Genetische Prionenerkrankung
Werbung

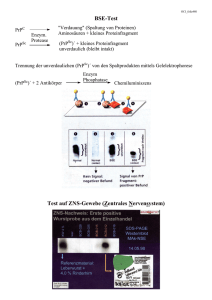
Prione und die Creutzfeld-Jakob Krankheit Creutzfeld-Jakob Eine beim Menschen bislang sehr selten auftretende, tödlich verlaufende und transmissible (übertragbare) spongiforme Enzephalopathie (TSE). Eine Hirnerkrankung, bei der es zu einer schwammartigen Veränderung des Gehirngewebes kommt (bei Mensch und bei Tier). Verursacher sind atypische Eiweiße die Prionen Alfons Maria Jakob (links) und Hans Gerhard Creutzfeldt Die Creutzfeldt-Jakob Krankheit (englisch: Creutzfeldt-Jakob Disease , CJD) wurde Anfang der 20er Jahre des letzten Jahrhunderts von den beiden Deutschen Neurologen Hans Gerhard Creutzfeldt und Neuropathologen Alfons Maria Jakob beschrieben. Prionkrankheiten beim Tier Krankheit: Wirt: Traberkrankheit (Scrapie) Schaf Übertragbare Nerz-Enzephalopathie (TME) Nerz Chronischer Kräftezerfall (CWD) Maultier, Hirsch, Reh Spongiforme Katzen-Enzephalopathie (FSE) Katzen Exotische Huftier-Enzephalopathie (EUE) Nyala, Kudu Spongiforme Rinder-Enzephalopathie (BSE) Rind Prionkrankheiten beim Tier Krankheit: Wirt: Traberkrankheit (Scrapie) Schaf Übertragbare Nerz-Enzephalopathie (TME) Nerz Chronischer Kräftezerfall (CWD) Maultier, Hirsch, Reh Spongiforme Katzen-Enzephalopathie (FSE) Katzen Exotische Huftier-Enzephalopathie (EUE) Nyala, Kudu Spongiforme Rinder-Enzephalopathie (BSE) Rind Prionkrankheiten beim Mensch Krankheit: Ätiologie: Gerstmann-Sträussler-ScheinkerSyndrom (GSS) Mutationen Fatale Familiäre Schlaflosigkeit (FFI) Mutationen Kuru Infektion durch Nahrung Creutzfeld-Jakob (CJD) Iatrogen Sporadisch Familiär Neue Variante (nvCJD) Infektion unbekannt Mutationen Infektion Prionkrankheiten beim Mensch Krankheit: Ätiologie: Gerstmann-Sträussler-ScheinkerSyndrom (GSS) Mutationen Fatale Familiäre Schlaflosigkeit (FFI) Mutationen Kuru Infektion durch Nahrung Creutzfeld-Jakob (CJD) Iatrogen Sporadisch Familiär Neue Variante (nvCJD) Infektion unbekannt Mutationen Infektion Kurze Wiederholung: Proteinstruktur Primärstruktur (Aminosäuresequenz, 1D) Sekundärstruktur (beta-Faltblatt; alpha-Helix) Tertiärstruktur (ein Polypeptid) Quartiärstruktur (mehrere Polypeptide) Was bestimmt die 3-dimensionale Struktur eines Proteins? Polarität Ladung Hydrophobie Größe Faltung an den Ribosomen und mittels Chaperonen! Methoden zur Aufklärung von Proteinstrukturen Kernresonanzspektroskopie (NMR); von nuclear magnetic resonance = kernmagnetische Resonanz Kristallstrukturanalyse Krankheitsbild BSE und Creutzfeldt-Jakob Krankheit Schreckhaftigkeit, Depressionen,Wahnvorstellungen Ataxie Gedächtnisstörungen Störungen der Wahrnehmung und Persönlichkeitsveränderungen Kreislaufprobleme und Verwirrtheit Demenz Krankheitsbild von BSE und CJD Krankheitsbild von BSE und CJD Das Prion-Protein Sruktur Vorkommen Funktion Eigenschaften C (PrP ) und SC PrP C PrP (natürlich) und Sc PrP (infektiös) Aus 254 AS bestehendes Glykoprotein Vorkommen des C PrP im lymphoretikulären Gewebe Oberfläche von Neuronen(Synapsen) Oberfläche von Astrozyten intrazellulär extrazellulär GPI Prion-Protein Funktion des zellulären Prion-Proteins Signalübertragung von Neuronen In der Bildung von Synapsen involviert Regulierung des Schlaf-Wach-Rhythmus sowie dem Überleben der Zellen PrPC bindet Kupfer und hat antioxidative Wirkung! Eigenschaften von PrPC (zellulär) und PrPSc (infektiös) Wann wirken Prion-Proteine toxisch? PrPC enthält zum großen Anteil a-Helices (43%) PrPSc mehr beta-Faltblattstrukturen, a-Helices (30%), b-sheets (43%) schlechter wasserlöslich hitzestabil Durch Proteasen nur schwer verdaulich Resistent gegen Proteolyse Aggregatbildung In Detergentien nicht lösbar Wann wirken Prion-Proteine toxisch? PrPC enthält zum großen Anteil alphaHelices (43%) PrPSc mehr beta-Faltblattstrukturen, a-Helices (30%), b-sheets (43%) schlechter wasserlöslich hitzestabil Durch Proteasen nur schwer verdaulich Resistent gegen Proteolyse Aggregatbildung In Detergentien nicht lösbar Beide enthalten die gleiche Aminosäuren-Primärsequenz Wann wirken Prion-Proteine toxisch? PrPC enthält zum großen Anteil alphaHelices (43%) PrPSc mehr beta-Faltblattstrukturen, a-Helices (30%), b-sheets (43%) schlechter wasserlöslich hitzestabil Durch Proteasen nur schwer verdaulich Resistent gegen Proteolyse Aggregatbildung In Detergentien nicht lösbar Beide enthalten die gleiche Aminosäuren-Primärsequenz Der genaue Vorgang der „Umfaltung“ von PrPC in PrPSc ist noch unbekannt. Theorie der Umfaltung PrP C PrP PrPSc Mutante PrPSc Infektion C PrPSc PrPSc PrPSc PrPSc Wie wirken Prion-Proteine toxisch? Wie wirken Prion-Proteine toxisch? Übertragungs-/ Infektionswege Die Initiation der Krankheit kann auf drei Weisen erfolgen: Sporadisch Prionenerkrankung Genetische Prionenerkrankung Infektion durch Ansteckung Übertragungs-/ Infektionswege Sporadisch Prionenerkrankung: zufällige Umfaltung eines PrPC in die PrPSc Form ohne erkennbare Ursache. Bildet die klassische Form der Creutzfeldt-Jakob-Krankheit (sCJD). Übertragungs-/ Infektionswege Sporadisch Prionenerkrankung: zufällige Umfaltung eines PrPC in die PrPSc Form ohne erkennbare Ursache. Bildet die klassische Form der Creutzfeldt-Jakob-Krankheit (sCJD). Genetische Prionenerkrankung: Das codierende Gen für PrPC das PRNP-Gen enthält einen Fehler (Mutation). Das fehlerhafte PrPC ist anfälliger für eine Umwandlung in PrPSc. Die fehlerhafte Information wird von Eltern auf die Kinder vererbt. - familiäre Form der Creutzfeldt-Jakob-Krankheit (fCJD) - Gerstmann-Sträussler-Scheinker-Syndrom (GSS) - tödliche familiäre Schlaflosigkeit (FFI) Genetische Ursachen der CreutzfeldtJakob Krankheit Es sind jeweils eine oder mehrere Mutationen bekannt, die zu fCJD, GSS oder FFI führen. Am Codon 129 besteht ein Methionin/Valin-Polymorphismus, der für Krankheitsausbruch und –verlauf mitentscheidend ist. Genetische Ursachen der CreutzfeldtJakob Krankheit Es sind jeweils eine oder mehrere Mutationen bekannt, die zu fCJD, GSS oder FFI führen. Am Codon 129 besteht ein Methionin/Valin-Polymorphismus, der für Krankheitsausbruch und –verlauf mitentscheidend ist. M129V E219K PRNP A117V M232R D178N V180I P105L P102L Y145* Q217R F198S V210I E200K fCJD Die familären Creutzfeldt-Jakob-Krankheiten (fCJD) sind umfassen ca. 10 bis 15 % aller CJD-Fälle im allgemeinen autosomal-dominant vererbt und sind eine Folge bestimmter Mutationen des Prion-Proteingens (PRNP) auf Chromosom 20. Gegenwärtig werden 23 verschiedene Mutationen des PrionProteingens beschrieben (Punktmutationen, Insertionen in der Repeat-Region oder Deletionen des Prion-Proteingens). spezifische Mutation (E200K-Mutation) des PRNP zurückführen. In Europa kommt neben der FFI-Mutation die E200K-Mutation als zweithäufigste CJD-Mutation vor. Genetische Ursachen des GSS Das Gerstmann-Sträussler-Scheinker-Syndrom (GSS) Eine seltene autosomal-dominant vererbbare neurodegenerative Krankheit durch die Mutationen P102L, P105L, A117V, F198S und Q217R des Prion-Proteingens (PRNP) auf Chromosom 20 hervorgerufen werden kann. GSS Das Gerstmann-Sträussler-Scheinker-Syndrom (GSS) Eine seltene autosomal-dominant vererbbare neurodegenerative Krankheit durch die Mutationen P102L, P105L, A117V, F198S und Q217R des Prion-Proteingens (PRNP) auf Chromosom 20 hervorgerufen werden kann. M129V E219K PRNP A117V M23R D178N V180I P105L P102L F198S Y145* Q217R V210I E200K Genetische Ursachen des FFI Die seltene letale familiäre Insomnie (englisch: Fatal Familial Insomnie, FFI), hereditären Prionerkrankungen eine autosomal-dominant vererbbare neurodegenerative Krankheit Mutation der von Aspartat (Asp) in Asparagin (Asn) am Codon 178 des Prion-Proteingens (PRNP) auf Chromosom 20 Gleichzeitig liegt Methionin (Met) im polymorphen Codon 129 des mutierten Allels vor. FFI Die seltene letale familiäre Insomnie (englisch: Fatal Familial Insomnie, FFI), hereditären Prionerkrankungen eine autosomal-dominant vererbbare neurodegenerative Krankheit Mutation der von Aspartat (Asp) in Asparagin (Asn) am Codon 178 des Prion-Proteingens (PRNP) auf Chromosom 20 Gleichzeitig liegt Methionin (Met) im polymorphen Codon 129 des mutierten Allels vor. M129V E219K PRNP A117V D178N M232R V180I P105L P102L Y145* Q217R F198S V210I E200K Übertragungs-/ Infektionswege „Ansteckung“ durch stark PrPSc-haltiges Material: Zufuhr von PrPSc von außerhalb in denn Organismus Körperfremde PrPSc wandelt körpereigene PrPC in die pathogene Form um Mengen abhängig Art des PrPSc Artenbarriere Inkubationszeit kann über Jahre erfolgen PrPSc-haltiges Material: über die Nahrung: Transplantation: von Kuh zu Kuh: 1g infiziertes Hirn (BSE) von Mensch zu Mensch: Kannibalismus Hirnhaut und Hornhaut der Augen (CJD) Hormonpräparate: Hypophysenextrakte (CJD) Blutprodukte: CJD: keine Fälle bekannt Therapieansätze? Therapieansätze bei Prionerkrankungen Eine Therapie aller Creutzfeldt-Jakob Krankheitsformen (sCJD, fCJD, iCJD, vCJD, GSS, FFI) gibt es bislang nicht. Die Erkrankungen endet immer mit dem Tode. Eine Behandlung der BSE oder Scrapie ist bisher nicht möglich. Die Isolation erkrankter Tiere ist wegen der langen Inkubationszeit und dem innerhalb des Krankheitsverlaufes späten Auftreten der typischen Symptome schwierig. Bei Schafen ist bekannt, dass ein Polymorphismus (natürliche Variabilität mehrere Aminosäuren in der Proteinkette) an drei Positionen des Prion-Proteins (136 A/HV; 154(R/H), 171 (R/Q) die Empfänglichkeit für Scrapie bestimmt. Es soll daher durch Zuchtprogramme der Anteil der Scrapie-resistenten Schafe erhöht werden. Quellen www.prionforschung.de www.Wikipedia.org Prions; Stanley B. Plusiner; Nobel Lecture; Proc. Natl. Acad. Sci. USA 95 (1998) The Ninth Datta Lecture; Molecular biology of transmissible spongiform encephalopathies; Charles Weissmann; 1996 Zürich, Schwitzerland Proin reseach: the next frontiers; Adriano Aguzzi and Charles Weissmann; Nature VOL 389 ; 23 October 1997