T - Fachrichtung Chemie TU Dresden
Werbung

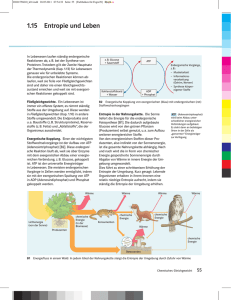
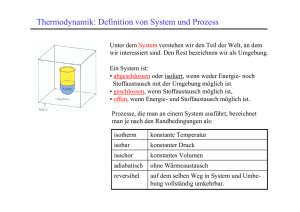
22 Kapitel 3: Thermodynamik Die historisch entwickelte Thermodynamik wird heute als phänomenologische Thermodynamik bezeichnet. Hierbei wird von 2 bis 3 Hauptsätzen ausgegangen, die sich aus der menschlichen Erfahrung ergeben haben und bis heute nicht widerlegt worden sind. Daraus wird alles andere streng abgeleitet. Dennoch bleibt wegen der Tatsache, dass man die Aussagen der Hauptsätze nicht direkt beweisen kann, für moderne Menschen ein ungutes Gefühl. Ein moderner Ansatz nennt sich statistische Thermodynamik und geht von den Eigenschaften des Moleküls aus, welches die phänomenologische Thermodynamik nicht kennt. Das Molekül hat diverse Freiheitsgrade zur Aufnahme von Energie (s. Kap. 2). Für viele (nicht zu große) Moleküle lässt sich eine Statistik über die Besetzung der Freiheitsgrade (ggf. Energieniveaus) bei gegebener Temperatur anstellen und man erhält als Summe die Gesamtenergie des betrachteten Systems, die der phänomenologischen inneren Energie u entspricht, jedoch ohne die Annahme der Gültigkeit von Hauptsätzen ermittelt wurde. In der Vorlesung wird zunächst ausschließlich die phänomenologische Thermodynamik behandelt. 3.1 Innere Energie und erster Hauptsatz Aus den Gasgesetzen folgt, dass die Energie eines geschlossenen Systems durch Zufuhr von Arbeit a erhöht werden kann (Gas in einseitig geschlossenem Zylinder mit beweglichem Kolben). Nach der kinetischen Gastheorie ist Energieerhöhung auch durch Temperaturerhöhung, d.h. durch Wärmezufuhr (Wärmemenge q) möglich. Man spricht daher auch von der inneren Energie u des (geschlossenen) Systems und kann Energieänderungen ausdrücken durch du = da + dq (3-1). Dies ist bereits eine Formulierung des ersten Hauptsatzes der Thermodynamik, der in Worten lautet: Die von einem geschlossenen System mit der Umgebung ausgetauschte Summe von Arbeit und Wärme ist gleich der Änderung der inneren Energie. u ist eine Zustandsgröße (a und q allein sind sog. Prozessgrößen, d.h. sie sind jeweils keine Zustandsgrößen, jedoch ihre Summe!). Der Zahlenwert von Änderungen der inneren Energie ∆u hängt nur von den Eigenschaften des Zustands ab, nicht aber vom Weg, auf welchem die Änderung herbeigeführt wurde. Anderenfalls könnte man Energie erzeugen oder vernichten, indem man zwei Zustände in einem Kreisprozess auf unterschiedlichen Wegen erreicht (vgl. mechanisches pepetuum mobile), was dem Prinzip der Energieerhaltung widerspricht. Der erste Hauptsatz ist so gesehen ein Energieerhaltungssatz für Chemiker. Arbeit und Wärme sind also Energieformen, aber keine Zustandsgrößen. Da da = –pdv (1-13) vom Weg abhängt, muss auch dq vom Weg abhängen, und zwar in einer zu da komplementären Weise, denn die Summe dieser beiden Energiebeträge ergibt eine Zustandsfunktion. Wärme(menge) kann durch Wärmeleitung von außen zugeführt werden. Arbeitszufuhr erfolgt z. B. durch Kompression des Systems, d.h. durch Verschieben von Wänden, die das Gefäß einschließen. Im isolierten System ist weder das eine noch das andere möglich. Dann gilt du = 0 (3-2), d.h. die innere Energie eines isolierten Systems ist konstant. u ist eine extensive Größe. Das Verhältnis U = u/n bezeichnet dagegen eine intensive Größe, die molare innere Energie (in Lehrbüchern oft anders symbolisiert, z.B. Um). Energiezufuhr ist daher in einem offenen System auch durch Stoffzufuhr möglich. Dafür gilt dann 23 du = da + dq + ΣUi dni (3-3). Die Ui sind partielle molare innere Energien der Komponenten. Der Ausdruck (3-3) wird auch kalorische Zustandsgleichung genannt. Einige Konsequenzen: (a) Wir betrachten zunächst Materie in einem geschlossen Behälter, also ein geschlossenes System, in dem weder Reaktionen ablaufen noch Kompressionsarbeit geleistet wird (dv = 0; pdv = 0 = da). Dann definiert der Ausdruck dq ∂u (3-4) = v = cv ∂T v dT die Wärmekapazität cv - hier eine extensive Zustandsgröße (intensiv Cp: spezifische Wärme); Zahlenwerte geben an, wieviel Energie (Wärme) benötigt wird, um die Materie um 1 K zu erwärmen. (b) Weil leichter messbar, benutzt man bei Flüssigkeiten und Festkörpern die Größe cp, die Wärmekapazität bei konstantem Druck (isobar, dp = 0): cp = (dqp/dT) (3-5). Unter dieser im Laboralltag häufiger gegebenen Bedingung (Atmosphärendruck) ist die Volumenarbeit nicht zu vermeiden und man verwendet zweckmäßigerweise anstelle der Zustandsgröße u eine andere, die Enthalpie h, definiert als h = u + pv (3-6), weil dann dh = du + pdv + vdp = dq + vdp (mit 1-13 und 3-1) sowie (∂h/∂T)p = (dqp/dT) = cp (3-7) (weil p = const.). Unterschiede in cp und cv sind leicht einzusehen, weil Volumenarbeit verrichtet wird, wenn der Druck konstant gehalten wird (vgl. Abb.); ∆q ist dann größer. Man erkennt daran, dass q (genau wie die Arbeit a, aber im Unterschied zu u, h, p, v und T) keine Zustandsfunktion ist: Um dieselbe Differenz der Zustandsgröße Temperatur, ∆T, zu erzeugen, werden unterschiedliche Wärmemengen ∆qv bzw. ∆qp benötigt. Sowohl die innere Energie als auch die Enthalpie eines idealen Gases hängen nur von der Temperatur ab: du = cvdT ; dh = cpdT (3-8). Es ist deshalb (exakt nur für ideale Gase) cp – cv = p(∂v/∂T)p = pnR/p = nR oder Cp – Cv = p(∂V/∂T)p = pR/p = R (3-9). (c) Für isotherme (dT = 0) Zustandsänderungen idealer Gase gilt nach (3-8) du = dh = 0 und dqv = pdv = nRT dv/v ; dqp = –vdp = –nRT dp/p (3-10). 24 (d) Zustandsänderungen, bei denen dq = 0 (kein Wärmeaustausch mit der Umgebung) heißen adiabatisch. Für diese gilt cv dT = – pdv ; cp dT = vdp (3-11). cp dT/T = nR dp/p (3-12). Division durch T = pv/nR liefert cv dT/T = –nR dv/v; Durch bestimmte Integration4 erhält man für eine Zustandsänderung von 1 nach 2 T c v ln 2 T1 T = ln 2 v T1 cv v = − nR ln 2 v1 v = ln 1 v2 nR (3-13). Wegen (3-9) und mit der Abkürzung κ = cp / cv kann man daher auch schreiben ( c p − cv ) / cv v T2 v1 = = 1 T1 v2 v2 und es gilt die Adiabatengleichung κ −1 p = 2 p1 ( c p − cv ) / c p p = 2 p1 ( κ −1) / κ (3-14) κ p 2 v1 = (oder p·vκ = konstant) p1 v 2 (3-15) (an Stelle von p·v = konstant für Isothermen). Entsprechend gibt es Unterschiede in den Arbeitsbeträgen, die ein System bei isothermer bzw. adiabatischer Prozessführung leisten kann (d.i. jeweils die Fläche unter den Isothermen bzw. Adiabaten): Für den isothermen Prozess ergibt sich –∆A = p∆V = (RT/V)∆V = RT ∫(dV/V) = RT ln(V2/V1) (3-16). Im adiabatischen Fall (∆Q = 0), bei welchem sich die Temperatur ändert, ist es einfacher, von dem auftretenden Temperaturunterschied auszugehen und den ersten Hauptsatz anzuwenden. dU = dA + dQ; 3.2 –∆A = ∆Q = Cv ∆T (3-1, 3-8). Zweiter Hauptsatz und Entropie 3.2.1 Irreversible Prozesse Es wird ein zweiter Hauptsatz der Thermodynamik benötigt, weil der erste eine Reihe von Beobachtungen nicht erfasst: 4 2 ln x 2 ∫ dx / x = ln x 2 − ln x1 = ln x1 1 25 1. Wärme wird stets vom wärmeren (1) auf den kälteren Körper (2) übertragen. Nach dem ersten Hauptsatz gilt dq1 + dq2 = 0, wobei die Erfahrung sagt, dass stets dq1 negativ ist. Der erste Hauptsatz würde jedoch auch das umgekehrte, also die spontane Wärmeübertragung vom kälteren auf den wärmeren Körper zulassen. 2. Die Expansion eines idealen Gases ins Vakuum erfolgt spontan und ohne Wärmeumsatz. Der Prozess könnte nach dem ersten Hauptsatz umkehrbar sein. 3. Die Energie einer Stahlkugel, die man aus einer gewissen Höhe in einen Sandhaufen fallen lässt, wird im Sand dissipiert. Die Umkehrung, also die Konzentration von Energie derart, dass eine Kugel aus einem Sandhaufen hochgeworfen wird, würde nicht im Widerspruch zum ersten Hauptsatz sein, wird aber nicht beobachtet. 1. bis 3. sind typische irreversible Prozesse, die nach dem ersten Hauptsatz durchaus umkehrbar sein könnten. Insbesondere wird die Richtung von Prozessen nicht berücksichtigt. 4. Auch spontan und freiwillig ablaufende chemische Reaktionen gehören den irreversiblen Prozessen (im thermodynamischen Sinn). Eine wichtige Beobachtung ist in diesem Zusammenhang zu nennen: Chemische Reaktionen laufen nicht nur dann freiwillig ab, wenn (wie bei Verbrennungen) Energie in Form von Wärme frei wird. Vor allem Reaktionen, bei denen große Mengen gasförmiger Produkte aus Flüssigkeiten oder Festkörpern entstehen, laufen auch dann spontan ab, wenn offensichtlich Wärmeenergie der Umgebung entzogen wird, wie an einer entsprechenden Abkühlung dieser Umgebung zu erkennen ist. Zwei Beispiele hierfür: 6 SOCl2 (flüssig) + CoCl2·6H2O (fest) → 12 HCl (gasf.) + 6 SO2 (gasf.) + CoCl2 (fest) CaF2 (fest) + H2SO4 (flüssig) → CaSO4 (fest) + 2 HF (gasf.) Offenbar gibt es sowohl exotherme Reaktionen, bei denen Wärme frei wird, als auch endotherme, bei denen Wärme verbraucht wird. Es stellt sich deshalb die Frage nach der Triebkraft von Reaktionen, die offensichtlich nicht allein mit dem Freiwerden von Wärme erklärbar ist (s. 3.2.4). 3.2.2 Entropie Es ist unmittelbar einleuchtend, dass beim Übergang vom Feststoff zur Flüssigkeit und weiter zum Gas die Unordnung vermehrt wird. Betrachtet man die oben genannten endothermen Reaktionen daraufhin, so stellt man fest, dass aus einer Flüssigkeit (SOCl2, H2SO4) und/oder einem Feststoff (CoCl2·6H2O; CaF2) große Mengen Gas entstehen (HCl, SO2; HF) und dass entsprechend die Unordnung drastisch größer wird. Diese Unordnungsvermehrung kommt daher neben dem Freiwerden von Wärme als Triebkraft für eine chemische Reaktion in Betracht. Ein Maß für die Unordnung ist die Entropie s, die mit der Unordnung ansteigt. Sie wird in J/K angegeben (bzw. als molare Entropie S in J mol–1K–1 ), und es wird später zu zeigen sein, dass es sich um eine Zustandsgröße handelt. Ein starres Festkörpergitter bei 0 K hat die Entropie 0. Die folgende Tabelle gibt einige molare Standard-Entropien S⊝ an. Ein Standardzustand liegt in der Thermodynamik bei p = 1 bar vor5 vor und wird meist für T = 298 K und n = 1 mol für die unter diesen Bedingungen stabile Modifikation (reine Phase) tabelliert. Substanz S⊝/J mol–1 K–1 5 H2O (gasf.) 189,1 H2O (fl) 70,1 C CO2 CO O2 CaCO3 CaO H2 5,7 214,1 198,1 206,3 92,9 39,8 130,7 In Lehrbüchern und Tabellenwerken wird manchmal p⊝ = 1 atm = 1,013 bar gesetzt, was zu kleinen Abweichungen in Tabellenwerten führt. 26 3.2.3 Reaktionsentropie und Reaktionsenthalpie Mit den Daten der letzten Tabelle lassen sich molare Standard-Reaktionsentropien ∆RS⊝ oft (auch anders symbolisiert, z.B. ∆RSm⊝) ausrechnen, indem die Summe der Standardentropien der Edukte von der der Produkte abgezogen wird [∆RS⊝ = Beispiele: 1.) ∆R S⊝ Σ ν i S⊝ (i) (mit Vorzeichen)]. C + O2 → CO2 = S ⊝ (CO2) – S⊝ (C) – S⊝(O2) = (214,1 – 206,3 – 5,7 = + 2,1) J mol–1 K–1 2.) H2 + 1/2 O2 → H2O (fl) ∆RS⊝= S⊝(H2O) – S⊝ (H2) – 1/2 S⊝ (O2) = (70,1 – 130,7 – 1/2 · 206,3 = –163,75) J mol–1 K–1. Im zweiten Beispiel ist die Reaktionsentropie negativ, jedoch gibt es einen deutlichen Wärmebzw. Enthalpiegewinn, wie bei Verbrennungen üblich. Die Triebkraft für eine Reaktion setzt sich also aus zwei Komponenten zusammen, einem Enthalpieanteil ∆RH⊝, der meistens der gewichtigere ist und in gleicher Weise berechnet werden kann wie die StandardReaktionsentropie, und einem Entropieanteil ∆RS⊝. Hinter dem Verfahren zur Berechnung von ∆RS⊝ oder ∆RH⊝ steckt der HESSsche Satz, eine frühe Formulierung der Zustandsfunktion und ein Spezialfall des 1. Hauptsatzes. Dieser Satz besagt, dass unabhängig vom Reaktionsweg in allen Fällen die gleiche Reaktionsenthalpie gemessen wird: A + B → C (Weg 1); A+B → D → C (Weg 2); ∆H⊝(Weg 1) = ∆H⊝(Weg 2) Dies versetzt uns in die Lage, Reaktionsenthalpien (und -entropien) auch für (ggf. hypothetische) unbekannte Reaktionen zu berechnen, wenn wir einen Reaktionsumweg über Reaktionen, deren Reaktionsenthalpien bekannt sind, konstruieren. Es wird z.B. die Reaktionsenthalpie für die Reaktion 2 C (fest) + O2 → 2 CO gesucht (∆RH⊝(I) = ?). Diese Reaktion lässt sich so nicht durchführen, sie führt auch bei Sauerstoffunterschuss immer zu einem Gemisch von CO und CO2. Bekannt sind folgende StandardReaktionsenthalpien, die in diesem Falle gleichzeitig Verbrennungsenthalpien ∆VH⊝ sind und für zahlreiche Reaktionen tabelliert vorliegen: C (fest) + O2 → CO2 ; ∆RH⊝ (II) = –393,5 kJ mol–1 2 CO + O2 → 2 CO2; ∆RH⊝(III) = –565,3 kJ FU–1 (s.u.) Es gibt jetzt zwei Wege, die zum CO2 führen: Weg 1: 2 C (fest) + 2 O2 → 2 CO2 ; (Hypothetischer) Weg 2: Folglich 2 ∆RH⊝(II) 2 C (fest) + O2 → 2 CO; ∆RH⊝(I) 2 CO + O2 → 2 CO2; ∆RH⊝(III) ∆RH⊝(I)+∆RH⊝(III) = 2∆RH⊝(II) ∆RH⊝(I) = 2 ∆RH⊝(II) – ∆RH⊝(III) = [2(–393,5) – (–565,3) = – 221,7] kJ FU–1. 27 Bei Berechnungen wie der obigen muss zur Einheit kJ mol–1 folgendes überlegt werden: Man kann die so angegebenen Messwerte als jeweils auf ein Mol O2 bezogen annehmen und hat dann keine Schwierigkeiten mit der Einheit. Da jedoch der Reaktionspartner oder das Produkt normalerweise mehr interessieren, müsste man berücksichtigen, dass von diesen ggf. 2 mol reagieren oder gebildet werden (Mol als Begriff groß, als Einheit klein geschrieben). Um dies Problem zu umgehen, gibt man das Ergebnis zweckmäßigerweise in kJ FU–1 an, wobei FU für einen molaren Formelumsatz steht. Bildungsenthalpien ∆BH⊝ Manchmal ist die Konstruktion eines Umwegs über Verbrennungsreaktionen etwas umständlich oder man findet die nötigen Daten nicht im Tabellenwerk. Deshalb werden öfter die sogenannten Bildungsenthalpien ∆BH⊝ herangezogen, die ebenfalls vielfach tabelliert vorliegen. Unter der Bildungsreaktion wird hier die (ggf. hypothetische) Bildung aus den Elementen des Periodensystems verstanden. Die Elemente kommen dabei so zur Reaktion, wie sie unter Standardbedingungen vorliegen, z. B. Br2 (fl), H2 (gasf), Fe (fest) usw. Elemente in dieser Form müssen nicht gebildet werden, deshalb ∆BH⊝ = 0. Zwei Beispiele für Bildungsreaktionen mit Tabellenwerten für die Standardbildungsenthalpie: bzw. C (fest) + 2 H2 → CH4; ∆BH⊝ = –79,6 kJ mol–1, 2 H2 + O2 → 2H2O (fl); ∆BH⊝ = –571,6 kJ/FU H2 + ½ O2 → H2O (fl); ∆BH⊝ = –285,8 kJ mol–1. (Im zweiten Beispiel ist die Bildungsenthalpie auch die Verbrennungsenthalpie.) Ein allgemeines Verfahren zur Berechnung von Reaktionsenthalpien (im Standard-Zustand) ergibt sich aus untenstehendem Schema. Daraus folgt: ∆RH⊝ = νC ∆BH⊝(C) +νD ∆BH⊝(D) –νA ∆BH⊝(A) – νB ∆BH⊝ (B) oder Σ νi ∆BH⊝ (i) (mit Vorzeichen) (3-17) Ganz analog sind auch Standardreaktionsentropien ∆RS⊝ aus tabellierten Standardbildungsentropien ∆BS⊝ auszurechnen. 3.2.4 Triebkraft von Reaktionen Die Zusammenfassung von Reaktionsenthalpie und Reaktionsentropie muss so erfolgen, dass jeweils das Freiwerden von Enthalpie und die Produktion von Entropie zur spontanen Reaktion führen. Die so definierte Größe ist ein Maß für die Triebkraft einer Reaktion. Sie heißt Freie (Standard-)Reaktionsenthalpie, basierend auf der Freien Enthalpie g (GIBBS-Energie) ∆G⊝ = ∆H⊝ – T ∆S⊝ GIBBS-HELMHOLTZ-Gleichung (3-18), 28 die mit zweimaliger Hilfe des HESSschen Satzes aus Enthalpie- und Entropiedaten zu berechnen ist. Ist ∆RG⊝ negativ, so läuft die Reaktion spontan ab. In machen Lehrbüchern findet man hierfür die Größe Affinität AR⊝ = –∆RG⊝, im allgemeinen als intensive Größe. Gleichung (318) gilt für konstanten Druck (in Tabellen meist Standarddruck 1bar) und konstante Temperatur (in Tabellen meist 298 K)6. Unter anderen Bedingungen ist die Berechnung der Triebkraft mit anderen Zustandsfunktionen zweckmäßiger: [∆H für p und S konstant; ∆U für V und S konstant; ∆F für V und T konstant („HELMHOLTZ-Energie“ oder „Freie Energie“7); vgl. Lehrbücher]. 3.2.5 CARNOT-Prozess und Definition der Entropie Während der erste Hauptsatz die innere Energie (und analoge thermodynamische Potenziale) definiert, drückt der zweite Hauptsatz die Rolle der Zustandsgröße Entropie bei irreversiblen Prozessen aus, erlaubt somit Aussagen zur Richtung von Prozessen. Es gibt je nach Problem zahlreiche verschiedene Formulierungen des 2. HS, von denen vier folgen: 1.) Bei irreversiblen Zustandsänderungen (Kugel fällt in Sandhaufen) nimmt die Entropie zu: s2 – s1 > 0 (3-19) (für isoliertes System). 2.) Wärme geht nicht spontan vom kälteren zum wärmeren Körper über. 3.) Es ist unmöglich, Wärme ohne Energieverlust in Arbeit zu verwandeln. 4.) Es gibt keine periodisch funktionierende Maschine, die nichts anderes tut, als Wärme in mechanische Arbeit zu verwandeln (Unmöglichkeit des perpetuum mobile zweiter Art). Die Aussage 4 soll quantitativ gezeigt werden. Dies wird i.a. an Hand des CARNOTschen8 Kreisprozesses vollzogen, der Zustandsänderungen einer hypothetischen Wärme-KraftMaschine, die abwechselnd reversible isotherme und adiabatische Schritte durchläuft, beschreibt (Andere Wärme-Kraft-Maschinen sind zwar der Praxis näher, jedoch wird in den entsprechenden Kreisprozessen die Zustandsgröße Entropie weniger deutlich). Eine alternative Formulierung des 2. Hauptsatzes nach CARATHEODORY9 erfordert mehr Mathematik. Die CARNOT-Maschine besteht aus einem idealen Gas in einem Zylinder mit beweglichem Kolben, der sich entweder in einem Wärmebad (für isotherme Prozesse) befindet oder mit einer Isolierung (für adiabatische Prozesse) versehen ist. Nebenstehend ist der CARNOT-Prozess in zwei verschiedenen Diagrammen dargestellt: Die (reversibel10 durchzuführenden) Schritte I bis IV sind: Schritt I: Isotherme Expansion eines (Mols eines) idealen Gases bei T = T1 von V1 nach V2; dabei erfolgt die Entnahme von Wärme ∆Qrev(I) aus dem Wärmebad und es wird Volumenarbeit ∆Arev(I) geleistet. 6 Ein Verfahren, um ∆RG⊝ auf andere Temperaturen (z.B. Siedetemperatur eines Reaktionsgemisches) umzurechnen, findet sich in Kap. 6.3.3. 7 Achtung: Im Englischen wird g als “free energy” oder „GIBBS-function“ und f als „work function“ (oft mit a symbolisiert) bezeichnet, nicht verwechseln! 8 Nicolas Léonard Sadi Carnot (1796-1832) 9 H. Margeneau und G. M. Murphy: The Mathematics of Physics and Chemistry; M. Born, Phys. Z. 22 (1921) 218, 249, 282 10 vgl. Kap. 3.2.7 29 Beim diesem Prozess ändert sich die innere Energie des idealen Gases nicht ∆U = ∆Q + ∆A = ∆Q + p∆V = 0 (vgl. Kap. 3.1).Es folgt ∆Q = ∆Qrev(I) = – ∆Arev(I) = RT1 ∫(dV/V) = RT1 ln(V2/V1) (3-20) Schritt II: Adiabatische Expansion von V2 auf V3; dazu wird das Wärmereservoir abgekoppelt und die Isolation angebracht. Die Folge ist eine Temperaturänderung von T1 nach T2. Es gilt ∆Qrev(II) = 0, so dass ∆U = ∆Arev (II) = Cv (T2–T1) (3-21) Schritt III: Isotherme Kompression; man muss dazu das Wärmebad wieder anschließen bei T2 und dann das Gas von V3 nach V4 komprimieren. Dabei ist wieder ∆U = 0, so dass ∆Qrev(III) = – ∆Arev(III) = RT2 ∫dV/V = – RT2 ln(V4/V3) (3-22) Schritt IV: Adiabatische Kompression; es sind wieder das Abkoppeln des Wärmebades und die Schaffung adiabatischer Bedingungen nötig. Sodann wird komprimiert, bis der Ausgangszustand erreicht ist. Es ist wieder ∆Qrev(IV) = 0, so dass ∆U = ∆Arev(IV) = Cv (T1 – T2) (3-23) In die Bilanz der Arbeit im CARNOTschen Kreisprozess gehen nur die isothermen Prozesse ein, da sich die Beiträge der adiabatischen aufheben. Es ergibt sich (als umschlossene Fläche im pV-Diagramm) ∆Ages = ∆Aisoth. = – R(T1 – T2) ln(V2/V1) (3-24), weil wegen der Adiabatengleichung (3-14,3-15) V2/V1 = V4/V3. Und da ∆Uges = 0, muss gelten: ∆Qrev, ges = – ∆Ages (3-25). Es zeigt sich, dass im Schritt III „nutzlos“ Wärme an das Reservoir abgegeben wurde. Deshalb kann für den CARNOTprozess ein Wirkungsgrad η= ∆Ages (3-26) ∆Qrev ( I) definiert werden,11 der zu berechnen ist nach η= ( ) ∆Q = RT ln ( ) R (T1 − T2 ) ln 1 V2 V1 V2 V1 rev (I) + ∆Qrev (III) T1 − T2 = ∆Qrev (I) T1 (3-27). Die Betrachtung des CARNOTschen Kreisprozesses liefert einen Zugang zur Zustandsfunktion Entropie: Wie oben (Kap. 3.1) erwähnt, ist q keine Zustandsfunktion. Betrachtet man jedoch die jeweiligen Quotienten ∆Q/T im CARNOTschen Kreisprozess, so stellt man fest, dass 11 Eine Möglichkeit; insbesondere technische Wirkungsgrade können auch anders definiert sein. 30 ∆Qrev (I) Q (III) V V ; = R ln 2 = − rev = − R ln 4 , V1 T2 V3 T1 d.h. dieser Quotient verhält sich wie eine Zustandsfunktion und diese definiert die Entropie: Σ(∆Qrev/T) = 0 dq rev ∆qrev ≡ ds ; = ∆s (für geschlossenes System und T konstant) (3-28) T T Für ein isoliertes System gilt ∆s ≥ 0 und zwar ∆s > 0 für irreversible und ∆s = 0 für reversible Prozesse (s. 3.2.7). Für offene Systeme müssen wieder Si·dni-Terme ergänzt werden. Obige Einführung der etwas abstrakten Zustandsfunktion Entropie, die mit "Maß für Unordnung" zwar anschaulich jedoch keineswegs umfassend rationalisiert werden kann, ist sehr gerafft, so dass sich die Lektüre einschlägiger Kapitel am besten in verschiedenen Lehrbüchern empfiehlt. 3.2.6 Ermittlung von Entropien und Enthalpien Offen geblieben war zu Beginn des Abschnitts 3.2, wie Entropien, insbesondere StandardEntropien für einzelne Stoffe bestimmt werden. In Kenntnis der thermodynamischen Definition (3-28) ist dies nicht mehr schwer. Bei konstantem Druck ergibt sich die Entropie nach Ermittlung der Wärmekapazität cp (bzw. der spezifischen Wärme Cp) eines Stoffes (dq in 3-28 ist dann hier cpdT) zu s= Tc ∫T p T dT = ∫ c p dln T 0 (3-29) 0 Bei Phasenumwandlungen treten Unstetigkeiten in cp-Temperatur- bzw. Entropie-TemperaturKurven auf, da sich die Entropie sprunghaft ändert (vgl. Bildung gasförmiger Produkte in Kapitel 3.2.1). Die sprunghaften Änderungen entsprechen (Phasen-)Umwandlungsentropien ∆sU und sind mit den entsprechenden Umwandlungsenthalpien (∆hU, Schmelzwärme, Verdampfungswärme) verknüpft: ∆Us = ∆Uh TU (3-30), so dass für den Entropieinhalt einer Substanz, die bei gegebener Temperatur T bereits eine Phasenumwandlung hinter sich hat, gilt TU s= ∫ 0 ∆ h c p dln T + U + TU T ∫ c ′p d ln T TU (3-31). Zur Illustration soll das Beispiel des Stickstoffs N2 dienen (s. Abbildung), der in zwei festen Modifikationen sowie flüssig und gasförmig existieren kann. Es treten deutliche Unterschiede in Cp für die verschiedenen Aggregatzustände zu Tage. Ganz analog ist die Enthalpie zu bestimmen: 31 TU ∆h = ∫ 0 T c p dT + ∆ U h + ∫ c ′p dT (3-32). TU Um ein Beispiel zu geben, soll die Frage behandelt werden, ob ein Mensch beim Verzehr von Speiseeis Energie gewinnt oder verbraucht. Die spezifische Wärme von Eis liegt bei 37,7, die von Wasser bei 75,5 J mol–1 K–1 (im betrachteten Temperaturbereich). Wir betrachten als Ausgangszustand eine 100 g - Portion Eis bei -10 °C entsprechend 5,55 mol. Diese Portion wird beim Verzehr auf 37 °C erwärmt. Dafür sind zunächst bis zum Schmelzpunkt 273 JmolK ∫ C p ndT = 37,7·5,55·10 molK = 2,09 kJ 263 aufzuwenden. Sodann wird die Schmelzenthalpie benötigt. Sie beträgt ∆UH = 6,008 kJ/mol, also [6,008 · 5,55 = 33,34] kJ für 100 g. Die weitere Erwärmung bis zur Körpertemperatur erfordert 310 molJK n ∫ C p dT = 5,55 ⋅ 75,5 ⋅ 37 = 15,50 kJ . molK 273 In der Summe müssen somit für Schmelzen und Erwärmen des Eises etwa 51 kJ pro 100 g aufgebracht werden. (Das Verfahren erlaubt generell die Ermittlung von Enthalpie- oder Entropiedifferenzen oder z.B. die Bestimmung des Enthalpie- oder Entropiegehalts einer Substanz, die nicht bei der in Tabellenwerken gegebenen Temperatur vorliegt.) Auf der anderen Seite enthält Speiseeis i.a. Zucker, woraus der Körper durch "Verbrennung" Energie gewinnen kann: C6H12O6 + 6 O2 → 6 CO2 + 6 H2O; ∆VH = -2800 kJ/FU Die molare Masse des Zuckers ist 180 g/mol; daraus folgt, dass die Verbrennung von 1 g Zucker 15,6 kJ Energie liefert. Das betrachtete Speiseeis kann demnach nur nahrhaft sein, wenn es mehr als 3,2 g Zucker pro 100 g Eis enthält. (Der Wert hängt natürlich von der Anfangstemperatur und weiteren verdaubaren Inhaltsstoffen des Eises ab, insbesondere bei Portionen mit Sahne.) Neben den Reaktionsenthalpien (die ggf. genauer, z.B. als Neutralisationsenthalpie, Verbrennungsenthalpie, usw. bezeichnet werden) und den bei Phasenumwandlungen auftretenden Umwandlungsenthalpien kennt man in der Chemie weitere Enthalpien12, die nach den damit verbundenen Prozessen als Verdünnungsenthalpie, Mischungs- bzw. Lösungsenthalpie, u.s.w. zu bezeichnen sind, und die positive oder negative Werte annehmen können. Gleiches gilt für die betreffenden Entropien. Ursache ist wie immer jeweils der Unterschied dieser Größen im Ausgangs- und im Endzustand. Es muss z.B. beim Auflösen eines Feststoffs in einem Lösemittel einerseits die Gitterenthalpie des Feststoffs aufgebracht werden, andererseits gewinnt das System beim Auflösen (Solvatisieren) der Moleküle oder Ionen die Solvatationsenthalpie. Die Bilanz bestimmt dann, ob ein Stoff in einem bestimmten Lösemittel gut oder schlecht löslich ist und ob dabei Wärme frei oder verbraucht wird. 12 In deutschsprachigen Lehrbüchern heißt es oft ...-wärme statt ...-enthalpie. - Man erkennt hier noch mal den praktischen Nutzen der Einführung der Enthalpie h (Gl. (3-6) anstelle der inneren Energie u), da alle hier genannten Prozesse normalerweise bei konstantem Druck (Atmosphärendruck) durchgeführt werden. 32 3.2.7 Entropieänderung bei nicht-cyclischen Prozessen Unter den Energieformen nimmt die Wärmeenergie q eine Sonderstellung ein (welche letztlich der 2. Hauptsatz beschreibt). Kinetische und potentielle, mechanische und elektrische Energie können jeweils (reversibel) vollständig ineinander und in Wärme umgewandelt werden, nicht jedoch Wärmeenergie in mechanische Energie, usw. Als Beispiel mag wieder die aus bestimmter Höhe im Schwerefeld der Erde fallende Stahlkugel dienen, deren potentielle und kinetische Energie beim Aufschlag auf den Untergrund vollständig in Wärme umgewandelt wird., d.h. in eine heftige Molekülbewegung, die durch Stöße rasch abgeleitet und insgesamt dissipiert wird. Der umgekehrte Prozess – Ausrichtung und Konzentration der molekularen Wärmebewegung an einem bestimmten Punkt, von dem aus die Kugel in die Höhe gehoben wird, ist äußerst unwahrscheinlich, würde allerdings dem 1. Hauptsatz nicht widersprechen. Spontane Prozesse wie Wärmeleitung, Diffusion, usw. verlaufen nach Beobachtung ausschließlich in der Richtung zunehmender Unordnung. Sie sind im thermodynamischen Sinn irreversibel, d.h. sie können nur unter Energieaufwand umgekehrt werden. Die Energie dazu muss der Umgebung des Systems entnommen werden. System und Umgebung zusammen bilden ein isoliertes Gesamtsystem, für das sowohl du = 0 als auch dq = 0 gilt. Jede reversible (umkehrbare) Wärmeaufnahme des von uns betrachteten geschlossenen (inneren) Systems muss daher von einer Wärmeabgabe der Umgebung (äußeres System) begleitet sein. Hier muss zwischen reversiblen und irreversiblen Prozessen unterschieden werden. Reversibel ist ein Prozess dann, wenn er jederzeit ohne zusätzlichen Energieaufwand umkehrbar ist und dabei keine „Spuren“ (Verformungen, u.ä.) hinterlässt. In der Praxis ist das nur näherungsweise erreichbar, indem man Prozesse langsam und nur in sehr kleinen Schritten durchführt (oder durchrechnet). Dagegen sind plötzliche Zustandsänderungen i.a. irreversibel, ebenso chemische Reaktionen. Beispiele: 1) Reversible Prozesse a) Reversible isotherme Ausdehnung eines idealen Gases von v1 auf v2. Wärmeaufnahme im inneren System ∆qi = nRT ln(v2/v1) = –∆a; die Umgebung verliert den gleichen Betrag, d.h. ∆qa = –∆qi, und die Entropieänderung des Gesamtsystems ist null: ∆s = ∆si + ∆sa = (∆qi + ∆qa)/T = 0. (3-33) b) Reversibler Phasenübergang (Schmelzen eines Festkörpers, Schmelzwärme ∆hfest-fl). Beim Schmelzvorgang erhöht sich die Entropie um ∆si = ∆hfest-fl/Tfest-fl, die Wärmemenge ∆hfest-fl wird dabei der Umgebung entzogen, deshalb ist ∆sa = –∆si und ∆s = 0. c) Im Grenzfall können wir ein System auch reversibel erwärmen, indem wir ständig für gleiche Temperatur im inneren und äußeren System sorgen. Dann gilt wieder ∆s = 0 und T2 T dq 2 cv dT ∆si = − ∆sa = ∫ = . T T∫1 T T1 (3-34) In nicht zu großen ∆T ist cv als unabhängig von T anzusehen (s. Abb. S. 30). Dann folgt ∆si = cv ln(T2/T1). Entsprechendes gilt für die reversible Erwärmung bei konstantem Druck. Obige Beispiele zeigen, dass die Entropie konstant bleibt, wenn im Gesamtsystem nur reversible Prozesse ablaufen. Für irreversible Prozesse gilt das nicht. Beipiele: 2) Irreversible Prozesse a) Plötzliche Expansion eines idealen Gases in ein Vakuum von v1 auf v2. In diesem Extremfall sind sowohl ∆u = 0 als auch ∆a = 0 (wegen p = 0), d.h. ∆qa = 0 und daher ist auch ∆sa = 0. Da die Entropie eine Zustandsfunktion ist, kann man die Entropievermehrung des Gases bzw. Sys- 33 tems aus einem reversiblen (Ersatz-)Prozess entnehmen, der zum gleichen Endzustand führt: ∆si = ∆qi/T = nR ln (v2/v1). ∆si ist jetzt > 0, daher ist auch ∆s =∆si + ∆sa = ∆si > 0. In realen Fällen ist p2 > 0 (aber < p1) und für die bei der irreversiblen Expansion abgegebene Arbeit (s. Abb.)13 gilt wegen ∆u = 0: –∆a(p2) = ∆qi(p2) < nRT ln (v2/v1). Die Zunahme der Entropie des inneren Systems ist nach wie vor ∆si , aber die mit der Umgebung ausgetauschte Wärmemenge ist nur ∆qa(p2) = –∆qi(p2). Infolgedessen ist ∆s = ∆si + ∆qa(p2)/T = nR ln (v2/v1) – ∆qi(p2)/T, so dass 0 ≤ ∆s < nR ln (v2/v1) (3-35), wobei das Gleichheitszeichen für den reversiblen Fall gilt. irreversibel reversibel b) Übertragung der Wärmemenge ∆q durch Wärmeleitung von einem Wärmereservoir bei der Temperatur T2 auf ein Reservoir bei niedrigerer Temperatur T1. Wenn ∆q hinreichend klein ist, bleiben die Temperaturen der beiden Reservoirs in diesem Prozess praktisch unverändert und die Entropieänderung ist insgesamt ∆s = –∆q/T2 + ∆q/T1 > 0, da T2 > T1 (3-36). c) Irreversibler Phasenübergang bei der Kristallisation von Wasser, das bei Normaldruck auf – 4 °C unterkühlt ist. ∆si kann wieder mit Hilfe eines reversiblen (Ersatz-) Prozesses ermittelt werden: 1 2 3 H2O(–4 °C) → H2O(0 °C) → Eis(0 °C) → Eis(–4 °C) Mit der Schmelzwärme von Wasser und den spezifischen Wärmen von Eis und Wasser ∆Hfest-fl = 6008 J/mol; Cp(Eis) = 37,7 J/mol, Cp(Wasser) = 75,5 J/mol (vgl. Abschnitt 3.2.6) erhält man ∆Si = ∆S1 + ∆S2 + ∆S3 = Cp(Wasser) ln (273/269) – ∆Hfest-fl/Tfest-fl – Cp(Eis) ln (273/269) = –21,4 J/(mol K). Da Wärme abgegeben wird, ist ∆Sa = ∆Hfest-fl/269 K = 22,3 J/(mol K), so dass insgesamt ∆S = ∆Si + ∆Sa = 0,9 J/(mol K) > 0 (3-37) Es zeigt sich, dass wie oben gesagt ∆s ≥ 0, wobei das Gleichheitszeichen für reversible, das Größerzeichen für irreversible Prozesse im Gesamtsystem gilt, welches als isoliertes System anzusehen ist. 13 aus Wedlers Lehrbuch