Frage:
Werbung

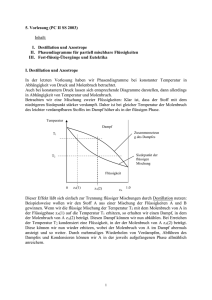
2. Thermodynamik der Mischphasen und Stoffgemischen 2.1 Phasenumwandlungen und chemisches Potential Treibende Kraft für den Übergang eines Stoffes von einer Phase in die andere (flüssig Dampf, fest flüssig, fest Dampf, fest fest’, usw.) ist die freie Enthalpie G = G (p, T, n1, n2 ,… ) die u.a. von der Zahl der Mole ni in Phase i abhängt. (Wenn nur wenige Teilchen in Phase i sind, ist die treibende Kraft dorthin besonders groß: Entropie-Beitrag.) Gibbsche Fundamentalgleichung k G G G dG dT dp p,n n T,p,n ji dni p T, n T i 1 i G H TS dG U pV dU pdV Vdp TdS SdT δQrev pdV dS δQ rev T dG = TdS – pdV + pdV + Vdp – TdS – SdT dG = Vdp – SdT G p = -S T G T = V p G Definition: Chemisches Potential i n i T , p ,n n j 1 i Temperaturabhängigkeit der chemischen Potentiale: i T p ,n j ni G ni T , p ,n j ni T G 2G 2 G T p Tni ni T ni p n j ni , p ,T S s i si = partielle molare Entropie n i S korreliert mit der Unordnung (S = kln(Ω)) Mit S(fest)<S(flüssig)<S(gasförmig) gilt dies auch für die Steigungen von µ der entsprechenden Phasen in Abhängigkeit von T. Folglich folgt qualitativ für µ(T): Aus obigem Graphen wird klar, dass im Phasengleichgewicht die Chemischen Potentiale gleich sein müssen also bei TSm: µfest = µflüssig und bei Ts: µflüssig = µgasf. 2 Druckabhängigkeit der chemischen Potentiale: i p Ti ,n j ni G ni T , p ,n j ni p G p T n i 2G 2G pni ni p T V vi n i T , p , n j ni partielles molares Volumen n j n i ,p,T Damit ist vorhersehbar wie sich eine Druckveränderung Schmelztemperatur oder die Siedetemperatur auswirkt. auf die Umgeformt gilt: Δµ = vmΔp Beispiel Gefrierpunktsveränderung bei Druckänderung (siehe Bild unten): Fall 1) alle Stoffe außer Wasser (Bild unten links): vm,fest <vm,flüssig also Δµfest < Δµflüssig und Gefriertemperaturerhöhung Fall 2) Wasser (Bild unten rechts): vm,fest > vm, flüssig also Δµfest > Δµflüssig und Gefriertemperaturerniedrigung 3 Beispiel Siedepunktsveränderung bei Druckänderung Allgemein gilt: vm,flüssig <vm,gasf. also Δµflüssig < Δµgasf. und Siedetemperaturerhöhung (Skizze analog Gefrierpunktserhöhung) i p Weiteres Beispiel für die Anwendung von 1. vm : Ti ,n j ni Ideales Gas p d p p2 2 1 id p2 p 2 dp vdp RT RT d ln p RT ln p1 ln p 2 p p1 p1 p1 p2 p1 RT ln p2 p1 mit p1 = p° (Standarddruck 1 bar) id RT ln p p vgl. hierzu auch die Ableitung der Nernstschen Gleichung! Man kann das chemische Potential bei Druck p aus µ° bei Standarddruck berechnen 2. Reales Gas Virialansatz p V = nRT + n B p (+ n C p2 + …) steigende Potenzen von p Für kleine Drücke ist der Abbruch nach dem 2. Glied möglich Somit: v = RT/p + B mit B als 2. Virialkoeffizienten Damit nun wie oben: p p 2 p2 p2 2 RT real d p B dp RT d ln p B dp p p1 p1 p1 1 v 4 real real p 2 p1 RT ln p2 B p 2 p1 p1 mit p1 = p° (Standarddruck 1 bar: hier noch Verhalten wie ideales Gas B p° = 0) p B p (B=0 für ideales Gas, siehe oben) p real RT ln Einfacher wären Rechnungen mit einer Gleichung, die formal der des idealen Gases entspricht: real RT ln fg p fg = p p = Druck nach idealem Gasgesetz fg = gemessener Druck ~ Fugazität = Fugazitätskoeffizient = f(p) Für kleine Drücke gilt: Wenn man fg kennt, kann man das chemische Potential des realen Gases bestimmen aus dem chemischen Potential des idealen Gases. Durch Vergleich der beiden Gleichungen R T ln p p RT ln B p p p RT ln B p ln B p R T Diese Beziehung gilt nur für kleine Drucke von wenigen bar, für die der Abbruch der Reihenentwicklung des Druckes (Virialansatz) nach dem 2. Glied gültig ist. 5 Beispiel: Werte von f für N2 p/bar ideales Gas 50 100 200 400 600 800 1000 0,98 0,97 0,97 1,06 1,22 1,47 1,81 Druck nach Gesetz des idealen Gases p = nRT/V berechnet aus gegebenem n,V,T fg/bar 49 97 194 424 732 1176 1810 gemessener Druck (nRT/V + Binnendruck) p klein ~ Anziehungskräfte). p groß ~ Abstoßung Für die hohen Drucke in dieser Tabelle hängt B von p ab. Hier müssten noch höhere Terme im Virialansatz berücksichtigt werden. erhält man durch graphische Integration aus den bei verschiedenen äußeren Drucken im Zylinderkolben gemessenen Volumina, siehe Übung. 6 2.2 Das chemische Potential einer Mischphase (quantitativ) A) Ideale Mischung in Gas- und kondensierter Phase: „Ideal“: In der Gasphase keine Wechselwirkung der Moleküle (ideales Gas/Gasmischung). In kondensierter Phase immer Wechselwirkung i i , j j und i j. In idealen Mischung ist die Wechselwirkungsenergie i j ungefähr gleich dem arithmetischen Mittel der i i , j j Wechselwirkungsenergien. Das chemische Potential der Komponente i in der Mischphase µi(p,T) ist gleich dem chemischen Potential der gleichen Komponente in der reinen Phase µi*(pi,T) bei dem Partialdruck pi, den die Komponente i in der Mischphase hat. µi(p,T) = *i (pi,T) im Gleichgewicht: gleiches chem. Potential + gleiche Partialdrücke µi = Chem. Potential der Mischphase *i = Chem. Potential der reinen Phase pi = Partialdruck von i in Mischphase Beispiel: Das chemische Potential von Substanz 1 in einer Mischung von 5 mbar Substanz 1 und 5 mbar Substanz 2 ist gleich dem chemischen Potential von 5 mbar der Substanz 1 alleine. μ i p, T μ i * p, T RTln pi p xi: Molenbruch der Substanz i xi µi(p,T) < µi*(p,T) dG < 0 und damit erfolgt spontane Mischung. Da xi druckunabhängig ist, gilt * vi i i vi * p T,x p T,x ähnlich Entropie, Wärmekapazität In einer idealen Mischung ändern sich Volumen, Enthalpie und Wärmekapazität nicht (relativ zur Summe der Einzelgrößen vor der Mischung): V=nivi*, H=nihi*, Cp=nicpi* usw. Die Entropie ändert sich jedoch bei einer idealen Mischung! 7 Beweis: μ μ * * si i i Rlnx i si Rlnx i T p,x T p,x 0 Also ist si > si*: die Entropie der idealen Mischphase ist größer als die Entropie der reinen Phase (Entropiezunahme ist treibende Kraft für die Vermischung). Mittlere Mischungsentropie S x i R ln x i x i R ln x i i i (-R ln xi ) = Entropiebeitrag der Komponente i in der Mischung xi = prozentualer Anteil des Beitrages (Molenbruch) B) Reale Mischungen In realen Mischungen (Gas oder kondensiert!) gilt: μ ireal p, T μ i * p, T RTln ai ai = Aktivität f i x i fi = Aktivitätskoeffizient (vgl. fg bei reinen realen Gasen) Hier ist also das chem. Potential der Substanz i von dem mit Faktor f i korrigierten Molenbruch xi abhängig. V=nivi*, H=nihi*, Cp=nicpi* usw. gilt in realen Mischungen nicht mehr! Zum Beispiel addieren sich beim Lösen von NaCl in Wasser die Volumina von Salz und Lösungsmittel nicht. Das Gesamtvolumen ist kleiner , da das in der Hydrathülle der Ionen befindliche Wasser viel dichter gepackt ist als im reinen Wasser. Beim Mischen zweier Substanzen kann Wärme freigesetzt oder muss aufgewendet werden: Mischungsenthalpie H = H - nihi* = nihi - nihi* . 8 2.3 Phasengleichgewichte P Phasen: Eine Phase zeigt keine sprunghafte Änderung irgendeiner physikalischen Größe (nur an der Phasengrenze). K Komponenten: Zusammensetzung Bestandteilen einer Phase mit K chemischen F Freiheitsgrade: Gibt die Dimension an, in der man sich bewegen kann, ohne dass eine Phase verschwindet. Gesamtzahl der Variablen, die den Zustand einer Phase eindeutig festlegen. K p, T, x1...x K : Bei K-Komponenten also K+2-Variable, aber xi 1, i 1 2 1 unabhängige Variable zur also K K 1 Charakterisierung einer Phase. Analyse: Gesamtzahl der Gleichgewichtsbedingungen T = T = T… p = p = p… P - 1 Gleichungen P - 1 Geichungen µi = µi = µi... P – 1 Gleichungen i=1…K ≙ Phase K + 2 Gleichungsbeziehungen mit jeweils P - 1 Gleichungen. Zahl der Freiheitsgrade = Gesamtzahl der Variablen – Gesamtzahl der Gleichgewichtsbedingungen F = (K + 1) P – (K + 2) (P – 1) F = KP + P – KP + K – 2P + 2 F = K – P + 2 Gibbsche Phasenregel F = 0 invariant (bei Variation nur eines Parameters verschwinden die Phasen) F = 1 univariant F = 2 divariant 9 2.3.1 Phasengleichgewichte in Einkomponentensystemen Beispiel 1: Dampf, ideales Gas K = 1, P = 1 F = K – P + 2 = 2 Von den Variablen p, T, v können 2 freigewählt werden (entspricht einer Fläche). Beispiel 2: Gleichgewicht Gas - Flüssigkeit K = 1, P = 2 daraus F = 1 Wahl von T legt p,v fest, usw. (entspricht einer Linie, siehe Phasentrennlinie im Diagramm Beispiel 3: Tripelpunkt Feststoff/Flüssigkeit/Dampf K = 1, P = 3 daraus F = 0 Dies entspricht einem Punkt, nur bei ein Wert von Temperatur, Druck, Volumen möglich p kritischer Punkt fest flüssig Dampf Tripelpunkt T Bemerkung: Die obige fest/flüssig-Gleichgewichtskurve gilt für Stoffe, die sich beim Schmelzen ausdehnen (nicht Wasser!): „Höherer Druck erschwert normalerweise das Schmelzen“. An den Phasengrenzen stehen die Phasen im Gleichgewicht (siehe oben): µflüssig = µfest und µflüssig=µgasförmig Natürlich ändern sich z.B. bei Temperaturänderung längs der Phasengrenzlinie die chemischen Potentiale in gleicher Weise) 10 Die Gleichungen von Clapeyron und von Clausius-Clapeyron Es gilt allgemein für Phase α und β T dT P p dp T T dT p P dp T -sdT + vdp = -sdT + vdp (s - s) dT = (v - v) dp dp s s a s dT v v a v Für eine reversible Schmelz-, Verdampfungs- oder Sublimationsvorgang: S Qrev H T T H p T Koex. Tv bezogen auf 1 mol Substanz Clapeyronsche Gleichung gibt die Steigung der Phasengrenzlinien an Beispiele: Feststoff/Dampf (Sublimation) } v positiv und groß → Steigung flach Flüssigk./Dampf (Verdampfung)} v positiv und groß → Steigung flach p p siehe Steigungen T Subl. T Verdampfun g der Gleichgewichtskurven nahe Tripelpunkt, also HSubl.>HVerd.; HSchmelze+HVerd.=HSubl. Feststoff/Flüssigk. (Schmelze) v hier klein ist → Steigung steil . Siehe Phasendiagramme unten: v negativ für Wasser (rechts) und positiv für alle anderen Stoffe (links) 11 Clausius Clapeyron: Spezialisierung für Koexistenz Flüssigk./Dampf und „ideales Gas“ v vgas RT p Näherung vflüssig << vgas und bei niedrigem Dampfdruck ist Gas ideal H p p T Koex. RT 2 lnp Gleichung nach Clausius Clapeyron ΔH ΔH T lnp s const. (Integration von Ps,T1 nach Ps,T2 2 RT RT ps = Gleichgewichtsdruck („Sättigungs“-Dampfdruck) ln ps,T2 ps,T1 ΔH verd. 1 1 R T1 T2 Augustsche Dampfdruckformel Hverd. = Verdampfungsenthalpie ps,T1 /H bekannt ps,T2 ps,T1 /p s,T2 bekannt ΔH verd. 12 T K 373 353 333 313 293 3,0 3,2 3,4 273 0 ln ps bar -1 -2 -3 -4 -5 2,6 2,8 3,6 -1 T 10-3 K-1 Die logarithmische Auftragung des Sättigungsdampfdrucks von Wasser in Abhängigkeit von 1/T zeigt, dass die Augustsche Dampfdruckformel gut erfüllt ist. 13 2.3.2 Phasengleichgewichte in Mehrkomponentensystemen Mischphase ⇋ reine Phase z.B. Salz/Wasser Wasserdampf Definition: Lös.: Lösungsmittel (besitzt Dampfdruck) Gelös: Gelöstes (kein Dampfdruck) Lös in Misch: Lösungsmittel in der Mischung Dampfdruckerniedrigung Der Dampfdruck eines Lösungsmittels (Lös) erniedrigt sich, wenn man einen Stoff im Lösungsmittel löst. Beispiel für das Lösungsmittel: Wasser Lös: = Wasser Das Chemische Potential in Lösung und Dampf sind gleich: µWasser in Misch = µDampf = µWasserdampf* Die Gasphase ist reine Phase (nur Wasser) 14 µWasser in Misch (p,T) = µWasser* (p,T) + R T ln aWasser in Misch µWasser* = chem. Potential des reinen, flüssigen Wassers aWasser in Misch = Aktivität mit aWasser in Misch = fWasser in Misch xWasser mit xwasser als Molenbruch des Lösungsmittels in der Lösung xW asser nW asser nj j Beachte: der Ausdruck RTln(aWasser in Misch) ist negativ Nun mit obigem: = µWasser in Misch (p,T) = µWasser* (p,T) + R T ln aWasser in Misch Da µWasser in Misch < µWasser* ist, ist der Dampfdruck über dem reinen Lösungsmittel größer als über Lösung (Grösseres Chem. Potential => Höherer Dampfdruck). µWasser* (p,T) + R T ln aWasser in Misch = µWasserdampf (p,T) Ableitung nach p W asserdampf * W asser* dp p dp RTd ln aW asserinMisch (T = constant) p T T * v1* v1 i p Mit v m folgt: vW asserdampf * v wasser * dp RTd ln aW asserinMisch Ti ,n j ni p ln aW asserinMisch RT T vW asserdampf * vW asser * Mit v Wasserdampf * v Wasser * und der Annahme ideales Gas (vDampf*=RT/p) p ln aW asserinMIsch p T p: Druch des Wasserdampfs d ln p = d ln aWasser in Misch. Zunahme des Wasserdampfdrucks bei Zunahme des Lösungsmittelanteils Druckänderung: Lösung reines Lösungsmittel Mit p1*, a1 =1 (reines Lösungsmittel) und p1, a1 (Lösung) folgt 15 pW asserüberMischug aW asserinMisch pW asserdampf * aW asser1 d ln p ln d ln a W asserinMisch pW asserdampfüberMisch pW asserdampf pW asserdampfüberMisch pW asserdampf * * ln a wasserinMisch aW asserinMisch f W asserinMisch xW asser Durch Messung der Erniedrigung des Dampfdruckes bei Zusatz des zu Lösenden zum reinen Lösungsmittel kann der Aktivitätskoeffizient fwasser des Wassers als Lösungsmittels bestimmt werden! In verdünnter Lösung ist fwasser ≃ 1 und pW asserdampfüberMisch pW asserdampf * xW asser 1 xGelös pW asserdampf * pW asserdampfüberMisch pW asserdampf * xGelöstes Obiges ist allgemein wenn am Wasser durch Lösungsmittel ersetztRaoult (1890): Die relative Erniedrigung des Lösungsmitteldampfdruckes ist gleich dem Molenbruch des gelösten Stoffes. (nicht von der Natur des gelösten Stoffes abhängig, nur von der Teilchenzahl: kolligative Eigenschaft. Bei Salzen in Lösung tragen Kationen und Anionen bei.) 16 Dampfdruck einer Glukoselösung in Wasser: 6,0 p (H20) mbar Ra ou ltsc he ess Ge un rad g e M 5,0 0,14 0 x2 Abweichung der Raoultschen Geraden von der Messkurve, da Raoultsches Gesetz nur für verdünnte Lösungen gilt. Bestimmung des Molenbruches des gelösten Stoffes Dampfdruckerniedrigung aus p 1,013 bar Tm TS* Tm* TS Phasendiagramm des Lösungsmittels; Phasendiagramm der Lösung 17 T Dampfdruckerniedrigung Der Dampfdruck von 1,013 bar wird erst bei höheren Temperaturen erreicht, also Siedepunktserhöhung Ts > Ts* TS TS* RTS*2 ΔvH x2 Siedepunktserhöhung Verdampfun gsenthalpie Formel wird abgeleitet aus chem. Potent. analog wie Clausius-Clapeyron Gleichung Gefrierpunktserniedrigung Tm < Tm* Tsm Tsm * RTsm *2 Δ sm H Gefrierpunktserniedrigung x2 Schmelzenthalpie „Salz im Winter streuen“ x2 n2 n2 M1n 2 n1 n 2 m1 n 1kg M1n 2 2 M1 1kg Lösungsmit el 1 1kg Lösungsmitel 1 m1 (Masse des Lösungsmittels) = 1 kg M1 (Molmasse des Lösungsmittels) m2 (Molalität: Molzahl des Gelösten/kg Lösungsmittel) x2 M1m 2 1 M1m 2 Bei hoher Verdünnung M1m2 << 1 folgt x2 M1m2 Ebulliosko pische Konstante RTS*2 M1 * TS TS m2 ΔvH 18 Tm Tm * RTm*2 M1 m2 ΔmH Kryoskopische Konstante Stoff H2O H2SO4 J2 Benzol Ebullioskopische Konstante kg mol-1 K 0,521 5,330 10,500 2,540 Kryoskopische Konstante kg mol-1 K 1,858 6,120 20,400 5,065 Osmotischer Druck (Lösungsmittel: Wasser) osmotischer Druck Lösungsmittel Lösung semipermeable Wand (nurfürfür Lösung, nicht nichtfür (nur Lösungsmittel, Gelöstes durchlässig) Gelöstes durchlässig) Das chemische Potential des Lösungsmittels ist in reiner Phase größer (positiver) als in Lösung: Diffusion von Lösungsmittel durch semipermeable Wand in die Lösung. Diffusion, bis der osmotische (hydrostatische) Druck π der Flüssigkeitssäule das chemische Potential der Lösung soweit erhöht, dass das chemische Potential des Lösungsmittels erreicht wird. Indizes: 1: Lösungsmittel, 2: gelöster Stoff µlinks = µrechts μ 1,flüss p, a 1 1 μ Gemisch,flüssig p π, a 1 reine Phase Lösung 19 Dann 1, Flüssigkeit 1, Dampf Und 1, Dampf 1, Dampf * (Dampfphase ist reine Phase) μ1,Dampf * p μ 1,flüssig * p π RTln a 1in Lös p π auch die Aktivität ist druckabhängig! (siehe Fugazitätskoeffizien t von N 2 ) W asserdampf * p W asser, fl * p p p wasser, fl p p * ln aW asserimGem dp RT ln aW asserimGem p RT p T p dp T Mit W asserdampf * p wasser, fl * p folgt RT ln aW asserimGem p vonrechts p p p W asser, fl * dp RT ln aW asserimGem dp p p p T T ln aWasserimGem WasserimGem Wasser , * (siehe oben) RT Mit p p p T T T folgt RT ln aW asserimGem p p p p p W asser, fl * wasser, fl * dp dp W asserimGem dp p p p T T T p p vwasserimGem p RT ln aW asserimGem p vW asserimGem dp vW asser, fl p v wasserimGem ≙ partielles molares Volumen des Lösungsmittels in der Lösung (wenig druckabhängig wegen geringer Kompressibilität von Lösungen) 20 Für eine ideal verdünnte Lösung ist aWasserimGem = xWasser = 1 – xGelöstes Mit ln(1 – xGelöstes) ≃ - xGelöstes RT x Gelöstes v WasserimGem π id vWasser,fl 1 VWasser,fl partielles molares Volumen des reinen Lösungsmittels id RT v m,W asser, fl xGelöstes Der osmotische Druck einer ideal verdünnten Lösung ist (wie die Dampfdruckerniedrigung, Siedepunktserhöhung und Gefrierpunktserniedrigung) dem Molenbruch des Gelösten proportional (nicht von Stoffart abhängig: kolligative Eigenschaft). Index 1: Wasser Index 2: gelöster Stoff id v1 n2 n RT 2 RT n1 n 2 n1 n1v1 V id V n2 RT Formale Analogie zum idealen Gasgesetz Nach van’t Hoff gilt: id c2 RT 1 m2 R T c2 = Molarität des Gelösten [mol/m3] 1 = Dichte des Lösungsmittels [kg/m3] m2 = Molalität des Gelösten [mol/kg] Experimentell: =gh = Dichte des Lösungsmittels [kg/m3] g = Erdbeschleunigung 9,81 [m/s2] h = Höhe der hydrostatischen Säule [m] Druck [N/m2] 21 /bar Experimentell nach van't Hoff Gleichung 20 10 0 0 0,2 0,4 0,8 0,6 1,0 m2 /mol kg-1 Osmotischer Druck einer wäßrigen Rohrzuckerlösung bei 273 K Bestimmung der Molekülmasse aus osmotischem Druck! (Makromoleküle) Löslichkeit fester Stoffe Schmelzent halpie des gelösten Stoffes ln x 2 T Molenbruch des gelösten Stoffes Δ Sm H 2 R 1 1 TSm T TSm=Schmelztemperatur [K] Löslichkeit von Gasen p2 = k x2 Henry – Dalton (1803): Ein Gas löst sich proportional zu seinem Druck p2 in einer Flüssigkeit (Molenbruch x2). Vergleiche „Sprudel“: CO2 in Wasser. Umgekehrt ist der Partialdruck eines gelösten Gases über der Lösung seinem Molenbruch in der Lösung proportional. 22 Siedediagramme (2 Komponenten in Flüssigkeit (hier α) und Dampf (hier β) Zusammensetzung des 2-Phasengebietes bei Temperatur T: ln x 2 v H 2 T T2* R T T2* x 2 N2 xF T/K 1 90 E D Dampf (p = 1.013 bar) T 85 Flüssigkeit/ Dampf B Flüssigkeit 80 C Zusammensetzung des Dampfes verdampfen (mehr leichter flüchtiges!) kondensieren G Ax x 0 0,2 0,4 0,6 0,8 2 1,0 x2,x2 Temperaturerhöhung: A B (Dampfbildung) E (alles verdampft) D (Zusammensetzung des verbliebenen Flüssigkeitstropfens) F (heißerer Dampf mit gleicher Zusammensetzung wie Flüssigkeit) A F: keine Trennung der 2 Komponenten Wenn aber Gleichgewichtsdampf (C) entfernt wird, erneut kondensiert, verdampft, kondensiert … wird, reichert sich die leichter flüchtige Komponente (2) im Kondensat an: destillative Trennung der 2 Komponenten. Idealisiertes Diagramm für den Fall, dass am Kolonnenkopf kein Destillat entnommen wird. 23 Kühler Kopf K Boden 1 2 3 4 Rücklauf Rücklauf Füllkörper Destillat Blase Die Qualität einer destillativen Trennung x 2K x1K Zahl der Böden und dem Rücklaufverhältnis v ab. 24 (am Kopf K) hängt von der Azeotrope Mischungen Siedepunktsminimum Siedepunktsmaximum T Azeotroper Punkt T 1 2 Azeotroper Punkt 2 Destillation 1 0 x 0 1,0 Anziehung 1 2 geringer als 1 2 x 1,0 Anziehung 1 2 größer als 1 2 Mittelwert 1 1 2 2 Mittelwert Am azeotropen Punkt ist eine weitere destillative Trennung nicht mehr möglich. 25