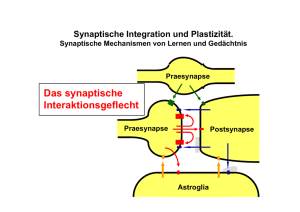
Die Freisetzung von BDNF an glutamatergen Synapsen und die
Werbung

Zusammenfassung 112 6 Zusammenfassung 6.1 Synaptische BDNF-Freisetzung nach Depolarisation glutamaterger Hippokampusneurone Die aktivitätsabhängige Modulation der synaptischen Transmission wird als das Korrelat für Lernen und Gedächtnis im Säuger-ZNS angesehen. Solche Effekte werden z.B. bei der Langzeitpotenzierung (LTP) beobachtet. Der allgemein anerkannte Startpunkt für LTP an vielen glutamatergen Synapsen beruht auf der Induktion der LTP durch Aktivierung der postsynaptischen NMDA-Rezeptoren. Für präsynaptische Veränderungen nach Induktion der LTP wird eine aktivitätsabhängige Freisetzung des brain-derived neurotrophic factor (BDNF) mit nachfolgender Wirkung auf die präsynaptische Zelle verantwortlich gemacht. Fungiert BDNF als retrograder Botenstoff, so würde es nach Induktion von LTP durch den NMDARezeptor postsynaptisch freigesetzt, um seine präsynaptische Wirkung zu entfalten. Zum Beweis der Hypothese von BDNF als retrogradem Botenstoff bei LTP fehlt jedoch der direkte Nachweis, dass BDNF aktivitätsabhängig an glutamatergen Synapsen freigesetzt wird. Dieser Nachweis sollte in der vorliegenden Arbeit erbracht werden. In den Experimenten wurde ein Fusionsprodukt aus BDNF und dem green fluorescent protein GFP verwendet (Haubensak et al., 1998). In der vorliegenden Arbeit konnte gezeigt werden, dass BDNF-GFP an glutamatergen, aber nicht an GABAergen Synapsen angereichert wird. Ein Hinweis auf ein präferentiell postsynaptisches Zusammenfassung 113 Vorkommen ist die Tatsache, dass 96 % des in Zellfortsätzen lokalisierten BDNF-GFP in Dendriten vorliegt. In lebenden Neuronen gelang die Zuordnung der BDNF-GFP-Cluster zu Synapsen durch eine Färbung mit dem Fluoreszenzfarbstoff FM 4-64. Mit zeitaufgelöster Videomikroskopie konnte gezeigt werden, dass die synaptischen BDNF-GFPAnsammlungen durch Depolarisation mit Kalium an Fluoreszenzintensität ver- lieren. Diese Abnahme war abhängig vom Vorhandensein extrazellulären Kalziums. Auch die Präinkubation der Neurone mit Tetanustoxin verhinderte die Fluoreszenzabnahme. Beide Ergebnisse sind Anzeichen für eine regulierte Exozytose aus sekretorischen Vesikeln. Zusätzlich gelang der Nachweis von freigesetztem BDNFGFP im Überstand stimulierter neuronaler Kulturen. Damit war die Ausschüttung von BDNF-GFP nach Ca2+-Influx die einzig überzeugende Erklärung für die Abnahme der Fluoreszenz. Zusammen mit dem Befund der präferentiell dendritischen Lokalisation ist damit das Modell, dass BDNF als retrograder Botenstoff postsynaptisch und aktivitätsabhängig an glutamatergen Synapsen freigesetzt wird, höchst plausibel. In über diese Arbeit hinausgehenden Experimenten gelang es, mittels hochfrequenter elektrischer Stimulation ebenfalls BDNF-GFP freizusetzen. Die in diesen Versuchen verwendeten Stimulationsparameter entsprachen denen, die zur Induktion von LTP verwendet werden. Es gelang der Nachweis, dass für die Ausschüttung von BDNFGFP die Depolarisation der postsynaptischen Membran durch die ionotropen Glutamatrezeptoren notwendig ist. Der direkte Nachweis einer postsynaptischen Freisetzung von BDNF ist damit zum ersten Mal glaubhaft gelungen (Hartmann et al., 2001). Zusammenfassung 114 6.2 Der trunkierte Tyrosinkinaserezeptor TrkB.T1 und seine Funktion in der Entwicklung dendritischer Filopodien hippokampaler Neurone Neben vielen anderen Faktoren spielen auch die Mitglieder der Neurotrophinfamilie eine Rolle in der Bildung und Modulation neuronaler Verschaltungen. Das Neurotrophin BDNF ist sowohl auf der Ebene der Synapsenentstehung und -modulation als auch auf der Ebene der Regulation der Neuritenbaumkomplexität involviert. Der BDNF-Rezeptor TrkB wird in verschiedenen Spleißvarianten exprimiert. Besonders die Funktionen der kinasedefizienten Isoform TrkB.T1 sind noch wenig erforscht. Neuere Daten weisen dem TrkB.T1 eine Rolle in der Entstehung von Filopodien nach Expression in sekundären Zelllinien zu (Hartmann, 1999; Haapasalo et al., 1999). Da Filopodien für die Vorstufen von Dendriten oder von spines (dem postsynaptischen Anteil glutamaterger Synapsen) gehalten werden, sollte dieser filopodieninduzierende Effekt des TrkB.T1 in Neuronen untersucht werden. Es gelang, durch die Expression von TrkB.T1 in Neuronen die Bildung solcher aktinhaltiger Fortsätze zu induzieren. Diese konnten durch ihr ausschließlich dendritisches Vorkommen als Filopodien eingeordnet werden. Die Expressionsmuster der TrkB-Rezeptorformen selbst zeigten keine Präferenz für eine axonale oder somatodendritische Lokalisation. Die TrkB.T1-exprimierende Hippokampusneurone wiesen gegenüber TrkB.FL- und GFP-Kontrollen eine statistisch signifikant erhöhte Filopodiendichte auf. Es konnte gezeigt werden, dass die Steigerung der Filopodiendichte kein verstärktes Dendritenwachstum und keine Erhöhung der Synapsendichte zur Folge hatte. Letzteres Ergebnis ist jedoch aufgrund des zu dichten Neuronenbesatzes der verwendeten Kulturen nur eingeschränkt aussagekräftig. Die Expression einer Deletionsmutante des TrkB.T1, der die intrazelluläre Domäne des trunkierten Rezeptors fehlte, hatte den gleichen Effekt wie der Wildtyp-TrkB.T1. Zusammenfassung 115 Folglich ist der intrazelluläre Anteil des TrkB.T1 nicht an der Vermittlung der Filopodienerhöhung beteiligt, und es musste nach einem für diesen Zweck geeigneten Interaktionspartner der extrazellulären Domäne gesucht werden. Ein geeigneter Kandidat für diese Funktion war die kinasepositive TrkB-Isoform (TrkB.FL). Die Koexpression von TrkB.FL und TrkB.T1 verringerte die Filopodiendichte jedoch auf basale Werte. Dieser dominant inhibitorische Effekt des TrkB.FL beruhte nicht auf seiner Tyrosinkinaseaktivität. Vermutlich interferiert die Interaktion zwischen TrkB.T1 und TrkB.FL mit der Bindung des TrkB.T1 an ein weiteres Membranprotein, dass die Signaltransduktion der Filopodienentstehung vermittelt. Der Nachweis, dass der niedrigaffine Neurotrophinrezeptor p75 hier eine Rolle spielt, gelang durch Experimente mit Inhibitoren der p75-Signaltransduktion. Die Verwendung von Spiroepoxid, einem Inhibitor des Ceramid-Signalweges, führte zu einer Reduktion der Erhöhung der Filopodiendichte in TrkB.T1-transfizierten Neuronen auf basale Werte. Ein ähnlicher Effekt stellte sich ein, wenn ein gegen den p75-Rezeptor gerichteter Antikörper (MC192) verwendet wurde. Dass die Aktivierung des p75-Rezeptors notwendig aber nicht hinreichend für die Erhöhung der Filopodiendichte ist, zeigte ein Experiment, in dem das Neurotrophin NGF zu kontrolltransfizierten Zellen gegeben wurde. Hierdurch wird zwar p75 aktiviert, aber der Effekt des TrkB.T1 konnte nicht nachgeahmt werden. Vermutlich ist der Effekt der Filopodienentstehung abhängig von der p75Stimulationsintensität oder von der Einbeziehung weiterer Signalwege, die in geeigneter Weise nur vom TrkB-T1-Rezeptor aktiviert werden. Die extrazelluläre Domäne des TrkB.T1 stellt wahrscheinlich einen Liganden für den p75 Rezeptor dar. Weitere Untersuchungen, insbesondere die Unterschiede zur Wirkung des p75Liganden NGF, könnten zusätzliche Erkenntnisse zu den überraschenden Effekten der TrkB.T1-Expression liefern.