedene Anwendungen zur Verfügung. Die Möglichkeit, mit einem Antiserum gegen Aldolase aus einer monokotylen Pflanze auch Aldolasen dikotyler Pflanzen zu detektieren, beruht auf der Sequenzhomologie cytosolischer Aldolasen, die zwischen 67% und 92% beträgt (PELZER-REITH et al., 1993). 4.4 Copolymerisationsversuche Bei der Untersuchung einer möglichen Bindung pflanzlicher, cytosolischer Aldolase an Aktinfilamente, die aufgrund der Sequenzhomologie eines zehn Aminosäuren umfassenden Sequenzabschnitts zu Aktin selbst und zu Aktinbinderegionen anderer ABPs zu vermuten war (vergl. Kap. 1.2.2; WINTER & HUBER, 2000), zeigte sich, dass eine Aktinbindung erfolgt (s. Abb. 3.11). 4.4.1 Einfluss von FBP, F-6P und F-2,6BP auf die Aldolase-Aktin-Bindung Die nähere Untersuchung der Bindung ergab Unterschiede im Hinblick auf den Einfluss der Metabolitkonzentration auf die Bindung pflanzlicher Aldolase im Vergleich zur gut untersuchten Bindung tierischer Aldolase an Aktinfilamente. So konnte eine vollständige Hemmung der Bindung nicht durch 1 mM des Substrates der Aldolase, FBP, hervorgerufen werden. Für tierische Aldolase ist eine vollständige Hemmung der Bindung durch 1 mM FBP bekannt (ARNOLD & PETTE, 1970). Auch eine zehnfache Erhöhung der Substratkonzentration führte nicht zu einer vollständigen Unterdrückung der Aktinbindung der pflanzlichen Aldolase. Unter Einfluss von 10 mM FBP wurde der Anteil an Aktin gebundener Aldolase im Vergleich zur Kontrolle um die Hälfte verringert (s. Abb. 3.12). Die Hemmung der AldolaseAktin-Bindung durch 1 mM FBP betrug 38% (s. Abb. 3.13). Die Substanzen F-2,6BP und F-6P wurden in derselben Konzentration, in der mit FBP eine deutliche Hemmung der Bindung hervorgerufen wurde (10 mM), auf ihren Einfluss auf die Aldolase-Aktin-Bindung untersucht. Das strukturell ähnliche F-2,6BP zeigte einen stärkeren Effekt als FBP. Es führte zur Hemmung der Bindung um 60% (s. Abb. 3.13). Die Aktinpolymerisation blieb dabei unbeeinflusst (s. Tab. 3.2). Eine noch deutlichere Reduktion Diskussion 118 der Bindung von Aldolase an Aktin ergab sich durch den Einfluss von F-6P. Die Aktinpolymerisation steigerte sich, jedoch sank der Anteil gebundener Aldolase auf 11% im Vergleich zu 34% in der Kontrolle (s. Abb. 3.12). Für die Berechnung des Anteils an Aktin gebundener Aldolase wurde das in der Probe polymerisierte Aktin gleich 100% gesetzt. Damit wurde die Bindung durch F-6P um knapp 70% gehemmt (s. Abb. 3.13). Dass bei den in dieser Arbeit eingesetzten Konzentrationen auch ein Einfluss von F-6P und F2,6BP auf die Bindung tierischer Muskel-Aldolase an Aktin nachweisbar wäre, ist aufgrund der Arbeiten von WANG et al. (1996) nicht auszuschließen. Weil Fructose-1-phosphat und FBP die Bindung tierischer Aldolase an Aktin unterdrückten, F-6P und F-2,6BP jedoch nicht, wurde vermutet, dass die Loslösung der Aldolase von F-Aktin durch die Konkurrenz zwischen Substrat und Aktin um die Bindung an Aldolase durch die Bindestelle des Phosphats in C1-Position stärker als in C6-Position ist (WANG et al., 1996). Im Gegensatz dazu war der hemmende Einfluss von F-6P und F-2,6BP bei pflanzlicher Aldolase stärker als der des Substrates FBP. Schließt man jeweils das Experiment, bei dem unter Einfluss von F6P (Exp. 4; s. Tab. 3.2) und F-2,6BP (Exp. 3, s. Tab. 3.2) eine ungewöhnlich große Menge Aktin pelletierte, aus, so ist dennoch ein deutlich hemmender Einfluss der beiden Substanzen auf die Aldolase-Aktin-Bindung nachzuweisen. Die Aktinpolymerisation würde dann der Kontrolle entsprechen, und die im Pellet nachgewiesene Aldolase entspräche dem Einfluss von FBP. Ein Einfluss der Position des Phosphates auf die Aldolase-Aktin-Bindung kann durch die vorliegende Untersuchung nicht nachgewiesen werden. Die eingesetzte Konzentration des F-2,6BP, das als Signalstoff in sehr geringen Konzentrationen wirkt, hat keine physiologische Relevanz. Ob F-2,6BP, das an der Regulation von Glykolyse und Guconeogenese beteiligt ist, auf die Bindung pflanzlicher Aldolase an Aktin auch in geringeren Konzentrationen einen Einfluss hat, ist nicht untersucht worden. Da es zur Kompetition zwischen der Substrat- und Aktinbindung in Anwesenheit des Substrates kommt, ist von einer Überlappung der Bindestellen auszugehen. Da im tierischen System eine Gewebespezifität der Bindung nachgewiesen wurde, wird diskutiert, ob durch die Bindung eines Teils der cytosolischen Aldolase an Aktin ein Gerüst oder Ankopplungsort für die Bindung anderer Enzyme in der Zelle aufgebaut wird (MOORHEAD & PLAXTON, 1992, WANG et al., 1997, KUSAKABE et al., 1997, KAO et al., 1999). Dadurch, dass die Bindung pflanzlicher Aldolase an Aktin durch ihr Substrat, auch in hohen Konzentrationen, nicht vollständig aufgelöst oder verhindert wurde, ist anzunehmen, dass die Positionierung der Aldolase in der Zelle in pflanzlichen Geweben eine noch wichtigere Rolle spielt als in tierischen Geweben. Diskussion 119 4.4.2 Einfluss des Redox-Milieus auf die Aldolase-Aktin-Bindung Bei der Untersuchung, welchen Einfluss das Redox-Milieu auf die Aldolase-Aktin-Bindung hat, wurde das natürliche Redoxreagenz Glutathion sowie als synthetisches Reagenz DTT eingesetzt. Glutathion ist für die aerobe Lebensweise wichtig zur Entgiftung von reaktiven Sauerstoffspezies und hat viele weitere Funktionen in Pflanzen. Beschrieben wurde z. B. ein Einfluss von Glutathion auf das Wurzelwachstum, die Blütenentwicklung, die Genexpression und bei der Entgiftung von Schwermetallen (SÁNCHEZ-FERNÁNDEZ et al., 1997; OGAWA et al., 2001; OGAWA et al., 2004; SEN, 2000; COBBETT, 2000). Glutathion kommt in reduzierter (GSH) und oxidierter Form (GSSG) vor. Die oxidierte Form inaktiviert Aldolase durch die Bildung gemischter Disulfide. Dadurch wird die Aldolase destabilisiert und ist bei geringeren Temperaturen denaturierbar sowie leichter angreifbar für Proteasen. Das Substrat der Aldolase schützt diese vor dem schädlichen Einfluss von Disulfiden (BOND & OFFERMANN, 1981; OFFERMANN et al., 1984). Die Bildung der Disulfidbrücken geht einher mit Veränderungen der Sekundär- oder Tertiärstruktur; das Molekulargewicht und die Quartärstruktur oder zumindest Teile davon bleiben jedoch erhalten (BOND & OFFERMANN, 1981). In Arabidopsis konnten plastidäre Aldolase und cytosolische Triosephosphat-Isomerase als Ziele für eine Glutathionylierung identifiziert werden (ITO et al., 2003). Neben Enzymen der Glykolyse wurden auch glutathionylierte Bestandteile des Cytoskeletts gefunden (GODON et al., 1998; FRATELLI et al., 2002, CHAI et al., 1994), bei einem davon handelt es sich um Aktin. Durch langsame Oxidation bilden sich Aktin-Dimere und Oligomere, die beim Einbau in Filamente zu deren Quervernetzung und zu Veränderungen der Elastizität der Filamente führen (TANG et al., 1999). Von DREWES & FAULSTICH (1993) wurde eine Destabilisierung von Aktinfilamenten durch gemischte Disulfide beschrieben. An der Disulfidbindung ist das C-terminale Cystein 374 beteiligt. Der C-Terminus schafft normalerweise den engen Kontakt zwischen den Aktin-Untereinheiten und stabilisiert so die Filamente. Ein weiterer interessanter Aspekt der Einflussnahme des Redox-Milieus auf Aktin ist die Untersuchung an A 431-Zellen (WANG et al., 2001). Dort wurde unter Einfluss des Wachstumsfaktors EGF, der das zelluläre Redox-Gleichgewicht in Richtung oxidativer Bedingungen verschiebt, die aktive Deglutathionylierung von Aktin beobachtet, wodurch die Polymerisationsrate stieg. Eine Zunahme der Polymerisation wurde zuvor unter oxidativen Bedingungen auch in einer anderen Zelllinie (P388D1) beobachtet (HINSHAW et al., 1991). Aufgrund dieser vielfältigen Einflüsse des Redox-Milieus auf Proteine des Cytoskeletts und der Glykolyse ist es denkbar, dass auch die Aldolase-Aktin-Bindung dadurch beeinflusst Diskussion 120 wird. Bei den Messungen zeigten sich jedoch im Vergleich zum Substrateinfluss (s. o.) nur geringfügige Abweichungen von der Kontrolle. Auch müssen bei den Ergebnissen die starken Schwankungen, die zwischen den Versuchen auftraten und die sich in den hohen Standardabweichungen widerspiegeln, berücksichtigt werden. Diese führten auch zu der recht geringen Signifikanz der Ergebnisse von = 0,5 (s. Kap. 3.2.2). Unter Einfluss von GSH wurden knapp 9% mehr Aldolase an Aktin gebunden als in der Kontrolle. Auch GSSG führte zu einer gesteigerten Bindung der Aldolase an Aktin. Hier lag der Unterschied zur Kontrolle bei 16% (s. Abb 3.14). Die Aktinpolymerisation wurde dabei unter reduzierenden Bedingungen leicht gesteigert und nahm bei oxidierenden Bedingungen ab. Da die von WANG et al. (2001) beschriebene Steigerung der Aktinpolymerisation unter oxidierenden Bedingungen auf aktiver Deglutathionylierung des Aktins beruht, stehen diese Ergebnisse im Einklang miteinander. Im Gegensatz zu Glutathion zeigte DTT in reduzierter Form kaum Einfluss auf die Aktinpolymerisation. In oxidierter Form führte es zu einer gesteigerten Aktinpolymerisation (s. Tab. 3.4). Der Anteil der an das polymerisierte Aktin gebundenen Aldolase wurde durch DTTred um knapp 6% gesteigert (s. Abb. 3.14). Damit fiel die Steigerung der Bindung von Aldolase an Aktin durch DTTred geringer aus als durch GSH. Eine Reduktion des Anteils gebundener Aldolase um 5% im Vergleich zur Kontrolle erfolgte durch DTTox. Der Einfluss des oxidierten DTT unterschiedet sich von dem des GSSG, durch das die Bindung gesteigert wurde (vergl. Abb. 3.15). Da Glutathion und DTT, vor allem bei Betrachtung der oxidierten Form, keinen gleichen Einfluss auf die Bindung zeigten, ist anhand der Ergebnisse nicht abzuschätzen, welchen Einfluss das Redox-Milieu auf die Bindung hat. Diskutiert wird eine Verlangsamung der Glykolyse und Verschiebung des Kohlenhydratflusses zur Regeneration von NADPH, das für die Entgiftungsreaktionen unter oxidativem Stress essentiell ist (GODON et al., 1998; CABISCOL et al., 2000). Ob dafür die Verlagerung der Enzyme innerhalb der Zelle, eine stärkere oder schwächere Bindung an Cytoskelettbestandteile oder andere Vorgänge notwendig sind, muss noch untersucht werden. Diskussion 121 4.5 Protein-Protein-Interaktionen im Hefe-2-Hybrid-System Die Suche nach Protein-Protein-Interaktionen erfolgte mit dem Hefe-2-Hybrid-System. Bei diesem System werden Fusionsproteinkonstrukte über Plasmide in Hefezellen eingeschleust. Dort findet im Cytosol an den Ribosomen die Synthese der Fremdproteine statt. Es handelt sich um Fusionsproteine aus einer Domäne eines Transkriptionsfaktors und einem anderen zu untersuchenden Protein. Die Fusionsproteine müssen, um als Transkriptionsfaktor zu wirken, in den Zellkern der Hefen transportiert werden. Cytosolische Aldolase aus Mais diente als Köder („bait ) für interagierende Proteine einer cDNA-Bank aus anaerobem Mais (Beute, „prey ). 4.5.1 Aldolase-Aldolase-Interaktion Nur ein Protein konnte in zwei unabhängig voneinander durchgeführten Hefe-2-HybridAnalysen als Interaktionspartner der Aldolase identifiziert werden (Hefekolonie 2.17 und 7.243, vgl. Tab. 3.11). Dabei handelte es sich um Aldolase. Da Aldolase Homotetramere bildet, wurde diese Interaktion erwartet. Alle anderen Interaktionspartner, die identifiziert wurden, konnten nicht im Rahmen weiterer Hefe-2-Hybrid-Versuche verifiziert werden. Eine mögliche Ursache hierfür liegt in der Toxizität des „Bait“-Konstruktes aus Aldolase und der Bindedomäne des Transkriptionsfaktors. Die Hefezellen mit diesem Konstrukt wuchsen sehr langsam, konnten aber mit Hilfe der Plattenanzucht in ausreichender Zelldichte für die Paarung der Hefezellen herangezogen werden. Die der Paarung folgende Anzucht der diploiden Hefezellen und die Selektion auf positive Interaktionspartner überlebten jedoch nur sehr wenige Kolonien, so dass alle anderen gefundenen Interaktionspartner der cytosolischen Mais-Aldolase in jeweils nur einem Versuch identifiziert werden konnten. Da kaum vollständige, für Proteine codierende Sequenzen identifiziert werden konnten, scheinen einige „Prey -Plasmide nur Bruchstücke der cDNA-Bank zu enthalten. 4.5.2 VDAC-Aldolase-Interaktion Einen interessanten Bindepartner der Aldolase stellt das mitochondriale spannungsabhängige Anionenkanalprotein VDAC dar. Die Übereinstimmung der im Hefe-2-Hybrid-System identifizierten Sequenz aus der cDNA-Bank mit der in der Datenbank GenBank veröffentlichten Sequenz beträgt 96% auf Nukleotidebene. Dabei handelt es sich um einen Teil der für VDAC kodierenden Sequenz, der vom N-terminalen Methionin ausgehend für108 Aminosäuren des Proteins kodiert. Das gesamte Protein setzt sich aus 276 Aminosäuren zusammen. Diskussion 122 VDAC, auch Porin genannt, befindet sich in der äußeren Mitochondrienmembran und stellt dort das dominierende Kanalprotein dar. Ein Modell über die Anordnung des Porins aus den Mitochondrien von Rinderherzen in der Membran (DE PINTO et al., 1991) zeigt 16 Transmembrandomänen und eine N-terminale -Helix, die cytosolisch exponiert ist. VDAC bildet eine wässrige Pore, durch die ein Austausch gelöster Stoffe zwischen Mitochondrien und Cytosol erfolgen kann. In der Literatur schwanken die Angaben über die Größe der Moleküle, die durch die Pore die Membran passieren können, zwischen 3 und 6 kDa. Für VDAC aus Mais geben ALJAMAL et al. (1993) einen Porendurchmesser von 1,6 nm an, der für Moleküle bis zu einer Masse von ca. 3 kDa permeabel ist. Bei geringem Membranpotential befindet sich VDAC im geöffneten Zustand und zeigt geringe Anionenselektivität. Bei Spannungen über 20 mV geht der Kanal in seinen wenig leitenden oder geschlossenen Zustand über. Der Fluss von Substraten wie Succinat, Citrat und Adeninnukleotiden ist dann stark reduziert. Die Spannungssensitivität resultiert aus einer Konformationsänderung und Bewegung geladener Domänen, vermutlich dienen positiv geladene Reste als Sensoren (THOMAS et al., 1993). In Anwesenheit von NADH wird der Kanal für Änderungen des Membranpotentials sensitiver und ist so wahrscheinlich besser in der Lage, sich den aktuellen Bedingungen des Stoffwechsels anzupassen (ZIZI et al., 1994). Die Regulation von VDAC ist ein effektiver Mechanismus, um den Fluss von Metaboliten zwischen Mitochondrien und Cytoplasma zu steuern. Auch die Funktionen der Mitochondrien können so reguliert werden. Durch das Schließen des Kanals wird die mitochondriale Respiration gehemmt (LIU & COLOMBINI, 1992). Eine direkte Verknüpfung zwischen den Mitochondrien und der Glykolyse war bis vor kurzem nur über die Interaktion der Hexokinase mit Mitochondrien bekannt (s. Kap. 1.2.1). Für zwei menschliche VDAC-Isoformen konnte man unterschiedliches Bindeverhalten für Hexokinase nachweisen (BLACHLY-DYSON et al., 1993). Die an VDAC gebundene Hexokinase nutzt für die Bildung von Glucose-6-phosphat ausschließlich ATP aus den Mitochondrien (BELTRANDELRIO & WILSON, 1992). Mit der Bindung an das Kanalprotein wird die Kinase aktiviert und verknüpft so die Verfügbarkeit von Glucose und den Glucose-Stoffwechsel mit den mitochondrialen Funktionen. Dies hat z. B. einen Einfluss auf die Apoptose, bei der die Mitochondrien eine wichtige Rolle durch die Freigabe von Cytochrom c spielen (GOTTLOB et al., 2001). Zusätzlich zur Hexokinase wurde kürzlich an isolierten Mitochondrien aus Arabidopsis die Assoziation von mindestens sieben Proteinen der Glykolyse an der äußeren mitochondrialen Membran entdeckt (GIEGÉ et al., 2003). Diese Assoziation ermöglichte den vollständigen Ablauf der Glykolyse an den Mitochondrien. Der Mechanismus der Assoziation blieb jedoch Diskussion 123 ungeklärt, und es wurde keine direkte Interaktion der Proteine mit dem Kanalprotein VDAC nachgewiesen. Die im Hefe-2-Hybrid-System gefundene Interaktion zwischen Aldolase und VDAC steht im Einklang mit der Assoziation der Glykolyse an Mitochondrien. Mit diesen Erkenntnissen erhärtet sich die Annahme, dass eine Mikrokompartimentierung der Glykolyse an Mitochondrien stattfindet und so eine direkte Verknüpfung zwischen dem Stoffwechsel der Mitochondrien und der im Cytosol der Zellen ablaufenden Glykolyse erfolgt. Neben dem Transfer von Metaboliten kann diese Interaktion auch regulatorische Aufgaben haben. Viele Proteine haben neben ihrer enzymatischen Aktivität weitere Funktionen, die häufig regulatorischer Art sind (JEFFREY, 1999). Die glykolytischen Enzyme können also auch eine Rolle in der Regulation mitochondrialer Funktionen ausüben. Hierüber müssen weitere Untersuchungen Aufschluss geben. 4.5.3 Interaktionen mit mRNA-Sequenzen unbekannter Funktion und einem Translationsinitiationsfaktor Den als Interaktionspartner der Aldolase identifizierten drei mRNA-Sequenzen (PC0102485, PC0138904 und CL3683_1) konnte auch mit Hilfe des Arabidopsis- oder Reis-Genoms keine Funktion zugewiesen werden. Die Interaktion eines Translations-Initiationsfaktors mit der Aldolase könnte auf die Regulation der Translation von mRNA, die mit der Glykolyse in Zusammenhang steht, hinweisen. Die Translation der Aldolase selbst könnte dadurch beeinflusst werden. Bei dem Translations-Initiationsfaktor GOS2 handelt es sich um den einzigen identifizierten Klon der cDNA-Bank, der die vollständige codierende Sequenz des Proteins enthält. Ob es sich bei diesen Interaktionen um tatsächlich vorhandene oder falsch positive handelt, müsste durch weitere Versuche geklärt werden. 4.5.4 Vermutlich falsch positive Interaktionen Bei der gefundenen Interaktion mit dem mitochondrialen Gen für 18S rRNA dürfte es sich um eine falsch positive Interaktion handeln, da nicht anzunehmen ist, dass in vivo eine Interaktion zwischen einem cytosolischen Protein und mitochondrialer ribosomaler RNA durch die räumliche Trennung der Kompartimente vorkommt. Ribosomale Proteine sind, wie auch Hitzeschockproteine, in der Lage, mit vielen verschiedenen Proteinen Komplexe zu formen, die stabil genug sind, um einen positiven Phänotyp hervorzubringen (HONG, 2000). Dies trifft vermutlich auch auf ribosomale RNA zu, so dass auch die Interaktion zwischen 25S rRNA und Aldolase ein falsch positives Ergebnis sein dürfte. Diskussion 124 Mit einem Anteil von zehn Prozent der Gesamtsequenz zeigte die identifizierte Sequenz des „Prey -Plasmids der Hefekolonie 2.43 nur einen kleinen Bereich an Identität zur MethioninAdenosyltransferase. In diesem Bereich lag die Übereinstimmung zwischen den beiden Sequenzen bei 88%. Die Methionin-Adenosyltransferase (oder auch S-Adenosyl-LMethionin-Synthetase, kurz SAM-S genannt) vermittelt die Synthese von S- Adenosylmethionin. Dieses entsteht durch den Transfer einer Adenosylgruppe von ATP auf das Schwefelatom des Methionins. Durch die positive Ladung des Schwefelatoms wird die Methylgruppe des Methionins aktiviert. Bei S-Adenosylmethionin handelt es sich um den wichtigsten Methylgruppendonor, der an der Methylierung von Nukleinsäuren, Proteinen, Kohlenhydraten, Membranlipiden und vielen anderen Substanzen in der Zelle beteiligt ist. Es ist außerdem das Ausgangsprodukt der Ethylensynthese (YANG & HOFFMAN, 1984). Somit stellt es eines der wichtigsten und zentralen biologischen Moleküle dar, es existiert in Organismen in vielfältigen Formen, die von verwandten Genen kodiert werden. Die SAM-SGene von Arabidopsis und Reis zeigen 80% Übereinstimmung, jedoch unterscheidet sich das Expressionsmuster in den Pflanzen (VAN BREUSEGEM et al., 1994). In Blättern von Arabidopsis konnte im Gegensatz zu Reis kaum SAM-S nachgewiesen werden. Eine individuelle Regulation der Gene oder Unterschiede in der Enzymkinetik der Isoenzyme wurden nur in wenigen Fällen beschrieben. Daher besteht die Möglichkeit, dass Assoziationen mit anderen Enzymen, die z. B. das Produkt S-Adenosylmethionin nutzen, Spezialisierungen hervorrufen (SCHRÖDER et al., 1997). Welche Rolle eine Assoziation der Aldolase mit SAM-S spielen könnte, ist jedoch unklar. Eine gewagte These wäre, dass diese Interaktion einen regulatorischen Einfluss auf die Richtung des Kohlenhydratflusses ausübt. Dieser kann in Richtung der Glykolyse stattfinden oder aber auch zur Synthese von Zellwandbestandteilen erfolgen. Weshalb die Regulation dann jedoch erst auf Ebene der Aldolase und nicht schon früher, z. B. bei der Saccharosespaltung oder der Bildung von Fructose-6-phosphat, erfolgen sollte, ist fraglich. Denkbar wäre auch eine Regulation auf Ebene der Enolase, durch die Phosphoenolpyruvat, das Ausgangsprodukt für die Bildung der Aminosäure Phenylalanin, gebildet wird. Daher scheint es realistischer, diese Interaktion als eine falsch positive einzustufen. Diskussion 125 4.6 Überprüfung der Protein-Protein-Interaktionen Die Interaktion von Aldolase und VDAC sollte mit Hilfe von Affinitätschromatographie überprüft werden. Dabei wurde das rekombinante His-tag-VDAC-Fusionsprotein an eine NiNTA-Säulenmatrix gekoppelt. Durch Zugabe rekombinanter Aldolase mit Strep-tag wurde eine Bindung zwischen den beiden Proteinen untersucht. Da jedoch das rekombinante Kanalprotein bei der Überexpression in E. coli in inclusion bodies“ eingeschlossen wurde, konnte nur unter denaturierenden Bedingungen gearbeitet werden. Dies schloss auch eine Untersuchung der Interaktion durch an Strep-Tactin gekoppelte Aldolase aus, da Strep-Tactin nicht für eine Behandlung mit Harnstoff geeignet ist. Diese Probleme und die unspezifische Bindung der Aldolase an die Ni-NTA-Matrix machen eine Aussage über eine Assoziation der Proteine innerhalb dieses Versuchsaufbaus schwierig. Es zeigte sich jedoch, dass sich die Elution der Aldolase von den Säulen mit und ohne VDAC unterscheidet. Die unspezifisch an die Matrix gebundene Aldolase in der Kontrolle konnte durch Imidazolkonzentrationen von 50 mM vollständig ausgewaschen werden. In Anwesenheit des VDAC dagegen wurde Aldolase auch noch in den Elutionsfraktionen mit 100-250 mM Imidazol nachgewiesen (s. Kap. 3.6). Diese Verschiebung der Elution der Aldolase könnte durch eine Interaktion mit dem Kanalprotein VDAC hervorgerufen worden sein. Eine denaturierende Behandlung der Proteine führt jedoch zur Zerstörung der nativen Konformation, die für die Bindung zwischen den Proteinen essentiell sein kann. Um die Interaktion zwischen VDAC und Aldolase eindeutig zu beweisen, müssten andere Methoden gewählt werden. Möglich wäre z. B. die Visualisierung der Protein-Protein-Interaktion in vivo durch „bimolecular fluorescence complementation“, bei der in Protoplasten zwei nichtfluoreszierende Fragmente des „yellow fluorescent protein“ (YFP) durch interagierende Proteine zu einem fluoreszierenden Komplex zusammengefügt werden (BRACHA-DRORI et al., 2004; WALTER et al., 2004). Obwohl Aldolase-YFP-Fusionsproteine durch die Verbreitung im Cytosol der Zellen zu einem cytosolischen Signal führen, konnte durch GIEGÉ et al. (2003) zusätzlich eine Konzentration des Fluoreszenssignals um Mitochondrien nachgewiesen werden. Voraussetzung für die Untersuchung der VDAC-Aldolase-Interaktion in Protoplasten wäre, dass VDAC mit dem nichtfluoreszierenden Fragment des YFP in Mitochondrien eingebaut wird und sich das YFPFragment im Cytosol der Zellen befindet. Diskussion 126 Die anhand der Hefe-2-Hybrid-Analysen und der Copolymerisationsversuche identifizierten Interaktionen der Aldolase mit Aktin und VDAC konnten mit Hilfe eines „Far-WesternBlots“ bestätigt werden (s Abb. 3.27). Es zeigte sich eine deutlich nachweisbare Interaktion zwischen den Proteinen. Dass eine Interaktion mit Tubulin, die von tierischer Aldolase bekannt ist (KARKHOFF-SCHWEIZER & KNULL, 1987; WALSH et al., 1989), mit Hilfe dieser Methode nicht möglich war, kann verschiedene Ursachen haben. Da Reste von Tubulin aus anderen Versuchen eingesetzt wurden, war es nicht möglich, die genaue Proteinmenge zu bestimmen. Es besteht daher die Möglichkeit, dass weniger als 2 µg Protein auf der Membran fixiert waren. VÉRTESSY et al. (1997) haben nachgewiesen, dass die Interaktion zwischen Aldolase und Tubulin konzentrationsabhängig und im Vergleich zur Bindung zwischen PFK und Tubulin schwächer ist. Die fehlgeschlagene Detektion kann somit auch auf einer für einen „Far-Western-Blot“ zu schwachen Interaktion zwischen Aldolase und Tubulin beruhen. In in vitro-Polymerisationsversuchen mit rekombinanter Strep-tag-Aldolase und tierischem Tubulin konnte jedoch in aktuellen Untersuchungen eine Interaktion pflanzlicher Aldolase mit Tubulin nachgewiesen werden (Daniela Holtgräwe, mündliche Mitteilung). Zusammenfassung 127 5 Zusammenfassung Um Beweise für eine mögliche Mikrokompartimentierung der Glykolyse im pflanzlichen System zu erhalten, sollten in der vorliegenden Arbeit Protein-Protein-Interaktionen der cytosolischen Mais-Aldolase mit anderen Proteinen experimentell nachgewiesen werden. Die in Tieren bekannte Interaktion des glykolytischen Enzyms Aldolase mit Aktin, einem Bestandteil des Cytoskeletts, wurde für Pflanzen in vitro durch Copolymerisationsversuche bestätigt. Die dafür benötigte katalytisch aktive Aldolase konnte nach heterologer Expression in Escherichia coli mit guter Ausbeute (0,02 mg/ml Zellkultur) gereinigt werden. Ihre Aktivität für Fructose-1,6-bisphosphat betrug 1,23 U/mg. Die Bindung pflanzlicher Aldolase an Aktinfilamente wurde – anders als im tierischen System – durch das Substrat Fructose-1,6-bisphosphat auch in hohen Konzentrationen (10 mM) nicht vollständig verhindert, sondern führte lediglich zu einer um 50% verringerten Bindung. Eine ebenfalls hemmende Wirkung auf die Bindung der Aldolase an Aktin wiesen Fructose-6-phosphat und Fructose-2,6-bisphosphat in Konzentrationen von 10 mM auf. Ein eindeutiger Einfluss des Redox-Milieus auf die Aldolase-Aktin-Bindung konnte nicht nachgewiesen werden. Glutathion führte in reduzierter und oxidierter Form zu einer geringen Zunahme der an Aktin gebundenen Aldolase. Unter Einfluss von reduziertem DTT zeigte sich ebenfalls eine erhöhte Aktinbindung. Dagegen sank der an Aktin gebundene Anteil der Aldolase unter Einfluss von oxidiertem DTT. Mit Hilfe des im Rahmen dieser Arbeit etablierten Hefe-2-Hybrid-Systems wurden weitere Interaktionspartner der Aldolase identifiziert. Insgesamt wurden neun mögliche ProteinProtein-Interaktionen nachgewiesen, bei denen es sich jedoch zum Teil um falsch-positive Interaktionen handeln kann. Neben einigen noch unbekannten Proteinen konnten Interaktionen mit einem Translations-Initiationsfaktor und dem spannungsabhängigen Anionenkanalprotein VDAC nachgewiesen werden. In Bindeversuchen auf Grundlage der Affinitätschromatographie mit den rekombinanten Proteinen VDAC und Aldolase wurde ein weiterer Hinweis auf eine Interaktion zwischen VDAC und Aldolase erhalten. Aufgrund unspezifischer Bindungen der Aldolase an die Affinitätsmatrix konnte mit dieser Methode jedoch keine eindeutige Verifizierung der Interaktion erzielt werden. Eine eindeutige Bestätigung der Interaktionen zwischen Aldolase und Aktin sowie zwischen Aldolase und VDAC erfolgte durch „Far-Western-Blots“. Zusammenfassung 128 Das im Rahmen dieser Arbeit hergestellte polyklonale Antiserum gegen cytosolische Aldolase aus Mais (Bioscience, Göttingen) steht für weitere immunologische Arbeiten zur Verfügung. Im Western-Blot wird damit spezifisch eine einzelne Bande von ca. 40 kDa in Rohextrakten von Maiskeimlingen und -wurzeln sowie von Arabidopsis-Blattgewebe und Kartoffelknollen erkannt. Literatur 129 6 Literatur ABE, S., YOU, W. & DAVIES, E. (1991) Protein bodies in corn endosperm are enclosed by and enmeshed in F-actin. Protoplasma 165, 139-149. ALJAMAL, J.A., GENCHI, G., DE PINTO, V., STEFANIZZI, L., DE SANTIS, A., BENZ, R. & PALMIERI, F. (1993) Purification and characterization of porin from corn (Zea mays L.) mitochondria. Plant Physiol. 102, 615-621. ALTSCHUL, S.F., GISH, W., MILLER, W., MYERS, E.W. & LIPMAN, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403-410. ALTSCHUL, S.F., MADDEN, T.L., SCHÄFFER, A.A., ZHANG, J., ZHANG, Z., MILLER, W. & LIPMAN, D.J. (1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389-3402. ANDERSON, L.E., GOLDHABER-GORDON, I.M., LI, D., TANG, X.-Y., XIANG, M. & PRAKASH, N. (1995) Enzyme-enzyme interaction in the chloroplast: Glyceraldehyde-3-phosphate dehydrogenase, triose phosphate isomerase and aldolase. Planta 196, 245-255. ARNOLD, H. & PETTE, D. (1969) Binding of glycolytic enzymes to structure proteins of the muscle. Eur. J. Biochem. 6, 163-171. Literatur 130 ARNOLD, H. & PETTE, D. (1970) Binding of aldolase and triosephosphate dehydrogenase to F-actin and modification of catalytic properties of aldolase. Eur. J. Biochem. 15, 360-366. AZAMA, K., ABE, S., SUGIMOTO, H. & DAVIES, E. (2003) Lysine-containing proteins in maize endosperm: A major contribution from cytoskeleton-associated carbohydrate-metabolizing enzymes. Planta 217, 628-638. BALU KA, F. & HASENSTEIN, K.H. (1997) Root cytoskeleton: Its role in perception of and response to gravity. Planta 203, S69-S78. BALU KA, F., SALAJ, J., MATHUR, J., BRAUN, M., JASPER, F., SAMAJ, J., CHUA, N.H., BARLOW, P.W. & VOLKMANN, D. (2000) Root hair formation: F-actin-dependent tip growth is initiated by local assembly of profiling-supported F-actin meshworks accumulated within expansin-enriched bulges. Dev. Biol. 227, 618-632. BARTEL, P.L., CHIEN, C.-T., STERNGLANZ, R. & FIELDS, S. (1993) Elimination of false positives that arise in using the two-hybrid system. BioTechniques 14, 920-924. BELTRANDELRIO, H. & WILSON, J.E. (1992) Coordinated regulation of cerebral glycolytic and oxidative metabolism, mediated by mitochondrially bound hexokinase dependent on intramitochondrially generated ATP. Arch. Biochem. Biophys. 296, 667-677. BERTHIAUME, L., BEAUDRY, D., LAZURE, C., TOLAN, D.R. & SYGUSCH, J. (1989) Recombinant anaerobic maize aldolase: Overexpression, characterization, and metabolic implications. Arch. Biochem. Biophys. 272, 281-289. Literatur 131 BERTHIAUME, L., LOISEL, T.P. & SYGUSCH, J. (1991) Carboxyl terminus region modulates catalytic activity of recombinant maize aldolase. J. Biol. Chem. 266, 17099-17105. BIRNBOIM, H.C. & DOLY, J. (1979) A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7, 1513-1523. BLACHLY-DYSON, E., ZAMBRONICZ, E.B., YU, W.H., ADAMS, V., MCCABE, E.R., ADELMAN, J., COLOMBINI, M. & FORTE, M. (1993) Cloning and functional expression in yeast of two human isoforms of the outer mitochondrial membrane channel, the voltage-dependent anion channel. J. Biol. Chem. 286, 1835-1841. BOND, J.S. & OFFERMANN, M.K. (1981) Initial events in the degradation of soluble cellular enzymes: Factors affecting the stability and proteolytic susceptibility of fructose-1,6-bisphosphate aldolase. Acta Biol. Med. Ger. 40, 1365-1374. BOWERMAN, B. & SEVERSON, A.F. (1999) Cell division: Plant-like properties of animal cell cytokinesis. Curr. Biol. 9, R658-R660. BRACHA-DRORI, K., SHICHRUR, K., KATZ, A., OLIVA, M., ANGELOVICI, R., YALOVSKY, S. & OHAD, N. (2004) Detection of protein-protein interactions in plants using bimolecular fluorescence complementation. Plant J. 40, 419-427. BRADFORD, M.M. (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248-254. Literatur 132 BULLOCK, W.O., FERNANDEZ, J.M. & SHORT, J.M. (1987) XL1-Blue: A high efficiency plasmid transforming recA Escherichia coli strain with beta-glucosidase selection. Biotechniques 5, 376-378. CABISCOL, E., PIULATS, E., ECHAVES, P., HERRERO, E. & ROS, J. (2000) Oxidative stress promotes specific protein damage in Saccharomyces cerevisiae. J. Biol. Chem. 275, 27393-27398. CARLIER, M.-F. & PANTALONI, D. (1997) Control of actin dynamics in cell motility. J. Mol. Biol. 269, 459-467. CAROTHERS CARRAWAY, C.A. (2000) The cytoskeleton in the transduction of signal and regulation of cellular function. In: CYTOSKELETON: SIGNALLING AND CELL REGULATION. (Hrsg.: Carraway, K.L. & Carothers Carraway, C.A.), pp. 1-7, Oxford University Press, Oxford, GB. CHAI, Y.-C., ASHRAF, S.S., ROKUTAN, K., JOHNSTON, R.B. JR., & THOMAS, J.A. (1994) S-thiolation of individual human neutrophil proteins including actin by stimulation of the respiratory burst: Evidence against a role for glutathione disulfide. Arch. Biochem. Biophys. 310, 273-281. CHEN, H., BERNSTEIN, B.W. & BAMBURG, J.R. (2000) Regulating actin-filament dynamics in vivo. Trends Biochem. Sci. 25, 19-23. CLARKE, F.M. & MASTERS, C.J. (1975) On the association of glycolytic enzymes with structural proteins of skeletal muscle. Biochim. Biophys. Acta 381, 37-46. CLARKE, F.M. & MORTON, D.J. (1976) Aldolase binding to actin-containing filaments. Biochem. J. 159, 797-798. Literatur 133 CLARKE, F.M. & MORTON, D.J. (1982) Glycolytic enzyme binding in fetal brain the role of actin. Biochem. Biophys. Res. Commun. 109, 388-393. CLORE, A.M., DANNENHOFFER, J.M. & LARKINS, B.A. (1996) EF-1 is associated with a cytoskeletal network surrounding protein bodies in maize endosperm cells. Plant Cell 8, 2003-20014. COBBETT, C.S. (2000) Phytochelatins and their roles in heavy metal detoxification. Plant Physiol. 123, 825-832. COLLINGS, D.A., WINTER, H., WYATT, S.E. & ALLEN, N.S. (1998) Growth dynamics and cytoskeleton organization during stem maturation and gravityinduced stem bending in Zea mays L. Planta 207, 246-258. COOPER, J.A. & SCHAFER, D.A. (2000) Control of actin assembly and disassembly at filament ends. Curr. Opin. Cell Biol. 12, 97-103. DAMERVAL, T., HOUMARD, J., GUGLIELMI, G., CSISZAR, K. & TANDEAU DE MARSAC, N. (1987) A developmentally regulated gvpABC operon is involved in the formation of gas vesicles in the cyanobacterium Calothrix 7601. Gene 54, 83-92. DA-SILVA, W.S., REZENDE, G.L. & GALINA, A. (2001) Subcellular distribution and kinetic properties of cytosolic and non-cytosolic hexokinases in maize seedling roots: Implications for hexose phosphorylation. J. Exp. Bot. 52, 1191-1201. Literatur 134 DAVIES, E., FILLINGHAM, B.D., OTO, Y. & ABE, S. (1991) Polyribosomes from peas. VII. Initial characterization of a population of polysomes associated with the cytoskeleton. Cell Biol. Int. Rep. 17, 331-340. DENNIS, D.T., HUANG, Y., NEGM, F.B. (1997) Glycolysis, the pentose phosphate pathway and anaerobic respiration. In: PLANT METABOLISM. 2nd edition, (Hrsg.: Dennis, D.T., Turpin, D.H., Lefebvre, D.D., Layzell, D.B.), pp. 105-109, Addison Wesley Logman Ltd., Essex, GB. DENNIS, E.S., GERLACH, W.L., WALKER, J.C., LAVIN, M. & PEACOCK, W.J. (1988) Anaerobically regulated aldolase gene of maize. A chimaeric origin? J. Mol. Biol. 202, 759-767. DE PINTO, V., PREZIOSO, G., THINNES, F., LINK, T.A. & PALMIERI, F. (1991) Peptide-specific antibodies and proteases as probes of the transmembrane topology of the bovine heart mitochondrial porin. Biochemistry 30, 10191-10200. DE RUIJTER, N.C.A. & EMONS, A.M.C. (1999) Actin-binding proteins in plant cells. Plant Biol. 1, 26-35. DON, M. & MASTERS, C. (1988) Chemical modification of the actin binding site of rabbit muscle aldolase by diethylpyrocarbonate. Mol. Cell. Biochem. 81, 145-153. DREWES, G. & FAULSTICH, H. (1993) Cooperative effects on filament stability in actin modified at the C-terminus by substitution or truncation. Eur. J. Biochem. 212, 247-253. Literatur 135 DURRIEU, C., BERNIER-VALENTIN, F., & ROUSSET, B. (1987) Microtubules bind glyceraldehyde 3-phosphate dehydrogenase and modulate its enzyme activity and quaternary structure. Arch. Biochem. Biophys. 252, 32-40. FOISSNER, I., LICHTSCHEIDL, I.K. & WASTENEYS, G.O. (1996) Actin-based vesicle dynamics and exocytosis during wound wall formation in characean internodal cells. Cell. Motil. Cytoskeleton 35, 35-48. FRATELLI, M., DEMOL, H., PUYPE, M., CASAGRANDE, S., EBERINI, I., SALMONA, M., BONETTO, V., MENGOZZI, M., DUFFIEUX, F., MICLET, E., BACHI, A., VANDEKERCKHOVE, J., GIANAZZA, E. & GHEZZI, P. (2002) Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. USA 99, 3505-3510. FULTON, A.B. (1982) How crowded is the cytoplasm? Cell 30, 345-347. GALINA, A., LOGULLO, C., SOUZA, E.F., REZENDE, G.L. & DA-SILVA, W.S. (1999) Sugar phosphorylation modulates ADP inhibition of maize mitochondrial hexokinase. Physiol. Plant. 105, 17-23. GIEGÉ, P., HEAZLEWOOD, J.L., ROESSNER-TUNALI, U., MILLAR, A.H., FERNIE, A.R., LEAVER, C.J. & SWEETLOVE, L.J. (2003) Enzymes of glycolysis are functionally associated with the mitochondrion in Arabidopsis cells. Plant Cell 15, 2140-2151. Literatur 136 GITLITS, V.M., TOH, B.H., LOVELAND, K.L. & SENTRY, J.W. (2000) The glycolytic enzyme enolase is present in sperm tail and displays nucleotidedependent association with microtubules. Eur. J. Cell Biol. 79, 104-111. GODON, C., LAGNIEL, G., LEE, J., BUHLER, J.-M., KIEFFER, S., PERROT, M., BOUCHERIE, H., TOLEDANO, M.B. & LABARRE, J. (1998) The H2O2 stimulon in Saccharomyces cerevisiae. J. Biol. Chem. 273, 22480-22489. GOTTLOB, K., MAJEWSKI, N., KENNEDY, S., KANDEL, E., ROBEY, R.B. & HAY, N. (2001) Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes & Dev. 15, 1406-1418. HAKE, S., KELLEY, P.M., TAYLOR, W.C. & FREELING, M. (1985) Coordinate induction of alcohol dehydrogenase 1, aldolase, and other anaerobic RNAs in maize. J. Biol. Chem. 260, 5050-5054. HANAHAN, D. (1983) Studies of transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557-580. HARPER, J.W., ADAMI, G.R., WEI, N., KEYOMARSI, K. & ELLEDGE, S.J. (1993) The p21 Cdk-interacting protein Cip 1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805-816. HARRIS, S.J. & WINZOR, D.J. (1990) Interactions of glycolytic enzymes with erythrocyte membranes. Biochim. Biophys. Acta 1038, 306-314. Literatur 137 HATANO, S. (1994) Actin-binding proteins in cell motility. Int. Rev. Cytol. 156, 199-273. HINSHAW, D.B., BURGER, J.M., BEALS, T.F., ARMSTRONG, B.C. & HYSLOP, P.A. (1991) Actin polymerisation in cellular oxidant injury. Arch. Biochem. Biophys. 288, 311-316. HONG, Y.-R. (2000) False positives: Detection and elimination. In: YEAST HYBRID TECHNOLOGIES (Hrsg.: Zhu, L. & Hannon, G.J.), pp. 101-113, Eaton Publishing BioTechniques Books Division, Natick, MA, USA. ITO, Y., ABE, S. & DAVIES, E. (1994) Co-localisation of cytoskeleton proteins and polysomes with a membrane fraction from peas. J. Exp. Bot. 271, 253-259. ITO, H., IWABUCHI, M. & OGAWA, K. (2003) The sugar-metabolic enzymes aldolase and triose-phosphate isomerase are targets of glutathionylation in Arabidopsis thaliana: Detection using biotinylated glutathione. Plant Cell Physiol. 44, 655-660. JAMES, P., HALLADAY, J. & CRAIG, E.A. (1996) Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144, 1425-1436. JANMEY, P.A. (1998) The cytoskeleton and cell signaling: Component localization and mechanical coupling. Physiol. Rev. 78, 763-781. Literatur 138 JEFFERY, C.J. (1999) Moonlighting proteins. Trends Biochem. Sci. 24, 8-11. KABSCH, W. & VANDEKERCKHOVE, J. (1992) Structure and function of actin. Annu. Rev. Biophys. Biomol. Struct. 21, 49-76. KÁLMÁN, M. & BOROSS, L. (1982) Characterization of enzyme-enzyme interaction using an affinity batch system. Biochim. Biophys. Acta 704, 272-277. KAO, A.W., NODA, Y., JOHNSON, J.H., PESSIN, J.E. & SALTIEL, A.R. (1999) Aldolase mediates the association of F-actin with the insulin-responsive glucose transporter GLUT4. J. Biol. Chem. 274, 17742-17747. KARKHOFF-SCHWEIZER, R. & KNULL, H.R. (1987) Demonstration of tubulin-glycolytic enzyme interactions using novel electrophoretic approach. Biochem. Biophys. Res. Commun. 146, 827-831. KELETI, T. & OVÁDI, J. (1988) Control of metabolism by dynamic macromolecular interactions. Curr. Top. Cell Reg. 29, 1-33 KELLEY, P.M. & FREELING, M. (1984a) Anaerobic expression of maize glucose phosphate isomerase I. J. Biol. Chem. 259, 673-677. KELLEY, P.M. & FREELING, M. (1984b) Anaerobic expression of maize fructose-1,6-diphosphate aldolase. J. Biol. Chem. 259, 14180-14183. Literatur 139 KELLEY, P.M. & TOLAN, D.R. (1986) The complete amino acid sequence for the anaerobically induced aldolase from maize derived from cDNA clones. Plant Physiol. 82, 1076-1080. KLYACHKO, N., AKSENOVA, L., DUNAEVA, M. & KULIKOVA, A. (2000) Interaction of plant polysomes with the actin cytoskeleton. Cell Biol. Int. 24, 351-358. KNULL, H.R., BRONSTEIN, W.W., DES JARDINS, P. & NIEHAUS, W.G. JR. (1980) Interaction of selected brain glycolytic enzymes with an F-actin-tropomyosin complex. J. Neurochem. 34, 222-225. KNULL, H.R. & WALSH, J.L. (1992) Association of glycolytic enzymes with the cytoskeleton. Curr. Top. Cell Regul. 33, 15-30. KÖHLER, W., SCHACHTEL, G. & VOLESKE, P. (2002) BIOSTATISTIK. 3. Auflage. Springer Verlag Berlin, Heidelberg, New York. KUSAKABE, T., MOTOKI, K. & HORI, K. (1997) Mode of interactions of human aldolase isozymes with cytoskeletons. Arch. Biochem. Biophys. 344, 184-193. LAEMMLI, U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680-685. LASZLO, A. & ST. LAWRENCE, P. (1983) Parallel induction and synthesis of PDC and ADH in anoxic maize roots. Mol. Gen. Genet. 192, 110-117. Literatur 140 LEGRAIN, P., WOJCIK, J. & GAUTHIER, J.-M. (2001) Protein-protein interaction maps: A lead towards cellular functions. Trends Genet. 17, 346-352. LI, B. & FIELDS, S. (1993) Identification of mutations in p53 that affect its binding to SV40 T antigen by using the yeast two-hybrid system. FASEB J. 7, 957-963. LICHTSCHEIDL, I.K. & URL, W.G. (1990) Organization and dynamics of cortical endoplasmic reticulum in inner epidermal cells of onion bulb seales. Protoplasma 157, 203-215. LIU, M.Y. & COLOMBINI, M. (1992) Regulation of mitochondrial respiration by controlling the permeability of the outer membrane through the mitochondrial channel, VDAC. Biochim. Biophys. Acta 1098, 255-260. LOW, P.S., RATHINAVELU, P. & HARRISON, M.L. (1993) Regulation of glycolysis via reversible enzyme binding to the membrane protein, band 3 J. Biol. Chem. 268, 14627-24631. LU, T., VAN DYKE, M. & SAWADOGO, M. (1993) Protein-protein interaction studies using immobilized oligohistidine fusion proteins. Anal. Biochem. 213, 318-322. LUBY-PHELBS, K. (1994) Constraints of diffusion in the cytoplasm of living cells. Comments Mol. Cell. Biophys. 8, 199-216. Literatur 141 LUNA, E.J. & HITT, A.L. (1992) Cytoskeleton-plasma membrane interactions. Science 258, 955-963. LUTHER, M.A. & LEE, J.C. (1986) The role of phosphorylation in the interaction of rabbit muscle phosphofructokinase with F-actin. J. Biol. Chem. 261, 1753-1759. MARSH, J.J. & LEBHERZ, H.G. (1992) Fructose-bisphosphate aldolases: An evolutionary history. Trends Biochem. Sci. 17, 110-113. MASTERS, C.J., REID S. & DON, M. (1987) Glycolysis new concept in an old pathway. Mol. Cell. Biochem. 76, 3-14. MATHEWS, C.K. (1993) The cell – bag of enzymes or network of channels? J. Bacteriol. 175, 6377-6381. MATHEWS, C.K. & SINHA, N.K. (1982) Are DNA precursors concentrated at replication sites? Proc. Natl. Acad. Sci. USA 79, 302-306. MCCARTY, D.R., SHAW, J.R. & HANNAH, L.C. (1986) The cloning, genetic mapping, and expression of the constitutive sucrose synthase locus of maize. Proc. Natl. Acad. Sci. USA 83, 9099-9103. MCDOWELL, J.M., HUANG, S., MCKINNEY, E.C., AN, Y.-Q. & MEAGEHER, R.B. (1996) Structure and evolution of the actin gene family in Arabidopsis thaliana. Genetics 142, 587-602. Literatur 142 MINASCHEK, G., GRÖSCHEL-STEWART, U., BLUM, S. & BREITER-HAHN, J. (1992) Microcompartmentation of glycolytic enzymes in cultured cells. Eur. J. Cell Biol. 58, 418-428. MOORHEAD, G.B.G. & PLAXTON, W.C. (1988) Binding of glycolytic enzymes to a particulate fraction in carrot and sugar beet storage roots. Plant Physiol. 86, 348-351. MOORHEAD, G.B.G. & PLAXTON, W.C. (1992) Evidence for an interaction between cytosolic aldolase and the ATP- and pyrophosphate-dependent phosphofructokinase in carrot storage roots. FEBS Lett. 313, 277-280. MOWBRAY, J. & MOSES, V. (1976) The tentative identification in Escherichia coli of a multienzyme complex with glycolytic activity. Eur. J. Biochem. 66, 25-36. MUENCH, D.G., WU, Y., COUGHLAN, S.J. & OKITA, T.W. (1998) Evidence for a cytoskeleton-associated binding site involved in prolamine mRNA localization to the protein bodies in rice endosperm tissue. Plant Physiol. 116, 559-569. NICK, P. (1999) Signals, motors, morphogenesis – the cytoskeleton in plant development. Plant Biol. 1, 169-179. OFFERMANN, M.K., MCKAY, M.J., MARSH, M.W. & BOND, J.S. (1984) Glutathione disulfide inactivates, destabilizes, and enhances proteolytic susceptibility of fructose-1,6-bisphosphate aldolase. J. Biol. Chem. 259, 8886-8891. Literatur 143 OGAWA, K., HATANO-IWASAKI, A., YANAGIDA, M. & IWABUCHI, M. (2004) Level of glutathione is regulated by ATP-dependent ligation of glutamate and cysteine through photosynthesis in Arabidopsis thaliana: Mechanism of strong interaction of light intensity with flowering. Plant Cell Physiol. 45, 1-8. OGAWA, K., TASAKA, Y., MINO, M., TANAKA, Y. & IWABUCHI, M. (2001) Association of glutathione with flowering in Arabidopsis thaliana. Plant Cell Physiol. 42, 524-530. O’REILLY, G. & CLARKE, F. (1993) Identification of an actin binding region in aldolase. FEBS Lett. 321, 69-72. OUPOROV, I.V., KNULL, H.R., LOWE, S.L. & THOMASSON, K.A. (2001) Interactions of glyceraldehyde-3-phosphate dehydrogenase with G- and F-actin predicted by Brownian dynamics. J. Mol. Recogn. 14, 29-41. OVÁDI, J. (1988) Old pathway – new concept: Control of glycolysis by metabolite-modulated dynamic enzyme associations. Trends Biochem. Sci. 13, 486-490. OVÁDI, J., ARAGÓN, J.J. & SOLS, A. (1986) Phosphofructokinase and fructosebisphosphatase from muscle can interact at physiological concentrations with mutual effects on their kinetic behavior. Biochem. Biophys. Res. Commun. 135, 852-856. OVÁDI, J. & KELETI, T. (1978) Kinetic evidence for interaction between aldolase and D-glyceraldehyde-3-phosphate dehydrogenase. Eur. J. Biochem. 85, 157-161. Literatur 144 OVÁDI, J. & SRERE, P.A. (1996) Metabolic consequences of enzyme interactions. Cell Biochem. Funct. 14, 249-258. PAGLIARO, L. & TAYLOR, D.L. (1988) Aldolase exists in both the fluid and solid phases of cytoplasm. J. Cell Biol. 107, 981-991. PAGLIARO, L. & TAYLOR, D.L. (1992) 2-Deoxyglucose und cytochalasin D modulate aldolase mobility in living 3T3 cells. J. Cell Biol. 118, 859-863. PELZER-REITH, B., PENGER, A. & SCHNARRENBERGER, C. (1993) Plant aldolases: cDNA and deduced amino-acid sequences of the chloroplast and cytosol enzyme from spinach. Plant Mol. Biol. 21, 331-340. PELZER-REITH, B., WIEGAND, S. & SCHNARRENBERGER, C. (1994) Plastid class I and cytosol class II aldolases of Euglena gracilis (purification and characterisation). Plant Physiol. 106, 1137-1144. PIERSON, E.S. & CRESTI, M. (1992) Cytoskeleton and cytoplasmic organization of pollen and pollen tubes. Int. Rev. Cytol. 140, 73-125. PLAXTON, W.C. (1996) The organization and regulation of plant glycolysis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47, 185-214. Literatur R S, 145 B., ORTEGA, F., PUIGJANER, J., COMIN, B., OROSZ, F., OVÁDI, J. & CASCANTE, M. (2000) Quantitative characterization of homo- and heteroassociations of muscle phosphofructokinase with aldolase. Biochim. Biophys. Acta 1479, 303-314. REN, H., GIBBON, B.C., ASHWORTH, S.L., SHERMAN, D.M., YUAN, M. & STAIGER, C.J. (1997) Actin purified from maize pollen functions in living plant cells. Plant Cell 9, 1445-1457. RYAZANOV, A.G., ASHMARINA, L.I. & MURONETZ, V.I. (1988) Association of glyceraldehyde-3-phosphate dehydrogenase with mono- and polyribosomes of rabbit reticulocytes. Eur. J. Biochem. 171, 301-305. SACHS, M.M. & FREELING, M. (1978) Selective synthesis of alcohol dehydrogenase during anaerobic treatment of maize. Mol. Gen. Genet. 161, 111-115. SACHS, M.M., FREELING, M. & OKIMOTO, R. (1980) The anaerobic proteins of maize. Cell 20, 761-767. SAMBROOK, J., FRITSCH, E.F. & MANIATIS, T. (1989) MOLECULAR CLONING: A LABORATORY MANUAL. 2nd edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA. SÁNCHEZ-FERNÁNDEZ, R., FRICKER, M., CORBEN, L.B., WHITE, N.S., SHEARD, N., LEAVER, C.J., VAN MONTAGU, M., INZÉ, D. & MAY, M.J. (1997) Cell proliferation and hair tip growth in the Arabidopsis root are under mechanistically different forms of redox control. Proc. Natl. Acad. Sci. USA 94, 2745-2750. Literatur 146 SANGER, F., FRITSCH, E.F. & COULSON, A.R. (1977) DNA sequencing with chain terminating inhibitors. Proc. Natl. Acad. Sci. USA 74, 5463-5467. SCHEIBE, R., WEDEL, N., VETTER, S., EMMERLICH, V. & SAUERMANN, S.-M. (2002) Co-existence of two regulatory NADP-glyceraldehyde 3-P dehydrogenase complexes in higher plant chloroplasts. Eur. J. Biochem. 269, 5617-5624. SCHLIWA, M. (1981) Proteins associated with cytoplasmic actin. Cell 25, 587-590. SCHRÖDER, G., EICHEL, J., BREINIG, S. & SCHRÖDER, J. (1997) Three differentially expressed S-adenosylmethionine synthetases from Catharantus roseus: Molecular and functional characterization. Plant Mol. Biol. 33, 211-222. SEDBROOK, J.C., CHEN, R. & MASSON, P.H. (1999) ARG1 (Altered response to gravity) encodes a DnaJ-like protein that potentially interacts with the cytoskeleton. Proc. Natl. Acad. Sci. USA 96, 1140-1145. SEN C.K. (2000) Cellular thiols and redox-regulated signal transduction. Curr. Top. Cell Regul. 36, 1-30. SPRINGER, B., WERR, W., STARLINGER, P., BENNETT, D.C., ZOKOLICA, M. & FREELING, M. (1986) The shrunken gene on chromosome 9 of Zea mays L. is expressed in various plant tissues and encodes an anaerobic protein. Mol. Gen. Genet. 205, 461-468. Literatur 147 SRERE, P.A. (1967) Enzyme concentrations in tissues. Science 158, 936-937. SRERE, P.A. (1987) Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 56, 21-56. SRERE, P.A. & KNULL, H.R. (1998) Location-location-location. Trends Biochem. Sci. 23, 319-320. SRIVASTAVA, D.K. & BERNHARD, S.A. (1986) Metabolite transfer via enzyme-enzyme complexes. Science 234, 1081-1086. STAIGER, C.J. (2000) Signaling to the actin cytoskeleton in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 51, 257-288. STANKOVI , B., ABE, S. & DAVIES, E. (1993) Co-localization of polysomes, cytoskeleton, and membranes with protein bodies from corn endosperm. Protoplasma 177, 66-72. STRYER, L. (1999) BIOCHEMIE. S. 507-531, 4. Auflage, Spektrum Akademischer Verlag GmbH, Heidelberg, Berlin, Oxford. STUDIER, F.W., ROSENBERG, A.H., DUNN, J.J. & DUBENDORFF, J.W. (1990) The use of T7 polymerase to direct expression of cloned genes. Methods Enzymol. 185, 60-89. Literatur 148 SYGUSCH, J. & BEAUDRY, D. (1997) Subunit interaction in mammalian aldolases. Biochem. J. 323, 671-676. SYGUSCH, J., BEAUDRY, D. & ALLAIRE, M. (1987) Molecular architecture of rabbit skeletal muscle aldolase at 2.7-Å resolution. Proc. Natl. Acad. Sci. USA 84, 7846-7850. TAIZ, L. & ZEIGER, E. (2000) PHYSIOLOGIE DER PFLANZEN. S. 24-33, Spektrum Akademischer Verlag GmbH, Heidelberg, Berlin. TAKAGI, S. (2000) Roles for actin filaments in chloroplast motility and anchoring. In: ACTIN: A DYNAMIC FRAMEWORK FOR MULTIPLE PLANT CELL FUNCTIONS (Hrsg.: Staiger, C.J., Balu ka, F., Volkmann, D. & Barlow, P.) pp. 203-212, Kluwer Academic Publishers, Dordrecht, NL TAN, Z. & BOSS, W.F. (1992) Association of phosphatidylinositol kinase, phosphatidylinositol monophosphate kinase, and diacylglycerol kinase with the cytoskeleton and F-actin fractions of carrot (Daucus carota L.) cells grown in suspension culture. Plant Physiol. 100, 2116-2120. TANG, J.X., JANMEY, P.A., STOSSEL, T.P. & ITO, T. (1999) Thiol oxidation of actin produces dimers that enhance the elasticity of the F-actin network. Biophys. J. 76, 2208-2215. TAYLOR, S.W., FAHY, E., ZHANG, B., GLENN, G.M., WARNOCK, D.E., WILEY, S., MURPHY, A.N., GAUCHER, S.P., CAPALDI, R.A., GIBSON, B.W. & GHOSH, S.S. (2003) Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 21, 281-286. Literatur 149 TERPE, K. (2003) Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 60, 523-533. THOMAS, L., BLACHLY-DYSON, E., COLOMBINI, M. & FORTE, M. (1993) Mapping of residues forming the voltage sensor of the voltage-dependent anionselective channel. Proc. Natl. Acad. Sci. USA 90, 5446-5449. TOMPA, P., BÄR, J. & BATKE, J. (1986) Interaction of enzymes involved in triosephosphate metabolism. Eur. J. Biochem. 159, 117-124. URETA, T. (1978) The role of isoenzymes in metabolism: A model of metabolic pathways as the basis for the biological role of isoenzymes. Curr. Top. Cell Reg. 13, 233-251. UYEDA, K. (1992) Interactions of glycolytic enzymes with cellular membranes. Curr. Top. Cell Reg. 33, 31-46. VAN BREUSEGEM, F., DEKEYSER, R., GIELEN, J., VAN MONTAGU, M. & CAPLAN, A. (1994) Characterization of a S-adenosylmethionine synthetase gene in rice. Plant Physiol. 105, 1463-1464. VAN GESTEL, K., KÖHLER, R.H. & VERBELEN, J.P. (2002) Plant mitochondria move on F-actin, but their positioning in the cortical cytoplasm depends on both F-actin and microtubules. J. Exp. Bot. 53, 659-667. Literatur 150 VÉRTESSY, B.G., OROSZ, F., KOVÁCS, J. & OVÁDI, J. (1997) Alternative binding of two sequential glycolytic enzymes to microtubules. J. Biol. Chem. 272, 25542-25546. VITAVSKA, O., WIECZOREK, H. & MERZENDORFER, H. (2003) A novel role for subunit C in mediating binding of the H+-V-ATPase to the actin cytoskeleton. J. Biol. Chem. 278, 18499-18505 VOLKER, K.W. & KNULL, H.R. (1993) Glycolytic enzyme-tubulin interactions: Role of tubulin carboxy terminals. J. Mol. Recogn. 6, 167-177. VOLKER, K.W. & KNULL, H.R. (1997) A glycolytic enzyme binding domain on tubulin. Arch. Biochem. Biophys. 338, 237-243. VOLKMANN, D. & BALU KA, F. (1999) The actin cytoskeleton in plants: From transport networks to signalling networks. Microsc. Res. Techn. 47, 135-154. WALKER, J.C., HOWARD, E.A., DENNIS, E.S. & PEACOCK, W.J. (1987) DNA sequence required for anaerobic expression of the maize alcohol dehydrogenase 1 gene. Proc. Natl. Acad. Sci. USA 84, 6624-6628. WALSH, T.P., CLARKE, F.M. & MASTERS, C.J. (1977) Modification of the kinetic parameters of aldolase on binding to actin containing filaments of muscle. Biochem. J. 165, 165-167. WALSH, J.L., KEITH, T.J. & KNULL, H.R. (1989) Glycolytic enzyme interactions with tubulin and microtubules. Biochim. Biophys. Acta 999, 64-70. Literatur 151 WALTER, M., CHABAN, C., SCHÜTZE, K., BATISTIC, O., WECKERMANN, K., NÄKE, C., BLASEVIC, D., GREFEN, C., SCHUMACHER, K., OECKING, C., HARTER, K. & KUDLA, J. (2004) Visualization of protein interactions in living plant cells using bimolecular fluorescence complementation. Plant J. 40, 428-438. WANG, J., BOJA, E.S., TAN, W., TEKLE, E., FALES, H.M., ENGLISH, S., MIEYAL, J.J. & CHOCK, P.B. (2001) Reversible glutathionylation regulates actin polymerisation in A431 cells. J. Biol. Chem. 276, 47763-47766. WANG, J., MORRIS, A.J., TOLAN, D.R. & PAGLIARO, L. (1996) The molecular nature of the F-actin binding activity of aldolase revealed with sitedirected mutants. J. Biol. Chem. 271, 6861-6865. WANG, J., TOLAN, D.R. & PAGLIARO, L. (1997) Metabolic compartmentation in living cells: Structural association of aldolase. Exp. Cell Res. 237, 445-451. WEGNER, A. (1982) Treadmilling of actin at physiological salt concentrations. J. Mol. Biol. 161, 607-615. WELCH, G.R. (1977) On the role of organized multienzyme systems in cellular metabolism: A general synthesis. Prog. Biophys. Molec. Biol. 32, 103-191. WIESE, A., GRÖNER, F., SONNEWALD, U., DEPPNER, H., LERCHL, J., HEBBEKER, U., FLÜGGE, U.-I. & WEBER, A. (1999) Spinach hexokinase I is located in the outer envelope membrane of plastids. FEBS Lett. 461, 13-18. Literatur 152 WILLIAMSON, R.E. (1993) Organelle movements. Annu. Rev. Plant Physiol. Plant Mol. Biol. 44, 181-202. WILSON, J.E. (1980) Brain hexokinase, the prototype ambiquitous enzyme. Curr. Top. Cell Reg. 16, 1-54. WINKEL, B.S.J. (2004) Metabolic channelling in plants. Annu. Rev. Plant Biol. 55, 85-107. WINTER, H. & HUBER, S.C. (2000) Sucrose metabolism and the actin cytoskeleton: SuSy as actin-binding protein. In: ACTIN: A DYNAMIC FRAMEWORK FOR MULTIPLE PLANT CELL FUNCTIONS (Hrsg.: Staiger, C.J., Balu ka, F., Volkmann, D. & Barlow, P.) pp. 119-128, Kluwer Academic Publishers Dordrecht, NL WU, Y., MUENCH, D.G., KIM, Y.-T., HWANG, Y.-S. & OKITA, T.W. (1998) Identification of polypeptides associated with an enriched cytoskeleton-protein body fraction from developing rice endosperm. Plant Cell Physiol. 39, 1252-1257. WU, J. & FILUTOWICZ, M. (1999) Hexahistidine (His6)-tag dependent protein dimerization: A cautionary tale. Acta Biochim. Pol. 46, 591-599. XIE, G. & WILSON, J.E. (1990) Tetrameric structure of mitochondrially bound rat brain hexokinase: A crosslinking study. Arch. Biochem. Biophys. 276, 285-293. Literatur YANG, S.F. & HOFFMAN, N.E. (1984) Ethylene biosynthesis and its regulation in higher plants. Annu. Rev. Plant Physiol. 35, 155-189. YANG, C., MIN, G.W., TONG, X.J., LUO, Z.E. & ZHAI, Z.H. (1995) The assembly of keratins from higher plant cells. Protoplasma 188, 128-132. YELTMAN, D. R. & HARRIS, B.G. (1980) Localisation and membrane association of aldolase in human erythrocytes. Arch. Biochem. Biophys. 199, 186-196. ZIZI, M., FORTES, M., BLACHLY-DYSON, E. & COLOMBINI, M. (1994) NADH regulates the gating of VDAC, the mitochondrial outer membrane channel. J. Biol. Chem. 269, 1614-1616. 153 Danksagung 154 Ich danke... für die freundliche Aufnahme in die Arbeitsgruppe, die interessante Themenstellung und Bereitstellung des Arbeitsplatzes sowie für die Hilfe bei auftretenden Problemen und die Diskussionsbereitschaft Frau Prof. Dr. R. Scheibe, für das Vertrauen, das sie in mich gesetzt hat, und die Betreuung der Arbeit Frau Dr. H. Winter, Prof. Dr. A. von Schaewen, Dr. J. Backhausen, Dr. S. Holtgrefe und Dr. H. Merzendorfer für die Hilfe in praktischen Dingen, Prof. Dr. O. Bakker-Grunwald für die Unterstützung, allen Mitgliedern der Arbeitsgruppe für eine schöne Zeit in kollegialer Umgebung, besonders hervorgehoben seien hier: Gudrun Kemper, die mich geduldig in die Laborarbeit eingeführt hat, Bianca Altmann, die immer zur Stelle war, wenn ich sie brauchte, Susanne Klocke, ohne die viele Dinge nicht so reibungslos gelaufen wären, Heike Wolf-Wibbelmann, die sich um die Pflanzen gekümmert hat, Daniela Holtgräwe, die mit mir gemeinsam diese Strecke gegangen ist, und Julia Pelz für die schöne gemeinsame Zeit und Unterstützung, für die ausdauernde private Unterstützung meinen Eltern Brigitte und Guntram Thilo, meiner Schwester Dörte Thilo und vor allem meinem Mann Thorsten Scholz, der Deutschen Forschungsgemeinschaft für die Gewährung eines Stipendiums im Rahmen des Graduiertenkollegs 612. Anhang 155 7 Anhang 7.1 Abkürzungsverzeichnis A Absorption ABP/ABPs Aktin-Bindeprotein/Aktin-Bindeproteine AD Aktivierungsdomäne Ade L-Adenin ADP Adenosin-5’-diphosphat AHT Anhydrotetracyclin-Hydrochlorid ALD Aldolase Amp Ampicillin ATP Adenosin-5’-triphosphat BD Bindedomäne BLAST „Basic Local Alignment Search Tool“ bp Basenpaar BSA Rinderserumalbumin CC kritische Konzentration cDNA komplementäre DNA Clm Chloramphenicol Da Dalton DEPC Diethyl-Pyrocarbonat DF Durchfluss DMF Dimethylformamid DMSO Dimethyl Sulfoxid DNA Desoxyribonucleinsäure ds doppelsträngig DTT Dithiothreitol E Elutionsschritt EDTA Ethylendiamintetraessigsäure EF-1 „elongation factor 1 “ ELISA „Enzyme linked immunosorbent assey“, Enzymimmunoassay Exp. Experiment FBP Fructose-1,6-bisphosphat F-6P Fructose-6-phosphat Anhang 156 F-2,6BP Fructose-2,6-bisphosphat g Erdbeschleunigung GAPDH Glycerinaldehyd-3-phosphat Dehydrogenase GDH Glycerin-3-phosphat Dehydrogenase gi GenBank-Identifizierungsnummer GSH reduziertes Glutathion GSSG oxidiertes Glutathion GTP Guanosintriphosphat His L-Histidin IPTG Isopropyl β-D-Thiogalaktopyranosid Kan Kanamycin KM Michaelis-Menten Konstante LD-PCR „long distance-PCR“ Leu L-Leucin Lys L-Lysin M Molar Ma Marker, Molekularmassenstandard MOPS 3-(N-Morpholino)-propan-sulfonat mRNA „messenger-RNA“, Boten-RNA MW Mittelwert n Nano NAD+ Nicotinamid-adenin-dinucleotid NADP Nicotinamid-adenin-dinucleotidphosphat Ni-NTA Nickel-Nitrilotriessigsäure OD optische Dichte ox oxidiertes p Piko P Pellet PAGE Polyacrylamid-Gelelektrophorese PCR „polymerase chain reaction“, Polymerasekettenreaktion PEP Phosphoenolpyruvat PFK Phosphofructokinase PGK Phosphoglycerat-Kinase Pi anorganisches Phosphat Anhang 157 QDO „Quadruple Dropout“ RE Rohextrakt red reduziertes RNA Ribonucleinsäure RT Raumtemperatur RT-PCR Reverse Transkriptase-PCR SAM-S S-Adenosyl-L-Methionin-Synthetase SD Minimalmedium Sd Standardabweichung SDS Natriumdodecylsulfat SNP „single nucleotid polymorphism“ TDO „Triple Dropout“ Tet Tetrazyclin TPI Triosephosphat-Isomerase Tris Tris-(hydroxymethyl)-amino-methan Trp L-Tryptophan ÜS Überstand Upm Umdrehungen pro Minute V Volt var. Varietät VDAC spannungsabhängiger Anionenkanal Vmax maximale Reaktionsgeschwindigkeit Vol Volumen v/v Volumen pro Volumen W Waschschritt w/v Gewicht pro Volumen X- -Gal 5-Bromo-4-chloro-3-indolyl- -D-Galaktopyranosid X-Gal 5-Bromo-4-chloro-3-indolyl-β-D-Galaktopyranosid Signifikanzniveau µ mikro unendlich sx2 Varianz Anhang 158 7.2 Ermittelte Sequenzdaten der Expressionskonstrukte 7.2.1 Nukleinsäuresequenz des Expressionskonstruktes ALD_pET-16b Dargestellt ist die Nukleinsäuresequenz des Konstruktes ALD_pET-16b mit N-terminalem His-tag (unterstrichen) und dem Start- und Stopcodon der für cytosolische Aldolase aus Mais kodierenden Sequenz (grau unterlegt). CATCATCATCATCATCATCATCATCATCACAGCAGCGGCCATATCGAAGGTCGTCATATG TCGGCCTACTGCGGAAAGTACAAGGATGAGCTCATCAAGAATGCTGCCTACATTGGCACC CCTGGCAAGGGTATCCTTGCTGCTGATGAGTCCACTGGCACCATTGGCAAGCGCCTTTCC AGCATCAATGTCGAGAACGTTGAGGAGAACCGCCGTGCCCTCCGTGAGCTCCTATTCTGC TGCCCTGGTGCTCTCCAGTACATCAGCGGTGTGATCCTCTTCGAGGAGACCCTGTACCAG AAGACCAAGGATGGCAAGCCTTTTGTTGATGTCCTCAAGGAGGGAGGCGTCCTCCCTGGC ATCAAGGTTGACAAGGGCACCATTGAGGTTGTTGGCACTGATAAGGAGACCACCACCCAA GGCCATGACGACCTTGGCAAGCGCTGCGCCAAGTACTACGAGGCAGGTGCCCGCTTTGCC AAGTGGCGCGCTGTTCTCAAGATTGGCCCTAATGAGCCATCACAGCTTGCCATCGACCTG AACGCTCAGGGTCTAGCTCGCTACGCCATCATCTGCCAGGAGAATGGTCTGGTGCCAATT GTTGAGCCTGAGATCCTTGTTGATGGCCCTCATGACATTGATCGCTGTGCTTACGTCACT GAGACCGTCCTTGCTGCCTGCTACAAGGCGCTCAACGAGCACCATGTCCTCCTGGAGGGT ACCCTCCTGAAGCCCAACATGGTGACTCCAGGCTCCGACTCCAAGAAGGTGACCCCTGAG GTGATTGCTGAGTACACCGTCCGTACCCTCCAGAGGACCGTACCTGCTGCTGTGCCTGCT GTTGTTTTCCTCTCTGGTGGACAGAGCGAGGAGGAGGCCACCCGCAACCTCAATGCCATG AACAAGCTCAGCACCAAGAAGCCGTGGTCCCTGTCTTTCTCCTTCGGCCGTGCCCTCCAG GCGAGCACCCTCAAGGCCTGGGCTGGCAAGGTGGAGAACTTGGAGAAGGCTAGAGCTGCC TTCCTCGCCAGGTGCAAGGCCAACTCTGAGGCTACCCTCGGCACCTACAAGGGTGATGCT GCCGCCGACACCGAGAGCCTCCACGTCAAGGACTACAAGTACTGA 7.2.2 Abgeleitete Aminosäuresequenz des Expressionskonstruktes ALD_pET-16b Dargestellt ist die Aminosäuresequenz des Konstruktes ALD_pET-16b mit N-terminalem His-tag (unterstrichen) und dem Start-Methionin (M) der cytosolische Aldolase aus Mais. H I N F K G R Y T A L L K H K V E G A Y V P V S A Y H H H N A A E N V E T L T I E R F A A I I T E T G S D V F L F S F R C K Stop H Y E Y V K C V S S G A H I E Q V W Q L K G R N H G N K G R E A K G A S H T R T T A N A V Q L E H P R K D V G C T S Q A S G A D K L L Y P E A T S K L G E K V K E E S L G G R K T I P A V E T G H I E P T G I L I A L T I L L F T P V N A T K Y E A L V Q N E E E R A K G A F D G E P H Y N W G R D C V H P E H T L A D H E C L D S I V V N G A M S P K D Q L L R A K A S T G E L L V L T M V A A G A G G A D E L N E D Y T L G K I G G Q K N T C I Q V R D P T R L L E G G Y L C L H L T S E S K K I P A N D L V T K L Y R S G K A I K P K A H K L G I Y Q D P A K R V D S V K Y G R N A P A K E S I V E L C M V W A D L I L D A A A V P S F Y Anhang 159 7.2.3 Nukleinsäuresequenz des Expressionskonstruktes ALD_pASK-IBA3 Dargestellt ist die Nukleinsäuresequenz des Konstruktes ALD_pASK-IBA3 mit C-terminalem Strep-tag (unterstrichen) und dem Startcodon der für cytosolische Aldolase aus Mais kodierenden Sequenz (grau unterlegt). ATGTCGGCCTACTGCGGAAAGTACAAGGATGAGCTCATCAAGAATGCTGCCTACATTGGC ACCCCTGGCAAGGGTATCCTTGCTGCTGATGAGTCCACTGGCACCATTGGCAAGCGCCTT TCCAGCATCAATGTCGAGAACGTTGAGGAGAACCGCCGTGCCCTCCGTGAGCTCCTATTC TGCTGCCCTGGTGCTCTCCAGTACATCAGCGGTGTGATCCTCTTCGAGGAGACCCTGTAC CAGAAGACCAAGGATGGCAAGCCTTTTGTTGATGTCCTCAAGGAGGGAGGCGTCCTCCCT GGCATCAAGGTTGACAAGGGCACCATTGAGGTTGTTGGCACTGATAAGGAGACCACCACC CAAGGCCATGACGACCTTGGCAAGCGCTGCGCCAAGTACTACGAGGCAGGTGCCCGCTTT GCCAAGTGGCGCGCTGTTCTCAAGATTGGCCCTAATGAGCCATCACAGCTTGCCATCGAC CTGAACGCTCAGGGTCTAGCTCGCTACGCCATCATCTGCCAGGAGAATGGTCTGGTGCCA ATTGTTGAGCCTGAGATCCTTGTTGATGGCCCTCATGACATTGATCGCTGCGCTTACGTC ACTGAGACCGTCCTTGCTGCCTGCTACAAGGCGCTCAACGAGCACCATGTCCTCCTGGAG GGTACCCTCCTGAAGCCCAACATGGTGACTCCAGGCTCCGACTCCAAGAAGGTGACCCCT GAGGTGATTGCTGAGTACACCGTCCGTACCCTCCAGAGGACCGTACCTGCTGCTGTGCCT GCTGTTGTCTTCCTCTCTGGTGGACAGAGCGAGGAGGAGGCCACCCGCAACCTCAATGCC ATGAACAAGCTCAGCACCAAGAAGCCGTGGTCCCTGTCTTTCTCCTTCGGCCGTGCCCTC CAGGCGAGCACCCTCAAGGCCTGGGCTGGCAAGGTGGAGAACTTGGAGAAGGCTAGAGCT GCCTTCCTCGCCAGGTGCAAGGCCAACTCTGAGGCTACCCTCGGCACCTACAAGGGTGAT GCTGCCGCCGACACCGAGAGCCTCCACGTCAAGGACTACAAGTACAGCGCTTGGAGCCAC CCGCAGTTCGAAAAATAA 7.2.4 Abgeleitete Aminosäuresequenz des Konstruktes ALD_pASK-IBA3 Dargestellt ist die Aminosäuresequenz des Konstruktes ALD_pASK-IBA3 mit C-terminalem Strep-tag (unterstrichen) und dem Start-Methionin (M) der cytosolische Aldolase aus Mais. M S P K D Q L L R A K A S T G E L L V L T M V A A G A G G A D E L N E D Y T L G K I G G Q K N T C I Q V R D P T R L L E G G Y L C L H L T S E S K K I P A N D L V T K L Y R S G K A I K P K A H K L G I Y Q D P A K R V D S V K Y G R N A P A K E S I V E L C M V W A D L I L D A A A V P S F Y I N F K G R Y T A L L K K V E G A Y V P V S A Y N E E T R A T G V F R S A N T I F I E S F S C A A V L E A I T D L F K W Y E Y V K C V S S G A S I E Q V W Q L K G R N H G N K G R E A K G A S P T R T T A N A V Q L E Q P R K D V G C T S Q A F G A D K L L Y P E A T E K L G E K V K E E S L K G I L R E L K P F T T T I G P P I V A L N V I A E A T T L K G T Y Stop A L V Q N E E E R A K A F D G E P H Y N W G D C V H P E H T L A D E C L D S I V V N G A Anhang 160 7.2.5 Schematische Dartsellung der Aldolase-Expressionskonstrukte Schematische Darstellung der Expressionskonstrukte ALD_pET-16b und ALD_pASK-IBA3 mit His- bzw. Strep-tag zur Reinigung nach heterologer Expression. His-tag ALD_pET-16b HHHHHHHHHH N C Aldolase Strep-tag ALD_pASK-IBA3 Aldolase WSHPQFEK 7.2.6 Nukleinsäuresequenz des Expressionskonstruktes VDAC_pRSET A Dargestellt ist die Nukleinsäuresequenz des Konstruktes VDAC_pRSET A mit N-terminalem His-tag (unterstrichen) und dem Start- und Stopcodon der codierenden Sequenz für VDAC aus Mais (grau unterlegt). CATCATCATCATCATCATGGTATGGCTAGCATGACTGGTGGACAGCAAATGGGTCGGGAT CTGTACGACGATGACGATAAGGATCGATGGGGATCCATGGTCGGGCCAGGCCTCTACACC GAGATCGGCAAGAAGACCAGGGATCTGCTGTACAAGGACTACCAGACTGACCACAAGTTC ACCCTCACTACCTACACCTCCAATGGCGTCGCTGTAACTGCTTCTGGCACAAAGAAAGCT GACCTGATCCTTGGCGAGATCCAATCACAGATAAAGAACAAGAACATGACCATAGATGTG AAAGCAAACTCGGAGTCAAATATCATTACGACAATTACTGTTGATGAGATTGCAACACCA GGGCTGAAGACCATCTTAAGTTTTGCTGTTCCTGATCAGAGATCTGGAAAGGTTGAGCTC CAGTATTTGCATGATTATGCTGGAGTTAATGCAAGCATCGGTCTGACAGCCAATCCTGTA GTCAACCTCTCTGCTGCCTTTGGTACTAAGGCTGTTGCAGTTGGTGCCGATATCTCACTT GATACTGCAACTGGGAACTTGACTAAATACAATGCTGCACTGAGGTTCACTCATGAAGAC CTTGTCGCATCCCTGAACCTGAACAACAAGGGAGACAGCCTTACTGGAGCCTACTATCAC AAGGTGAGCGAATTGACCAACACAGCTGTTGGGGCAGAGCTTACCCACAGCTTCTCGACC AATGAAAACACCCTCACCTTTGGCAGGCAGCACGCCCTGGACCCACTAACCGTCTTGAAG GCCCGCATTAACAACTCTGGCAAGGCCAGTGCCCTGATCCAGCACGAGTGGAGGCCAAAG TCGCTGGTCACCATCTCGGCCGAGGTCGACACAAAGACCATCGAGAAGAGCTCGAAGGTT GGGATTGCTGTGGCCCTCAAGCCCTGA 7.2.7 Abgeleitete Aminosäuresequenz des Konstruktes VDAC_pRSET A Dargestellt ist die Aminosäuresequenz des Konstruktes VDAC_pRSET A mit N-terminalem His-tag (unterstrichen) und dem Start-Methionin (M) von VDAC aus Mais. HHHHHHGMASMTGGQQMGRDLYDDDDKDRWGSMVGPGLY TEIGKKTRDLLYKDYQTDHKFTLTTYTSNGVAVTASGTKKA DLILGEIQSQIKNKNMTIDVKANSESNIITTITVDEIATPGLKT ILSFAVPDQRSGKVELQYLHDYAGVNASIGLTANPVVNLSA AFGTKAVAVGADISLDTATGNLTKYNAALRFTHEDLVASLN LNNKGDSLTGAYYHKVSELTNTAVGAELTHSFSTNENTLTF GRQHALDPLTVLKARINNSGKASALIQHEWRPKSLVTISAEV D T K T I E K S S K V G I A V A L K P Stop Anhang 161 7.2.8 Sequenzvergleiche der Expressionskonstrukte In den folgenden drei Abbildungen sind die Ergebnisse der Sequenzvergleiche der Expressionskonstrukte mit den in der Datenbank GenBank veröffentlichten Nukleinsäuresequenzen dargestellt. ALD 8 1 MZEADL 22 ALD 8 61 MZEADL 82 ALD 8 121 MZEADL 142 ALD 8 181 MZEADL 202 ALD 8 241 MZEADL 262 ALD 8 301 MZEADL 322 ALD 8 361 MZEADL 382 ALD 8 421 MZEADL 442 ALD 8 481 MZEADL 502 ALD 8 541 MZEADL 562 ALD 8 601 MZEADL 622 ALD 8 661 MZEADL 682 ALD 8 721 MZEADL 742 atgtcggcctactgcggaaagtacaaggatgagctcatcaagaatgctgcctacattggc 60 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| atgtcggcctactgcggaaagtacaaggatgagctcatcaagaatgctgcctacattggc 81 acccctggcaagggtatccttgctgctgatgagtccactggcaccattggcaagcgcctt 120 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| acccctggcaagggtatccttgctgctgatgagtccactggcaccattggcaagcgcctt 141 tccagcatcaatgtcgagaacgttgaggagaaccgccgtgccctccgtgagctcctattc 180 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| tccagcatcaatgtcgagaacgttgaggagaaccgccgtgccctccgtgagctcctattc 201 tgctgccctggtgctctccagtacatcagcggtgtgatcctcttcgaggagaccctgtac 240 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| tgctgccctggtgctctccagtacatcagcggtgtgatcctcttcgaggagaccctgtac 261 cagaagaccaaggatggcaagccttttgttgatgtcctcaaggagggaggcgtcctccct 300 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| cagaagaccaaggatggcaagccttttgttgatgtcctcaaggagggaggcgtcctccct 321 ggcatcaaggttgacaagggcaccattgaggttgttggcactgataaggagaccaccacc 360 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ggcatcaaggttgacaagggcaccattgaggttgttggcactgataaggagaccaccacc 381 caaggccatgacgaccttggcaagcgctgcgccaagtactacgaggcaggtgcccgcttt 420 ||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||| caaggccatgacgaccttggcaagcgctgcgccaagtactacgaggccggtgcccgcttt 441 gccaagtggcgcgctgttctcaagattggccctaatgagccatcacagcttgccatcgac 480 |||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||| gccaagtggcgcgctgttctcaagattggccccaatgagccatcacagcttgccatcgac 501 ctgaacgctcagggtctagctcgctacgccatcatctgccaggagaatggtctggtgcca 540 ||||||||||||||||| |||||||| ||||||||||||||||||||||||||||||||| ctgaacgctcagggtctggctcgctatgccatcatctgccaggagaatggtctggtgcca 561 attgttgagcctgagatccttgttgatggccctcatgacattgatcgctgtgcttacgtc 600 |||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||| attgttgagcctgagatccttgttgatggccctcatgacattgatcgctgcgcttacgtc 621 actgagaccgtccttgctgcctgctacaaggcgctcaacgagcaccatgtcctcctggag 660 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| actgagaccgtccttgctgcctgctacaaggcgctcaacgagcaccatgtcctcctggag 681 ggtaccctcctgaagcccaacatggtgactccaggctccgactccaagaaggtgacccct 720 |||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||| ggtaccctcctgaagcccaacatggtgactccaggctccgactccaagaaggtgactcct 741 gaggtgattgctgagtacaccgtccgtaccctccagaggaccgtacctgctgctgtgcct 780 |||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||| gaggtgattgctgagtacaccgtccgtaccctccagaggaccgtccctgctgctgtgcct 801 Anhang ALD 8 162 781 MZEADL 802 ALD 8 901 MZEADL 922 ALD 8 961 MZEADL 982 gctgttgttttcctctctggtggacagagcgaggaggaggccacccgcaacctcaatgcc 840 |||||| | ||||||||||||||||||||||||||||||||||||||||||||||||||| gctgttctcttcctctctggtggacagagcgaggaggaggccacccgcaacctcaatgcc 861 caggcgagcaccctcaaggcctgggctggcaaggtggagaacttggagaaggctagagct 960 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| caggcgagcaccctcaaggcctgggctggcaaggtggagaacttggagaaggctagagct 981 gccttcctcgccaggtgcaaggccaactctgaggctaccctcggcacctacaagggtgat 1020 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| gccttcctcgccaggtgcaaggccaactctgaggctaccctcggcacctacaagggtgat 1041 ALD 8 1021 gctgccgccgacaccgagagcctccacgtcaaggactacaagtactga 1068 |||||||||||||||||||||||||||||||||||||||||||||||| MZEADL 1042 gctgccgccgacaccgagagcctccacgtcaaggactacaagtactga 1089 Abb. 8.1: Sequenzvergleich der Aldolase des Expressionskonstruktes ALD_pET-16b mit der cytosolischen Aldolase aus Mais (MZEALD). Der Sequenzvergleich erfolgte mit dem BLAST-Programm gegen die Datenbank GenBank. Die grau unterlegten Bereiche zeigen ungleiche Nukleotide an. Durch das rot markierte Nukleotid kommt es zu einem Aminosäureaustausch in der Proteinsequenz. Anhang ALD-IBA3 163 1 MZEALD 22 ALD-IBA3 61 MZEALD 82 ALD-IBA3 121 MZEALD 142 ALD-IBA3 181 MZEALD 202 ALD-IBA3 241 MZEALD 262 ALD-IBA3 301 MZEALD 322 ALD-IBA3 361 MZEALD 382 ALD-IBA3 421 MZEALD 442 ALD-IBA3 481 MZEALD 502 ALD-IBA3 541 MZEALD 562 ALD-IBA3 601 MZEALD 622 ALD-IBA3 661 MZEALD 682 ALD-IBA3 721 MZEALD 742 ALD-IBA3 781 MZEALD 802 ALD-IBA3 841 MZEALD 862 atgtcggcctactgcggaaagtacaaggatgagctcatcaagaatgctgcctacattggc 60 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| atgtcggcctactgcggaaagtacaaggatgagctcatcaagaatgctgcctacattggc 81 acccctggcaagggtatccttgctgctgatgagtccactggcaccattggcaagcgcctt 120 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| acccctggcaagggtatccttgctgctgatgagtccactggcaccattggcaagcgcctt 141 tccagcatcaatgtcgagaacgttgaggagaaccgccgtgccctccgtgagctcctattc 180 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| tccagcatcaatgtcgagaacgttgaggagaaccgccgtgccctccgtgagctcctattc 201 tgctgccctggtgctctccagtacatcagcggtgtgatcctcttcgaggagaccctgtac 240 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| tgctgccctggtgctctccagtacatcagcggtgtgatcctcttcgaggagaccctgtac 261 cagaagaccaaggatggcaagccttttgttgatgtcctcaaggagggaggcgtcctccct 300 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| cagaagaccaaggatggcaagccttttgttgatgtcctcaaggagggaggcgtcctccct 321 ggcatcaaggttgacaagggcaccattgaggttgttggcactgataaggagaccaccacc 360 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ggcatcaaggttgacaagggcaccattgaggttgttggcactgataaggagaccaccacc 381 caaggccatgacgaccttggcaagcgctgcgccaagtactacgaggcaggtgcccgcttt 420 ||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||| caaggccatgacgaccttggcaagcgctgcgccaagtactacgaggccggtgcccgcttt 441 gccaagtggcgcgctgttctcaagattggccctaatgagccatcacagcttgccatcgac 480 |||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||| gccaagtggcgcgctgttctcaagattggccccaatgagccatcacagcttgccatcgac 501 ctgaacgctcagggtctagctcgctacgccatcatctgccaggagaatggtctggtgcca 540 ||||||||||||||||| |||||||| ||||||||||||||||||||||||||||||||| ctgaacgctcagggtctggctcgctatgccatcatctgccaggagaatggtctggtgcca 561 attgttgagcctgagatccttgttgatggccctcatgacattgatcgctgcgcttacgtc 600 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| attgttgagcctgagatccttgttgatggccctcatgacattgatcgctgcgcttacgtc 621 actgagaccgtccttgctgcctgctacaaggcgctcaacgagcaccatgtcctcctggag 660 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| actgagaccgtccttgctgcctgctacaaggcgctcaacgagcaccatgtcctcctggag 681 ggtaccctcctgaagcccaacatggtgactccaggctccgactccaagaaggtgacccct 720 |||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||| ggtaccctcctgaagcccaacatggtgactccaggctccgactccaagaaggtgactcct 741 gaggtgattgctgagtacaccgtccgtaccctccagaggaccgtacctgctgctgtgcct 780 |||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||| gaggtgattgctgagtacaccgtccgtaccctccagaggaccgtccctgctgctgtgcct 801 gctgttgtcttcctctctggtggacagagcgaggaggaggccacccgcaacctcaatgcc 840 |||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||| gctgttctcttcctctctggtggacagagcgaggaggaggccacccgcaacctcaatgcc 861 atgaacaagctcagcaccaagaagccgtggtccctgtctttctccttcggccgtgccctc 900 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| atgaacaagctcagcaccaagaagccgtggtccctgtctttctccttcggccgtgccctc 921 Anhang 164 ALD-IBA3 901 MZEALD 922 ALD-IBA3 961 MZEADL 982 ALD-IBA3 1021 MZEADL 1042 caggcgagcaccctcaaggcctgggctggcaaggtggagaacttggagaaggctagagct 960 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| caggcgagcaccctcaaggcctgggctggcaaggtggagaacttggagaaggctagagct 981 gccttcctcgccaggtgcaaggccaactctgaggctaccctcggcacctacaagggtgat 1020 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| gccttcctcgccaggtgcaaggccaactctgaggctaccctcggcacctacaagggtgat 1041 gctgccgccgacaccgagagcctccacgtcaaggactacaagtac 1065 ||||||||||||||||||||||||||||||||||||||||||||| gctgccgccgacaccgagagcctccacgtcaaggactacaagtac 1086 Abb. 8.2: Sequenzvergleich der Aldolase des Expressionskonstruktes ALD_pASK-IBA3 mit der cytosolischen Aldolase aus Mais (MZEALD). Der Sequenzvergleich erfolgte mit dem BLAST-Programm gegen die Datenbank GenBank. Die grau unterlegten Bereiche zeigen ungleiche Nukleotide an. Durch das rot markierte Nukleotid kommt es zu einem Aminosäureaustausch in der Proteinsequenz. Anhang vdac 165 1 vdac1a 93 vdac atggtcgggccaggcctctacaccgagatcggcaagaagaccagggatctgctgtacaag 60 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| atggtcgggccaggcctctacaccgagatcggcaagaagaccagggatctgctgtacaag 152 61 gactaccagactgaccacaagttcaccctcactacctacacctccaatggcgtcgctgta 120 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| vdac1a 153 gactaccagactgaccacaagttcaccctcactacctacacctccaatggcgtcgctgta 212 vdac 121 actgcttctagcacaaagaaagctgacctgatccttggcgagatccaatcacagataaag 180 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| vdac1a 213 actgcttctagcacaaagaaagctgacctgatccttggcgagatccaatcacagataaag 272 vdac 181 aacaagaacatgaccatagatgtgaaagcaaactcggagtcaaatatcattacaacaatt 240 ||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||| vdac1a 273 aacaagaacatgaccatagatgtgaaagcaaactcggagtcaaatatcattacgacaatt 332 vdac 241 actgttgatgagattgcaacaccagggctgaagaccatcttaagttttgctgttcctgat 300 |||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||| vdac1a 333 actgttgatgagattgcaacaccagggctgaagaccatcttaagctttgctgttcctgat 392 vdac 301 cagagatctggaaaggttgagctccagtatttgcatgattatgctggagttaatgcaagc 360 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| vdac1a 393 cagagatctggaaaggttgagctccagtatttgcatgattatgctggagttaatgcaagc 452 vdac 361 atcggtctgacagccaatcctgtagtcaacctctctgctgcctttggtactaaggctgtt 420 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| vdac1a 453 atcggtctgacagccaatcctgtagtcaacctctctgctgcctttggtactaaggctgtt 512 vdac 421 gcagttggtgccgatatctcacttgatactgcaactgggaacttgactaaatacaatgct 480 ||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||| vdac1a 513 gcacttggtgccgatatctcacttgatactgcaactgggaacttgactaaatacaatgct 572 vdac 481 gcactgaggttcactcatgaagaccttgtcgcatccctgaacctgaacaacaagggcgac 540 |||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||| vdac1a 573 gcactgaggttcactcatgaagaccttgtcgcatccctgaacctgaacaacaagggagac 632 vdac 541 agccttactggagcctactatcacaaggtgagcgaattgaccaacacagctgttggggca 600 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| vdac1a 633 agccttactggagcctactatcacaaggtgagcgaattgaccaacacagctgttggggca 692 vdac 601 gagcttacccacagcttctcgaccaatgaaaacaccctcacctttggcgggcagcacgcc 660 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| vdac1a 693 gagcttacccacagcttctcgaccaatgaaaacaccctcacctttggcgggcagcacgcc 752 vdac 661 ctggacccactaaccgtcttgaaggcccgcattaacaaatctggcaaggccagtgccctg 720 |||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||| vdac1a 753 ctggacccactaaccgtcttgaaggcccgcattaacaactctggcaaggccagtgccctg 812 vdac 721 atccagcacgagtggaggccaaagtcgctggtcaccatctcggccgaggtcgacacaaag 780 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| vdac1a 813 atccagcacgagtggaggccaaagtcgctggtcaccatctcggccgaggtcgacacaaag 872 vdac 781 accatcgagaagagctcgaaggttgggattggtgtggccctcaagccctga 831 ||||||||||||||||||||||||||||||| ||||||||||||||||||| vdac1a 873 accatcgagaagagctcgaaggttgggattgctgtggccctcaagccctga 923 Abb. 8.3: Sequenzvergleich des spannungsabhängigen Anionenkanalproteins VDAC (vdac) mit der Sequenz der Datenbank GenBank (vdac1a). Der Sequenzvergleich erfolgte mit dem BLAST-Programm. Die grau unterlegten Bereiche zeigen ungleiche Nukleotide an. Durch rot markierte Nukleotide kommt es zu einem Aminosäureaustausch in der Proteinsequenz. Anhang 166 7.3 Sequenzen der pGADT7-cDNA-Bank-Konstrukte Im Folgenden sind die ermittelten Sequenzen der pGADT7-cDNA-Bank-Konstrukte aus den blauen Hefekolonien aufgelistet. Sequenzen, die mehrfach identifiziert wurden, sind beispielhaft nur einmal dargestellt und zu jeder Sequenz ist die entsprechende BLASTAnalyse abgebildet. Unterstreichungen kennzeichnen Teile der Vektorensequenz. Sequenzen der pGADT7-cDNA-Bank-„Inserts der Hefekolonien 2.4, 2.5 und 2.7: CCATTATGGCCGGGAACAAAACCTGTCTTTAACGGGATGGTACTTACTTTCATACAGGTG CTGCATGGCTGTCGTCAGCTCGTGTCGTGAGATGTTTGGTCAAGTCCTATAACGAGCGAA ACCCTCGTTTTGTGTTGCTGAGACATGCGCCTAAAAAAAAAAAAAAAAAAAAAAAAAAAA >gi|13923|emb|X00794.1|MIZMRN01 Maize mitochondrial gene for 18S rRNA Length = 1968 Score = 278 bits (140), Expect = 2e-75 Identities = 140/140 (100%) Strand = Plus / Plus Query: 2 aacaaaacctgtctttaacgggatggtacttactttcatacaggtgctgcatggctgtcg |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 1053 aacaaaacctgtctttaacgggatggtacttactttcatacaggtgctgcatggctgtcg Query: 62 tcagctcgtgtcgtgagatgtttggtcaagtcctataacgagcgaaaccctcgttttgtg |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 1113 tcagctcgtgtcgtgagatgtttggtcaagtcctataacgagcgaaaccctcgttttgtg Query: 122 ttgctgagacatgcgcctaa 141 |||||||||||||||||||| Sbjct: 1173 ttgctgagacatgcgcctaa 1192 Sequenzen der pGADT7-cDNA-Bank-„Inserts der Hefekolonien 2.17 und 7.243: GTATGTGNTTTANCCCCTTTTCCTGCAGTACCCATACGACGTACCAGATTACGCTCATAT GGCCATGGAGGCCAGTGAATTCCACCCAAGCAGTGGTATCAACGCAGAGTGGCCATTATG GCCGGGGGCCTTAACCATTCCTCCCAACACCACCTCGCCGCGATCTCCGTAGCCTCCGTC GCGTCGAGCATCGATCATGTCGGCCTACTGCGGAAAGTACAAGGATGAGCTCATCAAGAA TGCTGCCTACATTGGCACCCCTGGCAAGGGTATCCTTGCTGCTGATGAGTCCACTGGCAC CATTGGCAAGCGCCTTTCCAGCATCAATGTCGAGAACGTTGAGGAGAACCGCCGTGCCCT CCGTGAGCTCCTATTCTGCTGCCCTGGTGCTCTCCAGTACATCAGCGGTGTGATCCTCTT CGAGGAGACCCTGTACCAGAAGACCAAGGATGGCAAGCCTTTTGTTGATGTCCTCAAGGA GGGAGGCGTCCTCCCTGGCATCAAGGTTGACAAGGGCACCATTGAGGTTGTTGGCACTGA TAAGGAGACCACCACCCAAGGCCATGACGACCTTGGCAAGCGCTGCGCCAAGTACTACGA GGCAGGTGCCCGCTTTGCCAAGTGGCGCGCTGTTCTCAAGATTGGCCCTAATGAGCCATC ACAGCTTGCCATCGACCTGAACGCTCANGGTCTAGCTCGCTACGCCATCATCTGCCAGGA GAATGGTCTGGTGCCAATTGTTGAGCCTGAGATCCTTGTTGATGGCCCTCATGACATTGA TCGTGTNTTACGTACTGANNCGTCTTGTGCTGTCAAGCGCTCAACAGCACCATGTNCTCT GAGGTCCCTCTGAGCCCACTGNGACTCAGCTCGNTCAAAAGNGACCCTGAGNGATGTGAT CACGNCNACCTCAAGACGACCTGTGTGGCTGTGTGTTCTTTGNGAAANCAGAGAGCCCCN CACTATGCTGACAC Anhang 167 >gi|168419|gb|M16220.1|MZEALD Maize (Z.mays) aldolase mRNA, complete cds Length = 1388 Score = 1168 bits (589), Expect = 0.0 Identities = 603/608 (99%) Strand = Plus / Plus Query: 176 ccgtcgcgtcgagcatcgatcatgtcggcctactgcggaaagtacaaggatgagctcatc 235 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 1 ccgtcgcgtcgagcatcgatcatgtcggcctactgcggaaagtacaaggatgagctcatc 60 Query: 236 aagaatgctgcctacattggcacccctggcaagggtatccttgctgctgatgagtccact 295 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 61 aagaatgctgcctacattggcacccctggcaagggtatccttgctgctgatgagtccact 120 Query: 296 ggcaccattggcaagcgcctttccagcatcaatgtcgagaacgttgaggagaaccgccgt 355 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 121 ggcaccattggcaagcgcctttccagcatcaatgtcgagaacgttgaggagaaccgccgt 180 Query: 356 gccctccgtgagctcctattctgctgccctggtgctctccagtacatcagcggtgtgatc 415 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 181 gccctccgtgagctcctattctgctgccctggtgctctccagtacatcagcggtgtgatc 240 Query: 416 ctcttcgaggagaccctgtaccagaagaccaaggatggcaagccttttgttgatgtcctc 475 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 241 ctcttcgaggagaccctgtaccagaagaccaaggatggcaagccttttgttgatgtcctc 300 Query: 476 aaggagggaggcgtcctccctggcatcaaggttgacaagggcaccattgaggttgttggc 535 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 301 aaggagggaggcgtcctccctggcatcaaggttgacaagggcaccattgaggttgttggc 360 Query: 536 actgataaggagaccaccacccaaggccatgacgaccttggcaagcgctgcgccaagtac 595 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 361 actgataaggagaccaccacccaaggccatgacgaccttggcaagcgctgcgccaagtac 420 Query: 596 tacgaggcaggtgcccgctttgccaagtggcgcgctgttctcaagattggccctaatgag 655 |||||||| |||||||||||||||||||||||||||||||||||||||||||| |||||| Sbjct: 421 tacgaggccggtgcccgctttgccaagtggcgcgctgttctcaagattggccccaatgag 480 Query: 656 ccatcacagcttgccatcgacctgaacgctcanggtctagctcgctacgccatcatctgc 715 |||||||||||||||||||||||||||||||| ||||| |||||||| |||||||||||| Sbjct: 481 ccatcacagcttgccatcgacctgaacgctcagggtctggctcgctatgccatcatctgc 540 Query: 716 caggagaatggtctggtgccaattgttgagcctgagatccttgttgatggccctcatgac 775 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 541 caggagaatggtctggtgccaattgttgagcctgagatccttgttgatggccctcatgac 600 Query: 776 attgatcg 783 |||||||| Sbjct: 601 attgatcg 608 Anhang 168 Sequenz des pGADT7-cDNA-Bank-„Inserts der Hefekolonie 2.43: CCATTATGGCCGGGCGGAACCTGCCCCTGGCTCAGGCCTGATGGGAAGACCCAGGTGACA GTCGAGTACCGCAATGAGGGTGGTGCCATGGTCCCCATCCGTGTCCACACCGTCCTCATC TCCACCCAGCACGACGAGACAGTGACCAATGATGAGATCGCTGCTGACCTGAAGGAGCAT GTCATCAAGCCTATCATCCCTGAGCAGTACCTTGACGAGAAGACCATCTTCCACCTTAAC CCATCCGGCCGCTTTGTCATTGGTGGACCTCACGGCGATGCTGGCCTCACTGGCCGCAAG ATCATCATTGACACCTACGGTGGCTGGGGAGCCCATGGTGGTGGCGCTTTNTNCGGCAAG GACCCAACCAAGGTTGACCGCAGCGGAGCCTATGTNGNGAGGCAGGCTGCCAAGAGCATC GTCGCCAGCGGCCTTGCTCGCCGCGCCATCGTNCAGGTGTNCTACGCCATCGGTGTGCCC NAGCCTCTCTCCGTGT >gi|17017258|gb|AF439721.1|AF439721 Zea mays methionine adenosyltransferase mRNA, partial cds Length = 760 Score = 87.7 bits (44), Expect = 1e-17 Identities = 71/80 (88%) Strand = Plus / Plus Query: 194 cagtaccttgacgagaagaccatcttccaccttaacccatccggccgctttgtcattggt 253 ||||||||||| ||||| |||||||||||||| |||||||| || ||||| ||||| ||| Sbjct: 634 cagtaccttgatgagaataccatcttccacctcaacccatcaggtcgcttcgtcatcggt 693 Query: 254 ggacctcacggcgatgctgg 273 ||||| ||||| |||||||| Sbjct: 694 ggaccacacggagatgctgg 713 Sequenz des pGADT7-cDNA-Bank-„Inserts der Hefekolonie 2.48: TCCACCCAAGCAGTGGTATCAACGCAGAGTGGCCATTATTGCCGGGGGCTTCGCCTCCAC CTATCCGCCGCCTCCCAATCATNCTGCCCCCAAACTCCCACCCCGCAGCATNCCCACGCC GCCGCACCTNGTCGCGTNGCGTCGTCACCGGCGTGGAGATGGTCGGGCCAGGCCTNTACA CCGAGATCGGCAAGAAGACCAGGGATCTGCTGTACAAGGACTACCAGACTGACCACAAGT TCACCCTCACTACCTACACCTNCAATGGCGTCGCTGTAACTGCTTNTAGCACAAAGAAAG CTGACCTGATCCTTGGCGAGATCCAATCACAGATAAAGAACAAGAACATGACCATAGATG TGAAAGCAAACTCGGAGTCAAATATNATTACAACAATTACTGNTGATGAGATTGCAACAC CAGGGCTGAAGACCATCTTAAGCTTTGCTGTTCCTGATCAGAGATCTGGAAAGGNTGAGC TNCANTATTTGCATGATTATGCTGGAGTTAATGCNAANCATTGGTCTGACNNNCNATCC >gi|5929927|gb|AF178950.1|AF178950 Zea mays voltage-dependent anion channel protein 1a (vdac1a) mRNA, complete cds; nuclear gene for mitochondrial product Length = 1179 Score = 731 bits (369), Expect = 0.0 Identities = 401/415 (96%) Strand = Plus / Plus Query: 82 Sbjct: 1 gccgcctcccaatcatnctgcccccaaactcccaccccgcagcatncccacgccgccgca 141 |||||||||||||||| |||||||||||||||||||||||||||| |||||||| || || gccgcctcccaatcatcctgcccccaaactcccaccccgcagcatccccacgccaccaca 60 Query: 142 cctngtcgcgtngcgtcgtcaccggcgtggagatggtcgggccaggcctntacaccgaga 201 ||| ||||||| ||||||||||||||||| ||||||||||||||||||| |||||||||| Sbjct: 61 cctcgtcgcgtcgcgtcgtcaccggcgtgaagatggtcgggccaggcctctacaccgaga 120 Query: 202 tcggcaagaagaccagggatctgctgtacaaggactaccagactgaccacaagttcaccc 261 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 121 tcggcaagaagaccagggatctgctgtacaaggactaccagactgaccacaagttcaccc 180 Anhang 169 Query: 262 tcactacctacacctncaatggcgtcgctgtaactgcttntagcacaaagaaagctgacc 321 ||||||||||||||| ||||||||||||||||||||||| |||||||||||||||||||| Sbjct: 181 tcactacctacacctccaatggcgtcgctgtaactgcttctagcacaaagaaagctgacc 240 Query: 322 tgatccttggcgagatccaatcacagataaagaacaagaacatgaccatagatgtgaaag 381 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 241 tgatccttggcgagatccaatcacagataaagaacaagaacatgaccatagatgtgaaag 300 Query: 382 caaactcggagtcaaatatnattacaacaattactgntgatgagattgcaacaccagggc 441 ||||||||||||||||||| ||||| |||||||||| ||||||||||||||||||||||| Sbjct: 301 caaactcggagtcaaatatcattacgacaattactgttgatgagattgcaacaccagggc 360 Query: 442 tgaagaccatcttaagctttgctgttcctgatcagagatctggaaaggntgagct 496 |||||||||||||||||||||||||||||||||||||||||||||||| |||||| Sbjct: 361 tgaagaccatcttaagctttgctgttcctgatcagagatctggaaaggttgagct 415 Sequenz des pGADT7-cDNA-Bank-„Inserts der Hefekolonie 4D: CCATTATGGCCGGGGAATCGGGCGGCCTCGCCGCCCGAATTGTAGTCTGGAGAGGCGTCC TCAGCGACGGACCGGGCCCAAGTTCTCTGGAAAGGGACGCCTGGGAGGGTGAGAGCCCCG TCCGGCCCGGACCCTGTCGCACCACGAGGCGCCGTCAACGAGTCGGGTTGTTTGGGAATG CAGCCCAAATCGGGCGGTAAACTCCGTCCAAGGCTAAATACAGGCGAGAGACCGATAGCG AACAAGTACCGCGAGGGAAAAAAAAAAAAAAAAAAAAAAAAAAAAA >gi|13397940|emb|AJ309824.2|ZMA309824 Zea mays 25S rRNA gene and transposon-like sequence Length = 10658 Score = 438 bits (221), Expect = e-123 Identities = 241/245 (98%), Gaps = 2/245 (0%) Strand = Plus / Plus Query: 1 Sbjct: 855 Query: 59 Sbjct: 915 Query: 119 Sbjct: 975 gaatcgggcggcct-cgccgcccgaattgta-gtctggagaggcgtcctcagcgacggac |||||||||||||| |||||||||||||||| |||||||||||||||||||||||||||| gaatcgggcggccttcgccgcccgaattgtaagtctggagaggcgtcctcagcgacggac cgggcccaagttctctggaaagggacgcctgggagggtgagagccccgtccggcccggac |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| cgggcccaagttctctggaaagggacgcctgggagggtgagagccccgtccggcccggac cctgtcgcaccacgaggcgccgtcaacgagtcgggttgtttgggaatgcagcccaaatcg ||| ||||||||||||||||| |||||||||||||||||||||||||||||||||||||| cctatcgcaccacgaggcgccatcaacgagtcgggttgtttgggaatgcagcccaaatcg Query: 179 ggcggtaaactccgtccaaggctaaatacaggcgagagaccgatagcgaacaagtaccgc |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 1035 ggcggtaaactccgtccaaggctaaatacaggcgagagaccgatagcgaacaagtaccgc Query: 239 gaggg 243 ||||| Sbjct: 1095 gaggg 1099 Anhang 170 Sequenz des pGADT7-cDNA-Bank-„Inserts der Hefekolonie 2.11a: GNGTATNANNCCTNCGACTGGCGTACCCATACGACGTACCAGATTACGCTCATATGGCCA TGGAGGCCAGTGAATTCCACCCAAGCAGTGGTATCAACGCAGAGTGGCCATTATGGCCGG GGGAGCACCAAGGACCTTCGGCGCCCGTTCAGGATCCACATCTTCCCGAGCGAGTTCTTG GTTGACCTCTTCCTCTTCGGACCACCTCCAAGGCTCTTGGTGTAGCTTGCCACTCTCACC AATCAAGTTTTCATGTCTGATCTCGACATTCAGATCCCAACTGCCTTCGATCCCTTCGCT GAGGCCAATGCTGGGGACTCTGGTGCGGCAGCAGGGTCAAAGGACTACGTACACGTGCGC ATCCAGCAGCGTAATGGTCGCAAGAGCCTGACCACCGTCCAGGGATTGAAGAAGGAGTTC AGCTACAGCAAGATCCTCAAAGATCTCAAGAAAGAGTTCTGCTGCAATGGTACAGTGGTC CAGGACCCAGAACTTGGACAGGTCATTCAGCTCCAGGGTGATCAGAGGAAGAACGTCTCG AATTTCCTCGTCCAGGCCGGCATTGTGAAGAAGGAGCACATCAAGATTCATGGTTTCTGA GCAACCGCCGGGTCCATAGCAAACCAACCCTGCATGCAGGGCCAGCTTATTATATATAAT ATCTGAGGTGGAGCATATCTATCTGGCTGTGACTGCAAGCAGCCTATACGCCGCTGGCGT TCCTTCTATTACACATATAGGCCCCTCCTGCTGATGCTTGTACCGTCGTGTCNACCTTGA CTACAAGCTCATCGACATCATGCTTGTTCTTATAAGACATGTTGATAACCTATTGGTAAA AAAAAAAAAAAAAAAAAAAACTGTGGCCGCTGGCTTAAGGGGCTCGTCGGANCTCACTCA CTGAAGATCGAATCTGAAACCCNAGTACTTACTGGATGGNCATTAATNTNATTTCTNTTG CTNTTTGACTTCTCTTAGTCACT >gi|2668739|gb|AF034944.1|AF034944 Zea mays translation initiation factor GOS2 (TIF) mRNA, complete cds Length = 759 Score = 1164 bits (587), Expect = 0.0 Identities = 625/631 (99%), Gaps = 5/631 (0%) Strand = Plus / Plus Query: 5 Sbjct: 6 Query: 65 Sbjct: 66 gagcaccaaggaccttcggcgcccgttcaggatccacatcttcccgagcgagttcttggt 64 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| gagcaccaaggaccttcggcgcccgttcaggatccacatcttcccgagcgagttcttggt 65 tgacctcttcctcttcggaccacctccaaggctcttggtgtagcttgccactctcaccaa 124 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| tgacctcttcctcttcggaccacctccaaggctcttggtgtagcttgccactctcaccaa 125 Query: 125 tcaagttttcatgtctgatctcgacattcagatcccaactgccttcgatcccttcgctga 184 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 126 tcaagttttcatgtctgatctcgacattcagatcccaactgccttcgatcccttcgctga 185 Query: 185 ggccaatgctggggactctggtgcggcagcagggtcaaaggactacgtacacgtgcgcat 244 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 186 ggccaatgctggggactctggtgcggcagcagggtcaaaggactacgtacacgtgcgcat 245 Query: 245 ccagcagcgtaatggtcgcaagagcctgaccaccgtccagggattgaagaaggagttcag 304 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 246 ccagcagcgtaatggtcgcaagagcctgaccaccgtccagggattgaagaaggagttcag 305 Query: 305 ctacagcaagatcctcaaagatctcaagaaagagttctgctgcaatggtacagtggtcca 364 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 306 ctacagcaagatcctcaaagatctcaagaaagagttctgctgcaatggtacagtggtcca 365 Query: 365 ggacccagaacttggacaggtcattcagctccagggtgatcagaggaagaacgtctcgaa 424 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 366 ggacccagaacttggacaggtcattcagctccagggtgatcagaggaagaacgtctcgaa 425 Query: 425 tttcctcgtccaggccggcattgtgaagaaggagcacatcaagattcatggtttctgagc 484 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 426 tttcctcgtccaggccggcattgtgaagaaggagcacatcaagattcatggtttctgagc 485 Query: 485 aaccgccgggtccatagcaaaccaaccctgcatgcagggccagcttattatatataatat 544 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 486 aaccgccgggtccatagcaaaccaaccctgcatgcagggccagcttattatatataatat 545 Anhang 171 Query: 545 ctgaggtggagcatatctatctggctgtg-actgcaagca-gccta-tacgccg-ctg-g 599 ||||||||||||||||||||||||||||| |||||||||| ||||| ||||||| ||| | Sbjct: 546 ctgaggtggagcatatctatctggctgtgtactgcaagcaggcctagtacgccgcctgcg 605 Query: 600 cgttccttctattacacatataggcccctcc 630 ||||||| ||||||||||||||||||||||| Sbjct: 606 cgttcctcctattacacatataggcccctcc 636 Sequenzen der pGADT7-cDNA-Bank-„Inserts der Hefekolonien 7.173 und 7.178: GTATGNGNTTTAAANCCTNTNACTGCAGTACCCATACGACGTACCAGATTACGCTCATAT GGCCATGGAGGCCAGTGAATTCCACCCAAGCAGTGGTATCAACGCAGAGTGGCCATTATG GCCGGGGACCCACTCCAGAAAACCTCACAAAAACCCTCGCTCCCCACAACCCGCCGCCGC CCACGCCGCCGCCTGTACCAGCGGCGTCAAGATGAGTCGGCACCCTGAAGTGAAGTGGGC TCAGAGGATCGACAAGGTTTACATCACTGTACAGTTACCTGATGCCAAGGACGCTAAGGT CAATTTGGAACCAGATGGTGTCTTCACATTCTCTGGCAGTGTTGGTACAAACTTATATGA ATTGAAACTAGATCTGAATGACAAAGTAAACGTTGAGGCTAGCAAAATAAGTGGGTGTCA GGTCCATATTCTGTATTGTAGAAAAAGCTGAGGCCAAATGGTGGAAGAAACTTGTTCGAG ATGATCAAAGGGCACCTCACTTTGTGAAAGTTGATTGGGACAAATGGGTTGACGAAGATG ATGATGGTGCTGATGTCAACTTGGATGGGATGGACTTCTCGAATTTTGGCGGTATGGGTG GTATGGGTGGCATGGGTGATATGGCTGGCTTGGGTGGTCTTGGTGGCATGGGTGGAATGG GTGGATGGGTGGTATGGGAATGGNTGATNCNNAGGTCATNNNGTNAANAC >gi|21206618|gb|AY103540.1| Zea mays PCO102485 mRNA sequence Length = 1129 Score = 680 bits (343), Expect = 0.0 Identities = 385/426 (90%), Gaps = 2/426 (0%) Strand = Plus / Plus Query: 184 cgccgccgcctgtaccagcggcgtcaagatgagtcggcaccctgaagtgaagtgggctca 243 ||||||||||||||||||| ||||||||||||||||||||||||| |||||||||||||| Sbjct: 119 cgccgccgcctgtaccagctgcgtcaagatgagtcggcaccctgaggtgaagtgggctca 178 Query: 244 gaggatcgacaaggtttacatcactgtacagttacctgatgccaaggacgctaaggtcaa 303 ||||||||||||||||||||||||||||||||||||||| || ||||| ||||||||||| Sbjct: 179 gaggatcgacaaggtttacatcactgtacagttacctgacgcaaaggatgctaaggtcaa 238 Query: 304 tttggaaccagatggtgtcttcacattctctggcagtgttggtacaaacttatatgaatt 363 |||||| ||||||||||||||||||||||||||||||| |||||||||||| |||||||| Sbjct: 239 tttggatccagatggtgtcttcacattctctggcagtgctggtacaaacttgtatgaatt 298 Query: 364 gaaactagatctgaatgacaaagtaaacgttgaggctagcaaaataa--gtgggtgtcag 421 ||||||||||||||||||||||||||||||||||||||||||||||| ||||| ||||| Sbjct: 299 gaaactagatctgaatgacaaagtaaacgttgaggctagcaaaataagtgtgggggtcag 358 Query: 422 gtccatattctgtattgtagaaaaagctgaggccaaatggtggaagaaacttgttcgaga 481 ||||||||||||||| |||||||||||||| |||||||||||||||||||| |||||||| Sbjct: 359 gtccatattctgtatcgtagaaaaagctgaagccaaatggtggaagaaactcgttcgaga 418 Query: 482 tgatcaaagggcacctcactttgtgaaagttgattgggacaaatgggttgacgaagatga 541 ||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||| Sbjct: 419 tgatcaaagggcacctcactttgtgaaagttgattgggacaaatgggttgatgaagatga 478 Query: 542 tgatggtgctgatgtcaacttggatgggatggacttctcgaannnnnnnnnnnnnnnnnn 601 ||||||||||||| ||||| |||||||||||||||||||||| Sbjct: 479 tgatggtgctgatatcaacatggatgggatggacttctcgaatttcggtggcatgggtgg 538 Anhang 172 Sequenz des pGADT7-cDNA-Bank-„Inserts der Hefekolonie 2.27: TTTTTAAAAAANCATCTTTAATATTNCTCACTATACGGCGAGCGCCGCCATGGAGTACCC ATACGACGTACCAGATTACGCTCATATGGCCATGGAGGCCAGTGAATTCCACCCAAGCAG TGGTATCAACGCAGAGTGGCCATTATGTCCGGGGACACAAGGCATTGTCAGCGGCGGCGG CGGCGGCGGGCCGGCGGAGACTCCCAGGCCAGGANCAGTCTGCCGATTGGAAGGGGAGAT GAAGGCCACGCTNAAGGGCCGNTACGAGGGCGACAAGGCCACGGCCGCCACCACCCTCGC CGCCCCCGNCGGCGAACCTTCGNCTCAAGGNCCTNCGCCACCGAAGGCCNCTTCACCAAC GGTNCTTTTCCTTGCGANGGCCTNACCCTTNACCCTTNAAAAAAGCCCGGGNGCTTTTCN TNNTTTGNACCNNTCAAGCCCCCNAANCCAAGAATGTTGCNCTTTTTAANNTTNATTAAA CTTTCCNNCCTTNNTNCNTTNAANAAANNNCCCTTAATNCTTTNNCCTAAATTTATTTCN AATTANCCTTTNCCCNNGGTNACC >gi|21209273|gb|AY106195.1| Zea mays PCO138904 mRNA sequence Length = 857 Score = 226 bits (114), Expect = 4e-56 Identities = 156/170 (91%), Gaps = 2/170 (1%) Strand = Plus / Plus Query: 198 agactcccaggccaggancagtctgccgattggaaggggagatgaaggccacgctnaagg 257 ||||||||||||||||| ||||||||||||||||||||||||||||||||||||| |||| Sbjct: 56 agactcccaggccaggagcagtctgccgattggaaggggagatgaaggccacgctcaagg 115 Query: 258 gccgntacgagggcgacaaggccacggccgccaccaccctcgccgcccccgncggcgaac 317 |||| |||||||||||||||||||||||||||||||||||||||||||||| ||||| || Sbjct: 116 gccgctacgagggcgacaaggccacggccgccaccaccctcgccgcccccgccggcg-ac 174 Query: 318 cttcgnctcaaggncctncgccaccgaaggccncttcaccaacggtnctt 367 || || ||||||| ||| ||||||||| | | ||||||||||||| ||| Sbjct: 175 ctccgcctcaagg-cctccgccaccgaggccgccttcaccaacggtcctt 223 Sequenz des pGADT7-cDNA-Bank-„Inserts der Hefekolonie 2.27: TAAAAAGCNNTCTTTAATACNACTCACTATANGGCGAGCGCCGCCATGGAGTACCCATAC GACGTACCAGATTACGCTCATATGGCCATGGAGGCCAGTGAATTCCACCCAAGCAGTGGT ATCAACGCAGAGTGGCCATTATGGCCGGGGACTCTACTCACCCACCCACTCACGGACTCA TCTACTGAGCAGCGCGGTGGGCATCCGAGCGAGAGGGCGAGCGCGGAAGGATCGGAATTC CGAAGGTTGGGCATGGAGGCGAGCGGCGAGAAGGCTGCGGTGGTGCGACGGNTGATGGAG GCGAANGAGGTGTTCGGGAAGACCTTTTTNNGGAATNCNCCGCCNANACCGGNTNTCACC ANCTNNTNCNTGGNNNCANNTNNTTGNNGNNNCCNNNNNCNNNNTNAANGGNCNNNCAAC NNNNNNCNNNNNCTTANNGNCNNNNNNTNNNCANNNCNNNCNNNCNNNNNNNT gi|21213628|gb|AY109778.1| Zea mays CL3683_1 mRNA sequence Length = 600 Score = 168 bits (85), Expect = 7e-39 Identities = 92/95 (96%) Strand = Plus / Plus Query: 232 tcggaattccgaaggttgggcatggaggcgagcggcgagaaggctgcggtggtgcgacgg 291 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 52 tcggaattccgaaggttgggcatggaggcgagcggcgagaaggctgcggtggtgcgacgg 111 Query: 292 ntgatggaggcgaangaggtgttcgggaagacctt 326 ||||||||||||| ||||||| |||||||||||| Sbjct: 112 ctgatggaggcgaaggaggtgtccgggaagacctt 146 Anhang Score = 44.1 bits (22), Expect = 0.28 Identities = 29/30 (96%), Gaps = 1/30 (3%) Strand = Plus / Plus Query: 180 atctactgagcagcgcggtgggcatccgag 209 |||||||||||||||| ||||||||||||| Sbjct: 1 atctactgagcagcgc-gtgggcatccgag 29 7.4 Tabelle zur statistischen Auswertung Tab. 8.1: Tabelle für die tTab-Werte zur statistischen Auswertung der Signifikanz zweier Proben. Aus: KÖHLER, SCHACHTEL & VOLESKE (2002). 173 Lebenslauf 174 Lebenslauf 12.05.1972 geboren in Berlin 1978-1980 Privatschule der Deutschen Botschaft Ankara, Zweigstelle Istanbul, Türkei 1980-1985 Evangelische Schule Steglitz, Grundschule, Berlin 1985-1989 Evangelische Schule Steglitz, Realschule, Berlin 1989-1992 Robert-Blum-Oberschule, Gymnasium, Berlin 1992-1999 Studium der Biologie an der Freien Universität Berlin Diplomarbeit bei Priv.-Doz. Dr. R. Schmidt am Institut für Gemüse- und Zierpflanzenbau Großbeeren/Erfurt e.V. mit dem Thema: „Einfluss einer pilzlichen Infektion der Wurzel auf die Assimilatverteilung der Tomate (Lycopersicon esculentum Mill.) in Abhängigkeit von der Strahlung“ Juni 2000- Oktober 2000 Praktikum zum Erlernen molekularbiologischer Methoden am Fachbereich Biologie/Chemie an der Universität Osnabrück in der Abteilung Pflanzenphysiologie bei Frau Prof. Dr. Renate Scheibe November 2000- Oktober 2003 Stipendiatin des Graduiertenkollegs: Physiologie, Wechselwirkungen „Molekulare zwischen zellulären Nanostrukturen“ Durchführung der experimentellen vorliegenden Dissertation in Arbeiten der zur Abteilung Pflanzenphysiologie der Universität Osnabrück November 2003- Dezember 2003 Wissenschaftliche Hilfskraft in der Pflanzenphysiologie der Universität Osnabrück Abteilung Eidesstattliche Versicherung 175 Erklärung über die Eigenständigkeit der erbrachten wissenschaftlichen Leistung Ich erkläre hiermit, dass ich die vorliegende Arbeit ohne unzulässige Hilfe Dritter und ohne Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe. Die aus anderen Quellen direkt oder indirekt übernommenen Daten und Konzepte sind unter Angabe der Quelle gekennzeichnet. Die Arbeit wurde bisher weder im In- noch im Ausland in gleicher oder ähnlicher Form einer anderen Prüfungsbehörde vorgelegt. Berlin, den