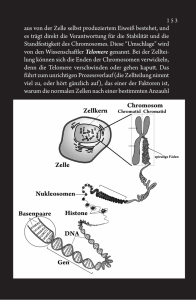
Dokument_48.
Werbung

Aus der Psychiatrischen und Psychotherapeutischen Klinik der Friedrich-Alexander-Universität Erlangen-Nürnberg Direktor: Prof. Dr. med. Johannes Kornhuber Untersuchung über den Einfluss der Telomerverkürzung auf die Schwere einer Depression und deren Therapierbarkeit bei Patienten mit niedrigen und hohen Antidepressiva-Dosen und Patienten mit Elektrokrampftherapie im Vergleich zu einer Kontrollgruppe Inaugural-Dissertation zur Erlangung der Doktorwürde der Medizinischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg vorgelegt von Marina Theresa Böhner aus Würzburg Gedruckt mit Erlaubnis der Medizinischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg Dekan: Prof. Dr. J. Schüttler Referent: Prof. Dr. R. Kalb Korreferent: Prof. Dr. J. Kornhuber Tag der mündlichen Prüfung: 16. März 2011 INHALTSVERZEICHNIS 1 Zusammenfassung ........................................................................... 5 1.1 Zusammenfassung............................................................................... 5 1.1.1 Hintergrund und Ziele ..................................................................... 5 1.1.2 Methoden ....................................................................................... 5 1.1.3 Ergebnisse und Beobachtungen .................................................... 5 1.1.4 Praktische Schlussfolgerungen ...................................................... 6 1.2 Summary ............................................................................................... 7 1.2.1 Background .................................................................................... 7 1.2.2 Methods.......................................................................................... 7 1.2.3 Results ........................................................................................... 7 1.2.4 Conclusion...................................................................................... 8 2 Einleitung .......................................................................................... 9 3 Theoretischer Hintergrund ............................................................ 11 3.1 Klassifikation affektiver Störungen .................................................. 11 3.1.1 Internationale statistische Klassifikation der Krankheiten und verwandter Gesundheitsprobleme (ICD-10)............................ 11 3.1.2 Diagnostisches und Statistisches Handbuch Psychischer Störungen (DSM-IV)................................................................ 13 3.1.3 Beck-Depressions-Inventar (BDI) ................................................. 14 3.1.4 Hamilton-Depressions-Skala (HAM-D, Hamilton rating scale for depression) ............................................................................. 15 3.2 Epidemiologie der Depression .......................................................... 15 3.3 Ätiologie der Depression ................................................................... 16 3.3.1 Familiäre und genetische Belastung ............................................ 16 3.3.2 Monoamin-Mangel-Hypothese ..................................................... 18 3.3.2.1 Störung der Monoamin-Konzentration .................................... 19 3.3.2.2 Störung der Rezeptorwirkung.................................................. 20 3.3.2.3 Störung in der Signaltransduktionskaskade ............................ 20 3.3.2.4 Störung im Dopaminstoffwechsel ............................................ 21 3.3.2.5 Zusammenfassung der Monoamin-Mangel-Hypothese........... 21 3.3.3 Störung in der Hypothalamus-Hypophysen-Nebennieren-Achse . 23 3.3.3.1 Die HHN-Achse und das Glukokortikoid Cortisol .................... 23 3.3.3.2 Die HHN-Achse und die Monoamine ...................................... 24 3.3.4 Störung in der Neurogenese und die Funktion des Brain-derived Neurotrophic Factor ................................................................ 25 3.3.5 Depression und kardiovaskuläre Erkrankungen: Folgen einer Entzündungsreaktion? ............................................................ 28 1 3.4 Therapiemöglichkeiten ...................................................................... 29 3.4.1 Psychopharmakotherapie ............................................................. 29 3.4.2 Psychotherapie............................................................................. 31 3.4.2.1 Psychoanalytische und psychodynamische Therapie ............. 31 3.4.2.2 Interpersonelle Psychotherapie ............................................... 33 3.4.2.3 Verhaltenstherapie .................................................................. 33 3.4.3 Elektrokrampftherapie .................................................................. 35 3.4.4 Transkranielle Magnetstimulation ................................................. 37 3.4.5 Vagusnervstimulation ................................................................... 37 3.4.6 Tiefe Hirnstimulation (deep brain stimulation, DBS) ..................... 38 3.4.7 Lichttherapie ................................................................................. 39 3.4.8 Schlafentzugstherapie .................................................................. 40 3.5 Telomere ............................................................................................. 41 3.5.1 Aufbau und Funktion der Telomere .............................................. 41 3.5.2 Telomere, Stammzellen und hämatopoetische Progenitorzellen.. 43 3.5.3 Die Bedeutung der Telomere bei der Entstehung von Krankheiten ............................................................................. 44 3.5.3.1 Kardiovaskuläre Erkrankungen ............................................... 44 3.5.3.2 Syndrome des vorzeitigen Alterns (Progerie-Syndrome) ........ 45 3.5.3.3 Tumorerkrankungen ................................................................ 47 3.5.3.4 Psychische Erkrankungen ....................................................... 48 4 Probanden und Methoden ............................................................. 50 4.1 Probanden .......................................................................................... 50 4.1.1 Kontrollgruppe .............................................................................. 50 4.1.2 Patientengruppe ........................................................................... 50 4.2 Bestimmung der Laborparameter ..................................................... 52 4.2.1 Bestimmung von adulten Stammzellen im peripheren Blut bei depressiven Patienten mit der Durchflusszytometrie .............. 53 4.2.1.1 Das Streulichtsignal ................................................................ 53 4.2.1.2 Das Fluoreszenzsignal ............................................................ 53 4.2.1.3 Bestimmung der CD34-, CD105-, CD133- und CD146-positiven Stammzellen in unserer Studie .............................................................. 54 4.2.2 Bestimmung der Telomerlänge im peripheren Blut bei depressiven und gesunden Probanden ....................................................... 56 4.2.2.1 Elektrophorese ........................................................................ 56 4.2.2.2 Southern Blot .......................................................................... 56 4.2.2.3 Bestimmung der Telomerlänge in unserer Studie ................... 57 4.2.3 Statistische Auswertung ............................................................... 59 5 Ergebnisdarstellung ....................................................................... 60 5.1 Deskription der Gesamtgruppe ......................................................... 60 5.1.1 Verteilung der Stichproben ........................................................... 60 5.1.2 Demographische Daten ................................................................ 61 5.1.3 Klinische Daten ............................................................................ 62 5.1.4 Labordaten ................................................................................... 63 2 5.2 Telomeranalyse .................................................................................. 64 5.2.1 Abhängigkeit von Alter und Geschlecht ........................................ 64 5.2.2 Vergleich der Gruppen ................................................................. 65 5.2.3 Analyse des Rauchverhaltens ...................................................... 70 5.2.4 Analyse der Erkrankungsdauer .................................................... 72 5.2.5 Analyse der Medikamentendosierung .......................................... 73 5.2.6 Analyse der Elektrokrampftherapie-Daten .................................... 76 5.2.7 Analyse der Transkraniellen Magnetstimulation ........................... 78 5.2.8 Analyse der Stammzelldaten ........................................................ 80 5.2.8.1 Analyse der Leukozyten bzw. Lymphozyten Gesamtzahl ....... 81 5.2.8.2 Analyse der Stammzellen ....................................................... 82 5.2.9 Analyse der psychischen Komorbiditäten ..................................... 83 5.2.9.1 Analyse der Stichprobenverteilung.......................................... 83 5.2.9.2 Persönlichkeitsstörung ............................................................ 85 5.2.10 Analyse der somatischen Komorbiditäten .................................. 86 5.2.10.1 Analyse der Stichprobenverteilung........................................ 86 5.2.11 Analyse des Suchtverhaltens ..................................................... 88 5.2.11.1 Analyse der Stichprobenverteilung........................................ 88 5.2.11.2 Benzodiazepin- und Alkoholabhängigkeit ............................. 89 5.2.12 Analyse der Familienanamnese ................................................. 91 6 Diskussion ...................................................................................... 94 7 Literaturverzeichnis ....................................................................... 98 8 Abkürzungsverzeichnis ............................................................... 114 9 Abbildungsverzeichnis ................................................................ 117 10 Tabellenverzeichnis ..................................................................... 118 11 Anhang .......................................................................................... 119 11.1 DSM-IV-Kriterien ............................................................................. 119 11.2 Beck Depressions-Inventar ........................................................... 120 11.3 Hamilton Depressionsskala .......................................................... 122 12 Danksagung .................................................................................. 124 3 4 1 Zusammenfassung 1.1 Zusammenfassung 1.1.1 Hintergrund und Ziele Bei einer Vielzahl von Erkrankungen wurden verkürzte Telomere beobachtet. Unser Ziel war es, die durchschnittliche Telomerlänge von Patienten mit einer Major Depression zu analysieren. Eine Schlüsselfrage war, ob sich verschiedene Gruppen von depressiven Patienten bezüglich der Telomerlänge unterscheiden. 1.1.2 Methoden Wir haben aus Blutproben von Patienten mit einer Major Depression (n = 54) und von gesunden Kontrollpersonen (n = 20) genomische DNA isoliert und mit Hilfe von Telomer-Restriktionsfragmenten und Southern Blot die durchschnittliche Telomerlänge bestimmt. Die depressiven Probanden wurden entsprechend ihrer Therapie in drei Gruppen eingeteilt. Dabei wurde einerseits die Totale Antidepressiva-Dosis und andererseits die Anwendung von Elektrokrampftherapien berücksichtigt. 1.1.3 Ergebnisse und Beobachtungen Die durchschnittliche Telomerlänge in der depressiven Gesamtgruppe (7.20 ± 0.61 kb) war signifikant kürzer als in der Kontrollgruppe (7.55 ± 0.54 kb). Wir konnten keinen signifikanten Unterschied bezüglich der Telomerlänge zwischen den verschiedenen Patientengruppen feststellen, aber jede einzelne Patientengruppe hatte signifikant kürzere Telomere als die gesunden Probanden. Weitere Analysen erbrachten weder signifikante Verbindungen zwischen der Telomerlänge und der Erkrankungsdauer noch zwischen der Telomerlänge und der Schwere der Erkrankung entsprechend der Hamilton Depression-Skala. 5 1.1.4 Praktische Schlussfolgerungen Diese Ergebnisse unterstützen erneut die Theorie, dass die Major Depression mit verkürzten Telomeren assoziiert ist. Jedoch scheinen sowohl die Therapie als auch die Erkrankungsdauer und die Schwere der Erkrankung keinen Einfluss auf die Telomerlänge zu haben. 6 1.2 Summary 1.2.1 Background Shortened telomere length has been observed in a variety of diseases. Our objective was to analyze mean telomere length of patients with major depressive disorder. A key question was whether telomere length varies in different groups of depressive patients. 1.2.2 Methods We obtained blood samples from patients with major depressive disorder (n = 54) and healthy subjects (n = 20). We isolated genomic DNA and measured mean telomere length using telomere restriction fragments and Southern blotting. We grouped patients according to the therapy they received including total antidepressant dose. 1.2.3 Results Mean telomere length of the entire patient group (7.20 ± 0.61 kb) was significantly shorter than in the control group (7.55 ± 0.54 kb). We observed no significant difference in telomere length among the different patient groups, but each of these different patient groups had significantly shorter telomeres than the healthy subjects. Further analysis revealed no significant association between telomere length and illness duration and between telomere length and the severity of depression as determined by Hamilton score. 7 1.2.4 Conclusion These results provide further evidence that major depressive disorder is associated with shortened telomeres. However, differences in the applied therapy, the duration of illness or the severity of depression do not seem to have any influence on telomere length. 8 2 Einleitung Depressive Erkrankungen nehmen in der heutigen Zeit eine immer bedeutendere Rolle ein. In den letzten Jahren ist zunehmend von der „Volkskrankheit Depression“ [188] die Rede – und die Folgen betreffen nicht nur die Erkrankten selbst, sondern stellen auch eine enorme Belastung für die Gesellschaft dar. Die Patienten leiden in erster Linie unter den direkten Symptomen einer Depression: Neben der Traurigkeit, Hoffnungslosigkeit und Antriebslosigkeit erschweren ein verändertes Schlaf-, Ess- und Sexualverhalten den Alltag. Vor allem im Rahmen rezidivierender Episoden ist häufig aber auch zwischen den depressiven Phasen kein geregeltes soziales Leben möglich, da Familien-, Privat- und Arbeitsleben immer wieder durch die Krankheit erschüttert werden. Hinzu kommt ein signifikant erhöhtes Risiko für somatische Erkrankungen [58]: unter anderem treten bei depressiven Menschen Alzheimer-Erkrankungen [103, 155], kardiovaskuläre Ereignisse [7, 88], Schlaganfälle [207] und vermutlich sogar Krebserkrankungen [157] häufiger auf. Dadurch erklärt sich auch der gigantische finanzielle Aufwand, der im Rahmen sozialer Leistungen für die Bundesrepublik entsteht: Einerseits müssen zahlreiche Medikamente und monatelange Krankenhausaufenthalte ermöglicht werden, andererseits kommen häufig noch Arbeitslosen- oder Frührentenansprüche hinzu. [22] Somit wird deutlich, wie wichtig eine effiziente Therapie von depressiven Erkrankungen ist. Aber obwohl uns heute eine Vielzahl von Medikamenten zur Verfügung steht, ist verhältnismäßig wenig über die molekularen Ursachen von Depressionen bekannt. Dies ist jedoch mit Sicherheit der wichtigste Ansatz, um insbesondere therapieresistenten Patienten zukünftig helfen zu können. Simon [186] beschreibt in diesem Zusammenhang einen sehr interessanten Befund. Er fand heraus, dass die Enden der Chromosomen, die so genannten 9 Telomere, bei Depressiven gegenüber Gesunden verkürzt sind. Das Interessante dabei ist, dass die Länge der Telomere Aufschluss über das Alter und die Teilungsfähigkeit einer Zelle gibt: Mit zunehmendem Alter einer Zelle verkürzen sich die Telomere und die Teilungsfähigkeit nimmt ab. Simon vermutet, dass es im Rahmen einer Depression zu chronischem Stress kommt, der die Zellteilung steigert und zu einer vorzeitigen Alterung der Zelle führt. In seiner Studie zeigte sich eine Voralterung von 10 Jahren bei Depressiven gegenüber einer gesunden Kontrollgruppe. Aber ist die Telomerverkürzung eine Folge der Depression oder begünstigen verkürzte Telomere die Entstehung einer Depression? Und was für eine Bedeutung hat das Ausmaß der Telomerverkürzung auf die Schwere der Depression und den Therapieerfolg? Einen weiteren interessanten Ansatzpunkt bietet Dome [48]. Er beschreibt eine verminderte Anzahl reifer und unreifer endothelialer Progenitorzellen bei Depressiven. Beruht dieses Defizit vielleicht auf der Telomerverkürzung? Verhindern die kürzeren Telomere eine suffiziente Stammzellbildung, die einer Depression entgegenwirken könnte? Diese Fragen sollen in der folgenden Studie genauer untersucht werden. Dabei werden einerseits Grundlagen aus Lehrbüchern und Fachbüchern zusammengefasst. Vor allem werden aber auch neueste Erkenntnisse aus Artikeln und Reviews berücksichtigt, um einen möglichst aktuellen Stand der Forschung wiederzugeben. 10 3 Theoretischer Hintergrund 3.1 Klassifikation affektiver Störungen Depressive Erkrankungen können anhand unterschiedlicher Klassifikationen eingeteilt werden. In klinischen Bereichen wird in der Regel auf die Internationale Klassifikation der Krankheiten (ICD-10) zurückgegriffen, im Zusammenhang mit Studien wird gerne auch das Diagnostische und Statistische Manual Psychischer Störungen (DSM-IV) angewandt. Zusätzlich kann anhand des Beck-Depressions-Inventars (BDI) mittels Selbstbeurteilung und anhand der Hamilton-Depressions-Skala (HAM-D) mittels Fremdbeurteilung die Schwere der Depression abgeschätzt werden. 3.1.1 Internationale statistische Klassifikation der Krankheiten und verwandter Gesundheitsprobleme (ICD-10) Das Kapitel V des ICD-10-GM 2010 (International Classification of Diseases 10th Revision – German modification 2010) befasst sich mit der Klassifikation psychischer und Verhaltensstörungen und zählt Depressionen neben den Manien zum Formenkreis der affektiven Störungen. Treten beide Krankheitsbilder kombiniert auf, spricht man von einer bipolaren Erkrankung. Tabelle 1 fasst die verschiedenen Diagnosen nach der Internationalen Klassifikation der Krankheiten in ihrer 10. Revision (ICD-10) zusammen: 11 TABELLE 1: Hauptgruppen der affektiven Störungen (F30-F39). [45] F30.- Manische Episode F30.0 Hypomanie F31.- Bipolare affektive Störung F32.- Depressive Episode F33.- Rezidivierende depressive Störung F34.- Anhaltende affektive Störungen F34.0 Zyklothymia F34.1 Dysthymia F38.- Andere affektive Störungen F39.- Nicht näher bezeichnete affektive Störungen Die Symptome in einer depressiven Episode sind vielfältig und können in drei Gruppen unterteilt werden (Tbl. 2): TABELLE 2: Typische somatische, sonstige und psychotische Symptome im Rahmen einer depressiven Episode. [137] Somatische Symptome • Interessenverlust oder Verlust der Freude an angenehmen Aktivitäten • mangelnde Fähigkeit, auf Ereignisse oder Aktivitäten emotional zu reagieren • Früherwachen • Morgentief • psychomotorische Hemmung oder Agitiertheit • Appetitverlust • Gewichtsverlust • Libidoverlust Sonstige Symptome • depressive Verstimmung • Verlust des Selbstvertrauens oder des Selbstwertgefühls • unbegründete Selbstvorwürfe oder Schuldgefühle • wiederkehrende Gedanken an den Tod, an Suizid oder suizidales Verhalten • vermindertes Denk- oder Konzentrationsvermögen, Unschlüssigkeit und Unentschlossenheit Psychotische Symptome • depressiver, Schuld-, hypochondrischer, nihilistischer, Beziehungs- oder Verfolgungswahn • Depressiver Stupor 12 Je nach Anzahl der Symptome wird zwischen leichten, mittelgradigen und schweren Episoden unterschieden, wobei letztere von psychotischen Krankheitszeichen begleitet werden können. Davon abgegrenzt gibt es noch sehr leichte und dafür lang andauernde affektive Störungen: Die Hypomanie wird zu der manischen Episode gezählt (F30.0) und bezeichnet die abgeschwächte Sonderform der Manie. Zyklothymie und Dysthymie sind das entsprechende Äquivalent bei bipolaren affektiven Störungen bzw. Depressionen und werden unter den anhaltenden affektiven Störungen aufgeführt (F34.0 bzw. F34.1). 3.1.2 Diagnostisches und Statistisches Handbuch Psychischer Störungen (DSM-IV) Im Zusammenhang mit Studien wird häufig auf die amerikanische DSM-IVKlassifikation zurückgegriffen. Eine Depression vom Major-Typ kann diagnostiziert werden, wenn folgende Kriterien erfüllt werden (siehe Anlage 1 und Tbl. 3): TABELLE 3: DSM-IV-Kriterien zur Diagnostizierung einer Depression vom Major-Typ. [174] A. Mindestens fünf Symptome, darunter mindestens ein Hauptsymptom, treten während einer zweiwöchigen Episode auf. Hauptsymptome • Depressive Verstimmung • Verlust an Interesse oder Freude Nebensymptome • Deutlicher Gewichtsverlust ohne Diät oder Gewichtszunahme • Vermehrter Schlaf oder Schlaflosigkeit • Psychomotorische Unruhe oder Verlangsamung • Müdigkeit und Energieverlust • Übermäßige, unangemessene Schuldgefühle oder Gefühle von Wertlosigkeit an fast allen Tagen • Subjektive oder beobachtbare verminderte Denk- und Entscheidungsfähigkeit • Suizidale Gedanken und/oder Handlungen 13 B. Es werden nicht die Kriterien für eine gemischte Episode erfüllt. C. Die Symptome verursachen in klinisch bedeutsamer Weise Leiden oder Beeinträchtigungen. D. Die Symptome können nicht durch die Einnahme von Substanzen (Medikamenten, Drogen etc.) oder Begleiterkrankungen (z. B. Hypothyreose) verursacht sein. 3.1.3 Beck-Depressions-Inventar (BDI) Das Beck-Depressions-Inventar wurde 1961 von Aaron T. Beck eingeführt und hilft dabei, die Schwere einer Depression abzuschätzen. Es setzt sich aus 21 Fragen zusammen, die vom Patienten selbst beantwortet werden (siehe Anlage 2). Auf jede Frage gibt es 4 Antwortmöglichkeiten, die mit 0-3 Punkten bewertet werden. Anhand der erreichten Punktzahl ergeben sich folgende Schweregrade (Tbl. 4): TABELLE 4: Schweregradeinteilung einer Depression gemäß Beck Depressionsinventar 1 (BDI). 0 10 19 30 - 9 - 18 - 29 - 63 Punkte: Punkte: Punkte: Punkte: keine Depression leichte bis mittelgradige Depression mittelgradige bis schwere Depression schwere Depression Der Nachteil des Beck-Depressions-Inventars liegt auf der Hand: da die Fragen von den Patienten selbst beantwortet werden, kann es leicht zu einer Unteroder Übertreibung der Symptome kommen. Da Patienten mit einer Depression jedoch neben einigen objektivierbaren Symptomen vor allem unter den subjektiven Beschwerden leiden, ermöglicht der BDI insbesondere in Kombination mit einem Fremdbeurteilungstest eine hilfreiche Einschätzung des Schweregrades. 1 http://en.wikipedia.org/wiki/Beck_Depression_Inventory 14 3.1.4 Hamilton-Depressions-Skala (HAM-D, Hamilton rating scale for depression) Die Hamilton-Skala wurde 1960 von Max Hamilton entwickelt. Der Frageboden umfasst 21 Fragen, die vom therapierenden Arzt bzw. vom Untersucher beantwortet werden (siehe Anlage 3). Auf jede Frage gibt es zwischen drei und fünf Antwortmöglichkeiten, so dass insgesamt zwischen 0 und 67 Punkte vergeben werden können. Die Gesamtpunktzahl wird folgendermaßen bewertet (Tbl. 5): TABELLE 5: Schweregradeinteilung einer Depression gemäß Hamilton-Depressionsskala (HAM-D). [13] 0 7 18 25 - 6 - 17 - 24 - 67 Punkte: Punkte: Punkte: Punkte: keine Depression leichte Depression mittelgradige Depression schwere Depression 3.2 Epidemiologie der Depression Wie bereits in der Einleitung erwähnt, sind die Folgen von Depressionen für die Betroffenen und die Bundesrepublik nicht zu unterschätzen. Bis zu ihrem 65. Lebensjahr erkranken ungefähr 25 % der Frauen und 12 % der Männer mindestens einmal in ihrem Leben an einer klinisch relevanten Depression. [209] Laut einer Studie von Günther [71] lag die 12-Monats-Prävalenz von depressiven Erkrankungen 2002 in Deutschland bei 3,62 %. Dies entspricht 2,3 Millionen Bürgern im Alter von 20 bis 98 Jahren, bei denen eine Depression vorlag. Jacobi [86] berechnet in seiner Studie aus den Jahren 1998/1999 sogar eine 12-Monats-Prävalenz von 10,7 % bei den 18- bis 65-Jährigen, was die Absolutzahl der betroffenen Bürger fast verdreifachen würde. Europaweit geht die Global Burden of Disease Study der Weltgesundheitsorganisation (WHO) [143] von 22,2 Millionen und weltweit sogar von 151,2 Millionen Menschen mit einer unipolaren Depression aus. Besonders interessant ist dabei, wie schwer die Behinderung, d. h. die Einschränkung der Gesundheit, von der WHO 15 bewertet wird. Eine schwere Depression beeinflusst die Gesundheit genauso wie Blindheit, die Lähmung aller vier Gliedmaßen oder Krebs im Endstadium. Zudem liegt die Depression weltweit an erster Stelle bei den „years lost due to disability“ (YLDs). YLDs bezeichnen die Anzahl der Lebensjahre, die nicht in vollkommener Gesundheit verbracht werden können, und berücksichtigt sowohl die Dauer und Schwere der Krankheit als auch die Anzahl der Erkrankten. Bei den Männern ist von 24,3 Mio. YLDs die Rede, bei Frauen sogar von 41 Mio. YLDs. Somit „kostet“ keine andere Krankheit so viele gesunde Jahre wie die Depression. Vor diesem Hintergrund überrascht es nicht, dass 2002 die jährlichen Ausgaben für depressive Erkrankungen in Deutschland bei 1.114 Mio. € für Männer und 2.801 Mio. € für Frauen lagen. 2004 konnte sogar noch ein Anstieg auf 1.216 Mio. € (Männer) und 2.971 Mio. € (Frauen) verzeichnet werden. [26] 3.3 Ätiologie der Depression Jahrzehnte lang versucht man schon zu verstehen, wie eine Depression entsteht. Zahlreiche Ansätze hinsichtlich Einflussfaktoren und molekularer Abläufe konnten dabei bis heute wissenschaftlich nachgewiesen werden. Häufig gibt es aber auch widersprüchliche Ergebnisse, die es unmöglich machen, die Ätiologie der Depression mit einem einzelnen Prinzip von Ursache und Wirkung zu erklären. Vielmehr muss von einem multifaktoriellen Geschehen ausgegangen werden, das sich individuell bei jedem Patienten unterscheiden kann. 3.3.1 Familiäre und genetische Belastung Affektive Störungen treten familiär gehäuft auf. Fasst man unipolare und bipolare Erkrankungen zusammen, so leiden ungefähr 20 % der Eltern und 30 % der Geschwister eines Betroffenen ebenfalls an einem der beiden Krankheitsbilder. Dabei fallen mit einer Erblichkeit von 73 – 93 % insbesondere 16 die bipolaren Störungen ins Gewicht. [37] Dennoch weisen auch im Falle einer Major Depression 9 % der Verwandten 1. Grades ebendiese Erkrankung auf im Gegensatz zu 3 % in der Normalbevölkerung. [8] Gegenwärtig schätzt man, dass die Krankheit zu 31 - 42 % vererbt wird. [37] Dieser Trend verstärkt sich, wenn man Patienten selektiert, bei denen sich die Depression besonders früh manifestiert hat. [150] Weitere Aspekte, die den Grad der Vererblichkeit beeinflussen, sind die Episodenanzahl, die Dauer der längsten Episode, wiederkehrende Gedanken an den Tod oder Selbstmord sowie der Grad des Leidens und das Ausmaß der Beeinträchtigung. [93] Fraglich ist allerdings, ob das vermehrte Auftreten dieser Krankheitsbilder innerhalb einer Familie auf vererbbare Faktoren zurückzuführen ist, oder ob das gemeinsame soziale Umfeld eine tragende Rolle spielt. Studien deuten darauf hin, dass beide Faktoren zusammenspielen. [29, 73, 205] So könnte eine genetische Variante die Grundlage für eine krankhafte Reaktion auf belastende Lebensereignisse bilden. Bislang konnten mehrere Polymorphismen beschrieben werden, die höchstwahrscheinlich die Entstehung einer Depression begünstigen: Eine genetische Auffälligkeit zeigt das Chromosom 15 an der Position 15q25-q26. Holmans [82] konnte einen Zusammenhang zwischen dieser Genvariante und rezidivierenden, früh beginnenden Depressionen feststellen. In weiteren Studien wurden spezifische genetische Auffälligkeiten in der Promotorregion SCL6A4 des Serotonintransportergens (5-HTTLPR) auf Chromosom 17 an Position 17q11.17-q12 analysiert. [123] Träger kurzer Allele (short allels = S) scheinen gegenüber Trägern langer Allele (long allels = L) ein erhöhtes Risiko zu haben, an einer Depression zu erkranken. [94] Caspi [29] vermutet hier eher einen indirekten Zusammenhang: Die Mutation begünstigt in erster Linie in Kombination mit belastenden Lebensereignissen die Manifestation einer Depression. Das führt er darauf zurück, dass das S-Allel nur zu einer verminderten Transkription fähig ist und somit eine suffiziente Serotonin-Antwort auf Stress verhindert. Allerdings konnten diese Ergebnisse nicht in einer umfassenden Metaanalyse von Risch [160] bestätigt werden: Unter über 14.000 Probanden zeigte sich zwar ein signifikanter Unterschied zwischen Depressiven und Kontrollpersonen 17 bezüglich belastender Lebensereignisse, aber es zeigte sich weder ein statistisch relevanter Zusammenhang zwischen dem Auftreten einer Depression und dem 5-HTTLPR-Genotyp noch zwischen belastenden Lebensereignissen und dem 5-HTTLPR-Genotyp. Darüber hinaus bewirkt eine Mutation in dieser Promotorregion bei depressiven und gesunden Personen wahrscheinlich eine Hyperreagibilität der Amygdala. [74, 78] Insbesondere nach emotionalen Stimuli soll es bei Short-Allel-Trägern zu einer übersteigerten Reaktionsbereitschaft kommen. [41, 42] Die Amygdala ist ein Teil des limbischen Systems und spielt eine wesentliche Rolle bei Emotionen, bei der Bildung des emotionalen Gedächtnisses und der Furchtkonditionierung. Das S-Allel im Serotonintransporter könnte in diesem Zusammenhang zu so genannten „negativen Bias“ führen, d. h. emotionale Erfahrungen werden tendenziell eher negativ bewertet. [206] Teilweise konnte diese gesteigerte Aktivität in Kernspin-Untersuchungen in Form einer Volumenzunahme insbesondere der linken Amygdala gegenüber gesunden Kontrollpersonen bestätigt werden. [72, 107, 126] Es gibt jedoch auch Studien, in denen eine signifikante Volumenminderung bei depressiven Probanden festgestellt wurde. [183] 3.3.2 Monoamin-Mangel-Hypothese Das Noradrenalin- und Serotoninsystem beeinflusst unter anderem unsere Gefühle, unsere Gedanken und unser Handeln. Insofern verwundert es heute nicht, dass diese Neurotransmitter vermutlich eine entscheidende Rolle bei der Pathologie der Depression spielen. Identifiziert wurde dieser Mechanismus Ende der 50er Jahre allerdings eher zufällig: Patientinnen, die wegen einer Tuberkulose mit Iproniazid behandelt wurden, hatten nach der Einnahme des MAO-Hemmers plötzlich merklich bessere Laune. [137] Zur gleichen Zeit entdeckte der Schweizer Psychiater Roland Kuhn aufgrund von sorgfältigen klinischen Beobachtungen 1957 das erste trizyklische Antidepressivum. Beim Versuch, eine nebenwirkungsärmere Variante des Neuroleptikums Chlorpromazin zu entwickeln, stieß er auf die antidepressive Wirkung von Imipramin. [12, 83] 18 In zahlreichen Studien konnten bei depressiven Patienten Dysregulationen im Monoaminstoffwechsel aufgezeigt werden. Dabei lassen sich drei Angriffspunkte unterscheiden: • Verminderung der Transmitterkonzentration in der präsynaptischen Zelle • Störung der Transmitterwirkung an den Rezeptoren im synaptischen Spalt • Unterbrechung der Signaltransduktion auf Ebene der Second Messenger in der postsynaptischen Zelle Darüber hinaus wird am Ende des Kapitels auf die besondere Rolle des Monoamins Dopamin eingegangen. 3.3.2.1 Störung der Monoamin-Konzentration Die Konzentration der Monoamine Serotonin und Noradrenalin kann durch eine verminderte Produktion oder einen gesteigerten Abbau der Transmitter herabgesetzt sein. Zhang [216] gelang der Nachweis einer Mutation in der gehirnspezifischen Tryptophan-Hydroxylase TPH-2 (siehe Abb. 1, Punkt ). Diese Mutation tritt gehäuft in der depressiven Bevölkerung auf und führt zu einer verminderten Serotonin-Syntheseleistung. Meyer [127] fand anhand von PETAufnahmen eine deutlich erhöhte MAO-A-Konzentration (siehe Abb. 1, Punkt ) in den Gehirnen von depressiven Personen, was zu einem vermehr- ten Abbau der Monoamine führt. Lambert [101] konnte unabhängig davon eine verminderte Produktion von Norepinephrin-Metaboliten (siehe Abb. 1, Punkt ) im Gehirn nachweisen. Auf der Basis dieser Erkenntnisse führte Ruhé [163] eine interessante Meta-Analyse durch. Zum einen testete er bei depressiven und gesunden Probanden die Auswirkung einer Tryptophan-Depletion (siehe Abb. 1, Punkt ), d. h. er entfernte das für die Synthese von Serotonin not- wendige Tryptophan aus den Körpern der Probanden. Des Weiteren beobachtete er die Folgen einer verminderten Noradrenalinsynthese, indem er die Tyrosin-Hydroxylase (siehe Abb. 1, Punkt ) inhibierte. Interessanterweise konnte er durch diese Maßnahmen bei Gesunden keine Depression hervorrufen, bei zuvor erfolgreich mit SSRIs therapierten depressiven Probanden 19 konnten jedoch Rückfälle provoziert werden. Diese Studie unterstreicht somit einmal mehr, dass die Ursache für eine depressive Erkrankung vermutlich aus einem Zusammenspiel mehrerer Faktoren resultiert. 3.3.2.2 Störung der Rezeptorwirkung In unterschiedlichen Studien fielen Rezeptor-Veränderungen im depressiven Probandenkollektiv auf. Einerseits findet man bei manchen Depressiven eine verminderte Konzentration des Proteins p11 (siehe Abb. 1, Punkt ). Dieses Protein ist für eine erhöhte Effektivität des präsynaptischen Serotonin-1BRezeptors (siehe Abb. 1, Punkt ) verantwortlich. [192] Andererseits scheint teilweise auch die Sensitivität des prä- und postsynaptischen Serotonin-1ARezeptors (siehe Abb. 1, Punkt ) herabgesetzt zu sein. [149] Darüber hinaus führt eine erhöhte präsynaptische α2-Noradrenalin-Rezeptor-Sensitivität (siehe Abb. 1, Punkt ) bei Depressiven über eine negative Rückkopplung wahr- scheinlich zu einer verminderten Norepinephrin-Freisetzung. [142] 3.3.2.3 Störung in der Signaltransduktionskaskade Serotonin und Noradrenalin lösen über postsynaptische Monoamin-Rezeptoren eine Second-Messenger-Kaskade aus. Dies führt entweder indirekt über Inositoltriphosphat (IP3) oder direkt über zyklisches Adenosinmonophosphat (cAMP) zu einer Aktivierung des cAMP response element-binding protein (CREB-1). CREB ist ein Transkriptionsfaktor, der die Expression bestimmter Gene reguliert, die unter anderem das Überleben von Neurotrophinen wie brain-derived neurotropic factor (BDNF) sichern. [49] (siehe auch Kapitel 2.3.4) In zwei Studien konnte eine verminderte IP3-Konzentration (siehe Abb. 1, Punkt ) im Gehirn nachgewiesen werden, zum einen direkt postmortem, [185] zum anderen indirekt anhand von MRT-Analysen. [40] Die Ursache könnte in einer erhöhten Serumkonzentration von IgM-Antikörpern gegen Inositol liegen. [112] Darüber hinaus wird eine verminderte cAMP-Antwort (siehe Abb. 1, Punkt ) bei Depressiven im Vergleich zu Kontrollpersonen diskutiert. [199] Die Bedeutung der CREB-Konzentration wird vor allem im Zusammenhang mit der Antidepressiva-Therapie interessant: So weisen Depressive ohne Antidepres20 siva-Therapie ein herabgesetztes CREB- und Phospho-CREB-Level (siehe Abb. 1, Punkt ) auf. [33] Unter einer Langzeittherapie mit Antidepressiva konnte jedoch im Tierversuch ein Anstieg von CREB im Gyrus dentatus beobachtet werden. [19] Dieser gehört als Teil des Hippocampus zum limbischen System und ist nicht nur für die Anlage neuer Erinnerungen verantwortlich, sondern spielt auch eine wesentliche Rolle bei der Stressverarbeitung und vermutilch auch bei der Neurogenese im Erwachsenenalter. [21, 24, 53, 166] 3.3.2.4 Störung im Dopaminstoffwechsel Die Modifikation des Dopaminstoffwechsels wird in erster Linie in der Antiparkinsontherapie genutzt. Da der Morbus Parkinson jedoch auffällig häufig mit einer Depression einhergeht, [91, 159] wurde auch die Wirksamkeit entsprechender Modulatoren in der Therapie von Depressionen untersucht. Sowohl das Antidepressivum Bupropion, ein Dopamin- Wiederaufnahmehemmer, als auch das ursprüngliche Antiparkinsonmittel Pramipexol, ein Dopaminrezeptoragonist, zeigen eine antidepressive Wirkung. [62] All diese Studien lassen eine zentrale Rolle des Monoaminstoffwechsels bei der Ätiologie der Depression vermuten. Man sollte jedoch bedenken, dass zwar ungefähr zwei Drittel auf diese Medikamente ansprechen, jedoch bessert sich auch ein Drittel auf Placebos. [115] Es ist also schwer einzuschätzen, wie viele Patienten eine Depression unabhängig von dem Monoamin-System entwickeln. 3.3.2.5 Zusammenfassung der Monoamin-Mangel-Hypothese In folgender Grafik werden noch einmal alle Angriffspunkte zur MonoaminMangel-Hypothese übersichtlich zusammengefasst. 21 ABBILDUNG 1: Veränderungen im Monoamin-Stoffwechsel und an der Synapse im Rahmen einer Depression. [9] Verminderte Serotonin-Syntheseleistung durch Mutation der TryptophanHydroxylase. Vermehrter Monoamin-Abbau durch eine erhöhte MAO-A- Konzentration. Verminderte Konzentration von Noradrenalin-Metaboliten. Provokation eines depressiven Rückfalls durch Tryptophan-Depletion. Ab- nahme der Noradrenalin-Synthese durch Inhibition der Tyrosin-Hydroxylase. Verminderte Protein p11-Konzentration bewirkt präsynaptischen Serotonin-1B-Rezeptors. eine Fehlsteuerung des Verminderte Sensitivität des prä- und postsynaptischen Serotonin-1B-Rezeptors. Verminderte Noradrenalin- Freisetzung über eine negative Rückkopplung durch die gesteigerte Sensitivität des präsynpatischen α2-Noradrenalin-Rezeptor. Konzentration. Verminderte cAMP-Antwort. Phopho-CREB-Level 22 Verminderte Inositol- Vermindertes CREB- und 3.3.3 Störung in der Hypothalamus-Hypophysen-NebennierenAchse 3.3.3.1 Die HHN-Achse und das Glukokortikoid Cortisol Eine weitere, zentrale Rolle bei der Depressionsforschung nimmt die Hypothalamus-Hypophysen-Nebennieren-Achse ein (HHN-Achse). Stress bewirkt über den Cortex im Hypothalamus die Freisetzung des Corticotropinreleasing hormone (CRH). Das führt in der Hypophyse zur CorticotropinAusschüttung, was wiederum die Sekretion von Cortisol in der Nebennierenrinde zur Folge hat. [8] Cortisol beeinflusst sowohl akut als auch langfristig das metabolische, kardiovaskuläre, zentralnervöse und das Immunsystem. McEwen [122] greift für diese Stressreaktion auf den Begriff „Allostase“ von Sterling [189] zurück, der die Fähigkeit beschreibt, Stabilität während einer Veränderung aufrechtzuerhalten. Konkret ist damit der Anpassungsvorgang des Organismus an Stress zugunsten Hypothalamus des verhindern Überlebens dabei gemeint. normalerweise Cortisolrezeptoren über eine im negative Rückkopplung eine zu hohe Cortisol-Ausschüttung. Durch häufigen Stress, eine fehlende Stressadaptation an einen repetitiven Stressor, eine verlängerte Stressantwort oder eine inadäquate Stressreaktion kann es jedoch zum so genannten „allostatic load“ kommen, so dass das physiologische Gleichgewicht nicht länger aufrecht erhalten werden kann und es zu einer chronischen Über- oder Unterreaktion kommt. Dies scheint auch bei Depressiven der Fall zu sein. Bei ihnen lassen sich unabhängig von gegenwärtigen, äußeren Stressfaktoren erhöhte Cortisol-Spiegel im Plasma, [27] eine erhöhte Cortisol-Ausscheidung im Urin, [129] ein gesteigertes CRHLevel im Liquor und vermehrt CRH-mRNA und -Proteine im limbischen Areal des Gehirns nachweisen. [124] Als mögliche Ursache kommt eine fehlende negative Rückkopplung in Frage, da bei jedem zweiten Depressiven keine Suppression des CRH durch das künstliche Glukokortikoid Dexamethason auslösbar ist. [28] Darüber hinaus ergaben weitere Untersuchungen interessante, direkte 23 Zusammenhänge zwischen Stress und Cortisol. Erwachsene, die in ihrer Kindheit körperlich oder psychisch missbraucht wurden, weisen ein erhöhtes CRH-Niveau im Liquor auf. [102] Dieses Resultat kann im Tierversuch indirekt mit dem so genannten „erzwungenen Schwimmtest“ genauer untersucht werden. Bei diesem Test befindet sich das Nagetier in einem mit Wasser gefüllten Behälter, in dem es bis zur totalen Erschöpfung schwimmen muss. In einer Studie wurden Jungtiere von ihrer Mutter getrennt. Im späteren Verlauf konnte bei den gleichen ausgewachsenen Versuchstieren eine deutlich verkürzte Schwimmzeit beobachtet werden, durch Gabe von Antidepressiva konnte die Schwimmzeit hingegen wieder verlängert werden. [14] Ein vergleichbarer Zusammenhang zwischen Stress, Cortisol und Antidepressiva war beim Menschen bislang jedoch nicht möglich. Eine Ursache dafür könnte sein, dass das Cortisolniveau zum einen zirkadianen Rhythmen unterliegt, und zum anderen von Individuum zu Individuum sehr unterschiedlich ist. Dadurch entstehen große Schnittmengen zwischen der Cortisolkonzentration bei Gesunden und Depressiven. Es scheint dennoch messbare Unterschiede im Verlauf zu geben. Setzt man eine Versuchsperson leichtem Stress aus, so zeigen sich bei der depressiven Gruppe in der anschließenden Erholungsphase noch deutlich höhere Cortisolspiegel als bei der Kontrollgruppe. [27] Dies könnte auf eine mangelnde Normalisierung des Hormonspiegels nach Stresssituationen hindeuten. 3.3.3.2 Die HHN-Achse und die Monoamine Holsboer [84] versuchte bereits im Jahr 2000 einen Zusammenhang zwischen der HHN-Achse und den Monoaminen herzuleiten. Ihm gelang es, die HHNAchse durch erhöhte Monoamin-Spiegel in der Synapse zu beeinflussen und Folgen des Langzeitstresses dadurch umzukehren. Dieser Befund lässt vermuten, dass Antidepressiva vielleicht gar nicht direkt eine Stimmungsbesserung bewirken, sondern nur indirekt die negativen Folgen der Niedergeschlagenheit vermindern und dadurch subjektiv zu einem besseren Befinden beitragen. [9] Der Versuch, eine Depression direkt durch Modulation der Cortisolausschüttung zu therapieren, erwies sich jedoch als schwierig. CRH-Rezeptorantagonisten 24 konnten lediglich im Tierversuch erfolgreich eingesetzt werden [108] und auch der Glukokortikoid-Rezeptorblocker Mifepriston führte nur bei sehr schweren, psychotischen Depressionen zu einer Besserung der Symptomatik. [60] Es bleibt jedoch die Frage bestehen, ob Stress eine Depression verursachen kann oder ob die Stressreaktion vielmehr Folge einer lang andauernden, belastenden Depression ist. 3.3.4 Störung in der Neurogenese und die Funktion des Brainderived Neurotrophic Factor Unabhängig von diesen Studien gibt es auch die Vermutung, dass die (gestörte) Neurogenese eine Rolle bei der Depressionsentstehung spielen könnte. Lange Zeit ging man davon aus, dass sich Nervenzellen nur bei Insekten, Fischen und Amphibien während des ganzen Lebens neu bilden können. Bei Säugetieren schien dies hingegen nicht der Fall zu sein. Neue Studien zweifeln diese Hypothese an. Denn zumindest in einzelnen Arealen des Gehirns, insbesondere im Hippocampus, konnte ein neurogenes Wachstum am adulten Gehirn von Säugetieren nachgewiesen werden – bislang jedoch vor allem bei Nagetieren und in geringerem Maße auch bei Primaten. [105, 153] Interessanterweise konnte in diesem Zusammenhang bei Studien an Nagetieren auch der Einfluss von Stress auf die Neurogenese genauer beschrieben werden: Die Gefangenhaltung in einem kleinen Behälter führt zu einer Unterdrückung der Neurogenese. Dieser Effekt kann jedoch durch die Gabe von Antidepressiva rückgängig gemacht werden. [114, 156] In Primaten konnte unter Antidepressiva- und Elektroschock-Therapie (analog zur Elektrokrampftherapie) eine verstärkte Neurogenese im Gyrus dentatus des Hippocampus beobachtet werden. [147] Die Übertragung auf den Menschen erweist sich jedoch als schwierig. Bislang konnte nur in einer Studie von Eriksson [56] post mortem die Neurogenese im menschlichen Hippocampus nachgewiesen werden. Zusätzlich gibt es Erkenntnisse, die indirekt darauf hindeuten, dass die adulte Neurogenese auch beim Menschen physiologisch vorkommt: 25 ein erhöhtes Glukokortikoid-Niveau ist bei Ratten und Primaten mit einer Atrophie des Hippocampus assoziiert. [169, 170] Eine Erklärung dafür könnte sein, dass das Hippocampusareal wesentlich an dem negativen Feedback der HHN-Achse beteiligt ist. Eine Dysregulation in der Glukokortikoidsekretion kann zunächst zu reversiblen, langfristig auch zu irreversiblen Zellschäden führen. [8] Betrifft dieser Zelluntergang den Hippocampus, so könnte dies zu einer verminderten Hemmung und somit zu einer gesteigerten Freisetzung von Glukokortikoiden führen, was schließlich in einen Teufelskreislauf mündet (Abb. 2). 1. Glukokortikoide ↑↑ 2. Hippocampusatrophie ↑ ABBILDUNG 2: 3. Negatives Feedback auf HHN-Achse ↓ Circulus vitiosus: Glukokortikoide. Ein zu hoher Glukokortikoidspiegel führt zur Hippocampusatrophie, wodurch die negative Rückkopplung herabgesetzt wird. Die dadurch bedingte erneute Cortisol-Ausschüttung führt zu einer weiteren Schädigung des Hippocampusgewebes. Neben der bereits erwähnten erhöhten Glukokortikoid-Freisetzung konnte in mehreren Studien bei depressiven Menschen interessanterweise auch eine deutliche Abnahme Magnetresonanztomographie des (MRT) Hippocampusvolumens nachgewiesen werden. in [111, der 190] Vermutlich besteht sogar ein Zusammenhang zwischen der Dauer der Depression und dem Ausmaß der Atrophie. [182, 184, 190] Alternativ zu dem Cortisol-Überschuss könnte jedoch auch ein Mangel des Neurotrophins Brainderived Neurotrophic Factor (BDNF) diese Größenminderung verursachen. Dieses Protein ist entscheidend für axonales Wachstum, neuronales Überleben und synaptische Plastizität und wird durch Stress und Cortisol supprimiert. [4, 99] Verstärkt wird diese Hypothese durch eine postsuizidal verminderte BDNF26 Konzentration im Hippocampus. [92] Duman [51] konnte eine Reduktion der BDNF-Expression unter anderem durch Stress nachweisen. Besonders hervorzuheben ist jedoch der Zusammenhang zwischen BDNF und der Therapie von Depressionen. Sowohl der regulierende Transkriptionsfaktor CREB als auch BDNF selbst steigen durch eine Therapie mit Antidepresssiva an. [19] Eine Zunahme des BDNF konnte auch durch die Behandlung mit Elektrokrampftherapie (EKT) erreicht werden. [35, 92] Dies könnte auf einen ursprünglichen BDNF-Mangel während einer depressiven Episode hindeuten. Durch eine verminderte Neurogenese, den Rückzug von Dendriten und einen vermehrten Zellverlust bzw. eine erhöhte Toxizität könnte dies ebenfalls zu einer Hippocampusatrophie führen. [111] Eine Studie deutet darauf hin, dass insbesondere Träger des met166-BDNFAllels häufiger einen verhältnismäßig kleinen Hippocampus haben. [61] Des Weiteren fand man heraus, dass eine epigenetische Histonacetylierung zu einer verminderten BDNF-Transkription führt. Dieser Effekt kann im Versuch durch Antidepressiva aufgehoben werden. [196] Es gibt allerdings auch Aspekte, die gegen eine ausschlaggebende Rolle des BDNF in der Ätiologie der Depression sprechen. Allem voran ist BDNF nicht sehr spezifisch für die Depression, sondern scheint vielmehr allgemein im Zusammenhang mit psychiatrischen Krankheiten zu stehen. [4] Diese Vermutung wird insbesondere durch BDNF-Knock-out-Mäuse bestätigt: sie zeigen kein spezifisch depressives Verhalten. [109] Hinzu kommt, dass eine erhöhte BDNF-Konzentration im Zusammenhang mit einer Inflammation im Gehirn und mit Neurotoxinen beobachtet wurde. Dieser Befund deutet auch auf negative Eigenschaften des Neurotrophins hin. [44] Andererseits ist es schwer zu unterscheiden, ob BDNF an der Entzündungsreaktion beteiligt ist oder der Entzündung entgegenwirken soll, da dieses Neurotrophin auch im Zusammenhang mit vaskulären Entzündungen wichtige Aufgaben erfüllt. 27 3.3.5 Depression und kardiovaskuläre Erkrankungen: Folgen einer Entzündungsreaktion? Die Depression geht mit einem erhöhten Risiko für verschiedene Krankheiten einher, unter anderem spielen kardiovaskuläre Krankheiten hier eine bedeutende Rolle. Bei Depressiven geht man von einem relativen Risiko von 1,5 bis 2 für die Manifestation einer koronaren Herzkrankheit aus, wenn die Person ansonsten physisch gesund ist. Existiert eine vorbestehende Herzerkrankung, so steigt das relative Risiko für die kardiale Morbidität und Mortalität sogar auf 1,5 bis 2,5 an. [104] Dome [48] vermutet, dass diese Komorbidität auf einer Suppression der Endothelialen Progenitorzellen (EPC) durch die Depression beruhen könnte. Dazu veranlasst hat ihn die Annahme, dass im Rahmen einer Depression eine Dysfunktion des Immunsystems und des Knochenmarks vorliegt. [151, 157] Interessanterweise spielen Endothelzellsignale wiederum eine wichtige Rolle für die Neurogenese im Gehirn. So beeinflusst der vaskuläre endotheliale Wachstumsfaktor (VEGF) in vitro nicht nur die Gefäß- sondern auch die Nervenzellneubildung. [203] Dies mündet erneut in einen Circulus vitiosus (Abb. 3). 1. Depression ↑↑ 5. KHK ↑ 2. Knochenmark, Immunsystem ↓ 4. Neurogenese ↓ 4. Angiogenese ↓ 3. VEGF ↓ ABBILDUNG 3: Circulus vitiosus: Knochenmark-/Immunsystem-Supression. Die Depression geht mit einer verminderten Leistung des Knochenmarks und Immunsystems einher, was zu einer VEGF-Suppression führt. Dadurch wird weniger Neurogenese und Angiogenese induziert und die Depression und vaskuläre Erkrankungen schreiten fort. 28 Das Immunsystem scheint jedoch in mehrfacher Hinsicht im Zusammenhang mit Depressionen relevant zu sein. Zahlreiche Studien sehen eine direkte Verbindung zwischen Depressionen und einer Entzündungsreaktion. Unter anderem kommt es zu einem Anstieg des C-reaktiven Proteins (CRP) und der Cytokine Interleukin-6 (IL-6) und Tumornekrosefaktor-alpha (TNF-α). Diese Entzündungsreaktion könnte wiederum ein kardiovaskuläres Geschehen beschleunigen. Möglicherweise spielen diese Parameter auch eine Rolle bei der Therapie von Depressionen. So deuten Studien darauf hin, dass Antidepressiva die Entzündungsreaktion abschwächen, Elektrokrampftherapien führen hingegen sogar zu einem Anstieg der Entzündungsparameter. [46] Umgekehrt scheint aber auch die Depression das Immunsystem zu beeinflussen. So konnte bei Stress, Angst und depressiver Symptomatik eine stärkere Progression von HIV/AIDS beobachtet werden. [65] 3.4 Therapiemöglichkeiten Zur Therapie der Depression stehen zahlreiche Möglichkeiten zur Verfügung. 3.4.1 Psychopharmakotherapie In Kapitel 2.3.2 wurde bereits darauf hingewiesen, dass die ersten Antidepressiva nur zufällig entdeckt wurden. Heute stehen dagegen unzählige Medikamente zur Therapie der Depression zur Verfügung. Die Antidepressiva können in drei Hauptgruppen unterteilt werden: Monoaminoxidase-Hemmer (MAO-Hemmer), unselektive tri- und tetrazyklische Antidepressiva und die nebenwirkungsärmeren selektiven Wiederaufnahmehemmer für Serotonin und/oder Noradrenalin (SSRI, SNRI, SSNRI). MAOHemmer setzen die präsynaptische Verstoffwechselung von Katecholaminen wie Noradrenalin und Serotonin herab und erhöhen somit deren Konzentration in der Synapse. Sowohl trizyklische Antidepressiva als auch Noradrenalin- und Serotonin-Wiederaufnahmehemmer verhindern die präsynaptische Wiederaufnahme dieser Substanzen, was ebenfalls zu einer erhöhten 29 Konzentration in der Synapse selbst führt. Durch beide Mechanismen sind eine vermehrte Rezeptorbindung und somit auch eine stärkere postsynaptische Wirkung möglich (Abb. 4). [9] Darüber hinaus gibt es noch atypische Antidepressiva. (Tbl. 6) Axon einer Nachbarzelle Dendriten Zellkern Zellkörper Myelinscheide Axon präsynaptische Zelle (Axon) Vesikel mit Neurotransmittern Synaptischer Spalt Impulsrichtung MAO Postsynaptische Zelle (Dendrit) ABBILDUNG 4: Abbildung einer Nervenzelle (Übersichtsbild) Axonende und Darstellung der Angriffspunkte von Antidepressiva an der Synapse (Detailbild): Tri/-Tetrazyklische Antidepressiva und selektive Aufnahmehemmer verhindern den Reuptake der Neurotransmittervesikel in die präsynaptische Zelle. MAO-Hemmer verhindert die präsynaptische Verstoffwechselung von Noradrenalin und Serotonin. 30 TABELLE 6: Einteilung der Antidepressiva. Gruppe Trizyklische AD Wirkmechanismus Nichtselektive SerotoninNoradrenalin-Reuptake-Inhibitoren Tetrazyklische AD Nichtselektive Reuptake-Inhibitoren und Rezeptorblocker Selektive Serotonin-ReuptakeInhibitoren SSRI SNRI SSNRI Atypische AD Selektive Noradrenalin-ReuptakeInhibitoren Selektive Serotonin-/NoradrenalinReuptake-Inhibitoren Ungeklärt Wirkstoffe Amitryptilin Desipramin Imipramin Mianserin Mirtazapin Fluoxetin Paroxetin Sertralin Reboxetin Venlafaxin Duloxetin Johanneskraut, Viloxazin Bis heute liegen jedoch die Ansprechraten der Antidepressiva nicht höher als 50 – 65 %, so dass unzähligen Patienten eine effektive medikamentöse Therapie verwehrt bleibt. [137] Hinzu kommt, dass die Wirkung der meisten Arzneimittel frühestens nach 2 - 3 Wochen eintritt, was die Compliance erschwert und den Zeitraum verlängert, bis die optimale Wirkungssubstanz gefunden wird. 3.4.2 Psychotherapie Neben den Medikamenten spielt die Psychotherapie eine wichtige Rolle bei der Behandlung depressiver Patienten. Diese umfasst drei Angriffspunkte. 3.4.2.1 Psychoanalytische und psychodynamische Therapie Die klassische Psychoanalyse wurde erstmals Ende des 19. Jahrhunderts von dem Wiener Nervenarzt Siegmund Freud erwähnt. Bis heute wurden diese Grundlagen weiterentwickelt zur psychoanalytischen Persönlichkeits- bzw. Krankheitslehre. [57] Das Ziel der Behandlung liegt darin, unbewusste Konflikte und fehlende Persönlichkeitsreife, die als Ursache für die depressive Symptomatik in Frage kommen, aufzudecken und zu beheben. Dies soll vorrangig durch das Bewusstmachen des Unbewussten geschehen. Dazu stehen dem 31 Therapeuten mehrere Mittel zur Verfügung. Die Therapiesituation entspricht der klassischen Vorstellung einer Psychotherapie: Der Analysand (der Patient) liegt auf einer Couch, der Analytiker (der Therapeut) sitzt passiv außerhalb des Blickfeldes des Patienten. Nun wird der Analysand zum freien Assoziieren angeregt. Er soll also seinen Gedanken freien Lauf lassen, ohne dass Themen tabuisiert werden. Der Analytiker soll sich zugleich in einer Haltung der gleichschwebenden Aufmerksamkeit befinden. Er soll also das Gehörte deuten und die unbewussten Wünsche, Emotionen, Ängste und Motive des Patienten erkennen. Als Letztes ist es noch Aufgabe des Therapeuten, sich ständig die Beziehung zum Patienten vor Augen zu halten und die damit verbundenen Übertragungen und Gegenübertragungen aufzugliedern, da daran typische emotionale Verhaltensmuster des Analysanden deutlich werden können. Dadurch soll der Patient erkennen, wo die bislang unerkannte eigentliche Ursache für sein Leiden liegt. [137, 181] Es ist jedoch zu bedenken, dass diese Art der Analyse des Gegenübers über Jahre hinweg mehrere Sitzungen pro Woche voraussetzt. Da dies unmöglich für jeden Patienten verwirklicht werden kann, entwickelten sich die psychodynamischen Therapien. [158] Der Therapeut nimmt dabei eine deutlich aktivere Rolle ein und sitzt dem Patienten gegenüber. Im Mittelpunkt steht diesmal das gegenwärtige Problem. Da hier nicht die gesamte Psyche des Patienten von Kindheit an aufgearbeitet werden soll, kann die Therapie in einem zeitlich begrenzten Rahmen stattfinden. [57] Dennoch ist in beiden Therapieansätzen eine uneingeschränkte Empathie und Verlässlichkeit des Therapeuten zwingende Voraussetzung. Vertrauen und Aufrichtigkeit sind die Basis eines Therapieerfolges, da sich der Patient andernfalls nicht öffnen und helfen lassen wird. Um zu entscheiden, welche Art der Behandlung für welchen Patienten geeignet ist, muss man sich veranschaulichen, wo das Problem verankert ist. Im Falle einer chronischen depressiven Störung verspricht eine lang andauernde, intensive Therapie in Form einer Psychoanalyse mehr Erfolg. Lässt sich die Depression jedoch auf eine konkrete Ursache zurückführen, bietet die psychodynamische Therapie einen schnelleren und effektiveren Weg zur Besserung der Symptomatik. [57] 32 3.4.2.2 Interpersonelle Psychotherapie Im Gegensatz zur Psychoanalytischen und Psychodynamischen Therapie wurde die Interpersonelle Psychotherapie spezifisch zur Therapie von Depressionen entwickelt und bedient sich dabei Techniken verschiedener Schulrichtungen. [97] Wie man dem Namen entnehmen kann, stehen bei dieser Therapie die zwischenmenschlichen Aspekte im Fokus der Aufmerksamkeit, da man annimmt, dass die Depression immer in einem psychosozialen und interpersonellen Kontext steht. [179] Sowohl die soziale Rolle als auch das zwischenmenschliche Verhaltensmuster ist also von Bedeutung. Unter Berücksichtigung dieses Zusammenhangs soll in 10 – 20 Einzelsitzungen nacheinander die depressive Symptomatik gelindert und die Krankenrolle gemindert werden. [137] 3.4.2.3 Verhaltenstherapie Der dritte Ansatz zur Behandlung von Depressionen umfasst die Verhaltenstherapie. Bei dieser Theorie geht man davon aus, dass die Ursache für die Depressionen durch fehlgeleitete Lernprozesse begründet ist. Die Grundlage bilden die Konditionierungsmodelle von Pawlow (1927) und Skinner (1953). Pawlow stellt durch die klassische Konditionierung den Zusammenhang zwischen einem zunächst neutralen Reiz und einer Emotion dar, wenn diese nur häufig genug zeitgleich ausgelöst werden. Skinner überträgt dieses Modell auf das Verhalten. Mit der instrumentellen/operanten Konditionierung erklärt er, wie das Verhalten durch die daraus resultierenden Konsequenzen beeinflusst wird. Bei der Verhaltensanalyse wird also untersucht, durch welche Bedingungen die Probleme der Patienten aufrechterhalten werden und welche Konsequenzen sich daraus ergeben. Den Mittelpunkt bildet das SORKC-Modell nach Kanfer (Abb. 5). [89] Es beschreibt, warum sich eine Person so verhält, wie sie sich verhält. 33 STIMULUS ORGANISMUS REAKTION KONTINGENZ CONSEQUENCE ABBILDUNG 5: SORKC-Modell nach Kanfer. Ein Stimulus (ein Reiz, eine Situation) wird von einem Organismus (einer Person) verarbeitet und bewirkt eine Reaktion, die wiederum eine Konsequenz (Belohnung, Bestrafung) zur Folge hat. Im Rahmen der Kontingenz werden die Zusammenhänge zwischen Stimulus, Reaktion und Konsequenz überprüft und führen gegebenenfalls beim nächsten Mal ein anderes Verhalten herbei. Der Stimulus steht für einen bestimmten Reiz bzw. eine Situation. Mit dem Organismus sind die individuellen biologisch-somatischen und kognitiven Charakteristika einer Person gemeint, die den Stimulus verarbeiten und dann in einer bestimmten Art und Weise eine Reaktion bewirken. Die Kontingenz beinhaltet die Überprüfung der regelhaften Zusammenhänge zwischen Stimulus, Reaktion und Consequenze, wobei Letztere die Folge des Verhaltens durch eine Belohnung oder Bestrafung beschreibt. Aus diesem Grund kann durch Manipulation der Variablen S, O, R und C mit unterschiedlichen Methoden das Verhalten verändert werden. Die Wirkung eines Stimulus kann durch Konfrontations- und Bewältigungstherapie modifiziert werden, der Organismus selbst wird durch kognitive Verfahren beeinflusst, die Reaktion wird mit Hilfe von Modelllernen verändert und die Konsequenz durch operante Methoden. [137] Insbesondere die kognitive Verhaltenstherapie ist bei Depressionen von entscheidender Bedeutung, da man bei diesem Ansatz davon ausgeht, dass sich Denken, Fühlen und Handeln gegenseitig stark beeinflussen. Dabei wird zunächst das Handeln, in einem weiteren Schritt das Denken positiv verändert, so dass sich der Patient letztendlich auch wieder besser fühlen soll. [78] 34 3.4.3 Elektrokrampftherapie Der ungarische Neuropsychiater Ladislas Meduna beobachtete 1934 erstmals, dass sich die Symptomatik von Psychosen mit künstlich durch Kampfer ausgelösten Krampfanfällen lindern ließ. Diese Methode optimierten 1938 die italienischen Wissenschaftler Cerletti und Bini [31] durch die etwas besser verträgliche Elektrokrampftherapie (EKT). Aufgrund fehlender Behandlungsalternativen und viel versprechender Therapieerfolge fand die EKT zunächst weltweit zahlreiche Anwendungsmöglichkeiten, unter anderem konnten auch bei Depressionen zahlreiche positive Ergebnisse verzeichnet werden. [59, 178] Mit der Einführung wirksamer Medikamente trat die Bedeutung der Krampftherapie jedoch wieder in den Hintergrund. Erst seit den 60er Jahren nimmt die Schocktherapie wieder an Beliebtheit zu, da die Krampfprovokation unter intensivmedizinischen Bedingungen, d. h. während einer Überwachung der Vitalparameter und nach Verabreichung von Narkotika und Muskelrelaxantien, eine wesentlich humanere Durchführung ermöglicht. [137, 178] Dennoch löst diese Art der Therapie bis heute bei vielen Menschen Unbehagen aus. Der Anblick eines bewusstlosen, wehrlosen Menschen, dem gezielt Elektroschocks verabreicht werden, scheint Erinnerungen an Menschenversuche insbesondere zu Zeiten des Dritten Reiches wachzurufen, die bedauerlicherweise auch unter dem Deckmantel der „medizinischen Forschung“ legitimiert wurden. Dabei sind die Nebenwirkungen verhältnismäßig selten und gering. In erster Linie ist das allgemeine Narkoserisiko von Belang. Die unerwünschten Folgen der EKT selbst sind meistens nur kurz andauernd (Stunden bis Tage). Verhältnismäßig oft kann es zu retro- oder anterograden Amnesien kommen und Verwirrungs- und Aufmerksamkeitsstörungen treten vermehrt auf. Aphasien, Agnosien und Apraxien treten dagegen nur selten auf. Verlängerte Krampfanfälle oder gar ein Status epilepticus werden nur äußerst selten ausgelöst. [137] Insgesamt nehmen diese Symptome jedoch nur bei wenigen Patienten einen so starken Charakter an, dass trotz einer Besserung der Depression zum Abbruch der Therapie geraten werden muss. Aus diesem Grund ist die Durchführung der EKT auch in der heutigen Zeit durchaus vertretbar und sinnvoll. Es gibt zwei wesentliche Gebiete, bei denen die Therapie mit einer Elektrokrampftherapie indiziert sein kann. Dazu zählen nach wie vor die Schizophrenien, aber auch Krankheitsbilder der 35 affektiven Störungen. Besonders gute Erfolge mit einer Ansprechrate von 70 – 90 % verspricht man sich bei schweren Depressionen, insbesondere wenn diese von psychotischen Symptomen begleitet werden. [148] Zusätzlich stellt die EKT bei medikamentös behandelten therapieresistenten Depressionen mit einer Erfolgswahrscheinlichkeit von 50 – 75 % eine sinnvolle Therapieergänzung dar. [95] Trotz unzähliger Studien ist der genaue Wirkmechanismus noch nicht gänzlich erforscht. Man geht jedoch davon aus, dass neben inhibitorischen Neurotransmittern sowohl Monoamine als auch Hormone beeinflusst werden. Zudem scheint sich die EKT positiv auf die Neurogenese auszuwirken. [125] Im Tierversuch konnte eine Proliferation von neuronalen Progenitorzellen im Hippocampus der Ratte in Abhängigkeit von VEGF nachgewiesen werden. [180] Mit Blick auf die Ätiologie der Depressionen (siehe Kapitel 2.3) scheinen also nahezu alle Angriffpunkte, die als Ursache für die Entstehung einer depressiven Episode in Betracht kommen, ausgenutzt zu werden. Unter anderem konnte in mehreren Studien ein Anstieg der inhibitorischen Gamma-Aminobuttersäure (GABA) gemessen werden. Dadurch kann eine Abnahme des Glutamat/GABA-Quotienten erzielt werden, der im Rahmen einer Depression häufig erhöht zu sein scheint. [165, 168, 173] Zusätzlich scheint die Sensitivität des Serotoninrezeptors 5-HT1A ebenso zuzunehmen wie die Bindung an die Dopamin-Rezeptoren D1 und D3 im Striatum. [79, 138, 191] Vermutlich steht insbesondere der Effekt auf die Dopamin-Wirkung in direktem Zusammenhang mit der Effizienz der EKT. [116, 117] Interessanterweise konnte im Tiermodell zudem neben einem Anstieg von neurotrophen Faktoren auch eine vermehrte Anzahl von Progenitorzellen im Hippocampus und eine Zunahme der Zellproliferation nachgewiesen werden. [120, 147] Darüber hinaus konnte nach der Therapie mit Elektrokonvulsion eine Normalisierung des Dexamethason-Tests erzielt werden. Diese Normalisierung der Hypothalamus-Hypophyse-Nebennieren-Achse scheint direkt mit der Wirksamkeit der EKT zu korrelieren. [100] Bislang konnten unzählige Angriffspunkte der EKT nachgewiesen werden, die genauen Wirkmechanismen sind jedoch noch unerforscht. Was man aber mit Sicherheit sagen kann, ist, dass die Elektrokrampftherapie die einzige hirnstimulierende Therapie ist, die so schnell und so häufig zu einer Besserung der Symptome führt. 36 Dennoch soll im Folgenden auch auf ähnliche Verfahren wie die Transkranielle Magnetstimulation, die Vagusnervstimulation und die Tiefe Hirnstimulation eingegangen werden. 3.4.4 Transkranielle Magnetstimulation Bei der Transkraniellen Magnetstimulation (TMS) werden mit Hilfe von starken Magnetfeldern bestimmte Regionen des Gehirns stimuliert oder gehemmt. Eine sinnvolle Applikationsnorm konnte bislang nicht erstellt werden, bei der Therapie von Depressionen scheinen jedoch repetitive Stimulationen im Bereich des präfrontalen Cortexes am effizientesten zu sein, da so langfristig Effekte an den Neuronen erzielt werden können. [137] Diese umfassen einerseits einen veränderten Blutfluss in den stimulierten Regionen, andererseits scheinen auch der Glukosestoffwechsel und Transkriptionsfaktoren für Neurotrophine und Transmitter beeinflusst zu werden. [39, 77] Die Nebenwirkungen sind aufgrund der nicht-invasiven Applikation gering. Am häufigsten ist mit Kopfschmerzen zu rechnen. Vereinzelt kann es zur Auslösung von hypomanen Phasen oder Manien kommen, selten wurde ein epileptischer Anfall provoziert. [38, 76] 3.4.5 Vagusnervstimulation Die Vagusnervstimulation (VNS) kam ursprünglich bei nicht beherrschbaren epileptischen Anfällen zum Einsatz. Erst später erkannte man den antidepressiven Effekt dieser Methode. Um den Vagus regelmäßig stimulieren zu können, muss operativ ein kleiner Schrittmacher an den linken Ast des X. Hirnnervs angebracht werden. Der rechte Ast sollte geschont werden, um eine Manipulation des Herz-Parasympathikus zu vermeiden. Ähnlich wie bei der TMS scheint der Blutfluss regional verändert zu werden. Vermutlich werden zusätzlich serotonerge und noradrenerge Systeme durch eine Aktivierung von Kerngebieten im Tractus solitarius beeinflusst, da dieser neben Projektionen zu limbischen und kortikalen Strukturen auch Bahnen zu noradrenergen Zentren wie dem Nucleus caeruleus und serotonergen Zentren wie den Raphekernen unterhält. [136] 37 Die Risiken der VNS beruhen in erster Linie auf lokalen Schädigungen im Operationsgebiet. So kann es verhältnismäßig oft zu Schädigungen des benachbarten Nervus recurrens kommen, was sich mit Heiserkeit, einer veränderten Stimmlage und Halsschmerzen äußern kann. Insbesondere zu Beginn ist auf Herzrhythmusstörungen zu achten. Bei regelmäßiger Stimulation scheint diese Nebenwirkung jedoch nicht mehr aufzutreten. Weiterhin konnte im Zusammenhang mit der Vagusnervstimulation ein vermindertes Appetitgefühl und somit eine Gewichtsabnahme beobachtet werden, [20, 145] was angesichts der zahlreichen appetitsteigernden Antidepressiva in den meisten Fällen jedoch eher eine gewünschte als eine unerwünschte Nebenwirkung ist. Da bislang jedoch die Wirksamkeit der Vagusstimulation in placebo-kontrollierten Studien sehr kontrovers diskutiert wird und das operative Risiko verhältnismäßig hoch einzuordnen ist, sollte diese Maßnahme den schwer depressiven Patienten vorbehalten bleiben. 3.4.6 Tiefe Hirnstimulation (deep brain stimulation, DBS) Die tiefe Hirnstimulation (DBS) wird vordergründig bei der Therapie von schweren Formen des Morbus Parkinson und bei starken Dystonien angewandt. [1, 10] Die Methode ist vergleichbar mit einem Schrittmacher für das Gehirn und setzt einen operativen Eingriff voraus. Durch Implantation einer Sonde kann die Funktion bestimmter Regionen moduliert werden. Zur Behandlung der Depression erbrachten unter anderem stimulierende Impulse im subgenualen Cingulum (Brodmann-Areal 25) und im Nucleus accumbens Erfolge, [63, 119, 176] in Einzelfallstudien führte auch die Stimulation des Epiphysenstiels zu einer Besserung der Depression. [171, 172] Der genaue Wirkmechanismus ist jedoch bislang nicht verstanden. Die Nebenwirkungen der Stimulation werden im Allgemeinen als gering eingeschätzt, bei Untersuchungen zur Therapie des Morbus Parkinson gab es jedoch Hinweise auf eine erhöhte Selbstmordrate nach einer DBS-Therapie. Weiterhin kann durch die Stimulation das Verhalten und Empfinden der Patienten verändert werden. [137] Darüber hinaus bestehen die üblichen Risiken eines Eingriffes am Gehirn (Blutungen, Infektionen, Hemiparese), die zwar selten auftreten, dafür aber umso weitreichendere Folgen 38 haben. [177] Aus diesem Grund wird die DBS gerade bei der Therapie sehr verzweifelter Menschen aus ethischer Sicht häufig als sehr fragwürdig eingestuft. Dennoch gibt es Fälle, in denen diese Therapieform gerechtfertigt sein kann. [193] 3.4.7 Lichttherapie Sonnenlicht wirkt sich positiv auf das Gemüt aus. Umgekehrt kann die Abwesenheit der Sonne jedoch auch eine so genannte saisonale Depression auslösen. Insbesondere bei dieser Untergruppe der affektiven Störungen wirkt daher die Therapie mit künstlichem Sonnenlicht besonders effektiv. Eine Kombination mit Medikamenten ist dabei ebenso denkbar wie eine alleinige Lichttherapie, falls Pharmazeutika nicht erwünscht oder kontraindiziert sind. In der Regel verwendet man eine Lampe, die das gesamte sichtbare Tageslichtspektrum mit einer Intensität von 10.000 Lux abstrahlt. Die klassische Therapie erfolgt täglich am Morgen für 30 Minuten, erste Erfolge können im Allgemeinen frühestens nach drei Tagen verzeichnet werden. [137] Besonders gut scheint diese Art der Therapie bei atypischen Depressionen mit erhöhtem Schlafbedürfnis und gesteigertem Appetit sowie bei psychischen Angstsymptomen zu wirken. Umgekehrt kommt es bei körperlichen Angstzuständen seltener zu einer deutlichen Symptombesserung. [110] Die Nebenwirkungen sind im Vergleich zu einer medikamentösen Therapie verhältnismäßig selten. Es kommt gelegentlich zu Augenbrennen und Kopfschmerzen aufgrund der Augenreizung durch das Licht. Diese Symptome können in äußerst geringen Fallzahlen durch Trizyklische Antidepressiva im Rahmen einer Photosensibilisierung noch verstärkt werden. [11] Zudem muss bei der Behandlung bipolarer Patienten mit der Provokation einer hypomanen Phase oder einer Manie gerechnet werden. In den letzten Jahren wurde viel an der Optimierung dieser Therapieform geforscht, bislang konnten aber in den meisten Fällen nur ähnliche Ziele wie bei der klassischen Anwendung erzielt werden. Die Kombination mit einem Schlafentzug scheint sich jedoch nach aktueller Studienlage zu bewähren. [11, 70] 39 3.4.8 Schlafentzugstherapie Wie schon mehrfach erwähnt, gehören Schlafstörungen zu den zentralen Symptomen einer depressiven Phase. Dies kann sich ebenso in Ein- und Durchschlafstörungen äußern wie in morgendlichem Früherwachen und ganztägiger Müdigkeit. Schlafstörungen können jedoch nicht nur Folge sondern auch Ursache einer Depression sein. In Studien fand man heraus, dass es im Rahmen eines chronischen Schlafdefizits zum einen zur Desensibilisierung von Serotonin-1A-Rezeptoren kommt, zum anderen scheint auch die Plastizität der Synapsen, die Gedächtnisleistung und die Funktion von NMDA-Rezeptoren beeinträchtigt zu werden. [162] Warum sich der intermittierende Schlafentzug dagegen positiv auf die Stimmung auszuwirken scheint, ist bislang ungeklärt. Es scheint einerseits durch die Aktivierung des Renin-Angiotensin-AldosteronSystems über eine gesteigerte Reninausschüttung eine Blutdrucksteigerung zu bewirken. [134] Andererseits wird die Aktivität der Hypophysen-NebennierenAchsen durch eine kurzfristig gesteigerte Cortisolsekretion über ein negatives Feedback herabgesetzt. [200] Zusätzlich scheint eine gesteigerte Neurogenese des Hippocampusareals angeregt zu werden. Insbesondere die beiden letzten Effekte scheinen mit der Ätiologie der Depression eng verknüpft zu sein (siehe Kapitel 2.3.3 bzw. 2.3.4). [64] Zusammenfassend kommt es bei annähernd 60 bis 70 % der Patienten zumindest zu einer kurzfristigen Symptomlinderung. [208, 211] Der klassische Schlafentzug ergänzt die medikamentöse Therapie und findet über eine komplette Nacht statt. Gegebenenfalls kann auch ein partieller Schlafentzug in der zweiten Nachthälfte, d. h. nach 1 Uhr, ausprobiert werden. Ein partieller Schlafentzug in der ersten Nachthälfte verspricht jedoch keinen Therapieerfolg. In jedem Fall ist ein verändertes Schlafverhalten an den Tagen vor und nach dem Entzug in Form von Vor- bzw. Nachschlafen kontraindiziert, ebenso verhindern bereits kurze Schlafepisoden während des Schlafentzugs ein Gelingen der Therapie. [137] Im Falle einer positiven Beeinflussung der Symptomatik kann der Schlafentzug ein- bis zweimal pro Woche wiederholt werden, es muss jedoch damit gerechnet werden, dass es nach kurzfristiger Symptomlinderung erneut zu einer Verschlechterung kommen kann. Die Nebenwirkungen des Schlafentzugs sind ähnlich wie bei der Lichttherapie selten, es kann jedoch bei bipolaren Patienten ebenfalls zur Auslösung hypomaner 40 Phasen und Manien kommen. Zusätzlich beobachtete Roehrs [161] eine verstärkte Schmerzwahrnehmung am Folgetag der Behandlung. Aus diesem Grund wird die Anwendung des Schlafentzugs bei Schmerzpatienten nur unter Vorbehalt empfohlen. 3.5 Telomere 3.5.1 Aufbau und Funktion der Telomere Als Telomere bezeichnet man die distalen Enden eukaryotischer Chromosomen. Diese Strukturen zeichnen sich bei Wirbeltieren durch hoch konservierte, doppelsträngige, repetitive TTAGGG-Nukleotidsequenzen aus. [128, 132] Den Abschluss der Telomere bildet der sogenannte G-tail bzw. G-overhang, [113] ein Guanin-reicher Einzelstrang am 3’-Ende mit angelagerten Proteinen, die als Telosom [106] oder Shelterin-Komplex [43] bezeichnet werden. Die Aufgabe der Telomere besteht darin, den Chromosomen Stabilität zu verleihen und die wertvolle Erbinformation vor diversen schädlichen Einflüssen zu schützen. Zum einen dürfen die einzelsträngigen Enden nicht mit Doppelstrangbrüchen verwechselt werden, weil dies zu einer irrtümlichem Antwort auf einen vermeintlichen DNA-Schaden führen würde. Weiterhin muss ein enzymatischer Abbau der Telomere verhindert werden und die Enden dürfen nicht miteinander verkleben. [15, 34] Deswegen bildet der G-overhang zusammen mit dem ShelterinKomplex eine Schlaufe, so dass eine Schutzkappe entsteht, die Griffith [66] als telomeric loop bzw. t-loop bezeichnete. 41 ABBILDUNG 6: Darstellung des t-loop und der angelagerten Proteine des Shelterin-Komplexes. [32] Darüber hinaus spielen die Telomere eine wichtige Rolle bei der Replikation, d. h. bei der Verdopplung der genetischen Information vor einer Zellteilung. Eine vollständige Replikation ist am 5’-Ende des diskontinuierlich replizierten Folgestrangs nicht möglich. [204] Stattdessen gehen vor jeder Mitose bzw. Meiose zwischen 30 und 250 Basenpaare verloren. [2] ABBILDUNG 7: DNA-Replikation. 2 Darstellung des Basenverlustes am 5’-Ende des Folgestrangs. 2 http://home.claranet.de/falkev/telo1.htm 42 Damit dies nicht unweigerlich zu einem Funktionsverlust durch Zerstörung von Genen führt, bestehen die Telomere aus unzähligen sich wiederholenden Basenfolgen, die keine Erbinformation enthalten. Die Telomere verlieren jedoch mit abnehmender Länge an Stabilität und die Gefahr von Mutationen und unkontrollierten Wachstumsprozessen nimmt zu. Aus diesem Grund wird ab einer bestimmten Telomerkürze entweder die Apoptose, d. h. der programmierte Zelltod, eingeleitet oder die Zelle differenziert sich, so dass eine erneute Teilung nicht mehr möglich ist. Dieser Weg wird als Seneszenz bzw. Zellzyklusarrest bezeichnet. [80, 210] Somit wird durch die Länge der Telomere wesentlich die Überlebensdauer einer Zelle bestimmt. Um die Teilungsfähigkeit dennoch zu erhöhen, können mittels reverser Transkription die TTAGGG-Sequenzen wieder verlängert werden. [96, 197] Dazu ist eine aktivierte Telomerase notwendig, die sich aus der reversen Transkriptase Tert und der RNA-Komponente Terc zusammensetzt. [69] Insbesondere in den Keimzellen wird so eine nahezu unbegrenzte Replikation ermöglicht. In etwas geringerem Maße trifft dies auch auf adulte Stammzellen in schnell regenerierbaren Geweben wie den Haarfollikeln [154], Darmkrypten [139], der Epidermis [75] und den Lymphozyten [81] zu. Man vermutet jedoch, dass die Telomeraseaktivität in diesen Zellen mit der Zeit abnimmt und somit zum Alterungsprozess des Körpers beiträgt. 3.5.2 Telomere, Stammzellen und hämatopoetische Progenitorzellen In besonderen Situationen, zum Beispiel bei einer schweren Erkrankung, wird im Körper die Teilung von Stammzellen angeregt, um einen Nachschub von gesunden, jungen Zellen zu gewährleisten. Aus embryonalen Stammzellen sind dabei zunächst Vorläuferzellen des Ektoderms (Haut, ZNS, Haare, Zähne), des Entoderms (Darm, Lunge, Pankreas, Leber) und des Mesoderms (Herz, Blut, Knochenmark, Muskel, Knochen) entstanden. Diese reifen bei Bedarf in mehreren Teilungen zu spezialisierten, differenzierten Zellen heran. Aus diesem Grund wurde in unserer Studie nicht nur die Telomerlänge in den Leukozyten bestimmt, sondern auch die Anzahl verschiedener Endothelialer Progenitorzellen (EPC) analysiert. Neben Hämatopoetischen Stammzellen 43 (HSC) (CD34, CD105) wurde dabei ein besonderes Augenmerk auf Neurale Stammzellmarker (CD133) gerichtet. [52] Da dieser Teil der Studie ausführlich in der Promotionsarbeit von Franziska Groenen [67] dargestellt wird, wird an dieser Stelle nicht näher auf die Details der Stammzellen eingegangen. 3.5.3 Die Bedeutung der Telomere bei der Entstehung von Krankheiten In zahlreichen Studien konnte bislang nachgewiesen werden, dass die Aktivität der Telomerase und somit auch die Länge der Telomere das Auftreten und den Verlauf diverser Krankheitsbilder beeinflussen. Dabei spielen drei Krankheitsgruppen eine entscheidende Rolle. Zum einen gibt es Krankheiten, die primär in höheren Lebensdekaden auftreten, wie zum Beispiel Erkrankungen des kardiovaskulären Systems. Zum anderen bewirken manche genetische Erkrankungen eine beschleunigte Alterung bestimmter Zellpopulationen. Diese werden auch als Syndrome des vorzeitigen Alterns oder Progerie-Syndrome bezeichnet. Im Rahmen dieser Erkrankungen treten zudem vermehrt Tumoren auf, die ebenfalls stark von der Aktivität der Telomerase abhängig sind. 3.5.3.1 Kardiovaskuläre Erkrankungen Viele Erkrankungen wie zum Beispiel die Herzinsuffizienz treten mit zunehmendem Alter deutlich häufiger auf als bei jungen Menschen. Da die Telomeraseaktivität im fortgeschrittenen Alter abnimmt und sich folglich die Telomere verkürzen, wurde vermutet, dass sich dies unter anderem auf die Funktion des Herzens negativ auswirken könne. In mehreren Studien konnte nachgewiesen werden, dass die Telomere beim Auftreten kardiovaskulärer Erkrankungen tatsächlich deutlich verkürzt sind. [23, 141, 167] Interessanterweise scheint auch die Mortalität im Rahmen dieser vornehmlich im höheren Alter auftretenden Erkrankungen mit der Telomerlänge zu korrelieren. Das heißt, je kürzer die Telomere sind, umso höher ist die Gefahr, an den Folgen einer Krankheit zu 44 versterben. [30] Darüber hinaus zeigen Studien, dass Faktoren, die den Alterungsprozess beschleunigen, wie beispielsweise Stress, Adipositas und Rauchen, ebenfalls zu einer Verkürzung der Telomere führen. [54, 198] 3.5.3.2 Syndrome des vorzeitigen Alterns (Progerie-Syndrome) Für die Entstehung dieser Erkrankungen kommen grundsätzlich zwei Ursachen in Frage. Zum einen kann eine Mutation der Telomerase zu einer verminderten Aktivität derselben führen und somit eine beschleunigte Zellalterung insbesondere von Telomerase-abhängigen Zellen induzieren. Nach Überschreiten des HayflickLimits, d. h. wenn sich die Telomere soweit verkürzt haben, dass eine weitere Zellteilung nicht ohne die erhöhte Gefahr einer Schädigung der Erbsubstanz gewährleistet werden kann, wird der vorzeitige Zelltod eingeleitet oder die Zellen differenzieren sich und verharren somit im Zellzyklusarrest. Teilen sich die Zellen dennoch weiter, so erhöht dies das Risiko einer Genmutation um ein Vielfaches. Zum anderen können Mutationen auch direkt durch defekte DNA-Reparaturproteine gehäuft auftreten. In beiden Fällen besteht die Gefahr der Deaktivierung von Tumorsuppressorgenen und Aktivierung von Onkogenen, was die Grundlage zur Entstehung von Tumoren bildet. [16] 45 TELOMERE KRANKHEITSBILDER DES NORMALE TELOMERVERKÜRZUNG VORZEITIGEN ALTERNS (~ 50 – 250 BP/Teilung) gestörte gestörte Telomerase- Reparatur- funktion proteine beschleunigte Zellalterung Unterschreiten der kritischen Telomerlänge APOPTOSE/ weitere SENESZENZ Zellteilungen erhöhtes Mutationsrisiko Reaktivierung der Telomerase + Verlust der Zellzykluskontrolle TUMORGENESE ABBILDUNG 8: Telomerlänge und Pathogenese. Veranschaulichung der Entstehung von Krankheiten in Abhängigkeit von der Telomerlänge. 46 Ein klassisches Beispiel für ein Progerie-Syndrom ist das Krankheitsbild der Dyskeratosis congenita (DC). Diese Erbkrankheit kann durch unterschiedliche Mutationen entstehen. So beruhen die autosomal-dominant vererbten Varianten direkt auf einem Defekt der Reversen Transkriptase oder der RNA-Komponente der Telomerase. [50, 202] Die häufiger auftretenden X-chromosomal vererbten Formen weisen Mutationen des Dyskerin-Gens DKC1 auf, was ebenfalls eine Komponente des Telomerasekomplexes darstellt. [50, 98] Die Klinik manifestiert sich daher vor allem in schnell regenerierenden Geweben. Äußerlich leiden die betroffenen Patienten unter schweren poikilodermatischen Haut- und Schleimhautveränderungen, dystrophem Nagelwachstum und Haarausfall. Besonders bedrohlich und potenziell tödlich sind jedoch die Manifestationen an inneren Organen in Form von Leber- und Lungenfibrosen, das gehäufte Auftreten von Malignomen auf dem Boden von Leukoplakien der Schleimhaut und schwere hämatologische Komplikationen in Folge einer Knochenmarkdysfunktion. [47]. Ebenso wie die Dyskeratosis congenita beruht die Aplastische Anämie auf einer gestörten Telomerasefunktion. [118, 212] Beispiele für Erkrankungen mit gestörten DNA-Reparatur-Proteinen und folglich einer erhöhten chromosomalen Instabilität sind das Nijmegen Breakage Syndrom, das Ataxie-TeleangiektasieSyndrom (Louis-Bar-Syndrom) und das Werner Syndrom. [18] 3.5.3.3 Tumorerkrankungen Durch eine verminderte Telomeraseaktivität altern Zellen und somit der ganze Körper, was das Entstehen verschiedenster Krankheiten begünstigt. Somit liegt der Gedanke nahe, durch eine dauerhafte Aktivierung der Telomerase nicht nur eine ewige Jugend und Unsterblichkeit erzielen zu können, sondern auch diverse Krankheiten heilen oder sogar deren Auftreten verhindern zu können. Die Länge der Telomere und die Aktivität der Telomerase sind jedoch Teil eines empfindlichen Gleichgewichts. Denn alle genannten Syndrome des vorzeitigen Alterns haben eine Gemeinsamkeit: Aufgrund des erhöhten Mutationsrisikos treten bei den betroffenen Patienten unverhältnismäßig häufig Malignome auf. [3, 55, 68, 194] Für Tumoren ist ein unkontrolliertes Wachstumsverhalten charakteristisch. Dies wird durch die Entkopplung des Zellzyklus von sämtlichen 47 Kontrollmechanismen ermöglichst. Ein Beispiel dafür ist das Tumorsuppressorgen p53, das im Falle einer DNA-Schädigung entweder die Apoptose oder den Zellzyklusarrest einleitet. [69] Wird p53 durch eine Mutation deaktiviert, können sich die entarteten Zellen weiter teilen. Da es im Rahmen der gesteigerten Zellteilung unweigerlich zu einer vorzeitigen Verkürzung der Telomere kommen würde und somit die Wachstumsfähigkeit limitiert wäre, weisen jedoch über 80 % der Tumoren beim Menschen eine dauerhafte Telomeraseaktivität auf. [87, 175] Höchstwahrscheinlich verfügen die restlichen Tumoren ebenfalls über ein Verfahren zur Telomerverlängerung, das als „alternative lengthening of telomeres“ (ALT) bezeichnet wird. [25, 133] Somit würde die künstliche Induktion der Telomeraseaktivität zur Therapie von Krankheiten immer das Risiko einer ungewollten Entartung des Gewebes erhöhen. 3.5.3.4 Psychische Erkrankungen Die Rolle der Telomeraseaktivität und der Telomerlänge im Zusammenhang mit somatischen Erkrankungen ist bereits seit Jahrzehnten bekannt. Erst in jüngster Vergangenheit fand man jedoch in Studien heraus, dass zum Teil auch bei psychischen Erkrankungen die Telomere gegenüber der Normalbevölkerung deutlich verkürzt sind. In einer Studie von 2006 stellte Simon et al [186] fest, dass die durchschnittliche Telomerlänge bei Patienten mit einer affektiven Störung (Major Depressionen und Bipolare Störungen) deutlich kürzer ist als in einer gesunden Kontrollgruppe (Any mood disorder: 6,98 kb ± 0,84 vs. Control: 7,64 kb ± 1,10). Die affektiven Störungen untereinander wiesen jedoch keinen Unterschied auf. Auch bei schizophrenen Erkrankungen konnte eine deutliche Verkürzung der Telomere nachgewiesen werden. [90] Yu et al [213] fand in seiner Studie zudem heraus, dass die Länge der Telomere bei schizophrenen Patienten scheinbar mit dem Therapieerfolg korreliert. Patienten, bei denen mit Antipsychotika eine deutliche Besserung der Symptomatik erzielt werden konnte, hatten dabei durchschnittlich nahezu genauso lange Telomere wie die Kontrollgruppe (Good Responders: 8,88 kb ± 0,90 vs. Control subjects: 8,91 kb ± 1,36). Die Probanden, die jedoch nicht von der medikamentösen Therapie profitieren 48 konnten, hatten dagegen deutlich kürzere Telomere (Poor Responders: 7,41 kb ± 0,97). Die Einteilung der Poor Responders und Good Responders wurde in der Studie durch mehrmalige Erhebung des BDI-Scores im Verlauf der Therapie objektiviert. Als mögliche Ursache für die kürzeren Telomere kommt bei beiden Erkrankungen eine generalisierte, dauerhafte, fehlregulierte Stressreaktion mit Ausschüttung der entsprechenden Mediatoren wie zum Beispiel Cortisol in Betracht. (vgl. Kapitel 2.3.3.1) [36, 152] Vermutlich verursacht der psychische Stress im Rahmen der Erkrankung langfristig oxidativen Stress, was wiederum biologischen Schaden in den Zellen anrichtet. [121, 130, 146] Vor dem Hintergrund dieser Erkenntnisse soll deswegen in dieser Studie erforscht werden, ob die Telomerlänge der depressiven Probanden wie bei Probanden mit einer Schizophrenie von dem Therapieerfolg abhängig ist oder ob die Länge der Telomere einen Einfluss auf die Schwere der Depression hat. 49 4 Probanden und Methoden 4.1 Probanden Die Studie wurde von der Ethik-Kommission der Friedrich-Alexander-Universität Erlangen-Nürnberg genehmigt. Alle Probanden nahmen freiwillig und nach ausführlicher Aufklärung sowohl über die Durchführung und Risiken der Untersuchung als auch über das Ziel der Studie teil. Die eingeschlossenen Patienten stammen alle aus der psychiatrischen und psychotherapeutischen Klinik der Universität Erlangen-Nürnberg, die Kontrollpersonen stammen aus dem Verwandten- und Bekanntenkreis der Doktoranden. 4.1.1 Kontrollgruppe Die Gruppe 1 entspricht der Kontrollgruppe. Sie umfasst 20 Probanden, die unter Berücksichtigung eines angepassten Geschlechterverhältnisses und eines annähernd gleichen Altersdurchschnitts ausgewählt wurden (Tbl. 7). Alle Kontrollpersonen verneinten eine Suchterkrankung, psychiatrische Erkrankungen oder die Einnahme von psychiatrischen oder neurologischen Medikamenten in der Vergangenheit oder Gegenwart. Zudem wurde eine gegenwärtige Infektion ausgeschlossen. TABELLE 7: Beschreibung der Kontrollgruppe (Gruppe 1). Männer Frauen Gesamt Anzahl 11 9 20 Alter ± SD 49,00 ± 14,7 49,22 ± 16,8 49,10 ± 15,2 4.1.2 Patientengruppe Die Patientengruppe umfasst 54 Probanden. Es wurden nur Probanden in die Studie eingeschlossen, bei denen die Kriterien für eine Major Depression 50 gemäß ICD-10- und DSM-IV erfüllt wurden. Dementsprechend wurden keine Patienten mit eine Schizophrenie zu der Studie zugelassen, andere psychiatrische, neurologische oder somatische Begleiterkrankungen waren jedoch aufgrund der geringen Probandenzahl kein Ausschlusskriterium. Zudem wurden Patienten mit missbräuchlichem Drogen- oder Medikamentenabusus in der Vergangenheit zu unserer Studie zugelassen. Die Gruppeneinteilung wurde aufgrund der Therapien, die die Patienten erhalten haben, vorgenommen. Dazu wurde zur Unterscheidung der Gruppe 2 und 3 die Totale Antidepressiva-Dosis (TAD) berechnet. Diese setzt sich wie folgt zusammen: Von jedem Antidepressivum wird die Tagesdosis als Anteil der zulässigen Tagesmaximaldosis angegeben. [164] Durch Addition aller Antidepressiva erhält man die totale Antidepressiva-Dosis. Ist zum Beispiel für das Medikament A eine Gesamtdosis von 200 mg zugelassen und ein Proband bekommt 100 mg täglich, so ergibt sich daraus der Wert 0,5. Nimmt er zusätzlich von Medikament B 75 mg bei einer zulässigen Gesamtdosis von 100 mg, so berechnet sich daraus ein Anteil von 0,75. Insgesamt hat dieser Proband also eine Totale Antidepressiva-Dosis von 0,5 + 0,75 = 1,25. Der Vorteil dieser Umrechnung besteht darin, dass die Dosierungen der unterschiedlichen Medikamente in Relation zueinander gesetzt werden und somit vergleichbar werden. Gruppe 2 umfasst alle Probanden, die eine TAD ≤ 1 haben, und wurde als „niedrig dosierte Antidepressiva-Gruppe“ bezeichnet. Dementsprechend wurden alle Probanden mit einer TAD > 1 der Gruppe 3 bzw. der „hoch dosierten Antidepressiva-Gruppe“ zugeteilt. Gruppe 4 umfasst unabhängig von der Medikamentendosierung alle Patienten, die jemals mit einer Elektrokrampftherapie behandelt wurden. (Tbl. 8) 51 TABELLE 8: Beschreibung der niedrig dosierten AntidepressivaGruppe, hoch dosierten Antidepressiva-Gruppe und Elektrokrampftherapie-Gruppe (Gruppen 2-4). Männer Frauen Gesamt Gruppe 2 (TAD ≤ 1) Anzahl 9 11 20 Alter ± SD 49,67 ± 15,1 48,55 ± 14,1 49,05 ± 14,2 Gruppe 3 (TAD > 1) Anzahl 7 9 16 Alter ± SD 49,00 ± 15,0 49,56 ± 16,1 49,31 ± 15,1 Gruppe 4 (Antidepressiva + EKT) Anzahl 5 13 18 Alter ± SD 50,20 ± 21,4 48,54 ± 10,8 49,00 ± 13,8 4.2 Bestimmung der Laborparameter Von jedem Probanden der depressiven Gruppen wurden zwei Blutröhrchen abgenommen. Ein EDTA-Röhrchen zur Bestimmung der Stammzellen und eines zur Bestimmung der Telomerlänge. Bei den Kontrollpersonen war leider eine sofortige Untersuchung des Blutes aus logistischen Gründen nicht möglich. Daher wurde in diesem Fall nur ein EDTA-Röhrchen zur Telomerlängen-Analyse entnommen. Die Stammzellen wurden zum Teil von uns selbst oder mit Unterstützung der Biologisch-Technischen Assistentin Ulrike Weinzierl im Forschungslabor Autoimmunität der Medizinischen Klinik 3 in Zusammenarbeit mit Prof. Dr. rer. nat. Dr. med. habil. Martin Herrmann bestimmt. Die Analyse der Telomerlänge konnte nicht vor Ort durchgeführt werden. Aus diesem Grund wurden die EDTA-Röhrchen unmittelbar nach der Entnahme tiefgefroren und zu einem späteren Zeitpunkt von Dr. Nils Hartmann im Leibniz Institut für Altersforschung, Fritz-Lipmann Institut in Jena, mit freundlicher Genehmigung von Dr. Christoph Englert bestimmt. 52 4.2.1 Bestimmung von adulten Stammzellen im peripheren Blut bei depressiven Patienten mit der Durchflusszytometrie Mit Hilfe der Durchflusszytometrie (FACS, Fluorescence Antibody Cell Sorter) ist es möglich, sowohl die Anzahl als auch die Eigenschaften bestimmter Zellen genau zu analysieren. Anhand der Zellgröße und charakteristischer Oberflächenmoleküle können so spezielle Zellpopulationen untersucht werden. 4.2.1.1 Das Streulichtsignal Anhand verschiedener Streulichter können verschiedene Zelltypen differenziert werden. Die einzelnen Zellen passieren in einer Spülflüssigkeit nacheinander den Lichtstrahl eines Argon-Ionen-Lasers. Dabei entstehen zwei Arten von Streulichtsignalen: (a) Das kaum abgelenkte Vorwärtsstreulicht (forward scatter, FSC) korreliert mit der Größe einer Zelle und dient in unserem Fall der Abgrenzung der Leukozyten von den Erythrozyten und Thrombozyten. (b) Das annähernd im 90° Winkel gebrochene Seitwärtsstreulicht (side scatter, SSC) gibt Auskunft über die Innenstruktur, z. B. die Granularität der Zelle. Dadurch ist eine Unterscheidung von Lymphozyten, Granulozyten und Monozyten möglich. Durch lineares Auftragen der beiden Größen in ein Diagramm können so die verschiedenen Zellpopulationen gut voneinander abgegrenzt werden. (Abb. 9) 4.2.1.2 Das Fluoreszenzsignal In einem weiteren Schritt werden mittels Fluoreszensmessung verschiedene Oberflächen-Antikörper detektiert. Dadurch kann die Anzahl bestimmter Zellen innerhalb einer Population ermittelt werden. Dazu werden spezifische Antikörper mit Fluoreszenzmarker wie dem grün fluoreszierenden Molekül FluoreszeinIsothiocyanat und dem gelbrot fluoreszierenden Phycoerythrin konjugiert. Bindet nun der Antikörper an ein Antigen, wird die Trägerzelle im Fluoreszenzlicht sichtbar. Durch Auftragen der verschieden Antikörper kann so eine weitere Zelldifferenzierung vorgenommen werden. [144, 214] (Abb. 10) 53 ABBILDUNG 9: Darstellung der Zellpopu- ABBILDUNG 10: Darstellung der Subpopu- lation. lationen. Differenzierung der Leuko- Differenzierung zyten-Populationen spezifischer Auftragen der durch Zellgröße Leukozyten durch Auftragen (FSC) und der Zellstruktur verschiedener Antikörper (SSC). (Markierung mit fluoreszierendem grünFluores- zein-Isothiocyanat und FITC gelbrot-fluoreszieren- dem Phycoerythrin PE. 4.2.1.3 Bestimmung der CD34-, CD105-, CD133- und CD146-positiven Stammzellen in unserer Studie Verwendete Geräte: • FACS-Gerät (Beckman Coulter GmbH, Instrumententyp: EPICS XL/MCL, Instance-Nummer: 823981, Deutschland) • Lyse-Gerät (Beckman Coulter GmbH, Instrumententyp: TQ-Prep, InstanceNr.: 821708, Deutschland) • FACS-Röhrchen (BD Falcon, 5ml Polystyrene Round- Bottom Tube, BD Biosciences, USA) • Pipetten (pipetman, Gilson, Nummer: L22842J; P200 und Nummer: R53514N; P20, Frankreich) • Pipettenspitzen • Multipette (Eppendorf, Deutschland) 54 • Voatex-Genie 2 (Modell-Nr.: G-560 E, Seriennummer: 2-127326, Scientific Industries, Inc., USA) • Combitips 12,5 ml (Eppendorf Biopur, Deutschland) Verwendete Materialien • 100 l EDTA-Vollblut • Jeweils 4 l Antikörper (für CD34/CD45Ro; CD105/CD45; CD133/CD146) • Mucadont • Coulter Isoton II Diluent • PBS • PBS-PFA1% Zur Bestimmung der adulten Stammzellen aus dem peripheren Blut wurden zunächst jeweils 100 l Blut mit 4 l verschiedener fluoreszierender Antikörper entsprechend Tabelle 9 für 30 Minuten im Kühlschrank inkubiert und anschließend die Erythrozyten lysiert. TABELLE 9: SET 1 2 3 Verwendete Antikörper zur Bestimmung der adulten Stammzellen. Antikörper CD34 CD105 CD133 Konjugat PE PE PE Antikörper CD45Ro CD45 CD146 Konjugat FITC FITC FTC Anschließend wurden die Proben mit 3 ml PBS (phosphatgepufferte Salzlösung, phosphate buffered saline) durch Zentrifugation bei 1800 UpM für 5 Minuten alkalisiert und in 500 l einer 1 %igen PFA-Lösung (Paraformaldehyd) in PBS fixiert. Zuletzt wurden die Zellen mit einem FACS-Gerät hinsichtlich ihrer Antikörper analysiert. Im Anschluss wurden die erhobenen Daten am Computer ausgewertet: Nach Festsetzen einer Bezugspopulationen (Leukozyten und Lymphozyten) wurde die absolute Anzahl der Zellen mit den spezifischen Antikörpern berechnet. Um diese Absolutwerte miteinander vergleichen zu können, wurde der Quotient aus dem Absolutwert und der Bezugspopulation gebildet. Beispiel: Ein Proband hat 200.000 Leukozyten, davon 50.000 Lymphozyten, und insgesamt 50 CD34+/CD45+-positive Zellen, so ergibt sich ein relativer 55 Anteil von 0,25 x 10-04 (Leukozyten) bzw. 1 x 10-03 (Lymphozyten) positiven Zellen. Um die Zahlen etwas umgänglicher zu machen, wurden die Leukozytenund Lymphozyten-Quotienten mit 100.000 multipliziert, so dass sich in unserem Beispiel bzgl. der Leukozyten ein relativer Wert von 25 CD34+/CD45+-positive Zellen ergibt, im Bezug auf die Lymphozyten errechnet sich die Zahl 100. 4.2.2 Bestimmung der Telomerlänge im peripheren Blut bei depressiven und gesunden Probanden Die gängigste und zuverlässigste Methode zur Bestimmung der Telomerlänge stellt die Elektrophorese mit anschließendem Southern Blot dar. 4.2.2.1 Elektrophorese Zunächst wird die genomische DNA aus der Zelle isoliert. Dies geschieht entweder manuell mittels Erythrozytenlyse und Deproteinisation oder, wie in unserer Studie, mit Hilfe eines DNA-Mini-Blood-Kits. Als nächstes wird die DNA von spezifischen Restriktionsenzymen in die gewünschten Abschnitte, die so genannten Telomerrestriktionsfragmente (TRF), unterteilt. Durch Auftragen auf ein Agarosegel trennen sich anschließend die DNA-Abschnitte durch Anlegen einer Spannung der Größe nach auf. 4.2.2.2 Southern Blot Das Ergebnis kann nach der Methode des Southern Blot sichtbar gemacht werden, indem man das entstandene Trennmuster auf eine Membran (z. B. Nylon oder Nitrocellulose) überträgt und fixiert. Zuletzt werden die gesuchten Sequenzen mit einer chemisch oder radioaktiv markierten, komplementären RNASonde gekennzeichnet, auf einem Röntgenfilm, Fotopapier oder PhosphoImager-Platten abgebildet und deren Länge mit Hilfe einer speziellen Software berechnet. 56 4.2.2.3 Bestimmung der Telomerlänge in unserer Studie Verwendete Geräte: • QIAmap kit (Qiagen, Hilden, Germany) • FLA-7000 PhosphoImager-System (Fujifilm, Düsseldorf, Germany) • MultiGauge Software (Fujifilm, Düsseldorf, Germany) Verwendete Materialien: • 1 ml EDTA-Vollblut • Restriktionsenzyme HinfI und RsaI • 0,7 % Agarosegel • Hybond-N+-Membran (Amersham) • [γ-32P]-ATP-markierte Telomersonde (TTAGGG)4 • 100,000 cpm/ml Rapid-hyb Buffer (GE Healthcare, Chalfont St.Giles, United Kingdom) • Waschlösung 3x SSC/0.1% SDS • Waschlösung 0.5x SSC/0.1% SDS Die Telomerlänge aller Probanden wurde aus Leukozyten bestimmt. Zunächst wurde unter Verwendung eines QIAmp kit (Qiagen, Hilden, Germany) genomische DNA aus 1 ml Blut isoliert. Aus dem gewonnenen Material wurden etwa 10 l DNA von den Restriktionsenzymen RsaI and HinfI über Nacht bei 37°C verdaut. Die so entstandenen Telomerrestriktionsfragmente wurden mit Ethylalkohol gefällt und 5 - 10 l DNA wurden auf ein 0,7 % Agarosegel aufgetragen. Die Fragmente wurden über mehrere Stunden bei einer Spannung von 150 Volt elektrophoretisch aufgetrennt. Das Gel wurde anschließend denaturiert und die DNA wurde mit einer Kapillare auf eine Nylonmembran (Hybond-N+, Amersham) übertragen und vernetzt (Southern Blot). Im nächsten Schritt wurde eine mit [γ-32P]-ATP radioaktiv markierte Telomersonde (TTAGGG) über Nacht bei 55°C an die Telomer-DNA hybridisiert. Dazu wurd e eine Sonde mit einer Konzentration von 100,000 cpm pro ml Rapid-hyp Buffer (GE Healthcare, Chalfont St.Giles, United Kingdom) verwendet. Zuletzt wurde die Membran einmal mit einer Lösung aus 3fach SSC (Saline-sodium citrate buffer) und 57 0.1 % SDS (Sodium dodecyl sulfate) und zweimal mit einer Lösung aus 0,5fach SSC und 0,1 % SDS bei einer Temperatur von 55°C gereinigt. Die Membran wurde mit einem FLA-7000 Scanner (Fujifilm, Düsseldorf, Germany, Abb. 11) zur Detektion und Visualisierung der radioaktiven Signale fotografiert und die durchschnittliche Telomerlänge wurde mit der Multi Gauge Software (Fujifilm, Düsseldorf, Germany) bestimmt. Um die Reproduzierbarkeit der verschiedenen Blots zu garantieren, wurden in jedem Durchlauf zwei Kontrollproben mit bekannter Telomerlänge mitgeführt. PPPPPC C PPCCCCPP P 10 kb 8 kb 10 kb 8 kb 6 kb 6 kb 4 kb 4 kb 2 kb 2 kb ABBILDUNG 11: Fotographie zwei verschiedener Southern Blots. Die Telomerlänge wurde von Leukozyten durch Restriktions-Fragmente Telomerund Southern Blotting bestimmt. Bei jedem Southern Blot wurden Proben von depressiven (P) und gesunden (C) Probanden bestimmt. 58 4.2.3 Statistische Auswertung Zur Auswertung der Statistik wurde das Statistical Package for Social Sciences (SPSS 17.0 und 18.0) angewandt. Da die Telomerlänge gemäß der Testung mit dem Shapiro-Wilk-Test nicht immer normalverteilt war, verglichen wir die Telomerlänge in den einzelnen Gruppen nicht-parametrisch mit dem Mann-Whitney U-Test. Der Spearman’s Rank Korrelationskoeffizient fand beim Vergleich zwischen der Telomerlänge und dem Alter Anwendung. Um den Zusammenhang zwischen zwei Variablen (zum Beispiel Telomerlänge und Erkrankungsdauer) unter Berücksichtigung weiterer Einflussvariablen wie Alter, Geschlecht und Rauchverhalten zu analysieren, wurde die partielle Korrelation angewandt. 59 5 Ergebnisdarstellung 5.1 Deskription der Gesamtgruppe Wie bereits in Kapitel 4.1.2.1 erwähnt, wurden die Probanden in vier Gruppen eingeteilt: Die gesunde Kontrollgruppe 1, die niedrig dosierte AntidepressivaGruppe 2, die hoch dosierte Antidepressiva-Gruppe 3 und die Elektrokrampftherapie-Gruppe 4. 5.1.1 Verteilung der Stichproben Insgesamt wurden 98 Probanden rekrutiert, davon 68 depressive Probanden und 32 Kontrollpersonen. Die Stichproben PT01 und PT02 wurden nachträglich ausgeschlossen, weil es sich nur um „Testproben“ zur Evaluation der Telomerbestimmung handelte und das Alter der Personen zu jung war. Die Stichprobe PT57 konnte nicht in die Studie mit eingeschlossen werden, weil die medikamentöse Entlassdosis des Probanden nicht eindeutig eruiert werden konnte. Außerdem wurden die Probanden PT31, PT32, PT53, PT74, PT76, PT103, PT104, PT107, PT109 und PT 110 aus der sehr jungen Kontrollgruppe 1 ausgeschlossen, um den Altersdurchschnitt auf 49,10 ± 15,25 Jahre anzuheben. Aus der EKT-Gruppe 4 wurde PT86 ausgeschlossen, um den Altersdurchschnitt auf 49,00 ± 13,8 Jahre zu senken. Im Anschluss wurden aus der niedrig dosierten Antidepressiva-Gruppe 2 die Probanden PT22, PT23, PT27, PT51 und PT63 sowie aus der hoch dosierten Antidepressiva-Gruppe 3 die Probanden PT12, PT16, PT28, PT37 und PT58 ausgeschlossen, um ebenfalls einen annähernd gleichen Altersdurchschnitt (Gruppe 2: 49,05 ± 14,19 Jahre bzw. Gruppe 3: 49,31 ± 15,09 Jahre) zu erhalten. Letztendlich wurden 20 Kontrollpersonen und 54 depressive Probanden in die Studie eingeschlossen. (Tbl. 10) 60 TABELLE 10: Verteilung der Stichproben auf die vier Probanden-Gruppen. Diagnose Kontrollgruppe 1 Niedrig dosierte AD-Gruppe 2 Hoch dosierte AD-Gruppe 3 EKT-Gruppe 4 Gesamtgruppe Häufigkeit 20 20 16 18 74 Prozent 27,0 27,0 21,7 24,3 100,0 5.1.2 Demographische Daten In die Studie wurden ausschließlich Kaukasier aufgenommen, um ein Verfälschen der Ergebnisse durch Rassenunterschiede auszuschließen. Zudem wurde auf ein annähernd gleiches Durchschnittsalter und ein ausgeglichenes Geschlechterverhältnis in den Untergruppen geachtet, da dies starke Auswirkungen auf die Telomerlänge haben könnte. Lediglich in der EKT-Gruppe sind deutlich mehr Frauen als Männer, was sich aufgrund der kleinen Gruppengröße nicht vermeiden ließ. (Tbl. 11) TABELLE 11: Demographische Daten der Probanden-Gruppen. n Geschlecht ♂/♀ n Alter, Jahre ± SD [Intervall] Nicht-/Raucher n Kaukasier n Gruppe 4 EKT-Gruppe Gruppe 3 Antidepressiva Hoch dosierte Gruppe 2 Antidepressiva Niedrig dosierte gruppe Gesamt- Depressive Gruppe 1 Kontrollgruppe Merkmale 20 54 20 16 18 11/9 21/33 9/11 7/9 5/13 49,10 ± 15,25 49,11 ± 14,07 49,05 ± 14,19 49,31 ± 15,09 49,00 ± 13,81 [24 ; 78] [19 ; 75] [19 ; 73] [24 ; 69] [21 ; 75] 17/3 38/16 13/7 10/6 15/3 20 54 20 16 18 61 5.1.3 Klinische Daten Die 3 depressiven Gruppen wurden zudem hinsichtlich ihrer klinischen Daten verglichen. (Tbl. 12) TABELLE 12: Klinische Daten der depressiven Gruppen. Gruppe 4 EKT-Gruppe Gruppe 3 Hoch dosierte Antidepressiva Gruppe 2 Antidepressiva Niedrig dosierte Gesamtgruppe Depressive Merkmale TAD bei Aufnahme ± SD 0,9 ± 0,8 0,6 ± 0,78 0,9 ± 1,0 1,1 ± 0,7 [Intervall] [0,0 ; 2,8] [0,0 ; 2,5] [0,0 ; 2,8] [0,0 ; 2,5] TAD bei Entlassung ± SD 1,1 ± 0,6 0,7 ± 0,2 1,6 ± 0,5 1,1 ± 0,7 [Intervall] [0,0 ; 2,8] [0,3 ; 1,0] [1,1 ; 2,8] [0,0 ; 2,5] HAM-D ± SD 23,9 ± 6,0 25,2 ± 5,7 23,6 ± 4,7 22,9 ± 7,2 [Intervall] [7 ; 36] [11 ; 36] [15 ; 31] [7 ; 36] BDI ± SD 26,9 ± 11,9 30,4 ± 13,9 24,2 ± 7,1 25,5 ± 12,7 [Intervall] [3 ; 59] [7 ; 59] [12 ; 42] [3 ; 50] 5,9 ± 1,0 5,7 ± 0,7 5,9 ± 1,0 6,2 ± 1,2 [5 ; 9] [5 ; 7] [5 ; 8] [5 ; 9] 1,6 ± 0,5 1,6 ± 0,5 1,6 ± 0,5 1,7 ± 0,5 DSM-IV ± SD [Intervall] Major-Kriterien ± SD [Intervall] Minor-Kriterien ± SD [Intervall] Erkrankungsdauer (Jahre) ± SD [Intervall] n stationäre Aufenthalte ± SD [Intervall] TCM n (Ja/Nein) Familienanamnese n (positiv/negativ) Internistische Erkrankungen n (Ja/Nein) Karzinom-Erkrankungen n (Ja/Nein) [1 ; 2] [1 ; 2] [1 ; 2] [1 ; 2] 4,3 ± 0,7 4,2 ± 0,4 4,4 ± 0,7 4,5 ± 0,9 [4 ; 7] [4 ; 5] [4 ; 6] [4 ; 7] 15,4 ± 14,4 14,6 ± 14,4 11,1 ± 12,6 20,3 ± 15,1 [0 ; 45] [0 ; 40] [0 ; 36] [1 ; 45] 2,9 ± 3,3 2,0 ± 2,0 2,2 ± 1,7 4,6 ± 4,8 [1 ; 17] [1 ; 8] [1 ; 6] [1 ; 17] 6/48 2/18 2/14 2/16 26/28 9/11 8/8 9/9 15/39 5/15 5/11 5/13 6/48 2/18 3/13 1/17 62 5.1.4 Labordaten Nachfolgend sind die wichtigsten Durchschnittswerte der Laborparameter im Gruppenvergleich aufgelistet (Tbl. 13): TABELLE 13: Laborparameter der Probanden-Gruppen. Telomerlänge [kb] ± SD [Intervall] Gruppe 4 EKT-Gruppe Gruppe 3 Antidepressiva Hoch dosierte Gruppe 2 Antidepressiva Niedrig dosierte Gesamtgruppe Depressive Gruppe 1 Kontrollgruppe Merkmale 7,54 ± 0,55 7,21 ± 0,61 7,22 ± 0,55 7,23 ± 0,78 7,17 ± 0,56 [6,3 ; 8,5] [5,8 ; 9,2] [6,3 ; 8,5] [5,8 ; 9,2] [6,6 ; 8,8] 20,00 21,00 19,00 22,00 22,50 CD34+/CD45+ (Leukos) 11,18 13,41 11,02 8,55 CD34+/CD45+ (Lymphos) 35,55 45,34 33,67 25,21 3,54 4,55 3,56 2,39 CD105+/CD45- (Lymphos) 12,65 16,40 11,83 9,23 CD105+/CD45+ (Leukos) 22,85 26,19 27,11 15,60 CD105+/CD45+ (Lymphos) 83,50 97,39 94,41 58,95 CD133+ (Leukos) 27,48 27,47 35,40 20,45 CD133+ (Lymphos) 85,59 94,83 100,28 62,27 4,39 4,82 6,15 2,32 13,39 15,71 17,23 7,15 CD146+ (Leukos) 199,36 210,11 230,24 159,97 CD146+ (Lymphos) 596,99 684,70 622,98 476,43 BP-Verlust/Jahr CD105+/CD45- (Leukos) CD133+/CD146+ (Leukos) CD133+/CD146+ (Lymphos) 63 5.2 Telomeranalyse 5.2.1 Abhängigkeit von Alter und Geschlecht In der Gesamtgruppe zeigt sich ein hochsignifikanter inverser Zusammenhang zwischen der Telomerlänge und dem Alter der Probanden (Korrelation nach Spearman, r = -,443, p < 0,01). Dieser setzt sich zusammen aus einer hochsignifikanten Korrelation in der Patientengruppe (Korrelation nach Spearman, r = -0,412, p = 0,02) und in der Kontrollgruppe (Korrelation nach Spearman, r = -0,439, p = 0,05). ABBILDUNG 12: Telomerlänge und Alter. Die Telomerlänge nimmt mit zunehmendem Alter sowohl in der depressiven Gesamtgruppe (r = -0,41, p < 0,01) als auch in der Kontrollgruppe (r = -0,44, p = 0,05) signifikant ab. Unter Einbeziehung aller Gruppen sind die Telomere der männlichen Probanden unter 50 Jahren signifikant länger als die Telomere der Probanden über 50 Jahre (Männer < 50 Jahre: 7,56 kb ± 0,61, Männer ≥ 50 Jahre: 7,06 kb ± 0,55, Mann-Whitney-U-Test, r = -2,15, p = 0,032, Tbl. 14). Ein ähnliches Bild zeigt sich in der Frauengruppe: In der Auswahl unter 50 Jahren liegt 64 die durchschnittliche Telomerlänge bei 7,47 kb ± 0,63, bei denen über 50 Jahren bei 7,09 kb ± 0,52 (Mann, Whitney-U-Test, r = -2,13 p = 0,034). Bei Gegenüberstellung der Telomerlänge in den beiden Geschlechtergruppen zeigt sich hingegen, kontrolliert nach dem Alter, kein signifikanter Unterschied. (Mann-Whitney-U-Test, r = -0,491, p = 0,623) TABELLE 14: Zusammenhang Alter – Telomerlänge und Geschlecht – Telomerlänge. Die p-Werte geben die Korrelation zwischen jungen und alten Männern (1), jungen und alten Frauen (2) und Männern und Frauen im Vergleich (3) an (MannWhitney-U-Test). Geschlecht Telomerlänge [kb] n Durchschnitt ± SD Intervall Männer (< 50 Jahre) 16 7,560 ± 0,61 6,641 – 8,796 Männer (≥ 50 Jahre) 16 7,064 ± 0,55 5,808 – 7,882 Frauen (< 50 Jahre) 21 7,473 ± 0,63 6,475 – 9,236 Frauen (≥ 50 Jahre) 21 7,089 ± 0,52 6,262 – 8,166 Männer 32 7,312 ± 0,63 5,808 – 8,796 Frauen 42 7,281 ± 0,60 6,262 – 9,236 Gesamt 74 7,294 ± 0,61 5,808 – 9,236 p-Wert (1) 0,032 - (2) 0,034 (3) 0,623 5.2.2 Vergleich der Gruppen Alle depressiven Gruppen haben im Durchschnitt deutlich kürzere Telomere als die gesunde Kontrollgruppe. (Abb. 13) 65 ABBILDUNG 13: Telomerlänge in den vier Gruppen. Die durchschnittlichen Telomerlängen der Patientengruppen (niedrig dosierte Antidepressiva-Gruppe: 7,215 ± 0,55 kb, hoch dosierte Antidepressiva-Gruppe: 0,75 kb, 7,223 ± Elektrokrampftherapie-Gruppe: 7,173 ± 0,56 kb) ist deutlich kürzer als die Telomerlänge der Kontrollgruppe (7,539 ± 0,55 kb). Dabei nimmt die Signifikanz mit Intensivierung der Therapie zu: Zwischen der niedrig dosierten Antidepressivagruppe und der Kontrollgruppe zeichnet sich lediglich ein Trend ab (Mann-Whitney-U-Test: r = -1,839, p = 0,068, Tbl. 15)), die hoch dosierte Antidepressivagruppe und die EKT-Gruppe unterscheiden sich dagegen signifikant von der Kontrollgruppe (Mann-Whitney-U-Test: Gruppe 3: r = -1,974, p = 0,049, Gruppe 4: r = -2,324, p = 0,019). Vergleicht man die depressive Gesamtgruppe mit der Kontrollgruppe bezüglich der Telomerlänge, berechnet sich sogar ein hochsignifikanter Unterschied (Mann-Whitney-U-Test: r = -2,550, p = 0,011). 66 TABELLE 15: Telomeranalyse im Gruppenvergleich. Die p-Werte geben den Telomerunterschied im Vergleich zur Kontrollgruppe an (Mann-Whitney-U-Test). Diagnose Telomerlänge, kb n Kontrollgruppe 1 Niedrig dosierte AD-Gruppe 2 Hoch dosierte AD-Gruppe 3 EKT-Gruppe 4 Depressive Gesamtgruppe p-Wert Durchschnitt ± SD Intervall 20 7,539 ± 0,55 6,262 – 8,535 20 7,215 ± 0,55 6,300 – 8,481 0,068 16 7,223 ± 0,75 5,808 – 9,235 0,049 18 7,173 ± 0,56 6,600 – 8,796 0,019 54 7,204 ± 0,61 5,808 – 9,236 0,011 Die einzelnen depressiven Gruppen unterscheiden sich untereinander jedoch nicht signifikant. (Abb. 14) ABBILDUNG 14: Telomerlänge und Alter (depressive Untergruppen). Die Telomerlängen der depressiven Untergruppen sind nahezu identisch. Ebenso ist der durchschnittliche Basenpaarverlust in allen vier Gruppen nahezu identisch (Abb. 15 – 18): 67 ABBILDUNG 15: Basenpaarverkürzung der Kontroll- gruppe. Durch graphische Ermittlung der exakten Telomerlänge im Alter von 40 bzw. 60 Jahren kann der jährliche Basenpaarverlust berechnet werden: (7720 BP – 7320 BP): 20 Jahre = - 20 BP/Jahr. ABBILDUNG 16: Basenpaarverkürzung der niedrig dosierten Antidepressiva-Gruppe. Durch graphische Ermittlung der exakten Telomerlänge im Alter von 40 bzw. 60 Jahren kann der jährliche Basenpaarverlust berechnet 7010 BP): 20 Jahre = - 19 BP/Jahr. 68 werden: (7390 BP – ABBILDUNG 17: Basenpaarverkürzung der hoch dosierten Antidepressiva-Gruppe. Durch graphische Ermittlung der exakten Telomerlänge im Alter von 40 bzw. 60 Jahren kann der jährliche Basenpaarverlust berechnet werden: (7430 BP – 6990 BP): 20 Jahre = - 22 BP/Jahr. ABBILDUNG 18: Basenpaarverkürzung der Elektrokrampftherapie-Gruppe. Durch graphische Ermittlung der exakten Telomerlänge im Alter von 40 bzw. 60 Jahren kann der jährliche Basenpaarverlust berechnet 6920 BP): 20 Jahre = 22,5 BP/Jahr. 69 werden: (7370 BP – Kontrollgruppe: - 20 BP/Jahr Niedrig dosierte Antidepressiva-Gruppe: - 19 BP/Jahr Hoch dosierte Antidepressiva-Gruppe: - 22 BP/Jahr Elektrokrampftherapie-Gruppe: - 22,5 BP/Jahr Betrachtet man die jährliche Basenpaarverkürzung der Kontrollgruppe als Norm, so ergeben sich folgende „Voralterungen“ der drei depressiven Gruppen: • Voralterung Gruppe 2: (7539 BP – 7215 BP)/20 BP/Jahr = 16,2 Jahre • Voralterung Gruppe 3: (7539 BP – 7223 BP)/20 BP/Jahr = 15,8 Jahre • Voralterung Gruppe 4: (7539 BP – 7173 BP)/20 BP/Jahr = 18,3 Jahre Durchschnittliche Voralterung der depressiven Gesamtgruppe: 16,75 Jahre 5.2.3 Analyse des Rauchverhaltens Das Patientenkollektiv wurde in eine Raucher- und eine Nichtrauchergruppe unterteilt. Nichtraucher war dabei per definitionem jeder, der seit 4 Wochen keine Zigarette geraucht hat. Die Rauchergruppe ist deutlich jünger als die Nichtrauchergruppe. Aus diesem Grund muss die Verteilung der durchschnittlichen Telomerlänge zusammen mit der Altersverteilung betrachtet werden. Da die Kontrollgruppe insgesamt nur drei Raucher umfasst, wurde die Analyse an der depressiven Gesamtgruppe durchgeführt (n Raucher/Nichtraucher = 16/38). Bei dem direkten Vergleich der Gruppen ohne Berücksichtigung des Alters ergibt sich keine Signifikanz (partielle Korrelation nach dem Alter kontrolliert, r = -0,010, p = 0,946, Tbl. 16). 70 TABELLE 16: Telomerlänge und Rauchverhalten. Der p-Wert gibt die Korrelation zwischen der Telomerlänge bei Rauchern und bei Nichtrauchern an. Es zeigt sich ohne Berücksichtigung des Altersunterschiedes kein signifikanter Unterschied (Mann-Whitney-U-Test, p > 0,05). Diagnose n Nichtraucher 38 Raucher Telomerlänge, kb ± SD Alter, Jahre ± SD [Intervall] [Intervall] 7,157 ± 0,55 51,13 ± 13,68 [5,8 ; 8,8] [21 ; 75] 7,313 ± 0,74 44,31 ± 14,24 [6,3 ; 9,2] [19 ; 68] 16 p-Wert 0,946 Stellt man jedoch die jährliche Basenpaarverkürzung in der Raucher- und Nichtrauchergruppe gegenüber, so zeigen sich deutliche Unterschiede (Abb. 19): Nichtraucher: (7,37 kb – 6,99 kb) / 20 Jahre = - 19 BP/Jahr Raucher: (7,43 kb – 6,90 kb) / 20 Jahre = - 26,5 BP /Jahr Bei den Rauchern verkürzen sich die Telomere demnach um ca. 7,5 BP pro Jahr mehr. ABBILDUNG 19: Telomerlänge und Rauchverhalten. Die jährliche Basenpaarverkürzung scheint bei Rauchern (- 26,5 BP/Jahr) deutlich höher als bei Nichtrauchern (- 19 BP/Jahr) zu sein. 71 5.2.4 Analyse der Erkrankungsdauer Als Nächstes wurden die drei Patientengruppen hinsichtlich der Erkrankungsdauer untersucht. Dabei zeigt sich folgendes Bild (Abb. 20): ABBILDUNG 20: Erkrankungsdauer in den depressiven Untergruppen. Die durchschnittliche Dauer der depressiven Erkrankung unterscheidet sich in den drei Untergruppen deutlich (niedrig 14,6 ± dosierte 14,4 Antidepressiva-Gruppe: Jahre, hoch dosierte Antidepressiva-Gruppe: 11,1 ± 12,6 Jahre, Elektrokrampftherapie-Gruppe: 20,3 ± 15,1 Jahre). Es zeigen sich kaum signifikante Unterschiede zwischen den drei Gruppen hinsichtlich der Erkrankungsdauer (Tbl. 17). Die hoch dosierte AntidepressivaGruppe hat dabei mit Abstand die kürzeste Erkrankungsdauer (11,1 ± 12,6 Jahre), die Dauer der Depression in der EKT-Gruppe ist dagegen fast doppelt so lang (20,3 ± 15,1 Jahre). Zwischen diesen beiden Gruppen zeigt sich ein gerade noch signifikanter Zusammenhang (Mann-Whitney-U-Test: r = -1,953, p = 0,050). Die niedrig dosierte Antidepressiva-Gruppe ordnet sich im Mittelfeld ein (14,6 ± 14,4 Jahre). 72 TABELLE 17: Telomerlänge und Erkrankungsdauer. Die drei Gruppen unterscheiden sich kaum, lediglich zwischen der hoch dosierten Antidepressiva-Gruppe und der EKT-Gruppe zeigt sich ein signifikanter Unterschied (partielle Korrelation, p = 0,050). Diagnose Erkrankungsdauer, Jahre ± SD p-Wert [Intervall] 14,6 ± 14,4 Niedrig dosierte AD-Gruppe [0 ; 40] 11,1 ± 12,6 Hoch dosierte AD-Gruppe ± SD [0 ; 36] 20,3 ± 15,1 EKT-Gruppe ± SD 0,050 [1 ; 45] In Bezug auf die Telomerlänge konnte weder in den Untergruppen noch in der depressiven Gesamtgruppe eine Korrelationen zwischen der Erkrankungsdauer und der Telomerlänge festgestellt werden (partielle Korrelation kontrolliert nach dem Alter, Gesamtgruppe: r = 0,011, p = 0,939, niedrig dosierte AntidepressivaGruppe: r = -0,073, p = 0,766, hoch dosierte Antidepressiva-Gruppe: r = 0,222, p = 0,427, EKT-Gruppe: r = -0,110, p = 0,673). 5.2.5 Analyse der Medikamentendosierung Die drei depressiven Gruppen wurden auf Zusammenhänge zwischen sämtlichen psychiatrischen und neurologischen Medikamentendosierungen und der Telomerlänge untersucht. Dabei wurden primär die Summenwerte einzelner Medikamentengruppen berücksichtigt, um die Fallzahlen möglichst groß zu halten. Es zeigen sich keine signifikanten Unterschiede zwischen den drei Gruppen. In den Medikamentengruppen kann jedoch eine inverse Korrelation zwischen der Medikamentendosis und der Telomerlänge beobachtet werden, insbesondere in der hoch dosierten Antidepressiva-Gruppe zeigt sich ein signifikanter Zusammenhang zwischen der Neuroleptika-Dosis bei Entlassung und der Telomerlänge (partielle Korrelation kontrolliert nach dem Alter, r = -0,586, p = 0,022, Tbl. 18). Dieser Befund zeigt sich in der EKT-Gruppe nicht. 73 TABELLE 18: Telomerlänge und Medikamentendosierungen. Die p-Werte geben die partiell korrelierte Signifikanz zwischen der jeweiligen [Intervall] (Entlassung), TMD ± SD Neuroleptika [Intervall] (Aufnahme), TMD ± SD Neuroleptika [Intervall] Antidepressiva (Entlassung), TMD ± SD [Intervall] (Aufnahme), TMD ± SD Antidepressiva [Intervall] Telomerlänge, kb ± SD Medikamentdosis und der zugehörigen Telomerlänge in einer Gruppe an. Niedrig dosierte 7,21 ± 0,6 0,6 ± 0,8 0,7 ± 0,2 0,1 ± 0,3 0,1 ± 0,3 AD-Gruppe [6,3 ; 8,5] [0,0 ; 2,5] [0,3 ; 1,0] [0,0 ; 1,1] [0,0 ; 1,2] p = ,072 p = ,658 p = ,856 p = ,521 Hoch dosierte 7,22 ± 0,8 0,9 ± 1,0 1,6 ± 0,5 0,1 ± 0,2 0,2 ± 0,3 AD-Gruppe [5,8 ; 9,2] [0,0 ; 2,8] [1,1 ; 2,8] [0,0 ; 0,7] [0,0 ; 1,0] p = ,934 p = ,078 p = ,285 p = ,022 7,17 ± 0,6 1,1 ± 0,7 1,1 ± 0,7 0,4 ± 0,5 0,4 ± 0,6 [6,6 ; 8,8] [0,0 ; 2,5] [0,0 ; 2,5] [0,0 ; 1,6] [0,0 ; 2,0] p = ,409 p = ,433 p = ,455 p = ,735 EKT-Gruppe Depressive 7,20 ± 0,6 0,9 ± 0,8 1,1 ± 0,6 0,2 ± 0,4 0,3 ± 0,4 Gesamtgruppe [5,8 ; 9,2] [0,0 ; 2,8] [0,0 ; 2,8] [0,0 ; 1,6] [0,0 ; 2,0] p = ,590 p = ,618 p = ,964 p = ,568 Niedrig dosierte Antidepressiva-Gruppe: Die alterskontrollierte inverse Korrelation zwischen der niedrig dosierten Antidepressiva-Dosis bei Aufnahme und der Telomerlänge (partielle Korrelation kontrolliert nach Alter, n (Personen, die Antidepressiva einnehmen) = 13, r = -0,422, p = 0,072, Abb.21 ) wird in erster Linie durch die Gabe von Selektiven Serotonin-Reuptake-Inhibitoren (SSRIs) bedingt (durchschnittliche Gesamtdosis: 0,44 TMD, partielle Korrelation kontrolliert nach n (Personen, die SSRIs einnehmen) = 8, r = -0,473, p = 0,041, Abb. 22). 74 Alter, ABBILDUNG 21: Telomerlänge und Totale Antidepressiva-Dosis bei ABBILDUNG 22: Telomerlänge stationärer Aufnahme. bei Mit abnehmender Telomerlänge Aufnahme. scheinen Mit die verabreichten und Gesamtdosis SSRIs stationärer abnehmender Antidepressiva-Dosen Telomerlänge anzusteigen. scheinen abreichten die verDosen selektiver SerotoninReuptake-Inhibitoren anzusteigen. Die Antidepressiva-Dosis bei Entlassung ist per definitionem ≤ 1 und somit nicht für eine Analyse dieser Art geeignet, da bei einer so geringen Dosisspanne mit keiner signifikanten Korrelation zu rechnen ist. Hoch dosierte Antidepressiva-Gruppe: In der hoch dosierten Medikamentengruppe zeigt sich, kontrolliert nach dem Alter, bei den Antidepressiva ein tendentieller (partielle Korrelation kontrolliert nach dem Alter, n (Personen, die Antidepressiva einnehmen) = 16, r = -0,468, p = 0,078, Abb. 23) und bei den Neuroleptika ein signifikanter (partielle Korrelation kontrolliert nach dem Alter, n (Personen die Neuroleptika einnehmen) = 11, r = -0,586, p = 0,022, Abb. 24) inverser Zusammenhang zwischen Medikamentendosis bei Entlassung und Telomerlänge. 75 und Totale ABBILDUNG 24: Telomerlänge Antidepressiva-Dosis bei Gesamtdosis ABBILDUNG 23: Telomerlänge und stationärer Entlassung. Neuroleptika Mit abnehmender Telomerlänge stationärer Entlas- scheinen sung. die verabreichten Antidepressiva-Dosen Mit anzusteigen. Telomerlänge abnehmender scheinen die abreichten von bei verDosen Neuroleptika anzusteigen. Die Analyse hinsichtlich der Medikamentendosierung bei Aufnahme ist in Gruppe 3 nicht sinnvoll, da hier gilt: n (TAD ≥ 1) = 5! (vgl. Analyse der Entlassdosis in Gruppe 2) 5.2.6 Analyse der Elektrokrampftherapie-Daten Bei Gruppe 4 wurde zudem der Einfluss der Elektrokrampftherapie auf die Telomere genauer analysiert. Es konnte kein Zusammenhang zwischen der Telomerlänge und der Anzahl von EKTs bzw. bipolaren EKTs, dem Zeitrahmen, in dem EKTs durchgeführt wurden und der Zeitspanne, die seit der letzten EKT vergangen war, festgestellt werden (partielle Korrelation, kontrolliert nach dem Alter, Tbl. 19). 76 TABELLE 19: Telomerlänge und EKT-Daten. Die p-Werte geben die partiell nach dem Alter korrelierten Zusammenhänge zwi- EKT-Gruppe 7,17 ± 0,6 [6,6 ; 8,8] 13,2 ± 7,3 0,9 ± 2,7 [kIntervall] EKT ± SD Latenz zur letzten [kIntervall] Dauer der EKT ± SD [kIntervall] EKTs) ± SD n (bitemporale [kIntervall] n (EKTs) ± SD [kIntervall] SD Telomerlänge, kb ± schen der Telomerlänge und den EKT-Daten an. 125,7 ± 113,0 ± 129,7 120,7 [2 ; 26] [0 ; 10] [4 ; 489] [1 ; 380] p = ,372 p = ,747 p = ,810 p = ,525 Zusätzlich wurde überprüft, ob ein Zusammenhang zwischen dem Erfolg einer Elektrokrampftherapie und der Telomerlänge besteht. Dazu wurden alle EKTPatienten in 2 Gruppen unterteilt. Zu den Respondern wurden alle Probanden gezählt, bei denen die EKT zu einer Besserung der EKT führte, ohne dass die Therapie aufgrund unangenehmer Nebenwirkungen abgebrochen werden musste. Als Non-Responder wurden alle Patienten gewertet, bei denen es zu keiner Linderung der Symptomatik kam oder bei denen – unabhängig vom Erfolg der Therapie – aufgrund der Nebenwirkungen die EKT-Sitzungen eingestellt werden mussten. Auch hier konnte ohne Berücksichtigung des Altersunterschiedes kein signifikanter Zusammenhang zwischen Response und Länge der Telomere festgestellt werden. (Mann-Whitney-U-Test, r = -1,626, p = 0,117, Tbl. 20) TABELLE 20: Telomerlänge und Response auf EKT. Die p-Werte geben den (nicht-signifikanten) Zusammenhang zwischen Telomerlänge und Alter bei Respondern und Non-Respondern auf EKT an. Diagnose n Responder 13 NonResponder 5 Telomerlänge, kb ± SD [Intervall] p-Wert 7,294 ± 0,61 Alter, Jahre ± SD p-Wert [Intervall] 45,46 ± 13,67 [6,6 ; 8,9] 0,059 6,859 ± 0,20 [6,6 ; 7,1] [21 ; 67] 58,20 ± 10,18 [49 ; 75] 77 0,117 Es zeigt sich jedoch ein deutlicher Unterschied hinsichtlich der jährlichen Basenpaarverkürzung: Bei den Non-Respondern scheinen die Telomere von Beginn an sehr kurz zu sein, dafür werden sie mit dem Alter nahezu nicht kürzer. Die Responder haben eine Basenpaarverkürzung, die dem durchschnittlichen Probandenkollektiv entspricht. ABBILDUNG 25: Telomerlänge und Response auf EKT. Die jährliche Basenpaarverkürzung scheint bei EKT-Respondern (- 23 BP/Jahr) deutlich höher als bei EKT-Non-Respondern (- 3,5 BP/Jahr) zu sein, wobei die Telomerlänge der Non-Responder scheinbar von Beginn an sehr kurz ist. Responder: (7,42 kb – 6,960 kb) / 20 Jahre = - 23 BP/Jahr Non-Responder: (6,91 kb – 6,84 kb) / 20 Jahre = - 3,5 BP/Jahr 5.2.7 Analyse der Transkraniellen Magnetstimulation Vorweg wurde überprüft, wie viele Probanden im Rahmen ihrer Depression mit einer transkraniellen Magnetstimulation (TMS) therapiert wurden. (Tbl. 21) 78 Transkranielle Magnetstimulation 2/18 2/14 2/16 4/32 samtgruppe Depressive Ge- Gruppe Antidepressiva- EKT-Gruppe AD-Gruppe (Ja/Nein) Hoch dosierte TMS AD-Gruppe Niedrig dosierte TABELLE 21: Verteilung der Stichproben (Transkranielle Magnetstimulation). 6/48 Eine transkranielle Magnetstimulation wurde nur bei sehr wenigen Patienten durchgeführt. Eine Analyse ist nur in der depressiven Gesamtgruppe möglich und die Aussagekraft ist stark eingeschränkt. Es zeigt sich alterskontrolliert kein signifikanter Unterschied hinsichtlich der Telomerlänge bei Patienten mit bzw. ohne eine Transkranielle Magnetstimulation (partielle Korrelation kontrolliert nach dem Alter, r = -0,075, p = ,594). In der Grafik (Abb. 26) deutet sich ein Unterschied in der Telomerverkürzung an. ABBILDUNG 26: Telomerlänge und Transkranielle Magnetstimulation. Die jährliche Basenpaarverkürzung scheint bei Patienten, die keine TMS erhalten haben (- 22,5 BP/Jahr), deutlich höher zu sein als bei Patienten mit einer TMS-Behandlung (- 9 BP/Jahr). 79 ohne TMS: (7,42 kb – 6,97 kb) / 20 Jahre = - 22,5 BP/Jahr mit TMS: (7,20 kb – 7,02 kb) / 20 Jahre = - 9 BP/Jahr TABELLE 22: Telomerlänge und transkranielle Magnetstimulation. Es gibt hinsichtlich der Telomerlänge keinen signifikanten Unterschied zwischen Patienten mit und ohne transkranieller Magnetstimulationen (partielle Korrelation, p > 0,05). Therapie n Ohne TMS 48 Mit TMS 6 Telomerlänge, kb ± SD Alter, Jahre ± SD [Intervall] [Intervall] 7,211 ± 0,63 49,40 ± 14,1 [5,8 ; 9,2] [19 ; 75] 7,141 ± 0,43 46, 83± 14,7 [6,6 ; 7,9] [30 ; 72] p-Wert (alterskontrolliert) ,594 5.2.8 Analyse der Stammzelldaten Zur Berechnung der Stammzellen waren drei Werte notwendig. 1. CD... Antikörper Lymphozyten Die CD... Antikörper Lymphozyten-Zahl gibt die absolute Anzahl der CD-positiven bzw. CD-negativen Stammzellen an. Sie sind nur in Relation zur Gesamt-Lymphozyten- bzw. Leukozytenzahl zu werten. Separat haben sie keine Aussagekraft, da ein Blutstropfen mit beispielsweise 9.000 Leukozyten statistisch mehr CD-positive bzw. -negative Stammzellen enthalten kann als ein Tropfen mit nur 3.000 Leukozyten. 2. CD... Leukozyten bzw. Lymphozyten Die CD... Leukozyten bzw. Lymphozyten-Zahl erfasst die gesamte Anzahl der Leukozyten bzw. Lymphozyten in dem untersuchten Blutstropfen. Da die Lymphozyten eine Untergruppe der Leukozyten sind, können beide Populationen als Bezugssystem fungieren. 80 3. CD...-Quotient (Leukozyten bzw. Lymphozyten) Der CD...-Quotient der Leukozyten bzw. Lymphozyten gibt das Verhältnis von der absoluten Zahl der Antikörper-positiven bzw. -negativen Zellen zur Summe der Leukozyten bzw. Lymphozyten: Zähler: CD... Antikörper Lymphozyten Nenner: CD... Leukozyten bzw. Lymphzyten Da der Absolutwert der CD...-positiven bzw. -negativen Stammzellen ausschließlich in Relation zur Leukozyten-/Lymphozytenzahl eine Aussagekraft besitzt, wurden nur die beiden anderen Werte hinsichtlich der Telomerlänge analysiert. 5.2.8.1 Analyse der Leukozyten bzw. Lymphozyten Gesamtzahl In der niedrig dosierten Antidepressiva-Gruppe korrelieren die Leukozyten mit der Telomerlänge invers (partielle Korrelation kontrolliert nach dem Alter, r = -0,456, p = 0,050, Abb. 27), in der EKT-Gruppe direkt (r = 0,418, p = 0,095, Abb. 28) miteinander. Diese Tendenz zeigt sich jedoch nur bei den Leukozyten, in denen die CD133-/CD146-positiven Stammzellen bestimmt wurden. ABBILDUNG 27: Telomerlänge und CD146-Leukozyten dosierte CD133/ (niedrig Antidepressiva- Gruppe). Mit abnehmender ABBILDUNG 28: Telomerlänge und CD133/ CD146-Leukozyten (ElektrokrampftherapieGruppe). Telo- Mit zunehmender Telo- merlänge nimmt die Anzahl merlänge nimmt die Anzahl der Leukozyten signifikant ab der Leukozyten tendentiell (r = -0,456, p = 0,050). zu (r = 0,418, p = 0,095). 81 5.2.8.2 Analyse der Stammzellen In der hoch dosierten Antidepressiva-Gruppe zeigt sich ein signifikanter Zusammenhang zwischen den CD34-/CD45-positiven Stammzellen (in Relation zur Leukozytengesamtzahl) und der Telomerlänge (partielle Korrelation kontrolliert nach dem Alter, r = 0,583, p = 0,023, Abb. 29). ABBILDUNG 29: Telomerlänge und (CD34+/CD45+)- Stammzellen (hoch dosierte Antidepressiva-Gruppe). Mit zunehmender Telomerlänge steigt die Zahl der (CD34+/CD45+)-Stammzellen signifikant an (r = 0,583, p = 0,023). Des Weiteren kann ebenfalls in der hoch dosierten Antidepressiva-Gruppe ein relativer Zusammenhang zwischen den CD105-positiven, aber CD45-negativen Stammzellen (in Relation zu den Lymphozyten) und der Telomerlänge beobachtet werden (r = -0,477, p = 0,072, Abb. 30). 82 ABBILDUNG 30: Telomerlänge und (CD105+/CD45-)- Stammzellen (hoch dosierte Antidepressiva-Gruppe). Mit abnehmender Telomerlänge nimmt die Zahl der (CD105+/CD45-)-Stammzellen tendenziell ab (r = -0,477, p = 0,072). ANMERKUNG: Die detaillierte Stammzellanalyse einschließlich der Gruppenunterschiede und Korrelationen mit anderen Parametern (abgesehen von der Telomerlänge) wird in einer separaten Promotionsarbeit von meiner Studienkollegin Franziska Groenen ausführlich dargestellt. [67] 5.2.9 Analyse der psychischen Komorbiditäten 5.2.9.1 Analyse der Stichprobenverteilung Bei der Analyse der psychischen Komorbidäten musste zunächst überprüft werden, in welchen Gruppen eine Auswertung überhaupt sinnvoll ist, da häufig die Fallzahlen zu gering waren (Tbl. 23). Signifikante Ergebnisse wurden deshalb nur berücksichtigt, wenn beide Gruppen die Bedingung n ≥ 5 erfüllten. 83 Gesamtgruppe Depressive Gruppe Antidepressiva- EKT-Gruppe AD-Gruppe (Ja/Nein) Hoch dosierte Komorbidität AD-Gruppe Psychische Niedrig dosierte TABELLE 23: Verteilung der Stichproben (Psychische Komorbiditäten). Bipolare Störung 2/18 1/15 3/15 3/33 6/48 Angst-/Panikstörung 6/14 7/9 2/16 13/23 15/39 Psychose 0/20 2/14 4/14 2/34 6/48 Persönlichkeitsstörung 5/15 1/15 0/18 6/30 6/48 Zwangserkrankung 1/19 0/16 2/16 1/35 3/51 Chron. Schmerzsyndrom 4/16 4/12 2/16 8/28 10/44 Essstörung 5/15 1/15 2/16 6/30 8/46 Unter der Rubrik Essstörung sind die Krankheitsbilder Anorexie, Adipositas per magna, Bulimia nervosa und sonstige Essstörungen zusammengefasst. Es gibt nur einen leichten Zusammenhang zwischen der Telomerlänge und dem Auftreten einer Persönlichkeitsstörung bei der niedrig dosierten Antidepressiva-Gruppe (Tbl. 24): TABELLE 24: Telomerlänge und psychische Komorbiditäten. In der niedrig dosierten Antidepressiva-Gruppe zeichnet sich ein tendenzieller Zusammenhang zwischen dem Auftreten von Persönlichkeitsstörungen und der Bipolare Störung Angst-/Panikstörung Gesamtgruppe ,436 ,869 ,528 ,699 Psychose Persönlichkeitsstörung Depressive Gruppe Antidepressiva- EKT-Gruppe AD-Gruppe (p-Wert) Hoch dosierte Komorbidität AD-Gruppe Psychische Niedrig dosierte Telomerlänge. (partielle Korrelation, p < 0,1). ,328 ,263 ,095 ,229 ,186 ,227 ,625 ,924 ,321 Zwangserkrankung Chron. Schmerzsyndrom Essstörung ,686 84 5.2.9.2 Persönlichkeitsstörung In der niedrig dosierten Antidepressiva-Gruppe ist die durchschnittliche Telomerlänge in dem Kollektiv mit Persönlichkeitsstörung deutlich länger als in der Gruppe ohne Persönlichkeitsstörungen (partielle Korrelation kontrolliert nach dem Alter, r = 0,394, p = 0,095, Tbl. 25). TABELLE 25: Telomerlänge und Persönlichkeitsstörung 1 (niedrig dosierte Antidepressiva-Gruppe). Beim Vergleich der Telomerlängen zwischen Patienten mit und ohne Persönlichkeitsstörung zeichnet sich der Trend ab, dass Probanden mit einer Persönlichkeitsstörung längere Telomere haben als Patienten ohne (partielle Korrelation, p < 0,1). Telomerlänge, Diagnose n kb ± SD Alter, Jahre ± SD [Intervall] Ohne Persönlichkeitsstörung Mit Persönlichkeitsstörung 15 5 [Intervall] 7,090 ± 0,48 50,00 ± 14,66 [6,3 ; 7,9] [19 ; 73] 7,588 ± 0,65 46,20 ± 13,81 [6,8 ; 8,5] [23 ; 58] p-Wert (alterskontrolliert) ,095 Zur Verdeutlichung wurde aufgrund des Altersunterschiedes zwischen den beiden Gruppen graphisch die durchschnittliche Telomerlänge im Alter von 49,00 Jahren ermittelt (Tbl. 26). TABELLE 26: Telomerlänge und Persönlichkeitsstörung 2 (niedrig dosierte Antidepressiva-Gruppe). Beim Vergleich der durchschnittlichen Telomerlänge im Alter von 49,00 Jahren haben die Probanden mit einer Persönlichkeitsstörung längere Telomere als die Probanden ohne diese Komorbidität. Diagnose Ohne Persönlichkeitsstörung Mit Persönlichkeitsstörung n Telomerlänge, kb Alter, Jahre 15 7,10 49,00 5 7,50 49,00 Graphisch veranschaulicht zeigt sich, dass sich die Telomere bei den Probanden mit einer Persönlichkeitsstörung jedoch scheinbar schneller verkürzen (Abb. 31). 85 ABBILDUNG 31: Telomerlänge und Persönlichkeits- störungen. Die jährliche Basenpaarverkürzung scheint bei depressiven Patienten mit einer Persönlichkeitsstörung (- 28 BP/Jahr) deutlich höher zu sein als bei Patienten ohne (- 15 BP/Jahr). ohne Persönlichkeitsstörung: (7,24 kb – 6,94 kb) / 20 Jahre = - 15 BP/Jahr mit Persönlichkeitsstörung: (7,76 kb – 7,20 kb) / 20 Jahre = - 28 BP/Jahr 5.2.10 Analyse der somatischen Komorbiditäten 5.2.10.1 Analyse der Stichprobenverteilung Zunächst wurden erneut alle Gruppen mit n < 5 nicht in die Auswertung mit eingeschlossen. (Tbl. 27) 86 Depressive Gesamtgruppe Gruppe Antidepressiva- EKT-Gruppe AD-Gruppe (Ja/Nein) Hoch dosierte Komorbidität AD-Gruppe somatische Niedrig dosierte TABELLE 27: Verteilung der Stichproben (Somatische Komorbiditäten). Diabetes mellitus Typ II 2/18 1/15 0/18 3/33 3/51 Internistische Erkrankung 5/15 5/11 5/13 10/26 15/39 Karzinom-Erkrankung 2/18 3/13 1/17 5/31 6/48 Unter dem Sammelbegriff „Internistische Erkrankungen“ wurden alle Krankheiten der Formenkreise „Metabolisches Syndrom“ und „Kardiovaskuläre Erkrankungen“ zusammengefasst. Es errechnen sich keine signifikanten Zusammenhänge zwischen der Telomerlänge und dem Auftreten internistischer Erkrankungen (partielle Korrelation kontrolliert nach dem Alter, Tbl. 28). TABELLE 28: Telomerlänge und somatische Komorbiditäten. Hinsichtlich der Telomerlänge gibt es keine signifikanten Unterschiede zwischen Probanden mit und ohne eine somatische Begleiterkrankung (partielle Korrela- Depressive Diabetes mellitus Typ II Internistische Erkrankung ,484 ,684 Karzinom-Erkrankung 87 ,277 ,826 ,449 ,891 ,756 Gesamtgruppe Gruppe Antidepressiva- EKT-Gruppe AD-Gruppe (p-Wert) Hoch dosierte Komorbidität AD-Gruppe somatische Niedrig dosierte tion, p > 0,05). 5.2.11 Analyse des Suchtverhaltens Die Auswirkungen des Rauchens wurden bereits ausführlich in Kapitel 4.2.3 dargestellt. An dieser Stelle soll nun noch der Einfluss einer Benzodiazepin-, Alkohol- und nicht näher bezeichneten Drogenabhängigkeit eruiert werden. 5.2.11.1 Analyse der Stichprobenverteilung Da die Anzahl der süchtigen Probanden sehr klein war, wurden unter der Annahme, dass sich ein exzessiver Drogenmissbrauch langfristig auf die Telomerlänge auswirken würde, gegenwärtig Süchtige mit in der Vergangenheit süchtigen Patienten in einer Gruppe zusammengefasst. Nach Ausschluss der Gruppen n < 5 ergibt sich folgende Stichprobenverteilung (Tbl. 29): Medikamentenabhängigkeit Gesamtgruppe Depressive Gruppe Antidepressiva- EKT-Gruppe AD-Gruppe Hoch dosierte (positiv/negativ) AD-Gruppe Suchtanamnese Niedrig dosierte TABELLE 29: Verteilung der Stichproben (Suchtanamnese). 3/17 1/15 2/16 4/32 6/48 Alkoholabhängigkeit 5/15 5/11 0/18 10/26 10/44 Drogenabhängigkeit 2/18 2/14 1/17 4/32 5/49 Suchtanamnese gesamt 8/12 6/10 3/15 14/22 17/37 (Benzodiazepine) In keiner der Gruppen zeigt sich ein signifikanter Zusammenhang zwischen der Suchtanamnese und der Länge der Telomere (partielle Korrelation kontrolliert nach dem Alter, Tbl. 30). 88 TABELLE 30: Telomerlänge und Suchtanamnese 1. Hinsichtlich der Telomerlänge gibt es keine signifikanten Unterschiede zwischen Probanden mit und ohne eine positive Suchtanamnese (partielle Korrelation, p > Medikamentenabhängigkeit Depressive Gesamtgruppe Gruppe Antidepressiva- EKT-Gruppe AD-Gruppe Hoch dosierte (p-Wert) AD-Gruppe Suchtanamnese Niedrig dosierte 0,05). ,849 (Benzodiazepine) Alkoholabhängigkeit ,795 ,521 ,491 Drogenabhängigkeit Suchtanamnese gesamt ,416 ,728 ,741 ,878 ,736 ,945 ABBILDUNG 32: Telomerlänge und Suchtverhalten. Es zeigt sich nahezu kein Unterschied hinsichtlich der Basenpaarverkürzung bei Patienten mit einer positiven und negativen Suchtanamnese. 5.2.11.2 Benzodiazepin- und Alkoholabhängigkeit Bei genauerer Analyse der einzelnen Grafiken in der depressiven Gesamtgruppe fällt jedoch auf, dass sich bei Benzodiazepin-abhängigen Probanden die 89 Telomere scheinbar etwas langsamer verkürzen, bei Alkoholabhängigen verkürzen sie sich offenbar deutlich schneller. ABBILDUNG 33: Telomerlänge und Benzo- ABBILDUNG 34: Telomerlänge diazepin-Abhängigkeit. Die jährliche verkürzung Alkohol-Abhängigkeit. Basenpaarscheint und Die jährliche Basenpaar- bei verkürzung scheint Patienten mit einer Benzo- Patienten mit diazepin-Abhängigkeit Alkohol-Abhängigkeit (- 11 bei einer (- BP/Jahr) deutlich geringer zu 37 BP/ sein als bei Patienten ohne Jahr) deutlich höher zu (- 21,5 BP/Jahr). sein als bei Patienten ohne (- 18 BP/Jahr). ohne Benzodiazepinabhängigkeit: (7,39 kb – 6,96 kb) / 20 J. = - 21,5 BP/Jahr mit Benzodiazepinabhängigkeit: (7,33 kb – 7,11 kb) / 20 J. = - 11 BP/Jahr ohne Alkoholabhängigkeit: (7,34 kb – 6,98 kb) / 20 J. = - 18 BP/Jahr mit Alkoholabhängigkeit: (7,69 kb – 6,95 kb) / 20 J. = - 37 BP/Jahr 90 TABELLE 31: Telomerlänge und Suchtanamnese 2. Beim Vergleich der durchschnittlichen Telomerlänge im Alter von 49,00 Jahren haben Probanden mit einer positiven Suchtanamnese (Benzodiazepin- und Alkoholabhängigkeit) längere Telomere als die Probanden ohne. Diagnose Ohne Benzodiazepinabhängigkeit Mit Benzodiazepinabhängigkeit Ohne Alkoholabhängigkeit Mit Alkoholabhängigkeit 5.2.12 n Telomerlänge, kb Alter, Jahre 48 7,19 49,00 6 7,23 49,00 44 7,18 49,00 10 7,35 49,00 Analyse der Familienanamnese Die Probanden mit einer positiven bzw. negativen Familienanamnese in Bezug auf psychiatrische Erkrankungen sind optimal auf beide Gruppen verteilt. (Tbl. 32) Gesamtgruppe 17/19 Depressive 9/9 Gruppe AD-Gruppe 8/8 Antidepressiva- 9/11 EKT-Gruppe Familienanamnese Hoch dosierte (positiv/negativ) AD-Gruppe Anamnese Niedrig dosierte TABELLE 32: Verteilung der Stichproben (Familienanamnese). 26/28 In unserer Studie gibt es keine signifikanten Korrelationen zwischen dem Auftreten einer psychischen Erkrankung in der Verwandtschaft ersten und zweiten Grades und der Länge der Telomere (partielle Korrelation kontrolliert nach dem Alter, Tbl. 33). 91 TABELLE 33: Telomerlänge und Familienanamnese 1. Hinsichtlich der Telomerlänge gibt es keinen signifikanten Unterschied zwischen Probanden mit und ohne eine positive Familienanamnese in Bezug auf psychia- Familienanamnese ,724 ,811 ,459 ,976 Gesamtgruppe Depressive Gruppe Antidepressiva- EKT-Gruppe AD-Gruppe (p-Wert) Hoch dosierte Anamnese AD-Gruppe Niedrig dosierte trische Erkrankungen (partielle Korrelation, p > 0,05). ,723 Betrachtet man die graphische Abbildung der depressiven Gesamtgruppe bzgl. der Familienanamnese, deutet sich der Trend an, dass Patienten mit einer positiven Familienanamnese zu einer schnelleren Telomerverkürzung neigen (Abb. 35). ABBILDUNG 35: Telomerlänge und Familienanamnese. Die jährliche Basenpaarverkürzung scheint bei depressiven Patienten mit einer positiven Familienanamnese (-24 BP/Jahr) deutlich höher zu sein als bei Patienten mit einer negativen Fa- milienanamnese (- 14 BP/Jahr). negative Familienanamnese: (7,29 kb – 7,01 kb ) / 20 J. = - 14 BP/Jahr positive Familienanamnese: (7,44 kb – 6,96 kb) / 20 J. = - 24 BP/Jahr 92 TABELLE 34: Telomerlänge und Familienanamnese 2. Beim Vergleich der durchschnittlichen Telomerlänge im Alter von 49,00 Jahren haben Probanden mit einer positiven Familienanamnese etwas kürzere Telomere als die Probanden mit einer negativen Familienanamnese. Diagnose Positive Familienanamnese Negative Familienanamnese n Telomerlänge [kb] Alter 28 7,16 49,00 26 7,22 49,00 93 6 Diskussion In unserer Studie konnten wir nachweisen, dass Patienten, die an einer Major Depression leiden, deutlich kürzere Telomere haben als gesunde Kontrollpersonen. Diese Ergebnisse bestätigen frühere Studien, in denen von kürzeren Telomeren bei Schizophrenien oder affektiven Störungen berichtet wird. [90, 186, 213] Neben psychiatrischen Erkrankungen sind die Telomere jedoch auch bei zahlreichen somatischen Erkrankungen wie Atherosklerose [167], Herzversagen [141], Demenz [85] und idiopathischer Lungenfibrose [195] deutlich verkürzt. Darüber hinaus vermutet man, dass die Telomerlänge auch von epidemiologischen Faktoren beeinflusst wird: Geschlecht, Alter, Rauchen, Adipositas, Stress, der sozioökonomische Status und sogar Pessimismus scheinen sich auf die Telomerlänge auszuwirken. [17, 140] Bislang ist es jedoch nicht möglich, den genauen Mechanismus zu erklären, der hinter der gesteigerten Telomerverkürzung steht. Hinsichtlich der Depressionsforschung konnte in einer kürzlich veröffentlichten Studie im Gehirn depressiver Patienten, und somit am Ort der Entstehung einer Depression, interessanterweise keine Telomerverkürzung nachgewiesen werden. [215] Darüber hinaus konnte in dieser Studie auch keine signifikante Telomerverkürzung mit zunehmendem Alter nachgewiesen werden, so dass beide Ergebnisse wahrscheinlich auf die niedrige Zellteilungsrate im Gehirn zurückzuführen sind, denn auf zellulärer Ebene weiß man, dass sich die Telomere in Abwesenheit des Enzyms Telomerase mit jeder Zellteilung verkürzen, bis nach Erreichen einer kritischen Länge die Apoptose oder der Zellzyklusarrest eingeleitet wird. Aus diesem Grund wurde die Telomerlänge von humanen Leukozyten als Marker für das mitotische Zellalter und als allgemeiner Index für das menschliche Altern vorgeschlagen. In unserer Studie konnte die deutliche Abhängigkeit der Telomerlänge vom Alter bestätigt werden (p ≤ 0,05). Unterschiede bezüglich des Geschlechts ergaben sich dagegen nicht (p = 0,623). Aus diesem Grund konnte auf eine Aufteilung in männliche und weibliche Probandengruppen verzichtet werden. 94 Ein weiterer Fokus unserer Studie stellte die Analyse des Rauchverhaltens dar. In zahlreichen Studien wurde von kürzeren Telomeren bei Rauchern berichtet. [6, 131, 135] In unserer Studie konnte keine signifikante Korrelation zwischen der Telomerlänge und dem Rauchverhalten nachgewiesen werden (p = 0,946), Die Ursache, warum diese Ergebnisse in unserer Studie nicht bestätigt wurden, könnte darin liegen, dass die gesunde Kontrollgruppe zu klein für eine Analyse ist und die Ergebnisse der depressiven Gesamtgruppe durch den „Störfaktor“ Depression verfälscht werden. Es zeichnet sich jedoch die Tendenz zu einer schnelleren Telomerverkürzung bei Rauchern als bei den Nichtrauchern (7,5 BP/Jahr) ab. Interessanterweise fand Valdes [198] in einer Studie heraus, dass bei Rauchern pro Packyear die Telomerverkürzung um ca. 5 BP pro Jahr zunimmt. Leider wurden in unserer Studie die Daten bezüglich der Packyears nicht erhoben, so dass ein direkter Vergleich mit diesen Daten nicht möglich ist. Neben dem Alter, Geschlecht und dem Rauchverhalten scheint auch Stress eine tragende Rolle bei der Telomerverkürzung zu spielen. Epel und Kollegen [54] fanden heraus, dass Stress zu kürzeren Telomeren führt und dass die Rate der Telomerverkürzung wiederum mit chronischem Vorhandensein von Stress korreliert. Diese Behauptung wird von Zellkulturexperimenten unterstützt, in denen die Telomerlänge schneller unter oxidativem Stress abnimmt, unter besseren Bedingungen dagegen länger aufrechterhalten bleiben kann. [201] Die Major Depression wird als dysregulierte Aktivierung einer chronischen Stressantwort bezeichnet. [121] Aus diesem Grund haben Simon und Kollegen [186] das Postulat aufgestellt, dass chronisch affektive Erkrankungen zu einem beschleunigten Voranschreiten der Zellalterung und somit auch zu einer Telomerverkürzung führen. Aviv [5] beschreibt in seiner Studie einen weiteren Ansatz zur Erklärung der Telomerverkürzung. Er sieht die Telomerlänge als eine Art Indikator für die körperliche Fitness, so dass man aus der Telomerlänge sowohl Information über das biologische Alter als auch über das Risiko für das Auftreten von Krankheiten erlangen kann. Um diese Ansätze hinsichtlich eines Zusammenhangs zwischen dem Auftreten einer Depression und einer vorzeitigen Telomerverkürzung zu vertiefen, haben wir die Patienten in drei Gruppen anhand der Behandlungsmethoden eingeteilt 95 (niedrig dosierte Antidepressiva, hoch dosierte Antidepressiva und Antidepressiva plus Elektrokrampftherapie). Unsere Hypothese war, dass das Ausmaß der Telomerverkürzung mit Intensivierung der Therapie oder mit zunehmender Schwere der Erkrankung zunimmt. Unsere Daten konnten diese Hypothese jedoch nicht unterstützen. Sowohl die depressive Gesamtgruppe (p = 0,011), als auch die depressiven Untergruppen hatten zwar deutlich kürzere Telomere als die gesunde Kontrollgruppe, Korrelationen zwischen der Telomerlänge und der Behandlungsart bzw. der Telomerlänge und der Schwere der Erkrankung konnten jedoch nicht nachgewiesen werden. Es zeichnet sich lediglich der Trend ab, dass mit zunehmender Antidepressiva-Dosis die Telomerlänge kürzer ist (niedrig dosierte Antidepressiva-Gruppe: p = 0,072, hoch-dosierte Antidepressiva-Gruppe: p = 0,078) und dass mit zunehmender Intensivierung der Therapie der Telomerunterschied zur Kontrollgruppe signifikanter wird (niedrig dosierte Antidepressiva-Gruppe: p = 0,068, hoch dosierte AntidepressivaGruppe: p = 0,049, EKT-Gruppe, p = 0,019). Interessanterweise nimmt die Rate der Basenpaarverkürzungen bei depressiven Probanden jedoch ebenfalls nicht gegenüber dem Kontrollkollektiv zu (Kontrollgruppe: -20 BP/Jahr, niedrig dosierte Antidepressiva-Gruppe: -19 BP/Jahr, hoch dosierte Antidepressiva-Gruppe: -22 BP/Jahr, EKT-Gruppe: 22,5 BP/Jahr). Insgesamt liegen jedoch alle Werte in der Größenordnung früheren Studien (-31 BP/Jahr [187] , -26 BP/Jahr [23] , -40 BP/Jahr [6]) und lassen vermuten, dass die Depression per se nicht zu einer verstärkten Telomerverkürzung führt. Unter diesem Gesichtpunkt weisen die Daten darauf hin, dass die Telomerlänge der Patienten schon vor dem Auftreten der Depression verkürzt ist und dass somit kürzere Telomere ein Risikofaktor für die Manifestation einer Depression sind. In unserer Studie errechnet sich eine Voralterung bei Depressiven um durchschnittlich 16,75 Jahre, was noch etwas über den berechneten 10 Jahren von Simon liegt. [186] Zusammenfassend muss jedoch berücksichtigt werden, dass die Gruppen eine verhältnismäßig kleine Anzahl von Probanden umfassen und insbesondere die Einteilung in Untergruppen oft eine sinnvolle Analyse nicht zuließ. Darüber hinaus stellt diese Studie nur eine Querschnittstudie dar, wohingegen eine 96 Verlaufsstudie, insbesondere in Hinsicht auf die Medikamentenanalyse und die Berechnung der jährlichen Basenpaarverkürzungen, mit Sicherheit sehr interessant wäre. Zuletzt kann auch die Einteilung der Gruppen sicherlich noch optimiert werden. Am sinnvollsten wäre mit Sicherheit die Erhebung des BDIs oder HAMDs zu Beginn der Therapie und die Durchführung einer Verlaufskontrolle nach 6 Wochen, um die Patienten standardgemäß in Responder und Non-Responder einteilen zu können. Alternativ könnten auch gezielte Studien zur Analyse einzelner Antidepressiva interessante Informationen zur Auswirkung von Medikamenten auf die Telomerlänge aufdecken. Dennoch bietet diese Studie interessante Ansatzpunkte für zukünftige Studien. Die verkürzten Telomere bei depressiven Patienten könnten zum Beispiel durch Traumata oder Stress in der Kindheit bedingt sein, da beide Faktoren als potentielle Risikofaktoren für eine Depression im Erwachsenenalter gewertet werden. Die kürzeren Telomere können auch genetisch bedingt sein. Studien haben gezeigt, dass die Telomerlänge zwischen Eltern und ihren Nachkommen miteinander korreliert. In zukünftigen Studien könnte dieser interessante Ansatzpunkt vertieft werden. Eine Möglichkeit besteht darin, die Telomerlänge von gesunden Verwandten depressiver Patienten zu bestimmen und diese sowohl mit den betroffenen Patienten selbst als auch mit gesunden, nichtverwandten Kontrollpersonen zu vergleichen. 97 7 Literaturverzeichnis 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. Alberts JL, Okun MS, Vitek JL. The persistent effects of unilateral pallidal and subthalamic deep brain stimulation on force control in advanced Parkinson's patients. Parkinsonism Relat Disord, 2008. 14(6): p. 481-8. Allsopp R. Telomere-Induced Senescence of Primary Cells. In: Lenhard Rudolph K, Hrsg. Telomeres and Telomerase in Ageing, Disease, and Cancer. 2008, Erste Auflage. Berlin, Heidelberg: Springer-Verlag. p. 2342. Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood, 2009. 113(26): p. 6549-57. Angelucci F, Brenè S, Mathé AA. BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry, 2005. 10(4): p. 34552. Aviv A. Telomeres and human somatic fitness. J Gerontol A Biol Sci Med Sci, 2006. 61(8): p. 871-3. Aviv A, Chen W, Gardner JP, Kimura M, Brimacombe M, Cao X, Srinivasan SR, Berenson GS. Leukocyte telomere dynamics: longitudinal findings among young adults in the Bogalusa Heart Study. Am J Epidemiol, 2009. 169(3): p. 323-9. Baune BT, Adrian I, Arolt V, Berger K. Associations between major depression, bipolar disorders, dysthymia and cardiovascular diseases in the general adult population. Psychotherapy and psychosomatics, 2006. 75(5): p. 319-26. Beblo T, Lautenbacher S. . Fortschritte der Neuropsychologie. 2006, Erste Auflage. Göttingen, Bern, Wien, Toronto, Seattle, Oxford, Prag: Hogrefe. Belmaker RH, Agam G. Major depressive disorder. N Engl J Med, 2008. 358(1): p. 55-68. Benabid AL, Chabardes S, Seigneuret E. Deep-brain stimulation in Parkinson's disease: long-term efficacy and safety - What happened this year? Curr Opin Neurol, 2005. 18(6): p. 623-30. Benedetti F, Barbini B, Fulgosi MC, Colombo C, Dallaspezia S, Pontiggia A, Smeraldi E. Combined total sleep deprivation and light therapy in the treatment of drug-resistant bipolar depression: acute response and longterm remission rates. J Clin Psychiatry, 2005. 66(12): p. 1535-40. Benkert O, Hautzinger M, Graf-Morgenstern M. Psychopharmakologischer Leitfaden für Psychologen und Psychotherapeuten. 2007, Erste Auflage. Berlin: Springer. Bermejo I, Komarahadi F. Instrumente zur Früherkennung und Wirkungsprüfung. In: Härter M, Bermejo I,Niebling W, Hrsg. Praxismanual Depression - Diagnostik und Therapie erfolgreich umsetzten.2007, Erste Auflage. Köln: Deutscher Ärzte-Verlag. 98 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. Bhansali P, Dunning J, Singer SE, David L, Schmauss C. Early life stress alters adult serotonin 2C receptor pre-mRNA editing and expression of the alpha subunit of the heterotrimeric G-protein G q. J Neurosci, 2007. 27(6): p. 1467-73. Blackburn EH. Switching and signaling at the telomere. Cell, 2001. 106(6): p. 661-73. Blasco MA. Telomere Binding Proteins and Disease. In: Rudolph KL, Hrsg. Telomeres and Telomerase in Ageing, Disease, and Cancer. 2007, Erste Auflage. Berlin, Heidelberg: Springer-Verlag. p. 229-244. Blasco MA. Telomere length, stem cells and aging. Nat Chem Biol, 2007. 3(10): p. 640-9. Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet, 2005. 6(8): p. 611-22. Blendy JA. The role of CREB in depression and antidepressant treatment. Biol Psychiatry, 2006. 59(12): p. 1144-50. Bodenlos JS, Kose S, Borckardt JJ, Nahas Z, Shaw D, O'Neil PM, George MS. Vagus nerve stimulation acutely alters food craving in adults with depression. Appetite, 2007. 48(2): p. 145-53. Bramham CR. Control of synaptic consolidation in the dentate gyrus: mechanisms, functions, and therapeutic implications. Prog Brain Res, 2007. 163: p. 453-71. Brieger P, Blöink R, Röttig S, Marneros A. [Disability payments due to unipolar depressive and bipolar affective disorders]. Psychiatrische Praxis, 2004. 31(4): p. 203-6. Brouilette S, Singh RK, Thompson JR, Goodall AH, Samani NJ. White cell telomere length and risk of premature myocardial infarction. Arterioscler Thromb Vasc Biol, 2003. 23(5): p. 842-6. Bruel-Jungerman E, Laroche S, Rampon C. New neurons in the dentate gyrus are involved in the expression of enhanced long-term memory following environmental enrichment. Eur J Neurosci, 2005. 21(2): p. 51321. Bryan TM, Reddel RR. Telomere dynamics and telomerase activity in in vitro immortalised human cells. Eur J Cancer, 1997. 33(5): p. 767-73. Bundesamt, Statistisches. Statistisches Jahrbuch. 2009, Destatis: Wiesbaden. Burke HM, Davis MC, Otte C, Mohr DC. Depression and cortisol responses to psychological stress: a meta-analysis. Psychoneuroendocrinology, 2005. 30(9): p. 846-56. Carroll BJ, Cassidy F, Naftolowitz D, Tatham NE, Wilson WH, Iranmanesh A, Liu PY, Veldhuis JD. Pathophysiology of hypercortisolism in depression. Acta psychiatrica Scandinavica Supplementum, 2007(433): p. 90-103. Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science, 2003. 301(5631): p. 386-9. Cawthon RM, Smith KR, O'Brien E, Sivatchenko A, Kerber RA. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet, 2003. 361(9355): p. 393-5. 99 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. Cerletti U, Bini L. Un nuevo metodo di shockterapie "L'elettroshock". Bolletino Accademia Medica Roma, 1938. 64: p. 136–138. Cesare AJ, Reddel RR. Alternative Lengthening of Telomeres in Mammalian Cells. In: Nosek J,Tomáska Lu, Editors. Origin and Evolution of Telomeres. 2008: Landes Bioscience. p. 194. Chang A, Li P, Warsh J. cAMP signal transduction abnormalities in the pathophysiology of mood disorders: contributions from postmortem brain studies. In: Agam G, Everall I,Belmaker R, Editors. The postmortem brain in psychiatric studies. 2002, Boston: Kluwer Academic. Chang S. Initiation of Genomic Instability, Cellular Senescence, and Organismal Aging by Dysfunctional Telomeres. In: Rudolph KL, Hrsg. Telomeres and Telomerase in Ageing, Disease, and Cancer. 2008, Erste Auflage. Berlin, Heidelberg: Springer-Verlag. p. 57 - 75. Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry, 2001. 50(4): p. 260-5. Chrousos GP. Stressors, stress, and neuroendocrine integration of the adaptive response. The 1997 Hans Selye Memorial Lecture. Ann N Y Acad Sci, 1998. 851: p. 311-35. Cichon S, Craddock N, Daly M, Faraone SV, Gejman PV, Kelsoe J, Lehner T, Levinson DF, Moran A, Sklar P, Sullivan PF. Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry, 2009. 166(5): p. 540-56. Conca A, Konig P, Hausmann A. Transcranial magnetic stimulation induces 'pseudoabsence seizure'. Acta Psychiatr Scand, 2000. 101(3): p. 246-8; discussion 248-9. Conca A, Peschina W, Konig P, Fritzsche H, Hausmann A. Effect of chronic repetitive transcranial magnetic stimulation on regional cerebral blood flow and regional cerebral glucose uptake in drug treatmentresistant depressives. A brief report. Neuropsychobiology, 2002. 45(1): p. 27-31. Coupland NJ, Ogilvie CJ, Hegadoren KM, Seres P, Hanstock CC, Allen PS. Decreased prefrontal Myo-inositol in major depressive disorder. Biol Psychiatry, 2005. 57(12): p. 1526-34. Dannlowski U, Ohrmann P, Bauer J, Deckert J, Hohoff C, Kugel H, Arolt V, Heindel W, Kersting A, Baune BT, Suslow T. 5-HTTLPR biases amygdala activity in response to masked facial expressions in major depression. Neuropsychopharmacology, 2008. 33(2): p. 418-24. Dannlowski U, Ohrmann P, Bauer J, Kugel H, Baune BT, Hohoff C, Kersting A, Arolt V, Heindel W, Deckert J, Suslow T. Serotonergic genes modulate amygdala activity in major depression. Genes Brain Behav, 2007. 6(7): p. 672-6. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev, 2005. 19(18): p. 2100-10. de Pablos RM, Villarán RF, Argüelles S, Herrera AJ, Venero JL, Ayala A, Cano J, Machado A. Stress increases vulnerability to inflammation in the rat prefrontal cortex. J Neurosci, 2006. 26(21): p. 5709-19. DIMDI. ICD-10-GM Version 2009: Systematisches Verzeichnis. Internationale statistische Klassifikation der Krankheiten und verwandter Gesundheitsprobleme. 2008. 10 (German Modification). 100 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. Dinan TG. Inflammatory markers in depression. Curr Opin Psychiatry, 2009. 22(1): p. 32-6. Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol, 2000. 110(4): p. 768-79. Dome P, Teleki Z, Rihmer Z, Peter L, Dobos J, Kenessey I, Tovari J, Timar J, Paku S, Kovacs G, Dome B. Circulating endothelial progenitor cells and depression: a possible novel link between heart and soul. Mol Psychiatry, 2008. Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased temporal cortex CREB concentrations and antidepressant treatment in major depression. Lancet, 1998. 352(9142): p. 1754-5. Du H-Y, Bessler M, Mason PJ. Telomerase Mutations and Premature Ageing in Humans. In: Rudolph KL, Hrsg. Telomeres and Telomerase in Ageing, Disease, and Cancer. 2008, Erste Auflage. Berlin, Heidelberg: Springer-Verlag. p. 77-107. Duman RS, Malberg J, Nakagawa S, D'Sa C. Neuronal plasticity and survival in mood disorders. Biol Psychiatry, 2000. 48(8): p. 732-9. eBioscience. Stem Cell Reagents: Your Research Starts Here (Stem Cell Guide 2009 – 2010), eBioscience, Hrsg. 2009, eBioscience: Hatfield. Eisch AJ, Cameron HA, Encinas JM, Meltzer LA, Ming GL, OverstreetWadiche LS. Adult neurogenesis, mental health, and mental illness: hope or hype? J Neurosci, 2008. 28(46): p. 11785-91. Epel ES, Blackburn EH, Lin J, Dhabhar FS, Adler NE, Morrow JD, Cawthon RM. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci USA, 2004. 101(49): p. 17312-5. Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner's syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore), 1966. 45(3): p. 177-221. Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat Med, 1998. 4(11): p. 1313-7. Ermann M. Psychosomatische Medizin und Psychotherapie: ein Lehrbuch auf psychoanalytischer Grundlage. 2007, Fünfte Auflage. Stuttgart: Kohlhammer. Evans D, Charney DS, Lewis L, Golden R, Gorman JM, Krishnan KRR, Nemeroff CB, Bremner J, Carney RM, Coyne JC, Delong MR, FrasureSmith N, Glassman A, Gold PW, Grant I, Gwyther L, Ironson G, Johnson RL, Kanner AM, Katon W, Kaufmann PG, Keefe FJ, Ketter T, Laughren TP, Leserman J, Lyketsos CG, McDonald WM, McEwen BS, Miller AH, Musselman D, O'Connor C, Petitto JM, Pollock BG, Robinson RG, Roose SP, Rowland J, Sheline Y, Sheps DS, Simon G, Spiegel D, Stunkard A, Sunderland T, Tibbits P, Valvo WJ. Mood disorders in the medically ill: scientific review and recommendations. Biol Psychiatry, 2005. 58(3): p. 175-89. Fink M. Meduna and the origins of convulsive therapy. Am J Psychiatry, 1984. 141(9): p. 1034-41. Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF. Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacology, 2006. 31(3): p. 628-36. 101 61. 62. 63. 64. 65. 66. 67. 68. 69. 70. 71. 72. Frodl T, Schüle C, Schmitt G, Born C, Baghai T, Zill P, Bottlender R, Rupprecht R, Bondy B, Reiser M, Möller H-J, Meisenzahl EM. Association of the brain-derived neurotrophic factor Val66Met polymorphism with reduced hippocampal volumes in major depression. Arch Gen Psychiatry, 2007. 64(4): p. 410-6. Gershon AA, Vishne T, Grunhaus L. Dopamine D2-like receptors and the antidepressant response. Biol Psychiatry, 2007. 61(2): p. 145-53. Giacobbe P, Kennedy SH. Deep brain stimulation for treatment-resistant depression: a psychiatric perspective. Curr Psychiatry Rep, 2006. 8(6): p. 437-44. Grassi Zucconi G, Cipriani S, Balgkouranidou I, Scattoni R. 'One night' sleep deprivation stimulates hippocampal neurogenesis. Brain Res Bull, 2006. 69(4): p. 375-81. Greeson JM, Hurwitz BE, Llabre MM, Schneiderman N, Penedo FJ, Klimas NG. Psychological distress, killer lymphocytes and disease severity in HIV/AIDS. Brain Behav Immun, 2008. 22(6): p. 901-11. Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T. Mammalian telomeres end in a large duplex loop. Cell, 1999. 97(4): p. 503-14. Groenen F. Untersuchung zur Zahl der hämatopoetischen Stammzellen bei Respondern, Non-Respondern und EKT-Patienten mit uni- oder bipolarer Depression [Dissertation]. 2010, Friedrich-AlexanderUniversität: Erlangen. Group TINBSS. Nijmegen breakage syndrome. Arch Dis Child, 2000. 82(5): p. 400-6. Guachalla Gutierrez L, Ju Z. Telomere Shortening Induces Cell Intrinsic Checkpoints and Environmental Alterations Limiting Adult Stem Cell Function. In: Rudolph KL, Hrsg. Telomere and Telomerase in Ageing, Disease, and Cancer. 2008, Erste Auflage. Berlin, Heidelberg: SpringerVerlag. p. 161 - 180. Guducu F, Caliyurt O, Vardar E, Tuglu C, Abay E. [Combination therapy using sertraline with sleep deprivation and light therapy compared to sertraline monotherapy for major depressive disorder]. Turk Psikiyatri Derg, 2005. 16(4): p. 245-51. Günther OH, Friemel S, Bernert S, Matschinger H, Angermeyer MC, König H-H. [The burden of depressive disorders in Germany - results from the European Study of the Epidemiology of Mental Disorders (ESEMeD)]. Psychiatrische Praxis, 2007. 34(6): p. 292-301. Hajek T, Kopecek M, Kozeny J, Gunde E, Alda M, Höschl C. Amygdala volumes in mood disorders--meta-analysis of magnetic resonance volumetry studies. Journal of affective disorders, 2009. 115(3): p. 395410. 102 73. 74. 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. 85. 86. Hammen C, Brennan PA, Keenan-Miller D, Hazel NA, Najman JM. Chronic and acute stress, gender, and serotonin transporter geneenvironment interactions predicting depression symptoms in youth. J Child Psychol Psychiatry, 2010. 51(2): p. 180-7. Hariri AR, Drabant EM, Munoz KE, Kolachana BS, Mattay VS, Egan MF, Weinberger DR. A susceptibility gene for affective disorders and the response of the human amygdala. Arch Gen Psychiatry, 2005. 62(2): p. 146-52. Harle-Bachor C, Boukamp P. Telomerase activity in the regenerative basal layer of the epidermis inhuman skin and in immortal and carcinoma-derived skin keratinocytes. Proc Natl Acad Sci U S A, 1996. 93(13): p. 6476-81. Hausmann A, Kramer-Reinstadler K, Lechner-Schoner T, Walpoth M, Rupp CI, Hinterhuber H, Conca A. Can bilateral prefrontal repetitive transcranial magnetic stimulation (rTMS) induce mania? A case report. J Clin Psychiatry, 2004. 65(11): p. 1575-6. Hausmann A, Weis C, Marksteiner J, Hinterhuber H, Humpel C. Chronic repetitive transcranial magnetic stimulation enhances c-fos in the parietal cortex and hippocampus. Brain Res Mol Brain Res, 2000. 76(2): p. 35562. Hautzinger M. Affektive Störungen. In: Förstl H, Hautzinger M, Roth G Hrsg. Neuropsychologie psychischer Störungen. 2005, Erste Auflage. Heidelberg: Springer. p. 421 - 480. Hayakawa H, Shimizu M, Nishida A, Motohashi N, Yamawaki S. Increase in serotonin 1A receptors in the dentate gyrus as revealed by autoradiographic analysis following repeated electroconvulsive shock but not imipramine treatment. Neuropsychobiology, 1994. 30(2-3): p. 53-6. Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell, 2004. 14(4): p. 501-13. Hiyama K, Hirai Y, Kyoizumi S, Akiyama M, Hiyama E, Piatyszek MA, Shay JW, Ishioka S, Yamakido M. Activation of telomerase in human lymphocytes and hematopoietic progenitor cells. J Immunol, 1995. 155(8): p. 3711-5. Holmans P, Weissman MM, Zubenko GS, Scheftner WA, Crowe RR, Depaulo JR, Knowles JA, Zubenko WN, Murphy-Eberenz K, Marta DH, Boutelle S, McInnis MG, Adams P, Gladis M, Steele J, Miller EB, Potash JB, Mackinnon DF, Levinson DF. Genetics of recurrent early-onset major depression (GenRED): final genome scan report. The American journal of psychiatry, 2007. 164(2): p. 248-58. Holsboer F. Biologie für die Seele: Mein Weg zur personalisierten Medizin. 2009: C. H. Beck. Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology, 2000. 23(5): p. 477-501. Honig LS, Schupf N, Lee JH, Tang MX, Mayeux R. Shorter telomeres are associated with mortality in those with APOE epsilon4 and dementia. Ann Neurol, 2006. 60(2): p. 181-7. Jacobi F, Wittchen HU, Holting C, Hofler M, Pfister H, Muller N, Lieb R. Prevalence, co-morbidity and correlates of mental disorders in the 103 87. 88. 89. 90. 91. 92. 93. 94. 95. 96. 97. 98. 99. general population: results from the German Health Interview and Examination Survey (GHS). Psychol Med, 2004. 34(4): p. 597-611. Ju Z, Rudolph KL. Telomeres and telomerase in cancer stem cells. Eur J Cancer, 2006. 42(9): p. 1197-203. Kamphuis MH, Kalmijn S, Tijhuis MAR, Geerlings MI, Giampaoli S, Nissinen A, Grobbee DE, Kromhout D. Depressive symptoms as risk factor of cardiovascular mortality in older European men: the Finland, Italy and Netherlands Elderly (FINE) study. European journal of cardiovascular prevention and rehabilitation : official journal of the European Society of Cardiology, Working Groups on Epidemiology & Prevention and Cardiac Rehabilitation and Exercise Physiology, 2006. 13(2): p. 199-206. Kanfer FH, Phillips JS. Lerntheoretische Grundlagen der Verhaltenstherapie. 1975. München: Kindler. Kao HT, Cawthon RM, Delisi LE, Bertisch HC, Ji F, Gordon D, Li P, Benedict MM, Greenberg WM, Porton B. Rapid telomere erosion in schizophrenia. Mol Psychiatry, 2008. 13(2): p. 118-9. Karceski S. Early Parkinson disease and depression. Neurology, 2007. 69(4): p. E2-3. Karege F, Vaudan G, Schwald M, Perroud N, La Harpe R. Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res Mol Brain Res, 2005. 136(1-2): p. 29-37. Kendler KS, Gardner CO, Prescott CA. Clinical characteristics of major depression that predict risk of depression in relatives. Arch Gen Psychiatry, 1999. 56(4): p. 322-7. Kendler KS, Kuhn JW, Vittum J, Prescott CA, Riley B. The interaction of stressful life events and a serotonin transporter polymorphism in the prediction of episodes of major depression: a replication. Arch Gen Psychiatry, 2005. 62(5): p. 529-35. Kho KH, van Vreeswijk MF, Simpson S, Zwinderman AH. A metaanalysis of electroconvulsive therapy efficacy in depression. J ECT, 2003. 19(3): p. 139-47. Kilian A, Bowtell DD, Abud HE, Hime GR, Venter DJ, Keese PK, Duncan EL, Reddel RR, Jefferson RA. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet, 1997. 6(12): p. 2011-9. Klerman GL, Weissman MM, Rounsaville BJ, Chevron ES. Interpersonal psychotherapie of depression. 1984, New York: Basic Books. Knight SW, Vulliamy TJ, Morgan B, Devriendt K, Mason PJ, Dokal I. Identification of novel DKC1 mutations in patients with dyskeratosis congenita: implications for pathophysiology and diagnosis. Hum Genet, 2001. 108(4): p. 299-303. Kozlovsky N, Matar M, Kaplan Z, Kotler M, Zohar J, Cohen H. Long-term down-regulation of BDNF mRNA in rat hippocampal CA1 subregion corelates with PTSD-like behavioural stress response. Int J Neuropsychopharmacol, 2007. 10(4): p. 345-352. 104 100. 101. 102. 103. 104. 105. 106. 107. 108. 109. 110. 111. Kunugi H, Ida I, Owashi T, Kimura M, Inoue Y, Nakagawa S, Yabana T, Urushibara T, Kanai R, Aihara M, Yuuki N, Otsubo T, Oshima A, Kudo K, Inoue T, Kitaichi Y, Shirakawa O, Isogawa K, Nagayama H, Kamijima K, Nanko S, Kanba S, Higuchi T, Mikuni M. Assessment of the dexamethasone/CRH test as a state-dependent marker for hypothalamic-pituitary-adrenal (HPA) axis abnormalities in major depressive episode: a Multicenter Study. Neuropsychopharmacology, 2006. 31(1): p. 212-20. Lambert G, Johansson M, Agren H, Friberg P. Reduced brain norepinephrine and dopamine release in treatment-refractory depressive illness: evidence in support of the catecholamine hypothesis of mood disorders. Arch Gen Psychiatry, 2000. 57(8): p. 787-93. Lee R, Geracioti TD, Kasckow JW, Coccaro EF. Childhood trauma and personality disorder: positive correlation with adult CSF corticotropinreleasing factor concentrations. The American journal of psychiatry, 2005. 162(5): p. 995-7. Leonard BE. Inflammation, depression and dementia: are they connected? Neurochem Res, 2007. 32(10): p. 1749-56. Lett HS, Blumenthal JA, Babyak MA, Sherwood A, Strauman T, Robins C, Newman MF. Depression as a risk factor for coronary artery disease: evidence, mechanisms, and treatment. Psychosomatic medicine, 2004. 66(3): p. 305-15. Li G, Pleasure SJ. Ongoing interplay between the neural network and neurogenesis in the adult hippocampus. Curr Opin Neurobiol, 2010. 20(1): p. 126-33. Liu D, O'Connor MS, Qin J, Songyang Z. Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins. J Biol Chem, 2004. 279(49): p. 51338-42. Lorenzetti V, Allen N, Whittle S, Yücel M. Amygdala volumes in a sample of current depressed and remitted depressed patients and healthy controls. Journal of affective disorders, 2009. Louis C, Cohen C, Depoortère R, Griebel G. Antidepressant-like effects of the corticotropin-releasing factor 1 receptor antagonist, SSR125543, and the vasopressin 1b receptor antagonist, SSR149415, in a DRL-72 s schedule in the rat. Neuropsychopharmacology, 2006. 31(10): p. 2180-7. Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L. Brain-derived neurotrophic factordeficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci USA, 1999. 96(26): p. 15239-44. MacKenzie B, Levitan RD. Psychic and somatic anxiety differentially predict response to light therapy in women with seasonal affective disorder. J Affect Disord, 2005. 88(2): p. 163-6. MacQueen GM, Campbell S, McEwen BS, Macdonald K, Amano S, Joffe RT, Nahmias C, Young LT. Course of illness, hippocampal function, and hippocampal volume in major depression. Proc Natl Acad Sci USA, 2003. 100(3): p. 1387-92. 105 112. 113. 114. 115. 116. 117. 118. 119. 120. 121. 122. 123. 124. 125. Maes M, Mihaylova I, Leunis JC. Increased serum IgM antibodies directed against phosphatidyl inositol (Pi) in chronic fatigue syndrome (CFS) and major depression: evidence that an IgM-mediated immune response against Pi is one factor underpinning the comorbidity between both CFS and depression. Neuro Endocrinol Lett, 2007. 28(6): p. 861-7. Makarov VL, Hirose Y, Langmore JP. Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell, 1997. 88(5): p. 657-66. Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci, 2000. 20(24): p. 9104-10. Mann JJ. The medical management of depression. N Engl J Med, 2005. 353(17): p. 1819-34. Mann JJ. Neurobiological correlates of the antidepressant action of electroconvulsive therapy. J ECT, 1998. 14(3): p. 172-80. Markianos M, Hatzimanolis J, Lykouras L. Relationship between prolactin responses to ECT and dopaminergic and serotonergic responsivity in depressed patients. Eur Arch Psychiatry Clin Neurosci, 2002. 252(4): p. 166-71. Marrone A, Stevens D, Vulliamy T, Dokal I, Mason PJ. Heterozygous telomerase RNA mutations found in dyskeratosis congenita and aplastic anemia reduce telomerase activity via haploinsufficiency. Blood, 2004. 104(13): p. 3936-42. Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, Schwalb JM, Kennedy SH. Deep brain stimulation for treatmentresistant depression. Neuron, 2005. 45(5): p. 651-60. McCormick LM, Boles Ponto LL, Pierson RK, Johnson HJ, Magnotta V, Brumm MC. Metabolic correlates of antidepressant and antipsychotic response in patients with psychotic depression undergoing electroconvulsive therapy. J ECT, 2007. 23(4): p. 265-73. McEwen BS. Mood disorders and allostatic load. Biol Psychiatry, 2003. 54(3): p. 200-7. McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med, 1998. 338(3): p. 171-9. Mendlewicz J, Massat I, Souery D, Del-Favero J, Oruc L, Nöthen MM, Blackwood D, Muir W, Battersby S, Lerer B, Segman RH, Kaneva R, Serretti A, Lilli R, Lorenzi C, Jakovljevic M, Ivezic S, Rietschel M, Milanova V, Van Broeckhoven C. Serotonin transporter 5HTTLPR polymorphism and affective disorders: no evidence of association in a large European multicenter study. Eur J Hum Genet, 2004. 12(5): p. 37782. Merali Z, Du L, Hrdina P, Palkovits M, Faludi G, Poulter MO, Anisman H. Dysregulation in the suicide brain: mRNA expression of corticotropinreleasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J Neurosci, 2004. 24(6): p. 1478-85. Merkl A, Heuser I, Bajbouj M. Antidepressant electroconvulsive therapy: Mechanism of action, recent advances and limitations. Exp Neurol, 2009. 106 126. 127. 128. 129. 130. 131. 132. 133. 134. 135. 136. 137. 138. 139. Mervaala E, Fohr J, Kononen M, Valkonen-Korhonen M, Vainio P, Partanen K, Partanen J, Tiihonen J, Viinamaki H, Karjalainen AK, Lehtonen J. Quantitative MRI of the hippocampus and amygdala in severe depression. Psychol Med, 2000. 30(1): p. 117-25. Meyer JH, Ginovart N, Boovariwala A, Sagrati S, Hussey D, Garcia A, Young T, Praschak-Rieder N, Wilson AA, Houle S. Elevated monoamine oxidase a levels in the brain: an explanation for the monoamine imbalance of major depression. Arch Gen Psychiatry, 2006. 63(11): p. 1209-16. Meyne J, Ratliff RL, Moyzis RK. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc Natl Acad Sci U S A, 1989. 86(18): p. 7049-53. Michelson D, Stratakis C, Hill L, Reynolds J, Galliven E, Chrousos G, Gold P. Bone mineral density in women with depression. N Engl J Med, 1996. 335(16): p. 1176-81. Moller P, Wallin H, Knudsen LE. Oxidative stress associated with exercise, psychological stress and life-style factors. Chem Biol Interact, 1996. 102(1): p. 17-36. Morla M, Busquets X, Pons J, Sauleda J, MacNee W, Agusti AG. Telomere shortening in smokers with and without COPD. Eur Respir J, 2006. 27(3): p. 525-8. Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, Meyne J, Ratliff RL, Wu JR. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci U S A, 1988. 85(18): p. 6622-6. Muntoni A, Reddel RR. The first molecular details of ALT in human tumor cells. Hum Mol Genet, 2005. 14 Spec No. 2: p. R191-6. Murck H, Uhr M, Ziegenbein M, Kunzel H, Held K, Antonijevic IA, Schussler P, Steiger A. Renin-angiotensin-aldosterone system, HPA-axis and sleep-EEG changes in unmedicated patients with depression after total sleep deprivation. Pharmacopsychiatry, 2006. 39(1): p. 23-9. Nawrot TS, Staessen JA, Gardner JP, Aviv A. Telomere length and possible link to X chromosome. Lancet, 2004. 363(9408): p. 507-10. Nemeroff CB, Mayberg HS, Krahl SE, McNamara J, Frazer A, Henry TR, George MS, Charney DS, Brannan SK. VNS therapy in treatmentresistant depression: clinical evidence and putative neurobiological mechanisms. Neuropsychopharmacology, 2006. 31(7): p. 1345-55. Nickel M. . 2009, Wien, New York: Springer. 200. Nowak G, Dulinski J. Effect of repeated treatment with electroconvulsive shock (ECS) on serotonin receptor density and turnover in the rat cerebral cortex. Pharmacol Biochem Behav, 1991. 38(3): p. 691-4. Nowak J, Januszkiewicz D, Lewandowski K, Nowicka-Kujawska K, Pernak M, Rembowska J, Nowak T, Wysocki J. Activity and expression of human telomerase in normal and malignant cells in gastric and colon cancer patients. Eur J Gastroenterol Hepatol, 2003. 15(1): p. 75-80. 107 140. 141. 142. 143. 144. 145. 146. 147. 148. 149. 150. 151. 152. 153. 154. O'Donovan A, Lin J, Dhabhar FS, Wolkowitz O, Tillie JM, Blackburn E, Epel E. Pessimism correlates with leukocyte telomere shortness and elevated interleukin-6 in post-menopausal women. Brain Behav Immun, 2009. 23(4): p. 446-9. Oh H, Wang SC, Prahash A, Sano M, Moravec CS, Taffet GE, Michael LH, Youker KA, Entman ML, Schneider MD. Telomere attrition and Chk2 activation in human heart failure. Proc Natl Acad Sci U S A, 2003. 100(9): p. 5378-83. Ordway GA, Schenk J, Stockmeier CA, May W, Klimek V. Elevated agonist binding to alpha2-adrenoceptors in the locus coeruleus in major depression. Biol Psychiatry, 2003. 53(4): p. 315-23. Organization WH. The Global Burden of Disease: Update 2004 2008. Othmer M, Zepp F. Flow cytometric immunophenotyping: principles and pitfalls. Eur J Pediatr, 1992. 151(6): p. 398-406. Pardo JV, Sheikh SA, Kuskowski MA, Surerus-Johnson C, Hagen MC, Lee JT, Rittberg BR, Adson DE. Weight loss during chronic, cervical vagus nerve stimulation in depressed patients with obesity: an observation. Int J Obes (Lond), 2007. 31(11): p. 1756-9. Passos JF, Saretzki G, von Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res, 2007. 35(22): p. 7505-13. Perera TD, Coplan JD, Lisanby SH, Lipira CM, Arif M, Carpio C, Spitzer G, Santarelli L, Scharf B, Hen R, Rosoklija G, Sackeim HA, Dwork AJ. Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J Neurosci, 2007. 27(18): p. 4894-901. Petrides G, Fink M, Husain MM, Knapp RG, Rush AJ, Mueller M, Rummans TA, O'Connor KM, Rasmussen KG, Jr., Bernstein HJ, Biggs M, Bailine SH, Kellner CH. ECT remission rates in psychotic versus nonpsychotic depressed patients: a report from CORE. J ECT, 2001. 17(4): p. 244-53. Pitchot W, Hansenne M, Pinto E, Reggers J, Fuchs S, Ansseau M. 5Hydroxytryptamine 1A receptors, major depression, and suicidal behavior. Biol Psychiatry, 2005. 58(11): p. 854-8. Plomin R DJ, McClear GE, Rutter M. . Gene, Umwelt und Verhalten – Einführung in die Verhaltenstherapie. 1999, Bern, Göttingen, Toronto, Seattle: Huber. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol, 2006. 27(1): p. 24-31. Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stressrelated disorders. The American journal of psychiatry, 2003. 160(9): p. 1554-65. Rakic P. Neurogenesis in adult primate neocortex: an evaluation of the evidence. Nat Rev Neurosci, 2002. 3(1): p. 65-71. Ramirez RD, Wright WE, Shay JW, Taylor RS. Telomerase activity concentrates in the mitotically active segments of human hair follicles. J Invest Dermatol, 1997. 108(1): p. 113-7. 108 155. 156. 157. 158. 159. 160. 161. 162. 163. 164. 165. 166. 167. 168. 169. 170. Rapp MA, Schnaider-Beeri M, Grossman HT, Sano M, Perl DP, Purohit DP, Gorman JM, Haroutunian V. Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch Gen Psychiatry, 2006. 63(2): p. 161-7. Reagan LP, Rosell DR, Wood GE, Spedding M, Muñoz C, Rothstein J, McEwen BS. Chronic restraint stress up-regulates GLT-1 mRNA and protein expression in the rat hippocampus: reversal by tianeptine. Proc Natl Acad Sci USA, 2004. 101(7): p. 2179-84. Reiche EMV, Nunes SOV, Morimoto HK. Stress, depression, the immune system, and cancer. Lancet Oncol, 2004. 5(10): p. 617-25. Reimer C, Rüger U. Psychodynamische Psychotherapien: Lehrbuch der tiefenpsychologisch fundierten Psychotherapieverfahren. 2006, Dritte Auflage. Heidelberg: Springer. Riedel O, Heuser I, Klotsche J, Dodel R, Wittchen HU. Occurrence risk and structure of depression in Parkinson disease with and without dementia: results from the GEPAD Study. J Geriatr Psychiatry Neurol, 2010. 23(1): p. 27-34. Risch N, Herrell R, Lehner T, Liang K-Y, Eaves L, Hoh J, Griem A, Kovacs M, Ott J, Merikangas KR. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA, 2009. 301(23): p. 2462-71. Roehrs T, Hyde M, Blaisdell B, Greenwald M, Roth T. Sleep loss and REM sleep loss are hyperalgesic. Sleep, 2006. 29(2): p. 145-51. Roman V, Walstra I, Luiten PG, Meerlo P. Too little sleep gradually desensitizes the serotonin 1A receptor system. Sleep, 2005. 28(12): p. 1505-10. Ruhe HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: a meta-analysis of monoamine depletion studies. Mol Psychiatry, 2007. 12(4): p. 331-59. Russ A. Drug Pocket 2008: Clinical Refernce Guide. 2007, Siebte Auflage. El Segundo, CA, USA Borm Bruckmeier Publishing. Sackeim HA, Decina P, Prohovnik I, Malitz S, Resor SR. Anticonvulsant and antidepressant properties of electroconvulsive therapy: a proposed mechanism of action. Biol Psychiatry, 1983. 18(11): p. 1301-10. Sahay A, Hen R. Adult hippocampal neurogenesis in depression. Nat Neurosci, 2007. 10(9): p. 1110-5. Samani NJ, Boultby R, Butler R, Thompson JR, Goodall AH. Telomere shortening in atherosclerosis. Lancet, 2001. 358(9280): p. 472-3. Sanacora G, Mason GF, Rothman DL, Hyder F, Ciarcia JJ, Ostroff RB, Berman RM, Krystal JH. Increased cortical GABA concentrations in depressed patients receiving ECT. Am J Psychiatry, 2003. 160(3): p. 577-9. Sapolsky RM. The possibility of neurotoxicity in the hippocampus in major depression: a primer on neuron death. Biol Psychiatry, 2000. 48(8): p. 755-65. Sapolsky RM, Uno H, Rebert CS, Finch CE. Hippocampal damage associated with prolonged glucocorticoid exposure in primates. J Neurosci, 1990. 10(9): p. 2897-902. 109 171. 172. 173. 174. 175. 176. 177. 178. 179. 180. 181. 182. 183. 184. 185. 186. Sartorius A, Henn FA. Deep brain stimulation of the lateral habenula in treatment resistant major depression. Med Hypotheses, 2007. 69(6): p. 1305-8. Sartorius A, Kiening KL, Kirsch P, von Gall CC, Haberkorn U, Unterberg AW, Henn FA, Meyer-Lindenberg A. Remission of major depression under deep brain stimulation of the lateral habenula in a therapyrefractory patient. Biol Psychiatry, 2010. 67(2): p. e9-e11. Sartorius A, Mahlstedt MM, Vollmayr B, Henn FA, Ende G. Elevated spectroscopic glutamate/gamma-amino butyric acid in rats bred for learned helplessness. Neuroreport, 2007. 18(14): p. 1469-73. Saß H, Wittchen H-U, Zaudig M, Houben I. Diagnostisches und Statistisches Manual Psychischer Störungen - Textrevision - DSM-IV-TR. 2003, Göttingen, Bern, Toronto, Seattle: Hogrefe. 1001. Satyanarayana A, Manns MP, Rudolph KL. Telomeres and telomerase: a dual role in hepatocarcinogenesis. Hepatology, 2004. 40(2): p. 276-83. Schlaepfer TE, Cohen MX, Frick C, Kosel M, Brodesser D, Axmacher N, Joe AY, Kreft M, Lenartz D, Sturm V. Deep brain stimulation to reward circuitry alleviates anhedonia in refractory major depression. Neuropsychopharmacology, 2008. 33(2): p. 368-77. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet, 2005. 366(9495): p. 1420-2. Schott KJ. Die Geschichte der Elektrokrampftherapie. In: Eschweiler GW, Hrsg. Elektromagnetische Therapien in der Psychiatrie : Elektrokrampftherapie (EKT), Transkranielle Magnetstimulation (TMS) und verwandte Verfahren. 2003, Darmstadt: Steinkopff p. 3-5. Schramm E. Interpersonelle Psychotherapie. 2009, Stuttgart: Schattauer. Segi-Nishida E, Warner-Schmidt JL, Duman RS. Electroconvulsive seizure and VEGF increase the proliferation of neural stem-like cells in rat hippocampus. Proc Natl Acad Sci USA, 2008. 105(32): p. 11352-7. Senf W, Broda M. Praxis der Psychotherapie: ein integratives Lehrbuch. 4 ed. 2007, Stuttgart, New York: Thieme. Shah SA, Doraiswamy PM, Husain MM, Escalona PR, Na C, Figiel GS, Patterson LJ, Ellinwood EH, Jr., McDonald WM, Boyko OB, et al. Posterior fossa abnormalities in major depression: a controlled magnetic resonance imaging study. Acta Psychiatr Scand, 1992. 85(6): p. 474-9. Sheline YI, Gado MH, Price JL. Amygdala core nuclei volumes are decreased in recurrent major depression. Neuroreport, 1998. 9(9): p. 2023-8. Sheline YI, Sanghavi M, Mintun MA, Gado MH. Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. J Neurosci, 1999. 19(12): p. 5034-43. Shimon H, Agam G, Belmaker RH, Hyde TM, Kleinman JE. Reduced frontal cortex inositol levels in postmortem brain of suicide victims and patients with bipolar disorder. The American journal of psychiatry, 1997. 154(8): p. 1148-50. Simon NM, Smoller JW, McNamara KL, Maser RS, Zalta AK, Pollack MH, Nierenberg AA, Fava M, Wong K-K. Telomere shortening and mood disorders: preliminary support for a chronic stress model of accelerated aging. Biol Psychiatry, 2006. 60(5): p. 432-5. 110 187. 188. 189. 190. 191. 192. 193. 194. 195. 196. 197. 198. 199. Slagboom PE, Droog S, Boomsma DI. Genetic determination of telomere size in humans: a twin study of three age groups. Am J Hum Genet, 1994. 55(5): p. 876-82. Spiessl H, Hubner-Liebermann B, Hajak G. [Depression, a widespread disease. Epidemiology, care situation, diagnosis, therapy and prevention]. Dtsch Med Wochenschr, 2006. 131(1-2): p. 35-40. Sterling P, Eyer J. Allostasis: a new paradigm to explain arousal pathology. In: Fisher S, Reason J,eds., Editors. Handbook of life stress, cognition and health. 1988, New York: John Wiley. p. 629-649. Strakowski SM, Adler CM, DelBello MP. Volumetric MRI studies of mood disorders: do they distinguish unipolar and bipolar disorder? Bipolar Disord, 2002. 4(2): p. 80-8. Strome EM, Zis AP, Doudet DJ. Electroconvulsive shock enhances striatal dopamine D1 and D3 receptor binding and improves motor performance in 6-OHDA-lesioned rats. J Psychiatry Neurosci, 2007. 32(3): p. 193-202. Svenningsson P, Chergui K, Rachleff I, Flajolet M, Zhang X, El Yacoubi M, Vaugeois J-M, Nomikos GG, Greengard P. Alterations in 5-HT1B receptor function by p11 in depression-like states. Science, 2006. 311(5757): p. 77-80. Synofzik M, Schlaepfer TE. Stimulating personality: ethical criteria for deep brain stimulation in psychiatric patients and for enhancement purposes. Biotechnol J, 2008. 3(12): p. 1511-20. Taylor AM, Metcalfe JA, Thick J, Mak YF. Leukemia and lymphoma in ataxia telangiectasia. Blood, 1996. 87(2): p. 423-38. Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A, 2007. 104(18): p. 7552-7. Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci, 2006. 9(4): p. 51925. Ulaner GA, Hu JF, Vu TH, Giudice LC, Hoffman AR. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res, 1998. 58(18): p. 4168-72. Valdes AM, Andrew T, Gardner JP, Kimura M, Oelsner E, Cherkas LF, Aviv A, Spector TD. Obesity, cigarette smoking, and telomere length in women. Lancet, 2005. 366(9486): p. 662-4. Valdizán EM, Gutierrez O, Pazos A. Adenylate cyclase activity in postmortem brain of suicide subjects: reduced response to betaadrenergic stimulation. Biol Psychiatry, 2003. 54(12): p. 1457-64. 111 200. 201. 202. 203. 204. 205. 206. 207. 208. 209. 210. 211. 212. 213. 214. 215. Voderholzer U, Hohagen F, Klein T, Jungnickel J, Kirschbaum C, Berger M, Riemann D. Impact of sleep deprivation and subsequent recovery sleep on cortisol in unmedicated depressed patients. Am J Psychiatry, 2004. 161(8): p. 1404-10. von Zglinicki T, Saretzki G, Ladhoff J, d'Adda di Fagagna F, Jackson SP. Human cell senescence as a DNA damage response. Mech Ageing Dev, 2005. 126(1): p. 111-7. Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature, 2001. 413(6854): p. 432-5. Warner-Schmidt JL, Duman RS. VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci USA, 2007. 104(11): p. 4647-52. Watson JD. Origin of concatemeric T7 DNA. Nat New Biol, 1972. 239(94): p. 197-201. Wichers M, Geschwind N, Jacobs N, Kenis G, Peeters F, Derom C, Thiery E, Delespaul P, van Os J. Transition from stress sensitivity to a depressive state: longitudinal twin study. Br J Psychiatry, 2009. 195(6): p. 498-503. Williams L, Gatt J, Schofield P, Olivieri G, Peduto A, Gordon E. 'Negativity bias' in risk for depression and anxiety: Brain-body fear circuitry correlates, 5-HTT-LPR and early life stress. Neuroimage, 2009. Williams LS. Depression and stroke: cause or consequence? Seminars in neurology, 2005. 25(4): p. 396-409. Wirz-Justice A, Van den Hoofdakker RH. Sleep deprivation in depression: what do we know, where do we go? Biol Psychiatry, 1999. 46(4): p. 445-53. Wittchen HU MN, Schmidkunz B, Winter S, Pfister H. Erscheinungsformen, Häufigkeit und Versorgung von Depressionen. Ergebnisse des bundesweiten Gesundheitssurveys 'Psychische Störungen'. Fortschr Med Orig, , 2000(118). Wright WE, Shay JW. The two-stage mechanism controlling cellular senescence and immortalization. Exp Gerontol, 1992. 27(4): p. 383-9. Wu JC, Bunney WE. The biological basis of an antidepressant response to sleep deprivation and relapse: review and hypothesis. Am J Psychiatry, 1990. 147(1): p. 14-21. Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, Young NS. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med, 2005. 352(14): p. 1413-24. Yu W-Y, Chang H-W, Lin C-H, Cho C-L. Short telomeres in patients with chronic schizophrenia who show a poor response to treatment. J Psychiatry Neurosci, 2008. 33(3): p. 244-7. Zeilinger M, Hübl W. Durchflusszytometrie. (Letzes Update: 2006.02.04) auf: http://www.med4you.at/laborbefunde/techniken/durchflusszytometrie/lbef _durchflusszytometrie.htm. (gesehen am 31.08.2010) Zhang D, Cheng L, Craig DW, Redman M, Liu C. Cerebellar telomere length and psychiatric disorders. Behav Genet, 2010. 40(2): p. 250-4. 112 216. Zhang X, Gainetdinov RR, Beaulieu J-M, Sotnikova TD, Burch LH, Williams RB, Schwartz DA, Krishnan KRR, Caron MG. Loss-of-function mutation in tryptophan hydroxylase-2 identified in unipolar major depression. Neuron, 2005. 45(1): p. 11-6. 113 8 Abkürzungsverzeichnis 5-HT 5-Hydroxytryptamin = Serotonin 5-HTTLPR serotonin-transporter-linked polymorphic region an den Serotonintransporter gebundere Polymorphismusregion AD Antidepressivum/Antidepressiva AIDS acquired immunodeficiency syndrome (erworbenes Immunschwächesyndrom) BDI Beck depression inventory (Beck-Depressionsinventar) BDNF brain-derived neurotrophic factor BP Basenpaar bzw. beziehungsweise cAMP zyklisches Adenosinmonophosphat chron. chronisch cpm counts per minute CREB cAMP response element-binding protein (Impulse pro Minute) (cAMP-Antwortelement-Bindeprotein) CRH Corticotropin-releasing hormone CRP C-reaktives Protein DBS deep brain stimulation Kortikotropin-Freisetzunghormon (Tiefe Hirnstimulation) DC Dyskeratosis congenita DKC1 Dyskerin DNA Deoxyribonucleic Acid (Desoxyribonukleinsäure) DSM-IV Diagnostic and Statistical Manual of Mental Disorders (Diagnostisches und Statistisches Handbuch Psychischer Störungen, 4. Revision) EPC endothelial progenitor cells EKT Elektrokrampftherapie etc. et cetera FACS Fluorescence Antibody Cell Sorter FITC Fluoreszenz-Isothiocyanat HAM-D Hamilton scale for depression (Endotheliale Progenitorzellen) (Durchflusszytometrie) (Hamilton-Depressionsskala) HHN Hypothalamus-Hypophyse-Nebennieren [-Achse] HIV human immunodeficiency virus (Menschliches Immunschwächevirus) 114 HSC hematopoietic stem cells Hämatopoetische Stammzellen ICD-10-GM International Statistical Classification of Diseases and Related Health Problems th 10 Revision – German Modification) (Internationale statistische Klassifikation der Krankheiten und verwandter Gesundheitsprobleme, 10. Revision, Deutsche Modifikation) IL-6 Interleukin-6 IP3 Inositoltriphosphat kb Kilobasenpaar MAO Monoaminoxidase MRT Magnetresonanztomographie n Anzahl p probability Wahrscheinlichkeit PBS phosphate buffered saline PCR polymerase chain reaction (Phosphatgepufferte Salzlösung) (Polymerase-Kettenreaktion) PE Phycoerythrin PET Positronenemissionstomographie PFA Paraformaldehyd r Rang RNA Ribonucleic Acid SD standard deviation (Ribonukleinsäure Standardabweichung SDS sodium dodecyl sulfate SNRI selective noradrenalin reuptake inhibitor SPSS Statistical Package for Social Sciences SSC saline-sodium citrate (Natriumdodecylsulfat) (Kochsalz-Natriumcitrat) (Selektiver Noradrenalin-Wiederaufnahmehemmer) SSNRI selective serotonin and noradrenalin reuptake inhibitor (Selektiver Serotonin- und Noradrenalin-Wiederaufnahmehemmer) SSRI selective serotonin reuptake inhibitor (Selektiver Serotonin-Wiederaufnahmehemmer) TAD Totale Antidepressiva-Dosis Terc telomerase RNA component Tert telomerase reverse transcriptase Telomerase RNA-Komponente Telomerase Reverse Transkriptase TMS Transkranielle Magnetstimulation TNF-α Tumornekrosefaktor alpha TPH Tryptophan-Hydroxylase 115 TRF telomere restriction fragments Telomerrestriktionsfragmente UpM Umdrehungen pro Minute usw. und so weiter VEGF vascular endothelial growth factor (vaskulärer endothelialer Wachstumsfaktor) vs. versus WHO World Health Organization gegen, gegenüber gestellt (Weltgesundheitsorganisation) VNS Vagusnervstimulation YLDs years lost due to disability (Jahre, die nicht in vollkommener Gesundheit gelebt werden konnten) z. B. zum Beispiel 116 9 Abbildungsverzeichnis ABBILDUNG 1: Veränderungen im Monoamin-Stoffwechsel und an der Synapse im Rahmen einer Depression. ABBILDUNG 2: Circulus vitiosus: Glukokortikoide. ABBILDUNG 3: Circulus vitiosus: Knochenmark-/Immunsystem-Supression. ABBILDUNG 4: Abbildung einer Nervenzelle (Übersichtsbild) und Darstellung der Angriffspunkte von Antidepressiva an der Synapse (Detailbild). ABBILDUNG 5: SORKC-Modell nach Kanfer. ABBILDUNG 6: Darstellung des t-loop und der angelagerten Proteine des Shelterin-Komplexes. ABBILDUNG 7: DNA-Replikation. ABBILDUNG 8: Telomerlänge und Pathogenese. ABBILDUNG 9: Darstellung der Zellpopulationen. ABBILDUNG 10: Darstellung der Subpopulationen. ABBILDUNG 11: Fotographie zwei verschiedener Southern Blots. ABBILDUNG 12: Telomerlänge und Alter. ABBILDUNG 13: Telomerlänge in den vier Gruppen. ABBILDUNG 14: Telomerlänge und Alter (depressive Untergruppen). ABBILDUNG 15: Basenpaarverkürzung der Kontrollgruppe. ABBILDUNG 16: Basenpaarverkürzung der niedrig dosierten Antidepressiva-Gruppe. ABBILDUNG 17: Basenpaarverkürzung der hoch dosierten Antidepressiva-Gruppe. ABBILDUNG 18: Basenpaarverkürzung der Elektrokrampftherapie-Gruppe. ABBILDUNG 19: Telomerlänge und Rauchverhalten. ABBILDUNG 20: Erkrankungsdauer in den depressiven Untergruppen. ABBILDUNG 21: Telomerlänge und Totale Antidepressiva-Dosis bei stationärer Aufnahme. ABBILDUNG 22: Telomerlänge und Gesamtdosis SSRIs bei stationärer Aufnahme. ABBILDUNG 23: Telomerlänge und Totale Antidepressiva-Dosis bei stationärer Entlassung. ABBILDUNG 24: Telomerlänge und Gesamtdosis Neuroleptika bei stationärer Entlassung. ABBILDUNG 25: Telomerlänge und Response auf EKT. ABBILDUNG 26: Telomerlänge und Transkranielle Magnetstimulation. ABBILDUNG 27: Telomerlänge und CD133/CD146-Leukozyten (niedrig dosierte Anti-depressivaGruppe). ABBILDUNG 28: Telomerlänge und CD133/CD146-Leukozyten (Elektrokrampftherapie-Gruppe). ABBILDUNG 29: Telomerlänge und (CD34+/CD45+)-Stammzellen (hoch dosierte Antidepressiva-Gruppe). ABBILDUNG 30: Telomerlänge und (CD105+/CD45-)-Stammzellen (hoch dosierte Antidepressiva-Gruppe). ABBILDUNG 31: Telomerlänge und Persönlichkeitsstörungen. ABBILDUNG 32: Telomerlänge und Suchtverhalten. ABBILDUNG 33: Telomerlänge und Benzodiazepin-Abhängigkeit. ABBILDUNG 34: Telomerlänge und Alkohol-Abhängigkeit. ABBILDUNG 35: Telomerlänge und Familienanamnese. 117 10 Tabellenverzeichnis TABELLE 1: Hauptgruppen der affektiven Störungen (F30-F39). TABELLE 2: Typische somatische, sonstige und psychotische Symptome im Rahmen einer depressiven Episode. TABELLE 3: TABELLE 4: DSM-IV-Kriterien zur Diagnostizierung einer Depression vom Major-Typ. Schweregradeinteilung einer Depression gemäß Beck Depressionsinventar (BDI). TABELLE 5: Schweregradeinteilung einer Depression gemäß Hamilton-Depressionsskala (HAM-D). TABELLE 6: Einteilung der Antidepressiva. TABELLE 7: Beschreibung der Kontrollgruppe (Gruppe 1). TABELLE 8: Beschreibung der niedrig dosierten Antidepressiva-Gruppe, hoch dosierten Antidepressiva-Gruppe und Elektrokrampftherapie-Gruppe (Gruppen 2-4). TABELLE 9: Verwendete Antikörper zur Bestimmung der adulten Stammzellen. TABELLE 10: Verteilung der Stichproben auf die vier Probanden-Gruppen. TABELLE 11: Demographische Daten der Probanden-Gruppen. TABELLE 12: Klinische Daten der depressiven Gruppen. TABELLE 13: Laborparameter der Probanden-Gruppen. TABELLE 14: Zusammenhang Alter – Telomerlänge und Geschlecht – Telomerlänge. TABELLE 15: Telomeranalyse im Gruppenvergleich. TABELLE 16: Telomerlänge und Rauchverhalten. TABELLE 17: Telomerlänge und Erkrankungsdauer. TABELLE 18: Telomerlänge und Medikamentendosierungen. TABELLE 19: Telomerlänge und EKT-Daten. TABELLE 20: Telomerlänge und Response auf EKT. TABELLE 21: Verteilung der Stichproben (Transkranielle Magnetstimulation). TABELLE 22: Telomerlänge und transkranielle Magnetstimulation. TABELLE 23: Verteilung der Stichproben (Psychische Komorbiditäten). TABELLE 24: Telomerlänge und psychische Komorbiditäten. TABELLE 25: Telomerlänge und Persönlichkeitsstörung 1 (niedrig dosierte AntidepressivaGruppe). TABELLE 26: Telomerlänge und Persönlichkeitsstörung 2 (niedrig dosierte AntidepressivaGruppe). TABELLE 27: Verteilung der Stichproben (Somatische Komorbiditäten). TABELLE 28: Telomerlänge und somatische Komorbiditäten. TABELLE 29: Verteilung der Stichproben (Suchtanamnese). TABELLE 30: Telomerlänge und Suchtanamnese 1. TABELLE 31: Telomerlänge und Suchtanamnese 2. TABELLE 32: Verteilung der Stichproben (Familienanamnese). TABELLE 33: Telomerlänge und Familienanamnese 1. TABELLE 34: Telomerlänge und Familienanamnese 2. 118 11 Anhang 11.1 DSM-IV-Kriterien Kriterien für eine Episode der Major Depression während derselben Zwei-Wochen-Periode: mindestens eines der Symptome der Kriterien 1-2 mindestens vier der Kriterien 3-9 1 2 3 4 5 6 7 8 9 Depressive Verstimmung Verlust an Interesse oder Freude Deutlicher Gewichtsverlust ohne Diät oder Gewichtszunahme Vermehrter Schlaf oder Schlaflosigkeit Psychomotorische Unruhe oder Verlangsamung Müdigkeit und Energieverlust Übermäßige, unangemessene Schuldgefühle oder Gefühle von Wertlosigkeit an fast allen Tagen Subjektive oder beobachtbare verminderte Denk- und Entscheidungsfähigkeit Suizidale Gedanken und/oder Handlungen Es werden nicht die Kriterien für eine gemischte Episode erfüllt. Die Symptome verursachen in klinisch bedeutsamer Weise Leiden oder Beeinträchtigungen. Die Symptome können nicht durch die Einnahme von Substanzen (Medikamenten, Drogen etc.) oder Begleiterkrankungen (z. B. Hypothyreose) verursacht sein. 119 11.2 Beck Depressions-Inventar Name: Geburtsdatum: Geschlecht: Ausfülldatum: Dieser Fragebogen enthält 21 Gruppen von Aussagen. Bitte lesen Sie jede Gruppe sorgfältig durch. Suchen Sie dann die eine Aussage in jeder Gruppe heraus, die am besten beschreibt, wie Sie sich in dieser Woche einschließlich heute gefühlt haben und kreuzen Sie die dazugehörige Ziffer (0, 1, 2 oder 3) an. Falls mehrere Aussagen einer Gruppe gleichermaßen zutreffen, können Sie auch mehrere Ziffern markieren. Lesen Sie auf jeden Fall alle Aussagen in jeder Gruppe, bevor Sie die Wahl treffen. 0 1 2 3 0 1 2 3 A Ich bin nicht traurig. Ich bin traurig. Ich bin die ganze Zeit traurig und komme nicht davon los. Ich bin so traurig oder unglücklich, dass ich es kaum noch ertrage. B Ich sehe nicht besonders mutlos in die Zukunft. Ich sehe mutlos in die Zukunft. Ich habe nichts, worauf ich mich freuen kann. Ich habe das Gefühl, dass die Zukunft hoffnungslos ist und dass die Situation nicht besser werden kann. 2 3 F Ich habe nicht das Gefühl, bestraft zu sein. Ich habe das Gefühl, vielleicht bestraft zu werden. Ich erwarte, bestraft zu werden. Ich habe das Gefühl, bestraft zu sein. 0 1 2 3 G Ich bin nicht von mir enttäuscht. Ich bin von mir enttäuscht. Ich finde mich fürchterlich. Ich hasse mich. 0 1 0 1 0 1 2 3 0 1 2 3 0 1 2 3 C Ich fühle mich nicht als Versager. Ich habe das Gefühl, öfter versagt zu haben als der Durchschnitt. Wenn ich auf mein Leben zurückblicke, sehe ich bloß eine Menge Fehlschläge. Ich habe das Gefühl, als Mensch ein völliger Versager zu sein. 2 3 0 1 D Ich kann die Dinge genauso genießen wie früher. Ich kann die Dinge nicht mehr so genießen wie früher. Ich kann aus nichts mehr eine echte Befriedigung ziehen. Ich bin mit allem unzufrieden oder gelangweilt. 2 3 0 1 2 3 E Ich habe keine Schuldgefühle. Ich habe häufig Schuldgefühle. Ich habe fast immer Schuldgefühle. Ich habe immer Schuldgefühle. 120 H Ich habe nicht das Gefühl, schlechter zu sein als alle anderen. Ich kritisiere mich wegen meiner Fehler und Schwächen. Ich mache mir die ganze Zeit Vorwürfe wegen meiner Mängel. Ich gebe mir für alles die Schuld, was schief geht. I Ich denke nicht daran, mir etwas anzutun. Ich denke manchmal an Selbstmord, aber ich würde es nicht tun. Ich möchte mich am liebsten umbringen. Ich würde mich umbringen, wenn ich die Gelegenheit hätte. J Ich weine nicht öfter als früher. Ich weine jetzt mehr als früher. Ich weine jetzt die ganze Zeit. Früher konnte ich weinen, aber jetzt kann ich es nicht mehr, obwohl ich es möchte. 0 1 2 3 K Ich bin nicht reizbarer als sonst. Ich bin jetzt leichter verärgert oder gereizt als früher. Ich fühle mich dauernd gereizt. Die Dinge, die mich früher geärgert haben, berühren mich nicht mehr. 0 1 2 3 0 0 1 2 3 0 1 2 3 0 1 2 3 0 1 2 3 0 1 2 3 L Ich habe nicht das Interesse an Menschen verloren. Ich interessiere mich jetzt weniger für Menschen als früher. Ich habe mein Interesse an anderen Menschen zum größten Teil verloren. Ich habe mein ganzes Interesse an anderen Menschen verloren. M Ich bin so entschlussfreudig wie immer. Ich schiebe Entscheidungen jetzt öfter als früher auf. Es fällt mir jetzt schwerer als früher, Entscheidungen zu treffen. Ich kann überhaupt keine Entscheidungen mehr treffen. N Ich habe nicht das Gefühl, schlechter auszusehen als früher. Ich mache mir Sorgen, dass ich alt oder unattraktiv aussehe. Ich habe das Gefühl, dass Veränderungen in meinem Aussehen eintreten, die mich hässlich machen. Ich finde mich hässlich. O Ich kann so gut arbeiten wie früher. Ich muss mir einen Ruck geben, bevor ich eine Tätigkeit in Angriff nehme. Ich muss mich zu jeder Tätigkeit zwingen. Ich bin unfähig zu arbeiten. P Ich schlafe so gut wie sonst. Ich schlafe nicht mehr so gut wie früher. Ich wache 1 bis 2 Stunden früher auf als sonst, und es fällt mir schwer, wieder einzuschlafen. Ich wache mehrere Stunden früher auf als sonst und kann nicht mehr einschlafen. 1 2 3 0 1 2 3 Q Ich ermüde nicht stärker als sonst. Ich ermüde schneller als früher. Fast alles ermüdet mich. Ich bin zu müde, um etwas zu tun. R Mein Appetit ist nicht schlechter als sonst. Mein Appetit ist nicht mehr so gut wie früher. Men Appetit hat sehr stark nachgelassen. Ich habe überhaupt keinen Appetit mehr. S Ich habe in letzter Zeit kaum abgenommen. Ich habe mehr als 2 Kilo abgenommen. Ich habe mehr als 5 Kilo abgenommen. Ich habe mehr als 8 Kilo abgenommen. Ich esse absichtlich weniger, um abzunehmen JA NEIN 0 1 2 3 0 1 2 3 T Ich mache mir keine größeren Sorgen um meine Gesundheit als sonst. Ich mache mir Sorgen über körperliche Probleme wie Schmerzen, Magenbeschwerden oder Verstopfung. Ich mache mir so große Sorgen über gesundheitliche Probleme, dass es mir schwer fällt, an etwas anderes zu denken. Ich mache mir so große Sorgen über gesundheitliche Probleme, dass ich an nicht anderes mehr denken kann. U Ich habe in letzter Zeit keine Veränderungen meines Interesses an Sex bemerkt. Ich interessiere mich jetzt weniger für Sex als früher. Ich interessiere mich jetzt viel weniger für Sex. Ich habe das Interesse an Sex völlig verloren. ________ Subtotal Seite 2 ________ Subtotal Seite 1 ________ Summenwert 121 11.3 Hamilton Depressionsskala CIPS HAMD Collegium Internationale Psychiatriae Scalarum Hamilton Depression Scale Anleitung Bitte jeweils nur die zutreffende Ziffer ankreuzen! Bitte alle Feststellungen beantworten! 1. Depressive Verstimmung (Gefühl der Traurigkeit, Hoffnungslosigkeit, Hilflosigkeit, Wertlosigkeit) Keine Nur auf Befragung geäußert Vom Patienten spontan geäußert Aus dem Verhalten zu erkennen (z.B. Gesichtsausdruck, Körperhaltung, Stimme, Neigung zum Weinen) Patient drückt FAST AUSSCHLIESSLICH diese Gefühlszustände in seiner verbalen und nicht verbalen Kommunikation aus 2. Schuldgefühle Keine Selbstvorwürfe, glaubt Mitmenschen enttäuscht zu haben Schuldgefühle oder Grübeln über frühere Fehler und „Sünden“ Jetzige Krankheit wird als Strafe gewertet, Versündigungswahn Anklagende oder bedrohende akustische oder optische Halluzinationen 3. Suizid Keiner Lebensüberdruss Todeswunsch, denkt an den eigenen Tod Suizidgedanken oder entsprechendes Verhalten Suizidversuche (jeder ernste Versuch) 4. Einschlafstörung Keine Gelegentliche Einschlafstörungen (mehr als ½ Stunde) Regelmäßige Einschlafstörungen 5. Durchschlafstörungen Keine Patient klagt über unruhigen oder gestörten Schlaf Nächtliches Aufwachen bzw. Aufstehen (falls nicht nur zur Harn- und Stuhlentleerung) 0 1 2 3 4 0 1 2 3 4 0 1 2 3 4 0 1 2 0 1 2 122 6. Schlafstörungen am Morgen Keine 0 Vorzeitiges Erwachen, aber nochmaliges Einschlafen 1 Vorzeitiges Erwachen ohne nochmaliges Einschlafen 2 7. Arbeit und sonstige Tätigkeiten Keine Beeinträchtigung 0 Hält sich für leistungsunfähig, erschöpft oder schlapp bei seinen Tätigkeiten (Arbeit oder Hobbys) oder fühlt sich entsprechend. 1 Verlust des Interesses an seinen Tätigkeiten (Arbeit oder Hobbys), muss sich dazu zwingen. Sagt das selbst oder lässt es durch Lustlosigkeit, Entscheidungslosigkeit und sprunghafte Entschlussänderungen erkennen. 2 Wendet weniger Zeit für seine Tätigkeiten auf oder leistet weniger. Bei stationärer Behandlung Ziffer 3 ankreuzen, wenn Patient weniger als 3 Stunden an Tätigkeiten teilnimmt. Ausgenommen Hausarbeiten auf der Station 3 Hat wegen der jetzigen Krankheit mit der Arbeit aufgehört. Bei stationärer Behandlung ist Ziffer 4 anzukreuzen, falls der Patient an keinen Tätigkeiten teilnimmt, mit Ausnahme der Hausarbeit auf Station, oder wenn der Patient die Hausarbeit nur unter Mithilfe leisten kann. 4 8. Depressive Hemmung (Verlangsamung von Denken und Sprache; Konzentrationsschwäche, reduzierte Motorik) Sprache und Denken normal 0 Geringe Verlangsamung bei der Exploration 1 Deutliche Verlangsamung bei der Exploration 2 Exploration schwierig 3 Ausgeprägter Stupor 4 9. Erregung Keine Zappeligkeit Spielen mit den Fingern, Haaren usw. Hin- und herlaufen, nicht still sitzen können Händeringen, Nägelbeißen, Haareraufen, Lippenbeißen usw. 10. Angst – psychisch Keine Schwierigkeit Subjektive Spannung und Reizbarkeit Sorgt sich um Nichtigkeiten Besorgte Grundhaltung, die sich im Gesichtsausdruck und in der Sprechweise äußert Ängste werden spontan vorgebracht 11. Angst – somatisch Körperliche Begleiterscheinungen der Angst wie: Gastrointestinale (Mundtrockenheit, Winde, Verdauungsstörungen, Durchfall, Krämpfe, Aufstoßen) – Kardiovaskuläre (Herzklopfen, Kopfschmerzen) – Respiratorische (Hyperventilation, Seufzen) – Pollakisurie – Schwitzen Keine Geringe Mäßige Starke Extreme (Patient ist handlungsunfähig) 12. Körperliche Symptome – gastrointestinale Keine Appetitmangel, isst aber ohne Zuspruch Schweregefühl im Abdomen Muss zum Essen angehalten werden. Verlangt oder benötigt Abführmittel oder andere Magen-Darmpräparate 13. Körperliche Symptome – allgemeine Keine Schweregefühl in Gliedern, Rücken oder Kopf. Rücken-, Kopf-, Muskelschmerzen. Verlust der Tatkraft, Erschöpfbarkeit Bei jeder deutlichen Ausprägung eines Symptoms 2 ankreuzen 14. Genitalsymptome wie etwa: Libidoverlust, Menstruationsstörungen etc. Keine Geringe Starke 15. Hypochondrie Keine Verstärkte Selbstbeobachtung (auf den Körper bezogen) Ganz in Anspruch genommen durch Sorgen um die eigene Gesundheit Zahlreiche Klagen, verlangt Hilfe etc. Hypochrondrische Wahnvorstellungen 16. Gewichtsverlust (entweder a oder b ankreuzen) a. Aus Anamnese Kein Gewichtsverlust 0 Gewichtsverlust wahrscheinlich im Zusammenhang mit jetziger Krankheit 1 Sicherer Gewichtsverlust laut Patient 2 b. Nach wöchentlichem Wiegen in der Klinik, wenn Gewichtsverlust Weniger als 0,5 kg/Woche 0 Mehr als 0,5 kg/Woche 1 Mehr als 1 kg/Woche 2 17. Krankheitseinsicht Patient erkennt, dass er depressiv und krank ist 0 Räumt Krankheit ein, führt sie aber auf schlechte Ernährung, Klima, Überarbeitung, Virus, Ruhebedürfnis etc. zurück 1 Leugnet Krankheit ab 2 18. Tagesschwankungen a. Geben Sie an, ob die Symptome schlimmer am Morgen oder am Abend sind. Sofern KEINE Tagesschwankungen auftreten, ist „Keine Tagesschwankungen“ anzukreuzen. Keine Tagesschwankungen 0 Symptome schlimmer am Morgen 1 Symptome schlimmer am Abend 2 b. Wenn es Schwankungen gibt, geben Sie die Stärke der SCHWANKUNGEN an. Falls es KEINE Schwankungen gibt, kreuzen sie „Keine“ an. Keine 0 Gering 1 Stark 2 19. Depersonalisation, Derealisation wie etwa: Unwirklichkeitsgefühl, nihilistische Ideen Keine 0 Gering 1 Mäßig 2 Stark 3 Extrem (Patient ist handlungsunfähig) 4 20. Paranoide Symptome Keine 0 Misstrauisch 1 Beziehungsideen 2 Beziehungs- und Verfolgungswahn 3 21. Zwangsymptome Keine 0 Gering 1 Stark 2 0 1 2 3 4 0 1 2 3 4 0 1 2 3 4 0 1 2 0 1 2 0 1 2 0 1 2 3 4 123 12 Danksagung An erster Stelle möchte ich mich bei meinem Doktorvater Prof. Roland Kalb bedanken. Durch seine Betreuung und vor allem Dank seiner Offenheit gegenüber Vorschlägen und Ideen konnte die Arbeit erst in der Form verwirklicht werden, wie sie jetzt ist. Ein ganz besonderer Dank geht auch an meine Mit-Doktorandin Franziska Groenen, die sich mit mir durch alle Höhen und Tiefen gekämpft hat (ohne Dich wäre ich mit Sicherheit so manches Mal verzweifelt und hätte einfach alles hingeschmissen... Ich hätte mir keine bessere Partnerin vorstellen können!). Daneben bin ich auch der Medizinischen Klinik für Immunologie und Rheumatologie der Uniklinik Erlangen verpflichtet, die nicht nur die Räumlichkeiten und Materialien zur Bestimmung der Stammzelldaten zur Verfügung stellte, sondern uns auch in die Kunst der FACS-Messung einwies und uns bei der Auswertung unterstütze (vielen Dank Ulrike Weinzierl und Prof. Martin Herrmann). Ebenso möchte ich mich bei dem Leibniz-Institut für Altersforschung in Jena bedanken, das auf einen simplen Anruf die Bestimmung der Telomere unentgeltlich übernahm und somit das frühe Ende unserer Studie verhinderte. Insbesondere vielen Dank an Dr. Christoph Englert für seine Unterstützung und die Hilfe bei dem Artikel. Mein größter Dank gilt Dr. Nils Hartmann, ohne den die Vollendung meiner Doktorarbeit und vor allem die Veröffentlichung unseres Artikels nie möglich gewesen wären! Er hatte immer eine Antwort auf meine unzähligen Fragen und machte sich durch seinen Einfallsreichtum und seine Ausdauer (insbesondere bei der Veröffentlichung des Artikels) unbezahlbar! Last but not least danke ich meinem Freund und meiner Familie von Herzen für die mentale Unterstützung und die nicht enden wollenden Motivationskünste. Insbesondere meiner Mutter und meinem Bruder danke ich auch für die unzähligen Korrekturlesungen der Arbeit und Verbesserungsvorschläge für den Artikel (Danke Oli, für Deine Ausdauer bei den nervtötenden Fragen des „Reviewer 2“!). Herzlichsten Dank an alle – Ihr seid die Besten! 124