Periphere Neuropathien
Werbung

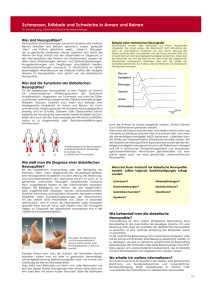
Medizinisch Genetisches Zentrum FACHINFORMATION ZU DEN ERBLICHEN, PERIPHEREN NEUROPATHIEN Die Charcot-Marie-Toothsche Erkrankung (CMT), auch hereditäre motorische C P und sensible Neuropathie (HMSN) genannt, stellt mit einer Prävalenz von 10– E A 40 / 100 000 die häufigste, erbliche periphere Neuropathie dar. Diese neurogenen Muskelatrophien sind sowohl klinisch als auch genetisch heteroP gen. Dyck und Lambert führten 1968 eine Klassifizierung der peri pherenMNeuD ropathien auf der Basis von genetischen, elektrophysiologischen und neuroT R S S pathologischen Kriterien ein, die bis heute Anwendung findet. P C Die HMSN Typ I (CMT1) ist durch deutlich reduzierte motorische und sensorisporadisch, mtDNA Deletion / e sche Nervenleitgeschwindigkeiten (NLG) und Zeichen einer De- und Remyelinisierung in N. suralis-Biopsien gekennzeichnet. Diese ErkrankungPwird zumeist autosomal dominant (AD) vererbt, aber auch X-gekoppelte und autosomal-rezessive (AR) Vererbung sowie Patienten mit sporadischen Formen bzw. ohne eindeutig erkennbaren Erbgang wurden beschrieben. Am häufigsten wird eine 1,4 Mb Duplikation, die das PMP22-Gen enthält, beobachtet M P (CMT1A-Duplikation). C C S Verwandte erbliche Neuropathien N E Die HMSN kann klinisch zumeist von den hereditären S motorischen Neuropathien (HMN) und den hereditären sensorischen undB autonomen Neuropathien (HSAN) abgegrenzt werden. Mittlerweile sind über M 40 Gene und Loci bekannt, die mit diesen Neuropathien assoziiert sind. S In der Abbildung auf der Rückseite dieser InformationMist eine Einteilung dargestellt, die die klinisch-genetische Zuordnung veranschaulicht. Wichtig für eine genetische Diagnosefindung ist neben der Zuordnung zu HMSN, HMN oder HSAN auch der Erbgang und die ethnischeT Zugehörigkeit der S Patienten. Bei einer Vererbung vom Vater zum Sohn ist beispielsweise eine XT chromosomale Erkrankung nahezu ausgeschlossen. E M Symptomatik C S S Phänotyp B Obwohl der in einem weiten Bereich auchE intrafamiliär variieren S kann, gibt es einige typische Gründe für Patienten, einen Arzt für eine DiaM gnosestellung zu konsultieren. Am meisten verbreitet sind Gangbeschwerden Eine HMSN Typ II (CMT2) ist durch verminderte Amplituden sowie normale und eine langsam progrediente Schwäche der Beine und/oder Hände, aber tRNALeu (m.3243A>G) andere t oder leicht reduzierte motorische und sensorische NLGs charakterisiert. Am P auch rekurrierende Lähmungen oder Fuß-Deformitäten sind Gründe für eine häufigsten werden Mutationen im Mitofusin 2 (MFN2)-Gen beobachtet, aber klinische Konsultation. Generell können das motorische und/oder das sensoritRNALywurden mit andere t auch Variationen in GJB1(Cx32) und MPZ sowie weiteren Genen sche System an einer Neuropathie beteiligt sein. Eine Untersuchung mittels dieser Neuropathie assoziiert. mtDNA Deletionen / Punktmuta Nervenleitgeschwindigkeit und Elektromyographie resultiert fast immer in auffälligen Befunden. Die Muskeleigenreflexe sind in der Regel abgeschwächt Die HMSN III oder Dejerine-Sottas-Syndrom (DSS) ist eine sehr schwer verlauoder erloschen. Eine N. suralis-Biopsie ist mitunter hilfreich, um die verschiefende Neuropathie mit extrem reduzierten NLGs und frühem Krankheitsbedenen Neuropathie-Typen zu differenzieren, z. B. um die chronische inflamginn. DSS-Patienten können Mutationen in PMP22, MPZ, EGR2 und weiteren E matorische demyelinisierende Polyneuropathy (CIDP) von einer HMSN/CMT Genen aufweisen. T M oder Subtypen der HMSN/CMT abzugrenzen. Die erblichen Neuropathien müssen sehr sorgfältig von den vielfältigen erworbenen (nicht-genetischen) NeuETFB E Patienten mit einer hereditären Neuropathie mit Neigung zu Druckläsionen ropathien, wie z. B. den immunvermittelten, toxischen und infektiösen Formen (hereditary neuropathy with liability to pressure palsies, HNPP) entwickeln unterschieden werden. gewöhnlich eine Mononeuropathie nach mildem Trauma. Einige Patienten zeiLebersche hereditäre optische Neuropath Eine molekulargenetische Analyse ist hier oft hilfreich und mit Blick auf gen eine generalisierte Neuropathie. HNPP wird autosomal dominant vererbt. m therapeutische Optionen bzw. Kontraindikationen, die weitere Diagnostik Am häufigsten wird eine Deletion reziprok zur CMT1A-Duplikation beobachm.14484T>C (ND6) N sowie die genetische Beratung von Bedeutung. tet. Autosomal dominante optische A Pearson-Syndrom: mtDNA Deleti Rhabdomyolyse, Belastungsintoleranz, CK-E C E Sensorische Ataxie, Neuropathie, D P P MGZ – Medizinisch Genetisches Zentrum, Bayerstraße 3– 5, 80335 München, Tel. 089 / 30 90 886-0, Fax 089 / 30 90 886-66, [email protected], www.mgz-muenchen.de KLINISCH-GENETISCHE ZUORDNUNG DER ERBLICHEN NEUROPATHIEN PERIPHERE NEUROPATHIE ELEKTROMYOGRAPHIE NERVENLEITGESCHWINDIGKEIT dHMN HSN/HSAN SPTLC1 SPTLC2 ATLASTIN1 WNK1/HSN2 FAM134B NTRK1 NGFB SCN9A HMSN/CMT HSPB8 GARS IGHMBP2 DCTN1 SETX BSCL2 N.medianus < 38 N.medianus > 38 Demyelinisierend: HMSN / CMT Typ 1 Intermediär dominant Cx32 MPZ DNM2 YARS ARHGEF10 PMP22 Dup. MPZ BSCL2 NEFL SIMPLE EGR2 SOX10 rezessiv SH3TC2 GDAP1 PRX MTMR2 SBF2 NDRG1 FGD4 FIG4 EGR2 CTDP1 X-gekoppelt Cx32 PRPS1 Medizinisch Genetisches Zentrum Axonal: HMSN /CMT Typ 2 dominant MFN2 RAB7A GARS HSPB1 NEFL HSPB8 TRPV4 AARS rezessiv LMNA SLC12A6 GAN MED25 X-gekoppelt Cx32