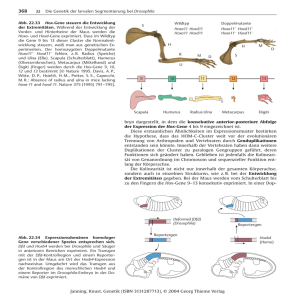
Biokatalyse
Werbung

Biokatalyse 46 S O N D E R A U S G A B E Abb.3: Elektronenmikroskopische Aufnahme des thermoalkaliphilen Bakteriums Anaerobranca gottschalkii mus aus dessen Genomsequenz heraus ist es überaus hilfreich, auch die in Spezialdatenbanken zusammengetragene Information über Stoffwechselwege einzubeziehen (Bsp. EcoCyc Datenbank (Pangea Systems) oder Kyoto Encyclopedia of Genes and Genomes, KEGG, www.tokyo-center.genome. ad.jp/kegg/). Bei einem gezielten Genomanalyseansatz zur Suche nach biotechnologisch wertvollen neuen Enzymen ist es nicht von Interesse, die Sequenz jedes Gens im Genom zu vervollständigen – die 20. Kopie eines ribosomalen Proteins oder eines Transkriptionsfaktors ist hier irrelevant. Nur die für weitere funktionelle Analysen ausgewählten Gene müssen vollständig aufgeklärt werden. Die Komplettierung dieser Gene aus Sequenzfragmenten ist meist einfach, da aus der shotgun-Sequenzierungsphase zahlreiche Plasmide vorliegen, welche die Sequenzlücken abdecken. Durch Sequenzierung mit spezifischen aus den bekannten Sequenzbruchstücken abgeleiteten Primern lassen sich die potenziell wertvollen Gene rasch und preiswert vervollständigen. Die Konstruktion einer Bibliothek mit großen DNA-Inserts (Cosmide oder BACs) – für die Vollsequenzierung von Genomen meist unverzichtbar – ist hier nicht erforderlich. Zwar kann man die Anordnung der Gene im partialsequenzierten Genom über Contigreihen nur bedingt und meist in über hundert Fragmenten bestimmen, doch ist die Information über großräumige Zusammenhänge der Genanordnung nur bedingt von Interesse, wenn ein Projekt primär die möglichst kostengünstige und rasche Identifizierung neuer enzymcodierender Gene zum Ziel hat, wie z.B. beim DBU-Projekt „Kohlenhydrat-prozessierende Enzyme aus dem thermoalkaliphilen Bakterium Anerobranca gottschalkii “. Bei Anwendung einer partiellen Genomanalyse zur Suche nach einem definierten Set interessanter Gene kann man also zu einem Bruchteil der Kosten einer Genomvollsequenzierung einen großen Teil aller interessanten Gene erfas- sen und einer raschen weiteren Verwertung zuführen. Nach partieller Sequenzierung des Genoms von Anaerobranca gottschalkii werden zur Zeit die Gene für die α-Amylase, das Verzweigungsenzym, die Pullulanase und die CGTase in den mesophilen Wirtsorganismen E. coli, B. subtilis und S. carnosus kloniert. Sekretion von Enzymen in Gram-positiven Wirtsorganismen Die Sekretion rekombinanter Enzyme in das Kulturmedium bakterieller Wirtsorganismen ist eine attraktive Alternative zur intrazellulären Überproduktion, da auf diesem Wege die Bildung von „inclusion bodies“, wie sie in E. coli beobachtet werden, stark reduziert werden kann. Darüber hinaus wird durch die Sekretion des Enzyms eine signifikante Produktanreicherung erzielt und eine kontinuierliche Herstellung ermöglicht. Aufgrund des Fehlens einer äußeren Membran sind Gram-positive Bakterien in der Lage, homologe und (einige) heterologe Proteine in großen Mengen direkt in den Kulturüberstand auszuscheiden und stellen daher potenziell vielversprechende mesophile Wirtsorganismen für die heterologe Expression von Enzymen aus dem thermoalkaliphilen Bakterium Anaerobranca gottschalkii dar. In den letzten Jahren wurden signifikante Fortschritte bei der Aufklärung des Mechanismus der Proteinsekretion bei Grampositiven Bakterien (insbesonders bei Bacillus subtilis als Gram-positivem Modellorganismus) erzielt (van Wely et al., 2001). Die Identifizierung und funktionelle Charakterisierung der Komponenten des generellen Proteinexportapparats (der sogenannten Sec-Translokase; Abb. 5 ) bei verschiedenen Gram-positiven Bakterien ergab, dass der Mechanismus der Proteintranslokation über die Cytoplasmamembran dieser Bakterienklasse weitgehend dem des am besten untersuchten Gram-negativen Modellbakteriums Escherichia coli (Manting und Driessen, D E R D B U 2000) entspricht: Sekretorische Proteine werden als höhermolekulare Vorläuferproteine mit einem aminoterminalen Signalpeptid synthetisiert. Während oder kurz nach ihrer Synthese werden die Vorläuferproteine durch spezifische Faktoren (wie zum Beispiel dem aus dem Ffh-Protein und der scRNA bestehenden bakteriellen Signalerkennungspartikel) erkannt und, vermittelt durch die Wechselwirkung mit einem Membranrezeptor (Srb), an die aus verschiedenen Sec-Proteinen aufgebaute Translokase herangeführt. Der membranintegrale Teil des Proteinexportapparats besteht aus fünf Proteinen (SecY, SecE, SecG, SecDF und YajC), die zusammen das Translokase-Holoenzym bilden. SecY und SecE stellen hierbei den Kernkomplex eines hydrophilen Kanals dar, durch den die Exportproteine über die Membran gelangen. SecG, SecDF sowie YajC stimulieren die Proteintranslokation am Kernkomplex. Eine weitere Untereinheit des Proteinexportapparats ist das SecA-Protein, welches als sogenannte TranslokationsATPase die Energie der ATP-Bindung und –Hydrolyse an die Translokation der Polypeptidkette über die Membran koppelt. Bei diesem Vorgang durchläuft SecA wiederholt Zyklen von ATP-getriebener Membraninsertion und –deinsertion, wobei bei jedem Zyklus etwa 20 bis 30 Aminosäuren der Polypeptidkette durch die Membran gefädelt werden (Nähnadelmodell). Während bzw. kurz nach der Translokation des Exportproteins über die Membran wird das Signalpeptid durch spezifische Signalpeptidasen abgespalten und das reife Protein auf der transSeite der Membran freigesetzt. Über die Vorgänge, die sich an den eigentlichen Membrantransport anschließen und die letztlich zur Freisetzung der korrekt gefalteten Proteine in den Kulturüberstand führen, ist bisher nur relativ wenig bekannt. Die Isolierung von B. subtilis -Mutanten, die eine reduzierte Sekretion einer überproduzierten α-Amylase aufwiesen, führte zur Identifizierung des PrsA-Proteins. PrsA ist ein an der Außenseite der Plasmamembran verankertes Lipoprotein, das eine gewisse Ähnlichkeit zur Parvulin-Klasse von Peptidyl-prolyl-cis/trans-Isomerasen aufweist. PrsA ist vermutlich an der extracytosolischen Faltung von Exportproteinen beteiligt und stellt, vor allem unter Überproduktionsbedingungen, einen limitierenden Faktor bei der Proteinsekretion in Gram-positiven Bakterien dar. So konnte gezeigt werden, dass die gleichzeitige Überexpression von sekretorischen Proteinen und PrsA zu einer signifikanten Steigerung der Ausbeute an korrekt gefalteten Proteinen im Überstand führen kann (Kontinen und Sarvas, 1993). Die Fähigkeit Gram-positiver Bakterien (zum Beispiel verschiedener Bacillus-Arten) zur effizienten Sekretion von Proteinen wird Biokatalyse S O N D E R A U S G A B E D E R 47 D B U auch schon seit langem für die industrielle Gewinnung einer Vielzahl von Enzymen eingesetzt (Harwood, 1992). Hierbei handelt es sich vor allem um hydrolytische Enzyme (Proteasen, Amylasen, Lipasen), die unter anderem in der Waschmittelindustrie und in der Lebensmittelindustrie zum Einsatz kommen. Die entsprechenden Enzyme stammen zumeist aus dem zelleigenen Enyzmrepertoire der jeweiligen Mikroorganismen und werden in vielen Fällen in Mengen von bis zu 20 g/l in das Kulturmedium sekretiert. Diese hohen Ausbeuten werden jedoch meist nur mit optimierten Produktionsstämmen erzielt, die durch mehrere Zyklen von ungerichteter Mutagenese und anschließendem Screening auf erhöhte Enzymausbeuten erhalten wurden. Die Ursache(n) für die erhöhten Ausbeuten an sekretiertem Enzym sind jedoch in den allermeisten Fällen nicht bekannt. Im Gegensatz dazu wurde bei Ansätzen zur Verwendung Gram-positiver Bakterien als Wirtsorganismen für die sekretorische Gewinnung von heterologen Proteinen (vor allem bei Proteinen eukaryontischen Ursprungs) in vielen Fällen beobachtet, dass die Mengen der gewünschten Proteine im Kulturüberstand deutlich geringer sind als die Mengen, die bei der Sekretion wirtseigener Enzyme erhalten werden können (Simonen und Palva, 1993). Für diesen Befund sind mehrere Engpässe im Sekretionsweg verantwortlich die, einzeln oder in Kombination, zu einer drastischen Verringerung der Ausbeute der gewünschten Proteine im Kulturüberstand führen können. Die bei der heterologen Proteinsekretion am häufigsten beobachteten Probleme sind hierbei (1) ein ineffizienter Transport des Proteins über die Cytoplasmamembran, (2) eine ineffiziente oder falsche Faltung des Proteins im Anschluss an den Membrantransport, (3) ein Abbau des Proteins durch zellwandassoziierte Proteasen sowie (4) der Abbau des Proteins durch lösliche Proteasen im Kulturüberstand. Der jeweilige Anteil der einzelnen Problemzonen an der Verminderung der Menge an sekretiertem Protein im Kulturüberstand ist dabei sowohl von den spezifischen Eigenschaften des heterologen Proteins wie auch von der Art des verwendeten Wirtsorganismus abhängig. So ist die Sekretion großer Mengen von Proteasen durch verschiedene Bacillus-Arten bei der industriellen Proteaseproduktion natürlich durchaus erwünscht, stellt aber gleichzeitig ein großes Problem beim Einsatz dieser BacillusStämme als Wirtsorganismen für die heterologe Proteinsekretion dar. Für die sekretorische Gewinnung von proteaselabilen heterologen Proteinen bieten sich daher vor allem Gram-positive Bakterien als Wirtssysteme an, die keinerlei (oder nur geringfügige) proteolytische Aktivitäten im Kulturüber- stand aufweisen. Einer der Organismen, der diese Voraussetzung erfüllt, ist das ursprünglich aus Trockenwurst isolierte Bakterium Staphylococcus carnosus. So erlaubte die Verwendung eines auf einer StaphylokokkenLipase basierenden Sekretionsvektorsystems in der Tat die effiziente Sekretion einer Vielzahl von heterologen Proteinen in den Kulturüberstand von S. carnosus (Meens et al., 1997; Sturmfels et al., 2001 und darin enthaltene Referenzen). Am Beispiel eines humanen Calcitonin-Vorläuferproteins wurde darüber hinaus gezeigt, dass mit diesem Wirtssystem Ausbeuten von bis zu 2 g sekretiertes Protein pro Liter Kulturmedium erzielt werden können (Dilsen et al., 2000). Basierend auf diesen vielversprechenden Eigenschaften soll daher bevorzugt S. carnosus als Wirtsorganismus für die sekretorische Gewinnung von Kohlehydrat-umsetzenden Enzymen aus A. gottschalkii im Rahmen des DBU-Verbundprojekts Biokatalyse eingesetzt werden. die Aminosäureaustausche den gewünschten Effekt hervorgerufen haben. Mit dieser Strategie ist es unter anderem gelungen, die Substratspezifitäten von Enzymen zu verändern bzw. ihre Thermostabilitäten zu erhöhen (Chen, 2001; Sterner und Liebl, 2001). In vielen anderen Fällen ist rationales Design jedoch gescheitert. Dies liegt vor allem daran, dass wegen der Komplexität von Enzymen auch bei genauer Kenntnis der Struktur die Auswirkung eines Aminosäureaustausches auf seine Funktion nicht exakt vorhergesagt werden kann. Diesem Problem trägt der Ansatz der gerichteten Evolution Rechnung, der sich am Prozess der natürlichen Evolution orientiert. Dabei ist eine genaue Kenntnis der Struktur-Funktionsbeziehung prinzipiell nicht erforderlich. Bei der gerichteten Evolution wird durch den zufälligen Einbau von Mutationen in das Wildtyp-Gen zunächst ein großes Repertoire an Genvarianten hergestellt. Anschließend werden die Genvarian- Veränderung der Funktion und Stabilität von Enzymen durch rationales Design und gerichtete Evolution Enzyme katalysieren metabolische Reaktionen mit hoher Effizienz und Spezifität und ihre Aktivitäten werden durch eine Reihe von Faktoren reguliert. Die ständige Anpassung der Aktivität, Regulation und Stabilität von Enzymen an sich verändernde Umweltbedingungen ist eine entscheidende Voraussetzung für das Überleben von Organismen. Seit einiger Zeit ist es möglich, die Eigenschaften von Enzymen gezielt im Labor zu verändern. Dies hat zum einen zu einem besseren Verständnis der natürlichen Evolutionsprozesse von Enzymen geführt (Babbitt und Gerlt, 2001). Zum anderen ist es möglich geworden, biotechnologisch relevante Enzymen zu erzeugen, die in industriellen Verfahren eingesetzt werden können (Petrounia und Arnold, 2000). Enzyme sind hier konventionellen chemischen Katalysatoren oft überlegen, da sie sehr substrat- und stereospezifisch sind und Reaktionen unter umweltschonenderen Bedingungen katalysieren (Arnold, 2001; Bornscheuer und Pohl, 2001). Es gibt zwei prinzipiell unterschiedliche Ansätze zur Veränderung von Enzymen, rationales Design und gerichtete Evolution (Chen, 2001). Rationales Design setzt die genaue Kenntnis der Struktur und Funktion eines Enzyms voraus. Aufbauend auf diesem Wissen, werden Aminosäuren an sorgfältig ausgewählten Positionen durch gerichtete Mutagenese gezielt ausgetauscht. Das mutierte Gen wird dann in einem geeigneten Wirtsorganismus – häufig Escherichia coli – exprimiert. Anschließend wird das Proteinprodukt gereinigt und in vitro getestet, ob Abb. 4 : Wachstum der in Genbanken abgelegten Sequenzen von 1982 bis Ende März 2001. Informationsquelle GenBank (NCBI). Y-Achse: Anzahl der sequenzierten Basen in Megabasenpaaren. X-Achse: Jahr. ten in Plasmide kloniert, die ihre Expression in einem passenden Wirtsorganismus ermöglichen; nach Transformation und Plattierung der Wirtszellen, werden diese nach den neuen Eigenschaften selektiert oder gescreent. Unter Selektion versteht man dabei die Kopplung des Überlebens der Wirtszelle an die Produktion des verbesserten Enzyms. In einem Screeningverfahren werden dagegen Wirtszellen, die ein Plasmid aufgenommen haben, einzeln auf die neue enzymatische Eigenschaft hin untersucht. Screening ist somit allgemeiner anwendbar als Selektion, jedoch in der Regel experimentell aufwändiger. Der Prozess von Mutation und Selektion/Screening kann über mehrere Runden durchgeführt und so lange wiederholt werden, bis weitere Verbesserungen nicht mehr möglich sind bzw. nicht mehr detektiert werden können. So können Biokatalyse 48 S O N D E R A U S G A B E Abb. 5 Sec-abhängiger Proteinexport bei Gram-positiven Bakterien. An Ribosomen im Cytosol synthetisierte Vorläuferproteine werden anhand ihres Signalpeptids als Exportprotein erkannt und an die Exportstellen in der Membran herangeführt. Das aus den Sec-Proteinen aufgebaute TranslokaseHoloenzym katalysiert den energieverbrauchenden (ATP und Membranpotential) Transport durch die Plasmamembran. β-SRP: bakterielles Signalerkennungspartikel bestehend aus scRNA und Ffh-Protein; Srb: membranassoziierter Rezeptor für β-SRP; SPase: Signalpeptidase; PrsA: an der Faltung exportierter Proteine beteiligtes Lipoprotein. Weitere Details siehe Text. durch schrittweise Erzeugung kleiner Verbesserungen über viele Generationen hinweg erhebliche Änderungen in Funktion und Stabilität von Enzymen erreicht werden. Während die Techniken zum zufälligen Einbau von Mutationen recht einheitlich sind – meist wird eine Polymerasekettenreaktion unter Bedingungen durchgeführt, die zum gehäuften Falscheinbau von Nukleotiden führen –, sind die verwendeten Screeningund Selektionssysteme spezifisch für das jeweilige Enzym und die zu verändernde Eigenschaft. Durch gerichtete Evolution wurden in jüngster Zeit die Löslichkeiten von Enzymen verbessert, Enantioselektivitäten gesteigert und erhöhte Stabilitäten gegenüber thermischer Inaktivierung erzielt (Petrounia und Arnold, 2001; Bornscheuer und Pohl, 2001; Sterner und Liebl, 2001). Als besonders erfolgreich bei der gerichteten Evolution neuer enzymatischer Eigenschaften hat sich der Einsatz kombinatorischer Methoden erwiesen, wie z.B. „DNAShuffling“ (Stemmer, 1994) oder „Staggered extension process“ (Zhao et al., 1998). Hierbei werden vorteilhafte Mutationen, die sich in verschiedenen Mitgliedern des Genrepertoires angesammelt haben, auf einem einzigen Gen miteinander kombiniert. Dies kann zu einer sehr schnellen und drastischen Verbesserung der Eigenschaften der gewünschten enzymatischen Eigenschaften führen (Ness et al., 2000). Unter Family-Shuffling versteht man eine vielversprechende Modifikation des ursprünglichen DNA-ShufflingAnsatzes (Crameri et al., 1998). Dabei werden Gene, die in unterschiedlichen Spezies für das gleiche Enzym kodieren, miteinander kombiniert. Auf diese Weise entstehen Hybridenzyme, die positive Eigenschaften verschiedener Eltern-Enzyme in sich vereinigen (Ness et al., 1999). Besonders instruktiv wird gerichtete Evolution dann, wenn die erzielte Veränderung einer enzymatischen Eigenschaft auf der Basis einer hochaufgelösten Struktur des Enzyms analysiert wird. Dies wurde kürzlich beispielhaft an der Verbesserung der katalytischen Aktivität eines thermostabilen Enzyms bei niedrigen Temperaturen gezeigt (Merz et al., 2000). Zusammenfassend bleibt zu sagen, dass die Eigenschaften von Enzymen sowohl durch rationales Design als auch durch gerichtete Evolution in gewünschter Weise verändert werden können. Während Design genaue Kenntnisse der Struktur und Funktion eines Enzyms voraussetzt, ist der Erfolg gerichteter Evolution an die Existenz eines effizienten Screening- oder Selektionssystems gebunden. Mit beiden Ansätzen wurden in der Vergangenheit beeindruckende Ergebnisse erzielt. Deshalb muss bei der Herstellung von maßgeschneiderten Enzymen aus dem extremophilen Mikroorganismus A. gottschalkii entschieden werden, welche Methode am erfolgversprechendsten ist oder ob die Kombination beider Ansätzen angeraten scheint. Literatur Arnold, F. H. (2001): Combinatorial and computational challenges for biocatalyst design. Nature 409: 253-257 Babbitt, P.C. , Gerlt, J. A. (2001): New functions from old scaffolds: how nature reengineers enzymes for new functions. Adv. Prot. Chem. 55: 1-28 Bornscheuer, U.T., Pohl, M. (2001): Improved biocatalysts by directed evolution and rational design. Curr. Opin. Chem. Biol. 5: 137-143 Chen, R. (2001): Enzyme engineering: rational design versus directed evolution. Trends Biochem. Sci. 19: 13-14 Crameri, A., Raillard, S.-A., Bermudez, E. & Stemmer W.P.C. (1998): DNA shuffling of a family of genes from diverse species accelerates directed evolution. Nature 391: 288-291 Dilsen, S.; Paul, W.; Sandgathe, A.; Tippe, D.; Freudl, R.; Thömmes, J.; Kula, M.-R.; Takors, R.; Wandrey, C.; D E R D B U Weuster-Botz, D. (2000): Fed-batch production of recombinant human calcitonin precursor fusion protein using Staphylococcus carnosus as an expressionsecretion system. Appl. Microbiol.Biotechnol. 54 : 361369 Harwood, C. R. (1992): Bacillus subtilis and its relatives: molecular biological and industrial workhorses. Trends Biotechnol. 10: 247-256 Manting, E. H. , Driessen, A. J. M., (2000): Escherichia coli translocase: the unravelling of a molecular machine. Mol.Microbiol. 37: 226-238 Kontinen, V. P.; Sarvas, M. (1993): The PrsA lipoprotein is essential for protein secretion in Bacillus subtilis and sets a limit for high-level secretion. Mol.Microbiol. 8: 727-737 Ladenstein, R., Antranikian, G., (1998): Proteins from hyperthermophiles: stability and enzymatic catalysis close to the boiling point of water, Adv. Biochem. Eng/ Biotechnol. 61: 37-85 Meens, J.; Herbort, M.; Klein, M.; Freudl, R. (1997): Use of the pre-pro part of Staphylococcus hyicus lipase as a carrier for secretion of Escherichia coli outer membrane protein A (OmpA) prevents proteolytic degradation of OmpA by cell-associated protease(s) in two different Gram-positive bacteria. Appl. Environ. Microbiol. 63: 2814-2820 Merz, A., Yee, M. C., Szadkowski, H., Pappenberger, G., Crameri, A., Stemmer, W. P., Yanofsky, C., and Kirschner, K. (2000): Improving the catalytic activity of a thermophilic enzyme at low temperatures. Biochemistry 39: 880-889 Ness, J.E., Welch, M., Giver, L., Bueno, M., Cherry, J.R., Borchert, T.V., Stemmer, W.P.C. & Minshull, J. (1999): DNA shuffling of subgenomic sequences of subtilisin. Nature Biotechnology 17: 893-896 Ness, J.E., del Cardayre, S.B., Minshull, J., Stemmer, WP.C. (2000): Molecular breeding-the natural approach to enzyme design. Adv. Prot. Chem. 55: 261-292 Niehaus, F., Bertoldo, C., Kähler, M., Antranikian, G., (1999): Extremophiles as a source of novel enzymes for industrial application, Appl. Microbiol. Biotechnol., 51: 711-729 Petrounia, I.P., Arnold, F.H. (2000): Designed evolution of enzymatic properties. Curr. Opin. Biotechnol. 11: 325330 Prowe, S. G., Antranikian, G (2001): Anerobranca gottschalkii sp. nov., a novel thermoalkaliphilic bacterium that grows anerobically at high pH and temperature, Int. J. Syst. Bacteriol., 51:457-465 Prowe, S.G., Characterization of extracellular amylolytic enzymes and sodium coupled energy transduction of a newly isolated, thermoalkaliphilic bacterium Anaerobranca gottschalkii, Dissertationsarbeit TU-HamburgHarburg Simonen, M.; Palva, I. (1993): Protein secretion in Bacillus species. Microbiol.Rev. 57: 109-137 Stemmer, W.P.C. (1994): Rapid evolution of a protein in vitro by DNA shuffling. Nature 370: 89-391 Sturmfels, A. ; Götz, F. ; Peschel, A. (2001): Secretion of human growth hormone by the food grade bacterium Staphylococcus carnosus requires a propeptide irrespective of the signal peptide used. Arch.Microbiol. 175: 295-300 Sterner, R., Liebl, W. (2001): Thermophilic adaptation of proteins. Crit. Rev. Biochem. Mol. Biol. 36: 39-106 van Wely, K. H. M.; Swaving, J.; Freudl, R.; Driessen, A. J. M.. (2001):Translocation of proteins across the cell envelope of Gram-positive bacteria. FEMS Microbiol.Rev. 25, “in press” Zhao, H., Giver, L., Shao, Z., Affholter, J.A., Arnold, F.H. (1998): Molecular evolution by staggered extension process (StEP) in vitro recombination. Nature Biotechnology 16: 258-261 Korrespondenzadresse: Prof. Dr. Garabed Antranikian TU Hamburg-Harburg Technische Mikrobiologie Kasernenstraße 12 21073 Hamburg Tel.: 040-42878 3117 Fax: 040-42878 2582 eMail: [email protected] Biokatalyse S O N D E R A U S G A B E D E R 49 D B U Dr. Elisabeth Heine1, Dr. Eva Schuh1, Dipl.-Chem. Nabil Daâloul1, Prof. Dr. Hartwig Höcker1, Rudi Breier2, Dipl.-Ing. Michael Schmidt2, Dr. Anke Apitz3, Dipl.Chem. Andrea Brünner3, Prof. Dr. KarlHeinz van Pée3, Dr. Katrin Scheibner4 1 Deutsches Wollforschungsinstitut an der RWTH Aachen e.V. (DWI) 2 Textilchemie Dr. Petry GmbH, Reutlingen 3 Institut für Biochemie, TU Dresden 4 JenaBios GmbH, Jena Oxidative Enzyme in der Textilindustrie Ziel des Verbundvorhabens ist die Entwicklung und industrielle Umsetzung alternativer Verfahren zur Carbonisur von Wolle und zur Baumwollbleiche unter Verwendung oxidativer Enzyme, die im Rahmen des Projektes für diese beiden Anwendungsbereiche produziert und optimiert werden. Konventionell kommen sowohl bei der Carbonisur als auch bei der Baumwollbleiche Chemikalien zum Einsatz, die Abwasser und Maschinenteile belasten. Das Ziel des 1. Teilbereichs ist es, mit Hilfe der enzymkatalysierten Oxidation von Vegetabilien in Wolle die bei der klassischen Carbonisur eingesetzten Chemikalienmengen zu reduzieren / zu ersetzen und die Schädigung der Wolle zu minimieren. Durch die Anwendung von Oxidoreduktasen soll ein in der Papierherstellung erprobtes Verfahren für die Wollindustrie nutzbar gemacht werden. Ziel des 2. Teilbereichs ist es, mit Hilfe von Oxidoreduktasen die farbigen Begleitstoffe der Baumwolle zu zerstören. Durch die enzymatische Bleiche sollen die Menge an Lauge, die bei der Peroxid-Bleiche benötigt wird und damit die Salzfracht im Abwasser reduziert werden. Weiterhin sollte dadurch eine Energie- und Chemikalieneinsparung erreicht werden. Einleitung und Fragestellung 쑺 Im Rahmen des Verbundvorhabens werden alternative Verfahren zur Carbonisur von Wolle und zur Bleiche von Baumwolle unter Einsatz oxidativer Enzyme entwickelt und im Pilotmaßstab etabliert. Obwohl diese beiden Verfahren gänzlich unterschiedliche textile Schwerpunkte darstellen, kommen in beiden Fällen die gleichen Enzyme zum Einsatz. Die Kombination dieser unterschiedlichen Themenkomplexe zu einem Verbund bietet daher den Vorteil, dass die notwendigen Entwicklungsarbeiten im Hinblick auf Enzymproduktion und -optimierung für beide Bereiche zum überwiegenden Teil durch den biotechnologisch orientierten Verbundpartner (JenaBios GmbH, Laccase, Mangan-Peroxidase) übernommen werden und ein Teilbereich (Peroxidase) durch das Forschungsinstitut (TU Dresden) abgedeckt wird. Die Realisierung der Verfahren im Labormaßstab erfolgt durch die Forschungsinstitute (DWI, Wollveredlung und TU Dresden, Baumwollveredlung) und die Umsetzung der Verfahren in den Pilotmaßstab im Hinblick auf den Transfer in die Textilveredlungsbetriebe durch den Hilfsmittelhersteller (Textilchemie Dr. Petry), der sowohl auf dem Sektor der Woll- als auch der Baumwollveredlung Expertise aufweist. Enzymkatalysierte Oxidation vegetabiler Bestandteile in Wolle als Alternative zur Carbonisur Als Vegetabilien werden pflanzliche Überreste in Wolle bezeichnet. Die Kontamination von Wolle mit Vegetabilien ist auf die Weidebedingungen und die Bedingungen, unter denen die Schafe gehalten werden, zurückzuführen. Menge und Art der Vegetabilien sind Faktoren, die zu Preisminderungen der Ware führen. Bei den Pflanzenresten handelt es sich um die klettenförmigen Verbreitungseinheiten bestimmter Futterpflanzen sowie Heu, Stroh und begrannte Grassamen. Die verholzten spiralbandigen Kletten der Pflanzengattung Medicago können bis zu 30% des Vegetabiliengehaltes von Waschwolle ausmachen [1] (Abb. 1). Im allgemeinen sind Kletten, insbesondere Ringelkletten, vorzugsweise bei Australwollen zu finden, Steinkletten bei Capwollen; bei beiden Provenienzen aber auch bestimmte Gräserarten (Ringelkletten: Xanthium spinosum, Medicago denticulata; Gräser: Barley grass (Hordeum lepinorum) [2]. Europäische Wollen sind weniger durch Kletten als durch Gräser und Stroh verunreinigt. Einige Kletten werden durch Krempeln und Kämmen erfolgreich entfernt, andere können so feinfaserig vorliegen, dass sie sich wie die Wollfasern selbst verhalten Abb. 1: Vegetabilien-belastete Rohwolle, Wollgewebe aus nicht-carbonisierter Wolle, aus Rohwolle isolierte Ringelkletten und Grassamen. Biokatalyse 50 S O N D E R A U S G A B E Abb. 2: Reaktionsmechanismus für den mikrobiellen Ligninabbau durch Manganperoxidase aus Nematoloma frowardii. und mit gekämmt werden. Diese Vegetabilien können bei der Verarbeitung und Veredlung der belasteten Wolle zu Problemen führen, z.B. verursachen Klettenfragmente beim Spinnen Fadenrisse oder unerwünsch- te Verdickungen im Faden. Bereits kleine Anteile an Vegetabilien in Tuchen verändern den Griff bzw. das Aussehen, da sie zu Fehlfärbungen führen. Krempel, Kämme und Spinnstrecken werden bei der Wollverarbei- Abb. 3: Fermentation von Bacillus sphaericus, dem Produzenten einer neuen intrazellulären Peroxidase mit ungewöhnlichen Eigenschaften D E R D B U tung durch Vegetabilien mechanisch belastet [3]. Die chemische Methode zur Entfernung der Vegetabilien ist die Carbonisur. Hierbei wird die gewaschene Wolle in ein Bad mit 6-8% Schwefelsäure gegeben, abgequetscht, vorgetrocknet und dann bei Temperaturen von über 100°C „gebrannt“. Die durch die Trocknung hervorgerufene Konzentrierung der Säure bewirkt eine Dehydratisierung (Verkohlung) der Cellulose. Die Reste der carbonisierten Vegetabilien können durch Walzen zerkleinert, der Klettenstaub anschließend ausgeklopft und die Wolle neutralisiert und gespült werden. Von harten Schalenteilen wird die Säure jedoch nicht gut aufgenommen, daher sind diese Stücke auch nach der Carbonisur kaum zu zertrümmern. Die Carbonisur ist nicht unproblematisch für die Wolle selbst. Neben den Vegetabilien kann auch die Wolle durch die Säure geschädigt werden. Die gesamten Wollverluste können je nach vorliegender Rohwolle 12% und mehr betragen. Bleichen von Baumwolle Die Baumwollfaser ist ein einzelliges, bandartig flaches Gebilde mit meist kräftiger Zellwand. Sie besteht zu 90-95% aus reiner Cellulose und erscheint unter dem Mikroskop als flaches Band mit korkenzieherartigen Drehungen. Diese Windungen greifen beim Verspinnen scharnierartig ineinander, so dass die Fasern gut im Garn haften. Baumwolle enthält außerdem Baumwollfette und –wachse, Ligninreste, Proteine, Pektine und Mineralstoffe [4]. Je nach Herkunftsland ist Rohbaumwolle weiß (Peru), gelb oder braun (ägyptische Baumwolle) gefärbt. Besonders die gelb- und braungefärbte Baumwolle, deren Farbe in erster Linie durch die anhaftenden Ligninreste verursacht wird, muss in der Vorbehandlung des Textilgutes gebleicht werden, um nachfolgend eine einheitliche Färbung der Garne erreichen zu können. Lange Zeit wurde Hypochlorit als bleichendes Agens eingesetzt. Aus Gründen der Umweltverträglichkeit ist es heute in Deutschland überwiegend durch Wasserstoffperoxid ersetzt [5]. Wasserstoffperoxid-Lösungen reagieren schwach sauer ud zerfallen allmählich in Wasser und Sauerstoff. Wird dieser wässrigen Lösung Alkali zugesetzt, entstehen Perhydroxidanionen, die als das wirksame Agens bei der Bleiche mit Peroxid angesehen werden. Die Zersetzung des Wasserstoffperoxids steigt mit zunehmender Temperatur, zunehmendem pH-Wert und wird durch Schwermetalle und leicht oxidierbare Substanzen katalysiert. Schwermetalle müssen daher vor dem Bleichprozess maskiert und die Eigenzersetzung bei der Dosierung des Wasserstoffperoxids unter Pro- Biokatalyse S O N D E R A U S G A B E D E R 51 D B U zessbedingungen berücksichtigt werden. Einen optimalen Bleicheffekt erzielt man bei pH 11, der mit einem Minimum an Faserschädigung zusammenfällt und daher meist zur Anwendung kommt. Andere eingesetzte Bleichmittel sind z.B. Peressigsäure als mildes oxidierendes Agens und Natriumdithionit als reduzierendes Bleichmittel [6]. Es ist günstig, vor der Bleiche die Faser alkalisch abzukochen. Dabei kommt gewöhnlich Natronlauge zum Einsatz. Durch jedes Bleichverfahren wird die Cellulose mehr oder weniger abgebaut, was sich in einer Abnahme des Durchschnittspolymerisationsgrades äußert. Es ist daher nicht beliebig oft zu wiederholen. Das Ziel einer guten Bleiche ist es, einen hohen Weißgrad, eine gute Haltbarkeit des Weiß und eine gute Saugfähigkeit der Ware mit geringstmöglicher Faserschädigung und einer hohen Wirtschaftlichkeit zu erreichen. Trotzdem erhält man mit der Bleiche allein in keinem Fall ein genügend reines Weiß, sondern die Faser besitzt noch immer einen gelblichen Stich. Daher schließt sich gewöhnlich an den Bleichprozess das optische Aufhellen an. Laccase Die Laccase (p-Benzendiol: O2-Oxidoreduktase, EC 1.10.3.2) ist eine KupferOxidase [10] und das verbreitetste Lignin modifizierende Enzym, das von fast allen Weißfäulepilzen sowie einer Reihe von Schimmelpilzen (Asco- und Deuteromyceten) und den höheren Pflanzen gebildet wird [11]. Laccasen sind glykosylierte Proteine mit einem Polysaccharidgehalt von 11-80% und Molekulargewichten zwischen 59 und 110 kDa, wobei dimere und tetramere Laccasen mit Molekulargewichten bis 390 kDa auftreten können [11]. Das katalytische Zentrum der Laccase enthält vier Kupferatome. Die Laccase katalysiert Ein-Elektron-Oxi- A Unterschied zur Meerrettich-Peroxidase vermag die LiP auch nicht-phenolischen Substraten mit hohem Redox-Potential (+1,5 V in Bezug auf die Standard-WasserstoffElektrode) ein Elektron zu entziehen, was zur Bildung von Arylkation-Radikalen und nachfolgend zu spontanem Einbau von Sauerstoff (O2) sowie Bindungsspaltungen führt [14]. Voraussetzung für die Wirksamkeit der LiP sind sehr niedrige pH-Werte; das Optimum des Enzyms liegt um pH 2,5 und bereits pH-Werte größer 4,5 unterbinden die Enzymaktivität vollständig. Die LiP ist ein wichtiges, jedoch nicht unbedingt erforderliches ligninolytisches Enzym, da es eine Reihe von Weißfäulepilzen gibt, denen die LiP fehlt und die eine B Der mikrobielle Ligninabbau Der mikrobielle Ligninabbau ist ein generell aerober, oxidativer Prozess. Eine Reihe von Mikroorganismen, sowohl Pilze als auch Bakterien (Actinomyceten), besitzen die Fähigkeit, die Struktur des Lignins chemisch zu modifizieren [7]; ein substantieller Abbau des Lignins verbunden mit seiner Mineralisierung ist allerdings auf eine einzige Gruppe von Mikroorganismen beschränkt - die Basidiomyceten (Ständerpilze). Innerhalb dieser artenreichen Pilzklasse haben sich im Laufe der Evolution zwei ökophysiologische Lebensweisen entwikkelt (Holzabbau und Streuzersetzung), die einen effektiven Angriff auf das Ligninpolymer außerhalb der Zelle (extrazellulär) gewährleisten [8]. Holzabbau und Streuzersetzung können zur sogenannten Weißfäule von Lignozellulosen führen, d.h. zu einem selektiven Verlust des Lignins, unter Zurücklassung weißgefärbter Zellulosefibrillen. Ander und Eriksson [9] wiesen nach, dass die Oxidation phenolischer Verbindungen durch holzabbauende Weißfäulepilze auf extrazelluläre Oxidoreduktasen (Laccasen, Peroxidasen) zurückzuführen ist. Aus Untersuchungen zur Spaltung von Modellverbindungen und zur Depolymerisation von Ligninpräparaten wurde deutlich, dass diese Enzyme verschiedene Bindungstypen im Lignin unspezifisch durch Radikalbildung (Ein-Elektron-Oxidationen) zu spalten vermögen; sie wurden deshalb als ligninolytische (oder Lignin modifizierende) Enzyme bezeichnet. Abb. 4: Rasterelektronenmikroskopische Aufnahmen A) von Grassamen und B) Ringelkletten, den Ausgangssubstraten für die Inkubation mit den Oxidoreduktasen. dationen einer Vielzahl phenolischer Substrate; gleichzeitig wird molekularer Sauerstoff (O2) durch Übertragung von vier Elektronen zu Wasser reduziert. Es kommt zur Bildung von Phenoxy-Radikalen, die nichtenzymatischen Folgereaktionen unterliegen (Chinonbildung, Polymerisation, Bindungsspaltung). In Gegenwart geeigneter RedoxMediatoren (niedermolekulare Vermittler von Oxidationsreaktionen) ist die Laccase auch in der Lage, besonders persistente nicht-phenolische Ligninstrukturen anzugreifen. Unter Verwendung synthetischer Mediatoren gelang es Call und Mücke [12] erstmals, native Lignozellulose in einem zellfreien System (in vitro) mittels Laccase zu delignifizieren. Lignin-Peroxidase (LiP) Die LiP (EC 1.11.1.14) ist ein H2O2-abhängiges Glykoprotein mit Protoporphyrin IX (Häm) im katalytischen Zentrum [13]. Der vermutlich am intensivsten untersuchte Weißfäulepilz Phanerochaete chrysosporium bildet das Enzym in multiplen Formen (maximal 15), die Molekulargewichte von 38 kD - 43 kD und isoelektrische Punkte (pI) von 3,2-4,7 besitzen. Die LiP ist eine echte Peroxidase (Donor: H2O2 Oxidoreduktase), die in ihrer Funktionsweise große Ähnlichkeit mit der Meerrettich-Peroxidase aufweist. Im andere Möglichkeit zur Spaltung nicht-phenolischer Ligninstrukturen entwickelt haben (z.B. Lentinus edodes, Ceriporiopsis subvermispora, Panus tigrinus [15]). Bei Vorhandensein der LiP hat diese wahrscheinlich die Aufgabe, bereits gebildete Ligninbruchstükke weiter abzubauen. Der primäre Angriff des Ligninmoleküls wird jedoch mit großer Sicherheit nicht durch die LiP katalysiert, da ein effektives Mediatorsystem bisher nicht nachgewiesen werden konnte und das Enzym selbst zu groß ist, um in den kompakten Lignozellulose-Komplex zu diffundieren. Diese Aufgabe kommt bei vielen Lignin abbauenden Pilzen der nachfolgend beschriebenen Mangan-Peroxidase zu. Mangan-Peroxidase (MnP) Die MnP (EC 1.11.1.13) ist extrazellulär, glykosyliert (10-39%), enthält Häm als prosthetische Gruppe im katalytischen Zentrum, besitzt Molekulargewichte von 40 - 54 kDa und pI-Werte zwischen 2,8 und 7,0 [16]. Im Unterschied zur LiP ist das Vorkommen der MnP nicht auf holzabbauende Pilze wie Trametes versicolor beschränkt, sondern sie wird auch von streuzersetzenden Basidiomyceten wie Agaricus bisporus gebildet. Reguliert wird die MnP-Bildung von der Konzentration der Mn(II)-Ionen des umgebenden Mediums [17]. Biokatalyse 52 Die „klassische“ MnP ist katalytisch abhängig von Mn(II)-Ionen und chelatisierenden organischen Säuren (Abb. 2); in jüngerer Zeit sind jedoch auch Mn-Peroxidasen beschrieben worden, die eine Mn-unabhängige Aktivtät besitzen und Übergangsformen zur LiP darstellen. Die MnP ist wie die LiP ausschließlich in sauren Medien wirksam, ihr pH-Optimum ist jedoch nicht so niedrig und liegt für die meisten Mn-Peroxidasen zwischen 4,0 und 4,5. Der pH-Bereich, in dem das Enzym arbeitet ist breiter als der der LiP und bewegt sich zwischen pH 2,0 und 7,5. A B S O N D E R A U S G A B E Die entstandenen Radikale unterliegen verschiedenen Folgereaktionen, bei denen es u.a. zur Addition von Sauerstoff und zu Bindungsspaltungen innerhalb des Moleküls kommt. Bevorzugte Substrate der Mn(III)-Ionen sind Phenole, jedoch werden auch nicht-phenolische Aromaten (z.B. Methoxybenzole, Veratrylalkohol) und aliphatische Carbonsäuren umgesetzt. Voraussetzung für die Wirksamkeit der Mn(III)-Ionen ist ihre Stabilisierung durch chelatisierende organische Säuren, andernfalls kommt es innerhalb weniger Sekunden zur Disproportionierung des Mn(III) und zur Bildung von Mn(II) und Mn(IV), wobei letzteres zu unlöslichem Braunstein (MnO2) reagiert und somit Mangan dem Redox-Zyklus entzogen wird. Geeignete Chelatoren der Mn(III)-Ionen sind Dicarbonsäuren (u.a. Malonat, Oxalat) oder α-Hydroxycarbonsäuren (u.a. Lactat, Tartrat). Die durch Chelatisierung stabilisierten Mn(III)-Ionen sind das eigentliche oxidative Agens während MnPkatalysierter Reaktionen und übertragen als niedermolekularer, diffundibler Redox-Mediator die Oxidationskraft des Enzyms auf die Folgesubstrate (Abb. 2). Auch die Chelatisierung der Mn(II)-Ionen ist für das Funktionieren der MnP notwendig, da nur in dieser Form eine hohe Affinität des Substrates zum Enzym besteht. Chelatisierende organische Säuren werden durch Pilze in Flüssigkultur ausgeschieden (u.a. Malat). Enzymproduktion und –optimierung Abb.5: A) Lichtmikroskopische Aufnahmen eines Querschnitts von Grassamen (Stärke- und Anteile mit I2/KI angefärbt) und B) eines Ausschnitts einer Ringelklette (Ligninanteile mittels Phloroglucin/HCl angefärbt). Alle scheinbar unspezifischen Reaktionen der MnP werden durch die primär gebildeten Mn(III)-Ionen katalysiert. Bei diesen handelt es sich um ein starkes Oxidationsmittel (Standardpotential 1,54 V), das eine große Zahl organischer Verbindungen anzugreifen vermag. Grundlage für diese Reaktionen ist der Entzug eines Elektrons oder die radikalische Abstraktion eines Wasserstoffatoms [18]. Die von Pilzen sekretierten Enzyme haben ihr pH-Optimum nahezu ausschließlich im sauren Bereich. Bleichprozesse mit Wasserstoffperoxid müssen dagegen im alkalischen Milieu bei 95°C durchgeführt werden, weil nur dann eine ausreichende Stabilität des bleichenden Agens H2O2 gewährleistet ist. Eine enzymatisch unterstützte Bleiche mit Peroxidasen (Donor: H2O2 Oxidoreduktasen) sollte daher ebenfalls im alkalischen Milieu und bei höheren Temperaturen durchführbar sein. Bisher bekannte Peroxidasen sind unter diesen Bedingungen allerdings nicht stabil oder nicht aktiv. Aus diesem Grund wurde am Institut für Biochemie ein Screening auf neue intrazelluläre Peroxidasen mit guter Stabilität unter Bleichbedingungen durchgeführt. Dabei wurde ein für den Einsatz in Bleichprozessen potenziell sehr interessantes Enzym gefunden. Die Peroxidase ist temperaturstabil bis 100°C und besitzt gute Aktivität bis zu pH 11,0. Das Enzym ist in der Lage, eine Reihe von Farbstoffen unterschiedlicher Struktur zu bleichen. Von besonderer Bedeutung ist hier, dass es gelungen ist, durch eine proteolytische Teilverdauung die Größe des noch immer aktiven Enzyms auf 6,5 kDa zu reduzieren [19]. Damit scheint die D E R D B U Peroxidase prinzipiell gut in der Lage zu sein, die zu bleichenden Substanzen zwischen den Zellstofffasern direkt anzugreifen. In der ersten Projektphase muss nun die Eignung dieses Enzyms für die Bleiche von Baumwolle unter den Bedingungen des technischen Prozesses überprüft werden. Bei Eignung des Enzyms für die Baumwollbleiche muss die Produktion des Enzyms optimiert werden. Bisher wird die Peroxidase aus dem Wildstamm eines Bodenbakteriums nach aufwändiger Reinigung gewonnen (Abb. 3). Es ist vorgesehen, durch Überexpression und Ausschleusen des Enzyms mit Hilfe eines geeigneten Wirtsstammes große Mengen der Peroxidase einfach zugänglich zu machen, so dass auf weitere Anreicherungsverfahren verzichtet werden kann und die Enzymlösung nach einfacher oder eventuell sogar ohne Vorbehandlung für die Bleiche einsetzbar ist. Gleichzeitig soll ein Screening nach neuen Peroxidasen durchgeführt werden. Dabei ist es schwierig, direkt nach Peroxidasen zu selektieren. Bei Zugabe von Wasserstoffperoxid zum Züchtungsmedium für Mikroorganismen muss in erster Linie mit einer erhöhten Produktion von Katalasen gerechnet werden. Diese stören die Aktivitätstests auf Peroxidasen erheblich und müssen daher abgetrennt werden. Ein einfaches Verfahren ist die Inaktivierung durch Hitze, ein weiteres die Abtrennung in einer nativen Polyacrylamidgelelektrophorese. Es hat sich bewährt, Bodenproben geeigneter Standorte für das Screening einzusetzen. Die autochthone Bodenflora enthält ein umfangreiches Spektrum interessanter Mikroorganismen, in denen gezielt nach Enzymen gesucht werden kann, die für die vorliegende Anwendung in Frage kommen. Es ist Wert zu legen auf eine gute Enzymaktivität im Alkalischen, gute Temperaturstabilität, Bleichvermögen und Verträglichkeit mit Bleichhilfsmitteln, so diese benötigt werden. Enzymkatalysierter Abbau von Vegetabilien in Wolle Es ist das Ziel dieses Teilbereichs, mit Hilfe der enzymkatalysierten Oxidation von Vegetabilien in Wolle, die bei der klassischen Carbonisur eingesetzten Chemikalienmengen zu reduzieren bzw. vollständig zu ersetzen. Durch die Anwendung von Oxidoreduktasen (vorwiegend Manganperoxidasen und Laccasen) wird ein bereits in der Papierherstellung erprobtes Verfahren für die Wollindustrie nutzbar gemacht. Unterschiedliche Vegetabilienarten (Grassamen und Ringelkletten) werden mit definierten Enzymen bzw. Enzymcocktails isoliert behandelt, um den Abbaugrad der einzelnen Vegetabilien Biokatalyse S O N D E R A U S G A B E D E R 53 D B U unter festgelegten Reaktionsbedingungen genau zu bestimmen. Vegetabilienbelastete Wollen und von einer Wollkämmstrecke stammende Vegetabilien wurden sortiert. Sowohl die anteilmäßig am stärksten vertretenen Grassamen als auch die Ringelkletten wurden als homogenes Ausgangssubstrat eingesetzt (Abb. 4). Im ersten Projektabschnitt kamen zum Abbau der Vegetabilien sowohl oxidative Enzyme der Projektpartner JenaBios GmbH und TU Dresden als auch kommerziell erhältliche hydrolytische Enzyme zum Einsatz. Die unterschiedlichen Enzyme wurden in einigen Fällen nacheinander bzw. in Kombination eingesetzt. Zur Beurteilung des enzymkatalytisch bewirkten Modifizierungsgrades der Vegetabilien wurden unterschiedliche analytische Methoden angewendet. Nach Inkubation der Vegetabilien mit hydrolytischen Enzymen wurde die Menge der in die Flotte freigesetzten reduzierenden Zucker [20] sowie der Gewichtsverlust der Vegetabilien und somit deren Abbaugrad bestimmt. Durch die kombinierte Anwendung von MnP und Xylanase wird eine Steigerung des Abbaugrades erzielt. Im Vergleich zur Puffer-vorbehandelten Referenz ist die Zugänglichkeit des Substrates nach MnP-Einwirkung für das hydrolytische Enzym höher. Die Morphologie der Quer- und Längsschnitte unterschiedlich behandelter Grassamen und Ringelkletten wurde mit Hilfe rasterelektronen- und lichtmikroskopischer Aufnahmen dokumentiert. Die Zuordnung der morphologischen Komponenten wurde bei den mit Hilfe des Lichtmikroskops untersuchten Proben durch verschiedene Anfärbetechniken erleichtert. In Abb. 5 sind die lichtmikroskopischen Aufnahmen von Grassamen und Ringelkletten gezeigt. Die Ligninanteile in den Vegetabilien wurden mittels Phloroglucin/HCl-Anfärbung sichtbar gemacht, Stärkeanteile mit I2/KI angefärbt. Enzymkatalysierte Baumwollbleiche Ziel dieses Teilbereiches ist es, die Eignung von Oxidoreduktasen für die Bleichung von Baumwolle zu testen. Dabei sollen sowohl kommerziell erhältliche, als auch neu isolierte Enzyme mit ungewöhnlichen Stabilitäten zum Einsatz kommen. Als erstes wurde die technische Bleiche unter Laborbedingungen durchgeführt als Vergleich für die Untersuchungen. Vor und nach der Behandlung wurden der Weißgrad und der Massenverlust bestimmt. Dann erfolgte die Modifizierung der Bleichbedingungen auf die Anforderungen der enzymatischen Behandlung. Stabilisatoren oder Benetzungsmittel können die Aktivität und Stabilität der Enzyme beeinflussen, daher wurde deren Einsatz im Bleichansatz vermieden. Statt dessen wurden die Stoffe vor dem enzymatischen Prozess so behandelt, dass eine gute Benetzbarkeit gewährleistet war. Erste Versuche zur enzymatischen Bleichung wurden mit isolierten Enzymen beim jeweiligen pH-Optimum durchgeführt. Zum Einsatz kamen die von der TU Dresden isolierte Peroxidase aus Bacillus sphaericus und die Perhydrolase aus Pseudomonas pyrrocinia sowie die von JenaBios zur Verfügung gestellten Manganperoxidasen aus Nematoloma frowardii und Clitobula sp. b1 und die Laccase aus Trametes versicolor TG1. Weiterhin kamen die kommerziell erhältlichen Peroxidasen aus Arthromyces ramosus und Meerrettich (HRP), Xylanase von Trichoderma viride und Cytochrom c aus Rinderherz und Cellulase (Perizym GBL, Fa. Dr. Petry) zum Einsatz. Es erwies sich als schwierig, mit isolierten Enzymen ein mit der industriellen Wasserstoffperoxidbleiche vergleichbares Ergebnis zu erreichen. Daher wurden anschließend mehrere Enzyme in Kombination eingesetzt. So kann ein Zusammenwirken von Cellulose abbauender Enzymaktivität und bleichender Enzymaktivität weit größere Effekte ergeben, als jedes der Enzyme allein. Mit diesen Kombinationen konnten bei passenden pH-Werten erste günstige Effekte erreicht werden. Gefördert durch die Deutsche Bundesstiftung Umwelt Literaturverzeichnis [1] Milnthorpe, E.J., 1943 Aust. Cent. Wool Committee Testing Ho. [2] Nitschke, G., 1977, Textilpraxis 32: 24-28. [3] Schiecke, H.E.: Wolle als textiler Rohstoff, Schiele & Schön GMBH, Berlin, 1979 [4] Bredereck,K., Gruber,M., Otterbach, A., Schulz,F. 1996, Die Hydrogelstruktur von Cellulosefasern und ihre Bedeutung für Fasereigenschaften und [10] Rydén, L.G., Hunt, L.T. 1993. Evolution of protein complexity: the blue copper-containing oxidases and related proteins. J. Mol. Evol. 36: 41-66. [11] Thurston, C.F. 1994. The structure and function of fungal laccases. Microbiol. 140: 19-26. [12] Call, H.P., Mücke, I. 1995. The laccase-mediator system (LMS) – a new concept. In: Abstr. 6th International Conference on Biotechnology in the Pulp and Paper Industry, Vienna, Austria, p.38. [13] Tien, M., Kirk, T.K. 1984. Lignin-degrading enzyme from Phanerochaete chrysosporium: Purification, characterization, and catalytic properties of a unique H2O2-requiring oxygenase. Proc. Natl. Acad. Sci. USA 81: 2280-2284. [14] Lundell, T., Wever, R., Floris, R., Harvey, P., Hatakka, A., Brunow, G. 1993. Lignin peroxidase L3 from Phlebia radiata. Pre-steady-state and steadystate studies with veratryl alcohol and a non-phenolic lignin model compound 1-(3,4-dimethoxyphenyl)-2(2-methoxyphenoxy)propane-1,3-diol. Eur. J. Biochem. 211: 391-402. [15] Akthar, M., Blanchette, R.A., Kirk, T.K. 1997. Fungal delignification and biomechanical pulping of wood. In: Eriksson, K.-E.L. (ed.) Biotechnology in the pulp and paper industry. Springer-Verlag, Berlin. [16] Vares, T. 1996. Ligninolytic enzymes and lignindegrading activity of taxonomically different white-rot fungi. PhD Thesis, Department of Applied Chemistry and Microbiology, University of Helsinki, Finland. [17] Hofrichter, M., Fritsche, W. 1997. Depolymerization of low-rank coal by extracellular fungal enzyme systems. II. The ligninolytic enzymes of the coalhumic-acid-depolymerizing fungus Nematoloma frowardii b19. Appl. Microbiol. Biotechnol. 47: 419424. [18] Hofrichter, M., Vares, T., Ziegenhagen, D., Friedrich, M., Jäger, M.G., Fritsche, W., Hatakka, A. 1998.Oxidative decomposition of malonic acid as basis for the action of Mn-peroxidase in the absence of hydrogen peroxide. FEBS Lett. 434: 362-366. [19] Apitz, A., van Pée, K. H. (2001). Isolation and charactization of a thermostable intracellular enzyme with peroxidase activity from Bacillus sphaericus. Arch. Microbiol. 175: 405-412 [20] Analytische Bestimmungsmethode: Betriebsinterner Nachweis reduzierender Zucker der Firma Extrakt Chemie GmbH & Co, Stadthagen. Textilveredlung, Textilveredlung 31: 194-200 [5] Stöhr, R., 1995, Enzyme – Biokatalysatoren in der Korrespondenzadresse Textilveredlung. Melliand Textilberichte 11: 1010. [6] Denter, U., Poulakis, K., Schollmeyer, E.,1996, Dr. Elisabeth Heine Peressigsäure in der Textilveredlung. Teil 2: Einsatz von Peressigsäure in der kombinierten Vorbehandlung von cellulosehaltigem Textilgut. Textilveredlung 31: 50-55 [7] Eriksson, K.-E. L., Enzyme application in fiber processing, ACS symposium series, ISSN 0097-6156; 687, San Francisco 1997, 2 - 14 [8] Tanesaka, E., Masuda, H., Kinugawa, K. 1993. Wood degrading ability of basidiomycetes that are wood decomposers, litter decomposers or mycorrhizal symbionts. Mycologia 85: 347-354. [9] Ander, P., Eriksson K-E. 1976. The importance of phenol oxidase activity in lignin degradation by the white-rot fungus Sporotrichum pulverulentum. Arch. Microbiol. 109: 1-8. Deutsches Wollforschungsinstitut an der RWTH Aachen e.V. Veltmanplatz 8 D-52062 Aachen Tel: +49 241 4469 129 Fax: +49 241 4469 100 e-mail: [email protected] URL: http://www.dwi.rwth-aachen.de Biokatalyse 54 S O N D E R A U S G A B E Svenja Weiß1, Gernot T. John1,5, Elmar Heinzle1,7, Eva Schultheiss2, Joachim Jose2, Sarina Arain, Christian Krause, Ingo Klimant6, Harald Waltenberger3, Josef Maier3, Thomas Räbiger4, 1Technische Biochemie, Universität des Saarlandes, 2Pharmazeutische und Medizinische Chemie, Universität des Saarlandes, Institut für Analytische Chemie, Chemo- und Biosensorik, Universität Regensburg, 3Microcoat, Bernried, 4BMG Labtechnologies, Offenburg, 5derzeitige Adresse: Presens GmbH, Regensburg, 6derzeitige Adresse: Institut für Analytische Chemie, Mikro- und Radiochemie, Technische Universität Graz, 7 Korrespondenzadresse Screening von Biokatalysatoren mit Sauerstoff- und pH-Mikrotiterplatten Ziel des vorgestellten Projektes ist die Entwicklung einer neuen Screeningmethode für Esterasen und Oxidasen mittels integrierter Mikrotiterplatten. Das neuentwickelte System soll anhand eines Beispiels auf Tauglichkeit für das HochDurchsatz-Screening geprüft werden. Als Beispiel dient eine Esterase, die durch Autodisplay an der Zelloberfläche exprimiert wird. Bisher wurden 96er Rundbodenplatten mit integrierten Sauerstoff- und pH-Sensoren entwickelt. Die pH-Platten können in einem pH-Bereich von 5 bis 8 eingesetzt werden. Die Routinemessung benötigt keine Kalibrierung jedes Wells. Die bisher entwickelten Sauerstoffplatten zeigen reproduzierbar die aktuelle Sauerstoffkonzentration an. Die technische Herstellung der Mikrotiterplatten wird zur Zeit etabliert. Die pHdyn-Methode zur Bestimmung von Enzymkinetiken ist etabliert und modelliert worden. Die Kinetikkonstanten stimmen gut mit denen mittels konventioneller Tests ermittelten überein. In Hinblick auf Einsatz einer pHstat-Methode wurde das Mischverhalten bei Zugabe kleiner Mengen Natronlauge experimentell untersucht und nach der Entwicklung geeigneter Modelle simuliert. Die Translokation einer Esterase mittels des Autotransportersystems AIDA-I ist durchgeführt worden. Differentielle Zellfraktionierung und Enzymaktivitätsbestimmungen belegen die Oberflächenexpression der Esterase in E. coli. 쑺 Zunehmender Wettbewerb und gleichzeitig steigende Anforderungen bezüglich des Umweltschutzes erfordern die Entwicklung neuer Prozesse und Produkte. Durch den Einsatz von chemischen und biochemischen Katalysatoren wird versucht Produktionsprozesse effizienter und nachhaltiger zu gestalten. Die moderne Molekularbiologie stellt eine Vielzahl von Werkzeugen zur Verfügung um kostengünstige Biokatalysatoren herzustellen (Schäfer und Dalboge, 1999) und ihre Eigenschaften durch evolutive und/oder rationale Variation zu optimieren (Arnold, 1996). Das Ziel dieses Projektes ist die Entwicklung einer neuen Methode zum quantitativen Screening von biokatalytischen Prozessen in Mikrotiterplatten und die Erprobung der Methode anhand evolutiv und rational veränderter Esterasevarianten, die mittels Autodisplay an die Oberfläche von E. coli transloziert werden. Die zu entwikkelnden Platten ermöglichen die kontinuierliche Messung der Sauerstoffkonzentration bzw. des pH-Wertes in jedem Well durch die Verwendung von immobilisierten Fluoreszenzoptoden. Dadurch wird auch die einfache Messung biokatalytischer Prozesse möglich, bei denen die Umsetzung nicht anhand der Produkte oder Edukte bestimmt werden kann. Sauerstoff und Protonen werden bei sehr vielen biokatalytischen Reaktionen umgesetzt und sind ebenso Indikatoren vieler Stoffwechselaktivitäten. Dadurch ergibt sich zukünftig ein breites Anwendungsgebiet für die beschichteten Mikrotiterplatten. Nach der Entwicklung und Optimierung der Sensorcocktails ist die Etablierung der technischen Beschichtung der Mikrotiterplatten geplant. Dafür ist der Auf- D E R D B U bau der dazu nötigen Produktionsstraßen nötig. Für die Mikrotiterplatten mit integrierten Optoden werden im Verlauf des Projektes Screeningmethoden für Enzymkinetiken entwickelt. Der Produktumsatz wird anhand der Änderung der Sauerstoffkonzentration bzw. der pH-Wertänderung (pHdyn-Methode) bestimmt. Als zweite Variante zur Bestimmung der Enzymkinetiken mittels der pH-Platten soll die pHstat-Methode etabliert werden. Dabei wird der pH-Wert konstant gehalten durch die Zugabe von Lauge bzw. Säure. Die Umsatzrate wird anhand des Säure- bzw. Laugeverbrauchs ermittelt. Zur pH-Regelung während der Messung in einem Reader ist der Einbau einer piezoelektrischen Nanoliterpumpe vorgesehen. Kenntnisse über das Mischverhalten bei Zugabe kleiner Volumina in Zusammenhang mit der pHstat-Methode sind interessant, da es durch örtlich hohe Konzentrationen zu reversiblen oder auch irreversiblen Schäden an Enzymen und Zellen kommen könnte. In einem weiteren Teilprojekt wird eine Esterase mit Hilfe des Autodisplay-Systems in aktiver Form an der Oberfläche von E. coli exprimiert und evolutiv an eine vorgegebene, enantioselektive Reaktion angepasst. Der Vorteil der Oberflächenexpression liegt darin, dass man mit ganzen Zellen arbeiten kann, d.h. dass zur Bereitstellung des Enzyms keine weitere Zellaufarbeitung notwendig ist und die Membrangängigkeit des verwendeten Substrates bei der Enzymmessung keine Rolle spielt. Das Screening der für den evolutiven Ansatz erzeugten Zufallsvarianten soll mit Hilfe der pH-sensitiven Mikrotiterplatten erfolgen. Beschichtete Mikrotiterplatten – Entwicklung und Charakterisierung Optoden Die bisher entwickelten Mikrotiterplatten sind handelsübliche 96er Rundbodenplatten aus Polystyren (Greiner, Frickenhausen, Deutschland), die am Boden mit pH- bzw. Sauerstoffsensoren beschichtet sind. Bei den eingesetzten Sensoren handelt es sich um Abb. 1: Mikrotiterplatte mit integriertem optischen pH-Sensor Biokatalyse S O N D E R A U S G A B E D E R sogenannte Optoden. Vermessen werden optische Parameter, wodurch Licht zum Träger der Information wird. Im Rahmen des Projektes wurde die Messung der Fluoreszenz gegenüber der Absorptionsmessung favorisiert, da diese Methode eine hohe Empfindlichkeit aufweist und eine Reihe von Variationen (Messung der Intensität, Abklingzeit, Polarisation) gestattet. Bei der Messung der Fluoreszenzintensität bereiten Schwankungen der Intensität der Lichtquelle und der Empfindlichkeit des Detektors, ungleichmäßige Spots, Lichtstreuung in trüben Proben und eine eventuell auftretende Eigenfluoreszenz der Probe Probleme. Um diese Nachteile zu vermeiden wird in den Platten das ratiometrische Prinzip genutzt und die Sensorschicht optisch isoliert. Für das ratiometrische Prinzip wird eine Mischung aus zwei Fluorophoren eingesetzt, deren Emissionen bei verschiedenen Wellenlängen vermessen werden. Der Indikator ändert die Fluoreszenzintensität in Abhängigkeit vom pH-Wert bzw. in Abhängigkeit von der Änderung der Sauerstoffkonzentration, der Referenzfarbstoff ist inert gegenüber dem jeweiligen Analyten. Durch die Messung des Intensitätsverhältnisses NEU 55 D B U Abb. 2: Modellreaktion der pHdyn-Methode, Hydrolyse von p-Nitrophenylacetat durch Penicillinacylase erhält man relativ stabile und kalibrationsfreie Signale (IR). Über eine Kalibrierung kann man von dem gemessenen Intensitätsverhältnis auf den jeweiligen pH-Wert bzw. die jeweilige Sauerstoffkonzentration schließen. Das Sensorsignal ist von der Temperatur abhängig, insbesondere der pH-Sensor, so dass die Temperatur während der Messung konstant gehalten werden muss. Die zu entwickelnden Optoden müssen verschiedenen Ansprüchen genügen. Gefordert sind ein schnelles Ansprechverhalten, ein dynamischer Messbereich, die Kalibrierfreiheit der Einzelsensoren, hohe Präzision, Sterilisierbarkeit, Stabilität, möglichst einfache und reproduzierbare Herstellung der Platten und die Möglichkeit des Auslesens der Platte mit herkömmlichen handelsüblichen Mikrotiterplattenreadern. Durch die Variation der Sensorzusammensetzung und –eigenschaften (Schichtdicke, usw.) erfolgt die Optimierung von Optoden für Mikrotiterplatten. Die verschiedenen Platten wurden hinsichtlich der Ansprechzeiten, der Reproduzierbarkeit und der Querempfindlichkeiten miteinander verglichen. Die Besonderheiten der verschiedenen bisher entwickelten integrierten Platten werden im Folgenden kurz besprochen. pH-Platten Optische Sensoren zur pH-Messung bieten gegenüber den herkömmlichen Elektroden eine Reihe von Vorzügen. Sie sind einfach zu handhaben und besitzen eine schnellere Ansprechzeit. Bei Lichtleitern gibt es die Möglichkeit viele Informationen gleichzeitig zu übertragen und somit eine Mehrkom- Technische Qualitätssicherung – von der Probenahme bis zum Ergebnis! Karl Cammann (Hrsg.) Instrumentelle analytische Chemie Nach einer neuen Erhebung des angesehenen Londoner „Laboratory of the Government Chemists“ werden – bedingt durch die Zunahme der High-TechProdukte – inzwischen ca. 10% des Bruttosozialproduktes der führenden Industrienationen für das Messen und Prüfen im Rahmen einer Qualitätssicherung ausgegeben! Eine Entwicklung, die auch für die Chemiker-Ausbildung an den deutschen Hochschulen nicht ohne Folgen bleiben wird. Das neuartige Lehrbuch Instrumentelle analytische Chemie stellt sich den neuen Herausforderungen. Der Schwerpunkt dieses Buches liegt daher auf der Qualitätssicherung, der in diesem Buch klar und aktuell wie in keinem anderen Analytik-Lehrbuch eine wichtige Bedeutung zukommt. 2000, 604 S., 300 Abb., geb. DM 159,90 · ISBN 3-8274-0057-0 Zeit sparen - bequem bestellen Telefon 07071-935369 Fax 07071-935393 e-mail: [email protected], http://www.spektrum-verlag.de Prof. Dr. Karl Cammann ist Dozent an der Universität Münster und Leiter des Instituts für Chemo- und Biosensorik in Münster. Biokatalyse 56 Abb. 3: Bestimmung der Kinetik der Hydrolyse von p-Nitrophenylacetat. A: Gemessene pHÄnderung und mit dem Modell berechnete Konzentration von p-Nitrophenylacetat. B: Vergleich der simulierten mit den gemessenen pH-Werten ponentenmessung zu ermöglichen. Weiterhin sind optische Sensoren unempfindlich gegenüber elektromagnetischen Einflüssen und sind gut miniaturisierbar und sterilisierbar. Nachteile sind vor allem der Störeinfluss von hohen und niedrigen Ionenstärken, organischen Lösungsmitteln und Proteinen, sowie der kleine dynamische Bereich von 24 pH-Einheiten im Gegensatz zu der Glaselektrode. In der Literatur beschriebene Fluoreszenz-pH-Indikatoren sind 8-Hydroxypyren3,6,8-trisulfonsäure (HPTS), Acridine, Naphthylderivate, Coumarine und Fluoresceinderivate. Die pH-abhängige Änderung der Intensität beruht auf den unterschiedlichen Fluoreszenzeigenschaften der deprotonierten und der protonierten Form des sensitiven Moleküls. Bei der bisherigen Entwicklung der Platten wurde Carboxyfluorescein verwendet, welches eine gute Abb. 4: Fließmuster bei Zugabe von 0.1N Natronlauge mittels einer piezoelektrischen Nanoliterpumpe S O N D E R A U S G A B E Einbindung des Fluorophors in das Polymer ermöglicht. Als Referenzsubstanzen wurden ein Ruthenium-Komplex bzw. LissaminTM eingesetzt. Beide wurden entweder eingebettet oder kovalent an ein Polymer gekoppelt. Die zur Zeit verwendeten Platten (Abb. 1) sind im Bereich von pH 5-8 einsetzbar. Durch die Verwendung des ratiometrischen Prinzips beträgt die durchschnittliche Abweichung 0.03 pH-Einheiten zwischen den einzelnen Wells. Zur Beschreibung der sigmoidalen Abhängigkeit der Fluoreszenzintensität vom pH-Wert eignet sich die Boltzmann-Näherung. D E R D B U (2) Dabei ist Ir,0 das Intensitätsverhältnis in Abwesenheit von Sauerstoff, IR das Intensitätsverhältnis bei der aktuellen Gelöstsauerstoffkonzentration [O 2]aq und K SV die Stern-Volmer-Konstante des Sensors. Zur Kalibrierung der Sensorplatten führt man eine Zwei-Punkt-Eichung mit sauerstofffreier ([O 2 ]aq=0) und gesättigter Lösung ([O2]aq=[O2]*aq) durch. Technische Herstellung (1) Der pH-Wert pH entspricht dabei dem x-Wert und das gemessene Intenstitätsverhältnis IR (Verhältnis zwischen Indikatorund Referenzintensität) dem y-Wert. A1 und A2 beschreiben die Intensitätswerte bei sehr niedrigen und sehr hohen pH-Werten, x0 ist der pH-Wert am Wendepunkt der Kurve, der ungefähr dem pKs-Wert entspricht. dx ist ein Maß für die Steigung der Kurve am Wendepunkt. Zur Kalibrierung verwendet man Puffer verschiedener pH-Werte in einem pH-Bereich, der die ganze Kurve umfasst. Eine umfassende Kalibrierung mit mindestens fünf Puffern ist nur zur Kalibrierung auf das Gerät nötig, zur weiteren Verwendung der Platte reicht die Kontrolle durch ein bis zwei Pufferlösungen pro Platte aus. Sauerstoffplatten Das Prinzip der Messung mittels der integrierten Mikrotiterplatten ist das der dynamischen Lumineszenzlöschung. Dabei nimmt die Fluoreszenzintensität in Abhängigkeit von der Zunahme der Sauerstoffkonzentration ab. Der im Grundzustand als Triplett vorliegende Sauerstoff wird durch strahlungslosen Energietransfer von dem fluoreszierenden Molekül auf das Sauerstoffmolekül in den energiereicheren Singulett-Zustand überführt. Das sensitive Fluorophor fällt dadurch in den Grundzustand zurück. Verwendet wurden verschiedene Ruthenium-Komplexe und Porphyrinderivat-Komplexe als Sauerstoffsensitive Indikatoren. Als Referenzfluorophor wurden verschiedene Rhodamine und LissaminTM verwendet. Der Nachteil der Optoden gegenüber der Clark-Elektrode, die in der heutigen Analytik vorwiegend eingesetzt wird, ist der nichtlineare Zusammenhang zwischen dem gemessenem Signal und der Sauerstoffkonzentration in der darüberstehenden Lösung. Die Abhängigkeit der Intensität von der Gelöstsauerstoffkonzentration kann jedoch anhand der Stern-Volmer-Gleichung beschrieben werden. Die Mikrotiterplatten wurden durch Aufbringen eines gelösten Cocktails beschichtet. Vorteil dieser Methode ist die Verwendbarkeit von herkömmlichen Mikrotiterplatten. Ein mögliches Problem stellt jedoch die mangelnde Kompatibilität von Platte und Sensormatrix oder des verwendeten Lösungsmittels dar. Der Sensorcocktail, bestehend aus einer Mischung aus je zwei Fluorophoren, der Substanz zur optischen Isolierung und des gelösten Polymer, wird mittels eines Pipettierroboters in jedes einzelne Well pipettiert. Zur Herstellung kleiner Spots müssen die Pipettiernadeln bei der Auftragung auf den Boden der Wells aufsetzen. Da jede Mikrotiterplatte kleine Unebenheiten aufweist, ist eine Federung dieser Nadeln nötig. Weiterhin kommt es, insbesondere bei den Sauerstoffplatten, durch Bestandteile des Sensorcocktails zu Dispensierproblemen. Durch eine Teflonbeschichtung der Pipettiernadel und mehrmaligen Spülen der Nadeln wird das Anhaften von Cocktailbestandteilen vermieden. Eine Sedimentation von Sensorbestandteilen kann durch ständiges Rühren im Vorlagengefäß vermieden werden. Auch während des Trocknungsprozesses kann es zu Sedimentationen von Cocktailbestandteilen kommen. Daher wurde zur Trocknung des applizierten Spots eine Apparatur entwickelt, die eine gleichmäßige und reproduzierbare Beschichtung der Platten gewährleistet. Enzymkinetik: Methoden, Modelle, Simulationen pHdyn-Methode Enzymkinetiken können über die Bestimmung der pH-Änderung in Abhängigkeit von der Produktumsetzung bestimmt werden. Spektroskopische Methoden zur Bestimmung von Enzymumsetzungen mittels löslicher pH-Indikatoren sind bereits in der Literatur beschrieben worden (Janes et al., 1998). Als Modellreaktion wurde die Verseifung von 4-Nitrophenylacetat mittels Penicillina- Biokatalyse S O N D E R A U S G A B E D E R 57 D B U cylase aus E. coli (EC 3.5.1.11) benutzt (Abb. 2). Zur Bestimmung wurden zum einen gelöster zum anderen immobilisierter Indikator benutzt. Bei der Bestimmung mit Hilfe von gelösten Indikator, Bromthymolblau, wird die pH-Änderung mittels Absorptionsmessung bestimmt (John und Heinzle, 2001). Bei der Verwendung der beschichteten Mikrotiterplatten wird die Änderung fluorimetrisch verfolgt. Der pH-Wert entspricht bei beiden Methoden einer Funktion von Umsetzung, Puffertyp und -konzentration. Der Assay wird analog den Küvettenassays mit kleineren Volumina und veränderten Pufferkonzentrationen durchgeführt. Mittels geeigneter Ionenbilanzen werden die aus den Intensitäten ermittelten pH-Werte in Produktkonzentrationen umgerechnet. Die Ergebnisse der Kinetikstudien stimmen gut mit denen konventioneller Assays überein. Zur Simulation der dynamischen Umsetzungen wurden Modelle entwickelt und mit Hilfe von MATLAB (Mathworks Software) durchgeführt. Die verwendete Modellreaktion folgt der MichaelisMenten-Kinetik. (3) Ionenbilanzen werden zur Berechnung des pH-Wertes verwendet. Durch Massenbilanzen und geschätzte Kinetikdaten werden die Konzentrationsverläufe von Substraten und Produkten berechnet. Bei der Modellierung wurde der Einfluss von Kohlendioxid durch Verwendung einer entsprechenden Bilanzgleichung berücksichtigt. Durch Variation von KM, kcat und S0 wird die Abweichung zwischen den experimentellen und den simulierten pH-Werten minimiert (Abb. 3). Die simulierten Kinetikdaten stimmen gut mit mittels klassischer Methoden gemessenen Werten überein (John, 2001). Mischverhalten Eine weitere Möglichkeit die Enzymaktivität zu bestimmen ist die pHstat-Methode. Zur Konstanthaltung des pH-Wertes müssen Lauge oder Säure zugegeben werden. Über die zugegebene Menge an Lauge oder Säure kann die Substratumsetzung ermittelt werden. Der sich nur in engen Grenzen verändernde pH-Wert ist ein Vorteil der pHstat-Methode, da sich die Enzymaktivität in Abhängigkeit vom jeweiligen pH-Wert ändert. Bei konventionellen Enzymassays ist dies nicht von Bedeutung, da dort Pufferkapazitäten eingesetzt werden, die ausreichen um den pH-Wert praktisch konstant zu halten. Zur Regelung des pH-Wertes ist die Zugabe kleiner Volumina nötig. Um die Mischzeiten und das Ansprechverhalten zu bestimmen wurden Vorversuche durchgeführt. Diese zeigten, dass das Mischen länger dauert als zunächst vermutet. Daher wurde das Mischverhalten experimentell untersucht. Visuell wurde das Mischverhalten mittels einer Videokamera beobachtet um das qualitative Fließmuster zu bestimmen und daraus erste Abschätzungen über örtlich hohe Konzentrationen und Mischzeiten zu erhalten. Die Natronlauge wurde hierbei mit Hilfe der piezoelektrischen Nanoliterpumpe zu einer schwach gepufferten Lösung gegeben. Das Muster konnte durch die Verfärbung des zugesetzten Thymolphthalein beobachtet werden. Ein Beispiel eines Fließmusters ist in Abb. 4 zu sehen. Hohe Konzentrationen, angezeigt durch dunkel gefärbte Regionen, konnten nur für wenige Sekunden beobachtet werden. Für den weiteren Verlauf der Vermischung waren leicht gefärbte Schlieren charakteristisch. Zur quantitativen Bestimmung der Mischzeiten wurde die Vermischung fluorimetrisch verfolgt. Diese wurden hierbei als t90 ermittelt und sind definiert als der Zeitpunkt ab dem alle weiteren Messwerte innerhalb des Intervalls 10 % des mittleren Endwertes Biokatalyse 58 S O N D E R A U S G A B E Sauerstofftransfer Abb. 5: Beispiel für die Intensitätskurven bei Zugabe kleiner Mengen Natronlauge und Detektion mittels gelösten und immobilisierten Indikator liegen. Zur Detektion wurde neben der Sensorplatte ein gelöster Indikator, Hydroxypyren-trisulfonsäure (HPTS), verwendet. Die Intensitätsmessung entspricht bei Verwendung von HPTS dem integralen pH-Wert über das ganze Well, bei Verwendung des Sensor entspricht sie dem pH-Wert am Boden des Wells. Oft konnte ein Überschwingen der Intensitätskurve beobachtet werden, insbesondere unter Verwendung des gelösten Indikators. Ein Beispiel für die beobachteten Intensitätskurven ist in Abb. 5 zu sehen. Die ermittelten Mischzeiten t90 hängen von der Zugabeart, dem Zugabevolumen und den Schüttelbedingungen (Frequenz und Dauer pro Zyklus) bzw. der Nachspülhäufigkeit ab und dauern bis zu über 10 min. Beobachtete Trends sind die Abnahme von t90 bei Zunahme der Zyklusschütteldauer bzw. Zunahme der Nachspülhäufigkeit. Die ermittelten Mischzeiten und die beobachteten Fließmuster deuten darauf hin, dass die pH-Regelung in Mikrotiterplatten bei entsprechend langsamen Prozessen möglich ist. Zur Etablierung eines Reglers wurde ein Mischmodell entwickelt. Zur Messung von Enzymkinetiken mittels Sauerstoffdetektion ist die Bestimmung des Sauerstofftransferkoeffizienten kLa wichtig. Zur Bestimmung der kLa-Werte wurde die dynamische Methode benutzt. Variiert wurden hierbei das Füllvolumen, die Schüttelzeit pro Zyklus, die Schüttelfrequenz und die Schüttelbewegung. Somit war es möglich den Massentransportkoeffizienten kL und die spezifische Austauschfläche a getrennt zu bestimmen. Zur Bestimmung der Sauerstoffaufnahmerate wird der Sauerstoff mit Hilfe von Natriumdithionit aus der Lösung entfernt und danach die Zunahme der Sauerstoffkonzentration beobachtet (Abb. 6a). Nach Integration der Sauerstoffbilanzierung kann man einen linearen Zusammenhang zwischen dem Logarithmus der Sauerstoffkonzentration und dem Sauerstoftransferkoeffizienten beobachten: (4) Bei der Auftragung von ln ([O 2]* aq– [O2]aq) gegen die Zeit t entspricht die Steigung dem negativen kLa (Abb. 6b). Bei der Untersuchung der Abhängigkeit des kLa-Wertes vom Füllvolumen konnte beobachtet werden, dass der Sauerstofftransferkoeffizient mit zunehmendem reziproken Volumen linear ansteigt. Die Auftragung des kLa gegen das reziproke Volumen zeigt ein direkt proportionales Verhalten zwischen Stofftransport und reziprokem Volumen, wie aufgrund der theoretischen Überlegungen zu erwarten war. Die Schüttelzeit pro Zyklus wurde variiert um den Sauerstoffeintrag bei ununterbrochenen Schütteln und die für die Kultivierung von Zellen wichtige OTR max (Oxygen Transfer Rate) abzuschätzen. Die ermittelten OTRmax-Werte betragen bis zu Abb. 6: Bestimmung des Sauerstoffeintrages in Mikrotiterplatten, links: Verlauf der Sauerstoffkonzentration nach Zugabe von Natriumthiosulfat, rechtes Bild: Auftragung der integrierten Sauerstoffbilanz: ln([O2] * aq – [O2]aq) gegen die Zeit, hierbei entspricht die Steigung der Geraden dem Sauerstofftransferkoeffizienten kLa D E R D B U ca. 7.5 mmol/l/h. Auch der Maximalwert liegt um ein 10- bis 150-faches unterhalb des in Bioreaktoren üblichen Wertes (Moser, 1988). Die zu erzielende Sauerstoffaufnahmerate ist nur bedingt ausreichend für die Kultivierung von Mikroorganismen. Für tierische Zellen ist der Sauerstoffeintrag ausreichend (Eyer et al., 1995). Autodisplay In diesem Teilprojekt wird mit Hilfe eines neuen, sehr effizienten Surface-DisplaySystems in Escherichia coli („Autodisplay„) eine bereits charakterisierte und kloniert vorliegende Esterase aus Burkholderia gladioli (Schlacher et al., 1998) evolutiv an eine gewünschte, enantioselektive Reaktion angepasst. Der evolutive Ansatz (Abb. 7) besteht darin, dass das Enzym in dem Bereich, der für die Substratspezifität verantwortlich ist, zufallsvariiert wird (Variation) und anschließend aus der Vielzahl an entstandenen Varianten diejenigen identifiziert werden, die eine vorgegebene Reaktion durchzuführen in der Lage sind (Selektion). Der innovative Ansatz besteht darin, diese Variation des Enzyms an der Oberfläche von Bakterienzellen durchzuführen, wobei jede Zelle eine definierte Variante in hoher Kopienzahl trägt. Dadurch ergibt sich der Vorteil, mit der korrekten Enzymvariante deren klonalen Produzenten zu isolieren, was u.a. ermöglicht über das entsprechende Gen Aufschluss über die Struktur zu erhalten. Ein weiterer Vorteil ist, dass man mit ganzen Zellen arbeiten kann. Dadurch entfällt zum einen die Präparation des Enzyms (also ein bis mehrere aufwändige Zwischenschritte) und zum anderen wird man unabhängig von der Membrangängigkeit des eingesetzten artifiziellen Substrates. Darüber hinaus bietet das hier angewendete Autodisplay System die zusätzliche Option, die neu erzeugte Variante anschließend durch Sekretion in sehr reiner Form produzieren zu können (Maurer et al., 1997). Eine besonders elegante Lösung zum Surface-Display und zur Sekretion von Proteinen in Gram-negativen Bakterien bietet die Familie der sogenannten Autotransporter Proteine (Jose et al., 1995). Es handelt sich dabei um sogenannte Ein-Komponenten-Transporter, d.h. diese Proteine werden von der Zelle als Vorläufermoleküle synthetisiert, denen alle Information zur Sekretion mitgegeben sind. Der Vorläufer besteht aus einem N-terminalen Signalpeptid, das beim Transport über die innere Membran abgespalten wird, dem natürlichen Passagier, einer Verbindungsregion („Linker„) und am C-Terminus aus der eigentlichen Autotransporterdomäne. Diese faltet sich als Porin-ähnliche Struktur, als sogenanntes β-Fass in die äußere Membran ein und Biokatalyse S O N D E R A U S G A B E D E R 59 D B U entlässt dadurch den Passagier an die Oberfläche (Abb. 8). Durch Ersatz der kodierenden Region für den natürlichen Passagier mit der für ein anderes Protein, kann dieses rekombinante Protein mit Hilfe des gleichen Mechanismus an die Zelloberfläche gebracht werden (Maurer et al., 1997, Lattemann et al., 2000, Jose et al., 2001). In Abhängigkeit des Protease-Repertoires der Wirtszelle wird dieser rekombinante Passagier dann in den Überstand entlassen (Sekretion) oder bleibt mit der Oberfläche verhaftet, was wir als „Autodisplay“ bezeichnet haben (Jose et al., 1996, Maurer et al., 1997). Besonders effizient – mit einem Anteil von bis zu 5 % des rekombinanten Proteins am Gesamtzellprotein oder mehr als 100.000 Kopien pro Zelle – funktioniert dieses System, wenn man zum Transport eine E. coli-Autotransporterdomäne in E. coliWirtsstämmen verwendet (Maurer et al., 1997, Jose et al., 2001). Darüber hinaus scheint die Größe des rekombinanten Passagiers keinen Einfluss auf die Effizienz der Oberflächenexpression zu haben, was ein Vorteil des „Autodisplays“ gegenüber dem für ähnliche Fragestellungen eingesetzten „Phage-Display“ sein kann. Erklärlich wird dies dadurch, dass es sich bei den natürlichen Passagieren von Autotransporterproteinen in der Regel um große Proteine (> 60 kDa) handelt (Jose et al., 1995). Im vorliegenden Projekt wird die kodierende Region für die Esterase aus B. gladioli über PCR mit denen für das Autodisplay notwendigen Genregionen verknüpft. Anschließend wird der Transport des dadurch kodierten Fusionsproteins an die Zelloberfläche von E. coli verfolgt und untersucht, ob die oberflächenständigen Enzymmoleküle katalytisch aktiv sind und ihre typischen Parameter zeigen. Im Anschluss daran wird über „error-prone“-PCR eine Zufallsvariation der oberflächenständigen Esterase durchgeführt. Die so entstandenen Enzymvarianten werden mit den entwickelten pH-sensitiven Mikrotiterplatten auf die Katalyse einer vorgegebenen Reaktion getestet. Der Vorteil der in unserem Fall eingesetzten Esterase ist zum einen ihre hohe Stabilität und zum anderen das schon von Natur aus breite Substratspektrum. Als Modellreaktionen dienen die Esterhydrolyse von Linalylacetat und die Acetatabspaltung von Cephalosporin C. Bei der Hydrolyse von Cephalosporin C entsteht das Ausgangsprodukt für semisynthetische Antibiotika. Ist diese Strategie zur „directed evolution“ eines Biokatalysators unter Vorgabe einer bestimmten Reaktion wie in dem vorliegenden Beispiel erfolgreich, so bietet sich die Option, sie generell und in vielfältiger Weise für ähnliche Problemlösungen einzusetzen. Ausblick Im weiteren Verlauf des Projektes soll die Produktion der Mikrotiterplatten auf die Beschichtung von Flachbodenplaten und 384er Mikrotiterplatten erweitert werden. Die pH-Regelung innerhalb eines Readers wird etabliert. Die bisher entwickelten Screeningmethoden werden erweitert und optimiert. Weiterhin steht die Optimierung des Autotransportersystems und der Modellesterase an. Literatur Arnold F. H. (1996): Directed Evolution. Creating Biocatalysis for the Future. Chem. Eng. Sci., 51: 5091-5102 Eyer K., Oeggerli A., Heinzle, E. (1995): On-Line Gas Analysis in Animal Cell Cultivation: II. Methods for Oxygen Uptake Rate Estimation and ist Application to Controlled Feeding of Glutamine. Biotechnol. Bioeng., 45(1): 54-62 Janes L.E., Löwendahl C., Kazlauskas R. J. (1998): Quantitative Screening of Hydrolase Libraries Using pH Indicators: Identifying Active and Enantioselective Hydrolases. Chem. Eur. J., 4: 2324-2331 John, G. T. (2001): Mikrotiterplatten mit integrierter pH- und Sauerstoffsensorik: Charakterisierung und biotechnologische Anwendungen. Diss., Universität des Saarlandes John G. T., Heinzle E. (2001): Quantitative Screening Method for Hydrolases in Microtiterplates Using pH Indicators: Determination of Kinetic Parameters by Dynamic pH Monitoring. Biotechnol. Bioeng., 72(6): 620-627 Maurer J., Jose J., Meyer T.F. (1997): Autodisplay: One-component-system for efficient surface diaplay and release of soluble recombinant proteins from Escherichia coli. J. Bacteriol., 179: 794-804 Maurer J., Jose J., Meyer T.F. (1999): Characterization of the Essential Transport Function of the AIDA-I Autotransporter and Evidence Supporting Structural Predictions. J. Bacteriol., 181(22): 7014-7020 Moser A.: Bioprocess Technology – Kinetics and Reactors. Springer-Verlag: New York, Wien, 1988, 1. Auflage Petersen E.I., Valinger G., Stubenrauch G., Schwab H. (2001): A novel esterase from Burkholderia gladioli which shows high deacetylation activity on Abb. 7: Directed Evolution von Enzymen unter Verwendung des Autodisplay Systems cephalosporins is related to β-lactamases and DDpeptidases. J. Biotechnol., 89: 11-25. Schäfer T., Dalboge H. (1999): Perspectives for Industrial Enzymes – The Key to Sustainable Solutions. In: Heiden S., Bock A.-K., Antanikian G. (Hrsg.) Industrielle Nutzung von Biokatalysatoren. Erich Schmidt Verlag, Berlin. Korrespondenzadresse Prof. Dr. E. Heinzle Universität des Saarlandes Technische Biochemie Im Stadtwald, Geb. 2 D-66123 Saarbrücken Tel.: 0681-302 2905 Fax: 0681-302 4572 eMail: [email protected]; http://www.uni-saarland.de/fak8/heinzle/ Abb. 8: Surface Display mit Hilfe des Autotransporter Sekretionsmechanismus Biokatalyse 60 S O N D E R A U S G A B E Tino Polen, Doris Rittmann, Dr. Volker F. Wendisch, Prof. Dr. Hermann Sahm, IBT I, Forschungszentrum Jülich Daniel Weber, Dr. Kerstin Sahm, Prof. Dr. Garabed Antranikian, Technische Mikrobiologie, TU Hamburg-Harburg Prof. Dr. Jörg Müller, Mikrosystemtechnik, TU Hamburg-Harburg Einsatz von DNA-Chips zur Optimierung biotechnologischer Prozesse Das hier beschriebene Projekt im Verbund Biokatalyse besteht aus vier Partnern, die es sich zum Ziel gesetzt haben, den Einsatz von DNA-Chips als unterstützendes Instrument zur Optimierung und Qualitätskontrolle biotechnologischer Prozesse zu etablieren. Als Modell für die Optimierung von Fermentationsprozessen mittels genomweiter Expressionsanalyse wurde der Acetat-Überflussmetabolismus in Escherichia coli ausgewählt, der sich störend auf alle biotechnologischen Prozesse mit E. coli auswirkt. Nach erfolgreicher Herstellung eines GenomChips für E. coli wurde dieser zur Validierung für die Untersuchung der Genexpression bei Phosphatmangel eingesetzt. Es zeigte sich, dass alle als phosphatestarvation-inducible beschriebenen Gene auch ein erhöhtes Signal auf dem Chip aufwiesen. Mit dem validierten Genomchip wurde weiterhin der Einfluss einer erhöhten Acetat-Konzentration auf das Genomexpressionsmuster bestimmt. Darüber hinaus wird eine Oligonukleotid-ChipHybridisierung zur Identifizierung von Bakterien etabliert und eine elektronische Kontrolleinheit zur Stringenzkontrolle der Hybridisierung entwickelt. Die Entwicklung eines kostengünstigen Druckers, der eine on-chip-Oligonukleotidsynthese erlaubt, soll die DNA-Chiptechnologie für einem breiten Kreis von Anwendern attraktiv machen. Einleitung und Fragestellung 쑺 Die DNA-Chip-Technologie steht noch am Anfang ihrer Entwicklung, aber schon heute wird prognostiziert, dass sie als Technik ähnlich revolutionäre Veränderungen bringen wird, wie z.B. die PCR-Technologie. DNA-Chips bieten mit der Parallelisierung, Miniaturisierung und Automation der für die Gentechnologie klassischen Hybridisierungsmethoden (Johnston, 1998) eine Vielzahl von Anwendungsmöglichkeiten. Mit DNA-Chips können in der für eine klassische Hybridisierungsanalyse benötigten Zeit parallel bis zu 10.000 solcher Analysen durchgeführt werden. Insbesondere die in immer kürzerer Zeit bereitgestellten Genomsequenzen schaffen einen hohen Bedarf an weiterführenden Hybridisierungsanalysen. Derzeit sind die Genome von mindestens 51 Mikroorganismen sequenziert (GenBank www.ncbi. nlm.nih.gov/PMGifs/Genomes/micr.html, Stand Mai 2001), darunter das Genom des bakteriellen Modellorganismus Escherichia coli. Die Genom-Sequenzierungsprojekte vervielfältigen das Wissen über die genetische Information von biotechnologisch relevanten Organismen rasant (Watson, 1993), lassen jedoch gleichzeitig einen erheblichen Bedarf an der funktionellen Charakterisierung der Gene entstehen, da aus der Struktur eines Gens in der Regel nicht auf dessen Funktion geschlossen werden kann. Die funktionelle Genom-Analyse ist eine Aufgabe für die Zukunft, zu deren Bewältigung es der Entwicklung und des Einsatzes von Technologien bedarf, die einen hohen Durchsatz und paralleles Arbeiten erlauben, Eigenschaften wie sie insbesondere von der DNA-Chip-Technologie geboten werden (Johnston, 1998). D E R D B U Neben den durch die Parallelisierung und Miniaturisierung bedingten rein quantitativen Vorteilen eröffnen die DNA-ChipAnalysen auch qualitativ eine neue Ebene bei der funktionellen Charakterisierung von Genen. Die Expression der Gene eines Organismus hängt auch von den Umwelteinflüssen, bei Mikroorganismen z. B. vom Nährstoffangebot, pH oder der Temperatur, ab. Während sich klassische Genexpressionsstudien modellhaft auf ein oder wenige Gene konzentriert haben, ermöglicht der Einsatz von DNA-Chips die Gewinnung von Genexpressionsdaten für das gesamte zelluläre Genom. Durch die ganzheitliche Analyse der Genexpression eines Organismus ergibt sich ein neues, im reduktionistischen Ansatz nicht erfassbares Verständnis der Regulation der Genexpression. Für biotechnologische Prozesse, in denen Zellen als Biokatalysatoren wirksam sind, vertieft der Einsatz der neuen DNA-Chip-Technik das Wissen über zelluläre Zusammenhänge und bietet damit ein großes Potenzial zur Optimierung dieser biotechnologischen Prozesse. Auch für andere Anwendungsbereiche bieten DNA-Chips entscheidende Vorteile. So wurden sie für Sequenzierungen, die Genotypisierung von Organismen und Genexpressionsversuche im F&E-Bereich sowie für ausgewählte Anwendungen im Bereich der medizinischen Diagnostik bzw. für das Screening nach neuen pharmazeutischen Wirkstoffen, für on-chip-PCR und zur Untersuchung von Biodiversität genutzt (Drmanac et al., 1998; Vahey et al., 1999; Holstege et al., 1998, Brandstetter et al., 2001, Guschin et al., 1997). Exemplarisch konnte damit die Eignung der DNA-Chips für verschiedenste Einsatzfelder gezeigt werden. Das große Potenzial dieses Hochdurchsatz-Screening-Instruments liegt jedoch besonders in der Routineanwendung. So bleibt es eine Herausforderung für die Zukunft, den DNA-Chip als Routineinstrument in biotechnologische Prozesse zu integrieren. Es gibt zwei verschiedene Arten von DNA-Chips, die Oligonukleotid-Chips und die DNA-Fragment-Chips. Beiden gemein ist die mikroskopische, feldförmige Anordnung von DNA-Molekülen auf inerten Oberflächen (z.B. Glas oder Plastik). Jedes DNA-Molekül leitet sich von einem Gen ab. Sind die auf dem Chip immobilisierten DNA-Moleküle kurz (15 bis 60 Nukleotide) und einzelsträngig, so spricht man von einem Oligonukleotid-Chip, wohingegen Chips mit langen, doppelsträngigen DNAMolekülen (100 bis 4.000 Basenpaare) als DNA-Fragment-Chips bezeichnet werden. Die Herstellung von Oligonukleotid-Chips kann über zwei Verfahren erfolgen, den Druck bereits synthetisierter Oligonukleotide auf die vorbereitete Glasoberfläche oder durch in situ -Synthese direkt auf dem Chip. Biokatalyse S O N D E R A U S G A B E D E R D B U Die in situ -Synthese der Oligonukleotide basiert im wesentlichen auf einer Kombination von Festphasen-Chemie, photolabilen Schutzgruppen und photolithographischen Masken (Fodor et al., 1991). Die Herstellung der photolithographischen Masken ist sehr kostenintensiv und dem Vorteil der Replizierbarkeit steht das Fehlen der Flexibilität bei der Umgestaltung von Oligochips gegenüber. Es ergibt sich für die Herstellung von Oligonukleotid-Chips der Bedarf an flexibleren und kostengünstigeren Herstellungsverfahren, gerade um Anwendungsbereiche außerhalb der Forschung und Entwicklung zu erschließen. Die Herstellung von DNA-Fragment-Chips umfasst keine in situSynthese, die ungefähr 100 bis 4.000 Basenpaare langen DNA-Moleküle werden zuerst synthetisiert (in der Regel über PCR) und dann auf die Trägeroberfläche transferiert und immobilisiert. Die Trennung des Synthese- vom Immobilisierungsschritt ermöglicht eine flexible Gestaltung der DNA-Fragment-Chips. Allerdings ist die Replizierbarkeit des Verfahrens begrenzt, die Herstellung erfolgt im Batch-Verfahren mit Stückzahlen von maximal 500 bis 1000. DNAFragment-Chips erlauben eine einfache und spezifische Hybridisierung, zu deren Optimierung im Vergleich zu Oligonukleotid-Chips nur wenig Aufwand betrieben werden muss. Allerdings limitiert die Kreuzhybridisierung sehr ähnlicher DNA-Sequenzen die Anwendung in der Genotypisierung z.B. von pathogenen Mikroorganismen (z.B. unterscheiden sich einige Antibiotika-resistente und -sensitive Stämme des Tuberkulose-Erregers Mycobacterium tuberculosis nur durch einzelne Basenpaaraustausche eines Gens; Piatek et al., 1998). Zur funktionellen Analyse eines komplexen Systems wie dem Genom einer mikrobiellen Zelle eignet sich der DNA-FragmentChip aufgrund von Spezifität und Robustheit sehr gut. Das hier vorgestellte Projekt wird von vier Partnern bearbeitet. Es widmet sich dem Einsatz von DNA-Chips zur Optimierung biotechnologischer Prozesse hinsichtlich ihrer Raum/Zeit-Ausbeute. Es werden einerseits DNA-Fragment-Chips für die genomweite Expressionsanalyse biotechnologisch interessanter Mikroorganismen insbesondere E. coli mit dem Ziel der Optimierung dieser Mikroorganismen für die Produktion eingesetzt (Forschungszentrum Jülich) und andererseits Oligonukleotid-Chips für die mikrobiologische Reinheitskontrolle in biotechnologischen Prozessen etabliert (Technische Mikrobiologie, TU Hamburg-Harburg). Des weiteren soll ein neuartiges Drucksystem entwickelt werden, um eine kostengünstige in situ-Synthese von Oligonukleotid-Chips zu ermöglichen (Mikrosystemtechnik, TU Hamburg-Harburg). Der Projektpartner SLS µ-Technologies wird die Industrialisierung des neuentwickelten Druckers übernehmen. 61 Abb. 1: Vorgehen bei der genomweiten Expressionsanalyse mit DNA-Chips. Im Beispiel wird RNA aus dem C. glutamicum-Wildstamm isoliert und in einer Reversen Transkriptionsreaktion mit Zufallsprimern und grünfluoreszenten Nukleotidanaloga markiert. Die RNA aus einem C. glutamicum Lysin-Produktionsstamm wird entsprechend rot-fluoreszierend markiert. Nach der Hybridisierung mit dem C. glutamicum-Genom-DNA-Chip zeigen rote Hybridisierungssignale erhöhte RNA-Spiegel im Lysinproduktionsstamm im Vergleich zum Wildstamm für dieses Gen an. Entsprechend zeigen in diesem Vergleich grüne Signale verringerte und gelbe Signale unveränderte RNA-Spiegel an. sitzt. Für ein Drittel davon lässt sich kein Funktionsvorschlag machen (Blattner et al.,1997). DNA-Chip-Analysen erlauben nun den Expressionsstatus all dieser Gene parallel zu bestimmen. Die Arbeitsgruppe von Prof Dr. F. Blattner hat Primer-Paare zur PCR-Amplifikation aller Gene von E. coli entworfen (kommerziell erhältlich bei Sigma-Genosys). In der Jülicher Arbeitsgruppe wurden die Gene des E. coli-Genoms mittels PCR amplifiziert. Für ca. 96% der Gene wurden in der sich anschließenden Gelelektrophorese Identität, Reinheitsgrad und eine für die DNA-Chip-Herstellung ausreichende Menge Genomweite Expressionsanalysen im Modellorganismus Escherichia coli Escherichia coli ist nicht nur ein bakterieller Modellorganismus, sondern wird auch in biotechnologischen Prozessen zur Produktion von z.B. Aminosäuren eingesetzt. Die aerobe, unter Überschuss von Kohlenstoff- und Energiequelle stattfindende Aminosäureproduktion kann als Überflussmetabolismus bezeichnet werden. Die DNAChip-Technik bietet die Möglichkeit, erstmals den Expressionsstatus aller Gene eines Organismus gleichzeitig zu bestimmen (DeRisi et al., 1997; Wodicka et al., 1997). Ein physiologischer Zustand, z.B. der Überflussmetabolismus, kann damit auch detailliert auf Ebene der Expression beschrieben werden. Solche genomweiten Expressionsanalysen (s. Abb. 1) gewähren neue Einblicke in zelluläre Regulationsvorgänge sowie neue Ansätze zur Optimierung biotechnologischer Prozesse. DNA-Chips für genomweite Expressionsanalysen in E. coli Die Sequenzierung und Annotation des Genoms von E. coli hat ergeben, dass dieser Organismus 4-290 Protein-kodierende Gene be- Abb. 2: Scatter-Plot-Darstellung der Hybridisierungssignale eines Vergleiches genomischer DNA (a) und einer genomweiten Expressionsanalyse (b). Genomische DNA aus den E. coli-Stämmen MG1655 und LJ110 wurden rotbzw. grünfluoreszierend markiert und mit dem E. coli-Genom-DNA-Chip hybridisiert (a). Für jedes Gen ist die normierte Rot- und Grün-Fluoreszenz dargestellt (schwarze Punkte). Gene mit gleichen Signalintensitäten finden sich auf der Winkelhalbierenden. Die schwarzen Linien zeigen zweifach erhöhte bzw. zweifach verringerte Signalintensitäten an. In einer genomweiten Expressionsanalyse wurde RNA aus E. coli MG1655 bei Anzucht unter Phosphatmangel isoliert und rotfluoreszierend markiert bzw. bei ausreichender Phosphatversorgung isoliert und grünfluoreszierend markiert und anschließend mit dem E. coli-Genom-DNA-Chip hybridisiert. Die Hybridisierungssignale (schwarze Rauten) von Genen, deren Expression sich nicht verändert hat, liegen auf der Winkelhalbierenden. Bei mehr als zweifach erhöhtem RNA-Spiegel finden sie sich im rotmarkierten Segment und bei mehr als zweifach verringertem RNA-Spiegel im grünmarkierten Segment. Biokatalyse 62 festgestellt. Nach anschließender Konzentration durch Ethanolpräzipitation wurden die PCR-Produkte mit dem Jülicher DNA-ChipRoboter in feldförmiger Anordnung auf modifizierte Glasoberflächen übertragen und immobilisiert. Dabei wurde nach dem an der Stanford University entwickelten Verfahren vorgegangen (Schena et al., 1995). Zur Verifizierung der E. coli-Genom-Chips wurde genomische DNA der nah verwandten Stämme MG1655 und LJ110 rot- bzw. grünfluoreszent markiert und mit dem E. coli-Genom-Chip hybridisiert (Abb. 2a). In dieser Analyse genomischer DNA kann für 96 bis 98% der Gene auf dem DNA-Chip ein Hybridisierungssignal bestimmt werden (Signal-Rausch-Verhältnis größer oder gleich 3). Für einige Gene wurde ein deutlich erhöhtes Hybridisierungs- S O N D E R A U S G A B E durchgeführt werden. Zur Validierung der genomweiten Expressionsanalyse mit E. coliGenom-DNA-Chips wurden die Expressionsmuster von E. coli MG1655-Zellen bestimmt, die entweder unter ausreichender Phosphatversorgung oder unter Phosphatmangelbedingungen kultiviert wurden (Abb. 2b). In dieser Analyse konnte für 74% der Gene ein Hybridisierungssignal ermittelt werden, diese Gene werden unter den oben genannten Bedingungen exprimiert. Für 36% der Gene lag das Signal mehr als dreifach über dem Hintergrund und für diese konnte ein Expressionsverhältnis quantifiziert werden. Bei Phosphatmangel wiesen im Vergleich zum Wachstum bei ausreichendem Phosphatangebot ca. 5% der Gene erhöhte und 6% der Gene verringerte RNA-Spiegel auf (s. Abb 2b). Gene bzw. Operons, die für E. coli als phosphatestarvation-inducible (psi) beschrieben wurden (Metcalf et al., 1990), zeigten in dieser Analyse ausnahmslos erhöhte RNA-Spiegel bei Phosphatmangelbedingungen. Dies wird in der Genome-Map-Image-Analyse der Expressionsdaten besonders deutlich (Abb. 3). Untersuchung differentieller Genexpression in Anwesenheit des Überflussmetaboliten Acetat Abb. 3: Ausschnitte der Genome-Map-ImageDarstellung einer DNA-Chip-Analyse. Die Hybridisierungssignale einer genomweiten Expressionsanalyse von E. coli (Rot-Fluoreszenz bei Phosphat-limitierenden Bedingungen und Grün-Fluoreszenz bei ausreichender Phosphatversorgung) wurden anhand der Anordnung der Gene im Genom rearrangiert (Gene, die nicht auf dem DNA-Chip vorhanden sind, sind grau dargestellt). Die Ausschnitte zeigen die Positionen auf dem E. coli-Chromosom, die Transkriptionsrichtung und die Hybridisierungssignale von Operons, die bei Phosphatmangel verstärkt exprimiert werden, und als Kontrolle die des nicht phosphat-abhängig regulierten rpsLG-fusAtufA Operons. signal im Stamm MG1655 bestimmt (Abb. 2a). Diese Gene sind wahrscheinlich im Stamm LJ110 deletiert oder im Stamm MG1655 amplifiziert (Ochman & Jones, 2000, haben beschrieben, dass 82 Gene im Stamm MG1655, nicht aber im Stamm W3110 vorkommen.). Genomweite Expressionsanalysen mit DNA-Fragment-Chips bzw. DNA microarrays sind für E. coli etabliert worden (Khodursky et al., 2000; Zimmer et al., 2000; Wendisch et al., 2001) und können auch mit Nylonmembranen (Richmond et al., 1999) oder Oligonukleotid-Chips (Selinger et al., 2001) In aerob wachsenden E. coli-Kulturen kommt es bei ausreichendem Angebot bezüglich der Kohlenstoff- und Energie-Quelle häufig zur Ausscheidung von Intermediaten des Zentralstoffwechsels, vor allem von Acetat. Dieser Acetat-Überflussmetabolismus wirkt sich störend auf alle biotechnologischen Prozesse mit E. coli aus, bei denen einerseits niedermolekulare Verbindungen wie die Aminosäure Threonin oder andererseits Proteine produziert werden. Der Bedeutung des Überflussmetabolismus in der Biotechnologie steht dessen noch unvollständiges Verständnis gegenüber. Zwar kann Überflussmetabolismus häufig stoffwechselphysiologisch erklärt werden, z.B. kann die Acetat-Produktion von E. coli durch eine Limitation der Tricarbonsäurecyclus-Aktivität erklärt werden. Jedoch bleibt im wesentlichen unklar, welche Regulationsmechanismen unter diesen Bedingungen aktiv bzw. außer Kraft gesetzt sind (Tempest & Neijssel, 1992). Die genomweite Charakterisierung von Veränderungen der Genexpression bei verschiedenen Bedingungen des Überflussmetabolismus bietet die Möglichkeit, solche Regulationsmechanismen zu identifizieren, die generell an der Regulation des Überflussmetabolismus beteiligt sind, sowie solche, die spezifisch für eine Form des Überflussmetabolismus (z.B. Acetat-Produktion bei Glucose-Überschuss und Magnesium-Mangel) sind. Überflussmetabolismus-Bedingungen sollen experimentell gezielt ausgelöst wer- D E R D B U Abb. 4: Mismatch Diskriminierung auf einem Oligonukleotid-Chip. Hybridisierung eines Oligonukleotid-Chips (Oligonukleotide jeweils 16fach repliziert) spezifisch für Bacteria mit markierter cDNA von E. coli. Durch Optimierung der Waschbedingungen konnten Bindungen mit einem Mismatch auf ein Minimum reduziert werden. 1: Sonde EUB338, 2: EUB338 mit einem zentralen Basenaustausch von G zu T, 3: EUB338 mit einem zentralen Basenaustausch von G zu C, 4: EUB338 mit einem zentralen Basenaustausch von G zu A den. Der Wildstamm von E. coli wird in einem definierten Mineralsalzmedium mit Temperatur- und pH-Statisierung kontinuierlich fermentiert. Als Referenzbedingung soll die Glucose-limitierte Fermentation dienen. Durch Sulfat-, Stickstoff-, Phosphat-, Magnesium- oder Kalium-Limitierung bei ausreichendem Glucose-Angebot sollen Überflussmetabolismus-Bedingungen ausgelöst werden. Fermentationsbegleitend werden Parameter wie Substratverbrauch, Biomassebildung, Produkt- und Nebenproduktbildung analytisch bestimmt (u.a. mittels NMR). Aus den Kulturen wird RNA isoliert und DNA-Chip-Analysen werden durchgeführt. Bei der Auswertung der genomweiten Expressionsdaten werden Genome-Map-Image- (s. Abb. 3) und Cluster-Verfahren benutzt. Die Analyse der stromaufwärts der Transkriptionsinitiationssignale liegenden Sequenzen soll die Identifizierung der cis-Elemente ermöglichen, die für eine gemeinsame Regulation maßgeblich sind. In geeigneten Transkriptionsfusionsexperimenten soll dann bestimmt werden, ob die identifizierten cis-Elemente für eine durch Überflussmetabolismus regulierte Expression hinreichend sind. Bei den Genexpressionsanalysen zum Überflussmetabolismus gilt es zwischen kausal für Überflussmetabolismus verantwortlichen Effekten im Gegensatz zu sekundären oder Folge-Effekten zu differenzieren. Es ist z.B. zu erwarten, dass allein die Anwesenheit des Überflussmetaboliten Acetat zu Veränderungen der Genexpression führt. Aus diesem Grunde wurde E. coli MG1655 auf LB-Komplexmedium mit bzw. ohne 20 mM Natriumacetat, pH 7, kultiviert, wobei auf eine Adaptation von mindestens 20 Generationen geachtet wurde. Außerdem unterschied sich die Wachstumsrate unter diesen Bedingungen nicht wesentlich. In genomweiten Expressionsanalysen konnte gezeigt werden, dass eine Reihe von Genen in Anwesenheit von Acetat erhöhte RNA-Spie- Biokatalyse S O N D E R A U S G A B E D E R 63 D B U gel aufwiesen. Dabei fiel auf, dass besonders Gene des Flagellenapparates betroffen waren (Polen et al., Manuskript in Vorbereitung). Darauf aufbauend wurden Expressionsanalysen in Ab- bzw. Anwesenheit von Natriumpropionat und nach Zugabe von Carbonyl-Cyanid-3-Chlorophenylhydrazon (CCCP) durchgeführt. Die Anwesenheit von Propionat führte im wesentlichen zu kongruenten Expressionsveränderungen wie die Anwesenheit von Acetat. Die Zugabe von CCCP, einem Entkoppler des transmembranen pH-Gradienten, beeinflusste die Genexpression kaum. Die Genexpressionsveränderungen durch Acetat oder Propionat, beides Salze schwacher Säuren, scheinen also nicht auf ihre entkoppelnde Wirkung zurückzuführen sein, da die Zugabe des Entkopplers CCCP nicht zu diesen Genexpressionsveränderungen führt (Polen et al., Manuskript in Vorbereitung). Etablierung von DNA-Oligonukleotid-Chips für die mikrobiologische Qualitätskontrolle Molekulare Hybridisierungsverfahren haben auch in der Identifizierung von Bakterien ihren festen Platz. Die Hybridisierung von DNA oder RNA, die direkt aus Umweltproben isoliert wird, mit Oligonukleotiden, die gegen diagnostische Bereiche der 16SrRNA gerichtet sind (Oligonukleotidsonden), erlaubt es heute die Zusammensetzung prokaryontischer Mischkulturen ohne vorherige Kultivierung zu bestimmen. In der biotechnologischen Produktion von Proteinen und niedermolekularen Wirkstoffen spielen mikrobiologische Reinkulturen eine entscheidende Rolle, während in der Lebensmittelproduktion häufig kontrollierte Mischkulturen von großer Bedeutung sind. Pharmazeutische Produkte und Lebensmittel werde erst dann zum Verkauf freigegeben, wenn ihre mikrobiologische Unbedenklichkeit bestätigt wurde, solange dies nicht der Fall ist, ist ein häufig großes Kapitalvolumen gebunden. Die effiziente und schnelle Überwachung der mikrobiologischen Reinheit von Produktionseinheit und Produkten ist daher nicht nur ein wesentlicher Bestandteil der Qualitätssicherung dieser Prozesse sondern auch von großer finanzieller Bedeutung für den Produzenten. Etablierte Methoden der mikrobiologischen Überwachung beruhen derzeit vorwiegend auf Kultivierung kombiniert mit physiologischen und biochemischen Tests. Die Dauer dieser Tests bewegt sich zwischen 48 h und 7 Tagen. Neben dem hohen Zeitaufwand haben diese Methoden den Nachteil, dass ein Teil der Mikroorganismen im Labor nicht kultivierbar ist und daher unerkannt bleibt. Häufig werden daher Kontaminationen, die einen Prozess stören und die mit hohem Zeit-, Energie- und Kostenaufwand hergestellten Produkte unbrauchbar machen können zu spät oder nicht erkannt. Ziel des Teilprojektes der Technischen Mikrobiologie ist die Etablierung einer Chip Hybridisierung, die es erlaubt, komplexe prokaryontische Mischkulturen, wie sie in biotechnologischen Prozessen vorkommen können, hinsichtlich ihrer Zusammensetzung zügig und umfassend ohne vorherige Kultivierung zu analysieren. Um entscheidende Vorteile gegenüber den klassischen Verfahren zu erzielen wird eine hohe Sensitivität angestrebt und eine Nachweisdauer von unter zwölf Stunden. Die Identifizierung der Bakterien erfolgt auf der Basis der Sequenz ihrer 16S ribosmalen RNA. Oligonukleotide, die gegen spezifische Bereiche der kodierenden Gene gerichtet sind (Sonden), werden auf den Chip gedruckt, und es erfolgt eine Hybridisierung mit fluoreszenzmarkierter RNA bzw. cDNA. Das Vorhandensein des spezifizierten Bakteriums wird anhand der nach der Hybridisierung auf dem Chip gebundenen Fluoreszenz gemessen. Entscheidend für einen Erfolg der Hybridisierung ist, dass nur Ziel-DNA binden kann, die zu 100% komplementär zur Sequenz des Oligonukleotids ist, und damit der gesuchten Art entspricht. Ziel DNA, die eine oder mehrere Basen Fehlpaarung aufweist, soll von der Bindung weitestgehend ausgeschlossen werden. Erreicht werden kann dieses durch Veränderung der Hybridisierungsstringenz, z.B. durch Veränderung der Salzkonzentration bzw. der Zugabe denaturierender Agenzien im Waschpuffer oder durch Veränderung der Waschtemperatur. Die benötigte Stringenz ist jedoch abhängig von der Sequenz des Oligonukleotids. Da es das Ziel ist, möglichst viele verschiedene Bakterien mit einem Chip zu identifizieren, wird auch bei sorgfältiger Auswahl der spezifischen Oligonukleotide nicht erreicht werden können, dass alle Sonden dieselben Stringenzbedingungen benötigen. Es werden daher unter- Abb. 5: Aufbau des DNA-Drucksystems schiedliche Verfahren getestet mit denen auf einem Chip verschiedene Stringenzbedingungen eingestellt werden können. Möglich ist eine zeitabhängige Stringenzveränderung gekoppelt mit der online-Detektion der Fluoreszenzsignale oder eine gleichzeitige lokale Stringenzveränderung mit anschließender Detektion. Dazu wird in Zusammenarbeit mit dem Projektpartner aus der Mikrosystemtechnik ein System entwickelt, das es erlaubt auf einem Chip gleichzeitig verschiedene Waschtemperaturen zu realisieren und somit die für jede Sonde erforderliche Temperatur einzustellen. Die hohe geforderte Sensivität macht den Einsatz der reversen Transkriptase PCR bzw. der PCR erforderlich. Hier bieten sich je nach Fragestellung auch spezifische PCRAmplifikationen an, wenn es um den Nachweis besonderer Indikatorstämme geht. Das System, ebenso wie die Auswahl der Sonden wird auf den jeweiligen Anwender abgestimmt. Da geringe Nachweiszeiten und eine möglichst einfache Durchführung des Nachweises entscheidende Faktoren für einen Einsatz des hier entwickelten Instruments sind, wird der Optimierung dieser Parameter besonderes Augenmerk geschenkt. Untersucht wird derzeit, ob es möglich ist, durch entsprechende Veränderungen der auf dem Chip vorgelegten Oligonukleotide eine vorherige Fluoreszenzmarkierung der Probe unnötig zu machen. DNA-Drucksystem Die in-situ-Synthese von DNA-Oligonukleotiden erfordert gegenwärtig noch einen relativ hohen zeitlichen und apparativen Aufwand, insbesondere wenn unterschiedliche Oligonukleotide parallel auf einem Chip hergestellt werden sollen. Um diesen Aufwand deutlich zu reduzieren und gleichzeitig ein System zu erzeugen, das sich ohne die Notwendigkeit, jeweils grundsätzliche Neuentwicklungen durchzuführen, auf im- Biokatalyse 64 S O N D E R A U S G A B E Abb. 7: PC-Steuerelektronik D E R D B U aufgebaut (Abb. 8), wobei sowohl Druckkopf wie Chip gegeneinander bewegt werden können und so auch größere Arraydimensionen als von der Spaltenlänge des Kopfes erfasst bzw. mehrere DNA-Chips nacheinander bedruckt werden können. Literatur mer größere Arrayfelder und kleinere Volumina übertragen lässt, wird in diesem Teilvorhaben in Mikrosystemtechnik ein „Druckkopf“ zu örtlich und zeitlich beliebiger Synthese frei wählbarer Sequenzen für die vier Komponenten der DNA entwickelt. Abb. 6: Reinigungskopf In der Technologie der Silizium-Glas-Mikromechanik werden in vier Reservoirs, die spaltenförmig nebeneinander im Druckkopf angeordnet sind und die jeweils für jede Zeile eine getrennt ansteuerbare Austrittsöffnung enthalten, die Komponenten in Kontakt zu dem lokal hydrophilen Trägersubstrat gebracht (Abb. 5). Unter Nutzung einer Kombination von hydrophoben und hydrophilen Oberflächenbereichen im Druckkopf und dem zu bedruckenden Chip, in einem weiteren Entwicklungsschritt zusätzlich durch Applikation elektrische Felder, wird der Chip lokal kurzzeitig mit dem Medium benetzt. Alternativ kann auch ein Tropfen abgelegt werden. Dazu wird eine dünne Glasmembran mit Hilfe eines pixelweise ansteuerbaren Arrays aus Piezokeramik über dem Druckreservoir oberhalb der Druckdüse ausgelenkt. Der DNA-Chip selbst kann z.B. aus einem hydrophilen Glassubstrat mit einer hydrophoben, z.B. teflonartigen Lochmatrix versehen, bestehen, oder aus einem hydrophoben Polymer, das mit einem hydrophilen Punktarray versehen wird. Zur Realisierung der Chips können je nach erforderlicher Strukturauflösung Druck- oder Dünnschichtund Lithographieprozesse benutzt werden. Je nach der erforderlichen Wechselwirkungszeit für die Bindung wird das Medium unmittelbar wieder im Druckkopf aufgenommen oder verbleibt auf dem Chip, um mit einem zweiten Kopf (Abb. 6) wieder entfernt zu werden. Dieser zweite Druckkopf wird auch für die Aktivierung der nächsten Bindung sowie die Trocknung des Chips genutzt. Der Druckkopf ist eingebettet in ein Rechner-gesteuertes System (Abb. 7), das sowohl die einzelnen Druckfelder der Köpfe aktiviert als auch die genaue Positionierung des Chips und seines Transports steuert. Transport wie Druckvorgang werden durch eine in das System integrierte und vom Rechner ausgelesene Kamera überwacht. Durch Integration einer Heizung und eines ebenfalls Rechner-gesteuerten zeitlich und örtlich variierbaren Temperaturprofils kann die Reaktionszeit zusätzlich beeinflusst werden, so dass ein vollständig automatisiertes Drucksystem für DNA-Sequenzen zunächst bis zu einer Komplexität von 30x30 Feldern und bis zu 20 Gliedern verfügbar wird. Das Gesamtsystem wird letztlich ähnlich wie ein Tintenstrahldrucker Blattner, F. R.; Bloch, C. A.; Perna, N. T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J. D.; Rode, C. K.; Mayhew, G. F.; Gregor, J.; Davis, N. W.; Kirkpatrick, H. A.; Goeden, M. A.; Rose, D. J.; Mau, B.; Shao, Y. (1997): The complete genome sequence of Escherichia coli K-12. Science 277: 1453-1474 Brandstetter, T., Zeltz, P., Pfeifer, D., Toder R. 2001. HLA-Chips – die HLA-Diagnostik der Zukunft. Bioforum 24: 212-213. DeRisi, J. L.; Iyer, V. R.; Brown, P. O. (1997): Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278: 680686 Khodursky, A.; Peter, B.; Cozzarelli, N.; Botstein, D., Brown, P. O.; Yanofsky, C. (2000): DNA microarray analysis of gene expression in response to physiological and genetic changes that affect tryptophan metabolism in Escherichia coli. Proc. Nat. Acad. Sc., USA 97: 12170-12175 Metcalf, W. W.; Steed, P. M.; Wanner, B. L. (1990): Identification of phosphate-starvation-inducible genes in Escherichia coli K-12 by DNA sequence analysis of psi::lacZ(Mud d1) transcriptional fusions. J. Bacteriol.172: 3191-3200 Ochman, H.; Jones, I. B. (2000) Evolutionary dynamics of full genome content in Escherichia coli. EMBO J. 2000 19: 6637-6643 Richmond, C. S.; Glasner, J. D.; Mau, R.; Hongfan, J.; Blattner, F. R. (1999): Genome-wide expression profiling in Escherichia coli K-12. Nucl. Acids Res. 27: 3821-3835 Schena, M.; Shalon, D.; Davis, R. W.; Brown, P. O. (1995): Quantitative monitoring of gene expression patterns with a complementary cDNA microarray. Science 270: 467-470 Selinger, D. W.; Cheung, K. J.; Mei, R.; Johansson, E. M.; Richmond, C. S.; Blattner, F. R.; Lockhart, D. J.; Church, G. M. (2000): RNA expression analysis using a 30 base pair resolution Escherichia coli genome array. Nature Biotechnol. 18: 1262-1268 Tempest, D. W.; Neijssel, O. M. (1992): Physiological and energetic aspects of bacterial metabolite overproduction. FEMS Microbiol. Lett. 100:169-176 Wendisch, V. F.; Zimmer, D. P.; Khodursky, A. B.; Peter, B. J.; Cozzarelli, N.; Kustu, S. (2001): Isolation of Escherichia coli mRNA and comparison of expression using mRNA and total RNA on DNA. Anal. Biochem. 290: 205-213 Wodicka, L.; Dong, H.; Mittmann, M.; Ho, M.; Lockhart, D. J. (1997): Genome-wide expression monitoring in Saccharomyces cerevisiae. Nature Biotechnol. 15: 1359-1367 Zimmer, D. P.; Soupene, E.; Lee, H. L.; Wendisch, V. F.; Khodursky, A. B.; Peter, B. J.; Bender, R. A.; Kustu, S. (2000): Nitrogen regulatory protein Ccontrolled genes of Escherichia coli: Scavenging as a defense against nitrogen limitation. Proc. Nat. Acad. Sc., USA 97: 14674-14679 Korrespondenzadresse: Dr. Volker F. Wendisch Abb. 8: Prinzip des DNA-Drucksystems Institut für Biotechnologie 1 Forschungszentrum Jülich, D-52425 Jülich Tel.: 02461-615169 Fax: 02461-612710 [email protected] http://www.fz-juelich.de/ibt/genomics/ genomics.html Biokatalyse S O N D E R A U S G A B E D E R 65 D B U Dr. Petra Peters-Wendisch, Dr. Lothar Eggeling, Dipl.-Biol. Roman Netzer und Prof. Dr. Hermann Sahm, Institut für Biotechnologie 1, Forschungszentrum Jülich Dipl.-Ök. Henning Serger und Prof. Dr. Udo Müller, Institut für Volkswirtschaftslehre, Universität Hannover, Dr. Birgit Willke und Dr. Robert Faurie, Amino GmbH, Frellstedt Mikrobielle L-Serinproduktion unter besonderer Berücksichtigung der Ökobilanzierung tiell. Da in den herkömmlichen Futtermitteln wie Soja und Getreideprodukten einige der essentiellen Aminosäuren aber nicht in ausreichender Menge vorhanden sind, ist die Zufütterung, insbesondere bei der Schweineund Geflügelmast, erforderlich. In der pharmazeutischen Industrie werden Aminosäuren höchster Reinheit benötigt. Ein sehr wichtiges Beispiel ist die Verwendung für die präund postoperative parenterale Ernährung. Eine Standard-Infusionslösung beinhaltet die für den Menschen essentiellen Aminosäuren. Zusätzlich enthält sie auch nicht-essentielle Aminosäuren, wie das L-Serin, dem hier eine besondere Bedeutung zukommt. Somit muss diese Aminosäure in ausreichender Menge und guter Qualität herzustellen sein. Neben diesem wichtigen Absatzmarkt für L-Serin wird es aber auch bei der Kosmetikherstellung z. B. von Hautschutzpräparaten verwendet. Die Produktion von L-Serin Die Aminosäure L-Serin stellt ein wirtschaftlich interessantes Produkt dar, das bisher nur durch saure Hydrolyse proteinogener Rohstoffe auf relativ umweltunverträgliche Weise hergestellt werden kann. Ziel ist es, ein neues Verfahren zur Produktion von L-Serin auf der Grundlage der Fermentation eines geeigneten Bakterienstammes zu entwickeln. Dieser Stamm soll mittels gentechnischer Methoden hergestellt werden. Integriert in den Forschungs- und Entwicklungsprozess werden das neue und das herkömmliche Verfahren basierend auf einer Ökobilanzierung und Wirtschaftlichkeitsanalyse bezüglich ihrer ökologischen und ökonomischen Eigenschaften miteinander verglichen. Als Grundlage des Forschungsprojektes dient der Wildtyp von Corynebacterium glutamicum, ein Organismus, der als guter Aminosäureproduzent bekannt ist. Um aus diesem einen L-Serinproduzenten herzustellen, werden verschiedene Ansätze parallel verfolgt. Neben einer Steigerung der Aktivität der L-Serinbiosyntheseenzyme, sowie einer Deregulation des Schlüsselenzyms, soll die Vorstufenbereitstellung und der Export von L-Serin erhöht sowie der mögliche Abbau verringert werden. In ersten Experimenten konnten die Gene serA, serC und serB, die für die L-Serinbiosynheseenzyme kodieren, aus C. glutamicum isoliert und kloniert werden. Die homologe Überexpression von serA ergab eine ca. 12-fache Steigerung der spezifischen Aktivität der 3-Phosphoglycerat-Dehydrogenase von 0,2 U/mg Protein auf 2,3 U/mg Protein. Das Enzym des Wildtyps wird durch 10 mM L-Serin zu 50 % gehemmt. Wurden serC bzw. serB im Wildtyp von C. glutamicum homolog überexprimiert, konnten bereits Spuren von L-Serin im Kulturüberstand nachgewiesen werden. Über die Arbeiten zur L-Serinbiosynthese hinaus wurde gezeigt, dass sich der L-Serinexport aus der Zelle, der durch das Exportprotein ThrE vermittelt wird, durch thrE-Überexpression steigern lässt. Die Verwendung von L-Serin 쑺 Hauptverwendungsgebiete für Aminosäuren sind die Nahrungsmittel-, Futtermittel- und Pharmaindustrie. Während die Gesamtproduktion an Aminosäuren 1986 weltweit etwa 650.000 t betrug (Enei et al., 1989) werden derzeit bereits mehr als 1,5 Millionen t Aminosäuren produziert (Eggeling et al. 2001). Dies zeigt welche wirtschaftliche Bedeutung der Produktion sowohl ökonomisch als auch ökologisch zukommt. Für die Nahrungsmittelindustrie sind Aminosäuren interessant, weil viele von ihnen geschmacksverstärkende Eigenschaften haben, wie z. B. das Natriumsalz des L-Glutamats. Die Futtermittelindustrie stellt Aminosäuren als Ergänzung zum Tierfutter her. Ein Teil der Aminosäuren ist für Tiere essen- Abb. 1: Strategien zur Entwicklung eines L-Serin produzierenden Bakterienstammes ausgehend vom Wildtyp von C. glutamicum Generell finden derzeit vier technische Verfahren Anwendung zur Produktion von Aminosäuren: die chemische Synthese, die enzymatische Katalyse, die saure Hydrolyse sowie die Fermentation geeigneter Mikroorganismen. Die großtechnische Herstellung von L-Serin erfolgt fast ausschließlich über die saure Hydrolyse und Extraktion aus Proteinhydrolysaten über Ionenaustauschchromatographie und Kristallisation. Durch diese Methode lassen sich beinahe alle Aminosäuren gewinnen. Rohstoffe hierfür sind proteinreiche Produkte wie z. B. Keratin, Federn oder Blutmehl aber auch Pflanzenproteine. Ein Nachteil dieses Verfahrens ist, dass hierbei nicht nur die gewünschten, sondern auch die anderen im Protein enthaltenen Aminosäuren anfallen. Dadurch entstehen bestimmte Aminosäuren, wie z. B. L-Alanin, im Überschuss. Aufgrund aktueller Akzeptanzprobleme tierischer Rohstoffe speziell von Wiederkäuern in Zusammenhang mit der BSE-Diskussion scheidet ein Großteil der o.g. Rohstoffe heute aus. Aus diesem Grund sind nicht mehr alle Aminosäuren (u.a. Prolin) extraktiv darstellbar. Ein weiterer wesentlicher Nachteil der sauren Hydrolyse ist die durch den Aufschluss bedingte hohe Umweltbelastung. Die Hydrolyse erfolgt unter Verwendung großer Mengen 5,8 M Salzsäure. Dadurch werden einige Aminosäuren, wie L-Glutamin, LAsparagin, L-Cystein und L-Tryptophan größtenteils zerstört und sind somit nicht zugänglich. Nicht verwertbare Hydrolyserückstände (Faserstoffe, Huminstoffe) müssen meist als Sondermüll verbrannt oder deponiert werden. Das flüssige Hydrolysat wird mit Natronlauge neutralisiert und durch Ionenaustauschchromatographie aufgetrennt. Neben der Aminosäurefraktion fallen so zusätzlich noch eine Salzfraktion sowie eine hohe Stickstofffracht an, die wiederum ins Abwas- Biokatalyse 66 ser entsorgt werden müssen und somit zu einer weiteren Belastung der Gewässer führen. Dies ist unter ökologischen Gesichtpunkten nicht mehr zeitgemäß. Fermentative Herstellungsprozesse von Aminosäuren lösen zunehmend die oben beschriebene Methode der sauren Hydrolyse ab. Das liegt vorallem daran, dass sich auf diesem Weg die gewünschte L-Aminosäure in enantiomerenreiner Form erzeugen lässt. Darüberhinaus fallen keine Begleitaminosäuren an, so dass die Aufreinigung wesentlich vereinfacht, und der Gesamtprozess damit ökologisch und ökonomisch erheblich verbessert wird. In der Literatur werden verschiedene Ansätze zur fermentativen Herstellung von LSerin beschrieben. Zumeist dient das relativ teure Glycin als Ausgangssubstrat in Kombination mit Glukose. So lassen sich z. B. mit einem Brevibacterium Stamm aus 20 g/l Glycin und 70 g/l Glukose bis zu 4,4 g/l L-Serin herstellen (Kubota et al. 1971) bzw. mit immobilisierten Zellen von Corynebacterium glycinophilum können in kontinuierlicher Kultur 6 g/l L-Serin aus 30 g/l Glycin und 100 g/l Glukose gewonnen werden. Höhere Ausbeuten werden mit dem methylotrophen Hyphomicrobium methylovorum aus Methanol und Glycin erzielt. Hier liefern 100 g/l Glycin und 48 g/l Methanol rund 33 g/l L-Serin (Izumi et al. 1993). Diese fermentativen Verfahren haben sich aufgrund der hohen Substratkosten (Glycin) als nicht wirtschaftlich erwiesen und werden nicht großtechnisch eingesetzt. Für die biotechnologische Aminosäureproduktion ausgehend von kostengünstigeren Substraten finden hauptsächlich Escherichia coli- und Corynebacterium glutamicum-Stämme Abb. 2: Die Reaktionen der Biosynthese und des Abbaus von L-Serin in C. glutamicum S O N D E R A U S G A B E Verwendung (Eggeling et al. 2001). Mit ihnen werden die Aminosäuren L-Threonin, L-Phenylalanin, L-Lysin und L-Glutamat aus Saccharose (aus Melasse) oder Glukose (aus Stärkehydrolysat) hergestellt. Während L-Glutamat schon mit dem Wildtyp von C. glutamicum synthetisiert werden kann, werden für die anderen drei Aminosäuren Stämme verwendet, die durch klassische Mutagenese verändert wurden (Aida et al. 1986; Eggeling et al. 2001). Entwicklung eines biotechnologischen Verfahrens zur L-Serin-Produktion mit C. glutamicum im Sinne eines produktionsintegrierten Umweltschutzes Unter der Leitung des Forschungszentrums Jülich, Institut für Biotechnologie 1 (Prof. Dr. Sahm) soll in Kooperation mit dem mittelständischen Unternehmen Amino GmbH in Frellstedt (Dr. Faurie) und der Abteilung Ordnungs- und Prozesspolitik der Universität Hannover (Prof. Dr. Müller) ein neues Verfahren zur L-Serin-Produktion mit C. glutamicum erarbeitet werden. Ziel ist es, dass sich dieses durch deutlich verminderte Umweltbelastungen klar von dem traditionellen Hydrolyseverfahren zur Herstellung von LSerin abhebt und sich auch unter den gegebenen Rahmenbedingungen des Marktes für Aminosäuren zeitnah umsetzen lässt. Im Rahmen des Projektes werden im Forschungszentrum Jülich und an der Universität Hannover problemorientierte wissenschaftliche Lösungsansätze erarbeitet. Diese werden bei der Amino GmbH in den Produktionsmassstab umgesetzt. In diesem Zusammenhang kommt der innovationsbegleitenden Betrachtung der ökologischen und ökonomischen Wirkungen des neu zu entwickelnden Verfahrens im Vergleich zum gegenwärtigen Stand der Technik eine besondere Bedeutung zu. Hierzu erstellt die Abteilung Ordnungs- und Prozesspolitik für das neue und das konventionelle Verfahren eine Ökobilanz und eine Analyse der Wirtschaftlichkeit unter den aktuellen Marktgegebenheiten. Aufgrund umfangreicher Kenntnisse zur Genetik und Physiologie von C. glutamicum am Institut für Biotechnologie (Eggeling et al. 2001) wurde dieses Bakterium als Modellorganismus ausgewählt, um das fermentative Verfahren zur Gewinnung der Aminosäure L-Serin zu entwickeln. Hierzu soll ausgehend vom Wildtyp von C. glutamicum durch gezielte gentechnische Veränderung des Stoffwechsels ein geeigneter L-Serin-Produktionsstamm erzeugt werden. Neben der Betrachtung des eigentlichen Biosyntheseweges liefert die Vorstufenbereitstellung ausgehend von Glukose als Substrat, der Export des L-Serins aus der Zelle sowie die Verhinderung des Abbaus durch die Serin-Dehydratase weitere Ansatzpunkte, die in die Entwicklung des Stammes miteinbezogen werden sollen (Abb. 1) D E R D B U Gene der L-Serinbiosynthese in C. glutamicum In Mikroorganismen sind zwei alternative L-Serinbiosynthesewege bekannt. Die meisten Bakterien nutzen den Weg über phosphorylierte Zwischenprodukte (Abb. 2), während in methylotrophen Bakterien nichtphosphorylierte Intermediate vorkommen. Da die weitere Umwandlung von L-Serin zu Glycin in einer reversiblen, durch das Enzym Serinhydroxymethyltransferase katalysierten Reaktion erfolgt (Abb. 2), kann die Umsetzung von Glycin zu L-Serin auch als dritter Syntheseweg von L-Serin aufgefasst werden. Aus GenBank-Analysen ergab sich, dass C. glutamicum über die Gene serA, serC und serB verfügt. Diese kodieren für die Enzyme 3-Phosphoglycerat-Dehydrogenase (serA), Phosphoserinaminotransferase (serC) und Phosphoserinphosphatase (serB). Die Anwesenheit der Gene serA, serC und serB legt nahe, dass der Weg zur L-Serinbildung in C. glutamicum vermutlich, wie in vielen anderen Organismen auch, über phosphorylierte Zwischenprodukte erfolgt. Das erste Enzym des L-Serinbiosyntheseweges, die von serA kodierte 3-Phosphoglycerat-Dehydrogenase setzt das Glykolyseintermediat 3-Phosphoglycerat zu 3-Phosphohydroxypyruvat um. Dabei entsteht NADH. Das serAGen von C. glutamicum besteht aus 1590 Basen, und die abgeleitete Peptidsequenz von 530 Aminosäuren weist ca. 45 % Ähnlichkeit zu bekannten 3-Phosphoglycerat-Dehydrogenasen, z. B. aus E. coli oder Hefe auf. Die größte Ähnlichkeit mit ca. 75 % besteht zu der putativen 3-Phosphoglycerat-Dehydrogenase aus dem zu C. glutamicum nahe verwandten Mycobacterium tuberculosis. Der zweite Schritt der L-Serinbiosynthese wird durch die Phosphoserinaminotransferase katalysiert, die durch das Gen serC kodiert ist. Bei dieser Transaminierung von 3Phosphohydroxypyruvat zu Phosphoserin fungiert L-Glutamat als Aminodonor und es entsteht Oxalacetat (Abb. 2). Das serC-Gen von C. glutamicum hat eine Länge von 1140 Basen und die abgeleitete Peptidsequenz beträgt 367 Aminosäuren. Die Ähnlichkeit zu bekannten Phosphoserinaminotransferasen liegt bei ca. 35 %. Für den letzten Schritt zum L-Serin ist die durch serB kodierte Phosphoserinphosphatase verantwortlich. Dieses Enzym hydrolysiert unter Freisetzung von anorganischem Phosphat Phosphoserin zu L-Serin (Abb. 2). Die von serB abgeleitete Peptidsequenz beinhaltet 448 Aminosäuren entsprechend einer Genlänge von 1347 Basen. Auffallend ist, dass das Polypeptid aus C. glutamicum länger ist, als das entsprechende aus den meisten anderen Organismen (Abb. 3). Im überlappenden Bereich sind die Ähnlichkeiten allerdings mit ca. 50 % sehr hoch. Welche Rolle die zusätzli- Biokatalyse S O N D E R A U S G A B E D E R 67 D B U chen Sequenzen z. B. für die Stabilität des Enzyms spielen ist unklar. Klonierung und homologe Überexpression des serA-Gens und Untersuchungen zur Regulation der 3-PhosphoglyceratDeydrogenase aus C. glutamicum Durch PCR wurde das serA-Gen aus chromosomaler DNA von C. glutamicum amplifiziert, isoliert und zunächst in einen E. coliVektor kloniert. Die anschließende Komplementation einer E. coli-Mutante, die einen Defekt in dem serA-Gen trägt, zeigte, dass es sich bei serA aus C. glutamicum um ein funktionelles, auch in E. coli aktives Gen handelt, das für eine aktive 3-Phosphoglycerat-Dehydrogenase kodiert. Anschließend wurde das Gen in einen Pendelvektor umkloniert, der sich für die homologe Expression von serA in C. glutamicum eignet. Das erhaltene Plasmid pZ1serA wurde in den Wildtyp von C. glutamicum eingebracht. Im Enzymtest wurde die Überexpression von serA bestimmt. Die spezifische Aktivität der 3-Phosphoglycerat-Dehydrogenase in dem entsprechenden Stamm betrug 2,3 U/mg Protein und war damit ca. 12-fach erhöht gegenüber dem Kontrollstamm, der nur den Leervektor enthielt (0,2 mU/mg Protein). Mit zellfreien Extrakten des Wildtyps von C. glutamicum wurden erste Untersuchungen zu den allosterischen Eigenschaften des Schlüsselenzyms 3-Phosphoglycerat-Dehydrogenase durchgeführt. Es konnte gezeigt werden, dass die 3-Phosphoglycerat-Dehydrogenase durch L-Serin „feedback“ gehemmt wird (s. Kasten 1), wobei die Zugabe von 10 mM L-Serin nach einer Inkubation von 5 min die Aktivität um 50 % inhibierte. Aus anderen Arbeiten ist bekannt, dass nach ungerichteter Mutagenese ein dereguliertes, nicht mehr Serin-feedback-inhibierbares 3Phosphoglycerat-Dehydrogenase-Enzym erhalten wurde. Diese Mutation beruhte auf einem Austausch der Aminosäure Glutamat an Position 325 zu Lysin (Suga et al. 1999). Aufbauend auf diesen Erkenntnissen sollen durch gengerichtete Mutagenese gezielt deregulierte 3-Phosphoglycerat-Dehydrogenase-Muteine konstruiert und für die Stammentwicklung des L-Serinproduzenten genutzt werden. Kasten 1: „Feedback“-Hemmung von Enzymaktivitäten Das Endprodukt einer Synthesekette hemmt in reversibler Weise die Aktivität des häufig ersten Enzyms des spezifischen Biosyntheseweges, des sog. Schlüsselenzyms. Dadurch ist es der Zelle möglich, die Syntheserate relativ schnell an den Bedarf an Endprodukt anzupassen, und so eine für die Zelle unnütze Anreicherung zu vermeiden. Abb. 3: Vergleich der Primärstruktur der Phosphoserinphosphatase von C. glutamicum mit der Primärstruktur von Phosphoserinphosphatasen aus anderen Organismen. Klonierung und homologe Überexpression der Gene serB und serC sowie deren Bedeutung für die Serinbildung in C. glutamicum Die Gene serB und serC wurden ebenfalls mittels PCR aus dem Genom von C. glutamicum amplifiziert, isoliert und kloniert. Die erhaltenen DNA-Fragmente wurden je in einen in C. glutamicum replizierbaren Vektor überführt und in den Wildtyp von C. glutamicum eingebracht. Während der Wildtyp kein L-Serin in den Kulturüberstand ausscheidet, führt die Überexpression von serB bzw. serC in C. glutamicum Wildtyp bereits zur Produktion geringer Mengen von L-Serin (PetersWendisch et al. 2000). In Zukunft soll nun die serB- und serC-Überexpression mit der Überexpression von serA und den entsprechenden Muteinen von serA kombiniert werden. Verbesserung der Vorstufenbereitstellung zur L-Serinbildung Das Glykolysezwischenprodukt 3-Phosphoglycerat ist direkter Vorläufer der L-Serinbiosynthese und aus diesem Grund soll die Bereitstellung dieses Metaboliten erhöht werden. Es ist bekannt, dass eine Pyruvatkinasemutante von C. glutamicum ssp. lactofermentum bei Wachstum auf Glukose bis zu 4,5 g/l Dihydroxyaceton bzw. Glycerinaldehyd in den Kulturüberstand ausscheidet (Gubler et al. 1994). Das deutet darauf hin, dass nicht nur PEP sondern auch frühere Glykolyseintermediate, wie z.B. Dihydroxyacetonphosphat und Glycerinaldehydphosphat in der Zelle anstauen und möglicherweise als nicht-phosphorylierte Substanzen ausgeschieden werden. Aufgrund dieser Ergebnisse war anzunehmen, dass eine Pyruvatkinasemutante bei Wachstum auf Glukose auch 3-Phosphoglycerat als L-Serin-Vorläufer anstaut, und dass nach Überexpression und Deregulation der LSerinbiosynthesegene der Kohlenstofffluss verstärkt in Richtung L-Serin geleitet werden kann. Es wurde eine definierte Pyruvatkinasemutante von C. glutamicum durch „gengerichtete Integrationsmutagenese“ (Kasten 2) konstruiert und bezüglich ihrer Wachstumseigenschaften analysiert. Die Mutante wächst zwar auf Glukose, kann aber auf Ribose als alleiniger Kohlenstoffquelle nicht wachsen. Das zeigt, dass der Pyruvat-generierende Schritt der Glykolyse wie gewünscht ausgeschaltet ist und das benötigte Pyruvat über die Reaktionen des Glukose-Aufnahmesystems (PTS) geliefert werden muss. Das Wachstum auf Substraten, wie Ribose, die nicht über ein solches PTS-System aufgenommen werden ist demnach nicht möglich. Der Stamm wird in Zukunft auf eine Bereitstellung vermehrter Glykolyseintermediate untersucht, und steht dann als Ausgangsstamm für die Überexpression der L-Serinbiosynthesegene zur Verfügung. Verbesserung des Transports von L-Serin aus der Zelle Neben einer erhöhten Bereitstellung von Vorstufen, sowie einem erhöhten Fluss in Richtung L-Serin, ist ein weiterer wesentlicher Aspekt, dass das intrazellulär gebildete L-Serin aus der Zelle ausgeschieden wird. Man weiss heute, dass die Ausschleusung der gewünschten Aminosäuren genauso wichtig ist wie deren intrazelluläre Synthese (Krämer, 1994; Vrljié et al. 1996, Simic et al. 2001). Kürzlich konnte das Exportprotein für LThreonin (ThrE) in C. glutamicum identifiziert und das entsprechende Gen isoliert werden (Simic et al., 2001). Aufgrund der strukturellen Ähnlichkeit von L-Serin und L-Threonin wurde auch der Transport von L-Serin aus der Zelle über diesen Exporter untersucht. Hierzu wurden Stämme von C. glutamicum hergestellt, die das Exportergen thrE plasmidkodiert überexprimiert bzw. chromosomal inaktiviert vorliegen hatten. Die Exportraten dieser Stämme für L-Serin wurden mit denen des Kontrollstamms, der das Leerplasmid trägt, verglichen (Abb. 4). Es zeigte sich, dass die Überexpression von thrE tatsächlich zu einem erhöhten Export von L-Serin führt, während die Inaktivierung eine Reduktion bewirkt. Die Raten betragen für den, das thrEGen überexprimierenden Stamm 1,9 nmol min-1 (mg Trockengewicht)-1 im Vergleich zum Kontrollstamm mit 1,4 und zum Stamm mit inaktiviertem thrE-Gen mit 0.6 nmol min-1 (mg Trockengewicht)-1. Bei diesem Experiment betrug die zellinterne L-Serinkonzentration mehr als 80 mM, um so eine Sättigung des Exportproteins sicher zustellen. Somit ist klar, dass durch den Exporter ThrE eine beschleunigte Ausschleusung von L-Serin erreicht werden kann. Da es auch in der Mutante mit inaktiviertem thrE-Gen zu einem Export kommt, gibt es noch weitere Wege für L-Serin aus der Zelle. Für L-Threonin wurde gezeigt, dass der Anteil des Exports durch Biokatalyse 68 Abb. 4: L-Serin-Exportratenverschiedener C. glutamicum-Stämme nach Zugabe von 1 mM L-Serin-Tripeptid ThrE nur 59 % beträgt, und das ein weiterer unbekannter Exporter sowie Diffusion ebenfalls am Export beteiligt sind (Simic et al., 2001). Damit sind prinzipiell die Wege auf denen L-Serin die Zelle verlässt bekannt, und es ist das Instrumentarium vorhanden um in Zukunft in einem Produzentenstamm zellintern synthetisiertes L-Serin auch beschleunigt auschleusen zu können. Vergleich des herkömmlichen Verfahrens zur L-Serinproduktion mit dem neuen fermentativen Verfahren auf der Basis einer Ökobilanzierung und Wirtschaftlichkeitsprüfung Die Ökobilanz Mit Hilfe von Ökobilanzen sollen über Vergleiche umweltrelevanter in- und outputs Erkenntnisse über die ökologische Kompatibilität unterschiedlicher Produkte bzw. Verfahren gewonnen werden. Ziel einer Ökobilanz ist letztendlich die Identifizierung umweltverträglicherer Produkte bzw. Verfahren bei bestehenden Alternativen. Im Rahmen dieses Forschungsprojekts wird ein Vergleich zwischen dem gegenwärtigen Stand der Technik in der Form des Verfahrens der sauren Hydrolyse und dem neuen fermentativen Verfahren durchgeführt. Die Grundlage für die umweltökonomischen Arbeiten bildet das Umweltinformationssystem Ökobilanz in Form des Ansatzes der Lebenszyklusanalyse, wie es von der ISO (1997 und 1998) vorgeschlagen wird (Abb. 5). Als Ziel wurde die Erstellung einer Produktökobilanz für die Herstellung der Aminosäu- S O N D E R A U S G A B E re L-Serin in Pharmaqualität zur Verwendung in Infusionslösungen definiert. Sie soll das Firmenmanagement bei der Entscheidungsfindung über die Implementierung neuer Produktionsverfahren unterstützen, indem sie das Set konventioneller betriebswirtschaftlicher Entscheidungsparameter um zusätzliche Informationen ergänzt, die diese nicht bereitstellen können. Die Abgrenzung des Bilanzraums und die Bestimmung der Bilanzgrenzen basiert auf dem „from the cradle to the gate“ Ansatz (Kasten 3). Die Erfassung der Daten für die Sachbilanz als Kernelement der Ökobilanz erfolgt aus einer Kombination von Datenermittlungen aus realen Produktionsumgebungen, upscales aus Versuchen, Expertenbefragungen sowie Literatur- und Internetrecherchen. Die Erstellung der Sachbilanz erfolgt mit Unterstützung der speziellen Ökobilanzsoftware Umberto, die eine integrierte Vorgehensweise von der qualitativen Darstellung der einzelnen Verfahrensschritte (Modellierung des Systems und Aufbau eines Stoffstromnetzes), über die quantitative Erfassung aller Parameter bis zur gegebenenfalls notwendigen Berechnung noch unbekannter Stoffströme in einem Stoffstromnetz ermöglicht. Bei der Wirkungsbilanz und der Bilanzbewertung kommen Datenbanken und Expertensysteme zum Einsatz, z.T. in Kombination mit Umberto. Die Erstellung der Ökobilanz erfolgt unmittelbar mit der Entwicklung der zu bewertenden Teilprozesse, wodurch eine zeitnahe prozessbegleitende Bewertung möglich wird, die eventuell ungünstige Entwicklungen bereits im Frühstadium erkennen lässt. So konnten erste Ergebnisse ökobilanzieller Untersuchungen (z.B. hinsichtlich des Einsatzes von Antibiotika) bereits frühzeitig in der Stammentwicklung berücksichtigt werden. Die derzeitigen Forschungsergebnisse deuten darauf hin, dass das neu zu entwickelnde fermentative Verfahren mit geringeren Umweltbelastungen verbunden ist als die saure Hydrolyse proteinogener Rohstoffe. Die Wirtschaftlichkeitsanalyse Da es das Ziel des Forschungsprojektes ist, dass der mittelständische Projektpartner D E R D B U Amino GmbH erfolgreich als Anbieter von LSerin am Markt auftreten kann, kommt der kostenrechnerischen Betrachtung eine erhebliche Bedeutung zu. Im Rahmen des Projektes wird der Ansatz verfolgt, die Ökobilanz mit modernen Kostenmanagementsystemen zu verbinden, um eine ökologisch und ökonomisch fundierte Entscheidungsfindung zu ermöglichen. In diesem Zusammenhang stellen sich besondere Herausforderungen, da es sich beim Markt für Aminosäuren um ein überaus intransparentes Marktumfeld handelt, durch welches Investitionsplanungen erschwert werden. Mit Hilfe von Ansätzen aus der Umweltkostenrechnung und des „Target Costing“ (Kasten 4) sollen kostenrechnerische Überlegungen und Marktparameter frühzeitig in den Entwicklungsprozess einfließen. Die Integration angepasster Kostenmanagementansätze in die Produkt- und Verfahrensentwicklung hat zum Ziel, die zukünftige Wettbewerbsfähigkeit des Produktes zu geKasten 2: „Integrationsmutagenese“ Eine Methode zur Geninaktivierung durch Integration eines Plasmids in das zu mutierende Gen. Da sich das Plasmid, das eine Antibiotikumresistenz trägt, im Zielorganismus nicht vermehren kann können nur solche Stämme überleben, die das Plasmid im Chromosom integriert haben. Die Integration erfolgt aufgrund homologer DNA-Sequenzen und natürlicher Rekombination. Es kommt daher zu einer Unterbrechung der Gensequenz durch das Plasmid, und damit zu der gewünschten Geninaktivierung. Kasten 3: „From the Cradle to the gate“-Ansatz Je nach Produkt ist bei einer Produktökobilanz eine unterschiedliche Abgrenzung des zu untersuchenden Bilanzraums sinnvoll. Beim „from the cradle to the gate“ Ansatz („von der Wiege bis zum (Betriebs)Tor“) erfolgt eine Dokumentation von der Rohstoffgewinnung über die Produktvorketten und die Produktion des Hauptproduktes bis zur Auslieferung an den Abnehmer. Bei Produkten mit intensiver Nutzung und Notwendigkeit der Entsorgung ist eine Ausdehnung des Bilanzraums um Produktverwendung und -entsorgung erforderlich („from the cradle to the grave“ Ansatz). Kasten 4: „Target Costing“ Abb. 5: Aufbau einer Ökobilanz, In Anlehnung an ISO (1997): ISO 14040: Environmental management Life cycle assessment - Principles and framework, Genève. Marktorientierter Kostenmanagementansatz, bei dem ausgehend vom Marktpreis zulässige Produktions- und Entwicklungskosten ermittelt werden. Biokatalyse S O N D E R A U S G A B E D E R währleisten und die Informationsbasis für Investitionsentscheidungen angesichts eines sehr komplexen Marktumfeldes zu verbessern. Durch den gewählten Ansatz konnte bereits frühzeitig ein Kostenmodell für die Produkt- und Verfahrensentwicklung erstellt werden. Es unterliegt einer laufenden Optimierung hinsichtlich der Projektfortschritte und veränderter Marktbedingungen und bildet die Basis für eine an den Markt- und Wettbewerbsbedingungen ausgerichtete Projektarbeit. Literaturangaben Aida K. (Hsrg.): Biotechnology of amino acids production. In: Progress in industrial microbiology. Vol. 24, Elsevier, Amsterdam, 1986 Eggeling, L., Pfefferle W. und Sahm H. (2001): Amino acids. In: Ratledge, C. und Kristiansen, B. (Hrsg.) Basic Biotechnology. Cambridge University Press, 2001 Enei, H., Yokozeki , K. und Akashi , K.: Recent progress in microbial production of amino acids. In: Japanese technology reviews Vol. V. Gordon and Breach Science Publishers, New York, 1989 Gubler, M., Jetten M., Lee S. H. und Sinskey A. J. (1994): Cloning of the pyruvate kinase gene (pyk) of Corynebacterium glutamicum and site-specific inactivation of pyk in a lysine-producing Corynebacterium lactofermentum strain. Appl. Env. Microbiol. 60: 2494-2500 Hodgson, J. (1994): Bulk amino-acid fermentation: technology and commodity trading. Biotechnology 12: 152-155 ISO (1997): ISO 14040: Environmental management – Life cycle assessment – Principles and framework, Genève. ISO (1998): ISO 14041: Environmental management – Life cycle assessment – Goal and scope definition and inventory analysis, Genève. Krämer, R. (1994): Secretion of amino acids: Physiology and mechanism. FEMS Microbiol. Rev. 13: 75-79. Kubota, K., Kageyama, K., Shiro, T. und Okumura, S. (1971): Fermantative production of L-serine. J. Gen. Appl. Microbiol. 17: 167-168 Izumi, Y., Yoshida, T., Miyazaki, S.S., Mitsunaga, T., Ohshiro, T., Shimao, M., Miyata, A. und Tanabe, T. (1993): L-Serine production by a methylotroph and ist related enzymes. Appl. Microbiol. Biotechnol. 39: 427432 Peters-Wendisch, P., Eggeling, L. und Sahm, H. (2000): Nukleotidsequenzen kodierend für Proteine beteiligt an der Biosynthese von L-Serin und Verfahren zu dessen Herstellung. Patentanmeldung PT 0.1960 Saski, R. und Pizer L. I. (1975): Regulatory properties of purified 3-Phosphoglycerat-Dehydrogenase from Bacillus subtilis. Eur. J. Biochem. 51: 415-427 Simic, P., Sahm, H., and Eggeling, L. (2001): LThreonine export: Use of peptides to identify a new translocator from Corynebacterium glutamicum. J. Bacteriol. (accepted for publication) Suga, M., Sugimoto M., Osumi T., Nakamatsu T., Hibino W. und Ito M. (1999): Method of producing Lserine by fermentation, Ajinomoto Co., Inc. Patent Veröffentlichungsnr. EP0931833 Tanaka, T., Yamamoto, K., Towprayoon, S., Nakajima, H., Sonomoto, K., Yokozeki, K., Kubota, K. und Tanaka, A. (1989): Continuous production of L-serine by immobilized growing Corynebacterium glycinophilum cells. Appl. Microbiol. Biotechnol. 30: 564-568 Vrljic ,M., Sahm, H., and Eggeling, L. (1996): A new type of transporter with a new type of cellular function: Llysine export from Corynebacterium glutamicum. Mol. Microbiol. 22:815-826 Korrespondenzadresse Prof. Dr. Hermann Sahm Institut für Biotechnologie 1 Forschungszentrum Jülich GmbH 52425 Jülich email: [email protected] 69 D B U Prof. Thomas Scheper, Dr. Roland Ulber, Dipl.-Chem. Tanja Koop, Dipl.-Chem. Dirk Kosemund, Institut für Technische Chemie der Universität Hannover Prof. Udo Müller, Dipl. Ök. Susanna Huebner, Dipl. Ök. Ralf Tostmann, Institut für Volkswirtschaftslehre Abt. Ordnungs u. Prozesspolitik Dr. Robert Faurie, Amino GmbH Frellstedt Entwicklung innovativer Aminosäureproduktion unter Berücksichtigung des Umweltkostenmanagements Im Rahmen des Projektes werden innovative enzymatische Aufarbeitungsverfahren für natürlich vorkommende Proteine konzipiert, getestet und hinsichtlich ihrer Wirtschaftlichkeit und Nachhaltigkeit genauer untersucht. Mit Hilfe dieses enzymatischen Verfahrens können ausgewählte Proteine effizient in ihre Einzelbestandteile (Aminosäuren, Peptide) zerlegt werden. Eine prozessintegrierte selektive Aufarbeitung macht die Abtrennung einzelner Aminosäuren oder Aminosäuregruppen wirtschaftlich interessant. Die genaue Abstimmung einzelner Prozessschritte führt zu einer effizienten Ausbeute und zu einer Reduktion von möglichen Verlusten. Alle Schritte im Forschungsbereich werden durch begleitende Wirtschaftlichkeitsberechnungen und Ökobilanzierungen hinsichtlich ihrer industriellen Verwirklichung untersucht. 쑺 Zur Zeit werden Aminosäuren durch Prozesse wie saure Hydrolyse, chemische Synthese oder Produktion mit Hilfe von Mikroorganismen gewonnen. Dabei fallen entweder hohe Salzfrachten an, was eine erhöhte Umweltbelastung zur Folge hat, oder die Verfahren liefern nur eine Aminosäure mit entsprechend hohem technischen Aufwand. Die enzymatische Hydrolyse proteinhaltiger, industrieller Nebenprodukte oder Abfallstoffe bietet die Möglichkeit, unter schonenden Bedingungen ein breites Spektrum an Aminosäuren aus diesen Reststoffen zu gewinnen. Nach der Hydrolyse müssen weitere Schritte, wie Ionenaustausch und Umkristallisation, zur Reinigung der Aminosäuren durchgeführt werden. Diese Aufreinigungsschritte verursachen oft zusätzlich hohe Energiekosten. Alternativ kann hier die selektive Adsorption von Aminosäuren aus den Aminosäuren/Proteingemischen an Zeolithe genutzt werden. Begleitend zu den geplanten technischen Prozessen werden vom Institut für Volkswirtschaftslehre Kostenaussagen für die einzelnen Module erstellt. Diese Daten liefern betriebswirtschaftliche Beurteilungskriterien für bestimmte Prozessgrößen, wie beispielsweise die Enzymmenge. Zusätzlich werden noch Umweltaspekte betrachtet, indem basierend auf den functional units der Module eine Ökobilanz erstellt wird. Ein Vergleich mit herkömmlichen Verfahren soll klarstellen, ob der geplante Prozess wirklich umweltschonender arbeitet. Enzymatische Hydrolyse und weitere Aufarbeitung A. Die enzymatische Hydrolyse Pflanzliche Proteinkonzentrate wie Maisoder Kartoffelkleber sind durch ihre Beschaffenheit oder durch mindergiftige Inhaltsstof- Biokatalyse 70 S O N D E R A U S G A B E Abb. 1: Geplanter Hydrolyseprozess für Hydrolysen im Technikum oder Industrie Maßstab. fe als menschliche Nahrung oftmals ungeeignet und werden daher nur als Tierfutter eingesetzt. In diesem Projekt werden gerade solche Proteinkonzentrate als mögliche Substrate untersucht. Sie werden durch Hitzedenaturierung gewonnen und sind daher meist schwer lösliche Substanzen. Daher muss die enzymatische Hydrolyse in zwei Schritten durchgeführt werden. Im ersten Schritt wird mit Hilfe einer Endoprotease das Protein aus dem jeweiligen Kleber in Form von Proteinfragmenten und Peptiden gelöst. Subtilisine, Proteasen der Serinklasse, erwiesen sich für diesen ersten Hydrolyseschritt als besonders gut geeignet. Die bisher getesteten Proteasen sind konventionell erhältlich, sollen aber später durch Extremozyme ersetzt werden. Der zweite Hydrolyseschritt erfolgt durch die Zugabe einer Exoprotease. Auch hier wurden konventionell erhältliche Exoproteasen (von Novo Nordisk, Röhm u.a. ) verwendet. Die eingesetzte Substratmenge kann nicht beliebig gewählt werden. Bei den Untersuchungen wurde gefunden, dass Substratkonzentrationen von 6 Gewichtsprozent als oberste Grenze anzusehen sind, wenn man einen hohen Hydrolysegrad erreichen will. Versuche zur Substratabhängigkeit des Prozesses zeigen, dass bei Enzym zu Proteinverhältnissen von 1 % für die Endoprotease und 5 % für die Exoprotease, Hydrolysegrade größer als 55 % in 18 Stunden erreichbar sind. Der Einsatz größerer Mengen an Exoprotease liefert noch höhere Hydrolysegrade, doch stellen die teuren Exoproteasen finanziell einen limitierender Faktor für den Prozess da. B. Abtrennung von Aminosäuren und Peptiden Die Aufarbeitung größerer Mengen an Hydrolysat soll mit industriell üblichen Verfahren erfolgen. Den geplanten Prozess zeigt Abbildung 1. Die Umsetzung wird derzeit mit dem Projektpartner Amino GmbH bearbeitet. Die der Filtration folgende Ionenausschlusschromatographie soll eine Auftrennung von Peptiden und Aminosäuren liefern. Da bei diesem Verfahren nur Wasser als Eluent verwendet wird, zählt es zu umweltschonenden Methoden der Aufarbeitung. Die Peptidfraktionen der Ionenausschlusschromatographie können als Nahrungsmittelzusätze genutzt werden. Die Aminosäurefraktion soll als Ausgangslösung für die schnelle Isolierung von Aminosäuren mit Zeolithen dienen. Adsorption von L-Aminosäuren an Zeolithen A. Screening nach geeigneten Zeolithen Zum Feinreinigen verschiedener Aminosäurefraktionen werden Zeolithe verwendet. In den Versuchen mit den synthetischen Zeolithen wurden wässrige Lösungen der proteinogenen L-Aminosäuren mit einer Konzentration von 2 g/L angesetzt. Der pH-Wert Abb. 2: Zeolithkaskade zur Abtrennung von L-Tryptophan und L-Phenylalanin aus einem Hydrolysegemisch D E R D B U wurde auf ca. 0,5 Einheiten unter den isoelektrischen Punkt der jeweiligen Aminosäure eingestellt. 20 mg des zu untersuchenden Zeolithen wurden mit jeweils 1 mL der Aminosäurelösungen versetzt und für 24 h bei RT und 1500 rpm geschüttelt. Anschließend wurde der Zeolith abzentrifugiert und die Aminosäurekonzentration des Überstandes wurde mit Hilfe einer HPLC bestimmt. Mit einem Zeolith des FAU-Typs konnten Adsorptionsgrade von 38 % für L-Phenylalanin und von 63 % für L-Tryptophan gemessen werden. Bei L-Histidin wurden 49% erreicht. L-Histidin kann über eine Änderung des pH-Wertes von den beiden aromatischen Aminosäuren abgetrennt werden. L-Phenylalanin und L-Tryptophan können jedoch von diesem Zeolithen nicht mehr getrennt eluiert werden (Trennfaktor = 0,6). Die Auftrennung dieser beiden Aminosäuren kann jedoch mit einem Zeolith des MFI-Typs erreicht werden. Er adsorbiert LPhenylalanin wesentlich besser (57 %) als L-Tryptophan (5 %). Dies entspricht einem Trennfaktor von 11,4. Dieser Zeolith ist auch für die Trennung von L-Leucin und L-Isoleucin geeignet. L-Leucin wird mit 39 % stärker adsorbiert als L-Isoleucin mit 9 %. B. Aufnahme von Ad- und Desorptionsisothermen Es wurden Ad- und Desorptionsisothermen für L-Tryptophan und L-Phenylalanin am Zeolith des FAU-Typs aufgenommen. Dazu wurden wässrige L-Tryptophan und L-Phenylalanin-Lösungen mit Konzentrationen zwischen 5 und 50 mmol/L hergestellt. Die Ansätze wurden 3 h bei RT und 1500 rpm geschüttelt. Anschließend wurde der Zeolith abzentrifugiert und die Aminosäurekonzentration im Überstand mittels HPLC bestimmt. Die Desorption erfolgte unter gleichen Bedingungen mit 0,1 M Natronlauge. Auch hier wurde nach der Zentrifugation die Aminosäurekonzentration im Überstand bestimmt. In den Versuchen zeigt sich ein linearer Zusammenhang zwischen Beladung und Konzentration der Ausgangslösung. Das weist darauf hin, dass die maximale Beladungskapazität noch nicht erreicht ist. Dieses Ergebnis findet man sowohl für L-Tryptophan als auch für L-Phenylalanin. Das wurde auch in weiteren Versuchen bestätigt, in denen die Adsorption von Gemische aus L-Tryptophan und L-Phenylalanin untersucht wurde. Dort zeigte sich dann auch keine Abhängigkeit der Beladung von der Konzentration der jeweils anderen L-Aminosäure. Außerdem wurde gezeigt, dass die Desorption fast quantitativ verläuft. Aufgrund dieser Ergebnisse ist der Aufbau folgender Zeolithkaskade zur Abtrennung von L-Tryptophan und L-Phenylalanin denkbar: Dieser Aufbau würde eine saubere