Die Diskussion um BSE (bovine spongiforme Enzephalopathie) und
Werbung
 und")
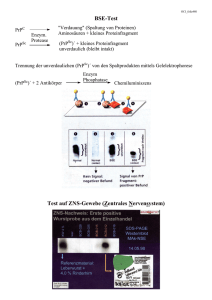
Ruth Baumann 12.11.2002 Referat: Prione/ BSE BSE = bovine spongiforme Enzephalopathie TSE = transmissible spongiform encephalopathies BSE gehört zur Gruppe der TSE-Erkrankungen ->Überbegriff für übertragbare spongiforme Encephalopathien; transmissible spongiform encephalopathies die verschiedene Säugerorganismen betreffen können und deren klinische Manifestation vor allem von einen Niedergang von Neuronen im Gehirn gekennzeichnet ist. Es gibt bis jetzt nur relativ wenige Erkenntnisse zum Pathogenitätsmechanismus der TSE-Erkrankungen, dennoch sind in den letzten Jahren beträchtliche Fortschritte in der Aufklärung von Funktion und Struktur des "Erregers" gemacht worden. Am Anfang der Erforschung der TSE-Erkrankungen stand eine sehr kontroverse Diskussion über den Charakter des Erregers. So wurde in erster Linie der Transfer viraler DNA als Ursache vermutet. Inzwischen ist doch allgemein akzeptiert, daß die Grundlage der TSE-Erkankungen in der Biochemie der Faltung und Funktion der Prionproteine liegt und hierzu sind in den letzten Jahren sehr viele neue Erkenntnisse erhalten worden. Prionproteine werden in allen höher entwickelten Organismen gebildet und bereits in den einfachsten eurkaryoten Zellen wie Hefen findet sich ein dem menschlichen Prion verwandtes Protein. Auch wenn nicht für alle speziesspezifischen Prionformen gezeigt, so kann doch davon ausgegangen werden, daß Prionen generell in wenigstens zwei Konformationen vorliegen können. D.h., trotz gleicher Aminosäurensequenz nehmen sie grundsätzlich unterschiedliche dreidimensionale 1 Strukturen ein. Die normalerweise in einem Organismus gebildeten, also körpereigenen Prion-proteine werden mit dem Kürzel PrPc versehen. Die in ihrer räumlichen Struktur veränderten pathogenen Varianten der Prionen, werden in Anlehnung an die Erregerform bei der Scrapieerkrankung des Schafes als PrPsc bezeichnet. Jedes Prionprotein kann also in einer normalen PrPc-Form und einer PrPsc-Variante vorkommen. Charakteristisch für die PrPsc-Form ist die außerordentliche Stabilität gegenüber denaturierenden Temperaturen, Chemikalien und gegenüber enzymatischer Spaltung durch Proteasen. Letztlich liegt in dieser "Umweltstabilität" auch die Ursache für seinen Eintrag in die Nahrungskette und seine Persistenz. Gleichzeitig bietet die Proteasenstabilität jedoch auch die Grundlage für den spezifischen Nachweis von PrPsc mittels Antikörper im WesternBlot oder im ELISA-Test, der in den Screeningprogrammen bei den Rindern eingesetzt wird. Durch spezifischen Verdau der Hirnproben mit Proteinase K kann die PrPsc -Form, da stabiler, erhalten werden und im Immunnachweis semiquantitativ bestimmt werden. Vorkommen und Struktur normaler Prionproteine Prionproteine sind extrazelluläre Sialoglycoproteine, die über einen sog. GPI-Anker (Glycosyl-Phosphatidylinositolanker) mit der Plasmamembran einer Zelle verbunden sind. Sie zeigen eine bemerkenswerte Heterogenität der Glycosylierung wobei die überwiegende Zahl der Proteine an zwei Asparaginresten, damit N-glycosidisch gebunden, antennenartige Oligosaccharidstrukturen tragen. Das Gen für das menschliche Protein ist auf Chromosom 20 lokalisiert und besteht aus zwei Exons und einem Intron. Der offene Leserahmen kodiert für ein Protein von 253 Aminosäuren. Die 5´nicht-translatierte Region des Gens zeigt eine bemerkenswerte Seqeunzvariabilität und auch innerhalb des kodierenden Bereichs, der in das Protein übersetzt wird, gibt es ebenso Unterschiede in der Aminosäurensequenz zwischen einzelnen Personen. Dies könnte für die vermutete unterschiedliche Empfänglichkeit von Individuen gegenüber TSE-Erregern von zentraler Bedeutung sein. 2 Charakteristisch für Prionproteine sind 4 aminoterminal gelegene sog. Octapeptidrepeats, d.h., sich wiederholende Peptidsequenzen, die vorwiegend aus Glycin-, Glutamin- und Prolinresten bestehen. Sie sind auch verantwortlich für die Ausprägung des pathogenen Konformers mit seinem höheren Anteil an ßFaltblattstruktur während das normale Prion einen höheren Anteil a-helikaler Strukturen aufweist. Die veränderte Proteinfaltung zu PrPsc führt auch zu den bei Creutzfeldt-Jakob und Scrapie-Erkrankungen nachweisbaren fibrillären Proteinstrukturen, die als Ablagerungen im neuronalen Gewebe nachweisbar sind. Die bereits zitierte proteolytische Stabilität des PrPsc beruht ebenfalls auf dieser ßFaltblattstruktur, die gleichzeitig verhindert, das einmal im Organismus gebildetes und falsch gefaltetes Prionprotein im Gegensatz zu allen normalen Proteinen nicht mehr abgebaut werden kann und akkumuliert, was die das scheinbar unaufhaltsame Fortschreiten der Erkrankung begünstigt. Es ist bewiesen, dass die Pathogenese einer infektiösen Prionener-krankung auf der direkten oder indirekten Wechselwirkung von PrPc mit PrPsc beruht. Experimente an sog. knock-out-Mäusen,bei denen das Gen für deren PrPc ausgeschaltet (PrPc-/--Tiere) worden ist (Tiere können kein köpereigenes Prion mehr bilden), haben gezeigt,dass diese Tiere gegenüber einer Infektion mit BSEErregern absolut geschützt sind. Bemerkenswerterweise entwickeln die Tiere ohne das PrPc keinen auffälligen Phänotyp, d.h., sie erscheinen völlig gesund. Bringt man in die PrPc-/--Mäuse anstelle des Mäusegens das humane Gen für PrPc ein, so können die Tiere wieder mit den Erregern vom Rind infiziert werden. Diese genetischen Modelle mit gezielter Expression von verschiedenen Prionformen in Mäusen waren von sehr großer Bedeutung für das Verständnis der Grundlagen von TSE-Erkrankungen und der Speziesspezifität der Transmission. Auf Grund dieser Experimente lässt sich also annehmen, daß das Prionprotein keine essentielle Körperfunktion unter normalen Bedingungen hat oder sein Verlust durch andere Proteine mit ähnlicher Funktion kompensiert wird. 3 Mittlerweile sind sehr viele solcher knock-out-Mäuselinien konstruiert worden, auch solche, die sog. negativ-dominate Formen von PrPc bilden. Diese Tiere sind gegen jede bekannte Form des Erregers resistent. (Dazu reichen einzelne Aminosäurenaustausche, wie sie auch als Polymorphismen des menschlichen prpc-Gens vorgefunden werden. Auch dies läßt vermuten, daß einzelne Individuen gegenüber pathogenen Prionen besser geschützt sein könnten) Da die Prionenerkrankungen die Präsenz eines körpereigenen Prions erfordern, stellt sich die Frage, wo im menschlichen oder tierischen Organismus die normalen Prionproteine gebildet werden. Früher ging man davon aus, dass nur neuronale Zellen dazu befähigt sind, heute vermutet man aber, dass sehr viele andere Zelltypen und damit auch andere Organe PrPc exprimieren. Dazu zählen u.a. Stammzellen im gastrointestinalen Epithel, die Lunge, die Milz sowie bestimmte Zellen des Reproduktionstraktes. Auch für Muskelzellen in Kultur ist die Expression von PrPc im Rahmen ihrer Differenzierung gezeigt worden. Von besonderer Relevanz ist auch die Bildung der Prionproteine in Blutzellen, da damit gegebenenfalls auch ein Risiko für die Übertragung bei Bluttransfusion (nicht beim Plasmatransfer) gegeben sein könnte. Es zeigt sich, daß sowohl Thrombocyten wie Lymphocyten und Leucocyten Prionproteine in ihrer Zellmembran aufweisen. Die Bildung in den immunkompetenten Zellen bzw. dem lymphoreticularen System lässt befürchten, dass auch hier eine Propagation, d.h., die Replikation pathogener PrPsc-Formen, ähnlich wie im Gehirn, stattfinden könnte. Ein Indiz dafür ist die Tatsache, dass bei den in England gestorbenen Menschen mit der neuen Variante der Creutzfeldt-Jakob-Erkrankung die PrPsc-Immunoreaktivität (d.h., die pathogene Proteinform) nicht nur im Gehirn sondern auch in lymphoiden Geweben wie Tonsillen bzw. Appendix nachgewiesen worden ist. Auf die Bedeutung der Expression von PrPc im peripheren Nerven und Immunzellen muß auch im Zusammenhang mit der Aufnahme des Erregers in den tierischen oder menschlichen Organismus und seines Transfers in das Gehirn hingewiesen werden. 4 Sowohl im intestinalen Nervensystem wie auch den immunkompetenten Zellen des Darmes lassen sich die Prionproteine nachweisen. Ob hier aber bereits eine starke Vermehrung der pathogenen Formen stattfindet, ist bisher nicht eindeutig belegt. Die Wanderung von PrPsc im Nervensystem aus dem Darm in das Gehirn ist experimentell dagegen gezeigt. Diese Wanderung ist kein aktiver Prozess bei dem das Protein sich selbständig bewegen würde sondern erfolgt im Rahmen normalen Zellmigrations- und -fusionsvorgänge. Welche Rolle den Immunzellen beim Transfer von pathogenen Prionen aus dem Darm in den Organismus zukommt, ist noch nicht abschließend geklärt. Diese Zellen wandern nämlich ebenfalls in einem physiologischen Prozess aus den Payerschen Platten der Darmschleimhaut in den Organismus aus und gelangen so in diverse Gewebe. Es steht jedoch außer Frage, daß nach der Aufnahme von erregerhaltigem Material mit der Nahrung die chemische und enzymatische Stabilität des PrPsc es stark begünstigt, dass das Protein in intakter Form in das Darmepithel aufgenommen wird und von dort über das Nervengewebe und/oder die intestinalen Immunzellen in periphere Gewebe und das Gehirn gelangen kann. In welchem Umfang dabei bereits eine Vermehrung von PrPsc durch Konformationsänderung des in diesen Zellen vorkommenden normalen PrPc stattfindet, ist weitgehend unbekannt. Proteinfaltung Prionproteine existieren in zwei Konformationen und können sich somit trotz gleicher Aminosäurensequenz in ihrer räumlichen Struktur und ihren physikochemischen Eigenschaften (u.a. enzymatischer Stabilität) unterscheiden. Die Grundlage der TSEErkrankungen beruht vorallem auf der Wechselwirkung von PrPc mit PrPsc. 5 Die Grundfrage aber, wie PrPsc aufgrund seiner spezifischen Form das normale PrPc in eine neue Form zwingen kann, ist damit aber noch immer nicht erklärt. . Jedes zelluläre Protein muss zur Wahrnehmung seiner Funktion eine bestimmte dreidimensionale Struktur einnehmen, die jedoch von seiner Umgebung abhängig ist. Die Herausbildung seiner räumlichen Struktur geschieht nach der Translation des Proteins, d.h., wenn die Polypeptidkette vorliegt unter Beteiligung von speziellen zellulären Proteinen und unter Beteiligung funktioneller Gruppen (z. Bsp. Cysteinresten) des jeweiligen Proteins. Proteinfaltungsprozesse unterliegen einer komplexen zellulären Kontrolle. So wird ein falsch gefaltetes Protein normalerweise durch ein "Qualitätssicherungssystem" erkannt und sofort einer Degradierung zugeführt. Da PrPsc aber nur geformt werden kann wenn normale PrPc-Proteine bereits vorhanden sind, wird hier die Proteinfaltungskontrolle der Zelle unterlaufen. Und wenn PrPsc erst einmal geformt ist, bleibt es durch seine recht hohe Proteasenresistenz auch (zumindest in Bruckstücken) erhalten und kann normale PrPc-Proteine durch Aufzwingen seiner Form transformieren. FRAGE: Woher kam das erste PrPsc-Molekül??? Nun, grundsätzlich könnte der Ursprung in einer spontanen Mutation im Priongen liegen, die für ebenso spontan auftretende Formen von Encephaolopathien bei Tier und Mensch verantwortlich gemacht werden. Eine Infektionskette kann sich daraus natürlich nur dann entwickeln, wenn die pathogene Form in eine Nahrungskette gelangt und aufgrund seiner besonderen Eigenschaften dort persistiert und propagiert werden kann. Da man heute PrPc recht leicht gentechnisch in größeren Mengen herstellen kann, sind Untersuchungen zur Transformation von PrPc in die PrPsc-Form auch in zellfreien Systemen möglich. Diese Studien belegen, daß die pathogene Form tatsächlich schon alleine durch die Protein-Protein-Wechselwirkung entstehen kann (Proteine only Theorie). 6 Auch die an der Proteinfaltung in der Zelle beteiligte Funktionsproteine der Chaperonfamilie (Timm) könnten eine Rolle in der Genese der PrPsc-Formen spielen. Dies lassen zumindest eine Reihe von Studien an neuronalen Zellen in Kultur vermuten. Ob genetische Unterschiede in diesen an der Faltung beteiligten Proteine ebenso wie unterschiedliche Glycosylierungsmuster von PrPc (aufgrund unterschiedlicher Ausstattung mit Glycosyltransferasen) zwischen Individuen das Erkrankungsrisiko mitbestimmen, läßt sich zum jetzigen Zeitpunkt noch nicht beantworten. Allerdings lässt die geringe Infektionsrate bei Rindern in großen Herden bei recht einheitlicher Ernährungsweise läßt ebenso wie die vergleichsweise geringe Zahl bisher diagnostizierter Fälle der neuen Creutzfeldt-Jakob-Variante beim Menschen trotz hohem Verzehr infiziertem Fleischs in England eine signifikante Beteiligung des genetischen Hintergrundes des Empfängers vermuten. Bindungsproteine des PrPsc Bindungsproteine könnten große Bedeutung sowohl für die Pathogenese der TSEErkrankungen als auch die Diagnostik haben. Bei der Suche nach einem solchen Bindungsproteinen für das PrPsc-Konformer wurde das Blutprotein Plasminogen identifiziert. Plasminogen ist die inaktive Vorstufe der u.a. am Gerinnungsprozess beteiligten Serinprotease Plasmin. Plasminogen bindet spezifisch die PrPsc-Form, nicht aber die PrPc-Variante der Prionen, wobei sich die Binding des PrPsc durch freies Lysin blockieren läßt. So vermutet man, dass auch ein im PrPsc auf der Oberfläche des Proteins liegender Lysinrest seine Binding an die sog. kringle-Domänen des Plasminogens vermittelt. Es ist bisher noch nicht klar, ob Plasminogen auch in vivo PrPsc-binden kann und damit möglicherweise seine Infektiösität beeinflusst. Allerdings ergeben sich aus der Bindungsspezifität neue Ansätze für diagnostische Verfahren. 7 Zelluläre Funktionen von PrPc Wenngleich die Befunde der PrPc-/--Mäuse, die kein körpereigenes Prion mehr bilden können, vermuten lassen, dass das Protein für die normale Zellfunktionen nicht bedeutend ist, steht außer Zweifel, dass es in der Pathophysiologie des Nervenzelluntergangs im Gehirn eine wichtige Rolle spielt. Der PrPsc-vermittelte Verlust von Neuronen ist offenbar die Folge einer erhöhten Apoptoserate (programmierter Zelltod). Bringt man PrPc in neuronalen Zellen zur verstärkten Expression, zeigen diese Zellen eine höhere Sensitivität gegenüber oxidativem Stress, erhöhte Raten der Lipidperoxidation, Zeichen von Apoptose und eine deutliche Reduktion der Aktivitäten glutathionabhängiger Enzyme sowie eine verminderte Aktivität der Kupfer- und Zink-abhängigen Superoxiddismutase (SOD) bei unverändertem Spiegel des Enzyms. Zu diesem Befund gesellt sich der Nachweis daß PrPc ein Kupfer-Ionen bindendes Protein ist, wobei die Bindungstellen für Kupfer in den bereits zitierten 4 Octapeptidrepeats liegen. Nach Bindung von Kupfer gewinnt PrPc auch selbst SOD-Aktivität und die Präsenz von PrPc in der Zellmembran beeinflußt offenbar auch den Kupfertransport in die Zelle bzw. den zellulären Kupferspiegel. Ebenso bemerkenswert ist die Beobachtung, dass die Kupferbindung an das membran-gebundene PrPc die Internalisierung des Prionproteins in die Zelle begünstigt. Wie viele andere Membranproteine unterliegt auch PrPc einer vom Clathrin abhängigen Endocytose, gelangt damit in ein frühes Endosomenkompartiment des Zellinneren und kann von dort wieder in die Zellmembran zurückkehren. Nach der endocytotischen Aufnahme des mit Kupfer beladenen PrPc in das Endosomen-kompartiment, könnte Kupfer dort freigesetzt und über Bindungsproteine in das Cytosol der Zelle entlassen werden. PrPc könnte nach seiner Rückkehr in die Zellmembran erneut extrazelluläres Kupfer binden und würde somit die Rolle eines Kupfertransporters wahrnehmen. Die Bindung von Kupfer oder von Mangan, das ebenso gut gebunden wird, begünstigt aber offenbar auch das Entstehen der PrPsc-Form und beeinflußt die Faltung des Prionproteins. Möglicherweise ist somit auch der Spurenelementstatus eines Individuums ein Faktor, der zur Pathogenese einer Prionenerkrankung beiträgt. 8 Neben dieser Rolle im Kupferstoffwechsel der Zelle ist für die PrPsc-Form auch eine direkte zytotoxische Funktion bekannt. So sind bereits die Octapeptidrepeats als Fragmente des PrPsc ab bestimmten konzentrationen toxisch für Neuronen. Auch gibt es Hinweise, daß diese Proteindomänen sich in die Zellmembran einlagern können und hier durch Oligomerisierung kanalähnliche Strukturen bilden, die die zelluläre Ionenhomöostase stören. Auch den Neuronen benachbarte Gliazellen sind offenbar am Nervenzelltod durch pathogene Prionen beteiligt. Bei ihnen scheint PrPsc eine verstärkte Glutamatfreisetzung zu bewirken, die, wenn sie länger anhält, toxisch auf Neuronen wirkt. Zusammenfassung Die Pathogenese der Prionenerkrankungen basiert auf einem bisher nicht bekannten biologischen Prinzip, bei dem ein Protein ein in der Primärstruktur identisches Protein in eine neue Konformation, d.h., andere räumliche Struktur zwingt. Es erlangt dadurch eine außerordentliche Stabilität gegenüber nahezu allen Umweltfaktoren, die sonst Proteine zu denaturieren oder degradieren vermögen. Diese Stabilität des Proteins ermöglicht seine Persistenz in der Nahrungskette und seine Aufnahme in den Organismus. Seine Replikationsfähigkeit ist im Empfängerorganismus dagegen von vielen weitgehend unbekannten Einflußgrößen abhängig. Menschliche Prionenerkrankungen sind also "Proteinfaltungskrankheiten". Sie können durch Mutationen in den Genen sporadisch entstehen, in autosomal-dominanter Form ererbt sein oder durch alimentäre Azfnahme der pathogenen Proteinform wie bei BSE bedingt sein. Die Bedeutung der Theorie, daß Prionen die Ursache der TSE-Erkrankungen sind, wurde entsprechend auch mit der Vergabe des Nobelpreises für Medizin an Stanley Prusiner 1997 gewürdigt. Er hat den Begriff Prion eingeführt und maßgebliche Studien zur Genese der TSE-Erkrankungen sind von seiner Arbeitgruppe vorgelegt worden 9 10