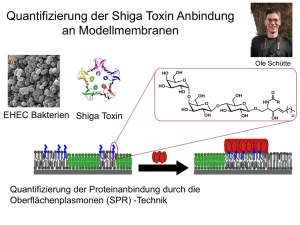
Institut für Experimentelle und Klinische
Werbung

Institut für Experimentelle und Klinische Pharmakologie und Toxikologie der AlbertLudwigs-Universität Freiburg Clostridiale Glukosylierende Toxine: Untersuchungen zur Autoprozessierung von Clostridium sordellii Letalem Toxin und Clostridium novyi α-Toxin sowie funktionelle Charakterisierung von Clostridium perfringens TpeL-Toxin INAUGURALDISSERTATION zur Erlangung des Doktorgrades der Fakultät für Chemie, Pharmazie und Geowissenschaften der Albert-Ludwigs-Universität Freiburg vorgelegt von Diplom-Pharmazeut und Apotheker Gregor Guttenberg geboren in Herbolzheim im Breisgau Februar 2012 Die Untersuchungen zur vorliegenden Arbeit wurden in der Zeit von Januar 2008 bis Dezember 2011 am Institut für Experimentelle und Klinische Pharmakologie und Toxikologie der Albert-Ludwigs-Universität Freiburg unter der Leitung von Prof. Dr. Dr. Klaus Aktories durchgeführt. Die Arbeit wurde der Fakultät für Chemie, Pharmazie und Geowissenschaften vorgelegt. Promotionsvorsitzender: Prof. Dr. Thorsten Koslowski Erstgutachter: Prof. Dr. Manfred Jung Zweitgutachter: Prof. Dr. Dr. Klaus Aktories Drittprüferin: Prof. Dr. Regine Süss Tag der mündlichen Prüfung: 06.03.2012 ORIGINALARBEITEN: Im Rahmen dieser Arbeit: Papatheodorou P., Zamboglou C., Genisyuerek S., Guttenberg G., Aktories K. Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS ONE (Mai 2010); 5:e10673 Genisyuerek S., Papatheodorou P., Guttenberg G., Schubert R., Benz R., Aktories K. Structural determinants for membrane insertion, pore formation and translocation of Clostridium difficile toxin B. Molecular Microbiology (März 2011); 79:1643-54 Guttenberg G., Papatheodorou P., Genisyuerek S., Lü W., Jank T., Einsle O., Aktories K. Inositol hexakisphosphate-dependent processing of Clostridium sordellii lethal toxin and Clostridium novyi α-toxin Journal of Biological Chemistry (April 2011); 286:14779-86 Guttenberg G., Hornei S., Jank T., Schwan C., Wilmes T., Lü W., Einsle O., Papatheodorou P., Aktories K. Molecular characteristics of Clostridium perfringens TpeL toxin and consequences of mono-O-GlcNAcylation of Ras in living cells Journal of Biological Chemistry (eingereicht), Januar 2012 Im Zeitraum dieser Arbeit: Giesemann T., Guttenberg G., Aktories K. Human α-Defensins inhibit Clostridium difficile Toxin B Gastroenterology (Juni 2008) 134:2049-2058 Papatheodorou P., Carette J., Bell G., Schwan C., Guttenberg G., Brummelkamp T., Aktories K. Lipolysis-stimulated lipoprotein receptor (LSR) is the host receptor for the binary toxin Clostridium difficile transferase (CDT) Proceedings of the National Academy of Sciences (September 2011) 108(39):16422-7 Papatheodorou P., Wilczek C., Nölke T., Guttenberg G., Hornuss D., Schwan C., Aktories K. Recombinant production of Clostridium spiroforme toxin and identification of its cellular receptor Infection and Immunity (Epub ahead of print, doi: 10.1128/IAI.06378-11), Januar 2012 I AUSGEWÄHLTE POSTERBEITRÄGE: Egerer M., Guttenberg G., Genisyürek S., Papatheodorou P., Aktories K. Auto-catalytic processing of Clostridium difficile toxin B – Binding of inositol hexakisphosphate 50. Jahrestagung der Deutschen Gesellschaft für Experimentelle und Klinische Pharmakologie und Toxikologie (DGPT), Mainz, 10.-12. März 2009 Poster-Abstract wurde veröffentlicht in: Naunyn-Schmiedeberg´s Archives of Pharmacology (2009) Egerer M., Guttenberg G., Genisyürek S., Papatheodorou P., Aktories K. Auto-catalytic processing of Clostridium difficile toxin B – Binding of inositol hexakisphosphate ETOX 14, Obernai, France, 28.Juni - 02.Juli 2009 Zamboglou C., Genisyuerek S., Guttenberg G., Hornei S., Aktories K., Papatheodorou P. Clathrin-dependent uptake of the clostridial glucosylating toxins 51. Jahrestagung der Deutschen Gesellschaft für Experimentelle und Klinische Pharmakologie und Toxikologie (DGPT), Mainz, 23.-25. März 2010 Sanofi-Aventis-Preis der Gesellschaft für Toxikologie (Bestes Poster im Bereich Toxikologie) Genisyuerek S., Papatheodorou P., Guttenberg G., Schubert R., Aktories K. New aspects in the structure-to-function relationship of Clostridium difficile toxin B 3rd International Clostridium difficile Symposium, Bled, Slovenia, 22.-24. September 2010 Guttenberg G., Papatheodorou P., Genisyuerek S., Lü W., Einsle O., Aktories K. Clostridial glucosylating toxins: Inositol hexakisphosphate-dependent processing of Clostridium sordellii lethal toxin and Clostridium novyi α-toxin 77. Jahrestagung der Deutschen Gesellschaft für Experimentelle und Klinische Pharmakologie und Toxikologie e. V. (DGPT), Frankfurt, 30. März – 01. April 2011 Papatheodorou P., Genisyuerek S., Zamboglou C., Guttenberg G., Benz R., Aktories K. Prerequisites for uptake of Clostridium difficile toxin B into host cells ETOX 15, Oslo, Norway, 18.Juni - 22.Juli 2011 II INHALTSVERZEICHNIS INHALTSVERZEICHNIS A: EINLEITUNG 1 1: Clostridien 1 2: Clostridium difficile 1 3: Clostridium perfringens, C. sordellii und C. novyi 2 4: Clostridiale Glukosylierende Toxine 3 4.1: Toxin A und Toxin B von Clostridium difficile 4 4.2: Letales Toxin von Clostridium sordellii 5 4.3: α-Toxin von Clostridium novyi 6 5: Das ABCD-Modell der CGTs 6 5.1: Glukosyltransferase-Domäne 6 5.1.1: Kleine GTPasen als Proteinsubstrate der CGTs 8 5.2: Rezeptorbindungs-Domäne 10 5.3: Cysteinprotease-Domäne 12 5.4: Porenbildende Region/Translokations-Domäne 12 6: Inositolhexakisphosphat-vermittelte Autoprozessierung 13 6.1: Prozessierung bakterieller AB-Toxine 13 6.2: Autoprozessierung 13 6.3: Intrinsische Cysteinprotease-Domäne 13 6.4: Inositolhexakisphosphat-induzierte Autoprozessierung 14 6.5: Kristallstruktur von Toxin A-CPD 14 B: ZIELSETZUNG DER ARBEIT 16 C: MATERIAL UND METHODEN 17 C.1: MATERIAL 17 1: Biologische Materialien 17 1.1: Bakterienstämme und Anzuchtmedien 18 1.1.1: Clostridien-Stämme 17 1.1.2: Bacillus megaterium-Stämme 17 1.1.3: Escherichia coli-Stämme 17 1.1.4: Nährmedien für Clostridien 17 1.1.5: Nährmedium für Bacillus megaterium und Escherichia coli 18 III INHALTSVERZEICHNIS 1.1.6: Antibiotika 18 1.2: Eukaryote Zelllinien 18 1.2.1: Kulturmedium 18 1.2.2: Kulturmedium-Zusätze 18 2: Plasmide und Vektoren 18 3: Verwendete Plasmide/Mutanten 19 4: Primer 19 5: Biochemikalien und Enzyme 19 5.1: DNA-modifizierende Enzyme 19 5.1.1: DNA-Polymerasen 19 5.1.2: DNA–Ligasen 19 5.1.3: Restriktionsendonukleasen 19 5.1.4: DNA-Phosphatasen 19 5.2: Protein-Marker 19 5.3: DNA-Leiter 19 5.4: Gelfiltration-Größenstandard 20 5.5: Antikörper und Antiseren 20 5.5.1: primäre Antikörper 20 5.5.2: sekundäre Antikörper 20 6: Kits 20 7: Radionuklide 20 8: besondere Chemikalien/Materialien 20 C.2: METHODEN 22 1: Molekularbiologische Methoden 22 1.1: Polymerase-Kettenreaktion 22 1.2: “touchdown-PCR” 22 1.3: Aufreinigung von PCR-Produkten 22 1.4: Alkohol-Fällung 23 1.5: Präparation von Plasmid-DNA 23 1.5.1: Präparation von Plasmid-DNA im kleinen Maßstab 23 1.5.2: Präparation von Plasmid-DNA im größeren Maßstab 24 1.6: Präparation von genomischer DNA 24 IV INHALTSVERZEICHNIS 1.7: Konzentrations- und Reinheitsbestimmung von DNA 25 1.8: Agarose-Gelelektrophorese 25 1.9: Isolierung von DNA aus Agarose-Gelen 26 1.10: Verdau von DNA mit Restriktionsendonukleasen 26 1.11: Vektor-Dephosphorylierung 26 1.12: Ligation von DNA-Fragmenten 27 1.13: Transformation chemisch-kompetenter Escherichia coli 27 1.14: Herstellung chemisch-kompetenter Escherichia coli 28 1.15: Elektroporation kompetenter Escherichia coli 28 1.16: Herstellung elektrisch-kompetenter Escherichia coli 28 1.17: Direkte Klonierung von PCR-Produkten 29 1.18: Blau-Weiß-Selektion 29 1.19: Zielgerichte Mutagenese 30 1.20: Sequenzierung von DNA 30 1.21: Transformation von Bacillus megaterium-Protoplasten 30 1.22: Herstellung von Bacillus megaterium-Protoplasten 31 2: Proteinbiochemische Methode 32 2.1: Aceton-Fällung 32 2.2: Ammoniumsulfat-Fällung 32 2.3: SDS-Polyacrylamid-Gelelektrophorese 32 2.4: Silberfärbung 33 2.5: Herstellung von Gesamtlysaten aus HeLa-Zellen 33 2.6.1: Western-Blot 34 2.6.2: „Stripping“ von Western-Blots 35 2.7: Proteinkonzentrationsbestimmung nach Bradford 35 2.8: Proteinkonzentrationsbestimmung mit BCA-Methode 36 2.9.1: Probenvorbereitung für Massenspektrometrie 36 2.9.2: Massenspektrometrie (MALDI-TOF) 36 2.9.3: Massenspektrometrie (LC-MS/MS) 37 2.10: Expression von rekombinanten Proteinen in Escherichia coli 38 2.11: Expression von rekombinanten Proteinen in Bacillus megaterium 38 2.12: Zellaufschluss 38 V INHALTSVERZEICHNIS 2.13: Proteinaufreinigung mittels Affinitätschromatographie 39 2.13.1: Proteinaufreinigung über 6xHistidin-tag 39 2.13.2: Proteinaufreinigung über GST-tag 39 2.13.3: Thrombinspaltung 40 2.14: Anionenaustauschchromatographie 40 2.15: Gelfiltration 40 2.16: Aufreinigung nativer Toxine aus Clostridien 41 2.16.1: Kultivierung von Clostridien 41 2.16.2: Aufreinigung nativer Toxine aus Kulturüberständen 41 2.16.3: Affinitätschromatographie von nativem Toxin A 42 2.17: Umpuffern von Proteinlösungen 42 2.18: GST-pull down 43 2.19: Glukosylierung kleiner GTPasen 43 2.20: Bestimmung der Glukohydrolase-Aktivität 44 2.21: In vitro-Bestimmung der autoproteolytischen Spaltung 44 2.22: InsP6-Filterbindung 45 2.23: Isothermale Titrationskalorimetrie 45 2.24: HPTS/DPX–Freisetzung aus Liposomen 46 2.25: Bestimmung der Fluoreszenz-Löschung von Tryptophanen nach Ligandenbindung („W-quenching“) 47 2.26: Immunopräzipitation von Ras 47 3: Zellbiologische Methoden 48 3.1: Zellvergiftungsversuche 48 3.2: Untersuchung der Porenbildung an biologischen Membranen 48 3.3: Fluoreszenz-Mikroskopie/DIC-Mikroskopie 49 D: ERGEBNISSE 50 1: Expression der rekombinanten CGTs in Bacillus megaterium 50 1.1: Klonierung der Gene der CGTs in den Bacillus megaterium Expressionsvektor pHis1522 50 1.1.1: Klonierung von pHis1522-Toxin B 50 1.1.2: Klonierung von pHis1522-Toxin A und pHis1522-Letales Toxin 51 1.1.3: Klonierung von pHis1522-α-Toxin 51 2: Expression und Aufreinigung rekombinanter CGTs 53 VI INHALTSVERZEICHNIS 2.1: Testexpression von rekombinantem Toxin B 53 2.2: Aufreinigung der rekombinanten CGTs 54 2.2.1: Ni2+-Affnitätsaufreinigung 54 2.2.2: Gelfiltration 54 2.3: Vergleich nativer und rekombinanter Aufreinigungen 55 3: Immunologische Charakterisierung der rekombinanten und nativen CGTs 57 4: Funktionelle Charakterisierungen der rekombinanten CGTs 58 4.1: Untersuchung zur in vivo-Aktivität rekombinanter CGTs in der Zellkultur 58 4.2: Untersuchung der in vitro-Aktivität der Glukosyltransferase-Domäne rekombinanter CGTs 59 4.3: Herstellung einer katalytisch inaktiven Toxin B-Mutante 60 4.4: Untersuchung der Porenbildung rekombinanter CGTs 61 5: Autokatalytsiche Prozessierung der CGTs 63 5.1.: CPD-vermittelte autokatalytische Prozessierung von Letalem Toxin und α-Toxin 63 Identifizierung der katalytischen Triade sowie der proteolytischen Schnittstelle von Letalem Toxin und α-Toxin 64 Bestimmung der Dissoziationskonstante der Bindung von InsP6 an die CPD von Letalem Toxin und α-Toxin 65 5.4.: Autokatalytische Prozessierung von Letalem Toxin und α-Toxin 67 5.5.: 5.6: Notwendigkeit funktioneller CPDs von Letalem Toxin und α-Toxin für eine effiziente Vergiftung von Vero-Zellen Bindung von InsP6 an Letales Toxin und α-Toxin 68 69 5.7: Autoprozessierung von Letalem Toxin bei saurem pH-Wert 70 5.8: Bindung von InsP6 an Letalem Toxin bei saurem pH-Wert 70 5.9: Charakterisierung der pH-abhängigen Bindung von InsP6 an Letales Toxin 71 5.10: Untersuchung zur Reversibilität der pH-abhängigen Bindung von InsP6 an Letales Toxin 72 Bestimmung der Dissoziationskonstante KD der Bindung von InsP6 an die CPD von Letalem Toxin bei saurem pH-Wert 73 6: Expression von rekombinantem TpeL in Bacillus megaterium 74 6.1: Klonierung von pHis1522-TpeL1-1651/1-1779 74 6.2: Aufreinigung von rekombinantem TpeL 74 6.3: Untersuchungen zur in vivo-Aktivität von TpeL1-1651 und TpeL1-1779 in der Zellkultur 75 5.2.: 5.3.: 5.11: VII INHALTSVERZEICHNIS 6.4: Durch TpeL verursachte Apoptose in HeLa-Zellen 76 6.5: Substratspektrum von TpeL 77 6.6: Akzeptoraminosäure in Ha-Ras 79 6.7: Kosubstratspektrum von TpeL 80 6.8: ANQ-Aminosäuren-Motiv bestimmt die Kosubstratspezifität von TpeL 82 6.9: Autokatalytische Prozessierung von TpeL 84 6.10: Vergleich der in vivo-Glukosylierung von Ras und Rac1 86 6.11: UDP-N-Acetylglukosamin ist das Kosubstrat für die in vivo-Glukosylierung von Ras 87 6.12: Funktionelle Konsequenz der Glukosylierung von Ras 88 6.13: Inhibition Ras-abhängiger Signaltransduktionswege durch TpeL 89 6.13.1: Inhibition der Neuritenbildung in PC12-Zellen 89 6.13.2: Inhibition der Phosphorylierung von Erk-1/2 90 E: DISKUSSION 92 F: ZUSAMMENFASSUNG 107 G: LITERATUR 110 H: ABKÜRZUNGSVERZEICHNIS 119 I: ANHANG 121 J: ERKLÄRUNGEN 128 K: DANKSAGUNGEN 129 VIII EINLEITUNG A: EINLEITUNG 1: Clostridien Die Gattung Clostridium („kloster (κλωστήρ)“ – griechisch für Spindel) umfasst eine Gruppe von stäbchenförmigen, grampositiven, anaeroben endosporenbildenden Bakterien (Hatheway, 1990). Im Gegensatz zur phylogenetisch verwandten Gattung Bacillus sind die Clostridien anaerob. Die Keime kommen ubiquitär vor, hauptsächlich in anaeroben Nischen im Erdboden, aber auch im Gastrointestinaltrakt von Säugetieren. Sie sind somit ein natürlicher Teil der Darmflora von Mensch und Tier. Die meisten Clostridien sind apathogen und stellen für den Menschen keine gesundheitliche Bedrohung dar. Es existieren jedoch Vertreter, die beim Menschen unter Umständen schwere Erkrankungen hervorrufen können. Zu diesen zählen Clostridium tetani (Erreger des Tetanus), Clostridium botulinum (Erreger des Botulismus) sowie verschiedene Gasbranderreger. Insbesondere Clostridium difficile, das eine Antibiotika-assoziierte Diarrhoe und eine Pseudomembranöse Colitis verursachen kann, ist von hoher medizinischer Bedeutung (Montecucco and Molgo, 2005; Hatheway, 1990; Voth and Ballard, 2005; Montecucco et al., 2004). 2: Clostridium difficile Clostridium difficile, ursprünglich Bacillus difficile benannt, wurde zum ersten Mal 1935 durch Hall und O`Toole beschrieben. Seinen Beinamen „difficile“ (lateinisch für schwierig) verdankt der Erreger dem Umstand, dass seine Anzucht recht anspruchsvoll ist. Clostridium difficile wurde zum ersten Mal im Stuhl Neugeborener als Bestandteil der Darmflora nachgewiesen (Hall and O'Toole, 1935; Voth and Ballard, 2005). Nach seiner Entdeckung wurde der Erreger zunächst als apathogen eingestuft, da die Neugeborenen keinerlei Anzeichen einer Krankheit entwickelten. Erst 1978 konnte Clostridium difficile mit dem Krankheitsbild der Antibiotika-assoziierten Diarrhoe und der Pseudomembranösen Colitis in Verbindung gebracht werden (Bartlett et al., 1978). Unter dem Begriff Antibiotika-assoziierte Diarrhoe werden Krankheitsbilder zusammengefasst, die durch eine Diarrhoe während oder wenige Tage nach der Gabe eines Antibiotikums charakterisiert sind. Die Antibiotika-assoziierte Diarrhoe nimmt in der Regel einen unkomplizierten, milden Verlauf, jedoch kann es gelegentlich zu schwereren Verlaufsformen kommen. Die Pseudomembranöse Colitis ist eine schwerwiegende Entzündung des Dickdarms. Charakteristisch ist das Auftreten von „Pseudomembranen“. Das sind punktförmige, gelbliche Plaques auf der Kolonschleimhaut, die aus Fibrin, neutrophilen Granulozyten und Zelldebris abgestorbener Zellen bestehen. Clostridium difficile ist in der Zwischenzeit zu einem der wichtigsten nosokomialen Erreger geworden. Nahezu jedes Antibiotikum kann eine Infektion mit Clostridium difficile nach sich 1 EINLEITUNG ziehen, wobei insbesondere Breitband-Antibiotika, wie Cephalosporine, Penicilline und Lincosamide besonders häufig Verursacher sind. Die Gabe des Antibiotikums bewirkt eine Schädigung der natürlichen Darmflora. Hierdurch kommt es zu einer verstärkten Vermehrung von Clostridium difficile, da eine wechselseitige Kontrolle im Wachstum durch die natürliche Darmflora entfällt. Die Therapie der Wahl bei einer Infektion mit Clostridium difficile ist die orale Gabe von Metronidazol oder von Vancomycin (Lyerly et al., 1988; Kelly and LaMont, 2008; Poxton et al., 2001; Rupnik et al., 2009) . Die Hauptvirulenzfaktoren von Clostridium difficile sind die beiden Exotoxine, Toxin A und Toxin B. Sie glukosylieren Vertreter der kleinen GTPasen der Rho-Unterfamilie, die dadurch inaktiviert werden. Dies führt zu dem Zusammenbruch des Aktin-Zytoskeletts, welches die Funktion der tight junctions von Darmepithelzellen und somit deren Barrierefunktion beeinflusst. Eine Öffnung der tight junctions erhöht die Permeabilität der Darmmukosa (Nusrat et al., 2001; Pothoulakis, 2000). Dies führt zur charakteristischen Diarrhoe (Poxton et al., 2001). Als ein weiteres Toxin bilden circa 35% der verschiedenen Clostridium difficileStämme, insbesondere der hypervirulente Nap1/027-Stamm, das binäre Toxin CDT (Clostridium difficile-Transferase) (McDonald et al., 2005; Kelly and LaMont, 2008). CDT ADP-ribosyliert G-Aktin an Arginin-177 und führt hierdurch zu einer Hemmung der AktinPolymerisation (Gülke et al., 2001). Dies induziert die Ausbildung Mikrotubuli-basierter Zellausstülpungen an der Oberfläche von intestinalen Epithelzellen. Hierdurch wird möglicherweise die Adhäsion der Bakterien im Dickdarm erhöht (Schwan et al., 2009). 3: Clostridium perfringens, C. sordellii und C. novyi Clostridium perfringens, Clostridium sordellii und Clostridium novyi sind typische Gasbranderreger. Sie können im menschlichen Darm gefunden werden, wo sie unter normalen Umständen keine Pathogenität aufweisen. Der Gasbrand ist eine schwerwiegende, lebensbedrohliche Wundinfektion. Gasbranderreger können durch Verschmutzungen in Wunden gelangen und diese infizieren. Grundlegend für eine Infektion ist eine lokale Hypoxie, da sich die Clostridien nur unter Luftabschluss vermehren können. Die Clostridien bilden CO2 (Gasbrand!) zudem eine Reihe für die Entstehung der Krankheit verantwortlich gemachten Toxine. Infolge von Gasbildung beginnt das Gewebe anzuschwellen. Die Wunde verfärbt sich und kann ein schmutzig erscheinendes und süßlich-übelriechendes Sekret absondern. Die Therapie des Gasbrands umfasst neben einer chirurgischen Entfernung infizierter Areale, hyperbare Oxygenierung, sowie den Einsatz von Antibiotika. Die Gasbranderreger können schwere Infektionen bei Nutztieren verursachen und dadurch hohe betriebswirtschaftliche Schäden auslösen (Hatheway, 1990). Clostridium perfringens ist das am weitesten verbreitete pathogene Bakterium und ist in 70 bis 80% der Fälle der Verursacher des Gasbrandes. Neben dem Gasbrand ist das Bakterium 2 EINLEITUNG häufiger Verursacher einer nekrotisierenden Pneumonie, gangränösen Cholezystitis oder einer Sepsis. Infektionen des Zentralen Nervensystems können zu einer Meningitis und in seltenen Fällen zu einer Enzephalitis führen. Der Erreger verfügt über ein breites Arsenal an unterschiedlichen Toxinen. Aufgrund der wichtigsten Toxine, α-, β-, ε- und ι-Toxin, wird Clostridium perfringens in die Serotypen A bis E unterteilt. In der Humanmedizin sind insbesondere Serotyp A und C von großer Bedeutung. Clostridium perfringens Serotyp A ist auch ein wichtiger „Lebensmittelvergifter“. Nach Verzehr von Nahrung, die mit Sporen kontaminiert ist, bilden die ausgekeimten Clostridien das Enterotoxin, das zu einer Lebensmittelvergiftung führen kann. Wesentlich schwerwiegender ist die durch Clostridium perfringens Typ C ausgelöste Enteritis necroticans, der sogenannte Darmbrand (Hatheway, 1990). Die Serotypen A, B und C von Clostridium perfringens bilden TpeL (toxin Clostridium perfringens large), dass vor kurzem erstmals beschrieben wurde. (Amimoto et al., 2007). Neben dem Gasbrand treten Infektionen mit Clostridium sordellii hauptsächlich nach der Geburt, gynäkologischen Routineeingriffen sowie nach intravenösem Drogenmissbrauch auf. Eine Infektion kann zu einer Pneumonie, Arthritis, Peritonitis sowie Endokarditis führen. In seltenen Fällen kann es in Folge einer Infektion zu einem toxischen Schock-Syndrom kommen. Pathogene Clostridium sordellii-Stämme bilden zahlreiche Virulenzfaktoren. Die beiden Toxine Letales Toxin sowie Hämorrhagisches Toxin sind die am besten untersuchten und werden als die eigentlichen Hauptvirulenzfaktoren des Bakteriums betrachtet (Hatheway, 1990; Genth and Just, 2010). Clostridium novyi bildet ebenfalls eine Reihe verschiedener Virulenzfaktoren aus. Einer dieser Virulenzfaktoren, α-Toxin, wird hauptsächlich für die Entstehung des Gasbrandes verantwortlich gemacht. Aufgrund der verschiedenen Virulenzfaktoren wird Clostridium novyi weiter in die drei Subtypen A, B und C unterteilt (Hatheway, 1990). 4: Clostridiale Glukosylierende Toxine Die Clostridialen Glukosylierenden Toxine (CGT) sind Mitglieder einer Familie bakterieller Glukosyltransferasen. Diese Familie umfasst Toxin A und B aus Clostridium difficile, Letales Toxin und Hämorrhagisches Toxin aus Clostridium sordellii sowie α-Toxin aus Clostridium novyi. Toxin A und B sind die am besten untersuchten Vertreter und werden als Prototypen dieser Toxinfamilie angesehen (Aktories and Just, 2005; Just and Gerhard, 2004). Des Weiteren konnten Amimoto und Mitarbeiter TpeL, das ein neues Mitglied dieser Toxinfamilie darstellt, im Überstand von Clostridium perfringens Typ C identifizieren (Amimoto et al., 2007). Die CGTs besitzen eine hohe Aminosäure-Sequenzidentität von 26,3 bis 75,8% (siehe Tab.A1), sowie eine hohe molekulare Masse, die von 250 bis 308 kDa reicht (Schirmer and Aktories, 2004; Busch and Aktories, 2000). 3 EINLEITUNG 100 43,0 42,5 27,3 27,7 Toxin B 61,8 100 75,8 29,7 30,0 Letales Toxin 61,8 87,8 100 29,8 30,5 α-Toxin 47,3 52,4 52,0 100 26,3 TpeL 41,6 46,4 46,6 44,9 100 Identität Toxin A Homologie Tab.A1: Identitäten und Homologien der Primärsequenz der CGTs (modifiziert aus Busch and Aktories, 2000)) Nach der selbst-vermittelten Aufnahme in die Wirtszelle modifizieren die Toxine kleine GTPase der Rho- und/oder Ras-Familie durch Übertragung von Zuckerresten (Just et al., 1995b). Die verschiedenen Vertreter haben ein unterschiedliches Substrat- und Kosubstratspektrum. Durch diese kovalente Modifikation werden die kleinen GTPasen inaktiviert. Dies führt zu einer Disaggregation des Aktin-Zytoskeletts, das sich phänotypisch in der Abrundung kultivierter Zellen bemerkbar macht (siehe Abb.A1) (Schirmer and Aktories, 2004). A B Abb.A1: Morphologische Veränderung von Ratten-Astrozyten nach Vergiftung mit Toxin B A: Kontrolle B: Zellen, die mit Toxin B vergiftet wurden, zeigen charakteristische Veränderungen in der Zellmorphologie aufgrund des Zusammenbruchs des Aktin-Zytoskeletts. Zu diesen Veränderungen zählen die Zellabrundung und die Ausbildung „Neuriten-ähnlicher“-Strukturen (modifiziert aus Barbieri et al., 2002). 4.1: Toxin A und Toxin B von Clostridium difficile Toxin A besteht aus 2710 Aminosäuren und hat eine molekulare Masse von 308 kDa. Toxin B ist etwas kleiner und besteht, bei einer molekularen Masse von 269 kDa, aus 2366 Aminosäuren. Toxin A und Toxin B besitzen ein identisches Substrat- und Kosubstratspektrum. Die beiden Toxine übertragen Glukose aus dem Zuckerdonor UDPGlukose ausschließlich auf Vertreter der Rho-GTPasen (siehe Tab.A2) (Just et al., 1995b; Just et al., 1995c). Gewisse Isoformen von Toxin B haben ein abweichendes 4 EINLEITUNG Substratspektrum und modifizieren zum Teil Ras-GTPasen (siehe Tab.A2) (Just and Gerhard, 2004; Voth and Ballard, 2005). Toxin Organismus MM Kosubstrat Substrat (RasProteine) Substrat (Rho-Proteine) Rho, Rac, Cdc42, RhoG, TC10 Rho, Rac, Cdc42 (kDa) Toxin A-VPI10463 Clostridium difficile VPI10463 308 UDP-Glukose (Rap) Toxin A-C34 Clostridium difficile C34 Clostridium difficile VPI10463 308 UDP-Glukose Rap 269 UDP-Glukose - Clostridium difficile C34 Clostridium difficile 1470 Clostridium sordellii VPI9048 Clostridium sordellii 6018 269 UDP-Glukose Ras, Ral, Rap Rho, Rac, Cdc42, RhoG, TC10 Rho, Rac, Cdc42 269 UDP-Glukose Ras, Ral, Rap Rac 270 UDP-Glukose Ras, Rap Ras, Rap 270 UDP-Glukose Ras, Ral, Rap Clostridium sordellii VPI9048 Clostridium novyi 19402 ~300 UDP-Glukose - Rac, (Cdc42), RhoG, Tc10 Rho, Rac, Cdc42 250 UDP-NAcetylglukosamin - Rho, Rac, Cdc42 Toxin B-VPI10463 Toxin B-C34 Toxin B-1470 Letales Toxin-VPI9048 Letales Toxin-6018 Hämorrhagisches ToxinVPI9048 α-Toxin-19402 Tab.A2: Kosubstrat- und Substratspezifität der CGTs In Klammer: Substrate aufgeführt, die nur schwach modifiziert werden bzw. wahrscheinlich keine in vivo Substrate darstellen. MM: molekulare Masse. (modifiziert aus Schirmer and Aktories, 2004; Just and Gerhard, 2004) Toxin B besitzt in den meisten Zellmodellen (Ausnahme intestinale Epithelzellen) eine 100 bis 1000-fach höhere zytotoxische Aktivität als Toxin A. Im Gegensatz zu Toxin B weist jedoch Toxin A eine deutlich höhere Toxizität in Tiermodellen auf. Aufgrund dieser beiden Beobachtungen wurde Toxin A zunächst als das Enterotoxin und Toxin B als das Zytotoxin bezeichnet (Lyerly et al., 1982; Lyerly et al., 1985; Triadafilopoulos et al., 1987). Das Auftreten von Toxin A-negativen/Toxin B-positiven Clostridium difficile-Stämmen, die das typische Krankheitsbild einer Pseudomembranösen Colitis auslösen können, lässt an diesem Bild zweifeln (Lyerly et al., 1992). Die Frage, welches Toxin in welchem Umfang zur Ausbildung des Krankheitsbildes beiträgt, wird widersprüchlich diskutiert und konnte bis heute nicht abschließend geklärt werden (Lyras et al., 2009; Kuehne et al., 2010) 4.2: Letales Toxin von Clostridium sordellii Letales Toxin besteht aus 2364 Aminosäuren und besitzt eine molekulare Masse von 270 kDa. Es hat eine hohe Sequenzähnlichkeit zu Toxin B (siehe Tab.A1). Obwohl Letales Toxin ebenfalls UDP-Glukose als Kosubstrat nutzt, weist es ein im Vergleich zu Toxin B abweichendes Substratspektrum auf. Zusätzlich glukosyliert es neben Rho-GTPasen auch Vertreter der Ras-GTPasen (siehe Tab.A2) (Just and Gerhard, 2004; Schirmer and Aktories, 2004; Just et al., 1996). 5 EINLEITUNG 4.3: α-Toxin von Clostridium novyi α-Toxin besteht aus 2178 Aminosäuren bei einer molekularen Masse von 250 kDa. Das Toxin bevorzugt als Kosubstrat UDP-N-Acetylglukosamin und überträgt den Zucker auf verschiedene Rho-GTPasen (siehe Tab.A2). Das Substratspektrum entspricht dem Spektrum von Toxin A und Toxin B (Just and Gerhard, 2004; Schirmer and Aktories, 2004; Selzer et al., 1996). 5: Das ABCD-Modell der CGTs Die Clostridialen Glukosylierenden Toxine sind typische monomere AB-Toxine und bestehen aus einer biologisch aktiven A-Komponente, der Glukosyltransferase-Domäne, und einer BKomponente, die die Aufnahme der Toxine in die Wirtszelle vermittelt (Giesemann et al., 2008; Von Eichel-Streiber et al., 1996). Die Multidomänen-Struktur der Toxine lässt sich am besten durch das modulare ABCD-Modell erklären (siehe Abb.A2) (Jank and Aktories, 2008). A B GTD N 1 CPD 543 PBR 830 TD 990 RBD 1500 1852 C RBD/CROP 2366 Abb.A2: Multidomänen-Struktur von Toxin A und Toxin B Oben: Toxin B ist ein typisches AB-Toxin mit der biologisch aktiven A-Komponente im N-Terminus, der Glukosyltransferase-Domäne (GTD) und der B-Komponente im C-Terminus, die die Aufnahme der AKomponente in die Zielzelle vermittelt. Die B-Komponente besteht aus einer Cysteinprotease-Domäne (CPD), einer Poren-bildenden Region (PBR)/Translokations-Domäne (TD) sowie einer Rezeptorbindungs-Domäne (RBD/CROP). Es wird vermutet, dass die CGTs eine weitere Rezeptorbindungs-Domäne (RBD) besitzen (Olling et al., 2011). Unten: Elektronenmikroskopische 3D-Rekonstitution von Toxin A (Pruitt et al., 2010). Die vier Module des ABCD-Modell sind in unterschiedlichen Farben dargestellt (rot: Glukosyltransferase-Domäne, blau: Cysteinprotease-Domäne, gelb: Poren-bildende Region/Translokations-Domäne, grün: RezeptorbindeDomäne/CROP). 5.1: Glukosyltransferase-Domäne Im N-Terminus der CGTs ist die enzymatisch aktive Glukosyltransferase-Domäne lokalisiert (Hofmann et al., 1997). Sie ist für die biologische Aktivität der Toxine durch Glukosylierung kleiner GTPasen verantwortlich (Selzer et al., 1996; Just et al., 1995c; Just et al., 1996; Just et al., 1995b). Anhand der Kristallstruktur der Glukosyltransferase-Domäne von Toxin B 6 EINLEITUNG (siehe Abb.A3) konnte diese der Glykosyltransferase A-Familie zugeordnet werden (Reinert et al., 2005). Aus der Kristallstruktur konnten einige für den Reaktionsmechanismus essentielle Aminosäuren identifiziert werden (siehe Abb.A4). Das charakteristische DXDMotiv (D286 und D288 für Toxin B) ist an der Bindung des Kosubstrats beteiligt (Busch et al., 1998). Die Aspartat-Reste binden direkt oder über ein Wassermolekül ein Mn2+-Ion, das seinerseits an der Bindung der Phosphate der UDP-Glukose beteiligt ist. Das Aspartat-286 ist weiterhin beteiligt an der Bindung der 3´-Hydroxylgruppe der UDP-Ribose sowie der Glukose (Jank et al., 2007a; Reinert et al., 2005). A B Abb.A3: Kristallstruktur der Glukosyltransferase-Domäne von Toxin B A: Darstellung der Kristallstruktur der Glukosyltransferase-Domäne von Toxin B als ribbon-Modell. Die Bereiche der typischen Glykosyltransferase Typ A-Faltung sind blau dargestellt, zusätzliche Bereiche der Glukosyltransferase-Domäne von Toxin B sind grün dargestellt. Im aktiven Zentrum ist als ball-and-stick-Modell 2+ UDP, α-D-Glukose und ein Mn -Ion (in Magenta) sowie Aspartat-286 und Aspartat-288 zu sehen. B: Darstellung der Kristallstruktur der Glukosyltransferase-Domäne von Toxin B als Oberflächen-Modell. Farbliche Darstellung entspricht der linken Abbildung. Zusätzlich in Gelb sind Aminosäuren 1 bis 18 dargestellt. Dieser Bereich hat wahrscheinlich Membran-bindende Eigenschaften (Mesmin et al., 2004). Im aktiven Zentrum sind als zwei Bälle γ1 die durchschnittliche Akzeptorsauerstoffposition (orange) sowie die Position des Threonin-37 O (schwarz) dargestellt. (modifiziert aus Reinert et al., 2005) Die Aminosäure Tryptophan-102 ist ebenfalls an der Bindung von UDP-Glukose beteiligt, in dem sie über π-π Wechselwirkungen mit dem Uracilring interagiert (Jank et al., 2007a; Busch et al., 2000). Die Aminosäuren Arginin-455, Aspartat-461, Lysin-463, Glutamat-472, Glutamat-449 der Helix 17 von Toxin B sind an der Bindung des Proteinsubstrates beteiligt (Jank et al., 2007a). Aminosäuren 510 bis 523 bilden die sogenannte „flexible Schleife“. Diese Struktur vollzieht vermutlich eine Konformationsänderung, wodurch nach Übertragung des Zuckers der Substratauswurf erfolgt. Tryptophan-520 ist nicht nur an der Koordination des UDP-Zuckers, sondern darüber hinaus an der Gewährleistung der richtigen Konformation und Position der flexiblen Schleife beteiligt (Jank and Aktories, 2008). 7 EINLEITUNG A B Abb.A4: Essentielle Aminosäuren für die Substrat- und Kosubstratbindung von Toxin B A: Darstellung für die Bindung des Kosubstrats essentieller Aminosäuren von Toxin B. W1 und W2 sind koordinierte Wassermoleküle. Die entsprechenden Abstände sind in Angström angeben. B: Darstellung für die Kosubstratspezifität entscheidender Aminosäuren von Toxin B (links) und α-Toxin (rechts). Toxin B benutzt als Kosubstrat UDP-Glukose, im Unterschied zu α-Toxin, welches UDP-N-Acetylglukosamin als Kosubstrat nutzt. Im α-Toxin ist Isoleucin-383 und Glutamin-385 von Toxin B durch Serin-383 und Alanin-385 ersetzt worden. Die sterisch anspruchsvollen Aminosäuren Isoleucin und Glutamin verhindern, dass UDP-N-Acetylglukosamin in der katalytischen Tasche von Toxin B binden kann (modifiziert aus Jank et al., 2005; Jank et al., 2007a). Bei der Übertragung des Zucker-Restes auf die GTPase bleibt die α-Konfiguration des anomeren Kohlenstoffes der Glukose erhalten (Vetter et al., 2000; Geyer et al., 2003). Der stereospezifische SN2-ähnliche Reaktionsmechanismus läuft wahrscheinlich über eine Oxocarbenium-Ion-Zwischenstufe ab (Reinert et al., 2005). Die Clostridialen Glukosylierenden Toxine variieren in ihrer Kosubstratspezifität. Im Gegensatz zu Toxin B nutzt α-Toxin UDP-N-Acetylglukosamin als Kosubstrat (Selzer et al., 1996). Die Aminosäuren Isoleucin-383 und Glutamin-385 in der katalytischen Tasche von Toxin B sind im α-Toxin in den äquivalenten Positionen durch Serin und Alanin ersetzt (siehe Abb.A4). Es wird diskutiert, dass die sterisch größeren Aminosäuren Isoleucin und Glutamin den Zugang von UDP-N-Acetylglukosamin in die katalytische Tasche erschweren und somit dafür verantwortlich sind, dass Toxin B UDP-Glukose als Kosubstrat nutzt (Jank et al., 2005). 5.1.1: Kleine GTPasen als Proteinsubstrate der CGTs Kleine GTPasen sind wichtige molekulare Schalter in einer Vielzahl von Signalprozessen. Rho-Proteine sind Bestandteile verschiedener Signaltransduktionswege zum Aktin- 8 EINLEITUNG Zytoskelett. Sie spielen eine wichtige Rolle bei der Zellmigration, Morphogenese und Zellpolarität. Ras-Proteine sind essentiell für die Regulation von Zelldifferenzierung und Proliferation (Takai et al., 2001). Die Aktivität der kleinen GTPasen wird in dem GTPaseZyklus durch die Bindung von GTP und GDP reguliert (siehe Abb.A5). CGTs inaktiv GDP GDI GTP GTP Glukose Rho GDP Rho GDP inaktiv inaktiv Glukose Pi GAP Rho GTP Effektor Glukose GEF Glukose Rho aktiv Abb.A5: Kleine GTPasen als Proteinsubstarte der CGTs Die kleinen GTPasen (hier: Rho) werden durch den dargestellten GTPase-Zyklus gesteuert. Im GTP-gebundenen Zustand sind die kleinen GTPasen aktiv bzw. im GDP-gebundenen Zustand inaktiv. In ihrem aktiven Zustand interagieren die GTPasen mit verschiedenen Effektoren und sind somit wichtige molekulare Schalter in zahlreichen Signaltransduktionswegen. Der GTPase-Zyklus wird durch GEF-, GAP- und GDI-Proteine beeinflusst. GEF-Proteine induzieren den Austausch von GDP zu GTP. GAP-Proteine steigern die intrinsische GTP-HydrolaseAktivität der kleinen GTPasen. GDI-Proteine halten inaktive GTPasen löslich im Zytosol. Die OMonoglukosylierung der kleine GTPasen durch die CGTs an einem Threonin-Rest (Position 35/37) führt zu einer Inaktivierung. Die GTPasen werden hauptsächlich in ihrem inaktiven GDP-gebundenen Zustand modifiziert. Die modifizierten GTPasen können nicht länger mit ihren Effektoren interagieren, durch GEF-Proteine aktiviert werden sowie mit GDI-Proteinen interagieren (modifiziert aus Jank and Aktories, 2008). Im GDP-gebundenen Zustand sind die kleinen GTPasen zunächst inaktiv (Jaffe and Hall, 2005). Der GTPase-Zyklus wird durch drei verschiedene Klassen von Proteinen beeinflusst. GEF-Proteine (guanine nucleotide exchange factor) induzieren den Austausch von GDP zu GTP (Rossman et al., 2005). Im GTP-gebundenen Zustand sind die GTPasen aktiv und können mit ihren Effektorproteinen interagieren. Die Aktivität wird durch die Hydrolyse von GTP zu GDP beendet. Die intrinsische GTP-Hydrolyse kann durch GAP-Proteine (GTPase activating protein) weiter gesteigert werden. Im inaktiven Zustand interagieren die kleinen GTPasen mit GDI-Proteinen (guanine nucleotide dissociation inhibitor) (Bernards and Settleman, 2004; Olofsson, 1999). In der aktivierten Form sind die GTPasen, vermittelt durch ihre Isoprenyl-Gruppe, an der Zytoplasmamembran verankert. GDI-Proteine halten inaktive GTPasen löslich im Zytosol. Sie sind somit an der subzellulären Lokalisation der kleinen GTPasen beteiligt (Etienne-Manneville and Hall, 2002). Die CGTs modifizieren verschiedene 9 EINLEITUNG Vertreter der Rho- sowie Ras-GTPasen durch eine O-Monoglukosylierung, wodurch diese inaktiviert werden (Just et al., 1995b; Just et al., 1996; Just et al., 1995c; Selzer et al., 1996). Die Übertragung der Zuckermoleküle erfolgt auf ein hoch konserviertes Threonin in Position 35 für Rac1 bzw. Position 37 für RhoA. Das Threonin-35/37 befindet sich in der switch-IRegion und ist über ein Mg2+-Ion an der Bindung der Nukleotide beteiligt. In dieser Region kommt es zu starken Konformationsänderungen nach Austausch der Nukleotide. Die Glukosylierung der kleinen GTPasen hat mehrere funktionelle Konsequenzen (siehe Abb.A5). Glukosylierte Rho/Ras-Proteine können nicht mehr durch GEF-Proteine aktiviert werden, nicht mehr mit ihren Effektoren interagieren sowie nicht mehr in ihre aktive, GTPgebunden Form überführt werden. Weiterhin können die GTPasen nicht länger mit GDI interagieren, das zu einer Relokalisation von inaktiven GTPasen an die Zytoplasmamembran führt (Sehr et al., 1998; Giesemann et al., 2008; Vetter et al., 2000; Geyer et al., 2003; Herrmann et al., 1998). Die GTPasen werden hauptsächlich in dem GDP-gebunden Zustand modifiziert, da das Threonin-35/37 im GTP-gebundenen Zustand für eine Glukosylierung schlechter zugänglich ist (Just et al., 1995b). 5.2: Rezeptorbindungs-Domäne Die Clostridialen Glukosylierenden Toxine werden gemäß dem short trip uptake-Modell aufgenommen (siehe Abb.A6) (Giesemann et al., 2008; Sandvig et al., 2004). Zunächst werden die Toxine nach Rezeptorbindung über Rezeptor-vermittelte Endozytose in Endosomen aufgenommen (Florin and Thelestam, 1983). Der Endozytose-Mechanismus wird hauptsächlich über Rezeptorbindungs-Domäne Clathrin gesteuert ist C-Terminus im (Papatheodorou lokalisiert und et al., 2010). Die besteht aus sich wiederholenden Oligopeptid-Einheiten (Frisch et al., 2003; Von Eichel-Streiber et al., 1996; Von Eichel-Streiber et al., 1992). Diese sogenannten CROPs (combined repetitive oligopeptides) bestehen für Toxin A und Toxin B aus 20 bis 22 oder 50 Aminosäuren. Die Oligopeptid-Wiederholungen sind in einer β-Haarnadel-Struktur angeordnet. Die einzelnen Haarnadeln sind um 120° gegeneinander verdreht. Die somit erzeugte, solenoid-ähnliche Struktur, weist eine große Oberfläche auf, welche die Ausbildung von Wechselwirkungen des Toxins mit Proteinen oder Kohlenhydrat-Strukturen der Wirtszelle ermöglicht (Greco et al., 2006; Ho et al., 2005). Die CROP-Bereiche weisen eine hohe Ähnlichkeit zu Kohlenhydratbindenden Proteinen aus Streptococcus mutans und Streptococcus downei auf (Von EichelStreiber and Sauerborn, 1990). 10 EINLEITUNG Dynamin saurer endosomaler pH-Wert Autoprozessierung Endosom Bindung von InsP6 Porenbildene Region/TranslokationsDomäne Glukosylierung aktiv Cysteinprotease-Domäne inaktiv -Glukose kleine GTPase + UDP-Glukose Glukosyltransferase-Domäne Rezeptorbinde-Domäne Rezeptor Clathrin clathrincoated pit Eps15 Abb.A6: Aufnahme der CGTs in Zielzellen Die CGTs werden gemäß des short trip uptake-Modells in die Zielzellen aufgenommen. Zunächst binden die Toxine über ihre Rezeptorbindungs-Domäne an bis heute noch nicht identifizierte, zelluläre Rezeptoren. Die Toxin-Rezeptor-Komplexe werden anschließend über Clathrin-vermittelte Endozytose in die Endosomen + aufgenommen. Die Endosomen werden schrittweise über eine membranständige, vesikuläre H -ATPase angesäuert. Der saure pH-Wert in den frühen Endosomen induziert eine Konformationsänderung der CGTs, wodurch zuvor verdeckte, hydrophobe Bereiche freigelegt werden. Dieser Anstieg in der Hydrophobizität erlaubt es den CGTs in die endosomale Membran zu inserieren und eine Pore auszubilden. Durch diese Pore findet wahrscheinlich die Translokation der Glukosyltransferase-Domäne ins Zytosol statt. Die Glukosyltransferase-Domäne wird autoproteolytisch durch eine intrinsische Cysteinprotease-Domäne abgespalten. Die Aktivität der Cysteinprotease-Domäne wird durch Inositolhexakisphosphat, einen niedermolekularer Bestandteil des Zytosols, induziert. Im Zytosol katalysiert die Glukosyltransferase-Domäne die Übertragung eines Zuckerrestes aus einem UDP-Zuckerdonor auf verschiedene Vertreter der Rho/RasUnterfamilie der kleinen GTPasen. Durch diese Modifikation werden die GTPasen inaktiviert (modifiziert aus Pruitt et al., 2010). Die Rezeptoren, die zur Aufnahme der Toxine in die Wirtszellen dienen, konnten bis heute nicht eindeutig identifiziert werden. Lediglich für Toxin A sind Lektin-ähnliche Eigenschaften beschrieben. Toxin A bindet an verschiedene, strukturell ähnliche Kohlenhydrat-Strukturen (Krivan et al., 1986; Tucker and Wilkins, 1991). Als Protein-Rezeptoren für Toxin A werden darüber hinaus die Sucrase-Isomaltase sowie das zytosolische Chaperon gp96 diskutiert (Na et al., 2008; Pothoulakis et al., 1996). Toxin A und Toxin B scheinen unterschiedliche Rezeptoren für ihre Aufnahme zu benutzen. In polarisierten Darmepithelzellen wird Toxin B verstärkt von der basolateralen Seite, Toxin A hingegen von der apikalen Seite 11 EINLEITUNG aufgenommen (Stubbe et al., 2000). In jüngster Zeit konnte gezeigt werden, dass das TpeLToxin sowie Mutanten von Toxin A und Toxin B, welche eine Deletion der CROP-Bereiche aufweisen, zytotoxisch für kultivierte Zellen sind. Aus diesem Umstand kann geschlossen werden, dass die Toxine eine weitere Rezeptorbindungs-Domäne aufweisen, die N-terminal zu dem CROP-Bereich lokalisiert ist (Genisyuerek et al., 2011; Amimoto et al., 2007; Olling et al., 2011). 5.3: Cysteinprotease-Domäne Für Toxin B wurde beschrieben, dass das Toxin während der Aufnahme eine Prozessierung erfährt. Lediglich die N-terminale Glukosyltransferase-Domäne wird aus dem Endosom transloziert und kann im Zytosol der Wirtszellen nachgewiesen werden (Pfeifer et al., 2003). Die Prozessierung wird durch das Toxin selbst vermittelt. Eine intrinsische CysteinproteaseDomäne vermittelt die Autoprozessierung durch Abspaltung der GlukosyltransferaseDomäne (Egerer et al., 2007). 5.4: Poren-bildende Region/Translokations-Domäne Die Translokation der Glukosyltransferase-Domäne in das Zytosol erfolgt wahrscheinlich aus den frühen Endosomen. Die Endosomen werden durch eine membranständige, vesikuläre H+-ATPase angesäuert. Der saure pH-Wert in den Endosomen induziert eine Konformationsänderung der Toxine, wodurch, zuvor verborgene, hydrophobe Bereiche freigelegt werden. Dies ermöglicht den Toxinen in die endosomale Membran zu inserieren und dort eine Pore zu bilden (Barth et al., 2001; Qa'Dan et al., 2000). Durch diese Pore findet vermutlich die Translokation der Glukosyltransferase-Domäne in das Zytosol statt. Für Toxin A und Toxin B konnte gezeigt werden, dass beide Proteine in der Lage sind, nach Ansäuern an der Oberfläche von kultivierten Zellen Poren zu bilden (Barth et al., 2001; Giesemann et al., 2006). Die Porenbildung von Toxin A scheint, im Gegensatz zu Toxin B, Cholesterolabhängig zu sein (Giesemann et al., 2006). Aufgrund von Hydrophobizitätsanalysen wurden die porenbildenden Eigenschaften zunächst einer hydrophoben Region zwischen Aminosäure 956 bis 1128 (für Toxin B) zugeschrieben (Von Eichel-Streiber et al., 1992; Jank and Aktories, 2008; Dove et al., 1990). Kürzlich konnte diese Poren-bildende Region auf Aminosäure 830 bis 990 (für Toxin B) eingrenzt werden (Genisyuerek et al., 2011). Innerhalb dieses Bereiches fungieren offenbar Glutamate (E970, E976) als pH-Sensoren. Die sauren Aminosäuren liegen in postulierten Schleifen-Bereichen von Helix-Schleifen-Helix-Strukturen. Bei neutralen pH-Werten sind sie zunächst deprotoniert und weisen eine negative Ladung auf. Nach dem Wechsel zu sauren pH-Werten liegen die Säuregruppen der Glutamate undissoziiert vor und ermöglichen eine Insertion hydrophober Schleifen-Bereiche in 12 EINLEITUNG biologische Membrane. Über den genauen Ablauf der Translokation der GlukosyltransferaseDomäne ist bis heute nur wenig bekannt. Es wird vermutet, dass die Translokation durch einen zusätzlichen Bereich zwischen Aminosäure 990 bis 1500 (für Toxin B) vermittelt wird (Genisyuerek et al., 2011). 6: Inositolhexakisphosphat-vermittelte Autoprozessierung 6.1: Prozessierung bakterieller AB-Toxine Typische bakterielle AB-Toxine bestehen aus einem Effektor (A-Komponente) und einer Rezeptorbindungs- und Translokations-Domäne (B-Komponente), die die Aufnahme des Effektors in die Wirtszelle vermittelt. Bei binären AB-Toxinen bilden die A- und BKomponenten entweder schon im Bakterium (z.B. Cholera-Toxin) oder erst nach Bindung der B-Komponente an den zellulären Rezeptoren (z.B. Anthrax-Toxine) einen Komplex (de Haan and Hirst, 2004; Collier and Young, 2003; Barth et al., 2004). Bei monomeren ABToxinen werden die beiden Komponenten als ein durchgängiges Oligopeptid gebildet. Sowohl binäre als auch monomere AB-Toxine können während der Aufnahme der Toxine in die Wirtszelle eine Prozessierung erfahren. Die Prozessierung kann durch zusätzliche bakterielle Proteasen oder durch endogene Wirtsproteasen erfolgen. Das Protektive Antigen (PA), die B-Komponente der binären Anthrax-Toxine, wird beispielsweise durch die Wirtsprotease Furin prozessiert (Collier and Young, 2003). Die Prozessierung bakterieller Toxine ermöglicht die gezielte Aufnahme kleiner, aktiver Effektor-Domänen (Egerer and Satchell, 2010). 6.2: Autoprozessierung Bakterielle Toxine können darüber hinaus auch mittels einer intrinsischen Protease prozessiert werden. Diese Autoprozessierung ist insbesondere für Toxine mit einer Multidomänenstruktur beschrieben worden (Egerer and Satchell, 2010; Reineke et al., 2007). Zu diesen Toxinen zählen MARTX-Toxine (multifunctional autoprocessing RTX-like) aus Vibrio cholerae, Vibrio vulnificus und Photorhabdus luminescens sowie die Clostridialen Glukosylierenden Toxine. 6.3: Intrinsische Cysteinprotease-Domäne Es wurde schon früh angenommen, dass die CGTs während der Aufnahme proteolytisch prozessiert werden (Henriques et al., 1987; Pfeifer et al., 2003). Zunächst wurde vermutet, dass eine endogene Wirtsprotease für die Prozessierung von Toxin B verantwortlich ist, da durch die Zugabe von Zelllysaten die Prozessierung eingeleitet werden konnte. Da jedoch 13 EINLEITUNG auch proteinfreier Zellextrakt die Prozessierung einleiten konnte, wurde von einer Autoprozessierung ausgegangen (Reineke et al., 2007). Die Autoprotease-Domäne konnte schließlich für das MARTXVc als eine intrinsische Cysteinprotease-Domäne (CPD) identifiziert werden (Sheahan et al., 2007). Aufgrund von Sequenzvergleichen zeigte sich, dass die Cysteinprotease-Domäne auch in den CGTs konserviert vorliegt (Egerer et al., 2007; Sheahan et al., 2007). Toxin A und Toxin B haben eine katalytische Triade, die neben den charakteristischen Cystein- (Toxin A: C700, Toxin B: C698) und Histidin-Resten (Toxin A: H655, Toxin B: H653) zusätzlich einen Aspartat-Rest (Toxin A: D589, Toxin B: D567) aufweist (Egerer et al., 2007; Pruitt et al., 2009). Die Peptidspaltung erfolgt stets nach einem Leucin-Rest. Für Toxin A und Toxin B konnten die Schnittstellen zwischen Leucin-542 und Alanin-543 bzw. zwischen Leucin-543 und Glycin-544 identifiziert werden (Rupnik et al., 2005; Egerer et al., 2007; Pruitt et al., 2009; Kreimeyer et al., 2011). 6.4: Inositolhexakisphosphat-induzierte Autoprozessierung Die Autoprozessierung wird durch Inositolphosphate aus dem Zytosol der Wirtszelle induziert. Für Toxin B wurde Inositolhexakisphosphat (InsP6) als der stärkste Aktivator beschrieben (Reineke et al., 2007). InsP6 bindet über ionische Wechselwirkungen an die Cysteinprotease-Domäne. Die Bindung erfolgt über die negativ geladenen Phosphatgruppen des InsP6 und über positiv geladene Arginin- und Lysin-Reste der CPD (Egerer et al., 2009; Pruitt et al., 2009). Für Toxin B konnte z.B. Lysin-600 als eine für die Bindung essentielle Aminosäure beschrieben werden (Egerer et al., 2009). Toxin B zeigt erst ab Konzentrationen von 1 µM InsP6 eine Autoprozessierung (Egerer et al., 2007). Die anderen Vertreter der CGTs, insbesondere Toxin A, benötigen millimolare InsP6-Konzentrationen für die Autoprozessierung (Reineke et al., 2007). Fragmente von Toxin A zeigen im Gegensatz zu dem Holotoxin bereits in niedrigen mikromolaren Konzentrationen eine Autoprozessierung (Pruitt et al., 2009). Es ist wichtig, darauf hinzuweisen, dass die physiologischen Konzentrationen an InsP6 in eukaryoten Zellen zwischen 10 und 60 µM liegen (Egerer and Satchell, 2010; Irvine and Schell, 2001). 6.5: Kristallstruktur von Toxin A-CPD Die Kristallstruktur der Cysteinprotease-Domäne von Toxin A und Toxin B (siehe Abb.A7) konnte kürzlich von verschiedenen Arbeitsgruppen gelöst werden (Pruitt et al., 2009; Puri et al., 2010). Die größte strukturelle Ähnlichkeit zeigt die Cysteinprotease-Domäne von Toxin A zu Caspase-7 (Pruitt et al., 2009). Beide CPD-Strukturen haben eine ähnliche Faltung mit einem zentralen, mehrsträngigem β-Faltblatt, flankiert von mehreren α-Helices. Am Cterminalen Ende verschiedener Stränge des β-Faltblattes sind die Aminosäuren der 14 EINLEITUNG katalytischen Triade lokalisiert. Das katalytische Cystein und Histidin sind über 6 Å voneinander entfernt (Pruitt et al., 2009). Dies lässt vermuten, dass das katalytische Cystein durch das Substrat aktiviert wird und nicht durch Protonierung des katalytischen Histidins. Das katalytische Histidin dient zur Protonierung der Abgangsgruppe. Das katalytische Aspartat ist bei der korrekten Anordnung des katalytischen Histidins beteiligt (Egerer and Satchell, 2010). Wichtige strukturelle Merkmale der CPD sind die InsP6-Bindetasche und die hydrophobe Substrat-Bindetasche. Ein Vielzahl der direkt mit InsP6 interagierenden Aminosäuren sind positiv geladene Arginin- und Lysin-Reste. Die Orientierung von InsP6 innerhalb dieser Tasche ist zwischen Toxin A- bzw. Toxin B-CPD und MARTXVc-CPD nicht konserviert (Pruitt et al., 2009; Lupardus et al., 2008). Die hydrophobe Substrat-Bindetasche bindet die P1-Aminosäure (N-terminale Aminosäure zur gespaltenen Bindung) der Schnittstelle. Im Unterschied zur Caspase 7 sind zwei C-terminale Helices durch eine dreisträngige Struktur, dem sogenannten β-flap, ersetzt. Der β-flap befindet sich zwischen der InsP6-Bindetasche und der hydrophoben Substrat-Bindetasche und trägt zu beiden Strukturmerkmalen essentielle Aminosäuren bei. InsP6 fungiert als ein allosterischer Aktivator der Cysteinprotease-Domäne. Nach Bindung von InsP6 erfährt der β-flap eine Konformationsänderung, durch welche es zu einer korrekten Orientierung der katalytischen Aminosäuren sowie der Substrat-Bindetasche kommt. Dieser allosterische Mechanismus ermöglicht somit eine ortsgebundene Autoprozessierung der Toxine und verhindert eine Prozessierung der Toxine vor dem Erreichen ihres Wirkungsortes (Egerer and Satchell, 2010; Pruitt et al., 2009). A B Abb.A7: Kristallstruktur der Toxin A-CPD A: Darstellung der Kristallstruktur der Toxin A-CPD als ribbon-Modell. B: Darstellung der Kristallstruktur der Toxin A-CPD als Oberflächen-Modell (modifiziert aus Shen, 2010). 15 ZIELSETZUNG DER ARBEIT B: ZIELSETZUNG DER ARBEIT Die Clostridialen Glukosylierenden Toxine (CGT) werden mittels Rezeptor-vermittelter Endozytose zunächst in die Endosomen ihrer Zielzellen aufgenommen. Die endosomalen Kompartimente erfahren eine Ansäuerung durch eine vesikuläre H+-ATPase. Die Erniedrigung des pH-Werts induziert eine Konformationsänderung der Toxine. Durch diese Konformationsänderung werden hydrophobe Proteinabschnitte nach außen exponiert, die die Insertion der Toxine in die endosomale Membran ermöglichen. Ein weiterer, wesentlicher Schritt im Intoxikationsvorgang dieser Toxine ist die Translokation und Freisetzung der Glukosyltransferase-Domäne aus dem Endosom Glukosyltransferase-Domäne autokatalytisch vom ins Zytosol. restlichen Hierbei Toxin wird die abgespalten. Untersuchungen zur Autoprozessierung der CGTs konzentrierten sich in der Vergangenheit zumeist auf die beiden Vertreter Toxin A und B aus Clostridium difficile. Es konnte gezeigt werden, dass eine intrinsische Cysteinprotease-Domäne (CPD) für die autokatalytische Abspaltung der Glukosyltransferase-Domäne von Toxin A und B verantwortlich ist. Die CPD von Toxin A und B wird durch die Bindung von Inositolhexakisphosphat, einem Bestandteil des Zytosols, aktiviert. In der vorliegenden Arbeit sollten Untersuchungen zur Autoprozessierung der CGTs Letales Toxin aus Clostridium sordellii und α-Toxin aus Clostridium novyi durchgeführt werden. Es sollte untersucht werden, inwiefern die autokatalytische Prozessierung mit Toxin A und B vergleichbar ist. Hierfür war es zunächst notwendig, die rekombinante Herstellung der CGTs zu etablieren und geeignete Toxinkonstrukte, die zur Untersuchung der autokatalytischen Prozessierung dieser beiden Toxine dienen, zu generieren. Die Etablierung eines geeigneten Expressionssystems für die CGTs sollte auch für TpeL aus Clostridium perfringens, einem neuen und bisher kaum untersuchten Mitglied dieser Toxinfamilie, genutzt werden. Rekombinantes TpeL sollte sowohl in vitro und in vivo funktionell charakterisiert werden. Für TpeL sollten die Fragen der Substrat- und Kosubstratspezifität sowie zur Autoprozessierung des Toxins beantwortet werden. 16 MATERIAL UND METHODEN C: MATERIAL UND METHODEN C.1: MATERIAL 1: Biologische Materialien 1.1: Bakterienstämme und Anzuchtmedien 1.1.1: Clostridien-Stämme Zur Gewinnung von chromosomaler DNA sowie zur Aufreinigung nativer CGTs wurden die unten aufgeführten Clostridien-Stämme benutzt. Clostridium difficile VPI 10463 Clostridium sordellii 6018 Clostridium novyi 19402 Clostridium perfringens CN3685 1.1.2: Bacillus megaterium-Stämme Zur Expression rekombinanter Proteine in Bacillus megaterium wurden W320-Stämme verwendet. Bacillus megaterium W320 (MoBiTec, Göttingen, Deutschland) lac- xyl+ 1.1.3: Escherichia coli-Stämme: Zur Expression rekombinanter Proteine sowie zur Plasmid-DNA-Vermehrung in Escherichia coli wurden BL21- bzw. TOP10-, XL1-Blue-, TG1-, Rosetta-Stämme verwendet. Escherichia coli BL21 (Stratagene, LaJolla, USA) E. coli B F- dcm ompT hsdS(rB- mB-) gal [malB+]K-12(λS) Escherichia coli TOP10 (Invitrogen, Darmstadt, Deutschland) F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 nupG recA1 araD139Δ(ara-leu)7697 galE15 galK16 rpsL(StrR)endA1 λEscherichia coli XL1-Blue (Stratagene, LaJolla, USA) recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F´ proAB lacIqZΔM15 Tn10 (Tetr)] Escherichia coli TG1 (Lucigen, Middleton, USA) supE, thi-1, Δ(lac-proAB), Δ(mcrB-hsdSM)5, (rK-mK-), [F´traD36, proAB, lacIq ΔM15] Escherichia coli Rosetta (Novagen, Darmstadt, Deutschland) F- ompT hsdSB(RB- mB-) gal dcm λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) pLysSRARE (Cam R) 1.1.4: Nährmedien für Clostridien BHI (brain heart infusion)-Medium (Difco, Augsburg, Deutschland) BHI-Agarplatte: zusätzlich 2% Agar 17 MATERIAL UND METHODEN 1.1.5: Nährmedium für Bacillus megaterium und Escherichia coli LB („Luria-Bertani“)-Medium (je 1 l): 10 g Bactotrypton, 10 g NaCl, 5 g Hefeextrakt, H2O LB-Agarplatte: zusätzlich 2% Agar 1.1.6: Antibiotika Kanamycin (30 µg/ml) (Roth, Karlsruhe, Deutschland) Ampicillin (100 µg/ml) (Roth, Karlsruhe, Deutschland) Tetrazyklin (100 µg/ml) (Sigma-Aldrich, Taufkirchen, Deutschland) 1.2: Eukaryote Zelllinien Die eukaryoten Zelllinien wurden in den angebenden Kulturmedien mit den entsprechenden Zusätzen bei 37°C und einem CO2-Gehalt von 5% kultiviert. Die Zellen wurden alle zwei bis drei Tage mit PBS gewaschen, trypsiniert und erneut in frischem Medium ausgesät. Vero (ATCC: CCL 81): Nierenzelllinie aus der grünen Meerkatze Kulturmedium: DMEM + 10% FCS, 1% Na-Pyruvat, 1% NEA, 100 U/ml Penicillin und 100 mg/ml Streptomycin CHO-K1 (ATCC: CCL 61): Ovariale Zelllinie aus dem chinesischen Hamster Kulturmedium: Ham´s F12 +10% FCS, 2 mM L-Glutamat, 100 U/ml Penicillin und 100 mg/ml Streptomycin HeLa (ATCC: CCL2): Humane epitheliale Zervix-Karzinomazelllinie Kulturmedium: DMEM +10% FCS, 1% Na-Pyruvat, 100 U/ml Penicillin und 100 mg/ml Streptomycin PC12 (ATCC: CRL-1721): Nebennierenzelllinie der Ratte aus einem Phäochromozytom Kulturmedium: RPMI-1640 +10% Pferdeserum, 5% FCS, 1% Na-Pyruvat, 100 U/ml Penicillin und 100 mg/ml Streptomycin 1.2.1: Kulturmedium: DMEM (Biochrom AG, Berlin, Deutschland) Ham´s F12 (Biochrom AG, Berlin, Deutschland) RPMI-1640 (Biochrom AG, Berlin, Deutschland) 1.2.2: Kulturmedium-Zusätze: Fetales Kälberserum (FCS) (Biochrom AG, Berlin, Deutschland) Pferdeserum (PAA Laboratories,Pasching, Österreich) L-Glutamat (PAN Biotech, Aidenbach, Deutschland) Penicillin/Streptomycin (PAN Biotech, Aidenbach, Deutschland) Na-Pyruvat (PAN Biotech, Aidenbach, Deutschland) Nicht-essentielle Aminosäuren (NEA) (PAA Laboratories,Pasching, Österreich) 2: Plasmide und Vektoren pHis1522 (MoBiTec, Göttingen, Deutschland) (zur Proteinexpression in Bacillus megaterium) pGEX2T/4T (GE Healthcare, München, Deutschland) (zur Proteinexpression in Escherichia coli) 18 MATERIAL UND METHODEN pCR-Blunt-2-TOPO (Invitrogen, Darmstadt, Deutschland) (zur Klonierung von PCR-Produkten) pET28a (Merck, Darmstadt, Deutschland) (zur Proteinexpression in Escherichia coli) 3: Verwendete Plasmide/Mutanten Eine detaillierte Auflistung aller in dieser Arbeit klonierter bzw. eingesetzter Konstrukte befindet sich im Anhang. 4: Primer Sämtliche in dieser Arbeit verwendeten Primer wurden von Apara Bioscience GmbH (Freiburg, Deutschland) erworben. Eine detaillierte Auflistung aller verwendeten Primer befindet sich im Anhang. 5: Biochemikalien und Enzyme 5.1: DNA-modifizierende Enzyme 5.1.1: DNA-Polymerasen Pfu-Ultra Fusion HS II (Stratgene, LaJolla, USA) Phusion (New England Biolabs, Schwalbach, Deutschland) 5.1.2: DNA–Ligasen T4-Ligase (Fermentas, St.Leon-Rot, Deutschland, New England Biolabs, Schwalbach, Deutschland) 5.1.3: Restriktionsendonukleasen BsrGI, DpnI, KpnI, PstI, SpeI, EheI, BamHI, XhoI, NdeI, XhoI (Fermentas, St.Leon-Rot, Deutschland, New England Biolabs, Schwalbach, Deutschland) 5.1.4: DNA-Phosphatasen Antarktische Phosphatase (New England Biolabs, Schwalbach, Deutschland) 5.2: Protein-Marker PageRuler Plus Prestained Protein Ladder (Fermentas, St.Leon-Rot, Deutschland) 10 – 15 – 27 – 37 – 55 – 70 – 100 – 130 – 250 kDa peqGOLD Protein-Marker I (Peqlab, Erlangen, Deutschland) 14 – 18 – 25 – 35 – 45 – 66 – 114 kDa Spectra Multicolor High Range Protein Ladder (Fermentas, St.Leon-Rot, Deutschland) 40 – 50 – 70 – 100 – 130 – 180 – 250 – 300 kDa 5.3: DNA-Leiter GeneRuler 1 kB DNA Ladder (Fermentas, St.Leon-Rot, Deutschland) 0.25 – 0.5 – 0.75 – 1 – 1.5 – 2 – 2.5 – 3 – 3.5 – 4 – 5 – 6 – 8 – 10 kb 19 MATERIAL UND METHODEN 5.4: Gelfiltration-Größenstandard Gelfiltrationsstandard (BioRad, München, Deutschland) 1,7 – 17 – 44 – 158 – 660 kDa 5.5: Antikörper und Antiseren 5.5.1: primäre Antikörper Anti-Toxin A (polyklonal, 1:10000, Kaninchen) (bereits vorhanden: Labor Prof.Aktories) Anti-Toxin B (polyklonal, 1:5000, Kaninchen) (bereits vorhanden: Labor Prof.Aktories) Anti-Letales Toxin (polyklonal, 1:5000, Maus) (bereits vorhanden: Labor Prof.Aktories) Anti-Toxin B1-546 (monoklonal, 1:10000, Maus) (bereits vorhanden: Labor Prof.Aktories) Anti-Rac1 Mab102 (monoklonal, 1:1000, Maus) (Millipore, Eschborn, Deutschland) Anti-Rac1 23A8 (monoklonal, 1:10000, Maus) (Cell Signalling, Danvers, USA) Anti-Ras Mab27H5 (monoklonal, 1:1000, Kaninchen) (Cell Signalling, Danvers, USA) Anti-v-H-Ras MabY13-259 (monoklonal, 1:40, Ratte) (Calbiochem, Darmstadt, Deutschland) Anti-PARP-1 (monoklonal, 1:1000, Kaninchen) (Cell Signalling, Danvers, USA) Anti-phospho-Erk1/2 T202/Y204 (monoklonal, 1:2000, Kaninchen) (Cell Signalling, Danvers, USA) Anti-GAPDH Mab374 (monoklonal, 1:2000, Maus) (Millipore, Eschborn, Deutschland) Anti-6xHisitidin (monoklonal, 1:1000, Maus) (BD, Franklin Lakes, USA) 5.5.2: sekundäre Antikörper Peroxidase-gekoppelte Anti-Maus IgG und IgM (1:3000) (Vector Laboratories, Burlingame, USA) Peroxidase-gekoppelte Anti-Kaninchen IgG und IgM (1:3000) (Rockland, Gilbertsville, USA) Peroxidase-gekoppelte Anti-Ratte IgG und IgM (1:500) (Santa Cruz Biotechnology, Santa Cruz, USA) 6: Kits GeneJET Plasmid Miniprep Kit (Fermentas, St.Leon-Rot, Deutschland) QIAGEN Plasmid Midi Kit (Qiagen, Hilden, Deutschland) QIAGEN Plasmid Maxi Kit (Qiagen, Hilden, Deutschland) QIAquick PCR Purification Kit (Qiagen, Hilden, Deutschland) QIAquick Gel Extraction Kit (Qiagen, Hilden, Deutschland) Zero Blunt TOPO PCR Cloning Kit (Invitrogen, Darmstadt, Deutschland) BCA-Assay-Protein Quantification Kit (Uptima, Montlucon, USA) 7: Radionuklide [3H]-Inositolhexakisphosphat (20 Ci/mmol) (Perkin Elmer Life Science, Boston, USA) UDP-[14C]-Glukose (0,5 mCi/ml) (Biotrend, Köln, Deutschland) UDP-[14C]-N-Acetylglukosamin (0,1 mCi/ml) (Biotrend, Köln, Deutschland) [86Rb+] (10 mCi/ml) (Perkin Elmer Life Science, Boston, USA) 8: sonstige Chemikalien/Materialien Benzamidin-Sepharose 6B (GE Healthcare, München, Deutschland) Glutathion-Sepharose 4B (GE Healthcare, München, Deutschland) Ni2+-IDA-Protino-Resin (Macherey-Nagel GmbH & Co. KG, Düren, Deutschland) CNBr-Sepharose 4B (GE Healthcare, München, Deutschland) Protein G-Sepharose Fast Flow (Sigma-Aldrich, Taufkirchen, Deutschland) 20 MATERIAL UND METHODEN Staurosporin (Sigma-Aldrich, Taufkirchen, Deutschland) EGF (rekombinant, Maus) (Bachem AG, Bubendorf, Schweiz) β-NGF (rekombinant, Maus) (Biomol GmbH, Hamburg, Deutschland) Konzentrator Vivaspin 6 ml, 20 ml (100 kDa) (VWR International GmBH, Darmstadt, Deutschland) Nukleotide (Roche Diagnostics, Basel, Schweiz) Inositolhexakisphosphat (Sigma-Aldrich, Taufkirchen, Deutschland) 21 MATERIAL UND METHODEN C.2: METHODEN 1: Molekularbiologische Methoden 1.1: Polymerase-Kettenreaktion Die Polymerase-Kettenreaktion (polymerase chain reaction [PCR]) ist eine Methode zur in vitro-Amplifizierung von DNA. Hierbei kommt es zu einer exponentiellen Vermehrung der zu amplifizierenden DNA-Fragmente. Ein typischer PCR-Reaktionansatz beinhaltet AusgangsDNA (Plasmid-DNA, PCR-Fragment oder genomische DNA), Oligonukleotid-Primern, Nukleotiden, eine DNA-abhängige DNA-Polymerase und Reaktionspuffer. Eine typische PCR-Reaktion Hybridisierungs- besteht aus (annealing) mehreren und Zyklen, die Elongationsschritt aus einem aufgebaut sind. Denaturierungs-, Während des Denaturierungsschrittes kommt es zur thermisch bedingten Trennung der DNA-Stränge. Nach Absenkung der Temperatur binden die Primer im Hybridisierungsschritt an die einzelsträngige DNA. Hierbei bindet das Primer-Paar (forward [fw]- und reverse [rev]-Primer) an den zu ihnen komplementären Sequenzabschnitten jeweils auf einem der beiden DNAStränge und definieren somit die zu amplifizierenden Bereiche. Die Elongation findet am Temperaturoptimum der DNA-Polymerase statt. Im Elongationsschritt werden die Primer verlängert bis eine exakte Kopie der Ausgangs-DNA entstanden ist. 1.2: “touchdown-PCR” „touchdown-PCR“ ist eine PCR-Variante, bei der schrittweise die Hybridisierungstemperatur von Zyklus zu Zyklus abgesenkt wird. Der Schmelzpunkt des Primers setzt hier das obere Temperaturlimit fest. Knapp unterhalb dieser Temperatur kommt es zu einer sehr spezifischen Basenpaarung des Primers mit dem DNA-Template. Durch diese Methode wird eine unspezifische Bindung der Primer und daraus folgend eine Amplifikation unspezifischer Bereiche vermieden. Die „touchdown-PCR“ ist besonders geeignet zur Amplifikation schwieriger Ausgangs-DNA. 1.3: Aufreinigung von PCR-Produkten Zur Aufreinigung von PCR-Produkten wurde das QIAquick PCR Purification Kit (Qiagen) eingesetzt. Mit diesem Kit ist es möglich, unerwünschte Bestandteile der PCR-Reaktion wie z.B. Primer oder Nukleotide, zu entfernen. Die Entfernung von Restriktionsenzymen nach erfolgtem Restriktionsverdau oder DNA-Polymerasen ist ebenfalls möglich. Zunächst wurde zu dem PCR-Produkt PB-Puffer (+pH-Indikator 1:250) in einem Volumenverhältnis 1:5 gegeben. Falls die entstandene Lösung eine orange bis rote Färbung aufwies, mussten 10 µl 22 MATERIAL UND METHODEN 3 M Na-Acetat (pH 5) zugegeben werden um den erwünschten pH-Wert zu erreichen. Anschließend wurde die Lösung auf ein Säulchen, bestehend aus einer Silikat-Membran, gegeben, an welche die DNA bindet. Nach Waschen mit 750 µl Ethanol-haltigem PE-Puffer und Trocknen des Säulchens durch Zentrifugation konnte die DNA mit 50 µl EB-Puffer eluiert werden. Material: EB-Puffer: 10 mM Tris/HCl (pH 8,5) Über die Zusammensetzung der anderen Puffer werden vom Hersteller keine genauen Angaben gemacht. 1.4: Alkohol-Fällung Durch Versetzen mit einem Alkohol in Gegenwart eines monovalenten Salzes ist es möglich Nukleinsäuren zu fällen. Zur Fällung mit Ethanol benötigt man ein Voulmenverhältnis 1:2,5. Zur Fällung mit Isopropanol braucht man lediglich ein Volumenverhältnis 1:0,6. Vor Zugabe des Alkohols wurde die DNA-Lösung mit Na-Acetat (pH 5) versetzt. Die Endkonzentration des Na-Acetats sollte final 0,3 M betragen. Anschließend wurde die DNA durch Zentrifugation pelletiert. Nach Waschen mit 70% (v/v) Ethanol konnte die DNA in Wasser bzw. 10 mM Tris/HCl-Puffer aufgenommen werden. 1.5: Präparation von Plasmid-DNA 1.5.1: Präparation von Plasmid-DNA im kleinen Maßstab Zur Aufreinigung von Plasmid-DNA im kleinen Maßstab wurden das GeneJET Plasmid Miniprep Kit (Fermentas) verwendet. Das Prinzip dieses Kits basiert auf der alkalischen Lyse, gefolgt von der Aufreinigung der DNA über eine Silikat-Matrix. Zunächst wurde aus einer Einzelkolonie eine 5 bis 10 ml Bakterien-Kultur (high copy- bzw. low copy-Plasmid) angeimpft und über Nacht bei 37°C inkubiert. Nach der Zellernte wurde das Pellet in 250 µl RNase A-haltigem Resuspendierungspuffer aufgenommen. Durch Zugabe von 250 µl LysePuffer wurde die Zelllyse gestartet. Anschließend wurden die geeigneten Bedingungen für die Bindung der Plasmid-DNA an die Silikat-Säulchen durch Zugabe von 350 µl Neutralisierungspuffer zum Zelllysat eingestellt. Es ist wichtig, zwischen den beschriebenen Schritten auf Vortexen der Lösungen zu verzichten. Das Mischen erfolgte durch vorsichtiges Invertieren. Nach Bindung an die Silikat-Matrix wurde die Plasmid-DNA mit jeweils 500 µl Waschpuffer zweimal gewaschen. Die Plasmid-DNA konnte schließlich nach Trocknen der Säule durch Zentrifugation mit 50 µl Elutionspuffer eluiert werden. 23 MATERIAL UND METHODEN Material: Elutionspuffer: 10 mM Tris/HCl (pH 8,5) Über die Zusammensetzung der anderen Puffer werden vom Hersteller keine genauen Angaben gemacht. 1.5.2: Präparation von Plasmid-DNA im größeren Maßstab Um hochreine Plasmid-DNA in größeren Mengen zu erhalten, wurden das QIAGEN Plasmid Midi Kit und das QIAGEN Plasmid Maxi Kit (Qiagen) eingesetzt. Aufgrund ihrer höheren Reinheit wurde diese Plasmid-DNA zur Transformation von Bacillus megateriumProtoblasten benutzt. Das Prinzip dieses Kits basiert ebenfalls auf der alkalischen Lyse, gefolgt von einer Aufreinigung der DNA über eine Silikat-Membran. Im Nachfolgenden ist der Ablauf für das QIAGEN Plasmid Midi Kit beschrieben. Nach der Zellernte der Bakterienkultur (25 ml high-copy- bzw. 100 ml low-copy-Plasmide) wurde das Zellpellet in 4 ml P1-Puffer resuspendiert, mit P2-Puffer versetzt und nach Invertieren für 5 Minuten bei Raumtemperatur inkubiert. Nach Zugabe von 4 ml P3-Puffer wurde nach Invertieren das Zelllysat bei 4°C für weitere 20 Minuten inkubiert. Die Zelldebris wurde durch Zentrifugation abgetrennt. Der Überstand, in dem sich die Plasmid-DNA befindet, wurde auf eine, zuvor mit 4 ml QBT-Puffer äquilibrierten Silikat-Säule gegeben. Die Säule wurde mit je zweimal 20 ml QC-Puffer gewaschen und anschließend die DNA mit 5 ml QF-Puffer von der Säule eluiert. Als zusätzlicher Reinigungsschritt wurde die DNA mit 3,5 ml Isopropanol gefällt. Nach Zentrifugation (15000 g, 30 min, 4°C) wurde das DNA-Präzipitat mit 2 ml 70% (v/v) Ethanol gewaschen und nach Lufttrocknung in Wasser bzw. EB-Puffer aufgenommen. Material: EB-Puffer: 10 mM Tris/HCl (pH 8,5) Über die Zusammensetzung der anderen Puffer werden vom Hersteller keine genauen Angaben gemacht. 1.6: Präparation von genomischer DNA Die Aufreinigung von genomischer DNA aus Clostridien wurde nach einer modifizierten Methode von Wren et al. durchgeführt (Wren and Tabaqchali, 1987). Zunächst wurden 50 ml BHI-Medium mit den entsprechenden Clostridien-Stämmen angeimpft und unter anaeroben Bedingungen (2 Platten Anaerocult A im 2,5 l Anaerobiertopf) über Nacht bei 37°C kultiviert. Nach der Zellernte wurde das Zellpellet mit TE-Puffer-1 gewaschen und anschließend in einem Lysozym-haltigen Resuspendierungspuffer aufgenommen und bei 37°C für eine Stunde inkubiert. Nach erneuter Zentrifugation wurde das Pellet in 2 ml TE-Puffer-2 resuspendiert. Der Ansatz wurde nach Zugabe von 125 µl Proteinase K (20 mg/ml) bei 37°C für weitere 30 Minuten inkubiert. Die Zelllyse wurde durch die Zugabe von 10% (m/v) NLauroylsarcosin-Lösung gestartet und das Lysat bei 37°C für 20 Minuten bis zur vollständigen Zelllyse inkubiert. Anschließend wurde zum Lysat eine Mischung aus 2,5 ml 24 MATERIAL UND METHODEN Wasser und 50 µl RNase A (10 mg/ml) zugegeben und bei 37°C erneut für 30 Minuten inkubiert. Die DNA wurde anschließend dreimal mit 5 ml einer Mischung aus Phenol, Isoamylalkohol und Chloroform ausgeschüttet. Final wurde eine Ethanol-Fällung der genomischen DNA (siehe Methoden 1.4) durchgeführt. Material: TE-Puffer-1: 10 mM Tris/HCl (pH 8), 1 mM EDTA Resuspendierungspuffer: 3 ml 1 M Saccharose, 3 ml TE-Puffer-1, 10 mg Lysozym TE-Puffer-2: 1,5 ml 50 mM Tris/HCl (pH 8), 0,5 ml 0,25 M EDTA Lysozym (Sigma-Aldrich, Taufkirchen, Deutschland) RNase A (Roche Diagnostics, Basel, Schweiz) Proteinase K (Sigma-Aldrich, Taufkirchen, Deutschland) Anaerocult A (Merck, Darmstadt, Deutschland) Roti Phenol/C/I (Phenol/Isoamylalkohol/Chloroform, Teile: 25/1/24) (Roth, Karlsruhe, Deutschland) N-Lauroylsarcosin (Sigma-Aldrich, Taufkirchen, Deutschland) 1.7: Konzentrations- und Reinheitsbestimmung von DNA Die Konzentrationsbestimmung von DNA erfolgte UV-photometrisch (NanoDrop 1000, Peqlab) über die Bestimmung der Absorption bei einer Wellenlänge von 260 nm. Ein OD260nm-Wert von 1,0 entspricht einer DNA-Konzentration von 50 ng/µL. Aus dem Vergleich der Werte OD260nm/OD280nm und OD260nm/OD230nm können Rückschlüsse über die Reinheit der DNA gezogen werden. Anhand dieser beiden Quotienten können Verunreinigungen durch Proteine bzw. Kohlenhydrate oder RNA bestimmt werden. Proteinfreie DNA-Proben sollten einen OD260nm/OD280nm-Wert zwischen 1,8 und 2,5 besitzen. Die OD260nm/OD230nm-Werte einer DNA-Probe sollten einen Wert von 0,6 nicht überschreiten. 1.8: Agarose-Gelelektrophorese DNA-Fragmente können nach ihrer Größe mittels Agarose-Gelelektrophorese aufgetrennt werden. Zur Trennung von DNA-Fragmenten einer Größe zwischen 0,5 bis 10 kB können 0,8% (m/v) Agarose-Gele verwendet werden. Die Gelelektrophorese wurde in TAE-Puffer bei einer konstanten Spannung von 100 V für eine Stunde durchgeführt. Zuvor wurden die DNAProben in Ladepuffer aufgenommen. Nach erfolgter Elektrophorese konnte die DNA durch Färbung mit Ethidiumbromid nachgewiesen werden. Alternativ können 5 µl Ethidiumbromid dem Agarose-Gel direkt zugegeben werden. Die DNA-Fragmente konnten nach Bestrahlung mit UV-Licht (ChemiDoc XR, Biorad) bei einer Wellenlänge von 254 nm anhand der intensiven Fluoreszenz des in die DNA interkalierten Ethidiumbromids sichtbar gemacht werden. 25 MATERIAL UND METHODEN Material: TAE-Puffer: 40 mM Tris-Acetat (pH 8), 1 mM EDTA Ladepuffer (6x): 0,25% (m/v) Bromphenolblau, 40% (m/v) Saccharose Agarose-Gel: 0,8% (m/v) Agarose, 50 ml TAE-Puffer, 5 µl Ethidiumbromid (final: 0,5 µg/ml) Ethidiumbromid (Sigma-Aldrich, Taufkirchen, Deutschland) Agarose (Sigma-Aldrich, Taufkirchen, Deutschland) 1.9: Isolierung von DNA aus Agarose-Gelen Zur Isolierung von DNA-Fragmenten aus Agarose-Gelen kam das QIAquick Gel Extraction Kit (Qiagen) zum Einsatz. Die zu isolierende DNA-Bande wurde nach der AgaroseGelelektrophorese unter UV-Licht mit einem Skalpell aus dem Gel geschnitten. Das Gelstück wurde mit 300 µl QG-Puffer je 100 mg Agarose versetzt und bei 50°C geschmolzen. Bei der Isolierungen von DNA-Fragmenten >0,5 kB oder <4,0 kB wurde zusätzlich 100 µl Isopropanol zugegeben. Nach vollständiger Solubilisierung des Gelstückes wurde die Lösung über ein Silikat-Säulchen gegeben. Zur vollständigen Entfernung von AgaroseResten wurde die Silikat-Säule mit 500 µl QG-Puffer nochmals gespült. Die gebundene DNA wurde anschließend mit 750 µl Ethanol-haltigem PE-Puffer gewaschen. Nach Trocknen der Silikat-Säule durch Zentrifugation konnte die DNA mit 50 µl EB-Puffer von der Säule eluiert werden. Material: EB-Puffer: 10 mM Tris/HCl (pH 8,5) Über die Zusammensetzung der anderen Puffer werden vom Hersteller keine genauen Angaben gemacht. 1.10: Verdau von DNA mit Restriktionsendonukleasen Mit Restriktionsendonukleasen vom Typ II ist es möglich DNA-Moleküle spezifisch an definierten palindromischen Sequenzen zu schneiden. Analytische und präparative Restriktionsansätze von DNA wurden mit 3 bis 20 U der jeweiligen Restriktionsendonukleasen (Fermentas, New England Biolabs) durchgeführt. Die Restriktion erfolgte bei 37°C für eine Stunde bzw. über Nacht in den entsprechenden von den Herstellern für die Restriktionsendonukleasen optimierten Puffern. Analytisch wurden 100 bis 300 ng sowie präparativ 5 bis 10 µg Plasmid-DNA verdaut. 1.11: Vektor-Dephosphorylierung Bei einem unvollständigen Verdau eines Vektors mit zwei Restriktionsendonukleasen können zwei kompatible Enden entstehen, die wieder miteinander ligieren können. Um eine Religation zu verhindern, können die 5´-Phosphatgruppen des Vektors enzymatisch mit einer alkalischen Phosphatase entfernt werden. Für diese Dephosphorylierung wurde die 26 MATERIAL UND METHODEN Antarktische Phosphatase (New England Biolabs) verwendet. Der verdaute Vektor wurde nach Agarose-Gelelektrophorese aus dem Agarose-Gel aufgereinigt und auf eine ungefähre Konzentration von 100 ng/µl eingestellt. Die Reaktion wurde bei 37°C für eine Stunde in APPuffer mit 5 U Phosphatase durchgeführt. Nach erfolgter Dephosphorylierung wurde die Phosphatase bei 65°C für 20 Minuten hitzeinaktiviert. 1.12: Ligation von DNA-Fragmenten Die Ligation von PCR-Produkten und DNA-Fragmenten in geöffnete Vektoren erfolgte mit der T4-DNA-Ligase (Fermentas, New England Biolabs). Die T4-DNA-Ligase katalysiert die Phosphodiesterbindung zwischen der 5´-Phosphat- und 3´-Hydroxylgruppe von DNASträngen. Bei der Ligation wurden 50 bis 100 ng Vektor eingesetzt. Die zu inserierende DNA sollte in einem etwa fünffachen Überschuss eingesetzt werden. Die einzusetzende Menge an Insert kann anhand einer Formel bestimmt werden. In diese Formel geht das Verhältnis der jeweiligen Größe des Vektors und Inserts ein. Die Ligation wurde mit 10 bis 20 U Ligase in T4-DNA-Ligase-Puffer bei 16°C über Nacht oder bei Raumtemperatur für 2 Stunden durchgeführt. Bei der Transformation durch Elektroporation wurde die Ligase zuvor für 10 Minuten bei 70°C hitzeinaktiviert. Formel: [ng DNA-Insert] = ([ng Vektor] x [kB Insert]x5)/( [kB Vektor]) 1.13: Transformation chemisch-kompetenter Escherichia coli Unter dem Begriff Transformation versteht man das Einschleusen von Plasmid-DNA in Bakterien. Die chemische Transformation erfolgte über die „Hitzeschock“-Methode. Chemisch-kompetente Escherichia coli (hier: BL21, XL1-Blue, TG1) wurden zunächst auf Eis aufgetaut. Ein Transformationsansatz bestand aus 10 bis 100 ng Plasmid-DNA und je 100 µl Bakteriensuspension. Nach Zugabe der DNA wurde der Ansatz für zunächst 30 Minuten auf Eis inkubiert. Anschließend erfolgte der eigentliche Hitzeschock. Hierzu wurden die Bakterien auf 42°C für 90 Sekunden erwärmt und anschließend für eine Minute lang auf Eis abgekühlt. Nach Zugabe von 800 µl SOC-Medium wurden die Bakterien unter Schütteln für eine Stunde bei 37°C inkubiert. Die Selektion der transformierten Bakterien erfolgte durch Ausstreichen auf LB-Selektionsmedium-Agarplatten. Nach einer erfolgreichen Transformation waren nach Inkubation der Agarplatten bei 37°C über Nacht einzelne Bakterien-Kolonien deutlich erkennbar. Material: SOC-Medium: 10 mM MgCl2, 20 mM Glucose, in LB-Medium 27 MATERIAL UND METHODEN 1.14: Herstellung chemisch-kompetenter Escherichia coli Escherichia coli können mit der Calciumchlorid-Methode chemisch-kompetent gemacht werden. Von einer frisch ausgestrichenen LB-Platte wurde eine Vorkultur angeimpft. Mit 1 ml dieser Vorkultur wurden 400 ml LB-Medium angeimpft und bei 37°C bis zu einer OD600nm von 0,7 bis 0,8 unter Schütteln inkubiert. Die Bakterien-Kultur wurde anschließend 15 Minuten lang auf Eis abgekühlt. Nach Abkühlen wurden die Bakterien geerntet und das Pellet in 100 ml 0,1 M CaCl2-Lösung resuspendiert. Die Bakterien-Suspension wurde auf Eis für 30 Minuten inkubiert und anschließend erneut zentrifugiert. Das Zellsediment wurde in 10 ml 0,1 M CaCl2/10% (m/v) Glycerin-Lösung resuspendiert. Je 100 µl der Bakterien-Suspension wurden aliquotiert und bei -80°C gelagert. 1.15: Elektroporation kompetenter Escherichia coli Elektroporation ist eine weitere Methode zur Transformation von Escherichia coli-Bakterien. Durch ein elektrisches Feld wird die Zellmembran aufgrund unterschiedlicher Effekte permeabilisiert und somit durchlässig für Plasmid-DNA. In einer Elektroporationsküvette wurden 50 µl elektrokompetente Escherichia coli (hier: XL1-Blue) sowie 2 µl eines Reaktionsansatzes (Ligation, zielgerichtete Mutagenese-PCR) zusammengegeben und leicht durchmischt. Die Elektroporation ist eine sehr salzempfindliche Methode. Bei zu hohen Salzkonzentrationen des Reaktionsansatzes kann es notwendig werden diesen umzupuffern (z.B. Gelfiltration, DNA-Fällung). Zur Elektroporation (Gene Pulser II, Biorad) prokaryoter Zellen wurden folgende Einstellungen benutzt; Spannung: 2,5 kV, Widerstand: 200 Ω, elektrische Kapazität: 25 µF. Die elektroporierten Bakterien-Zellen wurden nach Zugabe von 1 ml SOC-Medium bei 37°C für eine Stunde inkubiert und anschließend auf LBSelektionsmedium-Agarplatten ausgestrichen. Material: Elektroporationsküvette (2 mm) (Peqlab, Erlangen, Deutschland) SOC-Medium: 10 mM MgCl2, 20 mM Glucose in LB-Medium 1.16: Herstellung elektrisch-kompetenter Escherichia coli Mit Escherichia coli-Klonen von einer frisch ausgestrichenen LB-Platte wurde eine 5 ml Vorkultur angeimpft. Mit 1 ml dieser Vorkultur wurden 500 ml LB-Medium angeimpft. Diese Hauptkultur wurde bis zu einer OD600nm von 0,6 unter Schütteln bei 37°C inkubiert. Anschließend wurden die Bakterien-Zellen auf Eis für 30 Minuten abgekühlt und danach zentrifugiert. Das Bakterien-Pellet wurde zunächst in 500 ml eiskaltem Wasser resuspendiert. Nach erneuter Zentrifugation wurde das Pellet nun in 250 ml eiskaltem 28 MATERIAL UND METHODEN Wasser resuspendiert. Die Bakterien-Suspension wurde wiederholt zentrifugiert und das Pellet in 10 ml 10% (m/v) Glycerol aufgenommen. Letztendlich wurde das Pellet nach einer letzten Zentrifugation in 1,5 bis 2 ml 10% (m/v) Glycerol resuspendiert und nach Aliquotieren bei -80°C gelagert. 1.17: Direkte Klonierung von PCR-Produkten Eine direkte Klonierung von PCR-Produkten in einen Zwischenvektor ohne vorherigen Restriktionsverdau ist mit dem Zero Blunt TOPO PCR Cloning Kit (Invitrogen) möglich. Als Zwischenvektor dient hier der pCR-Blunt-2-TOPO-Vektor. An den 3´-Enden des Vektors ist eine Vaccinia-Virus DNA-Topoisomerase konjugiert. Dieses Enzym schneidet gezielt DNA nach dem 5´-CCCT-3´ Motiv eines Stranges. Die durch die Spaltung des Phosphodiesters freigesetzte Energie kann durch die erneute Ausbildung eines Phosphodiesters zwischen der 3´-Phosphat-Gruppe des Vektors Topoisomerase gespeichert und werden. der Diese Hydroxylgruppe Bindung des Tyrosin-274 kann schließlich an der der 5- Hydroxylgruppe des PCR-Fragments angegriffen werden und katalysiert hierdurch dessen Ligation. Diese Reaktion ist an das Vorhandensein von stumpfen 3´-Enden (blunt ends) des PCR-Produktes gebunden. Die Pfu-Polymerase erzeugt Amplifikate ohne Basenüberhänge und liefert somit DNA-Fragmente, die in dieser Reaktion sofort eingesetzt werden können. Bei der Durchführung wurden 0,5 bis 4 µl PCR-Produkt mit 1 µl Salz-Lösung und 1 µl pCRBlunt-2-TOPO-Vektor bei Raumtemperatur für 30 Minuten inkubiert. Anschließend wurden 2 µl des Ansatzes in chemisch-kompetente TOP-10-Zellen transformiert. Material: Über die Zusammensetzung der Salz-Lösung werden vom Hersteller keine genauen Angaben gemacht. 1.18: Blau-Weiß-Selektion Blau-Weiß-Selektion ist eine einfache Methode um Klone, die mit religierten Vektoren transformiert wurden, schnell und eindeutig zu identifizieren. Diese Selektion ist nur möglich mit Vektoren, die das Reportergen lacZ tragen (hier: pCR-Blunt-2-TOPO-Vektor). Das lacZGen codiert für das Enzym β-Galaktosidase. Das Gen wird nach Aufnahme des Vektors unterbrochen. β-Galaktosidase hydrolysiert als unphysiologisches Substrat 5-Brom-4-chlor3-indoxyl-β-D-galactopyranosid (X-Gal). Nach Dimerisierung entsteht ein blauer Indigofarbstoff. Klone, die mit einem Vektor ohne DNA-Insert transformiert wurden und somit ein ununterbrochenes lacZ-Gen tragen, können an der blauen Farbe dieser Klone erkannt werden. Nach der Transformation wurden die Bakterien auf Agarplatten ausgestrichen, die zuvor mit einer Mischung aus je 40 µl Isopropyl-β-D-thiogalactopyranosid (IPTG) (100 mM), X-Gal (40 mg/ml) und Wasser versetzt wurden. 29 MATERIAL UND METHODEN Material: Isopropyl-β-D-thiogalactopyranosid (IPTG) (Roth, Karlsruhe, Deutschland) 5-Bromo-4-chloro-3-indolyl-β-D-galactopyranosid (X-Gal) (Sigma-Aldrich, Taufkirchen, Deutschland) 1.19: Zielgerichte Mutagenese Durch zielgerichtete Mutagenese („Quik-Change“-Methode, Stratagene) ist es möglich, Punktmutationen in Plasmid-DNA einzuführen. In dieser, an einer PCR-angelehnten Methode, werden zueinander komplementäre Primer eingesetzt. Die Primer tragen in der Mitte die gewünschten Änderungen der Nukleotidsequenz, wodurch gezielte Mutationen eingefügt werden können. Während der zielgerechten Mutagenese wird das gesamte Plasmid amplifiziert, wobei ein linearisiertes Produkt entsteht. Die Ausgangs-DNA kann mit DpnI verdaut werden. DpnI erkennt seine Schnittstelle nur, wenn diese methyliert ist. Die neuentstandene Amplifikate sind nicht methyliert und werden somit nicht abgebaut. Als DNATemplate muss Plasmid-DNA verwendet werden, die zuvor aus einem dam+ Escherichia coliStamm aufgereinigt wurde, da lediglich die DNA dieser Stämme methyliert wird. Im Anschluss an die Quik-Change-PCR erfolgte der DpnI-Verdau mit 10 U bei 37°C über Nacht. 2 µl des Reaktionsansatzes wurden zur Transformation von Escherichia coli (hier: XL1-Blue) durch Elektroporation eingesetzt. 1.20: Sequenzierung von DNA Die in dieser Arbeit klonierten Sequenzabschnitte wurden durch eine DNA-Sequenzierung auf deren Richtigkeit überprüft. Die Sequenzierungen wurden im Auftrag von GATC-Biotech (Konstanz, Deutschland) durchgeführt. Für die Sequenzierung wurde die Kettenabbruchmethode nach Sanger in modifizierter Methode eingesetzt. Zu einem PCRReaktionsansatz, bestehend aus Primer, dNTPs und das zu sequenzierende DNA-Template, werden mit verschiedenen Fluoreszenzsonden markierte Didesoxynukleosidtriphosphate (ddNTPs) gegeben. Der Einbau eines solchen Nukleotides führt zu einem Kettenabbruch. Die unterschiedlich langen DNA-Fragmente werden mittels Kapillarelektrophorese aufgetrennt. Jedes DNA-Fragment trägt ein terminales Fluoreszenz-markiertes Nukleotid. Über die unterschiedlichen Fluoreszenzeigenschaften der Marker kann die DNA-Sequenz ermittelt werden. 1.21: Transformation von Bacillus megaterium-Protoplasten Die DNA-Transformation in gram-positive Bacilli bedarf besonderer Protokolle. Aufgrund ihrer Zellwand können manche gram-positive Bakterien externe DNA nicht aufnehmen. Bacilli können aber nach enzymatischem Verdau der Zellwand mit Lysozym DNA-kompetent 30 MATERIAL UND METHODEN gemacht werden. Hierbei entstehen zellwandlose Protoplasten, die nur noch von ihrer Zytoplasmamembran umgeben sind. Die Aufnahme der DNA wird durch Polyethylenglykol vermittelt. Zu 500 µl Protoplasten wurden 5 µg Plasmid-DNA gegeben. Anschließend wurde dieser Mischung 1,5 ml PEG-P-Lösung zugegeben. Nach zweiminütiger Inkubation bei Raumtemperatur wurden 5 ml SMMP-Lösung zugegeben. Der Ansatz wurde vorsichtig durchmischt und dann zentrifugiert (1300 g, 10 min, RT). Anschließend wurde zu dem Pellet 500 µl SMMP-Lösung gegeben und der gesamte Ansatz bei 37°C für 90 Minuten unter leichtem Schütteln (maximal 100 rpm) inkubiert. In einem Wasserbad (45°C) wurden 2,5 ml CR5-Topagar bis zur Verflüssigung erwärmt. Der Transformationsansatz wurde schließlich mit dem Topagar vorsichtig vermischt. Der Topagar wurde vorsichtig auf eine LBSelektionsmedium-Agarplatte gegeben und bei 37°C für 24 bis 48 Stunden inkubiert. Die entstanden Klone wurden innerhalb von zwei Tagen auf eine neue LB-SelektionsmediumAgarplatte umgestrichen. Material: 2x AB3 (Difco, Augsburg, Deutschland): 7 g in 200 ml H2O 2x SMM: 1 M Saccharose, 40 mM Na-Maleat, 40 mM MgCl2 (pH 6,5) SMMP: Mischung 1:1 aus 2x SMM und 2x AB3 PEG-P: 40% PEG 6000 in ein Volumen 1x SMM CR5-Topagar: besteht aus Komponente A, B und C A: 51,5 g Saccharose, 3,25 g MOPS, 0,33 g NaOH (pH 7,3) (Gesamtvolumen: 250 ml) B: 2 g Agar, 0,1 g Casaminosäuren (BD, Franklin Lakes, USA), 5 g Hefe-Extrakt (Roth, Karlsruhe, Deutschland) (Gesamtvolumen: 142,5 ml) C: 57,5 ml 8x CR5-Salze, 25 ml 12% (m/v) Prolin, 25 ml 20% (m/v) Glucose 8x CR5-Salz: 1,25 g K2SO4, 50 g MgCl2x6H2O, 0,25 g KH2PO4, 11 g CaCl2x2H2O (Gesamtvolumen: 625 ml) 1.22: Herstellung von Bacillus megaterium-Protoplasten Von einer mit Bacillus megaterium-Protoplasten ausgestrichenen LB-Agarplatte wurde zunächst eine 2 ml Vorkultur angeimpft. Mit 1 ml dieser Vorkultur wurde eine 50 ml LBMedium-Hauptkultur angeimpft und bei 37°C bis zu einer OD578nm von 1,0 inkubiert. Anschließend wurden die Bakterien pelletiert. Das Zellpellet wurde in 5 ml SMMP-Lösung resuspendiert und mit 50 µg Lysozym versetzt. Nach einstündiger Inkubation wurden die Zellen erneut zentrifugiert (1300 g, 5 min, 4°C). Das erhaltende Pellet wurde in 5 ml SMMPLösung resuspendiert. Nach Wiederholung dieses Schrittes wurde der Zellsuspension Glycerol bis zu einem Volumenverhältnis von 10% zugegeben. Nach dem Aliquotieren können Bacillus megaterium-Protoplasten bei -80°C gelagert werden. Material: SMMP: 1:1 Mischung aus 2x SMM und 2x AB3 Lysozym (Sigma-Aldrich, Taufkirchen, Deutschland) 31 MATERIAL UND METHODEN 2: Proteinbiochemische Methode 2.1: Aceton-Fällung Mit der denaturierenden Acetonfällung ist es möglich, Proteine quantitativ aus einer wässrigen Lösung zu fällen. Dies wird ausgenutzt um störende Agenzien abzutrennen oder das Protein zu konzentrieren. Hierzu wurde die Proteinlösung mit einem Volumenverhältnis von 1:5 mit eiskalten Aceton versetzt und gut durchmischt. Die Proteine wurden anschließend bei -20°C für 20 Minuten präzipitiert. Nach Zentrifugation wurde das Proteinpellet getrocknet und konnte dann in dem gewünschten Volumen aufgenommen werden. 2.2: Ammoniumsulfat-Fällung Im Gegensatz zur Aceton-Fällung bleibt bei der nicht-denaturierenden AmmoniumsulfatFällung die Konformation der Proteine erhalten. Die Proteine werden hierbei aus einer gesättigten Ammoniumsulfat-Lösung gefällt. Die für die Fällung benötigte Menge an Ammoniumsulfat kann anhand einer Formel bestimmt werden. Das Ammoniumsulfat wurde der Proteinlösung unter Rühren bei 4°C langsam zugeben und für mindestens 6 Stunden weitergerührt. Das Proteinpräzipitat wurde durch Zentrifugation abgetrennt und kann anschließend im gewünschten Volumen aufgenommen werden. Formel: [g (NH4)2SO4 = ml Proteinlösung x 492 g/1000 ml] 2.3: SDS-Polyacrylamid-Gelelektrophorese Mit der diskontinuierlichen SDS-Polyacrylamid-Gelektrophorese (SDS-PAGE) nach Lämmli ist es möglich, Proteine nach deren molekularen Massen aufzutrennen. SDS denaturiert Proteine und unterbindet Protein-Protein-Wechselwirkungen. Die Proteine binden Natriumdodecylsulfat (SDS) zu negativ geladenen SDS/Protein-Mizellen, die ein konstantes Masse-/Ladungs-Verhältnis aufweisen. Dithiothreitol (DTT) wurde eingesetzt, um vorhandene Disulfidbrücken zu reduzieren. Während der Gelelektrophorese wandern die Mizellen im elektrischen Feld zur Anode. Hierbei erzeugt die poröse Polyacrylamid-Matrix einen Molekularsiebeffekt. Zur Auftrennung von Proteinen bis 100 kDa wurden 12,5%-ige SDS-PAGE-Gele verwendet. Größere Proteine wurden mit 3 bis 12,5%-igen GradientenGelen aufgetrennt. Zur Herstellung eines Gradienten-Gels (Kammdicke: 0,75 mm) wurden zunächst 1,6 ml 3%-ige Gellösung und dann 1,6 ml 12,5%-ige Gellösung in einer Pipette aufgezogen. Zum Mischen des Gradienten wurden zwei bis drei Luftblasen durch die Pipette gezogen und anschließend das Gel gegossen. Vor der SDS-PAGE wurden die Proben mit 32 MATERIAL UND METHODEN SDS-Probenpuffer versetzt und anschließend bei 95°C für 5 Minuten erhitzt. Die Gelelektrophorese erfolgte bei einer konstanten Stromstärke von 30 mA je Gel in SDSLaufpuffer. Die aufgetrennten Proteine können mit Coomassie-Färbelösung gefärbt werden. Mit diesem Farbstoff ist es möglich 200 bis 300 ng Protein pro Bande sichtbar zu machen. Material: Acrylamid-Lösung Rotiphorese Gel 30 (Roth, Karlsruhe, Deutschland): 30% Acrylamid, 0,8% Bisacrylamid Sammel-Gel (5% Acrylamid) (10 ml): 1,7 ml Acrylamid-Lösung, 2,0 ml 0,625 M Tris/HCl (pH 6,8), 50 µl 20% (m/v) SDS, 100 µl 10% (m/v) TEMED (N, N, N’, N’-Tetramethylethylendiamin), 100 µl 10% APS (m/v) Ammoniumperoxodisulfat) (APS), 6,05 ml H2O Trenn-Gel (12,5% Acrylamid) (10 ml): 4,17 ml, Acrylamid-Lösung 5,0 ml 0,75 M Tris/HCl (pH 8,8), 45 µl 20% (m/v) SDS, 100 µl 10% (m/v) TEMED (N, N, N’, N’-Tetramethylethylendiamin), 40µl 10% APS (m/v) Ammoniumperoxodisulfat) (APS), 640 µl H2O Gradienten-Gel (3% Acrylamid) (10 ml): 1,2 ml, Acrylamid-Lösung 5,0 ml 0,75 M Tris/HCl (pH 8,8), 50 µl 20% (m/v) SDS, 50 µl 10% (m/v) TEMED (N, N, N’, N’-Tetramethylethylendiamin), 100 µl 10% APS (m/v) Ammoniumperoxodisulfat (APS), 3,8 ml H2O SDS-Probenpuffer (5x): 0,01% (m/v) Bromphenolblau, 125 mM Tris/HCl (pH 6,8), 0,1 mM DTT, 10% (m/v) SDS, 29% (m/v) Glycerol SDS-Laufpuffer: 25 mM Tris/HCl (pH 8,3), 192 mM Glycin, 0,1% (m/v) SDS Coomassie-Färbelösung: 0,025% (m/v) Coomassie Brillant Blue G-250, 0,025% (m/v) Coomassie Brillant Blue R-250, 45% (v/v) Methanol, 9% (v/v) Eisessig Entfärbelösung (1 l): 10% (v/v) Eisessig 2.4: Silberfärbung Mit der Silberfärbung können im Vergleich zur Coomassie-Färbung deutlich geringere Mengen Protein nachgewiesen werden. Ag+-Ionen binden an Glutamat-, Aspartat- und Cystein-Reste und werden während der Färbung durch alkalischen Formaldehyd zu elementarem Silber reduziert. Zunächst wurden die Proteine mit 30 ml 50% (v/v) Aceton für 10 Minuten fixiert und mehrmals mit Wasser gespült. Anschließend wurde das Gel mit 30 ml Wasser für 5 Minuten gewaschen. Das Gel wurde mit 30 ml 50% (v/v) Aceton für 5 Minuten sowie mit 50 µl Na2S2O3 in 30 ml Wasser für jeweils eine Minute vorbehandelt. Nach mehrmaligen Spülen mit Wasser wurde das Gel mit 0,4 ml AgNO 3, 0,3 ml Formalin (35% Formaldeyd) und 30 ml Wasser für 8 Minuten behandelt. Das Gel wurde vor dem nächsten Schritt erneut mehrmals mit Wasser gespült. Die Färbung wurde durch Zugabe von 0,6 g Na2CO3, 12,5 µl Formalin, 12,5 µl Na2S2O3 in 30 ml Wasser für 30 Sekunden bis 2 Minuten entwickelt. Nach gewünschter Färbung wurde die Reaktion durch Zugabe von 30 ml 6% (v/v) Essigsäure gestoppt. Material: AgNO3-Lösung: 20% (m/v) AgNO3 Na2S2O3-Lösung: 10% (m/v) Na2S2O3 33 MATERIAL UND METHODEN 2.5: Herstellung von Gesamtlysaten aus HeLa-Zellen Die HeLa-Zellen wurden zunächst mittels eines Zellschabers aus den Kulturgefäßen abgelöst. Die Zellsuspension wurde zweimal mit eiskaltem PBS gewaschen und anschließend in RIPA-Puffer resuspendiert. Der Zellaufschluss erfolgte über einen Potter Homogenisator auf Eis für 3 Minuten. Anschließend wurde das Zelllysat auf einem Überkopfschüttler bei 4°C für eine Stunde inkubiert. Die Zelldebris wurde durch Zentrifugation entfernt und die Proteinkonzentration mit der BCA-Methode (siehe Methoden 2.8) bestimmt. Für den Nachweis von Proteinen durch Western-Blot wurden 20 bis 50 µg Zelllysat eingesetzt. Material: RIPA-Puffer: 50 mM Tris/HCl (pH 7,5), 150 mM NaCl, 10% (m/v) Glycerol, 2 mM EGTA, 2 mM MgCl2, 1% (m/v) NP-40, 0,5% (m/v) Na-Desoxycholat, Protease-Inhibitor-Cocktail Complete (Roche Diagnostics, Basel, Schweiz), 1 mM Na3VO4 2.6.1: Western-Blot Proteine können mit einem für sie spezifischen Antikörper in einem Western-Blot nachgewiesen werden. Hierzu werden die Proteine zunächst durch SDS-PAGE nach Größe aufgetrennt. Bei dem Vorgang des sogenannten „Blottens“ werden die Proteine anschließend auf eine Membran (hier: PVDF) transferiert. Auf dieser Membran können die Proteine nun mit spezifischen Antikörpern nachgewiesen werden. Das Blotten erfolgte im semidry-Verfahren (SemiDry Transfer Cell Trans Blot SD, Biorad) bei einer konstanten Stromstärke von 100 mA je Gel für eine Stunde. Der „Blot-sandwich“ bestand aus drei Lagen Whatman-Papier, Polyvinylidendifluorid (PVDF) Blot-Membran, SDS-PAGE-Gel und wieder drei Lagen Whatman-Papier. Zur Kontrolle des Blotvorgangs wurde ein vorgefärbter Marker eingesetzt. Um eine unspezifische Bindung des ersten Antikörpers zu vermeiden, wurden freie Bindungsstellen des Blots durch Zugabe von 10 ml Block-Lösung für eine Stunde bei Raumtemperatur abgesättigt. Anschließend wurde der Blot bei 4°C über Nacht mit dem spezifischen Primär-Antikörper inkubiert. Nach dreimaligem Waschen des Blots mit je 10 ml TBS-T wurde der Blot mit einem Sekundär-Antikörper für eine Stunde bei Raumtemperatur inkubiert. Die Antikörper wurden in unterschiedlichen Verdünnungen (siehe Material 5.5.1/2) eingesetzt. Der Sekundär-Antikörper ist mit dem Enzym Meerrettich-Peroxidase gekoppelt und richtet sich gegen das fc-Fragment des Primär-Antikörpers. Anschließend wurden ungebundene Antikörper durch dreimaliges Waschen mit TBS-T entfernt. Der Blot wurde durch das Verfahren der enhanced chemiluminescence (ECL) entwickelt. Bei dieser Reaktion katalysiert die Meerrettich-Peroxidase die Umsetzung von Luminol zu 3Aminophthalsäure, wobei es zur Chemolumineszenz kommt. Das emittierte Licht kann mit 34 MATERIAL UND METHODEN Hilfe eines Chemilumineszenzdetektor (LAS-3000 mini, Fujifilm) detektiert werden. Optional wurde zur Sichtbarmachung der Proteine eine Ponceau S-Färbung der Blots durchgeführt. Material: Blot-Puffer: 25 mM Tris, 192 mM Glycin, 20% (v/v) Methanol Ponceau S-Lösung: 0,15% (m/v) Ponceau S in 0,5% (v/v) Essigsäure TBS: 150 mM NaCl, 50 mM Tris/HCl (pH 7,4) TBS-T: TBS mit 0,1% (v/v) Tween 20 Block-Lösung: 5% (m/v) Magermilchpulver in TBS-T oder 5% (m/v) BSA in TBS-T ECL-Lösung: 1 ml ECL-Lösung 1 und 1 ml ECL-Lösung 2 ECL-Lösung 1: 2,5 mM Luminol, 0,4 mM p-Cumarsäure, 100 mM Tris/HCl (pH 8,0) ECL-Lösung 2: 5 mM H2O2, 100 mM Tris/HCl (pH 8,0) Polyvinylidendifluorid (PVDF) Blot-Membran (Schleicher und Schüll, Dassel, Deutschland) 2.6.2: „Stripping“ von Western-Blots Bei dem ECL-Verfahren können bereits entwickelte Blots nach dem Vorgang des „Stripping“ erneut für eine weitere Immundetektion mit anderen Antikörpern benutzt werden. Hierbei wurden gebundene Antikörper durch einen stark sauren Stripping-Puffer von der BlotMembran abgelöst. Nach dreimaligem Waschen der Blots konnten diese erneut zur Detektion von Proteinen (siehe Methoden 2.6.1) eingesetzt werden. Material: Stripping-Puffer: 200 mM Glycin (pH 3) TBS: 150 mM NaCl, 50 mM Tris/HCl (pH 7,4) TBS-T: TBS mit 0,1% (v/v) Tween 20 2.7: Proteinkonzentrationsbestimmung nach Bradford Die Proteinbestimmungsmethode nach Bradford beruht auf der bathochromen Verschiebung des Absorptionsmaximums des Farbstoffes Coomassie Brilliant Blue G-250 von 465 nm auf 595 nm. Einer Mischung aus 200 µl Bradford-Reagenz (5x) und 800 µl Wasser wurden bestimmte Volumen einer Proteinlösung zugegeben. Die Proben wurden bei 595 nm gegen einen Leerwert ohne Protein vermessen. Der Bradford-Faktor wurde aus einer Eichkurve mit der Referenz BSA bestimmt (hier: final 1, 3, 5, 10 µg/ml). Die Proteinkonzentration in µg/µl kann anhand der angegebenen Formel bestimmt werden. Formel: Extinktion X Bradford-Faktor X Verdünnungsfaktor X 0,01 Material: Bradford-Reagenz (5x): 0,035% (m/v) Coomassie Brilliant Blue G-250, 25% (v/v) Ethanol, 50% (v/v) H3PO4, 25% H2O BSA-Standard (2 mg/ml) (Uptima, Montlucon, Frankreich) 35 MATERIAL UND METHODEN 2.8: Proteinkonzentrationsbestimmung anhand der BCA-Methode Die BCA-Methode basiert auf der Biuret-Reaktion. Proteine bilden mit Cu2+-Ionen in alkalischer Lösung einen Komplex. Während der Reaktion werden die Cu2+-Ionen wahrscheinlich zu Cu+-Ionen reduziert, die ihrerseits mit Bicinchoninsäure (BCA) einen violetten Farbkomplex bilden. Mit dieser Methode können Proteinkonzentrationen zwischen 20 µg/ml bis 2 mg/ml bestimmt werden. Zur Proteinkonzentrationsbestimmung wurde das BCA-Assay-Protein Quantification Kit (Uptima) eingesetzt. Zu 100 µl der Probe, des Leerwerts oder des BSA-Standards (hier: final 1, 5, 12,5, 25, 37,5, 50, 100 µg/ml) wurden 2 ml BCA-Reagenz (50:1 Mischung aus BCA-Reagenz 1 und 2) gegeben. Nach Durchmischen wurden die Ansätze bei 37°C für 30 Minuten inkubiert. Nach Abkühlen wurde die Absorption des entstandenen Farbkomplexes bei OD562nm gegen einen Leerwert vermessen. Die Proteinkonzentration der Probe kann durch Interpolation aus dem BSA-Standard-Graphen (hier: Absorption bei OD562nm gegen BSA-Konzentration) bestimmt werden. Material: BSA-Standard (2 mg/ml) (Uptima, Montlucon, Frankreich) Über die Zusammensetzung des BCA-Reagenz 1 und 2 werden vom Hersteller keine genauen Angaben gemacht. 2.9.1: Probenvorbereitung für Massenspektrometrie Die zu untersuchenden Proben wurden in SDS-Probenpuffer aufgenommen. Durch den Zusatz von 1 M DTT (final: 50 mM) wurden die Disulfidbrücken reduktiv geöffnet. Anschließend wurden die Proteine durch zehnminütiges Kochen bei 95°C vollständig denaturiert. Nach Abkühlen wurden, um die Thiolgruppen zu alkylieren, 1 M IodacetamidLösung (final: 120 mM) zugeben und der Reaktionsansatz für 20 Minuten im Dunkeln inkubiert. Das überschüssige Iodacetamid wurde durch die Zugabe von weiteren 1 M DTT (final: 50 mM) und einer Inkubation von 20 Minuten im Dunkeln abreagiert. Anschließend wurden die Proteine mittels SDS-PAGE (siehe Methoden 2.3) aufgetrennt. 2.9.2: Massenspektrometrie (MALDI-TOF) Zur eindeutigen Identifizierung von Proteinen wurde MALDI-TOF-Massenspektrometrie (Ionisationsmethode: MALDI [matrix-assisted laser desorption/ionisation], Detektionsmethode: TOF [time of flight]) eingesetzt. Die Messungen wurden mit dem Massenspektrometer Biflex II (Bruker), ausgerüstet mit einem Stickstoff-LASER (λ = 337 nm), durchgeführt. Die zu untersuchenden Proteine wurden als Banden aus getrockneten Coomassie-Gelen ausgeschnitten. Zur Entfernung des Coomassie-Farbstoffes sowie weiterer Verunreinigungen wurden die Proben mit Entfärbelösung für 3 Stunden bei 50°C 36 MATERIAL UND METHODEN behandelt. Nach vollständigem Trocknen des Gelstücks durch Vakuumzentrifugation wurden die Proteine mit 25 µl Trypsin-Lösung bei 37°C über Nacht proteolytisch verdaut. Jeweils 1 µl des Verdauansatzes sowie 1 µl gesättigte Matrix-Lösung wurden auf einen Probenträger aufgetragen. Bei dieser sogenannten dried droplet-Methode bildet die Probe mit der MatrixLösung Kokristalle aus. Die Messungen der Massen der einfach-positiv geladenen Peptide wurden im Reflektor-Modus mit dem Prinzip der verzögerten Extraktion (delayed extraction) durchgeführt. Die Toleranzgrenze wurde auf 0,2 Dalton festgelegt. Zur Kalibrierung der Massenspektren wurde der Matrix-Lösung als externer Standard eine Mischung von Peptiden mit bekannten Massen beigemischt. Die für die Identifizierung durchgeführte Zuordnung der Massen der Peptide zu einem Protein wurde mit Hilfe der Software ProFound sowie Mascot durchgeführt. Bei diesem sogenannten Verfahren des peptide mapping wurden die gemessenen Peptidmassen mit errechneten Peptidmassen theoretisch trypsinierter Proteine verglichen und können somit wie ein Fingerabdruck dem Protein zugeordnet werden. Material: Entfärbelösung: 1:1 Mischung aus Acetonitril und 50 mM Ammoniumhydrogencarbonat (pH 7,8) Trypsin-Lösung: 5 µg/ml Trypsin in Ammoniumhydrogencarbonat (pH 7,8) Matrix-Lösung: gesättigte Lösung α-Cyano-4-hydroxyzimtsäure in 1:1 Mischung aus Trifluoressigsäure und Acetonitril Peptid-Kalibrierungsstandard: (Bruker, Bremen, Deutschland): Angiotensin I (1046,5418), Angiotensin II (1296,6848), Substanz P (1347,7354), Bombesin (1619,8223), ACTH AS 1-17 (2093,1983), ACTH AS 18-39 (2465,1983), Somatostatin 28 (3147,4710). In Klammern sind die jeweiligen monoisotopischen Peaks ([M+H]+) der Peptide angeben. 2.9.3: Massenspektrometrie (LC-MS/MS) Zum massenspektrometrischen Nachweis der in vivo- bzw. in vitro-Glukosylierung von GTPasen durch TpeL wurde LC-MS/MS (Dr. Schlosser, ZBSA, Freiburg) eingesetzt. Die zu untersuchenden Proteinbanden wurden aus den Coomassie-gefärbten SDS-PAGE-Gelen ausgeschnitten. Die Proteinbanden wurden mit 30% (v/v) Acetonitril entfärbt, anschließend in reinem Acetonitril aufgenommen und schließlich über einen Vakuum-Konzentrator (Concentrator 5301, Eppendorf) getrocknet. Der Protease-Verdau (Trypsin, Thermolysin) wurde in 0,1 M NaHCO3 (pH 8) mit 0,1 μg Protease je Gelstück (Trypsin: bei 37°C über Nacht, Thermolysin: 60°C für 2,5 h) durchgeführt. Die Peptide wurden mit 5% (v/v) Ameisensäure extrahiert Für die MS-Messungen wurde ein Q-TOF-Massenspektrometer (Agilent 6520, Agilent Biotechnologies) mit nanoflow-System (Agilent 1200) und HPLC-Chip (Zorbax 300SB-C18)-ESI-interface eingesetzt. Die Peptide wurden mit einem linearen Acetonitril-Gradient (1%/min, bei 300 nl/min) eluiert. Die MS-Daten wurden mit Mascot Server 2.3 und SwissProt analysiert. 37 MATERIAL UND METHODEN 2.10: Expression von rekombinanten Proteine in Escherichia coli Die Expression der kleinen GTPasen erfolgte mit dem pGEX-Vektorsystem in Escherichia coli. Mit diesem Expressionssystem ist es möglich, Proteine als GST-Fusionsproteine zu exprimieren und aufzureinigen. Ein Liter Hauptkultur wurde mit 20 ml Vorkultur angeimpft und bei 37°C bis zu einer OD600nm von 0,6 bis 0,8 geschüttelt. Nach Induktion mit 1 M IPTGLösung (final: 200 µM) wurden die Bakterien bei 37°C für weitere 6 Stunden kultiviert. Die Expression der Glukosyltransferase-Domäne von TpeL erfolgte im pET-Vektorsystem ebenfalls in Escherichia coli. Mit dem pET-Expressionssystem (hier: pET28a) ist es möglich, Proteine fusioniert mit einem N-terminalen 6xHistidin-tag zu exprimieren und aufzureinigen. Im Gegensatz zum pGEX-Vektorsystem wurde die Induktion der Proteinexpression mit 1 M IPTG-Lösung (final: 1 mM) durchgeführt. Anschließend wurden die Bakterien bei 29°C über Nacht inkubiert. Material: Isopropyl-β-D-thiogalactopyranosid (IPTG) (Roth, Karlsruhe, Deutschland) 2.11: Expression von rekombinanten Proteinen in Bacillus megaterium Die Expression der Holotoxine und Toxinfragmente erfolgte mit dem pHis1522-Vektorsystem in Bacillus megaterium. Mit diesem Expressionssystem ist es möglich, Proteine mit einem Cterminalen 6xHistidin-tag zu exprimieren und aufzureinigen. Mit je 100 ml Vorkultur wurde ein Liter Hauptkultur angeimpft. Die Bakterien-Kultur wurde bei 37°C bis zu einer OD600nm von 0,3 inkubiert und anschließend durch Zugabe von 5% (m/v) Xylose die Proteinexpression induziert. Die Bacilli wurden entweder bei 37°C bis zu einer OD600nm von 1,5 (4 bis 6 h) oder bei 29°C über Nacht kultiviert. Material: Xylose (AppliChem, Darmstadt, Deutschland) 2.12: Zellaufschluss Nach der Zellernte wurde das Bakterien-Pellet in je 2 ml Lysepuffer je Gramm Pellet resuspendiert. Die Zelllyse erfolgte bei Escherichia coli-Kulturen mittels Ultraschall (Sonopuls HD60, Bandelin) (Einstellungen: 3x 30 s, jeweils 30 s Pause, 70% Leistung). Die Zelllyse von Bacillus megaterium-Kulturen erfolgte über einen dreimaligen Durchlauf über einen Mikrofluidizer (M110P, Microfluidics) bei 18000 bis 20000 psi. 38 MATERIAL UND METHODEN 2.13: Proteinaufreinigung mittels Affinitätschromatographie 2.13.1: Proteinaufreinigung über 6xHistidin-tag Nach erfolgtem Zellaufschluss wurde das Zelldebris durch Zentrifugation (bei Ni2+-Säule [HisTrap, GE-Healthcare]: Ultrazentrifugation [165000 g, 1h, 4°C], Ni2+-beads [Ni2+- IDAProtino-Resin, Macherey-Nagel GmbH & Co. KG]: Zentrifugation [48000g, 1h, 4°C]) entfernt. Anschließend wurde das klare Zelllysat mit Ni2+-beads inkubiert bzw. über eine Ni2+-Säule gegeben. Das gebundene Protein wurde mit 10 bis 20 Säulenvolumen einer Mischung aus Puffer A und B (Puffer A: 96%, Puffer B: 4%, entspricht 20 mM Imidazol) gewaschen und anschließend mit Puffer B eluiert. Der N-terminale 6xHistidin-tag der GlukosyltransferaseDomäne von TpeL (TpeL1-542) konnte optional durch eine Thrombinspaltung (siehe Methoden 2.13.3) abgetrennt werden. Material: Protokoll Nr.1 (für rekombinante CGT/-Fragmente): Lysepuffer: 20 mM Tris/HCl (pH 8,0), 300 mM NaCl, 20 mM Imidazol, 10% (m/v) Glycerol, 500 µM EDTA, ProteaseInhibitor-Cocktail Complete (Roche Diagnostics, Basel, Schweiz), DNAse Puffer A: 20 mM Tris/HCl (pH 8,0), 300 mM NaCl, 10% (m/v) Glycerol, Puffer B: 20 mM Tris/HCl (pH 8,0), 300 mM NaCl, 500 mM Imidazol, 10% (m/v) Glycerol Protokoll Nr.2 (für rekombinantes TpeL1-542): Lysepuffer: 50 mM HEPES (pH 8,0), 300 mM KCl, 20 mM Imidazol, 10% (m/v) Glycerol, 10 mM β-Mercaptoethanol, 500 µM EDTA, Protease-Inhibitor-Cocktail Complete (Roche Diagnostics, Basel, Schweiz), DNAse Puffer A: 50 HEPES (pH 8,0), 300 mM KCl, 10% (m/v) Glycerol, 10 mM β-Mercaptoethanol Puffer B: 50 HEPES (pH 8,0), 300 mM KCl, 500 mM Imidazol, 10% (m/v) Glycerol, 10 mM β-Mercaptoethanol 2.13.2: Proteinaufreinigung über GST-tag Das Zelldebris wurde durch Zentrifugation (bei Glutathion-Säule [GST-Trap, GE Healthcare]: Ultrazentrifugation [165000g, 1 h, 4°C], Glutathion-beads [Glutathion-Sepharose 4B, GE Healthcare]: Zentrifugation [48000g, 1 h, 4°C]) abgetrennt. Das so gewonnene Lysat wurde mit Glutathion-beads inkubiert bzw. über eine Glutathion-Säule geben. Nach Waschen mit je 5 bis 10 Säulenvolumen Waschpuffer-1 und -2 konnten die GST-Fusions-Proteine mit Elutionspuffer eluiert werden. Alternativ können die Proteine über eine Thrombinspaltung (siehe Methoden 2.13.3) von der Glutathion-Matrix abgelöst werden, wobei der GST-Anteil an der Matrix verbleibt. Material: Lysepuffer: 20 mM Tris/HCl (pH 7,4), 300 mM NaCl, 5 mM MgCl2 Waschpuffer-1: 20 mM Tris/HCl (pH 7,4), 300 mM NaCl Waschpuffer-2: 50 mM Tris/HCl (pH 8,0) 39 MATERIAL UND METHODEN Elutionspuffer: 50 mM Tris/HCl (pH 8,0), 10 mM Glutathion 2.13.3: Thrombinspaltung Die Thrombinspaltung erfolgte im Thrombinspaltpuffer über Nacht bei 4°C. Nach Spaltung wurde das Protein mit Thrombinspaltpuffer (ohne Thrombin) eluiert. Das in den Eluaten enthaltende Thrombin wurde über Benzamidin-beads (Benzamidin-Sepharose 6B, GE Healthcare) entfernt. Hierzu wurden 50 µl Benzamidin-beads je 500 µl Eluat zugegeben und bei 4°C für 10 bis 20 Minuten inkubiert. Die Benzamidin-beads wurden ihrerseits über Zentrifugation von der Proteinlösung abgetrennt. Material: Thrombinspaltpuffer-1 (für rekombinante kleine GTPasen): 50 mM Tris/HCl (pH 8,0), 150 mM NaCl, 2,5 mM CaCl2, 50 U Thrombin (Sigma-Aldrich, Taufkirchen, Deutschland) je 500 µl Thrombinspaltpuffer Thrombinspaltpuffer-2 (für rekombinantes TpeL1-542): 50 mM HEPES (pH 8), 150 mM KCl, 10% (m/v) Glycerol 2.14: Anionenaustauschchromatographie Bei der Anionenaustauschchromatographie binden Proteine durch elektrische Wechselwirkungen an eine Matrix, die aus positiv geladenen, quartären AmmoniumGruppen besteht. Die Bindung des Proteins hängt von dessen isoelektrischen Punkt sowie von der Ionenstärke und dem pH-Wert des Puffers ab. Durch eine Erhöhung der Ionenstärke des Puffers ist es möglich, an die Matrix gebundene Proteine gezielt wieder von dieser zu lösen. Zunächst wurden die Proteine durch Dialyse oder Umpuffern in Puffer A überführt. Die Proteinlösung wurde über eine zuvor mit jeweils fünf Säulenvolumen Puffer A, Puffer B und Puffer A äquilibrierten Säule (MonoQ 5/50 GL, Resource 6 ml, beide GE Healthcare) gegeben. Das gebundene Protein wurde mit 5 bis 10 Säulenvolumen Puffer A gewaschen und anschließend mit einem kontinuierlichen Gradienten an Puffer B eluiert. Material: Puffer A: 50 mM Tris/HCl (pH 7,5), Puffer B: 50 mM Tris/HCl (pH 7,5), 1 M NaCl 2.15: Gelfiltration Die Gelfiltration ist ein Verfahren, dass auf der Trennung von Proteinen nach Größe über ein Gelkompartiment basiert. Hierbei wandern größere Proteine schneller, da sie weniger in Wechselwirkung mit der Gelmatrix treten. Die stationäre Phase der eingesetzten Säulen (Superdex S200 10/300 GL, HiLoad Superdex S200 16/60, beide GE Healthcare), besteht aus einer Agarose/Dextran-Matrix mit definiertem Porendurchmesser. Diese Säulen sind geeignet zur Auftrennung von Proteinen mit einer Größe von 10 bis 600 kDa. Vor dem 40 MATERIAL UND METHODEN eigentlichen Lauf wurde die Säule mit jeweils einem Säulenvolumen Wasser und einem Säulenvolumen des gewünschten Gelfiltrationspuffers äquilibriert. Die Gelfiltration wurde zur Nachreinigung oder zum Umpuffern von Proteinlösungen benutzt. Material: Gelfiltrationspuffer-1 (für rekombinante CGT/-Fragmente): 20 mM Tris/HCl (pH 7,4), 150 mM NaCl, 10% (m/v) Glycerol Gelfiltrationspuffer-2 (für rekombinantes TpeL1-542): 50 mM HEPES (pH 7,4), 150 mM KCl, 10% (m/v) Glycerol 2.16: Aufreinigung nativer Toxine aus Clostridien Mit dem in den folgenden Abschnitten beschriebenen Verfahren ist es möglich, in ausreichenden Mengen native Toxin-Präparationen der CGTs aus Clostridien aufzureinigen. Hierzu zählten Toxin A und B aus Clostridium difficile VPI10463, Letales Toxin aus Clostridium sordellii 6018 und α-Toxin aus Clostridium novyi 19402. 2.16.1: Kultivierung von Clostridien Aus einer Stichkultur, einem Glycerolstock oder einer älteren Vorkultur wurden 80 ml BHIMedium angeimpft und unter anaeroben Bedingungen (zwei Platten Anaerocult A in 2,5 l Anaerobiertopf) bei 37°C über Nacht kultiviert. Die Hauptkultur bestand aus 4 l BHI-Medium in einem 5 l Erlenmeyer-Kolben. In diesen Kolben wurde ein am unteren Ende verschlossener 2,5 m langer Dialyse-Schlauch gegeben. Nach dem Autoklavieren der Hauptkultur wurde die Vorkultur mit Hilfe von 500 ml 0,9% (m/v) NaCl-Lösung in diesen Dialyse-Schlauch überführt. Anschließend wurde der Dialyse-Schlauch ebenfalls am oberen Ende verschlossen und die Bakterien-Kultur bei 37°C für bis zu 72 Stunden kultiviert. Der Dialyse-Schlauch besaß eine Porengröße von 14 kDa und bildet somit ein Reservoir für die CGTs sowie für die Clostridien. Material: Dialyse-Schlauch (cut-off: 14 kDa) (Kummer, Freiburg, Deutschland) 2.16.2: Aufreinigung nativer Toxine aus Kulturüberständen Nach Entfernen der Clostridien durch Zentrifugation wurden die nativen Toxine aus den Kulturüberständen mit einer Ammoniumsulfat-Fällung (siehe Methoden 2.2) ausgefällt. Nach Zentrifugation wurde das Präzipitat in 20 ml Puffer A aufgenommen und für 36 Stunden gegen Puffer A mit dreimaligem Pufferwechsel (alle 12 h) dialysiert. Die nativen Toxine wurden nachfolgend mit Anionenaustauschchromatographie (siehe Methoden 2.14) 41 MATERIAL UND METHODEN aufgereinigt. Gemäß ihrem isoelektrischen Punkt können die Toxine bei unterschiedlichen NaCl-Konzentrationen von der Säule eluiert werden. 2.16.3: Affinitätschromatographie von nativem Toxin A Die Lektin-ähnliche Eigenschaft von Toxin A versetzen es in die Lage, über seine im CTerminus befindlichen CROP-Bereiche Kohlenhydratstrukturen zu binden. Toxin A bindet bei 4°C hochaffin an das Glykoprotein Thyreoglobulin. Diese Bindung wird bei Erhöhung der Temperatur auf 37°C wieder gelöst. Dies ermöglicht es, Toxin A über eine Thyreoglobulinhaltige Matrix aufzureinigen. Hierzu wurden zunächst 1,6 g CNBr-aktivierte-beads (CNBraktivierte Sepharose 4B, GE Healthcare) in 1 mM HCl gequollen, anschließend mit 16 ml Thyreoglobulin-Lösung (60 mg in Kopplungspuffer) versetzt und bei 4°C über Nacht inkubiert. Ungebundenes Thyreoglobulin wurde durch dreimaliges Waschen mit Kopplungspuffer entfernt. Noch reaktive CNBr-Gruppen wurden durch die Zugabe von Glycin-Lösung abgesättigt. Die Matrix wurde je dreimal mit 30 ml Waschpuffer-1 und -2 sowie mit 20 ml TBS-Puffer gewaschen. Die Toxin-Lösung wurde mit Hilfe einer Peristaltikpumpe über Nacht bei 4°C über die Matrix zirkuliert. Nach Waschen mit kaltem TBS-Puffer wurde Toxin A bei 37°C nach 20 Minuten Inkubation mit TBS-Puffer eluiert. Material: Thyreoglobulin (Sigma-Aldrich, Taufkirchen, Deutschland) Kopplungspuffer: 200 mM NaHCO3 (pH 8,5), 500 mM NaCl Glycin-Lösung: 200 mM Glycin (pH 8,0) Waschpuffer-1: 100 mM Na-Acetat (pH 4,0), 500 mM NaCl Waschpuffer-2: 100 mM Tris/HCl (pH 8,0), 500 mM NaCl TBS-Puffer: 50 mM Tris/HCl (pH 7,5), 150 mM NaCl 2.17: Umpuffern von Proteinlösungen Die Umpufferung von Proteinlösung erfolgte über das Prinzip der Gelfiltration. Die hierzu eingesetzten Säulen (PD-10, HiTrap, beide GE-Healthcare) bestehen aus einer feinporigen Sephadex G-25 Matrix. Nur Moleküle mit einer molaren Masse unter 5 kDa können mit der Matrix interagieren. Da Proteine die Säule ungehindert passieren können, ist es möglich, diese von Puffer-Substanzen zu trennen. Nach Äquilibrieren mit 25 ml des gewünschten Puffers wurden 2,5 ml der Proteinlösung über die Säule gegeben. Anschließend wurde das Protein mit 3,5 ml Puffer eluiert. 42 MATERIAL UND METHODEN 2.18: GST-pull down Mit dem GST-pull down ist es möglich die Interaktion zwischen zwei Proteinen nachzuweisen. Hierbei wird ein Interaktionspartner als GST-Fusionsprotein in Bakterien exprimiert und über Glutathion-beschichtete beads immobilisiert. Anschließend wird der Interaktionspartner aus Zelllysaten bzw. dem Reaktionsansatz präzipitiert und durch Western-Blot nachgewiesen. Mit diesem Versuchsaufbau ist es möglich, aktives, GTP gebundenes Ras in Zelllysaten nachzuweisen. Hierzu wurde der Ras-Effektor Raf1B-RBD (Ras-binding-domain) als GST-Fusionsproteine mit pGEX-Vektorsystem in Escherichia coli Rosetta exprimiert und eingesetzt. Dazu wurden 400 ml Bakterienkultur bei 37°C bis zu einer OD550nm von 0,4 kultiviert und anschließend nach der Zugabe von PMSF (final: 400 µM) die Proteinexpression durch IPTG (final: 500 µM) induziert. Die Proteinexpression erfolgte bei 25°C für 4 Stunden. Das Bakterienpellet wurde in 5 ml Lysepuffer resuspendiert und für 15 Minuten bei 4°C auf einem Überkopfschüttler inkubiert. Nach der Zugabe von Lysozym (final: 1 mg/ml) und DNAse (0,5 mg/ml) wurde der Ansatz für weitere 15 Minuten inkubiert und anschließend die Zelldebris durch Zentrifugation (4000 g, 15 min, 4°C) entfernt. Je Reaktionsansatz wurden 10 µl Glutathion-beads (Glutathion-Sepharose 4B, GE Healthcare) zunächst äquilibriert und anschließend mit 10 µl Bakterienlysat in 600 µl IP-Puffer bei 4°C für 90 Minuten auf einem Überkopfschüttler inkubiert. Die beads-Suspension wurde anschließend dreimal mit IP-Puffer und einmal mit GST-fish-Puffer gewaschen. 1 mg des Gesamtlysats (siehe Methoden 2.5) wurde mit GST-Raf1B-RBD-beads in GST-fish-Puffer bei 4°C für 30 Minuten inkubiert. Nach viermaligem Waschen mit GST-fish-Puffer wurden die beads mit 10 µl SDS-Probenpuffer aufgekocht. Die Proteine wurde mittels SDS-PAGE (siehe Methoden 2.3) aufgetrennt und Ras im Western-Blot (siehe Methoden 2.6.1) detektiert. Material: Lysepuffer: 50 mM Tris/HCl (pH 7,5), 100 mM NaCl, 10% (m/v) Glycerol, 2 mM MgCl2, 5 mM DTT, 1% NP-40, 0,3% NaDesoxycholat, 100 µM PMSF IP-Puffer: 50 mM Tris/HCl (pH 7,5), 100 mM NaCl, 1 mM DTT GST-fish-Puffer: 50 mM Tris/HCl (pH 7,5), 100 mM NaCl, 10% (m/v) Glycerol, 2 mM MgCl2, 1% NP-40 SDS-Probenpuffer (5x): 0,01% (m/v) Bromphenolblau, 125 mM Tris/HCl (pH 6,8), 0,1 mM DTT, 10% (m/v) SDS, 29% (m/v) Glycerol 2.19: Glukosylierung kleiner GTPasen Die Enzymaktivität der CGTs kann in vitro bestimmt werden. Hierzu wurden die Toxine mit ihren Proteinsubstraten, den kleinen GTPasen, sowie [14C]-markiertem Uridinphosphatzucker (UDP-[14C]-Glukose bzw. UDP-[14C]-N-Acetylglukosamin) bei 37°C für unterschiedliche Zeitpunkte inkubiert. Die Glukosylierungsreaktion wurden mit 1 bis 100 nM Toxin, 5 bis 10 µM Proteinsubstrat sowie 10 µM [14C]-markiertem UDP-Zucker in Glukosylierungspuffer durchgeführt. Nach Trennung der Proteine durch SDS-PAGE wurde der Transfer des 43 MATERIAL UND METHODEN radioaktiven Zuckers auf die kleine GTPase mittels Autoradiographie (Phosphorimager SI System, GE Healthcare) detektiert. Die Daten wurden mit der Software ImageQuant analysiert. Material: Glukosylierungspuffer (10x): 500 mM HEPES (pH 7,5), 20 mM MgCl2, 10 mM MnCl2, 1 M KCl, 1 mg/ml BSA 2.20: Bestimmung der Glukohydrolase-Aktivität In Abwesenheit der Proteinsubstrate hydrolysieren die CGTs ihre Kosubstrate zu UDP und den entsprechenden Zuckern. Durch Papier-Dünnschichtchromatographie lassen sich die Hydrolyse-Produkte voneinander trennen. Unter der Verwendung von UDP-[14C]-Zuckern konnte das Verhältnis von UDP-Zucker zu Zucker ermittelt und hieraus die HydrolaseAktivität der Toxine bestimmt werden. Hierzu wurden 100 nM bis 1 µM Toxin mit 20 µM UDP[14C]-Zucker sowie 100 µM UDP-Zucker im Glukohydrolasepuffer bei 37°C für unterschiedliche Zeitpunkte inkubiert. Anschließend wurde 1 µl der Probe auf eine KieselgelPlatte (Silica-Gel 60 F254) aufgetragen und die Dünnschicht-Chromatographie durchgeführt. Als mobile Phase wurde n-Butanol/Essigsäure/Wasser-Mischung (Volumenverhältnis: 5/3/2) benutzt. Nach Trocknen der Platten konnten diese mittels Autoradiographie (Phosphorimager SI System, GE Healthcare) ausgewertet werden. Die Daten wurden mit der Software ImageQuant analysiert. Material: Glukohydrolasepuffer (10x): 500 mM HEPES (pH 7,5), 20 mM MgCl2, 1 mM MnCl2, 1 M KCl, 1 mg/ml BSA Silica-Gel 60 F254-Platten (Merck, Darmstadt, Deutschland) 2.21: In vitro-Bestimmung der autoproteolytischen Spaltung Die autoproteolytische Spaltung der Holotoxine und Toxinfragmente kann in vitro durch die Zugabe von Inositolhexakisphosphat eingeleitet werden. Hierzu wurden 2 µg Toxin mit unterschiedlichen Konzentrationen an InsP6 bei 37°C für die angegebenen Zeitpunkte in Spaltungspuffer inkubiert. Nach Auftrennung der Proteine durch SDS-PAGE konnte das Ausmaß der Spaltung anhand des Entstehens der beiden Spaltprodukte bewertet werden. Material: Spaltungspuffer: 100 mM Tris/HCl (pH 7,4) oder 100 mM Na-Acetat (pH 5) 44 MATERIAL UND METHODEN 2.22: InsP6-Filterbindung Die Bindung von Inositolhexakisphosphat (InsP6) an die Cysteinprotease-Domäne der Holotoxine bzw. Toxinfragmente kann über einen Filterbindungsversuch bestimmt werden. Es wurden 1 µM Toxin mit einer Mischung aus 200 nM [3H]-InsP6 und 4,8 µM InsP6 bei 4°C für 10 Minuten inkubiert. Um eine unspezifische Bindung von InsP6 an die Gefäßwände zu verhindern wurde den Reaktionsansätzen 50 µg BSA zugesetzt. Der gesamte Reaktionsansatz wurde über eine angefeuchtete Nitrocellulose-Membran gefiltert. Nach Waschen mit 3 ml Bindungspuffer wurde die Membran mit 3 ml Szintillationsflüssigkeit versetzt und die gebundene Radioaktivität mittels Szintillation (TriCarb 2900TR, Packard) gemessen. Die Messwerte entsprechender Kontrollexperimente ohne Toxin wurden von den jeweiligen Messwerten abgezogen. Material: Bindungspuffer: 100 mM Tris/HCl (pH 7 bzw. 7,4) oder 100 mM Na-Acetat (pH 5 bzw. 6) Nitrocellulose-Membran 0,45 µM (Whatman, Göttingen, Deutschland) Bovines Serum-Albumin (BSA) (Sigma-Aldrich, Taufkirchen, Deutschland) Ultima Gold (Perkin Elmer Life Science, Boston, USA) 2.23: Isothermale Titrationskalorimetrie Isothermale Titrationskalorimetrie (ITC) ist eine biophysikalische Methode, mit der es möglich ist, thermodynamische Parameter einer biochemischen Bindung zu untersuchen. Diese Methode wurde eingesetzt, um die Bindung von InsP6 an die Cysteinprotease-Domäne der verschiedenen CGTs zu charakterisieren. Das eingesetzte Kalorimeter (VP-ITC oder ITC 200, GE Healthcare) besteht aus zwei Zellen, einer Messzelle sowie Referenzzelle. Die Messzelle besitzt ein Fassungsvermögen von circa 1,8 bis 2,0 ml (für VP-ITC). In dieser Zelle findet die zu untersuchende Reaktion statt. Die Referenzzelle ist mit Wasser gefüllt. Beide Zellen sind von einem adiabatischen Schutzschild umgeben, wodurch der Wärmeaustausch mit der Umgebung unterbunden wird. Die Temperaturdifferenz zwischen den beiden Zellen wird durch kontinuierliches Heizen konstant gehalten. Durch Bindung eines Liganden an ein Protein kommt es zu einer exothermen bzw. endothermen Reaktion und somit zu einer Veränderung der Temperatur. Während der Messung werden konstante Volumen an Ligand zu der in der Messzelle befindlichen Proteinlösung gegeben. Bei einer exothermen Reaktion wird durch die Bindung des Liganden Wärme im Vergleich zur Referenzzelle in der Messzelle frei. Hierdurch kommt es zu einem Abfall der Heizleistung. Die Veränderung der Heizstromleistung (in µcal/s) kann in einem Rohdatendiagramm gegen die Zeit (in min) aufgetragen werden. Das Rohdatendiagramm enthält Zacken, sogenannte spikes, aus deren Fläche kann die Wärmemenge, die bei jeder Injektion freigesetzt wird, bestimmt werden. In einem sekundären Diagramm wird die berechnete Wärmemenge gegen 45 MATERIAL UND METHODEN das molare Verhältnis der beiden Bindepartner dargestellt. Aus dem Kurvenverlauf des Graphen können direkt die Bindekonstante KD sowie die Stöchiometrie der Bindung bestimmt werden. Die Messungen wurden mit 50 bis 100 µM Toxin und einer 10-fach höheren Konzentration an InsP6 durchgeführt. Die Messungen erfolgten bei einer konstanten Temperatur von 25°C. 2.24: HPTS/DPX–Freisetzung aus Liposomen Die Porenbildung der CGTs kann in vitro durch den Ausstrom eines Fluorophors aus Liposomen (LUV: large unilamellar vesicles) gemessen werden (Genisyuerek et al., 2011). Die Liposomen wurden aus Ei-Phosphatidylcholin und Cholesterol in einem Verhältnis 3:1 sowie 0,8% 1,2-Dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)-iminodiessigsäure)- 2+ succinyl] (Ni ) mit der Filmmethode hergestellt. Die Herstellung der Liposomen sowie deren Beladung mit einer Mischung aus dem Fluorophor 8-Hydroxypyren-1,3,6-trisulphonsäure (HPTS) und dem Quencher p-Xylen-bis(N-pyridiniumbromid) (DPX) wurde wie von PeschkaSüss et al. beschrieben durchgeführt (Peschka-Süss and Schubert, 2003). Zunächst wurden Liposomen (finale Konzentration: 5 µM) und die CGTs in TBS-Puffer in einer 3 ml Quarzküvette unter ständigem Rühren bei 25°C durchmischt. Die Porenbildung wurde durch das Ansäuern (pH 5) durch die Zugabe von HCl induziert. Durch die entstandene Pore können sowohl Fluorophor als auch Quencher aus den Liposomen ausströmen. Dieser Ausstrom ist durch eine Erhöhung der Fluoreszenz erkennbar, da in Lösung, außerhalb des Liposoms, die Fluoreszenz von HPTS nicht länger durch DPX gequencht werden kann. Die Veränderung der Fluoreszenz von HPTS kann fluoreszenz-spektroskopisch (Spektrometer LS-50B, Perkin Elmer), nach Anregung bei einer Wellenlänge von λ=403 nm, bei λ=510 nm gemessen werden. Zur Bestimmung der maximal möglichen Freisetzung an HPTS wurden die Liposomen durch Zugabe von Triton X-100 (finale Konzentration: 0,3% [m/v]) vollständig solubilisiert. Von diesem gemessenen Fluoreszenz-Wert wurde die Hintergrundfluoreszenz (Basislinie) abgezogen. Das Ausmaß der Porenbildung konnte nun prozentual zur maximalen Freisetzung angegeben werden. Material: Ei-Phosphatidylcholin (Lipoid, Ludwigshafen, Deutschland) Cholesterol (Sigma-Aldrich, München, Deutschland) 1,2-Dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)-iminodiessigsäure)-succinyl](Ni2+Salz) (Avanti Polar Lipids, Alabaster, USA) 8-Hydroxypyren-1,3,6-trisulphonsäure (HPTS) (Molecular Probes, Darmstadt, Deutschland) p-Xylen-bis(N-pyridiniumbromid) (DPX) (Molecular Probes, Darmstadt, Deutschland) 46 MATERIAL UND METHODEN 2.25: Bestimmung der Fluoreszenz-Löschung von Tryptophanen nach Ligandenbindung („W-quenching“) Mit dieser Methode ist es möglich, die Bindung eines niedermolekularen Liganden an ein Protein zu messen. Sowohl freie als auch proteingebundene Tryptophane besitzen Fluoreszenzeigenschaften. Nach Anregung der Tryptophane mit UV-Licht erfolgt eine Fluoreszenzemission zwischen 320 und 400 nm. Die Fluoreszenzemission ist abhängig von der Polarität der Umgebung der Tryptophane im Protein. Kommt es nun zu einer Bindung eines Liganden an das Protein, kann dies zu einer Konformationsänderung führen und somit zu einer Änderung der Fluoreszenzemission der Tryptophane. Die Erniedrigung der Emissionsintensität wird als Tryptophan-quenching bezeichnet. Zur Bestimmung der KDWerte wurden 1 µM TpeL1-542 mit steigenden Konzentrationen an UDP-N-Acetylglukosamin und UDP-Glukose (eingesetzte Konzentrationen: 1/10 KD bis 10x KD) im Reaktionspuffer in 96-well-Platten (schwarze 96-well-Platte, Greiner) zusammengegeben. Die Fluoreszenzemission der Tryptophane wurde nach Anregung bei 295 nM bei 340 nM (Infinite M200, Tecan) ausgelesen. Die Hintergrundfluoreszenz der UDP-Zucker wurde ebenfalls bestimmt und von den Messwerten abgezogen. Anschließend wurde das maximale quenching der Fluoreszenz bestimmt und relativ zur höchsten UDP-Zucker-Konzentration angegeben. Aus einem Diagramm, in dem das relative Fluoreszenz-quenching gegen die UDP-Zucker-Konzentration aufgetragen ist, kann die Dissoziationskonstante KD ermittelt werden. Material: Reaktionspuffer: 50 mM HEPES (pH 7,5), 150 mM KCl, 5 mM CaCl2 2.26: Immunopräzipitation von Ras Unter dem Begriff Immunopräzipitation versteht man das gezielte Ausfällen eines Proteins oder Peptids mit Hilfe eines spezifischen Antikörpers. Diese Methode wurde eingesetzt, um Ras aus HeLa-Zellen zu isolieren. Hierzu wurden HeLa-Zellen (vier 15 cm Schalen) zunächst lysiert (siehe Methoden 2.5) und anschließend das erhaltene Gesamtlysat auf eine Konzentration von 2 mg/ml eingestellt. Die Präzipitation von Ras erfolgte mit einem spezifischen Ras-Antikörper (anti-v-H-Ras MabY13-259). Hierzu wurden 20 µg Antikörper sowie 80 µl Protein G-Sepharose-beads (Sigma) dem Ansatz zugegeben und bei 4°C über Nacht inkubiert. Die Protein G-Sepharose-beads wurden zuvor mit RIPA-Puffer äquilibriert. Protein G bindet mit hoher Affinität die FC-Region von Immunglobulinen des IgG-Isotyps und ermöglicht somit die Abtrennung des Antikörpers aus dem Lysat. Nach viermaligem 47 MATERIAL UND METHODEN Waschen wurden schließlich die Protein-G-Sepharose-beads in 20 µl Lämmli-Puffer aufgekocht und die Proteine mittels SDS-PAGE (siehe Methoden 2.3) aufgetrennt. 3: Zellbiologische Methoden 3.1: Zellvergiftungsversuche Die Bestimmung der in vivo-Aktivität der CGTs kann anhand der charakteristischen morphologischen Veränderung eukaryoter Zellen nach Intoxikation erfolgen. Hierzu wurden die Toxine in unterschiedlichen Konzentrationen direkt nach Medium-Wechsel in das Kulturmedium der Zellen gegeben und bei 37°C inkubiert. Die Zellabrundung konnte mit einem invertiertem Lichtmikroskop (Axiovert 25, Zeiss) beobachtet und durch die Aufnahme von Bildern dokumentiert werden. 3.2: Untersuchung der Porenbildung an biologischen Membranen Die Porenbildung der CGTs nach deren Ansäuerung in die endosomale Membran kann an der Oberfläche von eukaryoten Zellen nachgeahmt und bestimmt werden. Zunächst wurden CHO-K1-Zellen (in 24-well-Kulturschalen) ausgesät, mit 1 µCi/ml [86Rb+]-Ionen versetzt und für 48 Stunden kultiviert. In dieser Zeit stellte sich ein Äquilibrium zwischen der intrazellulären und extrazellulären Konzentration an [86Rb+]-Ionen ein. Am Tag der Messung wurden durch den Wechsel des Mediums die extrazellulären [86Rb+]-Ionen entfernt. Um eine Aufnahme der Toxine während des Experiments zu verhindern, wurden dem Medium 100 µM Bafilomycin A1 zugegeben. Zunächst wurden die Ansätze bei 37°C für 20 Minuten inkubiert und anschließend auf Eis abgekühlt. Nach Abkühlen der Zellen wurden in 500 µl Medium die CGTs zugegeben und die Ansätze auf Eis für eine Stunde inkubiert. Bei 4°C findet lediglich eine Bindung der Toxine an die Zytoplasmamembran der Zelle statt. Durch zweimaliges Waschen mit kaltem PBS konnte ungebundenes Toxin entfernt werden. Die Porenbildung wurde durch Zugabe von 500 µl 37°C warmen Mediums, dessen pH-Wert durch Zugabe von HCl zuvor auf 5,0 eingestellt wurde, eingeleitet. Aus den durch die Toxine gebildeten Poren können nun die intrazellulären [86Rb+]-Ionen in den Extrazellulärraum ausströmen. Nach fünfminütiger Inkubation bei 37°C wurden die Zellen für weitere 40 Minuten auf Eis abgekühlt. Die ausgeströmten [86Rb+]-Ionen im Medium konnten nach Zugabe von 2 ml Szintillationsflüssigkeit durch Szintillationsmessung (TriCarb 2900TR, Packard) bestimmt werden. Aus dem Vergleich des [86Rb+]-Ausstroms bei den unterschiedlichen pH-Werten von 5,0 (Porenbildung) und 7,4 (Kontrollbedingungen) konnte das Ausmaß der Porenbildung bewertet werden. 48 MATERIAL UND METHODEN Material: PBS: 137 mM NaCl, 2,7 mM KCl, 10 mM Na2HPO4, 1,4 mM KH2PO4 (pH 7,4) Bafilomycin A1: (Biotrend, Köln, Deutschland) Ultima Gold (Perkin Elmer Life Science, Boston, USA) 3.3: Fluoreszenz-Mikroskopie/DIC-Mikroskopie Zur Durchführung von Immunfärbungen wurden die zu untersuchenden Zellen (auf coverslips in 6 bzw. 24-well-Kulturschalen, PC12-Zellen: beschichtet mit Polylysin [50 µg/ml], 2 h RT) zunächst dreimal mit warmen PBS gewaschen und mit 4% (m/v) p-Formaldehyd-Lösung bei Raumtemperatur für 20 Minuten fixiert. Anschließend wurden die Membranen der Zellen mit 0,15 % (v/v) Triton X-100 permeabilisiert. Das Blocken erfolgte mit 5% (m/v) BSA in PBST-Lösung für eine Stunde bei Raumtemperatur. Das filamentöse Aktin-Zytoskelett wurde durch TRITC-Phalloidin (1:200) bei Raumtemperatur für eine Stunde angefärbt. Nach dreimaligem Waschen mit PBS-T wurden die Zellen in eine Polymermatrix (Mowiol+DABCO) eingebettet. Die DNA des Zellkerns wurde mit dem Farbstoff DAPI gefärbt. Das AktinZytoskelett der fixierten Zellen wurden durch Fluoreszenz-Mikroskopie (Axiovert 200M, Zeiss) nach einer Anregung bei λ=554 nm, bei einer Emission bei λ=576 nM untersucht. Für live-cell-imaging wurden die Zellen (in Glasbodenschalen) in einer Kammer (6,5% CO2, 9% O2) bei 37°C inkubiert und durch DIC-Mikroskopie/Fluoreszenzmikroskopie (Axiovert 200M, Zeiss) beobachtet. Die mikroskopischen Daten wurden mit der Software Metamorph bearbeitet und ausgewertet. Material: PBS: 137 mM NaCl, 2,7 mM KCl, 10 mM Na2HPO4, 1,4 mM KH2PO4 (pH 7,4) PBS-T-Lösung: PBS + 0,05% (v/v) Tween 20 Mowiol (+DABCO) (Merck, Darmstadt, Deutschland bzw. Sigma-Aldrich, München, Germany) TRITC-Phalloidin (50 µg/ml) (Invitrogen, Karlsruhe, Deutschland) Polylysin (Biochrom AG, Berlin, Deutschland) 49 ERGEBNISSE D: ERGEBNISSE 1: Expression rekombinanter CGTs in Bacillus megaterium Die Expression rekombinanter Holotoxine in Escherichia coli lieferte nur geringe Ausbeuten (Tang-Feldman et al., 2002). Das gram-positive Bacillus megaterium- Proteinexpressionssystem stellt hier eine Alternative zur Expression der clostridialen Toxine dar. Yang et al. berichteten über die rekombinante Expression großer Mengen von Toxin A und Toxin B in Bacillus megaterium (Yang et al., 2008). Ausgehend von dieser Arbeit sollten nun neben Toxin A und Toxin B noch weitere CGTs in Bacillus megaterium exprimiert und aufgereinigt werden. 1.1: Klonierung der Gene der CGTs in den Bacillus megaterium Expressionsvektor pHis1522 Die Gene für die CGTs tcdA, tcdB aus Clostridium difficile und tcsL aus Clostridium sordellii liegen auf der chromosomalen DNA (Barroso et al., 1990; Sauerborn and Von EichelStreiber, 1990; Green et al., 1995). Das Gen tcnA aus Clostridium novyi ist prophagencodiert (Hofmann et al., 1995). Die entsprechende chromosomale DNA wurde aufgereinigt und diente als Matrize für die PCR-Amplifikation der Toxine. Zur Expression der Toxine wurde das Bacillus megaterium pHis1522-Vektorsystem verwendet. Mit diesem Vektor ist es möglich, Proteine fusioniert mit einem C-terminalen 6xHistidin-tag zu exprimieren. Es wurden für die Klonierung der Toxin-Gene die BsrGI- und KpnI-Restriktionsschnittstelle der MCS (multiple cloning site) benutzt. Die Benutzung der BsrGI-Schnittstelle unmittelbar vor dem ATG-Startcodon verhindert einen modifizierten N-Terminus des Proteins (Yang et al., 2008). Durch das Anfügen einer zusätzlichen Base (Guanin) am 3´-Ende der Gensequenz wird ein übereinstimmender Leserahmen mit dem vektorcodierten C-terminalen 6xHistidin-tag gewährleistet. Die einzelnen Klonierungsschritte innerhalb des pHis1522-shuttle- Vektorssystems wurden in Escherichia coli durchgeführt. Die Expression der Proteine fand anschließend in Bacillus megaterium statt. 1.1.1: Klonierung von pHis1522-Toxin B Das Gen tcdB wurde aus genomischer DNA des Clostridium difficile-Stammes VPI 10463 amplifiziert. Das PCR-Produkt (siehe Abb.D1A) wurde direkt in den pCR-TOPO-Blunt-2Vektor ligiert und die Richtigkeit mit einer analytischen Restriktion (siehe Abb.D1C) überprüft. Das resultierende Plasmid pCR-TOPO-Blunt-2-Toxin B wurde mit BsrGI und KpnI verdaut. Das Insert wurde anschließend aus einem Agarosegel aufgereinigt und in den bereits zuvor 50 ERGEBNISSE mit BsrGI und KpnI geschnittenen pHis1522-Vektor (siehe Abb.D1B) ligiert. Der Erfolg der Ligation wurde wiederum durch eine analytische Restriktion überprüft (siehe Abb.D1D). B A 8 000 6 000 1 8 000 6 000 3 000 3 000 1 000 C D 8 000 1 6 000 3 500 3 000 1 1 2 8 000 6 000 2 3 000 1 000 1 000 1 000 3 250 1 1 3 pCR-TOPOBlunt-2-Toxin B KpnI BsrGI pHis1522Toxin B 2 PstI 2 BsrGI BsrGI Abb.D1: Klonierung von pHis1522-Toxin B Agarose-Gelektrophorese: A: PCR-Amplifikation des Gens tcdB (7,1 kB), B: Restriktion von pHis1522 mit BsrGI und KpnI (7,4 kB), C: Restriktion von pCR-TOPO-Blunt-2-Toxin B mit BsrGI und KpnI (7,1/3,5/0,3 kB), D: Restriktion von pHis1522-Toxin B mit BsrGI und PstI (8,4/6,1 kB), Pfeilmarkierung der DNA-Fragmente, in Klammern: berechnete Größe der DNA-Fragmente, für C und D: schematische Darstellung der Plasmidkarte: Gen (rot), pCR-TOPO-Blunt-2 (blau), pHis1522 (schwarz), die nach analytischen Restriktion entstehenden DNAFragmente sind beziffert 1.1.2: Klonierung von pHis1522-Toxin A und pHis1522-Letales Toxin Zur Genamplifikation von Toxin A mittels PCR wurde genomische DNA aus Clostridium difficile VPI 10463 verwendet. Die Genamplifikation von Letalem Toxin erfolgte aus genomischer DNA aus Clostridium sordellii 6018. Die Herstellung der Konstrukte erfolgte entsprechend der Klonierung von pHis1522-Toxin B (siehe Ergebnisse 1.1.1), wobei die PCR-Produkte direkt, ohne eine Zwischenklonierung in den pCR-TOPO-Blunt-2-Vektor, in den pHis1522-Vektor kloniert wurden. 1.1.3: Klonierung von pHis1522-α-Toxin Das Gen tcnA enthält eine interne BsrGI- und SpeI-Schnittstelle. Aus diesem Grund ist es nicht möglich, das Gen direkt über die Schnittstellen BsrGI und KpnI zu klonieren. Um dennoch das Gen unter Verwendung dieser beiden Schnittstellen in den pHis1522-Vektor klonieren zu können, war es notwendig, das Gen zunächst in zwei Teilen zu amplifizieren und anschließend im Expressionsvektor zusammenzuführen. Hierzu wurde die interne SpeISchnittstelle verwendet, welche vor der internen BsrGI-Schnittstelle liegt und hierdurch eine Ligation beider Fragmente ermöglicht. Zur Amplifikation wurde genomische DNA aus 51 ERGEBNISSE Clostridium novyi 19402 verwendet. Der erste DNA-Abschnitt ist flankiert von einer 5´-BsrGIund der internen 3´-SpeI-Schnittstelle und umfasst Basenpaar 1/ATG bis 2965. Der zweite DNA-Abschnitt von Basenpaar 2971 bis 6534 ist von der internen 5´-SpeI- und 3´-KpnISchnittstelle flankiert. Nach der Restriktion wurden die PCR-Produkte zunächst in die entsprechend geschnittenen pHis1522-Vektoren ligiert. Anschließend wurde das Plasmid pHis1522-α-Toxin (Bp: 2965-6534) mit SpeI und KpnI verdaut und das DNA-Insert schließlich in das zuvor mit BsrGI und SpeI geöffnete Plasmid pHis1522-α-Toxin (Bp: 12970) kloniert Das Gen konnte somit über seine interne SpeI-Schnittstelle wieder fusioniert werden. 52 ERGEBNISSE 2: Expression und Aufreinigung rekombinanter CGTs Der pHis1522-Vektor trägt verschiedene Elemente des Bacillus megaterium-XyloseOperons. Der starke xylA-Promotor, dessen Transkription durch D-Xylose induzierbar ist, erlaubt es, Proteine bis zu 350-fach überzuexprimieren. Zunächst sollte für die Expression der CGTs eine Testexpression in kleinen Ansätzen durchgeführt werden. Im nächsten Abschnitt wird exemplarisch auf die Expression und Proteinaufreinigung von Toxin B näher eingegangen. 2.1: Testexpression von rekombinantem Toxin B Zunächst wurden Vorkulturen mit Bacillus megaterium-Klonen aus einer Transformation mit dem Plasmid pHis1522-Toxin B sowie der Negativkontrolle pHis1522 angeimpft. Die Hauptkulturen wurden bei einer OD600=0,3 mit Xylose induziert bzw. nicht induziert und bis zur Zellernte (bei einer OD600=1,5) bei 37°C geschüttelt. Die Bakterienpellets wurden anschließend durch Ultraschall aufgeschlossen und die Lysate durch Zentrifugation in eine lösliche Überstand- und eine unlösliche Pelletfraktion unterteilt. Die Proteine der Überstände sowie der Pellets wurden mittels SDS-PAGE (siehe Abb.D2A) aufgetrennt. Wie ersichtlich, erscheinen Proteine mit der entsprechenden molekularen Masse von Toxin B (siehe Pfeilmarkierung) im Überstand und in einem deutlich geringeren Ausmaß im Pellet induzierter Bakterienkulturen. Diese Proteine besitzen eine molekulare Masse von über 250 kDa. Sie sind weder im Lysat von Kontroll- noch im Lysat nicht induzierter Bakterienkulturen erkennbar. Dies legt nahe, dass es sich bei dem Protein um rekombinantes Toxin B handeln könnte. Um dies sicherzustellen, wurde ein Western-Blot (siehe Abb.D2B) durchgeführt. Rekombinantes Toxin B trägt einen C-terminalen 6xHistidin-tag und kann somit mit einem spezifischen Antikörper, der dieses Epitop erkennt, nachgewiesen werden. Wie in Abbildung D2B zu sehen ist, wird die entsprechende Proteinbande eindeutig von einem anti-6xHistidinAntikörper erkannt. Bei den zusätzlich erkannten Proteinbanden handelt es sich wahrscheinlich um bakterielle Proteine aus Bacillus megaterium oder Abbauprodukte des Toxins. Der 6xHistidin-tag erlaubt es weiterhin, eine Affininätsaufreinigung des Toxins aus dem Lysat mittels einer Ni2+-Matrix durchzuführen. In Abbildung D2C sind die einzelnen Schritte einer solchen Aufreinigung dargestellt. Es wird deutlich, dass es mit diesem Vorgehen möglich ist, das Protein weiter aufzureinigen und hierbei einen zufriedenstellenden Grad an Reinheit zu erreichen. 53 ERGEBNISSE A pHis1522 Ü C pHis1522-Toxin B P + Ü + P + + waschen 1 2 3 Elution 1 2 3 250kDa- 250kDa- pHis1522-Toxin B B Abb.D2: Testexpression von Toxin B SDS-PAGE (Coomassie-Gel): A: Lysate aus pHis1522-Toxin B bzw. pHis1522 transformierten Bacillus megaterium-Kultur. Ü: Überstand, P: Pellet, +: Induktion mit 5% Xylose, Western-Blot: B: von SDS-PAGE-Gel (aus A) mit anti-6xHistidin-Antikörper, SDS-PAGE (Coomassie-Gel): C: verschiedene Schritte der Aufreinigung 2+ von Toxin B über Ni -beads aus Lysaten induzierter Bacillus megaterium-Kulturen, jeweils dargestellt sind: Lysat (Überstand), Durchfluss, Waschfraktionen, Elutionen. Pfeilmarkierung von Toxin B 2.2: Aufreinigung der rekombinanten CGTs Die Aufreinigung der Toxine in größeren Maßstäben bestand aus zwei aufeinanderfolgenden Schritten und wurde mit Hilfe einer FPLC-Anlage (ÄKTA Prime/Purifier) durchgeführt. Die Aufreinigung umfasste zunächst eine Ni2+-Affinitätsaufreinigung (siehe Methoden 2.13.1) über eine Ni2+-Matrix mit darauf folgender Gelfiltration (siehe Methoden 2.15). 2.2.1: Ni2+-Affinitätsaufreinigung In einem ersten Affinitätsaufreinigungsschritt wurde Toxin B über eine Ni2+-Säule aus Zelllysaten aufgereinigt. Die an die Säule gebundenen Proteine wurden zunächst mit 10% Puffer A (50 mM Imidazol) gewaschen, um unspezifisch gebundene Verunreinigungen zu entfernen und anschließend mit einem kontinuierlichen Gradienten (50 bis 500 mM Imidazol) zwischen 20 bis 30% Puffer B (100 bis 150 mM Imidazol), von der Säule eluiert. 2.2.2: Gelfiltration Nachfolgend wurde mit Toxin B eine Gelfiltration durchgeführt. Diese Methode ermöglicht zusätzlich eine Bestimmung der molekularen Masse eines Proteins und hieraus dessen Dispersität. In Abbildung D3 ist das charakteristische Elutionsprofil von Toxin B zu sehen. 54 ERGEBNISSE Unter der Benutzung eines makromolekularen Größenstandards kann aus dem Elutionsvolumen eines Proteins dessen molekulare Masse in Lösung ermittelt werden. Die berechnete molekulare Masse von Toxin B beträgt etwa 265 kDa. Somit liegt das Toxin nach Gelfiltration größtenteils als Monomer in Lösung vor. Abb.D3: Elutionsprofil der Gelfiltration von Toxin B SDS-PAGE (Coomassie-Gel): Untersuchung entsprechenden peaks über Pfeilmarkierung der verschiedene Elutionsfraktionen, Zuordnung der 2.3: Vergleich nativer und rekombinanter Aufreinigungen Die CGTs konnten in größeren Mengen nur in der nativen Form aus den verschiedenen clostridialen Spezies aufgereinigt werden. Dieses Vorgehen umfasst ein mehrtägiges Wachstum der Bakterien unter anaeroben Bedingungen, einen Proteinfällungs- /Renaturierungsschritt sowie chromatographische Aufreinigungsschritte (Sullivan et al., 1982; Ball et al., 1993; Krivan and Wilkins, 1987; Martinez and Wilkins, 1992). Die Aufreinigung von nativen Toxinpräparationen ist somit im Vergleich zur Aufreinigung von rekombinanten Toxinpräparationen ein deutlich zeit- und arbeitsaufwendigeres Verfahren. Bacillus megaterium kann unter aeroben Bedingungen sowie unter dem Selektionsdruck eines Antibiotikums einfach kultiviert werden. Im Gegensatz hierzu sind die humanpathogenen Clostridium difficile, Clostridium sordellii und Clostridium novyi deutlich anspruchsvoller zu kultivieren. In Abbildung D4A sind als eine Übersicht repräsentative SDS-PAGE-Gele mit nativen und rekombinanten Proteinaufreinigungen liefern, Toxinpräparationen im Vergleich zu abgebildet. den nativen Die rekombinanten Proteinaufreinigungen, insbesondere für Letales Toxin und α-Toxin einen deutlich höheren Grad an Reinheit. Rekombinantes Letales Toxin baut sich während der Expression zum Teil ab. Hierbei entsteht ein etwa 200 kDa große Abbaubande (mit „*“ Markierung). Da der 6xHistidin-tag dieser Bande im Western-Blot (siehe Abb.D5) erkannt wird, handelt es sich um den CTerminus des Toxins. 55 ERGEBNISSE A B 250kDa- * nativ rekombinant Abb./Tab.D4: Vergleich von Proteinaufreinigungen nativer und rekombinanter CGTs SDS-PAGE (Coomassie-Gel): A: native Toxine (2 µg) wurden aus Clostridien aufgereinigt. Rekombinante Toxine (2 µg) wurden in Bacillus megaterium exprimiert und über Affinitätschromatographie und Gelfiltration aufgereinigt, Markierungen: Toxin-Banden. B: Proteinausbeute rekombinanter CGT-Aufreinigungen. Geschätzte Ausbeute (in ug/l Kultur) rekombinanter, in Bacillus megaterium exprimierter Toxine, nach Affinitätsaufreinigung und Gelfiltration, „*“-Markierung: C-terminales Abbauprodukt von Letalem Toxin. Es war nicht möglich diese Abbaubande durch weitere Aufreinigungsschritte (Gelfiltration, Anionenaustauschchromatographie) quantitativ vom Holotoxin abzutrennen. Proteinaufreinigungen von Toxin A und Toxin B liefern sowohl für native als auch für die rekombinanten Präparationen einen ähnlichen Grad an Reinheit. Die Proteinausbeuten der rekombinanten Toxine sind in einer Tabelle D4B zusammengefasst und lagen zwischen 100 bis 500 µg/l Bakterienkultur. 56 ERGEBNISSE 3: Immunologische Charakterisierung der rekombinanten und nativen CGTs Der Nachweis rekombinanter Proteine mit spezifischen Antikörpern ist ein wichtiger Teil ihrer Charakterisierung. Zum immunologischen Nachweis der CGTs stehen verschiedene monoklonale und polyklonale Antikörper (siehe Material 5.5.1) zur Verfügung. Die rekombinanten Toxine können über ihren 6xHistidin-tag mittels einem anti-6xHistidinAntikörper erkannt werden. Des Weiteren ist es möglich, Toxin A, Toxin B und Letales Toxin über toxinspezifische Antiseren zu identifizieren. Toxin B kann zusätzlich über einen monoklonalen Antikörper, der die Glukosyltransferase-Domäne erkennt, detektiert werden. In Abbildung D5 sind im Vergleich native und rekombinante Toxine im Western-Blot mit den unterschiedlichen Antikörpern dargestellt. Die nativen und die rekombinanten CGTs werden von den entsprechenden spezifischen Antikörpern erkannt. Toxin B und Letales Toxin stellen hier eine Ausnahme dar. Beide Toxine werden wechselseitig von Toxin B- oder Letales Toxin-Antiseren erkannt. Diese Kreuzreaktivität ist beschrieben und beruht auf der hohen Homologie der beiden Toxine (Chang et al., 1978; Martinez and Wilkins, 1992; Popoff, 1987). Die rekombinanten Toxine werden zusätzlich über den 6xHistidin-tag erkannt. Mit den zur Verfügung stehenden Antikörpern ist es möglich die verschiedenen native als auch rekombinanten Holotoxine voneinander zu unterscheiden. * * * * anti-Toxin A anti-Toxin B anti-Toxin B1-546 anti-Letales Toxin anti-6xHistidin Abb. D5: Immunologische Charakterisierung rekombinanter und nativer CGTs Western-Blot: Charakterisierung nativer („*“-Markierung) und rekombinanter Toxine (jeweils 250 bis 500 ng) mit unterschiedlichen Antikörpern: anti-Toxin A (pk), anti-Toxin B (pk), anti-Toxin B1-546 (mk), anti-Letales Toxin (pk), anti-6xHistidin (mk), n: natives Toxin, r: rekombinantes Toxin, pk: polyklonal, mk: monoklonal 57 ERGEBNISSE 4: Funktionelle Charakterisierungen der rekombinanten CGTs Die CGTs bewirken in vivo charakteristische zytopathische Veränderungen der Morphologie kultivierter Zellen (Chang et al., 1979; Lyerly et al., 1982; Bette et al., 1991). Toxin A und Toxin B induzieren eine Zellabrundung sowie die intermediäre Ausbildung Neuriten-ähnlicher Ausläufer (Fiorentini et al., 1990; Fiorentini and Thelestam, 1991). Letales Toxin induziert neben der Abrundung von Zellen zusätzlich eine massive Zellablösung (Chaves-Olarte et al., 2003). In vitro können verschiedene Aktivitäten der Toxine gemessen werden. Hierzu zählen die Enzymaktivität der Glukosyltransferase- (Just et al., 1995b) und der CysteinproteaseDomäne (Egerer et al., 2007; Egerer et al., 2009) sowie die Porenbildung (Barth et al., 2001; Genisyuerek et al., 2011), vermittelt durch eine Poren-bildende Region/TranslokationsDomäne. 4.1: Untersuchungen zur in vivo-Aktivität rekombinanter CGTs in der Zellkultur Die Überprüfung der in vivo-Aktivität der rekombinanten CGTs in der Zellkultur wurde mit Vero-Zellen durchgeführt. Hierzu wurden die Zellen mit den rekombinanten CGTs in unterschiedlichen Konzentrationen vergiftet. In Abbildung D6A sind lichtmikroskopische Aufnahmen vergifteter Vero-Zellen nach angegebenen Zeitpunkten zusammengefasst. A B Toxin A Toxin B Kontrolle Letales Toxin α-Toxin Toxin B Kontrolle Abb. D6: Zellvergiftung von Vero-Zellen mit rekombinanten CGTs Zellvergiftung: A: Vero-Zellen wurden mit unterschiedlichen Konzentrationen rekombinanter CGTs bei 37°C vergiftet. Mikroskopische Bilder wurden nach den angegebenen Zeitpunkten aufgenommen. Toxin A: 1 nM, 3 h; Toxin B: 100 pM, 1 h; Letales Toxin: 1 nM, 6 h; α-Toxin: 1 nM, 24 h; Kontrolle: 24 h Zellvergiftung (AktinFärbung): B: Vero-Zellen wurden mit Toxin B (100 pM) für eine 1 h vergiftet. Das Aktin-Zytoskelett wurde anschließend mit TRITC-Phalloidin angefärbt. In Abbildung D6B ist eine Färbung des Aktin-Zytoskeletts mit Toxin B vergifteten Vero-Zellen im Vergleich zu Kontrollzellen dargestellt. Aus dieser Abbildung ist ersichtlich, dass die 58 ERGEBNISSE rekombinanten CGTs die Abrundung der Zellen durch die Zerstörung des Aktin-Zytoskeletts induzieren können und somit eine in vivo-Aktivität aufweisen. Die CGTs sind in diesem Zellkulturmodell unterschiedlich aktiv. Toxin B zeigt deutlich die höchste Aktivität. 4.2: Untersuchungen zur in vitro-Aktivität der GlukosyltransferaseDomäne rekombinanter CGTs Die CGTs mono-O-glukosylieren verschiedene Vertreter kleiner GTPasen der Rho- und/oder Ras-Unterfamilie. Die Glukosyltransferase-Domäne katalysiert hierbei den Transfer einer Glukose-Einheit (bei α-Toxin: N-Acetylglukosamin) von den entsprechenden UDP-ZuckerDonoren auf das Zielsubstrat (Just et al., 1994; Just et al., 1995a; Just et al., 1996; Selzer et al., 1996). In Abwesenheit der Proteinsubstrate hydrolysieren die Toxine die entsprechenden UDP-Zucker. Die Glukosyltransferase- und Glukohydrolase-Aktivität der CGTs wurden in vitro untersucht. Zur eindeutigen Bestimmung der Substratspezifität wurden in vitroGlukosylierungen mit RhoA, Rac1 und Ha-Ras als Proteinsubstrat und UDP-Glukose oder UDP-N-Acetylglukosamin durchgeführt. In Abbildung D7 sind die erhaltenen Autoradiogramme dargestellt. Aus diesen Autoradiogrammen wird deutlich, dass Toxin A, Toxin B und α-Toxin RhoA und Rac1, aber nicht Ha-Ras modifizieren. Letales Toxin hingegen modifiziert Rac1, Ha-Ras, aber nicht RhoA. Das Kosubstratspektrum konnte unabhängig vom Substratspektrum über Messung der Glukohydrolase-Aktivität bestimmt und bestätigt werden (Daten nicht gezeigt). Die Ergebnisse mit rekombinanten CGTs entsprechen dem erwarteten Kosubstrat- und Substratspektrum der nativen Toxine und zeigen außerdem, dass die rekombinanten Toxine enzymatisch aktiv sind. Toxin A Toxin B Letales Toxin α-Toxin Abb.D7: Katalytische in vitro-Aktivität der rekombinanten CGTs In vitro-Glukosylierung (Autoradiogramm): Hierzu wurden 5 µM rekombinantes Rac1, Ha-Ras oder RhoA mit 14 unterschiedlichen Konzentrationen rekombinanter CGTs in Gegenwart von 10 µM UDP-[ C]-UDP-Glukose bzw. 14 UDP-[ C]-N-Acetylglukosamin (α-Toxin) inkubiert. Die Reaktion wurde für die angegebenen Zeitpunkte bei 37°C durchgeführt. Toxin A: 50 nM, 30 min; Toxin B: 10 nM, 20 min; Letales Toxin: 10 nM, 20 min; α-Toxin: 100 nM, 30 min 59 ERGEBNISSE 4.3: Herstellung einer katalytisch inaktiven Toxin B-Mutante Die rekombinante Expression der CGTs in Bacillus megaterium eröffnet die Möglichkeit, diese genetisch zu verändern oder zu modifizieren. Es ist nun möglich, Deletionsmutanten oder Punktmutanten zu generieren und zu exprimieren. Um dies zu verdeutlichen, sollte eine Holotoxin-Mutante von Toxin B hergestellt werden, deren Glukosyltransferase-Domäne enzymatisch inaktiv ist. Mittels zielgerichteter Mutagenese wurden Aspartat-286 und Aspartat-288 des DXD-Motivs in Alanine umgewandelt. Das charakteristische DXD-Motiv ist an der koordinativen Bindung der UDP-Glukose beteiligt (Busch et al., 1998). Die HolotoxinMutante wies, wie erwartet, eine deutlich verringerte in-vitro-Glukosylierungsaktivität (Abb.D8A) auf und entsprach der Aktivität der Negativkontrolle. A B Kontrolle (24 h) Toxin B (1 h) Rac1 Toxin B D286A/D288A (6 h) Toxin B D286A/D288A (24 h) Abb.D8: In vitro- und in vivo-Aktivität von Toxin B D286A/D288A In vitro-Glukosylierung (Autoradiogramm): A: 5 µM rekombinantes Rac1 wurden mit Toxin B1-546, Toxin B1-546 14 W520A, Toxin B D286A/D288A oder Toxin B mit 10 µM UDP-[ C]-UDP-Glukose für 30 min bei 37°C inkubiert. Toxin B1-546: 10 nM, Toxin B1-546 W520A: 10 nM, Toxin B D286A/D288A: 100 nM, Toxin B: 100 nM. Zellvergiftung: B: Vero-Zellen wurden mit Toxin B oder Toxin B D286A/D288A bei 37°C vergiftet. Mikroskopische Bilder wurden nach den angegebenen Zeitpunkten aufgenommen. Toxin B: 100 pM, 1 h; Toxin B D286A/D288A: 100 nM, 6 h bzw. 24 h; Kontrolle: 24 Als Negativkontrolle diente hierbei die Mutante Toxin B1-546 W520A, von welcher ebenfalls bekannt ist, dass sie eine deutlich verringerte Enzymaktivität aufweist (Jank et al., 2007a). In der Zellkultur zeigte Holotoxin B D286A/D288A gegenüber Toxin B eine verzögerte Abrundung von Vero-Zellen. Toxin B induziert die Zellabrundung schon in picomolaren Konzentrationen innerhalb von 30 bis 60 Minuten (siehe Abb.D8B). Im Gegensatz hierzu zeigte die D286A/D288A-Mutante selbst in hohen nanomolaren Konzentrationen (100 nM) nach 6 Stunden nur eine schwache Abrundung der Zellen. Eine deutliche Zellabrundung konnte erst nach 24 Stunden beobachtet werden. 60 ERGEBNISSE 4.4: Untersuchung der Porenbildung rekombinanter CGTs Die CGTs werden zunächst durch Rezeptor-vermittelte Endozytose in die Endosomen aufgenommen (Papatheodorou et al., 2010). In den Endosomen findet eine schrittweise Ansäuerung durch eine vesikuläre H+-ATPase statt. Diese Ansäuerung führt zu einer Konformationsänderung innerhalb der Toxine, wodurch hydrophobe Bereiche nach außen exponiert werden (Qa'Dan et al., 2001; Barroso et al., 1994). Der hieraus resultierende Anstieg in der Hydrophobizität ermöglicht es den Toxinen in die endosomale Membran zu inserierten und dort eine Pore auszubilden (Barth et al., 2001). Zur Untersuchung der Porenbildung stehen zwei unterschiedliche Versuchsanordnungen zur Verfügung: ein zellbasiertes System, in dem der [86Rb+]-Ionen Ausstrom durch die entstandenen Poren an der Oberfläche von Zellen gemessen werden kann (siehe Methoden 3.2) (Barth et al., 2001) sowie ein Liposomen-basiertes System, in dem der Ausstrom eines Fluoreszenzfarbstoffes nach Porenbildung aus den Liposomen gemessen werden kann (siehe Methoden 2.26) (Peschka-Süss and Schubert, 2003; Genisyuerek et al., 2011). Die CGTs wurden in beiden Systemen auf Porenbildungs-Aktivität untersucht (siehe Abb.9A/B). A B 80 7 60 pH=7,4 6 5 4 3 2 HPTS-Freisetzung (%) x-fache [86Rb+]-Freisetzung pH=5,0 40 Kontrolle Toxin A Toxin B Letales Toxin α-Toxin 20 1 0 0 0 10 20 30 40 50 60 Zeit (s) Abb. D9: Porenbildung in biologische Membrane/Liposomen durch rekombinante CGTs A: Porenbildung in biologische Membran von CHO-K1-Zellen: Hierzu wurden 10 nM rekombinantes Toxin A, Toxin B, Letales Toxin und Toxin B 1-546 eingesetzt. Die Porenbildung wurde durch Ansäuerung auf pH=5 86 + induziert. Die Freisetzung von [ Rb ]-Ionen unter Kontrollbedingungen (ohne Toxin: pH 7,4 und pH 5) wurde 86 + auf den Wert 1,0 gesetzt. Die Freisetzung von [ Rb ]-Ionen wurde für beide pH-Werte relativ zu den Kontrollwerten angegeben. B: Untersuchung der Porenbildung in Liposomen: Hierzu wurden 5 µM Liposomen sowie 1 nM der rekombinanten CGTs eingesetzt. Die Porenbildung wurde durch Ansäuerung auf pH=5 induziert. Zur Bestimmung der maximal möglichen HPTS-Freisetzung wurden die Liposomen durch die Zugabe von Triton X-100 solubilisiert. Von den erhaltenen Fluoreszenz-Werten wurde die Hintergrundfluoreszenz (vor Ansäuerung, pH=7,4) abgezogen. Die Porenbildung ist in Relation zur maximal möglichen Freisetzung angegeben. Toxin A (2-fache Freisetzung), Toxin B (6-fache Freisetzung) und Letales Toxin (5-fache Freisetzung) konnten hier eine deutliche Freisetzung von [86Rb+]-Ionen aus CHO-K1-Zellen 61 ERGEBNISSE induzieren (siehe Abb.D9A). Die Unterschiede im Ausmaß der Porenbildung kann möglicherweise auf die unterschiedliche Sensitivität der CHO-K1-Zellen gegenüber den unterschiedlichen Toxinen zurückgeführt werden. Im Liposomen-basierten System konnten alle vier Toxine eine vergleichbare Freisetzung des Fluoreszenzfarbstoffes verursachen (siehe Abb.D9B). Dieses System ist genauer definiert und bietet somit eine bessere Möglichkeit, die Toxine untereinander zu vergleichen. Zusammenfassend kann man aus diesen Daten schließen, dass die rekombinanten CGTs in der Lage sind Poren in Membranen zu bilden. 62 ERGEBNISSE 5: Autokatalytische Prozessierung der CGTs Die im N-Terminus des Toxins lokalisierte Glukosyltransferase-Domäne wird nach der Prozessierung in das Zytosol der Zelle abgegeben (Pfeifer et al., 2003). Diese Prozessierung findet bei vielen Vertretern der CGTs statt (Reineke et al., 2007; Rupnik et al., 2005; Egerer et al., 2007; Egerer and Satchell, 2010; Giesemann et al., 2008). Für Toxin A und B konnte gezeigt werden, dass die Spaltung durch eine intrinsische Cysteinprotease-Domäne katalysiert wird (Egerer et al., 2007; Egerer et al., 2009; Pruitt et al., 2009). Die Aktivität dieser Domäne wird durch die Bindung von Inositolhexakisphosphat an die CPD reguliert und somit die Prozessierung durch diese Interaktion eingeleitet (Pruitt et al., 2009; Egerer et al., 2009). Bis dato war über die Prozessierung des Letalen Toxins und des α-Toxins nur wenig bekannt. Es sollte nun die autokatalytische Prozessierung dieser beiden Toxine im Vergleich zu den besser charakterisierten CGTs, Toxin A und Toxin B, biochemisch untersucht werden. Die Expression in Bacillus megaterium ermöglichte es, für diese Untersuchungen ausschließlich rekombinante Toxine und Toxinfragmente einzusetzen. 5.1: CPD-vermittelte autokatalytische Prozessierung von Letalem Toxin und α-Toxin In vorherigen biochemischen Charakterisierungen der CPD von Toxin B konnte diese auf den Aminosäurenabschnitt 544-955 eingeschränkt werden (Egerer et al., 2007). In weiteren Untersuchungen von Pruitt et al. wurde die CPD von Toxin A auf die Aminosäuren 543-809 (entspricht bei Toxin B Aminosäure 544-807) weiter eingegrenzt (Pruitt et al., 2009). Durch Sequenzvergleiche (Abb.D10) mit Toxin A und B war es möglich, in Letalem Toxin und αToxin ebenfalls CPD-ähnliche Sequenzen zu identifizieren, die durch Aminosäuren 544-807 für Letales Toxin oder Aminosäuren 549-813 für α-Toxin bestimmt waren. DYTGGSL542↓SEDNGVD.....EVNLLGC700NMFSYDF Toxin A NYFEGSL543↓GEDDNLD.....EINLLGC698NMFSYSI Toxin B GYFEGAL543↓GEDDNLD.....EINLLGC698NMFSYSI Letales Toxin TYIGRTL548↓NYEDGLN.....KINILGC707YMFTPKV α-Toxin KYTNKSL542↓SENDKLN.....QIDLLGC696NMFSYNI TpeL Abb.D10: Ausschnitt von Sequenzvergleich der Primärsequenz der CGTs (↓) vermutete Schnittstelle der Prozessierung, (C) vermutetes katalytisches Cystein der CysteinproteaseDomäne. Das Sequenz-alignment wurde mit der Software ClustalW erstellt. Zur Untersuchung der autokatalytischen Prozessierung wurden Toxinfragmente, die aus der Glukosyltransferase- und Cysteinprotease-Domäne (GTD-CPD) (Toxin A1-809, Toxin B1-807, Letales Toxin1-807, α-Toxin1-813) bestanden, kloniert und aus Bacillus megaterium aufgereinigt. Diese Toxinfragmente wurden durch in vitro-Spaltungsversuche untersucht. Hierzu wurden die Toxinfragmente mit steigenden Konzentrationen an InsP6 inkubiert und die Spaltung 63 ERGEBNISSE anhand der entstandenen charakteristischen Spaltprodukte bewertet. Wie aus Abbildung D11 ersichtlich, kann die Prozessierung der Toxinfragmente durch die Zugabe von InsP6 induziert werden. Die Abspaltung der Glukosyltransferase-Domäne kann ab einer Konzentration von 0,1-1 µM InsP6 nachgewiesen werden. Es konnten keine wesentlichen Unterschiede der Prozessierung dieser Toxinfragmente zwischen den einzelnen CGTs beobachtet werden. Die Prozessierung ist auch für Letales Toxin und α-Toxin direkt abhängig von der CPD und somit unabhängig von anderen Domänen innerhalb der Toxine. InsP6 Toxin A1-809 Toxin A1-542 Toxin B1-807 Toxin B1-543 Letales Toxin1-807 Letales Toxin1-543 α-Toxin1-813 α-Toxin1-548 Abb. D11: InsP6-induzierte Autoprozessierung der GTD-CPD-Fragmente InsP6-induzierte Autoprozessierung (Coomassie-Gel): rekombinante GTD-CPD-Fragmente (2 µg) wurden mit ansteigenden Konzentration an InsP6 (10 nM bis 10 mM) bei 37°C für 1 h inkubiert. 5.2: Identifizierung der katalytischen Triade sowie der proteolytischen Schnittstelle von Letalem Toxin und α-Toxin Die katalytische Triade der Cysteinprotease-Domäne, die aus einem Aspartat, Histidin und Cystein (DHC-Motiv) besteht, ist innerhalb der CGTs stark konserviert (Egerer et al., 2007; Pruitt et al., 2009). Für Toxin A und B konnte das katalytische Cystein in Position-700 bzw. 698 identifiziert werden (Pruitt et al., 2009; Rupnik et al., 2005; Egerer et al., 2007). Die Schnittstelle liegt zwischen der Glukosyltransferase- und Cysteinprotease-Domäne. Die Spaltung erfolgt stets nach einem Leucin-Rest. Toxin A wird zwischen Leucin-542 und Serin543 gespalten (Pruitt et al., 2009). Die Schnittstelle für Toxin B liegt zwischen Leucin-543 und Glycin-544 (Rupnik et al., 2005; Egerer et al., 2007). Aus einem Sequenzvergleich der CGTs (Abb.D10) war es nun möglich, das katalytische Cystein des DHC-Motives sowie die Schnittstellen von Letalem Toxin und α-Toxin zu ermitteln. Die katalytischen Cystein-Reste konnten für Letales Toxin als Cystein-698 und für α-Toxin als Cystein-707 bestimmt werden. Als Schnittstellen ergaben sich die Positionen für das Letale Toxin zwischen Leucin-543 und Glycin-544 und für das α-Toxin zwischen Leucin-548 und Asparagin-549. Dies konnte durch Mutationen des katalytischen Cysteins bzw. der Schnittstelle zu Alanin nachgewiesen werden. Nach Einführung dieser Mutationen war es nicht mehr möglich, die 64 ERGEBNISSE Autoprozessierung 807L543A/G544A der Toxinfragmente Letales Toxin1-807C698A, Letales Toxin1- sowie α-Toxin1-813C707A und α-Toxin1-813L548A/N549A mit physiologisch relevanten Konzentrationen an InsP6 (100 µM) einzuleiten (siehe Abb.D12A). Zum Nachweis der richtigen Faltung der Toxinfragment-Mutanten wurden in vitro-Glukosylierungen von Rac1 durchgeführt (siehe Abb.D12B). Sowohl Wildtyp- als auch Toxinfragment-Mutanten waren in der Lage, Rac1 zu glukosylieren. A B wt - + C700A + L542A/ S543A + InsP6 (100µM) Toxin A1-809 Toxin A1-542 wt - + C698A + L543A/ G544A + Rac1 Toxin A1-809 InsP6 (100µM) Toxin B1-807 Toxin B1-543 wt - + C698A + L543A/ G544A + Rac1 Toxin B1-807 InsP6 (100µM) Letales Toxin1-807 Rac1 Letales Toxin1-543 Letales Toxin1-807 wt - + C707A + L548A/ N549A + InsP6 (100µM) α-Toxin1-813 α-Toxin1-548 Rac1 α-Toxin1-813 Abb. D12: Identifizierung der Schnittstelle und des katalytisches Cysteins der GTD-CPD-Fragmente InsP6-induzierte Autoprozessierung (Coomassie-Gel): A: GTD-CPD-Fragmente bzw. -Mutanten (2 µg) wurden mit 100 µM InsP6 bei 37°C für 1 h inkubiert. Dargestellt sind die GTD-CPD-Fragmente sowie die resultierende GTD-Spaltprodukte. In vitro-Glukosylierung (Autoradiogramm): B: 5 µM Rac1 wurden mit 10 nM GTD-CPD14 14 Fragmente bzw. -Mutanten mit 10 µM UDP-[ C]-Glukose bzw. UDP-[ C]-N-Acetylglukosamin (für α-Toxin) bei 37°C für 30 min inkubiert. 5.3: Bestimmung der Dissoziationskonstante der Bindung von InsP6 an die CPD von Letalem Toxin und α-Toxin Eine Bedingung für die autokatalytische Prozessierung der CGTs, ist die Bindung von InsP6 an die CPD. Zur Bestimmung der Dissoziationskonstanten der verschiedenen CPDs wurde die Isothermale Titrationskalorimetrie angewendet (siehe Abb.D13). 65 Toxin A543-809 Toxin B544-807 Letales Toxin544-807 α- Toxin549-813 ERGEBNISSE Abb.D13: Bindung von InsP6 an CPD-Fragmente der CGTs Isothermale Titrationskalorimetrie: Für die Bestimmung der KD-Werte wurde jede Titration zweimal durchgeführt. CPD-Fragmente wurden in einer Konzentration zwischen 50 µM bis 100 µM eingesetzt. InsP 6 wurde in einer zehnfach höheren Konzentration von 0,5 bis 1 mM eingesetzt. Aus den Messungen erhaltende Bindekonstante KD und Stöchiometriefaktor n: für Toxin A543-807: KD=4,8/5,3 µM; n=0,59/0,60; Toxin B544-807: KD= 4,4/5,1 µM; n=0,81/0,85; Letales Toxin544-807: KD=2,1/2,8 µM; n=0,64/0,64; α-Toxin549-813: KD=6,6/8,9 µM; n=0,59/0,60. SDS-PAGE (Coomassie-Gel): rekombinante CPD-Fragmente (2 µg) wurden in Bacillus megaterium exprimiert und mittels Affinitätschromatographie und Gelfiltration aufgereinigt. Mit dieser Methode ist es zusätzlich möglich, anhand der erhaltenen n-Werte, die Stöchiometrie der Bindung zu bestimmen. Zu diesem Zweck wurden die CPDToxinfragmente Toxin A543-809, Toxin B544-807, Letales Toxin544-807 und α-Toxin549-813 kloniert und aus Bacillus megaterium aufgereinigt. Die Titrationen wurden für jedes Toxinfragment jeweils zweimal durchgeführt. Die gemessenen KD-Werte lauteten für Toxin A: 4,8/5,3 µM, für Toxin B: 4,4/5,1 µM, für Letales Toxin: 2,1/2,8 µM und für α-Toxin: 6,7/8,9 µM. Aus den nWerten ergibt sich jeweils für die Stöchiometrie des Komplexes am ehesten ein Bindungsverhältnis von 1:1. Die KD-Werte zeigen, dass InsP6 nahezu an alle Toxinfragmente 66 ERGEBNISSE gleich stark bindet und somit keine Unterschiede in der Bindung an die verschiedenen CPDs besteht. 5.4: Autokatalytische Prozessierung von Letalem Toxin und α-Toxin In früheren Arbeiten wurden die Untersuchungen zur Autoprozessierung der CGTs in mit nativen Toxinpräparationen durchgeführt (Reineke et al., 2007; Egerer et al., 2007). Die Expression der Holotoxine in Bacillus megaterium ermöglicht es, nun ausschließlich rekombinante Proteine einzusetzen. Die Holotoxine wurden mit unterschiedlichen Konzentrationen an InsP6 (von 10 nM bis 10 mM) bei 37°C für eine Stunde inkubiert und anschließend die Spaltprodukte über SDS-PAGE aufgetrennt (siehe Abb.D14A). A B InsP6 InsP6 (100µM) Toxin A Toxin A Toxin B Toxin B Toxin B544-2366 Toxin B544-2366 Letales Toxin Letales Toxin α-Toxin α-Toxin α-Toxin549-2178 α-Toxin549-2178 Abb. D14: InsP6-induzierte Autoprozessierung der rekombinanten CGTs InsP6-induzierte Autoprozessierung (Coomassie-Gel): A: rekombinante CGTs (2 µg) wurden mit ansteigenden Konzentration an InsP6 (10 nM bis 10 mM) bei 37°C für 1 h inkubiert. B: rekombinante CGTs (2 µg) wurden mit 100 µM InsP6 für die angegeben Zeitpunkte bei 37°C inkubiert. Überraschenderweise zeigten nur Toxin B und α-Toxin eine Prozessierung bei physiologischen Konzentrationen von InsP6 (≤ 100 µM). Toxin A und Letales Toxin hingegen zeigten, selbst bei hohen InsP6-Konzentrationen (über 1 mM), eine deutlich reduzierte Autoprozessierung. Zur weiteren Charakterisierung dieser Unterschiede wurden Zeitreihen mit 100 µM InsP6 durchgeführt (siehe Abb.D14B). Im Vergleich zu Toxin B und α-Toxin zeigten Toxin A und Letales Toxin selbst nach Inkubation über Nacht keine nennenswerte Prozessierung. Offensichtlich existieren, im Gegensatz zu den Toxinfragmenten der CGTs, deutliche Unterschiede in der InsP6-vermittelten Autoprozessierung der Holotoxine. 67 ERGEBNISSE 5.5: Notwendigkeit funktioneller CPDs von Letalem Toxin und α-Toxin für eine effiziente Vergiftung von Vero-Zellen Letales Toxin zeigte unter in vitro-Bedingungen eine deutlich reduzierte Autoprozessierung. Zur Untersuchung der biologischen Bedeutung einer CPD-vermittelten Autoprozessierung wurden Toxin B-, Letales Toxin- und α-Toxin-Mutanten erzeugt, deren CPD durch zielgerichtete Mutagenese (Toxin B: C698A, Letales Toxin: C698A, α-Toxin C707A) inaktiviert wurde. Zunächst wurden die Toxin-Mutanten auf die InsP6-vermittelte Autoprozessierung getestet (siehe Abb.D15B). Letales Toxin C69A α- Toxin C707A Letales Toxin α- Toxin Toxin B C698A B Toxin B A C wt - + - C698A + InsP6 (100µM) Toxin B 544-2366 Toxin B wt - Rac1 Toxin B C698A + - + InsP6 (100µM) Letales Toxin Rac1 544-2364 Letales Toxin wt - Letales Toxin C707A + - + InsP6 (1mM) α-Toxin Rac1 549-2178 α-Toxin α-Toxin Abb.D15: Charakterisierung CPD-inaktiver-Mutanten von Letalem Toxin und α-Toxin: Zellvergiftung: A: Vero-Zellen wurden mit Wildtyp bzw. CPD-inaktiver-Mutanten von Toxin B, Letalem Toxin und α-Toxin bei 37°C vergiftet. Mikroskopische Bilder wurden nach den angegebenen Zeitpunkten aufgenommen. Toxin B/Toxin B C698A: 100 pM, 1 h; Letales Toxin/Letales Toxin C698A: 1 nM, 6 h; α-Toxin/αToxin C707A: 100 pM, 6h. InsP6-induzierte Autoprozessierung (Coomassie-Gel): B: Wildtyp bzw. CPD-inaktiveMutanten von Toxin B, Letales Toxin und α-Toxin wurden mit den angegebenen Konzentrationen von InsP 6 bei 37°C für 1 h inkubiert. In vitro-Glukosylierung (Autoradiogramm): C: 5 µM rekombinantes Rac1 wurden mit 14 Wildtyp bzw. CPD-inaktiver-Mutanten von Toxin B, Letales Toxin und α-Toxin und 10 µM UDP-[ C]-Glukose 14 bzw. UDP-[ C]-N-Acetylglukosamin (für α-Toxin) bei 37°C für 30 min inkubiert. Toxin B/Toxin B C698A: 10 nM, Letales Toxin/Letales Toxin C698A: 10 nM, α-Toxin/α-Toxin C707A: 100 nM Hierzu wurden die Toxin-Mutanten mit InsP6 inkubiert und die Autoprozessierung anhand des Entstehens charakteristischer Spaltprodukte bewertet. Die Toxin-Mutanten zeigten keinerlei Autoprozessierung nach InsP6-Zugabe. Zum Ausschluss einer generellen Fehlfaltung der Toxin-Mutanten wurden in vitro-Glukosylierungen mit Rac1 durchgeführt (siehe Abb.D15C). Eine vergleichbare Umsetzung von Rac1 durch die Wildtyp-Toxine oder Toxin-Mutanten ist ein Hinweis einer ordnungsgemäßen Faltung der Toxin-Mutanten. Der direkte Vergleich der Zytotoxizität von Wildtyp-Toxinen mit Toxin-Mutanten zeigte, dass bei gleichen Toxinkonzentrationen nur die Wildtyp-Toxine in der Lage sind, die Zellabrundung 68 ERGEBNISSE von Vero-Zellen zu induzieren (siehe Abb.D15A). Diese verzögerte Zellvergiftung der ToxinMutanten verglichen zu den Wildtyp-Toxinen konnten nicht nur für Toxin B und α-Toxin, sondern auch für Letales Toxin beobachtet werden. Hieraus lässt sich schließen, dass nicht nur für Toxin B und α-Toxin, sondern auch für Letales Toxin die CPD-vermittelte Autoprozessierung ein essentieller Schritt einer effizienten Zellvergiftung darstellt. 5.6: Bindung von InsP6 an Letales Toxin und α-Toxin Die rekombinanten CGTs zeigen eine unterschiedlich starke Autoprozessierung nach Zugabe von InsP6. Eine mögliche Ursache hierfür könnten Unterschiede in der Zugänglichkeit der Bindungsstelle für InsP6 an die Cysteinprotease-Domäne der verschiedenen Holotoxine sein. Zur Untersuchung der direkten Bindung von InsP6 an Toxin B, Letales Toxin und an α-Toxin wurden Filterbindungsversuche durchgeführt (siehe Abb.D16). Dieser Versuchsansatz war notwendig, da eine Bestimmung der KD-Werte durch Isothermale Titrationskalorimetrie aufgrund der hohen molekularen Masse, und hieraus resultierend der hohen notwendigen Mengen an Protein nicht möglich ist. Rekombinante Holotoxine wurden mit [3H]-InsP6 inkubiert und anschließend ungebundenes InsP6 durch Waschen über eine Nitrozellulose-Membran entfernt. Die Bindungsstudien ergaben, dass lediglich Toxin B und α-Toxin eine signifikante Bindung von InsP6 aufwiesen, wobei Toxin B eine höhere Bindung zeigte. Letales Toxin zeigte nur eine unzureichende InsP6-Bindung vergleichbar zur Negativkontrolle Toxin B1-546. gebundene Radioaktivität (cpm) 10000 pH 7.4 8000 2000 0 Abb.D16: InsP6-Bindung rekombinanter CGTs 3 InsP6-Filterbindungsversuch: 1 µM rekombinant CGTs und Toxin B1-546 wurden mit 200 nM [ H]-InsP6 und 4,8 µM InsP6 bei 4°C für 10 min inkubiert. Gebundene Radioaktivität wurde durch Szintillation bestimmt. 69 ERGEBNISSE 5.7: Autoprozessierung von Letalem Toxin bei saurem pH-Wert Letales Toxin zeigt in vitro keine nennenswerte Autoprozessierung oder Bindung von InsP6 unter physiologischen pH-Werten. Jedoch scheint es, dass für eine effiziente Zellvergiftung mit Letalem Toxin eine CPD-vermittelte Autoprozessierung notwendig ist. Dies könnte durch eine sterische Hinderung der Bindung von InsP6 an die CPD des Holotoxins verursacht sein. Die CGTs werden über Clathrin-vermittelte Endozytose aufgenommen und erreichen somit die sauren, frühen Endosomen. Es ist allgemein akzeptiert, dass der niedrige pH-Wert der Endosomen eine Konformationsänderung der CGTs induziert, wodurch diese in der Lage sind, in die endosomale Membran zu inserieren. Es kann spekuliert werden, dass erst diese Konformationsänderungen während der Aufnahme von Letalem Toxin den Zugang von InsP6 an die CPD des Holotoxins ermöglicht. Zur Überprüfung dieser Hypothese wurde das komplette Letale Toxin sowie ein Toxinfragment (GTD-CPD), bestehend aus der Glukosyltransferase- und Cysteinprotease-Domäne, mit steigenden Konzentrationen an InsP6 (10 nM bis 10 mM) bei saurem pH-Wert (pH 5) inkubiert (siehe Abb.D17). Interessanterweise zeigte nicht nur das Toxinfragment, sondern auch das Holotoxin von Letalem Toxin eine Autoprozessierung bei subphysiologischen InsP6-Konzentrationen (10 µM). Das GTD-CPD-Toxinfragment benötigt bei sauren pH-Werten, im Vergleich zu neutralen pH-Werten eine etwa hundertfach höhere InsP6-Konzentration für eine Prozessierung. Des Weiteren konnten bei sauren pH-Werten keinerlei Unterschiede mehr in der Autoprozessierung zwischen Holotoxin und Toxinfragment beobachtet werden. pH 5 InsP6 Letales Toxin Letales Toxin544-2364 Letales Toxin1-807 Letales Toxin1-543 Abb.D17: InsP6-induzierte Autoprozessierung von Letalem Toxin sowie GTD-CPD-Fragment von Letalem Toxin InsP6-induzierte Autoprozessierung (Coomassie-Gel): rekombinantes Letales Toxin (2 µg) und Letales Toxin GTD-CPD-Fragment (2 µg) wurden mit steigenden InsP6-Konzentrationen (10 nM bis 10 mM) bei pH 5 bei 37°C für 1 h inkubiert. 5.8: Bindung von InsP6 an Letalem Toxin bei saurem pH-Wert Offenbar ermöglicht eine durch den sauren pH-Wert induzierte Konformationsänderung die Autoprozessierung von Letalem Holotoxin. Eine Bedingung für die Autoprozessierung ist die Bindung von InsP6 an die CPD. Aus diesem Grund sollte nun in einem nächsten Schritt die Bindung von InsP6 an Letales Toxin bei sauren und neutralen pH-Werten direkt miteinander 70 ERGEBNISSE verglichen werden. Die Bindungsexperimente (siehe Abb.D18) ergaben, dass die Bindung von InsP6 an Letales Toxin unter sauren pH-Werten signifikant erhöht ist. Dies war nicht zutreffend für die Negativkontrolle Toxin B1-546. gebundene Radioaktivität (cpm) 4000 pH 7.4 pH 5 3000 1000 0 Abb.D18: Bindung von InsP6 an Letales Toxin bei pH=5 3 InsP6-Filterbindungsversuch: 1µM rekombinantes Letales Toxin und Toxin B1-546 wurden mit 200 nM [ H]-InsP6 und 4,8 µM InsP6 bei pH 7,4 oder pH 5 bei 4°C für 10 min inkubiert. Gebundene Radioaktivität wurde durch Szintillation bestimmt. 5.9: Charakterisierung der pH-abhängigen Bindung von InsP6 an Letales Toxin Zur Charakterisierung der pH-Abhängigkeit der Bindung von InsP6 wurden Filterbindungsexperimente bei unterschiedlichen pH-Werten mit Toxin B und Letalem Toxin durchgeführt (siehe Abb.D19). relativ gebundenes InsP6 (% vom höchsten Wert) 150 Letales Toxin Toxin B 125 100 75 50 25 0 5 6 7 7.4 pH Abb.D19: pH-abhängige Bindung von InsP6 an Letales Toxin 3 InsP6-Filterbindungsversuch: 1 µM rekombinantes Letales Toxin und Toxin B wurden mit 200 nM [ H]-InsP6 und 4,8 µM InsP6 bei pH 5, 6, 7 oder 7,4 bei 4°C für 10 Minuten inkubiert. Gebundene Radioaktivität wurde durch Szintillation bestimmt. 71 ERGEBNISSE Die untersuchten pH-Werte reichten von einem leicht sauren pH-Wert (pH 5, Endosomen) bis zu einem neutralen, physiologischen pH-Wert. Aus den Bindungsstudien wird deutlich, dass Letales Toxin eine erhöhte Bindung von InsP6 unter sauren pH-Bedingungen mit einem Optimum bei pH 5 aufweist. Toxin B hingegen zeigt ein gegensätzliches Bindungsverhalten mit einem Bindungsoptimum bei pH 7,4. 5.10: Untersuchung zur Reversibilität der pH-abhängigen Bindung von InsP6 an Letales Toxin Letales Toxin zeigt eine höhere Bindung von InsP6 bei saurem als bei neutralem pH. Diese Bindung wird wahrscheinlich erst durch eine pH-abhängige Konformationsänderung ermöglicht. Zur Untersuchung der Reversibilität dieser Bindung wurden Filterbindungsexperimente mit Letalem Toxin und Toxin B nach pH-Wechsel durchgeführt (siehe Abb.D20). Die Zugabe von InsP6 zu den Toxinen erfolgte entweder direkt bei pH 7,4, nach einem Wechsel von pH 7,4 zu pH 5 oder nach einem Wechsel von pH 7,4 über pH 5 wieder zu pH 7,4. Es konnte erneut bestätigt werden, dass Letales Toxin, im Gegensatz zu Toxin B, die höchste Bindung bei pH 5 aufweist. Des Weiteren wird deutlich, dass die pHabhängige Bindung von Letalem Toxin (bei pH 5) und ebenfalls von Toxin B (bei pH 7,4) reversibel ist. relativ gebundenes InsP6 (% vom höchsten Wert) 150 125 Letales Toxin Toxin B 100 75 50 25 0 Abb.D20: Reversibilität der pH-abhängigen Bindung von InsP6 an Letalem Toxin 3 InsP6-Filterbindungversuch: 1 µM rekombinantes Letales Toxin und Toxin B wurden mit 200 nM [ H]-InsP6 und 4,8 µM InsP6 bei 4°C nach den angegeben pH-Wechsel inkubiert. Nach jedem pH-Wechsel wurde der Reaktionsansatz für weitere 10 min inkubiert. 72 ERGEBNISSE 5.11: Bestimmung der Dissoziationskonstante der Bindung von InsP6 an die CPD von Letalem Toxin bei saurem pH-Wert Zur Untersuchung des Einflusses eines sauren pH-Wertes auf die Bindung von InsP6 an die CPD von Letalem Toxin wurde die Dissoziationskonstante bei pH 5 durch Isothermale Titrationskalorimetrie bestimmt (siehe Abb.D21). Die Dissoziationskonstante (KD=33,0/40,4 µM) zeigte eine zehnfach reduzierte Bindung von InsP6 an das CPD-Fragment bei sauren pH-Werten verglichen mit neutralen pH-Werten (siehe Abb.D13). Diese Daten deuten darauf hin, dass der saure pH-Wechsel lediglich die Bindung von InsP6 zum Holotoxin und nicht zum GTD-CPD-Fragment ermöglicht. Dies unterstützt die Vorstellung, dass Letales Toxin eine Konformationsänderung bei einem sauren pH-Wert erfährt, wodurch die Bindung von InsP6 erst ermöglicht wird. pH 5 Letales Toxin544-807 Abb.D21: Bindung von InsP6 an das CPD-Fragment von Letalem Toxin bei saurem pH-Wert Isothermale Titrationskalorimetrie: Für die Bestimmung der KD-Werte wurde die Titration zweimal durchgeführt. Letales Toxin544-807 wurde in einer Konzentration 70 µM eingesetzt. InsP6 wurde in einer zehnfach höheren Konzentration von 700 µM eingesetzt. Aus den Messungen erhaltene Bindekonstante KD und Stöchiometriefaktor n: KD=33,0/40,4 µM; n=0,64/0,15 73 ERGEBNISSE 6: Expression von rekombinantem TpeL in Bacillus megaterium Der Gasbranderreger Clostridium perfringens ist ein wichtiger Vertreter humanpathogener Clostridien. Der Erreger bildet eine Vielzahl verschiedener Toxine und wird nach deren Auftreten in die Untertypen A bis E weiter unterteilt (Hatheway, 1990). Kürzlich konnte in Kulturüberständen von Clostridium perfringens Typ C ein bis dato unbekanntes Zytotoxin nachgewiesen werden. Unter den in Clostridium perfringens Typ C beschrieben Toxinen ist es das Einzige, welches eine molekulare Masse von etwa 200 kDa aufweist. Aufgrund seiner Größe und der hohen Homologie zu Toxin A und Toxin B (siehe Einleitung Tab.A1) wurde TpeL von Amimoto et al. als ein weiterer Vertreter der Clostridialen Glukosylierenden Toxine eingestuft. Es wurde spekuliert, dass es sich um eine Glukosyltransferase handeln könnte, die kleine GTPasen der Rho- und Ras-Familien modifiziert (Amimoto et al., 2007). Zu Beginn dieser Arbeit waren weder das Proteinsubstrat- und Kosubstratspektrum der Glukosyltransferase-Domäne noch die Autoprozessierung des Toxins untersucht worden. Das Bacillus megaterium-Proteinexpressionssystem stellte nun eine Möglichkeit dar, TpeL erstmals rekombinant herzustellen und zu charakterisieren. 6.1: Klonierung von pHis1522-TpeL1-1651/1-1779 Das Gen tpeL wurde aus genomischer DNA von Clostridium perfringens Typ C (Stamm CN3685) mittels PCR amplifiziert. Die Primer wurden anhand von zwei verschiedenen Gensequenzen entworfen. Die Gensequenz 1 (tpeL-Gen 1: 5337 Bp/1779 As) entstammt aus einer shotgun-Sequenzierung des Genoms von Clostridium perfringens Typ C Stamm JGS1495 (unveröffentlicht: Paulsen und Sebastian, 2007). Die Gensequenz 2 (tpeL-Gen: 4953 Bp/1651 As) wurde durch eine direkte Gensequenzierung aus Clostridium perfringens Typ C Stamm MC18 ermittelt (Amimoto et al., 2007). Bei der Gensequenz 2 handelt es sich um eine 3´-Verkürzung von Gensequenz 1 (Verkürzung: 384 Bp/128 As). Die Klonierung der beiden Genabschnitte in den Vektor pHis1522 über die Schnittstellen BsrGI und KpnI wurde entsprechend zur Klonierung von pHis1522-Toxin B (siehe Ergebnisse 1.1.1) durchgeführt. 6.2: Aufreinigung von rekombinantem TpeL TpeL1-1651 (Gensequenz 2) und TpeL1-1779 (Gensequenz 1) wurden, wie bereits in dieser Arbeit für die anderen CGTs beschrieben, in Bacillus megaterium exprimiert und über Ni2+Affinitätschromatographie und Gelfiltration aufgereinigt. In Abbildung D22 ist ein SDS-PAGEGel der beiden Proteinaufreinigungen dargestellt. 74 ERGEBNISSE 250 kDa- 180 kDa- 130 kDa- Abb.D22: Aufreinigung von rekombinantem TpeL1-1651 und TpeL1-1779 SDS-PAGE (Coomassie): Rekombinantes TpeL1-1651 und TpeL1-1779 (2 µg) wurden in Bacillus megaterium 2+ exprimiert und über Ni -Affinitätschromatographie aufgereinigt (Markierungen: Toxin-Proteinbanden) TpeL1-1651 und TpeL1-1779 haben laut Berechnung eine molekulare Masse von 191 und 205 kDa. Beide TpeL-Varianten konnten erfolgreich in Bacillus megaterium exprimiert werden. Der Größenunterschied der beiden TpeL-Varianten von etwa 15 kDa ist deutlich sichtbar. 6.3: Untersuchungen zur in vivo-Aktivität von TpeL1-1651 und TpeL1-1779 in der Zellkultur Die Untersuchung der in vivo-Aktivität von TpeL1-1651 und TpeL1-1779 erfolgte in der Zellkultur mit HeLa-Zellen. In Abbildung D23 sind lichtmikroskopische Aufnahmen von HeLa-Zellen, die mit steigenden Konzentrationen an TpeL1-1779 vergiftet wurden, dargestellt. Es ist ersichtlich, dass lediglich die Variante TpeL1-1779, in der Lage ist, morphologische Veränderungen von HeLa-Zellen zu induzieren. Eine Veränderung der Zellmorphologie konnte erst ab einer Konzentration von 100 pM bis 1 nM beobachtet werden. Die kürzere Variante TpeL1-1651 bewirkte selbst in sehr hohen Konzentrationen (10 nM) nach Inkubation über Nacht keine Veränderungen der Zellmorphologie. TpeL1-1779 induzierte im Vergleich zu den anderen Vertretern der CGTs lediglich eine moderate Zellabrundung. Die morphologischen Veränderungen zeigten Hinweise auf die Einleitung von Apoptose. Eine deutliche Zellabrundung gefolgt von der Ausbildung Neuriten-artiger Strukturen, wie sie für Toxin A und B beschrieben sind, konnten bei Zellvergiftung von TpeL1-1779 nicht beobachtet werden (Fiorentini and Thelestam, 1991). 75 ERGEBNISSE TpeL1-1651 (10 nM) TpeL1-1779 (10 nM) TpeL1-1779 (1 nM) TpeL1-1779 (0,1 nM) TpeL1-1779 (0,01 nM) TpeL1-1779 (0,001 nM) Kontrolle Abb. D23: Zellvergiftung von HeLa-Zellen mit rekombinanten TpeL1-1651 und TpeL1-1779 Zellvergiftung: HeLa-Zellen wurden mit TpeL1-1651 (10 nM) und unterschiedlichen Konzentrationen an TpeL1-1779 (10 nM bis 1 pM) bei 37°C über Nacht vergiftet. 6.4: Durch TpeL induzierte Apoptose in HeLa-Zellen Die beobachteten Veränderungen der Zellmorphologie von HeLa-Zellen gaben erste Hinweise, dass TpeL1-1779 in der Lage ist, Apoptose einzuleiten. Zur Bestätigung dieser Vermutung wurden HeLa-Zellen mit einer hohen Konzentration von TpeL1-1779 (10 nM) vergiftet und die Veränderung der Morphologie der Zellen mikroskopisch beobachtet. Aus dem Zeitverlauf der Vergiftung (siehe Abb. D24) war zu erkennen, dass nach etwa sechs Stunden, die für eine Apoptose charakteristische Ausbildung von „apoptotic blebbs“ (dt. apoptotische Bläschen) an der Zytoplasmamembran beginnt. Nach acht Stunden (siehe Abb.D.24) sind insbesondere in der dargestellten Vergrößerung diese „apoptotic blebbs“ (siehe ◄Markierung) deutlich wahrnehmbar. 0h 4h 6h 8h VG Abb. D24: TpeL verursacht Apoptose in HeLa-Zellen Zellvergiftung: HeLa-Zellen wurden mit TpeL1-1779 (10 nM) bei 37°C vergiftet. Die Zellvergiftung wurde mit DICMikroskopie verfolgt. Mikroskopische Bilder wurden nach den angegebenen Zeitpunkten aufgenommen. Auswahlkasten: Vergrößerung (VG), ◄Markierung „apoptopic blebbs“, Maßstab entspricht 10 μm. 76 ERGEBNISSE Zum weiteren biochemischen Nachweis der Toxin-induzierten Apoptose wurde die PARP-1Fragmentierung in HeLa-Zellen nach TpeL1-1779-Vergiftung untersucht. PARP-1 (Poly-ADPRibose-Polymerase-1) wird während der Apoptose durch Caspase-3 proteolytisch gespalten (Nagata, 1997). Es zeigte sich (siehe Abb.D25), dass schon ab einer TpeL1-1779Konzentration von 100 pM eine zu Staurosporin (1 μM, Positivkontrolle) vergleichbar eintretende Apoptose nachweisbar war. Somit konnte sowohl mit zellbiologischen als auch biochemischen Methoden gezeigt werden, dass TpeL1-1779 in der Lage ist, Apoptose in eukaryoten Zellen auszulösen. PARP-1 PARP-1-Fragment GAPDH Abb. D25: PARP-1-Fragmentierung in HeLa-Zellen nach Vergiftung mit TpeL1-1779 Western-Blot: HeLa-Zellen wurden mit Staurosporin (1 μM) oder mit den angegebenen Konzentrationen von TpeL1-1779 bei 37°C über Nacht behandelt. 20 μg Gesamtlysat wurden mittels SDS-PAGE aufgetrennt und PARP-1 bzw. PARP-1-Fragment sowie GAPDH (Ladekontrolle) wurden mittels Western-Blot nachgewiesen. 6.5: Substratspektrum von TpeL Die CGTs modifizieren verschiedene Vertreter kleiner GTPasen der Rho- und RasUnterfamilie durch Übertragung von Zuckerresten. Als Kosubstrate wurden die beiden UDPZucker UDP-Glukose und UDP-N-Acetylglukosamin beschrieben (Just and Gerhard, 2004). Zur biochemischen Untersuchungen des Proteinsubstrat- und Kosubstratspektrums des TpeL-Toxins wurde lediglich die Glukosyltransferase-Domäne (TpeL1-542) rekombinant in Escherichia coli hergestellt. Es wurden in vitro-Glukosylierungen mit den Kosubstraten UDPN-Acetylglukosamin sowie UDP-Glukose mit verschiedenen kleinen GTPasen der Rho- und Ras-Familie durchgeführt. Das Proteinsubstratspektrum von TpeL wurde mit dem Spektrum von Letalem Toxin verglichen, da dieses Toxin sowohl Rho- als auch Ras-Proteine modifiziert (Just and Gerhard, 2004). Aus Abbildung D26 wird ersichtlich, dass TpeL die RasIsoformen Ha-Ras, K-Ras, N-Ras und R-Ras modifizierte, wobei sowohl UDP-NAcetylglukosamin als auch UDP-Glukose als Kosubstrat fungieren konnte. Des Weiteren wurden, im Vergleich zu Letalem Toxin, die Rac-Isoformen Rac1, Rac2 und Rac3 sowie die Rap-Isoformen Rap1B und Rap2A in einem deutlich geringeren Umfang modifiziert. 77 ERGEBNISSE BSA GTPase TpeL1-542/UDP-Glc-NAc TpeL1-542/UDP-Glc Letales Toxin1-546/UDP-Glc Abb. D26: Substratspektrum von TpeL1-542 In vitro-Glukosylierung (Autoradiogramm): Jeweils 3 µg der angegebenen rekombinanten GST-GTPasen wurden 14 mit TpeL1-542 (5 nM) oder Letalem Toxin1-546 (5 nM) in Gegenwart von 10 µM UDP-[ C]-UDP-Glukose (UDP-Glc) 14 bzw. UDP-[ C]-N-Acetylglukosamin (UDP-Glc-NAc) inkubiert. Die Reaktion wurde bei 37°C für 10 Minuten durchgeführt. Offenbar stellen in vitro die Ras-Isoformen die bevorzugten Proteinsubstrate von TpeL dar. Es wurde deutlich, dass im Gegensatz zu den Rac- und Rap-Isoformen Ha-Ras in der Anwesenheit beider UDP-Zucker gleichstark modifiziert werden konnte. Zur genaueren Charakterisierung des Proteinsubstratspektrums in vitro wurde die Umsetzung der weiteren identifizierten Proteinsubstrate Rac1 sowie Rap1B direkt mit Ha-Ras verglichen (siehe Abb.D27). UDP-Glc-NAc 5 5 5 50 UDP-Glc 500 Ha-Ras 5 500 TpeL1-542 (nM) Rac1 UDP-Glc 5 50 5 5 50 UDP-Glc-NAc 500 5 50 500 TpeL 1-542 (nM) RalA Rap1B Ha-Ras GTPase Abb.D27: In vitro-Glukosylierung verschiedener Proteinsubstrate im Vergleich zu Ha-Ras In vitro-Glukosylierung (Autoradiogramm): GST-Ha-Ras oder die angegebene GST-GTPase (jeweils 3 μg) wurden 14 mit TpeL1-542 (für Ha-Ras: 5 nM, für GST-GTPase: 5, 50 oder 500 nM) mit 10 μM UDP-[ C]-N-Acetylglukosamin 14 oder UDP-[ C]-UDP-Glukose bei 37°C für 10 min inkubiert. Hierzu wurden die GTPasen mit steigenden Konzentrationen an TpeL1-542 (5 nM bis 500 nM) in der Anwesenheit von UDP-Glukose oder UDP-N-Acetylglukosamin umgesetzt. Es zeigte sich, dass Rac1 ausschließlich in Anwesenheit von UDP-N-Acetylglukosamin modifiziert wurde und zwar etwa 10-fach schlechter als Ha-Ras. Rap1B wurde in Gegenwart von UDPGlukose etwa 100-fach und in Gegenwart von UDP-N-Acetylglukosamin etwa 10-fach 78 ERGEBNISSE schlechter als Ha-Ras modifiziert. Diese Daten bestätigten erneut, dass die Ras-Proteine (hier: Ha-Ras) in vitro die bevorzugten Proteinsubstrate von TpeL darstellen. 6.6: Akzeptoraminosäure in Ha-Ras Die CGTs O-monoglukosylieren unter anderem Ha-Ras, Rac1 und Cdc42 an Threonin-35 sowie Rho an Threonin-37 (Just and Gerhard, 2004). Dieser hochkonservierte Threonin-Rest befindet sich in der switch-I-Region der kleinen GTPasen (Wei et al., 1997). Zur Identifizierung der Akzeptoraminosäure der Glukosylierung wurde stellvertretend für die Proteinsubstrate von TpeL die Modifikation von Ha-Ras untersucht. Hierzu wurde Ha-Ras in Anwesenheit von UDP-N-Acetylglukosamin mit TpeL1-1651 in vitro glukosyliert. Sowohl Toxinbehandeltes als auch Toxin-unbehandeltes Ha-Ras wurden im Anschluss daran durch LCMS/MS analysiert. Das modifizierte Peptid (siehe Abb.D28A) wurde anhand einer Massenverschiebung von 203 Da aufgrund der Modifikation durch eine N-Acetyl-Hexose identifiziert. Dieses Peptid beinhaltete das bereits als Akzeptoraminosäure vermutete Threonin-35. Letales Toxin glukosyliert Ha-Ras ebenfalls an Threonin-35 (Just et al., 1996). Zur Bestätigung der MS-Daten wurden Kompetitionsexperimente zwischen Letalem Toxin und TpeL durchgeführt. Hierzu wurde Ha-Ras zunächst mit TpeL1-1651 oder mit Letalem Toxin jeweils mit unmarkiertem UDP-Zucker (bei TpeL1-1651: UDP-N-Acetylglukosamin, bei Letalem Toxin: UDP-Glukose) glukosyliert. Es zeigte sich (siehe Abb.D28B), dass hierdurch eine nachgeschaltete Glukosylierung mit [14C]-markierten UDP-Zuckern inhibiert werden konnte. Hieraus konnte geschlossen werden, dass TpeL und Letales Toxin in Ha-Ras die identische Aminosäure, Threonin-35, modifizieren. 79 ERGEBNISSE A T Peptid: SALTIQLIQNHFVDEYDP IEDSYR Modifikation mit N-Acetyl-Hexose: 1057,5103 m/z 3+ (33,7min) B mit TpeL Ha-Ras *TpeL *TcsL unmodifziert: 989,8172 m/z 3+ (34,3min) ohne TpeL Abb. D28: Akzeptoraminosäure der Übertragung von N-Acetylglukosamin durch TpeL auf Ha-Ras LC-MS/MS: A: Ha-Ras (5 µg) wurde mit oder ohne TpeL1-1651 (250 nM) mit UDP-N-Acetylglukosamin (100 µM) für 1 h bei 37°C inkubiert. Die Proteine wurden mittels SDS-PAGE aufgetrennt. Die Proteinbanden (Ha-Ras) wurden aus Coomassie-gefärbten SDS-PAGE-Gelen ausgeschnitten, mit Trypsin verdaut und durch LC-MS/MS vermessen. MS-Daten-Darstellung: y-Achse: Peptidionen-Intensität, x-Achse: Zählereignis (counts) gegen Erfassungszeit (acquisition time) in Minuten. Modifiziertes und unmodifiziertes Peptid sind mit entsprechenden Massen/Ladungsverhältnis und Erfassungszeit dargestellt. Das modifizierte Peptid weist im Vergleich zum unmodifizierten Peptid eine Massenverschiebung von 203 Da. Diese Massenverschiebung entspricht einer Modifikation durch eine N-Acetyl-Hexose. B: In vitro-Glukosylierung (Autoradiogramm): Ha-Ras (3 μg) wurde 14 mit TpeL1-1651 (100 nM) oder Letalem Toxin (100 nM) mit jeweils 10 μM UDP-[ C]-N-Acetylglukosamin (TpeL) 14 bzw. UDP-[ C]-UDP-Glukose (Letales Toxin) bei 37°C für 45 min inkubiert. Proben mit „*“-Markierung wurden zuvor mit dem angegebenen Toxin (jeweils 100 nM) mit jeweils 10 μM UDP-Zucker (TpeL: UDP-NAcetylglukosamin, Letales Toxin: UDP-Glukose) für 30 min bei 37°C inkubiert. TcsL: Letales Toxin 6.7: Kosubstratspektrum von TpeL Nach Berichten von Nagahama et al. nutzt TpeL sowohl UDP-N-Acetylglukosamin als auch UDP-Glukose als Kosubstrat zur Modifikation kleiner GTPasen (Nagahama et al., 2010). Bei der Umsetzung der Ras-Proteine, im Gegensatz zu den Rac- und Rap-Proteinen, zeigte TpeL unter den vorgegebenen Versuchsbedingungen keine Präferenz für eines der beiden Kosubstrate. Somit ist abschließend nicht geklärt, welcher UDP-Zucker in vitro durch das 80 ERGEBNISSE Toxin bevorzugt genutzt wird. Zur Beantwortung dieser Fragestellung wurden die k cat-Werte der Glukohydrolase-Reaktion in Abwesenheit der Proteinsubstrate sowie die KD-Werte der Bindung der beiden UDP-Zucker an die Glukosyltransferase-Domäne von TpeL bestimmt. Anhand der Zeitverläufe der Glukohydrolase-Reaktion (siehe Abb.D29A) und den daraus berechneten kcat-Werten (siehe Abb.D29B, UDP-GlcNAc: kcat=397,3 1/h, UDP-Glc: kcat=17,4 1/h) wurde ersichtlich, dass TpeL1-542 bevorzugt UDP-N-Acetylglukosamin hydrolysiert. Aus den KD-Werten (Abb.D30A/B: ermittelt durch W-quenching: UDP-GlcNAc: KD=9,6 µM; UDPGlc: KD=25,4 µM) wird ebenfalls eine geringe Präferenz für UDP-N-Acetylglukosamin deutlich. UDP-Glukose Glukose 35 hydrolysierter UDP-Zucker (%) N-Acetylglukosamin B UDP-N-Acetylglukosamin A UDP-Glukose UDP-N-Acetylglukosamin 30 kcat= 17,4 (+/- 1,4) 1/h kcat= 397,3 (+/- 13,9) 1/h 25 20 15 10 5 0 0 20 40 60 Zeit (min) 80 100 Abb. D29: Bestimmung der kcat-Werte der Glukohydrolase von UDP-N-Acetylglukosamin und UDP-Glukose durch TpeL1-542 In vitro-Glukohydrolase (Autoradiogramm): A: Zeitverläufe der Glukohydrolase von UDP-N-Acetylglukosamin 14 14 und UDP-Glukose. Hierzu wurden TpeL1-542 (100 nM) mit 20 µM UDP-[ C]-N-Acetylglukosamin oder UDP-[ C]Glukose und jeweils 100 µM nicht radioaktiv-markiertem UDP-Zucker für die angegeben Zeitpunkte bei 37°C inkubiert. Die Hydrolyse-Produkte wurden mittels Dünnschichtchromatographie aufgetrennt. B: Graphische Darstellung der Zeitverläufe. Aus den Zeitpunkten 5, 10 und 15 Minuten wurden die k cat-Werte für die Glukohydrolase-Reaktion von UDP-N-Acetylglukosamin und UDP-Glukose zu berechnet. 81 ERGEBNISSE B 1,2 1,0 1,0 relatives W-quenching relatives W-quenching A 1,2 0,8 0,6 0,4 0,2 0,8 0,6 0,4 0,2 KD= 9,6 (+/-2,6) µM 0,0 0 KD= 25,4 (+/-4,6) µM 0,0 100 200 300 400 UDP-N-Acetylglukosamin (µM) 500 0 100 200 300 UDP-Glukose (µM) 400 500 Abb. D30: Bestimmung der KD-Werte der Bindung von UDP-N-Acetylglukosamin und UDP-Glukose an TpeL1-542 W-quenching: A: UDP-N-Acetylglukosamin B: UDP-Glukose. Hierzu wurden jeweils 1 µM TpeL1-542 mit steigenden Konzentrationen an UDP-Zucker (von 195nM bis 400 µM) bei Raumtemperatur inkubiert. Die Fluoreszenzemission der Tryptophane wurde gemessen und anhand der Fluoreszenzabnahme nach Bindung der UDP-Zucker die KD-Werte bestimmt. 6.8: ANQ-Aminosäuren-Motiv bestimmt die Kosubstratspezifität von TpeL TpeL ist der einzige Vertreter der CGTs, der sowohl UDP-Glukose als auch UDP-NAcetylglukosamin als Kosubstrat nutzt (Nagahama et al., 2010). Zur Klärung der strukturellen Voraussetzungen für diese Kosubstratspezifität wurde zunächst ein Sequenzvergleich von Regionen, die für die Kosubstratspezifität bestimmend sind (Jank et al., 2005), mit verschiedenen Vertretern der CGTs durchgeführt (siehe Abb.D31). SELDTKIAFMFGK-----IA383NQ385VLISKKNSYSLNLIINQ SPLEVKIAFANNS-----VI383NQ385ALISLKDSYCSDLVINQ SDLEIKIAFALGS-----VI382NQ384ALISKQGSYLTNLVIEQ SPLEVKIAFNSKG-----II383NQ385GLISVKDSYCSNLIVKQ SQLEILLSRLKKATGKKTFS385NA387FIISNNDSLTLNNLISQ TpeL Letales Toxin Toxin B Toxin A α-Toxin Abb.D31: Ausschnitt von einem Sequenzvergleich der Primärsequenz der CGTs Rot: Aminosäuren die für die unterschiedliche Kosubstratspezifität der CGTs entscheidend sind. Das Sequenzalignment wurde mit der Software ClustalW erstellt. Toxine, die ausschließlich UDP-Glukose nutzen, besitzen ein INQ-(Isoleucin-AsparaginGlutamin)-Motiv. Im Gegensatz hierzu weist α-Toxin, das UDP-N-Acetylglukosamin nutzt, ein SNA-(Serin-Asparagin-Alanin)-Motiv auf. Interessanterweise besitzt TpeL in diesem Bereich ein abweichendes ANQ-(Alanin-Asparagin-Glutamin)-Motiv (Jank et al., 2005). Somit könnte dieses ANQ-Motiv mitentscheidend für die Kosubstratspezifität von TpeL sein. Zur Überprüfung dieser Hypothese wurde in die Glukosyltransferase-Domäne von TpeL das INQ-Motiv aus Letalem Toxin durch zielgerichtete Mutagenese eingebracht. Dieser Austausch sollte sich in einer Veränderung der Kosubstratspezifität hin zu UDP-Glukose 82 ERGEBNISSE bemerkbar machen. Glukosyltransferaseaktivität Durch und der Untersuchungen UDP-Zucker-Bindung der Glukohydrolase-, konnte eine veränderte Kosubstratspezifität der TpeL1-542A383I-Mutante nachgewiesen werden. Hierbei zeigte sich, dass diese Mutante eine deutlich erhöhte Glukohydrolyse (siehe Abb.D32A/B) von UDPGlukose (kcat=114,3 1/h) im Vergleich zu UDP-N-Acetylglukosamin (kcat=20,0 1/h) aufwies. 18 hydrolysierter UDP-Zucker (%) Glukose UDP-Glukose N-Acetylglukosamin B UDP-N-Acetylglukosamin A UDP-Glukose kcat= 114,3 (+/- 22,2) 1/h UDP-N-Acetylglukosamin kcat= 20,0 (+/- 4,0) 1/h 16 14 12 10 8 6 4 2 0 0 20 40 60 Zeit (min) 80 100 Abb. D32: Bestimmung der kcat-Werte der Glukohydrolase von UDP-N-Acetylglukosamin und UDP-Glukose durch TpeL1-542A383I In vitro-Glukohydrolase (Autoradiogramm): A: Zeitverläufe der Glukohydrolase von UDP-N-Acetylglukosamin 14 und UDP-Glukose. Hierzu wurden TpeL1-542A383I (100 nM) mit 20 µM UDP-[ C]-N-Acetylglukosamin oder UDP14 [ C]-UDP-Glukose und jeweils 100 µM nicht radioaktiv-markiertem UDP-Zucker für die angegeben Zeitpunkte bei 37°C inkubiert. Die Hydrolyse-Produkte wurden mittels Dünnschichtchromatographie aufgetrennt. B: Graphische Darstellung der Zeitverläufe. Aus den Zeitpunkten 5, 10 und 15 Minuten wurden die kcat-Werte für die Glukohydrolase-Reaktion von UDP-N-Acetylglukosamin und UDP-Glukose berechnet. Des Weiteren konnte eine erhöhte Bindung (siehe Abb.D33A/B) von UDP-Glukose (KD=7,6 µM) verglichen mit UDP-N-Acetylglukosamin (KD=32,1 µM) gemessen werden. Die in vitroGlukosylierung von Ha-Ras und Rac1 (siehe Abb.D34) zeigte, dass durch die A383IMutation das Proteinsubstratspektrum von TpeL nicht beeinflusst wurde. Jedoch konnte nun eine verstärkte Modifikation von Ha-Ras in Gegenwart von UDP-Glukose beobachtet werden. Die Modifikation von Rac1 konnte, im Gegensatz zum Wildtyp-Toxin, ausschließlich bei der Umsetzung mit UDP-Glukose festgestellt werden. In allen durchgeführten Experimenten konnte jedoch lediglich eine Präferenz der TpeL1-542A383I-Mutante für UDPGlukose festgestellt werden. Ein absoluter Wechsel der Kosubstratspezifität hin zu UDPGlukose konnte nicht beobachtet werden. UDP-N-Acetylglukosamin wurde immer noch, wenn auch als das „schlechtere“ Kosubstrat, akzeptiert. 83 ERGEBNISSE B 1,2 1,2 1,0 1,0 relatives W-quenching relatives W-quenching A 0,8 0,6 0,4 0,2 0,8 0,6 0,4 0,2 KD= 32,1 (+/- 19,9) µM KD= 7,6 (+/- 3,2) µM 0,0 0,0 0 200 400 600 800 1000 UDP-N-Acetylglukosamin (µM) 1200 0 200 400 600 800 UDP-Glukose (µM) 1000 1200 Abb. D33: Bestimmung der KD-Werte der Bindung von UDP-N-Acetylglukosamin und UDP-Glukose an TpeL1-542A383I W-quenching: A: UDP-N-Acetylglukosamin B: UDP-Glukose. Hierzu wurden jeweils 1 µM TpeL1-542 mit steigenden Konzentrationen an UDP-Zucker (von 0,195 nM bis 400 µM) bei Raumtemperatur inkubiert. Die Fluoreszenzemission der Tryptophane wurde gemessen und anhand der Fluoreszenzabnahme nach Bindung der UDP-Zucker die KD-Werte bestimmt. UDP-Glc 5 5 Ha-Ras 5 50 UDP-Glc-NAc 500 5 50 500 TpeL 1-542A383I (nM) Rac1 Abb.D34: Glukosylierung von Ha-Ras und Rac1 mit TpeL1-542A383I In vitro-Glukosylierung (Autoradiogramm): GST-Ha-Ras oder GST-Rac1 (3 μg) wurden mit TpeL1-542 A383I (für 14 14 Ha-Ras: 5 nM, für Rac1: 5, 50 oder 500 nM) mit 10 μM UDP-[ C]-N-Acetylglukosamin bzw. UDP-[ C]-Glukose bei 37°C für 10 min inkubiert. 6.9: Autokatalytische Prozessierung von TpeL TpeL weist eine hohe Homologie zu den CGTs auf (Amimoto et al., 2007). Für alle untersuchten Vertreter dieser Toxinfamilie konnte eine autokatalytische Autoprozessierung, wobei die Glukosyltransferase-Domäne durch eine intrinsische Cysteinprotease-Domäne abgespalten wird, beschrieben werden. Die Cysteinprotease-Domäne wird durch die Bindung von Inositolhexakisphosphat aktiviert (Egerer and Satchell, 2010). Zunächst wurden Spaltungsversuche mit TpeL1-1779 mit steigenden Konzentrationen Inositolhexakisphosphat durchgeführt (siehe Abb.D35A). 84 ERGEBNISSE A B InsP6 (100µM) InsP6 TpeL1-1779 TpeL1-1779 TpeL543-1779 TpeL1-542 Abb. D35: InsP6-induzierte Autoprozessierung von TpeL1-1779 InsP6-induzierte Autoprozessierung (Coomassie-Gel): A: TpeL1-1779 (2 µg) wurden mit ansteigenden Konzentration an InsP6 (10 nM bis 10 mM) bei 37°C für 1 h inkubiert. B: TpeL1-1779 (2 µg) wurden mit 100 µM InsP6 für die angegeben Zeitpunkte bei 37°C inkubiert. Es zeigte sich, dass nur durch Zugabe sehr hoher InsP6-Konzentrationen (ab 1 mM) eine Autoprozessierung zu beobachten war. Auch nach einer Inkubation über Nacht konnte keine nennenswerte Autoprozessierung von TpeL1-1779 bei physiologischen InsP6-Konzentrationen (100 µM) beobachtet werden (siehe Abb.D35B). Die vermutete Cysteinprotease-Domäne in TpeL (siehe Ergebnisse 5.1, Abb.D10) reicht von Aminosäure 543 bis 805. Des Weiteren konnte das katalytische Cystein in Position 696 identifiziert werden (siehe Ergebnisse 5.1, Abb.D10). Spaltungsversuche mit einem Toxinfragment, das die Glukosyltransferase- sowie die Cysteinprotease-Domäne (TpeL1-805) umfasst, zeigten eine Autoprozessierung bereits ab deutlich niedrigeren InsP6-Konzentrationen (ab 1 µM) (siehe Abb.D36A). A B C wt InsP6 TpeL1-805 TpeL1-542 - + C696A + InsP6 (100µM) TpeL1-805 TpeL1-542 Ha-Ras TpeL1-813 AbbD.36: InsP6-induzierte Autoprozessierung sowie Identifizierung des katalytischen Cysteins von TpeL-GTDCPD-Fragment InsP6-induzierte Autoprozessierung (Coomassie-Gel): A: TpeL1-805 (2 µg) wurden mit ansteigenden Konzentration an InsP6 (10 nM bis 10 mM) bei 37°C für 1 h inkubiert. InsP6-induzierte Autoprozessierung (Coomassie-Gel): B: TpeL1-805 bzw. TpeL1-805C696A-Mutante (2 µg) wurden mit 100 µM InsP6 bei 37°C für 1 h inkubiert. Dargestellt sind die GTD-CPD-Fragmente sowie die resultierende GTD-Spaltprodukte. In vitro-Glukosylierung (Autoradiogramm): C: 5 µM Ha-Ras wurden mit 10 nM TpeL1-805 und 14 TpeL1-805C696A-Mutante mit 10 µM UDP-[ C]-Glukose bei 37°C für 30 min inkubiert. Durch Mutationen des Cystein-696 konnte dessen katalytische Funktion innerhalb der Cysteinprotease-Domäne bestätigt werden. Die Toxinfragment-Mutante TpeL1-805 C696A konnte nicht mehr durch Zugabe physiologisch relevanter InsP6-Konzentrationen (100 µM) prozessiert werden (siehe Abb.D36B). Eine ordnungsgemäße Faltung konnte anhand der in vitro-Glukosylierung von Ha-Ras durch diese Toxin-Mutante sichergestellt werden (siehe Abb.D36C). Die Dissoziationskonstante der Bindung von InsP6 an die Cysteinprotease- 85 ERGEBNISSE Domäne wurde durch ITC bestimmt. Sie lag im niedrigen mikromolaren Bereich (KD=2,1/2,7 µM). Die Stöchiometrie der Bindung betrug ebenfalls 1:1 (siehe Abb.D37). TpeL543-805 Abb.D37: Bindung von InsP6 an CPD von TpeL Isothermale Titrationskalorimetrie: Für die Bestimmung des KD-Wertes wurde die Titration zweimal durchgeführt. TpeL543-805 wurden in einer Konzentration von 100 µM eingesetzt. InsP 6 wurde in einer Konzentration von 700 µM eingesetzt. Aus den Messungen erhaltene Dissoziationskonstante KD und Stöchiometriefaktor n: KD=2,1/2,7 µM; n=1,41/0,66. 6.10: Vergleich der in vivo-Glukosylierung von Ras und Rac1 Die Untersuchungen zum Proteinsubstratspektrum mit rekombinanten kleinen GTPasen zeigte, dass die Glukosyltransferase-Domäne von TpeL in vitro bevorzugt Ras-GTPasen und in einem geringeren Umfang Rac-GTPasen modifiziert. Mit Hilfe von Glukosylierungssensitiven Antikörpern gegen Ha-/K-/N-Ras und Rac1 war es nun möglich, die Modifikation dieser GTPasen nach Zellvergiftung mit TpeL1-1779-Holotoxin zu analysieren. Diese Antikörper können ihr Epitop, im Gegensatz zu herkömmlichen Antikörpern, nach Glukosylierung der kleinen GTPase nicht mehr erkennen (Genth et al., 2006; Huelsenbeck et al., 2009). Es wurden HeLa-Zellen mit ansteigenden Konzentrationen an TpeL1-1779 vergiftet und die Glukosylierung der Ras-GTPasen sowie von Rac1 mittels Western-Blot untersucht. Eine Modifikation von Ras (siehe Abb.D38) konnte schon ab einer Toxin-Konzentration von 10 pM nachgewiesen werden. Im Gegensatz hierzu konnte eine Glukosylierung von Rac1 (siehe Abb.D38) erst ab einer deutlich höheren TpeL1-1779-Konzentration von 1 nM beobachtet werden. Die Ras-Isoformen stellen somit, mit einer etwa 100-fachen Selektivität gegenüber Rac1, die präferierten zellulären Substrate von TpeL1-1779 dar. 86 ERGEBNISSE Rac1 nicht-glukosyliertes Rac1 Ras nicht-glukosyliertes Ras Abb. D38: In vivo-Glukosylierung von Ha-/K-/N-Ras und Rac1 in HeLa-Zellen Western-Blot: HeLa-Zellen wurden mit Letalem Toxin (Positivkontrolle: Ras, Rac1), Toxin A (Positivkontrolle: Rac1, Negativkontrolle: Ras) (jeweils 1 nM) oder den angegebenen Konzentrationen von TpeL 1-1779 bei 37°C über Nacht vergiftet. 20 μg Gesamtlysat wurden durch SDS-PAGE aufgetrennt. Die Glukosylierung von Rac1 sowie der Ras-Isoformen wurde durch Glukosylierungs-sensitive und -insensitive Antikörper (Ladekontrolle) mittels Western-Blot untersucht. 6.11: UDP-N-Acetylglukosamin ist das Kosubstrat für die in vivoGlukosylierung von Ras Die in vitro-Untersuchungen des Kosubstratspektrums der Glukosyltransferase-Domäne von TpeL zeigten eine Präferenz des Toxins für UDP-N-Acetylglukosamin. Jedoch konnte bei der Glukosylierung von Ha-Ras keine Unterschiede zwischen UDP-N-Acetylglukosamin und UDP-Glukose festgestellt werden. Somit ist nicht eindeutig geklärt, welcher UDP-Zucker in vivo als Kosubstrat nun fungiert. Zur eindeutigen Klärung dieser Frage wurde zelluläres Ras immunopräzipitiert und die Modifikation der GTPase durch LC-MS/MS analysiert. Hierzu wurden HeLa-Zellen mit 10 nM TpeL1-1779 über Nacht bei 37°C vergiftet. Nach der Immunopräzipitation wurden Proteinbanden in einem Größenbereich von 20 bis 25 kDa aus dem Coomassie-gefärbten SDS-PAGE ausgeschnitten und mit Thermolysin verdaut. Durch LC-MS/MS (siehe Abb.D39) konnte eindeutig ein Peptid von Ha-Ras identifiziert werden, dass aufgrund seiner Masse einen zusätzliche N-Acetyl-Hexose-Gruppe trug. Dieses Peptid beinhaltete die für Ha-Ras bereits bestimmte Akzeptoraminosäure Threonin-35. Ein mit einer Hexose modifiziertes Peptid konnte nur in Spuren nachgewiesen werden. Es konnte somit gezeigt werden, dass UDP-N-Acetylglukosamin in vivo für TpeL das bevorzugte Kosubstrat darstellt. 87 ERGEBNISSE Peptid: PTIEDSYRKQ Modifikation mit N-Acetyl-Hexose: 480, 5719 m/z 3+ Modifikation mit Hexose: 466,8964 m/z 3+ Abb. D39: Übertragung von N-Acetylglukosamin durch TpeL auf Ha-Ras LC-MS/MS: HeLa-Zellen wurden mit TpeL1-1779 (10 nM) bei 37°C über Nacht vergiftet. Aus dem Gesamtlysat wurde Ras immunopräzipitiert. Die Proteine wurden mittels SDS-PAGE aufgetrennt. Die Proteinbanden wurden aus einem Coomassie-gefärbten SDS-PAGE-Gel ausgeschnitten und durch LC-MS/MS vermessen. MS-DatenDarstellung: y-Achse: Peptidionen-Intensität, x-Achse: Zählereignis (counts) gegen Erfassungszeit (acquisition time) in Minuten. Mit Hexose oder N-Acetyl-Hexose modifizierte Peptide sind mit entsprechendem Massen/Ladungsverhältnis. 6.12: Funktionelle Konsequenz der Glukosylierung von Ras Die Glukosylierung von Rho- und Ras-GTPasen durch die Clostridialen Glukosylierenden Toxine führt zu deren funktionellen Inaktivierung. Unter anderem wird durch den übertragenen Zucker die Effektorbindung der kleinen GTPasen unterbunden (Sehr et al., 1998; Jank and Aktories, 2008). Zur Untersuchung der funktionellen Konsequenz einer in vivo-Glukosylierung von Ras wurden HeLa-Zellen mit TpeL1-1779 vergiftet und aktives, GTPgebundenes Ras mit einem GST-Effektor-pull down (hier: GST-Raf1B-Ras-Binde-Domäne) präzipitiert und mittels Western Blot detektiert (siehe Abb.D40). Ras GTP-Ras Abb. D40: Präzipitation von aktivem GTP-Ras nach Zellvergiftung mit TpeL1-1779 Western-Blot: HeLa-Zellen wurden mit Letalem Toxin (1 nM, Positivkontrolle), Toxin A (1 nM, Negativkontrolle) oder TpeL1-1779 (10 nM) bei 37°C über Nacht vergiftet. 20 µg Gesamtlysat wurden mit SDS-PAGE aufgetrennt und Ras im Western-Blot detektiert (Ladekontrolle). 1 mg Gesamtlysat wurden mit Glutathion-Sepharose-GSTRaf1B-RBD-beads für 30 min bei 4°C inkubiert. Präzipitiertes aktives Ras wurde mittels Western-Blot nachgewiesen. 88 ERGEBNISSE Die Glukosylierung der Ras-Isoformen in vivo durch TpeL1-1779 führte, wie zu erwarten, zu einer Inaktivierung der GTPasen. Es war nicht länger möglich, eine Interaktion von Ras mit seinem Effektor Raf1B in den mit TpeL1-1779 vergifteten HeLa-Zellen nachzuweisen. 6.13: Inhibition Ras-abhängiger Signaltransduktionswege durch TpeL Ras-Proteine fungieren in vielen Signaltransduktionswegen, die Wachstums- und Differenzierungsprozesse regulieren, als entscheidende molekulare Schalter. Die Rasabhängige Signaltransduktion ist am besten für RalGEF/Ral-, PI3K/AKT- sowie MAPKinase/Erk-Signalwege untersucht worden (Karnoub and Weinberg, 2008). In vitro- und in vivo-Untersuchungen in dieser Arbeit zeigten, dass TpeL1-1779 Ras-Proteine glukosyliert und hierdurch diese inaktiviert werden. Im nächsten Abschnitt sollte die Inhibition Ras-abhängiger Signalwege nach Zellvergiftung mit TpeL1-1779 näher untersucht werden. 6.13.1: Inhibition der Neuritenbildung in PC-12-Zellen Die PC12-Zelllinie ist abgeleitet aus einem Phäochromozytom der Ratte. NGF (neurite growth factor) bindet an die Rezeptor-Tyrosin-Kinase TrkA und aktiviert hierdurch Rasabhängige Signalwege, die zur Differenzierung der PC12-Zellen führen (Vaudry et al., 2002). Dieser Differenzierung-Prozess kann phänotypisch anhand der Ausbildung von Neuriten erkannt werden. Eine Inhibition von Ras sollte sich nun in einer fehlenden Neuritenbildung nach NGF-Stimulation bemerkbar machen. Etwa 40% der PC-12-Zellen bildeten Neuriten nach Stimulation mit NGF aus (siehe Abb.D41A/B). Diese Differenzierung der PC12-Zellen konnte in der Tat durch eine Behandlung der Zellen mit TpeL1-1779 aufgehoben werden. In einer TpeL1-1779-Konzentration von 100 pM, die noch nicht zytotoxisch und offenbar zur Modifikation von Ras-Proteinen führten (siehe Ergebnisse 6.10), bildeten lediglich noch etwa 4% der PC-Zellen Neuriten aus (siehe Abb.D41A/B). Dies weist darauf hin, dass die Glukosylierung von zellulärem Ras zur Hemmung Ras-abhängiger Signalwege führt. 89 ERGEBNISSE A B Kontrolle NGF relative Neuritenbildung zur Kontrolle 1,2 NGF 1,0 0,8 0,6 0,4 0,2 0,0 TpeL (0,1 nM)/NGF TpeL (0,001 nM)/NGF Abb. D41: Inhibition der NGF-vermittelte Neuritenbildung von PC-12-Zellen durch TpeL1-1779 Zellvergiftung (Aktin-Färbung): A: PC12-Zellen wurden mit 100 ng/ml NGF stimuliert und mit den angegebenen Konzentrationen an TpeL1-1779 bei 37°C für 2 Tage vergiftet. Das Aktin-Zytoskelett wurde anschließend mit TRITC-Phalloidin angefärbt. B: Quantifizierung der Neuritenbildung von PC12-Zellen nach NGF-Stimulation. 200 bis 400 Zellen wurden je Bedingung ausgezählt. Zellen mit Neuriten, die eine Mindestlänge des Zelldurchmessers aufwiesen wurden als positiv bewertet. 6.13.2: Inhibition der Phosphorylierung von Erk-1/2 Erk-1/2 (extracellular signal-regulated kinase-1/2) sind Proteinkinasen innerhalb der Proteinkinase-Kaskade des MAP-Kinase/Erk-Signalweges. Nach Ligandenbindung an eine Rezeptor-Tyrosin-Kinase (z.B epidermal growth factor-Rezeptor) durch Wachstumsfaktoren kommt es zu einer Aktivierung von Ras über Grb2 (growth factor receptor-bound protein 2) und dem Ras-GEF SOS (son of sevenless). GTP-Ras bindet und aktiviert als Effektoren RafKinasen, welche den Startpunkt der Phosphorylierungs-Kaskade dieses Signalweges darstellen. Innerhalb dieser Kaskade wird Erk-1/2 an Threonin-202 und Tyrosin-204 phosphoryliert (Karnoub et al., 2001; Downward, 2003). Es zeigte sich (siehe Abb.D42), dass in den mit TpeL1-1779 vergifteten HeLa-Zellen eine konzentrationsabhängige Inhibition der Erk1/2-Phosphorylierung zu beobachten war. Die Inhibition dieses Signalweges korrelierte mit der Glukosylierung von Ras und war zwischen einer Konzentration von 10 bis 100 pM TpeL11779 feststellbar. 90 ERGEBNISSE p-Erk-1 p-Erk-2 GAPDH Abb. D42: Inhibition der Erk-1/2-Phophorylierung nach Zellvergiftung mit TpeL 1-1779 Western-Blot: Gehungerte HeLa-Zellen (Kulturmedium ohne FCS) wurden mit Letalem Toxin (Positivkontrolle), Toxin A (Negativkontrolle) (jeweils mit 1 nM) oder den angegebenen Konzentrationen an TpeL 1-1779 bei 37°C über Nacht vergiftet. Anschließend wurden die Zellen mit EGF (10 ng/ml) bei 37°C für 10 min stimuliert. 50 µg Gesamtlysat wurden mittels SDS-PAGE aufgetrennt. Phosphoryliertes Erk-1/2 und GAPDH (Ladekontrolle) wurden mittels Western-Blot detektiert. 91 DISKUSSION E: DISKUSSION Die Clostridialen Glukosylierenden Toxine sind die wichtigsten Virulenzfaktoren zahlreicher humanpathogener Clostridien. Sie sind direkt beteiligt an der Ausbildung diverser Krankheitsbilder, wie z. B. dem Gasbrand oder der Pseudomembranösen Colitis (Hatheway, 1990). Untersuchungen zum Verständnis der Wirkungsweise dieser Toxine sind somit von hoher medizinischer Bedeutung. Des Weiteren werden die Toxine als zellbiologische Werkzeuge zur Untersuchung der Signaltransduktion von Rho- und Ras-GTPasen eingesetzt. Sie dienen hierbei zur gezielten Inaktivierung verschiedener kleiner GTPasen (Voth and Ballard, 2005; Just and Gerhard, 2004). 1: Expression und Aufreinigung der rekombinanten CGTs in Bacillus megaterium Um biochemische Untersuchungen zur Struktur-Funktions-Analyse der CGTs durchführen zu können, ist es wünschenswert, biologisch aktive Toxine mit einem höchstmöglichen Grad an Reinheit zu erlangen. Ursprünglich wurden die CGTs aus Kulturüberständen toxigener Clostridien aufgereinigt. Dies umfasst ein aufwendiges, mehrschrittiges Verfahren. Native Toxinpräparationen sind jedoch häufig kontaminiert mit weiteren clostridialen Produkten, die ebenfalls eine biologische Aktivität aufweisen können. So wurden ursprünglich Toxin A und Toxin B aus Clostridium difficile Mitochondrien-schädigende Eigenschaften zugeschrieben (He et al., 2000; He et al., 2002). Inzwischen wird jedoch eher angenommen, dass Proteinkontaminationen aus den nativen Toxinpräparationen, wie z. B. Toxin E, welches eine hohe Homologie zu phagencodierten Holinen aufweist, für die Schädigung der Mitochondrien verantwortlich ist (Gerhard et al., 2005). Die rekombinante Expression der Toxine stellt hierzu einen Lösungsansatz dar. Die Expression in Escherichia coli lieferte jedoch bisweilen nur unzureichende Ausbeuten mit einem niedrigen Grad an Reinheit. Es ist hingegen möglich, kleinere Toxinfragmente in größeren Mengen zu exprimieren. Die rekombinante Expression der GlukosyltransferaseDomäne bzw. Cysteinprotease-Domäne von Toxin B in Escherichia coli stellte die Grundlage zur Strukturauflösung dieser beiden Domänen durch Röntgenstrukturanalyse dar (Reinert et al., 2005; Shen et al., 2011). Proteinaufreinigungen aus Escherichia coli können jedoch störende Endotoxine oder Porenbildner enthalten (Burger et al., 2003). Eine Alternative zur rekombinanten Herstellung in Escherichia coli stellt das gram-positive Bacillus megateriumExpressionssystem dar. Dieses Expressionssystem weist zahlreiche Vorteile auf, wie beispielsweise das Fehlen von Endotoxinen und alkalischen Proteasen (Meinhardt et al., 1989). Burger et al. konnten zum ersten Mal rekombinantes Toxin A erfolgreich mit diesem System exprimieren. Die Ausbeute betrug 300 bis 500 µg/l Kultur (Burger et al., 2003). 92 DISKUSSION Deutlich höhere Ausbeuten an rekombinantem Toxin A und Toxin B von 5 bis 10 mg/l Kultur konnten Yang et al. berichten (Yang et al., 2008). Wahrscheinlich ermöglicht die enge phylogenetische Verwandtschaft zwischen der Gattung Clostridium und Bacillus und hieraus resultierend eine ähnliche codon-usage eine verbesserte Expression von clostridialen Proteinen. In Anlehnung an die beiden erwähnten Publikationen sollten nun in dieser Arbeit mit dem Bacillus megaterium-Expressionssystem neben Toxin A und B noch weitere Vertreter der CGTs rekombinant hergestellt werden. Nach der erfolgreichen Klonierung von pHis1522Toxin B und anschließender Transformation in Bacillus megaterium-Protoplasten wurde zunächst für Toxin B eine Expressionsstudie in einem kleinen Maßstab durchgeführt. Diese Expressionsstudie zeigte, dass nach Induktion eine Protein mit einer geschätzten molekulare Masse von über 250 kDa im Überstand von Zelllysaten nachweisbar war. Ein 6xHistidin-tag am C-Terminus des Toxins ermöglichte die Aufreinigung mittels Ni2+- Affinitätschromatographie und die Detektion mit einem spezifischen Antikörper. Es ist somit davon auszugehen, dass es sich bei diesem Protein um rekombinantes Toxin B handelte. Die Aufreinigung der rekombinanten CGTs in größeren Maßstäben umfasste neben Ni2+Affinitätschromatographie eine Gelfiltration. Mittels Gelfiltration ist es möglich, die molekulare Masse von Proteinen in Lösung zu bestimmen. Die berechnete molekulare Masse von Toxin B betrug 265 kDa. Es kann somit davon ausgegangen werden, dass Toxin B hauptsächlich als Monomer in Lösung vorlag. Es war insbesondere möglich, rekombinantes Letales Toxin und α-Toxin, verglichen mit nativen Toxinpräparationen, mit einem deutlich höheren Grad an Reinheit zu gewinnen. Die Ausbeute (nach Gelfiltration) für die rekombinanten CGTs lag zwischen 100 bis 500 µg/l Kultur (Toxin A: 250 µg/l, Toxin B: 500 µg/l) und somit zwanzigfach bis vierzigfach unter den von Yang et al. (Toxin A/B: 5-10 mg/l) berichteten Werten (Yang et al., 2008). Die Proteinausbeute von Toxin A war vergleichbar zu der von Burger et al. berichteten Proteinmengen (Toxin A: 300-500 µg/l) (Burger et al., 2003). Im Gegensatz zu Yang et al. wurden die Toxine in dieser Arbeit durch Gelfiltration anstelle von Anionenaustauschchromatographie nachgereinigt (Yang et al., 2008). Die Unterschiede in den Proteinausbeuten können teilweise durch die höheren Verluste während der Gelfiltration erklärt werden. Die Gelfiltration ist jedoch als Nachreinigungsmethode vorzuziehen, da dieses Verfahren eine klar definierte monodisperse Proteinpopulation gewährleistet. Die Identität der rekombinanten CGTs konnte über eine Detektion mittels spezifischer Antikörper bestätigt werden. Die funktionelle Charakterisierung der rekombinanten CGTs umfasste die Untersuchung der Zytotoxizität, die Messung der Enzymaktivität der Glukosyltransferase-Domäne sowie die Porenbildung. Es zeigte sich, dass die rekombinanten CGTs in der Lage sind, die Abrundung 93 DISKUSSION von Vero-Zellen zu induzieren. Die Abrundung der Zellen war eindeutig durch den Zusammenbruch des Aktin-Zytoskeletts verursacht. Die Untersuchung des Substrat- und Kosubstratspektrum zeigte, dass die rekombinanten CGTs ein zu den nativen Toxinen vergleichbares Spektrum aufweisen. Des Weiteren konnte für die rekombinanten CGTs eine Porenbildung in einem Zell-basierten- und Liposomen-basierten Meßsystem beobachtet werden. Mit dem Bacillus megaterium-Expressionssystem ist es nun möglich, rekombinante CGTs mit einem hohen Grad an Reinheit zu exprimieren und aufzureinigen. Die rekombinanten CGTs zeigten eine zu den nativen Toxinen vergleichbare biologische Aktivität sowohl in vivo als auch in vitro. Das Bacillus megaterium-Expressionssystem eröffnet nun nicht nur die Möglichkeit der Expression der Holotoxine oder längerer Toxinfragmente, sondern ermöglicht ebenfalls deren genetische Manipulation. Um diese Möglichkeit besser zu verdeutlichen, sollte in dieser Arbeit eine katalytisch inaktive Toxin B-Mutante generiert werden (Jank et al., 2007a). Aufgrund der Kristallstruktur der Glukosyltransferase-Domäne von Toxin B konnten zahlreiche für die Enzymaktivität essentielle Aminosäuren beschrieben werden. Durch zielgerichtete Mutagenese wurde die katalytisch inaktive Mutante Toxin B-D286A/D288A erzeugt. Hierzu wurden die Aspartat-Reste innerhalb des in den CGTs stark konservierten DXD-Motivs durch Alanine ersetzt. Das DXD-Motiv ist an der Bindung des Kosubstrats UDP-Glukose beteiligt. Die Bindung der Aspartate-286 und -288 an die Phosphat-Gruppen der UDP-Glukose erfolgt über ein Mn2+Ion. Dieses Aminosäuren-Motiv ist essentiell für die ordnungsgemäße Positionierung des Kosubstrats in der katalytischen Tasche (Jank et al., 2007b; Jank et al., 2007a). In vitroUntersuchungen der Enzymaktivität der Glukosyltransferase-Domäne von Letalem Toxin und Toxin B zeigten, dass eine Mutation eines dieser beiden Aspartat-Reste zu Alanin eine 5000fach reduzierte Enzymaktivität zur Folge hat. Der Austausch beider Aspartate zu Alanin resultiert in einem völligen Verlust der Enzymaktivität (Jank et al., 2007a; Busch et al., 1998). Die in dieser Arbeit durchgeführten Untersuchungen der in vitro-Enyzmaktivität von Holotoxin B-D286A/D288A zeigte, wie erwartet, eine deutlich reduzierte Glukosyltransferaseaktivität. In der Zellkultur wies die Toxin B-Mutante ebenfalls eine deutlich reduzierte Zytotoxizität auf. Toxin B induziert eine vollständige Abrundung von Vero-Zellen in picomolaren Konzentrationen (10-100 pM) schon nach 30 bis 60 Minuten. Im Gegensatz hierzu verursachte die Toxin B-Mutante eine Zellabrundung erst in einer 1000-fach höheren Konzentration (100 nM) nach Inkubation für 24 Stunden. 94 DISKUSSION Ausblick: Expression und Aufreinigung der rekombinanten CGTs in Bacillus megaterium Die CGTs üben eine Reihe biologischer Effekte auf Zellen aus, die scheinbar nicht im Zusammenhang mit der Glukosyltransferase-Aktivität stehen. So konnte berichtet werden, dass Toxin A verschiedene MAP-Kinasen (Erk, p38-Kinase oder JNK) aktivieren kann, welches die Freisetzung von Interleukin-8 induziert (Warny et al., 2000). Interleukin-8 wird eine wichtige Rolle bei der Entstehung von entzündlichen Prozessen während der Ausbildung der Pseudomembranösen Colitis zugeschrieben (Warny et al., 2000; Just and Gerhard, 2004). Frühere Untersuchungen von Glukosyltransferase-unabhängigen biologischen Effekten erwiesen sich als äußerst schwierig, da keine geeigneten Negativkontrollen zu Verfügung standen. Die im Rahmen dieser Arbeit generierte inaktive Holotoxin-Mutante kann hierbei von großem Nutzen sein und zur Untersuchung solcher Effekte eingesetzt werden. Die Erzeugung von Punktmutanten kann einen wichtigen Beitrag leisten, die Wirkungsweise der Holotoxine, insbesondere während der Aufnahme in die Zielzellen, besser zu verstehen. So konnten durch zielgerichtete Mutagenese saure Aminosäuren (Glutamat-970/976) in Toxin B identifiziert werden, die wahrscheinlich eine wichtige Rolle während der Porenbildung in die endosomale Membran haben (Genisyuerek et al., 2011). Die rekombinante Expression der CGTs stellt somit ein wertvolles Werkzeug zur Untersuchung von Strukturwirkungsbeziehungen der Toxine sowie zur Untersuchung von Rho/Ras-abhängigen Signaltransduktionswegen dar. Möglicherweise kann die rekombinante Expression in naher Zukunft einen wichtigen Beitrag bei der Ermittlung der Röntgenkristallstruktur der Holotoxine leisten. 2: Autokatalytische Prozessierung von Letalem Toxin und α-Toxin Während der zellulären Aufnahme erfahren die Toxine eine Prozessierung und lediglich die N-terminale Glukosyltransferase-Domäne erreicht das Zytosol der Zielzelle (Pfeifer et al., 2003). Für Toxin A und Toxin B konnte gezeigt werden, dass die proteolytische Abspaltung der Glukosyltransferase-Domäne durch eine benachbarte, intrinsische CysteinproteaseDomäne vermittelt wird (Pruitt et al., 2009; Egerer et al., 2007). Dieses Autoprozessierung wird durch die Bindung von Inositolhexakisphosphat, einem Bestandteil des Zytosols, induziert (Reineke et al., 2007). InsP6 bindet innerhalb der Cysteinprotease-Domäne und vermittelt eine allosterische Aktivierung der Autoprotease-Aktivität der Toxine (Egerer et al., 2009). Ein wichtiger Aspekt dieser Arbeit bestand in der biochemischen Charakterisierung der InsP6-vermittelten Autoprozessierung von Letalem Toxin und α-Toxin. Über die Autoprozessierung dieser beiden Toxine war zu Beginn dieser Arbeit nur wenig bekannt. Die 95 DISKUSSION rekombinante Expression der CGTs in Bacillus megaterium ermöglichte es, zu diesem Zweck ausschließlich rekombinante Proteine einzusetzen. In früheren biochemischen Untersuchungen konnte die Cysteinprotease-Domäne von Toxin A und Toxin B bereits eingegrenzt werden. Pruitt et al. konnten die Cysteinprotease-Domäne von Toxin A auf Aminosäure 543 bis 809 eingrenzen (Toxin B: Aminosäure 544 bis 807) (Pruitt et al., 2009). Aus Sequenzvergleichen mit Toxin A und Toxin B war es nun möglich, in der vorliegenden Arbeit eine vermutete Cysteinprotease-Domäne in Letalem Toxin und α-Toxin zu identifizieren. Die Cysteinprotease-Domäne konnte für Letales Toxin auf Aminosäure 544 bis 807 und für α-Toxin auf Aminosäure 548 bis 813 festgelegt werden. Erste in dieser Arbeit durchgeführten biochemische Untersuchungen mit kurzen Toxinfragmenten, die aus der Glukosyltransferase-Domäne und der CysteinproteaseDomäne (GTD-CPD) bestehen, zeigten, dass diese Fragmente alle eine vergleichbare Autoprozessierung durch subphysiologischen InsP6-Konzentrationen (0,1 bis 1 nM InsP6) erfahren. Dies ist im Einklang mit der Beobachtung, dass die Cysteinprotease-Domäne von Letalem Toxin und α-Toxin eine hohe Sequenzähnlichkeit zur Cysteinprotease-Domäne von Toxin A und Toxin B aufweist. Die katalytische Triade (für Toxin B: D587, H653, C698) sowie die für die Bindung von InsP6 verantwortlich gemachten basischen Aminosäuren (Toxin B: K600, R751, R752) sind innerhalb dieser Toxinfamilie hoch konserviert (Egerer et al., 2007; Egerer et al., 2009). Die Cysteinprotease-Domäne von Letalem Toxin und α-Toxin sind zu 78% bzw. 38% identisch zur Cysteinprotease-Domäne von Toxin B. Meine Befunde zeigen, dass die Autoprozessierung ausschließlich abhängig von der Cysteinprotease-Domäne und unabhängig von anderen C-terminal lokalisierten Domänen ist. Aus den durchgeführten Sequenzvergleichen war es ebenfalls möglich, das katalytische Cystein des DHC-Motivs sowie die Schnittstelle der Autoprozessierung für Letales Toxin und α-Toxin vorauszusagen. In dieser Arbeit konnte durch zielgerichtete Mutagenese der GTDCPD-Fragmente das katalytische Cystein (Letales Toxin: C698, α-Toxin: C707) sowie die Schnittstelle der Prozessierung für Letales Toxin und α-Toxin identifiziert werden. Die Prozessierung erfolgt stets nach einem Leucin-Rest (Letales Toxin: L543, α-Toxin: L548). Die Bestimmung der KD-Werte der Bindung von InsP6 an die Cysteinprotease-Domäne ergab ähnliche Bindekonstanten zwischen den untersuchten CPD-Fragmenten. Die KD-Werte lagen in einstelligen, mikromolaren Konzentrationsbereichen. Die Stöchiometrie der Bindung von InsP6 an die CPD betrug stets 1:1. Ausgehend von der Kristallstruktur der CPD von Toxin B konnte ein mögliches Strukturmodell der CPD von Letalem Toxin (Abb.E1) ermittelt werden. Aufgrund der Ähnlichkeit dieses Modells und der Kristallstruktur von Toxin B-CPD kann vermutet werden, dass keine wesentlichen Strukturunterschiede innerhalb der CPDs zu existieren scheinen. Dies alles legt nahe, dass die InsP6-vermittelte Aktivierung der Cysteinprotease-Domäne bei allen vier untersuchten CGTs über einen ähnlichen 96 DISKUSSION Mechanismus abläuft. Es ist jedoch bemerkenswert, dass, im Gegensatz zu den GTD-CPDFragmenten, deutliche Unterschiede in der Autoprozessierung der Holotoxine beobachtet werden konnten. So zeigten lediglich Toxin B und α-Toxin eine Autoprozessierung nach Zugabe von physiologisch relevanten InsP6-Konzentrationen (≥ 100 µM). Die für die Einleitung der Autoprozessierung der Holotoxine notwendigen InsP6-Konzentrationen lagen stets höher als die Konzentrationen, die für die Autoprozessierung der GTD-CPD-Fragmente notwendig waren. Für Toxin A und Letales Toxin war es selbst durch Zugabe hoher InsP6Konzentrationen (≥ 1 mM) nicht möglich, eine nennenswerte Prozessierung der Toxine einzuleiten. Abb.E1: Strukturmodell der CPD von Letalem Toxin Überlagerungsbild der Cysteinprotease-Domäne von Toxin B und Letalem Toxin. Rot: Kristallstruktur CPD-Toxin B (As.: 544 -797, 3PEE) mit gebundenen InsP6. Blau: Strukturmodell CPD-Letales Toxin. Das Strukturmodell der CPD von Letalem Toxin wurde mit der Software SWISS-MODEL im alignment mode erzeugt. Die beiden Strukturen wurden mit der Software PyMOL bearbeitet. Die CPDs sind als ribbon-Modell dargestellt. Das katalytische Cystein und Histidin sowie InsP6 sind als stick-Modell dargestellt. Filterbindungsexperimente zur InsP6-Bindung der Holotoxine ergaben, dass lediglich für die prozessierbaren Toxine, Toxin B und α-Toxin, eine signifikante Bindung messbar war. Das unterschiedliche Autoprozessierungsverhalten der CGTs kann somit auf eine veränderte Bindung von InsP6 zurückgeführt werden. Die deutlich reduzierte Bindung von InsP6 ist wahrscheinlich die Ursache für die verminderte Autoprozessierung von Toxin A und Letalem Toxin. Diese Unterschiede können nicht auf eine Fehlfaltung der in Bacillus megaterium exprimierten Proteine zurückgeführt werden, da sowohl Toxin A als auch Letales Toxin eine aktive Glukosyltransferase-Domäne und eine Zytotoxizität in der Zellkultur besaßen. Dies ist ein eindeutiges Anzeichen für eine ordnungsgemäße Faltung der beiden Toxine. Sowohl Reineke et al. als auch Kreimeyer et al. berichteten in früheren Arbeiten über eine, im Vergleich zu Toxin B, deutlich reduzierte InsP6-vermittelte Autoprozessierung von Toxin A (Reineke et al., 2007; Kreimeyer et al., 2011). Des Weiteren wurde von Egerer et al. ebenfalls eine stark verminderte InsP6-Bindung von Toxin A beschrieben (Egerer et al., 2007). Trotzdem scheint für eine effiziente Zellvergiftung durch Toxin A eine Cysteinprotease-abhängige Prozessierung notwendig zu sein. Durch Mutationen im Bereich 97 DISKUSSION der Schnittstelle (S541/L542/S543 zu A541/G542/A543) kann die Prozessierbarkeit von Toxin A verhindert werden. Kreimeyer et al. berichteten, dass diese Toxin A-Mutante verglichen zum Wildtyp eine 75-fach reduzierte Zytotoxizität aufweist (Kreimeyer et al., 2011). Zur Untersuchung des Einflusses der Autoprozessierung auf die Zytotoxizität von Letalem Toxin und α-Toxin wurden entsprechend nicht-prozessierbare Cysteinproteaseinaktive Mutanten dieser beiden Toxine (Letales Toxin C698A, α-Toxin C707A) generiert. Die Einführung dieser Mutation resultiert in einer deutlich reduzierten Zytotoxizität. Interessanterweise trifft dieser Sachverhalt auch für Letales Toxin zu, obwohl in vitro keine Autoprozessierung durch physiologisch relevante InsP6-Konzentrationen einzuleiten war. Somit scheint die Cysteinprotease-abhängige Autoprozessierung von Letalem Toxin ein essentieller Schritt für eine effiziente Zellaufnahme des Toxins zu sein. Dies legt nahe, dass noch zusätzliche Faktoren oder andere Bedingungen während der Autoprozessierung von Letalem Toxin eine wichtige Rolle spielen. Die Translokation der Glukosyltransferase-Domäne in das Zytosol erfolgt aus den frühen sauren Endosomen (Barth et al., 2001). Untersuchungen zur InsP6-abhängigen Autoprozessierung bei sauren pH-Werten zeigten, dass unter diesen Bedingungen Letales Toxin eine deutlich verstärkte autoproteolytische Spaltung erfährt. Bereits bei physiologisch relevanten InsP6-Konzentrationen (10-100 µM) war eine Autoprozessierung in vitro festzustellen. Ebenfalls konnte eine deutlich erhöhte InsP6-Bindung an Letales Toxin unter diesen Bedingungen (d. h. bei saurem pH-Wert) festgestellt werden. Es ist zu vermuten, dass dieser Effekt durch eine pH-abhängige Konformationsänderung innerhalb der Gesamtstruktur des Toxins bedingt ist. Eine Absenkung des pH-Wertes sollte eher die Bindung des sauren negativ-geladenen InsP6 zum Toxin reduzieren anstatt zu erhöhen. Dies konnte durch einen Vergleich der InsP6-Bindung von Letalem Toxin und Toxin B bei unterschiedlichen pH-Werten bestätigt werden. Es zeigte sich, dass die Bindung von InsP6 an Toxin B, im Gegensatz zu Letalem Toxin, mit abnehmenden pH-Werten ebenfalls abnimmt. Eine reduzierte Bindung von InsP6 an seine Bindestelle innerhalb der Cysteinprotease-Domäne von Letalem Toxin konnte durch eine etwa zehnfach reduzierte Bindekonstante unter sauren Bedingungen (KD pH=7,4: 2,1 µM, KD pH=5: 33 µM) bewiesen werden. Diese Daten weisen somit auf eine durch sauren pH-Wert induzierte reversible Konformationsänderung der Gesamtstruktur hin, wodurch die Cysteinprotease-Domäne erst für die Bindung von InsP6 zugänglich wird. In der Tat wurden schon zuvor pH-abhängige Konformationsänderungen von Letalem Toxin beschrieben. Qa`Dan et al. berichteten, dass eine kurzzeitige Absenkung des pH-Wertes zu einer deutlich erhöhten Zytotoxizität führt. Die Autoren beschreiben in ihrer Arbeit ebenfalls, dass die Absenkung des pH-Werts eine reversible Konformationsänderung des Letalen Toxins zur Folge hat, wodurch es zu einer Steigerung der Hydrophobizität des Toxins kommt 98 DISKUSSION (Qa'Dan et al., 2001). Natives, aus Clostridium sordellii aufgereinigtes, Letales Toxin bildet unter physiologischen Bedingungen höhermolekulare Proteinkomplexe aus, die reversibel bei sauren pH-Werten dissoziieren (Voth et al., 2004). Die erhöhte Zytotoxizität des Toxins wurde von Voth et al. zunächst durch die Auflösung dieser multimeren Proteinkomplexe erklärt. Die Verschiebung hin zu einer monodispersen Population sollte zu einer beschleunigten Aufnahme des monomeren Toxins führen (Voth et al., 2004). In der hier vorliegenden Arbeit wurden jedoch ausschließlich rekombinante in Bacillus megaterium exprimierte monodisperse Toxinpräparationen eingesetzt. Somit kann die pH-abhängige gesteigerte Zytotoxizität kaum durch die Dissoziation multimerer Toxinkomplexe erklärt werden. Da die Autoprozessierung von Letalem Toxin ein entscheidender Schritt der zellulären Aufnahme des Toxins ist, kann durch die pH-Abhängigkeit der Autoprozessierung die gesteigerte Zytotoxizität von Letalem Toxin bei sauren pH-Werten erklärt werden. Aufgrund der ähnlichen InsP6-Bindeeigenschaften der untersuchten Toxine kann geschlossen werden, dass weder in der Struktur der verschiedenen CysteinproteaseDomänen noch im Reaktionsablauf der Autoprozessierung wesentliche Unterschiede existieren. Somit müssen strukturelle, außerhalb der Cysteinprotease-Domäne liegende Eigenschaften die Bindung von InsP6 an die Cysteinprotease-Domäne beeinflussen. Kürzlich konnte basierend auf Elektronenmikroskopie ein 3D-Modell der Holotoxine A und B erstellt werden. Anhand dieses Modells wurde ebenfalls eine pH-abhängige Konformationsänderung (siehe Abb.E2) innerhalb der sauren Endosomen diskutiert. A B Abb.E2: 3D-Rekonstitutionsmodell der pH-abhängigen Konformationsänderung von Toxin A A: Toxin A bei neutralem pH-Wert (pH 7,4). B: Toxin A bei saurem pH-Wert (pH 5). Die vier Module des ABCDModell sind in unterschiedlichen Farben dargestellt; rot: Glukosyltransferase-Domäne, blau: CysteinproteaseDomäne, gelb: Translokations-Domäne, grün: Rezeptorbinde-Domäne/CROP (Pruitt et al., 2010). Besonders auffällig in diesem Modell sind die im C-Terminus befindlichen ausgedehnten CROP-Bereiche (Pruitt et al., 2010). Diese repetitiven Oligopeptid-Einheiten bilden eine solenoid-Struktur, die eine besonders große Oberfläche kreiert. (Ho et al., 2005). Insbesondere diese Bereiche variieren nun innerhalb der Familie der CGTs. Inwiefern der CTerminus der unterschiedlichen Toxine einen Einfluss auf die Bindung von InsP6 hat, ist bis heute nicht geklärt. Über die physiologische Bedeutung der pH-abhängigen Prozessierung von Letalem Toxin kann nur spekuliert werden. Möglicherweise hat sich Clostridium sordellii an eine Umgebung mit hohen extrazellulären InsP6-Konzentrationen angepasst. Als 99 DISKUSSION typischer Gasbranderreger infiziert Clostridium sordellii häufig verletztes Gewebe oder Wunden (Hatheway, 1990). Durch das Absterben von Zellen kann zytosolisches InsP6 möglicherweise extrazelluläre Kompartimente erreichen. Unter pH-neutralen Bedingungen kann extrazelluläres InsP6 aufgrund einer bestimmten Konformation des Letalen Toxins nicht an die Cysteinprotease-Domäne binden. Nach Clathrin-vermittelter Endozytose gelangt das Toxin zunächst in die Endosomen (Papatheodorou et al., 2010). Dort kommt es nach Ansäuerung zu einer partiellen Entfaltung und/oder einer Aktivierung von Domänen, die bei der Porenbildung und der Translokation der Glukosyltransferase-Domäne über die endosomale Membran beteiligt sind (Genisyuerek et al., 2011; Barth et al., 2001; Qa'Dan et al., 2000). Wahrscheinlich werden nach Insertion die Cysteinprotease-Domäne und die Glukosyltransferase-Domäne zusammen transloziert. In dieser Membran-inserierten Konformation des Toxins sind möglicherweise die C-terminalen Bereiche des Toxins von der Cysteinprotease-Domäne räumlich durch die endosomale Membran getrennt. Die eigentliche Autoprozessierung von Letalem Toxin erfolgt wahrscheinlich unter pH-neutralen Bedingungen im Zytosol. Dies kann dadurch erklärt werden, dass die InsP6-vermittelte Autoprozessierung der GTD-CPD-Fragmente von Letalem Toxin sich bei neutralen deutlich besser als bei sauren pH-Werten induzieren lässt. Durch diesen Mechanismus wird die Autoprozessierung von Letalem Toxin mit der Translokation aus den sauren Endosomen verknüpft. Die Prozessierung des Holotoxins vor der zellulären Aufnahme hätte einen völligen Wirkungsverlust des Toxins zur Folge. Somit stellen diese Beobachtungen einer pHabhängigen Autoprozessierung einen Anpassungsmechanismus des Letalen Toxins dar, um eine frühzeitige Autoprozessierung durch extrazelluläres InsP6, vor dem Erreichen des Wirkungsorts zu verhindern. 3: Charakterisierung von TpeL Die Familie der Clostridialen Glukosylierenden Toxine umfasst neben Toxin A und Toxin B aus Clostridium difficile, das Letale Toxin und das Hämorrhagische Toxin aus Clostridium sordellii sowie α-Toxin aus Clostridium novyi. Diese Toxine modifizieren verschiedene Vertreter kleiner GTPasen der Rho- und Ras-Unterfamilie durch die Übertragung von Glukose oder N-Acetylglukosamin aus den entsprechenden UDP-Zuckerdonoren (Selzer et al., 1996; Just et al., 1995b; Just et al., 1995c; Just et al., 1996). Kürzlich konnte von einer japanischen Arbeitsgruppe TpeL, ein weiteres, bis dato unbekanntes Toxin, in den Kulturüberständen von Clostridium perfringens Typ C (Stamm MC18) nachgewiesen und isoliert werden (Amimoto et al., 2007). Der offene Leserahmen des tpeL-Gens konnte durch N-terminale Aminosäuren-Sequenzierung des Proteins ermittelt und schließlich das Gen sequenziert werden. Das tpeL-Gen umfasst 4953 Basenpaare und codiert für ein 191 kDa großes Protein (Amimoto et al., 2007). Die ermittelte Gensequenz 100 DISKUSSION zeigt eine hohe Homologie (siehe Einleitung Tab.A1 [zwischen 41,6 bis 46,6%]) zu den Vertretern der Clostridialen Glukosylierenden Toxine. Jedoch fehlen im Vergleich zu den CGTs, die im C-Terminus lokalisierten CROP-Bereiche (Amimoto et al., 2007). Der NTerminus von TpeL entspricht der Glukosyltransferase-Domäne der CGTs. In diesem Bereich sind für den Reaktionsmechanismus der Glukosylierung essentielle Aminosäuren (für Toxin B: Aspartat-286, Aspartat-288 des DXD-Motivs, Tryptophan-102) innerhalb von TpeL konserviert. Aus diesen Gründen wurde von Amimoto et al. spekuliert, dass es sich bei TpeL um ein weiteres Mitglied der CGTs handeln könnte, welches Rho- und Ras-GTPasen modifiziert (Amimoto et al., 2007; Olling et al., 2011). Das tpeL-Gen kann in zahlreichen Clostridium perfringens Typ C-Stämmen nachgewiesen werden (Amimoto et al., 2007). Aus Sequenzvergleichen mit der Genomsequenz eines weiteren, sequenzierten Clostridium perfringens Typ C Stammes (JGS1495) zeigte sich, dass zwei verschiedene tpeL-Gensequenzen existieren. Das ursprüngliche tpeL-Gen stellt eine um 384 Basenpaare lange 3´-Verkürzung zu der aus dem Genom von Clostridium perfringens Typ C Stamm JGS1495 ermittelten Gensequenz dar. Zunächst wurden in der vorliegenden Arbeit beide Genabschnitte (codierend für: TpeL1-1651 und TpeL1-1779) in den pHis1522-Expressionsvektor kloniert und die Konstrukte in Bacillus megaterium erfolgreich exprimiert und aufgereinigt. Die Untersuchung der in vivo-Aktivität der beiden TpeL-Varianten erfolgte in der Zellkultur mit HeLa-Zellen. Lediglich die längere Variante TpeL1-1779 war in der Lage in der Zellkultur morphologische Veränderungen zu induzieren. Die kürzere Variante TpeL1-1651 besaß in vivo weder erkennbare zytopathische noch zytotoxische Aktivität. Durch Mikroinjektionsversuche (Daten nicht gezeigt) konnte jedoch bestätigt werden, dass TpeL1-1651 grundsätzlich aktiv ist und in der Lage, die Zellmorphologie von EBL-Zellen zu verändern. Im C-Terminus der Clostridialen Glukosylierenden Toxine sind Domänen lokalisiert, die für die Rezeptorbindung verantwortlich gemacht werden (Genisyuerek et al., 2011). Zu diesen Domänen zählt neben den CROP-Bereichen neuerdings eine weitere Rezeptorbinde-Domäne, die sich in Nterminaler Nachbarschaft befindet. Es ist anzunehmen, dass die Rezeptorbindung dieser Cterminal verkürzten TpeL-Variante aufgrund einer Deletion in der Rezeptorbindungs-Domäne gestört ist. Dies würde eine ordnungsgemäße Aufnahme des Toxins in die Zielzellen unterbinden. Ein weiteres Indiz für diese Hypothese ist, dass weitere TpeL-Varianten, die von Clostridium perfringens Serotypen A und B gebildet werden, der längeren TpeL-Variante entsprechen (Jiang et al., 2009; Chalmers et al., 2008). Es handelt sich bei der kürzeren tpeL-Gensequenz (aus Clostridium perfringens Typ C Stamm MC18) wahrscheinlich um eine fehlerhafte Datenbank-Sequenz, wobei es zu einem verfrühten Auftreten eines Stop-Codons aufgrund einer Baseninsertion (siehe Abb.E3) während der Gen-Sequenzierung, kam. 101 DISKUSSION As:E S S N N L I E Y N E N Y L :GAAAGTTCAAATAATTTAATTGA-ATATAATGAAAATATATTG :GAAAGTTCAAATAATTTAATTGA-ATATAATGAAAATATATTG DNA :GAAAGTTCAAATAATTTAATTGA-ATATAATGAAAATATATTG :GAAAGTTCAAATAATTTAATTGACATATAA------------As:E S S N N L I D I Stop Typ A Typ B Typ C (Sequenz 1) Typ C (Sequenz 2) Abb.E3: Ausschnitt von tpeL-Gensequenzen aus verschiedenen Clostridium perfringens Subtypen Vergleich der DNA-Sequenz von tpeL aus Clostridium perfringens Typ A, Typ B und Typ C. Gensequenz 1 entstammt einer shotgun-Sequenzierung des Genoms von Clostridium perfringens Typ C Stamm JGS1495. Gensequenz 2 entstammt einer Gen-Sequenzierung von tpeL aus Clostridium perfringens Typ C MC18. Die entsprechende Aminosäuren-Sequenz ist angegeben. Die Insertion einer zusätzlichen Cytosin-Basen (rot) in Sequenz 1 ist dargestellt und resultiert in einem verfrühten Stop-Codon (rot). Die Sequenz-alignments wurden mit der Software ClustalW erstellt. Die durch TpeL1-1779 induzierte Veränderung der Zellmorphologie von HeLa-Zellen ist durch eine schwache Zellabrundung sowie durch die Ausbildung apoptotischer Zellmerkmale gekennzeichnet. Die Veränderungen der Zellmorphologie konnten erst bei hohen Toxinkonzentrationen (100 pM bis 1nM) beobachtet werden. Eine starke Zellabrundung sowie die Ausbildung Neuriten-ähnlicher Strukturen, wie sie für Toxin A und B beschrieben wurden, konnten hingegen nicht beobachtet werden (Fiorentini and Thelestam, 1991). Die durch TpeL induzierte Apoptose konnte weiterhin biochemisch durch eine erhöhte Aktivität der Caspase-3, anhand der Fragmentierung von PARP-1, sowie durch apoptotische DNAFragmentierung (Daten nicht gezeigt) bestätigt werden. Interessanterweise zeigten mit TpeL vergiftete Zellen eine auffällige Ähnlichkeit zum Phänotyp von mit Letalem Toxin vergifteter Zellen. In der Literatur wurde für Letales Toxin ebenfalls beschrieben, dass es in der Lage ist, Apoptose einzuleiten. Es existieren zwei verschiedene Modellvorstellungen, wie dies zu Stande kommen kann (Genth and Just, 2010). Einmal wird diskutiert, dass Letales Toxin Apoptose auslösen kann, indem es, unabhängig von seiner Glukosyltransferase-Aktivität, direkt Mitochondrien schädigt (Petit et al., 2003). Naheliegender ist jedoch die zweite Vorstellung, dass Letales Toxin durch die Hemmung Ras-abhängiger Signaltransduktionswege Apoptose verursachen kann (Fiorentini et al., 2003). So wurde für Letales Toxin beschrieben, dass eine Glukosylierung von Ras die typischen Ras-Signalwege wie den MAP-Kinase-Weg (Raf/ERK), die Aktivierung von Ral (RalGEF/Ral) sowie PI3K/AktSignalwege hemmt (Just et al., 1996; Schmidt et al., 1998; Genth and Just, 2010). Die durch TpeL verursachten zellmorphologischen Veränderungen, die eindeutig auf Apoptose hindeuten, gaben im Rahmen dieser Arbeit erste Hinweise, dass TpeL wahrscheinlich RasProteine inaktiviert. Des Weiteren lässt die schwach ausgeprägte Zellabrundung vermuten, dass Rho-GTPasen wahrscheinlich in einem deutlich geringeren Ausmaß modifiziert werden. 102 DISKUSSION Diese Fragen sollten durch eine Analyse der Substratspezifität geklärt werden. Dazu erfolgte die Charakterisierung des Substratspektrums mit der in Escherichia coli rekombinant exprimierten Glukosyltransferase-Domäne von TpeL (Aminosäure 1 bis 542). Hierzu wurden in vitro-Glukosylierungen mit verschiedene kleinen GTPasen der Ras- und Rho-Unterfamilie mit den für die CGTs bereits beschriebenen Kosubstraten, UDP-N-Acetylglukosamin und UDP-Glukose, durchgeführt. Es zeigte sich, dass TpeL die Ras-Isoformen Ha-Ras, K-Ras, N-Ras und K-Ras in Anwesenheit beider Kosubstrate modifizierte. Bei der Modifikation von Ha-Ras konnten keine quantitativen Unterschiede zwischen der Umsetzung mit UDP-NAcetylglukosamin oder UDP-Glukose festgestellt werden. Hingegen fand die Umsetzung der Rac-Isoformen Rac1, Rac2 und Rac3 sowie der Rap-Isoformen Rap1B und Rap2A, die in einem deutlich geringeren Umfang durch TpeL modifiziert wurden, ausschließlich oder bevorzugt in der Anwesenheit von UDP-N-Acetylglukosamin statt. Zur Klärung der Frage, welcher UDP-Zucker in vitro nun das bevorzugte Kosubstrat darstellt, wurden in Abwesenheit der Proteinsubstrate die in vitro-Glukohydrolase sowie die Bindung der UDPZucker an die Glukosyltransferase-Domäne von TpeL bestimmt. Anhand der kcat-Werte der Glukohydrolase-Reaktion sowie der KD-Werte der UDP-Zucker-Bindung konnte ebenfalls eine Präferenz für UDP-N-Acetylglukosamin festgestellt werden. Durch massenspektrometrische Untersuchungen sowie Kompetitionsexperimente konnte gezeigt werden, dass die Übertragung von N-Acetylglukosamin auf Ha-Ras auf die Akzeptoraminosäure Threonin-35 erfolgte. Dies ist ebenfalls die beschriebe Akzeptoraminosäure für die Modifikation von Ha-Ras durch Letales Toxin (Just and Gerhard, 2004; Just et al., 1996). Parallel zur Durchführung dieser Arbeit wurden von Nagahama et al. Daten zum Substratspektrum der Glukosyltransferase-Domäne von TpeL veröffentlicht. Die Autoren konnten bestätigen, dass es sich bei TpeL tatsächlich um eine Glukosyltransferase handelt, die sowohl UDP-Glukose als auch UDP-N-Acetylglukosamin als Kosubstrat nutzt. Diese Forschergruppe konnte ebenfalls zeigen, dass die Ras-GTPasen Ha-Ras und Rap1B sowie Rac1 durch TpeL modifiziert werden. Die Umsetzung von Rac1 erfolgte ebenfalls ausschließlich in der Gegenwart von UDP-N-Acetylglukosamin. Die Übertragung des Zuckerrestes erfolgte bei der Modifikation von Rac1 auf Threonin-37, welches der Aminosäure Threonin-35 in Ha-Ras entspricht. Im Widerspruch zu der vorliegenden Arbeit standen jedoch die Beobachtungen, dass TpeL RalA modifizieren kann (Nagahama et al., 2010). Die Daten der vorliegenden Arbeit und von Nagahama et al. zeigen, dass TpeL als einziger bekannter Vertreter der CGTs, die beiden UDP-Zucker, UDP-N-Acetylglukosamin als auch UDP-Glukose, als Kosubstrat nutzen kann (Nagahama et al., 2010). Jank et al. konnten berichten, dass die Kosubstratspezifität der CGTs lediglich durch zwei Aminosäuren 103 DISKUSSION bestimmt wird. Toxin A, Toxin B und Letales Toxin, die UDP-Glukose nutzen, haben in den entsprechenden Aminosäure-Positionen (für Letales Toxin) Isoleucin-383 und Glutamin-385. Im Gegensatz hierzu sind in α-Toxin, das UDP-N-Acetylglukosamin nutzt, diese Aminosäuren durch Serin und Alanin ersetzt. Die sterisch deutlich größeren Aminosäuren Isoleucin und Glutamin ermöglichen lediglich den Zugang von UDP-Glukose in die katalytische Tasche (Jank et al., 2005). Interessanterweise besitzt TpeL hier eine weitere Variation innerhalb dieses Motives. TpeL besitzt in den entsprechenden Positionen Alanin und Glutamin (Nagahama et al., 2010). Dieses neue Motiv könnte möglicherweise die Tatsache erklären, dass TpeL beide UDP-Zucker als Kosubstrat nutzen kann. Anhand der Primärstruktur wird deutlich, dass TpeL und Letales Toxin sich innerhalb dieses Motives ausschließlich in Position 383 durch Alanin und Isoleucin unterscheiden. Der Vergleich der Kristallstruktur der Glukosyltransferase von Letalem Toxin, gebunden mit UDP-Glukose, und einem Proteinstruktur-Homologie-Modell der Glukosyltransferase-Domäne von TpeL (siehe Abb.E4) zeigt die unterschiedliche räumliche Positionierung dieser beider Aminosäuren. Q385 I383/A383 Abb. E4: Strukturmodell der Glukosyltransferase von TpeL Überlagerungsbild der Glukosyltransferase-Domäne von Letalem Toxin und TpeL. Rot: Kristallstruktur Glukosyltransferase-Domäne von Letalem Toxin (As.: 1-546, 2VKD) (Ziegler et al., 2008) gebunden mit UDP2+ Glukose und Mn -Ion. Blau: Strukturmodell der Glukosyltransferase-Domäne von TpeL. Das Strukturmodell der Glukosyltransferase-Domäne von TpeL wurde mit der Software SWISS-MODEL im alignment mode erzeugt. Die beiden Strukturen wurden mit der Software PyMOL bearbeitet. Die Glukosyltransferasen-Domänen sind als 2+ ribbon-Modell dargestellt. UDP-Glukose und Mn -Ion sind als stick-Modell dargestellt. Darstellung des für die Kosubstratspezifität entscheidenden Aminosäuren-Motivs. Letales Toxin: Isoleucin-383, Glutamin-385, TpeL: Alanin-383, Glutamin-385 Das Isoleucin-383 ragt in die Kosubstrat-Bindetasche von Letalem Toxin und zeigt in Richtung der UDP-Glukose. Diese Aminosäure könnte somit nun den Zugang von UDP-NAcetylglukosamin, aufgrund der zusätzlichen Acetylamino-Gruppe dieses UDP-Zuckers, in die Kosubstrat-Bindetasche blockieren. Die sterisch kleinere Aminosäure Alanin ermöglicht wahrscheinlich den Zugang beider UDP-Zucker. Zur Überprüfung dieser Hypothese wurde das INQ-Motiv in die Glukosyltransferase-Domäne von TpeL eingeführt. Dies sollte zu einer Veränderung der Kosubstratspezifität führen. In der Tat zeigte sich, dass die TpeL1-542A383I- 104 DISKUSSION Mutante nun UDP-Glukose, im Vergleich zu UDP-N-Acetylglukosamin, bevorzugt. Jedoch konnte diese TpeL-Mutante weiterhin, wenn auch in einem geringeren Umfang, UDP-NAcetylglukosamin nutzen. Es war durch diese Mutation nicht möglich, eine vollständige Änderung der Kosubstratspezifität hin zu UDP-Glukose zu erreichen. Es ist davon auszugehen, dass wahrscheinlich noch weitere Aminosäuren für die Kosubstratspezifität von TpeL und Letalem Toxin verantwortlich sind. Hierfür spricht ebenfalls, die Beobachtung von Jank et al., dass die Einführung des INQ-Motives in α-Toxin zwar ebenfalls zu einer Präferenz des Toxins für UDP-Glukose führte, jedoch diese Mutante noch in der Lage war, beide UDP-Zucker zu nutzen (Jank et al., 2005). Das Glutamin-385 kommt in beiden Strukturen vor und ist gleich positioniert. Somit kann diese Aminosäure keinen Einfluss auf die unterschiedliche Kosubstratspezifität von TpeL und Letalem Toxin ausüben. Die Clostridialen Glukosylierenden Toxine erfahren eine autoproteolytische Prozessierung während ihrer Aufnahme in die Zielzellen. Die Prozessierung erfolgt durch eine intrinsische Cysteinprotease-Domäne, deren Aktivität durch Inositolhexakisphosphat reguliert wird (Egerer and Satchell, 2010). Innerhalb der Primärsequenz von TpeL konnte ebenfalls eine Cysteinprotease-Domäne identifiziert werden. Eine erste biochemische Charakterisierung der CPD von TpeL mit Toxinfragmenten (GTD-CPD/CPD) zeigte eine zu den anderen Vertretern der CGTs vergleichbare Autoprozessierung sowie InsP6-Bindung. Ebenso konnte durch zielgerichtete Mutagenese das hochkonservierte katalytische Cystein-696 der Cysteinprotease-Domäne identifiziert werden. Spaltungsversuche mit dem Holotoxin TpeL1-1779 zeigten, dass dieses im Gegensatz zu dem GTD-CPD-Fragment nur in Anwesenheit sehr hoher InsP6-Konzentrationen (> 1mM) eine Autoprozessierung erfährt. Unter physiologischen InsP6-Konzentrationen (100 µM) konnte selbst nach Inkubation über Nacht keine nennenswerte Prozessierung beobachtet werden. TpeL1-1779 kann somit aufgrund seines Prozessierungsverhaltens Toxin A und Letalem Toxin zugeordnet werden, für die ebenfalls eine reduzierte Autoprozessierung der Holotoxine beschrieben werden konnte (Guttenberg et al., 2011; Egerer et al., 2007). Die Clostridialen Glukosylierenden Toxine dienen oft als wertvolle zellbiologische Werkzeuge, die gezielt als Inhibitoren der kleinen GTPasen der Rho- und Ras-Unterfamilie eingesetzt werden. Zum Einsatz kommen hierbei meistens Toxin B und Letales Toxin. Toxin B wird eingesetzt zur Inaktivierung von Rho-GTPasen (Voth and Ballard, 2005). Mit Letalem Toxin können neben Rac1 auch eine Vielzahl von kleinen GTPasen der Ras-Unterfamilie gehemmt werden (Just and Gerhard, 2004). Erste biochemische in vitro-Untersuchungen zum Substratspektrum von TpeL zeigten, dass dieses Toxin bevorzugt Ras-Proteine und in einem geringeren Umfang Rac und Rap-Proteine modifiziert. Im Vergleich zu Toxin B und Letalem Toxin besitzt TpeL1-1779 in vitro ein engeres Proteinsubstratspektrum. Somit könnte TpeL als zellbiologisches Werkzeug aufgrund der höheren Selektivität der Inhibierung der 105 DISKUSSION Ras-Isoformen Letalem Toxin überlegen sein. Mit Hilfe von Glukosylierungs-sensitiven Antikörpern gegen Ha-/K-/N-Ras und Rac1 sollte in HeLa-Zellen zunächst die Umsetzung dieser GTPasen durch TpeL1-1779 miteinander verglichen werden. Eine Modifikation von zellulärem Ras konnte schon ab einer TpeL1-1779-Konzentration von 10 pM festgestellt werden. Die Umsetzung von zellulärem Rac1 konnte hingegen erst ab einer deutlich höheren Konzentration von 1 nM nachgewiesen werden. Diese in vivo-Daten zeigen eine etwa 100fache Selektivität der Umsetzung der Ras-Isoformen verglichen zu Rac1. Die in vitroUntersuchungen des Kosubstratspektrum in Abwesenheit der Proteinsubstrate ergaben eine Präferenz von TpeL für UDP-N-Acetylglukosamin. Hingegen konnte bei der in vitroUmsetzung von Ha-Ras kein Unterschied zwischen den beiden UDP-Zuckern festgestellt werden. Durch die Immunopräzipitation von zellulärem Ras aus mit TpeL1-1779 vergifteten HeLa-Zellen wurde durch Massenspektrometrie jedoch eindeutig eine Modifikation der GTPase mit einer N-Acetyl-Hexose-Gruppe nachgewiesen werden. Eine Modifikation durch einen Hexose-Rest, wie sie bei einer Umsetzung mit UDP-Glukose vorliegen würde, war nicht nachweisbar. Diese weist daraufhin, dass TpeL in vivo UDP-N-Acetylglukosamin als Kosubstrat nutzt. Anhand von pulldown-Experimenten mit dem Ras-Effektor Raf1B konnte gezeigt werden, dass eine Modifikation von Ras zur Inaktivierung der GTPase führt. Dies sollte zur Inhibierung Ras-abhängiger Signaltransduktionswege führen. Ras spielt insbesonders eine entscheidende Rolle in Signaltransduktionswegen, die Differenzierungs- und Wachstumsprozesse kontrollieren (Karnoub and Weinberg, 2008; Downward, 2003). Aus diesem Grund sollten der Einfluss von TpeL1-1779 auf die Neuritenbildung von PC12-Zellen, einem Differenzierungsprozess, sowie der Einfluss auf den MAP-Kinase/Erk-Signalweg, einem Wachstumsprozess, untersucht werden. In der Tat war die Neuritenbildung von PC12Zellen nach NGF-Stimulation durch TpeL1-1779 hemmbar. Ebenfalls wurde eine Hemmung der Phosphorylierung von Erk-1/2 innerhalb der Phosphorylierungs-Kaskade des MAPKinase/Erk-Signalweges beobachtet. Die Inhibierung beider Signalwege erfolgte ab einer TpeL1-1779-Konzentration von 10 bis 100 pM. In diesen Konzentrationsbereichen konnten keine morphologischen Veränderungen der Zellen beobachtet werden, jedoch kann davon ausgegangen werden, dass zelluläres Ras vollständig modifiziert vorliegt. Somit scheint die Glukosylierung von Ras mit der Inhibierung der beiden untersuchten Signalwege zu korrelieren. Ausblick: Charakterisierung von TpeL Clostridium perfringens bildet eine Vielzahl verschiedener bakterieller Toxine, die entscheidend für die Ausbildung verschiedener Krankheitsbilder in Mensch und Tier sind (Hatheway, 1990). Über die Rolle von TpeL in der Pathogenese von Krankheitsbildern, die 106 DISKUSSION durch Clostridium perfringens Typ C verursacht sind, ist nur wenig bekannt. Es kann jedoch spekuliert werden, dass TpeL, entsprechend der Rolle der anderen CGTs, ein bedeutender Virulenzfaktor des Bakteriums darstellt. In der Tat konnte gezeigt werden, dass TpeL eine wichtige Rolle bei der Ausbildung von Läsionen während des Darmbrandes in Masthähnchen hat (Coursodon et al., 2011). Untersuchungen zum Verständnis der Wirkungsweise von TpeL sind somit von großer medizinischer Bedeutung. Des Weiteren könnte TpeL, aufgrund seiner Fähigkeit Ras-Proteine zu inaktivieren, als ein wichtiges pharmakologisches und zellbiologisches Werkzeug eingesetzt werden. So sind zahlreiche Krebserkrankungen beim Menschen durch eine fehlgesteuerte Ras- Signaltransduktion charakterisiert. Der häufigste und in 20% aller Krebserkrankungen ursächliche Grund ist eine direkte Mutation innerhalb des ras-Gens, die zu einer konstitutiven aktiven Ras-Form führt. Diese Mutationen führen in der Regel zu einer reduzierten intrinsischen oder GAP-vermittelten GTPase-Aktivität, wodurch es zu einer Akkumulation in der Zelle von aktivem GTPgebunden Ras kommt. Ras stellt somit als eines der wichtigsten Onkogene ein vielversprechendes potentielles „drug target“ dar. Die Entwicklung von FarnesyltransferaseInhibitoren, die die Aktivität von Ras hemmen konnten, stellte einem vielversprechenden Ansatz dar. Durch diese Stoffe wird die Farnesylierung der CAAX-Box der Ras-Proteine gehemmt. Aufgrund dieser fehlenden posttranslationalen Modifikation kann Ras nicht mehr länger an der Zytoplasmamembran verankert werden, welches essentiell für die biologische Aktivität der GTPase ist (Downward, 2003). Bis heute konnten jedoch - aus verschiedenen Gründen - keiner dieser Farnesyltransferase-Inhibitoren zur Marktreife gebracht werden (Karnoub and Weinberg, 2008; Downward, 2003). Ob TpeL möglicherweise eine Alternative zur Inhibierung von Ras in der Therapie unterschiedlicher Krebserkrankungen darstellt, bleibt zu prüfen. 107 ZUSAMMENFASSUNG F: ZUSAMMENFASSUNG Die Clostridialen Glukosylierenden Toxine sind von großer medizinischer Bedeutung, da sie die Hauptvirulenzfaktoren verschiedener humanpathogener Clostridien darstellen. Zunächst wurde in dieser Arbeit die rekombinante Expression und Aufreinigung dieser Toxine aus Bacillus megaterium etabliert. Funktionelle Charakterisierungen zeigten, dass die rekombinanten Toxine in allen untersuchten Eigenschaften den nativen Toxinpräparationen entsprachen. Auf Grundlage dieses Expressionssystems konnte anschließend die Autoprozessierung von von Letalem Toxin aus Clostridium sordellii und α-Toxin aus Clostridium novyi genauer untersucht werden. Sie ist ein essentieller Schritt in der zellulären Aufnahme dieser Toxine. Aus Sequenzvergleichen mit Toxin A und B konnte eine vermutete Cysteinprotease-Domäne in Letalem Toxin (Aminosäure 544 bis 807) und α-Toxin (Aminosäure 549 bis 813) identifiziert werden. Kurze Toxinfragmente (GTD-CPD), die aus der GlukosyltransferaseDomäne (GTD) und Cysteinprotease-Domäne (CPD) von Letalem Toxin und α-Toxin bestanden, zeigten unter subphysiologischen InsP6-Konzentrationen (<1 µM) eine Autoprozessierung. Durch Mutagenese konnten das katalytische Cystein (Letales Toxin: C698, α-Toxin: C707) der Protease-Domäne sowie die Schnittstelle (Letales Toxin: L543, αToxin: L548) der Autoprozessierung von Letalem Toxin und α-Toxin identifiziert werden. Die Bestimmung der Dissoziationskonstante der Bindung von InsP6 an die CysteinproteaseDomäne offenbarte ähnliche Bindungsaffinitäten für Letales Toxin (KD=2,1/2,8 µM) und αToxin (KD=6,6/8,9 µM). Es konnten keine nennenswerten Unterschiede in der Autoprozessierung der verkürzten GTD-CPD-Fragmente zwischen den einzelnen Toxinen beobachtet werden. Das vollständige Letale Toxin zeigte jedoch, im Unterschied zu Toxin B und α-Toxin, selbst in Gegenwart sehr hoher InsP6-Konzentrationen (>1 mM), keine nennenswerte Autoprozessierung. Diese Beobachtung konnte auf eine herabgesetzte InsP6Bindung des Holotoxins zurückgeführt werden. Nach Absenkung des pH-Wertes auf pH 5 (pH-Wert der frühen Endosomen) war es jedoch möglich, eine Autoprozessierung in Gegenwart physiologischer InsP6-Konzentrationen (10 µM) einzuleiten. Die GTD-CPD- bzw. CPD-Toxinfragmente von Letalem Toxin zeigten bei einem sauren pH-Wert eine reduzierte Autoprozessierung bzw. InsP6-Bindung. Die Autoprozessierung des kompletten Letalen Toxins ist offenbar von einer pH-Wert-induzierten Konformationsänderung, die eine InsP6Bindung an die Cysteinprotease-Domäne ermöglicht, abhängig. Die Autoprozessierung ist somit mit dem Translokationsprozess aus den sauren Endosomen verknüpft. Mit dem etablierten Bacillus megaterium-Proteinexpressionssystem wurde im zweiten Abschnitt dieser Arbeit das Clostridium perfringens TpeL-Toxin, ein bis dato kaum untersuchter Vertreter der Clostridialen Glukosylierenden Toxine, rekombinant hergestellt. Durch den Sequenzvergleich des Genoms eines bereits sequenzierten Clostridium 108 ZUSAMMENFASSUNG perfringens Typ C-Stamms konnten zwei unterschiedliche tpeL-Gensequenzen identifiziert werden. Danach stellt die bislang publizierte tpeL-Sequenz hierzu eine um 384 Basenpaare lange 3´-Verkürzung dar. Nur die längere Toxin-Variante TpeL1-1779, war in der Lage Veränderungen der Zellmorphologie sowie Apoptose in HeLa-Zellen zu induzieren. Die kürzere Toxin-Variante TpeL1-1651 kann offenbar aufgrund einer C-terminalen Deletion nicht in die Zielzellen eindringen. Die Charakterisierung des Substratspektrums von TpeL, die mit der isolierten Glukosyltransferase-Domäne durchgeführt wurde, zeigte, dass TpeL hauptsächlich RasIsoformen in Position Threonin-35 und in einem geringeren Umfang Rac- und Rap-Isoformen modifiziert. Für die Umsetzung konnten sowohl UDP-N-Acetylglukosamin als auch UDPGlukose als Kosubstrat fungieren. UDP-N-Acetylglukosamin scheint jedoch der bevorzugte UDP-Zucker zu sein. Diese Präferenz konnte ebenfalls in Abwesenheit der Proteinsubstrate anhand von Glukohydrolyse- (UDP-GlcNAc: kcat=397,3 1/h, UDP-Glc: kcat=17,4 1/h) sowie Bindungsexperimenten (UDP-GlcNAc: KD=9,6 µM, UDP-Glc: KD= 25,4 µM) bestätigt werden. Es konnte gezeigt werden, dass die einzigartige Kosubstratspezifität von TpeL von dem Aminosäure-Motiv 383-ANQ-385 abhängt. Dieses Motiv scheint sterisch den Zugang beider UDP-Zucker zur Kosubstrat-Bindetasche von TpeL zu ermöglichen. Ein Vergleich der in vivo-Glukosylierung von Ha-/K-/N-Ras und Rac1 in HeLa-Zellen zeigte, dass TpeL1-1779 eine etwa 100-fache Selektivität für die Ras-Proteine aufweist. Durch massenspektrometrische Untersuchungen von immunopräzipitiertem Ras konnte in vivo bestätigt werden, dass TpeL1-1779 UDP-N-Acetylglukosamin als Kosubstrat bevorzugt nutzt. Die Modifikation von Ras durch N-Acetylglukosamin führt zu dessen Inaktivierung und zur Blockade Ras-abhängiger Signaltransduktionswege. Stellvertretend konnte in dieser Arbeit die Inhibition der Neuritenbildung in PC12-Zellen sowie die Erk-1/2-Phophorylierung nach Stimulation mit Wachstumsfaktoren gezeigt werden. TpeL könnte somit in Zukunft eine wichtige Rolle als wertvolles zellbiologisches oder therapeutisches Werkzeug zur Inhibierung von Ras-Signalwegen einnehmen. 109 LITERATUR G: LITERATUR Aktories,K., and Just,I. (2005) Clostridial Rho-inhibiting protein toxins. Curr Top Microbiol Immunol 291: 113-145. Amimoto,K., Noro,T., Oishi,E., and Shimizu,M. (2007) A novel toxin homologous to large clostridial cytotoxins found in culture supernatant of Clostridium perfringens type C. Microbiology 153: 1198-1206. Ball,D.W., Van Tassell,R.L., Denton Roberts,M., Hahn,P.E., Lyerly,D.M., and Wilkins,T.D. (1993) Purification and characterization of alpha-toxin produced by Clostridium novyi type A. Infect Immun 61: 2912-2918. Barbieri,J.T., Riese,M.J., and Aktories,K. (2002) Bacterial toxins that modify the actin cytoskeleton. Annu Rev Cell Dev Biol 18: 315-344. Barroso,L.A., Moncrief,J.S., Lyerly,D.M., and Wilkins,T.D. (1994) Mutagenesis of the Clostridium difficile toxin B gene and effect on cytotoxic activity. Microb Pathog 16: 297-303. Barroso,L.A., Wang,S.-Z., Phelps,C.J., Johnson,J.L., and Wilkins,T.D. (1990) Nucleotide sequence of Clostridium difficile toxin B gene. Nucl Acids Res 18 (13): 4004. Barth,H., Aktories,K., Popoff,M.R., and Stiles,B.G. (2004) Binary bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol Mol Biol Rev 68: 373-402. Barth,H., Pfeifer,G., Hofmann,F., Maier,E., Benz,R., and Aktories,K. (2001) Low pH-induced formation of ion channels by Clostridium difficile toxin B in target cells. J Biol Chem 276: 10670-10676. Bartlett,J.G., Moon,N., Chang,T.W., Taylor,N., and Onderdonk,A.B. (1978) Role of Clostridium difficile in antibiotic-associated pseudomembranous colitits. Gastroenterology 75: 778-782. Bernards,A., and Settleman,J. (2004) GAP control: regulating the regulators of small GTPases. Trends Cell Biol 14: 377-385. Bette,P., Oksche,A., Mauler,F., Von Eichel-Streiber,C., Popoff,M.R., and Habermann,E. (1991) A comparative biochemical, pharmacological and immunological study of Clostridium novyi a-toxin, C. difficile toxin B and C.sordellii lethal toxin. Toxicon 29: 877-887. Burger,S., Tatge,H., Hofmann,F., Genth,H., Just,I., and Gerhard,R. (2003) Expression of recombinant Clostridium difficile toxin A using the Bacillus megaterium system. Biochem Biophys Res Commun 307: 584-588. Busch,C., and Aktories,K. (2000) Microbial toxins and the glucosylation of Rho family GTPases. Curr Opin Struct Biol 10: 528-535. Busch,C., Hofmann,F., Gerhard,R., and Aktories,K. (2000) Involvement of a conserved tryptophan residue in the UDP-glucose binding of large clostridial cytotoxin glycosyltransferases. J Biol Chem 275: 13228-13234. Busch,C., Hofmann,F., Selzer,J., Munro,J., Jeckel,D., and Aktories,K. (1998) A common motif of eukaryotic glycosyltransferases is essential for the enzyme activity of large clostridial cytotoxins. J Biol Chem 273: 19566-19572. 110 LITERATUR Chalmers,G., Bruce,H.L., Hunter,D.B., Parreira,V.R., Kulkarni,R.R., Jiang,Y.F. et al. (2008) Multilocus sequence typing analysis of Clostridium perfringens isolates from necrotic enteritis outbreaks in broiler chicken populations. J Clin Microbiol 46: 3957-3964. Chang,T.W., Gorbach,S.L., and Bartlett,J.B. (1978) Neutralization of Clostridium difficile toxin by Clostridium sordellii antitoxins. Infect Immun 22: 418-422. Chang,T.W., Lin,P.S., Gorbach,S.L., and Bartlett,J.G. (1979) Ultrastructural changes of cultured human amnion cells by Clostridium difficile toxin. Infect Immun 23: 795-798. Chaves-Olarte,E., Freer,E., Parra,A., Guzmán-Verri,C., Moreno,E., and Thelestam,M. (2003) R-Ras glucosylation and transient RhoA activation determine the cytopathic effect produced by toxin B variants from toxin A-negative strains of Clostridium difficile. J Biol Chem 278: 7956-7963. Collier,R.J., and Young,J.A. (2003) Anthrax toxin. Annu Rev Cell Dev Biol 19: 45-70. Coursodon,C.F., Glock,R.D., Moore,K.L., Cooper,K.K., and Songer,J.G. (2011) TpeLproducing strains of Clostridium perfringens type A are highly virulent for broiler chicks. Anaerobe 2011 Oct.17.: [Epub ahead of print] de Haan,L. and Hirst,T.R. (2004) Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms (Review). Mol Membr Biol 21: 77-92. Dove,C.H., Wang,S.Z., Price,S.B., Phelps,C.J., Lyerly,D.M., Wilkins,T.D., and Johnson,J.L. (1990) Molecular characterization of the Clostridium difficile toxin A gene. Infect Immun 58: 480-488. Downward,J. (2003) Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3: 11-22. Egerer,M., Giesemann,T., Herrmann,C., and Aktories,K. (2009) Autocatalytic processing of Clostridium difficile toxin B. Binding of inositol hexakisphosphate. J Biol Chem 284: 33893395. Egerer,M., Giesemann,T., Jank,T., Satchell,K.J., and Aktories,K. (2007) Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on a cysteine protease activity. J Biol Chem 282: 25314-25321. Egerer,M., and Satchell,K.J. (2010) Inositol hexakisphosphate-induced autoprocessing of large bacterial protein toxins. PLoS Pathog 6: e1000942. Etienne-Manneville,S., and Hall,A. (2002) Rho GTPases in cell biology. Nature 420: 629-635. Fiorentini,C., Falzano,L., Travaglione,S., and Fabbri,A. (2003) Hijacking Rho GTPases by protein toxins and apoptosis: molecular strategies of pathogenic bacteria. Cell Death Differ 10: 147-152. Fiorentini,C., Malorni,W., Paradisi,S., Giuliano,M., Mastrantonio,P., and Donelli,G. (1990) Interaction of Clostridium difficile toxin A with cultured cells: cytoskeletal changes and nuclear polarization. Infect Immun 58: 2329-2336. Fiorentini,C., and Thelestam,M. (1991) Clostridium difficile toxin A and its effects on cells. Toxicon 29: 543-567. Florin,I., and Thelestam,M. (1983) Internalization of Clostridium difficile cytotoxin into cultured human lung fibroblasts. Biochim Biophys Acta 763: 383-392. 111 LITERATUR Frisch,C., Gerhard,R., Aktories,K., Hofmann,F., and Just,I. (2003) The complete receptorbinding domain of Clostridium difficile toxin A is required for endocytosis. Biochem Biophys Res Commun 300: 706-711. Genisyuerek,S., Papatheodorou,P., Guttenberg,G., Schubert,R., Benz,R., and Aktories,K. (2011) Structural Determinants for Membrane Insertion, Pore Formation and Translocation of Clostridium difficile Toxin B. Mol Microbiol.: 1643-54 Genth,H., Huelsenbeck,J., Hartmann,B., Hofmann,F., Just,I., and Gerhard,R. (2006) Cellular stability of Rho-GTPases glucosylated by Clostridium difficile toxin B. FEBS Lett 580: 35653569. Genth,H., and Just,I. (2010) Functional implications of lethal toxin-catalysed glucosylation of (H/K/N)Ras and Rac1 in Clostridium sordellii-associated disease. Eur J Cell Biol.: 959-65 Gerhard,R., Burger,S., Tatge,H., Genth,H., Just,I., and Hofmann,F. (2005) Comparison of wild type with recombinant Clostridium difficile toxin A. Microb Pathog 38: 77-83. Geyer,M., Wilde,C., Selzer,J., Aktories,K., and Kalbitzer,H.R. (2003) Glucosylation of Ras by Clostridium sordellii lethal toxin: Consequences for the effector loop conformations observed by NMR spectroscopy. Biochemistry 42: 11951-11959. Giesemann,T., Egerer,M., Jank,T., and Aktories,K. (2008) Processing of Clostridium difficile toxins. J Med Microbiol 57: 690-696. Giesemann,T., Jank,T., Gerhard,R., Maier,E., Just,I., Benz,R., and Aktories,K. (2006) Cholesterol-dependent pore formation of clostridium difficile toxin A. J Biol Chem 281: 10808-10815. Greco,A., Ho,J.G., Lin,S.J., Palcic,M.M., Rupnik,M., and Ng,K.K. (2006) Carbohydrate recognition by Clostridium difficile toxin A. Nat Struct Mol Biol.:460-1 Green,G.A., Schué,V., and Monteil,H. (1995) Cloning and characterization of the cytotoxin Lencoding gene of Clostridium sordellii: Homology with Clostridium difficile cytotoxin B. Gene 161: 57-61. Gülke,I., Pfeifer,G., Liese,J., Fritz,M., Hofmann,F., Aktories,K., and Barth,H. (2001) Characterization of the enzymatic component of the ADP-ribosyltransferase toxin CDTa from Clostridium difficile. Infect Immun 69: 6004-6011. Guttenberg,G., Papatheodorou,P., Genisyuerek,S., Lu,W., Jank,T., Einsle,O., and Aktories,K. (2011) Inositol hexakisphosphate-dependent processing of Clostridium sordellii lethal toxin and Clostridium novyi alpha-toxin. J Biol Chem 286: 14779-14786. Hall,J.C., and O'Toole,E. (1935) Intestinal flora in new-born infants with a description of a new pathogenic anaerobe, Bacillus difficilis. Am J Diseases of Children 49: 390-402. Hatheway,C.L. (1990) Toxigenic clostridia. Clin Microbiol Rev 3: 66-98. He,D., Hagen,S.J., Pothoulakis,C., Chen,M., Medina,N.D., Warny,M., and LaMont,J.T. (2000) Clostridium difficile toxin A causes early damage to mitochondria in cultured cells. Gastroenterology 119: 139-150. He,D., Sougioultzis,S., Hagen,S., Liu,J., Keates,S., Keates,A.C. et al. (2002) Clostridium difficile toxin triggers human colonocyte IL-8 release via mitochondrial oxygen radical generation. Gastroenterology 122: 1048-1057. 112 LITERATUR Henriques,B., Florin,I., and Thelestam,M. (1987) Cellular internalisation of Clostridium difficile toxin A. Microb Pathogen 2: 455-463. Herrmann,C., Ahmadian,M.R., Hofmann,F., and Just,I. (1998) Functional consequences of monoglucosylation of H-Ras at effector domain amino acid threonine-35. J Biol Chem 273: 16134-16139. Ho,J.G., Greco,A., Rupnik,M., and Ng,K.K. (2005) Crystal structure of receptor-binding Cterminal repeats from Clostridium difficile toxin A. Proc Natl Acad Sci U S A 102: 1837318378. Hofmann,F., Busch,C., Prepens,U., Just,I., and Aktories,K. (1997) Localization of the glucosyltransferase activity of Clostridium difficile toxin B to the N-terminal part of the holotoxin. J Biol Chem 272: 11074-11078. Hofmann,F., Herrmann,A., Habermann,E., and Von Eichel-Streiber,C. (1995) Sequencing and analysis of the gene encoding the a-toxin of Clostridium novyi proves its homology to toxins A and B of Clostridium difficile. Mol Gen Genet 247: 670-679. Huelsenbeck,S.C., Klose,I., Reichenbach,M., Huelsenbeck,J., and Genth,H. (2009) Distinct kinetics of (H/K/N)Ras glucosylation and Rac1 glucosylation catalysed by Clostridium sordellii lethal toxin. FEBS Lett 583: 3133-3139. Irvine,R.F., and Schell,M.J. (2001) Back in the water: the return of the inositol phosphates. Nat Rev Mol Cell Biol 2: 327-338. Jaffe,A.B., and Hall,A. (2005) Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21: 247-269. Jank,T., and Aktories,K. (2008) Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol 16: 222-229. Jank,T., Giesemann,T., and Aktories,K. (2007a) Clostridium difficile glucosyltransferase toxin B - essential amino acids for substrate-binding. J Biol Chem 282: 35222-35231. Jank,T., Giesemann,T., and Aktories,K. (2007b) Rho-glucosylating Clostridium difficile toxins A and B: New insights into structure and function. Glycobiology 17: 15R-22R. Jank,T., Reinert,D.J., Giesemann,T., Schulz,G.E., and Aktories,K. (2005) Change of the donor substrate specificity of Clostridium difficile toxin B by site-directed mutagenesis. J Biol Chem 280: 37833-37838. Jiang,Y., Kulkarni,R.R., Parreira,V.R., and Prescott,J.F. (2009) Immunization of broiler chickens against Clostridium perfringens-induced necrotic enteritis using purified recombinant immunogenic proteins. Avian Dis 53: 409-415. Just,I., Fritz,G., Aktories,K., Giry,M., Popoff,M.R., Boquet,P. et al. (1994) Clostridium difficile toxin B acts on the GTP-binding protein Rho. J Biol Chem 269: 10706-10712. Just,I., and Gerhard,R. (2004) Large clostridial cytotoxins. Rev Physiol Biochem Pharmacol 152: 23-47. Just,I., Selzer,J., Hofmann,F., Green,G.A., and Aktories,K. (1996) Inactivation of Ras by Clostridium sordellii lethal toxin-catalyzed glucosylation. J Biol Chem 271: 10149-10153. 113 LITERATUR Just,I., Selzer,J., Von Eichel-Streiber,C., and Aktories,K. (1995a) The low molecular mass GTP-binding protein Rho is affected by toxin A from Clostridium difficile. J Clin Invest 95: 1026-1031. Just,I., Selzer,J., Wilm,M., Von Eichel-Streiber,C., Mann,M., and Aktories,K. (1995b) Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375: 500-503. Just,I., Wilm,M., Selzer,J., Rex,G., Von Eichel-Streiber,C., Mann,M., and Aktories,K. (1995c) The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem 270: 13932-13936. Karnoub,A.E., Der,C.J., and Campbell,S.L. (2001) The insert region of Rac1 is essential for membrane ruffling but not cellular transformation. Mol Cell Biol 21: 2847-2857. Karnoub,A.E., and Weinberg,R.A. (2008) Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 9: 517-531. Kelly,C.P., and LaMont,J.T. (2008) Clostridium difficile-more difficult than ever. N Engl J Med 359: 1932-1940. Kreimeyer,I., Euler,F., Marckscheffel,A., Tatge,H., Pich,A., Olling,A. et al. (2011) Autoproteolytic cleavage mediates cytotoxicity of Clostridium difficile toxin A. Naunyn Schmiedebergs Arch Pharmacol 383: 253-262. Krivan,H.C., Clark,G.F., Smith,D.F., and Wilkins,T.D. (1986) Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Gala1-3Galb1-4GlcNAc. Infect Immun 53: 573-581. Krivan,H.C., and Wilkins,T.D. (1987) Purification of Clostridium difficile toxin A by affinity chromatography on immobilized thyroglobulin. Infect Immun 55,No.8: 1873-1877. Kuehne,S.A., Cartman,S.T., Heap,J.T., Kelly,M.L., Cockayne,A., and Minton,N.P. (2010) The role of toxin A and toxin B in Clostridium difficile infection. Nature 467: 711-713. Lupardus,P.J., Shen,A., Bogyo,M., and Garcia,K.C. (2008) Small molecule-induced allosteric activation of the Vibrio cholerae RTX cysteine protease domain. Science 322: 265-268. Lyerly,D.M., Barroso,L.A., Wilkins,T.D., Depitre,C., and Corthier,G. (1992) Characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile. Infect Immun 60: 46334639. Lyerly,D.M., Krivan,H.C., and Wilkins,T.D. (1988) Clostridium difficile: its disease and toxins. Clin Microbiol Rev 1: 1-18. Lyerly,D.M., Lockwood,D.E., Richardson,S.H., and Wilkins,T.D. (1982) Biological activities of toxins A and B of Clostridium difficile. Infect Immun 35: 1147-1150. Lyerly,D.M., Saum,K.E., MacDonald,D.K., and Wilkins,T.D. (1985) Effects of Clostridium difficile toxins given intragastrically to animals. Infect Immun 47: 349-352. Lyras,D., O'Connor,J.R., Howarth,P.M., Sambol,S.P., Carter,G.P., Phumoonna,T. et al. (2009) Toxin B is essential for virulence of Clostridium difficile. Nature: 1176-9 Martinez,R.D., and Wilkins,T.D. (1992) Comparison of Clostridium sordellii toxins HT and LT with toxins A and B of C. difficile. J Med Microbiol 36: 30-36. 114 LITERATUR McDonald,L.C., Killgore,G.E., Thompson,A., Owens,R.C., Jr., Kazakova,S.V., Sambol,S.P. et al. (2005) An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med 353: 2433-2441. Meinhardt,F., Stahl,U., and Ebeling,W. (1989) Highly efficient expression of homologous and heterologous genes in Bacillus magaterium. Appl Microbiol Biotechnol 30: 343-350. Mesmin,B., Robbe,K., Geny,B., Luton,F., Brandolin,G., Popoff,M.R., and Antonny,B. (2004) A phosphatidylserine-binding site in the cytosolic fragment of Clostridium sordellii lethal toxin facilitates glucosylation of membrane-bound Rac and is required for cytotoxicity. J Biol Chem 279: 49876-49882. Montecucco,C., and Molgo,J. (2005) Botulinal neurotoxins: revival of an old killer. Curr Opin Pharmacol 5: 274-279. Montecucco,C., Rossetto,O., and Schiavo,G. (2004) Presynaptic receptor arrays for clostridial neurotoxins. Trends Microbiol 12: 442-446. Na,X., Kim,H., Moyer,M.P., Pothoulakis,C., and LaMont,J.T. (2008) gp96 is a human colonocyte plasma membrane binding protein for Clostridium difficile toxin A. Infect Immun 76: 2862-2871. Nagahama,M., Ohkubo,A., Oda,M., Kobayashi,K., Amimoto,K., Miyamoto,K., and Sakurai,J. (2010) Clostridium perfringens TpeL glycosylates the Rac and Ras subfamily proteins. Infect Immun. Nagata,S. (1997) Apoptosis by death factor. Cell 88: 355-365. Nusrat,A., Von Eichel-Streiber,C., Turner,J.R., Verkade,P., Madara,J.L., and Parkos,C.A. (2001) Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect Immun 69: 1329-1336. Olling,A., Goy,S., Hoffmann,F., Tatge,H., Just,I., and Gerhard,R. (2011) The Repetitive Oligopeptide Sequences Modulate Cytopathic Potency but Are Not Crucial for Cellular Uptake of Clostridium difficile Toxin A. PLoS One 6: e17623. Olofsson,B. (1999) Rho guanine dissociation inhibitors: pivotal molecules in cellular signalling. Cell Signal 11: 545-554. Papatheodorou,P., Zamboglou,C., Genisyuerek,S., Guttenberg,G., and Aktories,K. (2010) Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS One 5: e10673. Peschka-Süss,R., and Schubert,R. (2003) pH-sensitive liposomes. In Liposomes, a practical approach. Weissig,V., and Torchilin,V. (eds). Oxford: Oxford University Press, pp. 305-318. Petit,P., Breard,J., Montalescot,V., El Hadj,N.B., Levade,T., Popoff,M., and Geny,B. (2003) Lethal toxin from Clostridium sordellii induces apoptotic cell death by disruption of mitochondrial homeostasis in HL-60 cells. Cell Microbiol 5: 761-771. Pfeifer,G., Schirmer,J., Leemhuis,J., Busch,C., Meyer,D.K., Aktories,K., and Barth,H. (2003) Cellular uptake of Clostridium difficile toxin B: translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J Biol Chem 278: 44535-44541. Popoff,M.R. (1987) Purification and characterization of Clostridium sordellii lethal toxin and cross-reactivity with Clostridium difficile cytotoxin. Infect Immun 55: 35-43. 115 LITERATUR Pothoulakis,C. (2000) Effects of Clostridium difficile toxins on epithelial cell barrier. Ann NY Acad Sci 915: 347-356. Pothoulakis,C., Gilbert,R.J., Cladaras,C., Castagliuolo,I., Semenza,G., Hitti,Y. et al. (1996) Rabbit sucrase-isomaltase contains a functional intestinal receptor for Clostridium difficile toxin A. J Clin Invest 98: 641-649. Poxton,I.R., McCoubrey,J., and Blair,G. (2001) The pathogenicity of Clostridium difficile. Clin Microbiol Infect 7: 421-427. Pruitt,R.N., Chagot,B., Cover,M., Chazin,W.J., Spiller,B., and Lacy,D.B. (2009) Structurefunction analysis of inositol hexakisphosphate-induced autoprocessing in Clostridium difficile toxin A. J Biol Chem 284: 21934-21940. Pruitt,R.N., Chambers,M.G., Ng,K.K., Ohi,M.D., and Lacy,D.B. (2010) Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc Natl Acad Sci U S A 107: 13467-13472. Puri,A.W., Lupardus,P.J., Deu,E., Albrow,V.E., Garcia,K.C., Bogyo,M., and Shen,A. (2010) Rational design of inhibitors and activity-based probes targeting Clostridium difficile virulence factor TcdB. Chem Biol 17: 1201-1211. Qa'Dan,M., Spyres,L.M., and Ballard,J.D. (2000) pH-induced conformational changes in Clostridium difficile toxin B. Infect Immun 68: 2470-2474. Qa'Dan,M., Spyres,L.M., and Ballard,J.D. (2001) pH-enhanced cytopathic effects of Clostridium sordellii lethal toxin. Infect Immun 69: 5487-5493. Reineke,J., Tenzer,S., Rupnik,M., Koschinski,A., Hasselmayer,O., Schrattenholz,A. et al. (2007) Autocatalytic cleavage of Clostridium difficile toxin B. Nature 446: 415-419. Reinert,D.J., Jank,T., Aktories,K., and Schulz,G.E. (2005) Structural Basis for the Function of Clostridium difficile Toxin B. J Mol Biol 351: 973-981. Rossman,K.L., Der,C.J., and Sondek,J. (2005) GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 6: 167-180. Rupnik,M., Pabst,S., Rupnik,M., Von Eichel-Streiber,C., Urlaub,H., and Soling,H.D. (2005) Characterization of the cleavage site and function of resulting cleavage fragments after limited proteolysis of Clostridium difficile toxin B (TcdB) by host cells. Microbiology 151: 199208. Rupnik,M., Wilcox,M.H., and Gerding,D.N. (2009) Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7: 526-536. Sandvig,K., Spilsberg,B., Lauvrak,S.U., Torgersen,M.L., Iversen,T.G., and van,D.B. (2004) Pathways followed by protein toxins into cells. Int J Med Microbiol 293: 483-490. Sauerborn,M., and Von Eichel-Streiber,C. (1990) Nucleotide sequence of Clostridium difficile toxin A. Nucleic Acids Res 18: 1629-1630. Schirmer,J., and Aktories,K. (2004) Large clostridial cytotoxins: cellular biology of Rho/Rasglucosylating toxins. Biochim Biophys Acta 1673: 66-74. Schmidt,M., Vo,M., Thiel,M., Bauer,B., Grannass,A., Tapp,E. et al. (1998) Specific inhibition of phorbol ester-stimulated phospholipase D by Clostridium sordellii lethal toxin and Clostridium difficile toxin B-1470 in HEK-293 cells. J Biol Chem 273: 7413-7422. 116 LITERATUR Schwan,C., Stecher,B., Tzivelekidis,T., van,H.M., Rohde,M., Hardt,W.D. et al. (2009) Clostridium difficile toxin CDT induces formation of microtubule-based protrusions and increases adherence of bacteria. PLoS Pathog 5: e1000626. Sehr,P., Joseph,G., Genth,H., Just,I., Pick,E., and Aktories,K. (1998) Glucosylation and ADP-ribosylation of Rho proteins - Effects on nucleotide binding, GTPase activity, and effector-coupling. Biochemistry 37: 5296-5304. Selzer,J., Hofmann,F., Rex,G., Wilm,M., Mann,M., Just,I., and Aktories,K. (1996) Clostridium novyi a-toxin-catalyzed incorporation of GlcNAc into Rho subfamily proteins. J Biol Chem 271: 25173-25177. Sheahan,K.-L., Cordero,C.L., and Fullner Satchell,K.J. (2007) Autoprocessing of the Vibrio cholerae RTX toxin by the cysteine protease domain. EMBO J 26: 2552-2561. Shen,A. (2010) Autoproteolytic Activation of Bacterial Toxins. Toxins 2: 963-977. Shen,A., Lupardus,P.J., Gersch,M.M., Puri,A.W., Albrow,V.E., Garcia,K.C., and Bogyo,M. (2011) Defining an allosteric circuit in the cysteine protease domain of Clostridium difficile toxins. Nat Struct Mol Biol 18: 364-371. Stubbe,H., Berdoz,J., Kraehenbuhl,J.-P., and Corthésy,B. (2000) Polymeric IgA is superior to monomeric IgA and IgG carrying the same variable domain in preventing Clostridium difficile toxin A damaging of T84 monolayers. J Immunol 164: 1952-1960. Sullivan,N.M., Pellett,S., and Wilkins,T.D. (1982) Purification and characterization of toxins A and B of Clostridium difficile. Infect Immun 35: 1032-1040. Takai,Y., Sasaki,T., and Matozaki,T. (2001) Small GTP-binding proteins. Physiol Rev 81: 153-208. Tang-Feldman,Y.J., Ackermann,G., Henderson,J.P., Silva,J., Jr., and Cohen,S.H. (2002) One-step cloning and expression of Clostridium difficile toxin B gene (tcdB). Mol Cell Probes 16: 179-183. Triadafilopoulos,G., Pothoulakis,C., O'Brien,M.J., and LaMont,J.T. (1987) Differential effects of Clostridium difficile toxins A and B on rabbit ileum. Gastroenterology 93,No.2: 273-279. Tucker,K.D., and Wilkins,T.D. (1991) Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect Immun 59: 73-78. Vaudry,D., Stork,P.J., Lazarovici,P., and Eiden,L.E. (2002) Signaling pathways for PC12 cell differentiation: making the right connections. Science 296: 1648-1649. Vetter,I.R., Hofmann,F., Wohlgemuth,S., Herrmann,C., and Just,I. (2000) Structural consequences of mono-glucosylation of Ha-Ras by Clostridium sordellii lethal toxin. J Mol Biol 301: 1091-1095. Von Eichel-Streiber,C., Boquet,P., Sauerborn,M., and Thelestam,M. (1996) Large clostridial cytotoxins - a family of glycosyltransferases modifying small GTP-binding proteins. Trends Microbiol 4: 375-382. Von Eichel-Streiber,C., Laufenberg-Feldmann,R., Sartingen,S., Schulze,J., and Sauerborn,M. (1992) Comparative sequence analysis of the Clostridium difficile toxins A and B. Mol Gen Genet 233: 260-268. 117 LITERATUR Von Eichel-Streiber,C., and Sauerborn,M. (1990) Clostridium difficile toxin A carries a Cterminal repetitive structure homologous to the carbohydrate binding region of streptococcal glycosyltransferases. Gene 96: 107-113. Voth,D.E., and Ballard,J.D. (2005) Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18: 247-263. Voth,D.E., Qa'Dan,M., Hamm,E.E., Pelfrey,J.M., and Ballard,J.D. (2004) Clostridium sordellii lethal toxin is maintained in a multimeric protein complex. Infect Immun 72: 3366-3372. Warny,M., Keates,A.C., Keates,S., Castagliuolo,I., Zacks,J.K., Aboudola,S. et al. (2000) p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J Clin Invest 105: 1147-1156. Wei,Y., Zhang,Y., Derewenda,U., Liu,X., Minor,W., Nakamoto,R.K. et al. (1997) Crystal structure of RhoA-GDP and its functional implications. Nature Struct Biol 4: 699-703. Wren,B.W., and Tabaqchali,S. (1987) Restriction endonuclease DNA analysis of Clostridium difficile. J Clinic Microbiol 25: 2402-2404. Yang,G., Zhou,B., Wang,J., He,X., Sun,X., Nie,W. et al. (2008) Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol 8: 192. Ziegler,M.O., Jank,T., Aktories,K., and Schulz,G.E. (2008) Conformational changes and reaction of clostridial glycosylating toxins. J Mol Biol 377: 1346-1356. 118 ABKÜRZUNGSVERZEICHNIS H: ABKÜRZUNGSVERZEICHNIS A Abb. ADP APS ATP B BHI Bp BSA C. C CDT CGT CPD CROP dATP dCTP dGTP DNA dNTP DPX DTT dTTP ECL EDTA EGF ERK et al. FCS FPLC fw G GAP GAPDH GDI GDP GEF GST GTD Grb2 GTP HEPES HPTS IgG/M InsP6 IPTG JNK LB LC-MS/MS LUV MCS MALDI MARTX mk NEA NGF OD P PA PARP PBR PBS PCR PEG Pi pI pk PMSF PVDF RBD RBD (GST-Raf1B-RBD) rev RIPA Adenin Abbildung Adenosindiphosphat Ammoniumperoxodisulfat Adenintriphosphat Base engl. brain heart infusion Basenpaar engl. bovine serum albumine Clostridium Cytosin Clostridium difficile-Transferase Clostridiale Glukosylierende Toxine Cysteinprotease-Domäne engl. combined repetitive oligopeptide Didesoxyadenosintriphosphat Didesoxycytonosintriphosphat Didesoxyguanosintriphosphat Desoxyribonukleinsäure Desoxyribonukleosidtriphosphate p-Xylen-bis(N-pyridiniumbromid) Dithiothreitol Desoxythymidintriphosphat engl. enhanced chemiluminescence Ethylendiamintetraessigsäure engl. epidermal growth factor engl. extracellular signal-regulated kinase et alii engl. fetal calf serum engl. fast performance liquid chromatography engl. forward Guanin engl. GTPase activating protein Glycerinaldehyd-3-phosphat-Dehydrogenase engl. guanine nucleotide dissociation inhibitors Guanosindiphosphat engl. guanine nucleotide exchange factor Glutathion-S-Transferase Glukosyltransferase-Domäne engl. growth factor receptor-bound protein-2 Guanosintriphosphat 2-(4-(2-Hydroxyethyl)-1-piperazinyl)-ethansulfonsäure 8-Hydroxypyren-1,3,6-trisulphonsäure Immunglobulin G/M Inositolhexakisphosphat Isopropythiogalaktosid c-Jun N-terminale Kinase Luria Bertani engl. liquid chromatography–mass spectrometry/mass spectrometry engl. Large unilamellar vesicle engl. multiple cloning site engl. matrix-assisted laser desorption/ionisation engl. multifunctional autoprocessing RTX-like monoklonal nicht-essentielle Aminosäuren engl. neurite growth factor optische Dichte Pellet protektives Antigen Poly (ADP-ribose) polymerase Poren-bildende Region engl. phosphate buffered saline engl. polymerase chain reaction Polyethylenglykol Phosphat isoelektrischer Punkt polyklonal Phenylmethanesulfonylfluorid Polyvinylidendifluorid Rezeptorbinde-Domäne Ras-Binde-Domäne engl. reverse engl. radioimmunoprecipitation assay 119 ABKÜRZUNGSVERZEICHNIS RNA SDS SDS-PAGE SOS TRITC TAE Tab. TOF TBS TBS-T TD TEMED TpeL Tm Tris Ü UDP UDP-GlcNAc UDP-Glc UV X-Gal z.B. Ribonukleinsäure engl. sodium dodecyl sulfate engl. sodium dodecyl sulfate polyacrylamide gel electrophoresis engl. son of sevenless Tetramethylrhodamin-Isothiocyanat Tris-Acetat-EDTA Tabelle engl. time of flight engl. tris buffered saline engl. tris buffered saline & tween 20 Translokations-Domäne N,N,N′,N′-Tetramethylethan-1,2-diamin engl. toxin clostridium perfringens large Schmelztemperatur von Primern Tris-(hydroxymethyl)-aminomethan Überstand Uridindiphosphat UDP-N-Acetylglukosamin UDP-Glukose Ultra-Violett 5-Bromo-4-chloro-3-indolyl-β-D-galaktosid zum Beispiel EINHEITEN m µ n p A B Ci °C cal Da F g h m M min m/v psi s V v/v milli (1000) mikro (1:1000000) nano (1:1000000000) pico (1:1000000000000) Ampere Becquerel Curie Grad Celsius Kalorie Dalton Farad Gramm Stunde Meter molar Minute Masse zu Volumen-Verhältnis engl. pounds per square inch Sekunde Volt Volumen zu Volumen-Verhältnis AMINOSÄURENCODE Alanin A Arginin R Asparagin N Asparaginsäure D Cystein C Glutamin Q Glutaminsäure E Glycin G Histidin H Isoleucin I Leucin L Lysin K Methionin M Phenylalanin F Prolin P Serin S Threonin T Tryptophan W Tyrosin Y Valin V 120 ANHANG I: ANHANG 1: verwendete Plasmide/Mutanten Im Laufe dieser Arbeit wurden folgende Konstrukte kloniert bzw. waren aus früheren Arbeiten schon vorhanden. 1.1: Konstrukte in pHis-1522 pHis1522-Toxin A (BsrGI/KpnI) pHis1522-Toxin B (BsrGI/KpnI) pHis1522-Letales Toxin (BsrGI/KpnI) pHis1522-α-Toxin (BsrGI/SpeI/KpnI) pHis1522-TpeL1-1651 (BsrGI/KpnI) pHis1522-TpeL1-1779 (BsrGI/KpnI) pHis1522-Toxin B C698A (BsrGI/KpnI) pHis1522-Letales Toxin C698A (BsrGI/KpnI) pHis1522-α-Toxin C707A (BsrGI/KpnI) pHis1522-Toxin A1-809 (BsrGI/KpnI) pHis1522-Toxin B1-807 (BsrGI/KpnI) pHis1522-Letales Toxin1-807 (BsrGI/KpnI) pHis1522-α-Toxin A1-813 (BsrGI/KpnI) pHis1522-TpeL1-805 (BsrGI/KpnI) pHis1522-Toxin A1-809 L542/S543A (BsrGI/KpnI) pHis1522-Toxin B1-807 L543/G544A (BsrGI/KpnI) pHis1522-Letales Toxin1-807 L543/G544A (BsrGI/KpnI) pHis1522-α-Toxin A1-813 L548/N549A (BsrGI/KpnI) pHis1522-Toxin A1-809 C700A (BsrGI/KpnI) pHis1522-Toxin B1-807 C698A (BsrGI/KpnI) pHis1522-Letales Toxin1-807 C698A (BsrGI/KpnI) pHis1522-α-Toxin A1-813 C707A (BsrGI/KpnI) pHis1522-TpeL1-805 C696A (BsrGI/KpnI) pHis1522-Toxin A543-809 (BsrGI/KpnI) pHis1522-Toxin B544-807 (BsrGI/KpnI) pHis1522-Letales Toxin544-807 (BsrGI/KpnI) pHis1522-α-Toxin A548-813 (BsrGI/KpnI) pHis1522-TpeL543-805 (BsrGI/KpnI) 1.2: Konstrukte in pGEX4T/pGEX2T pGEX-Rac1 (bereits vorhanden: Labor Prof. Aktories) pGEX-Rac2(bereits vorhanden: Labor Prof. Aktories) pGEX-Rac3 (bereits vorhanden: Labor Prof. Aktories) pGEX-Cdc42 (bereits vorhanden: Labor Prof. Aktories) pGEX-RhoA (bereits vorhanden: Labor Prof. Aktories) pGEX-RhoB (bereits vorhanden: Labor Prof. Aktories) pGEX-RhoC (bereits vorhanden: Labor Prof. Aktories) pGEX-RhoD (bereits vorhanden: Labor Prof. Aktories) pGEX-RhoE (bereits vorhanden: Labor Prof. Aktories) 121 ANHANG pGEX-Rnd1 (bereits vorhanden: Labor Prof. Aktories) pGEX-Rnd2 (bereits vorhanden: Labor Prof. Aktories) pGEX-Ha-Ras (bereits vorhanden: Labor Prof. Aktories) pGEX-K-Ras(bereits vorhanden: Labor Prof. Aktories) pGEX-R-Ras (bereits vorhanden: Labor Prof. Aktories) pGEX-N-Ras (BamHI/XhoI) pGEX-RalA (bereits vorhanden: Labor Prof. Aktories) pGEX-RalB (bereits vorhanden: Labor Prof. Aktories) pGEX-Rap1B(bereits vorhanden: Labor Prof. Aktories) pGEX-Rap2A (bereits vorhanden: Labor Prof. Aktories) pGEX-Rheb (bereits vorhanden: Labor Prof. Aktories) pGEX-Raf1B-RBD (bereits vorhanden: Labor Prof. Aktories) pGEX-Letales Toxin1-546 (bereits vorhanden: Labor Prof. Aktories) pGEX-Toxin B1-546 W520A (bereits vorhanden: Labor Prof. Aktories) 1.3: Konstrukte in pET28a pET28a-TpeL1-542 (BamHI/XhoI) oder (NdeI/XhoI) pET28a-TpeL1-542 A383I (BamHI/XhoI) 2: Primer 2.1: Klonierungs-Primer Toxin A: BsrGI (1/ATG) fw 5´-GCGCTGTACAATGTCTTTAATATCTAAAGAAG-3´ BsrGI (1627/ATG) fw 5´-GCGCTGTACAATGTCTGAAGACAATGGGGTAG-3´ KpnI (+1) (2427) rev 5´-ATATATGGTACCCTAATGTTTTTATATCTTCTGA-3´ KpnI (+1) (8130) rev 5´-ATATATGGTACCCGCCATATATCCCAGGGGCTTT-3´ Toxin B: BsrGI (1/ATG) fw 5´-GCGCTGTACAATGAGTTTAGTTAATAGAAAAC-3´ BsrGI (1630/ATG) fw 5´-GCGCTGTACAATGGGTGAAGATGATAATCTTG-3´ BsrGI (3385/ATG) fw 5´-GCGCTGTACAATGTTAGTTGAAACTGAAGGAG-3´ KpnI (+1) (1638) rev 5´-ATATATGGTACCCATCTTCACCAAGAGAACCTTC-3´ KpnI (+1) (2421) rev 5´-ATATATGGTACCCATTATTTCTAATTTCTTGTAA-3´ KpnI (+1) (7098) rev 5´-ATATATGGTACCCTCACTAATCACTAATTGAGC-3´ Letales Toxin: BsrGI (1/ATG) fw 5´-GCGCTGTACAATGAACTTAGTTAACAAAGCCC-3´ 122 ANHANG BsrGI (1630/ATG) fw 5´-GCGCTGTACAATGGGAGAAGATGATAATCTTG-3´ KpnI (+1) (2421) rev 5´-ATATATGGTACCCATTATTCCTAATTTCTTGTAA-3´ KpnI (+1) (7092) rev 5´-ATATATGGTACCCTTCACTAACTACTAATTCAGC-3´ α-Toxin: Acc65I (1/ATG) fw 5´-GCGCGGTACCATGCTTATAACAAGAGAACAAT-3´ BsrGI (1/ATG) fw 5´-GCGCTGTACAATGCTTATAACAAGAGAACAAT-3´ BsrGI (1645/ATG) fw 5´-GCGCTGTACAATGAATTATGAAGATGGTTTAA-3´ SpeI (2961) fw 5´-GAATACTAGTTTAAAAGTACAAGC-3´ SpeI (2974) ev 5´-TTAAACTAGTATTCATAGTCA-3´ KpnI (+1) (2439) rev 5´-ATATATGGTACCCAGAATTTATATCTTTTGCAAA-3´ KpnI (+1) (6534) rev 5´-GCGCGGTACCCTACCAGGGTCAACTGTTCTCCTTTTTC-3´ TpeL: BsrGI (1/ATG) fw 5´-GCGCTGTACAATG GGGTTAATGTCAAAAGAACAAT-3´ BsrGI (1627/ATG) fw 5´-GCGCTGTACAATGGAAATGATAAACTAAATTTTA-3´ KpnI (+1) (2415) rev 5´-ATATATGGTACCCTTTTTTATCTAGACTATTTCTAAAAG-3´ KpnI (+1) (4953) rev 5´-ATATATGGTACCCCAATTAAATTATTTGAACTTTCACTTTCACCTGT-3´ KpnI (+1) (5337) rev 5´-GCGCGGTACCCATCTCTATATCCTATAATTGGTTCCATTTCAAAAATA-3´ BamHI (1/ATG) fw 5´-GCGCGGATCCATG GGGTTAATGTCAAAAGAACAAT-3´ XhoI (1626/STOP) rev 5´-ATATATCTCGAGTTATGATTTATTAGTATATTTTTCT-3´ N-Ras: BamHI (1/ATG) fw 5´-GCGCGGATCCATGACTGAGTACAAACTGGTGGTGGTT-3´ XhoI (513/STOP) rev 5´-ATATATCTCGAGTTAGAGTTTTTTCATTCGGTACTG-3´ 123 ANHANG 1.2: Mutagenese-Primer Mit Hilfe der Software Sitefind wurden zusätzliche stumme Mutationen eingefügt, wodurch neue Restriktionsschnittstellen entstehen. Anhand dieser Schnittstellen kann eine erfolgreiche Mutagenese nachgewiesen werden. Die verwandten Schnittstellen sind in Klammern angeben Toxin A: L542A/S543A fw (BamHI) 5´-CCATTGTCTTCAGCAGCGGCGGATCCACCAGTA-3´ L542A/S543A rev (BamHI) 5´-TACTGGTGGATCCGCTGCTGAAGACAATGG-3´ C700A fw (EheI) 5´-GTAGAAAACTTACTTGGCGCCAATATGTTTAGTTATGATTTTAATGTT-3´ C700A rev (EheI) 5´-AACATTAAAATCATAACTAAACATATTGGCGCCAAGTAAGTTTACTTCTAC-3´ Toxin B: L543A/G544A fw (PstI) 5´-TTGAAGGTTCTGCTGCAGAAGATGATAATC-3´ L543A/G544A rev (PstI) 5´-GATTATCATCTTCTGCAGCAGAACCTTCAA-3´ C698A fw (EheI) 5´-AATAAATTTATTAGGCGCCAATATGTTTAGCTAC-3´ C698A rev (EheI) 5´-GTAGCTAAACATATTGGCGCCTAATAAATTTATT-3´ D286A/D288A fw (EcoRI) 5´-GGTGGTATGTATTTAGCTGTTGCTATGTTACCAGGAATCCAACCAG-3´ D286A/D288A rev (EcoRI) 5´-CTGGGTGGATTCCTGGTAACATAGCAACAGCTAAATACATACCACC-3´ Letales Toxin: L543A/G544A fw (PstI) 5´-TTTGAAGGTGCAGCTGCAGAAGATGATAAT-3´ L543A/G544A rev (PstI) 5´-ATTATCATCTTCTGCAGCTGCACCTTCAAA-3´ C698A fw (EheI) 5´-ATTAAATTTACTGGGCGCCAACATGTTCAGCTAC-3´ C698A rev (EheI) 5´-GTAGCTGAACATGTTGGCGCCCAGTAAATTTATT-3´ α-Toxin: L548A/N549A fw (PstI) 5´-TAGGAAGAACTGCAGCTTATGAAGATGGTT-3´ L548A/N549A rev (PstI) 5´-AACCATCTTCATAAGCTGCAGTTCTTCCTA-3´ C707Afw (EheI) 5´-GAATAAATATTTAAAAATAAATATACTTGGCGCCTATATGTTTACTCCAAAGGTAGATATT-3´ C707A rev (EheI) 5´-AATATCTACCTTTGGAGTAAACATATAGGCGCCAAGTATATTTATTTTTAAATATTTATTC-3´ 124 ANHANG TpeL: C696A fw (EheI) 5´-ACATTTATATTATATGAAAACATATTGGCGCCTAACAAATCTATTTGAATTTCT-3´ C696A rev (EheI) 5´-AGAAATTCAAATAGATTTGTTAGGCGCCAATATGTTTTCATATAATATAAATGT-3´ A383I fw 5´-GCTTTTATGTTTGGGAAAATTATTAATCAAGTATTAATATCTAAAAAAAAT-3´ A383I rev 5´-ATTTTTTTTAGATATTAATACTTGATTAATAATTTTCCCAAACATAAAAGC-3´ 1.3: Sequenzier-Primer pHis1522 fw 5´-GGATTAACAAACTAACTC-3´ pHis1522 rev 5´-CTGCCAAGGGTTGGTTTGC-3´ Toxin A: 671 fw 5´-ACCTGTATTAGAATCATATA-3´ 1432 fw 5´-GTGGTCCAGGAGCTTATGCG-3´ 2091 fw 5´-AAAAATGTAGAAGTAAACTT-3´ 2791 fw 5´-GCGAACATATTACAAAAGAA-3´ 3491 fw 5´-AGATTTTAATAATAATTCGA-3´ 4191 fw 5´-GATTTTTCAGGCGATATAGA-3´ 4921 fw 5´-GTGAATGGAAAACATCGTCA-3´ 5623 fw 5´-CTGGAGCAGCTTTAACTAGT-3´ 6350 fw 5´-CTTTAATACTAACACTGCTG-3´ 7061 fw 5´-TGGATGGCAAACTATTGATG-3´ Toxin B: 784 fw 5´-AGGTGGAATTTAGCTGCTGC-3´ 1560 fw 5´-GTCATTTGACGATGCAAGAG-3´ 2115 fw 5´-CAACGTAGAGGAGACTTATC-3´ 267 1fw 5´-GAGACTGATGAGGGATTTAG-3´ 3493 fw 5´-GGTAAATGTGAAATCTGG-3´ 125 ANHANG 3952 fw 5´-GGTTCAGGAGGAACTTATGC-3´ 4501fw 5´-GATAGTAAGCCTTCATTTGG-3´ 4948 fw 5´-GGAAATAGACAAAATATGAT-3´ 5414 fw 5´-GGATGGATTGATTGGCTATG-3´ 5875 fw 5´-GCCCAGAAACAGGTAAAGCT-3´ 6270 fw 5´-GATGAAGATACAGCAGAAGC-3´ 6740 fw 5´-AGAAGGGCATAATGAGAACG-3´ Letales Toxin: 671 fw 5´-CCTAGAAGCCCTTAATAAGT-3´ 1391 fw 5´-ATTTGCTCCAGATGTTAGAA-3´ 2091 fw 5´-ATAGAAATAAATTTACTGGG-3´ 2791 fw 5´-ATATATCTAAAGAAATTTCT-3´ 3529 fw 5´-GGAGAGCTGAAGGTGGTTCA-3´ 4197 fw 5´-GGAGATATAAATGAAAGCAA-3´ 4951 fw 5´-CTAGCTATACCTTATATGTA-3´ 5622 fw 5´-GACCCAACGAAGAGTGGAGC-3´ 6339 fw 5´-GATTGTAAGTATTATTTTGA-3´ α-Toxin: 697 fw 5´-GCAATTGGAGCTACAGATAT-3´ 1397 fw 5´-CTGAAGTCAATTCAACTGTT-3´ 2127 fw 5´-GTTTACTCCAAAGGTAGATA-3´ 2869 w 5´-GAGATAGAACAAAATCCATC-3´ 3577 fw 5´-GATCACTATTTTAGTGCACC-3´ 4371 fw 5´-AGGATTAGGAAGTATTTCAG-3´ 5071 fw 5´-CCTATACCAAATAATCCACA-3´ 5771 fw 5´-ATGGAAATACAGGCGAAGCT-3´ 126 ANHANG TpeL: 581 fw 5´-CAAATGATAACTATACTAAATCTTTAAATG-3´ 1178 fw 5´-CATACTCATTAAATTTAATAATTAATC-3´ 1807 fw 5´-CCTTCAAATTCAATACTAGTTCAAG-3´ 2455 fw 5´-GTAAATGAAGTATATGAACAATTAAC-3´ 3052 fw 5´-CCATTAATAACAACAATAGTTGATGG-3´ 3613 fw 5´-GGTATGATGCTACCTAATGGCG-3´ 4301 fw 5´-GCTCTTCAAATATAAAATCAATACC-3´ 127 ERKLÄRUNG J: ERKLÄRUNG „Ich erkläre hiermit, dass ich die vorliegende Arbeit ohne unzulässige Hilfe Dritter und ohne Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe. Die aus anderen Quellen direkt oder indirekt übernommenen Daten und Konzepte sind unter Angabe der Quelle gekennzeichnet. Insbesondere habe ich hierfür nicht die entgeltliche Hilfe von Vermittlungs- bzw. Beratungsdiensten (Promotionsberater/Promotionsberaterin oder anderer Personen) in Anspruch genommen. Niemand hat von mir unmittelbar oder mittelbar geldwerte Leistungen für Arbeiten erhalten, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen. Die Arbeit wurde bisher weder im In- noch im Ausland in gleicher oder ähnlicher Form einer anderen Prüfungsbehörde vorgelegt.“ „Die Bestimmungen der Promotionsordnung der Fakultät für Chemie, Pharmazie und Geowissenschaften sind mir bekannt. Insbesondere weiß ich, dass ich vor Aushändigung der Promotionsurkunde zur Führung des Doktortitels nicht berechtigt bin.” 128 DANKSAGUNGEN K: DANKSAGUNGEN Zunächst danke ich Prof. Dr. Dr. Aktories für die Bereitstellung der interessanten Promotionsthematik, die engagierte Betreuung und die exzellenten Arbeitsbedingungen am Institut für Experimentelle und Klinische Pharmakologie und Toxikologie. Mein weiterer Dank gilt Prof. Dr. Manfred Jung für die Übernahme der Erstbegutachtung und Betreuung meiner Arbeit von Seiten der Fakultät Chemie, Pharmazie und Geowissenschaften. Prof. Dr. Regine Süss danke ich für ihre Tätigkeit als Drittprüferin. Dr. Panagiotis Papatheodorou danke ich für seine vielfältige und intensive Betreuung, seine ständige Ansprechbarkeit,.seine hilfreichen Ratschläge und seine Fähigkeit, mich stets zu motivieren und für die Wissenschaft zu begeistern. Sein Engagement und Interesse an dieser Arbeit waren mitentscheidend für ihr Gelingen. Dr. Wei Lü danke ich für seine umfangreichen und praktischen Ratschläge sowie seine Hilfe bei der Durchführung von ITC-Messungen. Dr. Andreas Schlosser und Ulrike Lanner danke ich für ihre Durchführung von LC-MS/MSMessungen. Sven Hornei und Otilia Wunderlich danke ich für ihre zuverlässige und treue Hilfe bei der Durchführung zahlreicher Experimente im Labor. Allen Mitarbeitern des Instituts für Experimentelle und Klinische Pharmakologie und Toxikologie danke ich für ihre große Hilfsbereitschaft und die durch sie geprägte Arbeitsatmosphäre, die ich stets als kollegial und wertschätzend wahrgenommen und empfunden habe. Selda, Tina, Thomas, Peter, Jan, Julia, Sven, Otilia und Dimi danke ich für ihr freundschaftliches Verhalten im Labor- und Büroalltag. Durch sie war es möglich, Rückschläge zuweilen nicht als Rückschritt anzusehen sondern auch in ihnen positive Aspekte zu erkennen. Ich danke meinen Freunden, die mir in der Weise geholfen haben, dass ich durch sie die Welt auch in anderen Perspektiven sehen konnte. Nicht zuletzt danke ich meiner Familie, die mein Studium und die Durchführung dieser Arbeit in vielfältiger Weise unterstützte und ermöglichte. 129