Prionenerkrankungen
Werbung

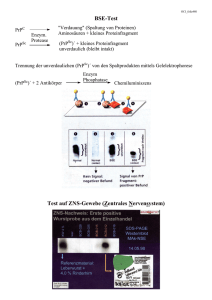
Prionenerkrankungen András Vincze-Nagy, 12.05.2010 Plau am See Prion • PRoteinaceous InfectiOus ageNt • nukleinsäurelose, faltungmodifizierte Proteine • in physiologischen und pathogenen Strukturen vorliegend • Physiologisch: 43 % Alpha-Helix • Pathogen: 30 % Alpha-Helix43 % Beta-Faltblatt • Umwandlung von physiologischen Prionen in pathogene Prionen möglich. PrPC – zelluläres Prion Protein • im peripheren und zentralen Nervensystem • kommt vor allem an der Zelloberfläche vor • schützt die Zellen vor zweiwertigen Kupfer-Ionen und freien Radikalen • einer der ersten Sensoren in der zellulären Abwehr von reaktivem O2 und freien Radikalen • Auswirkungen auf den enzymatischen Abbau von freien Radikalen • Fragliche Rolle in Myelinreparaturprozessen in Schwann-Zellen PrPC PrPSc – Scrapie Prion Protein • • • • wurde zuerst bei an Scrapie erkrankten Tieren gefunden PrPC in Kontakt mit PrPSc nimmt die Form von PrPSc an Große Mengen an PrPSc sind unlöslich Intrazelluläre Ablagerungen in Form von Amyloid Plaques Zelltod „Löcher im Gehirn“ schwammartige (spongiforme) Struktur Spongiforme Encephalopathie Spongiforme Encephalopathie gesicherte Prionkrankheiten • Creutzfeldt-Jakob-Krankheit – subakute spongiforme Enzephalopathie • Kuru – „Zittern“ – engl. „Laughing disease“ • Gerstmann-Sträussler-Scheinker-Syndrom – Mut. PrP-Gen - Codon 102, 105, 117, 198 oder 217 Chrom. 20 • Fatale familiäre Insomnie – Mut. PrP-Gen - Codon 178 - Chrom. 20 fragliche Prionkrankheiten • Familiäre progressive subkortikale Gliose – Prionen? • Multiple Sklerose – evtl. SVI, genetische Disposition • Pick-Atrophie und Alzheimer Krankheit – Prionen? • Amyotrophische Lateralsklerose – evtl. SVI, genetische Disposition Creutzfeldt-Jakob-Krankheit Ätiologie • Sporadische Form – Prionen – vCJD – Zusammenhang mit oralen Zufuhr von BSEinfiziertem Rindfleisch • Familiäre Häufnung – in 15% d.F., Mut. PrP-Gen auf Chrom 20 • Iatrogene Übertragung – Blut-, Gewebeinokulation, chirurgische Instrumente Creutzfeldt-Jakob-Krankheit Pathologie • PrPC wird durch posttranslationale Prozesse in das proteasenresistente und faltungsmodifizierte PrPSc umgewandelt • Histologisch – spongiforme Enzephalopathie mit Neuronenuntergang, Gliose und senilen Plaques • mäßige Hirnatrophie im Kortex, Striatum, Thalamus, gelegentlich Kleinhirn Creutzfeldt-Jakob-Krankheit Epidemiologie • Prädisp. alter: 50-80 J, Durch 65 J • F > M (1,5:1) • Inkubationszeit 6 Monate bis 30 Jahre • Inzidenz 1/1.000.000/Jahr in Deutschland • ca. 100 gesicherte Fälle/Jahr • Meldepflichtig! Creutzfeldt-Jakob-Krankheit Klinisch • Prodromalphase – Schlafstörungen, Schlaflosigkeit, Appetitlosigkeit, Verwirrtheit, verminderte Leistungsfähigkeit, abnormes Verhalten, evtl. Halluzinationen, Schwindel, Kopfschmerzen • Psychoorganische Veränderungen – Stimmungschwankungen, Konzentrationsstörung, Demenz, akinetischer Mutismus Creutzfeldt-Jakob-Krankheit Klinisch • Neurologische Funktionsstörungen – Kleinhirnsymptome mit Ataxie, extrapyramidalmotorische Störungen mit Ruhetremor, Chorea-artige Hyperkinesen, Paresen, Faszikulationen, Myoklonien, Muskelatrophie, Dysarthrie, Blickparesen, Nystagmus, zerebrale Krampfanfälle, Aphasie, Alexie • Koma und Dezerebration Tod Creutzfeldt-Jakob-Krankheit Diagnostik • Anamnese – familiäre Disposition, frühere neurochirurgische Operationen • Neurologische Untersuchung - Greifreflexe von Hand und Mund auslösbar, positive Pyramidenbahnzeichen • LP – klarer Liquor, Nachweis von NSE, S1000-betaProtein, Tau-Protein, 14-3-3-Protein Creutzfeldt-Jakob-Krankheit Diagnostik • CT/MRT - Normalbefund, in Spätstadium Hirnatrophie, Hyperintensität im Bereich der Stammganglien • EEG –triphasische spontane Aktivität (periodische Sharp-slow-wave-Komplexe) 1/sec. • Gehirnbiopsie, Autopsie – Nachweis des PrPSc im Gehirn Periodische Sharp-slow-wave-Komplexe Creutzfeldt-Jakob-Krankheit Therapie • keine kausale Therapie • Vermeidung sekundärer Komplikationen wie Inaktivitätspneumonie, Harnweginfekte • Clonazepam bei starken Myoklonien • genetische Beratung bei familiärer CJD • Selbsthilfegruppe/Angehörigegruppe Creutzfeldt-Jakob-Krankheit Prognose • infaust • schnell fortschreitende Demenz, tödlicher Ausgang nach 0,5-2 Jahren, meist durch Pneumonie, Exsikkose und nicht beherrschbare Hypotonien Kuru • „Zittern“ • engl. Lauching disease Ätiologie • Prionen, Verwandtschaft mit Creutzfeldt-Jakob-Krankheit wahrscheinlich Pathologie • Histologisch – Spongiforme Enzephalopathie Kuru Epidemiologie • endemisches Auftreten im östlichen Hochland von Papua-Neuguinea, früher bis zu 1% der Bewohner betroffen • Infektionsmodus – ritueller Bestattungs-Kannibalismus des Stammes der Fore (Essen von Hirngewebe der Verstorbenen) • Seit 1957 tritt diese Erkrankung nach Verbot des Kannibalismus praktisch nicht mehr auf • Inkubationszeit: 1-20 Jahre Kuru Klinik • • • • • • Kleinhirnataxie Gangunsicherheit Dysarthrie Paresen Blickparesen (Strabismus) schüttelfrostartiger Tremor Kuru Therapie • keine kausale Therapie bekannt, rascher Tod Prognose • sehr schlecht, Erkrankungsdauer mit Tod innerhalb von 6-12 Monaten Gerstmann-Sträussler-ScheinkerSyndrom • Zerebelläre Symptomatik im Vordergrund: – – – – Ataxie Gangunsicherheit Dysarthrie Nystagmus • keine oder erst sehr späte Demenz Fatale familiäre Insomnie • • • • • • • progrediente Schlaflosigkeit und Tagesschläfrigkeit Sympathikusaktivierung mit Hypertoniekrisen Tachykardie Hyperhidrose Tremor, Ataxie, Hyperreflexie, Myoklonien geringe Demenz, Halluzinationen Pathologie – Neuronenverlust in anterioren und mediodorsalen Thalamuskernen Literatur • Neurologie und Psychiatrie für studium und Praxis Gleixner, Müller, Wirth • eMedicine – http://emedicine.medscape.com/ • Wikipedia – en.wikipedia.org • Neurologie Psychiatrie – J. Klingelhöfer, M. Rentrop